CADTH Reimbursement Review
Trientine Hydrochloride (MAR-Trientine)
Sponsor: Marcan Pharmaceuticals Inc.
Therapeutic area: Wilson disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
CLF
Canadian Liver Foundation
DPA
d-penicillamine
HRQoL
health-related quality of life
MID
minimal important difference
SAP
Special Access Program
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Trientine hydrochloride (MAR-Trientine) 250 mg capsules, oral |
Indication | For the treatment of patients with Wilson disease who are intolerant to penicillamine |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 14, 2020 |
Sponsor | Marcan Pharmaceuticals Inc. |
NOC = Notice of Compliance.
Introduction
Wilson disease is a rare heterogenous autosomal recessive disease of copper metabolism that can present with hepatic, neurologic, or psychiatric involvement (or a combination of these), or it may be asymptomatic.1,2 Most patients with Wilson disease present at between 5 years and 35 years of age. In children and younger adults, the disease presentation is mainly hepatic, as neurologic manifestations tend to occur later as copper accumulates in the body.3-6 The global prevalence of Wilson disease has been estimated to be 1 in 30,000 in most populations, with a carrier gene frequency of 1 in 90 to 150 in the general population.7,8
Clinical manifestations of Wilson disease vary widely. Liver presentations can range from asymptomatic enzyme elevations to fulminant hepatic failure and other liver sequelae.1 Neurologic symptoms include movement disorders or rigid dystonia whereas psychiatric presentations may include depression, neurotic behaviour, or intellectual deterioration.1 Kayser-Fleischer rings, which are a yellow-brown discoloration of the cornea due to copper deposition, occur in 80% of all cases of Wilson disease.4 A diagnosis is most often made by biochemical findings which, if observed in combination with low ceruloplasmin levels (the major carrier for circulating copper in the blood), Kayser-Fleischer rings, and clinical signs, are usually definitive for Wilson disease; this is then ultimately confirmed by molecular genetic testing.1,3 The mainstays of treatment are dietary copper restriction, chelation therapy with either d-penicillamine (DPA) or trientine, and zinc salts.3 Limitations associated with chelation therapy include persistence or deterioration of neurologic symptoms (which may be irreversible) and intolerance to DPA in 20% to 40% of patients.9 Zinc therapy is associated with nausea and gastritis, which may be due to the salt form used.3,10 Treatment regimens for Wilson disease are complex and burdensome for patients as they require multiple dosing regimens to be appropriately spaced over the course of each day with regard to food and concomitant medication.9 Problems with adherence are observed in almost half of all patients with Wilson disease, which is a key concern given that treatment is lifelong.9 Untreated Wilson disease is ultimately fatal.10
Trientine is an oral copper chelating agent that forms a stable copper complex that is readily excreted in urine.11 MAR-Trientine (trientine hydrochloride) is available as 250 mg oral capsules and is approved by Health Canada for the treatment of patients 5 years of age and older with Wilson disease who are intolerant to penicillamine.11 The sponsor has requested reimbursement as per the indication. Prior to its approval by Health Canada on September 14, 2020, trientine hydrochloride was available to Canadian patients through a compassionate use program and later via the Health Canada Special Access Program (SAP). Due to the approval of MAR-Trientine by Health Canada, the SAP program has been terminated.
The objective of this systematic review is to evaluate the beneficial and harmful effects of trientine hydrochloride 250 mg oral capsules for the treatment of Wilson disease in patients who are intolerant to penicillamine.
Stakeholder Perspectives
The information in this section is a summary of input provided by 1 patient group that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient submission from the Canadian Liver Foundation (CLF) was received for this review. The CLF supports education and research into all forms of liver diseases and is committed to reducing the incidence and impact on Canadians at risk or living with liver diseases. The CLF gathered information through an online survey to which 8 patients and 5 caregivers responded, although additional input was collected from 2 health care professionals.
Patients described the negative impacts of Wilson disease on their day-to-day activities. These impacts were reiterated by caregivers, especially regarding the ability to work and travel. The emotional and psychological effects of living with and managing Wilson disease results in constant stress and fear as well as psychiatric symptoms such as anxiety and depression, which negatively affect patient and caregiver quality of life. Side effects of current treatments such as fatigue, appetite loss, nausea, and pain were described as completely to somewhat intolerable. The survey respondents felt it was important to have access and a choice of treatments for Wilson disease and for choices to be based on known side effects. The following outcomes were identified as being important to patients: the reduction of short-term and long-term side effects, overall quality of life, long-term disease stability, and adherence. Two patients and 2 caregivers had experience with trientine and relayed the challenges of accessing trientine via the SAP and obtaining private insurance coverage for it. If unable to access trientine, patients may have no choice but to use DPA, despite experiencing side effects, because they require chelation therapy to live. A benefit of trientine that patients highlighted was that it does not require refrigeration, thus making it more portable.
The responses from the 2 health care professionals firmly advocated for better access to medications for their patients with Wilson disease and described the difficulty in accessing trientine for their patients. Moreover, without reimbursement, trientine remains out of reach for many patients with Wilson disease, which is unacceptable in the views of the health care professionals, because these patients require effective and safe chelation therapy to live.
Clinician Input
Two clinical specialists with expertise in the diagnosis and management of Wilson disease in adult and pediatric patients, respectively, contributed to this review. The clinicians advised that not all patients will respond to, or tolerate, DPA or zinc. Further, Canadian patients who require chelation and cannot take DPA due to toxicity or intolerance (estimated to be 20% to 40% of patients) currently have no available chelation treatment options as, in the experts’ experience, zinc is inadequate in about 30% of patients and is relatively poorly tolerated. Available treatments have limited effect on acute liver failure, and none can reverse the neurologic or psychiatric manifestations of Wilson disease. A specific unmet need identified by the pediatric clinical expert was the lack of specific drug formulations (e.g., liquid formulations) to meet pediatric needs.
The current use of trientine after DPA treatment is mainly due to access issues. In the clinical experts’ opinion, if trientine were available as a first-line option, it would be preferred by many providers due to twice daily dosing, few adverse events (AEs), good tolerability, and solid efficacy. The clinical experts felt it was inappropriate for trientine to be limited only to patients who do not tolerate or fail DPA or zinc; however, if DPA and/or zinc must be tried before access to trientine is granted, intolerance or lack of efficacy should be based on subjective inability to tolerate the medication (AEs), poor adherence, and/or lack of efficacy based on symptom progression and/or inadequate de-coppering measured by non–ceruloplasmin-bound copper or 24-hour urinary copper excretion. Repeated trials of DPA or zinc should not be required before granting approval for trientine as toxicity with DPA may be worse upon re-challenge and some AEs associated with DPA are irreversible or slow to reverse and may be difficult, if not impossible, to predict. Significant delays in initiating therapy in patients with progressive disease can lead to irreversible impairment. This is particularly true with neurologic symptoms associated with Wilson disease.
The clinical experts advised that any patient with Wilson disease is expected to respond to trientine in terms of reducing overall body copper burden. Both adult and pediatric patients with hepatic-prominent Wilson disease are likely to have their hepatic symptoms respond to chelation therapy, including trientine. In contrast, patients with neurologic disease may have their neurologic symptoms worsen with the initiation of any chelator treatment, due to cerebral mobilization of copper that is too rapid.12 Some evidence and anecdotal reports suggest that neurologic worsening occurs more frequently with DPA than trientine, although this has not been rigorously evaluated. The experts felt that patients with advanced and progressive neurologic and/or psychiatric disease would be considered least suitable for trientine treatment, although trientine may still stabilize the disease and prevent further progression. Patients with acute liver failure often require an immediate liver transplant, so trientine is unlikely to benefit those presenting with an acute Wilsonian crisis. Patients without symptoms but with a confirmed diagnosis of Wilson disease should be treated; however, if the copper burden is not excessive, initial treatment with zinc is appropriate rather than chelation therapy.
According to the clinical experts, response to treatment is usually assessed by ceruloplasmin-bound copper, 24-hour urinary copper collection, and liver enzymes and function tests in both adult and pediatric patients. It is also important to assess neurologic and hepatic improvement following treatment. While some assessments are subjective, they can usually be supported by objective assessments. Treatment response should be subjectively evaluated monthly at treatment initiation (i.e., patient perspective on symptoms) and then every 6 months to 12 months once stable. Objective assessments such as neurologic assessment with or without brain MRI, laboratory improvement (non–ceruloplasmin-bound copper, 24-hour urinary copper excretion, or liver enzymes and function) should be evaluated at least annually but may require more frequent testing, especially at treatment initiation. In pediatric patients, response to treatment should be assessed more frequently (e.g., every 3 months to 6 months) due to the need for more frequent reassessment of dosage and treatment efficacy because of weight-based dosing.
The clinical experts reiterated that treatment of Wilson disease is lifelong and in all cases, if 1 chelator is stopped, an alternative treatment must be started immediately as patients cannot be left untreated. The main reason for treatment discontinuation would be inadequacy of treatment due to either lack of efficacy or tolerability issues. The experts agreed that while a specialist is required to diagnose Wilson disease and should be involved in the care of patients, they do not necessarily have to be the only prescriber of trientine. Once a diagnosis is established, patients can be followed locally because access to a specialty clinic or specialist with experience treating Wilson disease could be problematic for patients.
The clinical experts described the current situation with trientine in Canada as a risk to achieving desired patient outcomes. The approval of MAR-Trientine by Health Canada led to immediate suspension of the SAP without a plan in place to provide trientine to Canadian patients who were on treatment with imported product. This led to almost immediate loss of access to trientine, interruptions in treatment, and great anxiety for patients and providers. The clinical experts advocated for an improved process for bridging patients who are receiving a medication through the SAP to be able to retain access to the medication throughout the Health Canada approval and drug reimbursement review processes.
Drug Program Input
Key questions from the drug plans pertained to the reimbursement of trientine as a first-line treatment before DPA, its use in children younger than 5 years of age (as MAR-Trientine is approved only for patients ≥ 5 years), and restrictions on prescribers. The clinical experts advised that it is reasonable for trientine to be used before DPA in patients with a confirmed diagnosis of Wilson disease and that there is no compelling reason that trientine could not be used in children younger than 5 years, although the main limitation is the lack of a pediatric dosage form. While the clinical experts felt that a specialist is required to diagnose Wilson disease and should be involved in the care of patients, prescribing for adult patients should not be limited to only specialists. Due to the rarity of Wilson disease, there are few specialty clinics and specialists with experience treating patients with Wilson disease available. The experts also cautioned that it may be preferable not to place limitations on prescribers as it may lead to the undertreatment of adult patients who are not able to access an experienced prescriber. Regarding the care of pediatric patients, the pediatric clinical expert advised that pediatric patients in Canada are managed only by pediatric specialists and subspecialists and not by other care providers.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Two pivotal trials submitted by the sponsor were included in the systematic review. No additional trials from the literature search met the inclusion criteria for the systematic review and no indirect comparisons or other relevant evidence were identified. The first included study (Weiss et al. [2013]13) was a retrospective cohort analysis that evaluated the efficacy and safety of trientine compared to DPA in 405 patients with Wilson disease, based on hepatic and neurologic outcomes and treatment discontinuations due to AEs. The analysis included 380 patients who were examined at tertiary care centres in Germany (Heidelberg, Dresden, and Düsseldorf) and Austria (Vienna, Graz, and Linz) and 25 additional patients identified from the EuroWilson registry who had received trientine monotherapy. No patient inclusion criteria were stated and there was no information on the specific time frame of the study or the calendar years during which the patients were treated. It appears that efficacy outcomes were based on the latest available follow-up evaluation within a 6-month to 48-month period. Data on discontinuations and discontinuations due to AEs were collected over a median 13.3-year period, although no range of time was reported. The results of the analysis were reported by number of chelator treatments (i.e., 326 DPA treatments and 141 trientine treatments) rather than by the number of patients, and the researchers categorized the DPA and trientine treatments as first-line or second-line, but how this was determined is unknown. The second study (Study 16-VIN-0315) was an open-label, randomized, 2-period, 2-sequence, 2-treatment, crossover, single-dose, fasting bioequivalence study of MAR-Trientine 250 mg capsules compared to Syprine 250 mg capsules in 36 healthy adult volunteers. The objective of this study was to compare the rate and extent of absorption of trientine from the 2 formulations to determine if they were bioequivalent. As the purpose of Study 16-VIN-0315 was to assess bioequivalence in healthy volunteers and not the efficacy and safety of trientine in patients with Wilson disease, this study was not reviewed in detail in this report.
According to the clinical experts on the review team, the baseline characteristics of the patients in the Weiss et al. (2013) study are reasonably similar to those of Canadian patients who would be candidates for trientine, with the possible exception of pediatric patients (< 18 years of age). The median age of included patients at the time of diagnosis of Wilson disease (the only age parameter reported in the study) was 17 years to 19 years. Although patients younger than 18 years were included, no details on the number or the age of pediatric patients was provided. At initial presentation, about half of patients (207 [51.1%] patients) had only hepatic symptoms, 92 (22.7%) had only neurologic symptoms, 52 (12.8%) had mixed presentation (hepatic and neurologic symptoms), and 54 (13.3%) were asymptomatic, a similar distribution as expected in Canadian clinical practice.
Efficacy Results
Hepatic Impairment
In the Weiss et al. (2013) study, hepatic improvement scores after first-line treatment were comparable for all patients (25 out of 38 [65.8%] trientine treatments versus 185 out of 295 [62.7%] DPA treatments) and for symptomatic patients (25 out of 27 [92.6%] trientine treatments versus 185 out of 204 [90.7%] DPA treatments); these scores were not statistically significantly different. Following second-line treatment, hepatic improvement scores were generally lower than with first-line treatment for all patients (31 out of 103 [30.1%] trientine treatments and 12 out of 31 [38.7%] DPA treatments) and for symptomatic patients (31 out of 45 [68.9%] trientine treatments and 12 out of 16 [75.0%] DPA treatments). There were also no statistically significant differences between treatments. For symptomatic patients, stable hepatic disease categorized as unchanged hepatic symptoms was observed in 7.4% of first-line treatments for both groups (i.e., 2 out of 27 trientine treatments and 15 out of 204 DPA treatments). Stable hepatic disease after second-line therapy was reported in 10 out of 24 (22.2%) trientine treatments and 4 out of 16 (25%) DPA treatments. No statistical comparisons were reported for the number of treatments associated with stable or unchanged hepatic symptoms.
There were no first-line trientine treatments associated with hepatic worsening (i.e., defined as a decline in liver function or progression of chronic liver disease) compared to first-line DPA treatments for all patients (0 out of 38 [0%] trientine treatments versus 4 out of 295 [1.4%] DPA treatments) and for symptomatic patients (0 out of 27 [0%] trientine treatments versus 4 out of 204 [2.0%] DPA treatments). While second-line trientine treatment was associated with hepatic worsening, there were no second-line DPA treatments associated with hepatic worsening for all patients (4 out of 103 [3.9%] trientine treatments versus 0 out of 31 [0%] DPA treatments) and for symptomatic patients (4 out of 45 [8.9%] trientine treatments versus 0 out of 16 [0%] DPA treatments). The differences between trientine treatments and DPA treatments for hepatic worsening after either first-line or second-line treatments were not statistically significantly different. Overall, there were 12 treatments with an outcome of a liver transplant (i.e., 3 [2.1%] trientine treatments and 9 [2.7%] DPA treatments).
Neurologic Impairment
In the Weiss et al. (2013) study, neurologic improvement scores for first-line treatment were comparable between trientine treatments (11 out of 38 [28.9%]) and DPA treatments (77 out of 295 [26.1%]) for all patients but were numerically higher for DPA treatments (77 out of 114 [67.5%]) versus trientine treatments (11 out of 20 [55.0%]) in symptomatic patients, although the differences were not statistically significant. Following second-line therapy for all patients, neurologic improvement rates were comparable to those after first-line therapy for trientine treatments (26 out of 103 [25.2%]) but were numerically lower for DPA treatments (3 out of 31 [9.7%]). For symptomatic patients, neurologic improvement with second-line therapy after trientine treatments (26 out of 51 [51.0%]) was numerically higher than after DPA treatments (3 out of 13 [23.1%]). Nonetheless, all comparisons between trientine treatments and DPA treatments for all patients and for symptomatic patients for second-line therapy were not statistically significantly different. For symptomatic patients, stable neurologic disease, which was categorized as unchanged neurologic symptoms, was observed in 5 out of 20 (25.0%) trientine treatments and 31 out of 114 (27.2%) DPA treatments after first-line therapy and 17 out of 51 (33.3%) trientine treatments and 9 out of 13 (69.2%) DPA treatments after second-line therapy. No statistical comparisons were reported for stable or unchanged neurologic symptoms.
Rates of neurologic worsening after first-line therapy were statistically significantly higher for trientine treatments compared to DPA treatments for all patients (4 out of 38 [10.5%] trientine treatments versus 6 out of 295 [2.0%] DPA treatments) and for symptomatic patients (4 out of 20 [20.0%] trientine treatments and 6 out of 114 [5.3%] DPA treatments). For second-line therapy, rates of neurologic worsening were numerically higher with trientine treatments compared to DPA treatments for all patients (8 out of 103 [7.8%] and 1 out of 31 [3.4%], respectively) and symptomatic patients (8 out of 51 [15.7%] and 1 out of 13 [7.3%], respectively), although the differences were not statistically significant.
Harms Results
In the Weiss et al. (2013) study, the only harms outcomes reported were the proportions of chelator treatments with AEs that led to treatment discontinuation. Treatment discontinuations due to AEs were more common with DPA (94 out of 326 [28.8%] treatments) compared with trientine (10 out of 141 [7.1%] treatments). The difference between DPA treatments and trientine treatments was statistically significant (P = 0.039), as reported in the publication Clinical Gastroenterology and Hepatology.13 The frequency of AEs was higher with DPA treatments and the most common AEs that led to treatment discontinuation (≥ 5% frequency in either group) were arthralgia (29 out of 326 [8.9%] DPA treatments versus 4 out of 141 [2.8%] trientine treatments), an increase in antinuclear antibodies (22 out of 326 [6.7%] DPA treatments versus 1 out of 141 [0.7%] trientine treatments), and albuminuria/proteinuria (20 [6.1%] DPA treatments versus not reported for trientine treatments). Rates of discontinuations for any reason were not statistically significantly different between the chelator treatments (P = 0.360), as reported in the publication.13
Critical Appraisal
Key limitations of the Weiss et al. (2013) study pertaining to internal validity are the retrospective design, which is limited by lack of randomization and the non-prospective collection of efficacy and harms outcomes, and the unknown time frame of the study. The analysis was also not blinded, which may have introduced bias into the categorization of hepatic and neurologic outcomes and the identification of symptomatic patients, as all were subjectively assessed by the researchers. The reporting of results by number of chelator monotherapy treatments rather than by number of patients complicates the interpretation of baseline characteristics and efficacy and harms outcomes, as an individual patient may have been counted more than once in the results. This leads to double data counting, which compromises the validity of the dataset. For example, if an individual patient displays a specific characteristic such as hepatic presentation, this will result in more treatments being characterized as having hepatic presentation than if patients were randomly selected and counted only once in the dataset. There were no clear definitions or validation of the efficacy outcomes in terms of reliability, validity, responsiveness, or minimal important differences (MIDs), which makes interpretation difficult.
Key limitations relating to external validity in the Weiss et al. (2013) study are the lack of data for Canadian patients, the lack of evidence on combination use of trientine in with zinc, which is common in clinical practice, and the lack of evidence in pediatric patients. The diagnosis and treatment of Wilson disease can be challenging in children, as children may not display the same clinical and laboratory hallmarks of the disease as adults.14 No information on the dose and administration schedules of trientine or DPA used in the study were reported, so it is not known if the dosage regimens used in the study are in alignment with the Health Canada–approved dosages for trientine and DPA. There were also no data available for most efficacy outcomes identified in the review protocol, including outcomes of interest to patients, such as health-related quality of life (HRQoL) and adherence.
Conclusions
A retrospective cohort analysis of mainly adult patients with Wilson disease demonstrated that trientine has an efficacy comparable with DPA on improving hepatic and neurologic outcomes when used as first-line therapy, and as second-line therapy in patients who failed or were intolerant to DPA. First-line treatment with trientine was also associated with statistically significantly higher rates of neurologic worsening than DPA, but not when used as second-line treatment. More DPA treatments were discontinued due to AEs than trientine treatments, which was statistically significant. Due to the low quality of this study, there is considerable uncertainty in the relative estimates of efficacy and harms between trientine and DPA. Despite the limitations, this study comprises the largest body of evidence to date for use of trientine in Wilson disease. Although the evidence is very limited, this must be placed in the context of the long market history of trientine worldwide and experience gained in Canadian patients who received trientine via the SAP. The mechanism of action of trientine also represents a rational approach for treatment of a disease caused by excess copper accumulation. Despite the many limitations associated with the evidence, Canadian patients with Wilson disease who fail or cannot tolerate DPA currently have no alternative chelator option other than trientine, and left untreated, Wilson disease is associated with high morbidity and mortality.
Introduction
Disease Background
Wilson disease (hepatolenticular degeneration) is a rare heterogenous inherited disease of copper metabolism that can present with hepatic, neurologic, or psychiatric involvement (or a combination of these), or it may be asymptomatic.1,2 It is an autosomal recessive disorder associated with a mutation of the ATP7B gene, which encodes a metal-transporting P-type adenosine triphosphatase expressed primarily in hepatocytes that is involved in copper transport.3 Reduced activity or absence of the ATP7B protein causes impaired hepatocellular excretion of copper into bile, which in turn leads to copper accumulation and liver injury, and eventually to the accumulation of copper elsewhere in the body, such as the brain, kidneys, and cornea.3 An additional consequence of the loss of functional ATP7B protein is the inability to incorporate copper into ceruloplasmin, the major carrier protein for circulating copper in the blood. It accounts for 90% of the circulating copper in healthy individuals.3
Most patients with Wilson disease present between 5 years and 35 years of age, although patients are increasingly diagnosed in childhood or adolescence.3,5,6,10 In children and younger adults, Wilson disease most often presents with liver disease, as neurologic manifestations tend to occur later due to the accumulation of copper in other organs after the liver has been saturated.3-6 The clinical manifestations of Wilson disease vary widely. Liver presentations can range from asymptomatic enzyme elevations to fulminant hepatic failure (i.e., with severe coagulopathy, encephalopathy, acute Coombs-negative hemolysis, and rapidly progressive renal failure).1 Other liver sequelae can include recurrent jaundice, acute hepatitis-like illness, autoimmune hepatitis, chronic liver disease, fatty liver, and hemolytic anemia due to the destruction of erythrocytes by the high serum concentration of non–ceruloplasmin-bound copper.1 Neurologic symptoms include movement disorders (e.g., tremors, poor coordination, loss of fine motor control, chorea) or rigid dystonia (e.g., mask-like facies, rigidity, gait disturbance) whereas psychiatric presentations comprise depression, neurotic behaviour, personality disorder, affective changes, and intellectual deterioration.1 Kayser-Fleischer rings, which are a yellow-brown discoloration of the Descemet membrane in the cornea due to copper deposition, occurs in 98% of patients with neurologic disease and in 80% of all cases of Wilson disease.4
The global prevalence of Wilson disease has been estimated to be 1 in 30,000 in most populations, with a carrier gene frequency of 1 in 90 to 150 in the general population.7,8 Each sibling of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.1 First-degree relatives of a newly diagnosed patient must be screened for Wilson disease.1,3 Lower prevalence rates for Wilson disease have been reported in North America than in other parts of the world.15 Although Canadian-specific incidence and prevalence estimates are not available, a retrospective chart review of 48 ambulatory patients with Wilson disease from Toronto Western Hospital reported the following patient characteristics: median age at diagnosis was 17 years (range = 6 years to 63 years), 31.2% of patients presented with neurologic symptoms, 27.0% presented with hepatic symptoms, and 12.5% presented with mixed presentation. The remaining 29.2% of patients were asymptomatic, 50% of whom were diagnosed through family screening.16
Untreated Wilson disease is ultimately fatal, with most patients succumbing to liver disease and a minority from complications from progressive neurologic disease.10 Mortality has not been assessed prospectively in Wilson disease; however, in general, survival prognosis depends on the severity of liver and neurologic disease and on adherence to therapy (i.e., liver function can become normal after 1 year to 2 years of treatment in most patients who have no or compensated cirrhosis at presentation).10 Acute liver failure often requires a liver transplant. In patients with Wilson disease who undergo an orthotopic liver transplant, early survival may be slightly reduced, but is considered normal for a transplant population.10,17
A diagnosis of Wilson disease is most often established by biochemical findings which, if observed in combination with low ceruloplasmin levels, the presence of Kayser-Fleischer rings, and clinical signs, are usually definitive for Wilson disease.1,3 A diagnosis is confirmed by molecular genetic testing (i.e., single-gene testing, multigene panel, or more comprehensive genomic testing if required).1,3 Biochemical findings associated with Wilson disease include low serum ceruloplasmin concentration, subnormal serum concentration of copper and of non-ceruloplasmin-bound copper, high urinary copper, and increased hepatic copper concentration.3 For many patients, a combination of tests reflecting disturbed copper metabolism may be needed to make a diagnosis and a diagnostic scoring system (Leipzig score) based on available tests has been developed.18 The Leipzig scoring system considers typical clinical symptoms and signs of Wilson disease (i.e., presence or absence of Kayser-Fleischer rings and Coombs-negative hemolytic anemia, severity of neurologic symptoms, and serum ceruloplasmin levels) as well as the results of other tests (i.e., copper levels in the liver and urine, mutation analysis).18,19 Each factor is assigned a numerical score, with total score ranging from 0 to 22.18 A total Leipzig score of 4 or more establishes a diagnosis of Wilson disease, a score of 3 indicates that diagnosis is possible but more tests are needed, and a score of 2 or less implies diagnosis is very unlikely.10,18 Another scoring system (the Ferenci score) was proposed in 2001 to facilitate the diagnosis of Wilson disease in pediatric patients. It takes into consideration the presence of Kayser-Fleischer rings, Coombs-negative hemolytic anemia, neuropsychiatric symptoms, urinary and liver copper, rhodanine-positive hepatocytes (only if quantitative copper measurement is unavailable), and the detection of disease-causing mutations. A Ferenci score of 0 to 1 is unlikely for Wilson disease whereas a score of 2 to 3 is probable, and a score of 4 or more is highly likely for the disease.6
Standards of Therapy
The treatment of Wilson disease is dependent on the presence of clinically relevant disease or laboratory or histological evidence of aggressive inflammatory hepatic or neurologic injury, and whether the patient is identified before the onset of clinical symptoms.3 Current treatment options for Wilson disease such as chelation therapy or zinc salts were introduced into clinical practice more than 60 years ago, with the goal of achieving a negative copper balance in the body.3 Pharmacologic therapy for Wilson disease is lifelong and although a liver transplant — which corrects the underlying hepatic defects — is usually curative, it is generally reserved for patients with acute liver failure or decompensated liver cirrhosis.9
The mainstays of treatment of Wilson disease are dietary copper restriction, chelation therapy, and zinc salts.3 The first oral chelating agent for Wilson disease was DPA, which was introduced in 1956 and for which there is the most treatment experience worldwide.3 The major action of DPA is to chelate copper via its free sulfhydryl group and to promote urinary excretion of the chelated copper.3 DPA may also act by inducing metallothionein, an endogenous chelator of metals.3 The usual maintenance adult dose of DPA is 750 mg per day to 1,500 mg per day, whereas in children, DPA dosing is by body weight (i.e., 20 mg per kg per day), given in divided doses 2 to 3 times a day 1 hour before meals or 2 hours after meals or other medication as detailed in Table 2. As DPA also interferes with pyridoxine, supplemental pyridoxine (25 mg per day to 50 mg per day) should also be provided.3 Trientine, 1 of a family of chelators with a polyamine-like structure chemically distinct from DPA, was first introduced in 1969 as an alternative to DPA.3 Similar to DPA, trientine promotes the excretion of chelated copper by the kidneys.3 Trientine has typically been used in patients who are intolerant to DPA or have clinical features indicating potential intolerance to DPA (e.g., renal disease, congestive splenomegaly causing severe thrombocytopenia, autoimmune tendency).3 Typical doses range from 750 mg per day to 1,500 mg per day in adults and 20 mg per kg per day in children spaced appropriately from meals or other medication as with DPA. Zinc is also used to treat Wilson disease and was first introduced in the early 1960s.3 Zinc acts by interfering with the absorption of copper from the gastrointestinal tract by inducing enterocyte metallothionein, which has greater affinity for copper than zinc and so once bound, copper is not absorbed but eliminated in the fecal contents as enterocytes are shed in normal turnover.3 Zinc is usually reserved for maintenance therapy post de-coppering by chelation, although it may also be used in combination with chelation therapy.3 Zinc has also been used as a first-line monotherapy option for patients who are asymptomatic or pre-symptomatic.9 The usual dose is 150 mg per day elemental zinc in 3 divided doses for larger children and adults, 75 mg per day in 3 divided doses for children 6 years to 16 years of age weighing less than 50 kg, and 50 mg per day in 2 divided doses for children younger than 6 years, all taken 30 minutes before meals.9 Various zinc salts are available and while the actual salt used does not appear to affect efficacy, it may influence tolerability.9
There are various limitations associated with currently available treatments for Wilson disease.9 Although chelation therapy has been used for decades and is particularly beneficial in patients with hepatic symptoms, neurologic symptoms persist in about half of treated patients.9 Moreover, approximately 10% of patients deteriorate neurologically during treatment, and this is often irreversible.9 An estimated 20% to 40% of patients with Wilson disease cannot be maintained on DPA due to intolerance, as it is associated with many AEs such as immediate hypersensitivity reactions, rash, nephrotic syndrome, myasthenia-like or lupus-like syndromes, and bone marrow toxicity, as well as worsening neurologic symptoms that occur in approximately 10% to 50% of patients during the initial phase of treatment.3,9 In general, AEs associated with DPA resolve once trientine is substituted and do not recur during prolonged treatment.3,10 Although neurologic worsening with trientine has been reported, it is less common than with DPA.3,10 Treatment regimens for Wilson disease are complex and burdensome for patients as they require multiple dosing regimens to be appropriately spaced over the course of each day with regard to food and concomitant medication.9 Problems with adherence are observed in almost half of all patients with Wilson disease, which is a key concern given that treatment is lifelong.9
In describing the current treatment paradigm for Wilson disease, the clinical experts on the review team indicated that in Canada, chelation therapy is used as first-line treatment, with or without zinc. Zinc may be added when there is either concern of very excessive copper overload to accelerate copper reductions, or to minimize the dose of chelator required if there is intolerance or concern about worsening of neurologic symptoms. According to the clinical experts, most providers will initiate chelation therapy with DPA due historically to the limited access to trientine via Health Canada’s SAP. When trientine has been accessed through the SAP, the clinicians advised the brands used were Syprine and MAR-Trientine. Zinc is primarily used for maintenance therapy after successful chelation (typically after 1 year). It may also be used as initial monotherapy in people diagnosed through sibling screening who often have limited copper overload, or as a monotherapy in patients with a primary neurologic presentation in whom chelators may worsen disease, sometimes irreversibly. Both chelation and zinc must be given chronically to avoid disease progression and while both may improve symptoms over time, some symptoms — particularly neurologic symptoms — may show limited improvement even with effective therapy. The clinical experts described the main goals of treatment of Wilson disease to be to prevent death, organ failure (liver and brain), disease progression, disability (neurologic, psychiatric), and liver transplants; to reduce symptoms (hepatic, neurologic) to improve quality of life; to minimize AEs; and to maintain independence and employment, thereby reducing caregiver burden.
Drug
Trientine is an oral chelating agent with a polyamine-like structure that chelates copper by forming a stable complex with the 4 constituent nitrogens in a planar ring that is readily excreted in the urine.11 MAR-Trientine (trientine hydrochloride) is available as 250 mg oral capsules and is indicated for the treatment of patients with Wilson disease who are intolerant to penicillamine.11 MAR-Trientine is indicated only for the treatment of patients 5 years of age and older; based on the data submitted and reviewed by Health Canada, the safety and efficacy in patients younger than 5 years of age has not been established.11 The sponsor has requested reimbursement as per the Health Canada–approved indication. MAR-Trientine has not been previously reviewed by CADTH.
Table 2: Key Characteristics of Therapies for Wilson Disease
Characteristic | Trientine | DPA | Zinc salts |
|---|---|---|---|
Mechanism of action | Copper chelating agent | Copper and lead chelating agent | Interferes with intestinal absorption of copper and induces enterocyte metallothionein |
Indicationa | For the treatment of patients with Wilson disease who are intolerant to penicillamine Indicated for patients ≥ 5 years of age only | For the treatment of Wilson disease, chronic lead poisoning, cystinuria, and patients with severe active rheumatoid arthritis who have failed to respond to an adequate trial of conventional therapy | NA |
Route of administration | Oral (available as 250 mg capsules) | Oral (available as 250 mg capsules) | Oral (different zinc salts are used: sulphate, acetate, or gluconate) |
Recommended dose | 500 mg/day to 2,000 mg/day on an empty stomach at least 1 hour before meals or 2 hours after meals and at least 1 hour apart from any other drug, food, or milk given in divided doses, 2 times a day to 4 times a day | 750 mg/day to 1,500 mg/day on an empty stomach at least 1 hour before meals or 2 hours after meals and at least 1 hour apart from any other drug, food, or milk given in divided doses | 150 mg elemental zinc/day and for children < 50 kg body weight 75 mg/day administered as 3 divided doses, 30 minutes before meals |
Serious adverse effects or safety issues | Worsening of neurologic or neurocognitive functioning that may be irreversible in patients with pre-existing neurologic or neuro-psychiatric impairment, iron deficiency anemia | Hypersensitivity and immune reactions, serious hematological and renal adverse reactions, hypogeusia | Gastritis (may be dependent on the zinc salt used), immunosuppressant effects, elevations in serum lipase or amylase |
Other | Health Canada has not authorized an indication for children < 5 years as safety and efficacy have not been established in this population. | No age restriction. Due to interference with pyridoxine action, supplemental pyridoxine (25 mg/day to 50 mg/day) should be provided. | OTC therapy: If used in combination with chelators, dosing times must be spaced accordingly, which may be problematic for compliance with 3 times daily dosing. |
DPA = d-penicillamine; EASL = European Association for the Study of the Liver; NA = not applicable; OTC = over-the-counter.
aHealth Canada–approved indication.
Source: MAR-Trientine product monograph,11 Cuprimine product monograph,20 Weiss et al. (2011),21 and “EASL Clinical Practice Guidelines: Wilson’s disease” in the Journal of Hepatology.10
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Group(s) and Information Gathered
CADTH received 1 patient group submission from the CLF for this review. The CLF is dedicated to supporting education and research of all forms of liver diseases and has invested funding toward finding and understanding their causes, preventive measures, and treatments. The group is committed to reducing the incidence and impact on Canadians at risk or living with liver diseases, and reaches millions of people across Canada through educational programs and patient support programs, as well as awareness, fundraising, and outreach efforts. The CLF gathered information for this submission through an online questionnaire that was advertised on the foundation’s website via its social media platforms and to the group’s Canadian patient, caregiver, and health care professional contacts. Overall, 8 patients and 5 caregivers responded to the survey. Additional input was collected from 2 health care professionals aside from the direct call for patient input.
Disease Experience
Patients with Wilson disease felt that the day-to-day activities that were most impacted by the condition were their ability to exercise, work, travel, complete household activities, socialize, and fulfill family obligations. Caregivers reported that their ability to work and travel had been affected as well. One patient, who was also a student, noted that before chelation therapy, they had developed hand tremors that made writing difficult and caused them to withdraw socially. Another patient respondent even changed the copper pipes in their house to reduce the impact that they had on the copper levels in the tap water. Other patients described their experiences in the following quotes:
“It has caused me to have stage 4 liver cirrhosis and some neurological symptoms. My life has been drastically altered since my diagnosis.”
“Major depression, blood clotting impaired, skin rashes, etc. The depression I made it through, but that was very hard on everyone around me, and it almost cost me a job.”
Experiences With Currently Available Treatments
The CLF submission identified emotional and psychological effects associated with living with and managing the illness, in addition to currently available treatments. In particular, those living with Wilson disease expressed feelings of constant stress, fear, psychiatric symptoms, and cases of bipolar disorder. Psychiatric symptoms such as anxiety and depression were described as negatively affecting patient and caregiver quality of life and could “undermine the compliance needed to achieve disease regression.”
Respondents listed the following side effects to current treatments as being completely to somewhat intolerable: fatigue, appetite loss, nausea, and pain. Fever, dizziness, forgetfulness, and stomach irritation were described as being somewhat tolerable to very tolerable. Other symptoms that patients experienced with past treatments include lethargy, abnormal skin tightness, tingling hands, peripheral neuropathy, decreased platelets, constant muscle tension, and splenomegaly. Furthermore, respondents noted that these side effects were significant enough to lower their quality of life and impact their daily living. One caregiver described the issues with current treatment as follows:
Shelf life of her medication is only 10 days, and we have to order in advance as it has to made [sic] especially for us and it takes a few business days to get the order in. We risk running out before the new order comes in and we can’t go anywhere for long periods of time as we can’t bring extra refills with us.
Improved Outcomes
Respondents felt that it is very important both to be able to access and have choice among treatments for Wilson disease and for patients and health care providers to make treatment choices based on known side effects. The survey results also emphasized the importance of treatments being able to reduce short-term and long-term side effects, improve overall quality of life, and allow for long-term stability. Respondents were interested in new treatments that would allow patients to take less medication or take it less frequently and a caregiver suggested that a single, daily pill would be an improvement. Patients, caregivers, and health care professionals experience barriers and limitations in accessing treatment for Wilson disease. In this context, a patient stated that options should be readily available to Canadians “without all of the red tape that is currently experienced. It’s a lifesaving medication and we should not have to jump through hoops in order to receive it in Canada.” Patients were aware of trials for new drugs taking place in other countries and were interested in seeing “treatment options be more available in Canada, along with clinical trials. And affordability and coverage with existing govt health coverage.”
Experience With Drug Under Review
Four individuals — 2 patients and 2 caregivers — indicated having experience with trientine hydrochloride (MAR-Trientine) and all had gained access through private insurance, albeit after various challenges throughout the process. Respondents reported that trientine hydrochloride was either effective or very effective at managing Wilson disease and the side effects they experienced were mainly stomach irritation, fatigue, and minor pain.
Challenges to gaining access to trientine hydrochloride were described as obstacles with insurance companies, pharmacy channel gaps, issues with ongoing prescriptions, and medication contraindications. Patients who had previously gained access to the drug through the SAP indicated that obtaining insurance coverage had been a challenge, but generally felt that the process has since improved. A common theme among patients’ experiences was having to wait months for approval, encountering challenges with insurance coverage, being denied, and having to pay out-of-pocket for the medication, which could mean that individuals go untreated if costs are prohibitive. A caregiver made the following remarks regarding the cost of the medication: “…some folks have financial hardship and they have to forgo treatment altogether. The price of trientine is ridiculous…. No one should have to decide between paying their rent/groceries and taking a lifesaving medication.” If unable to access new medications, patients may have no choice in their treatment options or the associated side effects. One patient described being denied entry to a trientine hydrochloride SAP, and after discussing options with their hepatologist, stated that “…my only treatment option is solely penicillamine so there is nothing that can be done about my side effects because I am required to be on this medication for life in order to live.” One benefit that a couple of patients highlighted was that trientine hydrochloride doesn’t require refrigeration, making it more convenient for storage and everyday use, and allowing patients and caregivers to travel more easily with the medication. Both patients and caregivers expressed confidence that the drug would improve liver health, prevent further liver deterioration, and improve their quality of life.
Additional Information
Two health care providers contributed to the patient input submission and firmly advocated for better access to medications for their patients with Wilson disease. One stated:
Access to trientine for my Wilson disease patients has been extremely difficult. I applied for reimbursement for my patient but it was turned down. I tried again and have been waiting months for a response. One of my patients has developed cirrhosis and we are now planning for a liver transplant. This is not acceptable. Wilson disease patients NEED quick and affordable access to treatment – their lives depend on it.
Another highlighted the fact that trientine hydrochloride has gained approval from Health Canada and, thus, it “should essentially be available to Canadian patients, but without reimbursement, this treatment remains out of reach to Wilson disease patients. This systemic problem must be addressed in order to save lives.”
Clinician Input
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of Wilson disease in adult and pediatric patients, respectively.
Unmet Needs
Not all patients will respond to, or tolerate, DPA or zinc. Canadian patients who require chelation and cannot take DPA due to toxicity or intolerance (at least 30% of patients) currently have no available chelation treatment options. Moreover, zinc is inadequate in about 30% of patients and is relatively poorly tolerated due to significant nausea and the need for dosing 3 times daily. The currently available treatments have limited effect on acute liver failure (i.e., fulminant Wilson disease presentation) and none of the available treatments can reverse the neurologic or psychiatric manifestations of Wilson disease. A specific unmet need identified by the pediatric clinical expert was the lack of specific drug formulations (e.g., liquid formulations) to meet pediatric needs.
Place in Therapy
Due to the rarity of Wilson disease, it is unlikely that randomized data comparing trientine to DPA will ever be available. The current use of trientine after DPA is mainly due to access issues. In the expert’s opinion, if trientine were available as a first-line option, it would be preferred by many providers due to twice daily dosing, few AEs, good tolerability, and solid efficacy. Other considerations that can affect place in therapy are the relative costs of available therapies and the lack of specific drug formulations to meet pediatric needs.
If DPA and/or zinc must be tried before access to trientine is granted, the clinical experts advised that intolerance or lack of efficacy should be based on subjective inability to tolerate the medication (AEs), poor adherence, and/or lack of efficacy based on symptom progression and/or inadequate de-coppering measured by non–ceruloplasmin-bound copper or 24-hour urinary copper excretion. Repeated trials of DPA or zinc should not be required as toxicity with DPA may be worse upon re-challenge. Some AEs associated with DPA are irreversible or slow to reverse and may be difficult, if not impossible, to predict. Significant delays in initiating therapy in patients with progressive disease can lead to irreversible impairment and this is particularly true with neurologic Wilson disease.
Patient Population
The diagnosis of Wilson disease can be challenging due to the lack of a pathognomonic test. Multiple tests (e.g., biochemical blood tests, urine copper measurement, ophthalmologic examination, liver biopsy, quantitative liver copper content) are required and can be included in the Leipzig scoring system in adults or the Ferenci score for children to determine the likelihood of Wilson disease. Due to the rarity of the condition, poor recognition by many providers and an unclear diagnostic pathway mean many patients are still diagnosed late, often once irreversible damage (i.e., neurologic and/or hepatic) has occurred. Some patients present with acute liver failure and often require a transplant. Simplification of the diagnostic pathway and better access to genetic testing in Canada would improve diagnosis and allow earlier intervention before clinical presentation with advanced disease. Most patients are recognized when they present with symptomatic disease, although patients increasingly present with liver blood test abnormalities due to enhanced screening measures. More patients are now found at a pre-symptomatic stage (i.e., through sibling screening or due to increased awareness of Wilson disease and screening in the appropriate clinical context).
It is expected that any patient with Wilson disease would respond to trientine in terms of reducing overall body copper burden. Both adult and pediatric patients with hepatic-prominent Wilson disease are most likely to respond to chelation therapy, including trientine. Patients with neurologic disease may worsen with the initiation of any chelator and some evidence and anecdotal reports suggest that neurologic worsening occurs more frequently with DPA than with trientine, although this is controversial and has not been rigorously evaluated. Patients with advanced and progressive neurologic and/or psychiatric disease would be considered least suitable for trientine treatment, although trientine may still stabilize the disease and prevent further progression. Patients with acute liver failure often require an immediate transplant and so trientine is unlikely to benefit those presenting with an acute Wilsonian crisis (i.e., acute liver failure and hemolytic anemia). Patients without symptoms but with a confirmed diagnosis of Wilson disease should be treated; however, if the copper burden is not excessive, initial treatment with zinc is appropriate rather than chelation therapy.
Assessing Response to Treatment
In clinical practice (and in the clinical trial setting), response to treatment is assessed by ceruloplasmin-bound copper (calculated from serum copper and ceruloplasmin), 24-hour urinary copper collection, and liver enzymes and function tests in both adult and pediatric patients. It is also important to assess neurologic and hepatic improvement following treatment (e.g., objective neurologic improvement, brain MRI, liver enzymes, liver function) to monitor for ascites or jaundice. While some assessments are subjective, they can usually be supported by objective assessments. Treatment response should be subjectively evaluated monthly at treatment initiation (i.e., patient perspective on symptoms) and then every 6 months to 12 months once stable. Objective assessments such as neurologic assessment with or without brain MRI, laboratory improvement (non–ceruloplasmin-bound copper, 24-hour urinary copper excretion, or liver enzymes and function) should be evaluated at least annually but may require more frequent testing, especially at treatment initiation. In pediatric patients, response to treatment should be assessed more frequently (e.g., at least every 6 months).
A clinically meaningful response to treatment would be improved long-term survival, the prevention of a liver transplant, improved quality of life, the stabilization of symptoms and organ function, the normalization of liver tests, improvement in liver function (e.g., resolution of ascites or jaundice), neurologic and psychiatric symptom improvement, maintenance of independence, and improved adherence. There is no reason to expect that the magnitude of improvement would vary across physicians, except neurologists may see less improvement than hepatologists because liver involvement is more responsive to treatment than is neurologic Wilson disease.
Discontinuing Treatment
Treatment of Wilson disease is lifelong and in all cases, if 1 chelator treatment is stopped, an alternative chelator treatment must be started immediately as patients cannot be left untreated. The main reason for treatment discontinuation would be inadequacy of treatment due to either lack of efficacy or tolerability issues. For example, a patient started on chelation therapy for 1 year to 2 years may transition to zinc monotherapy for maintenance (this acts mainly by inhibiting intestinal absorption of copper); however, if there is evidence of rising copper levels (e.g., non–ceruloplasmin-bound copper, 24-hour urinary copper excretion), and/or liver injury (e.g., liver enzyme elevation), patients may require going back on chelation therapy for the long-term. Evidence of worsening neurologic function after chelator initiation should lead to prompt discontinuation of chelator therapy. Additionally, significant AEs known to be associated with DPA (e.g., rash, renal injury, neutropenia) should result in prompt discontinuation of DPA. While zinc can be used as a temporary therapy while waiting for approval of chelators, it is preferable to prevent delays in approval as even short periods of undertreatment can lead to significant disease progression.
Prescribing Conditions
Trientine can be safely prescribed in an outpatient clinic and/or specialty clinic setting, although due to the rarity of Wilson disease, there are few such specialty clinics. Once a clinical diagnosis of Wilson disease is established, patients can be followed locally because not all patients will be able to access a specialty clinic for continuous care. Periodic consultation or oversight via a specialized Wilson disease program can supplement local care but should not be a requirement for prescribing and/or approval of trientine. Close follow-up with patients and families (in the case of pediatric patients) is required to ensure adherence to therapy and to assess for sub-clinical disease or to undertake genetic testing in siblings.
The clinical experts agreed that a specialist is required to diagnose Wilson disease and should be involved in the care of patients, but not necessarily be the only prescriber of treatment. For pediatric patients, a specialist such as a pediatric hepatologist or metabolic disease specialist with experience in Wilson disease should be involved in the diagnosis, treatment, and monitoring of pediatric patients who would receive trientine. For adult patients, it may be preferable not to place limitations on prescribers as it may lead to undertreatment of patients who struggle to find an experienced provider. With training, primary care providers should be able to initiate and monitor adult patients, although it is still preferable that a specialist be involved in the care of adult patients. This would include hepatologists, gastroenterologists, and neurologists as the most relevant specialties, although general internal medicine and pediatrics have an important role, particularly in smaller communities and distant regions. Geneticists may be involved for diagnosis and possibly follow-up. Psychiatry provides ancillary support but is rarely the primary specialty involved.
Additional Considerations
The clinical experts described the current situation with trientine in Canada as a risk to achieving desired patient outcomes. The approval of MAR-Trientine by Health Canada led to immediate suspension of the SAP without a plan in place to provide trientine to Canadian patients who were already receiving treatment with imported product. This led to almost immediate loss of access to trientine, interruptions in treatment, and great anxiety for patients and providers. The clinical experts advised that it would have been preferable to have delayed the Health Canada approval of MAR-Trientine to allow ongoing use of trientine via the SAP until the CADTH review and provincial, territorial, and federal formulary decisions had been made. Alternatively, there could have been a program in place to bridge those patients who were already receiving trientine to enable uninterrupted access until a funding decision was made. Some public formularies have been able to facilitate access to trientine through the sponsor; however, the process is slow, non-standardized (as not all provinces have made trientine available), and distressing for patients and providers.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Clinicians may wish to access trientine before DPA due to its better tolerability profile. Is it reasonable to allow use as first-line treatment and if so, what criteria should apply? | Yes, it is reasonable for trientine to be used before DPA in patients with a confirmed diagnosis of Wilson disease as it has a better tolerability profile and is associated with fewer AEs. The use of trientine after DPA is not evidence-based and is largely due to access issues. |
Trientine is only approved for use in children ≥ 5 years of age. Clinicians may wish to use trientine in children < 5 years of age. Should this be allowed and if so, what criteria should apply? | Yes, there is no compelling reason not to use trientine in a child < 5 years of age; however, the main limitation is that it is not available in a dosage form amenable to dosing in children (e.g., liquid formulation as the current capsules should not be opened or chewed).11 It is expected that use in children < 5 years of age would be infrequent. |
The PM states that trientine should only be initiated by physicians experienced in the management of Wilson disease. How are these physicians identified? Do all jurisdictions have access to physicians with experience treating Wilson disease? | Please refer to the aforementioned Prescribing Conditions section for more detailed information in response to this question. A specialist is required to diagnose Wilson disease and should be involved in the care of patients but should not be the only prescriber of treatment. Due to the rarity of Wilson disease, there are few specialty clinics available. Once a patient is diagnosed, they can be followed locally; however, a specialist should provide oversight and ongoing support, as needed. |
Should prescribing be restricted only to certain specialties (e.g., gastroenterologists, hepatologists, internal medicine) or all practitioners? | Please refer to the aforementioned Prescribing Conditions section for more detailed information in response to this question. Prescribing should not be restricted only to specialists. For adult patients, it may be preferable not to place limitations on prescribers as it may lead to undertreatment of patients who are not able to access an experienced provider. |
AE = adverse event; DPA = d-penicillamine; PM = product monograph.
Clinical Evidence
The clinical evidence included in the review of MAR-Trientine is presented in the following section. The systematic review includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada. No additional studies that met the inclusion criteria as per the a priori protocol for the systematic review were identified in the literature. No indirect evidence was submitted by the sponsor or identified from the literature. Further, no sponsor-submitted long-term extension studies or additional relevant studies were considered to address important gaps in the evidence; therefore, no additional evidence was included in this review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of trientine hydrochloride 250 mg oral capsules for the treatment of Wilson disease in patients who are intolerant to penicillamine.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients ≥ 5 years of age with Wilson disease who are intolerant to penicillamine Subgroups:
|
Intervention | Trientine hydrochloride 500 mg/day to a maximum of 2,000 mg/day orally on an empty stomach in divided doses 2 times a day to 4 times a day |
Comparator |
|
Outcomes | Efficacy outcomes
Harms outcomes AEs, SAEs, WDAEs, mortality, AEs of special interest (e.g., rash, nephrotoxicity, polyneuropathy, pancytopenia, polymyositis, optic neuritis, iron deficiency anemia)a |
Study designs | Published and unpublished RCTs |
AE = adverse event; DPA = d-penicillamine; HRQoL = health-related quality of life; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.22
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were trientine and Wilson disease. Clinical trials registries were searched: the US National Institutes of Health’s ClinicalTrials.gov, the WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on May 17, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 15, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist.23 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
A focused literature search for network meta-analyses dealing with Wilson disease was run in MEDLINE All (1946–) on May 14, 2021. No limits were applied to the search.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
No studies were identified from the literature for inclusion in the systematic review (Figure 1). The only included studies are the pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, which are summarized in Table 5.
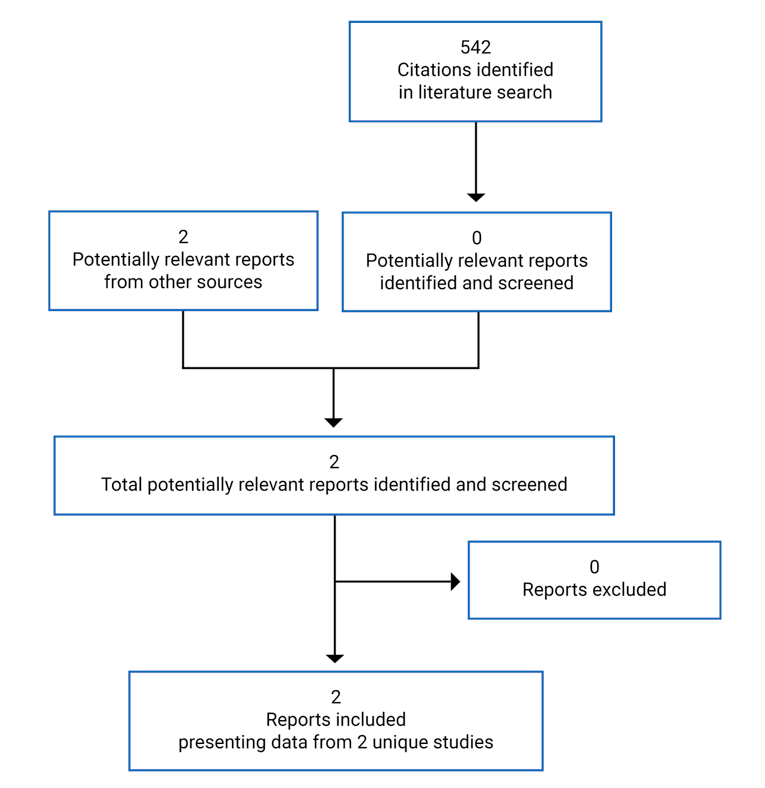
Table 5: Details of Included Studies
Detail | Weiss et al. (2013) | Study 16-VIN-0315a |
|---|---|---|
Designs and populations | ||
Study design | Retrospective cohort study | Open-label, randomized, 2-period, 2-sequence, crossover, oral fasting bioequivalence study |
Locations | Germany, Austria, EuroWilson registryb | India |
Patient enrolment dates | NR | December 26, 2016, to January 5, 2017 |
N | 405 (non-randomized) | 36 (randomized) |
Inclusion criteria | Diagnosis of Wilson disease and Leipzig score ≥ 4, ATP7B gene mutational status | Healthy male volunteers 18 to 45 years of age, BMI of 18.5 kg/m2 to 24.0 kg/m2, minimum weight of 45 kg, negative drug and alcohol screen |
Exclusion criteria | Leipzig score < 4, patients receiving only zinc salts or combination of zinc salts and chelator over the study treatment period, follow-up < 6 months | Hypersensitivity to trientine or related drug class, history of significant disease or disorder, concomitant enzyme inhibitor/inducer, history or presence of drug or alcohol abuse, significant smoking history |
Drugs | ||
Intervention | Trientine monotherapy (dose NR) | Trientine hydrochloride 250 mg capsules (manufactured by Emcure Pharmaceuticals, India) single dose of 250 mg, oral (test product) |
Comparator(s) | DPA monotherapy (dose NR) | Syprine (trientine hydrochloride) 250 mg capsules (manufactured by Pharmaceutics International, Inc, US, and distributed by Valeant Pharmaceuticals, US) single dose of 250 mg, oral (reference product) |
Duration | ||
Phase | ||
Run-in | NA | NA |
Open-label period | NA | Two 48-hour periods with 7-day washout between periods |
Follow-up | 48 monthsc | NA |
Outcomes | ||
Primary end point | Hepatic and neurologic outcomes (i.e., scored as “unchanged,” “improved to normal,” “improved but not normal,” “deteriorated,” or “asymptomatic”) | AUC0–t AUC0–inf Cmax |
Secondary and exploratory end points | NA | NA |
Notes | ||
Publications | Weiss et al. (2013) | None |
AUC0–t = area under the concentration-time curve to last quantifiable concentration; AUC0–inf = area under the concentration-time curve to infinity; BMI = body mass index; Cmax = peak concentration; DPA = d-penicillamine; NA = not applicable; NR = not reported.
Note: Two additional reports were included — the sponsor’s submission25 and the Health Canada Reviewer Report.26
aAs the purpose of Study 16-VIN-0315 was to assess bioequivalence in healthy volunteers and not the efficacy and safety of trientine in patients with Wilson disease, this study was not reviewed in detail in this report.
bOf the 405 total patients, 380 were enrolled from tertiary care centres in Germany and Austria and 25 were included from the EuroWilson registry, which enrols patients from Austria, Belgium, Croatia, the Czech Republic, Denmark, France, Germany, Greece, Hungary, India, Italy, the Netherlands, Norway, Pakistan, Poland, Portugal, Romania, Serbia, Spain, Switzerland, Turkey, and the UK.24
cThe duration of follow-up was not clearly defined; however, it appears that efficacy outcomes were based on the latest follow-up evaluation within a 6-month to 48-month follow-up period and harms outcomes were based on a median follow-up of 13.3 years (no range was reported).
Source: Weiss et al. (2013)13 and Study 16-VIN-0315 Clinical Study Report.27
Description of Studies
Two pivotal studies were submitted by the sponsor and included in the systematic review. The Weiss et al. (2013) study was a retrospective cohort analysis of 405 patients with Wilson disease who were treated with trientine or DPA and Study 16-VIN-0315 was an open-label, 2-period, 2-sequence, 2-treatment, crossover, single-dose, fasting bioequivalence study of trientine hydrochloride 250 mg capsules (test product) compared to Syprine (trientine hydrochloride) 250 mg capsules (reference product) in 36 healthy volunteers. As the purpose of Study 16-VIN-0315 was to assess bioequivalence in healthy volunteers and not the efficacy and safety of trientine in patients with Wilson disease, this study was not reviewed in detail in this report.
The objective of the Weiss et al. (2013) study was to evaluate the efficacy and safety of DPA compared to trientine therapy based on hepatic and neurologic outcomes and AEs that led to treatment discontinuation. Data on the initial presentation of patients and development of clinical and laboratory parameters under treatment with DPA or trientine were retrospectively collected from the records of 380 patients who were examined at an unspecified number of tertiary care centres in cities in Germany (Heidelberg, Dresden, and Düsseldorf) and Austria (Vienna, Graz, and Linz) and from 25 patients who were identified from the EuroWilson patient registry who had been treated with trientine monotherapy (i.e., a total of 405 patients). Patients with a stable disease course were seen at the tertiary centres approximately once per year, although patients were followed more closely if there was a change in medical therapy (e.g., at 3 months, 6 months, or 12 months after initiation of the change). Patients were included only if the duration of treatment was 6 months or more. The duration of follow-up was not clearly defined; however, it appears that efficacy outcomes were based on the latest follow-up evaluation within a 6-month to 48-month follow-up period and harms outcomes were based on a median follow-up of 13.3 years, although no range or time frame for collection of these data were specified.
Based on symptoms present at the time of diagnosis, patients were categorized into the following subgroups: asymptomatic, hepatic, neurologic, or mixed presentation (i.e., having both hepatic and neurologic symptoms). Hepatic and neurologic outcomes were also categorized according to first-line or second-line use of trientine or DPA. The results of the analysis were not reported by patient, but rather by the number of chelator monotherapies received. This totalled 467 chelator-based treatments (i.e., 326 DPA monotherapy treatments and 141 trientine monotherapy treatments). As there were 405 patients included in the analysis, an individual patient could have received both DPA and trientine in separate monotherapy regimens. No information was reported on the efficacy and safety of switching between DPA and trientine chelator therapies in individual patients. This study was supported by a grant from the Dietmar Hopp Foundation, a Young Investigator Grant from the Medical Faculty of the University of Heidelberg, and an unrestricted educational grant from the German Wilson disease patient organization Morbus Wilson e.V. EuroWilson was a Coordination Action funded by the 6th EU Research Framework Programme.
Populations
Inclusion and Exclusion Criteria
No inclusion criteria were specified in the Weiss et al. (2013) study. Rather, patients from an unspecified number of tertiary care centres in Germany and Austria or patients who were identified from the EuroWilson registry were included. The diagnosis of Wilson disease was based on the Leipzig score and patients who had a score of 4 or more were included. Patients who received only zinc salts or a combination of zinc and a chelator therapy were excluded from the analysis. Patients who had a follow-up after less than 6 months were also excluded; therefore, patients with acute liver failure who underwent a liver transplant soon after diagnosis of Wilson disease were excluded.
Baseline Characteristics
In the Weiss et al. (2013) study, baseline characteristics were recorded at the time of treatment initiation or change in the chelator-based treatment regimen. The presence of Kayser-Fleischer rings was established by slit-lamp examination and a diagnosis of cirrhosis was based on histology or on the presence of clinical signs of portal hypertension.
Baseline demographic and disease characteristics by chelator treatment were reported only by the number of chelator treatments, as detailed in Figure 2. Of the 326 DPA monotherapies, most (294 [90.2%] treatments) were first-line therapies whereas of the 141 trientine monotherapies, the majority (105 [74.5%] treatments) were second-line. The median age at diagnosis was 17.5 years for the DPA treatments compared with 19.5 years for the trientine treatments. Notable differences (i.e., ≥ 5%) between DPA and trientine treatments were the proportions of treatments categorized as being neurologic (72 [22.1%] versus 39 [27.7%]) or asymptomatic (52 [16%] versus 13 [9.2%]) at initial presentation, for DPA treatments and trientine treatments, respectively. All other baseline characteristics appeared to be similar between the treatments.
Baseline characteristics by number of patients was only available collectively for the whole study cohort (not by treatment). Of the 405 total included patients, 238 (58.8%) patients were female, and 167 (41.2%) patients were male. At initial presentation, 207 (51.1%) of all patients had only hepatic symptoms, 92 (22.7%) patients had only neurologic symptoms, 52 (12.8%) patients had hepatic and neurologic symptoms, and 54 (13.3%) patients were asymptomatic. There were 21 (5.2%) patients who presented with fulminant liver disease (acute liver failure) and 120 out of 399 (30.1%) patients who had cirrhosis at diagnosis. Overall, 64 (15.8%) of all patients were diagnosed by family screening and 205 out of 379 (54.1%) patients had Kayser-Fleischer rings present at the time of diagnosis.
Figure 2: Summary of Baseline Characteristics by Number of Chelator Treatments — Weiss et al. (2013)
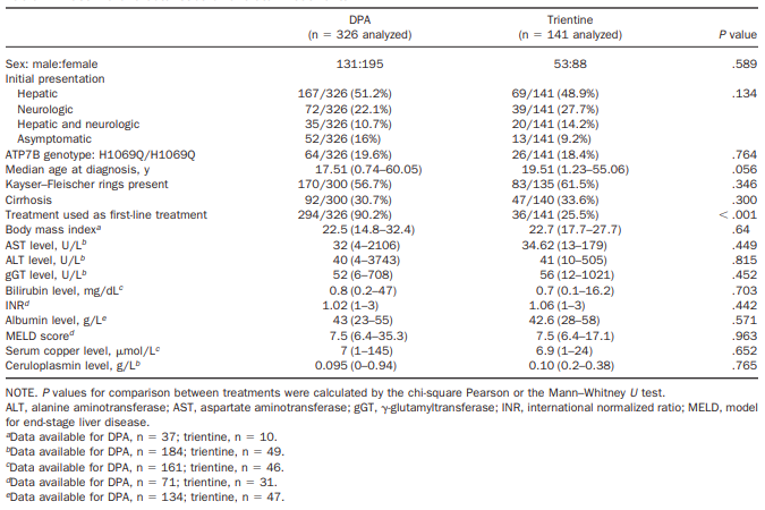
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from the American Gastroenterological Association.13
Interventions
In the Weiss et al. (2013) study, the rationale for selecting a chelator therapy (i.e., DPA or trientine) for a patient was not stated as the data were collected retrospectively. According to the researchers, patients generally started with chelation therapy when symptomatic. Further, no information on the dosage or treatment regimens used for either trientine or DPA was provided. The duration of treatment was also not clearly defined, nor was the time frame of the study or the calendar years over which time the patients were treated.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 6.
Table 6: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Weiss et al. (2013) | Study 16-VIN-0315 |
|---|---|---|
Hepatic impairment | Not specified as primary or secondary | NA |
Neurologic impairment | Not specified as primary or secondary | NA |
NA = not applicable.
In the Weiss et al. (2013) study, hepatic and neurologic outcomes were assessed at 6 months, 12 months, 24 months, 36 months, and 48 months after initiation of the current treatment regimen that the patient was on and the reported outcomes were based on the latest available follow-up evaluation within the 6-month to 48-month follow-up period. No rationale was provided for the choice of the 48-month follow-up period.
Hepatic outcome measures were derived from patient records and were based on clinical symptoms, course of liver enzymes, and liver function tests as assessed by the researchers. Patients with either clinical or biochemical signs of liver disease were considered symptomatic. Neurologic outcomes were also derived from patient records and were based on the course of neurologic disease as evaluated by the patient’s physician. Both hepatic and neurologic outcomes were categorized by the researchers as “unchanged,” “improved to normal,” “improved but not normal,” “deteriorated,” or “asymptomatic over duration.” For hepatic symptoms, the category of improved to normal implied normalized liver enzymes and liver function tests.
Harms were assessed as the number of discontinued treatments and the reasons for stopping or changing therapy were categorized by the researchers as due to AEs, orthotopic liver transplants, pregnancy, patient request, and other (not specified). The time period for collection of the harms outcomes was not stated and it was only reported that the median follow-up for the analysis of reasons for discontinuation of treatment and AEs was 13.3 years (no range was specified).
Statistical Analysis
Table 7: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Weiss et al. (2013) | |||
Hepatic and neurologic outcomes | Fisher exact test (2-tailed) | NA | NA |
Events leading to treatment discontinuation |
| NA | NA |
NA = not applicable.
Power Calculation
In the Weiss et al. (2013) study, no sample size or power calculation was reported. Of note, the efficacy outcomes reported in the study were not identified as either primary or secondary outcomes.
Statistical Tests
There was no statistical analysis plan available for the Weiss et al. (2013) study. The efficacy outcomes reported were the improvement or worsening of hepatic or neurologic outcomes, categorized as previously described, and counted as number of chelator monotherapy treatments. Comparisons of hepatic and neurologic outcomes between DPA and trientine monotherapy treatments were conducted using the 2-tailed Ficher exact test. A P value of less than 0.05 was considered statistically significant. No further information on the statistical analyses of the efficacy outcomes was reported. No data imputation methods were mentioned and no statistical adjustments were made for multiple comparisons to control the type I error rate.
Regarding harms outcomes, events leading to a change or discontinuation of a chelator monotherapy treatment were categorized by the authors as to the reasons for discontinuation and were analyzed using Kaplan–Meier estimation. P values were derived using the log-rank test (Mantel-Cox test) and a P value less than 0.05 was considered statistically significant. No further information on the statistical analyses of discontinuation or harms outcomes or on the Kaplan–Meier analysis was reported.
Subgroup Analyses
There were no pre-specified subgroups in the Weiss et al. (2013) study. Nonetheless, the efficacy outcomes of hepatic or neurologic worsening or improvement were reported for all patients and for a subpopulation of patients who were categorized as symptomatic and according to first-line treatment or second-line treatment with trientine or DPA. There did not appear to be an evaluation of the comparability of the apparent subgroups between the chelator monotherapies and there were no statistical adjustments made for multiple comparisons to control for type I error.
Analysis Populations
In the Weiss et al. (2013) study, the analysis population comprised the number of chelator monotherapy treatments (N = 467). The number of patients contributing to the analysis population included 380 patients who were examined at an unspecified number of tertiary care centres in Germany and Austria and 25 patients identified from the EuroWilson registry who were treated with trientine monotherapy.
Results
Treatment Disposition
For the Weiss et al. (2013) study, the disposition data pertains to the number of chelator monotherapy treatments (not the number of individual patients) as detailed in Figure 3. Overall, 142 out of 326 (43.6%) DPA treatments were discontinued compared with 36 out of 141 (25.5%) trientine treatments over the study duration; the specific time period for this is unknown. The main reason for discontinuation of DPA treatment was AEs (94 [28.8%] treatments) followed by “other” (23 [7.1%] treatments), which was not defined. For trientine, the main reason for treatment discontinuations was other (18 [12.8%] treatments) followed by AEs (10 [7.1%] treatments). Twelve treatments were discontinued due to liver transplants as a result of hepatic failure (i.e., 9 [2.8%] DPA treatments and 3 [2.1%] trientine treatments). Four (1.2%) DPA treatments were discontinued due to pregnancy and no trientine treatments were discontinued for this reason.
Figure 3: Treatment Disposition
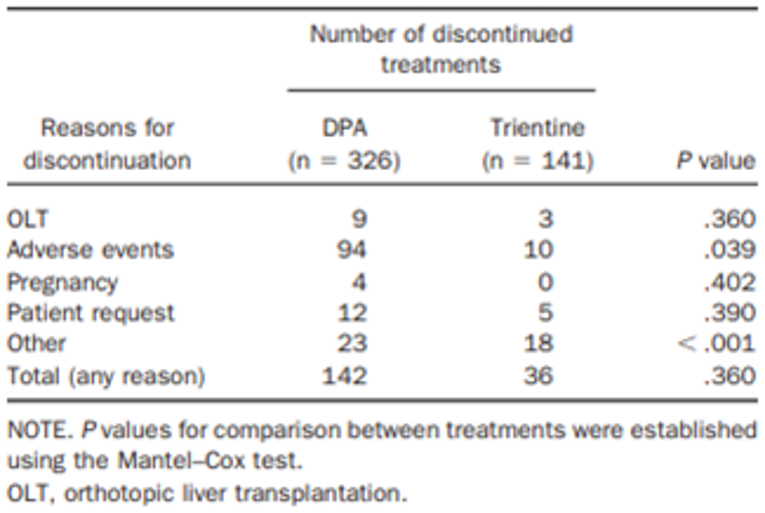
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from the American Gastroenterological Association.13
Exposure to Study Treatments
In the Weiss et al. (2013) study, efficacy outcomes were assessed from patient records for up to 48 months after initiation or a change in chelator monotherapy. For the analysis of treatment discontinuations, all that was provided was the statement that the median follow-up time for the Kaplan–Meier estimation was 13.3 years with no other explanation of the methodology used. No additional information was provided regarding exposure to either DPA or trientine treatment.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported as follows. No data were reported in the included studies for the following outcomes of interest as per the protocol for this systematic review: HRQoL, psychiatric manifestations, copper levels, adherence, or health care resource utilization.
Hepatic Impairment
Results of the detailed scoring of hepatic outcomes for all trientine and DPA chelator treatments included in the Weiss et al. (2013) study are provided in Figure 4. No statistical comparisons were conducted for these outcomes. Similar proportions of trientine and DPA monotherapy treatments were scored as unchanged (12 [8.5%] trientine treatments versus 19 [5.8%] DPA treatments) and improved but not normal (27 [19.1%] trientine treatments versus 58 [17.8%] DPA treatments). More than double the number of DPA treatments (139 [42.6%]) were scored as improved to normal compared with trientine treatments (29 [20.6%]). Further, a numerically larger proportion of trientine treatments (69 [48.9%]) were scored as asymptomatic over the study duration compared with DPA treatments (106 [32.5%]). Only a small proportion of both trientine (4 [2.8%]) and DPA (4 [1.2%]) treatments were scored as deteriorated.
Figure 4: Detailed Scoring of Hepatic or Neurologic Outcomes by Number of Chelator Treatments — Weiss et al. (2013)
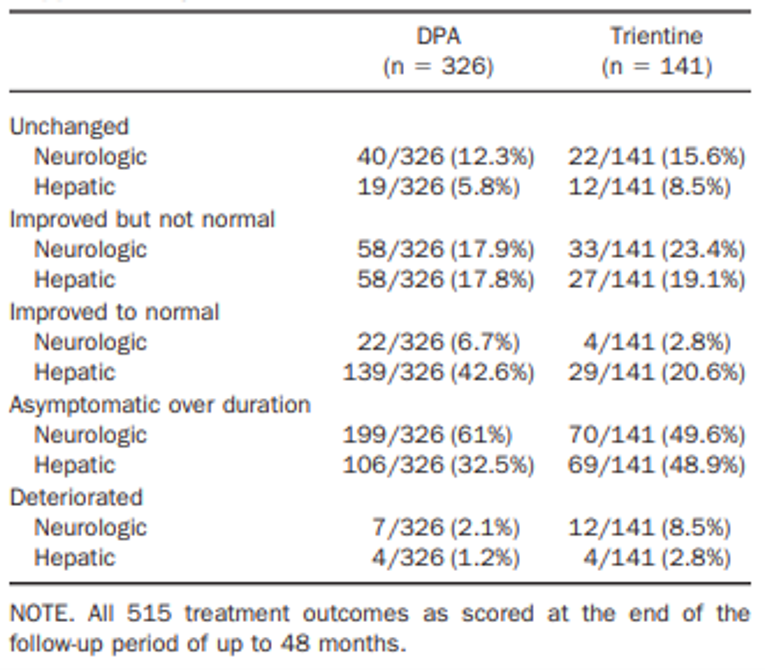
DPA = d-penicillamine.
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from the American Gastroenterological Association.13
Rates of hepatic improvement and worsening for trientine and DPA by line of treatment and reported for all patients or symptomatic patients, as reported by the researchers, are provided in Figure 5. Hepatic improvement scores after first-line treatment were comparable for all patients (25 out of 38 [65.8%] trientine treatments versus 185 out of 295 [62.7%] DPA treatments) and for symptomatic patients (25 out of 27 [92.6%] trientine treatments versus 185 out of 204 [90.7%] DPA treatments); these scores were not statistically significantly different (Figure 5). Following second-line treatment, hepatic improvement scores were generally lower than with first-line treatment for all patients (31 out of 103 [30.1%] trientine treatments and 12 out of 31 [38.7%] DPA treatments) and for symptomatic patients (31 out of 45 [68.9%] trientine treatments and 12 out of 16 [75.0%] DPA treatments). There were also no statistically significant differences between treatments. For symptomatic patients, stable hepatic disease categorized as unchanged hepatic symptoms was observed in 7.4% of first-line treatments for both groups (i.e., 2 out of 27 trientine treatments and 15 out of 204 DPA treatments). Stable hepatic disease after second-line therapy was reported in 10 out of 24 (22.2%) trientine treatments and 4 out of 16 (25%) DPA treatments. No statistical comparisons were reported for the number of treatments associated with stable or unchanged hepatic symptoms.
There were no first-line trientine treatments associated with hepatic worsening (i.e., defined as a decline in liver function or progression of chronic liver disease) compared to first-line DPA treatments for all patients (0 out of 38 [0%]) trientine treatments versus 4 out of 295 [1.4%] DPA treatments) and for symptomatic patients (0 out of 27 [0%] trientine treatments versus 4 out of 204 (2.0%) DPA treatments). (See Figure 5). While second-line trientine treatment was associated with hepatic worsening, there were no second-line DPA treatments associated with hepatic worsening for all patients (4 out of 103 [3.9%] trientine treatments versus 0 out of 31 [0%] DPA treatments) and for symptomatic patients (4 out of 45 [8.9%] trientine treatments versus 0 out of 16 [0%] DPA treatments). The differences between trientine and DPA treatments for hepatic worsening after either first-line or second-line treatments were not statistically significantly different. Overall, there were 12 treatments with an outcome of a liver transplant (i.e., 3 [2.1%] trientine treatments and 9 [2.7%] DPA treatments).
Figure 5: Rate of Hepatic or Neurologic Improvement and Worsening by Number of Chelator Treatments — Weiss et al. (2013)
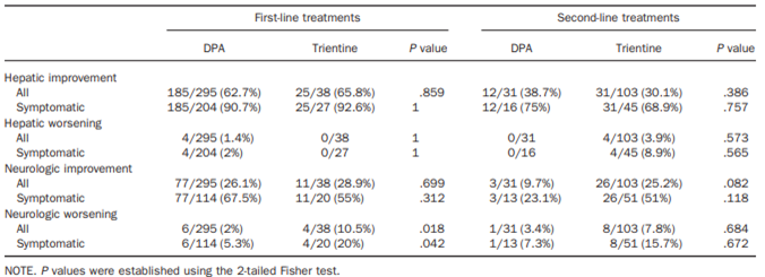
DPA = d-penicillamine.
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from the American Gastroenterological Association.13
Neurologic Impairment
Results of the detailed scoring of neurologic outcomes for all trientine and DPA chelator treatments included in the Weiss et al. (2013) study are provided in Figure 4. No statistical comparisons were conducted for these comparisons. Most treatments for either trientine (70 out of 141 [49.6%]) or DPA (199 out of 326 [61.0%]) were scored as asymptomatic over duration. There were numerically more DPA treatments (22 out of 326 [6.7%]) compared with trientine treatments (4 out of 141 [2.8%]) that were scored as improved to normal. The proportions of trientine and DPA monotherapy treatments that were scored as unchanged were 22 out of 141 (15.6%) and 40 out of 326 (12.3%) whereas those scored as improved but not normal were 33 out of 141 (23.4%) and 58 out of 326 (17.9%), respectively. There were numerically more treatments scored as deteriorated with trientine (12 out of 141 [8.5%]) compared with DPA (7 out of 326 [2.1%]).
Rates of neurologic improvement and worsening for trientine and DPA by line of treatment and for all patients or for symptomatic patients are provided in the bottom half of Figure 5. Neurologic improvement scores for first-line treatment were comparable between trientine treatments (11 out of 38 [28.9%]) and DPA treatments (77 out of 295 [26.1%]) for all patients but were numerically higher for DPA treatments (77 out of 114 [67.5%]) versus trientine treatments (11 out of 20 [55.0%]) in symptomatic patients, although the differences were not statistically significant (Figure 5). Following second-line therapy for all patients, neurologic improvement rates were comparable to those after first-line therapy for trientine treatments (26 out of 103 [25.2%]) but were numerically lower for DPA treatments (3 out of 31 [9.7%]). For symptomatic patients, neurologic improvement with second-line therapy after trientine treatments (26 out of 51 [51.0%]) was numerically higher than after DPA treatments (3 out of 13 [23.1%]). Nonetheless, all comparisons between trientine treatments and DPA treatments for all patients and for symptomatic patients for second-line therapy were not statistically significantly different. For symptomatic patients, stable neurologic disease, which was categorized as unchanged neurologic symptoms, was observed in 5 out of 20 (25.0%) trientine treatments and in 31 out of 114 (27.2%) DPA treatments after first-line therapy and in 17 out of 51 (33.3%) trientine treatments and 9 out of 13 (69.2%) DPA treatments after second-line therapy. No statistical comparisons were reported for stable or unchanged neurologic symptoms.
Rates of neurologic worsening after first-line therapy were statistically significantly higher for trientine treatments compared to DPA treatments for all patients (4 out of 38 [10.5%] versus 6 out of 295 [2.0%]; P = 0.018) and for symptomatic patients (4 out of 20 [20.0%] and 6 out of 114 [5.3%]; P = 0.042), respectively (Figure 5). For second-line therapy, rates of neurologic worsening were numerically higher with trientine treatments compared to DPA treatments for all patients (8 out of 103 [7.8%] and 1 out of 31 [3.4%], respectively) and symptomatic patients (8 out of 51 [15.7%] and 1 out of 13 [7.3%], respectively), although the differences were not statistically significant.
Harms
Only those harms identified in the review protocol are reported as follows. See Figure 6 for detailed harms data.
In the Weiss et al. (2013) study, the only harms outcomes that were reported were the proportions of chelator monotherapy treatments with AEs that led to treatment discontinuation.
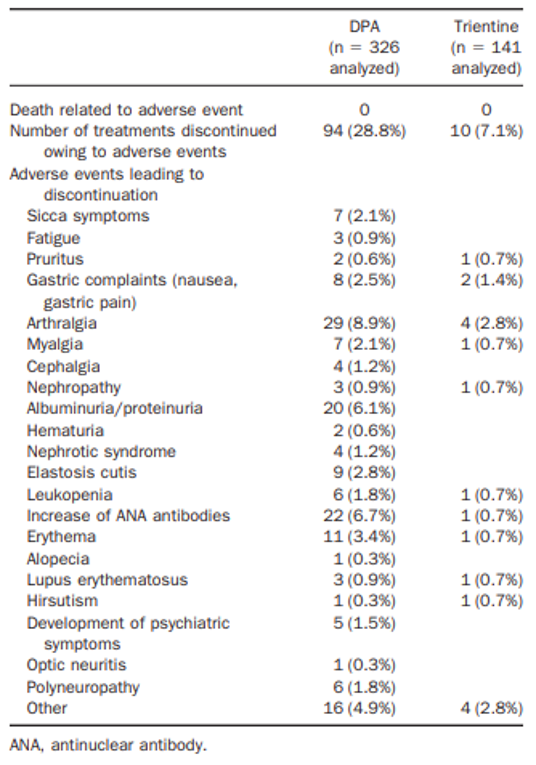
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from the American Gastroenterological Association.13
Adverse Events
No information is available from the Weiss et al. (2013) study on the overall frequency of treatment-emergent AEs with trientine or DPA monotherapy.
Serious Adverse Events
No information is available from the Weiss et al. (2013) study on the overall frequency of treatment-emergent serious AEs with trientine or DPA monotherapy.
Withdrawals Due to Adverse Events
In the Weiss et al. (2013) study, treatment discontinuations due to AEs were more common with DPA (94 out of 326 [28.8%] treatments) compared with trientine (10 out of 141 [7.1%] treatments) (Figure 6). The difference between DPA and trientine treatments was statistically significant (P = 0.039), as reported in the publication Clinical Gastroenterology and Hepatology.13 The frequency of AEs was higher with DPA treatments and the most common AEs (≥ 5% frequency in either group) that led to treatment discontinuation were arthralgia (29 out of 326 [8.9%] DPA treatments versus 4 out of 141 [2.8%] trientine treatments), an increase in antinuclear antibodies (22 out of 326 [6.7%] DPA treatments versus 1 out of 141 [0.7%] trientine treatments), and albuminuria/proteinuria (20 out of 326 [6.1%] DPA treatments versus not reported for trientine treatments). Rates of discontinuations for any reason were not statistically significantly different between the chelator treatments (P = 0.360), as reported in the publication.13 The results of the Kaplan–Meier estimation for discontinuation of treatment due to any cause (A) or due to AEs (B) are illustrated in Figure 7.
Figure 7: Discontinuation of Treatment Due to Any Cause (A) or Adverse Events (B) — Weiss et al. (2013)
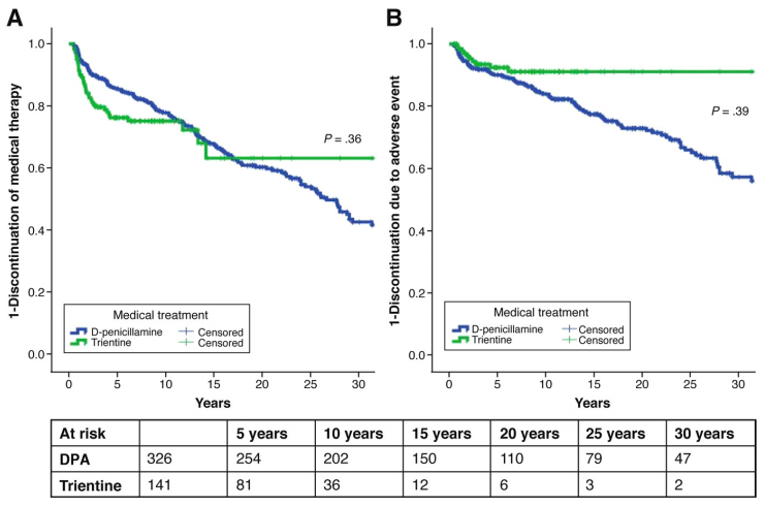
Note: There appears to be an error in the previously published Figure 2 (B) since in the text and tabular data of the Weiss et al. (2013) publication, the P value is reported as P = 0.039.
Source: Reprinted from Clinical Gastroenterology and Hepatology (“Efficacy and safety of oral chelators in treatment of patients with Wilson disease”), with permission from Elsevier.13
Mortality
No deaths were reported in the Weiss et al. (2013) study.
Notable Harms
The AEs of special interest identified in the protocol for the systematic review were rash, nephrotoxicity, polyneuropathy, pancytopenia, polymyositis, optic neuritis, and iron deficiency anemia. The only information available was from the Weiss et al. (2013) study, which reported the number of chelator monotherapy treatments discontinued due to AEs of special interest. Six (1.8%) DPA treatments were discontinued due to polyneuropathy compared to none reported for trientine treatments. There were also more DPA treatments discontinued due to nephrotic syndrome (4 [1.2%] treatments) and albuminuria/proteinuria (20 [6.1] treatments) compared to no trientine treatments discontinued for these reasons. One (0.7%) trientine treatment and 6 (1.8%) DPA treatments were discontinued due to leukopenia.
Critical Appraisal
Internal Validity
As a retrospective cohort analysis, the Weiss et al. (2013) study is limited by lack of randomization and the non-prospective collection of efficacy and harms outcomes. A major concern and source of bias with retrospective analyses is that patients were prescribed treatment based on their individual characteristics, rather than being randomly allocated to treatment.28 Another potential source of bias is that the researchers were reliant on past reporting of symptoms, clinical and laboratory parameters, and response to treatment by others who recorded information in patient records; thus, the researchers were unable to prospectively identify specific efficacy outcomes or to control for confounders. A potential confounder could have been the availability of DPA or trientine, which would have influenced the choice and sequence of therapy. There was more than a twofold greater number of DPA treatments (N = 326) compared to trientine treatments (N = 141), with most DPA treatments (90.2%) used as first-line treatment compared to trientine (25.5%). This could have been due to a lack of access to trientine and may have biased the harms results in favour of trientine. DPA is known to be poorly tolerated and since most patients in the study received DPA, there was likely a higher probability that DPA treatment would result in more treatment discontinuations due to AEs.
This study was also not blinded. This may have introduced bias into the categorization of hepatic and neurologic outcomes, which were all subjectively assessed. The authors relied on the reporting of symptoms and hepatic and neurologic outcomes in patient records, which they then subjectively categorized as improved or worsened. The authors also subjectively identified patients as having hepatic, neurologic, or mixed presentation and hepatic patients as being symptomatic based on clinical or biochemical signs of liver disease. No criteria or rules regarding how decisions were made to categorize patients based on relative differences were stated. This could have introduced substantial reporting bias, although the direction of such bias is unknown. Improvement or worsening of neurologic disease was based on the physician’s report in the patient record, so the researchers were dependent on the treating physician’s opinion of the outcome. The reporting of harms outcomes was also subject to reporting bias as the authors also subjectively categorized the reasons for treatment discontinuations due to AEs.
The specific time frame of the study or the calendar years over which time the patients were treated was not reported. If patients were treated during different chronologic time periods, it is possible that changes in clinical practice or availability of treatment guidelines could have influenced the choice of treatment. It was stated in the Weiss et al. (2013) study that outcomes were based on the latest available follow-up evaluation within a 6-month to 48-month period. It follows then that the cumulative hepatic and neurologic outcomes reported in the study could comprise different treatment intervals ranging anywhere between 6 months and 48 months for different patients. It is not known how this may have affected the study outcomes and if the cumulative treatment time was equal between trientine and DPA treatments. Further, it was reported that data on discontinuations and discontinuations due to AEs were collected over a median 13.3-year period to inform the Kaplan–Meier analysis, although no range of time over which these data were collected was reported. No further details were provided on the methodology of the Kaplan–Meier analysis, which makes these data difficult to interpret.
The reporting of results by number of chelator monotherapy treatments as opposed to the number of patients complicates the interpretation of the baseline patient characteristics and of the efficacy and harms outcomes. It is difficult to ascertain if the baseline characteristics are balanced between the chelator treatments or if differences in efficacy and harms outcomes are valid because an individual patient could have been counted more than once in the results. Double data counting compromises the validity of the dataset. For example, if an individual patient displays a specific characteristic such as hepatic presentation of Wilson disease and they are double-counted, this may result in more treatments being associated with hepatic presentation than if patients were randomly selected and counted only once in the dataset. There were 405 patients included in the study, but results are reported for 467 chelator monotherapy treatments. This infers that up to 62 patients were counted more than once (e.g., as first-line and second-line therapy).
There was no clear definition or validation of the efficacy outcomes in terms of reliability, validity, responsiveness, or MIDs, which makes interpretation of the efficacy results difficult. This is especially true regarding the relative differences between categories assigned by the authors (e.g., improved to normal versus improved but not normal).
The rate of treatment discontinuations was disproportionally higher with DPA treatments (43.6%) compared with trientine treatments (25.5%). No information was provided regarding patient follow-up after treatment discontinuation and there was no imputation of missing data. Treatment discontinuations were analyzed using Kaplan–Meier estimation. However, over time, the number of treatments at risk in the trientine group is considerably smaller relative to DPA (e.g., 150 DPA treatments versus 12 trientine treatments at 15 years), which makes interpretation of these data uncertain.
There were no pre-specified subgroups reported in the study methods. However, the authors subjectively categorized patients as being symptomatic and according to first-line or second-line therapy, and subsequently reported hepatic and neurologic outcomes according to these subpopulations. No statistical adjustments to control for type I error were made for the multiple comparisons of hepatic and neurologic outcomes between DPA and trientine or among the apparent subgroups. Moreover, the sample sizes of certain subgroups were small (i.e., N = 13 treatments or 20 treatments), which warrants caution in interpreting the results.
There were inconsistencies in the reporting of data in the publication of the study. For example, the abstract states that 471 chelator monotherapies were analyzed, although results are reported for 467 chelator treatments. The P value for the statistical assessment of the difference between trientine and DPA for discontinuations due to AEs was reported to be P = 0.039 in a table and the text of the publication; however, it was shown as P = 0.39 in the Kaplan–Meier curve depicting the same (Figure 7). Additionally, the time period for data on discontinuations and discontinuations due to AEs was stated to be a mean of 13.3 years and a median of 13.3 years in different sections of the publication and no measure of variability (e.g., standard deviation) or range was reported with these data.
External Validity
There were no Canadian patients included in the Weiss et al. (2013) study as patients were identified for inclusion from patient records of tertiary hospitals in Germany and Austria as well as the EuroWilson patient registry. The clinical experts involved in the review advised that the baseline characteristics of the patients in the study were reasonably similar to those of the population of Canadian patients who would be candidates for trientine, with the possible exception of pediatric patients. The only age characteristic that was reported in the Weiss et al. (2013) study was the median age at diagnosis, which was 19.51 years (range = 1.23 years to 55.06 years) for the trientine treatments and 17.51 years (range = 0.74 years to 60.05 years) for DPA treatments. As a result, it is not known how many pediatric patients younger than 18 years were included in the study.
Although patients were identified as being asymptomatic, or having hepatic, neurologic, or mixed presentation at initial diagnosis, the results were not reported according to initial presentation. Most patients (51.1%) presented with hepatic symptoms, followed by neurologic symptoms (22.7%); this is similar to what would be expected in Canadian patients, according to the clinical experts on the review team. It was noted that some specialized centres in Canada may predominantly treat patients with neurologic presentation because they are considered to be centres of excellence for neurologic care.
Dosage and administration schedules for trientine and DPA were not reported in the study, so it is not known if the doses used are in alignment with the Health Canada–approved doses of DPA and trientine. Additionally, it was not stated whether any patients who discontinued second-line therapy with DPA or trientine subsequently received a re-challenge with either DPA or trientine at a later time point, as only the first-line and second-line treatment approaches were reported. The clinical experts consulted on this review indicated that it may be possible to re-challenge a patient on chelator therapy following prior discontinuation of chelator treatment, especially if the reason for discontinuation was not due to serious intolerance or toxicity (e.g., if a patient discontinued chelation therapy due to gastrointestinal upset, it may be possible to re-try chelation therapy using a different dosage regimen). The clinical experts cautioned that toxicity with DPA may be worse upon re-challenge.
Patients who received combination therapy with zinc (i.e., a chelator plus zinc therapy) were excluded from the study. According to the clinical experts consulted for this review, upon diagnosis of Wilson disease, patients are usually treated with chelators with or without zinc as first-line therapy. Moreover, zinc may be added to chelation therapy when there is a concern of very excessive copper overload to accelerate copper reductions or to minimize the dose of a chelator if intolerance or concern about worsening of neurologic symptoms is an issue. Patients receiving only zinc monotherapy over the treatment period were also excluded. According to the clinical experts, zinc is primarily used as a maintenance therapy after a period (usually 1 year) of chelation therapy. Zinc monotherapy may also be used as initial therapy for people diagnosed through sibling screening who have very limited copper overload or in patients with a primary neurologic presentation in whom chelators may worsen neurologic symptoms, sometimes irreversibly. Patients who received 6 months or less of chelator treatment were also excluded from the study, so patients who immediately underwent a liver transplant soon after diagnosis of Wilson disease were not included in the analyses. Therefore, the results of this study may not be generalizable to these patients.
No data were reported for key outcomes of clinical relevance to patients such as HRQoL and adherence (based on the patient input received for this review). Additional outcomes included in the review protocol for which there also were no data were psychiatric manifestations, copper levels, and health care resource utilization. Thus, there is an evidence gap regarding many of the outcomes identified in the review protocol. For the outcomes that were reported in the study (i.e., hepatic and neurologic improvement and worsening), due to their subjective assessment by the authors of the study, it is not known if the same outcomes would be used, or the same subjective interpretation made, by physicians in Canadian clinical practice.
The duration of follow-up was 48 months whereas Wilson disease requires continuous lifelong treatment. As a result, the length of follow-up may not have been adequate to capture long-term efficacy and harms outcomes. A key outcome that was not reported but could be addressed by longer follow-up is patient adherence to treatment. Adherence with chelation therapy is vital to effective treatment of Wilson disease, as according to the clinical experts on the review team, poor adherence is the major reason for treatment failure in patients with Wilson disease.
Table 8 summarizes the generalizability of the evidence from the Weiss et al. (2013) study.
Table 8: Assessment of Generalizability of Evidence for MAR-Trientine
Domain | Factor | Evidence from Weiss et al. (2013) | CADTH’s assessment of generalizability |
|---|---|---|---|
Population | Age | The only age parameter reported was median age at diagnosis, which was 19.51 years (range = 1.23 years to 55.06 years) for trientine and 17.51 years (range = 0.74 years to 60.05 years) for DPA. It is not known how many pediatric patients (< 18 years) were included in the study. | Although it is not known how many pediatric patients were included, the study results are likely generalizable to pediatric patients (< 18 years) as the age range of patients at diagnosis implies pediatric patients were included. As further support, the clinical experts on the review team advised there are no compelling reasons why trientine cannot be used in patients < 5 years of age. |
Disease status at presentation | Patients were categorized as asymptomatic (13.3%), hepatic (51.1%), neurologic (22.7%), or mixed hepatic and neurologic (12.8%) at initial presentation; however, the results were not reported by disease status. | According to the clinical experts, the study included all presentations of Wilson disease in similar proportions as expected in Canadian patients and other baseline patient characteristics were similar to those of Canadian patients, so it is reasonable to assume that the results are generalizable to a wide range of patients with Wilson disease and varying presentations. | |
Intervention | Trientine 500 mg/day to a maximum of 2,000 mg/day orally on an empty stomach in divided doses 2 times a day to 4 times a day | No information on the dose of trientine or administration schedules was provided in the study. | In the absence of this information, it is not possible to determine if the dose and administration schedule of trientine was aligned with the Health Canada–approved dosage regimen for trientine. |
First-line use or second-line use | Treatments were categorized as first-line or second-line, although no systematic criteria for doing so were reported in the study. | Hepatic and neurologic outcomes were reported for trientine by first-line use and second-line use, so the results are generalizable to these lines of treatment. No information was provided on third-line use and beyond or re-challenge of chelator therapy. | |
Comparator | DPA 750 mg/day to 1,500 g/day on an empty stomach at least 1 hour before meals or 2 hours after meals and at least 1 hour apart from any other drug, food, or milk | No information on the dose of DPA or administration schedules was provided in the study. | In the absence of this information, it is not possible to determine if the dose and administration schedule of DPA was aligned with the Health Canada–approved dosage regimen for DPA. |
First-line use or second-line use | Treatments were categorized as first-line or second-line, although no systematic criteria for doing so were reported in the study. | Hepatic and neurologic outcomes were reported for DPA by first-line use and second-line use, so the results are generalizable to these lines of treatment. No information was provided on third-line use and beyond or re-challenge of chelator therapy. | |
Outcomes | Appropriateness of outcomes | Outcomes included hepatic and neurologic worsening/improvement; however, no details were provided regarding what comprised a hepatic or neurologic outcome. | The clinical experts advised that the outcomes were appropriate, although little detail was provided as to what comprised the hepatic and neurologic outcomes and what criteria or rules were considered in the assessment of improvement or worsening. |
Criteria used to define response | No criteria or rules to define response or relative differences in response were provided. | It is not possible to comment on the generalizability of the definition of response criteria as this information was not provided in the study. | |
Setting | Trial sites | The study included patients identified from tertiary care centres in Germany, Austria, and the EuroWilson patient registry. No Canadian patients were included. | Although no Canadian patients were included, the clinical experts consulted on the review agreed that the baseline patient characteristics of patients enrolled in the study were reasonably similar to the population of Canadian patients who would be candidates for trientine, with the possible exception of pediatric patients. |
Supportive medication | Patients who were on combination zinc plus chelator therapy were excluded from the study, as were patients who were on zinc monotherapy. | Canadian patients are usually treated with chelators with or without zinc as first-line therapy. Moreover, zinc may be added to chelation therapy when there is a concern of very excessive copper overload to accelerate copper reductions or to minimize the dose of a chelator if there is intolerance or concern about worsening of neurologic symptoms. As patients on combination chelator plus zinc or zinc monotherapy were excluded from the study, the results are not generalizable to these patients. |
DPA = d-penicillamine.
Discussion
Summary of Available Evidence
Two pivotal trials submitted by the sponsor were included in the systematic review. No additional trials from the literature search met the inclusion criteria for the systematic review and no indirect comparisons or other relevant evidence were identified. The first included study (Weiss et al. [2013]) was a retrospective cohort analysis that evaluated the efficacy and safety of trientine compared to DPA in 405 patients with Wilson disease based on hepatic and neurologic outcomes and treatment discontinuations over a 48-month period. The second study (Study 16-VIN-0315) was an open-label, 2-period, 2-sequence, 2-treatment, crossover, single-dose, fasting bioequivalence study of MAR-Trientine (trientine hydrochloride) 250 mg capsules compared to Syprine (trientine hydrochloride) 250 mg capsules in 36 healthy adult male and female volunteers. The objective of this study was to compare the rate and extent of absorption of trientine from the 2 formulations to determine if they were bioequivalent. As the purpose of Study 16-VIN-0315 was to assess bioequivalence in healthy volunteers and not efficacy and safety in patients with Wilson disease, it was not reviewed in detail in this report.
According to the clinical experts on the review team, the baseline characteristics of the patients in the Weiss et al. (2013) study are reasonably similar to Canadian patients who would be candidates for trientine, with the possible exception of pediatric patients (< 18 years of age). The median age of patients at the time of diagnosis of Wilson disease (the only age parameter reported in the study) was 17 years to 19 years. Although patients younger than 18 years were included, no details on the number or the age of pediatric patients were provided. The Weiss et al. (2013) study reported baseline characteristics and efficacy and safety outcomes by number of chelator treatments (and not by number of patients). At initial presentation, about half (207 [51.1%] treatments) were associated with only hepatic symptoms, 92 (22.7%) treatments with only neurologic symptoms, 52 (12.8%) treatments with mixed presentation (hepatic and neurologic symptoms), and 54 (13.3%) treatments were asymptomatic. If these proportions broadly reflect patient presentations, the clinical experts advised that a similar distribution would be seen in Canadian clinical practice. Key limitations of the Weiss et al. (2013) study are the retrospective design, the lack of randomization and blinding, no information on the time frame of the study, the subjective assessment of outcomes, the reporting of results by number of chelator treatments rather than by number of patients, and the exclusion of patients who received zinc monotherapy or combination chelator therapy with zinc. There were no data available for most efficacy outcomes identified in the review protocol, including outcomes of interest to patients such as HRQoL and adherence.
Interpretation of Results
Efficacy
The Weiss et al. (2013) study evaluated 467 chelator-based treatment regimens with a duration of between 6 months and 48 months, of which 141 (30.2%) were trientine treatments and 326 (69.8%) were DPA treatments. Of the trientine treatments, 36 (25.5%) were used as first-line treatment and 105 (74.5%) were used as second-line treatment, compared to 294 (90.2%) and 32 (9.8%) of DPA treatments, respectively. The disproportionate use of DPA as a first-line treatment may be due to lack of access to trientine, rather than trientine being reserved for use as a second-line option for patients who are intolerant to DPA. The clinical experts consulted for this review advised that there is no compelling reason that trientine cannot be used as a first-line therapy in patients with Wilson disease, and it may in fact be a preferred choice due to its better tolerability profile compared to DPA. The lack of randomization and stratification of patients according to first-line and second-line use of trientine or DPA at study entry resulted in a substantial baseline imbalance for the proportion of treatments used as first-line therapy, which was statistically significant (P < 0.001), as reported in the publication Clinical Gastroenterology and Hepatology.13 The reporting of baseline patient characteristics and efficacy and harms outcomes by number of chelator monotherapy treatments, as opposed to the number of patients, also complicates the interpretation of baseline imbalances and relative treatment effects because an individual patient may have been double-counted in the results (i.e., for first-line and second-line treatment), although the direction of bias is difficult to ascertain.
This study was also not blinded, which may have introduced bias into the assessment and categorization of the study outcomes (i.e., hepatic and neurologic outcomes and reasons for treatment discontinuations). As the data were not prospectively collected, the researchers relied on non-standardized reporting by others in patient records, from which they derived clinical and laboratory information. Using this information, they subjectively assessed and categorized patients as being symptomatic or having improved or worsening hepatic and neurologic outcomes. Hepatic outcomes were based on clinical symptoms, liver enzymes, and liver function tests, and patients with either clinical or biochemical evidence of liver disease were considered symptomatic. No details were provided as to what criteria or rules were followed to categorize patients as improved or worsened. This, coupled with a lack of validation of these outcomes or identification of MIDs, makes interpretation of the results challenging. Hepatic improvement scores following either first-line or second-line treatment were similar between trientine and DPA and no statistically significant differences were identified between the treatments either in all patients or in only symptomatic patients. When used as second-line treatment, the hepatic improvement scores were lower than for first-line use, but there were no statistically significant differences between trientine and DPA treatments. Similar proportions of trientine and DPA treatments (either first-line or second-line) were categorized as stable disease, which was defined as unchanged hepatic symptoms and was reported only for symptomatic patients. Hepatic deterioration or worsening was reported for 4 DPA first-line treatments and 4 trientine second-line treatments in all patients as well as in symptomatic patients, which suggests the 4 treatments may represent the same patients. There was no hepatic worsening in any patients (all patients or symptomatic patients) who received trientine as first-line therapy or DPA as second-line therapy. Improvement in hepatic symptoms with either trientine or DPA treatment is in line with the observations of the clinical experts involved in this review, as most patients with Wilson disease who present with hepatic symptoms are expected to have their hepatic symptoms respond well to chelation therapy.
The course of neurologic disease was based on physician evaluations of neurologic symptoms as reported in patient records. The researchers also categorized neurologic outcomes in a similar manner as hepatic outcomes, although no additional details were provided. Neurologic improvement scores were similar between trientine and DPA following first-line treatment in all patients and symptomatic patients, and the differences between treatments were not statistically significantly different. For second-line treatment, numerically fewer DPA treatments (3 out of 31 [9.7%] and 3 out of 13 [23.1%]) were associated with neurologic improvement compared with trientine treatments (26 out of 103 [25.2%] and 26 out of 51 [51.0%]) for all and symptomatic patients, respectively; however, the differences were not statistically significantly different. It must be noted that only 3 second-line DPA treatments contributed to these analyses, so these results warrant cautious interpretation. Rates of neurologic worsening were low and comparable between trientine and DPA treatments when used as second-line, with no statistically significant differences identified between the treatments, although numbers of patients included in these analyses were small. Importantly, the proportion of first-line treatments that resulted in neurologic worsening was statistically significantly higher with trientine than with DPA for all patients (4 out of 38 [10.5%] versus 6 out of 295 [2.0%] treatments; P = 0.018) and symptomatic patients (4 out of 20 [20.0%] versus 6 out of 114 [5.3%] treatments; P = 0.042), respectively. It is unclear why a higher proportion of trientine first-line treatments would have led to worsening of neurologic symptoms compared to DPA, although the clinical experts on the review team advised that a transient elevation of serum copper following initiation of any chelator treatment can potentiate neurologic symptoms in patients with Wilson disease and neurologic presentation, which may be irreversible. Stable neurologic disease was observed in similar proportions of trientine and DPA treatments after first-line treatment; however, following second-line treatment, a larger proportion of DPA treatments (9 out of 13 [69.2%]) were categorized as having stable disease than trientine treatments (17 out of 51 [33.3%]), although no statistical comparison was reported.
The results of Study 16-VIN-0315 demonstrated that MAR-Trientine (test product) is bioequivalent to Syprine (reference product) in accordance with Health Canada bioequivalence standards.29 Bioequivalence implies that the test product can be expected to have the same therapeutic effects and safety profile as the reference product when administered to patients under the conditions specified in the labelling.29 In their decision to approve MAR-Trientine, Health Canada considered that the results of Study 16-VIN-0315 supported that MAR-Trientine is representative of Syprine, which is authorized for use in the US and has been used in studies reported in the literature.26,30 Moreover, the clinical experts on the review team confirmed that Syprine (obtained via the Health Canada SAP) was used in Canada before termination of the SAP. Thus, Study 16-VIN-0315 provides evidence that MAR-Trientine is bioequivalent with a reference product that has been used in clinical practice in Canada for the treatment of Wilson disease.
It is evident that there is very low-quality evidence to support the efficacy of trientine for the treatment of Wilson disease. Although the literature search failed to identify any relevant studies for inclusion in this review, it did identify 2 systematic reviews of common therapies for Wilson disease.31,32 The first was a systematic review and meta-analysis that included 2 studies of trientine (i.e., the Weiss et al. [2013] study and a study by Członkowska et al. [2005] that compared trientine and tetrathiomolybdate, which is not approved in Canada).13,33 The studies were deemed to be of low quality and to have insufficient evidence to perform a meta-analysis; however, it was noted that the studies reported no difference in effectiveness of primary outcomes, although neurologic deterioration occurred more frequently with trientine as compared to DPA or tetrathiomolybdate, and the relative risk for AEs was lower under trientine therapy.31 The second systematic review sought to evaluate the clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease; however, no relevant studies of trientine were identified for inclusion.32 Despite the limited evidence, the use of trientine in Wilson disease must be considered in the context of the long market availability of trientine worldwide (i.e., since the 1960s) and the history of trientine use in Canada via the Health Canada SAP. As evidence of clinical efficacy of trientine, the Health Canada approval of MAR-Trientine relied on the published literature (most notably the Weiss et al. [2013] study) and market experience.30 An additional consideration is that the clinical pharmacology of trientine is relatively simple and its mechanism of action represents a rational approach for treatment of a disease caused by excess copper accumulation as it is a copper chelating agent that forms a stable copper complex that is readily eliminated by the kidneys. Moreover, evidence of its pharmacologic effect is directly measurable by urinary copper excretion.
There are numerous evidence gaps pertaining to the use of trientine for Wilson disease. No Canadian patients were included in the identified studies, no data were available for most of the efficacy outcomes identified in the review protocol (including those of interest to patients), and no studies were identified in pediatric patients despite the Health Canada approval of trientine for patients 5 years of age and older. A recent retrospective cohort analysis of 182 children with Wilson disease (mean age = 10.7 ± 4.2 years) included in a national Wilson disease registry in France from 1995 to 2019 reported that most children (84.6%) had hepatic presentation at diagnosis, whereas 10.4% had neurologic manifestations, and 4.9% were asymptomatic.14 Diagnosis and treatment of Wilson disease in children can be challenging as they often do not display the same clinical and laboratory hallmarks as adults, especially neurologic manifestations. Most children in the registry (72%) received DPA as first-line therapy followed by zinc (13%) and trientine (9%). Overall survival after 20 years of follow-up was 98%, supporting the notion that diagnosis at early stages of liver disease and proper treatment of Wilson disease result in excellent outcomes. There were also no data available for the use of trientine plus zinc, which is a frequent combination used in specific clinical conditions as described in the Clinician Input section of this report. These and other factors may impact the generalizability of the results of the Weiss et al. (2013) study to Canadian patients with Wilson disease, as detailed in Table 8. Despite the limitations, these evidence gaps must be considered in the context (as reiterated by the clinical experts) that currently Canadian patients with Wilson disease who fail or cannot tolerate DPA have no alternative chelator option. If these patients do not receive de-coppering treatment, they could die as their disease is ultimately fatal.
Harms
There are limited harms data available for trientine in patients with Wilson disease as the only source of harms data identified in patients was the Weiss et al. (2013) study. In this study, the only harms outcome reported was the proportions of trientine or DPA treatments that were discontinued due to AEs. Treatment discontinuations due to AEs were more common with DPA treatments (94 out of 326 [28.8%]) than with trientine treatments (36 out of 141 [7.1%]) and the difference was statistically significant (P = 0.039), as reported in the publication Clinical Gastroenterology and Hepatology.13 Although treatment discontinuations due to any cause were also higher with DPA treatments (142 out of 326 [43.6%]) than with trientine treatments (36 out of 141 [25.5%]), the difference was not statistically significantly different (P = 0.36).13 The main reasons for discontinuations due to AEs were arthralgia, increase in antinuclear antibodies, and albuminuria/proteinuria, which were all numerically higher with DPA treatments than with trientine treatments. The harms results may have been biased due to the disproportionate number of DPA treatments (N = 326) compared to trientine treatments (N = 141) and by most DPA treatments (294 out of 326 [90.2%]) being used as first-line treatment. DPA is known to be poorly tolerated and since most patients in the study received DPA, there was likely a higher probability that DPA treatment would result in more treatment discontinuations due to AEs than with trientine. Nonetheless, the clinical experts on the review team advised that the poor tolerability of DPA is well known and that between 20% to 40% of patients cannot take DPA due to toxicity or intolerance.
Although trientine is considered to be generally well tolerated, with the most common initial AE being self-limiting nausea in addition to occasional occurrences of skin rash and anemia, Health Canada considered the worsening of neurologic symptoms following the initiation of trientine in patients with pre-existing neurologic and/or psychiatric symptoms in their assessment of the safety of MAR-Trientine.30 Although this observation could be attributed in part to lack of efficacy, patients who have sustained damage to the blood-brain barrier from long-term exposure to elevated copper may be especially vulnerable to worsening neurologic effects of transient elevation of serum copper when it is liberated from central stores after the initiation of chelation therapy, and such deterioration may be irreversible.30 For this reason, MAR-Trientine is recommended only to be initiated by physicians experienced in the management of Wilson disease and a serious Warnings and Precautions section added to the MAR-Trientine product monograph to highlight this potential serious adverse reaction.30
Conclusions
A retrospective cohort analysis of mainly adult patients with Wilson disease demonstrated that trientine has an efficacy comparable with DPA on improving hepatic and neurologic outcomes when used as first-line therapy, and as second-line therapy in patients who failed or were intolerant to DPA. First-line treatment with trientine was also associated with statistically significantly higher rates of neurologic worsening than DPA, but not when used as second-line treatment. More DPA treatments were discontinued due to AEs than trientine treatments, which was statistically significant. Due to the low quality of this study, there is considerable uncertainty in the relative estimates of efficacy and harms between trientine and DPA. Despite the limitations, this study comprises the largest body of evidence to date for use of trientine in Wilson disease. Although the evidence is very limited, this must be placed in the context of the long market history of trientine worldwide and experience gained in Canadian patients who received trientine via the SAP. The mechanism of action of trientine also represents a rational approach for treatment of a disease caused by excess copper accumulation. Despite the many limitations associated with the evidence, Canadian patients with Wilson disease who fail or cannot tolerate DPA currently have no alternative chelator option other than trientine, and left untreated, Wilson disease is associated with high morbidity and mortality.
References
1.Weiss K. Wilson Disease. In: Adam MP, Ardinger HH, Pagon RA, Et al., eds. GeneReviews Seattle (WA): University of Washington, Seattle; 1993.
2.Stremmel W, Weiskirchen R. Therapeutic strategies in Wilson disease: pathophysiology and mode of action. Annals of Translational Medicine. 2021;9(8):732. PubMed
3.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47(6):2089-2111. PubMed
4.El-Youssef M. Wilson disease. Mayo Clin Proc. 2003;78(9):1126-1136. PubMed
5.Sturm E, Piersma FE, Tanner MS, Socha P, Roberts EA, Shneider BL. Controversies and variation in diagnosing and treating children with Wilson disease: results of an international survey. J Pediatr Gastroenterol Nutr. 2016;63(1):82-87. PubMed
6.Socha P, Janczyk W, Dhawan A, et al. Wilson's Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018;66(2):334-344. PubMed
7.Reilly M, Daly L, Hutchinson M. An epidemiological study of Wilson's disease in the Republic of Ireland. J Neurol Neurosurg Psychiatry. 1993;56(3):298-300. PubMed
8.Bachmann H, Lossner J, Kuhn HJ, et al. Occurrence, genetics, and epidemiology of Wilson's disease in East Germany. In: Czlonkowska, A, van der Hamer CJA, editors. Proc 5th. Intern. Symposium on Wilson's disease. . Delft (NL): Technical University; 1991:121-128.
9.Litwin T, Dziezyc K, Czlonkowska A. Wilson disease-treatment perspectives. Annals of Translational Medicine. 2019;7(Suppl 2):S68. PubMed
10.EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012;56(3):671-685. PubMed
11.MAR-Trientine (Trientine Hydrochloride): 250 mg capsules USP [product monograph]. Ottawa (ON): Marcan Pharmaceuticals Inc.; 2020 Sep 14.
12.Weiss KH, Stremmel W. Evolving Perspectives in Wilson Disease: Diagnosis, Treatment and Monitoring. Curr Gastroenterol Rep. 2011:1-7. PubMed
13.Weiss KH, Thurik F, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-1035.e1021-1022.
14.Couchonnal E, Lion-François L, Guillaud O, et al. Pediatric Wilson's disease: phenotypic, genetic characterization and outcome of 182 children in France. J Pediatr Gastroenterol Nutr. 2021. PubMed
15.Chu NS, Hung TP. Geographic variations in Wilson's disease. J Neurol Sci. 1993;117(1-2):1-7. PubMed
16.Moores A, Fox S, Lang A, Hirschfield GM. Wilson disease: Canadian perspectives on presentation and outcomes from an adult ambulatory setting. Can J Gastroenterol. 2012;26(6):333-339. PubMed
17.Arnon R, Annunziato R, Schilsky M, et al. Liver transplantation for children with Wilson disease: comparison of outcomes between children and adults. Clin Transplant. 2011;25(1):E52-60. PubMed
18.Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139-142. PubMed
19.Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010;52(6):1948-1956. PubMed
20.Cuprimine (penicillamine) 250 mg capsules USP [product monograph]. Ottawa (ON): Bausch Health Canada Inc.; 2019 Dec 12.
21.Weiss KH, Gotthardt DN, Klemm D, et al. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140(4):1189-1198. PubMed
22.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
23.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 May 4.
24.EuroWilson. European Reference Network for Wilson’s disease. http://www.eurowilson.org/en/about/project-summary/index.phtml. Accessed 2021 May 26.
25.Drug Reimbursement Review sponsor submission: MAR-trientine (trientine hydrochloride), 250 mg capsules [internal sponsor's package]. Ottawa (ON): Marcan Pharmaceuticals Inc.; 2021 May 4.
26.Health Canada reviewer's report: Mar-Trientine (trientine hydrochloride) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Mar-Trientine (trientine hydrochloride) 250 mg capsules[internal sponsor's package]. Ottawa (ON): Marcan Pharmaceuticals; 2021 May 4. City (PROV).
27.Clinical Study Report: 16-VIN-0315. An open label, balanced, randomized, single-dose, two-treatment, two-sequence, two-period, crossover oral bioequivalence study of Trientine Hydrochloride Capsules USP 250 mg of Emcure Pharmaceuticals Ltd., India and SYPRINE® (Trientine Hydrochloride) Capsules 250 mg of Valeant Pharmaceuticals North America LLC Bridgewater, NJ 08807 USA in healthy, adult, human subjects under fasting condition. [internal sponsor's report]. Ahmedabad (IN): Emcure Pharmaceuticals Ltd., India; 2017 Mar 21; 2017.
28.Stürmer T, Wang T, Golightly YM, Keil A, Lund JL, Jonsson Funk M. Methodological considerations when analysing and interpreting real-world data. Rheumatology (Oxford). 2020;59(1):14-25. PubMed
29.Comparative Bioavailability Standards: Formulations Used for Systemic Effects [Health Canada Guidance Document]. Ottawa (ON): Health Canada: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/bioavailability-bioequivalence/comparative-bioavailability-standards-formulations-used-systemic-effects.html. Accessed 2021 Jun 15.
30.Health Canada. Summary Basis of Decision. Mar-Trientine. Date: November 3, 2020. https://hpr-rps.hres.ca/reg-content/summary-basis-decision-detailTwo.php?lang=en&linkID=SBD00505. Accessed June 28, 2021.
31.Appenzeller-Herzog C, Mathes T, Heeres MLS, Weiss KH, Houwen RHJ, Ewald H. Comparative effectiveness of common therapies for Wilson disease: A systematic review and meta-analysis of controlled studies. Liver International. 2019;39(11):2136-2152. PubMed
32.Wiggelinkhuizen M, Tilanus ME, Bollen CW, Houwen RH. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther. 2009;29(9):947-958. PubMed
33.Członkowska A, Tarnacka B, Litwin T, Gajda J, Rodo M. Wilson's disease-cause of mortality in 164 patients during 1992-2003 observation period. J Neurol. 2005;252(6):698-703. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 17, 2021
Alerts: Weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: 1996-present
Humans
Language limit: English- and French-language
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for 1 character |
? | Truncation symbol for 1 or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase); |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
Database(s):
Embase 1974 to 2021 May 14
Ovid MEDLINE(R) ALL 1946 to May 14, 2021
Table 10: Multi-Database Search Strategy
# | Searches | Results |
|---|---|---|
1 | trientine/ | 2,132 |
2 | (trientine* or Cuprior* or Cufence* or Clovique* or Syprine* or SJ76Y07H5F or 7360ure56Q or hc3nx54582).ti,ab,rn,ot,nm,kf. | 2,135 |
3 | (AI3-24384 or Araldite* or "BRN 0605448" or BRN0605448 or CCRIS 6279 or CCRIS6279 or DEH 24 or DEH24 or EINECS 203-950-6 or EC 203-950-6 or HSDB 1002 or HSDB1002 or HY 951 or HY951 or NSC 443 or NSC443 or Tecza* or Trientina* or Trientinum* or Triethylene or cuprid or laszarin* or mk681 or mk 681 or mk0681 or "mk 0681").ti,ab,rn,ot,nm,kf. | 5,124 |
4 | 1 or 2 or 3 | 7,290 |
5 | 4 use medall | 3,560 |
6 | Hepatolenticular Degeneration/ | 12,036 |
7 | (hepatolenticular* or hepatocerebral* or neurohepat* or (wilson* adj3 (disease* or syndrome* or degenerat* or morbus)) or (westphall adj2 strumpell) or pseudoscleros* or pseudo-scleros* or (copper adj3 (storage or remov* or deplet* or chelat*)) or progressive lenticul*).ti,ab,kf. | 24,385 |
8 | 6 or 7 | 26,932 |
9 | 8 use medall | 12,111 |
10 | 5 and 9 | 350 |
11 | *trientine/ | 752 |
12 | (trientine* or Cuprior* or Cufence* or Clovique* or Syprine*).ti,ab,dq,kw. | 710 |
13 | (AI3-24384 or Araldite* or "BRN 0605448" or BRN0605448 or CCRIS 6279 or CCRIS6279 or DEH 24 or DEH24 or EINECS 203-950-6 or EC 203-950-6 or HSDB 1002 or HSDB1002 or HY 951 or HY951 or NSC 443 or NSC443 or Tecza* or Trien or Trientina* or Trientinum* or Triethylene* or cuprid or laszarin* or mk681 or mk 681 or mk0681 or "mk 0681").ti,ab,dq,kw. | 9,879 |
14 | 11 or 12 or 13 | 10,699 |
15 | 14 use oemezd | 5,970 |
16 | Wilson disease/ | 16,162 |
17 | (hepatolenticular* or hepatocerebral* or neurohepat* or (wilson* adj3 (disease* or syndrome* or degenerat* or morbus)) or (westphall adj2 strumpell) or pseudoscleros* or pseudo-scleros* or (copper adj3 (storage or remov* or deplet* or chelat*)) or progressive lenticul*).ti,ab,kw. | 24,480 |
18 | 16 or 17 | 27,956 |
19 | 18 use oemezd | 15,990 |
20 | 15 and 19 | 631 |
21 | conference abstract.pt. | 4,087,731 |
22 | conference review.pt. | 12,940 |
23 | 21 or 22 | 4,100,671 |
24 | 20 not 23 | 474 |
25 | 10 or 24 | 824 |
26 | remove duplicates from 25 | 542 |
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
Search -- Studies with results | [trientine AND Wilson’s Disease]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms -- | [trientine AND Wilson’s Disease]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms -- sleep apnea*, | [trientine AND Wilson’s Disease]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms -- sleep apnea*, | [trientine AND Wilson’s Disease]
Grey Literature
Search dates: May 4 – May11th, 2021
Keywords: trientine, Wilson’s Disease
Limits: No limits used.
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CHC
chronic hepatitis C
DPA
d-penicillamine
HCV
hepatitis C virus
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
SAP
Special Access Program
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Trientine hydrochloride (MAR-Trientine), oral capsules |
Submitted price | Trientine hydrochloride 250 mg: $20.00 per capsule |
Indication | For the treatment of patients with Wilson disease who are intolerant to penicillamine |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 14, 2020 |
Reimbursement request | As per indication |
Sponsor | Marcan Pharmaceuticals Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree |
Target population | Patients with Wilson disease who are intolerant to DPA |
Treatment | 75 mg daily oral zinc for 6 months, followed by trientine (1,000 mg daily) for patients who did not achieve stable hepatic symptoms on zinc |
Comparator | 75 mg daily oral zinc for 6 months, followed by no treatment for patients who did not achieve stable hepatic symptoms on zinc |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (41 years) |
Key data source | Retrospective cohort studies |
Submitted results | For zinc followed by trientine compared to zinc followed by no treatment: ICER = $46,160 per QALY ($68,000 = incremental costs; 1.47 = incremental QALYs) |
Key limitations |
|
CADTH reanalysis results |
|
DPA = d-penicillamine; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Conclusions
The clinical evidence regarding the use of trientine for the treatment of Wilson disease is associated with limitations due to the lack of randomized controlled trials or large observational studies. Despite this, trientine has been used for decades internationally and in Canadian patients who have received it through the Health Canada Special Access Program (SAP). For patients who fail or cannot tolerate d-penicillamine (DPA), there are currently no alternative chelation treatments other than trientine. According to experts consulted for this review, untreated Wilson disease will generally progress to irreversible and likely fatal hepatic and/or neurologic harm.
Due to the absence of evidence, CADTH was unable to determine a reliable base-case analysis. As an exploratory analysis, CADTH removed zinc from the treatment paradigm to align with the indicated Health Canada population, lowered the age of patients entering the model, reduced the proportion of patients whose worsening hepatic symptoms progress to acute liver failure, and increased the proportion of patients in liver failure who receive a liver transplant. In this exploratory analysis, the incremental cost-effectiveness ratio (ICER) associated with the use of trientine in patients with Wilson disease who do not respond to or cannot tolerate DPA was $87,676 per quality-adjusted life-year (QALY) at the submitted price of $20 per 250 mg capsule. At this ICER, a 27% price reduction would be required to achieve an ICER below $50,000 per QALY. Crucially, this analysis assumes that in the absence of trientine, there are no viable treatment alternatives available to patients who currently fail on DPA. As patients are left untreated, the majority will experience acute liver failure resulting in significant QALY losses, even under the most conservative scenarios explored by CADTH. Therefore, if patients are currently successfully managed on a non-chelation agent, such as zinc, this analysis offers no insight on the cost-effectiveness of switching patients to trientine instead. Annual treatment costs with trientine are high, up to $58,400 per patient, and lifelong due to the chronic nature of the condition. This makes an assessment of cost-effectiveness uncertain and heavily reliant on expert opinion due to the absence of reliable clinical evidence.
CADTH was unable to adjust for other limitations, including the paucity of clinical and economic evidence, the exclusion of neurologic symptoms in the model, the assumption of 100% adherence in a condition with a young population and difficult-to-tolerate treatment regimens, and the lack of disutilities assigned to patients experiencing acute liver failure and liver transplants.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from patient groups, registered clinician groups, and drug plans that participated in the CADTH review process. Feedback from clinician groups was not received for this submission.
Feedback was received from the Canadian Liver Foundation. It was collected through an online questionnaire with 8 patients and 5 caregivers responding, with additional information received from 2 health care providers. Wilson disease was associated with emotional and psychological effects impacting the ability to exercise, work, travel, complete household activities, socialize, and fulfill family obligations. Additionally, feelings of constant stress, fear, and psychiatric symptoms such as bipolar disorder, anxiety, and depression were described. Patients reported experience with zinc, d-penicillamine, and trientine. Respondents reported that fatigue, appetite loss, nausea, and pain were side effects that were completely to somewhat intolerable, while fever, dizziness, forgetfulness, and stomach irritation were described as being somewhat to very tolerable. Respondents noted these side effects were significant enough to impact their quality of life, and additional stress was associated with accessing medication, which reportedly could take months and denial of coverage could cause financial hardship. Two patients and 2 caregivers reported having experience with trientine, accessed through private insurance after a challenging process. All respondents reported trientine was either effective or very effective at managing Wilson disease and the side effects they experienced were mainly stomach irritation, fatigue, and minor pain. Patients also noted that unlike previous brands of trientine accessed through the SAP, MAR-Trientine does not require refrigeration, making storage and everyday use easier, and allowing patients and caregivers to travel more easily. One of the health care professionals emphasized the need for quick and affordable access to treatment for Wilson disease, reporting that 1 of their patients was denied access to trientine, developed cirrhosis, and is currently awaiting a liver transplant. The other indicated that without reimbursement, trientine remains out of reach for patients with Wilson disease.
Drug plans identified the following concerns related to the implementation of trientine: the inclusion of zinc as second-line therapy, the potential for physicians to want to access trientine as first-line therapy without a trial of DPA, the lack of evidence in patients under 5 years of age, a desire to see the price of MAR-Trientine compared to the confidential price of trientine previously accessed through the SAP, and the fact that the budget impact analysis (BIA) likely underestimates the number of eligible patients in Canada.
Several of these concerns were addressed in the sponsor’s model, namely:
the effect of treatment on hepatic outcomes and quality of life
the relative tolerability of trientine.
In addition, CADTH addressed some of these concerns as follows:
the removal of zinc from the modelled treatment pathway
increasing the number of patients eligible for trientine therapy in the BIA.
CADTH was unable to address the following concerns raised from stakeholder input:
the effect of trientine on psychological or neurologic outcomes in the assessment of cost-effectiveness
comparing the cost of trientine at the currently submitted price to that previously paid through the SAP.
Economic Review
The current review is for trientine hydrochloride (MAR-Trientine) for the treatment of patients with Wilson disease who are intolerant to penicillamine.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis in which zinc therapy followed by 250 mg trientine capsules for nonresponding patients was compared to zinc therapy followed by no treatment in patients (aged 42 years) with Wilson disease who are intolerant to d-penicillamine.1 This treatment sequence differs from the Health Canada indication, which specifies that trientine is indicated for the treatment of patients who are intolerant to DPA but does not suggest that zinc be used in between DPA and trientine.
The recommended starting dose of trientine for adult patients is 750 mg given in divided doses 2 times to 4 times daily. The daily dose may be gradually increased to a maximum of 2,000 mg daily, if required. The starting dose for patients aged 5 years to 17 years is 500 mg per day in 2 daily doses to 4 daily doses, with a maximum recommended dose of 1,500 mg per day for patients aged 12 or under. At the submitted price of $20 per 250 mg capsule, the annual cost of therapy with trientine ranges from $21,900 to $58,400 for adult patients, $14,600 to $58,400 for adolescents aged 13 years to 17 years, and $14,600 to $43,800 for children aged 5 years to 12 years.
For the base case, the sponsor estimated costs and QALYs for each treatment regimen from the perspective of a Canadian health care payer over a lifetime time horizon, using a 1.5% annual discount rate for both costs and QALYs.
Model Structure
The sponsor submitted a decision tree model where all patients enter the model after having become intolerant to DPA (see Figure 1 in Appendix 3). Patients then receive 6 months of zinc therapy, after which they may either be “responders,” defined as having stable hepatic symptoms, or be intolerant or nonresponsive to zinc. Responders continue on zinc therapy for the remainder of their lives, while nonresponders either receive 6 months of trientine or no treatment. Patients responding to trientine continue on trientine for the remainder of their lives. Those who do not respond to, are intolerant to, or who did not have the option of receiving trientine receive no further treatment. These patients may then live out their lives with worsened hepatic symptoms, or experience liver failure after 2 years and either die or receive a liver transplant, which then may be successful or fatal. Patients who have a successful liver transplant live out the remainder of their lives without further worsening of Wilson disease symptoms.
Model Inputs
Patients entered the model at 42 years of age, reported as being from a study by Weiss et al. in (2019),2 with 37.6% of patients being male, according to Weiss et al. (2013).3 Patients took a mean dose of 1,000 mg of trientine daily. Trientine efficacy in terms of the proportion of patients who do not experience hepatic worsening in the model is taken from the Wiess et al. (2013) study,3 a retrospective analysis where 4 out of 103 patients who received second-line trientine after DPA experienced a decline of liver function or progression of chronic liver disease. The efficacy of zinc was derived from the number of patients who discontinued zinc therapy after at least 6 months due to hepatic treatment failure in the assessment by Weiss and Stremmel (2011),4 which was also a retrospective analysis. Of patients who experience hepatic worsening and discontinue all therapy, 75% experience acute liver failure 2 years after discontinuation, based on clinical expert opinion elicited by the sponsor, with the remaining 25% having worsened symptoms for the remainder of their lives, but never experiencing liver failure. Of patients experiencing acute liver failure, 75% were assumed to receive a liver transplant, with the remaining 25% dying, based on a sample size of 4 patients from Wiess et al. (2013). Of note, the Weiss et al. (2013) study does not report any deaths. Finally, patients who receive a transplant are assumed to have a 90% chance of it being successful, based on a 2018 database study of 64,977 American patients undergoing a first-time liver transplant.5 Patients who achieved stable hepatic symptoms on treatment were assumed to have the life expectancy of the general population,6 weighted by gender (with a lifespan of 83 years), as were those who had worsened hepatic symptoms without liver failure. Patients who underwent a successful liver transplant were assumed to have their life expectancy reduced by 7 years, based on the estimated mean loss of life in a 2006 UK database study of adults who survived at least 6 months after a liver transplant.7
A health-related utility of 0.838 was assigned by the sponsor for patients with stable Wilson disease (i.e., those with stable hepatic symptoms), reportedly based on visual analogue scale scores from Weiss et al. (2019).2 Patients whose hepatic symptoms had worsened were assigned a health-related utility of 0.730 based on Wong et al. (2017),9 while those who received a successful transplant had a utility of 0.750. A continuing adverse event–related disutility of 0.0028 and 0.0100 was applied to patients using trientine and zinc, respectively, based on the proportion and severity of adverse events experienced while on treatment as reported in the published literature, including arthralgia, nausea or abdominal discomfort, skin rash, and fatigue.1 No disutility was applied for patients experiencing liver failure or undergoing a liver transplant.
Costs included the drug acquisition costs of trientine, while zinc, being an over-the-counter supplement, was assumed to not have a cost paid by publicly funded drug plans. Annual monitoring of stable Wilson disease was assigned $832 in annual costs and included 2 gastroenterology visits per year, urine and serum copper tests once yearly, urinalysis, a liver panel, a complete blood count, a liver ultrasound and an endoscopy twice per year, and a neurologic MRI scan every 2 years, based on expert consultation elicited by the sponsor and Schilsky (2017).1,8 Patients experiencing hepatic worsening had an additional $2,591 in annual monitoring costs, inflated from a 2017 study by Wong et al.,9 while patients in acute liver failure had a 1-time medical cost of $39,573 applied, those undergoing liver transplant had a 1-time cost of $135,575 applied, and those who had a successful liver transplant had a continuing annual cost of $27,597 applied based on inflating the 2017 costs estimated for patients suffering acute liver failure or transplant in an Ontario economic study of hepatitis A vaccinations.10
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted a probabilistic analysis of 2,000 iterations. The results of the deterministic analysis were very similar to the probabilistic analysis. The probabilistic findings are reported in the following sections.
Base-Case Results
When used after a trial of oral zinc therapy in patients with Wilson disease who are intolerant to DPA, when compared to a trial of zinc therapy followed by no treatment, the sponsor concluded that trientine was associated with $68,000 in increased costs, yielding an additional 1.47 QALYs, for an ICER of $46,160 per QALY (see Table 3). More details can be found in Table 10.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. zinc alone ($/QALY) |
|---|---|---|---|---|---|
Zinc alone | 96,114 | Reference | 24.16 | Reference | Reference |
Zinc followed by trientine | 164,115 | 68,000 | 25.63 | 1.47 | 46,160 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s Pharmacoeconomic Submission, Table 15.
Sensitivity and Scenario Analysis Results
The sponsor conducted a series of scenario analyses, including lowering the age at model entry to 20 years, varying the time taken to develop acute liver failure, varying the proportion of patients who develop acute live failure, and varying the mean daily dose of trientine to 750 mg or 1,250 mg. Of these, changing the proportion of patients experiencing liver failure and the mean daily dose of trientine had the largest impact on the ICER, with the ICER decreasing with higher risks of liver failure and increasing with higher daily doses. The sponsor also conducted an analysis from a societal perspective, which lowered the ICER.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
No treatment is likely not the current standard of care: The sponsor modelled the cost-effectiveness of trientine therapy compared to no treatment; however, the clinical experts consulted by CADTH indicated that patients with Wilson disease would never be left untreated. Patients who were intolerant to DPA or trientine would at least receive zinc. Both DPA and trientine may be reattempted at lower doses and with more adherence support, or compassionate access to tetrathiomolybdate,11 which is still considered experimental and not marketed in Canada, might be sought. Ideally, the cost-effectiveness of trientine compared to zinc therapy would be modelled. As zinc can be effective for some patients and would not incur drug plan costs due to being an over-the-counter product, the ICER for trientine when compared to zinc would be higher than that reported when trientine is compared to no treatment. However, only non-randomized data exists regarding the efficacy and tolerability of zinc therapy for Wilson disease. The sponsor submission assumes that 84% of patients who receive zinc respond and continue this therapy for the remainder of their life with stable hepatic symptoms.12 However, this evidence is based on a study that retrospectively analyzes the hepatic response rate for patients who were deemed appropriate for receiving zinc therapy. This includes patients who were asymptomatic or only had a neurologic presentation. This population is therefore not reflective of all patients with Wilson disease who are intolerant to DPA and the estimate of efficacy is not useful to the decision problem being analyzed in this submission.
CADTH was unable to adjust for this limitation in reanalysis. The ICER associated with the comparison of trientine to zinc therapy would be higher than that reported when trientine is compared to no treatment, but the magnitude of this difference is unknown. If a subset of patients who respond to zinc could be reliably identified, then it would not be cost-effective to use trientine on these patients, as suggested by the sponsor.
Clinical evidence is limited: The efficacy of trientine used in the model is derived from a retrospective cohort analysis of mainly adult patients with Wilson disease, where lack of response or tolerance to trientine was defined by the proportion of patients in the study who experienced hepatic worsening while on trientine as second-line therapy after previously using DPA (4 out of 103 patients [3.9%]).3 This study comprises the largest body of evidence for the use of trientine in Wilson disease. While clinical evidence is extremely limited and randomized studies do not exist, trientine has been used worldwide for patients with Wilson disease since the 1960s. According to the clinical experts consulted by CADTH, patients who are able to tolerate chelation therapy — either DPA or trientine — benefit. It is unlikely that high-quality clinical evidence in the form of a randomized controlled trial of trientine will ever be conducted due to ethical considerations, as untreated Wilson disease is typically fatal or requires a liver transplant.
CADTH was unable to compensate for this limitation in reanalysis. CADTH noted that changing the probability of hepatic worsening with trientine did not have a substantial impact on the model’s results as the model assumes that if a patient is intolerant to trientine, or it is ineffective, then they would only remain on therapy for 6 months. As the model assumes substantial health gains in the patients who do benefit, this compensates for the 6-month cost in the patients who do not benefit.
Proportion of patients with acute liver failure and transplant uncertain: The main benefit of trientine in the sponsor’s model is the offsetting of the costs and health consequences associated with acute liver failure and the subsequent need for liver transplant. The sponsor assumes that by preventing hepatic worsening, this will prevent acute liver failure from occurring. The experts consulted by CADTH agreed that the successful treatment of Wilson disease prevents such hepatic consequences; however, the magnitude of the risk of acute liver failure and the probability of receiving a transplant was perceived as highly uncertain. The sponsor assumed that 75% of patients with worsening hepatic symptoms would progress to acute liver failure 2 years after discontinuing therapy, based only on expert opinion. Additionally, the 75% probability of receiving a liver transplant was based on 3 patients on trientine in the Weiss et al. (2013) study discontinuing due to receiving a liver transplant. The assumption was that these 3 were part of the 4 whose hepatic symptoms worsened while on trientine. In contrast, while not specific to patients with Wilson disease, the Canadian Institute for Health Information reported that 464 liver transplants were performed in 2017, while a total of 64 patients died while waiting for a liver transplant (12%).15 The other 25% of patients in liver failure who did not receive a transplant in the model were assumed to have died, although no deaths were reported over the 48 months of follow-up in the Weiss et al. (2013) study.3
CADTH reanalyses explored the impact of assuming a more conservative assumption of 50% of patients with unstable Wilson disease progressing to acute liver failure, and a higher probability of those in acute liver failure receiving a transplant (88%). Scenario analyses were also conducted on the CADTH combined exploratory reanalysis assuming the sponsor’s input of 75% of unstable patients progressing to acute liver failure.
Modelled population is not consistent with the Health Canada indication or reimbursement request: The sponsor’s model assumes that all patients failing DPA due to intolerance or lack of efficacy would begin a 6-month trial of zinc. This treatment paradigm models trientine as a third-line therapy for Wilson disease rather than a second-line therapy following DPA as indicated in the product monograph and the reimbursement request. The clinical experts consulted by CADTH did not agree that the approximately 30% of patients who cannot tolerate DPA should be trialled on zinc before switching to trientine. According to the experts consulted by CADTH, patients with Wilson disease require chelation therapy (i.e., DPA or trientine) except in cases where the disease is asymptomatic or where the copper burden is already low. Additionally, the efficacy of trientine in the model is derived from patients studied in Weiss et al. (2013)3 who received trientine after DPA; these patients did not receive 6 months’ worth of zinc in between chelating therapies and, thus, efficacy of the treatment paradigm proposed by the sponsor is unknown.
CADTH removed the 6-month trial with zinc therapy from the model in exploratory reanalyses. This increased the incremental costs associated with trientine as more patients received it, but also increased incremental QALYs and the offsetting savings associated with fewer cases of acute liver failure and liver transplants.
Model does not account for neurologic symptoms: The sponsor’s model only considers hepatic symptoms associated with Wilson disease, assigning costs and quality of life based on hepatic health states and outcomes. However, the accumulation of copper associated with unstable Wilson disease often results in neurologic and psychiatric symptoms. Approximately 24% of patients in the Weiss et al. (2013) study had an initial presentation of neurologic symptoms, with a further 20% presenting with both hepatic and neurologic symptoms.3 The clinical experts consulted by CADTH advised that, untreated, the neurologic and psychological manifestations of Wilson disease will progress to irreversible impairment, having potentially profound impacts on both quality of life as well as on medical costs associated with psychiatric and long-term care.
CADTH was unable to adjust for this limitation in reanalyses. If the model was capable of representing the effects of trientine compared to no treatment on neurologic outcomes, the ICER associated with trientine could be lower than reported in current analyses given the evidence that, like hepatic outcomes, neurologic outcomes could be better for patients receiving trientine.3
Model structure does not reflect current practice: The sponsor’s model is in the form of a decision tree that does not reflect the complexity of the treatment or the natural history of Wilson disease. In the model, patients start treatment with trientine and either respond and tolerate it or are unresponsive or intolerant to it. Patients then continue treatment for the remainder of their lives or discontinue treatment forever and are left with lifelong hepatic consequences. All patients are fully adherent and gain the full benefit of treatment. In practice, the clinical experts consulted by CADTH indicated that treatment is likely to be iterative, with some patients fully adherent but others discontinuing treatment and then restarting once symptoms appear or worsen. Additionally, some responsive patients with minor symptoms may successfully be transitioned to zinc for maintenance therapy after chelation.13 Given the young age at which most patients with Wilson disease are diagnosed, often in adolescence or early adulthood, and the side effects and difficult-to-maintain regimens and dietary restrictions associated with treatment, nonadherence is particularly likely.13,14 Additionally, the clinical experts consulted by CADTH indicated that patients with Wilson disease would never be left untreated. A Markov model capable of representing time spent on and off treatments and the increased risks associated with a lack of adherence may have been more reflective of real-world experience. However, as Wilson disease is a rare condition, the evidence necessary to inform such a model is lacking.
CADTH was unable to adjust for this limitation in reanalysis. The incorporation of adherence may make trientine less cost-effective as, for a period, patients continue to pick up prescriptions — incurring a cost to the health service — but stop receiving benefits.
Impacts on costs and utilities do not change over time: In the model, if a patient receives a liver transplant, their costs are higher by $27,597 per year for the remainder of the patient’s life. This incremental cost likely includes costs related to liver transplant as well as general health care costs. General health care costs outside of monitoring are not included in the model for patients in other health states, so the incremental cost of post–liver transplant may be overestimated. Additionally, as the patient gets older, the incremental costs associated with having received a liver transplant decades prior may also decrease. The sponsor also assumes that utility values remain static over time when they will likely decrease as patients become older.
CADTH was unable to address the limitation of static utilities and therefore notes the model likely overestimates QALYs in both treatment groups. As there is a mortality benefit for patients who receive trientine, this will have a larger impact on that arm of the model.
The sponsor may have overestimated the cost savings associated with the avoidance of a liver transplant. A scenario analysis was conducted that reduced annual post–liver transplant costs by 50% to assess the impact of this value.
Health state utility values are uncertain: For the stable Wilson disease health state utility (0.838), the sponsor used the average of the 6-month and 12-month utilities measured by visual analogue scale reported in the 2019 study by Weiss et al. of patients who were using trientine after discontinuing DPA. As health utility data were not available specific to Wilson disease for patients with worsening hepatic symptoms or those who had undergone a liver transplant, utilities from a cost-effectiveness study in Canadian chronic hepatitis C (CHC) patients were used as a proxy, from patients with non-cirrhotic CHC (0.730) and post–liver transplant (0.750), respectively. In the absence of direct evidence in a Wilson disease population, extrapolating from another disease was necessary. CADTH noted other values used in the literature for chronic hepatitis C virus (HCV) from Saeed (2020)16; however, this study demonstrates the variability of utility estimates derived for HCV, which may be due to confounders associated with an HCV diagnosis.
CADTH performed a scenario analysis that used the highest and lowest utility estimates for non-cirrhotic CHC and post transplant as proxies for worsening hepatic symptoms and post transplant (Wilson disease), respectively.
Modelled population was too old: The sponsor’s model assumed a mean patient age of 42 years when entering the model, based on the mean baseline age of patients in a 2019 prospective study by Weiss et al.2 on the safety and efficacy of trientine in Wilson disease in patients withdrawn from DPA. However, most patients are diagnosed with Wilson disease at between 5 years and 35 years of age, often in adolescence, and the clinical experts consulted by CADTH did not consider it reasonable to assume it would take 7 years to 37 years to determine whether a patient would be able to tolerate treatment with DPA. The mean age at diagnosis reported in Weiss et al. (2013)3 was 17 years or 19 years, depending on the initial treatment group. Given that some delay is necessary to account for at least 1 trial of DPA therapy, CADTH believed the sponsor’s scenario analysis considering a mean starting age of 20 to be a more appropriate assumption. Given the variation in age of diagnosis seen in clinical practice, CADTH also considered it appropriate to model the starting age probabilistically.
CADTH assumed a mean starting age of 20 years in reanalyses, with a gamma distribution based on a standard error of 4 years.
Quality of life not considered for liver failure and transplant: The sponsor did not assign a disutility to modelled patients in acute liver failure or those undergoing a liver transplant, despite the inherent impact that liver failure and transplant have been shown to have on quality of life in other conditions.16 If such disutilities were included, the resulting ICER associated with trientine would be lower than estimated as it reduces the risk of liver failure and transplant.
CADTH was unable to add disutilities for the short-term impact on quality of life associated with acute liver failure or a liver transplant due to structural limitations of the model.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (see Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Successful liver transplants resolve Wilson disease symptoms | Acceptable. While neurologic effects of uncontrolled Wilson disease would not be reversed, a successful liver transplant would prevent further accumulation of copper and, thus, hepatic symptoms should resolve and further neurologic damage would not occur. |
Gender proportions based on patients who received trientine therapy in Weiss et al. (2013)a | Acceptable. The sponsor based the gender proportion of patients in the model on the proportion of women who received trientine therapy in Weiss et al. (2013).a While the generalizability of this proportion to the Canadian population of patients with Wilson disease is uncertain, the assumption does not have significant impact on model results. |
Utility decrements associated with AEs were assigned by treatment group | Inappropriate. Disutilities associated with adverse events are more appropriately modelled by event. However, this assumption does not have significant impact on model results. |
aWeiss et al. (2013).3
CADTH Reanalyses of the Economic Evaluation
Exploratory Results
Due to limitations in the body of clinical and economic evidence regarding the use of trientine for the treatment of Wilson disease, as well as limitations in the model structure, CADTH could not determine a base-case reanalysis. Instead, CADTH conducted a series of reanalyses exploring areas of uncertainty in the sponsor’s model and combined them into a merged exploratory reanalysis.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH exploratory reanalyses | ||
1. Trial with zinc removed | Zinc therapy is trialled for 6 months, and only patients who are intolerant or nonresponsive to zinc may receive trientine. | No patients respond to zinc, and the trial period is reduced to 0. Patients receiving trientine start it immediately, and hepatic consequences are moved up 6 months. |
2. Starting age of patient reduced and made probabilistic | Mean age: 42 years Distribution: Fixed | Mean age: 20 Distribution: Gamma, SE = 4 years (resulting range was 9 years to 36 years) |
3. Reduced probability of liver failure | 75% | 50% |
4. Increased probability of surviving to receive a liver transplant | 75% | 88% |
CADTH combined exploratory analysis | 1 + 2 + 3 + 4 | |
SE = standard error.
Due to limitations in the clinical evidence, CADTH was unable to determine a base-case reanalysis. Most of the changes made in exploratory analyses had only minimal impact on the ICER associated with trientine for the treatment of Wilson disease after intolerance or lack of response to DPA, due to the changes in assumption having similar impacts on the incremental costs and QALYs (see Table 6). The exploratory test of the assumption around the proportion of patients who progress to acute liver failure had the largest impact relative to the sponsor’s base case. When this was combined with the other exploratory analyses, it resulted in trientine being associated with $682,754 in incremental costs and 7.79 incremental QALYs, for a possible ICER of $87,676 per QALY gained.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Zinc + no treatment | 96,114 | 24.16 | Reference |
Zinc + trientine | 164,115 | 25.63 | 46,160 | |
CADTH reanalysis 1: Zinc removed | No treatment | 470,645 | 16.13 | Reference |
Trientine | 907,677 | 25.41 | 47,091 | |
CADTH reanalysis 2: Lower and probabilistic age | Zinc + no treatment | 129,535 | 32.30 | Reference |
Zinc + trientine | 218,223 | 34.13 | 48,583 | |
CADTH reanalysis 3: Lower risk of liver failure | Zinc + no treatment | 75,543 | 24.47 | Reference |
Zinc + trientine | 163,558 | 25.58 | 78,885 | |
CADTH reanalysis 4: Lower risk of death before transplant | Zinc + no treatment | 109,134 | 24.43 | Reference |
Zinc + trientine | 164,989 | 25.62 | 46,973 | |
CADTH combined exploratory reanalysis 1 through 4 | No treatment | 526,437 | 26.43 | Reference |
Trientine | 1,209,191 | 34.22 | 87,676 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: A minor error was corrected in the sponsor’s ICER formulas, which led to some scenarios reporting that trientine was “dominated” or “dominant” when it was not. This correction did not alter any model inputs or cost or QALY outputs.
Scenario Analysis Results
Scenario analyses were also conducted using the CADTH combined exploratory analyses to investigate the impact of assuming the risk of liver failure was as high as assumed by the sponsor, as well as reducing cost savings associated with post–liver transplant. Assuming a higher probability of acute liver failure lowered the ICER to just over $50,000 per QALY. Reducing post– liver transplant costs by 50% decreased the cost offsets associated with trientine by $196,349 per patient, resulting in a higher ICER of $112,103 per QALY. Additionally, the model was sensitive to the use of the highest and lowest utility values reported for non-cirrhotic and post-transplant health states in Saeed (2020) for patients with CHC, which were used as proxies for worsening hepatic symptoms and the post-transplant state, respectively, in patients with Wilson disease. When the highest utilities were used, the ICER increased to $120,514 per QALY. When the lowest utility values were used, the ICER decreased to $62,442 per QALY.
Price reduction analyses were conducted on the sponsor’s base case and the CADTH combined exploratory analysis. The sponsor’s base case was already cost-effective at a willingness-to-pay value of $50,000 per additional QALY, while a reduction of approximately 27% would increase the probability that trientine would be cost-effective under the CADTH exploratory assumptions.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for trientine vs. no treatment | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 46,160 | 87,676 |
10% | 37,533 | 73,903 |
20% | 28,120 | 59,902 |
30% | 18,939 | 44,136 |
ICER = incremental cost-effectiveness ratio; vs. = versus.
Issues for Consideration
Prior to the approval of MAR-Trientine in Canada on September 14, 2020, trientine was not available in Canada and patients requiring treatment with trientine accessed it through the Health Canada SAP. Patient group input indicated that this was burdensome and caused delays in access. Additionally, since the approval of MAR-Trientine, access has no longer been available through the SAP, leading patients to report further administrative burden in accessing private insurance, as well as delays and/or denials of coverage. As trientine was previously reimbursed through the SAP, a comparison of the $20 per 250 mg capsule cost in this submission to the confidential price previously being paid through the SAP may be appropriate.
A second trientine product, Waymade-Trientine, was recently approved by Health Canada17 and will be reviewed by CADTH in 2021.18 While not yet marketed, the relative price of Waymade-Trientine when it becomes available compared to that of MAR-Trientine may influence the cost-effectiveness of MAR-Trientine.
Overall Conclusions
The clinical evidence regarding the use of trientine for the treatment of Wilson disease is associated with limitations due to the lack of randomized controlled trials or large observational studies. Despite this, trientine has been used for decades internationally and in Canadian patients who have received it through the SAP. For patients who fail or cannot tolerate d-penicillamine, there are currently no alternative chelation treatments other than trientine. According to experts consulted for this review, untreated Wilson disease will generally progress to irreversible and likely fatal hepatic and/or neurologic harm.
Due to the absence of evidence, CADTH was unable to determine a reliable base-case analysis. As an exploratory analysis, CADTH removed zinc from the treatment paradigm to align with the indicated Health Canada population, lowered the age of patients entering the model, reduced the proportion of patients whose worsening hepatic symptoms progress to acute liver failure, and increased the proportion of patients in liver failure who receive a liver transplant. In this exploratory analysis, the ICER associated with the use of trientine in patients with Wilson disease who do not respond to or cannot tolerate DPA was $87,676 per QALY at the submitted price of $20 per 250 mg capsule. At this ICER, a 27% price reduction would be required to achieve an ICER below $50,000 per QALY. Crucially, this analysis assumes that in the absence of trientine, there are no viable treatment alternatives available to patients who currently fail on DPA. As patients are left untreated, the majority will experience acute liver failure, resulting in significant QALY losses, even under the most conservative scenarios explored by CADTH. Annual treatment costs with trientine are high, up to $58,400 per patient, and lifelong due to the chronic nature of the condition. This makes an assessment of cost-effectiveness uncertain and heavily reliant on expert opinion due to the absence of reliable clinical evidence.
CADTH was unable to adjust for other limitations, including the paucity of clinical and economic evidence, the exclusion of neurologic symptoms in the model, the assumption of 100% adherence in a condition with a young population and difficult-to-tolerate treatment regimens, and the lack of disutilities assigned to patients experiencing acute liver failure and liver transplants. The exclusion of these considerations has an uncertain impact on the overall ICER. Some exclusions would make trientine more cost-effective (the consideration of neurologic benefit), while others would make trientine less cost-effective (adherence). It is important to note that the evidence to support these additions would be reliant on assumptions and expert opinion.
CADTH explored uncertainties related to the probability of acute liver failure, utilities associated with hepatic worsening, and cost savings associated with liver transplants. The ICER ranged from $51,496 to $120,514 per QALY in these analyses. This further highlights the uncertainty and the reliance on expert opinion to inform cost-effectiveness.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: MAR-trientine (trientine hydrochloride), 250 mg capsules [internal sponsor's package]. Ottawa (ON): Marcan Pharmaceuticals Inc.; 2021 May 4. 2021.
2.Weiss KH, Pfeiffenberger J, Mohr I, et al. FRI-445-Safety and efficacy of trientine treatment in Wilson disease in patients withdrawn from d-Penicillamine: Final results from a prospective study. J Hepatol. 2019;70(1):e591-e592. PubMed
3.Weiss KH, Thurik F, Gotthardt DN, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-1035.e1021-1022.
4.Weiss KH, Stremmel W. Evolving Perspectives in Wilson Disease: Diagnosis, Treatment and Monitoring. Curr Gastroenterol Rep. 2011:1-7. PubMed
5.Baganate F, Beal EW, Tumin D, et al. Early mortality after liver transplantation: Defining the course and the cause. Surgery. 2018;164(4):694-704. PubMed
6.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 1800 Jan 1.
7.Barber K, Blackwell J, Collett D, Neuberger J, Group UKTLA. Life expectancy of adult liver allograft recipients in the UK. Gut. 2007;56(2):279-282. PubMed
8.Schilsky ML. Wilson Disease: Diagnosis, Treatment, and Follow-up. Clin Liver Dis. 2017;21(4):755-767. PubMed
9.Wong WW, Lee KM, Singh S, Wells G, Feld JJ, Krahn M. Drug therapies for chronic hepatitis C infection: a cost-effectiveness analysis. CMAJ Open. 2017;5(1):E97-E108. PubMed
10.Ramsay LC, Anyiwe K, Li M, Macdonald L, Coyte PC, Sander B. Economic evaluation of a publicly funded hepatitis A travel vaccination program in Ontario, Canada. Vaccine. 2019;37(11):1467-1475. PubMed
11.Weiss KH, Askari FK, Czlonkowska A, et al. Bis-choline tetrathiomolybdate in patients with Wilson's disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol. 2017;2(12):869-876. PubMed
12.Weiss KH, Gotthardt DN, Klemm D, et al. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140(4):1189-1198. PubMed
13.Roberts EA, Schilsky ML, American Association for Study of Liver D. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-2111. PubMed
14.European Association for Study of L. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012;56(3):671-685. PubMed
15.CIHI. Annual Statistics on Organ Replacement in Canada. Dialysis, Transplantation and Donation, 2008 to 20172018: https://secure.cihi.ca/free_products/snapshot-corr-2018-en.pdf?bcs-agent-scanner=313cd325-78a3-1045-ae7d-2b27ba98601f. Accessed 07 Jul 2021.
16.Saeed YA, Phoon A, Bielecki JM, et al. A Systematic Review and Meta-Analysis of Health Utilities in Patients With Chronic Hepatitis C. Value Health. 2020;23(1):127-137. PubMed
17.WAYMADE-Trientine (Trientine Hydrochloride): 250 mg capsules USP [product monograph]. Montreal, QC: Waymade Canada Inc.; 2021 Apr 19: https://pdf.hres.ca/dpd_pm/00060826.PDF. Accessed 2021 Jul 05.
18.CADTH. trientine hydrochloride. 2021; https://www.cadth.ca/trientine-hydrochloride-0. Accessed 2021 Jul 05.
19.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Jun 10.
20.Wong WWL, Haines A, Bremner KE, et al. Health care costs associated with chronic hepatitis C virus infection in Ontario, Canada: a retrospective cohort study. CMAJ Open. 2021;9(1):E167-E174. PubMed
21.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug name, doses and administration. City (PROV): Sponsor name; 1800 Jan 1.
22.Canadian Institute for Health Information. Prescribed Drug Spending In Canada, 2020: A Focus on Public Drug Programs. Ottawa, ON: CIHI; 2020: https://www.cihi.ca/sites/default/files/document/prescribed-drug-spending-in-canada-2020-report-en.pdf.
23.Choe EJ, Choi JW, Kang M, et al. A population-based epidemiology of Wilson's disease in South Korea between 2010 and 2016. Sci Rep. 2020;10(1):14041. PubMed
24.Sutherland GD, T. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. The Conference Board of Canada,; 2017: https://www.conferenceboard.ca/temp/758ed58f-2652-4178-9596-52557e8c3793/9326_Understanding-the-Gap__RPT.pdf?bcs-agent-scanner=079fbd59-37ed-dd44-9cdb-85b3ca35a1c0.
25.Table 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000) - using the M1 medium growth scenario. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 28-Jun-2021.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Wilson Disease
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Annual costa |
|---|---|---|---|---|---|---|
Trientine (MAR-Trientine) | 250 mg | Capsules | $20.0000 | Adult: 750 mg in 2 to 4 divided doses, may increase to a maximum of 2,000 mg if required Pediatric (5 to 17): 500 mg in 2 to 4 divided doses, may increase to a maximum of 1,500 mg for patients 12 and under if required.b | Adult: $60.00 to $160.00 Adolescent (13 to 17): $40.00 to $160.00 Child (5 to 12): $40.00 to $120.00 | Adult: $21,900 to $58,400 Adolescent (13 to 17): $14,600 to $58,400 Child (5 to 12): $14,600 to $43,800 |
Other Chelator | ||||||
D-penicillamine (Cuprimine) | 250 mg | Capsules | 3.9649 | Optimal dosage determined by measurement of copper excretion and the determination of free copper in the serum. A dose between 0.75 and 1.5g. It is seldom necessary to exceed 2g per day.b | $11.89 to $31.72 | $4,342 to $11,578 |
Zinc supplementation | ||||||
Zinc gluconate (over-the-counter brands) | 10 mg 25 mg 50 mg Elemental zinc | Tablets | 0.0465c 0.0875c 0.0509c | 50 mg 2 to 3 times dailyd | $0.18 to $0.26 | $64 to $96 |
aSponsor’s submitted price.1
bDosing as per product monographs. According the 2008 American Association for the Study of Liver Diseases Practice Guideline Diagnosis and Treatment of Wilson Disease, both d-penicillamine and trientine are dosed by weight, with a maximum dose of 20 mg/kg, reducing by 25% when clinically stable.13
cIQVIA Delta PA AQPP prices (accessed June 2021).19 Zinc gluconate tablets are over-the-counter products and not reimbursed by public plans.
dMinimum and usual dose as outlined in the 2008 American Association for the Study of Liver Diseases Practice Guideline.13
Note: All prices are from the Ontario Drug Benefit Formulary (accessed June 2021), unless otherwise indicated, and do not include dispensing fees.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The population modelled is third-line therapy after intolerance to d-penicillamine and zinc, rather than second-line therapy after intolerance to d-penicillamine as indicated. Patients are not likely to ever be left untreated in clinical practice. |
Model has been adequately programmed and has sufficient face validity | Yes | The economic model is transparently laid out. In the absence of evidence face validity is uncertain. |
Model structure is adequate for decision problem | No | The model is insufficiently structured to adequately reflect the complexity of the condition and treatment paradigm. A more complex model, such as a Markov model, would allow the impact of adherence and treatment cycling to be explored. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The model lacks the ability to assess uncertainty related to long-term treatment in particular issues with adherence. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Zinc alone | Zinc + trientine | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total life-years | 29.48 | 30.86 | 1.38 |
Stable Wilson disease | 26.00 | 30.65 | 4.65 |
Hepatic worsening | 1.51 | 0.14 | –1.37 |
Liver transplant | 1.97 | 0.08 | –1.90 |
Discounted QALYs | |||
Total QALYs | 24.16 | 25.63 | 1.47 |
Stable Wilson disease | 21.84 | 25.75 | 3.91 |
Hepatic worsening | 1.10 | 0.10 | –1.00 |
Liver transplant | 1.48 | 0.06 | –1.42 |
QALY loss due to AEs | 0.26 | 0.27 | 0.01 |
Total QALYs within trial period | 9.77 | 10.21 | 0.45 |
Total QALYs after trial period | 14.39 | 15.42 | 1.03 |
Discounted costs ($) | |||
Total costs | 96,114 | 164,115 | 68,000 |
Treatment acquisition costs | 0 | 135,702 | 135,702 |
Hepatic complications costs | 70,715 | 2,706 | –68,009 |
Monitoring costs | 25,344 | 25,649 | 305 |
AE costs | 55 | 57 | 3 |
ICER ($/LY) | 49,163 | ||
ICER ($/QALY) | 46,160 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Combined Exploratory Reanalysis
Table 11: Disaggregated Summary of CADTH’s Exploratory Economic Evaluation Results
Parameter | No treatment | Trientine | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total life-years | 35.82 | 40.99 | 5.17 |
Stable Wilson disease | 0.00 | 39.58 | 39.58 |
Hepatic worsening | 21.61 | 0.86 | –20.75 |
Liver transplant | 14.21 | 0.55 | –13.67 |
Discounted QALYs | |||
Total QALYs | 26.43 | 34.22 | 7.79 |
Stable Wilson disease | 0.00 | 33.29 | 33.29 |
Hepatic worsening | 15.75 | 0.62 | –15.21 |
Liver transplant | 10.68 | 0.41 | –10.27 |
QALY loss due to AEs | 0.00 | 0.11 | 0.11 |
Total QALYs within trial period | 8.22 | 10.26 | 2.04 |
Total QALYs after trial period | 18.21 | 23.96 | 5.75 |
Discounted costs ($) | |||
Total costs | 526,437 | 1,209,191 | 682,754 |
Treatment acquisition costs | 0 | 1,155,774 | 1,155,774 |
Hepatic complications costs | 470,434 | 18,209 | –452,226 |
Monitoring costs | 55,948 | 35,136 | –20,812 |
AE costs | 55 | 72 | 18 |
ICER ($/LY) | 132,166 | ||
ICER ($/QALY) | 87,676 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
As the CADTH combined exploratory analysis remains uncertain, a series of scenario analyses were conducted. Results of these analyses can be found in Table 12.
A) The risk of worsening hepatic symptoms progressing to liver failure is increased back to the sponsor’s assumption of 75%.
B) Post–liver transplant costs were reduced by 50% to account for potential waning over time and analyze the impact this variable has on the results.
C) The highest utilities reported by Saeed et al. (2020)16 for mild/moderate CHC (0.829) and post–liver transplant (0.75) health states were used as proxies for the worsening hepatic conditions and post–liver transplant health states in Wilson disease.
D) The lowest utilities reported by Saeed et al. (2020)16 for mild/moderate CHC (0.69) and post–liver transplant (0.57) health states were used as proxies for the worsening hepatic conditions and post–liver transplant health states in Wilson disease.
Table 12: Summary of CADTH Scenario Analysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Zinc + no treatment | 96,114 | 24.16 | Reference |
Zinc + trientine | 164,115 | 25.63 | 46,160 | |
CADTH Combined Exploratory Reanalysis | No treatment | 526,231 | 26.42 | Reference |
Trientine | 1,210,796 | 34.13 | 88,832 | |
CADTH Scenario A: Risk of acute liver failure in unstable WD is 75% | No treatment | 735,743 | 24.65 | Reference |
Trientine | 1,214,686 | 33.95 | 51,496 | |
CADTH Scenario B: reduced post-transplant costs by 50% | No treatment | 330,088 | 26.41 | Reference |
Trientine | 1,205,981 | 34.22 | 112,103 | |
CADTH Scenario C: higher alternate utilities | No treatment | 526,110 | 28.49 | Reference |
Trientine | 1,207,201 | 34.14 | 120,514 | |
CADTH Scenario D: lower alternate utilities | No treatment | 524,029 | 22.94 | Reference |
Trientine | 1,207,931 | 33.90 | 62,442 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; WD = Wilson disease.
Note: A minor error was corrected in the sponsor’s ICER formulas which led to some scenarios reporting that trientine was “dominated” or “dominant” when it was not. This correction did not alter any model inputs or cost or QALY outputs.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the sponsor-submitted BIA,21 the sponsor assessed the reimbursement of trientine for patients with Wilson disease who are intolerant to DPA compared to no treatment. The BIA was conducted from a Canadian public drug payer perspective over a 3-year time horizon using a claims-based approach and included only drug acquisition costs.
Data for the model were obtained from various sources including: internal communication between the sponsor and the SAP,21 the Canadian Institute for Health Information,22 and expert opinion.
Key inputs to the BIA are documented in Table 14.
State the key assumptions:
The number of patients who receive trientine will increase 20% per year from those who received it under the SAP.
Trientine will only displace no treatment rather than an active therapy.
Patients with Wilson disease are eligible for public coverage in the same proportions as the general population.
Downstream therapies such as those associated with liver transplants will not be impacted by the reimbursement of trientine.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Number of patients receiving trientine under SAP Number of patients receiving trientine under SAP outside Quebec Annual increase in patients using trientine from SAP Public coverage Patients aged < 65 years Percentage aged < 65 years covered by public plans Patients aged ≥ 65 years Percentage aged ≥ 65 years covered by public plans | 50 39 20%a 97.8%b 27.4%c 2.2%b 90.4%c |
Number of patients eligible for drug under review | 13/16/19 |
Market uptake (3 years) | |
Uptake (reference scenario) No treatment | 0%/0%/0% |
Uptake (new drug scenario) No treatment Trientine | 0%/0%/0% 100%/100%/100% |
Cost of treatment (per patient) | |
Cost of treatment over 1 month Trientine | $2,434.95 |
SAP = Special Access Program.
aAssumption based on clinical expert opinion.
bAppears to be based on the proportion of prevalent patients aged 60 and older in a recent Wilson disease epidemiological study from South Korea.23
cActive beneficiaries as a percentage of population, Figure B1, Canadian Institute for Health Information 2020.22
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s base-case BIA suggest that the yearly incremental expenditures associated with the reimbursement of trientine, including dispensing fees and markup, for patients intolerant to DPA were expected to be $456,482 in Year 1, $547,778 in Year 2, and $657,334 in Year 3, for a 3-year cumulative total of $1,661,595. When dispensing fees and markups are excluded, the sponsor’s model reports an incremental budget impact of $399,349 in Year 1, $479,219 in Year 2, and $575,062 in Year 3, for a 3-year cumulative total of $1,453,630. The sponsor conducted scenario analyses varying the proportion of patients younger than 65 years of age, the mean daily dose of trientine, and the annual growth rate in the number of patients on trientine by 25%. All scenarios had 3-year cumulative totals between $1.2 million and $2.6 million.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Eligible population underestimated: The sponsor’s model uses the number of patients in Canada (39 outside of Quebec) who were receiving MAR-Trientine through the SAP in the 2 months preceding the discontinuation of the SAP program in November of 2020 as the number of patients who would be eligible for trientine therapy in the base year. To acknowledge that the number of patients accessing trientine is likely to grow with its greater availability, the sponsor applied a 20% growth rate over the 3 years of the BIA. However, Wilson disease is estimated to affect 1 in 30,000 people,14 and 30% of Wilson disease patients have been estimated to be unable to tolerate DPA.13,14 Based on drug plan input, it was noted that more patients were on trientine than reported by the sponsor.
CADTH used an epidemiological approach to estimate the number of eligible patients in Canada based on prevalence of Wilson disease (1 in 30,000) and intolerance to DPA (30%). CADTH also assumed that not all prevalent patients experiencing intolerance to DPA would immediately receive trientine in its first year of reimbursement, and instead assumed a lag in access such that 50% of otherwise eligible patients would receive trientine in Year 1, 80% in Year 2, and 100% in Year 3.
Funding previously spent in SAP program not accounted for: Prior to November 2020, trientine was previously available to Canadian patients though the SAP program. The sponsor’s model does not account for funds previously used to acquire trientine before market authorization.
CADTH was unable to account for this limitation in reanalyses. The pan-Canadian (excl. Quebec) budget impact associated with reimbursing trientine through the public formularies would be lower than projected by the amount that was previously being spent to acquire trientine for patients accessing it through the SAP program.
Downstream medications were not considered: Treatment with trientine reduces the number of patients with Wilson disease who require a liver transplant, see the aforementioned Economic Evaluation section. Additionally, unstable Wilson disease is also associated with neurologic and psychiatric consequences. Due to this, the costs associated with reimbursing trientine are expected to be partially offset by the reduced number of patients requiring anti-rejection or other medications for hepatic consequences of unstable Wilson disease as well as medications associated with the neurologic and psychiatric worsening.
CADTH was unable to account for this limitation in reanalyses. The budgetary impact of reimbursing trientine would be lower than projected if downstream medications avoided were included.
Number of patients eligible for public funding is underestimated: The sponsor used the proportion of the population in each jurisdiction who are active beneficiaries of the public drug plans, stratified by age over and under 65 years,22 to determine the number of patients using trientine who would be publicly reimbursed. However, this likely underestimates the number of patients who would require public funding due to the high cost of treatment with trientine. As such, the proportion of patients who are reimbursed by the public plans for trientine may be better represented by the proportion of the population over and under age 65 who are eligible for public coverage,24 rather than only those who are currently active beneficiaries.
CADTH considered the proportion of the population who are eligible for public reimbursement in their base-case analysis.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by using an epidemiological approach to estimate the number of patients who would be eligible for treatment with trientine in Canada consistent with the Health Canada indication, and increasing the proportion of patients who are eligible for public reimbursement. Table 15 outlines the parameters used by the sponsor in comparison to those used by CADTH.
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH base case | ||
1. Eligible population | Eligible population in Canada in base year: 50 Annual growth rate: 20% Market share: 100% / 100% / 100% | Eligible population in Canada in base year: 380a Annual population growth rate: 1.06%b Access rate: 50% / 80% / 100% |
2. Percentage of the population covered by public drug plans | Active beneficiaries (28.8%)c | Eligible population (62.7%)d |
CADTH base case | 1 + 2 | |
aEligible population considers the Canadian population excluding Quebec, the prevalence of Wilson disease, and the proportion of patients who cannot tolerate d-penicillamine.
bAnnual growth rate is based on the growth rate projected by Statistics Canada in their M2 projection between 2020 and 2024.25
cCanadian Institute for Health Information (2020).22
dThe Conference Board of Canada (2017).24
Applying these changes increased the total 3-year budget impact of reimbursing trientine for patients with Wilson disease who are intolerant to DPA to $12,958,161 when dispensing fees and markups are excluded. The results of the CADTH step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
Table 16: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total |
|---|---|
Submitted base case | 1,661,595 |
CADTH reanalysis 1 | 5,956,137 |
CADTH reanalysis 2 | 3,612,976 |
CADTH base case | 12,958,161 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to address remaining uncertainty:
Dispensing fees and markups were included.
All patients who are intolerant to DPA can access trientine in the first and second years it is available.
The price of trientine is reduced by 27% as suggested previously in the Economic Evaluation section.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Sponsor’s base case | Budget impact | $0 | $456,482 | $547,778 | $657,334 | $1,661,595 |
CADTH base case | Budget impact | $0 | $2,780,886 | $4,496,691 | $5,680,583 | $12,958,161 |
CADTH scenario 1: dispensing fees and markups included | Budget impact | $0 | $2,936,178 | $4,747,798 | $5,997,801 | $13,681,776 |
CADTH scenario 2: No lag in access | Budget impact | $0 | $5,561,773 | $5,620,864 | $5,680,583 | $16,863,220 |
CADTH scenario 3: 27% price reduction | Budget impact | $0 | $2,030,047 | $3,282,585 | $4,146,826 | $9,459,458 |
BIA = budget impact analysis.
Note: As the reference scenario in all analyses was $0, only the budget impact is presented.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.