CADTH Reimbursement Review
Alpha1-Proteinase Inhibitor (Human) (Zemaira)
Sponsor: CSL Behring Canada, Inc.
Therapeutic area: Severe alpha1-proteinase inhibitor deficiency
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
A1-PI
alpha1-proteinase inhibitor
AAT
alpha1-antitrypsin
AATD
alpha1-antitrypsin deficiency
AE
adverse event
ANCOVA
analysis of covariance
CI
confidence interval
COPD
chronic obstructive pulmonary disease
DLCO
diffusing capacity of the lungs for carbon monoxide
FEV1
forced expiratory volume in the first second
FRC
functional residual capacity
FVC
forced vital capacity
ISWT
incremental shuttle walking test
ITT
intention to treat
OLE
open-label extension
P15
15th percentile of the lung density
PP
per protocol
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SGRQ
St. George’s Respiratory Questionnaire
TLC
total lung capacity
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Alpha1-proteinase inhibitor (human) (Zemaira), 60 mg/kg body weight administered by IV infusion once weekly |
Indication | Maintenance treatment of adults with severe alpha1-proteinase inhibitor deficiency and clinical evidence of emphysema |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 21, 2016 |
Sponsor | CSL Behring Canada, Inc. |
NOC = Notice of Compliance.
Introduction
Alpha1-proteinase inhibitor (A1-PI) deficiency, also known as alpha1-antitrypsin deficiency (AATD), is a genetic disorder characterized by low serum concentrations of A1-PI that has major physiologic consequences in the lower respiratory tract.1 A deficiency in endogenous A1-PI may subject an individual to a life-long, progressive loss of lung tissue and predisposes patients to early-onset emphysema.1 However, clinical expression is variable and not all individuals with A1-PI deficiency will develop overt disease. As is seen with chronic obstructive pulmonary disease (COPD) unrelated to this deficiency, patients may present with breathlessness, cough, wheeze, decreased exercise tolerance, and impactful exacerbations. Typically, the progression of lung disease in patients with A1-PI deficiency is gradual. Studies show there is often a delay of years before patients are diagnosed accurately. Delayed diagnosis of A1-PI deficiency leads to a worsening of symptoms and deterioration of functional status as well as a decreased life expectancy.1
The clinical experts consulted by CADTH noted that patients with A1-PI deficiency and emphysema receive standard therapies for COPD up to and including lung transplant for the most severely affected. However, a specific treatment strategy for A1-PI deficiency is to augment the patient’s native A1-PI with purified A1-PI from pooled donated blood. The goal of therapy with a protease inhibitor is to lessen the loss of lung tissue (as may be quantified by CT densitometry). This would be seen as a disease-modifying therapy that could be considered as a first-line treatment for patients with emphysema and A1-PI deficiency, administered in conjunction with the standard of care for COPD.
A1-PI (human) (Zemaira) is a lyophilized preparation of highly purified human A1-PI. Derived from pooled human plasma, it is administered intravenously once per week at the recommended dose of 60 mg/kg body weight.2 A1-PI (human) has a Health Canada indication for the maintenance treatment of adults with severe A1-PI deficiency and clinical evidence of emphysema. Severe A1-PI deficiency includes, but is not limited to, the PiZZ, PiZ(null), Pi(null,null), and PiSZ genotypes. Under the indication, patients are to be receiving optimal pharmacologic and non-pharmacologic treatment and showing evidence of lung disease, such as a forced expiratory volume in the first second (FEV1) below predicted, lower diffusion capacity, impaired walking capacity, or an increased number of exacerbations, as evaluated by a health care professional experienced in the treatment of A1-PI deficiency.2
The objective of this report is to perform a systematic review of the beneficial and harmful effects of Zemaira for the maintenance treatment of adults with severe A1-PI deficiency and clinical evidence of emphysema.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Alpha-1 Canada submitted the patient input for this review. Alpha-1 Canada is a national non-profit organization committed to advocating on behalf of Canadians affected by AATD and providing education and support to patients, caregivers, and the health care community. The submission was based on 2 virtual focus groups conducted in March 2021, 2 semi-structured interviews conducted over the phone in June 2021, 3 online surveys distributed between April and May 2021, and a single-question survey emailed to Canadian respirologists in May 2021. A total of 143 respondents (45 patients receiving A1-PI augmentation therapy, 53 patients not receiving therapy, 16 caregivers, and 29 Canadian respirologists) plus 2 families living with AATD were included in the patient input.
Respondents indicated that the physical manifestation of AATD impacts many aspects of their lives, ranging from their employment, relationships, extracurricular activities, and ability to do day-to-day tasks to their overall mental health. In areas where there is no publicly funded access to treatment, patients are weighing the steps they are willing to take to access therapy, such as continuing to work past retirement age to be eligible for private insurance, uprooting their lives to relocate to a province that offers coverage, or participating in clinical trials. Patients highlighted the costs to the health care system when they are unable to access treatment: they require inhalers to manage the symptoms of AATD, undergo frequent lung function tests, experience hospitalizations during exacerbations, and undergo lung transplant. The other major challenge for patients with A1-PI deficiency is the need to demonstrate deteriorated lung function before becoming eligible for augmentation therapy. Many felt they were doing additional damage to their lungs and compromising their quality of life while they waited to become eligible.
When patients are on A1-PI augmentation therapy, they are able to stabilize their lung function. Patients perceive this as the most important outcome in effective treatment because it is associated with their ability to perform activities of daily living and fully participate in their communities and with their families. Patients with A1-PI deficiency did not feel that any disadvantages were worth noting compared with the possibility of augmentation therapy improving their quality and longevity of life.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH for the purpose of this review indicated there is currently an unmet need, considering that no treatment other than A1-PI replacement can prevent the loss of lung tissue. A1-PI replacement is currently available in Canada mainly through private insurers and exceptional access programs. This treatment is considered disease modifying and would be a first-line treatment for any patient with emphysema and A1-PI deficiency and used in addition to standard of care for COPD. Since the drug is used to prevent the rapid progression of emphysema, there are no specific outcome parameters to measure to assess response to treatment, as there are no factors that would be used as a stopping rule, other than severe AEs. As the goal of augmentation therapy is to prevent or decrease the rate of further tissue damage, it is expected that some patients will keep deteriorating despite treatment. However, it is very likely that these patients would have deteriorated even more without A1-PI replacement, thus limiting the usefulness of assessing response to re-treatment using lung function or number of exacerbations in this particular instance.
Clinician Group Input
One clinician group, the Canadian Thoracic Society, provided input that is in line with the views from the input provided by the clinical experts consulted by CADTH. The meaningful impact of augmentation therapy and its potential role in clinical use has been acknowledged in statements by the Canadian Thoracic Society.
Drug Program Input
The drug program implementation questions were aimed at gaining insight from the clinical experts consulted by CADTH as to whether laboratory tests to check serum AATD level are accessible. The clinical experts consulted by CADTH indicated that these genetic tests are needed to confirm genotypes and should be done systematically, and that the technology is readily available. The clinical experts also noted that clinically important emphysema should be present before initiating treatment with A1-PI (human). It was also noted that there is no evidence that A1-PI (human) would work in the presence of continued smoking. The drug plans also asked whether patients with other (confirmed) genotypes receive a clinical benefit similar to the benefit for patients with the PiZZ genotype. The clinical experts noted there is a lack of data in patients with an SZ or MZ genotype, and that Zemaira should be made available for patients with a null, ZZ, or SZ genotype with evidence of lung disease. The drug plans also questioned whether the reimbursement criteria should be similar between Zemaira and Prolastin-C; the clinical experts noted that reimbursement criteria (initiation, discontinuation, and prescribing) should be identical for both Zemaira and Prolastin-C.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One published, manufacturer-sponsored, double-blind randomized controlled trial (RCT) was included in the systematic review: RAPID (n = 180).3,4 The trial evaluated the superiority of A1-PI (human) compared with placebo on the progression of disease in patients with emphysema with A1-PI deficiency and a reduced lung function. Disease progression was assessed by the decline of lung density, as measured by CT. A1-PI was administered at a dosage of 60 mg/kg through IV infusion once weekly for 24 months.
Patients in the trial had a mean age of 53 years. All patients were White. The mean FEV1 was 47% of predicted. Mean duration of disease was between 5 and 6 years. The majority of patients had a medical history before baseline as well as concurrent illness and concomitant medication.
Efficacy Results
A1-PI (human) was associated with a reduced rate of decline in lung density after 24 months compared with placebo in patients with emphysema with A1-PI deficiency and a reduced lung function when CT scans were taken at a full inspiration state (0.74 g/L; 95% confidence interval [CI], 0.06 to 1.42; 1-sided P = 0.017). CT lung densitometry measurements have been validated as a primary clinical end point for clinical study designs in monitoring emphysema progression in AATD. According to patient input, stabilizing lung function is perceived as the most important outcome in effective treatment because it is associated with the ability to perform activities of daily living. However, measurement of lung density is not used in clinical practice to assess disease progression; therefore, it is unknown how the slower decrease observed in RAPID in terms of lung density translates into better quality of life. In addition, the interpretation of the findings is affected by the fact that it was not clear at which specific inspiration state the measure was to be taken for the primary analysis. Results using other inspiration state measures also showed a slower decline in lung density with active treatment compared with placebo over a 24-month period, but the differences between groups were of smaller magnitude and did not reach statistical significance. From a statistical perspective, this is a major limitation, especially since the analysis was not controlled for multiplicity. However, according to the clinical experts consulted by CADTH, the most reliable way to measure lung density is at a full inspiration state, which is referred to as total lung capacity (TLC). When the lungs are full of air, more lung tissue is visible on the CT scan image and, therefore, the measurement obtained is considered more reliable.
Other important clinical outcomes such as exacerbations, symptoms, and function were reported as secondary outcomes; however, the differences between groups did not reach statistical significance for any of these outcomes other than the FEV1 to FVC ratio, where the treatment difference for the percentage change from baseline (day 1) to month 24 in the FEV1 to FVC ratio for observed values revealed a change of 4.24% (95% CI, −8.04 to −0.45; P = 0.029) in favour of placebo when compared with A1-PI (human); however, the FEV1 to FVC ratio results should be interpreted with caution due to the risk of inflated type I error.
Harms Results
Virtually all patients in both treatment groups experienced at least 1 adverse event (AE); however, discontinuation due to AEs was low, suggesting the harm profile might be considered acceptable. Respiratory-related AEs were commonly reported and, in some cases, were numerically higher with A1-PI (human) than with placebo; however, this might be a random fluctuation due to the small sample size. Serious AEs (SAEs) were frequently reported, and the incidence was similar between treatment groups. No cases of severe hypersensitivity were reported in the trial. One patient in the A1-PI (human) treatment arm died over the study period due to respiratory failure. In the placebo arm, 3 patients died over the study period from sepsis, pneumonia, and metastatic breast cancer.
Table 2: Summary of Key Results From the RAPID Trial
Key result | A1-PI (N = 93) | Placebo (N = 87) |
|---|---|---|
Change in the physiologically adjusted P15 lung density: Treatment comparison for annual rate of change (primary analysis in the trial)a | ||
Inspiration state: Mean of TLC and FRC | ||
Baseline, mean (SD) | 46.6 (15.6) | 49.8 (15.1) |
End of treatment time point (month 24), mean (SD) | 44.4 (15.5) | 45.5 (13.9) |
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.50 (0.22) | −2.12 (0.24) |
Difference between treatmentsb | 0.62 g/L | |
95% CI, 1-sided P value | −0.02 to 1.26; P = 0.029 | |
Inspiration state: TLC | ||
Baseline, mean (SD) | 45.5 (15.8) | 48.9 (15.5) |
End of treatment time point (month 24), mean (SD) | 43.6 (16.0) | 43.9 (13.8) |
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.45 (0.23) | −2.19 (0.25) |
Difference between treatmentsb | 0.74 g/L | |
95% CI, 1-sided P value | 0.06 to 1.42; P = 0.017 | |
Inspiration state: FRC | ||
Baseline, mean (SD) | 47.6 (15.7) | 50.7 (15.0) |
End of treatment time point (month 24), mean (SD) | 45.3 (15.3) | 46.8 (13.8) |
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.55 (0.24) | −2.02 (0.26) |
Difference between treatmentsb | 0.48 g/L | |
95% CI, 1-sided P value | −0.22 to 1.18; P = 0.090 | |
Pulmonary function: Treatment comparisons for percent change from baseline to month 24 | ||
FEV1 | ||
Difference between treatmentsc | −2.24 | |
95% CI, 2-sided P value | −5.73 to 1.26; P = 0.208 | |
FEV1% of predicted | ||
Difference between treatmentsc | −2.26 | |
95% CI, 2-sided P value | −5.79 to 1.26; P = 0.207 | |
FEV1 to FVC ratio | ||
Difference between treatmentsc | −4.24 | |
95% CI, 2-sided P value | −8.04 to −0.45; P = 0.029 | |
Exacerbations | ||
Rate of exacerbations per patient-year | ||
Estimated exacerbation ratiod | 1.26 | |
95% CI, 2-sided P value | 0.92 to 1.74; P = 0.152 | |
Symptoms and function | ||
Incremental shuttle walking test: Treatment comparison for change from baseline to month 24 | ||
Difference between treatmentse | −13.1 m | |
95% CI, 2-sided P value | −49.3 to 23.1; P = 0.477 | |
SGRQ symptom scale: Treatment comparison for change from baseline to month 24f | ||
Difference between treatmentsg | −1.11 | |
95% CI, 2-sided P value | −6.20 to 3.99; P = 0.669 | |
Harms | ||
AEs | 92 (98.9) | 86 (98.9) |
SAEs | 28 (30.1) | 28 (32.2) |
WDAEs | 1 (1.1) | 5 (5.7) |
Deaths | 1 (1.1) | 3 (3.4) |
A1-PI = alpha1-proteinase inhibitor; AE = adverse event; ANCOVA = analysis of covariance; CI = confidence interval; FEV1 = forced expiratory volume in the first second; FRC = functional residual capacity; FVC = forced vital capacity; ITT = intention to treat; P15 = 15th percentile of the lung density; SAE = serious adverse event; SD = standard deviation; SE = standard error; SGRQ = St. George’s Respiratory Questionnaire; TLC = total lung capacity; WDAE = withdrawal due to adverse event.
aThe annual rate of change in physiologically adjusted P15 was analyzed using CT scan data taken at both TLC and FRC inspiration states. For the combined analysis, both inspiration states were included as fixed effects in the primary efficacy model simultaneously (i.e., TLC and FRC states combined) as opposed to the separate analyses of the CT scans at TLC and FRC inspiration states, which were investigated by applying the primary model without the fixed effect for inspiration state.
bTreatment comparison for annual rate of change in physiologically adjusted P15 (g/L) at TLC and FRC states combined and separately based on a random regression model (ITT population). Statistical significance level of P = 0.025.
cTreatment comparison for percentage change from baseline to month 24 in key spirometry variables for observed values (ANCOVA) (ITT population).
dTreatment comparison for rate of exacerbations per patient-year (negative binomial regression model) (ITT population).
eTreatment comparison for change from baseline to month 24 in exercise capacity test – distance walked (m) for observed values (ANCOVA) (ITT population).
fHigher scores in the SGRQ indicate more limitations in terms of overall health, daily life, and perceived well-being in patients with obstructive airway disease.
gTreatment comparison for change from baseline to month 24 in SGRQ symptoms score for observed values (ANCOVA) (ITT population).
Source: Clinical Study Reports for the RAPID trial.5
Critical Appraisal
Though RAPID may be considered methodologically rigorous, the interpretation of the findings is affected by the small sample size and by the fact that it was not clear at which specific inspiration state the measure was to be taken for the primary analysis. From a statistical perspective, this is a major limitation, especially since the analysis was not controlled for multiplicity. However, according to the clinical experts consulted by CADTH, the most reliable way to measure lung density is at a full inspiration state, which is referred to as TLC, where statistical significance is reached. The trial population appeared similar to patients seen in clinical practice by the clinical experts consulted by CADTH; however, due to the limitations, such as small sample sizes, the real-world effectiveness of A1-PI (human) in Canadian patients may vary from what was observed in the trial. The strength of the evidence was reduced by the lack of controlled long-term data on efficacy and safety and the lack of trials comparing the clinical outcomes of A1-PI (human) with the other active treatment available.
Other Relevant Evidence
Additional relevant evidence addressing important gaps in the evidence was considered. One open-label extension (OLE) study from RAPID, RAPID-OLE (n = 140),6 was an extension study that collected long-term data on the safety and efficacy of A1-PI (human) on the progression of disease in patients with emphysema with A1-PI deficiency who had completed the 2-year treatment and observation periods in the RAPID study. Despite the limitations associated with the open-label, uncontrolled trial design, the findings suggested that the efficacy of A1-PI (human) was sustained in the long-term.
A noninferiority biochemical efficacy trial, Study 2002 (n = 44),7 suggested that Zemaira was considered to be noninferior to Prolastin throughout a 10-week blinded phase, based on the mean steady-state trough serum antigenic A1-PI levels in adult patients with a diagnosis of A1-PI deficiency and clinical evidence of emphysema.
One survival analysis8 evaluated the efficacy of A1-PI (human) plus standard therapy in the US compared with standard therapy alone in the UK on the outcomes of survival and lung transplant in adult patients with A1-PI deficiency and evidence of lung disease. Findings suggested that A1-PI (human) treatment was associated with benefits in terms of survival and time to lung transplant; however, the limitations inherent with the database study design and the differences between groups, especially in terms of the patient population included in the 2 treatment groups, highly affect our level of confidence in the evidence.
Conclusions
The results of RAPID suggest that A1-PI (human) was associated with a reduced rate in the validated primary outcome of decline in lung density after 24 months compared with placebo in patients with emphysema with A1-PI deficiency and a reduced lung function when CT scans were taken at a full inspiration state. This shows that treatment with A1-PI (human) might preserve lung tissue in these patients; however, as lung density is not used in clinical practice for the assessment of disease progression, the extent of how these findings translate into clinical benefits for patients in real life is unknown. RAPID was not informative regarding the efficacy of A1-PI (human) on the outcomes of survival and lung transplant because the sample size was relatively small and the follow-up was of limited duration for a slowly progressive disease. Other important clinical outcomes such as exacerbations and health-related quality of life were reported as secondary outcomes, for which no difference was seen. No major safety signal was identified. The additional evidence assessed to address important gaps in the evidence suggested a long-term maintenance of effect of more than 48 months as well as a similar biochemical efficacy compared with Prolastin; however, the level of confidence in the evidence is highly affected by several limitations, including the open-label uncontrolled trial design of the long-term extension study.
Introduction
Disease Background
A1-PI deficiency, also known as AATD, is a common genetic disorder; the prevalence of the genotypes associated with A1-PI deficiency is generally considered to be around 1 in 5,000 people.9,10 A1-PI deficiency is characterized by low serum concentrations of A1-PI, a serine anti-protease produced in the liver but that appears to have its most important physiologic role in the protection of the lung parenchyma from endogenous elastases released by the neutrophils.1,11 A deficiency in endogenous A1-PI may expose an individual to a life-long risk of lung tissue loss; thus, it predisposes patients to early-onset emphysema.1 As is also seen with COPD unrelated to this deficiency, patients present with breathlessness, cough, wheeze, decreased exercise tolerance, and impactful exacerbations. Exacerbations, most often triggered by infections, produce worsening of symptoms and may accelerate loss of lung tissue.
The progression of lung disease in patients with A1-PI deficiency is typically gradual. The clinical experts consulted by CADTH noted there is typically a delay in arriving at a specific diagnosis and patients are often treated as having asthma or non-alpha COPD, or are not recognized as having a significant pulmonary disorder. An appropriate diagnosis is achieved through genetic tests to confirm genotypes. Severe A1-PI deficiency includes, but is not limited to, the PiZZ, PiZ(null), Pi(null,null), and PiSZ genotypes. Failure to diagnose A1-PI deficiency in a timely manner may prevent initiation of appropriate therapies, and that delay can lead to a worsening of symptoms and deterioration of functional status as well as a decreased life expectancy.1
Standards of Therapy
Patients with A1-PI deficiency and emphysema receive standard therapies used for COPD that is unrelated to A1-PI deficiency. The clinical experts consulted by CADTH for the purpose of this review mentioned using treatment with short- and long-acting bronchodilators, pulmonary rehabilitation, antibiotics and steroids (as needed for exacerbation), inhaled corticosteroids in select individuals, and smoking cessation therapy for current smokers. Patients with severe symptoms may undergo evaluation for lung transplant.
Specific to A1-PI deficiency is the replacement of the protease inhibitor with an A1-PI. This treatment option is currently available in Canada mainly through private insurers and special access programs. The goal of therapy with a protease inhibitor is to lessen the loss of lung tissue, as assessed by CT densitometry. This would be considered disease modifying and would be considered as a first-line treatment for patients with emphysema and A1-PI deficiency and used in addition to standard of care for COPD.
Drug
A1-PI (human) is a lyophilized preparation of highly purified human A1-PI. Derived from pooled human plasma, it is administered intravenously once weekly at the recommended dose of 60 mg per kg of body weight.2
A1-PI (human) has a Health Canada indication for the maintenance treatment of adults with severe A1-PI deficiency and clinical evidence of emphysema (e.g., PiZZ, PiZ[null], Pi[null,null], and PiSZ genotypes) and clinical evidence of emphysema. Patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease, such as an lower FEV1 predicted, lower diffusion capacity, impaired walking capacity, or an increased number of exacerbations, as evaluated by a health care professional experienced in the treatment of A1-PI deficiency.2
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient advocacy group, Alpha-1 Canada, submitted the patient input for this review. Alpha-1 Canada is a national non-profit organization committed to advocating on behalf of Canadians affected by AATD and providing education and support to patients, caregivers, and the health care community. The submission was based on 2 virtual focus groups conducted in March 2021, 2 semi-structured interviews conducted over the phone in June 2021, 3 online surveys distributed between April and May 2021, and a single-question survey emailed to Canadian respirologists in May 2021. A total of 143 respondents (45 patients receiving A1-PI augmentation therapy, 53 patients not receiving therapy, 16 caregivers, and 29 Canadian respirologists) plus 2 families living with AATD were included in the patient input.
Respondents indicated that the physical manifestation of AATD impacts many aspects of their lives, ranging from employment, relationships, extracurricular activities, and day-to-day tasks to overall mental health. In areas where there is no publicly funded access to treatment, patients are weighing the steps they are willing to take to access therapy, such as continuing to work past retirement age to be eligible for private insurance, uprooting their lives to relocate to a province that offers coverage, or participating in clinical trials. Patients highlighted the costs to the health care system when they are unable to access treatment: they require inhalers to manage the symptoms of AATD, undergo frequent lung function tests, experience hospitalizations during exacerbations, and undergo lung transplant. The other major challenge patients with AATD face is the need to demonstrate deteriorated lung function before becoming eligible for augmentation therapy. Many felt they were doing additional damage to their lungs and compromising their quality of life while they waited to become eligible.
When patients with AATD are on augmentation therapy, they are able to stabilize their lung function. Patients perceive this as the most important outcome in effective treatment because it is associated with their ability to perform activities of daily living and fully participate in their communities and with their families. Patients did not feel that any disadvantages were worth noting in comparison with the possibility of augmentation therapy improving their quality and longevity of life.
A copy of the patient input from Alpha-1 Canada is presented in Appendix 4.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of A1-PI deficiency.
Unmet Needs
The clinical experts consulted by CADTH indicated that no treatments other than A1-PI replacement can prevent the progressive loss of lung function. A1-PI replacement can slow the rate of loss of lung tissue and may modify the severity of exacerbations through the anti-inflammatory properties of A1-PI treatment. The other symptoms that are similar to those of COPD are improved with the standard COPD therapies.
Place in Therapy
The clinical experts believed that an ideal treatment would reverse the loss of lung tissue destruction and noted that patients are typically not diagnosed until later in their disease course at a stage where there is established loss of lung tissue. Otherwise, the goals of treatment should include:
stabilization of the loss of lung function
improved survival
increased lung transplant–free survival
decreased exacerbation frequency and need for hospitalization
improved quality of life, including preservation or improvement of exercise capacity
stabilization or improvement of symptoms such as dyspnea
increased period of time before listing for lung transplant
reduction in the comorbidities common to COPD.
Patients with A1-PI deficiency and emphysema undergo standard therapy for COPD. The clinical experts mentioned using treatment with short- and long-acting bronchodilators, pulmonary rehabilitation, antibiotics and steroids as needed for exacerbation, inhaled corticosteroids in select individuals, and smoking cessation therapy for current smokers. Patients with severe symptoms may undergo evaluation for lung transplant.
Specific to A1-PI deficiency is the augmentation of the endogenous protease inhibitor with a purified exogenous A1-PI. This treatment option is currently available in Canada mainly through private insurers and special access programs. The goal of therapy with a protease inhibitor is to lessen the loss of lung tissue, as assessed by CT densitometry. The clinical experts noted that the treatment would be considered disease modifying and would be used as a first-line treatment for patients with emphysema and A1-PI deficiency, in addition to standard of care for COPD.
Patient Population
The clinical experts consulted by CADTH noted that the first step in the diagnosis of A1-PI deficiency is when COPD is diagnosed through clinical symptoms and spirometry (lung function tests). If a COPD patient is suspected of having A1-PI deficiency, then the alpha1-antitrypsin (AAT) blood level is measured and additional genetic testing is required to confirm the diagnosis and determine the specific genotype. In the absence of acute inflammation, levels above 1.1 g/L are normal and no further evaluation is needed, with few exceptions. If levels are below 1.1 g/L or if the clinician suspects that a rare non-functional genotype for AAT is present, then phenotyping or genotyping should be obtained. This process would establish the presence of common deficiency alleles (Z, S) or null in the absence of a normal M allele (e.g., ZZ, SZ, Z null). Carriers with 1 normal and 1 abnormal allele may have mildly reduced AAT serum levels, but they have a minimally increased risk of developing emphysema and are not regarded as candidates for augmentation therapy. Measurement of the serum AAT level is a widely available and inexpensive laboratory test. Nonetheless, there is significant under-diagnosis of AATD because the screening of AAT serum levels is not used routinely at the time of COPD diagnosis. In the presence of acute or chronic inflammation, AAT levels may be elevated and, if levels are borderline, phenotyping could be done. Patients with deficient serum levels but without evidence of lung disease should not be started on treatment but should be followed clinically to ensure they do not develop emphysema over time.
The clinical experts indicated that any patient with emphysema and A1-PI deficiency would be a candidate for treatment with the drug under review. Therefore, the presence of emphysema could be an indication to start A1-PI replacement therapy. Patients with very severe emphysema might receive little benefit from a treatment aimed at preventing the loss of lung tissue. FEV1 results may not accurately reflect the degree of emphysema. Functional status (exercise capacity) might be a better indicator for those with severe loss of FEV1, but there are no data to support this. The use of a multi-dimensional tool for assessment of COPD might be better used, but there are no data where these tools were used as starting or stopping parameters for treatment. The patients in need of intervention are those with a loss of lung tissue related to A1-PI deficiency. There are no clear disease phenotypes to help stratify therapy.
Any patient with A1-PI deficiency without lung disease or patients with a heterozygous genotype with 1 functional M allele would not be good candidates for treatment. Patients who are near the end stage of their disease would not be good candidates, but end-stage COPD is difficult to clinically establish with confidence.
Assessing Response to Treatment
The clinical experts consulted by CADTH indicated that, since the drug is used to prevent the rapid progression of emphysema, there are no specific outcome parameters to measure.
All standard pharmacological treatments for COPD (long-acting beta-agonist, long-acting muscarinic antagonist, and inhaled corticosteroids, where appropriate) should be initiated before considering A1-PI replacement therapy. These treatments may result in significant improvement of symptoms. Lung function may also improve if the reduction of FEV1 has a reversible component. Smoking is the primary driver of an accelerated loss of lung function, and there are minimal data regarding the efficacy of replacement therapy in current smokers. Pulmonary rehabilitation should be undertaken to determine if any loss of exercise capacity is secondary to deconditioning and therefore reversible.
Discontinuing Treatment
The clinical experts noted there are no factors that would be used as a stopping rule, other than SAEs such as hypersensitivity. Because the goal of augmentation therapy is to prevent or decrease the rate of further tissue damage, it is expected that some patients will keep deteriorating despite treatment. However, it is very likely that these patients would have deteriorated even more without A1-PI replacement; thus, using lung function or the number of exacerbations to assess response to treatment is inadequate in this particular case.
Prescribing Conditions
The clinical experts believed that A1-PI (human) can be given in any infusion centre. Although it is formulated for self-administration, the route (IV) and the complexity of reconstitution make it very unlikely to be given anywhere other than medical clinics or infusion centres.
The clinical experts suggested that A1-PI therapy should be started under the direction of a pulmonary medicine specialist. Once the patient is established on therapy, they could be co-managed with their family physician.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by 1 clinician group.
One clinician group provided input. The Canadian Thoracic Society is a national specialty society and membership-based professional association for health care providers working in respiratory care and research. It is recognized as an accrediting body of the Royal College of Physicians and Surgeons of Canada for specialist education and continuing professional development.
Place in Therapy
The clinician group providing input stated that standard COPD treatments are used concurrently with augmentation therapy; however, no other treatment has been shown to delay the progression of emphysema in these patients.
IV augmentation therapy is undertaken with purified AAT from blood donation. Worldwide, several companies market augmentation therapies derived from proprietary filtration and purification processes. It was noted in the clinician group input that, in Canada, only Prolastin and Prolastin-C (currently marketed by Grifols) have been available for clinical use. Single-supplier availability of any therapy is associated with a monopoly on prices and a risk of future shortfalls in availability. Availability of a new augmentation therapy product would mitigate those risks and potentially allow the health care system to re-examine the proposition that this blood product be made available through Canadian Blood Services, similar to other blood products. Other augmentation therapies, including Zemaira, have been used in a limited way in clinical trials. In the absence of head-to-head trials, the Canadian Thoracic Society input stated there was no reason to conclude that there are substantial differences in efficacy or safety among various purifications of what is a normal constituent of human blood.
The clinician group input also indicated that augmentation therapy for AATD is currently the only blood product not distributed by Canadian Blood Services. This anomaly has led to challenges for patients and caregivers who seek effective treatment for this genetic disorder. Paradoxically, as our understanding of the deficiency and its treatment has improved over the past 3 decades, the availability of augmentation therapy has diminished. Three decades ago, augmentation therapy was provided to patients through several provincial formularies. At present, only Quebec and British Columbia have included augmentation therapy support for patients meeting appropriate criteria. In other parts of Canada, augmentation therapy is available only to those with private health insurance. This has led to difficult lifestyle decisions for affected individuals. Individuals with significant disease and without private health insurance may be forced to consider relocation to another province and, in rare instances, relocation to other countries where augmentation therapy is routinely afforded to the genetically disadvantaged. Similar decisions face patients whose private health insurance is lost at the age of retirement.
Assessing Response to Treatment
It was stated in the clinician group input that, in the 3 decades since the Canadian Thoracic Society first suggested that a FEV1-based trial would be useful; this end point has been re-examined and its practical shortcomings noted. Although FEV1 remains essential in the diagnosis of common obstructive lung diseases, its role in day-to-day management is diminished. In the broad category of COPD, for example, pharmacologic interventions should be adjusted based on symptom burden and exacerbation tendency rather than spirometric cut points. The clinician group input suggested that, in the setting of emphysema caused by a severe deficiency of AAT, direct quantification of lung parenchymal density has been well studied and shown to be especially valuable. In a disease characterized by the loss of alveolar structure, lung density estimated objectively by CT scan techniques has proven to have better prognostic value than conventional measures of lung function.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Are laboratory tests to check serum AATD level available in all provinces? | Yes, the clinical experts confirmed that laboratory tests are easily available in all provinces. |
Are genetic tests to confirm genotypes such as PiZZ, PiZ,(null), Pi(null)(null) needed to confirm eligibility for treatment? Are these genetic tests available in all the provinces? | The clinical experts indicated that the genetic tests are needed to confirm genotypes and should be done systematically, and that the technology is readily available. The clinical experts also noted that genetic tests are available in most provinces but some provinces, such as Alberta, have continued to rely on outdated serum protein electrophoresis to determine, indirectly, the probable genotype. Buccal swab genotyping is readily available as sponsored by Grifols, the only company currently marketing augmentation therapy in Canada. The testing is done at Biocerna, a laboratory based in Maryland. |
What defines “optimal pharmacological and non-pharmacological treatment”? Is it practical to put this in the treatment eligibility criteria? | According to the clinical experts, “optimal pharmacological and non-pharmacological treatment” in A1-PI deficiency is the same as that for COPD. Sometimes, some interventions, such as pulmonary rehabilitation, may not be readily available to all patients. The clinical experts also noted that the only reasonable benchmark is to require that patients be cared for by a respiratory specialist and that they be non-smokers who ceased smoking at least 6 months before the date they start treatment with A1-PI (human). |
What if a patient has a confirmed genetic test suggesting severe AATD, but lung damage has not happened yet? Should such patients be eligible for treatment with Zemaira, or should they have to show clinical evidence of emphysema before being eligible? | The clinical experts’ opinion is that not all patients develop clinically important emphysema, despite having a severe deficiency of alpha1-antitrypsin. They also noted it is important to stipulate that clinically important emphysema should be present before initiating treatment with A1-PI (human). Clinically important emphysema is not defined by the presence of emphysema on a CT scan but by physiologically important emphysema, as determined by routine clinical pulmonary function tests. One benchmark suggested by the Canadian Thoracic Society is obstruction (FEV1 to FVC ratio below 0.70) and a FEV1 below 80% of predicted. |
Should smokers be eligible for A1-PI (human)? | The clinical experts suggested that smoking cessation is an essential part of the treatment. There is no evidence that Zemaira would work in the presence of continued smoking. |
What if a patient with severe AATD has received a lung transplant? How long does this patient have to wait before being eligible for treatment with an A1-PI? Should these patients be eligible for treatment with an A1-PI? | The clinical experts indicated there are no data to inform this question. There have been no studies of augmentation therapy following lung transplant. |
About 90% of patients had PiZZ genotype in the RAPID trial. Should patients with other genotypes be eligible for A1-PI (human)? Would patients with other (confirmed) genotypes receive a clinical benefit similar to patients with the PiZZ genotype? | It is the clinical experts’ opinion that there is a lack of data in patients with a SZ or MZ genotype, and that Zemaira should be made available to patients with a null, ZZ, or SZ genotype with evidence of lung disease. Patients with equivalent rare variant genotypes with evidence of lung disease should also be offered treatment. |
Should reimbursement for Zemaira be limited to certain genotypes or all? | The clinical experts suggested that reimbursement for Zemaira should be limited to SZ, ZZ, and null genotypes and to some of the rare variants that are considered equivalent. |
If a patient currently being treated with Prolastin-C needs to transition to Zemaira, would such a patient need to meet the eligibility criteria for Zemaira, or would they become eligible for Zemaira by default? | According to the clinical experts, reimbursement criteria should be identical for both Zemaira and Prolastin-C. Patients should become eligible for Zemaira by default. |
Does the evidence confirm that a slow decline in lung density translates into better clinical outcomes? | The clinical experts noted there are no direct clinical data on this; however, there are data extrapolated from observational studies of reduced mortality with augmentation therapy. In addition, there was a correlation between the preservation of lung density and maintenance of lung function test results, as measured by spirometry (in the RAPID open-label extension). |
How often should patients be followed up before they are approved to continue treatment? | According to the clinical experts, a follow-up should be done every 6 to 12 months; however, once the treatment has started, they suggest there is no reason to discontinue, except for issues around infusion problems or allergy. |
Do you expect that all patients receiving Prolastin-C will switch to Zemaira once it becomes available? | The clinical experts expect that many patients and physicians will stay with the current augmentation therapy used. There are no head-to-head studies to suggest superiority of any augmentation formulation over another. |
The primary end point in RAPID and the RAPID extension study was decline in lung density, as measured by CT scans. Keeping in mind the slow progression of AATD, the sponsor suggests that a CT scan is the only possible end point that can be assessed in a study and that would be acceptable to regulatory authorities. Is a CT scan a meaningful clinical outcome? What should be the frequency of CT scans? Would patients in rural areas be able to access CT scans for monitoring of therapeutic response? Are there any other tests or assessments required to monitor therapeutic effectiveness and safety? | The clinical experts noted there is good biologic plausibility that CT is an appropriate outcome, and that a CT scan is meaningful in this setting. While tobacco-related COPD is common, many pulmonary doctors know that the CT scan appearance is not helpful in many COPD patients who have other pathologic mechanisms that cause obstruction in the absence of important emphysema. AATD is unique in presenting a relatively homogenous emphysema. This makes lung density a useful end point. The clinical experts indicated that once treatment has started, there is no need for additional CT scans. CT scans are used to make a diagnosis of emphysema, not to follow clinical progression. CT scanning at baseline should be accessible to all Canadians with important lung disease. The clinical experts do not think there are any appropriate follow-up tests, since the objective of treatment is to prevent or delay the loss of lung tissue. |
Should the renewal criteria for Zemaira be similar to that of Prolastin-C? | According to the clinical experts, the renewal criteria for Zemaira should be similar to that of Prolastin-C. |
How do you define loss of response or absence of clinical benefit? | The clinical experts suggest there is no such thing as loss of response or absence of clinical benefit. Patients would be followed clinically with periodic assessment of symptoms and lung function. A treatment failure would be an accelerated loss of lung function. However, this would not prompt discontinuation of the augmentation therapy. This finding would prompt most clinicians to look for factors that account for the rapid decline. |
Should the discontinuation criteria for Zemaira be similar to that of Prolastin-C? | The clinical experts indicated that the discontinuation criteria for Zemaira can be similar to that of Prolastin-C; however, the clinical experts noted that they do not know of any sensible discontinuation criteria, except perhaps intolerance of or severe allergy to the therapy. They also noted that it may take up to several years to see the effects of the treatment and that, once the treatment has started, it should not be discontinued. |
Do you expect that clinicians would increase the dose of A1-PI (human) to 120 mg/kg or increase the frequency with 60 mg/kg dosing? Is there a need to put a cap on dosing and frequency? | According to the clinical experts, there might be some rare cases where the dosage would need to be increased, but only to compensate for missed doses or for travel purposes (120 mg/kg every 2 weeks). |
Infusion time for the 60 mg/kg body weight dose is 15 minutes. Are patients able to self-administer at home? Is there any training required? | The opinion of the clinical experts is that the requirements for home infusion seem to be beyond the capability of most people. It was also noted that while self-administration is possible with training, it is seldom done in Canada. |
Are there any concerns related to accessing specialists and laboratory requirements for therapeutic drug monitoring? | The clinical experts expressed no concerns. No special therapeutic monitoring is required. A blood sample can be drawn anywhere, and a telehealth appointment can also be done from anywhere now. The patient’s care should be guided by a respirologist. |
Should the prescribing criteria for Zemaira be similar to that of Prolastin-C? | According to the clinical experts, the prescribing criteria should be identical for both Zemaira and Prolastin-C. |
If patients were to switch from Prolastin-C to Zemaira, what should be the time frame for switching? | The clinical experts suggested there is no need for a washout period. Zemaira would be given at the time of the next scheduled dose of Prolastin. |
Zemaira can be stored in the refrigerator or at room temperature (at + 2°C to + 25°C). Do not freeze. Storage after reconstitution: Administer within 3 hours after reconstitution. Would ancillary supplies related to infusion be provided by a patient support program or is this expected to come from hospital transfusion services? | The clinical experts suggested that ancillary supplies related to infusion should be provided by a patient support program. The sponsor confirmed that ancillaries will be provided through the patient support program. |
Are there any concerns with the development and management of A1-PI antibodies? | The clinical experts expressed no concerns regarding such an issue. |
Would Zemaira reduce the use of other concomitant treatments required for the management of COPD or emphysema? | The clinical experts do not expect any change in the use of other concomitant treatments required for the management of lung disease. However, it should delay the introduction of expensive interventions, such as long-term home oxygen and transplant. |
A1-PI = alpha1-proteinase inhibitor; AATD = alpha1-antitrypsin deficiency; COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; FVC = forced vital capacity.
Clinical Evidence
The clinical evidence included in the review of Zemaira is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. No indirect evidence was submitted by the sponsor. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of A1-PI (human) 1,000 mg, 4,000 mg, and 5,000 mg for the maintenance treatment of adults with severe A1-PI deficiency and clinical evidence of emphysema.
Severe A1-PI deficiency includes, but is not limited to, the PiZZ, PiZ(null), Pi(null,null), PiSZ genotypes.
Patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease, such as lower predicted FEV1, lower diffusion capacity, impaired walking capacity, or an increased number of exacerbations, as evaluated by a health care professional experienced in the treatment of A1-PI deficiency.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with severe alpha1-proteinase inhibitor deficiency and clinical evidence of emphysema Subgroups: Severity of emphysema |
Intervention | Alpha1-proteinase inhibitor (human): 60 mg/kg body weight administered by IV infusion once weekly |
Comparator | Placebo Alpha1-proteinase inhibitor (human) (Zemaira) |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; FEV1 = forced expiratory volume in the first second; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.12
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Zemaira (A1-PI [human]) and A1-PI deficiency. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
CADTH-developed search filters were applied to limit retrieval to RCTs or controlled clinical trials. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on November 1, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Plasma Protein Product Expert Committee on February 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.13 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses dealing with A1-PIs or A1-PI deficiency was run in MEDLINE All (1946–) on November 1, 2021. No limits were applied to the search.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 5. A list of excluded studies is presented in Appendix 2.
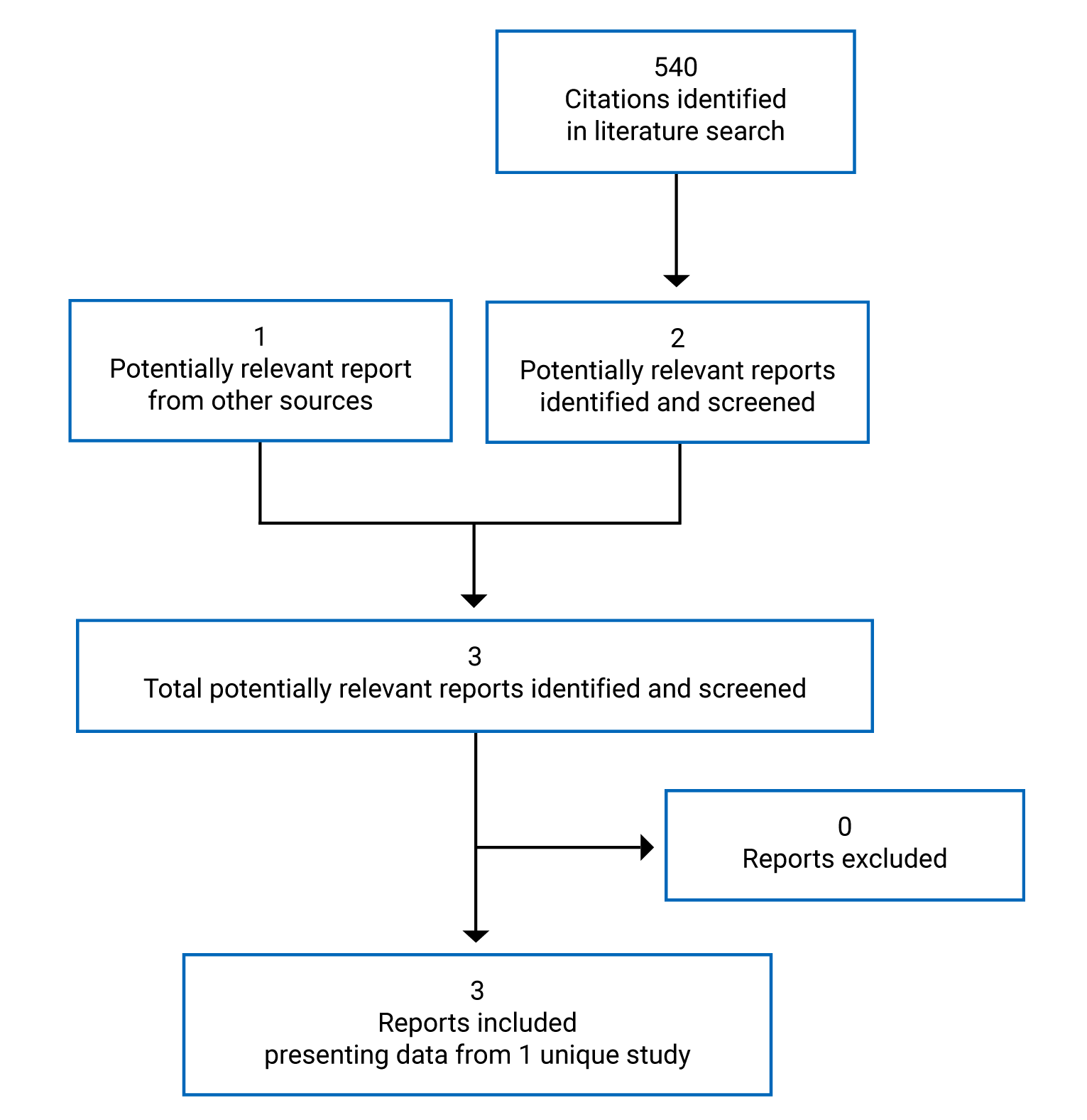
Table 5: Details of Included Studies
Detail | RAPID |
|---|---|
Designs and populations | |
Study design | Double-blind, placebo-controlled RCT |
Locations | Multi-centre: Australia, North America, Europe |
Patient enrolment dates | First patient enrolled: March 1, 2006 Last patient enrolled: September 26, 2012 |
Randomized (N) | N = 180 |
Inclusion criteria | Patients 18 to 65 years of age with:
|
Exclusion criteria |
|
Drugs | |
Intervention | A1-PI (human) (Zemaira), 60 mg/kg IV infusion once weekly for 24 months |
Comparator(s) | Double-blind placebo IV infusion once weekly for 24 months |
Duration | |
Phase | |
Double blind | 24 months |
Outcomes | |
Primary end point | Annual rate of decrease in lung density The primary objective is to evaluate the progression of emphysema, assessed by the decline of lung density as measured by CT. |
Secondary and exploratory end points |
|
Notes | |
Publications | Greulich et al. (2018)3 Chapman et al. (2015)4 |
A1-PI = alpha1-proteinase inhibitor; AE = adverse event; DLCO = diffusing capacity of the lungs for carbon monoxide; FEV1 = forced expiratory volume in the first second; IgA = immunoglobulin A; ISWT = incremental shuttle walking test; RCT = randomized controlled trial; SGRQ = St. George’s Respiratory Questionnaire.
Note: 1 additional report was included.14
Source: Clinical Study Reports for the RAPID trial.5
Description of Studies
One published, manufacturer-sponsored, double-blind RCT was included in the systematic review: RAPID (n = 180)3,4 evaluated the superiority of A1-PI (human) compared with placebo on the progression of disease in patients with emphysema with A1-PI deficiency and a reduced lung function. Disease progression was assessed by the decline of lung density, measured by CT. A1-PI (human) was administered at a dosage of 60 mg/kg through IV infusion once weekly for 24 months.
Populations
Inclusion and Exclusion Criteria
Patients were eligible for the trial if they were between 18 and 65 years of age with a diagnosis of A1-PI deficiency, defined as an A1-PI serum level lower than 11 µM or 50 mg/dL, and emphysema, defined as a FEV1 of between 35% and 70% of predicted. Patients with concomitant respiratory or liver disease could be included only if these were secondary to A1-PI deficiency. Key exclusion criteria included any other relevant chronic diseases as well as current tobacco smoking. Patients were also excluded if they had undergone lung transplant, lung volume reduction surgery or lobectomy, or were on a waiting list for such procedures.
Baseline Characteristics
Baseline characteristics are presented in Table 6. Patients enrolled in the trial had a mean age of approximately 53 years, with the trial population being fairly evenly distributed among men and women. All patients were White. The mean FEV1 was 47% of predicted. Mean duration of disease was between 5 and 6 years. The majority of patients had a medical history before baseline (72% and 63% in the active treatment group and placebo group, respectively), as well as concurrent illness (94% and 95%, respectively) and concomitant medication (99% and 98%, respectively). Overall, baseline characteristics appeared to be balanced between treatment groups. However, higher proportions of patients randomized to placebo experienced prior or concurrent respiratory, thoracic, and mediastinal disorders. Although this might be a random fluctuation due to the small sample size, potential imbalance cannot be excluded.
Table 6: Summary of Baseline Characteristics in the RAPID Trial
Characteristic | A1-PI N = 93 | Placebo N = 87 |
|---|---|---|
Age in years, mean (SD) | 53.8 (6.91) | 52.4 (7.81) |
Sex, n (%) | ||
Man | 48 (51.6) | 50 (57.5) |
Woman | 45 (48.4) | 37 (42.5) |
Race, n (%) | ||
White | 93 (100.0) | 87 (100.0) |
Body mass index in kg/m2, mean (SD) | 25.5 (4.79) | 26.6 (4.07) |
Disease characteristics, mean (SD) | ||
FEV1, L | 1.6 (0.51) | 1.6 (0.47) |
FEV1% predicted, % | 47.4 (12.1) | 47.2 (11.1) |
DLCO, mL/mmHg/min | 13.6 (5.31) | 15.0 (5.62) |
SGRQ total score | 44.3 (17.1) | 42.4 (18.0) |
Duration of disease, years | 5.6 (6.14) | 6.1 (6.56) |
Antigenic A1-PI levels, mg/mL | 0.3 (0.21) | 0.3 (0.1) |
Functional A1-PI levels, mg/mL | 0.2 (0.19) | 0.1 (0.07) |
A1-PI phenotype, n (%) | ||
ZZ | 83 (89.2) | 83 (95.4) |
SZ | 2 (2.2) | 0 |
Z or null | 2 (2.2) | 1 (1.1) |
Other | 6 (6.5) | 3 (3.4) |
Medical history before baseline, % | 72 | 63 |
Surgical and medical procedures | 42 | 43 |
Infections and infestations | 19 | 9 |
Gastrointestinal disorders | 12 | 10 |
Respiratory, thoracic, and mediastinal disorders | 9 | 12 |
Concurrent illness, % | 94 | 95 |
Severe concurrent illness | 22 | 28 |
Respiratory, thoracic, and mediastinal disorders | 63 | 72 |
Nervous system disorders | 31 | 29 |
Musculoskeletal and connective tissue disorders | 30 | 24 |
Gastrointestinal disorders | 28 | 25 |
Vascular disorders | 25 | 28 |
Congenital, familial, and genetic disorders | 22 | 16 |
Psychiatric disorders | 14 | 22 |
Concomitant medication, % | 99 | 98 |
Influenza vaccines | 55 | 55 |
Anilides | 55 | 50 |
Glucocorticoids | 47 | 39 |
Macrolides | 40 | 39 |
Selective beta-2-adrenoreceptor agonists | 36 | 37 |
Propionic acid derivatives | 31 | 39 |
Combinations of penicillins | 34 | 35 |
Fluoroquinolones | 33 | 33 |
Penicillins, extended spectrum | 34 | 30 |
Natural opium alkaloids | 26 | 28 |
Adrenergics and other drugs for obstructive airway | 24 | 28 |
A1-PI = alpha1-proteinase inhibitor; DLCO = diffusion capacity of carbon monoxide; FEV1 = forced expiratory volume in the first second; SD = standard deviation; SGRQ = St. George’s Respiratory Questionnaire.
Source: Clinical Study Reports for the RAPID trial.5
Interventions
Each patient received weekly IV infusions of A1-PI (human) at a dosage of 60 mg/kg, or equivalent volume of placebo, for 24 months. Both active treatment and placebo were packaged identically and identified only by patient number, as the study was double-blind. IV infusions were administered at the study centres at a rate of 0.08 mL/kg/min whenever possible, as determined by the response and comfort of the patient.
All medications being taken by patients upon study entry, and all medications given in addition to the study treatments during the study duration, were regarded as concomitant medications. Only other A1-PI products were considered prohibited throughout the study period.
Outcomes
A list of the efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 7. These end points are further summarized subsequently. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 7: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Outcome |
|---|---|
Survival | NR |
Time to lung transplant | NR |
Lung density | Primary |
Pulmonary function | Secondary |
Number and severity of exacerbations | Secondary |
Health-related quality of life | Secondary |
AEs | Secondary |
SAEs | Secondary |
WDAEs | Secondary |
Mortality | Secondary |
Severe hypersensitivity | Secondary |
AE = adverse event; NR = not reported; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Source: Clinical Study Reports for the RAPID trial.5
The primary efficacy outcome in RAPID was lung density, more specifically, the treatment comparison for annual rate of change in the physiologically adjusted 15th percentile of the lung density (P15), as measured by CT scan. CT lung densitometry measurements have been validated as a primary clinical end point for clinical study designs in monitoring emphysema progression in AATD. The clinical experts consulted by CADTH considered lung density to be a more sensitive indicator of disease progression than other outcomes, such as lung function tests.
A radiologist was designated as the sub-investigator and conducted training across sites. Multi-slice CT scanners were required with at least 4 detector rows and for standardized scanning protocols that defined the views and were optimized for density evaluation for each type of scanner. Sites had to submit calibration scans of phantoms (Perspex-foam models with standardized properties) before the scanning of the first patient and after upgrades and maintenance of the CT scanner. The density measurements were made by a central laboratory, with an independent quality-control check by a second technician and a process for querying inadequate scans.
Scans were obtained at both TLC and functional residual capacity (FRC) inspiration states. A range of approaches was selected to examine the primary efficacy variable, including analyses at different inspiration states and different adjustment models. However, the clinical experts consulted by CADTH agreed that the most reliable way to measure lung density is at a full inspiration state, which is referred to as TLC. Full inspiration provides a greater area for measurement on the CT scan image and therefore, the measurement obtained is considered more reliable.
Pulmonary function parameters were assessed as secondary outcomes and included FEV1, FEV1 percentage of predicted, the FEV1 to FVC ratio, and the diffusing capacity of the lungs for carbon monoxide (DLCO). Pulmonary function is regularly measured in clinical practice; however, it is not consistently representative of disease progression and severity of symptoms, according to the clinical experts consulted by CADTH.
Symptoms and function were assessed as a secondary outcome through the use of 2 validated questionnaires. The St. George's Respiratory Questionnaire (SGRQ), which is a disease-specific instrument, was originally designed to assess patient-related quality of life in patients with obstructive lung or airway disease. The questionnaire consists of 50 items with 76 weighted responses that measure health status across 3 domains: symptoms (frequency and severity), activity (activities that cause or are limited by breathlessness), and impact (social functioning, psychological disturbances resulting from airway disease). The symptom domain is scored using a 5-point Likert scale, while the activity and impact domains are scored in a dichotomous (yes or no) manner. The total score for each domain is derived by weighting the items in the domain based on empirical data. An overall total score is then computed, which ranges from 0 to 100. Higher scores from the questionnaire represent poor health status and lower scores represent good health status. A difference in the total SGRQ score of about 4 to 8 points would indicate clinically significant differences between populations.
The incremental shuttle walking test (ISWT) is a symptom test, developed to assess functional capacity in patients with COPD. It simulates an external field-walking exercise and is designed to incorporate incremental and progressive structures that stresses patients to respond in a symptom-limited maximal performance. The test is a modified version of a progressive externally paced 20 m shuttle-running test that has been widely used to assess functional capacities in athletes. The minimum clinically significant improvement in the ISWT identified in the study was 47.5 m. Further information regarding the measurement properties of the instruments is provided in Appendix 4.
Safety outcomes included AEs, SAEs, and discontinuation due to AEs (WDAEs) and mortality.
Statistical Analysis
The initial sample size calculation estimated that having 50 patients per treatment group would enable the study to achieve 80% power at a 1-sided level of significance of 0.20 to detect an effect size of 1 g/L/year on the decline in lung density, with a standard deviation (SD) of approximately 2.5 g/L/year. A blinded planned re-estimation of the study sample size was done while the trial was ongoing. The slope of the regression line for the decline in lung density based on this blinded sample size re-estimation indicated that the SD used in the original sample size estimation was appropriate. It also indicated that 180 patients would provide at least 88% power to detect the previous assumed effect size of a 1 g/L (SD = 2.5 g/L) difference in the yearly decline in lung density at a 1-sided significance level of 0.025, or would be sufficient to maintain at least 80% power to detect a 1.07 g/L (SD = 2.17 g/L) difference in the yearly decline according to the data used for the initial estimation.
The statistical analyses performed were initially specified in the study protocol. The study was designed to test for superiority. The primary end point was the annual rate of change in the adjusted P15, the volume-adjusted lung density, which was obtained from a linear random regression (mixed-effects) model and expressed as a slope of time and treatment interaction from the model. The null hypothesis for the treatment comparison was that the mean slope associated with the primary efficacy variable in patients treated with an A1-PI (beta 2) was numerically less than or equal to the mean slope in patients treated with placebo (beta 1). The alternative hypothesis was that the mean slope in the patients treated with A1-PI (human) was numerically greater than the mean slope in the patients treated with placebo. If beta 2 was greater than beta 1, the mean decline in adjusted lung density in the in the patients treated with A1-PI (human) was slower than in the placebo-treated patients. A 1-sided test derived from the linear random regression model on adjusted P15 with an alpha of 0.025 was used to test this hypothesis.
For the primary statistical analysis, no values were imputed. All available data were included in the analysis. All randomized patients with at least 1 valid CT scan (at any time point) were included. Missing values were assumed to be missing at random. Intention-to-treat (ITT) analyses were performed with and without (observed cases) imputation as outlined in the corresponding sections.
The primary efficacy variable, adjusted P15 values, was analyzed at the TLC and FRC inspiration states, meaning the TLC and FRC values were included in the primary model simultaneously. A likelihood-based mixed model (random regression model) was used in the primary analysis. In addition, the primary efficacy end point was analyzed by applying the primary efficacy analysis model (without the fixed effect for inspiration state) to the CT scans at TLC and FRC states separately. The principal interest was in the magnitude of the treatment-by-time interaction (difference of the regressions on time within treatment) and its level of significance because this indicated whether the 2 treatments differed in their effect on the rate of decline of the adjusted P15.
According to protocol, a sequential testing strategy for the treatment difference for A1-PI (human) versus placebo was to be used to support the regulatory strategy. The testing strategy was to be applied if a statistically significant treatment difference in favour for A1-PI (human) was found for the primary efficacy end point.
For the analysis of lung function parameters, the percentage change from baseline to month 24 (imputed and observed for the ITT population and observed for the per-protocol [PP] population) for each of the key spirometry variables was analyzed using an analysis of covariance (ANCOVA) with country, treatment, and the baseline value of the dependent variable as fixed covariates in the model. For the key spirometry variables, the difference between the slopes in patients treated with A1-PI (human) versus placebo was examined using a linear random regression model with country, time, treatment, and treatment-by-time interaction (a regression of time within treatment) as fixed effects and patient and patient-by-time interaction as random coefficients.
Pulmonary exacerbations for the ITT and PP populations were described as the number of patients with exacerbations and by calculating the annual rates of events that met the definition of an exacerbation. Treatments were compared for the number of exacerbations, adjusting for the treatment duration in years. A negative binomial regression was applied with country and treatment as fixed effects. Adjustment was made for the patient’s study duration by including log study duration as an offset variable in the model.
The analysis of the symptoms and function questionnaires was performed using the change from baseline to month 24 for the ITT population and was based on both the observed and imputed values. An ANCOVA with the country, treatment, and baseline value as fixed covariates was carried out. The estimated treatment difference derived from the ANCOVA, along with the 2-sided 95% CIs and 2-sided P values, is presented for the imputed and observed values in the ITT population and for the observed values in the PP population.
Analysis Populations
ITT population: This comprised all patients with A1-PI deficiency who were included in the study and randomized. In the ITT analysis, patients were assigned to the treatment to which they were randomized. The ITT population was the primary population for the analysis of the primary efficacy variable. For the end point analyses, the observed cases were patients with a baseline and at least 1 end point assessment available.
PP population: This was a subset of the ITT population from which the patients with a major protocol deviation were excluded (pre-specified definitions). Only those data affected by the protocol deviations were excluded. The final decision regarding which patients and which data would be excluded from the PP analysis was made before the data were unblinded. In the PP analysis, patients were assigned to the treatment groups to which they had been randomized. A sensitivity analysis of the primary efficacy variable was performed using the PP population.
Safety population: This comprised all patients who were included in the study and received at least 1 dose of A1-PI (human). In the safety analysis, patients were analyzed according to the treatment they actually received, which was defined as the treatment received most of the time during the study. The safety population was used for all safety analyses.
Patient Disposition
A total of 208 patients were screened; of these, 180 patients were randomized to the double-blind phase. More patients receiving placebo withdrew from the study than patients receiving active treatment. A total of 10% of patients randomized to A1-PI (human) discontinued from the study compared with 21% of patients receiving placebo. The most frequent reasons for discontinuation included death (1% in the active treatment group and 3% in the placebo group), AEs (1% and 5%, respectively) and consent withdrawn (5% and 8%, respectively).
Further details regarding patient disposition are provided in Table 8. The interpretation of these differences between treatment arms is limited by the small sample size; however, the clinical experts’ opinion suggests this does not diminish confidence in the study results.
Table 8: Patient Disposition in the RAPID Trial
Patient disposition | A1-PI | Placebo |
|---|---|---|
Screened, N | 208 | |
Randomized | 93 | 87 |
Discontinued from study, N (%) | 9 (9.7) | 18 (20.7) |
Reason for discontinuation, N (%) | ||
Adverse events | 1 (1.1) | 4 (4.6) |
Death | 1 (1.1) | 3 (3.4) |
Withdrawn consent | 5 (5.4) | 7 (8.0) |
Protocol violation | 0 | 1 (1.1) |
Missing reason | 1 (1.1) | 0 |
Other | 1 (1.1) | 3 (3.4) |
ITT, N | 93 | 87 |
PP, N | 83 | 76 |
Safety, N | 93 | 87 |
A1-PI = alpha1-proteinase inhibitor; ITT = intention to treat; PP = per protocol.
Source: Clinical Study Reports for the RAPID trial.5
Efficacy
Only those efficacy outcomes and analyses of the subgroups identified in the review protocol are reported subsequently. See Appendix 3 for detailed efficacy data.
Survival
No data were reported for the outcome of survival.
Time to Lung Transplant
No data were reported for the outcome of lung transplant.
Lung Density
The primary efficacy outcome in RAPID was lung density, more specifically, the treatment comparison for annual rate of change in the physiologically adjusted P15 lung density, as measured by CT scan. It was not clear at which specific inspiration state the measure was to be taken for the primary analysis. According to the clinical experts consulted by CADTH, the most reliable way to measure lung density is at the full inspiration state, which is referred to as TLC. When the lungs are full of air, more lung tissue is visible on the CT scan image and, therefore, the measurement obtained is considered more reliable. FRC refers to the end of a normal breath, when there is less lung visible on the CT scan image and, therefore, measurements at this inspiration state are considered less reliable and are used for volume correction.
The use of A1-PI (human) was associated with a reduced rate of decline in lung density over 24 months compared with placebo when measures were taken at the full inspiration state (0.74 g/L; 95% CI, 0.06 to 1.42; 1-sided P = 0.017). Results using other inspiration state measures also showed a slower decline in lung density with the active treatment compared with placebo over a 24-month period, but the differences between groups were of smaller magnitude and did not reach statistical significance.
Pulmonary Function
Pulmonary function is regularly measured in clinical practice; however, it is not consistently representative of disease progression and severity of symptoms according to the clinical experts consulted by CADTH. Pulmonary function parameters were assessed as secondary outcomes in RAPID and included FEV1, percentage of predicted FEV1, the FEV1 to FVC ratio, and DLCO. Overall, treatment comparisons for the percentage change after 24 months favoured A1-PI (human), but the results did not reach statistical significance for any of the parameters measured other than the FEV1 to FVC ratio (the trial was not powered to show statistical significance for secondary outcomes).
The mean FEV1 to FVC ratios with A1-PI (human) (0.45; SD = 0.11) and placebo (0.43; SD = 0.10) at day 1 were comparable. The mean percentage change in the FEV1 to FVC ratio from day 1 to month 24 was −2.77% (SD = 11.8) for A1-PI (human) and 1.07% (SD = 14.4) for placebo. The analysis of the treatment difference for the percentage change from baseline (day 1) to month 24 in the FEV1 to FVC ratio for observed values revealed a change of 4.24% (95% CI, −8.04 to −0.45; P = 0.029) in favour of placebo compared with A1-PI (human).
Number and Severity of Exacerbations
Throughout the trial duration of 24 months, patients receiving A1-PI (human) experienced a rate of 1.53 exacerbations per patient-year, while the result was 1.21 per patient-year in patients receiving placebo. The difference between treatments was not statistically significant (the trial was not powered to show statistical significance for secondary outcomes).
Symptoms and Function
Symptoms were assessed by the St. George's Respiratory Questionnaire (SGRQ) symptom scale, and function was assessed using the ISWT.
Treatment comparisons for percentage change after 24 months favoured A1-PI (human), but the results did not reach statistical significance (the trial was not powered to show statistical significance for secondary outcomes) and were not considered clinically meaningful.
Table 9: Summary of Key Efficacy Outcomes in the RAPID Trial
Outcome | A1-PI (N = 93) | Placebo (N = 87) |
|---|---|---|
Change in the physiologically adjusted P15 lung densitya | ||
Inspiration state: Mean of TLC and FRC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 46.6 (15.6) | 49.8 (15.1) |
End of treatment time point (month 24), mean (SD) | 44.4 (15.5) | 45.5 (13.9) |
Change from baseline, mean (SD) | −2.67 (4.30) | −3.93 (4.02) |
Percent change from baseline, mean (SD) | −6.06 (9.67) | −8.28 (8.89) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 67 |
Least squares mean (SE) | −2.33 (0.45) | −3.37 (0.50) |
Difference between treatmentsb | 1.04 g/L | |
95% CI, 1-sided P value | −0.26 to 2.34; P = 0.058 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.50 (0.22) | −2.12 (0.24) |
Difference between treatmentsc | 0.62 g/L | |
95% CI, 1-sided P value | −0.02 to 1.26; P = 0.029 | |
Inspiration state: TLC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 45.5 (15.8) | 48.9 (15.5) |
End of treatment time point (month 24), mean (SD) | 43.6 (16.0) | 43.9 (13.8) |
Change from baseline, mean (SD) | −2.60 (4.44) | −4.20 (4.50) |
Percent change from baseline, mean (SD) | −6.22 (9.66) | −8.97 (10.3) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 66 |
Least squares mean (SE) | −2.22 (0.47) | −3.54 (0.52) |
Difference between treatmentsb | 1.32 g/L | |
95% CI, 1-sided P value | −0.03 to 2.67; P = 0.028 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.45 (0.23) | −2.19 (0.25) |
Difference between treatmentsc | 0.74 g/L | |
95% CI, 1-sided P value | 0.06 to 1.42; P = 0.017 | |
Inspiration state: FRC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 47.6 (15.7) | 50.7 (15.0) |
End of treatment time point (month 24), mean (SD) | 45.3 (15.3) | 46.8 (13.8) |
Change from baseline, mean (SD) | −2.74 (4.75) | −3.73 (4.46) |
Percent change from baseline, mean (SD) | −5.81 (11.3) | −7.59 (9.64) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 67 |
Least squares mean (SE) | −2.44 (0.50) | −3.33 (0.56) |
Difference between treatments b | 0,89 g/L | |
95% CI, 1-sided P value | −0.57 to 2.34; P = 0.115 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | −1.55 (0.24) | −2.02 (0.26) |
Difference between treatmentsc | 0.48 g/L | |
95% CI, 1-sided P value | −0.22 to 1.18; P = 0.090 | |
Pulmonary function: Treatment comparisons for percent change from baseline to month 24 | ||
Number of patients contributing to the analysis | 89 | 84 |
FEV1 | ||
Difference between treatmentsd | −2.24 | |
95% CI, 2-sided P value | −5.73 to 1.26; P = 0.208 | |
FEV1% of predicted | ||
Difference between treatmentsd | −2.26 | |
95% CI, 2-sided P value | −5.79 to 1.26; P = 0.207 | |
FEV1 to FVC ratio | ||
Difference between treatmentsd | −4.24 | |
95% CI, 2-sided P value | −8.04 to −0.45; P = 0.029 | |
DLCO | ||
Difference between treatmentsd | −1.31 | |
95% CI, 2-sided P value | −6.80 to 4.19; P = 0.639 | |
Exacerbations | ||
Rate of exacerbations per patient-year | ||
Number of patients contributing to the analysis | 93 | 87 |
Estimated exacerbation ratioe | 1.26 | |
95% CI, 2-sided P value | 0.92 to 1.74; P = 0.152 | |
Duration in years | ||
Number of patients contributing to the analysis | 68 | 59 |
Mean (SD) | 0.26 (0.26) | 0.18 (0.19) |
Duration of hospitalizations due to exacerbations in years | ||
Number of patients contributing to the analysis | 19 | 16 |
Mean (SD) | 0.04 (0.03) | 0.02 (0.02) |
Duration of antibiotic treatment for exacerbations in years | ||
Number of patients contributing to the analysis | 59 | 52 |
Mean (SD) | 0.03 (0.01) | 0.02 (0.01) |
Symptoms and function | ||
Incremental shuttle walking test: Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 89 | 82 |
Difference between treatmentsf | −13.1 m | |
95% CI, 2-sided P value | −49.3 to 23.1; P = 0.477 | |
SGRQ symptom scale: Treatment comparison for change from baseline to month 24g | ||
Number of patients contributing to the analysis | 85 | 73 |
Difference between treatmentsh | −1.11 | |
95% CI, 2-sided P value | −6.20 to 3.99; P = 0.669 | |
A1-PI = alpha1-proteinase inhibitor; ANCOVA = analysis of covariance; CI = confidence interval; DLCO = diffusion capacity of carbon monoxide; FEV1 = forced expiratory volume in the first second; FRC = functional residual capacity; FVC = forced vital capacity; ITT = intention to treat; P15 = 15th percentile of the lung density; SD = standard deviation; SE = standard error; SGRQ = St. George’s Respiratory Questionnaire; TLC = total lung capacity.
aThe annual rate of change in physiologically adjusted P15 was analyzed using CT scan data taken at both TLC and FRC inspiration states. For the combined analysis, both inspiration states were included as fixed effects in the primary efficacy model simultaneously (i.e., TLC and FRC states combined) as opposed to the separate analyses of the CT scans at TLC and FRC inspiration states, which were investigated by applying the primary model without the fixed effect for inspiration state.
bAnalysis of the treatment difference for the change from baseline to month 24 in physiologically adjusted P15 for observed values using a mixed-effects model and ITT population. Statistical significance level of P = 0.025.
cTreatment comparison for annual rate of change in physiologically adjusted P15 (g/L) at TLC and FRC states combined and separately based on a random regression model (ITT population). Statistical significance level of P = 0.025 (uncontrolled for multiplicity).
dTreatment comparison for percentage change from baseline to month 24 in key spirometry variables for observed values (ANCOVA) (ITT population).
eTreatment comparison for rate of exacerbations per patient-year (negative binomial regression model) (ITT population).
fTreatment comparison for change from baseline to month 24 in exercise capacity test – distance walked (m) for observed values (ANCOVA) (ITT population).
gHigher scores in the SGRQ indicate more limitations in terms of overall health, daily life, and perceived well-being in patients with obstructive airway disease.
hTreatment comparison for change from baseline to month 24 in SGRQ symptoms score for observed values (ANCOVA) (ITT population).
Source: Clinical Study Reports for the RAPID trial.5
Harms
Only those harms identified in the review protocol are reported subsequently. See Table 10 for detailed harms data.
Adverse Events
The majority of patients experienced AEs throughout the trial duration (99% of patients in both treatment arms). The most frequently reported AE was headache (40% with A1-PI [human] and 38% with placebo). Other commonly reported AEs included respiratory-related AEs which, in some case, were numerically higher with A1-PI (human) than with placebo; however, these incidences are based on a limited number of patients. These AEs included COPD (32% and 23%, respectively), nasopharyngitis (32% and 30%), oropharyngeal pain (24% and 12%), condition aggravated (22% and 16%), cough (22% and 8%), lower respiratory tract infection (19% and 20%), dyspnea (18% and 12%), influenza (15% and 12%), upper respiratory tract infection (15% and 16%), pyrexia (14% and 7%), bronchitis (13% in each treatment arm), sinusitis (13% and 12%), and pneumonia (12% and 14%).
Serious Adverse Events
A total of 30% of patients in the A1-PI (human) treatment arm experienced SAEs compared with 32% of patients in the placebo arm. The most common SAEs reported in the A1-PI (human) arm were respiratory and included COPD (10% versus 2% in the placebo arm), pneumonia (3% versus 5%) and lower respiratory tract infection (1% versus 5%).
Withdrawals Due to Adverse Events
Discontinuations due to AEs were low. One patient withdrew from the study due to AEs in the A1-PI (human) treatment arm compared with 5 patients in the placebo arm.
Mortality
One patient in the A1-PI (human) treatment arm died over the study period due to respiratory failure. In the placebo arm, 3 patients died over the study period. The causes of death in the placebo group were sepsis (n = 1), pneumonia (n = 1), and metastatic breast cancer (n = 1).
Notable Harms
No cases of severe hypersensitivity were reported in the trial.
Table 10: Summary of Key Harms Outcomes in the RAPID Trial
Outcomes | A1-PI (N = 93) | Placebo (N = 87) |
|---|---|---|
Patients with ≥ 1 adverse events | ||
n (%) | 92 (98.9) | 86 (98.9) |
Most common events, n (%) | ||
Headache | 37 (39.8) | 33 (37.9) |
Chronic obstructive pulmonary disease | 30 (32.3) | 20 (23.0) |
Nasopharyngitis | 30 (32.3) | 26 (29.9) |
Oropharyngeal pain | 22 (23.7) | 10 (11.5) |
Condition aggravated | 20 (21.5) | 14 (16.1) |
Cough | 20 (21.5) | 7 (8.0) |
Lower respiratory tract infection | 18 (19.4) | 17 (19.5) |
Dyspnea | 17 (18.3) | 10 (11.5) |
Nausea | 15 (16.1) | 8 (9.2) |
Influenza | 14 (15.1) | 10 (11.5) |
Upper respiratory tract infection | 14 (15.1) | 14 (16.1) |
Pyrexia | 13 (14.0) | 6 (6.9) |
Bronchitis | 12 (12.9) | 11 (12.6) |
Sinusitis | 12 (12.9) | 10 (11.5) |
Pneumonia | 11 (11.8) | 12 (13.8) |
Patients with ≥ 1 serious adverse events | ||
n (%) | 28 (30.1) | 28 (32.2) |
Most common events, n (%) | ||
Chronic obstructive pulmonary disease | 9 (9.7) | 2 (2.3) |
Pneumonia | 3 (3.2) | 4 (4.6) |
Lower respiratory tract infection | 1 (1.1) | 4 (4.6) |
Patients who stopped treatment due to adverse events | ||
n (%) | 1 (1.1) | 5 (5.7) |
Events listing, n (%) | ||
Back pain | 1 (1.1) | 0 |
Asthenia | 0 | 1 (1.1) |
Balance disorder | 0 | 1 (1.1) |
Bronchitis | 0 | 1 (1.1) |
Deep vein thrombosis | 0 | 1 (1.1) |
Dizziness | 0 | 1 (1.1) |
Fatigue | 0 | 1 (1.1) |
Hypokinesia | 0 | 1 (1.1) |
Memory impairment | 0 | 1 (1.1) |
Neck pain | 0 | 1 (1.1) |
Sepsis | 0 | 1 (1.1) |
Tremor | 0 | 1 (1.1) |
Deaths | ||
n (%) | 1 (1) | 3 (3) |
Respiratory failure | 1 (1) | 0 |
Sepsis | 0 | 1 (1) |
Pneumonia | 0 | 1 (1) |
Breast cancer, metastatic | 0 | 1 (1) |
A1-PI = alpha1-proteinase inhibitor.
Source: Clinical Study Reports for the RAPID trial.5
Critical Appraisal
Internal Validity
Study Design, Intervention, and Comparator
RAPID was designed to evaluate the superiority of A1-PI (human) over placebo. The trial was randomized and double-blinded to minimize potential biases.
Selection, Allocation, and Disposition of Patients
Patients were appropriately randomized at a ratio of 1:1; randomization was stratified by centre. A randomization list containing the assignment of patient numbers to treatment groups was centrally generated by a computerized pseudo-random number generator.
RAPID was a double-blind study. A1-PI (human) and placebo were packaged identically. Individual packages were identified only by patient number. There is no indication that patients or the medical team could discover treatment allocation due to differential AEs or changes in disease progression. In addition, the primary outcome measure, lung density measured by CT scan, is considered objective and unlikely to be influenced by any suspicion of treatment allocation.
More patients receiving placebo withdrew from the study than patients receiving active treatment. The interpretation of these differences between treatment arms is limited by the small sample size; however, clinical experts’ opinion suggests this does not diminish confidence in the study results.
Outcome Measures
No adequate data were reported in RAPID for the outcomes of survival and lung transplant. This is to be expected however, considering the relatively short trial duration and slow disease progression.
The primary efficacy outcome in RAPID was lung density, as measured by CT scan. CT lung densitometry measurements have been validated as a primary clinical end point for clinical study designs in monitoring emphysema progression in AATD. The clinical experts consulted by CADTH considered lung density to be a more sensitive indicator of disease progression than other outcomes, such as lung function tests.
Scans were obtained at both TLC and FRC inspiration states. A range of approaches was selected to examine the primary efficacy variable, including analyses at different inspiration states and different adjustment models. However, the clinical experts consulted by CADTH agreed that the most reliable way to measure lung density is at a full inspiration state, which is referred to as TLC. When the lungs are full of air, more lung tissue is visible on the CT scan image and, therefore, the measurement obtained is considered more reliable. However, the interpretation of these findings is affected by the fact that it was not clear at which specific inspiration state the measure was to be taken for the primary analysis. As was the case for the TLC results, other inspiration state measures showed a slower decline in lung density with the active treatment compared with placebo over a 24-month period, but the differences between groups were of smaller magnitude and did not reach statistical significance. From a statistical perspective, this is a major limitation, especially since the analysis was not controlled for multiplicity.
The choice of secondary outcome measures was considered appropriate according to expert opinion. The 2 questionnaires used to assess symptoms and function were validated and have known minimally clinically important differences.
Statistical Analysis
Although RAPID had sufficient power for the analysis of the primary outcome, statistical significance was not reached for any of the secondary outcomes, suggesting a potential lack of power or sensitivity. The methods used for the analysis were appropriate for longitudinal data (mixed models and analysis of variance). There were predefined sensitivity analyses exploring different assumptions around the mechanism for missing data, as well as an exploration of the assumption of linearity of effect underlying their linear regression. The results for the sensitivity analyses were consistent with the main analyses, which is a strength. The changes around the interim analysis were not a threat to validity, as the unblinded analysis was not done and the blinded analysis was done late.
External Validity
Patient Selection
The inclusion and exclusion criteria appeared relevant and reasonable. Nevertheless, some categories of patients were excluded, such as elderly patients and those who underwent lung transplant. Therefore, the findings from RAPID are not generalizable to these categories of patients.
A majority of patients presented with a ZZ genotype. The level of disease progression appeared consistent with Canadian clinical practice, according to the clinical experts consulted by CADTH.
Treatment Regimen and Length of Follow-Up
The dose of A1-PI (human) used and IV method of administration were both in line with the Health Canada indication. However, the main issue is that RAPID does not inform on the comparative efficacy and safety of A1-PI (human) compared with any other active treatment. The 2-year double-blind treatment period was considered of appropriate duration to assess changes in lung density, but too short to see changes in relevant clinical outcomes, such as time to lung transplant or survival, considering the slow progression of emphysema in patients with A1-PI deficiency.
Outcome Measures
The primary outcome of lung density is a validated and reliable measure of disease progression in clinical trials. However, in terms of generalizability, clinical outcomes are usually preferred. As such, lung density is not used in clinical practice to assess disease progression and, therefore, it is unknown how the decrease observed in RAPID in terms of lung density translates into increases in patients’ symptoms and effects on daily activities. Pulmonary function, a secondary outcome in the trial, is regularly measured in clinical practice; however, it is not consistently representative of disease progression and severity of symptoms according to the clinical experts consulted by CADTH; significant emphysema may be present without marked loss of lung function. Symptoms and function were assessed using 2 questionnaires; however, the trial was not powered to show statistical significance for secondary outcomes.
Other Relevant Evidence
This section includes 1 submitted long-term extension study and 3 additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Long-Term Extension Study
One published, manufacturer-sponsored, open-label, long-term extension study has been summarized.
Methods
RAPID-OLE (n = 140),6 a long-term extension of RAPID, collected long-term data on the safety and efficacy of A1-PI (human) on the progression of emphysema in patients with emphysema with A1-PI deficiency who had completed the 2-year treatment and observation periods in the RAPID study. Patients residing in the US were excluded. Disease progression was assessed by the decline of lung density, measured by CT. The outcomes measured were also used to assess efficacy and safety in RAPID and have already been described in this report. All patients received A1-PI (human) administered at a dosage of 60 mg/kg IV infusion once weekly for an additional 24 months (total follow-up period of 48 months for RAPID plus RAPID-OLE). However, 2 treatment groups were defined based on the randomization assignment during the RAPID study: the early-start group included patients who had been randomized to active treatment, and the delayed-start group included patients who had been randomized to placebo and were reallocated to receive A1-PI (human).
Statistical Analysis
The ITT population was used for the analysis of the efficacy variables and comprised all patients included in the study. Patients may have not been included in all efficacy analyses due to missing efficacy assessments. However, all available data were analyzed.
The PP population was a subset of the ITT population, excluding patients and data affected by a major protocol deviation. The final decision regarding exclusion from the PP analysis was made before the database lock.
The completer population is a subset of the ITT population comprising patients who had a valid baseline lung density value (day 1 of the RAPID study) and a valid lung density value at month 48 from the RAPID-OLE study.
The safety population was used for the safety analyses and comprises all patients who were included in the study and who received at least 1 dose of A1-PI (human) during the RAPID-OLE study.
The primary efficacy analysis of repeated lung density measurements, pulmonary function data, and other longitudinal continuous measurements was based on linear random regression models. The change in the annual rate of lung density decline, as measured by adjusted P15, was calculated based on a mixed-effects model.
There was no formal hypothesis testing for confirmatory purposes. All analyses were exploratory. The statistics provided are descriptive.
Patient Disposition
Table 11: Patient Disposition, RAPID-OLE
Patient disposition | Early treatment | Delayed treatment |
|---|---|---|
Completed RAPID | 153 | |
Entered RAPID-OLE | 140 | |
76 | 64 | |
Completed study | 70 | 61 |
Discontinued study, n (%) | 6 (7.9) | 3 (4.7) |
Death | 1 (1.3) | 0 |
Adverse events | 0 | 1 (1.6) |
Withdrawn consent | 3 (3.9) | 1 (1.6) |
Other | 2 (2.6) | 1 (1.6) |
ITT, N | 76 | 64 |
PP, N | 72 | 60 |
Completer, N | 63 | 58 |
Safety, N | 76 | 64 |
ITT = intention to treat; OLE = open-label extension; PP = per protocol.
Source: Clinical Study Reports for the RAPID-OLE trial.15
Efficacy
The use of A1-PI (human) was associated with a change in lung density from month 24 to month 48 that was consistent with what was observed in patients treated with A1-PI (human) in RAPID. The change in lung density was also consistent between the early-start group (patients who had been randomized to active treatment) and the delayed-start group (patients who had been randomized to placebo and were reallocated to receive A1-PI [human]).
Harms
The majority of patients experienced AEs throughout the trial duration. A total of 36% of patients experienced SAEs; the most frequently reported SAE in all study patients was COPD (n = 17; 12.1%). Two patients discontinued due to AEs (COPD and lung transplant). One patient died; the patient was in the early treatment group and the cause of death was COPD.
Table 12: Summary of Key Results, RAPID-OLE
Result | Early treatment | Delayed treatment |
|---|---|---|
Change in the physiologically adjusted P15 lung density | ||
Inspiration state: TLC, ITT population | ||
Number of patients contributing to the analysis | 76 | 64 |
Baseline (month 24), mean (SD) | 42.2 (15.23) | 43.1 (14.03) |
End of treatment time point (month 48), mean (SD) | 38.1 (14.37) | 40.8 (15.19) |
Change from month 24 to 48, mean (SD) | −2.95 (5.343) | −2.52 (3.414) |
Percent change from month 24 to 48, mean (SD) | −6.81 (12.253) | −6.65 (9.449) |
Harms | ||
AEs, n (%) | 76 (100) | 62 (96.9) |
SAEs, n (%) | 28 (36.8) | 23 (35.9) |
WDAEs, n (%) | 1 (1.3) | 1 (1.6) |
Deaths, n (%) | 1 (1.3) | 0 |
AE = adverse event; ITT = intention to treat; P15 = 15th percentile of the lung density; SAE = serious adverse event; SD = standard deviation; TLC = total lung capacity; WDAE = withdrawal due to adverse event.
Source: Clinical Study Reports for the RAPID-OLE trial.15
Other Studies: Biochemical Efficacy
One manufacturer-sponsored, double-blind RCT has been summarized.
Methods
Study 2002 (n = 44)7 evaluated the noninferiority of Zemaira compared with Prolastin, another A1-PI product, on serum levels of A1-PI in adult patients with a diagnosis of A1-PI deficiency and clinical evidence of emphysema. All patients received an A1-PI product (Zemaira, n = 30; or Prolastin, n = 14) administered at a dosage of 60 mg/kg IV infusion once weekly for a 10-week blinded phase followed by a 14-week open phase.
Statistical Analysis
The following datasets were analyzed:
The safety (all treated) dataset, which comprised all patients enrolled into the study who received at least 1 dose of study medication, analyzed according to the treatment actually administered.
The PP dataset, which consisted of all patients with at least 1 evaluable trough serum A1-PI measurement in the time period between week 7 and week 11. The analysis of the PP dataset was performed according to the treatment actually administered.
The ITT dataset, which consisted of all patients with at least 1 follow-up data point, i.e., at least 1 evaluable trough serum A1-PI measurement in the time period between week 2 and week 11. The analysis of the ITT dataset was performed according to the randomized treatment.
The primary analysis was based on the PP dataset. Only serum antigenic A1-PI levels were considered. In a first step, the mean steady-state trough serum A1-PI level from week 7 to week 11 was calculated for each patient as a simple average of all evaluable measurements. In a second step, the per-patient means were considered to be “raw data” for the comparison between treatment groups. Each patient was weighted the same. It had to be demonstrated that the mean trough serum A1-PI level following Zemaira treatment was greater than 11 µM and not more than 3 µM lower than after Prolastin treatment. The noninferiority hypothesis was to be tested using a 2-group, 1-sided t-test at an alpha level of 5%. The second hypothesis was to be tested using a 1-group, 1-sided t-test at a level of 5%. Since both null hypotheses had to be rejected, an adjustment of the alpha level was not required. Two-sided 90% CIs were provided for the difference of mean steady-state trough serum antigenic A1-PI levels between treatment groups and for the mean steady-state trough serum antigenic A1-PI level in the Zemaira group (based on the data from week 7 to week 11).
A total of 45 patients were to be enrolled into this study, approximately 30 in the Zemaira group and approximately 15 in the Prolastin group. If the sample sizes of evaluable patients in the groups were 26 and 13 (corresponding to a dropout rate of 10% to 15%), a 2-group 1-sided t-test at an alpha level of 5% would have 95% power to reject the null hypothesis that Zemaira and Prolastin were not equivalent in favour of the alternative hypothesis that the means of the 2 groups were equivalent, assuming that the expected difference in means was 0.0 µM and the common SD was 2.6 µM.
Patient Disposition
One patient in each treatment group discontinued from the study.
Efficacy
The mean steady-state (weeks 7 to 11) trough antigenic A1-PI levels were 17.7 µM (SD = 2.50 µM; 90% CI, 16.88 to 18.45) in the Zemaira treatment group and 19.1 µM (SD = 2.20 µM; 90% CI, 18.07 to 20.15) in the Prolastin treatment group. The between–treatment groups difference was −1.45 µM (90% CI, −2.77 to −0.13). The predefined primary study objectives were met: the mean trough serum antigenic A1-PI level in the group treated with Zemaira was within 3 µM of the corresponding mean value in the group treated with Prolastin (noninferiority objective was met), and the mean trough serum antigenic A1-PI level in the group treated with Zemaira was greater than the therapeutic threshold of 11 µM. Consistency in the PP and ITT populations has been demonstrated by the alternative analysis of variance procedures as well as the planned statistical tests.
Harms
All patients reported at least 1 AE; however, only 1 patient in the Prolastin group discontinued the study treatment due to AEs. A total of 9 patients (20%) experienced at least 1 SAE. One patient in the Prolastin group died due to respiratory arrest with COPD.
Other Studies: Gaps in the Evidence
This section presents submitted additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Survival Analysis
One survival database study, Ellis (2019),8 available in a poster format only, evaluated the efficacy of A1-PI augmentation plus standard therapy in the US (active treatment arm) compared with standard therapy alone in the UK (control group) on the outcomes of survival and lung transplant in adult patients with A1-PI deficiency and evidence of lung disease.
Patients were matched based on age, sex, baseline year, and smoking status. The balance of covariates was assessed by calculation of standardized mean difference and by comparison of means. Kaplan–Meier survival curves were also performed.
Results from the survival analysis suggested a difference of 11.5% (95% CI, 3.6 to 19.2; P < 0.001) in favour of the US treatment group compared with UK matched controls. The proportions of patients who had a lung transplant within 5 years of baseline assessment was higher in the UK control group (58.5%; 95% CI, 40.3 to 71.2) compared with the US treatment group (13.3%; 95% CI, 7.54 to 18.7).
Several limitations highly affect our level of confidence in the evidence provided by Ellis (2019).8 The study was not randomized. The study results have only been reported in a poster; therefore, very little information is available to allow assessment of methodological quality. Despite matching efforts, the patient populations included in the 2 treatment groups were likely different, as they were from 2 different countries with 2 different health care systems. Patients receiving treatment in the US need to have private insurance while patients in the UK have access to a public health care system. There are therefore several potential confounding factors that cannot be entirely accounted for. In addition, differences between the 2 groups were noted in terms of data collection methods. The dosage and administration mode of the interventions were not specified, nor were concomitant medications. Immortal time bias needs to be considered in the US treatment group Considering the length of follow-up was different in the US versus the UK. There might also be differences in transplant waiting times between the 2 groups that are unrelated to treatment but are dependent on the health care system.
Discussion
Summary of Available Evidence
One published, manufacturer-sponsored, double-blind RCT was included in the systematic review: RAPID (n = 180).3,4 This trial evaluated the superiority of A1-PI (human) compared with placebo on the progression of disease in patients with emphysema with A1-PI deficiency and a reduced lung function. A1-PI (human) was administered at a dosage of 60 mg/kg through IV infusion once weekly for 24 months.
Though RAPID may be considered methodologically rigorous, the small sample sizes and lack of clarity regarding the choice of inspiration state for the primary statistical analyses of lung density limited interpretation of the findings. The trial population appeared similar to the patients seen in clinical practice by the clinical experts consulted by CADTH; however, the real-world effectiveness of A1-PI (human) in patients living in Canada may vary from what was observed in the trial. The strength of evidence was reduced by the lack of controlled long-term data on efficacy and safety, and the lack of trials comparing the clinical outcomes of A1-PI (human) with another active treatment.
Therefore, additional relevant evidence addressing important gaps in the evidence were considered. Additional evidence first included 1 open-label long-term extension study from RAPID. RAPID-OLE (n = 140)6 collected long-term data on the safety and efficacy of A1-PI (human) on the progression of disease in patients with emphysema with A1-PI deficiency who had completed the 2-year treatment and observation periods in the RAPID study (except for patients residing in the US). A1-PI (human) was administered at a dosage of 60 mg/kg through IV infusion once weekly. Disease progression was assessed by the decline of lung density, measured by CT, for an additional 24 months, for a total follow-up period of 48 months.
Another additional relevant study included in the sponsor’s submission to CADTH was a noninferiority biochemical efficacy trial: Study 2002 (n = 44)7 evaluated the noninferiority of Zemaira compared with Prolastin for a 10-week blinded phase on serum levels of A1-PI in adult patients with a diagnosis of A1-PI deficiency and clinical evidence of emphysema.
Finally, 1 survival analysis8 evaluated the efficacy of A1-PI (human) plus standard therapy in the US compared with standard therapy alone in the UK on the outcomes of survival and lung transplant in adult patients with A1-PI deficiency and evidence of lung disease. There were several limitations inherent with the database study design and the differences between groups, especially in terms of patient population, that highly affect our level of confidence in the evidence.
Interpretation of Results
Efficacy
A1-PI (human) was associated with a reduced rate of decline in lung density after 24 months compared with placebo in patients with emphysema with A1-PI deficiency and reduced lung function when CT scans were taken at a full inspiration state, and this measurement is considered more reliable. CT lung densitometry measurements have been validated as a primary clinical end point for clinical study designs in monitoring emphysema progression in AATD. According to patient input, stabilizing lung function is perceived as the most important outcome in effective treatment because it is associated with the ability to perform activities of daily living. However, lung density is not used in clinical practice to assess disease progression and, therefore, it is unknown how the slower decrease in lung density observed in RAPID translates into a better daily life for patients. RAPID does not inform on the efficacy of A1-PI (human) on the outcomes of survival and lung transplant, but this would have been hardly feasible, according to the clinical experts consulted by CADTH, considering the relatively short trial duration and slow disease progression. Other important clinical outcomes such as exacerbations and symptoms and function were reported as secondary outcomes; however, the differences between groups did not reach statistical significance for any of these outcomes.
The interpretation of the findings is affected by the fact that it was not clear at which specific inspiration state the measure was to be taken for the primary analysis. As was the case for TLC results, other inspiration state measures showed a slower decline in lung density with active treatment compared with placebo over a 24-month period, but the differences between groups were of smaller magnitude and did not reach statistical significance. From a statistical perspective, this is a major limitation, especially since the analysis was not controlled for multiplicity. However, according to the clinical experts consulted by CADTH, the most reliable way to measure lung density is at a full inspiration state, which is referred to as TLC. When the lungs are full of air, there is more lung tissue visible on the CT scan image and, therefore, the measurement obtained is considered more reliable. According to the clinical experts consulted by CADTH, other measurements of inspiration states are considered less reliable and are used mainly for volume correction.
To mitigate the lack of controlled long-term data on efficacy and safety and the lack of trials comparing the clinical outcomes of A1-PI (human) with other active treatments, additional evidence provided the following insights. Despite the limitations associated with the open-label uncontrolled trial design, findings from 1 long-term extension study suggested that the efficacy of A1-PI (human) was sustained in the long-term. As for comparative efficacy, Zemaira was considered to be noninferior to Prolastin in a biochemical efficacy study based on the mean steady-state trough serum antigenic A1-PI levels. In addition, findings from 1 survival analysis suggested that treatment with an A1-PI was associated with benefits in terms of survival and time to lung transplant; however, several limitations inherent with the database study design and the differences between groups, especially in terms of patient population, highly affect our level of confidence in these findings.
Harms
Virtually all patients in both treatment groups experienced at least 1 AE; however, discontinuation due to AEs was low, suggesting the harm profile might be considered acceptable. Respiratory-related AEs were commonly reported and, in some instances, were numerically higher with A1-PI (human) than with placebo; however, this might be an anecdotical observation due to the small sample size. SAEs were frequently reported, and their incidence was similar between treatment groups. No cases of severe hypersensitivity were reported in the trial. One patient in the A1-PI (human) treatment arm died over the study period due to respiratory failure. In the placebo arm, 3 patients died over the study period from sepsis, pneumonia, and metastatic breast cancer.
Additional data regarding A1-PI (human) safety included an OLE of RAPID that generally confirmed the safety profile observed in the double-blind study. No new safety signal was identified throughout the OLE.
Other Considerations
Based on the patient input received and clinical experts’ opinion, there is currently an unmet need in the treatment of A1-PI deficiency, a condition where lung tissue damage results in early emphysema and decreased life expectancy. Access to treatment is unequal and so, in areas where there are no publicly funded treatments available, patients have to weigh the steps they are willing to take to access therapy, including continuing to work past retirement age to be eligible for private insurance, uprooting their lives to relocate to a province that offers coverage, or participating in clinical trials. Untreated A1-PI deficiency results in use of supportive medications, frequent lung function tests, hospitalizations during exacerbations and, eventually, assessment for lung transplant. Patients reported that the need to demonstrate deteriorated lung function before becoming eligible for augmentation therapy results in additional stress, with a perception that their lungs are being further damaged, further compromising their quality of life, while waiting to become eligible. Stabilizing lung function is perceived as the most important outcome in effective treatment because it is associated with the ability to perform activities of daily living and fully participate in families and communities.
Conclusions
The results of RAPID suggest that A1-PI (human) was associated with a reduced rate in the validated primary outcome of decline in lung density after 24 months compared with placebo in patients with emphysema with A1-PI deficiency and a reduced lung function when CT scans were taken at a full inspiration state. This shows that treatment with an A1-PI might preserve lung tissue in these patients; however, as lung density is not used in clinical practice to assess disease progression, the extent of how these findings translate into clinical benefits for patients in real life is unknown. RAPID was not informative regarding the efficacy of A1-PI (human) on the outcomes of survival and lung transplant, the sample size being relatively small and follow-up of limited duration for a slowly progressive disease. Other important clinical outcomes such as exacerbations and health-related quality of life were reported as secondary outcomes, for which no difference was seen. No major safety signal was identified. Additional evidence assessed to address important gaps in the evidence suggested a long-term maintenance of effect of more than 48 months, as well as a similar biochemical efficacy compared with Prolastin; however, the level of confidence in the evidence is highly affected by several limitations, including the open-label uncontrolled trial design of the long-term extension study.
References
1.de Serres F, Blanco I. Role of alpha-1 antitrypsin in human health and disease. Journal of Internal Medicine. 2014;276(4):311-335. PubMed
2.Zemaira alpha1-proteinase inhibitor (human): powder and diluent for solution for injection [product monograph]. Ottawa (ON): CSL Behring Canada Inc.; 2019 Dec 16.
3.Greulich T, Chlumsky J, Wencker M, et al. Safety of biweekly alpha1-antitrypsin treatment in the RAPID programme. European Respiratory Journal. 2018;52(5):11. PubMed
4.Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360-368. PubMed
5.Clinical Study Report: CE1226_4001. A randomized, placebo-controlled, double-blind, multicenter Phase III/IV study to compare the efficacy and safety of 60 mg/kg body weight of Zemaira® weekly i.v. administration with placebo weekly i.v. administration in chronic augmentation and maintenance therapy in subjects with emphysema due to alpha1-proteinase inhibitor deficiency [internal sponsor's report]. Marburg (Germany): CSL Behring GmbH; 2015 Sep 10.
6.McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). The Lancet Respiratory Medicine. 2017;5(1):51-60. PubMed
7.Stocks JM, Brantly M, Pollock D, et al. Multi-center study: The biochemical efficacy, safety and tolerability of a new alpha1-proteinase inhibitor, Zemaira. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2006;3(1):17-23. PubMed
8.Ellis P, Holm K, Choate R, et al. Comparison of outcomes in augmentation naïve and augmented patients with alpha-1 antitrypsin deficiency related lung disease. European Respiratory Journal. 2019;54(suppl 63):PA3383.
9.Anonymous. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397-415. PubMed
10.Gøtzsche PC, Johansen HK. Intravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung disease. Cochrane Database Syst Rev. 2016;9(9):Cd007851. PubMed
11.Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respiratory Research. 2010;11:136. PubMed
12.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. Journal of clinical epidemiology. 2016;75:40-46. PubMed
13.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jan 26.
14.Drug Reimbursement Review sponsor submission: Zemaira, alpha1-protinase inhibitor (human) [internal sponsor's package]. Ottawa (ON): CSL Behring Canada; 2021 Sep 26.
15.Clinical Study Report: CE1226_3001. An open-label, non-controlled, multicenter, multinational study to evaluate the efficacy and safety of Zemaira® administration in chronic augmentation and maintenance therapy in subjects with emphysema due to alpha1-proteinase inhibitor deficiency who completed clinical study CE1226_4001. Marburg (Germany): CSL Behring GmbH; 2015 Jun 26.
16.Jones PW. Interpreting thresholds for a clinically significant change in health status in asthma and COPD. European Respiratory Journal. 2002;19(3):398. PubMed
17.Singh SJ, Jones PW, Evans R, Morgan MDL. Minimum clinically important improvement for the incremental shuttle walking test. Thorax. 2008;63(9):775. PubMed
18.Plasma Protein Therapeutics Association. PPTA Statement on Clinical and Surrogate Endpoints for Evaluating Efficacy of Alpha 1-Proteinase Inhibitor (Human) Augmentation Therapy. 2020; https://www.pptaglobal.org/.../954-ppta-statement-on-clinical-and-surrogate-endpoints.). Accessed 2021 Nov 14.
19.Shaker SB, Stavngaard T, Stolk J, Stoel B, Dirksen A. Alpha1-antitrypsin deficiency. 7: Computed tomographic imaging in alpha1-antitrypsin deficiency. Thorax. 2004;59(11):986-991. PubMed
20.Dawkins PA, Dowson LJ, Guest PJ, Stockley RA. Predictors of mortality in alpha1-antitrypsin deficiency. Thorax. 2003;58(12):1020-1026. PubMed
21.DenOtter TD, Schubert J. Hounsfield Unit. StatPearls. Treasure Island (FL): StatPearls Publishing; 2021: https://www.ncbi.nlm.nih.gov/books/NBK547721/. Accessed 2021 Nov 14.
22.Parr DG, Stoel BC, Stolk J, Stockley RA. Validation of computed tomographic lung densitometry for monitoring emphysema in alpha1-antitrypsin deficiency. Thorax. 2006;61(6):485-490. PubMed
23.Parr DG, Dirksen A, Piitulainen E, Deng C, Wencker M, Stockley RA. Exploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study of augmentation therapy in alpha 1-antitrypsin deficiency. Respiratory Research. 2009;10:75. PubMed
24.Crossley D, Renton M, Khan M, Low EV, Turner AM. CT densitometry in emphysema: a systematic review of its clinical utility. International Journal of Copd. 2018;13:547-563. PubMed
25.Jones PW, Quirk FH, Baveystock CM. The St George's Respiratory Questionnaire. Respir Med. 1991;85 Suppl B:25-31; discussion 33-27.
26.FDA. Chronic Obstructive Pulmonary Disease: Use of the St. George’s Respiratory Questionnaire as a PRO Assessment Tool Guidance for Industry. 2018; https://www.fda.gov/media/71121/download. Accessed 2021 Nov 14.
27.Stockley RA, Edgar RG, Starkey S, Turner AM. Health status decline in alpha-1 antitrypsin deficiency: a feasible outcome for disease modifying therapies? Respiratory Research. 2018;19(1):137. PubMed
28.Singh SJ, Morgan MD, Scott S, Walters D, Hardman AE. Development of a shuttle walking test of disability in patients with chronic airways obstruction. Thorax. 1992;47(12):1019-1024. PubMed
29.Lung Foundation Australia. Pulmonary rehabilitation: Incremental Shuttle Walking Test. 2016; https://pulmonaryrehab.com.au/patient-assessment/assessing-exercise-capacity/incremental-shuttle-walking-test/. Accessed 2021 Nov 14.
30.Singh SJ, Morgan MD, Hardman AE, Rowe C, Bardsley PA. Comparison of oxygen uptake during a conventional treadmill test and the shuttle walking test in chronic airflow limitation. Eur Respir J. 1994;7(11):2016-2020. PubMed
31.Hiller A-M, Piitulainen E, Jehpsson L, Tanash H. Decline in FEV(1) and hospitalized exacerbations in individuals with severe alpha-1 antitrypsin deficiency. International journal of chronic obstructive pulmonary disease. 2019;14:1075-1083. PubMed
32.Stockley RA, Edgar RG, Pillai A, Turner AM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? International journal of chronic obstructive pulmonary disease. 2016;11:1745-1756. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946–present)
Embase (1974–present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 1, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: RCTs; controlled clinical trials
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase); keyword (CDSR) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
alpha 1-antitrypsin/
((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one or a1) adj3 (antitrypsin or anti-trypsin or antiprotease or anti-protease or antiproteinase or anti-proteinase)).ti,ab,kf,ot,hw,rn,nm.
((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one or a1) adj4 (protease or proteinase or trypsin) adj4 (inhibitor* or inhibit or inhibits)).ti,ab,kf,ot,hw,rn,nm.
(alpha-1-PI or alfa-1-PI or Alpha1-PI or alfA1-PI or alpha-one-PI or alfa-one-PI).ti,ab,kf,ot,hw,rn,nm.
(prolastin* or aralast* or alfalastin* or pulmolast* or zemaira* or glassia* or trypsone* or respreeza* or respitin* or respikam* or infinia* or serpina1 or serpin A1 or A1PI or A1 PI or A1AT or F43I396OIS or Alpha1AT or CSL-964 or CSL964).ti,ab,kf,ot,hw,rn,nm.
(antitrypsin adj2 (Pittsburgh or Portland)).ti,ab,kf,ot,hw,rn,nm.
or/1-6
alpha 1-antitrypsin deficiency/
(((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one or a1) adj3 (antitrypsin or anti-trypsin or protease inhibitor* or proteinase inhibitor* or trypsin inhibitor*)) and deficien*).ti,ab,kf.
(mckusick 10740 or A1AD or AATD or A1ATD or A-1ATD or ((A1AT or A1AP or A1A or AAT) adj3 deficien*)).ti,ab,kf.
((inherit* or genetic or hereditary or alpha or alfa) and emphysema).ti,ab,kf.
((lung or pulmonary) adj3 deficien*).ti,ab,kf.
or/8-12
7 and 13
14 use medall
*alpha 1 antitrypsin/ or *alpha 1 antitrypsin concentrate/
((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one or a1) adj3 (antitrypsin or anti-trypsin or antiprotease or anti-protease or antiproteinase or anti-proteinase)).ti,ab,kf,dq.
((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one or a1) adj4 (protease or proteinase or trypsin) adj4 (inhibitor* or inhibit or inhibits)).ti,ab,kf,dq.
(alpha-1-PI or alfa-1-PI or Alpha1-PI or alfA1-PI or alpha-one-PI or alfa-one-PI).ti,ab,kf,dq.
(prolastin* or aralast* or alfalastin* or pulmolast* or zemaira* or glassia* or trypsone* or respreeza* or respitin* or respikam* or infinia* or serpina1 or serpin A1 or A1PI or A1 PI or A1AT or Alpha1AT or CSL-964 or CSL964).ti,ab,kf,dq.
(antitrypsin adj2 (Pittsburgh or Portland)).ti,ab,kf,dq.
or/16-21
alpha 1 antitrypsin deficiency/
(((alpha-1 or alfa-1 or Alpha1 or alfa1 or alpha-one or alfa-one) adj3 (antitrypsin or anti-trypsin or protease inhibitor* or proteinase inhibitor* or trypsin inhibitor*)) and deficien*).ti,ab,kf,dq.
(mckusick 10740 or A1AD or AATD or A1ATD or A-1ATD or ((A1AT or A1AP or A1A or AAT) adj3 deficien*)).ti,ab,kf,dq.
((inherit* or genetic or hereditary or alpha or alfa) and emphysema).ti,ab,kf,dq.
((lung or pulmonary) adj3 deficien*).ti,ab,kf,dq.
or/23-27
22 and 28
29 use oemezd
30 not (conference abstract or conference review).pt.
15 or 31
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
“Randomized Controlled Trial (topic)”/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
“Controlled Clinical Trial (topic)”/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf,kw.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf,kw.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf,kw.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf,kw.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(pragmatic study or pragmatic studies).ti,ab,hw,kf,kw.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf,kw.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(phase adj3 (III or “3”) adj3 (study or studies or trial*)).ti,hw,kf,kw.
or/33-63
32 and 64
(zemaira or respreeza).ti,ab,kf,ot,hw,rn,nm,dq.
64 and 66
67 not (conference abstract or conference review).pt.
65 or 68
remove duplicates from 69
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search – (Zemaira OR Respreeza OR Alfalastin OR Aralast OR Glassia OR Prolastin OR Prolastina OR Pulmolast OR Respikam OR Trypsone OR “Serpin A1” OR antitrypsin OR “alpha 1 Proteinase Inhibitor” OR “alpha 1 Protease Inhibitor” OR A1PI OR serpina1 OR Alpha1AT OR “anti-elastase” OR A1P1) AND (“Alpha1-antitrypsin Deficiency” OR Emphysema OR “Alpha-1 AT Deficiency” OR “Chronic Obstructive Pulmonary Disease” OR pulmonary OR “AAT Deficiency” OR AATD OR COPD OR “Alpha 1-proteinase Inhibitor Deficiency”)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms – with results – (Zemaira OR Respreeza OR Alfalastin OR Aralast OR Glassia OR Prolastin OR Prolastina OR Pulmolast OR Respikam OR Trypsone OR “Serpin A1” OR antitrypsin OR “alpha 1 Proteinase Inhibitor” OR “alpha 1 Protease Inhibitor” OR A1PI OR serpina1 OR Alpha1AT OR “anti-elastase” OR A1P1) AND (“Alpha1-antitrypsin Deficiency” OR Emphysema OR “Alpha-1 AT Deficiency” OR “Chronic Obstructive Pulmonary Disease” OR pulmonary OR “AAT Deficiency” OR AATD OR COPD OR “Alpha 1-proteinase Inhibitor Deficiency”)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – (Zemaira OR Respreeza OR Alfalastin OR Aralast OR Glassia OR Prolastin OR Prolastina OR Pulmolast OR Respikam OR Trypsone OR “Serpin A1” OR antitrypsin OR “alpha 1 Proteinase Inhibitor” OR “alpha 1 Protease Inhibitor” OR A1PI OR serpina1 OR Alpha1AT OR “anti-elastase” OR A1P1) AND (“Alpha1-antitrypsin Deficiency” OR Emphysema OR “Alpha-1 AT Deficiency” OR “Chronic Obstructive Pulmonary Disease” OR pulmonary OR “AAT Deficiency” OR AATD OR COPD OR “Alpha 1-proteinase Inhibitor Deficiency”)]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – (Zemaira OR Respreeza OR Alfalastin OR Aralast OR Glassia OR Prolastin OR Prolastina OR Pulmolast OR Respikam OR Trypsone OR “Serpin A1” OR antitrypsin OR “alpha 1 Proteinase Inhibitor” OR “alpha 1 Protease Inhibitor” OR A1PI OR serpina1 OR Alpha1AT OR “anti-elastase” OR A1P1) AND (“Alpha1-antitrypsin Deficiency” OR Emphysema OR “Alpha-1 AT Deficiency” OR “Chronic Obstructive Pulmonary Disease” OR pulmonary OR “AAT Deficiency” OR AATD OR COPD OR “Alpha 1-proteinase Inhibitor Deficiency”)]
Grey Literature
Search dates: October 18 to 26, 2021
Keywords: Zemaira OR Respreeza OR Alfalastin OR Aralast OR Glassia OR Prolastin OR Prolastina OR Pulmolast OR Respikam OR Trypsone OR “Serpin A1” OR antitrypsin OR “alpha 1 Proteinase Inhibitor” OR “alpha 1 Protease Inhibitor” OR A1PI OR serpina1 OR Alpha1AT OR “anti-elastase” OR A1P1) AND (“Alpha1-antitrypsin Deficiency” OR Emphysema OR “Alpha-1 AT Deficiency” OR “Chronic Obstructive Pulmonary Disease” OR pulmonary OR “AAT Deficiency” OR AATD OR COPD OR “Alpha 1-proteinase Inhibitor Deficiency”)
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
There were no excluded studies.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 14: Detailed Efficacy Outcomes in the RAPID Trial
Outcome | A1-PI N = 93 | PL N = 87 |
|---|---|---|
Change in the physiologically adjusted P15 lung densitya | ||
Inspiration state: Mean of TLC and FRC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 46.6 (15.6) | 49.8 (15.1) |
End of treatment time point (month 24), mean (SD) | 44.4 (15.5) | 45.5 (13.9) |
Change from baseline, mean (SD) | -2.67 (4.30) | -3.93 (4.02) |
Percent change from baseline, mean (SD) | -6.06 (9.67) | -8.28 (8.89) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 67 |
Least square means (SE) | -2.33 (0.45) | -3.37 (0.50) |
Difference between treatmentsb | 1.04 g/L | |
95% CI, 1-sided P value | -0.26 to 2.34; P = 0.058 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | -1.50 (0.22) | -2.12 (0.24) |
Difference between treatments c | 0.62 g/L | |
95% CI, 1-sided P value | -0.02 to 1.26; P = 0.029 | |
Inspiration state: TLC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 45.5 (15.8) | 48.9 (15.5) |
End of treatment time point (month 24), mean (SD) | 43.6 (16.0) | 43.9 (13.8) |
Change from baseline, mean (SD) | -2.60 (4.44) | -4.20 (4.50) |
Percent change from baseline, mean (SD) | -6.22 (9.66) | -8.97 (10.3) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 66 |
Least square means (SE) | -2.22 (0.47) | -3.54 (0.52) |
Difference between treatments b | 1.32 g/L | |
95% CI, 1-sided P value | -0.03 to 2.67; P = 0.028 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | -1.45 (0.23) | -2.19 (0.25) |
Difference between treatments c | 0.74 g/L | |
95% CI, 1-sided P value | 0.06 to 1.42; P = 0.017 | |
Inspiration state: FRC | ||
Number of patients contributing to the analysis | 90 | 83 |
Baseline, mean (SD) | 47.6 (15.7) | 50.7 (15.0) |
End of treatment time point (month 24), mean (SD) | 45.3 (15.3) | 46.8 (13.8) |
Change from baseline, mean (SD) | -2.74 (4.75) | -3.73 (4.46) |
Percent change from baseline, mean (SD) | -5.81 (11.3) | -7.59 (9.64) |
Treatment comparison for change from baseline to month 24 | ||
Number of patients contributing to the analysis | 80 | 67 |
Least square means (SE) | -2.44 (0.50) | -3.33 (0.56) |
Difference between treatments b | 0,89 g/L | |
95% CI, 1-sided P value | -0.57 to 2.34; P = 0.115 | |
Treatment comparison for annual rate of change (primary analysis in the trial) | ||
Number of patients contributing to the analysis | 92 | 85 |
Point estimate (SE) | -1.55 (0.24) | -2.02 (0.26) |
Difference between treatments c | 0.48 g/L | |
95% CI, 1-sided P value | -0.22 to 1.18; P = 0.090 | |
Pulmonary function (Treatment comparisons for percent change from baseline to month 24) | ||
FEV1 | ||
Number of patients contributing to the analysis | 89 | 84 |
Least square means (SE) | -4.29 (1.26) | -2.06 (1.30) |
Difference between treatments d | -2.24 | |
95% CI, 2-sided P value | -5.73 to 1.26; P = 0.208 | |
FEV1% of predicted | ||
Number of patients contributing to the analysis | 89 | 84 |
Least square means (SE) | -4.16 (1.27) | -1.90 (1.31) |
Difference between treatments d | -2.26 | |
95% CI, 2-sided P value | -5.79 to 1.26; P = 0.207 | |
FEV1/FVC ratio | ||
Number of patients contributing to the analysis | 89 | 84 |
Least square means (SE) | -2.68 (1.36) | 1.56 (1.40) |
Difference between treatments d | -4.24 | |
95% CI, 2-sided P value | -8.04 to -0.45; P = 0.029 | |
DLCO | ||
Number of patients contributing to the analysis | 89 | 84 |
Least square means (SE) | -3.16 (1.96) | -1.85 (2.03) |
Difference between treatments d | -1.31 | |
95% CI, 2-sided P value | -6.80 to 4.19; P = 0.639 | |
Exacerbations | ||
Rate of exacerbations per patient-year | ||
Number of patients contributing to the analysis | 93 | 87 |
Average exacerbation rate, patient-year | 1.53 | 1.21 |
Estimated exacerbation ratio e | 1.26 | |
95% CI, 2-sided P value | 0.92 to 1.74; P = 0.152 | |
Health-related quality of life | ||
Incremental shuttle walking test (Treatment comparison for change from baseline to month 24) | ||
Number of patients contributing to the analysis | 89 | 82 |
Least square means (SE) | 1.77 (13.0) | 14.9 (13.5) |
Difference between treatments f | -13.1 m | |
95% CI, 2-sided P value | -49.3 to 23.1; P = 0.477 | |
St. George’s Respiratory Questionnaire (SGRQ) Treatment comparison for change from baseline to month 24 g | ||
Number of patients contributing to the analysis | 85 | 73 |
Least square means (SE) | -1.19 (1.79) | -0.09 (1.93) |
Difference between treatments h | -1.11 | |
95% CI, 2-sided P value | -6.20 to 3.99; P = 0.669 | |
CI = confidence interval; DLCO = diffusion capacity of carbon monoxide; FEV1 = forced expiratory volume in the first second; FRC = functional residual capacity; FVC = forced vital capacity; P15 = 15th percentile of the lung density; SD = standard deviation; SE = standard error; TLC = total lung function.
a The annual rate of change in physiologically adjusted P15 was analyzed using CT scan data taken at both TLC and FRC inspiration states. For the combined analysis, both inspiration states were included as fixed effects in the primary efficacy model simultaneously (i.e., TLC and FRC states combined) as opposed to the separate analyses of the CT scans at TLC and FRC inspiration states which were investigated by applying the primary model without the fixed effect for inspiration state.
b Analysis of the treatment difference for the change from baseline to month 24 in physiologically adjusted P15 for observed values using a mixed-effects model and ITT population. Statistical significance level of P = 0.025.
c Treatment comparison for annual rate of change in physiologically adjusted P15 (g/L) at TLC and FRC states combined and separately based on a random regression model (ITT population) – point estimates. Statistical significance level of P = 0.025.
d Treatment comparison for % change from baseline to month 24 in key spirometry variables for observed values (ANCOVA) (ITT population).
e Treatment comparison for rate of exacerbations per subject year (negative binomial regression model) (ITT population).
f Treatment comparison for change from baseline to month 24 in exercise capacity test – distance walked (m) for observed values (ANCOVA) (ITT population).
g Higher scores in the SGRQ indicate more limitations in terms of overall health, daily life, and perceived well-being in subjects with obstructive airway disease.
h Treatment comparison for change from baseline to month 24 in SGRQ symptoms score for observed values (ANCOVA) (ITT population).
Source: Clinical Study Reports for the RAPID trial.5
Table 15: Detailed Harms Outcomes
Outcome | RAPID A1-PI N = 93 | RAPID Placebo N = 87 |
|---|---|---|
Patients with ≥ 1 adverse event | ||
n (%) | 92 (98.9) | 86 (98.9) |
Most common eventsa, n (%) | ||
Headache | 37 (39.8) | 33 (37.9) |
Chronic obstructive pulmonary disease | 30 (32.3) | 20 (23.0) |
Nasopharyngitis | 30 (32.3) | 26 (29.9) |
Oropharyngeal pain | 22 (23.7) | 10 (11.5) |
Condition aggravated | 20 (21.5) | 14 (16.1) |
Cough | 20 (21.5) | 7 (8.0) |
Lower respiratory tract infection | 18 (19.4) | 17 (19.5) |
Dyspnea | 17 (18.3) | 10 (11.5) |
Nausea | 15 (16.1) | 8 (9.2) |
Influenza | 14 (15.1) | 10 (11.5) |
Upper respiratory tract infection | 14 (15.1) | 14 (16.1) |
Pyrexia | 13 (14.0) | 6 (6.9) |
Back pain | 12 (12.9) | 10 (11.5) |
Bronchitis | 12 (12.9) | 11 (12.6) |
Sinusitis | 12 (12.9) | 10 (11.5) |
Pneumonia | 11 (11.8) | 12 (13.8) |
Fatigue | 8 (8.6) | 10 (11.5) |
Hypertension | 6 (6.5) | 9 (10.3) |
Edema peripheral | 6 (6.5) | 10 (11.5) |
Toothache | 5 (5.4) | 9 (10.3) |
Patients with ≥ 1 serious adverse event | ||
n (%) | 28 (30.1) | 28 (32.2) |
Most common events,b n (%) | ||
Chronic obstructive pulmonary disease | 9 (9.7) | 2 (2.3) |
Pneumonia | 3 (3.2) | 4 (4.6) |
Lung neoplasm | 2 (2.2) | 1 (1.1) |
Pneumothorax | 2 (2.2) | 1 (1.1) |
Back pain | 1 (1.1) | 1 (1.1) |
Bronchitis | 1 (1.1) | 1 (1.1) |
Diverticulitis | 1 (1.1) | 2 (2.3) |
Dyspnea | 1 (1.1) | 2 (2.3) |
Lower respiratory tract infection | 1 (1.1) | 4 (4.6) |
Patients who stopped treatment due to adverse events | ||
n (%) | 1 (1.1) | 5 (5.7) |
Events listing, n (%) | ||
Back pain | 1 (1.1) | 0 |
Asthenia | 0 | 1 (1.1) |
Balance disorder | 0 | 1 (1.1) |
Bronchitis | 0 | 1 (1.1) |
Deep vein thrombosis | 0 | 1 (1.1) |
Dizziness | 0 | 1 (1.1) |
Fatigue | 0 | 1 (1.1) |
Hypokinesia | 0 | 1 (1.1) |
Memory impairment | 0 | 1 (1.1) |
Neck pain | 0 | 1 (1.1) |
Sepsis | 0 | 1 (1.1) |
Tremor | 0 | 1 (1.1) |
Deaths | ||
n (%) | 1 (1) | 3 (3) |
Respiratory failure | 1 (1) | 0 |
Sepsis | 0 | 1 (1) |
Pneumonia | 0 | 1 (1) |
Breast cancer metastatic | 0 | 1 (1) |
a Experienced by ≥ 10% of patients in either group.
b Experienced by at least 1 patient in each group.
Source: Clinical Study Reports for the RAPID trial.5
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
The CT densitometry measurement in the lung
SGRQ
ISWT
Of note, the literature search focused on patients with AATD. Therefore, we did not investigate the validity of instruments in patients with other respiratory diseases (e.g., COPD, asthma).
Findings
Table 16: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
St. George’s Respiratory Questionnaire (SGRQ) | Disease-specific HRQoL questionnaire:
Domains:
|
| 4 points for patients with asthma (Total score)16 In COPD patients16:
|
Incremental shuttle walking test |
|
| In COPD:
MID 47.5 m (5 shuttles) in COPD17: No information for MID in AATD population. |
AATD = alpha1-antitrypsin deficiency; COPD = chronic obstructive pulmonary disease; MID = minimally important difference; HRQoL = health-related quality of life.
The CT Densitometry
CT lung densitometry measurements have been validated as a primary clinical end point for longitudinal and clinical study designs in monitoring emphysema progression in AATD. CT densitometry has been included in the guidance documents as a sensitive method for monitoring progressing emphysema in patients with AATD compared with routine pulmonary function tests or rate/severity of pulmonary exacerbations owing to the evidence generated from several randomized and longitudinal studies that have used CT densitometry to measure emphysema progression in patients with AATD.18,19
Shaker et al., 200419 provides a background overview of the use of CT in diagnosing and quantifying emphysema in general including emphysema associated with AATD. They discuss CT features in AATD and its correlation with pathology, lung function tests and quality of life in addition to the role of CT scanning in monitoring disease progression based on evidence generated from different study reports. They note that visual and quantitative CT scanning were closely correlated to the extent of emphysema and conclude that CT scanning is most sensitive in diagnosing emphysema, attributing subtypes, and determining severity. Most studies evaluated by Shaker et al.19 observed a good correlation between CT scanning with measures of lung function and health status and concluded that CT scanning may be more sensitive than lung function tests in monitoring emphysema in longitudinal studies.
CT Densitometry Validation Studies
Dawkins and colleagues (2003)20 prospectively assessed the predictive potential of physiological, radiological, and health status features to respiratory in relation to mortality in 256 patients with AATD (with the PiZ phenotype) using CT scanning. Follow-up data (including lung function tests, CT scans, post-bronchodilator flow rates post-measurements, and carbon monoxide uptake) for each patient was made available through the UK registry of patients with AATD. A total of 254 patients underwent lung function testing of whom 194 had undergone a thoracic CT scan. Lung function tests, CT scans, health status (measured using the St. George’s Respiratory Questionnaire, SGRQ), and other clinical data of both survivors and non-survivors were collected and compared in this study and these parameters were applied to survival analyses calculations. All deaths related to emphysema or an exacerbation of COPD (including bronchial infection or pneumonia leading to respiratory failure) were included as respiratory-related deaths. The predictive nature of the measurements was assessed using a forward stepwise regression analysis based on the Cox proportional hazard method. Only 170 (out of the 254) patients who had completed lung testing with complete follow-up data were included in the regression analysis. A total of 22 deaths were recorded in the study of which 10 were respiratory-related. The authors reported worse baseline status, poorer lung function, and poorer high-resolution CT (HRCT) scans in patients who died. They observed significantly lower scores in lung function parameters (which included FEV1, carbon monoxide transfer coefficient [KCO]), and CT scores in patients who died versus survivors. Patients that died also showed higher scores (worse) in the CT voxel index scores (defined as the proportion of highlighted voxels [volume element measured in mm3]) which are expressed as a percentage of the total voxel. According to the authors, this score is reflective of the proportion of emphysematous tissue) for scans made at both thoracic levels (the aortic arch (upper zone) and the level of the inferior pulmonary vein/right atrial confluence (lower zone) in inspiration and expiration and higher scores in the symptom questionnaire (SGRQ) used which indicated worse health status. Their univariate analysis showed that the upper zone expiratory scans had the best association with all-cause (P = 0.001) and respiratory mortality (P = 0.001). The FEV1 (P = 0.158 for all-cause, 0.015 for respiratory) and KCO (P = 0.002 for all-cause, 0.012 for respiratory) showed poorer associations with mortality. Only age provided more independent predictive information to all-cause mortality or respiratory mortality when CT scan data were included into their survival analysis. Their findings further confirm that FEV1 can predict respiratory mortality in patients with AATD.
The relationship between emphysema progression and disease stage in AATD has been explored using 2 CT approaches: the 15th percentile point (defined in this study as the cut-off value in Hounsfield units [HU] below which a specified percentage of all voxels are distributed), and voxel index (defined in the study as the proportion of lung voxels of low density below a specified threshold. The voxel is a volume element in 3-dimensional measurement expressed in mm3), threshold −950 HU (measurement unit/scale used in CT densitometry to interpret images. Calculations are based on a linear transformation baseline linear attenuation coefficient of the X-ray beam21) in 2 studies conducted by Parr et al., (2006).22 The validity of CT for monitoring emphysema was also assessed by evaluating the relationship between progression of CT lung densitometry and the rate of FEV1 decline in patients with AATD (PiZ). In the first study, the consistency of progression measured using densitometry and FEV1 in relation to disease stage was assessed in a group of 74 patients earlier recruited in a 2-year study. The relationship between CT densitometric parameters was based on a cross-sectional analysis of baseline images ((upper zone parameter + lower zone parameter)/2). The upper zone represents the area through the middle of the aortic arch and lower zone was defined as the area at the junction of inferior pulmonary veins and left atrium. The authors observed a close agreement between both densitometric parameters in the baseline measurements with a curvilinear relationship between Perc15 and VI −950 (rS = 0.994; P < 0.001). The 74 patients were further sub-grouped according to the Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease (GOLD) classification of disease stage (FEV1 > 80% predicted (group 1); FEV1 50% to 79% predicted (group 2); FEV1 30% to 49% predicted (group 3); FEV1 < 30% predicted (group 4)) and further analyses were performed. The authors found no significant trend in the rate of progression of Perc15 in association with disease stage, but the rate of progression of VI −950 was significantly associated with disease stage (a clear trend in progression rate was observed of VI −950, with a graded increase in rate in association with worsening disease stage that was observed in both the individual and combined upper and lower zone images (P = 0.004)). In second group of 34 patients who had completed 4 consecutive annual assessments, the annual measurements obtained in the 3-year period was summarized statistically. There was a correlation observed between the annual rate of CT progression in the upper zone images and an annual rate of decline in FEV1. FEV1 decline correlated with progression of lung densitometry (percentile point: rS = 0.527; P = 0.001; voxel index: rS = −0.398; P = 0.012). The authors concluded that the 15th percentile point CT method was consistent in measuring lung density compared with using the voxel method and are both valid to use in measuring emphysema longitudinal and interventional studies. Their study showed a relation between progression of CT densitometry and rate of decline in FEV1 in patients with AATD.
Parr and colleagues (2009)23 clarified the optimum approach for using CT densitometry data in assessing progression of emphysema in AAT deficiency (AATD) patients receiving alpha 1-antitrypsin (AAT) augmentation therapy in a randomized trial (EXACTLE). A total of 77 patients were randomly assigned to receive infusions of 60 mg/kg human AAT (Prolastin) or placebo over 2 to 2.5 years (Prolastin [n = 38] or placebo [n = 39]).Patients recruited in this study were 18 years and older, had a history of at least 1 exacerbation in the past 2 years, had a post-bronchodilator forced expiratory volume in 1 second (FEV1) ≥ 25% and ≤ 80% predicted and a ratio of post-bronchodilator FEV1 to slow vital capacity ≤ 0.70, or a carbon monoxide transfer coefficient /VA) of ≤ 0%. The primary outcome was progression of emphysema determined by change in lung density measured by CT scan of the whole lung (divided in 3 regions: pical, middle, and basal regions). The 15th percentile point was chosen as the parameter expressed as the 15th percentile lung density (P15), was defined in the study as the value (in Houndsfield units) below which the 15% of voxels with the lowest density are distributed. They expressed the PD15 value in grams per litre (PD15 [g/L] by adding 1,000 to the Hounsfield value obtained for the 15th percentile point). They determined sensitivity ratios for each densitometric index used in the trial by dividing the baseline value for the mean change from baseline in lung density by the standard error to obtain the sensitivity index. The study identified the most discriminative densitometric index that could be used as an outcome measure and compared regional densitometry with whole lung densitometry assessments. The CT scans were performed at baseline and at 12 and 24 months, with an option for continuation with an additional scan performed at 30 months. The mean decline in lung density was determined and adjusted for lung volume. The CT densitometric indices showed a significant decline in both groups studied throughout the trial (Prolastin and placebo). Findings from the regional densitometry measurements showed that the rate of emphysema in the placebo arm was similar between the apical, middle, and basal regions of the lung whereas in the active arm, the rate of emphysema progression was lower in basal regions compared with the apical and middle regions of the lung. The authors observed a significant treatment effect in the basal region which was also statistically significant (P = 0.04) compared with the other regions measured. Similar trends were also observed in the other regions of the lung but they were not statistically significant (middle [P = 0.155] and apical regions [P = 0.673]). The sensitivity ratios determined showed that the PD15 measurement of the basal region was more a sensitive than the analysis conducted in the apical region. The authors observed changes in PD15 from baseline to last CT scan (−2.645 g/L [Prolastin group] and −4.117 g/L [placebo group]), which were indicative of significant treatment effect (P = 0.049). Their analyses showed that the 15th percentile method (PD15) was a more sensitive measure (than the voxel method) based on the sensitivity ratios determined for all densitometry indices used in the study the study. The authors underline that the findings show that differences in the inspirational level may greatly influence the PD15 value obtained compared with other indices implemented in the study and they further highlight the importance of a method for correcting the differences observed in ling volume between scans when the PD15 method is used. After the authors adjusted density values to correct for differences in inspiration levels, the authors observed a decline in lung density using the indices included in the study. Decline in lung density observed in the measurements was consistent with emphysema progression. The article did not report comparisons between the CT measurements made and lung function measurements conducted.
In a systematic review with meta-analysis, Crossley and colleagues (2018) assessed the relationship between CT and routine clinical markers in patients with COPD and AATD.24 The authors summarized available studies presenting information related to the use of CT densitometry as a measure of severity and progression of lung disease in emphysema in relation to other clinical parameters such as lung function, mortality, hospital admissions and quality of life (QoL). A total of 36 studies were retrieved which compared CT densitometry with FEV1 percent predicted, 55 studies reported associations between the −950 HU (CT density) and clinical parameters, 23 studies comparing DLCO percent predicted to CT density, and 5 studies comparing the use of the SGRQ and CT density, 4 studies investigating CT density for COPD exacerbations, 6 studies evaluating CT density and mortality (3 of these studies provide a hazard ratio for all-cause mortality). The association between CT density and other clinical parameters were judged suitable as outcomes for airways disease trials (e.g., FEV1, SGRQ) and were consistently significant across the studies evaluated. They also observed a clear and consistent relationship between CT density and mortality suggesting that this measure is an appropriate surrogate outcome measure in studies of emphysema. The heterogeneity observed between the studies retrieved for the review was a major limitation precluding the authors from making further conclusions on the strength of each association. Notwithstanding, they observed a consistency in the direction of the relationship between density and lung function across patient groups corroborating that the CT density could be a valid surrogate outcome across a spectrum of diseases. Unfortunately, the wide range values for CT versus FEV1 correlations observed in their analyses made it difficult to identify an exact reference value for CT density that related to the minimally clinically important difference for the FEV1.
The St. George's Respiratory Questionnaire
The St. George’s Respiratory Questionnaire (SGRQ) is a disease-specific instrument, originally designed to assess patient-related quality of life in patients with obstructive lung/airway disease. Several versions have been developed to address specific disease areas, for example, the SGRQ-C (COPD-specific version of the SGRQ) and the SGRQ-I (interstitial lung disease). It can be self-administered or via face-to-face or telephone interviews and can be completed in 10 minutes.25,26
The questionnaire consists of 50 items with 76 weighted responses that measure health status across 3 domains: symptoms, which consists of 8 items (measures frequency and severity); activity, which consists of 16 items (measures activities that cause or are limited by breathlessness); and impact, which consists of 26 items (measures social functioning and psychological disturbances resulting from airways disease). The recall period varies depending on the instrument version used. It could be 1 year, 3 months or 4 weeks.26 During the development of the instrument, a 2-week recall period was implemented to assess repeatability in patients with stable asthma and, a year later, to assess responsiveness. The symptoms domain is scored using a 5-point Likert scale, while the activity and impact domains are scored in a dichotomous (yes or no) manner. Each domain score is calculated by weighting items in the domain. Individual scores are derived for each domain and a total score derived from the 3 domains (symptom, activity, and impacts) can also be computed ranging from 0 to 100. Higher scores from the questionnaire represent poor health status and lower scores represent good health status.25
Validity
During the development of the SGRQ instrument, the investigators collected weights from 140 patients across a wide range of asthma severity and across different countries (England, Finland, Holland, Italy, Thailand, and the US). Patients with stable asthma completed the questionnaires twice at 2-week intervals and the results showed good repeatability. They obtained a coefficient of variation of 19% for the total SGRQ score for the paired measurements. The respondents were also requested to complete other questionnaires for HRQoL (Medical Research Council [MRC] respiratory questionnaire and the Hospital Anxiety and Depression Scale [HAD] alongside the SGRQ). Spirometry and the 6-minute walking test (6MWD) were also administered to the patients. The study reported a correlation between the SGRQ and the reference measure in the other questionnaires that were relevant. A strong correlation was reported for cough and wheeze with the SGRQ symptoms score. The 6MWD and MRC dyspnea grade correlated with the SGRQ activity score, and the impact domain of the SGRQ showed more balanced distribution of associations, including anxiety, walking distance, dyspnea, and wheeze.25
The general health status was measured in a sample of 152 patients with airflow obstruction (composed of a mixed population of patients with asthma chronic obstructive airway disease). Patients completed the questionnaire with other HRQoL instruments (the Sickness Impact Profile (SIP), HAD, and MRC respiratory questionnaire, spirometry tests were conducted, and patients were also asked to complete the 6MWD test). The mean age of participant recruited was 63 years, and the mean FEV1 was 48% predicted (range: 11 to 114%). They observed a large measure of agreement between the total score obtained in the SGRQ and the SIP questionnaire although the SGRQ showed greater sensitivity to differences in disease severity in patients.
Responsiveness25
A total of 122 patients (of the 152 patients with chronic airflow limitation (chronic obstructive airway disease including asthma patients that were originally included during the development of the instrument)) that had completed questionnaires at baseline completed the questionnaire a year after the first administration. The authors reported correlations in the SGRQ total score and changes in the range of measures of activity (FVC, 6MWD, MRC dyspnea grade, frequency of wheeze, anxiety, and depression). The authors noted that changes in SGRQ score were significantly correlated with changes in FVC, anxiety, MRC dyspnea score and frequency of wheeze in their regression analysis (correlation coefficients obtained between SGRQ and of range of activity measurements were not presented in the paper for the 122 patients that completed the questionnaire 1 year later).
MID25
The authors conclude that Total SGRQ score of about 4 points would indicate clinically significant differences between populations (with chronic obstruct airway disease including asthma patients). Differences in scores greater than 7 points were considered quite large. The correlation (r2) values for the SGRQ total score in comparison with the spirometry tests and other questionnaires were not reported in the article.
Responsiveness
The responsiveness measurement showed small changes in the overall population over time. They obtained the following scores from the HRQoL investigated: SGRQ symptoms score, 1.1 ± 14.1; SGRQ total score, 1.3 ± 11.8; SF-36 physical component summary score, 21.0 ± 7.7; HAD depression score, 0.6 6 2.5; total fatigue score, 0.6 ± 2.7; and shuttle distance, 21.4 ± 121.1m. They observed a stronger correlation in change of SGRQ scores with changes in physical component measures than with changes in mental component measures.
MID
The clinical threshold for the St. George’s Respiratory Questionnaire has been assessed in patients with asthma and COPD. In patients with COPD, patient data were collected from a 16-week study that assessed the use of salmeterol in COPD. Patients were asked to describe their overall health based on a 5-point scale by choosing 1 of the following responses: “no effect,” “satisfactory,” “effective, or “very effective.” A score of 2.0 units (95% CI, 0.2 to 4.1 units; n = 87) was obtained from respondents who judged the treatment as “satisfactory.” In patients (n = 109) who judged the treatment as being “effective,” a mean improvement in the SGRQ score was 4.3 units (95% CI, 1.8 to 6.9 units) while a score of 8.1 units (95% CI, 4.7 to 11.4 units; n = 55) was obtained in patients that judged treatment to be “very effective.”16
Stockley and colleagues (2018)27 assessed decline in health using the St. Georges Respiratory Questionnaire (SGRQ) in a cohort of closely monitored patients with AATD who had never been treated with antitrypsin augmentation therapy to investigate if it could be implemented in new AATD trials. The study assessed the relationship between decline in lung physiology (structure) and SGRQ decline. Patients were recruited from the UK-AATD database and had follow-up data of up to 7 years. In total, 454 patients with the PiZZ genotype who had available data for at least 4 consecutive annual visits were assessed. A linear regression model was used to evaluate the decline in SGRQ total score and domains to obtain the change per year. The authors performed a comparison of the data with that from the Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease (GOLD) stages and subgroups defined as non and rapid decliners (≥ 1% predicted decline per year) for FEV1 and KCO. Their results showed a statistically significant (P < 0.0001) correlation of baseline SGRQ scores with baseline FEV1 although they obtained an r2 = 0.34 which indicated that that the variability in the group could be explained by FEV1 alone. They also found the statistically significant correlations (P< 0.0001) between the SGQR scores and patients with or without COPD although the variance for each group was small (r2 = 0.20 and 0.15 respectively). Statistically significant correlations were also observed between SGRQ total score and gas transfer for the whole group (r2 = 0.105; P < 0.0001) and those with COPD (r2 = 0.015; P = 0.008) but not those without COPD (r2 = 0.002; P = 0.35). A yearly increase in SGRQ scores was observed for the groups with and without COPD with a wide range seen. Stable scores were observed in the symptom and activity score in the non-COPD group, but the median total score showed gradual deterioration (+ 0.21 units per year). Worsening of symptoms and activity with time and an overall median increase in total score of + 0.66 units/year was observed in the COPD group although only the activity and total score deteriorated more rapidly than for those without COPD (P = 0.001 and P= 0.025 respectively).
No information available for MID, responsiveness, validity, and reliability in patients with AATD.
Incremental Shuttle Walking Test
The ISWT is a symptom test, developed to assess functional capacity in patients with COPD. It simulates an external field walking exercise with additional stress factors allowing patients to respond at a symptom-limited performance. The test is a modified version of a progressive externally paced 20 m shuttle running test that has been widely used to assess functional capacities in athletes.28,29
Test Procedure
During the test, patients are required to walk up and down a 10 m course identified by 2 cone insets, 0.5 m from either end. Patient walking speed is dictated with the help of an audio recorder signal played on a cassette tape. Accuracy of the timed signal is ensured by a calibration period of 1 minute. Patients are being asked to walk until they feel or are unable to maintain the required speed without becoming breathless. A standardized explanation to the test is played to the patient prior to commencing the test. The test is designed to begin after audio emits triple bleeps and participants are signalled by a bleep at different intervals to turn around the cone and proceed back down the course. The test design allows for small incremental increases in walking speed every minute. A change in speed to a next level is signalled by a triple bleep sound emitted by the audio recorder.
The walking speeds are assigned to levels, from level 1 onward, each lasting 1 minute. The distance a patient walks until they reach a cone is called a shuttle, representing 10 m. The number of shuttles in each level depends on the walking speed assigned at that level. The test is considered terminated if the patient is too breathless to maintain the required speed or if the operator ends the test based on the patient failure to complete the shuttle in the time allowed (that is, was more than 0 5 m away from the cone when the bleep sounded) or if the patient attains 85% of the predicted maximal heart rate (derived from the formula [210 – (0-65 × age)]).28
Standardization29
It is expected that investigators that intend to administer the test to patients standardize the procedure to ensure meaningful outcomes. It is recommended that the test be performed twice, and the best result recorded to account for the learning effect. Test administrators must allow a time lapse of at least 30 minutes if administered on the same day, or less than 1-week apart if administered in debilitated patients. Only instructions played from the recorder should be used during the test with no encouragements from investigators. The test area should have a comfortable ambient temperature throughout testing. Tracks must be similar for all tests administered to patient.
Validity
Singh et al. (1994)30 investigated the relationship between patients’ performance on the shuttle walking test and the maximum oxygen uptake (VO2max) during a conventional treadmill test in patients with chronic airflow limitation. The authors examined this relationship using 2 approaches: the conventional Douglas bag techniques (treadmill test) and a portable oxygen consumption meter (shuttle test). A total of 22 patients recruited into the study were placed in 2 separate experiments. Nineteen patients were assigned to the first experiment- the modified Balke treadmill walking test in a randomized and balanced design. This test evaluated the relationship between shuttle test performance and the VO2max. Patients underwent a single practice walk test and a maximal treadmill walking test, in a randomized, balanced design. Patients were required to perform a symptom-limited maximal exercise test during the treadmill test. The second experiment randomly assigned 10 patients in a shuttle test which was made up of a practice test, an unencumbered shuttle walking test and a test supporting the portable oxygen consumption meter. This test assessed physiological responses of patients. Patients paid 3 visits to the hospital at 1-week intervals, usually on the same day. Baseline measurements were recorded at all visits which included spirometry measurements (FEV1 and FVC), completion of the Chronic Respiratory Disease Questionnaire) in addition to height and weight measurements. The authors observed a strong relationship between patient performance in the shuttle test and the VO2max during the treadmill walking test (r = 0.88) and a consistent incremental increase in oxygen consumption and ventilation in response to the increasing intensity of the shuttle walking test (and observed strong relationship between VO2max and patient performance on the shuttle test ((r = 0.81)).
MID and Responsiveness in Patients With COPD
Singh et al., 200817 have established a minimally clinically important difference for the ISWT test in patients with COPD. A total of 372 patients recruited during the study underwent an initial tolerance test where they completed 2 shuttle ISWT tests with adequate rests of 20 to 30 minutes in between. These patients were subsequently enrolled into a rehabilitation program lasting for 7 weeks. Patients were asked to record exercise progress in diaries which were used to monitor compliance and progress. Another ISWT test was conducted in patients after 7 weeks after which subjects were asked to rate their exercise tolerance from a 5-response scale: “Compared to last time, how would you rate your exercise tolerance?” Responses were categorized as better (1), slightly better (2), about the same (3), slightly worse (4), or worse (5). Each response was assigned a value from 1 to 5. In their analysis, the authors reported a mean baseline age of 69.4 (SD = 8.4) years, a FEV1 of 1.06 (SD = 0.53), and a FEV1 to FVC ratio of 50.8 (18.1%). The mean baseline shuttle distance was 168.5 m (SD = 114.6 m), which increased to 234.7 m (SD = 125.3 m) after rehabilitation (mean improvement of 65.9 m [95% CI, 58.9 to 72.9]). They also report no relationship between baseline ISWT performance and the improvement in shuttle distance following rehabilitation based on the Bland-Altman plot constructed. They conducted a 1-way analysis of variance for the 5 response categories, which showed a significant difference in the mean distance achieved (P < 0.001). No differences were observed for responses in categories 1 (better), 2 (slightly better), and 3 (about the same) with a value of P > 0.05. The mean improvements observed in patients who responded, “slightly better” was 47.5 m (95% CI, 38.6 to 56.5) and in patients who responded “better,” mean improvements were 78.7 m (95% CI, 70.5 to 86.9), while those that responded “about the same” was 18.0 m (95% CI, 4.5 to 31.5). The authors concluded that for patients to rate their tolerance as “better,” they were required to improve by 8 shuttles in comparison with 5 shuttles, for them to rate tolerance as “slightly better,” and 2 shuttles to report “about the same.” The minimum clinically significant improvement in the ISWT identified in the study was 47.5 m (or 5 shuttles).
The authors also tested whether improvements reported by patients was independent of the observed baseline performance. Patients were sub-grouped into 4 quartiles (0 m to 80 m; 90 m to 150 m; 160 m to 250 m and ≥ 250 m) based on baseline ISWT scores. The authors observed that the increase in distance in patients that responded “slightly better” was not significantly different between the quartiles (P value = 0.9). In the group of patients (n = 55) that responded that they had no improvement, a change of 18 m was observed. As cited by the authors, the lower 95% CI, did not include no change. The authors concluded that “the patients had failed to rate small changes in the exercise performance.”
No information available on reliability, validity, and responsiveness in AATD population.
Appendix 5: Summary of Other Studies
Note that this appendix has not been copy-edited.
Aim
The aim of this section was to summarize and appraise evidence from Hiller et al., 201931 and Stockley et al., 201632 that were used to inform the pharmacoeconomic model.
Summary of Hiller et al., 201931
The economic model submitted by the sponsor assumed a starting value of 2.05 L for FEV1 upon entry into the model (baseline values obtained in the age group 40 to 59), this was based on the values reported in Hiller et al.
Hiller et al. (2019) estimated decline in FEV1 (ΔFEV1) in patients diagnosed with AAT deficiency (phenotypes PiZZ, PiZNull or PiNullNull) in a prospective registry and assessed factors that contributed to rapid decline in lung function in these patients. Patients 18 years and older diagnosed with severe disease (by isoelectric focusing testing) and currently enrolled in the Swedish National AAT Deficiency Registry, were included in the study. Patients participating in this registry have routine clinical and lung function examinations performed every 2 years, including the collection of other baseline data (smoking status, age, and augmentation therapy status) which were reported using questionnaires. The authors assessed decline in FEV1 based on spirometry results (at least 3) obtained at 2-year intervals during the defined follow-up period. Patients with COPD at baseline were sub-grouped into 4 categories according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines; GOLD I FEV1 ≥ 80% predicted, GOLD II 50% ≤ FEV1 < 80% predicted, GOLD III 30% ≤ FEV1 < 50% predicted and GOLD IV FEV1 < 30% predicted. Exacerbations were assessed using data retrieved from the Swedish National Patient Register. Data were obtained by crosslinking the AATD registry with the SNPR. Of the 1,640 patients enrolled in the registry, only 1,132 patients had data from at least 3 spirometry tests, thus were included in the study. Baseline characteristics were presented based on smoking status and age. The mean FEV1 value at baseline were 3.69 L (1.19), 2.05 L (1.12), and 1.72 L (0.83) in the age groups 18 to 39, 40 to 59, and at least 60, respectively.
Critical Appraisal of Hiller et al., 201931
Patient data were retrieved from the Swedish National AAT Deficiency Registry. Registry data were collected using questionnaires (physicians reported results of lung function tests and patients reported some baseline values such as smoking status and respiratory symptoms via questionnaires) which may have introduced reporting bias into the study. The authors did not report if the questionnaires had been previously validated and whether local hospitals used the same devices to measure lung function tests. This may have introduced variability in measurements for each patient and at each time frame. The spirometry measurements performed for each patient were conducted in accordance with regulatory (European) recommendations which increases the robustness of the data obtained. However, the authors highlight that only the limitation is that only pre-bronchodilator values were analyzed for the study. This may reduce the generalizability of the findings to only pre-bronchodilator measurements. Data related to exacerbations in patients was obtained from another registry (the Swedish National Patient Register). Although the registry is said to cover more than 99% of hospitalizations (since 1987) and 80% of hospital-based outpatient care since 2001, there is potential selection bias because some patients may have been excluded if they visited private clinics. There is also the risk of information bias (from misdiagnosis and non-differential bias based on differences in the ICD codes used). The authors did not mention whether the database has been validated prior to the study commencement. The study received ethical approval from the Lund University Regional Ethical Review Board prior to commencement. The authors clearly describe the statistical methods applied in the study to obtain the findings and defined the follow-up time for changes in FEV1 measurements, models used, and the variables included in the models fitted. The significance level was defined as alpha= 0.05. The authors present P values and corresponding confidence intervals. The baseline values (age, sex, including other variables such as smoking status, results of spirometry and lung functions tests) were collected and presented in the paper. The authors identify the strengths and limitations of the study.
Comparison of Hiller’s Study and the RAPID Trial
Patient population is similar to that of RAPID trial (PiZZ, PiZNull, or PiNullNull phenotypes). However, patients were identified through isoelectric focusing testing while in RAPID A1-PI deficiency was assessed by measuring functional A1-PI serum levels and was not based on genetic diagnosis.
The study population in the RAPID trial included newly diagnosed, previously untreated, currently treated patients, and patients who had previously received augmentation therapy while in Hiller’s study, patients included had not received augmentation therapy.
Baseline characteristics in terms of FEV1 differed in the 2 studies, where in the RAPID trial the baseline FEV1 was 1.58 L and 1.60 L in the alpha1-proteinase inhibitor and the placebo groups, respectively, while in Hiller et al., the mean FEV1 value at baseline were 3.69 L (1.19), 2.05 L (1.12), and 1.72 L (0.83) in the age groups 18 to 39, 40 to 59, and at least 60, respectively. Hence patients included in the Hiller et al. registry had higher FEV1 values at baseline than those enrolled in the RAPID trial, and therefore the average decline in the FEV1 reported in Hiller et al. might not be at the same rate as those enrolled in the RAPID trial. In the Hiller study, the authors report greater annual decline in FEV1 is participants that were grouped (based on baseline lung function values) as mild and moderate COPD (based on the GOLD I and II guidelines) compared with those with severe COPD.
The Health Canada indication of alpha1-proteinase inhibitor (Human) is for maintenance treatment in adults with severe A1-PI deficiency and clinical evidence of emphysema; however, it is not reported in Hiller et al.’s registry whether the patients had clinical evidence of emphysema.
Patients in Hiller were also placed in subgroups based on age (patients were at inclusion into registry into “young” (age = 18 to 39 years), “middle age” (age = 40-59 years), and “old” (> 60 years) and also sub-grouped based on smoking status. Stratification by age groups in RAPID trial is not similar to the groups created in Hiller’s study.
Respiratory symptoms experienced by patients were collected via questionnaires which may have introduced bias. Uncertainty related to validation of questionnaires prior to use. Uncertainty related to the validation of data from the registry prior to the study. In the RAPID trial, patients recorded exacerbations in patient diaries and were evaluated on every visit by investigator. Internal validity issues.
Frequency of assessments for lung functions were not performed in the same manner as in the RAPID trial. Decline in FEV1 was assessed based on results of 3 spirometry tests at 2-year intervals (or longer). In the RAPID trial, FEV1% predicted was assessed based on the Crapo criteria measured at baseline and in all visits. Results from both studies are not comparable.
In RAPID, reduced lung function in RAPID was defined as FEV1 ≥ 35% and ≤ 70% predicted according to the American Thoracic Society staging strata. In Hiller et al. (2019), lung measurements and definitions were made according to the European recommendations. Patients with COPD were sub-grouped into 4 at baseline based on the GOLD guidelines. Differences in guidelines used may have introduced bias which influences external validity of results.
Given the limitations mentioned above, it is uncertain whether Canadian patients with severe A1-PI deficiency and clinical evidence of emphysema who would be eligible for A1-PI (Human) would have a value of 2,050 mL for FEV1 when they start receiving treatment, as patients enrolled in the RAPID trial had a lower FEV1 at baseline.
Summary of Stockley et al. (2016)32
The economic model submitted by the sponsor indicated that there is heterogeneity in the rate of FEV1 decline among patients and using data from Stockley et al. the sponsor estimated that the weighted average of baseline FEV1 decline to be −49.03 mL/year.
Stockley et al. (2016) assessed decline in post-bronchodilator FEV1 and gas transfer (% predicted) in patients with AATD who had never received augmentation therapy. In total, 482 Patients (with PiZ phenotype) that were enrolled in the ADAPT (Antitrypsin Deficiency Assessment and Program for Treatment) registry with at least 4 annual lung function assessments were included in the analysis. Patients in the registry underwent post-bronchodilator lung function testing, including measurement of lung volumes and gas transfer (DLCO and KCO) in a local centre. Other baseline factors of patients were collected prospectively, and index cases were defined as those tested because of presentation with respiratory symptoms. COPD was defined as subjects with a post-bronchodilator FEV1/FVC ratio < 70%. Decline in lung function was determined by the change in % predicted for age, sex, height, and ethnicity. lung function decline was grouped into 4 categories groups based normal aging values: change (< −0.1% predicted per year), slow (−0.1% to < −0.5%), moderate (−0.5% to < −1.0%), and rapid decline (> −1.0%). The authors observed significant variations in individual rates of FEV1 decline from levels consistent with normal aging (23.5% of patients with established COPD, 57.5% of those without) to those of rapidly declining COPD. In total, 12.8% of non-smokers and 20.7% of ex-smokers with established COPD showed no decline in gas transfer while in patients with COPD, 33.3% of non-smokers and 25% of smokers showed no gas transfer decline. The authors observed no correlations between gas transfer and FEV1 in patients diagnosed with COPD while in non-COPD patients, a weak correlation was observed (r = 0.218; P < 0.025). These findings led to the conclusions that there exist differing rates of lung function decline in AATD.
Critical Appraisal of Stockley et al. (2016)32
The study by Stockley et al. (2016) was designed to collect data prospectively from patients referred to the ADAPT registry. All patients participating in the registry were assessed at a primary centre (the Birmingham centre), which eliminates variability in measurements between patients. However, there is potential assessment bias, given that the investigators were not blinded when examining patient data. Different spirometry tests were performed in the study (post-bronchodilator lung function testing, lung volume measurement, and gas transfer [DLCO and KCO]), based on acceptable guidelines, which increases the credibility and generalizability of the findings obtained. Baseline data were also collected (smoking history and current status, quality of life, exacerbation history and drug history) at the centre which increases the validity of the data. The presence of COPD was defined in the study as patients with post-bronchodilator FEV1/FVC ratio ,70%. This definition may be subjective and cannot be generalized to other practice settings. The author’s measured decline in lung function as % predicted based on published equations. Thus, the findings are generalizable to settings that use similar equations as those applied to derive the value. The statistical methods were appropriately described by the authors. Follow-up duration was also highlighted in the paper. The multivariate analysis was performed using (backwards) stepwise linear regression and the variable used in the model were defined a priori. The baseline characteristics of patients participating in the study were adequately captured in the paper. Correlation coefficients and P values were presented for the group comparisons conducted.
Comparison Between Stockley et al. and the RAPID Trial
Patients with the PiZ phenotype were included in the study (similar to RAPID). However, it is unclear from the paper what method of testing was used to confirm AATD prior to enrolling patient into the registry.
Lung function assessments were performed annually (patients included in the study had completed at least 4 annual assessments). This is different from RAPID where assessments were made at every visit. Results in both studies are not comparable.
Patients who had received augmentation therapy or any other potential disease-modifying therapy as part of a previous clinical trial were excluded from the analysis. This differs from RAPID in terms of patient populations included. Internal validity issues and may not be comparable with RAPID.
Assessments were conducted in a local hospital setting. Potential measurement and assessment bias by health care professionals and differences in instrument validation may introduce bias in results.
Guidelines used for the assessments may not be similar to those used in the RAPID trial (they do not specify the guidelines they used in the paper)- External validity since guidelines may not be the same.
Other baseline variables collected in the study were similar to those of in the RAPID trial.
Decline in lung function was calculated by determining the change in % predicted for age, sex, height, and ethnicity using published equations. Results were divided into those whose lung function decline was consistent with normal aging. In RAPID, lung function decline assessments were not made according to the subgroups used in Stockley. Results in the 2 studies are not comparable.
Patients in Stockley’s study were sub-grouped into those with COPD and non-COPD, which was not the case in RAPID. Difficult to assess similarities in baseline characteristics in Table 1 for both studies owing to the groups introduced in the analysis.
It was not reported whether patients had clinical evidence of emphysema.
The sponsor used weighted average to calculate FEV1 decline, however, there was no adjustment for confounding factor in this estimation especially given that lung function decline in AATD differs between patients.
The sponsor assumed that patients who receive augmentation therapy would achieve a 26% reduction in rate of decline of FEV1, however given the variability in rate of decline in FEV1 in patients who are not receiving augmentation therapy as reported, it is uncertain whether augmentation therapy would achieve that 26% in patients who have similar disease characteristics as those enrolled in Stockley et al., and adjusting for different effect modifiers should have been considered in order to produce a better estimate.
Pharmacoeconomic Review
Abbreviations
A1-PI
alpha1-proteinase inhibitor
AATD
alpha1-antitrypsin deficiency
CBS
Canadian Blood Services
COPD
chronic obstructive pulmonary disease
FEV1
forced expiratory volume in the first second
ICER
incremental cost-effectiveness ratio
LY
life-year
QALY
quality-adjusted life-year
SoC
standard of care
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | A1-PI (human) (Zemaira), lyophilized powder for IV injection |
Submitted price |
|
Indication | For maintenance treatment in adults with severe A1-PI deficiency (e.g., PiZZ, PiZ[null], Pi[null,null], PiSZ genotypes) (AATD) and clinical evidence of emphysema. Patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease (e.g., FEV1 lower than predicted, lower diffusion capacity, impaired walking capacity, or increased number of exacerbations) as evaluated by a health care professional experienced in the treatment of AATD. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | September 21, 2016 |
Reimbursement request | As per indication |
Sponsor | CSL Behring Canada, Inc. |
Submission history | Previously reviewed: No |
A1-PI = alpha1-proteinase inhibitor; AATD = alpha1-antitrypsin deficiency; FEV1 = forced expiratory volume in the first second; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Adults with severe alpha1-antitrypsin proteinase inhibitor deficiency and clinical evidence of emphysema |
Treatment | Alpha1-proteinase inhibitor (human) (Zemaira) plus SoC |
Comparators |
|
Perspectives |
|
Outcomes | QALYs, LYs |
Time horizon | Lifetime (50 years) |
Key data sources |
|
Submitted results | ICER = $272,225 per QALY for Zemaira plus SoC vs. SoC alone (incremental costs: $1,631,681; incremental QALYs: 6.09)a |
Key limitations |
|
CADTH reanalysis results |
|
COPD = chronic obstructive pulmonary disease; FEV1 = forced expiratory volume in the first second; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care.
aResults as obtained from the sponsor’s economic model.1
Conclusions
The results of RAPID suggest that Zemaira was associated with a statistically significantly reduced rate in the validated primary outcome of decline in lung density after 24 months compared with placebo in patients with alpha1-proteinase inhibitor (A1-PI) deficiency with emphysema and a reduced lung function when CT (CT) scans were taken at full inspiration state. This shows that treatment with Zemaira preserves lung function in these patients; however, as lung density is not used in clinical practice to assess disease progression, the extent to which these findings translate into clinical benefits for patients in real life is unknown. The results from RAPID were not informative regarding the efficacy of A1-PI on the outcomes of survival and lung transplant, the sample size being relatively small and the follow-up of limited duration for a slowly progressive disease. Other important clinical outcomes such as exacerbations and health-related quality of life were reported as secondary outcomes, for which the trial was not adequately powered.
On account of the discrepancies between the clinical and pharmacoeconomic data provided for this submission, along with the uncertainty and heterogeneity associated with the sources used to parameterize the model, CADTH was unable to derive a base case. The survival data used were uncertain and likely biased in favour of augmentation therapy, and the studies cited from the published literature were not representative of the Health Canada indication or the RAPID trial population. Thus, the results of the sponsor’s submitted model should be interpreted with extreme caution. CADTH did conduct a series of exploratory analyses in consultation with clinical experts to examine the implications of assumptions for which reliable evidence is not available. Changes made as part of the exploratory analysis include using a Gompertz function to extrapolate survival for both treatments, increasing the mortality after the follow-up period of the registry study, decreasing the health state utility values, decreasing the probability of lung transplant, and excluding the cost and disutilities associated with exacerbations. The CADTH results were similar to those of the sponsor: Zemaira is not cost-effective based on conventionally accepted incremental cost-effectiveness ratio (ICER) thresholds. From a Canadian Blood Services (CBS) perspective, these analyses resulted in an ICER of $664,549 per quality-adjusted life-year (QALY) for Zemaira plus standard of care (SoC) compared with SoC alone, with a 0% probability of cost-effectiveness at a $50,000 per QALY threshold. A price reduction of at least 93% would be necessary to achieve cost-effectiveness at this threshold. From a drug plan perspective, Zemaira is less expensive than the publicly available price for Prolastin-C, the only comparator, which is routinely available only in British Columbia.
The clinical uncertainty, as determined by the CADTH Clinical Review, as well as that introduced by the sponsor’s economic model, preempt CADTH from deriving a base case. The exploratory changes and other steps taken by CADTH do not eliminate the uncertainty, especially that pertaining to the impact of Zemaira on survival, but do serve to align most model inputs with clinical expert feedback to mitigate some uncertainty. The CADTH exploratory results are more likely to reflect reality than the sponsor’s base case; however, the impact of Zemaira on survival is unknown at this time.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
As part of the call for patient input, CADTH received feedback from Alpha-1 Canada, a national advocacy organization for Canadian patients with alpha1-antitrypsin deficiency (AATD). Alpha-1 Canada collected information from 14 patients and 2 families through focus groups and family-oriented interviews, and also distributed online surveys to 100 patients and caregivers with AATD. All interviewees and survey respondents resided in Canada. Most patients with AATD felt their condition impacted their ability to participate in moderate physical activity (90%), and many were no longer able to work (69%) or had to reduce their overall working hours (22%). About half of survey patients had been treated with augmentation therapy, with the most important goal of treatment being to stabilize lung function and prevent further deterioration. No side effects or other disadvantages of therapy were noted in the patient input.
CADTH received clinician input from the Canadian Thoracic Society, a national specialty organization for health care providers working in respiratory care and research. The input stated that the only (IV-administered) augmentation therapy currently available in Canada is Prolastin-C, but noted it is funded differently by different jurisdictions across Canada.
CADTH received drug plan input from CBS. The input discussed Prolastin-C extensively, specifically, its coverage and availability; the initiation, renewal, and stopping criteria of Prolastin-C and Zemaira; and whether patients currently on Prolastin-C would switch to Zemaira, if available. CBS was interested in whether clinicians would use a higher dose than recommended by the product monograph, namely, 120 mg/kg weekly. The input raised concerns relating to access to specialists and laboratory requirements for therapeutic drug monitoring. Questions were raised about the anticipated budget impact and sustainability if patients on Prolastin-C switched to Zemaira. The input noted the presence of confidential pricing for Prolastin-C.
Several of these concerns were addressed in the sponsor’s model:
No costs or disutilities were applied to the adverse events in the model (these events were not expected to be notable and the model was aligned with the patient group feedback).
In addition, CADTH addressed some of these concerns, as follows:
CADTH reduced the health state utility values to better reflect the impact of the disease on quality of life.
In the budget impact analysis (BIA), CADTH assumed a greater proportion of patients on Prolastin-C would switch to Zemaira.
CADTH was unable to address the following concerns raised from stakeholder input:
CADTH’s analyses are based on publicly available prices and do not incorporate the presence of a confidential price for Prolastin-C.
Economic Review
The current review is for A1-PI (human) (Zemaira) for adults with AATD and clinical evidence of emphysema.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing Zemaira plus SoC compared with SoC alone for maintenance treatment of adults with severe AATD and clinical evidence of emphysema. This modelled population aligned with the RAPID trial2 on which the Health Canada indication was based and which represents the reimbursement request.3 The product monograph further indicates that patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease (e.g., lower-than-predicted forced expiratory volume in the first second [FEV1], lower diffusion capacity, impaired walking capacity, or increased number of exacerbations) as evaluated by a health care professional experienced in the treatment of AATD.3
Zemaira is available in single-use vials containing lyophilized powder for reconstitution at 50 mg/mL for IV infusion. The recommended dosage of Zemaira is 60 mg/kg once weekly, administered over 15 minutes.3 The cost for Zemaira is $390 per 1,000 mg vial, $1,560 per 4,000 mg vial, and $1,950 per 5,000 mg vial1; the annual cost is $101,748 per patient based on the mean patient weight of 76 kg in the RAPID trial.2 Due to the rarity of the condition, wastage was assumed for all unused product. Zemaira is assumed to be given alongside SoC.
The comparator in the primary analysis, from the perspective of CBS, was SoC alone, which comprises treatments typically prescribed to emphysema and chronic obstructive pulmonary disease (COPD) patients: long-acting and short-acting beta2-agonists, long-acting muscarinic antagonists, inhaled corticosteroids, short-acting anticholinergics, xanthine bronchodilators, and phosphodiesterase type 4 inhibitors. The sponsor calculated an average weighted annual cost of $|||||||| for SoC based on publicly available prices. This cost for SoC was also added to the treatment costs of patients receiving Zemaira, as patients were assumed to continue receiving these medications concomitantly. The sponsor also submitted a key scenario analysis from the perspective of the Canadian public payer comparing Zemaira plus SoC with Prolastin-C plus SoC. The annual cost for Prolastin-C was $111,939, to which was also added the cost of SoC.
Outcomes of the model included QALYs and life-years (LYs) over a lifetime horizon of 50 years. Discounting (1.5% per annum) was applied to both costs and outcomes and an annual cycle length was used.
Model Structure
The sponsor submitted a Markov model consisting of 5 mutually exclusive health states. Three of the states pertained to a patient’s emphysema, which was defined according to FEV1, as follows: moderate (50% ≤ FEV1 ≤ 80% of predicted), severe (30% ≤ FEV1 ≤ 50% of predicted), and very severe (< 30% of predicted). The model also included states for post lung transplant and death. All patients began in the moderate emphysema health state to reflect the criteria in the RAPID trial,2 after which point they progressed to severe emphysema, very severe emphysema, post lung transplant, or death; it was assumed that no improvement in the patient’s clinical course could occur. In each health state, patients could either progress to the next state, remain in their current state, or move to the death state. Patients transitioning into the death state remained there until the end of the model time horizon. A figure of the sponsor’s model structure is available in Appendix 3 (Figure 1).
Model Inputs
The modelled population had baseline characteristics similar to the population enrolled in the RAPID trial (n = 180).2 Patients had a mean body weight of ||||| kg, and 54% were male. The starting age in the model was 52 years, based on the mean trial age.2
In the sponsor’s model, it was assumed that patients entered the moderate emphysema state and were receiving optimal pharmacological and non-pharmacological treatment for their emphysema. The baseline FEV1 was assumed to be 2,050 mL based on a retrospective Swedish study by Hiller et al. (2019) of patients with AATD.4 The annual rate of disease progression, as modelled by decline in FEV1, was based on a prospective UK study by Stockley et al. (2016) of patients with AATD who had never received augmentation therapy.5 A weighted average was calculated based on various categories of FEV1 decline experienced in the study, resulting in a per-year decline of 49 mL used in the model for SoC. To derive a rate of decline for Zemaira, a meta-analysis from Chapman et al. (2009) was cited, which reported that augmentation therapy was associated with a 26% reduction in the rate of FEV1 decline in patients with AATD.6 Thus, the rate of FEV1 decline in patients receiving Zemaira was assumed to be 26% lower than for patients receiving SoC, resulting in a decline of FEV1 of 36.3 mL per year.1 Of note, 4 of the 5 studies included in the meta-analysis used Prolastin-C as the augmentation therapy; the bioequivalence of Prolastin-C and Zemaira is assumed in the sponsor’s analysis.1
Patients progressed from the moderate, to severe, to very severe emphysema health states according to the rates described previously. From any of these emphysema states, patients could die based on survival curves from Ellis et al. (2019), who conducted a matched study of 2 registries, 1 from the US in which patients with AATD had received augmentation therapy, and the other from the UK in which patients with AATD had received SOC.7 The 16-year Kaplan–Meier data from the study were extrapolated using a Weibull function for both treatment arms for use in the sponsor’s model. These parametric functions were converted to per-cycle probabilities of death in the sponsor’s model; the probability of death was the same, regardless of emphysema health state.1 The model also provided an option to cap survival to that observed in the general population multiplied by the increased hazard experienced by COPD patients from a Canadian study.8
Once patients had progressed to the very severe health state, it was assumed they either received a lung transplant or died by the next model cycle. Thus, patients did not accumulate in the very severe emphysema health state because their transition probabilities to lung transplant and death equalled 1. Patients experienced a constant probability of death following lung transplant based on 5-year survival rates of bilateral lung transplant recipients from the Canadian Organ Replacement Register.9
The dosage of Zemaira used in the model was as described in the overview section: 60 mg/kg weekly, as per the RAPID trial and product monograph.2,3 As wastage was assumed, each administration required 5,000 mg of product, resulting in an annual cost of $101,748 per patient. The frequency of medication use in SoC was derived from the RAPID trial, and the sponsor calculated an average weighted annual cost of $|||||||||| for SoC, which was added to patients receiving Zemaira.
Health-related quality of life data were collected in RAPID through the St. George’s Respiratory Questionnaire instrument, but not via a generic, preference-based measure. Therefore, health state utilities were derived from a systematic review and meta-analysis of COPD patients: moderate emphysema, 0.821; severe emphysema, 0.741; very severe emphysema, 0.681; and, during an exacerbation, 0.519.10 The utility value following lung transplant was assumed to be 0.800.11
All costs used in the model, except drug prices, were inflated to 2021 Canadian dollars. The drug acquisition costs for Zemaira, Prolastin-C, and SoC have been described previously. Treatment administration costs were not included, based on the sponsor’s assumption that Zemaira could be administered in a company-sponsored infusion clinic or self-administered. Treatment with Zemaira is assumed to stop after lung transplant or death. Routine health care resource costs were included and consisted of chest X-rays and other scans, electrocardiograms, specialist visits, and long-term oxygen therapy. The total annual cost of health care resource use was calculated to be $915.63 per patient, with the majority of the cost coming from specialist visits.1 The sponsor also included the cost of exacerbations, which were calculated to be $775 and $11,553 for moderate and severe exacerbations, respectively, based on a prospective Canadian study.12 The frequency of such exacerbations was based on a US study,13 while the RAPID trial was used to determine that 19.7% more exacerbations occurred in the Zemaira arm compared with SoC.2 A weighted annual cost of moderate and severe exacerbations, which differed by emphysema state, was added to the costs for each treatment group. Adverse events were assumed to be minor and not requiring the use of health care resources or additional pharmacologic treatment. As such, no costs or disutilities were ascribed to these adverse events. A 1-time cost for lung transplant of $110,876 was added upon entry into this state, with post–lung transplant costs of $35,616 and $14,748 added in the first and second year following surgery.14
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
From the perspective of CBS, Zemaira plus SoC was associated with incremental costs of $1,657,409 and QALYs of 6.09 in comparison with SoC, resulting in an ICER of $272,225 per QALY (Table 3). The probability of being cost-effective at a $50,000 per QALY willingness-to-pay threshold was 0%. In the sponsor’s base case, 34.2% of the QALYs for Zemaira were gained beyond the 16-year follow-up of the Ellis et al. study.
From the perspective of the Canadian public payer, Zemaira plus SoC was less costly than Prolastin-C, based on the sponsor’s assumption of equal clinical effects (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Treatment | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
Canadian Blood Services perspective | |||||
SoC alone | 80,112 | Reference | 12.98 | Reference | Reference |
Zemaira | 1,737,521 | 1,657,409 | 19.06 | 6.09 | 272,225 |
Drug plan perspective | |||||
Prolastin-C | 1,903,682 | Reference | 18.99 | Reference | Reference |
Zemaira | 1,733,300 | −170,796 | 18.99 | 0 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Note that the submitted analyses are based on publicly available prices of comparators and may not reflect confidential, negotiated prices.
Sensitivity and Scenario Analysis Results
The sponsor conducted a number of sensitivity and scenario analyses involving the time horizon, discount rate, and the starting distribution of patients (equally split between the moderate and severe health states). In these analyses, the ICER was most sensitive to a shorter time horizon, with the ICER for Zemaira versus SoC being calculated as $2,091,242 and $551,294 per QALY with a 10- and 20-year time horizon, respectively.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
Uncertainty regarding the ability of outcomes from RAPID to predict clinically meaningful outcomes: The sponsor submitted results from the RAPID and RAPID open-label extension study. The CADTH Clinical Review concluded that, based on the results of RAPID, Zemaira was associated with a statistically significant reduction in the rate of decline in lung density compared with placebo in patients with AATD and emphysema. However, as lung density is not used in clinical practice to assess disease progression, the importance of these findings is unknown, and these results have little relevance to the pharmacoeconomic model, which was designed based on the expected progression of FEV1. The Clinical Review further noted that RAPID was not informative regarding the efficacy of Zemaira on the outcomes of survival and lung transplant on account of the small sample and limited follow-up.
To predict the impact of Zemaira on more meaningful and final outcomes such as survival, the sponsor consulted numerous, varied sources. While the RAPID trial was used to define the starting age, weight, frequency of SoC medication use, and relative difference in exacerbation rate, all other model parameters, including survival, disease progression, transition probabilities, absolute number of exacerbations, and health state utilities were derived from alternate published sources.4-7,13
As there is no evidence to support a survival benefit with Zemaira, and the information used by the sponsor to predict this association is associated with uncertainty, the results from the sponsor’s model should be interpreted with extreme caution. Given the nature of the model, even the CADTH exploratory analyses require that the assumption of a survival benefit with FEV1 improvement will hold, and the extent of this relationship is unknown.
Uncertainty and heterogeneity regarding the clinical inputs: Further to the previously noted limitation, there exists uncertainty and heterogeneity among the studies used to parameterize the economic model. The survival data included were from Ellis et al. (2019), who conducted a comparative analysis of 2 registries: a US registry was used to define the population receiving alpha1-antitrypsin augmentation therapy and a UK registry was used to define the control group of patients with AATD patients.7 Because the US health care system functions in a private, insurance-based manner, the patients receiving alpha1-antitrypsin augmentation therapy are those with insurance or of high socioeconomic status who can afford to pay out of pocket. These patients are compared with a general UK population that includes all patients, which introduces a bias in favour of augmentation therapy. While some matching was performed on age, sex, and smoking status, there was no effort to control for this socioeconomic and regional variation. Furthermore, the clinical experts consulted by CADTH reviewed this survival data and considered it to be highly uncertain and unreliable.
Further, the studies used from the published literature to parameterize the model were conducted in populations that are not necessarily representative of the RAPID trial population. The sponsor assumed a starting FEV1 value of 2,050 mL for patients included in the model, based on a prospective registry study from Sweden by Hiller et al. (2019).4 However, the baseline FEV1 value from the RAPID trial was 1,580 mL and 1,600 mL in the Zemaira and placebo groups, respectively.2 Furthermore, the emphysema status of patients was not reported in the Hiller et al. publication and, in fact, the baseline characteristics indicated that 39% of patients did not have respiratory symptoms at study enrolment.4 As the Health Canada indication for Zemaira stipulates that patients must have clinical evidence of emphysema, these discrepancies between the Hiller et al. registry and RAPID trial population contribute uncertainty to the analysis.
To define the rate of FEV1 progression, the sponsor used another published source, Stockley et al. (2016), who assessed FEV1 decline in patients with AATD prospectively enrolled in a UK registry.5 Based on a weighted average calculated from this publication, an annual rate of decline of 49 mL/year was used in the sponsor’s model for those on SoC. The CADTH Clinical Review noted issues with the validity of the Stockley et al. study regarding inclusion criteria, baseline characteristics, assessment timing and setting, and guidelines used. In addition, the publication did not explicitly report whether or not patients had clinical evidence of emphysema. The sponsor used a weighted average to calculate FEV1 decline; however, there was no adjustment for confounding, which is noteworthy, considering the heterogeneity in lung function decline in patients with AATD. The clinical experts noted that FEV1 values are often expressed in terms of a patient’s percent predicted value rather than an absolute measurement in millilitres, making it difficult to validate the sponsor’s assumptions, which are based on absolute values for annual decline.
The sponsor assumed that patients who receive Zemaira would achieve a 26% reduction in the rate of decline of FEV1, based on a meta-analysis of 5 studies.2 However, given the variability in the rate of decline of FEV1 in patients not receiving augmentation therapy, it is uncertain whether a 26% reduction in the rate of decline would be achieved by all patients. No adjustment for effect modifiers was considered in the Stockley et al. study, contributing additional uncertainty to the analysis.5
The sponsor’s base case predicted a discounted, incremental gain of 7.74 LYs based on biased and highly uncertain survival data. In addition, the studies used by the sponsor to parameterize certain aspects of their model pertaining to FEV1 decline were associated with uncertainty. On account of the clinical discrepancies, heterogeneity, and the uncertainties outlined in the first 2 limitations, CADTH was unable to derive a base case. And, while CADTH was unable to address the bias associated with the Ellis et al. survival data, further limitations were explored in exploratory and scenario analyses (described subsequently).
Overestimation of the survival extrapolation for patients with AATD: In addition to the aforementioned limitations of the published survival data, the extrapolation of these data does not meet face validity. In the sponsor’s base case, a Weibull model was used to extrapolate the survival data for both patients receiving Zemaira and SoC. This extrapolation resulted in the assumption that 10% of patients on Zemaira would still be alive at the end of the 50-year time horizon at the average age of 102. This assumed survival exceeds that of the general Canadian population, in which only 1.6% of people are expected to still be alive at age 102 (based on Statistics Canada data from 2017 to 2019).15 The clinical experts did not expect that treatment with Zemaira would extend the life of a patient with AATD beyond that observed in the general population. The clinical experts noted that the life expectancy of a person with AATD would be far less, in fact, with an average life expectancy of 65 years and an estimated survival rate among those not receiving a lung transplant of between 8 to 10 years from diagnosis.
Furthermore, the clinical experts noted additional aspects of the model that did not meet face validity, based on their clinical practice. They noted that, beyond approximately 75 years of age, the only patients still alive would be those who had received a successful lung transplant and, after 40 years in the model (average age of 92 years), almost no patients would be alive. In addition, the clinical experts noted that patients with AATD previously receiving SoC have often experienced prolonged exposure to corticosteroids, which are associated with cataracts, heart disease, gastrointestinal (GI) disease, osteoporosis, and other diseases and that, even with serum levels augmented with Zemaira, this population is not expected to be as healthy as the general population. In the sponsor’s base case, it was assumed that at age 75, 35% of patients treated with Zemaira would still be alive without a lung transplant and that, at age 92, 19% of patients would still be alive (10% without and 9% with lung transplant). The clinical experts noted that the sponsor’s extrapolation overestimates the survival of patients with AATD.
In consultation with clinical experts, CADTH extrapolated survival using a Gompertz function as part of the exploratory analysis. The extrapolated survival results were still overestimated for Zemaira, even when using the most conservative function. To further align the survival assumptions with clinical expert opinion, CADTH increased the mortality rate of Zemaira after 16 years such that less than 5% of patients were alive at age 75 without a lung transplant. Although the overall survival is likely still overestimated due to the bias associated with the Ellis et al. study, this approach was determined to align more closely with expert opinion than the sponsor’s original assumptions.
Overestimation of the health state utility of patients with severe and very severe emphysema: The sponsor derived utility values from a systematic review of patients with COPD.10 The clinical experts consulted by CADTH suggested that the utility values in the severe and very severe emphysema health states were likely overestimated relative to the value in the moderate state. It was noted that, especially for patients in the very severe state, their disease represents constant daily difficulty, often resulting in breathlessness, even at rest. The experts noted that these patients are often also out of the workforce and would have a severe reduction in their quality of life, a fact reflected in the patient input.
As part of the exploratory analysis, CADTH chose utility values from the published literature that aligned more closely with the clinical expert feedback.16
Uncertainty surrounding prevalence and availability of lung transplant: The clinical experts stated that of the patients progressing to less than a 30% FEV1, only a minority would receive lung transplants, due to a lack of availability. However, the sponsor assumed that all patients in the very severe state would receive a transplant (i.e., 1 - probability of death). This uses the death rate as the starting point and then assumes that all those remaining alive would be eligible for a lung transplant, which does not align with clinical expert opinion.
As part of the exploratory analysis, CADTH halved the probability of lung transplant. The remaining patients were assumed to die in the same cycle.
Improper inclusion of costs and disutilities associated with exacerbations: The sponsor incorporated the rates of exacerbations into their analyses, which were assumed to be associated with costs and a quality of life impact. To do so, they used the relative difference in exacerbation rate between Zemaira and placebo in RAPID, and the absolute rate of exacerbations from another published study.13 CADTH noted that exacerbation rates were not statistically different in RAPID, yet the sponsor assumed that the relative difference would be applicable to the rates observed in another study. This methodology is uncertain. The clinical experts highlighted the lack of significance for this outcome in RAPID, and stated further that Zemaira is not expected to influence the rate of exacerbations and that there was no biologic plausibility to suggest that it might.
As part of the exploratory analysis, CADTH excluded the costs associated with exacerbations but noted the sponsor failed to incorporate disutilities of exacerbations in their original model, even though their technical report suggests this was done. Therefore, the costs and disutilities associated with exacerbations were excluded from CADTH’s analysis.
One additional limitation was identified but was not considered to be a key limitation; the product monograph indicates that patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease (e.g., lower predicted FEV1, lower diffusion capacity, impaired walking capacity, or increased number of exacerbations), as evaluated by a health care professional experienced in the treatment of A1-PI deficiency.3 This element of the Health Canada indication was not considered in the economic analysis, as this stipulation does not seem to have been part of the inclusion criteria for the RAPID trial. Furthermore, given the heterogeneity between the sources outlined previously, it is unlikely that patients included in these retrospective studies would also meet these indicated criteria.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Bioequivalence of Zemaira and Prolastin-C was assumed. | Appropriate. This assumption was confirmed by clinical experts. |
Administration costs were excluded for Zemaira on the basis that the product may be administered in a company-sponsored infusion clinic or can be self-administered by the patient. | Not appropriate. Upon request, the sponsor provided further information on the support program, which consisted mainly of training and support services rather than dedicated infusion clinics. Furthermore, the clinical experts did not think self-administration of Zemaira would be reasonable, unless the patient had a permanent, IV access central line, which would be associated with insertion costs and risks of infection. |
The sponsor assumed a linear decline in FEV1 throughout the course of the model. | Not appropriate. The clinical experts noted that progression would occur more quickly with earlier-stage disease, where patients would experience a steady decline in FEV1. At the later stages of the disease, there is less lung capacity available to reduce and, therefore, the absolute decline in FEV1 tends to decrease over time. |
Patients experienced the same risk of death, regardless of health state. | Not appropriate. The clinical experts indicated that the risk of death would increase as a patient’s disease progressed. |
A1-PI = alpha1-proteinase inhibitor; FEV1 = forced expiratory volume in the first second.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Due to the choice of different evidence bases for the clinical and economic submissions, heterogeneity, and uncertainty within the clinical data used to parameterize the model, CADTH was unable to derive a base case. CADTH performed an exploratory analysis to explore the areas of uncertainty with the model. These changes, detailed in Table 5, included using a Gompertz function to extrapolate survival for both treatments, increasing the mortality after the 16-year follow-up from Ellis et al., decreasing the health state utility values, decreasing the probability of lung transplant, and excluding the cost and disutilities associated with exacerbations. The CADTH exploratory analysis represents the best estimate in light of the identified uncertainties.
Table 5: CADTH Exploratory Analyses on the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH exploratory case | ||
1. Parametric survival modelling | Weibull function for Zemaira and SoC | Gompertz function for Zemaira and SoC |
2. Mortality rate after 16 years | Consistent with extrapolation | Mortality increased after 16 years |
3. Utility values |
|
|
4. Probability of receiving a lung transplant | Equal to (1 - probability of death) | Equal to (1 - twice the probability of death) |
5. Cost of exacerbations | Included | Excluded |
CADTH exploratory analysis | — | Exploratory 1 + 2 + 3 + 4 + 5 |
A1-PI = alpha1-proteinase inhibitor; COPD = chronic obstructive pulmonary disease; SoC = standard of care.
In the CADTH exploratory analysis from the CBS perspective, Zemaira was associated with estimated total costs of $1,290,688 and total QALYs of 13.12, compared with total costs and QALYs of $53,356 and 11.26 for patients receiving SoC alone. The ICER for Zemaira plus SoC compared with SoC alone was $664,549 per QALY, and the probability of cost-effectiveness at a $50,000 per QALY willingness-to-pay threshold was 0%. Only 7.4% of the QALYs associated with Zemaira were accrued after the 16-year follow-up mark. CADTH noted that in the exploratory analysis, Zemaira was still associated with incremental LYs of 2.36, a result that remains highly uncertain, given the data sources used.
In the CADTH exploratory analysis from the payer perspective, Zemaira was less costly compared with Prolastin-C ($1,286,482 versus $1,410,153), based on the assumption of clinical similarity.
A detailed breakdown of the disaggregate results of the CADTH exploratory analysis is available in Table 13 and Table 14.
Table 6: Summary of the Stepped Analysis of the CADTH Exploratory Analysis (CBS Perspective; Deterministic)
Stepped analysis | Treatment | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | SoC | 80,836 | 12.94 | Reference |
Zemaira | 1,724,950 | 18.99 | 272,186 | |
CADTH reanalysis 1: Survival modelled using Gompertz | SoC | 74,589 | 12.18 | Reference |
Zemaira | 1,602,468 | 17.47 | 288,737 | |
CADTH reanalysis 2: Mortality rate increased after 16 years for Zemaira | SoC | 80,836 | 12.94 | Reference |
Zemaira | 1,351,408 | 14.40 | 872,329 | |
CADTH reanalysis 3: Utility values | SoC | 80,836 | 12.81 | Reference |
Zemaira | 1,724,950 | 18.77 | 275,510 | |
CADTH reanalysis 4: Halved the probability of lung transplant | SoC | 80,836 | 12.08 | Reference |
Zemaira | 1,724,950 | 18.03 | 276,214 | |
CADTH reanalysis 5: Exclusion of costs of exacerbations | SoC | 59,230 | 12.94 | Reference |
Zemaira | 1,692,103 | 18.99 | 270,325 | |
CADTH exploratory analysis (1 + 2 + 3 + 4 + 5) | SoC | 53,982 | 11.26 | Reference |
Zemaira | 1,286,482 | 13.13 | 660,309 | |
CADTH exploratory analysis (1 + 2 + 3 + 4 + 5): Probabilistic | SoC | 53,356 | 11.26 | Reference |
Zemaira | 1,290,688 | 13.12 | 664,549 |
CBS = Canadian Blood Services; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
Table 7: Summary of the Stepped Analysis of the CADTH Exploratory Analysis (Payer Perspective; Deterministic)
Stepped analysis | Drug | Total costs ($) | Incremental costs ($)a |
|---|---|---|---|
Sponsor’s base case | Prolastin-C | 1,886,511 | Reference |
Zemaira | 1,724,950 | −161,561 | |
CADTH reanalysis 1: Survival modelled using Gompertz | Prolastin-C | 1,666,425 | Reference |
Zemaira | 1,602,468 | −63,957 | |
CADTH reanalysis 2: Mortality rate increased after 16 years for Zemaira | Prolastin-C | 1,478,717 | Reference |
Zemaira | 1,351,408 | −127,309 | |
CADTH reanalysis 3: Utility values | Prolastin-C | 1,886,511 | Reference |
Zemaira | 1,724,950 | −161,561 | |
CADTH reanalysis 4: Halved the probability of lung transplant | SoC | 1,886,511 | Reference |
Zemaira | 1,724,950 | −161,561 | |
CADTH reanalysis 5: Exclusion of costs of exacerbations | Prolastin-C | 1,853,665 | Reference |
Zemaira | 1,692,103 | −161,562 | |
CADTH exploratory analysis (1 + 2 + 3 + 4 + 5) | Prolastin-C | 1,410,153 | Reference |
Zemaira | 1,286,482 | −123,671 |
A1-PI = alpha1-proteinase inhibitor; CBS = Canadian Blood Services; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
aBecause equal efficacy was assumed for all relevant parameters in this analysis, the only difference between treatments is the incremental cost; Prolastin-C is less expensive than Zemaira.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s and CADTH’s exploratory analysis. The CADTH exploratory analyses suggested a price reduction of at least 93% would be necessary to achieve cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY (Table 8).
Table 8: CADTH Price Reduction Analyses (CBS Perspective)
Analysis (price reduction) | ICERs for Zemaira vs. SoC ($/QALY) | |
|---|---|---|
Sponsor base case ($) | CADTH exploratory analysis ($) | |
No price reduction | 272,225 | 664,549 |
10% | 244,792 | 599,465 |
20% | 219,106 | 534,160 |
30% | 191,342 | 465,728 |
40% | 164,553 | 397,416 |
50% | 137,903 | 330,801 |
60% | 111,773 | 264,285 |
70% | 84,844 | 197,080 |
80% | 58,177 | 131,353 |
84% | 47,834 | 103,724 |
90% | 31,801 | 65,576 |
93% | 23,710 | 45,260 |
CBS = Canadian Blood Services; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
CADTH undertook several scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of Zemaira in the exploratory analysis, which are outlined as follows:
increased the mortality for patients receiving Zemaira during the first 16 years of the model by 50% (to explore the effect of the survival data from Ellis et al. on the model results)
shortened the time horizon to 20 years
shortened the time horizon to 10 years.
The results of these analyses are presented in Table 15 in Appendix 4. The scenario analysis where the time horizon was shortened to 10 years had the largest effect on the ICER, calculated to be $2,378,212 per QALY. The scenario in which the mortality was increased for the first 16 years resulted in an ICER of $1,412,393 per QALY.
Issues for Consideration
The clinical experts consulted by CADTH emphasized the high degree of phenotypic variability in this patient population, resulting in similarly high variability in disease progression and expected survival, both with and without lung transplant. This fact was evident in CADTH’s appraisal of the clinical evidence used to parameterize the model in which a high degree of heterogeneity was observed. This factor contributed to CADTH being unable to derive a base case.
Overall Conclusions
The results of RAPID suggest that Zemaira was associated with a statistically significantly reduced rate in the validated primary outcome of decline in lung density after 24 months compared with placebo in patients with A1-PI deficiency with emphysema and a reduced lung function when CT scans were taken at a full inspiration state. This shows that treatment with Zemaira preserves lung function in these patients; however, as lung density is not used in clinical practice to assess disease progression, the extent to which these findings translate into clinical benefits for patients in real life is unknown. The results from RAPID were not informative regarding the efficacy of A1-PI (human) on the outcomes of survival and lung transplant due to the sample size being relatively small and the follow-up of limited duration for a slowly progressive disease. Other important clinical outcomes such as exacerbations and health-related quality of life were reported as secondary outcomes, for which the trial was not adequately powered.
On account of the discrepancies between the clinical and pharmacoeconomic data provided for this submission, along with the uncertainty and heterogeneity associated with the sources used to parameterize the model, CADTH was unable to derive a base case. The survival data used were uncertain and likely biased in favour of augmentation therapy, and the studies cited from the published literature were not representative of the Health Canada indication or RAPID trial population. Thus, the results of the sponsor’s submitted model should be interpreted with extreme caution. CADTH did conduct a series of exploratory analyses in consultation with clinical experts to examine the implications of assumptions for which reliable evidence is not available. Changes made as part of the exploratory analysis include using a Gompertz function to extrapolate survival for both treatments, increasing the mortality after the follow-up period of the registry study, decreasing the health state utility values, decreasing the probability of lung transplant, and excluding the cost and disutilities associated with exacerbations. The CADTH results were similar to those of the sponsor: Zemaira is not cost-effective, based on conventionally accepted ICER thresholds. From a CBS perspective, these analyses resulted in an ICER of $664,549 per QALY for Zemaira plus SoC compared with SoC alone, with a 0% probability of cost-effectiveness at a $50,000 per QALY threshold. A price reduction of at least 93% would be necessary to achieve cost-effectiveness at this threshold. CADTH notes that in the exploratory analysis, Zemaira was still associated with incremental LYs of 2.36, a result that remains highly uncertain, given the data sources used. From a drug plan perspective, Zemaira is less expensive than the publicly available price for Prolastin-C, the only comparator, which is routinely available only in British Columbia. However, Prolastin-C has not been formally reviewed by CADTH through the formulary review process and, therefore, the cost-effectiveness of Prolastin-C versus SoC alone has not been assessed through a similar process.
The clinical uncertainty, as determined by the CADTH Clinical Review as well as that introduced by the sponsor’s economic model, preempt CADTH from deriving a base case. The exploratory changes and other steps taken by CADTH do not eliminate the uncertainty, especially that pertaining to the impact of Zemaira on survival, but do serve to align most model inputs with clinical expert feedback to mitigate some uncertainty. The CADTH exploratory results are more likely to reflect reality than the sponsor’s base case; however, the impact of Zemaira on survival is unknown at this time.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zemaira, 1000 mg, 4000 mg, 5000 mg, IV. CSL Behring Inc.: Amerisource Bergen Innomar Strategies; 2021 Sept 30.
2.Chapman K, Burdon J, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin defi ciency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386:360-368. PubMed
3.Zemaira® Alpha1-Proteinase Inhibitor (Human) Powder and Diluent for Solution for Injection [product monograph]. Ottawa (ON): CSL Behring Canada Inc; 2019 Dec 16.
4.Hiller A, Piitulainen E, Jehpsson L, Tanash H. Decline in FEV1 and hospitalized exacerbations in individuals with severe alpha-1 antitrypsin deficiency. International Journal of Chronic Obstructive Pulmonary Disease. 2019;14:1075-1083. PubMed
5.Stockley R, Edgar R, Pillai A, Turner A. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? International Journal of Chronic Obstructive Pulmonary Disease. 2016;11:1745-1756. PubMed
6.Chapman K, Stockley R, Dawkins C, Wilkes M, Navickis R. Augmentation Therapy for α1Antitrypsin Deficiency:A Meta-Analysis. Copd. 2009;6:177-184. PubMed
7.Ellis P, Holm K, Choate R, et al. Comparison of outcomes in augmentation naive and augmented patients with alpha-1 antitrypsin deficiency related lung disease [abstract]. European Respiratory Journal Conference: European Respiratory Society International Congress, ERS. 2019;54(Suppl. 63, PA3383).
8.Gershon A, Hwee J, Victor J, et al. Mortality trends in women and men with COPD in Ontario, Canada, 1996-2012. Thorax. 2015;70:121-126. PubMed
9.CIHI. Canadian Organ Replacement Register. 2016: https://www.cihi.ca/en/canadian-organ-replacement-register-corr. Accessed 2021 Aug 31.
10.Zhang Y, Morgan R, Alonso-Coello P, et al. A systematic review of how patients value COPD outcomes. Eur Respir J. 2018;52:1800222. PubMed
11.Menn P, Leidl R, Holle R. A lifetime Markov model for the economic evaluation of chronic obstructive pulmonary disease. Pharmacoeconomics. 2012;9:825-840. PubMed
12.Mittman N, Kuramoto L, Seung S, Haddon J, Bradley-Kennedy C, Fitzgerald J. The cost of moderate and severe COPD exacerbations to the Canadian healthcare system. Respiratory Medicine. 2008;102:413-421. PubMed
13.Campos M, Alazami S, Zhang G, et al. Exacerbations in subjects with alpha-1 antitrypsin deficiency receiving augmentation therapy. Respiratory Medicine. 2009;103:1532-1539. PubMed
14.Patient cost estimator. Ottawa (ON): Canadian Institute for Health Information; 2021: https://www.cihi.ca/en/patient-cost-estimator. Accessed 2021 Nov 15.
15.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2021 Nov 15.
16.Miravitlles M, Huerta A, Fernandez-Villar J, et al. Generic utilities in chronic obstructive pulmonary disease patients stratified according to different staging systems. Health Qual Life Outcomes. 2014;12:120. PubMed
17.PROLASTIN®-C Alpha1-Proteinase Inhibitor (Human), Highly Purified IV Injection, 1000 mg/vial [product monograph]. Clayton, North Carolina: Grifols Therapeutics Inc; 2017 Jun 30: https://pdf.hres.ca/dpd_pm/00039972.PDF. Accessed 2021 Nov 15.
18.Government of B. C. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2021 Nov 15.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CADTH Cost Comparison Table for Severe Alpha1-Proteinase Inhibitor Deficiency
Treatment | Strength or concentration | Form (vial size if single-use) | Price | Recommended dosagea | Average daily cost | Average annual cost |
|---|---|---|---|---|---|---|
Alpha1-proteinase inhibitor (Zemaira) | 50 mg/mL | 1,000 mg 4,000 mg 5,000 mg Powder for IV infusion | $390.0000b $1,560.0000 $1,950.0000 | 60 mg/kg once weekly | $278.57 | $101,748 |
Augmentation therapy | ||||||
Prolastin-C | 50 mg/mL | 1,000 mg Powder for IV infusion | $450.5130c | 60 mg/kg once weekly | $321.80 | $117,536 |
Note: Prices do not include dispensing fees. Costs are calculated based on a mean body weight of 76 kg as per the RAPID trial.2 Annual costs are based on 365.25 days per year.
a The recommended dosages are from the respective product monographs.3,17
b Sponsor’s submitted price.1
c Price taken from British Columbia drug formulary.18
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Several aspects of the sponsor’s model did not reflect clinical practice including using a linear rate of FEV1 decline and assuming the same probability of death regardless of health state. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Parameter inputs appeared in multiple places in the model, complicating the validation process. Many values were hard-coded rather than being explicitly calculated through model inputs. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Probabilistic distributions were applied to drug acquisition costs, which are known and not subject to uncertainty. The decision problem was not addressed as CADTH was unable to derive a base case. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Some differences between the technical report and model regarding both inputs and results (e.g., lung transplant costs, probabilistic base-case results). |
FEV1 = forced expiratory volume in the first second.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Results of the Sponsor’s Base-Case Analysis (CBS Perspective)
Parameter | Zemaira | SoC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 22.93 | 15.19 | 7.74 |
Discounted QALYs | |||
Total QALYs | 19.06 | 12.98 | 6.09 |
Moderate emphysema | 9.14 | 6.39 | 2.75 |
Severe emphysema | 6.75 | 3.86 | 2.88 |
Very severe emphysema | 0.48 | 0.38 | 0.09 |
Post lung transplant | 2.69 | 2.33 | 0.36 |
Discounted costs ($) | |||
Total costs | 1,737,521 | 80,112 | 1,657,409 |
Drug acquisition costs | 1,641,919 | 10,238 | 1,631,681 |
Health care resource use costs | 14,874 | 9,694 | 5,180 |
Exacerbation costs | 33,050 | 21,600 | 11,449 |
Lung transplant costs | 47,679 | 38,580 | 9,099 |
ICER ($/QALY) | 272,225 | ||
A1-PI = alpha1-proteinase inhibitor; CBS = Canadian Blood Services: ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Disaggregated Results of the Sponsor’s Base-Case Analysis (Payer Perspective)
Parameter | Zemaira | Prolastin-C | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 22.83 | 22.83 | 0.00 |
Discounted QALYs | |||
Total QALYs | 18.99 | 18.99 | 0.00 |
Moderate emphysema | 9.11 | 9.11 | 0.00 |
Severe emphysema | 6.72 | 6.72 | 0.00 |
Very severe emphysema | 0.48 | 0.48 | 0.00 |
Post lung transplant | 2.68 | 2.68 | 0.00 |
Discounted costs ($) | |||
Total costs | 1,733,300 | 1,903,682 | -170,382 |
Drug acquisition costs | 1,637,924 | 1,808,721 | -170,796 |
Health care resource use costs | 14,843 | 14,931 | -87 |
Exacerbation costs | 32,967 | 33,235 | -268 |
Lung transplant costs | 47,565 | 46,795 | 770 |
ICER ($/QALY) | Dominant | ||
A1-PI = alpha1-proteinase inhibitor; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Exploratory Analysis
Table 13: Disaggregated Results of the CADTH Exploratory Analysis (CBS Perspective)
Parameter | Zemaira | SoC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 15.57 | 13.21 | 2.36 |
Discounted QALYs | |||
Total QALYs | 13.12 | 11.26 | 1.86 |
Moderate emphysema | 8.04 | 6.40 | 1.64 |
Severe emphysema | 3.91 | 3.43 | 0.49 |
Very severe emphysema | 0.26 | 0.32 | -0.06 |
Post lung transplant | 0.90 | 1.11 | -0.21 |
Discounted costs ($) | |||
Total costs | 1,290,688 | 53,356 | 1,237,332 |
Drug acquisition costs | 1,251,470 | 9,741 | 1,241,728 |
Health care resource use costs | 11,314 | 9,208 | 2,105 |
Exacerbation costs | 0 | 0 | 0 |
Lung transplant costs | 27,905 | 34,406 | -6,502 |
ICER ($/QALY) | 664,549 | ||
A1-PI = alpha1-proteinase inhibitor; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SoC = standard of care.
Table 14: Disaggregated Results of the CADTH Exploratory Analysis (Payer Perspective)
Parameter | Zemaira | Prolastin-C | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 15.57 | 15.57 | 0.00 |
Discounted QALYs | |||
Total QALYs | 13.13 | 13.13 | 0.00 |
Moderate emphysema | 8.05 | 8.05 | 0.00 |
Severe emphysema | 3.92 | 3.92 | 0.00 |
Very severe emphysema | 0.26 | 0.26 | 0.00 |
Post lung transplant | 0.90 | 0.90 | 0.00 |
Discounted costs ($) | |||
Total costs | 1,286,482 | 1,410,153 | -123,671 |
Drug acquisition costs | 1,246,767 | 1,370,438 | -123,671 |
Health care resource use costs | 11,324 | 11,324 | 0 |
Exacerbation costs | 0 | 0 | 0 |
Lung transplant costs | 28,390 | 28,390 | 0 |
ICER ($/QALY) | Dominant | ||
A1-PI = alpha1-proteinase inhibitor; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 15: Summary of Scenario Analyses Conducted on CADTH Exploratory Analysis
Scenario | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH exploratory analysis | SoC | 53,356 | 11.26 | Reference |
Zemaira | 1,290,688 | 13.12 | 664,549 | |
1. Increased mortality by 50% first the first 16 years of the model | SoC | 53,159 | 11.27 | Reference |
Zemaira | 1,180,829 | 12.06 | 1,412,393 | |
2. Shortened time horizon to 20 years | SoC | 52,447 | 10.95 | Reference |
Zemaira | 1,268,392 | 12.75 | 675,538 | |
3. Shortened time horizon to 10 years | SoC | 34,382 | 8.10 | Reference |
Zemaira | 853,733 | 8.45 | 2,378,212 |
A1-PI = alpha1-proteinase inhibitor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SoC = standard of care.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the introduction of A1-PI (human) (Zemaira) for the treatment of adult patients with severe AATD and clinical evidence of emphysema. The analysis was taken from the perspective of the Canadian public drug plans using a registry-based approach, with only drug acquisition costs included. A 3-year time horizon was used, from 2023 to 2025, with 2022 as a base year. The population size was derived using information from Alpha-1 Canada, a national advocacy organization for Canadian patients with AATD.
The reference case scenario included only those patients being currently treated with Prolastin-C in Canada while the new drug scenario considered an expanded market which included Zemaira and Prolastin-C. The market size in the new drug scenario is expected to grow if Zemaira were introduced due to the removal of current reimbursement restrictions and other access issues for Prolastin-C which would not be applicable to Zemaira. Key inputs to the BIA are documented in Table 18.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target Population | |
Number of patients eligible for drug under review in reference scenario | ||||| / ||||| / ||||| |
Number of patients eligible for drug under review in new drug scenario | ||||| / ||||| / ||||| |
Market Uptake (3 years) | |
Uptake (reference scenario): Zemaira Prolastin-C | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) British Columbia: Zemaira Prolastin-C | |||||% / |||||% / |||||% |||||% / |||||% / |||||% |
All other jurisdictions: Zemaira Prolastin-C | |||||% / |||||% / |||||% 0% / 0% / 0% |
Cost of treatment (per patient) | |
Cost of treatment annually Zemaira Prolastin-C | $101,748 $111,938 |
Summary of the Sponsor’s BIA Results
The estimated budget impact of funding Zemaira for the treatment of patients with severe AATD was $24,307,821 in year 1, $34,538,348 in year 2, and $44,697,076 in year 3 for a 3-year total of $103,543,245.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Underestimation of the market uptake in British Columbia: As part of their base case, the sponsor assumed that Zemaira would capture |||||% of the market in British Columbia, if reimbursed. This assumption was based on the fact that British Columbia is the only province in which Prolastin-C is routinely available without the need for special access. However, this assumption did not align with feedback from CBS and the drug plans. It was noted that some provinces in which Prolastin-C is currently available through special access have already stopped approving new patients for Prolastin-C.
As part of the base case, CADTH assumed Zemaira would capture 100% of the market in British Columbia, as was already assumed for all other jurisdictions.
Uncertainty regarding the population size: While the sponsor used registry data from Alpha-1 Canada as a foundation from which to base their population size estimates, several further assumptions regarding population size were based on internal company projections not available to CADTH. This, in addition to hard coding of some values in the population sheet, made validation of the population size difficult, though CADTH notes that the estimates generally aligned with expert opinion.
CADTH was unable to address this is reanalysis.
Mark-up and dispensing fee assumptions have a large influence on the budget impact: CADTH noted that the sponsor’s mark-up and dispensing fee assumptions had a large influence on the overall budget impact. This was explored in scenario analysis.
As part of a scenario analysis, CADTH excluded dispensing fees and mark-ups.
An additional limitation was identified but was not considered to be a key limitation. The sponsor made a transcription error in their assessment of the patient population size in Newfoundland, based on the data from Alpha-1 Canada. CADTH corrected this as part of the base case.
CADTH Reanalyses of the BIA
CADTH corrected the population size in Newfoundland and assumed 100% market uptake for Zemaira in British Columbia for all 3 years of the BIA.
Table 18: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Newfoundland patients in 2021 | ||||| patients | 3 patients |
Changes to derive the CADTH base case | ||
1. Zemaira market uptake in British Columbia | |||||% in all 3 years | 100% in all 3 years |
CADTH base case | Reanalysis 1 | |
The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20. Based on the CADTH base case, the budget impact of the reimbursement of Zemaira for the treatment of severe AATD is expected to be $23,729,027 in year 1, $33,924,828 in year 2, and $44,046,744 in year 3, with a 3-year total of $101,700,599. A scenario analysis was conducted in which dispensing fees and mark-ups were excluded, which resulted in a 3-year budget impact of $92,045,135. From a CBS perspective, the estimated 3-year budget impact was $165,249,851. A scenario analysis considering the 93% price reduction suggested by the pharmacoeconomic analysis resulted in a 3-year cost savings of $50,746,087.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case (corrected) | $104,976,690 |
CADTH reanalysis 1 and base case | $101,700,599 |
BIA = budget impact analysis.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case (corrected) | Reference | $18,831,551 | $19,961,444 | $21,159,130 | $22,428,678 | $82,380,803 |
New drug | $18,831,551 | $44,719,524 | $56,174,754 | $67,631,665 | $187,357,493 | |
Budget impact | $0 | $24,758,080 | $35,015,623 | $45,202,987 | $104,976,690 | |
CADTH base case | Reference | $18,831,551 | $19,961,444 | $21,159,130 | $22,428,678 | $82,380,803 |
New drug | $18,831,551 | $43,690,471 | $55,083,958 | $66,475,422 | $184,081,402 | |
Budget impact | $0 | $23,729,027 | $33,924,828 | $44,046,744 | $101,700,599 | |
CADTH scenario analysis 1: mark-ups and dispensing fees excluded | Reference | $17,679,479 | $18,740,248 | $19,864,662 | $21,056,542 | $77,340,931 |
New drug | $17,679,479 | $40,190,545 | $50,568,863 | $60,947,180 | $169,386,066 | |
Budget impact | $0 | $21,450,297 | $30,704,200 | $39,890,638 | $92,045,135 | |
CADTH scenario analysis 2: CBS perspective (no Prolastin-C costs) | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $43,690,471 | $55,083,958 | $66,475,422 | $165,249,851 | |
Budget impact | $0 | $43,690,471 | $55,083,958 | $66,475,422 | $165,249,851 | |
CADTH scenario analysis 3: 93% price reduction from PE model | Reference | $18,831,551 | $19,961,444 | $21,159,130 | $22,428,678 | $82,380,803 |
New drug | $18,831,551 | $3,376,992 | $4,267,836 | $5,158,338 | $31,634,716 | |
Budget impact | $0 | -$16,584,452 | -$16,891,295 | -$17,270,340 | -$50,746,087 | |
CADTH scenario analysis 4: CBS perspective with 93% price reduction | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $3,376,992 | $4,267,836 | $5,158,338 | $12,803,165 | |
Budget impact | $0 | $3,376,992 | $4,267,836 | $5,158,338 | $12,803,165 |
BIA = budget impact analysis; PE = pharmacoeconomic.
Stakeholder Input
Patient Group Input
Alpha-1 Canada
About Alpha-1 Canada
Describe the purpose of your organization. Include a link to your website.
Response: Alpha-1 Canada (https://alpha1canada.ca/) is a national non-profit organization committed to advocating on behalf of Canadians affected by Alpha-1 Antitrypsin Deficiency (AATD). Alpha-1, a rare hereditary condition characterized by low circulating levels of alpha-1 antitrypsin (AAT) protein in the blood, leads to increased risk of lung and liver disease. Alpha-1 Canada provides education to patients and the health care community to increase awareness and testing for this genetic disease and supports the broad spectrum of patients and caregivers affected by AATD.
In addition to two decades of experience supporting and educating patients and health care providers, Alpha-1 Canada has emphasized its advocacy efforts the past four years. This work is on behalf of alpha- 1 patients across Canada who continue to experience declining lung function solely because they cannot access a plasma-derived replacement therapy that will provide the ATT protein which protects their lungs. Augmentation therapy is the only specific approved therapy to treat patients with severe alpha-1 deficiency and is currently only publicly funded for new patients in British Columbia and Quebec.
Alpha-1 Canada believes all alpha-1 patients across Canada should have equitable access to therapy. The organization’s position is in line with clinical practice guidelines in Canada and around the world, which have recognized that intravenous augmentation therapy can slow or even halt the accelerated progression of emphysema due to AATD, preventing exacerbations, hospitalizations, and avoid lung transplantation.
Alpha-1 Canada is the leading source of education and advocacy in Canada, with patient and caregiver survey respondents noting that of the few organizations supporting Canadians with alpha-1, the majority access information and receive support and guidance through Alpha-1 Canada. Alpha-1 Canada also works closely with AlphaNet Canada, a disease management organization which follows over 300 patients with AATD, those of whom manage their disease with treatment; however, Alpha-1 Canada advocates for just as many patients with AATD who would benefit from augmentation therapy according to Canadian guidelines, but lack access to this life-prolonging therapy.
Information Gathering
CADTH is interested in hearing from a wide range of patients and caregivers in this patient input submission. Describe how you gathered the perspectives: for example, by interviews, focus groups, or surveys; personal experience; or a combination of these. Where possible, include when the data were gathered; if data were gathered in Canada or elsewhere; demographics of the respondents; and how many patients, caregivers, and individuals with experience with the drug in review contributed insights. We will use this background to better understand the context of the perspectives shared.
Response: Alpha-1 Antitrypsin Deficiency (AATD) is a rare disease with limited data, especially Canadian data, due to the slow progression of the disease and the small number of individuals impacted by this condition. With this in mind, we used a mixed method approach to gathering patient and caregiver perspectives and experiences with alpha-1, providing as much rich and comprehensive data as possible.
Focus Group Sessions (Two Sessions: March 2021)
Two focus group sessions (2 hours each) were conducted virtually due to COVID-19 (via Zoom), the first with Alpha-1 patients receiving augmentation therapy (March 13, 2021) and the second with Alpha-1 patients not receiving augmentation therapy (March 20, 2021). The intention of the focus group sessions was to understand the disease experience from the individual’s perspective, how AATD impacts their quality of life, their experiences on currently available treatments, the improvement patients want to see when receiving treatment, their experiences accessing treatment, and their experiences with screening and testing.
Table 1: Focus Group Session Participants
Variable | Description |
|---|---|
Focus Group Session 1: Receiving Augmentation Therapy | |
Participants | 8 total participants (6 male and 2 female) |
Age Range | 1 - 70 years and older 5 - 60 - 69 years of age 0 - 50 - 59 years of age 2 - 40 - 49 years of age |
Locations Represented | 4 - ON 2 - BC 1 - AB 1 - SK/BC part-time |
Diagnosis / Treatment | Diagnosis - majority of participants were diagnosed 15-20 years ago, with 2 participants diagnosed approximately 5 years ago Treatment - all participants began augmentation therapy in the past 5 years |
Focus Group Session 2: NOT Receiving Augmentation Therapy | |
Participants | 6 total participants (4 male and 2 female) |
Age Range | 3 - 60 - 69 years of age 2 - 50 - 59 years of age 1 - 40 - 49 years of age |
Locations Represented | 5 - ON 1 – NL |
Lung Function Levels | 1 - 90% or higher |
1 - 80% - 89% | |
1 - 70% - 79% | |
0 - 60% - 69% | |
1 - 50% - 59% | |
0 - 40% - 49% | |
2 - 30% - 39% | |
Interviews (Two Family Sessions: June 2021)
As AATD is a genetic disease, many individuals in the focus group sessions discussed not only their concerns, but the concerns of their relatives as well. For example, an individual now diagnosed with AATD spoke about a sibling who died as a young adult from undiagnosed lung issues. To understand and showcase the experiences across families, we conducted two 1-hour semi-structured interviews over the phone with two families living with AATD. Consent was provided from each individual to use their name and any other identifiable information in this submission.
Table 2: Interviews (Two Family Sessions: June 2021)
Participant | Description |
|---|---|
Interview 1: Twin Sisters | |
Lori |
|
Lynn |
|
Interview 2: Hasulo Family | |
Steve |
|
Jeff |
|
Julie |
|
Survey (Distributed: Thursday April 1, 2021 – Monday, May 10, 2021)
Three separate surveys were developed and distributed:
Patients Receiving Augmentation Therapy (30 questions)
Patients Not Receiving Augmentation Therapy (30 questions)
Caregivers of Patients diagnosed with Alpha-1 (20 questions)
Surveys were developed and housed by a third-party organization. Sciteline is a Toronto-based SaaS company focused on supporting clinical research by connecting remotely to patients and ensuring patient data collection is in compliance with the Personal Health Information Protection Act 2004 (PHIPA) and Personal Information Protection and Electronic Documents Act (PIPEDA).
Surveys had a mixed approach to questions which ranged from multiple choice, rating scale, and open text. The surveys ran for 6 weeks and received a total of 100 completed responses (32 male and 68 female) all residing in Canada. Of the alpha-1 patients who responded, the majority (78%) were ZZ genotype with a few noted MZ and SZ alleles. The majority (44%) noted having symptoms for 7 years or longer, 9% had symptoms for 5 to 7 years, 13% had symptoms for 3 to 5 years, and 34% had symptoms for 3 or less years. Further respondent demographics and survey questions are outlined below.
Table 3: Respondent Demographics and Survey Responses
Variable | Description |
|---|---|
Survey Respondents Receiving Therapy | |
Number of respondents | 37 (18 male and 19 female) |
Age | Male Average age: 64 years (min: 48 / max: 78) Female Average Age: 61 years (min: 46 / max: 76) |
Location | 8 - AB 10 - BC 1 - NL 1 - NS 14 - ON 1 – SK 1 – QB 1 - Unknown |
Survey Respondents NOT Receiving Therapy | |
Number of respondents | 47 (9 male and 38 female) |
Age | Male Average age: 63 years (min: 45 / Max: 74) Female Average Age: 61 years (min: 22 / max: 79) |
Location | 9 - AB 4 - BC 2 - MB 4 - NB 1- NS 21 - ON 3 - QB 1 - SK 2 - Unknown |
Caregivers | |
Number of respondents | 16 (5 male and 11 female) |
Age | Male Average age: 52 years (min: 29 / Max: 71) Female Average Age: 49 years (min: 31 / max: 74) |
Location | 3 - AB 3 - ON 1 - SK 9 - N/A |
Canadian Respirologists (One Question Email Survey)
Many Alpha-1 patients who are not able to access augmentation therapy noted various other treatments to help manage the symptoms of AATD. To understand the top symptom management treatment prescribed, we sent a single question via email to 40 Canadian Respirologists on May 26, 2021. We received 29 responses.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
Response: Alpha-1 Antitrypsin (AAT) is an essential anti-inflammatory, anti-infective, immunomodulating protein, required for normal lung health. The rare genetic condition of Alpha-1 Antitrypsin Deficiency (AATD), leaves individuals without the essential protective protein, predisposing them to early onset genetic emphysema. This damages their lungs and makes it more difficult to breathe, with effects occurring as early as their 20s. If left untreated, affected individuals can expect a reduced life expectancy, as the lack of AAT makes the lungs more susceptible to bacteria and patients experience repeated lung damaged from proteases and inflammation.
This physical manifestation of AATD impacts individuals throughout many aspects of their lives ranging from employment, relationships, extracurricular activities, day-to-day tasks, and overall mental health. Of the individuals who responded to the survey, 70% noted that their or their loved one’s condition affects their mental health.
“.[..] it is very stressful on our family who are all very close to one another. This condition has been extremely debilitating for her both from a respiratory standpoint, and a mental health standpoint. It forces upon us a state of constant worry, and especially this past year with COVID-19. Their retirement plans have completely changed and they are now planning to potentially have to move to a province which covers her infusions as they cannot afford this treatment out-of-pocket. It is tough as a family member and caregiver to see someone debilitated and demoralized by such a harsh condition.” - Caregiver Survey Comment
Employment and ability to work are often affected by deteriorating lung function. The majority of Alpha-1 patients whose employment was affected reported that they were no longer able to work (69%) or had to reduce their overall working hours (22%).
“When I learned of my Alpha-1 deficiency I was concerned that my lifespan would be lessened as I grew older. I was also concerned with my lung function depleting because of the disease and not having access to [augmentation therapy]. It concerned me that my job as an ECE teacher was making me vulnerable to having exacerbations due to germs passed on to me during cold and flu season. I would have to take time off work due to chest infections and pneumonia.” - Survey Respondent
“It has been life changing for both of us affecting what we can do and can’t do because of extreme shortness of breath and need for oxygen therapy and any exertion limits abilities and activities possible. We had to retire at an early age to preserve the best quality of health and life possible.” - Survey Respondent
“...During the process due to my health, I lost my career, my wife, and my children, and any semblance of a reasonable quality of life. While on treatment my health remained stable. Once denied access, it spiraled out of control, stealing what little lung function I had left.” - Survey Respondent
Ninety-three percent (93%) of individuals whose condition affected social and community participation (n=53) mentioned this was due to their lack of physical stamina and endurance. Impact on physical ability was noted by all participants: over half (52%) felt their condition impacted their ability to participate in light physical activity, with over 90% feeling the impact during moderate (90%) and heavy (94%) physical activity.
“It’s an effort to do even the smallest things as I am always gasping for breath. For those of us who have been active all our life this is such a drastic change - socially and physically. It’s hard to cope as it encompasses everything you do, even walking in colder weather feels like someone is sitting on your chest. Walking up the stairs, I can’t seem to catch my breath, being completely winded.” - Focus Group Participant (Not Receiving Therapy)
“I’m a Kindergarten teacher, but I haven’t been able to work for the past year due to COVID-19 and because children pass on many different viruses and bacteria which I am highly susceptible to, risking further lung deterioration. I’ve quickly realized this past year that I couldn’t work in the school system anymore as I am putting myself at higher risk and have made up my mind to retire early.” - Lori (61), Interview participant, (Not Receiving Therapy)
The majority of participants in the survey, focus groups and interviews also articulated the importance of predictability in their lives as a benefit of receiving effective treatment. The opportunity to see a novel and effective treatment come to market is incredibly important given the limited coverage of current augmentation therapies for AATD. This is critical for alpha-1 patients as those not on treatment often mention the crippling anxiety they experience in regard to their inability to access treatment and individuals currently on treatment express the chronic fear and anxiety that their coverage may be terminated by a public or private payer at any time with little to no warning. Individuals with this condition are extremely aware that the only way to protect their lungs, improve their quality of life and reduce their chances of dying prematurely is through consistent augmentation therapy.
“When I was finally diagnosed with Alpha-1, I was at 50% lung capacity. I was initially on augmentation therapy, but lost it three years ago when my wife lost her insurance package at work. I did everything in my power to remain active, even though it causes me to be gasping for air, but I am extremely concerned about my lung capacity dropping each time I go for a breathing test check-up. I hope this [augmentation therapy] becomes a standard procedure that everyone who needs it has access to it and it doesn’t depend on whether you have a good insurance plan. It’s unfortunate to say, but I hope I don’t deteriorate as fast as others.” - Focus Group Participant (Not Receiving Therapy)
“I felt panic and unsure about the future. Dismay that there was no cure and disappointment that augmentation therapy was not available for me due to high cost and no coverage under health care. [...] My lung specialist has given me a prescription for treatment, but it is too expensive for me. I just hope to avoid any lung infections and avoid further deterioration. We have many grandchildren that we like to spend time with, but I do worry about picking up whatever bugs they get through interactions in school…” - Survey Participant (Not Receiving Therapy)
Experiences With Currently Available Treatments
CADTH examines the clinical benefit and cost-effectiveness of new drugs compared with currently available treatments. We can use this information to evaluate how well the drug under review might address gaps if current therapies fall short for patients and caregivers.
Describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
Response: The only treatment available for people living with AATD is augmentation therapy. It is the only form of treatment that stops lung destruction by restoring the protease / antiprotease balance required for lung health and maintenance. Without augmentation therapy, an individual’s lungs are left susceptible to infections and severe lung deterioration, leading to higher premature mortality rates. Lung deterioration is exacerbated by exposure to bacteria (e.g., mycobacterium avium complex and pseudomonas), increasing high utilization of antibiotics. This was clear in AlphaNet Canada’s Mortality study which demonstrated the following:
Reduction in health resource utilization between Year 1 and Year 2 for patients on augmentation therapy including reduced hospitalizations, lung-related hospitalizations, ER visits, primary physician visits, liver specialist visits, and other MD visits
Substantial reduction in the exacerbation rate and exacerbation severity for individuals on augmentation therapy after a year on therapy, with results sustained after two years
Slowed progression of declining quality-of-life through lung capacity maintenance.
Alpha-1 patients who are able to access augmentation therapy receive weekly infusions. Some patients noted that this schedule can interfere with other aspects of their life, like having to organize work schedules around their infusion schedule or the challenge of travelling abroad. But these “difficulties” are seen as extremely minor in exchange for improved quality and longevity of life. Some even noted flexible treatment options or being able to access augmentation therapy when travelling across Canada.
“Before COVID-19, my work was flexible, and I was able to have a nurse come in and give me my therapy in the conference room. Now she treats me at home.” - Focus Group Participant Receiving Therapy
“When I go to other provinces, I can arrange to get my treatments in those provinces. It is great to have the ability to travel freely within Canada.” - Lynn (61) - Interview participant receiving therapy
However, this lifesaving treatment is currently only publicly funded in British Columbia and Quebec. Other provinces are not able to consider including this treatment on their formularies as it is derived from human plasma. Offering this treatment across Canada requires review and approval by Canadian Blood Services and CADTH.
“…the Saskatchewan formulary does not list items that contain blood derivatives or sera. As [augmentation therapy] is derived from human plasma, it is not eligible for consideration of listing on the provincial formulary in Saskatchewan.” – Deputy Minister
Alpha-1 patients not living in British Columbia and Quebec have gone to great lengths to access augmentation therapy, often through private pay or clinical trials. They are motivated by slowing their decline in quality of life and reducing the number of severe exacerbations. On average each year, four to six patients registered with Alpha-1 Canada who are unable to access this life prolonging therapy where they currently live chose to relocate to either British Columbia or Quebec to access treatment. The ability to access treatment is seen as so essential that they are willing to uproot their lives and move away from their families and communities to live in a province that offers coverage.
“I did everything I could to get on augmentation therapy, but my benefits package is not the greatest and I have a lifetime cap of $100,000. That doesn’t even cover me for one year of augmentation therapy. This is why I decided for early retirement and will move to British Columbia,” - Focus Group Participant (Not Receiving Therapy)
“I was diagnosed 4 years ago when I had a bout of really bad pneumonia - walking pneumonia - and when I recovered, I had persistent, nagging chest pains and constant shortness of breath. At that time my company had a good plan that actually covered augmentation therapy; however, this all changed a year and a half later when my benefits package changed, and my new drug plan only covered me for 2 treatments. Since then, I have moved away from my family to British Columbia to be able to receive augmentation therapy.” - Jeff (54) - Hasulo Family
When alpha-1 patients are on augmentation therapy, they are able to stabilize their lung function, which is extremely important to alpha-1 patients and is seen as the main benefit of the treatment.
“I’ve had trouble breathing way back in 1998 and it took me almost 10 years to be diagnosed and I have been on it since. Augmentation therapy has kept me going and stable for the past 6 years.” Focus Group Participant Receiving Therapy
“I have been on 3 different antibiotics, on and off, over the past 10 years because of my condition. I kept on getting infections that I couldn’t get rid of, causing a steep decline in my lung function. Being on augmentation therapy has kept me stable.” - Focus Group Participant Receiving Therapy
“It would stop the depletion of my lung capacity, set it at a level. I have already lost some lung capacity, but I wouldn’t lose it as quickly over my lifetime.” - Lori (61 - ON) - Not Receiving Therapy
Many alpha-1 patients who are able to manage their lung function through augmentation therapy face fear and anxiety of what will happen if they are no longer able to access the treatment. Treatment is often terminated due to change in benefits packages, retirement causing a loss of benefits, reaching the life- time cap of a benefits plan, or the ending of a clinical trial.
“I was diagnosed back in 2009 but didn’t require augmentation therapy until 2017 when my lung capacity started declining rapidly. I have been on it since then and it has kept me stable, but my coverage will end when I retire at 65. I think about this all the time.” - Focus Group Participant Receiving Therapy
“I had a persistent bug over the winter that I couldn’t get rid of, and my lung function went off the charts from 100% to 50%. I am lucky to receive augmentation therapy, but my greatest anxiety is whether I can maintain my coverage and whether or not I can retire.” - Focus Group Participant Receiving Therapy
For those not able to access augmentation therapy, alpha-1 patients rely on prescriptions for a range of inhalers to manage the symptoms of AATD. Besides augmentation therapy, there are no other effective treatments to slow the progression of this disease.
“...There are no approved alternative treatments to augmentation therapy for patients with severe A1AT deficiency. We try to manage the symptoms by usual standard of care treatments that include respiratory vaccinations, encouraging physical activity, optimizing pharmacotherapy - largely inhaled medications although some oral therapies are used for those with frequent exacerbations, oxygen if stable severe hypoxemia, and management of other complications such as acute exacerbations.” - Canadian Respirologist
While inhalers can help patients manage their symptoms and may minimize the disruptions to their employment, social participation and day-to-day activities, they do not offer a long-term solution as patients lung function continually declines
Improved Outcomes
CADTH is interested in patients’ views on what outcomes we should consider when evaluating new therapies. What improvements would patients and caregivers like to see in a new treatment that is not achieved in currently available treatments? How might daily life and quality of life for patients, caregivers, and families be different if the new treatment provided those desired improvements? What trade-offs do patients, families, and caregivers consider when choosing therapy?
Response: The greatest challenge for those afflicted with AATD is lack of access to treatment that can greatly improve quality of life. Patients do not have the ability to consider trade-offs in choosing therapies, as the decision, where possible, is to access the only available therapy. In areas where there is not publicly funded access to treatment (see public payer graphic below from August 2021), patients are weighing the steps they are willing to take to access the therapy, such as continuing to work long past retirement age to be eligible for private insurance or uprooting their lives to move to a province that offers coverage.
Figure 1: Equitable Access to Augmentation Therapy Is the Primary Interest of Alpha-1 Patients: Alpha-1 Patients Receiving Treatment Through Public Payer
If able to access treatment, the most important indicator for patients is slowing the disease's progression, as measured by improving or stabilizing overall lung function. Zemaira is a highly purified alpha-1 protein, derived from human plasma and indicated to treat patients with Alpha 1 Antitrypsin Deficiency (AATD). It is the only Alpha 1 proteinase therapy that, in a randomized controlled trial, has been proven to be disease-modifying, by significantly reducing the loss of lung tissue, thereby slowing the progression of the disease.
The most important outcomes patients are looking for from treatment are improvements to the stability of their lung function and the associated impacts on their ability to perform activities of daily living and fully participate in their communities and with their families.
“I was in the army for 5 years right after high school and I would always be getting bronchitis and pneumonia. My last lung function test, I was at 37%. I used to be a big swimmer and now it’s embarrassing when I get out of breath from the smallest things. My family helps me with groceries and heavier stuff. I have 3 young girls and I can’t keep up with them, like hiking together. Thankfully, my partner is understanding of this disease and its effects. I’m trying to make the best out of this situation as I can but being on [augmentation therapy] can make sure I am around for longer and be with my family.” - Focus Group Respondent - Not Receiving Therapy.
Consistent coverage will ease anxiety for patients, providing them with options that do not require paying out-of-pocket or leave them vulnerable to changes in insurance coverage. Not only will this impact the individual’s physical health, but it will also greatly reduce chronic stress and anxiety.
“I was approved by my husband’s insurance company through work and was on [augmentation therapy] for 5 years. My husband passed away two years ago, and I was given a two-year grace period. That was up as of April 20, 2021, where I no longer have coverage for my [augmentation therapy]. I cannot get it through Alberta health care, I cannot get it anywhere unless I pay out-of-pocket, and I cannot afford it. So now I just get to sit here and let my lungs get worse until they don’t function anymore. The [augmentation therapy] that was saving my life has literally been yanked away from me and now I have to suffer. My children and my grandchildren will not only lose their grandfather but now their grandmother as well. What a sad situation” - Survey Participant
“I was devastated when I was diagnosed. My work health insurance covered my infusions in 2011, but the process was slow, 4 months from diagnosis to receiving [augmentation therapy]. I retired in 2019 as I am no longer able to work as an RN. My employment insurance benefits run until my 65th birthday, Sept 2023. Living in Ontario I have no other way of obtaining augmentation therapy. I am extremely anxious and uncertain regarding my health and the progression of A1AT when my insurance coverage ends. I feel that receiving augmentation therapy has slowed the progression of my disease and allowed me to have a good quality of life. I don't know what will happen when I no longer receive the therapy.” - Survey Participant
Currently, alpha-1 patients are forced to consider whether they are willing to leave their current place of residence for access to treatment, as many patients consider whether they are willing and able to permanently move to a province that provides public access to therapy. For many, living away from their families, communities and support is the only option to gain access to therapy, while for other patients it is impossible to uproot their lives because of employment or familial considerations.
“I would like to see national coverage to provide stability and choices in my life that I need and everyone else gets. I have to live away from my family so I can receive augmentation therapy and extend the quality and longevity of my life.” - Jeff (51) - Hasulo Family
The other major access challenge alpha-1 patients face is the need to demonstrate deteriorated lung function before being eligible to access augmentation therapy. Many felt that they were doing additional damage to their lungs - and compromising their quality of life - while they waited for their lung function to decline to the point that they were able to access therapy.
“I think it is ridiculous not to put the person with Alpha-1 Antitrypsin Deficiency in earlier at higher levels [of lung functioning]. Putting us through sooner would put overall less strain on the system. Yes, I know the drug is expensive, but if we are at higher levels while on augmentation therapy, it allows us to be more active and healthier for longer, reducing our likelihood of other comorbidities.” - Lori (61 - ON) Twin Sister
Experience With Drug Under Review
CADTH will carefully review the relevant scientific literature and clinical studies. We would like to hear from patients about their individual experiences with the new drug. This can help reviewers better understand how the drug under review meets the needs and preferences of patients, caregivers, and families.
How did patients have access to the drug under review (for example, clinical trials, private insurance)? Compared to any previous therapies patients have used, what were the benefits experienced? What were the disadvantages? How did the benefits and disadvantages impact the lives of patients, caregivers, and families? Consider side effects and if they were tolerated or how they were managed. Was the drug easier to use than previous therapies? If so, how? Are there subgroups of patients within this disease state for whom this drug is particularly helpful? In what ways? If applicable, please provide the sequencing of therapies that patients would have used prior to and after in relation to the new drug under review. Please also include a summary statement of the key values that are important to patients and caregivers with respect to the drug under review.
Response: As the alpha-1 patient group on augmentation therapy is small and Canadian patients are not currently able to access the drug under review, this section focuses on augmentation therapy overall.
As it has been noted throughout this submission, many alpha-1 patients face immense difficulty in accessing augmentation therapy and will go to extensive lengths to access the treatment. Examples of this include patients who have sold their homes and businesses to uproot their lives to move to a province with coverage, and others who have taken out reverse mortgages on their homes or launched GoFundMe campaigns to pay for therapy costs out-of-pocket. Other patients participate in clinical trials as a last resort, however it is a time-bounded solution.
“I approached the provincial government for funding but was denied. My insurance approved coverage but total payment would cover 1 infusion so I would be on the hook for the other 51 per year. I am currently in a clinical trial as my last resort. This plasma should be available through Canadian Blood Services because it is that important. I feel too young to die.” - Survey Respondent
“I felt my life in my latter years would be impacted. I do not have coverage for [augmentation therapy] in the future. The clinical trial ends in 18 months.” - Survey Respondent
The overall benefit of augmentation therapy is to slow or stabilize the disease and minimize its impact on lung function. Augmentation therapy is the only treatment that extends a patient’s life and supports better quality of life. Many Canadian respirologists surveyed repeatedly mentioned there is no other “treatment” besides augmentation therapy and other methods used are for symptom management only.
Alpha-1 patients rarely discussed disadvantages of augmentation therapy even when asked, as they did not feel that any disadvantages were worth noting in comparison to the possibility of maintaining their ability to breathe, living a longer and more active life and avoiding the impending loss of lung function
Patients highlighted the other costs to the health care system when they are not able to access treatment. They require other medications such as inhalers for symptom management, undergo frequently repeated tests of lung function, experience hospitalizations during exacerbations and severe lung challenges, and undergo lung transplant if their lung function has deteriorated to the point of eligibility.
“I have an older sister with alpha-1 who was not [able to access augmentation therapy] as she did not have funding and she received a double lung transplant at the age of 44 years…” - Survey Respondent
“...After six years my health, directly related to alpha-1, had degraded to the point where I had to go on disability. Eventually I found a job that allowed me to work from home tethered to my supplemental oxygen. Within a few years, I required a bi-lateral lung transplant.” - Survey Respondent
The key values that are important to patients and caregivers with respect to augmentation therapy are:
Equitable and wide-spread funded access: Patients across Canada should be able to receive augmentation therapy that is paid for by the government if they do not have private coverage
Improved longevity and reduced early mortality: Patients are focused on gaining access to a treatment that will extend their lives and minimize the chance of early mortality due to lung disease
Improved quality of life and participation in society: Patients are interested in treatment that will allow them to fully participate in their lives, including employment, engagement with family, activities of daily living, and active lifestyles
Improved mental health and reduced anxiety: Patients would like to see improved mental health and reduced anxiety, with less worry about loss of coverage, the consequences of continued disease progression and the impact on their families
Overall cost to the health care system: Patients see the overall cost of symptom management, repeated tests and the management of exacerbations as comparable to the cost of treatment
“It makes me angry that there is a treatment out there that is known to extend our lives and keep us in a healthy direction. Why do we have universal healthcare if you can’t protect every Canadian?” - Focus Group Participant Not Receiving Therapy
“I’ve worked all my life and I was looking forward to retiring, but not anymore. Retiring for what? To lose my coverage and spiral downwards to an earlier death?” Focus Group Participant Not Receiving Therapy
“It shouldn’t be imposed on us that the only way to access augmentation therapy is to move to another province - away from family, friends, the doctors we trust and everyone else - that’s a lot to ask.” - Focus Group Participant Not Receiving Therapy
Companion Diagnostic Test
If the drug in review has a companion diagnostic, please comment. Companion diagnostics are laboratory tests that provide information essential for the safe and effective use of particular therapeutic drugs. They work by detecting specific biomarkers that predict more favorable responses to certain drugs. In practice, companion diagnostics can identify patients who are likely to benefit or experience harms from particular therapies or monitor clinical responses to optimally guide treatment adjustments.
What are patient and caregiver experiences with the biomarker testing (companion diagnostic) associated with regarding the drug under review?
Consider: Access to testing for example, proximity to testing facility, availability of appointment. Testing: for example, how was the test done? Did testing delay the treatment from beginning? Were there any adverse effects associated with testing? Cost of testing: Who paid for testing? If the cost was out of pocket, what was the impact of having to pay? Were there travel costs involved? How patients and caregivers feel about testing: for example, understanding why the test happened, coping with anxiety while waiting for the test result, uncertainty about making a decision given the test result.
Response: Alpha-1 patients cannot be diagnosed by symptoms or by a medical examination alone. There are two ways of diagnosing Alpha-1 Antitrypsin Deficiency (AATD) and two additional approaches for further confirmation of AATD, which are commonly used and publicly available to Canadians.
Diagnosing AATD
Blood Test: Measures the level of alpha-1 antitrypsin in the bloodstream.
Genotype Test: Can be used to establish which SERPINA1 gene alleles are present, including the normal wild type M allele or variant alleles. This test does not identify every variant, but it will detect the most common ones (S and Z). Once the affected person’s SERPINA1 gene alleles have been identified, other family members may be tested to establish their own risk in developing emphysema and the likelihood of their children inheriting the disease (e.g., MZ- Normal, MZ - Carrier, while other alleles [ZZ, SZ] cause AATD)
“I first learned of Alpha-1 as my mom was a ZZ and my aunt was a SZ. My aunt died 4 years ago and my mom 3 years ago. I am just now learning about the MZ allele…” - Survey Respondent
“It's been tough in the sense that with a lot of doctors you’re getting different feedback depending on who you see. You do have to be your own health advocate in terms of learning about it. I’ve had to go to another clinic in order for me to push and say, Hey, I need my spirometry test and I need my fibro scans and all those kinds of things…It’s a genetic disease we didn’t really know about it and it’s not like we brought it upon ourselves.” Julie (43) - Hasulo Family
Confirming AATD
FEV1 testing: Initial evaluation with complete lung function testing is recommended and annual check-ups assessing their lung capacity levels aid doctors to monitor alpha-1 patients disease progression and guide decisions for further treatment and intervention. Most commonly used to understand severity and progression of disease.
CT Density: In newly diagnosed patients who are symptomatic and/or had abnormal pulmonary function testing, a baseline CT Density Scan is recommended.
“Every 6 months I have a respiratory test and levels now come back relatively the same because I am on augmentation therapy.” - Lynn (61 - BC) - Twin Sister
“When my brother was diagnosed and mentioned it was a genetic disease I immediately got tested. My blood work showed that my serum levels were pretty low so when I came back to British Columbia, I arranged a doctor's appointment and got the genetic test. I am SZ. I started the process of benchmarking my lungs and liver. I have very good lung capacity at the moment so I will continue to monitor with my doctor.” - Julie (43) - Hasulo Family
An accurate diagnosis facilitates the physician’s ability to actively intervene with measures to capture and maintain lung capacity (FEV1 levels) or to provide augmentation therapy to patients who need it when there is lung function decline. Of the alpha-1 patients not receiving augmentation therapy (survey participants), 57% mentioned not being prescribed the treatment. Throughout the focus group discussions and interviews, participants described managing and monitoring their condition with their doctor.
Diagnosis did not mean augmentation therapy would automatically be prescribed; prescriptions were only written if lung capacity levels were no longer stable. This demonstrates that Canadian physicians are properly monitoring alpha-1 patients and only prescribing treatment when it is truly required.
“Over the years, prior to my diagnosis, I was susceptible to pneumonia, colds that lasted longer than normal and a lot of bronchitis. I didn’t think too much about it at the time, but every time I had an infection my lungs were further damaged. I was finally diagnosed with Alpha-1 Antitrypsin Deficiency in 2011 and for the next 5 years I would keep a close eye on the condition with my pulmonary doctor and do breathing tests. My condition was stable at around 75% for those 5 years, but then my levels started to decline and continued to.
When I was at 68% lung function, and it looked like it was going to continue to decline I was put on augmentation therapy.” - Lynn (61 - BC) - Twin Sister
Alpha-1 patients did not express any concerns with respect to access and cost of testing.
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
Response: It was on the advice of the Provincial Territorial Blood Liaison Committee (PTBLC) and Canadian Blood Services (CBS) that we focus our efforts on advocating to manufacturers of drugs like Zemaira to go through this specific CADTH process.
On January 6, 2020, Mr. Max Hendriks, Deputy Minister of Health, Government of Saskatchewan informed Alpha-1 Canada that “the Saskatchewan formulary does not list items that contain blood derivatives or sera” Deputy Minister Hendriks noted that because augmentation therapy “is derived from human plasma, it is not eligible for consideration of listing on the provincial formulary in Saskatchewan”. So what are alpha-1 patients in Saskatchewan supposed to do? This sentiment was echoed among other provincial deputy health ministers, which is why it is imperative that augmentation therapy be added as a category with the nation’s blood operator, because augmentation therapy for severe alpha-1 antitrypsin deficiency is a plasma protein therapy and the only specific treatment for the disease.
Additionally, 12 other organizations - including national and international patient and clinical associations - have endorsed the listing of augmentation therapy with Canadian Blood Services (see organizations listed below and letters attached).
Endorsements received from:
The Canadian Association of Genetic Counsellors
Canadian Thoracic Society
The Canadian Lung Association
Canadian Registry for Alpha-1 Antitrypsin Deficiency
AlphaNet Canada
Canadian Vascular Access Association
Alpha-1 Foundation & Alpha-1 Global
Canadian Network for Respiratory Care
Canadian Society of Respiratory Therapists
Family Physician Airways Group of Canada
Plasma Protein Therapeutics Association
Network for Rare Blood Disorders Organization
Alpha-1 Canada has put together short videos showcasing some examples of the experiences of both people living with, and those treating patients with alpha-1 antitrypsin deficiency across Canada. Access to the videos for the can be found at https://drive.google.com/drive/folders/1yl46WzCN- 6nmuPaxM3Sw89dMnGvsxovo?usp=sharing
In conclusion, Alpha-1 Canada would like the CADTH reviewers and the expert committee to know that at the time of this submission, 27 Canadian alpha-1 patients passed away in the first ten months of 2021.
These Canadian alpha-1 patients were at various stages of their disease and they range in age from 49 to 84 years; however, what all of these patients had in common was the burden of the disease and a lifetime of chronic worry and stress, due to the unnecessary complexities of trying to secure (and maintain) consistent, uninterrupted access to treatment following their diagnosis. Unfortunately, for most of these patients, they had no access at all to the only specific treatment for their disease. This does not have to be the case for their extended family members who have also inherited the rare allele(s) that result in progressive lung decline, nor newly diagnosed Canadian patients, who should all have timely and equitable access to an alpha-1 protein replacement therapy. The nefarious nature of this disease has no bounds and therefore cannot be properly managed in a country where there is such an unpredictable patchwork of care. The inequities in access, both with public and private payers are dire for severely affected alpha-1 patient.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Support was provided by Santis Health, a health care consulting company, to write this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
While Alpha-1 Canada led the data collection, we also received support from Santis Health to record notes of focus groups and interviews. To ensure compliance with the Personal Health Information Protection Act 2004 (PHIPA) and Personal Information Protection and Electronic Documents Act (PIPEDA), Alpha-1 Canada contracted Sciteline, a Toronto-based SaaS company, to develop and house the survey.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 4: Conflict of Interest Declaration for Alpha-1 Canada
Company | Check Appropriate Dollar Range | ||||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
AlphaNet Canada | — | — | X | — | |
Innomar Strategies | X | — | — | — | |
CSL Behring | — | — | X | — | |
Grifols Canada | — | — | — | X | |
Takeda | — | — | X | — | |
Clinician Group Input
Canadian Thoracic Society
Lead Author: Kenneth R. Chapman
Co-Authors: Raymond Aceron, Joshua Wald, Darcy Marciniuk, Gail Dechman, Mohit Bhutani, Paul Hernandez, and Marla Beauchamp
About the Canadian Thoracic Society
The Canadian Thoracic Society (CTS) is a national specialty society and membership-based professional association for health care providers (HCPs) working in respiratory care and research. Our mission is to promote lung health by enhancing the ability of HCPs through leadership, collaboration, research, learning and advocacy, and providing the best respiratory practices in Canada. CTS is recognized as an accrediting body of the Royal College of Physicians and Surgeons for specialist education and continuing professional development. https://cts-sct.ca/
Information Gathering
The following document has been prepared as a submission to CADTH in response to its request for clinician input into the application by CSL Behring regarding its alpha-1 proteinase inhibitor, Zemaira®. The submission represents the viewpoint of the Canadian Thoracic Society with respect to alpha-1 anti-proteinase inhibitor therapy in general and with respect to this submission in particular. The first draft was prepared by ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The draft document was then submitted to the COPD Committee of the Canadian Thoracic Society for review, revision and approval prior to its submission.
The following material reviews the impact of Zemaira® and similar augmentation therapies on the progression of emphysema secondary to a severe deficiency of alpha-1 antitrypsin. It does not address pulmonary diseases other than emphysema (such as bronchiectasis) nor does it address other putative therapeutic roles of augmentation therapy in other contexts.
Current treatments
Describe the current treatment paradigm for the disease
The Canadian Thoracic Society (CTS) has considered augmentation therapy previously and has published several position statements. The first statement noted that such therapy had been approved for use in Canada based on its pharmacokinetic properties and safety as a blood product but in the absence of demonstrated impact on emphysema progression. This initial statement, published in 1992, called for further research. In particular, it sought data from a study using conventional measurements of spirometry in order to assess whether alpha-1 antitrypsin deficient individuals treated with purified alpha-1 antitrypsin intravenously had slower rates of FEV1 decline. It was subsequently recognized that the proposed randomized controlled trial was not feasible. It has been estimated that such a study would require a minimum intervention of five years with randomization of between 600 and 1,000 severely deficient individuals to augmentation therapy or placebo treatment arms. To date, the largest randomized and placebo-controlled trial that has been undertaken to test the efficacy of augmentation therapy recruited 180 patients treated for just two years. That initiative required the collaborative efforts of 24 international sites working for approximately seven years. It is generally agreed that trials based on FEV1 change over time are not feasible in this rare disease.
Subsequent CTS statements have been undertaken periodically to incorporate up-to-date information generated by ongoing research. The meaningful impact of augmentation therapy and its potential role in clinical use has been acknowledged by all subsequent Canadian Thoracic Society statements. The most thorough and specific statements in this regard were issued in 2012 and followed a rigorous GRADE guideline process. This statement dealt with two issues: screening (targeted testing) and the appropriate use of augmentation therapy. With respect to the latter, the statement concluded that: “The evidence…supports consideration of A1AT augmentation therapy in non-smoking or ex-smoking patients with COPD (forced expiratory volume in 1 s of 25% to 80% predicted) attributable to emphysema and documented A1AT deficiency (level ≤11 µmol/L) who are receiving optimal pharmacological and non-pharmacological therapies (including comprehensive case management and pulmonary rehabilitation) because of benefits in computed tomography scan lung density and mortality”. The CTS is currently updating this statement by incorporating additional scientific information gathered over the past decade. Although it is possible that the details of the CTS recommendations may change, the general recommendation is likely to stand and to be reinforced. Pending a systematic review of the literature, experts in this well-known rare disease field, who are represented in this submission, are confident that no discrepant data have been published. Moreover, the results of the RAPID trial concerning Zemaira® have provided the strongest and most comprehensive data so far published to support the role of augmentation therapy to prevent the progression of emphysema in alpha-1 antitrypsin deficiency.
In 1963, Laurell and Eriksson first reported that individuals with a severe deficiency of alpha-1 antitrypsin showed a propensity to develop emphysema early in life with less-than-expected exposure to tobacco smoking. Subsequent research has shown that inadequate levels of alpha-1 antitrypsin in the lung result in diminished ability to neutralize elastases, in particular neutrophil elastase. This protease-antiprotease imbalance results in the destruction of alveolar structure. The consequent pattern of emphysema is distinctive and is typically described as panlobular and basilar. That this mechanism of disease is of importance is by a natural history “dose response”. That is, carriers with only one abnormal gene of this protective protein have been shown to have very slightly increased risk of emphysema as compared to been individuals with two normal genes in normal serum levels. At the other end of the spectrum, rare individuals with two null genes who produce no detectable alpha-1 antitrypsin have a severely increased risk of premature emphysema development.
Given decreased levels of a normal blood constituent is associated with the development of emphysema, investigators worked to determine if it was feasible to administer this blood product in sufficient amounts to delay the progression of emphysema. The pivotal finding was published by Wewers and colleagues in 1987 when they reported that once per week intravenous infusions of purified A1AT at a dosage of 60 mg per kilogram could maintain alpha-1 antitrypsin serum levels above the protective threshold of 11 µMol and was associated with the appearance of infused alpha-1 antitrypsin in bronchoalveolar lavage fluid. Subsequent small cohort or randomized studies using clinical disease endpoints have shown supportive but inconclusive results. More information has been garnered from large natural history studies. The NHLBI registry reported in 1998 that of 927 individuals with severely deficient in alpha-1 antitrypsin deficiency, treatment with augmentation therapy was associated with a slower loss of lung function as measured by FEV1 and decreased mortality in those with moderate or severe airway obstruction. A subsequent meta-analysis of available data showed that intravenous augmentation therapy was associated with a slower rate of FEV1 decline approximating 17 mL per year (or 50% of the normal rate of FEV1 decline in healthy adult).
Treatment goals
What are the most important goals that an ideal treatment would address?
In the three decades since the CTS first suggested that an FEV1-based trial would be useful, this endpoint has been re-examined and its practical shortcomings noted. Although FEV1 remains essential in the diagnosis of common obstructive lung diseases, its role in day-to-day management is diminished. In the broad category of COPD, for example, the CTS and GOLD strategy both recommend adjusting pharmacologic interventions based on symptom burden and exacerbation tendency rather than spirometric cutpoints. In the setting of emphysema caused by a severe deficiency of alpha-1 antitrypsin, direct quantification of lung parenchymal density has been well-studied and shown to be especially valuable. In a disease characterized by the loss of alveolar structure, lung density estimated objectively by CT scan techniques has proven to have better prognostic value than conventional measures of lung function. This parameter has been adopted in clinical trials of augmentation therapy, proving to be more sensitive and specific than changes in FEV1. This was first documented by Dirksen’s study published in 1999. In that study of serial spirometry, the conventional lung function parameter proved insensitive while CT scan lung density identified a nearly significant trend in preserved lung structure for patients receiving intravenous augmentation therapy. In a later pilot trial known as the EXACTLE study, the technology and its analysis was explored further yielding similar results. The study results were subsequently pooled in a post hoc analysis and demonstrated highly significant results consistent with the protection of lung parenchyma by intravenous augmentation therapy. These were data available to the CTS for its position statement of 2012 and resulted in its recommendation concerning augmentation therapy and benefits in CT lung density. Since the statement of 2012, the most significant additional work has been the RAPID study considered to be a landmark in showing the efficacy of augmentation therapy in patients with a severe deficiency of alpha-1 antitrypsin. It was undertaken by CSL Behring using the Zemaira formulation currently being reviewed by CADTH. Published in the Lancet, this pivotal study confirmed that patients receiving augmentation therapy exhibited a slower rate of lung density loss than patients receiving matching placebo. In an extension of the double-blind trial, patients switched from placebo to augmentation therapy enjoyed a subsequent slowing of their lung density loss but never regained the lung density lost during the first 2 years of the trial prior to initiating augmentation. This study provided further data concerning potential benefits of augmentation therapy. In the initial analysis and publication, investigators were able to associate decreases in lung density with the risk of death or lung transplantation. In addition, the RAPID trial suggested a dose response to augmentation therapy. Although only one dosage of augmentation therapy was used during the trial (the dosage of 60 mg per kilogram per week), different volumes of distribution amongst subjects led to a post hoc analysis showing that higher serum levels were associated with a greater degree of protection against lung density loss as estimated by CT scan. In a subsequent publication of the extension study, changes in lung density were shown to be associated with changes in spirometry as measured by FEV1 and FVC, reassuring that CT density endpoint bears correlation with more familiar but slower clinical physiologic endpoints. Finally, the biochemical efficacy of augmentation therapy has been suggested by the analysis of elastin breakdown products. Serum desmosine and iso-desmosine levels were significantly lower in subjects receiving augmentation therapy than in subjects receiving placebo, a difference that also disappeared when the initial placebo recipients were treated with infusions of alpha-1 antitrypsin.
Treatment gaps (unmet needs)
Considering the previous treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Augmentation therapy for alpha-1 antitrypsin deficiency is currently the only blood product not distributed by Canadian Blood Services. This anomaly has led to challenges for patients and caregivers who seek effective treatment for this genetic disorder. Paradoxically, as our understanding of the deficiency and its treatment have improved over the past 3 decades, the availability of augmentation therapy has diminished. Three decades ago, augmentation therapy was provided to patients through several provincial formularies. At present, only Québec and British Columbia have included augmentation therapy support for patients meeting appropriate criteria. In other parts of Canada, augmentation therapy is available to only those with private health insurance. This has led to difficult lifestyle decisions for affected individuals. Individuals with significant disease and without private health insurance may be forced to consider relocation to another province and in rare instances relocation to other countries where augmentation therapy is routinely afforded the genetically disadvantaged. Similar decisions face patients whose private health insurance is lost at the age of retirement.
Which patients have the greatest unmet need for an intervention such as the drug under review?
The availability of augmentation therapy through private health insurance has also become increasingly difficult. Insurance companies at one time used relatively straightforward criteria for approval when augmentation therapy was prescribed. These included genetic evidence of deficiency and the diagnosis of significant obstructive pulmonary disease made by a respiratory specialist using measurements of lung function analogous to those used in clinical trial settings. However, since the introduction of legislation in Canada designed to prevent genetic discrimination, Canadian insurance companies have refused to accept genetic information. Patients with rare deficiency alleles may be disadvantaged if they have a dysfunctional alpha-1 antitrypsin variant which is quantified by serum level testing but serves no useful anti-protease function. Moreover, insurance companies have adopted the practice of introducing new criteria that are unrelated to product monograph instructions or guideline recommendations. The reality of these many and significant barriers is that patients who could benefit from receiving augmentation therapy are not able to realize this benefit.
A cost benefit analysis of augmentation therapy is beyond the scope of this submission and requires economic and clinical data not currently available. Nonetheless, the CTS understands that medical interventions may be costly and that is particularly true of “orphan” diseases requiring scarce therapies. It may be helpful to consider the likely economic impact of more widespread availability of augmentation therapy in Canada, a possible consequence of having a second entry augmentation product available. Although access to augmentation therapy in Canada is limited, best estimates suggest that approximately 250 Canadians are receiving augmentation therapy now. (The actual figures are unavailable for health privacy and commercial reasons). This leads to an estimate of 0.66 treated patients per 100,000 of the Canadian population. For the purposes of comparison, we can examine the prevalence of use in various European countries that offer unrestricted access to augmentation therapy through their public healthcare systems.
Table 5: Prevalence of Use in Various European Countries That Offer Unrestricted Access to Augmentation Therapy Through Their Public Healthcare Systems
Country | Patients Receiving Augmentation | Population | Augmentation Treatment/100,000 |
|---|---|---|---|
Canada | 250 | 38,000,000 | 0.66 |
Spain | 170 | 46,050,000 | 0.36 |
Italy | 115 | 59,800,000 | 0.19 |
Germany | 1,000 | 80,680,000 | 1.2 |
France | 300 | 64,730,000 | 0.46 |
The use of augmentation therapy may vary by population ethnic background (with severe deficiency being more common in those of northern European background), date of approval and other factors. Nonetheless, the figures show that treatment figures for deficient individuals are well below the gene prevalence for the disorder; not all patients with severe deficiency develop severe emphysema requiring augmentation therapy. If increased access to augmentation therapy allowed more frequent prescription and Canadian healthcare providers matched their European colleagues, the number of Canadians receiving therapy would likely remain below 500.
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Treatments for non-alpha-1 antitrypsin deficiency COPD are used concurrently with augmentation therapy.
Intravenous augmentation therapy is undertaken with alpha-1 antitrypsin purified from blood donation. Worldwide, several companies market augmentation therapies derived by proprietary filtration and purification processes. In Canada, only Prolastin and Prolastin-C (as currently marketed by Grifols) have been available for clinical use. Other augmentation therapies including Zemaira have been used in a limited way in clinical trials. In the absence of head-to-head trials, the CTS has no reason to conclude that there are substantial differences in efficacy or safety amongst various purifications of what is a normal constituent of human blood.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
No other treatment has been shown to delay the progression of emphysema in these patients.
How would this drug affect the sequencing of therapies for the target condition?
N/A
Which patients would be best suited for treatment with the drug under review?
“The evidence…supports consideration of A1AT augmentation therapy in non-smoking or ex-smoking patients with COPD (forced expiratory volume in 1 s of 25% to 80% predicted) attributable to emphysema and documented A1AT deficiency (level ≤11 µmol/L) who are receiving optimal pharmacological and non-pharmacological therapies (including comprehensive case management and pulmonary rehabilitation) …”
How would patients best suited for treatment with the drug under review be identified?
See above. No subset of responders has been identified.
Which patients would be least suitable for treatment with the drug under review?
Patients with minor or no deficiency of alpha-1 antitrypsin functionality. Patients without airflow limitation.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
No responder subset has been identified.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Patients are followed as per clinical routine for COPD. Lack of progression to decreased functionality, need for oxygen and transplantation are evidence of delayed disease progression.
What would be considered a clinically meaningful response to treatment?
See above.
How often should treatment response be assessed?
Patients should be cared for by specialists in respiratory medicine and monitored as per CTS COPD guidelines.
What factors should be considered when deciding to discontinue treatment?
There are no current stopping rules for augmentation therapy except in the case of severe allergic reaction (as may be seen in patients with IgA deficiency).
What settings are appropriate for treatment with the drug under review?
Infusions are safely done in outpatient infusion clinics and in patients’ homes by visiting healthcare personnel.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Respirologists should initiate and monitor this therapy.
Additional information
Is there any additional information you feel is pertinent to this review?
Executive Summary
The Canadian Thoracic Society (CTS) has considered augmentation therapy previously and has published several position statements supporting its use.
The most recent statement concluded that: “The evidence…supports consideration of A1AT augmentation therapy in non-smoking or ex-smoking patients with COPD (forced expiratory volume in 1 s of 25% to 80% predicted) attributable to emphysema and documented A1AT deficiency (level ≤11 µmol/L) who are receiving optimal pharmacological and non-pharmacological therapies (including comprehensive case management and pulmonary rehabilitation) because of benefits in computed tomography scan lung density and mortality”. This statement is being updated but the major conclusion is unlikely to be changed.
The only Intravenous augmentation therapy available clinically in Canada is Prolastin® and Prolastin-C®, with an additional product Zemaira®, available only through clinical trial use.
Augmentation therapy for alpha-1 antitrypsin deficiency is currently the only blood product not distributed by Canadian Blood Services. Furthermore, the availability of augmentation therapy has diminished over the past three decades.
Single supplier availability of any therapy is associated with a monopoly on prices, as well as risk of future shortfalls in availability. Availability of a new augmentation therapy product will mitigate these risks and potentially allow the healthcare system to re-examine the proposition that this blood product be made available through Canadian Blood Services similar to other blood products.
Process
The following document has been prepared as a submission to CADTH in response to its request for clinician input into the application by CSL Behring regarding its alpha-1 proteinase inhibitor, Zemaira®. The submission represents the viewpoint of the Canadian Thoracic Society with respect to alpha-1 anti-proteinase inhibitor therapy in general and with respect to this submission in particular. The first draft was prepared by ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The draft document was then submitted to the COPD Committee of the Canadian Thoracic Society for review, revision and approval prior to its submission.
Limitations
The following document reviews the impact of Zemaira® and similar augmentation therapies on the progression of emphysema secondary to a severe deficiency of alpha-1 antitrypsin. It does not address pulmonary diseases other than emphysema (such as bronchiectasis) nor does it address other putative therapeutic roles of augmentation therapy in other contexts.
Canadian Thoracic Society Statements
The Canadian Thoracic Society (CTS) has considered augmentation therapy previously and has published several position statements. The first statement noted that such therapy had been approved for use in Canada based on its pharmacokinetic properties and safety as a blood product but in the absence of demonstrated impact on emphysema progression. This initial statement, published in 1992, called for further research. In particular, it sought data from a study using conventional measurements of spirometry in order to assess whether alpha-1 antitrypsin deficient individuals treated with purified alpha-1 antitrypsin intravenously had slower rates of FEV1 decline. It was subsequently recognized that the proposed randomized controlled trial was not feasible. It has been estimated that such a study would require a minimum intervention of five years with randomization of between 600 and 1,000 severely deficient individuals to augmentation therapy or placebo treatment arms. To date, the largest randomized and placebo-controlled trial that has been undertaken to test the efficacy of augmentation therapy recruited 180 patients treated for just two years. That initiative required the collaborative efforts of 24 international sites working for approximately seven years. It is generally agreed that trials based on FEV1 change over time are not feasible in this rare disease.
Subsequent CTS statements have been undertaken periodically to incorporate up-to-date information generated by ongoing research. The meaningful impact of augmentation therapy and its potential role in clinical use has been acknowledged by all subsequent Canadian Thoracic Society statements. The most thorough and specific statements in this regard were issued in 2012 and followed a rigorous GRADE guideline process. This statement dealt with two issues: screening (targeted testing) and the appropriate use of augmentation therapy. With respect to the latter, the statement concluded that: “The evidence…supports consideration of A1AT augmentation therapy in non-smoking or ex-smoking patients with COPD (forced expiratory volume in 1 s of 25% to 80% predicted) attributable to emphysema and documented A1AT deficiency (level ≤11 µmol/L) who are receiving optimal pharmacological and non-pharmacological therapies (including comprehensive case management and pulmonary rehabilitation) because of benefits in computed tomography scan lung density and mortality”. The CTS is currently updating this statement by incorporating additional scientific information gathered over the past decade. Although it is possible that the details of the CTS recommendations may change, the general recommendation is likely to stand and to be reinforced. Pending a systematic review of the literature, experts in this well-known rare disease field, who are represented in this submission, are confident that no discrepant data have been published. Moreover, the results of the RAPID trial concerning Zemaira® have provided the strongest and most comprehensive data so far published to support the role of augmentation therapy to prevent the progression of emphysema in alpha-1 antitrypsin deficiency.
Zemaira® versus Alternatives
Intravenous augmentation therapy is undertaken with alpha-1 antitrypsin purified from blood donation. Worldwide, several companies market augmentation therapies derived by proprietary filtration and purification processes. In Canada, only Prolastin and Prolastin-C (as currently marketed by Grifols) have been available for clinical use. Other augmentation therapies including Zemaira have been used in a limited way in clinical trials. In the absence of head-to-head trials, the CTS has no reason to conclude that there are substantial differences in efficacy or safety amongst various purifications of what is a normal constituent of human blood.
Evidence to support the efficacy of augmentation therapy
In 1963, Laurell and Eriksson first reported that individuals with a severe deficiency of alpha-1 antitrypsin showed a propensity to develop emphysema early in life with less-than-expected exposure to tobacco smoking. Subsequent research has shown that inadequate levels of alpha-1 antitrypsin in the lung result in diminished ability to neutralize elastases, in particular neutrophil elastase. This protease-antiprotease imbalance results in the destruction of alveolar structure. The consequent pattern of emphysema is distinctive and is typically described as panlobular and basilar. That this mechanism of disease is of importance is by a natural history “dose response”. That is, carriers with only one abnormal gene of this protective protein have been shown to have very slightly increased risk of emphysema as compared to been individuals with two normal genes in normal serum levels. At the other end of the spectrum, rare individuals with two null genes who produce no detectable alpha-1 antitrypsin have a severely increased risk of premature emphysema development.
Given decreased levels of a normal blood constituent is associated with the development of emphysema, investigators worked to determine if it was feasible to administer this blood product in sufficient amounts to delay the progression of emphysema. The pivotal finding was published by Wewers and colleagues in 1987 when they reported that once per week intravenous infusions of purified A1AT at a dosage of 60 mg per kilogram could maintain alpha-1 antitrypsin serum levels above the protective threshold of 11 µMol and was associated with the appearance of infused alpha-1 antitrypsin in bronchoalveolar lavage fluid. Subsequent small cohort or randomized studies using clinical disease endpoints have shown supportive but inconclusive results. More information has been garnered from large natural history studies. The NHLBI registry reported in 1998 that of 927 individuals with severely deficient in alpha-1 antitrypsin deficiency, treatment with augmentation therapy was associated with a slower loss of lung function as measured by FEV1 and decreased mortality in those with moderate or severe airway obstruction. A subsequent meta-analysis of available data showed that intravenous augmentation therapy was associated with a slower rate of FEV1 decline approximating 17 mL per year (or 50% of the normal rate of FEV1 decline in healthy adult).
Relevant Endpoints
In the three decades since the CTS first suggested that an FEV1-based trial would be useful, this endpoint has been re-examined and its practical shortcomings noted. Although FEV1 remains essential in the diagnosis of common obstructive lung diseases, its role in day-to-day management is diminished. In the broad category of COPD, for example, the CTS and GOLD strategy both recommend adjusting pharmacologic interventions based on symptom burden and exacerbation tendency rather than spirometric cutpoints. In the setting of emphysema caused by a severe deficiency of alpha-1 antitrypsin, direct quantification of lung parenchymal density has been well-studied and shown to be especially valuable. In a disease characterized by the loss of alveolar structure, lung density estimated objectively by CT scan techniques has proven to have better prognostic value than conventional measures of lung function. This parameter has been adopted in clinical trials of augmentation therapy, proving to be more sensitive and specific than changes in FEV1. This was first documented by Dirksen’s study published in 1999. In that study of serial spirometry, the conventional lung function parameter proved insensitive while CT scan lung density identified a nearly significant trend in preserved lung structure for patients receiving intravenous augmentation therapy. In a later pilot trial known as the EXACTLE study, the technology and its analysis was explored further yielding similar results. The study results were subsequently pooled in a post hoc analysis and demonstrated highly significant results consistent with the protection of lung parenchyma by intravenous augmentation therapy. These were data available to the CTS for its position statement of 2012 and resulted in its recommendation concerning augmentation therapy and benefits in CT lung density.
Since the statement of 2012, the most significant additional work has been the RAPID study considered to be a landmark in showing the efficacy of augmentation therapy in patients with a severe deficiency of alpha-1 antitrypsin. It was undertaken by CSL Behring using the Zemaira formulation currently being reviewed by CADTH. Published in the Lancet, this pivotal study confirmed that patients receiving augmentation therapy exhibited a slower rate of lung density loss than patients receiving matching placebo. In an extension of the double-blind trial, patients switched from placebo to augmentation therapy enjoyed a subsequent slowing of their lung density loss but never regained the lung density lost during the first 2 years of the trial prior to initiating augmentation. This study provided further data concerning potential benefits of augmentation therapy. In the initial analysis and publication, investigators were able to associate decreases in lung density with the risk of death or lung transplantation. In addition, the RAPID trial suggested a dose response to augmentation therapy. Although only one dosage of augmentation therapy was used during the trial (the dosage of 60 mg per kilogram per week), different volumes of distribution amongst subjects led to a post hoc analysis showing that higher serum levels were associated with a greater degree of protection against lung density loss as estimated by CT scan. In a subsequent publication of the extension study, changes in lung density were shown to be associated with changes in spirometry as measured by FEV1 and FVC, reassuring that CT density endpoint bears correlation with more familiar but slower clinical physiologic endpoints. Finally, the biochemical efficacy of augmentation therapy has been suggested by the analysis of elastin breakdown products. Serum desmosine and iso-desmosine levels were significantly lower in subjects receiving augmentation therapy than in subjects receiving placebo, a difference that also disappeared when the initial placebo recipients were treated with infusions of alpha-1 antitrypsin.
The current use of augmentation therapy in Canada
Augmentation therapy for alpha-1 antitrypsin deficiency is currently the only blood product not distributed by Canadian Blood Services. This anomaly has led to challenges for patients and caregivers who seek effective treatment for this genetic disorder. Paradoxically, as our understanding of the deficiency and its treatment have improved over the past 3 decades, the availability of augmentation therapy has diminished. Three decades ago, augmentation therapy was provided to patients through several provincial formularies. At present, only Québec and British Columbia have included augmentation therapy support for patients meeting appropriate criteria. In other parts of Canada, augmentation therapy is available to only those with private health insurance. This has led to difficult lifestyle decisions for affected individuals. Individuals with significant disease and without private health insurance may be forced to consider relocation to another province and in rare instances relocation to other countries where augmentation therapy is routinely afforded the genetically disadvantaged. Similar decisions face patients whose private health insurance is lost at the age of retirement.
The availability of augmentation therapy through private health insurance has also become increasingly difficult. Insurance companies at one time used relatively straightforward criteria for approval when augmentation therapy was prescribed. These included genetic evidence of deficiency and the diagnosis of significant obstructive pulmonary disease made by a respiratory specialist using measurements of lung function analogous to those used in clinical trial settings. However, since the introduction of legislation in Canada designed to prevent genetic discrimination, Canadian insurance companies have refused to accept genetic information. Patients with rare deficiency alleles may be disadvantaged if they have a dysfunctional alpha-1 antitrypsin variant which is quantified by serum level testing but serves no useful anti-protease function. Moreover, insurance companies have adopted the practice of introducing new criteria that are unrelated to product monograph instructions or guideline recommendations. The reality of these many and significant barriers is that patients who could benefit from receiving augmentation therapy are not able to realize this benefit.
A cost benefit analysis of augmentation therapy is beyond the scope of this submission and requires economic and clinical data not currently available. Nonetheless, the CTS understands that medical interventions may be costly and that is particularly true of “orphan” diseases requiring scarce therapies. It may be helpful to consider the likely economic impact of more widespread availability of augmentation therapy in Canada, a possible consequence of having a second entry augmentation product available. Although access to augmentation therapy in Canada is limited, best estimates suggest that approximately 250 Canadians are receiving augmentation therapy now. (The actual figures are unavailable for health privacy and commercial reasons). This leads to an estimate of 0.66 treated patients per 100,000 of the Canadian population. For the purposes of comparison, we can examine the prevalence of use in various European countries that offer unrestricted access to augmentation therapy through their public healthcare systems.
Table 6: Prevalence of Use in Various European Countries That Offer Unrestricted Access to Augmentation Therapy Through Their Public Healthcare Systems
Country | Patients Receiving Augmentation | Population | Augmentation Treatment/100,000 |
|---|---|---|---|
Canada | 250 | 38,000,000 | 0.66 |
Spain | 170 | 46,050,000 | 0.36 |
Italy | 115 | 59,800,000 | 0.19 |
Germany | 1,000 | 80,680,000 | 1.2 |
France | 300 | 64,730,000 | 0.46 |
The use of augmentation therapy may vary by population ethnic background (with severe deficiency being more common in those of northern European background), date of approval and other factors. Nonetheless, the figures show that treatment figures for deficient individuals are well below the gene prevalence for the disorder; not all patients with severe deficiency develop severe emphysema requiring augmentation therapy. If increased access to augmentation therapy allowed more frequent prescription and Canadian healthcare providers matched their European colleagues, the number of Canadians receiving therapy would likely remain below 500.
We believe that the introduction of Zemaira® to the Canadian pharmacopeia will be valuable to our patients in several ways. First, augmentation therapy is unavoidably expensive given that it requires painstaking procedures to safely harvest blood product from pooled blood donations. Nonetheless, we believe that as in most areas, the availability of augmentation material from more than one supplier is likely to result in competition and price reduction. This should increase the availability of augmentation therapy to patients who will benefit from delayed loss of lung function, reduced disability and longer survival. Second, single supplier availability of any therapy is associated with the risk of future shortfalls in availability. This is not merely a theoretical risk. In the 1990s, one large supplier suffered a shortfall in production leading to worldwide shortages of augmentation therapy. Patients in Canada and elsewhere either failed to receive augmentation therapy or received it only at reduced dosages. The realities of the current COVID pandemic further emphasize this genuine limitation. Third, we understand that the availability of a new augmentation therapy product will allow the healthcare system to re-examine the proposition that this blood product be made available through Canadian Blood Services similar to other blood products. We believe that this national availability will be in the best interests of patients and patient well-being. Not only would interprovincial discrepancies in availability be resolved, but such availability could ensure that augmentation therapy is prescribed only in accordance with best medical practice much as is now seen with other valuable blood products such as intravenous immunoglobulin.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside help was sought or received.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside analysis was sought or received.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Kenneth R. Chapman
Position: Professor of Medicine, University of Toronto
Date: 20-October-2021
Table 7: Declaration for Canadian Thoracic Society Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Grifols (clinical trial payments) | — | — | — | X |
CSL Behring (consulting) | — | X | — | — |
Takeda | X | — | — | — |
Vertex (clinical trial payments) | — | — | X | — |
Mereo Biopharma (clinical trial payments) | — | — | X | — |
Declaration for Clinician 2
Name: Raymond Aceron
Position: Nurse Practitioner, Co-Chair CTS COPD Clinical Assembly
Date: Oct 20, 2021
Table 8: Declaration for Canadian Thoracic Society Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
Declaration for Clinician 3
Name Joshua Wald
Position Assistant Professor McMaster University
Date 21-10-2021
Table 9: Declaration for Canadian Thoracic Society Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | — | X | — | — |
Declaration for Clinician 4
Name: Gail Dechman
Position: Assistant Professor, School of Physiotherapy, Dalhousie University
Date: 20-10-2021
Table 10: Declaration for Canadian Thoracic Society Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
Declaration for Clinician 5
Name: Mohit Bhutani
Position: Professor of Medicine, University of Alberta, Past Chair of the CTS COPD Clinical Assembly
Date: 20-October-2021
Table 11: Declaration for Canadian Thoracic Society Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Astra Zeneca | — | X | — | — |
GSK | — | — | X | — |
Grifols | X | — | — | — |
Sanofi | — | — | X | — |
Declaration for Clinician 6
Name: Marla Beauchamp
Position: Assistant Professor, Canada Research Chair (tier 2) Mobility, Aging & Chronic Disease
Date: 20-10-2021
Table 12: Declaration for Canadian Thoracic Society Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
Declaration for Clinician 7
Name: Darcy Marciniuk
Position: Associate Vice-President Research, Professor of Medicine, University of Saskatchewan
Date: October 21, 2021
Table 13: Declaration for Canadian Thoracic Society Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Mereo BioPharma/Syneos Health LLC (research funding held and managed by the University of Saskatchewan) | — | — | — | X |
Declaration for Clinician 8
Name: Dr. Paul Hernandez
Position: President, Canadian Thoracic Society – Professor of Medicine, Dalhousie University
Date: 21-10-2021
Table 14: Declaration for Canadian Thoracic Society Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Grifols (fees paid to Dr. Hernandez’s institution – NS Health - for expenses related to conduct of a clinical trial) | — | — | — | X |
Declaration for Clinician 9
Name: Michael Stickland
Position: Professor, Department of Medicine, University of Alberta
Date: 22-10-2021
Table 15: Declaration for Canadian Thoracic Society Clinician 9
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.