CADTH Reimbursement Review
Estradiol and Progesterone (Bijuva)
Sponsor: Knight Therapeutics Inc.
Therapeutic area: Vasomotor symptoms associated with menopause
Clinical and Pharmacoeconomic Review
Abbreviations
AE
adverse event
ANCOVA
analysis of covariance
AUC0–inf
area under the concentration-time curve to infinity
AUC0–t
area under the concentration-time curve to last quantifiable concentration
BE
bioequivalence
CEE
conjugated equine estrogen
CGI
Clinical Global Impression
CI
confidence interval
Cmax
peak concentration
CVD
cardiovascular disease
E2
estradiol
EE
efficacy evaluable
EE-VMA
efficacy evaluable-vasomotor symptoms
ES
endometrial safety
HDL
high-density lipoprotein
HRQoL
health-related quality of life
HRT
hormone replacement therapy
LDL
low-density lipoprotein
LOCF
last observation carried forward
LSM
least squares mean
MENQOL
Menopause-specific Quality of Life Questionnaire
MID
minimal important difference
mITT
modified intention-to-treat
mITT-VMS
modified intention-to-treat–vasomotor symptoms
MMRM
mixed model of repeated measures
MOS
Medical Outcomes Study
MP
micronized progesterone
MPA
medroxyprogesterone acetate
NIHB
Non-Insured Health Benefits
PK
pharmacokinetic
SD
standard deviation
SE
standard error
TEAE
treatment emergent adverse event
VMS
vasomotor symptoms
VTE
venous thromboembolism
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Estrogen-progestogen (Bijuva) in oral capsules, 0.5 mg estradiol (as estradiol hemihydrate)/100 mg progesterone and 1 mg estradiol (as estradiol hemihydrate)/100 mg progesterone |
Indication | For the treatment of moderate-to-severe vasomotor symptoms associated with menopause in women with an intact uterus |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | September 17, 2020 |
Sponsor | Knight Therapeutics Inc. |
NOC = Notice of Compliance.
Introduction
Vasomotor symptoms (VMS) are the cardinal symptoms of menopause. They consist of profuse heat, sweating, and flushing around the neck, chest, and upper back, and are also commonly known as hot flashes (or hot flushes) and night sweats.1 VMS are experienced by the majority of women during the menopausal transition, with global reports ranging from 60% to 80%; approximately 20% of cases are deemed of severe intensity, with up to 20 to 30 episodes daily.2-5 In Canada, the prevalence of VMS is similar to other western countries, with hot flushes reported at between 68% to 78%, night sweats at 60% to 70%, and sleep disturbances in 67% to 77% of women in the menopausal transition.4,6 However, variation in the prevalence of VMS is reported due to differences in study design, selected populations, sample size, and screening and/or diagnostic tools, and prevalence is also variable within cultures and ethnic groups, based on some reports.7 Black and Hispanic women present with a higher incidence when compared to Asian and White women.4,7 Women with obesity have a higher incidence of VMS. VMS can last an average of 7.4 years.8 For many women, VMS have a significant impact on their quality of life, due to disturbances in mood and sleep quality.7 VMS can be a burden for the health care system due to higher medical care utilization costs, work productivity loss, and total costs.9
Pharmacological and non-pharmacological treatment options are available for clinicians and their patients to relieve VMS during menopause. Non-pharmacological therapies include lifestyle modifications — including smoking cessation and weight management, among others. Pharmacological options include hormone replacement therapy (HRT) targeting a replacement of estrogen levels; it encompasses estrogen therapy alone as well as combined estrogen plus progesterone therapy.10 HRT is considered the most effective option for the treatment of VMS.5,11 In clinical practice, estrogen alone with the lowest effective dose required is used in women without a uterus, while in those with an intact uterus, combinations of estrogens with progestogens are recommended. The addition of progesterone aims to protect the uterus from lining overgrowth of the endometrium that can lead to hyperplasia or carcinoma from unopposed exposure to estrogen alone.10 HRT should be individualized based on symptoms, family history, baseline risk assessment, a patient’s perspectives and preferences, and treatment goals. Current clinical guidelines from Canada recommend titrating doses of HRT, starting with low to standard doses of estrogens and adjusted based on symptoms.5 Symptoms can improve within 2 weeks of treatment but can take up to 6 weeks to show clinical benefits. Periodic re-evaluation is also recommended to assess a time frame of treatment. The duration of treatment varies among patients. Some patients (around 60%) will only require treatment for bothersome symptoms for less than 7 years, but up to 15% of patients will need treatment for up to 15 years or more.12
In Canada, pharmacological options to treat moderate-to-severe VMS include estrogens, either as conjugated equine estrogens (CEEs, with “conjugated” meaning a mixture of several equine estrogens), synthetic conjugated estrogens, 17beta-estradiol, and ethinyl estradiol. Estrogens are available as oral pills, transdermal patches, or vaginal applications.5 Transdermal patches of estrogen have the advantage of not needing to metabolize through the liver — hence, providing a more consistent level of estrogens, a situation that is preferred in some patients — and, based on observational data, have a lower risk of venous thromboembolism (VTE).5,13 Progestogens include micronized progesterone (MP) and synthetic progestogens (also called progestins), such as medroxyprogesterone acetate (MPA), norethindrone acetate, and drospirenone.5,10
The effects of HRT on cardiovascular disease (CVD) may vary, depending on when the therapy is initiated in relation to women’s age and/or time since menopause onset. In relation to progesterone risks, MP has the advantage of a better safety profile in terms of less risk of breast cancer and VTE when compared to synthetic progesterone.10,14-16
The goals of HRT are to achieve the minimal vasomotor symptomatology from hot flashes, improve health-related quality of life (HRQoL), including appropriate sleep patterns and mood, and improve productivity at work while addressing risks from VMS such as cardiovascular events and cancer.7
The objective of this tailored review is to assess the efficacy, safety, and cost related to the use of estradiol (E2)/MP (Bijuva) available as a new capsule combining fixed doses of 17beta-estradiol (estradiol hemihydrate) and MP in 2 presentations for oral administration: 0.5 mg/100 mg and 1 mg/100 mg. Bijuva has a Health Canada–approved indication for treating women with an intact uterus who are experiencing moderate-to-severe VMS during menopause. The estradiol-progesterone combination (Bijuva) has not been previously assessed in the CADTH reimbursement process. The sponsor’s reimbursement request is the same as the Health Canada–authorized indication that was approved in a standard review pathway as a new combination product (Notice of Compliance granted September 17, 2020). Estrogen replacement therapy has been used to reduce the number and intensity of hot flushes associated with menopause. The addition of progesterone for treating VMS opposes the development of endometrial hyperplasia, thought to be caused by estrogens. Bijuva is recommended for use only in patients with an intact uterus since the regimen includes a progestogen to assist in the prevention of endometrial hyperplasia. Currently, only individual formulations of estradiol and progesterone are available in Canada.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical expert(s) consulted by CADTH for the purpose of this review. No input was received from clinician groups for this review.
Patient Input
No patient groups submitted input for this review. However, to provide background on patients’ lived experiences, values, and preferences, and to build an understanding of what it is like to experience moderate or severe hot flushes, patient group websites were sought for original experiences from patients with VMS. Information from the website Healthtalk.org was obtained, assessed, and synthesized by the CADTH review team. Healthtalk.org is a non-profit organization in the UK containing hundreds of real people’s stories collected by academic researchers who interview people in their own homes.17 From this source, the topic of hot flushes and sweats (VMS) was searched and the results obtained were summarized.
Patients interviewed by experts expressed how symptoms of menopause affected their daily lives. Office and workplace rules and interactions are usually affected, as well as family life, as menopause does not occur in isolation. Family dynamics change and balancing work and life with moderate-to-severe symptoms represents an important challenge for patients and their families. Furthermore, concerns about family, relationships, work, and finances add uncertainty and anxiety to the burden and the stress that patients feel around the menopause. Many patients described how, on many occasions, it is impossible to get a good night’s sleep during the menopause, mainly due to hot flushes and sweats. Patients spoke about the “horrendous” effect of hot sweats on their sleep, of sleeping erratically and being awoken up to a “dozen times a night.” Waking up feeling hot 1 minute, cooling down, dozing off to sleep only to be woken up again by a hot sweat can be a vicious sleep-wake-sleep-wake cycle; 1 patient expressed that “you’re working nine to five and you need a good night’s sleep and it [night sweats] certainly did make me feel erratic.” Patients in the interviews described their hot flushes vividly, as a “creeping sensation” that rises from the feet through the whole body. It was an “explosion” in the chest and neck that goes “right up to your brow” — “a thermometer going up and down.”
There was no specific experience described on Healthtalk.org with the estrogen-progesterone (Bijuva) medication. However, patients who had been interviewed talked about their experiences of HRT of any kind, expressing feelings about its risks and benefits, and the concerns about long-term use. In the interviews, experts expressed how a proportion of patients decided not to take HRT due to the media coverage in 2009 and 2010 related to the risks of using HRT, particularly on the increased risk of breast cancer. Other interviewed patients felt like they had no choice but to take HRT. Among those who took HRT, they described the intervention as being “like a miracle,” “completely rejuvenating,” “unfailingly excellent,” and “the most wonderful drug in the whole wide world.” Deciding to take HRT and to stay on it long-term involves a careful weighing of risks versus benefits; patients emphasized the need for a complete shared decision-making process. When discussing the length of treatment with HRT, while some patients were willing to discontinue HRT, others were reluctant to stop taking the medication despite their doctor’s advice. Many patients were concerned with coming off “cold turkey” and returning to the undesirable features associated with stopping their medication. Other patients stated how weaning slowly over a period of time helped them to come off of the medication without any withdrawal symptoms.
Input From Clinical Experts Consulted by CADTH
The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of VMS associated with menopause.
An important unmet need expressed by the clinical expert consulted by CADTH was the lack of combination products available in Canada that can potentially increase adherence and ease of administration. Even when the expert considered that the new combination of estradiol-progesterone would not necessarily shift the treatment paradigm, it could provide a better option for some patients, especially with the convenience of using 1 tablet rather than taking the drug components separately; this may improve adherence and patient satisfaction. Currently, other available options include CEE, estradiol, MPA, and norethindrone. For patients with a higher risk for VTE or stroke, or those with high levels of triglycerides, a transdermal approach to estrogen therapy is preferred.
VMS are usually treated pharmacologically by trialing both hormonal and non-hormonal options. Combined estrogen and progesterone therapies (in 1 or 2 products) are considered by clinicians the most effective treatment options for VMS in women who have an intact uterus. HRT does not modify the underlying disease mechanism for VMS, but it provides symptomatic relief. This improves productivity at work and decreases the burden of disease for VMS and mood symptoms. The clinical expert agreed that treatment goals are mainly to reduce the severity of symptoms and improve quality of life. The clinical expert highlighted the importance of having a range of doses available to titrate appropriately for a patient to both improve symptoms and, again, when titrating down when appropriate (after a period of stabilization). Although estrogen therapy provides the majority of the treatment effect, a progesterone is required to protect the uterus from lining overgrowth that may lead to endometrial hyperplasia or carcinoma.
According to input from the clinical expert consulted by CADTH, the efficacy of Bijuva is expected to be similar to other HRT options in terms of reduction of VMS. Currently, the choice of Bijuva versus other HRT depends on access in terms of drug plan benefits and intolerance of higher doses of progesterone, as well as patient preference. The estradiol-progesterone combination may be more ideal for those patients with a higher risk for VTE or stroke, or those who wish to improve their glycemic profile, high-density lipoprotein (HDL) cholesterol, and low-density lipoprotein (LDL) cholesterol. Many clinicians prefer MP as this component is perceived to have a better safety profile than progesterone. It is always necessary to discuss with patients the possible benefits and risks of using HRT, including Bijuva, especially in those with high cardiovascular risk or diabetes, and in older populations.
The clinical expert considered that a reduction of at least 50% in the frequency and severity of VMS would be a meaningful effect. This includes improvements in sleepiness, work productivity, and mood. The reassessment of patients with VMS should be performed initially after 2 months to 4 months of initiating treatment, then again after 6 months, then every 1 year to 2 years; however, the frequency of assessments may vary among physicians. The factors that physicians should consider when deciding whether to discontinue treatment with estradiol-progesterone include side effects of the medication that can’t be improved by titrating doses, no significant improvement in symptoms despite adequate doses and adherence, and development of other disease process. Discontinuation will also be based on patient preferences and a shared decision process with their physicians.
Drug Program Input
Drug program officials provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The drug programs provided input and/or had questions pertaining to relevant comparators, initiation and discontinuation of therapy, and generalizability. The drug plans indicated that the efficacy measures used in review were the Clinical Global Impression (CGI), the Medical Outcomes Study (MOS) Sleep Scale, and the Menopause-specific Quality of Life Questionnaire (MENQOL), the latter of which has been validated in Canada. However, in Canadian clinical practice, family physicians typically use the Menopause Quick 6. The clinical expert consulted by CADTH indicated that the efficacy measures used in the pivotal study and identified by the drug plans are well known among clinicians and it is unlikely there will be difficulties in applying any of these tools in practice.
The drug plans noted that the inclusion and exclusion criteria and initiation criteria used in the pivotal study are similar to what is used in practice guidelines. This agrees with the input from the clinical expert consulted by CADTH.
No considerations for continuation, renewal, or discontinuation of therapy were identified. Outcomes were reported on symptom improvement and clinical impression. The clinical expert agreed that no specific criteria exist for the continuation or discontinuation of therapy. Discontinuation is usually based on clinical assessment and baseline risks (e.g., CVD risk, stroke, cancer) and a decision to stop therapy should be made on a case-by-case basis.
In terms of generalizability, the drug plans noted that there was a low representation of Asian populations in the pivotal study. Patients with CVD risk factors were excluded, although HRT is not necessarily contraindicated in these patients. The clinical expert consulted by CADTH agreed that although there was a low number Asian patients in the study, Asian ethnicity is not considered a significant modifier of the treatment effect.
Clinical Evidence
Pivotal Studies
Description of Studies
REPLENISH Trial
This CADTH clinical review was based on a summary of evidence provided by the sponsor. This included 1 randomized controlled trial that assessed the efficacy and safety of the combination of E2 and MP (Bijuva) for patients (40 years old to 65 years old) who are experiencing menopause with moderate-to-severe VMS. After screening 5,020 patients, a total of 1,845 patients were eligible for inclusion in the REPLENISH trial. Within this eligible population, patients could be included in 2 substudies based on further clinical eligibility criteria:
The VMS substudy included patients who, at enrolment, had moderate-to-severe VMS (i.e., a minimum daily frequency of ≥ 7 episodes per day [or ≥ 50 per week] of moderate-to-severe hot flushes of VMS). These patients were randomized 1:1:1:1 to 1 of 4 estradiol-progesterone doses (1 mg/100 mg, 0.5 mg/100 mg, 0.5 mg/50 mg, and 0.25 mg/50 mg) or placebo (5 arms in total) and participated in the VMS substudy for the first 12 weeks of treatment.
The non-substudy for VMS included patients who otherwise qualified for the REPLENISH study but did not report the required minimum daily frequency of moderate-to-severe hot flushes. They were randomized to 1 of 4 active treatment arms for 12 months and did not participate in the VMS substudy.
All patients in the REPLENISH trial, including the VMS substudy and the non-substudy patient populations, received blinded investigational product for 12 months.
The VMS substudy population, which provided the data used to support the Health Canada approval of Bijuva, is the focus of this report. This population included 766 patients randomized to 4 active treatment arms or placebo. An overall design and flow description of the REPLENISH study and VMS substudy is presented in Figure 1 in Appendix 1. Meanwhile, the non-substudy population included 1,079 patients randomly assigned to 4 active treatment arms, but not placebo. In this report, the VMS substudy population was used to analyze the co-primary efficacy end points using the data from doses approved in Canada (i.e., the 1 mg/100 mg and the 0.5 mg/100 mg doses). The REPLENISH study provides information about safety end points using the overall safety population and the endometrial safety (ES) population (N = 990 and 675, respectively, for the dosages addressed in this report). The ES population included all randomized patients who had taken at least 1 capsule of study treatment, had no major protocol violations, and had an acceptable biopsy at baseline and at month 12.
The VMS substudy includes the modified intention-to-treat (mITT) VMS population (known as mITT-VMS) with 141 patients in the estradiol-progesterone 1 mg/100 mg arm, 149 patients in the 0.5 mg/100 mg arm, and 135 patients in the placebo arm.
The co-primary efficacy end points evaluated in the VMS substudy included the mean change in frequency and severity of moderate-to-severe VMS from baseline to week 4 and baseline to week 12. Patients recorded hot flush frequency and severity up to week 12 in daily diaries. Hot flush severity was defined as mild, moderate, or severe. Secondary end points included the proportion of patients with a reduction of 50% or more in frequency of moderate-to-severe VMS from baseline at each week up to week 12; the CGI distribution (number and percentage of patients) at week 4, week 8, and week 12, with mean change in the frequency of moderate-to-severe VMS from baseline summarized by different categories of change based on the CGI; and a responder analysis based on responder groups obtained by an anchor based on the CGI. HRQoL was assessed using the change from baseline in MENQOL and the MOS Sleep Scale questionnaire.
Patients in the REPLENISH study were evaluated for safety end points for up to 360 days (double-blind phase). The primary safety end point was the incidence of endometrial hyperplasia with the estrogen-progesterone combination at 12 months. The secondary safety end point was the number of adverse events (AEs). Also, the ES population was evaluated. Although efficacy analyses were performed at 12 weeks, patients in the VMS substudy continued taking medication for 12 months for their potential inclusion in the ES population.
Bioequivalence
Two initial bioequivalence (BE) single-dose pharmacokinetic (PK) studies compared the bioavailability of estradiol-progesterone capsule 2 mg E2/200 mg MP with the same doses of Estrace (estradiol tablets) and Prometrium (progesterone) in healthy, adult, postmenopausal patients. In 1 of these studies, the administration of the study drug was under fasting conditions (Study 351); in the other study, the study drug was administered 30 minutes after the start of a high-fat, high-calorie meal (Study 352). Under fasting conditions (Study 351), the progesterone exposure for the estradiol-progesterone capsule was significantly lower than the reference for all primary PK parameters. However, under high-fat fed conditions (Study 352), all of the primary PK parameters for progesterone, as well as other parameters for estradiol and its metabolites, were higher for the estradiol-progesterone capsules than the reference in most cases.
Due to the intrasubject coefficient of variation being greater than 30% in many cases, a reference-replicated, reference-scaled, BE approach was taken in Study 459 (the main BE submission in this review) under high-fat, high-calorie fed conditions. Results showed that estradiol, estrone (free and total), and progesterone plasma concentrations had BE to the same doses of Estrace and Prometrium under high-fat fed conditions. While the 2 mg estradiol/200 mg progesterone capsule strength is not being proposed for marketing authorization, the capsule fill contains the same ingredients in the same proportions as the 1 mg/100 mg capsule strength and was manufactured using a comparable process. Therefore, the US FDA and Health Canada judged it to be representative of the commercial product. As such, the dosing recommendation included in the estrogen-progesterone product monograph is to take the capsule each evening with food.
REPLENISH Trial: Efficacy Results
For the weekly frequency of VMS episodes, both estradiol-progesterone doses (1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP) significantly reduced the number of moderate-to-severe VMS episodes when compared to placebo (Table 2). At week 4, the mean change (standard deviation [SD]) from baseline in the number of weekly moderate and severe VMS episodes was –40.6 (SD = 30.59) in the 1 mg E2/100 mg MP group, –35.1 (SD = 29.14) in the 0.5 mg E2/100 mg MP group, and –26.4 (SD = 27.05) in the placebo group. The least squares mean (LSM) change (standard error [SE]) versus placebo was statistically significant in both active treatment groups (–12.81 [SE = 3.30] and –8.07 [SE = 3.25] with P < 0.001 and 0.013, respectively). At week 12, the mean change (SD) from baseline was maintained with –55.1 (SD = 31.36), –53.7 (SD = 31.93), and –40.2 (SD = 29.79) fewer weekly VMS episodes, respectively, and the LSM change versus placebo was statistically significant in both active treatment groups at –16.58 (SE = 3.44) and –15.07 (SE = 3.39) in the 1 mg E2/100 mg MP group and the 0.5 mg E2/100 mg MP group, respectively (P < 0.001 for both comparisons).
Similarly, for the severity of VMS, the severity of symptoms decreased from baseline in both active treatment groups. At week 4, the mean change (SD) from baseline in the severity of symptoms was –0.48 (SD = 0.547), –0.51 (SD = 0.563), and –0.34 (SD = 0.386) in the 1 mg E2/100 mg MP group, the 0.5 mg E2/100 mg MP group, and the placebo group, respectively, and the LSM change (SE) versus placebo was statistically significant in both active treatment groups (–0.13 [SE = 0.06] and –0.17 [SE = 0.06] with P = 0.031 and 0.005, respectively). At week 12, the mean change (SD) from baseline was maintained with a reduction in the severity of symptoms of –1.12 (SD = 0.963), –0.90 (SD = 0.783), and –0.56 (SD = 0.603) in the 1 mg E2/100 mg MP group, the 0.5 mg E2/100 mg MP group, and the placebo group, respectively, and the LSM change (SE) versus placebo was statistically significant in both active treatment groups at –0.57 (SE = 0.10) and –0.39 (SE = 0.09) in the 1 mg E2/100 mg MP group and the 0.5 mg E2/100 mg MP group, respectively (P < 0.001 for both comparisons).
A responder was defined as a patient with a reduction of 50% or more from baseline and, separately, 75% or more from baseline in the number of moderate and severe VMS episodes, performed at week 4 and week 12. In both active treatment groups, a statistically significant difference was observed compared to placebo. At week 12, 79.0% of patients and 80.6% of patients in the 1 mg E2/100 mg MP group and the 0.5 mg E2/100 mg MP group, respectively, had a reduction of 50% or more in the number of moderate and severe VMS episodes compared with 58.3% in the placebo group, and 67.7% and 58.1% of patients in the 1 mg E2/100 mg MP group and the 0.5 mg E2/100 mg MP group, respectively, had a reduction of 75% or more compared with 32.2% of patients in the placebo group.
For the CGI analysis at week 12, the percentage of patients who reported “very much improved” or “much improved” was 82.1% and 72.9% in the 1 mg E2/100 mg MP group and the 0.5 mg E2/100 mg MP group, respectively, compared to 53.4% in the placebo group. At all time points, a statistically significant improvement in both active treatment groups was observed compared to placebo. Based on the nonparametric discriminant analysis, the threshold for reporting a meaningful decrease in weekly moderate-to-severe VMS, based on the best discrimination between patients who reported “minimally improved” and those patients who reported much or very much improved, was a decrease of 39 VMS episodes at week 12. Based on this definition, 91 (73.4%) patients, 94 (72.9%) patients, and 60 (52.2%) patients in the 1 mg E2/100 mg MP group, the 0.5 mg E2/100 mg MP group, and the placebo group were responders, respectively (P < 0.001).
At week 12, month 6, and month 12, statistically significant improvements (reductions) in the MENQOL total score were observed for both active treatment groups compared to placebo. For instance, at month 6, the MENQOL score mean change from baseline was –2.0 (1.22), –1.8 (1.22), and –1.6 (1.31) in the 1 mg E2/100 mg MP group, the 0.5 mg E2/100 mg MP group, and the placebo group, respectively (P < 0.001 for both comparisons to placebo).
When evaluating the MOS Sleep Scale score, at month 6 and month 12, statistically significant improvements were noted for both active treatment groups compared to placebo (P < 0.05), except for the 1 mg E2/100 mg MP group at month 12 (P = 0.058).
Harms Results
The safety population (N = 1,835) included randomized patients from the VMS substudy population and the non-substudy population (i.e., the overall study population) who took at least 1 dose of medication. An ES population (all patients randomized to active treatment who completed 12 treatment months and had evaluable baseline and 12-month biopsies) was also assessed for endometrial hyperplasia.
Overall, AEs of any kind were more common in the active treatment arms than in the placebo group, with AEs mostly consisting of headache, breast tenderness, nasopharyngitis, vaginal hemorrhage, vaginal discharge, abdominal pain, and dizziness. Most AEs were of mild to moderate severity. No cases of endometrial hyperplasia were observed during the trial in the 3 treatment groups over 12 months of follow-up, and there were 3 cases of breast cancer, all in the intervention arms and none in the placebo arm. The percentages of other AEs of special interest such as VTE, superficial thromboses, cardiovascular events, cerebrovascular events, syncope, and malignancies were low and these AEs did not occur with greater frequency in the intervention arms (1 mg E2/100 mg MP and 50 mg E2/100 mg MP) when compared to placebo.
Table 2: Summary of Key Results From the Included Study (REPLENISH)
Outcome | 1 mg E2/100 mg MP N = 141 | 0.5 mg E2/100 mg MP N = 149 | Placebo N = 135 |
|---|---|---|---|
Number of weekly moderate and severe VMS episodes, week 12: Co-primary end point | |||
N | 124 | 129 | 115 |
Baseline mean (SD) | 72.2 (25.04) | 72.8 (28.96) | 72.2 (22.66) |
Mean change from baseline (SD) | –55.1 (31.36) | –53.7 (31.93) | –40.2 (29.79) |
LSM change vs. placebo (SE) | –16.58 (3.44) | –15.07 (3.39) | Reference |
P value | < 0.001 | < 0.001 | Reference |
Weekly severity scores of VMS, week 12: Co-primary end point | |||
N | 124 | 129 | 115 |
Baseline mean (SD) | 2.55 (0.235) | 2.51 (0.248) | 2.52 (0.245) |
Mean change from baseline (SD) | –1.12 (0.963) | –0.90 (0.783) | –0.56 (0.603) |
LSM change vs. placebo (SE) | –0.57 (0.100) | –0.39 (0.099) | Reference |
P value | < 0.001 | < 0.001 | Reference |
Reduction of ≥ 50% in frequency of moderate and severe VMS from baseline to week 12 | |||
Total, N | 124 | 129 | 115 |
N (%) | 98 (79.0) | 104 (80.6) | 67 (58.3) |
P value (vs. placebo) | < 0.001 | < 0.001 | Reference |
Clinical Global Impression categories at week 12 | |||
Total, N | 123 | 133 | 116 |
Very much improved/much improved, n (%) | 101 (82.1) | 97 (72.9) | 62 (53.4) |
Minimally improved, n (%) | 17 (13.8) | 29 (21.8) | 26 (22.4) |
No change, n (%) | 5 (4.1) | 7 (5.3) | 28 (24.1) |
P value (vs. placebo) | < 0.001 | < 0.001 | Reference |
MENQOL score | |||
Total, N | 140 | 149 | 135 |
Baseline score, mean (SD) | 4.5 (1.17) | 4.3 (1.25) | 4.6 (1.34) |
N at week 12 | 124 | 135 | 116 |
Change from baseline to week 12, mean (SD) | –1.9 (–1.20) | –1.6 (1.23) | –1.4 (1.36) |
LSM change vs. placebo (SE) | –0.58 (0.145) | –0.34 (0.143) | Reference |
P value | < 0.001 | 0.016 | Reference |
MOS Sleep Scale total score | |||
Total, N | 140 | 148 | 134 |
Baseline score, mean (SD) | 48.0 (19.08) | 44.9 (17.43) | 47.3 (18.87) |
N at week 12 | 122 | 134 | 111 |
Change from baseline to week 12, mean (SD) | –16.7 (16.99) | –13.1 (16.22) | –11.5 (19.67) |
LSM change vs. placebo (SE) | –4.39 (2.059) | –2.54 (2.015) | Reference |
P value | 0.033 | 0.207 | Reference |
AEs (safety population), N | 415 | 424 | 151 |
Patients with at least 1 AE, n (%) | 297 (71.6) | 302 (71.2) | 78 (51.7) |
Patients with at least 1 SAE, n (%) | 9 (2.2) | 15 (3.5) | 2 (1.3) |
Patients with at least 1 SAE leading to discontinuation, n (%) | 45 (10.8) | 31 (7.3) | 10 (6.6) |
AEs of special interest at 12 months | — | — | — |
Breast cancer | 2 (0.5) | 1 (0.2) | 0 |
Breast tenderness | 45 (10.8) | 19 (4.5) | 1 (0.7) |
Coronary artery disease | 1 (0.2) | 0 | 0 |
Vaginal hemorrhage | 14 (3.4) | 10 (2.4) | 1 (0.7) |
Deep vein thrombosis or other VTEs | 0 | 0 | 0 |
AE = adverse event; E2 = 17beta-estradiol; LSM = least squares mean; MENQOL = Menopause-specific Quality of Life Questionnaire; MOS = Medical Outcomes Study; MP = micronized progesterone; SAE = serious adverse event; SD = standard deviation; SE = standard error; VMS = vasomotor symptoms; vs. = versus; VTE = venous thromboembolism.
Source: Clinical Study Report for REPLENISH (2021).18
Critical Appraisal
REPLENISH Study
Overall, prognostic variables were well balanced between the arms of the VMS substudy, with no major limitations in terms of the randomization process, allocation concealment, and outcome assessment. However, missingness of data was present due to the analysis of end points as complete cases available, leading to the possible imprecision of effect estimates and bias for the different end points. The magnitude and direction of this bias, however, are uncertain. The population included in the REPLENISH study was considered generalizable to the Canadian landscape; however, certain patient groups (e.g., those at high risk for CVD, VTE) were not included and generalizability of the study results to these groups is uncertain. Furthermore, longer-term follow-up is desirable to assess the risk of outcomes (harms) such as cancer or cardiovascular events.
Cost Information
The sponsor submitted a cost comparison evaluating the annual cost of estradiol-progesterone tablets compared to the combination of its individual components, as well as to combinations of other available estrogen and progesterone products. CADTH conducted a reanalysis of the sponsor-submitted cost comparison, considering only oral comparator products; a reanalysis of both recommended and daily dosing regimens, with costs based on the price of the least expensive interchangeable components; and a reanalysis of costs of each potential medroxyprogesterone dose separately. At the submitted price, the annual cost of estradiol-progesterone tablets is $327 per patient, which is less expensive than that of its individual components when used daily ($568 to $608 per patient annually) or cyclically ($444 to $551 per patient annually). Estradiol-progesterone tablets are also less expensive than combinations of conjugated estrogen and progesterone ($588 to $694 per patient annually) but more expensive than estradiol or conjugated estrogen plus medroxyprogesterone regimens ($74 to $202 per patient annually). The use of estradiol-progesterone tablets in place of combinations of individual estrogen and progesterone products would be associated with up to 12 fewer dispensing fees per year.
The cost comparison assumes clinical similarity between estradiol-progesterone and the included comparators. Evidence establishing the similarity of estradiol-progesterone to a combination of its individual components was available. However, there was no direct or indirect evidence submitted in comparison with the other comparators of interest and the cost-effectiveness of estradiol-progesterone to combinations of other estrogen and progesterone comparators is unknown.
Conclusions
Evidence from a single randomized placebo-controlled trial, the REPLENISH VMS substudy, showed that in women 40 years old to 65 years old with moderate-to-severe VMS during menopause and no cardiovascular, VTE, or cancer risk factors, E2-MP combination — either at 1 mg E2/100 mg MP or 0.5 mg E2/100 mg MP — improved the frequency and severity of VMS (co-primary end points) at 12 weeks compared to placebo. The improvements observed were considered clinically meaningful as were the results for secondary end points, such as the proportion of patients achieving a reduction of 50% or more and, separately, 75% or more in the frequency of moderate and severe VMS from baseline to week 12; the CGI score; HRQoL; and sleep quality. All of these results favoured treatment with the E2-MP combination. There was uncertainty in the evidence from imprecision of the treatment effect estimates obtained and the risk of bias due to missingness of data (analysis by available cases). One bioequivalence study using a reference-scaled bioequivalence approach demonstrated comparative bioavailability of the fixed-dose combination of estradiol-progesterone (Bijuva) to its individual components.
AEs were more frequent in the estradiol-progesterone combination treatment arms compared to the placebo arm, including breast tenderness, vaginal bleeding, headaches, and dizziness, although most of these AEs were well tolerated. No cases of endometrial hyperplasia were observed. Cardiovascular events, cerebrovascular events, cancer, and thrombosis were present in a small number of patients, with no important differences between intervention and placebo groups. However, longer follow-up is desirable to ascertain possible long-term effects and harms.
At the submitted price, the annual cost of estradiol-progesterone tablets is $327 per patient, which is less expensive than that of its individual components when used daily ($568 to $608 per patient annually) or cyclically ($444 to $551 per patient annually). Estradiol-progesterone tablets are also less expensive than combinations of conjugated estrogen and progesterone ($588 to $694 per patient annually) but more expensive than estradiol or conjugated estrogen plus medroxyprogesterone regimens ($74 to $202 per patient annually). These incremental costs or savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans.
Introduction
Disease Background
VMS are the cardinal, most commonly reported symptoms of menopause. These episodes of profuse heat, sweating, and flushing around the neck, chest, and upper back are also commonly known as hot flashes (or hot flushes) and night sweats.1 VMS are experienced by the majority of women during the menopausal transition, with global prevalence estimates ranging from 60% to 80%; approximately 20% of cases are deemed of severe intensity, with up to 20 to 30 episodes daily.2-5 In Canada, the prevalence is similar to other western countries with hot flushes reported at between 68% to 78%, night sweats at 60% to 70%, and sleep disturbances in 67% to 77% of women in the menopausal transition.4,6 However, differences in prevalence are reported due to study design, selected populations, sample size, and the use of different screening and/or diagnostic tools, and prevalence is also variable between and within cultures and ethnic groups, based on some reports.7 Black and Hispanic women present a higher incidence when compared to Asian and White women.4,7 Women with obesity have a higher incidence of VMS. A higher burden of symptoms has been reported in women of low socioeconomic status and education. VMS can last an average of 7.4 years.8
Several other symptoms can be present during this period, such as headaches, dizziness, rapid and/or irregular heartbeats, atrophic vaginitis, bladder irritability, mood changes, and general malaise.4 For many women, VMS have a significant impact on their quality of life, mainly from disturbances in mood and sleep quality. Furthermore, the menopausal transition represents a critical point in that it marks an increased risk of CVD, diabetes, higher bone turnover, and faster bone loss.7 When left untreated, VMS can be a burden for the health care system due to higher utilization costs, work productivity loss, and total costs.9
The diagnosis of VMS is usually straightforward, identified based on patients reporting symptoms to the primary care practitioner or specialist, who might then perform a baseline risk assessment for conditions such as cardiovascular events, VTE, or breast cancer.
Standards of Therapy
Pharmacological and non-pharmacological treatment options are available for clinicians and their patients to relieve VMS during menopause. Non-pharmacological therapies include lifestyle modifications — including smoking cessation and weight management, among others. Pharmacological options include HRT targeting a replacement of estrogen levels and encompasses estrogen therapy alone as well as combined estrogen plus progesterone therapy.10
HRT is considered the most effective option for the treatment of VMS.5,11 In clinical practice, estrogen alone with the lowest effective dose required is used in women without a uterus, while in women with an intact uterus, combinations of estrogens with progestogens are recommended. The addition of progesterone aims to protect the uterus from lining overgrowth of the endometrium that can lead to hyperplasia or carcinoma from unopposed exposure to estrogen alone.10 One systematic review found that, compared to placebo, estrogen alone or combined with progestogen reduced the weekly frequency of VMS by 75% as well as the severity of symptoms.11
HRT should be individualized based on symptoms, family history, baseline risk assessment, a patient’s perspectives and preferences, and treatment goals. Current clinical guidelines from Canada recommend titrating doses of HRT, starting with low to standard doses of estrogens and making dose adjustments based on symptoms.5 Symptoms can improve within 2 weeks of initiating treatment but can take up to 6 weeks to show benefits. Periodic re-evaluation is also recommended to assess a time frame of treatment. The duration of treatment varies among patients. Some patients (around 60%) will only require treatment for bothersome symptoms for less than 7 years, but up to 15% of patients will need treatment for up to 15 years or more.12
In the Canadian clinical landscape, pharmacological options to treat moderate-to-severe VMS include estrogens, either as CEE, synthetic conjugated estrogens, 17beta-estradiol, and ethinyl estradiol. Estrogens are available as oral pills, transdermal patches, or vaginal applications.5 Transdermal patches have the advantage of bypassing hepatic metabolism, hence providing a more stable level of estrogens, and — based on observational data — have a lower risk of VTE.5,13 Progestogens include MP and synthetic progestogens (also called progestins) such as MPA, norethindrone acetate, and drospirenone.5,10
Recent major clinical guidelines suggest that for healthy, recently menopausal women (meaning less than 10 years since their menopause began), the benefits of HRT (estrogen alone or with a progestogen) outweigh the risks of CVD and all-cause mortality, albeit with an increased risk of VTE — this in patients using oral CEE alone and CEE plus MPA therapy.10 However, the effects of HRT on CVD may vary, depending on when the therapy is initiated in relation to a woman’s age and/or time since menopause onset.
In relation to progesterone risks, observational evidence suggests that MP has the advantage of a better safety profile in terms of less risk of breast cancer and VTE when compared to synthetic progesterone (commonly preferred and used in Canada in combination with estrogens in separate formulations), at least for up to 5 years of treatment.10,14-16 Currently, there is only 1 formulation of MP available in Canada (Prometrium 100 mg), recommended to be taken sequentially at 200 mg daily for the last 14 days of estrogen treatment per cycle. According to the clinical expert consulted by CADTH, when the dose of estradiol or another estrogen product is higher or lower than the average dose that has been decided and indicated by the clinician, there is difficulty in titrating the progesterone dose; also, when higher doses of estrogen are required to target symptoms, higher progesterone dosing might be needed to protect the uterus, which highlights the importance of having lower dose preparations available.
The goals of HRT are to achieve minimal vasomotor symptomatology, improve HRQoL (including appropriate sleep patterns and mood), and improve productivity at work while addressing risks such as cardiovascular events and cancer.7
Drug
Estradiol-progesterone (Bijuva) is available as a fixed-dose combination of 17beta-estradiol (estradiol hemihydrate) and micronized progesterone in 2 capsule presentations for oral administration: 0.5 mg/100 mg and 1 mg/100 mg. It has a Health Canada–approved indication for treating women with an intact uterus who are experiencing moderate-to-severe VMS during menopause. This estradiol-progesterone combination has not been previously assessed by CADTH. The sponsor’s reimbursement request is the same as the Health Canada indication that was approved in a standard review pathway as a new combination product (Notice of Compliance granted September 17, 2020).
The mechanism of action of estradiol-progesterone stems from the deficiency of ovarian 17beta-estradiol production during and after menopause. This deficiency results in unstable thermoregulation, triggering hot flushes associated with sleep disturbance and excessive sweating. After menopause, most endogenous estrogen is produced by the conversion of androstenedione, secreted by the adrenal cortex, to estrone by peripheral tissues. Thus, estrone and the sulphate conjugated form, estrone sulphate, are the most abundant circulating estrogens in postmenopausal women. Estrogen replacement therapy has been used to reduce the number and intensity of hot flushes associated with menopause. The addition of progesterone for treating VMS opposes the development of endometrial hyperplasia thought to be caused by estrogens.
Estradiol-progesterone is recommended for use only in patients with an intact uterus since the regimen includes a progestogen whose role is to assist in the prevention of endometrial hyperplasia. Currently, only individual presentations of estradiol or progesterone are available in Canada.
The characteristics of the estradiol-progesterone combination are depicted in Table 3.
Table 3: Key Characteristics of Estradiol-Progesterone (Bijuva)
Characteristic | Estradiol-progesterone (Bijuva) |
|---|---|
Mechanism of action | Estradiol for replacement of circulating 17beta-estradiol and progesterone to oppose endometrial hyperplasia thought to be provoked by estrogens |
Indicationa | For the treatment of moderate-to-severe VMS associated with menopause in women with an intact uterus |
Route of administration | Oral |
Recommended dosage | 1 single capsule of either 1 mg/100 mg estradiol-progesterone or 0.5 mg/100 mg estradiol-progesterone, orally each evening |
Serious adverse effects or safety issues |
|
Other | Continuous combined HRT is intended for use in women with an intact uterus. Estradiol-progesterone should be used at the lowest effective dose and for a duration consistent with treatment goals and the benefits and risks for the individual woman. Postmenopausal women should be re-evaluated periodically as clinically appropriate to determine if treatment is still necessary. |
HRT = hormone replacement therapy; VMS = vasomotor symptoms.
aHealth Canada–approved indication.
Source: Bijuva product monograph (2021).19
Stakeholder Perspectives
Patient Group Input
No patient group submitted input for this review. However, to provide background on patients’ lived experiences, values, and preferences, and to build understanding of what it is like to experience moderate or severe hot flushes, patient groups’ websites were sought for original experiences from patients with VMS.
Information from the website Healthtalk.org was obtained, assessed, and synthesized by the CADTH review team. Healthtalk.org is a non-profit organization in the UK containing hundreds of real people’s stories collected by academic researchers who interview people in their own homes.17 Healthtalk.org is run by the Dipex Charity, a not-for-profit organization founded in 2001 with the aim of helping people to feel better prepared and informed in their clinical situations and conditions, in partnership with the Health Experiences Research Group at the University of Oxford. Healthtalk.org now has sister sites in more than 10 countries around the world, operating under the umbrella of DIPEx International.
From this source, the topic of hot flushes and sweats (VMS) was evaluated. Patients interviewed expressed the common symptoms of menopause and how these affect patients; they talked about their experiences with hot flushes and sweats, the effects on their life, and what they did to relieve the symptoms. Characterized by sudden feelings of heat that seem to come from nowhere and spread upwards through the body, chest, neck, and face, hot flushes and sweats are caused by changes in hormone levels that affect the body’s temperature control.
Quality of Life, Sleep, and Work With Menopausal Symptoms
Patients interviewed by experts from Healthtalk.org expressed how symptoms of menopause affect their daily lives. For instance, in a public location, women sometimes feel exposed by their inability to conceal the often unpredictable, unpleasant, and highly visible physical symptoms of the menopause, particularly when working with younger staff members, male colleagues, or clients. Office and workplace rules are rarely designed with the menopausal woman in mind. Wearing a compulsory uniform at work could pose difficulties, with 1 patient describing how she risked a “row with her boss” if she took her tie off at work when having a hot flush. Another, sharing an office with 9 people, found opening a window caused resentment.
Family life is often affected as menopause does not occur in isolation. Family dynamics change and balancing work and life with moderate-to-severe symptoms represent an important challenge for patients and their families. Furthermore, concerns about family, relationships, work, and finances add uncertainty and anxiety to the burden and the stress that patients feel around the menopause.
Many patients interviewed described how, on many occasions, it is impossible to get a good night’s sleep during the menopause, mainly due to hot flushes and sweats. Patients spoke about the “horrendous” effect of hot sweats on their sleep, of sleeping erratically and being awoken up to a “dozen times a night.” Waking up feeling hot 1 minute, cooling down, dozing off to sleep only to be woken up again by a hot sweat can be a vicious sleep-wake-sleep-wake cycle; 1 patient expressed that “you’re working nine to five and you need a good night’s sleep and it [night sweats] certainly did make me feel erratic.” Another patient remarked, “I think the biggest problem of all through the menopause was lack of sleep. A combination of night sweats, my own particular emotional turmoil but I was very badly lacking sleep.” Lack of sleep was noted as affecting almost every domain of patients’ lives through the day, often leaving them feeling “comatose,” snappy, and/or tearful, and many considered those symptoms as part of life now.
Hot Flushes and Sweat
Patients in the interviews described their hot flushes vividly. They depicted a “creeping sensation” that rises from the feet through the whole body. It was an “explosion” in the chest and neck that goes “right up to your brow” — “a thermometer going up and down.” One patient compared the warmth she feels to “going under a sun bed,” while another felt as if someone had opened a “little trap door” in her stomach and put a hot coal in. These hot flushes happen many times during the day without warning. At night, the sweats can also become disruptive. Patients describe tossing and turning and feeling hot “like a furnace,” waking up “soaking wet,” and experiencing “awful drenching sweats for about two years.” One patient’s night sweats felt like “a serious infection” that made her temperature “go haywire.” Others talked about searching for “cool parts” in the bed or getting up to change night clothes or bedding.
Experiences With HRT
There was no specific experience described with the estrogen-progesterone (Bijuva) medication on Healthtalk.org. However, patients who had been interviewed talked about their experiences of HRT of any kind, expressing feelings about its risks and benefits, and the concerns about long-term use. In the interviews, clinical experts described how a proportion of patients decided not to take HRT due to the media coverage in 2009 and 2010 related to the risks of using HRT, particularly on the increased risk of breast cancer.
Some patients, on the other hand, expressed feelings that they felt they had no choice but to take HRT. Among those who took HRT, they described the intervention as being “like a miracle,” “completely rejuvenating,” “unfailingly excellent,” and “the most wonderful drug in the whole wide world.” As hot flushes and night sweats eased, they noticed improvements in their sleep, concentration, and stamina. Some of the interviewed patients, while speaking positively about HRT and its effect on their quality of life, said that “at the back of your mind there’s a bit of a worry.” Deciding to take HRT and to stay on it long-term involves a careful weighing of risks versus benefits. Patients mentioned and emphasized the need for a complete shared decision-making process at the beginning of treatment and during the follow-up of symptoms, and how this should include continuous monitoring for cardiovascular risks and cancer screening.
Among the side effects of HRT, interviewed patients spoke of some discomfort such as nausea and diarrhea, facial hair, and weight gain.
When discussing the length of treatment with hormone replacement, some patients were willing to discontinue HRT while others were reluctant to stop taking the medication despite their doctor’s advice. Many patients were concerned with coming off “cold turkey” and returning to the undesirable features associated with stopping their medication. Other patients stated how weaning slowly over a period of time helped them to come off the medication without any withdrawal symptoms.
Clinician Input
Input From Clinical Experts Consulted By CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of VMS during menopause.
Unmet Needs
Patients with VMS during menopause are treated pharmacologically by trialling both hormonal and non-hormonal options. Combined estrogen and progesterone therapies (in 1 or 2 products) are the most effective treatment options for VMS in women who have an intact uterus. Estrogen alone is used in women without a uterus. The clinical expert described the Health Canada–approved bio-identical options (estradiol, MP) as providing the best safety profile, particularly with respect to the progesterone component. Further, the clinical expert consulted by CADTH indicated how MP is usually preferred due to having better safety data for the breast and when considering longer-term use (i.e., more than 3 years to 4 years), as well as for the risk for breast cancer. In regard to the duration of treatment, it is known that some patients will only require treatment for a few years for bothersome symptoms, but many patients may require it for longer than 5 years and even up to 15 years or more.
The clinical expert mentioned that hormonal treatments are not aimed at modifying the underlying disease mechanism for VMS. However, there is evidence for the treatment of perimenopausal depression with estrogen therapies; this acts by modulating the serotonergic pathways. Oral estrogen treatment can also improve LDL cholesterol and HDL cholesterol, which can decrease the risk for CVD.
According to the clinical expert, treatment goals are mainly aimed at reducing the severity of symptoms and improving quality of life. This may enhance productivity at work and decrease the burden of disease for VMS and mood symptoms. HRT may also improve bone health and delay disease progression of osteopenia and osteoporosis. Secondary gains are improved lipid profile and glycemic index, which delays disease progression in those at risk for CVD and diabetes mellitus. The clinical expert highlighted the importance of having a range of doses available to titrate appropriately for a patient to both improve symptoms and, again, when titrating down when appropriate (after a period of stabilization).
Although estrogen therapy provides the majority of the treatment effect for decreasing VMS, a progesterone-progestin is required to protect the uterus from lining overgrowth that may lead to endometrial hyperplasia or carcinoma. The clinical expert remarked that currently there is only 1 dose form of MP available on the Canadian market (Prometrium 100 mg), recommended to be taken sequentially at 200 mg daily for the last 14 days of estrogen treatment per cycle. When the dose of estradiol or another estrogen product is higher or lower than average, there is difficulty titrating the progesterone dose. The expert also shared that some patients are intolerant to the side effects of MP, and that it would be ideal to have access to doses other than 100 mg or 200 mg to alleviate this while protecting the uterus. Furthermore, after a period of stabilization, the patient may wish to decrease the dose or to stop HRT; it is important to have lower dose preparations available for this situation.
Place in Therapy
The clinical expert consulted by CADTH considers that Bijuva would not necessarily shift the treatment paradigm, nor is it the first treatment approved that will address the underlying disease process and symptoms, but it may provide a better option for some patients, especially with the convenience of using 1 tablet rather than taking estradiol and MP separately. This may improve adherence and increase patient satisfaction.
The clinical expert also considered that estradiol-progesterone could be a first-line treatment for VMS and perimenopausal mood symptoms. In addition, it would provide more options in terms of dosing for those who don’t tolerate MP at higher doses. Although estradiol-progesterone would not cause a major shift in the treatment choice paradigm, it would rather provide a better option for some patients with respect to dosing, ease of use, and improved adherence. Cheaper options include CEE, Estrace, MPA, and norethindrone. For patients with a higher risk for VTE or stroke, or those with high levels of triglycerides, a transdermal approach to estrogen therapy is preferred.
Patient Population
From the perspective of the clinical expert, the efficacy response to estradiol-progesterone for improving VMS is expected to be similar to that of other HRT options. Currently, the choice for estradiol-progesterone versus other HRT depends on access in terms of drug benefits and intolerance of higher doses of progesterone, as well as patient preference.
Estradiol-progesterone may be more ideal for those patients with a higher risk of VTE or stroke, or those who wish to improve their glycemic profile, HDL cholesterol, and LDL cholesterol. This is due to the safety profile of the MP component. The clinical expert highlighted that it is important to encourage patient-physician shared decision-making in clinical practice and discuss the possible benefits and risks of using Bijuva in those patients with high cardiovascular risk or diabetes, and in older populations.
The clinical expert considered that a reduction of at least 50% in the frequency and severity of VMS is considered a clinically meaningful treatment effect. This includes improvements in sleepiness, work productivity, and mood.
Assessing Response to Treatment and Discontinuation
The clinical expert recommended that reassessment of patients with VMS should be performed after 2 months to 4 months of initiating treatment, then again after 6 months, then every 1 year to 2 years; however, the frequency of assessments may vary among physicians. The factors that physicians should consider when deciding to discontinue estradiol-progesterone are side effects of the medication that can’t be improved by titrating dose; no significant improvement in symptoms despite adequate doses and adherence; development of other disease process that may increase the risk for CVD, VTE, stroke, or breast cancer; and changes in health that may represent a contraindication (e.g., active breast cancer, VTE).
Following a period of stabilization over several years, patients may wish to discontinue treatment or lower the dose, which would also be based on patient preferences and a shared decision-making process with their physicians. If symptoms do not recur with a lowered dose, then patients may wish to completely stop treatment.
Prescribing Conditions
The clinical expert did not think that specific conditions or clinical settings are required to diagnose and treat VMS, nor is there a need for subspecialist care for management of the condition.
Clinician group input
No input was received from clinician groups for the review of Bijuva.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
No head-to-head trials with standard of care were presented (i.e., Bijuva vs. placebo in all study designs). The drug plans would have liked to see:
| For consideration by CDEC. |
Considerations for initiation of therapy | |
The following efficacy measures were used in the pivotal study: Clinical Global Impression, MENQOL (validated in Canada), and MOS Sleep Scale. In Canadian clinical practice, Canadian family physicians use MQ6.20 | The clinical expert indicated the measures used in the pivotal study are validated, and those mentioned by the drug plans are also well known among Canadian physicians. It is unlikely there would be difficulties in applying any of these tools in practice. |
Eligibility criteria and treatment initiation criteria used in the pivotal study are similar to what is used in Canadian clinical practice:
| The clinical expert agreed that the inclusion and exclusion criteria used in the pivotal study were similar to what would be used in clinical practice. If needed, some patients outside the inclusion or exclusion criteria would require a case-by-case basis decision model. |
No considerations for continuation/renewal or discontinuation of therapy were identified. Prior therapies were not required for eligibility; however, the investigators had protocols to wean off therapies before initiating treatment. Outcomes were reported on symptom improvement and clinical impression. | The clinical expert agreed that no specific criteria exist for continuation or discontinuation of therapy. Discontinuation is usually based on clinical assessment and baseline risks (e.g., CVD risk, stroke, cancer) and decisions should be made on a case-by-case basis. |
Generalizability | |
In terms of generalizability, there was a low representation of Asian patients in the pivotal study. Patients in the study with CVD risk factors were excluded and HRT is not necessarily contraindicated in these patients. | The clinical expert agreed that the study included a low number of Asian patients, but this is unlikely to create any issues in decision-making. Similarly, patients with a high risk for CVD will generally not be considered for HRT. Only a minority of patients with CVD risk factors would be offered HRT and this should be based on an individual decision-making process between the physician and patient that considers benefits against risks of therapy. |
No concerns regarding budget impact assessment. Market share in a world with Bijuva will result in approximately 4.3% of the market share in year 3. Most BIAs of provinces and territories in Canada do not impact HRT therapy space significantly. The 3-year total budget is currently estimated at $92 million, with or without estradiol-progesterone. This should not impact jurisdictional budgets at the submitted price. The submitted cost is $0.8962 for both strengths of estradiol and progesterone at 0.5 mg/100 mg and 1 mg/100 mg. Also, there may be reductions in dispensing fees with the combination product. | For consideration by CDEC. |
BIA = budget impact analysis; CAD = coronary artery disease; CDEC = CADTH Canadian Drug Expert Committee; CVD = cardiovascular disease; CRF = chronic renal failure; FSH = follicle stimulating hormone; HRT = hormone replacement therapy; MENQOL = Menopause-specific Quality of Life Questionnaire; MOS = Medical Outcomes Study; MQ6 = Menopause Quick 6; vs. = versus; VTE = venous thromboembolism.
Sponsor’s Summary of the Clinical Evidence
Note that the clinical evidence summarized in this section was prepared by the sponsor in accordance with the CADTH tailored review process and has not been modified by CADTH.
Pivotal Study
Table 5: Details of Included Studies
Detail | REPLENISH |
|---|---|
Designs & Populations | |
Study Design | Phase 3, multi-center, randomized, double-blind, placebo-controlled trial |
Locations | 117 sites in the United States screened at least one patient; 111 centers randomized at least one patient into either the Vasomotor Symptoms (VMS) Sub-study (104 sites) or into the Non-Sub-study (98 sites) |
Randomized (N) | 1845 (1079 to the Non-Sub-study and 766 to the VMS Sub-study) |
Inclusion Criteria |
|
Exclusion Criteria |
|
(continued) |
|
(continued) |
|
Drugs | |
Intervention a | Combined estradiol 1 mg/progesterone 100 mg (large active, small placebo) Combined estradiol 0.5 mg/progesterone 100 mg (large active, small placebo) Combined estradiol 0.5 mg/progesterone 50 mg* (large placebo small active) Combined estradiol 0.25mg/ progesterone 50 mg* (large placebo, small active) All patients were to self-administer orally two capsules daily (one large, one small) at bedtime with food for 12 months |
Comparator(s) | Placebo (large placebo, small placebo) All patients were to self-administer orally two capsules daily at bedtime with food for 12 months |
Duration | |
Phase | |
Run-in | 60 days |
Double-blind | 360 days |
Follow-up | 15 days |
Outcomes | |
Primary End Point |
|
Secondary and Exploratory End Points | Secondary endpoints:
|
Notes | |
Publications |
|
(continued) |
|
CGI = clinical global impression MENQOL = menopause-specific quality of life questionnaire; MOS-sleep = medical outcomes study – sleep scale; VMS = vasomotor symptoms.
a Groups received the active treatment plus a placebo capsule to ensure adequate blinding.
Source: Clinical Study Report for REPLENISH.
Description of studies
The pivotal REPLENISH trial (N=1845) was a phase 3, multi-center, randomized, double-blind, placebo-controlled, parallel group trial to determine if estradiol (E2)/micronized progesterone (MP) combinations (BIJUVA™) given in a continuous fashion were effective at reducing the frequency and severity of VMS associated with menopause. Additionally, this trial was aimed at identifying an appropriate progesterone dose associated with a low incidence of endometrial hyperplasia.
The effect of BIJUVA on VMS was studied in the VMS Sub-study (N=766) with the specific aim of determining whether BIJUVA given in a continuous fashion is effective at reducing the frequency and severity of moderate to severe VMS associated with menopause when compared with placebo at Weeks 4 and 12. The VMS Sub-study supported the current Health Canada indication for BIJUVA and thus will be the population of interest for this review. During the Screening period, all patients were provided with a diary to self-assess the frequency and severity of their VMS. Patients who experienced a minimum daily frequency of ≥ 7 (or ≥ 50 per week) moderate to severe hot flushes participated in the VMS Sub-study for the first 12 weeks of treatment. Patients were randomized in to one of five treatment arms in a 1:1:1:1:1 allocation ratio [four active arms (Combined 1 mg E2/100 mg MP; Combined 0.5 mg E2/100 mg MP; Combined 0.5 mg/50 mg P; Combined 0.25 mg E2/50 mg MP) and one placebo arm). The combined 0.5 mg E2/ 50mg MP and the combined 0.25 mg E2/ 50 mg MP treatment arms were included in the REPLENISH trial, however data for these groups will not be presented as these doses are not approved in Canada.
The VMS Sub-study patients were stratified by treatment arm within the sites, and only VMS Sub-study patients had the possibility of being randomized to placebo. All patients, including VMS Sub-study participants, received blinded BIJUVA for 12 months. Approximately 1750 patients were planned for randomization into the study across an estimated 120 investigative sites in the United States (750 patients into the VMS Sub-study). A total of 5020 patients were screened for enrollment in this trial: 3175 were screen failures and 1845 were randomized into the trial (1079 to the Non-Sub-study and 766 to the VMS Sub-study). 111 centers randomized at least one patient into either the VMS Sub-study (104 sites) or into the Non-Sub-study (98 sites).
All patients (both VMS Sub-study and Non-Sub-study) completed hot flush diaries and bleeding and spotting diaries through Week 12. Patients in the VMS Sub-study completed Clinical Global Impression (CGI) questionnaires at Weeks 4, 8, and 12. The Menopause-Specific Quality of Life Questionnaire (MENQOL) and the Medical Outcomes Study-Sleep Questionnaire (MOS - Sleep) were administered at Randomization and Week 12.
The total duration of the study was approximately 14.5 months, which included a Screening period of approximately 60 days prior to Randomization, approximately 12 months of treatment, and a 15 day follow up period. As mentioned above, the VMS Sub-study was 12 weeks, but the patients continued the study after that timeframe.
Clinical evaluations were performed at the following time points:
Screening Period: Days −60 to 0
Visit 1 (Randomization): Week 0, Day 1
Visit 2 (Interim): Week 4, Day 28 (± 3 days)
Visit 3 (Interim): Week 8, Day 56 (± 3 days)
Visit 4 (Interim): Week 12, Day 84 (± 3 days)
Visit 5 (Interim): Month 6, Day 180 (± 4 days)
Visit 6 (Interim): Month 9, Day 270 (± 4 days)
Visit 7 (End of Treatment): Month 12, Day 360 (± 4 days)
Telephone Interview Approximately 15 days after last dose
Two different sizes of capsules were necessary to accommodate the different doses; thus, a double-dummy technique was used.
Treatment 1: Combined 1 mg E2/ 100 mg MP [large active; small placebo]
Treatment 2: Combined 0.5 mg E2/ 100 mg MP [large active; small placebo]
Treatment 3: Combined 0.5 mg E2/ 50 mg MP [large placebo; small active]
Treatment 4: Combined 0.25 mg E2/ 50 mg MP [large placebo; small active]
Two sizes of placebo capsules that were an identical match to the active study drug but without the E2/MP was taken orally by patients participating in the VMS Sub-study that were randomized to placebo. To maintain blinding, the study had a double-blind, double-dummy treatment. Patients randomized to active treatment took a placebo capsule matching the alternate capsule size from their active treatment. All patients took one large and one small capsule.
Treatment 5: Placebo [large placebo; small placebo]
Combined E2/MP (BIJUVA) and placebo were packaged in blisters/wallets, labeled, and sent to each site. Packaging was identical to maintain blinding of Investigators. Neither the patient nor the Investigator could identify the treatment from the packaging or label. The study staff, clinical research associates (CRAs), sponsor representatives, and all other study participants were blinded throughout the study as to the regimen the patient was receiving.
Populations
Inclusion and Exclusion Criteria
Patients enrolled in the REPLENISH trial were postmenopausal women between the ages of 40 to 65 with an intact uterus and a screening serum estradiol level of ≤ 50 pg/mL. Specifically, to participate in the VMS Sub-study, the patient must have also reported ≥ 7 moderate to severe hot flushes per day, or ≥ 50 per week, at the baseline assessment during Screening. Patients whose hot flashes were less frequent were still able to participate as Non-Sub-study patients.
Patients were excluded from the REPLENISH trial if they were currently hospitalized, had a history of: thrombosis of deep veins or arteries or a thromboembolic disorder; coronary artery or cerebrovascular disease; chronic liver or kidney dysfunction/disorder; malabsorption disorder; gallbladder dysfunction/disorders (unless gallbladder was removed); diabetes, thyroid disease or any other endocrinological disease; estrogen-dependent neoplasia; atypical ductal hyperplasia of the breast; undiagnosed vaginal bleeding; endometrial hyperplasia, melanoma, or uterine/endometrial, breast or ovarian cancer; other malignancy within the last 5 years, with the exception of basal cell (excluded if within 1 year) or non-invasive squamous cell (excluded if within 1 year) carcinoma of the skin; or has had a uterine ablation. Patients were also excluded if they had clinically significant uterine fibroids at Screening, were pregnant, had insufficient endometrial tissue for diagnosis, or endometrial polyps with atypical nuclei.
Patients were required to have appropriate washout periods for other estrogen alone or estrogen/progestin, selective estrogen receptor modulator (SERM), testosterone, or estrogen/testosterone products, other products expected to alter progesterone or estrogen activity or is being used to treat vasomotor symptoms or any medication known to induce or inhibit CYP3A4 enzyme activity.
For patients in the VMS Sub-study only, patients were excluded if there was use of medication that may have affected the outcome of the VMS endpoints within 28 days prior to Screening (eg, selective serotonin reuptake inhibitors [SSRIs], serotonin and norepinephrine reuptake inhibitors [SNRIs], aldomet, dopaminergic or antidopaminergic drugs, gabapentin, clonidine, or bellergal).
Analysis Populations
All patients who were randomly assigned and had taken at least one capsule of study drug formed the Safety population. Analysis was based on the actual treatment the patient took on Study Day 1. Patients who were found to have participated in the study twice with two separate randomization numbers were included in the adverse events (AEs) and endometrial safety summaries only.
The analysis population for endometrial safety (ES) is the ES population. An ES patient is all randomized patients who: 1) had taken at least one capsule of study treatment as documented (analysis was based on the actual treatment the patient took on Study Day 1); 2) had no major protocol violations; 3) had an acceptable biopsy at Baseline [ie, at least one biopsy with evaluable tissue and no read of endometrial hyperplasia or cancer, or endometrial polyp with either hyperplasia, glandular atypia of any degree (eg, atypical nuclei) or cancer]; 4) had a biopsy at Month 12 (defined as on or after Study Day 326) or had a diagnosis of endometrial hyperplasia prior to Month 12.
Patients who had an endometrial malignancy were not included in the numerator or denominator of the incidence calculation.
The modified intent to treat (mITT)– VMS population was the primary efficacy population. To be included in the mITT-VMS population, patients must have been randomized to the VMS Sub-study, had taken at least one dose (two capsules) of study drug, and: 1) had at least five (5) days of VMS diary data for Baseline measurement of frequency and severity of moderate to severe hot flushes; and 2) had at least four (4) days of VMS diary data for one on-treatment week of reporting of frequency and severity of hot flushes following initiation of study drug.
Analysis was based on the treatment group to which the patient was randomized. Patients who were found to have participated in the study twice with two separate randomization numbers were excluded.
Patients were included in the efficacy evaluable (EE)-VMS population if they were randomized to the VMS Sub-study, had taken at least one dose (two capsules) of study drug, and: 1) had at least seven per day or 50 per week moderate to severe hot flushes at Baseline; 2) had no major protocol violations that could have impacted the VMS endpoint (the Medical Monitor made the final decision on exclusion and the list was provided by the Sponsor prior to unblinding); 3) had at least four (4) days of VMS diary data for one on-treatment week of reporting of frequency and severity of hot flushes following initiation of study drug; and 4) had no dispensing error, defined as a patient who initiated the study with one arm but during the first 12 weeks of treatment inadvertently received an incorrect wallet from another randomization code.
Analysis was based on the actual treatment the patient took on Study Day 1. Patients who were found to have participated in the study twice with two separate randomization numbers were excluded.
Baseline Characteristics
Patients demonstrated similar demographics and gynecological histories across treatment arms for the safety population (Table 6). This is reflective of what is also observed in the mITT-VMS population as seen in Table 7. The average age of the patients was 54.7, 54.5, and 54.5 in the 1 mg E2/ 100 mg MP, 0.5 mg E2/ 100 mg MP, and placebo groups, respectively. Approximately two-thirds of patients were White, and the overall mean BMI ranged from 26.63 to 26.81 kg/m2. Baseline gynecological history was also consistent between the safety and mITT-VMS population and across treatment groups. Baseline values for the patients in the mITT-VMS population for the co-primary and select secondary efficacy endpoints were determined from the seven (7) consecutive days of VMS diary data collected prior to Randomization to study drug. Overall, the Baseline values were similar across treatment groups. Secondary efficacy endpoints included analyses of mild, moderate, and severe moderate VMS. Baseline mean weekly number of mild, moderate, and severe VMS ranged from 83.0 to 86.2, and Baseline mean severity ranged from 2.31 to 2.36 (see Primary Outcome(s) of the Studies for the definition of severity).
Table 6: Summary of Baseline Characteristics for the Safety Population
Characteristics | 1 mg E2/ 100 mg MP (N=415) | 0.5 mg E2/ 100 mg MP (N=424) | Placebo (N=151) |
|---|---|---|---|
Mean age, years (SD) | 54.7 (4.37) | 54.5 (4.52) | 54.5 (4.32) |
Race | |||
White, n (%) | 271 (65.3) | 281 (66.3) | 100 (66.2) |
Black or African American | 134 (32.3) | 136 (32.1) | 46 (30.5) |
Mean weight, kg (SD) | 72.1 (12.32) | 71.7 (13.07) | 71.4 (11.48) |
Mean height, cm (SD) | 163.7 (6.61) | 163.6 (6.95) | 163.5 (6.11) |
Mean BMI, kg/m2 (SD) | 26.81 (4.122) | 26.67 (4.344) | 26.63 (3.870) |
Mean time since last menstrual period, years (SD) | 5.8 (4.86) | 6.0 (5.10) | 6.0 (5.30) |
Bilateral Oophorecomy, n (%) | |||
No | 411 (99.0) | 418 (98.6) | 151 (100.0) |
Yes | 4 (1.0) | 6 (1.4) | 0 |
Parity, n (%) | |||
Nulliparous | 70 (16.9) | 66 (15.6) | 25 (16.6) |
Parous | 345 (83.1) | 358 (84.4) | 126 (83.4) |
Number of pregnancies, n (%) | |||
0 | 36 (8.7) | 38 (9.0) | 15 (9.9) |
≥ 1 | 379 (91.3) | 386 (91.0) | 136 (90.1) |
Number of vaginal births, n (%) | |||
0 | 34 (9.0) | 28 (7.3) | 10 (7.4) |
≥ 1 | 345 (91.0) | 358 (92.7) | 126 (92.6) |
E2 = 17β-estradiol; mITT-VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone; SD = standard deviation
Source: Clinical Study Report of REPLENISH
Table 7: Summary of Baseline Characteristics for the mITT-VMS Population
Characteristics | 1 mg E2/ 100 mg MP (N=141) | 0.5 mg E2/ 100 mg MP (N=149) | Placebo (N=135) |
|---|---|---|---|
Mean age, years (SD) | 54.7 (4.80) | 54.9 (4.45) | 54.3 (4.29) |
Race | |||
White, n (%) | 95 (67.4) | 99 (66.4) | 91 (67.4) |
Black or African American | 45 (31.9) | 48 (32.2) | 41 (30.4) |
Mean weight, kg (SD) | 71.7 (12.47) | 72.7 (13.19) | 71.7 (11.24) |
Mean height, cm (SD) | 164.3 (6.96) | 163.7 (7.49) | 163.8 (6.05) |
Mean BMI, kg/m2 (SD) | 26.45 (3.935) | 27.05 (4.333) | 26.64 (3.817) |
Mean time since last menstrual period, years (SD) | 6.1 (5.53) | 6.5 (5.43) | 5.7 (4.92) |
Bilateral Oophorecomy, n (%) | |||
No | 138 (97.9) | 146 (98.0) | 135 (100.0) |
Yes | 3 (2.1) | 3 (2.0) | 0 |
Parity, n (%) | |||
Nulliparous | 21 (14.9) | 25 (16.8) | 24 (17.8) |
Parous | 120 (85.1) | 124 (83.2) | 111 (82.2) |
Number of pregnancies, n (%) | |||
0 | 8 (5.7) | 16 (10.7) | 15 (11.1) |
≥ 1 | 133 (94.3) | 133 (89.3) | 120 (88.9) |
Number of vaginal births, n (%) | |||
0 | 13 (9.8) | 9 (6.8) | 9 (7.5) |
≥ 1 | 120 (90.2) | 124 (93.2) | 111 (92.5) |
Baseline Values for Co-Primary and Selected Secondary Endpoints | |||
Mean (SD) weekly number of moderate to severe VMS | 74.4 (35.26) | 72.1 (27.76) | 72.4 (23.26) |
Mean (SD) weekly severity score of moderate to severe VMS | 2.54 (0.320) | 2.51 (0.249) | 2.52 (0.246) |
Mean (SD) weekly number of mild, moderate, and severe VMS | 86.2 (40.61) | 85.1 (33.92) | 83.0 (26.47) |
Mean (SD) weekly severity of mild, moderate, and severe VMS | 2.36 (0.337) | 2.31 (0.333) | 2.34 (0.325) |
E2 = 17β-estradiol; mITT-VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone; SD = standard deviation; VMS = vasomotor symptoms
Source: Clinical Study Report of REPLENISH
Interventions
In the REPLENISH trial, the patients were to receive either 1 mg E2/100 mg MP or 0.5 mg E2/100 mg MP. Only those patients included in the VMS Sub-study could be randomized to the placebo group. Two different sizes of capsules were necessary to accommodate the different doses, including the lower doses of P that, as mentioned, are not approved for use in Canada. All patients were to self-administer orally two capsules daily at bedtime with food for 12 months. Each patient was dispensed enough study drug to last until the next scheduled visit, with allowance for visit windows. The patients were instructed to return the used and unused containers of study drug in the original packaging to the study site at Visits 2, 3, 4, 5, 6, and 7. Sites were to verify and document compliance based on counts of dispensed / returned study drug and any additional information reported by the patients (eg, lost capsules).
Treatments were recorded, including the drug or treatment name, start and stop dates, and indication of use. Patients were not to use estrogen, SERMs, progestin, or progesterone, other than study drug, in the specified timeframes prior to Screening nor during the study. The use of any medication known to induce or inhibit CYP3A4 enzyme activity that may also affect estrogen/progestin drug metabolism was prohibited within 28 days prior to Randomization (Visit 1) and throughout the study. The use of any medication, herbal products, or nutritional supplements known or suspected to interact with hormone therapy was prohibited within 28 days prior to Screening and throughout the study. Testosterone was prohibited within 8 weeks prior to Screening and during the study. For patients in the VMS Sub-study only, use of medication that may affect the outcome of the VMS endpoints within 28 days prior to Screening and during participation in the VMS Sub-study (through the first 12 weeks of treatment) were prohibited (eg, SSRIs, SNRIs, aldomet, dopaminergic or anti-dopaminergic drugs, gabapentin, clonidine, or bellergal). Patients were to report all concomitant medications, including OTC products and herbal or nutritional supplements/medications. Patients were instructed to report any changes in concomitant medications and were to be questioned by site personnel regarding concomitant medications at each visit and, when appropriate, if contacted between visits.
Patients were removed from the trial if any of the following circumstances occurred: withdrawal of consent; condition worsened to the degree that the Investigator felt it was unsafe for the patient to continue in the study; it was difficult/impossible to obtain laboratory samples; patient’s drug code was unblinded; an AE occurred for which the patient desired to discontinue treatment or the Investigator determined that it was in the patient’s best interest to be discontinued; significant protocol deviation/violation or a trend in deviations/violations; a concomitant therapy was reported or required which was likely to interfere with the results of the study or compromise patient safety; patient was lost to follow-up; patient became pregnant; or for administrative reasons.
Outcomes
The co-primary efficacy endpoints in the VMS Sub-study of the REPLENISH trial were: the mean change in frequency of moderate to severe VMS from Baseline to Week 4; frequency of moderate to severe VMS from Baseline to Week 12; severity of moderate to severe VMS from Baseline to Week 4; and severity of moderate to severe VMS from Baseline to Week 12.
The secondary endpoints in the VMS Sub-study were: mean change in frequency of moderate to severe VMS from Baseline to each week up to Week 12; mean change in severity of moderate to severe VMS from Baseline to each week up to Week 12; mean change in frequency of mild, moderate and severe VMS from Baseline to each week up to Week 12; mean change in severity of mild, moderate and severe VMS from Baseline to each week up to Week 12; Percentage of patients with 50% and, separately, 75% reduction in frequency of moderate to severe VMS from Baseline at each week up to Week 12; Percentage of patients with 50% and, separately, 75% reduction in frequency of mild, moderate and severe VMS from Baseline; CGI distribution (number and percentage of patients) at Week 4, Week 8, and Week 12, with mean change in the frequency of moderate to severe VMS from Baseline summarized within each CGI category at Weeks 4, 8, and 12 (Gerlinger method); Change from Baseline in menopause-specific quality of life questionnaire (MENQOL) evaluation parameters; and Change from Baseline in medical outcomes study (MOS) - Sleep evaluation parameters.
After completion of the initial Screening procedures, all patients who were eligible to continue Screening were provided with a Hot Flush diary that was to be completed for the remainder of the Screening. Patients were instructed to complete the diary daily by recording the number and severity of vasomotor symptoms (hot flushes) in their diaries. The severity of VMS was defined as: mild (sensation of heat without sweating); moderate (sensation of heat with sweating, able to continue activity); severe (sensation of heat with sweating, causing cessation of activity). All patients (both VMS Sub-study and Non-Sub-study) completed hot flush diaries through Week 12; however, the primary efficacy analysis was only conducted on patients in the VMS Sub-study. Patients were instructed to return their diary at each study visit (Visits 2 [Week 4], 3 [Week 8], and 4 [Week 12]).
At Weeks 4, 8, and 12, patients in the VMS Sub-study were asked to provide a CGI which is described below. Patients were instructed to answer the following question: “Rate the total improvement, whether or not in your judgment it is due entirely to drug treatment. Compared to your condition at admission to the study, how much has it changed?”
Patients were asked to answer the above question using the following options:
Very much improved
Much improved
Minimally improved
No change
Minimally worse
Much worse
Very much worse
Menopause-specific quality of life changes in study participants were assessed utilizing the MENQOL questionnaire. ((Hilditch 1996)) The MENQOL questionnaire is self-administered and assesses changes in quality of life over a one-month period. It is composed of 29 questions distributed across four domains: vasomotor, psychosocial, physical, and sexual. The MENQOL was administered at Visits 1 (Randomization), 4 (Week 12), 5 (Month 6), and 7 (Month 12) or Early Termination. The MENQOL questionnaire assessed changes in quality of life of study patients over a one month period. It was self-administered and was measured at Baseline, Week 12, Month 6 and Month 12 during the trial. It is composed of 29 questions distributed across four domains: vasomotor, psychosocial, physical and sexual. Change from Baseline in monthly scores were summarized and described within each treatment group for the mITT-VMS population.
The scoring algorithm was as follows:
Each domain was scored separately. The scale contained four domains:
Vasomotor- Items 1, 2, & 3
Psychosocial- Items 4-10
Physical- Items 11-26
Sexual- Items 27-29
For analyses, the original scores were converted to the analysis score ranging from 1 to 8 in the following manner: patient response can be one of eight from No to 6, where no corresponds to an analysis score of 1 and 6 corresponds to an analysis score of 8.
Since the domain subscales are not comprised of an equivalent number of items, the mean of the subscale was used as the overall subscale score. Each domain score ranged from 1 to 8
The MOS - Sleep questionnaire (Hays and Stewart, 1992) was utilized to assess changes in sleep for study participants. The questionnaire has 12 items that measure six dimensions of sleep over the past four weeks. It is self-administered and was provided to patients at Visits 1 (Randomization), 4 (Week 12), 5 (Month 6), and 7 (Month 12) or Early Termination.
Statistical analysis
All efficacy analyses were performed on the mITT-VMS and EE-VMS populations. The primary population was the MITT-VMS population and the secondary population for all efficacy analyses was the more restrictive EE-VMS population.
Continuous data were summarized with the following descriptive statistics: number of observations (n), mean, standard deviation (SD), median, minimum, and maximum. Categorical and ordinal data were summarized with frequencies (number of patients in category) and percentages. Percentages were computed using the number of patients with available data as the denominator, except for AEs, for which the denominator was the number of patients in each dose cohort, across all dose cohorts and for all patients in the Safety Population.
All attempts were made to prevent any missing values. Missing or invalid data was treated as missing and was not imputed for the primary analysis of the primary endpoint because the mixed model for repeated measures (MMRM) analysis was valid under the missing at random assumption. For the sensitivity analysis using analysis of covariance (ANCOVA), missing weekly data was imputed using last observation carried forward (LOCF).
Four doses of combination E2/MP (BIJUVA) were compared to placebo. Within each dose/placebo comparison, there were four co-primary endpoints: 1) mean change in frequency of moderate to severe VMS from Baseline to Week 4; 2) mean change in frequency of moderate to severe VMS from Baseline to Week 12; 3) mean change in severity of moderate to severe VMS from Baseline to Week 4; and 4) mean change in severity of moderate to severe VMS from Baseline to Week 12.
To account for the multiple comparisons of testing placebo to each of the four active doses of E2/MP and the multiple testing of the four co-primary endpoints, a gatekeeping testing procedure was followed. The testing started by examining the highest dose (combined estradiol 1 mg / progesterone 100 mg formulation) for the co-primary endpoints. If the four p-values for the co-primaries were significant (p ≤ 0.05) then the hypothesis testing continued to the next dose (combined estradiol 0.5 mg / progesterone 100 mg formulation) for each of the co-primaries, as described above. If at any point the hypothesis testing yielded a non-significant result, the testing was stopped. The gatekeeping procedure described was also followed for all secondary efficacy endpoint comparisons of each active treatment group with placebo. This maintained the consistency of approach for preservation of the familywise Type I error rate for each endpoint evaluation.
Primary Outcome(s) of the Studies
The most recent seven consecutive days of data prior to Randomization was used to determine the Baseline frequency and severity of hot flushes for each patient. The number of moderate to severe hot flushes from these seven days was also used to determine eligibility for the VMS Sub-study.
The weekly frequency of moderate to severe hot flushes was calculated from the daily diary records using a forward counting process of 7-day intervals beginning with the Baseline date. Diary data extending beyond 12 weeks (84 days) was excluded from this calculation. The weekly number of moderate to severe hot flushes for each assessment week (Baseline, and Weeks 1 through 12) was derived as:
Weekly Frequency = total number of moderate and severe hot flushes for the patient week
The weekly severity of hot flushes for the change in severity of moderate to severe vasomotor symptoms was derived as:
Baseline Weekly Severity Score = (number of moderate hot flushes for 7 days) x 2 + (number of severe hot flushes for 7 days) x 3] / (total number of moderate to severe hot flushes over 7 days).
On Treatment Weekly Severity Score = [(number of mild hot flushes for 7 days) x 1 + (number of moderate hot flushes for 7 days) x 2 + (number of severe hot flushes for 7 days) x 3] / (total number of mild, moderate and severe hot flushes over 7 days).
Absolute changes from baseline and respective differences from placebo in frequency and severity of VMS was listed and summarized. Means, SDs, minimum (MIN) and maximum (MAX) are provided for the co-primary efficacy endpoints.
A mixed model for repeated measures (MMRM) analysis was applied to the 12 weekly change scores. The model included Baseline as covariate, treatment, study week, and treatment-by-study week interaction as fixed factors, and patient as the repeated measure unit. Study week pertained to the 12-individual weekly hot flushes frequency derivations. The variance-covariance matrix of the change scores over time was assumed to be unstructured. If the computation did not converge, the covariance structure was reduced from, in the order of, “unstructured (UN)”, “Toeplity (TOEP)”, “autoregressive order 1 [AR(1)]” to “compound symmetry (CS)”.
Within each dose level/placebo comparison, there were four co-primary efficacy endpoints. The four co-primary endpoints were each tested at level alpha (0.05, two-tailed).
Ninety-five percent (95%), two-sided confidence intervals (CIs) were derived for least square (LS) mean changes from Baseline and respective differences from placebo for each dose and week. The gatekeeping procedure for the primary efficacy endpoints already described was used in the interpretation of p values and the confidence intervals.
In addition to the principal MMRM analysis of the four co-primary endpoints, a sensitivity evaluation was also conducted using an ANCOVA; SAS generalized linear model utilizing LOCF. For patients who discontinued the study prior to Week 12 or who had missing data at Week 4 or 12, the last observed weekly hot flush frequency or severity value was carried forward to all visits through Week 12. Patients who had no post-Baseline data were not included in the analysis (ie, there was no baseline observation carried forward application). The sensitivity evaluation was specifically designed to provide support for the MMRM; the primary MMRM approach was considered to have the most power for statistical inferences and was the principal a priori analysis method.
Power Calculation
The sample size for the VMS endpoint was based on the change in frequency and severity of hot flushes between the active treatment groups and placebo as outlined below. All attempts were made to prevent any missing values. Each of the four active treatment groups and the four co-primary outcomes was compared to the placebo group in a hierarchical order to preserve the test level of significance for each comparison at 5% (two-sided). Although a MMRM model was used for the final analysis, a two-group t-test was used to estimate sample size requirements for the VMS Sub-study.
Change in frequency: The mean change from Baseline in weekly frequency of moderate to severe hot flushes was assumed to be at least −56 for any given active treatment group and −35 for the placebo group at both Weeks 4 and 12. A common, between patient standard deviation of 35 across treatment groups and weeks was further assumed (Effect size = 60%).
Change in severity: The mean change in the severity score from Baseline for mild, moderate and severe hot flushes was assumed to be at least −0.7 for any given active treatment group and −0.4 for the placebo group at both Weeks 4 and 12. A common, between patient standard deviation of 0.6 across treatment groups and weeks was further assumed (Effect size = 50%).
Enrolling 150 patients in each treatment group provided at least 90% power to test the primary VMS hypotheses among all randomized patients in the mITT–VMS Population.
Secondary Outcomes of the Studies
Similar to the continuous co-primary endpoints for Weeks 4 and 12, the same MMRM model was applied to the changes in frequency and severity of mild, moderate and severe vasomotor symptoms for each assessment week up to Week 12. The calculation for frequency and severity of hot flushes remained the same, with the exception that hot flushes of all severities was included.
Responders were defined as the percent of patients with 50% and, separately, 75% reduction from Baseline in moderate to severe VMS at Week 12 compared between active and placebo treatments. These proportions were calculated and presented graphically. Simple comparisons of proportions using the Fisher’s exact test were made for each active treatment group compared to placebo. The gatekeeping approach for the primary efficacy endpoints previously described was employed for the formulation of inferences concerning each comparison.
The number and percentage of patients for each category of the CGI was summarized at Week 4, Week 8, and Week 12, with mean change in the frequency of moderate to severe VMS from Baseline summarized within each CGI category at Weeks 4, 8, and 12. ((Gerlinger, 2012; Revicki, 2008)) Descriptive analyses were conducted to show the mean changes in frequency of moderate to severe VMS at 12 weeks by different categories of change based on the CGI. The analysis focused on Baseline to Week 12 changes for estimating minimal important differences and responder groups. The minimal important difference was defined base on CGI ratings of ‘minimally improved’ category, and clinically meaningful responders were defined based on CGI ratings of ‘much improved’ or ‘very much improved’ combined. The worsen/no change group was defined as consisting of those women reporting CGI ratings of ‘no change’ to ‘very much worse’. Based on these CGI response groupings, a three-categorical variable was constructed, and a nonparametric discriminate analysis was conducted utilizing bootstrapping methods.
Change from Baseline for each of the four domains and overall scores of MENQOL were analyzed using ANCOVA with treatment group and region as factors, and Baseline score as covariate for the mITT-VMS.
The MOS - Sleep self-report questionnaire is composed of 12 items that measure six dimensions of sleep over the past four weeks. It was self-administered and was measured at Baseline, Week 12, Month 6, and Month 12 during the trial. Change in scores over the past four weeks (total and subscales) were summarized within each treatment group for the MITT-VMS population and MITT populations separately.
Most questions were scored with one of six numbers ranging from 1 (all of the time) to 6 (none of the time), indicating the frequency of various aspects of the disease-related sleep disruption over the preceding week. Patients also estimated the average amount of sleep per night during the past week. The SLP-9 scoring method was applied in the order as follows ((Spritzer, 2003)):
Answers to all questions, with the exception for Q1, Q2, Q4, and Q12, was reversed and rescaled to 0 to 100 such that 0 meant “best possible” and 100 meant “worst possible”
MOS_n_new ß (6-MOS_n_old) x 20, for n=3, 5, 6, 7, 8, and 9
Answers to Questions 1, 4, and 12 were rescaled as follows
MOS_1_new ß (MOS_1_old - 1) x 25
MOS_4_new ß (MOS_4_old - 1) x 20
MOS_12_new ß (MOS_12_old - 1) x 20
The SLP-9 total score was the average score of Q1, Q3, Q4, Q5, Q6, Q7, Q8, Q9, and Q12. If any of the individual questions used to obtain this total score was missing, the SLP-9 total score was set to a missing value. Additionally, items within each scale were averaged together to create the seven scale scores (sleep disturbance, snoring, sleep short of breath or headache, sleep adequacy, sleep somnolence, sleep problems index I, and sleep problems index II) as described in Spritzer 2003. ((Spritzer 2003)) Two additional measures based on the average number of hours sleep each night during the past 4 weeks was also determined.
Sponsor’s Summary of the Results
Patient Disposition
Across all treatment groups, a total of 5020 patients were screened for enrollment in this trial: 3175 were screen failures and 1845 were randomized into the trial (1079 to the Non-Sub-study and 766 to the VMS Sub-study). The most common primary reasons for screen failure included: inclusion criteria not met; exclusion criteria met; withdrew consent during screening; lost to follow-up; Investigator/Sponsor decision; and other.
The 1845 patients were randomized into five treatment groups: 1 mg E2/100 mg MP (N=415); 0.5 mg E2/100 mg MP (N=424); 0.5 mg/50 mg MP (N=421); 0.25 mg E2/50 mg MP (N=424); placebo (N=151). Data for the 0.5 mg E2/ 50mg MP and 0.25 mg E2/ 50 mg MP treatment arms are not presented as these doses are not approved in Canada.
In the 1 mg E2/100 mg MP, 0.5 mg E2/100 mg MP and placebo groups, 31.6%, 28.1%, and 38.4% of patients discontinued the study prior to the 12-month completion.
Of note, during Sponsor trial data review before database lock and unblinding, it was found that two (2) patients were screened and randomized at two separate sites but were confirmed to be the same patient. Both patients were removed from efficacy analyses and counted once in the Safety population; however, adverse events and endometrial biopsy results (if applicable) collected from the second randomization were included in the full safety profile of the first randomization.
Of the 766 patients randomized to the VMS Sub-study, 726 (94.8%) patients met the criteria to be included in the mITT-VMS population; 141 in the 1mg E2/100 mg MP group, 149 in the 0.5 mg E2/100 mg MP group and 135 in the placebo group. The most common reason patients were excluded was no post-Baseline VMS diary data. Overall, most patients completed the 52-week study. The most common reasons for discontinuation for the 52-weeks were: adverse event, lost to follow-up, patient withdrew consent, protocol deviation, lack of efficacy, other, and Investigator/Sponsor decision.
Table 8: Patient Disposition for the REPLENISH trial
Disposition | REPLENISH | ||
|---|---|---|---|
1 mg E2/ 100 mg MP | 0.5 mg E2/ 100 mg MP | Placebo | |
Screened, N | 5020a | ||
Randomized, N | 1845a | ||
Safety, N | 415 | 424 | 151 |
Discontinued, N (%) | 131 (31.6) | 119 (28.1) | 58 (38.4) |
Reason for discontinuation, N (%) | |||
Adverse events | 46 (11.1) | 33 (7.8) | 10 (6.6) |
Lost to follow-up | 27 (6.5) | 30 (7.1) | 17 (11.3) |
Patient withdrew consent | 36 (8.7) | 42 (9.9) | 13 (8.6) |
Protocol Deviation | 15 (3.6) | 6 (1.4) | 6 (4.0) |
Lack of efficacy | 5 (1.2) | 4 (0.9) | 12 (7.9) |
Investigator/sponsor decision | 1 (0.2) | 3 (0.7) | 0 |
Other | 1 (0.2) | 1 (0.2) | 0 |
Randomized to the VMS Sub-study | 766a | ||
mITT-VMS, N | 141 | 149 | 135 |
Discontinued, N (%) | 43 (30.5) | 31 (20.8) | 42 (31.1) |
Reason for discontinuation, N (%) | |||
Adverse events | 19 (13.5) | 5 (3.4) | 9 (6.7) |
Lost to follow up | 11 (7.8) | 7 (4.7) | 7 (5.2) |
Protocol deviation | 7 (5.0) | 3 (2.0) | 5 (3.7) |
Patient withdrew consent | 4 (2.8) | 15 (10.1) | 9 (6.7) |
Lack of efficacy | 2 (1.4) | 0 | 12 (8.9) |
Other | 0 | 1 (0.7) | 0 |
EE-VMS, N | 120 | 127 | 108 |
E2 = 17β-estradiol; EE = efficacy evaluable; mITT = modified intent to treat; MP = micronized progesterone; VMS = vasomotor symptoms
aIncludes patients screened and randomized for the 1 mg E2/100 mg MP, 0.5 mg E2/100 mg MP, 0.5 mg E2/50 mg MP and 0.25 mg/50 mg MP groups
Source: Clinical Study Report for REPLENISH
Exposure to study treatments
Study Treatments
In the safety population, the mean estradiol and progesterone exposures per group were consistent with the dosing regimens for that group. The mean durations of treatment were 259 days for the placebo group, 281 days for the 1 mg E2/ 100 mg MP group and 290 days in the 0.5 mg/ 100 mg MP group. Approximately 70% of patients in the active treatment groups and 62% in placebo had a duration of treatment of ≥ 326 days.
Concomitant Medications
During the 52-week course of the study, 89.4%, 91.5%, and 82.1% in the 1 mg E2/100 mg MP, 0.5 mg E2/100 mg MP and placebo group respectively, received a concomitant medication. The medications used were for other conditions and there were no imbalances with any individual medication use between treatments groups.
Efficacy
Frequency and Severity of Moderate to Severe VMS at Weeks 4 and 12
At Week 4, all treatment arms demonstrated a statistically significant reduction in the number of moderate and severe VMS compared to placebo. The mean change from Baseline for the active treatment groups ranged from −40.6 (1 mg E2/100 mg MP) to −35.1 (0.5 mg E2/100 mg MP) compared to −26.4 for placebo (Table 9). LS mean change from placebo for each treatment arm was: −12.81 for 1 mg E2/100 mg MP and −8.07 for 0.5 mg E2/100 mg MP.
By Week 12, all doses were statistically significantly different from placebo in reducing the number of moderate to severe VMS (p ≤ 0.002). The mean change from Baseline for the active treatment groups ranged from −55.1 (1 mg E2/100 mg MP) to −53.7 (0.5 mg E2/100 mg MP) compared to –40.2 for placebo. LS mean change from placebo for each treatment arm was: −16.58 for 1 mg E2/100 mg MP and −15.07 for 0.5 mg E2/100 mg MP.
At Week 4, both treatment arms demonstrated a statistically significant reduction in the severity of VMS compared to placebo (p = 0.031 and p = 0.005, respectively). The mean change from Baseline for the active treatment arms ranged from −0.51 (0.5 mg E2/100 mg MP) to −0.48 (1 mg E2/100 mg MP) compared to −0.34 for placebo. The LS mean change from placebo was: −0.13 for 1 mg E2/100 mg MP and −0.17 for 0.5 mg E2/100 mg MP (Table 9).
At Week 12, both doses remained statistically significantly different from placebo in reducing the severity of moderate to severe VMS. The mean change from Baseline ranged from −1.12 (1 mg E2/100 mg MP) to −0.90 (0.5 mg E2/100 mg MP) compared to −0.56 for placebo. The LS mean change from placebo was: −0.57 for 1 mg E2/100 mg MP, −0.39 for 0.5 mg E2/100 mg MP (Table 9).
The mean reduction in severity of moderate to severe VMS was statistically significantly different from placebo by Week 3 for both doses, 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP. For the overall 12 weeks, the mean change in severity was statistically significantly different from placebo for both doses.
Table 9: Change From Baseline and Placebo in the Mean Number of Weekly Moderate and Severe VMS at Week 4 and Week 12 (mITT-VMS population)
Drug or comparator | Total N | Baseline Mean (SD) | Mean change from baseline (SD) | LS Mean change from placebo (SE) | MMRM P value |
|---|---|---|---|---|---|
Number of weekly moderate and severe VMS, week 4 | |||||
1 mg E2/100 mg MP | 134 | 72.1 (27.80) | -40.6 (30.59) | -12.81 (3.30) | <0.001 |
0.5 mg E2/100 mg MP | 144 | 72.3 (28.06) | -35.1 (29.14) | -8.07 (3.25) | 0.013 |
Placebo | 126 | 72.3 (23.44) | -26.4 (27.05) | — | — |
Number of weekly moderate and severe VMS, week 12 | |||||
1 mg E2/100 mg MP | 124 | 72.2 (25.04) | -55.1 (31.36) | -16.58 (3.44) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 72.8 (28.96) | -53.7 (31.93) | -15.07 (3.39) | <0.001 |
Placebo | 115 | 72.2 (22.66) | -40.2 (29.79) | — | — |
Weekly severity scores of VMS, week 4 | |||||
1 mg E2/100 mg MP | 134 | 2.54 (0.325) | -0.48 (0.547) | -0.13 (0.061) | 0.031 |
0.5 mg E2/100 mg MP | 144 | 2.51 (0.248) | -0.51 (0.563) | -0.17 (0.060) | 0.005 |
Placebo | 126 | 2.52 (0.249) | -0.34 (0.386) | — | — |
Weekly severity scores of VMS, week 12 | |||||
1 mg E2/100 mg MP | 124 | 2.55 (0.235) | -1.12 (0.963) | -0.57 (0.100) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 2.51 (0.248) | -0.90 (0.783) | -0.39 (0.099) | <0.001 |
Placebo | 115 | 2.52 (0.245) | -0.56 (0.603) | — | — |
E2 = 17β-estradiol; mITT = modified intent to treat; MP = micronized progesterone; SD = standard deviation; SE = standard error; VMS = vasomotor symptoms
Source: Clinical Study Report for REPLENISH
Statistically significant reductions from placebo in the number of mild, moderate, and severe VMS were observed by Week 3 for the 1 mg E2/100 mg MP group, by Week 4 for the 0.5 mg E2/100 mg MP group and was maintained until Week 12. The mean change from baseline was −44.4 for the 1 mg E2/100 mg MP and −37.3 for the 0.5 mg E2/100 mg MP group compared with −26.8 for the placebo group at week 4 (Table 10). At Week 12, the change from baseline was −60.3 for the 1 mg E2/100 mg MP group and −58.8 for the 0.5 mg E2/100 mg MP group compared with −41.7 for the placebo group.
Statistically significant reductions from placebo in the severity of mild, moderate, and severe VMS were observed by Week 3 for the 1 mg E2/100 mg MP group. For the 0.5 mg E2/100 mg MP group, statistically significant reductions in severity were noted at various timepoints but were not consistent across the 12 weeks. At Week 12 both doses demonstrated a statistically significant reduction in the severity of mild, moderate, and severe VMS (Table 10).
Table 10: Change From Baseline and Placebo in the Mean Number of Weekly Mild, Moderate and Severe VMS for Week 1 Through Week 12 (mITT-VMS population)
Drug or comparator | Total N | Baseline Mean (SD) | Mean change from baseline (SD) | LS Mean change from placebo (SE) | MMRM P value |
|---|---|---|---|---|---|
Number of weekly mild, moderate and severe VMS for week 1 through week 4 | |||||
1 mg E2/100 mg MP | 134 | 86.2 (40.61) | -44.4 (34.53) | -15.32 (3.78) | <0.001 |
0.5 mg E2/100 mg MP | 144 | 85.1 (33.92) | -37.7 (35.38) | -8.92 (3.73) | 0.017 |
Placebo | 126 | 83.0 (26.47) | -26.8 (30.52) | — | — |
Number of weekly mild moderate and severe VMS for week 1 through week 12 | |||||
1 mg E2/100 mg MP | 124 | 86.2 (40.61) | -60.3 (36.42) | -20.61 (3.93) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 85.1 (33.92) | -58.8 (39.59) | -18.24 (3.87) | <0.001 |
Placebo | 115 | 83.0 (26.47) | -41.7 (36.35) | — | — |
Severity of weekly mild, moderate and severe VMS for week 1 through week 4 | |||||
1 mg E2/100 mg MP | 134 | 2.36 (0.337) | -0.31 (0.527) | -0.13 (0.058) | 0.027 |
0.5 mg E2/100 mg MP | 144 | 2.31 (0.333) | -0.31 (0.540) | -0.15 (0.057) | 0.011 |
Placebo | 126 | 2.34 (0.325) | -0.17 (0.368) | — | — |
Severity of weekly mild, moderate and severe VMS for week 1 through week 12 | |||||
1 mg E2/100 mg MP | 124 | 2.36 (0.337) | -0.94 (0.986) | -0.57 (0.101) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 2.31 (0.333) | -0.71 (0.784) | -0.37 (0.100) | <0.001 |
Placebo | 115 | 2.34 (0.325) | -0.39 (0.585) | — | — |
E2 = 17β-estradiol; mITT = modified intent to treat; MP = micronized progesterone; SD = standard deviation; SE = standard error; VMS = vasomotor symptoms
Source: Clinical Study Report for REPLENISH
Responder Analysis of Frequency and Severity of VMS at Weeks 4 and 12
A responder was defined as a patient with ≥ 50% reduction from Baseline in the number of moderate and severe VMS. An analysis of those with ≥ 75% reduction from Baseline in the number of moderate and severe VMS was also performed. The same analyses were performed for the reduction in the number of mild, moderate, and severe VMS. Assessment of responder rates was performed at Week 4 and Week 12.
A statistically significant difference between both treatment groups compared to placebo was observed at Weeks 4 and 12 (Table 11). At Week 4, 61.7% and 48.6% of patients in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively had a ≥ 50% reduction compared with 32.5% in the placebo group and 41.4% and 23.6% of patients in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively had a ≥ 75% reduction compared with 11.9% in the placebo group. At Week 12, 79.0% and 80.6% of patients in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively had a ≥ 50% reduction compared with 58.3% in the placebo group and 67.7% and 58.1% of patients in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively had a ≥ 75% reduction compared with 32.2% in the placebo group.
Table 11: Number (%) of Patients With ≥ 50% and ≥ 75% Reduction in Frequency of Moderate and Severe VMS From Baseline to Week 4 and Week 12 (mITT-VMS Population)
Drug or comparator | Total N | ≥50% Reduction | ≥75% Reduction | ||
|---|---|---|---|---|---|
n (%) | P value | n (%) | P value | ||
Week 4 | |||||
1 mg E2/100 mg MP | 133 | 82 (61.7) | <0.001 | 55 (41.4) | <0.001 |
0.5 mg E2/100 mg MP | 144 | 70 (48.6) | 0.009 | 34 (23.6) | 0.017 |
Placebo | 126 | 41 (32.5) | — | 15 (11.9) | — |
Week 12 | |||||
1 mg E2/100 mg MP | 124 | 98 (79.0) | <0.001 | 84 (67.7) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 104 (80.6) | <0.001 | 75 (58.1) | <0.001 |
Placebo | 115 | 67 (58.3) | — | 37 (32.2) | — |
E2 = 17β-estradiol; mITT = modified intent to treat; MP = micronized progesterone; VMS = vasomotor symptoms
Source: Clinical Study Report for REPLENISH
A responder analysis was also calculated for mild, moderate, and severe VMS. The number and percentage of patients with a decrease from Baseline of ≥ 50% and, separately, ≥ 75% in the mean weekly number of mild, moderate and severe VMS for Weeks 1 to 12 are shown in Table 12.
At Week 4, a statistically significantly difference vs placebo in the number of patients who had a ≥ 50% and a ≥ 75% reduction in the number of mild, moderate, and severe VMS was observed for both treatment groups with similar results were reported at Week 12.
Table 12: Number (%) of Patients With ≥ 50% and ≥ 75% Reduction in Frequency of Mild, Moderate and Severe VMS From Baseline to Week 4 and Week 12 (mITT-VMS Population)
Drug or comparator | Total N | ≥50% Reduction | ≥75% Reduction | ||
|---|---|---|---|---|---|
n (%) | P value | n (%) | P value | ||
Week 4 | |||||
1 mg E2/100 mg MP | 134 | 80 (59.7) | <0.001 | 44 (32.8) | <0.001 |
0.5 mg E2/100 mg MP | 144 | 62 (43.1) | 0.011 | 28 (19.4) | <0.001 |
Placebo | 126 | 35 (27.8) | — | 6 (4.8) | — |
Week 12 | |||||
1 mg E2/100 mg MP | 124 | 97 (78.2) | <0.001 | 73 (58.9) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 94 (72.9) | <0.001 | 64 (49.6) | <0.001 |
Placebo | 115 | 55 (47.8) | — | 32 (27.8) | — |
E2 = 17β-estradiol; mITT = modified intent to treat; MP = micronized progesterone; VMS = vasomotor symptoms
Source: Clinical Study Report for REPLENISH
Clinical Global Impression
For the CGI analysis, patients answered the following question: “Rate the total improvement, whether or not in your judgment it is due entirely to drug treatment. Compared to your condition at admission to the study, how much has it changed?” Potential responses included: very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse. The results for the top two responses for improvement (very much improved and much improved) and no change or worsening (minimally worse, much worse, or very much worse) were combined for each group and the active treatment groups were compared to placebo (Table 13). At Week 4, the percentage of patients who reported ‘very much improved’ or ‘much improved’ was 63.2% and 50.4% in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively compared to 32.8% in the placebo group. By Week 8, the percentage of patients who reported ‘very much improved’ or ‘much improved’ increased to 77.7% and 74.1% in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively 53.0% in the placebo group. At the last assessment (Week 12), the percentage of patients who reported ‘very much improved’ or ‘much improved was 82.1% and 72.9% in the 1 mg E2/100 mg MP and 0.5 mg E2/100 mg MP groups respectively compared to 53.4% in the placebo group. At all timepoints, a statistically significant improvement in both active treatment groups was observed compared to placebo.
Table 13: Clinical Global Impression for Weeks 4, 8, and 12 (mITT-VMS Population)
Drug or comparator | Total N | Very much improved/ much improved | Minimally improved | No change | P value |
|---|---|---|---|---|---|
n (%) | n (%) | n (%) | |||
Week 4 | |||||
1 mg E2/100 mg MP | 136 | 86 (63.2) | 37 (27.2) | 13 (9.6) | <0.001 |
0.5 mg E2/100 mg MP | 141 | 71 (50.4) | 49 (34.8) | 21 (14.9) | 0.005 |
Placebo | 125 | 41 (32.8) | 49 (39.2) | 35 (28.0) | — |
Week 8 | |||||
1 mg E2/100 mg MP | 130 | 101 (77.7) | 23 (17.7) | 6 (4.6) | <0.001 |
0.5 mg E2/100 mg MP | 139 | 103 (74.1) | 24 (17.3) | 12 (8.6) | <0.001 |
Placebo | 117 | 62 (53.0) | 25 (21.4) | 30 (25.6) | — |
Week 12 | |||||
1 mg E2/100 mg MP | 123 | 101 (82.1) | 17 (13.8) | 5 (4.1) | <0.001 |
0.5 mg E2/100 mg MP | 133 | 97 (72.9) | 29 (21.8) | 7 (5.3) | <0.001 |
Placebo | 116 | 62 (53.4) | 26 (22.4) | 28 (24.1) | — |
E2 = 17β-estradiol; mITT-VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone
Source: Clinical Study Report for REPLENISH
Based on the nonparametric discriminant analysis, the threshold for reporting a meaningful decrease in weekly moderate to severe VMS, based on the best discrimination between women who reported ‘minimally improved’ and those women who reported ‘much or very much improved’, was a decrease of 36 VMS at Week 4 and a decrease of 39 VMS at Week 12. Based on the CGI analyses, the responder definition should be based on criteria of a decrease of 36 to 39 moderate to severe VMS.
The number and percentage of patients who were responders, based on the above definition, are shown in Table 14. Statistically significant differences were observed for both active treatment groups when compared to placebo at Weeks 4 and 12.
Table 14: Number (%) of Patients With ≥ 36 and ≥ 39 Reduction in Frequency of Moderate and Severe VMS From Baseline to Week 4 and Week 12 (mITT-VMS Population)
Drug or comparator | Total N | n (%) | P value |
|---|---|---|---|
Week 4 (≥36 VMS Reduction) | |||
1 mg E2/100 mg MP | 134 | 79 (59.0) | <0.001 |
0.5 mg E2/100 mg MP | 144 | 66 (45.8) | 0.034 |
Placebo | 126 | 41 (32.5) | — |
Week 12 (≥39 VMS Reduction) | |||
1 mg E2/100 mg MP | 124 | 91 (73.4) | <0.001 |
0.5 mg E2/100 mg MP | 129 | 94 (72.9) | <0.001 |
Placebo | 115 | 60 (52.2) | — |
E2 = 17β-estradiol; mITT -VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone
Source: Clinical Study Report for REPLENISH
Menopause-specific quality of life (MENQOL)
Baseline scores, mean change from Baseline, and LS mean change from placebo results to Week 12, Month 6, and Month 12 in the MENQOL total score and the vasomotor domain score are shown in Table 15.
At Week 12 and Months 6 and 12, statistically significant improvements in the MENQOL Total Score was observed for both active treatment groups compared to placebo.
Table 15: Mean Change From Baseline and LS Mean Change From Placebo in the MENQOL Score at Week 12, Month 6, and Month 12 (mITT-VMS Population)
Drug or comparator | Total N | Baseline Score | N at time point | Mean change from baseline (SD) | LS Mean change from placebo (SE) | MMRM P value |
|---|---|---|---|---|---|---|
Week 12 | ||||||
1 mg E2/100 mg MP | 140 | 4.5 (1.17) | 124 | -1.9 (−1.20) | -0.58 (0.145) | <0.001 |
0.5 mg E2/100 mg MP | 149 | 4.3 (1.25) | 135 | -1.6 (1.23) | -0.34 (0.143) | 0.016 |
Placebo | 135 | 4.6 (1.34) | 116 | -1.4 (1.36) | — | — |
Month 6 | ||||||
1 mg E2/100 mg MP | 140 | 4.5 (1.17) | 116 | -2.0 (1.22) | -0.55 (0.150) | <0.001 |
0.5 mg E2/100 mg MP | 149 | 4.3 (1.25) | 130 | -1.8 (1.22) | -0.42 (0.146) | <0.001 |
Placebo | 135 | 4.6 (1.34) | 104 | -1.6 (1.31) | — | — |
Month 12 | ||||||
1 mg E2/100 mg MP | 140 | 4.5 (1.17) | 97 | -1.8 (1.45) | -0.43 (0.169) | 0.012 |
0.5 mg E2/100 mg MP | 149 | 4.3 (1.25) | 118 | -2.0 (1.27) | -0.73 (0.162) | <0.001 |
Placebo | 135 | 4.6 (1.34) | 93 | -1.5 (1.50) | — | — |
E2 = 17β-estradiol; mITT -VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone; SD = standard deviation; SE = standard error
Source: Clinical Study Report for REPLENISH
MOS-Sleep Questionnaire
Baseline, mean change from Baseline, and LS mean change from placebo for Week 12, Month 6, and Month 12 in MOS Total Sleep Scores are shown in Table 16. The Total Score is the average of nine of the twelve questions.
At Months 6 and 12, statistically significant improvements were noted for both active treatment groups compared to placebo (p < 0.05), except for the 1 mg E2/100 mg MP group at Month 12 (p = 0.058).
Table 16: Mean Change From Baseline and LS Mean Change From Placebo to Week 12, Month 6, and Month 12 in MOS Total Sleep Score (mITT-VMS Population)
Drug or comparator | Total N | Baseline Mean (SD) | N at time point | Mean change from baseline (SD) | LS Mean change from placebo (SE) | MMRM P value |
|---|---|---|---|---|---|---|
Week 12 | ||||||
1 mg E2/100 mg MP | 140 | 48.0 (19.08) | 122 | -16.7 (16.99) | -4.39 (2.059) | 0.033 |
0.5 mg E2/100 mg MP | 148 | 44.9 (17.43) | 134 | -13.1 (16.22) | -2.54 (2.015) | 0.207 |
Placebo | 134 | 47.3 (18.87) | 111 | -11.5 (19.67) | — | — |
Month 6 | ||||||
1 mg E2/100 mg MP | 140 | 48.0 (19.08) | 113 | -17.8 (17.28) | -5.48 (2.138) | 0.011 |
0.5 mg E2/100 mg MP | 148 | 44.9 (17.43) | 124 | -16.0 (16.60) | -5.25 (2.093) | 0.012 |
Placebo | 134 | 47.3 (18.87) | 101 | -11.7 (19.40) | — | — |
Month 12 | ||||||
1 mg E2/100 mg MP | 140 | 48.0 (19.08) | 96 | -14.9 (21.09) | -4.61 (2.427) | 0.058 |
0.5 mg E2/100 mg MP | 148 | 44.9 (17.43) | 117 | -15.8 (17.72) | -7.48 (2.322) | 0.001 |
Placebo | 134 | 47.3 (18.87) | 92 | -10.3 (21.78) | — | — |
E2 = 17β-estradiol; mITT -VMS = modified intent to treat – vasomotor symptom; MP = micronized progesterone; SD = standard deviation; SE = standard error
Source: Clinical Study Report for REPLENISH
Harms
Safety evaluation plan
The primary safety endpoint was the incidence of endometrial hyperplasia at 12 months (to demonstrate a hyperplasia proportion that was ≤ 1% with an upper bound of the one-sided 95 percent CI for that rate that does not exceed 4%) based on an a priori plan in which a consensus among two out of three pathologists was the final endometrial pathology diagnosis. For the primary endpoint, all endometrial biopsies were centrally read by three pathologists. Two pathologists, designated by the Sponsor prior to study start, were the primary pathologists (the pathologists were blinded to this designation).
The incidence rate of endometrial hyperplasia at Month 12 was calculated as follows:
I = A / B
Where I = incidence rate at Month 12 evaluation
A = all new patients with biopsies positive for endometrial hyperplasia during the study, but post-Baseline
B = all patients with biopsies following Month 11 meeting the criteria specified above, plus all patients with biopsies positive for endometrial hyperplasia by any of the pathologist before Month 11
An upper one-sided 95% confidence limit for the binomial proportion was calculated. In addition, 95% two-sided CIs were calculated for pairwise differences between groups in hyperplasia incidence.
A supplemental secondary analysis was performed based on the results from the three pathologists. In this supplemental analysis, the final diagnosis was based on agreement of two of the three pathologists reads. Consensus was reached when two of the three pathologist readers agreed on any of the above categories. For example, any two subcategories of “Non-endometrial malignancy/non-hyperplasia” will be classified as “Category 1: Non-endometrial malignancy/non-hyperplasia.” If all three readings were disparate (ie, each fell into a different category – Category 1, 2, or 3), the final 11 diagnosis was based on the-1 most severe of the three readings. A CI for the incidence proportion of hyperplasia was constructed in the same manner as for the primary safety analysis.
Other secondary endpoints included:
Proportion of patients with cumulative amenorrhea from Day 1 to Day 364
No bleeding: % by cycle and cumulative for consecutive 28-day cycles
Number of days with bleeding/spotting
Percent amenorrhea: Amenorrhea was defined as absence of bleeding or spotting. Within each treatment arm, the portion of patients with cumulative amenorrhea from Day 1 to Day 364 was calculated and compared between active and placebo treatments. Cumulative rates of amenorrhea were defined as the percentage of women who reported consecutive cycles of amenorrhea for a given cycle of time. For example, if a patient had no bleeding or spotting from Day 1 to Day 364, then this patient had cumulative amenorrhea from the 1st to 13th cycle. The number and percentage of patients with amenorrhea for each cumulative period was summarized separately for the 1st to 13th cycle, 2nd cycle to 13th cycle, …, and the 13th cycle.
Percent no bleeding: No bleeding was defined as absence of bleeding. Within each treatment arm, the percent of patients with no bleeding was calculated by cycle and for consecutive cycles and compared between active and placebo treatments.
Cumulative rates for no bleeding was defined as the percentage of women who reported consecutive cycles of no bleeding for a given cycle of time. For example, if a patient had no bleeding from Day 1 to Day 364, then this patient had no bleeding from the 1st to 13th cycle. The number and percentage of patients with no bleeding for each cumulative period was summarized separately for the 1st to 13th cycle, 2nd to 13th cycle, …, and the 13th cycle.
The number of days with bleeding/spotting, as reported on patient diaries, was summarized by cycle and treatment group.
Endometrial biopsy was performed at Baseline and Month 12 as part of the endometrial safety evaluation (primary safety endpoint). Bleeding data collected for the day on which an endometrial biopsy was performed, and for the six (6) days thereafter, was excluded for both cumulative and non-cumulative summaries. The last available data before the biopsy was performed was carried forward for those days (LOCF). Patients evaluated included the safety population less any patients who had no bleeding/spotting diary data.
Overall safety variable included incidence of AEs and serious adverse events (SAEs), incidence of endometrial polyps, and change from baseline in relevant clinical parameters. All AEs were listed by patient, including non-treatment emergent (i.e., pre-dosing or > 15 days after the last dose) AEs.
Overview of safety
No cases of endometrial hyperplasia were observed during the trial (Table 17) and the one-sided upper 95% confidence limit was less than 4% for all groups (1.06% for 1 mg E2/100 mg MP; 0.98% for 0.5 mg E2/100 mg MP; and 3.20% for the placebo group). All patients had a final diagnosis per the Pathology Charter of Category 1 (non-endometrial malignancy/non-hyperplasia). Similar results were noted for the overall Safety population. The results of the secondary analysis were the same as the primary analysis as there were no reports of endometrial hyperplasia.
Table 17: Incidence of Endometrial Hyperplasia at 12 Months (ES Population)
Adverse events | REPLENISH | ||
|---|---|---|---|
1 mg E2/100 mg MP N = 280 | 0.5 mg E2/100 mg MP N = 303 | Placebo N = 92 | |
Hyperplasia incidence rate, n (%) | 0 (0) | 0 (0) | 0 (0) |
One-sided upper 95%CI | 1.06% | 0.98% | 3.20% |
CI = confidence interval; E2 = 17β-estradiol; ES = endometrial safety; P = progesterone
Source: Clinical Study Report for REPLENISH
Cumulative amenorrhea from Cycle 1 to 13 was reported by 56.1% of patients in the 1 mg/100 mg group (p < 0.001), 67.6% in the 0.5 mg/100 mg group (p = 0.048), compared to 78.9% in placebo. At Cycle 13, cumulative amenorrhea rates were 90.2% in the 1 mg/100 mg group and 95.9% in the 0.5 mg/100 mg group, compared to 97.8% for placebo (there was a statistically significant difference between the 1 mg/100 mg group compared to placebo; p = 0.023). Similar data were reported for patients with cumulative amenorrhea from Day 1 to Day 364 in the Safety population.
Cumulative no bleeding from Cycle 1 to 13 was reported by 73.4% of patients in the 1 mg/100 mg group (p < 0.001) and 83.9% in the 0.5 mg/100 mg group, compared to 91.1% in placebo. At Cycle 13, rates of no bleeding were more than 97% in both treatment groups and not statistically different from placebo.
During the first trimester, the percentage of patients reporting spotting was 22.5% for the 0.5 mg/100 mg arm and 28.8% for the 1 mg/100 mg arm compared to 9.7% in placebo. By the fourth trimester, the percentage of patients with spotting decreased in all groups; 16.7% in the 1 mg/100 mg group, 6.9% in the 0.5 mg/100 mg group, and 4.3% in placebo.
Similar trends were observed in the percentage of patients with reported bleeding. During the first trimester, the percentage of patients reporting bleeding ranged from 8.8% in the 0.5 mg/100 mg arm and 15.4% in the 1 mg/100 mg arm compared to 3.9% in placebo. For the fourth trimester, the percentage of patients with bleeding decreased across all groups, with the highest percentage in the 1 mg/100 mg group (9.6%), followed by the 0.5 mg/100 mg group (5.3%), and placebo (2.2%).
The median number of days of bleeding and/or spotting for each cycle was zero (0) and the mean was less than one day (maximum number of days was 28 days). Study discontinuations due to bleeding-related AEs were 0.5% (0.5mg E2/50mg MP4 dose) and 1.4% (1mg E2/100mg MP4 dose). No women in the placebo group discontinued the study due to bleeding AEs.
See Table 18 for detailed harms data.
Table 18: Summary of Harms Data (Safety Population)
Adverse events | REPLENISH | ||
|---|---|---|---|
1 mg E2/100 mg MP N = 415 | 0.5 mg E2/100 mg MP N = 424 | Placebo N = 151 | |
Patients with at least one TEAE | |||
n (%) | 297 (71.6) | 302 (71.2) | 78 (51.7) |
Headache | 31 (7.5) | 24 (5.7) | 4 (2.6) |
Nasopharyngitis | 25 (6.0) | 41 (9.7) | 4 (2.6) |
Breast tenderness | 45 (10.8) | 19 (4.5) | 1 (0.7) |
Upper respiratory tract infection | 22 (5.3) | 26 (6.1) | 6 (4.0) |
Nausea | 20 (4.8) | 25 (5.9) | 2 (1.3) |
Back pain | 22 (5.3) | 11 (2.6) | 1 (0.7) |
Abdominal pain | 22 (5.3) | 10 (2.4) | 4 (2.6) |
Sinusitis | 20 (4.8) | 15 (3.5) | 3 (2.0) |
Dizziness | 17 (4.1) | 15 (3.5) | 3 (2.0) |
Pelvic pain | 17 (4.1) | 15 (3.5) | 0 |
Diarrhoea | 13 (3.1) | 13 (3.1) | 2 (1.3) |
Vulvovaginal mycotic infection | 14 (3.4) | 15 (3.5) | 4 (2.6) |
Abdominal distension | 15 (3.6) | 6 (1.4) | 1 (0.7) |
Vaginal haemorrhage | 14 (3.4) | 10 (2.4) | 1 (0.7) |
Vaginal discharge | 16 (3.9) | 13 (3.1) | 1 (0.7) |
Hypertension | 7 (1.7) | 13 (3.1) | 2 (1.3) |
Influenza | 4 (1.0) | 6 (1.4) | 2 (1.3) |
Patients with at least one serious TEAE | |||
n (%) | 9 (2.2) | 15 (3.5) | 2 (1.3) |
Coronary artery disease | 1 (0.2) | 0 | 0 |
Stress cardiomyopathy | 0 | 1 (0.2) | 0 |
Gastroduodenitis | 0 | 1 (0.2) | 0 |
Pancreatitis acute | 1 (0.2) | 0 | 0 |
Hepatic steatosis | 1 (0.2) | 0 | |
Bronchitis | 0 | 1 (0.2) | 0 |
Cholecystitis infective | 0 | 0 | 1 (0.7) |
Diverticulitis | 2 (0.5) | 0 | 0 |
Gastroenteritis viral | 1 (0.2) | 0 | 0 |
Pneumonia | 0 | 2 (0.5) | 0 |
Urosepsis | 0 | 1 (0.2) | 0 |
Ankle fracture | 0 | 1 (0.2) | 0 |
Foot fracture | 0 | 1 (0.2) | 0 |
Incisional hernia | 1 (0.2) | 0 | 0 |
Intervertebal disc protrusion | 0 | 1 (0.2) | 0 |
Osteoarthritis | 0 | 1 (0.2) | 0 |
Breast cancer | 2 (0.5) | 1 (0.2) | 0 |
Invasive ductal breast carcinoma | 0 | 1 (0.2) | 0 |
Myelodysplastic syndrome | 0 | 1 (0.2) | 0 |
Psychotic disorder | 1 (0.2) | 0 | 0 |
Uterine prolapse | 0 | 0 | 1 (0.7) |
Aortic aneurysm | 1 (0.2) | 0 | 0 |
Peripheral arterial occlusive disorder | 0 | 1 (0.2) | 0 |
Patients with at least one TEAE leading to study discontinuation | |||
n (%) | 45 (10.8) | 31 (7.3) | 10 (6.6) |
Most common events (2 or more patients) | |||
Nausea | 3 (0.7) | 1 (0.2) | 0 |
Vomiting | 2 (0.5) | 0 | 0 |
Diarrhoea | 2 (0.5) | 0 | 0 |
Fatigue | 0 | 2 (0.5) | 1 (0.7) |
Lung function test abnormal | 2 (0.5) | 0 | 0 |
Weight increased | 1 (0.2) | 2 (0.5) | 1 (0.7) |
Muscle spasms | 2 (0.5) | 0 | 0 |
Pain in extremity | 0 | 2 (0.5) | 0 |
Headache | 2 (0.5) | 0 | 1 (0.7) |
Agitation | 2 (0.5) | 0 | 0 |
Anxiety | 2 (0.5) | 0 | 0 |
Depression | 1 (0.2) | 2 (0.5) | 0 |
Breast tenderness | 6 (1.4) | 0 | 0 |
Uterine haemorrhage | 2 (0.5) | 0 | 0 |
Vaginal haemorrhage | 4 (1.0) | 4 (0.9) | 0 |
Alopecia | 2 (0.5) | 3 (0.7) | 0 |
E2 = 17β-estradiol; MP = micronized progesterone
Source: Clinical Summary Report for REPLENISH
Adverse events
Patients in the active treatment groups reported similar numbers of treatment emergent adverse events (TEAEs), ranging from 71.2% in the lowest dose (0.5 mg E2/100 mg MP) to 71.6% in the higher dose (1 mg E2/100 mg MP), while 51.7% of patients in the placebo group reported a TEAE.
The most frequently occurring TEAEs (occurring in ≥ 3% of patients in at least one active treatment group) and numerically more common than placebo were: headache, nasopharyngitis, breast tenderness, upper respiratory tract infection, nausea, back pain, abdominal pain, sinusitis, dizziness, pelvic pain, diarrhea, vulvovaginal mycotic infection, abdominal distension, vaginal discharge, hypertension, influenza, and vaginal hemorrhage. Most of the TEAEs were mild to moderate in severity.
Serious adverse events
Twenty-three (23) patients experienced a serious TEAE during the study (38 patients in the active treatment groups and two patients in the placebo group). TEAEs leading to study discontinuation were reported by 45 (10.8%) patients in the 1 mg E2/100 mg MP group, 31 (7.3%) patients in the 0.5 mg E2/100 mg MP group and 10 (6.6%) patients in the placebo group.
Adverse events of special interest
AEs of special interest that include venous thromboembolic (VTE) events; superficial thrombosis/phlebitis; cardiac AEs of interest; ECG reported AEs; cerebrovascular AEs of interest; chest pain AEs; syncope; breast cancer AEs; other breast AEs of interest; cervical AEs; AEs related to the endometrium; and malignancies. Generally, the percentages of AEs of special interest were low and did not occur with greater frequency in the active arms than placebo. Overall, the incidence and nature of the adverse events reported in this study are consistent with that expected for this population.
Bioequivalence
Summary of Studies in Clinical Pharmacology Program
A total of five Phase 1 studies and one Phase 3 study were conducted that included the assessment of estradiol, estrone, and progesterone blood levels (see Table 19). Exposures of subjects to estradiol, estrone, and progesterone after administration of Bijuva and the effect of food on these exposures is consistent with the effects seen with reference compounds Estrace® and Prometrium®.
Table 19: Summary of Studies in Clinical Pharmacology
Study Number | Dose (Estradiol/ Progesterone) | Description | Conclusions |
|---|---|---|---|
351 | 2 mg/200 mg | 24 subjects fasted, single dose, comparative BA, crossover with reference (Estrace, Prometrium) | E2 AUC showed BE while Cmax GM for Bijuva was higher than Reference. P AUC and Cmax GM values were slightly lower than Reference |
352 | 2 mg/200 mg | 24 subjects fed high fat, single dose, comparative BA, crossover with reference (Estrace, Prometrium) | Bijuva was similar to Reference under fed conditions, however, the high variability kept BE from being obtained |
459* | 2 mg/200 mg | 66 subjects fed high-fat, single dose, 3-way crossover, reference-replicated, reference-scaled BE (Estrace, Prometrium) | Bijuva showed BE to Reference under fed conditions |
TXC17-02 | 1 mg/100 mg | 24 subjects, single dose, two-treatment (fed and fasting), crossover, food effect | BA of P increased by high fat meal while E2 showed little to no food effect |
TXC16-01 | 1 mg/100 mg 0.5 mg/100 mg | 40 subjects fed moderate-fat, 1 and 7 daily doses, parallel group (20 subjects), PK | Steady state achieved within 7 days of once daily dosing. Expected accumulation for E2 and P and dose-related PK for E2. |
TXC12-05 | 1 mg/100 mg 0.5 mg/100 mg 0.5 mg/50 mg 0.25 mg/50 mg | >280 subjects/dose, single point >8 hr after dose, dose taken “at bedtime with food” and sample collected “next morning” as part of the long-term safety and efficacy study. | E2 and MP mean plasma concentrations were consistent throughout the duration of the study. Approximate dose proportionality was observed for E2 and MP. |
*Bioequivalence demonstrated
AUC = area under the serum/plasma concentration vs time curve, BA = bioavailability, BE = bioequivalence, Cmax = maximum concentration, E2 = estradiol, GM = geometric mean, MP = micronized progesterone, PK = pharmacokinetic, 351 = EPROG-1K-351-12, 352 = EPROG-1K-352-12, 459 = EPROG-1K-459-12
Bioequivalence
Comparative Bioavailability Studies
Studies 351 (EPROG-1K-351-12) and 352 (EPROG-1K-352-12), were initially conducted to compare the bioavailability of Bijuva 2 mg/200 mg (estradiol and progesterone capsule) with the same doses of Estrace (estradiol tablets) and Prometrium (progesterone) in healthy, adult, postmenopausal female subjects. Twenty-four subjects, age range 42 to 65 years, and 24 subjects, age range 45 to 65 years were enrolled and participated in Studies 351 and 352, respectively. Under fasting conditions (Study 351), Bijuva AUC0-t and AUC0-∞ for baseline-adjusted and unadjusted estradiol and total estrone showed bioequivalence to the Reference while progesterone exposure for Bijuva was significantly lower than the Reference for all primary PK parameters. Estrone showed bioequivalence between Bijuva and the Reference for Cmax but not AUC. Under high-fat fed conditions, Bijuva was bioequivalent to the Reference for unadjusted estradiol and estrone Cmax and AUC0-t. Baseline-adjusted Cmax and AUC0-∞ for estrone and AUC0-t for total estrone showed bioequivalence between Bijuva and the Reference. All of the primary PK parameters for progesterone as well as other parameters for estradiol and its metabolites were not bioequivalent, with the Bijuva levels being higher than the Reference in most cases. Due to the intrasubject coefficient of variation (CV) being > 30% in many cases, a reference-replicated, reference-scaled, bioequivalence approach was taken in Study 459.
Study 459 – Bioequivalence demonstrated
Study 459 (EPROG-1K-459-12) was an open-label, randomized, single-dose, reference-replicated, reference-scaled, crossover bioequivalence study that compared the bioavailability of Bijuva 2 mg/200 mg to the combined Reference consisting of 200 mg of Prometrium (progesterone) plus 2 mg of Estrace (estradiol tablets). Plasma levels of estradiol, progesterone, and unconjugated and total estrone were evaluated under fed conditions. In this study, a reference-replicated, reference-scaled, bioequivalence approach was taken to account for the high intra-patient variability observed in Study 351 and Study 352. ((Pickar 2015)).
Sixty-six healthy, postmenopausal female subjects were enrolled and administered the Test and Reference products using the same procedures as in Study 352 under high-fat, high-calorie fed conditions. Based on the randomized schedule, participants were assigned, in equal numbers, to one of three dosing sequences (TRR, RTR, or RRT, where T is the test drug and R is the reference product). In each sequence, participants received a single dose of Bijuva (2 mg/200 mg) in one study period and a single dose of estradiol plus a single dose of progesterone in each of the remaining two periods. The dose in each of the three study periods was separated by a 14-day washout to eliminate drug carryover effects.
For patients completing all three periods (N=62), PK parameters for baseline-adjusted and baseline-unadjusted levels of unconjugated estradiol, estrone, and progesterone, and total estrone were determined by performing a non-compartmental analysis. The scaled-average bioequivalence (SABE) method for highly variable drugs was used to compare Bijuva with the Reference products in cases where the within-subject CV for the reference product was 30% or more. A pharmacokinetic endpoint for an analyte was identified as bioequivalent when the 95% upper confidence bound on linearized SABE statistic was 0 or less. The unscaled average bioequivalence method was used to evaluate bioequivalence in cases where the within-subject coefficient of variation was less than 30%. A pharmacokinetic endpoint for an analyte was identified as bioequivalent when the 90% CI on the geometric mean ratio (GMR) fell between 0.80 and 1.25. Bioequivalence criteria had to be met for all three primary parameters (Cmax, AUC0-t, and AUC0-∞) in order to establish an analyte as bioequivalent.
The estradiol, estrone, and progesterone results obtained in Study 459 showed Bijuva 2 mg/200 mg to be bioequivalent to Prometrium 200 mg and Estrace 2 mg in healthy, adult, human, postmenopausal female subjects under fed conditions.
Table 20: Point Estimate, 95% Upper Confidence Bound, and Within-Subject SD (SWR) of Test Product Versus Averaged Reference Product (Baseline-Adjusted)
Pharmacokinetics | Point Estimate (%) | 95% Upper Confidence Bound | Within-subject SD (SWR) | Bioequivalent |
|---|---|---|---|---|
Unconjugated Estradiol | ||||
AUC0-t (pg.h/mL) | 93.14 | -0.0914 | 0.4109 | Yes |
AUC0-∞ (pg.h/mL) | 92.28 | -0.0312 | 0.3070 | Yes |
Cmax (pg/mL) | 88.24 | -0.0402 | 0.3435 | Yes |
Progesterone | ||||
AUC0-t (ng.h/mL) | 105.45 | -0.5429 | 0.9564 | Yes |
AUC0-∞ (ng.h/mL) | 99.13 | -0.5711 | 0.9779 | Yes |
Cmax (ng/mL) | 115.92 | -0.7850 | 1.1794 | Yes |
Unconjugated Estrone | ||||
AUC0-t (pg.h/mL) | 88.16 | 83.53 to 93.05 | 0.2037 | Yes |
AUC0-∞ (pg.h/mL) | 85.59 | 80.60 to 90.89 | 0.2875 | Yes |
Cmax (pg/mL) | 92.35 | 86.57 to 98.52 | 0.2558 | Yes |
Total Estrone | ||||
AUC0-t (ng.h/mL) | 104.81 | 96.36 to 113.99 | 0.2895 | Yes |
AUC0-∞ (ng.h/mL) | 103.95 | -0.0497 | 0.2999 | Yes |
Cmax (ng/mL) | 174.55 | 0.3351 | 0.3478 | No |
AUC0-∞ = Area under the concentration vs time curve extrapolated to infinity; AUC0-t = Area under the concentration vs time curve to the last measurable time point; CI = confidence interval; Cmax = maximum concentration; GM = geometric mean.
Test = BIJUVA (estradiol and progesterone capsules) 2 mg/200 mg; Reference = Prometrium 200 mg + Estrace 2 mg
Source: Common Technical Document 2.7.1 BIJUVA, Section 2.2.1
CADTH’s Critical Appraisal of the Clinical Evidence
CADTH conducted a critical appraisal of the clinical studies for estradiol-progesterone (Bijuva) based on the summary of the evidence provided by the sponsor.
Internal Validity
The pivotal study included in this CADTH-tailored review was the phase III multi-centre REPLENISH trial. REPLENISH was a randomized, double-blind, placebo-controlled, parallel trial including 1,845 patients to determine if the estradiol-progesterone combination (Bijuva) given in a continuous fashion was effective at reducing the frequency and severity of VMS. Of the 1,845 patients, 766 were included in the VMS substudy and 1,079 were included in the non-substudy for VMS. The focus of this submission for efficacy was the VMS substudy, which included 766 patients randomized to an active treatment arm at a dose of estrogen-progesterone approved in Canada (i.e., 1 mg E2/100 mg MP or 0.5 mg E2/100 mg MP) or placebo. For the assessment of safety, the sponsor included data for 675 patients from the ES population and 990 patients from the overall study safety population (i.e., substudy and non-substudy populations with the doses of estrogen-progesterone approved in Canada).
The objectives, end points, and interventions in the REPLENISH study were well described. The screening of patients for eligibility and screen failures were also well described. Patients in the VMS substudy were randomized in a 1:1:1:1:1 allocation ratio (5 groups if counting the doses not approved in Canada) within each study site to 1 of the treatment arms. Randomization was accomplished by using a reproducible, computer-generated block randomization schedule. Randomization codes were generated and held by the blinded team at the data management contract research organization. Subsequently, an interactive web response system was implemented where subjects were randomly assigned to receive 1 of the treatment regimens.
Blinding was adequate; the packaging of the interventions and placebo were identical to maintain the blinding of investigators. Neither the subject nor the investigator could identify the treatment from the packaging or label of the investigation product. The placebo used in the study was adequate in its similarity with the intervention, providing the reassurance of blinding. There is a possibility that patients in an intervention arm could have identified their assignment due to the higher frequency of AEs that were in the estradiol-progesterone arms, such as breast tenderness or vaginal discharge.
Overall, baseline characteristics were well balanced in the mITT-VMS population for demographics and baseline values of the co-primary and secondary end points of severity and number of VMS episodes, denoting that the randomization process achieved a proper balance of prognostic variables at baseline.
The primary end points evaluated in the REPLENISH trial VMS substudy were frequency and severity of VMS from baseline to week 4 and week 12, based on patients’ diaries for daily hot flushes, bleeding, and spotting. Key secondary end points were measured using the mean change in frequency and severity of moderate-to-severe VMS at each week from baseline to week 12, and as the proportion of patients with a reduction of 50% or more and, separately, 75% or more in frequency of moderate-to-severe symptoms. Also, the CGI scale and tools of HRQoL — specifically MENQOL— were used. For sleep changes, the authors of the study used the MOS Sleep Scale questionnaire to assess changes in sleep disturbances. MENQOL was developed on women 47 years of age to 62 years of age. It is a valid, reliable, and responsive self-administered quality-of-life questionnaire specific to the early postmenopausal period.21 MENQOL consists of 29 items divided into 4 validated domains (vasomotor, sexual, physical, and psychosocial). All primary and secondary end points were assessed in a double-blinded fashion by investigators and patients.
Data on efficacy end points were analyzed using the intention-to-treat principle (evaluating patients to the group to which they were initially randomly assigned). The investigators used measures to prevent missing data. These efficacy data points were treated as missing and not imputed for the primary analysis of the primary end points to ensure validity of the analysis of the mixed model of repeated measures (MMRM) under the missing at random assumption. For a sensitivity analysis, missing weekly data were imputed using the last observation carried forward (LOCF) method. Even with the efforts to manage missing data, analyses for the primary and key secondary end points were conducted as available cases (complete case analysis) at different time points. Although results demonstrated statistically significant changes from baseline when compared to placebo, evaluating only available cases leads to missingness of data and thus possible imprecision of effect estimates and bias for the different end points. The magnitude and direction of this bias, however, is uncertain.
Controlling for multiplicity was performed for all primary and secondary efficacy end point comparisons for each active treatment group with placebo. This maintained the approach for the preservation of the familywise type I error rate for each end point evaluation. The multiple testing of the 4 co-primary end points was performed using a gatekeeping testing procedure. The testing started by examining the highest dose (1 mg E2/100 mg MP) for the co-primary end points. If the P values for the co-primaries were significant (P ≤ 0.05), then the hypothesis testing continued to the next dose (0.5 mg E2/100 mg MP) for each of the co-primaries. If at any point the hypothesis testing yielded a non-significant result, the testing was stopped.
Sensitivity analyses were performed to support the MMRM and used ANCOVA for missing weekly data that were imputed using LOCF. Overall, the LOCF results were similar to those observed in the MMRM analyses for the mITT-VMS and EE-VMS populations demonstrating robustness of the primary results.
Overall, the study was a well-performed, randomized controlled trial with some issues related to the missingness of data (complete case analysis) and the potential for unblinding of participants due to patient awareness of AEs associated with their assignment to intervention groups, although the magnitude and direction of these biases are uncertain.
External Validity
According to input from the clinical expert consulted by CADTH, the patient population included in the REPLENISH trial is generally reflective of patients seen in Canadian clinical practice. The age of the patients included in the trial (40 years old to 65 years old) is reflective of the majority of patients who will seek treatment for ameliorating VMS in Canadian clinical practice. Since the REPLENISH trial excluded patients with different baseline characteristics (i.e., those with risk factors such as history of thrombosis, coronary artery disease, CVD, or cancer), the generalizability of the trial results in these groups of patients is uncertain. The number of patients of Asian ethnicity enrolled in the trial was small, although based on clinical expert input, this would not affect the applicability of the results.
The inclusion and exclusion criteria used in the REPLENISH trial were clearly defined and described. The instruments used for evaluation of the primary and secondary end points are familiar to clinicians in Canada, according to the clinical expert consulted by CADTH, and therefore there were no concerns related to their use and application in real-life clinical practice.
The time to follow-up was short for evaluating long-term outcomes, especially in relation to outcomes considered important to patients and clinicians, such as risk of breast, ovarian, or endometrial cancer, and risk of CVD and thrombosis in patients with longer durations of treatment. Table 21 summarizes the generalizability of the evidence from the REPLENISH trial.
Table 21: Assessment of Generalizability of Evidence for Estradiol-Progesterone (Bijuva)
Domain | Factor | Evidence | CADTH's assessment of generalizability |
|---|---|---|---|
Population | Patients with moderate-to-severe VMS, 40 years old to 65 years old and intact uterus No history of thrombosis, CVD, CAD, chronic liver or kidney disease, or endometrial, breast, or ovarian cancer | REPLENISH trial inclusion and exclusion criteria | The inclusion and exclusion criteria from the trial is similar to the indication submitted by the sponsor. However, certain groups of patients (e.g., high risk for cardiovascular disease) were excluded from the REPLENISH study and the generalizability of results to these patients is uncertain. |
Intervention | Estradiol (E2)-progesterone (micronized), 1 mg/100 mg or 0.5 mg/100 mg presentations | REPLENISH trial and Health Canada PM | There are no issues of generalizability since the intervention aligns with the Health Canada–approved doses for the indication under review. |
Comparator |
| Current database of Health Canada–approved drugs, sponsor clinical summary, and individual PMs | Current available HRTs for patients with VMS in menopause are applicable to the same patient population and reflect the clinical Canadian practice landscape, although with some variations in their availability in different jurisdictions. There are no direct head-to-head comparisons of estrogen-progesterone to other HRTs, either from superiority or noninferiority trials. |
Outcomes | Frequency and severity of symptoms, proportion of patients with at least 50% and, separately, 75% improvement in frequency of symptoms, and HRQoL | Trial protocol and trial publication of results | The outcomes assessed were considered relevant and important according to input from the clinical expert consulted by CADTH and patient interviews. There are some anticipated issues and uncertainties with the duration of treatment, as in clinical practice this is yet to be defined and varies among clinicians and in specific clinical situations. |
Setting | Outpatient setting | Trial sites | The administration of estradiol-progesterone does not require special inpatient settings or specialized care. |
CVD = cardiovascular disease; CAD = coronary artery disease; E2 = 17beta-estradiol; HRQoL = health-related quality of life; HRT = hormone replacement therapy; PM = product monograph; STEAR = selective tissue estrogenic activity regulator; VMS = vasomotor symptoms.
Sponsor-Submitted Cost Information
The sponsor submitted a cost comparison for the annual drug acquisition costs associated with estradiol-progesterone tablets (Bijuva) compared with other oral and transdermal regimens of 17beta-estradiol or conjugated estrogen in combination with progesterone or medroxyprogesterone taken as individual products.22 The sponsor’s estimated annual costs for comparator regimens can be found in Table 25 in Appendix 2. Markups and dispensing fees were not considered in the sponsor’s analysis.
The sponsor assumed that all patients would be 100% adherent, and that the proportion of patients using the different doses of medroxyprogesterone was equal. Only the recommended strengths of transdermal comparators were included or if no recommendation was made, the lowest strength was applied. All other health care costs were assumed to be equal between comparators and were thus not considered.
The sponsor’s analysis reports that at the submitted price of $0.8962 per tablet, estradiol-progesterone tablets cost $327 per patient per year. When compared to oral progesterone-based regimens, estradiol-progesterone tablets were $233 to $350 per patient per year less expensive, depending on the strength of estradiol used. When compared to medroxyprogesterone-based regimens, estradiol-progesterone tablets were $105 to $275 more expensive per patient per year. When considering transdermal regimens, estradiol-progesterone tablets were $415 to $547 less expensive per patient per year than progesterone-based regimens, and $92 less to $93 more costly per patient per year than medroxyprogesterone-based regimens. (Refer to Table 25 for more details.)
Critical Appraisal of Cost Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
Other included comparator regimens are not clinically equivalent: As not all jurisdictions reimburse progesterone, the sponsor also compared estradiol-progesterone tablets to combinations of other products that include oral and transdermal estradiol, oral conjugated estrogen, oral progesterone, and oral medroxyprogesterone. This requires the assumption of equal efficacy and safety for a cost comparison to be the appropriate form of analysis. According to the North American Menopause Society’s 2017 mission statement,10 transdermal hormone therapy may decrease the risk of VTE and stroke relative to oral hormone therapy, while some but not all observational evidence suggests that the use of medroxyprogesterone may be associated with a higher risk of breast cancer than MP. There is also some evidence suggesting oral estrogen may have beneficial effects on glycemic control as well as on HDL cholesterol and LDL cholesterol.23 While the sponsor also compared estradiol-progesterone tablets to combinations including transdermal estrogen products (Table 5), the clinical expert consulted by CADTH did not find it likely that patients who would otherwise receive transdermal products would receive estradiol-progesterone tablets, should they be available.
Due to the submission of a cost comparison, CADTH was unable to adjust for this limitation in reanalyses. While the relative costs of estradiol-progesterone tablets and combinations of other comparators can be assessed, the cost-effectiveness of estradiol-progesterone tablets relative to these comparators is unknown.
CADTH only considered comparisons of estradiol-progesterone to oral 17beta-estradiol or conjugated estrogen plus progesterone or medroxyprogesterone to be relevant, and thus limited its reanalyses to combinations of oral products.
Variability in comparator dosing: While most included comparator products have recommended dosing regimens that are cyclical (e.g., on day 1 to day 25 each month, for 14 days per month), according to the clinical expert consulted by CADTH, most patients use both the estrogen and progesterone components daily, similar to the recommended dosing of estradiol-progesterone tablets, to simplify the regimen and to prevent withdrawal bleeding.
CADTH reanalysis considered the costs of the recommended dose range of comparator regimens as well as daily dosing.
Interchangeable comparators: The sponsor considered all brands of comparators, including those that are legally interchangeable with less expensive products. However, the more expensive brands of legally interchangeable products are rarely reimbursed. For example, in the first quarter of 2021, less than 3% of publicly reimbursed claims across Canada for 17beta-estradiol tablets were for the Estrace brand, while the other 97% were for Lupin-Estradiol. Additionally, in most cases where the Estrace brand is reimbursed, claims data suggest that the cost paid per unit is similar to that of the generic brand.24
CADTH compared the cost of estradiol-progesterone tablets to combinations of the least expensive interchangeable components.
Medroxyprogesterone distribution is inappropriate: In calculating the cost of medroxyprogesterone-based regimens, the sponsor simplified the cost of the medroxyprogesterone component by averaging the cost of all applicable doses. This implicitly assumes that medroxyprogesterone doses are uniformly distributed, which is not consistent with 2020 public claims data reported in the IQVIA Pharmastat database (2021).24
CADTH reanalysis considered medroxyprogesterone dose levels separately.
Confidential pricing agreements: While the submitted price of estradiol-progesterone tablets is less than the cost of its individual components at publicly available list prices, these list prices are higher than the costs paid by jurisdictional drug plans due to confidential pricing agreements. Therefore, the submitted price of estradiol-progesterone tablets may require a price reduction to avoid incurring additional costs relative to its individual components.
CADTH was unable to address this limitation in reanalyses as the negotiated prices of comparators are unknown.
CADTH Reanalyses
CADTH conducted a reanalysis of the cost comparison addressing some of the identified limitations. CADTH reanalysis considered both recommended and daily dosing regimens, costs based on the price of the least expensive interchangeable components, and costs of each potential medroxyprogesterone dose separately.
When compared to its individual components, estradiol-progesterone tablets are $117 to $224 less costly per patient per year when 17beta-estradiol and progesterone are used as recommended in their product monographs, and $241 to $281 less costly per patient per year when the components are used daily (refer to Table 22). Additionally, the use of estradiol-progesterone tablets would result in savings due to reduced dispensing fees, requiring up to 12 fewer fees per year where reimbursed medications are dispensed monthly (e.g., up to $105.96 less per year when Ontario dispensing fees are assumed).
Table 22: CADTH Cost Comparison Table — New Combination Product and Individual Components
Drug or comparator | Strength | Dosage form | Price ($) | Recommended dosage | Average annual drug cost ($) | Incremental cost vs. Bijuva ($) |
|---|---|---|---|---|---|---|
Combination product | ||||||
Estradiol-progesterone (Bijuva) | 0.5 mg/100 mg 1 mg/100 mg | Capsule | 0.8962a | 1 tablet daily | 327 | Reference |
Individual components | ||||||
17beta-estradiol (generic) | 0.5 mg 1 mg 2 mg | Tablet | 0.1199 0.2313 0.4083 | 1 tablet daily from day 1 to day 21 up to day 25 each monthb | 30 to 36 58 to 69 103 to 122 | NA |
Progesterone (generic) | 100 mg | Capsule | 1.4358c | 200 mg daily for the last 12 days to 14 days of estrogen treatment per cycleb | 414 to 482 | NA |
Combination of individual components | ||||||
17beta-estradiol (generic) + progesterone (generic) | 0.5 mg 100 mg | Tablet Capsule | 0.1199 1.4358c | 0.5 mg daily for 21 days to 25 days per month 200 mg for 14 days per month | 444 to 518 | 117 to 191 |
0.5 mg/100 mg dailyd | 568 | 241 | ||||
1 mg 100 mg | Tablet Capsule | 0.2313 1.4358c | 1 mg daily for 21 days to 25 days per month 200 mg daily for 14 days per month | 472 to 551 | 145 to 224 | |
1 mg/100 mg dailyd | 608 | 281 | ||||
NA = not applicable; vs. = versus.
Note: All prices are from the Ontario Drug Benefit Formulary (September 2021)25 unless otherwise specified. All dosing is from the applicable product monographs. Positive incremental costs indicate that the comparator is more expensive than Bijuva. The strength, dose form, price, and dosage for each product within a combination are presented in the order listed in the Drug/comparator column.
aSponsor’s submitted price.
bAssumes 12 months per year.
cSaskatchewan Formulary (September 2021).
dDaily use of estrogen plus progesterone combinations is based on likely use in clinical practice rather than recommended dosing from product monographs.
While the comparison of estradiol-progesterone tablets to its individual components is the most relevant for a fixed-dose combination product, MP is not reimbursed in many jurisdictions. Should estradiol-progesterone tablets be reimbursed in such jurisdictions, they are likely to displace medroxyprogesterone-based regimens and may also displace conjugated estrogen-containing regimens. When all comparators are used daily, the annual cost of estradiol-progesterone tablets is $182 to $253 more costly per patient than that of estradiol plus medroxyprogesterone regimens and $125 to $162 more costly per patient than that of conjugated estrogen plus medroxyprogesterone regimens, but $326 to $338 less costly per patient than that of conjugated estrogen plus progesterone regimens (see Table 23).
Table 23: CADTH Cost Comparison Table — Other Oral Comparator Combinations
Drug or comparator | Strength | Dosage form | Price ($) | Dosage | Average annual drug cost ($) | Incremental cost vs. Bijuva |
|---|---|---|---|---|---|---|
Combination product | ||||||
Estradiol-progesterone (Bijuva) | 0.5 mg/100 mg 1 mg/100 mg | Capsule | 0.8962a | 1 tablet daily | 327 | Reference |
17beta-estradiol + medroxyprogesterone | ||||||
17beta-estradiol (generic) + medroxyprogesterone (generic) | 0.5 mg 2.5 mg | Tablet Tablet | 0.1199 0.1183 | 0.5 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 47 to 56 | –271 to –280 |
0.5 mg/2.5 mg dailyb | 87 | –240 | ||||
1 mg 2.5 mg | Tablet Tablet | 0.2313 0.1183 | 1 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 75 to 89 | –238 to –252 | |
1 mg/2.5 mg dailyb | 128 | –200 | ||||
0.5 mg 5 mg | Tablet Tablet | 0.1199 0.0823 | 0.5 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 42 to 50 | –277 to –285 | |
0.5 mg/5 mg dailyb | 74 | –253 | ||||
1 mg 5 mg | Tablet Tablet | 0.2313 0.0823 | 1 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 70 to 83 | –244 to –257 | |
1 mg/5 mg dailyb | 114 | –213 | ||||
0.5 mg 10 mg | Tablet Tablet | 0.1199 0.1670 | 0.5 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 54 to 64 | –263 to –273 | |
0.5 mg/10 mg daily | 105 | –222 | ||||
(continued) | 1 mg 10 mg | Tablet Tablet | 0.2313 0.1670 | 1 mg daily for 21 days to 25 days per month 1 tablet daily for 12 days to 14 days per month | 82 to 97 | –230 to –245 |
1 mg/10 mg daily | 145 | –182 | ||||
Conjugated estrogen + progesterone | ||||||
Conjugated estrogen (Premarin) + progesterone (generic) | 0.3 mg 100 mg | Capsule Capsule | 0.3533c 1.4358c | 0.3 mg daily or for 25 days per month 200 mg daily for 14 days per month | 588 | 261 |
0.3 mg/100 mg daily | 653 | 326 | ||||
0.625 mg 100 mg | Capsule Capsule | 0.3707 1.4358c | 0.625 mg daily or for 25 days per month 200 mg daily for 14 days per month | 594 | 267 | |
0.625 mg/100 mg daily | 694 | 332 | ||||
1.25 mg 100 mg | Capsule Capsule | 0.3865c 1.4358c | 1.25 mg daily or for 25 days per month 200 mg daily for 14 days per month | 598 | 271 | |
1.25 mg/100 mg daily | 665 | 338 | ||||
Conjugated estrogen + medroxyprogesterone | ||||||
Conjugated estrogen (Premarin) + medroxyprogesterone (generic) | 0.3 mg 2.5 mg | Capsule Tablet | 0.3533c 0.1183 | 0.3 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 123 to 126 | –201 to –204 |
0.3 mg/2.5 mg daily | 172 | –155 | ||||
(continued) | 0.625 mg 2.5 mg | Capsule Tablet | 0.3707 0.1183 | 0.625 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 128 to 131 | –196 to –199 |
0.625 mg/2.5 mg daily | 178 | –149 | ||||
1.25 mg 2.5 mg | Capsule Tablet | 0.3865c 0.1183 | 1.25 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 133 to 136 | –191 to –194 | |
1.25 mg/2.5 mg daily | 184 | –143 | ||||
0.3 mg 5 mg | Capsule Tablet | 0.3533c 0.0823 | 0.3 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 118 to 120 | –207 to –209 | |
0.3 mg/5 mg daily | 159 | –168 | ||||
0.625 mg 5 mg | Capsule Tablet | 0.3707 0.0823 | 0.625 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 123 to 125 | –202 to –204 | |
0.625 mg/5 mg daily | 165 | –162 | ||||
1.25 mg 5 mg | Capsule Tablet | 0.3865c 0.0823 | 1.25 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 128 to 130 | –197 to –199 | |
1.25 mg/5 mg daily | 171 | –156 | ||||
0.3 mg 10 mg | Capsule Tablet | 0.3533c 0.1670 | 0.3 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 130 to 134 | –193 to –197 | |
0.3 mg/10 mg daily | 190 | –137 | ||||
(continued) | 0.625 mg 10 mg | Capsule Tablet | 0.3707 0.1670 | 0.625 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 135 to 159 | –188 to –192 |
0.625 mg/10 mg daily | 196 | –131 | ||||
1.25 mg 10 mg | Capsule Tablet | 0.3865c 0.1670 | 1.25 mg daily or for 25 days per month 1 tablet daily for 12 days to 14 days per month | 140 to 144 | –183 to –187 | |
1.25 mg/10 mg daily | 202 | –125 | ||||
vs. = versus.
Note: All prices are from the Ontario Drug Benefit Formulary (September 2021)25 unless otherwise specified. All dosing is from the applicable product monographs. Positive incremental costs indicate the comparator is more expensive than Bijuva. The strength, dose form, price, and dosage for each product within a combination are presented in the order listed in the Drug/comparator column.
aSponsor’s submitted price.
bDaily use of estrogen plus progesterone combinations is based on likely use in clinical practice rather than recommended dosing from product monographs.
cSaskatchewan Formulary (September 2021).
While the sponsor also compared estradiol-progesterone tablets to combinations including transdermal estrogen products (Table 25), the clinical expert consulted by CADTH did not find it likely that patients who would otherwise receive transdermal products would receive estradiol-progesterone tablets should they be available. CADTH, therefore, did not consider regimens including transdermal products to be important comparators.
Price Reduction Analyses
At the submitted price, estradiol-progesterone tablets are less costly than the combination of their individual components at publicly available list prices. However, particularly in jurisdictions that do not reimburse progesterone, it is likely that estradiol-progesterone tablets will displace other oral combinations of estrogen and progesterone components — the least expensive of which is 17beta-estradiol plus medroxyprogesterone.
When dispensing fees and markups are not considered, a 56% to 77% price reduction would be required for the submitted price of estradiol-progesterone tablets to be equivalent to the cost of daily estradiol plus medroxyprogesterone, depending on the doses of the individual component products considered. When Ontario-specific markups and monthly dispensing fees are included, a 26% to 47% reduction in the submitted price of estradiol-progesterone would be required for its annual cost to be equivalent to that of daily estradiol plus medroxyprogesterone (refer to Table 24).
Table 24: CADTH Price Reduction Analyses
Scenario | Submitted price ($) | Reduction needed (%) | Reduced price ($) | Savings relative to submitted pricea ($) |
|---|---|---|---|---|
Price reduction required to equal least expensive comparator excluding markups and dispensing fees (17beta-estradiol plus medroxyprogesterone)b | 0.8962 | 56% to 77%d | 0.2022 to 0.3496d | 182 to 253 |
Price reduction required to equal least expensive comparator including markups and dispensing fees (17beta-estradiol plus medroxyprogesterone)b, e | 0.8962 | 26% to 47%d | 0.4714 to 0.6677 | 90 to 168 |
aSavings from the sponsor list price per patient per year.
bAssumes daily use of comparator regimen.
dThe exact reduction required depends on the dose of 17beta-estradiol and medroxyprogesterone selected. The most frequently reimbursed dose of 17beta-estradiol tablets is 1 mg, while the most frequently prescribed dose of medroxyprogesterone is 2.5 mg, which together have a daily cost of $0.3496.24
eAnalysis assumes a markup of 8% and dispensing fees of $8.83 per claim. Twelve claims per year are assumed for estradiol-progesterone tablets, while 24 claims per year are assumed for the combination of 17beta-estradiol plus progesterone.
Issues for Consideration
Possibility of cyclical treatment: While the recommended dosing of estradiol-progesterone tablets is once daily, according to the clinical expert consulted by CADTH, where it is deemed appropriate for a patient to receive progesterone for only part of a month (e.g., for 14 days per month), these patients could receive estradiol-progesterone tablets for these days and estradiol alone for the remaining portion of the cycle. While incremental drug costs or savings associated with estradiol-progesterone tablets relative to other regimens would be lessened in these circumstances compared to a daily dosing schedule, such a regimen would no longer lead to a savings in dispensing fees.
Discussion
Summary of Available Evidence
Clinical evidence from 1 clinical study submitted and summarized by the sponsor evaluates the efficacy and safety of the combination of E2 and MP (Bijuva) for women (40 years old to 65 years old) who are experiencing menopause with moderate-to-severe VMS. The REPLENISH trial originally randomized 1,845 patients; of these, the investigators evaluated the safety of estradiol-progesterone in 1,835 patients. The trial included a VMS substudy population to evaluate efficacy in 766 patients who were randomized 1:1:1:1:1 to estradiol-progesterone 1 mg/100 mg, 0.5 mg/100 mg, 0.5 mg/50 mg, 0.25 mg/50 mg, or placebo in a double-blind, parallel design. This tailored CADTH review includes the sponsor-submitted efficacy evaluation of the 1 mg/100 mg group and the 0.5 mg/100 mg group (relative to placebo), since these are the doses approved in Canada. Safety data from the overall study was also submitted by the sponsor and presented in this report as the safety population and ES population.
Eligible patients were women in menopause with moderate-to-severe VMS and an intact uterus. Patients must have had reported 7 or more episodes of moderate-to-severe hot flushes, or 50 or more episodes per week at baseline to enter the VMS substudy, have a body mass index of 34 kg/m2 or less, and no history of thromboembolic disorders; coronary or cerebrovascular disease; undiagnosed vaginal bleeding; endometrial, breast, or ovarian cancer; or any estrogen-dependent neoplasia.
The co-primary end points evaluated were the mean change in frequency and severity of moderate-to-severe VMS from baseline to week 4 and week 12. Secondary end points included the mean change in frequency and severity of moderate-to-severe VMS from baseline to each week up to week 12, as well as mild symptoms evaluated separately. The proportion of patients with a reduction of 50% or more and, separately, 75% or more in frequency of moderate-to-severe VMS from baseline at each week up to week 12 were also evaluated. Furthermore, investigators evaluated the CGI distribution (number and percentage of patients) at week 4, week 8, and week 12, with mean change in the frequency of moderate-to-severe VMS from baseline summarized by different categories of change based on the CGI, with focus of change at week 12 to estimate a minimal important difference (MID) and responder groups. HRQoL was also assessed as a secondary end point using the change from baseline in MENQOL and the MOS Sleep Scale (sleep parameters). Patients were followed up and evaluated for up to 360 days (double-blind phase).
Overall, no major limitations in terms of the randomization process, allocation concealment, and outcome assessment were detected, with an appropriate balance of baseline prognostic variables between the arms of the VMS substudy. Some limitations included missingness of data due to assessment of the end points as complete cases available, which leads to imprecision and risk of bias. In general, the population included is reflective of a majority of women seeking treatment for VMS in Canadian clinical practice, but some issues of external validity were identified given the exclusion of certain high-risk patient groups from the trial (e.g., VTE, CVD) and uncertainty about longer-term outcomes (harms) such as cancer or cardiovascular risks.
The submitted information for this review included a bioequivalence study (Study 459), which was assessed by Health Canada to compare the bioavailability of Bijuva (estradiol-progesterone) 2 mg/200 mg to Estrace (estradiol tablets) 2 mg and Prometrium (progesterone capsules) 200 mg in healthy postmenopausal patients under high-fat and high-calorie fed conditions.26 Due to large intrasubject variability in the pharmacokinetics of progesterone in previous pharmacologic studies, a scaled average bioequivalence method for highly variable drugs was used to assess the pharmacokinetics and oral bioavailability of the estradiol-progesterone combination capsule (Bijuva). In this open-label randomized bioequivalence study, the combination of estradiol-progesterone 2 mg/200 mg demonstrated bioequivalence to Prometrium 200 mg and Estrace 2 mg among 62 healthy postmenopausal patients.27 While the 2 mg estradiol/200 mg progesterone capsule strength is not being proposed for marketing authorization, the capsule fill contains the same ingredients in the same proportions as the 1 mg/100 mg capsule strength and was manufactured using a comparable process. Therefore, the US FDA and Health Canada judged it to be representative of the commercial product.26
Interpretation of Results
Efficacy
The REPLENISH trial evaluated frequency and severity of VMS as co-primary end points. As secondary end points, investigators performed responder analyses of the number of patients with a reduction of 50% or more and, separately, 75% or more in frequency of symptoms, based on thresholds anchored to the CGI (thresholds were “very much” and “much improved”), and HRQoL (MENQOL, MOS Sleep Scale). All end points were evaluated through to 12 weeks, a time point considered of clinical relevance and likely to be important to patients, according to the clinical expert consulted by CADTH.
For the co-primary end points, the number of moderate-to-severe VMS events were reduced from baseline at 12 weeks by an average of 55.1, 53.7, and 40.2 events per week in the estradiol-progesterone 1 mg/100 mg arm, 0.5 mg/100 mg arm, and placebo arm, respectively (from an average baseline of 72 episodes per week at baseline in all arms). While there is no consensus on how to define a clinically relevant decrease in VMS, the clinical expert consulted for this review believed that a 50% reduction in weekly VMS episodes is clinically meaningful. For the same end point of VMS frequency of symptoms, a responder analysis based on the number of patients with a reduction of 50% or more and, separately, 75% or more in frequency of moderate-to-severe symptoms from baseline at week 12 was performed. The results showed a clinically meaningful reduction, with 79% of patients and 80.6% of patients in the 1 mg E2/100 mg MP group and 0.5 mg E2/100 mg MP groups, respectively, having a 50% or more reduction in frequency of moderate-to-severe symptoms.
This implied an absolute increase (improvement) in 20.7% of patients in the 1 mg E2/100 mg MP group and 22.3% of patients in the 0.5 mg E2/100 mg MP group when compared to placebo (58.3% event rate). It is important to highlight the improvement in the placebo group at week 12, although this number is not high when using the 75% or more reduction definition (32.2% of patients in the placebo group). This placebo effect was expected and, in the opinion of the clinical expert consulted by CADTH, is commonly observed in clinical practice. It can be due to several factors, such as placebo effects per se, regression to the mean, or natural evolution of the disease.
Similarly, an anchor-based (discriminant analysis) method using the CGI was used to obtain the threshold for a meaningful decrease in weekly moderate-to-severe VMS, based on the best discrimination between patients who reported being minimally improved and those patients who reported being much improved or very much improved, with a decrease of 36 VMS episodes at week 4 and a decrease of 39 VMS episodes at week 12 as the threshold to define responders versus nonresponders. Based on this definition, 52.2% of patients in the placebo group improved, and 23% more patients improved by using estradiol-progesterone 1 mg/100 mg and 0.5 mg/100 mg. The clinical expert considered that this difference observed at 12 weeks was meaningful.
HRQoL is considered a critical outcome from both the patient and the clinician perspectives. The estradiol-progesterone combination demonstrated improvements that will likely have impact on HRQoL domains. When interpreting the results based on MENQOL, a MID of 1 point change was considered a meaningful clinical result.21,28 All 3 arms of the REPLENISH study reached the MID at week 12 and up to 12 months of follow-up with lower (better) values in the intervention arms when compared to placebo. These results are in agreement with previous literature28 on the use of estrogens and its effect on HRQoL, as well as with the expectations from the clinical expert consulted by CADTH.
Sleeping disturbances are also important from the patient and clinician perspectives. Previous evidence suggests that estrogens improve measures of sleep when compared to placebo.28 In the evaluation of sleep disturbances, investigators used the MOS Sleep Scale total sleep score measured at week 12, month 6, and month 12, and found a statistically significant decrease in sleep disturbances at month 6 and month 12 compared to placebo (except for the 1 mg E2/100 mg MP dose at month 6 and the 0.5 mg E2/100 mg MP dose at week 12). As with other analyses, this analysis was based on complete cases, and some imprecision and uncertainty in estimates was observed.
One bioequivalence study using a reference-scaled bioequivalence approach demonstrated comparative bioavailability of the fixed-dose combination of estradiol-progesterone (Bijuva) to its individual components.
Harms
Potential harms are critical factors for decision-making for patients and clinicians, particularly those related to the increased risk of cancer, CVD, and thrombosis.
AEs were more frequent in the 2 intervention arms compared to the placebo arm. The most common AEs were headache, breast tenderness, nasopharyngitis, vaginal hemorrhage, vaginal discharge, abdominal pain, and dizziness. Most AEs were of mild to moderate severity.
No cases of endometrial hyperplasia were observed during the trial in all 3 groups over 12 months of follow-up, and there were 3 cases of breast cancer, all in the intervention arms and none in the placebo arm. The percentages of other AEs of special interest such as VTE, superficial thromboses, cardiovascular events, cerebrovascular events, syncope, and malignancies were low and did not occur with greater frequency in the intervention arms when compared to placebo. These numbers were consistent with what was expected for this population, according to the clinical expert consulted by CADTH.
The length of follow-up for assessing AEs was a total of 12 months for the safety population. However, longer follow-up is desirable to address long-term occurring harms, especially those correlated with time of treatment and doses.
Cost
At the submitted price of $0.90 per 0.5 mg/100 mg or 1 mg/100 mg tablet, the annual cost of estradiol-progesterone tablets is $327 per patient. This annual cost is less than that of its individual components when used daily ($568 to $608 per patient annually) or cyclically ($444 to $551 per patient annually). However, estradiol-progesterone tablets may displace other oral combination regimens, particularly in jurisdictions that do not reimburse progesterone. Estradiol-progesterone tablets are less expensive than combinations of conjugated estrogen and progesterone ($588 to $694 per patient annually) but more expensive than combinations of estradiol or conjugated estrogen plus medroxyprogesterone ($74 to $202 per patient annually). Additionally, the use of estradiol-progesterone tablets would be associated with up to 12 fewer dispensing fees per year compared to combinations of estrogen and progesterone individual components. These incremental costs or savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans.
Conclusions
Evidence from a single randomized placebo-controlled trial, the REPLENISH VMS substudy, showed that in women 40 years old to 65 years old with moderate-to-severe VMS during menopause and no cardiovascular, VTE, or cancer risk factors, E2-MP combination — either at 1 mg E2/100 mg MP or 0.5 mg E2/100 mg MP — improved the frequency and severity of VMS (co-primary end points) at 12 weeks compared to placebo. The improvements observed were considered clinically meaningful as were the results for secondary end points, such as the proportion of patients achieving a reduction of 50% or more and, separately, 75% or more in the frequency of moderate and severe VMS from baseline to week 12; the CGI score; HRQoL; and sleep quality. All of these results favoured treatment with the E2-MP combination. There was uncertainty in the evidence from imprecision of the treatment effect estimates obtained and the risk of bias due to missingness of data (analysis by available cases). One bioequivalence study using a reference-scaled bioequivalence approach demonstrated comparative bioavailability of the fixed-dose combination of estradiol-progesterone (Bijuva) to its individual components.
AEs were more frequent in the estradiol-progesterone combination treatment arms compared to the placebo arm, including breast tenderness, vaginal bleeding, headaches, and dizziness, although most of these AEs were well tolerated. No cases of endometrial hyperplasia were observed. Cardiovascular events, cerebrovascular events, cancer, and thrombosis were present in a small number of patients, with no important differences between intervention and placebo groups. However, longer follow-up is desirable to ascertain possible long-term effects and harms.
At the submitted price, the annual cost of estradiol-progesterone tablets is $327 per patient, which is less expensive than that of its individual components when used daily ($568 to $608 per patient annually) or cyclically ($444 to $551 per patient annually). Estradiol-progesterone tablets are also less expensive than combinations of conjugated estrogen and progesterone ($588 to $694 per patient annually) but more expensive than estradiol or conjugated estrogen plus medroxyprogesterone regimens ($74 to $202 per patient annually). These incremental costs or savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans.
References
1.Thurston RC, Joffe H. Vasomotor symptoms and menopause: findings from the Study of Women's Health across the Nation. Obstet Gynecol Clin North Am. 2011;38(3):489-501. PubMed
2.Woods NF, Mitchell ES. Symptoms during the perimenopause: prevalence, severity, trajectory, and significance in women's lives. Am J Med. 2005;118 Suppl 12B:14-24. PubMed
3.Freeman EW, Sammel MD, Sanders RJ. Risk of long-term hot flashes after natural menopause: evidence from the Penn Ovarian Aging Study cohort. Menopause. 2014;21(9):924-932. PubMed
4.Freeman EW, Sherif K. Prevalence of hot flushes and night sweats around the world: a systematic review. Climacteric. 2007;10(3):197-214. PubMed
5.Yuksel N, Evaniuk D, Huang L, et al. Guideline No. 422a: Menopause: Vasomotor Symptoms, Prescription Therapeutic Agents, Complementary and Alternative Medicine, Nutrition, and Lifestyle. J Obstet Gynaecol Can. 2021. PubMed
6.Minkin MJ, Reiter S, Maamari R. Prevalence of postmenopausal symptoms in North America and Europe. Menopause. 2015;22(11):1231-1238. PubMed
7.Archer DF, Sturdee DW, Baber R, et al. Menopausal hot flushes and night sweats: where are we now? Climacteric. 2011;14(5):515-528. PubMed
8.Avis NE, Crawford SL, Greendale G, et al. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med. 2015;175(4):531-539. PubMed
9.Sarrel P, Portman D, Lefebvre P, et al. Incremental direct and indirect costs of untreated vasomotor symptoms. Menopause. 2015;22(3):260-266. PubMed
10.The NHTPSAP. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24(7):728-753. PubMed
11.Maclennan AH, Broadbent JL, Lester S, Moore V. Oral oestrogen and combined oestrogen/progestogen therapy versus placebo for hot flushes. Cochrane Database Syst Rev. 2004(4):CD002978. PubMed
12.Kronenberg F. Hot flashes: epidemiology and physiology. Ann N Y Acad Sci. 1990;592:52-86; discussion 123-133. PubMed
13.Oliver-Williams C, Glisic M, Shahzad S, et al. The route of administration, timing, duration and dose of postmenopausal hormone therapy and cardiovascular outcomes in women: a systematic review. Hum Reprod Update. 2019;25(2):257-271. PubMed
14.Fournier A, Mesrine S, Dossus L, Boutron-Ruault MC, Clavel-Chapelon F, Chabbert-Buffet N. Risk of breast cancer after stopping menopausal hormone therapy in the E3N cohort. Breast Cancer Res Treat. 2014;145(2):535-543. PubMed
15.Cordina-Duverger E, Truong T, Anger A, et al. Risk of breast cancer by type of menopausal hormone therapy: a case-control study among post-menopausal women in France. PLoS ONE. 2013;8(11):e78016. PubMed
16.Stute P, Wildt L, Neulen J. The impact of micronized progesterone on breast cancer risk: a systematic review. Climacteric. 2018;21(2):111-122. PubMed
17.Charity D. Menopause: Hot flushes and sweats. healthtalk 2018; https://healthtalk.org/menopause/hot-flushes-and-sweats. Accessed 2021 Sep 13, 2021.
18.Drug Reimbursement Review sponsor submission: Bijuva (estradiol and progesterone), 0.5 mg estradiol/100 mg progesterone and 1 mg estradiol/100 mg progesterone, oral capsules [internal sponsor's package]. Montreal (QC): Knight Therapeutics Inc.; 2021 Jun 30.
19.Bijuva (estradiol and progesterone), 0.5 mg estradiol/100 mg progesterone and 1 mg estradiol/100 mg progesterone, oral capsules [product monograph]. Montreal (QC): Knight Therapeutics Inc.; 2021 Jun 30.
20.Goldstein S. An efficient tool for the primary care management of menopause. Can Fam Physician. 2017;63(4):295-298. PubMed
21.Lewis JE, Hilditch JR, Wong CJ. Further psychometric property development of the Menopause-Specific Quality of Life questionnaire and development of a modified version, MENQOL-Intervention questionnaire. Maturitas. 2005;50(3):209-221. PubMed
22.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Bijuva (estradiol and progesterone), 0.5 mg estradiol/100 mg progesterone and 1 mg estradiol/100 mg progesterone, oral capsules Montreal (QC): Knight Therapeutics. 2021.
23.Mauvais-Jarvis F, Manson JE, Stevenson JC, Fonseca VA. Menopausal Hormone Therapy and Type 2 Diabetes Prevention: Evidence, Mechanisms, and Clinical Implications. Endocr Rev. 2017;38(3):173-188. PubMed
24.PharmaStat. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Aug 22.
25.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Aug 24.
26.DBE2 Comprehensive Summary: Bioequivalence (CS-BE) Review, dated July 17, 2020. 2020.
27.Pickar JH, Bon C, Amadio JM, Mirkin S, Bernick B. Pharmacokinetics of the first combination 17beta-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause. 2015;22(12):1308-1316. PubMed
28.Grant MD, Marbella A, Wang AT, et al. Menopausal Symptoms: Comparative Effectiveness of Therapies [Internet]. Agency for Healthcare Research and Quality (US). 2015:03.
29.Stankiewicz AP, L; Popadiuk, C;. Prevalence of self-reported hysterectomy among Canadian women, 2000/2001–2008. Chronic Diseases and Injuries in Canada, 2014; https://www.canada.ca/en/public-health/services/reports-publications/health-promotion-chronic-disease-prevention-canada-research-policy-practice/vol-34-no-1-2014/prevalence-self-reported-hysterectomy-among-canadian-women-20002001-2008.html. Accessed 2021 09 15, 2021.
30.Deecher DC, Dorries K. Understanding the pathophysiology of vasomotor symptoms (hot flushes and night sweats) that occur in perimenopause, menopause, and postmenopause life stages. Arch Womens Ment Health. 2007;10(6):247-257. PubMed
31.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2018 to 2019. Ottawa (ON): Indigenous Services Canada,; 2020: https://www.sac-isc.gc.ca/eng/1584392581890/1584393350542. Accessed 2021 08 16.
32.Sutherland GD, T. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. The Conference Board of Canada,; 2017: https://www.conferenceboard.ca/temp/758ed58f-2652-4178-9596-52557e8c3793/9326_Understanding-the-Gap__RPT.pdf?bcs-agent-scanner=079fbd59-37ed-dd44-9cdb-85b3ca35a1c0.
33.Table: 17-10-0057-01. Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2019: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2021 Aug 22.
34.Chen I, Choudhry AJ, Tulandi T. Hysterectomy Trends: A Canadian Perspective on the Past, Present, and Future. Journal of Obstetrics and Gynaecology Canada. 2019;41:S340-S342. PubMed
Appendix 1: Study Design Description
Note that this appendix has not been copy-edited.
Figure 1: Overall Design of the REPLENISH Study
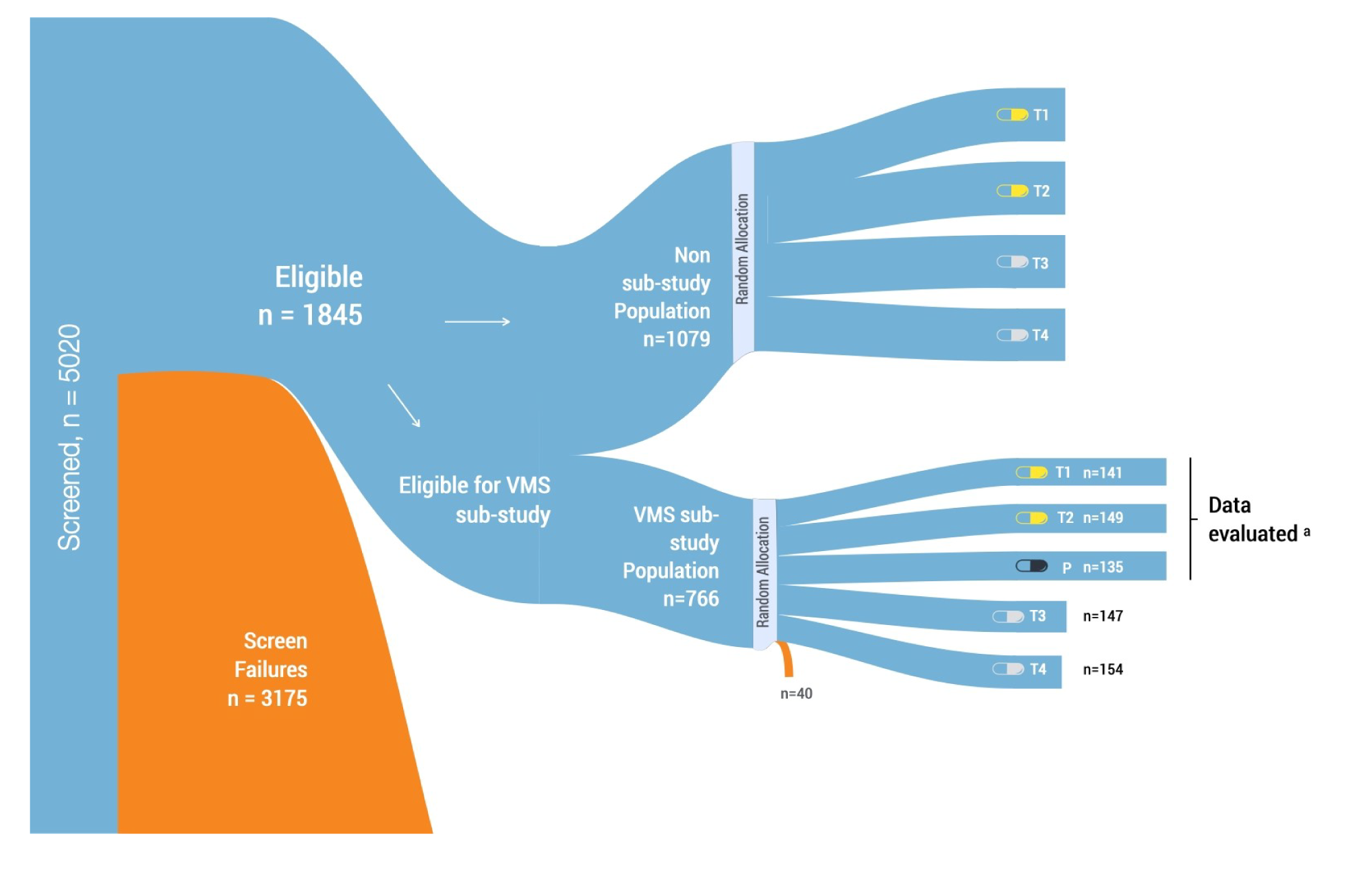
P = placebo; T1 = estradiol-progesterone 1 mg /100 mg; T2 = estradiol-progesterone 0.5 mg/100 mg; T3 = estradiol-progesterone 0.5 mg /50 mg; T4 = estradiol-progesterone 0.25 mg/50 mg; VMS = vasomotor symptoms.
a Efficacy end points were assessed in the VMS substudy. For this CADTH-tailored review, only the dosages approved in Canada are presented (i.e., T1 = estradiol-progesterone 1 mg/100 mg; T2 = estradiol-progesterone 0.5 mg/100 mg).
Source: Information from the Clinical Study Report for REPLENISH (2021).18
Appendix 2: Additional Economic Information
Note that this appendix has not been copy-edited.
Additional Details on the Sponsor’s Submission
Table 25: Sponsor’s Comparative Treatment Costs
Generic name (brand name) | Annual drug cost ($) | Difference in drug acquisition costs per year | Difference in total costs per year |
|---|---|---|---|
Estradiol-progesterone (Bijuva; 0.5 mg/100 mg) | $327 | — | — |
Estradiol-progesterone (Bijuva; 1 mg/100 mg) | $327 | — | — |
Oral comparators | |||
Estrace | |||
17beta-estradiol (Estrace; 0.5 mg) + progesterone (Prometrium) | $599 | –$271 | –$271 |
17beta-estradiol (Estrace; 0.5 mg) + progesterone (Teva-Progesterone) | $573 | –$246 | –$246 |
17beta-estradiol (Estrace; 0.5 mg) + medroxyprogesterone (Provera) | $144 | $184 | $184 |
17beta-estradiol (Estrace; 0.5 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $66 | $261 | $261 |
17beta-estradiol (Estrace; 1 mg) + progesterone (Prometrium) | $644 | –$317 | –$317 |
17beta-estradiol (Estrace; 1 mg) + progesterone (Teva-Progesterone) | $619 | –$292 | –$292 |
17beta-estradiol (Estrace; 1 mg) + medroxyprogesterone (Provera) | $189 | $138 | $138 |
17beta-estradiol (Estrace; 1 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $112 | $215 | $215 |
17beta-estradiol (Estrace; 2 mg) + progesterone (Prometrium) | $717 | NA* | NA* |
17beta-estradiol (Estrace; 2 mg) + progesterone (Teva-Progesterone) | $692 | NA* | NA* |
17beta-estradiol (Estrace; 2 mg) + medroxyprogesterone (Provera) | $262 | NA* | NA* |
17beta-estradiol (Estrace; 2mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $185 | NA* | NA* |
Generic estradiol | |||
17beta-estradiol (Lupin-Estradiol; 0.5 mg) + progesterone (Prometrium) | $585 | –$258 | –$258 |
17beta-estradiol (Lupin-Estradiol; 0.5 mg) + progesterone (Teva-Progesterone) | $560 | –$233 | –$233 |
17beta-estradiol (Lupin-Estradiol; 0.5 mg) + medroxyprogesterone (Provera) | $130 | $197 | $197 |
17beta-estradiol (Lupin-Estradiol; 0.5 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $52 | $275 | $275 |
17beta-estradiol (Lupin-Estradiol; 1 mg) + progesterone (Prometrium) | $619 | –$291 | –$291 |
17beta-estradiol (Lupin-Estradiol; 1 mg) + progesterone (Teva-Progesterone) | $593 | –$266 | –$266 |
17beta-estradiol (Lupin-Estradiol; 1 mg) + medroxyprogesterone (Provera) | $164 | $164 | $164 |
17beta-estradiol (Lupin-Estradiol; 1 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $86 | $241 | $241 |
17beta-estradiol (Lupin-Estradiol; 2 mg) + progesterone (Prometrium) | $672 | NA* | NA* |
17beta-estradiol (Lupin-Estradiol; 2 mg) + progesterone (Teva-Progesterone) | $646 | NA* | NA* |
17beta-estradiol (Lupin-Estradiol; 2 mg) + medroxyprogesterone (Provera) | $217 | NA* | NA* |
17beta-estradiol (Lupin-Estradiol; 2 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $139 | NA* | NA* |
Premarin | |||
Conjugated estrogen (Premarin; 0.3 mg) + progesterone (Prometrium) | $671 | –$344 | –$344 |
Conjugated estrogen (Premarin; 0.3 mg) + progesterone (Teva-Progesterone) | $646 | –$319 | –$319 |
Conjugated estrogen (Premarin; 0.3 mg) + medroxyprogesterone (Provera) | $216 | $111 | $111 |
Conjugated estrogen (Premarin; 0.3 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $139 | $189 | $189 |
Conjugated estrogen (Premarin; 0.625 mg) + progesterone (Prometrium) | $677 | –$350 | –$350 |
Conjugated estrogen (Premarin; 0.625 mg) + progesterone (Teva-Progesterone) | $652 | –$325 | –$325 |
Conjugated estrogen (Premarin; 0.625 mg) + medroxyprogesterone (Provera) | $222 | $105 | $105 |
Conjugated estrogen (Premarin; 0.625 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $145 | $183 | $183 |
Conjugated estrogen (Premarin; 1.25 mg) + progesterone (Prometrium) | $683 | NA* | NA* |
Conjugated estrogen (Premarin; 1.25 mg) + progesterone (Teva-Progesterone) | $658 | NA* | NA* |
Conjugated estrogen (Premarin; 1.25 mg) + medroxyprogesterone (Provera) | $228 | NA* | NA* |
Conjugated estrogen (Premarin; 1.25 mg) + medroxyprogesterone (Teva-Medroxyprogesterone) | $150 | NA* | NA* |
Transdermal comparators | |||
Climara | |||
17beta-estradiol (Climara) + progesterone (Prometrium) | $837 | –$510 | –$510 |
17beta-estradiol (Climara) + progesterone (Teva-Progesterone) | $812 | –$484 | –$484 |
17beta-estradiol (Climara) + medroxyprogesterone (Provera) | $382 | –$55 | –$55 |
17beta-estradiol (Climara) + medroxyprogesterone (Teva-Medroxyprogesterone) | $304 | $23 | $23 |
Sandoz Estradiol Derm | |||
17beta-estradiol (Sandoz Estradiol Derm) + progesterone (Prometrium) | $767 | –$440 | –$440 |
17beta-estradiol (Sandoz Estradiol Derm) + progesterone (Teva-Progesterone) | $742 | –$415 | –$415 |
17beta-estradiol (Sandoz Estradiol Derm) + medroxyprogesterone (Provera) | $312 | $15 | $15 |
17beta-estradiol (Sandoz Estradiol Derm) + medroxyprogesterone (Teva-Medroxyprogesterone) | $234 | $93 | $93 |
Estradot | |||
17beta-estradiol (Estradot) + progesterone (Prometrium) | $874 | –$547 | –$547 |
17beta-estradiol (Estradot) + progesterone (Teva-Progesterone) | $849 | –$522 | –$522 |
17beta-estradiol (Estradot) + medroxyprogesterone (Provera) | $419 | –$92 | –$92 |
17beta-estradiol (Estradot) + medroxyprogesterone (Teva-Medroxyprogesterone) | $342 | –$15 | –$15 |
Estrogel | |||
17beta-estradiol (Estrogel) + progesterone (Prometrium) | $872 | –$545 | –$545 |
17beta-estradiol (Estrogel) + progesterone (Teva-Progesterone) | $847 | –$519 | –$519 |
17beta-estradiol (Estrogel) + medroxyprogesterone (Provera) | $417 | –$90 | –$90 |
17beta-estradiol (Estrogel) + medroxyprogesterone (Teva-Medroxyprogesterone) | $339 | –$12 | –$12 |
Divigel | |||
17beta-estradiol (Divigel) + progesterone (Prometrium) | $848 | –$521 | –$521 |
17beta-estradiol (Divigel) + progesterone (Teva-Progesterone) | $823 | –$496 | –$496 |
17beta-estradiol (Divigel) + medroxyprogesterone (Provera) | $393 | –$66 | –$66 |
17beta-estradiol (Divigel) + medroxyprogesterone (Teva-Medroxyprogesterone) | $315 | $12 | $12 |
Estalis | |||
17beta-estradiol–norethindrone acetate (Estalis) | $364 | –$37 | –$37 |
Oesclim | |||
17beta-estradiol (Oesclim) + progesterone (Prometrium) | $862 | –$535 | –$535 |
17beta-estradiol (Oesclim) + progesterone (Teva-Progesterone) | $837 | –$510 | –$510 |
17beta-estradiol (Oesclim) + medroxyprogesterone (Provera) | $407 | –$80 | –$80 |
17beta-estradiol (Oesclim) + medroxyprogesterone (Teva-Medroxyprogesterone) | $329 | –$2 | –$2 |
Oral combination | |||
17beta-estradiol–drospirenone (Angeliq) | Not listed | — | — |
Estradiol-norethindrone acetate (Activelle) | Not listed | — | — |
Conjugated estrogens-bazedoxifene acetate (Duavive) | Not listed | — | — |
Selective tissue estrogenic activity regulator (STEAR) | |||
Tibolone (Tibella) | Not listed | — | — |
NA = not applicable.
*As mentioned in the preceding assumptions table, since there isn’t a dose of Bijuva clinically equivalent to Estrace 2 mg, Lupin-Estradiol 2 mg and Premarin 1.25 mg, Bijuva is not expected to displace these treatment options. Therefore, annual cost differences were not calculated for these comparators.
Appendix 3: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 26: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted an epidemiology-based model29 to estimate the budgetary impact of reimbursing estradiol-progesterone tablets for the treatment of moderate-to-severe VMS associated with menopause in patients with an intact uterus.19 The sponsor’s analysis was conducted over a 3-year time period (2023 to 2025) where the total number of people at risk of VMS was estimated by first identifying the total number of female persons aged 45 to 64 in each provincial jurisdiction using Statistics Canada projections. It was then estimated that 85% would experience VMS during menopause based on values from the literature,2,20,30 and of these, 40% would experience moderate-to-severe symptoms, with 25% seeking out and receiving treatment, both cited by the sponsor as based on clinical expert opinion. Similarly, the Non-Insured Health Benefits (NIHB) 2019 annual report31 was used to estimate the number of patients who would be reimbursed by NIHB, with the same aforementioned assumptions applied to estimate the population of interest. NIHB recipients residing within the borders of 1 of the included provinces were subtracted from the total population of that province to avoid double counting.
The number of patients who would be reimbursed by the public drug plans was then estimated by multiplying the proportion of the population within each jurisdiction considered eligible for public coverage by the proportion who do not have private insurance.32 Finally, an annual growth rate was applied based on Statistics Canada’s projected change in the population of people aged 45 years to 64 years.33
The reference scenario (World Without in Figure 2) included oral and transdermal estradiol and conjugated estrogen products in combination with oral progesterone or medroxyprogesterone products. The 2 components of each regimen were assumed to be selected independently. Market shares within each jurisdiction were derived from 2020 IQVIA claims data for the provincial jurisdictions, and 2019 data for NIHB. List prices as well as markups and dispensing fees were also applied as appropriate to each jurisdiction to calculate the total cost of the reference scenario.
Assumed displacement rates when estradiol-progesterone tablets are introduced were complex, jurisdiction-specific, and relied on the sponsor’s internal forecasts. The majority of estradiol-progesterone market share was assumed to come from combinations of the individual component products where those products are already reimbursed, and from oral estradiol plus medroxyprogesterone and conjugated estrogen plus medroxyprogesterone where progesterone is not reimbursed. In jurisdictions which reimburse transdermal products, 20% of the market share captured by estradiol-progesterone tablets was assumed to come from combinations including transdermal products. Overall uptake was assumed to be higher in jurisdictions which already reimburse progesterone, and ranged from 0.6% to 1.7% of the overall market share of VMS treatments in year 1, rising to 2.7% to 6% by year 3.
State the key assumptions:
The number of people who will experience menopause can be proxied by the number of female persons reported by Statistics Canada in the 45-year-old to 64-year-old age bracket.
Patient eligibility for public drug plan coverage is independent of access to a private drug plan.
Thirteen cycles of treatment per year were assumed.
Adherence was assumed to be 100%.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
Source: Figure 1 from Sponsor’s submitted Budget Impact Analysis report.8
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s base case suggest that the incremental budget impact associated with the reimbursement of estradiol-progesterone tablets would be a savings of $111,767 in year 1, $258,208 in year 2, and $386,108 in year 3, for a cumulative 3-year budgetary savings of $756,083 when including markups and dispensing fees every 28 days. When dispensing fees and markups are excluded, the 3-year budgetary impact is an increase in costs of $28,075.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Proportion of patients with VMS overestimated: The sponsor estimated that 85% of patients would experience VMS symptoms in menopause, based on values identified in the literature.2,20,30 However, the studies cited by the sponsor report that up to 80% of patients will experience VMS,20,30 or that 85% of patients will experience at least 1 of any type of symptom associated with menopause, inclusive of non-VMS symptoms.2 The clinical expert consulted by CADTH estimated that 75% of patients in menopause experience VMS, which is consistent with the literature values.
CADTH reanalyses assumed 75% of patients in menopause will experience VMS.
Population not limited to those with an intact uterus: The indication for estradiol-progesterone tablets is limited to patients with an intact uterus, however in deriving the eligible population, the sponsor included all patients experiencing VMS, regardless of uterine status. A 2014 prevalence study reported that between 2000 and 2008, 13.9% of women in Canada aged 40 years to 49 years, 29.4% of women aged 50 years to 59 years, and 38.1% of women aged 60 years to 69 years had undergone a hysterectomy.29 However, the prevalence of hysterectomy has been decreasing in Canada over time as new therapies and practices become available.34 The clinical expert consulted by CADTH estimated that of patients requiring treatment for VMS, at the current time, approximately 25% would not have an intact uterus.
CADTH reanalyses assumed that 75% of patients requiring treatment for VMS would have an intact uterus.
Public drug plan beneficiaries underestimated: The sponsor calculated the number of beneficiaries within each jurisdiction by multiplying the proportion of people who are eligible for each public plan by the proportion of people who do not have private insurance, as reported by the Conference Board of Canada in 2017.32 This method does not account for variability in the way each jurisdiction reports eligibility (e.g., whether personal income or medical care costs are considered before determining ‘eligibility’ or after). Additionally, the method assumes that eligibility for public reimbursement is independent of having access to a private insurance plan, an unlikely scenario given that private insurance is most frequently tied to employment in Canada, and thus to higher income levels, while reimbursement by public drug plans is correlated to lower levels of income in patients under 65 years of age.
CADTH reanalyses considered the proportion of the population aged 25 years to 64 years who are enrolled in each public plan to be a better approximation of the proportion of patients who would be publicly reimbursed for VMS-related treatments.32
Comparator dosing inappropriately estimated: The sponsor’s analysis estimated the dosing of estrogen and progesterone comparator components by using the middle value of dose ranges recommended in the appropriate product monographs (e.g., if the recommended dose was 21 to 25 days per cycle, the sponsor assumed usage 23 days out of 28). However, the clinical expert consulted by CADTH estimated that approximately 70% of patients use daily dosing, similar to the recommended dose schedule of estradiol-progesterone tablets. Additionally, the sponsor’s analysis assumes that patients are using a 28-day cycles, whereas patients primarily use calendar months to simplify regimens. Finally, the sponsor estimated a uniform distribution of use across medroxyprogesterone doses, which is not consistent with 2020 public claims data from the IQVIA Pharmastat database.24 As medroxyprogesterone is used for multiple indications, the exact distribution associated with its use for patients with VMS is uncertain, however this method of estimation is more likely to be accurate than a uniform distribution as assumed by the sponsor.
CADTH reanalyses assumed that 70% of patients were receiving daily doses of their oral combination therapies, while the remaining 30% were using 30-day cycles. Dispensing fees were adjusted to be applied every 30 days rather than every 28 days. Medroxyprogesterone doses were assumed be used in the proportions reported for each jurisdiction in public claims data from 2020.
Transdermal products are unlikely to be displaced: The sponsor’s analysis assumed that in jurisdictions which reimburse transdermal estrogen products, 20% of the market share capture of estradiol-progesterone tablets would come from the displacement of combinations which include transdermal products. The clinical expert consulted by CADTH indicated that the use of transdermal estrogen therapy was typically based on patient preference or on clinical factors such as increased risk of VTE or stroke, and thus it was unlikely that there would be much switching from regimens including a transdermal estrogen product to estradiol-progesterone tablets.
CADTH did not include transdermal comparators in its base-case reanalysis.
Uncertainty in market share uptake: The sponsor’s estimates that estradiol-progesterone tablets would capture between 2.7% and 6% of the market of patients using estrogen and progesterone combinations for the treatment of moderate-to-severe VMS by the third year of its reimbursement is based on unspecified internal forecasts and is therefore highly uncertain. In jurisdictions which reimburse progesterone, the clinical expert consulted by CADTH indicated that estradiol-progesterone tablets would displace most use of its individual components, with lower displacement of other oral combinations. Should jurisdictions which do not currently reimburse progesterone reimburse estradiol-progesterone tablets, the clinical expert consulted by CADTH predicted that estradiol-progesterone tablets would eventually displace the majority of medroxyprogesterone-based regimens, and as such, may increase budgetary costs rather than resulting in savings given the lower price of medroxyprogesterone-based regimens.
Due to a lack of alternate data to inform market uptake, CADTH was unable to address this limitation in its base-case analysis. As an exploratory scenario analysis, CADTH assumed that in jurisdictions which reimburse progesterone, estradiol-progesterone tablets would capture a market share equal to 20%, 50%, and 70% of its individual components plus 5%, 7.5%, and 10% of other oral combinations in year 1, year 2, and year 3, respectively. In jurisdictions which do not currently reimburse progesterone, CADTH assumed a market capture of 20%, 35%, and 50% of oral comparators in year 1, year 2, and year 3, respectively. This scenario is speculative in nature and demonstrates uncertainty in the budgetary impact of reimbursing estradiol-progesterone tablets due to uncertainty in its potential uptake.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s submitted analysis by decreasing the proportion of people in menopause who experience VMS, removing patients without an intact uterus from the population of interest, increasing the proportion of public drug plan beneficiaries, altering assumptions around comparator dosing, and excluding transdermal estrogen products from the analysis. (Refer to Table 27.)
Table 27: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Proportion of population with VMS | 85% | 75% |
2. Proportion of patients with intact uterus | Not considered (100%) | 75% |
3. Public beneficiaries | Proportion of population eligible for a public plan x proportion of population without private insurance | Proportion of population enrolled in a public plan |
4. Comparator dosing | All patients use cyclical dosing where recommended for comparators 28-day cycles Uniform medroxyprogesterone dosing | 70% of patients use daily dosing, 30% use cyclical for comparators Monthly cycles Medroxyprogesterone dosing based on 2020 public claims proportions |
5. Removal of transdermal products | Transdermal products included where reimbursed, making up 20% of regimens displaced by estradiol-progesterone tablets | Transdermal products excluded |
CADTH base case | Reanalyses 1 through 5 | |
The results of the CADTH step-wise reanalysis are presented in summary format in Table 28 and a more detailed breakdown is presented in Table 29. Applying these changes resulted in a 3-year budgetary savings of $358,330.
Table 28: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total |
|---|---|
Submitted base case | –$756,083 |
CADTH reanalysis 1: Lower VMS proportion | –$667,132 |
CADTH reanalysis 2: Patients without intact uteruses removed | –$567,062 |
CADTH reanalysis 3: Proportion population publicly reimbursed | –$997,258 |
CADTH reanalysis 4: Comparator dosing | –$717,165 |
CADTH reanalysis 5: transdermal products excluded | –$447,628 |
CADTH base case | –$358,330 |
VMS = vasomotor symptoms.
Of note, in both the sponsor’s and CADTH’s analyses, the exclusion of dispensing fees and markups from the analysis resulted in increased budgetary spending, indicating that overall estimated savings were due to the reduction of 12 or 13 dispensing fees per patient per year in the CADTH and sponsor’s base cases, respectively.
CADTH also conducted an exploratory scenario analysis increasing the assumed market share capture of estradiol-progesterone tablets to highlight uncertainty in the budgetary impact of the sponsor’s market share assumptions.
Table 29: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $33,616,050 | $33,639,796 | $33,660,660 | $33,716,259 | $101,016,716 |
New drug | $33,616,050 | $33,528,030 | $33,402,452 | $33,330,151 | $100,260,633 | |
Budget impact | $0 | –$111,767 | –$258,208 | –$386,108 | –$756,083 | |
Submitted scenario analysis: no fees or markups | Reference | $14,020,803 | $14,036,218 | $14,050,657 | $14,079,248 | $42,166,124 |
New drug | $14,020,803 | $14,032,444 | $14,065,413 | $14,096,342 | $42,194,200 | |
Budget impact | $0 | –$3,774 | $14,756 | $17,094 | $28,075 | |
CADTH base case | Reference | $26,236,845 | $26,179,984 | $26,120,400 | $26,084,136 | $78,384,520 |
New drug | $26,236,845 | $26,123,779 | $25,999,863 | $25,902,548 | $78,026,190 | |
Budget impact | $0 | –$56,206 | –$120,537 | –$181,588 | –$358,330 | |
CADTH scenario analysis A: no fees or markups | Reference | $10,497,769 | $10,476,153 | $10,453,410 | $10,439,649 | $31,369,212 |
New drug | $10,497,769 | $10,491,243 | $10,521,649 | $10,533,593 | $31,546,485 | |
Budget impact | $0 | $15,090 | $68,239 | $93,943 | $177,273 | |
CADTH scenario B: increased market uptake of estradiol- progesterone | Reference | $26,236,845 | $26,179,984 | $26,120,400 | $26,084,136 | $78,384,520 |
New drug | $26,236,845 | $26,582,452 | $26,792,673 | $27,068,496 | $80,443,621 | |
Budget impact | $0 | $402,468 | $672,273 | $984,360 | $2,059,101 |
Appendix 4: Sponsor References
Note that this appendix has not been copy-edited.
Constantine GD, Simon JA, Kaunitz AM, et al. TX-001HR is associated with a clinically meaningful effect on severity of moderate to severe vasomotor symptoms in the REPLENISH trial. Menopause (New York, NY). 2020;27(11):1236-1241. PubMed
Kagan R, Constantine G, Kaunitz AM, Bernick B, Mirkin S. Improvement in sleep outcomes with a 17β-estradiol-progesterone oral capsule (TX-001HR) for postmenopausal women. Menopause. 2018;26(6):622-628. PubMed
Kaunitz AM, Bitner D, Constantine GD, Bernick B, Graham S, Mirkin S. 17β-estradiol/progesterone in a single, oral, softgel capsule (TX-001HR) significantly increased the number of vasomotor symptom-free days in the REPLENISH trial. Menopause (New York, NY). 2020;27(12):1382-1387. PubMed
Lobo RA, Archer DF, Kagan R, et al. A 17β-Estradiol-Progesterone Oral Capsule for Vasomotor Symptoms in Postmenopausal Women: A Randomized Controlled Trial. Obstet Gynecol. 2018;132(1):161-170. PubMed
Mirkin S, Amadio JM, Bernick BA, Pickar JH, Archer DF. 17β-Estradiol and natural progesterone for menopausal hormone therapy: REPLENISH phase 3 study design of a combination capsule and evidence review. Maturitas. 2015;81(1):28-35. PubMed
Mirkin S, Goldstein SR, Archer DF, Pickar JH, Graham S, Bernick B. Endometrial safety and bleeding profile of a 17β-estradiol/progesterone oral softgel capsule (TX-001HR). Menopause. 2020;27(4):410-417. PubMed
Mirkin S, Graham S, Revicki DA, Bender RH, Bernick B, Constantine GD. Relationship between vasomotor symptom improvements and quality of life and sleep outcomes in menopausal women treated with oral, combined 17β-estradiol/progesterone. Menopause (New York, NY). 2019;26(6):637-642. PubMed
Hilditch JR, Lewis J, Peter A, et al. A menopause-specific quality of life questionnaire: development and psychometric properties. Maturitas. 1996;24(3):161-175. PubMed
Gerlinger C, Gude K, Hiemeyer F, Schmelter T, Schäfers M. An empirically validated responder definition for the reduction of moderate to severe hot flushes in postmenopausal women. Menopause. 2012;19(7):799-803. PubMed
Pickar JH, Bon C, Amadio JM, Mirkin S, Bernick B. Pharmacokinetics of the first combination 17β-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause (New York, NY). 2015/12// 2015;22(12):1308-1316. doi:10.1097/gme.0000000000000467
Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. Journal of clinical epidemiology. 2008;61(2):102-109. PubMed
Spritzer KL, Hays RD. MOS Sleep Scale: A manual for use and scoring. Los Angeles, CA Nov 2003.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.