Drugs, Health Technologies, Health Systems
Reimbursement Review
Etrasimod (Velsipity)
Sponsor: Pfizer Canada
Therapeutic area: Ulcerative colitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
5-ASA
5-aminosalicyclic acid
AE
adverse event
AESI
adverse event of special interest
CD
Crohn disease
CI
confidence interval
CrI
credible interval
DIC
deviance information criterion
FAS
full analysis set
GI
gastrointestinal
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
ITC
indirect treatment comparison
JAK
Janus kinase
JAKi
Janus kinase inhibitor
LS
least squares
MID
minimal important difference
MMS
modified Mayo score
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
OLE
open-label extension
RCT
randomized controlled trial
S1P
sphingosine 1-phosphate
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TMS
total Mayo score
TNF
tumour necrosis factor
TNFi
tumour necrosis factor inhibitor
UC
ulcerative colitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Etrasimod (Velsipity), 2 mg film-coated tablet |
Sponsor | Pfizer Canada |
Indication | The treatment of adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment |
Reimbursement request | As per indication |
Health Canada approval status | Post NOC |
Health Canada review pathway | Standard review |
NOC date | January 8, 2024 |
Recommended dose | 2 mg once daily, orally |
NOC = Notice of Compliance; UC = ulcerative colitis.
Introduction
Ulcerative colitis (UC) is a chronic form of inflammatory bowel disease (IBD) that affects the mucosal layer of the large intestine. It almost invariably involves the rectum and frequently extends continuously into the proximal colon.1 UC is characterized by blood in the stool with mucus, frequent diarrhea, loss of appetite, and tenesmus (severe rectal cramp or spasm).2 Extraintestinal manifestations may also occur, such as arthritis.3 About 10% to 15% of patients with UC experience an aggressive course.4 Relapse is common, with the cumulative risk of relapse being 70% to 80% at 10 years.4 UC has a considerable impact on patients’ health-related quality of life (HRQoL)5,6 and their ability to perform their regular daily routines such as jobs or domestic chores,7-10 and it impacts their caregivers and family, workplace, and community.11 Although the risk of mortality from UC itself is low, the disease is associated with an increased risk of other complications (e.g., respiratory diseases, colorectal cancer, lymphoma, and skin cancer) that result in higher mortality compared with the general population.11 The prevalence for UC in 2023 in Canada was estimated to be 414 per 100,000.12 It is estimated that among the patients in Canada with UC, 32% to 46% have moderate disease and 13% to 14% have severe disease.13
The clinical expert consulted by CADTH pointed out that the treatment goals for patients with UC are to achieve rapid symptomatic relief and to induce and maintain clinical, serological, biomarker, and endoscopic remission in both the short and long-term. In patients with moderately to severely active UC, oral corticosteroids are typically the first-line therapy, but are used only for inducing remission due to their adverse effects.14,15 Thiopurines (e.g., azathioprine, 6-mercaptopurine), 5-aminosalicylic acid (5-ASA), antitumour necrosis factor (anti-TNF) therapy, or vedolizumab can be used to maintain remission.14,15 For patients for whom 5-ASAs, corticosteroids, or thiopurines are unable to induce or maintain remission or are not tolerated, advanced therapies are used.14,15 Of note, most Canadian drug plans require a patient with moderately to severely active UC to have experienced failure of steroid tapering with azathioprine or 6-mercaptopurine before being eligible for a biologic. As such, advanced therapies are typically not used for first-line maintenance of steroid-induced remission.15 Under circumstances where medical therapy fails, colectomy (which is associated with risks of complications and additional procedures) may be required.11 The clinical expert consulted by CADTH noted that early introduction of effective advanced therapy is important for patients’ benefit, particularly in avoiding repeated courses of corticosteroids, as recurrent use of corticosteroids to control UC symptoms is not ideal due to their multiple adverse effects. The clinical expert consulted by CADTH and the sponsor indicated there is limited robust evidence and thus no recent Canadian guidelines on the preferred sequencing (that is, which drug is optimally used first) for advanced therapies in UC.16
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of etrasimod (Velsipity) 2 mg tablet taken orally once daily in the treatment of adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Gastrointestinal (GI) Society and Crohn and Colitis Canada (CCC), provided input for this review. The GI Society’s input was informed by surveys conducted between 2015 and 2023 (N = 54 to 579), focus groups, and 1-to-1 interviews with patients with IBD. CCC’s input was compiled from 2 online surveys conducted in 2022. The first captured the experience of 354 patients with moderate to severe UC, and the second received responses from 4 patients with UC.
From the patient’s perspective, UC has a profound effect on daily life — physically, emotionally, and socially — at home, school, or in the workplace. Symptoms can be relentless, embarrassing, and scary. Sustained remission and/or treatment response is important. The concern of future flares — possibly worse than the last and occurring at unpredictable times — remains constant among patients with UC. Patients noted the most important aspects around UC management include having enough treatment options, having treatments that are well tolerated, and minimizing steroid use.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert noted that a significant portion of patients have disease that does not respond to the available advanced therapies, and some UC becomes refractory over time. The clinical expert indicated that multiple drug failures and ongoing progressive disease activity may lead to adverse consequences, including surgery to remove the entire colon. Moreover, there is a lack of available oral therapies, as most are delivered intravenously or subcutaneously.
The clinical expert indicated that a clear sequence of medications that is optimal for treating moderate to severe UC is not yet established. The clinical expert noted that in an outpatient context, etrasimod could be introduced early, in the course of 5-ASA failure, as it may induce remission and thus would not be reserved for patients for whom other drugs are contraindicated or other access limitations. The clinical expert noted that the evidence suggests the efficacy of etrasimod diminishes with more drug failures. Therefore, the clinical expert suggested that to optimize its efficacy, etrasimod should be considered for and administered to patients with UC earlier in their disease course.
The clinical expert indicated that patients with a confirmed pathologic or histologic diagnosis of moderate to severe UC are typically diagnosed by a gastroenterologist and, occasionally, by a surgeon in more rural parts of the country. Misdiagnosis is infrequent. The clinical expert noted that, although some clinical risk factors such as early age of onset (younger than 40 years), extensive colitis, and need for corticosteroids at diagnosis may be associated with a more complex course, there are currently no available predictors of disease response to a therapy (e.g., generic profile or available blood tests).
The clinical expert indicated the most important patient outcomes at various stages are as follows: first, in the short-term, clinical response is important to ensure patients are responding in terms of a reduction in symptoms, including extreme stool frequency, diarrhea, rectal bleeding, tenesmus, nighttime defecation, and urgency. Next, the main target in the intermediate term is symptom improvement or remission and the resolution of both blood-based (C-reactive protein) and stool-based biomarkers (fecal calprotectin). Finally, usually within 6 months, the goal is ideally to exhibit endoscopic healing or at least significant improvement. The clinical expert indicated that for UC, the goal of exhibiting histologic healing is not currently considered a robust accepted treatment target, although there is evidence to suggest histologic healing does predict improved outcomes; however, histologic healing is not used as a clinical target in routine clinical practice. The clinical expert also noted that etrasimod would not likely be used in an acute hospitalized setting for acute severe UC, as this is a unique context with standard of care, with IV anti-TNF alpha drugs used predominantly. The clinical expert noted that after the initiation of medication, a check-in within the first 1 to 2 weeks is essential to verify some clinical improvement. Another check-in around 4 to 6 weeks is appropriate, followed by a full assessment with blood work and stool studies completed at 12 weeks. It is preferred to have an endoscopic exam within 6 to 12 months of treatment initiation. The clinical expert indicated that treatment discontinuation of etrasimod should be considered in a manner similar to other advanced therapies for adults with moderate to severe UC, with factors that include:
an inability to decrease the oral corticosteroid dose despite treatment with etrasimod (steroid dependence)
the early recurrence of symptoms despite the full 12 weeks of initial therapy with etrasimod
a persistent elevation of biomarkers, especially fecal calprotectin, and limited or no improvement of symptoms after 12 weeks of initial treatment with etrasimod
evidence of persistent disease activity after initial therapy (12 weeks) or signs of progression during maintenance therapy based on endoscopy.
The clinical expert noted that the prescribing of etrasimod should be limited to gastroenterologists who treat IBD, with the exception of internal medicine physicians or surgeons in rural settings.
Clinician Group Input
One clinician group provided input, the Canadian IBD Interest Group, which is an assembly of gastroenterologists from across Canada with subspecialty expertise in IBD management. The group’s input was informed by 12 specialists.
In general, the input from the clinician group is in alignment with the input from the clinical expert consulted by CADTH. The clinician group noted that treatment for UC is influenced by disease severity and may involve medications, including oral and/or rectal 5-ASA, systemic corticosteroids, advanced biologics (adalimumab, infliximab, golimumab, vedolizumab, ustekinumab, mirikizumab), and advanced small-molecule drugs (tofacitinib, upadacitinib, ozanimod). The clinician group indicated there is a need for oral therapies that are well tolerated and provide durable disease control.
In alignment with input from the clinical expert consulted by CADTH, the clinician group anticipated that etrasimod is likely to be used as a first-line advanced therapy and could also be used in selected cases as a second- or third-line drug for UC treatment, based on several advantages of etrasimod, including its:
oral delivery
once-daily dosing regimen
efficacy in all patient subgroups, including those with limited proctitis (the clinician group noted that the patients with UC with ulcerative proctitis have been excluded from previous clinical trials, but they represent up to 30% of the overall population with UC)
favourable long-term safety compared with existing oral alternatives, including ozanimod, upadacitinib, and tofacitinib.
The clinician group noted that etrasimod would be unlikely to be used in patients with fulminant UC or who were hospitalized due to UC, as this therapy has not been evaluated in that setting. The clinician group noted that discontinuation with etrasimod could be considered when there is an inadequate clinical response (assessment of both symptoms and objective biomarkers of disease activity) within 12 to 16 weeks of treatment, or a significant adverse effect occurs.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for etrasimod:
relevant comparators
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
care provision issues
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
The 2 multicentre, phase III, double-blind, randomized, placebo-controlled trials, ELEVATE UC 12 (N = 354) and ELEVATE 52 (N = 433) that were submitted by the sponsor compared etrasimod (2 mg daily oral) with placebo in patients with moderately to severely active UC. In both trials, randomization was done using a 2:1 ratio where patients received either etrasimod or placebo for 12 weeks and 52 weeks, respectively. Clinical remission was defined as patients having a stool frequency subscore of 0 (or a score of 1 with a ≥ 1 point decrease from baseline), a rectal bleeding subscore of 0, and an endoscopic score of 1 or less (excluding friability). These were the primary outcomes in both protocols. The key secondary outcomes were similar in both protocols, including endoscopic improvement, symptomatic remission, and mucosal healing. Corticosteroid-free clinical remission at week 52 and sustained clinical remission at both week 12 and week 52 were reported as the secondary outcomes in the ELEVATE UC 52 trial. HRQoL was assessed using the Inflammatory Bowel Disease Questionnaire (IBDQ) and compared (i.e., study drug versus placebo). Harms were also reported.
Patients in the trial populations had an approximate mean age of 40.5 years and a mean UC duration of 6.0 to 7.9 years. There were slightly more male (range, 53% to 63%) than female (range, 38% to 47%) patients. Most enrolled patients were white (range, 75% to 89%), followed by Asian, Black or African American, American Indian or Alaska Native, and multiple. At baseline, approximately 27% to 32% of the patients were receiving corticosteroid, and 78% to 84% were receiving oral 5-ASA. Approximately one-third of the enrolled patients reported prior use of at least 1 biologic or Janus kinase (JAK) inhibitor (JAKi) (range, 29% to 34%).17-19
Efficacy Results
The key efficacy results from the ELEVATE UC 12 and ELEVATE UC 52 trials are summarized in Table 2 and listed in order from the most important to the least important outcomes, as suggested by the clinical expert consulted by CADTH. According to the statistical analysis plans for both trials,19-21 the primary analysis of the efficacy end points was conducted in the full analysis set (FAS) among patients with a baseline modified Mayo score (MMS) of 5 to 9 (N = 334 in the ELEVATE UC 12 trial and N = 409 in the ELEVATE UC 52 trial).
Endoscopic Improvement
In both the ELEVATE UC 12 and ELEVATE UC 52 trials, a greater proportion of patients in the etrasimod group compared with the placebo group had endoscopic improvement at week 12 and week 52. The between-group common risk differences were 12.1% (95% confidence interval [CI], 3.0% to 21.2%; P = 0.009) in the ELEVATE UC 12 trial and 21.2% (95% CI, 13.0% to 29.3%; P < 0.001) in the ELEVATE UC 52 trial at week 12, and 26.7% (95% CI, 19.0% to 34.4%; P < 0.001) in the ELEVATE UC 52 trial at week 52. Greater between-group risk differences were observed for patients treated with etrasimod versus placebo in the subgroup of patients who were naive to any prior biologic or JAKi therapy compared with those who were not, and in the subgroup of patients who had received only 1 prior biologic or JAKi compared with those who had received more than 1 (no interaction P values were provided).
Mucosal Healing
At week 52, a greater proportion of patients in the etrasimod group (26.6%) compared with the placebo group (8.1%) had mucosal healing, with a between-group common risk difference of 18.4% (95% CI, 11.4% to 25.4%; P < 0.001) in the ELEVATE UC 52 trial.
Clinical Remission
In both pivotal trials, a greater proportion of patients in the etrasimod group compared with the placebo group had clinical remission at week 12 and week 52. The between-group common risk differences were 9.7% (95% CI, 1.1% to 18.2%; P = 0.026) in the ELEVATE UC 12 trial and 19.8% (95% CI, 12.9% to 26.6%; P < 0.001) in the ELEVATE UC 52 trial at week 12, and 25.4% (95% CI, 18.4% to 32.4%; P < 0.001) in the UC 52 trial at week 52.
Sustained Clinical Remission
A greater proportion of patients in the etrasimod group (17.9%) compared with the placebo group (2.2%) had sustained clinical remission at both week 12 and week 52, with a between-group common risk difference of 15.8% (95% CI, 10.7% to 21.0%; P < 0.001), based on the results from the ELEVATE UC 52 trial.
Corticosteroid-Free Clinical Remission
At week 52, a greater proportion of patients in the etrasimod group (32.1%) compared with the placebo group (6.7%) achieved clinical remission and were corticosteroid-free for at least 12 weeks, with a common risk difference of 25.4% (95% CI, 18.4% to 32.4%; P < 0.001). Similarly, at week 52, among the patients who were receiving oral corticosteroids for UC at baseline, a greater proportion of patients in the etrasimod group (31.0%) compared with the placebo group (7.5%) achieved clinical remission and were corticosteroid-free for at least 4 weeks, with a common risk difference of 23.1% (95% CI, 10.2% to 35.9%; P < 0.001).
Clinical Response
In both pivotal trials, a greater proportion of patients in the etrasimod group compared with placebo had clinical response with a between-group common risk difference of 21.2% (95% CI, 10.2% to 32.3%; P < 0.001) in ELEVATE UC 12 and 28.3% (95% CI, 18.5% to 38.0%; P < 0.001) in ELEVATE UC 52 at week 12 and 24.9% (95% CI, 15.8% to 34.1%; P < 0.001) in the UC 52 trial at week 52.
Symptomatic Remission
At week 52, a greater proportion of patients in the etrasimod group (43.4%) compared with the placebo group (18.5%) had mucosal healing, with a between-group common risk difference of 24.9% (95% CI, 16.2% to 33.6%; P < 0.001) in the ELEVATE UC 52 trial.
HRQoL Assessed With the IBDQ Total Score
In both pivotal trials, the IBDQ total scores in the etrasimod group showed patients experienced a greater increase in mean change from baseline compared with those in the placebo group at week 12 and week 52. The least squares (LS) mean differences between the 2 groups were 17.33 points (95% CI, 8.50 to 26.16; P < 0.001) in the ELEVATE UC 12 trial and 15.44 points (95% CI, 6.54 to 24.35; P < 0.001) in the ELEVATE UC 52 trial at week 12, and 17.70 points (95% CI, 6.64 to 28.76; P = 0.002) in the ELEVATE UC 52 trial at week 52.
Harms Results
The analysis of harms was conducted in the FAS among patients with a baseline MMS of 4 to 9 (N = 354 in the ELEVATE UC 12 trial and N = 433 in the ELEVATE UC 52 trial). Evidence from the pivotal trials showed etrasimod was generally safe and well tolerated.
Treatment-emergent adverse events (TEAEs) were experienced by approximately 47% of patients in the ELEVATE UC 12 study, and from 56% to 71% of patients in the ELEVATE UC 52 study. The most common TEAEs in the 2 pivotal trials were anemia (range, 6% to 10% across the different study groups), headache (2% to 8%), nausea (2% to 4%), UC (1% to 9%), and pyrexia (3% to 5%). In both trials, serious TEAEs occurred in approximately 2% to 7% of patients across the different treatment arms and were approximately similar between the 2 groups. The most frequently reported serious TEAEs and TEAEs leading to discontinuation of treatment in both trials was UC (not more than 2.5% across the study groups).
Across both trials, a greater proportion of patients in the etrasimod group reported cardiovascular-related adverse events of special interest (AESIs) than in the placebo group, whereas there was a greater proportion of patients in the placebo group experiencing infection-related AESIs than in the etrasimod group. No AESIs related to pulmonary disorders, macular edema, posterior reversible encephalopathy syndrome, or malignancy were reported in the ELEVATE UC 12 trial. Similar findings were demonstrated in the ELEVATE UC 52 trial, except for 1 patient (0.3%) in the etrasimod group who reported macular edema, and 1 patient in each treatment group (0.3% in the etrasimod group and 0.7% in the placebo group) who reported pulmonary disorders.
Critical Appraisal
Both trials used appropriate randomization methods, allocation concealment, randomization stratification, double-blind approaches, and statistical methods for the primary and key secondary outcomes. Both trials used the placebo as the comparator, and there is a lack of head-to-head direct evidence comparing etrasimod against other active pharmacotherapies that are relevant to clinical practice in Canada. It is notable that the FDA guidance to industry for conducting interventional trials in patients with UC22 encourages sponsors to use active treatments as controls. To align with the regulatory body’s guidance on moderate to severe UC22 that became available during or after the trials, the sponsor amended its statistical analysis plans and performed the primary efficacy analysis in the FAS of patients with a baseline MMS of 5 to 9 (excluding a total of 44 patients with a baseline MMS of 4 in the 2 trials),19 although the patients who were randomized were those with a baseline MMS of 4 to 9. In general, the CADTH review team and the clinical expert consulted by CADTH did not identify major issues with such a change in the efficacy analysis that would impact the study results, based on the patient characteristics that appeared to be reasonably balanced between the treatment groups, and the findings in the supplementary analyses of the same outcomes using the entire FAS for both studies were similar.
Some efficacy end points (e.g., MMS subscore of stool frequency and rectal bleeding, and the HRQoL outcome assessed with the IBDQ) were recorded and reported by patients. Although these subjective outcomes may be influenced by knowledge of treatment assignment, the double-blind design of the trials likely mitigated this risk. The CADTH review team noted that in the ELEVATE UC 52 trial, a higher proportion of patients in the placebo group (50.7%) discontinued the treatment due to disease worsening compared with the etrasimod group (27.3%) during the 52-week trial period. Withdrawal by patient as a reason for discontinuing the study or treatment was higher in the placebo group in both trials, except among those who discontinued from the ELEVATE UC 52 trial, where a higher percentage of patients treated with etrasimod discontinued the study by patient choice. Also, for the IBDQ total score at week 52 in the ELEVATE UC 52 trial, the missing data rate was higher in the placebo group than in the etrasimod group. There was no concrete evidence beyond these points that clearly showed unblinding due to patients’ inferences on treatment assignment based on symptom changes or the occurrence of other factors. Thus, the extent to which this could have affected the efficacy and HRQoL outcome results, particularly the outcomes at week 52, is unclear. Overall, no important imbalances in baseline patient characteristics, concomitant medications, or drop-outs of prognostic importance between the 2 study groups were identified. The overall concomitant use of systemic corticosteroids appeared similar between groups in each study, although the reported use of budesonide by patients was 3% to 6% more in the etrasimod groups versus the placebo groups in both studies. As well, more patients treated with etrasimod (5.9% and 3.5%) compared with placebo (1.7% and 1.4%) concurrently received immunomodulators. While these are notable differences, the relatively small percentages (< 10%) and small between-group differences (< 5%) mean these were unlikely to have been important confounders of the results in both trials. Overall, the statistical methods used in both trials were appropriate. The HRQoL assessed with the IBDQ (an efficacy-related outcome) at week 52 was most likely underpowered, as its outcome data were only available for fewer than half of the patients assessed with the IBDQ at baseline. The subgroup analyses were also likely underpowered to identify subgroup differences. An appropriate method for adjusting for multiplicity was used for the primary and secondary outcomes, but there was no multiplicity control for the subgroup analyses. The interaction P values for the subgroup analyses were not provided.
While the indication for etrasimod is for the treatment of moderately to severely active UC in adults, patients aged 16 to 80 years were eligible for both trials, yet a relatively small proportion of the enrolled patients (5.0% to 7.4%) were aged 65 years or older, and only 1 person in each study was younger than 18 years. No patients in the ELEVATE UC 12 trial and only 0.7% of the patients in the ELEVATE UC 52 trial were aged 75 years or older at baseline. These small population results limit the trial’s generalizability among older patients. The clinical expert consulted by CADTH noted the need for some caution when using etrasimod in patients who are 65 years and older because there is a higher likelihood of concomitant diseases and/or multiple medications (polypharmacy), as well as a higher potential for decreased hepatic, renal, cardiac, or pulmonary function. Patients in both trials were recruited from multiple countries, including Canada. The clinical expert did not raise any major concerns in the generalizability of the results of the trials to clinical practice in Canada, based on the eligibility criteria of patients, the demographic characteristics of the patients from the diversity aspect, and the etrasimod dose in the 2 trials. The clinical expert pointed out that the inclusion of patients with UC with isolated proctitis, a subgroup of patients with UC that is most often excluded from clinical trials, is helpful for clinical practice, contributing evidence for the efficacy and safety of etrasimod in this specific patient group. The clinical expert noted the importance of monitoring patients using biomarker examinations (e.g., fecal calprotectin) during treatment with etrasimod. The placebo-controlled period of the ELEVATE UC 52 trial was 1 year, which aligns with current regulatory guidance. However, given patients and clinicians often report the waning of treatment effect with advanced therapies for UC, longer-term comparative evidence on the durability of the effectiveness of etrasimod would be informative. The occurrence of some adverse events (AEs), especially rare ones, may take longer than 52 weeks to be identified. Longer-term follow-up to assess safety and a direct comparison between etrasimod versus other advanced therapies would be preferred.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) framework was used to assess the certainty of the evidence for outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.23,24 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with the clinical expert, and input received from the patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: endoscopic improvement, mucosal healing, clinical remission, sustained clinical remission, corticosteroid-free clinical remission, clinical response, symptomatic remission, change in IBDQ, and serious TEAEs.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on the minimal important difference (MID) thresholds identified in the literature for the IBDQ total score. The target of the certainty-of-evidence assessment was the presence or absence of an important effect, based on thresholds informed by the clinical expert consulted for this review, in terms of endoscopic improvement, mucosal healing, clinical remission, sustained clinical remission, corticosteroid-free clinical remission, clinical response, and symptomatic remission.
Findings from the ELEVATE UC 12 and ELEVATE UC 52 trials were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for etrasimod versus placebo in adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.
Long-Term Extension Studies
There are currently no results available from any long-term extension studies of etrasimod in moderately to severely active UC. One ongoing, single-arm, long-term open-label extension (OLE) study (ELEVATE UC OLE25) of etrasimod 2 mg/day taken orally has an estimated primary completion date of February 6, 2027.
Indirect Comparisons
One indirect treatment comparison (ITC) was submitted by the sponsor to estimate the relative efficacy and safety of etrasimod versus advanced therapies for the treatment of adult patients with moderately to severely active UC.26,27
Description of Studies
The trials included in the ITC enrolled adult patients with moderately to severely active UC and studied the following advanced therapies for these patients: adalimumab, infliximab, golimumab, tofacitinib, ustekinumab, vedolizumab, upadacitinib, mirikizumab, and ozanimod. Efficacy outcomes included clinical outcomes (remission and response) and safety outcomes (serious infections, serious adverse events [SAEs], any AE, and treatment discontinuations due to AEs), which generally aligned with the outcomes that were important to patients and clinicians.
Efficacy Results
The results of the network meta-analysis (NMA) █████████ ████ █████████ ███ ███ ████████████ █████████ ████ █████ ████████ █████████ ███ ████████ ████████ ███ ████████ █████████ ██ █████████ ███ ███████████ ███████ ██ ██████ █████ ███████████ ███ ████ █████ ██ ██ ███ ███████████ ███ ████████ ██████████ ██ █ ███ ████████████ ███ ███ ████████ █████████ ██████ ███ ███ ██████████ ███████ ███ ██████████ ████████ ███ █████ █████████ ███ ██████████ ████████ ██ ██████████ ████ ████ ██ ███████ ███ ███████████ ███ ████████████ ████████ ████ █████████ █████ ████████ █████████████ ████████ ███ ████████ ████████ ███ ████████ █████████ ██ ███ █████████ ██████ ████████████ ████ ████████ ████ █████████ █████ ████████ ███████████████████ ████████ ██████ ████ ██████████ █████ ███ ███████████ ██████ █████ ████ ██ ███████████ ███████ █████████ ███ ███ ████ ██ ███ ███████████ ██████████ █████████ ███ ████████ ████████ ██ ███ ████
Table 2: Summary of Findings for Etrasimod Versus Placebo for Adults With Moderately to Severely Active UC
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
Endoscopic improvement | ||||
Proportion of patients with endoscopic improvement Follow-up: 12 weeks | 743 (2 RCTs) | ELEVATE UC 12 trial:
ELEVATE UC 52 trial:
| Higha | Etrasimod results in a clinically important increase in the proportion of patients with endoscopic improvement at 12 weeks when compared with placebo |
Proportion of patients with endoscopic improvement Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Higha | Etrasimod results in a clinically important increase in the proportion of patients with endoscopic improvement at 52 weeks when compared with placebo |
Mucosal healing | ||||
Proportion of patients with mucosal healing Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52
| Highb | Etrasimod results in a clinically important increase in the proportion of patients with mucosal healing at 52 weeks when compared with placebo |
Clinical remission | ||||
Proportion of patients with clinical remission Follow-up: 12 weeks | 743 (2 RCTs) | ELEVATE UC 12 trial:
ELEVATE UC 52 trial:
| Highc | Etrasimod results in a clinically important increase in the proportion of patients with clinical remission at 12 weeks when compared with placebo |
Proportion of patients with clinical remission Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Highc | Etrasimod results in a clinically important increase in the proportion of patients with clinical remission at 52 weeks when compared with placebo |
Sustained clinical remission | ||||
Proportion of patients with sustained clinical remission at both week 12 and week 52 Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Highd | Etrasimod results in a clinically important increase in the proportion of patients with sustained clinical remission at both week 12 and week 52 when compared with placebo |
Corticosteroid-free clinical remission | ||||
Proportion of patients with clinical remission at week 52 and were corticosteroid-free for ≥ 12 weeks Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Highe | Etrasimod results in a clinically important increase in the proportion of patients with clinical remission at 52 weeks and were corticosteroid-free for at least 12 weeks when compared with placebo |
Proportion of patients (who were receiving oral corticosteroids for UC at baseline) with clinical remission at week 52 and were corticosteroid-free for ≥ 4 weeks Follow-up: 52 weeks | 127 (1 RCT) | ELEVATE UC 52 trial:
| Highf | Etrasimod results in a clinically important increase in the proportion of patients (who were receiving oral corticosteroids for UC at baseline) with clinical remission at 52 weeks and were corticosteroid-free for at least 4 weeks when compared with placebo |
Clinical response | ||||
Proportion of patients with clinical response Follow-up: 12 weeks | 743 (2 RCTs) | ELEVATE UC 12 trial:
ELEVATE UC 52 trial:
| Highg | Etrasimod results in a clinically important increase in the proportion of patients with clinical response at 12 weeks when compared with placebo |
Proportion of patients with clinical response Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Highg | Etrasimod results in a clinically important increase in the proportion of patients with clinical response at 52 weeks when compared with placebo |
Symptomatic remission | ||||
Proportion of patients with sustained symptomatic remission Follow-up: 52 weeks | 409 (1 RCT) | ELEVATE UC 52 trial:
| Highh | Etrasimod results in a clinically important increase in the proportion of patients with symptomatic remission at 52 weeks when compared with placebo |
HRQoL (IBDQ) | ||||
Change from baseline in IBDQ total score, ranging from a score of 32 (worst HRQoL) to 224 (best HRQoL), LS mean change (SE) Follow-up: 12 weeks | 592 (2 RCTs) | ELEVATE UC 12 trial:
ELEVATE UC 52 trial:
| Moderatei | Etrasimod likely results in little to no difference in IBDQ improvement at 12 weeks when compared with placebo |
Change from baseline in IBDQ total score, ranging from a score of 32 (worst HRQoL) to 224 (best HRQoL), LS mean change (SE) Follow-up: 52 weeks | 168 (1 RCT) | ELEVATE UC 52 trial:
| Lowj | Etrasimod may result in little to no difference in IBDQ improvement at 52 weeks when compared with placebo |
Harms | ||||
Proportion of patients with serious TEAEs Follow-up: 12 weeks | 354 (1 RCT) | ELEVATE UC 12 trial:
| Moderatek | Etrasimod likely results in little to no difference in serious TEAEs at 12 weeks when compared with placebo |
Proportion of patients with serious TEAEs Follow-up: 52 weeks | 433 (1 RCT) | ELEVATE UC 52 trial:
| Moderatek | Etrasimod likely results in little to no difference in serious TEAEs at 52 weeks when compared with placebo |
CI = confidence interval; ES = endoscopic score; FAS = full analysis set; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; LS = least squares; MID = minimal important difference; MMS = modified Mayo score; NR = not reported; RB = rectal bleed; RCT = randomized controlled trial; SE = standard error; SF = stool frequency; TEAE = treatment-emergent adverse event; UC = ulcerative colitis.
Note: The primary analysis of the efficacy end points was conducted in the FAS among patients with a baseline MMS of 5 to 9 (N = 334 in the ELEVATE UC 12 trial and N = 409 in the ELEVATE UC 52 trial). Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aEndoscopic improvement was defined as patients with an ES of ≤ 1 (excluding friability). An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 5% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome. Although the lower boundary of the 95% CI for the between-group difference in the ELEVATE UC 12 trial was 3%, which could be considered as a source of serious imprecision, this did not result in the level of certainty of the overall evidence for this outcome being rated down because the evidence from the ELEVATE UC 52 trial was also taken into consideration.
bMucosal healing was defined as patients who have an ES of ≤ 1 (excluding friability) with histologic remission measured by a Geboes Index score < 2.0. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 5% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
cClinical remission was defined as patients who have an SF subscore of 0 (or a score of 1 with a ≥ 1 point decrease from baseline), an RB subscore of 0, and an ES ≤ 1 (excluding friability). An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 7.5% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome. Although the lower boundary of the 95% CI for the between-group difference in the ELEVATE UC 12 trial was 1.14%, which could be considered as a source of serious imprecision, this did not result in the level of certainty of the overall evidence for this outcome being rated down because the evidence from the ELEVATE UC 52 trial was also taken into consideration.
dSustained clinical remission was defined as patients with an SF subscore of 0 (or a score of 1 with a ≥ 1-point decrease from baseline), an RB subscore of 0, and an ES of ≤ 1 (excluding friability) at both week 12 and week 52. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 10% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
eCorticosteroid-free for ≥ 12 weeks and achieved clinical remission at week 52 was defined as patients with an SF subscore of 0 (or a score of 1 with a ≥ 1-point decrease from baseline), an RB subscore of 0, and an ES of ≤ 1 (excluding friability), and who had not received corticosteroids for at least 12 weeks in the 40-week treatment period. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 7.5% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
fCorticosteroid-free for ≥ 4 weeks and achieved clinical remission at week 52 was defined as patients with an SF subscore of 0 (or a score of 1 with a ≥ 1-point decrease from baseline), an RB subscore of 0, and an ES of ≤ 1 (excluding friability), and who had not received corticosteroids for at least 4 weeks in the 40-week treatment period. Results of this outcome include those who were receiving oral corticosteroid for UC at baseline. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 7.5% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
gClinical response was defined as patients with a ≥ 2-point improvement and a ≥ 30% decrease from baseline in MMS, and a ≥ 1-point decrease from baseline in RB subscore or an absolute RB subscore ≤ 1. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 10% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
hSymptomatic remission was defined as patients with an SF subscore of 0 (or a score of 1 with a ≥ 1 point decrease from baseline) and an RB subscore of 0. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 10% between the groups was identified by the clinical expert consulted by CADTH as a threshold of clinical importance for this outcome.
iThe level of evidence was rated down 1 level for serious imprecision. Based on the MID identified in the literature (≥ 15 points above placebo based on between-group data), the point estimate suggested little to no difference, and the 95% CI for the between-group difference crossed the MID threshold. The impact of missing outcome data was unclear (less than 10% of the patients in both the ELEVATE UC 12 and ELEVATE UC 52 trials had IBDQ results available at baseline) and no notable between-group imbalances in missing data were identified.
jThe level of evidence was rated down 1 level for serious risk of bias and was rated down 1 level for serious imprecision. More than half of the patients with IBDQ results available at baseline did not respond at week 52, and there was a higher proportion of patients with missing data in the placebo group than in the etrasimod group. No sensitivity analyses were done to assess the impact of the missing data for this outcome. While the exact impact of such missing outcome data on the results is unclear, the CADTH review team considered that the risk of bias for this outcome was high. Based on the MID identified in the literature (≥ 15 points above placebo based on between-group data), the point estimate suggested little to no difference, and the 95% CI for the between-group difference crossed the MID threshold.
kThe level of evidence was rated down 1 level for serious imprecision due to the small number of events.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and sponsor’s submissions.16,19
The networks of efficacy analyses were ██████ ██ █ ████ ████████ ████ ██████████ ███ █████ ████████ ██████ █ ██████ ██████████ ██████████ ███ ████ ███ ████████ ██████ █████ ██████████ █████████ █████ ████ ████████ ███ █████ ████ ██████████ ████ ██████████ █████████ █████████ ██████ ██████████ █████ ████ ██████████ █████████████ ██████ ██████████ ███ ████ ██████████ ██████████ ███████████ ███████ █████████ ██ ████████ ██ ██████████ ████ ███ ███████████ ████ █████ ████ ████ ███████████
Harms Results
Safety results among the overall and advanced therapy–naive trial population during the induction phase and maintenance phase ███ ████ ████ ███ ███ ███████ ███ ████ ███ ███████████ ██ █████████ ██████ █████ ████████ █████████ ███ ███ ██████ ███ ███ ████████ █████████ ████████████████ ███ ██ ████ ███ █████ ███ █████ ███████ █████ ███ ████████ █████████ ██ ██████████ ██ ███████████ ██ ██ █████ █ █████ ███ █ █████ ███ ████ █ ██ █████ ██ ███ ███████████ ██████ ████████ ████ ██████ ██████ ██ ███████████ ████ ███████ ██ █████ ███ █ ████ ███████ ████████ ██ ███ ████████████ ███ ███████████ ███████ ██████ █████████ ██ ███ ███████████ █████████████
██ ████████ █████████████ ███ ████ ████ █████████ ███████████ ██████ ███████████ ████ █████ ████ ████ ███████████
Critical Appraisal
The networks were sparsely populated with relatively few nodes centred around a single connection (placebo) in a star geometry. Furthermore, most closed loops were between different doses of individual drugs and, consequently, the evidence was essentially all indirect, increasing uncertainty in the estimates for each outcome, and the consistency assumption could not be assessed. Additionally, most nodes were informed by only 1 or 2 trials, increasing the chance the comparisons were underpowered, which impacted model selection and the types of adjustments that could be done. These factors mean there was imprecision in many of the estimates (as evidenced by relatively wide 95% credible intervals [CrIs] for many pairwise comparisons) and validating the key assumptions for the NMA is difficult, thereby increasing the uncertainty surrounding the results.
Overall, the clinical expert did not expect any major issues with the representativeness of the study populations enrolled in the RCTs included in the ITC in relation to those who may also be eligible for treatment with etrasimod in Canada. However, there was variability in patient characteristics (also potential treatment-effect modifiers) across the studies, such as for the definition of severity of UC based on Mayo score, disease duration, and concomitant medication use. It is also likely that there were differences in patients’ experience with previous treatments (number and type) and differences in eligibility criteria regarding intolerance to or failure of at least 1 of the conventional therapies or biologics, further introducing bias into the analysis. There was heterogeneity in the treatment regimens among treatments, the duration of the induction and maintenance phases, and the methods used for rerandomization into the maintenance phase. Moreover, the included studies used different definitions for the efficacy and safety outcomes, creating issues when analyzing efficacy and harms results.
The dissimilarity among patients, important heterogeneity across studies, and wide CrIs (in safety outcomes) made it challenging to draw definitive efficacy and safety conclusions as to whether etrasimod was superior or inferior to other advanced therapies, including ozanimod, in adult patients with moderately to severely active UC.
Studies Addressing Gaps in the Evidence From the Systematic Review
No studies addressing gaps in the pivotal and RCT evidence were identified for this review.
Conclusions
Two phase III, multicentre, double-blind RCTs evaluated the efficacy and safety of etrasimod compared with placebo in adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. Compared with placebo at 12 and 52 weeks, etrasimod results in a clinically important increase in the proportion of patients who have endoscopic improvement, clinical remission, and clinical response. At 52 weeks, etrasimod results in a clinically important increase in the proportion of patients who have mucosal healing, sustained clinical remission, corticosteroid-free clinical remission in the overall population as well as in those who were receiving oral corticosteroids for UC at baseline, and symptomatic remission compared with placebo. Etrasimod likely results (at 12 weeks) or may result (at 52 weeks) in little to no difference in improvement in HRQoL based on the IBDQ, and likely results in little to no difference in the proportion of patients who have serious TEAEs at 12 weeks and 52 weeks compared with placebo. AEs were common but no particular concerns were identified beyond those noted in the product monograph or what is expected for sphingosine 1-phosphate (S1P) receptor modulators. However, the frequencies of AEs are based on relatively short observation periods and younger patient populations than would be included in real-world practice.
There is a data gap in the head-to-head direct evidence between etrasimod and other advanced therapies for moderately to severely active UC. Indirect evidence submitted by the sponsor ████████ ████ █████ ███ ██ ██ ██████████ ██████████ ██ ████████ ██ █████ ████████ ███████ █████████ ███ █████ ████████ █████████ ██████ █████████ ██ ███████████.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of etrasimod (Velsipity) 2 mg film-coated tablet taken orally once daily in the treatment of adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Overview of the Condition
IBD is a group of diseases characterized by chronic recurrent, progressive inflammation of the GI tract.28 There are 2 main types of IBD: Crohn disease (CD) and UC. UC is a chronic disease characterized by inflammation predominantly of the mucosal layer of the large intestine (colon) most often involving the rectum, and frequently extends continuously into the proximal colon.1 The cause of UC remains uncertain, but a combination of genetic and environmental factors contributes to immune dysregulation and upregulation in response to micro-organisms in the GI tract.29 UC is characterized by blood in the stool with mucus, frequent diarrhea, urgency, loss of appetite, and tenesmus (severe rectal cramp or spasm).2 Although UC principally affects the GI tract, extraintestinal manifestations may also occur, such as arthritis.3 There is no notable difference in the frequency of UC among males and females.30 Although the risk of mortality from UC itself is low, the disease is associated with an increased risk of other complications (e.g., respiratory diseases, colorectal cancer, lymphoma, and skin cancer) that result in higher mortality compared with the general population.11 About 30% to 60% of patients with UC first present with isolated proctitis (involvement is limited to the rectum).31,32 Patients with proctitis are more prone to proximal extension (i.e., more colon becomes involved in active disease), higher colectomy rates, an increased need for advanced therapy, and higher hospitalization rates than patients who start with extensive colitis.31,33,34 Among patients with isolated proctitis who are untreated for 1 year, the relapse rate is between 47% and 86%.35
While most patients have a mild or moderate disease course, about 10% to 15% experience an aggressive course of UC.4 Relapse is common, with a cumulative risk of relapse of 70% to 80% at 10 years.4 Achieving endoscopic healing earlier may be associated with a reduced risk of future colectomy.4 The chronic nature of UC has a considerable impact on a patient’s HRQoL, including psychological, physical, sexual, and social domains of HRQoL due to chronicity of symptoms such as urgency, frequency, and incontinence.5,6 The medical and surgical treatments for UC (e.g., colectomies) and their potential accompanying complications can also negatively impact HRQoL and productivity.7,36-39 Individuals with UC are at greater risk of comorbid anxiety, depression, and impaired social interactions.5,6,40,41 Patients with UC frequently report fatigue and sleep disturbance as well as an inability to perform regular daily routines such as jobs or domestic chores.7-10 Furthermore, the disease can impact the patient’s caregivers and family, workplace, and community.11
Estimated Disease Prevalence
The prevalence of UC in Canada in 2023 was estimated to be 414 per 100,000.12 It is estimated that 32% to 46% of patients in Canada with UC have moderate disease, and 13% to 14% have severe disease.13
Diagnosis of the Condition
Diagnosis of UC includes laboratory and stool testing to rule out bacterial, viral, or parasitic infection, as well as tests to rule out CD.42,43 Endoscopic examination with a tissue biopsy is required to make a diagnosis of UC, combined with adjunctive clinical manifestations, a review of medical history, physical exams, laboratory tests, and histological and radiological examinations.42-44 Tests are also conducted to assess the extent and severity of disease, with the endoscopic Mayo score reflecting disease severity, which is required to determine eligibility for reimbursement for certain treatments in Canada.45
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following have been summarized and validated by the CADTH review team.
The clinical expert consulted by CADTH pointed out that the treatment goals for patients with UC are to achieve rapid symptomatic relief and to induce and maintain clinical, serological, biomarker, and endoscopic remission in both the short and long-term. Per the clinical practice guidelines, the primary goal for UC treatment is the improvement of symptoms, i.e., the induction of clinical remission (normal, nonbloody stools at a frequency of ≤ 3 per day).14 The ultimate goal for long-term management of UC is complete remission, involving both clinical remission and endoscopic remission with mucosal healing.14 Endoscopic remission is associated with decreased hospitalizations, decreased use of or freedom from corticosteroids, and a decreased need for colectomy.46-49 The SPIRIT (Selecting End PoInts foR Disease-ModIfication Trials) consensus from the International Organization for the Study of Inflammatory Bowel Disease identified improvement in patients’ symptoms (e.g., reduction in stool frequency) and in HRQoL, prevention of disability, and reduction in UC-related morbidity (e.g., surgery, hospitalizations, disease extension, and GI and extraintestinal dysplasia or cancer) and mortality as key therapeutic goals.50
Pharmacologic treatment is the mainstay of therapy for UC. Various drugs and drug classes are now available to treat UC. In patients with moderately to severely active UC, oral corticosteroids are typically the first-line therapy, but are used only for inducing remission due to their adverse effects.14,15 In patients who respond to corticosteroids, thiopurines (e.g., azathioprine, 6-mercaptopurine) can be used to maintain remission.14,15 Other recommendations include 5-ASA therapy in patients naive to 5-ASA drugs and to anti-TNF therapy or vedolizumab.14 However, most Canadian drug plans require a patient with moderately to severely active UC to have failed to respond steroid tapering using azathioprine or 6-mercaptopurine as a replacement before being eligible for a biologic. As such, advanced therapies are typically not used for first-line maintenance of steroid-induced remission.15
For patients for whom 5-ASAs, corticosteroids, or thiopurines are unable to induce or maintain remission or are not tolerated, advanced therapies are used. Anti-TNF therapy followed by vedolizumab was recommended for second- and third-line induction and maintenance of remission by the Toronto consensus guidelines in 2015 (which are the most recent Canadian guidelines for the treatment of UC).14,15 However, clinical practice has evolved since then, with the introduction of new advanced therapies.42 The clinical expert consulted by CADTH noted that early introduction of effective advanced therapy is important for patient benefit, particularly for avoiding the adverse effects of recurrent or prolonged courses of steroids. The choice of which advanced therapy to prescribe is ideally a decision shared with the patient that considers the disease characteristics, comorbidities, and patient preferences as well as a given drug’s efficacy, safety, and mode of administration.16 Patients whose disease fails to respond or loses response, or who are intolerant to 1 advanced treatment would be offered a different one, potentially in a different medication class with a different mechanism of action.16 The clinical expert consulted by CADTH and the sponsor indicated there are limited extant data and no recent Canadian guidelines available to specifically guide the sequencing of advanced therapies in UC.16
Under circumstances where medical therapy fails to keep a patient’s UC in complete remission, colectomy (removal of all or part of the colon) may be required.11 However, colectomy carries a risk of serious potential complications, including bleeding and infections,51 and physicians and surgeons work together closely to try to achieve disease control medically if at all possible. The clinical expert noted that emergent subtotal colectomy (taking the entire colon except for the rectum) remains a real and worrisome risk for patients with severe UC, which may be followed by additional surgical procedures, including the creation of an ileoanal pouch anastomosis. Either a temporary or permanent ileostomy requires specialized care and ostomy devices.51
Drug Under Review
Indication and Reimbursement Request
Etrasimod is indicated for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.52 The sponsor’s reimbursement request is in line with the Health Canada indication.
Dosing and Administration
The recommended dosage is 2 mg taken orally once daily.52
Mechanism of Action
Etrasimod is a highly selective S1P receptor modulator. The S1P receptor is involved in the regulation of multiple immune-inflammatory pathways, regulating lymphocyte egress from lymph nodes and into the blood.53,54 S1P signals through 5 different receptor subtypes (S1P1, S1P2, S1P3, S1P4, and S1P5), where each receptor subtype has a different cell specificity.55 Etrasimod exhibits selectivity to S1P1, S1P4, and S1P5 receptors, and minimal activation of S1P3 receptors, but has no activation of S1P2 receptors.52,56,57
Main Comparators
Table 3 summarizes the key characteristics of etrasimod and the 9 other advanced therapies available in Canada for the treatment of patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.
Table 3: Key Characteristics of Etrasimod and Main Comparators
Drug name (brand name) | Mechanism of action | Indicationa | Route of administration and recommended dosage | Serious adverse effects and/or safety issues |
|---|---|---|---|---|
Drug under review | ||||
Etrasimod (Velsipity)52 | Selective S1P receptor modulator. It may reduce lymphocyte migration into inflammation sites and reduce cytokine response. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. | 2 mg orally once daily. |
|
Comparators | ||||
S1P receptor modulators | ||||
Ozanimod (Zeposia)58 | S1P receptor modulator. Binds to the S1P1 receptors on lymphocytes, preventing egress from lymph nodes. It may reduce lymphocyte migration into the CNS and intestine. | For the treatment of adult patients with moderately to severely active UC who had an inadequate response, loss of response, or were intolerant to either conventional therapy or a biologic drug. |
| Malignancies, particularly of the skin. Initiation of ozanimod may result in transient reductions in heart rate and atrioventricular delays. |
Anti-TNF biologics | ||||
Adalimumab (Humira)59 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. |
|
| Serious infections, malignancies, and neurologic events. The most common adverse reaction in rheumatoid arthritis patients treated with Humira was injection-site reactions. |
Adalimumab (biosimilars: Abrilada, Amgevita, Hulio, Hyrimoz)60-63 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. |
|
| Serious infections (pneumonia), malignancies, and neurologic events. |
Adalimumab (biosimilars: Hadlima, Idacio, Simlandi, Yuflyma)64-67 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy including corticosteroids and/or azathioprine or 6-MP or who are intolerant to such therapies. | 160 mg in week 0, 80 mg in week 2, then 40 mg every other week thereafter as monotherapy or in combination with conventional therapies. Administered by SC injection. | Serious infections (pneumonia), malignancies, and neurologic events. |
Golimumab (Simponi)68 | Anti-TNF. Human monoclonal antibody that binds with p55 or p75 human TNF receptors. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to or have medical contraindications for conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6-MP for inducing and maintaining a clinical response, inducing clinical remission, achieving sustained clinical remission in induction responders, or improving endoscopic appearance of the mucosa during induction. | 200 mg administered by SC injection at week 0 followed by 100 mg at week 2, and then 50 mg every 4 weeks thereafter. For maintenance, a dose of 100 mg every 4 weeks can be considered at the discretion of the treating physician. | Upper respiratory tract infection. |
Infliximab (Remicade)69 | Anti-TNF. IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors. |
| IV infusion of 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter, for the treatment of adult and pediatric patients (aged ≥ 6 years). Doses up to 10 mg/kg may be used in some adult patients. | Infections and malignancies. |
Anti-TNF. IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors. |
| Adults and pediatric patients (aged ≥ 6 years): IV infusion 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter. Doses up to 10 mg/kg may be used. | Infections and malignancies. | |
Anti-integrin | ||||
Vedolizumab (Entyvio)73 | IgG1 monoclonal antibody. Binds to the human alpha-4 beta-7 integrin, acting as a gut-selective anti-inflammatory biologic. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loss of response to, or were intolerant to either conventional therapy or infliximab, a TNF alpha antagonist. | 300 mg administered by IV infusion at 0, 2, and 6 weeks and then every 8 weeks thereafter. The SC maintenance dose is 108 mg every 8 weeks. | Infections and malignancies. |
Anti-IL 12, anti-IL 23 | ||||
Mirikizumab (Omvoh)74 | Humanized IgG4 monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human IL-23 cytokine to inhibit its interaction with the IL-23 receptor. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. |
| Upper respiratory tract infection, headache, and site injection reactions (e.g., rash, maculopapular rash, papular rash, and pruritic rash). |
Ustekinumab (Stelara)75 | Human IgG1 monoclonal antibody. Neutralizes cellular responses mediated by IL-12 and IL-23. | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic or have medical contraindications to such therapies. | Single weight-based IV infusion (approximating 6 mg/kg) followed by a 90 mg SC dose 8 weeks later, then 90 mg SC every 8 weeks thereafter for maintenance. In patients with low inflammatory burden, a single IV dose followed 8 weeks later by 90 mg SC, then every 12 weeks thereafter may be considered at the discretion of the treating physician. | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. |
JAK inhibitors | ||||
Selective JAK inhibitor. Blocks several cytokine pathways and lymphocyte activation. | For the treatment of adult patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to either conventional UC therapy or a TNF alpha inhibitor. | 10 mg orally b.i.d. for induction for at least 8 weeks and 5 mg given b.i.d. for maintenance. Depending on therapeutic response, 10 mg b.i.d. may also be used for maintenance in some patients. However, the lowest effective dose possible should be used for maintenance therapy to minimize adverse effects. | A Health Canada warning indicated an increased risk of thromboses (pulmonary and deep vein thrombosis) and death, and increased risk of serious infection, including herpes zoster infections. Of note, tofacitinib is not recommended in combination with biological UC therapies or with potent immunosuppressants such as azathioprine and cyclosporine. | |
Upadacitinib (Rinvoq)80 | Selective JAK inhibitor. Demonstrates activity against JAK1, JAK2, JAK3, and TYK2. | For the treatment of adult patients with moderately to severely active UC who have experienced prior treatment failure, i.e., an inadequate response to, loss of response to, or intolerance to at least 1 conventional and/or biologic therapy. |
| Upper respiratory tract infection. Of note, upadacitinib should not be used in combination with other JAK inhibitors, immunomodulating biologics (e.g., biologic DMARDs), or with potent immunosuppressants such as azathioprine, 6-MP, and cyclosporine. |
6-MP = 6-mercaptopurine; AE = adverse event; b.i.d. = twice a day; CNS = central nervous system; CV = cardiovascular; DMARD = disease-modifying antirheumatic drug; IgG1 = immunoglobulin G1; IgG1k = immunoglobulin G1 kappa; IgG4 = immunoglobulin G4; IL = interleukin; ITC = indirect treatment comparison; JAK = Janus kinase; PRES = posterior reversible encephalopathy syndrome; S1P = sphingosine 1-phosphate; S1P1 = sphingosine 1-phosphate receptor subtype 1; SC = subcutaneous; TNF = tumour necrosis factor; TYK2 = tyrosine kinase 2; UC = ulcerative colitis.
Note: All the comparators in this table were included in the ITC as well as the pharmacoeconomic analyses. Adalimumab was included in the ITC, but it was not broken down by adalimumab biosimilars vs. the reference biologic drug. Infliximab was included in the ITC, but it was not broken down by infliximab biosimilars vs. the reference biologic drug. Tofacitinib was included in the ITC, but it was not broken down by tofacitinib generic vs. the reference biologic drug.
aHealth Canada–approved indication.
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the Stakeholder section of this report.
CADTH received 2 patient group input submissions, 1 from the GI Society and the other from CCC. The GI Society is a national charity that is committed to improving the lives of people with GI and liver conditions, supporting research, and advocating for appropriate patient access to health care. CCC is a national, volunteer-based health charity focused on finding treatments and improving the lives of people with CD and UC.
The GI Society collected patient input using a series of surveys conducted between 2015 and 2023 (N = 54 to 579), focus groups, and 1-to-1 interviews with patients with IBD. Patient input from CCC was compiled from 2 online surveys conducted in 2022. In the first survey, the number of respondents with moderate to severe UC was 354, and the second survey captured the experiences of 4 patients with UC.
Both patient groups noted that UC has a profound effect on daily life — physically, emotionally, and socially — at home, school, or in the workplace. Many patients surveyed by CCC revealed they hid aspects of their diagnosis from their friends, coworkers, and classmates. Nearly two-thirds (63%) of respondents agreed that their family and friends do not understand what they are going through. Patients noted that symptoms can be relentless, embarrassing, and scary. Based on the surveys conducted by CCC, the most frequently reported UC-related complications were mental health and stress (65%), joint inflammation and arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), skin conditions (about 30%), and malnutrition and weight loss (about 30%). Patients stated that sustained remission and/or treatment response is more important than relieving any 1 symptom. The patients with UC expressed their constant concern that there would be future flares, possibly worse than the last and occurring at unpredictable times, which is disastrously disruptive.
Regarding current treatments for UC, it was noted that although there are several available options, most patients have difficulty obtaining remission or adequate symptom relief. Based on survey data from the GI Society, approximately a quarter (24%) of patients with IBD found available medications to be adequate, 56% found them to be only somewhat adequate, and 20% found them to be not at all adequate. About half of the patients (56%) surveyed by CCC believed that different treatment options could make them feel better. According to the input from CCC, although steroid use is an important part of symptom management for UC, patients reported not being particularly supportive of this treatment option and had concerns about side effects from systemic steroid use. Patient input from the GI Society emphasized the importance of having a variety of treatment options available because UC is a chronic disease and there is no cure. According to the patient input, a patient typically needs to change the type(s) of treatment when there is an inadequate response to the initial treatment. Patient respondents expressed a need for new and effective options to achieve mucosal healing and reduce the symptoms of UC. The input from CCC noted that patients seek any treatments that can relieve UC symptoms to protect their ability to work, study, and care for family. The patients interviewed by CCC noted the most important aspects around UC management include having enough treatment options, understanding side effects, and minimizing steroid use. Neither patient group was able to identify a patient to interview who had experience with etrasimod therapy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of UC.
Unmet Needs
The clinical expert consulted by CADTH considered the symptomatic relief with clinical, serological, biomarker, and endoscopic remission through Health Canada–approved therapies that are effective at induction and during maintenance as the goals of UC treatment. The clinical expert noted several challenges among the currently available medical therapies for UC in Canada. First, a significant portion of patients will not respond to advanced therapy (most therapies exhibit endoscopic healing rates below 50% at 1 year). Second, it is common for patients who respond initially to lose response after a period of symptom relief. For those medications where a loss of response occurs, dose escalation is common. Therefore, in many patients with UC, multiple options, including increasing medication dosage and trying several types, are needed to maintain response and meet longer-term treatment goals. The clinical expert pointed out that multiple drug failures and ongoing progressive disease activity may lead to adverse consequences, including surgery to remove the entire colon. Finally, there is also a gap in available oral therapies, as most drugs used currently are administered intravenously or subcutaneously.
Place in Therapy
The clinical expert indicated that a clear sequence of medications that is optimal for treating moderate to severe UC is not yet established. The clinical expert noted that in an outpatient context, etrasimod (an S1P receptor modulator) could be introduced primarily in patients with moderate UC following the failure of a 5-ASA treatment. The clinical expert’s opinion was that etrasimod might be better placed earlier in treatment sequencing. The clinical expert noted the evidence suggests the efficacy of etrasimod may be lower when used after more drug failures, based on the subgroup results in patients in both etrasimod trials who had previously received more than 1 advanced therapy or who had experienced the failure of an anti-TNF treatment. Therefore, the clinical expert suggested that, to optimize efficacy, etrasimod should be considered for and administered to patients with UC earlier in their disease course. However, the clinical expert acknowledged that optimal sequencing of medications for moderate to severe UC is unclear across all available products, and the trials for etrasimod were not designed to specifically address the sequencing question.
Patient Population
The clinical expert described that patients with a confirmed pathologic or histologic diagnosis of moderate to severe UC are typically diagnosed by a gastroenterologist and sometimes, in more rural parts of the country, by a surgeon. Misdiagnosis is infrequent. The clinical expert noted that although some clinical risk factors such as early age of onset (younger than 40 years), extensive colitis, and need for corticosteroids at diagnosis may be associated with a more complex course, there are currently no available predictors of disease response to a therapy (e.g., generic profile or available blood tests). The clinical expert also noted that etrasimod would not likely be used in the acute, hospitalized setting for acute severe UC, as this is a unique context with standard of care with IV anti-TNF alpha drugs used predominantly.
Assessing the Response to Treatment
The clinical expert indicated that outcome assessments are guided by international recommendations regarding treatment goals for IBD, i.e., the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE)-II guidelines published in 2021.81 The clinical expert indicated the outcomes that are most important for patients at various stages. First, in the short-term, early in the disease course, clinical response is important to ensure patients are responding in terms of their symptoms, including extreme stool frequency, diarrhea, rectal bleeding, nighttime defecation, and urgency. The relief of these symptoms is important for patients in terms of their daily function, return to work or school, and ability to sleep through the night. Next, the main intermediate-term target is symptom improvement or remission and resolution of both blood-based (C-reactive protein) and stool-based biomarkers (fecal calprotectin). Finally, usually within 6 months, the goal is ideally to exhibit endoscopic healing or at least significant improvement. The clinical expert indicated that for UC, the goal of exhibiting histologic healing is not currently considered a robust accepted treatment target, although there is evidence to suggest histologic healing does predict improved outcomes; however, it is not used as a clinical target in routine clinical practice. The clinical expert noted that after the initiation of medication, a check-in within the first 1 to 2 weeks is essential to verify some clinical improvement. Once an early response is established, another check-in at around 4 to 6 weeks is appropriate, followed by a full assessment with blood work and stool studies completed at 12 weeks. An endoscopic exam at between 6 and 12 months of treatment is preferred with etrasimod.
Discontinuing Treatment
The clinical expert indicated that the discontinuation of treatment with etrasimod should be considered in a manner similar to other advanced therapies for adults with moderate to severe UC, with factors that include:
an inability to decrease the oral corticosteroid dose despite treatment with etrasimod (steroid dependence)
the early recurrence of symptoms despite the full 12 weeks of initial therapy with etrasimod
a persistent elevation of biomarkers, especially fecal calprotectin, and limited or no improvement of symptoms after 12 weeks of initial treatment with etrasimod
evidence of persistent disease activity after initial therapy with etrasimod (12 weeks) or signs of progression during maintenance therapy based on endoscopy.
Prescribing Considerations
The clinical expert indicated that, ideally, the prescription of etrasimod should be limited to gastroenterologists who treat IBD (e.g., in the community or in academic centres); however, prescribing by expert internal medicine physicians or surgeons in rural settings would be an exception.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by the clinician group. The full original clinician group input received by CADTH has been included in the Stakeholder section of this report.
CADTH received 1 clinician group input submission from the Canadian IBD Interest Group, which is an assembly of gastroenterologists from across Canada with subspecialty expertise in IBD management. Input from the clinician group was based on a meeting in December 2023 among 12 clinicians (members of the group) to discuss UC treatment and review the literature and data from the etrasimod clinical development program.
Based on the input from the clinician group, the treatment goals for UC are in alignment with those of the clinical expert consulted by CADTH and include controlling symptoms as well as improving objective markers of disease activity, i.e., endoscopic assessment, histologic assessment, and biomarkers such as fecal calprotectin and serum C-reactive protein. The clinician group noted that treatment for UC is influenced by disease severity and may involve medications that include oral and/or rectal 5-ASA, systemic corticosteroids, advanced biologics (adalimumab, infliximab, golimumab, vedolizumab, ustekinumab, mirikizumab) and advanced small-molecule drugs (tofacitinib, upadacitinib, ozanimod). The clinician group pointed out that there is a need for oral therapies that are well tolerated and provide durable disease control.
The clinician group anticipated that etrasimod is likely to be used as a first-line advanced therapy and could also be used in selected cases as a second- or third-line drug for UC treatment, based on several potential advantages of etrasimod that, from their perspective, include:
oral delivery
a once-daily dosing regimen
efficacy in all patient subgroups, including those with limited proctitis (the clinician group noted that the patients with UC with ulcerative proctitis have been excluded from previous clinical trials, but they represent up to 30% of the overall UC population)
favourable long-term safety compared with existing oral alternatives, including ozanimod, upadacitinib, and tofacitinib.
The clinician group noted that etrasimod would be unlikely to be used in patients with fulminant UC or who were hospitalized due to UC, as this therapy has not been evaluated in that setting. The clinician group pointed out that discontinuation with etrasimod can be considered when there is an inadequate clinical response (assessment of both symptoms and objective biomarkers of disease activity) within 12 to 16 weeks of treatment, or a significant adverse effect occurs.
In alignment with the input from the clinical expert consulted by CADTH, the clinician group anticipated that etrasimod will be prescribed by physicians experienced in the management of UC, most often by gastroenterologists or general internists with specific training and experience. The clinician group indicated that etrasimod would be administered at home and, for the rare patients who require first-dose monitoring, its administration should be under supervision in an ambulatory clinic setting.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There are many conventional and advanced treatments in this space. Additionally, there is 1 other approved drug (ozanimod) that can be used as a comparator for etrasimod. The clinical trials compared etrasimod with placebo. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
UC is diagnosed definitively through endoscopy. Other differentials can be ruled out through lab testing of blood or fecal matter and testing for infectious causes. Scoring (staging):
The indication for etrasimod is for moderate to severe UC. This is in line with the comparator (ozanimod) and other advanced biologic and nonbiologic treatments. | This is a comment from the drug plans to inform CDEC deliberations. The clinical expert consulted by CADTH confirmed that UC is definitively diagnosed endoscopically through endoscopic assessment and histologic confirmation (establishing chronicity). The clinical expert noted that the ESR is no longer used in UC diagnosis. |
Ozanimod is approved for patients aged between 18 and 64 years. Etrasimod is seeking funding for patients aged 18 years and older. Should etrasimod be approved for patients who are older than 64 years, or should it be in line with ozanimod? (The risk of bradycardia and reflex hypertension is greater with ozanimod than with etrasimod, but still seems to be a risk.) | The clinical expert noted that older patients (e.g., older than 64 years) are a more vulnerable population due to comorbidities and the potential for multiple prescribed additional medications. Harms associated with S1P receptor modulators like etrasimod include cardiac dysfunction, especially dysrhythmias. Currently, there are limited safety data in older patients with UC and, even if these AEs turn out to occur infrequently, they could have important health consequences. Therefore, until there are more long-term harms data, clinicians would likely be cautious in starting etrasimod in older patients with UC and would prefer to prescribe other advanced therapies that have well-established harms profiles and those therapies with which clinicians have years of experience (e.g., vedolizumab, ustekinumab, and mirikizumab). |
The drug plans noted that 20% to 40% of patients on conventional therapy do not respond to treatment. Should patients require a trial of conventional therapy (5-ASAs, thiopurines, sulfasalazine, corticosteroids) before initiation of etrasimod? Or should a diagnosis of moderate or severe UC give them access to etrasimod? | The clinical expert indicated that based on evidence from the ELEVATE trials, etrasimod would not be reserved for patients for whom there are other contraindicated drugs or other access limitations. The clinical expert noted the evidence suggests the efficacy of etrasimod diminishes with more drug failures. Therefore, to optimize the efficacy, the clinical expert suggested that etrasimod should be considered and administered to patients with UC earlier in their disease course (i.e., a trial of conventional therapy before initiation of treatment for moderate to severe UC would not be required). |
Should patients who develop AEs such as transaminitis or lymphopenia be eligible for re-treatment once their lab values normalize? | The clinical expert pointed out that re-treatment would depend on the severity of abnormality in the patients’ lab values (e.g., the level of liver enzyme to monitor the AEs of liver injury), which may preclude the re-introduction of etrasimod. |
Would patients with fulminant UC be eligible for treatment? Question to expert: Do you expect etrasimod to be used in Crohn disease? | The clinical expert pointed out that patients with fulminant UC would not be candidates for etrasimod. The clinical expert noted that etrasimod is unlikely to be used in patients with Crohn disease. |
Ozanimod initiation criteria: Mesalamine 4 g/day for 4 weeks AND corticosteroid (failure to respond to prednisone 40 mg for 2 weeks or steroid-dependent and unable to taper off). Proposed etrasimod criteria: Failure of 5-ASA and/or corticosteroid. There is a discrepancy in the proposed initiation criteria for etrasimod and the current criteria for ozanimod. To CADTH: Consider alignment with initiation criteria for ozanimod, if appropriate. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Reassessment is based on the Mayo score, which includes endoscopic findings. Will patients be required to have an endoscopy done yearly to show remission? Or will a partial Mayo score suffice? | The clinical expert noted that patients should neither be expected nor required to undergo endoscopic examinations annually and noted that there can be challenges with access to regular endoscopies. The clinical expert pointed out that surrogate measures, including the biomarker (level of fecal calprotectin) that is accurate in the detection of colonic inflammation, are used to determine the state of disease activity. The clinical expert noted that a partial Mayo score is also important in consideration for continuation or renewal of etrasimod. |
Ozanimod was recently negotiated with a successful LOI. The renewal criteria require reassessment by a specialist within 10 to 12 months, and confirmation of a decrease in a partial Mayo score of greater than or equal to 2. Consider alignment with renewal criteria for ozanimod, if appropriate. | This is a comment from the drug plans to inform CDEC deliberations. |
Considerations for discontinuation of therapy | |
What are the parameters for the discontinuation criteria to be considered? Should an increase in Mayo score be considered a discontinuation criterion? | The clinical expert indicated the treatment discontinuation of etrasimod should be considered in a manner similar to other advanced therapies for adults with moderate to severe UC, with factors including:
The clinical expert noted that an increase in Mayo score alone is unlikely, but when it is used in combination with an increase in fecal calprotectin, it can be considered a discontinuation criterion for etrasimod. |
Considerations for prescribing of therapy | |
The drug plans noted that etrasimod is given once daily by mouth. Unlike ozanimod, etrasimod does not require induction and can be started at a therapeutic dose of 2 mg daily. The drug plans also noted that etrasimod is administered orally with no handling precautions. | This is a comment from the drug plans to inform CDEC deliberations. |
There may be difficulties in accessing gastroenterologists in rural settings. Virtual assessment could be an option; however, there is still the requirement for endoscopy to ensure that diagnosis and, potentially, renewal criteria are met. Endoscopy may not be readily available to patients. | This is a comment from the drug plans to inform CDEC deliberations. |
Will patients on etrasimod be eligible for additional treatment with biologics or JAK inhibitors? Criteria for ozanimod do not allow for additional treatment but do allow for change in therapy to biologics or JAK inhibitors. | The clinical expert noted that it is not likely that etrasimod to be used in combination with other advanced treatments or JAK inhibitors. |
The drug plans asked for CDEC to consider aligning prescribing criteria for etrasimod with ozanimod, as appropriate. | This is a comment from the drug plans to inform CDEC deliberations. |
Care provision issues | |
The drug plans noted that bradycardia, hypertension, transaminitis, and lymphopenia are expected adverse effects. | This is a comment from the drug plans to inform CDEC deliberations. |
Should immunization be a requirement for prescribing etrasimod? If so, what vaccines (e.g., childhood vaccines, vaccines for pneumonia, RSV, shingles)? | The clinical expert pointed out that it would be safest to have immunization before prescribing etrasimod; however, mandating this is unlikely to be feasible. |
The drug plans noted there is a need for initial assessment and monitoring, including endoscopy, ECG to monitor QTc prolongation and evidence of second-degree AV block (should be readily available), fundoscopy in diabetics, lab work for initial access and monitoring (LFTs, CBC). Question for clinical expert: Do you foresee access delays due to endoscopies? Do you expect issues with endoscopy being a criterion for renewal? | The clinical expert noted that the challenge in accessing to endoscopic examination is universal across Canada for patients with UC (i.e., not unique to the administration of etrasimod). The clinical expert noted that the requirement of an endoscopic examination for etrasimod renewal would be prohibitive for the use of etrasimod, and an endoscopic examination is not commonly applied to the renewal of other UC medications. The clinical expert suggested that, alternatively, a partial Mayo score could be used in determining etrasimod renewal. |
System and economic issues | |
There would be no concern if criteria and pricing were in line with the recently negotiated criteria and price for ozanimod. The intention is for this to be an additional treatment tool for moderate to severe UC. | This is a comment from the drug plans to inform CDEC deliberations. |
Ozanimod has recently completed negotiations, and all jurisdictions participated in the LOI. Etrasimod would need confidential pricing equal to ozanimod, as they are both in the same class of drug (S1P inhibitors). | This is a comment from the drug plans to inform CDEC deliberations. |
5-ASA = 5-aminosalicyclic acid; AE = adverse event; AV = atrioventricular; CBC = complete blood count; CDEC = Canadian Drug Expert Committee; CRP = C-reactive protein; ECG = electrocardiogram; ESR = erythrocyte sedimentation rate; JAK = Janus kinase; LFT = liver function test; LOI = letter of intent; MD = medical doctor; RSV = respiratory syncytial virus; S1P = sphingosine 1-phosphate; UC = ulcerative colitis.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of etrasimod (2 mg taken orally once daily) in the treatment of adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. The focus will be placed on comparing etrasimod with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of etrasimod is presented in 4 sections, with CADTH’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. CADTH’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes the sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence. There were no long-term extension studies (section 2) nor additional studies to address important gaps in the systematic review evidence (section 4) submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
2 pivotal placebo-controlled RCTs
1 NMA.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
ELEVATE UC 12 and ELEVATE UC 52 are pivotal phase III RCTs evaluating the safety and efficacy of etrasimod for moderately to severely active UC when administered for 12 weeks (ELEVATE UC 12 trial) and 52 weeks (ELEVATE UC 52 trial).
ELEVATE UC 12 Trial
ELEVATE UC 12 (N = 354) was a multicentre, randomized, placebo-controlled, double-blind study. The primary objective of the ELEVATE UC 12 trial was to assess the efficacy of etrasimod in clinical remission at week 12 in patients with moderately to severely active UC. The ELEVATE UC 12 trial consisted of a 28-day screening period, a 12-week randomized treatment period, and 2-week and 4-week follow-up periods. At the end of the 12-week treatment period, patients underwent efficacy and safety assessments and were evaluated for clinical response or remission. At the end of the 12-week treatment period, patients had the choice to enter the OLE study (ELEVATE OLE) if they met the eligibility criteria. Patients who did not participate in the ELEVATE OLE study had follow-up visits at weeks 2 and 4 after the last dose of the study treatment. A study design flow diagram of the ELEVATE UC 12 trial is outlined in Figure 1.
Table 5: Details of Studies Included in the Systematic Review
Detail | ELEVATE UC 12 trial | ELEVATE UC 52 trial |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, randomized, double-blind, placebo-controlled study. | Phase III, multicentre, randomized, double-blind, placebo-controlled study. |
Locations | This study was conducted at 407 centres in 39 countries from continents of Africa, the Americas, Asia, Australia, and Europe. | This study was conducted at 315 centres in 37 countries from continents of Africa, the Americas (Canada = 1 site with 1 patient), Asia, Australia, and Europe. |
Patient enrolment dates | Start date: September 2020. End date: December 2021. | Start date: June 2019. End date: February 2022. |
Randomized (N) | N = 354:
| N = 433:
|
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention | Etrasimod 2 mg tablet taken orally once daily for up to 12 weeks of treatment. | Etrasimod 2 mg tablet taken orally once daily for up to 52 weeks of treatment. |
Comparator | Placebo tablet taken orally once daily for 12 weeks of treatment. | Placebo tablet taken orally once daily for 52 weeks of treatment. |
Study duration | ||
Screening phase | 28 days | 28 days |
Treatment phase | 12 weeks | 52 weeks:
|
Follow-up phase | Follow-up visits at weeks 2 and 4 after the last dose of study drug if the patient was not enrolled in the long-term extensions (ELEVATE OLE study). | Follow-up visits at weeks 2 and 4 after the last dose of study drug if the patient was not enrolled in the long-term extensions (ELEVATE OLE study). |
Outcomes | ||
Primary end point | The proportion of patients achieving clinical remission at week 12. |
|
Secondary and exploratory end points | Key secondary:
Other secondary:
Exploratory:
Safety: AEs, TEAEs, serious TEAEs, TEAEs leading to discontinuation of treatment, notable harms, including cardiovascular events, infections, liver injury, and pulmonary disorders. | Key secondary:
Other secondary:
Exploratory:
Safety: AEs, TEAEs, serious TEAEs, TEAEs leading to discontinuation of treatment, and notable harms, including cardiovascular events, infections, liver injury, and pulmonary disorders. |
Publication status | ||
Publications | Sandborn, W.J. et al. (2023)82 | |
5-ASA = 5-aminosalicylic acid; AE = adverse event; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; BP = blood pressure; CD = Crohn disease; ECG = electrocardiogram; EIM = extraintestinal manifestation; ES = endoscopic score; HR = heart rate; JAK = Janus kinase; MMS = modified Mayo score; NHI = Nancy histological index; OLE = open-label extension; RB = rectal bleeding; RHI = Robarts histopathology index; SF = stool frequency; TEAE = treatment-emergent adverse event; TMS = total Mayo score; UC = ulcerative colitis; ULN = upper limit of normal.
aInadequate response, loss of response, and intolerance are defined as: 1) Inadequate response: Signs and symptoms of persistently active disease despite a history of completing a dosing regimen; 2) Loss of response: Recurrence of symptoms of active disease during treatment following prior clinical benefit (discontinuation despite clinical benefit does not qualify as treatment failure or patient being intolerant to UC biologic therapy); 3) Intolerance: Including, but not limited to, infusion- or injection-related reaction, demyelination, congestive heart failure, infection, or any other related AE that led to a reduction in dose or discontinuation of the medication.
bAdequate hematological function defined by white blood cell count ≥ 3.5 × 109/L with ANC ≥ 1.5 × 109/L, lymphocyte count ≥ 0.8 × 109/L, platelet count ≥ 100 × 109/L, and hemoglobin ≥ 8 g/dL.
cAdequate hepatic function defined by a total bilirubin level ≤ 1.5 × ULN range and AST and ALT levels ≤ 2.0 × ULN. Patients with an isolated total bilirubin elevation and normal AST and ALT diagnosed with Gilbert syndrome could participate.
dAdequate renal function defined by an estimated glomerular filtration rate ≥ 30 mL/min/1.73 m2 by the Chronic Kidney Disease Epidemiology Collaboration equation at screening.
eSevere extensive colitis as evidenced by physician judgment that the patient is likely to require hospitalization for medical care or surgical intervention of any kind for UC (e.g., colectomy) within 12 weeks following randomization; current evidence of fulminant colitis, toxic megacolon, or recent history (within last 6 months) of toxic megacolon or bowel perforation; and previous total or partial colectomy.
fHave any of the following conditions or receiving treatments that may affect cardiovascular function: myocardial infarction, unstable angina, stroke or transient ischemic attack, decompensated heart failure requiring hospitalization or Class III or IV heart failure ≤ 6 months before or during the screening period; history or presence of second-degree or third-degree atrioventricular block, sick sinus syndrome, or periods of asystole for > 3 seconds without a functional pacemaker; history or presence of recurrent symptomatic bradycardia or recurrent cardiogenic syncope; screening or week 0 (day 1) prerandomization vital sign readings (taken in the sitting position) showing an HR < 50 bpm, or systolic BP < 90 mm Hg, or diastolic BP < 55 mm Hg. The taking of vital signs may be repeated up to 3 times during a visit to confirm abnormal readings; screening or week 0 (day 1) prerandomization ECG with a PR interval > 200 ms or Fridericia’s corrected QT interval (QTcF) ≥ 450 ms in males or ≥ 470 ms in females; start, stop, change, or planned change in dosage of any antiarrhythmic drug (class I to IV) ≤ 1 week before screening or within 1 week before or after randomization.
gA 40-week maintenance treatment period with a treat-through design.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and the sponsor’s summary of clinical evidence.16
Patients were enrolled across 407 sites in 39 countries, including 2 sites in Canada. Eligible patients were randomized in a 2:1 ratio to receive either blinded etrasimod at a dose of 2 mg (n = 238) or matching placebo (n = 116) for 12 weeks of treatment. Patients in the ELEVATE UC 12 trial were centrally assigned to randomized study treatment using an interactive web response system. Randomization was stratified by previous biologic or JAKi therapy exposure (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). The ELEVATE UC 12 study was completed on December 7, 2021.
Figure 1: Study Design of the ELEVATE UC 12 Trial
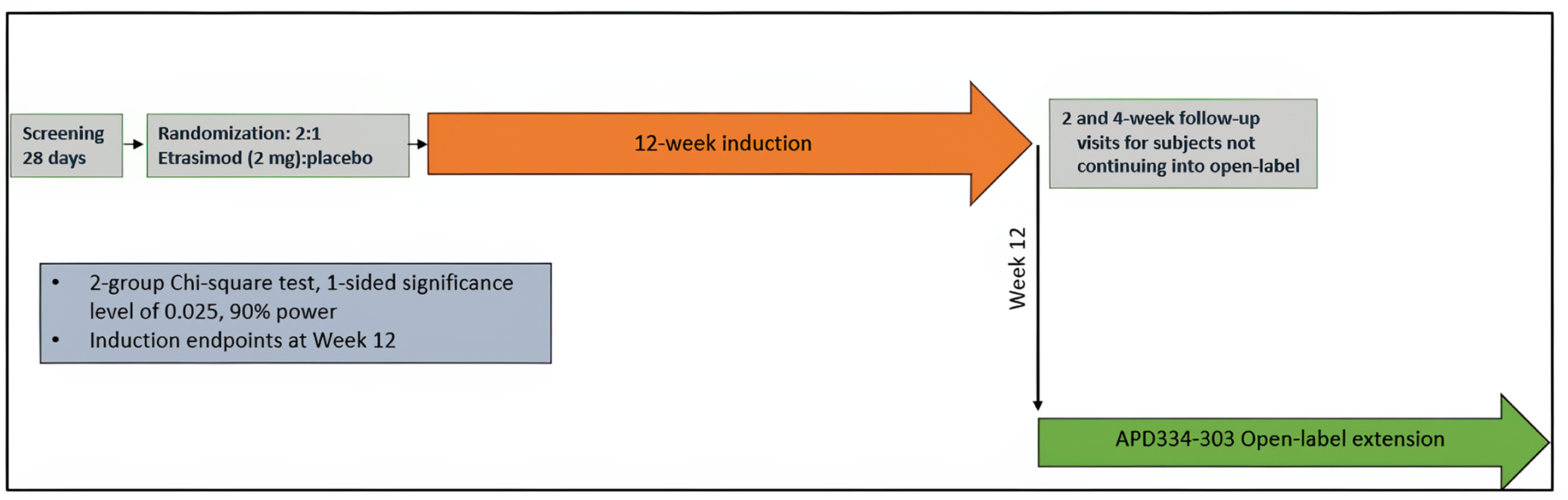
OLE = open-label extension.
Source: Clinical Study Report for ELEVATE UC 12.18
ELEVATE UC 52 Trial
ELEVATE UC 52 (N = 433) was a multicentre, randomized, placebo-controlled, double-blind study. The primary objective of the ELEVATE UC 52 trial was to assess the efficacy of etrasimod in clinical remission after 12 and 52 weeks of treatment in patients with moderately to severely active UC. The ELEVATE UC 52 trial comprised a 28-day screening period and a 12-week induction treatment period followed by a 40-week maintenance treatment period with a treat-through design (i.e., patients continued their double-blind study treatment), and 2-week and 4-week follow-up periods for patients who did not enrol in the ELEVATE OLE study. At the end of the 12-week treatment period, patients underwent efficacy and safety assessments and were evaluated for clinical response or remission as well as UC disease worsening. Patients who experienced disease worsening after 12 weeks of treatment and met the predefined eligibility criteria could participate in the ELEVATE OLE study. Patients who did not meet disease worsening criteria, including those achieving clinical response or clinical remission at week 12, continued into the 40-week treatment period and continued their double-blind treatment (per the treat-through design). Patients who either experienced disease worsening during the 40-week treatment period or completed all study procedures at week 52 had the choice to enrol into the ELEVATE OLE study if they met the predefined eligibility criteria. Patients who did not participate in the ELEVATE OLE study had follow-up visits at weeks 2 and 4 after the last dose of the study treatment. A study design flow diagram of the ELEVATE UC 12 trial is outlined in Figure 2.
Patients were enrolled across 315 sites in 37 countries, including 1 site in Canada. Eligible patients were randomized in a 2:1 ratio to receive either a blinded etrasimod oral tablet at a dose of 2 mg (n = 289) or a matching placebo (n = 144) for up to 52 weeks of treatment. Patients were centrally assigned to randomized study treatment using an interactive web response system. Randomization was stratified by previous exposure to a biologic or JAKi therapy (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). The ELEVATE UC 52 study was completed on February 22, 2022.
Figure 2: Study Design of the ELEVATE UC 52 Trial
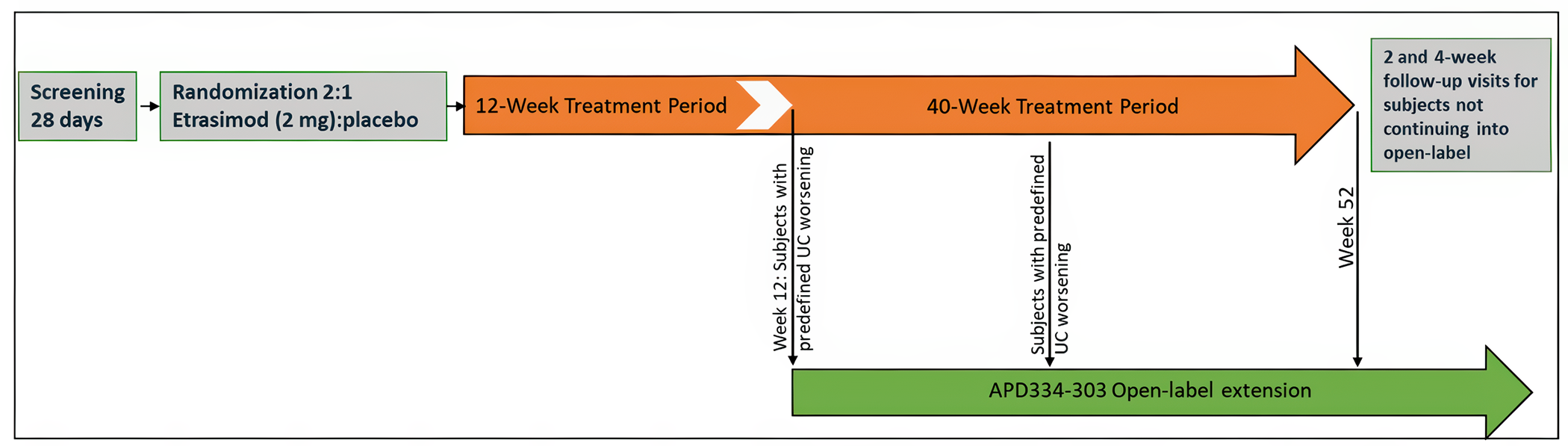
OLE = open-label extension; UC = ulcerative colitis.
Source: Clinical Study Report for the ELEVATE UC 52 trial.17
Populations
Inclusion and Exclusion Criteria
In both the ELEVATE UC 12 and ELEVATE UC 52 studies, eligible patients were aged 16 to 80 years and had moderately to severely active UC. Active UC was confirmed by endoscopy with rectal involvement equal to or greater than 10 cm. Moderate to severe UC was defined as an MMS of 4 to 9, which included an endoscopic subscore of 2 or greater, and a rectal bleeding subscore of 1 or greater. Patients had to be diagnosed with UC at least 3 months before the study screening period. Patients also had to have demonstrated an inadequate response, loss of response, or an intolerance to at least 1 conventional therapy (e.g., corticosteroid, thiopurines) or a biologic or JAKi therapy (e.g., anti-TNF alpha antibodies) approved for the treatment of UC. Inadequate response to at least 1 conventional biologic or JAKi therapy was defined as having signs and symptoms of persistently active disease despite a history of completing a dosing regimen. Loss of response was defined as recurrence of symptoms of active disease during treatment following prior clinical benefit (discontinuation despite clinical benefit does not qualify as treatment failure or the patient being intolerant to UC biologic therapy). Intolerance was defined as including, but not limited to, infusion-related or injection-related reactions, demyelination, congestive heart failure, infection, or any other related AE that led to a reduction in dose or discontinuation of the medication. Patients with isolated proctitis (less than 10 cm rectal involvement) at baseline who met other eligibility criteria could enrol in both studies, with enrolment capped at 15% of total patients.
Patients were excluded from both the ELEVATE UC 12 and ELEVATE UC 52 studies if they had severe extensive colitis, a diagnosis of CD or indeterminate colitis, or the presence or history of a fistula consistent with CD, or a diagnosis of microscopic colitis, ischemic colitis, or infectious colitis. Patients were excluded from both studies if they previously received 3 or more biologic drugs, 2 or more biologics and a JAKi approved for the treatment of UC, or any S1P receptor modulator. Patients who had a clinically relevant cardiac condition (a history of myocardial infarction, stroke, or second-degree or third-degree atrioventricular block), a history of opportunistic infections, macular edema, or a history of or currently active primary or secondary immunodeficiency were also excluded from both the ELEVATE UC 12 and ELEVATE UC 52 studies.
Interventions
Study treatment for both the etrasimod and placebo groups consisted of 1 tablet taken once daily by mouth for a period of 12 weeks in the ELEVATE UC 12 trial, and 52 weeks in the ELEVATE UC 52 trial.
Starting with the week 12 assessment in both studies, patients whose disease had not improved or had worsened compared with baseline could be eligible to participate in the ELEVATE OLE study provided their endoscopic score was 2 or greater and they met 1 of the following eligibility criteria:
a rectal bleeding subscore of 2 or greater at 2 time points at least 7 days and no more than 14 days apart
rectal bleeding plus stool frequency subscores of 4 or greater at 2 time points at least 7 days and no more than 14 days apart
a rectal bleeding subscore of 2 or greater, or rectal bleeding plus stool frequency subscores of 4 or greater (in any order) at 2 time points at least 7 days and no more than 14 days apart.
Concomitant Medications
The following concomitant UC therapies were permitted in both the ELEVATE UC 12 and ELEVATE UC 52 studies; however, these products could not be started during screening or during the treatment period in patients who were not already receiving them:
immunosuppressive drugs, such as oral azathioprine or 6-mercaptopurine had to be discontinued at least 2 weeks before randomization
oral 5-ASA compounds, provided the dose had been stable for at least 2 weeks immediately before randomization
oral corticosteroid therapy (prednisone at a stable dose of 20 mg/day or less, budesonide at a stable dose of 9 mg/day or less, or an equivalent steroid), provided the dose had been stable for the 4 weeks immediately before the screening endoscopy assessment
probiotics (e.g., Culturelle, Saccharomyces boulardii) provided the dose had been stable for the 2 weeks immediately before randomization.
During the 12-week treatment period in both studies, patients were to maintain their stable baseline corticosteroid dose.
In the ELEVATE UC 52 trial, following the week 12 assessment, corticosteroids were tapered for patients entering the 40-week treatment period. The recommended tapering schedule for oral corticosteroids (other than budesonide extended-release tablets) was as follows:
Dose of prednisone or equivalent is greater than 10 mg/day: Taper daily dose by 5 mg/week until receiving 10 mg/day, and then continue tapering at 2.5 mg/week until 0 mg/day.
Dose of prednisone or equivalent is 10 mg/day or less: Taper daily dose by 2.5 mg/week until 0 mg/day.
Live vaccines and medications that were expected to cause clinically important drug–drug interactions (e.g., moderate or strong inhibitors of CYP2C8 or CYP2C9) were prohibited during the ELEVATE UC 12 and ELEVATE UC 52 studies.
Outcomes
A list of efficacy end points assessed in the Clinical Study Reports for the ELEVATE UC 12 and ELEVATE UC 52 trials is provided in Table 6, followed by descriptions of the outcome measures. The summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review, according to the clinical expert consulted by CADTH, and stakeholder input from patient and clinician groups and public drug plans. Using the same considerations, the CADTH review team selected end points considered to be most relevant to inform CADTH’s expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. The proportion of patients with serious TEAEs was also assessed using GRADE.
Efficacy Outcomes
The ELEVATE UC 12 and ELEVATE UC 52 trials investigated the same outcomes at either week 12 only (ELEVATE UC 12 trial) or both week 12 and week 52 (ELEVATE UC 52 trial). Sustained clinical remission at both weeks 12 and 52 was investigated only in the ELEVATE UC 52 study.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | ELEVATE UC 12 trial | ELEVATE UC 52 trial |
|---|---|---|---|
Endoscopic improvement | At week 12 | Key secondarya | Key secondarya |
At week 52 | NA | Key secondarya | |
Mucosal healing | At week 12 | Key secondarya | Key secondarya |
At week 52 | NA | Key secondarya | |
At both weeks 12 and 52 | NA | Secondary | |
Clinical remission | At week 12 | Primarya | Primarya |
At week 52 | NA | Primarya | |
Sustained clinical remission | At both weeks 12 and 52 | NA | Key secondarya |
Corticosteroid-free for ≥ 12 weeks before week 52 and achieved clinical remission | At week 52 | NA | Key secondarya |
Corticosteroid-free for ≥ 4 weeks before week 52 and achieved clinical remissionb | At week 52 | NA | Secondary |
Clinical response | At week 12 | Secondary | Secondary |
At week 52 | NA | Secondary | |
At both weeks 12 and 52 | NA | Secondary | |
Symptomatic remission | At week 12 | Key secondarya | Key secondarya |
At week 52 | NA | Key secondarya | |
IBDQ total score | From baseline to week 12 | Other efficacy end point | NA |
From baseline to weeks 12 and 52 | NA | Other efficacy end point |
GRADE = Grading of Recommendations Assessment, Development and Evaluation; IBDQ = Inflammatory Bowel Disease Questionnaire; NA = not applicable; UC = ulcerative colitis.
aStatistical testing for these end points was adjusted for multiple comparisons (i.e., Hochberg procedure was used to adjust for multiple comparisons).
bThe results of this outcome among the patients who were receiving corticosteroids for UC at baseline were included in the GRADE assessment.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and the sponsor’s summary of clinical evidence.16
A description of the efficacy outcome measures and their measurement properties that were used in both the ELEVATE UC 12 and ELEVATE UC 52 trials are presented in Table 7.
Endoscopic Improvement
Endoscopic improvement was defined as achieving an endoscopic subscore of 0 or 1, excluding friability.
Mucosal Healing
Mucosal healing was defined as achieving an endoscopic subscore of 1 or less, with histologic remission measured by a Geboes Index score of less than 2.
The Geboes Index score assesses features associated with histological inflammation in UC.83,84 This histologic grading system evaluates all aspects of mucosal injury seen in UC, including crypt architecture, lamina propria chronic inflammation, lamina propria eosinophils, lamina propria neutrophils, intraepithelial neutrophils, crypt destruction, and surface epithelial injury.
Clinical Remission
Clinical remission was the primary end point in the ELEVATE UC 12 trial at week 12 and in the ELEVATE UC 52 trial at weeks 12 and 52. Clinical remission was based on the MMS as opposed to the total Mayo score (TMS). The MMS excludes the Physician’s Global Assessment to reduce subjectivity in the assessment, in accordance with the regulatory guidance for trials in UC.22,85 Clinical remission was defined as:
a stool frequency subscore of 0 or 1 with at least a 1-point decrease from baseline
a rectal bleeding subscore of 0
an endoscopic subscore of 0 or 1.
Regulatory guidance22,85 has indicated that the presence of any friability is not consistent with clinical remission, which requires an endoscopic subscore of 0 or 1. To align with this guidance, the endoscopic score was increased to 2. Scoring of the MMS subscores is further detailed in Table 7.
Rectal bleeding and stool frequency subscores were from patient-derived assessments and were captured in an e-diary that was recorded in real time and not subject to change. The rectal bleeding subscore reports the most severe amount of blood passed by rectum in a 24-hour period. The stool frequency subscore reflects the number of stools in a 24-hour period relative to the normal number of stools for that individual. The total number of stools passed in a 24-hour period was recorded by the patient in a daily e-diary. On visits when MMS was calculated, these subscores were derived using the scores from the last 3 consecutive days of diary entries or 4 nonconsecutive days within 7 days before the assessment date. Otherwise, the rectal bleeding and stool frequency subscores were considered missing and the patient was considered a nonresponder.
Endoscopy images were reviewed by a local endoscopist and central laboratory reader. In the case of a score discrepancy, a second read by a central adjudication reader was performed. The endoscopic subscore reports the worst appearance of the mucosa on flexible sigmoidoscopy or colonoscopy on a 4-point scale. Endoscopy was performed during screening at week 12 and week 52. Biopsies were obtained at each endoscopy to support assessment of the histopathology end points.
For patients who have had pancolitis for more than 8 years or patients who have had left-sided colitis for more than 12 years without a surveillance colonoscopy within 12 months of baseline, a colonoscopy and biopsies were performed at screening in accordance with the local standard of care to rule out dysplasia. Any adenomatous polyps were to be removed before a patient’s first dose of the study treatment.
Sustained Clinical Remission
Sustained clinical remission was defined as achieving clinical remission at both weeks 12 and 52 and was assessed in the ELEVATE UC 52 trial only.
Corticosteroid-Free Clinical Remission
Corticosteroid-free clinical remission was defined as achieving clinical remission at week 52 and no corticosteroid use for 12 or more weeks before week 52. This outcome was assessed in the ELEVATE UC 52 trial only. The proportion of patients who had not received corticosteroids for at least 4 weeks immediately before week 52 and achieved clinical remission at week 52 was also reported as another end point in the ELEVATE UC 52 trial.
Clinical Response
Clinical response was defined as a decrease of at least 2 points and at least 30% from baseline in MMS, and a decrease of at least 1 point from baseline in the rectal bleeding subscore, or an absolute rectal bleeding subscore of 0 or 1.
Symptomatic Remission
Symptomatic remission was defined as achieving a stool frequency subscore of 0 (or a subscore of 1 with at least a 1-point decrease from baseline) and a rectal bleeding subscore of 0.
Inflammatory Bowel Disease Questionnaire Total Score
The IBDQ was used to assess disease-specific HRQoL in the ELEVATE UC 12 trial at week 12 and the ELEVATE UC 52 trial at week 52. The IBDQ consists of a 32-item list, subdivided into 4 dimensions, including systemic symptoms, bowel symptoms, emotional function, and social function. Total scores range from 32 to 224, with a higher score indicating a better HRQoL. The IBDQ has been shown to have good internal consistency and test–retest reliability and shown to be responsive to change in IBD.86-88 Available studies have suggested that an improvement of 30 points from baseline or an improvement of at least 15 points or greater above placebo may constitute an MID.89-94
Harms Outcomes
Harms were assessed using monitoring of AEs, clinical laboratory findings, 12-lead electrocardiographs, physical examinations, vital signs, pulmonary function tests, ophthalmoscopy, and optical coherence tomography.
AEs were predefined and reported in both the ELEVATE UC 12 and ELEVATE UC 52 studies, including AEs, serious TEAEs, TEAEs leading to discontinuation, death, and TEAEs of special interest, including progressive multifocal leukoencephalopathy, cardiovascular events, macular edema, pulmonary disorders, infections, and liver injury.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
MMS | In the MMS, the definition of an ES of 1 no longer includes mucosal friability and the PGA is excluded. The components of the MMS include:
Scale components are scored on a 4-point scale from 0 to 3, with a score of 0 indicative of normal and a higher score indicative of more severe symptoms. The MMS is a sum of the Mayo SF, RB, and ES, giving a maximum score of 9. | Validity: In a cross-sectional survey of 2,608 patients with UC and their treating gastroenterologist, increases in the MMS were associated with increased odds of adverse outcomes, including a current flare (OR = 1.52; SE = 0.10), a higher number of flares in the past year (OR = 1.17; SE = 0.03), deterioration in clinical status (OR = 1.48; SE = 0.10) and patient-reported overall WPAI (score = 6.94; SE = 0.888).95 A 1-point increase in the MMS was associated with a 0.02-unit decrease in EQ-5D and a 2.73-point decrease in the SIBDQ, suggesting a change in score of > 4 points might be associated with a clinically meaningful reduction in HRQoL.95 Reliability and responsiveness: No studies of the reliability and responsiveness of the MMS were identified. | Evidence of an MID for the MMS in patients with UC was not identified. |
IBDQ | The IBDQ is a disease-specific questionnaire used to assess disease-specific HRQoL in patients with IBD.96 The IBDQ is a 32-item Likert-based questionnaire divided into 4 dimensions:
Responses are graded on a scale from 1 (worst situation) to 7 (best situation). Total IBDQ score ranges from 32 to 224, with higher scores representing better HRQoL. Scores ranging from 170 to 190 are indicative of remission. | Validity: The emotional function dimension of the IBDQ was found to be strongly correlated with the Rand questionnaire (r −0.76; P < 0.001); the systemic symptoms dimension of the IBDQ was weakly correlated to change in the disease activity index (r = 0.036; P = 0.442); and patients’ global rating of change in emotional function was moderately correlated to the emotional function dimension (r = 0.52; P < 0.001) and the bowel symptom dimension (r = 0.42; P = 0.003) of the IBDQ.96 The IBDQ was found to detect changes in the social and emotional state of patients.86 Reliability and responsiveness: The IBDQ was shown to be highly reliable through evaluation of internal consistency (Cronbach alpha 0.7) and test–retest assessment (ICC = 0.9 to 0.99 or Pearson r ≥ 0.8). The IBDQ was also shown to be responsive to change in patients with IBD (P < 0.05).87,88 | While some suggest that an increase of 15 to 32 points may be considered a clinically relevant improvement in HRQoL for patients with CD and UC, evidence from clinical trials suggests that a change of more than 30 points is associated with clinical benefits and an improvement of 15 points or greater above placebo is required among patients with IBD, including those with UC.89-94 |
CD = Crohn disease; ES = endoscopic score; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation; MID = minimal important difference; MMS = modified Mayo score; OR = odds ratio; PGA = Physician’s Global Assessment; RB = rectal bleed; SE = standard error; SF = stool frequency; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; UC = ulcerative colitis; WPAI = Work Productivity and Activity Impairment.
Protocol Amendments
Several amendments were made to the protocols for the ELEVATE UC 12 and ELEVATE UC 52 trials. The major changes included amending the terminology of the first stratification factor, from patients having experienced failure with previous biologic or JAKi therapy to patients being naive to such therapy at study entry; modifying the documentation of response to prior UC therapy as “an adequate response to, loss of response to, or intolerance to prior therapy”; and modifying eligibility criteria (e.g., list of prior therapy failures or nonresponse, contraception use, cardiovascular disease history, and prior therapy washout period). An exclusion criterion was modified to having received treatment with at least 3 biologic drugs or at least 2 biologics plus a JAKi approved for treatment of UC, and the washout period for methotrexate was changed from within 16 weeks to 8 weeks of screening. Experiencing an AE and noncompliance with the protocol or study treatment were added as reasons for treatment discontinuation, and an update was made to define adequate hepatic function as having alanine aminotransferase and aspartate aminotransferase levels that are twice the upper limit of normal or less (reduced from 3 times the upper limit of normal or less). The final amendment of the study protocol was dated February 22, 2021, for the ELEVATE UC 12 trial (version 3; patients were enrolled between September 2020 and August 2021), and December 22, 2020, for the ELEVATE UC 52 trial (version 4; patients were enrolled between June 2019 and January 2021).19
Changes to Statistical Analysis
On January 22, 2022, the primary analysis set for the efficacy end points was updated to the FAS in patients with an MMS of 5 to 9 at baseline, and the subgroup definition related to MMS score was updated accordingly. The sponsor noted that the change in the FAS was made to align with the changes to FDA guidance22 received after the study protocols were finalized. The revised FDA guidance states that, “For clinical trials for drugs intended to treat moderately to severely active UC: patients should have a score of 5 to 9 on the MMS, including an endoscopy subscore of at least 2.” The sponsor also noted that the statistical analysis plan for both pivotal trials was finalized on January 19, 2022, when early draft guidance was available from the FDA.22 This change excluded approximately 44 patients who had a baseline MMS of 4 from the primary efficacy analysis. The full FAS (patients with a baseline MMS of 4 to 9) was used in supplementary analyses of efficacy end points and safety assessment.
Statistical Analysis
The statistical analysis of the efficacy end points in the pivotal trials assessed in this Clinical Review Report is summarized in Table 8.
Sample Size and Power Calculation
ELEVATE UC 12 Trial
In the ELEVATE UC 12 trial, based on a 2-group Fisher exact test, a 1-sided significance level of 0.025, and a randomization ratio of 2:1, a total of 330 patients (220 patients in the etrasimod group and 110 patients in the placebo group) were required to achieve at least 90% power to detect a difference of 12.5% in the primary end point of clinical remission between the etrasimod (18.5%) and placebo (6.0%) groups.
ELEVATE UC 52 Trial
Based on a 2-group Fisher exact test, a 1-sided significance level of 0.025, and a randomization ratio of 2:1, a total of 420 patients (280 patients in the etrasimod group and 140 patients in the placebo group) were required to achieve 93.4% power to detect a difference of 13.5% in the primary end point of clinical remission at week 52 between the etrasimod (23.5%) and placebo (10.0%) group. With this sample size, there was 96% power to detect a difference of 12.5% in the other primary end point of clinical remission at week 12, assuming a placebo rate of 6.0%. The lower bound of overall power for the coprimary end points (i.e., clinical remission at weeks 12 and 52) was at least 90%; since the coprimary end points were expected to be at least moderately positively correlated, the actual overall power to reject both null hypotheses was likely greater than 90%.
Statistical Testing
The primary efficacy analysis in the ELEVATE UC 12 and ELEVATE UC 52 trials was performed for the FAS and a baseline MMS score of 5 to 9. All primary and key secondary end points were statistically analyzed using a Cochran-Mantel-Haenszel test, stratified by naive to biologic or JAKi therapy at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). Reported randomization stratum was used in the model. The analysis results were presented as the number and proportion of responders in the treatment groups, difference in proportions and 95% CI, and odds ratio and 95% CI. Risk differences in the end points between the etrasimod and placebo groups were calculated using the Wilson score method.
Change from baseline in an MMS was summarized by visit using descriptive statistics (n, mean, standard deviation [SD], median, minimum, and maximum), and also using a mixed model for repeated measures (MMRM) adjusted for naive to biologic or JAKi therapy at study entry (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (MMS of 4 to 6 or 7 to 9), treatment, visit, treatment by visit interaction, a covariate of the corresponding baseline MMS, and a random patient effect. LS means, standard errors, CIs, and P values by visit were reported. For HRQoL measurements (i.e., the Short Form [36] Health Survey), comparisons were made using MMRM analysis, adjusted for naive to biologic or JAKi therapy at study entry (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (MMS of 4 to 6 or 7 to 9), treatment, visit, treatment by visit interaction, a covariate of the corresponding baseline MMS, and a random patient effect.
Multiple Comparisons and Multiplicity
ELEVATE UC 12 Trial
To control for multiplicity for the primary and key secondary end points, the familywise type I error was controlled at a fixed 2-sided significance level of 0.05 using the parallel gatekeeping procedure, that is, the truncated Hochberg procedure. First, the whole significance level of 0.05 was spent on testing the primary end point. Only if the primary null hypothesis was rejected at the significance level could testing proceed for the 3 key secondary end points. Any key secondary end point that failed to be significant at the significance level was considered exploratory. All other end points were evaluated without multiplicity adjustment (Figure 3).
ELEVATE UC 52
To control for multiplicity for the primary and key secondary end points, the familywise type I error was controlled at a fixed 2-sided significance level of 0.05 using the parallel gatekeeping procedure, i.e., traditional Hochberg and truncated Hochberg procedures. First, the whole significance level was spent on testing coprimary end points (clinical remission at weeks 12 and 52). This study was considered an overall success only if both of the primary null hypotheses were rejected, each at the significance level (as coprimary hypotheses). The study was considered a partial success if only 1 of the 2 primary null hypotheses were rejected. Only if both of the primary null hypotheses were rejected could testing proceed for the 8 key secondary end points. Any key secondary end point using this method to control multiplicity that failed to be significant at the significance level was considered exploratory. All other end points were evaluated without multiplicity adjustment (Figure 4).
Subgroup Analyses
The following subgroups, planned a priori in the statistical analyses plans, aligned with the subgroups identified as relevant by the clinical expert consulted by CADTH for this review: extent of disease (left-sided colitis or proctosigmoiditis, pancolitis, or isolated proctitis based on the electronic case report form), isolated proctitis based on central read (yes or no), prior UC treatment with oral 5-ASA only (yes or no), prior UC treatment failure of oral 5-ASA only (yes or no), naive to biologic or JAKi therapy at study entry (yes or no), prior UC treatment failure with an anti-TNF alpha (yes or no), and number of prior biologic or JAKi therapies (1 or > 1), among others. Only the subgroups identified as relevant are reported herein. Subgroup analyses were done only for the FAS population with a baseline MMS of 4 to 9. Subgroup analyses were not powered to detect differences between treatment groups. No interaction P value was provided for the subgroup analyses.
Figure 3: Gatekeeping Procedure Summary for the ELEVATE UC 12 Trial
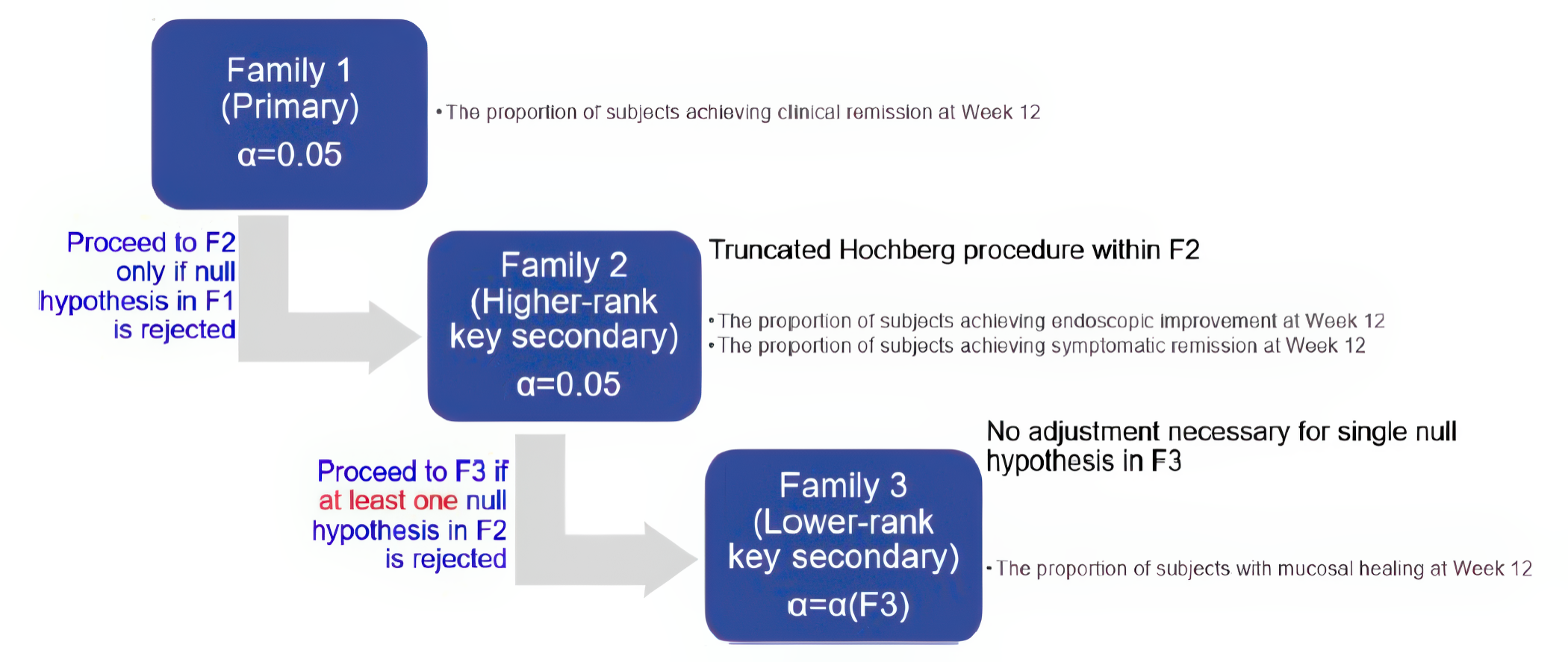
F1 = family 1; F2 = family 2, F3 = family 3.
Source: Clinical Study Report for ELEVATE UC 12.18
Sensitivity Analyses
The following sensitivity analyses were implemented to explore different approaches to handle missing data: multiple imputation under missing at random, tipping point analysis, multiple imputation with copy reference under missing not at random, and multiple imputation under missing at random or using a nonresponder imputation. In this hybrid imputation, a multiple imputation approach was used to handle endoscopy data that were missing due to the impact of the COVID-19 pandemic, and a nonresponder imputation was used for data that were missing for reasons other than the impact of the COVID-19 pandemic. All sensitivity analyses were performed in the FAS among patients with an actual baseline MMS of 5 to 9.
Figure 4: Gatekeeping Procedure Summary for the ELEVATE UC 52 Trial
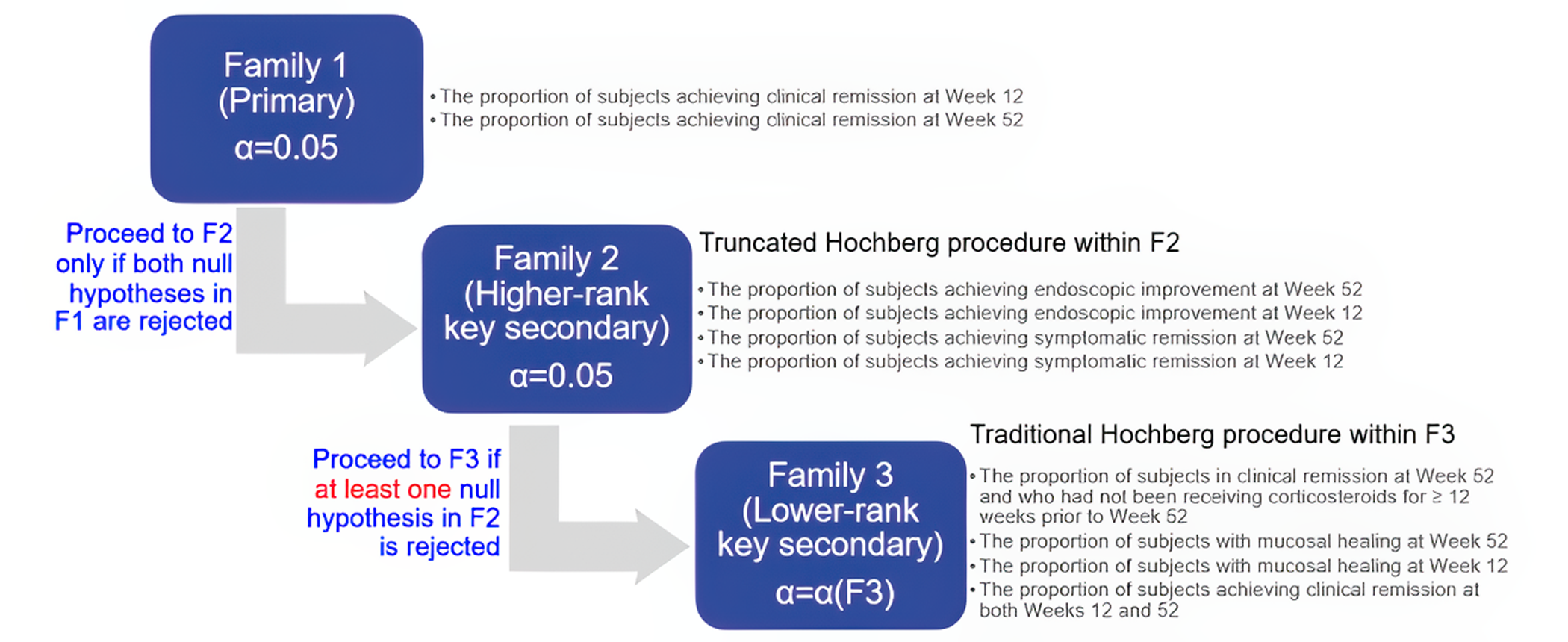
F1 = family 1; F2 = family 2, F3 = family 3.
Source: Clinical Study Report for ELEVATE UC 52.17
Missing Data
In the ELEVATE UC 12 and ELEVATE UC 52 trials, all patients who had missing data during the study were considered nonresponders in the analysis of all efficacy end points at any subsequent time points, including those who:
discontinued the study due to a lack of efficacy or an AE related to UC
initiated a rescue medication for UC
had an increase in the dose of their existing UC medication over baseline levels
had undergone a rescue medical procedure (e.g., colectomy, ileostomy, or sigmoidectomy).
Rescue medications included any exposure after the first dose of the study drug, including biologics with immunomodulatory properties (e.g., anti-TNF alpha antibodies), nonbiologic medicines with immunomodulatory properties (e.g., immunosuppressant drugs), 5-ASA compounds, systemic glucocorticoids, and topical glucocorticoids. Rescue medical procedure included any exposure after the first dose of the study drug, including leukocyte apheresis, other apheresis, and plasma exchange. If the medication or procedure was used in the follow-up period, then it was not considered to be a rescue therapy. The impact of rescue therapy use in the analysis was timing-dependent, e.g., if a patient started a rescue therapy between the outcome assessments at week 12 and week 52, then it could have a potential impact on the end point analysis at week 52 but would have no impact on the end point assessment at week 12.
In the main analysis of the continuous or scored end points, such as biomarker measures, urgency, numerical rating scale (NRS), abdominal pain NRS, and HRQoL measures, patients with missing data were handled using observed cases only or using an MMRM.
Supplementary Analyses
In the ELEVATE UC 12 and ELEVATE UC 52 trials, supplementary analyses of the primary and key secondary end points were conducted using the FAS of patients with a baseline MMS of 4 to 9, the modified FAS (with data as observed), and the per-protocol set in patients with a baseline MMS of 5 to 9.
Table 8 summarizes the statistical analysis for each outcome reported in the systemic review for the ELEVATE UC 12 and ELEVATE UC 52 trials.
Table 8: Statistical Analysis of Efficacy End Points: ELEVATE UC 12 and ELEVATE UC 52 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
ELEVATE UC 12 trial primary end point | ||||
Proportion of patients achieving clinical remission at week 12. | Cochran-Mantel-Haenszel | Stratified by naive to biologic or JAK inhibitor therapy status at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). | Patients with missing efficacy outcome data will be included in the primary analysis using the following methods:
|
These sensitivity analyses used the FAS with a baseline MMS of 5 to 9. |
ELEVATE UC 12 trial key secondary end points | ||||
The proportion of patients achieving endoscopic improvement at week 12. | Cochran-Mantel-Haenszel | Stratified by naive to biologic or JAK inhibitor therapy status at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). | Patients with missing efficacy outcome data will be included in the primary analysis using the following methods:
|
FAS with a baseline MMS of 5 to 9 was used for sensitivity analyses. |
The proportion of patients achieving symptomatic remission at week 12. | ||||
The proportion of patients with mucosal healing at week 12. | ||||
ELEVATE UC 52 trial primary end points | ||||
The proportion of patients who achieved clinical remission at week 12. | Cochran-Mantel-Haenszel | Stratified by naive to biologic or JAK inhibitor therapy at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). | Subjects with a missing efficacy outcome were included in the primary analyses using the following missing data methods:
|
FAS with a baseline MMS of 5 to 9 was used for sensitivity analyses. |
The proportion of patients who achieved clinical remission at week 52. | ||||
ELEVATE UC 52 trial key secondary end points | ||||
The proportion of patients achieving endoscopic improvement at weeks 12 and 52. | Cochran-Mantel-Haenszel | Stratified by naive to biologic or JAK inhibitor therapy at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). | Subjects with a missing efficacy outcome were included in the primary analyses using the following missing data methods:
|
FAS with a baseline MMS of 5 to 9 was used for sensitivity analyses. |
The proportion of patients achieving symptomatic remission at weeks 12 and 52. | ||||
The proportion of patients achieving corticosteroid-free clinical remission at week 52. | ||||
The proportion of patients achieving sustained clinical remission at week 52. | ||||
The proportion of patients with mucosal healing at weeks 12 and 52. | ||||
HRQoL outcome in the ELEVATE UC 12 trial and ELEVATE UC 52 | ||||
IBDQ scores and change from baseline at weeks 12 and 52. | MMRM | Adjusted for naive to biologic to JAK inhibitor therapy at study entry (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (MMS of 4 to 6 or 7 to 9), treatment, visit, treatment by visit interaction. | NA | NA |
CR = copy reference; FAS = full analysis set; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; JAK = Janus kinase; MAR = missing at random; MMRM = mixed model for repeated measures; MMS = modified Mayo score; MNAR = missing not at random; NA = not applicable; NRI = nonresponder imputation.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
Analysis Populations
The ELEVATE UC 12 and ELEVATE UC 52 trials used the same definitions for the different analysis sets (Table 9).
Table 9: Analysis Populations of the ELEVATE UC 12 and ELEVATE UC 52 Trials
Population | Definition | Application |
|---|---|---|
FAS and with a baseline MMS of 5 to 9 | The FAS consists of all randomized patients who received at least 1 dose of the study treatment with a baseline MMS of 5 to 9. Patients were analyzed according to the treatment to which they were randomized, regardless of the treatment actually received. | Primary efficacy analyses were based on the FAS and a baseline MMS of 5 to 9. |
FAS | The FAS consists of all randomized patients who received at least 1 dose of the study treatment. Patients were analyzed according to the treatment to which they were randomized, regardless of the treatment actually received. | Supplementary efficacy analyses were based on the FAS and a baseline MMS of 4 to 9. |
mFAS | The mFAS consists of all randomized patients who received at least 1 dose of the study treatment and had a baseline and at least 1 post-randomization measurement. Patients were summarized by the treatment to which they were randomized, regardless of the treatment received. | The mFAS can vary between end points, since some patients may have the data needed for inclusion in the mFAS for some end points, but not for other end points. |
Per-protocol set | The per-protocol set consists of all patients in the FAS who adhered to the protocol. | This set was used in sensitivity analyses of the primary and key secondary end points to evaluate the influences of important protocol deviations on the primary results. |
Safety analysis set | The safety analysis set includes all randomized patients who received at least 1 dose of the study treatment and was used for all safety analyses. Patients were analyzed according to the treatment received, regardless of randomization. | Safety analyses were based on the FAS and a baseline MMS of 4 to 9. |
BAS | The BAS comprises all randomized patients who received at least 1 dose of the study treatment with a baseline MMS of 5 to 9. | For biomarker analysis. |
BAS = biomarker analysis set; FAS = full analysis set; mFAS = modified full analysis set; MMS = modified Mayo score.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
Results
Patient Disposition
Patient disposition for the ELEVATE UC 12 and ELEVATE UC 52 trials among the overall population of randomized patients is summarized in Table 10.
ELEVATE UC 12 Trial
Of the 606 patients screened in the ELEVATE UC 12 trial, 252 patients (41.6%) failed screening. The main reasons for screening failure included failure to meet eligibility criteria (37.0%) and withdrawal by patient (2.5%). In the ELEVATE UC 12 trial, 238 patients were randomized to the etrasimod arm and 116 patients were randomized to the placebo arm. A total of 38 patients (10.7%) discontinued from the study. The most common reasons for study discontinuation included withdrawal by patient (4.0%), AE (2.5%), and physician decision (1.7%).
Study treatment discontinuation occurred in 24 patients (10.1%) in the etrasimod group and 11 patients (9.5%) in the placebo group. The most common reasons for treatment discontinuation in the etrasimod and placebo groups included AE (4.6% and 0.9%, respectively), withdrawal by patient (2.1% and 5.2%, respectively), physician decision (1.3% and 1.7%, respectively), and lack of efficacy (1.3% and 0%, respectively). A total of 214 patients (89.9%) in the etrasimod group and 105 patients (90.5%) in the placebo group completed the study treatment.
ELEVATE UC 52 Trial
Of the 821 patients screened in the ELEVATE UC 52 trial, 388 patients (47.3%) failed screening. The main reasons for screening failures included failure to meet eligibility criteria (41.0%) and withdrawal by patient (2.3%). In the ELEVATE UC 52 trial, 289 patients were randomized to the etrasimod arm and 144 patients were randomized to the placebo arm. A total of 226 patients (52.2%) discontinued the study. The most common reasons for study discontinuation included disease worsening (35.1%), withdrawal by patient (7.9%), and AE (3.5%).
Study treatment discontinuation occurred in 123 patients (42.6%) in the etrasimod group and 98 (68.1%) in the placebo group. The most common reasons for treatment discontinuation in the etrasimod and placebo groups included disease worsening (27.3% and 50.7%, respectively), withdrawal by patient (5.9% and 6.9%, respectively), AE (3.5% in both groups), and lack of efficacy (2.4% and 2.8%, respectively). A total of 166 patients (57.4%) in the etrasimod group and 46 patients (31.9%) in the placebo group completed the study treatment.
In both the ELEVATE UC 12 and ELEVATE UC 52 trials, patient disposition among those with a baseline MMS of 5 to 919 was similar to the overall data presented in Table 10.
Baseline Characteristics
A summary of baseline patient demographics, disease characteristics, and medication history in the pivotal trials is shown in Table 11. These characteristics are limited to those considered most relevant to this review or might affect the outcomes or interpretation of the study results.
Table 10: Summary of Patient Disposition From the Studies Included in the Systematic Review
Patient disposition | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Screened, N | 606 | 821 | ||
Failed screening, n (%) | 252 (41.6) | 388 (47.3) | ||
Reason for screening failure, n (%) | ||||
Failure to meet eligibility criteria | 224 (37.0) | 337 (41.0) | ||
Withdrawal by patient | 15 (2.5) | 19 (2.3) | ||
Adverse event | 3 (0.5) | 1 (0.1) | ||
Physician decision | 2 (0.3) | 1 (0.1) | ||
Lost to follow-up | 2 (0.3) | 2 (0.2) | ||
Protocol deviation | 0 | 1 (0.1) | ||
Others | 6 (1.0) | 26 (3.2) | ||
Missing | 0 | 1 (0.1) | ||
Randomized, N | 238 | 116 | 289 | 144 |
Discontinued from study, n (%) | 25 (10.5) | 13 (11.2) | 128 (44.3) | 98 (68.1) |
Reason for discontinuation, n (%) | ||||
Disease worsening | 0 | 0 | 79 (27.3) | 73 (50.7) |
Withdrawal by patient | 6 (2.5) | 8 (6.9) | 24 (8.3) | 10 (6.9) |
Adverse events | 9 (3.8) | 0 | 10 (3.5) | 5 (3.5) |
Lack of efficacy | 4 (1.7) | 0 | 7 (2.4) | 4 (2.8) |
Physician decision | 4 (1.7) | 2 (1.7) | 2 (0.7) | 2 (1.4) |
Lost to follow-up | 0 | 1 (0.9) | 1 (0.3) | 2 (1.4) |
Protocol deviation | 1 (0.4) | 1 (0.9) | 1 (0.3) | 0 |
Pregnancy | 0 | 0 | 2 (0.7) | 0 |
Other | 1 (0.4) | 1 (0.9) | 2 (0.7) | 2 (1.4) |
Discontinued treatment, n (%) | 24 (10.1) | 11 (9.5) | 123 (42.6) | 98 (68.1) |
Reasons for discontinuing treatment, n (%) | ||||
Disease worsening | 0 | 0 | 79 (27.3) | 73 (50.7) |
Withdrawal by patient | 5 (2.1) | 6 (5.2) | 17 (5.9) | 10 (6.9) |
Adverse event | 11 (4.6) | 1 (0.9) | 10 (3.5) | 5 (3.5) |
Physician decision | 3 (1.3) | 2 (1.7) | 3 (1.0) | 2 (1.4) |
Lack of efficacy | 3 (1.3) | 0 | 7 (2.4) | 4 (2.8) |
Lost to follow-up | 0 | 1 (0.9) | 1 (0.3) | 2 (1.4) |
Pregnancy | 0 | 0 | 2 (0.7) | 0 |
Protocol deviation | 1 (0.4) | 1 (0.9) | 1 (0.3) | 0 |
Other | 1 (0.4) | 0 | 3 (1.0) | 2 (1.4) |
Completed study treatment, n (%) | 214 (89.9) | 105 (90.5) | 166 (57.4) | 46 (31.9) |
Entered ELEVATE OLE, n (%) | 208 (87.4) | 102 (87.9) | 231 (79.9) | 115 (79.9) |
FAS with baseline MMS of 5 to 9, Na | 222 | 112 | 274 | 135 |
Patients with baseline MMS of 4, nb | 16 | 4 | 15 | 9 |
FAS with baseline MMS of 4 to 9, N | 238 | 116 | 289 | 144 |
Safety, N | 238 | 116 | 289 | 144 |
FAS = full analysis set; MMS = modified Mayo score; OLE = open-label extension.
aPatients with a baseline MMS of 5 to 9 were considered for the primary analysis of efficacy end points in the ELEVATE UC 12 and ELEVATE UC 52 trials.
bPatients with a baseline MMS of 4 were not included in the primary analysis of efficacy end points in the ELEVATE UC 12 and ELEVATE UC 52 trials.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
Table 11: Summary of Baseline Characteristics From Studies Included in the Systematic Review — FAS and Baseline MMS of 5 to 9
Characteristic | ELEVATE UC 12 | ELEVATE UC 52 | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 222) | Placebo (N = 112) | Etrasimod 2 mg (N = 274) | Placebo (N = 135) | |
Age, years | ||||
Mean (SD) | 40.4 (13.7) | 40.7 (13.2) | 41.6 (14.0) | 38.6 (14.0) |
Median (range) | 38.0 (16 to 73) | 38.5 (18 to 72) | 40.0 (18 to 78) | 35.0 (17 to 78) |
< 18 years, n (%) | 1 (0.5) | 0 | 0 | 1 (0.7) |
18 to 64 years, n (%) | 210 (94.6) | 106 (94.6) | 257 (93.8) | 124 (91.9) |
≥ 65 years, n (%) | 11 (5.0) | 6 (5.4) | 17 (6.2) | 10 (7.4) |
≥ 75 years, n (%) | 0 | 0 | 2 (0.7) | 1 (0.7) |
Sex, n (%) | ||||
Male | 125 (56.3) | 70 (62.5) | 144 (52.6) | 80 (59.3) |
Female | 97 (43.7) | 42 (37.5) | 130 (47.4) | 55 (40.7) |
Race, n (%) | ||||
American Indian or Alaska Native | 5 (2.3) | 1 (0.9) | 1 (0.4) | 3 (2.2) |
Asian | 42 (18.9) | 22 (19.6) | 19 (6.9) | 9 (6.7) |
Black or African American | 2 (0.9) | 2 (1.8) | 6 (2.2) | 3 (2.2) |
Multiple | 1 (0.5) | 0 | 0 | 0 |
White | 166 (74.8) | 87 (77.7) | 244 (89.1) | 120 (88.9) |
Not reported | 6 (2.7) | 0 | 4 (1.5) | 0 |
Region, n (%) | ||||
North America | 20 (9.0) | 9 (8.0) | 47 (17.2) | 29 (21.5) |
Western Europe | 17 (7.7) | 5 (4.5) | 23 (8.4) | 12 (8.9) |
Eastern Europe | 124 (55.9) | 61 (54.5) | 171 (62.4) | 81 (60.0) |
Other | 61 (27.5) | 37 (33.0) | 33 (12.0) | 13 (9.6) |
BMI, kg/m2 | ||||
Mean (SD) | 24.4 (4.9) | 25.4 (4.4) | 25.4 (5.6) | 25.2 (5.3) |
Disease duration, years | ||||
Mean (SD) | 7.2 (6.5) | 7.9 (7.4) | 7.6 (8.1) | 6.0 (5.6) |
MMS score, mean (SD) | 6.7 (1.1) | 6.7 (1.1) | 6.9 (1.0) | 6.8 (1.0) |
RB subscore, mean (SD) | 1.7 (0.5) | 1.7 (0.6) | 1.8 (0.6) | 1.7 (0.5) |
Baseline SF score, mean (SD) | 2.5 (0.7) | 2.4 (0.7) | 2.5 (0.6) | 2.5 (0.6) |
ES subscore, n (%) | ||||
2 | 95 (42.8) | 52 (46.4) | 112 (40.9) | 47 (34.8) |
3 | 127 (57.2) | 60 (53.6) | 162 (59.1) | 88 (65.2) |
Extent of disease, n (%) | ||||
Left-sided colitis or proctosigmoiditis | 135 (60.8) | 61 (54.5) | 161 (59.2) | 81 (60.4) |
Pancolitis | 75 (33.8) | 40 (35.7) | 92 (33.8) | 47 (35.1) |
Proctitis | 12 (5.4) | 11 (9.8) | 19 (7.0) | 6 (4.5) |
High-sensitivity C-reactive protein, mg/L, mean (SD) | 7.9 (12.9) | 8.2 (15.9) | 10.0 (15.8) | 11.3 (18.6) |
Fecal calprotectin, mg/kg, mean (SD) | ||||
n | 222 | 110 | 271 | 134 |
Mean (SD) | 2,544.1 (5,480.0) | 2,108.0 (4,316.0) | 2,527.7 (4,627.7) | 2,690.3 (5,450.5) |
Baseline corticosteroid use, n (%) | ||||
Yes | 60 (27.0) | 32 (28.6) | 87 (31.8) | 40 (29.6) |
No | 162 (73.0) | 80 (71.4) | 187 (68.2) | 95 (70.4) |
Baseline oral 5-ASA use, n (%) | ||||
Yes | 187 (84.2) | 91 (81.3) | 214 (78.1) | 105 (77.8) |
No | 35 (15.8) | 21 (18.8) | 60 (21.9) | 30 (22.2) |
Prior use of biologic or JAK inhibitor therapy,a n (%) | ||||
0b | 148 (66.7) | 74 (66.1) | 194 (70.8) | 93 (68.9) |
1 | 34 (15.3) | 20 (17.9) | 41 (15.0) | 24 (17.8) |
2 or more drugs | 40 (18.0) | 18 (16.1) | 39 (14.2) | 18 (13.3) |
Prior TNF failure and baseline corticosteroid use, n (%) | ||||
Prior TNF failure and baseline corticosteroid use | 21 (9.5) | 10 (8.9) | 13 (4.7) | 8 (5.9) |
Prior TNF failure and no baseline corticosteroid use | 27 (12.2) | 14 (12.5) | 33 (12.0) | 21 (15.6) |
No prior TNF failure but had baseline corticosteroid use | 39 (17.6) | 22 (19.6) | 74 (27.0) | 32 (23.7) |
5-ASA = 5-aminosalicylic acid; BMI = body mass index; ES = endoscopic score; FAS = full analysis set; JAK = Janus kinase; MMS = modified Mayo score; RB = rectal bleeding; SD = standard deviation; SF = stool frequency; TNF = tumour necrosis factor; UC = ulcerative colitis.
aAs reported by investigators during screening.
bThis row represented the number of patients (%) who were naive to biologics at the baseline of the study.
Source: Sponsor’s additional information.19
ELEVATE UC 12 Trial
The study population in the ELEVATE UC 12 trial had a mean age of 40.5 years (SD = 13.47 years), and 94.6% of the study population was aged between 18 and 64 years. A total of 195 patients (58.4%) were male, and 139 patients (41.6%) were female. The majority of patients were white (75.7%), followed by Asian (19.2%), American Indian or Alaska Native (1.8%), Black or African American (1.2%), multiple (0.3%), and unknown (1.8%). In the ELEVATE UC 12 trial, 55.4% of patients were from eastern Europe and 8.7% of patients were from North America.19 The mean baseline MMS in both the etrasimod and placebo groups was 6.7 (SD = 1.1), and the mean disease duration was 7.2 years (SD = 6.5) in the etrasimod group and 7.9 years (SD = 7.4) in the placebo group. Baseline corticosteroid and 5-ASA use in the etrasimod and placebo groups was present in 27.0% and 28.6% of patients, and 84.2% and 81.3% of patients, respectively. In terms of prior UC therapies, the proportion of patients in the etrasimod and placebo groups reporting prior use of biologics was 33.3% and 33.9%, respectively. The proportion of patients in the etrasimod and placebo groups reporting prior use of at least 1 biologic or JAKi was 33.3% and 33.9%, respectively. The proportion of patients reporting prior anti-TNF failure and baseline corticosteroid use was similar between treatment groups (9.5% and 8.9% of patients in the etrasimod and placebo groups, respectively). The proportion of patients reporting prior failure of anti-TNF therapy and no baseline corticosteroid use was 12.2% in the etrasimod group and 12.5% in the placebo group. No information was reported for the proportion of patients with failure of 5-ASA therapy. Overall, the baseline characteristics were well balanced between treatment arms.
ELEVATE UC 52 Trial
The study population in the ELEVATE UC 52 trial had a mean age of 40.6 years (SD = 14.1), and 93.2% of the study population was aged between 18 and 64 years. The median age in the etrasimod group was higher than in the placebo group. A total of 224 patients (54.8%) were male and 185 patients (45.2%) were female. There was a larger proportion of males in the placebo group than in the etrasimod group. The majority of patients were white (89.0%), followed by Asian (6.8%), Black or African American (2.2%), American Indian or Alaska Native (1.0%), and unknown (1.0%). In the ELEVATE UC 52 trial, 61.6% of patients were from eastern Europe and 18.6% of patients were from North America.19 The mean baseline MMS was 6.9 (SD = 1.0) and 6.8 (SD = 1.0) in the etrasimod and placebo groups, respectively. The mean disease duration was 7.6 years (SD = 8.1) in the etrasimod group and 6.0 years (SD = 5.6) in the placebo group. Baseline corticosteroid use in the etrasimod and placebo groups was present in 31.8% and 29.6% of patients, respectively; baseline 5-ASA use was present in 78.1% and 77.8% of patients, respectively. In terms of prior UC therapies, the proportion of patients in the etrasimod and placebo groups reporting prior use of at least 1 biologic was 29.2% and 31.1%, respectively. The proportion of patients reporting prior anti-TNF failure and baseline corticosteroid use was similar between treatment groups: 4.7% in the etrasimod group and 5.9% in the placebo group. The proportion of patients reporting prior failure of anti-TNF therapy and no baseline corticosteroid use was 12.0% in the etrasimod group and 15.6% in the placebo group. No information was reported for the proportion of patients with failure of 5-ASA therapy. Overall, the baseline characteristics were well balanced between treatment arms.
Concomitant Medications for UC
Concomitant medication use in the ELEVATE UC 12 and ELEVATE UC 52 studies is summarized in Table 12. Concomitant medication use for UC was generally similar across treatment groups in the pivotal trials. A total of 91.5% of patients in the ELEVATE UC 12 trial and 88.5% of patients in the ELEVATE UC 52 trial received at least 1 concomitant medication for UC during the trial. The most common concomitant medications were drugs for antidiarrheals, intestinal anti-inflammatory or anti-infective drugs, and corticosteroids for systematic use.
Table 12: Summary of Concomitant Medications in the ELEVATE UC 12 and ELEVATE UC 52 Trials — Safety Analysis Set
Concomitant medications | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Patients with ≥ 1 concomitant medication, n (%) | ||||
Patients with ≥ 1 concomitant medication for UC, n (%) | 221 (92.9) | 103 (88.8) | 257(88.9) | 126 (87.5) |
Antidiarrheals, intestinal anti-inflammatory, or anti-infective drugs, n (%) | 208 (87.4) | 96 (82.8) | 236 (81.7) | 113 (78.5) |
Mesalazine | 185 (77.7) | 89 (76.7) | 196 (67.8) | 91 (63.2) |
Sulfasalazine | 17 (7.1) | 5 (4.3) | 34 (11.8) | 18 (12.5) |
Budesonide | 17 (7.1) | 5 (4.3) | 27 (9.3) | 4 (2.8) |
Loperamide hydrochloride | 5 (2.1) | 4 (3.4) | NR | NR |
Loperamide | 0 | 2 (1.7) | 6 (2.1) | 4 (2.8) |
Bacillus mesentericus, Clostridium butyricum, Enterococcus faecalis | 5 (2.1) | 1 (0.9) | NR | NR |
Bifidobacterium breve, Bifidobacterium infantis, Bifidobacterium longum, Lactobacillus acidophilus, Lactobacillus bulgaricus, Lactobacillus paracasei, Lactobacillus plantarum, Streptococcus thermophilus | 2 (0.8) | 1 (0.9) | 5 (1.7) | 1 (0.7) |
Corticosteroids for systemic use, n (%) | 56 (23.5) | 32 (27.6) | 43 (29.9) | 79 (27.3) |
Prednisone | 24 (10.1) | 11 (9.5) | 22 (15.3) | 27 (9.3) |
Prednisolone | 20 (8.4) | 12 (10.3) | 11 (7.6) | 27 (9.3) |
Methylprednisolone | 13 (5.5) | 7 (6.0) | 10 (6.9) | 26 (9.0) |
Deflazacort | 0 | 1 (0.9) | NR | NR |
Hydrocortisone | NR | NR | 0 | 4 (1.4) |
Deflazacort | 0 | 1 (0.9) | 1 (0.7) | 1 (0.3) |
Dexamethasone | 0 | 1 (0.9) | 1 (0.7) | 1 (0.3) |
Hydrocortisone sodium succinate | 1 (0.4) | 0 | NR | NR |
Methylprednisolone sodium succinate | 1 (0.4) | 0 | NR | NR |
Immunosuppressants, n (%) | 14 (5.9) | 2 (1.7) | 10 (3.5) | 2 (1.4) |
Drugs for functional gastrointestinal disorders, n (%) | 12 (5.0) | 3 (2.6) | 14 (4.8) | 6 (4.2) |
Drugs for acid-related disorders, n (%) | 7 (2.9) | 6 (5.2) | 3 (1.0) | 2 (1.4) |
Drugs for constipation, n (%) | 6 (2.5) | 2 (1.7) | 3 (1.0) | 2 (1.4) |
Antianemic preparations, n (%) | 4 (1.7) | 3 (2.6) | 5 (1.7) | 3 (2.1) |
NR = not reported; UC = ulcerative colitis.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and sponsor’s additional information.19 Details included in the table are from the sponsor’s summary of clinical evidence.16
Exposure to Study Treatments
Treatment exposure from the pivotal trials is summarized in Table 13.
ELEVATE UC 12 Trial
Treatment exposure time was similar between the etrasimod and placebo groups: 12.1 weeks (SD = 3.0) and 12.2 weeks (SD = 2.8), respectively. In the ELEVATE UC 12 trial, the proportion of patients who had at least 12 weeks of treatment exposure was similar in the etrasimod and placebo groups (85.3% and 84.5%, respectively). Treatment compliance was also similar across groups.
ELEVATE UC 52 Trial
Treatment exposure time was higher in the etrasimod group (38.0 weeks; SD = 19.3) compared with the placebo group (27.5 weeks; SD = 18.9). In the ELEVATE UC 52 trial, the proportion of patients who had 12 to 26 weeks of treatment exposure was 28.0% and 50.0% in the etrasimod and placebo groups, respectively. Fewer patients had a treatment exposure of at least 52 weeks, with a greater proportion in the etrasimod group (45.7%) than in the placebo group (27.8%). Treatment compliance was similar across groups.
Table 13: Summary of Patient Exposure in the ELEVATE UC 12 and ELEVATE UC 52 Trials — Safety Analysis Set
Exposure | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Study treatment exposure, n (%) | ||||
< 4 weeks | 13 (5.5) | 5 (4.3) | 10 (3.5) | 3 (2.1) |
4 to < 8 weeks | 9 (3.8) | 2 (1.7) | 6 (2.1) | 7 (4.9) |
8 to < 12 weeks | 13 (5.5) | 11 (9.5) | 7 (2.4) | 6 (4.2) |
≥ 12 weeks | 203 (85.3) | 98 (84.5) | NA | NA |
12 to < 26 weeks | NA | NA | 81 (28.0) | 72 (50.0) |
26 to < 39 weeks | NA | NA | 6 (2.1) | 8 (5.6) |
39 to < 52 weeks | NA | NA | 47 (16.3) | 8 (5.6) |
≥ 52 weeks | NA | NA | 132 (45.7) | 40 (27.8) |
Duration of treatment exposure, weeksa | ||||
Mean (SD) | 12.1 (3.0) | 12.2 (2.8) | 38.0 (19.3) | 27.5 (18.9) |
Median (range) | 12.6 (0 to 19) | 12.9 (0 to 16) | 51.4 (0 to 59) | 15 (2 to 58) |
Overall complianceb | ||||
Patients contributing to the analysis, N | 238 | 116 | 288 | 142 |
Mean (SD) | 100.0 (2.9) | 99.5 (4.7) | 100.5 (11.8) | 99.9 (9.6) |
< 80%, n (%) | 0 | 1 (0.9) | 1 (0.3) | 2 (1.4) |
80% to 120%, n (%) | 237 (99.6) | 114 (98.3) | 282 (97.9) | 136 (95.8) |
> 120%, n (%) | 1 (0.4) | 1 (0.9) | 5 (1.7) | 4 (2.8) |
NA = not applicable; SD = standard deviation.
Note: Percentages are based on the number of patients in the analysis set.
aExposure for the entire treatment period was calculated as (end date of study treatment minus start date of study treatment plus 1) divided by 7.
bOverall compliance is calculated as total number of tablets taken divided by total number of tablets expected during the treatment period.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and the sponsor’s summary of clinical evidence.16
Treatment Modifications
Table 14 summarizes the treatment modifications in the ELEVATE UC 12 and ELEVATE UC 52 trials.
ELEVATE UC 12 Trial
A total of 7 patients (2.9%) in the etrasimod group and 5 patients (4.3%) in the placebo group had at least 1 treatment interruption, mostly due to AEs (2.5% and 3.4% in the etrasimod and placebo groups, respectively). Treatment overdoses were reported in 2 patients (0.6%).
ELEVATE UC 52 Trial
A total of 35 patients (12.1%) in the etrasimod group and 12 patients (8.3%) in the placebo group had at least 1 treatment interruption, mostly due to AEs (6.9% and 4.2% in the etrasimod and placebo groups, respectively). Treatment overdose was reported in 1 patient (0.3%) in the etrasimod group.
Table 14: Study Treatment Modifications in the ELEVATE UC 12 and ELEVATE UC 52 Trials — Safety Analysis Set
Exposure, n (%) | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Patients with ≥ 1 treatment interruption | 7 (2.9) | 5 (4.3) | 35 (12.1) | 12 (8.3) |
Adverse event | 6 (2.5) | 4 (3.4) | 20 (6.9) | 6 (4.2) |
Other | 1 (0.4) | 1 (0.9) | 17 (5.9) | 6 (4.2) |
Patients with ≥ 1 treatment interruption of > 7 days | 0 | 1 (0.9) | 10 (3.5) | 2 (1.4) |
Patients with ≥ 1 treatment interruption of > 14 days | 0 | 0 | 7 (2.4) | 2 (1.4) |
Patients with ≥ 1 treatment overdose | 1 (0.4) | 1 (0.9) | 1 (0.3) | 0 |
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
Protocol Deviations
Protocol deviations in the pivotal trials for the full FAS population are summarized in Table 15.
In the ELEVATE UC 12 trial, a total of 54 patients (22.7%) in the etrasimod group and 22 patients (19.0%) in the placebo group had at least 1 important protocol deviation. The most frequent protocol deviations in the etrasimod and placebo groups were study procedure (13.4% and 7.8%, respectively), eligibility and entry criteria (4.2% and 3.4%, respectively), visit schedule (2.5% and 3.4%, respectively), and laboratory assessment (4.2% and 1.7%, respectively). A total of 10 protocol deviations (2.8%) were impacted by the COVID-19 pandemic.
In the ELEVATE UC 52 trial, a total of 150 patients (51.9%) in the etrasimod group and 76 patients (52.8%) in the placebo group had at least 1 important protocol deviation. The most frequent protocol deviations in the etrasimod and placebo groups were study procedure (29.8 and 29.2%, respectively), laboratory assessment (17.6% and 13.2%, respectively), eligibility and entry criteria (5.5% and 6.9%, respectively), and visit schedule (5.2% and 4.9%, respectively). A total of 61 protocol deviations (14.1%) were impacted by the COVID-19 pandemic.
Table 15: Summary of Important Protocol Deviations From the Pivotal Studies
Protocol deviations, n (%) | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Patients with ≥ 1 important protocol deviation | 54 (22.7) | 22 (19.0) | 150 (51.9) | 76 (52.8) |
Informed consent | 1 (0.4) | 1 (0.9) | 7 (2.4) | 0 |
Eligibility and entry criteria | 10 (4.2) | 4 (3.4) | 16 (5.5) | 3 (2.1) |
Concomitant medication | 2 (0.8) | 0 | 13 (4.4) | 4 (2.8) |
Randomization | 0 | 1 (0.9) | 0 | 3 (2.1) |
Serious adverse event | 0 | 0 | 2 (0.7) | 4 (2.8) |
Laboratory assessment | 10 (4.2) | 2 (1.7) | 51 (17.6) | 19 (13.2) |
Study procedures | 32 (13.4) | 9 (7.8) | 86 (29.8) | 42 (29.2) |
Efficacy | 1 (0.4) | 1 (0.9) | 6 (2.1) | 3 (2.1) |
Investigational product compliance | 1 (0.4) | 1 (0.9) | 2 (0.7) | 3 (2.1) |
Visit schedule | 6 (2.5) | 4 (3.4) | 15 (5.2) | 7 (4.9) |
Deviations impacted by COVID-19 pandemic | 6 (2.5) | 4 (3.4) | 39 (13.5) | 22 (15.3) |
Deviations not impacted by COVID-19 pandemic | 49 (20.6) | 20 (17.2) | 134 (46.4) | 64 (44.4) |
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
Efficacy
A summary of the key efficacy results is shown in Table 16. The primary analysis of the efficacy end points was conducted in the FAS among patients with a baseline MMS of 5 to 9.
Endoscopic Improvement
ELEVATE UC 12 Trial
In the ELEVATE UC 12 trial, a greater proportion of patients in the etrasimod group (30.6%) compared with the placebo group (18.8%) had endoscopic improvement at week 12, with a common risk difference of 12.1% (95% CI, 3.0% to 21.2%; P = 0.009).
In the subgroups identified as relevant by the clinical expert consulted by CADTH — including the extent of disease (proctosigmoiditis or left-sided colitis, pancolitis, and proctitis), isolated proctitis, prior UC treatment with oral 5-ASA only, prior UC treatment failure of oral 5-ASA only, and prior UC treatment failure of anti-TNF — more patients achieved endoscopic improvement at week 12 with etrasimod than with placebo (Appendix 1 Detailed Outcome Data[REMOVED REF FIELD]). There was a greater between-group difference for patients treated with etrasimod versus placebo in the subgroup of patients who were naive to biologic or JAKi therapy at baseline (18.9%; 95% CI, 7.9% to 29.8%) than those who were not (5.6%; 95% CI, −10.13% to 21.2%). There was a greater between-group difference for patients treated with etrasimod versus placebo in the subgroup of patients who had received 1 prior biologic or JAKi therapy (−4.13%; 95% CI, −25.6% to 17.3%) than the patients who had received more than 1 prior biologic or JAKi therapy (13.3%; 95% CI, −10.2% to 36.8%).19
ELEVATE UC 52 Trial
In the ELEVATE UC 52 trial, a greater proportion of patients in the etrasimod group (35.0%) compared with the placebo group (14.1%) had endoscopic improvement at week 12, with a common risk difference of 21.2% (95% CI, 13.0% to 29.3%; P < 0.001). A greater proportion of patients in the etrasimod group (37.2%) compared with the placebo group (10.4%) had endoscopic improvement at week 52, with a common risk difference of 26.7% (95% CI, 19.0% to 34.4%; P < 0.001).
In the prespecified subgroups — including the extent of disease (proctosigmoiditis or left-sided colitis, pancolitis, and proctitis), isolated proctitis, prior UC treatment of oral 5-ASA only, prior UC treatment failure of oral 5-ASA only, and prior UC treatment failure of anti-TNF — more patients achieved endoscopic improvement at weeks 12 and 52 on etrasimod than on placebo (Appendix 1 Detailed Outcome Data, Figure 9 and Figure 10). For patients treated with etrasimod versus placebo, there were greater between-group differences at baseline in the subgroup of patients who were naive to biologic or JAKi therapy, with a between-group difference of 22.1% at week 12 (95% CI, 12.1% to 32.2%) and 26.8% at week 52 (95% CI, 17.3% to 36.4%) compared with patients who were not naive to such therapy, with between-group differences of 16.5% (95% CI, 3.9% to 29.2%) at week 12 and 23.5% (95% CI, 11.1% to 35.9%) at week 52. There were greater between-group differences for patients treated with etrasimod versus placebo in the subgroup of patients who had received 1 prior biologic or JAKi therapy, with a between-group difference of 24.2% (95% CI, 9.3% to 39.0%) at week 12 and 24.8% (95% CI, 7.9% to 41.8%) at week 52, whereas the between-group difference among patients who had received more than 1 prior biologic or JAKi therapy was 7.7% (95% CI, −12.6% to 28.0%) at week 12 and 7.7% (95% CI, −12.6% to 28.0%) at week 52.19
Mucosal Healing
ELEVATE UC 12 Trial
In the ELEVATE UC 12 trial, a greater proportion of patients in the etrasimod group (16.2%) compared with the placebo group (8.9%) had mucosal healing at week 12, with a common risk difference of 7.4% (95% CI, 0.5% to 14.4%; P = 0.036).
ELEVATE UC 52 Trial
In the ELEVATE UC 52 trial, a greater proportion of patients in the etrasimod group (21.2%) compared with the placebo group (4.4%) had mucosal healing at week 12, with a common risk difference of 16.9% (95% CI, 10.8% to 23.0%; P < 0.001). A greater proportion of patients in the etrasimod group (26.6%) compared with the placebo group (8.1%) had mucosal healing at week 52, with a common risk difference of 18.4% (95% CI, 11.4% to 25.4%; P < 0.001).
For mucosal healing, only the outcome results at week 52 were considered important for informing the deliberations of CADTH’s expert committee and were assessed using GRADE.
Clinical Remission
ELEVATE UC 12 Trial
A greater proportion of patients in the etrasimod group (24.8%) compared with the placebo group (15.2%) achieved clinical remission at week 12, with a common risk difference of 9.7% (95% CI, 1.1% to 18.2%; P = 0.026).
ELEVATE UC 52 Trial
A greater proportion of patients in the etrasimod group (27.0%) compared with the placebo group (7.4%) achieved clinical remission at week 12, with a common risk difference of 19.75% (95% CI, 12.9% to 26.6%; P < 0.001). A greater proportion of patients in the etrasimod group (32.1%) compared with the placebo group (6.7%) achieved clinical remission at week 52, with a common risk difference of 25.4% (95% CI, 18.4% to 32.4%; P < 0.001).
Sustained Clinical Remission
ELEVATE UC 52 Trial
At both weeks 12 and 52, a greater proportion of patients in the etrasimod group (17.9%) compared with the placebo group (2.2%) achieved sustained clinical remission, with a common risk difference of 15.8% (95% CI, 10.7% to 21.0%; P < 0.001).
Corticosteroid-Free Clinical Remission
ELEVATE UC 52 Trial
At week 52, 32.1% of patients in the etrasimod group and 6.7% of patients in the placebo group achieved clinical remission and were corticosteroid-free for at least 12 weeks, with a common risk difference of 25.4% (95% CI, 18.4% to 32.4%; P < 0.001). At week 52 and among the overall patient population, 32.1% of patients in the etrasimod group and 6.7% of patients in the placebo group achieved clinical remission and were corticosteroid-free for at least 4 weeks, with a common risk difference of 25.1% (95% CI, 18.1% to 32.1%; P < 0.001). At week 52 among the patients who were receiving oral corticosteroids for UC at baseline, 27 patients (31.0%) in the etrasimod group and 3 patients (7.5%) in the placebo group achieved clinical remission and were corticosteroid-free for at least 4 weeks, with a common risk difference of 23.1% (95% CI, 10.2% to 35.9%; P < 0.001).
Clinical Response
ELEVATE UC 12 Trial
At week 12, a greater proportion of patients in the etrasimod group experienced clinical response compared with the placebo group (62.2% and 41.1%, respectively), with a common risk difference of 21.2% (95% CI, 10.2% to 32.3%; P < 0.001).
ELEVATE UC 52 Trial
At week 12, a greater proportion of patients in the etrasimod group had a clinical response compared with the placebo group (62.4% and 34.1%, respectively), with a common risk difference of 28.3% (95% CI, 18.5% to 38.0%; P < 0.001). At week 52, 48.2% of patients in the etrasimod group and 23.0% of patients in the placebo group had a clinical response, with a common risk difference of 24.9% (95% CI, 15.8% to 34.1%; P < 0.001). At both weeks 12 and 52, a greater proportion of patients in the etrasimod group had a clinical response compared with the placebo group (44.9% and 18.5%, respectively), with a common risk difference of 26.2% (95% CI, 17.5% to 34.8%; P < 0.001).
Symptomatic Remission
ELEVATE UC 12 Trial
A greater proportion of patients in the etrasimod group (46.8%) compared with the placebo group (29.5%) achieved symptomatic remission at week 12, with a common risk difference of 17.5% (95% CI, 6.8% to 28.2%; P = 0.001).
ELEVATE UC 52 Trial
At week 12, a greater proportion of patients in the etrasimod group (46.0%) compared with the placebo group (21.5%) achieved symptomatic remission at week 12, with a common risk difference of 24.6% (95% CI, 15.5% to 33.6%; P < 0.001). At week 52, 43.4% of patients in the etrasimod group and 18.5% of patients in the placebo group achieved symptomatic remission, with a common risk difference of 24.9% (95% CI, 16.2% to 33.6%; P < 0.001).
For symptomatic remission, only the outcome results at week 52 were considered important for informing the deliberations of CADTH’s expert committee and were assessed using GRADE.
Based on the results from the ELEVATE UC 12 and ELEVATE UC 52 trials, in the prespecified subgroups — including the extent of disease (proctosigmoiditis or left-sided colitis, pancolitis, and proctitis), isolated proctitis, prior UC treatment of oral 5-ASA only, prior UC treatment failure of oral 5-ASA only, and prior UC treatment failure of anti-TNF — more patients on etrasimod than on placebo had mucosal healing and clinical remission at week 12 and week 52. In both studies, the results of the prespecified subgroup analyses were consistent with the primary analysis results at weeks 12 and 52 for sustained clinical remission, corticosteroid-free clinical remission, clinical response, and symptomatic remission.16,19
In the ELEVATE UC 12 and ELEVATE UC 52 trials, the results of sensitivity analyses of endoscopic improvement, mucosal healing, clinical remission, and symptomatic remission were consistent with the primary analysis results at weeks 12 and 52.
HRQoL Assessed With the IBDQ Total Score
ELEVATE UC 12 Trial
At week 12, in the IBDQ total score, patients in the etrasimod group experienced a mean change of 45.5 points from baseline compared with a change of 30.4 points in the placebo group, with an LS mean difference between groups of 17.3 (95% CI, 8.5 to 26.2; P < 0.001).
ELEVATE UC 52 Trial
At week 12, in the IBDQ total score, patients in the etrasimod group experienced a mean change of 44.3 points from baseline compared with a change of 26.7 points in the placebo group, with an LS mean difference between groups of 15.44 (95% CI, 6.5 to 24.4; P < 0.001). At week 52, in the IBDQ total score, patients in the etrasimod group experienced a mean change of 66.6 points from baseline compared with a change of 52.5 points in the placebo group, with an LS mean difference between groups of 17.7 (95% CI, 6.6 to 28.8; P = 0.002).
Table 16: Summary of Key Efficacy Results of the ELEVATE UC 12 and ELEVATE UC 52 Trials — FAS and a Baseline MMS of 5 to 9
Outcome | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 222) | Placebo (N = 112) | Etrasimod 2 mg (N = 274) | Placebo (N = 135) | |
Endoscopic improvement | ||||
Week 12 | ||||
Patients with endoscopic improvement,a n (%) | 68 (30.6) | 21 (18.8) | 96 (35.0) | 19 (14.1) |
Percentage difference from placebo, % | 11.88 | 20.96 | ||
Odds ratio (95% CI) | 2.03 (1.14 to 3.60) | 3.33 (1.93 to 5.76) | ||
Common risk difference, % (95% CI)b | 12.11 (3.00 to 21.23) | 21.18 (13.03 to 29.32) | ||
P valueb | 0.009 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients with endoscopic improvement,a n (%) | NA | NA | 102 (37.2) | 14 (10.4) |
Percentage difference from placebo, % | NA | NA | 26.86 | |
Odds ratio (95% CI) | NA | NA | 5.10 (2.77 to 9.37) | |
Common risk difference, % (95% CI)b | NA | NA | 26.69 (18.99 to 34.39) | |
P valueb | NA | NA | < 0.001 | Reference |
Mucosal healing | ||||
Week 12 | ||||
Patients with mucosal healing,c n (%) | 36 (16.2) | 10 (8.9) | 58 (21.2) | 6 (4.4) |
Percentage difference from placebo, % | 7.29 | 16.72 | ||
Odds ratio (95% CI) | 2.09 (0.97 to 4.50) | 5.38 (2.32 to 12.45) | ||
Common risk difference, % (95% CI)b | 7.44 (0.50 to 14.39) | 16.88 (10.78 to 22.98) | ||
P valueb | 0.036 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients with mucosal healing,c n (%) | NA | NA | 73 (26.6) | 11 (8.1) |
Percentage difference from placebo, % | NA | 18.49 | ||
Odds ratio (95% CI) | NA | 4.05 (2.07 to 7.92) | ||
Common risk difference, % (95% CI)b | NA | 18.39 (11.39 to 25.39) | ||
P valueb | NA | NA | < 0.001 | Reference |
Clinical remission | ||||
Week 12 | ||||
Patients with clinical remission,d n (%) | 55 (24.8) | 17 (15.2) | 74 (27.0) | 10 (7.4) |
Percentage difference from placebo, % | 9.60 | 19.60 | ||
Odds ratio (95% CI) | 1.9 (1.03 to 3.52) | 4.68 (2.32 to 9.44) | ||
Common risk difference, % (95% CI)b | 9.69 (1.14 to 18.23) | 19.75 (12.88 to 26.63) | ||
P valueb | 0.026 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients with clinical remission,d n (%) | NA | NA | 88 (32.1) | 9 (6.7) |
Percentage difference from placebo, % | NA | NA | 25.45 | |
Odds ratio (95% CI) | NA | NA | 6.54 (3.18 to 13.44) | |
Common risk difference, % (95% CI)b | NA | NA | 25.39 (18.42 to 32.36) | |
P valueb | NA | NA | < 0.001 | Reference |
Sustained clinical remission at both weeks 12 and 52 | ||||
Patients with sustained clinical remission,e n (%) | NA | NA | 49 (17.9) | 3 (2.2) |
Percentage difference from placebo, % | NA | NA | 15.66 | |
Odds ratio (95% CI) | NA | NA | 9.81 (2.98 to 32.36) | |
Common risk difference, % (95% CI)b | NA | NA | 15.84 (10.66 to 21.03) | |
P valueb | NA | NA | < 0.001 | Reference |
Corticosteroid-free for ≥ 12 weeks and achieved clinical remission at week 52 | ||||
Patients with corticosteroid-free clinical remission,f n (%) | NA | NA | 88 (32.1) | 9 (6.7) |
Percentage difference from placebo, % | NA | NA | 25.45 | |
Odds ratio (95% CI) | NA | NA | 6.54 (3.18 to 13.44) | |
Common risk difference, % (95% CI)b | NA | NA | 25.39 (18.42 to 32.36) | |
P valueb | NA | NA | < 0.001 | Reference |
Corticosteroid-free for ≥ 4 weeks and achieved clinical remission at week 52 | ||||
Patients with corticosteroid-free clinical remission,g n (%) | NA | NA | 88 (32.1) | 9 (6.7) |
Percentage difference from placebo, % | NA | NA | 25.45 | |
Odds ratio (95% CI) | NA | NA | 6.38 (3.11 to 13.10) | |
Common risk difference, % (95% CI)b | NA | NA | 25.11 (18.10 to 32.13) | |
P valueb | NA | NA | < 0.001 | Reference |
Corticosteroid-free for ≥ 4 weeks and achieved clinical remission at week 52 | ||||
Patients receiving oral corticosteroids for UC at baseline, n | NA | NA | 87 | 40 |
Patients with corticosteroid-free clinical remission,g n (%) | NA | NA | 27 (31.0) | 3 (7.5) |
Percentage difference from placebo, % | NA | NA | 23.53 | |
Odds ratio (95% CI) | NA | NA | 5.44 (1.53 to 19.39) | |
Common risk difference, % (95% CI)b | NA | NA | 23.05 (10.20 to 35.90) | |
P valueb | NA | NA | < 0.001 | Reference |
Clinical response | ||||
Week 12 | ||||
Patients with clinical response,h n (%) | 138 (62.2) | 46 (41.1) | 171 (62.4) | 46 (34.1) |
Percentage difference from placebo, % | 21.09 | 28.33 | ||
Odds ratio (95% CI) | 2.40 (1.50 to 3.83) | 3.29 (2.12 to 5.10) | ||
Common risk difference, % (95% CI)b | 21.23 (10.18 to 32.29) | 28.27 (18.51 to 38.02) | ||
P valueb | < 0.001 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients with clinical response,h n (%) | NA | NA | 132 (48.2) | 31 (23.0) |
Percentage difference from placebo, % | NA | NA | 25.21 | |
Odds ratio (95% CI) | NA | NA | 3.17 (1.97 to 5.10) | |
Common risk difference, % (95% CI)b | NA | NA | 24.93 (15.79 to 34.07) | |
P valueb | NA | NA | < 0.001 | Reference |
Both weeks 12 and 52 | ||||
Patients with clinical response,h n (%) | NA | NA | 123 (44.9) | 25 (18.5) |
Percentage difference from placebo, % | NA | NA | 26.37 | |
Odds ratio (95% CI) | NA | NA | 3.71 (2.24 to 6.15) | |
Common risk difference (95% CI), %b | NA | NA | 26.16 (17.48 to 34.84) | |
P valueb | NA | NA | < 0.001 | Reference |
Symptomatic remission | ||||
Week 12 | ||||
Patients with symptomatic remission,i n (%) | 104 (46.8) | 33 (29.5) | 126 (46.0) | 29 (21.5) |
Percentage difference from placebo, % | 17.38 | 24.50 | ||
Odds ratio (95% CI) | 2.13 (1.31 to 3.46) | 3.14 (1.95 to 5.06) | ||
Common risk difference, % (95% CI)b | 17.48 (6.81 to 28.15) | 24.55 (15.46 to 33.63) | ||
P valueb | 0.001 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients with symptomatic remission,i n (%) | NA | NA | 119 (43.4) | 25 (18.5) |
Percentage difference from placebo, % | NA | NA | 24.91 | |
Odds ratio (95% CI) | NA | NA | 3.46 (2.09 to 5.72) | |
Common risk difference, % (95% CI)b | NA | NA | 24.89 (16.17 to 33.60) | |
P valueb | NA | NA | < 0.001 | Reference |
IBDQj total score and change from baseline | ||||
Baseline | ||||
Patients contributing to the analysis, N | 191 | 96 | 237 | 112 |
Mean (SD) | 124.5 (35.26) | 120.5 (33.45) | 115.3 (33.16) | 117.4 (33.89) |
Week 12 | ||||
Patients contributing to the analysis, N | 175 | 91 | 220 | 106 |
Mean (SD) | 169.5 (38.20) | 150.8 (38.67) | 158.7 (42.94) | 145.0 (41.45) |
Change from baseline, mean (SD) | 45.5 (40.03) | 30.4 (38.62) | 44.3 (43.00) | 26.7 (36.80) |
Change from baseline, LS mean (SE)k | 47.49 (2.87) | 30.16 (3.78) | 42.79 (2.77) | 27.35 (3.88) |
LS mean difference (95% CI)k | 17.33 (8.50 to 26.16) | 15.44 (6.54 to 24.35) | ||
P valuek | < 0.001 | Reference | < 0.001 | Reference |
Week 52 | ||||
Patients contributing to the analysis, N | NA | NA | 132 | 36 |
Mean (SD) | NA | NA | 181.3 (34.69) | 174.3 (34.69) |
Change from baseline, mean (SD) | NA | NA | 66.6 (38.97) | 52.5 (34.30) |
Change from baseline, LS mean (SE)k | NA | NA | 55.78 (2.96) | 38.08 (4.95) |
LS mean difference (95% CI)k | NA | NA | 17.70 (6.64 to 28.76) | |
P valuek | NA | NA | 0.002 | Reference |
CI = confidence interval; ES = endoscopic score; FAS = full analysis set; IBDQ = Inflammatory Bowel Disease Questionnaire; JAK = Janus kinase; LS = least squares; MMRM = mixed model for repeated measures; MMS = modified Mayo score; NA = not applicable; RB = rectal bleeding; SD = standard deviation; SE = standard error; SF = stool frequency; UC = ulcerative colitis.
Note: Percentages are based on the number of patients in the analysis set.
aDefined as patients with an ES of ≤ 1 (excluding friability).
bEstimates are from a Cochran-Mantel-Haenszel test stratified by naive to biologic or JAK inhibitor therapy at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). Ratio is for etrasimod over placebo. Difference (%) is for etrasimod minus placebo and is based on estimated common risk difference using the Cochran-Mantel-Haenszel weights.
cDefined as patients who have an ES of ≤ 1 (excluding friability) with histologic remission measured by a Geboes Index score of < 2.0.
dDefined as patients who have an SF subscore of 0 (or 1 with a ≥ 1 point decrease from baseline), RB subscore of 0, and ES of ≤ 1 (excluding friability).
eDefined as patients with an SF subscore of 0 (or 1 with a ≥ 1-point decrease from baseline), RB subscore of 0, and ES of ≤ 1 (excluding friability) at both week 12 and week 52.
fDefined as patients with an SF subscore of 0 (or 1 with a ≥ 1-point decrease from baseline), RB subscore of 0, ES of ≤ 1 (excluding friability), and who have not received corticosteroids for ≥ 12 weeks in the 40-week treatment period.
gDefined as patients with an SF subscore of 0 (or 1 with a ≥ 1 point decrease from baseline), RB subscore of 0, ES of ≤ 1 (excluding friability), and who have not received corticosteroids for ≥ 4 weeks in the 40-week treatment period.
hDefined as patients with a ≥ 2-point and ≥ 30% decrease from baseline in MMS, and a ≥ 1-point decrease from baseline in RB subscore or an absolute RB subscore ≤ 1.
iDefined as patients with an SF subscore of 0 (or 1 with a ≥ 1 point decrease from baseline) and an RB subscore of 0.
jIBDQ scores and change from baseline were reported using a modified FAS and an actual baseline MMS of 5 to 9.
kEstimates are from an MMRM model for change from baseline with a covariate for baseline score, and factors for naive to biologic or JAK inhibitor therapy at study entry (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (MMS 4 to 6 or 7 to 9), treatment, visit, and treatment by visit interaction.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 5217 and sponsor’s additional information.19 Details included in the table are from the sponsor’s summary of clinical evidence.16
Harms
A summary of harms reported in the safety analysis sets in the ELEVATE UC 12 and ELEVATE UC 52 trials is provided in Table 17.
Adverse Events
In the ELEVATE UC 12 trial, 47.1% and 46.6% of patients reported a TEAE in the etrasimod and placebo groups, respectively. During the 12 weeks of treatment in the ELEVATE UC 12 trial, the most common TEAEs in the etrasimod and placebo groups were anemia (5.9% and 6.9%, respectively), headache (4.6% and 1.7%, respectively), nausea (4.2% and 1.7%, respectively), UC (3.8% and 0.9%, respectively), pyrexia (3.4% and 2.6%, respectively), and arthralgia (1.7% and 2.6%, respectively). A total of 2.5% and 1.7% of patients in the etrasimod and placebo groups, respectively, reported serious TEAEs.
Table 17: Summary of Harms Results in the ELEVATE UC 12 and ELEVATE UC 52 Trials — Safety Analysis Set
Adverse event | ELEVATE UC 12 trial | ELEVATE UC 52 trial | ||
|---|---|---|---|---|
Etrasimod 2 mg (N = 238) | Placebo (N = 116) | Etrasimod 2 mg (N = 289) | Placebo (N = 144) | |
Patients with ≥ 1 TEAE, n (%) | 112 (47.1) | 54 (46.6) | 206 (71.3) | 81 (56.3) |
TEAEs by severity, n (%) | ||||
Grade 1 | 62 (26.1) | 31 (26.7) | 101 (34.9) | 40 (27.8) |
Grade 2 | 42 (17.6) | 21 (18.1) | 84 (29.1) | 30 (20.8) |
Grade 3 | 7 (2.9) | 2 (1.7) | 20 (6.9) | 10 (6.9) |
Grade 4 | 1 (0.4) | 0 | 1 (0.3) | 1 (0.7) |
TEAEs in ≥ 1% of patients in either treatment group, n (%) | ||||
Anemia | 14 (5.9) | 8 (6.9) | 24 (8.3) | 14 (9.7) |
Headache | 11 (4.6) | 2 (1.7) | 24 (8.3) | 7 (4.9) |
Nausea | 10 (4.2) | 2 (1.7) | 9 (3.1) | 2 (1.4) |
Colitis ulcerative | 9 (3.8) | 1 (0.9) | 22 (7.6) | 13 (9.0) |
Pyrexia | 8 (3.4) | 3 (2.6) | 14 (4.8) | 6 (4.2) |
Arthralgia | 4 (1.7) | 3 (2.6) | 13 (4.5) | 3 (2.1) |
Back pain | 4 (1.7) | 0 | 7 (2.4) | 3 (2.1) |
Abdominal pain | 3 (1.3) | 3 (2.6) | 11 (3.8) | 5 (3.5) |
Abdominal distension | 5 (2.1) | 0 | 4 (1.4) | 3 (2.1) |
Gamma-glutamyl transferase increased | 5 (2.1) | 0 | 5 (1.7) | 2 (1.4) |
Vomiting | 5 (2.1) | 2 (1.7) | 5 (1.7) | 0 |
Urinary tract infection | 4 (1.7) | 0 | 6 (2.1) | 3 (2.1) |
Alanine aminotransferase increased | 3 (1.3) | 0 | 8 (2.8) | 2 (1.4) |
Hypophosphatemia | 4 (1.7) | 0 | NR | NR |
Sinus bradycardia | 4 (1.7) | 0 | NR | NR |
COVID-19 | 3 (1.3) | 3 (2.6) | 20 (6.9) | 9 (6.3) |
Blood creatine phosphokinase increased | 3 (1.3) | 1 (0.9) | 5 (1.7) | 1 (0.7) |
Nasopharyngitis | 3 (1.3) | 2 (1.7) | 3 (1.0) | 4 (2.8) |
Migraine | 2 (0.8) | 4 (3.4) | 3 (1.0) | 0 |
Tachycardia | 2 (0.8) | 2 (1.7) | NR | NR |
Asthenia | NR | NR | 7 (2.4) | 2 (1.4) |
Dizziness | 3 (1.3) | 0 | 15 (5.2) | 1 (0.7) |
Fatigue | 3 (1.3) | 0 | 5 (1.7) | 2 (1.4) |
Hypertension | 3 (1.3) | 1 (0.9) | 8 (2.8) | 1 (0.7) |
Iron deficiency anemia | 3 (1.3) | 3 (2.6) | NR | NR |
Hemorrhoids | NR | NR | 7 (2.4) | 0 |
Flatulence | NR | NR | 6 (2.1) | 0 |
Hypercholesterolemia | NR | NR | 6 (2.1) | 0 |
Respiratory tract infection viral | NR | NR | 6 (2.1) | 2 (1.4) |
Muscle spasms | NR | NR | 5 (1.7) | 0 |
Rash | NR | NR | 5 (1.7) | 3 (2.1) |
Conjunctivitis | NR | NR | 1 (0.3) | 4 (2.8) |
Serious TEAEs reported in ≥ 1 patient in either treatment group, n (%) | ||||
Patients with ≥ 1 serious TEAE | 6 (2.5) | 2 (1.7) | 20 (6.9) | 9 (6.3) |
Colitis ulcerative | 3 (1.3) | 0 | 6 (2.1) | 3 (2.1) |
Abdominal pain | 0 | 1 (0.9) | 1 (0.3) | 0 |
Anemia | 0 | 1 (0.9) | 2 (0.7) | 1 (0.7) |
Mucosal prolapse syndrome | NR | NR | 1 (0.3) | 0 |
Proctitis | NR | NR | 1 (0.3) | 0 |
Large intestine perforation | NR | NR | 0 | 1 (0.7) |
Coronary artery disease | 1 (0.4) | 0 | NR | NR |
COVID-19 | NR | NR | 1 (0.3) | 1 (0.7) |
COVID-19 pneumonia | NR | NR | 1 (0.3) | 1 (0.7) |
Pneumonia bacterial | NR | NR | 1 (0.3) | 0 |
Campylobacter infection | NR | NR | 0 | 1 (0.7) |
Cellulitis | NR | NR | 0 | 1 (0.7) |
Peritonitis | NR | NR | 0 | 1 (0.7) |
Arthralgia | NR | NR | 1 (0.3) | 0 |
Musculoskeletal chest pain | NR | NR | 1 (0.3) | 0 |
Intracranial pressure increased | NR | NR | 1 (0.3) | 0 |
Migraine | 1 (0.4) | 0 | 1 (0.3) | 0 |
Allergy to arthropod bite | NR | NR | 1 (0.3) | 0 |
Hepatobiliary procedural complication | 1 (0.4) | 0 | NR | NR |
Hepatic enzyme increased | NR | NR | 1 (0.3) | 0 |
Anembryonic gestation | NR | NR | 1 (0.3) | 0 |
Breast-conserving surgery | NR | NR | 1 (0.3) | 0 |
TEAEs leading to discontinuation of treatment, n (%) | ||||
Patients with ≥ 1 TEAE leading to study treatment discontinuation | 13 (5.5) | 1 (0.9) | 12 (4.2) | 7 (4.9) |
Colitis ulcerative | 6 (2.5) | 0 | 4 (1.4) | 2 (1.4) |
Sinus bradycardia | 2 (0.8) | 0 | NR | NR |
Atrioventricular block first degree | 1 (0.4) | 0 | 1 (0.3) | 0 |
Bradycardia | 1 (0.4) | 0 | 1 (0.3) | 0 |
Diarrhea | 1 (0.4) | 0 | NR | NR |
Liver function test abnormal | 1 (0.4) | 0 | NR | NR |
Weight decreased | 1 (0.4) | 0 | NR | NR |
Abdominal pain upper | 0 | 1 (0.9) | NR | NR |
Alanine aminotransferase increased | NR | NR | 1 (0.3) | 1 (0.7) |
Blood alkaline phosphatase increased | NR | NR | 1 (0.3) | 0 |
Clostridium difficile infection | NR | NR | 1 (0.3) | 0 |
COVID-19 | NR | NR | 1 (0.3) | 0 |
Macular edema | NR | NR | 1 (0.3) | 0 |
Pyrexia | NR | NR | 1 (0.3) | 0 |
Anemia | NR | NR | 0 | 1 (0.7) |
Large intestine perforation | NR | NR | 0 | 1 (0.7) |
Malaise | NR | NR | 0 | 1 (0.7) |
Tuberculosis | NR | NR | 0 | 1 (0.7) |
Selected adverse events of special interest, n (%) | ||||
Cardiovascular events | 8 (3.4) | 2 (1.7) | 12 (4.2) | 0 |
Liver injury | 3 (1.3) | 0 | 4 (1.4) | 2 (1.4) |
Infections | 2 (0.8) | 2 (1.7) | 8 (2.8) | 7 (4.9) |
Opportunistic infections | 1 (0.4) | 0 | 0 | 1 (0.7) |
Severe infections | 0 | 0 | 3 (1.0) | 5 (3.5) |
Macular edema | 0 | 0 | 1 (0.3) | 0 |
Pulmonary disorders | 0 | 0 | 1 (0.3) | 1 (0.7) |
AE = adverse event; MedDRA = Medical Dictionary for Regulatory Activities; NR = not reported; SMQ = Standardised MedDRA Query; SOC = system organ class; TEAE = treatment-emergent adverse event.
Note: TEAEs are defined as any AE that started or worsened in severity on or after the first dose of the study treatment. Terms are coded using MedDRA Version 24.1. Percentages are based on the number of patients in the analysis set. AE categories are sorted in order of decreasing total frequency in the etrasimod treatment group and then sorted within the category in order of decreasing frequency of the subcategory and preferred term. Severe infections are defined as events in the “Infections and Infestations” MedDRA SOC, with severity reported as grade 3 (severe), grade 4 (life-threatening), or grade 5 (death). Opportunistic infections (narrow) are based on an SMQ. Patients are counted only once per summarization level.
Sources: Clinical Study Reports for ELEVATE UC 1218 and ELEVATE UC 52.17 Details included in the table are from the sponsor’s summary of clinical evidence.16
In the ELEVATE UC 52 trial, 71.3% and 56.3% of patients reported a TEAE in the etrasimod and placebo groups, respectively. Over 52 weeks of treatment in the ELEVATE UC 52 trial, the most common TEAEs in the etrasimod and placebo groups were anemia (8.3% and 9.7%, respectively), headache (8.3% and 4.9%, respectively), UC (7.6% and 9.0%, respectively), COVID-19 infection (6.9% and 6.3%, respectively), dizziness (5.2% and 0.7%, respectively), and pyrexia (4.8% and 4.2%, respectively).
Serious Adverse Events
In the ELEVATE UC 12 trial, 2.5% and 1.7% of patients reported serious TEAEs in the etrasimod and placebo groups, respectively. The most common serious TEAEs in the etrasimod and placebo groups included UC (1.3% and 0%, respectively), abdominal pain (0% and 0.9%, respectively), and anemia (0% and 0.9%, respectively).
In the ELEVATE UC 52 trial, 6.9% and 6.3% of patients reported serious TEAEs in the etrasimod and placebo groups, respectively. The most common serious TEAEs in the etrasimod and placebo groups included UC (2.1% in both groups), anemia (0.7% in both groups), and COVID-19 pneumonia (0.3% and 0.7%, respectively).
Withdrawals Due to Adverse Events
In the ELEVATE UC 12 trial, 5.5% and 0.9% of patients withdrew from the trial due to an AE in the etrasimod and placebo groups, respectively. In the ELEVATE UC 52 trial, 4.2% and 4.9% of patients withdrew from the trial due to a TEAE in the etrasimod and placebo groups, respectively. In both the ELEVATE UC12 and ELEVATE UC 52 studies, UC was the most common reason for withdrawal (not more than 2.5% across the study groups).
Mortality
No deaths were reported in either the ELEVATE UC 12 or ELEVATE UC 52 trial.
Notable Harms
In the ELEVATE UC 12 trial, 3.4% of patients in the etrasimod group experienced cardiovascular events compared with 1.7% of patients in the placebo group. In the ELEVATE UC 12 trial, in the etrasimod and placebo groups, liver injury was reported in 1.3% and 0% of patients, respectively, and infections were reported in 0.8% and 1.7% of patients, respectively. No AESIs in the categories of pulmonary disorders, macular edema, posterior reversible encephalopathy syndrome, progressive multifocal leukoencephalopathy, or malignancy were reported in the ELEVATE UC 12 trial.18 Of note, 1 patient in the etrasimod group and 1 patient in the placebo group reported AEs of macular edema. Neither of these were identified as AESIs per the definition of macular edema by preferred term, which is confirmed by an increase in central foveal thickness of 40 μm or greater or a central foveal thickness of 40 μm or greater, with associated symptoms and clinically significant abnormal findings.18
In the ELEVATE UC 52 trial, 4.2% of patients in the etrasimod group experienced cardiovascular events compared with 0% of patients in the placebo group. In the ELEVATE UC 52 trial, in the etrasimod and placebo groups, liver injury was reported in 1.4% of patients in both groups, and infections were reported in 2.8% and 4.9% of patients, respectively. In the ELEVATE UC 52 trial, 1 patient (0.3%) in the etrasimod group and no patients (0%) in the placebo group reported macular edema. There were no AESIs of posterior reversible encephalopathy syndrome, progressive multifocal leukoencephalopathy, or malignancy reported during the ELEVATE UC 52 study.17
Critical Appraisal
Internal Validity
Based on the CADTH review team’s assessment, randomization in both the ELEVATE UC 12 and ELEVATE UC 52 trials was performed using an appropriate methodology with adequate allocation concealment, i.e., an interactive web response system. Randomization stratification was prespecified and was based on relevant prognostic factors, i.e., naive to biologic or JAKi therapy at study entry (yes or no), baseline corticosteroid use (yes or no), and baseline disease activity (MMS of 4 to 6 or 7 to 9). For both pivotal trials, patients who had a baseline MMS of 4 to 9 were eligible and enrolled in the studies; however, while safety outcomes were presented in the FAS of patients with a baseline MMS of 4 to 9 (i.e., among all patients who were randomized), a primary efficacy analysis was performed in the FAS of patients with a baseline MMS of 5 to 9, in accordance with the trials’ statistical analysis plans.17-21 As a result, in the 2 trials, a total of 44 patients (20 of 354 patients in the ELEVATE UC 12 trial and 24 of 433 patients in the ELEVATE UC 52 trial) with a baseline MMS of 4 were excluded from the primary efficacy analysis, which might compromise randomization.19 According to the sponsor, this amendment to the statistical analysis plan (i.e., changing the FAS to comprise patients with an actual baseline MMS of 4 to 9 to patients with an MMS of 5 to 9) for both trials was made in January 2022 to align with the regulatory body’s feedback on what is considered a moderate to severe UC population, which was outlined in the FDA draft guidance (Ulcerative Colitis: Developing Drugs for Treatment — Guidance for Industry)22 but which became available after the finalization of study protocols and the initiation of patients recruitment (ELEVATE UC 12 enrolled patients between September 2020 and August 2021, and ELEVATE UC 52 enrolled patients between June 2019 and January 2021). In general, the CADTH review team and the clinical expert consulted by CADTH did not identify any major issues with excluding patients with a baseline MMS of 4 for the efficacy analysis that would impact the study results, based on the patient characteristics that appeared to be reasonably balanced between the treatment groups, and given the similar findings in the supplementary analyses of the same outcomes that used the entire FAS for both studies.
In both trials, the double-blind approaches that masked participants as well as investigators (including the outcome assessors) regarding treatment allocation, from the time of random assignment until the time of unblinding per the study protocols, were appropriate. The study drugs were identical in physical appearance and packaging. Some efficacy end points (clinical remission and the related outcomes, clinical response, and symptomatic remission) were composite outcomes that included MMS subscores for stool frequency and rectal bleeding, which were recorded and reported by patients. The HRQoL outcome that was assessed with the IBDQ was also a patient-reported measure. Although these subjective (components of) outcomes may be influenced by knowledge of treatment assignment, the double-blind design of the trials likely mitigated this risk. The CADTH review team noted that in the ELEVATE UC 52 trial, a higher proportion of patients in the placebo group discontinued the treatment due to disease worsening (50.7%) compared with the etrasimod group (27.3%) during the 52-week trial period. Withdrawal by patient as a reason for discontinuing the study or treatment was higher in the placebo group in both trials, except among those who discontinued the study in the ELEVATE UC 52 trial, where a higher percentage of patients treated with etrasimod discontinued the study by patient choice. Also, for the IBDQ total score at week 52 in the ELEVATE UC 52 trial, the rate of missing data was higher in the placebo group than in the etrasimod group. There was no concrete evidence beyond these points that clearly showed unblinding due to patients’ inferences on treatment assignment based on symptom changes or the occurrence of other factors. Thus, the extent to which this could have affected the efficacy and HRQoL outcome results, particularly the outcomes at week 52, is unclear.
Both the ELEVATE UC 12 and ELEVATE UC 52 trials used placebo as the comparator instead of an advanced therapy for UC that is relevant to clinical practice in Canada. The clinical expert consulted by CADTH pointed out that relevant pharmacotherapies for patients with moderately to severely active UC in clinical practice in Canada include adalimumab, golimumab, infliximab, mirikizumab, ozanimod, tofacitinib, upadacitinib, ustekinumab, and vedolizumab. The sponsor submitted evidence regarding comparative effectiveness, which is summarized in the ITC section of this report. It is notable that the FDA guidance to industry for conducting interventional trials in patients with UC22 encourages sponsors to use active treatments as controls.
Overall, the baseline demographic and disease characteristics appeared to be reasonably balanced between the etrasimod and placebo groups in the ELEVATE UC 12 and ELEVATE UC 52 trials. At baseline, 29.2% to 33.9% of the patients across the different groups in the 2 trials reported the use of at least 1 prior biologic or JAKi; the percentages were similar between groups within the studies. The clinical expert did not identify any important imbalance in the baseline characteristics of prognostic importance between the 2 groups within each study.
In the ELEVATE UC 12 and ELEVATE UC 52 trials, most patients received 1 or more concomitant drugs for UC, including antidiarrheals, intestinal anti-inflammatory or anti-infective drugs (mainly mesalazine), corticosteroids for systemic use, and immunosuppressants, among others. The overall concomitant use of systemic corticosteroids appeared similar between groups in each study, although the reported use of budesonide by patients was 3% to 6% higher in the etrasimod groups versus the placebo groups in both studies (Table 12). As well, more patients treated with etrasimod (5.9% and 3.5%) compared with placebo (1.7% and 1.4%) received immunomodulators concurrently. While these are notable differences, the relatively small percentages (< 10%) and small between-group differences (< 5%) mean these differences were unlikely to have been important confounders of the results in both trials.
Overall, the statistical methods used in both trials were appropriate. The trials were powered on their primary and key secondary end points for comparison between the treatment groups. The HRQoL assessed with the IBDQ (an efficacy-related outcome) at week 52 was most likely underpowered, as its outcome data were only available for fewer than half of those with an IBDQ result assessed at baseline. The subgroup analyses were also likely underpowered to identify subgroup differences. An appropriate method for adjusting for multiplicity was used for the primary and secondary outcomes, but there was no multiplicity control for the subgroup analyses. The interaction P values for subgroup analyses were not provided.
In the ELEVATE UC 52 trial, the rates of treatment discontinuation (42.6% in the etrasimod group and 68.1% in the placebo group) and study discontinuation (44.3% in the etrasimod group and 68.1% in the placebo group) were high, mainly due to disease worsening and withdrawal by patients. Nonresponder imputation was used per FDA guidance for missing data in the base case; as well, supportive preplanned sensitivity analyses were done under different assumptions (missing at random, missing not at random). Nonresponder imputation is generally conservative, but it assumes that missing data occur randomly and are unrelated to unobserved variables. However, this assumption is often unrealistic and, if violated, the imputed values may be biased, especially when differences between groups are pronounced.97 However, appropriate methods (e.g., tipping point and multiple imputation) for the sensitivity analyses were used. These analyses confirmed that the results of the trial remained robust to the differential discontinuations between groups.
External Validity
While the indication for etrasimod is for the treatment of moderately to severely active UC in adults, patients aged 16 to 80 years were eligible for the ELEVATE UC 12 and ELEVATE UC 52 trials, yet a relatively small proportion of the enrolled patients were aged 65 years or older (5.0% to 7.4% across the different groups in both trials) and 1 person in each study was younger than 18 years. No patients in the ELEVATE UC 12 trial and only 0.7% of the patients in the ELEVATE UC 52 trial were aged 75 years or older at baseline. These small populations limit the trials’ generalizability among older patients. The clinical expert consulted by CADTH noted that clinicians would be cautious about using etrasimod in patients aged 65 years and older because there is a higher likelihood of comorbidity and/or multiple medications (polypharmacy), as well as a higher potential for decreased hepatic, renal, cardiac, or pulmonary function.
Patients in the ELEVATE UC 12 and ELEVATE UC 52 trials were recruited from multiple countries, including Canada. Approximately 9% of patients in the ELEVATE UC 12 trial and 19% of patients in ELEVATE UC 52 were from North America. The majority of patients enrolled in these trials (55% to 62% across the treatment groups) were from eastern Europe. Also, there were relatively high rates of screening failure in both the ELEVATE UC 12 (41.6%) and ELEVATE UC 52 (47.3%) trials, mainly due to failure to meet eligibility criteria. The clinical expert did not regard these failures as factors that might essentially influence the generalizability of the studies’ results. The clinical expert noted that the eligibility criteria for patients in both trials generally aligned with the diagnosis standard and treatment indication for moderately to severely active UC in clinical settings, and the demographic characteristics of the patients from a diversity aspect in the 2 trials were mostly in line with the patients seen in clinical practice in Canada. Moreover, the clinical expert pointed out that the inclusion of patients with UC with isolated proctitis (< 10 cm rectal involvement) with a limitation of not more than 15% of the total number of the included patients (which is a subgroup of patients with UC that is most often excluded from clinical trials), is helpful for clinical practice, contributing evidence for the efficacy and safety of etrasimod in this specific patient group.
The clinical expert considered the recommended dosage for etrasimod (2 mg taken orally once daily) to be adequate and reasonable for clinical practice in Canada. The clinical expert pointed out the importance of monitoring patients using biomarker examinations (e.g., fecal calprotectin) during treatment with etrasimod.
The trials included outcomes that were important to patients, including sustained clinical remission, corticosteroid-free clinical remission, symptomatic remission, and HRQoL. These outcomes, together with other outcomes reported in this review (i.e., endoscopic improvement, mucosal healing at the longer follow-up time point, and clinical response) were considered appropriate by the clinical expert and the clinician group. The placebo-controlled period of the ELEVATE UC 52 trial was 1 year, which aligns with current regulatory guidance. However, given that patients and clinicians often report a waning of treatment effect with advanced therapies for UC, longer-term comparative evidence on the durability of the effectiveness of etrasimod would be informative, especially in the context of a health technology assessment on comparative effectiveness for purposes of reimbursement. Likewise, the occurrence of some AEs, especially rare ones, may take longer than 52 weeks to be identified. Longer-term follow-up to assess safety and a direct comparison between etrasimod with other advanced therapies would be preferred.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CADTH’s expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:23,24
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on thresholds (MIDs) identified in the literature for the IBDQ total score. The target of the certainty-of-evidence assessment was the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for endoscopic improvement, mucosal healing, clinical remission, sustained clinical remission, corticosteroid-free clinical remission, clinical response, and symptomatic remission.
For the GRADE assessments, findings from the ELEVATE UC 12 and ELEVATE UC 52 trials were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for etrasimod versus placebo in adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Studies
There are currently no results available from any long-term extension studies of etrasimod in moderately to severely active UC.
The sponsor did note there is an ongoing, single-arm, long-term extension study (ELEVATE UC OLE25) that evaluates the safety and efficacy of etrasimod in patients with moderately to severely active UC who previously received double-blind treatment (etrasimod 2 mg/day or placebo) during inclusion in 1 of the phase III or phase II double-blind, placebo-controlled parent studies.16 These studies include but are not limited to ELEVATE UC 52 (APD334 to 301/NCT03945188),98 ELEVATE UC 12 (APD334 to 302/NCT03996369),99 and GLADIATOR UC (APD334 to 210/NCT04607837).100 The ELEVATE UC OLE trial includes patients aged 16 to 80 years with study locations in 39 countries, including Canada (Toronto and Montreal).25 The estimated primary completion date for the ELEVATE UC OLE trial is February 6, 2027.25 It is anticipated that this trial will ultimately provide up to 8 years of efficacy and safety follow-up data for etrasimod in adult patients with moderately to severely active UC.16
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
In the absence of direct head-to-head trials evaluating the comparative efficacy and safety of etrasimod versus relevant comparators for moderately to severely active UC in adult patients, the sponsor submitted a systematic review with an NMA. The sponsor-conducted NMA was used to inform the sponsor-submitted economic model for etrasimod.
Description of Indirect Treatment Comparison
The sponsor submitted 1 NMA that comprises the ██████ ███ █████████ ████ ███ ███ ████████████████████ ██████ ███ ██ ██ ████████ ████████████ ███ █████ ███████████ ████████████ ██████ ███ ██ ██ ██████ █████ ███████ █ ██████████ ███████ █████████ █████████ ████████ █████████████████ ███ ███ █████████ ██ ████████ ████ ████████ ██ ██████ ██████ ██ █████ ██ █████████ ████████ █████ ████████.26,27,101
Indirect Treatment Comparison Design
Objectives
The objective of the sponsor-submitted NMA was to evaluate the relative efficacy and safety of etrasimod versus relevant advanced therapies for moderately to severely active UC in adults.
Study Selection Methods
The NMA followed the guidelines published by the National Institute for Health and Care Excellence (NICE) in NICE DSU [Decision Support Unit] Technical Support Document 2.102 A systematic literature search was conducted in November 2022 and updated in August 2023 for the submission to CADTH to identify RCTs. Multiple electronic databases, including MEDLINE, Embase, and the Cochrane Library, were searched, supplemented with searches of conference proceedings and other sources to identify evidence available from RCTs on the efficacy and safety of interventions used to treat moderate to severe UC.103 Study selection from the initial literature search was conducted by 2 independent reviewers and data extraction was also conducted independently by 2 reviewers. Study selection criteria are summarized in Table 18. The quality of the selected studies was assessed by 2 independent reviewers using the NICE checklist.103 A single reviewer was involved in study selection, data extraction, and quality appraisal, with an independent reviewer overseeing quality checks at each stage following the updated literature search.
The NMA made comparisons between etrasimod versus infliximab, adalimumab, golimumab, vedolizumab, ustekinumab, tofacitinib, filgotinib, upadacitinib, mirikizumab, and ozanimod.26
Table 18: Study Selection Criteria and Methods for ITC Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients with moderately to severely active UC who have previously had an inadequate response, loss of response, or were intolerant to either conventional or advanced therapy. |
Intervention | Etrasimod 2 mg taken orally once daily |
Comparator | The interventions compared with etrasimod included in this analysis are as follows, together with their licensed daily dose ranges according to the EMA summary of product characteristics:
The dose range was divided into recommended induction and maintenance doses if specified in the respective EMA summaries of product characteristics.104-113 |
Outcome | The relevant outcomes included in the analyses for the induction phase were:
For the maintenance phase, the relevant outcomes included in the analyses were:
It should be noted that outcome definitions varied across trials due to the scales used (i.e., either the TMS or MMS were used). |
Study designs | RCTs were of interest for this analysis. The reference lists of relevant systematic literature reviews and meta-analyses were manually searched to identify further relevant publications |
Publication characteristics | Journal articles published from inception until November 15, 2022, and conference abstracts published from 2020 onward were included. Publications in the English language were eligible for inclusion. The systematic search was updated with a targeted search, including articles published from inception until August 7, 2023, and abstracts from conferences proceedings published from November 2022 through August 2023. |
Exclusion criteria | The following were not eligible for inclusion:
|
Databases searched | Databases included in the literature search were MEDLINE, Embase, and the Cochrane Library. To supplement the literature identified from the electronic database search, grey literature searches were carried out across conferences of interest and clinical trial registries (NIH trial registry, EU Clinical Trials Register, International Clinical Trials Registry Platform). Google Scholar searches were additionally conducted. Bibliographies from relevant systematic reviews were also cross-checked. The systematic search was updated with a targeted search of the same electronic databases, clinical trial registries, and supplemental searches. |
Selection process | All records were screened based on a review of the title and abstract by 2 different reviewers, then further screened and selected based on the full text by 2 independent reviewers. Any disagreements were resolved through discussion and review with a third reviewer. A single reviewer was involved in the study selection for the TLR. |
Data extraction process | Two independent reviewers extracted the data into a predesigned Microsoft Excel template. The extracted data were then compared and collated. In case of discrepancies, a third reviewer was involved. A single reviewer conducted the data extraction for the TLR. |
Quality assessment | For the quality assessment, the NICE checklist was used.103 Two independent reviewers conducted the assessment. A single reviewer conducted the quality assessment for the TLR. |
AE = adverse event; EMA = European Medicines Agency; EU = European Union; ITC = indirect treatment comparison; MMS = modified Mayo score; NICE = National Institute of Health and Care Excellence; NIH = National Institutes of Health; NMA = network meta-analysis; RCT = randomized controlled trial; SC = subcutaneous; TLR = targeted literature review; TMS = total Mayo score; UC = ulcerative colitis.
aNot given daily; given on specific weeks.
bBased on dose weight of patient at the time of dosing.
cThe initial induction dose for children and adolescents is 80 mg.
dSuggested dose for patients with renal impairment.
eMirikizumab was included in the Canadian NMA only.
fOutcomes were not assessed in the Canadian base-case analyses.
Sources: Sponsor-submitted NMAs.26,27,101,114
ITC Analysis Methods
Statistical Model
The NMAs were conducted under a Bayesian framework102 using a Markov chain Monte Carlo, in accordance with NICE technical support documents.102,115 Placebo was selected as the reference treatment for all analyses.26 Results were generated using both random-effects and fixed-effects models when possible and compared for goodness of fit to the data, calculated as the total residual deviance.26 The fixed-effects models were the most appropriate model approach in the majority of cases.26 Typically, only 1 study informed each comparison and, as a result, it was inappropriate to use random-effects models. The deviance information criterion (DIC) was reported for both models to inform the model fit, with lower values of DIC indicating a better model fit. Typically, differences of 5 for the DIC would be considered an important difference.26 The multinomial approach was the preferred analysis approach where outcomes were correlated, as it allows for the inclusion of all available information and ensures coherence across the outcomes. This approach was applied where appropriate and possible. The probit link function was used for the multinomial modelling.16
Prior Distribution
Since the test-specific baselines, 𝜇𝑖, are regarded as nuisance parameters (i.e., they are estimated in the model but are not of interest), they were given vague priors, where 𝜇𝑖 is asymptotic to 𝒩(0, 104).26 Furthermore, under the consistency assumptions of the NMA and the assumption that consistency equations can be written generally as 𝑑𝑡𝑖1,𝑡𝑖𝑘 = 𝑑1,𝑡𝑖𝑘 − 𝑑1,𝑡𝑖1, the parameters 𝑑12 and 𝑑13 were given vague prior distributions: 𝒩 (0, 104).26 These noninformative priors applied to both the fixed-effects and the random-effects models.26 In addition, the random-effects model required priors for the variance of parameters 𝛿𝑖,𝑘.26 A vague prior was set for its SD: 𝑠𝑑 is asymptotic to 𝒰niform (0, 5).26 This prior was adjusted where necessary to improve model convergence.26 In addition, for the meta-regression coefficient, a vague prior was given, 𝒩 (0, 104). For the ordinal category cut-offs, a vague prior was also used, 𝒰niform(0, 2).26
Model Selection
As mentioned, as only 1 study informed each comparison, it was inappropriate to use random-effects models.26 A fixed-effects model was the most appropriate model approach in the majority of cases.26 Where possible, results were generated using both random-effects and fixed-effects models and compared for goodness of fit to the data, calculated as the total residual deviance.26 The DIC, a measure of model fit, was reported for both models, with lower values of DIC indicating a better model fit (typically differences of 5 would be considered an important difference ).16
Assessment of Homogeneity
Homogeneity was assessed by performing a pairwise meta-analysis for each comparison informing the network (e.g., etrasimod versus placebo) or, if there were multiple studies informing the comparison, using the I2 statistic and the P value from the chi-square test to assess potential heterogeneity.26,101 This type of analysis may provide more robust comparisons against these comparators and give an indication of the potential for bias across the standard NMAs due to the concomitant use of medications.26
The level of significance used was 0.1, since the chi-square statistic is typically underpowered. To interpret the I2 value, the following was used:116
0% to 40%: Might not be important.
30% to 60%: May represent moderate heterogeneity.
50% to 90%: May represent substantial heterogeneity.
75% to 100%: Considerable heterogeneity.
The trials included in the NMA were carefully assessed for possible sources of heterogeneity in a feasibility assessment in collaboration with experts in either UC or in the statistical or economic component of the development of these analyses.26 Feedback, which was obtained through a sponsor-conducted virtual advisory board meeting, was sought on the potential sources of heterogeneity in the patient and study characteristics of the included studies, treatments, and analysis approaches.26
Assessment of Inconsistency
Given the structure of the network and the structure for any closed loop, all nodes are linked by the same trial; no inconsistencies were expected. Nonetheless, to test the consistency assumption, an unrelated mean effects (UME) model was implemented for the primary analyses (global [AL] analyses AL1 to AL9, fixed-effects models without baseline risk adjustment).117 Primary NMA models and inconsistency models (UME models) were compared according to the recommendations of Daly et al. (2022).118
Construction of Nodes
All nodes were connected by the same trial.26 The analyses did not pool doses or regimens.26
Outcomes
Clinical efficacy outcomes analyzed include:
clinical response
clinical remission
sustained clinical response among induction responders
sustained clinical remission among induction responders.
The safety outcomes analyzed include:
serious infections
SAEs
overall AEs
treatment discontinuations due to AEs.
No preplanned outcomes were excluded or not analyzed.16
Of note, there were differing definitions of the outcomes used across trials due to the scales used, i.e., the TMS or the MMS. Where a trial reported both the TMS and the MMS, the TMS was used, given that the TMS was reported more frequently.26 The MMS was used for the ELEVATE trials since this was the primary outcome measure.82
Analyses
The primary analyses conducted are summarized in Table 19. Trials with a maintenance phase of less than 38 weeks were excluded to align with the primary efficacy analyses, and only trials from the overall population were included, so trials that were “naive only” were excluded.26 Parts of studies or entire studies were excluded from the review because their doses were outside the range authorized by the European Medicines Agency.26
The population of biologics- and JAKi-naive, biologics-naive, TNF inhibitor (TNFi)-naive, and TNFi- and biologics-naive patients is referred to as the advanced therapy–naive population. The population of biologics- and JAKi-exposed, biologics-exposed, and/or TNFi-exposed patients is referred to as the advanced therapy–experienced population.26
Table 19: Summary of Main Analyses for ITC Submitted by the Sponsor
Analysis name | Population | Outcome | Phase | Model | Fixed or random effects | Baseline risk adjustment |
|---|---|---|---|---|---|---|
CA1 | Advanced therapy–naive | Clinical response and clinical remission | Induction | Multinomial | Fixed and random | No |
CA2 | Advanced therapy–experienced | Clinical response and clinical remission | Induction | Multinomial | Fixed | No |
CA3 | Advanced therapy–naive | Sustained clinical response and clinical remission among induction responders | Maintenance | Multinomial | Fixed | No |
CA4 | Advanced therapy–experienced | Sustained clinical response and clinical remission among induction responders | Maintenance | Multinomial | Fixed | No |
CA5 | Overall and advanced therapy–naive | Serious infections | Induction | Binomial | Fixed and random | Yes |
CA8 | Overall and advanced therapy–naive | Serious infections | Maintenance | Binomial | Fixed and random | No |
AL1a | Advanced therapy–naive | Clinical response | Induction | Multinomial | Fixed | No |
AL1b | Advanced therapy–naive | Clinical remission | Induction | Multinomial | Fixed | No |
AL2a | Advanced therapy–experienced | Clinical response | Induction | Multinomial | Fixed | No |
AL2b | Advanced therapy–experienced | Clinical remission | Induction | Multinomial | Fixed | No |
AL3a | Advanced therapy–naive | Sustained clinical response among induction responders | Maintenance | Multinomial | Fixed | No |
AL3b | Advanced therapy–naive | Sustained clinical remission among induction responders | Maintenance | Multinomial | Fixed | No |
AL4a | Advanced therapy–experienced | Sustained clinical response among induction responders | Maintenance | Multinomial | Fixed | No |
AL4b | Advanced therapy–experienced | Sustained clinical remission among induction responders | Maintenance | Multinomial | Fixed | No |
AL5 | Overall | Serious infections | Induction | Binomial | Fixed | No |
AL6 | Overall | Serious infections | Maintenance | Binomial | Fixed | No |
AL7a | Overall | Serious AEs | Maintenance | Binomial | Fixed | No |
AL8a | Overall | Overall AEs | Maintenance | Binomial | Fixed | No |
AL9a | Overall | Discontinuation due to AEs | Maintenance | Binomial | Fixed | No |
TT1 | Advanced therapy–naive | Clinical response and clinical remission | Treatment | Multinomial | Fixed | No |
TT2 | Advanced therapy–experienced | Clinical response and clinical remission | Treatment | Multinomial | Fixed | No |
AE = adverse event; ITC = indirect treatment comparison.
Note: Global analyses are labelled AL, while analyses conducted for Canada are labelled CA. Global analyses that included only treat-through trials are labelled TT. Analyses conducted for Canada include mirikizumab as a comparator, which was not included in the global analyses.
aThis analysis was not included in the Canadian model.
Sources: Sponsor-submitted network meta-analyses.26,101
Subgroup Analysis
The focus of this analysis was on the biologics-naive (i.e., advanced therapy–naive) and biologics-exposed (i.e., advanced therapy–exposed or –experienced) subgroups, since there was precedent to split the population due to prior treatment being an effect modifier. Consideration was also given to other subgroups and the overall population.26
Sensitivity Analysis
A summary of the sensitivity analyses conducted is presented in Table 20. No sensitivity analyses were conducted for the Canadian base-case analyses.119 Several sensitivity analyses were conducted for the global analyses to test assumptions of the data generated by the ELEVATE trials.26
Sensitivity analyses SA1 to SA4 were performed to investigate the robustness of the primary analyses by using TMS outcomes instead of MMS outcomes for the ELEVATE trials, as most trials included in the networks report outcomes using the TMS rather than the MMS, despite the general conclusion that there is a strong correlation between the 2 score measures.
Sensitivity analyses SA5 to SA8 used the FAS in the ELEVATE trials. In the primary analyses, the population in the ELEVATE trials was restricted to patients who had an MMS of between 5 and 9, as this aligned with the definition that other trials have used to define moderately to severely active UC. The FAS population from the ELEVATE trials with a baseline MMS of 4 to 9 was used to examine the impact of including patients with less severe disease.
As only failure and no-failure data were available for upadacitinib and there were no therapy–naive or therapy–exposed data available during the time of the analysis, sensitivity analyses SA9 to SA12 were run, excluding upadacitinib from the network to assess the impact. Upadacitinib failure data were used for sensitivity analyses SA5 to SA12 rather than exposure data, as this was the only data available at the time of the analysis.
To assess the impact that studies with Asian-only populations have on the analyses, sensitivity analyses SA13 and SA14 were conducted and applied only to analysis networks that included these studies (AL1 and AL3).
Sensitivity analyses SA15 to SA18 were conducted to assess the impact of stratifying patients in the ELEVATE trials by biologic status only because most trials included in the analysis networks stratified patients by prior biologic status at baseline rather than prior biologic or JAKi treatment status.
The main analyses included trials with maintenance periods of at least 38 weeks; however, there were several trials with shorter maintenance periods, particularly infliximab trials. Sensitivity analysis SA19 was conducted to investigate the impact of including trials with a shorter maintenance period. Sensitivity analysis treat-through trial 1 (SATT1) also assessed the impact of including treat-through trials with a shorter maintenance period.
In the main safety analyses, only the overall (mixed advanced therapy–naive and advanced therapy–exposed) populations in the trials were included; however, there were several trials that included only advanced therapy–naive populations. These trials were included in sensitivity analyses SA24 to SA28 to investigate the impact of the included trials and comparators on outcomes.
Two trials, Sandborn (2012) and VISIBLE 1,120,121 were excluded from the main efficacy analyses, as they reported only 1 of the 2 efficacy outcomes for the multinomial model. The trials were included in sensitivity analyses SA29 to SA32, with the additional assumption that the cut-offs for the missing categories (in the multinomial model) were sampled from the distribution informed by other trials.
Summary of ITC Analysis Methods
The ITC analysis methods are summarized in Table 21.
Table 20: Summary of Sensitivity Analyses for ITC Submitted by the Sponsor
Sensitivity analysis name | Sensitivity variable | Description |
|---|---|---|
SA1 | Outcome based on TMS | Analysis of AL1 using efficacy data based on TMS, rather than MMS, for the ELEVATE trials (using binomial models) |
SA2 | Outcome based on TMS | Analysis of AL2 using efficacy data based on TMS, rather than MMS, for the ELEVATE trials (using binomial models) |
SA3 | Outcome based on TMS | Analysis of AL3 using efficacy data based on TMS, rather than MMS, for the ELEVATE trials |
SA4 | Outcome based on TMS | Analysis of AL4 using efficacy data based on TMS, rather than MMS, for the ELEVATE trials |
SA5a | MMS 5 to 9 population | Analysis of AL1 using the FAS in the ELEVATE trials (patients with MMS 4 to 9 instead of restricting to patients with an MMS of 5 to 9 to match other trials) |
SA6a | MMS 5 to 9 population | Analysis of AL2 using the FAS in the ELEVATE trials (patients with MMS 4 to 9 instead of restricting to patients with an MMS of 5 to 9 to match other trials) |
SA7a | MMS 5 to 9 population | Analysis of AL3 using the FAS in the ELEVATE trials (patients with MMS 4 to 9 instead of restricting to patients with an MMS of 5 to 9 to match other trials) |
SA8a | MMS 5 to 9 population | Analysis of AL4 using the FAS in the ELEVATE trials (patients with MMS 4 to 9 instead of restricting to patients with an MMS of 5 to 9 to match other trials) |
SA9a,b | Exclude failure populations | Analysis of AL1 excluding no prior biologics failure and no prior TNFi failure populations |
SA10a,b | Exclude failure populations | Analysis of AL2 excluding prior biologics failure and prior TNFi failure populations |
SA11a,b | Exclude failure populations | Analysis of AL3 excluding no prior biologics failure and no prior TNFi failure populations |
SA12a,b | Exclude failure populations | Analysis of AL4 excluding prior biologics failure and prior TNFi failure populations |
SA13 | Exclude Asian-only populations | Analysis of AL1 excluding trials with Asian-only study populations |
SA14 | Exclude Asian-only populations | Analysis of AL3 excluding trials with Asian-only study populations |
SA15 | Biologics only | Analysis of AL1 with the population defined by biologics only in the ELEVATE studies |
SA16 | Biologics only | Analysis of AL2 with the population defined by biologics only in the ELEVATE studies |
SA17 | Biologics only | Analysis of AL3 with the population defined by biologics only in the ELEVATE studies |
SA18 | Biologics only | Analysis of AL4 with the population defined by biologics only in the ELEVATE studies |
SA19 | Maintenance period length | Analysis of AL3, including trials with a maintenance period of less than 38 weeks |
SA24 | Include naive trials | Analysis of AL5, including trials that are wholly naive |
SA25 | Include naive trials | Analysis of AL6, including trials that are wholly naive |
SA26 | Include naive trials | Analysis of AL7, including trials that are wholly naive |
SA27 | Include naive trials | Analysis of AL8, including trials that are wholly naive |
SA28 | Include naive trials | Analysis of AL9, including trials that are wholly naive |
SA29 | Include single-outcome trials | Analysis of AL1, including trials that reported only 1 outcome |
SA30 | Include single-outcome trials | Analysis of AL2, including trials that reported only 1 outcome |
SA31 | Include single-outcome trials | Analysis of AL3, including trials that reported only 1 outcome |
SA32 | Include single-outcome trials | Analysis of AL4, including trials that reported only 1 outcome |
SATT1 | Maintenance period length | Analysis of TT1, including trials with maintenance period less than 38 weeks |
FAS = full analysis set; ITC = indirect treatment comparison; MMS = modified Mayo score; TMS = total Mayo score; SA = sensitivity analysis; TNFi = tumour necrosis factor inhibitor; TT = treat through.
Note: Global analyses are labelled AL.
aModels were conducted before the inclusion of the HIBISCUS studies.
bSAs were not conducted, as they were relevant when only failure and non-failure subgroup data were available for upadacitinib; however, given more recent reporting on the upadacitinib trial (which reported on naive and exposed subgroups), these SAs are no longer required but are included in the table for completeness.
Sources: Sponsor-submitted network meta-analyses.26,101
Table 21: ITC Analysis Methods
Methods | Description |
|---|---|
Analysis methods | The NMAs were conducted under a Bayesian framework using MCMC sampling. |
Priors | Test-specific baselines were given vague priors. Furthermore, under the consistency assumptions, parameters 𝑑12 and 𝑑13 in the consistency equation were given vague prior distribution. These noninformative priors applied to both the fixed-effects and random-effects models. In addition, the random-effects model required priors for the variance of parameters 𝛿𝑖,𝑘. A vague prior was set for its standard deviation: 𝑠𝑑 ~ 𝒰niform(0, 5). This prior was adjusted where necessary to improve model convergence. In addition, for the meta-regression coefficient, a vague prior was given, 𝒩(0, 104). For the ordinal category cut-offs, a vague prior was also used, 𝒰niform(0, 2). |
Assessment of model fit | The fixed-effects models were the most appropriate model approach in the majority of cases. Where possible, results were generated using both random-effects and fixed-effects models and compared for goodness of fit to the data, calculated as the total residual deviance. The DIC, a measure of model fit, was reported for both models. |
Assessment of consistency | Given the structure of the network and that for any closed loops, all nodes are connected by the same trial; no inconsistency was expected. A UME was implemented for the primary analyses (AL1 to AL9, fixed-effects models with no baseline risk adjustment) to assess the consistency assumption. The primary NMA models and the inconsistency models (UME models) were compared following the guidance by Daly et al. (2022). |
Assessment of convergence | Comparative models were fitted using 3 MCMC chains. Convergence was assessed by reviewing trace plots and assessing the Gelman-Rubin statistics, and correlation was assessed by reviewing autocorrelation plots. If correlation was present, an appropriate thinning factor was applied. The number of iterations and burn-in iterations are reported alongside the results. The complexity of the models combined with the limited data informing the networks resulted in convergence issues, particularly in estimating the baseline risk adjustment and the random-effects models. Attempts were made to overcome convergence issues, for example, by using less informative priors. |
Outcomes | Outcomes include clinical response, clinical remission, and serious infections. Analyses were divided between the outcomes analyzed during the induction period and the outcomes analyzed during the maintenance period. |
Follow-up time points | Separate analyses were conducted for outcomes reported over the induction or maintenance period. The main analyses excluded studies with a maintenance period of less than 38 weeks to improve homogeneity in the end points. A sensitivity analysis (global model) included trials with a maintenance period of less than 38 weeks (this applied to AL3 only). |
Construction of nodes | All nodes are connected by the same trial. The analyses did not pool doses or regimens. |
Sensitivity analyses (conducted only for the global model) |
|
Subgroup analysis | Separate analyses were conducted for 3 populations: advanced therapy–naive, advanced therapy–experienced, and overall population. |
Methods for pairwise meta-analysis | To assess the assumption of homogeneity, pairwise meta-analyses for each comparison informing the network were performed. This type of analysis may provide more robust comparisons against these comparators and give an indication of the potential for bias across the standard NMAs due to the concomitant use of medications. |
DIC = deviance information criterion; ITC = indirect treatment comparison; MCMC = Markov chain Monte Carlo; MMS = modified Mayo score; NMA = network meta-analysis; TMS = total Mayo score; TNFi = tumour necrosis factor inhibitor; UME = unrelated mean effects.
Sources: Sponsor-submitted NMAs.26,101
Table 22: Assessment of Homogeneity for ITC
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Patient characteristics | An assessment of within-trial baseline characteristics was conducted across the various populations. Based on the baseline characteristics assessed, the within-trial differences were minimal and generally balanced across arms. One of the characteristics where there was some variation across studies was the use of concomitant medications. The most common concomitant medications were glucocorticoids and corticosteroids, immunosuppressants and, to a lesser extent, budesonide. Corticosteroid use varied among studies. In the overall population, the proportion varied from 13.4% to more than 60%. It is possible that corticoid use may be an effect modifier; therefore, the variation of corticoid use across studies may influence outcomes. |
Treatment history | A number of studies have conducted analyses in subgroup populations. The most common subgroups assessed included prior exposure to advanced therapies. Some of the trials included a wholly naive patient population. Subgroup analyses were performed that stratified patients by advanced therapy exposure status within the NMAs. |
Trial eligibility criteria | Overall, the inclusion criteria were similar across studies. Some of the most prevalent inclusion criteria comprised the following:
The most prevalent exclusion criteria included excluding patients with toxic megacolon, a diagnosis of Crohn disease, indeterminate colitis, Clostridioides difficile, a prior colectomy (which in some studies varied between an extensive resection or partial or total colectomy) and fulminant colitis. Other studies excluded patients if the UC was limited to the rectum only or to less than a specified amount of the colon. Several studies also excluded patients based on prior therapies. |
Dosing of comparators | There were some differences in the treatment regimens among the trials. For example, 2 vedolizumab studies had slightly varying dosing regimens: GEMINI 1 had 300 mg doses at weeks 0 and 2, whereas VARSITY had an additional dose at week 6. Similarly, in the trials of infliximab, over the induction period, the Probert (2003) trial had doses at weeks 0 and 2, whereas the ACT-1, ACT-2, NCT01551290, Jiang (2015), and Kobayashi (2016) studies had doses at weeks 0, 2, and 6. The Järnerot (2005) study did not report the dosing regimen in that trial. In addition, the LIBERTY UC trial had a subcutaneous injection formulation for infliximab compared with all other trials, which had IV formulations of infliximab. |
Placebo response | Placebo response varied among studies. Given the clear heterogeneity among placebo responses across studies and the treatments included in the feasibility assessment, combined with the correlation between placebo response and observed treatment effect, placebo was adjusted for through meta-regression. |
Definitions of end points | There were 2 outcome definitions used to define clinical response, namely, the TMS (full Mayo score) and the adapted or modified MCS, also called the MMS. However, among the studies that reported the adapted or modified MCS, there was some variation. Mucosal healing was defined in most studies as endoscopic improvement plus histologic remission. For these studies, endoscopic improvement was defined as an endoscopic subscore of ≤ 1, with the exception of the U-ACHIEVE phase III maintenance study, which specified an endoscopic subscore of 0. Histologic remission was defined as a Geboes score of < 2. Some studies (e.g., SELECTION trial) added further requirements. Endoscopic improvement was described similarly for all studies and was defined as an endoscopic subscore of ≤ 1. Sustained clinical remission was defined in all studies as clinical remission at the end of induction (or at baseline for maintenance-only studies) and at the end of maintenance. Similar to the clinical response and clinical remission outcomes described above, most of the studies define clinical remission using the TMS and some use the MMS. Corticosteroid-free clinical remission was defined as the number of patients who were corticosteroid-free at the end of maintenance and had achieved clinical remission. Some studies specified a time period during which patients should have remained corticosteroid-free to achieve the outcome. This ranged from 4 weeks before the end of the maintenance period for the OCTAVE Sustain trial to 6 months before the end of the maintenance period for the SELECTION study. |
Timing of end point evaluation | The timing of outcomes over both the induction and maintenance periods varied somewhat. Of the trials that included an induction period, the length of these induction periods generally varied between 6 and 12 weeks. The Järnerot (2005) and VARSITY trials had a 13- and 14-week induction period, respectively. The Sands (2001) trial had only a 2-week induction period. For trials that included a maintenance period, the length of the maintenance period also varied between studies and generally ranged from 38 to 52 weeks. However, there were several studies with shorter maintenance periods, for example, the ACT-2, TOUCHSTONE, and Jiang (2015) studies had maintenance durations of 16, 22, and 24 weeks, respectively. |
Clinical trial setting | Some trials were conducted in a wholly Asian population, i.e., Jiang (2015), PURSUIT-J, and Kobayashi (2016). |
Study design | There are 2 trial designs in the included studies, namely, the treat-through trial design and the responder rerandomized trial design. The differing trial designs cannot be synthesized in the same meta-analysis without adjustment. The feasibility assessment examined 2 approaches to adjust the outcomes of 1 trial design to match the other. Mimicking the rerandomized trials was considered the most appropriate approach, given the majority of trials were rerandomized, and this approach would allow an estimate of outcomes for the induction and maintenance periods separately. |
ITC = indirect treatment comparison; MCS = Mayo Clinic score; MMS = modified Mayo score; NMA = network meta-analysis; TMS = total Mayo score; UC = ulcerative colitis.
Sources: Sponsor-submitted NMAs.122
Results of ITC
Summary of Included Studies
A total of 28 trials were included in the Canadian NMA. The majority of the comparisons from the included studies retained for analysis were phase III or IIIb trials (n = 25), while the remaining were phase II or III, phase IIb or III (n = 2), phase II or IIb (n = 3), or not reported (n = 1).122 Thirteen comparisons reported data for the induction period only, 3 comparisons reported data for the maintenance period only, and 15 comparisons reported data for both the induction and maintenance periods.122 Among the trials with a maintenance period, 4 trials had a treat-through study design and 10 trials had a rerandomized study design.122 An assessment of homogeneity is provided in Table 22. █████ █████ ████ █ ███ ███████████ ████ ██████ ████ ████████ ██████████████ ████ ████ ███ █████████████ ████████████
Results
Evidence Networks
The networks of evidence for the Canadian base-case analysis of clinical response and clinical remission, as well as serious infections during the induction or maintenance phase (CA1, CA2, CA3, CA4, CA5, and CA8) are presented in Figure 5, Figure 6, and Figure 7.
Figure 5: Network Diagram for the Multinomial Analysis of Clinical Response and Clinical Remission (in Induction Phase)
ADA = adalimumab; ETS = etrasimod; FIL = filgotinib; GOL = golimumab; IFX = infliximab; MIR = mirikizumab; OZN = ozanimod; PBO = placebo; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VDZ = vedolizumab.
Note: Figure A illustrates the network diagram for the multinomial analysis of clinical response and clinical remission during the induction phase among the advanced therapy–naive population (analysis CA1). Figure B illustrates the network diagram for the multinomial analysis of clinical response and clinical remission during the induction phase among the advanced therapy–experienced population (analysis CA2).
Sources: Sponsor-submitted network meta-analyses.27
Figure 6: Network Diagram for the Multinomial Analysis of Sustained Clinical Response and Clinical Remission Among Induction Phase Responders (in Maintenance Phase)
ADA = adalimumab; ETS = etrasimod; FIL = filgotinib; GOL = golimumab; IFX = infliximab; MIR = mirikizumab; OZN = ozanimod; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VDZ = vedolizumab.
Note: Figure A illustrates the network diagram for the multinomial analysis of sustained clinical response and clinical remission among induction phase responders during the maintenance phase among the advanced therapy–naive population (analysis CA3). Figure B illustrates the network diagram for the multinomial analysis of sustained clinical response and clinical remission among induction phase responders during the maintenance phase among the advanced therapy–experienced population (analysis CA4).
Sources: Sponsor-submitted network meta-analyses.27
Figure 7: Network Diagram for the Binomial Analysis of Serious Infections in Overall and Advanced Therapy–Naive Populations
ADA = adalimumab; ETS = etrasimod; FIL = filgotinib; GOL = golimumab; IFX = infliximab; MIR = mirikizumab; OZN = ozanimod; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VDZ = vedolizumab.
Note: Figure A illustrates the network diagram for the binomial analysis of serious infections during the induction phase among the overall and advanced therapy–naive populations (analysis CA5). Figure B illustrates the network diagram for the binomial analysis of serious infections during the maintenance phase among the overall and advanced therapy–naive populations (analysis CA8).
Sources: Sponsor-submitted network meta-analyses.27
Efficacy
The Canadian NMA results for efficacy outcomes (etrasimod versus the relevant comparators) at induction and maintenance are summarized in Table 23 and Table 24. In general, the results of the sensitivity analyses conducted for the global NMA were consistent with the base-case analyses.16
Table 23: Summary of Canadian NMA Results for Efficacy Outcomes (Clinical Response, and Clinical Remission) at Induction, Etrasimod Versus Comparators)
Population | Treatment | Clinical response, median RR (95% CrI) | Clinical remission, median RR (95% CrI) |
|---|---|---|---|
Advanced therapy–naive | Ozanimod 1 mg | ████ █████ | ████ █████ |
Tofacitinib 10 mg | ████ █████ | ████ █████ | |
Infliximab 5 mg/kg | ████ █████ | ████ █████ | |
Ustekinumab 6 mg/kg | ████ █████ | ████ █████ | |
Golimumab 200 mg and 100 mg | ████ █████ | ████ █████ | |
Adalimumab 160 mg and 80 mg | ████ █████ | ████ █████ | |
Adalimumab 80 mg and 40 mg | ████ █████ | ████ █████ | |
Vedolizumab 300 mg (weeks 0, 2, 6) | ████ █████ | ████ █████ | |
Vedolizumab 300 mg (weeks 0, 2) | ████ █████ | ████ █████ | |
Upadacitinib 45 mg | ████ █████ | ████ █████ | |
Mirikizumab 300 mg | ████ █████ | ████ █████ | |
Advanced therapy–experienced | Ozanimod 1 mg | ████ █████ | ████ █████ |
Tofacitinib 10 mg | ████ █████ | ████ █████ | |
Ustekinumab 6 mg/kg | ████ █████ | ████ █████ | |
Adalimumab 160 mg and 80 mg | ████ █████ | ████ █████ | |
Vedolizumab 300 mg (weeks 0, 2, 6) | ████ █████ | ████ █████ | |
Vedolizumab 300 mg (weeks 0, 2) | ████ █████ | ████ █████ | |
Upadacitinib 45 mg | ████ █████ | ████ █████ | |
Mirikizumab 300 mg | ████ █████ | ████ █████ |
CrI = credible interval; NMA = network meta-analysis; RR = relative risk.
Note: This table presents the results of pairwise comparisons among all treatments for the fixed-effects model without the baseline risk adjustment.
Sources: Sponsor-submitted NMA.27
Table 24: Summary of Canadian NMA Results for Efficacy Outcomes (Sustained Clinical Response and Clinical Remission) at Maintenance Among Induction Phase Responders, Etrasimod Versus Comparator
Population | Treatment | Sustained clinical response, median RR (95% CrI) | Clinical remission among induction phase responders, median RR (95% CrI) |
|---|---|---|---|
Advanced therapy–naive | Ozanimod 1 mg | ████ █████ | ████ ██████ █████ |
Tofacitinib 5 mg | ████ █████ | ████ ██████ █████ | |
Tofacitinib 10 mg | ████ █████ | ████ ██████ █████ | |
Infliximab 5 mg/kg | ████ █████ | ████ ██████ █████ | |
Ustekinumab 90 mg q.12.w. | ████ █████ | ████ ██████ █████ | |
Ustekinumab 90 mg q.8.w. | ████ █████ | ████ ██████ █████ | |
Golimumab 50 mg | ████ █████ | ████ ██████ █████ | |
Golimumab 100 mg | ████ █████ | ████ ██████ █████ | |
Adalimumab 40 mg | ████ █████ | ████ ██████ █████ | |
Vedolizumab 300 mg q.8.w. | ████ █████ | ████ ██████ █████ | |
Vedolizumab 300 mg q.4.w. | ████ █████ | ████ ██████ █████ | |
Upadacitinib 15 mg | ████ █████ | ████ ██████ █████ | |
Upadacitinib 30 mg | ████ █████ | ████ ██████ █████ | |
Mirikizumab 200 mg | ████ █████ | ████ ██████ █████ | |
Advanced therapy–experienced | Ozanimod 1 mg | ████ █████ | ████ ██████ █████ |
Tofacitinib 5 mg | ████ █████ | ████ ██████ █████ | |
Tofacitinib 10 mg | ████ █████ | ████ ██████ █████ | |
Ustekinumab 90 mg q.12.w. | ████ █████ | ████ ██████ █████ | |
Ustekinumab 90 mg q.8.w. | ████ █████ | ████ ██████ █████ | |
Adalimumab 40 mg | ████ █████ | ████ ██████ █████ | |
Vedolizumab 300 mg q.8.w. | ████ █████ | ████ ██████ █████ | |
Vedolizumab 300 mg q.4.w. | ████ █████ | ████ ██████ █████ | |
Upadacitinib 15 mg | ████ █████ | ████ ██████ █████ | |
Upadacitinib 30 mg | ████ █████ | ████ ██████ █████ | |
Mirikizumab 200 mg | ████ █████ | ████ ██████ █████ |
CrI = credible interval; NMA = network meta-analysis; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; RR = relative risk.
Note: This table presents the results of pairwise comparisons among all treatments for the fixed-effects model without the baseline risk adjustment.
Sources: Sponsor-submitted NMAs.27
Clinical Response
Results from the fixed-effects model for clinical response demonstrated █████████ ██ ██ ███████ ████████████████ ██████ ██ ███ ██████ ████ ███ ████████████ ███████ ████ █████████ ██ ███ ████████ ███████ █████ ███████████ █████ ████ ██ ████ ███ ███ ████ ██ ███ ███████████Table 23██ ██████ ███████ █████ ██████ ████ ██████████ ██████ ███ ███ ████████ ██ █████████ ██ ███ ████████████████ ███████████ ███ █████ ████ ██ ███████████ █ ███ ███ █████ ███████████ ██ ████████████████ ████████ ████ ██████████ ██ ████████ ██████ ██ ██ ███ █████ ██ █████ ██ █████████
███████ ████ ███ █████ ███████ █████ ██ ████████ ███████████████████ ██████████ ███████ ████████████ ███ ████████ ████████ ██ █████████Table 23█████ ███ ██ ██████████ ██████████ ███ ███ █████ █████████ ██ ████████ ████████ ██ ███ ████████ ███████████████████ ████████ ████ ██ ██ ██ █████ ██ █████████
Clinical Remission
███████ ████ ███ █████ ███████ █████ ████████████ ████ █████████ ████████ ████ ██████████ ██████ ██ ███ ██████ ██ ██████ ██ ███ ████████ █████████████ ██████████Table 23████████████ ██████ ████████ ████████ ██████ █████████ ███ ████████ █████████ ██ ████ ███████████████ ███ ████████ ███████████████████ ███████████Table 23██ ███ ████ ██████ ██ ███████ ██ ████ ███ ████████ █████████. Results from the random-effects model for this population found that the 95% CrI did not exclude the null for some comparisons between active treatments and placebo.27 ███ ██████ ███████ █████ ██████ ███████ ██████ ███ ███ ████████ ██ █████████ ██ ███ ████████ ████████████████████████ ██████████ ███ █████ ██ █████ ██ ██████████████ ███ ████████ █████████████ ████ ████████ ███████████ ████ ████████ ██ ████████ ███ ██████████ ██ ███ █████ ██████████ ██████ ████████████
Sustained Clinical Response Among Induction Phase Responders
For the maintenance phase, both random-effects and fixed-effects models were attempted; however, since only the fixed-effects model converged, that approach was used.27 The 95% CrI did not exclude the null for some comparisons between active treatments and placebo in the advanced therapy–naive population.27 In the advanced therapy–naive population, █████ ███ ██████████ ███ █████████ ████████ ████████ ██ ███ ███████████ █████Table 24███ ███ ██████████████████ ███████████ ███ █████████ ████████ ██████ ██ ██████████ ██ █████ ███████████ █████████ ███████Table 24██. Of note, for etrasimod, the number of patients on which these analyses are based was low.27 In the placebo arm of the ELEVATE UC 52 trial, there were only 11 patients in the denominator of the placebo arm, of which 6 were responders.27 Furthermore, the results for this population are not reflective of data from the ELEVATE UC 12 and 52 phase III RCTs, which demonstrated that etrasimod provided improvement in sustained clinical response.
Clinical Remission Among Induction Phase Responders
For the maintenance phase, results from the fixed-effects model demonstrated that the 95% CrI did not exclude the null for some comparisons between active treatments and placebo in the advanced therapy–naive population.27 Among this population, for clinical remission among induction phase responders, █████████ ██████ ██ ██████████ ██ ███ █████ █████████ ██████ ███████ (Table 24).27 Among the advanced therapy–experienced population, ███ ████████ █████████ █████ █████████ █████ ███████████ █████████ ███ ████████ ██ ████████████ ███ ██ ███ ██ ██ ███████ ███ ██████ ██ ██████████ ██ █████ ███████████ █████████ ███████ (Table 24).27 The results of this analysis are based on only 11 patients in the denominator for the placebo arm, and among them, 6 patients had a placebo response.27 Furthermore, the results for this population are not reflective of data from the ELEVATE UC 12 and 52 phase III RCTs, which demonstrated that etrasimod provided improvement in sustained clinical remission.16
Harms
Serious Infections
Results of the Canadian NMA from the fixed-effects model and random-effects model with baseline risk adjustment ████████████ ██ ██████████ ██ ███████ ██████████ ███████ █████████ ███ ███ ████████████ ██ ███████ ██ ███ ███████ ███ ████████ █████████████ █████ ███████████ ███ ██████ ███ █████████ █████ ██ ███ ███████████ ██████
For the other harms outcomes (SAEs, AEs, and discontinuation due to AEs), the results were only available from the global NMA (excluding mirikizumab from networks) and over the maintenance period for the overall and advanced therapy–naive population.16,26
Serious Adverse Events
█████ ████ ██ ███████████ ███████ █████████ █ ██ ███ ███ ██████ ██████████ ██ ███████ ██ ██████ ███ █████ ██████ ██ ██████ ███████ ██████ ██ ███
Overall Adverse Events
██ ███ █████ ██████ ██████ █████ ████ ██ ███████████ ███████ █████████ █ ██ ███ ███ ██ ███ ███████████ ██ █████ ██ ███████ ████ ██ ███ ██████ ███████ █████ █████ ████ ██ ███████████ ████████ ██████████
Discontinuation Due to Adverse Events
There were zero events in the placebo arms for the adalimumab 40 mg and etrasimod 2 mg studies; hence, continuity corrections were applied.26 The continuity correction as described in the statistical analysis plan was to add 0.5 to the numerator and 1 to the denominator for all arms of the relevant study.102,123 This resulted in wide CrIs, which should be interpreted with caution.26 ██ ███ █████ ███████ ██████ ██ ██████████ ██████████ ████ ████ ██████ ████ █████████ ███████████ ███ ███████ ██████ ██ ███████████ ████ ███████ ██ █████ ███ █ ████ ███████ ████████ ██ ███ ████████████ ███ ███████████ ███████ ██████ █████████ ██ ███████ ███████ ███████ ███████ ███████████ ██ ████ ██ █████████ ███ █████ ███████████ ██ ███ ██████ ███████ ██████
Critical Appraisal of ITC
In the sponsor-submitted ITC, studies were identified by searching multiple databases based on inclusion and exclusion criteria defined a priori. A quality assessment of the included studies was conducted using the NICE checklist. In general, all studies in the NMAs were found to have a low risk of bias, with details provided in the sponsor’s systematic literature review technical report.114 Based on the ITC feasibility assessment, 1 study was not included in the Canadian NMA that aligned with the Health Canada–approved dosage regimen. There was only a single reviewer for screening, data extraction, and study quality assessment for the targeted literature review (Canadian NMA), although 2 independent reviewers conducted these processes in duplicate (disagreement was resolved in discussion with a third reviewer) for the global NMA. The CADTH review team considered this to be acceptable, as the targeted literature review aimed to search for any eligible trials for mirikizumab only and an independent reviewer performed quality checks at each stage.
Overall, the patient, intervention, comparator, and outcome (PICO) approach that was used to identify studies for inclusion was consistent with the review objective, and the studies included in the ITC were appropriate. Overall, the clinical expert did not expect any major issues regarding the representativeness of the study populations enrolled in the RCTs that were included in the ITC in relation to the populations in Canada that may be eligible for treatment with etrasimod. However, the NMA in the technical report provided limited information on all trial and patient details and the potentially important confounders and effect modifiers from the individual trials. In consultation with the clinical expert, except for filgotinib, which is not used in Canada, all of the other comparator treatments that provided information to the network are relevant to clinical practice in Canada. The impact of the filgotinib nodes is difficult to determine because sensitivity analyses, such as removing the nodes from the network, were not done for the Canadian NMA.
The networks were sparsely populated, with relatively few nodes centred around a single connection (placebo) in a star geometry. Furthermore, most closed loops were between different doses of individual drugs and, consequently, all of the evidence was essentially indirect, increasing uncertainty in the estimates for each outcome, and the consistency assumption could not be assessed. Additionally, most nodes were informed by only 1 or 2 trials, increasing the chance the comparisons were underpowered, which impacted model selection (e.g., fixed effects versus random effects), model stability (convergence), and the types of adjustments that could be done (e.g., meta-regression to account for placebo response rates). These factors mean there was imprecision in many of the estimates (as evidenced by relatively wide 95% CrIs for many pairwise comparisons) and validating the key assumptions for the NMA was difficult, thereby increasing the uncertainty surrounding the results.
While the NMA technical report provided select details about the included trials and trial populations, it was clear regarding the approach used for assessing similarity and homogeneity between trials. Also, the review report based on the global model submitted to NICE124 for its reimbursement review of etrasimod provided additional information for assessing these key assumptions for NMAs. Multiple sources of differences in trial designs, patient characteristics, and outcome assessments indicated potentially important heterogeneity across the included studies. This has been noted in previous CADTH reimbursement reviews of drugs for the treatment of moderate to severe UC and, therefore, is not unique to this NMA. Using the information reported in the NMA technical report (Table 22), there were notable differences in the trial eligibility criteria and patient characteristics between studies, including potential confounders and treatment-effect modifiers. For example, disease severity was defined using different scoring tools and ranges, including a TMS of 6 to 12, an MMS (which omits the Physician’s Global Assessment from the TMS) of 4 to 9, an MMS of 5 to 9, a partial Mayo score (which omits the endoscopic score from the TMS) of 1 to 7, and a partial Mayo score of 5 to 9. Disease duration before enrolment ranged from 3 months to 6 months for the studies that reported a minimum duration of a confirmed UC diagnosis in the study inclusion criteria. The proportion of patients using concomitant medications was inconsistent across studies, including for glucocorticoids and corticosteroids, immunosuppressants, and budesonide, among others. For example, the proportion of concomitant corticosteroid use ranged from 13.4% to more than 60%. The clinical expert noted that corticoid use might be an effect modifier; therefore, it could have influenced the results. Treatment history is a known potential effect modifier. Some studies enrolled only treatment-naive patients while others enrolled patients who were treatment-naive or treatment-experienced. Reporting of treatment exposure (type and number of previous therapies) was not consistent. Some studies used 1 subgroup (e.g., treatment-naive versus -exposed), while others reported outcomes for multiple subgroups based on the type of therapy exposure or number of previous therapies. Prior treatment exposure was sometimes defined as exposure to a TNFi or biologic therapy, or as treatment failure of a TNFi, or having an inadequate response to biologic therapy. As described previously, the ELEVATE UC trials defined patients with prior exposure as having received or not received a biologic therapy or JAKi. It was unclear whether the definition regarding intolerance to or failure of at least 1 of the conventional therapies or biologics or JAKi drugs was reasonably similar across studies and in how they were assessed. The NMAs used subgroup analyses that stratified patients by advanced therapy exposure status, which was an appropriate approach to reduce heterogeneity in the dataset by treatment history. Of note, most of the included trials did not report AEs by treatment history subgroups. The NMA for serious infections used the nonstratified (full trial) populations. This approach seems reasonable, as the prior advanced therapy exposure status is unlikely to be an effect modifier of AE outcomes.
In addition to patient characteristics, heterogeneity related to study design, treatment regimens, and outcomes was an issue in the ITC. For example, the length of the induction periods in most studies ranged from 6 to 12 weeks, with the exception of 2 studies with a longer induction duration (13 and 14 weeks, respectively) and 1 study with a shorter (2-week) induction period. The duration of the maintenance phases in most studies ranged from 38 to 52 weeks, with the exception of 3 studies with a shorter maintenance duration (16, 22, and 24 weeks). The clinical expert did not expect the variability in the induction and maintenance periods to introduce major bias in the interpretation of results. Two different designs (treat-through and responder rerandomization) were used in the included studies. The patients who entered the maintenance phases in these designs would be different, which would likely lead to biased estimates of the relative treatment effects. Also, the placebo arms in randomized responder maintenance trials are not true placebo arms because (in most trials) patients received active induction treatment, and the carry-over effect would make the placebo groups not comparable. Therefore, using the reported maintenance phase outcomes from these trial designs in the same network is not recommended because it would violate the similarity and homogeneity assumptions. The sponsor used the approach of mimicking the rerandomized trials for data synthesis (most trials were rerandomized and this approach allowed an estimate of outcomes for the induction and maintenance periods separately), which was regarded as acceptable by the CADTH review team, although the potential for introducing bias into the analysis from unresolved differences in the studies remains. Moreover, as was observed with the ELEVATE UC studies, placebo response varied among the included studies. The methods for the NMA specified accounting for this by meta-regression (refer to the next paragraph), which was considered appropriate by the CADTH reviewers and aligned with recommended methods guidance.102 Moreover, there were some differences in the treatment regimens among treatments that were treated as different nodes in the NMA network diagrams, particularly the dosing regimens for the same drug (e.g., vedolizumab 300 mg at weeks 0, 2, and 6 only; vedolizumab 300 mg at weeks 0 and 2 only; ustekinumab 90 mg every 8 weeks; and ustekinumab 90 mg every 12 weeks, among others), for both efficacy and safety outcomes. Lastly, there was variability in the definition of outcomes. For example, both the TMS and MMS were used to define clinical response as well as clinical remission, and there was variation in the criteria for achieving these end points across studies. There was also variation in whether endoscopic components were centrally read or done locally. Specific AEs, SAEs, or serious infections were not reported, and it is likely that these safety outcomes were variably defined and recorded across studies.
Overall, the statistical methods, including model selection, adjustment methods (including the adjustment for placebo rates using a meta-regression), subgroup analyses (patients who were advanced therapy–naive versus those who were advanced therapy–exposed), sensitivity analyses (although these were done only for the global NMA), and the methods used to try to account for sources of heterogeneity, were acceptable. However, not all sources of heterogeneity could be adjusted due to the lack of information from the included studies and the complexity of the sources of heterogeneity. Some efficacy outcomes at the maintenance phase and most safety outcomes had wide CrIs due to small sample sizes, further affecting the precision of the estimates for harms.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
No studies addressing gaps in the pivotal phase III evidence were submitted.
The sponsor did note that there are 3 ongoing or completed studies investigating the effects of etrasimod in the treatment of moderately to severely active UC. However, the populations in those studies do not address any gaps in the evidence for the present review.16 Specifically, NCT05287126 (ongoing)125 is recruiting adolescent patients aged from 12 years to younger than 18 years, but the indication for etrasimod in the present review is for adults (the pivotal phase III ELEVATE UC 12 and 52 trials include participants aged 16 to 80 years).82 The NCT05061446 (ongoing)126 and ELEVATE UC 40 JAPAN (NCT04706793, completed) studies127 are focused on the Japanese population residing in Japan. The data from the NCT05061446 and ELEVATE UC 40 JAPAN studies are not considered to address any gaps relevant to the population of interest in Canada.16
Discussion
Summary of Available Evidence
Two phase III, double-blind, placebo-controlled, multicentre, international RCTs, ELEVATE UC 12 (N = 354) and ELEVATE UC 52 (N = 433), were included in this review to investigate the efficacy and safety of etrasimod versus placebo in adult patients with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. Patients in both trials were randomized in a 2:1 ratio to receive 2 mg etrasimod or placebo taken orally once daily for 12 weeks in the ELEVATE UC 12 trial, and for 52 weeks in the ELEVATE UC 52 trial. Clinical remission was the primary outcome in both protocols. Key secondary outcomes were similar in both trials, including endoscopic improvement, symptomatic remission, and mucosal healing. Corticosteroid-free clinical remission at week 52 and sustained clinical remission at both weeks 12 and 52 were also reported as secondary outcomes in the ELEVATE UC 52 trial. HRQoL assessed with the IBDQ and harms (TEAEs, serious TEAEs, TEAEs leading to discontinuation of treatment, and AESIs) were also reported.
Patients in both trials had an approximate mean age of 40.5 years and a mean UC duration of 6.0 to 7.9 years. There were slightly more male (53% to 63% across the treatment groups) than female (38% to 47%) patients. In the 2 trials, the majority of enrolled patients were white (75% to 89%, followed by Asian, Black or African American, American Indian or Alaska Native, and multiple). At baseline, approximately 27% to 32% of the patients were receiving corticosteroids, and 78% to 84% were receiving an oral 5-ASA. An approximate one-third of the enrolled patients reported prior use of at least 1 biologic or JAKi (29% to 34%). There were relatively small proportions of patients who reported prior anti-TNF failure with baseline corticosteroid use (5% to 10%), or prior anti-TNF failure with no baseline corticosteroid use (12% to 16%) in the 2 trials.
One sponsor-performed ITC was submitted to ████████ ███ ████████ ████████ ███ ██████ ██ ████████████ ████████ █████████ ███ ███ █████████ ██ █████ ████████ ████ ██████████ ██ ████████ ██████ ███ ███████ ████████ █████ ████████ ████ ██████████ ██ ████████ ██████ ██ ███ ███████████ ███ ██ ████ ███ ████████ ██ ████████ ████████ ██ ██████ ███████████ █████████████████████ ██████████ ████████████ ████████████ ████████████ █████████████ █████████ ████████████ ████████ ████████ ████████ ████████ ██████████ ███ ██████████ ███ ██████ ██████████████ ████ █████ ███████ ███████████ ███ ███████████████ ███ ██ █████ █████ █████████ ███████ ████ ███ ████ █████████ ████████ ██ ████████ ███ ███████████
Interpretation of Results
Efficacy
Etrasimod belongs to a class of small-molecule S1P receptor modulators that target immune-mediated inflammatory diseases, including UC.16,52 The other drug of the same class for UC in Canada is ozanimod, which is also taken orally but requires an induction period of about 7 days.42,58 Despite a wider range of treatment options after conventional therapies, an unmet need was still identified by the clinical expert consulted by CADTH, as well as by patient and clinician groups, as a sizable portion of patients does not respond or cannot maintain a response to the therapies for UC that are currently available in Canada. The clinical expert pointed out that multiple drug failures and ongoing disease activity may lead to adverse consequences, including surgery. Thus, there is interest in providing patients with moderate to severe UC with a treatment that demonstrates efficacy in the short and long-term, with fewer adverse effects.
Endoscopic improvement and mucosal healing at a longer follow-up time point have become the target goals in the treatment of patients with moderately to severely active UC, according to the clinical expert. There is high-certainty evidence in the 2 pivotal studies that etrasimod results in a clinically important increase in the proportion of patients with endoscopic improvement at 12 weeks and 52 weeks compared with placebo. No ITC evidence for endoscopic improvement was submitted by the sponsor; therefore, no conclusions could be drawn on the relative efficacy of etrasimod compared with other active therapies for this important outcome. There is high-certainty evidence in the ELEVATE UC 52 study that etrasimod results in a clinically important increase in the proportion of patients with mucosal healing at 52 weeks; no ITC evidence was reported for this outcome. Other efficacy end points from the pivotal trials that are regarded as important for this review were composite outcomes, mainly incorporating subscores in the MMS. Likewise, there was high-certainty evidence that a clinically important increase in the proportions of patients achieving clinical remission (the primary objective for both studies), sustained clinical remission, corticosteroid-free remission, clinical response, and symptomatic remission was achieved at 12 or 52 weeks with etrasimod compared with placebo in the ELEVATE UC 52 study. Corticosteroid-free clinical remission was regarded as important due to AEs associated with systemic steroid use and risk of steroid resistance. Based on the results from the ELEVATE UC 52 trial at week 52, among the patients who were receiving oral corticosteroids for UC at baseline, the patients in the etrasimod group were more likely to achieve corticosteroid-free clinical remission for 4 weeks or longer (odds ratio = 5.4; 95% CI, 1.5 to 19.4). Of note, for most of the efficacy outcomes listed earlier, there were greater between-group risk differences for patients treated with etrasimod versus placebo in the subgroup of patients who had received 1 prior biologic or JAKi compared with those in the other subgroup of patients who had received more than 1 prior biologic or JAKi. A similar observation was made for the subgroups of patients in the ELEVATE UC 52 trial whose condition had previously failed to respond to anti-TNF therapy compared with those whose condition had responded. In addition, the lower bound of the 95% CI crossed the null value. However, the trials were not designed to draw causative inferences based on subgroup results, and no tests for interaction for these subgroup analyses were reported, leaving uncertainties in drawing any definitive conclusion of such a subgroup effect. Based on the findings of the pivotal trials, the clinical expert noted that to optimize efficacy, clinicians would likely prefer to use etrasimod in patients with moderate to severe UC earlier in the advanced therapy pathway.
The patient group and clinical expert regarded HRQoL as an outcome of importance. Based on the IBDQ — a validated measurement comprising domains of bowel symptoms (including abdominal cramps, abdominal pain, feeling of defecation need, and abdominal bloating, among others), systemic symptoms, social function, and emotional function — patients in the etrasimod group reported greater improvement from baseline than patients in the placebo group at 12 weeks and 52 weeks. The between-group differences in IBDQ change at both time points were not regarded as clinically meaningful, as the 95% CI included the MID identified from the literature. The point estimate for the IBDQ at 52 weeks was slightly above the MID, and its imprecise result could be related to a high rate of missing outcome data (more than half of patients had missing data).
Moreover, the clinical expert noted that the inclusion of patients with UC with isolated proctitis, a patient population that is often excluded from clinical trials, is helpful for clinical practice, as it adds evidence on the efficacy and safety of etrasimod in this specific patient group. The clinical expert and the clinician group both noted that the IV and subcutaneous administration of most of the other drugs for UC is a barrier to treatment for some patients. The once-daily oral administration of etrasimod may therefore be a preferred option for patients with UC, a position that aligns with the input from the patient group. Also, the fact that etrasimod does not require an induction dosing regimen and is therefore simpler in this way versus other treatments is also potentially relevant to patients. However, there is no direct evidence that etrasimod leads to better treatment adherence, reduced health resource use, or improved outcomes versus other advanced therapies. The clinical expert noted the importance of monitoring patients by examining biomarkers (e.g., fecal calprotectin) during the treatment of etrasimod, particularly among older patients, due to the higher frequency and likelihood of comorbidities and concomitant medications in that population.
Head-to-head trial data are not available to compare etrasimod with the other relevant active therapies used in this population. Based on the indirect evidence submitted by the sponsor, █████████ ██████ ███████ ████████ ██ ████████████ ████████ ███ ████████ █████████ ████████ ████ █████ ██████████ ██████ ██ ██████████████ ███████ █████████ ███ ██████████ █████████ ██████████ █████ ███ ████████ █████████████ ███████████ ██ █████████ █████ ███ ███████ █████████ ███ ████████████ ██████████████████ █████ ███ ████████ █████████████ ██ ████ ██ ███ ████████ ███████████████████ ██████████ ██ ████ █████████ ███ ████████████. Of note, the clinical expert stated that in their clinical experience, adalimumab does not achieve clinical response at induction as well as other therapies. There was no statistically significant difference between adalimumab and placebo for the proportion of patients demonstrating clinical responses at week 8 in the ULTRA 1 trial.128 Upadacitinib is a JAKi that is also administered orally. JAKi drugs like upadacitinib have important warnings and precautions associated with their use stemming from their mechanism of action, according to a post-marketing study of tofacitinib in the treatment of rheumatoid arthritis.129 Whether upadacitinib is associated with similar safety concerns in patients with UC is unclear, owing to the lack of longer-term evidence. When interpreting the results of indirect comparison ███████ █████████ ███ ████████████ ██ ███████████ ███ ███████ ███ █████ ██ █ ██████████ ███████ ██████ ████. In general, while appropriate methods and approaches were used in conducting the NMA, the comparative efficacy results are influenced by the lack of direct comparisons in the networks, few trials per comparator, high imprecision, heterogeneity, and other unresolvable uncertainties in the ITC evidence.
Harms
TEAEs were common but the frequency of AEs, SAEs, withdrawals due to AEs, and notable harms was generally comparable between etrasimod-treated patients and placebo-treated patients at 12 weeks (ELEVATE UC 12 trial) and at 52 weeks (ELEVATE UC 52 trial). In consultation with the clinical expert, the frequencies of the notable harms reported in the 2 trials (cardiovascular events, liver injuries, opportunistic infections, macular edema, and so forth) were similar to those associated with other advanced therapies for UC, and there were no new concerns related to etrasimod beyond the AEs already identified and noted in its product monograph. However, the clinical expert agreed with CADTH reviewers that because of the duration of the studies and relatively younger patient population enrolled compared with the population of patients with UC in Canada, any conclusions surrounding longer-term harms in a broader population are restricted. The estimated primary completion date for the ELEVATE UC OLE trial is February 6, 2027.
Head-to-head trial data are not available to compare the safety of etrasimod with other active therapies. Based on indirect evidence submitted by the sponsor, the ████ ███ ███ ████ ███ █████████ ██████████ ██ ███████ ██████████ ████ █████████ ████████ ████ █████ ██████████ ███ ██████ ███ ████████████ ███ ███ ████████ ██ ███ █████████ █████████ ████ ███ ██████████ ██ ████████ ████ ███ ████ █████ ██ █████████ ████████████████ ███ ██ ███ ████ █████████ █████████ ███ ███ █████ █████████ █████████ ███ ████████. However, these analyses were done on smaller sample sizes and few events. When added to the aforementioned limitations of the NMA, the comparative harms results are considered highly uncertain.
Conclusion
Two phase III, multicentre, double-blind RCTs evaluated the efficacy and safety of etrasimod compared with placebo in adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. Compared with placebo at 12 weeks and 52 weeks, etrasimod results in a clinically important increase in the proportions of patients who have endoscopic improvement, clinical remission, and clinical response. At 52 weeks, etrasimod results in a clinically important increase in the proportions of patients who have mucosal healing, sustained clinical remission, and corticosteroid-free clinical remission in the overall trial population as well in patients who were receiving oral corticosteroids for UC at baseline, and symptomatic remission compared with placebo. Etrasimod likely results (at 12 weeks) or may result (at 52 weeks) in little to no difference in improvement in HRQoL based on the IBDQ, and likely results in little to no difference in the proportion of patients who have serious TEAEs at 12 weeks and 52 weeks compared with placebo. AEs were common but no particular concerns were identified beyond those noted in the product monograph or what is expected for S1P receptor modulators. However, the frequencies of AEs are based on relatively short observation periods and on younger patient populations than would be included in real-world practice.
There is a data gap in head-to-head, direct evidence between etrasimod and other advanced therapies for moderately to severely active UC. Indirect evidence submitted by the sponsor ████████ ████ █████ ███ ██ ██ ██████████ ██████████ ██ ████████ ██ █████ ████████ ███████ █████████ ███ █████ ████████ █████████ ██████ █████████ ██ ████████████.
References
1.Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am J Gastroenterol. 2019;114(3):384-413.PubMed
2.Longo D, Fauci A, Kasper D, Hauser S, Jameson J, Loscalzo J. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill Education; 2011.
3.Greuter T, Vavricka SR. Extraintestinal manifestations in inflammatory bowel disease - epidemiology, genetics, and pathogenesis. Expert Rev Gastroenterol Hepatol. 2019;13(4):307-317.PubMed
4.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural History of Adult Ulcerative Colitis in Population-based Cohorts: A Systematic Review. Clin Gastroenterol Hepatol. 2018;16(3):343-356.e3.PubMed
5.Armuzzi A, Liguori G. Quality of life in patients with moderate to severe ulcerative colitis and the impact of treatment: A narrative review. Dig Liver Dis. 2021;53(7):803-808.PubMed
6.Benchimol EI, Bernstein CN, Bitton A, et al. The Impact of Inflammatory Bowel Disease in Canada 2018: A Scientific Report from the Canadian Gastro-Intestinal Epidemiology Consortium to Crohn’s and Colitis Canada. J Can Assoc Gastroenterol. 2019;2(suppl 1):S1-S5.PubMed
7.Armuzzi A, Tarallo M, Lucas J, et al. The association between disease activity and patient-reported outcomes in patients with moderate-to-severe ulcerative colitis in the United States and Europe. BMC Gastroenterol. 2020;20(1):18.PubMed
8.Sikirica M, Lynch J, Hoskin B, Kershaw J, Wild R, Lukanova R. P0412 Humanistic burden and impact of the disease in moderate to severe patients with Crohn’s disease (CD) and ulcerative colitis (UC): findings from a cross sectional study in the US and France, Germany, Italy, Spain, UK (EU5). United European Gastroenterol J. 2020;8(suppl 1):348.
9.Burisch J, Zhao M, Odes S. P860 The Costs of Inflammatory Bowel Diseases in High-Income Settings. J Crohns Colitis. 2023;17(suppl 1):i983.
10.Ruiz-Casas L, Evans J, Rose A, et al. The LUCID study: living with ulcerative colitis; identifying the socioeconomic burden in Europe. BMC Gastroenterol. 2021;21(1):456.PubMed
11.Crohn’s and Colitis Canada (CCFC). Impact of inflammatory bowel disease in Canada [Internet]. Toronto (ON): Crohn’s and Colitis Canada; 2018: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf. Accessed 2022 Oct 5.
12.Crohn’s and Colitis Canada (CCFC). Impact of inflammatory bowel disease in Canada [Internet]. Toronto (ON): Crohn’s and Colitis Canada; 2023: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2023-IBD-Report-English-LR.pdf. Accessed 2023 Aug 8.
13.Schreiber S, Panés J, Louis E, Holley D, Buch M, Paridaens K. National differences in ulcerative colitis experience and management among patients from five European countries and Canada: an online survey. J Crohns Colitis. 2013;7(6):497-509.PubMed
14.Bressler B, Marshall JK, Bernstein CN, et al; Toronto Ulcerative Colitis Consensus Group. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035-1058.e3.PubMed
15.CADTH technology review: appropriate pharmacotherapy for inflammatory bowel disease [Internet]. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/hta-he/ho0003-he0018-ibd-ou360.pdf. Accessed 2023 Aug 17.
16.Velsipity (etrasimod) for the treatment of adult patients with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment: summary of clinical evidence. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer Canada ULC; 2024 Feb 5.
17.Clinical Study Report: APD334-302. A Phase 3, Randomized, Double-Blind, Placebo-Controlled, 52-week Study to Assess the Efficacy and Safety of Etrasimod in Subjects with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. San Diego (CA): Arena Pharmaceuticals; 2022 Sept 9.
18.Clinical Study Report: APD334-301. A Phase 3, Randomized, Double-Blind, Placebo-Controlled, 12-week Study to Assess the Efficacy and Safety of Etrasimod in Subjects with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. San Diego (CA): Arena Pharmaceuticals; 2022 Sept 12.
19.Pfizer Canada response to March 1, 2024 CADTH request for additional information regarding Velsipity (etrasimod) review [internal additional sponsor's information]. Kirkland (QC): Pfizer Canada; 2024 Mar 12.
20.Pharmaceuticals IA. Statistical Analysis Plan. A Phase 3, Randomized, Double-Blind, Placebo-Controlled, 52-week Study to Assess the Efficacy and Safety of Etrasimod in Subjects with Moderately to Severely Active Ulcerative Colitis. Version 1.0. 2022: https://clinicaltrials.gov/ProvidedDocs/88/NCT03945188/SAP_001.pdf. Accessed 2022 Jan 24.
21.Pharmaceuticals IA. Statistical Analysis Plan. A Phase 3, Randomized, Double-Blind, Placebo-Controlled, 12-week Study to Assess the Efficacy and Safety of Etrasimod in Subjects with Moderately to Severely Active Ulcerative. Version 1.0. 2022: https://clinicaltrials.gov/ProvidedDocs/69/NCT03996369/SAP_001.pdf. Accessed 2022 Jan 24.
22.U.S. Food & Drug Administration. Ulcerative Colitis: Developing Drugs for Treatment Guidance for Industry. 2022; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ulcerative-colitis-developing-drugs-treatment. Accessed 2024 Mar 12.
23.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-406.PubMed
24.Santesso N, Glenton C, Dahm P, et al; GRADE Working Group. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135.PubMed
25.Pharmaceuticals A. NCT03950232: An Extension Study for Treatment of Moderately to Severely Active Ulcerative Colitis (ELEVATE UC OLE) [Internet] ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/ct2/show/NCT03950232#eligibility. Accessed 2023 Sep 29.
26.Global ITC Technical Report. Etrasimod for the treatment of patients with moderately to severely active ulcerative colitis, Version 2.1. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer Canada ULC; 2023 Oct 19.
27.Canadian Final ITC Technical Report. Etrasimod for the treatment of patients with moderately to severely active ulcerative colitis: Canadian base case analyses including mirikizumab. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer Canada ULC; 2023 Sept 15.
28.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(suppl 1):S3-S9.PubMed
29.Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-321.e2.PubMed
30.Crohn’s and Colitis Foundation of Canada (CCFC). The impact of inflammatory bowel disease in Canada 2012 Final Report and Recommendations [Internet]. Toronto (ON): Crohn’s and Colitis Foundation of Canada; 2012: http://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/ccfc-ibd-impact-report-2012.pdf. Accessed 2023 May 10.
31.Torres J, Billioud V, Sachar DB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis as a progressive disease: the forgotten evidence. Inflamm Bowel Dis. 2012;18(7):1356-1363.PubMed
32.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389(10080):1756-1770.PubMed
33.Roda G, Narula N, Pinotti R, et al. Systematic review with meta-analysis: proximal disease extension in limited ulcerative colitis. Aliment Pharmacol Ther. 2017;45(12):1481-1492.PubMed
34.Ungar B, Kopylov U. Long-term outcome of ulcerative proctitis. United European Gastroenterol J. 2020;8(8):847-848.PubMed
35.Regueiro MD. Diagnosis and treatment of ulcerative proctitis. J Clin Gastroenterol. 2004;38(9):733-740.PubMed
36.Casellas F, Arenas JI, Baudet JS, et al. Impairment of health-related quality of life in patients with inflammatory bowel disease: a Spanish multicenter study. Inflamm Bowel Dis. 2005;11(5):488-496.PubMed
37.Sarid O, Slonim-Nevo V, Schwartz D, et al; Israel IBD Research Nucleus (IIRN). Differing Relationship of Psycho-Social Variables with Active Ulcerative Colitis or Crohn’s Disease. Int J Behav Med. 2018;25(3):341-350.PubMed
38.Ghosh S, Sanchez Gonzalez Y, Zhou W, et al. Upadacitinib Treatment Improves Symptoms of Bowel Urgency and Abdominal Pain, and Correlates With Quality of Life Improvements in Patients With Moderate to Severe Ulcerative Colitis. J Crohns Colitis. 2021;15(12):2022-2030.PubMed
39.Peyrin-Biroulet L, Germain A, Patel AS, Lindsay JO. Systematic review: outcomes and post-operative complications following colectomy for ulcerative colitis. Aliment Pharmacol Ther. 2016;44(8):807-816.PubMed
40.Rubin DT, Dubinsky MC, Panaccione R, et al. The impact of ulcerative colitis on patients’ lives compared to other chronic diseases: a patient survey. Dig Dis Sci. 2010;55(4):1044-1052.PubMed
41.Becker HM, Grigat D, Ghosh S, et al. Living with inflammatory bowel disease: A Crohn’s and Colitis Canada survey. Can J Gastroenterol Hepatol. 2015;29(2):77-84.PubMed
42.Drug Reimbursement Review clinical guidance report: ozanimod (Zeposia) for the treatment of adult patients with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, loss of response, or were intolerant to either conventional therapy or a biologic agent. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/ozanimod-0. Accessed 2023 Oct 5.
43.Crohn’s and Colitis Canada. Getting Diagnosed [Internet]. Crohn’s and Colitis Canada; 2019: https://crohnsandcolitis.ca/About-Crohn-s-Colitis/IBD-Journey/Diagnosis-and-Testing/Getting-Diagnosed. Accessed 2023 Jan 18.
44.Kucharzik T, Koletzko S, Kannengiesser K, Dignass A. Ulcerative Colitis-Diagnostic and Therapeutic Algorithms. Dtsch Arztebl Int. 2020;117(33-34):564-574.PubMed
45.Pant S, Le K, Vannabouathong C, Dyrda P. Formulary Management of Targeted Immune Modulators in Ulcerative Colitis [Internet]. Vol 3: Can J Health Technol; 2023: https://canjhealthtechnol.ca/index.php/cjht/article/download/ES0366/1262?inline=1. Accessed 2023 Aug 15.
46.Ardizzone S, Cassinotti A, Duca P, et al. Mucosal healing predicts late outcomes after the first course of corticosteroids for newly diagnosed ulcerative colitis. Clin Gastroenterol Hepatol. 2011;9(6):483-489.e3.PubMed
47.Colombel JF, Rutgeerts P, Reinisch W, et al. Early mucosal healing with infliximab is associated with improved long-term clinical outcomes in ulcerative colitis. Gastroenterology. 2011;141(4):1194-1201.PubMed
48.Frøslie KF, Jahnsen J, Moum BA, Vatn MH, Group IBSEN; IBSEN Group. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology. 2007;133(2):412-422.PubMed
49.Laharie D, Filippi J, Roblin X, et al. Impact of mucosal healing on long-term outcomes in ulcerative colitis treated with infliximab: a multicenter experience. Aliment Pharmacol Ther. 2013;37(10):998-1004.PubMed
50.Le Berre C, Peyrin-Biroulet L; SPIRIT-IOIBD study group. Selecting End Points for Disease-Modification Trials in Inflammatory Bowel Disease: the SPIRIT Consensus From the IOIBD. Gastroenterology. 2021;160(5):1452-1460.e21.PubMed
51.Mayo Clinical Staff. Colectomy. 2022: https://www.mayoclinic.org/tests-procedures/colectomy/about/pac-20384631. Accessed 2023 Oct 2.
52.Velsipity (etrasimod): 2 mg etrasimod (as etrasimod L-arginine) oral tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2024 Jan 30.
53.Danese S, Furfaro F, Vetrano S. Targeting S1P in Inflammatory Bowel Disease: New Avenues for Modulating Intestinal Leukocyte Migration. J Crohns Colitis. 2018;12(suppl_2).
54.Blaho VA, Hla T. Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev. 2011;111(10):6299-6320.PubMed
55.Grassi S, Mauri L, Prioni S. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front Pharmacol, 2019 Jan 18 [internet]. https://www.frontiersin.org/articles/10.3389/fphar.2019.00807. Accessed 2023 Jan 18.
56.Buzard DJ, Kim SH, Lopez L, et al. Discovery of APD334: Design of a Clinical Stage Functional Antagonist of the Sphingosine-1-phosphate-1 Receptor. ACS Med Chem Lett. 2014;5(12):1313-1317.PubMed
57.Al-Shamma H, Lehmann-Bruinsma K, Carroll C, et al. The Selective Sphingosine 1-Phosphate Receptor Modulator Etrasimod Regulates Lymphocyte Trafficking and Alleviates Experimental Colitis. J Pharmacol Exp Ther. 2019;369(3):311-317.PubMed
58.Product monograph including patient medication information PrZeposia®. ozanimod (as ozanimod hydrochloride). Saint-Laurent (QC): Bristol-Myers Squibb Canada; 2023 Dec 19: https://www.bms.com/assets/bms/ca/documents/productmonograph/ZEPOSIA_EN_PM.pdf. Accessed 2023 Aug 11.
59.Product monograph including patient medication information. PrHUMIRA® adalimumab injection. St-Laurent (QC): AbbVie Corporation; 2022 Sep 16: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf. Accessed 2023 Aug 10.
60.Product monograph including patient medication information Amgevita® adalimumab injection. Mississauga (ON): Amgen Canada Inc.; 2022 Sep 9: https://pdf.hres.ca/dpd_pm/00067468.PDF. Accessed 2023 Aug 17.
61.Product monograph including patient medication information PrHulio® adalimumab injection. Etobicoke (ON): BGP Pharma ULC; 2023 Jul 20: https://pdf.hres.ca/dpd_pm/00071858.PDF. Accessed 2023 Aug 17.
62.Product monograph including patient medication information PrAbrilada® Adalimumab injection. Kirkland (QC): Pfizer Canada ULC; 2022 Nov 14: https://pdf.hres.ca/dpd_pm/00068228.PDF. Accessed 2023 Aug 21.
63.Product monograph including patient medication information PrHyrimoz® adalimumab injection. Boucherville (QC): Sandoz Canada Inc.; 2022 Oct 11: https://pdf.hres.ca/dpd_pm/00067757.PDF. Accessed 2023 Aug 21.
64.Product monograph including patient medication information. PrHADLIMA® adalimumab injection. PrHADLIMA® PushTouch® adalimumab injection. Kirkland (QC): Merck Canada Inc.; 2020 Nov 26: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/HADLIMA-PM_E.pdf. Accessed 2023 Aug 21.
65.Product monograph including patient medication information PrYuflyma TM adalimumab injection. Toronto (ON): Celltrion Healthcare Canada Limited; 2023 Feb 3: https://pdf.hres.ca/dpd_pm/00069429.PDF. Accessed 2023 Aug 21.
66.Product monograph including patient medication information PrIdacio® Adalimumab Injection. Toronto (ON): Fresenius Kabi Canada Ltd.; 2020 Oct 30: https://pdf.hres.ca/dpd_pm/00058541.PDF. Accessed 2023 Aug 21.
67.Product monograph including patient medication information PrSimlandi TM Adalimumab injection. Boucherville (QC): JAMP Pharma Corporation; 2022 Dec 9: https://pdf.hres.ca/dpd_pm/00068814.PDF. Accessed 2023 Aug 21.
68.Product monograph Simponi® (golimumab) and SIMPONI® I.V (golimumab). Toronto (ON): Janssen Inc.; 2018 Jan 15: https://pdf.hres.ca/dpd_pm/00043422.PDF. Accessed 2023 Aug 11.
69.Product monograph Remicade® Infliximab. Toronto (ON): Janssen Inc.; 2017 Aug 4: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF. Accessed 2023 Aug 11.
70.Product monograph including patient medication information. PrAVSOLA® infliximab for injection. Mississauga (ON): Amgen Canada Inc.; 2022 May 17: https://pdf.hres.ca/dpd_pm/00065940.PDF. Accessed 2023 Aug 17.
71.Product monograph including patient medication information PrRENFLEXIS® Infliximab for Injection. Kirkland (QC): Organon Canada Inc.; 2023 Feb 9: https://pdf.hres.ca/dpd_pm/00069528.PDF. Accessed 2023 Aug 21.
72.Product monograph PrInflectra® (infliximab for Injection). Kirkland (QC): Pfizer Canada Inc.; 2017 Nov 7: https://pdf.hres.ca/dpd_pm/00042037.PDF. Accessed 2023 Aug 11.
73.Product monograph Entyvio TM. vedolizumab powder for concentrate for solution for infusion. Oakville (ON): Takeda Canada Inc.; 2016 Mar 22: https://pdf.hres.ca/dpd_pm/00034239.PDF. Accessed 2023 Aug 15.
74.Product monograph including patient medication information Omvoh mirikizumab injection. Toronto (ON): Eli Lilly Canada, Inc.; 2023 Jul 20: https://pdf.hres.ca/dpd_pm/00071771.PDF. Accessed 2023 Sep 19.
75.Product monograph including patient medication information PrStelara® ustekinumab injection. Toronto (ON): Janssen Inc.; 2008 Dec 12: https://pdf.hres.ca/dpd_pm/00069002.PDF. Accessed 2023 Aug 15.
76.Product monograph including patient medication information. PrXELJANZ® tofacitinib, tablets, oral. PrXELJANZ® XR tofacitinib extended-release, tablets, oral. Kirkland (QC): Pfizer Canada ULC; 2023 Aug 11: https://pdf.hres.ca/dpd_pm/00071982.PDF. Accessed 2023 Aug 21.
77.Product monograph including patient medication information Pr pms-TOFACITINIB Tofacitinib Tablets. Montreal (QC): Pharmascience Inc.; 2022 Aug 23: https://pdf.hres.ca/dpd_pm/00069222.PDF. Accessed 2023 Aug 15.
78.Product monograph including patient medication information PrTaro-tofacitinib tofacitinib tablets, oral. Brampton (ON): Taro Pharmaceuticals Inc.; 2022 Jul 13: https://pdf.hres.ca/dpd_pm/00068405.PDF. Accessed 2023 Aug 18.
79.Product monograph including patient medication information. PrAuro-tofacitinib tofacitinib tablets. Woodbridge (ON): Auro Pharma Inc.; 2022 Aug 19: https://pdf.hres.ca/dpd_pm/00068455.PDF. Accessed 2023 Sep 22.
80.Product monograph including patient medication information. PrRinvoq® upadacitinib extended-release tablets. St-Laurent (QC): AbbVie Corporation; 2023 Oct 31: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf. Accessed 2023 Dec 11.
81.Turner D, Ricciuto A, Lewis A, et al; International Organization for the Study of IBD. STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology. 2021;160(5):1570-1583.PubMed
82.Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. 2023;401(10383):1159-1171.PubMed
83.Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47(3):404-409.PubMed
84.Bryant RV, Winer S, Travis SP, Riddell RH. Systematic review: histological remission in inflammatory bowel disease. Is ‘complete’ remission the new treatment paradigm? An IOIBD initiative. J Crohns Colitis. 2014;8(12):1582-1597.PubMed
85.Guideline on the development of new medicinal products for the treatment of Ulcerative Colitis [CHMP/EWP/18463/2006 Rev.1]. Amsterdam (NL): European Medicines Agency; 2022: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-newmedicinal-products-treatment-ulcerative-colitis-revision-1_en.pdf. Accessed 2024 Feb 28.
86.Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-269.PubMed
87.Chen X-L, Zhong LH, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177.PubMed
88.Alrubaiy L, Rikaby I, Dodds P, Hutchings HA, Williams JG. Systematic review of health-related quality of life measures for inflammatory bowel disease. J Crohns Colitis. 2015;9(3):284-292.PubMed
89.Irvine EJ, Feagan B, Rochon J, et al; Canadian Crohn’s Relapse Prevention Trial Study Group. Quality of life: a valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Gastroenterology. 1994;106(2):287-296.PubMed
90.Pallis AG, Vlachonikolis IG, Mouzas IA. Assessing health-related quality of life in patients with inflammatory bowel disease, in Crete, Greece. BMC Gastroenterol. 2002;2:1.PubMed
91.Gregor JC, McDonald JW, Klar N, et al. An evaluation of utility measurement in Crohn’s disease. Inflamm Bowel Dis. 1997;3(4):265-276.PubMed
92.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-S27.PubMed
93.Irvine EJ. Quality of life of patients with ulcerative colitis: past, present, and future. Inflamm Bowel Dis. 2008;14(4):554-565.PubMed
94.LeBlanc K, Mosli MH, Parker CE, MacDonald JK. The impact of biological interventions for ulcerative colitis on health-related quality of life. Cochrane Database Syst Rev. 2015;2015(9):CD008655.PubMed
95.Naegeli AN, Hunter T, Dong Y, et al. Full, Partial, and Modified Permutations of the Mayo Score: Characterizing Clinical and Patient-Reported Outcomes in Ulcerative Colitis Patients. Crohns Colitis 360. 2021;3(1):otab007.PubMed
96.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-810.PubMed
97.Kim M, Merrill JT, Wang C, et al. SLE clinical trials: impact of missing data on estimating treatment effects. Lupus Sci Med. 2019;6(1):e000348.PubMed
98.Pharmaceuticals A. NCT03945188: Etrasimod Versus Placebo for the Treatment of Moderately to Severely Active Ulcerative Colitis (ELEVATE UC 52) [Internet] ClinicalTrials.gov. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03945188?term=ELEVATE+UC&draw=2&rank=2. Accessed 2023 Jan 11.
99.Pharmaceuticals A. NCT03996369: Etrasimod Versus Placebo as Induction Therapy in Moderately to Severely Active Ulcerative Colitis (ELEVATE UC 12) [Internet] ClinicalTrials.gov. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03996369?term=ELEVATE+UC&draw=2&rank=1. Accessed 2023 Jan 11.
100.Pfizer. NCT04607837: Etrasimod Versus Placebo for the Treatment of Moderately Active Ulcerative Colitis (GLADIATOR UC). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/ct2/show/NCT04607837. Accessed 2023 Feb 23.
101.Etrasimod for the treatment of patients with moderately to severely active ulcerative colitis (statistical analysis plan); Canadian analyses, Version 1.0. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer; 2023.
102.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials [Internet]. London (UK): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/books/NBK310366/.
103.National Institute for Health Care Excellence. The Guidelines Manual [Internet]. London (UK): NICE: https://www.nice.org.uk/process/pmg6/resources/the-guidelines-manual-pdf-2007970804933. Accessed 2023 Aug 3.
104.European Medicines Agency. HUMIRA (adalimumab) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf. Accessed 2023 Jul 31.
105.European Medicines Agency. SIMPONI (golimumab) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/simponi-epar-product-information_en.pdf. Accessed 2023 Jul 31.
106.European Medicines Agency. ENTYVIO (vedolizumab) SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/entyvio-epar-product-information_en.pdf. Accessed 2023 Jul 31.
107.European Medicines Agency. STELARA (ustekinumab) SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/stelara-epar-product-information_en.pdf. Accessed 2023 Jul 31.
108.European Medicines Agency. XELJANZ (tofacitinib) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/xeljanz-epar-product-information_en.pdf. Accessed 2023 Jul 31.
109.European Medicines Agency. JYSELECA (filgotinib) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/jyseleca-epar-product-information_en.pdf. Accessed 2023 Jul 31.
110.European Medicines Agency. RINVOQ (upadacitinib) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/rinvoq-epar-product-information_en.pdf. Accessed 2023 Jul 31.
111.European Medicines Agency. ZEPOSIA (ozanimod) SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. Available from: https://www.ema.europa.eu/en/documents/product-information/zeposia-epar-product-information_en.pdf.
112.European Medicines Agency. REMICADE (infliximab) - SUMMARY OF PRODUCT CHARACTERISTICS [Internet] Eur. Med. Agency. 2023. Available from: https://www.ema.europa.eu/en/documents/product-information/remicade-epar-product-information_en.pdf. Accessed 2023 Jul 31.
113.European Medicines Agency. OMVOH (mirikizumab) SUMMARY OF PRODUCT CHARACTERISTICS [Internet]. Eur. Med. Agency. 2023. Available from: https://www.ema.europa.eu/en/documents/product-information/omvoh-epar-product-information_en.pdf. Accessed 2023 Sep 19.
114.Report of a systematic literature review of efficacy and safety of etrasimod and other advanced therapies in patients with moderately to severely active ulcerative colitis (UC). [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer Canada ULC; 2023 Aug 4.
115.Dias S, Ades T, Welton N, Jansen J, Sutton A. Network Meta-Analysis for Decision-Making [Internet]. Wiley; 2018: https://www.wiley.com/en-gb/Network+Meta+Analysis+for+Decision+Making-p-9781118951729. Accessed 2018 Aug 3.
116.Higgins J, James J. Cochrane Handbook for Systematic Reviews of Interventions, Version 6.3, 2022 [Internet]. https://training.cochrane.org/handbook. Accessed 2023 Jun 26.
117.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 5: Evidence Synthesis in the Baseline Natural History Model [Internet]. National Institute for Health and Clinical Excellence. London (UK): National Institute for Health and Care Excellence (NICE); 2012: . Accessed 2024 Mar 1.
118.Daly C, Downing B, Welton N. A practical guide to inconsistency checks in Bayesian network meta-analysis [Internet]. https://www.bristol.ac.uk/media-library/sites/social-community-medicine/documents/mpes/Guide%20to%20Checking%20for%20Inconsistency%20in%20NMA_TSU.pdf. Accessed 2023 Aug 11.
119.Pfizer. Etrasimod for the treatment of patients with moderately to severely active ulcerative colitis (interim ITC report); Version 1.0. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer Canada ULC; 2023.
120.Sandborn WJ, Ghosh S, Panes J, et al; Study A3921063 Investigators. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616-624.PubMed
121.Sandborn WJ, Baert F, Danese S, et al. Efficacy and Safety of Vedolizumab Subcutaneous Formulation in a Randomized Trial of Patients With Ulcerative Colitis. Gastroenterology. 2020;158(3):562-572.e12.PubMed
122.Report FA. Etrasimod for the treatment of patients with moderately to severely active ulcerative colitis; Version 2.1. [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Velsipity (etrasimod) 2 mg oral tablets. Kirkland, QC: Pfizer; 2023.
123.Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data. Stat Med. 2004;23(9):1351-1375.PubMed
124.Single Technology Appraisal. Etrasimod for treating moderately to severely active ulcerrative colitis [ID5091]. Committee Papers. London (UK): National Institute for Health Care Excellence; 2023: https://www.nice.org.uk/guidance/ta956/evidence/committee-papers-pdf-13318100317. Accessed 2024 Mar 1.
125.Pfizer. NCT05287126: A Study to Evaluate Etrasimod Treatment in Adolescents With Ulcerative Colitis. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2023: https://clinicaltrials.gov/ct2/show/NCT05287126. Accessed 2023 Dec 20.
126.Pfizer. NCT05061446: Etrasimod Dose-Ranging Versus Placebo as Induction Therapy Study in Adult Japanese Subjects With Moderately to Severely Active Ulcerative Colitis. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine: https://clinicaltrials.gov/ct2/show/NCT05061446. Accessed 2024 Mar 1.
127.Pharmaceuticals A. NCT04706793: A Phase 3, Double-Blind, Placebo-Controlled, 40-week Extension Study to Assess the Efficacy and Safety of Etrasimod in Japanese Subjects With Moderately to Severely Active Ulcerative Colitis (ELEVATE UC 40 JAPAN). ClinicalTrials.gov. Bethesda, MD: U.S. National Library of Medicine; 2023, https://clinicaltrials.gov/study/NCT04706793. Accessed March 1, 2024.
128.Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780-787.PubMed
129.Ytterberg SR, Bhatt DL, Mikuls TR, et al; ORAL Surveillance Investigators. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N Engl J Med. 2022;386(4):316-326.PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 8: Forest Plot for Endoscopic Improvement at Week 12 for Overall and Subgroups of the ELEVATE UC 12 Trial — FAS and a Baseline MMS of 4 to 9
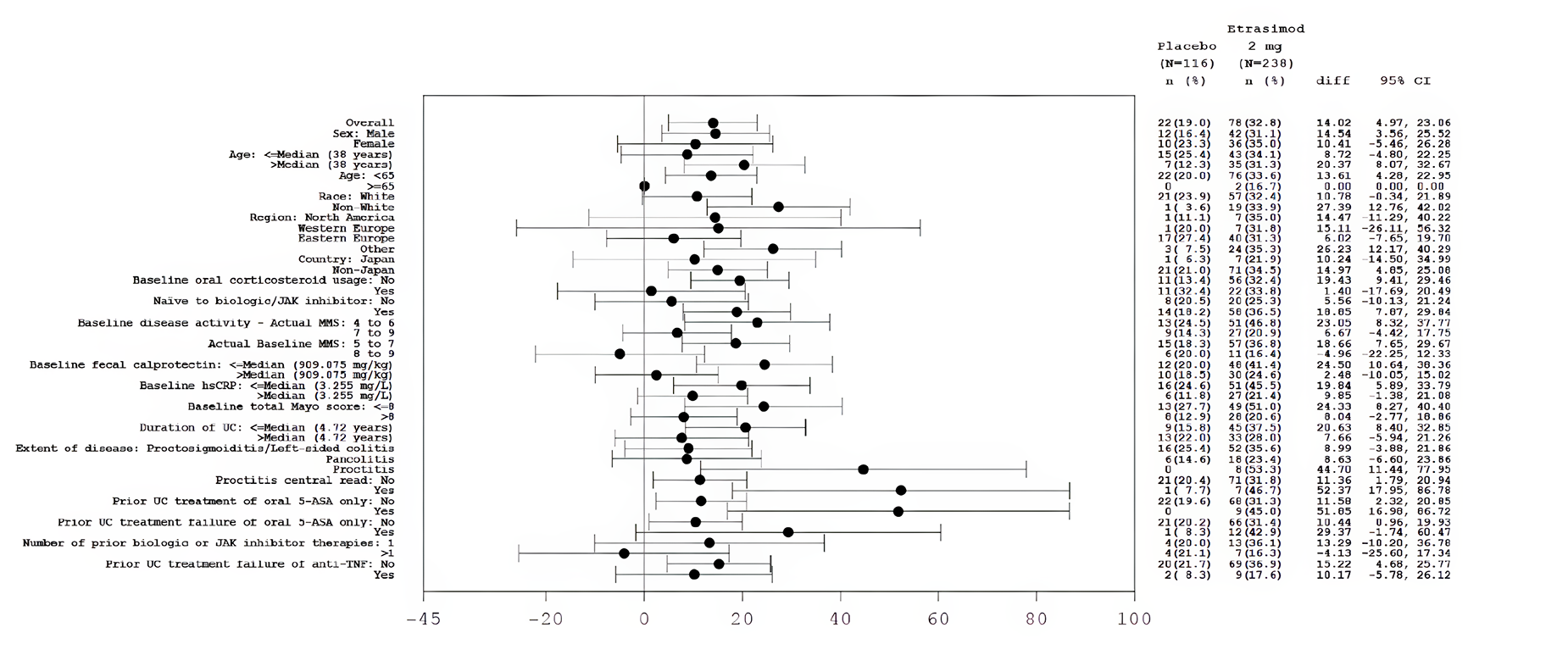
5-ASA = 5-aminosalicylic acid; anti-TNF = antitumour necrosis factor; CI = confidence interval; ES = endoscopic score; FAS = full analysis set; hsCRP = high-sensitivity C-reactive protein; JAK = Janus kinase; MMS = modified Mayo score; UC = ulcerative colitis.
Note: Endoscopic improvement is defined as patients with an ES subscore ≤ 1 (excluding friability). Not reported or missing race are excluded.
Source: Sponsor’s submission.19
Figure 9: Forest Plot for Endoscopic Improvement at Week 12 for Overall and Subgroups of the ELEVATE UC 52 Trial — FAS and a Baseline MMS of 4 to 9
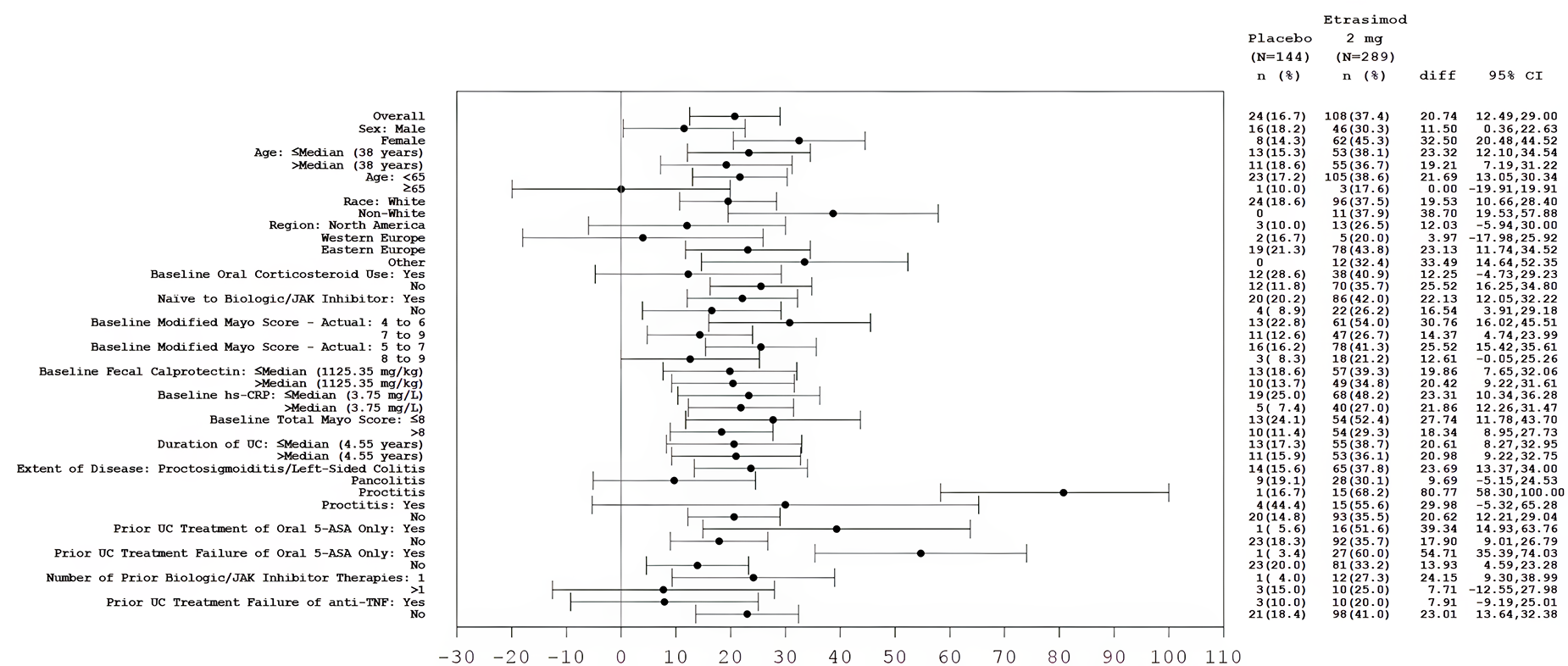
5-ASA = 5-aminosalicylic acid; anti-TNF = antitumour necrosis factor; CI = confidence interval; ES = endoscopic score; FAS = full analysis set; hsCRP = high-sensitivity C-reactive protein; JAK = Janus kinase; MMS = modified Mayo score; UC = ulcerative colitis.
Note: Endoscopic improvement is defined as patients with an ES subscore ≤ 1 (excluding friability). Not reported or missing race are excluded.
Source: Sponsor’s submission.19
Figure 10: Forest Plot for Endoscopic Improvement at Week 52 for Overall and Subgroups of the ELEVATE UC 52 Trial — FAS and a Baseline MMS of 4 to 9
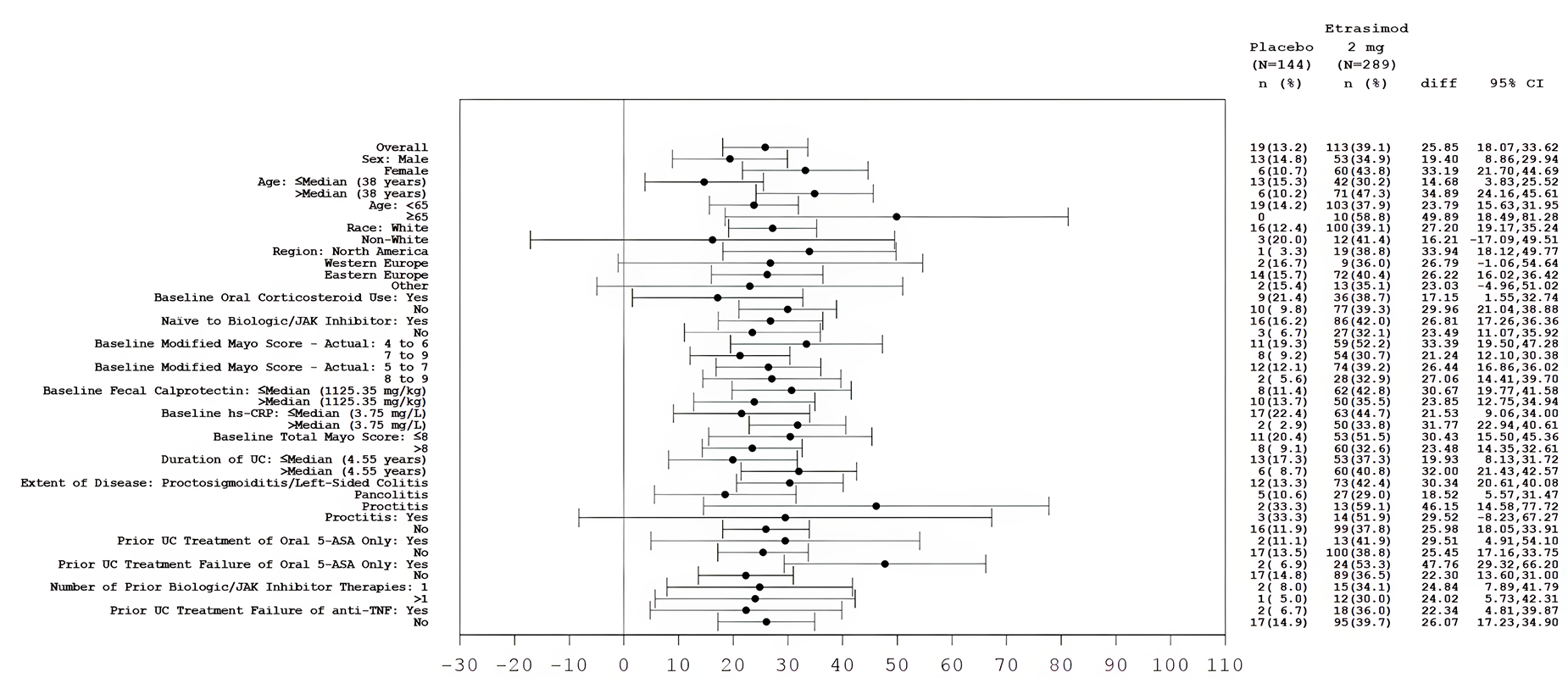
5-ASA = 5-aminosalicylic acid; anti-TNF = antitumour necrosis factor; CI = confidence interval; ES = endoscopic score; FAS = full analysis set; hsCRP = high-sensitivity C-reactive protein; JAK = Janus kinase; MMS = modified Mayo score; UC = ulcerative colitis.
Note: Endoscopic improvement is defined as patients with an ES subscore ≤ 1 (excluding friability). Not reported or missing race are excluded.
Source: Sponsor’s submission.19
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
HRQoL
health-related quality of life
MMS
modified Mayo score
NMA
network meta-analysis
pCPA
pan-Canadian Pharmaceutical Alliance
QALY
quality-adjusted life-year
UC
ulcerative colitis
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Etrasimod (Velsipity), 2 mg oral tablets |
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 31, 2024 |
Reimbursement request | As per indication |
Sponsor | Pfizer Canada ULC |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy (advanced therapy–naive) or an advanced treatmenta (advanced treatment–experienced). |
Treatment | Etrasimod |
Dose regimen | 2 mg once daily |
Submitted price | $43.10 per 2 mg tablet |
Submitted treatment cost | $15,688 per patient per year |
Comparatorsb |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (60 years) |
Key data source | NMAs; effectiveness of etrasimod informed by the ELEVATE UC 12 and ELEVATE UC 52 trials |
Submitted results | Advanced therapy–naive subgroup:
Advanced therapy–experienced subgroup:
|
Key limitations |
|
CDA-AMC reanalysis results |
|
ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; SC = subcutaneous; UC = ulcerative colitis.
aAdvanced therapies were assumed by the sponsor to include adalimumab (branded and biosimilar), golimumab, infliximab (branded and biosimilar), mirikizumab, ozanimod, tofacitinib (branded and generic), upadacitinib, and vedolizumab.
bThe comparators included by the sponsor were the same for both subgroups, with the exception that golimumab and infliximab were excluded from the advanced therapy–experienced subgroup.
Conclusions
Based on the CDA-AMC Clinical Review, etrasimod results in an increase in the proportion of patients with moderately to severely active ulcerative colitis (UC) who have a clinical response and clinical remission compared with placebo, based on data from the ELEVATE UC 12 and ELEVATE UC 52 trials. There are no direct head-to-head trials comparing etrasimod with other advanced therapies for the indicated population, and the indirect evidence submitted by the sponsor suggests that █████ ███ ██ ██ ██████████ ██████████ in efficacy or safety between etrasimod and other advanced treatments during either the induction or maintenance phases. Thus, there is insufficient evidence to suggest that etrasimod should be priced higher than currently reimbursed treatments for moderately to severely active UC. To ensure cost-effectiveness, etrasimod should be priced no more than the least costly advanced therapy that is funded in the population to be reimbursed.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Crohn and Colitis Canada and the Gastrointestinal Society, which collected perspectives of patients with UC through surveys and interviews. Patients with UC reported diarrhea, bowel urgency, incontinence, abdominal pain, fever, rectal bleeding, nausea, weight loss, and a negative impact of UC on their mental health. Respondents described experience with a variety of treatments, which included systemic steroids, sulfasalazine, 5-aminosalicylates, biologics, Janus kinase inhibitors, sphingosine 1-phosphate inhibitors, immunomodulators, antibiotics, and nonsystemic steroids. Patients reported experiencing recurrent flares, loss of treatment response over time, and the need to continuously switch treatments until all options had been exhausted. The treatment goals described by patients included sustained remission and response as well as improved quality of life. Patients also expressed the need to have a range of treatment options. None of the respondents had experience with etrasimod.
Clinician input was received from a group of gastroenterologists specializing in the management of inflammatory bowel disease in Canada. The clinicians noted that the goals of treatment are to control the symptoms of UC and to prevent disease progression, surgery, and disability. Clinicians anticipate that etrasimod will likely be used as a first-line advanced therapy (i.e., after failure of conventional therapies) or as a second- or third-line advanced therapy option for some patients. The group indicated that response to treatment should be assessed 3 to 4 months after treatment initiation using a combination of symptoms and objective measures, and that endoscopic disease activity should be assessed at 6 to 12 months. Treatment should be discontinued if the patient experiences adverse events (AEs), has no response to treatment, or loses an initial response.
CDA-AMC-participating drug plans noted concerns with the appropriateness of placebo as a comparator in the ELEVATE UC clinical trials because there are multiple advanced treatments approved for the treatment of UC. Drug plan input also noted concerns about the comparative safety of advanced therapies, especially in terms of AEs such as bradycardia, hypertension, transaminitis, and lymphopenia.
Several of these concerns were addressed in the sponsor’s model:
Treatment response (clinical response and remission) was incorporated into the sponsor’s model through the use of the modified Mayo score (MMS).
CDA-AMC was unable to address the following concerns raised from stakeholder input:
CDA-AMC was unable to consider endoscopic response owing to the structure of the sponsor’s model, although endoscopic score is considered within the MMS.
The time at which initial response to treatment is assessed in clinical practice (3 to 4 months) differs from that used in the sponsor’s model (6 to 12 weeks).
Treatment sequencing could not be addressed owing to a lack of clinical data.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of etrasimod compared with advanced therapies, which were assumed by the sponsor to include adalimumab (branded and biosimilar), infliximab (branded and biosimilar), golimumab, tofacitinib (branded and generic), upadacitinib, vedolizumab (IV and subcutaneous), ozanimod, and mirikizumab.1 The model population comprised adult patients with moderately to severely active UC who had an inadequate response, lost response, or were intolerant to either conventional or an advanced therapy. The sponsor conducted separate analyses for the advanced therapy–naive and –experienced subgroups, with the same comparators in both subgroups except for golimumab and infliximab, which were excluded from the advanced therapy–experienced subgroup. The modelled population is aligned with the Health Canada indication and the reimbursement request.
Etrasimod is available as a 2 mg tablet at a submitted price of $43.10 per tablet.2 The recommended dose is 2 mg once daily,3 which the sponsor estimated would result in an annual per-patient cost of $15,688. The annual maintenance costs for comparators in the sponsor’s model ranged from $4,361 for tofacitinib to $30,871 for mirikizumab.
The economic evaluation was conducted over a lifetime time horizon (60 years, 8-week cycle length), from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and quality-adjusted life-years [QALYs]) were discounted at a rate of 1.5% per annum.
Model Structure
The sponsor’s analysis included a short-term induction phase and a longer-term maintenance phase (Figure 1),1 with the same model structure for both advanced therapy–naive and –experienced patient subgroups. Patients entered the induction phase with moderately to severely active UC and received etrasimod or a comparator. During the induction period, patients could experience 1 of the following outcomes: response, response without remission, remission, or surgery. The length of induction differed across treatments (12 weeks for etrasimod and 6 to 12 weeks for comparators). Following the induction phase, patients who had achieved remission or response entered the maintenance phase in the corresponding health state and remained on their initial treatment. Each cycle, a proportion of patients in these states were assumed to be at risk of loss of response. Patients who did not have a treatment response at the end of the induction period or lost response to treatment were assumed to receive subsequent therapy, which consisted of a basket of advanced therapies. In the base case, the sponsor assumed that patients who do not respond to initial treatment or who lose their initial response will try up to 2 lines of subsequent therapy, with a portion of patients transitioning to best supportive care (BSC) each cycle, which was assumed by the sponsor to be 5-aminosalicylic acid, corticosteroids, and conventional immunomodulators. The model included a state for surgery (emergency and elective), and 2 post-surgery health states (with and without complications). Patients who underwent surgery were assumed to remain in the post-surgery health states for the remainder of the model’s time horizon and to be at risk of post-surgical complications. Patients could transition to death from any health state.
Model Inputs
Baseline patient characteristics in the model were based on the ELEVATE UC 52 trial for patients with an MMS of 5 to 9 at baseline (advanced therapy–naive: aged 40 years, 52% male, 73 kg; advanced therapy–experienced: aged 43 years, 62% male, 77 kg).4
Clinical efficacy and safety inputs in the model (i.e., clinical response, clinical remission, loss of response, serious infection rates) were obtained from the sponsor-submitted network meta-analyses (NMAs) for the advanced therapy–naive and advanced therapy–experienced subgroups conducted at the end of the induction (at 8 to 12 weeks) and maintenance (at 38 to 52 weeks) phases.5 The definition of response varied across the trials included in the NMA. The probability of response without remission was assumed by the sponsor to be the difference between the probability of clinical response and remission. The sponsor assumed equal efficacy and safety for biosimilars and branded drugs (i.e., adalimumab, infliximab), as well as for vedolizumab IV and subcutaneous formulations. The probability of loss of treatment response was estimated by the sponsor among overall responders (identified as those having a clinical response and response without remission) and responders (identified as those having a response without remission) as 1 minus the probability of response at the end of the maintenance phase. The sponsor assumed that loss of response would be consistent over the lifetime time horizon. The sponsor estimated the effectiveness of subsequent therapy (i.e., probability of clinical response, remission, loss of response) based on a weighted average of the effectiveness of treatments in the basket and informed by the results of the sponsor’s NMA. The distribution of patients on each treatment in the basket of subsequent treatments was informed by expert opinion obtained by the sponsor. The risk of surgery and post-surgical complications was obtained from the literature.678 The sponsor assumed no additional UC-related risk of death; however, the general population mortality risk was increased by 1.3% for patients who underwent surgery.9,10
Health state utility values for active UC (0.41), response without remission (0.76), remission (0.87), surgery (0.61), and post-surgery states (without complications: 0.72; with complications: 0.34) were obtained from the literature.11,12 The sponsor assumed that patients in the induction phase have active UC, and that patients who later transition to BSC have the same utility as patients during induction (i.e., active UC). Utilities were age- and sex-adjusted. The disutility associated with serious infections (0.16) was obtained from the literature.13
The economic model included costs related to drug acquisition and administration, disease management, AEs, and surgery. Drug acquisition costs for etrasimod were based on the sponsor-submitted price.1 The recommended dosing regimen of comparators was sourced from respective product monographs, and their acquisition costs were obtained from the Ontario Drug Benefit Formulary, the Régie de l’assurance maladie du Québec formulary, and previous CDA-AMC reports.14-17 Unit dose and dosing frequency of treatments in the induction and maintenance phase were obtained from the ELEVATE UC trials for etrasimod and the respective product monographs for comparators. For treatments that have a standard and a high dose recommendation, the sponsor assumed that 30% of patients would receive the high dose in the maintenance period. Administration costs were included for drugs administered by IV infusion but not for those administered orally or by subcutaneous injection. Health care resource use by health state was obtained from the literature.18 Disease management costs included costs of outpatient visits to gastroenterologists, blood tests, emergency and elective endoscopy, hospitalization and stoma care (post colectomy), with unit costs obtained from the Ontario Health Insurance Plan database, Canadian Institute for Health Information (CIHI), and the literature.19,20 The cost of managing AEs was assumed by the sponsor to comprise the average cost of treating 5 types of serious infections (sepsis, pneumonia, urinary tract infection, respiratory infection, bronchitis) based on data from CIHI.21 The cost of elective and emergency surgery was obtained from the literature.22
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The sponsor submitted 2 subgroup analyses (advanced therapy–naive, advanced therapy–experienced) to reflect the overall indicated population. The deterministic and probabilistic incremental cost-effectiveness ratios (ICERs) were similar, and the probabilistic findings are presented subsequently.
Base-Case Results
Among patients in the advanced therapy–naive subgroup, etrasimod was associated with an estimated cost of $838,905 and 22.63 QALYs over the 60-year horizon (Table 3). In sequential analysis, etrasimod was associated with an incremental cost-effectiveness ratio of $40,215 per QALY gained versus tofacitinib (incremental costs: $27,281; incremental QALYs: 0.68), with a 43% probability of being considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Of the 22.63 QALYs predicted by the sponsor’s model to be gained with etrasimod, approximately 97% were accrued after the first year of treatment (i.e., beyond the duration of the ELEVATE UC 52 trial) and approximately 12% were accrued by patients receiving induction or maintenance treatment, with the remainder (88%) gained by patients receiving subsequent therapy or BSC, or after surgery.
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Advanced Therapy–Naive Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Tofacitinib | 811,624 | 21.95 | Reference |
Etrasimod | 838,905 | 22.63 | 40,215 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the efficiency frontier are reported.
Source: Sponsor’s pharmacoeconomic submission.1
Among patients in the advanced therapy–experienced subgroup, etrasimod was associated with an estimated cost of $791,796 and 21.16 QALYs over the 60-year horizon. In sequential analysis, etrasimod was extendedly dominated by tofacitinib and upadacitinib. There was a 7% probability that etrasimod would be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Of the 21.61 QALYs predicted by the sponsor’s model to be gained with etrasimod, approximately 97% were accrued after the first year of treatment (i.e., beyond the duration of the ELEVATE UC 52 trial) and 10% were gained by patients receiving induction or maintenance treatment, with the remainder gained by patients receiving subsequent therapy or BSC, or post surgery.
Table 4: Summary of the Sponsor’s Economic Evaluation Results — Advanced Therapy–Experienced Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Tofacitinib | 721,886 | 20.96 | Reference |
Upadacitinib | 807,120 | 21.91 | 89,186 |
Etrasimod | 791,796 | 21.16 | Extendedly dominated by tofacitinib and upadacitinib |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only results for etrasimod and treatments that are on the efficiency frontier are reported.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity and scenario analyses adopting alternative discounting rates and utility estimates derived from the ELEVATE UC 52 trial data. The sponsor additionally considered a scenario exploring the impact of adopting a societal perspective, which included additional costs associated with out-of-pocket costs, transportation costs (travel and parking or travel fares), and patient and caregiver lost income. However, no sequential analyses were provided (i.e., etrasimod was compared with each of the other advanced therapies in a pairwise fashion), limiting the interpretation of the findings.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative clinical efficacy of etrasimod is uncertain: There is a lack of direct head-to-head evidence comparing etrasimod with other advanced therapies. To inform efficacy in the economic model (i.e., clinical response and remission), the sponsor conducted an NMA to estimate the relative efficacy of etrasimod in the advanced therapy–naive and –experienced subgroups in the induction and maintenance phases. As noted in the CDA-AMC Clinical Review, the indirect evidence submitted by the sponsor suggests ████ █████ ███ ██ ██ ██████████ ███████████ in efficacy between etrasimod and relevant comparators in either the induction or maintenance phases, although the presence of unresolved heterogeneity (e.g., in patient characteristics, treatment history, and outcome definitions), and substantial imprecision precludes meaningful conclusions from being made. CDA-AMC notes that health-related quality of life (HRQoL) was not assessed in the sponsor’s NMA.
Given the lack of direct evidence and limitations with the sponsor’s NMA, the clinical efficacy of etrasimod compared with other advanced therapies is uncertain. As the sponsor-submitted NMA suggests that █████ ███ ██ ██ ██████████ in efficacy between etrasimod and other advanced therapies, it is uncertain whether etrasimod provides a benefit relative to other advanced therapies currently funded for this indication. In the CDA-AMC base case, CDA-AMC assumed equal clinical efficacy among all advanced therapies.
Failure to adequately characterize decision uncertainty: Consistent with CDA-AMC economic guidelines, the sponsor’s base case used a Monte Carlo simulation to characterize the uncertainty of most input parameters, with the values for each iteration drawn from a random sampling of values between a lower and upper bound. However, for the efficacy inputs (clinical response, remission), the sponsor implemented hard-coded relative risks based on externally derived iterations using the Convergence Diagnostic and Output Analysis (CODA) output of the sponsor’s NMA. This limits the usefulness of the sponsor’s probabilistic model, as the efficacy values were predefined and hard coded within the model, and CDA-AMC was unable to alter the efficacy inputs for probabilistic reanalyses. As such, when CDA-AMC adopted alternative assumptions about efficacy within the model, these changes were reflected in the deterministic model but not in the probabilistic analysis.
CDA-AMC could not address this limitation, and the probabilistic output could not be used to derive the CDA-AMC base case, given the uncertainties associated with the results. All CDA-AMC reanalyses are thus based on the sponsor’s deterministic model.
Uncertainty in the long term effectiveness of etrasimod: Evidence of the long-term effectiveness of etrasimod beyond 52 weeks is not available. In the pharmacoeconomic model, the sponsor incorporated the potential for loss of treatment effectiveness, derived from the results of the sponsor’s NMA for response and response without remission at the end of the maintenance phase, and assumed that this loss of response will remain constant over the model’s lifetime horizon. The use of data from the end of the maintenance period (i.e., at 52 weeks) to inform long-term loss of effectiveness is associated with substantial uncertainty, including whether the rate of waning is consistent over time for etrasimod and comparators. Given that the majority of the incremental QALYs (97%) predicted by the sponsor’s model to be gained with etrasimod in both subgroups were derived on the basis of extrapolated findings rather than observed benefit, the lack of long-term data introduces considerable uncertainty into the analysis.
This limitation could not be addressed by CDA-AMC, owing to a lack of long-term clinical data. The direction and magnitude of the impact of this limitation are unknown, given that the comparative rate of potential effectiveness waning with etrasimod versus other advanced therapies for UC is unknown.
The impact of AEs on costs and QALYs was not adequately captured: The sponsor’s model incorporated costs and utility decrements related to serious infections, the rates of which were derived from the sponsor’s NMA. The sponsor did not justify modelling only serious infections instead of considering the broader category of serious adverse events (SAEs), which were included as an outcome in the NMA. For serious infections, the results of the sponsor’s NMA suggest that █████ ███ ██ ██ ██████████ in serious infections between etrasimod and other advanced therapies in the overall population and advanced therapy–naive subgroup from the pivotal trials; however, as noted earlier, the CDA-AMC Clinical Review identified several limitations with the sponsor-submitted NMA that preclude meaningful conclusions from being made. CDA-AMC additionally notes that the sponsor did not specify the type of serious infections included in the NMA. In the calculation of costs for treating AEs, the sponsor considered 5 types of infection (sepsis, pneumonia, urinary tract infection, respiratory infection, and bronchitis); however, it is unknown whether these are the serious infections included within the basket of “serious infections” in the NMA. Finally, the sponsor incorporated a utility decrement of 0.156 for serious infections, assuming that this value represents the average decrease in HRQoL for those with a serious infection, despite not specifying the types of infections contributing to this disutility.
CDA-AMC notes that, although the pivotal etrasimod trials showed few SAEs, the product monograph for etrasimod includes a serious warnings and precautions note that includes malignancies, cardiovascular events, and liver injury. As noted in the CDA-AMC Clinical Review, cardiovascular events and liver injury were experienced by 4.2% and 1.4%, respectively, of patients who received etrasimod. CDA-AMC further notes that additional AEs noted to be of special interest to clinicians (e.g., progressive multifocal leukoencephalopathy, macular edema, pulmonary disorders) were not considered in the sponsor’s model and the impact of these AEs on costs and QALYs is unknown.
The comparative safety of etrasimod versus other advanced therapies available for the treatment of UC is uncertain. Because the sponsor-submitted NMA suggests that █████ ███ ██ ██ ██████████ in serious infections between etrasimod and other advanced therapies, CDA-AMC assumed an equal risk of serious infections for all advanced therapies in the CDA-AMC base case. CDA-AMC could not address the impact of AEs beyond serious infections on the estimated cost-effectiveness of etrasimod.
The impact of subsequent therapy is uncertain: The sponsor modelled the effectiveness and costs of subsequent therapy for patients who do not respond to initial treatment or later lose response, assuming that subsequent therapy would comprise a basket of advanced therapies. In the base case, the sponsor assumed that patients would try up to 2 lines of subsequent therapy, with a portion of patients transitioning to BSC each model cycle. The clinical expert opinion obtained by CDA-AMC for this review indicated it is unlikely that patients would try only 2 lines of subsequent therapy. Instead, patients are likely to continue to try additional treatments until all options are exhausted. Clinical expert feedback also indicated that patients would be more likely to undergo surgery rather than transition to BSC.
The sponsor estimated the effectiveness of subsequent therapy, based on data from the submitted NMA and weighted by the proportion of patients assumed to receive each advanced therapy as part of subsequent treatment (informed by clinical expert opinion). As noted earlier, the sponsor’s NMA suggested ██ ██████████ ███████████ in efficacy between etrasimod and relevant comparators. CDA-AMC notes that, in the sponsor’s base case, 88% to 90% of QALYs gained with etrasimod were accrued after the discontinuation of initial treatment, based on the effectiveness of subsequent therapy, rather than with etrasimod as initial treatment, in both the advanced therapy–naive and advanced therapy–experienced subgroups.
In the CDA-AMC base case, CDA-AMC assumed equal efficacy of all advanced therapies, which impacted both initial and subsequent treatment because of the structure of the sponsor’s model.
Health state utility values adopted by the sponsor are uncertain: The sponsor’s base case adopted health state utility values for nonsurgical health states from a published abstract (Woehl et al., 2008).11 There are multiple utility values reported in the literature for health states related to moderate to severe UC, with estimates differing markedly from those reported by Woehl et al. For example, the utility value for active moderate to severe UC reported by Woehl et al.11 (0.41) is considerably lower than that reported by Vaizey et al. and Swinburn et al., which ranged from 0.68 to 0.77 for moderate disease and 0.45 to 0.66 for severe disease.23,24 CDA-AMC has critiqued the reliability of the utility estimates from Woehl in multiple previous reviews.25-27
CDA-AMC notes that the sponsor additionally estimated health state utility values from data collected in the ELEVATE UC 52 trial (Short Form [36] Health Survey data mapped to EQ-5D-3L28). The utility value (0.78) estimated by the sponsor for the active UC state using these data are relatively consistent with the utility value (0.73) estimated using the EQ-5D-5L data collected in the TRUENORTH trial for ozanimod.16 CDA-AMC acknowledges that the pivotal trial-based values may overestimate utility due to the trial design (i.e., only patients who had achieved at least a response in the induction phase were permitted to continue into maintenance for assessment of HRQoL) in both the ELEVATE UC 52 and TRUENORTH trials.
In the CDA-AMC base case, utility values from Swinburn et al. (2012) were adopted, consistent with prior CDA-AMC reviews for this indication.
Relevant comparators are not included for the advanced therapy–experienced subgroup: The sponsor omitted golimumab and infliximab as comparators in the advanced therapy–experienced subgroup, which the sponsor justified based on the exclusion of patients with exposure to previous advanced treatments in the respective pivotal trials. As noted in the CDA-AMC economic guidelines, the selection of comparators should be conceptually driven and should not be determined by the availability of data.29 The clinical expert input received by CDA-AMC indicated that golimumab and infliximab would be used in this subgroup. While CDA-AMC acknowledges that data gaps may exist, the economic guidelines recommend that this should be addressed in scenario analyses or discussed with respect to potential implications for decision-making.
CDA-AMC was unable to address this limitation in reanalyses. The cost-effectiveness of etrasimod versus golimumab and infliximab in the advanced therapy–experienced subgroup is unknown.
Inappropriate comparator price: The sponsor’s model included both branded and generic tofacitinib. Because the perspective of the public drug plan is adopted in the base case, the price of comparators should be based on the amount reimbursed by the public drug plan.
The price of generic tofacitinib was used to derive the CDA-AMC base case.
Additionally, the following key assumptions were made by the sponsor and appraised by CDA-AMC (Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The time horizon was assumed to be 60 years based on a starting age of 40 years for the advanced therapy–naive group and 43 years for the advanced therapy–experienced group. | Uncertain. Approximately 4% of the advanced therapy–naive cohort remained alive at 110 years and 1% of the advanced therapy–experienced cohort remained alive at 103 years, which exceeds the average life expectancy in Canada.9 |
The sponsor assumed that 30% of patients receiving adalimumab (brand and biosimilar), golimumab, infliximab (brand and biosimilar), tofacitinib (brand and generic), upadacitinib, and vedolizumab (IV) would receive an escalated dose during the maintenance phase. | Uncertain. The clinical expert input received by CDA-AMC indicated that dose escalation may occur if a patient has a disease flare or a nonresponse, and rarely if a patient is stable and responding well to treatment. |
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC undertook reanalyses that addressed key limitations within the submitted model, as summarized in Table 6. The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts.
Table 6: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
Price of tofacitinib | Both branded and generic tofacitinib were included by the sponsor, with the price of branded tofacitinib assumed to be $23.9589 per 5 mg tablet and $42.3436 per 10 mg tablet. | The price of tofacitinib in the model was based on the amount paid by public drug plans (5 mg: $5.9897; 10 mg: $21.1718).a |
Changes to derive the CDA-AMC base case | ||
1. Comparative efficacy | The risk ratios for response and remission were derived from the sponsor’s NMA. | The risk ratios for response and remission were assumed to be equivalent for all advanced therapies, including etrasimod.b,c |
2. Comparative safety | The risk ratios for serious infections were derived from the sponsor’s NMA. | The risk ratios for serious infections were assumed to be equivalent for all advanced therapies, including etrasimod. |
3. Health state utility values | From Woehl (2008):
| From Swinburn (2012):
|
CDA-AMC base case | ― | Reanalysis 1 + 2 + 3 |
NMA = network meta-analysis; UC = ulcerative colitis.
aAll patients in the sponsor’s corrected base case and in CDA-AMC’s reanalyses were thus assumed to receive generic tofacitinib because the price of the branded drug has been replaced with the generic price and the sponsor assumed equal efficacy between branded and generic forms (i.e., equal costs and benefits for both forms).
bThe lengths of the induction and maintenance periods were additionally set to be equal in the CDA-AMC reanalysis. While, in clinical practice, the length of the induction period varies across advanced therapies, this change was implemented to address a structural limitation in the sponsor’s model.
cThis change imposes an equal probability of loss of treatment response for all advanced therapies owing to the structure of the sponsor’s model. Whether treatment effectiveness waning is equivalent for all treatments is unknown owing to a lack of long-term clinical data.
In the CDA-AMC base case for both the advanced therapy–naive and advanced therapy–experienced subgroups, etrasimod was more costly and equally effective compared with adalimumab biosimilar (Table 7, Table 8). CDA-AMC was unable to determine the probability that etrasimod is cost-effective at a willingness-to-pay threshold (e.g., of $50,000 per QALY), owing to the structural limitations of the sponsor’s model (that is, all CDA-AMC analyses are deterministic and do not reflect uncertainty). Additional results are provided in Appendix 4.
Table 7: Summary of the CDA-AMC Reanalysis Results — Advanced Therapy–Naive Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor base case (corrected) | |||
Infliximab biosimilar | 742,213 | 20.70 | Reference |
Adalimumab biosimilar | 742,847 | 20.86 | 3,973 |
Etrasimod | 746,145 | 21.31 | 7,383 |
CDA-AMC base case | |||
Adalimumab biosimilar | 748,944 | 21.47 | Reference |
Etrasimod | 760,759 | 21.47 | Dominated by adalimumab biosimilar |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Deterministic analyses are presented for the sponsor’s base case (corrected) and the CDA-AMC base case.
Table 8: Summary of the CDA-AMC Reanalysis Results — Advanced Therapy–Experienced Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor base case (corrected) | |||
Adalimumab biosimilar | 700,519 | 19.67 | Reference |
Upadacitinib | 721,904 | 21.38 | 12,557 |
Etrasimod | 709,424 | 19.81 | Extendedly dominated by adalimumab biosimilar and upadacitinib |
CDA-AMC base case | |||
Adalimumab biosimilar | 717,458 | 19.94 | Reference |
Etrasimod | 725,230 | 19.94 | Dominated by adalimumab biosimilar |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Deterministic analyses are presented for the sponsor’s base case (corrected) and the CDA-AMC base case.
Scenario Analysis Results
As the CDA-AMC base case assumes equal comparative efficacy and safety across treatments, CDA-AMC considered price reductions based on the submitted price for etrasimod and the publicly accessible list prices of all other advanced therapies on a yearly basis. Compared with other treatments for moderately to severely active UC, the annual per-patient drug acquisition cost of etrasimod is anticipated to be higher than for tofacitinib, adalimumab biosimilars, and infliximab biosimilars (12,862 to 13,697) (Appendix 1). A price reduction of approximately 68% would be needed for the annual acquisition cost of etrasimod to be equivalent to that of the lowest-cost comparator (i.e., tofacitinib) in the first year of treatment (72% in subsequent years). This analysis assumed that all patients received 5 mg tofacitinib twice a day in the maintenance phase, which was consistent with assumptions in the sponsor’s and CDA-AMC’s base case.
Issues for Consideration
Upadacitinib and mirikizumab are currently under active negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA). If these negotiations conclude with a letter of intent, the price paid by the drug plans may be lower than the price incorporated in the sponsor’s pharmacoeconomic model. The sponsor for ozanimod has successfully completed negotiations with the pCPA30 and the drug is listed on some public drug formularies, and the price paid by jurisdictional drug plans is likely to be lower than the publicly available list price.
Ustekinumab previously received a positive recommendation from the Canadian Drug Expert Committee for this indication; however, negotiations with pCPA concluded without agreement, and CDA-AMC accepted a deviation request by the sponsor to exclude ustekinumab from its analyses. CDA-AMC notes that biosimilars for ustekinumab have recently been approved by Health Canada and are reimbursed on some public formularies. The cost-effectiveness of etrasimod versus ustekinumab (branded or biosimilar) is unknown.
There are policies in place in some jurisdictions promoting the use of biosimilars over originator products. While the sponsor’s analysis included both biosimilar and originator products, the relative market share of each is expected to change over time.
Clinician input received by CDA-AMC indicated that some patients may prefer an oral treatment over an injection or infusion. Etrasimod is 1 such option that would be available to patients should it be reimbursed (along with ozanimod, tofacitinib, and upadacitinib).
Overall Conclusions
Based on the CDA-AMC Clinical Review, etrasimod results in an increase in the proportion of patients with moderately to severely active UC who have a clinical response and clinical remission compared with placebo based on data from the ELEVATE UC 12 and ELEVATE UC 52 trials; however, there may be little to no difference in HRQoL based on the Inflammatory Bowel Disease Questionnaire. There are no direct head-to-head trials comparing etrasimod with other advanced therapies for the indicated population. Indirect evidence submitted by the sponsor suggests that █████ ███ ██ ██ ██████████ ██████████ in efficacy or safety between etrasimod and other advanced treatments during either the induction or maintenance phases. The CDA-AMC Clinical Review identified substantial imprecision and unresolved heterogeneity in the sponsor’s NMA, owing primarily to differences in the source trials. HRQoL was not assessed in the sponsor-submitted NMA.
The sponsor submitted an economic analysis comparing the cost-effectiveness of etrasimod with other advanced therapies in the advanced therapy–naive and advanced therapy–experienced subgroups, based on data from the sponsor’s NMA. In addition to the uncertainty in the clinical evidence, CDA-AMC identified several additional sources of uncertainty in the sponsor’s economic submission, including uncertainty related to the programming of the model, the long-term effectiveness of etrasimod, the impact of AEs on costs and QALYs, the impact of subsequent therapies, and the validity of the adopted health state utility values.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis, which included adopting equivalent efficacy and safety for all advanced therapies, including etrasimod, and adopting alternative health state utility values. In the CDA-AMC base case for both the advanced therapy–naive and advanced therapy–experienced subgroups, etrasimod was equally effective but more costly than adalimumab biosimilar. Given that indirect evidence submitted by the sponsor suggests that █████ ███ ██ ██ ██████████ ██████████ in efficacy or safety between etrasimod and other advanced treatments during either the induction or maintenance phases, there is insufficient evidence to suggest that etrasimod should be priced higher than currently reimbursed treatments for moderately to severely active UC. Thus, to ensure cost-effectiveness, etrasimod should be priced no more than the least costly advanced therapy that is funded in the population to be reimbursed.
References
1.Weinstein MC, Torrance G, McGuire A. QALYs: the basics. Value Health. 2009;12 Suppl 1:S5-9. PubMed
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Etrasimod, 2 mg tablet (oral). Kirkland Quebec: Pfizer Canada ULC; 2024 Feb 05. 2024.
3.Velsipity (etrasimod): tablet, 2 mg etrasimod (as etrasimod L-arginine), oral [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2024 Jan 30.
4.Sandborn WJ, Vermeire S, Peyrin-Biroulet L. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. The Lancet. 2023;401(10383):1159-1171. PubMed
5.Chen G, Ratcliffe J. A Review of the Development and Application of Generic Multi-Attribute Utility Instruments for Paediatric Populations. Pharmacoeconomics. 2015;33(10):1013-1028. PubMed
6.Misra R, Askari A, Faiz O, Arebi N. Colectomy Rates for Ulcerative Colitis Differ between Ethnic Groups: Results from a 15-Year Nationwide Cohort Study. Can J Gastroenterol Hepatol. 2016;2016:1-7. PubMed
7.National clinical audit of inpatient care for adults with ulcerative colitis. Inflammatory Bowel Disease programme. 2014; https://www.rcplondon.ac.uk/file/1165. [sponsor submitted reference].
8.Segal JP, McLaughlin SD, Faiz OD, Hart AL, Clark SK. Incidence and Long-term Implications of Prepouch Ileitis: An Observational Study. Dis Colon Rectum. 2018;61(4):472-475. PubMed
9.Statistics Canada. Life expectancy and other elements of the complete life table, three-year estimates. 2023; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.3&pickMembers%5B2%5D=4.3&cubeTimeFrame.startYear=2019+%2F+2021&cubeTimeFrame.endYear=2019+%2F+2021&referencePeriods=20190101%2C20190101. Accessed 2023 Nov 27.
10.Jess T, Gamborg M, Munkholm P, Sørensen TIA. Overall and Cause-Specific Mortality in Ulcerative Colitis: Meta-analysis of Population-Based Inception Cohort Studies. Am J Gastroenterol. 2007;102(3):609-617. PubMed
11.The relation between disease activity, quality of life and health utility in patients with ulcerative colitis. Gut. 2008.
12.Arseneau KO, Sultan S, Provenzale DT. Do Patient Preferences Influence Decisions on Treatment for Patients With Steroid-Refractory Ulcerative Colitis? Clin Gastroenterol Hepatol. 2006;4(9):1135-1142. PubMed
13.Stevenson M, Archer R, Tosh J. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess. 2016;20(35):1-610. PubMed
14.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2024 May 6.
15.Régie de l’assurance maladie du Québec. List of Medications. 2023; https://www.ramq.gouv.qc.ca/sites/default/files/documents/non_indexes/liste_med_2023-07-06_en.pdf [sponsor submitted reference].
16.CADTH Reimbursement Review: ozanimod (Zeposia). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/SR0714-Zeposia-meta.pdf. [sponsor submitted reference].
17.CADTH Reimbursement Review: CADTH Reimbursement Recommendation mirikizumab (Omvoh). Ottawa (ON): CADTH; 2023: https://www.cadth.ca/sites/default/files/DRR/2023/SR0773%20Omvoh%20-%20Draft%20CADTH%20Recommendation_For%20posting%20August%2017%2C%202023.pdf. [sponsor submitted reference].
18.Tsai HH, Punekar YS, Morris J, Fortun P. A model of the long-term cost effectiveness of scheduled maintenance treatment with infliximab for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2008;28(10):1230-1239. PubMed
19.Ontario Health Insurance Plan. Physician Services Under the Health Insurance Act. 2023; https://health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. [sponsor submitted reference].
20.Canadian Institute for Health Information. Cost of a Standard Hospital Stay. 2022; https://yourhealthsystem.cihi.ca/hsp/inbrief?lang=en#!/indicators/015/cost-of-a-standard-hospital-stay-cshs/;mapC1;mapLevel2;provinceC9001;trend(C1,C5001,C4000);/. [sponsor submitted reference].
21.Canadian Institute for Health Information. Patient Cost Estimator. 2020; https://www.cihi.ca/en/patient-cost-estimator. [sponsor submitted reference].
22.CADTH Common Drug Review: Pharmacoeconomic Review Report: ustekinumab. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0627-stelara-pharmacoeconomic-review-report.pdf. [sponsor submitted reference].
23.Vaizey CJ, Gibson PR, Black CM. Disease status, patient quality of life and healthcare resource use for ulcerative colitis in the UK: an observational study. Frontline Gastroenterol. 2014;5(3):183-189. PubMed
24.Swinburn P, Elwick H, Bean K. PTU-127 The impact of surgery on health related quality of life in ulcerative colitis: Abstract PTU-127 Figure 1. Gut. 2012;61(Suppl 2).
25.Pharmacoeconomic Review Report: tofacitinib (Xeljanz). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0572-xeljanz-pharmacoeconomic-report.pdf. [sponsor submitted reference].
26.Common Drug Review Pharmacoeconomic Review Report: vedolizumab (Entyvio). Ottawa (ON): CADTH; 2015: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0421_Entyvio_PE_Report.pdf. [sponsor submitted reference].
27.Pharmacoeconomic Review Report: adalimumab (Humira). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0450_Humira_UC_PE_Report.pdf. Accessed 2024 May 8.
28.Rowen D, Brazier J, Roberts J. Mapping SF-36 onto the EQ-5D index: how reliable is the relationship? Health Qual Life Outcomes. 2009;7(1). PubMed
29.Canadian Agency for Drugs and Technologies in Health (CADTH). Guidelines for the Economic Evaluation of Health Technologies: Canada. 4th ed. 2017; https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-0. Accessed 2024 May 8.
30.pan-Canadian Pharmaceutical Alliance. Zeposia (ozanimod). 2024; https://www.pcpacanada.ca/negotiation/22096. Accessed 2024 Apr 24.
31.Government of Alberta. Interactive drug benefit list. 2024; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2024 May 6.
32.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2024 May 6.
33.CADTH Reimbursement Review: upadacitinib (Rinvoq). https://www.cadth.ca/sites/default/files/DRR/2023/SR0730-Rinvoq-UC-Combined-Report.pdf. Accessed 2024 May 10.
34.Saskatchewan Drug Plan: search formulary. 2024; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2024 May 6.
35.Rowen D, Azzabi Zouraq I, Chevrou-Severac H, van Hout B. International Regulations and Recommendations for Utility Data for Health Technology Assessment. Pharmacoeconomics. 2017;35(Suppl 1):11-19. PubMed
36.PharmaStat. Ottawa (ON): IQVIA; 2023: https://www.iqvia.com/. [sponsor submitted reference].
37.pan-Canadian Pharmaceutical Alliance. Rinvoq (upadacitinib). 2024; https://www.pcpacanada.ca/negotiation/22462. Accessed 2024 Apr 24.
38.pan-Canadian Pharmaceutical Alliance. Omvoh (mirikizumab). 2024; https://www.pcpacanada.ca/negotiation/22481. Accessed 2024 Apr 24.
Appendix 1: Cost Comparison Table
Table 9: CDA-AMC Cost Comparison Table for Moderately to Severely Active Ulcerative Colitis
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Etrasimod (Velsipity) | 2 mg | Tablet | 43.1000a | 2 mg dailyb | 43.10 | 15,742 |
SP1 receptor agonists | ||||||
Ozanimod (Zeposia) | 0.23 mg 0.46 mg 0.92 mg | Capsule | 68.4929c 68.4929c 68.4932c | Induction: 0.23 mg daily on days 1 to 4, 0.46 mg daily on days 5 to 7 Maintenance: 0.92 daily | 68.49 | 25,017 |
JAK inhibitors | ||||||
Tofacitinib (generics) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | Induction: 10 mg twice daily for at least 8 weeks Maintenance: 5 mg or 10 mg twice daily | Year 1: 13.82 to 23.96 Year 2+: 11.98 to 23.96 | Year 1: 5,046 to 8,750h Year 2+: 4,375 to 8,750h |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Extended-release tablet | 51.6810 76.9600d 101.8100e | Induction: 45 mg once daily for 8 weeks Maintenance: 15 mg or 30 mg once daily | Year 1: 59.37 to 80.77 Year 2+: 51.68 to 76.96 | Year 1: 21,671 to 29,482 Year 2+: 18,864 to 28,090 |
Biologics | ||||||
Adalimumab (Humira) | 20 mg/0.2 mL 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 397.0500d 794.1000d | Induction: 160 mg at week 0, then 80 mg at week 2 Maintenance: 40 mg every other week | Year 1: 65.42 Year 2+: 56.72 | Year 1: 23,880 Year 2+: 20,703 |
Adalimumab biosimilars | 20 mg/0.4 mL 40 mg/0.4 mL 40 mg/0.8 mL 80 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 235.6350 471.2700 471.2700 942.5400 | Induction: 160 mg at week 0, then 80 mg at week 2 Maintenance: 40 mg every other week | Year 1: 38.82 Year 2+: 33.66 | Year 1: 14,172 Year 2+: 12,287 |
Golimumab (Simponi) | 50 mg/0.5 mL 100 mg/1 mL | Prefilled syringe or autoinjector for SC injection | 1,555.1700d 1,555.1700d | Induction: 200 mg at week 0, then 100 mg at week 2 Maintenance: 50 mg every 4 weeks | Year 1: 61.93 Year 2+: 55.54 | Year 1: 22,606 Year 2+: 20,273 |
Infliximab (Remicade) | 100 mg | Vial for IV infusion | 987.5600d | Induction: 5 mg/kg at week 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 84.07 Year 2+: 70.54 | Year 1: 30,685 Year 2+: 25,747 |
Infliximab biosimilar (Inflectra) | 100 mg | Vial powder for IV infusion | 525.0000 | Induction: 5 mg/kg at week 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 44.69 Year 2+: 37.50 | Year 1: 16,313 Year 2+: 13,688 |
Infliximab biosimilar (Avsola, Renflexis) | 100 mg | Vial powder for IV infusion | 493.0000 | Induction: 5 mg/kg at week 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 41.96 Year 2+: 35.21 | Year 1: 15,318 Year 2+: 12,853 |
Mirikizumab (Omvoh) | 300 mg/15 mL | Vial for IV infusion | 2,374.6600f | Induction: 300 mg IV infusion at weeks 0, 4, and 8 Maintenance: 200 mg SC injection every 4 weeks | 68.49 | 25,000 |
100 mg/1 mL | Autoinjector pen for SC injection or prefilled syringe for SC injection | 1,187.3300f | ||||
Ustekinumab (Stelara) | 130 mg/26.0 mL | Vial for IV infusion Prefilled syringe for SC injection | 2,080.000g | Induction (IV infusion): 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereafter Maintenance (SC injection): 90 mg every 8 weeks | Year 1: 86.53 Year 2+: 82.02 | Year 1: 31,584 Year 2+: 29,937 |
45 mg/0.5 mL 90 mg/1.0 mL | 4,593.1400 | |||||
Ustekinumab (Jamteki, Wezlana) | 45mg/0.5mL 90mg/mL 130mg/26mL | Prefilled syringe or vial for SC injection Vial for IV infusion | 2,755.8840 2,755.8840 1,248.0000 | Induction (IV infusion): 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereafter Maintenance (SC injection): 90 mg every 8 weeks | Year 1: 58.53 Year 2+: 49.21 | Year 1: 21,362 Year 2+: 17,962 |
Vedolizumab (Entyvio) | 300 mg | Vial for IV infusion | 3,571.9500d | Induction: 300 mg at weeks 0, 2, and 6 Maintenance: 300 mg every 8 weeks | Year 1: 72.40 Year 2+: 60.75 | Year 1: 26,425 Year 2+: 22,173 |
Vedolizumab SC (Entyvio) | 108 mg/0.68 mL | Prefilled syringe or pen for SC injection | 892.9800d | Induction: 300 mg by IV infusion at weeks 0 and 2 Maintenance: 108 mg by SC injection every 2 weeks | Year 1: 74.73 Year 2+: 60.75 | Year 1: 27,276 Year 2+: 22,173 |
b.i.d. = twice a day; SC = subcutaneous injection.
The comparators presented in the following table have been deemed to be appropriate based on feedback from a clinical expert and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 6, 2024),14 unless otherwise indicated, and do not include dispensing fees. Annual period assumes 365.25 days. Average weight is assumed to be 75 kg. Recommended doses are based on the respective product monographs.
Note: This table has not been copy-edited.
aSponsor’s submitted price.1
bEtrasimod product monograph.3
cAlberta Drug Benefit List.31
dOntario Exceptional Access Program.32
eUpadacitinib CDA-AMC Reimbursement Review.33
fMirikizumab CDA-AMC Reimbursement Review.17
gSaskatchewan Drug Plan formulary.34
hAssumes that CDA-AMC-participating drug plans would reimburse the lowest-cost option (i.e., patients who require 10 mg b.i.d. would receive 2 × 5 mg b.i.d.). If drug plans reimburse the higher-cost 10 mg tablet, the annual cost of generic tofacitinib would be $15,455 per patient (daily cost: $42.34).
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comment |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | No | The model excluded comparators relevant to the advanced therapy–experienced subgroup. |
Model has been adequately programmed and has sufficient face validity. | No | When all efficacy and safety inputs were set to be equivalent for all treatments, the sponsor’s model predicted an incremental difference in QALYs, which lacks face validity. These differences were due to the programmed length of the induction and maintenance phases in the sponsor’s model. The sponsor describes the induction phase as being modelled using a decision tree; however, the induction phase was programmed within the Markov trace, with an assigned output at the end of the induction period. The sponsor’s submitted model included numerous IFERROR statements, which makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors. |
Model structure is adequate for decision problem. | No | The relapsing-remitting nature of the disease and treatment paradigm with subsequent therapy is not accurately captured. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | The efficacy inputs in the model were hard coded and presampled from the sponsor’s NMA output. As such, CDA-AMC was unable to make changes to the efficacy inputs used in the probabilistic analyses. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | Because of the structure of the sponsor’s model, CDA-AMC was unable to use the sponsor’s model for probabilistic reanalyses. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details). | No | The sponsor’s budget impact analysis model included multiple sheets that reported different results, with no explanation provided to explain these differences. The lack of clear and transparent reporting impeded a thorough understanding of the model’s intricacies and findings. |
NMA = network meta-analysis; QALY = quality-adjusted life-year.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Model Structure, Maintenance Phase
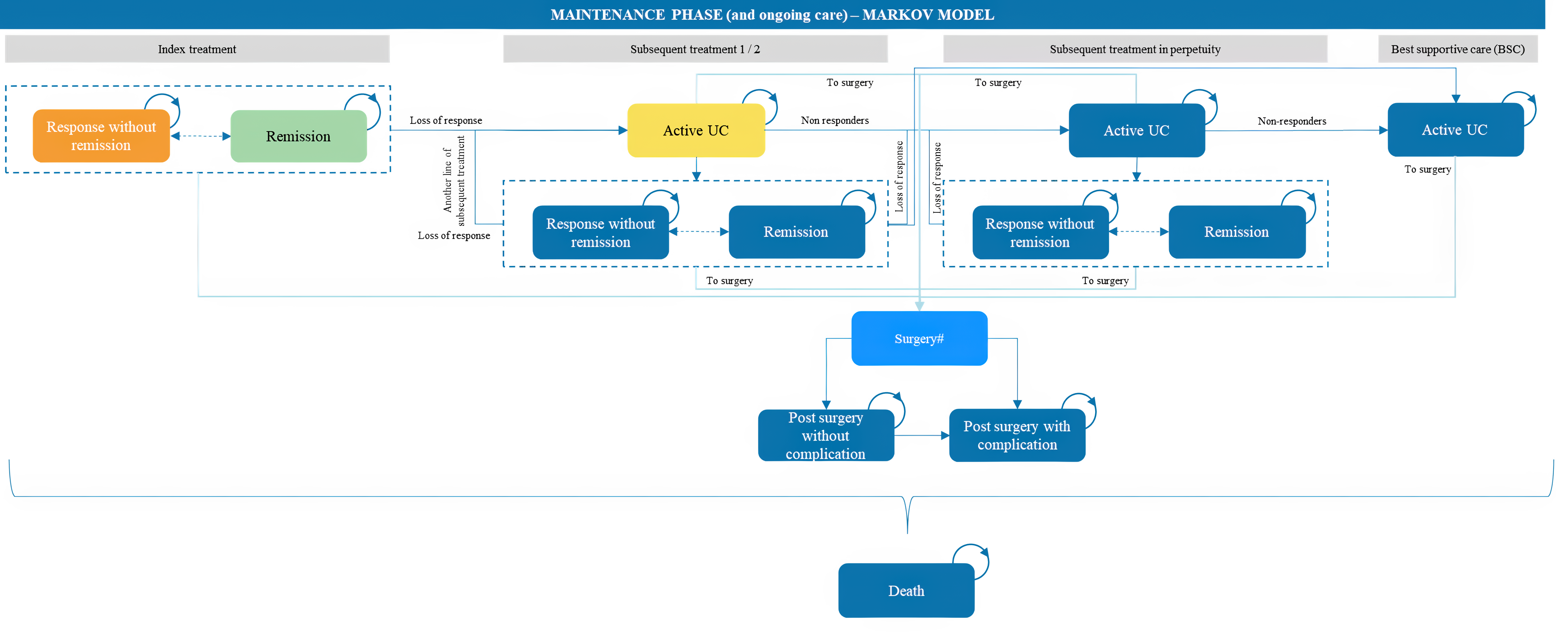
UC = ulcerative colitis.
Note: The sponsor’s model additionally included an induction phase (not pictured here).
Note: This figure has not been copy-edited.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Table 11: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results, Advanced Therapy–Naive Subgroup
Parameter | ETR | ADA | ADA-b | GOL | IFX | IFX-b | OZA | TOF | TOF-g | UPA | VED (IV) | VED (SC) | MIR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | |||||||||||||
Total | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 | 31.57 |
Discounted QALYs | |||||||||||||
Total | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 | 21.47 |
Discounted costs ($) | |||||||||||||
Total | 760,759 | 773,449 | 748,944 | 772,389 | 790,600 | 751,224 | 769,334 | 779,460 | 779,460 | 761,481 | 766,379 | 765,111 | 775,088 |
Drug costs | 333,789 | 346,479 | 321,974 | 345,419 | 363,629 | 324,253 | 342,363 | 352,490 | 352,490 | 334,511 | 339,409 | 338,141 | 348,118 |
Drug acquisition | 328,008 | 340,698 | 316,193 | 339,759 | 356,477 | 317,101 | 336,583 | 346,573 | 346,573 | 328,730 | 332,816 | 332,816 | 342,402 |
Administration | 5,781 | 5,781 | 5,781 | 5,659 | 7,152 | 7,152 | 5,781 | 5,917 | 5,917 | 5,781 | 6,592 | 5,325 | 5,716 |
Adverse event management | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 | 50,692 |
Disease management | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 | 373,565 |
Surgery | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 | 2,714 |
ADA = adalimumab; ADA-b = adalimumab biosimilar; BSC = best supportive care; ETR = etrasimod; GOL = golimumab; IFX = infliximab; IFX-b = infliximab biosimilar; LY = life-year; MIR = mirikizumab; OZA = ozanimod; QALY = quality-adjusted life-year; TOF = tofacitinib (brand); TOF-g = tofacitinib; UC = ulcerative colitis; UPA = upadacitinib; VED (IV) = vedolizumab (IV); VED (SC) = vedolizumab (subcutaneous).
Note: Deterministic analyses.
Note: This table has not been copy-edited.
Table 12: Disaggregated Summary of CDA-AMC’s Economic Evaluation Results, Advanced Therapy–Experienced Subgroup
Parameter | ETR | ADA | ADA-b | OZA | TOF | TOF-g | UPA | VED (IV) | VED (SC) | MIR |
|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | ||||||||||
Total | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 | 29.75 |
Discounted QALYs | ||||||||||
Total | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 | 19.94 |
Discounted costs ($) | ||||||||||
Total | 725,230 | 730,971 | 717,458 | 721,379 | 759,020 | 759,020 | 721,149 | 721,022 | 720,398 | 718,696 |
Drug costs | 315,060 | 320,801 | 307,287 | 311,209 | 348,850 | 348,850 | 310,979 | 310,851 | 310,228 | 308,526 |
Drug acquisition | 309,429 | 315,169 | 301,656 | 305,577 | 343,081 | 343,081 | 305,347 | 305,061 | 305,061 | 302,963 |
Administration | 5,632 | 5,632 | 5,632 | 5,632 | 5,769 | 5,769 | 5,632 | 5,790 | 5,167 | 5,563 |
Adverse event management | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 | 48,531 |
Disease management | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 | 359,011 |
Surgery | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 | 2,628 |
ADA = adalimumab; ADA-b = adalimumab biosimilar; BSC = best supportive care; ETR = etrasimod; LY = life-year; MIR = mirikizumab; OZA = ozanimod; QALY = quality-adjusted life-year; TOF = tofacitinib (brand); TOF-g = tofacitinib; UC = ulcerative colitis; UPA = upadacitinib; VED (IV) = vedolizumab (IV); VED (SC) = vedolizumab (subcutaneous).
Note: Deterministic analyses.
Note: This table has not been copy-edited.
Table 13: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results, Advanced Therapy–Naive Subgroup
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Tofacitinib | 811,624 | 21.95 | Reference |
Etrasimod | 838,905 | 22.63 | 40,215 | |
Infliximab biosimilar | 828,858 | 21.96 | Extendedly dominated by tofacitinib, etrasimod | |
Ozanimod | 835,270 | 21.79 | Dominated by tofacitinib | |
Mirikizumab | 838,371 | 21.88 | Dominated by tofacitinib | |
Adalimumab biosimilar | 840,522 | 22.01 | Dominated by etrasimod | |
Upadacitinib | 841,828 | 21.98 | Dominated by etrasimod | |
Golimumab | 849,180 | 21.91 | Dominated by tofacitinib | |
Vedolizumab SC | 849,555 | 22.26 | Dominated by etrasimod | |
Vedolizumab IV | 852,085 | 22.25 | Dominated by etrasimod | |
Adalimumab | 853,492 | 22.01 | Dominated by adalimumab biosimilar | |
Infliximab | 867,796 | 21.92 | Dominated by infliximab biosimilar | |
Tofacitinib (brand) | 875,701 | 21.95 | Dominated by tofacitinib | |
Sponsor’s base case, corrected (deterministic) | Infliximab biosimilar | 742,213 | 20.70 | Reference |
Adalimumab biosimilar | 742,847 | 20.86 | 3,973 | |
Ozanimod | 742,811 | 20.58 | Dominated by infliximab biosimilar | |
Mirikizumab | 743,866 | 20.66 | Dominated by infliximab biosimilar | |
Etrasimod | 746,145 | 21.31 | 7,383 | |
Adalimumab | 755,435 | 20.86 | Dominated by adalimumab biosimilar | |
Upadacitinib | 755,605 | 20.66 | Dominated by infliximab biosimilar | |
Vedolizumab (SC) | 758,259 | 21.02 | Dominated by etrasimod | |
Vedolizumab (IV) | 760,609 | 21.01 | Dominated by etrasimod | |
Tofacitinib | 761,077 | 20.74 | Dominated by adalimumab biosimilar | |
Golimumab | 762,040 | 20.68 | Dominated by infliximab biosimilar | |
Infliximab | 778,301 | 20.70 | Dominated by infliximab biosimilar | |
CDA-AMC base case: 1 + 2 + 3 (deterministic) | Adalimumab biosimilar | 748,944 | 21.47 | Reference |
Infliximab biosimilar | 751,224 | 21.47 | Dominated by adalimumab biosimilar | |
Etrasimod | 760,759 | 21.47 | Dominated by adalimumab biosimilar | |
Upadacitinib | 761,481 | 21.47 | Dominated by adalimumab biosimilar | |
Vedolizumab (SC) | 765,111 | 21.47 | Dominated by adalimumab biosimilar | |
Vedolizumab (IV) | 766,379 | 21.47 | Dominated by adalimumab biosimilar | |
Ozanimod | 769,334 | 21.47 | Dominated by adalimumab biosimilar | |
Golimumab | 772,389 | 21.47 | Dominated by adalimumab biosimilar | |
Adalimumab | 773,449 | 21.47 | Dominated by adalimumab biosimilar | |
Mirikizumab | 775,088 | 21.47 | Dominated by adalimumab biosimilar | |
Tofacitinib | 779,460 | 21.47 | Dominated by adalimumab biosimilar | |
Infliximab | 790,600 | 21.47 | Dominated by adalimumab biosimilar |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. Refer to Table 6 for the steps taken to derive the CDA-AMC base case.
Table 14: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results, Advanced Therapy–Experienced Subgroup
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Tofacitinib | 721,886 | 20.96 | Reference |
Etrasimod | 791,796 | 21.16 | Extendedly dominated by tofacitinib and upadacitinib | |
Adalimumab biosimilar | 782,036 | 20.89 | Dominated by tofacitinib | |
Ozanimod | 800,667 | 20.84 | Dominated by tofacitinib | |
Vedolizumab SC | 802,758 | 20.97 | Dominated by etrasimod | |
Adalimumab | 805,055 | 20.89 | Dominated by adalimumab biosimilar | |
Vedolizumab IV | 805,541 | 20.97 | Dominated by vedolizumab SC | |
Upadacitinib | 807,120 | 21.91 | 89,186 | |
Tofacitinib (branded) | 818,546 | 20.96 | Dominated by tofacitinib | |
Mirikizumab | 824,883 | 20.93 | Dominated by tofacitinib | |
Sponsor’s base case, corrected (deterministic) | Adalimumab biosimilar | 700,519 | 19.67 | Reference |
Ozanimod | 706,179 | 19.54 | Dominated by adalimumab biosimilar | |
Etrasimod | 709,424 | 19.81 | Extendedly dominated by adalimumab biosimilar and upadacitinib | |
Vedolizumab (SC) | 712,834 | 19.62 | Dominated by adalimumab biosimilar | |
Adalimumab | 714,038 | 19.67 | Dominated by adalimumab biosimilar | |
Vedolizumab (IV) | 714,513 | 19.62 | Dominated by vedolizumab SC | |
Mirikizumab | 715,681 | 19.63 | Dominated by adalimumab biosimilar | |
Tofacitinib | 715,724 | 19.57 | Dominated by adalimumab biosimilar | |
Upadacitinib | 721,904 | 21.38 | 12,557 | |
CDA-AMC base case: 1 + 2 + 3 (deterministic) | Adalimumab biosimilar | 717,458 | 19.94 | Reference |
Mirikizumab | 718,696 | 19.94 | Dominated by adalimumab biosimilar | |
Vedolizumab (SC) | 720,398 | 19.94 | Dominated by adalimumab biosimilar | |
Vedolizumab (IV) | 721,022 | 19.94 | Dominated by vedolizumab (SC) | |
Upadacitinib | 721,149 | 19.94 | Dominated by adalimumab biosimilar | |
Ozanimod | 721,379 | 19.94 | Dominated by adalimumab biosimilar | |
Etrasimod | 725,230 | 19.94 | Dominated by adalimumab biosimilar | |
Adalimumab | 730,971 | 19.94 | Dominated by adalimumab biosimilar | |
Tofacitinib | 759,020 | 19.94 | Dominated by adalimumab biosimilar |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. Refer to Table 6 for the steps taken to derive the CDA-AMC base case.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 15: Summary of Key Take Aways
Key take aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor’s submitted budget impact analysis (BIA),35 intended to assess the expected incremental budget impact of reimbursing etrasimod for the treatment of moderately to severely active UC in patients who are eligible for treatment with advanced therapies, while the Health Canada indication for etrasimod is for moderately to severely active UC who had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment. The BIA was conducted from the public drug program perspective over a 3-year time horizon (2025 to 2027).
A claims-based approach was used by the sponsor to estimate the number of annual claims for the eligible population. Claims data were obtained from IQVIA PharmaStat database36 (June 2019 to July 2023) for advanced therapies (adalimumab [brand and biosimilar], infliximab [brand and biosimilar], golimumab, tofacitinib [brand name and generic], vedolizumab). The proportion of claims specific to UC for each treatment was based on data internal to Pfizer (12% of adalimumab claims, 32% of infliximab, 2% of golimumab, 6% of tofacitinib, 45% of vedolizumab).35 The sponsor standardized the duration of all claims to a common duration (30 days). Market share for ozanimod, upadacitinib, and mirikizumab were based on sponsor assumption. The sponsor assumed that most of the market share of etrasimod will be captured from infliximab biosimilars, adalimumab biosimilars, and tofacitinib. The BIA included costs related to drug acquisition, dispensing fees, and pharmacy markups. The sponsor estimated the cost per 30-day claim using the cost information obtained from the IQVIA PharmaStat database for adalimumab [brand and biosimilar], infliximab [brand and biosimilar], golimumab, tofacitinib [brand and generic], vedolizumab. For etrasimod, ozanimod, upadacitinib, and mirikizumab, the sponsor estimated the cost per 30-day claim using publicly available list prices and recommended dosages obtained from respective monographs. Key inputs to the BIA are documented in Table 16.
Table 16: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) |
|---|---|
Target population | |
Number of claims for moderately to severely active UCa | 63,861 / 74,066 / 84,271 |
Market uptake (3 years) | |
Uptake (reference scenario) Etrasimod Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Tofacitinib (brand) Tofacitinib Vedolizumab Upadacitinib Ozanimod Mirikizumab | 0.0% / 0.0% / 0.0% 0.0% / 0.0% / 0.0% 23.6% / 23.2% / 21.4% 0% / 0% / 0% 28.2% / 26.4% / 23.5% 1.0% / 0.9% / 0.7% 0.1% / 0.1% / 0.1% 23.1% / 22.7% / 20.9% 22.4% / 20.4% / 17.8% 0.6% / 2.1% / 5.2% 0.6% / 2.1% / 5.2% 0.6% / 2.1% / 5.2% |
Uptake (new drug scenario) Etrasimod Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Tofacitinib (brand) Tofacitinib Vedolizumab Upadacitinib Ozanimod Mirikizumab | 0.6% / 2.1% / 5.2% 0.0% / 0.0% / 0.0% 23.5% / 22.7% / 20.3% 0.0% / 0.0% / 0.0% 28.0% / 25.9% / 22.3% 1.0% / 0.9% / 0.7% 0.1% / 0.1% / 0.1% 23.0% / 22.2% / 19.8% 22.2% / 20.0% / 16.8% 0.6% / 2.1% / 4.9% 0.6% / 2.1% / 4.9% 0.6% / 2.1% / 4.9% |
Cost of treatment (per patient, annual)b | |
Etrasimod Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Tofacitinib (branded) Tofacitinib Vedolizumab Upadacitinib Ozanimod Mirikizumab | $16,822 $25,369 $23,016 $47,783 $44,385 $32,658 $17,185 $5,015 $41,134 $19,193 $26,657 $30,780 |
UC = ulcerative colitis.
aThe sponsor estimated the number of annual claims, not the number of eligible patients.
bCosts estimated by the sponsor using cost per 30-day claim and includes dispensing fees and pharmacy markup.
Summary of the Sponsor’s BIA Results
The sponsor estimated that reimbursing etrasimod for the treatment of moderately to severely active UC in patients who are eligible for treatment with advanced therapies will be cost-saving to the public drug plans, with an estimated savings of $5,953,968 over the first 3 years (year 1: $361,421; year 2: $1,519,959; year 3: $4,072,588).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of a claims-based approach to estimate market size and treatment costs is uncertain: The sponsor estimated the market size in terms of the number of claims using historic public claims data for most comparators. However, the included comparators are also indicated for the treatment of conditions such as rheumatoid arthritis, ankylosing spondylitis, Crohn disease, and plaque psoriasis, and the information about the indication is not available. It is also unclear how the claims align with the Health Canada indicated population (i.e., moderately to severely active UC who had an inadequate response, lost response, or were intolerant to either conventional therapy or an advanced treatment). CDA-AMC notes that previous submissions for this indication have adopted an epidemiologic approach,16 which may overcome some of the limitations associated with a claims-based approach. The rationale behind the sponsor's chosen approach was not provided.
The sponsor similarly estimated treatment costs using claims data, via a cost-per-claim approach, and assumed claim-to-claim displacement between etrasimod and comparators. CDA-AMC notes that, because the treatments have different dosage frequencies and durations, this approach introduces additional uncertainty. The sponsor did not estimate treatment costs separately for induction and maintenance phases, and it is unclear if patients who received an induction dose also accrued the cost of maintenance treatment in the sponsor’s adopted approach. There are also treatments (i.e., upadacitinib, ozanimod, mirikizumab) that currently do not have claims data available, and the sponsor estimated market shared based on assumption; this introduces additional uncertainty into the estimated population. Finally, CDA-AMC notes that the sponsor estimated the number of claims per year, not the number of patients eligible for treatment. It would be more appropriate to estimate of the number of patients eligible for the drug under review than the number of annual claims.
CDA-AMC was unable to address the limitations of a claims-based approach.
Additional limitations were identified but were not considered to be key limitations. Given the limitations associated with the use of a claims-based approach by the sponsor, CDA-AMC was unable to address these limitations.
Market uptake of etrasimod is uncertain: The sponsor’s submitted base case assumed that etrasimod would capture 5.2% of the market share by year 3 in patients with moderate to severe UC. Clinical expert feedback obtained by CDA-AMC for this review noted the potential for more rapid uptake of etrasimod among the advanced therapy–naive subgroup. As the sponsor’s budget impact was not estimated separately for advanced therapy–naive and –experienced subgroups, the sponsor’s estimates of market displacement are uncertain.
Market share of comparators is uncertain: The sponsor allocated market share to upadacitinib, mirikizumab, and ozanimod. Upadacitinib and mirikizumab are currently under ongoing negotiations with pCPA for UC37,38 and it is unknown whether or when they will be reimbursed by public formularies. While ozanimod negotiations with pCPA concluded with a letter of intent, it has yet to be reimbursed across all CDA-AMC-participating jurisdictions.30 The clinical expert consulted for this review by CDA-AMC that the estimated market share for these treatments by the sponsor may have been underestimated.
The price of drugs paid by public drug plans is uncertain: The sponsor’s analysis was based on publicly available list prices and sponsor-submitted prices from previous CDA-AMC reviews for all comparators. Adalimumab biosimilar, infliximab biosimilar, and vedolizumab have gone through negotiations at pCPA, and the prices paid by public drug plans are not known.
Dispensing and markup fees are included: The sponsor included a dispensing fee of $10.56 and a pharmacy markup of 7.6% for all treatments. Dispensing fees and markups vary across jurisdictions; however, the sponsor did not incorporate jurisdiction-specific fees. Consequently, the estimated budget impact may not accurately reflect the actual costs incurred by public health care payers.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CDA-AMC expects that the budget impact of reimbursing etrasimod for the treatment of moderate to severe UC will be sensitive to more reliable inputs which may affect the market size calculation, uptake and displacement of comparators by etrasimod, and the prices of advanced therapies for UC paid for by the public drug plans. Whether the introduction of etrasimod will be cost savings and the extent of any savings realized by the drug plans is therefore highly uncertain.
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 134,566,773 | 154,655,371 | 175,995,554 | 196,350,785 | 527,001,709 |
New drug | 134,566,773 | 154,293,949 | 174,475,595 | 192,278,197 | 521,047,741 | |
Budget impact | 0 | −361,421 | −1,519,959 | −4,072,588 | −5,953,968 |
BIA = budget impact analysis.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.