CADTH Reimbursement Review
Prasterone (Intrarosa)
Sponsor: Lupin Pharma Canada Ltd.
Therapeutic area: Post-menopausal vulvovaginal atrophy
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
CI
confidence interval
CrI
credible interval
CTCAE
Common Terminology Criteria for Adverse Events
DB
double blind
DHEA
dehydroepiandrosterone
FSFI
Female Sexual Function Index
GSM
genitourinary syndrome of menopause
ITC
indirect treatment comparison
ITT
intention to treat
LOCF
last observation carried forward
MD
mean difference
MENQOL
Menopause-Specific Quality of Life questionnaire
MID
minimal important difference
NMA
network meta-analysis
Pap
Papanicolaou
PP
per protocol
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SOGC
Society of Obstetricians and Gynaecologists of Canada
VAS
visual analogue scale
VASQ
Vaginal Atrophy Symptom Questionnaire
VHI
Vaginal Health Index
VVA
vulvovaginal atrophy
WHC
Women’s Health Coalition of Alberta
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Prasterone (Intrarosa) 6.5 mg ovule, administered intravaginally |
Indication | Prasterone is indicated for the treatment of post-menopausal VVA |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 1, 2019 |
Sponsor | Lupin Pharma Canada Ltd. |
NOC = Notice of Compliance; VVA = vulvovaginal atrophy.
Introduction
The vagina, vulva, lower urinary tract, and pelvic floor of patients contain hormone receptors to estrogen, androgen, or both. Genitourinary syndrome of menopause (GSM) describes the consequences of hormone deficiency which affect urogenital tissues and result in genitourinary symptoms that patients with menopause experience.1,2 GSM is a broader term for vulvovaginal atrophy (VVA),the latter of which focuses on changes in the genital tissues and related symptoms.3 Symptoms of GSM can be grouped as genital, including dryness, burning, and irritation; sexual, including lack of lubrication, discomfort or pain, and impaired function; and urinary, including urgency, dysuria, and recurrent urinary tract infections. Signs of GSM can be observed through physical examination, as there may be changes in the colour, size, and integrity of the anatomy of the vagina. There may also be signs of decreased lubrication and an increase in vaginal pH; typically a pH of greater than 5.0 would be considered abnormal.1
The prevalence of patients who experience VVA or GSM is uncertain, as estimates are largely dependent on reporting of symptoms from patients. A recent study of 4,246 women from Sweden, Finland, the US, the UK, and Canada reported varying prevalence of VVA; in Canada, 34% of post-menopausal women reported having VVA.4 It is suggested that many aging patients will experience GSM, with vaginal dryness being the most commonly reported symptom. Estimates of prevalence are likely underestimates, as menopausal and post-menopausal patients attribute their symptoms to changes associated with normal aging; these patients may be hesitant to report symptoms to their treating health care providers. Previous literature estimates that 60% to 90% of post-menopausal patients may suffer from VVA, and experience important deficits in their quality of life because of it.5 Due to the potential for underreporting, and consequently undertreatment, it may be important for health care providers to take the initiative and ask post-menopausal patients about symptoms related to GSM to identify the condition as early as possible and provide optimal care.
Prasterone is a synthetic form of dehydroepiandrosterone (DHEA), which is a natural steroid compound with no estrogenic, androgenic, or other hormonal activity. When prasterone is administered intravaginally, the cells in the vagina convert it into estrogen and androgens.6 The recommended dosage is 1 ovule, containing 6.5 mg prasterone, administered intravaginally once a day.6 Prasterone is indicated for the treatment of post-menopausal VVA. The sponsor has requested the reimbursement of prasterone as per the Health Canada indication. Prasterone has not been previously reviewed by CADTH.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Input was received from 1 patient group, the Women’s Health Coalition of Alberta (WHC). The WHC advocates, raises awareness, and educates about uro-gynecological and reproductive health of patients of all ages. The WHC noted the overall lack of awareness and understanding of uro-gynecological health, the limited therapeutic options for peri- and post-menopausal conditions (e.g., post-menopausal VVA), and potential inequity in accessing preferred treatments when they are not reimbursed by public drug plans. The WHC emphasized that clinical and psychological impacts caused by untreated menopausal conditions are often overlooked and dismissed and expressed the expectation that a positive reimbursement recommendation for prasterone would improve treatment options for patients and potentially raise clinician awareness of the importance of treating menopausal conditions.
To provide additional background on lived experience, values, and preferences of patients with VVA, patient group websites were sought for original experiences of patients with VVA. Healthtalk.org is a non-profit organization that has collected hundreds of stories from patients with any health condition.7 Information from Healthtalk.org pertaining to VVA was obtained, assessed, and synthesized by the CADTH review team. This included video interviews with 13 British patients. Among the interviewed patients, vaginal dryness, decline in libido, and urinary problems were reported as some of the complications experienced after entering menopause. Most patients reported a decline in sexual activity due to loss of libido. Vaginal dryness was another issue patients reported encountering during menopause. Comments also acknowledged the importance of sex in a marriage and the important complications that can happen within a relationship over time due to decreased sexual activity and symptoms of VVA. During the interview, 1 woman indicated that she was not aware of the effects of hormone replacement therapies, and that treatment with hormone replacement therapies may not prevent the “thinning of the vaginal wall.” The thinning of vaginal tissue was stated to cause severe discomfort for many of these patients resulting in tears and bleeding. Patients also commented on how the decline in their estrogen levels affected the pelvic floor, bladder, uterus, vagina, and bowel leading to urinary and bowel problems. Comments on difficulty with incontinence, and the impacts on quality of life were echoed many other patients.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
One clinical specialist with expertise in the diagnosis and management of VVA provided input for this review.
The clinical expert consulting with CADTH for this review indicated that as many as 70% of women are expected to have GSM by the age of 70. Over-the-counter moisturizers and lubricants may help provide patients with some symptomatic relief, but these treatments do not affect the underlying disease mechanism and can also be expensive for patients. Vaginal estrogen treatment was identified as being the most effective treatment option for patients. However, all estrogen-based products (despite being systemic or local) have a black box warning issued from Health Canada for several disease risks, limiting its use for some patients. The clinical expert consulting with CADTH for this review stated that prasterone would provide patients with another treatment option as it can help to improve their physiology and sexual function. In addition, prasterone could be an option for patients with contraindications to estrogen therapies, including patients with breast cancer or other estrogen-based cancers, and patients with cardiovascular disease risk.
The dosing schedule of prasterone was acknowledged to be different compared to other estrogen-based therapies; other therapies are prescribed to patients at an interval of twice weekly which some patients may easily forget, compared to prasterone which is administered daily. Patients who would benefit from treatment with prasterone would be identified by an experienced clinician both by a physical exam and by asking patients about symptoms of GSM and sexual function. According to the clinical expert, a patient’s response to treatment can be assessed through self-reported symptoms and a clinical examination of vaginal colour, lubrication, sensation, and pain. Any reduction on GSM symptoms (for example, dyspareunia, dryness, pain, discomfort, burning, itch, dysuria) was stated to be considered a clinically meaningful response to treatment. Response to treatment was stated by the expert to be assessed 3 to 4 months following treatment initiation, although some studies suggest that patients may improve dramatically within the first month of treatment. After an initial assessment of treatment, it may not be necessary to continue assessing patient’s response to treatment unless a new symptom occurs, or symptoms worsen again. Adverse events (AEs) were stated to be of little worry as the clinical expert believed that prasterone is a very well-tolerated treatment. The clinical expert confirmed that prasterone may be prescribed by family physicians or at specialty clinics including gynecology, urology, or urogynecology clinics. Diagnosis of post-menopausal VVA can be made by a family physician, nurse practitioner, or a specialist if the patient is referred to 1 (i.e., gynecologist or urologist).
Clinician Group Input
Input was received from 2 clinician groups: Cleopatra (prepared by 2 registered nurses) and the Society of Obstetricians and Gynaecologists of Canada (SOGC; prepared by 1 physician). No significant contrary views were presented. Both clinician groups highlighted that many patients may suffer from VVA and that prasterone may provide a useful treatment option for patients to treat symptoms and the underlying condition.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for prasterone:
Considerations for initiation of therapy
Considerations for prescribing of therapy
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
A total of 3 studies were summarized and critically appraised in this CADTH report: the ERC-238, ERC-231, and ERC-230 trials. The ERC-238 trial was a phase III, double-blind (DB), placebo-controlled, multi-centre trial that aimed to confirm the efficacy of 12 weeks of treatment with once-daily administration of an intravaginal 0.5% prasterone ovule (N = 374) compared to once-daily administration of an intravaginal placebo ovule (N = 180) on pain with sexual activity (dyspareunia) among post-menopausal patients aged 40 to 80 years who had dyspareunia as their most bothersome symptom of VVA. The ERC-231 trial was a phase III, DB, placebo-controlled, multi-centre trial assessing the efficacy of intravaginal prasterone 6.5 mg (N = 87), or 3.25 mg compared to placebo (N = 80) among post-menopausal patients having moderate to severe dyspareunia as their most bothersome symptom of VVA at baseline. Only the prasterone (6.5 mg) group was considered relevant for this CADTH review as this is the dose approved by Health Canada. The duration of the trial was 12 weeks. The ERC-230 trial was a phase III, open-label, single-group study (N = 521) which examined the long-term safety of daily treatment with intravaginal prasterone (6.5 mg). The trial duration was 52 weeks.
The 4 coprimary end points of the ERC-238 and ERC-231 trials included percentage of parabasal cells, percentage of superficial cells, vaginal pH, and severity score of dyspareunia. Secondary end points included sexual function (measured using the Female Sexual Function Index [FSFI]), vaginal dryness, vaginal irritation or itching, and safety. The primary objective of the ERC-230 trial was to evaluate the long-term safety of prasterone among post-menopausal patients with VVA; safety was assessed through AEs, mammography, Papanicolaou (Pap) smear, endometrial biopsy, and other outcomes. Secondary end points of the ERC-230 trial included percentage of parabasal cells, percentage of superficial cells, vaginal pH, severity score of dyspareunia, sexual function (measured using the FSFI), vaginal dryness, and vaginal irritation or itching.
Baseline characteristics were similar between the prasterone and placebo groups in the ERC-238 and ERC-231 trials as well as across the 2 trials. In all 3 trials, the median age of patients was between 57 years and 59 years. Overall, the majority of patients were White (> 85%) and non-Hispanic or Latino (≥ 88%). Patients reported both natural and surgical causes of their last menstruation, which occurred at a mean age of between 44 and 50 years. Previous hormone therapy was reported by approximately half of patients across all trials. Key differences across trials included mean years since last menstruation (between 13 and 14 years for patients in the ERC-238 and ERC-231 trials, versus approximately 8 years in the ERC-230 trial), proportion of patients reporting a hysterectomy (38% in the ERC-238 trial versus 61% in the ERC-231 trial; patients in ERC-230 were non-hysterectomized), and oophorectomy (26% to 33% in the ERC-238 and ERC-231 trials, respectively, compared to 5% in the ERC-230 trial).
Efficacy Results
Key efficacy results are summarized in Table 2. A summary of the 4 coprimary end points of the ERC-238 and ERC-231 trials are summarized here, along with the corresponding results for the ERC-230 trial.
Dyspareunia
In the ERC-238 trial, the mean change from baseline in severity score of dyspareunia was greater for the prasterone group (–1.42; standard deviation [SD] = 1.00) compared to the placebo group (–1.06; SD = 1.02) at 12 weeks; the mean difference [MD] for the prasterone group versus the placebo group was –0.35 (SD for MD not reported; P = 0.0002) in favour of prasterone. In the ERC-231 trial, the mean change from baseline in severity score of dyspareunia was greater for the prasterone group (–1.27; SD = 0.99) compared to the placebo group (–0.87; SD = 0.95) at 12 weeks; the MD for the prasterone group versus the placebo group was –0.40 (SD for MD not reported; P < 0.0001) in favour of prasterone. In the ERC-230 trial, the mean severity score of dyspareunia was reported for patients who had moderate to severe dyspareunia as their most bothersome symptom at baseline while also meeting VVA criteria for superficial cells (≤ 5%) and vaginal pH (> 5.0) (n = 183). The severity score of dyspareunia was 2.57 (SD = 0.50) at baseline and 0.87 (SD = 0.96) at week 52; the mean change from baseline was –1.69 (SD = 0.97). The mean severity score of dyspareunia was also reported for patients who had moderate to severe dyspareunia at baseline while also meeting VVA criteria for superficial cells (≤ 5%) and vaginal pH (> 5.0) (n = 240). The severity score of dyspareunia was 2.53 (SD = 0.50) at baseline and 0.85 (SD = 0.95) at week 52; the mean change from baseline was –1.68 (SD = 0.95).
Vaginal Cell Maturation
Vaginal cell maturation was assessed using the change from baseline in the percentages of parabasal and superficial cells. In the ERC-238 trial, the mean change from baseline in the percentage of parabasal cells was greater for the prasterone group (–41.51%; SD = 36.26%) compared to the placebo group (–11.98%; SD = 29.58) at 12 weeks; the MD for the prasterone group versus the placebo group was –29.53 (SD for MD not reported; P < 0.001) in favour of prasterone. In the ERC-231 trial, the mean change from baseline in the percentage of parabasal cells was greater for the prasterone group (–47.40%; SD = 42.50) compared to the placebo group (–1.62%; SD = 28.22) at 12 weeks; the MD for the prasterone group versus the placebo group was –45.77% (SD for MD not reported; P < 0.0001) in favour of prasterone. In the ERC-230 trial, the mean change from baseline to week 52 in percentage of parabasal cells among all patients who were treated with prasterone was –42.67 (SD = 39.23). The mean change in percentage of parabasal cells was also analyzed in a group of 292 patients who had dyspareunia, vaginal dryness, or vaginal irritation or itching as their most bothersome symptom. The mean change from baseline to week 52 in percentage of parabasal cells among all patients treated with prasterone was –49.14 (SD = 37.91).
The mean change from baseline in the percentage of superficial cells was greater for the prasterone group (10.20%; SD = 10.35) compared to the placebo group (1.75%; SD = 3.33) at 12 weeks in the ERC-238 trial; the MD for the prasterone group versus the placebo group was 8.46% (SD for MD not reported; P < 0.001) in favour of prasterone. In the ERC-231 trial, the mean change from baseline in the percentage of superficial cells was greater for the prasterone group (5.62%; SD = 5.49) compared to the placebo group (0.91%; SD = 2.69) at 12 weeks; the MD for the prasterone group versus the placebo group was 4.71% (SD for MD not reported; P < 0.0001) in favour of prasterone. In the ERC-230 trial, the mean change from baseline to week 52 in percentage of superficial cells among all patients who were treated with prasterone was 7.41% (SD = 8.06). The percent change in superficial cells waw also analyzed in a group of 292 patients who had dyspareunia, vaginal dryness, or irritation/itching as their most bothersome symptom. The mean change from baseline of superficial cells among all patients treated with prasterone was 7.85% (SD = 7.15).
Vaginal pH
In the ERC-238 trial, the mean change from baseline in vaginal pH was greater for the prasterone group (–0.94; SD = 0.94) compared to the placebo group (–0.27; SD = 0.74) at 12 weeks; the MD for prasterone versus placebo was –0.67 (SD for MD not reported; P < 0.0001) in favour of prasterone. In the ERC-231 trial, the mean change from baseline in vaginal pH was greater for the prasterone group (–1.04; SD = 1.00) compared to the placebo group –0.21; SD = 0.69) at 12 weeks; the MD for prasterone versus placebo was –0.83 (SD for MD not reported; P < 0.0001) in favour of prasterone. In the ERC-230 trial, the mean chance from baseline to week 52 in vaginal pH among all patients who were treated with prasterone was –1.14 (SD = 0.96). The percent change in parabasal cells were also analyzed in a group of 293 patients who had dyspareunia, vaginal dryness, or irritation/itching as their most bothersome symptom. The mean change from baseline to week 52 of parabasal cells among all patients treated with prasterone for this subgroup was –1.27 (SD = 0.90).
Harms Results
Adverse Events
The proportions of patients reporting at least 1 AE in the ERC-238 trial were similar between treatment groups, with 179 patients (47.9%) in the prasterone group and 77 patients (42.8%) in the placebo group reporting at least 1 AE. There was a higher proportion of patients in the prasterone group with at least 1 AE than the placebo group in the ERC-231 trial; 46 patients (52.9%) in the prasterone group and 35 patients (43.8%) in the placebo group reported at least 1 AE. A greater proportion of AEs were reported in the ERC-230 trial with 418 patients (80.2%) experiencing AEs. In general, application site discharge (ERC-238: 6.1% in the prasterone group versus 5.6% in the placebo group; ERC-231: 5.7% versus 6.3%, respectively; ERC-230: 14.0% in the prasterone group) and urinary tract infections (4.5% in the prasterone group versus 2.8% in the placebo group; ERC-231: 5.7% versus 5.0%, respectively; ERC-230: 10.2% in the prasterone group) were the most commonly reported AEs across all trials.
Serious AEs
Serious AEs (SAEs) were infrequently reported across trials. In the ERC-238 trial, 1.6% of patients in the prasterone group experienced an SAE compared to 0 patients in the placebo group. In the ERC-231 trial, 1.1% of patients in the prasterone group experienced an SAE compared to 0 patients in the placebo group. In the ERC-230 trial, SAEs occurred in 3.5% of patients.
Discontinuations Due to AEs
Few patients discontinued treatment due to an AE across all trials and reporting of these AEs was generally consistent across treatment groups. In the ERC-238 trial, 1.3% of patients in the prasterone group versus 2.8% of patients in the placebo group discontinued treatment due to an AE. In the ERC-231 trial, 1.1% of patients in the prasterone group and 1.3% of patients in the placebo group discontinued treatment due to an AE. In the ERC-230 trial, 6.0% of patients discontinued treatment due to an AE.
Mortality
There were no deaths in any of the trials.
Notable Harms
Notable harms identified in the CADTH systematic review protocol included vaginal hemorrhage, endometrial dysplasia, cervical dysplasia, and breast mass. In general, few patients experienced notable harms reported as AEs across the ERC-238, ERC-231, and ERC-230 trials, and there was little-to-no difference in reporting of notable harms across treatment groups. Vaginal hemorrhage was reported among 1.1% of patients in both the prasterone and placebo groups in the ERC-238 trial, 0 patients and 2.5% of patients in the prasterone and placebo groups, respectively, in the ERC-231 trial, and 2.5% of patients in the ERC-230 trial. Cervical dysplasia was reported among 1.9% of patients in the prasterone group versus 0 patients in the placebo group in the ERC-238 trial, 3.4% of patients in the prasterone group versus 2.5% of patients in the placebo group in the ERC-231 trial, and 3.8% of patients in the ERC-230 trial. Breast mass was reported in 0.3% of patients in the prasterone group versus 0 patients in the placebo group of the ERC-238 trial, 0.4% of patients in the ERC-230 trial, and 0 patients in the ERC-231 trial.
The ERC-230 trial also reported on breast, endometrial, and cervical safety. Endometrial safety was also reported in the ERC-231 trial. Breast examinations were conducted using mammograms at screening and at week 52. A total of 451 patients (98%) had a mammogram; of these patients 455 patients (99%) showed normal or no significant findings. Significant breast pathology was observed among 2 patients which included 1 case each of atypical ductal hyperplasia and infiltrating carcinoma. Undetermined status was reported among 2 patients; 1 patient refused follow-up and findings from the other patient were reported as being probably benign. The results of the remaining 15 patients were reported to be benign. In general, normal breast findings were observed for patients who received long-term treatment with prasterone. In general, long-term administration of prasterone in the ERC-230 trial was not associated with cervical dysplasia. Papanicolaou (Pap) smears were conducted for patients who received prasterone for at least 26 weeks. A Pap smear was conducted for 430 of 432 patients who received prasterone for 52 weeks (90%). A total of 13 patients yielded results of atypical squamous cells of uncertain significance (ASCUS), low grade squamous intraepithelial lesion, or high grade squamous intraepithelial lesion. Of these 13 patients, 7 had a negative HPV test or colposcopy.8 In the ERC-231 trial, approximately 40% of patients were non-hysterectomized and underwent an endometrial biopsy at screening (31 to 25 patients per treatment group). Almost all non-hysterectomized patients (99%), including 28 patients in the prasterone group and 27 patients in the placebo group, underwent an endometrial biopsy at week 12; 5 patients in the prasterone group and 2 patients in the placebo group did not have sufficient tissue for biopsy at this time. At week 12, the endometrium of all evaluable patients was atrophic, and the sponsor reported no clinically significant results.9 In the ERC-230 trial, endometrial biopsies were performed for patients who received prasterone for at least 3 months. For patients with unevaluable endometrial biopsies or who refused endometrial biopsies at the end of treatment, transvaginal ultrasounds were performed; these were performed for 43 patients. In total, 457 patients (94%) had a biopsy at the end of the 52-week study period. Among patients who underwent a biopsy, the endometrium of most patients (91%) was atrophic. Among the 43 patients who underwent a transvaginal ultrasound, the average endometrial thickness was 2.2 mm (SD = 1.4). There were no clinically significant histological findings in the ERC-230 trial with long-term use of prasterone.8
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Study detail | ERC-238 | ERC-231 | ERC-230a | ||
|---|---|---|---|---|---|
Prasterone N = 374 | Placebo N = 180 | Prasterone N = 87 | Placebo N = 80 | All patients N = 521 | |
Dyspareunia symptom scoreb | 325 | 157 | 81 | 77 | 183 |
Baseline, mean (SD) | 2.54 (0.50) | 2.56 (0.50) | 2.63 (0.49) | 2.58 (0.50) | 2.57 (0.50) |
Week 12, mean (SD) | 1.13 (0.98) | 1.50 (1.05) | 1.36 (1.10) | 1.71 (1.00) | NA |
Mean change (SD) | –1.42 (1.00) | –1.06 (1.02) | –1.27 (0.99) | –0.87 (0.95) | NA |
Mean difference from placebo (SD) | –0.35 (NR) | — | –0.40 (NR) | — | NA |
P valuec | 0.0002 | — | 0.0132 | — | NA |
Week 52, mean (SD) | — | — | — | — | 0.87 (0.96) |
Change from baseline, mean (SD) | — | — | — | — | –1.69 (0.97) |
P valued | — | — | — | — | < 0.0001 |
Percentage of parabasal cells | 325 | 157 | 81 | 77 | 454 |
Baseline, mean (SD) | 54.25 (38.64) | 51.66 (37.60) | 65.05 (41.69) | 68.48 (38.66) | 55.49 (43.30) |
Week 12, mean (SD) | 12.74 (18.44) | 39.68 (33.57) | 17.65 (25.87) | 66.86 (38.32) | — |
Mean change (SD) | –41.51 (36.26) | –11.98 (29.58) | –47.40 (42.50) | –1.62 (28.22) | — |
Mean difference from placebo (SD) | –29.53 (NR) | — | –45.77 (NR) | — | — |
P valuec | < 0.0001 | — | < 0.0001 | — | — |
Week 52, mean (SD) | — | — | — | — | 12.81 (20.57) |
Mean change from baseline to week 52 (SD) | — | — | — | — | –42.67 (39.23) |
P valued | — | — | — | — | < 0.0001 |
Percentage of superficial cells | 325 | 157 | 81 | 77 | 454 |
Baseline, mean (SD) | 1.02 (1.44) | 1.04 (1.40) | 0.68 (1.10) | 0.73 (1.33) | 2.02 (3.96) |
Week 12, mean (SD) | 11.22 (10.18) | 2.78 (3.37) | 6.30 (5.33) | 1.64 (2.88) | NA |
Mean change (SD) | 10.20 (10.35) | 1.75 (3.33) | 5.62 (5.49) | 0.91 (2.69) | NA |
Mean difference from placebo (SD) | 8.46 (NR) | — | 4.71 (NR) | — | NA |
P valuec | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | NA |
Week 52, mean (SD) | — | — | — | — | 9.42 (7.60) |
Mean change from baseline to week 52 (SD) | — | — | — | — | 7.41 (8.06) |
P valued | — | — | — | — | < 0.0001 |
Vaginal pH, N | 325 | 157 | 81 | 77 | 457 |
Baseline, mean (SD) | 6.34 (0.65) | 6.32 (0.66) | 6.47 (0.64) | 6.51 (0.59) | 6.23 (0.79) |
Week 12, mean (SD) | 5.39 (0.94) | 6.05 (0.89) | 5.43 (0.94) | 6.31 (0.81) | — |
Mean change (SD) | –0.94 (0.94) | –0.27 (0.74) | –1.04 (1.00) | –0.21 (0.69) | — |
Mean difference from placebo (SD) | –0.67 (NR) | — | –0.83 (NR) | — | — |
P valuec | < 0.0001 | — | < 0.0001 | — | — |
Week 52, mean (SD) | — | — | NA | NA | 5.09 (0.82) |
Mean change from baseline (SD) | — | NA | NA | NA | –1.14 (0.96) |
P valued | — | NA | NA | NA | < 0.0001 |
Harms, n (%) (safety population) | |||||
AEs | 179 (47.9) | 77 (42.8) | 46 (52.9) | 35 (43.8) | 418 (80.2) |
SAEs | 6 (1.6) | 0 (0.0) | 1 (1.1) | 0 (0.0) | 18 (3.5) |
WDAE (from study treatment) | 5 (1.3) | 5 (2.8) | 1 (1.1) | 1 (1.3) | 31 (6.0) |
Deaths | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | |||||
Vaginal hemorrhage | 4 (1.1) | 2 (1.1) | 0 (0.0) | 2 (2.5) | 13 (2.5) |
Endometrial dysplasia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Cervical dysplasia | 7 (1.9) | 0 (0.0) | 3 (3.4) | 2 (2.5) | 20 (3.8) |
Breast mass | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.4) |
AE = adverse event; ANCOVA = analysis of covariance; NA = not applicable; SAE = serious adverse event; SD = standard deviation; WDAE = withdrawal due to adverse event.
aSeverity score of dyspareunia, percentage of parabasal and superficial cells, and vaginal pH were secondary end points in the ERC-230 trial and were not adjusted for multiplicity.
bDyspareunia was measured as part of the Vaginal Atrophy Symptom Questionnaire. The severity of each symptom assessed using the visual analogue scale was recorded as none, mild, moderate, or severe and analyzed using the scores of 0, 1, 2, or 3, respectively. Therefore, lower scores indicated improved symptom scores. Change from baseline in symptom score of dyspareunia was 1 of 4 coprimary end points in the ERC-238 and ERC-231 trials. Each coprimary end point was not adjusted for multiplicity as statistical significance of each coprimary end point was required for conclusion of superiority of prasterone over the placebo group. In the ERC-230 trial, change from baseline in symptom score of dyspareunia was a secondary end point and was not adjusted for multiplicity.
cANCOVA test with treatment group as the main factor and baseline value as the covariate (P value vs. placebo).
dP value from a paired t-test (P value vs. baseline).
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Critical Appraisal
The ERC-230 trial used an open-label single-group design to evaluate treatment with prasterone among post-menopausal patients with VVA. Since this study lacks a comparison (control) group and there is no control for potential confounding variables, causal relationships cannot be established. All efficacy end points in the ERC-230 trial were secondary and no adjustments were made for multiplicity. In addition, the placebo used in the trials may have had an effect on patients. Placebo ovules were administered in a capsule which may have had some moisturizing effects for patients. Therefore, the treatment effects of the prasterone ovule compared to a true placebo may be underestimated. In fact, results from efficacy analyses did reveal that patients in the placebo groups also experienced some benefit from the placebo as patients in the placebo group also reported improved symptoms, albeit not as great as patients in the prasterone group.
Both the ERC-238 and ERC-231 trials were placebo-controlled trials. Although it was stated by the sponsor that placebo was an appropriate comparator, the CADTH team noted that estrogen-based therapies would have been available during the inception of these trials and the clinical expert consulted by CADTH for this review agreed that a comparison with a vaginal estrogen therapy would have been of value. It is typical that trial eligibility criteria can be restrictive and, ultimately, not representative of all patients in clinical practice. That eligibility criteria were overly restrictive is likely evident from the large number of patients who were considered screen failures across the trials; the high rate of screen failures may partially be due to guidance from the FDA recommending that patients adhere to a number of clinical criteria. Patients with comorbidities were excluded from the ERC-238, ERC-231, and ERC-230 trials. In particular, patients with a history of cancer were excluded from the trials; this was considered a population of interest as post-menopausal women with history of cancer are still at risk for VVA and may benefit from non-hormonal therapies such as prasterone. The impact of treatment on patients with comorbidities is not clear. The duration of the ERC-238 and ERC-231 trials was only 12 weeks. As the trial durations were short, the long-term benefits and harms of prasterone on patients is uncertain, and patients who are prescribed prasterone in clinical practice are likely to take this treatment for longer than 12 weeks. The ERC-230 trial was conducted for 52 weeks; however, the study is lacking a control group which does now allow for definitive conclusions to be drawn about the effects of longer-term treatment.
Indirect Comparisons
Description of Studies
The CADTH literature search identified 1 network meta-analysis (NMA) publication by Li et al.11 Li et al. conducted several NMAs to indirectly compare treatment with prasterone to other treatments for VVA among people with menopause. A total of 29 trials which incorporated 8,311 patients were included in the indirect treatment comparison (ITC) by Li et al. evaluating the following treatments: laser therapy, vaginal estrogen, ospemifene, vaginal DHEA, and moisturization/lubrication.11
Characteristics of study design revealed inclusion of both open-label and blinded randomized controlled trials (RCTs). Trials were published between 1992 and 2020. All patients included in the trials had a mean age between 58 years and 60 years of age. All trials except 312-14 excluded patients with breast or gynecological cancers. Treatment duration was heterogeneous, with most trials assessing treatment for 12 weeks. Outcomes assessed included urinary and sexual outcomes (i.e., dryness, itching, dyspareunia, urinary tract infections), AEs, and health-related quality of life assessed through various tools. Different doses of treatments were also used in the 29 trials; specifically regarding DHEA, studies assessing doses of 0.5% (6.5 mg) and 0.25% (3.25 mg) were included.11 The authors did not report on the number of studies included for the NMAs conducted for each end point (vaginal dryness, vaginal burning and itching, dyspareunia, sexual function, vaginal pH, proportion of parabasal cells, and AEs) nor on their risk of bias. It is not clear how the nodes were created, though it appears that similar treatments were merged regardless of dose and duration. The tool used to measure the end points across the included trials was not specified. The network structure was not described. The authors indicated that the model converged “adequately” but relevant data were not provided to support this assertion.
Efficacy Results
Vaginal Dryness
No differences were observed between DHEA and vaginal estrogen therapy (MD = 0.32; 95% credible interval [CrI], −8.54 to 8.77). The I2 value for heterogeneity was 0%, but the pairwise frequentist analyses showed high heterogeneity. Subgroup analyses did not seem to explain the heterogeneity for the comparisons of interest (DHEA versus other treatments). There did not appear to be any sensitivity analyses performed for this comparison. Publication bias was not detected.
Dyspareunia
Little-to-no difference was observed between DHEA and vaginal estrogen therapy (−4; 95% CrI, −13.88 to 4.46). The I2 value for heterogeneity was 11%.
Sexual Function (FSFI)
No differences were observed between DHEA and vaginal estrogen therapy (MD = 1.04; 95% CrI, –1.99 to 3.93). The I2 value for heterogeneity was 0%.
Vaginal pH
The I2 value for heterogeneity was 4%. Vaginal estrogen therapy (MD = 0.4; 95% CrI, 0.11 to 0.69) was favoured over DHEA.
Proportion of Parabasal Cells
No differences were observed between DHEA and vaginal estrogen therapy (MD = 1.6; 95% CrI, −12.45 to 13.84). The I2 value for heterogeneity was 9%.
Harms Results
No difference was found between DHEA and vaginal estrogen therapy (odds ratio = 1.54; 95% CrI, 0.91 to 2.62). The I2 value for heterogeneity was less than 25% among treatments.
Critical Appraisal
Though several databases were searched for the systematic review, the authors did not search other sources (e.g., clinical trial registries) so it is possible that some relevant studies were missed. Methods of data extraction were not described, so error within the findings is possible. Studies were assessed for risk of bias, but it is not clear how this assessment was carried out, so it is difficult to assess the validity of these assessments. Differences in trial and baseline characteristics are likely to have impacted the indirect comparisons, although the exact effect of these difference is unclear. An assessment of similarity across trials in each NMA was not conducted; therefore, whether underlying assumptions of the NMAs (i.e., homogeneity and transitivity) have been met are uncertain. There was a lack of clear reporting regarding the construction of nodes in the NMAs. However, based on reported information it was assumed that treatment doses, durations, and outcomes measures for single treatments were combined into single nodes. The combination of different doses, durations, and outcomes measures for treatments is likely to have introduced bias, as the efficacy and safety of treatments which may not have been administered or measured in the same way is uncertain. CrIs were also wide, indicating the potential for substantial uncertainty between treatment comparisons, including the comparisons between DHEA and vaginal estrogen therapies. Subgroup and sensitivity analyses revealed sources of variation for each end point. It is probable that heterogeneity across trials affects the confidence of results of the NMA.
Other Relevant Evidence
The following studies were included as additional evidence: the ERC-210 trial,15 the Estip-Es study,16 and a study by Barton et al.12 The ERC-210 trial was a multi-centre, DB, randomized, placebo-controlled, phase III trial to determine the dose response of prasterone on symptoms and vaginal mucosa parameters in post-menopausal women with VVA. The Estip-Es study was an observational study conducted in Spain which evaluated the effectiveness and safety of prasterone in a real-world clinical setting.16 The study by Barton et al.12 examined the use prasterone for treatment of post-menopausal symptoms of VVA in patients with a history of breast or gynecological cancer.
Description of Studies
The ERC-210 trial, which started in June 2007 and was completed in October 2008, was a multi-centre (US and Canada), prospective, DB, randomized, parallel assignment, placebo-controlled, phase III trial to determine the dose response of prasterone on symptoms and vaginal mucosa parameters in post-menopausal women with VVA. The study informed the dose of prasterone to use for the subsequent phase III studies. Patients were randomized to receive prasterone at 3.25 mg (n = 53), 6.5 mg (n = 56), or 13.0 mg (n = 54), or placebo (n = 53). Only the 6.5 mg dose of prasterone was relevant to this review.
The Estip-Es study was a multi-centre, prospective, noncomparative, observational study with 184 adult post-menopausal patients who were routinely seen in medical centres throughout Spain for GSM. Patients had used vaginal moisturizers/lubricants and/or vaginal hormone therapy and switched to intravaginal prasterone without a washout period.
The study by Barton et al.12 was a multi-centre (US and Canada), 3-group, DB, parallel group RCT where 443 patients were randomized to receive either 3.25 mg (n = 147) or 6.5 mg of prasterone (n = 149) in a plain bioadhesive moisturizer, or a plain bioadhesive moisturizer alone (n = 147). Only the 6.5 mg dose of prasterone was relevant to this review.
Efficacy Results
ERC-210
The percentage of superficial cells were measured to be 0.62% (SD = 1.02) at baseline and 0.54% (SD = 0.95) at week 12 (P = 0.7460 versus baseline) for the placebo group. The percentage of superficial cells were measured to be 0.40% (SD = 0.62) at baseline and 5.20% (SD = 6.54) at week 12 for the prasterone group. The MD in change was 4.88% (P = 0.0111) of superficial cells in prasterone group compared to the placebo group at week 12.
The percentage of parabasal cells were measured to be 46.73% (SD = 44.05) at baseline and 47.81% (SD = 38.36) at week 12 (P = 0.7686 versus baseline) for the placebo group. The percentage of parabasal cells were measured to be 53.40% (SD = 41.01) at baseline and 11.00% (SD = 18.77) at week 12 for the prasterone group. The MD in change was 43.48% (P < 0.0001) of parabasal cells in the prasterone group compared to the placebo group at week 12.
In the placebo group, the mean vaginal pH was 6.49 (SD = 0.69) at baseline and 6.01 (SD = 1.12) at week 12 (P = 0.005 versus baseline). In the prasterone group, the mean vaginal pH was 6.64 (SD = 0.51) at baseline and 5.17 (SD = 0.91) at week 12. At week 12, there was a mean 0.99 greater change (P = 0.0001) in pH in the prasterone group compared to the placebo group.
The mean severity score of dyspareunia was 2.77 (SD = 0.43) at baseline and 2.35 (SD = 0.94) at week 12 (P = 0.0132 versus baseline) for the placebo group. The mean severity score of dyspareunia was 2.73 (SD = 0.45) at baseline and 1.10 (SD = 1.18) at week 12. There was a mean 1.21 greater change (P < 0.0001) in symptom score in prasterone group compared to the placebo group at week 12.
Estip-Es Study
In the overall study population, the total FSFI score increased from 15.7 (SD = 6.3) to 19.9 (SD = 5.38) with the mean change of 4.2 over 30 days. Increased scores from baseline to post-treatment with prasterone were observed in all the FSFI domains with variable magnitudes. A visual analogue scale (VAS) was administered to assess the self-reported impact on GSM across 19 items, encompassing symptoms including dryness, dyspareunia, bleeding, burning, itching, urinary problems and infections, and abdominal pain. There was a numerical decrease (improvement) in all symptoms assessed using the VAS except for vaginal discharge; however, it should be noted that application site discharge is an expected AE related to use of prasterone.
Barton et al.
The primary end point was self-rated severity of patients’ most bothersome symptom, either dryness or dyspareunia using an ordinal scale of none, mild, moderate, severe, or very severe. There was no difference (P = 0.08) between the 6.5 mg prasterone (mean = 1.8; 95% confidence interval [CI], –1.97 to –1.54) and plain moisturizer (mean = –1.5; 95% CI –1.74 to –1.27) groups in changes of either dryness or dyspareunia at week 12.
Harms Results
ERC-210
Of patients who received prasterone, 47 (84%) patients experienced at least 1 AE, compared to 35 (65%) patients in the placebo group. The most common AEs (≥ 5%) reported in the prasterone group were cough (11%), headache (9%), and vaginal discharge (9%). The percentage of patients who withdrew from treatment due to an AE was 4% for both the placebo and prasterone groups. For the prasterone group, 1 (2%) patient had cervical dysplasia, and none had vaginal discharge.
Estip-Es Study
In the overall population, 6.5% of patients reported AEs (e.g., blisters on the face, hair loss, constipation, leukorrhea, and dizziness) during follow-up at 30 ± 7 days. No further detail regarding these AEs was provided in the published paper.
Barton et al.
The most common clinician-graded AEs (reported in > 5% of any treatment group) included headache and breast pain, which were not different between treatment groups.
Critical Appraisal
ERC-210
The plan for the primary analysis was amended following feedback from the FDA to restrict to the subgroup of patients who identified dyspareunia as their most bothersome symptom at baseline. This revision was post hoc and in a subgroup of patients, thereby breaking randomization. The direction and extent of any selection bias related to imbalances in characteristics is unclear because updated baseline characteristics for the subgroup were not reported. However, the Bonferroni adjustment for the coprimary analyses was a conservative approach to help mitigate the potential bias introduced by the revised analysis. The differences between prasterone 0.5% and placebo groups were statistically significant following the Bonferroni adjustment. The sample sizes of patients randomized to the prasterone and placebo groups were 56 and 54, respectively. The amendment of the analysis to a subgroup of these patients means that the sample sizes were reduced to 30 patients and 26 patients, respectively, with no information regarding baseline characteristics of this subgroup population provided. Since moisturizer (placebo) may have some effect on vaginal parameters and symptoms, the treatment effect of the prasterone ovule may have been smaller versus the placebo ovule than it would have been versus a true placebo. The relatively short follow-up and small number of patients in the ERC-210 study are inadequate to confirm the long-term benefits of prasterone beyond 12 weeks and assess rare, long-term harms.
Estip-Es Study
The Estip-Es study was an observational study with the objective of evaluating the efficacy, safety, and tolerability of prasterone for the treatment of post-menopausal women with GSM in clinical practice. As there was no comparator group, the efficacy of prasterone relative to other therapies was not clear based on data from this study. In addition, the nature of this study design may introduce bias due to the inability to control for confounding patient characteristics. The lack of blinding to treatment allocation and the subjective nature of all of the outcomes could have contributed to patients reporting greater improvements with a switch to prasterone than they would have in a DB RCT. Patients enrolled in the Estip-Es study were not subject to a washout period; therefore, it is possible that residual effects from previous treatments may have carried over and affected patient outcomes while receiving treatment with prasterone. The study used “a validated short version with 7 items” for FSFI; however, no references were provided related to the validity and reliability of the short form. Due to the lack of detailed information on patient’s baseline characteristics, it is difficult to ascertain to what extent the enrolled population reflects the Canadian population who are eligible for treatment with prasterone. The small sample size further limits generalizability of this study to the Canadian population. The Estip-Es study enrolled women from medical centres throughout Spain for GSM; therefore, these women were seeking medical intervention for symptoms related to VVA; due to this, there is a possibility for selection bias, as patients who were dissatisfied with their previous treatments were likely to have been enrolled in the Estip-Es trial and may view treatment with prasterone more positively. Follow-up visits for patients were conducted approximately 1 month after recruitment into the Estip-Es study. This short-term follow-up may not be an optimal time frame to capture benefits and harms related to treatment with prasterone.
Barton et al.
The study was conducted for a period of 12 weeks. The 12-week duration may not be ideal for capturing the efficacy and safety and treatment with prasterone among post-menopausal women with history of breast and gynecological cancers. Treatment with prasterone may occur for longer periods of time, and longer-term data would be necessary for understanding the long-term impact of treatment in this patient population with a history of hormone-dependent cancers. For the primary outcome, approximately 20% and 25% patients discontinued before completion of the study in the plain moisturizer and 6.5 mg prasterone groups, respectively. Primary analysis was based on a completed analysis set (“primary end point” data) and was not done in an intention-to-treat (ITT) method. Therefore, the high rate of study discontinuations (missing data of ≥ 20% in each group) introduces uncertainty in the results and it is unclear how the last value carried forward missing data imputation method may have biased the results. Also, it is unclear if the last value carried forward missing data imputation method was used for all the other analyses besides primary outcomes (i.e., FSFI and quality of life). After all the losses to follow-up, the “primary end point data” set did not meet its intended sample size (i.e., 145 patients in each arm), so the study is at risk of being underpowered. This study specified that patients administer compounded intravaginal prasterone in a gel formulation using a syringe (without a needle) whereas the Health Canada product monograph specifies that prasterone be administered as an ovule and inserted using an applicator. Therefore, comparability across other studies which assess prasterone as an ovule versus the gel is limited.
Conclusions
Three trials, including 2 multi-centre, randomized, DB, placebo-controlled trials (ERC-238 and ERC-231), and 1 multi-centre, single-group, open-label study (ERC-230) provided evidence on the safety and efficacy of prasterone for post-menopausal patients with VVA. Compared to placebo, prasterone 6.5 mg showed greater improvements after 12 weeks of treatment in percentage of parabasal cells, percentage of superficial cells, vaginal pH, dyspareunia, and vaginal dryness that were clinically meaningful according to the clinical expert consulted by CADTH. The ERC-230 trial provided long-term data on the use of prasterone; however, the lack of a comparator precluded the ascertainment of causal relationships, and the study did not adjust for multiple comparisons. The findings over a treatment period of 52 weeks; however, seemed similar to the findings of the 2 shorter-term trials, suggesting that it is possible that the benefits would be sustained with continued treatment. While the results for sexual function measured using the FSFI in the ERC-238 trial also favoured the prasterone group compared to the placebo group, these results were unadjusted for multiplicity and should be considered exploratory. Safety data from the ERC-238 and ERC-231 trials showed similar proportions of patients with AEs between the prasterone and placebo groups; the ERC-230 trial reported higher proportions of patients of AEs compared to the ERC-238 and ERC-231 trials, which may be expected due to the longer exposure to prasterone. However, due to the lack of a control group, it is unclear whether AEs may be associated with prasterone itself. AEs identified in the ERC-230 trial were mostly similar to those identified in the ERC-238 and ERC-231 trials; all trials reported application site discharge and urinary tract infections as the most commonly reported AEs. Comparison of prasterone to other therapies was assessed through a published ITC. In general, the ITC did not provide evidence for a difference in efficacy between prasterone and vaginal estrogen therapies (grouped as a single comparator), though there was considerable uncertainty in the treatment effect estimates. Limitations related to reporting of the NMA and heterogeneity across the included studies that could not be resolved precluded drawing strong conclusions about the comparative effectiveness and safety of prasterone versus other treatments for VVA. Additional evidence was identified to inform on the safety and efficacy of prasterone: the ERC-210 trial, the Estip-Es study, and a study by Barton et al. While the results of the ERC-210 trial were supportive of those from the 3 pivotal trials mentioned above, limitations of the design and analyses of the Estip-Es and Barton et al. studies precluded drawing concrete conclusions regarding the efficacy or safety of prasterone versus vaginal estrogen therapies or moisturizers/lubricants. The study by Barton et al. enrolled patients with history of breast and gynecological cancers, but did not find a difference between DHEA 6.5 mg daily and placebo in most bothersome symptom (dyspareunia or vaginal dryness) and did not assess the notable harms identified for this review outside of AE reporting over the 12-week treatment period.
Introduction
Disease Background
Due to aging and changes during menopause, some patients may experience symptoms of VVA. Hormonal changes, particularly the decrease in estrogen, may result in symptoms such as vaginal dryness, irritation, dyspareunia, and recurrent urinary tract infections. The pelvic floor is particularly susceptible to changes related to menopause as there are estrogen receptors in the vulva, vaginal, bladder, urethra, and pelvic floor musculature.1 VVA may refer more specifically to symptoms of dyspareunia and may be limited to patients who are sexually active. GSM is a broader term which encompasses other genitourinary symptoms and may not be limited to patients are sexually active.1 Symptoms of GSM can be grouped as genital, including dryness, burning, and irritation; sexual, including lack of lubrication, discomfort or pain and impaired function; and urinary, including urgency, dysuria, and recurrent urinary tract infections. Signs of GSM can be observed through physical examination conducted by an experienced health care provider, as there may be changes in colour, size, and integrity of the anatomy of the vagina. There may also be signs of decreased lubrication and an increase in vaginal pH; typically a pH of greater than 5.0 would be considered abnormal.1
While no official estimates of VVA among Canadians are available, a study which included 1,016 Canadians reported a prevalence of VVA of 34%.4 However, estimates regarding the prevalence of patients who experience VVA or GSM may be underreported. Many patients will not report changes they experience as they will associate changes with normal aging. Previous literature suggests that 60% to 90% of post-menopausal patients may suffer from VVA, and experience significant deficits in their quality of life because of it.5 Due to underreporting, it may be important for health care providers to take the initiative and ask post-menopausal patients about symptoms related to GSM to identify the condition as early as possible and provide optimal care.
Standards of Therapy
The treatment goals for post-menopausal VVA are to improve patients’ symptoms and sexual function with minimal side effects and thereby improve patients’ quality of life. Current treatments for post-menopausal VVA currently include moisturizers, lubricants, and vaginal estrogen. According to the clinical expert consulted by CADTH for this review, moisturizers and lubricants may benefit patients in providing them with some relief from symptoms; however, moisturizers and lubricants do not affect the underlying disease mechanism and can also be costly for patients. Estrogen treatment favourably alters patients’ physiology to treat the underlying disease and targets disease symptoms. However, vaginal estrogen therapies also have a black box warning issued from Health Canada for several disease risks (myocardial infarction, stroke, invasive breast cancer, pulmonary emboli, and deep vein thrombosis for most products) based on evidence for oral estrogen plus progestin therapy and oral estrogen-alone therapy. They are also contraindicated in patients with known or suspected estrogen-dependent malignant neoplasia and patients with known, suspected, or past history of breast cancer, also based on evidence for systemic therapies. According to the clinical expert, it is possible for some patients with these contraindications to be treated with vaginal estrogen, though the product monograph warnings can lead to hesitancy among these patients to use vaginal estrogen. Estrogen hormonal therapies may be administered to patients vaginally as creams, tablets, capsules, or a ring, or as an oral therapy. Some patients may prefer products other than vaginal creams as they can be messy. Due to patient needs that are unmet by currently available treatment options, some patients face a great impact on their quality of life.
Drug
Prasterone is a synthetic form of DHEA, which is a natural steroid compound inactive by itself. DHEA has no estrogenic, androgenic, or other hormonal activity. When prasterone is administered intravaginally as an ovule, the cells in the vagina transform it into estrogen and androgens.6 The recommended dose of prasterone is 0.5%, or 6.5 mg, approved by Health Canada.6 Prasterone has not been previously reviewed by CADTH. The sponsor has requested the reimbursement of prasterone as per the Health Canada indication.
Table 3: Key Characteristics of Treatments for Vulvovaginal Atrophy
Study detail | Prasterone | Estradiol vaginal insert | 17 Beta-estradiol vaginal ring | Conjugated estrogen vaginal cream |
|---|---|---|---|---|
Mechanism of action | Prasterone is a compound which is inactive by itself having no estrogenic, androgenic, or hormonal activity. It is a natural steroid compound. Prasterone is transformed inside the vaginal cells into estrogens and androgens when administered intravaginally | Estrogen therapy for estrogen deficiency. | Estrogen therapy for estrogen deficiency. | Estrogen therapy for estrogen deficiency. |
Indicationa | For the treatment of post-menopausal VVA | For the treatment of the symptoms of vaginal atrophy due to estrogen deficiency | For post-menopausal urogenital complaints due to estrogen deficiency such as feeling of dryness in the vagina (atrophic vaginitis) with or without pruritus vulvae, dyspareunia, dysuria, and urinary urgency (atrophic mucosa in the urethra and trigonum) | For the treatment of atrophic vaginitis, dyspareunia, and kraurosis vulvae |
Route of administration | Intravaginal (suppository) | Vaginal tablet | Vaginal ring | Vaginal cream |
Recommended dose | 6.5 mg ovule once a day | Initial dose: start with 4 mcg dose, insert 1 tablet daily for 2 weeks Maintenance dose: insert 1 tablet twice weekly, every 3 to 4 days | The ring (2 mg) is to remain in place continuously for 3 months, after which it is to be removed and, if continuation of therapy is deemed appropriate, replaced by a new ring; the need to continue treatment should be assessed at 3- or 6-month intervals | The lowest dose that will control symptoms should be chosen Low dose: 0.5 g administered intravaginally or topically twice weekly (for example, Monday and Thursday) Maximum recommended dose: administered intravaginally or topically |
(continued) | in a cyclic regimen (daily for 21 days and then off for 7 days). Generally, patients should be started at 0.5 g daily; dosage adjustments (0.5 g to 2 g) may be made based on individual response | |||
Serious adverse effects or safety issues | Contraindications:
Estrogen is a metabolite of prasterone. Use of exogenous estrogen is contraindicated in patients with a known history of breast cancer. In addition, prasterone has not been studied in patients with a history of breast cancer | Serious warnings:
Contraindications:
| Serious warnings:
Contraindications:
| Serious warnings:
Contraindications:
|
(continued) |
• Lactation |
CEE = conjugated equine estrogens; CHD = coronary heart disease; MI = myocardial infarction; MPA = medroxyprogesterone acetate; VVA = vulvovaginal atrophy.
aHealth Canada–approved indication.
Source: Intrarosa product monograph,6 Vagifem product monograph,17 Estring product monograph,18 and Premarin product monograph.19
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups and on information from a patient group website that was sought for original experiences from patients with VVA. The original patient group submission can be found in the Stakeholder Input section at the end of this report.
Input was received from 1 patient group, the Women’s Health Coalition of Alberta (WHC). The WHC advocates, raises awareness, and educates about uro-gynecological and reproductive health of patients of all ages. WHC is committed to empowering people to speak openly about patients’ reproductive and sexual health, as well as encouraging people to address barriers, gaps, policies, and unconscious bias impacting this population. WHC is also committed to ensuring access to the right treatment at the right time to improve patients’ health outcomes.
The WHC noted the overall lack of awareness and understanding of uro-gynecological health, the limited therapeutic options for peri- and post-menopausal conditions (e.g., post-menopausal VVA), and potential inequity in accessing preferred treatments when they are not reimbursed by public drug plans. The WHC emphasized that clinical and psychological impacts caused by untreated menopausal conditions are often overlooked and dismissed and expressed the expectation that a positive reimbursement recommendation for prasterone would improve treatment options for patients and potentially raise clinician awareness of the importance of treating menopausal conditions.
To provide additional background on lived experience, values, and preferences of patients with VVA, patient group websites were sought for original experiences of patients with VVA. Healthtalk.org is a non-profit organization that has collected hundreds of stories from patients with any health condition.7 Information from Healthtalk.org pertaining to VVA were obtained, assessed, and synthesized by the CADTH review team. This included video interviews with 13 British patients.
Among the interviewed patients, vaginal dryness, decline in libido, and urinary problems were reported as some of the complications experienced after entering menopause. Patients reported mixed reactions regarding the decline in libido after menopause. While 1 patient reported that the changes in the libido did not impact her sexual activity, others reported a decline in sexual activity due to loss of libido. Vaginal dryness was another issue patients reported encountering during menopause. One patient reported negative impacts on her sexuality due to decreased vaginal lubrication. She also pointed out the importance of sex in a marriage and the important complications that can happen within a relationship over time due to decreased sexual activity and symptoms of VVA. During the interview, 1 woman indicated that she was not aware of the effects of hormone replacement therapies, and that treatment with hormone replacement therapies may not prevent the “thinning of the vaginal wall.” The patient had expected that use of hormone replacement therapy “would protect from vaginal problems.” Another woman commented on the discomfort experienced during a vaginal smear, and the need for use of an estrogen cream to “get a correct reading from the cervix.” The thinning of vaginal tissue was stated to caused severe discomfort for many of these patients. One patient mentioned about her vaginal tissue getting “very, very thin,” resulting in tears and bleeding. Another patient described the chain of effects created by the lack of estrogen in the whole body after menopause, affecting the collagen, the pelvic floor, bladder, uterus, vagina, and bowel leading to urinary and bowel problems. The patient stated, “So you can get fecal incontinence, which is mortifying, bladder irritability or incontinence, and also your womb can come down, or your vagina can come down and that’s called a prolapse.” Comments on difficulty with incontinence, and the impacts on quality of life were echoed many other patients; patients also reported that they were likely to attribute this problem to the onset of menopause. Patients also expressed the importance of the support they get from their spouses during the difficult times, which helps them tackle the everyday difficulties and brings them hope.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of post-menopausal VVA.
Unmet Needs
The clinical expert consulting with CADTH for this review indicated that recognition of symptoms and identifying VVA or GSM is paramount for patients as it is underreported among patients, but very common as 70% of patients are expected to have GSM by the age of 70 years. Over-the-counter moisturizers and lubricants may help provide patients with some symptomatic relief, but these treatments do not affect the underlying disease mechanism and can also be expensive for patients. Vaginal estrogen treatment was identified as being the most effective treatment option currently available for patients. However, all estrogen-based products (despite being systemic or local) have a black box warning issued from Health Canada for several disease risks, limiting its use for some patients. The clinical expert stated that estrogen treatment favourably alters patients’ physiology to treat the underlying disease and target disease symptoms; however, the clinical expert did not consider it to be effective for improving some aspects related patient’s sexual function.
The clinical expert stated that there were unmet needs for patients for whom estrogen therapy is contraindicated, or who do not want to take this treatment. In addition, some patients do not like using creams as they can be very messy. Tablets or suppositories can be less messy compared to currently available therapies and may help to improve patient compliance and convenience. Currently available treatment options also do not address the androgen component which is provided with DHEA.
Place in Therapy
The clinical expert consulting with CADTH for this review stated that prasterone would provide patients with another treatment option as it can help to improve their physiology and sexual function. In addition, prasterone could be an option for patients with contraindications to estrogen therapies, including patients with breast cancer or other estrogen-dependent cancers, and patients with cardiovascular disease risk.
The clinical expert indicated that prasterone would be suitable as a first-line treatment for patients with post-menopausal VVA. Since prasterone is converted into estrogens and androgens, it is believed to target both GSM and sexual function; based on this, the clinical expert suggested that prasterone may be preferred over currently available estrogen-based treatments. The dosing schedule of prasterone was acknowledged to be different compared to other estrogen-based therapies; other therapies are prescribed to patients at an interval of twice weekly which some patients may easily forget, compared to prasterone which is administered daily.
Patients may consider trying other therapies first primarily due to cost; conjugated estrogen cream (Premarin cream) and vaginal estradiol inserts (Vagifem) are treatment options which are currently covered under public drug plans and accessible to many patients. However, should prasterone be covered and accessible to patients, then it was stated to be an appropriate first-line therapy for patients.
Patient Population
Patients who would benefit from treatment with prasterone would be identified by an experienced clinician both by a physical exam and by asking patients about symptoms of GSM and sexual function. The clinical expert stated that post-menopausal VVA is likely underdiagnosed as approximately 20% to 25% of affected patients tend to seek treatment. The clinical expert stated that all patients who are post-menopausal should be asked about their genitourinary health and sexual health; this is something that is often overlooked among this population of patients. Patients with a history of breast cancer or estrogen-dependent cancers were identified as being a group of interest for treatment with prasterone. As the vulva and vagina have androgen receptors, the clinical expert believed that patients with vulvodynia who are refractory to other treatments may benefit from prasterone as an additional treatment option.
The clinical expert stated that patients who are pre-menopausal and patients who do not complain of GSM symptoms should not be offered treatment with prasterone.
The clinical expert stated that, while it is not possible to identify the patients who would be most likely to respond to treatment with prasterone, most patients who experience symptoms of GSM would be expected to experience some improvement.
Assessing Response to Treatment
According to the clinical expert, a patient’s response to treatment can be assessed through self-reported symptoms and a clinical examination of vaginal colour, lubrication, sensation, and pain. Any reduction in GSM symptoms (e.g., dyspareunia, dryness, pain, discomfort, burning, itch, dysuria) was stated to be considered a clinically meaningful response to treatment.
Response to treatment was stated by the expert to be assessed 3 months to 4 months following treatment initiation, although some studies suggest that patients may improve dramatically within the first month of treatment. After an initial assessment of treatment, if patients are happy with their treatment, then it may not be necessary to continue assessing patient’s response to treatment unless a new symptom occurs, or symptoms worsen again.
Discontinuing Treatment
AEs related to prasterone were stated to be of little worry as the clinical expert believed that prasterone is a very well-tolerated treatment. The clinical expert noted that some patients may find the discharge associated with treatment with prasterone to be bothersome; these patients may wish to discontinue treatment. Some patients may also choose to discontinue treatment if they stop having sexual intercourse and no longer experience dyspareunia.
Prescribing Conditions
The clinical expert confirmed that prasterone may be prescribed by family physicians or at specialty clinics including gynecology, urology, or urogynecology clinics. Diagnosis of post-menopausal VVA can be made by a family physician, nurse practitioner, or a specialist if the patient is referred to 1 (i.e., gynecologist or urologist).
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The original clinician group submissions can be found in the Stakeholder Input section at the end of this report.
Input was received from 2 clinician groups: Cleopatra (prepared by 2 registered nurses) and the SOGC (prepared by 1 physician).
Cleopatra is a virtual clinic with the goal of raising awareness about patients’ intimate health issues, including, but not limited to, vaginal dryness, painful sex, and urinary tract infections. The SOGC is 1 of Canada’s oldest national specialty organizations. The goal of the SOGC is to lead the advancement of patients’ health, working with obstetricians, gynecologists, family physicians, nurses, midwives, and allied health professionals working in the field of patients’ sexual and reproductive health.
Unmet Needs
According to the clinician groups, approximately 70% of women will experience some degree of VVA. Symptoms of post-menopausal VVA were stated to lead to recurrent urinary tract infections and low sexual desire. Once dermatological conditions have been ruled out, both medical and non-medical treatment choices were stated to be used as treatments for VVA. Non-medical treatment options were stated to include good hydration, avoidance of irritants, and correction of contributing conditions (e.g., urinary incontinence). Pelvic floor physiotherapy was also stated to be another treatment option, especially if patients are experiencing dyspareunia or incontinence. While physiotherapy can be effective, it can also be costly for patients if they are not covered by insurance.
According to the SOGC, medical interventions can include vaginal lubricants, vaginal moisturizers (i.e., polycarbophil, polyacrylic aid, hyaluronic acid with or without vitamin E), vaginal estrogen, prasterone, ospemifene, or laser therapy. Cleopatra also listed vaginal moisturizer, prasterone, vaginal estrogen, and laser therapy as treatment options. Vaginal lubricants were stated by the SOGC as being useful for patients whose predominant symptom was dyspareunia. Some vaginal moisturizers, such as polycarbophil or polyacrylic acid, were stated to be unpleasant to patients. Vaginal estrogens were identified as being a common treatment for patients as they correct both symptoms of VVA, and can reverse underlying atrophic changes. When vaginal estrogens are used as directed, they were stated by the SOGC as being safe with minimal systemic absorption of estrogen. Prasterone and ospemifene were listed by the SOGC as new therapies in Canada, with no experience with them outside of clinical trials for prasterone. Vaginal laser therapy was also mentioned by both groups as a therapy for VVA; however, the SOGC noted that RCT evidence suggests that it is not a suitable treatment option for VVA.
Treatment goals for this patient population include improvement in patients’ overall health, bladder health, relationships, and overall quality of life. As many patients will experience vaginal dryness and recurrent urinary tract infections, the condition may impact a patient’s self-esteem and is a significant contributor to a diminished quality of life.
Both the SOGC and Cleopatra stated that not all patients with VVA will respond to available treatments. Many treatment options are estrogen based. The clinician groups highlighted that many patients express trepidation about taking estrogen treatment. Cleopatra stated that some patients with a history of breast cancer may not be comfortable taking estrogen-based treatments. Vaginal laser therapy, while being experimental and potentially damaging to patients, is an expensive therapy and unavailable in certain geographic areas and was stated by the SOGC to be gaining popularity due to fears related to estrogen. In addition, vaginal moisturizers are available to patients as over-the-counter medications but may contain some ingredients that are not suggested for patients with sensitive vaginal issues.
Overall, the SOGC stated that there may be unexpected treatment benefits related to prasterone which are not currently offered to patients through available treatments for VVA.
Place in Therapy
Before seeking intervention from a medical physician, the SOGC stated that it is common for many patients to try lubricants and moisturizers. The SOGC stated that prasterone could be used as a first-line treatment in patients who seek medical help. Cleopatra stated that prasterone or vaginal estrogen are appropriate treatment options if over-the-counter products do not help. Prasterone was also stated to address the underlying disease condition and have a minimal toxicity profile. In particular, prasterone would be preferred for patients who complain of VVA side effects related to sexual desire, and who prefer not to take estrogen-based therapies. In general, SOGC stated that patient preference plays an integral role in patients’ health care. The SOGC also stated that concomitant lifestyle advice would also be recommended to patients.
Patient Population
Prasterone would be used for post-menopausal patients with VVA whose quality of life is impacted due to symptoms of VVA. In particular, the SOGC highlighted patients who are unable to have sexual intercourse due to symptoms of VVA, patients who are unable to have intercourse because of partner issues or are at risk of having vaginal strictures and adhesions which can lead to permanent loss of use of the vagina; these patients should be treated to restore the vaginal mucosa to a state of health.
Patients suitable for treatment with prasterone were stated to be identified by their treating physicians or nurses during an assessment. Both Cleopatra and the SOGC highlighted the underdiagnosis of patients with this condition and stated that more patients need to be treated for VVA and GSM as many patients suffer from this condition needlessly; some patients may be reluctant to seek help, putting the onus on the health care providers to enquire about VVA and identifying patients who could benefit from treatment. SOGC stated that a clinical examination will usually reveal the problem; however, decreased frequency of Pap screening and a shift away from annual clinical examinations may result in symptoms being ignored until patients become highly symptomatic. By the time a diagnosis of VVA is made for patients, there may be sexual health and relationship issues for patients to deal with. SOGC highlighted a need for early recognition of symptoms for patients to have optimal care.
Patients least suitable for treatment with prasterone were stated to include pregnant patients or those who have a sensitivity to prasterone or any ingredient in the product. The SOGC also acknowledged that patients using systemic menopausal hormone treatment may also be candidates for local treatment as the doses of systematic treatment currently used are not always adequate to reverse VVA.
Assessing Response to Treatment
Assessing a patient’s response to treatment was stated to be based on symptom reporting or clinical examination. For example, reductions in urinary tract infections, painless sex, and vaginal comfort were identified as being markers for treatment response. A clinically meaningful response to treatment would include improvement in intimate health, ability to resume sexual relations, and reduction in symptoms during daily living. Both Cleopatra and SOGC stated that an annual assessment of patient’s response would be sufficient after diagnosis.
Discontinuing Treatment
Discontinuation of treatment would be based on patients’ self-monitoring their symptoms and responses to treatment. If patients experience a reaction or lack of response to treatment, they may discontinue treatment. If patients no longer have a need for addressing symptoms, they may also stop treatment for VVA.
Prescribing Conditions
Both clinician groups acknowledged that prasterone could be prescribed to patients either in primary or specialty clinics.
Additional Considerations
Both clinician group inputs stated that prasterone would be a welcome addition to the current treatment options available to patients.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Studies only compared Intrarosa to placebo. Vagifem is a possible comparator and is covered in most provinces. Vagifem is a low-dose twice weekly estrogen insert and was not directly compared to Intrarosa in the submitted trials. | For CDEC consideration. |
Considerations for initiation of therapy | |
The patients included had moderate to severe symptoms. There is uncertainty around whether severity matters and if it can be measured. | The clinical expert commented that severity can be measured using a Likert scale, or by using subscales from the FSFI. In clinical practice, severity of symptoms may be assessed by determining the impacts of symptoms on patients. For example, patients who experience dyspareunia to the point of being unable to engage in sexual activity may be considered to have moderate to severe dyspareunia. |
Moisturizers and lubricants should remain as first-line treatment. Next, Intrarosa and low-dose estrogen topicals and inserts are possible options. Intrarosa was not directly compared to Vagifem; therefore, there is no evidence to say one is more efficacious than the other. Would a patient need to say one prefers a nonestrogen therapy to receive this? Or are there other factors like cancer treatment that would necessitate nonestrogen therapy? | The clinical expert commented that some patients are hesitant to try estrogen therapies due to fears related to increased risk of cancers, blood clots, and/or stroke. Despite education which indicates that risk to patients is low, patients remain hesitant to try these therapies. Prasterone would be a useful treatment option for patients who would prefer not to take or have a contraindication to treatment with estrogen-based therapies. However, such reasons are not necessary to prescribe prasterone and it is appropriate to consider prasterone as a first-line treatment option for patients seeking medical care. In general, patient preference should be considered when prescribing therapies for post-menopausal patients with VVA. |
Considerations for prescribing of therapy | |
Possible use with other topical or vaginal insert treatments, or other hormonal therapies should be a consideration. There is potential for androgen and estrogen levels to rise with Intrarosa as well. With additional treatments, this increase could be exponential. | The clinical expert stated that use of 2 local therapies would not be recommended. Use of prasterone may impact circulating androgen levels, although the clinical expert commented that levels would not be so significantly increased to which warrant concern. Prasterone may be used in combination with testosterone therapy. Clinical guidelines for testosterone treatment recommend that androgen levels be assessed every 6 months. Therefore, any additive effects of treatments used in combination with prasterone would likely be observed during a patient’s assessment. |
CDEC = CADTH Canadian Drug Expert Committee; FSFI = Female Sexual Function Index; VVA = vulvovaginal atrophy.
Clinical Evidence
The clinical evidence included in the review of prasterone (Intrarosa) is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that met a priori selection criteria according to the protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the protocol. The third section includes additional relevant studies that did not meet the a priori selection criteria but were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of prasterone at 6.5 mg per day administered as an ovule intravaginally for the treatment of post-menopausal vulvovaginal atrophy.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients with post-menopausal vulvovaginal atrophy |
Interventiona | Prasterone 6.5 mg ovule administered intravaginally once a day |
Comparatora | Local hormonal therapy
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThese treatments may be administered with or without vaginal lubricants or moisturizers.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.20
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were prasterone and vulvovaginal atrophy. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on October 29, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on February 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist. Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the pre-determined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for NMAs dealing with prasterone was run in MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid on October 29, 2021. No limits were added to limit the search.
Findings From the Literature
A total of 193 records were identified from the literature and screened for relevance; of these 15 were reviewed by full text and 9 reports of 3 unique studies were included in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
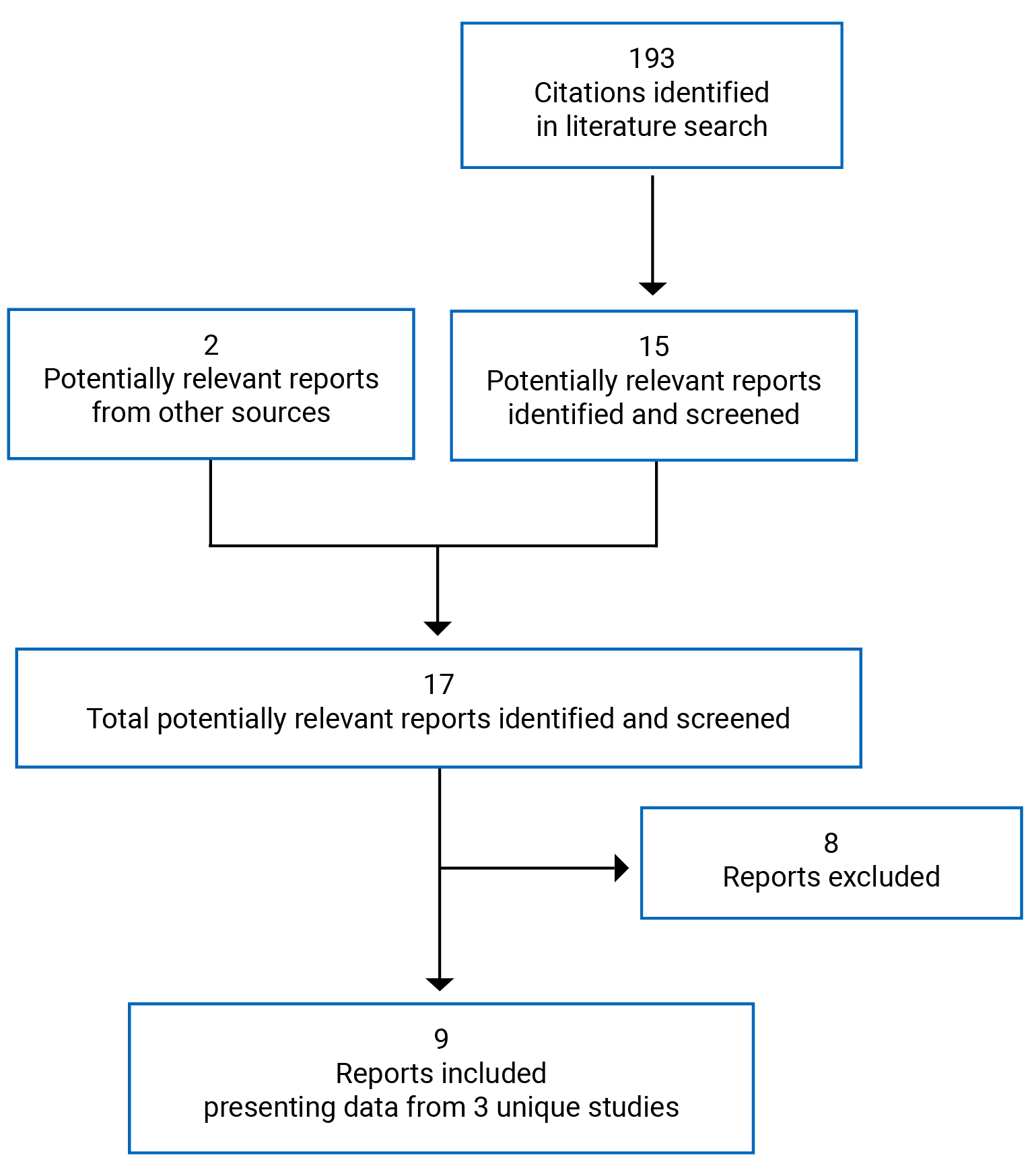
Table 6: Detail of Included Studies
Study detail | ERC-238 | ERC-231 | ERC-230 |
|---|---|---|---|
Designs and populations | |||
Study design | Multi-centre, phase III, DB RCT | Multi-centre, phase III, DB RCT | Multi-centre, phase III, OL single-group trial |
Locations | Canada (14 study sites) and the US (24 study sites) | Canada (9 study sites) and the US (24 study sites) | Canada (10 study sites) and the US (31 study sites) |
Patient enrolment dates | First patient visit: February 11, 2014 Last patient visit: January 6, 2015 | First patient visit: November 30, 2010 Last patient visit: July 29, 2011 | First patient visit: November 30, 2010 Last patient visit: July 16, 2012 |
Randomized (N) | 558 | 255 (87 for 0.25% DHEA not relevant to this report) | 530 |
Inclusion criteria |
All studies:
ERC-238 and ERC-231 also required:
ERC-238 also required:
ERC-230 also required:
| ||
Exclusion criteria |
| ||
|
|
| |
Drugs | |||
Intervention | Prasterone ovule (suppository) 0.5% (6.5 mg) inserted intravaginally daily | ||
Comparators | Placebo ovule (suppository) inserted intravaginally daily | Placebo ovule (suppository) inserted intravaginally dailyc | None |
Duration | |||
Phase | |||
Screening | To a maximum of 8 weeks | To a maximum of 6 weeks | To a maximum of 6 weeks |
Double blind | 12 weeks | 12 weeks | 52 weeks |
Outcomes | |||
Primary end point | Change from baseline to week 12 in:
| Change from baseline to week 12 in:
|
|
Secondary and exploratory end points |
|
|
|
Notes | |||
Publications | Labrie et al. (2018)21 Labrie et al. (2016)22 Labrie et al. (2015)23 | Archer et al. (2017)24 Archer et al. (2015)25 | Bouchard et al. (2016)26 Labrie et al. (2015)27 |
DB = double blind; DHEA = dehydroepiandrosterone; FSH = follicle-stimulating hormone; OL = open label; Pap = Papanicolaou; RCT = randomized controlled trial.
Note: Two additional reported were included.28,29
aThe ERC-230 trial enrolled only non-hysterectomized patients, while the ERC-238 and ERC-231 trials enrolled both hysterectomized and non-hysterectomized patients.
bAmerican College of Radiology Breast Imaging-Reporting and Data System (BI-RADS) category 1 or 2.
cThe ERC-231 trial included a third treatment group of patients who were treated with a prasterone ovule (suppository) of 0.25% (3.25 mg) inserted intravaginally daily before bedtime (usually evening) for 12 weeks. This treatment group was not relevant for this CADTH review as this dose is not recommended by Health Canada; therefore, this group was not reported in this table.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Description of Studies
ERC-238
The ERC-238 trial was a phase III, DB, placebo-controlled, multi-centre (38 sites in Canada and US) trial that aimed to confirm the efficacy of 12 weeks of treatment with once-daily intravaginal DHEA at 0.5% (6.5 mg) compared to once-daily intravaginal placebo ovule for pain at sexual activity (dyspareunia) among post-menopausal patients aged 40 years to 80 years who had moderate to severe dyspareunia as their most bothersome symptom of VVA. A total of 558 patients were randomized 2:1 to prasterone (6.5 mg) or matching placebo centrally using a permuted block design. Main outcomes included change from baseline in percentage of superficial and basal cells, vaginal pH, and severity score of dyspareunia.
ERC-231
The ERC-231 trial was a phase III, placebo-controlled, multi-centre trial assessing the efficacy of intravaginal prasterone at 6.5 mg or 3.25 mg compared to placebo among post-menopausal patients experiencing moderate to severe dyspareunia as their most bothersome symptom of VVA at baseline. The trial was conducted in 33 sites including 24 sites in the US and 9 sites in Canada. The trial consisted of a screening period lasting between 4 weeks to 6 weeks, followed by a 12-week treatment period. Patients were randomized in 1:1:1 ratio to receive each treatment. Randomization was conducted centrally by Veristat Inc. using permuted block design. Blocks of 3 patients were generated with each block sequence randomly allocated. Main outcomes included change from baseline in percentage of superficial and basal cells, vaginal pH, and severity score of dyspareunia. For this CADTH review, only the prasterone group treated at 6.5 mg (0.5%) was considered relevant as this is the Health Canada–approved dose; data from patients who received prasterone at 3.25 mg are not summarized in this report.
ERC-230
The ERC-230 trial was a phase III, open-label, single-group study which examined the long-term safety of daily treatment with intravaginal prasterone (6.5 mg). The duration of the trial was 12 months (52 weeks). The study was conducted in 10 sites in Canada and 31 sites in the US. There were 2 phases of the trial, including a screening period of 4 weeks to 6 weeks, and a treatment period of 52 weeks. All patients received treatment with DHEA. As the main purpose of this trial was to evaluate the long-term safety of prasterone, the main outcomes included AEs, SAEs, and mortality.
Populations
Inclusion and Exclusion Criteria
Inclusion criteria across the ERC-238, ERC-231, and ERC-230 trials included post-menopausal patients with VVA who self-identified dyspareunia as moderate to severe and as their most bothersome symptom. Patients also had 5% of less superficial cells on vaginal smear, a vaginal pH greater than 5, a normal mammogram and physical breast examination, and a normal Pap smear. The ERC-238 and ERC-231 trials enrolled patients regardless of whether they were hysterectomized and non-hysterectomized, while the ERC-230 trial enrolled only non-hysterectomized patients. Patients in the ERC-230 trial could also report moderate to severe vaginal dryness or irritation/itching in addition to dyspareunia.
Exclusion criteria included a previous diagnosis of cancer and having history of significant or uncontrolled comorbidities (i.e., thromboembolic disease, endocrine disease, diabetes mellitus, depression). In addition, patients were excluded if they received vaginal hormonal products, DHEA, or estrogen or progestin therapies (local or systemic) within 8 weeks or within 6 months of study entry, depending on the specific type of product.
Baseline Characteristics
A summary of baseline characteristics is shown in Table 7. Baseline characteristics were similar between the prasterone and placebo groups in the ERC-238 and ERC-231 trials as well as across the 2 trials. In all 3 trials, patients had a mean age of between 58 years and 60 years and were mostly White (> 85%) and non-Hispanic or Latino (≥ 88%). Patients reported both natural and surgical causes of their last menstruation, which occurred at a mean age of 44 years to 50 years. Previous hormone therapy was reported by approximately half of patients across all trials.
There were a few key differences across trials. The mean years since last menstruation was 13 years to 14 years for patients in the ERC-238 and ERC-231 trials, versus approximately 8 years in the ERC-230 trial. More than 1-third of patients in the ERC-238 trial reported having a hysterectomy, versus 2-thirds of patients in the ERC-231 trial; patients in the ERC-230 trial were non-hysterectomized. Oophorectomy was performed in approximately 1-quarter to 1-third of patients in ERC-238 and ERC-231, compared to 5% in the ERC-230 trial.
Table 7: Summary of Baseline Characteristics (Safety Population)
Characteristic | ERC-238a | ERC-231a | ERC-230b | ||
|---|---|---|---|---|---|
Prasterone N = 374 | Placebo N = 180 | Prasterone N = 87 | Placebo N = 80 | Prasterone N = 521 | |
Age (years) | |||||
Mean (SD) | 59.5 (6.78) | 59.6 (5.75) | 57.5 (5.63) | 58.8 (5.89) | 57.9 (5.65) |
Race | |||||
White | 338 (90) | 163 (91) | 83 (95) | 69 (86) | 478 (92) |
Black or African American | 13 (7) | 28 (7) | 3 (3) | 9 (11) | 31 (6) |
Asian | 2 (1) | 4 (1) | 1 (1) | 1 (1) | 3 (1) |
American Indian or Alaskan Native | 0 (0) | 1 (0) | 0 (0) | 0 (0) | 3 (1) |
Native Hawaiian or Other Pacific Islander | 0 (0) | 1 (0) | 0 (0) | 0 (0) | 0 (0) |
Other | 2 (1) | 2 (1) | 0 (0) | 1 (1) | 4 (1) |
Ethnicity | |||||
Not Hispanic or Latina | 330 (88) | 166 (92) | 78 (90) | 79 (99) | 497 (95) |
Hispanic or Latina | 44 (12) | 14 (8) | 9 (10) | 1 (1) | 24 (5) |
Reproductive history | |||||
Years since last menses, mean (SD) | 14.14 (9.48) | 13.40 (8.55) | 14.02 (8.79) | 13.88 (8.75) | 8.37 (5.62) |
Cause of menopause | |||||
Natural | 237 (63) | 120 (67) | 43 (49) | 38 (48) | 521 (100) |
Surgical | 137 (37) | 60 (33) | 44 (51) | 42 (53) | 0 (0) |
Age (years) at last menses, mean (SD) | |||||
All patients | 45.37 (7.28) | 46.15 (6.73) | 43.48 (8.50) | 44.94 (7.46) | 49.58 (4.62) |
Natural menopause | 48.62 (4.34) | 48.93 (4.55) | 48.40 (5.02) | 47.42 (6.34) | 49.58 (4.62) |
Surgical menopause | 39.75 (7.90) | 40.60 (6.98) | 38.68 (8.50) | 42.69 (7.74) | 0 (0.00) |
Hysterectomy | 144 (39) | 64 (36) | 52 (60) | 49 (61) | 521 (100) |
Oophorectomy | |||||
Any oophorectomy | 102 (27) | 44 (24) | 26 (30) | 29 (36) | 28 (5) |
Bilateral oophorectomy | 72 (19) | 31 (17) | 19 (22) | 20 (25) | 3 (1) |
Prior therapy | |||||
Previous hormone replacement therapy, n (%) | 158 (42) | 75 (42) | 48 (55) | 49 (61) | 268 (51) |
SD = standard deviation.
Note: Values are presented as n (%) unless otherwise indicated.
aIntention-to-treat population.
bSafety population.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Interventions
In the ERC-238 and ERC-231 trials, patients received either prasterone 0.5% (6.5 mg) solubilized in Witepsol (hard fat) as a vaginal ovule (suppository) or matching placebo (Witepsol only). Patients in the ERC-230 trial all received prasterone 0.5% (6.5 mg) solubilized in Witepsol (hard fat) as a vaginal ovule (suppository); there was no comparison group. In all trials, patients inserted their assigned therapy intravaginally using a single-use applicator daily before bedtime (usually in the evening) for 12 weeks. Instructions and a demonstration regarding proper administration of study treatment were given to patients before their first administration. Patients were instructed not to take a bath or shower for at least 1 hour after application for adequate absorption, and to not use any vaginal care product on the vagina or vulva during the treatment period. If a day of therapy was forgotten, patients were instructed to insert it immediately, but a minimum interval of 8 hours should be observed between doses. Patients were required to return all boxes at week 6 and 12 to count the vaginal ovules and assess adherence. Treatment adherence was also assessed using a diary card where patients would record the frequency of treatment application.
Concomitant treatments which were necessary for the patient’s well-being were allowed during the study. The following concomitant medications were not permitted in all trials: hormone replacement therapy, progestogen medication, natural oral “estrogenic” products, vaginal cream or gel, vaginal lubricant, or vaginal douching.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized below. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
The 4 coprimary end points of the ERC-238 trial and the ERC-231 trial were change from baseline to week 12 in each of the following: percentage of parabasal cells, percentage of superficial cells, vaginal pH, and severity score of dyspareunia as the most bothersome self-reported VVA symptom.10 The main purpose of the ERC-230 trial was to evaluate the long-term (52 week) safety of intravaginal DHEA; however, efficacy variables were evaluated as secondary end points.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | ERC-238 | ERC-231 | ERC-230 |
|---|---|---|---|
Severity score of dyspareunia | Primary | Primary | Secondary |
Vaginal dryness, vaginal or vulvar irritation/itching | Secondary | Secondary | Secondary |
Sexual function (FSFI) | Secondary | Secondary | Secondary |
Percentage of superficial cells | Primary | Primary | Secondary |
Percentage of parabasal cells | Primary | Primary | Secondary |
Vaginal pH | Primary | Primary | Secondary |
Safety (i.e., AEs, SAEs, WDAEs, mortality) | Secondary | Secondary | Primary |
AE = adverse event; FSFI = Female Sexual Function Index; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Efficacy End Points
Dyspareunia
Dyspareunia was assessed as part of the Vaginal Atrophy Symptom Questionnaire (VASQ) which consists of items evaluating dryness, soreness, irritation, dyspareunia, and vaginal discharge (detailed description and appraisal in Appendix 3). The VASQ was used to evaluate the severity score of symptoms of VVA associated with menopause in all 3 trials and was administered at screening, baseline, week 6, and week 12 in the ERC-238 and ERC-231 trials and at screening, baseline, week 12, week 26, week 39, and week 52 in the ERC-230 trial. Patients who discontinued from the trial were assessed at their discontinuation visit. Data from the VASQ were self-reported by patients. The severity of each VVA symptom was recorded as none, mild, moderate, or severe and were analyzed using scores of 0, 1, 2, or 3, respectively. No minimal important difference (MID) has been established for the VASQ. Construct validity was demonstrated with the VASQ; however, populations assessed included patients treated with systemic hormonal therapy, and did not include post-menopausal patients.30 Reliability of the VASQ has not been well established.
Vaginal Dryness
Vaginal dryness was assessed as 1 of the symptoms in the VASQ.
Vaginal and/or Vulvar Irritation or Itching
Vulvovaginal irritation or itching was assessed as 1 of the symptoms in the VASQ.
Sexual Function
Sexual function was measured using the FSFI which contains 19 items and can be used to assessed 6 key domains, including arousal/lubrication, subjective arousal (psychological), satisfaction, desire, pain at sexual activity, and orgasm, over the past 4 weeks (detailed description and appraisal in Appendix 3). Patients’ total FSFI scores could range from 2.0 to 36.0 with higher scores indicating greater sexual function. Female sexual dysfunction is defined in the FSFI as a total score of less than 26.5. For the ERC-238 and ERC-231 trials, the FSFI questionnaire was completed by patients at their screening visit, baseline, week 6, and week 12 (or discontinuation visit). For the ERC-230 trial, the FSFI was completed at screening, baseline, week 26, and week 52 (or discontinuation visit). Under the original protocols of the ERC-231 and ERC-230 trials, patients were to complete the Menopause-Specific Quality of Life questionnaire (MENQOL). In updated versions of these trials’ protocols patients were to complete the FSFI instead of the MENQOL. Validity and reliability of the FSFI has been demonstrated in peri- and post-menopausal women. No MIDs have been established for the FSFI.
Vaginal Cell Maturation
In the ERC-238 and ERC-231 trials, the vaginal cell maturation index was determined from vaginal smears collected at baseline, week 6, and week 12 (or discontinuation visit) of the trial. In the ERC-230 trial the vaginal cell maturation index was determined at screening, baseline, week 26, and week 52. Patients who discontinued therapy were to complete their vaginal smears at their discontinuation visit. Vaginal smear samples were examined by experienced cytopathologists who were blinded to treatment assignment of patients. A 100-cell count was performed to classify cells as parabasal (including basal), intermediate, and superficial squamous cell types. A greater number of cells were usually counted, and the numbers obtained for each of the 3 cell populations were divided to be reported as a total of 100.
Vaginal pH
Vaginal pH was determined by using a pH strip which was applied to an Ayre spatula (or equivalent) to the opposite lateral wall of the vagina. The change in colour on the indicator strip was compared to a colour chart to determine the pH. The pH numbers were read on the ColourpHast, an indicator with a range from pH 4.0 to 7.0. In the ERC-238 and ERC-231 trials, vaginal pH of patients was determined at screening, baseline, week 6, and week 12. In the ERC-230 trial, vaginal pH was measured at screening, baseline, week 26, and week 52. For patients who discontinued the trial, vaginal smears were conducted at their discontinuation visit.
Harms
Adverse Events
In all trials, harms outcomes included reporting of all AEs (including SAEs, unexpected AEs, withdrawal due to AEs, and mortality). AEs included all and any medical experiences, regardless of their relationship to the study treatment. SAEs included any occurrences that resulted in death, were immediately life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, were a congenital anomaly or birth defect, or were an important medical event that the investigator considered serious.10 All AEs were graded using the Common Terminology Criteria for Adverse Events (CTCAE, version 4.03). AEs were tabulated if they started or worsened after the start of treatment (after first dose) through 30 days after the last dose of the study treatment.
Notable Harms
Regarding the ERC-230 trial, breast, endometrial, and Pap smear safety were assessed. Breast safety was assessed through mammograms at screening and at week 52 in the ERC-230 trial. Pap smears were conducted for patients who received prasterone for 26 weeks or longer. Endometrial biopsies were performed for patients who received prasterone for 3 months or longer. Endometrial safety was also assessed in the ERC-231 trial among patients who were non-hysterectomized and underwent an endometrial biopsy at screening and at week 12. For all trials, Pap spears and endometrial biopsy samples were assessed by a central laboratory.
Statistical Analysis
In general, for the ERC-238, ERC-231, and ERC-230 trials, observations conducted at baseline (day 1) were used for analyses related to change from baseline parameters. If no sexual activity occurred between screening and baseline, the screening value for vaginal pain associated with sexual activity was used for the baseline value. For other analyses, the screening value was used as the baseline value if there were missing data at baseline.
ERC-238
Sample Size
Determination of sample size for the ERC-238 trial was based on results from the ERC-231 trial. The sample size required to detect the same differences between DHEA 0.5% and placebo as seen in the ERC-231 trial was based on a 1-sided alpha of 0.025 and an overall power of 97.7% across all end points, or greater than 99.99% for each coprimary end point. Based on a randomization of 2:1 for prasterone to placebo, the largest required sample size out of all coprimary efficacy end points was 274 patients in the prasterone group and 137 patients in the placebo group, based on the end point of severity of dyspareunia. An allowance for 15% loss to follow-up resulted in an expected 322 patients in the prasterone group and 161 patients in the placebo group. In addition, the sample size of 322 patients treated with prasterone allowed for the sponsor to achieve the total number of 1,500 patients exposed to intravaginal prasterone across all trials, as in accordance with the International Conference on Harmonization E1 guidelines.
Analysis of Efficacy End Points
The ITT populations were used for analyses of efficacy variables, with the per-protocol (PP) population also analyzed as supportive evidence. The primary analysis of the 4 coprimary end points (change from baseline to week 12 in vaginal pH, percent parabasal cells, percent superficial cells, and symptom score of dyspareunia) was based on data pooled across all sites with results presented separately for each treatment group. An analysis of covariance model was used, with the treatment group as the main factor and the baseline value as the covariate. The P value for the baseline adjusted least squares MD between groups was presented for prasterone versus placebo. The P values of the coprimary end points were not adjusted for multiplicity as statistical significance of each coprimary end point was required to form a conclusion of superiority of prasterone over placebo. No other approaches to adjust for multiplicity were applied to the coprimary end points. Each of the coprimary end points were analyzed as continuous variables. Statistical analyses were performed at a 2-sided significance level of 0.05 unless otherwise specified.
The symptom scores of vaginal dryness and vaginal irritation or itching were tested as second- and third-order end points, respectively, and were tested among patients who identified these symptoms as being moderate or severe at baseline. To control for the potential for type I error due to multiple end points, the primary symptom score of dyspareunia had to be statistically significant before testing of vaginal dryness, which had to be statistically significant before testing of vulvovaginal irritation or itching.
In the ERC-238 trial, to assess change in sexual function, the mean changes from baseline to week 12 in the following sexual domains of the FSFI were evaluated: arousal/lubrication, subjective arousal (arousal/psychological), desire, satisfaction, and orgasm. No adjustments for multiplicity were conducted for analyses of the FSFI. While the pain at sexual activity domain was used for calculation of the total score of the FSFI, the individual domain of pain at sexual activity was not a secondary end point since pain was evaluated as a coprimary end point using the VASQ. For FSFI analyses, a computational formula reported in Rosen et al.31 was used to derive the individual domain scores and full-scale scores of the FSFI. Individual domain scores were obtained by adding the scores of the individual items that comprise the domain and multiplying the sum by the domain factor (a detailed description and appraisal of the FSFI is provided in Appendix 3). The total score was obtained by summing the 6 domain scores. A score of 0 for each domain indicated that no sexual activity was reported during the past month. When an individual domain question had “missing” as the reported value at baseline, both the domain score and the total score were not calculated for this patient at baseline. The individual domain scores and the global score of the FSFI questionnaire were analyzed using analysis of covariance for the difference in change from baseline between prasterone treatment and placebo.
Handling of Missing Data
Patients who were lost to follow-up or discontinued were not replaced. Missing safety data were not imputed.
The last observation carried forward (LOCF) approach was used for efficacy end points in the ITT population when there were missing time points due to discontinuation of a patient, or from missing samples or data for efficacy parameters. In patients who reported pain at sexual activity as their most bothersome symptom at baseline but did not engage in sexual activity after baseline, the baseline value for severity of dyspareunia was carried forward through week 12 and was used for analysis. Also, the evaluation of dyspareunia at week 6 was carried forward if not available at week 12 or discontinuation. Similarly, if the sample for pH, parabasal cells, or superficial cells was missing at day 1, the screening value was used for baseline.
For the FSFI questionnaire, if a patient did not answer 1 or more questions at baseline, the answer(s) given at screening was used for baseline calculations. If a patient had missing answers at both screening and baseline, then the domain score (if applicable) containing a missing answer(s) as well as the full-scale score (total score) of the patient were not calculated and not included in the analysis for the baseline and post-baseline time points.
ERC-231
Sample Size
The sample size calculations for the ERC-231 trial were based on results of the ERC-210 trial,15 which demonstrated a statistically significant improvement in all prasterone groups compared to placebo for change from baseline to 12 weeks in change in severity score of dyspareunia, vaginal dryness, vaginal irritation or itching, and vaginal pH. The sample size requirements for tests of the coprimary outcomes were based on comparisons to placebo for both prasterone groups (0.25% and 0.5%); a 2-sided alpha level of 0.025 was used to control for the 2 comparisons versus placebo. The power for all tests was set at 95%. The largest required sample size out of all coprimary efficacy end points was 60 per treatment group; the sponsor planned to have 70 patients to be enrolled into each treatment group to account for drop-outs and to provide increased safety data.
Analysis of Efficacy End Points
The ITT populations were used for analyses of efficacy variables, with the PP population also analyzed as supportive evidence. The analyses of efficacy end points, including adjustments for covariates and handling of missing data, for the ERC-231 trial were conducted in the same manner as the ERC-238 trial which are reported above (in the ERC-238 section). Adjustments to multiplicity were the same as those used in ERC-238, except that ERC-231 also adjusted for the multiple doses of prasterone assessed using the Hochberg modification of the Bonferroni procedure. This procedure was used to preserve the overall type I error of 0.05 for testing of the 2 doses of prasterone versus placebo.
ERC-230
Sample Size
There were 450 patients enrolled to have data from 300 or more patients at 12 months; this was conducted in agreement with International Conference on Harmonization E1 recommendations and requirements for regulatory agencies.
Analyses of Efficacy End Points
For the ERC-230 trial, efficacy variables were analyzed using the “Safety Population for Efficacy Analysis” Set. Data from across all study sites was pooled for safety and efficacy analyses. No adjustments for multiplicity to P values were used for analyses of secondary efficacy outcomes. For efficacy end points, mean values at each visit and changes from baseline were reported using continuous descriptive statistics where appropriate, and t-tests were used to assess the change from baseline to each visit. Vaginal cell maturation index change was analyzed in the following manner: change from baseline to week 26 and week 52 (in percentage of basal or parabasal and superficial cells). The vaginal pH change was evaluated as follows: change from to week 26 and week 52.
Regarding the FSFI, the FSFI parameters were tested using change from baseline to week 26 and week 52. Actual values were also summarized. For FSFI analyses, a computational formula reported in Rosen et al.31 was used to derive the individual domain scores and full-scale scores of the FSFI. Individual domain scores were obtained by adding the scores of the individual items that comprise the domain and multiplying the sum by the domain factor. The total score was obtained by adding the 6 domain scores. A score of 0 for each domain indicated that no sexual activity was reported during the past month. When a question for an individual domain had missing as the reported value at baseline, both the domain score and the total score were not calculated for this patient at baseline. A statistical hierarchy was not implemented for the ERC-230 trial, as the main objective of the study was to evaluate the safety of prasterone. As the original protocol of the trial, dated October 25, 2020, specified use of the MENQOL, only patients enrolled after a change was made to the protocol, dated March 7, 2011, which switched to use of the FSFI, were included in these analyses.
Handling of Missing Data
Patients who were lost to follow-up or discontinued were not replaced. Missing safety data were not imputed. For efficacy outcomes with missing data, the last observation was carried forward for the analysis only if patients had at least 1 post-baseline measurement. When no post-baseline efficacy data were evaluated for an end point, the patient was not included in the analysis of that end point. If an efficacy parameter was missing at baseline, the screening value was used as the baseline value.
Analysis Populations
A summary of the analysis populations used in the ERC-238, ERC-231, and ERC-230 trials are reported in Table 9.
Table 9: Summary of Analysis Populations in the ERC-238, ERC-231, and ERC-230 Trials
Analysis set | N | Definition |
|---|---|---|
ERC-238 | ||
ITT population | 482 | The ITT population consisted of all patients who received at least 1 dose of study drug (according to patient diaries) with a baseline evaluation meeting the study eligibility criteria. Patients were analyzed as randomized. The ITT population was considered the primary analysis population. Patients in the ITT population must have met all of the following inclusion criteria:
|
mITT population | 448 | The mITT population consisted of patients in the ITT population who had post-baseline sexual activity at least once before the evaluation of dyspareunia at week 6 and week 12, or discontinuation of the treatment during the 12-week trial period. The coprimary end point of dyspareunia was analyzed in the mITT population in addition to the ITT population. |
Per-protocol population | 373 | The per-protocol population consisted of a subset of patients in the ITT population who completed all 12 weeks of the study with no major protocol violations considered to compromise the efficacy data. Major protocol violations were determined before the study blind was broken, and patients must have received at least 90% of the required number of applications of study treatment. |
Safety population | 554 | The safety population was defined as all patients who received an administration of any amount of either study treatment (DHEA or placebo), and who had any post-baseline safety information available. All safety parameters were analyzed using this population, and patients were analyzed according to actual treatment received. |
ERC-231 | ||
ITT population | 237 | The ITT population was defined in the same manner as the ERC-238 trial. |
Per-protocol population | 204 | The per-protocol population was defined in the same manner as the ERC-238 trial, except for the following: patients in the per-protocol population must meet inclusion criteria at screening and baseline and have efficacy evaluations at week 12. |
Safety population | 253 | The safety population was defined in the same manner as the ERC-238 trial. |
ERC-230 | ||
Safety population | 521 | The safety population was defined as all patients who received an administration of any amount of DHEA and who have any safety information available. All analyses of safety parameters were based on this population. Analyses on this population were performed separately for the following subgroups:
|
Safety population for efficacy analysis | 487 | The “safety population for efficacy analysis” was defined as all patients who received an administration of any amount of DHEA and who have at least 1 post-baseline valid data entry for the efficacy parameter being evaluated. Analyses on this population were performed separately for the same subgroups specified in the safety population. |
DHEA = dehydroepiandrosterone; ITT = intention to treat; mITT = modified intention to treat.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Results
Patient Disposition
In general, disposition of patients did not differ between treatment groups within trials. Disposition of patients was generally consistent across trials, though the percentages of randomized patients discontinuing the study were highest in the ERC-230 trial (18%), followed by the ERC-231 trial (11% to 13%), and then the ERC-238 trial (5% to 6%). While almost all randomized patients in the ERC-231 trial were included in the ITT population, a notable proportion of randomized patients in the ERC-238 trial were not included in the ITT population. In the ERC-238 trial, there were 23 patients in the placebo group and 49 patients in the prasterone group who were excluded from the ITT population because at least 1 study eligibility criterion was not met at baseline.
ERC-238
A total of 1,226 patients were screened for eligibility; of these, 558 were enrolled and randomized into the trial with 376 patients in the prasterone group and 182 patients in the placebo group.10 Screen failures were mainly due to |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| (||||||%).32 Most patients (94%) completed the study. Few patients discontinued from the trial (5% in the prasterone group and 6% in the placebo group); reasons for discontinuation from the trial included patient’s withdrawal of consent (40% versus 18% of patients who discontinued in the prasterone and placebo groups, respectively), AEs (25% versus 45%, respectively), lost to follow-up (15% versus 18%, respectively), non-compliance (5% versus 9%, respectively), investigators’ decision (5% versus 0%, respectively), or other (10% versus 9%, respectively). In general, no major differences in patient disposition were noted between treatment groups.10
ERC-231
A total of 464 patients were screened for eligibility; of these patients, 87 patients were randomized into the DHEA 0.5% group and 81 were randomized into the placebo group.9 Screen failures were mainly due to ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| (||||||%).32 Most patients completed the trial, including 87% of patients in the prasterone group and 89% of patients in the placebo group. The primary reasons for discontinuing were due to “other” (64% versus 33% of patients who discontinued in the prasterone and placebo groups, respectively), patients withdrawing consent (18% versus 44%, respectively), and AEs (18% versus 11%, respectively).9
ERC-230
A total of 798 patients were screened for eligibility; of these patients, 530 were enrolled into the study.8 Screen failures were primarily due to |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| (||||||%).32 Few patients discontinued from the ERC-230 trial; reasons for discontinuation were mostly due to patients withdrawing consent (33% of discontinuations) and AEs (31%).8
Classifications | ERC-238 | ERC-231 | ERC-230 | ||
|---|---|---|---|---|---|
Prasterone | Placebo | Prasterone | Placebo | Prasterone | |
Screened, N | 1,226 | 464 | 798 | ||
Screen failures, N (%) | 668 (54) | 209 (45) | 268 (34) | ||
Randomized, N (%) | 376 | 182 | 87a | 81a | 530b |
Completed study, N (% of randomized) | 356 (95) | 171 (94) | 76 (87) | 72 (89) | 435 (82) |
Discontinued from study, N (% of randomized) | 20 (5) | 11 (6) | 11 (13) | 9 (11) | 95 (18) |
Reason for discontinuation, N (% of randomized) | |||||
Adverse events | 5 (1) | 5 (3) | 2 (2) | 1 (1) | 29 (6) |
Disease progression | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Nonadherence | 1 (< 1) | 1 (< 1) | 0 (0) | 1 (1) | 4 (1) |
Lost to follow-up | 3 (1) | 2 (1) | 0 (0) | 0 (0) | 16 (3) |
Patient withdrew consent | 8 (2) | 2 (1) | 2 (2) | 4 (5) | 31 (6) |
Investigator’s decision | 1 (< 1) | 0 (0) | 0 (0) | 0 (0) | 3 (1) |
Death | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Other | 2 (< 1) | 1 (< 1) | 7 (8) | 3 (4) | 12 (2) |
ITT population, N | 325 | 157 | 81 | 77 | NA |
PP population, N | 254 | 119 | 70 | 65 | NA |
Safety, N | 374 | 180 | 87 | 80 | 521 |
Safety population for efficacy analysis, N | NA | NA | NA | NA | 487 |
ITT = intention to treat; PP = per protocol.
aThe ERC-231 trial randomized patients to 3 treatment groups: prasterone (0.5% or 0.25%) or placebo. Patients randomized to the prasterone 0.25% group are not relevant to this CADTH report and are not reported here.
bThe number of patients reflects enrolled patients as there was no randomization in the ERC-230 trial.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 ERC-230 Clinical Study Report.8
Protocol Deviations
ERC-238
Major protocol deviations were reported in 35 patients (9.3%) in the prasterone group and 11 patients (6.0% in the placebo group). The sponsor reported these deviations not to have affected the integrity of the study data. There was no premature unblinding of patients in the study.10
ERC-231
There were 4 (4.6%) major protocol deviations reported in the prasterone 0.5% group and 5 (6.2%) protocol deviations in the placebo group. These deviations were reported by the sponsor not to have affected the integrity of the study data, and there was no premature unblinding of patients in the study.9
ERC-230
There was a total of 36 patients with major protocol deviations in the ERC-230 trial. Major protocol deviations were reported by the sponsor not to have affected the integrity of the trial data.8
Exposure to Study Treatments
A summary of patients’ exposure to study treatments is reported in Table 11. The mean duration of treatment was similar between the treatment with prasterone and placebo groups in the ERC-238 trial (82.3 days versus 82.4 days, respectively) and the ERC-231 trial (76.2 days versus 76.6 days, respectively) trials, that is, approximately 11 to 12 weeks. The mean total exposure to treatment was also similar between the prasterone and placebo groups in the ERC-238 (79.9 days versus 80.4 days, respectively) and ERC-231 (74.5 days versus 74.5 days, respectively) trials. As the ERC-230 trial was conducted over a longer period of time (52 weeks), the overall duration of exposure to DHEA was greater. The mean duration of treatment for patients in the ERC-230 trial was 325.0 days with a mean total exposure of 316.0 days, corresponding to over 45 weeks.
Table 11: Exposure to Study Treatments (Safety Population)
ERC-238 | ERC-231 | ERC-230 | |||
|---|---|---|---|---|---|
DHEA 0.5% (N = 374) | Placebo (N = 180) | DHEA 0.5% (N = 87) | Placebo (N = 78) | All patients (N = 521) | |
Duration of treatment (days)a | |||||
N | 373 | 178 | 87 | 80 | 521 |
Mean (SD) | 82.29 (11.98) | 82.35 (10.87) | 76.24 (16.38) | 76.58 (16.29) | 325.01 (89.11) |
Total exposure (days)b | |||||
N | 373 | 178 | 87 | 80 | 518 |
Mean (SD) | 79.90 (12.29) | 80.42 (11.79) | 74.49 (16.90) | 74.50 (16.71) | 315.95 (89.10) |
Total exposure, week 12%c | |||||
N | 374 | 180 | 87 | 80 | 518 |
Mean (SD) | 97.03 (14.75) | 96.61 (15.80) | 88.31 (19.91) | 88.41 (19.75) | 95.07 (12.94) |
Total exposure, week 52%c | |||||
N | — | — | — | — | 518 |
Mean (SD) | — | — | — | — | 86.69 (24.43) |
DHEA = dehydroepiandrosterone; SD = standard deviation.
aBased on diary data, duration of treatment is the last date of drug administration minus the first date plus 1.
bBased on diary data, the total number of days the subject applied study medication.
cBased on medication count; 100 multiplied by (number of applications done divided by number of expected applications) during this time period.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following.
Health-Related Quality of Life
Health-related quality of life was not assessed in the ERC-238, ERC-231, and ERC-230 trials.
Dyspareunia
A summary of results for change from baseline to week 12 of dyspareunia in the ERC-238 and ERC-231 trials is reported in Table 12. The summary of results for change from baseline to the end of treatment period of dyspareunia in the ERC-230 trial is reported in Table 13. It should be noted that in the ERC-238 and ERC-231 trials, eligibility criteria specified that patients report having pain at sexual activity (dyspareunia) of moderate to severe intensity being perceived as the most bothersome symptom of VVA both at the time of screening and at baseline. Change from baseline in symptom score of dyspareunia was a coprimary end point in the ERC-238 and ERC-231 trials, and a secondary end point in the ERC-230 trial. The PP analyses results were similar to those for the ITT analyses in the ERC-238 and ERC-231 trials.
ERC-238
The mean change from baseline in severity score of dyspareunia was greater for the prasterone group (–1.42; SD = 1.00) compared to the placebo group (–1.06; SD = 1.02) at 12 weeks; the MD for prasterone versus placebo was –0.35 (SD for MD not reported; P = 0.0002). The results in the modified ITT population were similar to those in the ITT population.
ERC-231
The mean change from baseline in severity score of dyspareunia was greater for the prasterone group (–1.27; SD = 0.99) compared to the placebo group (–0.87; SD = 0.95) at 12 weeks; the MD for prasterone versus placebo was –0.40 (SD for MD not reported; P < 0.0132).
ERC-230
The mean severity score of pain at sexual activity was reported for patients who had moderate to severe dyspareunia as their most bothersome symptom at baseline while also meeting VVA criteria for superficial cells (≤ 5%) and vaginal pH (> 5.0) (n = 183). The baseline score severity score of dyspareunia was 2.57 (SD = 0.50) at and 0.87 (SD = 0.96) at week 52; the mean change from baseline was –1.69 (SD = 0.97).
The mean severity score of dyspareunia was reported for patients who had moderate to severe dyspareunia at baseline while also meeting VVA criteria for superficial cells (≤ 5%) and vaginal pH (> 5.0) (n = 240). The severity score of dyspareunia was 2.53 (SD = 0.50) at baseline and 0.85 (SD = 0.95) at week 52; the mean change from baseline was –1.68 (SD = 0.95).
Table 12: Dyspareunia Symptom Score — ERC-238 and ERC-231 (ITT Population)
Study detail | ERC-238 | ERC-231 | ||
|---|---|---|---|---|
Prasterone N = 325 | Placebo N = 157 | Prasterone N = 81 | Placebo N = 77 | |
Baseline, mean (SD) | 2.54 (0.50) | 2.56 (0.50) | 2.63 (0.49) | 2.58 (0.50) |
Week 12, mean (SD) | 1.13 (0.98) | 1.50 (1.05) | 1.36 (1.10) | 1.71 (1.00) |
Mean change (SD) | –1.42 (1.00) | –1.06 (1.02) | –1.27 (0.99) | –0.87 (0.95) |
Mean difference from placebo (SD) | –0.35 (NR) | — | –0.40 (NR) | — |
P valuea | 0.0002 | — | 0.0132 | — |
ANCOVA = analysis of covariance; ITT = intention to treat; SD = standard deviation.
Note: Dyspareunia was measured as part of the VASQ. The severity of each symptom assessed using the VAS was recorded as none, mild, moderate, or severe and analyzed using the scores of 0, 1, 2, or 3, respectively. Therefore, lower scores indicated improved symptom scores.
Change from baseline in symptom score of dyspareunia was 1 of 4 coprimary end points. Each coprimary end point was not adjusted for multiplicity as statistical significance of each coprimary end point was required for conclusion of superiority of prasterone over the placebo group.
aANCOVA test with treatment group as the main factor and baseline value as the covariate (P value vs. placebo).
Source: ERC-238 Clinical Study Report,10 and ERC-231 Clinical Study Report.9
Table 13: Dyspareunia Symptom Score — ERC-230
Study detail | MBS pain with sexual activity subgroupa N = 183 | MS pain with sexual activity subgroupb N = 240 |
|---|---|---|
Baseline | ||
Mean (SD) | 2.57 (0.50) | 2.53 (0.50) |
Week 52 | ||
Mean (SD) | 0.87 (0.96) | 0.85 (0.95) |
Change from baseline, mean (SD) | –1.69 (0.97) | –1.68 (0.95) |
P valuec | < 0.0001 | < 0.0001 |
MBS = most bothersome symptom; MS = moderate to severe; SD = standard deviation.
Note: Dyspareunia was measured as part of the VASQ. The severity of each symptom assessed using the VAS was recorded as none, mild, moderate, or severe and analyzed using the scores of 0, 1, 2, or 3, respectively. Therefore, lower scores indicated improved symptom scores. Change from baseline in symptom score of dyspareunia was a secondary end point and was not adjusted for multiplicity.
a-b Subjects from the Safety Population who have at least one post-baseline efficacy assessment and who have a baseline (Day 1) evaluation meeting study entry criteria and VVA criteria, namely < = 5% of superficial cells on vaginal smear, a vaginal pH above 5, and who have self-identified:
amoderate to severe (MS) pain at sexual activity as their most bothersome symptom (MBS)
bmoderate to severe (MS) pain at sexual activity (MBS or not MBS)
cP value from a paired t-test.
Source: ERC-230 Clinical Study Report.8
Vaginal Dryness
Vaginal dryness was tested as a second-order end point dependent on statistical significance of change in symptom score of dyspareunia in the ERC-238 and ERC-231 trials. As both the ERC-238 and ERC-231 trials demonstrated statistically significant improvement with prasterone in symptom score for dyspareunia, vaginal dryness was tested. A summary of change from baseline in vaginal dryness among patients who had moderate to severe dyspareunia and who also had moderate to severe vaginal dryness at baseline for the ERC-238 and ERC-231 trials is reported in Table 14; the corresponding data for the ERC-230 trial are reported in Table 15. The PP analyses results were similar to those for the ITT analyses in the ERC-238 and ERC-231 trials.
ERC-238
The mean change from baseline in severity score of vaginal dryness was greater for the prasterone group (–1.44; SD = 0.93) compared to the placebo group (–1.17; SD = 0.99) at 12 weeks; the MD for prasterone versus placebo was –0.27 (SD for MD not reported; P = 0.004). The PP analyses results were similar to those for the ITT analyses.
ERC-231
The mean change from baseline in severity score of vaginal dryness was similar for the prasterone group (–1.45; SD = 0.95) compared to the placebo group (–1.02; SD = 1.08) at 12 weeks; the MD for prasterone versus placebo was –0.43 (SD for MD not reported; P = 0.0128). The PP analyses results were similar to those for the ITT analyses.
ERC-230
The severity score of vaginal dryness among patients who reported moderate to severe vaginal dryness at baseline while also meeting criteria for superficial cells (≤ 5%) and vaginal pH (> 5.0) and who reported vaginal dryness as their most bothersome symptom were summarized (n = 81). The severity score of vaginal dryness at baseline was 2.19 (SD = 0.39) and 0.67 (SD = 0.81) at week 52; the mean change from baseline was –1.52 (SD = 0.78).
The severity score of vaginal dryness among patients who reported moderate to severe vaginal dryness at baseline were summarized (n = 251). The severity score of vaginal dryness at baseline was 2.22 (SD = 0.42) and 0.59 (SD = 0.74) at week 52; the mean change from baseline was –1.63 (SD = 0.79).
Table 14: Vaginal Dryness (ITT Population With Moderate to Severe Vaginal Dryness at Baseline) — ERC-238 and ERC-231
Study detail | ERC-238 | ERC-231 | ||
|---|---|---|---|---|
Prasterone N = 273 | Placebo N = 132 | Prasterone N = 62 | Placebo N = 60 | |
Baseline, mean (SD) | 2.30 (0.46) | 2.30 (0.46) | 2.37 (0.49) | 2.33 (0.48) |
Week 12, mean (SD) | 0.86 (0.87) | 1.13 (0.88) | 0.92 (0.80) | 1.32 (0.95) |
Mean change from baseline (SD) | –1.44 (0.93) | –1.17 (0.99) | –1.45 (0.95) | –1.02 (1.08) |
Mean difference from placebo (SD) | –0.27 (NR) | — | –0.43 (NR) | — |
P valuea | 0.004 | — | 0.0128 | — |
ANCOVA = analysis of covariance; ITT = intention to treat; SD = standard deviation.
Note: Vaginal dryness was tested as a second-order end point dependent on the statistical significance of change in symptom score of dyspareunia. Therefore, the primary symptom score parameter of dyspareunia demonstrated statistical significance in favour of prasterone before testing for vaginal dryness.
aANCOVA test with treatment group as the main factor and baseline value as the covariate (P value vs. placebo).
Source: ERC-238 Clinical Study Report10 and ERC-231 Clinical Study Report.9
Table 15: Vaginal Dryness — ERC-230
Study detail | ERC-230 | |
|---|---|---|
MBS vaginal dryness subgroupa N = 81 | MS vaginal dryness subgroupb N = 251 | |
Baseline | ||
Mean (SD) | 2.19 (0.39) | 2.22 (0.42) |
Week 52 | ||
Mean (SD) | 0.67 (0.81) | 0.59 (0.74) |
Change from baseline, mean (SD) | –1.52 (0.78) | –1.63 (0.79) |
P valuec | < 0.0001 | < 0.0001 |
MBS = most bothersome symptom; MS = moderate to severe; SD = standard deviation.
Note: Change from baseline in symptom score of vaginal dryness was a secondary end point and was not adjusted for multiplicity.
a, bPatients from the safety population who have at least 1 post-baseline efficacy assessment and who have a baseline (day 1) evaluation meeting study entry criteria and vulvovaginal atrophy criteria, namely superficial cells on vaginal smear were 5% or less, vaginal pH was greater than 5, and who have self-identified:
aMS vaginal dryness as their MBS.
bMS vaginal dryness (MBS or not MBS).
cP value from a paired t-test.
Source: ERC-230 Clinical Study Report.8
Vaginal and/or Vulvar Irritation or Itching
In the ERC-238 and ERC-231 trials, vaginal irritation or itching was tested as a third-order end point dependent on statistical significance of vaginal dryness; as vaginal dryness demonstrated statistical significance in these trials, analyses of vaginal irritation or itching were conducted. This was a secondary end point in the ERC-230 trial and was not adjusted for multiplicity. A summary of change from baseline in vaginal irritation or itching for the ERC-238 and ERC-231 trials is reported in Table 16; the corresponding data for the ERC-230 trial are reported in Table 17. The PP analyses results were similar to those for the ITT analyses in the ERC-238 and ERC-231 trials.
ERC-238
The mean change from baseline in moderate to severe vulvovaginal irritation or itching was greater for the prasterone group (–1.56; SD = 0.99) compared to the placebo group (–1.36; SD = 1.11) at 12 weeks; the MD for prasterone versus placebo was –0.06 (SD for MD not reported; P = 0.6404). The PP analyses results were similar to those for the ITT analyses.
ERC-231
The mean change from baseline in moderate to severe vulvovaginal irritation or itching was greater for the prasterone group (–1.36; SD = 1.11) compared to the placebo group (–1.09; SD = 0.90) at 12 weeks; the MD for prasterone versus placebo was –0.27 (SD for MD not reported; P = 0.1976). The PP analyses results were similar to those for the ITT analyses.
ERC-230
A total of 23 patients reported moderate to severe irritation or itching at baseline as the most bothersome symptom at baseline. The mean severity score was 2.13 (SD = 0.34) at baseline and 0.74 (SD = 0.69) at week 52; the mean change from baseline was –1.39 (SD = 0.78).
A total of 86 patients reported moderate to severe irritation or itching at baseline while meeting the criteria for superficial cells (≤ 5.0%) and vaginal pH (> 5.0) at baseline. The mean severity score of moderate to severe itching was 2.10 (SD = 0.31) units at baseline and 0.60 (SD = 0.74) at week 52; the mean change from baseline was –1.50 (SD = 0.82).
There was a total of 63 patients who had moderate to severe irritation or itching at baseline but who did not consider moderate to severe itching as their most bothersome symptom at baseline. The mean severity score of moderate to severe itching was 2.10 (SD = 0.76) at baseline and 0.56 (SD = 0.84) at week 52; the mean change from baseline was –1.54 (SD = 0.84).
Table 16: Vulvovaginal Irritation or Itching (ITT Population With Moderate to Severe Vulvovaginal Irritation or Itching at Baseline) — ERC-238 and ERC-231
Study detail | ERC-238 | ERC-231 | ||
|---|---|---|---|---|
Prasterone | Placebo | Prasterone | Placebo | |
Moderate to severe vulvovaginal irritation or itching | ||||
N | 126 | 64 | 25 | 23 |
Baseline, mean (SD) | 2.33 (0.47) | 2.20 (0.41) | 2.24 (0.44) | 2.35 (0.49) |
Week 12, mean (SD) | 0.78 (0.90) | 0.70 (0.83) | 0.88 (0.88) | 1.26 (0.96) |
P valuea | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
Mean change from baseline (SD) | –1.56 (0.99) | –1.50 (0.93) | –1.36 (1.11) | –1.09 (0.90) |
Mean difference from placebo (SD) | –0.06 (NR) | — | –0.27 (NR) | — |
P valueb | 0.6404 | — | 0.1976 | — |
ANCOVA = analysis of covariance; ITT = intention to treat; SD = standard deviation.
Note: Vaginal irritation or itching was tested as a third-order end point dependent on statistical significance of vaginal dryness. Therefore, the test for vaginal dryness demonstrated statistical significance in favour of prasterone before testing for vaginal irritation or itching.
aP value from a paired t-test (P value vs. baseline).
bANCOVA test with treatment group as the main factor and baseline value as the covariate (P value vs. placebo).
Source: ERC-238 Clinical Study Report10 and ERC-231 Clinical Study Report.9
Table 17: Vulvovaginal Irritation or Itching — ERC-230
Study detail | ERC-230 | ||
|---|---|---|---|
MBS VVA irritation/itching subgroupa (N = 23) | MS VVA irritation/itching subgroupb (N = 86) | MS VVA irritation/itching subgroup without MBS VVA irritation/itching at baselinec (N = 63) | |
Baseline | |||
Mean (SD) | 2.13 (0.34) | 2.10 (0.31) | 2.10 (0.30) |
Week 52 | |||
Mean (SD) | 0.74 (0.69) | 0.60 (0.74) | 0.56 (0.76) |
Change from baseline, mean (SD) | –1.39 (0.78) | –1.50 (0.82) | –1.54 (0.84) |
P valued | < 0.0001 | < 0.0001 | < 0.0001 |
MBS = most bothersome symptom; MS = moderate to severe; SD = standard deviation; VVA = vulvovaginal atrophy.
Note: Change from baseline in symptom score of dyspareunia was a secondary end point and was not adjusted for multiplicity.
a-c Patients from the safety population who have at least 1 post-baseline efficacy assessment and who have a baseline (day 1) evaluation meeting study entry criteria and VVA criteria, namely superficial cells on vaginal smear were 5% or less, vaginal pH was greater than 5, and who have self-identified:
aMS vulvovaginal irritation or itching as their MBS.
bMS vulvovaginal irritation or itching (MBS or not MBS).
cMS vulvovaginal irritation or itching as not being MBS.
dP value from a paired t-test.
Source: ERC-230 Clinical Study Report.8
Sexual Function
A summary of results for the ERC-238 and ERC-230 trials are reported in Table 18 and Table 19, respectively. Sexual function was considered a secondary outcome and was not controlled for multiplicity.
ERC-238
The mean baseline scores for the FSFI total score were 14.29 (SD = 6.49) and 14.25 (SD = 6.50) in the prasterone and placebo groups, respectively. Both groups showed improvement with increased total scores of 23.14 (SD = 8.11) in the prasterone group and 20.53 (SD = 8.43) in the placebo group at week 12. The mean change from baseline was greater in the prasterone group (8.85; SD = 7.85) than the placebo group (6.28; SD = 8.31). The trend of improvement from baseline to week 12 was consistent for both treatment groups for all domains of the FSFI; in addition, the mean change from baseline was greater for the prasterone group than the placebo group for all domains. The PP analyses results were similar to those for the ITT analyses.
ERC-230
The mean baseline score for the FSFI total score was 13.43 (SD = 7.54) and 21.50 (SD = 9.96) at week 52; the mean change from baseline was 8.08 (SD = 8.84). Findings for individual domains of the FSFI are shown in Table 26.
Table 18: FSFI (ITT Population) — ERC-238
Study detail | Prasterone (N = 300) | Placebo (N = 149) |
|---|---|---|
Total score | ||
Baseline, mean (SD) | 14.29 (6.49) | 14.25 (6.50) |
Week 12, mean (SD) | 23.14 (8.11) | 20.53 (8.43) |
Change from baseline, mean (SD) | 8.85 (7.85) | 6.28 (8.31) |
P valuea | 0.0006 | — |
Desire domain | ||
N | 325 | 157 |
Baseline, mean (SD) | 2.58 (1.15) | 2.64 (1.15) |
Week 12, mean (SD) | 3.32 (1.09) | 3.11 (1.17) |
Change from baseline, mean (SD) | 0.74 (1.11) | 0.47 (1.06) |
P valuea | 0.0105 | — |
Arousal domain | ||
N | 325 | 157 |
Baseline, mean (SD) | 2.57 (1.42) | 2.59 (1.48) |
Week 12, mean (SD) | 3.74 (1.56) | 3.33 (1.55) |
Change from baseline, mean (SD) | 1.17 (1.54) | 0.73 (1.72) |
P valuea | 0.0022 | — |
Lubrication domain | ||
N | 325 | 157 |
Baseline, mean (SD) | 2.00 (1.80) | 1.91 (1.23) |
Week 12, mean (SD) | 4.13 (1.72) | 3.53 (1.76) |
Change from baseline | 2.12 (1.82) | 1.61 (1.88) |
P valuea | 0.0005 | — |
Orgasm domain | ||
N | 325 | 157 |
Baseline, mean (SD) | 2.53 (1.74) | 2.41 (1.71) |
Week 12, mean (SD) | 3.80 (1.81) | 3.42 (1.89) |
Change from baseline, mean (SD) | 1.27 (1.85) | 1.01 (1.97) |
P valuea | 0.0470 | — |
Satisfaction domain | ||
N | 300 | 149 |
Baseline, mean (SD) | 2.84 (1.46) | 2.92 (1.46) |
Week 12, mean (SD) | 4.21 (1.50) | 3.80 (1.63) |
Change from baseline, mean (SD) | 1.37 (1.54) | 0.89 (1.51) |
P valuea | 0.0012 | — |
Pain domain | ||
N | 325 | 157 |
Baseline, mean (SD) | 1.61 (1.24) | 1.68 (1.28) |
Week 12, mean (SD) | 3.82 (2.03) | 3.24 (2.06) |
Change from baseline, mean (SD) | 2.21 (2.03) | 1.56 (1.88) |
P valuea | 0.001 | — |
ANCOVA = analysis of covariance; FSFI = Female Sexual Function Index; ITT = intention to treat; SD = standard deviation.
aANCOVA test with treatment group as the main factor and baseline value as the covariate (P value vs. placebo). Analyses of the FSFI were not adjusted for multiplicity in the overall statistical hierarchy of the trials.
Source: ERC-238 Clinical Study Report.10
Table 19: Female Function Sexual Index — ERC-230
Study detail | Prasterone |
|---|---|
All patients (N = 154) | |
Total score | |
N | 148 |
Baseline, mean (SD) | 13.43 (7.54) |
Week 52, mean (SD) | 21.50 (9.96) |
Change from baseline, mean (SD) | 8.08 (8.84) |
P valuea | < 0.0001 |
Desire domain | |
N | 154 |
Baseline, mean (SD) | 2.39 (1.07) |
Week 52, mean (SD) | 3.06 (1.20) |
Change from baseline, mean (SD) | 0.67 (1.06) |
P valuea | < 0.0001 |
Arousal domain | |
N | 154 |
Baseline, mean (SD) | 2.22 (1.65) |
Week 52, mean (SD) | 3.32 (1.96) |
Change from baseline, mean (SD) | 1.10 (1.71) |
P valuea | < 0.0001 |
Lubrication domain | |
N | 154 |
Baseline, mean (SD) | 1.69 (1.45) |
Week 52, mean (SD) | 3.63 (2.20) |
Change from baseline, mean (SD) | 1.94 (2.08) |
P valuea | < 0.0001 |
Orgasm domain | |
N | 154 |
Baseline, mean (SD) | 2.33 (1.91) |
Week 52, mean (SD) | 3.53 (2.22) |
Change from baseline, mean (SD) | 1.20 (2.07) |
P valuea | < 0.0001 |
Satisfaction domain | |
N | 148 |
Baseline, mean (SD) | 2.81 (1.43) |
Week 52, mean (SD) | 3.95 (1.76) |
Change from baseline, mean (SD) | 1.15 (1.76) |
P valuea | < 0.0001 |
Pain domain | |
N | 154 |
Baseline, mean (SD), | 1.70 (1.80) |
Week 52, mean (SD) | 3.53 (2.49) |
Change from baseline, mean (SD) | 1.83 (2.28) |
P valuea | < 0.0001 |
SD = standard deviation.
Note: Analyses conducted using the safety population for efficacy analyses.
aP value from a paired t-test. A statistical hierarchy was not implemented for analyses of the Female Sexual Function Index in the ERC-230 trial.
Source: ERC-230 Clinical Study Report.8
Urinary Symptoms
Urinary symptoms were not assessed in the ERC-238, ERC-231, and ERC-230 trials.
Depression
Depression was not assessed in the ERC-238, ERC-231, and ERC-230 trials.
Anxiety
Anxiety was not assessed in the ERC-238, ERC-231, and ERC-230 trials.
Vaginal Cell Maturation
Vaginal cell maturation was assessed using the change from baseline in percentages of parabasal and superficial cells. These end points were 2 of 4 coprimary end points of the ERC-238 and ERC-231 trials. A summary of results for change from baseline to the end of treatment period (week 12) of parabasal cells in the ERC-238 and ERC-231 trials is reported in Table 20. The summary of results for change from baseline to the end of treatment period of parabasal and superficial cells in the ERC-230 trial is reported in Table 21. The PP analyses results were similar to those for the ITT analyses in the ERC-238 and ERC-231 trials.
ERC-238
The mean change from baseline in the percentage of parabasal cells was greater for the prasterone group (–41.51%; SD = 36.26%) compared to the placebo group (–11.98%; SD = 29.58) at 12 weeks; the MD for prasterone versus placebo was –29.53 (SD for MD not reported; P < 0.001).
The mean change from baseline in the percentage of superficial cells was greater for the prasterone group (10.20%; SD = 10.35) compared to the placebo group (1.75%; SD = 3.33) at 12 weeks; the MD for prasterone versus placebo was 8.46% (SD for MD not reported; P < 0.001).
ERC-231
The mean change from baseline in the percentage of parabasal cells was greater for the prasterone group (–47.40%; SD = 42.50) compared to the placebo group (–1.62%; SD = 28.22) at 12 weeks; the MD for prasterone versus placebo was –45.77% (SD for MD not reported; P < 0.0001).
The mean change from baseline in the percentage of superficial cells was greater for the prasterone group (5.62%; SD = 5.49) compared to the placebo group (0.91%; SD = 2.69) at 12 weeks; the MD for prasterone versus placebo was 4.71% (SD for MD not reported; P < 0.0001).
ERC-230
The mean chance from baseline to week 52 in percentage of parabasal cells among all patients who were treated with prasterone was –42.67% (SD = 39.23). The percent change in parabasal cells were also analyzed in a group of 292 patients who had dyspareunia, vaginal dryness or irritation, or itching as their most bothersome symptom. The mean change from baseline to week 52 of parabasal cells among all patients treated with prasterone was –49.14% (SD = 37.91).
The mean change from baseline to week 52 in percentage of superficial cells among all patients who were treated with prasterone was 7.41% (SD = 8.06). The percent change in superficial cells was also analyzed in a group of 292 patients who had dyspareunia, vaginal dryness or irritation, or itching as their most bothersome symptom. The mean change from baseline of superficial cells among all patients treated with prasterone was 7.85% (SD = 7.15).
Table 20: Percentage of Parabasal and Superficial Cells (ITT Population) — ERC-238 and ERC-231
Study detail | ERC-238 | ERC-231 | ||
|---|---|---|---|---|
Prasterone (N = 325) | Placebo (N = 157) | Prasterone (N = 81) | Placebo (N = 77) | |
Percentage of parabasal cells | ||||
Baseline, mean (SD) | 54.25 (38.64) | 51.66 (37.60) | 65.05 (41.69) | 68.48 (38.66) |
Week 12, mean (SD) | 12.74 (18.44) | 39.68 (33.57) | 17.65 (25.87) | 66.86 (38.32) |
Mean change from baseline to week 12 (SD) | –41.51 (36.26) | –11.98 (29.58) | –47.40 (42.50) | –1.62 (28.22) |
Mean difference from placebo (SD) | –29.53 (NR) | — | –45.77 (NR) | — |
P valuea | < 0.0001 | — | < 0.0001 | — |
Percentage of superficial cells | ||||
Baseline, mean (SD) | 1.02 (1.44) | 1.04 (1.40) | 0.68 (1.10) | 0.73 (1.33) |
Week 12, mean (SD) | 11.22 (10.18) | 2.78 (3.37) | 6.30 (5.33) | 1.64 (2.88) |
Mean change (SD) | 10.20 (10.35) | 1.75 (3.33) | 5.62 (5.49) | 0.91 (2.69) |
Mean difference from placebo (SD) | 8.46 (NR) | — | 4.71 (NR) | — |
P valuea | < 0.0001 | — | < 0.0001 | — |
ANCOVA = analysis of covariance; DHEA = dehydroepiandrosterone; ITT = intention to treat; SD = standard deviation.
aANCOVA test with treatment group as the main factor and baseline value as the covariate. The P values of the 4 coprimary end points (vaginal pH, percent parabasal cells, percent superficial cells, and symptom score of dyspareunia) were not adjusted for multiplicity as statistical significance of each coprimary end point was required to form a conclusion of superiority of DHEA over placebo.
Source: ERC-238 Clinical Study Report10 and ERC-231 Clinical Study Report.9
Table 21: Percentage of Parabasal and Superficial Cells — ERC-230
Study detail | ERC-230 | |
|---|---|---|
All safety population N = 487 | Population with MBS of dyspareunia, vaginal dryness, or irritation/itching N = 293 | |
Percentage of parabasal cells | 454 | 292 |
Baseline, mean (SD) | 55.49 (42.30) | 63.95 (41.16) |
Week 52, mean (SD) | 12.81 (20.57) | 14.80 (21.73) |
Mean change from baseline to week 52 (SD) | –42.67 (39.23) | –49.14 (37.91) |
P valuea | < 0.0001 | < 0.0001 |
Percentage of superficial cells | 454 | 292 |
Baseline, mean (SD) | 2.02 (3.96) | 0.96 (1.38) |
Week 52, mean (SD) | 9.42 (7.60) | 8.81 (7.07) |
Mean change from baseline (SD) | 7.41 (8.06) | 7.85 (7.15) |
P valuea | < 0.0001 | < 0.0001 |
MBS = most bothersome symptom; SD = standard deviation.
Note: Change from baseline in percentage of parabasal cells was a secondary end point and was not adjusted for multiplicity.
aP value from a paired t-test.
Source: ERC-230 Clinical Study Report.8
Vaginal pH
A summary of results for change from baseline to week 12 of vaginal pH in the ERC-238 and ERC-231 trials is reported in Table 22. The summary of results for change from baseline to the end of treatment period of parabasal cells in the ERC-230 trial is reported in Table 23. The PP analyses results were similar to those for the ITT analyses in the ERC-238 and ERC-231 trials.
ERC-238
The mean change from baseline in vaginal pH was greater for the prasterone group (–0.94; SD = 0.94) compared to the placebo group (–0.27; SD = 0.74) at 12 weeks; the MD for prasterone versus placebo was –0.67 (SD for MD not reported; P < 0.001).
ERC-231
The mean change from baseline in vaginal pH was greater for the prasterone group (–1.04; SD = 1.00) compared to the placebo group (–0.21; SD = 0.69) at 12 weeks; the MD for prasterone versus placebo was –0.83 (SD for MD not reported; P < 0.001).
ERC-230
The mean chance from baseline to week 52 in vaginal pH among all patients who were treated with prasterone was –1.14 (SD = 0.96). The percent change in parabasal cells were also analyzed in a group of 293 patients who had dyspareunia, vaginal dryness or irritation, or itching as their most bothersome symptom. The mean change from baseline to week 52 of parabasal cells among all patients treated with prasterone for this subgroup was –1.27 (SD = 0.90).
Table 22: Vaginal pH (ITT Population) — ERC-238 and ERC-231
Study detail | ERC-238 | ERC-231 | ||
|---|---|---|---|---|
DHEA 0.5% N = 325 | Placebo N = 157 | DHEA 0.5% N = 81 | Placebo N = 77 | |
Vaginal pH | ||||
Baseline, mean (SD) | 6.34 (0.65) | 6.32 (0.66) | 6.47 (0.64) | 6.51 (0.59) |
Week 12, mean (SD) | 5.39 (0.94) | 6.05 (0.89) | 5.43 (0.94) | 6.31 (0.81) |
Mean change (SD) | –0.94 (0.94) | –0.27 (0.74) | –1.04 (1.00) | –0.21 (0.69) |
Mean difference from placebo (SD) | –0.67 (NR) | — | –0.83 (NR) | — |
P valuea | < 0.0001 | — | < 0.0001 | — |
ANCOVA = analysis of covariance; DHEA = dehydroepiandrosterone; ITT = intention to treat; SD = standard deviation.
aANCOVA test with treatment group as the main factor and baseline value as the covariate. The P values of the 4 coprimary end points (vaginal pH, percent parabasal cells, percent superficial cells and symptom score of dyspareunia) were not adjusted for multiplicity as statistical significance of each coprimary end point was required to form a conclusion of superiority of DHEA over placebo.
Source: ERC-238 Clinical Study Report10 and ERC-231 Clinical Study Report.9
Table 23: Vaginal pH — ERC-230
Study detail | ERC-230 | |
|---|---|---|
DHEA 0.5% All safety population N = 487 | MBS dyspareunia, vaginal dryness, or irritation/itching N = 293 | |
Vaginal pH | 457 | 293 |
Baseline, mean (SD) | 6.23 (0.79) | 6.40 (0.65) |
Week 52, mean (SD) | 5.09 (0.82) | 5.13 (0.83) |
Mean change from baseline (SD) | –1.14 (0.96) | –1.27 (0.90) |
P valuea | < 0.0001 | < 0.0001 |
DHEA = dehydroepiandrosterone; MBS = most bothersome symptom; SD = standard deviation.
Note: Change from baseline in vaginal pH was a secondary end point and was not adjusted for multiplicity.
aP value from a paired t-test.
Source: ERC-230 Clinical Study Report.8
Harms
Only those harms identified in the review protocol are reported below. See Table 24 for detailed harms data.
Table 24: Summary of Harms (Safety Population)
Study detail | ERC-238 | ERC-231 | ERC-230 | ||
|---|---|---|---|---|---|
DHEA 0.5% N = 374 | Placebo N = 180 | DHEA 0.5% N = 87 | Placebo N = 80 | All patients N = 521 | |
Patients with ≥ 1 adverse event | 179 (47.9) | 77 (42.8) | 46 (52.9) | 35 (43.8) | 418 (80.2) |
Most common eventsa | |||||
Application site discharge | 23 (6.1) | 10 (5.6) | 5 (5.7) | 5 (6.3) | 73 (14.0) |
Urinary tract infection | 17 (4.5) | 5 (2.8) | 5 (5.7) | 4 (5.0) | 53 (10.2) |
Weight increased | 15 (4.0) | 4 (2.2) | < 1% | < 1% | 11 (2.1) |
Weight decreased | 11 (2.9) | 6 (3.3) | < 1% | < 1% | 20 (3.8) |
Nasopharyngitis | 8 (2.1) | 1 (0.6) | 1 (1.1) | 5 (6.3) | 51 (9.8) |
Cervical dysplasia | 7 (1.9) | 0 (0.0) | 2 (2.5) | 2 (2.5) | 20 (3.8) |
Abdominal pain | 6 (1.6) | 2 (1.1) | 0 (0.0) | 0 (0.0) | 16 (3.1) |
Headache | 6 (1.6) | 3 (1.7) | 5 (5.7) | 1 (1.3) | 19 (3.6) |
Hot flush | 6 (1.6) | 7 (3.9) | 1 (1.3) | 2 (2.3) | 23 (4.4) |
Nausea | 3 (0.8) | 4 (2.2) | 4 (4.6) | 1 (1.3) | 16 (3.1) |
Arthralgia | < 1% | < 1% | 2 (2.3) | 3 (3.8) | 14 (2.7) |
Back pain | < 1% | < 1% | 0 (0.0) | 1 (1.3) | 22 (4.2) |
Sinusitis | < 1% | < 1% | 1 (1.1) | 2 (2.5) | 20 (3.8) |
Vulvovaginal candidiasis | < 1% | < 1% | 3 (3.4) | 0 (0.0) | 7 (1.3) |
Vulvovaginal burning sensation | < 1% | < 1% | 1 (1.1) | 3 (3.8) | 6 (1.2) |
Influenza | < 1% | < 1% | < 1% | < 1% | 16 (3.1) |
Patients with ≥ 1 SAE | 6 (1.6) | 0 (0.0) | 1 (1.1) | 0 (0.0) | 18 (3.5) |
Patients who stopped treatment due to adverse events | 5 (1.3) | 5 (2.8) | 1 (1.1) | 1 (1.3) | 31 (6.0) |
Discontinuation due to SAE | 2 (0.5) | 0 (0.0) | 1 (1.1) | 0 (0.0) | 9 (1.7) |
Deaths | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms | |||||
Vaginal hemorrhage | 4 (1.1) | 2 (1.1) | 0 (0.0) | 2 (2.5) | 13 (2.5) |
Endometrial dysplasia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Cervical dysplasia | 7 (1.9) | 0 (0.0) | 3 (3.4) | 2 (2.5) | 20 (3.8) |
Breast mass | 1 (0.3) | 0 (0.0) | NR | NR | 2 (0.4) |
DHEA = dehydroepiandrosterone; SAE = serious adverse event.
Note: Values are presented as n (%).
aAdverse events reported in 3% or more by patients in any group.
Source: ERC-238 Clinical Study Report,10 ERC-231 Clinical Study Report,9 and ERC-230 Clinical Study Report.8
Adverse Events
The proportion of patients reporting at least 1 AE between treatment groups in the ERC-238 trial were similar between treatment groups, with 179 patients (47.9%) in the prasterone group and 77 patients (42.8%) in the placebo group reporting at least 1 AE of any grade. There was a higher proportion of patients in in the prasterone group with at least 1 AE than in the placebo group in the ERC-231 trial; 46 patients (52.9%) in the prasterone group and 35 patients (43.8%) in the placebo group reported at least 1 AE of any grade. A greater proportion of AEs were reported in the ERC-230 trial with 418 patients (80.2%) experiencing AEs of any grade.
The most commonly reported (≥ 3% in any treatment group) AEs in the ERC-238 trial included application site discharge (6.1% of patients in the prasterone group and 5.6% in the placebo group), urinary tract infection (4.5% versus 2.8%, respectively), weight gain (4.0% versus 2.2%, respectively), weight loss (2.9% versus 3.3%, respectively), and hot flush (1.6% versus 3.9%, respectively). The most commonly reported AEs in the ERC-231 trial included application site discharge (5.7% versus 6.3% in the prasterone and placebo groups, respectively), urinary tract infection (5.7% versus 5.0%, respectively), nasopharyngitis (1.1% versus 6.3%, respectively), headache (5.7% versus 1.3%, respectively), nausea (4.6% versus 1.3%, respectively), arthralgia (2.3% versus 3.8%, respectively), and vulvovaginal candidiasis (3.4% versus 0%, respectively). The most commonly reported AEs in the ERC-230 trial included application site discharge (14.0%), urinary tract infection (10.2%), weight loss (3.8%), nasopharyngitis (9.8%), cervical dysplasia (3.8%), abdominal pain (3.1%), headache (3.6%), hot flush (4.4%), nausea (3.1%), back pain (4.2%), and sinusitis (3.8%). In general, application site discharge and urinary tract infections were the most commonly reported AEs across all trials.
Serious Adverse Events
SAEs were infrequently reported across trials. In the ERC-238 trial, 1.6% of patients in the prasterone group experienced an SAE compared to 0 patients in the placebo group. In the ERC-231 trial, 1.1% of patients experienced an SAE compared to 0 patients in the placebo group. In the ERC-230 trial, SAEs occurred in 3.5% of patients.
Discontinuations Due to AEs
Few patients discontinued treatment due to an AE across all trials. In the ERC-238 trial, 1.3% of patients in the prasterone group versus 2.8% of patients in the placebo group discontinued treatment due to an AE. Of these patients, 0.5% in the prasterone group discontinued treatment due to an SAE. In the ERC-231 trial, 1.1% of patients in the prasterone group and 1.3% of patients in the placebo group discontinued treatment due to an AE. In the ERC-230 trial, 6.0% of patients discontinued treatment due to an AE.
Mortality
There were no deaths in any of the trials.
Notable Harms
Notable harms identified in the CADTH systematic review protocol included vaginal hemorrhage, endometrial dysplasia, cervical dysplasia, and breast mass. In general, few patients experienced notable harms reported as AEs across the ERC-238, ERC-231, and ERC-230 trials, and there was little-to-no difference in reporting of notable harms across treatment groups. Vaginal hemorrhage was reported among 1.1% of patients in the prasterone and placebo groups in the ERC-238 trial, 0 patients and 2.5% of patients in the prasterone and placebo groups, respectively, in the ERC-231 trial, and 2.5% of patients in the ERC-230 trial. Cervical dysplasia was reported among 1.9% of patients in the prasterone group versus 0 patients in the placebo group in the ERC-238 trial, 3.4% of patients in the prasterone group versus 2.5% of patients in the placebo group in the ERC-231 trial, and 3.8% of patients in the ERC-230 trial. Breast mass was reported in 0.3% of patients in the prasterone group versus 0 patients in the ERC-238 trial, 0.4% of patients in the ERC-230 trial, and 0 patients in the ERC-231 trial.
The ERC-230 trial also reported on breast, endometrial, and Pap smear safety. Endometrial safety was also reported in the ERC-231 trial.
Breast Safety
Breast examinations were conducted using mammograms at screening and at week 52 in the ERC-230 trial. A total of 451 patients (98%) had a mammogram; of these patients, 455 patients (99%) showed normal or no significant findings. Significant breast pathology was observed among 2 patients which included 1 case each of atypical ductal hyperplasia and infiltrating carcinoma. Undetermined status was reported among 2 patients; 1 patient refused follow-up and findings from the other patient were reported as being probably benign and this patient was instructed to have a follow-up exam 6 months later. The results of the remaining 15 women were reported to be benign. In general, normal breast findings were observed for women who received long-term treatment with prasterone.
Pap Smear Safety
In general, long-term administration of prasterone in the ERC-230 trial was not associated with cervical dysplasia. Pap smears were conducted for patients who received prasterone for 26 weeks or longer. A terminal Pap smear was conducted for 430 of 432 patients who received prasterone for 52 weeks (90%). A total of 13 patients yielded results of ASCUS, low grade squamous intraepithelial lesion, or high grade squamous intraepithelial lesion. Of these 13 patients, 7 had a negative HPV test or colposcopy.8
Endometrial Safety
In the ERC-231 trial, approximately 40% of patients were non-hysterectomized and underwent an endometrial biopsy at screening (31 to 25 patients per treatment group). Almost all non-hysterectomized patients (99%), including 28 patients in the prasterone group and 27 patients in the placebo group, underwent an endometrial biopsy at week 12; 5 patients in the prasterone group and 2 patients in the placebo group did not have sufficient tissue for biopsy at this time. At week 12, the endometrium of all evaluable patients was atrophic, and the sponsor reported no clinically significant results.9 In the ERC-230 trial, endometrial biopsies were performed for patients who received prasterone for 3 months or longer. For patients with unevaluable endometrial biopsies or who reused endometrial biopsies at the end of treatment, transvaginal ultrasounds were performed; this was performed for 43 patients. In total, 457 patients (94%) had a biopsy at the end of the 52-week study period. The endometrium of most patients (91%) was atrophic. Among the 43 patients who underwent a transvaginal ultrasound, the average endometrial thickness was 2.2 mm (SD = 1.4). There were no clinically significant histological findings in the ERC-230 trial with long-term use of prasterone.8
Critical Appraisal
Internal Validity
Both the ERC-238 and ERC-231 trials were DB, randomized, phase III trials; features of the randomization (computer-generated permuted block design) and central allocation mean that the trials are unlikely to be affected by selection bias. Additionally, the lack of important differences in baseline characteristics across groups signal that the randomization was successful. The DB (patients, personnel and investigators) nature of the trials is advantageous in reducing the risk of performance and detection bias related to knowledge of treatment assignment from patients and investigators. The sponsor reported that no unintentional unblinding occurred for either the ERC-238 or ERC-231 trials, and since the treatment administration of both prasterone and placebo were identical and the AEs were similar between treatment groups, it is unlikely that unblinding occurred. The ERC-230 trial used an open-label, single-group design to evaluate treatment with prasterone among post-menopausal patients with VVA. Since this study lacks a comparison (control) group and there is no control for potential confounding variables, causal relationships cannot be established. The trial was open label; therefore, all patients were aware of treatment assignment, and all patients received the same therapy. It is possible that knowledge of the treatment would lead patients to overestimate both its potential benefits and potential harms. A greater proportion of patients in the ERC-230 trial reported AEs as compared to the other 2 trials, but it is not clear whether this is related to the open-label design, increased duration of exposure to placebo as compared to the ERC-231 and ERC-238 trials, or other factors.
Eligibility criteria specified that patients were to be excluded if they received hormonal therapy within 6 months of the start of the trial. This was considered appropriate given the potential for residual treatment effects from hormonal therapy to influence trial results; however, the clinical expert consulted by CADTH for this review stated that a length of 3 months may have been sufficient to prevent any residual effects of prior hormonal therapies on patient outcomes in these trials.
The 4 coprimary end points of the ERC-238 and ERC-231 trials were change from baseline to 12 weeks in the following: percentage of parabasal cells, percentage of superficial cells, vaginal pH, and severity score of dyspareunia. No adjustments were made for multiple testing of these coprimary end points; this was appropriate because end points were required to be statistically significant in favour of DHEA to conclude superiority of DHEA over placebo. Further, secondary end points assessing vaginal dryness and vaginal irritation or itching were tested as second- and third-order end points, respectively; testing vaginal dryness was specified to occur only if the primary symptom score of dyspareunia was statistically significant, and testing of vaginal irritation or itching was specified to occur only if testing of vaginal dryness was statistically significant. This testing hierarchy allowed for control of type I error for these secondary end points. However, sample size calculations did not consider secondary end points of vaginal dryness and vaginal irritation or itching, these end points were only measured in patients with these symptoms as moderate to severe at baseline, and randomization methods did not incorporate stratification by these symptoms. The results of these end points should be interpreted with caution.
In efficacy analyses which compared changes from baseline to later time points, missing data were handled by using the LOCF method. It is possible that use of this method for handling of missing data may over- or underestimate the effectiveness of therapies. The exact impact of this potential source bias is uncertain, though the use of LOCF is less likely to lead to bias in favour of prasterone if treatment benefit is expected to improve over the course of the treatment period.
The FSFI was used to analyze sexual function in post-menopausal patients with VVA. In both the ERC-231 and ERC-230 trials, patients were to complete the MENQOL under the original protocol of these trials. After protocol amendments, patients were to instead complete the FSFI. No justification was provided as to why protocols were amended to use the FSFI instead of the MENQOL. Results were reported for patients in the ERC-230 trial; as only 29% of enrolled patients provided data for analyses related to the FSFI, these results are likely not powered and may be biased as data from most patients were unavailable. Results from the ERC-230 trial provide an indication of impacts of long-term treatment with DHEA on patients; however, the ERC-230 trial did not have a comparator group making interpretation of results difficult. Due to the limited number of patients who completed the FSFI in the ERC-231 trial, the results for these data were not provided. While the results of the ERC-231 trial were not available, results of the ERC-238 trial (for which the FSFI was a part of the original protocol) were reported and provide an indication of how treatment with prasterone may impact patients’ sexual function compared to placebo. In addition, no adjustments for multiplicity for these analyses were made as part of the overall testing scheme in any of the trials and results should be considered exploratory.
In addition, the placebo used in the trials may have had a beneficial effect on patients. Placebo ovules were administered in a capsule which may have had some moisturizing effects for patients. Therefore, the treatment effects of the prasterone ovule compared to a true placebo may be underestimated. In fact, results from efficacy analyses did reveal that patients in the placebo groups also experienced some benefit from the placebo as patients in the placebo group also reported improved symptoms, albeit not as great as patients in the prasterone group.
External Validity
Both the ERC-238 and ERC-231 trials were placebo-controlled trials. It was stated by the sponsor that placebo was an appropriate comparator as there is no standard funded therapy for treatment of post-menopausal VVA. Patients may use over-the-counter therapies, such as moisturizers and lubricants, to combat symptoms of VVA. However, the CADTH team noted that estrogen-based therapies are also available for post-menopausal patients with VVA. Specifically, Vagifem, an estradiol vaginal insert, would have been available during the inception of these trials. The clinical expert consulted by CADTH for this review agreed that there may have been other appropriate comparators that would have been of value to use in the trials, to allow for a better understanding of the efficacy of prasterone compared to other therapies that are available to patients.
The eligibility criteria of the trial were considered to be mostly appropriate and reflective of post-menopausal patients with VVA. However, it is typical that trial eligibility criteria can be restrictive and, ultimately, not representative of all patients in clinical practice. That eligibility criteria were overly restrictive is likely evident by the large number of patients who were considered screen failures; the high rate of screen failures may partially be due to guidance from the FDA recommending that enrolled patients identify at least 1 moderate to severe symptom that is most bothersome, have 5% or less superficial cells on a vaginal smear, and a vaginal pH of greater than 5.0. Patients with comorbidities were excluded from the ERC-238, ERC-231, and ERC-230 trials. In particular, patients with a history of cancer were excluded from the trials; this was considered a population of interest as post-menopausal women with history of cancer are still at risk for VVA and may benefit from non-hormonal therapies such as prasterone. The impact of treatment on patients with comorbidities is not clear. Although, based on comments from the clinical expert consulted by CADTH for this review, the findings are likely to be generalizable to patients seen in Canadian clinical practice.
In general, baseline characteristics of the trials were considered to be reflective of post-menopausal patients with VVA. However, it was noted that the majority of patients enrolled in all trials were mostly White and non-Hispanic or Latino. The clinical expert consulted by CADTH for this review commented that, while it is typical for clinical trials to enroll mostly White patients, this may not be reflective of all patients who may also be diagnosed with VVA. In particular, Canada is a multicultural and diverse population. Many Canadian patients who suffer from VVA and who are not White are likely not represented by the patients enrolled in the ERC-238, ERC-231, and ERC-230 trials.
The interventions assigned in the trials included prasterone and placebo. The protocols of the trials specified that patients should not take concurrent treatment with other moisturizers or lubricants. However, it was noted by the clinical expert consulted by CADTH for this review that patients may use these therapies in combination in the real world. The efficacy of prasterone in combination with moisturizers or lubricants is unknown.
The duration of the ERC-238 and ERC-231 trials was only 12 weeks. As the trial durations were short, the long-term benefits and harms of prasterone on patients is uncertain, and patients who are prescribed prasterone in clinical practice are likely to take this treatment for longer than 12 weeks. The ERC-230 trial was conducted for 52 weeks; however, the study is lacking a control group and thus does not allow for definitive conclusions about the effects of longer-term treatment to be drawn.
The 4 coprimary end points of the ERC-238 and ERC-231 trials, and secondary end points of the ERC-230 trial were decrease in percentage of parabasal cells, increase in the percentage of superficial cells, decrease in vaginal pH, and improvement in the severity score of dyspareunia as the most bothersome self-reported VVA symptom. The 4 coprimary end points also align with recommendations from the FDA which specify that these outcomes should be used for studies of this indication.33 Other secondary end points included analyses of vaginal dryness, vaginal irritation or itching, and sexual function assessed using the FSFI. The clinical expert consulting with CADTH for this review agreed that these outcomes were important for consideration to patients and clinicians in the treatment of post-menopausal VVA. However, end points such as the change of parabasal and superficial cells may not typically be assessed in clinical practice. End points which assess symptoms and severity of symptoms may be more relevant to patients.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise available indirect evidence comparing DHEA to other relevant treatments (identified in the protocol) for post-menopausal women experiencing VVA.
A focused literature search for NMAs dealing with prasterone was run in MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid on October 29, 2021. No limits were added to limit the search. Of 8 records identified by the CADTH literature search, 1 published NMA by Li et al.11 was included.
Description of Indirect Comparison
Li et al. conducted several NMAs to indirectly compare treatment with prasterone to other treatments for VVA among post- menopausal people. Selection criteria for studies to be included in the NMAs are described in Table 31.
Table 25: Study Selection Criteria and Methods for ITC
Characteristics | Li et al. (2021) |
|---|---|
Population | Women presenting with some of the signs, or all of the signs and symptoms associated with GSM
Excluded: Patients with underlying comorbidities such as pelvic organ prolapse, bacterial vaginosis, Trichomonas vaginalis, and Candidal vaginitis |
Intervention | Any treatment for VVA |
Comparator | None specified |
Outcome | Primary:
Secondary:
|
Study design | Randomized controlled trials |
Publication characteristics | No limits for language were imposed when searching PubMed. Retrieved citations were from database inception to date of the search (March 2020). |
FSFI = Female Sexual Function Index; GSM = genitourinary syndrome of menopause; ITC = indirect treatment comparison; VHI = Vaginal Health Index; VVA = vulvovaginal atrophy.
Source: Li et al. (2021).11
Methods of the ITC
Objectives
The aim of the NMA conducted by Li et al.11 was to identify the safest and most effective treatments for post-menopausal women having symptoms of GSM.
Study Selection Methods
The NMA by Li et al. was informed by an a priori registered protocol. A description of the eligibility criteria used to identify relevant studies in each of the NMAs is reported in Table 25. To identify relevant articles for inclusion in their NMA, Li et al.11 conducted a literature search in the following electronic databases from inception to March 2020: PubMed, Embase, Scopus, Cochrane Library, Web of Science, and ScienceDirect. The target population included women with symptoms of GSM. Interventions and comparators were not specified in by the authors, but it appears that any treatment for VVA was eligible. Included study designs were limited to RCTs published in any language.
A literature search was conducted based on criteria reported in Table 25. Studies were screened by title and abstract by 2 independent reviewers, and then considered further for inclusion into the NMA. A third independent reviewer conducted arbitration.11 Methods for data extraction were not reported. Risk of bias was appraised using the Cochrane Risk of Bias tool; the methods (e.g., number of reviewers involved) were not described.
ITC Analysis Methods
Details of the methodology used for the sponsor’s ITC are provided in Table 26. NMAs were undertaken using Bayesian random effects models, employing non-informative priors for the overall mean effect and between-study SDs. It is not clear how the authors assessed clinical and methodological heterogeneity across the studies within the NMAs. Nodes appear to have included similar treatments regardless of dose and duration. Model geometry was not shown and assessment of model fit was not reported; convergence was assessed using the Brooks-Gelman-Rubin method. Node splitting was used to assess consistency between direct and indirect effect estimates. Statistical heterogeneity was assessed using the I2 statistic: I2 less than 25% was considered by the authors to indicate no heterogeneity, 25% to 50% to indicate moderate heterogeneity, 50% to 75% to indicate high heterogeneity; and greater than 75% to indicate extremely high heterogeneity. Heterogeneity was explored using subgroup analyses and meta-regressions. The authors indicate that sensitivity analyses were performed to estimate the stability of the meta-analyses.
Table 26: ITC Analysis Methods
Detail | Li et al. (2021) |
|---|---|
ITC methods | An NMA was conducted using a Bayesian approach using random effect models. The arm-based approach was used to impute information, which was modelled by use of binomial data for AEs (binomial likelihood, logit link) or sample means for other outcomes (normal likelihood, identity link) with normal distribution. Markov chain Monte Carlo simulations with 50,000 iterations were conducted with a burn-in of 20,000 iterations. In addition, the DerSimonian and Laird random effect models with inverse-variance weights were used to complement the estimates obtained after Bayesian pairwise analysis. |
Priors | Non-informative or vague priors for the overall mean effect (θ ~ N [0, 1002]) and the between-study standard deviation (τ ~ uniform [0, 2]) were given. |
Assessment of model fit | Not reported. |
Assessment of consistency | The node-splitting model was used to assess consistency. Inconsistencies were assessed by comparing the between-study variance. A Bayesian P value was calculated to estimate the measure of conflict between direct and indirect evidence by evaluating the proportion of times the direct treatment effect exceeded the indirect treatment effect. |
Assessment of convergence | Convergence was assessed using the Brooks-Gelman-Rubin method. |
Outcomes | Vaginal dryness, vaginal burning and itching, dyspareunia, sexual function, vaginal pH, proportion of parabasal cells, and AEs. |
Construction of nodes | Treatments which were the same (regardless of dose or duration) were pooled together. |
Sensitivity analyses | Sensitivity analyses were performed to estimate the stability of the meta-analysis. The nature of these sensitivity analyses was not described. Sensitivity analyses which demonstrated a fundamental change in the heterogeneity of the findings of the NMA were considered to show poor stability. |
Subgroup analysis | Subgroup analyses and meta-regressions were conducted on demographic variables (year of publication, region/country, age of patients, history of breast or gynecological cancers) and for doses of treatments and treatment duration. |
Methods for pairwise meta-analysis | For comparative variables, the mean difference or odds ratio were computed where appropriate at a 95% confidence interval. The included treatments were ranked to define the probability associated with their effectiveness. |
Assessment of publication bias | Explored visually using funnel plots and statistically using Egger and Begg regression tests. |
AE = adverse event; ITC = indirect treatment comparison; NMA = network meta-analysis.
Source: Li et al. (2021).11
Results of the ITC
Summary of Included Studies
The literature search identified 29 trials which incorporated 8,311 patients evaluating the following treatments: laser therapy, vaginal estrogen, ospemifene, vaginal DHEA, and moisturization/lubrication.11 Vaginal DHEA and vaginal estrogen were relevant to this review.
Characteristics of study design revealed inclusion of both open-label and blinded RCTs. Trials were published between 1992 and 2020. All patients included in the trials had a mean age of 58 years to 60 years. All trials except 312-14,34 excluded patients with breast or gynecological cancers. Treatment duration was heterogeneous, with most trials assessing treatment for 12 weeks. Outcomes assessed included urinary and sexual outcomes (i.e., dryness, itching, dyspareunia, urinary tract infections), AEs, and health-related quality of life assessed through various tools. Different doses of treatments were also used in the 29 trials; specifically regarding DHEA, studies assessing doses of 0.5% (6.5 mg) and 0.25% (3.25 mg) were included.11
The authors did not report on the number of studies included for the NMA of any end points assessed (vaginal dryness, vaginal burning and itching, dyspareunia, sexual function, vaginal pH, proportion of parabasal cells, and AEs) nor on their risk of bias. It is not clear how the nodes were created, though it appears that similar treatments were merged regardless of dose and duration (including all vaginal estrogen therapies). The tool used to measure the end points across the included trials was not specified. The network structure was not described. The authors indicated that the model converged “adequately” but relevant data were not provided to support this assertion.
Risk of Bias
The risk of bias assessment revealed that most trials showed low or unclear risk of bias in the domains assessed (random sequence generation, allocation concealment, blinding of patients and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other bias). There was high risk of bias reported for random sequence generation for 4 studies, allocation concealment for 3 studies, blinding of patients for 6 studies, blinding of outcome assessment for 1 study, and incomplete outcome data for 3 studies.11
Results
The following treatments were considered in the NMA by Li et al.11: laser therapy, vaginal estrogen, ospemifene, vaginal DHEA, and moisturization/lubrication. As DHEA (prasterone) is the main treatment of consideration for this CADTH review, only results of comparisons between DHEA and other relevant treatments specified in the protocol (i.e., vaginal estrogen therapies) are presented.
Vaginal Dryness
No differences were observed between DHEA and vaginal estrogen therapy (MD = 0.32; 95% CrI, −8.54 to 8.77). The I2 value for heterogeneity was 0%, but the pairwise frequentist analyses showed high heterogeneity. Subgroup analyses did not seem to explain the heterogeneity for the comparisons of interest (DHEA versus other treatments). There did not appear to be any sensitivity analyses performed for this comparison. Publication bias was not detected.
Dyspareunia
Little-to-no difference was observed between DHEA and vaginal estrogen therapy (−4.00; 95% CrI, −13.88 to 4.46). The I2 value for heterogeneity was 11%.
Sexual Function (FSFI)
No differences were observed between DHEA and vaginal estrogen therapy (MD = 1.04; 95% CrI, −1.99 to 3.93). The I2 value for heterogeneity was 0%.
Vaginal pH
The I2 value for heterogeneity was 4%. Vaginal estrogen therapy (MD = 0.4; 95% CrI, 0.11 to 0.69) was favoured over DHEA.
Proportion of Parabasal Cells
No differences were observed between DHEA and vaginal estrogen therapy (MD = 1.6; 95% CrI, −12.45 to 13.84). The I2 value for heterogeneity was 9%.
Adverse Events
No difference was found between DHEA and vaginal estrogen therapy (odds ratio = 1.54; 95% CrI, 0.91 to 2.62). The I2 value for heterogeneity was less than 25% among treatments.
Critical Appraisal of the ITC
The systematic review to locate studies for inclusion in the NMAs followed an a priori developed protocol. Though several databases were searched up to March 2020, the authors did not search other sources (e.g., clinical trial registries) so it is possible that some relevant studies were missed, and those published after March 2020 would not have been included. Methods of data extraction were not described, so error within the findings is possible. Studies were assessed for risk of bias, but it is not clear how this assessment was carried out, so it is difficult to assess the validity of these assessments.
The eligibility criteria for inclusion of studies in the NMAs appears relevant, though these are described in minimal detail within the publication. All studies included in the NMA were RCTs. Characteristics of studies included in the NMA were somewhat varied as some trials were open-label while others were blinded. In addition, few trials also included patients who had history of breast and gynecological cancers; the clinical expert consulted by CADTH for this review suggested that patients with history of cancers may have worse outcomes compared to patients without history of cancers, which could potentially underestimate treatment effects. In addition, there may be additional concerns for safety due to contraindications with estrogen-based therapies among cancer patients, especially if they are hormone dependent. Treatment duration varied across trials with durations between 4 weeks and 52 weeks; most trials were 12 weeks. It is possible that patients who underwent longer durations of therapy experienced greater benefit from treatment. However, the main eligibility criteria of the trials suggest that, in general, all trials enrolled post-menopausal women with symptoms of GSM and that populations of women across trials may generally be consistent. Differences in trial and baseline characteristics are likely to have impacted the indirect comparisons; although, the exact effect of these difference is unclear. An assessment of similarity across trials in each NMA was not conducted; therefore, whether underlying assumptions of the NMAs (i.e., homogeneity and transitivity) have been met are uncertain.
The risk of bias assessment conducted by the authors suggested that most trials included in the NMA had low or unclear risk of bias. Of the 29 trials, few of them revealed high risk of bias; domains which were determined to have high levels of bias were regarding random sequence generation, allocation concealment, blinding of patients and personnel, blinding of outcome assessment, and incomplete outcome data among 11 of the trials. As it was not always clear which studies and how many patients were included within each NMA, it was not always possible to fully quantify the extent of potential bias within the analyses. It is possible in these cases that analyses were affected by substantial bias. Publication bias was not detected in any of the NMAs.
There was a lack of clear reporting regarding the construction of nodes in the NMAs. However, based on reported information, it was assumed that treatment doses, durations, and outcomes measures for single treatments were combined into single nodes. The combination of different doses, durations, and outcomes measures for treatments is likely to have introduced bias, as the efficacy and safety of treatments which may not have been administered or measured the same is uncertain. In particular, formulations of placebo were not consistent across trials and may not be considered equivalent. Combination of different placebo groups across different trials is likely not appropriate given the potentially different effects they may have in treating symptoms of VVA. No sensitivity analyses were conducted to explore the effects of treatments at different formulations, doses, durations, or measurements. The effect of this bias is unclear.
A number of outcomes were assessed in the NMAs including vaginal dryness, vaginal burning, vaginal itching, dyspareunia, FSFI, urinary incontinence, pH, vaginal health index (VHI), AEs, and endometrial thickness. However, DHEA was not incorporated in the analyses for some of these outcomes, including vaginal burning, vaginal itching, urinary incontinence, VHI, and endometrial thickening, likely due to lack of available data. The outcomes assessed in the NMA which included DHEA in the analyses are useful for patients and clinicians, as they captured efficacy, safety, and health-related quality of life. Conclusions cannot be drawn for comparisons between DHEA and other therapies for end points which did not include DHEA in the analyses.
The NMA by Li et al.11 was conducted using a random effects models. The random effects model is likely to be appropriate as it incorporates the assumption that studies are measuring different but related treatment effects. Because the authors provided no measures of model fit, it is unclear whether the random effects models would have been preferred over fixed effects models.
In many instances, the results of the NMAs showed a lack of difference between treatments, as CrI included values of 0 for associated MDs. CrIs were also wide, indicating the potential for substantial uncertainty between treatment comparisons. In general, the results of the NMA suggested that DHEA was favoured over placebo; these results are aligned with results from the ERC-238 and ERC-231 trials which are discussed in the main body of this CADTH report. Comparisons of DHEA to other therapies should be interpreted with caution, as it is uncertain how DHEA may compare to vaginal estrogen therapies if directly compared.
Statistical heterogeneity was assessed using an I2 statistic. I2 values less than 25% were pre-specified by the authors to indicate no heterogeneity. For analyses of end points which included DHEA, the I2 values were less than 25%, suggesting little or no heterogeneity. All models were also reported to have adequate convergence; however, data regarding convergence was not provided. The authors also conducted meta-regressions on demographic variables and study characteristics to identify sources of variation. The meta-regression revealed that dryness, pH, VHI, and proportion of parabasal cells showed high heterogeneities among frequentist pairwise comparisons. Subgroup and sensitivity analyses revealed sources of variation for each end point. It is probable that heterogeneity across trials may affect the confidence of results of the NMA.
Other Relevant Evidence
This section includes additional relevant studies included in the sponsor’s submission to CADTH, and additional studies identified in the CADTH literature search that were considered to address important gaps in the evidence included in the systematic review. The following studies were included as additional evidence: the ERC-210 trial,15 the Estip-Es study,16 and a study by Barton et al.12 The ERC-210 trial was a multi-centre, DB, randomized, placebo-controlled, phase III trial to determine the dose response of prasterone on symptoms and vaginal mucosa parameters in post-menopausal women with VVA. The Estip-Es study was an observational study conducted in Spain which evaluated the effectiveness and safety of prasterone in a real-world clinical setting.16 The study by Barton et al.12 examined the use prasterone for treatment of post-menopausal symptoms of VVA in patients with a history of breast or gynecological cancer. A summary and critical appraisal of these studies are provided below.
ERC-210 Trial
The ERC-210 trial is summarized here as supportive evidence for the efficacy and safety of prasterone 0.5% as it included patients with symptoms other than dyspareunia as the most bothersome symptom and had a placebo control group (unlike the ERC-230 trial).
Description of Study
The ERC-210 trial, which started in June 2007 and was completed in October 2008, was a multi-centre (US and Canada), prospective, DB, randomized, parallel assignment, placebo-controlled, phase III trial to determine the dose response of prasterone on symptoms and vaginal mucosa parameters in post-menopausal women with VVA. The study informed the dose of prasterone to use for the subsequent phase III studies. Post-menopausal women who self-identified as having at least 1 moderate to severe symptom of VVA were enrolled. A total of 217 patients were centrally randomized using a permuted block randomization scheme to receive prasterone at 1% (13 mg), 0.5% (6.5 mg), 0.25% (3.25 mg), or placebo. Only the data for prasterone at a dose of 0.5% are relevant to this review and reported here. The study was divided into 2 phases: a screening period of 4 weeks to 6 weeks followed by a treatment period of 12 weeks.
Populations
The inclusion and exclusion criteria for the ERC-210 trial were mostly the same as those for the ERC-231 and ERC-238 trials, which are described in Table 6. Briefly, the inclusion criteria of the ERC-210 trial specified post-menopausal women aged 40 years to 75 years who self-identified to have at least 1 moderate to severe symptom of VVA. Women had to have a low maturation index (≤ 5% of superficial cells on a vaginal smear) and a vaginal pH of greater than 5 at baseline. Exclusion criteria included undiagnosed abnormal genital bleeding, uncontrolled hypertension, endometrial hyperplasia, endometrial cancer, and use of estrogen or progesterone products in the 4 weeks to 6 weeks before study entry.
Baseline characteristics of randomized patients are presented in Table 27. Characteristics were similar between the prasterone 0.5% and placebo groups. Most patients were White (96%), most were non-hysterectomized (59%), most were non-ovariectomized (70%), and most had received previous hormone replacement therapy (72%). The most bothersome symptom that was most commonly self-identified by patients at the start of treatment was dyspareunia (61%).
Table 27: Summary of Baseline Characteristics (ITT Population) — ERC-210
Characteristics | Placebo N = 53 | Prasterone 0.5% N = 56 |
|---|---|---|
Age (years), mean (SD) | 59 (5.29) | 58 (5.62) |
Race | ||
White | 49 (92.5) | 54 (96.4) |
Black or African American | 3 (5.7) | 1 (1.8) |
Other | 1 (1.9) | 1 (1.8) |
Number of years since last menstruationa, mean (SD) | 13 (8.24) | 13 (8.21) |
Hysterectomy | ||
No | 31 (58.5) | 33 (58.9) |
Yes | 22 (41.5) | 23 (41.1) |
Oophorectomy | ||
No | 38 (71.7) | 38 (67.9) |
Yes | 15 (28.3) | 18 (32.1) |
HRT | ||
No | 17 (32.1) | 13 (23.2) |
Yes | 36 (67.9) | 43 (76.8) |
ITT = intention to treat; HRT = hormone replacement therapy; SD = standard deviation.
Note: Values are presented as n (%) unless otherwise indicated.
aNumber of years since last menses is the patient’s current age (years) at study entry minus her age (years) at last menstrual cycle.
Source: ERC-210 Clinical Study Report.15
Interventions
Patients received a daily dose of 1 1.3 mL vaginal suppository (ovule) of the following prasterone concentrations: 0.0% (placebo or 0 mg), 0.25% (3.25 mg), 0.5% (6.5 mg), or 1.0% (13 mg). Treatments were applied daily intravaginally, in the evening or at bedtime. The prasterone ovules contained a lipophilic base as non-active ingredient.
Concomitant medications necessary for the patients’ well-being were allowed during the study except for vaginal creams, gels, or lubricants, vaginal douching, and natural “estrogenic” products (unless they were already on the natural “estrogenic” products before study initiation, in which case they were allowed to continue).
Outcomes
The 4 coprimary end points were change from baseline to week 12 in percent parabasal cells, percent superficial cells, vaginal pH, and most bothersome symptom based on patients’ self-assessment. These outcomes were assessed as previously described for the ERC-238 and ERC-231 trials. Assessments of these outcomes occurred at screening, baseline, as well as weeks 2, 4, 8, and 12.
Health-related quality of life was assessed using the MENQOL questionnaire and Psychological General Well-Being questionnaire and sexual function was assessed using the Abbreviated Sexual Function questionnaire. End points based on these outcomes were secondary end points.
Tolerability and AEs were assessed at baseline and weeks 2, 4, 8, and 12, or discontinuation visit following at least 1 month of treatment and at discretion of physician. Endometrial biopsy was examined at central lab at screening, week 12, and discontinuation visit following at least 1 month of treatment and at discretion of physician.
Statistical Analysis
The trial was designed for 50 patients per treatment group for a total of 200 patients.
Efficacy analyses were pre-specified to be performed on the ITT population, defined as treated patients who had a baseline and at least 1 post-baseline efficacy assessment. Missing follow-up observations for the coprimary end points among patients included in the ITT analysis were imputed using the LOCF method, in which missing efficacy data for patients who discontinued the study were imputed using the last non-missing evaluation before discontinuation. The primary statistical analyses were performed using analysis of covariance, with the baseline value used as the covariate and a 2-sided significance level of 0.05. There was no P value adjustment for multiplicity, as significance of all of the 4 primary end points was needed to demonstrate efficacy.
The study was originally designed to evaluate the aggregate most bothersome symptoms of VVA in addition to the other 3 coprimary end points. However, following feedback from the FDA, the primary analyses were redone (post hoc) on a subgroup of patients (n = 114; 64% of those randomized) who had a vaginal pH of greater than 5 and superficial cells of 5% or less and who self-identified as having dyspareunia as their most bothersome symptom at baseline. Given this amendment, an adjustment for multiplicity was performed to account for the choice of dyspareunia out of the 3 potential most bothersome symptoms of vaginal dryness, vaginal itching or irritation, and dyspareunia. A Bonferroni correction was applied, where the P value for statistical significance for each of the 4 coprimary end points was 0.05 divided by 3, equalling 0.0167.
Safety analyses were performed on the safety population, defined as all patients who received any of the study treatments and had safety information available. Analysis was based on the treatment actually received and missing safety data were not imputed.
Patient Disposition
A total of 403 patients were screened and 217 (54%) were randomized to treatment groups; 54 and 56 patients were randomized to the placebo and prasterone 0.5% groups, respectively (Table 28).
Table 28: Patient Disposition in the ERC-210 Trial
Detail | Placebo (N) | Prasterone 0.5% (N) |
|---|---|---|
Total screened | 403 | |
Screen failure | 185 | |
Randomizeda | 54 | 56 |
Reason for discontinuation (% of discontinuations) | 6 | 4 |
Adverse event | 2 (33) | 2 (50) |
Patient withdrew consent | 3 (50) | 0 (0) |
Loss to follow-up | 0 (0) | 1 (25) |
Other | 1 (17) | 1 (25) |
ITT analysis setb | 53 | 56 |
ITT population meeting baseline criteriac | 41 | 50 |
Safety analysis setd | 54 | 56 |
ITT = intention to treat.
aPatients who had a randomization number and used at least 1 application.
bTreated patients who had a baseline and at least 1 post-baseline efficacy assessment.
cPatients who satisfy all 3 criteria for entry at day 1: pH greater than 5, superficial cells of 5% or less, and self-identified most bothersome symptom of at least moderate severity.
dAll patients who receive an administration of either test article (prasterone at any dose or placebo), and who have any safety information available.
Source: ERC-210 Clinical Study Report.15
Exposure to Study Treatments
Patients received treatment for a mean of 76 days (range: 15 to 91 days) in the placebo group and 80 days (range: 14 to 86 days) in the prasterone group. Adherence to treatment was a mean of 99% (SD = 2.35) based on patient diary, or 100% (SD = 2.97) based on medication count for the placebo group. Similarly, adherence was 99% (SD = 3.88) based on patient diary, or 100% (SD = 5.18) based on medication count for the prasterone group.
Efficacy
This report will focus on the primary outcomes due to the dose response design, limited sample size, and the amendments which restricted the primary end point analyses to a subgroup of patients with dyspareunia as the most bothersome symptom (Table 29).
The percentage of superficial cells were measured to be 0.62% (SD = 1.02) at baseline and 0.54% (SD = 0.95) at week 12 (P = 0.7460 versus baseline) for the placebo group. The percentage of superficial cells were measured to be 0.40% (SD = 0.62) at baseline and 5.20% (SD = 6.54) at week 12 for the prasterone group. The MD in change was 4.88% (P = 0.0111) of superficial cells in prasterone group compared to the placebo group at week 12.
The percentage of parabasal cells was measured to be 46.73% (SD = 44.05) at baseline and 47.81% (SD = 38.36) at week 12 (P = 0.7686 versus baseline) for the placebo group. The percentage of parabasal cells was measured to be 53.40% (SD = 41.01) at baseline and 11.00% (SD = 18.77) at week 12 for the prasterone group. The MD in change was 43.48% (P < 0.0001) of parabasal cells in the prasterone group compared to the placebo group at week 12.
In the placebo group, the mean vaginal pH was 6.49 (SD = 0.69) at baseline and 6.01 (SD = 1.12) at week 12 (P = 0.005 versus baseline). In the prasterone group, the mean vaginal pH was 6.64 (SD = 0.51) at baseline and 5.17 (SD = 0.91) at week 12. At week 12, there was a mean 0.99 greater change (P = 0.0001) in pH in the prasterone group compared to the placebo group.
The mean severity score of dyspareunia was 2.77 (SD = 0.43) at baseline and 2.35 (SD = 0.94) at week 12 (P = 0.0132 versus baseline) for the placebo group. The mean severity score of dyspareunia was 2.73 (SD = 0.45) at baseline and 1.10 (SD = 1.18) at week 12. There was a mean 1.21 greater change (P < 0.0001) in symptom score in prasterone group compared to the placebo group at week 12.
Table 29: Summary of Coprimary End Points From ERC-210 (ITT Population Meeting Baseline Criteria With Dyspareunia as MBS)
Study detail | Placebo N = 41 | Prasterone 0.5% N = 50 |
|---|---|---|
Percentage of superficial cells | N = 26 | N = 30 |
Baseline, mean (SD) | 0.62 (1.02) | 0.40 (0.62) |
Week 12, mean (SD) | 0.54 (0.95) | 5.20 (6.54) |
Mean change from baseline (SD) | –0.08 (1.20) | 4.80 (6.58) |
Difference from placebo | Reference | 4.88 |
P value | Reference | 0.0111 |
Percentage of parabasal cells | N = 26 | N = 30 |
Baseline, mean (SD) | 46.73 (44.05) | 53.40 (41.01) |
Week 12, mean (SD) | 47.81 (38.36) | 11.00 (18.77) |
Mean change from baseline (SD) | 1.08 (18.46) | –42.40 (40.31) |
Difference from placebo | Reference | –43.48 |
P value | Reference | < 0.0001 |
Vaginal pH (units) | N = 26 | N = 30 |
Baseline, mean (SD) | 6.49 (0.69) | 6.64 (0.51) |
Week 12, mean (SD) | 6.01 (1.12) | 5.17 (0.91) |
Mean change from baseline (SD) | –0.48 (0.80) | –1.47 (1.01) |
Difference from placebo | Reference | –0.99 |
P value | Reference | 0.0001 |
Dyspareunia (score) | N = 26 | N = 30 |
Baseline, mean (SD) | 2.77 (0.43) | 2.73 (0.45) |
Week 12, mean (SD) | 2.35 (0.94) | 1.10 (1.18) |
Mean change from baseline (SD) | –0.42 (0.81) | –1.63 (1.16) |
Difference from placebo | Reference | –1.21 |
P value | Reference | < 0.0001 |
ANCOVA = analysis of covariance; MBS = most bothersome symptom; SD = standard deviation.
Note: These results are for the subgroup of patients who at baseline had a pH of greater than 5, superficial cells of 5% or less, and self-identified moderate to severe dyspareunia as their most bothersome symptom. Results from this post hoc analysis consider the adjustment for multiplicity in the choice of dyspareunia among the 3 most bothersome symptoms. Applying the Bonferroni correction, the P value for the statistical significance for each of the 4 coprimary end points was assessed against 0.05 divided by 3, which equals 0.0167. ANCOVA models were used with the treatment group as the main factor and baseline value as the covariate.
Source: ERC-210 Clinical Study Report.15
Harms
Of patients who received prasterone, 47 (84%) patients experienced at least 1 AE, compared to 35 (65%) patients in the placebo group. The most common AEs (≥ 5%) reported in prasterone group were cough (11%), headache (9%), and vaginal discharge (9%). The percentage of patients who withdrew from treatment due to an AE was 4% for both the placebo and prasterone groups. For the prasterone group, 1 (2%) patient had cervical dysplasia and none had vaginal discharge. Adverse events of endometrial dysplasia and breast mass were not reported (Table 30).
Detail | Placebo N = 54 | Prasterone 0.5% N = 56 |
|---|---|---|
Patients with ≥ 1 adverse event, n (%) | 35 (65) | 47 (84) |
Cough | 2 (4) | 6 (11) |
Headache | 3 (6) | 5 (9) |
Vaginal discharge | 3 (6) | 5 (9) |
Nasopharyngitis | 4 (7) | 4 (7) |
Hot flush | 4 (7) | 4 (7) |
Vulvovaginal pruritus | 3 (6) | 4 (7) |
Nausea | 1 (2) | 4 (7) |
Vulvovaginal burning sensation | 4 (7) | 3 (5) |
Abdominal pain | 4 (7) | 3 (5) |
Abdominal distension | 3 (6) | 2 (4) |
Patients with ≥ 1 serious adverse event | 0 (0) | 1 (2) |
Appendicectomy | 0 (0) | 1 (2) |
Withdrawal due to adverse events | 2 (4) | 2 (4) |
Cervical dysplasia | ||||| (|||||) | ||||| (|||||) |
MSK injury | ||||| (|||||) | ||||| (|||||) |
Migraine | ||||| (|||||) | ||||| (|||||) |
Deaths | ||
n (%) | 0 (0) | 0 (0) |
Notable harms, n (%) | ||
Cervical dysplasia | 2 (4) | 1 (2) |
Vaginal hemorrhage | 1 (2) | 0 (0) |
Endometrial dysplasia | NR | NR |
Breast mass | NR | NR |
MSK = musculoskeletal; NR = not reported.
Source: ERC-210 Clinical Study Report.15
Critical Appraisal
Internal Validity
The plan for the primary analysis was amended following feedback from the FDA to restrict to the subgroup of patients who identified dyspareunia as their most bothersome symptom at baseline. This revision was post hoc and in a subgroup of patients, thereby breaking randomization (i.e., the characteristics of the treatment groups may no longer have been similar with respect to known and unknown confounding and prognostic variables). The direction and extent of any selection bias related to imbalances in characteristics is unclear because updated baseline characteristics for the subgroup were not reported. However, the Bonferroni adjustment for the coprimary analyses was a conservative approach to help mitigate the potential bias introduced by the revised analysis. The differences between the prasterone 0.5% and placebo groups were statistically significant following the Bonferroni adjustment.
External Validity
The sample sizes of patients randomized to the prasterone and placebo groups were 56 and 54, respectively. The amendment of the analysis to a subgroup of these patients means that the sample sizes were reduced to 30 patients and 26 patients, respectively, with no information regarding baseline characteristics of this subgroup population provided. In addition, greater than 90% of patients were White. Taken together, it is unclear whether the results are generalizable to post-menopausal patients with VVA in Canada due to limited representativeness of the sample population.
Since moisturizer (placebo) may have some effect on vaginal parameters and symptoms, it is possible that the treatment effect of the prasterone ovule was smaller versus the placebo ovule than it would have been versus a true placebo.
Finally, the clinical expert consulted by CADTH for this review expected that patients would be on prasterone treatment indefinitely unless they wish to discontinue and/or significant harms occur. A relatively short follow-up and small number of patients in the ERC-210 trial are inadequate to confirm the long-term benefits of prasterone beyond 12 weeks and assess rare, long-term harms.
Observational Study (Estip-Es Study)
The objective of the observational “Estip-Es” study from Spain was to evaluate the effectiveness, safety, and tolerability of prasterone in a real-world clinical setting.16 This study is summarized here for supportive evidence as it evaluated relevant outcomes in patients who switched from previous vaginal hormonal therapy to prasterone.
Description of Study
The Estip-Es study was a multi-centre prospective, noncomparative, observational study with 184 adult post-menopausal patients who were routinely seen in medical centres throughout Spain for GSM. The enrolment period was from August 2019 to December 2019.
Populations
Patients included post-menopausal women with GSM confirmed by a gynecological examination (including determination of vaginal pH and cytology, if necessary). Exclusion criteria included starting other treatments for GSM during the study; previous treatment for GSM with an expected long-term residual effect (except for vaginal hormonal therapies); starting therapies that may worsen GSM; and contraindication for the use of intravaginal prasterone.
The mean age of patients at baseline was 57.98 (SD = 6.06) years and the mean time since menopause was 7.56 years (SD = 6.23). Of 184 patients, 42.9% were using vaginal hormonal therapy and 32.1% were using vaginal moisturizers or lubricants. The overall FSFI score for the total population (n = 184) at baseline was 15.7 (SD = 6.3).
Interventions
Patients had used vaginal moisturizers or lubricants and/or vaginal hormone therapy and switched to intravaginal prasterone without a washout period. No information about specific prasterone doses or frequency of administration was provided.
Outcomes
Outcomes were assessed at baseline and after 30 ± 7 days of treatment with prasterone.
Sexual function was self-reported on a validated short version of the FSFI with 7 items. Each item ranged from never (score 0) to always (score 5). Higher scores indicate better sexual function. A score between 0 and 20 suggests that sexual dysfunction may be present. An increase of greater than 3 points was interpreted as considerable clinical improvement, an increase between 2 and 3 points as moderate, and an increase of 1 point as mild clinical improvement.
A VAS was administered to assess the self-reported impact on GSM across 19 items, encompassing symptoms including dryness, dyspareunia, bleeding, burning, itching, urinary problems and infections, and abdominal pain. Each item ranged from 0 (absence of discomfort) to 10 (extreme discomfort). Higher scores indicate more discomfort. A decrease of greater than 3 points was interpreted as considerable improvement, 2 to 3 points as moderate improvement, 1 point as mild clinical improvement, and less than 1 point as absence of clinical improvement.
Statistical Analysis
There was no a priori sample size reported. Categorical variables were expressed as frequencies (n, %) and changes from baseline to follow-up were assessed using the chi-square test or the Fisher exact test when appropriate. Continuous variables were expressed as mean (SD) and changes between baseline and follow-up were assessed using the Student t-test or the Mann–Whitney U test, as applicable. Missing data or lost values were not imputed. The authors mentioned that missing data for important variables were controlled for using filters when collecting data from the case report form. However, it is unclear exactly how they performed this filtering to control for missing data. Statistical significance was set at P less than 0.05 compared to baseline. No information regarding adjustment for multiplicity was found in the published paper.
Exposure to Study Treatments
At the end of the study, 78.4% of patients remained on prasterone. The primary reason for patients withdrawing from the trial was AEs (6.5%).
Results
Baseline Characteristics
A total of 184 post-menopausal women with GSM were enrolled and treated with intravaginal prasterone, including 59 women who were taking vaginal moisturizers or lubricants and 79 women taking vaginal hormonal therapy. The mean age of women was 57.98 years (SD = 6.06) with a mean time since onset of menopause of 7.56 years (SD = 6.23). Few women (6.1%) were smokers or had diabetes (6.1%). Just under half of patients (42.9%) were taking vaginal hormonal therapy and 32.1% were taking vaginal moisturizers or lubricants. The authors noted that there was less frequent use of vaginal hormonal therapy among the subgroup of patients who were taking vaginal moisturizers or lubricants (27.1%) compared to the overall patient population (42.9%); no other differences were noted.
Efficacy
In the overall study population, the total FSFI score increased from 15.7 (SD = 6.3) to 19.9 (SD = 5.38) with the mean change of 4.2 (P < 0.01) over 30 days (Table 31). Increased scores from baseline to post-treatment with prasterone were observed in all the FSFI domains with variable magnitudes.
There was a numerical decrease (improvement) in all symptoms assessed using the VAS except for vaginal discharge; however, it should be noted that application site discharge is an expected AE related to use of prasterone. Likewise, there was a numerical increase (improvement) in all FSFI domain scores.
Table 31: Short FSFI and Symptoms (VAS) at Baseline and After Treatment With Prasterone
Study detail | Overall study populationa (N = 184) | ||
|---|---|---|---|
FSFI | Baseline | After treatment | Pb |
Desire for or interest in sexual activity | 1.78 ± 0.99 | 2.49 ± 0.92 | < 0.01 |
Unhappy with low Interest in sexual activity | 2.34 ± 1.22 | 2.70 ± 1.25 | < 0.01 |
Take a long time to become aroused | 2.24 ± 1.24 | 2.98 ± 0.94 | < 0.01 |
Feel indifferent regarding sex | 2.64 ± 1.11 | 3.20 ± 1.00 | < 0.01 |
Low sexual desire | 2.29 ± 1.15 | 3.03 ± 1.03 | < 0.01 |
Disappointed with my interest in sex | 2.52 ± 1.49 | 3.05 ± 1.26 | < 0.01 |
Reach orgasm easily | 1.92 ± 1.19 | 2.42 ± 1.12 | < 0.01 |
Overall | 15.7 ± 6.3 | 19.9 ± 5.38 | < 0.01 |
VAS | Baseline | After treatment | Pb |
Dryness of the vagina | 8.07 ± 1.88 | 4.73 ± 2.41 | < 0.01 |
Dryness of the external genitalia | 7.20 ± 2.23 | 4.16 ± 2.32 | < 0.01 |
Dyspareunia of the vagina | 8.23 ± 2.32 | 5.09 ± 2.65 | < 0.01 |
Dyspareunia during penetration | 7.95 ± 2.14 | 5.60 ± 2.73 | < 0.01 |
Discomfort during exercise | 3.80 ± 3.61 | 1.71 ± 2.21 | < 0.01 |
Vaginal bleeding during sexual intercourse | 1.43 ± 2.25 | 0.74 ± 1.56 | < 0.01 |
Vaginal bleeding during sexual contact | 1.17 ± 2.04 | 0.56 ± 1.30 | < 0.01 |
Burning or irritation of the vagina | 5.41 ± 3.54 | 2.79 ± 3.00 | < 0.01 |
Burning or irritation of the external genitalia | 5.02 ± 3.43 | 2.63 ± 2.79 | < 0.01 |
Itching of the vagina | 3.40 ± 3.23 | 1.36 ± 1.90 | < 0.01 |
Itching of the external genitalia | 3.05 ± 3.47 | 1.65 ± 2.16 | < 0.01 |
Vaginal discharge | 2.31 ± 2.73 | 2.98 ± 2.99 | < 0.01 |
Urinary incontinence | 2.72 ± 3.11 | 1.78 ± 2.65 | < 0.01 |
Urinary urgency | 2.89 ± 2.97 | 2.09 ± 2.56 | < 0.01 |
Urinary frequency | 3.73 ± 2.72 | 2.91 ± 2.70 | < 0.01 |
Urinary difficulties | 1.56 ± 2.40 | 1.09 ± 1.91 | < 0.01 |
Recurrent urinary tract infection | 2.29 ± 3.29 | 1.11 ± 2.32 | < 0.01 |
Urinary tract infection associated with sexual intercourse | 1.55 ± 2.99 | 0.76 ± 1.97 | < 0.01 |
Abdominal pain | 2.42 ± 2.98 | 1.47 ± 2.21 | < 0.01 |
FSFI = Female Sexual Function Index; VAS = visual analogue scale.
aAll-enrolled patients were analyzed. Categorical variables were expressed as frequencies (n [%]) and changes from baseline to follow-up were assessed using the chi-square test or the Fisher exact test when appropriate. Continuous variables were expressed as mean and standard deviation and changes between baseline and follow-up were assessed using the Student t-test or the Mann–Whitney U test, as applicable.
bP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: González SP, Mainar LB, Campo LR. Effectiveness, Safety and Tolerability of Intravaginal Prasterone for the Treatment of Genitourinary Syndrome in Postmenopausal Women in Spain: The Estip-Es Study. Am J Biomed Sci and Res. 2021 to 126. Copyright by Silvia P González. Reproduced according to the Creative Commons License CC BY version 4.0.
Harms
In the overall population, 6.5% of patients reported AEs (e.g., blisters on the face, hair loss, constipation, leukorrhea, and dizziness) during follow-up at 30 ± 7 days. No further detail regarding these AEs was provided in the published paper.
Critical Appraisal
Internal Validity
The Estip-Es study was an observational study with the objective of evaluating the efficacy, safety, and tolerability of prasterone for the treatment of post-menopausal women with GSM in clinical practice. As there was no comparator group, the efficacy of prasterone relative to other therapies was not clear based on data from this study. The lack of blinding to treatment allocation and the subjective nature of all of the outcomes could have also contributed to patients reporting greater improvements with a switch to prasterone than they would have in a DB RCT. Patients enrolled in the Estip-Es study were not subject to a washout period; therefore, it is possible that residual effects from previous treatments may have carried over and affected patient outcomes while receiving treatment with prasterone.
Greater than 29% of patients discontinued treatment and an ITT approach was not used. Therefore, the analysis is based on data from a select patient population. There is a potential for selection bias since the authors do not report anything about using a consecutive sample. Since there is not a prior protocol available, there is a potential for selective reporting bias as well. Moreover, numerous statistical analyses were conducted without adjustment for multiple comparisons.
There was limited reporting of safety data for this study. It was unclear how safety data were collected (i.e., solicitation from each patient or voluntary reporting).
Lastly, the study used “a validated short version with 7 items” for FSFI; however, no references were provided related to the validity and reliability of the short form. Use of this questionnaire may also be subject to bias as this was an open-label trial without a comparator group.
External Validity
Due to the lack of detailed information on patient’s baseline characteristics, it is difficult to ascertain to what extent the enrolled population reflects the Canadian population who are eligible for treatment with prasterone. Also, this study was performed in Spain where cultural perception of VVA may differ from those in Canada. The small sample size further limits generalizability of this study to the Canadian population. The Estip-Es study enrolled women from medical centres throughout Spain for GSM; therefore, these women were seeking medical intervention for symptoms related to VVA. For this reason, there is a possibility for selection bias, as patients who were dissatisfied with their previous treatments were likely to have been enrolled in the Estip-Es trial and may view treatment with prasterone more positively.
The Estip-Es study failed to report details about the intervention, such as the dosage and frequency of administration of prasterone, and whether they aligned with those specified in the product monograph for prasterone.
Follow-up visits for patients were conducted approximately 1 month after recruitment into the Estip-Es study. This short-term follow-up may not be an optimal time frame to capture benefits and harms related to treatment with prasterone. In particular, as patients may be on treatment with prasterone for long periods of time, the 1-month follow-up may not be reflective of true patient experiences.
Trial in Post-Menopausal Cancer Survivors (Barton, et al.)
Barton et al.12 conducted a study in post-menopausal women with a history of breast or gynecological cancer. Post-menopausal women with a history of breast or gynecological cancer who have VVA are an important subgroup with the unmet need for VVA therapies that are not estrogen based. Patients with a previous diagnosis of cancer were excluded from the ERC-231, ERC-238, and ERC-230 trials.
Methods
This study was a multi-centre (US and Canada), 3-group, DB, parallel group RCT where 443 patients were randomized to receive either 3.25 mg or 6.5 mg of DHEA in a plain bioadhesive moisturizer, or a plain bioadhesive moisturizer alone (placebo). Randomization was accomplished using the Pocock-Simon dynamic allocation procedure stratified by hysterectomy status (yes or no), cigarette smoking history (current, past, or never), use of aromatase inhibitors (anastrozole, letrozole, exemestane, or none), and the current use of tamoxifen (yes or no). The primary aim of this study was to compare 3.25 mg or 6.5 mg prasterone contained in plain moisturizer versus plain moisturizer alone over 12 weeks for the alleviation of the most bothersome vaginal symptom. This summary focuses on the comparison between the approved prasterone 6.5 mg dose and placebo.
Populations
Patients eligible for inclusion were post-menopausal women with a history of breast or gynecologic cancer and no current evidence of disease, who had completed chemotherapy and/or radiation. Patients had to report either dyspareunia or vaginal dryness of at least moderate severity and this symptom had to have been present for at least 2 months. Patients could be receiving treatment with tamoxifen or an aromatase inhibitor for at least 8 weeks before enrolment, without plans to change treatment during the study to minimize any changes in important variables. Patients could not use vaginal products other than water-based lubricants during intercourse.
Patients were not eligible for the study if they had prior pelvic surgery resulting in anatomic changes, had prior radiation to the pelvis, active vaginal infections, or had used any (systemic or local) hormonal product (including soy or any compounded hormones) in the preceding 4 weeks.
Interventions
The active ingredient, DHEA, was added to a bioadhesive base to result in either 3.25 mg/0.4 mL or 6.5 mg/0.4 mL gel. The bioadhesive base was used as the plain moisturizer in the control group. Study patients inserted 1 syringe (without needle) of gel daily right before going to bed and after any sexual activity for 12 weeks.
Outcomes
The primary end point was self-rated severity of patients’ most bothersome symptom, either dryness or dyspareunia using an ordinal scale of none, mild, moderate, severe, or very severe.
Secondary outcomes included AEs measured by provider-reported CTCAE version 4.0. In addition, an investigator-developed, patient self-reported questionnaire asked patients to report on 8 potential toxicities and overall quality of life, using a numeric analogue scale from 0 (“not at all”) to 10 (“as bad as it can be”). For the analysis, these numbers were inverted such that negative numbers indicate worsening of symptoms and positive numbers indicate improvement. Another secondary outcome was overall sexual function, measured using the FSFI.
Statistical Analysis
A total of 145 patients in each group were required to provide 80% power to detect a difference of 0.36 SD with a type I error of 2.5%. An effect size of 0.36 SD was selected based on previous work by Labrie et al. which demonstrated safety and improved symptoms among post-menopausal women treated with prasterone for VVA.35,36
For the primary analysis (“primary end point set”), the mean change from baseline to week 12 in the severity of vaginal dryness or dyspareunia for each prasterone dose was considered a continuous variable (derived from an ordinal scale). Doses of prasterone were compared to plain moisturizer using 2 independent t-tests with a Bonferroni correction due to having 2 correlated primary hypothesis tests, 3.25 mg prasterone versus plain moisturizer and 6.5 mg prasterone versus plain moisturizer. The study results were also analyzed using logistic regression when considering the end point as ordinal in nature and using the LOCF missing data imputation method.
Other analyses were not adjusted for multiplicity, including analyses conducted at multiple time points and questionnaires. Also, information regarding how the authors treated missing data with respect to secondary outcome analyses were missing.
Patient Disposition and Exposure to Study Treatments
A total of 464 patients were screened for eligibility; of these 21 patients withdrew consent or failed to meet eligibility criteria. A total of 149 patients were randomized into the 6.5 mg DHEA group and 147 patients were randomized into the plain moisturizer group. Most patients completed the study. A total of 35 patients (24%) in the DHEA group discontinued the study, mainly due to AEs (11%), refusal to continue (11%), and unspecified reasons (0.01%). In the plain moisturizer group, 29 patients (20%) discontinued from the study due to AEs (10%), refusal to continue (10%), and receiving an alternative treatment (0.01%). Patient who discontinued were not included in the sample of patients used for primary and secondary analyses, leaving 112 patients in the DHEA group and 118 patients in the plain moisturizer group. It should be noted that 2 additional patients in the DHEA group were excluded from the analyses; reasons for exclusion were not provided.
Results
Baseline Characteristics
A total of 464 patients were enrolled, with 149 patients enrolled to the DHEA 6.5 mg group and 147 enrolled in the plain moisturizer group. Baseline characteristics of the patients were similar between the DHEA and placebo groups (Table 32). Dryness was reported as the most bothersome symptom for 42% of patients in the prasterone group and 49% in the plain moisturizer group and dyspareunia was reported as the most bothersome symptom for 58% and 51% of patients in the DHEA and plain moisturizer groups, respectively. The mean severity score of the most bothersome symptom for either dryness or dyspareunia was 4 out of 5 for the DHEA group, and 4.1 for patients in the plain moisturizer group; the severity of dryness or dyspareunia was considered severe at baseline for patients in this study.
Most patient had history of breast cancer (97% in each group); few patients had history of ovarian (2% in each group) or endometrial (1% in each group) cancers. The mean age was 57.3 years (SD = 8.2) in the DHEA group and 58.0 years (SD = 7.3) in the plain moisturizer group. Most patients were White (95% versus 93% in the DHEA and plain moisturizer groups, respectively). More than half of patients had naturally occurring menopause (59% in the DHEA group versus 65% in the plain moisturizer group). Patients reported concomitant aromatase inhibitor use including anastrozole or letrozole (48% versus 49% in the DHEA and plain moisturizer groups, respectively) and exemestane (7% versus 6%). A proportion of 16% in each group reported currently taking tamoxifen therapy. Patients reported a mean time of 24.5 months (SD = 18) and 22.0 months (SD = 18.7) of current therapy in the DHEA and plain moisturizer groups, respectively.
Table 32: Baseline and Treatment Characteristics
Demographic characteristics | Plain moisturizer N = 147 | 3.25 mg DHEA N = 147 | 6.5 mg DHEA N = 149 | P value |
|---|---|---|---|---|
Age in years (SD) | 58 (7.3) | 56.8 (6.7) | 57.3 (8.2) | 0.63 |
Race (%) | ||||
White | 137 (93) | 142 (97) | 142 (95) | 0.63 |
Black/African American | 7 (5) | 3 (2) | 5 (4) | |
Asian | 1 (1) | 0 (0) | 0 (0) | |
Missing | 2 (1) | 2 (1) | 2 (1) | |
Menopause—natural | 95 (65) | 98 (67) | 88 (59) | 0.37 |
Bilateral oophorectomy | 48 (33) | 43 (30) | 55 (37) | 0.38 |
Weight in kg (SD) | 75 (16.6) | 77 (14.7) | 73 (14.8) | 0.06 |
Height in cm (SD) | 163.2 (6.6) | 164.4 (6.0) | 163.1 (6.8) | 0.20 |
Treatment characteristics | ||||
Breast | 142 (97) | 143 (97) | NR | 0.70 |
Ovarian | 3 (2) | 4 (3) | 3 (2) | |
Endometrial | 2 (1) | 0 (0) | 2 (1) | |
Tamoxifen (current) | 23 (16) | 22 (15) | 24 (16) | 0.96 |
AI current: | NA | NA | NA | NA |
Anastrozole/letrozole | 72 (49) | 71 (48) | 72 (48) | 1.0 |
Exemestane | 9 (6) | 11 (8) | 10 (7) | |
Months on current therapy (SD) | 22 (18.7) | 21.1 (17.1) | 24.5 (18) | 0.31 |
AI = aromatase inhibitor; DHEA = dehydroepiandrosterone; SD = standard deviation.
Reprinted by permission from Springer: Barton DL, Sloan JA, Shuster LT, et al. Evaluating the efficacy of vaginal dehydroepiandrosterone for vaginal symptoms in post-menopausal cancer survivors: NCCTG N10C1 (Alliance). Supportive Care in Cancer. 2018;26(2):643 to 650. [Copyright 2018].
Efficacy
There was no difference (P = 0.08) between the DHEA (mean = –1.8; 95% CI –1.97 to –1.54) and plain moisturizer (mean = –1.5’ 95% CI –1.74 to –1.27) groups in changes of either dryness or dyspareunia at week 12.
Patients using DHEA reported numerically improved scores for all domains of FSFI and total FSFI score at week 12 compared to baseline and the improved scores were numerically greater compared to those in plain moisturizer group. For quality of life, the score increased at week 12 compared to baseline in DHEA group; however, it decreased at week 12 compared to baseline in the plain moisturizer group (Table 33).
Table 33: FSFI and Quality of Life
Study detail | Plain moisturizer N = 118 | 3.25 mg DHEA N = 123 | 6.5 mg DHEA N = 112 |
|---|---|---|---|
Arousala | |||
Baseline | 2.1 (1.8) (1.8 to 2.3) | 2.1 (1.7) (1.8 to 2.4) | 1.9 (1.7) (1.7 to 2.2) |
12 weeks | 2.5 (1.8) (2.2 to 2.9) | 2.8 (1.9) (2.5 to 3.2) | 3.0 (1.7) (2.7 to 3.3) |
Change from baseline | 0.4 (1.6) (0.1 to 0.7) | 0.7 (1.4)* (0.4 to 1.0) | 1.0 (1.6)** (0.7 to 1.2) |
Lubricationa | |||
Baseline | 1.4 (1.2) (1.2 to 1.6) | 1.5 (1.3) (1.3 to 1.7) | 1.4 (1.3) (1.1 to 1.6) |
12 weeks | 2.5 (1.9) (2.1 to 2.8) | 2.8 (2.0) (2.5 to 3.2) | 3.0 (2.0) (2.6 to 3.4) |
Change from baseline | 1.1 (1.7) (0.7 to 1.4) | 1.3 (1.8) (1.0 to 1.6) | 1.6 (1.7)* (1.3 to 1.9) |
Satisfactiona | |||
Baseline | 2.9 (1.6) (2.6 to 3.1) | 2.7 (1.7) (2.4 to 2.9) | 2.7 (1.6) (2.4 to 3.0) |
12 weeks | 3.4 (1.8) (3.1 to 3.7) | 3.6 (1.8) (3.3 to 3.9) | 3.8 (1.6) (3.5 to 4.1) |
Change from baseline | 0.5 (1.5) (0.2 to 0.8) | 0.9 (1.5) (0.7 to 1.2) | 1.1 (1.6)* (0.8 to 1.4) |
Paina | |||
Baseline | 1.6 (1.4) (1.4 to 1.9) | 1.6 (1.5) (1.4 to 1.9) | 1.4 (1.3) (1.2 to 1.6) |
12 weeks | 2.5 (2.0) (2.1 to 2.8) | 3.0 (2.0) (2.7 to 3.4) | 3.5 (2.0) (3.1 to 3.9) |
Change from baseline | 1.0 (1.8) (0.6 to 1.3) | 1.4 (1.7)* (1.1 to 1.7) | 2.0 (1.6)*** (1.7 to 2.3) |
Overall totala | |||
Baseline | 12.2 (7.5) (11.0 to 13.5) | 12.5 (7.9) (11.2 to 13.8) | 11.6 (7.3) (10.4 to 12.8) |
12 weeks | 16.2 (9.3) (14.5 to 17.9) | 17.9 (9.6) (16.2 to 19.7) | 19.1 (8.7) (17.5 to 20.7) |
Change from baseline | 3.8 (7.4) (2.4 to 5.1) | 5.5 (7.5) (4.2 to 6.8) | 7.1 (7.3) (5.8 to 8.5) |
Quality of lifea,, mean (SD) | |||
Baseline | 7.6 (2.0) | 7.3 (1.8) | 7.5 (1.8) |
12 weeks | 7.4 (2.3) | 7.5 (1.7) | 7.8 (1.7) |
Change from baseline | –0.3 (2.2) | 0.2 (1.7) | 0.3 (1.9) |
DHEA = dehydroepiandrosterone; FSFI = Female Sexual Function Index; SD = standard deviation.
Note: Values are presented as mean (SD) and 95% confidence interval unless otherwise indicated.
Significantly different vs. plain moisturizer: *P < 0.05, **P ≤ 0.01, and *** P ≤ 0.001.
aSummary statistics including means and frequencies were calculated, and end points analyzed with the appropriate statistical methods including chi-square tests, Fisher exact tests, and t-tests.
Reprinted by permission from Springer: Barton DL, Sloan JA, Shuster LT, et al. Evaluating the efficacy of vaginal dehydroepiandrosterone for vaginal symptoms in post-menopausal cancer survivors: NCCTG N10C1 (Alliance). Supportive Care in Cancer. 2018;26(2):643 to 650. [Copyright 2018].
Harms
The most common clinician-graded CTCAE side effects (> 5%) included headache and breast pain, which were not different between study groups. Providers graded hirsutism and specific components of hirsutism (voice alteration, increased body hair and acne) primarily as grade 1 (98%) and there were no differences between study groups. No further data regarding clinician-graded CTCAE side effects were reported in the published paper.
Self-reported side effect changes were not different between the plain moisturizer and DHEA groups (Table 34). However, statistically significant (P ≤ 0.02) voice change was observed in patients receiving DHEA (change from baseline: –0.2, 95% CI, –0.4 to 0.0) compared with plain moisturizer (change from baseline: 0.2, 95% CI, 0.0 to 0.4).
Self-reported side effects | Plain moisturizer N = 118 | 3.25 mg DHEA N = 123 | 6.5 mg DHEA N = 112 |
|---|---|---|---|
Vaginal discharge | –0.7 (2.6) (–1.1 to –0.2) | −0.8 (2.3) (−1.3 to −0.4) | –0.7 (2.3) (–1.2 to –0.3) |
Rash in vaginal area | 0.1 (1.6) (–0.2 to 0.4) | 0.1 (1.3) (−0.1 to 0.3) | –0.1 (0.8) (–0.3 to 0) |
Unwanted hair growth | 0.7 (2.4) (0.2 to 1.1) | 0.4 (2.1) (0.1 to 0.8) | 0.3 (1.7) (0.0 to 0.6) |
Unwanted hair loss | 0.3 (1.9) (–0.1 to 0.6) | 0.0 (1.5) (−0.2 to 0.3) | 0.2 (1.5) (0.0 to 0.5) |
Change in voice | 0.2 (1.2) (0.0 to 0.4) | −0.1 (0.7)* (−0.3 to 0) | –0.2 (1.1)* (–0.4 to 0) |
Acne | 0.1 (1.5) (–0.2 to 0.4) | −0.2 (1.8) (−0.5 to 0.1) | –0.3 (1.9) (–0.6 to 0.1) |
Headaches | 0.5 (2.2) (0.1 to 0.9) | −0.2 (2.0)** (−0.5 to 0.2) | –0.1 (2.2) (–0.5 to 0.3) |
Breast pain | 0.3 (1.6) (0.0 to 0.6) | 0.3 (1.5) (0.0 to 0.5) | 0.4 (1.8) (0.0 to 0.7) |
DHEA = dehydroepiandrosterone; SD = standard deviation.
Note: Values are presented as mean (SD) and 95% confidence interval.
Significantly different vs. plain moisturizer: * P ≤ 0.02 and **P < 0.05.
Reprinted by permission from Springer: Barton DL, Sloan JA, Shuster LT, et al. Evaluating the efficacy of vaginal dehydroepiandrosterone for vaginal symptoms in post-menopausal cancer survivors: NCCTG N10C1 (Alliance). Supportive Care in Cancer. 2018;26(2):643 to 650. [Copyright 2018].
Critical Appraisal
Internal Validity
For the primary outcome, approximately 20% and 25% patients discontinued before completion of the study in the plain moisturizer and DHEA groups, respectively. Primary analysis was based on a completed analysis set (“primary end point” data) and was not done in an ITT method. Therefore, the high rate of study discontinuations (missing data of ≥ 20% in each group) introduces uncertainty in the results and it is unclear how the last value carried forward missing data imputation method may have biased the results. Also, it is unclear if the last value carried forward missing data imputation method was used for all the other analyses besides primary outcomes, (i.e., FSFI and quality of life). After all the losses to follow-up, the “primary end point data” set did not meet their intended sample size, (i.e., 145 patients in each arm), so the study is at risk of being underpowered.
For secondary outcomes of self-reported side effects and quality of life, the study used investigator-developed questionnaires. No information was provided that the questionnaires have been validated for their psychometric properties.
Moreover, Barton, et al. stated that most bothersome symptom, either dryness or dyspareunia, was rated as none, mild, moderate, severe, or very severe with no specific indication as to how this descriptive rating scale corresponds to the numerical rating scale as reported in the results (i.e., the study group treated most bothersome symptom as continuous, not ordinal data when reporting results). Considering that the study group performed logistic regression to confirm the results, it seems appropriate to treat data as continuous. However, the primary approach makes inappropriate assumption about the data from a relatively small sample size.
External Validity
The study was conducted for a period of 12 weeks. The 12-week duration may not be ideal for capturing the efficacy and safety and treatment with DHEA among post-menopausal women with history of breast and gynecological cancers. Treatment with DHEA may occur for longer periods of time, and longer-term data would be necessary to understand the long-term impact of treatment in this patient population with history of hormone-dependent cancers.
One could assume that based on the inclusion criteria requiring a history of breast cancer or gynecological cancer (and protocol adherence), most patients would have a history of breast cancer. However, with data missing in the percentage of patients with a history of breast cancer in the intravaginal DHEA group, it is difficult to generalize the finding to a broad population composed of post-menopausal breast cancer survivors. Also, since there is no protocol available, there is a potential for selective outcome reporting.
Even though the FSFI is widely used and assessed in cancer survivors, the MID has not been established. Therefore, determining meaningful improvement or deterioration of sexual function based on changes in the FSFI total or subscale scores remains unclear.
This study specified that patients administer compounded intravaginal DHEA in a gel formulation using a syringe (without a needle) whereas the Health Canada product monograph specifies that prasterone be administered as an ovule and inserted using an applicator. According to the clinical expert consulted by CADTH for this review, the difference between gel, which is similar to creams, and ovule formulations is significant enough to affect adherence and satisfaction rates in patients. Therefore, comparability across other studies which assess DHEA as an ovule versus the gel is limited.
Discussion
Summary of Available Evidence
Two multi-centre, randomized, DB, placebo-controlled trials (the ERC-238 and ERC-231 trials), 1 multi-centre single-group, open-label study (the ERC-230 trial), 1 published ITC, and 3 studies providing supplemental information (the ERC-210 trial15 [N = 217], the Estip-Es study16 [N = 184, and another study by Barton et al.12 [N = 464]) contributed evidence to this report. Patients assessed included post-menopausal women with VVA. The ERC-210 trial also enrolled patients whose most bothersome symptom was not necessarily dyspareunia, the Estip-Es study evaluated the efficacy and safety of prasterone in a real-world clinical setting, and the study by Barton et al.12 included post-menopausal women who had a history of breast or gynecological cancer while having VVA symptoms. The coprimary end points of the ERC-238 and ERC-231 trials included change in percentage of parabasal cells, change in the percentage of superficial cells, change in vaginal pH, and change in the severity score of dyspareunia as the most bothersome self-reported VVA symptom. The primary objective of the ERC-230 trial was to assess the long-term safety of treatment with DHEA.
Patients included in the ERC-238, ERC-231, and ERC-230 trials were of mostly White or non-Hispanic or Latino ethnicity and had a mean of 13 years since their last menses. Patients enrolled also self-identified with moderate to severe dyspareunia (or, in the ERC-230 trial, self-identified at least 1 of dyspareunia, vaginal dryness, or vaginal and/or vulvar irritation or itching as moderate to severe) as their most bothersome VVA symptom at baseline. The major limitation of the trials included a lack of an appropriate comparator, as the ERC-238 and ERC-231 trials included comparison to placebo and the ERC-230 trial did not include a comparator group at all. A published ITC by Li et al.11 was identified which compared prasterone to vaginal estrogen. However, the overall quality of the ITC was considered poor and had statistical limitations which are likely to introduce uncertainty in the treatment comparisons. Additional statistical limitations of the ERC-238, ERC-231, and ERC-230 trials should be considered, including a lack of adjustment of multiplicity and end points which were not included in statistical hierarchies and the results of which should be considered descriptive.
Interpretation of Results
Efficacy
The ERC-238 and ERC-231 trials demonstrated a statistically significant improvement with DHEA over placebo in the 4 coprimary end points after 12 weeks of treatment including a: decrease in percentage of parabasal cells, increase in the percentage of superficial cells, decrease in vaginal pH, and improvement in the severity score of dyspareunia as the most bothersome self-reported VVA symptom. These end points were considered clinically meaningful for patients, and the clinical expert consulted by CADTH for this review confirmed that the magnitude of benefits observed were clinically meaningful. However, it should be noted that no formal values (e.g., MIDs) have been validated to confirm efficacy of end points assessed in this CADTH report beyond clinical expert opinion. In addition, efficacy end points assessing cytology (change in parabasal and superficial cells) and vaginal pH are not outcomes which are used to guide treatment practices or assess patient’s responses to treatment in clinical settings; such end points may be more useful in settings of clinical trials. Rather, treatment of patients is more likely to rely on assessment of symptoms and physical examination for colour, size, and integrity of the anatomy of the vagina. Further, it is likely that only symptomatic patients are likely to be considered for treatment; these patients may have initially started with over-the-counter therapies before seeking intervention from a health care practitioner as their symptoms worsened.
The clinical expert consulted by CADTH indicated that in clinical practice, patients would be expected to use prasterone for longer than 12 weeks. Comparative evidence was unavailable to understand the potential beneficial effects of longer-term treatment with confidence. Longer-term data were available from the single-arm ERC-230 trial which was conducted over a 52-week period. Efficacy end points were secondary in the ERC-230 trial and the lack of comparison group or control for confounding variables, in addition to the absence of consideration for multiple testing, limits the conclusions that can be drawn from this study. The treatment group showed improvements at 52 weeks that seemed to align with the treatment groups in the ERC-238 and ERC-231 trials, which may provide some support to the idea that the benefits of prasterone would be sustained over longer periods of time with consistent administration.
Sexual function results were summarized for the ERC-238 and ERC-230 trials, based on the FSFI. The FSFI is a commonly used and validated tool for measurement of women’s overall sexual function. Results for the ERC-238 trial suggested improvement in sexual function domains, including desire, arousal, lubrication, orgasm, satisfaction, and pain. Longer-term data from the ERC-230 trial suggested that improvements in sexual function may continue to be experienced by patients with continued use of prasterone. Although, it should be noted that the week 12 score for the total score of the FSFI was 23.14 in the prasterone group in the ERC-238 trial, and the week 52 total score was 21.50 in the ERC-230 trial. While these scores are improvements compared to baseline scores, which were 14.29 in the ERC-238 trial and 13.43 in the ERC-230 trial, they are both below the score of 26.55; a value of less than 26.55 is considered to indicate sexual dysfunction in women who complete the questionnaire. Therefore, women may continue to experience symptoms related to VVA even while being treated with prasterone. However, the clinical expert consulted by CADTH for this review agreed that improvements were observed compared to baseline which suggest that treatment with prasterone is likely beneficial in helping to ease symptoms related to VVA and treating the underlying condition to slow its progression. However, statistical considerations should be made when interpreting data pertaining to the FSFI, as assessment of sexual function was a secondary end point in all trials and was not adjusted for multiplicity. In addition, long-term data from the ERC-230 trial was likely underpowered as only data from patients who enrolled after an amendment to the protocol were included. Data pertaining to the FSFI and the impact of prasterone on sexual function should be interpreted with caution.
The comparator group in the ERC-238 and ERC-231 trials included placebo. However, a number of therapies are available to patients, including estrogen therapies. In particular, Vagifem was identified as being available to Canadian patients and is covered under most public insurance plans. The lack of head-to-head comparative evidence is a key limitation of the evidence available for prasterone. No indirect comparisons were submitted by the sponsor. One published ITC was identified in the CADTH literature search that compared DHEA to laser therapy, vaginal estrogen, ospemifene, and moisturizers and lubricants. Efficacy end points included changes in vaginal dryness, dyspareunia, health-related quality of life (assessed through the FSFI), vaginal pH, and proportion of parabasal cells. The results did not clearly favour DHEA over other therapies other than placebo. In particular, neither DHEA nor estrogen therapies, which may be more commonly used among Canadian patients, were favoured between each other. In general, CrIs were wide which may suggest considerable uncertainty between treatment comparison estimates. A number of methodological concerns were identified for the NMA presented, including in some cases violations to the underlying assumptions of transitivity and coherence. It is difficult to interpret the results of the NMA because of important limitations related to reporting of methods and other information needed to appraise the NMA, and unaccounted for heterogeneity across the 29 trials included in the analyses. The limitations of the currently available comparative evidence make it unclear how DHEA may compare to other therapies post-menopausal patients may use for treatment of VVA.
The purpose of the ERC-230 trial was to inform on the long-term harms of treatment with prasterone, as the trial was conducted for a period of 12 months versus the ERC-238 and ERC-231 trials which were conducted for a period of 12 weeks. The clinical expert consulted by CADTH for this review expected that the benefit of treatment with prasterone would continue to be observed over time, and that the results of the ERC-230 trial were supportive of this. While results of the ERC-230 trial appear to support improved symptoms related to VVA for post-menopausal women, the lack of a control group and the statistical considerations of efficacy end points should be considered.
Additional evidence was also summarized in the CADTH report related to the ERC-210 trial, the Estip-Es study, and the study by Barton et al. Although the results of the ERC-210 trial, the observational Estip-Es study, and the trial by Barton et al. were supportive of treatment with prasterone (or DHEA), and aligned with results from the ERC-238, ERC-231, and ERC-230 trials, limitations of the design and analyses of the studies precluded drawing concrete conclusions regarding the efficacy or effectiveness of prasterone.
Harms
AEs were reported in approximately half of patients in the ERC-238 and ERC-231 trials and did not suggest major differences between the prasterone or placebo groups. Application site discharge was 1 of the most commonly reported AEs in the ERC-238, ERC-231, and ERC-230 trials. This AE was stated to be an expected outcome of treatment with the ovule containing prasterone, as the intravaginal administration of the therapy is expected to result in some discharge at the application site. The clinical expert consulted by CADTH confirmed that this AE is likely not due to the prasterone itself and is most likely related to the hard fat composition of the vehicle which is expected to cause discharge when administered intravaginally. Other commonly reported AEs included urinary tract infections, hot flush, and nasopharyngitis; the clinical expert consulted by CADTH confirmed that the frequency of these AEs was as expected and are likely to be manageable by patients and clinicians. The reporting of AEs was greater in the ERC-230 trial compared to the ERC-238 and ERC-231 trials. However, the trial duration of the ERC-238 and ERC-231 trials was 12 weeks, while the ERC-230 trial was conducted over a 52-week period. It was expected that reporting of AEs would be greater for the ERC-230 trial as the trial duration is longer, and patients would have an overall greater exposure to prasterone. In particular, the proportion of urinary tract infections reported in the ERC-230 trial was almost double what was reported for patients who also received prasterone in the ERC-238 and ERC-231 trials (10.2% versus 4.5% and 5.7%, respectively). As the ERC-230 trial did not have a comparison group, it is unclear whether this increase in urinary tract infections was related to treatment with prasterone.
Notable harms pre-specified in the CADTH systematic review protocol included vaginal hemorrhage, endometrial dysplasia, cervical dysplasia, and breast mass. In general, the proportion of patients with these harms as AEs was low (< 4%); for the ERC-238 and ERC-231 trials, the proportions of patients with these harms were similar between the prasterone and placebo groups. The ERC-230 trial also reported on additional breast, cervical, and endometrial safety data which were obtained from patients who underwent mammograms, Pap smears, and endometrial biopsies, respectively, at baseline and at the end of the study. Breast safety data showed that almost all (99%) patients who underwent a mammogram had normal or no significant findings. Pap smear safety data revealed that of the 430 patients who underwent a Pap smear, only 13 patients revealed atypical results of ASCUS, low-grade squamous epithelial lesion, or high-grade squamous epithelial lesion; of these 13 patients, 7 had a negative HPV test. Regarding endometrial safety, the endometrium of most patients who were able to undergo an endometrial biopsy (86%) was considered atrophic with no clinically significant histological findings; the 43 patients who instead underwent a transvaginal ultrasound had an average endometrial thickness of 2.2 mm (SD = 1.4). Additional endometrial safety data were reported for non-hysterectomized patients enrolled in the ERC-231 trial, which reported similar results as the ERC-230 trial where all patients were found to have an atrophic endometrium after biopsy. In general, while no evidence suggested an increased risk of breast, cervical, or endometrial dysplasia, the short trial duration of the ERC-231 trial and the lack of a comparator in the ERC-230 trial should be taken into consideration when interpreting these safety data.
Additional safety data were identified for post-menopausal patients with history of breast and gynecological cancers in a study by Barton et al.12; this was identified as a population of interest as many therapies for VVA rely on estrogen-based therapies which are contraindicated for patients with history of breast and gynecological cancers. The most commonly reported AEs (> 5) in the study by Barton et al.12 included headache and breast pain; there were no differences in the proportions of these AEs between patients who were treated with prasterone and plain moisturizers. While there were no indications of serious harms with treatment of prasterone in this patient population, the study was limited by a short follow-up time of 12 weeks. Longer-term data would be valuable for understanding the effects of prasterone treatment in this population of patients who are sensitive to hormonal therapies and are having VVA.
AEs were assessed in the NMA published by Li et al.11 and no evidence was found for a difference in the odds of AEs occurring with treatment between prasterone and vaginal estrogen therapy. As mentioned previously, the NMA had some limitations which may have introduced uncertainty into estimates and results should be interpreted with caution.
Conclusions
Three trials, including 2 multi-centre, randomized, DB, placebo-controlled trials (the ERC-238 and ERC-231 TRIALS), and 1 multi-centre, single-group, open-label study (the ERC-230 study) provided evidence on the safety and efficacy of prasterone for post-menopausal patients with VVA. Compared to placebo, prasterone 6.5 mg showed greater improvements after 12 weeks of treatment in percentage of parabasal cells, percentage of superficial cells, vaginal pH, dyspareunia, and vaginal dryness that were clinically meaningful according to the clinical expert consulted by CADTH. The ERC-230 trial provided long-term data on the use of prasterone; however, the lack of a comparator precluded the ascertainment of causal relationships, and the study did not adjust for multiple comparisons. The findings over a treatment period of 52 weeks, however, seemed similar to the findings of the 2 shorter-term trials, suggesting that it is possible that the benefits would be sustained with continued treatment. While the results for sexual function measured using the FSFI in the ERC-238 trial also favoured the prasterone group compared to the placebo group, these results were unadjusted for multiplicity and should be considered exploratory. Safety data from the ERC-238 and ERC-231 trials showed similar proportions of patients with AEs between the prasterone and placebo groups; the ERC-230 trial reported higher proportions of patients with AEs compared to the ERC-238 and ERC-231 trials, which may be expected due to the longer exposure to prasterone. However, due to the lack of a control group, it is unclear whether AEs may be associated with prasterone itself. AEs identified in the ERC-230 trial were mostly similar to those identified in the ERC-238 and ERC-231 trials; all trials reported application site discharge and urinary tract infections as the most commonly reported AEs. Comparison of prasterone to other therapies was assessed through a published ITC. In general, the ITC did not provide evidence for a difference in efficacy between prasterone and vaginal estrogen therapies (grouped as a single comparator), though there was considerable uncertainty in the treatment effect estimates. Limitations related to reporting of the NMA and heterogeneity across the included studies that could not be resolved precluded drawing strong conclusions about the comparative effectiveness and safety of prasterone versus other treatments for VVA. Additional evidence was identified to inform on the safety and efficacy of prasterone: the ERC-210 trial, the Estip-Es study, and a study by Barton et al. While the results of the ERC-210 trial were supportive of those from the 3 pivotal trials mentioned above, limitations of the design and analyses of the Estip-Es and Barton et al. studies precluded drawing concrete conclusions regarding the efficacy or safety of prasterone versus vaginal estrogen therapies or moisturizers and lubricants. The study by Barton et al. enrolled patients with history of breast and gynecological cancers but did not find a difference between DHEA 6.5 mg daily and placebo for the most bothersome symptom (dyspareunia or vaginal dryness) and did not assess the notable harms identified for this review outside of AE reporting over the 12-week treatment period.
References
1.Johnston S, Bouchard C, Fortier M, Wolfman W. Guideline No. 422b: Menopause and Genitourinary Health. J Obstet Gynaecol Can. 2021;07:07.
2.Portman DJ, Gass MLS. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women's Sexual Health and The North American Menopause Society. Climacteric. 2014;17(5):557-563. PubMed
3.The North American Menopause Society. The 2020 genitourinary syndrome of menopause position statement of The North American Menopause Society. Menopause. 2020;27(9):976-992. PubMed
4.Nappi RE, Kokot-Kierepa M. Women's voices in the menopause: results from an international survey on vaginal atrophy. Maturitas. 2010;67(3):233-238. PubMed
5.Nappi RE, Palacios S, Bruyniks N, Particco M, Panay N, on behalf of the ESi. The burden of vulvovaginal atrophy on women's daily living: implications on quality of life from a face-to-face real-life survey. Menopause. 2019;26(5). PubMed
6.Intrarosa (prasterone vaginal ovules): Ovules, 6.5 mg prasterone, vaginal [product monograph]. In: Quebec: (QC): Endoceutics, Inc.; 2019.
7.Menopause. Dipex Charity. Healthtalk Web site. https://healthtalk.org/. Published 2019. Accessed 23 Dec 2021.
8.EndoCeutics Inc. DHEA Against Vaginal Atrophy - Safety Study of 12 Months [internal sponsor's report]. In: Quebec, Canada EndoCeutics Inc.; 2015.
9.EndoCeutics Inc. Clinical Study Report: ERC-231. DHEA Against Vulvovaginal Atrophy (Placebo-Controlled, Double-Blind and Randomized Phase III Study of 3-Month Intravaginal DHEA) [internal sponsor's report]. In: Quebec, Canada EndoCeutics Inc.; 2015.
10.EndoCeutics Inc. Clinical Study Report: ERC-238. Intravaginal Prasterone (DHEA) Against Vulvovaginal Atrophy Associated with Menopause (Placebo Controlled, Double Blind and Randomized Phase III Study) [internal sponsor's report]. In: Quebec, Canada EndoCeutics Inc.; 2015.
11.Li B, Duan H, Chang Y, Wang S. Efficacy and safety of current therapies for genitourinary syndrome of menopause: A Bayesian network analysis of 29 randomized trials and 8311 patients. Pharmacological Research. 2021;164 (no pagination). PubMed
12.Barton DL, Sloan JA, Shuster LT, et al. Evaluating the efficacy of vaginal dehydroepiandosterone for vaginal symptoms in postmenopausal cancer survivors: NCCTG N10C1 (Alliance). Support Care Cancer. 2018;26(2):643-650. PubMed
13.Biglia N, Peano E, Sgandurra P, et al. Low-dose vaginal estrogens or vaginal moisturizer in breast cancer survivors with urogenital atrophy: a preliminary study. Gynecol Endocrinol. 2010;26(6):404-412. PubMed
14.Loprinzi CL, Abu-Ghazaleh S, Sloan JA, et al. Phase III randomized double-blind study to evaluate the efficacy of a polycarbophil-based vaginal moisturizer in women with breast cancer. Journal of Clinical Oncology. 1997;15(3):969-973. PubMed
15.EndoCeutics Inc. Clinical Study Report: ERC-210. [internal sponsor's report]. In: Quebec, Canada EndoCeutics Inc.
16.Gonzalez SP, Mainar LB, Campo LR. Effectiveness, Safety and Tolerability of Intravaginal Prasterone for the Treatment of Genitourinary Syndrome in Postmenopausal Women in Spain: The Estip-Es Study. American Journal of Biomedical Science & Research. 2021;12(6).
17.Vagifem 10 (Estradiol vaginal inserts USP): 10 µg estradiol, Vaginal inserts with applicators [product monograph]. In: Mississauga (ON): Novo Nordisk Canada Inc.; 2017: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/Vagifem_10_PM_English.pdf. Accessed 12 Jan 2021.
18.ESTRING (17 β-Estradiol) Vaginal Ring, 2 mg estrogen [product monograph]. In: Kirkland (QC)2017: https://www.pfizer.ca/sites/default/files/201711/Estring_PM_E_208652_16Nov2017.pdf. Accessed 12 Jan 2021.
19.PREMARIN (conjugated estrogens sustained release tablets) 0.3 mg, 0.625 mg, and 1.25 mg, ESTROGENIC HORMONES [product monograph]. In: Kirkland (QC): Wyeth Canada 2014: https://www.pfizer.ca/sites/default/files/201710/Premarin_PM_177429_1Dec2014_EN.pdf. Accessed 12 Jan 2021.
20.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
21.Labrie F, Archer DF, Koltun W, et al. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause. 2018;25(11):1339-1353. PubMed
22.Labrie F, Archer DF, Koltun W, et al. Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause. 2016;23(3):243-256. PubMed
23.Labrie F, Derogatis L, Archer DF, et al. Effect of Intravaginal Prasterone on Sexual Dysfunction in Postmenopausal Women with Vulvovaginal Atrophy. J Sex Med. 2015;12(12):2401-2412. PubMed
24.Archer DF, Labrie F, Montesino M, Martel C. Comparison of intravaginal 6.5mg (0.50%) prasterone, 0.3mg conjugated estrogens and 10mug estradiol on symptoms of vulvovaginal atrophy. J Steroid Biochem Mol Biol. 2017;174:1-8. PubMed
25.Archer DF, Labrie F, Bouchard C, et al. Treatment of pain at sexual activity (dyspareunia) with intravaginal dehydroepiandrosterone (prasterone). Menopause. 2015;22(9):950-963. PubMed
26.Bouchard C, Labrie F, Derogatis L, et al. Effect of intravaginal dehydroepiandrosterone (DHEA) on the female sexual function in postmenopausal women: ERC-230 open-label study. Horm. 2016;25(3):181-190. PubMed
27.Labrie F, Archer DF, Bouchard C, et al. Prasterone has parallel beneficial effects on the main symptoms of vulvovaginal atrophy: 52-week open-label study. Maturitas. 2015;81(1):46-56. PubMed
28.Center for Drug Evaluation R. Summary Review. In: Intrarosa (prasterone) Vaginal inserts. EndoCeutics Inc.: Manufacturer. Application No.:208470. Approval date: 11/16/2016 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2016.
29.Assessment Report. In: Estradiol-containing (0.01% w/w) medicinal products for topical use, Procedure no: EMEA/H/A-31/1482. Amsterdam (NL): European Medicines Agency (EMA); 2020.
30.Davila GW, Singh A, Karapanagiotou I, et al. Are women with urogenital atrophy symptomatic? Am J Obstet Gynecol. 2003;188(2):382-388. PubMed
31.Rosen R, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther. 2000;26(2):191-208. PubMed
32.Endoceutics, Inc. response to November 23, 2021 DRR request for additional information regarding Intrarosa DRR review: [internal additional sponsor's information]. In: Quebec (QC): Endoceutics, Inc.; 2021.
33.Guidance for Industry. In: Estrogen and Estrogen/Progestin Drug Products to Treat Vasomotor Symptoms and Vulvar and Vaginal Atrophy Symptoms — Recommendations for Clinical Evaluation. Rockville (MD): Center for Drug Evaluation and Research; 2003
34.Simon JA, Lin VH, Radovich C, Bachmann GA, The Ospemifene Study G. One-year long-term safety extension study of ospemifene for the treatment of vulvar and vaginal atrophy in postmenopausal women with a uterus. Menopause. 2013;20(4). PubMed
35.Labrie F, Archer D, Bouchard C, et al. Effect of intravaginal dehydroepiandrosterone (Prasterone) on libido and sexual dysfunction in postmenopausal women. Menopause. 2009;16(5):923-931. PubMed
36.Labrie F, Archer D, Bouchard C, et al. Intravaginal dehydroepiandrosterone (Prasterone), a physiological and highly efficient treatment of vaginal atrophy. Menopause. 2009;16(5):907-922. PubMed
37.Meston CM. Validation of the Female Sexual Function Index (FSFI) in women with female orgasmic disorder and in women with hypoactive sexual desire disorder. J Sex Marital Ther. 2003;29(1):39-46. PubMed
38.Wiegel M, Meston C, Rosen R. The female sexual function index (FSFI): cross-validation and development of clinical cutoff scores. J Sex Marital Ther. 2005;31(1):1-20. PubMed
39.Reed SD, Mitchell CM, Joffe H, et al. Sexual Function in Women on Estradiol or Venlafaxine for Hot Flushes: A Randomized Controlled Trial. Obstet Gynecol. 2014;124(2 PART 1). PubMed
40.Stephenson KR, Toorabally N, Lyons L, C MM. Further Validation of the Female Sexual Function Index: Specificity and Associations With Clinical Interview Data. J Sex Marital Ther. 2016;42(5):448-461. PubMed
41.Neijenhuijs KI, Hooghiemstra N, Holtmaat K, et al. The Female Sexual Function Index (FSFI)-A Systematic Review of Measurement Properties. J Sex Med. 2019;16(5):640-660. PubMed
42.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
43.Leiblum S, Bachmann G, Kemmann E, Colburn D, Swartzman L. Vaginal atrophy in the postmenopausal woman. The importance of sexual activity and hormones. Jama. 1983;249(16):2195-2198. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: October 27, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit retrieval by study type
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
exp Dehydroepiandrosterone/
(prasteron* or dehydroepiandrosterone or dehydro-epi-androsterone or DHEA or DHEAS or dehydroisoandrosterone or androstenolone or andrestenol or anastar or aslera or astenile or chetovis or diandron* or gynodian or prestara or psicoterone or siscelar plus or dehydroandrosterone or BRN 2058110 or BRN2058110 or "caswell no. 051F" or CCRIS 3277 or "5-Androsten-3-ol-17-one" or "hydroxyandrost-5-en-17-one" or "hydroxy-5-androsten-17-one" or Chetovis or hormoforin or 53-43-0 or 459AG36T1B or 105597-37-3 or 108673-53-6 or 9013-35-8 or 2283-82-1 or 25375-38-6 or EM-760 or EM760 or GL 701 or GL701or NSC 9896 or NSC9896 or EL-10 or EL10 or "PB 005" or PB005).ti,ab,kf,ot,hw,rn,nm.
or/1-2
Dyspareunia/
(Vagina/ or Vaginal Diseases/ or Vulva/ or Vulvan Diseases/) and Atrophy/
administration, vaginal/
((vulvovagina* or vagina*) adj3 (atroph* or erosion)).ti,ab,kf.
(VVA or genitourinary syndrome or genito urinary syndrome or GSM or dyspareunia* or atrophic vaginitis).ti,ab,kf.
or/4-7
and/3,9
intrarosa*.ti,ab,kf,rn,nm.
or/10-11
12 use medall
*Prasterone/
*Prasterone sulfate/
(prasteron* or dehydroepiandrosterone or dehydro-epi-androsterone or DHEA or DHEAS or dehydroisoandrosterone or androstenolone or andrestenol or anastar or aslera or astenile or chetovis or diandron* or gynodian or prestara or psicoterone or siscelar plus or dehydroandrosterone or BRN 2058110 or BRN2058110 or "caswell no. 051F" or CCRIS 3277 or "5-Androsten-3-ol-17-one" or "hydroxyandrost-5-en-17-one" or "hydroxy-5-androsten-17-one" or Chetovis or hormoforin or EM-760 or EM760 or GL 701 or GL701or NSC 9896 or NSC9896 or EL-10 or EL10 or "PB 005" or PB005).ti,ab,kf,dq.
or/14-16
Dyspareunia/
Vagina atrophy/
((vulvovagina* or vagina*) adj3 (atroph* or erosion)).ti,ab,kf,dq.
(VVA or genitourinary syndrome or genito urinary syndrome or GSM or dyspareunia* or atrophic vaginitis).ti,ab,kf,dq.
or/18-21
and/17,22
intrarosa*.ti,ab,kf,dq.
or/23-24
25 use oemezd
(conference abstract or conference review).pt.
26 not 27
or/13,28
remove duplicates from 29
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Intrarosa*, prasterone, menopaus*, DHEA, dehydroepiandrosterone]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- Intrarosa*, prasterone, menopaus*, DHEA, dehydroepiandrosterone]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Intrarosa*, prasterone, menopaus*, DHEA, dehydroepiandrosterone]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Intrarosa*, prasterone, menopaus*, DHEA, dehydroepiandrosterone]
Grey Literature
Search dates: October 21, 2021 – October 25, 2021
Keywords: Intrarosa*, prasterone, menopaus*, DHEA, dehydroepiandrosterone
Limits: No limits were applied
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Barton DL, Shuster LT, Dockter T, et al. Systemic and local effects of vaginal dehydroepiandrosterone (DHEA): NCCTG N10C1 (Alliance). Support Care Cancer. 2018;26(4):1335-1343. | Population |
Bouchard C, Labrie F, Archer DF, et al. Decreased efficacy of twice weekly intravaginal dehydroepiandrosterone on vulvovaginal atrophy. Climacteric. 2015;18(4):590-607. | Intervention |
Labrie F. DHEA as physiological replacement therapy at menopause. Journal of Endocrinological Investigation. 1998;21(6):399-401. | Intervention |
Labrie F, Archer D, Bouchard C, et al. Lack of influence of dyspareunia on the beneficial effect of intravaginal prasterone (dehydroepiandrosterone, DHEA) on sexual dysfunction in postmenopausal patients. J Sex Med. 2014;11(7):1766-1785. | Comparator |
Labrie F, Archer DF, Bouchard C, et al. Intravaginal dehydroepiandrosterone (prasterone), a highly efficient treatment of dyspareunia. Climacteric. 2011;14(2):282-288. | Comparator |
Pieta W, Smolarczyk R. Vaginal dehydroepiandrosterone compared to other methods of treating vaginal and vulvar atrophy associated with menopause. Przeglad Menopauzalny. 2020;19(4):195-199. | Review article |
Sauer U, Talaulikar V, Davies MC. Efficacy of intravaginal dehydroepiandrosterone (DHEA) for symptomatic patients in the peri- or postmenopausal phase. Maturitas. 2018;116:79-82. | Review article |
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
FSFI was included in the ERC-230, ERC-231, and ERC-238 trials as secondary end points.
VASQ was included in the ERC-230 trial as a secondary end point and in the ERC-231, and ERC-238 trials as primary end points.
Findings
Table 37: Summary of outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
FSFI | A self-completed, 19-item Likert-type scale that consists of 6 domains measuring female sexual function: desire, arousal, lubrication, orgasm, satisfaction, and pain. Score ranges for each domain and full scale:
Higher score indicates better sexual function | Validity: Content validity has been ensured by panel selection of initial items, pre-testing with healthy volunteers, and consultation with experts.31 Discriminant validity has been demonstrated between control groups and patients with various sexual dysfunctions, e.g., FSAD, FSOD, HSDD, and other types (all P ≤ 0.001).31,37,38 Divergent validity has been shown between FSFI and Locke-Wallace Marital Adjustment Test in control groups and patients with FSAD, FSOD, HSDD, in which Pearson correlation coefficients (r) ranged from 0.03 – 0.72, where satisfaction domain showed relatively higher correlation compared to other domains.31,37 Reliability: Pearson correlation coefficients (r) for test-retest reliability were high for all domains (r = 0.79 – 0.86) and full scale (r = 0.88) in a sample population composed of controls and patients with FSAD (n = 259).31 Internal consistency was acceptable (Cronbach α ≥0.7) for all 6 domains and full scale in control and various patient groups, e.g., FSAD, FSOD, HSDD, and other types, except for the desire domain in patients with HSSD (α = 0.58).31,37,38 Responsiveness to change: No data were located. | Unknown FSFI total score of 26.55 is a cut-off to differentiate patients with or without sexual dysfunction |
VASQ | A self-completed questionnaire that consists of 5 items, i.e., dryness, soreness, irritation, dyspareunia (to be completed only if sexually active), and vaginal discharge. Each item can be answered according to severity levels, i.e., none (0), mild (1), moderate (2), and severe (3). The total score is calculated as the sum of individual symptom scores divided by 5, for the sexually active patients who completed the dyspareunia question and divided by 4 if answered “N/A” to the dyspareunia question. Higher score represents more severe atrophic symptom burden. | Validity: Construct validity was assessed by Wilcoxon rank sum tests in female volunteers (mean age = 68.55, range 23-89 years) undergoing systemic hormonal therapy and those not being treated (n = 135, P = 0.0007) based on atrophy symptom scores and maturation values.30 Based on Spearman correlations, little-to-no correlation was observed with maturation value (r = 0.061), age (r = -0.004), vaginal pH (r = 0.031). A weak correlation was found with vaginal health score (r = 0.139), which became weak-moderate in a subgroup of women > 65 years (r = 0.250).30 Reliability: No data were located. Responsiveness to change: No data were located. | Unknown |
FSAD = female sexual arousal disorder; FSFI = Female Sexual Function Index; FSOD = female sexual orgasm disorder; HSDD = hypoactive sexual desire disorder; MID = minimal important difference; VASQ = vaginal atrophy symptom questionnaire.
Female Sexual Function Index
The FSFI is a self-reported, multidimensional, 19-item measure of female sexual function consisting of 6 domains: desire, arousal, lubrication, orgasm, satisfaction, and pain. Following initial usability testing among 30 female volunteers, the FSFI was tested for validity and reliability with a sample of 259 control females from the general population (age 21–68 years) and an age-matched clinical sample of patients who met DSM-IV-TR criteria for Female Sexual Arousal Disorder (FSAD).31 Later, the instrument was tested among females with other sexual dysfunction diagnoses, e.g., Female Sexual Orgasm Disorder (FSOD), Hypoactive Sexual Desire Disorder (HSDD), dyspareunia / vaginismus (pain), and multiple sexual dysfunctions.37,38 The questionnaire was designed to be used to assess female sexual function and QoL in clinical trials or epidemiological studies.31
FSFI provides an overall sexual function score on a Likert-type scale. Specifically, each item is scored from 0 to 5 except for questions 1 and 2 (for desire domain), 15-16 (in satisfaction domain), which are scored from 1 to 5. The individual domain scores and full-scale (overall) score of the FSFI can be derived from the computational formula. For individual domain scores, scores of the individual items that comprise the domain are added and multiplied by the corresponding domain factor. Full-scale score is obtained by adding 6 domain scores together. Score ranges for the arousal, lubrication, orgasm, and pain domains are from 0 to 6.0; for desire and satisfaction domains ranges are from 1.2 to 6.0 and from 0.8 to 6.0, respectively. The total FSFI score ranges from 2.0 to 36.0. Higher scores indicate a greater level of sexual function. The recall period is the past 4 weeks. A domain score of zero indicates that the subject reported having no sexual activity during the past month.39 An FSFI total score of 26.55 is taken to be the cut score for differentiating women with and without sexual dysfunction.38
The FSFI has been translated into more than 20 languages and has been adapted in more than 30 countries.40,41 Also, it has been studied for use with multiple populations, including women from different age groups, with diverse medical conditions, and with various sexual dysfunctions.40,41
Reliability
Rosen et al.31 assessed test-retest reliability at 2 separate visits 2 to 4 weeks apart, with 131 general population controls and 128 age-matched patients diagnosed with FSAD at 5 research centres in the US. It is unclear if the post-menopausal patients included in the study had symptoms attributable to VVA. Overall test-retest reliability was acceptable (r ≥ 0.70)42 for all of the domains (Pearson’s product-moment correlation [r] = 0.79 – 0.86) and for the total scale (r = 0.88) among the full sample. In general, higher test-retest reliability of domain scores was obtained for the control group than for the FSAD group. For the FSAD group, the domain of desire showed the highest test-retest reliability (r = 0.80), with the other domains showing moderate to high correlations (r = 0.62 to 0.71).
In addition, Rosen et al.31 demonstrated acceptable internal consistency (Cronbach alpha ≥ 0.70)42 for all 6 domains (Cronbach alpha ≥ 0.82 for all 6 domains). Meston37 conducted further internal consistency testing among women with FSOD (n = 71) or HSDD (n= 44), as well as control patients (n = 71) who were age-matched to FSOD patients. Meston found that inter-item correlations remained high for all the domain scores among women with FSOD (Cronbach alpha ≥ 0.84), control women (alpha ≥ 0.83), and women with HSDD (alpha ≥ 0.74), except for the desire domain in patients with HSSD (alpha = 0.58). The moderate Cronbach alpha value of 0.58 suggests that the 2-item FSFI desire composite may not be a reliable indicator of sexual desire among this population.
Lastly, Wiegel et al.38 combined data from Rosen et al. (n = 255) and Meston (n = 138) with an additional sample population (n = 134) to assess internal consistency in women with or without sexual dysfunction. Internal consistency for subscales and total score was acceptable (alpha ≥ 0.7)42 for all sample populations and all domains regardless of sexual dysfunction status indicating that questionnaire items remained highly related within each domain or all scale in women with or without sexual dysfunction.
Validity
Validity reflects the degree to which the instrument measures what it aims to measure.
Content validity has been ensured by Rosen et al.31 in the population as described in the reliability section above. Briefly, the FSFI was developed in a series of stages, including panel selection of the initial items, pre-testing with 30 healthy, female volunteers at 3 investigational sites, followed by linguistic and conceptual validation with a panel of expert consultants.
Discriminant validity was assessed by Rosen et al.31 by comparing the mean responses of patients with FSAD (n = 126-128) with those of the controls without FSAD (n = 129-131). Significant differences (for all P ≤ 0.001) between the groups were observed for all 6 domains (not included in this report) and the full-scale score (19.2 ± SD 6.63 in 126 patients with FSAD vs. 30.5 ± SD 5.29 in 129 control patients). Moreover, Meston37 assessed the discriminant validity in additional clinical samples by comparing the mean responses of women with FSOD (n = 71) and HSDD (n = 44) to those of the age-matched control women (n = 71 and 44, respectively). The results from between groups analyses of variance revealed significant differences between women with sexual dysfunction (FSOD or HSDD) and their controls on each of the FSFI domain and total scores (for all P ≤ 0.001). As expected, the largest differences between women with FSOD and controls were noted for orgasm and arousal domains (effect size estimated using Cohen D = 1.69, 1.58, respectively), and the largest differences between women with HSDD and controls were seen for the arousal and desire domains (ES estimated using Cohen D = 1.85, 1.69, respectively). Lastly, discriminant validity has been confirmed by Wiegel et al.38 with the sample populations described above in reliability section. Evidence for discriminant validity was observed for the total score (MANOVA and univariate tests, p < 0.001) and individual domain scores (univariate tests, P < 0.001) between patients with and without sexual dysfunction diagnoses.
Divergent validity was tested by Rosen et al.31 by specifically measuring the construct under study (i.e., sexual function) compared to an instrument that assesses a different, albeit partially related, construct (e.g., marital satisfaction). The Pearson product-moment correlation between the Locke-Wallace Marital Adjustment Test and the total FSFI score was moderate for the control group (r = 0.53) and low for the FSAD group (r = 0.22). This result indicates that for subjects with sexual dysfunction, FSFI scores appeared to have a greater degree of independence with the marital adjustment effect compared to control group. To extend divergent validity testing in other clinical samples, Meston37 calculated Pearson correlations between the FSFI scores and the Locke-Wallace Marital Adjustment Test score in patients with FSOD or HSDD. Correlations between the Locke-Wallace Marital Adjustment Test and total FSFI scores were low for women with FSOD (r = 0.22) or HSDD (r = 0.16), and moderate for controls (r = 0.52). Of note, the satisfaction domain showed moderate to high level of correlation (r = 0.40 – 0.72) between FSFI and Locke-Wallace across all the samples.
Responsiveness to Change
There was no evidence located to support the FSFI’s responsiveness to clinical or health status changes among post-menopausal patients over time.
MID
MID for FSFI has not been estimated in post-menopausal patients with VVA-associated symptoms.
Other Considerations and Limitations
The 3 studies, i.e., Rosen et al., Meston, Wiegel et al., assessed validity in control and sexually dysfunctional populations that contain various numbers of post-menopausal women. Based on the age of populations included in these studies, the number of post-menopausal women seems small, For example, Rosen et al.31 study population had mean age ± SD ages of the FSAD group (n = 128) and control group (n = 131) 40.5 ± 12.98 years and 39.7 ± 13.15 years, respectively. Meston study37 included women between 18 and 53 years of age with mean ± SD for patients with FSOD (n = 71) 29.4 ± 8.76 years, HSDD (n = 44) 33.0 ± 10.42 years, and controls (n = 71) 29.2 ± 7.9 years, respectively. Lastly, in Wiegel et al., the mean age of the combined sample was 36.2 ±13.2 years and ranged from 18-72 years. Only 3.6% (n = 20) patients in Wiegel et al. study were peri-or post-menopausal. Taken together, no study is specifically designed to test psychometric properties of FSFI in post-menopausal women.
Also, these studies were conducted with patients diagnosed with sexual dysfunctions rather than those with symptoms of post-menopausal VVA. Some manifestations of sexual dysfunctions and post-menopausal VVA may overlap, for example, dyspareunia. However, the extent of overlap is unclear. It makes it difficult to apply the validity results to population other than those with sexual dysfunction diagnoses.
Vaginal Atrophy Symptom Questionnaire
The sponsor used their own term, VASQ, to refer to the atrophy symptom questionnaire, which was modified from previous reported form, Vaginal Atrophy Index (VAI).43
The VASQ that was to be completed by patients in the sponsor’s trials consists of 5 items: dryness, soreness, irritation, dyspareunia, and vaginal discharge. The dyspareunia item is only to be completed if patients are sexually active. Each item can be answered among 4 choices, i.e., none, mild, moderate, and severe, according to the symptom severity the patients are experiencing. No information regarding recall period was available.
The presence and/or severity of each recorded symptom on the patient form was assigned a score from 0 (none/no atrophy) to 3 (severe). The total atrophy symptoms (or VASQ) score is calculated as the sum of individual symptom scores divided by 5, for the sexually active patients who completed the question regarding dyspareunia. For those respondents who were not sexually active and who answered “N/A” to the dyspareunia question, the sum of individual symptom scores is divided by 4. Higher scores represent more severe atrophic symptom burden.
Reliability
There was no evidence located to support the reliability of the VASQ.
Validity
Davila et al.30 assessed construct validity of the VASQ in 135 female volunteers who were patients being seen for gynecologic care (mean age = 68.55 years, range 23-89 years). Results showed that women who were receiving systemic estrogen therapy had significantly higher atrophy symptoms scores (mean =0.53; SD = not reported) than women who were not on systemic estrogen treatment (mean = 0.27; SD = not reported; P = 0.0007).
In addition, Davila et al.30 assessed the strength of relationships between several variables and atrophy symptom score. Vaginal atrophy symptom scores had little-to-no correlation with maturation value (r = 0.061), age (r = -0.004), or vaginal pH (r = 0.031).
Responsiveness to Change
There was no evidence located to support the responsiveness of the VASQ to changes in vaginal symptom in post-menopausal patients during treatments over time.
MID
MIDs for the VASQ has not been estimated in post-menopausal patients with VVA-associated symptoms.
Other Considerations and Limitations
Even though VAI has been used in previous urogenital atrophy study,43 it has not been rigorously tested for psychometric properties.
Even though Davila et al. study provides some validity evidence for the VASQ, the population chosen did not select for patients who were post-menopausal or who had symptoms of VVA.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
AHT
alternative hormone therapy
BIA
business impact analysis
CE
conjugated estrogen
DHEA
dehydroepiandrosterone
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
QALY
quality-adjusted life-year
VVA
vulvovaginal atrophy
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Prasterone (Intrarosa) vaginal ovules |
Submitted price | Prasterone, 6.5 mg, $1.46 per ovule ($40.78 per box of 28 ovules) |
Indication | For the treatment of post-menopausal vulvovaginal atrophy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 1, 2019 |
Reimbursement request | As per indication |
Sponsor | Lupin Pharma Canada Ltd. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Post-menopausal patients with VVA who exhibited moderate to severe dyspareunia as their MBS |
Treatment | Prasterone |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | 30 years |
Key data source | ERC-231 and ERC-238 clinical trials for prasterone and no treatment; studies from the literature informed key data for CE cream, estrone cream, and estradiol ring |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
CE = conjugated estrogen; ICER = incremental cost-effectiveness ratio; MBS = most bothersome symptom; QALY = quality-adjusted life-year; VVA = vulvovaginal atrophy.
Conclusions
Based on the CADTH clinical review, prasterone is an effective therapy for vulvovaginal atrophy (VVA), when compared to placebo, in post-menopausal patients whose most bothersome symptom is dyspareunia . The trials do not provide direct evidence on the relative efficacy and safety of prasterone versus other available local hormone-based therapies. An indirect treatment comparison (ITC) comparing prasterone with vaginal estrogen therapies was identified; however, key limitations with the evidence make it unclear how prasterone may compare to other therapies post-menopausal patients may use for the treatment of VVA. The relative effectiveness of prasterone to estrogen-based vaginal inserts or creams is therefore unknown, as is the magnitude of any potential safety differences.
Due to the extent of uncertainty with the clinical evidence in the model, a CADTH base case could not be derived. CADTH undertook exploratory reanalyses to address limitations identified in the sponsor’s submission. The CADTH combined exploratory analysis considered all comparators relevant to Canadian clinical practice, assumed equal response to treatment, adverse event (AE), and discontinuation rates due to the absence of direct or robust indirect comparative evidence, and altered the dosing assumptions for estradiol vaginal tablets and conjugated estrogen (CE) cream to align with the dosing most commonly used in clinical practice. In CADTH’s combined exploratory analysis, prasterone was more costly ($462 incremental costs) and equally effective in comparison with CE cream, leading to prasterone being dominated by CE cream. A price reduction of 89% would be required for prasterone to be cost-neutral to CE cream. These results are driven primarily by differences in drug acquisition costs, with prasterone being more costly than most other available hormone therapies for local application used to treat symptoms of VVA, with the exception of estradiol vaginal tablets.
The results of the CADTH reanalyses are similar when compared to the sponsor’s. In the sponsor’s scenario, which included additional comparators, prasterone was found to be dominated by CE cream (i.e., more costly, and slightly less effective) given the sponsor’s analysis included better response rates at 12 weeks with CE cream in comparison with prasterone. In comparison, CADTH assumed equal efficacy in the exploratory analysis due to lack of robust evidence to indicate differential treatment efficacy among all comparators. Based on the economic analysis, there is no evidence to support a price premium for prasterone in comparison with other available local hormone therapies used to treat symptoms of VVA. Likewise, the sponsor’s analysis suggests cost savings with prasterone may be necessary should it be inferior to its less expensive comparators.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Feedback was received from the Women’s Health Coalition of Alberta, which highlighted the underserved nature of uro-gynecological health in the health care system, the limited therapeutic options for conditions associated with menopause, and the often overlooked or dismissed clinical and psychological effects of untreated menopausal conditions. The source of this information was unclear.
Three clinician groups submitted input: Cleopatra, a private virtual clinic; the Society of Obstetricians and Gynaecologists of Canada, and the Mount Sinai Division of Menopause and Mature Women’s Health. All organizations indicated that over-the-counter lubricants and moisturizers are generally used first to treat VVA, often before a patient seeks medical help, though 2 groups specified they contain ingredients that may be harmful or unpleasant to sensitive vaginal tissues. All groups saw prasterone as being an alternative to vaginal estrogen inserts or creams, usable in place of estrogen or in cases where estrogen therapy had failed or was not tolerated. Prasterone was seen by the responding clinician groups as associated with fewer side effects and as less concerning to patients than estrogen-based therapies.
Drug plan input noted that prasterone was only compared to placebo in clinical trials; therefore, its efficacy relative to topical or inserted estrogen products is unknown. Also mentioned was the potential for combination therapy with vaginal estrogen products, and the consideration that moisturizers and lubricants should remain first-line treatments, with this product becoming a second-line option like low-dose estrogen topicals or inserts.
Several of these concerns were addressed in the sponsor’s model.
Differences in potential side effect profiles between comparators were considered.
Other available vaginal estrogen inserts and creams frequently used in practice were included in an additional analysis.
In addition, CADTH addressed some of these concerns as follows.
In the absence of direct or indirect comparative evidence, CADTH reanalyses assumed equal probability of response and discontinuation between active comparators.
Increased market size in the business impact analysis (BIA) for VVA treatments once prasterone is available was included to account for patients who would not or could not otherwise be treated with estrogen-based therapies.
CADTH was unable to address the following concerns raised from stakeholder input.
The sponsor’s model could not account for the use of additional treatments after initial failure to respond to or being unable to tolerate prasterone or the comparators of interest.
Economic Review
The current review is for prasterone (Intrarosa) for the treatment of post-menopausal VVA.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of prasterone ovules compared to estradiol vaginal tablets (Vagifem) and to no treatment in post-menopausal adults with VVA who exhibited moderate to severe dyspareunia as their most bothersome symptom.1 This population is different than that specified in the Health Canada–approved indication, which is for the treatment of post-menopausal VVA and was not specific to moderate to severe dyspareunia as the most bothersome symptom.2 The sponsor also submitted an additional comparator scenario including estrone cream (Estragyn), the estradiol ring (Estring), and CE cream (Premarin) upon request from CADTH.
The recommended dose of prasterone is 1 6.5 mg ovule inserted once a day at bedtime, with patients re-evaluated every 6 months or as clinically appropriate to determine if treatment is still necessary. At the submitted price of $1.46 per 6.5 mg ovule, the annual cost of therapy with prasterone is $532 per patient.
For the base case and additional comparator scenario, the sponsor estimated costs and quality-adjusted life-years (QALYs) for each treatment regimen from the perspective of a Canadian health care payer, over a 30-year time horizon, using a 1.5% annual discount rate for both costs and QALYs.
Model Structure
The sponsor submitted a Markov model with 3-month cycles where all patients entered the model in an initial “12-week assessment” health state. After the assessment period, patients in any of the active treatment groups could then transition to the response, non-response, or death health states (Figure 1) while patients in the no treatment group transitioned to non-response or death.1 Patients who did not respond by 3 months (12 weeks in the trials) were assumed to discontinue therapy and remain in the non-response state for the remainder of the model time horizon. Patients who did respond were assumed to continue to respond until they discontinued therapy, at which point they transitioned to the non-response state and remained there until death.
Model Inputs
Patients entered the model at a mean age of 59 years, based on the mean age of patients in the prasterone clinical trials. Patients were assigned to either: 6.5 mg prasterone ovules inserted daily, 10 mcg estradiol vaginal tablets, where 80% of patients were assumed to use them cyclically3 (21 days on treatment, 7 days off treatment) and 20% used them daily, or no treatment.4-6 Response to treatment was defined as the proportion of patients in clinical trials who had reported dyspareunia as their most bothersome symptom who had moved from moderate to severe symptoms at baseline to mild or none by week 12, as measured by a 4-point dyspareunia symptom severity scale. In the base case, transition probabilities for prasterone were derived from weighted, pooled data from 2 trials, ERC-2315 and ERC-238,6 and were placebo-adjusted by subtracting the absolute probability of response for placebo patients from the absolute probability of response for prasterone patients, due to the moisturizing qualities of the placebo inserts used in clinical trials. Patients in the prasterone group were thus assigned a ||||| probability of responding (||||| response rate in placebo arm of trial subtracted from ||||| response rate from prasterone arm of trial), and the no treatment arm was set to 0% response. As similar response data for estradiol vaginal tablets was not available, the sponsor instead compared the placebo-adjusted severity score improvement reported in a clinical trial of the tablets (–0.36)7 to that of prasterone (–0.39),5,6 resulting in a relative treatment effect of 0.92, which led to an assumed probability of response of ||||| (||||| × 0.92) with estradiol vaginal tablets. Similar calculations were conducted in the additional comparator scenario for estrone and CE cream, while the estradiol ring was assumed to have the same effectiveness as estradiol tablets.8-15 Estrone cream and the estrone ring were assumed to be used as recommended in their product monographs,16,17 while 30% of patients using CE cream were assumed to use 0.5 g daily for 21 days with 7 days off18 and the remaining 70% were assumed to use 0.5 g twice weekly.1
Patients on treatment who had not responded by 3 months were assumed to discontinue therapy and remain non-responsive for the remainder of their lives, receiving no further therapy. Patients who did respond were assumed to continue responding until they discontinued therapy, the rate of which was based on the proportion of patients who withdrew during clinical trials of prasterone5,6 or estradiol vaginal tablets,7,11 calculated as a probability per 3-month cycle (6.7% for prasterone, 9.5% for estradiol vaginal tablets) and extended out over the full time horizon. Patients in all health states had the same risk of mortality as the age- and gender-matched general population.19
Health utilities for the response and non-response states were derived from a 2015 quality of life study of patients with VVA,20 with response assigned the weighted average utility of those with no or mild VVA symptoms (0.844) and non-response assigned the weighted average of patients with moderate or severe VVA symptoms (0.779). Patients could also experience the comorbidities of depression, anxiety, or urinary tract infections, with age-adjusted disutility scores derived from previous cost-effectiveness analyses of treatments for endometriosis21 and uterine fibroids22 and applied per cycle. The probability of a non-responding patient having a comorbidity was derived by taking the incremental probability of women with VVA having the comorbidity minus the probability of women without VVA having it.23,24 Responders were assumed to have half the probability of having each comorbidity that non-responders were assigned. Patients could also experience 1-time AEs, including cervical dysplasia, diarrhea, pain, sinusitis, vaginal discharge, vaginal hemorrhage, vaginal odour, ventricular extrasystoles, vulvovaginal pruritus, or weight gain. The rates for each event were placebo-adjusted as reported in the clinical trials for prasterone or in the applicable product monographs for estradiol vaginal tablets and the other active comparators.3,16-18
Costs included the drug acquisition costs of prasterone and estradiol vaginal tablets,1,25 costs associated with treating comorbidities and AEs,19,25,26 and physician visits,19 where responders were assumed to visit their general practitioner and gynecologist once annually each, and non-responders were assumed to visit their gynecologists 1 additional time per year.
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted a probabilistic analysis of 5,000 iterations. The results of the deterministic analysis were very similar to the probabilistic analysis. The probabilistic findings are reported below.
Base-Case Results
The sponsor’s base-case results are presented in Table 3. Prasterone was associated with 0.04 incremental QALYs at an additional cost of $376 when compared to no treatment, resulting in an incremental cost-effectiveness ratio (ICER) of $9,861 per QALY gained. Estradiol vaginal tablets were associated with fewer QALYs and a higher cost than prasterone in the sponsor’s analysis and were therefore dominated by prasterone.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
No treatment | 6,759 | 15.850 | Reference |
Prasterone (Intrarosa) | 7,137 | 15.888 | $9,861 |
Estradiol (Vagifem) | 7,197 | 15.875 | Dominated by prasterone |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Costs of comparators are based on publicly available list prices and may not reflect costs actually paid by public drug plans.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case and additional comparator scenario are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
Upon request from CADTH, the sponsor submitted an updated economic model and report which included a scenario analysis comparing prasterone to other available comparators used to treat post-menopausal VVA, including topical CE cream, estrone (Estragyn cream), and an estradiol vaginal ring (Estring). Results are presented in Table 4. Under the sponsor’s assumptions, CE cream was the least expensive comparator, even compared to no treatment, and provided the most QALYs; thus, all other comparators were dominated by CE cream, including prasterone.
Table 4: Summary of the Sponsor’s Additional Comparator Scenario Analysis
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
CE (Premarin cream) | 6,731 | 15.917 | Reference |
No treatment | 6,752 | 15.855 | Dominated by CE |
Estrone (Estragyn) | 6,852 | 15.880 | Dominated by CE |
Estradiol ring (Estring) | 7,049 | 15.892 | Dominated by CE |
Prasterone (Intrarosa) | 7,134 | 15.893 | Dominated by CE |
Estradiol (Vagifem) | 7,190 | 15.880 | Dominated by CE |
CE = conjugated estrogen; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Costs of comparators are based on publicly available list prices and may not reflect costs actually paid by public drug plans.
Source: Sponsor’s pharmacoeconomic submission.1
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
Key comparators were excluded from the sponsor’s base case: The sponsor’s base-case analysis only considered prasterone in comparison with estradiol vaginal tablets (Vagifem) and no treatment. The sponsor argued that of the available vaginal estrogen products, only the estradiol vaginal tablet was a relevant comparator to prasterone as they considered the estradiol vaginal tablet to be the dominant local estrogen treatment in Canada. Additionally, the reluctance of some women to use estrogen treatment was cited as the reason no treatment was also considered a relevant comparator. However, according to the clinical expert consulted by CADTH as well as IQVIA Pharmastat data,27 CE cream, estrone, and the estradiol ring are all treatments in use and publicly reimbursed in at least some jurisdictions in Canada and are therefore relevant to include. Of note, estradiol vaginal tablets are not reimbursed in all jurisdictions (e.g., British Columbia), and thus are not a comparator of interest to all jurisdictions.
CADTH considered the sponsor’s scenario which included the additional comparators to be most relevant. All other CADTH exploratory reanalyses were conducted on this scenario rather than on the sponsor’s base case.
Comparative clinical efficacy and safety of prasterone with relevant comparators is unknown: No comparative clinical evidence was submitted comparing prasterone to any active comparator; the included clinical trials compared prasterone only to placebo.5,6 In the absence of comparative evidence, the sponsor’s method of determining placebo-adjusted response probabilities across different trials, and even outcomes, and calculating a relative ratio of response in comparison with prasterone constitutes a naive comparison between treatments, with limited attempt to adjust for trial or patient characteristic differences. The sponsor’s approach to determining relative response rates in the submitted model, and the results predicted by the model, are associated with considerable uncertainty.
CADTH searched for relevant ITCs and identified a 2021 ITC by Li et al.28 that included a comparison of vaginal dehydroepiandrosterone (DHEA; prasterone) and vaginal estrogen. While vaginal DHEA was favoured over placebo in terms of efficacy outcomes, consistent with the direct evidence, no significant differences were found between vaginal DHEA and vaginal estrogen for vaginal dryness, dyspareunia, pH, the Female Sexual Function index, or odds of an AE occurring. However, due to a number of methodological concerns it is difficult to interpret the results of the ITC with any certainty in determining how prasterone may compare to vaginal estrogen therapies used to treat VVA in post-menopausal patients. As a result, the CADTH clinical review was unable to comment on the comparative efficacy of prasterone in relation to any comparator besides placebo. In the absence of direct or indirect evidence supporting differences between active treatments, it is inappropriate to assume differential efficacy in the base case.
CADTH took a conservative approach and assumed equal response to treatment at 3 months among all active comparators and assumed AEs were equal as well. No treatment continued to have a 0% response rate.
Model does not reflect treatment pathway of post-menopausal VVA: In the sponsor’s model, patients received treatment for their VVA once and either discontinued due to non-response after 3 months or discontinued later over time. Modelled patients then remained untreated for the remainder of their lives. In clinical practice, patients who do not respond to 1 treatment are likely to receive another. Additionally, according to the clinical expert consulted by CADTH, while many patients who initially respond well discontinue treatment over time due to personal circumstances, life events, or changing priorities; they often return to therapy as those circumstances resolve. Treatment is therefore often iterative, and patients may discontinue and restart the same product or switch to other products over time.
CADTH was unable to adjust for this limitation due to the structure of the model.
Treatment discontinuation rates are highly uncertain and may be overestimated: To estimate long-term treatment discontinuation rates over time for patients who initially responded to therapy, the sponsor calculated the weighted proportion of patients who dropped out of each treatment group of the applicable clinical trials,5-15 calculated the rate of discontinuation over the length of each trial, and used that rate to derive the probability of discontinuation over each 3-month cycle. It is unlikely that dropout rates within a clinical trial are generalizable to those in long-term clinical practice, nor is it appropriate to model discontinuation rates in a group of patients who have responded to their therapy using discontinuation data from patients who generally did not yet know if they would respond or not. As prasterone has not been directly compared or indirectly compared in a robust manner to other active treatments, the relative treatment discontinuation rates between prasterone and other active treatments are unknown. The clinical expert consulted by CADTH estimated that approximately 20% of patients responding to local hormone therapy would discontinue each year, corresponding to a 5.43% probability per 3-month cycle.
CADTH assumed a 5.43% probability of discontinuation per 3-month cycle for all active therapies in the base case. A scenario analysis was conducted where CE cream was assumed to have a 6.94% probability of discontinuation rate per cycle (25% per year) to account for a potentially higher discontinuation rate for that treatment due to patient preferences.
Comparator dosing is overestimated: The sponsor’s analysis assumes that 80% of patients using estradiol vaginal tablets will use them according to the product monograph recommendation (i.e., daily for 2 weeks followed by twice weekly) with the other 20% of patients continuing with daily use for the duration of treatment. While the clinical expert consulted by CADTH agreed that 15% to 20% of patients would use estradiol tablets at an increased dose, the increased dose would be 3 times weekly rather than daily use, which was not deemed relevant to Canadian practice. Likewise, the sponsor estimated that 70% of patients would use CE cream twice weekly with the remaining 30% using it cyclically (21 days on, 7 days off), while the clinical expert consulted by CADTH did not agree that the latter was used for long-term treatment, instead estimating that 80% of patients would use 0.5 g of CE cream twice weekly after 2 weeks of daily use, while the remaining 20% would use 1 g twice weekly after 2 weeks of daily use.
CADTH assumed 20% of patients using estradiol tablets would use them 3 times weekly after the first 2 weeks of daily use, and that 20% of patients using CE cream would use a 1 g dose rather than 0.5 g twice weekly after 2 weeks of daily use.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted As Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The population of patients with VVA who will seek treatment can be represented by a population whose most bothersome VVA symptom is moderate to severe dyspareunia. | Uncertain. This population aligns with those of the prasterone clinical trials but does not encompass the entirety of the indicated population. However, according to the clinical expert consulted by CADTH, patients seeking treatment beyond vaginal moisturizers and lubricants would most often have moderate to severe dyspareunia and find it to be their most bothersome symptom, and thus this discrepancy was not considered a key limitation of the submitted analysis. |
Response to therapy reduces by half the increased rates of depression, anxiety, and UTIs associated with VVA. | Possible. It is likely that the excess rate of these comorbidities associated with VVA would be reduced with successful treatment for VVA, although the magnitude of this reduction is highly uncertain. However, altering this assumption has little effect on the model. |
Response and discontinuation rates from trials of 12 weeks’ (84 days) duration can be generalized to a cycle length of 3 months (91.25 days). | Inappropriate; however, relative response and discontinuation rates measured at 91 days were not expected to be substantially different from those measured at 84 days and thus this was not considered a key limitation. |
UTI = urinary tract infection; VVA = vulvovaginal atrophy.
CADTH Reanalyses of the Economic Evaluation
Exploratory Results
Due to limitations with the comparative clinical efficacy and safety of prasterone with relevant comparators, CADTH could not determine a base-case reanalysis. Instead, CADTH conducted exploratory reanalyses. CADTH reanalyses addressed key limitations of the submitted model as outlined in Table 6 and included all comparators commonly used for VVA in Canada, assuming equal probability of response, AEs, and discontinuation for all active comparators, and adjusting the dosing of estradiol tablets and CE cream. CADTH could not address the issue of the submitted model not reflecting the treatment pathway for post-menopausal VVA.
Table 6: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH exploratory reanalysis | ||
1. Comparators | No treatment and estradiol tablets only were the comparators included in sponsor’s base case. | No treatment, estradiol tablets, estradiol ring, estrone, and CE cream were included. |
2. Probability of response at 3 months | Prasterone: |||||| Estradiol tablets: |||||| Estrone: |||||| Estradiol ring: |||||| CE cream: 30.46% No treatment: 0.00% | All active comparators: |||||| No treatment: 0.00% |
3. Probability of AEs | Based on individual product monographs | All active comparators were assumed equal to prasterone No treatment: NA |
4. Probability of discontinuation per 3-month cycle | Prasterone: 6.70% Estradiol tablets: 9.52% Estrone: 9.52%% Estradiol ring: 6.95% CE cream: 7.39% No treatment: NA | All active comparators: 5.43% No treatment: NA |
5. Vagifem dosing | 80% of patients: daily for 2 weeks, then twice weekly 20% of patients: daily | 80% of patients: daily for 2 weeks, then twice weekly 20% of patients: daily for 2 weeks, then 3 times weekly |
6. CE dosing | 70% of patients: 0.5 g twice weekly 30% of patients: 0.5 g daily for 21 days, then 7 days off | 80% of patients: 0.5 g daily for 2 weeks, then twice weekly 20% of patients: 1 g daily for 2 weeks, then twice weekly |
CADTH combined exploratory reanalysis | 1 through 6 | |
AE = adverse events; CE = conjugated estrogen; NA = not applicable.
CADTH’s exploratory analyses, including a combined reanalysis, are presented in Table 7. Disaggregated results are presented in Appendix 4.
When all comparators of interest were included, response, AEs, and discontinuation were assumed equal between active comparators, and when the dosing of estradiol tablets and CE cream were adjusted, the use of prasterone was associated with $467 in increased costs and equal QALYs when compared with CE cream, leading to it being dominated (i.e., costing more than CE cream and equally effective). All other comparators were also similarly dominated by CE.
Table 7: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs)a |
|---|---|---|---|---|
Sponsor’s additional comparator scenario (reanalysis 1) | CE (Premarin cream) | 6,737 | 15.914 | Reference |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,858 | 15.876 | Dominated | |
Estradiol ring (Estring) | 7,054 | 15.889 | Dominated | |
Prasterone (Intrarosa) | 7,140 | 15.891 | Dominated | |
Estradiol (Vagifem) | 7,192 | 15.876 | Dominated | |
CADTH reanalysis 2: equal probability of response | CE (Premarin cream) | 6,757 | 15.886 | Reference |
Prasterone (Intrarosa) | 7,140 | 15.891 | 91,189 | |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,862 | 15.879 | Dominated | |
Estradiol ring (Estring) | 7,051 | 15.888 | Extendedly dominated | |
Estradiol (Vagifem) | 7,212 | 15.879 | Dominated | |
CADTH reanalysis 3: equal AEs | CE (Premarin cream) | 6,731 | 15.915 | Reference |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,847 | 15.877 | Dominated | |
Estradiol ring (Estring) | 6,991 | 15.890 | Dominated | |
Prasterone (Intrarosa) | 7,140 | 15.891 | Dominated | |
Estradiol (Vagifem) | 7,181 | 15.877 | Dominated | |
CADTH reanalysis 4: equal discontinuation | CE (Premarin cream) | 6,722 | 15.935 | Reference |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,884 | 15.894 | Dominated | |
Estradiol ring (Estring) | 7,090 | 15.899 | Dominated | |
Prasterone (Intrarosa) | 7,192 | 15.899 | Dominated | |
Estradiol (Vagifem) | 7,342 | 15.894 | Dominated | |
CADTH reanalysis 5: Vagifem dosing | CE (Premarin cream) | 6,737 | 15.914 | Reference |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,858 | 15.876 | Dominated | |
Estradiol ring (Estring) | 7,054 | 15.889 | Dominated | |
Estradiol (Vagifem) | 7,084 | 15.876 | Dominated | |
Prasterone (Intrarosa) | 7,140 | 15.891 | Dominated | |
CADTH reanalysis 6: CE cream dosing | CE (Premarin cream) | 6,730 | 15.914 | Reference |
No treatment | 6,764 | 15.852 | Dominated | |
Estrone (Estragyn) | 6,858 | 15.876 | Dominated | |
Estradiol ring (Estring) | 7,054 | 15.889 | Dominated | |
Prasterone (Intrarosa) | 7,140 | 15.891 | Dominated | |
Estradiol (Vagifem) | 7,192 | 15.876 | Dominated | |
CADTH combined exploratory reanalysis (1 through 6) | CE (Premarin cream) | 6,710 | 15.910 | Reference |
No treatment | 6,741 | 15.864 | Dominated | |
Estrone (Estragyn) | 6,856 | 15.910 | Dominated | |
Estradiol ring (Estring) | 7,001 | 15.910 | Dominated | |
Prasterone (Intrarosa) | 7,172 | 15.910 | Dominated | |
Estradiol (Vagifem) | 7,177 | 15.910 | Dominated |
AE = adverse event; CE = conjugated estrogen; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The combined CADTH exploratory reanalysis is reported as a probabilistic analysis. All other reanalyses are reported deterministically.
aReference product is the least costly alternative.
Scenario Analysis Results
CADTH conducted several scenario analyses to explore uncertainty in the combined exploratory analysis, including resetting the probabilities of response, AEs, and discontinuation to the sponsor’s assumptions, and increasing the annual discontinuation rate of CE cream to 25%. Results of these scenarios are presented in Appendix 4.
Price reduction analyses were conducted on the sponsor’s additional comparator scenario and the CADTH combined exploratory analysis. Under the CADTH assumptions, with equal efficacy and safety between comparators, a price reduction of 89% would be required for prasterone to be cost-neutral when compared to CE cream.
Table 8: CADTH Price Reduction Analyses
Analysis | ICERs for prasterone vs. CE cream ($/QALY) | |
|---|---|---|
Price reduction | Sponsor’s additional comparator scenario (ICER, deterministic) | CADTH combined reanalysis (ICER, deterministic) |
No price reduction | Dominated | Dominated (more costly than CE cream) |
10% | Dominated | Dominated |
20% | Dominated | Dominated |
30% | Dominated | Dominated |
40% | Dominated | Dominated |
50% | Dominated | Dominated |
60% | Dominated | Dominated |
70% | Dominated | Dominated |
80% | Dominated | Dominated |
90% | Less effective and less costly than CE cream (CE cream ICER is $43 per QALY compared to prasterone) | Dominant (less costly than CE cream) |
CE = conjugated estrogen; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
Additional comparators will soon be available: Two additional comparators, ospemifene (Osphena)29 and estradiol hemihydrate (Imvexxy),30 have recently been approved by Health Canada and are currently under review by CADTH. The CADTH Canadian Drug Expert Committee recommended that Imvexxy be reimbursed for the treatment of post-menopausal moderate to severe dyspareunia in a similar manner to currently funded vaginal estrogen products if the cost could be negotiated to provide savings relative to the least costly local hormone therapy reimbursed for the indication.31
Potential safety advantage for some patients: According to the clinical expert consulted by CADTH, the use of prasterone may be safer for patients with a history of estrogen-dependent cancers when compared to estrogen-based products. As such, it may become the treatment of choice for these patients, who may previously have been excluded from hormone-based VVA treatment.
CE cream is not favoured by patients: While CE cream is dominant in both the sponsor’s and CADTH’s analyses, it is messier to use than the other comparators and, according to the clinical expert consulted by CADTH, has a scent which patients often find off-putting, affecting adherence and treatment continuation. Some patients may also find it undesirable to use a product manufactured from equine urine. CADTH considered a higher discontinuation rate in a scenario analysis to account for this potential issue.
Equity concerns: VVA and associated sexual health concerns are considered an often overlooked and undertreated condition by the patient and clinical groups who provided input for this review, contributing to gender inequities in health care. This sentiment was echoed by the clinical expert consulted by CADTH. Consideration of effective and cost-effective treatments which provide an alternate treatment to estrogen-based vaginal inserts and creams may help address inequities related to this indication.
Overall Conclusions
Based on the CADTH clinical review, prasterone is an effective therapy for VVA in post-menopausal patients whose most bothersome symptom is dyspareunia when compared to placebo. The trials do not provide direct evidence on the relative efficacy and safety of prasterone versus other available local hormone-based therapies. An ITC comparing prasterone with vaginal estrogen therapies was identified; however, key limitations with the evidence make it unclear how prasterone may compare to other therapies post-menopausal patients may use for the treatment of VVA. The relative effectiveness of prasterone to estrogen-based vaginal inserts or creams is therefore unknown, as is the magnitude of any potential safety differences.
According to the clinical expert consulted by CADTH, treatment of VVA is often iterative in nature, with patients remaining on therapy for a time and then discontinuing due to lack of response, inconvenience, or change in lifestyle or priorities. The sponsor’s model allowed for 1 treatment period only, with discontinuing patients remaining untreated for the remainder of their lives, whereas patient in clinical practice may restart a previous therapy or switch to a new 1 if needed or desired. Also, according to the clinical expert consulted by CADTH, estradiol tablets and CE cream are typically used twice weekly after an initial period of daily dosing, rather than daily or cyclically as considered by the sponsor.
Due to the extent of uncertainty with the clinical evidence in the model, a CADTH base case could not be derived. CADTH undertook exploratory reanalyses to address limitations identified in the sponsor’s submission. The CADTH combined exploratory analysis considered all comparators relevant to Canadian clinical practice, assumed equal response, AEs, and discontinuation rates due to the absence of direct or robust indirect comparative evidence, and altered the dosing assumptions for estradiol vaginal tablets and CE cream to align with the dosing most commonly used in clinical practice. In CADTH’s combined exploratory analysis, prasterone was more costly ($462 incremental costs) and equally effective in comparison with CE cream, leading to prasterone being dominated by CE cream. A price reduction of 89% would be required for prasterone to be cost-neutral to CE cream. These results are driven primarily by differences in drug acquisition costs, with prasterone being more costly than most other available local hormone therapies used to treat symptoms of VVA, with the exception of estradiol vaginal tablets.
The results of the CADTH reanalyses are similar when compared to the sponsor’s. In the sponsor’s scenario which included additional comparators, prasterone was found to be dominated by CE cream (i.e., more costly, and slightly less effective) given the sponsor’s analysis included better response rates at 12 weeks with CE cream in comparison with prasterone. In comparison, CADTH assumed equal efficacy in the exploratory analysis due to lack of robust evidence to indicate differential treatment efficacy among all comparators. Based on the economic analysis, there is no evidence to support a price premium for prasterone in comparison with other available local hormone therapies used to treat symptoms of VVA. Likewise, the sponsor’s analysis suggests cost savings with prasterone may be necessary should it be inferior to its less expensive comparators.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Intrarosa, prasterone 6.5 mg vaginal ovules. Quebec (QC): Endoceutics, Inc.; 2021 Oct 1.
2.Intrarosa (prasterone vaginal ovules): Ovules, 6.5 mg prasterone, vaginal [product monograph]. Quebec: (QC): Endoceutics, Inc.; 2019.
3.Vagifem 10: Estradiol 10 mcg, vaginal tablets with applicators [product monograph]. Mississauga (ON): Novo Nordisk Canada Inc; 2016: https://pdf.hres.ca/dpd_pm/00035045.PDF. Accessed 2022 Jan 27.
4.Clinical Study Report: ERC-210. Topical DHEA Against Vaginal Atrophy (3-Month Placebo-Controlled Double-Blind Randomized Study) [internal sponsor's report]. Quebec City (QC): EndoCeutics Inc.; 2008.
5.Clinical Study Report: ERC-231. DHEA Against Vaginal Atrophy (Placebo-Controlled, Double-Blind and Randomized Phase III Study of 3-Month Intravaginal DHEA) [internal sponsor's report]. Quebec City (QC): EndoCeutics Inc.; 2015.
6.Clinical Study Report: ERC-238. Intravaginal Preasterone (DHEA) Against Vulvovaginal Atrophy Associate With Menopause (Placebo-Controlled, Double-Blind Randomized Phase III Study) [internal sponsor's report]. Quebec City (QC): EndoCeutics Inc.; 2015.
7.Simon J, Nachtigall L, Gut R, Lang E, Archer DF, Utian W. Effective treatment of vaginal atrophy with an ultra-low-dose estradiol vaginal tablet. Obstet Gynecol. 2008;112(5):1053-1060. PubMed
8.Ayton RA, Darling GM, Murkies AL, et al. A comparative study of safety and efficacy of continuous low dose oestradiol released from a vaginal ring compared with conjugated equine oestrogen vaginal cream in the treatment of postmenopausal urogenital atrophy. Br J Obstet Gynaecol. 1996;103(4):351-358. PubMed
9.Bachmann G, Notelovitz, M., Nachtigall, L., Birgerson, L. A comparative study of low-dose estradiol vaginal ring and conjugated estrogen cream for postmenopausal urogenital atrophy. Primary Care Update of OB/GYN. 1997;4(3):109-115.
10.Bachmann G, Bouchard C, Hoppe D, et al. Efficacy and safety of low-dose regimens of conjugated estrogens cream administered vaginally. Menopause. 2009;16(4):719-727. PubMed
11.Bachmann G, Lobo RA, Gut R, Nachtigall L, Notelovitz M. Efficacy of low-dose estradiol vaginal tablets in the treatment of atrophic vaginitis: a randomized controlled trial. Obstet Gynecol. 2008;111(1):67-76. PubMed
12.Casper F, Petri E. Local treatment of urogenital atrophy with an estradiol-releasing vaginal ring: a comparative and a placebo-controlled multicenter study. Vaginal Ring Study Group. Int Urogynecol J Pelvic Floor Dysfunct. 1999;10(3):171-176. PubMed
13.Henriksson L, Stjernquist M, Boquist L, Alander U, Selinus I. A comparative multicenter study of the effects of continuous low-dose estradiol released from a new vaginal ring versus estriol vaginal pessaries in postmenopausal women with symptoms and signs of urogenital atrophy. Am J Obstet Gynecol. 1994;171(3):624-632. PubMed
14.Smith P, Heimer G, Lindskog M, Ulmsten U. Oestradiol-releasing vaginal ring for treatment of postmenopausal urogenital atrophy. Maturitas. 1993;16(2):145-154. PubMed
15.Weisberg E, Ayton R, Darling G, et al. Endometrial and vaginal effects of low-dose estradiol delivered by vaginal ring or vaginal tablet. Climacteric. 2005;8(1):83-92. PubMed
16.Estragyn Vaginal Cream (Estrone vaginal cream, 0.1% w/w) [product monograph]. Montreal (QC): Searchlight Pharma Inc.; 2016: https://pdf.hres.ca/dpd_pm/00036132.PDF. Accessed 2022 Jan 27.
17.Estring: 17B-Estradiol: Vaginal Ring, 2 mg [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2017: https://pdf.hres.ca/dpd_pm/00042172.PDF. Accessed 2022 Jan 27.
18.Premarin Vaginal Cream (Conjugated Estrogens CSD, 0.625 mg/g) [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2018: https://pdf.hres.ca/dpd_pm/00045945.PDF. Accessed 2022 Jan 27.
19.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2022 Jan 27.
20.DiBonaventura M, Luo X, Moffatt M, Bushmakin AG, Kumar M, Bobula J. The Association Between Vulvovaginal Atrophy Symptoms and Quality of Life Among Postmenopausal Women in the United States and Western Europe. J Womens Health (Larchmt). 2015;24(9):713-722. PubMed
21.Wang ST, Johnson SJ, Mitchell D, Soliman AM, Vora JB, Agarwal SK. Cost-effectiveness of elagolix versus leuprolide acetate for treating moderate-to-severe endometriosis pain in the USA. J Comp Eff Res. 2019;8(5):337-355. PubMed
22.Geale K, Saridogan E, Lehmann M, Arriagada P, Hultberg M, Henriksson M. Repeated intermittent ulipristal acetate in the treatment of uterine fibroids: a cost-effectiveness analysis. Clinicoecon Outcomes Res. 2017;9:669-676. PubMed
23.Moyneur. S-7 Depression and Anxiety Relative Risks in Women Newly Diagnosed with Vulvovaginal Atrophy and Dyspareunia. Abstract Presentations at North American Menopause Society (NAMS) Annual Meeting. Philadelphia (PA)2017.
24.Moyneur. S-8 Urinary Track Infection Relative Risk in Women Newly Diagnosed with Vulvovaginal Atrophy and Dyspareunia. Abstract Presentations at North American Menopause Society (NAMS) Annual Meeting. Philadelphia (PA)2017.
25.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jan 27.
26.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2022 Jan 27.
27.PharmaStat. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Jan 27.
28.Li B, Duan H, Chang Y, Wang S. Efficacy and safety of current therapies for genitourinary syndrome of menopause: A Bayesian network analysis of 29 randomized trials and 8311 patients. Pharmacol Res. 2021;164 (no pagination). PubMed
29.Osphena: Ospemifene tablets, 60 mg oral selective estrogen receptor modulator [product monograph]. Blainville (QC): Duchesnay Inc.; 2021: https://pdf.hres.ca/dpd_pm/00062157.PDF. Accessed 2022 Jan 27.
30.Imvexxy: Estradiol vaginal inserts, 4 mcg and 10 mcg estradiol, vaginal [product monograph]. Montreal (QC): Knight Therapeutics Inc.; 2020: https://pdf.hres.ca/dpd_pm/00057922.PDF. Accessed 2022 Jan 27.
31.CADTH Canadian Drug Expert Committee (CDEC) draft recommendation: [Imvexxy]. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/SR0694%20Imvexxy%20-%20CADTH%20Draft%20Recommendation%20for%20posting%20December%2016%2C%202021.pdf. Accessed 2021 Jan 04.
32.Government BC. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2022 Jan 27.
33.Drug Product Database online query. Government of Canada; 2021. https://health-products.canada.ca/dpd-bdpp/index-eng.jsp. Accessed 2021 Jan 04.
34.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Intrarosa, prasterone 6.5 mg vaginal ovules. Quebec (QC): Endoceutics, Inc.; 2021 Oct 1.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CADTH Cost Comparison Table for Topical Treatments Used for VVA
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Course or annual costa |
|---|---|---|---|---|---|---|
Prasterone (Intrarosa) | 6.5 mg | Vaginal ovule | $1.4566a | Insert 1 ovule daily at bedtime | $1.46 | $532 |
Vaginal estrogens | ||||||
Conjugated estrogen cream (Premarin) | 0.625 mg/g | Vaginal cream | $0.8423 | 0.5 g twice weekly to 2 g daily for 21 of 28 days | $0.12 to $1.26 | $44 to $461 |
17β-Estradiol (Vagifem) | 10 mcg | Vaginal tablet | $4.4089 | Initially insert 1 tablet daily for 2 weeks, followed by 1 tablet twice weeklyb | First 2 weeks: $4.41 Thereafter: $1.26 | First year: $503 Thereafter: $460 |
Estradiol (Estring) | 2 mg | Vaginal ring | $89.2100 | Insert one ring every 3 months, removing previous ring | $0.98 | $357 |
Estrone (Estragyn) | 0.1% | Vaginal cream | $0.8182c | 0.5 to 4 g daily, 3 weeks on and 1 week off, adjusted to the lowest dose that controls symptoms | $0.31 to $2.46 | $112 to $896 |
17β-Estradiol (Imvexxy) | 4mcg 10 mcg | Tab insert | $3.6288d | Initially insert 1 tablet daily for 2 weeks, followed by 1 tablet twice weekly | First 2 weeks: $3.63 Thereafter: $1.04 | First year: $415 Thereafter: $378 |
VVA = vulvovaginal atrophy.
All prices are from the Ontario Drug Benefit Formulary (accessed November 2021),25 unless otherwise indicated, and do not include dispensing fees. Dosages are from the applicable product monographs unless otherwise indicated.
aSponsor-submitted price.1
bThe clinical expert consulted by CADTH indicated that approximately 20% of patients using estradiol vaginal tablets may be on a maintenance dose of 3 times weekly, which would increase the daily and annual costs for those patients to $1.89 and $689, respectively.
cBritish Columbia Formulary list price.32
dPrice submitted to CADTH during Imvexxy review.31 At the time of this review, Imvexxy was approved but not yet marketed, according to Health Canada.33
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The inclusion of only patients whose most bothersome symptom is moderate to severe dyspareunia is not reflective of the Health Canada indication, which is for the treatment of post-menopausal VVA without limitation. However, it is unlikely this difference would have a substantial impact on the model. |
Model has been adequately programmed and has sufficient face validity | Yes | |
Model structure is adequate for decision problem | No | Important comparators in use in Canadian practice were excluded from the base-case analysis. Sponsor did provide updated model considering additional comparators upon request. Additionally, the model structure does not allow for multiple therapies or restarting therapy after discontinuation. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes |
VVA = vulvovaginal atrophy.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Note: Model cycles were 3 months in duration rather than 12 weeks.
Source: Sponsor’s Pharmacoeconomic evaluation submission, Figure 5.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of Sponsor’s Base Case Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. reference)a | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
No treatment | Assessment | 0.10 | — | — |
Response | 0.00 | — | — | |
Non-Response | 15.75 | — | — | |
Total | 15.85 | — | — | |
Estradiol (Vagifem) | Assessment | 0.10 | 0 | — |
Response | 0.33 | 0.33 | — | |
Non-Response | 15.44 | -0.31 | — | |
Total | 15.87 | 0.02 | — | |
Prasterone (Intrarosa) | Assessment | 0.10 | 0 | 0 |
Response | 0.50 | 0.50 | 0.17 | |
Non-Response | 15.29 | -0.46 | -0.16 | |
Total | 15.89 | 0.04 | 0.01 | |
Discounted costs ($) | ||||
No treatment | Drug Acquisition | 0 | — | — |
Tests & Medical Visits | 6,286.02 | — | — | |
Comorbidities | 473.27 | — | — | |
Adverse Events | 0 | — | — | |
Total | 6,759.29 | — | — | |
Prasterone (Intrarosa) | Drug Acquisition | 450.57 | 450.57 | — |
Tests & Medical Visits | 6,208.31 | -77.71 | — | |
Comorbidities | 473.02 | -0.25 | — | |
Adverse Events | 5.53 | 5.53 | — | |
Total | 7,137.42 | 378.14 | — | |
Estradiol (Vagifem) | Drug Acquisition | 471.61 | 471.61 | 21.04 |
Tests & Medical Visits | 6,235.56 | –50.45 | 27.26 | |
Comorbidities | 473.10 | –0.17 | 0.08 | |
Adverse Events | 16.60 | 16.60 | 11.07 | |
Total | 7,196.87 | 437.58 | 59.44 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
No treatment | Ref. | Ref. | ||
Prasterone (Intrarosa) | 9,860.63 | 9,860.63 | ||
Estradiol (Vagifem) | 17,670.21a | Dominated by prasterone | ||
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
aCalculated by CADTH.
Table 12: Disaggregated Summary of Sponsor’s All-Comparator Scenario Results
Treatment | Component | Value | Incremental (vs. reference)a | Incremental (sequential)a |
|---|---|---|---|---|
Discounted QALYs | ||||
No treatment | Assessment | 0.10 | — | — |
Response | 0.00 | — | — | |
Non-Response | 15.76 | — | — | |
Total | 15.85 | — | — | |
Estrone (Estragyn) | Assessment | 0.10 | 0a | — |
Response | 0.33 | 0.33 | — | |
Non-Response | 15.45 | –0.31 | — | |
Total | 15.88 | 0.02 | — | |
Estradiol tablets (Vagifem) | Assessment | 0.10 | 0 | 0.00 |
Response | 0.33 | 0.33 | 0.00 | |
Non-Response | 15.45 | –0.31 | 0.00 | |
Total | 15.88 | 0.02 | 0.00 | |
Estradiol ring (Estring) | Assessment | 0.10 | 0.00 | 0.00 |
Response | 0.50 | 0.50 | 0.16 | |
Non-Response | 15.30 | –0.46 | –0.15 | |
Total | 15.89 | 0.04 | 0.01 | |
Prasterone (Intrarosa) | Assessment | 0.10 | 0.00 | 0.00 |
Response | 0.50 | 0.50 | 0.01 | |
Non-Response | 15.29 | –0.46 | –0.01 | |
Total | 15.89 | 0.00 | 0.00 | |
CE cream (Premarin) | Assessment | 0.10 | 0.00 | 0.00 |
Response | 0.82 | 0.82 | 0.32 | |
Non-Response | 15.00 | –0.76 | –0.29 | |
Total | 15.92 | 0.06 | 0.02 | |
Discounted costs ($) | ||||
CE cream (Premarin) | Drug Acquisition | 80 | — | — |
Tests & Medical Visits | 6,165 | — | — | |
Comorbidities | 472 | — | — | |
Adverse Events | 14 | — | — | |
Total | 6,731 | — | — | |
No treatment | Drug Acquisition | 0 | –80 | — |
Tests & Medical Visits | 6,286 | 115 | — | |
Comorbidities | 473 | 0 | — | |
Adverse Events | 0 | –14 | — | |
Total | 6,752 | 21 | — | |
Estrone (Estragyn) | Drug Acquisition | 134 | 54 | 134 |
Tests & Medical Visits | 6,230 | 64 | –50 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 17 | 3 | 17 | |
Total | 6,852 | 121 | 100 | |
Estradiol ring (Estring) | Drug Acquisition | 300 | 219 | 165 |
Tests & Medical Visits | 6,208 | 43 | –21 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 70 | 56 | 53 | |
Total | 7,049 | 318 | 196 | |
Prasterone (Intrarosa) | Drug Acquisition | 450 | 370 | 151 |
Tests & Medical Visits | 6,206 | 41 | –2 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 6 | –8 | 11 | |
Total | 7,134 | 403 | 56 | |
Estradiol tablets (Vagifem) | Drug Acquisition | 472 | 391 | 21 |
Tests & Medical Visits | 6,229 | 64 | 23 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 17 | 3 | 11 | |
Total | 7,190 | 459 | 56 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
CE cream (Premarin) | Ref. | Ref. | ||
No treatment | Dominated | Dominated | ||
Estrone (Estragyn) | Dominated | Dominated | ||
Estradiol ring (Estring) | Dominated | Dominated | ||
Prasterone (Intrarosa) | Dominated | Dominated | ||
Estradiol tablets (Vagifem) | Dominated | Dominated | ||
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
aCalculated by CADTH.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 13: Disaggregated Summary of CADTH Combined Exploratory Reanalysis
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
No treatment | Assessment | 0.10 | — | — |
Response | 0.00 | — | — | |
Non-Response | 15.77 | — | — | |
Total | 15.86 | — | — | |
CE cream (Premarin) | Assessment | 0.10 | 0 | — |
Response | 0.61 | 0.61 | — | |
Non-Response | 15.20 | –0.56 | — | |
Total | 15.91 | 0.05 | — | |
Estrone (Estragyn) | Assessment | 0.10 | 0 | 0 |
Response | 0.61 | 0.61 | 0 | |
Non-Response | 15.20 | –0.56 | 0 | |
Total | 15.91 | 0.05 | 0 | |
Estradiol ring (Estring) | Assessment | 0.10 | 0 | 0 |
Response | 0.61 | 0.61 | 0 | |
Non-Response | 15.20 | –0.56 | 0 | |
Total | 15.91 | 0.05 | 0 | |
Estradiol tablet (Vagifem) | Assessment | 0.10 | 0 | 0 |
Response | 0.61 | 0.61 | 0 | |
Non-Response | 15.20 | –0.56 | 0 | |
Total | 15.91 | 0.05 | 0 | |
Prasterone (Intrarosa) | Assessment | 0.10 | 0 | 0 |
Response | 0.61 | 0.61 | 0 | |
Non-Response | 15.20 | –0.56 | 0 | |
Total | 15.91 | 0.05 | 0 | |
Discounted costs ($) | ||||
CE cream (Premarin) | Drug Acquisition | 56 | — | — |
Tests & Medical Visits | 6,176 | — | — | |
Comorbidities | 472 | — | — | |
Adverse Events | 6 | — | — | |
Total | 6,710 | — | — | |
No treatment | Drug Acquisition | 0 | –56 | — |
Tests & Medical Visits | 6,269 | 92 | — | |
Comorbidities | 472 | 0 | — | |
Adverse Events | 0 | –6 | — | |
Total | 6,741 | 31 | — | |
Estrone (Estragyn) | Drug Acquisition | 202 | 146 | 202 |
Tests & Medical Visits | 6,176 | 0 | –92 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 6 | 0 | 6 | |
Total | 6,856 | 146 | 115 | |
Estradiol ring (Estring) | Drug Acquisition | 348 | 291 | 146 |
Tests & Medical Visits | 6,176 | 0 | 0 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 6 | 0 | 0 | |
Total | 7,001 | 291 | 146 | |
Prasterone (Intrarosa) | Drug Acquisition | 519 | 462 | 171 |
Tests & Medical Visits | 6,176 | 0 | 0 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 6 | 0 | 0 | |
Total | 7,172 | 462 | 171 | |
Estradiol tablets (Vagifem) | Drug Acquisition | 523 | 467 | 5 |
Tests & Medical Visits | 6,176 | 0 | 0 | |
Comorbidities | 472 | 0 | 0 | |
Adverse Events | 6 | 0 | 0 | |
Total | 7,177 | 467 | 5 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
CE cream (Premarin) | Ref. | Ref. | ||
No treatment | Dominated | Dominated | ||
Estrone (Estragyn) | Dominated | Dominated | ||
Estradiol ring (Estring) | Dominated | Dominated | ||
Prasterone (Intrarosa) | Dominated | Dominated | ||
Estradiol tablets (Vagifem) | Dominated | Dominated | ||
CE = conjugated estrogen; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
Scenario Analyses
Table 14: CADTH Scenario Analyses
Scenarios | CADTH Base Case | CADTH Scenario |
|---|---|---|
Scenario Analyses | ||
Scenario A: Differential response as submitted by sponsor | All active comparators: |||||||||| No treatment: NA | Prasterone: |||||||||| Estradiol tablets: |||||||||| Estrone: |||||||||| Estradiol ring: |||||||||| CE cream: 30.46% No treatment: 0% |
Scenario B: Differential discontinuation as submitted by sponsor | All active comparators: 5.43% No treatment: NA | Prasterone: 6.70% Estradiol tablets: 9.52% Estrone: 9.52%% Estradiol ring: 6.95% CE cream: 7.39% No treatment: NA |
Scenario C: Differential AEs as submitted by sponsor | All active comparators assumed equal to prasterone. No treatment: NA. | Based on product monographs as per sponsor’s base case. |
Scenation D: CE cream discontinuation | 20% annually | 25% annually |
CE = conjugated estrogen.
Table 15: Summary of Scenario Analyses Around the CADTH Base Case (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH deterministic combined reanalysis | CE cream | 6,733 | 15.899 | Ref. |
Prasterone | 7,192 | 15.899 | Dominated | |
Scenario A: Differential response | CE cream | 6,700 | 15.936 | Ref. |
Prasterone | 7,192 | 15.899 | Dominated | |
Scenario B: Differential discontinuation | CE cream | 6,744 | 15.887 | Ref. |
Estradiol ring | 6,987 | 15.889 | 118,381 | |
Prasterone | 7,140 | 15.891 | 114,694 (sequential) | |
Scenario C: Differential AEs | CE cream | 6,739 | 15.898 | Ref |
Prasterone | 7,192 | 15.899 | 558,231 | |
Scenario D: higher CE cream discontinuation | CE cream | 6,742 | 15.889 | Ref |
Estrone cream | 6,878 | 15.899 | 14,433 | |
Prasterone | 7,192 | 15.899 | Dominated |
AE = adverse event CE = conjugated estrogen.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
In the sponsor-submitted budget impact analysis,34 the sponsor assessed the reimbursement of prasterone for the treatment of post-menopausal VVA. The BIA was conducted from a Canadian public drug payer perspective over a 3-year time horizon using a claims-based approach and included only drug acquisition costs (and dispensing fees). The main comparators considered were prasterone and estradiol vaginal tablets. Other comparators (i.e., CE cream, estrone cream, and the estradiol ring) were combined under the term alternative hormonal therapy (AHT).
Data for the model was obtained from: IQVIA claims data,27 internal forecasts, and formulary-specific costs. Key inputs to the BIA are documented in Table 17.
Key assumptions included:
Prasterone will only displace market share from estradiol vaginal tablets (Vagifem). No additional patients will be treated, nor will other comparators be displaced, even in jurisdictions which do not reimburse Vagifem.
Dispensing fees will occur at similar frequencies for both prasterone and estradiol vaginal tablets.
The average days per claim reported in IQVIA Pharmastat is an accurate reflection of the average number of days dispensed claims last for a given product.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Forecasted claims (standardized to 28 days) | |
Number of forecasted claims for estradiol vaginal tablets | 365,675 / 389,736 / 413,797a |
Number of forecasted claims for AHT | 146,846 / 142,641 / 138,423a |
Market Uptake, Reference Scenario (3 years) | |
Estradiol vaginal tablets (Vagifem) | 71% / 73% / 75%a |
AHT | 29% / 27% / 25%a |
Market Uptake, New Drug Scenario (3 years) | |
Prasterone (Intrarosa) | 7% / 15% / 22%b |
Estradiol vaginal tablets (Vagifem) | 64% / 59% / 52%ab |
AHT | 29% / 27% / 25%a |
Cost of treatment (per patient per standardized 28 days, with M&DF / without M&DF)c | |
Prasterone | $40.78 / range: $52.88 to $72.82 |
Estradiol vaginal tablets | range: $28.93 to $45.52 / range: $41.24 to $76.89 |
AHT | range: $17.36 to $22.87 / range: $30.03 to $54.36 |
AHT = alternate hormonal therapy consisting of conjugated equine estrogen cream (Premarin), estrone cream (Estragyn), and the estradiol vaginal ring (Estring). M&DF = markups and dispensing fees.
aForecasted from 2017 through 2020 IQVIA Pharmastat data.27
bBased on Sponsor’s internal market approximations, equivalent to 10%, 20%, and 30% of estradiol tablet claims in the reference scenario in Years 1, 2, and 3.34
cCalculated using the days per claim reported in IQVIA Pharmastat for jurisdictions which report such data. Costs for estradiol tablets and AHT differ between jurisdictions, as do markups and dispensing fees.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of prasterone for the treatment of VVA would be associated with a savings of $69,298 in year 1, $148,004 in year 2, and $236,146 in year 3, for a total 3-year incremental savings of $453,447.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Prasterone is likely to displace all available comparators: The sponsor has assumed that prasterone will displace 10%, 20%, and 30% of claims for estradiol vaginal tablets over the first 3 years of its availability and that it will not displace the other available AHTs (i.e., CCE cream, estrone cream, or the estradiol vaginal ring) prescribed for the indication under review in Canada. This assumption also leads to the implicit assumption that prasterone will not be reimbursed in jurisdictions which do not reimburse estradiol vaginal tablets (e.g., British Columbia). In contrast, the clinical expert consulted by CADTH indicated that prasterone would likely displace the products comprising AHT in all jurisdictions in a similar proportion to its displacement of estradiol vaginal tablets.
CADTH reanalyses assumed that prasterone will gain 10%, 20%, and 30% of the total market share of estradiol vaginal tablets and AHT, and that it would do so in the proportion those categories are projected to be reimbursed in each jurisdiction. E.g., if estradiol tablets make up 75% of projected claims in the reference scenario for a jurisdiction in a given year, then 75% of prasterone claims will displace estradiol tablets in that year.
The availability of prasterone is likely to increase total market size: In their pharmacoeconomic submission, the sponsor indicated that some patients are reluctant to take an estrogen-based treatment, and thus no treatment was a relevant comparator to prasterone, implying these patients would not otherwise be treated for VVA. Additionally, the clinical expert consulted by CADTH indicated that clinicians are less likely to treat patients with a history of estrogen-dependent cancers with estrogen-based therapy and that these patients would likely receive prasterone once it is available. As such, it is likely that the reimbursement of prasterone will increase the total number of claims made for VVA therapies in Canada. The clinical expert consulted by CADTH estimated that approximately 1 in 12 patients requiring VVA treatment would not be prescribed or would not use estrogen therapy but would be prescribed and use prasterone therapy. Including these patients would increase the total number of claims by 9%.
CADTH reanalyses assumed the total number of claims would increase by 3% in year 1, 6% in year 2, and 9% in year 3 with all such additional claims being for prasterone.
Market uptake of prasterone is uncertain: The sponsor’s estimate that prasterone would displace 10%, 20%, and 30% of comparator claims in Years 1, 2, and 3 of its availability, respectively, is uncertain. According to the clinical expert consulted by CADTH, these proportions are plausible but may be an underestimate as clinicians become more familiar with prasterone use, predicting that prasterone may take up to 50% of total claims by the third year of availability.
CADTH conducted a scenario analysis where prasterone was assumed to displace 10%, 25%, and 50% of estradiol vaginal tablet and AHT claims in Years 1, 2, and 3, respectively.
Dispensing fees overestimated for comparators: In calculating the average cost per claim, the sponsor’s model standardizes all claims reported in IQVIA Pharmastat to 28 days for ease of calculation within the model, as prasterone is available in 28-tablet (i.e., 28-day) packages. However, using Ontario as an example, the mean days of treatment per claim for estradiol vaginal tablets was 58 days, while the weighted average for AHT was 50 days. Despite this difference, the sponsor’s model adds a dispensing fee every 28 days for each comparator, thus overestimating the dispensing fees that will be paid for comparator products.
CADTH reanalyses applied the multiplier the sponsor used to standardize drug costs and markups to a 28-day claim for each comparator to the dispensing fee as well, better modelling the reduced frequency at which these comparators are dispensed according to the provided claims data. A scenario was conducted in which dispensing fees for prasterone were halved under the assumption that 2 packages (56 days of treatment) would be dispensed at a time, bringing prasterone dispensing patterns closer to those of its comparators, though this is associated with uncertainty.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by assuming all available comparators used to treat VVA in Canada will be displaced, assuming the availability of a non-estrogen-based treatment option will increase the market size of available VVA therapies, and reducing the number of dispensing fees applied to available comparators. Table 18 outlines the parameters used by the sponsor in comparison to those used by CADTH.
Table 18: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None. | None. | None. |
Changes to derive the CADTH base case | ||
1. Products displaced | Prasterone displaces 10% / 20% / 30% of estradiol vaginal tablet claims in Y1 / Y2 / Y3. AHT is not displaced. | Prasterone displaces 10% / 20% / 30% of estradiol vaginal and AHT claims in Y1 / Y2 / Y3 in the proportions those claims were forecast for each jurisdiction. |
2. Market size expansion | No increase in market size | Market size increases by 3% in year 1, 6% in year 2, and 9% in year 3, with all such claims attributed to prasterone. |
3. Dispensing fees | A full dispensing fee was applied every 28 days for all comparators | A full dispensing fee was applied every 28 days for prasterone, while the fee applied per cycle was adjusted to reflect the mean length of claim for each comparator in the provided claims data. |
CADTH base case | Reanalyses 1 through 3 | |
AHT = alternate hormonal therapy consisting of conjugated equine estrogen cream (Premarin), estrone cream (Estragyn), and the estradiol vaginal ring (Estring).
Applying these changes resulted in the total 3-year budget impact of reimbursing prasterone for the treatment of post-menopausal VVA to $14,019,986.The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | –$453,447 |
CADTH reanalysis 1: All estrogen-based comparators are displaced | $2,640,310 |
CADTH reanalysis 2: Expanded market size | $8,271,363 |
CADTH reanalysis 3: Dispensing fees adjusted to claim length | $1,567,897 |
CADTH base case (Reanalyses 1 through 3) | $14,019,986 |
BIA = budget impact analysis.
CADTH also conduced additional scenario analyses to address remaining uncertainty:
Prasterone market share assumed to be 10%, 25%, and 50% in years 1, 2, and 3, respectively.
Assumed market share expansion due to prasterone availability is halved.
Dispensing fees associated with prasterone are halved to assume 56 days of treatment are dispensed each refill.
The price of prasterone was reduced by 86% as suggested in the economic evaluation.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $39,876,345 | $41,676,618 | $43,473,285 | $45,266,903 | $130,416,807 |
New drug | $39,876,345 | $41,607,320 | $43,325,282 | $45,030,758 | $129,963,360 | |
Budget impact | $0 | –$69,298 | –$148,004 | –$236,146 | –$453,447 | |
CADTH base case | Reference | $35,831,327 | $37,468,564 | $39,102,568 | $40,733,847 | $117,304,979 |
New drug | $35,831,327 | $39,741,244 | $43,744,062 | $47,839,659 | $131,324,965 | |
Budget impact | $0 | $2,272,680 | $4,641,494 | $7,105,812 | $14,019,986 | |
CADTH scenario analysis A: Larger prasterone uptake | Reference | $35,831,327 | $37,468,564 | $39,102,568 | $40,733,847 | $117,304,979 |
New drug | $35,831,327 | $39,741,244 | $44,185,474 | $49,603,641 | $133,530,359 | |
Budget impact | $0 | $2,272,680 | $5,082,905 | $8,869,794 | $16,225,380 | |
CADTH Scenario Analysis B: Market share expansion halved | Reference | $35,831,327 | $37,468,564 | $39,102,568 | $40,733,847 | $117,304,979 |
New drug | $35,831,327 | $39,046,682 | $42,306,138 | $45,609,740 | $126,962,560 | |
Budget impact | $0 | $1,578,118 | $3,203,570 | $4,875,893 | $9,657,581 | |
CADTH Scenario Analysis C: Prasterone dispensing fees halved | Reference | $35,831,327 | $37,468,564 | $39,102,568 | $40,733,847 | $117,304,979 |
New drug | $35,831,327 | $39,188,384 | $42,599,636 | $46,065,063 | $127,853,083 | |
Budget impact | $0 | $1,719,820 | $3,497,068 | $5,331,216 | $10,548,103 | |
CADTH Scenario Analysis D: 89% prasterone price reduction | Reference | $35,831,327 | $37,468,564 | $39,102,568 | $40,733,847 | $117,304,979 |
New drug | $35,831,327 | $35,382,107 | $34,718,137 | $33,840,369 | $103,940,613 | |
Budget impact | $0 | –$2,086,457 | –$4,384,431 | –$6,893,478 | –$13,364,366 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Women’s Health Coalition of Alberta Society
About the Women’s Health Coalition of Alberta Society
The Women’s Health Coalition of Alberta Society (WHC) is committed to creating a movement that empowers people to speak openly, learn and engage with purpose to address barriers, gaps, policies and unconscious- bias, that impact women’s menstrual, reproductive, and sexual health. We are enabling advocacy, awareness and education in gynecological, uro-gynecological, menstrual, uterine, and reproductive health, through all the ages and stages of a woman’s life.
The WHC is in support of CADTH recommending access and reimbursement for Pasterone/Intrarosa as a therapeutic option for postmenopausal vulvovaginal atrophy.
The WHC is highly committed to ensuring that women have access to the right treatment and support at the right time, for improved health outcomes. Uro-gynecological health is not well understood, is underserved in the health system and offers very limited therapeutic options for conditions associated with menopause (peri and post). In addition, when reimbursement is not available, it can result in preferred treatments only being accessible to women with private health coverage and/or personal wealth.
The clinical and psychological effects of untreated menopausal conditions are overlooked and commonly dismissed. Any improvement in therapeutic choice/access, in the treatment of menopausal conditions, will benefit women physically and wholistically. Recommendation of Pasterone/Intrarosa will not only improve treatment options, choice, and access for women facing menopausal conditions, it may raise clinician awareness of the importance of treating menopausal conditions.
We welcome the opportunity to address this matter with you in greater detail. Should you wish to speak with us, please e-mail us at info@theWHC.ca.
The Women’s Health Coalition of Alberta Society (WHC) is committed to creating a movement that empowers people to speak openly, learn and engage with purpose to address barriers, gaps, and policies that impact women’s menstrual, reproductive, and sexual health. The WHC is raising awareness, conducting research and advocating to address gender bias in health delivery. We are a network of women who have faced health challenges, people who care about, and for women, health care professionals, and business leaders motivated to improving women’s health.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for the Women’s Health Coalition of Alberta Society
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Allergan | — | X | — | — |
Hologic | — | X | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Women’s Health Coalition of Alberta Society
Date: October 27, 2021
Clinician Input
Cleopatra
About Cleopatra
Please describe the purpose of your organization. Include a link to your website (if applicable).
Virtual clinic to raise awareness, about women’s intimate health issues including vaginal dryness, painful sex, urinary tract infections amongst other health issues. https://getcleopatra.com/
Information Gathering
Please describe how you gathered the information included in the submission.
This is an area of health care that I have been working in for many years. I have done research, education, and prepared many presentations. I knew this medication was available in the US and have been waiting for it to be available for women in Canada. This is an issue that affects women’s overall health, bladder health and relationships.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: This issue affects upwards of 70% of women and it very common in postmenopausal women. It may lead to recurrent urinary tract infections and low sexual desire.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: This is an issue that impacts a woman’s overall health, bladder health and relationships. Many women will experience vaginal dryness and recurrent urinary tract infections. The condition may impact a woman’s self-esteem. It is important that this condition be
Treatment Gaps (unmet needs)
Considering the treatment goals in the previous section, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatments are needed to improve compliance. Formulations are needed to improve convenience.
Response: There are a number of treatments available, but they don’t all work for all women. Most are estrogen based. Prasterone is DHEA and would be a great new option for women. Many of the treatments are low dose localized estrogen therapy and there are women who have had a history of breast cancer who may not be comfortable using estrogen. Personal moisturizers are over the counter, but some contain ingredients that are not health for women’s sensitive vaginal tissues. CO2 laser therapy is another treatment for VVA and GSM but it is very expensive and not available in certain geographic areas.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Post-menopausal women are a very large group as women are living longer. Treating VVA or GSM is beneficial for quality of live. Women who have gone through menopause and are no longer fertile. Prasterone would address this need and would provide another treatment option for women. It would also be a safer option for many women as it does not contain estrogen.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Prasterone will be another tool in the toolbox for women’s intimate health post menopause. Yes, it would address the underlying disease condition. It may be a first line treatment for some women.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: Always start with conservative measures so an OTC personal moisturizer that is hormone free should be tried first, then DHEA should be next or estrogen. DHEA has a minimal side effect profile.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Women could try low dose localized estrogen therapy or laser treatment.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Post-menopausal women.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Patients would be identified by their physicians or their nurse during an assessment. There is under diagnosis for sure. More women need to be treated for VVA and GSM. Many women suffer needlessly
Which patients would be least suitable for treatment with the drug under review?
Response: Pregnant women or those who have a sensitivity to DHEA or any ingredients in the product. Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: Most women would likely respond to treatment, but they would be assessed 4-6 weeks after treatment starts.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Reduction in urinary tract infections, painless sex, vaginal comfort.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms.
Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Improvement in intimate health.
How often should treatment response be assessed?
Response: Annually
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify, e.g., loss of lower limb mobility. Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Response: Reaction or lack of response
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Outpatient clinic, speciality clinic also.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: No.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: Women will benefit with by adding this to the armamentarium of treatment options.
Conflict of Interest Declarations for Cleopatra
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
None.
Declaration for Clinician 1
Name: Maureen McGrath
Position: RN
Table 2: Declaration for Cleopatra Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: PleaTracy Purcello
Table 3: Declaration for Cleopatra Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Society of Obstetricians and Gynaecologists of Canada
About the Society of Obstetricians and Gynaecologists of Canada
Please describe the purpose of your organization. Include a link to your website (if applicable). The SOGC is one of Canada’s oldest national specialty organizations. Established in 1944, the Society’s mission is to lead the advancement of patients’s health through excellence and collaborative professional practice. The SOGC has grown to over 4,200 members, comprised of obstetricians, gynaecologists, family physicians, nurses, midwives, and allied health professionals working in the field of patients's sexual and reproductive health.
Information Gathering
Please describe how you gathered the information included in the submission.
As a gynecologist with expertise in menopause I have been aware of the development of Prasterone since it’s earliest days, and have attended presentations of the emerging data at scientific meetings, viewed abstracts, and followed the published literature
Current Treatments
Describe the current treatment paradigm for the disease. Focus on the Canadian context. Please include drug and non-drug treatments. Current treatment of VVA, once dermatologic conditions have been ruled out, includes both medical and non-medical interventions. Non-medical includes maintaining good hydration, avoiding irritants. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Current treatment of VVA, once dermatologic conditions have been ruled out, includes both medical and non-medical interventions.
Non medical includes maintaining good hydration, avoiding irritants and correcting contributing conditions, such as urinary incontinence.
Pelvic floor physiotherapy may be of value, especially where there is dyspareunia or incontinence, if the patient can afford this un-insured but very effective therapy
Drug treatments include:
Vaginal lubricant, if dyspareunia is the predominant symptoms. Some of thee contain parabens as preservatives, so caution is required to avoid these agents.
Vaginal moisturizers:
Polycarbophil, polyacrylic acid, (many patients find these unpleasant)
Hyaluronic acid with or without vitamin E
Vaginal Estrogen: Long the mainstay, these correct both symptoms and reverse the underlying atrophic changes. Used as directed they can be safely used with minimal systemic absorption of estrogen.
DHEA/ Prasterone: This is new to Canda, so my knowledge comes solely from the research evidence, and experience of the clinics who participated in the clinical trials
Ospemiphene: This is new to Canda. I do not have clinical experience with it, but it is an oral selective estrogen receptor modulator, for VVVA and dyspareunia.
Vaginal laser: This is an experimental therapy. Recent higher quality data suggests we should not be using this for VVA (JAMA. 2021;326(14):1381-1389. doi:10.1001/jama.2021.14892)
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Improve quality of life. 80% of post-menopausal patients will experience some degree of VVA. It is a significant contributor to a diminished quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in the previous section, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatments are needed to improve compliance. Formulations are needed to improve convenience.
Response: Not all patients respond to available treatments. There is considerable fear of estrogen among the population, which seems quite refractory to evidence. Laser is an experimental and potentially damaging therapy that is gaining popularity because of the fears of estrogen. There may be unexpected treatment benefits of Prasterone, that are not offered by currently available treatments for VVA
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Post-menopausal patients, who make up the fastest growing segment of the Canadian population.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Many patients have tried lubricants and moisturizers before seeking medical help. Prasterone would therefore be a secondary treatment, but primary treatment option for health care providers. Patient choice plays an essential role in patients’s health care.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: Patients typically have tried lubricants and moisturizers. We would ensure that the diagnosis is correct before prescribing, and wold give concomitant lifestyle advice. Prasterone wold be indicated for VVA, it could be a preferred option for the partial side effect on desire for those patients for whom this is a concern, it would be preferred by patients who prefer not to use estrogen.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Vagainl estrogen or Ospemiphene would be alternatives
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Prasterone would be a good choice for any post-menopausal patients with VVA. Patients where VVA is having an impact on the quality of their life would be ideal candidates. Patients who have VVA to the point that they are unable to have intercourse; or who are unable to have intercourse because of partner issues, are at risk of having vaginal strictures and adhesions, which can lead to permanent loss of use of the vagina. These patients should be treated to restore the vaginal mucosa to a state of health
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: There is good evidence that patients are reluctant to seek help, and so there is an onus on health care providers to enquire about VVA. Clinical examination will reveal the problem, but with the decreased frequency of papa screening, and the shift away from the annual clinical examination, these changes are often missed, until they become highly symptomatic. By the time the diagnosis is made, there may now be sexual health and relationship issues to content with. Early recognition and treatment if much the preferred care
Which patients would be least suitable for treatment with the drug under review?
Response: n/a
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: Any. It is important to note that patients who are using systemic menopausal hormone treatment will also be candidates for local treatment, as the doses currently employed are not always adequate to reverse VVA
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Symptom reporting, or clinical examination
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Reduction in symptoms during daily living. Ability to resume sexual relations
How often should treatment response be assessed?
Response: Once a diagnosis is made and an effective treatment is found, annual is sufficient
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify, e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Response: Typically, patients self-monitor, and will stop treatment for VVA when no longer required
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Primary of specialist ambulatory setting
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: No.
Additional information
Is there any additional information you feel is pertinent to this review?
Response: Feel very proud that this product is Canadian.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
I did not.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
I did not. I did view a presentation for the medical liaison from Lupin but have had no contact with their marketing personnel. I did not use any materials from Lupin.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
No conflict.
Declaration for Clinician 1
Name: Jennifer Blake
Position: Because of my position with SOGC I do not have any role with any industry.
Table 4: Declaration for the Society of Obstetricians and Gynaecologists of Canada Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.