CADTH Reimbursement Review
Estradiol (Imvexxy)
Sponsor: Knight Therapeutics Inc.
Therapeutic area: Dyspareunia
Clinical and Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
Budget Impact Analysis
CADTH
Canadian Agency for Drugs and Technologies in Health
CI
confidence interval
DB
double blind
FSFI
Female Sexual Function Index
IP
investigational product
ITT
intention-to-treat population
LOCF
last observation carried forward
LS
least square
mITT
modified intention to treat
MMRM
Mixed Model Repeated Measures
PP
per-protocol
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
VVA
vulvovaginal atrophy
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Estradiol (Imvexxy), 4 mcg and 10 mcg, vaginal inserts |
Indication | For the treatment of postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy |
Reimbursement request | As per indication |
Health Canada Approval Status | NOC |
Health Canada Review Pathway | Standard |
NOC date | August 17, 2020 |
Sponsor | Knight Therapeutics Inc. |
NOC = Notice of Compliance.
Introduction
Menopause is associated with both systemic and genital changes related to the progressive reduction and loss of estrogen production. Vulvovaginal atrophy (VVA) is among the most prevalent and concerning clinical condition of menopause.1 The self-reported prevalence of VVA symptoms varies from 4% in early postmenopausal years to higher than 80% in later years.1,2 The most frequently reported vaginal symptoms were vaginal dryness, dyspareunia, and decreased sexual interest. VVA-related symptoms negatively impair womens’ quality of life.1,3,4 VVA is a chronic condition that typically does not improve if left untreated.1
The first-line treatments for VVA symptoms are nonhormonal vaginal moisturizers and lubricants. For postmenopausal women who do not respond well to these, estrogen therapy or other hormonal medications can be prescribed when there are no contraindications.5,6 Vaginal estrogen therapy, such as topical creams, intravaginal tablets and/or estradiol-releasing ring, is preferred to manage the symptoms of VVA over systemic estrogen therapy, when only genitourinary symptoms are present.
The estradiol vaginal insert (Imvexxy) is a softgel formulation containing estradiol. It is available as 4 mcg and 10 mcg 17 beta-estradiol and is used intravaginally. It received Health Canada approval on August 17, 2020 for the treatment of postmenopausal moderate to severe dyspareunia, 1 of the key symptoms of VVA.7 The product monograph recommends starting at the 4 mcg dosage strength, with dosage adjustment guided by the clinical response. The initial dose is 1 vaginal insert daily at approximately the same time for 2 weeks. The maintenance dose is 1 vaginal insert twice weekly, every 3 to 4 days.8
The objective of the current review was to perform a review of the beneficial and harmful effects of the estradiol vaginal inserts in postmenopausal women with moderate to severe dyspareunia.
The clinical and pharmacoeconomic evidence for the review were provided through the CADTH tailored review process. A tailored review consists of CADTH conducting an appraisal of the clinical evidence and pharmacoeconomic evaluation filed by the sponsor using a CADTH-provided review template that is specific to the type of drug product to be reviewed.
Stakeholder Perspectives
The information in this section is a summary of input provided by the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
No input was provided by patient groups.
Clinician input
Input From Clinical Experts Consulted by CADTH
The clinical expert indicated that not all patients respond to the available treatments for dyspareunia. Some treatment options are difficult, uncomfortable, or messy to administer. Some women are reluctant to initiate hormonal treatment due to the safety concerns regarding exogenous hormone therapy.
In the clinical expert’s opinion, the estradiol vaginal insert is another form of existing medication for treatment of VVA, including dyspareunia. It would be used as a first-line treatment or after failure on other treatments for women who are suitable to receive estrogen replacement for VVA.
Most menopausal women with VVA-related symptoms are likely to benefit from vaginal estrogen therapy, such as estradiol inserts.
The clinical expert also indicated that in clinical practice, treatment response is assessed based on patient’s self report of improvement in symptoms. This is a clinically meaningful outcome measure. The expert suggested treatment response be assessed at 3 to 6 months following initiation of treatment, and again at 6 to 12 months, then yearly thereafter if continued treatment is required.
Estradiol vaginal inserts are likely prescribed in an outpatient ambulatory clinic setting by a family physician or gynecologist. The drug can be self-administered by the patient in her own home.
Clinician Group Input
No input was provided by the clinician groups.
Drug Program Input
The Pharmaceutical Advisory Committee Formulary Working Group identified the following jurisdictional implementation issues: relevant comparators, consideration for initiation of therapy, consideration for prescribing of therapy, generalizability, system issues, and economic considerations. The clinical expert consulted by CADTH weighed evidence from the REJOICE trial and other clinical considerations to provide responses to drug program implementation questions. Refer to Table 4 for more information.
Clinical Evidence
Pivotal Studies
Description of Studies
One phase III study (REJOICE, N = 574) was submitted to support the clinical benefit of estradiol vaginal inserts.9 The trial enrolled postmenopausal women with moderate to severe symptoms of vaginal pain associated with sexual activity.
The REJOICE study was a double-blind, placebo-controlled RCT that assessed the efficacy and safety of the estradiol vaginal insert for the treatment of postmenopausal moderate to severe dyspareunia. Eligible patients were randomized to receive the estradiol vaginal insert 4 mcg, 10 mcg, or 25 mcg, or placebo for 12 weeks. The results for the estradiol vaginal insert 25 mcg group are not reported in this report because this dose is not approved for use. The coprimary efficacy end points were change from baseline to week 12 in percent change in superficial cells compared to placebo, change from baseline to week 12 in percent change in parabasal cells compared to placebo, change from baseline to week 12 in percent change in pH compared to placebo, and change from baseline to week 12 on the severity of the most bothersome symptoms (MBS) of dyspareunia (vaginal pain associated with sexual activity) associated with VVA (using the VVA Symptoms Self-Assessment Questionnaire) compared to placebo. The average age of the women participating in REJOICE was 59 to 60 years. The majority of the women were White (86% to 88%). Gynecological history was similar across treatment groups, except that more patients in the estradiol 4 mcg or 10 mcg groups had prior hysterectomy (46% to 47% with estradiol versus 39% with placebo), bilateral oophorectomy (26% to 27% versus 21%) and surgical menopause (39% to 40% versus 34%). The mean time since menopause was 13.9 to 14.2 years and prior hormone replacement therapy was used in 17.6% to 19.3% of women. Baseline assessments of parabasal cells, superficial cells, vaginal pH and severity of MBS of dyspareunia were similar across treatment groups. For study participation, patients needed to identify that their MBS was moderate to severe dyspareunia. The mean baseline severity score for dyspareunia across treatment groups was 2.6 to 2.7.
Efficacy Results
After 3 months treatment, the REJOICE study met its objective by demonstrating improvement in favour of both doses of the estradiol vaginal inserts versus placebo on the 4 coprimary end points: change from baseline to week 12 in the percentage of parabasal cells, superficial cells, vaginal pH, and severity of dyspareunia. One of the outcomes was the change from baseline in patient-reported severity of dyspareunia, which was consistent with clinical practice, according to the clinical expert consulted by CADTH.
At week 12, vaginal dryness was improved with both doses of the estradiol vaginal insert compared with placebo, while only the estradiol 10 mcg group had improved vulvar and/or vaginal itching or irritation versus placebo. The expert indicated that the results of these secondary efficacy outcomes were consistent with the primary outcomes, which favoured estradiol over placebo; however, the differences between estradiol and placebo may not be considered clinically important.
According to the clinical expert, patient-reported symptom relief is a clinically relevant outcome in the study population. In REJOICE, a VVA Symptoms Self-Assessment Questionnaire was used to self-assess patient’s symptoms of VVA, including vaginal pain associated with sexual activity, vaginal dryness, and vulvar and/or vaginal itching or irritation. However, no information was provided in the submission describing the validity and reliability of this questionnaire, and there was no minimal clinically meaningful difference (MCID) reported in the indicated population. Although estradiol vaginal inserts appeared to be efficacious versus placebo, it is difficult to determine whether the magnitude of benefit observed is clinically significant.
Severity of VVA (no atrophy, mild, moderate and severe atrophy) was evaluated using a vaginal mucosa assessment scale, which examines vaginal secretions, epithelial integrity, epithelial surface thickness and colour during pelvic examination. Normal vaginal secretions, epithelial integrity, epithelial surface thickness and colour at week 12 were more likely to be observed in patients treated with estradiol (4 mcg and 10 mcg) compared to placebo.
Treatment with the estradiol vaginal insert was associated with improved sexual function in postmenopausal women, measured by the Female Sexual Function Index (FSFI). The 10 mcg of estradiol showed statistically significant improvements in Total Score, Lubrication and Pain. There were no statistically significant differences between estradiol 4 mcg and placebo.
Harms Results
During the 3-month study period, the frequency of treatment-emergent adverse events (AEs) was similar between 2 doses of the estradiol vaginal insert and placebo: estradiol 4 mcg 50.8%, estradiol 10 mcg 49.2% and placebo 57.8%. Commonly reported AEs were nasopharyngitis, upper respiratory tract infection, back pain, headache, vaginal discharge, and vulvovaginal pruritus. Patients in the placebo group were more likely to report vaginal discharge and vulvovaginal pruritus compared to the estradiol groups. Three patients in the estradiol 10 mcg group reported serious adverse events (SAEs), while no SAEs were reported in the estradiol 4 mcg group. There was 1 case of cervical myelopathy reported in the placebo group. The frequency of withdrawal due to adverse events (WDAEs) was 1.0%, 1.6% and 2.6% in the estradiol 4 mcg group, estradiol 10 mcg group and placebo, respectively. In terms of AEs of particular interest for the review, the frequency of vaginal hemorrhage, cervical dysplasia, and breast mass was numerically higher in the placebo group compared with estradiol groups.
Table 2: Summary of Key Results from REJOICE
Outcomes | Total N | Baseline Mean (SD) | N | Mean (SD) at week 12 | LS mean change from baseline (SE) at week 12 | P value vs. placebo | |
|---|---|---|---|---|---|---|---|
Efficacy (mITT population) | |||||||
Change in severity of dyspareunia | |||||||
Imvexxy 4 mcg | 186 | 2.7 (0.48) | 151 | 1.1 (0.98) | –1.52 (0.071) | 0.0149 a | |
Imvexxy 10 mcg | 188 | 2.6 (0.48) | 154 | 0.9 (0.92) | –1.69 (0.071) | < 0.0001 a | |
Placebo | 187 | 2.7 (0.46) | 163 | 1.4 (1.02) | –1.28 (0.070) | NA | |
Change in severity of vaginal dryness | |||||||
Imvexxy 4 mcg | 186 | 2.3 (0.68) | 171 | 1.1 (0.98) | –1.27 (0.068) | 0.0014 | |
Imvexxy 10 mcg | 188 | 2.4 (0.65) | 173 | 0.9 (0.89) | –1.47 (0.067) | < 0.0001 | |
Placebo | 187 | 2.4 (0.68) | 174 | 1.4 (0.98) | –0.97 (0.067) | NA | |
Change in severity of vulvar and/or vaginal itching or irritation | |||||||
Imvexxy 4 mcg | 186 | 1.2 (1.08) | 171 | 0.5 (0.71) | –0.75 (0.055) | 0.0503 | |
Imvexxy 10 mcg | 188 | 1.3 (1.01) | 173 | 0.4 (0.7) | –0.81 (0.055) | 0.0055 | |
Placebo | 187 | 1.1 (0.99) | 174 | 0.6 (0.84) | –0.60 (0.055) | NA | |
Change in FSFI total score | |||||||
Imvexxy 4 mcg | 173 | 14.8 (6.13) | 153 | 22.6 (8.4) | 7.909 (SE NR) | 0.9075 | |
Imvexxy 10 mcg | 172 | 15.8 (6.24) | 152 | 24.8 (7.59) | 9.431 (SE NR) | 0.0492 | |
Placebo | 175 | 14.4 (6.61) | 158 | 22 (8.54) | 7.458 (SE NR) | NA | |
Harms (safety population) | |||||||
Adverse events, n (%) | |||||||
Imvexxy 4 mcg | 191 | 97 (50.8) | |||||
Imvexxy 10 mcg | 191 | 94 (49.2) | |||||
Placebo | 192 | 111 (57.8) | |||||
Serious adverse events, n (%) | |||||||
Imvexxy 4 mcg | 191 | 0 (0.0) | |||||
Imvexxy 10 mcg | 191 | 3 (1.6) | |||||
Placebo | 192 | 1 (0.5) | |||||
Withdrawal due to adverse events, n (%) | |||||||
Imvexxy 4 mcg | 191 | 2 (1.0) | |||||
Imvexxy 10 mcg | 191 | 3 (1.6) | |||||
Placebo | 192 | 5 (2.6) | |||||
Adverse events of special interest, n (%) | |||||||
Imvexxy 4 mcg | 191 | 3 (1.6) Vaginal hemorrhage 2 (1.0) Cervical dysplasia 1 (0.5) Breast mass 0 | |||||
Imvexxy 10 mcg | 191 | 2 (1.0) Vaginal hemorrhage 1 (0.5) Cervical dysplasia 1 (0.5) Breast mass 0 | |||||
Placebo | 192 | 5 (2.6) Vaginal hemorrhage 3 (1.6) Cervical dysplasia 1 (0.5) Breast mass 1 (0.5) | |||||
FSFI = Female Sexual Function Index; NA = not applicable; NR = not reported; SD = standard deviation; SE = standard error.
aMixed model repeated measures vs. placebo.
Source: Clinical Study Report for REJOICE.9
Critical Appraisal
In the REJOICE study, differences were noted in the patients’ baseline characteristics between 4 mcg and 10 mcg estradiol inserts and the placebo group. The data suggest that more patients in the estradiol inserts groups had a hysterectomy and bilateral oophorectomy, therefore a higher proportion of these patients were surgically menopausal, compared to those in the placebo group. It is unknown whether patients with surgical menopause will respond differently than those with natural menopause, and whether these imbalances would affect interpretation of the results.
Both subjective (e.g., self-reported symptom relief or change in sexual function) and objective efficacy outcomes (e.g., change in percentage of superficial cells, vaginal pH) were evaluated in the REJOICE study. Although self-reported outcomes are considered clinically relevant in practice to measure treatment response according to the clinical expert, there are no published MCIDs identified for such outcome measures in postmenopausal women. Therefore, it is unclear whether the scales used and the reported between-group differences are clinically meaningful.
Multiplicity was controlled for in REJOICE based on a closed fixed sequence serial testing procedure, with the 4 coprimary end points being included. Outcomes outside of the testing hierarchy, such as HRQoL (measured with FSFI), should be viewed as supportive evidence for the overall effects of estradiol vaginal inserts and need to be interpreted with caution, due to the possible inflated type I error.
This was a 3-month study, therefore long-term safety (on endometrium and breast, or in general) and efficacy data are unavailable for the 2 doses of estradiol vaginal inserts. There is a lack of direct or indirect evidence from the included evidence to demonstrate comparative efficacy and safety of the estradiol vaginal insert versus other local hormonal therapy in the study population.
Indirect Comparisons
No indirect treatment comparisons were identified for this review.
Other Relevant Evidence
No other relevant studies were identified for this review.
Other Considerations
A bioavailability study compared 10 mcg dose of the estradiol vaginal insert with another vaginal estrogen therapy (10 mcg dose of Vagifem) in healthy postmenopausal women. The results suggested that the extent of systemic exposure of estradiol 10 mcg was statistically significantly lower than that of Vagifem 10 mcg. The lack of comparative safety data between these makes it unknown at present whether there are differences in the safety profiles in the indicated population.
Cost Information
At the submitted price, the estradiol vaginal insert (Imvexxy) costs $414 per patient annually in the first year of use and $377 in subsequent years of use. CADTH conducted a reanalysis of the sponsor submitted cost comparison, considering: all relevant local hormone therapies; costs in the first and subsequent years of use; and, the lowest available list price for conjugated estrogen cream and the estradiol ring. The annual cost or cost savings with Imvexxy depend on the choice of comparator. Compared with the existing estradiol vaginal insert (Vagifem), annual cost savings with Imvexxy were $78 per person in the first year and $71 per person in subsequent years of use. Compared with cream-based comparators, annual per person incremental costs ranged from cost savings of $450 to increased costs of $338, depending on the dose of the cream-based comparators. The incremental cost compared with the estradiol ring was $115 in first year and $79 in subsequent years of use. The incremental costs were calculated based on publicly available list prices of comparators and may not reflect actual prices paid by Canadian public drug plans. Additionally, the price of conjugated estrogen (Premarin cream) and the estradiol ring comparator (Estring) varies across jurisdictions, and as such, incremental costs will vary across jurisdictions.
The cost comparison assumes clinical similarity between Imvexxy and the other local hormone therapies included in the analysis. Based on a sponsor submitted bioequivalence study, the 10 mcg dose of Imvexxy is likely clinically similar to Vagifem at the same dose in healthy postmenopausal women. The clinical review conducted by CADTH noted that there was a lack of direct or indirect clinical evidence comparing Imvexxy to local hormone therapies in the indicated population (menopausal women with dyspareunia). As a result, the cost comparison with Vagifem is likely appropriate, while the appropriateness of the cost comparison with the cream and ring based local hormone therapies is associated with uncertainty.
Conclusions
Evidence from 1 RCT supported the efficacy of the estradiol vaginal insert (4 mcg and 10 mcg) for the treatment of postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy. Compared to placebo, patients who were treated with the estradiol vaginal insert for 12 weeks showed benefits in symptom relief (vaginal pain associated with sexual activity, vaginal dryness, vulvar and/or vaginal itching or irritation). Improvements in sexual function were observed with the estradiol vaginal insert 10 mcg versus placebo, but not with the 4 mcg dose. The frequency of adverse events, serious adverse events and treatment discontinuation due to adverse events (AEs) were similar across treatment groups and were consistent with the expected adverse event profile for an estradiol-containing product.
Longer-term (beyond 12 weeks) efficacy and safety of the estradiol vaginal insert is unknown. There is a lack of comparative evidence between the estradiol vaginal insert and the other vaginal estrogen therapies in postmenopausal women with moderate to severe dyspareunia. One bioequivalence study suggested the 10 mcg dose of the estradiol vaginal insert is similar to the estradiol vaginal tablet (Vagifem) at the same dose, but there are no clinical studies to confirm outcomes are similar between the 2 estradiol products.
At the submitted price, the estradiol vaginal insert (Imvexxy) costs $414 per patient annually in the first year of use and $377 in subsequent years of use. Imvexxy is cost saving compared with the other available estradiol vaginal insert (Vagifem), but is associated with higher costs compared with the estradiol ring. The cost (or savings) with Imvexxy varies by cream-based comparators depending on the dose. Based on the submitted clinical evidence, the cost comparison with the estradiol vaginal insert (Vagifem) is likely appropriate, while the appropriateness of the cost comparison with the cream and ring based local hormone therapies is associated with uncertainty.
Introduction
Disease Background
Menopause is associated with both systemic and genital changes related to the progressive reduction and loss of estrogen production. Vulvovaginal atrophy (VVA) is among the most prevalent and concerning clinical condition of menopause.1 In 2014, the new term genitourinary syndrome of menopause (GSM) was introduced by the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. This term is more generalizable and inclusive than VVA, and describes the genital, sexual and urinary changes (e.g., recurrent urinary tract infections, dysuria, urinary frequency and urgency) in the lower genital tract associated with menopause.5,10
The development and the severity of VVA depends mainly on the duration of hypoestrogenism. The decline in circulating estrogen associated with menopause strongly correlates with decreased vaginal lactobacilli, increased vaginal pH (range of 6.0 to 8.0), thinned vaginal epithelium, reduced vascular flow and reduced fluid secretion in the vagina.4 The postmenopausal vagina is also at risk of infections and inflammation.4
Many postmenopausal women complain of discomfort associated with hormonal changes, including vaginal dryness and sexual pain disorder, or dyspareunia. The self-reported prevalence of VVA symptoms varies from 4% in early postmenopausal years to over 80% in later years.1,2 In large observational studies of postmenopausal women in Europe, North America, and Asia, the most frequently reported vaginal symptoms were vaginal dryness (85% to 100%), dyspareunia (52% to 78%), and decreased sexual interest (93%). Other commonly reported symptoms associated with VVA include burning, itching, and dysuria.2,3,11 These symptoms negatively impact their lifestyle and/or social factors, produce anxiety or depressive symptoms, and impair women’s quality of life.1,3,4
VVA is a chronic condition that typically does not improve if left untreated.1
Standards of Therapy
According to the clinical expert consulted by CADTH, an ideal treatment for vulvovaginal disease would provide complete symptom relief from the vulvovaginal and/or urogenital changes experienced by women in menopause.
The first-line treatments for VVA symptoms are nonhormonal vaginal moisturizers and lubricants. They may increase vaginal moisture and improve vaginal dryness and dyspareunia. These products do not reverse most atrophic vaginal changes; therefore they may be useful for patients with mild symptoms. In addition, phytoestrogenic preparation, vitamin E, and topical anesthetics may increase vaginal lubrication or have painful atrophy; however, their efficacy and safety in patients with VVA-related symptoms have not been evaluated in well-designed clinical trials.5,6
For postmenopausal women who do not respond well to moisturizers and lubricants, estrogen therapy or other hormonal medications can be prescribed when there are no contraindications.5,6 The effect of systemic hormone therapy on urogenital symptoms have been demonstrated in practice and clinical trials. Exogenous estrogen restores normal vaginal pH levels, thickens and revascularizes the epithelium and increases vaginal lubrication, thus alleviates VVA-related symptoms including dryness, irritation, pruritus, dyspareunia and urinary urgency, and may also lower the incidence of lower urinary tract infections.4 Findings from clinical research suggested that 10% to 25% of women using systemic hormonal therapy still experienced VVA symptoms. Safety concerns related to oral or transdermal hormone replacement therapy have been raised by clinicians and patients.4,12
Vaginal estrogen therapy, such as creams, intravaginal tablets and/or estradiol-releasing ring, is preferred to manage the symptoms of VVA over systemic therapy, when only genitourinary symptoms are present. With vaginal therapy, only small dosages are normally needed to treat vaginal compared to systemic symptoms, also low-potency estrogens can be used.4 In this way, local hormone therapies result in less systemic estrogen absorption and therefore decrease the risk of endometrial stimulation, uterine bleeding, and breast tenderness.6,12
Results of a Cochrane review involving over 6,000 postmenopausal women suggested that various intravaginal estrogenic preparations had similar effect in relieving VVA symptoms and comparable safety profile when compared with each other, although treatment with creams may be associated with more AE such as vaginal irritation or itchiness, vaginal discharge, vaginal bleeding or pelvic pain, compared with tablets and the ring. This may be due to greater absorption or to higher-than-recommended doses being inadvertently inserted into the vagina.4,13 In general, serious adverse events are uncommon with the use of vaginal estrogen therapy.4
Drug
The estradiol vaginal insert (Imvexxy) is a softgel formulation containing estradiol. It is available as 4 mcg and 10 mcg 17 beta-estradiol and is used intravaginally. It received Health Canada approval on August 17, 2020 for the treatment of postmenopausal moderate to severe dyspareunia, 1 of the key symptoms of VVA.7
The recommended starting dose is 4 mcg, with dosage adjustment guided by the clinical response. The initial dose is 1 vaginal insert daily at approximately the same time for 2 weeks. The maintenance dose is 1 vaginal insert twice weekly, every 3 to 4 days.8
In the current review, the sponsor is seeking reimbursement as per the indication for the estradiol vaginal insert, which is for the treatment of postmenopausal moderate to severe dyspareunia.
Table 3: Key Characteristics of Vaginal Estrogen Therapies
Key characteristics | Imvexxy, softgel | Vagifem, tablet | Estring, ring | Premarin, cream |
|---|---|---|---|---|
Mechanism of action | Estrogen therapy for estrogen deficiency | |||
Indicationa | Treatment of postmenopausal moderate to severe dyspareunia. | Treatment of the symptoms of vaginal atrophy due to estrogen deficiency. | For postmenopausal urogenital complaints due to estrogen deficiency such as feeling of dryness in the vagina with or without pruritus vulvae, dyspareunia, dysuria, and urinary urgency. | Treatment of atrophic vaginitis, dyspareunia, and kraurosis vulvae. |
Route of administration | Vaginal | |||
Recommended dose | Initial dose: start with 4 mcg dose, 1 insert daily for 2 weeks. Maintenance dose: 1 insert twice weekly, every 3 to 4 days. Dosage adjustments should be guided by clinical response. The gel should be inserted by manual placement without an applicator, by inserting the smaller end up for a depth of about 2 inches into the vaginal canal. | Initial dose: 10 mcg dose, 1 insert daily for 2 weeks. Maintenance dose: 1 insert twice weekly with a 3 to 4 day interval between doses. Tablet is inserted into the vagina as far as it can comfortably go without force, using an applicator. | The ring (2 mg) should be left in place continuously for 90 days and if continuation of therapy is deemed appropriate, replace by a new ring. The ring should be inserted into the upper third of the vaginal vault. | The cream should be administered cyclically for short-term use only. Low dose: 0.5 g is administered intravaginally or topically twice weekly. Max. dose: women should be started at 0.5 g daily. Dosage adjustment (0.5 to 2) may be made based on individual response. |
Serious adverse effects or safety issues | Estrogens with or without progestins should not be prescribed for primary or secondary prevention of CV diseases. Estrogens with or without progestins should be prescribed at the lowest effective dose for the approved indication. Estrogens with or without progestins should be prescribed for the shortest period possible for the approved indication. | |||
CV = cardiovascular.
aHealth Canada–approved indication.
Source: Product monographs of Imvexxy,8 Vagifem,14 Estring,15 and Premarin.16
Stakeholder Perspectives
Patient Group Input
No input was provided by patient groups.
Clinician Input
Input From Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 gynecologist with expertise in the diagnosis and management of vaginal pain symptoms.
Unmet Needs
Not all patients respond to the available treatments for dyspareunia. Some treatment options are difficult, uncomfortable, or messy to administer, which negatively affects treatment adherence. Due to the safety concerns regarding exogenous hormone therapy on tissues other than urogenital tract (e.g., breast), some women may be reluctant to initiate hormonal treatment.
Place in Therapy
The clinical expert indicated that the estradiol vaginal insert is another form of existing medication for treatment of VVA. It would be used as a first-line treatment or after failure on other treatments for women who are suitable to receive estrogen replacement for VVA.
Patient Population
The clinical expert indicated that most menopausal women with VVA-related symptoms are likely to benefit from vaginal estrogen therapy, such as estradiol inserts. These women can be identified through clinical history and visual inspection of the vulva on physical examination.
Women with contraindications to estrogen therapy, such as active liver disease, hormone sensitive malignancy (breast or endometrium), elevated risk for venous thromboembolism, are not eligible for vaginal estradiol inserts. The expert noted that, given evidence of less systemic absorption of the estradiol insert versus other formulations, clinicians may consider its use in women with certain contraindications but who have persistent moderate or severe VVA symptoms and are refractory to other nonhormonal treatments. It should be noted that the list of contraindications and warnings and precautions in the product monograph for the estradiol vaginal inserts is the same or similar to other available products.
Assessing Response to Treatment
Treatment response is assessed based on patient’s self report of improvement in symptoms. A clinically meaningful response to treatment includes patient report of decrease in sensation of vaginal dryness, decreased vaginal burning or pain, decreased frequency of urinary tract infections or bladder urgency or irritation, and decreased dryness and pain during intercourse. The expert consulted by CADTH for this review suggested treatment response be assessed at 3 to 6 months following initiation of treatment, and again at 6 to 12 months, then yearly thereafter if continued treatment is required.
In clinical practice, histologic examination is generally not performed or required.
Discontinuing Treatment
Patient can discontinue treatment if she wishes, or for any AEs related to the treatment, though symptoms may return thereafter.
Prescribing Conditions
Estradiol vaginal inserts are likely prescribed in an outpatient ambulatory clinic setting by a family physician or gynecologist. The drug can be self-administered by the patient in her own home.
Clinician Group Input
No input was provided by clinician groups.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Implementation issues | Clinical expert response |
|---|---|
Relevant comparators | |
No evidence comparing efficacy and safety vs. currently funded vaginal estrogen products. Comparisons to placebo (Phase 3, REJOICE) and Vagifem 10 mcg (in a PK study) only. | No response. For CDEC consideration. |
Other vaginal estrogen products (e.g., Vagifem 10 mcg tablet, Premarin vaginal cream) are listed as an open benefit under most public plans, except Vagifem is not funded in BC. | No response. For CDEC consideration. |
Considerations for initiation of therapy | |
Other vaginal estrogen products (e.g., Vagifem 10 mcg tablet, Premarin vaginal cream) were not reviewed by CADTH but are listed as an open benefit under most public plans; therefore, consider criteria which indicates to “reimburse in a similar manner to currently funded vaginal estrogen products.” | No response. For CDEC consideration. |
Considerations for prescribing of therapy | |
In the product monograph of Imvexxy, it indicates that “generally, women should be started at the 4 mcg dosage strength. Dosage adjustment should be guided by clinical response.”
| The clinical expert estimated that half of the patients need to escalate to the 10 mcg dose. The 4 mcg dose would be used for 3 to 4 months before escalating to the 10 mcg dose if symptoms have not improved at that time. The clinical expert indicated that the currently available vaginal estrogen tablets are not used in this manner, although this may be considered in women who do not benefit from the treatment at lower doses. Dose escalation (from 4 mcg to 8 mcg or 14 mcg) would only be used in women unresponsive to usual dosage, and following a discussion of risk and benefits. The expert estimated that only a small proportion of these patients would use higher dose (8 mcg or 14 mcg), as this is not the standard treatment regimen with uncertain benefit. |
Generalizability | |
Is there any reason to believe that Imvexxy could not be used more broadly, for example, in patients with other causes of estrogen deficiency and/or for symptoms of vaginal atrophy, other than dyspareunia?
Given the differences in Health Canada–approved indication between Imvexxy and Vagifem, and other vaginal estrogen products are listed as an open benefit under most public plans, consider criteria which indicates to “reimburse in a similar manner to currently funded vaginal estrogen products.” | Dyspareunia is one of the VVA-related symptoms in postmenopausal women. Although the Health Canada–approved indication for Imvexxy is “for the treatment of postmenopausal moderate to severe dyspareunia,” the clinical expert indicated that this drug would be considered for use in a broader population – postmenopausal women with other VVA-related symptoms, such as vaginal dryness, are likely to benefit from vaginal estrogen therapy (e.g., estradiol insert) in clinical practice. |
System and economic issues | |
The sponsor expects that Imvexxy will displace market share primarily from Vagifem, as it’s the most similar comparator used to treat dyspareunia in terms of formulation and administration. Compared to available treatments, the cumulative 3-year budget impact was savings of $649,340. Vagifem is not funded in BC. | The clinical expert indicated it is reasonable to assume that of the market Imvexxy captures, 99% is from Vagifem, given the similarity in formulation and administration. |
Confidential negotiated prices may exist for Vagifem, Premarin vaginal cream and Estring. If there is a lack of evidence to demonstrate superiority of Imvexxy vs. comparators, consider pricing condition that drug plan cost for Imvexxy not exceed the drug plan cost of least costly vaginal estrogen product. | No response. For CDEC consideration. |
Sponsor’s Summary of the Clinical Evidence
Note that the clinical evidence summarized in this section was prepared by the sponsor in accordance with the CADTH tailored review process and has not been copy-edited.
Pivotal Studies
Table 5: Details of Included Studies
Detail | REJOICE (TXV14 to 01) |
|---|---|
Designs and Populations | |
Study Design | Phase 3, multicenter, randomized, double blind, placebo-controlled trial |
Locations | 100 centers (89 centers randomized at least one patient in the United States and Canada) |
Randomized (N) | 574 |
Inclusion Criteria |
|
Exclusion Criteria |
|
Drugs | |
Intervention | IMVEXXY 4 mcg estradiol vaginal insert (N = 191) IMVEXXY 10 mcg estradiol vaginal insert (N = 191) IMVEXXY 25 mcg estradiol vaginal insert (N = 190)a Participants self-administered 1 softgel capsule into the vagina at approximately the same time daily for 2 weeks, then twice weekly (~3 to 4 days apart) for 10 weeks for a total of 12 weeks |
Comparator(s) | Placebo vaginal insert (N = 192) Participants self- administered 1 softgel capsule into the vagina at approximately the same time daily for 2 weeks, then twice weekly (~3 to 4 days apart) for 10 weeks for a total of 12 weeks |
Duration | |
Phase | |
Run-in | 14 weeks (8 weeks of washout and 6 weeks of screening) |
Double-blind | 12 weeks |
Follow-up | 2 weeks |
Outcomes | |
Primary End Point |
|
Secondary and Exploratory End Points | Secondary end points:
|
Notes | |
Publications | Constantine et al. The REJOICE trial: a phase 3 randomized, controlled trial evaluating the safety and efficacy of a novel vaginal estradiol soft-gel capsule for symptomatic vulvar and vaginal atrophy. Menopause. 2017; 24(4):409 to 416. doi: 10.1097/GME.0000000000000786. |
aDose not approved by Health Canada, results for this treatment group are not presented
BMI = body mass index; FSFI = female sexual function index; MBS = most bothersome symptoms; VVA = vulvar and vaginal atrophy
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Description of studies
The pivotal REJOICE trial (N = 764) was a phase 3, multicenter, randomized, double-blind, placebo-controlled trial that compared the efficacy and safety of a 12-week treatment with 1 of 3 doses of estradiol vaginal inserts (IMVEXXY 4 mcg, IMVEXXY 10 mcg, IMVEXXY 25 mcg) with placebo in postmenopausal women with moderate to severe symptoms of vulvar and vaginal atrophy (VVA). The main primary objective was to assess the efficacy on vaginal superficial cells, vaginal parabasal cells, vaginal pH, and the symptom of moderate to severe dyspareunia (vaginal pain associated with sexual activity) as the most bothersome symptom (MBS) associated with VVA. Approximately 700 patients were planned for randomization into the study across an estimated 100 investigative sites in the United States and Canada. A total of 764 patients across 89 sites were randomized. Patients were randomly assigned in a 1:1:1:1 ratio to receive 1 of 4 treatment regimens: IMVEXXY 4 mcg, IMVEXXY 10 mcg, IMVEXXY 25 mcg, or placebo. The IMVEXXY 25 mcg treatment arm was included in the REJOICE trial, however data for this group will not be presented as Health Canada approval was not requested for this dose.
The total duration of the study was approximately 20 to 22 weeks. This time included a 6 to 8-week Screening Period (six weeks for patients without an intact uterus and 8 weeks for patients with an intact uterus), 12 weeks on investigational product, and follow-up approximately 15 days after the last dose of investigation product. The patient’s involvement may have been up to 30 weeks if an 8-week wash-out period was necessary. Clinical evaluations were performed at the following time points:
Washout: Week −14 to −6
Visit 1A: Screening Period (Week −6 to 0)
Visit 1B: Screening Period (Week −4 to 0)
Visit 2: Randomization/Baseline (Week 0, Day1)
Visit 3: Interim (Week 2, Day 14 ± 3 days)
Visit 4: Interim (Week 6, Day 42 ± 3 days)
Visit 5: Interim (Week 8, Day 56 ± 3 days)
Visit 6: End of Treatment or Early Termination (Week 12, Day 84 ± 3 days)
Telephone Interview: Week 14 (approximately 15 days after last dose of study drug)
The study staff, clinical research associates (CRAs), sponsor representatives, and all other study participants were blinded throughout the study as to the regimen the patient was receiving. The packaging and label of IMVEXXY and placebo was identical to maintain adequate blinding of investigators and patients. No stratification of randomized treatment was performed.
Populations
Inclusion and exclusion criteria
Patients enrolled in the REJOICE trial were postmenopausal women, 40 to 75 years of age, with a diagnosis of VVA. Specifically, patients must have ≤ 5% superficial cells on a vaginal cytological smear, vaginal pH > 5.0, and moderate to severe dyspareunia associated with sexual activity considered the MBS that had an onset in the postmenopausal years.
Patients were excluded from the REJOICE trial if they did not complete appropriate washouts from previous treatments, hypersensitivity to estrogens, endometrial hyperplasia, undiagnosed vaginal bleeding, chronic liver or kidney dysfunction, thromboembolic disorders, previous stroke or myocardial infarction, cancer within the past 5 years, alcohol or drug abuse, heavy smoking, history of sexual abuse, use of marijuana or any clinically important abnormalities on Screening physical exam, assessments, ECG, or laboratory tests.
Analysis populations
All patients who were randomly assigned and had at least 1 dose of study drug formed the intent to treat (ITT) and safety population. Patients were summarized by using the treatment they were randomized to. The modified intent to treat (mITT) population was the primary efficacy population. It was defined as all ITT patients who received the treatment to which they were randomized, had baseline values for all co-primary variables, and had at least 1 post-baseline value for any of following 4 co-primary variables at any visit: parabasal cells; superficial cells; vaginal pH; and MBS of dyspareunia. The efficacy evaluable (EE) population excluded those in the mITT population who did not meet key study inclusion/exclusion criteria, used prohibited medications, and/or reported ≤ 80% overall study drug compliance based on diary.
Baseline characteristics
The patients’ baseline characteristics for the mITT population appeared to be balanced across treatment groups, as seen in Table 6. The average age of the women was 59.8, 58.6 and 59.4 years in the IMVEXXY 4 mcg, 10 mcg and placebo group, respectively. The majority of the women were White (85.6% to 87.8%) and had a mean body mass index (BMI) of 26.6 to 26.8 kg/m2.
Gynecological history was also similar across treatment groups in the mITT population. A slightly lower percentage of women in the IMVEXXY groups had natural menopause (59.7% and 60.6%) compared with the placebo group (66.3%). The mean time since menopause was 13.9 to 14.2 and prior hormone replacement therapy was used in 17.6% to 19.3% of women.
Baseline assessments of parabasal cells, superficial cells, vaginal pH and severity of MBS of dyspareunia were similar across treatment groups. Mean percentage of parabasal cells in the IMVEXXY 4 mcg, 10 mcg and placebo groups were 52.3%, 51.3%, and 52.0%, respectively. Mean percentage of superficial cells in the IMVEXXY 4 mcg, 10 mcg, and placebo groups were 1.3%, 1.2%, and 1.3%, respectively. Mean pH was 6.27 to 6.34 which is consistent with the values of postmenopausal vaginal pH. For study participation, patients needed to identify that their MBS was moderate to severe dyspareunia. The mean baseline severity score for dyspareunia across treatment groups was 2.6 to 2.7.
Table 6: Summary of Baseline Characteristics for the mITT Population
Characteristics | IMVEXXY 4 mcg (N = 186) | IMVEXXY 10 mcg (N = 188) | Placebo (N = 187) |
|---|---|---|---|
Mean age, years (SD) | 59.8 (5.95) | 58.6 (6.30) | 59.4 (5.99) |
Race, n (%) | |||
White | 162 (87.1) | 165 (87.8) | 160 (85.6) |
Black or African American | 20 (10.8) | 21 (11.2) | 21 (11.2) |
Asian | 3 (1.6) | 2 (1.1) | 1 (0.5) |
Other | 1 (0.5) | 0 (0) | 5 (2.7) |
Mean height, cm (SD) | 162.9 (6.79) | 162.9 (6.96) | 162.1 (6.17) |
Mean weight, kg (SD) | 70.7 (14.23) | 71.1 (13.55) | 70.1 (13.33) |
Mean BMI, kg/m2 (SD) | 26.6 (4.91) | 26.8 (4.71) | 26.6 (4.58) |
Had a hysterectomy, n (%) | 87 (46.8) | 86 (45.7) | 73 (39.0) |
Have an intact cervix, n (%) | 112 (60.2) | 112 (59.6) | 127 (67.9) |
Natural menopause, n (%) | 111 (59.7) | 114 (60.6) | 124 (66.3) |
Surgical menopause, n (%) | 75 (40.3) | 74 (39.4) | 63 (33.7) |
Reported bilateral oophorectomy, n (%) | 49 (26.3) | 51 (27.1) | 40 (21.4) |
Mean years since menopause (SD) | 14.2 (8.92) | 14.3 (9.43) | 13.9 (9.44) |
Mean number of pregnancies, n (SD) | 2.3 (1.67) | 2.4 (1.55) | 2.4 (1.66) |
Mean number of vaginal births, n (SD) | 1.8 (1.16) | 1.7 (1.30) | 1.7 (1.40) |
Prior hormone replacement therapy, n (%) | 34 (18.3) | 33 (17.6) | 36 (19.3) |
Mean percentage of parabasal cells, (SD) | 52.3 (39.21) | 51.3 (37.96) | 52.0 (39.22) |
Mean percentage of superficial cells, (SD) | 1.3 (1.24) | 1.2 (1.23) | 1.3 (1.31) |
Mean vaginal pH, (SD) | 6.34 (0.871) | 6.27 (0.832) | 6.33 (1.042) |
Mean severity of MBS of dyspareunia, (SD) | 2.7 (0.48) | 2.6 (0.48) | 2.7 (0.46) |
BMI = body mass index; MBS = most bothersome symptoms; mITT = modified intent to treat; SD = standard deviation.
Source: Clinical Study Report for REJOICE (Clinical Study Report).
Interventions
In the REJOICE trial, the patients received either IMVEXXY 4 mcg, IMVEXXY 10 mcg or placebo as 1 softgel capsules daily for 2 weeks, then 1 softgel capsule twice weekly for 10 weeks. IP was dispensed to all eligible patients at Visit 2. Each patient was provided a total of 30 softgel capsules of IP in a labelled bottle, allowing for extra capsules for accidental loss or damage. A second bottle containing 30 softgel capsules was dispensed at Visit 5. Each patient was trained by the clinical site to self-administer intravaginally 1 capsule daily at approximately the same hour for 2 weeks (14 days). Starting on Day 15, each patient administered 1 capsule twice weekly for the remaining 10 weeks. Twice weekly dosing should have been approximately 3 to 4 days apart and should not have exceeded more than twice in a 7-day period. Patients received the following oral and written instructions for IP administration: Remove vaginal capsule from the bottle. Find a position most comfortable for you. Insert the capsule with the smaller end up into vaginal canal for about 2 inches.
Concomitant medications/treatments could be used to treat chronic or intercurrent medical conditions at the discretion of the Investigator. All concomitant medications/treatments (prescription as well as over-the-counter non-prescription), including the drug or treatment name, start and stop dates and indication of use were to be recorded in the patient diary and the electronic case report form (eCRF). The following medications were prohibited for the duration of the study: investigational drugs other than IMVEXXY; estrogen-, progestin-, androgen (ie, dehydroepiandrosterone [DHEA]) or selective estrogen receptor modulator (SERM)-containing medications other than the IP; medications, remedies, and supplements known to treat VVA; vaginal lubricants and moisturizers (eg, Replens) had to be discontinued 7 days before the Visit 1B vaginal pH assessment; and all medications excluded before the study.
Patients were removed from the trial if any of the following circumstances occurred: withdrawal of consent for any reason, patient’s condition worsened to the degree that the investigator felt it was unsafe to continue the study, patient’s drug code was unblinded, adverse event occurred that the patient desired to discontinue treatment or investigator determined it was in their best interest to discontinue, significant protocol deviation/violation, concomitant therapy was likely to interfere with rests of the study or compromise safety, patient lost to follow-up, patient became pregnant, or administrative reasons.
Outcomes
The 4 co-primary efficacy end points in the REJOICE trial are change from baseline to week 12 in: the percentage change of vaginal superficial cells; the percentage change of vaginal parabasal cells; vaginal pH; and severity of MBS of dyspareunia associated with VVA.
The secondary end points are change from baseline to Weeks 2, 6, and 8 in: percentage of vaginal superficial cells, percentage of vaginal parabasal cells; vaginal pH; severity of the MBS of dyspareunia associated with VVA; severity of vaginal dryness and vulvar and/or vaginal itching or irritation associated with VVA; and visual evaluation of the vaginal mucosa. Additionally, the change from baseline in the Female Sexual Function Index (FSFI) at week 12 was also measured.
Vaginal cytological smears were collected from the lateral vaginal walls according to standard procedures at Screening 1B and Weeks 2, 6, 8, and 12 (or Early Termination). The percentage of superficial, parabasal, and intermediate cells were determined for each sample. Vaginal pH was measured at Screening Visit 1B and Weeks 2, 6, 8, and 12 (or Early Termination) with a pH indicator strip to the lateral vaginal wall. The colour of the strip was compared immediately with a colourimetric scale and the measurement was recorded.
The VVA Symptoms Self-Assessment Questionnaire is an instrument that patients utilize to self-assess their symptoms of VVA, including vaginal pain associated with sexual activity, vaginal dryness, and vulvar and/or vaginal itching or irritation. Each item was rated on a 4-point severity scale from 0 (none), 1 (mild), 2 (moderate), and 3 (severe). Patients were asked at Screening Visit 1A and 1B to complete the Questionnaire and identify their MBS which determined their eligibility for the study. Screening Visit 1B evaluation results were considered as Baseline data for the statistical analyses. Randomized patients were asked to complete the VVA Symptoms Self-Assessment Questionnaire at Weeks 2, 6, 8, and 12 (or Early Termination).
The FSFI is a brief, multidimensional questionnaire for assessing sexual function in women. The questionnaire consists of 19 items that assess sexual function over the past 4 weeks and yield domain scores in 6 areas: sexual desire, arousal, lubrication, orgasm, satisfaction, and pain. Further validation of the instrument was conducted to extend the validation to include dyspareunia/vaginismus (pain), and multiple sexual dysfunctions.(Wiegel et al, 2005) The FSFI questionnaire was administered at Randomization and Week 12.
Statistical analysis
Analysis of Primary and Secondary Outcomes
Continuous data was summarized with the following descriptive statistics: number of observations (n), mean, standard deviation (SD), median, minimum, and maximum. Categorical and ordinal data was summarized with frequencies (number of patients in category) and percentages. Percentages were computed using the number of patients with available data as the denominator, except for AEs, for which the denominator was the number of patients in each dose cohort, across all dose cohorts and for all patients in the Safety Population.
Three doses of IMVEXXY were compared to placebo. Within each dose/placebo comparison, there were 4 co-primary end points: (1) vaginal parabasal cells, (2) vaginal superficial cells, (3) vaginal pH, and (4) severity of MBS of dyspareunia (vaginal pain associated with sexual activity). The 4 co-primary end points were tested using a closed fixed sequence serial testing procedure, in which each primary end point was tested at level alpha (0.025, 1-tailed) until no hypothesis was rejected and then all subsequent hypotheses were also accepted.
To account for the multiple comparisons of testing placebo to each of the 3 doses of IMVEXXY (4 mcg, 10 mcg, and 25 mcg) and the multiple testing of the 4 co-primary end points, the procedural testing started by examining the highest dose (25 mcg) for each of the co-primary end points in the following order: 1) vaginal superficial cells, 2) vaginal parabasal cells, 3) vaginal pH, and 4) severity of the MBS of dyspareunia. If all of the p-values for each of the 4 co-primaries were significant (P ≤ 0.05) then the hypothesis testing continued on to the next lowest dose (10 mcg) for each of the co-primaries, as described above. If all of the 4 co-primaries were significant (P ≤ 0.05 for IMVEXXY 10 mcg, then the hypothesis testing continued for the next lowest dose (4 mcg). If at any point the hypothesis testing yielded a non-significant result, the testing was to be stopped.
Primary and secondary efficacy end points were measured at Baseline and at Weeks 2, 6, 8 and 12. The analysis examined change from baseline. Therefore, analysis of covariance (ANCOVAs) were based on mixed model repeated measures MMRM where the random effect was patient and the 2 fixed effects were treatment group and Visit (2, 6, 8 and 12 weeks). Baseline measures and age were used as covariates. ANCOVAs were therefore not calculated independently for each study collection period.
The change in mean from baseline of each active treatment group from the placebo group for each numerical efficacy end point ([1] vaginal parabasal cells, [2] vaginal superficial cells, [3] vaginal pH, and [4] severity of the MBS of dyspareunia) associated with VVA was defined as treatment Least Square (LS) Mean change – placebo LS Mean change. The 95% confidence interval (CI) for the difference in LS Mean changes between treated and placebo is also displayed. A comparison of treated to placebo at each baseline visit using the same methodology was performed.
The change from baseline for each of the 3 VVA Symptoms Self-Assessment Questionnaire items had 7 possible values (−3, −2, −1, 0, + 1, + 2, + 3) at each post-baseline visit (Weeks 2, 6, 8 and 12) where −3 represents a change from severe to none and + 3 represents the change from none to severe. The change from Baseline to each post-baseline visit for actively treated patients was compared to placebo and tested using the MMRM.
The FSFI total summary score is a numerically continuous measure that was descriptively summarized at Visit 2 (Randomization) and Visit 6 (Week 12/End of Treatment) and the change in the total summary score (Visit 6 minus Visit 2) was also descriptively summarized. Summaries were by treatment arm, and all active treatment arms combined. In addition, the change in mean from baseline of each active treatment group from the placebo group for each numerically continuous end point was evaluated. The LS mean changes and the 95% CI for the difference in LS mean changes between treated and placebo are provided.
Power Calculation
The sample size needed per dose versus placebo for each test of hypothesis in the mITT population to achieve a given power were calculated using the data from available literature. Table 7 below provides the effect sizes, power and sample size determinations for each of the primary end points. Based on the power analysis and the design considerations, approximately 175 patients per treatment arm were enrolled.
Table 7: Power Analysis and Sample Size Determinations
Primary End point | Effect Size (%)a | Power Based upon N = 140 per group per mITT |
|---|---|---|
% Parabasal cells | 150.3% | > 0.999 |
% Superficial cells | 115.3% | > 0.999 |
Vaginal pH | 77.4% | > 0.999 |
Severity of Dyspareuniab | 30.0%, 41.2%, 70.5% | 0.50, 0.80, > 0.999 |
aEffect Size is calculated for all primary end points as 100% times difference (treated minus placebo) in mean changes divided by standard deviation at Week 12 from baseline.
bRange from 30% (Vagifem 10 mcg) (Simon et al, 2008), 41.2% (Vagifem 25 mcg) (FDA Medical Officer’s Review of Vagifem NDA 20 to 908, 1999), 70.5% (Premarin cream 2/week) (Bachmann et al, 2009)
Data Imputation Methods
All attempts were made to prevent any missing values. Missing or invalid data was treated as missing and was not imputed. No last observation carried forward (LOCF) methods for efficacy were used as repeated measures mixed effects model (MMRM) methods were used where applicable for efficacy.
Sensitivity Analyses
The Mantel Haenszel test was used as a sensitivity analysis to examine the change from baseline to Week 12 of the severity of the MBS of dyspareunia associated with VVA compared to placebo. For the sensitivity analyses, the following 3 pair-wise comparisons were performed for Week 12 (primary) and Weeks 2, 6 and 8 (secondary) change from Baseline: active treatment, high dose group versus placebo; active treatment, middle dose group versus placebo; active treatment, low dose group versus placebo. Sensitivity analysis using the Mantel-Haenszel test was also used for the change from baseline for each of the 3 VVA symptoms Self-Assessment Questionnaire compared to placebo, and change from baseline for the visual evaluation of vaginal mucosa compared to placebo.
Additional efficacy analyses including sensitivity analyses were conducted based on age (age tertiles), BMI (tertiles), uterine status, pregnancy status, and parity (vaginal births).
Sponsor’s Summary of the Results
Patient Disposition
In the REJOICE trial, a total of 2183 patients were screened. A total of 574 patients were randomized to either the IMVEXXY 4 mcg, IMVEXXY 10 mcg, or placebo. Of those, 47 patients were prematurely withdrawn: 16 (8.4%) in the IMVEXXY 4 mcg group, 17 (8.9%) in the IMVEXXY 10 mcg group, and 14 (7.3%) in the placebo group. The most common reasons for early discontinuation were withdrawal of consent, adverse event, lost to follow up, and lack of efficacy. The completion and discontinuation rates, and reasons for discontinuation, were similar across the IMVEXXY groups and the placebo group. Complete details are presented in Table 8.
REJOICE | |||
|---|---|---|---|
IMVEXXY 4 mcg | IMVEXXY 10 mcg | Placebo | |
Screened, N | 2183* | ||
Randomized, N | 191 | 191 | 192 |
Discontinued, N (%) (safety population) | 16 (8.4) | 17 (8.9) | 14 (7.3) |
Reason for discontinuation, N (%) | |||
Withdrawal of consent | 6 (3.1) | 7 (3.7) | 5 (2.6) |
Adverse events | 2 (1.0) | 3 (1.6) | 5 (2.6) |
Lost to follow-up | 3 (1.6) | 3 (1.6) | 4 (2.1) |
Lack of efficacy | 2 (1.0) | 2 (1.0) | 0 (0.0) |
Investigator decision | 2 (1.0) | 2 (1.0) | 0 (0.0) |
Protocol violation | 2 (1.0) | 1 (0.5) | 0 (0.0) |
mITT, N | 186 | 188 | 187 |
Safety population, N | 191 | 191 | 192 |
EE, N | 172 | 171 | 176 |
*Includes all patients screened for IMVEXXY 4, 10, and 25 mcg and placebo
mITT = modified intent to treat; EE = efficacy evaluable
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Exposure to study treatments
Study Treatments
The overall mean number of doses taken by patients in the study was 33 and was consistent across groups. The mean estradiol exposure during the study was 131 mcg for IMVEXXY 4 mcg, and 325 mcg for IMVEXXY 10 mcg. A patient must have used at least 80% of the IP to be considered compliant with IP administration. Capsule count and diary cards were used to determine patient compliance at each study visit. Compliance rate in the safety population was 92.7% in the IMVEXXY 4 mcg group, 90.1% in the IMVEXXY 10 mcg group and 92.7% in the placebo group.
Concomitant Medications
During the course of the study, 89.5%, 93.2% and 91.6% of the safety population patients in the IMVEXXY 4 mcg, IMVEXXY 10 mcg and placebo groups, respectively, took a concomitant medication during the course of the study. The medications used were for other conditions and there were no imbalances with any individual medication use between treatments groups. Overall, 47.1%, 46.1%, and 46.9% of the safety population patients in the IMVEXXY 4 mcg, IMVEXXY 10 mcg and placebo groups, respectively, took a concomitant medication for an adverse event. The most commonly used concomitant medication used for an adverse event was ibuprofen, which was used by 42.9%, 45.5%, and 49% of the IMVEXXY 4 mcg, IMVEXXY 10 mcg and placebo groups, respectively.
Efficacy
Percent Change in Parabasal Cells
Statistically significant differences (P < 0.0001) were observed in the change from Baseline to Week 12 in the percentage of parabasal cells for both IMVEXXY groups (4 mcg, 10 mcg) compared to placebo (−40.63, −44.07 vs −6.73, respectively) (Table 9).
The change from Baseline to Weeks 2, 6, and 8 in percentage of parabasal cells was statistically improved at each time point and for both doses of IMVEXXY compared to placebo (P < 0.0001) (Table 10). A statistically significant decrease in parabasal cells occurred by Week 2 and was sustained through Week 12.
Percent Change in Superficial Cells
Statistically significant differences (P < 0.0001) were noted in the change from Baseline to Week 12 in the percentage of superficial cells for both IMVEXXY groups (4 mcg, and 10 mcg) compared to placebo (17.50, 16.72 vs 5.63, respectively) (Table 9).
For each time point and for both doses of IMVEXXY, a statistically significant increase was observed in the change from Baseline to Weeks 2, 6, and 8 in the percentage of superficial cells compared to placebo (P < 0.0001) (Table 10). A statistically significant increase in superficial cells occurred by Week 2 and was sustained through Week 12
Change in vaginal pH
The mean vaginal pH at Baseline was more than 1 unit above the inclusion criteria of greater than 5.0 (6.34, 6.27 and 6.33 for IMVEXXY 4 mcg, 10 mcg and placebo, respectively). By Week 12, there was a decrease of at least 1 unit for all doses of IMVEXXY compared to a decrease of 0.28 unit for placebo. The change from Baseline to Week 12 in vaginal pH was statistically significant (< 0.0001) for both IMVEXXY groups compared to placebo (−1.32, −1.42 vs −0.28, respectively) (Table 9).
For each time point and for both doses of IMVEXXY, a statistically significant decrease in pH was observed in the change from Baseline to Weeks 2, 6, and 8 at each time point (by at least 1 unit) compared to placebo (P < 0.0001) (Table 10). A statistically significant decrease in vaginal pH occurred by Week 2 and was sustained through Week 12.
Change in Severity of Dyspareunia
A statistically significant reduction in the severity of dyspareunia, change from Baseline to Week 12, was found for both doses of IMVEXXY compared to placebo (Table 27). The MMRM p-value for the comparison between IMVEXXY 4 mcg and placebo was 0.0149 (−1.52 vs −1.28); and the p-value for the IMVEXXY 10 mcg comparison to placebo was < 0.0001 (−1.69 vs −1.28) (Table 9). Of note, at Week 12, a total of 9.1% of women had no sex with vaginal penetration and 7.0% had missing data, and thus were not included in the efficacy analysis as change from Baseline could not be determined.
The percentage of patients reporting no dyspareunia at the end of the study was 25.8%, and 32.4% vs 19.8% in IMVEXXY 4 mcg, and 10 mcg and placebo, respectively. Change of 3 severity levels was reported in 17.2%, 18.1%. and 12.8%, respectively. Additionally, the severity of dyspareunia at the end of the study improved by 2 to 3 levels in 41.4% of patients in the IMVEXXY 4 mcg group, and 47.4% in the IMVEXXY 10 mcg group compared to 35.8% in the placebo group.
Improvement in the change from Baseline to Weeks 2, 6, and 8 in the severity of dyspareunia was statistically significant at each time point and for both doses of IMVEXXY compared to placebo (Table 10). A significant reduction in dyspareunia was noted as early as Week 2 for both doses of IMVEXXY and was sustained through Week 12.
Table 9: Change from Baseline to Week 12 for the Four Co-primary End points; Parabasal Cells, Superficial Cells, Vaginal pH and Dyspareunia (mITT Population)
Total N | Baseline Mean (SD) | N | Mean (SD) at Week 12 | LS Mean change from baseline (SE) at Week 12 | P value vs placebo | |
|---|---|---|---|---|---|---|
Percent change in Parabasal cells a | ||||||
IMVEXXY 4 mcg | 186 | 52.3 (39.21) | 170 | 12.0 (22.32) | −40.63 (1.755) | < 0.0001 a |
IMVEXXY 10 mcg | 188 | 51.3 (37.96) | 171 | 7.8 (18.51) | −44.07 (1.751) | < 0.0001 a |
Placebo | 187 | 52.0 (39.22) | 174 | 45.2 (40.27) | −6.73 (1.750) | - |
Percent change in superficial cells | ||||||
IMVEXXY 4 mcg | 186 | 1.3 (1.24) | 170 | 18.7 (19.54) | 17.50 (1.542) | < 0.0001 a |
IMVEXXY 10 mcg | 188 | 1.2 (1.23) | 171 | 18.5 (19.95) | 16.72 (1.540) | < 0.0001 a |
Placebo | 187 | 1.3 (1.31) | 174 | 7.0 (14.70) | 5.63 (1.537) | - |
Change in vaginal pH | ||||||
IMVEXXY 4 mcg | 186 | 6.34 (0.871) | 170 | 5.03 (0.961) | −1.32 (0.066) | < 0.0001 a |
IMVEXXY 10 mcg | 188 | 6.27 (0.832) | 171 | 4.86 (0.737) | −1.42 (0.066) | < 0.0001 a |
Placebo | 187 | 6.33 (1.042) | 174 | 6.07 (1.373) | −0.28 (0.066) | - |
Change in severity of dyspareunia | ||||||
IMVEXXY 4 mcg | 186 | 2.7 (0.48) | 151 | 1.1 (0.98) | −1.52 (0.071) | 0.0149 a |
IMVEXXY 10 mcg | 188 | 2.6 (0.48) | 154 | 0.9 (0.92) | −1.69 (0.071) | < 0.0001 a |
Placebo | 187 | 2.7 (0.46) | 163 | 1.4 (1.02) | −1.28 (0.070) | - |
mITT = modified intention-to-treat; SD = standard deviation; SE = standard error.
aMixed model repeated measures vs. placebo
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Table 10: Change from Baseline to Weeks 2, 6, and 8 in Percentage of Parabasal and Superficial Cells, Vaginal pH, and MBS of Dyspareunia (mITT Population)
N | LS Mean change from baseline (SE) | P value a | |
|---|---|---|---|
Parabasal cells, Week 2 | |||
IMVEXXY 4 mcg | 186 | −40.23 (1.720) | < 0.0001 |
IMVEXXY 10 mcg | 188 | −44.42 (1.710) | < 0.0001 |
Placebo | 185 | −7.00 (1.720) | - |
Parabasal cells, Week 6 | |||
IMVEXXY 4 mcg | 172 | −39.36 (1.750) | < 0.0001 |
IMVEXXY 10 mcg | 170 | −43.55 (1.752) | < 0.0001 |
Placebo | 176 | −9.23 (1.741) | - |
Parabasal cells, Week 8 | |||
IMVEXXY 4 mcg | 164 | −41.87 (1.768) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −43.78 (1.764) | < 0.0001 |
Placebo | 167 | −7.86 (1.760) | - |
Superficial cells, Week 2 | |||
IMVEXXY 4 mcg | 186 | 31.35 (1.496) | < 0.0001 |
IMVEXXY 10 mcg | 188 | 31.93 (1.488) | < 0.0001 |
Placebo | 185 | 6.05 (1.498) | - |
Superficial cells, Week 6 | |||
IMVEXXY 4 mcg | 172 | 18.41 (1.536) | < 0.0001 |
IMVEXXY 10 mcg | 170 | 16.88 (1.543) | < 0.0001 |
Placebo | 176 | 5.43 (1.525) | - |
Superficial cells, Week 8 | |||
IMVEXXY 4 mcg | 164 | 19.04 (1.561) | < 0.0001 |
IMVEXXY 10 mcg | 165 | 17.41 (1.558) | < 0.0001 |
Placebo | 167 | 5.98 (1.551) | - |
Vaginal pH, Week 2 | |||
IMVEXXY 4 mcg | 186 | −1.23 (0.064) | < 0.0001 |
IMVEXXY 10 mcg | 188 | −1.37 (0.064) | < 0.0001 |
Placebo | 186 | −0.28 (0.064) | - |
Vaginal pH, Week 6 | |||
IMVEXXY 4 mcg | 172 | −1.32 (0.066) | < 0.0001 |
IMVEXXY 10 mcg | 170 | −1.40 (0.066) | < 0.0001 |
Placebo | 176 | −0.30 (0.065) | - |
Vaginal pH, Week 8 | |||
IMVEXXY 4 mcg | 164 | −1.35 (0.067) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −1.46 (0.067) | < 0.0001 |
Placebo | 167 | −0.38 (0.066) | - |
Dyspareunia, Week 2 | |||
IMVEXXY 4 mcg | 145 | −0.99 (0.072) | 0.0260 |
IMVEXXY 10 mcg | 147 | −1.08 (0.072) | 0.0019 |
Placebo | 141 | −0.76 (0.072) | - |
Dyspareunia, Week 6 | |||
IMVEXXY 4 mcg | 148 | −1.30 (0.072) | 0.0069 |
IMVEXXY 10 mcg | 150 | −1.37 (0.072) | 0.0009 |
Placebo | 159 | −1.03 (0.070) | - |
Dyspareunia, Week 8 | |||
IMVEXXY 4 mcg | 140 | −1.52 (0.073) | 0.0003 |
IMVEXXY 10 mcg | 136 | −1.64 (0.074) | < 0.0001 |
Placebo | 143 | −1.15 (0.072) | - |
mITT = modified intention-to-treat; SE = standard error.
aMixed model repeated measures vs. placebo.
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Vaginal Dryness
At Baseline, 92.4% of patients in the IMVEXXY 4 mcg group, 94.1% in the IMVEXXY 10 mcg group and 93.6% in the placebo group, respectively, reported either moderate or severe vaginal dryness. The change from Baseline to Weeks 2, 6, 8, and 12 in the severity of vaginal dryness, regardless of Baseline severity is shown in Table 11. The LS Mean change was statistically significantly different at each time point for the IMVEXXY 10 mcg compared to placebo. For IMVEXXY 4 mcg, there was a statistical improvement starting at Week 6 through Week 12.
The percentage of patients reporting no dryness at the end of the study was 31.2%, 36.7%, vs 17.1% in IMVEXXY 4 mcg, 10 mcg, and placebo, respectively. Change of 3 severity levels was reported in 12.4%, 14.4% and 9.6%, respectively. Additionally, the severity of dryness at the end of the study improved by 2 to 3 levels in 38.2% of patients in the IMVEXXY 4 mcg group, and 47.4% in the IMVEXXY 10 mcg group compared to 28.9% in the placebo group.
Vulvar and/or vaginal itching or irritation
At Baseline, patients that reported either moderate to severe vulvar and/or vaginal itching or irritation occurred in 45.2%, 48.4% and 34.8% in the IMVEXXY 4 mcg, IMVEXXY 10 mcg and placebo groups, respectively. Overall, the mean Baseline severity was 1.2. A summary of the LS mean change from Baseline to Weeks 2, 6, 8, and 12 in the severity of vulvar and/or vaginal itching or irritation is shown in Table 11. At Week 12, p-values for IMVEXXY 4 mcg, and 10 mcg compared to placebo were 0.0503, and 0.0055, respectively.
The percentage of patients (mITT) reporting no vulvar and/or vaginal itching or irritation at the end of the study was 59.7%, and 65.4% vs 59.4% in IMVEXXY 4 mcg, and 10 mcg and placebo, respectively. Change of 3 severity levels was reported in 5.9%, 4.8% and 3.7%, respectively. Additionally, the severity of vulvar and/or vaginal itching or irritation at the end of the study improved by 2 to 3 levels in 22.6% of patients in the IMVEXXY 4 mcg group, and 26.6% in the IMVEXXY 10 mcg group compared to 17.6% in the placebo group.
Table 11: Change from Baseline to Weeks 2, 6, 8 and 12 in Severity of Vaginal Dryness, and Vulvar and/or Vaginal Itching or Irritation (mITT Population)
N | LS Mean change from baseline (SE) | P value a | |
|---|---|---|---|
Severity of vaginal dryness, Week 2 | |||
IMVEXXY 4 mcg | 186 | −0.86 (0.066) | 0.1269 |
IMVEXXY 10 mcg | 188 | −1.01 (0.065) | 0.0019 |
Placebo | 185 | −0.72 (0.066) | - |
Severity of vaginal dryness, Week 6 | |||
IMVEXXY 4 mcg | 172 | −1.14 (0.067) | 0.0094 |
IMVEXXY 10 mcg | 170 | −1.27 (0.068) | 0.0001 |
Placebo | 176 | −0.90 (0.067) | - |
Severity of vaginal dryness, Week 8 | |||
IMVEXXY 4 mcg | 163 | −1.25 (0.069) | 0.0128 |
IMVEXXY 10 mcg | 165 | −1.44 (0.068) | < 0.0001 |
Placebo | 167 | −1.01 (0.068) | - |
Severity of vaginal dryness, Week 12 | |||
IMVEXXY 4 mcg | 171 | −1.27 (0.068) | 0.0014 |
IMVEXXY 10 mcg | 173 | −1.47 (0.067) | < 0.0001 |
Placebo | 174 | −0.97 (0.067) | - |
Severity of vulvar and/or vaginal itching or irritation, Week 2 | |||
IMVEXXY 4 mcg | 186 | −0.47 (0.054) | 0.9616 |
IMVEXXY 10 mcg | 188 | −0.56 (0.053) | 0.2439 |
Placebo | 184 | −0.47 (0.054) | - |
Severity of vulvar and/or vaginal itching or irritation, Week 6 | |||
IMVEXXY 4 mcg | 172 | −0.57 (0.055) | 0.7829 |
IMVEXXY 10 mcg | 170 | −0.64 (0.055) | 0.2328 |
Placebo | 176 | −0.55 (0.055) | - |
Severity of vulvar and/or vaginal itching or irritation, Week 8 | |||
IMVEXXY 4 mcg | 163 | −0.74 (0.056) | 0.0639 |
IMVEXXY 10 mcg | 165 | −0.76 (0.056) | 0.0356 |
Placebo | 167 | −0.59 (0.056) | - |
Severity of vulvar and/or vaginal itching or irritation, Week 12 | |||
IMVEXXY 4 mcg | 171 | −0.75 (0.055) | 0.0503 |
IMVEXXY 10 mcg | 173 | −0.81 (0.055) | 0.0055 |
Placebo | 174 | −0.60 (0.055) | - |
aMixed model repeated measures vs. placebo
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Vaginal Mucosa
Visual evaluation was performed during pelvic examination at Weeks 2, 6, 8 and 12 using the vaginal mucosa assessment scale shown in Figure 1. The mean change from baseline to Weeks 2, 6, 8, and 12 in the visual evaluation of the vagina is shown in Table 12.
At Baseline, normal vaginal colour was assessed in less than 3% of patients (2.7% in IMVEXXY 4 mcg; 1.6% in IMVEXXY 10 mcg; and 2.7% in placebo) with a mean severity of 1.8. Statistically significant improvement in colour compared with placebo was noted as early as 2 weeks for both IMVEXXY groups and was sustained through Week 12 (P ≤ 0.0001 for all). At Week 12, normal vaginal colour was assessed in over 30% of patients in the TX-004HR groups (30.1% in IMVEXXY 4 mcg; 36.7% in IMVEXXY 10 mcg) compared to 18.7% in placebo.
At Baseline, normal vaginal epithelial integrity was assessed in 11.3% of patients in IMVEXXY 4 mcg; 16.5% in IMVEXXY 10 mcg; and 12.3% in placebo with a mean severity of 1.5. Statistically significant reductions in severity compared to placebo were noted as early as 2 weeks for all groups and was sustained throughout the study. At Week 12, normal epithelial integrity was assessed in 51.1% of patients in the TX-004HR 4 mcg group; and 61.2% in TX-004HR 10 mcg compared to 38.0% in placebo.
At Baseline, normal vaginal epithelial surface thickness was noted in less than 3% of patients (2.7% in IMVEXXY 4 mcg; 1.1% in IMVEXXY 10 mcg; and 2.7% in placebo), with a mean severity of 1.9 across all groups. Statistically significant improvements were noted as early as 2 weeks for all groups and were sustained through Week 12 (P < 0.0001 for all). At Week 12, normal epithelial surface thickness was reported in 22.0% of patients in the IMVEXXY 4 mcg group; and 27.7% in IMVEXXY 10 mcg; compared to 18.7% in placebo.
At Baseline, normal vaginal secretion was noted in less than 5% of patients 4.3% in IMVEXXY 4 mcg; 4.8% in IMVEXXY 10 mcg; and 1.6% in placebo) with an overall mean severity score of 1.7. Statistically significant improvement in secretions were noted as early as 2 weeks for all groups and was sustained through Week 12. At Week 12, normal vaginal secretions were reported in 34.9% of patients in the IMVEXXY 4 mcg group; and 43.1% in IMVEXXY 10 mcg compared to 24.1% in placebo.
Figure 1: Vaginal Mucosa Assessment Scale
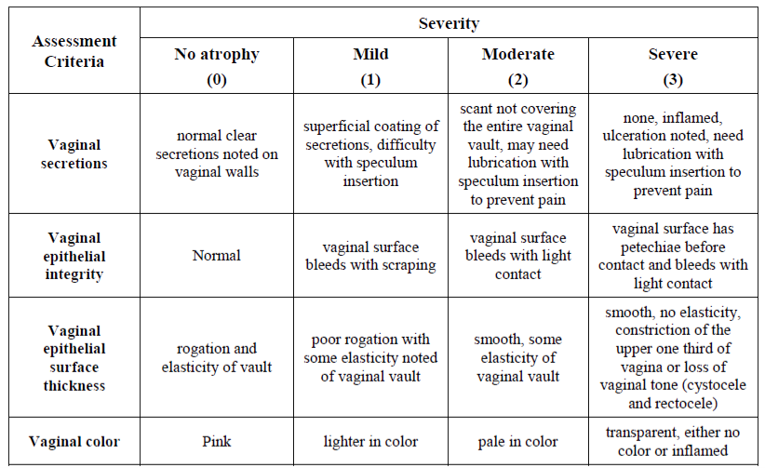
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Table 12: LS Mean Change from Baseline to Weeks 2, 6, 8, and 12 in Vaginal Colour, Vaginal Epithelial Integrity, Vaginal Epithelial Surface Thickness, and Vaginal Secretions
N | LS Mean change from baseline (SE) | P value a | |
|---|---|---|---|
Vaginal Colour, Week 2 | |||
IMVEXXY 4 mcg | 185 | −0.69 (0.048) | < 0.0001 |
IMVEXXY 10 mcg | 187 | −0.77 (0.047) | < 0.0001 |
Placebo | 186 | −0.40 (0.047) | - |
Vaginal Colour, Week 6 | |||
IMVEXXY 4 mcg | 172 | −0.82 (0.049) | < 0.0001 |
IMVEXXY 10 mcg | 170 | −0.93 (0.049) | < 0.0001 |
Placebo | 176 | −0.50 (0.048) | - |
Vaginal Colour, Week 8 | |||
IMVEXXY 4 mcg | 164 | −0.98 (0.050) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −1.04 (0.050) | < 0.0001 |
Placebo | 167 | −0.50 (0.049) | - |
Vaginal Colour, Week 12 | |||
IMVEXXY 4 mcg | 171 | −0.97 (0.049) | < 0.0001 |
IMVEXXY 10 mcg | 173 | −1.06 (0.049) | < 0.0001 |
Placebo | 175 | −0.60 (0.049) | - |
Vaginal epithelial integrity, Week 2 | |||
IMVEXXY 4 mcg | 185 | −0.85 (0.049) | < 0.0001 |
IMVEXXY 10 mcg | 187 | −0.87 (0.049) | < 0.0001 |
Placebo | 186 | −0.53 (0.049) | - |
Vaginal epithelial integrity, Week 6 | |||
IMVEXXY 4 mcg | 172 | −0.97 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 170 | −1.02 (0.051) | < 0.0001 |
Placebo | 176 | −0.61 (0.050) | - |
Vaginal epithelial integrity, Week 8 | |||
IMVEXXY 4 mcg | 164 | −1.03 (0.052) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −1.08 (0.051) | < 0.0001 |
Placebo | 167 | −0.66 (0.051) | - |
Vaginal epithelial integrity, Week 12 | |||
IMVEXXY 4 mcg | 171 | −0.97 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 173 | −1.07 (0.051) | < 0.0001 |
Placebo | 175 | −0.60 (0.050) | - |
Vaginal epithelial surface thickness, Week 2 | |||
IMVEXXY 4 mcg | 185 | −0.76 (0.049) | < 0.0001 |
IMVEXXY 10 mcg | 187 | −0.76 (0.049) | < 0.0001 |
Placebo | 186 | −0.40 (0.049) | - |
Vaginal epithelial surface thickness, Week 6 | |||
IMVEXXY 4 mcg | 172 | −0.85 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 170 | −0.93 (0.051) | < 0.0001 |
Placebo | 176 | −0.53 (0.050) | - |
Vaginal epithelial surface thickness, Week 8 | |||
IMVEXXY 4 mcg | 164 | −0.96 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −1.04 (0.051) | < 0.0001 |
Placebo | 167 | −0.59 (0.051) | - |
Vaginal epithelial surface thickness, Week 12 | |||
IMVEXXY 4 mcg | 171 | −0.98 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 173 | −1.03 (0.051) | < 0.0001 |
Placebo | 175 | −0.61 (0.050) | - |
Vaginal secretions, Week 2 | |||
IMVEXXY 4 mcg | 185 | −0.79 (0.050) | 0.0004 |
IMVEXXY 10 mcg | 187 | −0.83 (0.050) | < 0.0001 |
Placebo | 186 | −0.54 (0.050) | - |
Vaginal secretions, Week 6 | |||
IMVEXXY 4 mcg | 172 | −0.90 (0.051) | 0.0001 |
IMVEXXY 10 mcg | 170 | −0.95 (0.051) | < 0.0001 |
Placebo | 176 | −0.60 (0.051) | - |
Vaginal secretions, Week 8 | |||
IMVEXXY 4 mcg | 164 | −1.00 (0.052) | < 0.0001 |
IMVEXXY 10 mcg | 165 | −1.04 (0.052) | < 0.0001 |
Placebo | 167 | −0.63 (0.052) | - |
Vaginal secretions, Week 12 | |||
IMVEXXY 4 mcg | 171 | −1.01 (0.051) | < 0.0001 |
IMVEXXY 10 mcg | 173 | −1.06 (0.051) | < 0.0001 |
Placebo | 175 | −0.64 (0.051) | - |
aMixed model repeated measures vs. placebo
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Female Sexual Function Index
The FSFI Questionnaire consists of 19 questions divided among 6 domains and has a minimum total score of 2.0 and a maximum score of 36.0 points. At Baseline, the overall mean Total Score was 14.8 for IMVEXXY 4 mcg; 15.8 for IMVEXXY 10 mcg; and 14.4 for placebo. The LS mean change in the FSFI Total Score and domain scores from Baseline to Week 12 are summarized in Table 13. IMVEXXY 10 mcg showed statistically significant improvements compared to placebo for: Total Score, Lubrication, and Pain. There were no statistically significant differences between the total and domain scores for IMVEXXY 4 mcg and placebo. The minimal clinically meaningful differences (MCIDs) for the total score and each domain score in FSFI have not been established in postmenopausal women with VVA.
Table 13: Female Sexual Function Index Total and Domain Scores: LS Mean Change from Baseline to Week 12
N | LS Mean change from baseline (p-value) at Week 12 | |
|---|---|---|
Total Score | ||
IMVEXXY 4 mcg | 153 | 7.909 (0.9075) |
IMVEXXY 10 mcg | 152 | 9.431 (0.0492)a |
Placebo | 158 | 7.458 |
Arousal | ||
IMVEXXY 4 mcg | 154 | 0.875 (0.9719) |
IMVEXXY 10 mcg | 152 | 1.287 (0.0614) |
Placebo | 159 | 0.930 |
Desire | ||
IMVEXXY 4 mcg | 154 | 0.625 (0.9999) |
IMVEXXY 10 mcg | 152 | 0.800 (0.2855) |
Placebo | 159 | 0.630 |
Lubrication | ||
IMVEXXY 4 mcg | 153 | 1.834 (0.4162) |
IMVEXXY 10 mcg | 152 | 2.242 (0.0013)a |
Placebo | 159 | 1.595 |
Orgasm | ||
IMVEXXY 4 mcg | 153 | 1.162 (0.9929) |
IMVEXXY 10 mcg | 152 | 1.274 (0.9634) |
Placebo | 159 | 1.202 |
Pain | ||
IMVEXXY 4 mcg | 154 | 2.173 (0.5146) |
IMVEXXY 10 mcg | 152 | 2.548 (0.0099)a |
Placebo | 159 | 1.930 |
Satisfaction | ||
IMVEXXY 4 mcg | 154 | 1.257 (0.9039) |
IMVEXXY 10 mcg | 152 | 1.384 (0.3751) |
Placebo | 158 | 1.174 |
aindicates a statistically significant difference
LS = least square
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Harms
Safety evaluation plan
Safety assessments included: AEs (including SAEs), endometrial biopsy, gynecological examination (pelvic examination, Pap smear, and breast examination), clinical laboratory testing, physical examination findings, vital signs, 12-lead ECG, pregnancy test, medical/gynecological history and prior medications. As part of the study procedures and AE monitoring, women were provided diaries at each visit to record any symptoms or complaints throughout the study which were then reviewed with the women at each visit by the clinical staff. Vital signs, physical and breast examinations, safety laboratory measurements and ECGs were performed as well. The collection of endometrial biopsies was also required at Baseline and at Week 12, or end of treatment in women with an intact uterus (n = 321).
Assessment of all safety data was performed by the Investigator or a designated Sub-Investigator with appropriate medical training. In this study, an AE included an undesirable medical condition occurring at any time, including Baseline or Washout periods, even if no study treatment had been administered. For each AE, the Investigator evaluated and reported the onset date, resolution date, intensity, causality, action taken, serious outcome (if applicable), and whether or not it caused the patient to discontinue the study.
An AE was considered treatment emergent if the onset time was after administration of study drug through the final follow-up visit, or if a pre-existing AE increased in severity during the 120-day post first dose follow-up period compared to the pre-dose severity. If the start date/time of the AE was unknown, it was assumed to be after the start of study drug.
Overview of safety
See Table 14 for detailed harms data.
Table 14: Summary of Harms, Safety population
Adverse events | REJOICE | ||
|---|---|---|---|
IMVEXXY 4 mcg N = 191 | IMVEXXY 10 mcg N = 191 | Placebo N = 192 | |
Patients with at least one adverse event a | |||
n (%) | 97 (50.8) | 94 (49.2) | 111 (57.8) |
Most common events | |||
Nasopharyngitis | 5 (2.6) | 6 (3.1) | 10 (5.2) |
Upper respiratory tract infection | 5 (2.6) | 6 (3.1) | 5 (2.6) |
Back pain | 9 (4.7) | 1 (0.5) | 8 (4.2) |
Headache | 12 (6.3) | 14 (7.3) | 15 (7.8) |
Vaginal discharge | 5 (2.6) | 6 (3.1) | 13 (6.8) |
Vulvovaginal pruritus | 4 (2.1) | 3 (1.6) | 10 (5.2) |
Patients with at least one serious adverse event | |||
n (%) | 0 (0) | 3 (1.6) | 1 (0.5) |
Sinus node dysfunction | 0 (0) | 1 (0.5) | 0 (0) |
Ankle fracture | 0 (0) | 1 (0.5) | 0 (0) |
Arthralgia | 0 (0) | 1 (0.5) | 0 (0) |
Malignant melanoma | 0 (0) | 1 (0.5) | 0 (0) |
Cervical myelopathy | 0 (0) | 0 (0) | 1 (0.5) |
Adverse events leading to discontinuation | |||
n (%) | 2 (1.0) | 3 (1.6) | 5 (2.6) |
Most common events | |||
Edema peripheral | 1 (0.5) | 0 (0) | 0 (0) |
Muscle spasms | 0 (0) | 1 (0.5) | 0 (0) |
Muscle twitching | 1 (0.5) | 0 (0) | 0 (0) |
Malignant melanoma | 0 (0) | 1 (0.5) | 0 (0) |
Headache | 1 (0.5) | 0 (0) | 0 (0) |
Affect lability | 0 (0) | 0 (0) | 1 (0.5) |
Dysuria | 0 (0) | 1 (0.5) | 0 (0) |
Vulvovaginal burning sensation | 0 (0) | 0 (0) | 2 (1.0) |
Alopecia | 0 (0) | 0 (0) | 1 (0.5) |
Chloasma | 1 (0.5) | 0 (0) | 0 (0) |
Aortic calcification | 0 (0) | 0 (0) | 1 (0.5) |
Adverse events of special interest | |||
n (%) | 3 (1.6) | 2 (1.0) | 5 (2.6) |
Vaginal hemorrhage | 2 (1.0) | 1 (0.5) | 3 (1.6) |
Proliferative endometrium and disordered proliferative pattern b | 0 | 1 (1.1) | 0 |
Cervical dysplasia | 1 (0.5) | 1 (0.5) | 1 (0.5) |
Breast mass | 0 (0) | 0 (0) | 1 (0.5) |
aTreatment emergent adverse events and occurred in ≥ 3% in any treatment arm
bEndometrial safety population (n = 91 in the 10 mcg arm)
Source: Clinical Study Report for REJOICE (Clinical Study Report)
Adverse events
A similar proportion of patients reported TEAEs following treatment with either IMVEXXY or placebo. In the IMVEXXY 4 mcg, 10 mcg, and placebo groups, 97 (50.8%), 94 (49.2%), and 111 (57.8%) of patients reported at least 1 TEAE, respectively. None of the reported TEAEs occurred more frequently in the IMVEXXY groups compared with the placebo groups. Most of the TEAEs were considered mild to moderate in severity. The majority of the TEAEs were classified as not related to study drug.
Serious adverse events
There were a total of 4 serious AEs during the study, none in the IMVEXXY 4 mcg group, 3 in the IMVEXXY 10 mcg group and 1 in the placebo group. None of them were considered related to study drug.
Withdrawals due to adverse events
Ten patients discontinued from the study due to a TEAE, 2 in the IMVEXXY 4 mcg group, 3 in the IMVEXXY 10 mcg group and 5 in the placebo group.
Adverse events of special interest
TEAEs related to the reproductive system and other areas of special interest (vascular arterial disorders, selected cardiovascular safety, and malignancy/cancer) were noted. There were no AEs of special interest that occurred with greater frequency in the IMVEXXY groups than in placebo. Reported AEs of special interest included vaginal hemorrhage, cervical dysplasia and breast mass.
Endometrial Safety
Of 321 women with an intact uterus, 285 women qualified for endometrial safety evaluations. No endometrial hyperplasia or malignancies were diagnosed from endometrial biopsy samples at Week 12. One woman in the IMVEXXY 10 mcg group had 2 out of 3 reads of proliferation on her endometrial biopsy sample (one read of proliferative endometrium, 1 read of benign/inactive/atrophic, and 1 reader reported disordered proliferative pattern); she had a report of vaginal bleeding 1 day post endometrial biopsy that self-limited and lasted 10 days.
Endometrial Polyps
Benign polyps were observed in 11 women at Baseline (4 each in the IMVEXXY 4 and 10 mcg groups and 3 in the placebo group). At Week 12, 1 woman in the IMVEXXY 4 mcg group had an endometrial polyp. All polyps were benign and asymptomatic.
Vaginal Spotting/Bleeding
Of all randomized women, vaginal bleeding and/or spotting were reported by 2 women each in the IMVEXXY 4 and 10 mcg groups (1.0%) and by 3 women in the placebo group (1.6%). All were self-limited and resolved. Six women who reported vaginal bleeding or spotting had endometrial biopsies consistent with benign or insufficient tissue; these were all self-limited and assessed as mild.
Pap Smear
At Baseline, 1 woman in the placebo group had atypical cells of undetermined significance. At 12 weeks, 2 women each in IMVEXXY 4 and 10 mcg groups had a report of atypical cells of undetermined significance, and 1 woman each in IMVEXXY 4 and 10 mcg groups and 2 in placebo had reports of low-grade squamous intraepithelial lesions. All women with low-grade squamous intraepithelial lesions had colposcopies that were negative for dysplasia or malignancy.
Breast Examination
Breast examinations were performed at Screening and Week 12, end of treatment, or discontinuation. At Week 12, there were no clinically significant abnormal breast examinations. Of 7 breast-related TEAEs, 6 were in the placebo group. One woman in the IMVEXXY 10 mcg group had breast tenderness. All TEAEs were assessed as mild in severity. All patients completed the study.
Bioequivalence
In a randomized, 2-way crossover, open-label, single-dose study of the relative bioavailability of a 10 mcg dose of IMVEXXY (Test) and a 10 mcg dose of Vagifem (Reference), plasma concentrations of estradiol, estrone and estrone sulphate in 36 healthy postmenopausal women were compared. A total of 13 blood samples were taken at prescribed time points per the study protocol: −1.00, −0.50, 0.00, 1.00, 2.00, 4.00, 6.00, 8.00, 10.00, 12.00, 14.00, 18.00 and 24.00 hours with reference to the time of insertion into the vagina. Each patient was required to remain in a supine position for 4 hours after dosing and to refrain from strenuous activity until they were checked out of the clinic. After completion of Period I of the study, patients entered a 14-day washout period before crossing over to Period II. All procedures in Period II were identical to those described for Period I with patients receiving the alternative treatment. The statistical analyses to compare treatments were conducted using the general linear model (GLM). PK parameters AUC0 to 24 and Cmax were evaluated after natural logarithmic transformation. Summary statistics, descriptive statistics, analysis of variance (ANOVA), and 90% confidence intervals (CI) were calculated for baseline-adjusted and baseline-unadjusted data for estradiol, estrone and estrone sulphate for all PK parameters for both Test and Reference products. One patient did not complete the study for reasons unrelated to the study drug and was excluded from the PK analysis. One patient’s baseline adjusted values for estradiol were below zero and was excluded from that analysis.
Low levels of estradiol, estrone and estrone sulphate were absorbed systemically following vaginal administration. Treatment with IMVEXXY resulted in significantly lower estradiol, estrone, and estrone sulphate levels than with 10 mcg Vagifem in healthy, postmenopausal female patients. Baseline adjusted values and test/reference (T/R) ratios can be found in Table 15. The extent of systemic exposure of IMVEXXY 10 mcg was statistically significantly lower than that of Vagifem 10 mcg in healthy, postmenopausal females. Estradiol concentrations are only modestly higher than baseline, endogenous concentrations.
Table 15: Statistical Results of IMVEXXY versus Vagifem for Estradiol, Estrone, Estrone Sulphate – Baseline Adjusted (N = 34)
Pharmacokinetics | IMVEXXY 10 mcg (test) | Vagifem 10 mcg (reference) | T/R Ratio, % (90% CI) p-value |
|---|---|---|---|
Estradiol | |||
AUC0 to 24 (pg.h/mL) | 49.73 | 131.04 | 37.95 (29.21 to 49.31) < 0.0001 |
Cmax (pg/mL) | 14.45 | 20.20 | 71.54 (56.82 to 90.08) 0.0194 |
Estrone | |||
AUC0 to 24 (pg.h/mL) | 24.20 | 47.90 | 50.51 (38.37 to 66.50) 0.0002 |
Cmax (pg/mL) | 5.16 | 6.93 | 74.50 (61.69 to 89.97) 0.0127 |
Estrone Sulphate | |||
AUC0 to 24 (pg.h/mL) | 68.5 | 118.4 | 57.87 (41.68 to 80.35) 0.0091 |
Cmax (pg/mL) | 12.3 | 16.5 | 74.55 (59.43 to 93.51) 0.0366 |
AUC0 to 24 – area under the concentration-time curve 24 hours; Cmax = peak concentration; T/R = test/reference
Source: Common Technical Document 2.7.1
CADTH’s Critical Appraisal of the Clinical Evidence
Internal Validity
Appropriate methods of randomization, blinding and allocation concealment were reported in the REJOICE study. However, no stratification was performed for randomization. Time since menopause and severity of dyspareunia were similar across treatment groups at baseline, while there were differences noted in other patients’ baseline characteristics between 4 mcg and 10 mcg estradiol inserts and the placebo group, such as history of hysterectomy, percentage of natural menopause and surgical menopause, and percentage of bilateral oophorectomy. The data suggest that more patients in the estradiol inserts groups had a hysterectomy and bilateral oophorectomy, therefore a higher proportion of these patients were surgically menopausal, compared to those in the placebo group. These differences were numerically small, and it is unknown whether patients with surgical menopause respond differently than those with natural menopause, and whether these imbalances would affect interpretation of the results.
The percentage of patients who discontinued prematurely in this study ranged from 7% to 9%, and the reasons for study discontinuation were similar between treatment groups, suggesting that blinding was maintained throughout the study period.
Both subjective (e.g., self-reported symptom relief or change in sexual function) and objective efficacy outcomes (e.g., change in percentage of superficial cells, vaginal pH) were evaluated in the REJOICE study. Although self-reported outcomes are considered clinically relevant in practice to measure treatment response according to the clinical expert, there are no published MCIDs identified for such outcome measures in postmenopausal women. Therefore, it is unclear whether the scales used and the reported between-group differences are clinically meaningful.
All patients who were randomly assigned and had at least 1 dose of study drug formed the ITT population. This was not a true ITT population, but a modified ITT (mITT) population, and it was the primary efficacy population in the REJOICE study. The mITT population was defined as all ITT patients who received the treatment to which they were assigned, had baseline values for all 4 coprimary variables, and had at least 1 post-baseline value for any of the 4 coprimary variables at any visit. In this trial, study participants who either reported no sexual activity at week 12 (9.1%) or missing data on dyspareunia at week 12 (7.0%) were excluded from MMRM analysis of MBS dyspareunia, and missing data were not imputed, there is a concern about the robustness of these study results. The MMRM analysis assumes the data are missing at random and although dropouts were not differential in REJOICE, it is unclear if this major assumption for MMRM analysis is met within the data and how it may have biased the study results. Additional sensitivity analysis using last observation carried forward to handle the missing data was requested by the US FDA, and the results showed a similar statistically significant reduction in the severity of dyspareunia for the 2 doses of estradiol vaginal inserts, therefore supported the primary efficacy analysis.17 For secondary analysis, missing data would have been a larger concern and could have biased the observed estimates (e.g., HRQoL where more missing data, approximately 10%, was noted) when a complete case analysis was performed.
Multiplicity was controlled for in REJOICE based on a closed fixed sequence serial testing procedure, with the 4 coprimary end points being included. Outcomes outside of the testing hierarchy such as HRQoL (measured with FSFI), should be viewed as supportive evidence for the overall effects of estradiol vaginal inserts and need to be interpreted with caution due to the possible inflated type I error.
External Validity
According to the clinical expert consulted by CADTH, the inclusion and exclusion criteria for REJOICE were generally consistent with clinical practice. Based on patients’ baseline characteristics, the study populations reflect a typical Canadian population that would receive vaginal estradiol therapy in practice. Both subjective and objective outcomes were measured in REJOICE. In postmenopausal women with VVA symptoms, and the subjective self-reported symptom relief is a clinically meaningful outcome.
REJOICE was a 3-month study, therefore long-term safety (on endometrium and breast, or in general) and efficacy data are unavailable for the 2 doses of estradiol vaginal inserts.
There is a lack of direct or indirect evidence from the included study to demonstrate comparative efficacy and safety of the estradiol vaginal insert versus other local hormonal therapy in the study population.
Indirect Evidence
A focused literature search for network meta-analyses (NMAs) dealing with Imvexxy (estradiol vaginal inserts) and menopause (including dyspareunia, a symptom of vulvar and vaginal atrophy) was run in MEDLINE All (1946-) on July 12, 2021.
No relevant indirect treatment comparisons were identified for this review.
Other Relevant Evidence
No other relevant studies were identified for this review.
Sponsor Submitted Cost Comparison
Estradiol vaginal insert (Imvexxy) is available in 4 mcg and 10 mcg strengths. The sponsor submitted a cost comparison of treatments for postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, in which Imvexxy was compared with other local hormone therapies currently reimbursed for this indication.18 This included other estradiol products (including Vagifem, the other available estradiol vaginal insert, and Estring, an insertable ring comparator), as well as creams (estrone (EstraGyn) and conjugated estrogen [Premarin cream]). Only drug acquisition costs were considered, and the sponsor assumed a 100% treatment adherence rate, dosing as per product monographs,8,14-16,19 and equal use of accompanying progestin therapies for all included comparators (i.e., no cost implications).
The recommended dose of Imvexxy is 1 tablet inserted daily for 2 weeks, followed by a maintenance dose of 1 tablet inserted twice weekly every 3 to 4 days. At the submitted price of $3.63 per tablet insert, the annual cost of treatment was estimated to be $413.62 per person regardless of strength. Compared with Vagifem, Imvexxy was associated with annual savings of $77.53 per patient. In comparison with the cream-based comparators, the sponsor estimated the cost impact per patient of Imvexxy ranges from cost savings of $413.62 to a cost increase of $369.88, depending on the prescribed dose of the cream-based treatment. When compared to the estradiol ring comparator, the sponsor estimated an increased annual cost of $56.84 per patient.
CADTH’s Critical Appraisal of Cost Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the cost comparison:
Annual drug costs of the estradiol vaginal inserts (Imvexxy and Vagifem) varies from first year to second year of use, which affects potential cost savings: The sponsor submitted a cost comparison for the first year of treatment. Given the initial dosing with Imvexxy and Vagifem differs from the maintenance dosing, the costs associated with treatment with estradiol inserts, along with incremental costs relative to the other comparators, would differ from the first year of use in comparison with subsequent years.
In the CADTH reanalysis, annual drug costs and incremental costs are presented for both the first year of treatment and subsequent years of treatment.
List price of conjugated estrogen cream and the estradiol ring varies across jurisdictions: The sponsor’s submitted cost comparison based the unit price of conjugated estrogen cream ($0.84 per 0.625 mg) and the estradiol ring ($89.21 per 2 mg ring) on the Ontario Drug Benefit formulary.20 However, the cost of these comparators varies across jurisdictions; the lowest listed price for conjugated estrogen cream is $0.76 per 0.625 mg21 and for the estradiol ring is $74.67 per 2 mg ring.22 As such, estimated incremental costs from the reimbursement of Imvexxy will vary across jurisdictions.
In a CADTH reanalysis, the incremental costs associated with the Imvexxy were estimated based on the lowest publicly available list prices for conjugated estrogen cream and the estradiol ring.
CADTH Reanalyses
The CADTH reanalysis considered the average annual drug cost and difference in annual costs for both the first and subsequent years of use, as well as the lowest available list prices for conjugated estrogen cream and the estradiol ring, Table 16. The annual treatment cost with Imvexxy was estimated to be lower in subsequent years ($377 per patient per year) than the first year of treatment. In comparison with Vagifem, Imvexxy was associated with cost savings of $78 per patient in the first year and $71 in subsequent years. In comparison with the cream-based comparators, the difference in annual incremental costs per person ranged from cost savings of $450 to increased costs of $338, depending on the dosage regimen with the cream and year of treatment. The annual difference in costs in comparison with the estradiol ring comparator rose to $115 in the first year and $79 in subsequent years, per patient.
Table 16: CADTH Cost Comparison Table for Treatment of Post-Menopausal Moderate to Severe Dyspareunia
Generic name (brand name) | Strength | Dosage form | Price ($) | Recommended dosage regimen | Annual drug cost ($) | Difference in annual cost |
|---|---|---|---|---|---|---|
17 beta-estradiol (Imvexxy) | 4 mcg | Tablet insert | $3.6288a | Initial dose: 1 vaginal insert daily at the approximately the same time for 2 weeks. Maintenance dose: 1 vaginal insert twice weekly, every three to four days.f | First year: $414 Subsequent years: $377 | Reference |
17 beta-estradiol (Imvexxy) | 10 mcg | Tablet insert | $3.6288a | Initial dose: 1 vaginal insert daily at the approximately the same time for 2 weeks. Maintenance dose: 1 vaginal insert twice weekly, every three to four days.f | First year: $414 Subsequent years: $377 | Reference |
Insert comparator | ||||||
17 beta-estradiol (Vagifem) | 10 mcg | Tab insert | $4.3089b | Initial dose: 1 vaginal insert daily for 2 weeks. Maintenance dose: 1 vaginal insert twice weekly with 3- or 4-day interval between doses.g | First year: $491 Subsequent years: $448 | First year: –$78 Subsequent years: –$71 |
Cream comparators | ||||||
Estrone (EstraGyn) | 0.1% w/w | Gram | $0.7576c | 0.5 to 4 g per day taken intravaginally, adjusted to the lowest amount that controls symptoms. EstraGyn is intended for short-term use. Administration should be cyclic (e.g., three weeks on one week off).h | $103 to $827 | First year: –$414 to $310 Subsequent years: –$450 to $274 |
Conjugated estrogen (Premarin cream) | 0.625 mg/g | Gram | $0.7510d | Low dose: Premarin cream (0.5g) is administered intravaginally or topically twice weekly. Maximum Recommended Dose: Premarin cream is administered intravaginally or topically in a cyclic regimen (daily for 21 days and then off for 7 days). Generally, women should be started at the 0.5g daily dosage strength. Dosage adjustments (0.5 to 2 g) may be made based on individual response.i | $39 to $410 | First year: –$4 to $375 Subsequent years: –$33 to $338 |
Ring comparator | ||||||
17 beta-estradiol (Estring) | 2 mg | Ring | $ 74.6655e | One Estring is to remain in place continuously for three months, after which it is to be removed and, if continuation of therapy is deemed appropriate, replaced by a new ring. The need to continue treatment should be assessed at 3 or 6 month intervals.j | $299 | First year: $115 Subsequent years: $79 |
Note: Reanalyses are based on publicly available prices of the comparator treatments. Annual period assumes 365 days or 52 weeks for all comparators.
aSponsor’s submission.18
bOntario Drug Benefit Formulary,20 accessed August 27, 2021.
cNova Scotia Drug Formulary,23 accessed August 27, 2021.
dSaskatchewan Drug Formulary,21 accessed August 27, 2021.
eAlberta Drug Formulary,22 accessed August 27, 2021.
fKnight Therapeutics Inc. IMVEXXY Product Monograph,8 accessed August 27, 2021.
gNovo Nordisk Canada Inc. Vagifem 10 Product Monograph,14 accessed August 27, 2021.
hSearchlight Pharma Inc. EstraGyn Vaginal Cream Product Monograph,19 accessed August 27, 2021.
iPfizer Canada Inc. Premarin Vaginal Cream Product Monograph,16 accessed August 27, 2021.
jPfizer Canada Inc. Estring Product Monograph,15 accessed August 27, 2021.
Price Reduction Analyses
CADTH conducted exploratory price reduction analyses estimating the percentage reduction to the sponsor’s submitted price required for Imvexxy to be cost-neutral to the least expensive comparators available, Table 17. These scenarios only considered cost-neutrality in subsequent years of use. The submitted price of Imvexxy would have to be reduced by 90% for the annual treatment acquisition cost to be equivalent to that of the lowest dose (0.5 g) of conjugated estrogen cream. When considering the estradiol ring comparator, the submitted price of Imvexxy would need to be reduced by 21% for treatment acquisition costs to be cost neutral.
Table 17: CADTH Price Reduction Analyses
Scenario | Submitted price ($) | Reduction needed (%) | Reduced price ($) | Savings relative to submitted pricea ($) |
|---|---|---|---|---|
Price reduction required to equal least expensive cream (conjugated estrogen) at lowest dose | 3.6288 | 90 | 0.3755 | 338 |
Price reduction required to equal estradiol ring | 3.6288 | 21 | 2.87175 | 79 |
aSavings from the sponsor list price per patient per year for year 2 and onward. Relative to publicly available list prices of comparators
Issues for Consideration
Comparative efficacy and safety of Imvexxy to other local hormone therapies is uncertain: An assumption of clinical equivalence is required for a cost comparison to be considered an appropriate form of analysis to assess the cost-effectiveness of Imvexxy. The CADTH clinical review noted that there is a lack of direct and indirect evidence from the sponsor’s submitted study to demonstrate the comparative efficacy and safety of the Imvexxy with the other local hormone therapies considered in the sponsor’s cost comparison. A bioequivalence study submitted by the sponsor suggests the assumption of clinical equivalence between both estradiol vaginal inserts (Imvexxy and Vagifem) is likely to be appropriate at the 10 mcg dose in healthy postmenopausal women, with less certainty with regard to the 4 mcg dose of Imvexxy in comparison with Vagifem at 10 mcg. As a result, the cost comparison with Vagifem is likely appropriate, while the appropriateness of the cost comparison with the cream and ring based local hormone therapies is associated with uncertainty given the lack of evidence to inform the assumption of equivalent efficacy and safety.
Increases in dosing of Imvexxy may affect relative drug costs: Drug plan input noted concerns regarding the potential for the use of multiple tablet inserts when escalating the dose to address a lack of response to initial treatment with 4 mcg or 10 mcg of Imvexxy. Should multiple tablets be used, drug costs would double, reducing or eliminating cost savings depending on the comparator of interest. Such situations were thought to be unlikely according to the clinical expert consulted by CADTH for this review.
Analysis based on publicly available list prices: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
Discussion
Summary of Available Evidence
One phase III study (REJOICE, N = 574) submitted by the sponsor was summarized and appraised in this review. The trial enrolled postmenopausal women with moderate to severe symptom of vaginal pain associated with sexual activity.
REJOICE was a double-blind, placebo-controlled RCT that assessed the efficacy and safety of the estradiol vaginal insert for the treatment of postmenopausal moderate to severe dyspareunia. Eligible patients were randomized to receive the estradiol vaginal insert 4 mcg or 10 mcg, or placebo for 12 weeks. The coprimary efficacy end points were change from baseline to week 12 in percent change in superficial cells compared to placebo, change from baseline to week 12 in percent change in parabasal cells compared to placebo, change from baseline to week 12 in percent change in pH compared to placebo, and change from baseline to week 12 on the severity of the MBS of dyspareunia (vaginal pain associated with sexual activity) associated with VVA compared to placebo.
The key limitations of the REJOICE study were a lack of an active comparator arm of other vaginal estrogen therapies, and a lack of longer-term efficacy and safety data for the drug in study population.
Interpretation of Results
Efficacy
After 3 months treatment, the REJOICE study met its objective by demonstrating improvement in favour of both doses of the estradiol vaginal inserts versus placebo on the 4 coprimary end points: change from baseline to week 12 in the percentage of parabasal cells, superficial cells, vaginal pH, and severity of dyspareunia. One of the outcomes was the change from baseline in patient-reported severity of dyspareunia, which was consistent with clinical practice, according to the clinical expert consulted by CADTH.
At week 12, vaginal dryness was improved with both doses of estradiol vaginal insert compared with placebo, while only the estradiol 10 mcg group had improved vulvar and/or vaginal itching or irritation versus placebo. The expert indicated that the results of these secondary efficacy outcomes were consistent with the primary outcomes, which favoured estradiol over placebo; however, the differences between estradiol and placebo may not be considered clinically important.
According to the clinical expert, patient-reported symptom relief is a clinically relevant outcome in the study population, while histologic examination is generally not performed in practice. In REJOICE, a VVA Symptoms Self-Assessment Questionnaire was used to self-assess patient’s symptoms of VVA, including vaginal pain associated with sexual activity, vaginal dryness, and vulvar and/or vaginal itching or irritation. However, no information was provided in the submission describing the validity and reliability of this questionnaire, nor was a MCID reported in the indicated population. Although estradiol vaginal inserts appeared to be efficacious versus placebo, it is difficult to determine whether the magnitude of benefit observed is clinically significant.
Severity of VVA (no atrophy, mild, moderate and severe atrophy) was evaluated using a vaginal mucosa assessment scale, which examines vaginal secretions, epithelial integrity, epithelial surface thickness and colour during pelvic examination. Normal vaginal secretions, epithelial integrity, epithelial surface thickness and colour at week 12 were more likely to be observed in patients treated with estradiol (4 mcg and 10 mcg) compared to placebo.
Treatment with the estradiol vaginal insert was associated with improved sexual function in postmenopausal women, measured by the FSFI. The 10 mcg of estradiol insert showed statistically significant improvements in Total Score, Lubrication and Pain of FSFI; however, it is unclear whether the between-group differences were clinically meaningful. MCIDs of the total score and other domain scores for FSFI have not been established in women with postmenopausal dyspareunia. Furthermore, there were no statistically significant differences between estradiol 4 mcg and placebo, which suggests less benefit with the lower dose estradiol therapy. Of note, patients who need the estradiol vaginal inserts should start with the 4 mcg dose first, based on the product monograph. This implies that while the 4 mcg dose is effective on other efficacy outcomes compared to placebo, it was not effective in improving sexual function, an important clinical outcome in the study population. When treated with 4 mcg estradiol vaginal inserts, women may not have adequate response to the lower dose of the drug and may need dose escalation. From a cost perspective, there will be no cost increase for women who need to increase their dose from 4 mcg to 10 mcg, which will apply to the majority of patients. However, in the rare occasions where the dose must be increased from 10 mcg to 14 mcg, there will be an impact on costs. The clinical expert consulted by CADTH estimated that only a small proportion of these patients would use a higher dose (followed by a discussion of risks and benefits), as this is not the standard treatment regimen with uncertain benefit.
No longer-term data beyond the 12-week REJOICE study were provided on the efficacy of estradiol vaginal inserts. Therefore, estradiol vaginal insert is efficacious compared with placebo in improving symptoms related to VVA in short-term.
No comparative effectiveness evidence was submitted. As presented in Table 3, there are various estrogen containing products with the same chemical entity and a similar indication as estradiol vaginal insert. The bioequivalence data between estradiol vaginal insert 10 mcg and estradiol vaginal tablets 10 mcg (Vagifem) suggest these are similar products, albeit with seemingly less systemic absorption with the former product. These data suggest that estradiol administered in the softgel formulation should have similar effect as estradiol administered in the tablet form; though the lack of comparative data, especially on patient-important outcomes like reduced symptoms, sexual health, and quality of life is a key limitation. This limitation also applies to comparisons between estradiol vaginal insert and other available products.
Harms
During the 3-month study period of REJOICE, the frequency of AEs and WDAEs was similar between the 2 doses of estradiol and placebo. Three patients in the estradiol 10 mcg group reported SAEs, while no SAEs were reported in the estradiol 4 mcg and 1 SAE was reported in the placebo group. The data are limited in terms of frequency and duration of follow-up to determine whether a dose response and frequency of SAEs is present.
The submission highlighted the following as AEs of interest: AEs related to the reproductive system, vascular arterial disorders, selected cardiovascular events, and malignancy. These are consistent with reported safety concerns related to estrogen and progesterone products, input from clinical experts, and harms of interest in regulatory evaluations. The report on these outcomes provided to CADTH suggested no clear differences between treatment groups or serious events for these AEs of interest. In its safety assessment, Health Canada noted that initiating therapy with the lowest dosage strength of estradiol vaginal insert, 4 mcg, would be consistent with the clinical approach to treating VVA symptoms and that patients and clinicians would have the lowest dosage strength of any estradiol-alone product approved for this indication, providing treatment options.24 The recommendation was based on the REJOICE study and “Higher doses of vaginally administered estrogens, and oral and topically applied estrogens, are well studied and have been found to be safe and effective when used appropriately.” However, the Health Canada reviewer’s report also noted the following:
No long-term general and endometrial safety data or chronic use drug exposure data, of at least 12-months duration, is available in the NDS submission for the 4 mcg and 10 mcg estradiol vaginal insert.
The finding of proliferative endometrium and disordered proliferative endometrium at only 12-weeks of drug exposure in phase III Trial TXV14 to 01 confirm that long-term safety data were necessary to support the long-term general and endometrial safety and chronic use drug exposure of the 4 mcg and 10 mcg estradiol vaginal inserts for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause.
The sponsor will conduct a class post-marketing requirement long-term (3 to 5 years) observational study to identify the incidence of endometrial cancer associated use of unopposed low dose vaginal estrogen products in postmenopausal women.
The product monograph for the estradiol vaginal insert contains the same contraindications (which includes endometrial hyperplasia) and warnings and precautions as the other available products with the same or similar indication.
No comparative safety data were submitted. A randomized, open-label crossover, single-dose study of the relative bioavailability of a 10 mcg dose of estradiol vaginal insert compared with another vaginal estrogen therapy (estradiol vaginal tablet 10 mcg dose [Vagifem]) was conducted in healthy postmenopausal women. The results suggested that the extent of systemic exposure of estradiol vaginal insert 10 mcg was statistically significantly lower than that of estradiol vaginal tablet 10 mcg. It is unclear based on the existing data whether the lower systemic absorption will lead to fewer AEs compared with comparators in practice.
Cost
At the submitted price, Imvexxy costs $414 per patient annually in the first year of use and $377 in subsequent years of use. CADTH conducted a reanalysis of the sponsor submitted cost comparison, considering: all relevant local hormone therapies; costs in the first and subsequent years of use; and the lowest available list price for conjugated estrogen cream and the estradiol ring. The annual cost or cost savings with Imvexxy depend on the choice of comparator. Compared with the existing estradiol vaginal insert (Vagifem), annual cost savings with Imvexxy were $78 per person in the first year and $71 per person in subsequent years of use. Compared with cream-based comparators, annual per person incremental costs ranged from cost savings of $450 to increased costs of $338, depending on the dose of the cream-based comparators. The incremental cost compared with the estradiol ring was $115 in first year and $79 in subsequent years of use. The incremental costs were calculated based on publicly available list prices of comparators and may not reflect actual prices paid by Canadian public drug plans. Additionally, the price of conjugated estrogen cream (Premarin cream) and the estradiol ring comparator (Estring) varies across jurisdictions, and as such, incremental costs will vary across jurisdictions.
The cost comparison assumes clinical similarity between Imvexxy and the other local hormone therapies included in the analysis. Based on a sponsor’s submitted bioequivalence study, the 10 mcg dose of Imvexxy is likely clinically similar to Vagifem at the same dose in healthy postmenopausal women. The clinical review conducted by CADTH noted that there was a lack of direct or indirect clinical evidence comparing Imvexxy to local hormone therapies in the indicated population (menopausal women with dyspareunia). As a result, the cost comparison with Vagifem is likely appropriate, while the appropriateness of the cost comparison with the cream and ring based local hormone therapies is associated with uncertainty.
Conclusions
Evidence from 1 RCT supported the efficacy of estradiol vaginal insert (4 mcg and 10 mcg) for the treatment of postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy. Compared to placebo, patients who were treated with estradiol vaginal insert for 12 weeks showed benefits in symptom relief (vaginal pain associated with sexual activity, vaginal dryness, vulvar and/or vaginal itching or irritation). Improvements in sexual function were observed with the estradiol vaginal insert 10 mcg versus placebo, but not with the 4 mcg dose. The frequency of AEs, serious adverse events and treatment discontinuation due to AE were similar across treatment groups and were consistent with the expected adverse event profile for an estradiol-containing product.
Longer-term (beyond 12 weeks) efficacy and safety of estradiol vaginal insert is unknown. There is a lack of comparative evidence between estradiol vaginal insert and the other vaginal estrogen therapies in postmenopausal women with moderate to severe dyspareunia. One bioequivalence study suggested the 10 mcg dose of the estradiol vaginal insert is similar to the estradiol vaginal tablet (Vagifem) at the same dose, but there are no clinical studies to confirm outcomes are similar between the 2 estradiol products.
At the submitted price, the estradiol vaginal insert costs $414 per patient annually in the first year of use and $377 in subsequent years of use. Estradiol vaginal insert is cost saving compared with the other available estradiol vaginal tablet (Vagifem), but is associated with higher costs compared with the estradiol ring. The cost (or savings) with estradiol vaginal insert varies by cream-based comparators depending on the dose. Based on the submitted clinical evidence, the cost comparison with the estradiol vaginal tablet (Vagifem) is likely appropriate, while the appropriateness of the cost comparison with the cream and ring based local hormone therapies is associated with uncertainty.
References
1.Perez-Lopez FR, Vieira-Baptista P, Phillips N, Cohen-Sacher B, Fialho S, Stockdale CK. Clinical manifestations and evaluation of postmenopausal vulvovaginal atrophy. Gynecol Endocrinol. 2021;37(8):740-745. PubMed
2.Palma F, Volpe A, Villa P, Cagnacci A. Vaginal atrophy of women in postmenopause. Results from a multicentric observational study: the AGATA study. Maturitas. 2016;83:40-44. PubMed
3.Simon JA, Kokot-Kierepa M, Goldstein J, Nappi RE. Vaginal health in the United States: results from the Vaginal Health: Insights, Views & Attitudes survey. Menopause. 2013;20(10):1043-1048. PubMed
4.Sturdee DW, Panay N. Recommendations for the management of postmenopausal vaginal atrophy. Climacteric. 2010;13(6):509-522. PubMed
5.Bachmann G, Santen RJ. Genitourinary syndrome of menopause (vulvovaginal atrophy): treatment. In: TW P, ed. UpToDate. Waltham (MA): UpToDate; 2021. Accessed 2021 Oct 28.
6.The 2020 genitourinary syndrome of menopause position statement of The North American Menopause Society. Menopause. 2020;27(9):976-992. PubMed
7.Health Canada. Notice of Compliance (NOC) for Imvexxy. 2020; https://health-products.canada.ca/noc-ac/index-eng.jsp. Accessed 2021 Aug 27.
8.Imvexxy (estradiol vaginal inserts): 4 mcg and 10 mcg estradiol, vaginal [product monograph]. Montreal (QC): Knight Therapeutics Inc.; 2021 Aug 13.
9.Clinical Study Report: TXV14-01 (REJOICE). A phase 3, randomized, double-blind, placebo-controlled, multi-center trial to evaluate the safety and efficacy of TX-004HR in postmenopausal women with moderate to severe symptoms of vulvar and vaginal atrophy [internal sponsor's report]. Montreal (QC): Knight Therapeutics Inc; 2015 Jun 9.
10.Santoro. Genitourinary syndrome of menopause: underdiagnosed and undertreated. Contemp Ob Gyn. 2018;64(7).
11.Ruan X, Zhang L, Cui Y, Gu M, Mueck AO. Genitourinary syndrome of menopause in Chinese perimenopausal and postmenopausal women. Climacteric. 2021;24(3):297-304. PubMed
12.e-CPS. Ottawa (ON): Canadian Pharmacists Association; 2021: https://www.e-therapeutics.ca/. Accessed 2021 Sep 13.
13.Lethaby A, Ayeleke RO, Roberts H. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2016;2016(8):Cd001500. PubMed
14.Vagifem (estradiol vaginal inserts USP): 10 ug estradiol [product monograph]. Mississauga (ON): Novo Nordisk Canada Inc; 2017 Mar 20: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/Vagifem_10_PM_English.pdf. Accessed 2021 Aug 27.
15.Estring (17 β-estradiol): 2 mg vaginal ring [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2017 Nov 16: https://pdf.hres.ca/dpd_pm/00042172.PDF. Accessed 2021 Aug 27.
16.Premarin (conjugated estrogens): 0.3 mg, 0.625 mg, and 1.25 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada Inc., Licensee; 2012 Jan 10: https://pdf.hres.ca/dpd_pm/00015274.PDF. Accessed 2021 Aug 27.
17.Center for Drug Evaluation Research. Statistical review(s). Imvexxy (estradiol vaginal inserts). Company: TherapeuticsMD. Application No.: 208564. Approval date: 05/29/2018 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/208564Orig1s000TOC.cfm. Accessed 2021 Jul 12.
18.Drug Reimbursement Review sponsor submission: Imvexxy (estradiol), 4 mcg and 10 mcg vaginal inserts capsules [internal sponsor's package]. Montreal (QC): Knight Therapeutics Inc.; 2021 Jun 23.
19.EstraGyn (esterified estrogen): 0.3 mg & 0.625 mg tablets [product monograph]. Montreal (QC): Searchlight Pharma Inc; 2015 Apr 24: https://pdf.hres.ca/dpd_pm/00030970.PDF. Accessed 2021 Aug 27.
20.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Aug 27.
21.Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2021 Aug 27.
22.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020 Mar 31: https://idbl.ab.bluecross.ca/idbl/drugsList;jsessionid=KT3IcpZMfWVQZVSr2Dd5im7OkwO5l7gh9TaX2c4U1xIuTYkq5xZH!-1427890521?searchTerm=estradiol&category=&genericName=&ptc=&mfgCode=. Accessed 2021 Aug 27.
23.Nova Scotia formualry. Halifax (NS): Nova Scotia Department of Health; 2021: https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf. Accessed 2021 Aug 27.
24.Health Canada reviewer's report: Imvexxy (estradiol vaginal inserts) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imvexxy (estradiol), 4 mcg and 10 mcg vaginal inserts capsules. Montreal (QC): Knight Therapeutics Inc; 2021 Jun 23.
25.Lima MJF, Valim MD, de Medeiros SF. Construct and criterion validity of the Postmenopause Sexuality Questionnaire - PMSQ. Rev Bras Ginecol Obstet. 2020;42(1):26-34. PubMed
26.Baser RE, Li Y, Carter J. Psychometric validation of the Female Sexual Function Index (FSFI) in cancer survivors. Cancer. 2012;118(18):4606-4618. PubMed
27.Perez-Herrezuelo I, Hita-Contreras F, Martinez-Amat A, et al. The female sexual function index: reliability and validity in Spanish postmenopausal women. Menopause. 2019;26(4):401-408. PubMed
28.Wylomanski S, Bouquin R, Philippe HJ, et al. Psychometric properties of the French Female Sexual Function Index (FSFI). Qual Life Res. 2014;23(7):2079-2087. PubMed
Appendix 1: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Female Sexual Function Index (FSFI)
Findings
FSFI
The FSFI is a multidimensional questionnaire for assessing sexual function in women. The questionnaire consists of 19 items that assesses sexual function over the past 4 weeks and yield domain scores in 6 areas: sexual desire, arousal, lubrication, orgasm, satisfaction, and pain.25 The items are answered on an ordinal Likert scale (scored from 0 or 1 to 5). The scoring algorithm sums items on each domain and then scales the sums so that each subscale has a maximum score of 6. The FSFI total score is the sum of the 6 domain scores and has a maximum score of 36. Higher scores indicate better functioning.25,26 Based on validation studies, a cutoff point of 26.5 was proposed. However, its cutoff point to discriminate menopause women with or without sexual dysfunction was established as 23, such that any menopause women with a FSFI total scores of less than 23 are considered at risk of sexual dysfunction.25 FSFI has been validated in various populations, including healthy women, postmenopausal women, dyspareunia or vaginismus (pain), multiple sexual dysfunctions, and cancer survivors.26 Reliability and validity of FSFI used in different languages have also been examined.27,28 The MCIDs for the FSFI total score and domain scores in postmenopausal women with VVA have not been identified in the literature.
In clinical research, FSFI is usually used as a gold standard questionnaire to validate other instruments.
Appendix 2: Additional Economic Information
Note that this appendix has not been copy-edited.
Additional Details on the Sponsor’s Submission
Table 18: Sponsor’s Drug Acquisition Cost Comparison
Generic Name (Brand Name) | Strength | Dosage Form | Price ($) | Recommended Dosage Regimen | Annual Drug Cost ($) | Difference in Annual Cost |
|---|---|---|---|---|---|---|
17 Beta-estradiol (IMVEXXY) | 4mcg | Tab insert | $3.6288a | Initial dose: 1 vaginal insert daily at the approximately the same time for 2 weeks Maintenance dose: 1 vaginal insert twice weekly, every three to four days d | $413.68 | — |
17 Beta-estradiol (IMVEXXY) | 10mcg | Tab insert | $3.628 a | Initial dose: 1 vaginal insert daily at the approximately the same time for 2 weeks Maintenance dose: 1 vaginal insert twice weekly, every three to four days d | $413.68 | — |
Insert comparator | ||||||
17 Beta-estradiol (Vagifem) | 10mcg | Tab insert | $4.3089 b | Initial dose: 1 vaginal insert daily for 2 weeks Maintenance dose: 1 vaginal insert twice weekly with 3- or 4-day interval between doses e | $491.21 | –$77.53 |
Cream comparators | ||||||
Estrone (Estragyn) | 0.1% w/w | gram | $0.7576 c | 0.5 to 4 g per day taken intravaginally, adjusted to the lowest amount that controls symptoms. Estragyn is intended for short-term use. Administration should be cyclic (e.g., three weeks on one week off) f | $103.41 to $827.30 | -$413.62 to $310.27 |
Conjugated estrogen (Premarin cream) | 0.625mg/g | gram | $0.8423 b | Low dose: Premarin cream (0.5g) is administered intravaginally or topically twice weekly. Maximum Recommended Dose: Premarin cream is administered intravaginally or topically in a cyclic regimen (daily for 21 days and then off for 7 days). Generally, women should be started at the 0.5 g daily dosage strength. Dosage adjustments (0.5 to 2 g) may be made based on individual response. g | $43.80 to $459.90 | -$46.21 to $369.88 |
Ring comparator | ||||||
17 Beta-estradiol (Estring) | 2mg | Ring | $89.2100 b | One Estring is to remain in place continuously for three months, after which it is to be removed and, if continuation of therapy is deemed appropriate, replaced by a new ring. The need to continue treatment should be assessed at 3- or 6-month intervals. h | $356.84 | $56.84 |
Tab = tablet.
a. Provided by manufacturer.
b. Ontario Drug Benefit Formulary.
c. Nova Scotia Drug Formulary.
d. Knight Therapeutics Inc. IMVEXXY Product Monograph. August 13, 2020.
e. Novo Nordisk Canada Inc. Vagifem 10 Product Monograph. May 18, 2016.
f. Searchlight Pharma Inc. EstraGyn Vaginal Cream Product Monograph. August 17, 2016.
g. Pfizer Canada Inc. Premarin Vaginal Cream Product Monograph. June 7, 2018.
h. Pfizer Canada Inc. Estring Product Monograph. November 16, 2017.
Additional Details on the CADTH Reanalyses and Additional Analyses
CADTH did not conduct any additional pharmacoeconomic analyses in the review of estradiol vaginal insert (Imvexxy).
Appendix 3: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 19: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the expected budget impact of reimbursing the estradiol vaginal insert (Imvexxy) for the treatment of postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy. The BIA was from the perspective of the public drug plan, over a 3-year time horizon, and included drug acquisition costs, pharmacy markup and dispensing fees. The sponsor submitted a base-case analysis that considered local hormone therapies, which included the other available estradiol vaginal insert (Vagifem), estrone cream (Estragyn), conjugated estrogen cream (Premarin cream) and the estradiol ring insert (Estring).
The sponsor estimated the market size via a claims-based approach, using historical provincial public drug plan claims data from IQVIA PharmaStat (Q1 2016 to Q4 2020) to forecast the number of claims and units for drugs currently reimbursed beyond Q1 2022, assuming a linear trend. The sponsor included public claims made across all indications for the comparators of interest to determine the reference scenario. In a scenario with Imvexxy entering the market, the sponsor assumed it would have a total market share of 1.0% in year 1, 2.4% in year 2 and 4.2% in year 3 in all jurisdictions except British Columbia. The sponsor assumed a market share of 0.1% in year 1, 0.2% in year 2 and 0.3% in year 3 for Imvexxy in British Columbia where Vagifem is not currently funded by the public formulary. The sponsor assumed Imvexxy would obtain its market share primarily from the displacement of Vagifem (99% of Imvexxy market share listed above from Vagifem), as the 2 drugs have a similar formulation and administration, with the remaining 1% taken from conjugated estrogen cream.
The sponsor used jurisdiction-specific unit costs for included comparators. The sponsor estimated annual drug acquisition costs by applying the drug unit price and jurisdiction-specific markup on the number of units, and jurisdiction-specific dispensing fees on the number of claims. Then, results for each jurisdiction were aggregated for the pan-Canadian budget impact estimate.
Summary of the Sponsor’s BIA Results
The sponsor estimated that funding Imvexxy for the treatment of postmenopausal moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, would result in budgetary savings of $78,400 in Year 1, $200,008 in Year 2, and $370,932 in Year 3, for total savings of $649,340 over 3-years. Results remained robust to sensitivity analyses testing alternative inputs for the anticipated uptake of estradiol tablet inserts, the number of units per claim of estradiol tablet inserts, the number of units in the entire market, and exclusion of markups and dispensing fees.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of claims-based approach to estimate market size introduces uncertainty with the anticipated budget impact of Imvexxy: The sponsor estimated market size for the indication of Imvexxy was based on claims data for the relevant comparators. CADTH confirmed with the sponsor that the claims for Vagifem, Estrone, conjugated estrogen cream and the estradiol ring correspond to public claims made across all indications. The listed products are all used in the treatment of vulvar and vaginal atrophy, and not necessarily for dyspareunia specifically. CADTH confirmed with the sponsor that approximately 90% of women with vulvar and vaginal atrophy experience dyspareunia. The clinical expert consulted by CADTH also noted that while the indication for Imvexxy is specific to dyspareunia, it would likely be prescribed more broadly, in the same population as Vagifem. Based on the sponsor’s approach, the market size may have been overestimated given the lack of indication specific claims data. However, given the proportion of patients with vulvar and vaginal atrophy who experience dyspareunia, and that the likely clinical use of estradiol vaginal inserts (Imvexxy and Vagifem) is in the same populations, this limitation is likely to have a limited impact. Decreases to the market size would diminish the anticipated cost savings with the introduction of Imvexxy.
Further, the sponsor does not convert the number of claims into the number of users; instead, the sponsor assumes unit to unit and claim to claim displacement between estradiol tablet inserts (Imvexxy and Vagifem). Given both estradiol tablet inserts (Imvexxy and Vagifem) have the same dosing regimen and 99% of the market uptake of Imvexxy is assumed to come from displacing Vagifem, this is unlikely to have had a great impact on results. However, for transparency and completeness, claims data-based models should provide an estimate of the number of active beneficiaries based on the number of claims.11
CADTH explored the impact of a reduced market size in a scenario analysis, assuming a 10% reduction in the total market size.
There is uncertainty in the anticipated uptake of Imvexxy as well as the products displaced: The sponsor assumes Imvexxy has a total market share of 1.0% in year 1, 2.4% in year 2 and 4.2% in year 3 in all jurisdiction except for British Columbia, and that 99% of the uptake of Imvexxy comes from displacing Vagifem. These estimates were deemed to be reasonable by the clinical expert consulted by CADTH for this review, but are nonetheless associated with some uncertainty. Should Imvexxy capture a greater market share or displace more cream or ring comparators (e.g., in jurisdictions where Vagifem is not currently funded), then the budget impact would likely differ.
CADTH explored the impact of uncertainty in the anticipated uptake of Imvexxy in a scenario analysis, assuming Imvexxy captures an arbitrary 90% from Vagifem and 10% from conjugated estrogen cream.
Actual price of drugs paid for by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. Actual costs paid by public drug plans are unknown.
This limitation could not be addressed by CADTH. Confidential negotiated prices for the included local hormone therapy comparators may lead to budgetary savings being limited or eliminated
CADTH Reanalyses of the BIA
CADTH did not undertake a base case reanalysis, as CADTH could not identify more appropriate assumptions to the sponsor’s base case to address uncertainty in the estimated market size and the market uptake of Imvexxy.
CADTH conducted the following scenario analyses:
Assuming a 10% reduction in total market size.
Assuming Imvexxy captures 90% of its market share from Vagifem and 10% from conjugated estrogen cream.
Results of these analyses are presented in Table 20, along with a detailed breakdown of the sponsor’s base case results. The reimbursement of Imvexxy was associated with cost savings in both scenario analyses. Savings decreased as the estimated market size and market share captured from Vagifem decreased.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Appraisal of the sponsor Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year totala |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $37,887,790 | $39,633,807 | $41,440,883 | $43,247,960 | $162,210,440 |
New drug | $37,887,790 | $39,555,407 | $41,240,875 | $42,877,027 | $161,561,100 | |
Budget impact | $0 | -$78,400 | -$200,008 | -$370,932 | -$649,340 | |
CADTH scenario analysis: 10% reduction in market size | Reference | $37,887,790 | $39,633,807 | $41,440,883 | $43,247,960 | $162,210,440 |
New drug | $37,887,790 | $39,563,247 | $41,260,876 | $42,914,121 | $161,626,034 | |
Budget impact | $0 | -$70,560 | -$180,007 | -$333,839 | -$584,406 | |
CADTH scenario analysis: 90% market share from Vagifem and 10% from Premarin cream | Reference | $37,887,790 | $39,633,807 | $41,440,883 | $43,247,960 | $162,210,440 |
New drug | $37,887,790 | $39,596,863 | $41,346,345 | $43,072,090 | $161,903,088 | |
Budget impact | $0 | -$36,944 | -$94,539 | -$175,869 | -$307,352 |
aIncludes jurisdiction-specific markups and dispensing fees
Appendix 4: Sponsor’s References
Note that this appendix has not been copy-edited.
Bachmann G, Bouchard C, Hoppe D, et al. Efficacy and safety of low-dose regimens of conjugated estrogens cream administered vaginally. Menopause. 2009;16(4):719-727. PubMed
Clinical Study Report. A Phase 3, Randomized, Double-Blind, Placebo-Controlled, Multi-Center Trial to Evaluate the Safety and Efficacy of TX-004HR in Postmenopausal Women with Moderate to Severe Symptoms of Vulvar and Vaginal Atrophy. Boac Raton, FL: TherapeuticsMD; 09 Jun 2015 2015.
Common Technical Document 2.7.1 Summary of Biopharmaceutics Studies for Joyesta (estradiol vaginal inserts). Knight Therapeutics Inc.
Constantine GD, Simon JA, Pickar JH, et al. The REJOICE trial: a phase 3 randomized, controlled trial evaluating the safety and efficacy of a novel vaginal estradiol soft-gel capsule for symptomatic vulvar and vaginal atrophy. Menopause. 2017;24(4):409-416. PubMed
FDA Medical Officer’s Review of Vagifem. 1999; https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/020908_s000_MedR.pdf.
Novo Nordisk Canada Inc. Product Monograph Vagifem (Estradiol vaginal inserts USP, 10 μg estradiol). 2017.
Nova Scotia Pharmacare. Nova Scotia Formulary 2020. [updated 2-12-2020; cited 2-12-2020]. Available from: https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf.
Ontario Ministry of Health. Ontario Drug Benefit Formulary/Comparative Drug Index 2020. [updated 29 November 2019; cited 5 February 2020]. Available from: https://www.formulary.health.gov.on.ca/formulary/.
Pfizer Canada. Premarin (conjugated estrogens vaginal cream) [product monograph]. Revised June 07, 2018. Pfizer website. https://www.pfizer.ca/sites/default/files/202005/Premarin-VC_PM_E_12-May-2020_OSIP_N.pdf. Accessed April 10, 2021.
Pfizer Canada Inc. Product Monograph Estring (17 β-Estradiol) Vaginal Ring, 2 mg. 2017.
Searchlight Pharma Inc. Product Monograph ESTRAGYN® VAGINAL CREAM (Estrone vaginal cream). 2016.
Simon J, Nachtigall L, Gut R, Lang E, Archer DF, Utian W. Effective treatment of vaginal atrophy with an ultra-low-dose estradiol vaginal tablet. Obstet Gynecol. 2008;112(5):1053-1060. PubMed
Wiegel M, Meston C, Rosen R. The female sexual function index (FSFI): cross-validation and development of clinical cutoff scores. J Sex Marital Ther. 2005;31(1):1-20. PubMed
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.