CADTH Reimbursement Review
Givosiran (Givlaari)
Sponsor: Alnylam Netherlands B.V.
Therapeutic area: Acute hepatic porphyria in adults
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAR
annualized attack rate
ADP
aminolevulinic acid dehydratase-deficient porphyria
AE
adverse event
AHP
acute hepatic porphyria
AIP
acute intermittent porphyria
ALA
aminolevulinic acid
ALAS1
5'-aminolevulinate synthase 1
ALT
alanine transaminase
ANCOVA
analysis of covariance
APF
American Porphyria Foundation
AUC
area under the curve
BFI
Brief Fatigue Inventory
BFI-SF
Brief Fatigue Inventory – Short Form
BIPNET
British and Irish Porphyria Network
BP
bodily pain
BPI
Brief Pain Inventory
BPI-SF
Brief Pain Inventory – Short Form
CAP
Canadian Association for Porphyria
CHE
chronic high excreter
CI
confidence interval
Cr
creatinine
DB
double blind
EAP
expanded access program
ECOG
Eastern Cooperative Oncology Group
EQ-5D
EuroQol 5-Dimensions questionnaire
EQ-5D-5L
EuroQol 5-Dimensions 5-Levels questionnaire
EQ VAS
EuroQol Visual Analogue Scale
FAS
full analysis set
GnRH
gonadotropin-releasing hormone
GH
general health
HCP
hereditary coproporphyria
HRQoL
health-related quality of life
IMMPACT
Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials
IQR
interquartile range
LS
least squares
MCS
Mental Component Summary
mFAS
modified full analysis set
MH
mental health
MID
minimally important difference
MMRM
mixed-effects model for repeated measures
mRNA
messenger ribonucleic acid
NRS
numeric rating scale
OLE
open-label extension
PBG
porphobilinogen
PCS
Physical Component Summary
PD
pharmacodynamic
PF
physical functioning
PGIC
Patient Global Impression of Change
PI-NRS
pain-intensity numeric rating scale
PK
pharmacokinetic
POMS
Profile of Mood States
PPEQ
Porphyria Patient Experience Questionnaire
RCT
randomized controlled trial
RE
role emotional
RNA
ribonucleic acid
RP
role physical
SAE
serious adverse event
SD
standard deviation
SEM
standard error of the mean
SF
social functioning
SF-12
12-item Short Form Health Survey
ULN
upper limit of normal
VAS
Visual Analogue Scale
VP
variegate porphyria
VT
vitality
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Givosiran (Givlaari), 189 mg/mL, solution for subcutaneous injection |
Indication | For the treatment of acute hepatic porphyria in adults |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 9, 2020 |
Sponsor | Alnylam Netherlands B.V. |
NOC = Notice of Compliance.
Introduction
The porphyrias are a group of metabolic disorders caused by altered activities of enzymes within the heme biosynthetic pathway. The acute hepatic porphyrias (AHPs) include acute intermittent porphyria (AIP), aminolevulinic acid dehydratase-deficient porphyria (ADP), hereditary coproporphyria (HCP), and variegate porphyria (VP). Each type of AHP is characterized by a specific genetic mutation involved in the synthesis of heme in the liver; however, these types are clinically indistinguishable.1 Information about the prevalence and incidence of AHP that is specific to Canadians is not available. The estimated prevalence of AHP in the Canadian provinces (excluding Quebec) is |||| per million population2; however, it was noted by both the sponsor and the clinical experts consulted for this review that patients with AHP are underdiagnosed due to a lack of available treatment options and poor awareness of the disease.
In patients with AHP, the altered enzymatic activity within this pathway results in an excessive accumulation of the intermediate porphyrin precursors, aminolevulinic acid (ALA) and porphobilinogen (PBG), which are neurotoxins that can precipitate an acute attack.1,3 During an acute attack, patients report a significant increase in pain that can gradually build over hours and last for a number of days.3 Some patients report pain in the chest, back, or extremities; however, severe abdominal pain is more typical. Long-term complications with recurrent acute attacks may include chronic pain, chronic kidney failure, and liver damage.
Treatment for AHP is mainly targeted at preventing acute attacks. The British and Irish Porphyria Network (BIPNET) recommends that recurrent acute attacks be managed through general measures and the avoidance of precipitating factors, gonadotropin-releasing hormone (GnRH) analogues, prophylactic heme arginate, and/or liver transplantation.4 The clinical experts consulted for this review indicated that the avoidance of triggers is sufficient for many patients. One of the biggest contributors to an acute attack is a change in progesterone levels during the menstrual cycle. In some cases, GnRH analogues may be used (with expert guidance) to suppress the menstrual cycle; however, the clinical experts consulted for this review reported that long-term use of hormone therapy is typically not an option due to the severity of the side effects. Despite the narrow indication for the treatment of acute attacks, hemin is often considered for prophylactic use outside of the indication in patients who exhibit recurrent attacks with the preventive measures described previously; however, the use of hemin is also associated with complications resulting from the requirement for venous access, iron overload, and difficulty withdrawing from treatment.4 Liver transplantation may be considered in patients with recurrent attacks who are frequently hospitalized and exhibit diminishing health-related quality of life (HRQoL).
The drug under review, givosiran, is a double-stranded, small, interfering RNA that results in a reduction of liver 5'-aminolevulinate synthase 1 (ALAS1) mRNA. In Canada, givosiran is indicated for the treatment of AHP in adults.5 It is available as a solution for subcutaneous injection (189 mg givosiran/mL) and does not require additional reconstitution or dilution before administration. The recommended dose is 2.5 mg/kg once monthly, based on body weight. The objective of this review is to perform a systematic review of the beneficial and harmful effects of givosiran 2.5 mg/kg once monthly for the treatment of AHP in adults.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received 2 patient group submissions for this review from the Canadian Association for Porphyria/Association Canadienne de Porphyrie (CAP) and the American Porphyria Foundation (APF). CAP is a national voluntary charity whose mission is to deliver evidence-based information and support to patients with porphyria, their families, health care providers, and the general public. APF provides programs to raise awareness and educate health care professionals and the general public in 76 countries around the world. Of its international members, more than 300 are located in Canada. To obtain input for this review, CAP distributed a survey to its members in February 2021. The survey was restricted to Canadian patients and caregivers with experience with AHP. In total, 22 patients and 4 caregivers responded to the survey. CAP also requested support from the British Porphyria Association, which shared 3 interviews from individuals who had received givosiran. APF used its social media platforms and online newsletters to connect with Canadian patients about their experiences with porphyria. It also collected responses by telephone and email. Some of the responses in the APF submission were collected during an Alnylam Patient Advisory Board meeting. Twelve individual patient submissions were collected from Canadians.
In both submissions, respondents noted that they had experienced the following symptoms, among others: pain, fatigue, nausea, weakness, paralysis, neuropathy, seizures, anxiety, and depression. More than 80% of patients from the CAP survey had experienced symptoms at least once a month, with many reporting that these symptoms occurred more than 20 days per month. The group also reported that 86% of respondents had at least 1 attack in the past year and 36% had at least 10. Furthermore, 55% of patients had gone to the emergency room at least once in the past year due to an attack, while 18% had gone at least 10 times. Porphyria attacks can prevent patients and caregivers from being able to work, lead to poorer quality of life, and negatively affect relationships. The patient input submissions described how symptoms and efforts to avoid triggers could strain social relationships and make it difficult to care for their families. Both groups emphasized the negative effects that porphyria had on their daily lives and mental health.
Respondents would like a cure for porphyria, but many believe a more realistic goal is to have a treatment that prevents attacks and reduces symptoms, particularly pain, nerve damage, and paralysis. Patients and caregivers would like to see additional options that are more effective, have fewer side effects, offer an easier mode of administration, can be administered outside of a hospital, and will lead to improvements in quality of life. Other limitations to accessing treatments that were identified include the need for travel, the requirement for venous access, and the lack of access to specialists and proper diagnostic testing.
Clinician Input
Input From Clinical Experts Consulted by CADTH
One of the major goals in the management of AHP is to reduce the frequency of AHP attacks. According to the clinical experts consulted for this review, most patients with recurrent attacks will continue to have recurrent attacks with currently available treatment strategies. The experts noted that while prophylactic hemin can be used to reduce the rate of AHP attacks, with case reports of improvement, the use of prophylactic hemin is outside of the Health Canada–approved indication and has not been studied well. GnRH may also be used to prevent AHP attacks, but it is not approved for prolonged use and is associated with side effects and loss of bone mineral density.
As per feedback from the clinical experts consulted for this review, givosiran would be used in patients that have recurrent attacks because there is no evidence to support its use in asymptomatic individuals or acute attacks. The clinical experts felt that givosiran would not be used as a first-line treatment or to treat the first AHP attack. They recommended that patients with AHP try other approaches to treatment, such as avoidance of triggers, before givosiran. The experts expected givosiran to provide an alternative therapy for a small subset of patients with frequent or recurrent attacks who would otherwise require hospitalization and hemin administration. The experts recommended that givosiran be reserved for patients with recurrent symptoms or flares that are consistently affecting their HRQoL. Givosiran was also described by the experts as an appropriate treatment for patients who qualify for hemin prophylaxis but cannot adhere to treatment due to toxicity or lack of convenience.
The following outcomes were noted by the clinical experts as those that are used to determine response to treatment in clinical practice: reduced attack rate, reduced hospitalization, reduced need for hemin, frequency of neurovisceral flares, and improved patient-reported outcomes, such as daily symptoms, HRQoL, and work-life productivity. The clinical experts suggested that patients be assessed for response to treatment every 6 months or annually. All of the experts agreed that 1 year would be a sufficient amount of time to assess a patient’s response to treatment; however, the variable presentation of the disease—as evidenced by yearly fluctuations in attack frequency—was noted as a limitation in this assessment.
In general, the clinical experts felt that patients treated with givosiran would continue with treatment until there was a reason for discontinuation, such as safety concerns or an increase (or lack of improvement) in rate of attacks with treatment, which may indicate that treatment is not working. The clinical experts also indicated that menopause would be a potential reason to trial treatment discontinuation in patients with stable disease. However, it was challenging for the clinicians to specifically define response to treatment due to the heterogeneous nature of AHP among patients. The clinical experts also noted that if attacks recurred following discontinuation, restarting treatment with givosiran would be a possibility.
Clinician Group Input
CADTH did not receive any input from clinician groups for this review.
Drug Program Input
The drug programs inquired about the requirements for diagnosis of types of AHP, the use of givosiran outside of the criteria used in Study 003 (see Description of Studies in the next section), discontinuation of therapy, the use of givosiran in combination with hemin for an acute attack, and generalizability issues for non-AIP types of AHP. The clinical experts noted that the biochemical tests for urinary ALA and PBG are specific to AHP and, along with clinical evidence consistent with porphyria attacks, are sufficient to make a diagnosis; genetic tests are not required. The clinical experts indicated that treatment decisions would be made on a case-by-case basis using clinical judgment, but would generally be guided by the criteria outlined in the pivotal trial. The clinical experts did not express concern about the use of givosiran in combination with hemin. The results of the trial in patients with AIP were considered generalizable to all patients with AHP.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
One multi-centre, placebo-controlled, double-blind (DB), phase III study (Study 003) was included in the CADTH systematic review. Study 003 was designed to evaluate the efficacy and safety of givosiran administered once monthly in patients with AHP. Included patients had to be at least 12 years old with a documented diagnosis of AIP, HCP, VP, or ADP, had experienced at least 2 composite porphyria attacks within 6 months before screening, and had to be willing to abstain from prophylactic use of hemin during the trial. The primary objective was to evaluate the effect of subcutaneous givosiran compared to placebo in terms of the rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home over 6 months in patients with AIP. The annualized rate of porphyria attacks in patients with AHP and the following assessments in patients with AIP were included as secondary outcomes: urinary ALA and PBG levels, hemin use, daily worst scores for symptoms (including pain, fatigue, and nausea), and HRQoL as measured using the 12-item Short Form Health Survey (SF-12). Opioid use, the Porphyria Patient Experience Questionnaire (PPEQ), and the ability to work or attend school, as well as the secondary end points analyzed in patients with AHP, were included as exploratory outcomes. Study 003 implemented a statistical hierarchy to control for multiple testing, where the first outcome to be tested was the annualized attack rate (AAR) in patients with AIP over the 6-month DB period followed by the following outcomes (conducted in patients with AIP unless indicated otherwise): urinary ALA levels at 3 months; urinary ALA levels at 6 months; urinary PBG levels at 6 months; annualized rate of administered hemin doses over the 6-month DB period; AAR in patients with AHP over the 6-month DB period; daily worst pain score; fatigue score; nausea score over the 6-month DB period; and, change from baseline in the Physical Component Summary (PCS) of the SF-12 at 6 months.
A total of 94 patients were randomized in Study 003, 89 (95%) of whom had AIP. Patients with AIP were between the ages of 19 years and 65 years (mean = 37.3 years to 40.7 years); 89% to 91% were female; and 35% to 40% resided in North America. Between 40% and 44% of patients had prior experience with prophylactic hemin, and based on the composite definition of porphyria attacks, the median historical AAR was 8 attacks (range = 4 to 34) and 8 attacks (range = 0 to 46) in the givosiran and placebo treatment groups, respectively. While not having a porphyria attack, 48% to 56% of patients reported having chronic symptoms, and 28% to 30% reported chronic opioid use. Baseline characteristics in patients with AHP were similar to those reported for patients with AIP.
Efficacy Results
A summary of key results from the pivotal trial is provided in Table 2. The description of results provided here will focus on analyses conducted in the modified full analysis set (mFAS) for patients with AIP. Results based on the full analysis set (FAS) in patients with all types of AHP will only be described if there is a notable difference from the results based on the mFAS.
The primary end point of the pivotal trial was the annualized rate of porphyria attacks in patients with AIP over the 6-month DB period, where porphyria attacks were defined as events requiring hospitalization, urgent health care visits, or IV hemin administration at home. The mean AAR based on the composite end point was 3.22 (95% confidence interval [CI], 2.25 to 4.59) and 12.52 (95% CI, 9.35 to 16.76) for patients in the givosiran treatment group and placebo treatment group, respectively. This corresponded to a 74% reduction in the rate of porphyria attacks for patients in the givosiran group relative to patients receiving placebo (rate ratio = 0.26; 95% CI, 0.16 to 0.41; P < 0.001). The number of attacks for each component of the primary outcome was also reported. Treatment with givosiran corresponded to a 49% rate reduction in attacks that required hospitalization [rate ratio = 0.51 (95% CI, 0.25 to 1.04)], and an 84% rate reduction in attacks requiring an urgent health care visit (rate ratio = 0.16; 95% CI, 0.09 to 0.31). A total of 3 attacks required IV hemin administration at home for patients in the givosiran group, and 32 attacks required IV hemin administration at home for patients in the placebo group (rate ratio was not assessed due to n < 10 in the givosiran group).
HRQoL was evaluated using the SF-12, the EuroQol 5-Dimensions 5-Levels questionnaire (EQ-5D-5L), and the Patient Global Impression of Change (PGIC). Each of these HRQoL outcomes is widely used in clinical trials; however, evidence of validity, reliability, and responsiveness, or a minimally important difference (MID) in patients with AHP, were not identified. All of the HRQoL outcomes were reported as exploratory except for the PCS of the SF-12, which was a secondary outcome in Study 003. At month 6, the least squares (LS) mean change from baseline in the PCS score was 5.37 (standard error of the mean [SEM] = 1.17) for the givosiran treatment group and 1.43 (SEM = 1.22) for the placebo treatment group. The between-groups difference in the LS mean PCS score for givosiran compared to placebo was 3.94 (95% CI, 0.59 to 7.29; P = 0.0216). Due to a failure higher in the statistical testing hierarchy, the reported P value cannot be interpreted as statistically significant. The results of the change from baseline in the domains scores for the SF-12 suggest that the PCS score was driven by the bodily pain (BP) and role physical (RP) domains. The Mental Component Summary (MCS) score of the SF-12 was reported descriptively. At month 6, the mean changes from baseline in MCS score were 3.55 (standard deviation [SD] = 10.08) and 1.30 (SD = 8.54) for patients receiving givosiran and placebo, respectively. For the EQ-5D-5L index, the LS mean changes from baseline at month 6 were |||||||||||||||||||||||||||||||||| and |||||||||| |||||||||||||||||| for the givosiran and placebo treatment groups, respectively. For the EQ-5D-5L Visual Analogue Scale (VAS), the LS mean change from baseline at month 6 was |||||||||||||||||||||||||||| and |||||||||||||||||||||||||||||| for the givosiran and placebo treatment groups, respectively. At month 6, the percentages of patients who reported that their status had improved from the start of the study through the PGIC were 88.9% and 37.1% for those in the givosiran and placebo treatment groups, respectively.
In terms of management of symptoms related to porphyria, the change in self-reported assessments of pain, fatigue, and nausea based on a numeric rating scale (NRS) were reported in Study 003. Post hoc non-parametric tests were used to evaluate daily worst pain following demonstration of a deviation from normality and failed statistical test using the analysis of covariance (ANCOVA) model. The medians of the area under the curve (AUC) for the change from baseline in the weekly mean score for daily worst pain over the 6-month treatment period were –11.5 (interquartile range [IQR] = –29.2 to 3.0) and 5.3 (IQR = –23.1 to 11.2) for the givosiran and placebo treatment groups, respectively. This indicated a decrease in the rating of daily worst pain for patients receiving givosiran and an increase for those receiving placebo. The treatment-group difference for rating of daily worst pain was –10.1 (95% CI, –22.8 to 0.9; P = 0.0455) for givosiran compared to placebo. At month 6, the changes from baseline in daily worst fatigue and daily worst nausea were also evaluated; a difference between treatment groups was not observed.
In Study 003, hemin was permitted only as a rescue medication for the treatment of acute porphyria attacks and was reported as days of hemin use. In patients with AIP, 54% of those in the givosiran treatment group and 23% of those in the placebo treatment group reported 0 days of hemin use over the 6-month treatment period. When compared to placebo, treatment with givosiran corresponded to a 77% rate reduction in days of hemin use based on a rate ratio of 0.23 (95% CI, 0.11 to 0.45; P < 0.001). Reported hemin use is consistent with the reduction in AAR reported for the primary outcome. The results for urinary levels of ALA and PBG were also consistent with the primary outcome. At month 6, urinary levels of ALA and PBG were lower among patients receiving givosiran than among those receiving placebo. This corresponded to a between-group difference of –19.14 mmol/mol creatinine (Cr) (95% CI, –26.04 to –12.24; P < 0.001) for ALA levels and –36.20 mmol/mol Cr (95% CI, – 49.71 to – 22.70; P < 0.001) for urinary PBG levels, both in favour of givosiran.
Opioid use, the PPEQ, and days of missed work or school were also reported as exploratory efficacy outcomes in Study 003. Reduced complications of AHP, hospitalization and health care use, and mortality were included in the systematic review protocol, but were not reported in the pivotal trial. However, attacks requiring hospitalization and health care use were incorporated in the composite definition of acute porphyria attacks, and mortality was reported as a safety outcome.
The primary and key secondary outcomes — AAR and change in urinary ALA levels — were analyzed by subgroups. The only subgroup analysis of interest to this review was by high or low historical AAR. The subgroup analyses were consistent with the results in the overall population. Additionally, a number of sensitivity analyses were conducted to account for variation in the primary end point based on reporting of porphyria attacks, which were all consistent with the primary analysis.
Harms Results
A summary of key safety results is provided in Table 2. In Study 003, 85% of patients with AIP experienced at least 1 adverse event (AE), with nausea, injection-site reaction, chronic kidney disease, fatigue, increase in alanine transaminase (ALT), and decrease in glomerular filtration rate more commonly reported among patients who received givosiran. Serious adverse events (SAEs) were reported more frequently among patients in the givosiran group (17%) than in patients in the placebo group (9%). Specific SAEs were infrequent, with the only SAEs reported by more than 1 person being chronic kidney disease (2 patients in the givosiran treatment group, 0 of those receiving placebo) and device-related infection (2 patients in the placebo treatment group, 1 of those receiving givosiran). A single patient randomized to receive givosiran withdrew from treatment due to an AE. The patient |||||||||||| discontinued treatment due to ALT elevation. No deaths were reported during the 6-month DB period of Study 003.
Motor neuropathy, hepatocellular carcinoma, injection-site reactions, transaminase elevation, and progression of renal impairment were included in the CADTH systematic review protocol as notable harms. As previously described, injection-site reactions and transaminase elevation were more common among patients receiving givosiran. Nerve compression and peripheral neuropathy were reported for motor neuropathy and were more common in the placebo group. There were no cases of hepatocellular carcinoma reported during the 6-month treatment period; however, 6 months may have not been a sufficient amount of time to observe this safety outcome.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Result | Patients with AIP (mFAS) | Patients with AHP (FAS) | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Annualized rate of porphyria attacka | ||||
Total number of attacks | 83 | 284 | 90 | 297 |
Mean AAR (95% CI) | 3.22 (2.25 to 4.59) | 12.52 (9.35 to 16.76) | 3.35 (2.37 to 4.74) | 12.26 (9.22 to 16.29) |
Rate ratio (95% CI), givosiran vs. placebo | 0.26 (0.16 to 0.41) | 0.27 (0.17 to 0.43) | ||
P value | < 0.001 | < 0.001 | ||
PCS of SF-12b | ||||
Number of patients contributing to the analysis | 45 | 42 | 47 | 45 |
Baseline, mean (SD) | 39.43 (9.61) | 38.42 (9.45) | 39.47 (9.83) | 38.10 (9.82) |
Change from baseline, mean (SEM) | 5.37 (1.17) | 1.43 (1.22) | 5.15 (1.16) | 1.46 (1.19) |
Treatment-group difference vs. control (95% CI) | 3.94 (0.59 to 7.29) | 3.69 (0.41 to 6.96) | ||
P value | 0.0216f | 0.0280f | ||
BPI-SF: Pain numerical rating score, daily worst pain scorec | ||||
n | 46 | 43 | 48 | 46 |
Baseline weekly mean score, mean (SD) | 2.93 (2.34) | 3.64 (2.23) | 2.97 (2.30) | 3.74 (2.23) |
AUC of change from baseline in weekly mean score | ||||
Median (IQR) | –11.51 (–29.18 to 3.04) | 5.29 (–23.05 to 11.15) | –7.80 (–28.329 to 3.583) | 2.31 (–19.505 to 10.512) |
Median of treatment difference (95% CI), givosiran – placebo | –10.07 (–22.83 to 0.94) | –9.39 (–21.02 to 1.22) | ||
P value | 0.0455f | 0.0613f | ||
Annualized days of hemin used | ||||
Total number of days of hemin use | 195 | 583 | 227 | 587 |
Patients with 0 days of hemin use, n (%) | 25 (54.3) | 10 (23.3) | 26 (54.2) | 12 (26.1) |
Mean annualized days of hemin use (95% CI) | 6.8 (4.2 to 10.9) | 29.7 (18.4 to 47.9) | 7.4 (4.5 to 12.0) | 28.4 (17.4 to 46.2) |
Rate ratio of annualized days of hemin use (95% CI), givosiran vs. placebo | 0.23 (0.11 to 0.45) | 0.26 (0.13 to 0.52) | ||
P value | < 0.001 | 0.0002f | ||
Urinary ALA levels (mmol/mol Cr)e | ||||
n | 46 | 43 | 48 | 46 |
Baseline, mean (SD) | 19.97 (16.80) | 17.52 (10.89) | 19.65 (16.61) | 17.27 (10.80) |
Month 3 | ||||
LS mean (SEM), change from baseline at month 3 | 1.76 (1.41) | 19.97 (1.48) | 1.72 (1.38) | 19.36 (1.41) |
Difference in LS mean (95% CI), givosiran – placebo | –18.21 (–22.26 to –14.16) | –17.64 (–21.55 to –13.73) | ||
P value | < 0.001 | < 0.001b | ||
Month 6 | ||||
LS mean (SEM), change from baseline at month 6 | 4.01 (2.35) | 23.15 (2.53) | 3.93 (2.27) | 22.28 (2.39) |
Difference in LS mean (95% CI), givosiran – placebo | –19.14 (–26.04 to –12.24) | –18.35 (–24.92 to –11.78) | ||
P value | < 0.001 | < 0.001f | ||
Urinary PBG levels (mmol/mol Cr)e | ||||
n | 46 | 43 | 48 | 46 |
Baseline, mean (SD) | 50.36 (34.33) | 46.80 (24.32) | 49.00 (34.41) | 45.39 (24.52) |
Month 6 | ||||
LS mean (SEM), change from baseline at month 3 | 12.91 (4.64) | 49.11 (4.96) | 12.45 (4.50) | 47.70 (4.69) |
Difference in LS mean (95% CI), givosiran – placebo | – 36.20 (– 49.71 to – 22.70) | –35.25 (–48.13 to –22.36) | ||
P value | < 0.001 | < 0.001f | ||
Harms, n (%) (SAS) | ||||
AEs | 41 (89.1) | 35 (81.4) | 43 (89.6) | 37 (80.4) |
SAEs | 8 (17.4) | 4 (9.3) | 10 (20.8) | 4 (8.7) |
WDAEs (from study treatment) | 0 | 0 | 1 (2.1) | 0 |
Deaths | 0 | 0 | 0 | 0 |
Notable harms | ||||
Motor neuropathyg | 0 | 3 (7.0) | 0 | 3 (6.5) |
Hepatocellular carcinoma | 0 | 0 | 0 | 0 |
Injection-site reactions | 8 (17.4) | 0 | 8 (16.7) | 0 |
Transaminase elevation | ||||
ALT increased | 4 (8.7) | 1 (2.3) | 4 (8.3) | 1 (2.2) |
AST increased | 3 (6.5) | 1 (2.3) | 3 (6.3) | 1 (2.2) |
Progression of renal impairmenth | 4 (8.7) | 0 | 5 (10.4) | 0 |
AAR = annualized attack rate; AE = adverse event; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; ALT = alanine transaminase; AST = aspartate transaminase; AUC = area under the curve; BPI-SF = Short Form Brief Pain Inventory; CI = confidence interval; CKD = chronic kidney disease; Cr = creatinine; FAS = full analysis set; IQR = interquartile range; LS = least squares; mFAS = modified full analysis set; MMRM = mixed-effects model for repeated measures; PBG = porphobilinogen; PCS = Physical Component Score; SAE = serious adverse event; SAS = safety analysis set; SD = standard deviation; SEM = standard error of the mean; SF-12 = 12-item Short Form Health Survey; vs. = versus ; WDAE = withdrawal due to adverse event.
aThe rates, rate ratio, corresponding 95% CI, and P value for comparing givosiran 2.5 mg/kg vs. placebo were derived using the negative binomial regression model with treatment-group and stratification factors (prior hemin prophylaxis status and historical attack rates) as fixed effects and the logarithm of the follow-up time as an offset variable. The negative binomial regression analysis was not performed when fewer than 10 patients in a treatment group reported an attack.
bThe LS means, treatment differences in LS means, their corresponding SEMs, 95% CIs, and P values for comparing 2.5 mg/kg givosiran vs. placebo were derived using the MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect.
cEstimated using the Hodges-Lehmann method. The P value was estimated from a stratified Wilcoxon test with stratification factors, prior hemin prophylaxis status, and historical attack rates. Note that normality was assessed using a Q-Q plot and the Shapiro-Wilk test, which indicated that the data had a significant deviation from a normal distribution. Therefore, a non-parametric analysis was conducted (i.e., a stratified Wilcoxon test).
dAnalyzed using the negative binomial regression model with treatment group and stratification factors (prior hemin prophylaxis status and historical attack rates) as fixed effects and the logarithm of the follow-up time as an offset variable.
eAnalyzed using an MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect.
fP value has not been adjusted for multiple testing or was calculated following a statistical testing failure earlier in the hierarchy, and should be interpreted as nominal.
gNerve compression and peripheral neuropathy were included under this notable harm. For patients with AIP or AHP, 0 patients in the givosiran treatment group and 3 patients in the placebo treatment group (1 with nerve compression, 2 with peripheral neuropathy) reported motor neuropathy.
hProgression of renal impairment was not specifically reported; the AE in this category corresponds to CKD, reported as an AE.
Source: Clinical Study Report.6
Critical Appraisal
One of the key limitations of the internal validity of the study was the use of a composite outcome for the primary end point, which was the AAR based on attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home. A specific MID was not identified for the AAR, although the clinical experts indicated that in general, a reduction in attacks is clinically meaningful. The frequency of attacks was reported descriptively for each of the individual components, which highlighted some variability in the treatment benefit associated with givosiran compared to the composite outcome. Variation in clinical practice and potential for unblinding or deduction of treatment allocation may have also biased treatment, which would affect the results of the individual components. As a result, there is notable uncertainty regarding the ability to interpret the individual components of the composite end point; however, the estimates of effect for each of the components were in the same direction and were not expected to have affected the overall composite outcome. A number of secondary outcomes were included and controlled for multiplicity using a statistical testing hierarchy; however, a failed statistical test for the change from baseline in daily worst pain rendered all subsequent secondary outcomes unadjusted for multiple testing. This included the evaluation of nausea, fatigue, and HRQoL through the PCS of the SF-12, which were all outcomes that were clinically relevant and important to patients. Further, all other HRQoL outcomes were exploratory and without an identified disease-specific MID, which hindered the interpretability of the results. Regarding the generalizability of the pivotal trial results, 95% of the study population were patients with AIP (1 of the 4 types of AHP); however, according to the clinical experts, there is no biological a priori to expect that the observed results would not be generalizable to different AHP types. According to the sponsor, “the study was enriched for attack frequency to ensure the ability to measure a difference in treatment effect on the primary composite porphyria attack end point.”6 The higher historical frequency of attacks at baseline for patients included Study 003; the inclusion criterion of at least 2 attacks in the past 6 months at baseline may limit the generalizability of the results to patients with less frequent attacks. These patients represent most of the patients in clinical practice, according to the clinical experts consulted for this review.
Indirect Comparisons
Indirect treatment evidence for givosiran was not identified in this review.
Other Relevant Evidence
Study 001 and Study 002
Description of Studies
Study 001 was a 3-part, multi-centre, placebo-controlled, phase I study of the safety and tolerability of subcutaneous givosiran for the treatment of adults with AIP. Parts A, B, and C were single-ascending dose, multiple-ascending dose, and multidose in design, respectively. The adaptive design allowed for different dosing regimens and dose levels to be assessed based on new safety, tolerability, and pharmacodynamic (PD) data. In total, 40 patients with AIP who are chronic high excreters (CHEs) were randomized to parts A and B (n = 23), while those with AIP who had recurrent attacks were randomized to part C (n = 17). Data were summarized for patients who received givosiran 2.5 mg/kg (part A, n = 3; part C, n = 3). Patients in the 2.5 mg/kg cohort of part C had a mean of 14.7 attacks (SD = 18.9 attacks) in the 12 months before the study, and one-third of patients were on prophylactic hemin.
Study 002 (N = 16) is a multi-centre, open-label, phase I/II study of the long-term safety and tolerability of subcutaneous givosiran for treatment of adults with AIP who completed part C of Study 001. The results of an interim analysis of Study 002 were summarized for this review. Patients received givosiran 2.5 mg/kg monthly or 5.0 mg/kg monthly or every 3 months until the safety review committee assessed safety, tolerability, and efficacy data, and agreed that all patients would be transitioned to receive a 2.5 mg/kg dose. Treatment duration is estimated to be up to 36 months; the estimated total time in study with screening and baseline will be up to 44 months. Nearly all patients (93.8%) in Study 002 had at least 1 porphyria attack in the 12 months preceding the study, with a mean of 13.0 porphyria attacks (SD = 13.1 attacks) attacks during that time period. All patients had used hemin during an acute attack, and half had used it prophylactically.
Efficacy Results
In Study 001, patients had fewer attacks during the treatment and follow-up phase compared to the run-in of part C for all attacks, attacks requiring hospitalization, and attacks requiring urgent health care visits. The cohort receiving givosiran 2.5 mg/kg monthly had a mean AAR of 2.9 (SEM = 1.91) for composite attacks and a mean annualized rate of hemin use of 2.9 days (SEM = 1.44 days) during the treatment and follow-up period. The placebo group had a mean AAR of 16.7 (SEM = 4.97) for composite attacks and mean annualized rate of hemin use of 23.4 days (SEM = 9.9 days) during the treatment and follow-up period.
In Study 002, patients had fewer composite attacks during the treatment period compared to the run-in (n = 9 and n = 72, respectively) and fewer attacks requiring hospitalization, urgent health care visits, and treatment with hemin at home. The mean composite AARs were 17.0 (SEM = 3.5) and 1.2 (SEM = 0.4) for the run-in period of Study 001, part C and the treatment period, respectively. The mean rate for annualized hemin use was 33.1 days (SEM = 7.0 days) during the run-in period compared to 1.1 days (SEM = 0.6 days) during the treatment period of Study 002.
HRQoL was also assessed using the EQ-5D-5L in Study 001 and Study 002.
Harms Results
Most patients (66.7%) in part A of Study 001 and 100% of patients in both part C of Study 001 and in Study 002 experienced at least 1 AE. In part C, the most frequently reported AEs were abdominal pain, abdominal distension, nausea, and injection-site reaction. In Study 002, the most commonly reported AEs were abdominal pain, fatigue, nausea, and injection-site reaction. SAEs were reported in 100% of patients who received givosiran 2.5 mg/kg in part C of Study 001 and in 25% of those in Study 002. SAEs included functional gastrointestinal disorder, pyrexia, anaphylactic reaction, Clostridium difficile colitis, sinusitis bacterial infection, mental status changes, dyspnea, and deep vein thrombosis. There was 1 withdrawal due to adverse event (WDAE) in Study 002, and no deaths reported in the cohorts of interest.
Critical Appraisal
A key limitation of Study 001 was the single-blind, adaptive study design. Study 002 was limited by an open-label study design that selected for patients who were able to tolerate and adhere to treatment, which may bias the results in favour of givosiran. Both studies had small sample sizes; only a couple of patients were randomized to receive givosiran 2.5 mg/kg, the intended commercial dose, for a short duration of time.
Study 003 Open-Label Extension
Description of Studies
The 6-month, DB, placebo-controlled Study 003 was followed by an ongoing, open-label extension (OLE) period (Study 003 OLE). The OLE is expected to continue for 29 months7 and was designed to evaluate the long-term efficacy and safety of givosiran for treatment of adults with AIP. Patients who completed the DB portion of Study 003 (N = 94) were eligible to participate in the OLE phase. The baseline characteristics of the patients included in the OLE were similar to those reported for the DB treatment period, with a slightly higher mean historical AAR of 11.6 (SD = 9.0); prior prophylactic hemin use was reported by 44.2% of patients. Initially, patients received either 1.25 mg/kg or 2.5 mg/kg givosiran, but with protocol amendment 5 (after the cut-off date for the interim report), all patients received the latter dose.
Efficacy Results
After 18 months, the median follow-up, the mean number of attacks during givosiran treatment was 3.4 (SEM = 0.7), and appeared to be stable over time following treatment with givosiran. Mean (SEM) AARs for attacks requiring hospitalization, urgent health care visit, treatment with IV hemin at home, and treatment without IV hemin at home were |||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||| respectively. The mean (SEM) number of days of hemin use was |||||||||||||||| Urinary levels of ALA decreased from baseline by a mean (SD) of |||||||||||||||| mmol/mol and |||||||||||||||||| mmol/mol at month 12 and month 18, respectively. Urinary levels of PBG also decreased by an average (SD) |||||||||||||||||| mmol/mol and |||||||||||||||||| mmol/mol for the same time points from baseline. Patient-reported outcomes — including the SF-12, EQ-5D-5L, PGIC, and PPEQ, as well as daily worst symptom scores — were also reported during the OLE and were consistent with the results described in the DB treatment period.
Harms Results
Nearly all patients (94.8%) experienced at least 1 AE, with 32.5% reporting nausea, 27.3% injection-site reaction, 22.1% fatigue, 22.1% nasopharyngitis, and 19.5% headache. SAEs occurred in 24.7% of patients, with chronic kidney disease, device breakage, and urinary tract infection being reported by 2.6% of patients for each SAE. There was 1 WDAE. No deaths were reported.
Critical Appraisal
The OLE was subject to most of the limitations associated with the DB treatment period. Additional limitations of the extension period of Study 003 include the lack of a randomized comparison group and the open-label design, which may have influenced patients’ and clinicians’ perception of improvement (which, in turn, may be reflected in the patient-reported and safety outcomes). It is also worth noting that there was a dose change for those who initially enrolled under protocol amendment 3 and received givosiran 1.25 mg/kg. At month 13, patients who had inadequate disease control were able to increase their dose to 2.5 mg/kg from 1.25 mg/kg, and with protocol amendment 5, all patients were to receive givosiran 2.5 mg/kg (the intended commercial dose).
Study 005
Study 005 is an international program that will provide expanded access to givosiran to patients 12 years and older with AHP.8 It is ongoing. No additional information was available for this review.
Conclusions
One DB, placebo-controlled, phase III randomized controlled trial (RCT), Study 003, evaluated the efficacy and safety of givosiran compared to placebo. Included patients had a diagnosis of AHP and had experienced at least 2 porphyria attacks in the 6 months before screening. Over the 6-month treatment period, a 74% reduction in the rate of acute porphyria attacks was demonstrated with givosiran compared to placebo in patients with AIP based on a rate ratio of 0.26 (95% CI, 0.16 to 0.41; P < 0.001). Similar results were reported in all patients with AHP. The primary outcome of porphyria attacks was a composite outcome that included attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home. This outcome did not have a defined minimum clinically important difference. However, the clinical experts identified that in general, a reduction in acute attacks was clinically relevant. A treatment difference in favour of givosiran was also reported for a reduction in annualized days of hemin use and change from baseline in urinary ALA and PBG levels. This difference supports the beneficial direction of the primary outcome. Management of pain was an outcome important to patients, but the results did not demonstrate a clinically meaningful difference in daily worst pain scores. Reported HRQoL outcomes were also important to patients but were subject to limitations that hindered the interpretability of the results. With regards to the safety assessment, the majority of patients in the trial experienced at least 1 AE, with nausea, injection-site reaction, chronic kidney disease, fatigue, increase in ALT, and decrease in glomerular filtration rate more commonly reported among patients who received givosiran. Reported SAEs and treatment discontinuations due to AEs were infrequent, and no deaths were reported. Although the DB treatment period was limited to 6 months on treatment, evidence from the OLE of Study 003 demonstrated maintenance of treatment effect for up to 18 months and did not detect any new safety concerns. Gaps in the evidence of the efficacy and safety of givosiran were identified in patients with concomitant prophylactic hemin use and in long-term safety and efficacy data beyond 18 months of treatment.
Introduction
Disease Background
The porphyrias are a group of metabolic disorders caused by altered activities of enzymes within the heme biosynthetic pathway. Altered enzyme activity is usually due to an inherited mutation in the gene for that enzyme. Porphyrias are classified as hepatic or erythropoietic based on whether pathway intermediates first accumulate in the liver or in the bone marrow, respectively.9 The types of AHP include AIP, ADP, HCP, and VP. Each is characterized by a specific genetic mutation involved in the synthesis of heme in the liver; however, the types are clinically indistinguishable.1
In the liver, ALAS1 is the first, as well as a rate-limiting, enzyme in the heme biosynthetic pathway.1,10 Upregulation of hepatic ALAS1 is an important feature during exacerbations of AHPs because it leads to an increase of intermediates in the heme biosynthetic pathway, such as ALA and PBG.3,10 In patients with AHP, the altered enzymatic activity within this pathway results in an excessive accumulation of the intermediate porphyrin precursors (ALA and PBG), which are neurotoxins that can precipitate an acute attack.1,3 AHP can be exacerbated through induction of the ALAS1 gene by drugs and other factors, such as stress, fasting, alcohol use, smoking, and hormones. Induction of hepatic heme oxygenase, which degrades heme, can also lead to induction of ALAS1.10 During an acute attack, patients report a significant increase in pain that can gradually build over hours and last for a number of days.3 Some patients report pain in the chest, back, or extremities; however, severe abdominal pain is more typical. Long-term complications with recurrent acute attacks may include chronic pain, chronic kidney failure, and liver damage.
Overall, these porphyrias cause acute and chronic symptoms due to effects on the nervous system, with the most common being neuropathic abdominal pain.9 The motor, sensory, and autonomic nervous systems are often affected, resulting in autonomic changes (e.g., tachycardia, hypertension), muscle weakness, sensory loss, and pain in the back, chest, and extremities. Linenberger and Fertrin (2020) refer to severe abdominal pain, peripheral neuropathy, and central or autonomic nervous system manifestations as the “classic triad,”11 but even these severe symptoms may be discounted because they mimic other diseases, and physical findings are often minimal.9
AIP is the most common AHP prototype, although the symptoms are common to all AHPs. HCP and VP may also present with blistering skin lesions. AIP, HCP, and VP are autosomal dominant, inherited disorders with low penetrance and female predominance. One review notes that at least 90% or more of heterozygotes for disease-causing mutations remain asymptomatic for life.1 ADP is autosomal recessive and extremely rare, usually with an onset of attacks in the early teenage years, and all cases exhibit elevated erythrocyte zinc protoporphyrin. Very rare cases of homozygous AIP, HCP, and VP have a completely different phenotype from the other AHPs.9 AIP occurs in all races, but may be most common in northern Europeans; males and females are equally likely to inherit a PBGD gene mutation, but AIP is more likely to manifest in women than men; AIP affects adults and typically presents in a patient’s thirties or forties10 (although the clinical expert said twenties or thirties).
In the absence of information about prevalence and incidence of AHP that is specific to Canadians, the sponsor reported an estimate of the prevalence of AHP in Canada based on European data from Elder et al. (2013).12 The estimated prevalence of AHP in the Canadian provinces (excluding Quebec) was |||| per million population2; however, it was noted by both the sponsor and the clinical experts consulted for this review that patients with AHP are underdiagnosed due to a lack of available treatment options and poor awareness of the disease. Of note, endemic populations have been described in British Columbia, Manitoba, and Nova Scotia. The combined prevalence of these disorders is estimated to be approximately 5 per 100,000, with instances of higher prevalence due to founder effects.1
Diagnosis
Patients with AHP exhibit nonspecific and variable symptoms, which may result in them being seen by a number of specialists before receiving a diagnosis. The challenge in diagnosing patients with AHP is arriving at the point where one suspects the disease. The clinical experts consulted for this review stated that ultimately, expert clinician judgment in the context of laboratory and clinical reviews is the best way to identify AHP in patients. The clinical experts also noted that once AHP is suspected, confirming a diagnosis is relatively straightforward. A diagnosis is made based on serum, urinary, and fecal biochemical tests, which includes testing for elevated levels of PBG, ALA, and other porphyrins.3 Urinary ALA, PBG, and porphyrin excretion is notably increased during porphyria attacks; therefore, it is recommended that a patient presenting with acute symptoms without a history of acute porphyria have urinary PBG tested as soon as possible.10 The caveat is that the biochemical tests are rarely performed in-house, and obtaining results may take time. False-negative biochemical tests are also common; thus, genetic studies with symptoms in keeping with AHP and documented responsiveness to first-line therapy are sufficient to support the diagnosis, as noted by the clinical experts. Genetic studies may also be conducted. These are not required, but are often done.
Further testing is required to distinguish AIP, HCP, VP, and ADP. Substantial elevation of plasma or urinary PBG and ALA during an attack is common to AIP, HCP, and VP, but elevated levels of PBG are not observed with ADP.10
Standards of Therapy
According to the clinical experts consulted for this review, the goals of treatment for Canadian patients living with AHP are to reduce the frequency of acute porphyria attacks and hospitalizations and improve HRQoL and work-life productivity. Currently, there are no specific Canadian clinical practice guidelines for the treatment AHP; therefore, expert opinion is primarily used to guide treatment, as per feedback from the clinical experts consulted for this review. Of note, the clinical experts reported that currently in Canada, there is a lack of sufficient specialists with extensive experience in managing patients with AHP. As such, some patients may lack access to optimal management.
Treatment for AHP is targeted mainly at preventing acute attacks. The BIPNET recommends that recurrent acute attacks be managed through general measures and avoidance of precipitating factors, through treatment with GnRH analogues and prophylactic heme arginate, and/or through liver transplantation.4 Patient education about avoidance of triggering factors is recommended for the prevention of acute attacks. Triggers can include certain medications, smoking, alcohol, illicit drugs, and periods of fasting or low carbohydrate intake.4,13 The clinical experts consulted for this review indicated that avoiding triggers enables patients to mitigate or self-manage porphyria attacks, and is sufficient for many patients. Changes in the level of progesterone during the menstrual cycle are 1 of the biggest contributors to an acute attack; therefore, the use of GnRH analogues to suppress the menstrual cycle can be an option for treatment in some patients, along with expert guidance. Side effects are significant with hormone therapy and may include depression, hot flushes, reduced libido, osteoporosis, and other menopausal symptoms. The BIPNET recommends that patients using hormone therapy be reviewed often and receive regular gynecological and annual bone-density exams.4 The clinical experts consulted for this review reported that long-term use of hormone therapy is typically not an option due to the severity of the side effects.
In Canada, hemin for injection is indicated for the amelioration of recurrent episodes of AIP temporally related to the menstrual cycle in susceptible women after initial carbohydrate therapy is known or suspected to be inadequate. Limitations of use include consideration of an appropriate period of carbohydrate loading (i.e., 400 g of glucose per day for 1 to 2 days) before use.14 Despite the narrow indication for the treatment of acute attacks, hemin is often considered for prophylactic use outside of the indication in patients who exhibit recurrent attacks with the preventive measures described earlier. The clinical experts consulted for this review indicated that most patients do not require treatment between attacks, but that a small number who have recurrent attacks will require prophylactic hemin weekly. Hemin is also associated with complications resulting from the requirement for venous access, iron overload, and difficulty withdrawing from treatment.4 For patients who are unresponsive to treatments for recurrent attacks, are frequently hospitalized, and exhibit diminishing HRQoL, a liver transplant may be considered, and is highly effective in many cases without advanced motor neuropathy.15
Despite the preventive measures described, acute porphyria attacks can still occur. Typically, mild attacks may be managed at home with increased carbohydrate intake and analgesic medication, but moderate to severe attacks require hospitalization so that IV hemin can be administered. According to the clinical experts, IV glucose can be used if hemin is not available, but it is not as effective. Additionally, patients in hospital are treated medically for symptoms (pain, nausea, and vomiting) and are closely observed, with their salt and water balance monitored. The prognosis is usually good if the disease is recognized and if treatment and preventive measures are started before severe nerve damage has occurred.13 Hemin is the most effective treatment for acute attacks. As described earlier, it is administered intravenously. Intravenous glucose is usually given as a 10% solution while hemin is being prepared, but should not delay the administration of hemin.
Drug
Givosiran is a double-stranded, small, interfering RNA that causes the degradation of ALAS1 mRNA in hepatocytes through RNA interference, reducing elevated levels of liver ALAS1 mRNA. This leads to reduced circulating levels of neurotoxic intermediates ALA and PBG, 2 factors associated with attacks and other disease manifestations of AHP.5
In Canada, givosiran is indicated for the treatment of AHP in adults.5 It is available as a solution for subcutaneous injection (189 mg givosiran/mL) and does not require additional reconstitution or dilution before administration. The recommended dose is 2.5 mg/kg once monthly, based on body weight.
Givosiran underwent an expedited review at Health Canada and has been requested for reimbursement as per the approved Health Canada indication.
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Group and Information Gathered
CADTH received 2 patient group submissions for this review: 1 from CAP and 1 from APF.
CAP is a national, voluntary charity whose mission is to deliver evidence-based information and support to patients with porphyria, their families, health care providers, and the general public. The group also aims to achieve standards and evidence-based comprehensive care for all people with porphyria throughout their lifespans.
APF is dedicated to improving the health and well-being of individuals and families affected by porphyrias. The group provides programs to raise awareness and educate health care professionals and the general public in 76 countries around the world. APF also helps to establish support groups and mentorship of the next generation of experts and supports porphyria research. Of its international members, more than 300 are located in Canada. More information about APF can be found at http://www.porphyriafoundation.org/.
CAP created and distributed a survey to its members via email and social media platforms for a 2-week period in February 2021. The survey was available in English and French and was restricted to Canadian patients and caregivers who had experience with AHP. In total, 22 patients and 4 caregivers responded with diagnoses of AIP (n = 20), HCP (n = 3), and VP (n = 3). Of the patients represented in the survey, 23 were female and 3 were male; respondents were from Alberta (n = 10), British Columbia (n = 7), Ontario (n = 6), Manitoba (n = 2), and Quebec (n = 1). CAP also requested support from the British Porphyria Association, which shared interviews from 3 individuals who had received givosiran. While preparing this submission, CAP discussed and reviewed it with members of the Canadian Hemophilia Society and Network of Rare Blood Disorder Organizations.
APF used its social media platforms and online newsletters to connect with Canadian patients about their experiences with porphyria and collected responses by telephone and email. Some of the responses in the APF submission were collected during an Alnylam Patient Advisory Board meeting. Twelve individual patient submissions were collected from Canadians in Ontario (n = 6), British Columbia (n = 2), Quebec (n = 2), Manitoba (n = 1), and Nova Scotia (n = 1).
Disease Experience
Respondents in both submissions reported experiencing the following symptoms, among others: pain, fatigue, nausea, weakness, paralysis, neuropathy, seizures, anxiety, and depression. More than 80% of patients who responded to the CAP survey had experienced symptoms at least once a month, with many reporting that these symptoms occurred more than 20 days per month. The group also reported that 86% of respondents had at least 1 attack in the past year and 36% had at least 10. Furthermore, 55% of patients had gone to the emergency room at least once in the past year due to an attack, while 18% had gone at least 10 times. One of the most frequently reported symptoms is pain. An APF survey respondent stated:
I have to have a lot of pain meds. My desire is to free from them but I cannot be free from pain meds without being free of attacks. The pain is too much for a person to endure. A doctor with AIP said that it was not compatible with life to have attacks with no pain treatment.
Porphyria attacks can also prevent patients and caregivers from being able to work, lead to poorer quality of life, and negatively affect relationships. For instance, 59% of CAP respondents stated that it had affected their career and financial well-being. Respondents said, “It has destroyed and robbed me of my education, livelihood, finances and health” and “I usually have to make arrangements at work on a short notice when my wife is in attack and have to take her to the hospital and have to work from hospitals many times.”
A respondent who is both a patient and caregiver described their experience as follows:
Because of AIP, I had to give up my career as a Registered Nurse. The symptoms, including the anxiety, was so severe and debilitating that I could not longer function well in daily life. …now I am the caretaker advocate for my brother. He has been on disability for 15 years and is worsening. Two lives are unable to be productive because of porphyria.
Both groups emphasized the negative effect that porphyria had on their daily lives and mental health. In the CAP survey, 64% reported that the disease had significantly affected their mental health, while 82% said they experienced anxiety or depression monthly. Patients described their situations:
“There is a lot of grieving, loss of work, social losses and having to accept the illness.”
“The psychiatric symptoms are debilitating. Attacks start with a distinct foggy head difficulty processing thought, inability to make decisions, memory issues, high anxiety, restlessness withdrawal, obsessive thoughts paranoia unable to stop mind from racing causes insomnia.”
Moreover, 94% of patients and 100% of caregivers from the CAP group felt that AHP had affected their overall well-being, while significant impacts on family life and social life were noted by 50% and 41% of respondents, respectively. The patient input submissions described how experiencing symptoms and trying to avoid triggers could strain social relationships and make it difficult to care for families. Respondents were also concerned about the possibility of passing the condition on to their children. The APF submission stated: “Porphyrias are known as the ‘little imitator’ as it has a host of generic symptoms making it hard to identify, particularly because it cannot be diagnosed without porphyria specific tests. It will not show on normal tests.”
Consequently, patients often face difficulties when trying to obtain a diagnosis or access treatment for their attacks. One caregiver wished that emergency room doctors were more aware of the disease:
This could have helped doctors to come to right diagnosis earlier, and to avoid unnecessary tests and treatments. Especially in AIP, once an episode or attack starts, if the right medications/treatment are not given soon in the crises (and unsafe medications are not avoided), the consequences are devastating for the patient.
The caregiver also noted that the patient was initially misdiagnosed and given unnecessary tests and treatments.
Experiences With Currently Available Treatments
Most patients from the CAP survey have attempted to manage porphyria by avoiding attack triggers (86%), adopting a high-glucose diet (82%), using hematin (hemin, Panhematin) (27%), using GnRH analogues (9%), or taking additional medications to treat impacts like pain (50% opioid, 41% non-opioid), sleep problems (50%), anxiety (41%), and depression (32%). Although respondents felt that these treatments could help with symptoms, they noted that the treatments are not without side effects, such as weight gain, diabetes, iron overload, induced menopause, and others specific to the additional medications.
Both groups described hematin as being effective at reducing the length and/or severity of intermittent attacks, but they also acknowledged its drawbacks. For example, it has been associated with platelet aggregation, and frequent use can cause iron overload and damage to veins. Furthermore, it is often infused through a peripherally inserted central catheter line or port, which can become blocked, requiring surgical replacement. Other side effects that were mentioned include headaches and low-grade fever that resolve within 2 days. One individual said, “Panhematin saved my life. But as patients, it is much more than just saving our lives…There is not quality unless attacks are prevented.”
A caregiver described how weekly hemin treatments have helped their wife:
[S]he is in fact feeling better in terms of gaining her strength doing everyday activities. It has also reduced the amount of pain killer by 25% got rid of most of the nerve medications. It has also reduced the number of hospitalizations from 12 a years to 2-3 and has given her some independence….
This caregiver noted that while there were benefits, they felt as though their wife could be developing tolerance to the treatment and that the weekly schedule made working and planning around infusions somewhat challenging.
Patients described facing difficulties when trying to access treatment in hospitals. They reported that there is a lack of awareness of AIP among health care workers, and that the recommended treatment is not readily available. The patient groups also noted that delays and non-optimal treatments can exacerbate attacks and lead to permanent damage.
Improved Outcomes
Ideally, respondents would like a cure for porphyria. However, they believe a realistic goal is a treatment that prevents attacks and reduces symptoms, particularly pain, nerve damage, and paralysis. Patients and caregivers would like to see additional options that are more effective, cause fewer side effects, present an easier mode of administration, can be administered outside of a hospital, and lead to improvements in quality of life. Other limitations to treatment access that were identified include the need for travel, the requirement for venous access, and the lack of access to specialists and proper diagnostic testing. Respondents raised concerns about financial assistance and insurance coverage, and said they would like health care providers to have greater knowledge of the condition. They felt strongly that there should be financial assistance to help alleviate the burden of treatment costs.
A parent stated:
Hematin made a difference in the life of my daughter in that it stopped her attacks after they began, but it did not stop her attacks from happening. We need a treatment to stop the attacks from occurring, because attacks are so excruciating and can lead to paralysis and death.
When asked what would be important to discuss with their physicians about treatment options, APF survey respondents mentioned available treatments, benefits and drawbacks of treatments, safety and efficacy, side effects, patient monitoring, impact on quality of life, frequency of administration, and treatment cost.
Experience With Drug Under Review
Although none of the CAP survey respondents had received givosiran (Givlaari), with help from the British Porphyria Association, the group was able to share 3 patients’ experiences. Two had accessed the drug for 2 years through the ENVISION trial. All 3 patients described givosiran as “life-changing.” More specifically, the drug had been able to reduce or eliminate attacks, pain and pain medications, fatigue, nausea, and anxiety. It improved patients’ physical health, appetite, sleep, and concentration. In terms of social and work impacts, the respondents highlighted greater independence, the ability to attend and contribute more at school and work and toward family life, and the tendency to be more optimistic about planning their futures.
Because givosiran is a monthly injection, APF suggested that it is an easier treatment to receive compared to hemin infusions. The group noted a few side effects, such as injection-site reactions, allergic reactions, and nausea, while those reported by Alnylam include liver and kidney problems. APF also noted that patients may be able to access givosiran through the Alnylam Assist program. Successful treatment can reduce costly hospital admissions and the burden on patients and families. Respondents who had received the medication felt that their attacks were treated; they reported both fewer attacks and less severe attacks. Moreover, despite still experiencing attacks and side effects while receiving the drug, many wanted to continue with it. APF also stated that patients receiving givosiran may be prescribed hematin to stop an attack quickly as it is occurring. However, there may be exceptions to this approach that the patient group was not aware of.
The following quotes illustrate patients’ experiences with givosiran:
“My greatest hope was to return to work. That dream is coming true as I’m almost finished school, a feat I could never have done without Givlaari. It gave me my life back.”
“I was frightened for my life, because I was at the point that I no longer had veins for Panhematin infusions. Givlaari was my last chance. I was on the Givlaari trials and had immediate success. Some people say it takes a while to gain effect, but for me, I stopped having attacks after my first shot of Givlaari. Life changed. I could return to work. I could return to life.”
“I was totally debilitated. I suffered terribly and was repeatedly hospitalized. I finally lost my job as I was unable to go to work. My life was a shambles until I was given Givlaari. Life has turned around for me. I want other people to have my experience.”
In its submission, APF stated that Alnylam provides free diagnostic DNA testing for many patients who fit the criteria, and that insurance may also cover testing.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). In addition, as part of the givosiran review, a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion is presented in this section.
Unmet Needs
One of the major goals in the management of AHP is to reduce the frequency of AHP attacks. According to the clinical experts consulted for this review, currently, most patients with recurrent attacks will continue to have these because there is no indicated treatment for prevention. The experts noted that while prophylactic hemin can be used to reduce the rate of AHP attacks (with case reports of improvement), the use of prophylactic hemin is outside of the Health Canada–approved indication and has not been studied well. The experts described further issues with prophylactic hemin. These include a serious side effect of iron overload, some minor but nuanced infusion reactions, and the need for a central venous catheter for regular prophylactic use. The experts stated that there is a need for treatments that are better tolerated due to the substantial risk and requirement for central venous access associated with the current IV treatments. Further, formulations are needed to improve convenience and compliance because IV access requires hospital-based care. The experts noted that this is particularly problematic for patients who do not live near large health care centres, which are often the only centres with access to specialized care. GnRH may also be used to prevent AHP attacks, but is not approved for prolonged use and is associated with side effects and loss of bone mineral density (given that it leads to a drug-induced menopause state).
Place in Therapy
The clinical experts described givosiran as the first treatment approved for prevention or prophylaxis against acute attacks; however, givosiran is not the first treatment that addresses the underlying disease process, given that hemin also achieves this goal. As described, while hemin is used as a regular weekly treatment to prevent attacks, this is not an approved indication, as noted by the clinical experts. The mechanisms of action for givosiran and hemin are different, but both decrease ALA and PBG, which are the intermediates that cause AHP attacks and symptoms, as described by the clinical experts. The experts felt that the 2 treatments can complement each other; however, givosiran is used to prevent AHP attacks rather than to treat acute attacks, as hemin does. The clinical experts also noted that if an attack occurred despite treatment with givosiran, then hemin could still be used, and its effectiveness is unlikely to be diminished.
As per feedback from the clinical experts on this review, givosiran would be used in people who have recurrent attacks; there is no evidence to support its use in asymptomatic individuals or during acute attacks. Also, the experts stated that givosiran would not be used as a first-line treatment or to treat the first AHP attack. The clinical experts indicated that other approaches to treatment should be tried for patients with AHP before givosiran is recommended. These approaches include avoidance of triggers, such as smoking or fasting, administration of IV dextrose 10% in water, and/or hemin use; however, the experts also reiterated the limitations of hemin use described under unmet needs, particularly in the prevention of attacks. According to the clinical experts, most patients living with AHP will either have mild symptoms or 1 attack without frequent recurrence; prevention and avoidance of triggers is helpful for these cases.
Therefore, the experts expected givosiran to provide an alternative therapy for a small subset of patients with frequent or recurrent attacks who would otherwise require hospitalization and hemin administration. They recommended that givosiran be reserved for patients with recurrent symptoms or flares that consistently affect their HRQoL. Givosiran was also described by the experts as an appropriate treatment for patients who qualify for hemin prophylaxis but cannot adhere to treatment due to toxicity or lack of convenience.
Patient Population
The clinical experts described the diagnosis of AHP as relatively straightforward. It is obtained through biochemical tests for levels of urinary ALA, PBG, and other porphyrins. These tests were described by the experts as being very specific, but subject to false negatives, which makes clinician judgment an important factor in the diagnosis. The experts noted that AHP can be confirmed through genetic testing, but this testing is not required. They noted that the challenge with identifying patients with AHP is arriving at a suspicion of AHP because of the variable presentation of disease and symptoms that are not specific to the disease. The clinical experts also stated that it is highly unlikely that a clinician would incorrectly diagnose a patient with AHP. The greater concern is underdiagnosis of the disease due to the previously described challenges associated with identifying patients based on physiological and pre-analytical variables.
The clinical experts indicated that the group of patients most in need would be those with frequent or recurrent severe attacks, and that the frequency and severity of attacks are the most important factors when considering the use of givosiran. They noted that severity is heavily based on the number of severe attacks in a given period, but that it was challenging to further define frequency of attacks because the presentation of disease varies among patients. Therefore, the clinical experts felt it was most appropriate to assess the severity of disease on a case-by-case basis using clinical judgment. In addition to frequency and severity of attacks, hemin use and comorbidities were also considered important factors in the decision to use givosiran for patients. More specifically, the clinical experts noted that hemin use was an indicator of disease that was not well-controlled, and that the presence of comorbidities resulting from AHP would be considered a priority, with the goal of preventing further complications. According to the clinical experts, laboratory porphyria levels, subtypes and genetic testing do not predict disease severity because there is variable penetrance and variation in clinical course.
The clinical experts also stated that givosiran is least suitable — and would not be considered — for patients who are asymptomatic, are identified with AHP through family history and never develop clinical symptoms, have not been therapeutically challenged with initial lines of therapy, or whose clinical symptoms are mild or characterized by infrequent attacks. Additionally, the experts felt that givosiran would not be suitable in populations that have not been studied, including children, adults aged 65 years and older, pregnant or breastfeeding individuals, or patients with moderate to severe liver disease or significant elevation in transaminases.
Assessing Response to Treatment
The clinical experts stated that it is not possible to identify the patients most likely to exhibit a response to treatment with givosiran based on the current evidence, biochemical tests, or patient characteristics.
The clinical experts described the following outcomes as those that are used to determine response to treatment in clinical practice: reduced attack rate, reduced hospitalization, reduced need for hemin, frequency of neurovisceral flares, and improved patient-reported outcomes, such as daily symptoms, HRQoL, and work-life productivity.
An improvement in HRQoL and a reduction in frequency of attacks, hospitalizations, and attacks requiring hospitalization or hemin use would be considered clinically meaningful responses to treatment by the clinical experts consulted on this review, and would be considered when making a decision to continue treatment with givosiran. One of the experts also noted that the following would be characterized as a clinically meaningful response to treatment: a reduction in, improvement of, or stabilization of psychiatric manifestations and neurovisceral symptoms (abdominal or back pain, axonal neuropathy) in addition to the attainment of major motor milestones and the ability to perform activities of daily living.
The clinical experts suggested that patients be assessed for response to treatment every 6 months or annually. One suggested that patients be assessed more frequently (monthly) during the acute phase (described as the first 3 months), followed by evaluations every 3 months, then every 6 months once stabilization is achieved. All of the experts agreed that 1 year would be a sufficient amount of time in which to assess response to treatment; however, the variable presentation of disease (such as yearly fluctuations in attack frequency) was noted as a limitation to the certainty of this assessment.
Discontinuing Treatment
All of the clinical experts agreed that the frequency of attacks is the most important factor in the decision to continue treatment with givosiran. HRQoL and the frequency of attacks leading to hospitalization were also noted by the experts as being important for consideration. In general, the clinical experts felt that patients treated with givosiran would continue with treatment until they encountered a reason for discontinuation, such as safety concerns, including severe local reactions, anaphylaxis, severe gastrointestinal symptoms, severe fatigue, or increase in liver function tests and/or creatinine. Discontinuation of treatment may also be considered with deterioration of HRQoL, safety concerns that outweigh the perceived benefit due to reduced symptoms or attacks, or a lack of response to treatment. Of note, it was challenging for the clinicians to specifically define response to treatment due to the heterogeneous nature of AHP among patients, but an increased or similar rate of attacks while on treatment was noted as a sign that treatment may not be working. Feedback from the clinical experts also indicated that menopause would be a potential reason to trial treatment discontinuation in patients with stable disease. They also noted that if attacks recurred following discontinuation, restarting treatment with givosiran would be a possibility.
Prescribing Conditions
The clinical experts indicated that givosiran is administered by a subcutaneous injection and can be given in a community nursing clinic, hospital outpatient clinic, or any infusion clinic, whether it is a community or specialty clinic. The experts felt that ideally, administration of givosiran should be overseen by a specialist responsible for the diagnosis, treatment, and monitoring of patients receiving givosiran. According to the clinical experts, patients with AHP should be monitored by a clinic specialized in the management of porphyria, but because it is a rare condition, this is not always available. General internal medicine and hematology specialists would be most likely to manage these patients, but specialists in hepatology, nephrology, and neurology were also mentioned as relevant.
Clinician Group Input
CADTH did not receive any input from clinician groups for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Givosiran is indicated for only 4 types of AHP, not the types that manifest cutaneously. Urine testing identifies key indicators of this condition and types, but once confirmed biologically, the specific type must be confirmed with genetic tests. How widely available are these tests, given that the condition is so rare? | The clinical experts indicated that the availability of genetic testing may vary among jurisdictions. As a genetic disease, 1 of the contributing factors is the existence of genetic clusters across Canada. In areas with evidence of hereditary AHP, a genetic panel of known porphyria genes is more likely to be available for suspected patients to be tested against. The clinical experts also noted that the biochemical tests for urinary ALA and PBG are specific to AHP and, along with clinical evidence consistent with porphyria attacks, are sufficient to make a diagnosis. Due to the lack of available genetic tests, this is not a requirement for making a diagnosis, but it would be required to confirm one. |
The approved Health Canada indication for givosiran is for adults only. Citing a broader indication for givosiran in Europe and the inclusion criteria in the ENVISION trial, could givosiran be used in patients aged 12 years and older in Canada? Patients included in the ENVISION trial must have had 2 or more attacks in the last 6 months. Could givosiran be used with patients who experienced fewer? | Despite the inclusion criteria for the ENVISION trial, the patients who were enrolled in the trial were between the ages of 19 years and 65 years at baseline. As such, givosiran is not indicated by Health Canada for pediatric patients. Similarly, the clinical experts do not foresee its use in pediatric patients until evidence in this population in available. The clinical experts would consider using givosiran in patients with fewer than 2 attacks in 6 months depending on disease-related complications resulting from the severity of an attack. It was noted that this would be considered on a case-by-case basis. Otherwise, the experts would follow the criteria outlined in the ENVISION trial. |
Should therapy end if attacks cease to occur, or only if patients experience serious side effects? How serious? Can this be defined? | The clinical experts did not think that therapy should end if attacks ceased to occur. As noted in the clinician input section, a trial of discontinuation may be considered around menopause, when attacks tend to become less frequent. Safety concerns, such as serious side effects, would be a reason to consider discontinuation of treatment. The clinical experts noted that this would depend on the nature of the side effect and whether the benefits of treatment outweigh the safety concerns. The clinical experts could not define a specific safety situation that would lead to treatment discontinuation, stating that it would need to be evaluated on a case-by-case basis. |
Given that hemin may be needed as rescue therapy for an acute attack, is the use of givosiran in combination with hemin a concern? | The clinical experts did not express concern with the use of givosiran in combination with hemin. In fact, the 2 were expected to be complementary because givosiran should be used to prevent acute porphyria attacks, while hemin is used as a treatment for acute attacks. |
Are there generalizability issues in populations that match the indication but for whom there are insufficient data (for types of AHP other than AIP)? | Based on the mechanism of action of givosiran and the pathogenesis of the different types of AHP, the clinical experts would treat patients with AIP, ADP, HCP, or VP the same. |
As per the monograph, the drug needs to be given by a health care practitioner in the home. Can it be self-injected? | The clinical experts indicated that givosiran is administered according to the product monograph; therefore, it should not be self-injected. |
It is likely that patients will need to be monitored for chronic kidney disease and elevated liver function tests, given that these were significant adverse effects noted in the trials. Is management only to stop the drug if severe? Can it be restarted? | The clinical experts reported that assessment of chronic kidney disease and elevated liver function tests would be a factor in the consideration of treatment discontinuation. As previously noted, safety concerns would be evaluated on a case-by-case basis, and treatment decisions would be up to the treating clinician. Further, the clinical experts noted that in general, treatment could be restarted if the safety issue was no longer expected to be a concern. |
ADP = aminolevulinic acid dehydratase-deficient porphyria; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; HCP = hereditary coproporphyria; PBG = porphobilinogen; VP = variegate porphyria.
Clinical Evidence
The clinical evidence included in the review of givosiran is presented in 2 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes sponsor-submitted, long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of givosiran 2.5 mg/kg once monthly for the treatment of AHP in adults.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect those considered to be important to patients, clinicians, and drug plans.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with acute hepatic porphyria Subgroups: By subtype (AIP, ADP, HCP, VP) Frequency/history of attacks |
Intervention | Givosiran 2.5 mg/kg once monthly by subcutaneous injection |
Comparator | Prophylactic hemin Gonadotropin-releasing hormone analogue (in women only) Standard of care or placebo |
Outcomes | Efficacy outcomes: Frequency of attacksa HRQoLa Management of symptomsa (e.g., gastrointestinal, cardiovascular, neurologic) Reduced complications of AHPa (e.g., hypertension, CKD, liver cancer, neuropathy) Rescue medication use Opioid use Activities of daily living Hospitalization and health care usea Ability to work or attend school ALA, PBG, and porphyrin levels Mortality Harms outcomes: AEs, SAEs, WDAEs, mortality, motor neuropathy, hepatocellular carcinoma, injection-site reactions, transaminase elevation, progression of renal impairment |
Study designs | Published and unpublished phase III and IV RCTs |
ADP = aminolevulinic acid dehydratase-deficient porphyria; AE = adverse event; AHP = acute haptic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; CKD = chronic kidney disease; HCP = hereditary coproporphyria; HRQoL = health-related quality of life; PBG = porphobilinogen; RCT = randomized controlled trial; SAE = serious adverse event; VP = variegate porphyria; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search was performed by an information specialist using a peer-reviewed search strategy. The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist (https://www.cadth.ca/resources/finding-evidence/press).16
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) through Ovid and Embase (1974‒) through Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Givlaari (givosiran). Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on March 23, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on July 21, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist (https://www.cadth.ca/grey-matters).17 Included in this search were the websites of regulatory agencies (US FDA and the European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 5. A list of excluded studies is presented in Appendix 2.
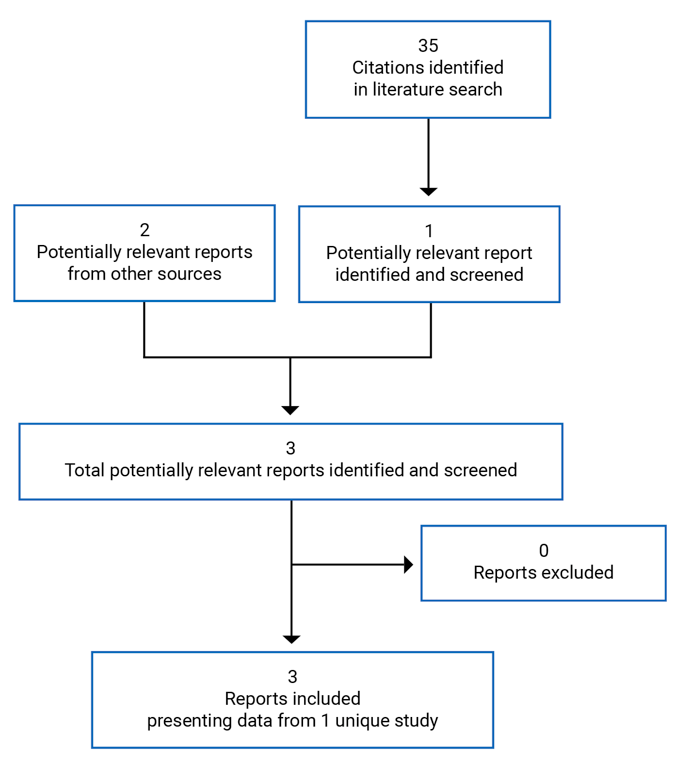
Table 5: Details of Included Studies
Details | Study 003 |
|---|---|
Designs and populations | |
Study design | Phase III, DB, placebo-controlled RCT with OLE |
Locations | 36 centres in 18 countries: Canada, US, Mexico, Europe, Australia, and Asia |
Patient enrolment dates | November 16, 2017, through June 27, 2018 |
Randomized (N) | 94 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 2.5mg/kg givosiran once monthly by SC injection |
Comparator(s) | Placebo (sodium chloride 0.9% w/v) for SC administration |
Duration | |
Phase | |
| 60 days |
| 6 months |
| Up to 29 months |
| 1 month |
Outcomes | |
Primary end point | Annualized rate of porphyria attack (attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home) in patients with AIP over the initial 6-month DB period |
Secondary and exploratory end points | Secondary
Over the 6-month DB period:
Exploratory Measured in patients with AIP and in patients with AHP over the 6-month treatment period or over the OLE period:
|
Notes | |
Publications | Balwani (2020)18 |
AAR = annualized attack rate; ADA = antidrug antibody; ADP = aminolevulinic acid dehydratase-deficient porphyria; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; ALT = alanine aminotransferase; BFI-SF = Brief Fatigue Inventory – Short Form; BPI-SF = Brief Pain Inventory – Short Form; DB = double blind; eGFR = estimated glomerular filtration rate; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; HCP = hereditary coproporphyria; INR = international normalized ratio; NRS = numeric rating scale; OLE = open-label extension; PBG = porphobilinogen; PCS = Physical Component Summary; PGIC = Patient Global Impression of Change; PK = pharmacokinetic; PPEQ = Porphyria Patient Experience Questionnaire; RCT = randomized controlled trial; SC = subcutaneous; SF-12 = 12-item Short Form Health Survey; TBIL = total bilirubin; ULN = upper limit of normal; VP = variegate porphyria.
Note: Two additional reports were included (Balwani18 and Health Canada Reviewers Report19).
aDefined as any of the following: AIP = mutation in the HMBS gene (also referred to as the PBGD gene); HCP = mutation in the CPOX gene; VP = mutation in the PPOX gene; ADP = mutation in the ALAD homozygous or compound heterozygous genes.
bIf a patient’s genetic testing did not identify a mutation in a porphyria-related gene (< 5% of cases), a patient may have been eligible for the study if they had both clinical features and diagnostic biochemical criteria consistent with AHP.
cUsing the Modification of Diet in Renal Disease formula.
Description of Studies
Figure 2 outlines the study design. Study 003 was a multi-centre, placebo-controlled, DB phase III study designed to evaluate the efficacy and safety of givosiran administered once monthly in patients with AHP. Its primary objective was to evaluate the effect of givosiran compared to placebo in terms of the rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP. Study 003 was conducted internationally, and 1 of the 36 participating study centres was located in Canada.
An interactive response system was used to assign patients to a treatment and to maintain blinding; members of the study team did not have access to unblinded data until the end of the 6-month DB period. A total of 94 patients were randomized in a 1:1 ratio to receive 2.5 mg/kg givosiran or placebo administered subcutaneously. Randomization was stratified by AHP type: AIP with genetic evidence of mutation in the HMBS gene versus non-AIP (HCP, VP, ADP, or any AHP without an identified mutation in a porphyria-related gene). Patients with AIP were further stratified by prophylactic hemin use at screening as well as by each patient’s historical AAR. More specifically, patients who used hemin prophylactically before study entry were stratified by their historical porphyria AAR over the 12 months before randomization, based on having fewer than 7 attacks versus 7 attacks or more. Patients who were not using hemin prophylactically before study entry were stratified by their historical porphyria AAR in the previous 12 months up until randomization, based on fewer than 12 attacks versus 12 attacks or more.
Patients participated in a screening period for up to 2 months before randomization. During this period, patient history and ALA and PBG levels were collected for study eligibility purposes. Patients who did not meet the study eligibility criteria were able to undergo rescreening. The 6-month DB treatment period followed, with an OLE period of up to 29 months (summarized in Other Relevant Evidence later in this section). A 1-month follow-up period concluded the study.
The study was enriched for attack frequency to ensure the ability to measure a difference in treatment effect on the primary composite porphyria attack end point.
Patients who discontinued from the study drug or withdrew from the study were not replaced.
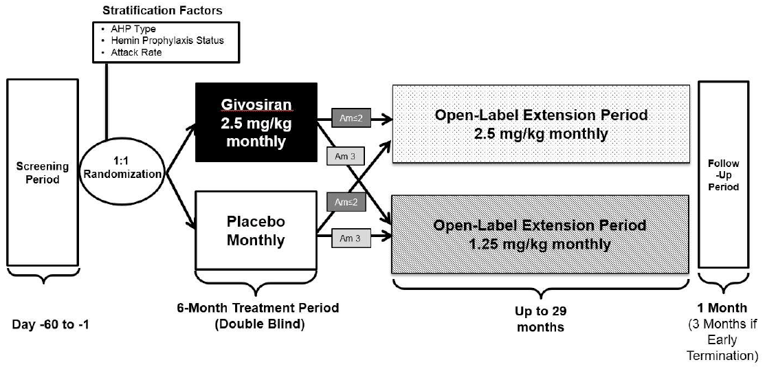
AHP = acute hepatic porphyria; Am ≤ 2 = original protocol, protocol amendment 1, and protocol amendment 2; Am 3 = protocol amendment 3.
Source: Clinical Study Report.6
Populations
Inclusion and Exclusion Criteria
To be eligible for inclusion in Study 003, patients had to be at least 12 years old with a documented diagnosis of AIP, HCP, VP, or ADP. The diagnosis was based on clinical features, evidence of elevated urinary or plasma PBG or ALA (at least 4 × the upper limit of normal [ULN]) within the year before screening, and genetic evidence of mutation in a porphyria-related gene (AIP = mutation in the HMBS gene; HCP = mutation in the CPOX gene; VP = mutation in the PPOX gene; ADP = mutation in the ALAD homozygous or compound heterozygous genes). Patients with genetic testing that did not identify a mutation in a porphyria-related gene were considered eligible if they had both clinical features and diagnostic biochemical criteria consistent with AHP, as outlined in Figure 3. Patients were also required to have active disease (at least 2 composite porphyria attacks; i.e., attacks requiring hospitalization, urgent health care visit, or treatment with IV hemin at home) within 6 months before screening and be willing to abstain from prophylactic use of hemin during the trial.
Patients were excluded from Study 003 if they had elevated levels of ALT (≥ 2 × ULN), total bilirubin (> 1.5 × ULN), or an international normalized ratio greater than 1.5; impaired renal function (eGFR < 30 mL/min/1.73 m2); or a history of allergies, infections, or malignancy. Additionally, patients were excluded if they had a history of recurrent pancreatitis or a major surgery planned within 6 months, or if they were pregnant, breastfeeding, or planning to become pregnant.
Figure 3: Biochemical Diagnosis of AHP in the Absence of Identified Mutation in a Porphyria-Related Gene
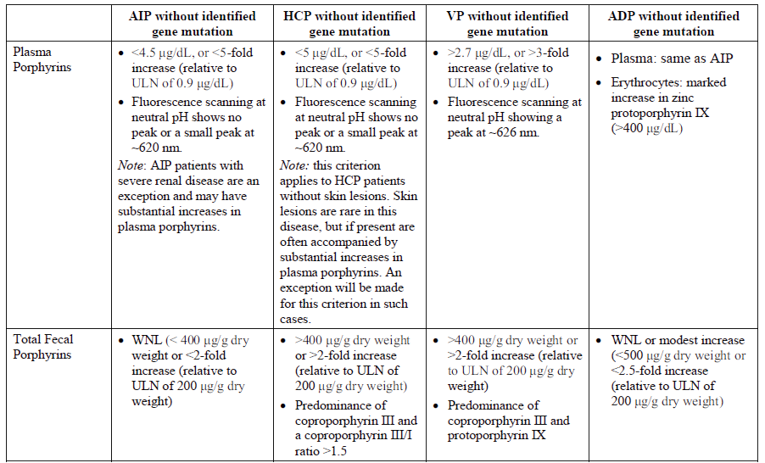
ADP = aminolevulinic acid dehydratase-deficient porphyria; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; HCP = hereditary coproporphyria; ULN = upper limit of normal; VP = variegate porphyria; WNL = within normal limits.
Source: Clinical Study Report.6
Baseline Characteristics
A summary of baseline characteristics for Study 003 is provided in Table 6. The data from Study 003 are presented using 2 datasets: 1 for patients with AIP and 1 for all patients with AHP. The dataset for patients with AIP included only patients who were identified with the AIP subtype. All patients with AHP, regardless of subtype (i.e., patients with AIP, ADP, HCP, VP, and patients with AHP without an identified mutation in a porphyria-related gene) were included in the “all patients with AHP” dataset. Where relevant, the data from Study 003 will be described using these 2 datasets for the remainder of the report.
Patients with AIP who were randomized in Study 003 were between the ages of 19 years and 65 years, with a mean age of 37.3 years to 40.7 years. Most were female (89% to 91%) and White (77% to 80%); 35% to 40% resided in North America. The mean number of years since diagnosis was 8 years to 11 years, and between 40% and 44% of patients had prior experience with prophylactic hemin. During the 6 months before screening for Study 003, the median number of porphyria attacks requiring hospitalization, urgent health care visit, or hemin use at home was between 3 and 4, with a range of 0 to 25. Using the composite definition of porphyria attacks, the median historical AAR was 8 attacks (range = 4 to 34) and 8 attacks (range = 0 to 46) in the givosiran and placebo treatment groups, respectively. Detail regarding the reason for including a patient with a minimum AAR of 0 in the 6 months before screening in the placebo treatment group was not provided. While not having a porphyria attack, between 48% and 56% of patients reported having chronic symptoms, and 28% to 30% reported chronic opioid use.
In general, the baseline characteristics were similar between treatment groups. Among patients with AIP, the givosiran treatment group had a greater number of years since their diagnosis, a greater proportion of patients with a prior hemin prophylaxis regimen, and a smaller proportion of patients with prior chronic symptoms when not having attacks.
Baseline characteristics in patients with AHP were similar to those reported for patients with AIP. Patient comorbidities are summarized in Table 7.
Table 6: Summary of Baseline Characteristics (Study 003, 6-Month Double-Blind Period, SAS)
Characteristic | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Demographic characteristics | ||||
Age (years), mean (SD) | 40.7 (12.0) | 37.3 (10.5) | 40.1 (12.1) | 37.4 (10.5) |
Age (years), range | 19 to 65 | 20 to 60 | 19 to 65 | 20 to 60 |
Gender (% female), n (%) | 41 (89.1) | 39 (90.7) | 43 (89.6) | 41 (89.1) |
Body weight (kg), mean (SD) | 65.71 (15.91) | 68.50 (16.69) | 65.85 (15.63) | 67.88 (16.82) |
BMI (kg/m2), mean (SD) | 24.27 (5.24) | 25.66 (6.34) | 24.31 (5.15) | 25.49 (6.38) |
Race, n (%) | ||||
White | 37 (80.4) | 33 (76.7) | 39 (81.3) | 34 (73.9) |
Black | 0 | 0 | 0 | 1 (2.2) |
Asian | 8 (17.4) | 6 (14.0) | 8 (16.7) | 7 (15.2) |
Other | 1 (2.2) | 4 (9.3) | 1 (2.1) | 4 (8.7) |
Region, n (%) | ||||
North America | 16 (34.8) | 17 (39.5) | 16 (33.3) | 18 (39.1) |
Europe | 22 (47.8) | 18 (41.9) | 23 (47.9) | 19 (41.3) |
Other (Asia, Australia, Mexico) | 8 (17.4) | 8 (18.6) | 9 (18.8) | 9 (19.6) |
Disease characteristics | ||||
Years since diagnosis, mean (SD) | 11.47 (11.27) | 8.44 (8.69) | 11.09 (11.18) | 8.25 (8.47) |
Years since diagnosis, range | 0.2 to 43.3 | 0.1 to 38.5 | 0.2 to 43.3 | 0.1 to 38.5 |
Patients with prior hemin prophylaxis regimen, n (%) | 20 (43.5) | 17 (39.5) | 20 (41.7) | 18 (39.1) |
Number of attacks requiring hospitalization, urgent health care visit or hemin use at home during the 6 months before screening, median (range) | 4 (2 to 24) | 3 (0a to 25) | 4 (2 to 24) | 3 (0a to 25) |
Historical AARb | ||||
High, n (%) | 23 (50.0) | 20 (46.5) | 24 (50.0) | 21 (45.7) |
Low, n (%) | 23 (50.0) | 23 (53.5) | 24 (50.0) | 25 (54.3) |
Median (range) | 8 (4 to 34) | 8 (0 to 46) | 8 (4 to 34) | 7 (0 to 46) |
Patients with prior chronic symptoms when not having attacks,c n (%) | 22 (47.8) | 24 (55.8) | 23 (47.9) | 26 (56.5) |
Patients with prior chronic opioid use when not having attacks,d n (%) | 14 (30.4) | 12 (27.9) | 14 (29.2) | 13 (28.3) |
Urinary ALA (mmol/mol Cr), mean (SD) | 19.97 (16.80) | 17.52 (10.89) | 19.65 (16.61) | 17.27 (10.79) |
Cr normalized urinary ALA (× ULN), mean (SD) | 13.59 (11.43) | 11.92 (7.41) | 13.37 (11.30) | 11.75 (7.34) |
Urinary PBG (mmol/mol Cr), mean (SD) | 50.36 (34.33) | 46.80 (24.32) | 49.00 (34.41) | 45.39 (24.52) |
Cr normalized urinary PBG (× ULN), mean (SD) | 367.56 (250.55) | 341.62 (177.54) | 357.67 (251.17) | 331.34 (178.98) |
AAR = annualized attack rate; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; BMI = body mass index; Cr = creatinine; PBG = porphobilinogen; SAS = Safety Analysis Set; SD = standard deviation; ULN = upper limit of normal.
aOne patient was included in the primary efficacy analysis but was excluded from the sensitivity analysis based on the per-protocol set.
bHistorical composite AAR was calculated based on the number of attacks requiring hospitalization, health care facility visit, or hemin use at home during the 6 months before randomization. For patients on a hemin prophylaxis regimen before the study, AAR is “high” if the historical AAR is greater than or equal to 7 and “low” if less than 7. For patients who were not on a prior hemin prophylaxis regimen, AAR is “high” if historical AAR is greater than or equal to 12 or “low” if less than 12.
cYes, if patients experienced symptoms of porphyria when not having an attack daily or on most days before the study.
dYes, if patients took opioids for porphyria when not having an attack daily or on most days.
Table 7: Medical History in Common With More Than 15.0% of Patients With AHP (Study 003, 6-Month Double-Blind Period, SAS)
Characteristic | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Surgical and medical procedures | 40 (87.0) | 38 (88.4) | 42 (87.5) | 40 (87.0) |
Central venous catheterization | 33 (71.7) | 31 (72.1) | 35 (72.9) | 32 (69.6) |
Nervous system disorders | 31 (67.4) | 21 (48.8) | 32 (66.7) | 24 (52.2) |
Neuropathy peripheral | 20 (43.5) | 14 (32.6) | 20 (41.7) | 16 (34.8) |
Metabolism and nutrition disorders | 25 (54.3) | 25 (58.1) | 25 (52.1) | 27 (58.7) |
Iron overload | 16 (34.8) | 14 (32.6) | 16 (33.3) | 15 (32.6) |
Investigations | 21 (45.7) | 25 (58.1) | 22 (45.8) | 25 (54.3) |
Transaminases increased | 16 (34.8) | 18 (41.9) | 17 (35.4) | 18 (39.1) |
Gastrointestinal disorders | 22 (47.8) | 20 (46.5) | 23 (47.9) | 22 (47.8) |
Constipation | 11 (23.9) | 6 (14.0) | 12 (25.0) | 8 (17.4) |
Gastroesophageal reflux disease | 11 (23.9) | 6 (14.0) | 11 (22.9) | 6 (13.0) |
Nausea | 7 (15.2) | 10 (23.3) | 7 (14.6) | 10 (21.7) |
Psychiatric disorders | 25 (54.3) | 17 (39.5) | 26 (54.2) | 18 (39.1) |
Depression | 16 (34.8) | 8 (18.6) | 17 (35.4) | 8 (17.4) |
Anxiety | 13 (28.3) | 9 (20.9) | 13 (27.1) | 9 (19.6) |
Insomnia | 8 (17.4) | 7 (16.3) | 9 (18.8) | 8 (17.4) |
Renal and urinary disorders | 12 (26.1) | 17 (39.5) | 13 (27.1) | 17 (37.0) |
Chronic kidney disease | 8 (17.4) | 9 (20.9) | 8 (16.7) | 9 (19.6) |
Vascular disorders | 14 (30.4) | 15 (34.9) | 14 (29.2) | 15 (32.6) |
Hypertension | 13 (28.3) | 11 (25.6) | 13 (27.1) | 11 (23.9) |
Blood and lymphatic system disorders | 12 (26.1) | 16 (37.2) | 12 (25.0) | 17 (37.0) |
Anemia | 6 (13.0) | 13 (30.2) | 6 (12.5) | 13 (28.3) |
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; SAS = Safety Analysis Set.
Source: Clinical Study Report.6
Interventions
Patients were randomized to givosiran or placebo. Those randomized to givosiran received 2.5 mg/kg once monthly during the 6-month DB period of Study 003. Givosiran was supplied as a sterile solution in water for subcutaneous injection and administered by a qualified and authorized health care professional into patients’ abdomens, upper arms, or thighs. Patients were observed for at least 20 minutes following the injection. Patients randomized to placebo received sodium chloride 0.9% weight/volume for subcutaneous injection. The placebo was administered using identical packaging and product volume. To maintain blinding, syringes were masked before the withdrawal of givosiran or placebo from a masked vial.
Modifications to the dose of givosiran were not permitted during the 6-month DB period, with the exception of changes due to ALT elevation. A downward titration to 1.25 mg/kg once monthly (from givosiran 2.5 mg/kg once monthly) for patients who were withheld from treatment due to elevated ALT was introduced in accordance with protocol amendment number 2.
Patients were permitted to miss 1 dose of the study drug within the 6-month treatment period. If more than 1 dose was missed, a decision regarding continuation of treatment was made at the discretion of the investigator and medical monitor.
Patients recorded concomitant medication use through an electronic case report form. A list of concomitant medications reported during the 6-month DB period of the ENVISION trial is available in Table 12. Hemin prophylaxis was not permitted during the study, but patients were allowed to receive hemin for the treatment of acute attacks if clinically indicated. Hemin used for acute attacks was reported as a concomitant medication. Patients who were receiving treatment with a GnRH analogue at screening were permitted to enrol if they remained on GnRH treatment throughout the 6-month DB period. The sponsor reported that initiation of treatment with GnRH during the study was discouraged, but the extent of GnRH use was not reported. Analgesic usage for the management of porphyria and porphyria attacks was permitted based on clinical judgment and either reported in the patient’s e-diary or as a concomitant medication, if taken at a health care facility. Lastly, standard vitamins and topical medications were permitted; however, topical steroids were not allowed to be applied anywhere near the injection site unless medically indicated.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized here. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 003 |
|---|---|
Annualized rate of porphyria attack | Primary/secondary |
HRQoL by SF-12a, EQ-5D-5L, PGIC | Secondary/exploratory |
Daily worst pain score, nausea score, and fatigue score | Secondary/exploratory |
Annualized rate of hemin administration | Secondary/exploratory |
PPEQ | Exploratory |
Analgesic usage (opioid and non-opioid) | Exploratory |
Missed days of work | Exploratory |
Urinary ALA levels | Secondary/exploratory |
Urinary PBG levels | Secondary/exploratory |
ALA = aminolevulinic acid; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; HRQoL = health-related quality of life; PBG = porphobilinogen; PGIC = Patient Global Impression of Change; PPEQ = Porphyria Patient Experience Questionnaire; SF-12 = 12-item Short Form Health Survey.
Note: The outcomes described as “secondary/exploratory” were analyzed as secondary outcomes in patients with AIP and exploratory outcomes in patients with AHP.
aThe PCS of the SF-12 analyzed in patients with AIP was a secondary outcome; all other outcomes related to the SF-12 were exploratory.
Porphyria Attacks
In Study 003, the primary composite outcome was the annualized rate of porphyria attacks, which included attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP during the 6-month DB period of the study. All porphyria attacks, which included all 4 definitions of porphyria-related attacks, were also reported.
Porphyria attacks were defined as acute episodes of neurovisceral pain in the abdomen, back, chest, extremities, or limbs with no other medically determined cause that required treatment with IV dextrose, hemin, carbohydrates, analgesics, or other medications (e.g., antiemetics at a dose frequency beyond that used in the patient’s usual daily management). Patients or caregivers were to record any potential porphyria attacks for the duration of the study when they occurred using the provided electronic diary, by telephone, or by email to the study site or investigator. Of note, patients or caregivers were trained on the use of the electronic diary at screening, and complete instructions were available at study sites in a study manual. Health care professionals were also able to notify the study centre of porphyria attacks. All potential porphyria attacks were adjudicated by the investigator. If an event was determined to be inconsistent with a protocol-defined attack, an alternative reason was recorded, such as duplicate entry, AE, or other.
Further, the ENVISION trial used the following definitions to identify non-overlapping components of events relating to porphyria attacks:
attack requiring hospitalization: admission to an inpatient unit or a visit to an emergency department that resulted in a stay of at least 24 hours
attack requiring urgent health care visit: urgent, unscheduled visit to a physician’s office or practice, an infusion centre, or an emergency department visit that did not meet the criteria for hospitalization
attack requiring IV hemin administration at home: home was any location that did not meet the criteria for hospitalization or an urgent health care visit
attack at home not requiring IV hemin.
No literature was identified that assessed the annualized rate of porphyria attack for validity, reliability, or responsiveness in patients with porphyria. No MID was identified in populations with AHP.
Health-Related Quality of Life
In the ENVISION trial, HRQoL was assessed through the SF-12, EQ-5D-5L, and PGIC.
12-Item Short Form Health Survey
The SF-12 is a generic, patient-reported measure of HRQoL based on the 36-item version of the survey (SF-36). Patients answer based on a 4-week recall period. Each of the 12 items fall into 1 of 8 health scales, including: physical functioning (PF) (2 items), RP (2 items), BP (1 item), general health (GH) (1 item), vitality (VT) (1 item), social functioning (SF) (1 item), role emotional (RE) (2 items), and mental health (MH) (2 items).20 The first 4 scales (PF, RP, BP, and GH) make up the PCS, while the latter 4 (VT, SF, RE, and MH) fall under the MCS. The PCS and MCS correspond to the physical and psychological burden of disease, respectively. The component summaries are standardized to have a mean of 50 and an SD of 10 based on the general US population, and scores can range from 0 to 100, with higher scores reflecting better HRQoL.20 Validity and reliability have been demonstrated in a diverse population; however, no literature was identified that assessed the SF-12 for validity, reliability, or responsiveness in patients with porphyria. An MID was not identified for this population, either; however, MIDs for the SF-12 PCS and MCS have been estimated based on a study of 458 patients with lower back pain.21 Overall, an improvement of at least 3.29 on the PCS and 3.77 on the MCS would be clinically meaningful in patients with low back pain.21 In the ENVISION trial, the component summary scores and individual item scores were reported as a change from baseline.
EuroQol 5-Dimensions 5-Levels Questionnaire
The EQ-5D-5L is a generic, self-reported, HRQoL instrument developed by the EuroQol Group that is applicable to a wide range of health conditions and treatments.22 The EQ-5D-5L consists of a descriptive system and the EuroQol VAS.
The descriptive system comprises 5 dimensions: mobility, self-care, usual activities, pain or discomfort, and anxiety or depression. Each dimension is answered based on 5 levels (1 = no problems; 2 = slight problems; 3 = moderate problems; 4 = severe problems; and 5 = extreme problems or unable to perform, the worst response in the dimension).22 Respondents choose the level that reflects their health state for each of the 5 dimensions. In total, there are 3,125 possible unique health states defined by the EQ-5D-5L, with 11111 and 55555 representing the best and worst health states, respectively. Results from the EQ-5D-5L descriptive system are converted into a single index score using a scoring algorithm that takes local patient and population preferences into account. Therefore, the index score is a country-specific value and a major feature of the EuroQol 5-Dimensions (EQ-5D) instrument.23 The range of index scores will differ according to the scoring algorithm used; however, in all scoring algorithms of the EQ-5D-5L, a score of 0 represents the health state “dead” and 1.0 reflects “perfect health.”
The VAS records the respondent’s self-rated health on a vertical VAS where the end points are labelled 0 (“the worst health you can imagine”) and 100 (“the best health you can imagine”). Respondents are asked to mark an X on the point of the VAS that best represents their health on that day. The EQ-5D index and VAS scores can be summarized and analyzed as continuous data.22,23
The validity and reliability of the EQ-5D-5L have been demonstrated in a diverse population, but no literature was identified that assessed the EQ-5D-5L for validity, reliability, or responsiveness in patients with porphyria. An MID was not identified in populations with AHP; however, the MID for the index score was estimated to range from 0.037 to 0.056 in the general Canadian population. Both the index score and VAS were reported in the ENVISION trial. Index scores were described as a change from baseline, and domain scores were reported categorically. The VAS was reported as a change from baseline.
Patient Global Impression of Change
The PGIC is a widely used, validated outcome measure for clinical pain trials.24,25 It is a single question answered on a 7-point numerical scale to indicated perceived change in overall health status since the study began. Patients are asked to consider their overall health since the start of the trial by choosing a response (1 = very much improved; 2 = much improved; 3 = minimally improved; 4 = no change; 5 = minimally worse; 6 = much worse; 7 = very much worse).26 The PGIC questionnaire has been recommended for use in chronic pain clinical trials by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) as a core outcome measure of global improvement with treatment.27 No literature was identified that assessed the PGIC for validity, reliability, or responsiveness in patients with porphyria. No MID was identified in populations with AHP. In the ENVISION trial, the PGIC was reported descriptively. It was also reported as 2 categories: “improved,” including responses for very much improved, much improved, and minimally improved; and “no change or worsening,” including responses for no change, minimally worse, much worse, and very much worse. The proportion of patients who improved was compared between treatment groups.
Management of Symptoms
Patients or caregivers were asked to record assessments of pain, nausea, and fatigue in the patient’s electronic diary daily throughout the ENVISION trial. As noted under the description of outcomes for porphyria attacks, patients or caregivers were trained in the use of the electronic diary, and instructions were provided in a study manual. The questionnaires used to evaluate each of these symptoms are described in this section. For each symptom, a weekly (7-day) score was derived from an average of daily symptom measurements reported in the electronic diary. The change from baseline in the weekly average score was reported in the ENVISION trial. The AUC of the change from baseline was also reported to account for peaks in symptoms during attacks and lingering chronic symptoms.
Pain
The Brief Pain Inventory (BPI) is a self-reported, 11-item questionnaire designed to provide information on 2 subscales: pain intensity (the sensory dimension, 4 items) and the degree to which pain interferes with functioning in daily living (the reactive dimension, 7 items). The ENVISION trial used a single question from the BPI Short Form (BPI-SF) to assess the severity of daily pain.6 Patients were asked to rate the worst level of pain they experienced during the past 24 hours on an NRS from 0 = “no pain” to 10 = “as bad as you can imagine.” As a single item, it has shown internal consistency (0.70 < Cronbach alpha < 0.90) and reliability (between 0.8 and 0.96) among various populations.28 Construct, convergent, and discriminative validity, internal consistency, and test-retest reliability have been demonstrated for the BPI-SF28,29; however, no literature was identified that assessed the BPI-SF for validity, reliability, or responsiveness in patients with porphyria. Although an overall MID of the BPI has not been identified from the literature, a 2-point change has been suggested as a reasonable estimate of the MID for worst pain among breast cancer patients with metastatic disease.30 An MID specific to patients with AHP was not identified.
Fatigue
The Brief Fatigue Inventory (BFI) is a self-reported questionnaire to assess the severity and impact of fatigue on daily functioning. The items of the BFI are measured on a 0-to-10 NRS. In the ENVISION trial, a single question was used from the Brief Fatigue Inventory – Short Form (BFI-SF) to assess the severity of daily fatigue.6 Patients were asked to rate the worst level of fatigue they experienced during the past 24 hours on a scale from 0 = “no fatigue” to 10 = “as bad as you can imagine.”
Validity and reliability of the whole scale was assessed in samples of patients with cancer, rheumatoid arthritis, and community-dwelling adults and older adults.31 Construct validity, concurrent validity, and discriminant validity of the BFI have been demonstrated in cancer patients, as well as reliability, which was acceptable (Cronbach alpha values were 0.95 to 0.96).32 Further, both the Chinese and Taiwanese versions of the BFI were previously validated in patients with cancer and provided evidence of validity of the single item for worst fatigue compared to the Chinese version of the SF-36 and Eastern Cooperative Oncology Group (ECOG) performance status, as well as the Taiwanese Profile of Mood States (POMS) short-form questionnaire.33 An MID for the worst fatigue item was estimated to be 1.5 using the 0.5 SD method,33 but no MID was identified in populations with AHP. Moreover, no literature was identified that assessed the BFI-SF for validity, reliability, or responsiveness in patients with porphyria. Nausea
The nausea NRS was used to assess nausea in Study 003. It is a self-reported, single question used to assess a patient’s level of nausea in the past 24 hours on a scale from 0 to 10, where 0 represents no nausea and 10 represents “nausea as bad as you can imagine.” Construct validity and reliability were demonstrated for the instrument.34 No literature was identified that assessed the Nausea Numerical Rating Scale for validity, reliability, or responsiveness in patients with porphyria. Further, an MID was not identified in populations with AHP.
Rescue Medication Use
Measures of rescue medication in Study 003 included an evaluation of hemin use for acute attacks. This was reported as the annualized rate of hemin use over the 6-month DB period.
Opioid Use
Opioid usage was reported under analgesic usage in Study 003. Analgesic usage at home by patients throughout the study was self-reported through the electronic diary that was also used to report porphyria attacks and symptoms. Analgesic usage was reported using daily questions regarding type, dose, and frequency. Analgesic medications that were taken at a health care facility, such as during an attack, were captured as concomitant medication usage. Analgesic medication usage was reported descriptively by medication category (opioid, non-opioid, either opioid or non-opioid). This included the proportion of patients reporting analgesic medication use and the proportion of days with analgesic use during the 6-month DB period.
Activities of Daily Living
In the ENVISION trial, activities of daily living were reported using the PPEQ. The PPEQ was described by the sponsor as a custom tool consisting of 8 questions used to assess a patient’s ability to perform daily living activities (questions 1 to 5), treatment experience (questions 6 and 7), and functional status (question 8). Questions are answered on a 5-point global rating of change scale.6 More specifically, patients select from the following options for questions 1 to 7: much better, minimally better, no change, minimally worse, or much worse, based on their current experience compared to before the start of the study. Question 8 is answered by choosing 1 of the following: always, most of the time, sometimes, rarely, or never, based on the last 4 weeks. Responses for each of the 8 questions of the PPEQ were reported descriptively in the ENVISION trial. No literature was identified that assessed the PPEQ for validity, reliability, or responsiveness in patients with porphyria. No MID was identified in populations with AHP.
Hospitalization and Health Care Use
Hospitalization and health care use were reported indirectly through reporting of porphyria attacks, namely those leading to hospitalizations or urgent health care visits. These outcomes were previously described under frequency of porphyria attacks.
Ability to Work or Attend School
In Study 003, the ability to work or attend school was reported as the number of missed days of work or school due to porphyria symptoms or attacks in the past 4 weeks and summarized descriptively.
ALA, PBG, and Porphyrin Levels
Study 003 reported urinary ALA levels and PBG levels during the study. Spot urine samples for ALA and PBG measurement were collected at study visits before treatment dosing; however, if hemin was used for an attack, scheduled urinary ALA and PBG were collected 4 days after the patient’s last hemin dose. If feasible and permitted by local regulations, urine samples for ALA and PBG assessments may have been collected by a home health care professional, sent to the study centre by mail, or brought to the study centre at the next visit. For analysis in the trial, urinary ALA levels were reported at 3 months and 6 months, and PBG was reported at 6 months. Measurements of ALA or PBG that were taken up to 3 days following hemin use were treated as missing and excluded from analysis.
Mortality
Mortality was not reported as an efficacy outcome in Study 003.
Harms
Safety outcomes reported included the incidence, severity, seriousness, and relatedness of AEs over the 6-month DB period in patients with any AHP. Porphyria attacks were reported as efficacy outcomes; therefore, they were not treated as AEs or SAEs. However, non–porphyria-related AEs during porphyria attacks were reported. Other AEs of special interest were reported, and these included ALT elevations greater than 3 times the ULN, lipase or amylase greater than 3 times the ULN, severe or serious injection-site reactions, and anaphylactic reactions.
Statistical Analysis
Primary Outcome
The primary outcome of the pivotal trial was annualized rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP over the 6-month DB period. The definitions used for the 3 types of attacks included in the primary outcome are summarized in the Outcomes section. The AAR was calculated using the following formula:
AAR = (total number of porphyria attacks ÷ total number of days in the treatment period) × 365.25
Power Calculation
The study planned to enrol at least 74 patients, including 70 with AIP. It was estimated that 70 patients would yield 90% power to detect a 45% reduction in the AAR at a 2-sided 5% significance level, with the following assumptions: a mean AAR of 8, an SD of 5 in the placebo group, and a mean AAR of 4.4 with an SD of 3 in the givosiran group. The study was also designed to have at least 80% power with a dropout rate of up to 15% under the same assumptions that have been described.
Statistical Test or Model
A summary of the statistical testing methodology is presented in Table 9. The primary analysis was conducted in the mFAS, which included only patients with AIP from the FAS. A negative binomial regression model was used for the primary analysis, which included stratification factors for patients with AIP as fixed effects — namely, the use of a hemin prophylaxis regimen before the study (yes versus no) and historical AAR (high versus low). The logarithm of the amount of time (in units of years) that each patient spends in the 6-month DB period was included in the model as an offset variable to account for the different lengths of follow-up time among patients. An estimated ratio of mean AARs between the givosiran and placebo groups with corresponding 95% CI based on the negative binomial regression model was reported. Data were not imputed for the primary analysis of AAR. Descriptive statistics for the AAR were also reported.
Each of the 3 components of the primary outcome or AAR were analyzed similarly to the primary analysis. A zero-inflated negative binomial regression model was also used to analyze each component to address the potential issue of an “excessive” number of patients with zero attacks. A model-based analysis would not be conducted if fewer than 10 patients experienced an attack.
Secondary Outcomes
Secondary outcomes were conducted in the mFAS unless otherwise specified.
Annualized event rate type end points were analyzed in the same way as the primary analysis of AAR. This included the annualized rate of administered hemin doses in patients with AIP and AAR in patients with any type of AHP (in the FAS) over the 6-month DB treatment period. Data were not imputed for any of these outcomes.
The biomarker outcomes (urinary ALA and PBG) were analyzed and reported as the LS mean difference between treatment groups at 3 months and 6 months for urinary ALA, and at 6 months for urinary PBG. The analysis of biomarker end points involved a comparison between treatment groups using a mixed-effects model for repeated measures (MMRM), with the baseline biomarker level as a continuous covariate, stratification factors, visit, treatment, and visit by treatment as fixed effects, and patient as a random effect. Measurements of urinary ALA or PBG within 3 days after hemin use were treated as missing and excluded from analysis. The SF-12 scores were also analyzed as a change from baseline at month 6 using an MMRM model with baseline score as a continuous covariate, and treatment group, stratification factors, visit (month 3 or month 6), and visit by treatment interaction as fixed effects. Missing data for biomarker outcomes and the SF-12 were implicitly imputed through the use of the MMRM model.
Daily worst scores for pain, fatigue, and nausea were measured using the BPI-SF NRS, BPI-SF NRS, and an NRS, respectively, and reported through electronic patient diaries. The changes from baseline in weekly mean scores were reported, as well as the AUC from baseline over 6 months. An ANCOVA model was used to compare the AUC change from baseline between treatment groups. The ANCOVA model included fixed effects of treatment arms and the 2 stratification factors. Weekly mean scores were derived from an average of daily scores for a completed week, defined by the completion of at least 4 daily diary entries. A missing week was defined as a week with at least 4 missing entries. If an attack occurred during the missing week, a mean weekly score was computed if there was at least 1 entry. If no entries were reported, the weekly mean score was imputed using the average of all non-missing attack-week weekly mean scores if there were available data; if not, the weekly mean score was imputed by the worst weekly mean scores within the same treatment period for the patient. In a missing week without any attack days, the weekly mean score was imputed using the last observed non-missing week without any attack days, if available; if not, the baseline observation carried forward was imputed.
Exploratory Outcomes
All secondary end points defined for the patients with AIP were analyzed for the patients with AHP using similar methods. Continuous exploratory end points in patients with AIP included the EQ-5D-5L index score, which was analyzed using an MMRM model similar to the model used for the PCS of the SF-12. The remainder of the outcomes were reported descriptively, which included the PGIC at month 6 and month 12, missed days of work or school at month 6, PPEQ, patient responses to each of the EQ-5D domains, and analgesic medication use.
Testing Strategy
The primary analysis was conducted at a 2-sided significance level of 0.049, reflecting a penalty of 0.001 for an unblinded interim analysis. To control for the overall type I error rate, a fixed-sequence, hierarchical testing strategy was implemented. If the primary analysis was statistically significant, then the secondary end points were tested at the same significance level of 0.049 in the order-specified testing hierarchy. Statistical testing was not conducted following failure to reject the null hypothesis at an early end point within the hierarchy; however, nominal P values were reported. Statistical tests were conducted according to the following hierarchy:
urinary ALA levels in patients with AIP at 3 months
urinary ALA levels in patients with AIP at 6 months
urinary PBG levels in patients with AIP at 6 months
annualized rate of administered hemin doses (evaluated by annualized days of hemin use) in patients with AIP over the 6-month DB period
annualized rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with any AHP over the 6-month DB period
daily worst pain score as measured by BPI-SF NRS in patients with AIP over the 6- month DB period
daily worst fatigue score as measured by BFI-SF NRS in patients with AIP over the 6-month DB period
daily worst nausea score as measured by NRS in patients with AIP over the 6-month DB period
change from baseline in the PCS of the SF-12 in patients with AIP at 6 months.
Table 9: Statistical Analysis of Efficacy End Points (Study 003, 6-Month DB Treatment Period)
End pointa | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Primary analysis: AAR | Negative binomial regression, including amount of time spent in the 6-month DB period as an offset variable | Fixed effects: stratification factors | Scenario-based sensitivity analyses:
Per-protocol analysis |
Biomarker end points
| MMRM with corresponding biomarker level at baseline as a continuous covariate | Fixed effects: stratification factors, visit, treatment, and visit by treatment Random effects: patient | For ALA-related end points, if the dropout rate is 5% or greater in the givosiran treatment group, sensitivity analyses will be performed using a pattern mixture model approach |
Annualized-event-rate type end points
| Same as primary analysis; i.e., negative binomial regression, including amount of time spent in the 6-month DB period as an offset variable | Same as primary analysis | NA |
Daily worst scores for
| ANCOVA | Fixed effects: treatment arms and stratification factors | NA |
SF-12, PCS | MMRM with baseline score as a continuous covariate | Fixed effects: treatment group, stratification factors, visit, and visit by treatment interaction | NA |
EQ-5D-5L | Similar to the SF-12, PCS | Similar to the SF-12, PCS | NA |
AAR = annualized attack rate; AHP = acute hepatic porphyria; ALA = aminolevulinic acid; ANCOVA = analysis of covariance; BFI-SF = Brief Fatigue Inventory – Short Form; BPI-SF = Brief Pain Inventory – Short Form; DB = double blind; EQ-5D-5L = EuroQol 5-dimension questionnaire; FAS = full analysis set; MMRM = mixed-effects model for repeated measures; NA = not applicable; NRS = numeric rating scale; PBG = porphobilinogen; PCS = Physical Components Summary; SF-12 = 12-item Short Form Health Survey.
aEnd points were analyzed in patients with AIP (mFAS) unless otherwise indicated.
Source: Clinical Study Report.6
Interim Analysis
A preplanned, unblinded interim analysis of patients’ ALA levels at 3 months was conducted when approximately 30 patients with AIP had completed at least 3 months of the treatment period. The significance level for the comparison at the interim was 0.001 (2-sided).
Subgroup Analyses
The following subgroups were included in the CADTH systematic review protocol: AHP subtype (AIP, ADP, HCP, VP) and frequency and history of attacks. Study 003 included pre-specified subgroup analyses of the primary outcome, using similar methods as the primary analysis, for the following subgroups:
age at screening (< or ≥ median age in the overall population)
race (White or non-White)
sex (female or male)
region group 1: North America (including the US and Canada) or other (outside North America)
region group 2: Europe or other (outside Europe)
baseline body mass index (< 25 or ≥ 25)
prior hemin prophylaxis status (yes or no)
historical attack rates before randomization based on the hemin prophylaxis status before the study (high or low)
for patients on a hemin prophylaxis regimen at the time of screening, if AAR ≥ 7, the patient is considered having high attack rates before the study; for patients who were not on a hemin prophylaxis regimen at screening, AAR ≥ 12 is considered with high attack rates
prior chronic opioid use when not having attacks (yes or no)
prior chronic symptoms when not having attacks (yes or no).
Anticipating that there would be only a few non-AIP patients, the sponsor planned to report only descriptive statistics for these patients. The sponsor also reported that other subgroups, or subgroup analyses of other secondary end points, might be conducted.
For the purpose of this review, subgroup analyses by historical attack rates were presented as per the systematic review protocol. Additionally, all outcomes presented in this review were reported in the mFAS where available, in alignment with the subgroup for AHP subtypes outlined in the systematic review protocol.
Sensitivity Analyses
The following sensitivity analyses were conducted on the primary end point:
To account for undercounting attacks, a sensitivity analysis counting all discrete attacks was conducted. For investigator-confirmed attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home, all discrete attacks were counted even if they overlapped during a day.
To account for overcounting attacks, a sensitivity analysis was conducted where attacks were counted using a 2-day window. More specifically, the attack-counting window was extended from a 1-day to a 2-day window for investigator-confirmed attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home. This meant that attacks that occurred on the same calendar day, or were separated by 1 calendar day, were counted as 1 attack.
A sensitivity analysis was conducted using all investigator-confirmed attacks, which included all attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in addition to attacks treated at home not requiring hemin use.
A sensitivity analysis was conducted that included all porphyria attacks, counting potential attacks. More specifically, both investigator-confirmed attacks and attacks that were not confirmed by the investigator but were deemed to be potential attacks were included. Duplicates and attacks due to entry errors were excluded.
A sensitivity analysis was conducted in which attacks were treated as recurrent events through the Andersen-Gill model. The primary porphyria attack composite end point was analyzed using an Andersen-Gill model by treatment group and stratification factors (prior hemin prophylaxis status and historical attack rates).
Lastly, the primary porphyria attack composite end point was analyzed based on the per-protocol analysis set.
Protocol Deviations
Major protocol deviations were deviations that were considered to have potentially had a significant impact on the data collected in the study, or to have significantly affected a patient’s rights, safety, or well-being. The sponsor reported a designated plan for handling protocol deviations. Briefly, whether or not to exclude a patient from the per-protocol set was based on the sponsor’s judgment of the potential impact on the primary efficacy results.
Analysis Populations
The FAS included all randomized patients who received at least 1 dose of study drug. Patients were grouped by the treatment group to which they had been randomized.
An mFAS was used in Study 003, which followed the same definition as the FAS except that only randomized patients with AIP identified by a mutation in the HMBS gene were included.
The per-protocol set included all randomized patients with AIP (with identified mutation in the HMBS gene) who received at least 4 doses (> 60%) of the study drug during the 6-month DB period, were followed for the collection of attack data through 6 months (≥ 162 days) and did not experience major protocol deviations that could affect the primary efficacy results (e.g., not meeting the key inclusion or exclusion criteria). Patients were analyzed according to their randomly assigned treatment groups.
The safety analysis set included all patients who received at least 1 dose of the study drug, grouped according to the treatment actually received. Of note, patients who received any amount of givosiran during the 6-month DB period were included in the givosiran arm.
Results
Patient Disposition
A summary of the patient disposition in Study 003 is available in Table 10. A total of 109 patients were screened, and 94 were randomized to receive either givosiran or placebo. A total of 89 (95%) of randomized patients had AIP, and 5 (5%) had non-AIP types of AHP. One patient had HCP and was randomized to givosiran; 2 patients had VP, with 1 randomized to givosiran and 1 randomized to placebo; and 2 patients had AHP without an identified mutation. Both were randomized to placebo. No patients with ADP were enrolled in the study. All randomized patients completed the 6-month DB treatment period. Overall, 1 patient discontinued from study due to an AE. This patient was randomized to the placebo treatment group and had non-AIP AHP.
Nineteen major protocol deviations among 6 patients were reported. The study drug was administered with unmasked syringes at 1 study site, resulting in protocol deviations for 4 patients. Two patients did not meet the inclusion criteria, despite enrolment in the study, which also resulted in protocol deviations.
Table 10: Patient Disposition (Study 003, 6-Month Double-Blind Period, FAS)
Disposition | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Screened, N | NR | 109 | ||
Randomized, Na (%) | 46 (100) | 43 (100) | 48 (100) | 46 (100) |
Discontinued from study, N (%) | 0 | 0 | 0 | 1 (2.1) |
Reason for discontinuation, N (%) | ||||
Adverse events | 0 | 0 | 0 | 1 (2.1) |
FAS, N | NA | NA | 48 | 46 |
mFAS, N | 46 | 43 | NA | NA |
PPS, N | NA | NA | 46 | 42 |
Safety, N | NA | NA | 48 | 46 |
ADP = aminolevulinic acid dehydratase-deficient porphyria; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; FAS = full analysis set; HCP = hereditary coproporphyria; NR = not reported; mFAS = modified full analysis set; NA = not applicable; PPS = per-protocol set; VP = variegate porphyria.
aRandomization was stratified by AHP type: AIP with genetic evidence of mutation in the HMBS gene vs. non-AIP (HCP, VP, ADP, or any AHP without an identified mutation in a porphyria-related gene). Patients with AIP were further stratified by prophylactic hemin use at screening and historical annualized attack rate (low vs. high).
Source: Clinical Study Report.6
Exposure to Study Treatments
Exposure to study treatments is summarized in Table 11. The mean duration of exposure to treatment was 5.5 months for patients with AHP in the givosiran and placebo treatment groups. For patients with AIP, the mean durations of exposure to treatment were 5.6 months and 5.5 months for patients in the givosiran and placebo treatment groups, respectively. In patients with AIP, 8% and 2% of patients in the givosiran and placebo treatment groups, respectively, missed 1 treatment dose, and 1 patient in the givosiran treatment group missed 2 doses; the rest of the patients with AIP did not miss any doses. Reasons for missed doses were summarized by the sponsor. Three patients in the givosiran treatment group and 1 patient in the placebo treatment group missed their doses because the study visit could not be scheduled within the dose window. The fourth patient in the givosiran treatment group missed 1 dose because their week 2 study visit was documented as month 1. One patient in the givosiran treatment group missed 2 doses because givosiran was withheld due to transaminase elevations after they had received 4 doses.
The proportion of patients with missing doses was similar for patients with AHP.
Table 11: Exposure to Study Treatments (Study 003, 6-Month Double-Blind Period, SAS)
Parameter | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Total duration of exposure (months) | ||||
Mean (SD) | 5.57 (0.20) | 5.51 (0.15) | 5.51 (0.45) | 5.50 (0.15) |
Median (range) | 5.52 (5.3 to 6.4) | 5.52 (5.3 to 6.0) | 5.52 (2.7 to 6.4) | 5.52 (5.3 to 6.0) |
Patients with missing doses, n (%) | ||||
No missing dose | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
1 missing dose | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
2 missing doses | v vvvvv | v | v vvvvv | v |
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; SD = standard deviation.
Source: Clinical Study Report.6
Concomitant medication use during the 6-month DB period is summarized in Table 12. Concomitant medication use was reported by almost all patients with AHP (i.e., 95% of patients receiving givosiran and 100% of patients receiving placebo). Among the most commonly reported concomitant medications used during the study (by therapeutic class), a difference in concomitant use of at least 10% between treatment groups was reported for the following classes of medications (in patients treated with givosiran versus placebo, respectively): natural opium alkaloids (46% versus 70%), other heme products (46% versus 74%), electrolyte solutions (10% versus 24%), fluoroquinolones (8% versus 20%), other opioids (8% versus 22%), solutions affecting the electrolyte balance (6% versus 17%), corticosteroids (4% versus 15%), vitamin D and analogues (25% versus 13%), and other antidepressants (19% versus 9%). Overall and differential concomitant medication use among patients with AIP was similar to the overall patient population (patients with AHP). Additionally, concomitant use of GnRH was reported by 2 patients in the givosiran treatment group and 2 patients in the placebo treatment group. All patients who used GnRH concomitantly were patients with AIP.
Table 12: Concomitant Medications (6-Month Double-Blind Period, SAS)
Concomitant medication | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Concomitant medication use | ||||
Patient-reported use of at least 1 concomitant medication, n (%) | 44 (95.7) | 43 (100.0) | 46 (95.8) | 46 (100.0) |
Most commonly reported concomitant medications (by anatomical therapeutic class),a n (%) | ||||
Anilides | 24 (52.2) | 24 (55.8) | 26 (54.2) | 25 (54.3) |
Natural opium alkaloids | 21 (45.7) | 32 (74.4) | 22 (45.8) | 32 (69.6) |
Other heme productsb | 21 (45.7) | 33 (76.7) | 22 (45.8) | 34 (73.9) |
Serotonin (5HT3) antagonists | 17 (37.0) | 19 (44.2) | 18 (37.5) | 20 (43.5) |
Benzodiazepine derivatives | 16 (34.8) | 14 (32.6) | 16 (33.3) | 14 (30.4) |
Propionic acid derivatives | 12 (26.1) | 14 (32.6) | 13 (27.1) | 15 (32.6) |
Proton pump inhibitors | 12 (26.1) | 9 (20.9) | 13 (27.1) | 10 (21.7) |
Other analgesics and antipyretics | 11 (23.9) | 12 (27.9) | 12 (25.0) | 12 (26.1) |
Vitamin D and analogues | 11 (23.9) | 6 (14.0) | 12 (25.0) | 6 (13.0) |
Solutions for parenteral nutrition | 9 (19.6) | 14 (32.6) | 10 (20.8) | 16 (34.8) |
Heparin group | 8 (17.4) | 9 (20.9) | 9 (18.8) | 9 (19.6) |
Osmotically acting laxatives | 7 (15.2) | 5 (11.6) | 9 (18.8) | 7 (15.2) |
Other antidepressants | 8 (17.4) | 4 (9.3) | 9 (18.8) | 4 (8.7) |
Contact laxatives | 7 (15.2) | 6 (14.0) | 8 (16.7) | 6 (13.0) |
Other antiemetics | 6 (13.0) | 8 (18.6) | 7 (14.6) | 8 (17.4) |
Other antihistamines for systemic use | 5 (10.9) | 6 (14.0) | 6 (12.5) | 7 (15.2) |
Electrolyte solutions | 4 (8.7) | 10 (23.3) | 5 (10.4) | 11 (23.9) |
Phenylpiperidine derivatives | 5 (10.9) | 8 (18.6) | 5 (10.4) | 8 (17.4) |
Propulsives | 4 (8.7) | 9 (20.9) | 5 (10.4) | 9 (19.6) |
Fluoroquinolones | 4 (8.7) | 7 (16.3) | 4 (8.3) | 9 (19.6) |
Other opioids | 4 (8.7) | 10 (23.3) | 4 (8.3) | 10 (21.7) |
Solutions affecting the electrolyte balance | 3 (6.5) | 8 (18.6) | 3 (6.3) | 8 (17.4) |
Corticosteroids | 2 (4.3) | 5 (11.6) | 2 (4.2) | 7 (15.2) |
5HT3 = 5-hydroxytryptamine; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; SAS = safety analysis set.
aFrequency ≥ 15% in any treatment group.
bIncluded the use of heme arginate, hematin, and hemin.
Source: Clinical Study Report.6
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in this section. See Appendix 3 for detailed efficacy data.
Frequency of AHP Attacks
A summary of outcomes related to the frequency of porphyria attacks is provided in Table 13.
The results described herein are for patients with AIP using the mFAS unless otherwise stated. Based on the definition of porphyria attacks used for the primary end point, which included attacks that required hospitalization, an urgent health care visit, or IV hemin administration at home, patients with AIP reported a total of 83 and 284 porphyria attacks in the givosiran group and placebo group, respectively, over the 6-month treatment period. Among patients in the givosiran group, 50.0% did not report a porphyria attack with the 6-month treatment period, compared to 16.3% of patients receiving placebo. The mean AAR based on the composite end point was 3.22 (95% CI, 2.25 to 4.59) and 12.52 (95% CI, 9.35 to 16.76) for patients in the givosiran group and placebo group, respectively. This corresponded to a rate ratio of 0.26 (95% CI, 0.16 to 0.41; P < 0.001) or a 74% reduction in the rate of porphyria attacks for patients in the givosiran group relative to patients receiving placebo.
When only considering attacks requiring hospitalization, the mean AAR was 1.65 (95% CI, 0.98 to 2.78) and 3.21 (95% CI, 0.98 to 5.20) for patients in the givosiran and placebo groups, respectively. This corresponded to a rate ratio of 0.51 (95% CI, 0.25 to 1.04) or a 49% rate reduction in porphyria attacks requiring hospitalization. The mean AAR for attacks requiring an urgent health care visit was 1.22 (95% CI, 0.73 to 2.05) and 7.53 (95% CI, 5.13 to 11.05) for patients in the givosiran and placebo groups, respectively, corresponding to an 84% rate reduction (rate ratio = 0.16, 95% CI, 0.09 to 0.31). Lastly, a total of 3 attacks required IV hemin administration at home for patients in the givosiran group, while 32 attacks required this for patients in the placebo group.
The AAR for patients with at least 1 attack and the number of porphyria attacks treated at home without IV hemin were also reported (Table 13). Porphyria attacks that occurred at home that did not require IV hemin accounted for 19 of the 102 porphyria attacks reported in the givosiran group and for 14 of the 298 attacks reported in the placebo group for patients with AIP. The numbers of porphyria attacks that occurred at home that did not require IV hemin were similar between treatment groups for all patients with AHP (19 for givosiran, 20 for placebo).
All of the outcomes described previously were also measured for all patients with AHP (FAS), which yielded results similar to those reported for patients with AIP.
Table 13: Porphyria Attacks (6-Month Double-Blind Period, FAS and mFAS)
Porphyria attacks | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Annualized rate of porphyria attacka | ||||
Composite end point: attacks requiring hospitalization, an urgent health care visit, or IV hemin administration at home | ||||
Total number of attacks | 83 | 284 | 90 | 297 |
Total follow-up time (years) | 21.5 | 19.9 | 22.4 | 21.2 |
Number of patients with 0 attacks, n (%) | 23 (50.0) | 7 (16.3) | 24 (50.0) | 8 (17.4) |
Median AAR (IQR) | 1.04 (0 to 6.23) | 10.68 (2.24 to 26.09) | 1.04 (0 to 6.35) | 10.65 (2.24 to 25.93) |
Mean AAR (95% CI) | 3.22 (2.25 to 4.59) | 12.52 (9.35 to 16.76) | 3.35 (2.37 to 4.74) | 12.26 (9.22 to 16.29) |
Rate ratio (95% CI), givosiran vs. placebo | 0.26 (0.16 to 0.41) | — | 0.27 (0.17 to 0.43) | — |
P value | < 0.001 | — | < 0.001 | — |
Attacks requiring hospitalization | ||||
Total number of attacks | 43 | 68 | 50 | 69 |
Mean AAR (95% CI) | 1.65 (0.98 to 2.78) | 3.21 (1.98 to 5.20) | 1.74 (1.04 to 2.92) | 3.06 (1.90 to 4.94) |
Rate ratio (95% CI), givosiran vs. placebo | 0.51 (0.25 to 1.04) | — | 0.57 (0.28 to 1.15) | — |
Attacks requiring urgent health care visit | ||||
Total number of attacks | 37 | 184 | 37 | 196 |
Mean AAR (95% CI) | 1.22 (0.73 to 2.05) | 7.53 (5.13 to 11.05) | 1.19 (0.72 to 1.97) | 7.51 (5.21 to 10.83) |
Rate ratio (95% CI), givosiran vs. placebo | 0.16 (0.09 to 0.31) | — | 0.16 (0.08 to 0.30) | — |
Attacks requiring IV hemin administration at home | ||||
Total number of attacksb | 3 | 32 | 3 | 32 |
AAR for patients with at least 1 attack | ||||
n | 23 | 35 | 24 | 38 |
Median AAR (IQR) | 6.23 (2.17 to 11.20) | 13.28 (6.56 to 26.89) | 6.35 (2.95 to 13.17) | 13.28 (6.64 to 26.73) |
Mean AAR (SEM) | 7.78 (1.26) | 17.11 (2.20) | 8.09 (1.25) | 16.96 (2.11) |
All porphyria attacks,c n | ||||
All attacks | vvv | vvv | vvv | vvv |
Attacks treated at home without IV hemin | vv | vv | vv | vv |
AAR = annualized attack ratio; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; CI = confidence interval; IQR = interquartile range; SEM = standard error of the mean; vs. = versus .
aThe rates, rate ratio, corresponding 95% CI, and P value for comparing givosiran 2.5 mg/kg vs. placebo were derived using the negative binomial regression model with treatment group and stratification factors (prior hemin prophylaxis status and historical attack rates) as fixed effects and the logarithm of the follow-up time as an offset variable. The negative binomial regression analysis was not performed when fewer than 10 patients in a treatment group reported an attack.
bAs outlined in the statistical analysis plan, a model-based analysis was not conducted because fewer than 10 patients experienced an attack.
cIncludes porphyria attacks included in the composite end point (attacks requiring hospitalization, urgent health care visit, IV hemin administration at home) in addition to attacks treated at home without IV hemin.
Source: Clinical Study Report.6
The average number of attacks based on the composite end point, per patient per month over the 6-month treatment period, is presented in Figure 4 and Figure 5 for all patients with AHP and for patients with AIP, respectively. Briefly, the average number of attacks per patient per month was approximately 1 for patients receiving placebo and fewer than 0.6 for patients receiving givosiran over the 6-month treatment period in patients with AIP (Figure 4). The results for all patients with AHP were similar to the results reported for patients with AIP (Figure 5).
Figure 4: Average Number of Attacks (Composite End Point) per Patient per Month (6-Month Double-Blind Period, mFAS)
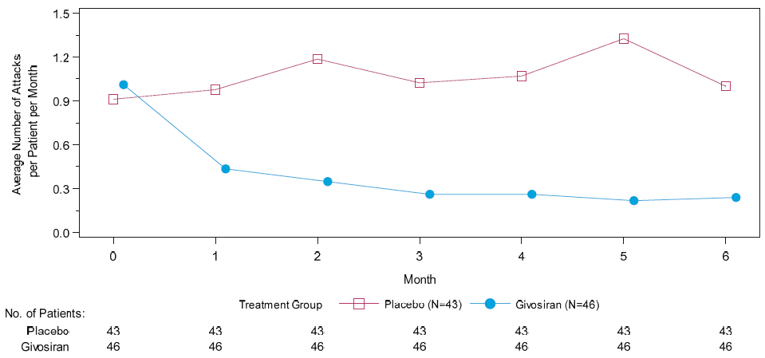
mFAS = modified full analysis set.
Source: Clinical Study Report.6
Figure 5: Average Number of Attacks (Composite End Point) per Patient per Month (6-Month Double-Blind Period, FAS)
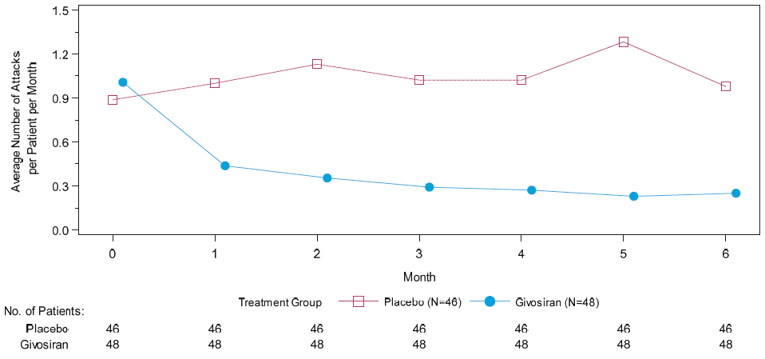
FAS = full analysis set.
Source: Clinical Study Report.6
Sensitivity Analyses
Sensitivity analyses were only conducted on the primary analysis, annualized rate of porphyria attack in patients with AIP (Table 14).
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 14: Sensitivity Analysis of Porphyria Attacks (6-Month Double-Blind Period, FAS)
Analysis | Patients with AIP | |
|---|---|---|
Givosiran N = 46 | Placebo N = 43 | |
Annualized rate of porphyria attack (composite end point) | ||
vvvvvvvv vvvvvvvvvvv vvvv | ||
vvvvvvvv vvv vvvvvvvv vvvvvvvvvv | ||
vvvvv vvvvvv vv vvvvvvvvvvvvvv vv vvvvvvvv vvvv vv vvvvv v vvvvvvv v vvv | vvvvv vvvvvv | vvvvvv vvvvvv |
vvvv vvv vvvv vvv | vvvv vvvvv vv vvvvv | vvvvv vvvvv vv vvvvvv |
vvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvv vvvvv vv vvvvv | |
| v vvvvvv | |
vvvvvvvv vvvvvvv vvvvv v vvvvv vvvvvvvvv | ||
vvvvv vvvvvv vv vvvvvvvvvvvvvv vv vvvvvvvv vvvv vv vvvvv v vvvvvvv v vvv | vvvvv vvvvvv | vvvvvv vvvvvv |
vvvv vvv vvvv vvv | vvvv vvvvv vv vvvvv | vvvvv vvvvv vv vvvvvv |
vvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvv vvvvv vv vvvvv | |
| v vvvvvv | |
vvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvvvvv | ||
vvvvv vvvvvv vv vvvvvvvvvvvvvv vv vvvvvvvv vvvv vv vvvvv v vvvvvvv v vvv | vvvvvv vvvvvv | vvvvvv vvvvvv |
vvvv vvv vvvv vvv | vvvv vvvvv vv vvvvv | vvvvv vvvvvv vv vvvvvv |
vvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvv vvvvv vv vvvvv | |
| v vvvvvv | |
vvv vvvvvvvvv vvvvvvvv vvvvvvvv vvvvvvvvv vvvvvvvvvv | ||
vvvvv vvvvvv vv vvvvvvvvvvvvvv vv vvvvvvvv vvvv vv vvvvv v vvvvvvv v vvv | vvvvvv vvvvvv | vvvvvv vvvvvv |
vvvv vvv vvvv vvv | vvvv vvvvv vv vvvvv | vvvvv vvvvvv vv vvvvvv |
vvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvv vvvvv vv vvvvv | |
| v vvvvvv | |
vvvvvvvvvvvvv vvvvvv vvvvv | ||
vvvvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvvv vvvvvv vv vvvvvv | |
| v vvvvvv | |
vvvvvvvv vvvvvvvv vvvvvvvvvvv vvvv | ||
v | vv | vv |
vvvvv vvvvvv vv vvvvvvvvvvvvvv vv vvvvvvvv vvvv vv vvvvv v vvvvvvv v vvv | vvvvv vvvvvv | vvvvvv vvvvvv |
vvvv vvv vvvv vvv | vvvv vvvvv vv vvvvv | vvvvv vvvvv vv vvvvvv |
vvvv vvvvv vvvv vvvv vvvvvvvvv vvv vvvvvvv | vvvv vvvvv vv vvvvv | |
| v vvvvvv | |
vvv v vvvvvvvvvv vvvvvv vvvvv AIP = acute intermittent porphyria; FAS = full analysis set. vv v vvvvvvvvvv vvvvvvvvv vv v vvvvv vvvvvvvv vvvv v vvvvvvvv vvvv vvvvvvvv vvvv vvv v vvv vvvvvvvv vvvv
v vvv vvvvvv vvvv vvvvvv vvv vvvvvvvvvvvvv vvv vv vvv vvvvvvv vvv vvvvvvvvv vvvvvvvvv vvv vvvvv vvvvvv vvvvvvv vvvv vvvvvvv vvvvv vvv vvvvvvvv vvvvvvvv vvvvvvvvvv vvvvv vvvv vvvvvvvvv vvvvv vvv vvvvvvvvvvvvvv vvvvvvv vvvvvv vvvvv vvvvvvvvvvv vvvvvv vvv vvvvvvvvvv vvvvvv vvvvvv vv vvvvv vvvvvvvv vvv vvv vvvvvvvvv vv vvv vvvvvvvvv vvvv vv vv vvvvvv vvvvvvvvv
v vvvvvvvv vvv vvvvvvvv vvvvvvvv vvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvv vvvvvvvvv vvvvvvvvvvvvvvvv vvvvvv vvvvvvvvvv vvvvvv vv vv vvvvv vvvvvvvvvvvvvv vv vvvvv vvv vvvvvvvv vvvvvvv vvvv vvvvvvv vvvv vv vvvv vvvvvvv vvvvvv v vvvv
v vvvvvvvv vvvvvvv vvvvv v vvvvv vvvvvvv vvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvv vvvvvvvvv vvvvvvvvvvvvvvvv vvvvvv vvvvvvvvvv vvvvvv vv vv vvvvv vvvvvvvvvvvvvv vv vvvvv vvvvvv vvv vvvvvv vvvvvvvv vvvvvv vvvv vvvvv vv v vvvvv vvvvvvv vvv vvvvvvv vvvv vvvvvvvv vv vvv vvvv vvvvvvvv vvvv vv vvvv vvvvvvvvv vv v vvvvvvvv vvvv vvvv vvvvvvv vv v vvvvvvv
v vvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvvv vvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvv vvvvvvv vvv vvvvvvvvvvvvvvvv vvvvvvvvv vvvvvvvvv vvvvvvv vvvvvvvvv vvvvvvvvvvvvvvvv vvvvvv vvvvvvvvvv vvvvvv vv vv vvvvv vvvvvvvvvvvvvv vv vvvvv vv vvvvvvv vv vvvv vvv vvvvvvvvv vvvvv vvvv
v vvv vvvvvvvvv vvvvvvvv vvvvvvvv vvvvvvvvv vvvvvvvv vvvv vvvvvvvvvvvvvvvvvvvvvv vvvvvvv vvv vvvvvvv vvv vvvvvvvvv vv vvvvvvvvvvvv vvv vvvv vvvvvv vv vvvvvvvvv vvvvvvv vvvv vvvvvvvvv vvvvvvvvvv vvv vvvvvvv vvv vv vvvvv vvvvvv vvvv vvvvvvvvv
v vvvvvvvv vvvvvvv vv vvvvvvvvv vvvvvvv vvvvvvvvvvvvv vvvvvv vvv vvvvvvv vvvvvvvvv vvvvvv vvvvvvvvv vvvvvvvv vvv vvvvvvvv vvvvv vv vvvvvvvvvvvvv vvvvv vvvv vvvvvvvvv vvvvv vvv vvvvvvvvvvvvvv vvvvvvv vvvvvv vvvvv vvvvvvvvvvv vvvvvv vvv vvvvvvvvvv vvvvvv vvvvvv vv vvvvvvvv v vvvvvv vvvvv vv vvvvvvvvvv v vvvvvvvvv vvvvvvv vvv vvv vvvvv vvvvvvvvvv
v vvv vvvvvvvv vvvvvvvvv vvv vvvvvvv vvvvvvvvv vvvvvv vvvvvvvvv vvvvvvvv vvv vvvvvvvv vvvvv vv vvv vvv vvvvvvvv vvvvvvvv vvvv
v vvvvvvv vvv vvv vvvvvvvv vvv vvvvvvvv vvvvvvv vvv vvvvvvvvvv vvvvvv vv vvvvvvvvvvv vvvvvvvvvv
Source: Clinical Study Report.6
Subgroup Analyses
The AAR based on the composite end point was reported by historical attack rates as a subgroup (Appendix 3, Figure 21 and Figure 22). Based on the mFAS, for patients with AIP and a high historical attack rate (n = 43), the rate ratio for givosiran compared to placebo was 0.27 (95% CI, 0.16 to 0.46). For patients with AIP and a low historical attack rate (n = 46), the rate ratio for givosiran compared to placebo was 0.23 (95% CI, 0.09 to 0.56). The comparison of givosiran to placebo in the FAS corresponded to a rate ratio of 0.29 (95% CI, 0.18 to 0.47) for patients with a high historical attack rate (n = 45) and a rate ratio of 0.23 (95% CI, 0.09 to 0.56) for patients with a low historical attack rate (n = 49).
Health-Related Quality of Life
Study 003 evaluated HRQoL using the SF-12, EQ-5D-5L, and PGIC. The results for these outcomes are available in Table 15; categorical domain-level results for the EQ-5D-5L are available in Appendix 3.
The PCS of the SF-12 was the only HRQoL outcome included as a secondary outcome in Study 003; the rest were exploratory outcomes. In patients with AIP, the change from baseline in the PCS score at month 6 was an LS mean of 5.37 (95% CI, 3.05 to 7.69) for the givosiran treatment group and 1.43 (95% CI, –1.00 to 3.86) for the placebo treatment group. The between-groups difference in the LS mean for givosiran compared to placebo was 3.94 (95% CI, 0.59 to 7.29; P = 0.0216); however, the P value cannot be interpreted due to a prior failure in the hierarchical testing strategy. The change from baseline in the MCS was reported descriptively. At month 6 in patients with AIP in the givosiran treatment group, the mean change from baseline was 3.66 (SD = 10.08), and for patients in the placebo treatment group, it was 1.30 (SD = 8.54). A between-groups comparison was not reported. The change from baseline to month 6 in each of the domains of the SF-12 is provided in patients with AIP in Figure 6. The PF, RP, BP, and GH domains contribute to the PCS. The LS mean differences between givosiran and placebo was 1.4 (95% CI, –2.0 to 4.7), 4.4 (95% CI, 1.3 to 7.5), 7.2 (95% CI, 3.2 to 11.2), and 3.3 (95% CI, –0.7 to 7.2) for each of these domains, respectively. VT, SF, RE, and MH contribute to the MCS, and had an LS mean difference of 17 (95% CI, –2.0 to 5.5), 5.1 (95% CI, 1.6 to 8.7), 1.4 (95% CI, –2.5 to 5.2), and 2.8 (95% CI, –0.9 to 6.4), respectively.
In terms of the EQ-5D-5L index for patients with AIP, the LS mean change from baseline at month 6 was |||||||||||||||||||||||||| |||| for the givosiran treatment group, and |||||||||||||||||||||||||||||||| for the placebo treatment group, which corresponded to a between-groups difference of ||||||||||||||||||||||||||||||||||. Using the VAS of the EQ-5D-5L, the LS mean change from baseline at month 6 was |||||||||||||||||||||||||||| and |||||||||||||||||||||||||||||| for the givosiran and placebo treatment groups, respectively. The between-groups difference at month 6 in the EQ-5D-5L VAS was ||||||||||||||||||||||||||||||.
Lastly, the PGIC was assessed at month 6 as the change in patient status since the start of the study. For patients with AIP, 88.9% of those in the givosiran group and 37.1% of those in the placebo group reported their status as improved since the start of the study. Further, in the givosiran treatment group, 1 (2.8%) patient reported no change, 2 (5.6%) patients reported that their status was minimally worse, and 1 (2.8%) patient reported that their status was very much worse. In the placebo treatment group, 42.9% of patients reported no change in their status, 6 (17.1%) reported that their status was minimally worse, and 1 (2.9%) reported that their status was much worse. The proportion of patients who reported an improvement in their status was compared between the 2 treatment groups. An odds ratio of 13.5 (95% CI, 3.90 to 47.03) for the givosiran group compared to placebo was reported for patients with AIP.
All of the outcomes described previously were also reported for all patients with AHP and were consistent with the results for patients with AIP.
Table 15: HRQoL Outcomes at Month 6 (6-Month Double-Blind Period, mFAS and FAS)
HRQoL outcome | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
PCS of SF-12a | ||||
n at baseline | 46 | 42 | 48 | 45 |
Baseline, mean (SD) | 39.43 (9.61) | 38.42 (9.45) | 39.47 (9.83) | 38.10 (9.82) |
n at month 6 | 45 | 42 | 47 | 46 |
LS mean (SEM) change from baseline at month 6 | 5.37 (1.17) | 1.43 (1.22) | 5.15 (1.16) | 1.46 (1.19) |
95% CI | 3.05 to 7.69 | –1.00 to 3.86 | 2.85 to 7.45 | –0.90 to 3.83 |
Difference in LS mean (SEM), givosiran – placebo | 3.94 (1.68) | — | 3.69 (1.65) | — |
95% CI | 0.59 to 7.29 | — | 0.41 to 6.96 | — |
P value | 0.0216d | — | 0.0280e | — |
MCS of SF-12 | ||||
n at baseline | 46 | 42 | 48 | 45 |
Baseline, mean (SD) | 40.41 (8.10) | 41.04 (10.08) | 39.90 (8.30) | 41.75 (10.29) |
n at month 6 | 45 | 42 | 47 | 45 |
Mean (SD) change from baseline at month 6 | 3.66 (10.08) | 1.30 (8.54) | 3.57 (9.87) | 0.42 (9.54) |
EQ-5D-5L indexa | ||||
n at baseline | vv | vv | vv | vv |
Baseline, mean (SD) | vvvv vvvvvv | vvvv vvvvvv | vvvv vvvvvv | vvvv vvvvvv |
n at month 6 | vv | vv | vv | vv |
LS mean (SEM) change from baseline at month 6 | vvvv vvvvvv | v vvvv vvvvvv | vvvv vvvvvv | vvvvv vvvvvv |
95% CI | vvvvv vv vvvv | vvvvv vv vvvv | vvvvv vv vvvv | vvvvv vv vvvv |
Difference in LS mean (SEM), givosiran – placebo | vvvv vvvvvv | — | vvvv vvvvvv | — |
95% CI | vvvvv vv vvvv | — | vvvvv vv vvvv | — |
P value | vvvvvvv | — | vvvvvvv | — |
EQ-5D-5L VASb | ||||
n at baseline | vv | vv | vv | vv |
Baseline, mean (SD) | vvvv vvvvvv | vvvv vvvvvv | vvvv vvvvvv | vvvv vvvvvv |
n at month 6 | vv | vv | vv | vv |
LS mean (SEM) change from baseline at month 6 | vvv vvvvv | v vvv vvvvv | vvv vvvvv | v vvv vvvvv |
95% CI | vvv vv vvvv | v vvv vv vvv | v vvv vv vvvv | v vvv vv vvv |
Difference in LS mean (SEM), givosiran – placebo | vvv vvvvv | — | vvv vvvvv | — |
95% CI | vvvv vv vvvv | — | vvvv vv vvvv | — |
P value | vvvvvvv | — | vvvvvvv | — |
PGIC score at month 6c | ||||
Patient status since the study start | ||||
n | 36 | 35 | 37 | 38 |
Improved, n (%) | 32 (88.9) | 13 (37.1) | 33 (89.2) | 14 (36.8) |
Very much improved | 10 (27.8) | 0 | 10 (27.0) | 0 |
Much improved | 12 (33.3) | 7 (20.0) | 12 (32.4) | 7 (18.4) |
Minimally improved | 10 (27.8) | 6 (17.1) | 11 (29.7) | 7 (18.4) |
No change or worsening, n (%) | 4 (11.1) | 22 (62.9) | 4 (10.8) | 24 (63.2) |
No change | 1 (2.8) | 15 (42.9) | 1 (2.7) | 16 (42.1) |
Very much worse | 1 (2.8) | 0 | 1 (2.7) | 0 |
Much worse | 0 | 1 (2.9) | 0 | 2 (5.3) |
Minimally worse | 2 (5.6) | 6 (17.1) | 2 (5.4) | 6 (15.8) |
Odds ratio (95% CI), givosiran vs. placebo | 13.5 (3.90 to 47.03) | 14.1 (4.14 to 48.35) | ||
P value | < 0.001e | < 0.001e | ||
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; CI = confidence interval; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; FAS = full analysis set; HRQoL = health-related quality of life; LS = least squares; MCS = Mental Component Score; mFAS = modified full analysis set; MMRM = mixed-effects model for repeated measures; PCS = Physical Component Score; PGIC = Patient Global Impression of Change; SD = standard deviation; SEM = standard error of the mean; SF-12 = 12-item Short Form Health Survey; VAS = Visual Analogue Scale.
aThe LS means, treatment differences in LS means, corresponding SEMs, and 95% CIs and P values for comparing 2.5 mg/kg givosiran vs. placebo were derived using the MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect.
bThe LS means, treatment differences in LS means, their corresponding SEMs, and 95% CIs and P value for comparing givosiran 2.5 mg/kg vs. placebo were derived using the MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect. Visit was fitted as a categorical variable, and the variance covariance matrix was assumed to be unstructured. A Kenward Roger approximation was used to estimate denominator degrees of freedom.
cThe P value was calculated from a Mantel-Haenszel chi-square test.
dThe P value cannot be interpreted due to a failure at a prior step in the hierarchical testing strategy.
eReported P values should be interpreted as nominal.
Source: Clinical Study Report.6
Figure 6: Difference Between Groups in Change From Baseline to Month 6 in SF-12 Domain Scores (6-Month Double-Blind Period, mFAS)
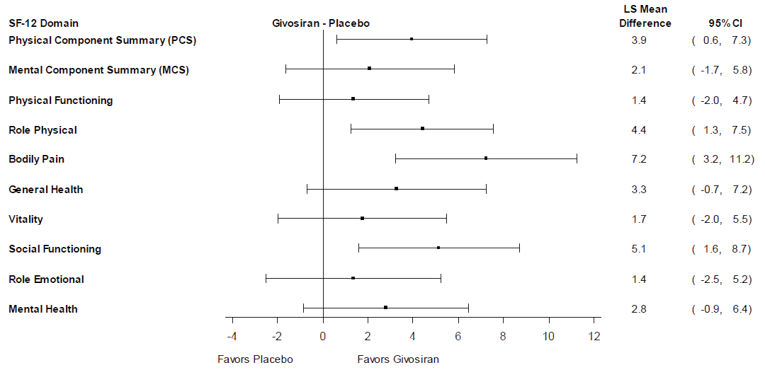
CI = confidence interval; LS = least squares; MCS = Mental Component Summary; mFAS = modified full analysis set; PCS = Physical Component Summary; SF-12 = 12-item Short Form Health Survey.
Source: Clinical Study Report.6
Management of Symptoms
The change in self-reported assessments of pain, fatigue, and nausea based on an NRS were secondary outcomes in Study 003. The results are presented in Figure 16.
Based on the pre-specified ANCOVA model, the LS mean for the AUC of change from baseline in weekly mean daily worst pain scores at 6 months in patients with AIP corresponded to a treatment-group difference of –12.7 (95% CI, –25.5 to 0.2; P = 0.053). A post hoc reanalysis of this outcome using a non-parametric test was conducted following a demonstration of deviation from normality. In patients with AIP, the medians of the AUC of change from baseline in weekly mean score for daily worst pain over the 6-month treatment period were –11.5 (IQR = –29.2 to 3.0) and 5.3 (IQR = –23.1 to 11.2) for the givosiran and placebo treatment groups, respectively. The median of the treatment-group difference for givosiran compared to placebo was –10.1 (95% CI, –22.8 to 0.9; P = 0.0455). The average change from baseline in weekly mean score for daily worst pain over the 6-month treatment period was –0.5 (95% CI, –1.0 to 0.1). Both the median of the AUC and the median of the average change from baseline in the weekly mean score for daily worst pain correspond to a reduction in pain for the givosiran treatment group and an increase in pain for the placebo treatment group.
For the assessment of daily worst fatigue score over the 6-month treatment period in patients with AIP, the means of the AUC of the change from baseline were –11.2 (95% CI, –20.1 to –2.2) and –4.2 (95% CI, –13.5 to 5.1) for patients in the givosiran and placebo treatment groups, respectively. These corresponded to a treatment-group difference for givosiran compared to placebo of –6.4 (95% CI, –19.8 to 6.0; P = 0.2876). The treatment-group difference in LS mean for the average change from baseline in weekly mean score was –0.32 (95% CI, –0.90 to 0.25; P = 0.2698).
In patients with AIP, the AUC of the change from baseline in weekly mean scores for daily worst nausea over the 6-month treatment period were an LS mean of 1.5 (95% CI, –5.1 to 8.1) in the givosiran group and –4.0 (95% CI, –10.9 to 2.9) in the placebo group. The between-groups difference in LS mean for givosiran compared to placebo was 5.5 (95% CI, –4.0 to 15.0; P = 0.2532). Based on the average change from baseline in weekly mean scores for daily worst nausea, a treatment-group difference of 0.3 (95% CI, –0.2 to 0.7; P = 0.2459) was reported for the givosiran treatment group compared to placebo.
Assessments of pain, fatigue, and nausea over the 6-month treatment period were also reported for all patients with AHP and were consistent with the results for patients with AIP.
Table 16: Pain, Fatigue, and Nausea Over 6 Months (6-Month DB Period, mFAS and FAS)
Scores | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
BPI-SF: Pain numerical rating score, daily worst pain scorea,b | ||||
n | 46 | 43 | 48 | 46 |
Baseline weekly mean score, mean (SD) | 2.93 (2.34) | 3.64 (2.23) | 2.97 (2.30) | 3.74 (2.23) |
AUC of change from baseline in weekly mean score | ||||
Median (IQR) | –11.51 (–29.18 to 3.04) | 5.29 (–23.05 to 11.15) | –7.80 (–28.329 to 3.583) | 2.31 (–19.50 to 10.512) |
Median of treatment difference (95% CI), givosiran – placebo | –10.07 (–22.83 to 0.94) | –9.39 (–21.02 to 1.22) | ||
P value | 0.0455 | 0.0613d | ||
Average change from baseline in weekly mean score | ||||
Median (IQR) | –0.51 (–1.31 to 0.14) | 0.25 (–1.02 to 0.47) | –0.34 (–1.25 to 0.19) | 0.10 (–0.89 to 0.46) |
Median of treatment difference (95% CI), givosiran – placebo | –0.45 (–1.00 to 0.06) | –0.41 (–0.92 to 0.08) | ||
P value | 0.0493d | 0.0658d | ||
BFI-SF: Fatigue numerical rating score, daily worst fatigue scorec | ||||
n | 46 | 43 | 48 | 46 |
Baseline weekly mean score, mean (SD) | 4.02 (2.55) | 4.68 (2.33) | 4.13 (2.58) | 4.70 (2.34) |
AUC of change from baseline in weekly mean score | ||||
LS mean (SEM) | –11.15 (4.50) | –4.21 (4.69) | –10.46 (4.35) | –3.68 (4.46) |
95% CI | –20.10 to –2.20 | –13.53 to 5.12 | –19.10 to –1.83 | –12.54 to 5.18 |
Difference in LS mean (SEM), givosiran – placebo | –6.94 (6.49) | –6.79 (6.19) | ||
95% CI | –19.84 to 5.96 | –19.09 to 5.51 | ||
P value | 0.2876e | 0.2759d | ||
Average change from baseline in weekly mean score | ||||
LS mean (SEM) | –0.50 (0.20) | –0.18 (0.21) | –0.47 (0.19) | –0.16 (0.20) |
95% CI | –0.90 to –0.10 | –0.60 to 0.23 | –0.86 to –0.09 | –0.55 to 0.24 |
Difference in LS mean (SEM), givosiran – placebo | –0.32 (0.29) | –0.31 (0.28) | ||
95% CI | –0.90 to 0.25 | –0.86 to 0.24 | ||
P value | 0.2698d | 0.2590d | ||
Nausea numerical rating score, daily worst nausea scorec | ||||
n | 46 | 43 | 48 | 46 |
Baseline weekly mean score, mean (SD) | 1.51 (1.70) | 1.99 (1.85) | 1.57 (1.70) | 1.91 (1.84) |
AUC of the change from baseline in weekly mean score | ||||
LS mean (SEM) | 1.48 (3.31) | –4.01 (3.45) | 1.60 (3.27) | –3.00 (3.36) |
95% CI | –5.10 to 8.06 | –10.88 to 2.86 | –4.90 to 8.09 | –9.67 to 3.68 |
Difference in LS mean (SEM), givosiran – placebo | 5.49 (4.77) | 4.59 (4.66) | ||
95% CI | –4.00 to 14.98 | –4.66 to 13.84 | ||
P value | 0.2532e | 0.3266d | ||
Average change from baseline in weekly mean score | ||||
LS mean (SEM) | 0.07 (0.15) | –0.18 (0.15) | 0.07 (0.15) | –0.14 (0.15) |
95% CI | –0.23 to 0.36 | –0.49 to 0.12 | –0.22 to 0.36 | –0.43 to 0.16 |
Difference in LS mean (SEM), givosiran – placebo | 0.25 (0.21) | 0.21 (0.21) | ||
95% CI | –0.17 to 0.67 | –0.20 to 0.62 | ||
P value | 0.2459d | 0.3192d | ||
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; AUC = area under the curve; BFI-SF = Brief Fatigue Inventory – Short Form; BPI-SF = Brief Pain Inventory – Short Form; CI = confidence interval; DB = double blind; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; FAS = full analysis set; IQR = interquartile range; LS = least squares; mFAS = modified full analysis set; SD = standard deviation; SEM = standard error of mean.
aEstimated using the Hodges-Lehmann method. The P value was estimated from stratified Wilcoxon test with stratification factors, prior hemin prophylaxis status, and historical attack rates.
bNormality was assessed using a Q-Q plot and the Shapiro-Wilk test, which indicated that the data had a significant deviation from a normal distribution. Therefore, a non-parametric analysis was conducted (stratified Wilcoxon test).
cAnalyzed using the ANCOVA model with treatment and stratification factors (prior hemin prophylaxis status and historical attack rates) as fixed effects, and the corresponding weekly mean score at baseline as a covariate. Missing weekly mean scores were imputed.
dThe P value has not been adjusted for multiple testing and should be interpreted as nominal.
eThe P value cannot be interpreted due to a failure at a prior step in the hierarchical testing strategy.
Source: Clinical Study Report.6
Reduced Complications of AHP
Outcomes related to reduced complications of AHP were not reported in the included studies.
Rescue Medication Use
In Study 003, hemin was only permitted as a rescue medication for the treatment of acute porphyria attacks and was reported as days of hemin use (Table 17). In patients with AIP, 54% of patients in the givosiran group and 23% of patients in the placebo group reported zero days of hemin use over the 6-month treatment period. The means of annualized days of hemin use in the givosiran and placebo groups were 6.8 days (95% CI, 4.2 to 10.9) and 29.7 days (95% CI, 18.4 to 47.9), respectively. This corresponded to a 77% rate reduction in days of hemin use for givosiran compared to placebo based on a rate ratio of 0.23 (95% CI, 0.11 to 0.45; P < 0.001).
In patients with AHP, the reported use of hemin for the treatment of acute porphyria attacks was similar to that for patients with AIP. In the givosiran and placebo groups, 54% and 26% of patients had zero days of hemin use in the 6-month treatment period. The rate ratio for annualized days of hemin use for givosiran compared to placebo was 0.26 (95% CI, 0.13 to 0.52; P = 0.0002).
Table 17: Hemin Use (6-Month Double-Blind Period, mFAS and FAS)
Hemin use | Annualized days of hemin usea | |||
|---|---|---|---|---|
Patients with AIP | All patients with AHP | |||
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Total number of days of hemin use | 195 | 583 | 227 | 587 |
Mean (SEM) | 4.2 (1.1) | 13.6 (2.1) | 4.7 (1.2) | 12.8 (2.0) |
Total follow-up time (years) | 21.5 | 19.9 | 22.4 | 21.2 |
Mean (SEM) | 0.47 (0.0) | 0.46 (0.0) | 0.47 (0.0) | 0.46 (0.0) |
Patients with 0 days of hemin use, n (%) | 25 (54.3) | 10 (23.3) | 26 (54.2) | 12 (26.1) |
Patients with ≥ 1 days of hemin use, n (%) | 21 (45.7) | 33 (76.7) | 22 (45.8) | 34 (73.9) |
Mean annualized days of hemin use (95% CI) | 6.8 (4.2 to 10.9) | 29.7 (18.4 to 47.9) | 7.4 (4.5 to 12.0) | 28.4 (17.4 to 46.2) |
Rate ratio of annualized days of hemin use (95% CI), givosiran vs. placebo | 0.23 (0.11 to 0.45) | 0.26 (0.13 to 0.52) | ||
P value | < 0.001 | 0.0002b | ||
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; CI = confidence interval; SEM = standard error of the mean; vs. = versus .
aAnalyzed using the negative binomial regression model with treatment group and stratification factors (prior hemin prophylaxis status and historical attack rates) as fixed effects and the logarithm of the follow-up time as an offset variable.
bThe P value has not been adjusted for multiple testing and should be interpreted as nominal.
Source: Clinical Study Report.6
Opioid Use
Opioid use was reported descriptively under analgesic medication use in Study 003 and has been summarized in Table 18. In patients with AIP, the mean proportion of days with opioid use was 23.3 days (SD = 35.3 days) for patients in the givosiran treatment group and 38.0 days (SD = 39.4 days) for patients in the placebo treatment group. The median proportion of days with opioid use was 3.0 (Q1 to Q3: 0 to 38.5) and 10.8 (Q1 to Q3: 2.4 to 83.3) for patients in the givosiran and placebo groups, respectively.
Over the 6-month treatment period, 67% and 88% of patients in the givosiran and placebo groups, respectively, reported opioid use. Opioid use was also reported as the number of patients with analgesic opioid use during months 1 to 3 and months 4 to 6. In the givosiran treatment group, 63% of patients used opioids for pain between months 1 and 3, and 52% used opioids for pain between months 4 and 6. In the placebo treatment group, 81% of patients used opioids for pain between months 1 and 3, and 84% used opioids for pain between months 4 and 6.
Analgesic use of opioids in patients with AHP were similar to those for patients with AIP (Table 18).
Table 18: Opioid Use (6-Month Double-Blind Period, mFAS and FAS)
Opioid use | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Proportion of days with opioid use | ||||
n | 46 | 43 | 48 | 46 |
Mean (SD) | 23.31 (35.25) | 38.02 (39.44) | 23.06 (34.69) | 35.54 (39.27) |
Median (IQR) | 3.01 (0.00 to 38.46) | 10.78 (2.35 to 83.33) | 3.01 (0.00 to 36.49) | 8.48 (1.78 to 72.29) |
Number of patients with opioid use, n (%) | ||||
Months 1 to 3 | 29 (63.0) | 35 (81.4) | 30 (62.5) | 35 (76.1) |
Months 4 to 6 | 24 (52.2) | 36 (83.7) | 25 (52.1) | 36 (78.3) |
Over the 6-month DB period | 31 (67.4) | 38 (88.4) | 32 (66.7) | 38 (82.6) |
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; DB = double blind; IQR = interquartile range; SD = standard deviation.
Source: Clinical Study Report.6
Activities of Daily Living
Data for the PPEQ were reported descriptively.
Responses to the PPEQ at month 6 were reported in patients with AIP only (Figure 7) and in all patients with AHP (Figure 8). Questions 1 through 7 of the PPEQ reflect the patient’s response compared to before the start of study, and question 8 is based on the previous 4 weeks. Overall, a greater proportion of patients receiving givosiran answered “much better” or “always” to all questions included the PPEQ compared to those receiving placebo. More specifically, 33% to 36% of patients with AIP in the givosiran group (versus 6% to 14% of patients in the placebo group) rated the following activities as being much better at month 6 compared at to the start of the study: travelling for more than 1 day for work or pleasure, participating in social activities, planning future events, doing household chores, and exercising moderately. The convenience of current porphyria treatment and patients’ overall satisfaction with porphyria treatment were both rated as much better at month 6 compared to the start of study by 74% of patients in the givosiran group compared to 9% and 15% of patients in the placebo group for each of the 2 questions, respectively. For question 8, 43% and 6% of patients in the givosiran and placebo groups, respectively, responded “always” to whether the study drug helped them live more normal lives.
The proportion of patients who responded “much better” or “always” was similar between patients with AIP and AHP.
Figure 7: Response to the PPEQ at Month 6 (6-Month Double-Blind Period, mFAS)
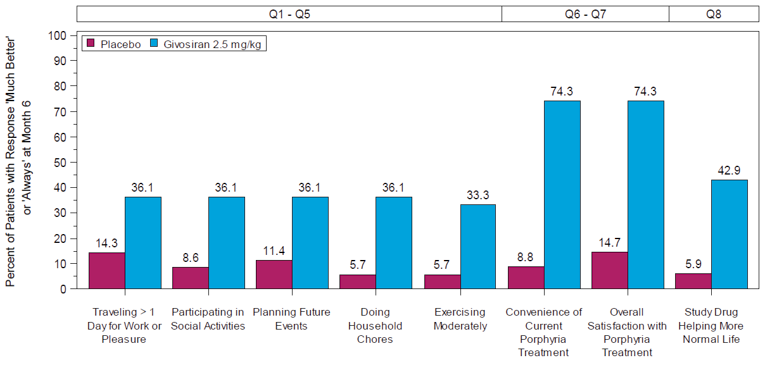
mFAS = modified full analysis set; PPEQ = Porphyria Patient Experience Questionnaire; Q = question.
Note: Questions 1 to 7 contained response options of much better, minimally better, no change, minimally worse, and much worse. Question 8 had response options of always, most of the time, sometimes, rarely, and never.
Source: Clinical Study Report.6
Figure 8: Response to the PPEQ at Month 6 (6-Month Double-Blind Period, FAS)
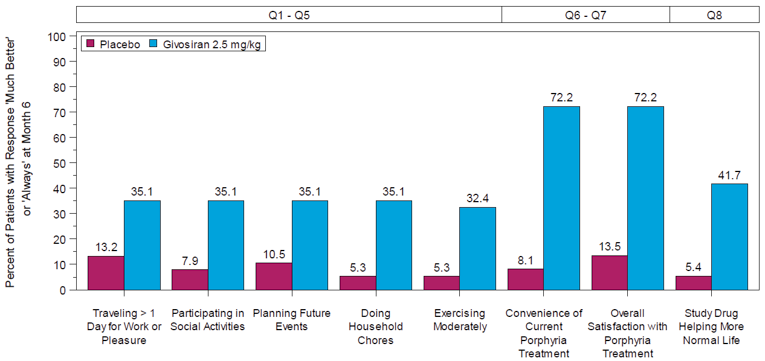
FAS = full analysis set; PPEQ = Porphyria Patient Experience Questionnaire; Q = question.
Source: Clinical Study Report.6
Hospitalization and Health Care Use
Attacks requiring hospitalization or an urgent health care visit were reported in the Frequency of AHP Attacks section.
Ability to Work or Attend School
Study 003 reported on days of missed work or school. The results for the number of missed days in the past 4 weeks are presented in Table 19. In patients with AIP at baseline, the mean reported numbers of missed days of work were 3.3 days (SD = 3.5 days) and 6.4 days (SD = 6.5 days) for patients in the givosiran and placebo groups, respectively. At month 3, the mean numbers of missed days of work were 2.4 days (SD = 3.9 days) and 4.8 days (SD = 6.8 days) for givosiran and placebo, respectively. At month 6, a mean of 2.4 days (SD = 6.8 days) of missed work was reported in the givosiran treatment group, and a mean of 6.9 days (SD = 8.0 days) was reported for placebo.
Among patients with AIP, 5 contributed to the evaluation of missed days of school. For all patients with AHP, 6 patients contributed to this evaluation. In the givosiran group, the mean numbers of missed days of school at baseline, month 3, and month 6 were 7.3 days (SD = 11.8 days), 0 days, and 2.8 days (SD = 4.9 days), respectively, for patients with AIP. In the corresponding placebo group, the mean numbers of missed days of school at baseline, month 3, and month 6 were 3.5 days (SD = 0.7 days), 0.5 days (SD = 0.7 days), and 0 days, respectively.
The results for the overall population of all patients with AHP were similar to those for patients with AIP (Table 19).
Table 19: Missed Work or School (6-Month Double-Blind Period, mFAS and FAS)
Missed days | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Missed days of work for patients who had been employed in the past 4 weeks | ||||
Baseline | ||||
n | 20 | 20 | 20 | 21 |
Mean (SD) | 3.3 (3.5) | 6.4 (6.5) | 3.3 (3.5) | 6.1 (6.5) |
Median (range) | 2.5 (0 to 10) | 5.0 (0 to 28) | 2.5 (0 to 10) | 5.0 (0 to 28) |
Month 3 | ||||
n | 19 | 20 | 19 | 21 |
Mean (SD) | 2.4 (3.9) | 4.8 (6.8) | 2.4 (3.9) | 4.9 (6.7) |
Median (range) | 0 (0 to 13) | 2.5 (0 to 28) | 0 (0 to 13) | 3.0 (0 to 28) |
Month 6 | ||||
n | 17 | 19 | 17 | 20 |
Mean (SD) | 2.4 (6.8) | 6.9 (8.0) | 2.4 (6.8) | 6.7 (7.8) |
Median (range) | 0 (0 to 28) | 5.0 (0 to 28) | 0 (0 to 28) | 5.0 (0 to 8) |
Missed days of school for patients who had been attending school in the past 4 weeks | ||||
Baseline | ||||
n | 3 | 2 | 4 | 2 |
Mean (SD) | 7.3 (11.8) | 3.5 (0.7) | 6.3 (9.9) | 3.5 (0.7) |
Median (range) | 1.0 (0 to 21) | 3.5 (3 to 4) | 2.0 (0 to 21) | 3.5 (3 to 4) |
Month 3 | ||||
n | 2 | 2 | 3 | 2 |
Mean (SD) | 0 | 0.5 (0.7) | 2.7 (4.6) | 0.5 (0.7) |
Median (range) | 0 | 0.5 (0 to 1) | 0.0 (0 to 8) | 0.5 (0 to 1) |
Month 6 | ||||
n | 4 | 1 | 5 | 1 |
Mean (SD) | 2.8 (4.9) | 0 | 2.8 (4.2) | 0 |
Median (range) | 0.5 (0 to 10) | 0 (0 to 0) | 1.0 (0 to 10) | 0 (0 to 0) |
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; SD = standard deviation.
Source: Clinical Study Report.6
ALA, PBG, and Porphyrin Levels
Biomarkers for AHP, namely ALA and PBG, were analyzed in Study 003; the results are presented in Table 20.
For patients with AIP at baseline, the mean levels of urinary ALA were 19.97 mmol/mol Cr (SD = 16.80 mmol/mol Cr) and 17.52 mmol/mol Cr (SD = 10.89 mmol/mol Cr) in the givosiran and placebo groups, respectively. At month 3, the LS mean change from baseline in urinary ALA levels were 1.76 mmol/mol Cr (95% CI, –1.05 to 4.57) for givosiran and 19.97 mmol/mol Cr (95% CI, 17.03 to 22.90) for placebo, which corresponded to a between-groups difference of –18.21 (95% CI, –22.26 to –14.16; P < 0.001) in favour of givosiran. At month 6, the LS mean of the change from baseline in urinary ALA levels was 4.01 mmol/mol Cr (95% CI, –0.69 to 8.72) for givosiran and 23.15 mmol/mol Cr (95% CI, 18.09 to 28.21) for placebo, corresponding to a between-groups difference of –19.14 mmol/mol Cr (95% CI, –26.04 to –12.24; P < 0.001) in favour of givosiran as well.
In patients with AIP, urinary levels of PBG at baseline were 50.36 mmol/mol Cr (SD = 34.33 mmol/mol Cr) and 46.80 mmol/mol Cr (SD = 24.32 mmol/mol Cr) for patients in the givosiran and placebo groups, respectively. At month 6, the LS mean of the change from baseline in urinary PBG was 12.91 mmol/mol Cr (95% CI, 3.66 to 22.15) for the givosiran group and 49.11 mmol/mol Cr (95% CI, 39.24 to 58.98) for the placebo group. The between-groups difference in urinary PBG at month 6 for givosiran compared to placebo was –36.20 mmol/mol Cr (95% CI, – 49.71 to – 22.70; P < 0.001) in favour of givosiran.
Post hoc non-parametric tests were conducted for urinary ALA and PBG levels following demonstration of a significant deviation from the normal distribution. The median urinary ALA levels and PBG levels for patients in the givosiran and placebo groups are presented by month in patients with AIP (Figure 9 and Figure 10). Based on the median measured at each month, levels of urinary ALA and PBG were consistent over the 6-month treatment period following month 1. The results of the non-parametric tests were consistent with the results of the pre-specified analyses presented in Table 20.
Overall, the results of urinary ALA and PBG levels in patients with AIP were similar for all patients with AHP.
Table 20: AHP Biomarkers (6-Month Double-Blind Period, mFAS and FAS)
AHP Biomarker | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Urinary ALA levels (mmol/mol Cr)a | ||||
n | 46 | 43 | 48 | 46 |
Baseline, mean (SD) | 19.97 (16.80) | 17.52 (10.89) | 19.65 (16.61) | 17.27 (10.80) |
Month 3 | ||||
LS mean (SEM) change from baseline at month 3 | 1.76 (1.41) | 19.97 (1.48) | 1.72 (1.38) | 19.36 (1.41) |
95% CI | –1.05 to 4.57 | 17.03 to 22.90 | –1.03 to 4.46 | 16.55 to 22.16 |
Difference in LS mean (SEM), givosiran – placebo | –18.21 (2.04) | –17.64 (1.97) | ||
95% CI | –22.26 to –14.16 | –21.55 to –13.73 | ||
P value | < 0.001 | < 0.001b | ||
Month 6 | ||||
LS mean (SEM) change from baseline at month 6 | 4.01 (2.35) | 23.15 (2.53) | 3.93 (2.27) | 22.28 (2.39) |
95% CI | –0.69 to 8.72 | 18.09 to 28.21 | –0.61 to 8.46 | 17.51 to 27.04 |
Difference in LS mean (SEM), givosiran – placebo | –19.14 (3.45) | –18.35 (3.29) | ||
95% CI | –26.04 to –12.24 | –24.92 to –11.78 | ||
P value | < 0.001 | < 0.001b | ||
Urinary PBG levels (mmol/mol Cr)a | ||||
n | 46 | 43 | 48 | 46 |
Baseline, mean (SD) | 50.36 (34.33) | 46.80 (24.32) | 49.00 (34.41) | 45.39 (24.52) |
Month 6 | ||||
LS mean (SEM) change from baseline at month 6 | 12.91 (4.64) | 49.11 (4.96) | 12.45 (4.50) | 47.70 (4.69) |
95% CI | 3.66 to 22.15 | 39.24 to 58.98 | 3.51 to 21.39 | 38.38 to 57.01 |
Difference in LS mean (SEM), givosiran – placebo | –36.20 (6.79) | –35.25 (6.48) | ||
95% CI | –49.71 to –22.70 | –48.13 to –22.36 | ||
P value | < 0.001 | < 0.001b | ||
AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; CI = confidence interval; Cr = creatinine; FAS = full analysis set; LS = least squares; mFAS = modified full analysis set; MMRM = mixed-effects model for repeated measures; PBG = porphobilinogen; SD = standard deviation; SEM = standard error of the mean.
aAnalyzed using an MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect.
bThe P value has not been adjusted for multiple testing and should be interpreted as nominal.
Source: Source: Clinical Study Report.6
Figure 9: Median (IQR) Urinary ALA Levels Over Time During the 6-Month Double-Blind Period in Patients With AIP (mFAS)
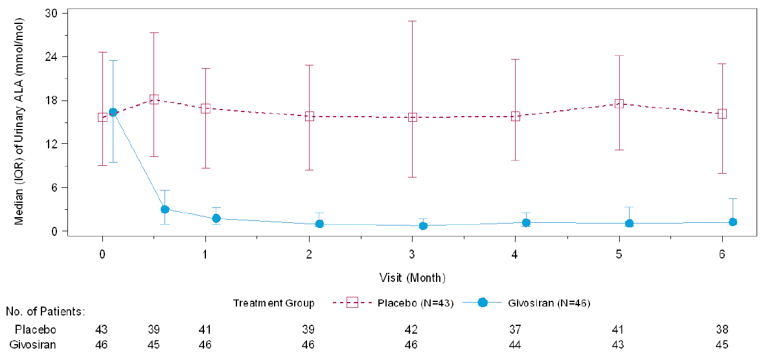
AIP = acute intermittent porphyria; ALA = aminolevulinic acid; IQR = interquartile range; mFAS = modified full analysis set.
Source: Source: Clinical Study Report.6
Figure 10: Median (IQR) Urinary PBG Levels Over Time During the 6-Month Double-Blind in Patients With AIP (mFAS)
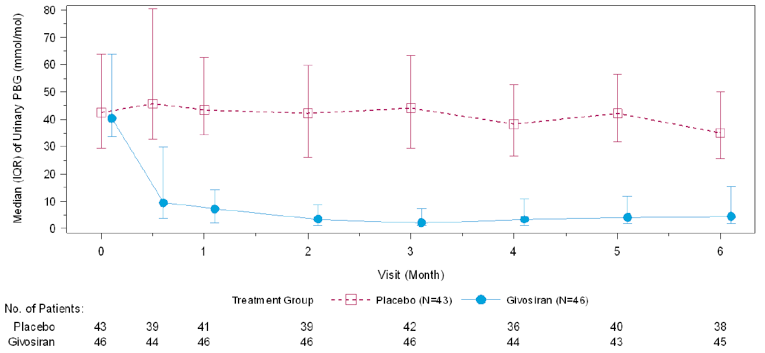
AIP = acute intermittent porphyria; IQR = interquartile range; mFAS = modified full analysis set; PBG = porphobilinogen.
Source: Source: Clinical Study Report.6
Subgroup Analyses
Assessments of urinary ALA levels were analyzed by subgroups reflecting the historical frequency of attacks, or by patients with a high AAR versus a low AAR.
In patients with AIP and a high AAR, baseline levels of ALA were 24.35 mmol/mol Cr (SD = 21.47 mmol/mol Cr) and 19.67 mmol/mol Cr (SD = 8.10 mmol/mol Cr) in the givosiran and placebo groups, respectively. The changes from baseline at months 3 and 6 were 2.47 mmol/mol Cr (95% CI, –1.74 to 6.68) and 6.43 mmol/mol Cr (95% CI, –2.37 to 15.22), respectively, for the givosiran group and 23.58 mmol/mol Cr (95% CI, 19.06 to 28.09) and 28.84 mmol/mol Cr (95% CI, 18.81 to 38.87), respectively, for the placebo group. This corresponded to a between-groups difference of –21.11 (95% CI, –27.31 to –14.90) at month 3 and–22.41 (95% CI, –35.87 to –8.96) at month 6.
In patients with AIP and a low AAR, baseline levels of ALA were 15.60 mmol/mol Cr (SD = 8.74 mmol/mol Cr) and 15.65 mmol/mol Cr (SD = 12.71 mmol/mol Cr) for patients in the givosiran and placebo groups, respectively. The changes from baseline at month 3 and month 6 were 1.32 mmol/mol Cr (95% CI, –2.27 to 4.91) and 1.84 mmol/mol Cr (95% CI, –0.84 to 4.53), respectively, for the givosiran group and 15.50 mmol/mol Cr (95% CI, 11.84 to 19.17) and 14.06 mmol/mol Cr (95% CI, 11.25 to 16.87), respectively, for the placebo treatment group. This corresponded to a between-groups difference of –14.19 (95% CI, –19.32 to –9.06) at month 3 and –12.22 (95% CI, –16.10 to –8.33) at month 6.
The subgroup results for patients with AHP were similar to those discussed for patients with AIP only (Table 21).
Table 21: Subgroup Analyses by Historical High or Low AAR — Urinary ALA Levels by Frequency of Attacks (6-Month Double-Blind Period, mFAS and FAS)
Subgroup | Patients with AIP | All patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
High AARa | ||||
n | 23 | 20 | 24 | 21 |
Baseline, mean (SD) | 24.35 (21.47) | 19.67 (8.10) | 23.47 (21.43) | 19.83 (7.93) |
Month 3 | ||||
LS mean (SE) change from baseline at month 3 | 2.47 (2.08) | 23.58 (2.23) | 2.44 (2.02) | 22.96 (2.16) |
95% CI | –1.74 to 6.68 | 19.06 to 28.09 | –1.63 to 6.52 | 18.61 to 27.32 |
Difference in LS mean (SE), givosiran – placebo | –21.11 (3.07) | –20.52 (2.96) | ||
95% CI | –27.31 to –14.90 | (–26.50 to –14.54) | ||
Month 6 | ||||
LS mean (SE) change from baseline at month 6 | 6.43 (4.34) | 28.84 (4.95) | 6.246 (4.157) | 27.82 (4.71) |
95% CI | –2.37 to 15.22 | 18.81 to 38.87 | –2.17 to 14.66 | 18.30 to 37.35) |
Difference in LS mean (SE), givosiran-placebo | –22.41 (6.63) | –21.58 (6.32) | ||
95% CI | –35.87 to –8.96 | –34.36 to –8.79 | ||
Low AARa | ||||
n | 23 | 23 | 24 | 25 |
Baseline, mean (SD) | 15.60 (8.74) | 15.65 (12.71) | 15.83 (8.63) | 15.11 (12.47) |
Month 3 | ||||
LS mean (SE) change from baseline at month 3 | 1.32 (1.78) | 15.50 (1.82) | 1.23 (1.75) | 15.08 (1.72) |
95% CI | –2.27 to 4.91 | 11.84 to 19.17 | –2.30 to 4.77 | 11.62 to 18.54 |
Difference in LS mean (SE), givosiran – placebo | –14.19 (2.54) | –13.84 (2.46) | ||
95% CI | –19.32 to –9.06 | –18.79 to –8.90 | ||
Month 6 | ||||
LS mean (SE) change from baseline at month 6 | 1.84 (1.33) | 14.06 (1.39) | 1.79 (1.34) | 13.73 (1.34) |
95% CI | –0.84 to 4.53 | 11.25 to 16.87 | –0.90 to 4.49 | 11.03 to 16.42 |
Difference in LS mean (SE), givosiran – placebo | –12.22 (1.92) | –11.94 (1.89) | ||
95% CI | –16.10 to –8.33 | –15.75 to –8.12 | ||
AAR = annualized attack rate; AHP = acute hepatic porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; CI = confidence interval; FAS = full analysis set; LS = least squares; mFAS = modified full analysis set; MMRM = mixed-effects model for repeated measures; SD = standard deviation; SE = standard error; vs. = versus .
aThe LS means, treatment differences in LS means, their corresponding SEMs, and 95% CIs and P values for comparing 2.5 mg/kg givosiran vs. placebo were derived using the MMRM model with the corresponding value at baseline as a continuous fixed covariate, stratification factors (prior hemin prophylaxis status and historical attack rates), visit, treatment, and treatment-by-visit interaction as fixed effects, and patient as a random effect. A difference of < 0 represents a favourable outcome for 2.5 mg/kg givosiran.
Source: Source: Clinical Study Report.6
Mortality
Mortality was not reported as an efficacy outcome in Study 003.
Harms
Only those harms identified in the review protocol are reported here. See Table 22 for detailed harms data.
Adverse Events
Over the 6-month DB treatment period in patients with AIP, 89% of patients receiving givosiran reported at least 1 AE, as did 81% of patients receiving placebo. Overall, the most frequently reported AEs in patients receiving givosiran versus placebo were nausea (28% versus 12%), injection-site reaction (17% versus 0), headache (13% versus 16%), and urinary tract infection (7% versus 14%). The following AEs were reported at least 5% more frequently in patients treated with givosiran: nausea, injection-site reaction, chronic kidney disease, fatigue, increase in ALT, decrease in glomerular filtration rate, and rash. The following AEs were reported at least 5% more frequently in patients in the placebo group: urinary tract infection, vomiting, back pain, pyrexia, and hypoesthesia. Reporting of AEs was comparable between patients with AIP and AHP.
Serious Adverse Events
In patients with AIP, at least 1 SAE was reported by 17% (n = 8) of patients in the givosiran group and 9% (n = 4) of patients in the placebo group over the 6-month treatment period. For patients with AHP, 21% (n = 10) and 9% (n = 4) of patients in the givosiran and placebo groups, respectively, reported at least 1 SAE. Among patients with AIP, infections and infestations were reported more frequently in the placebo group (n = 3) than in the givosiran group (n = 1). Chronic kidney disease was reported by 2 patients in the givosiran group and by no patients in the placebo treatment group. Additionally, hypoglycemia, major depression, asthma, and a surgical or medical procedure for pain management were each reported by 1 patient in the givosiran group and by no patients in the placebo treatment group.
The frequencies of specific SAEs in patients with AHP was similar to those reported in patients with AIP.
Withdrawals Due to Adverse Events
One patient from the givosiran group withdrew from treatment due to an AE. The patient |||||||||||| discontinued treatment because of ALT elevation.
Mortality
No deaths were reported during the 6-month DB period of Study 003.
Notable Harms
Motor neuropathy, hepatocellular carcinoma, injection-site reactions, transaminase elevation, and progression of renal impairment were included in the CADTH systematic review protocol as notable harms. In patients with AIP, injection-site reactions and progression of renal impairment were reported in 8 (17%) and 4 (9%) patients, respectively, in the givosiran group, and by 0 patients in the placebo group. Increased ALT was reported by 4 (9%) patients receiving givosiran and by 1 (2%) patient receiving placebo, while increased AST was reported in 3 (7%) patients and 1 (2%) patient receiving givosiran and placebo, respectively. Three (7%) patients in the placebo group reported motor neuropathy as an AE; none reported this in the givosiran group. There were no cases of hepatocellular carcinoma reported during the 6-month treatment period. The frequency of notable harms was the same for patients with AHP, with the exception of progression of renal impairment, which was reported in 5 (10%) patients in the givosiran group.
Table 22: Summary of Harms (6-Month Double-Blind Period, SAS)
Harms | Patients with AIP | Patients with AHP | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Patients with ≥ 1 AE | ||||
Patients with ≥ 1 AE, n (%) | 41 (89.1) | 35 (81.4) | 43 (89.6) | 37 (80.4) |
Most common events,a n (%) | ||||
Nausea | 13 (28.3) | 5 (11.6) | 13 (27.1) | 5 (10.9) |
Injection-site reaction | 8 (17.4) | 0 | 8 (16.7) | 0 |
Headache | 6 (13.0) | 7 (16.3) | 6 (12.5) | 7 (15.2) |
Chronic kidney disease | 4 (8.7) | 0 | 5 (10.4) | 0 |
Fatigue | 4 (8.7) | 2 (4.7) | 5 (10.4) | 2 (4.3) |
Abdominal pain | 4 (8.7) | 3 (7.0) | 4 (8.3) | 3 (6.5) |
ALT increased | 4 (8.7) | 1 (2.3) | 4 (8.3) | 1 (2.2) |
Nasopharyngitis | 4 (8.7) | 3 (7.0) | 4 (8.3) | 3 (6.5) |
Upper respiratory tract infection | 4 (8.7) | 3 (7.0) | 4 (8.3) | 3 (6.5) |
AST increased | 3 (6.5) | 1 (2.3) | 3 (6.3) | 1 (2.2) |
Asthenia | 3 (6.5) | 4 (9.3) | 3 (6.3) | 4 (8.7) |
Constipation | 3 (6.5) | 2 (4.7) | 3 (6.3) | 2 (4.3) |
Device occlusion | 3 (6.5) | 1 (2.3) | 3 (6.3) | 1 (2.2) |
Device-related infection | 3 (6.5) | 3 (7.0) | 3 (6.3) | 3 (6.5) |
Decreased glomerular filtration rate | 3 (6.5) | 0 | 3 (6.3) | 0 |
Rash | 3 (6.5) | 0 | 3 (6.3) | 0 |
Tooth infection | 3 (6.5) | 1 (2.3) | 3 (6.3) | 1 (2.2) |
Urinary tract infection | 3 (6.5) | 6 (14.0) | 3 (6.3) | 6 (13.0) |
Dizziness | 2 (4.3) | 3 (7.0) | 2 (4.2) | 3 (6.5) |
Vomiting | 2 (4.3) | 5 (11.6) | 2 (4.2) | 5 (10.9) |
Anxiety | 1 (2.2) | 3 (7.0) | 1 (2.1) | 3 (6.5) |
Back pain | 1 (2.2) | 4 (9.3) | 1 (2.1) | 4 (8.7) |
Increased lipase | 1 (2.2) | 3 (7.0) | 1 (2.1) | 3 (6.5) |
Myalgia | 0 | 2 (4.7) | 1 (2.1) | 3 (6.5) |
Pyrexia | 1 (2.2) | 6 (14.0) | 1 (2.1) | 6 (13.0) |
Dyspepsia | 0 | 4 (9.3) | 0 | 4 (8.7) |
Hypoesthesia | 0 | 4 (9.3) | 0 | 4 (8.7) |
Patients with ≥ 1 SAE | ||||
Patients with ≥ 1 SAE, n (%) | 8 (17.4) | 4 (9.3) | 10 (20.8) | 4 (8.7) |
SAE by system organ class, n (%)b | ||||
General disorders and administration site conditions | 1 (2.2) | 1 (2.3) | 1 (2.1) | 1 (2.2) |
Pyrexia | 1 (2.2) | 1 (2.3) | 1 (2.1) | 1 (2.2) |
Infections and infestations | 1 (2.2) | 3 (7.0) | 2 (4.2) | 3 (6.5) |
Device-related infection | 1 (2.2) | 2 (4.7) | 1 (2.1) | 2 (4.3) |
Escherichia urinary tract infection | 0 | 1 (2.3) | 0 | 1 (2.2) |
Gastroenteritis | 0 | 0 | 1 (2.1) | 0 |
Sepsis | 0 | 1 (2.3) | 0 | 1 (2.2) |
Septic shock | 0 | 1 (2.3) | 0 | 1 (2.2) |
Injury, poisoning and procedural complications | 0 | 1 (2.3) | 0 | 1 (2.2) |
Fractured sacrum | 0 | 1 (2.3) | 0 | 1 (2.2) |
Investigations | 0 | 0 | 1 (2.1) | 0 |
Abnormal liver function test | 0 | 0 | 1 (2.1) | 0 |
Metabolism and nutrition disorders | 1 (2.2) | 0 | 1 (2.1) | 0 |
Hypoglycemia | 1 (2.2) | 0 | 1 (2.1) | 0 |
Psychiatric disorders | 1 (2.2) | 0 | 1 (2.1) | 0 |
Major depression | 1 (2.2) | 0 | 1 (2.1) | 0 |
Renal and urinary disorders | 2 (4.3) | 0 | 2 (4.2) | 0 |
Chronic kidney disease | 2 (4.3) | 0 | 2 (4.2) | 0 |
Respiratory, thoracic, and mediastinal disorders | 1 (2.2) | 0 | 1 (2.1) | 0 |
Asthma | 1 (2.2) | 0 | 1 (2.1) | 0 |
Surgical and medical procedures | 1 (2.2) | 0 | 1 (2.1) | 0 |
Pain management | 1 (2.2) | 0 | 1 (2.1) | 0 |
Patients who stopped treatment due to AEs | ||||
n (%) | 0 | 0 | 1 (2.1) | 0 |
Deaths | ||||
n (%) | 0 | 0 | 0 | 0 |
Notable harms, n (%) | ||||
Motor neuropathyc | 0 | 3 (7.0) | 0 | 3 (6.5) |
Hepatocellular carcinoma | 0 | 0 | 0 | 0 |
Injection-site reactions | 8 (17.4) | 0 | 8 (16.7) | 0 |
Transaminase elevation | ||||
Increased ALT | 4 (8.7) | 1 (2.3) | 4 (8.3) | 1 (2.2) |
Increased AST | 3 (6.5) | 1 (2.3) | 3 (6.3) | 1 (2.2) |
Progression of renal impairmentd | 4 (8.7) | 0 | 5 (10.4) | 0 |
AE = adverse event; AIP = acute intermittent porphyria; AHP = acute hepatic porphyria; ALT = alanine transaminase; AST = aspartate transaminase; SAE = serious adverse event; SAS = Safety Analysis Set.
aFrequency ≥ 5% in any treatment group.
bReported by number of events rather than number of patients experiencing a SAE; therefore, the total number of events may be greater than the number of patients that experienced at least 1 SAE.
cNerve compression and peripheral neuropathy were included under this notable harm. For patients with AIP and patients with AHP, 0 patients in the givosiran group and 3 patients in the placebo group reported motor neuropathy (1 with nerve compression and 2 with peripheral neuropathy).
dProgression of renal impairment was not specifically reported; AEs in this category correspond to chronic kidney disease, reported as an AE.
Source: Clinical Study Report.6
Critical Appraisal
Internal Validity
Randomization was conducted using an interactive response system, which is an adequate method. Randomization was stratified by type of AHP (AIP versus non-AIP). Patients with AIP were further stratified by prophylactic hemin use and high or low historical attack rate in the 12 months before randomization. More specifically, patients who used hemin prophylactically before study entry were stratified by a historical AAR of fewer than 7 attacks versus 7 attacks or more. Patients who were not using hemin prophylactically before study entry were stratified by a historical AAR of fewer than 12 attacks versus 12 attacks or more. Although the sponsor reported that the cut-offs used for high and low historical AAR were based on the EXPLORE study, the criteria used to define high and low historical annualized attack rates were fairly arbitrary, according to the clinical experts; therefore, it is unclear how meaningful the stratification was. Study 003 was a DB study, and efforts were made to maintain blinding; however, injection-site reactions (which were reported solely in the givosiran group) offered the potential for unblinding, as did higher rates of nausea. Additionally, a major protocol deviation was reported for 4 patients who experienced a total of 17 instances of study drug administration (givosiran or placebo) with a syringe that was not masked with a blinding strip before study drug administration. However, this affected the givosiran and placebo groups equally and is not expected to bias the results.
Despite a small sample size, the treatment groups were well balanced overall. Differences to note include a slight difference in race, although feedback from the clinical experts indicated that AHP does not present differently by race. On average, patients in the placebo group had received a diagnosis of AHP more recently than patients in the givosiran group, by 3 years. A greater proportion of patients receiving givosiran had a history of prophylactic hemin use compared to those in the placebo group. Whether there would be a difference in the level of disease control between the 2 treatment groups is uncertain. The treatment groups differed in their history of medical conditions, including their history of peripheral neuropathy, gastrointestinal disorders (constipation, gastroesophageal reflux disease, and nausea), psychiatric disorders (depression and anxiety), and anemia. Feedback from the clinical experts indicated that patients’ history of medical conditions may not be related to AHP. The impact of these on outcomes such as daily worst nausea and HRQoL is unknown.
Overall dropout rates were extremely low. One patient with a non-AIP type of AHP who was randomized to placebo withdrew from the study due to an AE (elevated ALT). Missing data were not an issue for outcomes, and adherence was strong throughout the study. Among patients with AIP, 89% and 98% of those randomized to the givosiran and placebo groups did not miss a dose during the 6-month treatment period; adherence was similar among patients with AHP. All of the reported missed doses were due to administrative and visit scheduling issues, with the exception of 2 doses missed by 1 patient in the givosiran group, whose treatment was withheld due to transaminase elevations after 4 doses.
The primary outcome in Study 003 was the annualized rate of porphyria attacks, which was a composite end point that included attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP during the 6-month DB period. Of note, the sponsor indicated that “the analysis of the primary end point in patients with AIP was to allow evaluation of efficacy in a homogenous population and because of the scarcity of non-AIP subtypes compared to AIP.”6 The 3 components are objective measures of attacks; therefore, they are unlikely to contribute to the misidentification of a porphyria attack. Further, the sponsor noted that the 3-component primary end point was supported by results from the EXPLORE natural history study,25 which demonstrated that the majority of porphyria attacks were aligned with this definition.35 The primary end point excludes porphyria attacks that occur at home that do not require IV hemin; however, a sensitivity analysis was conducted that included all porphyria attacks, which was consistent with the primary analysis.
The components of the composite end point were reported descriptively. According to the clinical experts, the definition used for a porphyria attack requiring hospitalization was suggestive of an attack that is fairly severe. However, the experts also noted that hospitalization of a patient and options for are determined by a physician and clinical practice, which may have varied across study sites. The possibility of unblinding (previously described) also had the potential to influence patient or physician beliefs around treatment. Despite the observed variation in the rate reduction of attacks for each component, the ability to interpret the individual components of the composite end point is limited by potential bias. However, the estimates of effect for each of the components were in the same direction and not expected to have substantially affected the overall composite outcome.
A number of patient-reported outcomes were included in Study 003, including the SF-12, EQ-5D-5L, and PGIC to evaluate HRQoL; a single item of the BPI-SF, a single item of the BFI-SF, and NRS for daily worst nausea to evaluate symptoms of AHP; and the PPEQ to evaluate activities of daily living. With the exception of the PPEQ, patient-reported outcomes were not validated in patients with AHP, and disease-specific MIDs were not available for most of the outcomes; thus, the ability to appropriately evaluate the treatment effect based on these outcomes was limited. The patient-reported outcomes are broadly used in clinical trials and well validated for other populations, but evidence of validity of these for the use of the single item for daily worst pain from the BPI-SF was not identified. The PPEQ is specific to patients with porphyria; however, evidence of validity, reliability, responsiveness, or an MID was not identified for this outcome. The remaining outcomes included in this review — namely, the assessments of rescue medication use, opioid use, ability to attend work or school, and urinary ALA and PBG — were objective and subject to few limitations.
Study 003 was adequately powered to detect a change in the primary outcome, and a hierarchical testing procedure was employed to control for multiplicity among the secondary end points. Most of the secondary end points included in the testing hierarchy were specific to patients with AIP, with the exception of AAR in patients with AHP. Secondary outcomes were also evaluated in all patients with AHP, but included as exploratory outcomes and subject to substantial uncertainty. The statistical testing strategy stopped testing of the remaining end points in the hierarchy following a test that was not statistically significant. In Study 003, the results for daily worst pain were not statistically significant; therefore, nominal P values whose significance cannot be interpreted were reported for daily worst fatigue, daily worst nausea, and the change in PCS of the SF-12, which are clinically relevant outcomes considered important to patients.
There were no missing data for the following outcomes: composite AAR, annualized rate of hemin use, opioid usage. For ALA and PBG levels, 6% to 7% of data were missing; less than 3% of data were missing for the SF-12 and EQ-5D-5L. Two patient-reported exploratory outcomes, PGIC and PPEQ, were missing data for 20% of patients; according to the sponsor, this was because these outcomes were added through an amendment to the protocol. Data were imputed for missing patient-reported outcomes of pain, fatigue, and nausea following a pre-specified algorithm describing the methods used to account for missing data. Of note, missing data for up to 8% of patients for most of the DB period were unlikely to bias the results, but data were missing for 30% of patients at the week 24 visit (month 6). A post hoc reanalysis of end points using a non-parametric Wilcoxon test was conducted for outcomes that demonstrated a significant deviation from a normal distribution, namely daily worst pain, urinary ALA levels, and urinary PBG levels. Although this was performed post hoc, a non-parametric test was considered appropriate. Most of the outcomes included in the trials were reported as a rate ratio or mean change from baseline. The assessments of pain, fatigue, and nausea were conducted using the AUC to account for variability in symptoms for patients with AHP, which was considered appropriate.
Additionally, a preplanned, unblinded interim analysis was conducted to ensure that the study was adequately powered for the primary end point comparison at the final analysis. The primary analysis and subsequent secondary analyses were conducted at a 2-sided significance level of 0.049, reflecting a penalty of 0.001 for an unblinded interim analysis.
Subgroup analyses of the primary end point and urinary ALA secondary end point were pre-specified. As noted, patients with AIP were stratified by historical AAR at baseline, although the cut-offs used were arbitrary, according to the clinical experts. Rate ratios were calculated for the subgroup analyses, but statistical tests were not conducted. Subgroup analyses were not adjusted for multiplicity, and the trial was not powered to detect a difference based on the subgroups. CIs were wide, but did not cross 0.
External Validity
Givosiran is indicated for the treatment of adults with AHP in Canada. The diagnostic criteria used in the clinical trial were consistent with clinical practice, according to feedback from the clinical experts. The pivotal trial was composed of patients with AIP and non-AIP types of AHP, and was analyzed as such. In total, 95% of the study population were patients with AIP. However, feedback from the clinical experts on this review indicated that the trial data applied to all patients with AHP because of the mechanisms of action of givosiran and disease pathology, which were not expected to result in a differential treatment effects by AHP type. In terms of the study population being reflective of Canadian patients with AHP, the mean age of patients in the pivotal trial was higher than that of patients seen in clinical practice; the clinical experts described the average age of their patient population as between 20 years and 30 years. Of note, patients at least 12 years of age were eligible to enrol in Study 003; however, the patients who actually participated in Study 003 were 19 years or older and, as a result, generalizable to the patient population under review. Another consideration regarding the generalizability of the study population was that 41% to 48% of patients resided in Europe, where there is a higher prevalence (and therefore greater awareness) of this rare disease. The clinical experts relayed that because of the lack of available treatments for AHP, standard of care for a patient with a diagnosis of AHP is unlikely to vary by region. However, this is something to consider. The additional baseline characteristics and medical histories of the study population were typical of Canadian patients living with AHP.
Based on the composite definition of porphyria attacks used for the primary end point, at baseline, the median numbers of porphyria attacks in the 6 months before screening were 4 (range = 2 to 24) and 3 (range = 0 to 25) for patients with AIP randomized to givosiran and patients with AIP randomized to placebo, respectively. According to the clinical experts, patients in the study had a history of more frequent porphyria attacks than most patients seen in clinical practice in Canada. The sponsor acknowledged this, stating that “the study was enriched for attack frequency to ensure the ability to measure a difference in treatment effect on the primary composite porphyria attack end point.”6 As a result, the change in the rate of porphyria attacks observed in each of the treatment groups in the trial may not be generalizable to what would be expected in clinical practice.
The use of givosiran in Study 003 was aligned with the Health Canada–approved indication. Although the indication for givosiran is broad, the clinical experts anticipated that it would be used to prevent porphyria attacks rather than to treat acute attacks in clinical practice; this aligns with its use in Study 003. Patients receiving prophylactic hemin were required to discontinue its use before the study; screening for this occurred at least 4 days following discontinuation. Patients who were receiving treatment with a GnRH analogue at screening were permitted to enrol if they remained on GnRH treatment throughout the 6-month DB period, but the number of patients using GnRH before enrolment was not reported. The use of hemin (to treat acute attacks) and GnRH in the trial was consistent with standard of care in clinical practice. The duration of treatment evaluated in the DB treatment period was 6 months, followed by an open-label treatment period of up to 29 months. The clinical experts indicated that they would expect patients to remain on treatment unless there was a specific safety or biological reason to discontinue it; therefore, a 6-month evaluation period is not representative of the long-term use of givosiran in clinical practice. The duration of the trial may limit the ability to evaluate the impact of givosiran on HRQoL and safety outcomes.
The outcomes of greatest importance to clinicians and patients were frequency of AHP attacks, management of symptoms, and HRQoL, in addition to a reduction in side effects from treatment. All of these outcomes were included in the pivotal trial in some capacity. The clinical experts indicated that formal evaluation of HRQoL— through the use of questionnaires, for example — is not done in clinical practice, where HRQoL is evaluated informally. In the study, patient-reported symptoms, such as pain, fatigue, and nausea, were recorded using an electronic diary and measured using an NRS, which is also unlikely in clinical practice but important to patients. The definitions used for AHP attacks were consistent with those used in clinical practice.
Additionally, the clinical experts indicated that patients with a history of attacks related to AHP are accustomed to facing scarce treatment options for an acute attack, and often tend to treat their attacks symptomatically at home. Therefore, it is plausible that participation in the clinical trial would increase patients’ access and attention to care and, in turn, their likelihood of seeking care for acute attacks. This could affect the generalizability of the trial results to clinical practice and has the potential to further inflate the reported attack rate.
Indirect Evidence
A focused literature search for network metanalyses dealing with porphyrias was run in MEDLINE All (1946–) and Embase (1974‒) through Ovid on March 24, 2021. No limits were applied to the search. Indirect treatment evidence for givosiran was not identified in this review.
Other Relevant Evidence
A phase I study, a phase I/II study, a phase III OLE study, and an expanded access program (EAP) were included in the sponsor’s submission to CADTH and were considered to address important gaps in the evidence included in the systematic review.
Study ALN-AS1-001 (001) and Study ALN-AS1-002 (002) provided additional information and long-term data on the safety and tolerability of givosiran in adult patients with AIP. The ENVISION ALN-AS1-003 study (Study 003 OLE) provides additional long-term evidence on the safety and efficacy of givosiran in adult patients with AHP. Study ALN-AS1-005 (005) is an EAP of givosiran for patients with AHP. Study 002 and Study 003 OLE were ongoing at the time of this review.
Phase I (Completed) and Phase I/II (Ongoing) Clinical Trials: Study ALN-AS1-001 and Study ALN-AS1-002
Methods
Study 001 was a 3-part, multi-centre, placebo-controlled, phase I study of the safety and tolerability of subcutaneous givosiran for the treatment of adults with AIP. Parts A, B, and C were single-ascending dose, multiple-ascending dose, and multidose in design, respectively. The adaptive design allowed for different dosing regimens and dose levels to be assessed based on new safety, tolerability, and PD data. Study 001 was conducted at 6 sites in Great Britain, Sweden, and the US. The study was initiated on May 6, 2015, and completed on September 6, 2017. In total, 40 patients with AIP who are CHEs were randomized to parts A and B (n = 23), while those with AIP who had recurrent attacks were randomized to part C (n = 17).
In all 3 parts, patients were randomized 3:1 to receive givosiran or placebo. Parts A and B had 60-day screening periods before treatment was administered. Part A was single-blind, single-ascending dose with 5 possible cohorts, where each cohort received a different dose of givosiran. Patients in a cohort received a single administration of 1 dose of givosiran or placebo. The following doses were assessed: 0.035 mg/kg, 0.10 mg/kg, 0.35 mg/kg, 1.0 mg/kg, and 2.5 mg/kg, each administered to a different cohort. Patients who completed part A could take part in a subsequent part A cohort or in part B if they met the eligibility criteria. Part B was a single-blind, multiple-ascending dose, and patients received 2 injections over a 28-day period of 1 of the following doses: 0.35 mg/kg givosiran, 1.0 mg/kg givosiran, or placebo. Patients in parts A and B were followed for up to 6 weeks after the treatment period. Part C was DB and multidose, and after the 168-day run-in period, patients received doses of 2.5 mg/kg givosiran, 5.0 mg/kg givosiran, or placebo once monthly (4 injections total) or once every 3 months (2 injections total) for a 3-month period. Patients in part C were followed for up to 3 months after the treatment period. Patients who completed part C were able to participate in Study 002, an OLE.
Study 002 was an ongoing, multi-centre, open-label, phase I/II study of the long-term safety and tolerability of subcutaneous givosiran for the treatment of adults with AIP who completed part C of Study 001. Study 002 (N = 16) was conducted at 5 sites in Great Britain, Sweden, and the US.36 The study began on October 24, 201636; the data presented in this summary are based on the interim report, with a cut-off date of December 13, 2018. Patients received givosiran 2.5 mg/kg monthly or 5.0 mg/kg monthly or every 3 months until the safety review committee assessed safety, tolerability, and efficacy data and agreed that all patients would transition to receive the 2.5 mg/kg dose. Treatment duration was estimated to be up to 36 months, with the estimated total time in study with screening and baseline up to 44 months.
Figure 11, Figure 12, Figure 13, and Figure 14 outline the study designs for parts A, B, and C of Study 001 and for Study 002, respectively.
Figure 11: Design of Study 001, Part A
ALA = aminolevulinic acid; ALN-AS1 = givosiran; D = day; ET = early termination; PBG = porphobilinogen; Q2W = every 2 weeks; Q4W = every 4 weeks; W = week.
Source: Clinical Study Report for Study ALN-AS1-001.37
Figure 12: Design of Study 001, Part B
ALA = aminolevulinic acid; ALN-AS1 = givosiran; D = day; ET = early termination; PBG = porphobilinogen; Q2W = every 2 weeks; Q4W = every 4 weeks; W = week.
Source: Clinical Study Report for Study ALN-AS1-001.37
Figure 13: Design of Study 001, Part C
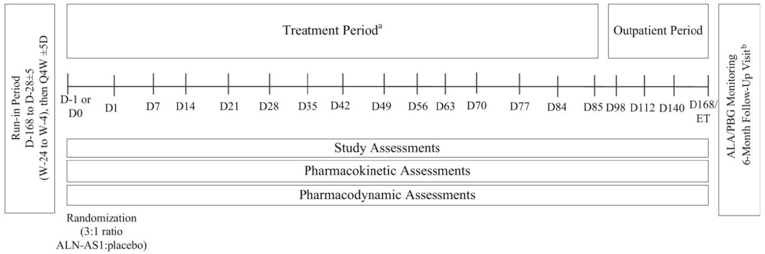
ALA = aminolevulinic acid; ALN-AS1 = givosiran; D = day; ET = early termination; PBG = porphobilinogen; Q4W = every 4 weeks; W = week.
Source: Clinical Study Report for Study ALN-AS1-001.37
Figure 14: Design of Study 002
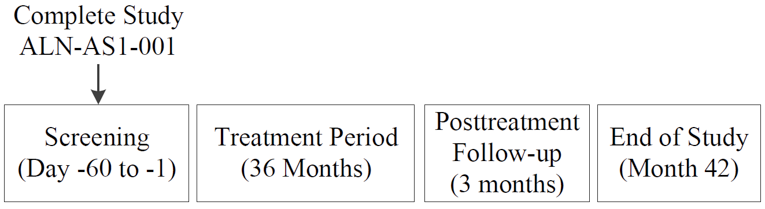
ALN-AS1-001 = Study 001.
Source: Clinical Study Report of ALN-AS1-002.38
Populations
The inclusion and exclusion criteria for studies 001 and 002 are outlined in Table 23. In short, adults between 18 years and 65 years were eligible if they had a diagnosis of AIP. More specifically, patients in parts A and B were CHEs (and had experienced no porphyria attacks within 6 months of the study), while those in part C had to have experienced at least 2 porphyria attacks in the 6 months before the run-in period. Patients in Study 002 must have completed and tolerated the study drug dosing in Study 001, part C. Furthermore, patients were to stop receiving prophylactic hemin during either Study 001 or Study 002. Table 24 summarizes the baseline characteristics of patients who received either placebo or givosiran 2.5 mg/kg in Study 001 and all patients in Study 002.
In Study 001, part A, 3 patients received givosiran 2.5 mg/kg and 6 received placebo, while in part C, 3 patients received givosiran 2.5 mg/kg and 4 received placebo. Briefly, patients who were treated with givosiran 2.5 mg/kg had mean ages of 42 years (SD = 11.6 years) and 30 years (SD = 10.7 years) for Study 001, parts A and C, respectively. The majority of patients in the 2.5 mg/kg cohort of Study 001 were female and White, with a mean body mass of 82.5 kg (SD = 10.45 kg) in part A and 84.5 kg (SD = 16.86 kg) in part C. Patients in the 2.5 mg/kg cohort of part C had a mean of 14.7 attacks (SD = 18.9 attacks) in the 12 months before the study. One-third of patients in the study were on prophylactic hemin.
Initially, patients could receive 2.5 mg/kg or 5.0 mg/kg of givosiran. Based on a review of the safety data, it was later decided that all 16 patients were to receive 2.5 mg/kg. Baseline characteristics for Study 002 were similar. Patients had a mean age of 37.4 years (SD = 12.0 years); 87.5% were female; 81.3% were White; and patients had a mean body mass of 75.84 kg (SD = 18.46 kg). Nearly all patients (93.8%) in Study 002 had experienced at least 1 porphyria attack in the 12 months preceding the study, with a mean of 13.0 attacks (SD = 13.1 attacks) during that time period. All had used hemin during an acute attack, and half had used it prophylactically.
Table 23: Summary of Inclusion and Exclusion Criteria for Studies 001 and 002
Study 001, parts A and B | Study 001, part C | Study 002 |
|---|---|---|
Inclusion criteria | ||
|
|
|
Exclusion criteria | ||
|
|
|
AIP = acute intermittent porphyria; ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CHE = chronic high excreter; Cr = creatinine; ECG = electrocardiogram; eGFR = estimated glomerular filtration rate; GalNAc = N-acetylgalactosamine; GGT = gamma glutamyl transpeptidase; GnRH = gonadotropin-releasing hormone; HBV = hepatitis B virus; HCV = hepatitis C virus; HMBS = hydroxymethylbilane synthase; MDRD = Modification of Diet in Renal Disease; PBG = porphobilinogen, PBGD = porphobilinogen deaminase; SC = subcutaneous; TBIL = total bilirubin; ULN = upper limit of normal.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Table 24: Summary of Baseline Characteristics in Studies 001 and 002 — Safety Population
Characteristic | Study 001, part A | Study 001, part C | Study 002 | ||
|---|---|---|---|---|---|
Placebo (N = 6) | Givosiran 2.5 mg/kg (N = 3) | Placebo (N = 4) | Givosiran 2.5 mg/kg QM (N = 3) | All givosiran (N = 16) | |
Demographics | |||||
Age (years), mean (SD) | 48 (11.6) | 42 (11.6) | 43 (13.6) | 30 (10.7) | 37.4 (12.0) |
Age (years), median (range) | 49 (35 to 64) | 36 (34 to 55) | 42 (27 to 60) | 28 (21 to 42) | 39.5 (21 to 60) |
Female, n (%) | 5 (83.3) | 3 (100) | 2 (50.0) | 3 (100) | 14 (87.5) |
Body mass (kg), mean (SD) | 80.9 (10.52) | 82.5 (10.45) | 91.4 (20.82) | 84.5 (16.86) | 75.84 (18.46) |
Race, n (%) | |||||
White | 5 (83.3) | 2 (66.7) | 4 (100) | 3 (100) | 13 (81.3) |
Black | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (12.5) |
Asian | 1 (16.7) | 1 (33.3) | 0 (0) | 0 (0) | 1 (6.3) |
Disease historya | |||||
Patients with porphyria attacks in past 12 months, n (%) | NA | NA | 4 (100) | 2 (66.7) | 15 (93.8) |
Required hospitalization, n (%) | NA | NA | 2 (50.0) | 1 (33.3) | 8 (50.0) |
Treated at outpatient clinic or infusion centre, n (%) | NA | NA | 4 (100) | 1 (33.3) | 9 (56.3) |
Treated at home, n (%) | NA | NA | 0 (0) | 1 (33.3) | 5 (31.3) |
Number of attacks in past 12 months, mean (SD) | NA | NA | 18.8 (21.0) | 14.7 (18.9) | 13.0 (13.1) |
Number of attacks in past 12 months, median (range) | NA | NA | 10.0 (5 to 50) | 8.0 (0 to 36) | 10.0 (0 to 50) |
Hemin use during attack, n (%) | NA | NA | 4 (100) | 3 (100) | 16 (100) |
Hemin prophylaxis, n (%) | NA | NA | 2 (50.0) | 1 (33.3) | 8 (50.0) |
Other porphyria treatments, n (%) | |||||
Hormone suppression | NA | NA | 0 (0) | 1 (33.3) | 4 (25.0) |
High-carbohydrate diet | NA | NA | 2 (50.0) | 1 (33.3) | 7 (43.8) |
Glucose infusions | NA | NA | 2 (50.0) | 2 (66.7) | 10 (62.5) |
Other | NA | NA | 0 (0) | 2 (66.7) | 4 (25.0) |
Self-treated at home, n (%) | |||||
Sugar water | NA | NA | 0 (0) | 0 (0) | 2 (12.5) |
High carbohydrates | NA | NA | 2 (50.0) | 1 (33.3) | 9 (56.3) |
Opioid or narcotic analgesic medications | NA | NA | 2 (50.0) | 1 (33.3) | 9 (56.3) |
Other | NA | NA | 2 (50.0) | 1 (33.3) | 9 (56.3) |
NA = not applicable; QM = once monthly; SD = standard deviation.
aFor Study 002, before enrolment in Study 001 part C.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Interventions
In Study 001, patients received subcutaneous givosiran or placebo on an inpatient basis at the study centre. For part A, a single administration of givosiran (0.035 mg/kg, 0.1 mg/kg, 0.35 mg/kg, 1.0 mg/kg, or 2.5 mg/kg) or placebo was given. In part B, patients received 2 monthly doses of givosiran (0.35 mg/kg or 1.0 mg/kg) or placebo. For part C, patients received doses of 2.5 mg/kg givosiran, 5.0 mg/kg givosiran, or placebo once monthly (4 injections total) or once every 3 months (2 injections total) for a 4-month period.
In Study 002, patients received subcutaneous givosiran 2.5 mg/kg monthly or 5.0 mg/kg monthly or every 3 months. Data from Study 001 indicated that the 2.5 mg/kg and 5.0 mg/kg doses both demonstrated acceptable safety and more stability in lowering delta-ALA when administered monthly versus every 3 months. It was also noted that the higher dose of givosiran did not confer any additional benefit in terms of lowering delta-ALA levels. As a result, all patients receiving 5.0 mg/kg (monthly or every 3 months; n = 7) were transitioned to the lower dose, such that all study patients were receiving 2.5 mg/kg monthly. This was the dose used in the phase III ENVISION study and the intended commercial dose.
Prophylactic hemin use was not permitted during either study, though hemin use was allowed during acute attacks.
Outcomes
The primary outcomes of Study 001 were the safety and tolerability of givosiran for the treatment of patients with AIP who are CHEs or have recurrent attacks. Secondary end points included pharmacokinetic (PK) and PD evaluations. Exploratory end points included additional PD investigations and, for part C only, the number of porphyria attacks and hemin doses, among other clinical activity measures. Porphyria attacks were reported as exploratory efficacy end points rather than as AEs. Attacks were analyzed based on 2 definitions: all attacks (comprising attacks requiring hospitalization, urgent health care visit, or hemin administration at home, or attacks at home that did not require hemin administration); and composite attacks (comprising attacks requiring hospitalization, urgent health care visit, or hemin administration at home). The individual components of the attack rate were also analyzed. The EQ-5D-5L was captured as an exploratory outcome, and harms data for AEs, SAEs, WDAEs, and deaths were collected.
The primary outcomes of Study 002 were the long-term safety and tolerability of givosiran for the treatment of patients with AIP and recurrent attacks. Secondary end points included PD evaluations and clinical activity assessments. Exploratory end points included PK analyses, HRQoL assessments, and biochemical investigations. The clinical activity of givosiran was assessed by the frequency and characteristics of porphyria attacks and doses of hemin administered. HRQoL was assessed using the EQ-5D-5L and BPI-SF.
Statistical Analysis
The sample size for Study 001 was not based on power calculations. The study planned to enrol up to 64 patients. Summary statistics were presented for continuous data, and event count data were summarized as annualized rates and SEM estimates. No inferential statistical analyses were planned for safety assessments in the study. No imputations were made for missing data. The safety population was the main analysis set used for evaluating safety and clinical activity.
The sample size for Study 002 was not based on power calculations. It was made up of patients who had completed Study 001, part C. No formal statistical analyses were performed. Descriptive statistics were presented for continuous variables, while frequencies and percentages were presented for categorical and ordinal variables. In Study 002, missing continuous and categorical variables were not imputed unless otherwise specified, and no imputations were made for missing or partially missing dates. Interim analyses were descriptive and did not involve formal hypothesis testing. The safety population was the main analysis set used for evaluating safety and clinical activity.
Patient Disposition
The patient disposition for studies 001 and 002 is summarized in Table 25.
In Study 001, 49 individuals were screened, 40 of whom were randomized. Parts A and B had a total of 23 unique patients where 20 patients and 16 patients made up the givosiran and placebo groups, respectively. Three patients participated in both parts A and B, and 2 patients took part in 2 different dose cohorts of part A. Each unique patient was only counted once in each column of the summaries. Part C consisted of 17 patients, where 13 patients and 4 patients made up the givosiran and placebo groups, respectively. All patients completed treatment in parts A and B, while 94.1% of patients in part C completed treatment. In the latter, there was 1 WDAE when a patient in the givosiran 5.0 mg/kg group had a fatal SAE.
All 16 patients who completed Study 001, part C took part in Study 002. At the time of the interim report, no patients had completed treatment, and 1 patient (6.3%) had discontinued due to an AE.
The safety populations for studies 001 and 002 included all patients who received at least 1 dose of the study drug.
Table 25: Patient Disposition for Studies 001 and 002
Disposition | Study 001, parts A and B | Study 001, part C | Study 002 |
|---|---|---|---|
Screened | 49 | NAa | |
Randomized, N (%) | 23 (100) | 17 (100) | 16 (100) |
Completed treatment, n (%) | 23 (100) | 16 (94.1) | 0 (0) |
Withdrew from study, n (%) | 0 | 1 (5.9) | 1 (6.3) |
Fatal SAE | 0 | 1 (5.9) | 0 |
AE | 0 | 0 | 1 (6.3) |
Safety analysis sets, n (%) | 23 (100) | 17 (100) | 16 (100) |
AE = adverse event; NA = not applicable; SAE = serious adverse event.
aParticipants in Study 002 were patients who had completed Study 001, part C.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Exposure to Study Treatments
Table 26 summarizes the exposure to givosiran for Study 001, parts A and C and Study 002 for patients who received givosiran 2.5 mg/kg or placebo.
In Study 001, part A, patients received a single 2.5 mg/kg dose of either givosiran (n = 3) or placebo (n = 5).
In Study 001, part C, patients received between 2 and 4 doses of either givosiran 2.5 mg/kg monthly (n = 3) or placebo (n = 4), resulting in a mean exposure of 3.04 months (SD = 0.0) for the cohort receiving givosiran 2.5 mg/kg monthly.
Figure 15 outlines patients’ transitions from Study 001, part C to Study 002. Of the 16 patients who rolled over from Study 001, part C, 12 had received givosiran and 4 had been given placebo. Upon enrolment into Study 002, 9 patients were started on 2.5 mg/kg givosiran monthly, while the other 7 patients began with either 5.0 mg/kg monthly (n = 3) or every 3 months (n = 4). After a review of the safety, tolerability, and efficacy data, it was agreed that all patients would be transitioned to receive a 2.5 mg/kg dose. Overall, patients in Study 002 had a mean of 16.29 months (SD = 5.06) of exposure to givosiran 2.5 mg/kg monthly and a mean of 16 doses (SD = 5.7). Two patients (12.5%) missed 1 dose during the study.
Table 26: Exposure to Givosiran 2.5 mg/kg in Studies 001 and 002 — Safety Population
Exposure | Study 001, part A | Study 001, part C | Study 002 | ||
|---|---|---|---|---|---|
Placebo (N = 5) | 2.5 mg/kg (N = 3) | Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | 2.5 mg/kg QMa (N = 16) | |
Duration of study drug exposure (months)b | |||||
Mean (SD) | NA | NA | 3.29 (0.27) | 3.04 (0.00) | 16.29 (5.06) |
Median (range) | NA | NA | 3.27 (3.0 to 3.6) | 3.04 (3.0 to 3.0) | 15.59 (2.1 to 23.1) |
Total number of doses | |||||
Mean (SD) | 1.0 (0.0) | 1.0 (0.0) | 3.0 (1.2) | 4.0 (0.0) | 16 (5.7) |
Median (range) | 1.0 (1 to 1) | 1.0 (1 to 1) | 3.0 (2 to 4) | 4.0 (4 to 4) | 16.5 (1 to 24) |
Patients with missing doses | |||||
None, n (%) | 5 (100) | 3 (100) | 4 (100) | 3 (100) | 14 (87.5) |
1, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (12.5) |
NA = not applicable; QM = once monthly; SD = standard deviation.
aIncludes only exposure to 2.5 mg/kg QM.
bDuration of treatment period (months) = (last dosing date minus first dosing date plus 1) divided by 28. One month = 28 days.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Figure 15: Transition From Study 001, Part C to Study 002 for Patients Who Received Givosiran 2.5 mg/kg or Placebo
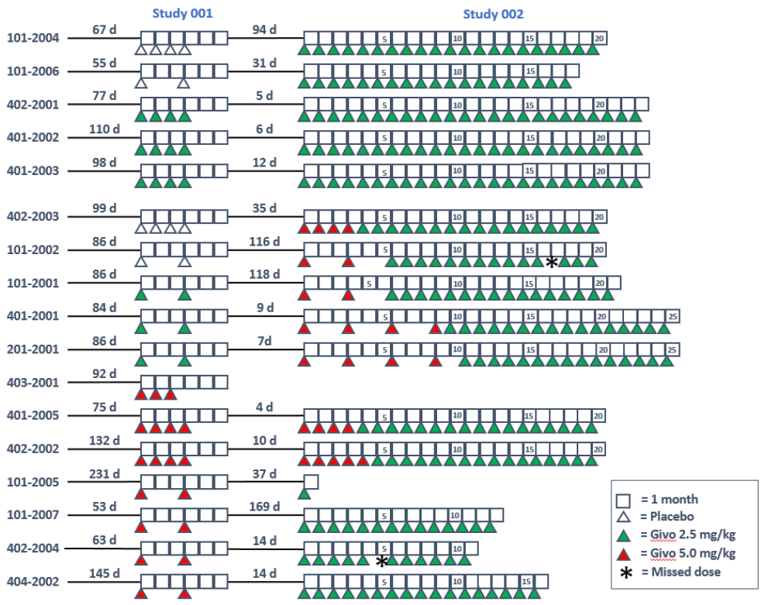
d = day; Givo = givosiran.
Source: Clinical Study Report of ALN-AS1-002.38
Efficacy
PK and PD efficacy outcomes will not be discussed in this summary.
Table 27, Table 28, and Table 29 summarize porphyria attack outcomes, hemin use, and HRQoL outcomes, respectively, in Study 001, part C and Study 002, with a focus on results for patients who received givosiran 2.5 mg/kg monthly.
Patients had fewer attacks during the treatment and follow-up phases compared to the run-in of Study 001, part C for all attacks, attacks requiring hospitalization, and attacks requiring urgent health care visits. The cohort receiving givosiran 2.5 mg/kg monthly had a mean AAR of 2.9 (SEM = 1.91) for composite attacks and a mean annualized rate of hemin use of 2.9 days (SEM = 1.44) during the treatment and follow-up periods. The placebo group had a mean AAR of 16.7 (SEM = 4.97) for composite attacks and a mean annualized rate of hemin use of 23.4 days (SEM = 9.9) during the treatment and follow-up periods. The mean change from baseline to last post-dose assessment for EQ-5D-5L index scores were 0.0738 (SD = 0.0637) and 0.0103 (SD = 0.1141) for the groups receiving placebo and givosiran 2.5 mg/kg monthly, respectively. The mean change from baseline to last post-dose assessment EQ VAS scores were 26.3 (SD = 11.1) and –2.7 (SD = 8.7) for the groups receiving placebo and givosiran 2.5 mg/kg monthly, respectively.
During Study 002, patients had fewer composite attacks during the treatment period compared to the run-in period (n = 9 and n = 72, respectively) and fewer attacks requiring hospitalization, urgent health care visits, or treatment with hemin at home. There was a greater number of instances where patients treated attacks at home without hemin during the treatment period (n = 32) versus during the run-in period (n = 9). The mean composite AAR was 17.0 (SEM = 3.5) and 1.2 (SEM = 0.4) for the run-in period of Study 001, part C and the treatment period, respectively. The mean rate for annualized hemin use was 33.1 days (SEM = 7.0 days) during the run-in period compared to 1.1 days (SEM = 0.6 days) during the treatment period of Study 002. In terms of HRQoL, mean EQ-5D-5L index and VAS scores increased by a mean of 0.0580 (SD = 0.1120) and 11.6 (SD = 17.8), respectively, from baseline to the last post-dose assessment.
Table 27: Summary of Porphyria Attack Outcomes in Study 001, Part C and Study 002 — Safety Population
Outcome | Study 001, part C | Study 002 | ||||
|---|---|---|---|---|---|---|
Run-in period | Treatment and follow-up periods | Run-in period for Study 001, part C | Treatment period | |||
Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | 2.5 mg/kg QM (N = 16) | 2.5 mg/kg QM (N = 16) | |
Composite attacks,a n | 17 | 8 | 32 | 4 | 72 | 9b |
AAR, mean (SEM) | 20.2 (5.66) | 10.3 (4.17) | 16.7 (4.97) | 2.9 (1.91) | 17.0 (3.5) | 1.2 (0.4)b |
Percent change from run-in (%) | NA | NA | –17.6 | –71.9 | NA | –93.0b |
Percent difference from placebo (%) | NA | NA | NA | –82.6 | NA | NA |
All attacks,c n | 17 | 12 | 32 | 9 | 81 | 44 |
AAR, mean (SEM) | 20.2 (5.66) | 15.4 (1.56) | 16.7 (4.97) | 6.5 (0.00) | 19.1 (3.3) | 2.0 (0.7) |
Percentage change from run-in (%) | NA | NA | –17.6 | –57.8 | NA | –89.3 |
Percentage difference from placebo (%) | NA | NA | NA | –61.1 | NA | NA |
Attacks requiring hospitalization, n | 2 | 0 | 3 | 0 | 16 | 4 |
AAR, mean (SEM) | 2.4 (1.51) | 0.0 (0.00) | 1.6 (0.50) | 0.0 (0.00) | 3.8 (2.2) | 0.2 (0.1) |
Percentage change from run-in (%) | NA | NA | –34.3 | NA | NA | –95.1 |
Percentage difference from placebo (%) | NA | NA | NA | –100.0 | NA | NA |
Requiring urgent health care visit, n | 15 | 8 | 29 | 4 | 35 | 7 |
AAR, mean (SEM) | 17.8 (5.54) | 10.3 (4.17) | 15.1 (5.00) | 2.9 (1.91) | 8.3 (2.5) | 0.3 (0.1) |
Percentage change from run-in (%) | NA | NA | –15.3 | –71.9 | NA | –96.1 |
Percentage difference from placebo (%) | NA | NA | NA | –80.8 | NA | NA |
Treated at home with hemin, n | 0 | 0 | 0 | 0 | 21 | 1 |
AAR, mean (SEM) | 0.0 (0.00) | 0.0 (0.00) | 0.0 (0.00) | 0.0 (0.00) | 5.0 (3.2) | 0.0 (0.00) |
Percentage change from run-in (%) | NA | NA | NA | NA | NA | –99.1 |
Percentage difference from placebo (%) | NA | NA | NA | NA | NA | NA |
Treated at home without hemin, n | 0 | 4 | 0 | 5 | 9 | 32 |
AAR, mean (SEM) | 0.0 (0.00) | 5.1 (5.62) | 0.0 (0.00) | 3.6 (1.91) | 2.1 (1.2) | 1.5 (0.6) |
Percentage change from run-in (%) | NA | NA | NA | –29.7 | NA | –29.9 |
Percentage difference from placebo (%) | NA | NA | NA | NA | NA | NA |
AAR = annualized attack rate; NA = not applicable; QM = once monthly; SEM = standard error of the mean.
aInclude attacks requiring hospitalization, urgent health care visit, and IV hemin treatment at home.
bData for first 6 months of the treatment period.
cIncludes attacks requiring hospitalization, urgent health care visit, IV hemin treatment at home, and attacks treated at home without hemin.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Table 28: Summary of Hemin Use in Study 001, Part C and Study 002 — Safety Population
Hemin use | Study 001, part C | Study 002 | ||||
|---|---|---|---|---|---|---|
Run-in | Treatment and follow-up periods | Run-in period for Study 001, part C | Treatment period | |||
Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | 2.5 mg/kg QM (N = 16) | 2.5 mg/kg QM (N = 16) | |
Annualized hemin doses (days) | 30 | 13 | 45 | 4 | 140 | 24 |
Annualized rate, mean (SEM) | 35.7 (5.49) | 16.7 (6.77) | 23.4 (9.88) | 2.9 (1.44) | 33.1 (7.0) | 1.1 (0.6) |
Percentage change from run-in to treatment and follow-up (%) | NA | NA | –34.3 | –82.7 | NA | –96.6 |
Percentage difference from placebo (%) | NA | –53.2 | NA | –87.6 | NA | NA |
NA = not applicable; QM = once monthly; SEM = standard error of the mean.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Table 29: Summary of HRQoL Outcomes in Study 001, Part C and Study 002 — Safety Population
Outcomes | Study 001, part C | Study 002 | |
|---|---|---|---|
Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | 2.5 mg/kg QM (N = 16) | |
EQ-5D-5L index score | |||
Baseline, mean (SD) | 0.8440 (0.1191) | 0.7460 (0.1202) | 0.8140 (0.1050) |
Last post-dose assessment, n | 4 | 3 | 15 |
Last post-dose assessment, mean (SD) | 0.9178 (0.0950) | 0.7563 (0.1145) | 0.8600 (0.1320) |
Change from baseline at last post-dose assessment, mean (SD) | 0.0738 (0.0637) | 0.0103 (0.1141) | 0.0580 (0.1120) |
EQ VAS score | |||
Baseline, mean (SD) | 56.3 (26.9) | 74.3 (13.7) | 68.9 (20.9) |
Last post-dose assessment, n | 4 | 3 | 15 |
Last post-dose assessment, mean (SD) | 82.5 (28.7) | 71.7 (10.4) | 78.8 (19.4) |
Change from baseline at last post-dose assessment, mean (SD) | 26.3 (11.1) | –2.7 (8.7) | 11.6 (17.8) |
EQ-5D-5L = EuroQoL 5-Dimensions 5 Levels questionnaire; EQ VAS = EuroQoL Visual Analogue Scale; HRQoL = health-related quality of life; QM = once monthly; SD = standard deviation.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Harms
Table 30 summarizes the harms outcomes in Study 001, parts A and C and Study 002, focusing on results for patients who received givosiran 2.5 mg/kg monthly where data were available. Most patients (66.7%) in part A of Study 001 and 100% of patients in both part C of Study 001 and in Study 002 experienced at least 1 AE. In Study 001, part C, the most frequently reported AEs were abdominal pain, abdominal distension, nausea, and injection-site reaction (66.7% of patients). In Study 002, the most common AEs were abdominal pain, fatigue, and nausea (43.8% of patients), while 37.5% of patients had an injection-site reaction. SAEs were reported in 100% of patients who received givosiran 2.5 mg/kg in Study 001, part C and in 25% of patients in Study 002. SAEs included functional gastrointestinal disorder, pyrexia, anaphylactic reaction, Clostridium difficile colitis, sinusitis bacterial infection, mental status changes, dyspnea, and deep vein thrombosis. There was 1 WDAE in Study 002. No deaths were reported in the cohorts of interest.
Table 30: Summary of Harms Outcomes in Study 001, Parts A and C and Study 002 — Safety Population
Outcomes | Study 001, part A | Study 001, part C | Study 002 | ||
|---|---|---|---|---|---|
Placebo (N = 5) | 2.5 mg/kg (N = 3) | Placebo (N = 4) | 2.5 mg/kg QM (N = 3) | All givosiran (N = 16) | |
Patients with ≥ 1 AE, n (%) | 5 (100) | 2 (66.7) | 4 (100) | 3 (100) | 16 (100) |
AE,a n (%) | |||||
Abdominal pain | 0 (0) | 0 (0) | 1 (25.0) | 2 (66.7) | 7 (43.8) |
Fatigue | NR | NR | 0 (0) | 0 (0) | 7 (43.8) |
Nausea | NR | NR | 1 (25.0) | 2 (66.7) | 7 (43.8) |
Injection-site reaction | NR | NR | 0 (0) | 2 (66.7) | 6 (37.5) |
Erythema | NR | NR | NR | NR | 6 (37.5) |
Pruritis | NR | NR | NR | NR | 4 (25.0) |
Headache | NR | NR | 1 (25.0) | 0 (0) | 5 (31.3) |
Myalgia | NR | NR | NR | NR | 5 (31.3) |
Nasopharyngitis | 0 (0) | 0 (0) | 1 (25.0) | 1 (33.3) | 5 (31.3) |
Diarrhea | 0 (0) | 0 (0) | 1 (25.0) | 0 (0) | 4 (25.0) |
INR increase | NR | NR | NR | NR | 4 (25.0) |
Back pain | 2 (40.0) | 0 (0) | 0 (0) | 1 (33.3) | 3 (18.8) |
Migraine | NR | NR | 0 (0) | 1 (33.3) | 3 (18.8) |
Paresthesia | NR | NR | 0 (0) | 1 (33.3) | 3 (18.8) |
Vomiting | NR | NR | 2 (50.0) | 1 (33.3) | 3 (18.8) |
Abdominal distension | NR | NR | 0 (0) | 2 (66.7) | NR |
Peripheral swelling | NR | NR | 0 (0) | 1 (33.3) | NR |
Patients with ≥ 1 SAE, n (%) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 4 (25.0) |
SAE,b n (%) | |||||
Functional gastrointestinal disorder | 0 (0) | 0 (0) | 0 (0) | 1 (33.3) | NR |
Pyrexia | NR | NR | NR | NR | 1 (6.3) |
Anaphylactic reaction | NR | NR | NR | NR | 1 (6.3) |
Clostridium difficile colitis | NR | NR | NR | NR | 1 (6.3) |
Sinusitis bacterial infection | NR | NR | NR | NR | 1 (6.3) |
Mental status changes | NR | NR | NR | NR | 1 (6.3) |
Dyspnea | NR | NR | NR | NR | 1 (6.3) |
Deep vein thrombosis | NR | NR | NR | NR | 1 (6.3) |
WDAE, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (6.3) |
Death, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
AE = adverse event; INR = international normalized ratio; NR = not reported; QM = once monthly dose; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aFrequency ≥ 2 patients in Study 001 or ≥ 4 patients in Study 002.
bFrequency ≥ 1 patient.
Source: Clinical Study Report for studies ALN-AS1-001 and ALN-AS1-002.37,38
Critical Appraisal
Internal Validity
Part A of Study 001 was single-blinded, while in part C, patients, investigators, and study centre staff were blinded. Although it was stated that the drug product could potentially be visually distinguished from placebo, syringes were masked in the pharmacy before being transferred to the clinic. All parts of Study 001 were placebo-controlled, which may have helped to reduce bias. There were 9 individuals who failed the screening process, but the reasons for this were not described. There was no imputation for missing data in Study 001. For part C, HRQoL was measured using the EQ-5D at baseline, during the treatment period only for instances where the patient was hospitalized for an attack, and twice in the follow-up period at scheduled visits. Therefore, HRQoL while on treatment but not when hospitalized for an attack is unknown for these patients. However, if it were reported, the data would be limited to only 3 individuals who received givosiran 2.5 mg/kg for a total of 3 months. Study 001 was adaptive in nature and allowed for dose level and dose regimen changes to occur based on safety, tolerability, and PD data. More specifically, dose increases were based on only 3 patients in each cohort of part A, all of whom were CHEs and did not have an acute porphyria attack in the 6 months leading up the to the study. While this design may be reasonable for a phase I safety and tolerability study, the results should be used to inform treatment in this setting only and not generalized beyond this.
Study 002 had an open-label design. Given that patients were aware they were receiving active treatment, this may have affected their perception of improvement during the study, which could have influenced patient-reported and HRQoL outcomes. All patients who completed part C of Study 001 rolled over into Study 002, and no new patients were enrolled. Therefore, the results reflect patients who were able to tolerate and adhere to treatment for the 3 months of the parent study. There was no imputation for missing data in Study 002. EQ-5D scores were taken at baseline and every 6 months for the first 18 months of treatment, including if a patient was hospitalized for an attack. However, these data are limited to the 16 patients in Study 002. Furthermore, 7 patients initially received givosiran 5.0 mg/kg monthly or every 3 months, while the other 9 patients were treated with givosiran 2.5 mg/kg monthly. Although all patients eventually received the 2.5 mg/kg dose, it is unknown if the 5.0 mg/kg treatment (with duration ranging from 4 months to 11 months) had any impact on study outcomes for the 7 patients.
Studies 001 and 002 both had small sample sizes, and in Study 001, only 3 patients in each of parts A and C were randomized to receive monthly doses of givosiran 2.5 mg/kg, the intended commercial dose, for a short duration. Consequently, any conclusions on HRQoL (based on the EQ-5D) and safety are limited by the small number of patients, the small number of doses, the short exposure time, and the populations described in the baseline characteristics.
External Validity
Most patients in studies 001 and 002 were female and White. The clinical experts consulted by CADTH for this review noted that the majority of patients treated in clinical practice are female and they did not expect there to be any differences among races. Although hemin was allowed for the treatment of acute attacks, prophylactic use was discontinued before the start of the studies. This may not reflect clinical practice, and the safety of the concurrent use of givosiran and prophylactic hemin is unknown. Patients in Study 001, part A were CHEs (i.e., they did not have recurring acute attacks), and the clinical experts consulted for this review would not expect to treat this group of patients with givosiran, a fact that limits the utility of the data from part A. Additionally, the inclusion criteria for Study 001, part C stated that patients must have experienced at least 2 porphyria attacks in the 6 months preceding the start of the study. The clinical experts felt that this criterion could be too restrictive, and said they would use 12 months for assessment in clinical practice.
Phase III Open-Label Extension Study (Ongoing): Study 003 OLE
Methods
Study 003 was a 6-month, DB, placebo-controlled study that began on December 7, 2017, and has been described in the preceding sections. It was followed by an ongoing OLE period (Study 003 OLE) that was expected to continue for 29 months and be completed in May 20217; however, only an the interim report (cut-off date of January 10, 2020) was available at the time of this review. Study 003 OLE was a phase III study of the long-term efficacy and safety of givosiran for the treatment of adults with AIP. It was conducted at 36 sites in 18 countries (1 site in Canada). Patients who completed the DB portion of Study 003 (N = 94) were eligible to participate in the OLE phase. This summary focuses on patients who received givosiran 2.5 mg/kg during the OLE.
Figure 16 outlines the study design for the Study 003 DB and OLE phases.
Figure 16: Design of Study 003 Double-Blind and Open-Label Extension Phases
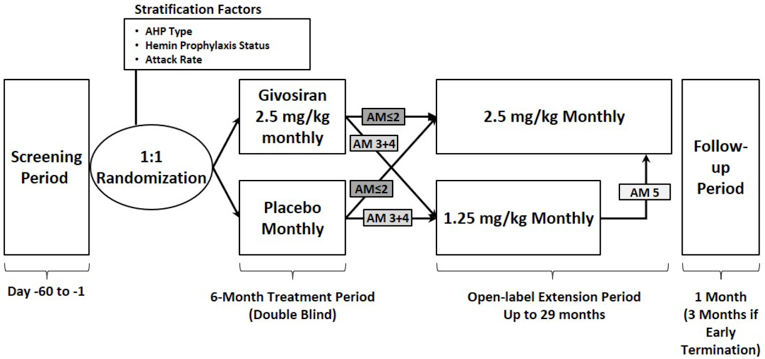
AHP = acute hepatic porphyria; AM ≤ 2 = original protocol (September 6, 2017), protocol amendment 1 (May 4, 2018), and protocol amendment 2 (July 26, 2018); AM 3 + 4 = protocol amendment 3 (September 21, 2018) and protocol amendment 4 (May 28, 2019); AM 5 = protocol amendment 5 (February 12, 2020).
Note: Patients entering the OLE period under the original protocol, protocol amendment 1, or protocol amendment 2 (AM ≤ 2) received givosiran 2.5 mg/kg monthly in the OLE period, while those entering under protocol amendments 3 or 4 (AM 3 + 4) received givosiran 1.25 mg/kg monthly in the OLE period. Upon implementation of protocol amendment 5 (after the data cut-off of January 10, 2020), all patients receiving givosiran 1.25 mg/kg monthly had their dose increased to 2.5 mg/kg monthly.
Source: Clinical Study Report for Study ALN-AS1-003.39
Populations
Patients were eligible to participate in Study 003 OLE after completing the DB phase. Briefly, they must have met the following inclusion criteria:
female or male, aged 12 years or older
documented diagnosis of AIP, HCP, VP, or ADP based on clinical features; at least 1 documented urinary or plasma PBG or ALA value greater than or equal to 4 times the ULN within the past year before or during screening, AND either documented genetic evidence of mutation in a porphyria-related gene, OR had both clinical features and diagnostic biochemical criteria consistent with AHP if genetic testing did not identify a mutation in a porphyria-related gene
active disease with at least 2 porphyria attacks in the 6 months before screening
not receiving prophylactic hemin during screening or during the study
adequate venous access.
Patients were excluded if they met any of the following exclusion criteria:
any of the following lab parameters: ALT greater than 2 times the ULN, total bilirubin greater than 1.5 times the ULN, INR greater than 1.5, eGFR less than 30 mL/min/1.73 m2
anticipating a liver transplant during the study period or other major surgery during the first 6 months of the study
history of multiple drug allergies, allergic reaction to an oligonucleotide or to N-acetylgalactosamine, intolerance to subcutaneous injection
known HIV, HCV, or HBV infection
enrolled in or recently participated in another investigational device or drug study
females who were pregnant, breastfeeding, or planning to become pregnant during the study
history of pancreatitis, serious infection, or malignancy
any condition that would make the patient unsuitable for dosing, could interfere with the study compliance, the patient’s safety, and/or the patient’s participation in the 6-month treatment period of the study.
Table 31 summarizes the baseline characteristics of patients in Study 003 OLE, focusing on patients who received givosiran 2.5 mg/kg. Patients ranged in age from 19 years to 65 years, with a mean age of 39.4 years (SD = 11.6). Most were female (89.6%), White (76.6%), and had a mean body mass of 66.39 kg (SD = 15.47 kg). The mean time since diagnosis was 10.78 years (SD = 9.98), and patients had experienced an average of 5.8 attacks (SD = 4.5) in the 6 months before randomization. The mean historical AAR was 11.6 (SD = 9.0), and 44.2% of patients had previously used hemin prophylactically. Patients had other diagnosed comorbidities, such as neuropathy (37.7%), increased transaminases (36.4%), iron overload (33.8%), hypertension (27.3%), renal failure and impairment (27.3%), and chronic kidney disease (20.8%).
Table 31: Summary of Baseline Characteristics in Study 003 OLE — All Givosiran-Treated Set
Summary | Placebo and givosiran 2.5 mg/kg (N = 29)a | Givosiran 2.5 mg/kg and givosiran 2.5 mg/kg (N = 27)b | Givosiran 2.5 mg/kg and givosiran 1.25 mg/kg (N = 20)c | All givosiran 2.5 mg/kgd (N = 77) |
|---|---|---|---|---|
Demographics | ||||
Age (years), mean (SD) | 38.3 (10.7) | 41.7 (12.3) | 38.5 (12.0) | 39.4 (11.6) |
Age (years), median (range) | 38.0 (20 to 60) | 44.0 (23 to 65) | 37.5 (19 to 58) | 38.0 (19 to 65) |
Female, n (%) | 26 (89.7) | 24 (88.9) | 18 (90.0) | 69 (89.6) |
Race, n (%) | ||||
White | 20 (69.0) | 21 (77.8) | 17 (85.0) | 59 (76.6) |
Asian | 5 (17.2) | 5 (18.5) | 3 (15.0) | 13 (16.9) |
Black | 1 (3.4) | 0 (0) | 0 (0) | 1 (1.3) |
Native Hawaiian or other Pacific Islander | 1 (3.4) | 0 (0) | 0 (0) | 1 (1.3) |
Other | 2 (6.9) | 1 (3.7) | 0 (0) | 3 (3.9) |
Body mass (kg), mean (SD) | 67.29 (15.45) | 67.01 (18.07) | 64.40 (12.37) | 66.39 (15.47) |
Disease history | ||||
Years since diagnosis, mean (SD) | 10.28 (7.77) | 12.94 (11.85) | 8.98 (10.20) | 10.78 (9.98) |
Number of attackse during the 6 months before randomization, mean (SD) | 5.4 (4.6) | 5.8 (4.3) | 6.6 (4.8) | 5.8 (4.5) |
Historical AAR,f mean (SD) | 10.9 (9.3) | 11.6 (8.6) | 13.1 (9.7) | 11.6 (9.0) |
Prior hemin prophylaxis, n (%) | 14 (48.3) | 13 (48.1) | 7 (35.0) | 34 (44.2) |
Comorbidities | ||||
Transaminases increased, n (%) | 11 (37.9) | 11 (40.7) | 5 (25.0) | 28 (36.4) |
Renal failure and impairment HLT,g n (%) | 11 (37.9) | 6 (22.2) | 3 (15.0) | 21 (27.3) |
Chronic kidney disease, n (%) | 8 (27.6) | 5 (18.5) | 3 (15.0) | 16 (20.8) |
Hypertension, n (%) | 7 (24.1) | 9 (33.3) | 5 (25.0) | 21 (27.3) |
Neuropathy, n (%) | 9 (31.0) | 12 (44.4) | 8 (40.0) | 29 (37.7) |
Iron overload, n (%) | 10 (34.5) | 10 (37.0) | 6 (30.0) | 26 (33.8) |
AAR = annualized attack rate; DB = double blind; Givo = givosiran; HLT = high-level term; OLE = open-label extension; SD = standard deviation.
aIncludes patients who received placebo in the DB period and givosiran in the OLE, and included data only post-givosiran treatment.
bIncludes patients who received givosiran in the DB and OLE periods. (Patient 101-3001, who discontinued in the DB period, was counted in the “givosiran and givosiran” and “all givosiran” groups only).
cPatients who dose-escalated due to inadequate disease control during the OLE at or after the Month 13 visit are counted in the treatment group according to the dose assigned at the beginning of the OLE period.
dIncludes all patients who received givosiran 2.5 mg/kg in either the DB and/or OLE periods.
eIncludes attacks requiring hospitalization, health care facility visit, or hemin use at home.
fThe historical AAR was calculated by annualizing the number of attacks requiring hospitalization, health care facility visit, or hemin use at home during the 6 months before randomization.
gIncludes acute kidney injury, chronic kidney disease, renal failure, renal impairment, and renal injury.
Source: Clinical Study Report of ALN-AS1-003.39
Interventions
During the OLE period, patients received givosiran 2.5 mg/kg monthly if they were enrolled under protocol amendment 1 or 2 and received givosiran 1.25 mg/kg monthly if they were enrolled under protocol amendment 3. The 1.25 mg/kg dose was introduced under protocol amendment 2 in response to patients with elevated transaminase levels. At month 13, patients who had been given givosiran 1.25 mg/kg monthly and were experiencing inadequate disease control could have their dose increased to 2.5 mg/kg monthly if they demonstrated tolerability to the study drug and if they met the ALA and clinical criteria. With protocol amendment 5 (after the cut-off date for the interim report), all patients were transitioned to receive givosiran 2.5 mg/kg monthly based on tolerability to the study drug.
Prophylactic hemin use was not permitted during the study, but hemin use to treat acute attacks was allowed.
Outcomes
The end points of the OLE phase were exploratory and included the rate of porphyria attacks (a composite outcome composed of need for hospitalization, urgent health care visit, or IV hemin administration at home) in patients with AIP and AHP; the rate of administered hemin; and urinary ALA, PBG, and ALAS1 mRNA levels. Patient-reported outcomes were also captured. These included daily worst pain, nausea, and fatigue scores over 12 months; PCS score on version 2 of the SF-12 v2; EQ-5D-5L index score; PGIC score; and PPEQ score. Harms data for AEs, SAEs, WDAEs, and deaths were also summarized.
Statistical Analysis
Both patients and investigators remained blinded to treatment assignments beyond the 6-month DB period until the database lock for the month 12 interim analysis. For the OLE period, categorical variables were summarized using counts and percentages while continuous variables for efficacy outcomes were summarized using descriptive statistics. Data were summarized as “during givosiran treatment” for patients who received at least 1 dose of givosiran after their first dose. The AAR and annualized days of hemin use were calculated for patients who had at least 85 days of follow-up during the OLE phase to avoid unstable estimations. No imputation was performed for missing data if patients provided attack data for the entire 6-month DB period. No inferential statistical analyses were planned for safety assessments in the study.
Analysis sets were presented as follows:
givosiran and givosiran: patients who received givosiran during the DB and OLE phases, including patients who discontinued treatment during the DB period
placebo and givosiran: patients who received placebo during the DB and givosiran during the OLE phases
all givosiran: patients who received at least 1 dose of givosiran during either DB or OLE phases.
Analysis sets were further separated by doses received during the DB and OLE phases (e.g., givosiran 2.5 mg/kg and givosiran 1.25 mg/kg).
Patient Disposition
Of the 94 patients who completed the 6-month DB phase, 93 (98.9%) entered the OLE phase. In total, 5 patients (5.3%) withdrew from the study due to AE (n = 1), pregnancy (n = 1), or patient decision (n = 3).
The “all givosiran” set was the main analysis set for long-term efficacy and safety and included all randomized patients who received at least 1 dose of givosiran during the DB or OLE phases. The FAS included all randomized patients who received at least 1 dose of the study drug and were grouped according to their randomly assigned treatment group. The safety analysis set was made up of all patients who received at least 1 dose of the study drug and were grouped according to the treatment actually received. All 3 analysis sets included 94 patients.
Table 32: Patient Disposition in Study 003 Open-Label Extension
Disposition | Placebo and givosiran 2.5 mg/kg (N = 29) | Givosiran 2.5 mg/kg and givosiran 2.5 mg/kg (N = 27) | Givosiran 2.5 mg/kg and givosiran 1.25 mg/kg (N = 20) | All givosiran 2.5 mg/kga (N = 77) | All givosiran (N = 94) |
|---|---|---|---|---|---|
Treated, N (%) | 29 (100.0) | 27 (100.0) | 20 (100.0) | 77 (100.0) | 94 (100) |
Entered OLE period, n (%) | 29 (100.0) | 27 (100.0) | 20 (100.0) | 76 (98.7) | 93 (98.9) |
Withdrew from study, n (%) | 3 (10.3) | 1 (3.7) | 0 (0) | 5 (6.5) | 5 (5.3) |
AE | 0 (0) | 0 (0) | 0 (0) | 1 (1.3) | 1 (1.1) |
Pregnancy | 1 (3.4) | 0 (0) | 0 (0) | 1 (1.3) | 1 (1.1) |
Patient decision | 2 (6.9) | 1 (3.7) | 0 (0) | 3 (3.9) | 3 (3.2) |
All givosiran treated, n (%) | 29 (100.0) | 27 (100.0) | 20 (100.0) | 77 (100.0) | 94 (100) |
Full analysis set, n (%) | 29 (100.0) | 27 (100.0) | 20 (100.0) | 77 (100.0) | 94 (100) |
Safety analysis set, n (%) | 29 (100.0) | 27 (100.0) | 20 (100.0) | 77 (100.0) | 94 (100) |
AE = adverse event; DB = double blind; OLE = open-label extension.
Source: Clinical Study Report for Study ALN-AS1-003.39
Exposure to Study Treatments
Patients in the all givosiran 2.5 mg/kg group (n = 77) of the OLE had a mean exposure of 16.77 months (SD = 4.69 months) and a median of 17.38 months (range = 1.8 months to 25.1 months). During the OLE period, 6 patients missed 1 dose of givosiran 2.5 mg/kg and 4 missed 2 doses.
No patients who started on givosiran 2.5 mg/kg during the OLE phase had a dose reduction. Eleven patients who initially received givosiran 1.25 mg/kg had a dose increase to 2.5 mg/kg during month 13 to month 15 due to inadequate disease control, as per the protocol dose modification rules.
Efficacy
Table 33 summarizes the efficacy outcomes for patients who received givosiran 2.5 mg/kg during the DB and OLE phases. The mean number of attacks during givosiran treatment was |||||| with a mean patient-level AAR of ||||||||||. Mean AARs for attacks requiring hospitalization, urgent health care visit, treatment with IV hemin at home, and treatment without IV hemin at home were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, respectively. The average number of days of hemin use was |||||| days. Creatinine-normalized urinary ALA decreased by a mean of |||||||||||||||||| mmol/mol and |||||| |||||||| mmol/mol at month 12 and month 18, respectively, from Study 003 baseline measures. Creatinine-normalized urinary PBG values decreased by an average |||||||||||||||||| mmol/mol and |||||||||||||||||| mmol/mol for the same time points from baseline.
Figure 17, Figure 18, and Figure 19 compare the “placebo and givosiran” and the “givosiran and givosiran” groups for key efficacy outcomes during the DB and OLE phases. Figure 17 shows the average number of attacks per patient per month decreasing in the first couple of months after starting givosiran for either group; attack numbers remained low over time. Median urinary ALA and PBG levels show a similar decreasing and levelling trend in Figure 18 and Figure 19, respectively.
Table 33: Summary of Efficacy Outcomes in Study 003 OLE — All Givosiran-Treated Set
Outcomes | Givosiran 2.5 mg/kg (N = 77) |
|---|---|
Porphyria attack composite end pointa (n = 76)b | |
Total number of attacks during givosiran treatment, mean (SEM) | 3.4 (0.7) |
Patient-level AAR, mean (SEM) | 2.42 (0.43) |
AAR analysis of porphyria attack components (n = 76)b | |
All attacks,c patient-level AAR, mean (SEM) | 3.02 (0.46) |
Attacks requiring hospitalization, mean (SEM) | 1.12 (0.31) |
Attacks requiring urgent health care visit, mean (SEM) | 1.25 (0.33) |
Attacks requiring treatment with IV hemin at home, mean (SEM) | 0.06 (0.04) |
Attacks requiring treatment without IV hemin at home, mean (SEM) | 0.60 (0.14) |
Hemin use (n = 76)b | |
Total days, mean (SEM) | 6.8 (1.7) |
Annualized days of hemin use, mean (SEM) | 4.77 (1.14) |
Creatinine-normalized urinary ALA levels (mmol/mol), mean (SD) | |
Baseline (n = 77) | 18.535 (14.220) |
Month 6, change from baseline (n = 71) | –14.646 (14.778) |
Month 12, change from baseline (n = 68) | –14.455 (14.107) |
Month 18, change from baseline (n = 48) | –16.924 (15.154) |
Creatinine-normalized urinary PBG levels (mmol/mol), mean (SD) | |
Baseline (n = 77) | 46.746 (29.371) |
Month 6, change from baseline (n = 71) | –35.047 (31.015) |
Month 12, change from baseline (n = 68) | –35.923 (28.470) |
Month 18, change from baseline (n = 48) | –39.172 (31.048) |
AAR = annualized attack rate; ALA = aminolevulinic acid; OLE = open-label extension; PBG = porphobilinogen; SD = standard deviation; SEM = standard error of mean.
Notes: Givosiran 2.5 mg/kg includes patients who received givosiran 2.5 mg/kg as the first dose in either the double-blind or OLE study.
aIncludes attacks that require hospitalization, urgent health care visit, and treatment with IV hemin at home.
bOne patient whose follow-up duration after taking givosiran was < 85 days was excluded from the analysis.
cIncludes attacks that require hospitalization, urgent health care visit, treatment with IV hemin at home, and treatment without IV hemin at home.
Source: Clinical Study Report for Study ALN-AS1-003.39
Figure 17: Average Number of Attacks per Patient per Month; Porphyria Attack Composite End Point — Full Analysis Set
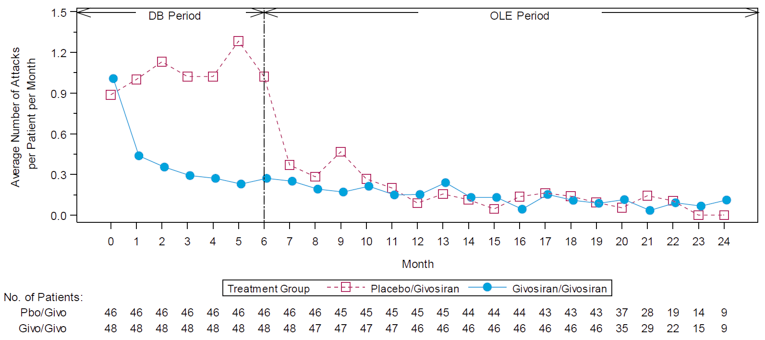
DB = double blind; Givo = givosiran; OLE = open-label extension; Pbo = placebo.
Note: Month 0 represents 6 months before randomization, and the estimate is calculated as total number of attacks divided by the total duration in months. Month 1 and beyond are categorized relative to the first dose of the study drug, and the estimate is calculated as the total number of attacks divided by the total number of patients that remaining in the study at that time point. One month equals 28 days for the purposes of these calculations. A few attacks occurring after the first dose of the OLE period were counted into month 6 if patients entered the OLE period earlier than study day 168. A few attacks occurring before the first dose of the OLE period were counted into month 7 if patients entered the OLE period later than study day 168.
Source: Clinical Study Report for Study ALN-AS1-003.39
Figure 18: Median Urinary ALA Levels During Givosiran Treatment — Full Analysis Set
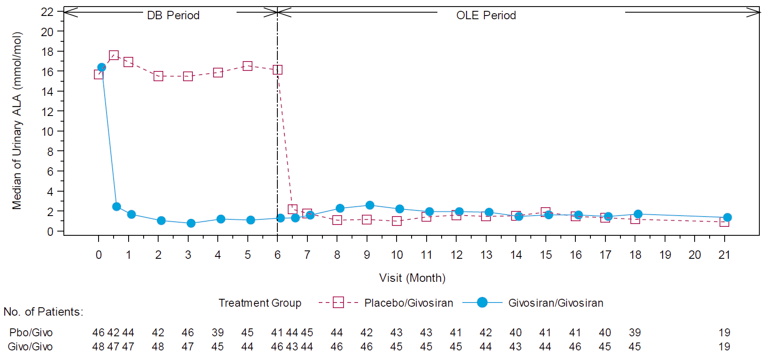
ALA = aminolevulinic acid; DB = double blind; Givo = givosiran; OLE = open-label extension; Pbo = placebo.
Source: Clinical Study Report for Study ALN-AS1-003.39
Figure 19: Median Urinary PBG Levels During Givosiran Treatment — Full Analysis Set
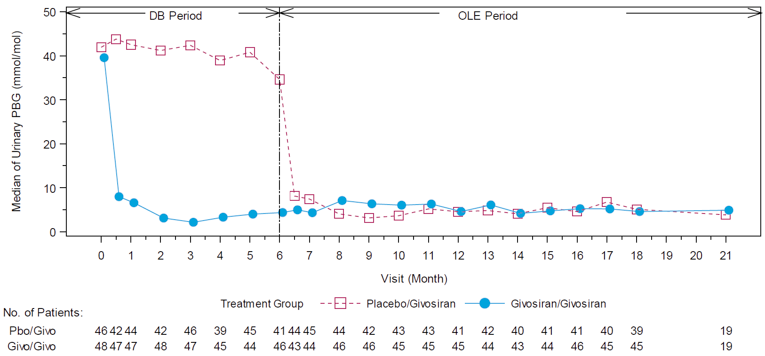
DB = double blind; Givo = givosiran; OLE = open-label extension; PBG = porphobilinogen; Pbo = placebo.
Source: Clinical Study Report of ALN-AS1-003.39
Table 34 summarizes HRQoL outcomes during the OLE phase. The PCS and MCS scores of the SF-12 both showed an increase from baseline to 12 months and 18 months. For the PCS, the mean change from baseline was |||| |||||| points at month 12 and |||||||||||||| points at month 18, while the MCS scores showed an average change from baseline of |||||||||||||||| points and |||||||||||||||| points at the same time points. Mean daily worst pain and fatigue scores were lower by |||||||||| points and |||||||||| points, respectively, during givosiran treatment compared to baseline, while the daily worst nausea score was |||||||||| points higher compared to baseline. EQ-5D-5L index and VAS scores increased from baseline: the former score changed by a mean of |||||||||||||||||| points at month 12 and |||||||||||||||||| points at month 18, while the VAS score changed by |||||||| and |||||||||| for the same time points. At month 6, 90.5% of patients (57 of 63) noted an improvement on the PGIC (includes categories of minimally improved, much improved, and very much improved), while 97.8% of patients (45 of 46) felt improved at month 12. Figure 20 shows the percentage of patients who indicated “much better” ability on the PPEQ. There was an increase in the proportion of placebo and givosiran patients who responded “much better” or “always” from month 6 to month 12. The number of patients who responded “much worse” or “never” remained low (0 patients to 2 patients) at each assessment.
Table 34: Summary of HRQoL, Symptom, and Utility Outcomes in Study 003 OLE — All Givosiran-Treated Set
Outcome | Givosiran 2.5 mg/kg (N = 77) |
|---|---|
PCS of SF-12, mean (SD) | |
Baseline (n = 77) | vvvvvv vvvvvvv |
Month 6, change from baseline (n = 73) | vvvvv vvvvvvv |
Month 12, change from baseline (n = 71) | vvvvv vvvvvvv |
Month 18, change from baseline (n = 47) | vvvvv vvvvvvv |
MCS of SF-12, mean (SD) | |
Baseline (n = 77) | vvvvvv vvvvvvv |
Month 6, change from baseline (n = 73) | vvvvv vvvvvvvv |
Month 12, change from baseline (n = 71) | vvvvv vvvvvvvv |
Month 18, change from baseline (n = 47) | vvvvv vvvvvvvv |
Daily worst pain score, mean (SD) | |
Baseline | vvvv vvvvvv |
During givosiran treatment, change from baseline | vvvvv vvvvvv |
Daily worst fatigue score, mean (SD) | |
Baseline | vvvv vvvvvv |
During givosiran treatment, change from baseline | vvvvv vvvvvv |
Daily worst nausea score, mean (SD) | |
Baseline | vvvv vvvvvv |
During givosiran treatment, change from baseline | vvvv vvvvvv |
EQ-5D-5L index score, mean (SD) | |
Baseline (n = 76) | vvvvvv vvvvvvvv |
Month 6, change from baseline (n = 73) | vvvvvv vvvvvvvv |
Month 12, change from baseline (n = 69) | vvvvvv vvvvvvvv |
Month 18, change from baseline (n = 47) | vvvvvv vvvvvvvv |
EQ VAS score, mean (SD) | |
Baseline (n = 77) | vvvv vvvvvv |
Month 6, change from baseline (n = 74) | vvv vvvvvv |
Month 12, change from baseline (n = 70) | vvv vvvvvv |
Month 18, change from baseline (n = 47) | vvvv vvvvvv |
PGIC score, n/N (%) | |
Month 6, improveda | vvvvv vvvvvv |
Month 12, improveda | vvvvv vvvvvv |
EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; EQ VAS = Euro Quality of Life Visual Analogue Scale; HRQoL = health-related quality of life; MCS = Mental Component Summary; OLE = open-label extension; PCS = Physical Component Summary; PGIC = Patient Global Impression of Change; SD = standard deviation; SF-12 = 12-item Short Form Health Survey, version 2.
Note: Givosiran 2.5 mg/kg includes patients who received givosiran 2.5 mg/kg as the first dose in either the double-blind or OLE study.
aImproved includes minimally improved, much improved, and very much improved.
Source: Clinical Study Report for Study ALN-AS1-003.39
Figure 20: Redacted by Sponsor

This figure has been redacted at the request of the sponsor.
Source: Clinical Study Report for Study ALN-AS1-003.39
Harms
Harms outcomes for Study 003 OLE are summarized in Table 35. Nearly all patients (94.8%) experienced at least 1 AE, with 32.5% reporting nausea, 27.3% reporting injection-site reaction, 22.1% reporting fatigue, 22.1% reporting nasopharyngitis, and 19.5% reporting headache. SAEs occurred in 24.7% of patients, with chronic kidney disease, device breakage, and urinary tract infection reported by 2.6% of patients for each. There was 1 WDAE. No deaths were reported.
Table 35: Summary of Harms Outcomes in Study 003 Open-Label Extension
Harms | Givosiran 2.5 mg/kg (N = 77) |
|---|---|
Patients with ≥ 1 AE, n (%) | 73 (94.8) |
AE,a n (%) | |
Nausea | 25 (32.5) |
Injection-site reaction | 21 (27.3) |
Fatigue | 17 (22.1) |
Nasopharyngitis | 17 (22.1) |
Headache | 15 (19.5) |
Urinary tract infection | 13 (16.9) |
Abdominal pain | 13 (16.9) |
Upper respiratory tract infection | 12 (15.6) |
Vomiting | 12 (15.6) |
Diarrhea | 10 (13.0) |
Lipase increase | 8 (10.4) |
Patients with ≥ 1 SAE, n (%) | 19 (24.7) |
SAE,b n (%) | |
Chronic kidney disease | 2 (2.6) |
Device breakage | 2 (2.6) |
Urinary tract infection | 2 (2.6) |
WDAE, n (%) | 1 (1.3) |
Death, n (%) | 0 (0) |
Notable harms, n (%) | |
Injection-site reactions | 21 (27.3) |
Transaminases increase | 2 (2.6) |
ALT increase | 7 (9.1) |
AST increase | 6 (7.8) |
Chronic kidney disease | 6 (7.8) |
Iron overload | 3 (3.9) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Note: “All givosiran 2.5 mg/kg” includes patients receiving givosiran 2.5 mg/kg as the first dose once monthly in either the DB or OLE studies.
aFrequency ≥ 10% of all patients.
bFrequency > 1 patient.
Source: Clinical Study Report for Study ALN-AS1-003.39
Critical Appraisal
Internal Validity
The OLE period of Study 003 had a number of design limitations. For example, it lacked a randomized comparison group to provide context and control for potential confounders during the OLE period. Furthermore, the open-label design and unblinding 12 months after the start of the study (or 6 months into the OLE phase) may have influenced some patients’ and clinicians’ perceptions of improvement, and this could be reflected in the patient-reported outcomes. Amendment 3 was introduced in September 2018 so that patients with elevated transaminase levels could receive a lower dose of givosiran (i.e., 1.25 mg/kg). Unblinding had taken place in the following months (12 months from the start of the study), near the time when patients with inadequate control on 1.25 mg/kg were able to increase their dose to 2.5 mg/kg. It is uncertain if the implementation of the lower dose, the unblinding, and the dose increase due to inadequate control had an impact on the study outcomes. Patients kept electronic diary records (reporting on pain, fatigue, nausea, and analgesic use) daily for the first 12 months of the study, and then only when potential attacks occurred from month 13 onwards. It is unknown if the change in reporting frequency would affect the accuracy of patient-reported outcomes, although these were exploratory in the OLE phase. No imputation was performed for missing data if patients provided attack data for the entire 6-month DB period. Attacks were considered for efficacy assessment and not counted as AEs. Additional issues have been described earlier in the appraisal of the DB portion of Study 003.
External Validity
The study inclusion criteria specified that patients must have had at least 2 porphyria attacks in the 6 months leading up to the Study 003. The study population was considered to be enriched for attack frequency because patients were required to have experienced at least 2 attacks in the 6 months before entering the study. This may limit the generalizability of the study to patients who have less frequent attacks. The clinical experts consulted for this review suggested that 12 months was a more reasonable time frame for clinical practice. Furthermore, it is possible that patients who could benefit from givosiran were excluded based on the requirement for the number of attacks. Although the minimum age for enrolment was 12 years, no patients were younger than 19 years or older than 65 years; thus, there are no data from this study for the use of givosiran in adolescent or geriatric patients. Overall, the clinical experts consulted by CADTH for this review noted that the study population appeared to represent patients who would be treated in practice. Hemin was allowed for the treatment of acute attacks, but its prophylactic use was discontinued before the start of the study, which may not reflect real-world use. Additionally, the safety of concurrent givosiran and prophylactic hemin use is unknown. It is also worth noting that there was a dose change for those who initially enrolled under protocol amendment 3 and received givosiran 1.25 mg/kg due to elevated transaminase levels. At month 13, patients who had inadequate disease control were able to increase their dose to 2.5 mg/kg from 1.25 mg/kg, and with protocol amendment 5, all patients were to receive givosiran 2.5 mg/kg (the intended commercial dose). The product monograph indicates that patients who have a dose interruption due to severe or clinically significant transaminase levels and who then improve may resume givosiran at 1.25 mg/kg or 2.5 mg/kg monthly, although it also notes that there are limited safety and efficacy data around dose changes following transaminase elevations.
Expanded Access Program (Ongoing): Study ALN-AS1-005
Study ALN-AS1-005 is an international program that will provide expanded access to givosiran to patients 12 years and older with AHP.8 It is ongoing; no additional information is available.
Discussion
Summary of Available Evidence
One multi-centre, placebo-controlled, DB, phase III study, Study 003, was included in the CADTH systematic review. Study 003 was designed to evaluate the efficacy and safety of givosiran administered once monthly in patients with AHP. Included patients had to be at least 12 years old with a documented diagnosis of AIP, HCP, VP, or ADP, have experienced at least 2 composite porphyria attacks within 6 months before screening, and be willing to abstain from prophylactic use of hemin during the trial. The primary objective was to evaluate the effect of subcutaneous givosiran compared to placebo in terms of the rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home over 6 months in patients with AIP. The annualized rate of porphyria attacks in patients with AHP and the following assessments in patients with AIP were included as secondary outcomes: urinary ALA and PBG levels, hemin use, daily worst scores for symptoms of pain, fatigue, and nausea, and HRQoL through the SF-12. Opioid use, the PPEQ, and ability to work or attend school — as well as the secondary end points analyzed in patients with AHP — were included as exploratory outcomes.
A total of 94 patients were randomized in Study 003, 89 (95%) of whom had AIP. Patients with AIP were between the ages of 19 years and 65 years (with a mean age of 37.3 years to 40.7 years); 89% to 91% were female, and 35% to 40% resided in North America. Between 40% and 44% of patients had prior experience with the use of prophylactic hemin; based on the composite definition of porphyria attacks, the median historical AAR was 8 attacks (range = 4 to 34) and 8 attacks (range = 0 to 46) in the givosiran and placebo treatment groups, respectively. When not having a porphyria attack, 48% to 56% of patients reported having chronic symptoms, and 28% to 30% of patients reported chronic opioid use. Baseline characteristics in patients with AHP were similar to those reported for patients with AIP.
Four other relevant studies were summarized for this review: phase I of Study 001, phase I/II of Study 002, the OLE of Study 003, and EAP Study 005. No indirect treatment comparisons were identified for this review.
Interpretation of Results
Efficacy
Current treatment for AHP is limited to preventing attacks through avoidance of triggers, hormone therapy, and prophylactic hemin use (an indication not approved by Health Canada). Preventive measures are often successful in limiting the frequency of attacks in the majority of symptomatic patients with AHP, according to the clinical experts consulted for this review; however, there is a subset of patients living with AHP who experience severe, recurrent attacks that are not avoided using these measures. Recurrent attacks are associated with poor health outcomes due to porphyria-related complications, and these have a significant impact on patients’ quality of life. Therefore, the prevention of porphyria attacks or reduction in the frequency of acute attacks was described as a critical outcome by patients and clinicians.
At baseline, the median AAR for patients with AIP was 8 attacks (range = 0 to 46). After 6 months of treatment, the median AAR was 1 attack (IQR = 0 to 6) for patients treated with givosiran and 11 attacks (IQR = 2 to 26) for patients receiving placebo. The primary end point of the pivotal trial was the annualized rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP over the 6-month DB period, which corresponded to a mean 74% reduction in the rate of porphyria attacks (rate ratio = 0.26; 95% CI, 0.16 to 0.41; P < 0.001) for patients in the givosiran treatment group relative to patients receiving placebo. Similarly, a mean 73% reduction in the rate of porphyria attacks (rate ratio = 0.27; 95% CI, 0.17 to 0.43; P < 0.001) was reported for all patients with AHP, which included patients with both AIP and non-AIP types of AHP. The clinical experts consulted for this review were unable to quantify a clinically meaningful reduction in acute attacks, given that the clinical presentation and severity of attack varies between patients; however, there was consensus regarding the fact that in general, a reduction in the number of attacks was meaningful and clinically relevant. Thus, givosiran demonstrates a benefit in terms of a reduction in the frequency of attacks.
However, there are a few limitations to consider. As previously described, the study was enriched for attack frequency to ensure the ability to measure a difference in treatment effect on the primary composite porphyria attack end point. This was done by selecting patients with a higher historical AAR before randomization. This is supported by the results of the EXPLORE natural history study that reported a median AAR of 2.0 (range = 0.0 to 37.0)35 for patients with AHP as well as the experience of the clinical experts consulted for this review. This impairs the generalizability of the primary end point and the reduction in attack rate observed in the individual treatment arms to all patients with AHP, although it is relevant to a subset patients who experience frequent attacks. Additionally, the trial is placebo-controlled, and prophylactic hemin was not permitted, but it is used in practice to prevent acute attacks in patients with recurrent attacks and frequent hospitalizations. Therefore, the treatment effect observed in the trial was potentially greater than what would be expected for comparison to best supportive care in clinical practice.
The primary end point was also based on a composite end point where the components correspond to varying degrees of treatment effect, but the results for the components were reported descriptively. The clinical experts consulted for this review indicated that porphyria attacks that result in hospitalization are potentially more severe than other attacks, but this also depends on factors such as access to alternative care settings that are equipped to handle patients with AHP. Overall, the components of the composite outcome for AAR each represent important measures of attacks for both patients and clinicians, particularly attacks leading to hospitalizations, given that a reduction in these is a major treatment goal for patients with AHP. However, the results of each of the components are subject to bias and were reported descriptively, which limits the ability to draw concrete conclusions from them. Despite this, the frequency of attacks reported for each of the components was consistent with the direction observed for the composite outcome, and none of the components appeared to be driving the effect, thereby supporting the primary analysis.
Study 003 employed sensitivity analyses for the primary end point to: account for undercounting and overcounting attacks, assess the AAR for all investigator-included attacks (including those treated at home without IV hemin use), and assess the AAR including all potential attacks. Additionally, the primary analysis was conducted using an alternative statistical model that treated attacks as recurrent events. It was also conducted using the per-protocol set. All of the sensitivity analyses were consistent with and supportive of the primary analysis. A number of pre-specified subgroup analyses were employed as well, with historical attack rates (high versus low) meeting the CADTH systematic review protocol. The treatment effect observed in patients with a high or low historical AAR was consistent with the primary analysis. Of note, the criteria used to define these subgroups were considered arbitrary by the clinical experts consulted for this review; therefore, there is uncertainty with how meaningful this subgroup analysis was.
Study 003 included urinary levels of ALA and PBG at month 3 and/or month 6, the annualized rate of administered hemin doses over the 6-month treatment period, the assessment of the change of daily worst pain over the 6-month treatment period, and opioid use, and the results supported a reduced frequency of attacks. For context, measures of ALA and PBG are highly variable in patients with AHP, but the clinical experts indicated that porphyrin levels are substantially higher (up to 4 times greater than normal levels) during an acute attack. At month 3 and month 6, urinary ALA increased in both the givosiran and placebo treatment groups; however, the difference between treatment groups favoured givosiran (P < 0.001). Over the 6-month treatment period, 54.3% of patients in the givosiran treatment group and 23.3% of patients in the placebo treatment group did not require hemin use. Further, this corresponded to a 77% rate reduction in days of hemin use for patients receiving givosiran compared to those receiving placebo, based on a rate ratio of 0.23 (95% CI, 0.11 to 0.45; P < 0.001). Subgroup analyses of urinary ALA levels were conducted by high versus low historical AAR. The change in urinary ALA levels from baseline measured at month 3 and month 6 in patients with AIP was similar for those in the high AAR subgroup compared to the overall population. A smaller change from baseline was reported for those with a low AAR at baseline compared to the overall population.
According to patient groups’ input, a reduction in symptoms, particularly pain, is of significant importance to patients. Patients enrolled in the trial evaluated pain by rating the worst level of pain experienced each day using an NRS ranging from “no pain” (0) to pain that was “as bad as you can imagine” (10). This standalone question was derived from the 11-item BPI-SF, which is a widely used, well-validated assessment of pain in clinical trials28,29,40; however, no evidence was identified that assessed the overall BPI-SF for validity, reliability, or responsiveness in patients with porphyria. Pain was reported as a weekly mean score for daily worst pain. An MID was not identified for the assessment of patients with porphyria, but a 2-point change has been suggested as a reasonable estimate of the MID for worst pain among breast cancer patients with metastatic disease.30 Using this 2-point MID for reference, the average change from baseline in the weekly mean score was not clinically meaningful in either treatment group. Given that AHP is often characterized by painful acute attacks, pain was also assessed using the AUC of change from baseline in weekly mean score to account for the expected variations in daily pain throughout the trial. The pre-specified analysis of daily worst pain was analyzed using an ANCOVA model, which did not demonstrate a statistically significant difference between givosiran and placebo (LS mean difference = –12.7; 95% CI, –25.5 to 0.2; P = 0.053). The assessment of pain was reanalyzed using non-parametric methods following the demonstration of a significant deviation from the normal. The non-parametric test corresponded to a treatment-group difference in favour of givosiran (median of treatment difference = –10.1; 95% CI, –22.8 to 0.9; P = 0.0455); however, this analysis was conducted post hoc and at risk of type I error, which introduces uncertainty into the results. Moreover, the treatment-group difference reported in all patients with AHP was –9.4 (95% CI, –21.0 to 1.2; P = 0.0613). Based on the available evidence, givosiran was not associated with an improvement in pain compared to placebo.
Analgesic use of opioids at home was patient-reported and summarized descriptively in Study 003. Similar use was reported in patients with AIP and all patients with AHP. In general, patients in the givosiran group reported less use of opioids during the trial (based on the proportion of days with opioid use) compared to patients in the placebo group; however, the ability to interpret this data is limited without a comparison to opioid use at baseline. Over the 6-month DB treatment period, 67% and 88% of patients receiving givosiran and placebo, respectively, reported use of opioids. A reduction in opioid use was also noted as an outcome that was important to patients; however, without further assessments, it can only be concluded that opioid use was high in both treatment groups throughout the trial.
Fatigue and nausea are symptoms of porphyria that were also evaluated in Study 003 using similar methods to those used to evaluate the daily worst pain score. The severity of fatigue was assessed using a single question from the BFI-SF, analogous to the item of the BPI-SF used for daily worst pain score. The single item was validated using the Chinese and Taiwanese versions of the BFI-SF, and an MID for the worst fatigue item was estimated to be 1.5 using the 0.5 SD method33; however, no MID was identified in populations with AHP. Nausea was assessed using a 10-point NRS. Due to the failure of the statistical test for the higher-ranked daily worst pain outcome, P values for nausea and fatigue outcomes were reported as nominal. Briefly, daily worst fatigue scores at month 6 had decreased from baseline in both treatment groups, but did not meet the MID threshold of a change by 1.5 units. The treatment-group difference in the LS mean for AUC of change from baseline in daily worst fatigue score was –6.9 (95% CI, –19.8 to 6.0; P = 0.2876). The daily worst nausea score at month 6 increased in the givosiran group and decreased in the placebo group, with a treatment-group difference in the LS mean for AUC of change from baseline of 5.5 (95% CI, –4.0 to 15.0; P = 0.2532). In summary, givosiran does not offer a benefit in terms of fatigue or nausea compared to placebo.
Living with AHP was reported by patients and clinicians as having a substantial impact on HRQoL; an improvement in HRQoL was an important outcome to patients. Study 003 included 3 HRQoL outcomes: the SF-12, EQ-5D-5L, and PGIC, evaluated as a change from baseline at month 6. Each of these outcomes is well validated and widely used in clinical trials; however they are generic HRQoL measures that have not been evaluated in patients with porphyria. Also, an MID specific to porphyria was not identified for these outcomes. An MID of 3.29 on the SF-12 PCS was reported for patients with low back pain,21 but whether this is applicable to patients with AHP is unknown. The HRQoL outcomes were not controlled for multiplicity, with the exception of the PCS for SF-12; however, due to a failure to reject the null for an analysis conducted earlier in the testing hierarchy, the reported P value must be interpreted nominally. The treatment-group difference reported for the PCS of the SF-12 was 3.94 (95% CI, 0.59 to 7.29; P = 0.0216). As noted in the FDA review, “the PCS score included concepts that may not be content relevant for the target population (general health, moderate activities, climbing stairs) based on cited qualitative data in the Patient Symptom and Experience report.”41 This is reflected in the domain scores of the PCS, in which the direction of the treatment-group difference for the BP and RP domains appear to drive the overall PCS. The MCS was reported descriptively and appears to be influenced by the SF domain. The CIs for the difference in the mean change of the rest of the domains of the SF-12 cross 0. The within-group changes in HRQoL, as measured by the EQ-5D-5L index score, were not clinically meaningful using the MID estimated for the general population. Lastly, the PGIC was reported descriptively and was an exploratory outcome. The majority of patients in the givosiran group (89%) reported an improvement in their overall status after 6 months of treatment; however, this outcome was related to overall health and was not specific to AHP. Overall, the evidence was not supportive of an added benefit in HRQoL from treatment with givosiran compared to placebo.
Other efficacy outcomes included in Study 003 that were also important to patients include the PPEQ, which evaluated activities of daily living and treatment-related questions, and days of missed work or school. Both were exploratory outcomes and reported descriptively. The PPEQ is an outcome that was created for patients living with porphyria, but it has not been externally validated, and an MID was not identified. Overall, the percentages of patients responding “much better” to questions about performing actions or tasks, “much better” to questions about treatment satisfaction, and “always” to the question about treatment helping them to live a normal life were higher in the givosiran group than in the placebo group for each question. A conclusion regarding missed work or school was limited by missing data for more than 50% of patients included in the trial and by lack of statistical testing.
An important consideration for the interpretation of the evidence that has been discussed for givosiran is that 95% of the patient population in the pivotal trial, Study 003, are patients with AIP. Only 5 non-AIP patients were included in the study. However, based on feedback from the clinical experts and on the pathophysiology of all types of AHP, the results in patients with AIP are generalizable to patients with non-AIP types of AHP. This was supported by the FDA review, the European Medicines Agency review, and the Health Canada Review of the evidence for givosiran.19,42,43
Four other relevant studies were summarized for this review: Study 001, Study 002, the OLE of Study 003, and Study 005. Study 001 was a 3-part, multi-centre, placebo-controlled, phase I study of the safety and tolerability of subcutaneous givosiran for the treatment of adults with AIP. Parts A, B (n = 23), and C (n = 17) were single-ascending dose, multiple-ascending dose, and multidose in design, respectively. Study 002 was an ongoing (at the time of this review), multi-centre, open-label, phase I/II study of the long-term safety and tolerability of subcutaneous givosiran for treatment of adults with AIP who completed Study 001, part C (N = 16). Study 001, part C and Study 002 provided evidence for porphyria attack outcomes, hemin use, and HRQoL through the EQ-5D-5L in patients who received givosiran 2.5 mg/kg once monthly that was consistent with (and therefore supportive of) the evidence summarized for the pivotal trial.
One of the major limitations of Study 003 was the 6-month duration of treatment. Considering that treatment with givosiran is expected to continue long-term until there is a need for discontinuation, 6 months is an insufficient duration of time to assess long-term efficacy and safety outcomes. Study 003 OLE (N = 77) is a phase III extension study of the long-term efficacy and safety of givosiran for the treatment of adults with AIP that helps to fill the gap in the knowledge of long-term outcomes. The OLE is expected to provide data for treatment with givosiran up to 29 months, but information up to only 18 months was available for this review. In short, the OLE provided evidence for the maintenance of treatment effect for an additional 12 months following the DB treatment period (18 months on treatment in total) in terms of frequency of acute attacks and urinary ALA and PBG levels, as well as evidence of patient-reported outcomes of symptoms, HRQoL, and the PPEQ that were consistent with the DB treatment period. Further, patients who switched from placebo in the DB treatment period to givosiran in the OLE showed similar efficacy results over the 18 months to patients who stayed on givosiran throughout the study.
Lastly, Study 005 is an international program that will provide expanded access to givosiran to patients 12 years and older with AHP.8 It is ongoing, and no additional information is available.
Harms
In Study 003, 85% of patients with AIP experienced at least 1 AE, with nausea, injection-site reaction, chronic kidney disease, fatigue, increase in ALT, and decrease in glomerular filtration rate more commonly reported among patients who received givosiran. SAEs were reported more frequently among patients in the givosiran group (17%) than among patients in the placebo group (9%). Specific SAEs were infrequent, with the only SAEs reported by more than 1 person being chronic kidney disease (2 patients in the givosiran group, 0 receiving placebo) and device-related infection (2 patients in the placebo group, 1 receiving givosiran). The proportion of patients reporting AEs and SAEs among patients with all types of AHP were similar to those reported in the AIP population. A single patient randomized to receive givosiran withdrew from treatment due to AEs. The patient |||||||||||| discontinued treatment because of ALT elevation. No deaths were reported during the 6-month DB period of Study 003.
In the 18 months reported on for the OLE of Study 003, nearly all patients (94.8%) experienced at least 1 AE. Consistent with the 6-month DB treatment period, the most commonly reported AEs were nausea (32.5%), injection-site reaction (27.3%), and fatigue (22.1%), in addition to nasopharyngitis (22.1%) and headache (19.5%). SAEs occurred in 24.7% of patients, with chronic kidney disease, device breakage, and urinary tract infection being reported by 2.6% (n = 2) of patients for each. There was 1 WDAE, and no deaths were reported. The patient who withdrew from treatment received placebo during the DB treatment period and givosiran 1.25 mg/kg in the OLE, and withdrew due to a drug hypersensitivity.
Motor neuropathy, hepatocellular carcinoma, injection-site reactions, transaminase elevation, and progression of renal impairment were included in the CADTH systematic review protocol as notable harms. As previously described, injection-site reactions and transaminase elevation were more common among patients receiving givosiran. Nerve compression and peripheral neuropathy were reported for motor neuropathy and were more common in the placebo treatment group. Progression of renal impairment was more common in the givosiran group, and is included as a renal warning in the Health Canada label for givosiran along with a hepatic warning for transaminase elevations.5 There were no cases of hepatocellular carcinoma reported during the 6-month treatment period, which may not have been a sufficient amount of time during which to observe this safety outcome. The frequency of notable harms was similar for patients with AHP.
Considering that the available phase III evidence for the assessment of givosiran is limited to a single placebo-controlled RCT with up to 18 months on treatment, a longer duration of follow-up on givosiran treatment is required to determine the safety of the medication in the longer term to reflect the anticipated chronic use of givosiran in clinical practice.
Conclusions
One DB, placebo-controlled, phase III RCT, Study 003, evaluated the efficacy and safety of givosiran compared to placebo. Included patients had a diagnosis of AHP and had experienced at least 2 porphyria attacks in the 6 months before screening. Over the 6-month treatment period, a 74% reduction in the rate of acute porphyria attacks was demonstrated with givosiran compared to placebo in patients with AIP based on a rate ratio of 0.26 (95% CI, 0.16 to 0.41; P < 0.001). Similar results were reported in all patients with AHP. The primary outcome of porphyria attacks was a composite outcome that included attacks requiring hospitalization, an urgent health care visit, or IV hemin administration at home. This outcome did not have a defined minimum clinical important difference. However, the clinical experts identified that in general, a reduction in acute attacks was clinically relevant. A treatment difference in favour of givosiran was also reported for reduction in annualized days of hemin use and change from baseline in urinary ALA and PBG levels. This difference supports the beneficial direction of the primary outcome. Management of pain was an outcome important to patients, but the results did not demonstrate a clinically meaningful difference in daily worst pain scores. Reported HRQoL outcomes were also important to patients, but subject to limitations that hindered the interpretability of the results. With regards to the safety assessment, the majority of patients in the trial experienced at least 1 AE, with nausea, injection-site reaction, chronic kidney disease, fatigue, increase in ALT, and decrease in glomerular filtration rate more commonly reported among patients who received givosiran. Reported SAEs and treatment discontinuations due to AEs were infrequent, and no deaths were reported. Although the DB treatment period was limited to 6 months on treatment, evidence from the OLE of Study 003 demonstrated maintenance of treatment effect for up to 18 months and did not detect any new safety concerns. Gaps in the evidence of the efficacy and safety of givosiran were identified in patients with concomitant prophylactic hemin use and long-term safety and efficacy data beyond 18 months of treatment.
References
1.Balwani M, Wang B, Anderson KE, et al. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology. 2017;66(4):1314-1322. PubMed
2.Drug Reimbursement Review sponsor submission: Givlaari (givosiran injection) [internal sponsor's package]. Mississauga (ON): Alnylam Pharmaceuticals Canada ULC; 2021 Feb 22.
3.Stolzel U, Doss MO, Schuppan D. Clinical Guide and Update on Porphyrias. Gastroenterology. 2019;157(2):365-381 e364. PubMed
4.Stein P, Badminton M, Barth J, et al. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem. 2013;50(Pt 3):217-223. PubMed
5.Givlaari™ givosiran injection solution, 189 mg/mL givosiran (as givosiran sodium), subcutaneous injection [product monograph]. Amsterdam: Alnylam Netherlands B.V.; 2020 Oct 8.
6.Clinical study report: Primary Analysis for ALN-AS1-003 (Givosiran) ENVISION: A Phase 3 Randomized, Doubleblind, Placebo-Controlled, Multicenter Study with an Open-label Extension to Evaluate the Efficacy and Safety of Givosiran in Patients with Acute Hepatic Porphyrias [internal sponsor's report]. Cambridge (MA): Alnylam Pharmaceuticals, Inc.; 2019 May 16.
7.Alnylam Pharmaceuticals. ENVISION: A Study to Evaluate the Efficacy and Safety of Givosiran (ALN-AS1) in Patients With Acute Hepatic Porphyrias (AHP). ClinicalTrials.gov; 2020 [update 2021 Jun 22]: https://www.clinicaltrials.gov/ct2/show/NCT03338816. Accessed 2021 Jun 28.
8.Alnylam Pharmaceuticals. Expanded Access Protocol of Givosiran for Patients With Acute Hepatic Porphyria. ClinicalTrials.gov; 2019 [updated 2021 Jun 15]: https://www.clinicaltrials.gov/ct2/show/NCT04056481. Accessed 2021 Jun 28.
9.Anderson KE. Porphyrias: An overview. Waltham (MA): UpToDate; 2020 Dec 4.
10.Sood GK. Acute intermittent porphyria: Pathogenesis, clinical features, and diagnosis. Waltham (MA): UpToDate; 2020 Jun 19.
11.Linenberger M, Fertrin KY. Updates on the diagnosis and management of the most common hereditary porphyrias: AIP and EPP. Hematology. 2020;2020(1):400-410. PubMed
12.Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36(5):849-857. PubMed
13.Rare Diseases Clinical Research Network: The Porphyrias Consortium. The Acute Porphyrias. 2020; https://www.rarediseasesnetwork.org/cms/porphyrias/Healthcare-Professionals/Disorder-Definitions. Accessed 2021 Jun 28.
14.Panhematin® Hemin for Injection 268 mg Hemin per Vial Sterile Powder for Reconstitution for Injection [product monograph]. Toronto: Recordati Rare Diseases Canada Inc.; 2018 Jul 13: https://pdf.hres.ca/dpd_pm/00046317.PDF. Accessed 2021 Jun 28.
15.Sood GK. Acute intermittent porphyria: Management. Waltham (MA): UpToDate; 2020 Jun 23.
16.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
17.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/finding-evidence-literature-searching-tools-support-systematic-reviews. Accessed 2021 Jun 28.
18.Balwani M, Sardh E, Ventura P, et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med. 2020;382(24):2289-2301. PubMed
19.Health Canada reviewer's report: Givosiran (as givosiran sodium) [internal sponsor's report]. Amsterdam: Alnylam Netherlands B.V.; 2020 Sep.
20.Ware J, Kosinski M, Keller S. A 12-item short-form health survey: construction of scales and preliminary tests of reliability and validity. Medical Care. 1996;34(3):220-233. PubMed
21.Díaz-Arribas MJ, Fernández-Serrano M, Royuela A, et al. Minimal Clinically Important Difference in Quality of Life for Patients With Low Back Pain. Spine (Phila Pa 1976). 2017;42(24):1908-1916. PubMed
22.van Reenen M, Janssen B. EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument version 2.1. Rotterdam (NL): The EuroQol Research Foundation; 2015: https://euroqol.org/wp-content/uploads/2016/09/EQ-5D-5L_UserGuide_2015.pdf. Accessed 2021 Jun 28.
23.2014 Alberta population norms for EQ-5D-5L. Calgary (AB): Health Quality Council of Alberta; 2014: https://d10k7k7mywg42z.cloudfront.net/assets/542f01f2edb2f37083002e54/2014_EQ_5D_5L_report_FINALFINAL.pdf. Accessed 2021 Jun 28.
24.Dunkl PR, Taylor AG, McConnell GG, Alfano AP, Conaway MR. Responsiveness of fibromyalgia clinical trial outcome measures. J Rheumatol. 2000;27(11):2683-2691. PubMed
25.Farrar JT, Young JP, Jr., LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94(2):149-158. PubMed
26.(NIMH) NIoMH. Patient Global Impressions scale - Change, Improvement, Severity (PGI-C, PGI-I, PGI-S). https://eprovide.mapi-trust.org/instruments/patient-global-impressions-scale-change-improvement-severity. Accessed 2021 Jun 28.
27.Dworkin RH, Turk DC, Trudeau JJ, et al. Validation of the Short-form McGill Pain Questionnaire-2 (SF-MPQ-2) in acute low back pain. J Pain. 2015;16(4):357-366. PubMed
28.Atkinson TM, Mendoza TR, Sit L, Passik S, Cleeland C, Basch E. The Brief Pain Inventory and its “pain at its worst in the last 24 hours” item: clinical trial endpoint considerations. Pain Med. 2010;11(3):337-346. PubMed
29.Song CY, Lin SF, Huang CY, Wu HC, Chen CH, Hsieh CL. Validation of the Brief Pain Inventory in patients with low back pain. Spine (Phila Pa 1976). 2016;41(15):E937-942. PubMed
30.Mathias SD, Crosby RD, Qian Y, Jiang Q, Dansey R, Chung K. Estimating minimally important differences for the worst pain rating of the Brief Pain Inventory-Short Form. The Journal of Supportive Oncology. 2011;9(2):72-78. PubMed
31.Shuman-Paretsky MJ, Belser-Ehrlich J, Holtzer R. Psychometric properties of the Brief Fatigue Inventory in community-dwelling older adults. Arch Phys Med Rehabil. 2014;95(8):1533-1539. PubMed
32.Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer. 1999;85(5):1186-1196. PubMed
33.Wang XS, Hao XS, Wang Y, et al. Validation study of the Chinese version of the Brief Fatigue Inventory (BFI-C). J Pain Symptom Manage. 2004;27(4):322-332. PubMed
34.Meek R, Egerton-Warburton D, Mee MJ, Braitberg G. Measurement and monitoring of nausea severity in emergency department patients: a comparison of scales and exploration of treatment efficacy outcome measures. Acad Emerg Med. 2015;22(6):685-693. PubMed
35.Gouya L, Ventura P, Balwani M, et al. EXPLORE: A Prospective, Multinational, Natural History Study of Patients with Acute Hepatic Porphyria with Recurrent Attacks. Hepatology. 2020;71(5):1546-1558. PubMed
36.Alnylam Pharmaceuticals. A Study to Evaluate Long-term Safety and Clinical Activity of Givosiran (ALN-AS1) in Patient With Acute Intermittent Porphyria (AIP). ClinicalTrials.gov; 2016 [updated 2021 Jun 15]: https://www.clinicaltrials.gov/ct2/show/NCT02949830. Accessed 2021 Jun 28.
37.Clinical study report: ALN-AS1-001 Givosiran (ALN-AS1) A Phase 1, Single Ascending Dose, Multiple Ascending Dose, and Multidose Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics Study of Subcutaneously Administered ALN-AS1 in Patients with Acute Intermittent Porphyria (AIP) [internal sponsor's report]. Cambridge (MA): Alnylam Pharmaceuticals, Inc.; 2018 Aug 31.
38.Clinical study report: ALN-AS1-002 Interim 1 Givosiran (ALN-AS1) A Multicenter, Open-label Extension Study to Evaluate the Long-term Safety and Clinical Activity of Subcutaneously Administered ALN-AS1 in Patients with Acute Intermittent Porphyria who have Completed a Previous Clinical Study with ALN-AS1 [internal sponsor's report]. Cambridge (MA): Alnylam Pharmaceuticals, Inc.; 2019 Apr 9.
39.Clinical Study Report: Interim analysis for ALN-AS1-003 (GIVOSIRAN) ENVISION: A Phase 3 Randomized, Double-blind, Placebo-Controlled, Multicenter Study with an Open-label Extension to Evaluate the Efficacy and Safety of Givosiran in Patients with Acute Hepatic Porphyrias [internal sponsor's report]. Cambridge (MA): Alnylam Pharmaceuticals, Inc.; 2020 Jun 2.
40.Poquet N, Lin C. The Brief Pain Inventory (BPI). J Physiother. 2016;62(1):52. PubMed
41.Center for Drug Evaluation Research. Other Review(s). Drug Approval Package: GIVLAARI (givosiran) Injection Company: Alnylam Pharmaceuticals Inc. Application Number: 212194 Approval Date: 11/20/2019 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2019 Nov 20: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212194Orig1s000OtherR.pdf. Accessed 2021 Jun 28.
42.Center for Drug Evaluation Research. Multi-discipline review. Drug Approval Package: GIVLAARI (givosiran) Injection Company: Alnylam Pharmaceuticals Inc. Application Number: 212194 Approval Date: 11/20/2019 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2019 Nov 20: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212194Orig1s000TOC.cfm. Accessed 2021 Mar 16.
43.Committee for Medicinal Products for Human Use (CHMP). Givlaari: Assessment Report. Amsterdam: European Medicines Agency; 2020 Jan 30: https://www.ema.europa.eu/en/documents/assessment-report/givlaari-epar-public-assessment-report_en.pdf. Accessed 2021 Jan 30.
44.Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113(1-2):9-19. PubMed
45.Westenberg RF, Zale EL, Heinhuis TJ, et al. Does a Brief Mindfulness Exercise Improve Outcomes in Upper Extremity Patients? A Randomized Controlled Trial. Clin Orthop Relat Res. 2018;476(4):790-798. PubMed
46.Brief Fatigue Inventory. Sydney (AU): The Moving Ahead Centre of Research Excellence (CRE) in Brain Recovery; [Date unknown]: http://movingahead.psy.unsw.edu.au/documents/research/outcome%20measures/adult/TBI%20Related%20Symptoms/Website%20Brief%20Fatigue%20Inventory.pdf. Accessed 2019 Oct 7.
47.Lin CC, Chang AP, Chen ML, Cleeland CS, Mendoza TR, Wang XS. Validation of the Taiwanese version of the Brief Fatigue Inventory. J Pain Symptom Manage. 2006;32(1):52-59. PubMed
48.Wikström L, Nilsson M, Broström A, Eriksson K. Patients' self-reported nausea: Validation of the Numerical Rating Scale and of a daily summary of repeated Numerical Rating Scale scores. J Clin Nurs. 2019;28(5-6):959-968. PubMed
49.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-defined estimates of the minimally important difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of Search: March 23, 2021
Alerts: Bi-weekly search updates until project completion
Study types: No search filters used
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for 1 character |
? | Truncation symbol for 1 or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase); |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(givlaari* or givosiran* or ALN-AS1 or ALNAS1 or WHO 10280 or WHO10280 or ROV204583W).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*Givosiran/
(givlaari* or givosiran* or ALN-AS1 or ALNAS1 or WHO 10280 or WHO10280).ti,ab,kw,dq.
3 or 4
5 use oemezd
2 or 6
7 not (conference abstract or conference review).pt.
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- givlaari OR givosiran OR “ALN-AS1” OR ALNAS1 OR “WHO 10280” OR WHO10280]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- givlaari OR givosiran OR “ALN-AS1” OR ALNAS1 OR “WHO 10280” OR WHO10280]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- givlaari OR givosiran OR “ALN-AS1” OR ALNAS1 OR “WHO 10280” OR WHO10280]
Grey Literature
Search dates: March 11 to 16, 2021
Keywords: givlaari OR givosiran OR “ALN-AS1” OR ALNAS1 OR “WHO 10280” OR WHO10280
Limits:
Updated: None
Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool For Searching Health-Related Grey Literature (https://www.cadth.ca/grey-matters) were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
No studies were excluded at the full-text screening stage.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Figure 21: Forest Plot of AAR During the 6-Month Double-Blind; Subgroup Analysis; AIP Patients (mFAS)
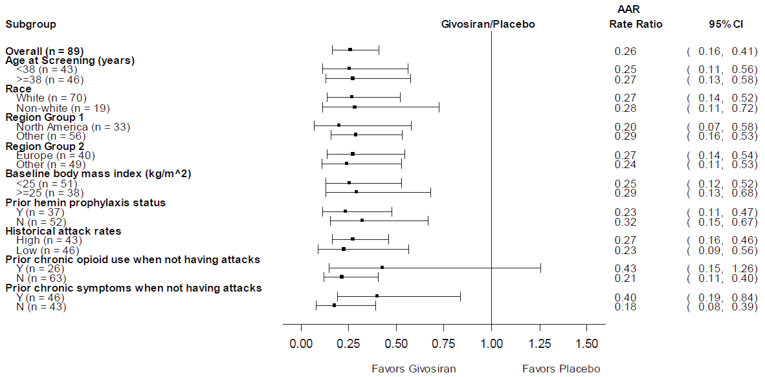
AAR = annualized attack rate; AIP = acute intermittent porphyria; CI = confidence interval; DB = double blind; FASAIP = mFAS = AIP patients in full analysis set; N = no; Y = yes.
Rate ratio and corresponding CIs are derived using negative binomial regression model with the logarithm of the follow-up time as an offset variable.
Source: Clinical Study Report.6
Figure 22: Forest Plot of AAR During the 6-Month Double-Blind; Subgroup Analysis; AIP Patients (FAS)
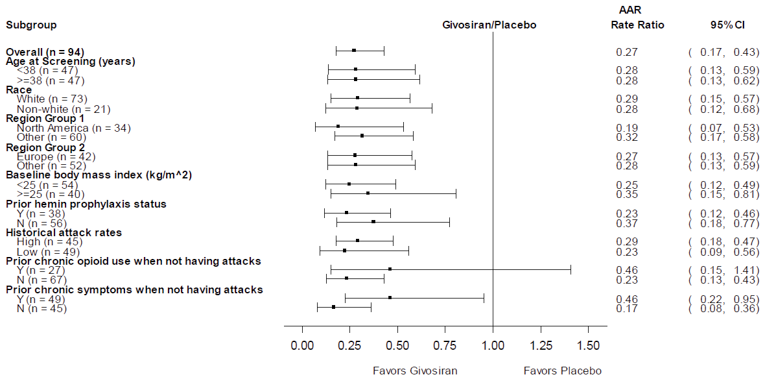
AAR = annualized attack rate; AIP = acute intermittent porphyria; CI = confidence interval; DB = double blind; N = no; Y = yes.
Rate ratio and corresponding CIs are derived using negative binomial regression model with the logarithm of the follow-up time as an offset variable.
Source: Clinical Study Report.6
Table 37: HRQoL Outcomes — EQ-5D-5L Individual Domain Data (6-Month DB Period, FAS)
Outcome | AIP patients, n (%) | All AHP patients, n (%) | ||
|---|---|---|---|---|
Givosiran N = 46 | Placebo N = 43 | Givosiran N = 48 | Placebo N = 46 | |
Mobility | ||||
I have no problems in walking | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have slight problems in walking | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have moderate problems in walking | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
I have severe problems in walking | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
I am unable to walk | v vvvvv | v | v vvvvv | v vvvvv |
Self-Care | ||||
I have no problems washing or dressing myself | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have slight problems washing or dressing myself | v vvvvv | v vvvvvv | v vvvvv | v vvvvvv |
I have moderate problems washing or dressing myself | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
I have severe problems washing or dressing myself | v vvvvv | v | v vvvvv | v vvvvv |
I am unable to wash or dress myself | v | v | v | v |
Usual Activities | ||||
I have no problems doing my usual activities | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have slight problems doing my usual activities | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have moderate problems doing my usual activities | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have severe problems doing my usual activities | v | v vvvvv | v vvvvv | v vvvvv |
I am unable to do my usual activities | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
Pain/Discomfort | ||||
I have no pain or discomfort | vv vvvvvv | v vvvvvv | vv vvvvvv | v vvvvvv |
I have slight pain or discomfort | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have moderate pain or discomfort | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I have severe pain or discomfort | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
I have extreme pain or discomfort | v | v vvvvv | v | v vvvvv |
Anxiety/Depression | ||||
I am not anxious or depressed | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I am slightly anxious or depressed | vv vvvvvv | vv vvvvvv | vv vvvvvv | vv vvvvvv |
I am moderately anxious or depressed | v vvvvvv | v vvvvvv | v vvvvvv | v vvvvvv |
I am severely anxious or depressed | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
I am extremely anxious or depressed | v vvvvv | v vvvvv | v vvvvv | v vvvvv |
CI = confidence interval; DB = double blind; EQ-5D-5L = EuroQol 5 dimensions; FAS = full analysis set; SD = standard deviation.
Source: Clinical Study Report.6
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Annualized rate of porphyria attack (primary composite outcome): porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home
Brief Fatigue Inventory – Short Form (BFI-SF)
Brief Pain Inventory – Short Form (BPI-SF)
Nausea Numerical Rating Scale
European Quality of Life Scale – 5 Dimensions – 5 Levels (EQ-5D-5L)
PGIC
PPEQ
SF-12 v.2
Findings
Table 38: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Annualized rate of porphyria attack (primary composite outcome) | Consists of porphyria attacks requiring:
| No literature was identified that assessed the annualized rate of porphyria attack for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
BFI-SF | Self-reported, 9-item instrument for assessing fatigue. A single question was used in the ENVISION trial. | Construct, concurrent, and discriminant validity; internal consistency; and reliability were demonstrated for the instrument. No literature was identified that assessed the BFI-SF for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
BPI-SF | Self-reported, 11-item instrument for assessing pain intensity and pain interference. A single question was used in the ENVISION trial. | Construct, convergent, and discriminative validity; internal consistency; and test-retest reliability were demonstrated for the instrument. No literature was identified that assessed the BPI-SF for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
Nausea Numerical Rating Scale | Self-reported, single question for assessing a patient’s level of nausea in the past 24 hours on a scale from 0 to 10. | Construct validity and reliability were demonstrated for the instrument. No literature was identified that assessed the Nausea Numerical Rating Scale for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
EQ-5D-5L | EQ-5D-5L index: Generic, preference-based measure of HRQoL consisting of 5 domains: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Scores range from 0 to 1 with higher scores indicating better health status. EQ VAS: Generic, preference-based measure of HRQoL presented as a scale from 0 to 100 with 0 anchored as the worst possible health state and 100 as the best. | Validity and reliability have been demonstrated in a diverse population. No literature was identified that assessed the EQ-5D-5L for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. MID for the index score was estimated to range from 0.037 to 0.056 in the general Canadian population. |
PGIC | A single question answered based on a 7-point numerical scale used for rating global improvement with treatment. | No literature was identified that assessed the PGIC for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
PPEQ | Eight questions used to assess daily living activities, treatment experience, and functional status on a 5-point rating of change scale. | No literature was identified that assessed the PPEQ for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
SF-12 v.2 | Patient-reported measure of HRQoL based on a 4-week recall period. 12-item version of the Short Form Health Survey composed of 8 concepts belonging to either the PCS or MCS. The PCS and MCS range from 0 to 100, where higher scores indicate better HRQoL. | Validity and reliability have been demonstrated in a diverse population. No literature was identified that assessed the SF-12 for validity, reliability, or responsiveness in patients with porphyria. | Not identified in populations with AHP. |
AHP = acute hepatic porphyria; BFI-SF = Brief Fatigue Inventory – Short Form; BPI-SF = Brief Pain Inventory – Short Form; EQ-5D-5L = European Quality of Life Scale – 5 Dimensions – 5 Levels; HRQoL = health-related quality of life; MCS = Mental Component Summary; MID = minimal important difference; PCS = Physical Component Summary; PGIC = Patient Global Impression of Change; PPEQ = Porphyria Patient Experience Questionnaire; SF-12 v.2 = 12-item Short Form Health Survey, version 2; VAS = visual analogue scale.
Annualized Rate of Porphyria Attack (Primary Composite Outcome): Porphyria Attacks Requiring Hospitalization, Urgent Health Care Visit, or IV Hemin Administration at Home
In the ENVISION trial, the primary composite outcome was the annualized rate of porphyria attacks requiring hospitalization, urgent health care visit, or IV hemin administration at home in patients with AIP during the 6-month DB period of the study. Patients or caregivers were to record any potential porphyria attacks for the duration of the study when they occurred using the provided electronic diary, by telephone, or email to the study site or investigator. Health care professionals could also notify the study centre of porphyria attacks and all attacks were confirmed by the investigator. If an event was determined to be inconsistent with a protocol-defined attack, an alternative reason was recorded.
Porphyria attacks were defined as meeting all of the following criteria:
Acute episode of neurovisceral pain in abdomen, back, chest, extremities, or limbs
No other medically determined cause than a porphyria attack
Requiring treatment with IV dextrose, hemin, carbohydrates, analgesics, or other medications (e.g., antiemetics at a dose frequency beyond the patient’s usual daily porphyria management)
The 3 components of the composite outcome were considered non-overlapping and porphyria attacks were defined as follows:
Requiring hospitalization: admission to an inpatient unit or a visit to an emergency department that resulted in an at least 24-hour stay
Requiring urgent health care visit: urgent, unscheduled office/practice, infusion centre, or emergency department visit that did not meet the criteria for hospitalization
Requiring IV hemin administration at home: home was any location that did not meet the criteria for a hospitalization or urgent health care visit
No literature was identified that assessed the annualized rate of porphyria attack for validity, reliability, or responsiveness in patients with porphyria.
No MID was identified in populations with AHP.
Brief Pain Inventory – Short Form (BPI-SF)
The BPI is a questionnaire designed to provide information on 2 subscales: pain intensity (the sensory dimension, 4 items) and the degree to which pain interferes with functioning in daily living (the reactive dimension, 7 items). It is recommended by the IMMPACT as a core outcome measure of pain.44 Four items assess pain intensity: 1) at its worst in the last 24 hours, 2) at its least in the last 24 hours, 3) average pain, and 4) pain right now, using a 0 to 10 NRS, with 0 representing “no pain” and 10 representing “pain as bad as you can imagine.” For the 7 items assessing pain interference with functioning, patients are asked to rate how pain interferes with 7 life domains, including general activity, mood, walking ability, normal work, relations with others, sleep, and enjoyment of life, on a similar 11-point NRS. The anchor points for each item of the interference scale are 0 = “not interfered” and 10 = “completely interfered.” The scores for the 2 subscales (pain intensity and pain interference) range from 0 to 10 and are calculated using the mean of their corresponding items’ scores. The total score of BPI is the mean of the subscale scores. A higher score represents a higher pain intensity or pain interference. The BPI also contains supplemental items that allow a patient to indicate treatments they are receiving for their pain, the percentage of relief obtained in the past 24 hours from the treatments, and the anatomic location of their pain on a body diagram.28,29
Originally developed for evaluation of cancer pain (e.g., breast, prostate, colon, rectum, or gynecologic cancer), it has also been shown to be a reliable (e.g., internal consistency and test-retest reliability) and valid (e.g., construct, convergent, and discriminative validity) instrument for evaluation of non-malignant chronic pain (e.g., low back pain, osteoarthritis, rheumatoid arthritis or multiple sclerosis) across various languages.28,29 For the 2 subscales, test-retest reliability (r = 0.8 for either subscale) and internal consistency (Cronbach alpha ranged from 0.81 to 0.89 and from 0.88 to 0.95, respectively) have been reported.40 No literature was identified that assessed the overall BPI-SF for validity, reliability, or responsiveness in patients with porphyria.
In the ENVISION trial, a single question was used from the BPI-SF to assess the severity of daily pain.6 Patients were asked to rate their worst level of pain experienced during the past 24 hours on a scale from 0 = “no pain” to 10 = “as bad as you can imagine.” As a single item, it has shown internal consistency (0.70 < Cronbach alpha < 0.90) and reliability (between 0.8 and 0.96) among various populations.28
Although an overall MID of the BPI has not been identified from the literature, a 2-point change has been suggested as a reasonable estimate of the MID for worst pain item among breast cancer patients with metastatic disease30 while a 1-point change was suggested for pain intensity in patients with chronic pain and musculoskeletal pain.45 Farrar et al. looked at 10 placebo-controlled studies investigating the use of pregabalin in patients with chronic pain (N = 2,724).25 Outcomes of the trials included both the pain intensity numeric rating scale (PI-NRS), which asks patients to rate their pain in the past 24 hours from 0 to 10) and PGIC. The investigators defined clinical importance a priori to be “much improved” or better on the PGIC. They estimated that a change score of approximately –2 points or percent change of –30% on the PI-NRS from baseline represented clinical improvement.25 No MID was identified in populations with AHP.
Brief Fatigue Inventory – Short Form
The BFI is a self-reported questionnaire to assess the severity and impact of fatigue on daily functioning. Two dimensions are measured in the 9-item instrument: fatigue (3 items on current, usual, and worst levels) and the interference of fatigue on daily life (6 items on general activity, mood, walking ability, normal work, relations with others, and enjoyment of life).31 The items are measured on a 0 to 10 NRS. For the severity of fatigue dimension, 0 represents “no fatigue” and 10 represents “fatigue as bad as you can imagine.” For the interference from fatigue dimension, 0 represents “does not interfere” and 10 represents “completely interferes.” A score of 7 to 10 is considered severe fatigue.32 A global fatigue score can be obtained by averaging all the items on the BFI.46
Validity and reliability have been assessed in samples of patients with cancer, rheumatoid arthritis, and community-dwelling adults and older adults.31 Construct validity, concurrent validity, and discriminant validity of the BFI have been demonstrated in cancer patients. Reliability of the BFI was acceptable (Cronbach alpha values were 0.95 to 0.96) based on a study of 305 adult patients with cancer.32 Reliability and internal consistency were demonstrated in a sample of 302 community-dwelling older adults when the BFI-SF was divided into 2 subscales, severity and interference, with Cronbach alpha values of 0.818 and 0.869, respectively.31 No literature was identified that assessed the BFI-SF for validity, reliability, or responsiveness in patients with porphyria.
In the ENVISION trial, a single question was used from the BFI-SF to assess the severity of daily fatigue6 Patients were asked to rate their worst level of fatigue experienced during the past 24 hours on a scale from 0 = “no fatigue” to 10 = “as bad as you can imagine.”
Both Chinese33 and Taiwanese47 versions of the BFI (BFI-C and BFI-T, respectively) have been previously validated. The BFI-C demonstrated validity and reliability in a sample of 249 patients with cancer.33 As predicted by the investigators, the worst fatigue item correlated with the fatigue-related items of the Chinese version of the SF-36 indicating convergent validity. The Pearson correlation coefficients were –0.51, –0.49, 0.44, and –0.52 for the PCS, MCS, physical functioning subscale, and vitality subscale of the Chinese SF-36, respectively. The mean scores for the worst fatigue item differed among patients with different ECOG performance status levels (0 versus 1 versus 2 to 4) demonstrating known-group validity. Patients with a poorer ECOG performance status (2 to 4) showed significantly higher worst fatigue scores (r = 0.421, P < 0.001). An MID for the worst fatigue item was estimated to be 1.5 using the0.5 SD method.33 The BFI-T showed validity and reliability in a sample of 235 inpatients and 186 outpatients from oncology clinics.47 Convergent validity was demonstrated between the worst fatigue item and fatigue-related subscales of the Taiwanese POMS short form questionnaire. Pearson correlation coefficients were 0.82 and –0.69 between the BFI-T worst fatigue item and POMS vigour and fatigue subscales, respectively. The investigators also noted that inpatients and patients with a lower Karnofsky Performance Status had significantly higher worst fatigue scores demonstrating known-group validity.
No MID was identified in populations with AHP.
Nausea Numerical Rating Scale
The Nausea Numerical Rating Scale is a self-reported, single question used to assess the severity of nausea a patient experiences.2 In the ENVISION trial, patients were asked to rank the worst nausea they felt in the past 24 hours on a 0 to 10 NRS where 0 represents “no nausea” and 10 represents “nausea as bad as you can imagine.”
In a study by Meek et al., the nausea NRS was compared to a VAS for nausea, the latter of which has been previously validated in patients visiting the emergency department.34 Meek et al. used data from a convenience sample of 258 patients who visited the emergency department in Australia.34 Patients rated their nausea on 3 scales: (1) 100 mm VAS where the far left represented “no nausea” and the far right “unbearable nausea;” (2) NRS where 0 indicated “no nausea” and 10 “unbearable nausea;” and (3) adjectival scale consisting of descriptors “none,” “mild,” “moderate,” and “severe.” The instruments were administered both at enrolment and 30 minutes after treatment. The results showed that the VAS and NRS correlated well with 1 another (Spearman rank correlation coefficient = 0.83) and that the scales could discriminate between descriptors of the adjectival scale. For the NRS, “severe,” “moderate,” “mild,” and “none” had median (interquartile range) scores of 9 (8 to 9), 6 (5 to 7), 4 (3 to 5), and 0 (0 to 1), respectively. On the VAS, the same categories had medians (interquartile range) of 90.5 mm (79 to 97 mm), 59 mm (48 to 71 mm), 34 mm (25 to 49 mm). and 5 mm (3 to 5 mm), respectively. The differences between each adjective category were found to be statistically significant for both numerical scales. The NRS and a verbal form of the adjectival scale have been compared in 479 Swedish patients before and after major surgery and showed similar correlation.48 The Spearman rank correlation coefficient was 0.79 between the verbal scale (also using “none,” “mild,” “moderate,” and “severe”) and the NRS. In these studies, patients were asked for their current level of nausea rather than their worst level over a specified time period.
No literature was identified that assessed the Nausea Numerical Rating Score specifically for worst nausea experienced.
No MID was identified in populations with AHP.
European Quality of Life Scale – 5 Dimensions – 5 Levels (EQ-5D-5L)
The EQ-5D is a generic, self-reported, HRQoL instrument developed by the EuroQol Group that is applicable to a wide range of health conditions and treatments.22 As a generic measure of HRQoL that can capture the net effect of treatment benefits and harms, the EQ-5D provides valuable information from the patient perspective. The original 3-level version of the EQ-5D (EQ-5D-3L) was introduced in 1990 and was composed of 5 dimensions pertaining to HRQoL.22 Respondents indicate their health status in terms of 5 dimensions based on 3 levels of severity. To improve sensitivity and reduce ceiling effects, the EQ-5D-3L was updated in 2005 and expanded to 5 levels for respondents to answer each dimension with, thus creating the EQ-5D-5L, which was used in the ENVISION trial.22
The EQ-5D-5L consists of a descriptive system and the EQ VAS. The descriptive system comprises 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension is answered based on 5 levels, where 1 = “no problems,” 2 = “slight problems,” 3 = “moderate problems,” 4 = “severe problems,” and 5 = “extreme problems” or “unable to perform,” which is the worst response in the dimension.22 Respondents choose the level that reflects their health state for each of the 5 dimensions. In total, there are 3,125 possible unique health states defined by the EQ-5D-5L, with 11111 and 55555 representing the best and worst health states, respectively. The numerical values assigned to levels 1 to 5 for each dimension reflect rank order categories of function. In terms of measurement properties, these are ordinal data and do not have interval properties, therefore, they should not be summed or averaged to, for example, produce a single dimension score. Results from the EQ-5D-5L descriptive system can be converted into a single index score using a scoring algorithm taking the local patient and population preferences into account. Therefore, the index score is a country-specific value and a major feature of the EQ-5D instrument.23 The range of index scores will differ according to the scoring algorithm used; however, in all scoring algorithms of the EQ-5D-5L, a score of 0 represents the health state “dead” and 1.0 reflects “perfect health.” Negative scores are also possible for those health states that society (not the individual patient) considers to be “worse than dead.”
The EQ VAS records the respondent’s self-rated health on a vertical VAS where the end points are labelled 0 (“the worst health you can imagine”) and 100 (“the best health you can imagine”). Respondents are asked to mark a X on the point of the VAS that best represents their health on that day. The EQ-5D index and VAS scores can be summarized and analyzed as continuous data.22,23 Overall, the EQ-5D produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121 or 21143,
a population preference-weighted health index score based on the descriptive system,
a self-reported assessment of health status based on the EQ VAS.
The EQ-5D-5L has been validated in terms of feasibility, ceiling effects, discriminatory power, and convergent validity in a diverse patient population from 6 countries with chronic conditions.22 No literature was identified that assessed the EQ-5D-5L for validity, reliability, or responsiveness in patients with porphyria.
In EXPLORE, a prospective, multinational, natural history study, patients with AHP and recurring attacks were enrolled if they had at least 3 attacks in the 12 months before the start of the study or were receiving prophylactic treatment.35 In total, 112 patients were followed for at least 6 months with an optional extension to 12 months. EQ-5D-5L was used to assess HRQoL at baseline, 6, and 12 months during EXPLORE. Overall, mean baseline index scores were 0.78 (SD = 0.15) which the authors noted to be lower than a typical score of 0.92 from the European population of with the same age range (median age in EXPLORE = 38 years, reference population age 35 to 44 years). More than half of patients in the study reported having at least some problem with pain/discomfort (64%), anxiety/depression (51%), and ability to perform usual activities (51%). The study population’s baseline VAS score was 66 which was maintained throughout EXPLORE. The authors observed no notable difference in EQ-5D-5L scores between those who were receiving prophylactic treatment and those who were not.
A Canadian-specific estimate of an MID for the EQ-5D-5L was generated by simulating the effects of single level transitions in each dimension.49 The results yielded MIDs with a summarized mean of 0.056 (SD = 0.011), and a summarized median of 0.056 (interquartile range = 0.049 to 0.063).49 No MID was identified in populations with AHP.
Patient Global Impression of Change
The PGIC is a widely used, validated outcome measure for clinical pain trials.24,25 It is a single question answered on a 7-point numerical scale to indicated perceived change in overall health status since the study began. Patients are asked to consider their overall health since the start of the trial by choosing a response from the following: 1 = “very much improved,” 2 = “much improved,” 3 = “minimally improved,” 4 = “no change,” 5 = “minimally worse,” 6 = “much worse,” 7 = “very much worse.”26
In a dataset of 2,724 patients who received pregabalin for diabetic neuropathy, postherpetic neuralgia, chronic low back pain, fibromyalgia, and osteoarthritis from 10 placebo-controlled clinical trials, the 11-point PI-NRS and PGIC were used as determinants of a clinically important difference and the relationship between the 2 outcome measures was explored. A consistent relationship between the change in 11-point PI-NRS and the PGIC was demonstrated regardless of study, disease type, age, sex, study result, or treatment group.25 The PGIC questionnaire has been recommended for use in chronic pain clinical trials by IMMPACT as a core outcome measure of global improvement with treatment.27
No literature was identified that assessed the PGIC for validity, reliability, or responsiveness in patients with porphyria.
No MID was identified in populations with AHP.
Porphyria Patient Experience Questionnaire
The PPEQ consists of 8 questions used to assess a patient’s ability to perform daily living activities (questions 1 to 5), treatment experience (questions 6 and 7), and functional status (question 8) on a 5-point global rating of change scale.6 The first 5 items deal with travel, social activities, planning for the future, completing household tasks, and exercising. Items 6 and 7 ask about convenience and satisfaction with treatment, while the last question asks about the medication’s ability to help the patient achieve a more normal life compared to before the study. For the first 7 items, patients select from the following options: “much better,” “minimally better,” “no change,” “minimally worse,” or “much worse” based on their current experience compared to before the start of the study. The last item is answered by choosing 1 of the following: “always,” “most of the time,” “sometimes,” “rarely,” or “never” based on the last 4 weeks. Responses for each of the 8 questions of the PPEQ were reported descriptively in the ENVISION trial.
No literature was identified that assessed the PPEQ for validity, reliability, or responsiveness in patients with porphyria.
No MID was identified in populations with AHP.
12-item Short Form Health Survey, version 2 (SF-12 v.2)
The SF-12 is a generic, patient-reported measure of HRQoL based on the 36-item version of the survey (SF-36). Patients answer based on a 4-week recall period. Each item falls into 1 of 8 health scales, including:
physical functioning (PF), 2 items
role physical (RP), 2 items
bodily pain (BP), 1 item
general health (GH), 1 item
vitality (VT), 1 item
social functioning (SF), 1 item
role emotional (RE), 2 items
and mental health (MH), 2 items.20
The “physical functioning” scale assesses the extent to which daily life is affected, “role physical” measures limitations in roles due to problems with physical health, “bodily pain” measures the frequency of pain and how much pain interferes with normal functioning, “general health” measures the patient’s perception of their overall health, “vitality” assesses fatigue and energy levels, “social functioning” measures how much a patient’s illness affects social functioning, “role emotional” assesses role limitation due to emotional issues, and “mental health” assesses psychological distress.20 The first 4 scales (PF, RP, BP, and GH) make up the PCS while the latter 4 (VT, SF, RE, and MH) fall under the MCS. The PCS and MCS correspond to the physical and psychological burden of disease, respectively. The component summaries are standardized to have a mean of 50 and SD of 10 based on the general US population and higher scores reflect better HRQoL.20 In the ENVISION trial, the PCS was a secondary outcome while the MCS was an exploratory outcome.
Test-retest reliability of the SF-12 summary scores have been demonstrated in the general US and UK populations with coefficients of 0.890 and 0.864, respectively, for the PCS, and 0.760 and 0.774 for the MCS.20 Discriminant validity was demonstrated for groups known to differ in physical and mental conditions, and cross-validation was assessed between the SF-12 and SF-36 with correlations of 0.951 and 0.969 for the PCS and MCS, respectively. No literature was identified that assessed the SF-12 for validity, reliability, or responsiveness in patients with porphyria.
MIDs for the SF-12 PCS and MCS have been estimated based on a study of 458 patients with lower back pain.21 The 4 methods used to calculate the MID included the minimum detectable change (MDC), average change, change difference (CD), and receiver operating characteristic curve, which allowed for calculation of AUC. MID estimates ranged from 0.56 to 3.29 based on the MDC and CD methods, respectively, for the PCS. The MID for the MCS was suggested to range from 1.13 to 3.77 based on the CD and MDC methods, respectively. Overall, the authors suggested that an improvement of at least 3.29 on the PCS and 3.77 on the MCS would be clinically meaningful to patients with low back pain.21 No MID was identified in populations with AHP.
Pharmacoeconomic Review
Abbreviations
AAR
annualized attack rate
AHP
acute hepatic porphyria
AIP
acute intermittent porphyria
BIA
budget impact analysis
BSC
best supportive care
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
OLE
open-label extension
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Givosiran (Givlaari), 189 mg/mL solution for subcutaneous injection |
Submitted price | Givosiran, 189 mg/mL, solution for subcutaneous injection: $64,454.30 (price per carton containing 1 single-use 2 mL vial, which holds 1 mL givosiran sodium in solution) |
Indication | For the treatment of acute hepatic porphyria in adults |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | October 9, 2020 |
Reimbursement request | As per indication |
Sponsor | Alnylam Netherlands B.V. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with a documented diagnosis of acute hepatic porphyria |
Treatment | Givosiran |
Comparator | BSC: no treatment |
Perspective | Canadian publicly funded health care payer |
Outcome | QALY; life-years |
Time horizon | Lifetime (defined as 59 years) |
Key data source | ENVISION trial |
Submitted results | Compared with BSC, givosiran was dominant (associated with more QALYs, with a gain of 13.23) and less costly (savings of $8,658,644) |
Key limitations |
|
CADTH reanalysis results |
|
AHP = acute hepatic porphyria; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; OLE = open-label extension; vs. = versus.
Conclusions
The ENVISION trial demonstrated a clinically meaningful reduction in the rate of acute porphyric attacks in adult patients with acute intermittent porphyria (AIP) compared to placebo. The findings are generalizable to all patients with acute hepatic porphyria (AHP), based on the primary composite end point, which included attacks requiring hospitalization, an urgent health care visit, or IV hemin administration. The comparative evidence on the rate of attacks between treatment arms was available for 6 months; data on givosiran that were available for an additional 12 months based on an open-label extension (OLE) period showed a maintenance of treatment effect. However, the clinical efficacy and safety of givosiran beyond 18 months are unknown. There was also no evidence available on the impact of givosiran on AHP-related chronic conditions or on any reduction in complications of AHP. The CADTH clinical review further noted several limitations with the evaluation of health-related quality of life (HRQoL) data, which prevented the interpretation of whether there was an added benefit in this outcome measure from treatment with givosiran compared with placebo in the trial.
The CADTH base-case reanalysis included removing the severe health state; revising health state utility values to reflect a similar impact of AHP-related chronic conditions for all patients; assuming that all patients remained on treatment from 18 months until death (lifetime time horizon); assuming that all female AHP patients who experienced attacks in the symptomatic and recurrent health states before menopause onset became asymptomatic at the onset of menopause; assuming that only 1 vial of hemin was used for a single urgent health care visit; and changing the mean duration of an acute attack, among other changes. In the CADTH base case, givosiran was associated with an incremental cost-effectiveness ratio (ICER) of $17,928,198 per quality-adjusted life-year (QALY) gained (incremental costs of $6,205,467 and an incremental benefit of 0.35 QALYs) compared with best supportive care (BSC). A price reduction of at least 63% would be required for givosiran to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
CADTH was unable to address important limitations related to how the model was conceptualized with regards to AHP disease severity and uncertainties with the long-term efficacy of givosiran. The cost-effectiveness of givosiran was primarily driven by drug acquisition costs and assumptions affecting the number of, and costs associated with, acute porphyric attacks requiring IV hemin.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process. Specifically, it contains information that pertains to the economic submission.
Two patient groups, the American Porphyria Foundation and the Canadian Association for Porphyria, provided input. Patient input indicated that AHP is a life-threatening group of diseases characterized by attacks that cause excruciating, burning pain, nerve damage, nausea, a rapid pulse, and paralysis, among other symptoms. Patients reported that the nature of the condition and seriousness of the attacks resulted in a life of extreme suffering because the attacks had major impacts on their daily lives and affected their ability to work, have a career, and sustain relationships with family and friends. Patients reported that attacks were associated with hospitalizations and could lead to death. Patients indicated that hemin administered by infusion through a central line or peripheral vein is the only treatment prescribed to treat attacks, but it does not prevent them. They also stated that even if givosiran were available, access to hemin would still be critical to stopping attacks once they occur (however, patients may be less likely to have attacks while on treatment with givosiran). Patients who had received treatment with givosiran indicated that it was life-changing because it was able to significantly reduce the overall number of attacks, or nearly eliminate attacks, almost immediately. They reported that givosiran was associated with some side effects, including injection-site reactions, allergic reactions, and nausea; however, they also indicated that givosiran was an easier treatment to take than hemin infusions and could be administered on a monthly basis.
No registered clinician input was received.
Feedback from the drug plans noted that there were no available treatment options for the indicated population to prevent attacks, only to treat attacks. These options included carbohydrate loading or hemin, particularly for recurrent AIP among women, in whom carbohydrate therapy is inadequate. Preventive measures include avoidance of triggers. Drug plans had concerns related to combining givosiran with hemin. They were also concerned about the anticipated budget impact of reimbursing givosiran but noted that it may offset the costs associated with treating attacks.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s base-case analysis compared givosiran to BSC (i.e., no active treatment to prevent attacks), which is reflective of current clinical practice, based on the patient and drug plan input.
The sponsor modelled HRQoL by assigning a temporary decline in quality of life associated with acute attacks every cycle to capture the impact of attacks on a patient’s quality of life, as well as a disutility associated with the long-term impacts of underlying chronic conditions by health state; however, the latter was associated with considerable uncertainty.
In addition to the base-case analysis conducted from the public health care payer perspective, the sponsor also included a scenario analysis from the societal perspective to capture the costs associated with patients’ and caregivers’ lost productivity. CADTH similarly reported a scenario analysis under a societal perspective.
CADTH was able to address the following concerns:
In addition to the budget impact analysis (BIA) base case conducted from the drug program plan perspective, CADTH reported base-case results from the broader public health care payer perspective to address drug plans’ concerns about the anticipated budget impact and highlight which drug costs would be incurred by drug plans versus the health care payer.
CADTH was unable to address the following concerns raised from stakeholder input:
Combination usage with IV hemin outside of its use during acute attacks.
Economic Review
The current review is for givosiran (Givlaari) for adult patients with AHP.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing givosiran versus BSC (i.e., no active treatment to prevent attacks) for the treatment of adults with AHP. The modelled population was aligned with the Health Canada–indicated population.
The recommended dose of givosiran is 2.5 mg/kg once monthly through subcutaneous injection. At a submitted cost of $64,454 per single-use 2 mL vial (189 mg/mL), the average annual cost of givosiran is approximately $773,448 for a patient weighing 67 kg.1 The economic analysis was conducted from the perspective of the public health care payer over a lifetime time horizon (defined as 59 years). Costs and clinical outcomes (i.e., QALYs) were discounted at a rate of 1.5% per annum.1 Drug wastage was assumed.
Model Structure
A Markov model structure was employed to capture the long-term costs and effects of AHP, a debilitating disease that is characterized by intermittent attacks causing a range of nervous system symptoms.1,2 The disease course was modelled through 4 health states defined by the mean number of attacks requiring treatment with IV hemin experienced in a given year (asymptomatic: 0; symptomatic: > 0 to ≤ 4; recurrent: > 4 to ≤ 24; severe: > 24) and an absorbing death state.1,3 With the exception of the severe health state (which was based on an arbitrary assumption), all health states were conceptualized according to an AHP disease severity classification framework identified in the literature.3 Each model cycle was 6 months in duration to align with the double-blind period of the ENVISION trial. All patients entered the model in either the symptomatic, recurrent, or severe health state, according to the distribution of baseline disease severity in the ENVISION trial,4 and could experience a number of acute porphyria attacks every 6 months based on the state they occupied. Patients who received givosiran and BSC could either worsen by moving from the health state of entry to a health state of greater disease severity (e.g., recurrent or severe), or improve by moving to a less severe health state. Patients could move to the absorbing death state during any cycle.1
Model Inputs
The patient cohort comprised AHP patients (the majority with AIP, the most common type) whose baseline characteristics mainly reflected those outlined in a study by Roblin et al. (2020)5 describing Canadian physicians’ experiences in diagnosing AHP. Specifically, the age of the modelled population was 41 years, and 61% of the population was female.5 The mean patient weight of the modelled population was 66.84 kg, which aligned with the patient population in the ENVISION trial.4
To derive patient transition probabilities between health states, the sponsor pooled individual efficacy data on the number of acute attacks from the ENVISION trial at different time points. For patients on givosiran, separate transition matrices were derived from observations at 6 months, 12 months, and 18 months (reflecting the double-blind and OLE periods), while for patients receiving BSC, a single transition matrix at 6 months was derived. For each transition matrix, the sponsor determined the mean annualized attack rate (AAR) of each patient at the beginning of the relevant time period, then determined their mean AAR at the end of each time period. Patients were then categorized into the 4 possible health states based on their mean AAR; the likelihood of moving between each state over the course of that time period was derived by combining the data for all patients for each possible transition (e.g., recurrent at the beginning of the cycle to symptomatic). After the first 6 months, patients who had received BSC were assumed to remain in the same health state, such that they could neither improve nor worsen (i.e., no further transitions between alive states, only transition to death) for the remainder of the model time horizon. This assumption was due to a lack of natural history data on the likelihood of disease worsening or improvement. Patients who received givosiran could continue to move between the AHP disease severity health states based on the transition probabilities derived at the end of the 18-month OLE period for an additional 3.5 years (up to year 5), unless treatment was discontinued for any reason, or until death. From year 5, patients on givosiran were also assumed to remain in the same health state for the remainder of the model time horizon until their death. The mean number of attacks experienced in each health state was based on the mean pooled AAR for all patients within each disease severity classification. Time to off-treatment with givosiran was based on a parametric extrapolation of time to discontinuation data obtained from the 18-month observation period for patients on givosiran and extrapolated for the entire model time horizon. Based on the best-fitting curve, a probability of treatment discontinuation averaging approximately 2.5% per cycle (every 6 months) was applied to patients receiving givosiran, who could discontinue treatment at any time during the model time horizon.2 Upon discontinuation, patients who originally received givosiran were assumed to remain in their existing health state and to have the same transition probabilities as patients receiving BSC. The sponsor’s model also included the assumption that female patients who were asymptomatic and receiving givosiran at the age at which they experienced menopause would discontinue treatment and remain in the asymptomatic health state, given that the likely trigger for their attacks was now absent.
Treatment-related adverse events associated with givosiran and BSC in the model were based on the incidence of severe adverse events that occurred during the 6-month, double-blind period of the ENVISION trial.2 The proportion of patients with AHP-related chronic conditions by health state was based on a study by Neeleman et al. (2018).3 Background mortality was modelled using all-cause age- and sex-specific mortality rates from the general population. Based on a Norwegian study of patients with AHP, an increased risk of mortality was applied for patients with AHP compared to the general population.6 The model assumed no differences in mortality between AHP health states and no survival benefit of givosiran over BSC.2
Health state utility values were applied to each AHP disease severity health state, beginning with an age-specific utility value for the general population (between age groups 40 years to 44 years and 45 years to 49 years) and subtracting AHP-related utility decrements. Specifically, a temporary disutility associated with each acute attack (assumed to last 7.29 days, on average, according to the EXPLORE trial7) and a long-term utility decrement associated with the presence of AHP-related chronic conditions was applied at every model cycle. Caregiver disutilities were applied to each AHP disease severity health stated based on the stage of Multiple Sclerosis severity as a proxy, based on an observational study by Ancaster et al. (2013).8
Costs captured in the economic model included those associated with acute attacks, AHP-related chronic conditions, treatment-related adverse events, opioid addiction, and end-of-life care. A micro-costing approach of resource use for the treatment of AHP attacks was undertaken to derive the costs of acute porphyric attacks treated in hospital or at urgent health care facilities using published literature, Canadian databases, and expert opinion.1 As part of this approach, the sponsor assumed an average length of hospital stay of 7.29 days for attacks requiring hospitalization to align with the assumption about the average duration of an attack. Costs associated with IV hemin treatment and administration were based on a previous CADTH Technology Review for Panhematin.9 The number of vials of hemin required to treat an attack differed by location of treatment, with 2.5 vials required in an urgent health care facility and 4 vials required for treatment in hospital. The costs of opioid addiction were based on a study that examined the societal costs of untreated opioid dependence in Canada by Wall et al. (2000).10 These were applied to 82% of patients, in the recurrent and severe health states, based on a study by Neeleman et al.3 The sponsor also included costs associated with managing AHP-related pain and chronic neurologic and psychiatric conditions. These were based on a variety of sources in the literature.
Summary of Sponsor’s Economic Evaluation Results
The sponsor presented both deterministic and probabilistic analyses for its base case. The results of these analyses were not aligned, given that the total costs obtained with givosiran in both analyses differed by more than 15%. CADTH could not determine the source of this discrepancy. As a result, the deterministic findings are presented in Table 3, and the probabilistic results are commented on in comparison.
Base-Case Results
In the sponsor’s deterministic base case, givosiran was found to be less costly (–$8,658,644) and more effective (13.23 incremental QALYs) over a lifetime time horizon than BSC. As a result, it was dominant over BSC.1 The majority of costs associated with givosiran are from drug acquisition, with the drug’s incremental cost savings in comparison with BSC derived almost entirely from the avoidance of acute attacks requiring IV hemin. The model results indicated that 1.4% of the incremental benefit of givosiran compared with BSC was derived from the 6-month period for which there are trial data, with the remaining 98.6% from the period for which there are no available data.
Givosiran was dominant in the sponsor’s probabilistic analysis as well. The main difference between the probabilistic and deterministic results is a variation in the total costs associated with givosiran, which were $9.3 million in the deterministic base case and $11 million in the probabilistic base case.
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Deterministic)
Drug | Total costs ($) | Incremental costs of givosiran ($) | Total QALYs | Incremental QALYs of givosiran | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
BSC | 17,987,732 | NA | 5.81 | NA | NA |
Givosiran | 9,329,088 | –8,658,644 | 19.04 | 13.23 | Dominant |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity and scenario analyses. These included adopting the societal perspective (i.e., including productivity costs); varying the discount rate (0% and 3%); extrapolating the treatment efficacy of givosiran up to 3 years and assuming that patients would remain in their respective health states at that time point for the remainder of the model time horizon; setting the hospital length of stay to be equal to the number of hemin administrations received per attack treated in hospital (i.e., 4); assuming that patients who discontinued treatment with givosiran would have a |% per-cycle probability of disease worsening; and assuming that patients who received BSC would have a |% per-cycle probability of progressive disease after the first 6 months, up to year 5. In all scenarios, givosiran remained dominant compared with BSC.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Conceptualization of the model based on the frequency of acute porphyric attacks may not appropriately capture disease severity: Several issues were identified with the conceptualization of the sponsor’s submitted model structure in terms of how disease severity was captured. First, the sponsor modelled the disease course with AHP based on the mean number of acute attacks per year requiring IV hemin administration (asymptomatic: 0; symptomatic: > 0 to ≤ 4; recurrent: > 4 to ≤ 24) according to an AHP disease classification framework proposed by Neeleman et al. (2018).3 The sponsor also added a severe health state (> 24 attacks per year) based on an arbitrary assumption. The clinical experts consulted by CADTH indicated that the sponsor’s conceptualization of the model was limited by this classification because disease severity is not solely defined by frequency of attacks. It is also defined by the nature and durations of attacks and the level of intervention required. However, the sponsor’s model was conceptualized on the basis that each attack was the same, with frequency driving severity.
Additionally, the severe health state classification (defined by a mean AAR of |) was not reflective of Canadian clinical practice, according to the clinical experts consulted by CADTH. They indicated that this mean AAR was unrealistic. They further noted that the number of attacks requiring hospitalization or an urgent health care visit with IV hemin administration was unlikely to be more than 10 per year in Canadian clinical practice, with an average of 6 attacks per year being more common. The sponsor claimed that the mean AAR for the severe health state was derived from the ENVISION trial; however, this information could not be verified by the CADTH clinical review team. The CADTH clinical experts speculated that if these attacks were, in fact, obtained from trial data, then it is likely that the number of attacks was overestimated because it included mild attacks not requiring IV hemin use, as per the trial’s definition of an attack. Consequently, this may have driven the frequency of attacks in the model to a number higher than expected in practice. Because more patients receiving BSC were in the severe health state, this led to an overestimation of the total number of attacks requiring IV hemin for patients receiving BSC, biasing results in favour of givosiran.
CADTH was unable to address the limitations related to the sponsor’s conceptualization of the model structure, which did not appropriately capture disease severity. CADTH removed the severe health state and reallocated patients originally assigned to the severe health state at baseline to the recurrent health state while keeping the mean AAR of | in the recurrent health state.
Uncertainty associated with the impact of AHP-related chronic conditions on health state utility values: The sponsor included the occurrence of AHP-related chronic conditions (i.e., chronic pain and/or neurologic and/or psychiatric conditions) in the model, with prevalence varying by health state. For instance, the recurrent AHP state had the highest prevalence of a combination of these conditions compared with symptomatic and asymptomatic AHP states. The impact of these chronic conditions on HRQoL was then included as a health state disutility, with the highest disutility for patients in the severe health state and the lowest for patients in the asymptomatic state. In the absence of relevant data, the sponsor’s model assumes that givosiran is not associated with increased survival, that any differences in QALYs between givosiran and BSC in the model are based strictly on HRQoL improvements, and that the sponsor has also included a utility decrement associated with the occurrence of porphyric attacks. Given that more patients on givosiran in the sponsor’s model achieved the asymptomatic health state, they did not experience as great a disutility due to AHP-related chronic conditions as did patients receiving BSC. Two issues were identified with the inclusion of the impact of chronic conditions on health state utility values. Most notably, there is no clinical evidence from the ENVISION trial to support the assumption of an improvement in AHP-related chronic conditions with givosiran that would result in the QALY gains observed in the economic model. The trial did not report the impact of givosiran on chronic conditions or any reduced complications of AHP. The CADTH clinical review further noted several limitations with the evaluation of HRQoL data that prevented the interpretation of whether there was an added benefit in this outcome measure from treatment with givosiran compared with placebo in the trial.
In addition to a lack of evidence in support of this impact, the health state utility values used to determine the disutilities associated with AHP-related chronic conditions in the economic model were likely inappropriate. The disutilities were derived from various published literature sources that were not specific to AHP, and there was likely overlap in the values used for the different chronic conditions that led to double counting of the impacts of chronic condition when these values were combined. In the sponsor’s base case, the inclusion of the impact of chronic conditions on health state utilities led to substantially underestimated total QALYs for patients receiving BSC, which overestimated the QALY gains associated with givosiran.
In the absence of any clinical evidence to support the assumptions that givosiran is associated with substantial gains in QALYs compared to BSC (moderated by the prevalence of chronic conditions), and in the absence of condition-specific utility values for AHP, CADTH addressed this limitation by assuming that the utility decrement associated with chronic conditions would be the same for all health states, such that the disutility would be equal for all patients regardless of treatment. CADTH applied the utility decrement for the asymptomatic health state to all AHP health states. In a scenario analysis, CADTH applied the utility decrements assumed in the sponsor’s base case.
The long-term comparative efficacy of givosiran versus BSC is uncertain: In the economic model, the comparative efficacy of givosiran versus BSC was based on the 6-month, double-blind trial period, with an additional 12 months of data covering the OLE period, for a total of 18 months of data for givosiran. In the submitted model, for patients receiving givosiran, the sponsor assumed that patients continued to have a likelihood of improving until the end of 18 months, even though the OLE period in the trial only demonstrated a maintenance of effect from the end of 6 months to the end of 18 months, such that patients neither improved nor worsened. Additionally, in the sponsor’s base case, the likelihood of moving between health states (i.e., the probability of moving to a health state with fewer attacks or a greater number of attacks) observed at the end of 18 months was assumed to last for another 3.5 years (up to year 5), at which point patients remained in their respective health states and maintained the clinical benefits accrued for the remainder of their lifetime. With these assumptions, the vast majority of patients on givosiran entered the asymptomatic health state by year 5 and remained attack-free for nearly the entirety of the time horizon. However, there is no long-term clinical evidence to support these assumptions, and these assumptions likely led to a vast overestimation of the reduction in rate of attacks for patients on givosiran compared with BSC in the model versus estimates from the trial. Only 1.4% of the QALY benefits observed with givosiran in the sponsor’s base case were from the period for which there are observed data, with the remaining 98.6% based on assumptions in the period for which there are no data. As a result, the benefit observed with givosiran is likely to be substantially overestimated, biasing results in favour of givosiran.
CADTH was unable to address this limitation.
Patients who discontinued givosiran were assumed to maintain clinical benefit from treatment over the lifetime time horizon: In its base case, the sponsor assumed that patients who discontinued treatment on givosiran at any point in time would experience disease progression similarly to patients who received BSC. Patients receiving BSC did not experience any disease progression beyond the first 6 months in the model. This was determined to be a reasonable assumption for patients receiving BSC, reflecting the natural history of AHP in the absence of natural history data, but it was not a reasonable assumption for those discontinuing givosiran. The sponsor assumed that patients who discontinued givosiran remained in the same health state (in which they discontinued treatment) until death. These patients did not experience future attacks and maintained the clinical benefits of treatment for the remainder of the lifetime time horizon without accruing treatment costs. CADTH’s clinical experts indicated that this clinical assumption did not align with their expectations, because patients who receive givosiran are expected to experience future attacks following treatment discontinuation unless treatment discontinuation occurs at menopause for women. Additionally, the sponsor assumed that the discontinuation rate (approximately 2.5% per cycle) for givosiran over 18 months of trial data could be extrapolated for the remainder of the time horizon (up to 30 years). However, it is unlikely that the discontinuation rate observed in the trial would hold over the course of a lifetime time horizon. A greater rate of discontinuation is likely to occur during the trial rather than beyond the trial period, given that only patients demonstrating good tolerability and response will remain on treatment. These issues with the implementation of treatment discontinuation in the model likely underestimated the total costs associated with givosiran (i.e., both treatment- and attack-related costs), and overestimated the total QALYs associated with givosiran.
In its base case, CADTH assumed that all patients remained on givosiran for the remainder of the model time horizon. Due to structural constraints, CADTH was unable to incorporate more appropriate transition probabilities for patients who discontinued due.
The impact of menopause on AHP disease severity in the model does not capture the likely disease course: The sponsor assumed that female patients with AHP who achieved the asymptomatic health state before menopause would remain asymptomatic for the remainder of their lifetimes, or until death, while those who continued to experience attacks before menopause onset were assumed to continue to experience attacks post-menopause. At menopause onset, patients on givosiran and in the asymptomatic health state were assumed to discontinue treatment (i.e., did not incur any further costs associated with givosiran) and remain asymptomatic, while patients in the symptomatic or recurrent health states were assumed to remain in their health states and on treatment (i.e., continued to experience attacks and incur costs associated with givosiran after menopause) for the remainder of their lifetimes. All patients receiving BSC were assumed to remain in their respective health states. However, the clinical experts consulted by CADTH noted that the disease is expected to improve at the time of menopause for all female patients with AHP (i.e., severity and frequency of attacks is expected to decrease) regardless of which treatment is administered, given that the likely trigger for the attacks (hormone levels at the time of menstruation) would no longer be present. The sponsor’s assumption likely led to an overestimation of the costs and disutility associated with AHP attacks for patients receiving BSC because the majority of these patients were in the recurrent or severe health states at the time of menopause.
CADTH addressed this limitation by assuming that all female patients with AHP transition to the asymptomatic health state at the onset of menopause.
The implementation of efficacy data from the composite end point in the model is associated with uncertainty and may not align with trial results: Clinical efficacy in the trial was based on the mean AAR, a composite end point that was defined according to the level of intervention required to treat an attack (i.e., requiring hospitalization, urgent health care visit, or IV hemin use at home). As noted in the clinical review, givosiran was associated with a 49% rate reduction in porphyria attacks requiring hospitalization and a 74% rate reduction in attacks requiring an urgent health care visit, respectively, compared to patients who received BSC. The economic model did not consider the potential for a different rate reduction adapted to the location where an attack was treated. The sponsor applied the distribution of attacks requiring hospitalization (82%) or urgent health care visit (18%) regardless of which treatment was received. Given that attacks treated in hospital require greater resources, differences in the relative reduction of attacks requiring hospitalization or an urgent health care visit could lead to differences in the level of health care resources used to treat attacks, particularly due to differences in the amount of IV hemin required for each type of attack. The lack of consideration for the potential differential impact of givosiran treatment based on whether an attack was treated in a hospital or urgent care clinic introduces meaningful uncertainty into the model results and may bias the cost-effectiveness of givosiran.
CADTH was unable to address this limitation.
The vials of hemin required to treat an acute porphyric attack in urgent care did not reflect usage in clinical practice: In the economic model, the sponsor assumed that the amount of hemin required to treat an acute porphyric attack differed based on treatment setting (i.e., 2.5 vials of hemin for an urgent health care visit versus 4 vials for a hospitalization). The number of vials of hemin per attack requiring hospitalization was based on an earlier CADTH report on IV hemin,9 while no source was provided for vials used during urgent health care visits. The clinical experts consulted by CADTH indicated that hemin was administered at approximately 3 mg/kg per day, aligned with the standard dosing schedule noted in the product monograph,9 such that approximately 1 vial of hemin would be required per administration (i.e., per day of treatment) in either setting for an average patient weighing approximately 67 kg. CADTH’s clinical experts also noted that an urgent health care visit would not last more than a day, and therefore, the assumption that a single urgent health care visit required 2.5 vials was an overestimate. Since a greater proportion of patients who received BSC experienced attacks, the sponsor’s base-case assumption biased the total costs in favour of givosiran.
CADTH addressed this limitation by adjusting the number of vials of hemin per urgent health care visit to 1 vial.
The mean duration of an acute porphyric attack assumed in the model is likely overestimated: The sponsor assumed that the mean duration of an acute porphyric attack was 7.29 days, based on the EXPLORE study.7 However, in the ENVISION trial, the mean duration of an acute porphyric attack was 5.73 days for patients in the placebo arm and 5.45 days for patients who received givosiran. CADTH’s clinical experts indicated that the average duration of an attack in the ENVISION trial aligned with their expectations. Because more patients receiving BSC experienced attacks, the longer length of stay increased the costs and disutilities associated with acute porphyric attacks for these patients, biasing the results in favour of givosiran.
CADTH addressed this limitation by changing the mean duration of an acute porphyric attack to that observed in the ENVISION trial for patients in the placebo arm (5.73). In a scenario analysis, CADTH explored the impact of a higher mean duration of an acute porphyric attack (7.29 days), as assumed in the sponsor’s base case.
The proportion of patients assumed to have an opioid addiction in the recurrent and severe health states was not appropriate: The sponsor assumed that 82% of patients with AHP in the recurrent and severe health states had an opioid addiction. This was based on a study in the literature by Neeleman et al. (2018),3 but the assumption was inappropriate, according to CADTH’s clinical experts. Expert feedback indicated that the inclusion of opioid addiction within the model implies inappropriate use of opioids among patients with AHP. Such patients experience chronic pain that requires ongoing use of opioids, but the clinical experts indicated that they would not classify such patients as addicted. Additionally, the sponsor applied costs associated with opioid dependence to this proportion of patients based on a study by Wall et al. (2000)10 that examined the societal costs of untreated opioid dependence in Canada. However, societal costs are not relevant to the public health care payer perspective. The inclusion of these costs in the sponsor’s base case overestimated the total costs associated with BSC and biased the results in favour of givosiran because a greater proportion of patients who received BSC were in the recurrent and severe health states and were assumed to have an opioid addiction.
CADTH addressed this limitation by setting the proportion of patients with an opioid addiction in the recurrent and severe health states to 0%.
The patient characteristics in the modelled population were not representative of the patient population expected to be treated in practice or in the pivotal trial, informing the cost-effectiveness analysis: In the economic model, the mean age (41 years) and the proportion of females in the modelled cohort (61%) did not reflect the characteristics of patients in the ENVISION trial. CADTH’s clinical experts affirmed that the patient characteristics in the ENVISION trial aligned with their expectations of patient characteristics in clinical practice (i.e., a mean age of 38 years and 89% female). This had a limited impact on the sponsor’s base case, but it did have an impact when considering the assumptions about treatment discontinuation related to menopause.
CADTH addressed this limitation by revising patient characteristics (i.e., initial age, and the proportion of females) of the modelled cohort to reflect the characteristics of the patient population in the ENVISION trial.
The caregiver impacts incorporated into the base case were not appropriate for the public payer perspective: Utility impacts on caregivers were applied within the sponsor’s base case to all AHP disease severity health states. The CADTH submission requirements indicate that the target population in the base-case analysis must reflect the Health Canada–indicated population, which does not include caregivers. The inclusion of caregiver disutilities led to an overestimate of the benefit associated with givosiran in the sponsor’s base case because it increased the QALY gains received by patients in the form of caregiver benefits.
CADTH addressed this limitation by removing caregiver disutilities in the base case and considered their inclusion as part of a scenario that also included costs from the societal perspective.
The model’s deterministic and probabilistic results did not align: As noted in the summary of the sponsor’s base-case results, the deterministic and probabilistic base-case results were not aligned. Although both sets of results indicated that givosiran was dominant, the disaggregate results showed that the total costs for givosiran differed by approximately $1.74 million in the sponsor’s base case, while the total costs for BSC were similar. Additionally, discrepancies between total QALYs, in addition to costs, were identified when assessing the CADTH base-case results both probabilistically and deterministically. The sponsor’s model did not provide disaggregate costs or QALYs probabilistically, which made it difficult to determine the source of the misalignment between probabilistic and deterministic results, and CADTH was unable to identify the source of the discrepancy. As a result, CADTH presented all analyses deterministically, and the impact of uncertainty on the estimated costs and outcomes could not be assessed in the CADTH base case.
CADTH addressed this limitation by presenting deterministic reanalyses. The probabilistic base case is presented for comparison.
Additional limitations were identified but were not considered to be key limitations.
The relative dose intensity used to adjust the total dose of givosiran may be slightly underestimated: The sponsor claimed to adjust the total dose of givosiran by the relative dose intensity (0.99) for patients in the ENVISION trial. This assumes that drug costs would reflect the expected amount of drug received by patients. However, the relative dose intensity could not be verified in the clinical study reports for the ENVISION trial. CADTH adjusted the relative dose intensity to reflect patients receiving a full dose of givosiran, administered based on patient weight, as noted in the product monograph.11
The CADTH base-case reanalysis applied the conservative assumption that patients would receive a full dose (100%).
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Assumes that acute attacks have no effect on mortality. | Appropriate according to the clinical experts consulted by CADTH. Further, mortality was not reported as an efficacy outcome in the ENVISION trial. |
Mortality hazard ratio of 1.3 vs. the general population, was assumed to be the same for patients despite disease severity health state. | Uncertain; however, unlikely to affect the model results. |
Assumes givosiran has no survival benefit over BSC. | Appropriate because there is no evidence of survival benefit. |
Menopause assumed to occur at the age of 51 years among female patients with AHP. | Appropriate. |
Prevalence of chronic conditions among a proportion of patients in any health state and the prevalence of chronic conditions for patients in the severe state were assumed to be equal to those in the recurrent state. | Appropriate according to the clinical experts consulted by CADTH. |
The length of stay of hospitalization to treat acute attacks was assumed to be equal to the average duration of an attack used in the model and obtained from the natural history study, EXPLORE (i.e., 7.3 days). | Appropriate according to the clinical experts consulted by CADTH because it is likely that patients would be hospitalized for the entire duration of an attack. While the sponsor assumed an average duration of attack that was higher than that expected by the clinical experts, it is plausible that some attacks may last longer than others. In a scenario analysis, CADTH explored the impact of an attack lasting an average of 7.3 days. |
The sponsor assumed the standard error of the mean was 10% of the mean value for most parameters in the model. | Not appropriate. This selection was arbitrary, with no appropriate justification provided. This approach to defining probability distributions is inappropriate because parameters with low sensitivity (i.e., those that are least responsible for driving the model’s results) and higher uncertainty may affect the model’s output more than parameters that have high sensitivity (i.e., are most responsible for driving the model’s results) but are estimated with greater precision. |
AHP = acute hepatic porphyria; BSC = best supportive care; vs. = versus.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook the reanalyses outlined in Table 5 to address, where possible, limitations in the sponsor’s submitted economic model. CADTH was unable to address the limitations related to the sponsor’s definition of AHP disease severity in conceptualizing the model and uncertainty in the long-term efficacy of givosiran. Additionally, CADTH was unable to address the impact of parameter uncertainty, due to issues with the sponsor’s probabilistic analysis. All analyses presented are deterministic.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Severe health state | The recurrent and severe health states were mutually exclusive. The transition matrices reflected movement between 4 AHP health states (asymptomatic, symptomatic, recurrent, and severe). | The severe health state was removed. The number of patients in the severe health state was added to the number of patients in the recurrent health state at baseline. The transition matrices were updated to reflect movement between the asymptomatic, symptomatic, and recurrent health states only. |
2. Health state utility value decrements associated with chronic conditions | Different utility decrements were applied to each health state: Asymptomatic: –0.173 Symptomatic: –0.418 Recurrent: –0.555 Severe: –0.555 | The same utility decrement was applied to all health states, based on the asymptomatic state: Asymptomatic: –0.173 Symptomatic: –0.173 Recurrent: –0.173 Severe: –0.173 |
3. Time on treatment with givosiran | The per-cycle discontinuation rate beyond 18 months was based on extrapolated time-on-treatment data from the ENVISION trial. | All patients remain on treatment from 18 months until the remainder of their lifetime. |
4. Impact of menopause on disease natural history | No female patients with AHP experiencing attacks in the symptomatic and recurrent health states become asymptomatic at the onset of menopause. | All female patients with AHP experiencing attacks in the symptomatic and recurrent health states become asymptomatic at the onset of menopause. |
5. Vials of hemin required to treat an acute porphyric attack for a single urgent health care visit | 2.5 vials of hemin | 1 vial of hemin |
6. Mean duration of attack | 7.29 days | 5.73 days |
7. Proportion of patients with an opioid addiction in the recurrent and severe health states | 82% | 0% |
8. Baseline patient characteristics (mean age and proportion of females) | Mean age: 41 years Proportion of females: 61% | Mean age: 38 years Proportion of females: 89% |
9. Caregiver disutilities | Included | Excluded |
10. Relative dose intensity | 99% | 100% |
CADTH base case | Reanalyses 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8 + 9 | |
AHP = acute hepatic porphyria; BSC = best supportive care.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or standard errors in probabilistic analyses) that are not identified as limitations.
The results for the stepwise analyses can be found in Table 6. Results from the deterministic CADTH base case found that givosiran was associated with incremental costs of $6,205,467 and an incremental benefit of 0.35 QALYs compared with BSC over the lifetime time horizon. The ICER for givosiran versus BSC was $17,928,198 per QALY gained. Approximately 0.71% of the incremental benefit was from the period for which there were observed data (i.e., 6 months).
The results of the CADTH base case differed substantially from those of the sponsor’s submitted base case (i.e., givosiran dominant) when accounting for clinical uncertainties, such as the appropriateness of a severe health state, assumptions related to treatment discontinuation and the impact of menopause on AHP, and the long-term QALY benefits associated with givosiran due to impacts of AHP-related chronic conditions. Individually, each stepped analysis resulted in small changes to the sponsor’s results; however, when accounting for all aspects of clinical uncertainty, the ICER increased substantively, given the cost of treatment and high clinical uncertainty. The number of attacks experienced by patients receiving BSC, the long-term QALY benefits associated with givosiran due to the impacts of AHP-related chronic conditions, and the drug acquisition costs of givosiran were the key drivers of the analysis.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base casea | BSCa | 17,987,732 | 5.81 | Reference |
Givosiran | 9,329,088 | 19.04 | Dominant | |
CADTH reanalysis 1 (removal of severe state) | BSCa | 10,151,600 | 6.86 | Reference |
Givosiran | 8,491,871 | 19.38 | Dominant | |
CADTH reanalysis 2 (health state utility value decrements associated with chronic conditions) | BSCa | 17,987,732 | 14.63 | Reference |
Givosiran | 9,329,088 | 19.64 | Dominant | |
CADTH reanalysis 3 (time on treatment) | BSCa | 17,987,732 | 5.81 | Reference |
Givosiran | 13,670,851 | 19.05 | Dominant | |
CADTH reanalysis 4 (impact of menopause on natural history of disease) | BSCa | 10,404,887 | 11.76 | Reference |
Givosiran | 9,329,064 | 19.04 | Dominant | |
CADTH reanalysis 5 (vials of hemin) | BSCa | 16,989,049 | 5.81 | Reference |
Givosiran | 9,257,032 | 19.04 | Dominant | |
CADTH reanalysis 6 (mean duration of attack) | BSCa | 17,291,970 | 6.21 | Reference |
Givosiran | 9,278,888 | 19.07 | Dominant | |
CADTH reanalysis 7 (proportion of patients with opioid addiction) | BSCa | 17,625,529 | 5.81 | Reference |
Givosiran | 9,310,092 | 19.04 | Dominant | |
CADTH reanalysis 8 (patient characteristics) | BSCa | 19,278,740 | 6.23 | Reference |
Givosiran | 8,766,086 | 20.43 | Dominant | |
CADTH reanalysis 9 (caregiver disutilities) | BSCa | 17,987,732 | 9.32 | Reference |
Givosiran | 9,329,088 | 19.31 | Dominant | |
CADTH reanalysis 10 (relative dose intensity) | BSCa | 17,987,732 | 5.81 | Reference |
Givosiran | 9,410,536 | 19.04 | Dominant | |
CADTH base case (reanalyses 1 through 10) | BSCa | 4,212,515 | 21.12 | Reference |
Givosiran | 10,417,981 | 21.46 | 17,928,198 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aReference product is the least costly alternative.
Scenario Analysis Results
CADTH undertook a series of price reduction analyses of the price of givosiran based on the CADTH base-case reanalyses. In the sponsor’s base case, givosiran remains dominant at all price reductions. In the CADTH base case, givosiran is cost-effective at a willingness-to-pay threshold of $50,000 per QALY with a 63% price reduction. When considering a scenario that includes the impacts of AHP-related chronic conditions on quality of life, as per the sponsor’s base-case assumption, a 61% price reduction is still required for givosiran to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY. When considering a population of patients who experience recurrent attacks only at baseline, a price reduction of 57% is required for givosiran to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
Table 7: CADTH Price Reduction Analyses (Deterministic)
Analysis | ICERs for givosiran vs. BSC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor’s base case | CADTH reanalysis |
No price reduction | Dominant | 17,928,198 |
10% | Dominant | 15,058,721 |
20% | Dominant | 12,189,243 |
30% | Dominant | 9,319,766 |
40% | Dominant | 6,450,288 |
50% | Dominant | 3,580,811 |
60% | Dominant | 711,334 |
63% | Dominant | Dominant |
70% | Dominant | Dominant |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CADTH also undertook several scenario analyses of the deterministic CADTH base case to determine the impact of alternative assumptions on the cost-effectiveness of givosiran versus BSC. These included the following:
changing the mean AAR in the recurrent health state to 6
applying the alternative assumption that all patients are classified as having recurrent AHP (i.e., 100% of patients are in the recurrent health state)
applying the alternative assumption that 80% of attacks requiring hospitalization or an urgent health care visit require treatment with hemin
assuming that the mean duration of an attack is 7.29 days and including a similar length of stay for attacks treated in hospital
adopting the societal perspective to incorporate the financial burden borne by patients as a result of the costs of patients’ and caregivers’ lost productivity due to the frequency of attacks and the presence of chronic conditions, as well as the quality-of-life impacts on caregivers
applying health state utility decrements associated with the occurrence of chronic conditions associated with AHP, as per the sponsor’s base-case assumption
changing the number of vials of hemin per urgent health care visit to 3
assuming that no patients experienced disease improvement at menopause.
The results of these analyses are presented in Appendix 4, Table 11. The results were most sensitive to a lower mean AAR for recurrent AHP (i.e., mean AAR = 6) and the impact of chronic conditions on HRQoL; however, the latter scenario relies on assumptions that are associated with significant uncertainty.
Issues for Consideration
Place in therapy: The expert panel consulted by CADTH noted that givosiran is most likely to be prescribed to patients who experience recurrent attacks. The cost-effectiveness of givosiran differs according to the baseline distribution of patients by AHP severity; the ICER is $14,211,820 when only patients with recurrent disease at baseline are treated. The overall conclusions do not change, but the magnitude of the price reduction required for givosiran to be cost-effective may be lower.
Off-label hemin use: Prophylactic hemin may be used in some settings. However, IV hemin is predominantly administered to patients with AHP in Canada as a rescue medication, with the aim of stopping an attack for those whose attacks require hospitalization or urgent health care. The cost-effectiveness of givosiran compared with prophylactic hemin for the prevention of AHP-related attacks is unknown.
Overall Conclusions
In adult patients with AIP, a clinically meaningful reduction in the rate of acute porphyric attacks was demonstrated with givosiran compared to placebo in the ENVISION trial. The findings are generalizable to all patients with AHP, based on the primary composite end point, which included attacks requiring hospitalization, urgent health care visit, or IV hemin administration. Comparative evidence on the rate of attacks between treatment arms was available for 6 months, and an additional 12 months of data for givosiran were available based on an OLE period, which showed a maintained treatment effect. However, the clinical efficacy and safety of givosiran beyond 18 months is unknown. There was also no evidence available on the impact of givosiran on chronic conditions or on any reductions in complications of AHP. The CADTH clinical review further noted several limitations with the evaluation of HRQoL data that prevented the interpretation of whether there was an added benefit in this outcome measure from treatment with givosiran compared with placebo in the trial.
CADTH identified several major limitations with the submitted economic evaluation beyond those related to the clinical evidence. These included issues related to the sponsor’s conceptualization of the model and inappropriate capture of AHP disease severity; uncertainty associated with the health state utility impacts of AHP-related chronic conditions; inappropriate assumptions around treatment discontinuation for patients who received givosiran; inappropriate assumptions related to the impact of menopause on patients with symptomatic or recurrent disease; and assumptions regarding the duration of an attack and amount of IV hemin required per urgent health care visit. Additionally, the sponsor’s probabilistic analysis was not aligned with the deterministic results. Due to the discrepancies, CADTH presented deterministic analyses. Parameter uncertainty within the sponsor’s model could not be fully explored.
The CADTH base-case reanalysis included removing the severe health state; revising health state utility values to reflect a similar quality-of-life impact of AHP-related chronic conditions for all patients; assuming all patients remained on treatment from the end of 18 months until death; assuming that all female patients with AHP who experienced attacks in the symptomatic and recurrent health states before menopause onset became asymptomatic at the onset of menopause; requiring only 1 vial of hemin for a single urgent health care visit; and changing the mean duration of an acute attack, among other changes. In the CADTH base case, givosiran was associated with an ICER of $17,928,198 per QALY gained (incremental costs of $6,205,467 and an incremental benefit of 0.35 QALYs) compared with BSC. Under the CADTH base-case assumptions, a price reduction of at least 63% would be required for givosiran to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
The cost-effectiveness of givosiran was driven primarily by drug acquisition costs, assumptions that affected the mean number of attacks for patients receiving BSC, and costs associated with acute porphyric attacks that require IV hemin. CADTH was unable to address limitations related to the sponsor’s conceptualization of the economic model, uncertainties with the long-term efficacy of givosiran, or issues around the implementation of the clinical evidence based on a composite end point. As a result, the ICER and subsequent price reduction are likely to be underestimated.
References
1.Pharmacoeconomic Evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Givlaari (givosiran injection) [internal sponsor's package]. Mississauga (ON): Alnylam Pharmaceuticals Canada ULC. 2021 Feb 22.
2.Pharmacoeconomic evaluation. In: CDR submission: Vonvendi (von Willebrand factor), lyophilized powder for solution 650 and 1300 IU VWF:RCo / vial intravenous injection [CONFIDENTIAL sponsor's submission]. Toronto (ON): Shire Pharma Canada ULC; 2020 May.
3.Neeleman RA, Wagenmakers M, Koole-Lesuis RH, et al. Medical and financial burden of acute intermittent porphyria. Journal of inherited metabolic disease. 2018;41(5):809-817. PubMed
4.Clinical study report: Primary Analysis for ALN-AS1-003 (Givosiran) ENVISION: A Phase 3 Randomized, Doubleblind, Placebo-Controlled, Multicenter Study with an Open-label Extension to Evaluate the Efficacy and Safety of Givosiran in Patients with Acute Hepatic Porphyrias [internal sponsor's report]. Cambridge (MA): Alnylam Pharmaceuticals, Inc.; 2019 May 16.
5.Roblin S, Meninger S, Murray S, et al. A166 Analysis of symptoms, diagnostic patterns, and Canadian provider perspective of acute hepatic porphyria. J Can Assoc Gastroenterol. 2020;3(Suppl 1):31-32.
6.Baravelli CM, Aarsand AK, Sandberg S, Tollånes MC. Sick leave, disability, and mortality in acute hepatic porphyria: a nationwide cohort study. Orphanet journal of rare diseases. 2020;15(1):56. PubMed
7.Gouya L, Ventura P, Balwani M, et al. EXPLORE: A Prospective, Multinational, Natural History Study of Patients with Acute Hepatic Porphyria with Recurrent Attacks. Hepatology. 2020;71(5):1546-1558. PubMed
8.Acaster S, Perard R, Chauhan D, Lloyd AJ. A forgotten aspect of the NICE reference case: an observational study of the health related quality of life impact on caregivers of people with multiple sclerosis. BMC Health Serv Res. 2013;13:346. PubMed
9.CADTH Technology Review Hemin for Injection (Panhematin): Budget Impact Analysis. Ottawa: CADTH; 2019: https://www.cadth.ca/sites/default/files/hta-he/ob0006_porphyria-report-redacted-final.pdf. Accessed 2021 May 28.
10.Wall R, Rehm J, Fischer B, et al. Social costs of untreated opioid dependence. J Urban Health. 2000;77(4):688-722. PubMed
11.Givosiran Injection solution, 189 mg/mL givosiran (as givosiran sodium), subcutaneous injection [product monograph]. Amsterdam: Alnylam Netherlands B.V.; 2020 Oct 8.
12.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Givlaari (givosiran injection) [internal sponsor's package]. Mississauga (ON): Alnylam Pharmaceuticals Canada ULC. 2021 Feb 22. 2021 Feb.
13.Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. Journal of inherited metabolic disease. 2013;36(5):849-857. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Acute Hepatic Porphyria
Treatment | Strength | Form | Price ($) | Recommended dosage | 28-day cycle cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Givosiran (Givlaari) | 189 mg/mL | 1 mL single-use vial | 64,454.2979 a | 2.5 mg/kg monthly | 64,454 | 773,448b |
Note: CADTH calculations assume a patient weight of approximately 67 kg based on the ENVISION trial.
aSponsor-submitted price.
bAssumed 12 doses per year based on monthly administration.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | See key limitations section. |
Model structure is adequate for decision problem | No | See key limitations section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | See key limitations section and key assumptions table. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | See key limitations section. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results (Deterministic)
Health state | Givosiran | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 32.03 | 32.03 | 0 |
Asymptomatic | 30.74 | 20.04 | 10.70 |
Symptomatic | 0.68 | 1.61 | −0.9 |
Recurrent | 0.62 | 10.38 | −9.80 |
Severe | 0.00 | 0.00 | 0.00 |
Discounted QALYs | |||
Total | 21.46 | 21.12 | 0.35 |
Asymptomatic | 20.58 | 13.16 | 7.42 |
Symptomatic | 0.47 | 1.11 | −0.64 |
Recurrent | 0.41 | 6.85 | −6.44 |
Severe | 0 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 10,417,981 | 4,212,515 | 6,205,467 |
Acquisition | 9,932,089 | 0 | 9,932,089 |
Administration | 3,985 | 0 | 3,985 |
Chronic Symptoms | 133,715 | 289,798 | −156,084 |
Attacks | 347,551 | 3,922,115 | −3,574,564 |
Adverse events | 63 | 23 | 40 |
End of life | 579 | 579 | 0 |
ICER ($/QALY) | 17,928,198 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
Scenario Analyses
Table 11: Scenario Analyses for Givosiran Versus BSC (Deterministic)
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Mean annualized attack rate in the recurrent AHP health state is 6 | BSCa | 2,653,892 | 21.27 | Ref. |
Givosiran | 10,299,894 | 21.47 | 37,001,517 | |
All patients have recurrent AHP | BSCa | 4,854,339 | 21.06 | Ref. |
Givosiran | 10,506,023 | 21.45 | 14,211,820 | |
80% of attacks require hospitalization or an urgent health care visit | BSCa | 3,562,744 | 21.12 | Ref. |
Givosiran | 10,360,403 | 21.46 | 19,639,100 | |
Mean duration of attack is 7.29 days | BSCa | 4,389,054 | 21.01 | Ref. |
Givosiran | 10,433,625 | 21.45 | 13,726,340 | |
Societal perspective (including caregiver disutilities) | BSCa | 4,212,515 | 19.53 | Ref. |
Givosiran | 10,380,277 | 21.30 | 3,473,786 | |
Utility impact of chronic diseases included | BSCa | 4,212,515 | 16.89 | Ref. |
Givosiran | 10,417,981 | 21.06 | 1,487,023 | |
Mean annualized attack rate in the recurrent AHP health state is 6 and all patients have recurrent AHP | BSCa | 3,027,886 | 21.23 | Ref. |
Givosiran | 10,349,866 | 21.47 | 31,033,462 | |
Changing the number of vials of hemin per urgent health care visit to 3 | BSCa | 4,550,546 | 21.12 | Ref. |
Givosiran | 10,447,935 | 21.46 | 17,038,133 | |
No patients improve at menopause (i.e., 0%) | BSCa | 8,973,028 | 19.97 | Ref. |
Givosiran | 22,150,277 | 20.75 | 16,803,730 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference.
aReference product is least costly alternative.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 12: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor assessed the budget impact of the introduction of givosiran for patients with AHP, from the perspective of the public health care payer in the Canadian setting (excluding Quebec) over a 3-year time horizon. The sponsor included drug acquisition and administration costs, in addition to costs associated with acute attacks and treatment with IV hemin. In the reference scenario, the sponsor assumed that patients would be eligible to receive BSC, which was associated with no costs. In the new drug scenario, givosiran was assumed to displace market shares of BSC.12
The sponsor estimated the eligible population size using an epidemiological approach, by leveraging data from multiple sources in the literature and assumptions based on clinical expert input.12 Key inputs to the BIA are documented in Table 13.12
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Annual population growth | 0.59% |
Prevalence of AHP patients | 15.13 per million |
Percentage of AHP patients diagnosed | ||% |
Incidence of AHP | 0.12 per million |
Proportion of patients requiring hospitalization | 81.82% |
Number of patients eligible for drug under review | 106 / 106 / 107 |
Market uptake (3 years) | |
Uptake (reference scenario) Best supportive care | ||% / ||% / ||% |
Uptake (new drug scenario) Givosiran BSC | | % / | % / | % | % / | % / | % |
Cost of treatment (per patient) | |
Cost of treatment, per year Givosirana BSC | $696,385.73 $0 |
Cost per attack, per year Givosiran BSC Cost of hemin treatment Givosiranb BSC | $7,670 $0 $31,481 $0 |
BSC = best supportive care.
aAssumes treatment adherence rate of | % for givosiran.
bAssumes 2.5 vials per urgent health care visit and 4 vials per hospitalization.
Source: Sponsor’s submitted budget impact analysis report.12
Summary of the Sponsor’s BIA Results
The sponsor presented a base case under the broader health care system perspective that included drug acquisition and administration costs, as well as costs associated with the treatment of acute attacks which included IV hemin treatment costs. Results of the sponsor’s base-case analysis under the health care system perspective revealed that the introduction of givosiran in patients with AHP would result in an incremental budget impact of $14,726,100 in Year 1, $22,288,151 in Year 2, and $32,238,218 in Year 3, for a total budget impact of $69,252,468 over the 3-year time horizon.
According to CADTH submission requirements, the sponsor’s base case should have reflected the public drug program perspective, which is to include only drug acquisition costs, with the broader health care payer perspective assessed in a scenario analysis. As such, the 3-year budget impact when considering only the cost of pharmacologic treatment with givosiran and excluding the cost of acute attacks from the public drug payer perspective under the sponsor’s base-case assumptions was $121,171,117.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the estimated eligible population size: The sponsor undertook an epidemiological approach to estimate the size of the population eligible for givosiran. This required assessing the published literature13 to derive estimates for the prevalence and incidence of AHP, however, Canadian-specific values could not be identified. The sponsor further used internal data to derive the rate of diagnosis for AHP patients, a value which could not be appraised by CADTH. The clinical experts consulted by CADTH indicated that the prevalence and incidence of AHP has not been well-established in Canada and while European literature may be a good source for the epidemiology of AHP, they noted that the estimate of the target population derived from the sponsor’s assumptions and inputs may be associated with some uncertainty.
CADTH could not address this limitation due to the absence of more appropriate data. A scenario analysis was conducted increasing the rate of diagnosis to 30%, to determine the impact of increasing the eligible population size on the potential budget impact.
The uptake of givosiran in the new drug scenario is likely underestimated: The sponsor anticipated that givosiran would capture |%, |% and |% of the market share distribution in Years 1, 2, and 3. The clinical experts consulted by CADTH described that givosiran’s place in therapy is intended for patients with recurrent AHP and that the sponsor’s anticipated uptake is likely underestimated over the 3-year time horizon when compared with the baseline distribution of patients in the pharmacoeconomic model. To align the proportion of patients with recurrent AHP in the BIA with the baseline distribution of AHP disease severity in the CADTH pharmacoeconomic analysis, CADTH revised the market share uptake of givosiran across Years 1, 2, and 3 to 73%.
CADTH addressed this limitation by revising the market share uptake of givosiran in the new drug scenario to 73% across Years 1, 2, and 3. A scenario analysis was conducted using the sponsor’s base-case assumption.
Proportion of patients assumed to adhere to treatment uncertain: The sponsor adjusted drug acquisition costs of givosiran by an assumed adherence rate of |%. It is expected that patients with AHP will fully adhere to treatment (i.e., 100%) to ensure they prevent acute porphyric attacks, and there is limited evidence in support of the rate assumed by the sponsor. This assumption likely led to an underestimation of the budget impact associated with givosiran.
CADTH addressed this limitation by revising the treatment adherence rate to 100%.
CADTH Reanalyses of the BIA
A table noting the changes made to the sponsor’s BIA as part of the CADTH reanalysis is available in Table 16. All analyses are from the public drug plan perspective, unless otherwise noted.
Table 14: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correction to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH base casea | ||
1. Market share estimates in the new drug scenario (across Years 1 to 3) | | % / | % / | % | 73% / 73% / 73% |
2. Treatment adherence | | % | 100% |
CADTH base case | Reanalyses 1 + 2 | |
aChanges to derive the CADTH base case under the drug program plan perspective.
The sponsor’s 3-year costs under the drug plan perspective were $121,171,117. Applying the changes in Table 15 resulted in an increase in budget impact under the drug plan perspective to $181,761,126 over 3 years. The results of the CADTH stepwise reanalyses are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16.
Table 15: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case – health care payer perspective | $69,252,468 |
Submitted base case - drug plan perspective | $121,171,117 |
CADTH reanalysis 1 – drug plan perspective | $163,650,646 |
CADTH reanalysis 2 – drug plan perspective | $128,444,479 |
CADTH base case – drug plan perspective | $181,761,126 |
CADTH conducted a scenario analysis from the broader public health care payer perspective (Scenario 1 - Table 17) including the above changes made to the sponsor’s base case from the drug plan perspective, along with the following additional changes specific to the health care payer perspective to align with the CADTH pharmacoeconomic base case:
a. revising the amount of IV hemin used for treatment of an urgent health care visit to 1 vial per urgent health care visit.
b. setting the proportion of patients with severe AHP to 0% and changing the proportion of patients with symptomatic and recurrent AHP to 27% and 73%, respectively.
CADTH conducted the following additional scenario analyses from the drug plan perspective (Scenarios 2 to 4, Table 17):
2. Assumed an increase in the rate of diagnosis among AHP patients to 30%.
3. Assumed the sponsor’s market share estimates in the new drug scenario, across the 3-year time horizon: | % / | % / | %.
4. Applied a 63% reduction in the price of givosiran to align with the point at which the ICER is within the willingness-to-pay threshold of $50,000 per QALY in the CADTH pharmacoeconomic base case.
The scenario analysis from the broader health care payer perspective resulted in an increase from the sponsor’s submitted base case from $69,252,468 over 3 years to $129,996,431. Assuming an increase in the diagnosis rate of AHP from ||||% to 30% led to an increase in the expected budget impact up to $235,129,286 over 3 years.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Sponsor-submitted base case (health care system perspective) | Reference | $42,338,134 | $42,741,354 | $42,741,354 | $43,144,575 | $128,627,283 |
New drug | $42,338,134 | $57,467,454 | $65,029,505 | $75,382,792 | $197,879,751 | |
Budget impact | $0 | $14,726,100 | $22,288,151 | $32,238,218 | $69,252,468 | |
Sponsor’s base case (drug plan program perspective) | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $25,766,272 | $38,997,601 | $56,407,244 | $121,171,117 | |
Budget impact | $0 | $25,766,272 | $38,997,601 | $56,407,244 | $121,171,117 | |
CADTH base case (drug plan perspective) | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $60,329,225 | $60,329,225 | $61,102,676 | $181,761,126 | |
Budget impact | $0 | $60,329,225 | $60,329,225 | $61,102,676 | $181,761,126 |
BIA = budget impact analysis.
Table 17: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1 | Reference | $31,255,279 | $31,552,948 | $31,552,948 | $31,850,617 | $94,956,514 |
New drug | $31,255,279 | $74,700,700 | $74,700,700 | $75,551,545 | $224,952,945 | |
Budget impact | $0 | $43,147,752 | $43,147,752 | $43,700,928 | $129,996,431 | |
CADTH scenario analysis 2 | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $78,118,612 | $78,118,612 | $78,892,063 | $235,129,286 | |
Budget impact | $0 | $78,118,612 | $78,118,612 | $78,892,063 | $235,129,286 | |
CADTH scenario analysis 3 | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $29,391,161 | $44,086,741 | $63,423,031 | $136,900,933 | |
Budget impact | $0 | $29,391,161 | $44,086,741 | $63,423,031 | $136,900,933 | |
CADTH scenario analysis 4 | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $22,321,813 | $22,321,813 | $22,607,990 | $67,251,617 | |
Budget impact | $0 | $22,321,813 | $22,321,813 | $22,607,990 | $67,251,617 |
Note: All scenario analyses are conducted based on the CADTH base case undertaken from the drug program plan perspective.
BIA = budget impact analysis.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.