CADTH Reimbursement Review
Ravulizumab (Ultomiris)
Sponsor: Alexion Pharma Canada Corp.
Therapeutic area: Paroxysmal nocturnal hemoglobinuria
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ADA
anti-drug antibody
AE
adverse event
CI
confidence interval
DLBCL
diffuse large B-cell lymphoma
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
FACIT-F
Functional Assessment of Chronic Illness Therapy–Fatigue
FAS
full analysis set
GEE
generalized estimating equation
HRQoL
health-related quality of life
LDH
lactate dehydrogenase
MAVE
major adverse vascular event
MID
minimal important difference
OR
odds ratio
PNH
paroxysmal nocturnal hemoglobinuria
PP
per protocol
RBC
red blood cell
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Ravulizumab (Ultomiris), 10 mg/mL concentrate for solution for infusion |
Indication | For the treatment of adult patients with paroxysmal nocturnal hemoglobinuria |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 28, 2019 |
Sponsor | Alexion Pharma Canada Corp. |
NOC = Notice of Compliance.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is an extremely rare, chronic disease characterized by intravascular hemolysis and heterogenous signs and symptoms that include hemoglobinuria, anemia, abdominal pain, fatigue, dysphagia, and erectile dysfunction. Complications of PNH include thrombosis, chronic kidney disease, and pulmonary hypertension. Although the incidence of PNH has not been extensively characterized, 1 study in the UK estimated an annual incidence of clinical PNH of approximately 0.13 per 100,000 persons. PNH is a consequence of an acquired genetic mutation leading to clonal expansion of hematopoietic stem cells that produce abnormal blood cells that are susceptible to complement-mediated intravascular hemolysis.
Prior to the approval of ravulizumab, the terminal complement inhibitor eculizumab was the only Health Canada–approved drug indicated for the treatment of PNH. According to a 2019 Canadian consensus statement, it is recommended that eculizumab be initiated in patients with a leukocyte PNH clone of greater than 10%, significant intravascular hemolysis, and at least 1 of: symptomatic anemia, thrombosis, renal insufficiency, pulmonary insufficiency or hypertension, or severe abdominal pain, and these criteria correspond to the reimbursement criteria for Canadian public drug plans. Even in patients receiving eculizumab, breakthrough disease with elevated lactate dehydrogenase (LDH) and signs or symptoms can occur and supportive care may be necessary with or without eculizumab treatment. The following supportive treatment may also be required: folic acid and other hematinic support, transfusions (mostly red blood cells [RBCs]), analgesia for abdominal pain or esophageal spasm, and anticoagulation.
Ravulizumab 10 mg/mL concentrate for solution for infusion is indicated for the treatment of adult patients with PNH. The recommended dosing regimen consists of a single loading dose followed 2 weeks later by the first maintenance dose, and maintenance doses are administered every 8 weeks. The loading and maintenance doses are weight-based according to 3 different body weight ranges. Ravulizumab is a terminal complement inhibitor that specifically binds to the complement protein C5 and inhibits terminal complement-mediated intravascular hemolysis.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of ravulizumab 10 mg/mL for the treatment of adult patients with PNH.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient group submission was received from the Canadian Association of PNH Patients. Information was gathered through 1-on-one interviews with individuals living with PNH in Canada and from the scientific literature. The negative impacts of PNH described were dependence on frequent transfusions and difficulty in maintaining school attendance or employment for patients and caregivers due to frequent clinic visits, blood transfusions, and hospitalizations. According to the patient input, patients want treatment options and the less burdensome treatment regimen of ravulizumab (every 8 weeks) compared with eculizumab (every 2 weeks) represents to them an improvement in quality of life and the opportunity to travel for longer periods of time. It was also noted that patients with PNH who are immunocompromised would prefer to visit the clinic for infusions less frequently in the context of the COVID-19 pandemic occurring during the time of the patient interviews.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of PNH.
One unmet need of patients with PNH is that the quality of life of patients being treated with eculizumab could be improved by modifying the treatment schedule or ease of treatment administration. Additionally, at the dosage recommended by the Health Canada–approved product monograph for eculizumab in PNH, approximately 20% of patients do not have complete control of signs and symptoms. This is associated with incomplete pharmacologic C5 inhibition and could be addressed by administering higher doses of eculizumab. Finally, patients who have clinically significant anemia secondary to eculizumab treatment have an unmet need for extravascular hemolysis control; however, extravascular hemolysis would not be expected to improve with ravulizumab treatment.
Ravulizumab has the same mechanism of action as eculizumab and if funded, would be considered first-line therapy in place of eculizumab for most patients. Patients in need of anti-complement therapy include those with evidence of a PNH clone (usually white blood cell clone size > 10%), hemolysis (i.e., LDH > 1.5 × the upper limit of normal [ULN]), and symptoms. Almost all, if not all, patients with hemolytic PNH who would qualify for eculizumab would similarly be expected to respond to ravulizumab. Neither treatment would be effective in the small proportion of patients of Japanese (approximately 3%) and Han-Chinese (approximately 1%) descent who have a polymorphism which negates the effect of eculizumab as there is no effective target on C5. Currently, eculizumab may be preferred over ravulizumab during pregnancy given the available efficacy and safety data for eculizumab although this may change as more clinical experience with ravulizumab accumulates.
A clinically meaningful response to treatment would include improved symptoms and signs (e.g., fatigue, dyspnea, kidney function, abdominal pain, erectile dysfunction) and/or reduced transfusion demands. Response is assessed by review of the signs and symptoms and mapping onto biochemical evidence of reduced intravascular hemolysis (LDH < 1.5 × ULN) and improved blood counts (e.g., hemoglobin), and other parameters (e.g., creatinine, echocardiogram). Discontinuation of anti-complement therapy would rarely be considered and relevant situations would include: nonresponse (almost always associated with the polymorphism that negates the effect of C5 inhibition); persistent, severe adverse reactions (very rare); progression to severe bone marrow failure requiring bone marrow transplant; and regression of PNH clone to less than 10%, if associated with resolution of clinically significant hemolysis.
Clinician Group Input
Clinician group submissions were not received for this review.
Drug Program Input
The drug programs were interested in aligning the initiation, renewal, and discontinuation criteria for ravulizumab with the existing criteria for eculizumab should it be recommended for reimbursement. There was a question for the clinical expert regarding the appropriate cut-off for clone size for the diagnosis of PNH. Noting that the eculizumab maintenance dose can be escalated to 1,200 mg or more every 2 weeks, the drug plans asked the clinical expert whether dose escalation could occur with ravulizumab. The drug plans also noted that the sponsor estimated that ravulizumab would be cost saving from year 4 onward and had a question for the expert committee as to whether this statement was accurate given that biosimilars could enter the market in the future.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Two relevant studies, the ALXN1210-PNH-301 and ALXN1210-PNH-302 studies (referred to here as Study 301 and Study 302, respectively), were selected for inclusion in the CADTH systematic review. Both studies were open-label, active-controlled, parallel-group, noninferiority, randomized controlled trials (RCTs). Both studies were sponsored by Alexion Pharmaceuticals, Corp. and the primary evaluation periods of both studies took place from 2016 to 2018. Study 301 (N = 246) enrolled adult patients with PNH who were treatment-naive, whereas Study 302 (N = 197) enrolled adult patients with PNH who had been receiving eculizumab. Patients were randomized 1:1 to ravulizumab or eculizumab. Noninferiority of ravulizumab compared with eculizumab was assessed for transfusion avoidance, fatigue, breakthrough hemolysis, LDH normalization, and hemoglobin stabilization during a 26-week primary evaluation period.
Patients in both studies were required to have a PNH diagnosis confirmed by flow cytometry (granulocyte or monocyte clone size of at least 5%) and patients in Study 301 were required to have an LDH level of at least 1.5 × ULN and at least 1 PNH-related sign or symptom in the past 3 months. Patients in Study 302 were required to have received eculizumab and have controlled LDH for at least the 6 months before the study. Across both studies, approximately half of patients were male, most were either Asian or White, and mean age was 45 years to 49 years. In Study 301, most patients had an LDH level of 3 × ULN or greater and had received at least 1 transfusion in the past year. Patients in Study 302 had a mean LDH of 228 U/L to 235 U/L (with the ULN for LDH considered to be 246 U/L), with 12.2% to 13.4% of patients having received at least 1 transfusion in the past year. Patients in Study 301 had a shorter mean disease duration (6.4 years to 6.7 years) than patients in Study 302 (11.9 years to 12.4 years), who had been receiving eculizumab for a mean of 5.6 years to 6.0 years. There were lower percentages of patients in Study 301 who had experienced a major adverse vascular event (MAVE; 13.6% to 20.7%) than in Study 302 (22.4% to 28.9%).
Efficacy Results
The results for transfusion avoidance, LDH normalization, and percentage change in LDH level, which were the primary and coprimary end points in the studies, are presented in Table 2. Results for other clinically important outcomes, health-related quality of life (HRQoL) and fatigue, are also presented. The results for the per-protocol (PP) analyses for all primary and key secondary end points were consistent with the primary analyses.
Transfusion Avoidance
Transfusion avoidance was a coprimary end point in Study 301 and a key secondary end point in Study 302 that was tested for noninferiority in both studies according to the closed testing procedure. The mean difference in the percentage of patients achieving transfusion avoidance in the ravulizumab versus the eculizumab group was 6.8% (95% confidence interval [CI], –4.66% to 18.14%) in Study 301 and 5.5% (95% CI, –4.27% to 15.68%) in Study 302. Noninferiority was met in both studies as the lower bounds of the 95% CIs were higher than –20%.
Intravascular Hemolysis
LDH normalization was a coprimary end point in Study 301 and a secondary end point in Study 302. The odds ratio (OR) for the proportion of patients achieving LDH normalization from day 29 to day 183 in Study 301 was 1.187 (95% CI, 0.796 to 1.769) for ravulizumab versus eculizumab. Noninferiority was met as the lower bound of the 95% CI was greater than 0.39. In Study 302, the OR for the proportion of patients achieving LDH normalization from baseline to day 183 was 0.801 (95% CI, 0.500 to 1.282) and the outcome was not part of the statistical testing hierarchy.
Mean percent change in LDH level from baseline to day 183 was the primary end point in Study 302 and a key secondary end point in Study 301. It was tested for noninferiority in both studies and for superiority in Study 302 in accordance with the closed testing procedure. In Study 302, the least squares mean difference in percent change in LDH level was –9.21% (95% CI, –18.84% to 0.42%) for ravulizumab versus eculizumab. Noninferiority was met as the upper bound of the 95% CI was lower than 15%. Percent change in LDH was the first outcome in the Study 302 testing hierarchy for superiority. The significance level was not met for superiority and no further testing was performed. In Study 301, the least squares mean difference in percent change in LDH level was –0.83% (95% CI, –5.21% to 3.56%) for ravulizumab versus eculizumab. Noninferiority was met as the upper bound of the 95% CI was lower than 20%.
Health-Related Quality of Life
Change in the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status score from baseline to week 26 was a secondary end point and not part of the closed testing procedure in either study. Increase in global health status score corresponds to improvement. In Study 301, patients in the ravulizumab and eculizumab group had a change in global health status score of 13.17 (standard deviation [SD] = 21.44) and 12.85 (SD = 21.83), respectively. In Study 302, baseline and week 26 scores were similar to each other within each group, with a change in global health status score of 1.15 (SD = 16.51) in the ravulizumab group and –1.93 (SD = 15.34) in the eculizumab group.
Symptoms of PNH
The change in Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) total score was a key secondary end point and was tested for noninferiority in accordance with the closed testing procedure in both studies. The mean difference in change from baseline to week 26 in FACIT-F total score in the ravulizumab versus the eculizumab group was 0.67 (95% CI, –1.21 to 2.55) in Study 301 and 1.47 (95% CI, –0.21 to 3.15) in Study 302. Noninferiority was met in both studies as the lower bounds of the 95% CIs were higher than –5 and –3 in Study 301 and Study 302, respectively.
Harms Results
The results for adverse events (AEs) are presented in Table 2. Most patients (86.8% to 88.0%) in both treatment groups in both studies reported at least 1 AE. The most common AE was headache and there were no notable imbalances in AEs. Serious AEs (SAEs) were reported in 4.1% to 8.8% of each treatment group in both studies. The most common SAEs were hemolysis and pyrexia, which occurred in 3.1% or less of each treatment group. There were no withdrawals due to AEs in either study. One patient in the eculizumab group in Study 301 died due to lung adenocarcinoma during the extension phase of the study.
In terms of notable harms, serious infections were reported in 1.0% to 3.3% of each treatment group in both studies. Infusion reactions were reported in 3.1% to 8.8% of patients across each treatment group in both studies. As for treatment-emergent anti-drug antibody (ADA)–positive samples, there was 1 in each treatment group in Study 301 and 1 in the eculizumab group in Study 302 and titres were considered to be low.
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Key results | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Transfusion avoidance | ||||
Patients achieving transfusion avoidancea (coprimary end point in Study 301), n (%) | 92 (73.6) | 80 (66.1) | 85 (87.6) | 81 (82.7) |
Mean difference, % (95% CI) | 6.8 (–4.7 to 18.1) | Reference | 5.5 (–4.3 to 15.7) | Reference |
LDH normalization and percent change in LDH level | ||||
Proportion of patients achieving LDH normalizationb (coprimary end point in Study 301) (95% CI) | 0.536 (0.459 to 0.612) | 0.494 (0.417 to 0.570) | 0.660 (0.561 to 0.747) | 0.708 (0.613 to 0.788) |
OR (95% CI) | 1.187 (0.796 to 1.769) | Reference | 0.801 (0.500 to 1.282)c | Reference |
Mean LDH leveld (primary end point in Study 302), U/L (SD) | NA | NA | NA | NA |
Baseline | 1,633.53 (778.75) | 1,578.30 (727.06) | 228.01 (48.71) | 235.22 (49.71) |
Week 26 | 277.96 (102.88) | 330.45 (480.80) | 224.11 (51.72) | 244.11 (70.29) |
LSM % change (SE) | –76.84 (1.58) | –76.02 (1.62) | –0.82 (3.03) | 8.39 (3.04) |
Mean difference in change (95% CI) | –0.83 (–5.21 to 3.56) | Reference | –9.21 (–18.84 to 0.42)e | Reference |
Harms, n (%) (safety set) | ||||
AEs | 110 (88.0) | 105 (86.8) | 85 (87.6) | 86 (87.8) |
SAEs | 11 (8.8) | 9 (7.4) | 4 (4.1) | 8 (8.2) |
WDAE (from study treatment) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Deaths | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Notable harms | ||||
Serious infections | 2 (1.6) | 4 (3.3) | 2 (2.1) | 1 (1.0) |
Infusion reactions | 11 (8.8) | 10 (8.3) | 8 (8.2) | 3 (3.1) |
AE = adverse event; CI = confidence interval; LDH = lactate dehydrogenase; LSM = least squares mean; NA = not applicable; OR = odds ratio; SAE = serious adverse event; SD = standard deviation; SE = standard error; WDAE = withdrawal due to adverse event.
aTransfusion avoidance from baseline through week 26 in the full analysis set. In accordance with the closed testing procedures in both studies, noninferiority testing was conducted. Difference in transfusion avoidance was calculated as a weighted combination of differences in each randomization stratum using Mantel-Haenszel weights. The 95% CI was computed using the stratified Newcombe CI method. Patients who fulfilled the protocol-specified transfusion criteria were analyzed as having received a transfusion, regardless of whether the patients had actually received a transfusion.
bLDH normalization from day 29 through week 26 (Study 301) or from baseline through week 26 (Study 302) in the full analysis set. In accordance with the closed testing procedure in Study 301, noninferiority testing was conducted. A generalized estimating equation was used with the following terms: treatment group, history of transfusion, and baseline LDH level. A first-order autoregressive structure was assumed for within-patient correlation.
cOutcome was outside of the statistical testing hierarchy.
dA mixed-effects model for repeated measures was used in the full analysis set, which included the following terms: treatment group, randomization factors, baseline LDH level, study visit, and study visit by treatment group interaction. An unstructured covariance structure was used. In accordance with the closed testing procedures in both studies, noninferiority testing was conducted in both studies and superiority testing was conducted in Study 302.
eP = 0.0583; approximate P value for superiority associated with the upper bound provided.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Critical Appraisal
The pre-specified noninferiority margins for the primary and key secondary end points (aside from percent change in LDH, potentially) were based on a magnitude of loss of benefit that may not be clinically acceptable. However, there are several factors that mitigate the risk of unacceptable loss of benefit with ravulizumab versus eculizumab. These include that all of the primary and key secondary end points met their respective noninferiority margins, there were minimal missing data, the PP analyses were consistent with the primary analyses for all end points, and a more conservative margin would have been met for all end points.
The open-label nature of the studies means that outcomes relying on subjective reporting, such as the EORTC QLQ-C30 and the FACIT-F, could have been biased with potential for bias in favour of ravulizumab. Additionally, the reliability, validity, and responsiveness of the EORTC QLQ-C30 and the FACIT-F have yet to be characterized in patients with PNH. Statistical testing was only performed for the FACIT-F score and not for other symptom assessments or for the EORTC QLQ-C30 scales.
The criteria for Study 302 were chosen in such a way that patients requiring a higher dose or more frequent dosing of eculizumab beyond the product monograph-recommended dosage would have been excluded. While these patients were included in Study 301, the studies did not allow for deviation from the labelled dosage of eculizumab (900 mg maintenance dose) and this may have biased the efficacy results in favour of ravulizumab relative to how eculizumab is dosed in clinical practice.
Indirect Comparisons
No relevant indirect comparisons were identified.
Other Relevant Evidence
Description of Studies
Safety and efficacy results from the respective extension periods for Study 301 (N = 243) and Study 302 (N = 191), during which all patients received ravulizumab, were also submitted by the sponsor and are presented in this report for the 26-week period following the randomized treatment period. Also included in the sponsor’s submission was a patient preference substudy (N = 95) which allowed patients to enrol from Study 302 who enrolled in the extension period and had received at least 2 doses of ravulizumab during the extension period. A novel patient preference questionnaire was developed for the study and the objective of the study was to assess patient preferences for ravulizumab or eculizumab and to identify the key factors influencing preference.
Efficacy Results
The results from the extension periods of Study 301 and Study 302 were reported as summary statistics and indicated that efficacy as assessed through transfusion avoidance, FACIT-F score, breakthrough hemolysis, LDH normalization, and hemoglobin stabilization was generally maintained with ravulizumab treatment for another 26 weeks following the randomized treatment period.
According to the results from the questionnaire administered in the patient preference substudy, 93% of patients preferred ravulizumab overall with 43% of patients choosing frequency of infusions and 23% of patients choosing overall quality of life as the most important treatment factor when deciding preference.
Harms Results
The AE profiles in the extension periods of Study 301 and Study 302 were similar to those in the randomized treatment periods, with no new safety signals identified. The frequency of headaches numerically decreased between the 2 periods in both treatment groups in both studies.
Critical Appraisal
The extension periods of Study 301 and Study 302 do not provide evidence for the comparative efficacy of ravulizumab versus eculizumab because all patients who continued in the extension periods received ravulizumab. As well, reductions in sample size in periods beyond the first 52 weeks of study treatment precluded the ability to assess results beyond 1 year of treatment, which is a concern given the chronic nature of the disease.
There were several limitations identified in the patient preference substudy that introduce substantial uncertainty in the results. These include the lack of evidence for the reliability and responsiveness of the questionnaire, the potential for recall bias given that ravulizumab was the most recent treatment for all patients, the small sample size relative to the population of Study 302, and uncertainty surrounding reasons for the reduction in sample size.
Conclusions
Ravulizumab is noninferior to eculizumab in transfusion avoidance, occurrence of breakthrough hemolysis, LDH normalization, and hemoglobin stabilization over 26 weeks of treatment in adult patients with PNH, with maintenance of efficacy up to 52 weeks of treatment. Evidence regarding comparative efficacy in symptom control, such as improvement of fatigue, is supportive of noninferiority but is associated with some uncertainty given that the study was open-label, the patient-reported outcomes have not been validated in patients with PNH, and statistical testing was not performed for outcomes other than FACIT-F score. Conclusions cannot be drawn for HRQoL due to the same limitations. The efficacy of ravulizumab versus eculizumab is less certain for the scenario in which the maintenance dose of eculizumab increases beyond what is specified in the product monograph for PNH, as is the case with clinical practice in Canada. Results from a patient preference study demonstrated that most patients who had experienced treatment with both drugs preferred ravulizumab over eculizumab with frequency of infusions being the dominant deciding factor, but serious limitations in the study contribute much uncertainty to the estimated proportion of patients who preferred ravulizumab. The safety profiles of ravulizumab and eculizumab were similar to each other with no new safety concerns.
Introduction
Disease Background
PNH is an extremely rare, chronic disease characterized by intravascular hemolysis and heterogenous signs and symptoms that include hemoglobinuria, anemia, abdominal pain, fatigue, dysphagia, and erectile dysfunction.3 Complications of PNH include thrombosis, chronic kidney disease, and pulmonary hypertension.4 In Canada, the median age of disease onset has been estimated at 43 years.3 Although the incidence of PNH has not been extensively characterized, 1 study in the UK5 estimated an annual incidence of clinical PNH of approximately 0.13 per 100,000 persons. PNH is a consequence of an acquired genetic mutation leading to clonal expansion of hematopoietic stem cells that produce abnormal RBCs, leukocytes, and platelets deficient in glycophosphatidylinositol anchor proteins.4 These abnormal RBCs are susceptible to complement-mediated intravascular hemolysis. PNH may develop as hemolytic PNH, typically with a white blood cell clone size of greater than 10%, or in association with a bone marrow disorder such as aplastic anemia or myelodysplastic syndrome, which is often accompanied by smaller clone sizes.3 Studies examining survival of patients with PNH following diagnosis found a range of median survival from 14.6 years to 32 years,6,7 while results from a study in patients with PNH treated with eculizumab suggested that their survival was similar to that of age-matched controls.8
Delays in diagnosing PNH are common due to the non-specific nature of the signs and symptoms. In cases of suspected PNH, diagnostic testing with flow cytometry for PNH clones in RBCs and in neutrophils and monocytes can be performed and is available at most academic centres and via community lab services, according to the clinical expert consulted by CADTH. The clinical expert also stated that intravascular hemolysis can be identified by testing for elevated LDH, undetectable haptoglobin, and a negative direct antibody test.
Standards of Therapy
Prior to the approval of ravulizumab, the terminal complement inhibitor eculizumab was the only Health Canada–approved drug indicated for the treatment of PNH. According to the 2019 Canadian consensus statement by the Canadian PNH Network, eculizumab should be initiated in patients with “a leukocyte PNH clone greater than 10%, laboratory evidence of significant intravascular hemolysis, and at least 1 of: symptomatic anemia (regardless of transfusion dependence), thrombosis, renal insufficiency, pulmonary insufficiency or hypertension, or abdominal pain requiring administration of opioid analgesia”3 These criteria correspond to the reimbursement criteria for Canadian public drug plans.3 There is also a suggestion that eculizumab should be considered in patients meeting the same PNH clone and intravascular hemolysis criteria and who have disabling fatigue or who are pregnant.3 Patients receiving complement inhibitor should also have an up-to-date meningococcal vaccination.3 Even in patients receiving eculizumab, breakthrough disease with elevated LDH and signs or symptoms can occur and supportive care may be necessary with or without eculizumab treatment.3 According to the clinical expert consulted by CADTH, the following supportive treatment may be required: folic acid and other hematinic support, transfusions (mostly RBCs), analgesia for abdominal pain or esophageal spasm, and anticoagulation (prophylactically if the patient has elevated LDH but is not eligible for eculizumab, or therapeutically if the patient has a history of thrombosis). While bone marrow transplant is a curative therapy for PNH, the clinical expert noted that it is only considered in patients with primary bone marrow failure (e.g., aplastic anemia) or PNH that is unresponsive to complement blockade, which is rare.
According to the clinical expert consulted by CADTH, the most important treatment goals for PNH are the inhibition of complement-mediated intravascular hemolysis, reduction in risk of thrombosis, improvement in quality of life via symptom reduction and minimizing impact on patients’ activities, and overall extended survival.
Drug
Ravulizumab 10 mg/mL concentrate for solution for infusion is indicated for the treatment of adult patients with PNH and has not been previously reviewed by CADTH. The recommended dosing regimen consists of a single loading dose followed 2 weeks later by the first maintenance dose, and maintenance doses are then subsequently administered every 8 weeks. The loading and maintenance doses are weight-based according to 3 different body weight ranges (Table 3 for details). Ravulizumab is a terminal complement inhibitor that specifically binds to the complement protein C5 and inhibits terminal complement-mediated intravascular hemolysis. The sponsor’s reimbursement request is identical to the Health Canada–approved indication.
Table 3: Key Characteristics of Ravulizumab and Eculizumab
Key characteristics | Ravulizumab | Eculizumab |
|---|---|---|
Mechanism of action | Ravulizumab is a terminal complement inhibitor with high specificity and affinity to the complement protein C5; it antagonizes terminal complement-mediated inflammation, cell activation, and cell lysis. Ravulizumab is recycled from the early endosome back into the vascular compartment, resulting in an extended terminal elimination half-life. | Eculizumab is a terminal complement inhibitor that specifically binds to the complement protein C5 with high affinity; it inhibits terminal complement-mediated intravascular hemolysis in patients with PNH. |
Indicationa | For the treatment of adult patients with PNH | For the treatment of patients with PNH to reduce hemolysis. Eculizumab was studied in clinical trials in patients with a history of at least 1 transfusion during the past 2 years. |
Route of administration | IV infusion | IV infusion |
Recommended dose |
| • 600 mg every 7 days for the first 4 weeks, followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter |
Serious adverse effects or safety issues |
|
|
PNH = paroxysmal nocturnal hemoglobinuria.
aHealth Canada–approved indication.
Stakeholder Perspectives
Patient Group Input
One patient group submission was received from the Canadian Association of PNH Patients. Information was gathered through 1-on-one interviews with individuals living with PNH in Canada and from the scientific literature. The negative impacts of PNH described were dependence on frequent transfusions and difficulty in maintaining school attendance or employment for patients and caregivers due to frequent clinic visits, blood transfusions, and hospitalizations. According to the patient input, patients want treatment options and the less burdensome treatment regimen of ravulizumab (every 8 weeks) compared with eculizumab (every 2 weeks) represents to them an improvement in quality of life and the opportunity to travel for longer periods of time. It was also noted that patients with PNH who are immunocompromised would prefer to visit the clinic for infusions less frequently in the context of the COVID-19 pandemic occurring during the time of the patient interviews.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of PNH.
Unmet Needs
Not all patients with PNH have complete control of intravascular hemolysis with eculizumab. Approximately 20% of patients require higher doses of eculizumab or more frequent doses than recommended in the Health Canada–approved product monograph for the PNH indication to properly control symptoms and signs associated with pharmacokinetic breakthrough (i.e., insufficient pharmacologic C5 inhibition). Higher doses (i.e., the recommended 1,200 mg maintenance dose for other indications) or more catered doses (e.g., weight-based doses) are needed to more fully suppress C5.
In patients receiving eculizumab, quality of life and impact on patients’ lives could be improved by modifying the treatment schedule or ease of treatment administration. Some examples would be less frequent infusions than every 2 weeks, self-administered subcutaneous injections, or oral therapies.
Extravascular hemolysis control remains an unmet need for those who have clinically significant anemia secondary to eculizumab treatment, which is caused by C5 blockade and would not be expected to improve with ravulizumab treatment. Proximal complement inhibitors have the potential to minimize this complication in PNH, with pegcetacoplan having received FDA approval and various trials currently under way. However, these are not clinically available yet in Canada and C5 blockade is still of utmost importance to control intravascular hemolysis.
Place in Therapy
Ravulizumab has the same mechanism of action as eculizumab. Its modifications allow more profound C5 blockade and greater recycling of the molecule, thereby extending the treatment cycle from every 2 weeks to every 8 weeks. Although ravulizumab is not the first treatment to address the disease process, it is expected to improve some aspects of treatment, including pharmacokinetic breakthrough risk and patient convenience. If funded, ravulizumab would be considered first-line therapy in place of eculizumab for most patients and other supportive therapies would remain the same. Patients already being treated with eculizumab can be directly transitioned from eculizumab to ravulizumab.
Patient Population
PNH is an ultra-rare disease (annual incidence of approximately 5 to 10 cases per million persons) and manifests usually in vague and general or common symptoms (e.g., fatigue, thrombosis, iron deficiency anemia). As such, it can take years for the diagnosis to be made (usually following referral to a hematologist). Testing is made by high-sensitivity flow cytometry, which is available in most, if not all, academic centres. Some community labs have agreements to send samples to academic labs for processing. Along with PNH flow cytometry, the identification of intravascular hemolysis (with elevated LDH, undetectable haptoglobin, and negative direct antiglobulin test) is fairly straightforward as long as the physician thinks to test for it.
Patients without evidence of hemolysis with small PNH clones (usually < 10%, and more often < 1%) would not usually require treatment with complement blockade. Their PNH clones would be most commonly associated with a diagnosis of aplastic anemia or myelodysplastic syndrome.
Patients in need of anti-complement therapy include those with evidence of a PNH clone (usually white blood cell clone size > 10%), hemolysis (i.e., LDH > 1.5 x ULN), and symptoms. The current criteria for reimbursement of eculizumab in Canadian jurisdictions, which have not changed since eculizumab’s approval in 2009, stipulate a diagnosis of PNH based on PNH clone size, LDH level, and at least 1 of the following signs or symptoms of PNH: a thrombotic or embolic event, at least 4 units of RBCs transfused in the previous 12 months, chronic or recurrent anemia where causes other than hemolysis have been excluded, pulmonary insufficiency, renal insufficiency, or smooth muscle spasm. The severity of the criteria surrounding signs or symptoms are challenging, particularly the smooth muscle spasm criterion which requires hospitalization and/or use of narcotic analgesia. Other symptoms should also be considered as these are all signs of systemic complement dysregulation and intravascular hemolysis: thrombosis, anemia, dyspnea, pulmonary hypertension, kidney failure, abdominal pain, dysphagia, refractory erectile dysfunction, and persistent fatigue. Adding refractory erectile dysfunction and persistent fatigue to the list of signs and symptoms specifically could be beneficial for patient access. Another issue with the current reimbursement criteria is that most jurisdictions only consider granulocytes for PNH clone (granulocyte clone size > 10%) when white blood cells clone size greater than 10% would be a more appropriate criterion. Although uncommon, some patients with PNH have a monocyte clone size of greater than 10% and a neutrophil clone size of less than 10%.
Almost all, if not all, patients with hemolytic PNH who would qualify for eculizumab would similarly be expected to respond to ravulizumab. Neither treatment would be effective in the small proportion of patients of Japanese (approximately 3%) and Han-Chinese (approximately 1%) descent who have a polymorphism which negates the effect of eculizumab as there is no effective target on C5. As such, ravulizumab would be a replacement for eculizumab in most situations. One area where current perspective suggests eculizumab is preferred is in the context of pregnancy given the currently available efficacy and safety data.11 Patients who are pregnant would be treated with eculizumab during pregnancy and perhaps for 4 weeks to 6 weeks postpartum; however, before and after, they could be treated with ravulizumab. This may change as more clinical experience with ravulizumab accumulates.
Assessing Response to Treatment
The following outcomes are used in clinical trials and in clinical practice: LDH normalization (LDH < 1.5 × ULN) as a surrogate for hemolysis (associated with a reduction in risk of thrombosis and other symptoms), transfusion independence in patients who required transfusions before treatment, hemoglobin stabilization (in concert with LDH normalization), improvement in PNH symptoms and signs, absence of thrombosis, and improvement in quality of life. A clinically meaningful response to treatment would include improved symptoms and signs (e.g., fatigue, dyspnea, kidney function, abdominal pain, erectile dysfunction) and/or reduced transfusion demands. There is evidence for improved survival with eculizumab treatment and the same is also anticipated for ravulizumab.
Patients with evidence of hemolysis with LDH are the easiest to monitor for a response as there would be rapid LDH reduction after the drug is started (within 4 weeks in most cases, if not faster). No patients have been identified who would not respond (unless it is known that they hold the C5 polymorphism described above). Suspicion for this would come when there is no improvement in LDH and symptoms after starting treatment and in the right ethnic context (Japanese or Han-Chinese).
Patients are typically followed every 2 weeks to 4 weeks at the time of treatment initiation to follow symptoms, laboratory evidence of improvement (e.g., LDH reduction), and to monitor for safety signals or AEs. Response is anticipated within the first 2 months of starting therapy. Patients are then monitored approximately every 3 months to 6 months once they are felt to be stable with fully suppressed intravascular hemolysis and terminal complement activity. Response is assessed by review of the signs and symptoms and mapping onto biochemical evidence of reduced intravascular hemolysis (LDH < 1.5 × ULN) and improved blood counts (e.g., hemoglobin), and other parameters (e.g., creatinine, echocardiogram, and so on). Patients are also monitored with PNH flow cytometry every 6 months to 12 months for changes in their clone size.
Discontinuing Treatment
There are a few situations where discontinuation of complement blockade would be considered:
instances of nonresponse, which are very rare and almost always associated with the C5 polymorphism that negates the effect of C5 inhibition
instances of persistent, severe adverse reactions, which are also very rare
progression to severe marrow failure that would require bone marrow transplant
regression of the PNH clone to less than 10% which would usually correspond to resolved or resolving hemolysis
Prescribing Conditions
Hematologists typically diagnose, treat, and monitor PNH patients in Canada. As the disease is extremely rare, referral to Canadian PNH Network Centres is encouraged, and these centres assume care of these patients or offer a shared-care model with the community physician.
Treatment is usually given in the community, either infused in an infusion clinic or at the patient’s home. A hospital setting is uncommon for treatment except in situations where admission is otherwise required (e.g., elective surgery). In those situations, it is desirable to have the drug available in the event of an acute exacerbation.
Clinician Group Input
There were no clinician group submissions received for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Questions From the Drug Programs and Responses From Clinical Experts
Questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Eculizumab is listed on most public drug plans; however, the criteria are not publicly available for most plans. The clinical trials for ravulizumab required that diagnosis of PNH be confirmed by flow cytometry with a granulocyte or monocyte clone size of at least 5% before initiation. Current criteria for eculizumab requires a granulocyte clone size of ≥ 10%. For consistency, alignment with initiation criteria for eculizumab should be considered. Question for the clinical expert: What is the appropriate cut-off for clone size for the diagnosis of PNH? | A threshold of 10% for PNH clone size is likely appropriate as long as either a granulocyte or monocyte clone size of at least 10% is accepted. The current criteria in Ontario only allow for granulocyte clone size to be considered and occasionally there are patients with active disease and with monocyte clone size much greater than 10% but granulocyte clone size of approximately 9%. Therefore, clone type should be considered along with clone size for the initiation criteria. |
Considerations for continuation or renewal of therapy | |
Consider alignment with renewal criteria for eculizumab. | No response expected from the clinical expert. |
Considerations for discontinuation of therapy | |
Consider alignment with renewal criteria for eculizumab. | No response expected from the clinical expert. |
Considerations for prescribing of therapy | |
The recommended dose of eculizumab is 900 mg IV every 2 weeks. However, if breakthrough hemolysis occurs, the sponsor noted that the dose could be escalated to 1,200 mg or more every 2 weeks. Ravulizumab is dosed by weight and given IV every 8 weeks. Question for the clinical expert: Could dose escalation occur with ravulizumab? | There is very limited experience with ravulizumab in Canada, but experience in the US suggests that there are some patients who experience persistent breakthrough hemolysis with the recommended dosage. In such cases, ravulizumab is dosed every 7 weeks or even every 6 weeks if necessary. While the tendency with eculizumab would be to increase the dose to maintain the same dosing schedule, it is unclear whether the dose for ravulizumab can be increased. |
System and economic issues | |
The submitted price for ravulizumab is $7,296.67 per vial and the annual cost is $561,841. It is expected that patients will transition from eculizumab to ravulizumab. Patent expiry for eculizumab is 2027 and for ravulizumab 2035. If patients transition to the new, more convenient C5 inhibitor then savings that could be obtained by the entry of biosimilars will be lost. Question for CDEC: The budget impact analysis report estimates that Ravulizumab would be cost saving from year 4 onward. Is this accurate given that biosimilars could enter the market in the future? | No response expected from the clinical expert. |
CDEC = CADTH Canadian Drug Expert Committee; PNH = paroxysmal nocturnal hemoglobinuria.
Clinical Evidence
The clinical evidence included in the review of ravulizumab is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of ravulizumab 10 mg/mL for the treatment of adult patients with PNH.
Methods
Studies selected for inclusion in the systematic review will include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients aged ≥ 18 years with PNH Subgroups: disease severity (e.g., PNH clone size, serum LDH, history of thrombotic event) |
Intervention | Ravulizumab IV infusion with the following dosage schedule:
|
Comparator | Eculizumab |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs |
|
AE = adverse event; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; PNH = paroxysmal nocturnal hemoglobinuria; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.12
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Ultomiris (ravulizumab). Clinical trials registries were searched — the US National Institutes of Health’s clinicaltrials.gov and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on August 20, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on December 15, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.13 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
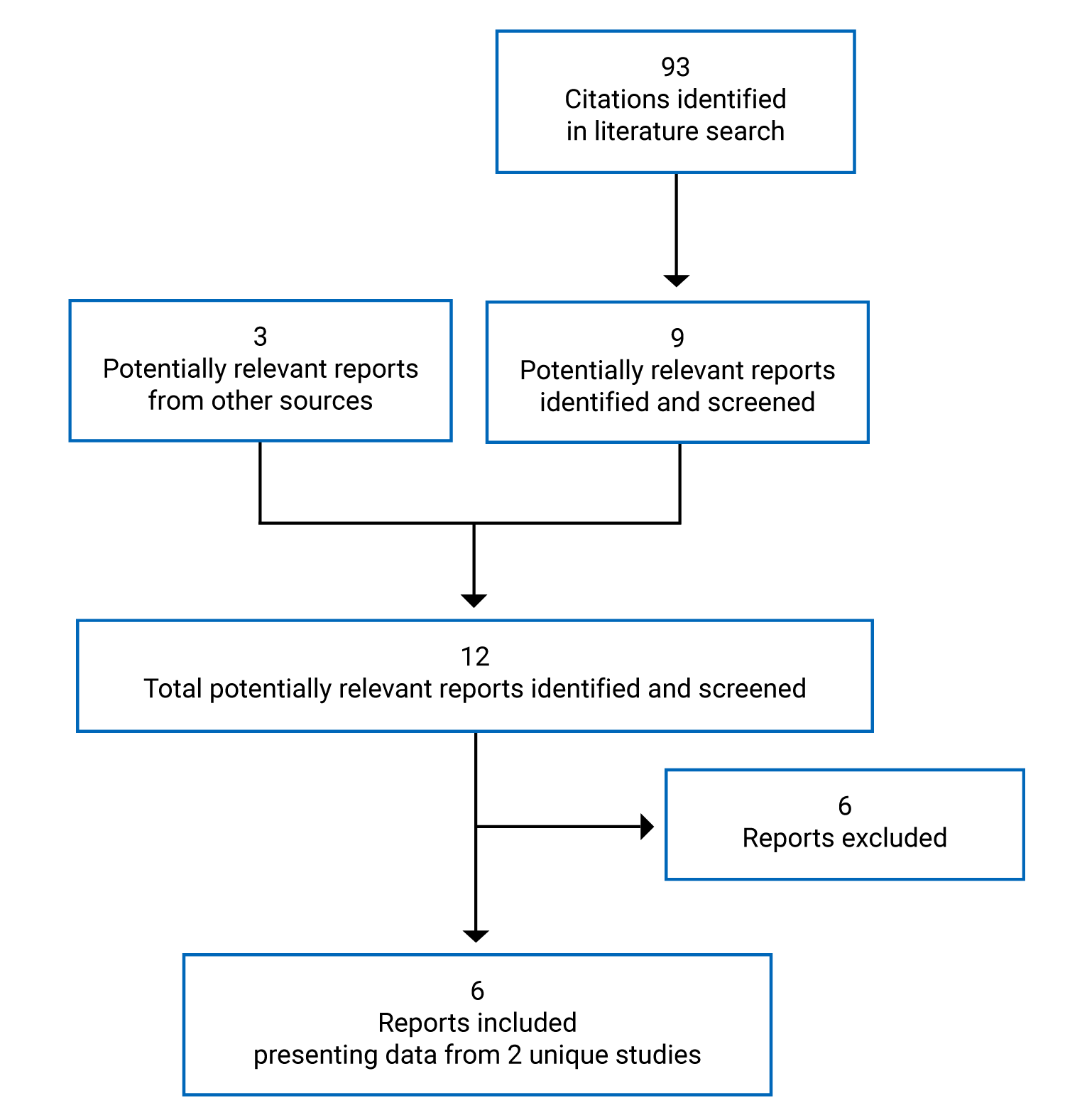
Table 6: Details of Included Studies
Study details | Study 301 | Study 302 |
|---|---|---|
Study design | Phase III, open-label, active-controlled, parallel-group, noninferiority RCT | Phase III, open-label, active-controlled, parallel-group, noninferiority RCT |
Locations | 123 sites in 25 countries in South America, Australia, Europe, North America, and Asia (including 2 sites in Canada) | 49 sites in 11 countries in Australia, North America, Europe, and Asia (including 3 sites in Canada) |
Patient enrolment dates | First patient treated on December 20, 2016 and last patient completed 26-week primary evaluation period on January 25, 2018 | First patient treated on December 20, 2016 and last patient completed 26-week primary evaluation period on March 8, 2018 |
Randomized (N) | 246 | 197 |
Inclusion criteria (unique) |
|
|
Inclusion criteria (common) |
| Same as for Study 301 |
Exclusion criteria (unique) | Current or previous treatment with a complement inhibitor | LDH level > 2 × ULN or MAVE in the 6 months before first day of study treatment |
Exclusion criteria (common) |
| Same as for Study 301 |
(continued) |
| — |
Intervention | Ravulizumab IV infusion with weight-based doses
| Same as for Study 301 |
Comparator(s) | Eculizumab IV infusion as follows:
| Eculizumab IV infusion 900 mg q.2.w. |
Phase | ||
Screening | Up to 4 weeks | Up to 4 weeks |
Randomized treatment | 26 weeks | 26 weeks |
Ravulizumab extension | Up to 2 years | Up to 2 years |
Primary end point | Coprimary end points:
| LDH % change from baseline to week 26 (noninferiority) |
Secondary and exploratory end points | Key secondary end point hierarchy Tests for noninferiority:
| Secondary end point hierarchy Tests for noninferiority:
|
(continued) |
To be followed by tests for superiority:
Other secondary end points
Safety end points
• immunogenicity |
To be followed by tests for superiority:
Other secondary end points
Safety end points
• immunogenicity |
Publications | Lee et al. (2019)14 | |
AE = adverse event; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; LDH = lactate dehydrogenase; MAVE = major adverse vascular event; PNH = paroxysmal nocturnal hemoglobinuria; q.2.w. = every 2 weeks; q.8.w. = every 8 weeks; RBC = red blood cell; RCT = randomized controlled trial; SAE = serious adverse event; ULN = upper limit of normal.
Note: Three additional reports were included.17-19
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Description of Studies
Two relevant studies, the ALXN1210-PNH-301 and ALXN1210-PNH-302 studies (referred to in the present report as Study 301 and Study 302, respectively), were selected for inclusion in the CADTH systematic review. Both studies were open-label, active-controlled, parallel-group RCTs identified as pivotal studies and also identified in the CADTH systematic literature search. Both studies were sponsored by Alexion Pharmaceuticals, Corp. The primary study objectives were to assess the noninferiority of ravulizumab compared with eculizumab in adult patients with PNH who had never been treated with a complement inhibitor (Study 301) and in adult patients with PNH who were clinically stable after having been treated with eculizumab for at least the previous 6 months (Study 302).
Study 301 (N = 246; primary evaluation phase from 2016 to 2018; 2 sites in Canada) randomized patients 1:1 to ravulizumab IV infusion with a weight-based loading dose on day 1 followed by weight-based maintenance doses every 8 weeks starting on day 15 or eculizumab IV infusion with 600 mg induction doses on days 1, 8, 15, and 22 followed by 900 mg every 2 weeks starting on day 29. Randomization in Study 301 was stratified by transfusion history and screening LDH level. Study 302 (N = 197; primary evaluation period from 2016 to 2018; 3 sites in Canada) randomized patients 1:1 to the same interventions as in Study 301, with randomization stratified by transfusion history.
In the 4-week screening period for both studies, hemoglobin level was evaluated before randomization and within 5 days of study drug administration. Patients who met the protocol-specified transfusion criteria received packed RBC transfusion so that hemoglobin level was above the protocol-specified threshold for transfusion, as confirmed by central or local laboratory.
In both studies, study visits occurred weekly starting on day 1, followed by study visits every 2 weeks starting on day 29. Efficacy and safety were evaluated over 26 weeks of treatment, after which patients could enter the extension period in which all patients received ravulizumab. The extensions period in both studies was planned for 2 years, according to the Clinical Study Reports. Results from the extension period from week 26 to week 52 are presented in the other relevant evidence section of this report.
Populations
Inclusion and Exclusion Criteria
Details on key inclusion and exclusion criteria for the studies are presented in Table 6. Patients in both studies had to have a PNH diagnosis confirmed by flow cytometry (granulocyte or monocyte clone size of at least 5%). Patients in Study 301, the treatment-naive population, had to have an LDH level of 1.5 × ULN or greater and at least 1 of the following PNH-related signs or symptoms in the past 3 months: fatigue, hemoglobinuria, abdominal pain, dyspnea, anemia (hemoglobin < 10 g/dL), history of a MAVE, dysphagia, erectile dysfunction, or history of packed RBC transfusion due to PNH. Patients in Study 302, the eculizumab-treated population, had to have received eculizumab according to the labelled dosing recommendation for PNH and have controlled LDH (< 2 × ULN) and no MAVE for at least 6 months and a screening LDH of 1.5 × ULN or greater. According to the clinical expert consulted by CADTH for the review, these criteria would have been sufficient to exclude patients who would receive an eculizumab dosage beyond the Health Canada–approved PNH dosage due to consistent pharmacokinetic-related breakthrough hemolysis. In both studies, patients with platelet count of less than 30,000/mm3 or absolute neutrophil count of less than 500/µL were excluded. The clinical expert noted that these criteria may have been implemented to exclude patients with frank bone marrow failure, though patients with these counts in clinical practice can still receive treatment if they otherwise have evidence of hemolytic PNH.
Baseline Characteristics
Across Study 301 and Study 302, approximately half of patients were male, most were either Asian or White, and mean age was 45 years to 49 years. Although the distribution of patients among races in Study 301 was not reflective of Canadian patients and was not balanced between the ravulizumab and eculizumab groups, the clinical expert consulted by CADTH confirmed that differences in race would not notably impact outcomes. In Study 301, most patients (85.6% to 86.8%) had an LDH level of 3 × ULN or greater and had received at least 1 transfusion in the past year (82.4% to 82.6%). Patients in Study 302 had a mean LDH of 228 U/L to 235 U/L (with ULN for LDH considered to be 246 U/L), with 12.2% to 13.4% of patients having received at least 1 transfusion in the past year. Patients in Study 301 had a shorter mean disease duration (6.4 years to 6.7 years) than patients in Study 302 (11.9 years to 12.4 years), who had been receiving eculizumab for a mean of 5.6 years to 6.0 years. There were lower percentages of patients in Study 301 who had experienced a MAVE (13.6% to 20.7%) than in Study 302 (22.4% to 28.9%). Percentages of patients with a history of anemia and hematuria or hemoglobinuria were notably higher in Study 301 compared with Study 302. The clinical expert consulted by CADTH considered the percentages in Study 301 to reflect patients in the international PNH registry while a possible explanation of the lower percentages in Study 302 is recall bias due to proximity to experience with untreated disease. Although there were some imbalances between groups within each trial, they were not expected by the clinical expert to contribute any bias to the efficacy or safety results.
Table 7: Summary of Baseline Characteristics
Baseline characteristic | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Sex, n (%) | NA | NA | NA | NA |
Male | 65 (52.0) | 69 (57.0) | 50 (51.5) | 48 (49.0) |
Female | 60 (48.0) | 52 (43.0) | 47 (48.5) | 50 (51.0) |
Race, n (%) | NA | NA | NA | NA |
Asian | 72 (57.6) | 57 (47.1) | 23 (23.7) | 19 (19.4) |
White | 43 (34.4) | 51 (42.1) | 50 (51.5) | 61 (62.2) |
Black or African American | 2 (1.6) | 4 (3.3) | 5 (5.2) | 3 (3.1) |
American Indian or Alaska Native | 1 (0.8) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Other | 4 (3.2) | 4 (3.3) | 2 (2.1) | 1 (1.0) |
Not reported | 3 (2.4) | 4 (3.3) | 13 (13.4) | 13 (13.3) |
Unknown | 0 (0.0) | 0 (0.0) | 3 (3.1) | 1 (1.0) |
Age, years | NA | NA | NA | NA |
Mean (SD) | 44.8 (15.2) | 46.2 (16.2) | 46.6 (14.4) | 48.8 (14.0) |
Median (minimum, maximum) | 43 (18, 83) | 45 (18, 86) | 45 (18, 79) | 49 (23, 77) |
Age > 65 years, n (%) | 14 (11.2) | 18 (14.9) | 12 (12.4) | 14 (14.3) |
Mean disease duration, years (SD) | 6.7 (8.1) | 6.4 (7.5) | 12.4 (8.4) | 11.9 (9.4) |
Mean PNH clone size, % (SD) | NA | NA | NA | NA |
RBC type II | 12.36 (20.54) | 13.70 (17.67) | 14.90 (19.55) | 16.33 (23.64) |
RBC type III | 26.29 (17.25) | 25.21 (16.94) | 44.58 (30.52) | 43.47 (29.71) |
Total RBC | 38.40 (23.75) | 38.74 (23.19) | 60.63 (32.52) | 59.47 (31.41) |
Granulocyte | 84.22 (20.96) | 85.28 (18.98) | 82.63 (23.60) | 83.95 (21.38) |
Monocyte | 86.86 (18.08) | 89.15 (15.19) | 85.64 (20.45) | 86.07 (19.74) |
Mean duration on eculizumab before study treatment, years (SD) | NA | NA | 6.0 (3.5) | 5.6 (3.5) |
Mean LDH, U/L (SD) | NR | NR | 228.01 (48.71) | 235.22 (49.71) |
LDH ≥ 1.5 × ULN and < 3 × ULNa, n (%) | 18 (14.4) | 16 (13.2) | NR | NR |
LDH ≥ 3 × ULNa, n (%) | 107 (85.6) | 105 (86.8) | NR | NR |
pRBC and whole blood transfusions within 12 months before first study dose | NA | NA | NA | NA |
Patients with transfusionsb, n (%) | 103 (82.4) | 100 (82.6) | 13 (13.4) | 12 (12.2) |
Mean number of transfusions (SD) | 6.6 (6.0) | 5.7 (5.5) | 4.9 (5.5) | 2.5 (2.3) |
Mean units transfused (SD) | 9.0 (7.7) | 8.6 (7.9) | 7.9 (8.8) | 4.2 (3.8) |
Patients with 1 to 14 unitsa, n (%) | 80 (64.0) | 76 (62.8) | NR | NR |
Patients with > 14 unitsa, n (%) | 23 (18.4) | 24 (19.8) | NR | NR |
Patients with a history of PNH conditions, n (%) | 121 (96.8) | 120 (99.2) | 90 (92.8) | 96 (98.0) |
Anemia | 103 (82.4) | 105 (86.8) | 64 (66.0) | 67 (68.4) |
Hematuria or hemoglobinuria | 81 (64.8) | 75 (62.0) | 47 (48.5) | 48 (49.0) |
Aplastic anemia | 41 (32.8) | 38 (31.4) | 34 (35.1) | 39 (39.8) |
Renal failure | 19 (15.2) | 11 (9.1) | 11 (11.3) | 7 (7.1) |
Myelodysplastic syndrome | 7 (5.6) | 6 (5.0) | 3 (3.1) | 6 (6.1) |
Pregnancy complication | 3 (2.4) | 4 (3.3) | 4 (4.1) | 9 (9.2) |
Other | 27 (21.6) | 13 (10.7) | 14 (14.4) | 14 (14.3) |
Patients with a history of MAVE, n (%) | 17 (13.6) | 25 (20.7) | 28 (28.9) | 22 (22.4) |
LDH = lactate dehydrogenase; MAVE = major adverse vascular event; NA = not applicable; NR = not reported; PNH = paroxysmal nocturnal hemoglobinuria; pRBC = packed red blood cell; RBC = red blood cell; SD = standard deviation; ULN = upper limit of normal.
Note: Baseline characteristics are presented for the full analysis set.
aA randomization stratification group in Study 301. Results presented are those for observed values rather than those used for randomization.
bA randomization stratification group in Study 302. Results presented are those for observed values rather than those used for randomization.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Interventions
In both studies, patients were assigned to treatment groups using a computer-generated random sequence via an interactive voice- or web-response system. Randomization was stratified in Study 301 by transfusion history in the past 1 year in units of packed RBCs (0 units, 1 unit to 14 units, or > 14 units) and by screening LDH level (1.5 to < 3 × ULN or ≥ 3 × ULN). There were 5 instances of a patient being stratified to a transfusion history category that did not match their observed category. Randomization in Study 302 was stratified by whether or not patients had any history of transfusion in the past 1 year. There were 3 instances of patients being stratified to a transfusion history category that did not match their observed category.
Ravulizumab and eculizumab were supplied as sterile, preservative-free 10 mg/mL solutions in single-use vials to be diluted in saline for IV infusion. The loading dose for ravulizumab was 2,400 mg for patients weighing 40 kg to less than 60 kg, 2,700 mg for patients weighing 60 kg to less than 100 kg, and 3,000 mg for patients weighing 100 kg and greater. The maintenance dose for ravulizumab, given every 8 weeks starting 2 weeks after the loading dose was
3,000 mg for patients weighing 40 kg to less than 60 kg, 3,300 mg for patients weighing 60 kg to less than 100 kg, and 3,600 mg for patients weighing 100 kg and greater. In Study 301, patients in the eculizumab group received 600 mg of eculizumab every 7 days for the first 4 doses, following by 900 mg 1 week after the fourth dose and every 2 weeks afterwards. In Study 302, patients in the eculizumab group continued with eculizumab 900 mg every 2 weeks. For all patients in Study 302, the first dose of study drug was administered 2 weeks after the last dose of eculizumab before study treatment.
The permitted duration of infusion for eculizumab was 25 minutes to 45 minutes (excluding IV flush), with the exception of a maximum of 120 minutes when managing an AE. The minimum infusion duration for ravulizumab ranged from 102 minutes to 114 minutes for the loading doses and 120 minutes to 140 minutes for the maintenance doses, depending on body weight category.
Concomitant medications necessary for patients’ care were permitted during both studies, with the exception of anticoagulants in patients not on a stable dose regimen for at least 2 weeks before initiation of study treatment.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized below. A detailed discussion and critical appraisal of the outcome measures are provided in Appendix 3.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 301 | Study 302 |
|---|---|---|
Thrombotic events | Proportion of patients experiencing MAVEs (secondary) | Proportion of patients experiencing MAVEs (secondary) |
Health-related quality of life | Change in EORTC QLQ-C30 global health status (secondary) | Change EORTC QLQ-C30 global health status (secondary) |
Transfusions | Proportion of patients achieving transfusion avoidance (coprimary) Units transfused (secondary) | Proportion of patients achieving transfusion avoidance (key secondary) Units transfused (secondary) |
Symptoms of PNH | Percent change in FACIT-F total score (key secondary) EORTC QLQ-C30 symptom scales (secondary) Shift in clinical manifestations of PNH (secondary) | Percent change in FACIT-F total score (key secondary) EORTC QLQ-C30 symptom scales (secondary) Shift in clinical manifestations of PNH (secondary) |
Breakthrough hemolysis events | Proportion of patients with breakthrough hemolysis (key secondary) | Proportion of patients with breakthrough hemolysis (key secondary) |
Intravascular hemolysis | Proportion of patients with LDH normalization (coprimary) Percent change in LDH (key secondary) | Percent change in LDH (primary) Proportion of patients with normal LDH at week 26 (secondary) |
Hemoglobin stabilization | Proportion of patients with hemoglobin stabilization (key secondary) | Proportion of patients with hemoglobin stabilization (key secondary) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; LDH = lactate dehydrogenase; MAVE = major adverse vascular event; PNH = paroxysmal nocturnal hemoglobinuria.
Note: Key secondary end points were included in the statistical testing hierarchy for the closed testing procedure. Secondary end points were not included in the closed testing procedure.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Thrombotic Events
In both studies, the occurrence of MAVEs during the primary evaluation period was assessed as an exploratory end point, along with the method of diagnosis, date of diagnosis, and date of resolution.
Health-Related Quality of Life
The EORTC QLQ-C30, a multidimensional, cancer-specific, self-administered questionnaire for assessing HRQoL, was administered in both studies at screening and on days 1, 8, 29, 71, 127, and 183 (or at the early termination visit if applicable). The questionnaire includes a global health status score and change in the global health status score from baseline to week 26 was a secondary end point in both studies. Scores range from 0 to 100 and a higher global health status score indicates better HRQoL. Estimates of the minimal important difference (MID) for the scale scores in patients with cancer are 10 points to 20 points for moderate changes and greater than 20 points for large change. Estimates of MIDs for the scale scores were not identified in patients with PNH. More details on the EORTC QLQ-C30 and its properties can be found in Appendix 3.
Transfusions
Sample collection for laboratory assessments occurred at each study visit and at visits where study treatment was administered, samples were collected before the dose and not from a heparinized line. Hemoglobin was assessed at screening and at each study visit (weekly until day 29, followed by visits every 2 weeks). In both studies, patients were to receive a packed RBC transfusion when they met either of the following criteria: hemoglobin value of 9 g/dL or less with signs or symptoms of sufficient severity to warrant a transfusion, or hemoglobin value of 7 g/dL or less regardless of presence of clinical signs or symptoms. The number of units to be transfused was determined by the investigator and the transfusion was recommended to take place within 48 hours of the hemoglobin determination. Patients received a transfusion before randomization and within 5 days before day 1 if they met either of the transfusion criteria. The proportion of patients achieving transfusion avoidance from day 1 through day 183 was a coprimary end point in Study 301 and a key secondary end point in Study 302. The total number of units transfused was a secondary end point in both studies.
Symptoms of PNH
Also in both studies, the FACIT-F was administered to patients at screening and on days 1, 8, 29, 71, 127, and 183 (or at the early termination visit if applicable). Percent change in FACIT-F score from baseline to week 26 was a key secondary end point in both studies. The FACIT-F scale is a 13-item questionnaire used to assess patient fatigue and energy levels. Scores range from 0 to 52, with lower scores indicating greater fatigue. Estimates of the MID for the FACIT-F score were not identified in patients with PNH. More details on the FACIT-F scale and its properties are presented in Appendix 3.
In both studies, the EORTC QLQ-C30 symptom scales were also assessed. The fatigue, dyspnea, and pain scale scores results are included in the present report and changes in the scores from baseline to week 26 were secondary end points. Scores range from 0 to 100 and higher symptom scale scores indicate a higher level of symptomology. Estimates of the MID for the scale scores in patients with cancer are 10 points to 20 points for moderate changes and greater than 20 points for large change. Estimates of MIDs for the scale scores were not identified in patients with PNH. More details on the EORTC QLQ-C30 and its properties can be found in Appendix 3.
In both studies, the presence or absence of the following signs and symptoms of PNH were recorded at each study visit: fatigue, chest pain, abdominal pain, dyspnea, dysphagia, erectile dysfunction, and red or dark urine or hemoglobinuria. Shifts in these signs and symptoms during the primary evaluation period were reported as secondary end points.
Breakthrough Hemolysis Events
If a suspected breakthrough hemolysis event occurred, LDH and other central laboratory assessments were to occur at an unscheduled visit (if the event did not occur at a scheduled visit). Breakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, dyspnea, anemia [hemoglobin < 10 g/dL], MAVE, dysphagia, or erectile dysfunction) in the presence of LDH 2 × ULN or greater following prior reduction of LDH to less than 1.5 × ULN. Free C5 of 0.5 mcg/mL or greater was defined as suboptimal C5 inhibition and concomitant infections and complement-amplifying conditions were also assessed to further characterize breakthrough hemolysis events. Proportion of patients with breakthrough hemolysis during the primary evaluation period was a key secondary end point in both studies.
Intravascular Hemolysis
Serum LDH was assessed at screening and at each study visit in both studies (weekly until day 29, followed by visits every 2 weeks). Samples were collected before each study drug administration and sent for testing at a central laboratory. Samples with a serum potassium value of 6 mmol/L or greater and an LDH value of 2 × ULN or greater were considered to have undergone ex vivo hemolysis and were excluded from efficacy analyses, although such LDH values could be used for determining breakthrough hemolysis. LDH normalization was defined as an LDH level less than or equal to the ULN (246 U/L). Proportion of patients with LDH normalization from day 29 through day 183 was a coprimary end point in Study 301, percent change in LDH from baseline to day 183 was a key secondary end point in Study 301 and the primary end point in Study 302, and proportion of patients with LDH normalization at day 183 was a secondary end point in Study 302.
The relationship between the LDH threshold of 1.5 × ULN and the key PNH clinical outcomes of mortality and thromboembolism has been described in multiple publications reporting on patients in a national South Korean PNH registry who had not received eculizumab. Details on the evidence supporting 1.5 × ULN as a clinically meaningful threshold for LDH are presented in Appendix 3.
Hemoglobin Stabilization
Hemoglobin was assessed at screening and at each study visit in both studies. As with serum LDH, samples were collected before each study drug administration and sent for testing at a central laboratory. Proportion of patients with hemoglobin stabilization was a key secondary end point in both studies with stabilization defined as avoidance of a 2 g/dL or greater decrease in hemoglobin level in the absence of transfusion from baseline through day 183.
Adverse Events
AEs were assessed through continuous AE monitoring; laboratory results for hematology, blood chemistry, coagulation, and urinalysis measurements at each study visit; vital signs measurements at each study visit; and abbreviated physical examination and electrocardiograms on days 1, 71, and 183.
Immunogenicity
Samples for assessing presence and titre of ADAs were collected before dosing on days 1, 71, 127, and 183 in both studies.
Statistical Analysis
Primary End Points of the Studies
The coprimary end points of Study 301 were percentage of patients with transfusion avoidance throughout the primary evaluation period (assessed by the difference between groups) and proportion of patients with LDH normalization from day 29 through day 183 (assessed by the OR). Ravulizumab had to meet noninferiority for both end points to meet the primary objective of the study. The primary end point of Study 302 was percent change in LDH from baseline to day 183 and, similarly, ravulizumab was tested for noninferiority to eculizumab.
For transfusion avoidance, the lower bound of the 95% CI for the OR of ravulizumab versus eculizumab had to be greater than 0.39 to meet the noninferiority margin. For LDH normalization, the lower bound of the 95% CI had to be higher than –20% for the difference in percentage of patients with LDH normalization in the ravulizumab group versus the eculizumab group to meet the noninferiority margin. For percent change in LDH, the upper bound of the 95% CI had to be lower than 15% for the mean percent change in LDH for ravulizumab versus eculizumab.
Statistical Models
A summary of the statistical models used for coprimary and primary and key secondary end points in both studies is presented in Table 10. In both studies, main analyses were performed in the full analysis set (FAS) and sensitivity analyses were conducted in the PP set for the primary and coprimary and key secondary end points.
In Study 301, the between-group difference in percentage of patients achieving transfusion avoidance was calculated as a weighted combination of the differences in each randomization stratum (based on transfusion history and screening LDH level) using Mantel-Haesnzel weights. The 95% CI was computed using the stratified Newcombe method with exact methods used if CIs could not be estimated using the Newcombe method due to small cell sizes. The OR for patients with LDH normalization in the ravulizumab group versus the eculizumab group was estimated using a generalized estimating equation (GEE) with treatment, transfusion history (categorical), and baseline LDH level (continuous) as explanatory variables and a first-order autoregressive structure for within-patient correlation between visits in time. All available LDH assessments from day 29 through day 183 were used.
In Study 302, the between-group difference in percent change in LDH level was estimated using a mixed-effects model for repeated measures with treatment, study visit, study visit by treatment interaction, and transfusion history (yes or no in the past 1 year) as fixed, categorial effects and baseline LDH as a fixed, continuous covariate. An unstructured covariance matrix was assumed for within-patient errors and the Kenward-Roger approximation was used to estimate denominator degrees of freedom.
Statistical Testing
If the coprimary and primary end points in Study 301 and Study 302, respectively, were met, then the key secondary end points were also tested for noninferiority of ravulizumab using a closed testing procedure. If all key secondary end points demonstrated noninferiority, a hierarchy of end points was to be tested for superiority using a closed testing procedure and a significance level of 0.05. For the noninferiority and superiority testing hierarchies, please refer to Table 6. In each hierarchy, the next end point could only be tested if statistical significance was met in the previous end point.
Sample Size Calculations
In Study 301, the required sample size was based on the calculations for the coprimary end point requiring more patients. For the LDH normalization and transfusion avoidance end points, it was estimated that at least 142 patients and 193 patients would be required, respectively, to provide 80% power to demonstrate noninferiority. The estimated required sample size for Study 301 was 214, based on the transfusion avoidance end point and an assumed 10% dropout rate.
In Study 302, it was estimated that 192 patients would be required to provide 90% power to demonstrate noninferiority in percent change in LDH, assuming a SD of 30% in both treatment groups (based on the TRIUMPH study21), and a dropout rate of 10%.
There were no power calculations reported for key secondary or secondary end points in either study.
Noninferiority Margins
For all noninferiority tests, the lower bound of the 95% CI of the estimate of the difference for ravulizumab versus eculizumab had to be higher than the specified noninferiority margin (or the upper bound had to be lower than the margin). As presented in Table 9, most of the noninferiority margins were based on a 50% or less loss of benefit from eculizumab versus placebo in the TRIUMPH study20 or a 50% loss of benefit based on patients treated with eculizumab versus untreated patients (for the Study 301 margins) or patients who discontinued eculizumab (for the Study 302 margins) in the sponsor’s global PNH registry. Some margins were slightly more conservative compared to a margin determined by a 50% or less loss of benefit. For percent change in LDH, the margins were based on a 25% or less loss of benefit in Study 301 and a 11% or less loss of benefit in Study 302. Although it was acknowledged that more conservative noninferiority margins could have been selected, it was determined that the required sample size for more conservative margins would not have been feasible give the rarity of PNH. In particular, it would have been difficult to enrol sufficient numbers of treatment-naive patients for Study 301.
Table 9: Noninferiority Margins
Outcome measure | Study 301 | Study 302 |
|---|---|---|
Proportion of patients achieving transfusion avoidance (difference in percentage) | 20% difference, based on ≤ 50% loss of benefit from the global PNH registry, adjusted for history of transfusions | 20% difference (25% rounded to 20%), based on ≤ 50% loss of benefit from the global PNH registry |
Proportion of patients with LDH normalization (odds ratio) | 0.39, based on ≤ 50% loss of benefit from the TRIUMPH study (in patients with baseline LDH of < 2,400 U/L) | NA |
Percent change in LDH (difference in percentage) | 20% difference (22% rounded to 20%), based on ≤ 25% loss of benefit from the TRIUMPH study | 15% difference, based on ≤ 11% loss of benefit from the global PNH registry |
Change in FACIT-F (difference in change in score) | 5-point difference, based on ≤ 50% loss of benefit in difference in change from baseline from the TRIUMPH study | 3-point difference, based on ≤ 50% loss of benefit in change from baseline with eculizumab from the TRIUMPH study |
Proportion of patients with breakthrough hemolysis (difference in percentage) | 20% difference, which is conservative compared to a 35% difference which is based on ≤ 50% loss of benefit from the TRIUMPH study | 20% difference (23% rounded to 20%), based on ≤ 50% loss of benefit between patients on eculizumab in the TRIUMPH study and patients who discontinued eculizumab in the global PNH registry |
Proportion of patients with stabilized hemoglobin (difference in percentage) | 20% difference, based on ≤ 50% loss of benefit from the TRIUMPH study | 20% difference (22.5% rounded to 20%), based on ≤ 50% loss of benefit from the global PNH registry |
FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; LDH = lactate dehydrogenase; NA = not applicable; PNH = paroxysmal nocturnal hemoglobinuria.
Note: Benefit of eculizumab estimated from the global PNH registry was relative to untreated patients for Study 301 outcomes and relative to patients who discontinued eculizumab for Study 302 outcomes.
Note: The TRIUMPH Study21 compared eculizumab with placebo in patients with PNH.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Subgroup Analyses
There was no pre-specified statistical testing for differences in efficacy by subgroups of patients. The following subgroups were analyzed for primary, coprimary, and key secondary end points in both studies: randomization stratification variables (transfusion history for both studies and screening LDH level for Study 301), sex, race, region, and age. The only relevant subgroups in the studies, according to the CADTH systematic review protocol, were those categorized according to screening LDH level. In Study 301, the categories were screening LDH level of 1.5 to less than 3 × ULN and 3 × ULN or greater. There were no subgroups in Study 302 categorized by LDH level.
Data Imputation Methods
Data imputation methods for each outcome were the same across both studies. For percent change in LDH, proportion of patients with LDH normalization, and FACIT-F score there was no imputation for a patient at a particular visit. For proportion of patients with transfusion avoidance, breakthrough hemolysis, or hemoglobin stabilization, patients who discontinued the study due to lack of efficacy during the primary evaluation period were considered nonresponders. Patients who discontinued the study for other reasons were included, but assessments following discontinuation were not included.
For the EORTC QLQ-C30 subscale scores and the FACIT-F subscale score, scores were computed if more than 50% of the items were completed, in accordance with their scoring guidelines. There was no imputation for missing subscale scores.
Sensitivity Analyses
In addition to PP analyses, additional sensitivity analyses were conducted for the coprimary and primary end points. In Study 301, both coprimary end points were analyzed using a finer categorization of transfusion history (0, 1 to 4, > 4 to 14, or > 14 units), as well as no adjustment factors. Transfusion avoidance was also analyzed with response being defined as not actually receiving a transfusion as opposed to not meeting the transfusion criteria. LDH normalization was analyzed with transfusion history as a continuous variable as well as with a weighted GEE to account for drop-outs under the missing at random assumption. As well, the proportion of patients with a median LDH from day 29 through day 183 of below ULN was compared between treatment groups. In Study 301, the primary end point was also analyzed without adjustment for transfusion history and baseline LDH.
Statistical Models for Key Secondary End Points
A summary of the statistical models used for the primary and key secondary end points is presented in Table 10. Percent change in LDH in Study 301 and percent change in FACIT-F score in both studies were analyzed using the same methods as for the primary end point in Study 302 (with both randomization strata in Study 301 used in place of transfusion history as fixed, categorical effects). In Study 301, breakthrough hemolysis and hemoglobin stabilization were analyzed using the same methods as for transfusion avoidance. In Study 302, transfusion avoidance, breakthrough hemolysis, and hemoglobin stabilization were analyzed using the methods as for transfusion avoidance in Study 301, except that the Study 302 randomization stratum (transfusion history) replaced the Study 301 randomization strata as a fixed, categorical effect.
LDH normalization in Study 302, a secondary end point, was analyzed using the same model and adjustment factors as for LDH normalization in Study 301, except that LDH normalization from baseline to day 183 was considered (rather than starting at day 29).
In both studies, the EORTC QLQ-C30 subscale scores (secondary end points) were reported as summary statistics (mean, median, and change from baseline) at each assessment. Shifts from baseline in clinical manifestations of PNH in both studies, also secondary end points, were summarized for each study visit.
Table 10: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Study 301 | |||
Proportion of patients achieving transfusion avoidance (coprimary) |
|
|
|
Proportion of patients with LDH normalization from day 29 to day 183 (coprimary) |
|
|
|
|
|
| PP analysis |
| Same as for transfusion avoidance | Same as for transfusion avoidance | PP analysis |
Study 302 | |||
% change in LDH (primary) |
|
|
|
|
| pRBC transfusion history (yes or no within 1 year before day 1) | PP analysis |
% change in FACIT-F score (key secondary) | Same as for % change in LDH | Same as for % change in LDH | PP analysis |
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GEE = generalized estimating equation; LDH = lactate dehydrogenase; MMRM = mixed-effects model for repeated measures; PP = per protocol; pRBC = packed red blood cell; ULN = upper limit of normal.
Note: Transfusion avoidance is defined as patients who did not receive a transfusion and did not meet the protocol-specified guidelines for transfusion up to day 183.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Analysis Populations
In both studies, the primary efficacy analyses were conducted in the FAS and patients were analyzed according to the treatment group to which they were randomized. In Study 301, the FAS included all patients who received at least 1 dose of study drug and had at least 1 efficacy assessment following the first dose of study drug. In Study 302, the FAS included all patients who received at least 1 dose of study drug.
The PP set included all patients in the FAS who missed no doses of ravulizumab or no more than 1 dose of eculizumab during the primary evaluation period, never received the wrong study treatment, followed the protocol-specified transfusion criteria, and met key study selection criteria.
The safety set in both studies was used for safety analyses and included all patients who received at least 1 dose of study drug. Patients were analyzed according to the study treatment they received.
Results
Patient Disposition
All of the randomized patients in Study 301 received study treatment and were included in the FAS. Two patients in the eculizumab group (1.7%) discontinued the study; 1 due to physician decision and 1 due to withdrawal by patient. In Study 302, all but 1 patient in each treatment group received study treatment and were included in the FAS. One patient in the ravulizumab group (1.0%) withdrew from the study and 3 in the eculizumab group (3.0%) discontinued the study (1 patient each for withdrawal by patient, lack of efficacy, and pregnancy). The FAS and safety set were identical in both studies.
The following numbers of patients were excluded from the PP set: 1 patient from each treatment group in Study 301 and 4 and 5 patients were excluded from the ravulizumab and eculizumab groups, respectively, in Study 302. One patient in each treatment group in Study 301 and 2 patients in the ravulizumab group and 3 patients in the eculizumab group in Study 302 met the transfusion criteria but did not receive a transfusion during the primary evaluation period. In Study 302, 2 patients in the ravulizumab group had received some eculizumab doses in the 6 months before study treatment that fell outside of the ± 2-day window for planned infusions. Also, 2 patients in the eculizumab group had an LDH value of greater than 2 × ULN in those 6 months.
Category or analysis set | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Screened, N | 285 | 208 | ||
Randomized, N (%) | 125 (100.0) | 121 (100.0) | 98 (100.0) | 99 (100.0) |
Withdrawal before study treatment, N | 0 | 0 | 1 | 1 |
Received study treatment, N (%) | 125 (100.0) | 121 (100.0) | 97 (99.0) | 98 (99.0) |
Discontinued study, N | 0 | 2 (1.7) | 1 (1.0) | 3 (3.0) |
Reason for study discontinuation, N | NA | NA | NA | NA |
Physician decision | 0 | 1 | 0 | 0 |
Withdrawal by patient | 0 | 1 | 1 | 1 |
Lack of efficacy | 0 | 0 | 0 | 1 |
Pregnancy | 0 | 0 | 0 | 1 |
Full analysis set, N | 125 (100.0) | 121 (100.0) | 97 (99.0) | 98 (99.0) |
PP, N | 124 (99.2) | 120 (99.2) | 93 (94.9) | 93 (93.9) |
Safety, N | 125 (100.0) | 121 (100.0) | 97 (99.0) | 98 (99.0) |
NA = not applicable; PP = per protocol.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Protocol Deviations
A predefined list of select protocol deviations to be summarized was provided in each study’s statistical analysis plan. Major protocol deviations were deviations considered to potentially impact the rights, welfare, or safety of the patients and/or the integrity of study data. These deviations did not appear to be predefined and it was unclear how they were adjudicated.
The percentage of patients with at least 1 major protocol deviation in each treatment group in each study ranged from 10.4% to 16.5% (Table 12). In both studies, there were no protocol deviations that were considered to impact the safety of the patients or the interpretation of the efficacy and safety results. Aside from patients who met the transfusion criteria and did not receive a timely transfusion, there were 2 patients and 1 patient in the eculizumab group of Study 301 and Study 302, respectively, who received a transfusion despite not meeting the criteria.
Table 12: Major Protocol Deviations
Protocol deviation | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Patients with ≥ 1 major protocol deviation, n (%) | 13 (10.4) | 20 (16.5) | 16 (16.5) | 14 (14.3) |
Failure to follow transfusion guidelines | 4 (3.2) | 8 (6.6) | 3 (3.1) | 5 (5.1) |
Other study procedures/tests | 0 (0.0) | 0 (0.0) | 5 (5.2) | 1 (1.0) |
Informed consent | 4 (3.2) | 3 (2.5) | 2 (2.1) | 0 (0.0) |
Delayed safety reporting | 3 (2.4) | 3 (2.5) | 0 (0.0) | 2 (2.0) |
Assigned to incorrect stratification group | 1 (0.8) | 4 (3.3) | 1 (1.0) | 2 (2.0) |
Single dose missed or single dose was less than planned dose | 1 (0.8) | 2 (1.7) | 0 (0.0) | 0 (0.0) |
Did not meet eligibility criteriaa | 0 (0.0) | 2 (1.7) | 5 (5.2) | 2 (2.0) |
Laboratory assessment criteria (test not performed or sample cancelled) | 0 (0.0) | 0 (0.0) | 2 (2.1) | 2 (2.0) |
LDH = lactate dehydrogenase.
Note: Results are presented for the full analysis set.
aReasons: untreated basal cell carcinoma, randomization before documentation of HIV results, prior doses of eculizumab administered outside ± 2-day window, no vaccination for meningococcal infection within 3 years, randomization without screening LDH value, and prior LDH value greater than 2 × ULN in patients who had been receiving eculizumab.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Exposure to Study Treatments
Details on treatment exposure are presented in Table 13. With the exception of 1 patient in the eculizumab group of Study 301 who missed 1 planned infusion, all patients in both studies had treatment adherence of 100% during their time in the study in the primary evaluation period. In Study 301, 3 patients in the ravulizumab group and 1 patient in the eculizumab group each had 1 infusion in which the full dose was not administered. In Study 302, 1 patient in the eculizumab group received 1 extra dose due to breakthrough hemolysis following acute pyelonephritis.
Percentages of patients with an infusion interruption were higher in Study 301 (8.0% and 9.9% for the ravulizumab and eculizumab groups, respectively) compared with Study 302 (1.0% and 5.1% for the ravulizumab and eculizumab groups, respectively), though reasons other than AEs were not reported. Two patients in the ravulizumab group and 1 patient in the eculizumab group in Study 301 and 2 patients in the eculizumab group in Study 301 had an infusion interruption due to AE (Table 22 for specific AEs).
Treatment exposure | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Mean duration from first to last study drug infusion, days (SD) | 126.9 (0.98) | 166.8 (17.79) | 125.9 (12.84) | 166.1 (19.14) |
Number of infusions per patient | NA | NA | NA | NA |
Mean (SD) | 4.0 (0.00) | 14.8 (1.38) | 4.0 (0.30) | 12.8 (1.37) |
Median (minimum, maximum) | 4.0 (4, 4) | 15.0 (2, 15) | 4.0 (1, 4) | 13.0 (1, 14) |
Patients with an infusion interruption, n (%) | 10 (8.0)a | 12 (9.9)b | 1 (1.0) | 5 (5.1)a |
Treatment adherencec, n (%) | NA | NA | NA | NA |
100% | 125 (100.0) | 120 (99.2) | 97 (100.0) | 98 (100.0) |
≥ 90% to < 100% | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
NA = not applicable; SD = standard deviation.
Note: Results are presented for the full analysis set.
aOf these, 2 patients had at least 1 infusion interruption due to an adverse event.
bOf these, 1 patient had 1 infusion interruption due to an adverse event.
cTreatment adherence is defined as the number of infusions divided by number of expected infusions for entire randomized treatment period (excluding day 183 infusion). For patients who discontinued the study, the denominator was the number of expected infusions up to the date of discontinuation.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below.
Survival
Survival was not assessed in Study 301 and Study 302 as an efficacy outcome.
Thrombotic Events
Treatment-emergent MAVEs were assessed in Study 301 and Study 302. In Study 301, 2 patients in the ravulizumab group each experienced 1 event of deep vein thrombosis and 1 patient in the eculizumab group experienced a mesenteric venous thrombosis event. There were no MAVEs in Study 302.
Health-Related Quality of Life
The results for the EORTC QLQ-C30 global health status score are presented in Table 14. Change in the global health status score from baseline to week 26 was a secondary end point and not part of the closed testing procedure in both studies. In Study 301, patients in the ravulizumab and eculizumab groups had a change in global health status score of 13.17 (SD = 21.44) and 12.85 (SD = 21.83), respectively. In Study 302, baseline and week 26 scores were similar to each other within each group, with a change in global health status score of 1.15 (SD = 16.51) in the ravulizumab group and –1.93 (SD = 15.34) in the eculizumab group. Increase in global health status score corresponds to improvement in health status.
Table 14: EORTC QLQ-C30 Global Health Status Score
EORTC QLQ-C30 subscales | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Mean global health status score, N | 124 | 118 | 95 | 95 |
Baseline (SD) | 56.1 (20.3) | 57.5 (20.3) | 75.3 (17.2) | 69.5 (16.5) |
Week 26 (SD) | 69.5 (20.1) | 69.8 (16.9) | 76.6 (15.6) | 67.7 (22.1) |
Change from baseline to week 26 (SD) | 13.17 (21.44) | 12.85 (21.83) | 1.15 (16.51) | –1.93 (15.34) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; SD = standard deviation.
Note: Outcome was outside of the statistical testing hierarchy and was evaluated in the full analysis set. Baseline was defined as the last non-missing value before the first dose of study drug.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Transfusions
The results for transfusion avoidance and transfusions are presented in Table 15. Transfusion avoidance was a coprimary end point in Study 301 and a key secondary end point in Study 302 that was tested for noninferiority in both studies according to the closed testing procedure. According to the protocol-specified transfusion criteria, the mean difference in percentage of patients achieving transfusion avoidance in the ravulizumab versus the eculizumab group was 6.8% (95% CI, –4.66% to 18.14%) in Study 301 and 5.5% (95% CI, –4.27% to 15.68%) in Study 302. Noninferiority was met in both studies as the lower bounds of the 95% CIs were higher than –20%. The results for the PP analyses, which excluded patients who met the transfusion criteria but did not receive a transfusion during the primary evaluation period, were consistent with the FAS results. The results from other sensitivity analyses using a different categorization of transfusion history, defining transfusion avoidance as achieved only by patients who did not receive a transfusion, and removing adjustment for randomization factors were also consistent with the FAS results.
In the subgroups categorized by screening LDH level in Study 301, mean difference in percentage of patients achieving transfusion avoidance in the ravulizumab versus the eculizumab group was 20.1% (95% CI, –13.37% to 51.04%) in patients with LDH of 1.5 to 3 × ULN and 5.3% (95% CI, –8.28% to 18.52%) in patients with LDH of 3 × ULN and greater.
In patients who received at least 1 transfusion, the mean number of transfusions in the ravulizumab and eculizumab groups was 3.3 (SD = 4.15) and 3.6 (SD = 3.06) in Study 301 and 2.7 (SD = 2.75) and 2.0 (SD = 1.29) in Study 302. The mean number of units transfused in the ravulizumab and eculizumab groups was 4.8 (SD = 5.06) and 5.6 (SD = 5.93) in Study 301 and 4.3 (SD = 4.76) and 3.4 (SD = 3.01) in Study 302.
Table 15: Transfusions and Transfusion Avoidance
Transfusions | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Patients achieving transfusion avoidancea (coprimary end point in Study 301), n (%) | 92 (73.6) | 80 (66.1) | 85 (87.6) | 81 (82.7) |
Mean difference, % (95% CI) | 6.8 (–4.7 to 18.1) | Reference | 5.5 (–4.3 to 15.7) | Reference |
Patients receiving transfusion, n (%) | 32 (25.6) | 40 (33.1) | 10 (10.3) | 14 (14.3) |
Mean number of transfusionsb (SD) | 3.3 (4.2) | 3.6 (3.1) | 2.7 (2.8) | 2.0 (1.3) |
Mean number of units transfusedb (SD) | 4.8 (5.1) | 5.6 (6.0) | 4.3 (4.8) | 3.4 (3.0) |
In patients with LDH of 1.5 to 3 × ULN, n | 18 | 16 | NA | NA |
Patients achieving transfusion avoidance, n (%) | 16 (88.9) | 11 (68.8) | NA | NA |
Mean difference, % (95% CI) | 20.1 (–13.4 to 51.0) | Reference | NA | NA |
In patients with LDH ≥ 3 × ULN, n | 107 | 105 | NA | NA |
Patients achieving transfusion avoidance, n (%) | 76 (71.0) | 69 (65.7) | NA | NA |
Mean difference, % (95% CI) | 5.3 (–8.3 to 18.5) | Reference | NA | NA |
CI = confidence interval; LDH = lactate dehydrogenase; NA = not applicable; SD = standard deviation; ULN = upper limit of normal.
aTransfusion avoidance from baseline through week 26 in the full analysis set. In accordance with the closed testing procedures in both trials, noninferiority but not superiority testing for transfusion avoidance was conducted. Difference in transfusion avoidance was calculated as a weighted combination of differences in each randomization stratum using Mantel-Haenszel weights. The 95% CI was computed using the stratified Newcombe CI method. Patients who fulfilled the protocol-specified transfusion criteria were analyzed as having received a transfusion, regardless of whether the patients had actually received a transfusion.
bTransfusions and units received were evaluated per patient who received at least 1 transfusion in the full analysis set during the 26-week treatment period and were outside of the statistical testing hierarchy.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Symptoms of PNH
The results for the FACIT-F score are presented in Table 16. The change in FACIT-F total score was a key secondary end point and was tested for noninferiority in accordance with the closed testing procedure in both studies. The mean difference in change from baseline to week 26 in FACIT-F total score in the ravulizumab versus the eculizumab group was 0.67 (95% CI, –1.21 to 2.55) in Study 301 and 1.47 (95% CI, –0.21 to 3.15) in Study 302. Noninferiority was met in both studies as the lower bounds of the 95% CIs were higher than –5 and –3 in Study 301 and Study 302, respectively. The results for the PP analyses were consistent with the FAS results.
Table 16: Fatigue Measured by FACIT-F Total Score
FACIT-F | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Mean FACIT-F total scorea (SD) | NA | NA | NA | NA |
Baseline | 36.7 (9.68) | 36.9 (10.34) | 42.5 (9.42) | 40.7 (9.49) |
Week 26 | 44.1 (7.84) | 43.6 (7.42) | 44.1 (8.49) | 41.5 (10.19) |
LSM change from baseline to week 26 (SEM) | 7.07 (0.773) | 6.40 (0.789) | 2.01 (0.697) | 0.54 (0.704) |
Mean difference in change (95% CI) | 0.67 (–1.21 to 2.55) | Reference | 1.47 (–0.21 to 3.15) | Reference |
CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; LDH = lactate dehydrogenase; LSM = least squares mean; NA = not applicable; SD = standard deviation; SEM = standard error of the mean.
Note: In accordance with the closed testing procedures in both trials, noninferiority but not superiority testing for FACIT-F total score was conducted.
aA mixed-effects model for repeated measures was used in the full analysis set, which included the following terms: treatment group, transfusion history, baseline LDH level (Study ALXN1210-PNH-301 only), baseline FACIT-F total score, study visit, and study visit by treatment group interaction. An unstructured covariance structure was used.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
The results for the EORTC QLQ-C30 symptom subscale scores are presented in Table 17. Changes from baseline to week 26 in the subscale scores were secondary end points and were not part of the closed testing procedure in both studies. Decreases in symptom subscale scores correspond to improvement in symptoms. Changes in the treatment-naive patients of Study 301 were consistently greater in magnitude than changes in patients in Study 302. In Study 301, change for the ravulizumab and eculizumab groups, respectively, was –20.2 (SD = 24.51) and –18.6 (SD = 24.49) for the fatigue scale, –11.3 (SD = 23.43) and –7.6 (SD = 23.41) for the pain scale, and –14.1 (SD = 24.78) and –17.1 (SD = 25.99) for the dyspnea scale. In Study 302, change for the ravulizumab and eculizumab groups, respectively, was –4.97 (SD = 17.260) and –0.71 (SD = 15.271) for the fatigue scale, 0.87 (SD = 12.722) and 1.42 (SD = 19.959) for the pain scale, and –1.74 (SD = 18.950) and –1.06 (SD = 20.417) for the dyspnea scale.
Table 17: EORTC QLQ-C30 Symptom Subscale Scores
EORTC QLQ-C30 subscales | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Mean fatigue symptom scale score, n | 125 | 119 | 94 | 94 |
Baseline (SD) | 39.3 (22.75) | 37.3 (23.42) | 25.3 (22.63) | 26.3 (21.73) |
Week 26 (SD) | 19.1 (21.33) | 19.1 (18.57) | 20.2 (20.68) | 25.4 (23.37) |
Change from baseline to week 26 (SD) | –20.2 (24.51) | –18.6 (24.49) | –4.97 (17.260) | –0.71 (15.271) |
Mean pain symptom scale score, n | 124 | 117 | 95 | 94 |
Baseline (SD) | 18.7 (23.44) | 15.3 (20.92) | 6.2 (15.84) | 8.3 (14.99) |
Week 26 (SD) | 7.3 (15.93) | 8.1 (16.46) | 7.2 (17.47) | 9.9 (18.82) |
Change from baseline to week 26 (SD) | –11.3 (23.43) | –7.6 (23.41) | 0.87 (12.722) | 1.42 (19.959) |
Mean dyspnea symptom scale score, n | 125 | 119 | 96 | 94 |
Baseline (SD) | 27.2 (25.54) | 29.5 (25.89) | 11.7 (19.86) | 19.4 (25.29) |
Week 26 (SD) | 13.1 (19.80) | 12.6 (19.87) | 10.1 (18.80) | 18.1 (26.63) |
Change from baseline to week 26 (SD) | –14.1 (24.78) | –17.1 (25.99) | –1.74 (18.950) | –1.06 (20.417) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; SD = standard deviation.
Note: Outcome was outside of the statistical testing hierarchy and was evaluated in the full analysis set. Baseline was defined as the last non-missing value before the first dose of study drug.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
The results for shifts in signs and symptoms of PNH recorded at baseline and at day 183 are presented in Table 18. In both studies, shifts during the primary evaluation period were secondary end points. For each of the signs and symptoms in the treatment-naive patients of Study 301, most of the patients with the sign or symptom at baseline had a shift such that they did not have the clinical manifestation at day 183. Shifts in signs and symptoms absent at baseline in Study 301 to being present at day 183 were reported in 5.0% of patients or less in each treatment group. For each of the signs and symptoms in the previously treated patients of Study 302, most patients did not report a shift from baseline to day 183. For fatigue, 20.8% of patients in the ravulizumab group reported no fatigue at baseline with a shift to fatigue being present at day 183 compared with 8.4% in the eculizumab group. All other clinical manifestations had 10.5% of patients or less with a shift from absent to present or from present to absent in each treatment group.
Table 18: Shifts in Sign and Symptoms of PNH
Signs and symptoms | Study 301 RAV “yes” at day 183 | Study 301 RAV “no” at day 183 | Study 301 ECU “yes” at day 183 | Study 301 ECU “no” at day 183 | Study 302 RAV “yes” at day 183 | Study 302 RAV “no” at day 183 | Study 302 ECU “yes” at day 183 | Study 302 ECU “no” at day 183 |
|---|---|---|---|---|---|---|---|---|
Patients, by sign or symptom and status at baseline, n (%) | NA | NA | NA | NA | NA | NA | NA | NA |
Fatigue, yes | 30 (24.0) | 50 (40.0) | 31 (26.1) | 45 (37.8) | 22 (22.9) | 7 (7.3) | 28 (29.5) | 10 (10.5) |
Fatigue, no | 6 (4.8) | 39 (31.2) | 5 (4.2) | 38 (31.9) | 20 (20.8) | 47 (49.0) | 8 (8.4) | 49 (51.6) |
Abdominal pain, yes | 3 (2.4) | 14 (11.2) | 4 (3.4) | 11 (9.2) | 2 (2.1) | 3 (3.1) | 4 (4.2) | 2 (2.1) |
Abdominal pain, no | 3 (2.4) | 105 (84.0) | 2 (1.7) | 102 (85.7) | 3 (3.1) | 88 (91.7) | 8 (8.4) | 81 (85.3) |
Dyspnea, yes | 14 (11.2) | 28 (22.4) | 11 (9.2) | 27 (22.7) | 3 (3.1) | 3 (3.1) | 8 (8.4) | 2 (2.1) |
Dyspnea, no | 4 (3.2) | 79 (63.2) | 6 (5.0) | 75 (63.0) | 3 (3.1) | 87 (90.6) | 9 (9.5) | 76 (80.0) |
Dysphagia, yes | 1 (0.8) | 12 (9.6) | 1 (0.8) | 15 (12.6) | 2 (2.1) | 0 (0.0) | 2 (2.1) | 0 (0.0) |
Dysphagia, no | 2 (1.6) | 110 (88.0) | 0 (0.0) | 103 (86.6) | 3 (3.1) | 91 (94.8) | 3 (3.2) | 90 (94.7) |
Chest pain, yes | 1 (0.8) | 4 (3.2) | 5 (4.2) | 12 (10.1) | 0 (0.0) | 0 (0.0) | 1 (1.1) | 0 (0.0) |
Chest pain, no | 2 (1.6) | 118 (94.4) | 2 (1.7) | 100 (84.0) | 2 (2.1) | 94 (97.9) | 4 (4.2) | 90 (94.7) |
Red/dark urine or hemoglobinuria, yes | 12 (9.6) | 59 (47.2) | 8 (6.8) | 48 (40.7) | 4 (4.2) | 0 (0.0) | 1 (1.1) | 6 (6.3) |
Red/dark urine or hemoglobinuria, no | 1 (0.8) | 53 (42.4) | 3 (2.5) | 59 (50.0) | 4 (4.2) | 88 (91.7) | 8 (8.4) | 80 (84.2) |
Erectile dysfunction, yes | 6 (4.8) | 10 (8.0) | 3 (2.5) | 18 (15.1) | 3 (3.1) | 2 (2.1) | 5 (5.3) | 2 (2.1) |
Erectile dysfunction, no | 4 (3.2) | 45 (36.0) | 2 (1.7) | 45 (37.8) | 3 (3.1) | 42 (43.8) | 1 (1.1) | 39 (41.1) |
Erectile dysfunction, NA, n(%) | 60 (48.0) | NA | 51 (42.9) | NA | 47 (49.0) | NA | 48 (50.5) | NA |
ECU = eculizumab; NA = not applicable; RAV = ravulizumab.
Note: Outcome was outside of the statistical testing hierarchy and was evaluated in the full analysis set (125 and 121 in the ravulizumab and eculizumab groups of Study 301, respectively, and 97 and 98 in the ravulizumab and eculizumab groups of Study 302, respectively). Baseline was defined as the last non-missing value before the first dose of study drug. Baseline and day 183 assessments were reported for 125 and 119 in the ravulizumab and eculizumab groups of Study 301, respectively, and 96 and 95 in the ravulizumab and eculizumab groups of Study 302, respectively.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Breakthrough Hemolysis Events
The results for breakthrough hemolysis are presented in Table 19. The proportion of patients with breakthrough hemolysis during the primary evaluation period was a key secondary end point in both studies. It was tested for noninferiority in both studies and for superiority in Study 301 in accordance with the closed testing procedure. The mean difference in the percentage of patients with breakthrough hemolysis in the ravulizumab versus the eculizumab group was –6.7% (95% CI, –14.21% to 0.18%) in Study 301 and –5.1% (95% CI, –18.99% to 8.89%) in Study 302. Noninferiority was met in both studies as the upper bounds of the 95% CIs were lower than 20%. The results for the PP analyses were consistent with the FAS results. Breakthrough hemolysis was the first outcome in the Study 301 testing hierarchy for superiority. The significance level was not met for superiority and no further superiority testing was performed.
A breakdown of events by whether they were concurrent with suboptimal C5 inhibition (free C5 ≥ 0.5 mcg/mL) and/or a complement-amplifying condition (i.e., infection) is provided in Table 19. Patients receiving ravulizumab in both studies did not experience a breakthrough hemolysis event concurrent with suboptimal C5 inhibition, while 7 and 4 patients receiving eculizumab in Study 301 and Study 302, respectively, did experience such events (with 2 of those patients in Study 301 and 1 of those patients in Study 301 having a concomitant infection).
Table 19: Breakthrough Hemolysis
Breakthrough hemolysis | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Patients with breakthrough hemolysis, n (%) | 5 (4.0) | 13 (10.7) | 0 (0.0) | 5 (5.1) |
Mean difference, % (95% CI) | –6.7 (–14.21 to 0.18)a | Reference | –5.1 (–18.99 to 8.89) | Reference |
Breakthrough hemolysis events, n | 5 | 15 | 0 | 7 |
Free C5 ≥ 0.5 mcg/mL alone | 0 | 5 | 0 | 3 |
Complement-amplifying condition (i.e., infection) alone | 4 | 4 | 0 | 2 |
Free C5 ≥ 0.5 mcg/mL and concomitant infection | 0 | 2 | 0 | 1 |
Undeterminedb | 1 | 4 | 0 | 1 |
CI = confidence interval; LDH = lactate dehydrogenase; ULN = upper limit of normal.
Note: Patients with breakthrough hemolysis, evaluated in the full analysis set, were those with 1 or more worsening symptoms or signs of intravascular hemolysis in the presence of LDH 2 or more × ULN following prior reduction of LDH to less than 1.5 × ULN. In accordance with the closed testing procedures, the outcome was tested for noninferiority in both studies and for superiority in Study 301. Difference in percentage of patients with breakthrough hemolysis was calculated as a weighted combination of differences in each randomization stratum using Mantel-Haenszel weights. The 95% CI was computed using the stratified Newcombe CI method.
aP = 0.0558.
bUndetermined breakthrough hemolysis events were those without free C5 of 0.5 mcg/mL or greater and without an identified concomitant infection.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Complications of PNH
Complications of PNH were not assessed in Study 301 and Study 302 as an efficacy outcome.
Intravascular Hemolysis
The results for LDH normalization and mean LDH level are presented in Table 20. LDH normalization was a coprimary end point in Study 301 and a secondary end point in Study 302. The OR for the proportion of patients achieving LDH normalization from day 29 to 183 in Study 301 was 1.187 (95% CI, 0.796 to 1.769) for ravulizumab versus eculizumab. Noninferiority was met as the lower bound of the 95% CI was greater than 0.39. The results for the PP analysis and the other sensitivity analyses using different categorization of transfusion history, removing adjustment for randomization factors, and weighted GEE to account for drop-outs under the missing at random assumption were consistent with the main results. In patients with a screening LDH of 1.5 to 3 × ULN (N = 34), the OR was 1.71 (95% CI, 0.56 to 5.23) and in patients with a screening LDH of ≥ 3 × ULN (N = 212), the OR was 1.13 (95% CI, 0.73 to 1.73). In Study 302, the OR for the proportion of patients achieving LDH normalization from baseline to day 183 was 0.801 (95% CI, 0.500 to 1.282).
Mean percent change in LDH level from baseline to day 183 was the primary end point in Study 302 and a key secondary end point in Study 301. It was tested for noninferiority in both studies and for superiority in Study 302 in accordance with the closed testing procedure. In Study 302, the least squares mean difference in percent change in LDH level was –9.21% (95% CI, –18.84% to 0.42%) for ravulizumab versus eculizumab. Noninferiority was met as the upper bound of the 95% CI was lower than 15%. Percent change in LDH was the first outcome in the Study 302 testing hierarchy for superiority. The significance level was not met for superiority and no further testing was performed. In Study 301, the least squares mean difference in percent change in LDH level was –0.83% (95% CI, –5.21% to 3.56%) for ravulizumab versus eculizumab. Noninferiority was met as the upper bound of the 95% CI was lower than 20%.
Table 20: LDH Normalization and LDH Level
LDH | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Proportion of patients achieving LDH normalizationa (coprimary end point in Study 301) (95% CI) | 0.536 (0.459 to 0.612) | 0.494 (0.417 to 0.570) | 0.660 (0.561 to 0.747) | 0.708 (0.613 to 0.788) |
OR (95% CI) | 1.187 (0.796 to 1.769)b | Reference | 0.801 (0.500 to 1.282)c | Reference |
Mean LDH leveld (primary end point in Study 302), U/L (SD) | NA | NA | NA | NA |
Baseline | 1,633.53 (778.752) | 1,578.30 (727.061) | 228.01 (48.712) | 235.22 (49.710) |
Week 26 | 277.96 (102.879) | 330.45 (480.796) | 224.11 (51.719) | 244.11 (70.292) |
LSM % change (SE) | –76.84 (1.582) | –76.02 (1.617) | –0.82 (3.033) | 8.39 (3.041) |
Mean difference in change (95% CI) | –0.83 (–5.21 to 3.56)b | Reference | –9.21 (–18.84 to 0.42)e | Reference |
In patients with LDH of 1.5 to 3 × ULN, n | 18 | 16 | NA | NA |
Proportion of patients achieving LDH normalization, adjusted prevalence (95% CI) | 0.595 (0.380 to 0.779) | 0.462 (0.255 to 0.683) | NA | NA |
OR (95% CI) | 1.71 (0.56 to 5.23) | Reference | NA | NA |
In patients with LDH ≥ 3 × ULN, n | 107 | 105 | NA | NA |
Proportion of patients achieving LDH normalization, adjusted prevalence (95% CI) | 0.506 (0.422 to 0.591) | 0.477 (0.393 to 0.562) | NA | NA |
OR (95% CI) | 1.13 (0.73 to 1.73) | Reference | NA | NA |
CI = confidence interval; LDH = lactate dehydrogenase; LSM = least squares mean; NA = not applicable; OR = odds ratio; SD = standard deviation; SE = standard error; ULN = upper limit of normal.
Note: In accordance with the closed testing procedure for Study 301, superiority testing for this outcome was not conducted.
aLDH normalization from day 29 through week 26 (Study 301) or from baseline through week 26 (Study 302) in the full analysis set. A generalized estimating equation was used with the following terms: treatment group, history of transfusion, and baseline LDH level. A first-order autoregressive structure was assumed for within-patient correlation.
bIn accordance with the closed testing procedure, superiority testing was not conducted.
cOutcome was outside of the statistical testing hierarchy.
dA mixed-effects model for repeated measures was used in the full analysis set, which included the following terms: treatment group, randomization factors, baseline LDH level, study visit, and study visit by treatment group interaction. An unstructured covariance structure was used.
eP = 0.0583; an approximate P value for superiority associated with the upper bound.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Hemoglobin Stabilization
The results for hemoglobin stabilization are presented in Table 21. Hemoglobin stabilization throughout the primary evaluation period was a key secondary end point for both studies and was tested for noninferiority in both studies according to the closed testing procedure. The mean difference in percentage of patients with hemoglobin stabilization in the ravulizumab versus the eculizumab group was 2.9% (95% CI, –8.80% to 14.64%) in Study 301 and 1.4% (95% CI, –10.41% to 13.31%) in Study 302. Noninferiority was met in both studies as the lower bounds of the 95% CIs were higher than –20%. The results for the PP analyses, which excluded patients who met the transfusion criteria but did not receive a transfusion during the primary evaluation period, were consistent with the FAS results.
Table 21: Hemoglobin Stabilization
Hemoglobin stabilization | Study 301 ravulizumab N = 125 | Study 301 eculizumab N = 121 | Study 302 ravulizumab N = 97 | Study 302 eculizumab N = 98 |
|---|---|---|---|---|
Patients with hemoglobin stabilization, n (%) | 85 (68.0) | 78 (64.5) | 74 (76.3) | 74 (75.5) |
Mean difference, % (95% CI) | 2.9 (–8.8 to 14.6) | Reference | 1.4 (–10.4 to 13.3) | Reference |
CI = confidence interval.
Note: Patients with hemoglobin stabilization, evaluated in the full analysis set, were those who did not have a greater than 2 g/dL decrease in hemoglobin throughout the primary evaluation period and did not have a transfusion. In accordance with the closed testing procedures, superiority testing for this outcome was not conducted in either trial. Difference in percentage of patients with hemoglobin stabilization was calculated as a weighted combination of differences in each randomization stratum using Mantel-Haenszel weights. The 95% CI was computed using the stratified Newcombe CI method.
Source: Clinical Study Reports for Study 301 and Study 302.1,2
Health Care Resource Utilization
Health care resource utilization was not assessed in Study 301 and Study 302 as an efficacy outcome.
Harms
Only those harms identified in the review protocol are reported below. Table 22 contains detailed harms data.
Adverse Events
Most patients (86.8% to 88.0%) in both treatment groups in both studies reported at least 1 AE. The most common AE was headache, which was reported in 17.3% to 36.0% of patients in each treatment group. In Study 302, headaches were reported by 26.8% of the ravulizumab group and 17.3% of the eculizumab group. According to the clinical expert consulted by CADTH, it is possible that the transition from pre-study eculizumab to ravulizumab accounted for this difference due to the more profound C5 blockade with ravulizumab. Headaches were numerically more common in both treatment groups in the treatment-naive patients of Study 301 compared with the ravulizumab group of Study 302. Other AEs that were reported in at least 10% of any treatment group were upper respiratory tract infection, nasopharyngitis, pyrexia, and cough. There were no notable imbalances in AEs.
Serious AEs
SAEs were reported in 4.1% to 8.8% of each treatment group in both studies. The most common SAEs were hemolysis and pyrexia, which occurred in 3.1% or less of each treatment group.
Withdrawals Due to AEs
There were no withdrawals due to AEs in either study.
Mortality
One patient in the eculizumab group in Study 301 died due to lung adenocarcinoma during the extension phase of the study.
Notable Harms
Serious infections were reported in 1.0% to 3.3% of each treatment group in both studies. Each specific SAE was reported in 1 patient. Infusion reactions were reported in 3.1% to 8.8% of patients across each treatment group in both studies. In Study 301, the AEs leading to infusion interruption were 1 infusion-related reaction (lower back pain) and 1 patient with muscle spasms during each infusion in the ravulizumab group and 1 patient with recurrent headache in the eculizumab group. In Study 302, 1 patient had back pain (3 occurrences) and 1 patient had flank pain, both in the eculizumab group.
In Study 301, ADAs were present in 12 patients (9.6%) in the ravulizumab group and in 6 patients (5.0%) in the eculizumab group at baseline and there was 1 treatment-emergent ADA-positive sample in each treatment group. In Study 302, ADAs were present in 4 patients (4.1%) in the ravulizumab group at baseline and there was 1 treatment-emergent ADA-positive sample in the eculizumab group. All ADA titres in the positive samples were considered to be low.
Adverse events | ALXN1210-PNH-301 | ALXN1210-PNH-302 | ||
|---|---|---|---|---|
Ravulizumab N = 125 | Eculizumab N = 121 | Ravulizumab N = 97 | Eculizumab N = 98 | |
Patients with ≥ 1 AE | ||||
n (%) | 110 (88.0) | 105 (86.8) | 85 (87.6) | 86 (87.8) |
Most common eventsa | NA | NA | NA | NA |
Headache | 45 (36.0) | 40 (33.1) | 26 (26.8) | 17 (17.3) |
Upper respiratory tract infection | 13 (10.4) | 7 (5.8) | 18 (18.6) | 10 (10.2) |
Nausea | 11 (8.8) | 10 (8.3) | 8 (8.2) | 9 (9.2) |
Nasopharyngitis | 11 (8.8) | 18 (14.9) | 21 (21.6) | 20 (20.4) |
Diarrhea | 10 (8.0) | 5 (4.1) | 9 (9.3) | 7 (7.1) |
Viral upper respiratory tract infection | 9 (7.2) | 10 (8.3) | 1 (1.0) | 0 (0.0) |
Pain in extremity | 9 (7.2) | 7 (5.8) | 5 (5.2) | 4 (4.1) |
Dizziness | 9 (7.2) | 7 (5.8) | 3 (3.1) | 7 (7.1) |
Arthralgia | 8 (6.4) | 8 (6.6) | 3 (3.1) | 4 (4.1) |
Oropharyngeal pain | 8 (6.4) | 6 (5.0) | 4 (4.1) | 9 (9.2) |
Abdominal pain | 7 (5.6) | 7 (5.8) | 6 (6.2) | 9 (9.2) |
Myalgia | 7 (5.6) | 9 (7.4) | 2 (2.1) | 4 (4.1) |
Back pain | 7 (5.6) | 6 (5.0) | 4 (4.1) | 4 (4.1) |
Pyrexia | 6 (4.8) | 13 (10.7) | 9 (9.3) | 5 (5.1) |
Hypokalemia | 6 (4.8) | 6 (5.0) | 0 (0.0) | 1 (1.0) |
Vomiting | 5 (4.0) | 4 (3.3) | 6 (6.2) | 4 (4.1) |
Fatigue | 5 (4.0) | 4 (3.3) | 6 (6.2) | 6 (6.1) |
Dyspepsia | 4 (3.2) | 6 (5.0) | 0 (0.0) | 2 (2.0) |
Musculoskeletal pain | 4 (3.2) | 2 (1.7) | 2 (2.1) | 5 (5.1) |
Cough | 4 (3.2) | 8 (6.6) | 5 (5.2) | 10 (10.2) |
Anemia | 3 (2.4) | 5 (4.1) | 6 (6.2) | 3 (3.1) |
Influenza-like illness | 3 (2.4) | 1 (0.8) | 7 (7.2) | 8 (8.2) |
Dyspnea | 3 (2.4) | 2 (1.7) | 0 (0.0) | 6 (6.1) |
Chest pain | 2 (1.6) | 5 (4.1) | 3 (3.1) | 9 (9.2) |
Rhinitis | 2 (1.6) | 3 (2.5) | 5 (5.2) | 4 (4.1) |
Insomnia | 2 (1.6) | 6 (5.0) | 1 (1.0) | 1 (1.0) |
Constipation | 1 (0.8) | 3 (2.5) | 7 (7.2) | 5 (5.1) |
Infusion-related reactionb | 1 (0.8) | 0 (0.0) | 3 (3.1) | 1 (1.0) |
Patients with ≥ 1 SAE | ||||
n (%) | 11 (8.8) | 9 (7.4) | 4 (4.1) | 8 (8.2) |
Most common eventsc | NA | NA | NA | NA |
Hemolysis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (2.0) |
Pyrexia | 1 (0.8) | 2 (1.7) | 0 (0.0) | 3 (3.1) |
Patients who stopped treatment due to AEs | ||||
n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Deaths | ||||
n (%) | 0 (0.0) | 1 (0.8)d | 0 (0.0) | 0 (0.0) |
Patients with notable AEs, n (%) | ||||
Serious infections | 2 (1.6) | 4 (3.3) | 2 (2.1) | 1 (1.0) |
Infusion reactions | 11 (8.8) | 10 (8.3) | 8 (8.2) | 3 (3.1) |
Patients with notable SAEs, n (%) | ||||
Infections and infestations | NA | NA | NA | NA |
Abscess limb | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Cellulitis | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Infection | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Influenza | 0 (0.0) | 0 (0.0) | 1 (1.0) | 0 (0.0) |
Leptospirosis | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Lower respiratory tract infection | 0 (0.0) | 0 (0.0) | 1 (1.0) | 0 (0.0) |
Pneumonia | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Pyelonephritis acute | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.0) |
Systemic infection | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Viral upper respiratory tract infection | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
AE = adverse event; NA = not applicable; SAE = serious adverse event.
Note: AEs are reported for the safety set.
aFrequency of 5% or greater in any treatment group.
bIdentified as a notable harm in the systematic review protocol.
cFrequency of greater than 1% in any treatment group.
dDeath due to lung adenocarcinoma occurred in the extension phase.
Source: Clinical Study Reports for Stud 301 and Study 302.1,2
Critical Appraisal
Internal Validity
Study 301 and Study 302 had appropriate randomization and allocation methods and there were no notable imbalances in baseline characteristics between the treatment groups. Study discontinuations were 3% or less in each treatment group and treatment adherence was 100% for all but 1 patient across both studies. Therefore, there are no concerns of bias due to study drop-outs or imbalanced treatment exposure.
The statistical analysis methods used for the efficacy end points were appropriate. The closed testing procedure in both studies was appropriate for controlling the type I error rate and ensuring an accurate interpretation of statistical significance for tests of superiority.
Justification for the chosen noninferiority margins was provided based on prior RCT and registry data; however, the margin for all end points aside from percent change in LDH was based on a 50% or less loss of benefit or similar loss of benefit from eculizumab. The rationale for not selecting more conservative noninferiority margins was that the required sample size would not have been feasible given the rarity of the disease. The clinical expert consulted by CADTH for this review noted that a 50% loss of benefit would not be clinically acceptable, but also recognized that the rarity of the disease does have implications for clinical trial recruitment. The margin for percent change in LDH was based on a 25% or less loss of benefit in Study 301 and a 11% or less loss of benefit in Study 302 and the clinical expert consulted by CADTH found these margins more clinically acceptable. Concerns about the relatively generous noninferiority margins are alleviated by the fact that all primary and key secondary end points met the noninferiority margin in both the primary and PP analyses and that in all cases a more conservative margin would have been met.
Although the subgroup analyses in the studies were preplanned, there were no sample size considerations for them, no control for type I error rate, and no statistical testing for treatment-by-subgroup interaction. Therefore, conclusions cannot be drawn regarding analyses by subgroup.
The open-label nature of the studies means that outcomes relying on subjective reporting, such as the EORTC QLQ-C30 and the FACIT-F could have been biased, with potential for bias in favour of ravulizumab. Determination of the presence of PNH-related signs or symptoms was not considered by the clinical expert to be prone to bias from lack of blinding. Therefore, end points based on laboratory values and/or presence of PNH-related signs or symptoms (i.e., all other primary and key secondary end points) had a low likelihood of bias from lack of blinding. However, reporting of AEs may have been susceptible to bias from lack of blinding.
While the EORTC QLQ-C30 and the FACIT-F have been used extensively in other disease areas, their reliability, validity, and responsiveness have yet to be characterized in patients with PNH. Although estimates for MIDs for these instruments were not identified in this patient population, changes in the relevant scores were not tested for superiority.
External Validity
The patient populations in the studies were overall representative of patients seen in Canadian clinical practice, according to the clinical expert consulted by CADTH. However, there were some exclusions of small subpopulations of patients due to the study selection criteria. The clinical expert noted that patients with frank bone marrow failure would likely have been excluded due to the criteria on platelet and absolute neutrophil count, though these patients in clinical practice can still receive treatment. The criteria for Study 302 were chosen in such a way that patients requiring a higher dose or more frequent dosing of eculizumab beyond the product monograph-recommended dosage would have been excluded, according to the clinical expert. Also, patients who were pregnant or breastfeeding were excluded.
As outlined by the clinical expert in the clinical input section earlier in the present report, approximately 20% of patients require higher doses of eculizumab or more frequent doses than recommended in the Health Canada–approved product monograph for the PNH indication (900 mg maintenance dose every 2 weeks) to properly control symptoms and signs associated with pharmacokinetic breakthrough. Therefore, a similar portion of patients in Study 301 would have been expected to experience pharmacokinetic breakthrough in the eculizumab group since the studies did not allow for deviation from the labelled dosage of eculizumab. This is contrary to Canadian clinical practice, in which dosing would be adjusted to address pharmacokinetic breakthrough. For Study 301, the lack of permitted dosage adjustments may have biased the efficacy results in favour of ravulizumab relative to how eculizumab is dosed in clinical practice.
Finally, the efficacy results do not address important outcomes such as survival and complications of PNH other than thrombotic events (not assessed in either study) and MAVEs (no statistical testing).
Indirect Evidence
CADTH performed a literature review to identify any relevant indirect comparisons that could supplement the available direct evidence. A focused literature search for network meta-analyses dealing with PNH was run in MEDLINE All (1946–) on August 20, 2021. No limits were applied to the search. The search yielded 1 result and its full text was reviewed for relevance using the criteria in Table 5. The network meta-analysis was excluded due to its use of a mixture of RCTs and observational studies. The CADTH systematic review protocol includes eculizumab as the only relevant comparator and ravulizumab and eculizumab were directly compared in Study 301 and Study 302.
Other Relevant Evidence
This section includes 2 long-term extension studies and 1 additional relevant substudy included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Efficacy and safety at time points beyond the primary evaluation period in pivotal trials Study 301 and Study 302, as well as patient preferences regarding treatment, were evaluated within the sponsor’s submission. A summary and critical appraisal of the additional evidence is presented in this section.
Long-Term Extension Studies
Pivotal studies, Study 301 and Study 302 included in the sponsor’s submission included a 26-week primary evaluation period. Presented in this section are the efficacy and safety outcomes for patients in both studies during the extension period, up to the 52-week time point post-baseline. All patients, regardless of the drug received in the primary evaluation period, were transitioned to ravulizumab for the extension period.
Methods
Presented are the results from the extension periods of both Study 301 and Study 302. Following the 26-week randomized treatment period, patients had the option to continue into the extension period where all patients received ravulizumab for an additional 26 weeks. Patients in the ravulizumab treatment arm continued receiving ravulizumab maintenance dosing every 8 weeks, while patients in the eculizumab arm were transitioned to receive a ravulizumab loading dose followed by ravulizumab maintenance dosing every 8 weeks for an additional 26 weeks.
Populations
All patients who entered the extension period of both studies were originally enrolled in the randomized treatment period of Study 301 and Study 302. As such, the inclusion and exclusion criteria are consistent with what is reported in earlier sections. Of the 246 patients who received ravulizumab or eculizumab in Study 301, 243 (98.8%) patients entered the extension period, and of the 192 patients who received ravulizumab or eculizumab in Study 302, 191 (99.4%) patients entered the extension period. Therefore, baseline characteristics are expected to be similar as reported in the systematic review section.
Interventions
All patients either maintained their treatment with ravulizumab or were transitioned from eculizumab to ravulizumab. Use of the study drug was consistent with the primary evaluation period.
Outcomes
The primary and key secondary end points assessed in the primary evaluation period of both studies were also assessed during the extension period of both studies. Briefly, they were transfusion avoidance, FACIT-F scores, breakthrough hemolysis events, LDH normalization and change from baseline, hemoglobin stabilization, as well as safety outcomes.
Statistical Analysis
There was no comparator arm in the extension period as all patients either maintained or were transitioned to treatment with ravulizumab. As such, there was no formal statistical testing conducted on the results. All results presented are for the FAS of patients who entered the extension period and descriptive statistics were used to summarize the results rather than the statistical models used in the primary evaluation period. The initial 26-week primary evaluation period will be referred to as Period 1 and the following 26-week extension period will be referred to as Period 2.
Patient Disposition
In Study 301, 246 total patients were treated with either ravulizumab or eculizumab in the 26-week primary evaluation period. Of these, 99.2% of the patients in the ravulizumab group and 98.3% of the patients in the eculizumab group entered the extension period with 2.4% and 4.1%, respectively, discontinuing treatment at some point during the extension period. Two patients from the eculizumab to ravulizumab group discontinued due to an AE. Other reasons for discontinuation included pregnancy, physician decision, as well as 1 patient who died while on treatment.
In Study 302, 195 total patients were treated with either ravulizumab or eculizumab in the 26-week primary evaluation period. Of these, 99.0% of the patients in the ravulizumab group and 96.9% of the eculizumab group entered the extension period, with 1% of patients in each group discontinuing at some point during the extension period due to either physician decision or withdrawal by patient. Table 23 below reports patient disposition results for both Study 301 and Study 302.
Table 23: Patient Disposition in Extension Studies
Category or analysis set | Study 301 RAV–RAV N = 125 | Study 301 ECU–RAV N = 121 | Study 302 RAV–RAV N = 97 | Study 302 ECU–RAV N = 98 |
|---|---|---|---|---|
Completed 26-week primary evaluation period | 125 (100.0) | 119 (98.3) | 96 (99.0) | 95 (96.9) |
Entered into extension period | 124 (99.2) | 119 (98.3) | 96 (99.0) | 95 (96.9) |
Ongoing in extension period at data cut-off | 121 (96.8) | 114 (94.2) | 95 (97.9) | 94 (95.9) |
Discontinued extension period | 3 (2.4) | 5 (4.1) | 1 (1.0) | 1 (1.0) |
Adverse event | 0 (0.0) | 2 (1.7) | 0 (0.0) | 0 (0.0) |
Death | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Pregnancy | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Physician decision | 1 (0.8) | 2 (1.7) | 0 (0.0) | 1 (1.0) |
Withdrawal by patient | 0 (0.0) | 0 (0.0) | 1 (1.0) | 0 (0.0) |
Other | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
ECU = eculizumab; RAV = ravulizumab.
Note: Values are n (%).
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Exposure to Study Treatments
As Study 301 and Study 302 continued to monitor patients beyond the 52-week time point presented in this review, mean treatment duration is greater than noted during the primary evaluation period with a mean duration of 290.7 days for both groups in Study 301. In Study 302 there was shorter follow-up available with a mean duration of 216.5 days (SD = 31.06) in the ravulizumab to ravulizumab group and 216.0 days (SD = 27.83) in the eculizumab to ravulizumab group. A similar difference is seen between Study 301 and Study 302 with regards to number of infusions per patient. Infusion interruptions were more commonly seen in Study 301 with a total of 17 across both groups, of these 4 were reported to be due to AEs. Treatment adherence was consistent across studies and treatment groups. Full treatment exposure results are shown in Table 24.
Table 24: Treatment Exposure in the Extension Period
Treatment exposure | Study 301 RAV–RAV Period 2 N = 124 | Study 301 ECU–RAV Period 2 N = 119 | Study 302 RAV–RAV Period 2 N = 96 | Study 302 ECU–RAV Period 2 N = 95 |
|---|---|---|---|---|
Mean duration from first to last study drug infusion, days (SD) | 290.7 (66.85) | 290.7 (70.40) | 216.5 (31.06) | 216.0 (27.83) |
Number of infusions per patient | NA | NA | NA | NA |
Mean (SD) | 5.8 (1.17) | 6.4 (1.33) | 4.3 (0.59) | 5.2 (0.53) |
Median (minimum, maximum) | 6.0 (1, 8) | 6.0 (2, 9) | 4.0 (1, 5) | 5.0 (2, 6) |
Patients with an infusion interruption, n (%) | 8 (6.5) | 9 (7.6) | 1 (1.0) | 3 (3.2) |
Treatment adherencea, n (%) | NA | NA | NA | NA |
100% | 123 (99.2) | 119 (100.0) | 96 (100.0) | 95 (100.0) |
≥ 80% to < 100% | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
ECU = eculizumab; NA = not applicable; RAV = ravulizumab; SD = standard deviation.
Note: Treatment duration for extension period is defined as the end of the extension period reached or discontinuation date in extension period-first extension infusion date plus 1 day. Period 1 refers to the primary evaluation period of 0 to 26 weeks and Period 2 refers to the extension period of > 26 weeks to 52 weeks.
aTreatment adherence is defined as the number of infusions divided by number of expected infusions for entire randomized treatment period (excluding day 183 infusion).
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Efficacy
Survival
Survival was not assessed in the Study 301 and Study 302 extensions as an efficacy outcome.
Thrombotic Events
In Study 301, there were 2 MAVEs reported during the extension period up to the 52-week time point. One event was an arterial embolism and the second was a jugular vein thrombosis. In Study 302, there were 2 MAVEs reported during the extension period, 1 of thrombophlebitis and 1 cerebral infarction.
Results for MAVEs occurring during the 2 years before ravulizumab initiation and up to 2 years following ravulizumab initiation in Study 301 are reported in an abstract presentation from Latour et al.24 The results are not included here due to the post hoc nature of the analysis and the immature follow-up at the 2-year extension time point.
Health-Related Quality of Life
HRQoL as measured by EORTC QLQ-C30 was not reported in the Study 301 and Study 302 extensions.
Transfusions
Table 25 presents the transfusion avoidance results for Study 301 and Study 302. The number of patients achieving transfusion avoidance in the extension period remained consistent with the results from the primary evaluation period in both studies. In Study 301, 90.2% of patients in the ravulizumab to ravulizumab group and 87.3% of patients in the eculizumab to ravulizumab group maintained response, while 62.5% and 72.5% of patients in each group who did not respond in the primary evaluation period maintained no response, with regards to transfusion avoidance, during the extension period.
Table 25: Transfusion Avoidance in the Extension Period
Transfusions | Study 301 RAV–RAV N = 124 | Study 301 ECU–RAV N = 119 | Study 302 RAV–RAV N = 96 | Study 302 ECU–RAV N = 95 |
|---|---|---|---|---|
Patients achieving transfusion avoidancea in Period 1, n (%) | 92 (74.2) | 79 (66.4) | 85 (87.6) | 81 (82.7) |
Patients achieving transfusion avoidancea in Period 2, n (%) | 95 (76.6) | 80 (67.2) | 83 (86.5) | 79 (83.2) |
Period 2 compared to Period 1 | NA | NA | NA | NA |
Patients with response in Period 1, N | 92 | 79 | NA | NA |
Maintaining response, n (%) | 83 (90.2) | 69 (87.3) | NA | NA |
95% CI | 84.15% to 96.29% | 80.01% to 94.67% | NA | NA |
Losing response, n (%) | 9 (9.8) | 10 (12.7) | NA | NA |
95% CI | 3.71% to 15.85% | 5.33% to 19.99% | NA | NA |
Patients without response in Period 1, N | 32 | 40 | NA | NA |
Maintaining no response, n (%) | 20 (62.5) | 29 (72.5) | NA | NA |
95% CI | 45.73% to 79.27% | 58.66% to 86.34% | NA | NA |
Gaining response, n (%) | 12 (37.5) | 11 (27.5) | NA | NA |
95% CI | 20.73% to 54.27% | 13.66% to 41.34% | NA | NA |
CI = confidence interval; ECU = eculizumab; NA = not applicable; RAV = ravulizumab.
Note: Period 1 refers to the primary evaluation period of 0 to 26 weeks and Period 2 refers to the extension period of more than 26 weeks to 52 weeks. Transfusion refers to packed red blood cell or whole blood transfusion. Transfusion avoidance was defined as the proportion of patients who remained transfusion-free and did not require a transfusion per protocol-specified guidelines through each period. Patients who withdrew from the study due to lack of efficacy were considered as nonresponders and were counted in the group requiring transfusions.
aPatients who fulfilled the protocol-specified transfusion criteria were analyzed as having received a transfusion, regardless of whether the patients had actually received a transfusion.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Symptoms of PNH
Fatigue as measured by FACIT-F score is shown below in Figure 2 for Study 301, and Figure 3 for Study 302. FACIT-F scores observed at the end of the primary evaluation period appeared to be maintained throughout the extension period in both Study 301 and Study 302.
Figure 2: Fatigue Measured by FACIT-Fatigue (Study 301 Extension)
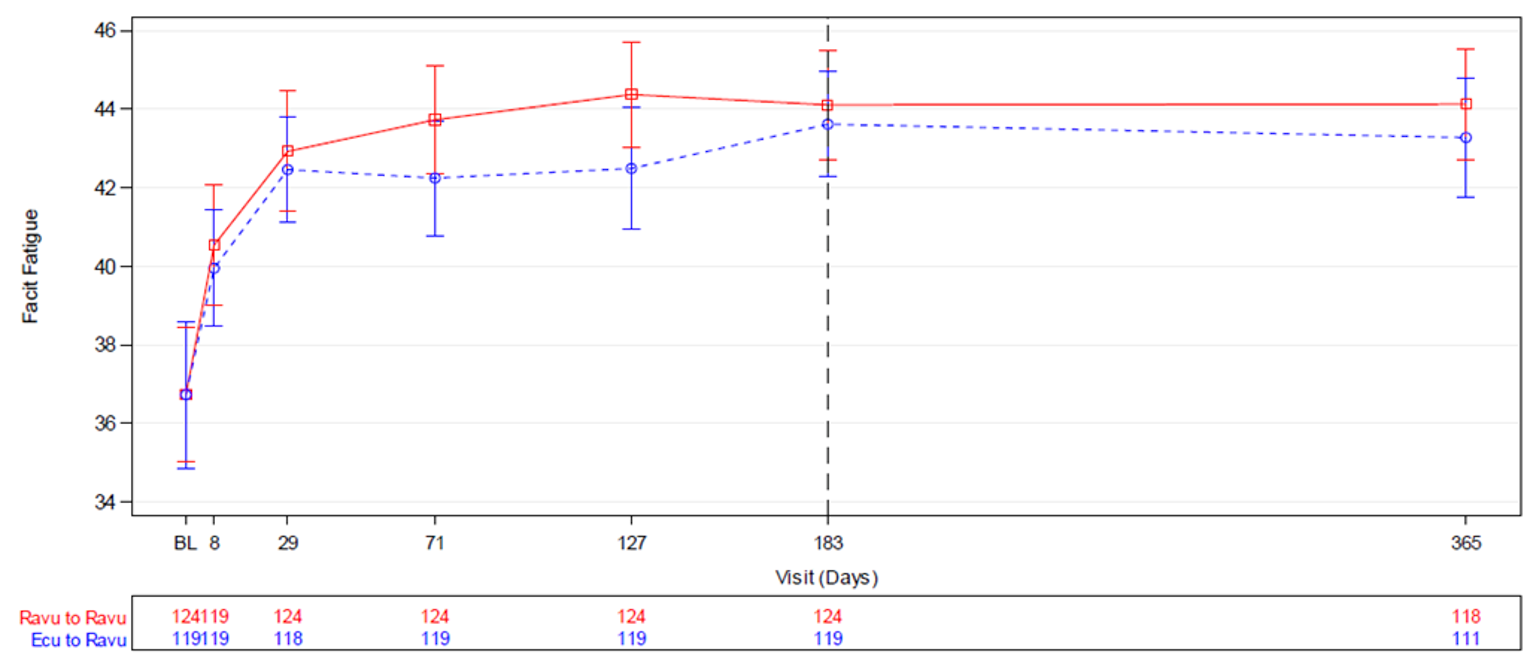
BL = Period 1 baseline; Ecu = eculizumab; FACIT = Functional Assessment of Chronic Illness Therapy; Ravu = ravulizumab.
Note: FACIT-Fatigue scale ranges from 0 to 52, with a higher score indicating less fatigue. Period 1 baseline was defined as the last non-missing assessment value before first study drug dose. Day 183 represents the start of the extension period. Patients in the Ecu to Ravu group received Ecu before day 183 and received Ravu from day 183 onwards.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Figure 3: Fatigue Measured by FACIT-Fatigue (Study 302 Extension)
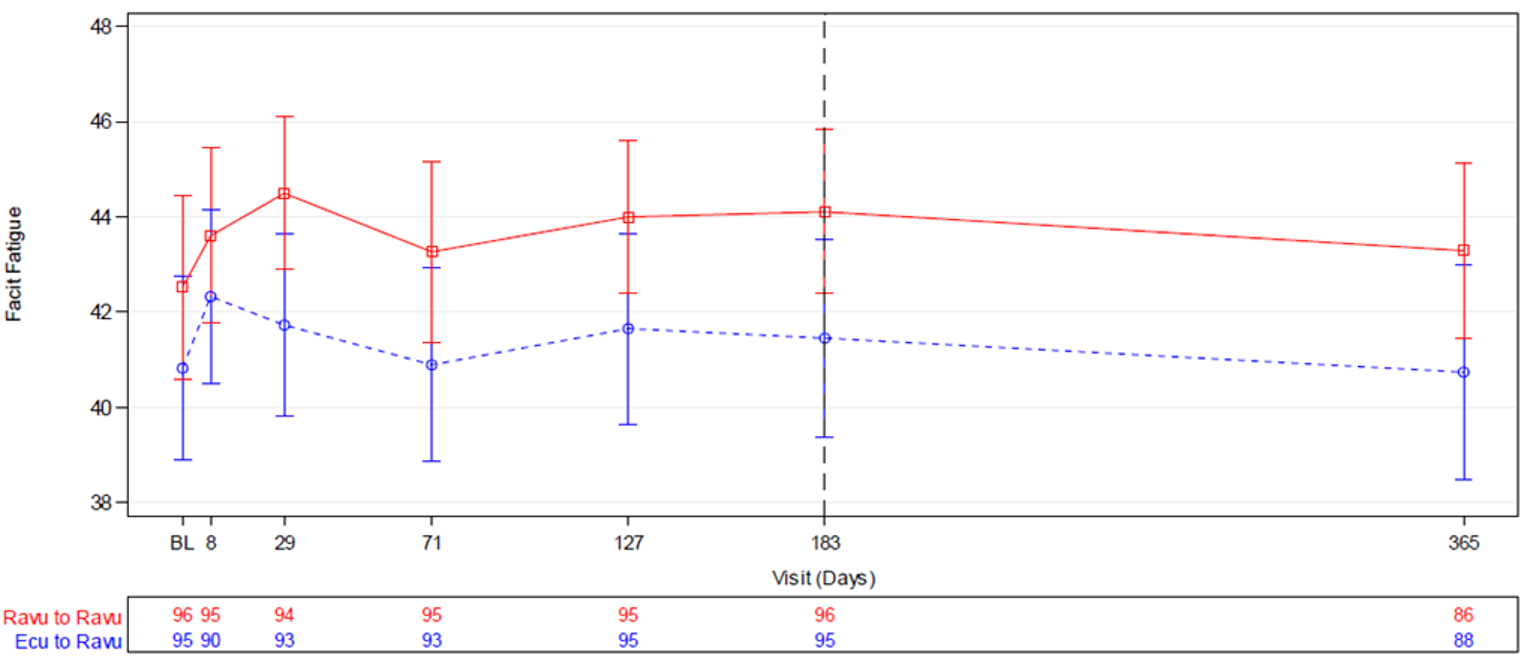
BL = Period 1 baseline; Ecu = eculizumab; FACIT = Functional Assessment of Chronic Illness Therapy; Ravu = ravulizumab.
Note: FACIT-Fatigue scale ranges from 0 to 52, with a higher score indicating less fatigue. Period 1 baseline was defined as the last non-missing assessment value before first study drug dose. Day 183 represented the start of the extension period. Patients in the Ecu to Ravu group received Ecu before day 183 and received Ravu after day 183.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Breakthrough Hemolysis
Table 26 presents the breakthrough hemolysis results for Study 301 and Study 302. The results for breakthrough hemolysis in the extension period were similar to the results from the primary evaluation period in both studies. The group that transitioned from eculizumab to ravulizumab saw a numeric reduction in proportion of patients with breakthrough hemolysis from 10.9% in Period 1 to 1.6% in Period 2 in Study 301 and from 4.2% in Period 1 to 1.1% in Period 2 in Study 302. The group that maintained treatment with ravulizumab into Period 2 saw similar proportions of patients with breakthrough hemolysis from 4.0% in Period 1 and 3.2% In Period 2 in Study 301, and a slight numeric increase from 0% in Period 1 to 3.1% in Period 2 in Study 302. In Study 301, 99.2% of patients in both the ravulizumab to ravulizumab group and eculizumab to ravulizumab group who did not experience a breakthrough hemolysis event in Period 1, maintained no breakthrough hemolysis events in Period 2. Similarly, in Study 302, 96.9% of patients in the ravulizumab to ravulizumab group and 100% of patients in the eculizumab to ravulizumab arm, maintained a no breakthrough hemolysis event response.
Table 26: Breakthrough Hemolysis in the Extension Period
Transfusions | Study 301 RAV–RAV N = 124 | Study 301 ECU–RAV N = 119 | Study 302 RAV–RAV N = 96 | Study 302 ECU–RAV N = 95 |
|---|---|---|---|---|
Patients with breakthrough hemolysis in Period 1, n (%) | 5 (4.0) | 13 (10.9) | 0 (0.0) | 4 (4.2) |
Patients with breakthrough hemolysis in Period 2, n (%) | 4 (3.2) | 2 (1.6) | 3 (3.1) | 1 (1.1) |
Period 2 compared to Period 1 | NA | NA | NA | NA |
Patients with no BTH in Period 1, N | 119 | 106 | 96 | 91 |
Maintaining no BTH, n (%) | 118 (99.2) | 105 (99.2) | 93 (96.9) | 91 (100.0) |
95% CI | 97.52% to 100.00% | 97.22% to 100.00% | 93.39% to 100.00% | 100.00% to 100.00% |
With new BTH, n (%) | 1 (0.8) | 1 (0.9) | 3 (3.1) | 0 (0.0) |
95% CI | 0% to 2.48% | 0% to 2.78% | 0% to 6.61% | NA |
Patients with BTH in Period 1, N | 5 | 13 | 0 (0.0) | 4 |
Repeating BTH, n (%) | 3 (60.0) | 1 (7.7) | 0 (0.0) | 1 (25) |
95% CI | 17.06% to 100.00% | 0.00% to 22.18% | NA | 0.00% to 67.43% |
With no new, n (%) | 2 (40.0) | 12 (92.3) | 0 (0.0) | 3 (75.0) |
95% CI | 0.00% to 82.94% | 77.82% to 100.00% | NA | 32.57% to 100.00% |
BTH = breakthrough hemolysis; CI = confidence interval; ECU = eculizumab; LDH = lactate dehydrogenase; MAVE = major adverse vascular events; NA = not applicable; RAV = ravulizumab; ULN = upper limit of normal.
Note: Period 1 refers to the primary evaluation period of 0 to 26 weeks and Period 2 refers to the extension period of more than 26 weeks to 52 weeks. Breakthrough hemolysis was defined as at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin < 10 g/dL], MAVE [including thrombosis], dysphagia, or erectile dysfunction) in the presence of elevated LDH 2 × ULN or greater.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Intravascular Hemolysis
Table 27 presents the LDH normalization results for Study 301. The results for LDH normalization in the extension period remained consistent with the results from the primary evaluation period. In Study 301, 75% of patients in the ravulizumab to ravulizumab group and 72% of patients in the eculizumab to ravulizumab group who had achieved LDH normalization in Period 1 maintained normalization in Period 2. Conversely, 85% of patients in the ravulizumab to ravulizumab group and 80.6% of patients in the eculizumab to ravulizumab group who did not achieve LDH normalization in Period 1 maintained a lack of response throughout Period 2. Figure 4 below shows the LDH levels visually throughout Study 301, indicating that mean LDH levels at the end of the primary evaluation period were maintained in both groups throughout the extension period.
Table 27: LDH Normalization in the Extension Period
Transfusions | Study 301 (RAV–RAV) N = 124 | Study 301 (ECU–RAV) N = 119 |
|---|---|---|
Patients achieving LDH normalization in Period 1, n (%) | 60 (48.4) | 50 (42.0) |
Patients achieving LDH normalization in Period 2, n (%) | 54 (43.5) | 48 (40.3) |
Period 2 compared to Period 1 | NA | NA |
Patients with response in Period 1, N | 60 | 50 |
Maintaining response, n (%) | 45 (75.0) | 36 (72.0) |
95% CI | 64.04% to 85.96% | 59.55% to 84.45% |
Losing response, n (%) | 15 (25.0) | 14 (28.0) |
95% CI | 14.04% to 35.96% | 15.55% to 40.45% |
Patients with no response in Period 1, N | 64 | 69 |
Maintaining no response, n (%) | 51 (85.0) | 50 (80.6) |
95% CI | 75.97% to 94.03% | 70.81% to 90.48% |
Gaining response, n (%) | 9 (15.0) | 12 (19.4) |
95% CI | 5.97% to 24.03% | 9.52% to 29.19% |
CI = confidence interval; ECU = eculizumab; LDH = lactate dehydrogenase; NA = not applicable; RAV = ravulizumab.
Note: Period 1 refers to the primary evaluation period of 0 to 26 weeks and Period 2 refers to the extension period of 26 weeks or more to 52 weeks. Patients with non-missing LDH normalization status at the end of Period 1 and Period 2 are compared.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Figure 4: LDH Levels Over Time (Study 301 Extension)
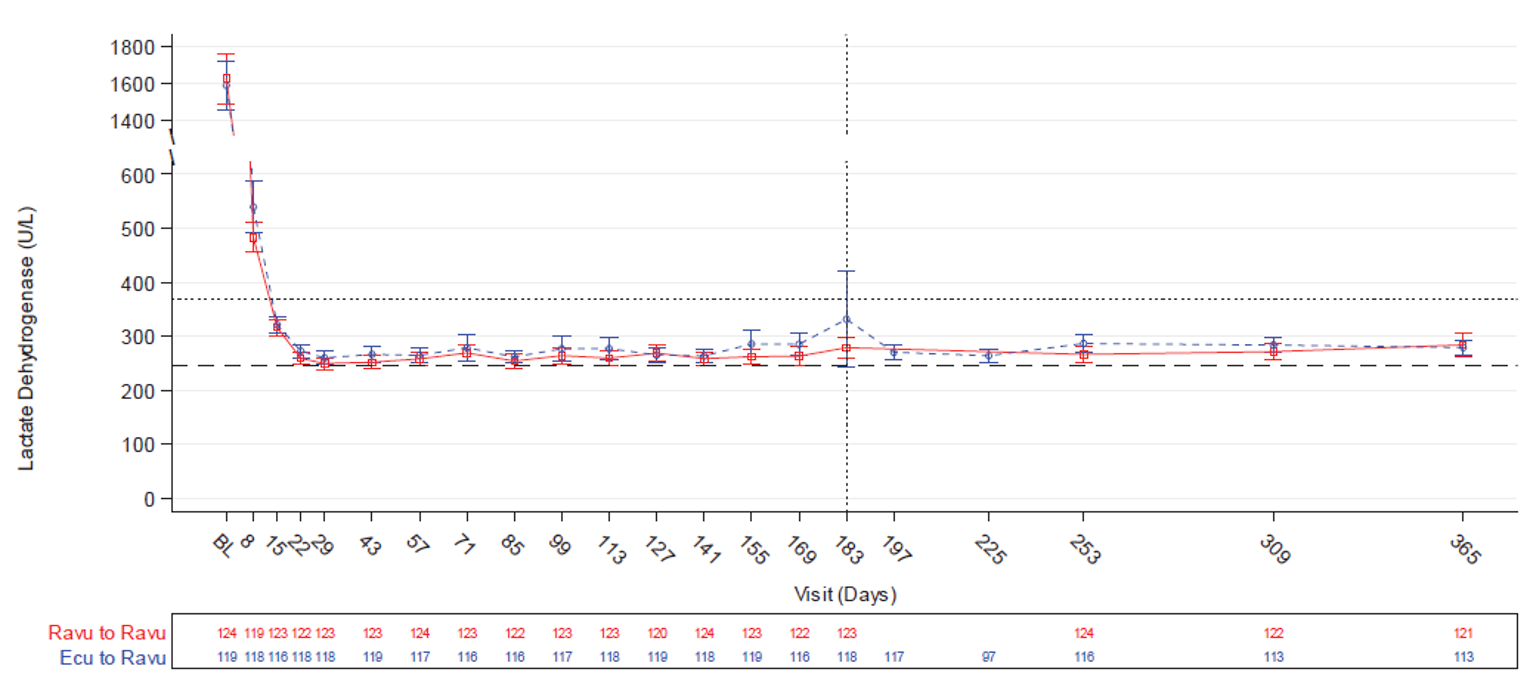
Ecu = eculizumab; LDH = lactate dehydrogenase; Ravu = ravulizumab.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Figure 5 shows the mean LDH levels in Study 302 for both groups throughout the primary evaluation period and extension period. LDH levels in the extension period remained consistent with the results found in the primary evaluation period.
Figure 5: LDH Levels Over Time (Study 302 Extension)
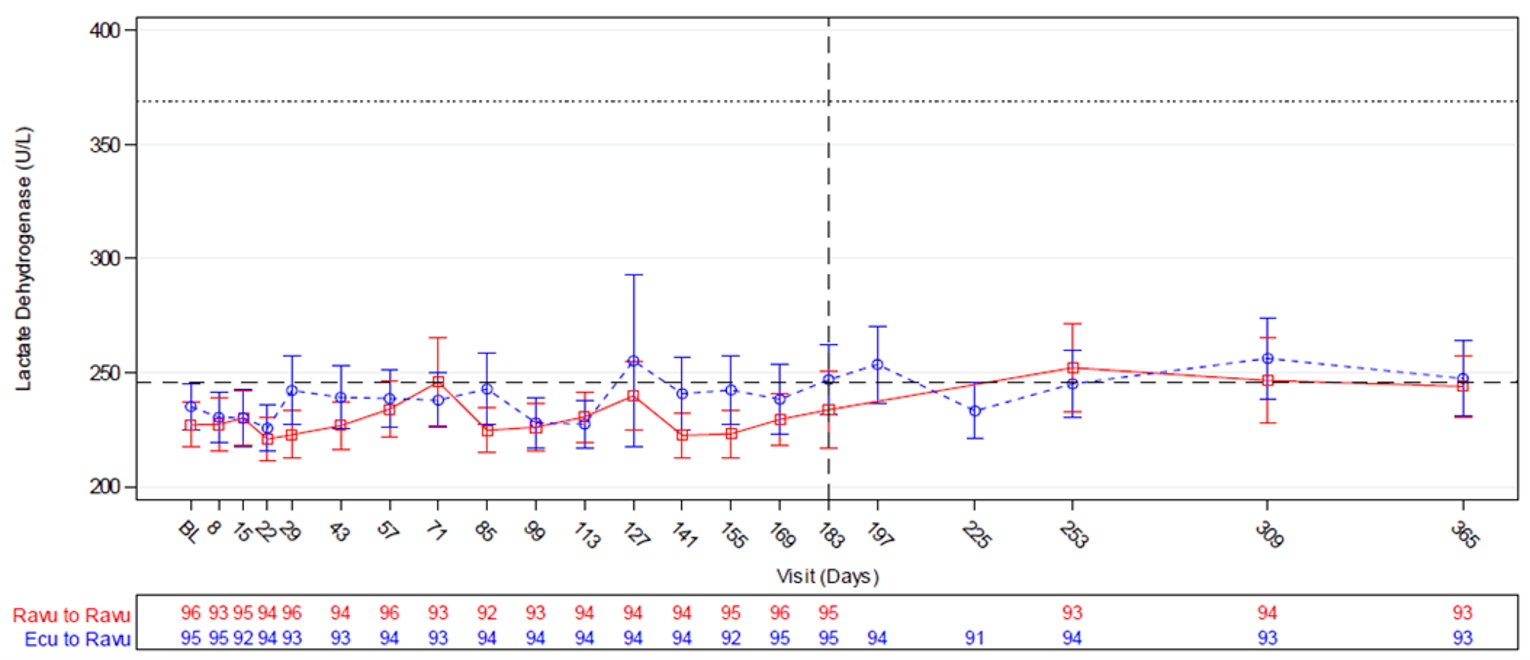
Ecu = eculizumab; LDH = lactate dehydrogenase; Ravu = ravulizumab.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Hemoglobin Stabilization
Table 28 presents the hemoglobin stabilization results for Study 301 and Study 302. The results for hemoglobin stabilization in the extension period remained consistent with the results from the primary evaluation period in both studies. In Study 301, 89.4% of patients in the ravulizumab to ravulizumab group and 85.7% of patients in the eculizumab to ravulizumab group who achieved hemoglobin stabilization in Period 1 maintained their response in Period 2. Similarly, in Study 302, 81.2% of patients in the ravulizumab to ravulizumab group and 81.1% of patients in the eculizumab to ravulizumab arm who achieved hemoglobin stabilization in Period 1 maintained their response in Period 2.
Table 28: Hemoglobin Stabilization in the Extension Period
Transfusions | Study 301 RAV–RAV N = 124 | Study 301 ECU–RAV N = 119 | Study 302 RAV–RAV N = 96 | Study 302 ECU–RAV N = 95 |
|---|---|---|---|---|
Patients with hemoglobin stabilization in Period 1, n (%) | 85 (68.5) | 77 (64.7) | 72 (75.0) | 72 (75.8) |
Patients with hemoglobin stabilization in Period 2, n (%) | 91 (73.4) | 78 (65.5) | 78 (81.2) | 77 (81.1) |
Period 2 compared to Period 1 | NA | NA | NA | NA |
Patients with response in Period 1, N | 85 | 77 | 72 | 72 |
Maintaining response, n (%) | 76 (89.4) | 66 (85.7) | 65 (90.3) | 66 (91.7) |
95% CI | 82.9% to 96.0% | 77.9% to 93.5% | 83.4% to 97.1% | 85.3% to 98.1% |
Losing response, n (%) | 9 (10.6) | 11 (14.3) | 7 (9.7) | 6 (8.3) |
95% CI | 4.0% to 17.1% | 6.5% to 22.1% | 2.9% to 16.6% | 1.9% to 14.7% |
Patients without response in Period 1, N | 39 | 42 | 24 | 23 |
Maintaining no response, n (%) | 24 (61.5) | 30 (71.4) | 11 (45.8) | 12 (52.2) |
95% CI | 46.3% to 76.8% | 57.8% to 85.1% | 25.9% to 65.8% | 31.8% to 72.6% |
Gaining response, n (%) | 15 (38.5) | 12 (28.6) | 13 (54.2) | 11 (47.8) |
95% CI | 23.2% to 53.7% | 14.9% to 42.2% | 34.2% to 74.1% | 27.4% to 68.2% |
CI = confidence interval; ECU = eculizumab; NA = not applicable; RAV = ravulizumab.
Note: Period 1 refers to the primary evaluation period of 0 to 26 weeks and Period 2 refers to the extension period of 26 weeks or more to 52 weeks. Stabilized hemoglobin was defined as avoidance of a 2 g/dL or greater decrease in hemoglobin level from each period’s baseline in the absence of transfusion in that period.
Source: Clinical Study Report for Study 301 and Study 302 (52-week data update).22,23
Harms
Presented in Table 29 is the summary of harms from the extension period up to 52 weeks for Study 301 and Study 302. No additional safety signals were identified when compared to the results presented for the 26-week primary evaluation period. In Period 2 of Study 301, 63.7% of patients in the ravulizumab to ravulizumab group and 74.8% of patients in the eculizumab to ravulizumab group experienced 1 or more AEs. In Period 2 of Study 302, 79.2% of patients in the ravulizumab to ravulizumab group and 74.7% of patients in the eculizumab to ravulizumab group experienced 1 or more AEs. Similar to what was seen in the primary evaluation period, headaches and nasopharyngitis were the most common AEs reported; however, the frequency was consistently lower in Period 2 compared to the primary evaluation period across all groups. Patients experiencing 1 or more SAEs in both the Study 301 and Study 302 extension periods was more consistent with the results from the primary evaluation period. In Study 301, 7.3% of patients in the ravulizumab to ravulizumab group and 5.9% of patients in the eculizumab to ravulizumab group experienced an SAE, while in Study 302 the percentages of patients were 8.3% and 5.3%, respectively.
Of the notable harms identified, serious infections appeared to remain consistent with the results from the primary evaluation period while infusion reactions were slightly lower in Period 2 compared to the primary evaluation period. In Study 301, 1.6% of patients in the ravulizumab to ravulizumab group and 3.4% of patients in the eculizumab to ravulizumab group experienced a serious infection, while in Study 302 the percentages of patients were 3.1% and 2.1%, respectively. In Study 301, 4.0% of patients in the ravulizumab to ravulizumab group and 5.9% of patients in the eculizumab to ravulizumab group experienced an infusion reaction, while in Study 302 the percentages of patients were 2.1% and 1.1%, respectively.
The sponsor included safety data for 2 additional 6-month periods beyond the 52-week data cut-off; however, due to the immature follow-up for these time points, the results were not included in this review. From the sample size available, there were no additional noteworthy safety signals identified in these additional periods.
Table 29: Summary of Harms in the Extension Period
Adverse events | Study 301 | Study 302 | ||
|---|---|---|---|---|
RAV–RAV Period 2 N = 125 | ECU–RAV Period 2 N = 121 | RAV–RAV Period 2 N = 96 | ECU–RAV Period 2 N = 95 | |
Patients with ≥ 1 AE | ||||
n (%) | 79 (63.7) | 89 (74.8) | 76 (79.2) | 71 (74.7) |
Most common eventsa | ||||
Anemia | 0 (0.0) | 6 (5.0) | 1 (1.0) | 5 (5.3) |
Nausea | 2 (1.6) | 6 (5.0) | 3 (3.1) | 3 (3.2) |
Diarrhea | 2 (1.6) | 4 (3.4) | 6 (6.3) | 5 (5.3) |
Abdominal pain | 3 (2.4) | 6 (5.0) | 4 (4.2) | 4 (4.2) |
Constipation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.1) |
Dyspepsia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Vomiting | 0 (0.0) | 0 (0.0) | 3 (3.1) | 0 (0.0) |
Influenza-like illness | 0 (0.0) | 0 (0.0) | 1 (1.0) | 4 (4.2) |
Pyrexia | 7 (5.6) | 0 (0.0) | 6 (6.3) | 6 (6.3) |
Fatigue | 0 (0.0) | 0 (0.0) | 13 (13.5) | 13 (13.7) |
Chest pain | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.1) |
Nasopharyngitis | 8 (5.6) | 15 (12.6) | 6 (6.3) | 7 (7.4) |
Upper respiratory tract infection | 10 (8.1) | 5 (4.2) | 9 (9.4) | 8 (8.4) |
Viral upper respiratory tract infection | 3 (2.4) | 2 (1.7) | 0 (0.0) | 0 (0.0) |
Rhinitis | 0 (0.0) | 0 (0.0) | 4 (4.2) | 0 (0.0) |
Arthralgia | 3 (2.4) | 5 (4.2) | 0 (0.0) | 0 (0.0) |
Myalgia | 1 (0.8) | 3 (2.5) | 0 (0.0) | 0 (0.0) |
Pain in extremity | 0 (0.0) | 3 (2.5) | 4 (4.2) | 5 (5.3) |
Back pain | 1 (0.8) | 0 (0.0) | 0 (0.0) | 6 (6.3) |
Musculoskeletal pain | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.1) |
Nervous system disorders | ||||
Headache | 6 (4.8) | 10 (8.4) | 6 (6.3) | 10 (10.5) |
Dizziness | 0 (0.0) | 0 (0.0) | 0 (0.0) | 6 (6.3) |
Oropharyngeal pain | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (2.1) |
Cough | 0 (0.0) | 4 (3.4) | 3 (3.1) | 4 (4.2) |
Dyspnea | 0 (0.0) | 1 (0.8) | 0 (0.0) | 3 (3.2) |
Patients with ≥ 1 SAE | ||||
n (%) | 9 (7.3) | 7 (5.9) | 8 (8.3) | 5 (5.3) |
Patients who discontinued study drug due to AE | ||||
n (%) | 0 (0.0) | 2 (1.7)b | 0 (0.0) | 0 (0.0) |
Deaths | ||||
n (%) | 0 (0.0) | 0 (0.0)c | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | ||||
Serious infections (other than meningococcal and aspergillus) | 2 (1.6) | 4 (3.4) | 3 (3.1) | 2 (2.1) |
Infusion reactions | 5 (4.0) | 7 (5.9) | 2 (2.1) | 1 (1.1) |
AE = adverse event; ECU = eculizumab; RAV = ravulizumab; SAE = serious adverse event.
Note: Period 2 refers to the extension period of 26 weeks or greater to 52 weeks.
aFrequency is reported if 5% or greater in any treatment group or presented as part of the primary evaluation period in an earlier section of this review.
bOne patient developed a lung adenocarcinoma and discontinued the study drug, this patient later died and is discussed in footnote C. A second patient developed myelodysplastic syndrome and discontinued study drug.
cOne death that occurred during the extension period was reported in Period 1 because onset of the lung adenocarcinoma occurred during Period 1. A second death due to pulmonary sepsis occurred during Period 3, beyond the 52-week data cut-off.
Source: Clinical Study Reports for Study 301 and Study 302 (52-week data update).22,23
Critical Appraisal
Internal Validity
The long-term extension period of Study 301 and Study 302 was conducted to evaluate the long-term efficacy and safety of ravulizumab for the treatment of PNH, beyond the 26-week randomized primary evaluation period. The number of patients that discontinued treatment during the extension period of Study 301 and Study 302 was acceptable and consistent with the number of patients that discontinued during the primary evaluation period. Therefore, there is little concern that patients discontinuing treatment would bias the results in favour of ravulizumab. Unfortunately, due to the nature of the long-term extension design, all patients were transitioned to ravulizumab. This breaks randomization and the comparative primary evaluation period becomes instead a single-arm non-comparative extension. As such, it is difficult to make any claim about the comparative efficacy of ravulizumab and eculizumab beyond the 26-week primary evaluation period and potential for confounding factors in a single-arm setting make all conclusions significantly more uncertain. The extension period allows a comparison of safety and efficacy during the primary evaluation period and the extension period; however, there was no statistical analysis conducted and therefore all conclusions must be qualitative in nature.
External Validity
The inclusion of a long-term extension study in the sponsor’s submission allows for greater generalizability of the efficacy data to patients who will be on treatment for longer than the 26-week period that was included in the primary evaluation period. Extending the efficacy data to 52 weeks increases confidence that efficacy will be maintained long-term; however, as is inherent in all studies of chronic disease, it is difficult to conclude with certainty beyond the presented data throughout the lifetime of a patient receiving ravulizumab.
Patient Preference Substudy
Included in the sponsor’s submission was a patient preference substudy conducted by a team from Northwestern University Center for Outcomes Research and Education for the sponsor in a subset of patients from Study 302. The study included treatment-experienced patients who had been treated with eculizumab for at least 6 months before Study 302 enrolment and subsequently received ravulizumab in either the primary evaluation period or extension period. The goal of the substudy was to assess patient preferences for treatment with ravulizumab or eculizumab and to identify the key factors influencing preference.
Methods
The substudy investigators composed a novel patient preference survey for the purposes of this study. To inform the creation of the survey, patient concept elicitation interviews were conducted in patients diagnosed with PNH who had previously received either ravulizumab or eculizumab, with preference for patients who were already participating in Study 302. The investigators finalized the questionnaire to include 11 questions in total; these include 1 overall preference question, 1 question evaluating preference according to 9 treatment characteristics, 1 question evaluating the most important characteristic impacting preference, 4 questions evaluating specific aspects of treatment with ravulizumab, and 4 questions evaluating specific aspects of treatment with eculizumab. The final 4 questions for each treatment used an agreement scale with 0 indicating no agreement with a given statement and 4 indicating full agreement with a given statement. All other preference questions were asked with the available responses of prefer ravulizumab, prefer eculizumab, or no preference.
Patients were given the opportunity to enrol in the patient preference substudy if they had also entered the extension period of Study 302 and had received a minimum of 2 doses of ravulizumab during the extension period. There were no exclusion criteria and the patient preference survey was administered to each participant at a single point in time.
Statistical Analysis
A planned sample size of 95 was calculated to have at least 80% power to detect a 65% or greater observed proportion of patients preferring ravulizumab under a null hypothesis of 50%. A 2-sided exact binomial test allowing for type I error of 0.05 was used in these calculations. Tests for mean differences in response were conducted for the final 4 questions for each treatment, P values were calculated from a paired t-test as well as the Wilcoxon signed rank test. There was no statistical hierarchy established and the analysis was not adjusted for multiplicity.
Results
Of the 98 patients that enrolled in the substudy, 3 did not respond to the first question of the survey, leaving 95 evaluable patients. The characteristics of the surveyed patients are shown in Table 30. Patients were evenly represented from both treatment arms with 53% of patients having received ravulizumab in the primary evaluation period and 47% having received eculizumab. The mean number of days between the last randomized study treatment and administration of the survey was 306 (SD = 55).
Table 30: Patient Characteristics in Study 302 Substudy
Characteristics | Study 302 substudy (N = 95) |
|---|---|
Age in years, mean (SD) | 50 (13) |
Received ravulizumab in the primary evaluation period, n (%) | 50 (53) |
Received eculizumab in the primary evaluation period, n (%) | 45 (47) |
Male, n (%) | 53 (56) |
Female, n (%) | 42 (44) |
Years since diagnosis, mean (SD) | 14 (10) |
Days between last randomized study treatment and survey, mean (SD) | 306 (55) |
History of major adverse vascular events, n (%) | 24 (25) |
SD = standard deviation.
Source: Patient and Health Care Provider Preference for the Treatment of Paroxysmal Nocturnal Hemoglobinuria: Final Report.25
Table 31 shows the results from the overall preference question as well as questions relating to specific aspects of treatment. When asked for overall preference, 93% of patients indicated that they preferred ravulizumab. For the treatment aspect questions, patients responded with either no preference, ranging from 2% to 53%, or a preference for ravulizumab, ranging from 45% to 98%. Two questions resulted in 98% of patients preferring ravulizumab — the questions regarding frequency of infusions and being able to plan activities.
Table 31: Treatment Preference in Study 302 Substudy
Treatment details | Study 302 substudy (N = 95) | ||
|---|---|---|---|
Prefera ravulizumab, n (%) | No preference, n (%) | Prefera eculizumab, n (%) | |
Overall preference | 88 (93) | 6 (6) | 1 (1) |
Controlling fatigue | 61 (64) | 30 (32) | 4 (4) |
Controlling symptoms other than fatigueb | 57 (61) | 34 (36) | 3 (3) |
Frequency of infusions | 93 (98) | 2 (2) | 0 (0) |
Side effects of treatment | 43 (45) | 50 (53) | 2 (2) |
Convenience of receiving treatment | 81 (85) | 9 (9) | 5 (5) |
Being able to plan activitiesb | 92 (98) | 2 (2) | 0 (0) |
Effectiveness of the medication until the next infusionb | 73 (78) | 17 (18) | 4 (4) |
Anxiety related to the infusion | 46 (48) | 45 (47) | 4 (4) |
Your overall quality of lifec | 82 (88) | 10 (11) | 1 (1) |
aDefined as responding “Strongly” or Somewhat” prefer respective drug.
bMissing 1 response.
cMissing 2 responses.
Source: Patient and Health Care Provider Preference for the Treatment of Paroxysmal Nocturnal Hemoglobinuria: Final Report.25
Figure 6 shows which factors patients indicated were the most important for deciding treatment preference. Most patients chose frequency of infusions as the most important factor with overall quality of life the second most common factor cited by patients in the substudy.
Figure 6: Patients’ Most Important Treatment Factor for Deciding Preference in Study 302 Substudy
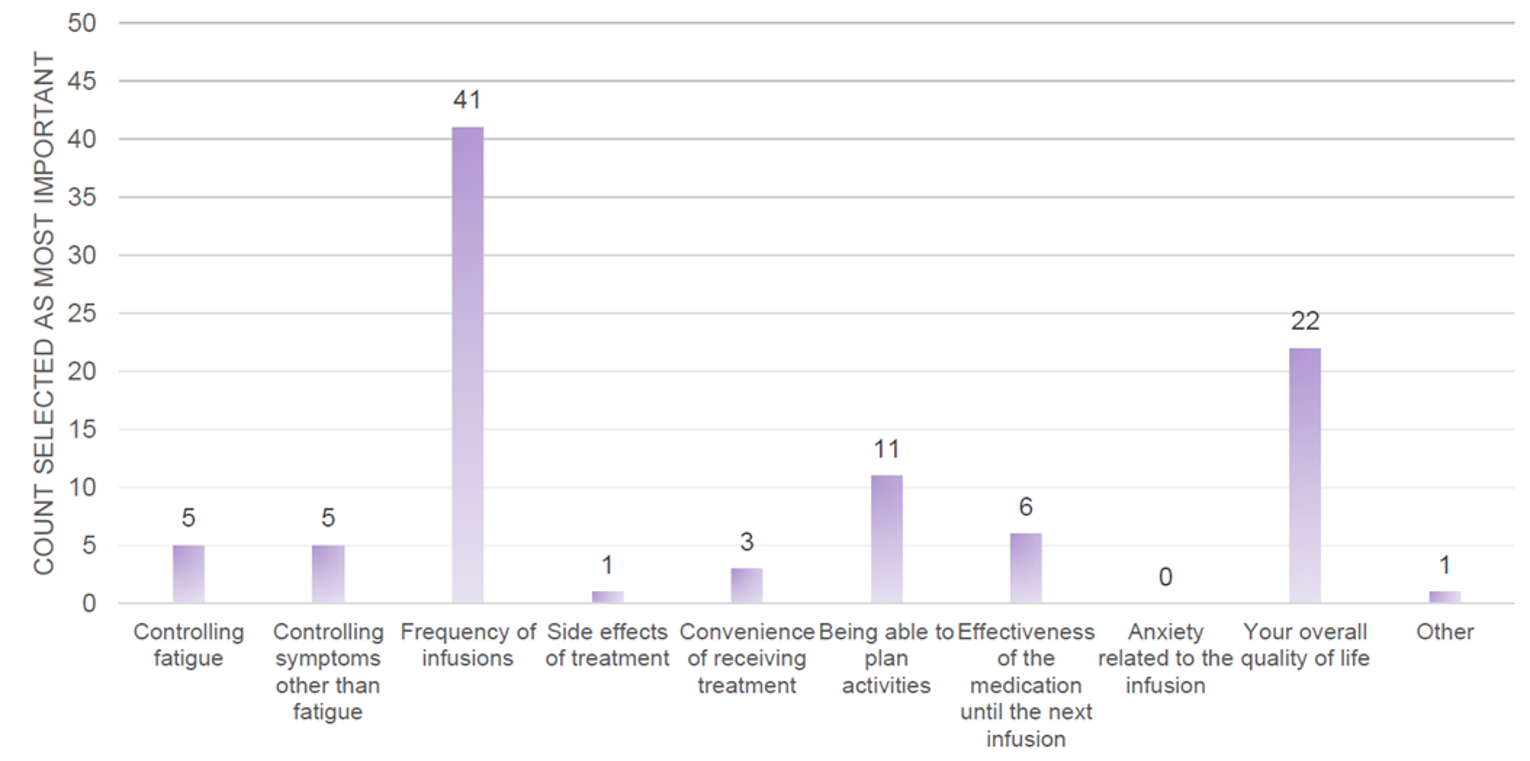
Source: Patient and Health Care Provider Preference for the Treatment of Paroxysmal Nocturnal Hemoglobinuria: Final Report.25
Shown in Table 32 are 4 questions that were asked of patients regarding their experience receiving ravulizumab and eculizumab therapy. Patients generally favoured ravulizumab with the greatest absolute difference between the mean response scores observed in the question regarding frequency of infusions.
Table 32: Ratings on Treatment-Related Factors in Study 302 Substudy
Treatment-related concerns | Study 302 substudy (N = 95) | ||||
|---|---|---|---|---|---|
Ravulizumab meana | Eculizumab meana | Ravulizumab–eculizumab mean (SD) | P (paired t-test)b | Effect sizec | |
The frequency of infusions disrupted my life | 0.39 | 2.21 | –1.82 | < 0.001 | –1.46 |
After receiving infusions, I had fatigue | 0.62 | 1.21 | –0.59 | < 0.001 | –0.56 |
Effective in treat symptoms of PNH | 3.59 | 3.36 | 0.23 | 0.01 | 0.27 |
While I was receiving treatments, I was able to enjoy life | 3.62 | 2.81 | 0.81 | < 0.001 | 0.88 |
PNH = paroxysmal nocturnal hemoglobinuria.
aMean of responses on an agreement scale of 0 indicating “Not at all” to 4 indicating “Very much.” Higher means indicate greater agreement.
bStatistical testing was not adjusted for multiplicity and should be considered as nominal P values.
cEffect sizes calculated as the difference in mean scores divided by the SD of the mean differences.
Source: Patient and Health Care Provider Preference for the Treatment of Paroxysmal Nocturnal Hemoglobinuria: Final Report.25
Critical Appraisal
Internal Validity
The patient preference substudy was conducted to evaluate patient preference between ravulizumab and eculizumab for the treatment of PNH. Patient preference was evaluated using subjective, patient-reported outcomes within an open-label study design, which, though necessary to evaluate frequency of infusions as a factor affecting patient preferences, could have biased the evaluation of other treatment aspects in favour of the study treatment. Furthermore, the questionnaire used was developed by the study investigators specifically for the purposes of this trial with no evidence of reliability, responsiveness, or an MID presented. Patient interviews to inform the development of the survey were preferentially recruited from those already taking part in Study 302 and the content validity of the questionnaire is unclear for the broader population outside of the Study 302 population. The statistical analysis presented includes P values that are not controlled for multiplicity and are therefore at increased risk of type I error.
There were 98 total patients that were initially enrolled in the substudy, while a total of 191 patients in Study 302 entered the extension period and would therefore be eligible to be included in the substudy. With no breakdown of the number of patients who were offered the survey but declined, it is difficult to rule out significant selection bias which could favour ravulizumab. Furthermore, given the length of time elapsed from the last randomized study treatment was an average of 306 days and that roughly half of the included patients received ravulizumab in the randomized period, there is a substantial gap in time for many patients since their last eculizumab dose. Thus, there is the potential for recall bias to influence the survey responses, although the direction of this bias is unclear.
External Validity
The patient preference substudy was conducted during the long-term extension period of Study 302. As PNH is a lifelong disease, patient preference conclusions cannot be extrapolated with certainty over the lifetime of a patient being treated for PNH.
Discussion
Summary of Available Evidence
Two studies, both of them open-label, active-controlled, parallel-group, noninferiority RCTs identified as pivotal studies, were selected for inclusion in the CADTH systematic review. Study 301 (N = 246) enrolled adult patients with PNH who were treatment-naive while Study 302 (N = 197) enrolled adult patients with PNH who had been receiving eculizumab. Patients were randomized 1:1 to ravulizumab or eculizumab and noninferiority of ravulizumab compared with eculizumab was assessed for transfusion avoidance, fatigue, breakthrough hemolysis, LDH normalization, and hemoglobin stabilization during a 26-week randomized treatment period.
Safety and efficacy results from the respective extension periods for Study 301 and Study 302, during which all patients received ravulizumab, were also submitted by the sponsor and are presented in this report for the 26-week period following the randomized treatment period. Also included in the sponsor’s submission was a patient preference substudy which allowed patients to enroll from Study 302 who enrolled in the extension period and had received at least 2 doses of ravulizumab during the extension period. A novel patient preference questionnaire was developed for the study and the objective of the study was to assess patient preferences for ravulizumab or eculizumab and to identify the key factors influencing preference.
Interpretation of Results
Efficacy
Overall, evidence from the 2 pivotal trials for ravulizumab (Study 301 and Study 302) supports the noninferiority of ravulizumab to eculizumab when both are administered according to the product monograph over 26 weeks in adult patients with PNH in terms of transfusion avoidance, occurrence of breakthrough hemolysis, LDH normalization, and hemoglobin stabilization. In accordance with the closed testing procedures in the studies, conclusions regarding superiority of ravulizumab over eculizumab in terms of any of the efficacy outcomes cannot be drawn. Although the pre-specified noninferiority margins for the primary and key secondary end points (aside from percent change in LDH, potentially) were based on a magnitude of loss of benefit that may not be clinically acceptable, there are several factors that substantially mitigate the risk of unacceptable loss of benefit with ravulizumab versus eculizumab. All of the primary and key secondary end points met their respective noninferiority margins, there was minimal missing data, the PP analyses were consistent with the primary analyses for all end points, and a more conservative margin would have been met for all end points. The efficacy results for eculizumab treatment in both studies were generally as expected by the clinical expert consulted by CADTH.
For the treatment-naive patients of Study 301, there was likely a subset of patients who would have needed more intensive dosing of eculizumab than recommended in the Health Canada–approved product monograph for the treatment of PNH to provide more complete complement blockade and prevent pharmacokinetic breakthrough hemolysis. According to the clinical expert, these patients comprise approximately 20% of Canadian patients with PNH and treatment with eculizumab 1,200 mg every 2 weeks is common for them. The studies did not allow for dosage adjustments, which is contrary to Canadian clinical practice for the treatment of PNH with eculizumab and the efficacy results may have been more favourable for the eculizumab group had dosage adjustments of eculizumab according to clinical practice been permitted. Since the study selection criteria for Study 302 likely excluded these patients, the issue of potential bias is likely not a concern. However, the generalizability of the Study 302 efficacy findings to this subpopulation is not entirely clear.
The clinical expert consulted by CADTH noted that the requirement of an LDH level at or below the ULN throughout treatment for the outcome of LDH normalization was conservative. Given the strong evidence for the clinical meaningfulness of the 1.5 × ULN threshold for LDH (Appendix 3 for details), the fact that the mean LDH level after treatment initiation was maintained well below the 1.5 × ULN threshold provides reassurance that most patients displayed a clinically meaningful response to treatment in terms of LDH level.
The open-label nature of the studies means that there was potential for bias in favour of ravulizumab for end points relying on subjective reporting, such as the EORTC QLQ-C30 scales and the FACIT-F. While ravulizumab was found to be noninferior to eculizumab for FACIT-F score in both studies, this potential for bias contributes some uncertainty to the finding. Bias from lack of blinding was not a concern for the remaining primary and key secondary end points, although it is possible that AE reporting was affected by knowledge of treatment assignment.
Aside from potential bias from lack of blinding, the EORTC QLQ-C30 scales were secondary end points and therefore not included in the statistical testing hierarchies. As well, the psychometric properties and estimated MID have yet to be established in patients with PNH. Shifts in signs and symptoms of PNH were reported, but these were secondary end points and also excluded from statistical testing. Therefore, no conclusions could be drawn regarding HRQoL or symptoms of PNH. Survival, complications of PNH other than thrombotic events, and health care resource utilization were not assessed in the studies and the studies were not designed to compare the incidence of MAVEs between the treatment groups. Therefore, conclusions could not be drawn regarding survival, complications of PNH (including thrombotic events), or health care resource utilization. Given that ravulizumab was noninferior to eculizumab in terms of control of intravascular hemolysis, transfusion avoidance, occurrence of breakthrough hemolysis, and hemoglobin stabilization, as well as the identical mechanism of action between the 2 drugs, the clinical expert anticipated that the results for these outcomes would translate into noninferior efficacy of ravulizumab for survival and prevention of thrombotic events.
Transfusion avoidance, FACIT-F score, breakthrough hemolysis events, LDH normalization, LDH level, and hemoglobin stabilization were analyzed with summary statistics in the extension period of both studies. Similar to the primary evaluation, study discontinuations were minimal and the results from the 26-week period following the primary evaluation period support the maintenance of efficacy with ravulizumab treatment. Reductions in sample size in subsequent 26-week periods precluded the ability to assess results beyond 1 year of treatment, which is a concern given the chronic nature of the disease.
Finally, patient preference for treatment with ravulizumab versus eculizumab was assessed in a substudy of Study 302. According to the results from a novel questionnaire administered once to each patient, 93% of patients preferred ravulizumab overall with 43% of patients choosing frequency of infusions and 23% of patients choosing overall quality of life as the most important treatment factor when deciding preference. However, there were several limitations identified in the study that introduce substantial uncertainty in the results. These include the lack of evidence of reliability and responsiveness of the questionnaire, the potential for recall bias given that ravulizumab was the most recent treatment for all patients, the small sample size relative to the population of Study 302, and uncertainty surrounding reasons for the reduction in sample size. Overall, the substudy results are consistent with the clinical expert’s expectations that most patients receiving eculizumab would prefer to switch to ravulizumab if available and that the frequency of infusions is an important factor to patients. In addition, the patient input submission indicated that patients expect the less burdensome treatment regimen of ravulizumab to translate to an improvement in quality of life and the ability to be away from home for longer periods of time and to travel.
Harms
Most patients in both studies reported at least 1 AE, with the most common AE being headache. There were no notable differences in AEs between the treatment groups and the clinical expert did not find any of the AEs particularly concerning. Serious infections and infusion reactions were not notably different between treatment groups and were each reported in low enough frequencies to not be of concern. The occurrence of AEs and SAEs during the extension period of both studies was similar to the primary evaluation period and no new safety signals were identified.
Patients in both studies were required to have had recent vaccination against meningococcal infections and no such infections were observed in the studies. The product monographs of both drugs contain a serious warning about life-threatening meningococcal infections stating that patients must be vaccinated against meningococcal infection before or at the time of initiating treatment.
Conclusions
Ravulizumab is noninferior to eculizumab in transfusion avoidance, occurrence of breakthrough hemolysis, LDH normalization, and hemoglobin stabilization over 26 weeks of treatment in adult patients with PNH, with maintenance of efficacy up to 52 weeks of treatment. Evidence regarding comparative efficacy in symptom control, such as improvement of fatigue, is supportive of noninferiority but is associated with some uncertainty given that the study was open-label, the patient-reported outcomes have not been validated in patients with PNH, and statistical testing was not performed for outcomes other than FACIT-F score. Conclusions cannot be drawn for HRQoL due to the same limitations. The efficacy of ravulizumab versus eculizumab is less certain for the scenario in which the maintenance dose of eculizumab increases beyond what is specified in the product monograph for PNH, as is the case with clinical practice in Canada. Results from a patient preference study demonstrated that most patients who had experienced treatment with both drugs preferred ravulizumab over eculizumab with frequency of infusions being the dominant deciding factor, but serious limitations in the study contribute much uncertainty to the estimated proportion of patients who preferred ravulizumab. The safety profiles of ravulizumab and eculizumab were similar to each other with no new safety concerns.
References
1.Clinical Study Report: ALXN1210-PNH-301. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in complement inhibitor-naïve adult patients with paroxysmal nocturnal hemoglobinuria (PNH) [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc.; 2018.
2.Clinical Study Report: ALXN1210-PNH-302. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in adult patients with paroxysmal nocturnal hemoglobinuria (PNH) currently treated with eculizumab [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc.; 2018.
3.Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: a consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36-52. PubMed
4.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804-2811. PubMed
5.Hill A, Platts PJ, Smith A, et al. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood. 2006;108(11):985-985.
6.Nishimura JI, Kanakura Y, Ware RE, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore). 2004;83(3):193-207. PubMed
7.Socié G, Mary JY, de Gramont A, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348(9027):573-577. PubMed
8.Kelly RJ, Hill A, Arnold LM, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786-6792. PubMed
9.Ultomiris (ravulizumab): 10 mg/mL concentrate for solution for infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2019 Aug 28.
10.Soliris (eculiuzmab): 30 mL parenteral solution (10 mg/mL) for injection [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2021 Mar 25.
11.Kelly RJ, Höchsmann B, Szer J, et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2015;373(11):1032-1039. PubMed
12.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
13.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Aug 9.
14.Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530-539. PubMed
15.Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540-549. PubMed
16.Brodsky RA, Peffault de Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230-237. PubMed
17.Health Canada reviewer's report: Ultomiris (ravulizumab) [internal sponsor's report]. Vaughan (ON): Alexion Pharm Canada Corp.; 2019.
18.Center for Drug Evaluation Research. Multidiscipline review(s). Ultomiris (ravulizumab) IV administration. Company: Alexion Pharmaceuticals Inc. Application No.: 761108. Approval date: 12/21/2018 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018 Dec 20: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761108Orig1s000MultidisciplineR.pdf. Accessed 2021 Aug 9.
19.EPAR - Public assessment report: Ultomiris (ravulizumab). Amsterdam (NL): European Medicines Agency; 2019: https://www.ema.europa.eu/en/documents/assessment-report/ultomiris-epar-public-assessment-report_en.pdf. Accessed 2021 Aug 6.
20.Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-1243. PubMed
21.Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233-1243. PubMed
22.Addendum to Clinical Study Report (52-week data update): ALXN1210-PNH-301. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in complement inhibitor-naïve adult patients with paroxysmal nocturnal hemoglobinuria (PNH) [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc.; 2019.
23.Addendum to Clinical Study Report (52-week data update): ALXN1210-PNH-302. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in adult patients with paroxysmal nocturnal hemoglobinuria (PNH) currently treated with eculizumab [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc.; 2019.
24.Peffault de Latour R, Hill A, Fureder W, et al. Ravulizumab reduces the risk of thrombosis in adult patients with paroxysmal nocturnal hemoglobinuria and high disease activity: 2-year data from a phase 3, open-label study [abstract]. Presented at: EHA Virtual Congress; 2021 Jun 9-17. https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/324709/regis.peffault.de.latour.ravulizumab.reduces.the.risk.of.thrombosis.in.adult.html?f=menu%3D14%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Aspeaker%3D615281. Accessed 2021 Sep 30.
25.Patient and health care provider preference for the treatment of paroxysmal nocturnal hemoglobinuria: final report [internal sponsor's report]. Chicago (IL): Northwestern University Center for Outcomes Research and Education (NUCORE); 2019.
26.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
27.Georgakopoulos A, Kontodimopoulos N, Chatziioannou S, Niakas D. EORTC QLQ-C30 and FACT-Lym for the assessment of health-related quality of life of newly diagnosed lymphoma patients undergoing chemotherapy. Eur J Oncol Nurs. 2013;17(6):849-855. PubMed
28.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
29.Cella D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) Scale: a new tool for the assessment of outcomes in cancer anemia and fatigue. Semin Hematol. 1997;34(3 Suppl 2):13-19. PubMed
30.Chandran V, Bhella S, Schentag C, Gladman DD. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66(7):936-939. PubMed
31.Bessler M, Hiken J. The pathophysiology of disease in patients with paroxysmal nocturnal hemoglobinuria. Hematology. 2008:104-110. PubMed
32.Jang JH, Kim JS, Yoon SS, et al. Predictive factors of mortality in population of patients with paroxysmal nocturnal hemoglobinuria (PNH): results from a Korean PNH registry. J Korean Med Sci. 2016;31(2):214-221. PubMed
33.Lee JW, Jang JH, Kim JS, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean registry. Int J Hematol. 2013;97(6):749-757. PubMed
34.Fayers P, Bottomley A. Quality of life research within the EORTC-the EORTC QLQ-C30. European Organisation for Research and Treatment of Cancer. Eur J Cancer. 2002;38(Suppl 4):S125-133. PubMed
35.Osoba D, Aaronson N, Zee B, Sprangers M, te Velde A. Modification of the EORTC QLQ-C30 (version 2.0) based on content validity and reliability testing in large samples of patients with cancer. The Study Group on Quality of Life of the EORTC and the Symptom Control and Quality of Life Committees of the NCI of Canada Clinical Trials Group. Qual Life Res. 1997;6(2):103-108. PubMed
36.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-1807. PubMed
37.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
38.Musoro JZ, Coens C, Fiteni F, et al. Minimally important differences for interpreting EORTC QLQ-C30 scores in patients with advanced breast cancer. JNCI Cancer Spectr. 2019;3(3):pkz037. PubMed
39.Reddy SM, Bingham IC. Outcome measures in psoriatic arthritis clinical trials. Curr Rheumatol Rep. 2005;7(4):299-305. PubMed
40.Cella D, Lai JS, Chang CH, Peterman A, Slavin M. Fatigue in cancer patients compared with fatigue in the general United States population. Cancer. 2002;94(2):528-538. PubMed
41.Acaster S, Dickerhoof R, DeBusk K, Bernard K, Strauss W, Allen LF. Qualitative and quantitative validation of the FACIT-fatigue scale in iron deficiency anemia. Health Qual Life Outcomes. 2015;13:60. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: August 20, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
No date or language limits were used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(ravulizumab* or Ultomiris* or ALXN1210 or ALXN-1210 or ALXN1810 or ALXN-1810 or C3VX249T6L).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*ravulizumab/
(ravulizumab* or Ultomiris* or ALXN1210 or ALXN-1210 or ALXN1810 or ALXN-1810).ti,ab,kw,dq.
3 or 4
5 use oemezd
6 not (conference review or conference abstract).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Ultomiris or ravulizumab]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Ultomiris or ravulizumab]
Grey Literature
Search dates: August 9, 2021–August 16, 2021
Keywords: Ultomiris or ravulizumab, paroxysmal nocturnal hemoglobinuria
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Ishiyama K, Nakao S, Usuki K, et al. Results from multinational phase III studies of ravulizumab (ALXN1210) versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria: subgroup analysis of Japanese patients. Int J Hematol. 2020;112(4):466-476. | Study design |
Peffault de Latour R, Brodsky RA, Ortiz S, et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase III randomised, multicentre studies. Br J Haematol. 2020;191(3):476-485. | Outcomes |
Alashkar F, Rottinghaus S, Vance C, et al. No evidence for hypogammaglobulinemia in patients with paroxysmal nocturnal hemoglobinuria (PNH) chronically treated with ravulizumab. PLoS ONE [Electronic Resource]. 2020;15(3):e0230869. | Study design |
Peipert JD, Kulasekararaj AG, Gaya A, et al. Patient preferences and quality of life implications of ravulizumab (every 8 weeks) and eculizumab (every 2 weeks) for the treatment of paroxysmal nocturnal hemoglobinuria. PLoS ONE [Electronic Resource]. 2020;15(9):e0237497. | Study design |
Schrezenmeier H, Kulasekararaj A, Mitchell L, et al. One-year efficacy and safety of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria naive to complement inhibitor therapy: open-label extension of a randomized study. Ther Adv Hematol. 2020;11:2040620720966137. | Study design (open-label extension study presented under Other Relevant Evidence) |
Kulasekararaj AG, Hill A, Langemeijer S, et al. One-year outcomes from a phase III randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol. 2021;106(3):389-397. | Study design (open-label extension study presented under Other Relevant Evidence) |
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-C30
FACIT-Fatigue
LDH ≥1.5 x ULN
Findings
Table 35: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | 30-item, patient-reported, cancer-specific, quality of life questionnaire using 4- and 7- point Likert scales.26 It consists of 5 multi-item functional scales (physical, role, emotional, cognitive, and social), 3 multi-item symptom scales (fatigue, nausea-vomiting, and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial impact), and a 2-item GHS/QoL scale. | Reliability of the EORTC QLQ-C30 in HL and DLBCL patients undergoing chemotherapy measured by Cronbach alpha was 0.79 for GHS/QoL, 0.51 to 0.85 for functional scales, and 0.82 to 0.86 for symptom scales/items.27 No evidence of validity, reliability, or responsiveness in patients with PNH. | Patients with cancer28: • 5 to 10 points: small change • 10 to 20 points: moderate change • > 20 points: large change No MID identified in patients with PNH. |
FACIT-Fatigue | 13-item, patient-reported, fatigue-specific, quality of life questionnaire using a 5-point Likert scale.29 | Internal consistency by Cronbach alpha of 0.9529 and test-retest by intraclass correlation coefficient of 0.95.30 No evidence of validity, reliability, or responsiveness in patients with PNH. | No MID identified in patients with PNH. |
LDH ≥ 1.5 x ULN | Laboratory test used in diagnosis and monitoring of disease activity.31 | Associated with 4.8-fold increase in risk of mortality32 and increased risk of thromboembolism (OR 7.0; P = 0.013) in patients with PNH.33 | NA |
DLBCL = diffuse large B-cell lymphoma; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; HL = Hodgkin’s lymphoma; HRQoL = health-related quality of life; LDH = lactate dehydrogenase; MID = minimal important difference; OR = odds ratio; PNH = paroxysmal nocturnal hemoglobinuria; QoL = quality of life; ULN = upper limit of normal.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
Description and Scoring
The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) is one of the most used patient-reported outcome measures in oncology clinical trials. It is a multidimensional, cancer-specific, self-administered measure of HRQoL.26
The EORTC QLQ-C30 is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), a global health status/HRQoL scale, and 6 single items assessing additional symptoms commonly reported by cancer patients (dyspnea, loss of appetite, insomnia, constipation and diarrhea) as well as perceived financial impact of the disease.26
The EORTC QLQ-C30 uses a 1-week recall period to assess functional status and symptoms. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global HRQoL scale, the response format is a 7-point Likert-type scale with anchors at 1 = “very poor” and 7 = “excellent.” Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A higher score on the functional scales represents better functioning, a higher score on the symptom scales represents a higher level of symptomatology, and a higher score on the global health status/HRQoL scale represents a higher HRQoL.34
According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least half of the items. The values for missing items are interpolated with the average of the respondent-completed items.34
Assessment of Validity and Reliability
In its initial development, the EORTC QLQ-C30 underwent an evaluation of its psychometric properties and demonstrated reliability and validity in cancer patients in an international field trial of 305 patients in 13 multicultural clinical research settings.26 A revision of the EORTC QLQ-C30 was undertaken to improve low internal consistency, content validity for the role functional scale, and a conceptual difficulty (undue emphasis on physical function in the global HRQoL scale).35 The original and new versions were applied in a total of 1,181 patients with cancer in Canada and the Netherlands. Internal consistency improved for the role functioning scale in the new version (Cronbach alpha ranging from 0.78 to 0.88 in the 2 country samples), and substitution of the new item for the previous version did not alter internal consistency (Cronbach alpha ranging from 0.81 to 0.92).35
The EORTC QLQ-C30 (version 3.0) is the version currently in use. Version 3.0 differs from the previous version 2.0 in that the number of response options for the first 5 items of the questionnaire comprising the physical function scale was increased from 2 options (yes/no in version 2.0) to 4 options (not at all, a little, quite a bit, very much). Internal consistency, reliability, construct validity, criterion validity, and responsiveness of the EORTC QLQ-C30 version 3.0 was assessed in 622 patients with head and neck cancer from 12 countries. Version 3.0 was more reliable than previous versions.36 Internal consistency of the multi-item scales was assessed using Cronbach alpha, with a value of 0.70 being considered adequate.37 The internal consistency of the new physical function scale of the EORTC QLQ-C30 version 3.0 was 0.84 compared with 0.66 in version 1.0. The EORTC QLQ-C30 version 3.0 was able to discriminate between head and neck cancer patients who were disease-free, who were newly diagnosed, and who had recurrent disease. As well, differences were noted between patients with different stages of disease and according to Karnofsky performance status (KPS): the new scale had a stronger association with KPS. Furthermore, there was a strong correlation observed between all subscale scores on the EORTC QLQ-C30 version 3.0 and symptom/treatment toxicity scores. Responsiveness to change was assessed using the standardized response mean (SRM), with an SRM of 0.20 considered small, 0.50 considered medium, and 0.80 considered large. The changes in the scores of QLQ-C30 demonstrated a small to medium SRM in response to treatment over time with scores mostly changing between 5 and 10 points.36
In the Georgakopoulos et al. (2013) study, the validity of the EORTC QLQ-C30 was assessed in 80 newly diagnosed patients with Hodgkin lymphoma and diffuse large B-cell lymphoma (DLBCL) undergoing chemotherapy (Adriamycin, Bleomycin, Vinblastine, Dacarbazine [ABVD] for Hodgkin lymphoma, and R-CHOP for DLBCL).27 Data were collected from the clinical research section of the Biomedical Research Foundation of the Academy of Athens in patients who had completed their chemotherapy (4-8 ABVD cycles or 6-8 R-CHOP cycles). The QLQ-C30 and other questionnaires were administered for self-completion, and the researcher was present for any clarifications. A difference of more than 10 units was considered significant for the 0-100 scales. Reliability as measured by Cronbach alpha for the EORTC QLQ-C30 was 0.79 for global health status/QoL, ranged from 0.51 to 0.85 for functional scales, and 0.82 to 0.86 for symptom scales/items indicating acceptable internal consistency for most dimensions. However, in the 2 functional scales of the QLQ-C30 instrument (emotional and cognitive functioning) the threshold of 0.70 was not met (0.63 and 0.51), demonstrating concerns around the internal consistency and reliability for these domains. No statistically significant differences between patients with HL and those with DLBCL were recorded, with exception in the symptom scale of the QLQ-C30 “appetite loss,” where a statistically significantly higher score for patients with HL was observed.27
Evidence of validity and reliability of the EORTC QLQ-C30 was not identified in the literature for patients with PNH.
Minimal Important Difference
A study by Osoba and colleagues, conducted in patients with breast cancer and small-cell lung cancer, estimated that a change in score on any scale of the EORTC QLQ-C30 of 10 points would be clinically significant. This estimate was based on an anchor-based approach to estimate the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported “very much” change had corresponding changes in the EORTC QLQ-C30 of more than 20 points, though this was conducted in a previous version of the measure.28
A more recent study from 2019 aimed to describe the MID for interpreting the EORTC QLQ-C30 measure in patients with advanced breast cancer patients. This study used an anchor-based approach utilizing performance status and chosen selected AEs as the clinical anchoring variables. The authors found that MIDs for within-group changes ranged from 5 to 14 points for improvements and from -14 to -4 points for deterioration across the individual scales. For between- group differences, the MIDs ranged from 4 to 11 points for improvements and from -18 to -4 points for deterioration across the individual scales.38
No studies reporting MID in PNH were identified.
FACIT-Fatigue
Description and Scoring
The Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-Fatigue) scale, previously known as the Functional Assessment of Cancer Therapy – Fatigue (FACT-F), is used to assess patient fatigue and energy levels and has been adapted for use in a number of chronic diseases.39 It is a 13-item questionnaire and scores questions on a 5-point Likert scale. The total 13-item scale ranges from zero (extreme fatigue) to 52 (no fatigue).29
Assessment of Validity and Reliability
In the initial development of the FACIT-Fatigue scale the questionnaire showed good stability (r = 0.87) along with strong internal consistency (Cronbach alpha = 0.95).29 When analyzed for test-retest reliability coefficient the measure again showed good stability (r = 0.84 to 0.90) and strong internal consistency (Cronbach alpha = 0.93 to 0.95).29 A more recent study from 2007 confirmed these results in a population of 135 patients with psoriatic arthritis that showed the FACIT-Fatigue questionnaire to have strong internal validity (Cronbach alpha = 0.95) and test-retest reliability (intraclass correlation coefficient = 0.95).30 In study of 3,492 participants, the FACIT-Fatigue questionnaire was able to successfully discriminate anemic cancer patients from the general population with high sensitivity and reasonable specificity.40 In a study of iron deficient anemia patients, FACIT-Fatigue showed convergence (r = 0.74) as well as responsiveness when compared to other relevant measures such as the SF-36 vitality subscale.41
No studies assessing the validity and reliability of the FACIT-Fatigue questionnaire in PNH were identified.
Minimal Important Difference
There were no identified studies that reported an MID for the FACIT-Fatigue scale in PNH.
LDH Levels
Description
The screening and diagnosis of PNH relies on flow cytometry to identify PNH clones, presentation of clinical symptoms, and supportive laboratory tests. These laboratory tests can include hemoglobin, RBC counts, LDH levels.31 LDH normalization and change from baseline LDH are used as a primary surrogate outcome in both the Study 301 and Study 302 presented in the current review. Specifically, the threshold of LDH ≥1.5 x ULN is used as a key indicator for disease control.
Evidence of Relationship to Clinical Outcomes
The relationship between the LDH threshold of ≥1.5 x ULN and the key PNH clinical outcomes of mortality and thromboembolism has been described in multiple publications reporting on a national South Korean PNH registry covering 41 years of data and 301 patients who had not received eculizumab.32,33 The Jang et al. (2016) study reported that, when compared to age- and gender-matched controls in the general population, a diagnosis of PNH resulted in a 3.9-fold increased risk of mortality.32 One of the key risk factors identified by the authors was patients with LDH ≥1.5 x ULN, which resulted in a 4.8-fold increase in risk of mortality compared to age- and gender-matched controls in the general population (p <0.001). PNH patients below this threshold showed no difference in mortality compared to healthy age- and gender-matched controls.32 The most important risk factor for mortality identified by the Jang et al. (2016) study was thromboembolism, which was associated with a 14-fold increased risk of mortality (p <0.001).32 Another publication using the same South Korean PNH registry reported on risk factors for thromboembolism in PNH patients.33 Multivariate analysis of the dataset found that PNH patients with LDH levels ≥1.5 x ULN were at significantly increased risk of thromboembolism compared to patients below this LDH threshold (OR = 7.0; p = 0.013).33 Patients with elevated LDH levels combined with clinical symptoms were associated with an even greater risk of thromboembolism than either single risk factor alone.33 The results from these registry-based publications, along with general acceptance from Canadian clinicians, further support the use of LDH ≥1.5 x ULN as a relevant outcome in PNH.3
Pharmacoeconomic Review
Abbreviations
BIA
budget impact assessment
BTH
breakthrough hemolysis
CAC
complement-amplifying condition
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ICER
incremental cost-effectiveness ratio
LDH
lactate dehydrogenase
PNH
paroxysmal nocturnal hemoglobinuria
pRBCs
packed red blood cells
QALY
quality-adjusted life-year
ULN
upper limit of normal
Executive Summary
The Executive Summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ravulizumab (Ultomiris), 10 mg/mL concentrate for solution for infusion |
Submitted price | Ravulizumab, 10 mg/mL, 30 mL vial: $7,296.67 |
Indication | For the treatment of adult patients with paroxysmal nocturnal hemoglobinuria |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 28, 2019 |
Reimbursement request | As per indication |
Sponsor | Alexion Pharma Canada Corp. |
Submission history | Previously reviewed: no |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov cohort model |
Target population | Adult patients with PNH, stratified by those who are treatment-naive to complement inhibitor therapy and those who are stable on eculizumab |
Treatment | Ravulizumab |
Comparator | Eculizumab |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (up to 100 years of age) |
Key data source | Clinical Study 301 for patients who are treatment-naive (Cohort 1) and Study 302 for patients who are stable on the labelled recommended dose for eculizumab for at least 6 months (Cohort 2) |
Submitted results | Ravulizumab dominated eculizumab (i.e., ravulizumab was more effective and less costly [incremental QALYs = 0.92; incremental costs = –$42,858]) |
Key limitations | The CADTH clinical review concluded that ravulizumab was noninferior to eculizumab, for all outcomes, including those used in the sponsor’s model, and that no conclusions may be made with regards to superiority. The sponsor’s model, however, suggests that patients receiving ravulizumab will experience better outcomes than patients taking eculizumab; for example, they will never experience incomplete C5 inhibition breakthrough hemolysis events over the entire model horizon. Health states used in the model did not capture all important aspects of the condition affecting patient quality of life, health care resource utilization, and/or mortality, such as thrombosis. The sponsor used treatment-specific utilities values, which is inappropriate, as utility values should reflect health states, not specific treatments. Additionally, the sponsor incorporated a utility increment related to the frequency of administration visits with ravulizumab which was not appropriately implemented in the submitted model, and the increment itself was based on assumption and is associated with uncertainty. The likelihood of up-dosing associated with both eculizumab and ravulizumab is highly uncertain, as the relationship between a higher dose and drug efficacy has not been established in clinical studies. Additionally, how patients receiving ravulizumab or eculizumab may be up-dosed (e.g., higher doses at the same administration frequency, reducing administration frequency, or up-dosing patients taking ravulizumab with a dose of eculizumab) is uncertain. |
CADTH reanalysis results | CADTH undertook reanalyses to address limitations, which included: assuming equal efficacy of ravulizumab and eculizumab, making health state utility values equal for ravulizumab and eculizumab, and removing the utility increment due to frequency of health care visit for patients receiving ravulizumab. When assuming equal clinical efficacy, ravulizumab compared to eculizumab is associated with lower total costs, resulting in cost savings of $13,386. Whether cost savings will be realized is highly uncertain as cost savings are only realized much later in the time horizon (i.e., higher first year or loading doses costs with ravulizumab are only offset by lower maintenance dose costs after 26 and 34 years in the treatment-naive and treatment-experienced populations, respectively) and should the actual cost of eculizumab be even 1% less than current list price ravulizumab would be more costly. |
LY = life-year; PNH = paroxysmal nocturnal hemoglobinuria; QALY = quality-adjusted life-year.
Conclusions
The CADTH clinical review concluded ravulizumab is noninferior to eculizumab in transfusion avoidance, occurrence of breakthrough hemolysis (BTH), lactate dehydrogenase (LDH) normalization, and hemoglobin stabilization over 26 weeks of treatment in adult patients with paroxysmal nocturnal hemoglobinuria (PNH), with maintenance of efficacy up to 52 weeks of treatment.
Based on the findings of the CADTH clinical review, CADTH assumed equal efficacy of ravulizumab and eculizumab. Further, CADTH undertook reanalyses to address limitations which included removing treatment-specific utility differences within health states for ravulizumab and eculizumab and removing the utility increment due to frequency of health care visits for patients receiving ravulizumab. Based on the CADTH reanalysis, conclusions remain similar to the sponsor’s: ravulizumab is equally as effective and potentially less costly compared with eculizumab when patients are treated for a lifetime (up to 100 years of age).
These results are driven by the lower drug acquisition cost of ravulizumab maintenance doses, when considering the publicly available list price of eculizumab over a lifetime time horizon (Table 7 for cost comparison). Because loading dose costs are higher for ravulizumab than eculizumab, cost savings with ravulizumab are realized much further into the time horizon. Patients who are treatment-naive would need to receive ravulizumab for more than 26 years before cost savings are realized or 34 years for patients who are treatment-experienced.
There is further uncertainty associated with the potential cost savings with ravulizumab, as eculizumab was previously reviewed by CADTH for the same indication and received a “do not list” recommendation, in part because it was found to have an incremental cost-effectiveness ratio (ICER) of $2.4 million per quality-adjusted life-year (QALY) gained compared with supportive care and would require a substantial reduction in price to be considered cost-effective. While participating drug plans may list eculizumab, the actual price may be lower than the current list price. Based on a CADTH threshold analysis, should the actual price of eculizumab for the participating drug plans be 1% less than the current list price, ravulizumab would be more costly than eculizumab. Therefore, ravulizumab is unlikely to be cost-effective at its submitted price and a price reduction is likely required, the magnitude of which is unknown.
Further uncertainty remains which could not be addressed by CADTH: up-dosing with either treatment would add drug costs and have an uncertain effect on clinical outcomes; reduced infusion frequency associated with ravulizumab could be preferred by patients, the impact (utility) of which was not appropriately captured in the analysis; and, as best supportive care was not included in the analysis as a comparator, the cost-effectiveness of ravulizumab compared to no active comparator is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups and drug plans that participated in the CADTH review process. No registered clinician input was received for this review.
Patient input for this review was received from the Canadian Association of PNH Patients. Patient input was gathered through 1-on-one interviews with individuals living with PNH in Canada. The submission also made reference to scientific literature to describe the impacts of living with PNH. The submission reported that the most devastating symptom associated with hemolysis is thrombosis, which can cause organ damage and death. Other symptoms patients with PNH experience include fatigue, difficulty swallowing, pulmonary hypertension, chronic kidney disease, shortness of breath, abdominal pain, erectile dysfunction, dark-coloured urine, anemia, and reduced quality of life. Patients who have experience with the current treatment (eculizumab) reported a burden associated with biweekly infusions. They also noted a difficulty securing public drug plan access to eculizumab treatment. Patients who have used ravulizumab reported it to significantly change their lives, allowing them to live a full life, resume work, and participate in society. The most important benefit identified as associated with ravulizumab was the reduced infusion frequency, allowing for a fuller life. Another benefit identified was the specific target of the complement protein to prevent hemolysis. Expectations for new treatments included hopes for an improved quality of life relating to reduced infusion frequency and prevention of BTH to avoid the return of PNH symptoms.
Drug plan input noted that the most relevant comparator, eculizumab, is listed on most public drug plans but that reimbursement criteria are not transparently published. They noted a consideration for alignment of initiation, continuation, and discontinuation criteria between eculizumab and ravulizumab. Drug plans also noted the potential for up-dosing with eculizumab and questioned whether dose escalation could also occur with ravulizumab.
Several of these concerns were addressed in the sponsor’s model: BTH events were modelled, health state utility values capturing PNH symptoms were applied, and the sponsor’s budget impact analysis (BIA) was aligned with eculizumab reimbursement criteria.
In addition, CADTH addressed some of these concerns by increasing the proportion of patients who switch from eculizumab to ravulizumab in the BIA to reflect patient input.
CADTH was unable to address the following concerns raised from stakeholder input.
Thrombosis was not considered as a model outcome.
The pharmacoeconomic analysis population is aligned with the clinical trial inclusion and exclusion criteria and may not reflect reimbursement criteria of eculizumab.
The decrement to quality of life associated with medication administration could not be incorporated using the sponsor’s existing model structure.
Potential up-dosing could not be addressed in the pharmacoeconomic analysis due to the limited evidence for both ravulizumab and eculizumab and the uncertainty in the proportion of patients receiving each treatment who would require up-dosing. An eculizumab up-dosing scenario was considered in the BIA.
Economic Review
The current review is for ravulizumab (Ultomiris) for adult patients with PNH.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of ravulizumab compared with eculizumab in adult patients with PNH, aligned with the Health Canada indication for ravulizumab.
Ravulizumab is a single-dose vial for IV infusion available in 300 mg/30 mL vials. The recommended dose for ravulizumab is weight based and consists of a loading dose (2,400 mg, 2,700 mg, and 3,000 mg for body weights ≥ 40 kg to < 60 kg, ≥ 60 kg to < 100 kg, and ≥ 100 kg, respectively) followed by maintenance dosing (3,000 mg, 3,300 mg, and 3,6000 mg for body weights ≥ 40 kg to < 60 kg, ≥ 60 kg to < 100 kg, and ≥ 100 kg, respectively).1 Maintenance doses are initiated 2 weeks after the loading dose and then administered every 8 weeks thereafter. For patients switching to ravulizumab from eculizumab, the ravulizumab loading dose should be administered 2 weeks after the last eculizumab infusion. At the sponsor’s submitted price of $7,296.67 per 300 mg vial, the cost per maintenance dose administration is $72,967, $80,263, and $87,560 for body weights of 40 kg or greater to less than 60 kg, 60 kg or greater to less than 100 kg, and 100 kg or greater, respectively. Assuming patients receive 6.5 administrations annually after the first year, the estimated annual cost of ravulizumab treatment ranges between $474,284 and $569,140, depending on patient weight (Table 7). Annual costs for treatment with ravulizumab are higher in the first year owing to loading dose administrations and patients receiving 7 maintenance doses in their first year of treatment (Table 7 for year 1 annual costs). The cost of eculizumab reflected the public list price ($6,7422 per 300 mg vial) and was administered at a loading dose of 600 mg every 7 days for the first 4 weeks, then 900 mg for the fifth dose 1 week later, followed by a maintenance dose of 900 mg every 2 weeks thereafter. At the wholesale price for eculizumab, the cost per 900 mg maintenance administration is $20,226, leading to an annual cost in subsequent years of $525,876. Annual costs for treatment with eculizumab are also higher in the first year ($559,586) owing to the loading dose administrations (Table 10 for year 1 annual costs).
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime time horizon (up to 100 years of age) from the perspective of a Canadian public health care payer. Discounting (1.5% per annum) was applied to both costs and outcomes. The model was run separately for patients who are treatment-naive and those who are experienced and stable on eculizumab, with final results weighted by the proportion of each population that was assumed to make up the treatment population.
Model Structure
The sponsor submitted a Markov state transition model with 2-week cycle lengths, corresponding to the lowest dosing frequency for eculizumab. The model included 11 health states and was primarily based on the presence of BTH with additional states related to a history of BTH and the need for continuous up-dosing (Figure 1). All patients began treatment in the no BTH state. From there, patients could remain having no BTH events, or could transition to having a complement-amplifying condition (CAC)-related BTH event or an incomplete C5 inhibition-related BTH event. Patients with CAC BTH events returned to the no BTH health state. Once patients had an incomplete C5 inhibition-related BTH event, they could transition to a history of incomplete C5 inhibition BTH-related health states: 1 where they had a history of incomplete C5 inhibition-related BTH events but no BTH, or, where they had a history of incomplete C5 inhibition-related BTH events and had ongoing incomplete C5 inhibition-related BTH events. People with a history of incomplete C5 inhibition-related BTH events who were in the no BTH state could also transition to having a CAC-related BTH event. Only patients receiving eculizumab could move to the history of incomplete C5 inhibition-related BTH events health states as the sponsor assumed no patients on ravulizumab experienced incomplete C5 inhibition BTH based on no events being observed in the clinical trials.3,4 In a scenario, the sponsor’s model structure allows for those with ongoing incomplete C5 inhibition-related BTH events to transition to a scenario of receiving a continuous up-dose of eculizumab. All patients had an equal probability of experiencing spontaneous remission regardless of treatment and the model also included a background mortality state and an optional PNH-related mortality state that only patients experiencing BTH events were at risk of, but this was not in use in the base case. The model also accounted for the requirement of blood transfusions based on health state and treatment. Additionally, the model included the flexibility to include 1-off up-dosing for CAC with eculizumab, although this was not considered in the sponsor’s base case.
Model Inputs
The model’s baseline population characteristics and clinical efficacy parameters were characterized according to Study 301 for patients who were treatment-naive (Cohort 1) and Study 302 for patients who were clinically stable on eculizumab (Cohort 2). The sponsor also included an optional third cohort of patients who were continually up-dosed on eculizumab, though this cohort was not considered in the sponsor’s base case. Study 301 and Study 302 were both multi-national, randomized, open-label trials. Study 301 compared ravulizumab to eculizumab in adult patients with PNH who were complement inhibitor-naive; whereas, Study 302 compared ravulizumab to eculizumab in patients with PNH who were clinically stable on eculizumab. The sponsor assumed that the Study 301 and Study 302 populations, including baseline age (45.5 and 47.7, respectively)3,4 reflected the Canadian population. The sponsor assumed that 5% of patients using ravulizumab are treatment-naive and 95% are stable on eculizumab.5
Patient movement in the model was primarily based on BTH event data from Study 301 and Study 302 for Cohort 1 and Cohort 2, respectively. BTH events in Study 301 and Study 302 were classified as incomplete C5 inhibition, CAC, or undetermined. Incomplete C5 inhibition events were defined as individual free C5 greater than or equal to 0.5 mcg/mL.5 CAC events occurred if there was any known condition that could increase complement activity, with infection being the most common cause observed in Study 301 and Study 302. Undetermined events had neither incomplete C5 inhibition nor concomitant infection. In the sponsor’s model, undetermined BTH events were considered to be CAC events as it was assumed there was a possibility that an underlying CAC cause may not have been adequately captured. The duration of an incomplete C5 inhibition BTH event was assumed to be 2 days, based on literature specifying that these events usually occur within 2 days before the next infusion of eculizumab in a 14-day dosing schedule.6,7 CAC BTH events were assumed to last for a full cycle length.5 The probability of requiring a transfusion of packed red blood cells (pRBCs) in the model, along with the mean number of pRBCs required, varied by treatment and whether patients experienced a BTH event, and was populated using patient-level data from Study 301 and Study 302 for Cohort 1 and 2, respectively.
A constant, per cycle probability of entering the spontaneous remission health state was incorporated into the model by fitting an exponential survival function to patients’ spontaneous remission-free survival times observed in a 1995 study of the natural history of PNH.5,8 Patients experiencing spontaneous remission were assumed to no longer require complement inhibitor therapy.5 Background mortality across all health states was assumed to be equal to that of the general Canadian population and based on age-adjusted mortality from Canadian life tables.9 While the model incorporated the functionality to assign an excess mortality risk by CAC or incomplete C5 inhibition BTH events, the sponsor’s base case assumed no excess mortality risk was associated with BTH events.5 Drug-related adverse events were not incorporated in the sponsor’s model.
Health state utility values were estimated separately for Cohort 1 and 2 and by complement inhibitor therapy based on the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) data from Study 301 and Study 302.3,4 EORTC QLQ-C30 data were mapped to EQ-5D 3-Levels utilities.10 Utility decrements associated with the experience of a BTH event and blood transfusions were also sourced from clinical trials. Taken together, the base utilities by treatment status were combined with the probability of receiving a transfusion to derive utility values for no BTH health states; these plus BTH-related decrements were applied to derive values associated with BTH events and all patients receiving ravulizumab received the associated utility increment across all health states sourced from the literature.11 The utility for patients experiencing spontaneous remission was that associated with the highest health state utility values across treatments and cohorts and was not treatment specific.
No administration costs were included in the model given the sponsor currently funds the administration of both ravulizumab and eculizumab.5 Costs related to meningococcal vaccines, assumed to be provided 2 weeks before treatment initiation to mitigate the risk of meningococcal infection, were included. The model also included event costs associated with BTH and blood transfusions. The resource utilization for BTH events was obtained from clinical expert opinion, while the number of transfusions associated with each health state by treatment arm was obtained from clinical Study 301 and Study 302.3,4 In both cases, the unit costs were obtained from the Ontario Schedule of Benefits.12 No health care management costs associated with PNH aside from transfusions and BTH events were included.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented below.
Base-Case Results
Ravulizumab was associated with a QALY gain of 0.92 at a cost that was $42,858 less than eculizumab, resulting in ravulizumab dominating (i.e., being more effective and less costly) eculizumab (Table 3). Ravulizumab was dominant in 95% of iterations. At a willingness-to-pay threshold of $50,000 per QALY gained, there was a 97% probability of ravulizumab being cost-effective compared to eculizumab.
Drug costs accounted for more than 99% of total costs for both treatments. There was no life-year gain associated with ravulizumab. All of the QALY gain for ravulizumab compared with eculizumab was accrued in the no BTH health state. This occurred because 0 ravulizumab life-years were spent in any of the BTH-related health states; all ravulizumab life-years accrued in the no BTH and spontaneous remission states.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. eculizumab ($/QALY) |
|---|---|---|---|---|---|
Eculizumab | 11,216,092 | Reference | 23.34 | Reference | Reference |
Ravulizumab | 11,173,235 | –42,858 | 24.26 | 0.92 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.5
Sensitivity and Scenario Analysis Results
The same conclusions made in the aggregate population were also made in the treatment-naive and treatment-experienced cohorts, with ravulizumab dominating eculizumab. A breakdown of incremental costs and QALYs by cohort and for the aggregate population is provided in Table 9.
The sponsor conducted a number of scenario analyses examining uncertainty. The only scenario whereby ravulizumab was not dominant occurred when the time horizon was shortened to 10 years. This occurred because it takes greater than 10 years to recuperate the additional costs of the ravulizumab loading dose for patients who are switching from eculizumab and because loading dose costs for ravulizumab are higher than those for eculizumab.
While cost-effectiveness conclusions associated with other scenarios remained unchanged from the sponsor’s base case, a few resulted in substantial changes to incremental costs and QALYs. The scenarios exploring up-dosing of eculizumab for incomplete inhibition of C5-related BTH events led to greater cost savings (meaning additional eculizumab costs relative to ravulizumab). Having an increased background mortality risk for patients with PNH led to fewer incremental costs and similar QALYs.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The CADTH clinical review concluded that ravulizumab is noninferior to eculizumab and conclusions on superiority of ravulizumab to eculizumab could not be drawn. The data used to inform treatment efficacy parameters in the sponsor’s model were derived from 2 noninferiority phase III trials comparing the effectiveness of ravulizumab with eculizumab across a variety of outcomes. According to the CADTH clinical review, the noninferiority margins for ravulizumab were met for all outcomes; however, superiority was not reached. The CADTH clinical review report therefore concluded that ravulizumab was noninferior to eculizumab in transfusion avoidance, occurrence of BTH, LDH normalization, and hemoglobin stabilization. Given this, the sponsor’s base case, which used trial data to inform BTH-related transition probabilities, transfusion probabilities, and transfusion volumes, does not align with the CADTH clinical review conclusions, as the transition probabilities for ravulizumab were set to perform better than eculizumab across all outcomes, based on rates observed in the clinical trials. In particular, the model predicted no incomplete C5 inhibition-related events for patients receiving ravulizumab over the entire time horizon, which is uncertain given the conclusions of the clinical review on the short-term evidence, as well as a lack of long-term evidence in support. According to the clinical expert consulted for this review, there are anecdotal cases in jurisdictions where ravulizumab is available where incomplete C5 inhibition has led to a shorter dosing interval than the labelled 8-week interval to maintain C5 inhibition. It is therefore unreasonable to assume there will be no instances of incomplete C5 inhibition with ravulizumab for the entire model time horizon. While there is potential for ravulizumab to perform better than eculizumab based on its pharmacokinetic profile, CADTH concluded that the sponsor’s base-case transition probabilities were inappropriate and not supported by clinical evidence given the trial’s inability to demonstrate that ravulizumab is superior to eculizumab.
In addition to the conclusions related to BTH events, ravulizumab did not demonstrate superiority to eculizumab for other efficacy outcomes parameterized in the model, including the probability of transfusion and volume required. According to the clinical expert consulted by CADTH for this review, outside of the context of BTH events, transfusions would not be expected to differ between treatment populations. As CADTH concluded noninferiority of ravulizumab to eculizumab, transfusions related to BTH events were also assumed to be similar across comparators. Finally, according to the clinical expert consulted for this review, among those requiring transfusions, the volume of pRBCs received is not expected to differ between treatments.
In CADTH reanalyses, to align with the finding of ravulizumab being noninferior to eculizumab, CADTH set efficacy for ravulizumab to be equal to eculizumab.
To explore uncertainty surrounding the comparative clinical effectiveness between treatments, CADTH conducted several scenarios: 1 where eculizumab efficacy was set to be equal to ravulizumab, and 1 using the sponsor’s efficacy assumptions which assume ravulizumab is more efficacious than eculizumab.
Health states used in the model did not capture all aspects of the condition. To capture the costs and health-related quality of life associated with PNH disease progression and the impacts of treatment, the sponsor’s model was based on BTH events. BTH was defined as having at least 1 new or worsening symptom or sign of intravascular hemolysis (fatigue, hemoglobinuria, abdominal pain, shortness of breath [dyspnea], anemia [hemoglobin < 10 g/dL], major adverse vascular event [including thrombosis], dysphagia, or erectile dysfunction) in the presence of elevated LDH 2 or more × the upper limit of normal (ULN), after prior LDH reduction to less than 1.5 × ULN on therapy.3,4 While prevention of BTH events is noted to be an important component of the treatment of PNH, both clinical expert feedback and patient input received noted thrombosis to be the most devastating consequence of disease, which was not explicitly modelled. Other symptoms noted to be important in the patient input received by CADTH included fatigue, difficulty swallowing, pulmonary hypertension, chronic kidney disease, and shortness of breath, none of which were explicitly modelled. While the clinical expert noted that some symptoms such as improvements in fatigue are related to BTH events, this was not explicitly modelled, and overall BTH it is a poor proxy for many other outcomes important to patients and which affect health system costs.
CADTH was unable to incorporate other important aspects of PNH in the model. Given that ravulizumab demonstrated noninferiority to eculizumab across most outcomes, the influence of not including all aspects of the condition in the model on cost-effectiveness results should be limited.
Health state utility values and approach to their implementation w inappropriate. Several issues were identified with the approach to identifying and incorporating patient utility values in the submitted model. The sponsor conducted a regression analysis on data from Study 301 and Study 302 to estimate utility impacts associated with BTH events and transfusions. A treatment indicator was included to explore a difference in utility between ravulizumab and eculizumab, independent of BTH events and transfusions and led to treatment-specific utility values which were used in the sponsor’s base case.5 The sponsor derived ravulizumab base utility values by adding the treatment indicator for ravulizumab onto eculizumab utilities, meaning the utility for ravulizumab patients was fixed to be 0.01 and 0.02 better than for patients receiving eculizumab in Cohort 1 and Cohort 2, respectively.5 The use of treatment-specific utility values is contradictory to CADTH guidelines that specify that utilities be associated with health states.13 All outcomes associated with treatment, along with their impact on patient utility, should be explicitly modelled, rather than captured using a treatment-specific utility value. Including a treatment indicator to capture a difference in utility between treatments that has not been modelled is therefore inappropriate.
Second, to capture the utility difference associated with reduced visit frequency due to less frequent treatment administration with ravulizumab, the sponsor added a utility benefit for ravulizumab of 0.02. The value was based on an assumption and informed by a study demonstrating a higher utility for cardiovascular patients who had 7 or less general practice visits compared to those who were seen more than 7 times annually.11 First, whether a utility benefit captured in this patient population and visit setting would apply to patients with PNH receiving infusions is uncertain. Second, a more appropriate way of explicitly incorporating the quality of life benefits associated with infusions would have been to incorporate an administration disutility for all patients and apply it during cycles that they received an infusion. As patients receiving ravulizumab would receive fewer infusions, this would result in an overall higher QALY gain, but it is uncertain whether the difference in QALYs between treatments due to fewer treatment administrations would be accurately represented by the sponsor’s assumed utility increment for ravulizumab. Finally, whether reduced infusion frequency would improve health-related quality of life or quality of life in general is uncertain as mentioned by the clinical expert consulted by CADTH. From a health care system perspective, health-related quality of life benefits should be captured, whereas general quality of life benefits would be captured from a broader societal perspective which is not considered in the CADTH base case.
Finally, the sponsor estimated utility values in the model by mapping clinical trial EORTC QLQ-C30 data to EQ-5D values using an algorithm published by Longworth at al.10 According to CADTH Guidelines for Economic Evaluation, mapping as a means of deriving health utilities is not recommended.13 Instead, CADTH prefers the use of a generic indirect utility measure to obtain utility scores for the economic model.13 This is because the utility values garnered through mapping can vary dramatically depending on instruments being mapped, the algorithm used for mapping, and the severity of the included health states. As noted in the guidelines and by the sponsor, there are several published algorithms available to map EORTC QLQ-C30 data to EQ-5D values.5 As demonstrated by the sponsor’s scenario analyses exploring the use of a different mapping algorithm by McKenzie et al.,14 not only do the utility values derived vary from those derived using the Longworth algorithm, so too do the resulting total QALYs estimated in the model. Whereas the sponsor’s base case led to 0.92 incremental QALYs between ravulizumab and eculizumab, when the McKenzie algorithm was used it led to 1.59 incremental QALYs, a 53% difference.5 This demonstrates the uncertainty that mapping introduces into the analysis and highlights its inappropriateness.
CADTH was unable to address the uncertainty associated with deriving utility estimates using mapping. In the CADTH reanalysis, both the treatment indicator and the utility increment for reduced treatment administration visits with ravulizumab were removed as they do not explicitly model outcomes associated with complement inhibitor treatment. Additionally, the value of the reduced visit increment is uncertain.
Up-doses associated with BTH events are highly uncertain and may affect total treatment-related costs. The sponsor’s model allowed for the flexibility to consider an increased dose in response to incomplete C5 inhibition or CAC-related BTH events. No up-dosing was considered in the sponsor’s base case. According to the clinical expert consulted by CADTH for this review, some up-dosing for patients receiving eculizumab is occurring in Canadian clinical practice, with a proportion of patients receiving a maintenance dose of 1,200 mg rather than 900 mg to maintain C5 inhibition. However, in Study 301 and Study 302, only the 900 mg dose of eculizumab was allowed; therefore, the effect of up-dosing on BTH events is not captured by the trial. Consequently, while the option to up-dose in the model adds costs and removes the disutility associated with a BTH event, it does not influence other efficacy outcomes such as BTH transitions, transfusions, or thrombosis. Further, while Study 301 and Study 302 did not capture incomplete C5 inhibition for patients receiving ravulizumab, it is uncertain whether C5 inhibition would be sustained in the long-term. According to the clinical expert consulted for this review, there are anecdotal cases in jurisdictions where ravulizumab is available where incomplete C5 inhibition has led to a shorter dosing interval than the labelled 8-week interval. Taken together, due to the uncertainty regarding the form up-dosing would take should it occur (higher dose or reduced intervals), the proportion of patients receiving eculizumab and ravulizumab who would require 1-off or continuously higher levels of drug to maintain C5 inhibition and the influence of higher doses on drug efficacy, CADTH agrees with the sponsor’s base case which excludes the consideration of up-dosing for both treatments. If increased doses are required of either drug, it will add costs and have an uncertain influence on outcomes.
Due to the uncertainty regarding the proportion of patients on ravulizumab and eculizumab who would require 1-off or continuous up-dosing, how up-dosing would occur (e.g., higher dose given at the same administration intervals, shortening the time between administrations, whether ravulizumab patients would be up-dosed on ravulizumab or receive a dose of eculizumab), and the influence of higher doses on model efficacy parameters, CADTH did not incorporate up-dosing in the reanalysis. If only patients receiving eculizumab require a higher dose to maintain C5 inhibition for the model time horizon, ravulizumab will dominate as it will be associated with lower costs. If patients receiving ravulizumab also require a higher dose, the influence on the cost-effectiveness results are uncertain.
The cost-effectiveness of ravulizumab compared with best supportive care is unknown. In the sponsor’s submission, ravulizumab was compared with eculizumab. Best supportive care was not included as a comparator. According to the clinical expert consulted by CADTH for this review, eculizumab is the most relevant comparator to ravulizumab. However, as eculizumab received a “do not list” recommendation during its review by CADTH for PNH, for participating plans that do not reimburse eculizumab, the cost-effectiveness of ravulizumab is unknown. The CADTH review of eculizumab estimated an ICER of $2.4 million per QALY gained with eculizumab compared with supportive care and noted that without a substantial reduction in price, it would not be considered cost-effective.15 This is noteworthy as participating drug plans that do reimburse eculizumab may have negotiated some price reduction. Based on a CADTH threshold analysis, should the price of eculizumab be 1% less than the current list price, ravulizumab will be more costly.
CADTH was unable to address this limitation. As such, the cost-effectiveness of ravulizumab relative to best supportive care is unknown.
The following limitations were identified but were not deemed key limitations:
The rate of spontaneous remission incorporated in the model may be an overestimate. In the sponsor’s model, spontaneous remission was informed by a study by Hilleman et al. which followed patients with PNH for 30 years and found approximately 15% of patients had a spontaneous clinical recovery. As noted by the sponsor, the rate of spontaneous remission is uncertain, and, according to the clinical expert consulted by CADTH for this review, the rate may be closer to 5%. However, as spontaneous remission is assumed to be equal in both arms, it is not expected that the rate of spontaneous remission will drive results.
CADTH conducted a scenario exploring the impact of no spontaneous remission as a scenario analysis.
The difference in infusion times between comparators was not explored. As noted in the CADTH clinical review report, eculizumab and ravulizumab administration differs in both frequency and duration of the infusion., Eculizumab is infused over 35 minutes,1 whereas ravulizumab maintenance doses are infused over 120 to 140 minutes. The sponsor did not include administration costs as part of the submission as it was assumed that these would be covered for the duration of the model time horizon. It would have been more accurate to include the option to explore administration frequencies and durations as a model parameter to explore the scenario where the infusions differ in time should funding through the patient support program change in the future.
CADTH was unable to address this limitation. The cost per infusion would be expected to be higher for ravulizumab than eculizumab because of the duration; however, overall infusions costs would be lower for ravulizumab as there are fewer annual infusions.
Mortality associated with PNH is uncertain. In the sponsor’s base case, background mortality was assumed to be equal to that of the general population and the sponsor assumed that there was no excess mortality risk associated with BTH events. According to the clinical expert consulted by CADTH for this review, it is expected that the mortality of patients treated with complement inhibitors will follow that of the general population for the first 10 years, and then will become higher than that of the general population due to factors such as bone marrow failure, which is not prevented by complement inhibitor treatment. Second, the clinical expert consulted for this review also noted that there is a mortality risk associated with experiencing a BTH event; however, the magnitude of the risk is uncertain.
As background mortality was assumed to be equal across treatments, an increasing mortality risk over time for all patients would not be expected to influence incremental life-years between treatments. Additionally, as the CADTH base case has assumed the risk of experiencing a BTH event would be equal across treatments, mortality associated with BTH events will also not influence incremental life-years.
CADTH was unable to address this limitation; however, it is not expected to influence cost-effectiveness results.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH’s comment |
|---|---|
Rates of spontaneous remission were assumed to be the same among patients receiving ravulizumab and eculizumab. | Appropriate according to the clinical expert consulted by CADTH for this review. |
The occurrence and management of iron overload was assumed to be the same among patients receiving ravulizumab and eculizumab, and therefore was not incorporated in the economic analysis. | Appropriate according to the clinical expert consulted by CADTH for this review. |
Adverse events were not incorporated in the model. | Appropriate. According to the CADTH clinical review report there were no notable differences in adverse events between the treatment groups and the clinical expert did not find any of the adverse events particularly concerning. |
In the sponsor’s model, 5% of the population was expected to be treatment-naive, and 95% were expected to be experienced and stable receiving eculizumab. | Appropriate according to the clinical expert consulted by CADTH for this review. The sponsor’s model included the option to model a third population, which consisted of patients on a continuously increasing dose of eculizumab but did not include such patients in the base case as such patients were not anticipated to be switched to ravulizumab. Similarly, there may be patients who are not stable on eculizumab, and such patients were also assumed to not be eligible for a switch to ravulizumab. The clinical expert consulted by CADTH for this review agreed that such patients would not be treated with ravulizumab. |
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH reanalyses addressed several limitations within the economic model, summarized in Table 5. CADTH was unable to address limitations regarding health states not fully reflecting all key aspects of PNH, the approach to incorporating treatment frequency impact on utility and the magnitude of health-related quality of life benefit from administration frequency, the difference in infusion times between comparators, and uncertainty surrounding mortality assumptions.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH’s value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Ravulizumab utility for incomplete C5 inhibition of BTH-related health states | Not available | Set equal to eculizumab |
Changes to derive the CADTH base case | ||
1. Ravulizumab efficacy | Superior to eculizumab | Equal to eculizumab |
2. Base utilities values | Treatment specific | Treatment indicator removed |
3. Ravulizumab administration visit utility increment | 0.02 | 0.00 |
CADTH base case | — | 1 + 2 + 3 |
BTH = breakthrough hemolysis.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or standard errors in probabilistic analyses, and so forth) that are not identified as limitations.
The results of CADTH’s stepped analysis are presented in Table 6. CADTH’s base-case reanalysis demonstrates that, compared with eculizumab, ravulizumab yields the same number of QALYs and is associated with lower costs (–$13,386), resulting in ravulizumab dominating eculizumab (Table 6). Ravulizumab was dominant in all steps of the analysis. Removing the utility increment related to fewer treatment administrations for ravulizumab resulted in the greatest decrease in total QALYs with ravulizumab. The majority (56%) of the QALYs for both comparators was accrued in the no BTH health state (Table 10). Similar to the sponsor’s base case, nearly all (99.7%) of total costs for both comparators were drug costs (Table 10). When considering drug costs alone, in the treatment-naive population, patients must remain on treatment for 26.75 years before overall drug costs with ravulizumab become lower than eculizumab. In the treatment-experienced population, it took 34.73 years of treatment for ravulizumab to become less costly than eculizumab.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Eculizumab | 11,216,092 | 23.34 | Reference |
Ravulizumab | 11,173,235 | 24.26 | Dominant | |
Sponsor’s corrected base case | Eculizumab | 11,216,092 | 23.34 | Reference |
Ravulizumab | 11,173,235 | 24.26 | Dominant | |
Sponsor’s base case (deterministic) | Eculizumab | 11,186,517 | 23.23 | Reference |
Ravulizumab | 11,139,677 | 24.18 | Dominant | |
CADTH reanalysis 1 — ravulizumab efficacy | Eculizumab | 11,186,517 | 23.23 | Reference |
Ravulizumab | 11,173,349 | 23.98 | Dominant | |
CADTH reanalysis 2 — remove treatment indicator for utilities | Eculizumab | 11,186,517 | 23.37 | Reference |
Ravulizumab | 11,139,677 | 23.91 | Dominant | |
CADTH reanalysis 3 — remove ravulizumab utility increment | Eculizumab | 11,186,517 | 23.11 | Reference |
Ravulizumab | 11,139,677 | 23.63 | Dominant | |
CADTH base case (1 + 2 + 3) (deterministic) | Eculizumab | 11,186,517 | 23.25 | Reference |
Ravulizumab | 11,173,349 | 23.25 | Dominant | |
CADTH base case (1 + 2 + 3) (probabilistic) | Eculizumab | 11,200,869 | 23.30 | Reference |
Ravulizumab | 11,187,484 | 23.30 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Results of all steps are presented deterministically. The cumulative CADTH base case is presented probabilistically as well.
Scenario Analysis Results
To address remaining uncertainty regarding parameterization of the model, CADTH conducted several scenario analyses. Full results are presented in Table 11. Ravulizumab remained dominant in all scenarios apart from a scenario exploring its cost-effectiveness in patients weighing more than 100 kg. In this scenario, eculizumab dominated ravulizumab, meaning that ravulizumab is not cost-effective compared to eculizumab in this patient population. Using the sponsor’s base-case efficacy parameters led to a 0.10 QALY gain for ravulizumab compared to eculizumab, highlighting that the improvements in BTH event frequencies and transfusions used in the sponsor’s analysis do not drive the cost-effectiveness of ravulizumab. Applying the sponsor’s utility increment associated with fewer visits for ravulizumab led to a QALY gain of 0.39 for ravulizumab. This scenario is uncertain as the value of the increment used was based on assumption, and whether reduced visit frequencies influence health-related quality of life rather than general quality of life is uncertain. CADTH conducted a threshold analysis using the CADTH base case to examine the price for eculizumab at which ravulizumab would be considered cost-effective. If the price per vial of eculizumab is $6,734 or more, ravulizumab results in cost savings over a lifetime time horizon; however, if the confidentially negotiated price of eculizumab is $6,733 per vial or less, ravulizumab will not be considered cost-effective compared to eculizumab and eculizumab will dominate ravulizumab.
Issues for Consideration
Should eculizumab biosimilars become available, and should these biosimilars be considered equivalent to eculizumab, ravulizumab is unlikely to remain less costly than eculizumab biosimilars, and eculizumab biosimilars would be considered optimal.
Administration fees for complement inhibitor therapies were not included in the sponsor’s model as they were assumed to be covered by the sponsor’s patient support program for the entirety of the model time horizon. If this were to change, it is expected that administration fees will be greater for eculizumab compared with ravulizumab due to the greater frequency of administration; however, chair time for a given administration will be longer for patients receiving ravulizumab.
Overall Conclusions
The CADTH clinical review found that ravulizumab is noninferior to eculizumab in transfusion avoidance, occurrence of BTH, LDH normalization, and hemoglobin stabilization over 26 weeks of treatment in adult patients with PNH, with maintenance of efficacy up to 52 weeks of treatment.
In the submitted model, ravulizumab was set to perform better across relevant outcomes for the entire model time horizon, which is uncertain in the long-term. Additional identified limitations include that the health states used in the model did not fully capture all aspects of PNH that may impact patient health-related quality of life, mortality, or health system costs. Several limitations were also identified with the utility values, including the use of treatment-specific utility values and the sponsor added a utility increment to ravulizumab associated with fewer administration visits, which was inappropriate because the utility increment value was uncertain, and it is uncertain if reduced visit frequency would be associated with improved health-related quality of life specifically. CADTH also noted that up-dosing practices in the real-world setting are highly uncertain but may affect the estimated cost-effectiveness of ravulizumab in comparison with eculizumab.
Given the findings of the CADTH clinical review, CADTH assumed equal efficacy for ravulizumab and eculizumab. Further, CADTH undertook reanalyses to address limitations which included removing treatment-specific utility differences within health states for ravulizumab and eculizumab and removing the utility increment due to frequency of health care visits for patients receiving ravulizumab. Based on the CADTH reanalysis, conclusions remain similar to the sponsor’s: ravulizumab is equally as effective and potentially less costly compared with eculizumab when patients are treated for a lifetime (up to 100 years of age).
These results are driven by the lower drug acquisition cost of ravulizumab maintenance doses, when considering the publicly available list price of eculizumab over a lifetime time horizon (Table 7 for cost comparison). Because loading dose costs are higher for ravulizumab than eculizumab, cost savings with ravulizumab are realized much further into the time horizon. Patients who are treatment-naive would need to receive ravulizumab for more than 26 years before cost savings are realized or 34 years for patients who are treatment-experienced.
There is further uncertainty associated with the potential cost savings with ravulizumab, as eculizumab was previously reviewed by CADTH for the same indication and received a “do not list” recommendation, in part because it was found to have an ICER of $2.4 million per QALY gained compared with supportive care and would require a substantial reduction in price to be considered cost-effective. While participating drug plans may list eculizumab, the actual price may be lower than the current list price. Based on a CADTH threshold analysis, should the actual price of eculizumab for the participating drug plans be 1% less than the current list price, ravulizumab would be more costly than eculizumab. Therefore, ravulizumab is unlikely to be cost-effective at its submitted price and a price reduction is likely required, the magnitude of which is unknown.
Further uncertainty remains which could not be addressed by CADTH: up-dosing with either treatment would add drug costs and have an uncertain effect on clinical outcomes; reduced infusion frequency associated with ravulizumab could be preferred by patients, the impact (utility) of which was not appropriately captured in the analysis; and, as best supportive care was not included in the analysis as a comparator, the cost-effectiveness of ravulizumab compared to no active comparator is unknown. Though the model did not capture outcomes identified as important based on patient and clinician input such as thrombosis, this is not expected to influence conclusions as ravulizumab was found to be noninferior to eculizumab for most outcomes.
References
1.Ultomiris (ravulizumab): 10 mg/mL concentrate for solution for infusion [product monograph]. Zürich (CH): Alexion Pharma GmbH; 2019 Aug 28.
2.Government of Alberta. Interactive drug benefit list. 2021; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2021 Oct 15.
3.Clinical Study Report: ALXN1210-PNH-301. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in complement inhibitor-naïve adult patients with paroxysmal nocturnal hemoglobinuria (PNH) [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc; 2018 May 23.
4.Clinical Study Report: ALXN1210-PNH-302. A phase 3, randomized, open-label, active-controlled study of ALXN1210 versus eculizumab in adult patients with paroxysmal nocturnal hemoglobinuria (PNH) currently treatment with eculizumab [internal sponsor's report]. New Haven (CT): Alexion Pharmaceuticals, Inc; 2018 May 30.
5.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughn (ON): Alexion Pharma Canada Corp; 2021 Jul 22.
6.Kelly R, Arnold L, Richards S, et al. Modification of the eculizumab dose to successfully manage intravascular breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;112(11):3441.
7.Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood, The Journal of the American Society of Hematology. 2014;124(18):2804-2811. PubMed
8.Hillmen P, Lewis S, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253-1258. PubMed
9.Statistics Canada. Life Tables, Canada, Provinces and Territories. https://www150.statcan.gc.ca/n1/en/catalogue/84-537-X. Accessed Sept 12 2021.
10.Louise Longworth ea. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. 2014.
11.Ludt S, Wensing M, Szecsenyi J, et al. Predictors of health-related quality of life in patients at risk for cardiovascular disease in European primary care. PLoS One. 2011;6(12):e29334. PubMed
12.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Sep 24.
13.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2021 Oct 6.
14.McKenzie L, Van Der Pol M. Mapping the EORTC QLQ C-30 onto the EQ-5D instrument: the potential to estimate QALYs without generic preference data. Value Health. 2009;12(1):167-171. PubMed
15.CADTH. CEDAC FINAL RECOMMENDATION ECULIZUMAB (Soliris – Alexion Pharmaceuticals, Inc.) Indication: Paroxysmal Nocturnal Hemoglobinuria 2010. Accessed Oct 5, 2021.
16.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughn (ON): Alexion Pharma Canada Corp; 2021 Jul 22.
17.PNH–ravulizumab advisory board meeting [internal sponsor's report]. In Drug Reimbursement Review sponsor submission: Ultomiris (ravulizumab), 10 mg/mL concentrate for solution for infusion. Vaughn (ON): Alexion Pharma Canada Corp; 2019 Apr 5.
18.Patriquin CJ, Kiss T, Caplan S, et al. How we treat paroxysmal nocturnal hemoglobinuria: a consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol. 2019;102(1):36-52. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for Paroxysmal Nocturnal Hemoglobinuria
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Ravulizumab (Ultomiris) | 10 mg/mL | 30 mL single-dose vial of concentrate for solution for IV infusion | $7,296.6700 | Loading dose, with maintenance doses given starting 2 weeks after, then administered every 8 weeks thereafter, based on weight as followsa: ≥ 40 kg to < 60 kg Loading: 2,400 mg Maintenance: 3,000 mg ≥ 60 kg to < 100 kg Loading: 2,700 mg Maintenance: 3,300 mg ≥ 100 kg Loading: 3,000 mg Maintenance: 3,600 mg | ≥ 40 kg to < 60 kg: Year 1b: 1,559.29 Subsequent years c: 1,299.41 ≥ 60 kg to < 100 kg: Year 1 b: 1,719.22 Subsequent years c: 1,429.35 ≥ 100 kg: Year 1 b: 1,879.14 Subsequent years c: 1,559.29 | ≥ 40 kg to < 60 kg: Year 1 b: 569,140 Subsequent years c: 474,284 ≥ 60 kg to < 100 kg: Year 1 b: 627,514 Subsequent years c: 521,712 ≥ 100 kg: Year 1 b: 685,887 Subsequent years c: 569,140 |
Compliment inhibitor | ||||||
Eculizumab (Soliris) | 10 mg/mL | 300 mg single-use vial | $6,742.0000d | Loading: 600 mg every 7 days for the first 4 weeks, then 900 mg for the fifth dose 1 week later Maintenance: 900 mg every 2 weeks thereafter | Year 1e: 1,533.11 Subsequent yearsf: 1,440.76 | Year 1 e: 559,586 Subsequent yearsf: 525,876 |
aFor patients switching from eculizumab, the loading dose of ravulizumab is given 2 weeks after the last eculizumab infusion. Maintenance doses are then given every 8 weeks, starting 2 weeks after the loading dose.
bYear 1 costs assume 1 loading dose and 7 maintenance doses.
cSubsequent year dosing are based on an average of 6.5 administrations (52/8) per year.
dAlberta drug formulary (accessed October 2021).2
eYear 1 costs assume four 600 mg doses and 25 900 mg doses.
fSubsequent year costs assume 26 administrations per year.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Thrombosis has been identified as an important outcome by patients and clinical experts, however this was not modelled. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | It was not possible to consider administration disutility or administration costs using the sponsor’s submitted model. Complete reporting of probabilistic results by cohort was not provided (i.e., total costs and QALYs were not reported by cohort, only incremental) |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
BTH: breakthrough hemolysis; CAC: complement-amplifying conditions; Hx: history of; IncC5Inhib: incomplete C5 inhibition; PNH: paroxysmal nocturnal hemoglobinuria.
Source: Sponsor’s pharmacoeconomic submission5
Detailed Results of the Sponsor’s Base Case
Table 9: Summary of Results of the Sponsor’s Base Case by Treatment Status
Cohort | Percentage of population | Incremental cost of ravulizumab ($) | Incremental QALYs of ravulizumab | Incremental cost per QALY |
|---|---|---|---|---|
Cohort 1-Treatment-naive | 5% | −66,425 | 0.78 | Ravulizumab dominates eculizumab |
Cohort 2-Treatment-experienced | 95% | −41,617 | 0.93 | Ravulizumab dominates eculizumab |
Aggregate population | 100% | −42,858 | 0.92 | Ravulizumab dominates eculizumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission5
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Ravulizumab | Eculizumab | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 27.39 | 27.39 | 0.00 |
No BTH | 15.23 | 15.23 | 0.00 |
CAC BTH | 0.05 | 0.05 | 0.00 |
IncC5Inhib BTH | 0.01 | 0.01 | 0.00 |
History of IncC5Inhib BTH, no BTH | 4.14 | 4.14 | 0.00 |
Subsequent IncC5Inhib BTH | 1.71 | 1.71 | 0.00 |
History of IncC5Inhib BTH, CAC BTH | 0.02 | 0.02 | 0.00 |
History of IncC5Inhib BTH, continuous up-dose | 0.00 | 0.00 | 0.00 |
Cont. up-dose, CAC BTH | 0.00 | 0.00 | 0.00 |
Spontaneous remission | 6.23 | 6.23 | 0.00 |
Discounted QALYs | |||
Total | 23.30 | 23.30 | 0.00 |
No BTH | 12.98 | 12.98 | 0.00 |
CAC BTH | 0.03 | 0.03 | 0.00 |
IncC5Inhib BTH | 0.01 | 0.01 | 0.00 |
History of IncC5Inhib BTH, no BTH | 3.52 | 3.52 | 0.00 |
Subsequent IncC5Inhib BTH | 1.37 | 1.37 | 0.00 |
History of IncC5Inhib BTH, CAC BTH | 0.01 | 0.01 | 0.00 |
History of IncC5Inhib BTH, continuous up-dose | 0.00 | 0.00 | 0.00 |
Cont. up-dose, CAC BTH | 0.00 | 0.00 | 0.00 |
Spontaneous remission | 5.37 | 5.37 | 0.00 |
Discounted costs ($) | |||
Total | 11,187,484 | 11,200,869 | −13,386 |
Drug acquisition | 11,152,148 | 11,165,533 | −13,386 |
Medical | 35,336 | 35,336 | 0.00 |
ICER ($/QALY) | Ravulizumab dominates | ||
BTH: breakthrough hemolysis; CAC: complement-amplifying conditions; ICER = incremental cost-effectiveness ratio; IncC5Inhib: incomplete C5 inhibition; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 11: CADTH Scenario Analyses
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH base case | Eculizumab | 11,200,869 | 23.30 | Reference |
Ravulizumab | 11,187,484 | 23.30 | Dominant | |
Subgroup analysis: Cohort 1 (treatment-naive) | Eculizumab | 11,607,420 | 23.50 | Reference |
Ravulizumab | 11,578,304 | 23.50 | Dominant | |
Subgroup analysis: Cohort 2 (treatment-experienced) | Eculizumab | 11,233,063 | 23.28 | Reference |
Ravulizumab | 11,220,092 | 23.28 | Dominant | |
Sponsor’s efficacy assumptions | Eculizumab | 11,226,476 | 23.33 | Reference |
Ravulizumab | 11,184,188 | 23.43 | Dominant | |
Eculizumab equal to ravulizumab in terms of BTH events, transfusion probabilities and volumes | Eculizumab | 11,202,079 | 23.39 | Reference |
Ravulizumab | 11,188,479 | 23.39 | Dominant | |
Ravulizumab utility increment: 0.02 | Eculizumab | 11,212,734 | 23.43 | Reference |
Ravulizumab | 11,199,267 | 23.82 | Dominant | |
No spontaneous remission | Eculizumab | 14,497,790 | 23.19 | Reference |
Ravulizumab | 14,458,441 | 23.19 | Dominant | |
All patients greater than 100 kg | Ravulizumab | 12,212,766 | 23.30 | Reference |
Eculizumab | 11,210,258 | 23.30 | Dominant |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 12: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA estimating the budget impact of introducing ravulizumab for the treatment of adult patients with PNH. The BIA base case was undertaken from a publicly funded drug plan perspective considering only drug costs over a 3-year time horizon. The sponsor’s patient support plan data were used to estimate the number of individuals being treated for PNH and discontinuation rates (Table 13).
The sponsor assumed that no new patients were expected to start complement inhibitor therapy over the 3-year time horizon.16
The sponsor compared a reference scenario, where only eculizumab was available to treat adult patients with PNH with a new drug scenario where ravulizumab is also funded according to current eculizumab reimbursement criteria (i.e., 1) granulocyte clone > 10%; 2) LDH greater than 1.5 × ULN; and 3) at least 1 of the following: thrombosis, transfusions, anemia, pulmonary insufficiency, renal insufficiency, and smooth muscle spasms). Eculizumab costs included in the analyses accounted for up-dosing by assuming that 10% of patients were maintained at a dose of 1,200 mg every 2 weeks, and 90% were maintained at 900 mg.17 Ravulizumab costs assumed that 91% of patient weights fell between 60 to less than 100 kg; 7% were between 40 to less than 60 kg, and 2% were greater than 100 kg, based on Canadian PNH registry data.18
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Year 0 numbers (baseline) Projected number of existing cases of treated PNH in Canadian population at Year 0 Projected new cases of treated PNH in Canadian population in Year 0 Discontinuation rate Total PNH patients on eculizumab Year 0 Subsequent year numbers Current/continuing cases from previous year Annual rate of switching to ravulizumab New patients Discontinuation rate | |||| |||| 2.6%a |||| |||| 15%b 0 2.6%a |
Number of patients eligible for drug under review (reference scenario)c Number of patients eligible for drug under review (new drug scenario) | |||| / |||| / |||| |||| / |||| / |||| |
Market uptake (3 years) | |
Uptake (reference scenario) Eculizumab | 100% / 100% / 100% |
Uptake (new drug scenario) Ravulizumab Eculizumab | 15% / 28% / 39% 85% / 72% / 61% |
Cost of treatment (per patient) | |
Cost of treatment over one year Ravulizumab (New patients-treatment-naive) Ravulizumab (New patients-switch from eculizumab) Ravulizumab (Continuing patients) Eculizumab (New patients) Eculizumab (Continuing patients) | $565,007 $565,007 $519,340 $539,696 $543,405 |
Abbreviations; PNH = paroxysmal nocturnal hemoglobinuria
aApplied to number of patients currently using eculizumab or ravulizumab in that year (e.g., = ||||*2.6% = ||||)
bApplied to the number of patients currently using eculizumab in that year.
cThe number of patients in the reference and new drug scenarios differed in the sponsor’s base case because of how the discontinuation rate was implemented. Refer to the CADTH Appraisal of the Sponsor’s BIA’ for additional details
Summary of the Sponsor’s BIA Results
The sponsor estimated the net budget impact of introducing ravulizumab for the treatment of adult patients with PNH would be $601,724 in Year 1, $335,187 in Year 2 and $118,759 in Year 3 for a total budget impact $1,055,670 over 3 years.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The BIA population is not aligned with the Health Canada indication. Rather than using an epidemiological based approach and applying filters to derive the size of the eligible population, the sponsor estimated the market size of ravulizumab based on their confidential patient support program for eculizumab. As there may be reimbursement criteria for eculizumab that are narrower than the Health Canada indication, this means that the population estimated by the sponsor is smaller than the total Health Canada indicated eligible population size. According to the clinical expert consulted by CADTH for this review, there is a proportion of patients in clinical practice who meet the Health Canada indication for eculizumab but do not meet jurisdictional reimbursement criteria. These patients would therefore also be eligible for ravulizumab but would not be captured in the sponsor’s estimates.
Additionally, using confidential data from the patient support program meant that CADTH was unable to validate key input parameters including population size, the number of new patients per year and discontinuation rate. This means there is uncertainty regarding the size of the eligible population for ravulizumab.
CADTH was unable to estimate the total potential size of the eligible population for ravulizumab using the sponsor’s existing model structure. Should ravulizumab be reimbursed in a manner similar to eculizumab, this is expected to have minimal influence on the budget impact estimate. If ravulizumab is reimbursed according to its Health Canada indication, the budget impact will be greater than estimated by the sponsor and CADTH.
CADTH explored a scenario analysis that increased the total size of the eligible patient population by 5% to explore the impact of reimbursement for a broader population.
The anticipated uptake of ravulizumab is not aligned with clinical expert expectations. In the sponsor’s base case it was assumed that 15% of patients current receiving eculizumab would switch to ravulizumab each year should ravulizumab become available. Based on the number of patients in the BIA, this works out to ravulizumab having a market share of 15% / 28% / 39% in Year 1, 2, and 3, respectively. According to the clinical expert consulted by CADTH for this review, approximately 50% of eligible patients would be expected to uptake ravulizumab upon availability and this was expected to increase to nearly 100% by Year 3. This is echoed by patient feedback received as part of this review, which indicated that they would expect a large proportion of patients to switch to ravulizumab should it be reimbursed by public payers.
In CADTH reanalysis, the proportion of eligible patients who will use ravulizumab in Year 1, 2, and 3 were changed to 50% / 75% / 95%, respectively.
The number of new PNH patients eligible for complement inhibitor therapy was lower than clinical expert expectations. The sponsor estimated the number of new PNH patients who would start complement inhibitor therapy for the baseline year to be |||| based on projections of the number of new PNH patients historically from 2016 to 2020. However, during Years 1, 2, and 3 of the BIA, zero new patients are assumed to start complement inhibitor therapy, despite the patient support program data showing greater than |||| new patients initiating treatment with eculizumab over the past 4 years. Additionally, feedback from the clinical expert consulted by CADTH for this review indicated that while there may be a low incidence of PNH, if patients who are diagnosed meet the reimbursement criteria for complement inhibitor therapy, they will initiate therapy. Finally, the sponsor assumed that 92.6% of naive patients will initiate ravulizumab in the new drug scenario, but clinical expert feedback indicated nearly all new patients would initiate ravulizumab instead of eculizumab should both be available.
In CADTH reanalyses, the number of new patients each year was based on the average number of new patients in the sponsor’s patient support program between 2016 and 2020. Additionally, it was assumed that 97% of new patients would start ravulizumab.
The implementation of patient discontinuation in the model was inappropriate. The sponsor’s analysis assumed that 2.6% of patients currently receiving eculizumab and ravulizumab would discontinue therapy based on their patient support program. When CADTH validated this rate with the clinical expert consulted for this review, discontinuation was deemed to be higher than that observed in clinical practice.
In addition, the sponsor’s implementation of the discontinuation rate led to their being different numbers of eligible PNH patients between the reference and new drug scenario, which is inappropriate and not expected. This is because the number of patients who discontinue is calculated by multiplying the number of current patients receiving eculizumab or ravulizumab by the discontinuation rate and leads to a larger total number of patients discontinuing in the reference rather than the new drug scenario (8.68 vs 7.59, respectively).
To address this, CADTH corrected the sponsor’s base case by setting the sponsor’s discontinuation rate parameter to 0%. This led to the same number of patients in the reference and new drug scenario.
To reflect feedback that a proportion of patients will discontinue, CADTH used the discontinuation rate in the ravulizumab arm from Study 302. To incorporate this, CADTH calculated the total number of patients who would discontinue each year, and weighted that value by the comparator’s respective annual market share to ensure that the number of patients discontinuing was the same in the reference and new drug scenarios.
The dosing for eculizumab is not aligned with the pharmacoeconomic analysis. In the sponsor’s BIA, 90% of patients receiving eculizumab received a 900 mg dose, and 10% were assumed to be up-dosed and receiving the 1,200 mg dose. According to the clinical expert consulted by CADTH for this review, there are proportion of patients receiving eculizumab, however it is uncertain whether a proportion of patients receiving ravulizumab will also require up-dosing to maintain C5 inhibition. As the sponsor’s and CADTH’s pharmacoeconomic report assumes no up-dosing across comparators, to align with this, CADTH has assumed no up-dosing will occur in the BIA.
In CADTH reanalyses, 100% of eculizumab patients receive the 900 mg dose. To explore the impact of eculizumab up-dosing, in a scenario analysis, the sponsor’s assumption of 10% of patients receiving the 1,200 mg dose was explored.
The number of ravulizumab administrations in the first year of treatment was underestimated. To calculate ravulizumab costs for patients who initiate treatment, the sponsor assumed that patients would receive 6.25 of maintenance administrations in the first year of treatment. However, CADTH calculated that patients will receive a loading dose administration, followed by 7 maintenance administrations in the first year of treatment. The following year, patients would receive 6 administrations, followed by 7 administrations in their third year of treatment, which is reflected in the sponsor’s rate of 6.5 annual administrations in subsequent years of therapy.
CADTH’s reanalysis assigned ravulizumab patients to receive 7 maintenance administrations in their first year of treatment to accurately capture the costs of switching from eculizumab to ravulizumab.
The distribution of patient weights in the BIA are uncertain. In the sponsor’s analysis, the distribution of patients across weight dosing categories was based on Canadian PNH registry data.16 However, the clinical expert consulted for this review felt that the proportion of patients greater than 100 kg in his clinical practice was greater than the proportion in the registry. As ravulizumab dosing is weight based and those greater than 100 kg require a higher dose that leads to greater subsequent year annual costs than patients who receive eculizumab, this could add costs to the budget impact of reimbursing ravulizumab.
As a scenario analysis, CADTH explored the impact of assuming10% of the patient population was greater than 100 kg.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by increasing the uptake of ravulizumab, changing the number of new patients eligible for complement inhibitor therapy from 0 to 14, increasing the proportion of new patients who initiate ravulizumab, using the discontinuation rate for ravulizumab observed in Study 302, assuming no up-dosing with eculizumab, and assuming that ravulizumab patients receive 7 maintenance dose administrations in the first year of treatment. Table 14 notes the assumptions used by the sponsor in comparison to those used by CADTH in its reanalysis.
Table 14: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Discontinuation rate for eculizumab and ravulizumab | 2.6% | 0% |
Changes to derive the CADTH base case | ||
1. Ravulizumab uptake | 15% / 28% / 39%b | 50% / 75% / 95% |
2. Number of new patients annually | 0 | 14 |
3. Percentage of naive patients initiating ravulizumab | 92.6% | 97% |
4. Total percentage of PNH patients discontinuing therapy | Not applicable | 1.03% |
5. Distribution of patients across eculizumab dosing | 900 mg: 90% 1,200 mg: 10% | 100% receiving 900 mg dose |
6. Ravulizumab maintenance administrations in year 1 | 6.25 | 7 |
CADTH base case | 1 + 2 + 3 + 4 + 5 + 6 | |
Abbreviations; PNH = paroxysmal nocturnal hemoglobinuria.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses, and so forth) that are not identified as limitations.
bMarket shares for ravulizumab in the sponsor’s analysis were calculated by CADTH since the sponsor programmed a switching rate of 15% to calculate the number of patients receiving ravulizumab.
Applying these changes increased the total 3-year budget impact to $13,180,849. The results of the CADTH step-wise reanalyses are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16.
Table 15: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $1,055,670 |
Corrected base case-making discontinuation rate 0% | -$228,374 |
CADTH reanalysis 1- Uptake 50% / 75% / 95% | -$1,118,379 |
CADTH reanalysis 2 + 3a- 97% of new patients start ravulizumab | -$175,235 |
CADTH reanalysis 4- Discontinuation rate 1.03% | -$117,238 |
CADTH reanalysis 5 - No eculizumab up-dosing | $1,439,790 |
CADTH reanalysis 6 to 7 ravulizumab doses in first year of treatment | $2,477,033 |
CADTH base case | $13,180,849 |
BIA = budget impact analysis.
aStepped reanalyses 2 and 3 were combined because in the sponsor’s base case, there were no new patients, therefore, the proportion of new patients starting ravulizumab resulted in no change to the submitted base case when implemented alone. Only when new patients are added to the BIA does step 3 apply
CADTH also conducted additional scenario analyses to address remaining uncertainty:
Increase the total eligible population size by 5% to account for the proportion of patients who may be eligible for complement inhibitor therapy according to the Health Canada indication for complement inhibitors but not meet jurisdictional reimbursement criteria for eculizumab.
Assuming 10% of eculizumab patients receive the 1,200 mg dose of eculizumab.
Assuming 10% of patients are greater than 100 kg.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $60,971,013 | $59,421,891 | $57,876,922 | $56,372,122 | $173,670,935 |
New drug | $60,971,013 | $60,023,615 | $58,212,109 | $56,490,881 | $174,726,606 | |
Budget impact | $0 | $601,724 | $335,187 | $118,759 | $1,055,670 | |
CADTH base case | Reference | $61,679,516 | $68,449,508 | $75,108,735 | $81,699,372 | $225,257,614 |
New drug | $61,679,516 | $75,405,672 | $78,368,071 | $84,664,720 | $238,438,464 | |
Budget impact | $0 | $6,956,164 | $3,259,336 | $2,965,349 | $13,180,849 | |
CADTH scenario analysis 1: Population size increase by 5% | Reference | $61,679,516 | $71,871,983 | $78,864,172 | $85,784,340 | $236,520,495 |
New drug | $61,679,516 | $79,175,956 | $82,286,475 | $88,897,956 | $250,360,387 | |
Budget impact | $0 | $7,303,973 | $3,422,303 | $3,113,616 | $13,839,892 | |
CADTH scenario analysis 2: Eculizumab up-dosing | Reference | $63,537,611 | $70,479,300 | $77,360,501 | $84,170,826 | $232,010,627 |
New drug | $63,537,611 | $76,420,568 | $78,931,013 | $84,788,293 | $240,139,874 | |
Budget impact | $0 | $5,941,268 | $1,570,512 | $617,467 | $8,129,247 | |
CADTH scenario analysis 3: 10% of patients greater than 100 kg | Reference | $61,679,516 | $68,449,508 | $75,108,735 | $81,699,372 | $225,257,614 |
New drug | $61,679,516 | $75,835,259 | $78,949,741 | $85,429,910 | $240,214,910 | |
Budget impact | $0 | $7,385,751 | $3,841,006 | $3,730,538 | $14,957,295 |
BIA = budget impact analysis.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.