CADTH Reimbursement Review
Luspatercept (Reblozyl)
Sponsor: Celgene Inc., a Bristol Myers Squibb company
Therapeutic area: Myelodysplastic syndromes-associated anemia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AAMAC
Aplastic Anemia and Myelodysplasia Association of Canada
AML
acute myeloid leukemia
ATB-MPG
Alberta Tumour Board Myeloid Physicians Group
ATG
anti-thymocyte globulin
CBC
complete blood count
CI
confidence interval
CSA
cyclosporine
ECOG
Eastern Cooperative Oncology Group
DAC
Drug Advisory Committee
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ESA
erythropoiesis-stimulating agent
FAB
French-American-British
FACT-G
Functional Assessment of Cancer Therapy–General
G-CSF
granulocyte colony-stimulating factor
HMA
hypomethylating agent
HRQoL
health-related quality of life
IPSS
International Prognostic Scoring System
IPSS-R
Revised International Prognostic Scoring System
ICT
iron chelation therapy
ITT
intention to treat
IWG
International Working Group
KM
Kaplan-Meier
LLSC
Leukemia & Lymphoma Society of Canada
LS
least squares
MDS
myelodysplastic syndromes
mHI-E
modified hematologic improvement–erythroid
MID
minimal important difference
MPN
myeloproliferative neoplasms
QoL-E
quality of life questionnaire for patients with myelodysplastic syndromes
RBC
red blood cell
RBC-TI
red blood cell–transfusion independence
SD
standard deviation
SE
standard error
SOC
system organ class
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Luspatercept (Reblozyl), 25 mg per vial, 75 mg per vial, powder for solution for SC injection |
Indication | Treatment of adult patients with transfusion-dependent anemia requiring at least 2 RBC units over 8 weeks resulting from very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 11, 2021 |
Sponsor | Celgene Inc., a Bristol Myers Squibb company |
NOC = Notice of Compliance; MDS = myelodysplastic syndromes; RBC = red blood cell; SC = subcutaneous.
Introduction
The myelodysplastic syndromes (MDS) encompass a spectrum of hematopoietic stem cell malignancies that are characterized by ineffective hematopoiesis and a propensity to evolve to acute myeloid leukemia (AML). They are clinically recognized as cytopenia(s) and dysplasia(s) in at least 1 major myeloid lineage with no other attributable causes. Optimal evaluation of patients involves integration of morphologic (e.g., according to WHO criteria), cytogenetic, and molecular characterization to facilitate diagnosis and prognostic stratification via the Revised International Prognostic Scoring System (IPSS-R) for MDS. Ring sideroblasts are erythroid precursors in which, after Prussian blue staining (Perls reaction), a minimum of 5 siderotic granules cover at least a third of the nuclear circumference.1
A diagnostic evaluation of MDS in a patient with unexplained persistent cytopenia(s) requires a bone marrow biopsy and aspiration to detect dysplasia and assess marrow cellularity. Cytogenetic testing is a standard of care and there is often a need to exclude other causes of cytopenias. Anemia is the most common cytopenia observed, and frequently associated symptoms are fatigue, weakness, exercise intolerance, angina, and cognitive impairment. The initial evaluation of anemia in MDS seeks to identify alternative etiologies, such as iron deficiency, nutrient deficiencies, hypothyroidism, renal disease, or gastrointestinal bleeding.2-4
Therapeutic approaches for MDS include those directed at ameliorating the underlying bone marrow disease or managing the resulting cytopenias. These options include growth factors such as erythropoiesis-stimulating agents (ESAs) or granulocyte colony-stimulating factor (G-CSF), hypomethylating agents (HMAs) such as azacitidine, immunosuppression or immunomodulation (e.g., lenalidomide), chemotherapy, and allogeneic hematopoietic stem cell transplantation, which is the only current curative option. Many patients with MDS, particularly those with lower-risk disease, are managed with supportive care alone, including red blood cell (RBC) transfusion, often for months to years.
Use of growth factors, as with RBC transfusions and adjunctive iron chelation, can be considered a form of supportive therapy, whereas medications such as lenalidomide, HMAs, cyclosporine (CSA), anti-thymocyte globulin (ATG), and stem cell transplantation are better classified as disease-modifying therapies. Available guidelines differ in their preferences for each of these agents, but all are based on patient scores on the International Prognostic Scoring System (IPSS) or IPSS-R and do not contain specific recommendations for the management of patients with refractory anemia with ring sideroblasts.5 In Canada, the most relevant treatment guideline for very low- to intermediate-risk MDS is the 2018 publication Systemic therapy for the treatment of adult patients with lower-risk myelodysplastic syndromes.6
In 1 of the few Canadian studies, the Calgary metropolitan area had a total incidence rate of 2.60 MDS cases per 100,000 person-years, corresponding to an age-standardized incidence of 3.69 for Canada. The study period was from January 1, 2011, to December 31, 2015. The male-to-female sex ratio was 1.35, and the median age at diagnosis was 75 years. With these results, 1,295 new annual cases of MDS were predicted in Canada.7
The objective of this report is to perform a systematic review of the beneficial and harmful effects of luspatercept (25 mg per vial or 75 mg per vial) powder for solution for subcutaneous injection for the treatment of RBC transfusion-dependent anemia associated with very low- to intermediate-risk MDS in adult patients with ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy. The recommended starting dose of luspatercept is 1 mg per kilogram of body weight up to a maximum of 1.75 mg/kg administered by a subcutaneous injection every 3 weeks.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from 2 clinical experts and 2 clinician groups consulted by CADTH for the purpose of this review.
Patient Input
One joint submission from 2 patient groups, the Leukemia and Lymphoma Society of Canada (LLSC) and the Aplastic Anemia and Myelodysplasia Association of Canada (AAMAC), was received in response to CADTH’s call for patient input. The LLSC is a national organization with a mission to cure leukemia, lymphoma, Hodgkin disease, and myeloma, and improve the quality of life of Canadians affected by all 137 different types of blood cancer. The AAMAC is a national organization with a mission of providing a seamless support network for every Canadian patient, family member, friend, and concerned health care provider dealing with aplastic anemia, myelodysplasia, and paroxysmal nocturnal hemoglobinuria.
The LLSC created an online survey to gather input from patients on the treatments for MDS and luspatercept, if applicable. The online survey was available in French and English via Survey Monkey and was open to respondents from December 7, 2020, to January 4, 2021. It was promoted by the LLSC and an organization devoted to supporting those with myeloproliferative neoplasms (MPNs), the Canadian MPN Network, through social media channels and directly by email. A total of 20 respondents completed the survey, including 18 who identified as patients, 1 who identified as a caregiver, and 1 who identified as a friend or family member answering on behalf of a patient with MDS.
According to the patient input received for this review, 17 respondents identified symptoms of MDS affecting quality of life, with fatigue and infections mentioned repeatedly, as well as the transfusion schedule. Transfusion schedules were mentioned as affecting quality of life, with 1 patient stating, “I have weekly transfusions and my life revolves around that.”
Respondents to the survey identified several frontline treatments they received for MDS after their diagnosis. These included blood transfusions, chemotherapy, drug therapy, stem cell or bone marrow transplant, blood cell growth factor therapy, watch-and-wait approach, ATG therapy, and immunoglobulin therapy. Respondents reported both positive and negative experiences with these therapies. The survey asked participants which factors are the most important to consider when making decisions about a new cancer treatment. The most common response was the possible impact on disease. Other factors to consider cited by participants included physician recommendation, quality of life, outpatient treatment, and closeness of home.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts stated that no funded or approved treatments are available to address key outcomes for patients with transfusion-dependent anemia associated with MDS. They added that not all patients respond to or tolerate these treatments, even if they are obtained (privately or through a compassionate access program). The only therapeutic intervention for the treatment of lower-risk MDS that has demonstrated an association with improvement in overall survival is iron chelation therapy (ICT). Of the disease-modifying therapies used for low-risk MDS, lenalidomide has been shown to improve health-related quality of life (HRQoL) in patients both with and without the del(5q) cytogenetic abnormality. However, lenalidomide has been associated with causing significant neutropenia or thrombocytopenia.
The clinical experts anticipated that luspatercept would be used as a second-line treatment following ESA failures or a first-line treatment in patients not expected to respond to ESAs. The clinical experts noted that therapies that increase hemoglobin and decrease RBC-transfusion dependence cannot be assumed to improve patient symptoms or HRQoL, particularly when those therapies themselves can have adverse effects.
The clinical experts noted that luspatercept has only been studied in low-risk MDS patients with ringed sideroblasts who have failed ESA therapy, and there is no evidence that it is in fact superior to ESA therapy in this setting. To be a preferred treatment for symptomatic anemia, luspatercept would either need to establish superiority through a direct comparison with ESAs (i.e., via a randomized controlled trial), or establish a stronger evidence base (through direct comparison with a control) that it can directly improve a patient-related outcome such as HRQoL. The clinical experts consulted by CADTH were of the opinion that patients with low-risk IPSS scores and ringed sideroblasts are the most likely to respond to therapy with luspatercept. The patients who require regular RBC transfusions are the ones most in need of this intervention as transfusion dependency is associated with shorter overall survival, more cardiac events, and inferior HRQoL. The clinical experts further noted that patients who are most likely to exhibit a response to treatment with luspatercept would be identified by their IPSS score, endogenous erythropoietin level, and monthly transfusion needs. A variety of scoring systems are available for this purpose. The clinical experts noted that a clinically meaningful response to treatment would be an improvement in HRQoL using a validated scoring system (e.g., Functional Assessment of Cancer Therapy–General [FACT-G] or EuroQol 5-Dimensions questionnaire). They also noted that a reduction in or elimination of transfusions would be clinically meaningful.
The clinical experts were of 2 opinions regarding the timing of assessments. One expert expressed that, because luspatercept is administered as a subcutaneous injection every 3 weeks, reviewing quality of life and/or a complete blood count (CBC) at each visit would be an appropriate interval. Transfusion independence may be evaluated every 8 weeks (with a review conducted at the 9-week visit). The second expert expressed that treatment responses should be assessed every month for 6 months and then every 3 months.
The clinical experts agreed that disease progression, intolerable adverse events not responding to dose reduction, and failure to achieve a response criterion after 9 weeks despite dose escalation to 1.75 mg/kg could be reasonably interpreted as a lack of meaningful response and treatment would be discontinued.
The clinical experts noted that, while many patients will likely receive their first subcutaneous injection in a medical setting and it would be administered by a health care professional (on either an inpatient or outpatient basis) as per the product monograph,8 the majority should be able to self-administer in a community setting. They added that diagnosis of low-grade MDS requires a specialist consultation, and the ability to prescribe luspatercept should therefore be restricted to individuals with special training in managing the diagnosis (typically a hematologist or oncologist), although once initiated it would be reasonable for non-specialists to continue prescribing and monitoring.
Clinician Group Input
Clinician input on the review of luspatercept for the treatment of adult patients with very low- to intermediate-risk MDS-associated anemia who have ring sideroblasts and require RBC transfusions was received from 2 groups: the Ontario Health (Cancer Care Ontario) Hematology Disease Site Drug Advisory Committee (DAC) and the Alberta Tumour Board Myeloid Physicians Group (ATB-MPG).
Both groups agreed that the current treatment for patients involves support with RBC transfusions and ESAs. The clinicians from Alberta noted that ESAs are most effective in patients with low transfusion requirements and erythropoietin levels and are funded to various degrees across the country. They added that there is currently no funding for ESAs in Alberta, although they are commonly used and considered standard of care, and that erythropoietin is recommended in Alberta clinical practice guidelines for patients with lower-risk MDS. Both groups agreed that the 10% of MDS patients with del(5q) mutations may be treated with lenalidomide. With respect to needs that are not being met with the currently available treatments, both clinician groups agreed that no other treatment options are currently available, other than transfusion, ESAs for some patients, and, for a small subset of patients, HMAs such as azacitidine or a combination of decitabine and cedazuridine.
Both clinician groups agreed that luspatercept would be an additional line of therapy for patients with symptomatic anemia who have progressed on ESAs, have not responded to ESAs, or have a high erythropoietin level that precludes a response to ESA therapy to reduce transfusions and their consequences (i.e., iron overload). The groups agreed that patients best suited for treatment with luspatercept are lower-risk MDS patients with symptomatic anemia who have failed ESAs or for whom ESA therapy is inappropriate. The clinicians from Alberta added that patients in this group have no other effective treatment options other than long-term transfusions and ICT to help manage the related iron overload associated with the side effects of chelation.
Both clinician groups agreed that transfusion frequency (reduction in transfusion requirements) and improvement in hemoglobin levels are outcomes used in clinical practice to determine whether a patient is responding to treatment. Both groups also agreed that a clinically meaningful response to treatment would be a reduction in transfusions.
With respect to factors that should be considered when deciding to discontinue treatment, the clinicians from Ontario noted that worsening of MDS, progression to a higher-risk category, or transformation to AML should be considered. The clinicians from Alberta stated that a decrease in hemoglobin without an alternative cause, an increase in transfusion requirements, or a need to introduce regular transfusions in patients who have been transfusion-independent would be factors to consider.
According to both clinician groups, the most appropriate settings for treatment are community settings such as pharmacies, outpatient clinics, and specialty clinics. The clinicians from Alberta added that a hematology or medical oncology specialist would be required to diagnose, treat, and monitor patients who might receive the drug under review. The clinicians from Alberta noted that the benefit to patients who can become transfusion-independent (or remain so after developing symptomatic anemia) is significant and can reduce the burden both to patients and the health care institutions that provide regular transfusion support over extended periods to these patients.
Drug Program Input
The drug plans stated that ESA treatment should be considered before funding. They also noted that the trial was limited to patients who had failed a prior course of ESA therapy. However, they added that it would be reasonable to initiate treatment directly with luspatercept in patients predicted to have less than a 25% chance of responding to ESA therapy (based on the Nordic or similar prognostic scoring system). The drug plans had questions regarding the appropriate place in therapy for luspatercept, and whether previous treatment with ESAs should be required. The plans requested information as to when treatment with luspatercept should be discontinued. The plans also sought the clinical experts’ opinions regarding administration of luspatercept, specifically around monitoring hemoglobin levels and ensuring equal access.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
One pivotal trial (MEDALIST; N = 229) was included in the CADTH systematic review. MEDALIST is an ongoing phase III, randomized, double-blind, placebo-controlled study of the efficacy and safety of luspatercept in adult patients for the treatment of RBC transfusion-dependent anemia associated with very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy. The MEDALIST trial was conducted at 65 sites globally. Four sites in Canada enrolled 14 patients.
Eligible patients were randomized (2:1) to receive either luspatercept or placebo along with best supportive care. The randomized double-blind phase of the study was divided into a 24-week primary treatment phase, a week-25 assessment phase, and a 24-week extension phase. Patients received a starting dose of 1 mg of the study drug per kilogram of body weight administered by subcutaneous injection every 3 weeks. During the treatment period the dose levels were titrated (increased) stepwise to a maximum of 1.75 mg/kg or reduced based on a clinical response. The maximum total dose per administration was not to exceed 168 mg. Randomization was stratified based on RBC transfusion burden at baseline (≥ 6 units over 8 weeks versus < 6 units over 8 weeks) and IPSS-R score at baseline (very low or low versus intermediate).
For patients to continue the double-blind treatment beyond the first 24 calendar weeks, the following criteria had to be confirmed by the investigator at the week 25 visit: evidence of clinical benefit (e.g., decrease in RBC transfusion requirement compared with baseline requirement or hemoglobin increase compared with baseline) and absence of disease progression according to criteria established by the MDS International Working Group (IWG) for altering the natural history of MDS. Based on the outcome of the week-25 MDS disease assessment visit, patients were either discontinued from treatment and entered the post-treatment follow-up period or continued the double-blind treatment with the same study drug in the extension phase of the treatment period. As of the May 8, 2018, data cut-off date, 128 (83.7%) and 68 (89.5%) of the patients had completed 24 weeks of treatment in the luspatercept and placebo treatment groups, respectively. In addition, 78 (51%) and 12 (15.8%) of the patients had completed 48 weeks of treatment in the luspatercept and placebo treatment groups, respectively.
The primary outcome of the study was the proportion of patients treated with luspatercept versus placebo who achieved red blood cell–transfusion independence (RBC-TI) for at least 8 weeks (any consecutive 56-day period) from week 1 to week 24. The measure upon which the 2 key secondary outcomes was based was the proportion of patients who achieved RBC-TI for at least 12 weeks (any consecutive 84-day period) from week 1 to week 48 and the proportion of patients who achieve RBC-TI for at least 12 weeks (any consecutive 84-day period) from week 1 to week 24.
Overall, the baseline characteristics of the patients enrolled in the MEDALIST study were well balanced. Approximately 2-thirds of the patients in the MEDALIST study were male and White. The mean weight was 76.2 kg and 77.4 kg in the luspatercept and placebo treatment groups, respectively. The mean age of the patients was 70.5 (standard deviation [SD] = 8.68) and 70.7 (SD = 10.88) in the luspatercept and placebo treatment groups, respectively. Of the patients in the luspatercept and placebo treatment groups, 94.8% and 97.4%, respectively, were classified as having refractory cytopenia with multilineage dysplasia, according to the WHO classification. In the luspatercept and placebo treatment groups, 71.2% and 75% of patients, respectively, were classified as at low risk according to the IPSS-R; 59.5% of the patients in the luspatercept treatment group and 42.1% of the patients in the placebo treatment group had an Eastern Cooperative Oncology Group (ECOG) performance status of 1; and 5.2% of the patients in the luspatercept treatment group and 14.5% of the patients in the placebo treatment group had an ECOG performance status of 2.
Efficacy Results
In the MEDALIST study, the efficacy outcomes identified in the protocol were hematologic response, HRQoL, overall survival, iron accumulation, ICT use, progression to AML, and health care resource utilization. The primary and 2 key secondary efficacy outcomes were analyzed using an intention-to-treat (ITT) population.
At week 24, a greater proportion of patients in the luspatercept treatment group (37.9%) achieved the primary outcome of RBC-TI for at least 8 weeks (any consecutive 56-day period) compared with the placebo group (13.16%), with a common risk difference in the response rate of 24.56 (95% confidence interval [CI], 14.48 to 34.64). The odds ratio of 5.06 (95% CI, 2.28 to 11.26; P < 0.0001) favoured luspatercept treatment over placebo. However, according to the clinical experts consulted by CADTH, the results were not clinically meaningful as 8 weeks is too short a duration to assess response.
At week 48 and week 24, a greater proportion of patients in the luspatercept treatment group achieved the 2 key secondary outcomes of RBC-TI for at least 12 weeks (any consecutive 84-day period) compared with the placebo group. From week 1 to week 48, in the luspatercept treatment group 33.3% of the patients responded to the treatment, and in the placebo group 11.84% of the patients responded to the treatment, with a common risk difference in the response rate of 21.37 (95% CI, 11.23 to 31.51). The odds ratio of 4.04 (95% CI, 1.83 to 8.96; P = 0.0003) favoured the luspatercept treatment over placebo. From week 1 to week 24, in the luspatercept treatment group 28.1% of the patients responded to the treatment and in placebo group 7.89% of the patients responded to the treatment, with a common risk difference in the response rate of 20.0 (95% CI, 10.92 to 29.08). The odds ratio of 5.07 (95% CI, 2.00 to 12.84; P = 0.0002) favoured luspatercept treatment over placebo.
Other efficacy outcomes identified in the CADTH review protocol were reported descriptively, including the number of RBC units transfused, duration of RBC-TI, time to RBC-TI, mean change in hemoglobin, modified hematologic improvement (mHI-E), overall survival, iron accumulation (through serum ferritin levels), ICT use, progression to AML, and health care resource utilization. In the absence of any formal statistical testing, whether luspatercept had an effect on any of these outcomes remains unknown. The HRQoL was a secondary and exploratory outcome in the MEDALIST study and was measured using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-30) and quality of life questionnaire for patients with myelodysplastic syndromes (QoL-E) instruments; however, none of these outcomes were controlled for multiplicity. For HRQoL outcomes, no difference in the treatment groups was observed and no minimal important difference (MID) for patients with transfusion-dependent anemia associated with MDS was identified from literature.
Subgroup analyses identified in the CADTH review protocol for which results were available in the MEDALIST study included IPSS-R scores (very low risk or low risk versus intermediate risk), and baseline hematological status. The results of the subgroup analysis aligned with the results of the full study population.
Harms Results
In the MEDALIST trial, 98.0% and 92.1% of the patients in the luspatercept and placebo groups, respectively, reported at least 1 adverse event. The most commonly occurring adverse events were fatigue (26.8% and 13.2% of the patients in luspatercept and placebo group, respectively), diarrhea (22.2% and 9.2%, respectively), nausea (20.3% and 7.9%, respectively), and dizziness (19.6% and 5.3%, respectively).
Serious adverse events were reported by 31.4% of patients in the luspatercept treatment group and 30.3% of patients in the placebo group. The most common serious adverse event was pneumonia, which was reported by 2% of the patients in the luspatercept group and 2.6% of the patients in the placebo group. The proportion of patients who stopped treatment due to an adverse event was 8.5% and 7.9% in the luspatercept and placebo treatment groups, respectively. The most common reason for stopping treatment was benign, malignant, and unspecified neoplasms (including cysts and polyps).
During the treatment period, 3.3% of patients (n = 5) in the luspatercept treatment group and 5.3% of patients (n = 4) in the placebo treatment group died. In the luspatercept treatment group 1 patient died due to multiple organ dysfunction syndrome, 2 patients died of sepsis, 1 patient died due to renal failure, and 1 patient died of hemorrhagic shock. In the placebo treatment group, 2 patients died due to general disorders and administration site conditions, 1 patient died of sepsis, and 1 patient died of respiratory failure. In the post-treatment period, an additional 4.6% of the patients (n = 7) in the luspatercept treatment group and 6.6% of the patients (n = 5) in the placebo treatment group died.
Notable harms identified in the CADTH review protocol included thromboembolic events; hypertension, hepatic, and renal events; hypersensitivity reactions; and malignancies. In the luspatercept treatment group 2.6% of patients (n = 4) and in the placebo treatment group 3.9% of patients (n = 3) experienced a thromboembolic or thrombophlebitis event. Under the system organ class (SOC) of hepatobiliary disorders, 5.2% of patients in the luspatercept treatment group and 5.3% of patients in the placebo group reported at least 1 associated adverse event. Under the SOC of renal and urinary disorders, 18.3% of patients in the luspatercept treatment group and 13.2% of patients in the placebo group reported at least 1 associated adverse event. Hypertension was reported as an adverse event in 8.5% of patients in the luspatercept treatment group and 7.9% of patients in the placebo group.
Table 2: Summary of Key Results from Pivotal and Protocol-Selected Studies
MEDALIST study results | Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) |
|---|---|---|
RBC-TI of 8 weeks or more during week 1 through week 24 (ITT population) | ||
Number of responders, n (%) | 58 (37.9) | 10 (13.2) |
Common risk difference on response rate, % (95% CI) | 24.56 (14.48 to 34.64) | |
Odds ratio (95% CI)a | 5.06 (2.28 to 11.26) | |
P value | < 0.0001 | |
RBC-TI of 12 weeks or more during week 1 through week 48 (ITT population) | ||
Number of responders, n (%) | 51 (33.3) | 9 (11.84) |
Common risk difference on response rate, % (95% CI) | 21.37 (11.23 to 31.51) | |
Odds ratio (95% CI)a | 4.045 (1.827 to 8.956) | |
P value | 0.0003 | |
RBC-TI of 12 weeks or more during week 1 through week 24 (ITT population) | ||
Number of responders, n (%) | 43 (28.10) | 6 (7.89) |
Common risk difference on response rate, % (95% CI) | 20.00 (10.92 to 29.08) | |
Odds ratio (95% CI)a | 5.07 (2.00 to 12.84) | |
P value | 0.0002 | |
Harms, n (%) (safety population) | ||
AEs | 150 (98.0) | 70 (92.1) |
SAEs | 48 (31.4) | 23 (30.3) |
WDAE (from study treatment) | 13 (8.5) | 6 (7.9) |
Deaths (during treatment period) | 5 (3.3) | 4 (5.3) |
Notable harms (all grades, reported in at least 5% of the patients in either treatment group) (safety population) | ||
Fatigue, n (%) | 41 (26.8) | 10 (13.2) |
Diarrhea, n (%) | 34 (22.2) | 7 (9.2) |
Asthenia, n (%) | 31 (20.3) | 9 (11.8) |
Nausea, n (%) | 31 (20.3) | 6 (7.9) |
Dizziness, n (%) | 30 (19.6) | 4 (5.3) |
Back pain, n (%) | 29 (19.0) | 5 (6.6) |
Cough, n (%) | 27 (17.6) | 10 (13.2) |
Edema peripheral, n (%) | 25 (16.3) | 13 (17.1) |
Headache, n (%) | 24 (15.7) | 5 (6.6) |
Hypertension, n (%) | 13 (8.5) | 6 (7.9) |
Fall, n (%) | 15 (9.8) | 9 (11.8) |
Neutropenia, n (%) | 7 (4.6) | 7 (9.2) |
Notable harms (grade 3 or higher, reported in at least 5% of the patients in either treatment group) (safety population) | ||
Patient with at least 1 TEAE, n (%) | 65 (42.5) | 34 (44.7) |
Anemia, n (%) | 10 (6.5) | 5 (6.6) |
Hypertension, n (%) | 5 (3.3) | 3 (3.9) |
Iron overload, n (%) | 3 (2.0) | 1 (1.3) |
AE = adverse event; BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
Note: Patients who discontinued from the study before week 48 without achieving at least 56 consecutive days (8 weeks) of RBC-TI were counted as nonresponders.
aCochran-Mantel-Haenszel test stratified for average baseline RBC transfusion requirement, and baseline Revised IPSS score.
Source: Clinical Study Report for MEDALIST.9
Critical Appraisal
The MEDALIST study was a randomized, placebo-controlled, double-blind study. Overall randomization (using an interactive response technology system) and treatment allocation, as stratified by transfusion burden at baseline (≥ 6 units of RBCs over 8 weeks versus < 6 units over 8 weeks) and IPSS-R score at baseline (very low or low versus intermediate) were conducted appropriately. However, as noted by the FDA, blinding in the study may have been inadequate due to the production of the placebo control syringe on site and the lack of specific instructions to mask the product, increasing the risk of accidental unblinding unacceptably,10 which may have introduced bias in the results.
The baseline patient, disease and MDS treatment-history characteristics were generally well balanced. A higher number of patients in the luspatercept treatment group experienced transformation to AML, nervous system disorders, and fatigue leading to study drug discontinuation.
The clinical experts consulted by CADTH were of the opinion that the duration of hematologic response of the primary end point, i.e., at least 8 weeks (any consecutive 56 days), was not clinically meaningful and the appropriate measure for clinical meaningfulness would be for patients to be transfusion-independent for at least 16 weeks, which is in accordance with the proposed IWG 2018 hematological response criteria. A hematologic response of transfusion independence for 12 weeks (any consecutive 84 days) is more clinically meaningful compared with 8 weeks. The effect size of the primary end point of transfusion independence for 8 weeks in the study was small, with a transfusion independence of 8 weeks being obtained in only about 38% of patients experiencing a differential response compared about 25% in the placebo group. Only about 1-quarter of the patients exposed to luspatercept had any apparent benefit, assuming that fulfillment of the primary objective represents a benefit to the patient.10
Only a subset of patients who initially responded in the first 24 weeks were eligible for inclusion in the extension phase. The interpretation of this end point is therefore problematic as few patients were eligible for the extension phase and therefore could not achieve the end point of 12 weeks of response due to the study design.
The clinical experts noted that, based on baseline demographic and disease characteristics, the study population was representative of Canadian patients with transfusion-dependent anemia associated with MDS. In Canada the mean age of an MDS patient is 74 years, which is similar to that of the study population, which was 70.5 years.
Conclusions
One phase III randomized controlled trial (MEDALIST; N = 229) was included in the CADTH systematic review of luspatercept for adult patients with transfusion-dependent anemia associated with MDS. The study demonstrated that treatment with luspatercept was superior to placebo in terms of achieving transfusion independence for at least 8 weeks (any consecutive 56 days) from week 1 through week 24. Further, luspatercept was superior to placebo in achieving transfusion independence for at least 12 weeks (any consecutive 84 days) from week 1 through week 48 and week 1 through week 24. Results of the primary end point were not deemed clinically meaningful by the clinical experts consulted by CADTH, and results of the 48-week secondary end point were difficult to interpret due to study design. The other end points of the study that were evaluated were HRQoL, overall survival, progression to AML, iron accumulation, ICT use, and health care resource utilization. However, none of these outcomes were controlled for multiplicity and, due to limitations associated with statistical methodology, the effect of luspatercept on these outcomes is currently unknown. During the trial, the median overall survival had not been achieved. Key evidence gaps include the short duration of transfusion independence for the primary outcome of 8 weeks, study design, and no improvement in HRQoL.
Key safety issues with luspatercept include the rate of occurrence of thromboembolic events, which was lower in the luspatercept treatment arm compared to the placebo group. A higher number of patients in the luspatercept treatment group experienced fatigue, diarrhea, asthenia, nausea, and dizziness.
Introduction
Disease Background
Myelodysplastic syndromes encompass a spectrum of hematopoietic stem cell malignancies that are characterized by ineffective hematopoiesis and a propensity to evolve to AML.11 They are clinically recognized as cytopenia(s) and dysplasia(s) in at least 1 major myeloid lineage with no other attributable causes.7 Optimal evaluation of patients involves integration of morphologic (e.g., according to WHO criteria), cytogenetic, and molecular characterization to facilitate diagnosis and prognostic stratification via the IPSS-R for MDS.12 Ring sideroblasts are erythroid precursors in which, after Prussian blue staining (Perls reaction), a minimum of 5 siderotic granules cover at least a third of the nuclear circumference. The detection of bone marrow ring sideroblasts can be seen in a variety of clonal hematological and non-clonal disorders. Clonal conditions associated with ring sideroblasts include myeloid neoplasms, synonymous with the presence of bone marrow ring sideroblasts, which includes refractory anemia with ring sideroblasts, classified under MDS with ring sideroblasts, and refractory anemia with ring sideroblasts with thrombocytosis, called MDS or MPN with ring sideroblasts and thrombocytosis.1
The presentation of disease is heterogeneous, but patients often manifest with symptoms related to cytopenias such as fatigue, infections, or hemorrhagic complications. A diagnostic evaluation of MDS in a patient with unexplained persistent cytopenia(s) requires a bone marrow biopsy and aspiration to detect dysplasia and assess marrow cellularity. Well-established diagnostic tools for MDS with widespread availability are peripheral and differential blood counts, cytomorphology of peripheral blood and bone marrow smears, and cytogenetics of bone marrow cells. Cytogenetic testing is a standard of care and there is often a need to exclude other causes of cytopenias. Anemia is the most common cytopenia observed, and frequently associated symptoms are fatigue, weakness, exercise intolerance, angina, or cognitive impairment. The various risk identification and classification tools categorize disease risk based on cytogenetic abnormalities, the degree of cytopenias, and the percentage of bone marrow blasts. The initial evaluation of anemia in MDS seeks to identify alternative etiologies, such as iron deficiency, nutrient deficiencies, hypothyroidism, renal disease, or gastrointestinal bleeding.2-4,13
In 1 of the few Canadian studies, the Calgary metropolitan area had a total incidence rate of 2.60 MDS cases per 100,000 person-years, corresponding to an age-standardized incidence of 3.69 for Canada. The study period was from January 1, 2011, to December 31, 2015. The male-to-female sex ratio was 1.35, and the median age at diagnosis was 75 years. With these results, 1,295 new annual cases of MDS were predicted in Canada.7
Standards of Therapy
The presence of ringed sideroblasts, which constitute a histologic subgroup in the French-American-British (FAB) classification system of MDS and in the WHO system that replaced it, has been found to convey a lower likelihood of response to ESA therapy. This was noted by the clinical experts consulted by CADTH. However, because ringed sideroblasts do not themselves appear to carry prognostic significance within the WHO classification system,1 they are not acknowledged in either the IPSS or the IPSS-R currently used to stage MDS.
Therapeutic approaches for MDS include those directed at ameliorating the underlying bone marrow disease or managing the resulting cytopenias. These options include growth factors such as ESAs or G-CSF, HMAs such as azacitidine, immunosuppression or immunomodulation (e.g., lenalidomide), chemotherapy, and allogeneic hematopoietic stem cell transplantation, which is the only current curative option. Many patients with MDS, particularly those with lower-risk disease, are managed with supportive care alone, including transfusion, often for months to years. Transfusions of RBCs are given primarily to prevent serious complications of both acute and chronic anemia. Transfusions are also used to manage the broader consequences of bone marrow failure, including fatigue and other symptoms related to anemia, to improve patient HRQoL.14 Symptomatic anemia, which is the most frequent cytopenia exhibited by patients with lower-risk MDS, may be reduced via RBC transfusions or ESAs. However, chronic RBC transfusions are associated with fluctuating levels of hemoglobin, iron overload, and dependence on hospitals and caregivers.12
Use of growth factors, as with RBC transfusions and adjunctive iron chelation, can be considered a form of supportive therapy, whereas medications such as lenalidomide, HMAs, CSA, ATG, and stem cell transplantation are generally classified as disease-modifying therapies. Available guidelines differ in their preferences for each of these agents,5 but all are based on patient IPSS or IPSS-R scores and do not contain specific recommendations for the management of patients with refractory anemia with ringed sideroblasts. In Canada, the most relevant treatment guideline for very low- to intermediate-risk MDS is the 2018 publication Systemic therapy for the treatment of adult patients with lower-risk myelodysplastic syndromes.6 The scope of this guideline is patients 18 years of age or older with an IPSS score of no more than 1 or an IPSS-R score of no more than 3.5. In such patients with a hemoglobin level of less than 100 g/L and symptoms of anemia, first-line therapy is an ESA with or without G-CSF, with the addition of ICT in patients with signs of transfusional iron overload. The only exception to this recommendation is patients with del(5q) syndrome who are receiving 2 or more units of RBCs per month and have a serum erythropoietin level greater than 500 U/L. In these patients, lenalidomide is considered first-line therapy. For all other patients, failure to respond to ESA with or without G-CSF should be managed by a trial of ATG and CSA, if the patient is 65 years of age or younger, and with azacytidine in all others.
Drug
Luspatercept (Reblozyl) is a recombinant fusion protein of 2 identical chains, each consisting of a modified form of the extracellular domain of human activin receptor type IIB linked to the human immunoglobulin G1 Fc domain, that binds select endogenous transforming growth factor beta superfamily ligands to inhibit Smad2/3 signalling.8
Luspatercept is indicated for the treatment of adult patients with transfusion-dependent anemia requiring at least 2 units of RBCs over 8 weeks resulting from very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy.8 Luspatercept was granted a standard review by Health Canada and received a Notice of Compliance on February 11, 2021.
The sponsor’s reimbursement request is as per the indication under review.
Luspatercept is a lyophilized powder for reconstitution available in 2 strengths: 25 mg per vial and 75 mg per vial. The Health Canada–recommended starting dose is 1 mg/kg every 3 weeks by subcutaneous injection. Prior to each administration hemoglobin levels need to be assessed and reviewed. If an RBC transfusion occurred before dosing, the pre-transfusion hemoglobin needs to be considered for dosing purposes. If the pre-dose hemoglobin is greater than or equal to 115 g/L and the hemoglobin level is not influenced by a recent transfusion, dosing should be delayed until hemoglobin is less than or equal to 110 g/L. The Health Canada–recommended dose adjustments are summarized in Table 3. Based on the Health Canada product monograph, luspatercept should be discontinued if a patient does not achieve a response after 9 weeks of treatment (administration of 3 doses) at the maximum dose level if no other causes are found, or if unacceptable toxicity occurs at any time. Luspatercept should be reconstituted and administered by a health care professional.8 Table 4 presents the key characteristics of luspatercept.
Table 3: Recommended Dose Titration, Dose Modifications, and Treatment Discontinuation of Luspatercept
Parameters | Luspatercept dosing recommendation |
|---|---|
Insufficient response | |
Not RBC transfusion–free after at least 2 consecutive doses (6 weeks) at the 1 mg/kg starting dose | Increase dose to 1.33 mg/kg every 3 weeks |
Not RBC transfusion–free after at least 2 consecutive doses (6 weeks) at 1.33 mg/kg | Increase dose to 1.75 mg/kg every 3 weeks |
No reduction in RBC transfusion burden after at least 3 consecutive doses (9 weeks) at 1.75 mg/kg | Discontinue luspatercept |
Pre-dose hemoglobin ≥ 115 g/L or rapid hemoglobin rise | |
Pre-dose hemoglobin is ≥ 115 g/L in the absence of transfusions | Delay dose and restart only when hemoglobin is ≤ 110 g/L |
Increase in hemoglobin > 20 g/L within 3 weeks in the absence of transfusion and
|
|
Adverse events | |
Any grade 2 adverse reaction | Delay dose until resolved to ≤ grade 1 |
Grade 3 or 4 hypersensitivity reactions | Discontinue luspatercept |
Grade 3 or 4 leukocytosis (> 100,000 WBC/μL) or hematologic malignancy is suspected |
|
Other grade 3 or 4 adverse reactions | Delay dose until resolved to ≤ grade 1 |
RBC = red blood cell; WBC = white blood cell.
Note: Grades as per National Cancer Institute Common Terminology Criteria for Adverse Events or, when not defined, grade 1 is mild, grade 2 is moderate, grade 3 is severe, and grade 4 is life-threatening.
Source: Product monograph for Reblozyl.8
Table 4: Key Characteristics of Luspatercept
Characteristics | Luspatercept |
|---|---|
Mechanism of action | Luspatercept is a r-Fc protein of 2 identical chains, each consisting of a modified form of the extracellular domain of human activin receptor type IIB linked to the human immunoglobulin G1 Fc domain, that binds select endogenous transforming growth factor beta superfamily ligands to inhibit Smad2/3 signalling |
Indication under reviewa | Treatment of adult patients with transfusion-dependent anemia requiring at least 2 units of RBCs over 8 weeks resulting from very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy |
Route of administration | Subcutaneous injection |
Recommended dose | Recommended starting dose of 1.0 mg/kg, maximum dose 1.75 mg/kg |
Serious adverse effects or safety Issues |
|
Other |
|
MDS = myelodysplastic syndromes; RBC = red blood cell; r-Fc = recombinant fusion protein.
aHealth Canada–approved indication. Reblozyl is also indicated for the treatment of adult patients with RBC transfusion-dependent anemia associated with beta-thalassemia.
Source: Clinical Study Report for MEDALIST.9
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Group and Information Gathered
One joint submission from 2 patient groups, the LLSC and AAMAC, was received in response to CADTH’s call for patient input.
The LLSC is a national organization with a mission of curing leukemia, lymphoma, Hodgkin disease, and myeloma, and improving the quality of life of Canadians affected by all the 137 different types of blood cancer.
The AAMAC is a national organization with a mission of providing a seamless support network for every Canadian patient, family member, friend, and concerned health care provider dealing with aplastic anemia, myelodysplasia, and paroxysmal nocturnal hemoglobinuria.
The LLSC created an online survey to gather input from patients on the treatments for MDS and luspatercept, if applicable. The online survey was available in French and English via Survey Monkey and was open to respondents from December 7, 2020, to January 4, 2021. It was promoted by the LLSC and the Canadian MPN Network through social media channels and directly by email. Twenty respondents completed the survey, including 18 who identified as patients, 1 who identified as a caregiver, and 1 who identified as a friend or family member answering on behalf of a patient with MDS. Thirteen respondents identified as female, 6 identified as male, and 1 respondent did not provide a gender. All respondents were from Canada, with 10 from British Columbia, 4 from Ontario, 3 from Alberta, 2 from Quebec, and 1 from Nova Scotia. The respondents ranged in age between 45 and 84 years, with 1 between the ages 45 and 54, 7 between 55 to 64, 5 between 65 to 74, and 7 between 75 to 84. Respondents were asked to identify the year they were diagnosed with MDS, and this ranged from 2000 to 2020.
Disease Experience
According to the patient input received for this review, 17 respondents identified symptoms of MDS affecting quality of life, with fatigue and infections mentioned repeatedly, as well as the transfusion schedule. One respondent reported no impact. When asked if any aspects or symptoms are easier to control, fatigue was commonly mentioned.
In response to how symptoms of MDS affect their quality of life, 1 patient noted that, “Fatigue prevents me from doing as much as I would like to,” and another patient stated, “I get tired a lot. Also, I have to watch out for infections, e.g.,: not visit grandchildren when they have a cold, etc.” The impact of transfusion schedules was mentioned as an impact on quality of life, with 1 patient stating, “I have weekly transfusions and my life revolves around that.”
Another patient reported:
A lot of fatigue during the day, little energy to do activity, i.e., short periods a day. Numbness in the hands (weak) and Permanent neuropathy in both feet (deep pain, painful numbness and swelling.) Difficulty concentrating and speaking at times, poor memory at times, inattention. Almost impossible to do a simple sporting activity such as walking for more than 30 minutes without a break.
Respondents also noted how the quality of life of family members and friends was affected by their symptoms, with 1 patient stating, “My husband is my caregiver and he spends a lot of time on my appointments and care,” and another stating, “Need support from family to help me do things and drive me to my many appointments/hospital.”
Experiences With Currently Available Treatments
Respondents to the survey identified several frontline treatments they received for MDS after their diagnosis. Eleven received blood transfusions, 9 received chemotherapy, 8 received drug therapy, 6 received a stem cell or bone marrow transplant, 6 received blood cell growth factor therapy, 5 took a watch-and-wait approach, 1 received ATG therapy, and 1 received immunoglobulin therapy. Respondents reported both positive and negative experiences with these therapies. One patient stated, “The drug put my red blood count up so I no longer needed transfusions. Unfortunately I developed neuropathy in my feet, legs and hands, which has impacted by life a lot as I cannot walk as much as I used to.” Another respondent described the treatment effects as “Positive…it is controlling MDS so far. Negative…is not knowing how long the treatments will work for. Injections can be painful, the side effects.”
When respondents were asked to identify the MDS treatment side effects that had a large or extremely large impact on their quality of life, the most commonly cited symptoms were low blood cell counts (10 respondents), extreme fatigue (9 respondents), anemia (7 respondents), and infection (5 respondents). Other symptoms identified by 2 or 3 respondents included graft-versus-host disease, diarrhea, rashes, hair loss, mouth sores, nausea and vomiting, constipation, tingling sensations, and lung, heart, kidney, or nerve problems. These side effects affected respondents’ lives in various ways, and those with a large or extremely large impact included changes to physical activity (9 respondents), anxiety (5 respondents), mental health and overall happiness (4 respondents), and eating challenges (4 respondents). These quality-of-life issues experienced due to MDS diagnosis and treatment were articulated by respondents, with 1 patient stating:
MDS affects all aspects of my life…I was told the treatments would give me a quality of life…it does for 2 weeks out of the monthly treatments…my life has changed completely…COVID-19 has not helped” and another stating “being tired a lot means not able to do any extensive travelling, glad we did a fair amount prior to diagnosis of MDS.
Respondents were asked to identify any challenges accessing treatment for MDS or health care services, and, generally, respondents did not identify issues or challenges accessing treatment. Proximity to treatment and wait times to access specialist care were mentioned. One patient stated, “I have to drive almost 30 minutes to and from the hospital for treatments but this is not a huge challenge.”
Improved Outcomes
The survey asked participants the factors most important to consider when making decisions about a new cancer treatment. The most common response was the possible impact on disease (11 respondents), with 1 participant stating, “An increase in hemoglobin without an initial reduction!” Other factors to consider cited by participants included physician recommendation (9 respondents), quality of life (6 respondents), outpatient treatment (4 respondents), and closeness of home (3 respondents). Respondents also shared the improvements they would like to see in new treatments that are not currently available and 1 respondent stated, “Oral versions or ability to administer at home rather than hospital setting” and another stated, “I would like to see more new treatments become available to cure MDS, but I realise that is likely not possible at the moment. New treatments should not cause neuropathy which does impact quality of life.”
Experience With Drug Under Review
None of the respondents to the survey indicated they have taken luspatercept.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process, providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy. The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of transfusion-dependent anemia associated with very low- to intermediate-risk MDS.
Unmet Needs
The clinical experts stated that no funded or approved treatments are available to address key outcomes for patients with transfusion-dependent anemia associated with MDS. They added that not all patients respond to or tolerate these treatments even if they are obtained (privately or through a compassionate access program). The only therapeutic intervention for the treatment of lower-risk MDS that has been demonstrated to improve overall survival is ICT. The effect of ESAs on HRQoL is unclear. Of the disease-modifying therapies used for low-risk MDS, lenalidomide has been shown to improve HRQoL in patients both with and without the del(5q) cytogenetic abnormality. However, lenalidomide has been associated with neutropenia and thrombocytopenia. Of the HMAs or immunomodulatory drugs in use, a beneficial effect from decitabine has been noted. Evidence that available treatments for low-grade MDS are meeting patient-related outcomes is limited. However, this is primarily due to the poor quality of evidence available for changes in HRQoL.
Place in Therapy
The clinical experts noted that luspatercept would be used as a second-line treatment following ESA failure or a first-line treatment in patients not expected to respond to ESA. However, they added that intolerance to ESA is uncommon. Luspatercept has been shown to effectively increase hemoglobin and decrease transfusion dependence in patients with low-risk MDS and ringed sideroblasts who have failed ESA therapy. As opposed to ESA therapy, which augments the existing dyserythropoiesis of MDS, luspatercept may correct the shortened circulating lifespan of dysplastic erythrocytes. However, as it does not change the actual dysplasia intrinsic to MDS, it should still be considered symptomatic management therapy rather than a disease-modifying drug. The clinical experts noted that therapies that increased hemoglobin and decreased RBC transfusion dependence cannot be assumed to improve patient symptoms or HRQoL, particularly when those therapies themselves can have adverse effects.
Patient Population
The clinical experts noted that luspatercept has only been studied in patients with low-risk MDS with ringed sideroblasts and who have failed ESA therapy, and there is no evidence that it is superior to ESA therapy in this setting. Luspatercept would either need to establish superiority, through a direct comparison with ESAs (i.e., via a randomized controlled trial), or establish a stronger evidence base (through direct comparison with a control) that it can directly improve a patient-related outcome such as HRQoL, to be a preferred treatment for symptomatic anemia. The clinical experts anticipated that patients with low-risk IPSS scores and ringed sideroblasts are the most likely to respond to therapy with luspatercept. It is likely that patients with other forms of MDS (i.e., without ringed sideroblasts) will also show some improvement in their hemoglobin levels, although these populations have not been studied to the same degree. Patients with ringed sideroblasts, very low-, low-, and intermediate-risk disease as measured by the IPSS-R, and who are RBC transfusion-dependent with up to 6 units every 8 weeks are most likely to respond, although even the most heavily transfused may still benefit with respect to reduction in transfusion frequency and number of units transfused. The patients who require regular RBC transfusions are those most in need of this intervention as transfusion dependency is associated with shorter overall survival, more cardiac events, and inferior HRQoL. The clinical experts noted that, while the reported adverse events in the phase III trial of luspatercept did not suggest that the medication should be avoided in patients with certain disease characteristics, a large number of patients were never eligible to enrol due to underlying medical conditions. In patients with such exclusions (e.g., those with renal insufficiency or hepatic injury, a history of cancer or recent thrombotic episode, or ongoing uncontrolled infection), caution with this medication would be advised.
The clinical experts noted that it is not challenging to identify which patients would be best suited for treatment with luspatercept. Any patient with ringed sideroblasts exceeding 5% should be eligible. Anyone with ringed sideroblasts who is dependent on RBC transfusion due to symptomatic anemia should also be eligible. The clinical experts noted that identifying patients who are at risk of failing ESA therapy and eligible for luspatercept would be straightforward if a weighted scoring algorithm such as the Nordic scoring method was used, and it might be reasonable to attempt a trial of luspatercept in these patients without first attempting ESA therapy.
The clinical experts further noted that identification of patients who are most likely to exhibit a response to treatment with luspatercept would be on the basis of their IPSS score, endogenous erythropoietin level, and monthly transfusion needs. A variety of scoring systems are available for this purpose.
The clinical experts noted that a clinically meaningful response to treatment would be an improvement in HRQoL using a validated scoring system (e.g., FACT-G or EuroQol 5-Dimensions). If a change in hemoglobin specifically induced by luspatercept could be validated as a surrogate marker of improved HRQoL, then a change or stabilization of hemoglobin would also be an appropriate metric to monitor treatment response. They also noted that a reduction in or elimination of transfusions would be clinically meaningful.
Assessing Response to Treatment
The clinical experts were of 2 opinions regarding how to assess response to treatment. One expert expressed that, because luspatercept is administered as a subcutaneous injection every 3 weeks, reviewing the quality of life and/or CBC at each visit would be appropriate. Transfusion independence may be evaluated every 8 weeks (with a review conducted at the 9-week visit). The second expert supported monthly assessments of treatment response for 6 months, and then every 3 months.
Discontinuing Treatment
The clinical experts agreed that disease progression, intolerable adverse events not responding to dose reduction, and failure to achieve a response criterion after 9 weeks despite dose escalation to 1.75 mg/kg, could be reasonably interpreted as a lack of meaningful response, and treatment would be discontinued.
Prescribing Conditions
The clinical experts noted that, while many patients will likely receive their first subcutaneous injection in a medical setting and it would be administered by a health care professional (on either an inpatient or outpatient basis) as per the product monograph,8 the majority should be able to self-administer in a community setting. A diagnosis of low-grade MDS requires a specialist consultation (i.e., pathology) and, given the many treatment options available for patients with MDS (including enrolment in clinical trials), the ability to prescribe luspatercept should be reserved for individuals with special training in managing the diagnosis (typically a hematologist or oncologist), although once initiated it would be reasonable for non-specialists to continue prescribing and monitoring.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Clinician input on the review of luspatercept for the treatment of adult patients with very low- to intermediate-risk MDS-associated anemia who have ring sideroblasts and require RBC transfusions was received from 2 groups: the DAC and the ATB-MPG.
The DAC provides evidence-based clinical and health system guidance on drug-related issues, including provincial drug reimbursement programs and the Systemic Treatment Program. The group collected information for this review through discussions at monthly DAC meetings.
The ATB-MPG consists of physicians within the Alberta Hematology Tumour Group who treat myeloid malignancies and acute leukemias (MDS, MPN, AML, and acute lymphoblastic leukemia). They meet provincially every 3 months and provide annual updates to treatment guidelines for Alberta. Input for this review was collected by reviewing data in publications to develop guidelines that are then reviewed in a group setting, modified based on discussion, and approved by the group before publication on its website.
Unmet Needs
Both groups agreed that the current treatment for patients involves transfusion support with RBC transfusions, and ESAs. The clinicians from Alberta noted that ESAs are most effective in patients with low transfusion requirements and erythropoietin levels and are variably funded across the country. They added that funding for ESAs is currently not available in Alberta, although ESA therapy is commonly used and considered standard of care, and erythropoietin is recommended in the Alberta clinical practice guidelines for patients with lower-risk MDS.
The clinicians from Ontario noted that, in cases with low endogenous erythropoietin levels (< 500 U/L), patients will receive ESA injections; however, these patients fail ESAs and become transfusion-dependent again and do not have good options besides disease-modifying therapies (e.g., HMAs or lenalidomide). The clinicians added that some intermediate-risk patients may be treated with azacitidine based on an IPSS score, and that oral azacitidine plus decitabine can potentially be accessed compassionately or self-paid by some patients.
Both groups agreed that patients with the presence of del(5q) MDS (approximately 10% of all MDS patients) may be treated with lenalidomide.
The clinicians from Alberta noted that transfusions for these patients can be lifesaving and improve quality of life; however, the hemoglobin levels of patients can vary dramatically over weeks depending on where patients have recently had a transfusion. Additionally, the clinicians noted that patients will become transfusion-overloaded and many will require chelation therapy, which has associated costs and side effects. The group also noted that the Canadian guidelines recommend chelation for patients who have a life expectancy of at least a year, 20 units of blood, or ferritin levels exceeding 1,000 mcg/L. The clinicians added that ESAs can keep patient hemoglobin levels stable (avoiding large fluctuations) and are well tolerated; however, the response with ESAs is normally lost at a median of about 18 months, and no other treatment options are available. The group commented that current treatments do not affect the underlying disease mechanism or prevent progression of disease, and there is significant evidence that patients with higher transfusion needs have increased mortality. The clinicians noted that this can be related to the differences in disease pathology and increases in ferritin and iron load, which are associated with increased mortality and possible cardiac iron loading, and should be considered for patients with lower-risk disease who have relatively longer median survival times.
Both clinician groups agreed that transfusion independence, reduction in transformation to AML, and improving HRQoL are the most important treatment goals. The clinicians from Alberta added that prolonging life, delaying disease progression, reducing severity of symptoms, reducing burden on caregivers (including bringing patients to frequent and lifelong transfusion support visits) and the health care system and facilities are also important treatment goals. With respect to the latter, the clinicians added that they hope to improve symptoms, reduce the need for frequent lab work (CBCs, type and screens, and iron monitoring) and length of times in infusion chairs receiving blood transfusions at hospitals and cancer centres.
With respect to needs that are not being met with the currently available treatments, both clinician groups agreed that no other treatment options are currently available other than transfusion, ESAs for some patients, and, for a small subset of patients, HMAs such as azacitidine or decitabine plus cedazuridine. The clinicians from Alberta added that not all patients respond to ESAs and patients eventually progress on ESA therapy and become transfusion-dependent again if they have an initial response. An additional treatment option is needed to avoid or reduce RBC transfusions and concomitant iron loading, as well as provide stable hemoglobin levels, which reduces the times of major anemia symptoms as well as visits to health care facilities for transfusions. The clinicians from Alberta also noted that many patients in Alberta live in rural areas where travelling to labs and health facilities for transfusions is difficult, particularly for elderly patients. The Alberta clinicians also noted that, although the current review does not provide additional benefits to patients with lower-risk MDS without ring sideroblasts, patients with ring sideroblasts tend to have the highest transfusion needs and the longest overall survival, and it is therefore important to avoid iron overload.
The clinicians from Ontario noted that azacitidine-ineligible patients, who constitute the majority of patients with a lower-risk (≤ intermediate-1) IPSS score, are those with the greatest unmet needs. According to the clinicians from Alberta, the patients with the greatest unmet need are those who have not responded to ESAs or have lost their response to ESAs, and those who have a higher erythropoietin level and are unlikely to respond to ESAs.
Place in Therapy
Both clinician groups agreed that luspatercept would be an additional line of therapy for symptomatic anemia for patients who have progressed on ESAs, have not responded to ESAs, or have a high erythropoietin level that precludes a response to ESA therapy to reduce transfusion and their consequences (i.e., iron overload). The clinicians from Alberta added that this would be expected to cause a shift in the current treatment paradigm and keep a significant number of people from requiring regular transfusion support at their health care facilities.
With respect to whether patients should try other treatments before initiating treatment with the drug under review, the clinicians from Ontario noted that no other treatments are available other than azacitidine, decitabine plus cedazuridine, or ESAs for eligible patients. The clinicians from Alberta added that the drug indication is for patients who have failed or are not suitable for ESAs, and this is the appropriate order of treatments in clinical practice. The Alberta group added that, for patients with higher erythropoietin levels and more than 2 units of RBC transfusions per month, response rates to ESAs are extremely low, and these patients should be targeted appropriately to receive luspatercept. For patients with fewer than 2 units of RBC transfusions per month and low erythropoietin levels, the Alberta clinicians noted that ESAs have a good response rate and would be an appropriate first-line therapy with luspatercept being available if there is no response or progression.
Both clinician groups agreed that this treatment provides an additional therapeutic option for patients who have failed ESAs to become transfusion-independent. They also noted that this allows for an effective therapy for anemia in MDS. The clinicians from Alberta added that no other treatment options are available for patients who do not respond to luspatercept after either progressing on ESAs or being inappropriate for ESA therapy; therefore, patients who do not respond to luspatercept or who progress would require long-term transfusion support.
Patient Population
Both clinician groups agreed that patients best suited for treatment with luspatercept are lower-risk MDS patients with symptomatic anemia who have failed ESAs or are inappropriate for ESA therapy. These patients have very low, low, and intermediate IPSS-R scores. The clinicians from Ontario added that the forest plot from the primary publication showed all groups benefit, irrespective of age, degree of transfusion dependence, gender, and time since diagnosis. The clinicians from Alberta added that patients in this group have no other effective treatment options other than long-term transfusions and iron chelation to help manage the side effects associated with iron overload. The Alberta clinicians added that patients with higher-risk MDS would be better served with HMAs and are not included in this reimbursement review.
With respect to how to identify patients best suited for treatment with the drug under review, the clinicians from Ontario noted that the MEDALIST trial enrolled patients with very low, low or intermediate IPSS-R scores who have ringed sideroblasts, failed erythropoietin, and have erythropoietin levels below 500 U/L. They added that the study also used the IPSS-R, whereas azacitidine eligibility is based on the IPSS. The clinicians added that the trial excluded patients with del(5q) genetic abnormalities or secondary MDS.
The clinicians from Alberta added that patients are assigned an IPSS-R score at diagnosis based on bone marrow aspirate results (i.e., blast count, cytogenetics risk, and degree of cytopenias), and this identifies the presence of ringed sideroblasts along with next-generation sequencing of DNA for the SRSF1 mutation seen in ringed sideroblast disease. The clinicians noted that patients are identified by the morphologic diagnosis, results of the scoring scale for those patients, and commonly available lab tests, such as erythropoietin levels. They also added that most patients requiring regular transfusions would be investigated and have a clear diagnosis as long as they are willing to undergo bone marrow aspiration. The Alberta group added that next-generation sequencing is available in Alberta and would be required more frequently to confirm a ringed sideroblast diagnosis in patients with 5% to 15% ringed sideroblasts. The clinicians added that they currently do this on all newly diagnosed MDS patients in Alberta, and they anticipate this will become standard of care as funding for testing becomes available across the country.
With respect to which patients would be least suitable for treatment with the drug under review, the Ontario clinicians noted that patients with low erythropoietin levels or higher-risk patients would be least suitable. The clinicians from Alberta noted that patients with allergies to the medications would be least suitable for treatment with the drug under review but added that the drug is otherwise appropriate for all patients as per the reimbursement request.
Both clinician groups agreed that it is not possible to identify patients who are most likely to exhibit a response to treatment with the drug under review. The clinicians from Alberta noted that rates of response can be improved with lower erythropoietin levels; however, this is not highly discriminative and would not warrant excluding patients from treatment eligibility.
Assessing Response to Treatment
Both clinician groups agreed that transfusion frequency (reduction in transfusion requirements) and improvement in hemoglobin levels are outcomes used to determine whether a patient is responding to treatment in clinical practice. Both groups also agreed that a reduction in transfusions would be a clinically meaningful response to treatment. The clinicians from Alberta noted that an improvement in hemoglobin levels by 15 g/L and a reduction in transfusion requirements of at least 25% would be meaningful response measures. The Ontario clinicians noted that response should be assessed every 3 to 4 weeks, while the clinicians from Alberta added that CBCs should be performed monthly; initially they would be performed weekly, by type and screen in patients who are currently transfusion-dependent, and if they are stable off of transfusions, CBC can be performed less frequently.
Discontinuing Treatment
The clinicians from Ontario noted that worsening of MDS, progression to a higher-risk category, or transformation to AML should be considered when deciding whether to discontinue treatment. The clinicians from Alberta noted that a decrease in hemoglobin without an alternative cause, an increase in transfusion requirements, or a need to introduce regular transfusions in patients who have been transfusion-independent should be considered. The clinicians from Alberta added that, if a patient becomes ill for other reasons (i.e., infection or bleeding, both of which are more common in MDS patients), they may transiently require transfusion again while the cause of the deterioration is treated. However, the clinicians added that this should not preclude ongoing therapy if it is effective, except for the effect of intercurrent illness.
Prescribing Conditions
According to both clinician groups, the most appropriate settings for treatment are community settings such as pharmacies, outpatient clinics, and specialty clinics.
The clinicians from Alberta added that a hematology or medical oncology specialist would be required to diagnose, treat, and monitor patients who might receive the drug under review.
Additional Considerations
The clinicians from Alberta noted that there are limited treatment options in this group of patients with otherwise good-risk MDS and often long-life expectancies. They added that the benefit to patients who can become transfusion-independent (or remain so after developing symptomatic anemia) is significant and can reduce burdens on both patients and the health care institutions that provide regular transfusion support over long time periods to these patients.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
1. Should ESA agents be considered in the comparators? | The study population is aligned with the Health Canada–approved indication, which limits the use of luspatercept to patients who have failed or are not suitable for erythropoietin-based therapy. All patients enrolled in the MEDALIST trial had previously failed on treatment with ESAs. |
2. Of patients in the pivotal study (MEDALIST), 95% had received ESA previously and 48% had received iron chelation. Patients in the study were intolerant of or ineligible for (serum EPO > 200 U/L) ESA treatment. Should previous ESA treatment be a consideration before funding? | One of the clinical experts was of the opinion that some patients may not respond to treatment with ESAs, and such patients could be identified using scoring algorithms such as the Nordic method. Such patients should not be required to try ESAs before luspatercept. The second clinical expert was of the opinion that previous ESA treatment should be considered before funding, and that the inclusion criteria of the trial would be used to determine which patients would be suitable for treatment with luspatercept. |
3. The pivotal trial (MEDALIST) enrolled a total of 229 patients. Approximately 66% of those in the treatment arm (N = 153) were 74 years of age or younger. Based on the age in the study, are the results applicable to the usual population expected to be treated? | Both clinical experts agreed that the results of the MEDALIST trial would be applicable to the usual population expected to be treated in Canada. |
4. Need to define absence of clinical benefit (e.g., not RBC transfusion–free after 9 weeks of treatment at the maximum dose) and disease progression (e.g., progression to AML). | One of clinical experts defined the absence of clinical benefit as no hematologic response as per IWG criteria and failure to demonstrate a clinical meaningful improvement in quality of life. The second clinical expert defined an absence of clinical benefit as a failure to exhibit transfusion independence over an 8-week period or no 50% reduction in transfusions achieved compared to pre–drug trial after a 6-month treatment course. Neither expert commented on disease progression. |
5. Should therapy end if patient does not experience clinical benefit after 9 weeks of treatment (after 3 doses) at the maximum dose level if no other causes are found or if unacceptable toxicity occurs at any time (as per product monograph)? | Both clinical experts agreed with the discontinuation stipulation in the product monograph and concurred that therapy would be ended if a patient does not experience clinical benefit after 9 weeks of treatment (after 3 doses) at the maximum dose level if no other causes are found or if unacceptable toxicity occurs at any time. |
6. Must be administered by a health care professional (product monograph). Hgb must be reviewed before each administration and dose adjusted if required. If the pre-dose Hgb is greater than or equal to 11.5 g/dL (115 g/L) and the Hgb level is not influenced by recent transfusion, delay dosing until Hgb is less than or equal to 11.0 g/dL (110 g/L). | The clinical experts indicated that luspatercept could potentially be self-administered by the patient, despite the stipulation in the product monograph that luspatercept should be administered by a health care professional. The clinical experts did not necessarily agree with delaying luspatercept in patients with elevated Hgb levels and that administration of luspatercept in such patients should be at the physician’s discretion. |
7. Access to Hgb monitoring and access to health care professionals for every-3-weeks administration in rural communities may need to be considered. | The clinical experts agreed that all patients who are eligible for treatment with luspatercept should have equal access to the treatment and that there should be, flexibility as to where the treatment is administered. |
AML = acute myeloid leukemia; ESA = erythroid-stimulating agents; EPO = erythropoietin; Hgb = hemoglobin; RBC = red blood cell.
Clinical Evidence
The clinical evidence included in the review of luspatercept is presented in the systematic review. Which included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. No additional evidence to fill any evidence gaps or indirect evidence was identified for this review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of luspatercept (25 mg per vial or 75 mg per vial) powder for solution for subcutaneous injection for the treatment of RBC transfusion-dependent anemia associated with very low- to intermediate-risk MDS.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adult patients with RBC transfusion-dependent anemia associated with very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy Subgroups:
|
Intervention | Luspatercept powder for solution for subcutaneous injection
|
Comparators |
|
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, notable harms (e.g., thromboembolic events, hypertension, hepatic and renal events, hypersensitivity reactions, malignancies) |
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; AML = acute myeloid leukemia; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; ICT = iron chelation therapy; IPSS-R = Revised International Prognostic Scoring System; MDS = myelodysplastic syndromes; QoL-E = quality of life questionnaire for patients with myelodysplastic syndromes; RBC = red blood cell; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the Peer Review of Electronic Search Strategies checklist (https://www.cadth.ca/resources/finding-evidence/press).15
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid and Embase (1974‒) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Reblozyl (luspatercept). Clinical trials registries searched: included the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on March 29, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on July 21, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist (https://www.cadth.ca/grey-matters).16 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings from the Literature
One study was identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 7. A list of excluded studies is presented in Appendix 2.
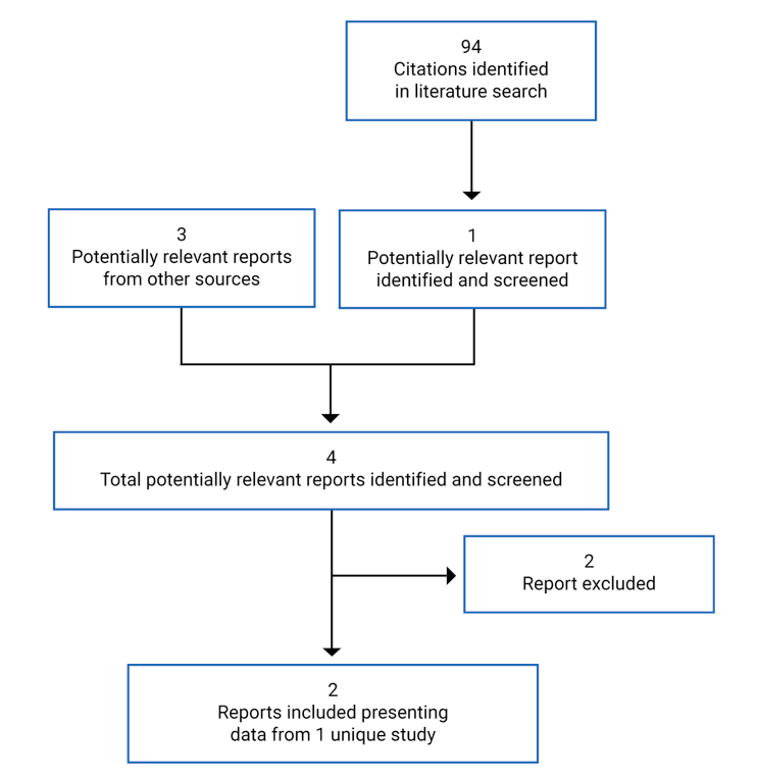
Table 7: Details of Included Studies
MEDALIST | |
|---|---|
Designs and populations | |
Study design | Double-blind RCT, phase III, placebo-controlled |
Locations | 65 centres: France, UK, US, Canada, Spain, Italy, Turkey, Germany, Belgium, Netherlands, and Sweden |
Patient enrolment dates | February 9, 2016, to May 8, 2018 |
Randomized (N) | 229 |
Inclusion criteria |
|
Exclusion criteria |
|
Exclusion criteria (continued) |
|
Drugs | |
Intervention | Luspatercept starting dose of 1.0 mg/kg subcutaneously once every 3 weeks to a maximum dose of 1.75 mg/kg |
Comparator(s) | Placebo |
Duration | |
Phase | |
Screening | 5 weeks |
Primary treatment | 24 weeks |
Assessment period | Week 25 |
Extension treatment | Until patients experienced unacceptable toxicities, disease progression; withdrew consent or met any other discontinuation criteria |
Post-treatment follow-up | 42 days |
Long-term follow-up | At least 3 years |
Outcomes | |
Primary end point | RBC-TI with a duration of ≥ 8 weeks measured at 24 weeks. |
Secondary and exploratory end points | Key Secondary:
Secondary:
|
Secondary and exploratory end points (continued) |
Exploratory:
|
Notes | |
Publications | Fenaux (2020)17 |
AML = acute myeloid leukemia; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EPO = erythropoietin; ESA = erythroid-stimulating agents; FAB = French-American-British; G-CSF = granulocyte colony-stimulating factor; HBV = hepatitis B virus; HCV = hepatitis C virus; HI-N = hematologic improvement–neutrophils; HI-P = hematologic improvement–platelets; HMA = hypomethylating agent; ICT = iron chelation therapy; IMiD = immunomodulatory imide drug; IPSS-R = Revised International Prognostic Scoring System; MDS = myelodysplastic syndromes; mHI-E = modified hematologic improvement–erythroid; QoL-E = quality of life questionnaire for patients with myelodysplastic syndromes; RBC = red blood cell; RBC-TI = red blood cell–transfusion independence.
Source: Clinical Study Report for MEDALIST.9
Description of Studies
One pivotal trial (MEDALIST; N = 229) was included in the CADTH systematic review. Details of MEDALIST are provided in Table 7 and Figure 2.
The MEDALIST trial is an ongoing phase III, randomized, double-blind, placebo-controlled study of the efficacy and safety of luspatercept in adult patients with RBC transfusion-dependent anemia associated with very low- to intermediate-risk MDS who have ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy. It was conducted at 65 sites globally. Four sites in Canada enrolled 14 patients.
The MEDALIST trial consisted of a 5-week screening period, which assessed patients for eligibility into the study. Prior to randomization, patients had to have had at least 16 weeks of transfusion history available. Central reviews of bone marrow aspirate smear and biopsy, peripheral blood smears, and cytogenetics were used to confirm MDS diagnosis according to WHO and/or FAB classification, and to determine the baseline IPSS-R risk classification. The classification used in the sponsor submission is shown in Appendix 3 (Figure 5, and Figure 6).
Following the 5-week screening period, eligible patients were randomized (2:1) to receive either luspatercept or placebo along with best supportive care. Patients were randomized by using interactive response technology and no crossover between the treatment groups was permitted. Randomization was stratified based on RBC transfusion burden at baseline (≥ 6 units over 8 weeks versus < 6 units over 8 weeks, based on the mean of the 2 consecutive 8-week periods immediately before randomization) and IPSS-R score at baseline (very low or low versus intermediate). Patients must have had at least 16 weeks of transfusion history available immediately preceding and including the date of randomization. Prior transfusion data included the type of transfusion (e.g., RBC, platelets), number of units, and date of transfusion. Data on RBC transfusions included the hemoglobin value for which the transfusion was administered (i.e., pre-transfusion hemoglobin value). The randomized double-blind phase of the study was divided into a 24-week primary treatment phase, a week-25 assessment phase, and a 24-week extension phase. Patients received a starting dose of 1 mg of the study drug per kilogram of body weight administered by a subcutaneous injection every 3 weeks. During the double-blind period the dose levels were titrated (increased) stepwise up to a maximum of 1.75 mg/kg or reduced based on a clinical response. The maximum total dose per administration was not to exceed 168 mg. Luspatercept prepackaged in 3 mL glass vials at 25 mg per vial and 75 mg per vial was provided by the sponsor. Placebo used in the study was sterile normal saline (0.9% sodium chloride for injection) prepared in syringes by the investigational site’s designated individuals to match the active syringe and administered as a subcutaneous injection. The blind was not to be broken during the study unless, in the opinion of the investigator, it was necessary to safely treat the patient. However, the investigator could contact the medical monitor before breaking the blind to discuss whether the unblinding would be in the best interest of the patient.
The week-25 assessment phase had to be completed 24 calendar weeks after the date of the first dose, regardless of dose delays. A 14-day window was allowed for the week-25 visit. For patients to continue the double-blind treatment beyond the first 24 calendar weeks, the following criteria had to be confirmed by the investigator at the week-25 visit: evidence of clinical benefit (e.g., decrease in RBC transfusion requirement compared with baseline requirement or hemoglobin increase compared with baseline) and absence of disease progression as per IWG-MDS criteria for altering the natural history of MDS. Based on the outcome of the week-25 MDS disease assessment visit, patients were either discontinued from treatment and entered the post-treatment follow-up period or continued the double-blind treatment with the same study drug in the extension phase of the treatment period.
Patients who met the criteria to remain on double-blind treatment after completion of the week-25 assessment could continue their originally assigned treatment in the extension phase until the patient experienced unacceptable toxicities, disease progression per IWG criteria for altering the natural history of MDS, withdrew consent, or met any other discontinuation criteria. The MDS disease assessment was performed on day 1 of extension cycle 8 and was to be repeated on day 1 of every eighth extension cycle thereafter (i.e., extension cycles 8, 16, and 24+, or approximately every 24 weeks) until the patient was discontinued from treatment. Results of the double-blind treatment period of 48 weeks are presented in this report.
The primary objective of the study was to measure the proportion of patients treated with luspatercept versus placebo who achieved RBC-TI for at least 8 weeks (any consecutive 56-day period) from week 1 to week 24. The 2 key secondary outcomes were the proportion of patients who achieved RBC-TI for at least 12 weeks (any consecutive 84-day period) from week 1 to week 48 and the proportion of patients who achieved RBC-TI for at least 12 weeks (any consecutive 84-day period) from week 1 to week 24.
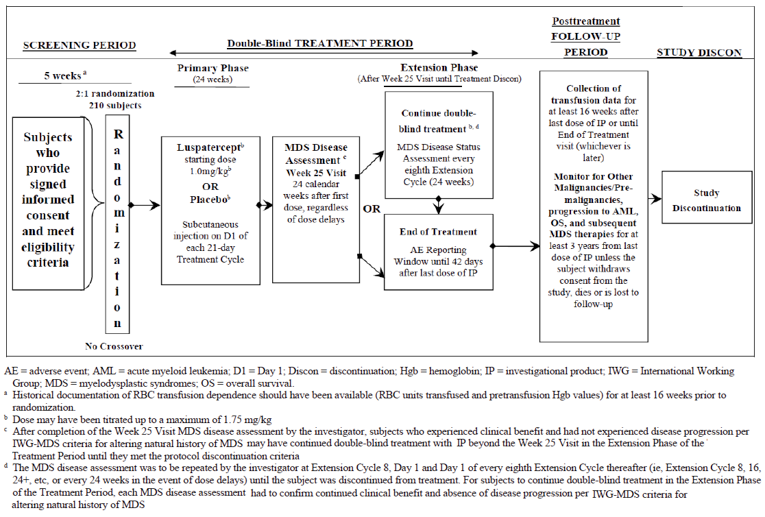
Populations
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria for MEDALIST are presented in Table 7. Adult patients 18 years of age and older with a documented diagnosis of MDS according to WHO and/or FAB classification who met the IPSS-R classification of very low-, low- or intermediate-risk disease and were refractory or intolerant to or ineligible for prior ESA treatment were eligible for the study. Patients with an average transfusion requirement of at least 2 units over 8 weeks of packed RBCs and hemoglobin levels of no more than 10.0 g/dL at the time of, or within 7 days before, an RBC transfusion were included in the study. Patients were excluded from the MEDALIST study if they had a prior allogeneic or autologous stem cell transplant, therapy with disease-modifying drugs such as lenalidomide, HMAs, or immunosuppressive therapy. Patients with MDS associated with the del(5q) cytogenetic abnormality, secondary MDS or a known history of diagnosis of AML were excluded.
Baseline Characteristics
The baseline characteristics, disease characteristics, baseline transfusion burden, and MDS treatment history are summarized in Table 8, Table 9, Table 10, and Table 11, respectively. Approximately 2-thirds of the patients in the MEDALIST study were male and White. The mean weights were 76.2 kg and 77.4 kg in the luspatercept and placebo treatment groups, respectively. The mean age of the patients at baseline was 70.5 years (SD = 8.68) and 70.7 years (SD = 10.88) in the luspatercept and placebo treatment groups, respectively. Of the patients in the luspatercept and placebo treatment groups, 94.8% and 97.4%, respectively, were classified as having refractory cytopenia with multilineage dysplasia, according to the WHO classification. In addition, 71.2% and 75% of the patients were classified as at low risk under the IPSS-R classification in the luspatercept and placebo treatment groups, respectively. Patients with the SF3B1 mutation accounted for 92.2% of the luspatercept treatment group and 85.5% of the placebo treatment group. Of the patients in the MEDALIST study, 59.5% in the luspatercept treatment group and 42.1% in the placebo treatment group had an ECOG performance status of 1, and 5.2% of the patients in the luspatercept treatment group and 14.5% of the patients in the placebo treatment group had an ECOG performance status of 2. In addition, 96.7% of the patients in the luspatercept treatment group and 92.1% of the patients in the placebo treatment group had been previously treated with an ESA; 3.3% of the patients in the luspatercept treatment group and 7.9% in the placebo treatment group were ESA-naive; 97.3% of the patients in the luspatercept treatment group and 98.6% of the patients in the placebo treatment group discontinued an ESA due to being refractory to the treatment; and 2.7% and 1.4% of the patients were intolerant to an ESA treatment in the luspatercept and placebo treatment groups, respectively. Anti-anemic preparations were the most used prior medications, with 96.1% of the total study population having used them. Of the total study population, 32.3%, 31.9%, and 27.5% had previously used drugs for acid-related disorders, immunostimulants, and antithrombotic drugs, respectively.
Table 8: Summary of Baseline Characteristics in the MEDALIST Trial (ITT Population)
Characteristics | Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) |
|---|---|---|
Age, years | ||
Mean (SD) | 70.5 (8.68) | 70.7 (10.88) |
Median (minimum to maximum) | 71.0 (40 to 95) | 72.0 (26 to 91) |
Gender, n (%) | ||
Male | 94 (61.4) | 50 (65.8) |
Female | 59 (38.6) | 26 (34.2) |
Race, n (%) | ||
Black or African-American | 1 (0.7) | 0 |
White | 107 (69.9) | 51 (67.1) |
Not collected or reported | 44 (28.8) | 24 (31.6) |
Other | 1 (0.7) | 1 (1.3) |
Ethnicity, n (%) | ||
Hispanic or Latino | 3 (2.0) | 4 (5.3) |
Not Hispanic or Latino | 115 (75.2) | 52 (68.4) |
Not reported | 35 (22.9) | 20 (26.3) |
Weight, kg | ||
Mean (SD) | 76.2 (15.07) | 77.4 (15.78) |
Median (Min, Max) | 76.0 (46, 124) | 75.0 (51, 153) |
BMI,a kg/True | ||
n (%) | 152 (99.3) | 75 (98.6) |
Mean (SD) | 26.6 (4.19) | 27.0 (4.58) |
Median (minimum to maximum) | 26.2 (17 to 40) | 27.1 (20 to 48) |
BMI = body mass index; BSC = best supportive care; ITT = intention to treat; SD = standard deviation
Source: Clinical Study Report for MEDALIST.9
Table 9: Summary of Baseline Disease Characteristics in the MEDALIST Trial (ITT Population)
Characteristics | Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) |
|---|---|---|
Time since original MDS diagnosis, months | ||
Mean (SD) | 57.8 (56.59) | 52.7 (42.29) |
Median (minimum to maximum) | 44.0 (3 to 421) | 36.1 (4 to 193) |
Time since original MDS diagnosis categories,a n (%) | ||
≤ 2 years | 40 (26.1) | 19 (25.0) |
> 2 to 5 years | 62 (40.5) | 34 (44.7) |
> 5 years | 51 (33.3) | 23 (30.3) |
Ring sideroblasts, n (%) | ||
≥ 15% | 153 (100) | 76 (100) |
MDS WHO classification, n (%) | ||
MDS RARS | 7 (4.6) | 2 (2.6) |
MDS RCMDb | 145 (94.8) | 74 (97.4) |
Otherc | 1 (0.7) | 0 |
IPSS-R classification risk category, n (%) | ||
Very low | 18 (11.8) | 6 (7.9) |
Low | 109 (71.2) | 57 (75.0) |
Intermediate | 25 (16.3) | 13 (17.1) |
High | 1 (0.7) | 0 |
Serum EPO (U/L) | ||
n (%) | 152 (99.3) | 76 (100) |
Mean (SD) | 279.6 (361.33) | 284.5 (433.84) |
Median (minimum to maximum) | 156.9 (12 to 2,454) | 130.8 (29 to 2,760) |
Serum EPO (U/L) categories, n (%) | ||
< 100 | 51 (33.3) | 31 (40.8) |
100 to < 200 | 37 (24.2) | 19 (25.0) |
200 to 500 | 43 (28.1) | 15 (19.7) |
> 500 | 21 (13.7) | 11 (14.5) |
Missing | 1 (0.7) | 0 |
SF3B1, n (%) | ||
Mutated | 141 (92.2) | 65 (85.5) |
Non-mutated | 12 (7.8) | 10 (13.2) |
Missing | 0 | 1 (1.3) |
ECOG performance status, n (%) | ||
0 | 54 (35.3) | 33 (43.4) |
1 | 91 (59.5) | 32 (42.1) |
2 | 8 (5.2) | 11 (14.5) |
BSC = best supportive care; ECOG = Eastern Cooperative Oncology Group; EPO = erythropoietin; IPSS-R = Revised International Prognostic Scoring System; ITT = intention to treat; MDS = myelodysplastic syndromes; RARS = refractory anemia with ring sideroblasts; RBC = red blood cell; RCMD = refractory cytopenia with multilineage dysplasia; SD = standard deviation.
aDefined as the number of years from the date of original diagnosis to the date of informed consent.
bAll patients were classified as RCMD with ring sideroblasts as they were required to have ring sideroblasts per inclusion criteria.
cLocally diagnosed MDS with ring sideroblasts and multilineage dysplasia.
Source: Clinical Study Report for MEDALIST.9
Table 10: Summary of Baseline Transfusion Burden in the MEDALIST Trial (ITT Population)
Characteristics | Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) |
|---|---|---|
RBC transfusions per last 8 weeks | ||
Mean (SD) | 5.9 (2.97) | 6.2 (2.99) |
Median (minimum to maximum) | 6.0 (2 to 16) | 6.0 (0 to 16) |
RBC transfusions per last 8 weeks categories, n (%) | ||
≥ 6 units | 78 (51.0) | 46 (60.5) |
< 6 units | 75 (49.0) | 30 (39.5) |
≥ 4 and < 6 units | 47 (30.7) | 19 (25.0) |
< 4 units | 28 (18.3) | 11 (14.5) |
RBC transfusions per 8 weeks over 16 weeks, n (%) | ||
Mean (SD) | 5.5 (2.76) | 5.8 (2.95) |
Median (minimum to maximum) | 5.0 (1 to 15) | 5.0 (2 to 20) |
RBC transfusions per 8 weeks over 16 weeks categories, n (%) | ||
≥ 6 units | 66 (43.1) | 33 (43.4) |
< 6 units | 87 (56.9) | 43 (56.6) |
≥ 4 and < 6 units | 41 (26.8) | 23 (30.3) |
< 4 units | 46 (30.1) | 20 (26.3) |
Hemoglobin, (g/dL) | ||
Mean (SD) | 7.7 (0.84) | 7.6 (0.77) |
Median (minimum to maximum) | 7.6 (6 to 10) | 7.6 (5 to 9) |
ITT = intention to treat; RBC = red blood cell; SD = standard deviation.
Source: Clinical Study Report for MEDALIST.9
Table 11: Summary of Baseline MDS Treatment History in the MEDALIST Trial (ITT Population)
Characteristics | Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) |
|---|---|---|
Prior ESA, n (%) | ||
Yes | 148 (96.7) | 70 (92.1) |
No | 5 (3.3) | 6 (7.9) |
Reasons for prior ESA discontinuation,a n (%) | ||
Refractoryb | 144 (97.3) | 69 (98.6) |
Intolerantc | 4 (2.7) | 1 (1.4) |
Time from end of prior ESA to start of study,d months | ||
ne | 148 | 70 |
Mean (SD) | 14.79 (28.824) | 11.18 (13.553) |
Median (minimum to maximum) | 5.26 (0.9 to 257.9) | 5.13 (0.2 to 64.9) |
Time from end of prior ESA to start of study categories,d n (%)a | ||
< 6 months | 82 (55.4) | 37 (52.9) |
6 to 12 months | 21 (14.2) | 13 (18.6) |
> 12 to 24 months | 19 (12.8) | 7 (10.0) |
> 24 months | 26 (17.6) | 13 (18.6) |
Longest duration of prior ESA treatment, (months) | ||
ne | 148 | 70 |
Mean (SD) | 17.83 (22.415) | 19.51 (20.202) |
Median (minimum to maximum) | 10.48 (1.2 to 143.2) | 13.17 (1.4 to 90.9) |
Longest duration of prior ESA treatment, n (%)a | ||
< 6 months | 48 (32.4) | 20 (28.6) |
6 to 12 months | 34 (23.0) | 9 (12.9) |
> 12 to 24 months | 35 (23.6) | 21 (30.0) |
> 24 months | 31 (20.9) | 20 (28.6) |
Prior ICT use, n (%) | ||
Yes | 71 (46.4) | 40 (52.6) |
No | 82 (53.6) | 36 (47.4) |
Prior G-CSF/GM-CSF usage,f n (%) | ||
Yes | 51 (33.3) | 22 (28.9) |
No | 102 (66.7) | 54 (71.1) |
ESA = erythropoiesis-stimulating agent; G-CSF = granulocyte colony-stimulating factor; GM-CSF = granulocyte macrophage colony-stimulating factor; ICT = iron chelation therapy; ITT = intention to treat; MDS = myelodysplastic syndromes; SD = standard deviation.
aPercentages calculated relative to the number of patients with prior ESA use.
bDefined as documentation of nonresponse or response that is no longer maintained to prior ESA-containing regimen.
cDefined as documentation of discontinuation of prior ESA-containing regimen at any time after introduction due to intolerance or an adverse event.
dTime from end of prior ESA to start of study was defined as the number of months from the date of the end of prior ESA to the date of day 1 of cycle 1. When cycle 1, day 1 was missing, the randomization date was used.
eNumber of patients with prior ESA use.
fAny drugs with Anatomic Therapeutic Chemical code L or L03 for G-CSF/GM-CSF usage.
Source: Clinical Study Report for MEDALIST.9
Interventions
Patients eligible for the MEDALIST study were randomized in a 2:1 ratio to receive either luspatercept or placebo along with BSC every 3 weeks during the treatment period. Both treatments were administered as a subcutaneous injection in the patient’s upper arm, thigh, and/or abdomen. Doses were administered by the study staff at the clinical site and treatment administrations were documented. Patients in the luspatercept group received the study drug once every 3 weeks at a starting dose level of 1 mg/kg up to a maximum dose of 1.75 mg/kg. The maximum volume per subcutaneous injection was not to exceed 1.2 mL. The maximum total dose per administration was not to exceed 168 mg, which resulted in a maximum total volume of 3.36 mL after reconstitution. Best supportive care included RBC transfusions; iron chelation, antibiotic, antiviral, and/or antifungal therapies; and nutritional support as needed. The use of ESAs as BSC was excluded from the study.
The dose titration criteria are presented in Table 12. Dose delay of luspatercept was allowed due to increased hemoglobin or adverse events. If there was an insufficient response for 21 weeks or longer from the previous dose administered, treatment was to be discontinued.8 Dose delays due to an adverse event were at the discretion of the investigator.
Table 12: Starting Dose Level with Dose Reductions and Dose Titration
Third dose reduction (approximately 25%) | Second dose reduction (approximately 25%) | First dose reduction (approximately 25%) | Starting dose level | First dose titration | Second dose titration | |
|---|---|---|---|---|---|---|
0.45 mg/kg | 0.6 mg/kg | 0.8 mg/kg | 1 mg/kg | 1.33 mg/kg | 1.75 mg/kg | |
Source: Clinical Study Report for MEDALIST.9
Best supportive care specifically excluded cancer surgery, immunotherapy, biologic therapy, radiotherapy, and systemic chemotherapy in which the goal was to eradicate or slow the progression of the disease. Cytotoxic, chemotherapeutic, targeted, or investigational agents; azacitidine, decitabine, or other HMAs; lenalidomide, thalidomide, and other immunomodulatory imide drugs; ESAs; and other RBC hematopoietic growth factors and hydroxyurea were specifically excluded as concomitant medications during the study.
At least 1 concomitant medication was used by 98.7% of the patients in the luspatercept treatment group and 94.7% of the patients in the placebo treatment group, with 51.1% of the total population having used analgesics. Use of all other therapeutic products was reported by 48.4% of the patients in the luspatercept treatment group and 48.7% of the patients in the placebo treatment group. Antithrombotic drugs were used by 34% of the patients in the luspatercept treatment group and 38.2% of the patients in the placebo treatment group. The proportions of patients who initiated other therapies to treat MDS were 15% in the luspatercept treatment group and 29% in the placebo treatment group. Azacitidine, epoetin, lenalidomide, and daratumumab were the most commonly used therapies for subsequent MDS treatment.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 13. These end points are summarized below. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
Table 13: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measures | MEDALIST |
|---|---|
RBC-TI ≥ 8 weeks from week 1 through week 24 | Primary |
RBC-TI ≥ 12 weeks from week 1 through week 48 | Key secondary |
RBC-TI ≥ 12 weeks from week 1 through week 24 | Key secondary |
RBCs units transfused | Secondary |
Duration of RBC-TI | Secondary |
Time to RBC-TI | Secondary |
Mean change in hemoglobin | Secondary |
Mean change in modified hematologic improvement–erythroid | Secondary |
HRQoL | Secondary and exploratory |
Overall survival | Secondary |
Iron accumulation | Secondary |
ICT use | Secondary |
Progression to acute myeloid leukemia | Secondary |
Health care resource utilization | Exploratory |
HRQoL = health-related quality of life; ICT = iron chelation therapy; RBC-TI = red blood cell–transfusion independence.
Hematologic Response
Hematologic response was assessed through RBC-TI, RBC units transfused, duration of RBC-TI, time to RBC-TI, mean change in hemoglobin and modified hematologic improvement–erythroid (mHI-E). The primary end point of MEDALIST was based on an RBC-TI of at least 8 weeks (RBC transfusion–free over any consecutive 56-day period, from week 1 through week 24). A transfusion-independence response is defined as the absence of any RBC transfusion during any consecutive 56-day period during the primary phase of the treatment period (first 24 weeks of double-blind treatment), such as days 1 to 56, days 2 to 57, days 3 to 58, and so on.
Two key secondary outcomes assessed a hematologic response based on an RBC-TI of at least 12 weeks, i.e., RBC transfusion–free over any consecutive 84-day period, from week 1 through week 24 and week 1 through week 48.
Units of RBCs transfused was defined as the mean change in total number of RBC units transfused over a fixed period of 16 weeks (weeks 9 through 24 and weeks 33 through 48) compared to the total number of RBC units transfused in the 16 weeks immediately on or before the first dose date. Duration of RBC-TI was defined as longest duration of RBC-TI for at least 8 weeks, i.e., any consecutive 56-day period during the treatment period (week 1 to week 24 and week 1 to week 48). Patients who maintained RBC-TI at the analysis were censored.
Time to RBC-TI was defined as the time between the first dose date and the date of onset of transfusion independence first observed (i.e., day 1 of 56 days without any RBC transfusions). Time to RBC-TI was reported only for patients who achieved an RBC-TI of at least 8 weeks.
Mean change in hemoglobin was defined as the proportion of patients with a mean hemoglobin increase of at least 1.0 g/dL after applying a 14/3-day rule. Under the 14/3-day rule, only hemoglobin values that are measured at least 14 days after a transfusion can be used unless there is another transfusion within 3 days after the hemoglobin assessment. If this occurs, the second hemoglobin value may be used, despite being recorded less than 14 days after the previous transfusion.
The mHI-E measure was defined as the proportion of patients meeting the mHI-E criteria set by the IWG and sustained over any consecutive 56-day period during the treatment period (week 1 to week 24 and week 1 to week 48). Patients meeting the mHI-E criteria were defined as those receiving fewer than 4 units over 8 weeks at baseline; a responder had to satisfy the 2 conditions that there was no RBC transfusion within the response interval and a mean of hemoglobin increase of at least 1.5 g/dL from baseline. For patients who received at least 4 units over 8 weeks units at baseline, a responder must satisfy the condition that there was a decrease of at least 4 units of RBCs from baseline over any consecutive 56-day period.
HRQoL
Assessments of HRQoL used the QoL-E and EORTC QLQ-C30 instruments. The HRQoL determined the effects of luspatercept and placebo on mean change from baseline HRQoL at assessment week 25, as assessed with the QoL-E and EORTC QLQ-C30. The domains assessed from the EORTC QLQ-C30 were global health status/quality of life, physical functioning, emotional functioning, fatigue, and dyspnea. All domains of the QoL-E questionnaire were considered exploratory. In patients with low- to intermediate-MDS, data on validity, reliability, and responsiveness were not identified in the literature for the EORTC QLQ-C30. Earlier versions of the QoL-E that compared responses to the FACT-G questionnaire confirmed good clinical validity and reliability18; however, responsiveness of the instrument was not identified from literature.
The QoL-E is a 29-item, self-administered, multi-dimensional questionnaire that evaluates patient-reported outcomes in patients with MDS.19 The goal is to assess the effect of MDS on patients’ lives. The questionnaire is scored on a standardized scale, with values ranging from 0 to 100, using Likert-scale questions and dichotomous response questions. Each domain is scored separately, with a higher score indicating a better quality of life. It includes questions on physical, functional, social, and sexual well-being, focusing on fatigue and MDS-specific items. The recall period for temporal items is 1 week, and 1 month for the general health item.18 Three versions (1.0, 2.0, and 3.0) have been examined in the literature. Version 3.0 was used in the MEDALIST study. An MID for patients with transfusion-dependent low- to intermediate-MDS was not identified in the literature.
The EORTC QLQ-C30 is a 30-item, self-administered questionnaire designed to evaluate the quality of life of patients with cancer. The questionnaire contains items relevant to several domains, including function (physical, role, cognitive, emotional, and social), symptoms (fatigue, pain, and nausea and vomiting), quality of life, and several single items commonly associated with cancer (dyspnea, loss of appetite, insomnia, constipation, and diarrhea). Each domain is scored separately on a scale from 0 to 100, with a higher score indicating a higher response for that domain. For example, a high score in the symptom domain indicates a patient is dealing with many symptoms (i.e., possibly poorer conditions), whereas a high score in the functional domain indicates a patient has a high level of functioning (i.e., managing the disease well).20 An MID for patients with transfusion-dependent low- to intermediate-risk MDS was not identified in the literature. Appendix 5 provides a description and appraisal of these outcome measures.
Patients were considered compliant with the EORTC QLQ-C30 assessment if at least half (i.e., ≥ 15 out of 30) of the EORTC QLQ-C30 items are non-missing at a given assessment visit. The comparability of the HRQoL-evaluable and non-evaluable populations was assessed at baseline to determine whether the primary outcome was generalizable to the entire ITT population. The HRQoL-non-evaluable population was defined as those patients in the ITT population who were not included in the HRQoL-evaluable population.
Overall Survival
Overall survival was defined as the time between randomization and death or censored date. Patients who died, regardless of the cause of death, were considered to have had an event. Patients who were alive at the time of analysis were censored at the last assessment date at which the patient was known to be alive. All patients who were lost to follow-up were also censored at the time of last contact.
Iron Accumulation
Iron accumulation was measured through serum ferritin levels. Mean change in serum ferritin was calculated as the difference of change in mean serum ferritin at week 9 to week 24 and week 33 to week 48 from baseline.
ICT Use
Iron chelation therapy use was defined as the mean change in mean daily dose of ICT. It was the change in daily dose of ICT for each patient and was calculated as the difference of the post-baseline mean daily dose and baseline mean daily dose. Two comparisons were performed between the luspatercept treatment group and placebo treatment group. Mean changes from baseline for mean daily dose of ICT were averaged over week 9 to week 24 and mean daily doses of ICT were averaged over week 33 to week 48.
Progression to Acute Myeloid Leukemia
Time to progression to AML was defined as the time between randomization and first diagnosis of AML as per WHO classification of at least 20% blasts in peripheral blood or bone marrow. Patients with a diagnosis of AML were considered to have had an event. Patients who had not progressed to AML at the time of analysis were censored at the last assessment date, which did not indicate progression to AML.
Health Care Resource Utilization
Health Care Resource Utilization was assessed as the number of patients who had a doctor office visit or emergency room visit, or a hospitalization ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Statistical Analysis
In the MEDALIST study an estimated sample size of 210 patients (140 in the luspatercept treatment group and 70 in the placebo treatment group) was required to achieve at least 90% power to detect the difference between the treatment groups with a 1-sided 0.025 level of significance. Test statistics on difference of proportions used a pooled estimate of variance and assumed a 10% dropout rate for each treatment group. The assumed targeted response rate for the primary end point was 30% in the luspatercept treatment group and 10% for the placebo group.
All efficacy outcomes were to be evaluated using the ITT population, except for the HRQoL analyses, which used the HRQoL-evaluable population (defined in the following section). For the statistical plan, the sponsor defined the primary outcome as the proportion of patients who achieve RBC-TI of at least 8 weeks (i.e., RBC transfusion–free over any consecutive 56-day period) from week 1 through week 24. Patients had to have at least 56 days of transfusion independence before (and including) the week-24 cut-off date to be qualified as responders. Patients who failed to achieve at least 56 days of transfusion independence before or on the cut-off date were counted as nonresponders. For the primary efficacy end point, 56 days of RBC-transfusion independence, the response rate was calculated using the number of responders divided by the number of patients (responders plus nonresponders in the ITT population). Patients who discontinued from the primary phase of the treatment period without achieving at least 56 days consecutive of RBC transfusion independence were included as nonresponders.
The secondary efficacy outcomes were tested in the following pre-specified sequential order:
Proportion of patients achieving RBC-TI with a duration of at least 12 weeks is the absence of any RBC transfusion during any consecutive 84-day period during the treatment period (week 1 to week 48)
Proportion of patients achieving RBC-TI with a duration of at least 12 weeks is the absence of any RBC transfusion during any consecutive 84-day period during the treatment period (week 1 to week 24)
To control the overall type I error rate of 0.025 due to multiplicity, the end points were tested using a sequential gate-keeping method. Only if the result from the primary efficacy analysis (RBC-TI of ≥ 8 weeks [week 1 to week 24]) in the ITT population showed statistical significance was the secondary end point tested next (RBC-TI ≥ 12 weeks analysis [first tested for week 1 to week 48 and then week 1 to week 24]). The Cochran-Mantel-Haenszel test was used to compare the 2 treatment groups and estimate the common risk difference accounting for the stratification variables.
All other end points analyzed in the study were not included in the gate-keeping procedures and did not control for type I errors (i.e., RBC units transfused; duration of RBC-TI, time to RBC-TI, mean change in hemoglobin and mHI-E, HRQoL, overall survival, iron accumulation, ICT use, progression to AML, and health care resource utilization).
Units of RBCs transfused was summarized for each treatment group using descriptive statistics. Duration of RBC-TI was estimated using Kaplan–Meier methods and the 2 treatment groups were compared using a stratified log-rank test. The KM estimates for median duration of RBC-TI as well as 2-sided 95% CIs were summarized for each treatment group, adjusted for the stratification variables (RBC transfusion burden at baseline [≥ 6 units over 8 weeks versus < 6 units over 8 weeks, based on the mean of the 2 consecutive 8-week periods immediately before randomization] and IPSS-R score at baseline [very low or low versus intermediate]). Cox models were used to calculate the hazard ratios. The proportion of patients achieving a mean hemoglobin increase of ≥ 1.0 g/dL, and proportion of patients achieving mHI-E were summarized using the Cochran-Mantel-Haenszel test to compare the 2 treatment groups. Time to RBC-TI (in days) was summarized using descriptive statistics by treatment group.
To assess the effect of luspatercept versus placebo on HRQoL, an analysis of change from week 1 was performed to compare the scores of the questionnaires at week 25, week 48, and end of treatment using analysis of variance models adjusted for baseline domain scores and randomization stratification factors. The least squares (LS) means (95% CIs and P values) for changes from baseline at each post-baseline visit for all domains within each treatment group, and the difference in the LS means (95% CIs and P value) between treatment groups at each post-baseline visit, at week 25 visit, were estimated. The HRQoL end points and all assessments were only descriptive analyses.
Mean serum ferritin change was summarized using an analysis of variance model, which compared the treatment difference between groups, with the stratification factors and baseline serum ferritin value as covariates.
Overall survival was estimated using KM methods and compared using a stratified log-rank test, stratifying by average baseline RBC transfusion requirement (≥ 6 units versus < 6 units of RBCs per 8 weeks) and baseline IPSS-R (very low or low versus intermediate risk). A stratified log-rank test was used for the confirmatory P value. The KM estimates for median overall survival and associated 2-sided 95% CIs were summarized for each treatment group. At the time of the final overall survival analysis, a stratified Cox proportional hazards model was used to estimate the corresponding hazard ratio and 2-sided 95% CI for luspatercept relative to placebo.
Progression to AML was estimated using KM methods and treatment groups were compared using a log-rank test. The KM estimates for median time to AML progression and 2-sided 95% CIs were summarized for each treatment group. At the time of the final analysis, a Cox proportional hazards model was used to estimate the corresponding hazard ratio and 2-sided 95% CI. Time to AML progression from initial MDS diagnosis is defined by the WHO classification as greater than 20% blasts in peripheral blood or bone marrow and was summarized using descriptive statistics for each treatment group.
Handling of Missing Data or Dropout
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Subgroups
Although a number of subgroups were identified by the sponsor, only those subgroups identified in the protocol will be presented (i.e., SF3B1 status, IPSS-R score, and baseline hematologic status). The baseline hematologic status was not part of the pre-planned subgroup analyses. The subgroup analyses by SF3B1 status and IPSS-R score status did not account for multiplicity of testing.
Analysis Populations
The ITT population consisted of all randomized patients regardless of whether the patient received study treatment. The ITT population was the primary analysis population for all efficacy outcomes.
The safety population consisted of all patients who were randomized and received at least 1 dose of the study treatment.
The HRQoL-evaluable population comprised all patients in the ITT population who completed the HRQoL assessment at baseline (screening) and at least 1 post-baseline assessment visit. The completion of an HRQoL assessment was defined for EORTC QLQ-C30 as greater than or equal to 50% of all items that were answered (i.e., greater than or equal to 15 items of the 30 items or a non-missing total score).
Results
Patient Disposition
In MEDALIST, 229 patients were randomized: 153 to the luspatercept treatment group and 76 to the placebo group. The proportion of patients who completed 24 weeks of treatment was similar in both treatment groups. The proportion of patients who completed 48 weeks of treatment was higher in the luspatercept treatment group. Only patients who were responders in the primary phase were eligible for the extension phase following the week-25 assessment. The percentage of patients who withdrew from the treatment due to lack of efficacy was 65.8% in the placebo treatment group and 33.3% in the luspatercept treatment group. As of the May 8, 2018, cut-off date, 12 and 9 deaths were reported in the luspatercept and placebo treatment groups, respectively.
Table 14 presents the patient disposition of the MEDALIST study.
Table 14: Patient Disposition (ITT Population) in the MEDALIST Trial
Patient disposition | Luspatercept + BSC | Placebo + BSC |
|---|---|---|
Screened, N | 290 | |
Randomized, N (%) | 153 | 76 |
Patients completing treatment, n (%) | ||
Completed 24 weeks of treatment, n (%) | 128 (83.7) | 68 (89.5) |
Completed 48 weeks of treatment, n (%) | 78 (51.0) | 12 (15.8) |
Patients remaining as of data cut-off date | 70 (45.8) | 6 (7.9) |
Discontinued from treatment, n (%) | 83 (54.2) | 70 (92.1) |
Reason for discontinuation, n (%) | ||
Lack of efficacy | 51 (33.3) | 50 (65.8) |
Withdrawal by patient | 14 (9.2) | 10 (13.2) |
Adverse Event | 10 (6.5) | 4 (5.3) |
Progressive diseasea | 3 (2.0) | 2 (2.6) |
Protocol violation | 1 (0.7) | 0 |
Otherb | 4 (2.6) | 4 (5.3) |
Discontinued from study, n (%) | 30 (19.6) | 19 (25.0) |
Reason for discontinuation, n (%) | ||
Withdrawal by patient | 13 (8.5) | 8 (10.5) |
Death | 12 (7.8) | 9 (11.8) |
Lost to follow-up | 2 (1.3) | 0 |
Otherc | 3 (2.0) | 2 (2.6) |
ITT, N | 153 | 76 |
HRQoL-evaluable population, N | 149 | 76 |
Safety, N | 153 | 76 |
BSC = best supportive care; HRQoL = health-related quality of life; ITT = intention to treat.
aProgressed to high-risk myelodysplastic syndromes or AML.
bOther reasons for treatment discontinuation in the included investigator’s decision, lack of efficacy, and pre-existing condition precluding continued dosing following protocol amendment.
cOther reasons for study discontinuation included withdrawn consent and worsening of existing clinical condition.
Source: Clinical Study Report for MEDALIST.9
Protocol Violation
At least 1 protocol violation was reported in 13.5% of the patients in the total population: 15.7% in the luspatercept treatment group, and 9.2% in the placebo group. Inclusion criteria were not met by 5.9% and 3.9% of the patients in the luspatercept and placebo treatment arms, respectively. Protocol violations related to good clinical practice were reported for 5.2% and 3.9% of the patients in the luspatercept and placebo treatment arms, respectively. Patients who did not meet all the inclusion criteria most frequently had failed to meet the transfusion requirements, ESA refractory, intolerance, and/or ineligibility status, or less than 4 weeks had elapsed between the last dose of prior ESA or G-CSF therapy and randomization. The protocol violations in the trial are listed in Table 15.
Table 15: Protocol Violations (ITT Population) in the MEDALIST Trial
Protocol violations | Luspatercept + BSC N = 153 | Placebo + BSC N = 76 |
|---|---|---|
Number of patients with ≥ 1 protocol violation, n (%) | 24 (15.7) | 7 (9.2) |
Entered study but patient did not meet inclusion criteria, n (%) | 9 (5.9) | 3 (3.9) |
Good clinical practice issues, n (%) | 8 (5.2) | 3 (3.9) |
Investigational product issues, n (%) | 6 (3.9) | 0 |
Developed study drug withdrawal criteria but were not withdrawn from treatment, n (%) | 2 (1.3) | 0 |
Concomitant medication and/or procedure, n (%) | 1 (0.7) | 0 |
Missing visit or assessment/procedure, n (%) | 0 | 1 (1.3) |
ITT = intention to treat.
Source: Clinical Study Report for MEDALIST.9
Exposure to Study Treatments
A summary of exposure to study treatment was conducted in the safety population. The mean treatment duration was 46.6 weeks (SD = 24.14) and 30.6 weeks (SD = 15.19) in the luspatercept and placebo treatment groups, respectively. The mean number of doses (each dose was administered every 3 weeks) that each patient received was 15.2 (SD = 8.10) and 10.1 (SD = 5.03) in the luspatercept and placebo treatment groups, respectively. In the luspatercept and placebo treatment groups, 30.7% and 9.2% of the patients, respectively, received between 17 to 24 doses per patient; 40.3% and 32.3% of the patients, respectively, received dose levels of 1.0 mg/kg; and 35% and 43.6% of the patients, respectively, received the maximum dose level of 1.75 mg/kg.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below. As of the May 8, 2018, data analysis cut-off date, not all patients had completed 24 weeks of treatment. Appendix 3 provides detailed efficacy data. Results of the ad hoc analysis summarizing data from July 1, 2019, are presented in Appendix 4.
Hematologic Response
The primary outcome of the MEDALIST study was the proportion of patients who achieved RBC-TI for at least 8 weeks (any consecutive 56-day period) from week 1 to week 24. The primary end point was achieved by 37.9% of the patients in the luspatercept treatment group and 13.16% of the patients in placebo group, with a common risk difference in the response rate of 24.56 (95% CI, 14.48 to 34.64). The odds ratio of 5.06 (95% CI, 2.28 to 11.25; P < 0.0001) favoured the luspatercept treatment over placebo. The results of the primary efficacy end point are presented in Table 16.
Table 16: RBC-TI of 8 Weeks or More During Week 1 Through Week 24 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Number of responders, n (%) | 58 (37.9) | 10 (13.2) |
Response rate, % (95% CI) | 37.91 (30.20 to 46.10) | 13.16 (6.49 to 22.87) |
Common risk difference on response rate, % (95% CI) | 24.56 (14.48 to 34.64) | |
Odds ratio (95% CI)a | 5.06 (2.28 to 11.26) | |
P value | < 0.0001 | |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
aCochran-Mantel-Haenszel test stratified for average baseline red blood cell–transfusion requirement, and baseline Revised International Prognostic Scoring System score.
Source: Clinical Study Report for MEDALIST.9
The 2 secondary outcomes of the MEDALIST study were the proportion of patients who achieved RBC-TI for at least 12 weeks (any consecutive 84-day period) from week 1 to week 48 and week 1 to week 24. During week 1 to week 48, 33.3% of the patients in the luspatercept treatment group and 11.84% of the patients in the placebo group responded to the treatment, with a common risk difference in the response rate of 21.37 (95% CI, 11.23 to 31.51). The odds ratio of 4.04 (95% CI, 1.82 to 8.96; P = 0.0003) favoured the luspatercept treatment over placebo. The results of this secondary efficacy end point are presented in Table 17.
Table 17: RBC-TI of 12 Weeks or More During Week 1 Through Week 48 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Number of responders, n (%) | 51 (33.3) | 9 (11.84) |
Response rate, % (95% CI) | 33.33 (25.93 to 41.40) | 11.84 (5.56 to 21.29) |
Common risk difference on response rate, % (95% CI) | 21.37 (11.23 to 31.51) | |
Odds ratio (95% CI)a | 4.045 (1.827 to 8.956) | |
P value | 0.0003 | |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
aCochran-Mantel-Haenszel test stratified for average baseline red blood cell–transfusion requirement, and baseline Revised International Prognostic Scoring System score.
Source: Clinical Study Report for MEDALIST.9
During week 1 to week 24, 28.1% of the patients in the luspatercept treatment group and 7.89% of the patients in placebo group responded to the treatment, with a common risk difference in the response rate of 20.0 (95% CI, 10.92 to 29.08). The odds ratio of 5.07 (95% CI, 2.00 to 12.84; P = 0.0002) favoured the luspatercept treatment over placebo. The results of this secondary efficacy end point are presented in Table 18.
Table 18: RBC-TI of 12 Weeks or More During Week 1 through Week 24 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Number of responders, n (%) | 43 (28.10) | 6 (7.89) |
Response rate, % (95% CI) | 28.10 (21.14 to 35.93) | 7.89 (2.95 to 16.40) |
Common risk difference on response rate, % (95% CI) | 20.00 (10.92 to 29.08) | |
Odds ratio (95% CI)a | 5.07 (2.00 to 12.84) | |
P value | 0.0002 | |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
aCochran-Mantel-Haenszel test stratified for average baseline red blood cell–transfusion requirement, and baseline Revised International Prognostic Scoring System score.
Source: Clinical Study Report for MEDALIST.9
Assessments were also performed for the proportion of patients who achieved RBC-TI for at least 8 weeks (any consecutive 56-day period) from week 1 to week 48. This end point was outside the statistical testing hierarchy. The assessments found that 45.1% of the patients in the luspatercept treatment group and 15.8% of the patients in the placebo group responded to the treatment, with a common risk difference in the response rate of 29.55 (95% CI, 18.73 to 40.36). The odds ratio was 5.31 (95% CI, 2.53 to 11.15) between the luspatercept treatment group and the placebo treatment group. The results of this end point are presented in Table 19.
Table 19: RBC-TI of 8 Weeks or More During Week 1 Through Week 48 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Number of responders, n (%) | 69 (45.10) | 12 (15.79) |
Response rate, % (95% CI) | 45.10 (37.05 to 53.34) | 15.79 (8.43 to 25.96) |
Common risk difference on response rate, % (95% CI) | 29.55 (18.73 to 40.36) | |
Odds ratio (95% CI)a | 5.31 (2.53 to 11.15) | |
P value | < 0.0001 | |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
aCochran-Mantel-Haenszel test stratified for average baseline red blood cell–transfusion requirement, and baseline Revised International Prognostic Scoring System score.
Source: Clinical Study Report for MEDALIST.9
RBC Units Transfused
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The mean ||||||| change from baseline of RBC units transfused over week 9 to week 24 was −3.0 (||) and 0.4 (||) in the luspatercept treatment group (n = 128) and placebo treatment group (n = 68), respectively. The median (minimum to maximum) change from baseline of RBC units transfused over week 9 to week 24 was −4.0 (−18 to 14) and 0 (−15 to 9) in the luspatercept treatment group and placebo treatment group, respectively. The mean ||||||| change from baseline of RBC units transfused over week 33 to week 48 was −4.9 (||) and −3.9 (||) in the luspatercept treatment group (n = 78) and placebo treatment group (n = 12), respectively. The median (minimum to maximum) change from baseline of RBC units transfused over week 33 to week 48 was −5.0 (−21 to 7) and −2.5 (−21 to 3) in the luspatercept treatment group and placebo treatment group, respectively.
Duration of Red Blood Cell–Transfusion Independence
The median (minimum to maximum) durations of longest single episodes of RBC-TI of 8 weeks or longer based on KM estimates were 30.6 (20.6 to 40.6) weeks and 13.6 (9.1 to 54.9) weeks in the luspatercept treatment group and placebo group, respectively. Of the patients who responded to treatment and achieved RBC-TI of at least 8 weeks from week 1 through week 24, 34.5% in the luspatercept treatment group and 20.0% in the placebo group maintained response as of the cut-off date. In addition, 62.07% of the patients who responded to luspatercept treatment had more than 1 episode of response during the treatment period of week 1 to week 24, and 22% of the patients who responded to luspatercept treatment had 3 or more episodes of RBC-TI of at least 8 weeks during the entire treatment phase. Four of the patients who were responders in the placebo treatment group had more than 1 episode of response. No patients in the placebo treatment group had more than 2 episodes of RBC-TI of at least 8 weeks. These results are presented in Table 20 and Figure 3.
Table 20: Duration of RBC-TI of 8 Weeks or More in Patients Who Responded During Week 1 Through Week 24 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 58) | Placebo + BSC (N = 10) | |
Median duration, weeks (95% CI)a | 30.6 (20.6 to 40.6) | 13.6 (9.1 to 54.9) |
Patients who maintained response (censored), n (%) | 20 (34.5) | 2 (20.0) |
Patients who lost response, n (%) | 38 (65.5) | 8 (80.0) |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
Note: Patients who lost response are those who received red blood cell transfusions following an RBC-TI period. Patients who maintained RBC-TI at the time of the analysis were censored.
aMedian and 95% CI was from the unstratified Kaplan–Meier method.
Source: Clinical Study Report for MEDALIST.9
Figure 3: Kaplan–Meier Curve of Duration of RBC-TI of 8 Weeks or More in Patients Who Responded During Weeks 1 Through 24 (ITT Population)
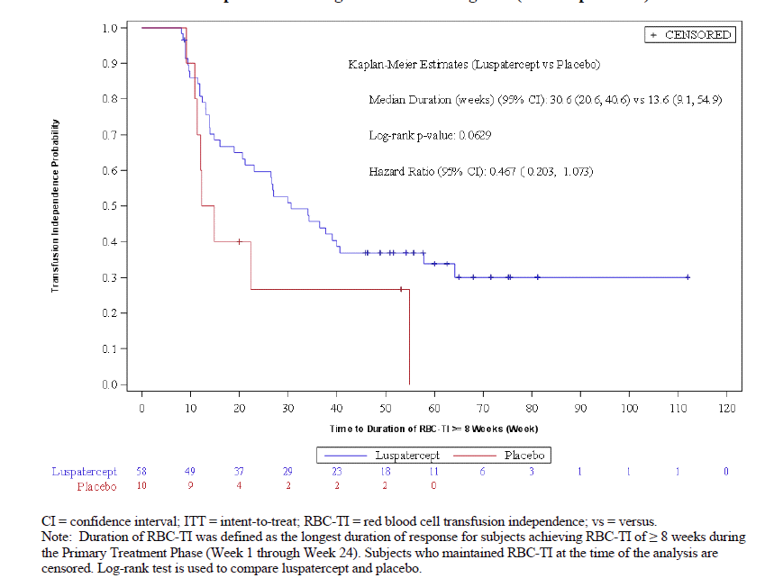
CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence; vs = versus.
Note: Duration of RBC-TI was defined as the longest duration of response for subjects achieving RBC-TI of at least 8 weeks during the primary treatment phase (week 1 through week 24). Subjects who maintained RBC-TI at the time of the analysis were censored. A log-rank test was used to compare luspatercept and placebo.
Source: Clinical Study Report for MEDALIST.9
During week 1 through week 48 the median durations (minimum to maximum) of the longest single episode of RBC-TI of at least 8 weeks based on KM estimates were 30.6 weeks (20.6 to 57.9) and 18.6 weeks (10.9 to 54.9) in the luspatercept treatment group and placebo group, respectively. The proportion of patients who responded to treatment and achieved RBC-TI of at least 8 weeks from week 1 through week 48 was 39.1% in the luspatercept treatment group (n = 69), while 33.3% of the placebo group (n = 12) maintained response as of the cut-off date. These results are presented in Table 21 and Figure 4.
Table 21: Duration of RBC-TI of 8 Weeks or More in Patients Who Responded During Week 1 Through Week 48 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 69) | Placebo + BSC (N = 12) | |
Median duration, weeks (95% CI)a | 30.6 (20.6 to 57.9) | 18.6 (10.9 to 54.9) |
Patients who maintained response (censored), n (%) | 27 (39.1) | 4 (33.3) |
Patients who lost response, n (%) | 42 (60.9) | 8 (66.7) |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
Note: Patients who lost response are those who received RBC transfusion following an RBC-TI period. Patients who maintained RBC-TI at the time of the analysis were censored.
aMedian was from the unstratified Kaplan–Meier method.
Source: Clinical Study Report for MEDALIST.9
Figure 4: Kaplan–Meier Curve of Duration of RBC-TI of 8 Weeks or More in Patients Who Responded During Weeks 1 Through 48 (ITT Population)
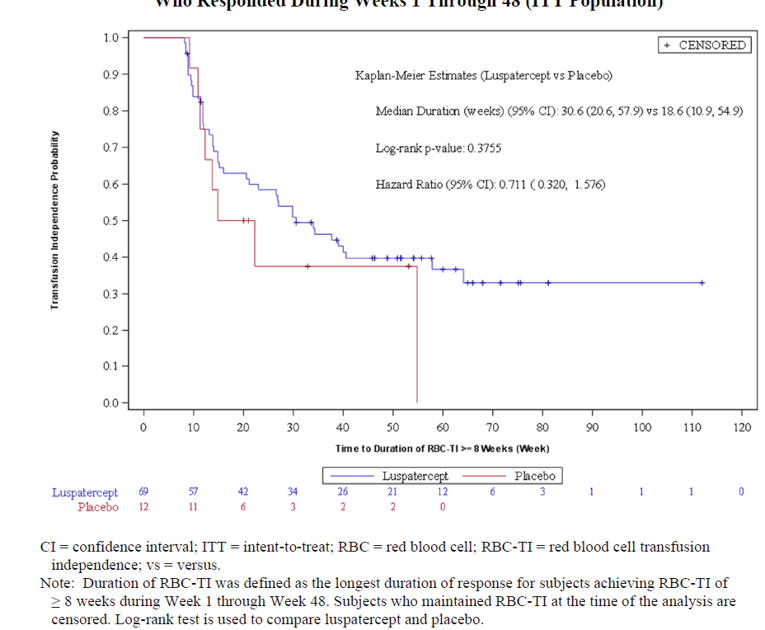
CI = confidence interval; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence; vs = versus.
Note: Duration of RBC-TI was defined as the longest duration of response for subjects achieving RBC-TI of at least 8 weeks during the primary treatment phase (week 1 through week 24). A log-rank test was used to compare luspatercept and placebo. Patients who maintained RBC-TI at the time of the analysis were censored.
Source: Clinical Study Report for MEDALIST.9
Analysis of RBC-TI of more than 8 weeks and transfusion reduction by baseline transfusion burden during weeks 1 through week 24 is provided in Table 22.
Table 22: RBC-TI of 8 Weeks or More and Transfusion Reduction by Baseline Transfusion Burden During Weeks 1 to 24 (ITT Population)
Characteristics | MEDALIST | ||
|---|---|---|---|
Luspatercept + BSC | Placebo + BSC | P value | |
Transfusion independence, n/N (%) | |||
≥ 8 weeks | 58/153 (37.91) | 10/76 (13.16) | < 0.0001 |
≥ 6 weeks | 6/66 (9.1) | 1/33 (3.0) | 0.2699 |
≥ 4 and < 6 weeks | 15/41 (36.6) | 1/23 (4.3) | 0.0046 |
< 4 weeks | 37/46 (80.4) | 8/20 (40.0) | 0.0013 |
Transfusion reduction, n/N (%) | |||
Reduction of 4 RBC units over 8 weeks | 52/107 (48.6) | 8/56 (14.3) | < 0.0001 |
≥ 4 and < 6 RBC units over 8 weeks | 16/41 (39.0) | 1/23 (4.3) | 0.0028 |
≥ 6 RBC units over 8 weeks | 36/66 (54.5) | 7/33 (21.2) | 0.0017 |
BSC = best supportive care; ITT = intention to treat; RBC = red blood cell; RBC-TI = red blood cell–transfusion independence.
aFrom a Cochran-Mantel-Haenszel test.
Source: Clinical Study Report for MEDALIST.9
Time to Red Blood Cell–Transfusion Independence
The mean for the time to RBC-TI of 8 weeks or more for responses achieved during week 1 through week 24 was 17.2 days (SD = 29.4) and 26.0 days (SD = 31.83) in the luspatercept and placebo treatment groups, respectively. The mean time to RBC-TI of 8 weeks or more for responses achieved during week 1 through week 48 was 40.3 days (SD = 61.03) and 57.2 days (SD = 79.18) in the luspatercept and placebo treatment groups, respectively. Multiple responses occurred, with 62.07% of the patients who responded to luspatercept treatment experiencing more than a single episode of response during the treatment period of week 1 to week 24. In addition, 22% of the patients who responded to luspatercept treatment had 3 or more episodes of RBC-TI of at least 8 weeks during the entire treatment phase. Four of the patients who were responders in the placebo treatment group had more than 1 episode of response. No patients in the placebo treatment group had more than 2 episodes of RBC-TI of a least 8 weeks. These results are shown in Table 23.
Table 23: Time to RBC-TI of 8 Weeks or More for Responses Achieved During Week 1 to Week 24 and Week 1 to Week 48 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC | Placebo + BSC | |
Time to RBC-TI of 8 weeks or more for responses achieved during week 1 to week 24 (days) | ||
n (%) | 58 (37.9) | 10 (13.1) |
Mean (SD) | 17.2 (29.40) | 26.0 (31.83) |
Median (minimum to maximum) | 1.0 (1.0 to 106.0) | 17.0 (1.0 to 100.0) |
Time to RBC-TI of 8 weeks or more for responses achieved during week 1 to week 48 (days) | ||
n (%) | 69 (45.0) | 12 (15.8) |
Mean (SD) | 40.3 (61.03) | 57.2 (79.18) |
Median (minimum to maximum) | 2.0 (1.0 to 232.0) | 22.5 (1.0 to 241.0) |
BSC = best supportive care; ITT = intention to treat; RBC-TI = red blood cell–transfusion independence; SD = standard deviation.
Source: Clinical Study Report for MEDALIST.9
Change in Hemoglobin
The proportions of patients who had a mean change in hemoglobin of at least 1.0 g/dL during week 1 through week 24 were 35.3% (95% CI, 27.75 to 43.42) in the luspatercept treatment group and 7.9% (95% CI, 2.95 to 16.40) in the placebo treatment group. The proportions of patients who had a mean change in hemoglobin of at least 1.0 g/dL during week 1 through week 48 were 41.2% (95% CI, 33.29 to 49.41) in the luspatercept treatment group and 10.5% (95% CI, 4.66 to 19.69) in the placebo treatment group. These results are shown in Table 24.
Table 24: Mean Hemoglobin Increase of 1.0 g/dL or Greater (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Week 1 to week 24 | ||
n (%) | 54 (35.3) | 6 (7.9) |
95% CI | 27.75 to 43.42 | 2.95 to 16.40 |
P valuea | < 0.0001 | |
Week 1 to week 48 | ||
n (%) | 63 (41.2) | 8 (10.5) |
95% CI | 33.29 to 49.41 | 4.66 to 19.69 |
P valuea | < 0.0001 | |
CI = confidence interval; ITT = intention to treat.
aFrom Fisher’s exact test.
Source: Clinical Study Report for MEDALIST.9
Modified Hematologic Improvement–Erythroid
The proportions of patients who had an mHI-E during week 1 through week 24 were 52.9% (95% CI, 44.72 to 61.05) in the luspatercept treatment group and 11.8% (95% CI, 5.56 to 21.29) in the placebo treatment group. The proportions of patients who had an mHI-E during week 1 through week 48 were 58.8% (95% CI, 50.59 to 66.71) in the luspatercept treatment group and 17.1% (95% CI, 9.43 to 27.47) in the placebo treatment group. These results are shown in Table 25.
Table 25: Patients Who Achieved mHI-E (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Week 1 to week 24 | ||
n (%) | 81 (52.9) | 9 (11.8) |
95% CI | 44.72 to 61.05 | 5.56 to 21.29 |
P valuea | < 0.0001 | |
RBC transfusion reduction of 4 units over 8 weeks, n (%) | 52/107 (48.6) | 8/56 (14.3) |
Mean hemoglobin increase of ≥ 1.5 g/dL for 8 weeks, n (%) | 29/46 (63.0) | 1/20 (5.0) |
Week 1 to week 48 | ||
n (%) | 90 (58.8) | 13 (17.1) |
95% CI | 50.59 to 66.71 | 9.43 to 27.47 |
P valuea | < 0.0001 | |
RBC transfusion reduction of 4 units over 8 weeks, n (%) | 58/107 (54.2) | 12/56 (21.4) |
Mean hemoglobin increase of ≥ 1.5 g/dL for 8 weeks, n (%) | 32/46 (69.6) | 1/20 (5.0) |
BSC = best supportive care; CI = confidence interval; ITT = intention to treat; mHI-E = modified hematologic improvement–erythroid; RBC = red blood cell.
aFrom a Cochran-Mantel-Haenszel test.
Source: Clinical Study Report for MEDALIST.9
HRQoL
In the MEDALIST study, HRQoL was measured using 2 instruments, the EORTC QLQ-C30 and QoL-E. All domains of the QoL-E were considered exploratory, and these results are provided in Appendix 3.
EORTC QLQ-C30 – Global Health Status
In the luspatercept treatment group the mean change from baseline at week 25 (Table 26) in the QLQ-C30 for global health status was |||||||||||||| and in the placebo treatment group it was ||||||||||||||.
Table 26: Summary of EORTC QLQ-C30 Change from Baseline in Global Health by Visit at Week 25 (HRQoL-Evaluable Population) — Redacted
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
|||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||
EORTC QLQ-C30 – Physical Functioning Status
In the luspatercept treatment group the mean change from baseline at week 25 (Table 27) in the QLQ-C30 for physical functioning status was |||||||||||||||| and in the placebo treatment group it was ||||||||.
Table 27: Summary of EORTC QLQ-C30 Change from Baseline in Physical Functioning by Visit at Week 25 (HRQoL-Evaluable Population) — Redacted
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||
EORTC QLQ-C30 – Fatigue Status
In the luspatercept treatment group the mean change from baseline at week 25 (Table 28) in the QLQ-C30 for fatigue status was ||||||||||||||||||||| and in the placebo treatment group it was ||||||||||||||||||.
Table 28: Summary of EORTC QLQ-C30 Change From Baseline in Fatigue by Visit at Week 25 (HRQoL-Evaluable Population) — Redacted
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||
EORTC QLQ-C30 – Emotional Functioning Status
In the luspatercept treatment group the mean change from baseline at week 25 (Table 29) in the QLQ-C30 for emotional functioning status was |||||||||||| and in the placebo treatment group it was ||||||||||||||.
Table 29: Summary of EORTC QLQ-C30 Change From Baseline in Emotional Functioning by Visit at Week 25 (HRQoL-Evaluable Population) — Redacted
||||||||||||||||||||||||||||| | MEDALIST | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
Week 25 | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
Change from baseline at week 25 | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||
EORTC QLQ-C30 – Dyspnea Status
In the luspatercept treatment group the mean (SD) change from baseline at week 25 (Table 30) in the QLQ-C30 for dyspnea status was |||||||||| and in the placebo treatment group it was ||||||||||||||
Table 30: Summary of EORTC QLQ-C30 Change From Baseline in Dyspnea by Visit at Week 25 (HRQoL-Evaluable Population) — Redacted
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||
Overall Survival
At the time of the data cut-off date (May 8, 2018), 7.8% of patients in the luspatercept treatment group and 11.8% of patients in the placebo group had died. Censored patients accounted for 92.2% of the patients in the luspatercept treatment group and 88.2% patients in the placebo group. The median (minimum to maximum) follow-up times for overall survival were 13.9 (2.8 to 26.2) and 14.3 (1.7 to 21.8) months in the luspatercept and placebo treatment groups, respectively. The estimated hazard ratio based on KM estimates was 0.763 (95% CI, 0.318 to 1.829; P = 0.5427). The median overall survival was not reached in either treatment group. These results are presented in Table 31.
Table 31: Overall Survival (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Patients alive (censored), n (%) | 141 (92.2) | 67 (88.2) |
Patients died, n (%) | 12 (7.8) | 9 (11.8) |
Kaplan–Meier estimates | ||
Median (months) (95% CI)a | NE | NE |
P valueb | 0.5427 | |
Hazard ratio (95% CI)c | 0.763 (0.318 to 1.829) | |
Summary of follow-up time (months) | ||
n (%) | 153 (100) | 76 (100) |
Mean (SD) | 14.1 (4.62) | 14.1 (4.28) |
Median (minimum to maximum) | 13.9 (2.8 to 26.2) | 14.3 (1.7 to 21.8) |
CI = confidence interval; ITT = intention to treat; NE = not evaluable.
aMedian was from the Kaplan–Meier method stratified by average baseline red blood cell–transfusion requirement and baseline Revised IPSS score.
bP value from log-rank test.
cHazard ratio from Cox proportional hazards model with red blood cell–transfusion requirement and baseline Revised IPSS score as covariates.
Source: Clinical Study Report for MEDALIST.9
Iron Accumulation
Iron accumulation was measured through mean change in mean serum ferritin levels, which were calculated during weeks 9 through 24 and weeks 33 through 48. Among those with a baseline and follow-up measurement, the mean changes in serum ferritin level from baseline (Table 32) were −2.7 mcg/L (SD = 54.05) in the luspatercept treatment group and 226.5 mcg/L (68.02) in the placebo treatment group. The LS mean difference for the luspatercept treatment group versus placebo treatment group was −229.1 (standard error [SE] = 74.43; 95% CI, −375.8 to −82.4) for week 9 through week 24. For weeks 33 through 48 the mean changes in serum ferritin level from baseline (Table 32) were −72.0 mcg/L (SE = 74.76) in the luspatercept treatment group and 247.4 mcg/L (SE = 140.96) in the placebo treatment group. The LS mean difference for the luspatercept treatment group versus placebo treatment group was −319.5 (SE = 144.57; 95% CI, −606.3 to −32.7).
Table 32: Mean Change in Mean Serum Ferritin Level (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Mean change from baseline averaged over week 9 through week 24 (mcg/L) | ||
n (%) | 148 (96.7) | 74 (97.3) |
LS mean (SE) | −2.7 (54.05) | 226.5 (68.02) |
LS mean of difference (SE) | −229.1 (74.43; 95% CI, −375.8 to −82.4) | |
P value a | 0.0024 | |
Mean change from baseline averaged over week 33 through week 48 (mcg/L) | ||
n (%) | 89 (58.1) | 16 (21.0) |
LS mean (SE) | −72.0 (74.76) | 247.4 (140.96) |
LS mean of difference (SE) | −319.5 (144.57; 95% CI, −606.3 to −32.7) | |
P valuea | 0.0294 | |
BSC = best supportive care; CI = confidence interval; LS = least squares; SE = standard error.
aEstimates were based on an analysis of covariance model, with the change in value as the dependent variable, treatment group (2 levels) as a factor, and baseline serum ferritin value as covariates, stratified by average baseline red blood cell–transfusion requirement, and baseline Revised IPSS.
Source: Clinical Study Report for MEDALIST.9
ICT Use
During week 9 through week 24, the LS mean changes from baseline in average ICT dose were 10.0 mg/day (SE = 29.25; 95% CI, −47.7 to 67.7) in the luspatercept treatment group (n = 128) and 51.0 mg/day (SE = 35.92; 95% CI, −19.9 to 121.8) in the placebo group (n = 68). The LS mean difference between luspatercept and placebo was −1.0 (SE = 40.18; 95% CI, −120.3 to 38.2). From week 33 through week 48, the LS mean changes from baseline in average ICT dose were −148.8 mg/day (SE = 46.13; 95% CI, −240.5 to −57.1) in the luspatercept treatment group (n = 78) and −123.8 mg/day (SE = 92.19; 95% CI, −307.1 to 59.5) in the placebo group (n = 12). The LS mean difference between luspatercept and placebo was −24.9 (SE = 93.42; 95% CI, −210.7 to 160.8). These results are shown in Table 33.
Table 33: Change in Mean Daily Dose (mg) of ICT (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Mean change from baseline for mean daily dose (mg/day) of ICT averaged over weeks 9 through 24 | ||
n (%) | 128 (83.6) | 68 (89.4) |
LS mean (SE) | 10.0 (29.25) | 51.0 (35.92) |
95% CI for LS mean | (−47.7 to 67.7) | (−19.9 to 121.8) |
LS mean difference (SE) | −41.0 (40.18) | |
95% CI for LS mean difference | (−120.3 to 38.2) | |
P valuea | 0.3087 | |
Mean change from baseline for mean daily dose (mg/day) of ICT averaged over weeks 33 through 48 | ||
n (%) | 78 (50.9) | 12 (15.7) |
LS mean (SE) | −148.8 (46.13) | −123.8 (92.19) |
95% CI for LS mean | (−240.5 to −57.1) | (−307.1 to 59.5) |
LS mean difference (SE) | −24.9 (93.42) | |
95% CI for LS mean difference | (−210.7 to 160.8) | |
P valuea | 0.7903 | |
BSC = best supportive care; CI = confidence interval; ICT = iron chelation therapy; ITT = intention to treat; LS = least squares; SE = standard error.
aFrom analysis of covariance, with the change in daily dose as the dependent variable, treatment group (2 levels) as a factor, and baseline ICT value as covariates, stratified by average baseline red blood cell–transfusion requirement, and baseline IPSS-R = Revised International Prognostic Scoring System.
Source: Clinical Study Report for MEDALIST.9
Progression to AML
Three patients (2%) in the luspatercept treatment group and 1 patient (1.3%) in the placebo group experienced disease progression to AML during the treatment period. The median time to progression to AML was not reached in either treatment group.
Health Care Resource Utilization
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| || |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| These results are shown in Table 34.
Table 34: Health Care Resource Utilization (ITT Population) — Redacted
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
|---|---|---|
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||| |
||||||||||||||||||||
||||||||||||||||||||||||||||
Subgroup Analyses
Subgroup analyses conducted in the MEDALIST study that were identified as being of interest in the CADTH review protocol were IPSS-R score (very low risk or low risk versus intermediate risk), and baseline hematological status. Treatment response occurred in 40% of the IPSS-R intermediate-risk patients in the luspatercept treatment group versus 7.7% of the IPSS-R patients at intermediate risk in the placebo group (odds ratio = 8.00; 95% CI, 0.89 to 71.6; P = 0.0398). In the low-risk patients, a treatment response was observed in 37.6% of the patients in the luspatercept group and 14.0% of the patients in the placebo group (odds ratio = 3.69; 95% CI, 1.59 to 8.57; P = 0.0016). In the patients at very low risk, a treatment response was observed in 38.9% in the luspatercept group and 16.7% in the placebo group (odds ratio = 3.18; 95% CI, 0.30 to 33.3; P = 0.3276).
Harms
Only those harms identified in the review protocol are reported below. Table 35 provides detailed harms data.
Adverse Events
In the MEDALIST trial, 98.0% and 92.1% of the patients in the luspatercept and placebo groups reported at least 1 adverse event, respectively. The most commonly occurring adverse events were fatigue (26.8% and 13.2%, respectively), diarrhea (22.2% and 9.2%, respectively), nausea (20.3% and 7.9%, respectively), and dizziness (19.6% and 5.3%, respectively).
Serious Adverse Events
In the MEDALIST trial, serious adverse events were reported by 31.4% of the patients in the luspatercept treatment arm and 30.3% of the patients in the placebo group. The most common serious adverse event was pneumonia, which was reported by 2% of the patients in the luspatercept group and 2.6% of the patients in the placebo group. Four patients (2.6%) reported a thromboembolic event in the luspatercept treatment group, and 3 patients (3.9%) reported a thromboembolic event in the placebo treatment group.
Withdrawals Due to Adverse Events
The proportion of patients who stopped treatment due to an adverse event was 8.5% and 7.9% in the luspatercept and placebo treatment groups, respectively. The most common reason for stopping treatment was benign, malignant, and unspecified neoplasms (including cysts and polyps), which were reported for 3 patients (2%) in the luspatercept treatment group and 2 patients (2.6%) in the placebo treatment group. Nervous system disorders, which included headache, memory impairment and Parkinson disease, led to cessation of treatment for 3 patients (2%) in the luspatercept treatment group, and general disorders and administration site conditions, which included fatigue and general physical health deterioration, caused 2 patients (1.3%) in the luspatercept treatment group and 1 patient (1.3%) in the placebo treatment group to cease treatment. Two patients (1.3%) in the luspatercept treatment group withdrew from the study due to renal and urinary disorders, with 1 each (0.7%) withdrawing due to chronic kidney disease and renal failure.
Mortality
During the treatment period 3.3% of patients (n = 5) in the luspatercept treatment group and 5.3% of patients (n = 4) in the placebo treatment group died. In the luspatercept treatment group 1 patient died due to multiple organ dysfunction syndrome (classified as general disorders and administration site condition), 2 patients died of sepsis (classified as infections and infestations), 1 patient died due to renal failure (classified as a renal or urinary disorder), and 1 patient died of a shock hemorrhagic (classified as a vascular disorder). In the placebo treatment arm 1 patient died due general physical health deterioration, 1 patient died due to urosepsis (both classified as general disorders and administration site conditions), 1 patient died of sepsis (classified as infection or infestation), and 1 patient died of respiratory failure (classified as respiratory, thoracic, or mediastinal disorder).
In the post-treatment period, an additional 4.6% (n = 7) and 6.6% (n = 5) of patients in the luspatercept treatment group and the placebo treatment group, respectively, died. Two patients died in the luspatercept treatment group due to infections and infestations (1 each due to sepsis and soft tissue infection). Two patients died due to sepsis in the placebo treatment group. One patient in the luspatercept treatment group died due to disease progression (to high-risk MDS). One patient in the luspatercept treatment group died due to progression to AML, and 1 patient died due to MDS. One patient in the luspatercept treatment group died due to a myocardial infarction. One patient in the luspatercept treatment group died due to intestinal ischemia. One patient in the placebo treatment group died due to hepatic failure.
Notable Harms
The notable harms identified in the CADTH review protocol included the following: thromboembolic events, hypertension, hepatic and renal events, hypersensitivity reactions, and malignancies. In the luspatercept treatment group 2.6% of patients (n = 4) and in the placebo treatment group 3.9% patients (n = 3) experienced a thromboembolic and thrombophlebitis event. Under the SOC of hepatobiliary disorders, 5.2% of patients in the luspatercept treatment group and 5.3% of patients in the placebo group reported at least 1 associated adverse event. Under the SOC of renal and urinary disorders, 18.3% of patients in the luspatercept treatment group and 13.2% of patients in the placebo group reported at least 1 associated adverse event. Hypertension was reported as an adverse event in 8.5% patients in the luspatercept treatment group and 7.9% patients in the placebo group. Malignancies (transformation to AML, basal cell carcinoma, or squamous cell carcinoma of skin) were reported in 3.3% of patients in the luspatercept treatment group, and 1.3% of patients in the placebo group reported at least 1 associated adverse event. No systemic hypersensitivity reactions were reported in either treatment group.
Table 35: Summary of Harms (Safety Population)
Harms | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Patients with ≥ 1 adverse event | ||
n (%) | 150 (98.0) | 70 (92.1) |
NCI CTCAE Grade ≥ 3 TEAE, n (%) | 65 (42.5) | 34 (44.7) |
TEAE leading to dose interruption, n (%) | 23 (15.0) | 4 (5.3) |
TEAE leading to dose reduction, n (%) | 7 (4.6) | 0 |
Patients with at least 1 thromboembolic event, n (%) | 4 (2.6) | 3 (3.9) |
Patients with ≥ 1 SAE | ||
n (%) | 48 (31.4) | 23 (30.3) |
Pneumonia, n (%) | 3 (2.0) | 2 (2.6) |
Patient with at least 1 TEAE leading to study drug discontinuation | ||
n (%) | 13 (8.5) | 6 (7.9) |
Neoplasms benign, malignant, and unspecified, n (%) | 3 (2.0) | 2 (2.6) |
Nervous system disorders, n (%) | 3 (2.0) | 0 |
General disorders and administration site conditions, n (%) | 2 (1.3) | 1 (1.3) |
Infections and infestations, n (%) | 2 (1.3) | 0 |
Renal and urinary disorders, n (%) | 2 (1.3) | 0 |
Investigations, n (%) | 1 (0.7) | 0 |
Musculoskeletal and connective tissue disorders, n (%) | 1 (0.7) | 1 (1.3) |
Respiratory, thoracic, and mediastinal disorders, n (%) | 1 (0.7) | 1 (1.3) |
Vascular disorders, n (%) | 1 (0.7) | 0 |
Blood and lymphatic system disorders, n (%) | 0 | 1 (1.3) |
Deaths during treatment period | ||
n (%) | 5 (3.3) | 4 (5.3) |
General disorders and administration site conditions | 1 (0.7) | 2 (2.6) |
Infections and infestations, n (%) | 2 (1.3) | 1 (1.3) |
Renal and urinary disorders, n (%) | 1 (0.7) | 0 |
Respiratory, thoracic, and mediastinal disorders | 0 | 1 (1.3) |
Vascular disorders | 1 (0.7) | 0 |
Notable harms (all grades, reported in at least 5% of the patients in either treatment group) | ||
Fatigue, n (%) | 41 (26.8) | 10 (13.2) |
Diarrhea, n (%) | 34 (22.2) | 7 (9.2) |
Asthenia, n (%) | 31 (20.3) | 9 (11.8) |
Nausea, n (%) | 31 (20.3) | 6 (7.9) |
Dizziness, n (%) | 30 (19.6) | 4 (5.3) |
Back pain, n (%) | 29 (19.0) | 5 (6.6) |
Cough, n (%) | 27 (17.6) | 10 (13.2) |
Edema peripheral, n (%) | 25 (16.3) | 13 (17.1) |
Headache, n (%) | 24 (15.7) | 5 (6.6) |
Hypertension, n (%) | 13 (8.5) | 6 (7.9) |
Fall, n (%) | 15 (9.8) | 9 (11.8) |
Neutropenia, n (%) | 7 (4.6) | 7 (9.2) |
Notable harms (grade 3 or higher, reported in at least 5% of the patients in either treatment group) | ||
Patient with at least 1 TEAE, n (%) | 65 (42.5) | 34 (44.7) |
Anemia, n (%) | 10 (6.5) | 5 (6.6) |
Hypertension, n (%) | 5 (3.3) | 3 (3.9) |
Iron overload, n (%) | 3 (2.0) | 1 (1.3) |
AE = adverse event; BSC = best supportive care; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Source: Clinical Study Report for MEDALIST.9
Critical Appraisal
Internal Validity
The MEDALIST trial was a randomized placebo-controlled, double-blind study. Overall randomization (using an interactive response technology system) and treatment allocation, as stratified by RBC transfusion burden at baseline (≥ 6 units over 8 weeks versus < 6 units over 8 weeks) and IPSS-R score at baseline (very low or low versus intermediate) were conducted appropriately. The baseline patient, disease, and MDS treatment-history characteristics were generally well balanced.
The study was double-blinded, but the study design allowed for unblinding at the discretion of the investigator and it was unclear how many patients were unblinded. In addition, it is unclear if blinding was maintained for patients who discontinued the study drug but remained in the study, or how many patients elected to be unblinded. A total of 31 protocol violations were reported and 1 patient withdrew from treatment due to a protocol violation. There were more violations in the luspatercept treatment arm than in the placebo arm and this could have biased the efficacy results in favour of the treatment arm. While issues involving good clinical practice of the protocol violation were reported to the sponsor within 24 hours, how the blinding was handled during this time is unclear. Lack of efficacy within the placebo group and occurrence of more adverse events in the luspatercept group could have unblinded patients. Although it is unlikely to affect the primary or key secondary end points of the study, which were objective measures, it could result in bias with respect to the self-reporting of adverse events and subjective end points, including changes in HRQoL. The FDA noted that the blinding in the study is inadequate, increasing the risk of bias in a trial that uses an end point that may be manipulated due to conscious or unconscious bias. Specifically, the production of the placebo control syringe on site and the lack of specific instructions to mask the product increase the risk of accidental unblinding unacceptably,10 and could affect all outcomes, but particularly self-reported outcomes such as HRQoL.
The primary and key secondary end points were appropriately controlled for multiplicity and a hierarchical statistical plan was followed. The FDA similarly noted that the effect size of the primary end point of transfusion independence for 8 weeks in the study was small, with RBC-TI of 8 weeks obtained in only about 38% of patients with a differential response compared to placebo of about 25%. In other words, only about 1-quarter of the patients exposed to luspatercept had any apparent benefit, assuming that fulfillment of the primary objective represents a benefit to the patient.10 All other end points (other hematologic responses, HRQoL, overall survival, progression to AML, iron accumulation, ICT use, and health care resource utilization) were not part of the statistical testing plan and were not controlled for multiplicity. All significant P values are therefore at risk of a type I error and should be interpreted as supportive evidence for the overall efficacy of luspatercept.
An MID for patients with transfusion-dependent anemia associated with MDS could not be identified from the literature for either instrument used to assess HRQoL (EORTC QLQ-C30 and QoL-E). Moreover, the case analysis was completed for this data only with different subsets of patients at each time point. As it is not a true ITT population, the HRQoL would be subjected to an increased risk of bias due to the complete-case analysis approach.
Only a subset of patients who initially responded in the first 24 weeks were eligible for the extension phase. Interpretation of this end point is therefore problematic, as few patients were eligible for the extension phase and therefore could not achieve the end point of 12 weeks of response due to the study design. For patients missing data, imputation assuming the patients were nonresponders was used appropriately for the primary and key secondary end points. However, for other end points in the study, analyses were only completed in those patients with both baseline and follow-up measurements (complete-case analysis). As more than 10% of the data were missing for many of these end points, these assessments are likely to be biased. As of the data cut-off date there were patients who were continuing treatment and were censored from the analyses, making interpretation of evidence difficult.
The clinical experts consulted by CADTH were of the opinion that serum ferritin levels correlate weakly with total body iron, and an appropriate measure for total body iron would be liver or myocardial MRI.
Although numerous subgroup analyses were presented for the primary and key secondary efficacy end points, the subgroups of interest for this review according to the protocol were SF3B1 mutation status, IPSS-R status, and baseline hematologic status. Importantly, these subgroup (SF3B1 mutation status and IPSS-R status) analyses, which were among numerous subgroups tested, are at risk of a type I error and were not included in the randomization scheme. Imbalances in characteristics between luspatercept and placebo would therefore be expected and could affect the results observed within the subgroup. For the primary and key secondary efficacy end points, SF3B1 mutation status, although part of the pre-planned subgroup analyses, was not provided for this subgroup. Although the results of the subgroups were largely consistent with the overall findings, the FDA noted that subgroup analysis demonstrated that the effect was indistinguishable from placebo for numerous populations, including people with high transfusion burden, patients with high baseline erythropoietin, and patients with low platelets.10 The clinical experts were also of the opinion that the WHO and FAB classifications used for MDS were not current, and these classifications should be based on the IPSS-R and IWG 2018 criteria.
External Validity
The clinical experts noted that, based on baseline demographic and disease characteristics, the study population was representative of Canadian patients with transfusion-dependent anemia associated with MDS. In Canada the mean age of an MDS patient is 74 years, which is similar to the mean age of the study population, which was 70.5 years. The clinical experts were also of the opinion that, because patients with a del(5q) cytogenetic abnormality represent a small proportion of patients, excluding them from the trial may have been unnecessary and could have restricted access to luspatercept for these patients. The experts also noted that most patients in their registries were White; however, in the MEDALIST trial ethnicity was captured as Hispanic or Latino (or not) and this provides insufficient information to comment on the generalizability of ethnicity to the Canadian population. The clinical experts consulted by CADTH were of the opinion that the exclusion of patients with a history of thrombosis from the trial may have biased the results for the risk of thrombosis.
With regards to the choice of comparators used in the MEDALIST study, the clinical experts consulted by CADTH were of the opinion that the comparators listed in the protocol were all potential second-line treatment options for patients with transfusion-dependent anemia associated with MDS from a global perspective; the sponsor’s choice of comparator therefore may not be fully appropriate as the comparators listed in the protocol have restricted access in Canada.
The clinical experts consulted by CADTH were of the opinion that the duration of hematologic response of the primary end point, i.e., at least 8 weeks (any consecutive 56 days), was not clinically meaningful, and that an appropriate measure for clinical meaningfulness would be for patients to be transfusion-independent at least for 16 weeks, which is in accordance with the proposed IWG 2018 hematological response criteria.21 A hematologic response of transfusion independence for 12 weeks (i.e., any consecutive 84 days) is more clinically meaningful compared with 8 weeks. The other end points described by the clinical experts as clinically meaningful were improvement in HRQoL, reduction in number of units transfused, change in mHI-E, and change in hemoglobin.
Discussion
Summary of Available Evidence
One ongoing phase III study, MEDALIST (N = 229), was included in the systematic review. The trial included adult patients 18 years and older who had transfusion-dependent anemia associated with very low- to intermediate-risk MDS with ring sideroblasts and who have failed or are not suitable for erythropoietin-based therapy.
The MEDALIST trial is a multi-centre, randomized, double-blind, placebo-controlled study of the efficacy and safety of luspatercept in adult patients with transfusion-dependent anemia associated with very low- to intermediate-risk MDS with ring sideroblasts. Four sites in Canada enrolled 14 patients in the trial.
Eligible patients were randomized in a 2:1 double-blind manner to receive either luspatercept or placebo along with best supportive care for 24 weeks, followed by a week-25 assessment. Patients who had evidence of clinical benefit and absence of disease progression as per the IWG at the week-25 assessment continued into the extension phase. Patients received a starting dose of 1 mg of the study drug per kilogram of body weight administered by a subcutaneous injection every 3 weeks for 24 weeks and up to 48 weeks for those entering the extension. During these periods the dose levels were titrated (increased) stepwise to a maximum of 1.75 mg/kg.
The measure upon which the primary end point of the MEDALIST trial was based was a hematologic response of RBC-TI for at least 8 weeks (i.e., any consecutive 56 days) during week 1 through week 24. The key secondary end points were the proportion of patients with hematologic response of RBC-TI for at least 12 weeks (i.e., any consecutive 84 days) during week 1 through week 48 and week 1 through week 24. Other efficacy outcomes identified in the review protocol were other hematologic responses, HRQoL, overall survival, progression to AML, iron accumulation, ICT use, and health care resource utilization.
The MEDALIST trial used appropriate randomization methods and the primary and the key secondary end points accounted for multiplicity of testing using gate-keeping approaches. The main limitations were the lack of multiplicity controls for all other end points, potential for unblinding due to concerns of masking with the administration of the drug, and lack of efficacy or adverse events, which could have affected all end points but particularly the self-reported measures (e.g., HRQoL, adverse events). Moreover, according to the clinical experts, substantial data were missing for some end points (i.e., HRQoL), and the duration of response of the primary outcome was not long enough to estimate clinical meaningfulness. Last, only patients who responded in the first 24 weeks were allowed to enter the extension phase, making interpretation of the data for weeks 1 to 48 difficult.
Interpretation of Results
Efficacy
In MEDALIST, the primary end point of a hematological response demonstrated transfusion independence and was statistically significant in favour of the luspatercept treatment group. The primary efficacy end point demonstrated that a significantly greater number of patients responded to the luspatercept treatment compared to placebo and achieved transfusion independence for at least 8 weeks (i.e., any consecutive 56 days) during week 1 through week 24. However, the clinical importance of these changes is unclear as only an additional 25% of patients responded compared to placebo. Furthermore, the clinical experts consulted by CADTH were of the opinion that transfusion independence of 8 weeks is not clinically meaningful and that an appropriate measure of the duration of response for transfusion independence would be 16 weeks.
The first of the 2 key secondary outcomes of the MEDALIST study demonstrated that a significantly higher proportion of patients responded to the luspatercept treatment compared to placebo and achieved transfusion independence for at least 12 weeks (i.e., any consecutive 84 days) during weeks 1 through 48. Only a subset of patients who initially responded in the first 24 weeks were eligible for the extension phase. Interpretation of this end point is therefore problematic as few patients were eligible for the extension phase and therefore could not achieve the end point of a 12-week response due to the study design.
The second of the 2 key secondary outcomes of the MEDALIST study demonstrated that a significantly higher proportion of patients responded to luspatercept treatment compared to placebo and achieved transfusion independence for at least 12 weeks (i.e., any consecutive 84 days) during weeks 1 week 24. However, compared to placebo, only 20% of the patients in the luspatercept treatment group responded for this end point.
Other efficacy end points were also reported descriptively and should be interpreted as supportive evidence. The clinical experts consulted by CADTH acknowledged that HRQoL is considered important to patients, followed by overall survival. The MEDALIST trial analyzed the HRQoL using 2 instruments (EORTC QLQ-C30 and QoL-E). No major differences in HRQoL were noted between the treatment groups, with several domains being numerically lower in the luspatercept treatment group compared to the placebo treatment group; however, interpretation is limited by substantial missing data due to the complete-case design for the end points and the lack of an MID for the measures. All domains of the QoL-E were exploratory in the study. The median overall survival of the study had not been achieved, even by the July 1, 2019, data cut-off date, which limited interpretation of effect of luspatercept on overall survival. No difference was evident in the median time to RBC-TI during week 1 through 24 and week 1 through week 48. For the HRQoL end point, the FDA noted that there was no improvement in quality of life for patients who received luspatercept or who responded to luspatercept. The benefit of decreased transfusion requirement was therefore not correlated with any improvement in quality of life for patients.10
The outcomes of progression to AML, iron accumulation (as presented through serum ferritin levels), ICT use, and health care resource utilization do not show any meaningful changes. The clinical experts consulted by CADTH agreed that change in hemoglobin, mHI-E, and reduction in number of units of RBCs transfused would be clinically meaningful. The clinical experts consulted by CADTH suggested that serum ferritin levels were not a reliable indicator of total body iron and that there are frequently large fluctuations with this measurement. The clinical experts suggested that MRI of liver iron concentration and myocardial iron concentration were more reliable indicators of total body iron.
Harms
In the MEDALIST trial, 98% of the patients in the luspatercept safety population had at least 1 adverse event and 2.6% of the patients had at least 1 thromboembolic event. The most common treatment-emergent adverse events were fatigue, diarrhea, asthenia, and nausea, all of which occurred more frequently in patients treated with luspatercept than in those treated with placebo.
Patients with at least 1 serious adverse event accounted for 31.4% of the luspatercept group. The most commonly reported serious adverse event was pneumonia, with 2% of the patients in the luspatercept group and 2.6% of the patients in the placebo group reporting it.
The proportion of patients who stopped treatment due to an adverse event was 8.5% in the luspatercept treatment group and 7.9% in the placebo treatment group. The most common reason for stopping treatment was transformation to AML (2 patients in the luspatercept treatment group); nervous system disorders including headache, memory impairment, and Parkinson disease (1 patient each in the luspatercept treatment group); and fatigue (2 patients in the luspatercept treatment group). The notable harms identified in the CADTH review protocol included thromboembolic events, hypertension, hepatic and renal events, hypersensitivity reactions, and malignancies. Under the SOC of renal and urinary disorders, 18.3% of patients in the luspatercept treatment group and 13.2% of patients in the placebo group reported at least 1 associated adverse event. Hypertension was reported as an adverse event in 8.5% of the patients in the luspatercept treatment group and 7.9% of the patients in the placebo group. Malignancies (transformation to AML, basal cell carcinoma, squamous cell carcinoma of skin) were reported in 3.3% of patients in the luspatercept treatment group while 1.3% of patients in the placebo group reported at least 1 associated adverse event. No systemic hypersensitivity reactions were reported.
Conclusions
One phase III randomized controlled trial (MEDALIST; N = 229) was included in the CADTH systematic review of luspatercept for adult patients with transfusion-dependent anemia associated with MDS. The study demonstrated that treatment with luspatercept was superior to placebo in terms of achieving transfusion independence for at least 8 weeks (i.e., any consecutive 56 days) from week 1 through week 24. Luspatercept was superior to placebo in achieving transfusion independence for at least 12 weeks (i.e., any consecutive 84 days) from week 1 through week 48 and week 1 through week 24. Results of the primary end point were deemed not clinically meaningful by the clinical experts consulted by CADTH, and results of the 48-week secondary end point are difficult to interpret due to study design. The other end points of the study that were evaluated were HRQoL, overall survival, progression to AML, iron accumulation, ICT use, and health care resource utilization. However, none of these outcomes were controlled for multiplicity and, due to limitations associated with statistical methodology, the effect of luspatercept on these outcomes is currently unknown. During the trial the median overall survival had not been achieved. Key evidence gaps include the short duration of transfusion independence for the primary outcome of 8 weeks, study design, and no improvement in HRQoL.
Key safety issues with luspatercept include the rate of occurrence of thromboembolic events, which was lower in the luspatercept treatment arm compared to the placebo group. More patients in the luspatercept treatment group experienced fatigue, diarrhea, asthenia, nausea, and dizziness.
References
1.Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis: “2019 Update on Diagnosis, Risk-stratification, and Management”. Am J Hematol. 2019;94(4):475-488. PubMed
2.Escalante CP, Chisolm S, Song J, et al. Fatigue, symptom burden, and health-related quality of life in patients with myelodysplastic syndrome, aplastic anemia, and paroxysmal nocturnal hemoglobinuria. Cancer Med. 2019;8(2):543-553. PubMed
3.Platzbecker U. Treatment of MDS. Blood. 2019;133(10):1096-1107. PubMed
4.Lewis R, Bewersdorf JP, Zeidan AM. Clinical Management of Anemia in Patients with Myelodysplastic Syndromes: An Update on Emerging Therapeutic Options. Cancer Manag Res. 2021;13:645-657. PubMed
5.Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(1):60-87. PubMed
6.Buckstein R, Baldassarre F, Maze D, Schuh A, Cheung M. Systemic therapy for the treatment of adult patients with lower-risk myelodysplastic syndromes. 2018.
7.Slack J, Nguyen L, Naugler C, Rashid-Kolvear F. Incidence of Myelodysplastic Syndromes in a Major Canadian Metropolitan Area. J Appl Lab Med. 2018;3(3):378-383. PubMed
8.Reblozyl (luspatercept): 25 mg, 75 mg/vial of lyophilized powder for solution for subcutaneous injection [product monograph]. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2020 Sep 25.
9.Clinical Study Report: ACE-536-MDS-001 (MEDALIST). A phase 3, double-blind, randomized study to compare the efficacy and safety of luspatercept (ACE-536) versus placebo for the treatment of anemia due to IPSS-R very low, low, or intermediate risk myelodysplastic syndromes in subjects with ring sideroblasts who require red blood cell transfusions [internal sponsor's report]. Summit (NJ): Celgene Corporation; 2019 Mar 13.
10.Center for Drug Evaluation Research. Medical review(s). Reblozyl (luspatercept) subcutaneous injection. Company: Celgene Corporation. Application No.: 761136. Approval date: 04/03/2020 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2020 Apr 3: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761136Orig2s000TOC.cfm. Accessed 2021 Mar 22.
11.Raza A, Reeves JA, Feldman EJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. 2008;111(1):86-93. PubMed
12.Santini V, Valcarcel D, Platzbecker U, et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin Cancer Res. 2019;25(23):6976-6985. PubMed
13.Fenaux P, Haase D, Sanz GF, Santini V, Buske C, Group EGW. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25 Suppl 3:iii57-69. PubMed
14.Wood EM, McQuilten ZK. Outpatient transfusions for myelodysplastic syndromes. Hematology Am Soc Hematol Educ Program. 2020;2020(1):167-174. PubMed
15.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
16.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Mar 31.
17.Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N Engl J Med. 2020;382(2):140-151. PubMed
18.Oliva E, Nobile F, Dimitrov BD. Development and validation of QOL-E© instrument for the assessment of health-related quality of life in myelodysplastic syndromes. Central European Journal of Medicine. 2013;8.
19.Oliva ED, Borislav D. QoL-E. 2001: https://qol-e.it/team/. Accessed April 20 2021.
20.EORTC QLQ-C30 Scoring Manual. 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed April 20 2021.
21.Platzbecker U, Fenaux P, Ades L, et al. Proposals for revised IWG 2018 hematological response criteria in patients with MDS included in clinical trials. Blood. 2019;133(10):1020-1030. PubMed
22.Drug Reimbursement Review sponsor submission: Reblozyl (luspatercept) for myelodysplastic syndromes (MDS)-associated anemia, 25 mg, 75 mg/vial of lyophilized powder for solution for subcutaneous injection [internal sponsor's package]. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2021 Jan 28.
23.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
24.Weitz I, Meyers G, Lamy T, et al. Cross-sectional validation study of patient-reported outcomes in patients with paroxysmal nocturnal haemoglobinuria. Intern Med J. 2013;43(3):298-307. PubMed
25.Groenvold M, Klee MC, Sprangers MA, Aaronson NK. Validation of the EORTC QLQ-C30 quality of life questionnaire through combined qualitative and quantitative assessment of patient-observer agreement. J Clin Epidemiol. 1997;50(4):441-450. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of Search: March 29, 2021
Alerts: Weekly search updates until project completion
Study Types: No filters were applied to limit the retrieval by study type
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
Strategy Search
(luspatercept* or reblozyl* or ACE-536 or ACE536 or AQK7UBA1LS).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*luspatercept/ or (luspatercept* or Reblozyl* or ACE-536 or ACE536).ti,ab,kw,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms – Reblozyl/luspatercept; myelodysplastic syndromes
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms – Reblozyl/luspatercept; myelodysplastic syndromes
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms – Reblozyl/luspatercept; myelodysplastic syndromes
EU Clinical Trials
Register European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms – Reblozyl/luspatercept; myelodysplastic syndromes
Grey Literature
Search dates: March 22 – April 1, 2021
Keywords: Reblozyl/luspatercept; myelodysplastic syndromes
Limits: Publication years: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool For Searching Health-Related Grey Literature (https://www.cadth.ca/grey-matters) were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
1. Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N Engl J Med. 2020;382(2):140-151. | Duplicate study – Publication of MEDALIST trial. |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Please see Table 15 in the following publication: Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
Figure 5: FAB Myelodysplastic Syndromes Classification System
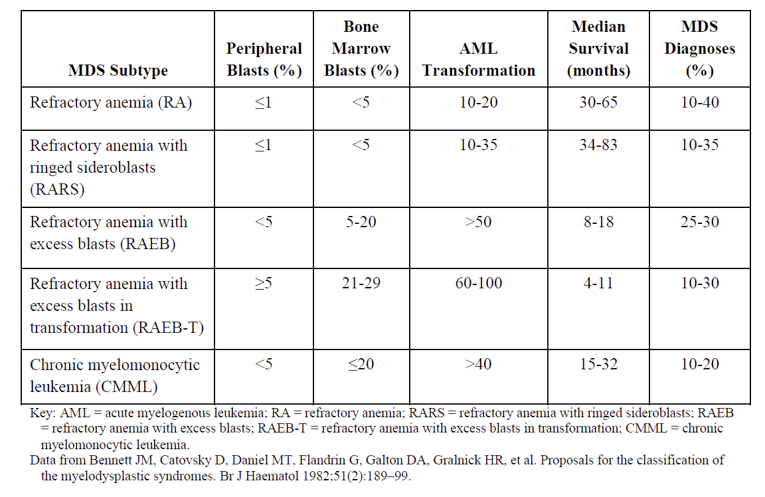
Source: Clinical Study Report for MEDALIST.9
Figure 6: International Prognostic Scoring System Score — Revised (IPSS-R)
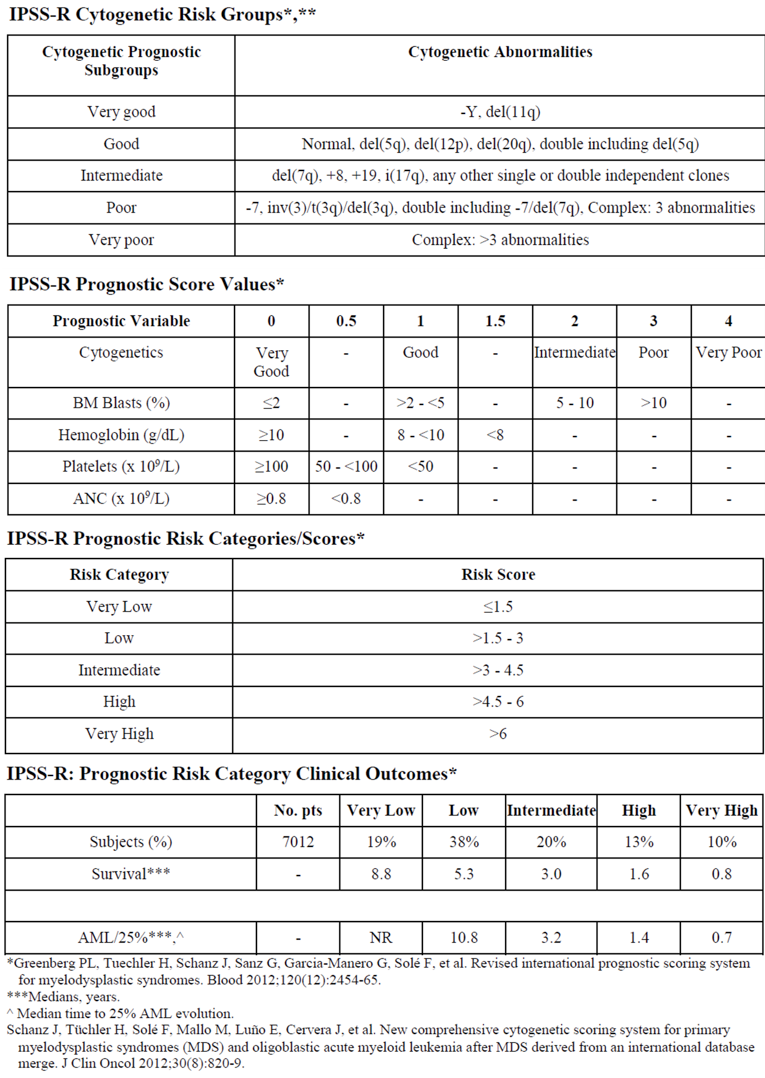
Source: Clinical Study Report for MEDALIST.9
HRQoL — QoL-E
Figure 7: Observed Median Scores and Median Changes from Baseline in Physical Well-Being Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted

Figure 8: Observed Median Scores and Median Changes from Baseline in Functional Well-Being Domain of the QoL-E =Over 25-Week Study Period (HRQoL-Evaluable Population) —Redacted

Figure 9: Observed Median Scores and Median Changes from Baseline in Social- and Family-Life Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted

Figure 10: Observed Median Scores and Median Changes from Baseline in Sexual Well-Being Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted

Figure 11: Observed Median Scores and Median Changes from Baseline in the Fatigue Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted

Figure 12: Observed Median Scores and Median Changes from Baseline in MDS-specific Disturbances Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted

Figure 13: Observed Median Scores and Median Changes from Baseline in the QoL-E General Domain of the QoL-E Over 25-Week Study Period (HRQoL-Evaluable Population) — Redacted



Appendix 4: Ad hoc Efficacy and Safety Update
Note that this appendix has not been copy-edited.
Objective
The pharmacoeconomic model submitted by the sponsor is based on the July 2019 data cut from the MEDALIST study. This was an ad hoc efficacy and safety analysis that was outside of the statistical analysis plan. The ad hoc analysis was the basis for some of the analyses submitted in the pharmacoeconomic model of luspatercept submitted by the sponsor. Results of this analysis are provided in order to additional context to the pharmacoeconomic evaluation.
Methods
The study design, statistical analysis plan, analysis population, primary and secondary efficacy end points are as previously described in the main body of the report. Figure 17 shows the updated patient disposition as of July 1, 2019.

Results
Efficacy
Updated results as of July 1, 2019, are reported here.
As of July 1, 2019, an additional 15 patients in the luspatercept treatment group and 2 patients in the placebo treatment group achieved RBC-TI of at least 8 weeks after the primary phase of the trial. Accounting for the additional responses 47.7% and 15.8% of patients achieved RBC-TI ≥ 8 weeks over the entire treatment period in the luspatercept and placebo treatment groups, respectively. Of the 15 patients who responded to luspatercept the response occurred after the primary phase (weeks 1 to 24), |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Of the total 73 patients who responded to luspatercept, 51 (69.9%) experienced ≥ 2 episodes of RBC-TI ≥ 8 weeks during the entire treatment phase and 28 (38.4%) had ≥ 3 episodes. Of the 12 patients who responded to placebo, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. This is reported in Table 37.
Table 38: RBC-TI of 8 Weeks or More During Entire Treatment Phase, as of July 1, 2019 (ITT Population)
Characteristics | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC ||||||||| | |
Number of responders, n (%) | 73 (47.7) | ||||||||| |
Number of responders with 1 response, n (%) | 22 (14.4) | ||||||||| |
Number of responders with 2 responses, n (%) | 23 (15.0) | ||||||||| |
Number of responders with 3 responses, n (%) | 28 (18.3) | ||||||||| |
ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
aDefined as the absence of any RBC transfusion during any consecutive 56-day period during the primary phase of the treatment period (first 24 weeks of double-blind treatment)
Source: Ad hoc Report for MEDALIST22
Analysis of RBC-TI of more than 8 weeks and transfusion reduction by baseline transfusion burden during the entire treatment period (week 1 through week 24, week-25, week 1 through week 48) as of July 01, 2019 is reported in Table 38.
Table 39: RBC-TI of 8 Weeks or More During Entire Treatment Phase by Baseline Transfusion Burden, as of July 1, 2019 (ITT Population)
RBC-TI ≥ 8 Weeks Over the Entire Treatment Period | MEDALIST | |
|---|---|---|
Luspatercept + BSC (N = 153) | Placebo + BSC (N = 76) | |
Average Baseline RBC Transfusion Requirement, n/N (%) | ||
≥ 6 Units/8 weeks | 14/66 (21.2) | 2/33 (6.1) |
≥ 4 to < 6 Units/8 weeks | 20/41 (48.8) | 2/23 (8.7) |
< 4 Units/8 weeks | 39/46 (84.8) | 8/20 (40.0) |
ITT = intention to treat; RBC-TI = red blood cell–transfusion independence.
Source: Ad hoc Report for MEDALIST22
As of May 08, 2018, the median duration of the longest single episode of RBC transfusion-independence was 30.6 weeks in the luspatercept treatment group and 13.6 weeks in the placebo treatment group. The cumulative duration of RBC transfusion-independence ≥ 8 weeks was calculated by summing the durations of all RBC transfusion-independence ≥ 8 weeks occurring at any time during treatment. In the |||||||||| luspatercept treatment responders, the median cumulative duration of RBC transfusion-independence ≥ 8 weeks was |||||||||||||||||||| The median cumulative duration of RBC transfusion-independence ≥ 8 weeks in the |||||||||| placebo responders was ||||||||||. As of July 01, 2019, in the 73 luspatercept treatment responders who achieved RBC transfusion-independence ≥ 8 weeks at any time during the treatment phase, the median cumulative duration of response was 79.9 weeks. The median cumulative duration of RBC transfusion-independence ≥ 8 weeks at any time during the treatment phase in the 12 responders in the placebo treatment group was 21.0 weeks (Figure 18). As this was an ad hoc analysis and was not part of a formal statistical testing plan the evidence cannot be critically appraised and is limited in interpretation.
Figure 17: Cumulative Duration of RBC-TI of At Least 8-Week Responses in Primary End Point Responders as of July 1, 2019 — Redacted


As of ||||||||||||||||||||||||||||| and |||||||||| of the patients demonstrated a reduction of at least |||||||||||||||||||| in RBC transfusion burden for at least |||||||||| over the entire treatment period, in the luspatercept treatment group and placebo treatment group respectively. In the luspatercept treatment group, the median duration to reduction of at least |||||||||| in RBC transfusion burden for at least |||||||||| was ||||||||||||||||||||. As this was an ad hoc analysis and was not part of a formal statistical testing plan the evidence cannot be critically appraised and is limited in interpretation.
Appendix 5: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-C30
QoL-E
Findings
Table 40: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | EORTC QLQ-C30 is a questionnaire used to assess HRQoL in cancer patients. It includes 5 functional scales, 3 symptom scales, a global health status/QoL scale, and 6 single items. Scores range from 0 – 100.20 | EORTC QLQ-C30 has been validated in several disease-specific populations23-25 Validity, reliability, and responsiveness for patients with low- to intermediate-MDS was not identified in the literature. | A MID for patients with low- to intermediate-MDS was not identified in the literature. |
QoL-E | QoL-E is a questionnaire specific to patients with MDS and is used to evaluate the impact of disease and treatment. The questionnaire examines 5 dimensions, including one specific MDS-related dimension. It is scored on a standardized scale using Likert-scale or dichotomous response options, with a range score of 0 -100. Better HRQoL correlates to a higher score.19 | Validity: Earlier versions of the QoL-E were compared to responses in the FACT-G questionnaire and demonstrated a correlation coefficient of ≥0.71. Construct validity between the 2 instruments was supported by factor analysis, showing many domains formed clusters. Stepwise regression analysis confirmed good clinical validity.18 Reliability: Test-retest analysis demonstrated intra-class correlation coefficients between 0.65 and 0.80, and stability of scores over time.18 Responsiveness: Responsiveness was not reported. | A MID for patients with low- to intermediate-MDS was not identified in the literature. |
HRQoL = health-related quality of life; MID = minimal important difference; MDS = myelodysplastic syndromes.
EORTC QLQ-C30
The EORTC QLQ-C30 is a 30-item, self-administered questionnaire designed to evaluate the quality of life of cancer patients. The questionnaire contains items relevant to function (physical, role, cognitive, emotional, and social), symptoms (fatigue, pain, and nausea and vomiting), quality of life, and several single items commonly associated with cancer (dyspnea, loss of appetite, insomnia, constipation, and diarrhea). Items are scored on a scale from 0 – 100.20
Validity, reliability, and responsiveness for patients with low- to intermediate-MDS was not identified in the literature, nor was a MID.
The validity and reliability of the EORTC QLQ-C30 was tested in an international cohort of 305 patients with lung cancer.23 Patients completed the questionnaire twice, once before treatment started and once after initiation of treatment. On average the questionnaires were completed 28 days apart, in either an interview format (if patients required assistance) or via self-administration. Analysis of covariance was completed to determine if method of administration impacted responses, and no statistically significant differences were found. Responses from participants were well distributed along the scales for each item with reliability for the multi-item scales ranging from 0.54 to 0.86 prior to treatment, and 0.52 to 0.89 post-treatment. Scale reliability was stable regardless of age, education level, performance status, and mode of delivery. Inter-scale correlations were calculated as part of the validity analysis, with all results statistically significant (P<0.01). Validity was further tested by comparing patient subgroups with varying clinical status and responses and yielded consistent results. Function and symptom measures distinguished between patient performance status, weight loss, and treatment toxicity, and suggest the questionnaire is responsive to changes in patient condition.23
The validity, reliability, and responsiveness of the EORTC QLQ-C30 was examined in 29 patients with paroxysmal nocturnal hemoglobinuria.24 Patient-reported feedback confirmed the content validity and relevance of the questionnaire items to measure symptom severity in patients with paroxysmal nocturnal hemoglobinuria.
The EORTC QLQ-C30 was examined using a mixed-methods approach to determine the extent of agreement between patient responses to the questionnaire and observer’s rating of patient responses in an interview format with 95 patients with breast or gynecological cancer.25 No statistically significant differences were found between the patients’ mean scores and the interviewers’ means scores for any items in the questionnaire, with a median overall agreement of 0.85. 27 of the 30 items in the questionnaire had at least substantial agreement (Kappa ≥ 0.61), with the remaining 3 items in the moderate agreement range (Kappa 0.41-0.60). These results may suggest the responses to the EORTC QLQ-C30 are interpreted similarly between respondent and interviewer in patients with breast or gynecological cancer.25
QoL-E
The QoL-E is a 29-item, self-administered, multi-dimensional questionnaire which evaluates patient-reported outcomes in patients with MDS.19 The goal is to assess the effect of MDS on patients’ lives. It includes questions on physical, functional, social, and sexual well-being, plus specific focus on fatigue and MDS-specific items.18
QoL-E was initially pilot-tested in a cohort of 52 patients with MDS.18 Patients concurrently completed the FACT-G questionnaire, and several items between the instruments were statistically significant with a correlation coefficient of ≥0.71. Strong correlations were noted between the physical well-being, emotional well-being, functional well-being, and treatment outcome scores for the FACT-G. Stepwise regression analysis demonstrated good clinical validity, with hemoglobin levels being an independent predictor of physical well-being, fatigue, and general well-being. Re-testing for reliability was completed with 39 of the original 52 patients within 2 months of the initial evaluation, a time period considered short enough for minimal changes in patient condition to occur. Intra-class coefficients for the 6 domains were between 0.65 and 0.80, demonstrating stability of scores over time.18
A descriptive analysis was performed on the revised, finalized version of the QoL-E in 147 patients with MDS. Feasibility and time to complete were confirmed and reliability analysis demonstrated an coefficient of ≥0.70 in all domains, demonstrating good internal validity.18 Instrument responsiveness was not reported and a MID for patients with MDS was not identified in the literature.
Pharmacoeconomic Review
Abbreviations
AML
acute myeloid leukemia
BIA
budget impact analysis
ESA
erythropoiesis-stimulating agent
HR MDS
high-risk myelodysplastic syndrome
HTB
high transfusion burden
ICER
incremental cost-effectiveness ratio
ICT
iron chelation therapy
IPSS-R
Revised International Prognostic Scoring System
ITB
intermediate transfusion burden
LTB
low transfusion burden
MDS
myelodysplastic syndromes
QALY
quality-adjusted life-year
RBC
red blood cell
RBC-TI
red blood cell–transfusion independence
TD
transfusion-dependent
TI
transfusion-independent
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Luspatercept (Reblozyl), lyophilized powder for solution for subcutaneous injection |
Submitted price | Luspatercept, 25 mg vial: $2,189.00 Luspatercept, 75 mg vial: $6,567.00 |
Indication | For the treatment of adult patients with very low- to intermediate-risk myelodysplastic syndromes (MDS)-associated anemia who have ring sideroblasts and require RBC transfusions |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | February 11, 2021 |
Reimbursement request | As per indication |
Sponsor | Celgene Inc., a Bristol Myers Squibb company |
Submission history | Previously reviewed: Yes Indication: For the treatment of adult patients with RBC transfusion-dependent anemia associated with beta-thalassemia Recommendation date: Under review Recommendation: Embargoed |
MDS = myelodysplastic syndromes; NOC = Notice of Compliance; RBC = red blood cell.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with very low- to intermediate-risk MDS-associated anemia who have ring sideroblasts requiring RBC transfusions and have received or are not eligible for ESAs |
Treatment | Luspatercept + best supportive care |
Comparator | BSC alone, comprising regular RBC transfusions and ICT |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (10 years) |
Key data sources |
|
Submitted results | ICER = $206,439 per QALY for luspatercept + BSC vs. BSC (incremental QALYs: 0.79; incremental costs: $162,196) |
Key limitations |
|
CADTH reanalysis results |
|
BSC = best supportive care; ESA = erythropoietin-stimulating agent; HR MDS = high-risk myelodysplastic syndrome; ICER = incremental cost-effectiveness ratio; ICT = iron chelation therapy; MDS = myelodysplastic syndrome; QALY = quality-adjusted life-year; RBC = red blood cell; TI = transfusion-independent; TD = transfusion-dependent.
Conclusions
The CADTH Clinical Review based on the MEDALIST trial found that luspatercept was superior to placebo in terms of achieving red blood cell–transfusion independence (RBC-TI) for 8 weeks or longer, between week 1 to week 24, which was the primary end point for the study. The study also demonstrated the superiority of luspatercept to placebo with respect to the secondary end points of achieving RBC-TI for at least 12 weeks between weeks 1 and 24 or between weeks 1 and 48. Other end points were not included in the statistical testing hierarchy and are subject to type I error, and the median overall survival was not reached in either group.
The CADTH reanalysis of the sponsor’s base case included an alternative assumption around overall survival extrapolation to mitigate the overall survival benefits that were observed outside the trial period. Based on the CADTH reanalyses, the incremental cost-effectiveness ratio (ICER) of luspatercept + best supportive care (BSC) versus BSC alone for patients with myelodysplastic syndromes (MDS)-associated anemia was $623,219 per quality-adjusted life-year (QALY), with a 0% chance of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY. For luspatercept to be cost-effective at a threshold of $50,000 per QALY, a price reduction of 85% would be required.
Key drivers of the analysis are the transition probabilities between transfusion-independent (TI) and transfusion-dependent (TD) health states during and after luspatercept treatment as observed in MEDALIST, and the cost of luspatercept. Uncertainty remains surrounding the overall survival modelling assumptions, as clinical outcomes from MEDALIST focused primarily on hematological response as defined by transfusion independence for 8 or 12 weeks over the 48-week trial period. The effect of luspatercept on patient quality of life remains uncertain as an improvement in transfusion burden does not necessarily improve quality of life.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
The Leukemia and Lymphoma Society of Canada and the Aplastic Anemia and Myelodysplasia Association of Canada provided a joint response to CADTH’s call for patient input. The Leukemia and Lymphoma Society created an online survey and received responses from 18 patients with MDS, 1 caregiver, and 1 family member of a patient with MDS. All patients were Canadian. Patients had received a variety of frontline treatments for their MDS, including blood transfusions, chemotherapy, or stem cell or bone marrow transplant. The most common impactful treatment side effects were low blood cell counts, extreme fatigue, anemia, and infection. The most important outcomes for patients trying a new treatment were disease impact, quality of life, and closeness to home. No surveyed respondents had experience with luspatercept.
CADTH received clinical group input from 2 groups: the Ontario Health (Cancer Care Ontario) Hematology Disease Site Drug Advisory Committee and the Alberta Tumour Board Myeloid Physicians Group. Clinicians described the current treatment paradigm for patients with lower-risk MDS as transfusion support characterized by red blood cell (RBC) transfusions and erythropoiesis-stimulating agent (ESAs). For patients who lose response to ESAs (the indication under review), no treatment options are available beyond transfusion support with RBC transfusion and iron chelation therapy (ICT), except lenalidomide in the approximately 10% of MDS patients with the del(5q) mutation. Azacitidine may be used for some intermediate-risk patients based on their International Prognostic Scoring System status. Clinicians would consider luspatercept as an additional line of therapy for patients who have progressed on ESAs. The expectation is that luspatercept would reduce the number of patients requiring regular transfusion support and therefore reduce the nursing, facility, and time burdens for treating facilities.
Feedback from the drug plans emphasized the requirements for hemoglobin monitoring and for luspatercept to be administered by a health care professional, access to whom may be difficult in rural communities. The drug plans noted that 95% of patients in MEDALIST had received ESAs previously and that such a history may be a requirement for funding. The plans expressed uncertainty around the criteria for discontinuation of luspatercept cited in the product monograph, in which it is stated that treatment should be discontinued if the patient does not experience clinical benefit after 9 weeks.
Several of these concerns were addressed in the sponsor’s model:
The costs of azacitidine and allogeneic hematopoietic stem cell transplant were included for patients who progressed to high-risk myelodysplastic syndrome (HR MDS).
Costs of hemoglobin monitoring and administration costs were included.
CADTH addressed some of these concerns by including only ESA-refractory patients in the base case of the budget impact analysis (BIA) to better reflect the funding request.
CADTH was unable to address the following concerns raised from stakeholder input:
Direct incorporation of patient quality of life was not included in the pharmacoeconomic model. While the sponsor collected quality-of-life information in MEDALIST (through the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 and the QoL-E questionnaire for patients with myelodysplastic syndromes) the results were considered exploratory and, as a result, the sponsor modelled the impact of luspatercept based on changes in transfusion burden.
In light of the scope of the sponsor’s model, a broader perspective that considers travel time and other societal costs related to MDS is warranted.
Economic Review
The current review is for luspatercept (Reblozyl) for adult patients with MDS-associated anemia who have ring sideroblasts requiring RBC transfusions and have received or are not eligible for ESAs.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing luspatercept with BSC compared to BSC alone for the treatment of adult patients with MDS-associated anemia requiring RBC transfusions and who have received or are not eligible for ESAs. The modelled population aligned with the Health Canada indication and reimbursement request1 for adults with very low-, low-, or intermediate-risk MDS-associated anemia who have ring sideroblasts who require RBC transfusions and have received or are not eligible for ESAs. The ESA regimen must have been either at least 40,000 IU of recombinant human erythropoietin per week for at least 8 doses (or equivalent) or at least 500 mcg of darbepoetin-alpha every 3 weeks for at least 4 doses (or equivalent).
Luspatercept is available as a powder that must be reconstituted and administered by a health care professional as a subcutaneous injection of 50 mg/mL. The recommended starting dosage of luspatercept for MDS is 1.0 mg/kg once every 3 weeks but may be increased to 1.33 mg/kg every 3 weeks if the patient is not RBC transfusion–free after at least 2 consecutive doses (6 weeks) at the 1.0 mg/kg starting dose. If the patient is not RBC transfusion–free after at least 2 consecutive doses (6 weeks) at the 1.33 mg/kg dose, the dose may be increased to 1.75 mg/kg. The dose should not be increased more frequently than every 6 weeks (2 doses) or beyond the maximum dose of 1.75 mg/kg.1 Patients should continue luspatercept for as long as they experience a clinical benefit, and discontinue if there is no clinical benefit after 9 weeks (3 doses) at the maximum dose level of 1.75 mg/kg. Hemoglobin results should be assessed before each administration of luspatercept, and the drug should only be given if the hemoglobin is no more than 110 g/L.1 The cost for luspatercept is $2,189 per 25 mg vial and $6,567 per 75 mg vial2; the annual cost of luspatercept ranges from $152,188 to $228,281 per patient, as calculated by CADTH (Table 8), based on the mean patient weight of 76.0 kg used in the MEDALIST trial.3
In the model, the annual cost of luspatercept was calculated by the sponsor to be $179,442, based on individual patient weights from MEDALIST.3 An administration cost of $54.25 per dose was applied based on the Ontario Schedule of Benefits for a standard chemotherapy administration, for an additional annual cost of $944.2 No vial sharing was assumed in the base case, and luspatercept was assumed to be given alongside BSC. The comparator for this economic analysis was BSC alone, consisting of regular RBC transfusions and ICT to prevent chronic iron overload due to regular RBC transfusions. The unit cost of an RBC unit was $422, and the cost per transfusion visit was $264.2 The costs of routine monitoring by physicians and specialists, which ranged from $516 to $3,457 per annum depending on patient health state, were also included. The ICTs included in this analysis were deferoxamine and deferasirox, with doses varying from low to very high, depending on patient-level data from MEDALIST.3 The annual costs for deferoxamine and deferasirox ranged from $4,527 to $13,581 and $16,803 to $50,409, respectively, depending on the dose received. An annual administration cost of $7,488 was applied to deferoxamine as it is an IV therapy,4 and costs for monitoring ICT-related adverse events were also included.
For patients progressing to high or very HR MDS it was expected that they could receive either azacitidine or allogenic hematopoietic stem cell transplantation; the 28-day cost of the former was $26,376 and the average 1-time cost of the latter was $62,927. Patients progressing to acute myeloid leukemia (AML) incurred a per-cycle cost of $5,995.
Outputs of the model included QALYs and life-years over a lifetime horizon of 10 years. The base-case analysis was conducted from the perspective of the Canadian public health care system, with an annual discount of 1.5% applied to both costs and outcomes. The cycle length was 4 weeks during the first year and 3 months thereafter as the sponsor indicated that progression of the disease evolves more slowly after 1 year. A half-cycle correction was applied.
Model Structure
The sponsor submitted a Markov model consisting of 7 mutually exclusive health states. Four were characterized by transfusion burden: low transfusion burden (LTB), intermediate transfusion burden (ITB), high transfusion burden (HTB), or TI. The model included 3 additional health states associated with HR MDS, AML, or death (Figure 1). At model entry, all patients began in the LTB, ITB, or HTB health states, and could transition to the TI state if they achieved a response to treatment. At any time, patients could transition to any other health state.
Model Inputs
Baseline characteristics for the patient cohort were informed by the intention-to-treat population in MEDALIST (N = 229).3 The median age and weight of the population was 71 years and 76 kg, respectively, and 63% of participants were male. The baseline distribution of patients in transfusion-burden health states was based on values established in MEDALIST: LTB = 28.8%, ITB = 27.9%, and HTB = 43.3%. Tranfusion independence was defined as 0 RBC units over 8 weeks, LTB was more than 0 to fewer than 4 RBC units over 8 weeks, ITB was at least 4 to fewer than 6 RBC units over 8 weeks, and HTB was at least 6 RBC units over 8 weeks.
In MEDALIST, response was defined as achieving TI for at least 8 consecutive weeks in the first 24 weeks. In the model, patients who achieved TI for at least 8 weeks were considered responders and were moved to the TI state. From the TI state, patients could lose response and transition back to other health states, the risk of which was modelled using a duration-of-response curve. Parametric curves were fit to MEDALIST duration-of-response data from weeks 1 to 48 in the trial. For the luspatercept + BSC arm, log-normal curves were used based on goodness-of-fit statistics and, for the BSC arm, Weibull was selected based on visual inspection of plausibility. In each arm, the maximum duration of response was 110 weeks based on MEDALIST, after which point no patients were classified as responders.3 Responders to luspatercept are assumed to remain on treatment until they lost response. In the base case, nonresponders were assumed to discontinue treatment with luspatercept after week 25.
In the first year of the model, the transition probabilities between states were constant according to probability matrices derived from MEDALIST, but transitions to AML and death after 1 year were modelled using survival curves. Because only 4 patients in MEDALIST developed AML during the study, estimates of AML incidence curves were derived from the literature based on Revised International Prognostic Scoring System (IPSS-R) risk groups and fit with a Gompertz curve to estimate the transition to AML.5 For overall survival, Gompertz curves were fit to extrapolate the data from MEDALIST beyond the median follow-up of 26.25 months.3 The model also allowed for the possibility of calculating overall survival based on a TI reference curve (Weibull fit to data from a Spanish study6), to which hazard ratios were applied for TD and HR MDS patients.7,8 Survival for patients with AML was calculated based on Oran et al.,9 who evaluated patients from the Surveillance, Epidemiology, and End Results (SEER) database.
Utility values for the TI and TD states (LTB to HTB) were derived from a time trade-off study of primarily UK patients that estimated utility values as follows: 0.85 for TI, 0.77 for LTB, 0.71 for ITB, and 0.65 for HTB.10 The utilities for the HR MDS and AML states were also 0.65, based on an assumption and another AML study, respectively.11 Last, a disutility of −0.23 was applied to patients receiving deferoxamine as it is administered as a subcutaneous injection rather than orally.12
Dosing of luspatercept was based on the product monograph as outlined in the Overview section of the sponsor’s economic evalution. Most patients received a dose ranging from 1.00 mg/kg to 1.75 mg/kg every 3 weeks as outlined by the monograph, but 3.6% of patients received a lower dose (0.60 mg/kg or 0.80 mg/kg). The numbers of 25 mg and 75 mg vials were calculated based on individual patient weights in MEDALIST, and no vial sharing was assumed in the base case.2
In the model, the annual cost of luspatercept was calculated based on individual patient weights from MEDALIST.3 An administration cost based on the Ontario Schedule of Benefits for a standard chemotherapy administration was added, for an additional annual cost of $944.2 The unit cost of an RBC unit was based on clinical expert feedback, and the cost per transfusion visit was derived from Canadian literature.13 The costs of routine monitoring by physicians and specialists ranged from $516 to $3,457 per annum depending on patient health state. The drug acquisition costs for ICT were obtained from the Ontario Drug Benefit formulary14 and Saskatchewan Drug Formulary.15 An annual administration cost of $7,488 was applied to deferoxamine as it is an IV therapy,4 and modest costs for monitoring ICT-related adverse events were also included. The cost of azacitidine was sourced from the product monograph16 and the cost of allogenic hematopoietic stem cell transplantation was sourced from the Canadian Institute for Health Information patient cost calculator.17
Summary of Sponsor’s Economic Evaluation Results
The sponsor’s model allowed for consideration of different data cut-offs. To align with the clinical review, the scenario considering the May 2018 data cut-off was chosen for presentation of the base case. The sponsor submitted probabilistic analyses based on 1,000 iterations. Deterministic and probabilistic results were similar, but probabilistic results were consistently slightly higher than what was reported by the sponsor. CADTH used 2,000 iterations in all cases due to issues with model convergence. Probabilistic findings are presented below.
Base-Case Results
Luspatercept + BSC was associated with an incremental cost of $162,196 and 0.79 QALYs in comparison with BSC, for an ICER of $206,439 per QALY (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results (May 2018 data cut-off)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($ per QALY) |
|---|---|---|---|---|---|
BSC | $157,068 | Reference | 2.19 | Reference | Reference |
Luspatercept + BSC | $319,264 | $162,196 | 2.98 | 0.79 | $206,439 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analyses are based on the publicly available prices of the concomitant treatments (e.g., ICT).
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted a number of sensitivity and scenario analyses. In these analyses, the ICERs for luspatercept + BSC compared to BSC alone were most sensitive to the chosen time horizon and assumptions surrounding how overall survival was incorporated into the model (i.e., based on treatment received, based on transfusion burden health states, or based on IPSS-R risk scores).
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Overestimation of the survival benefit associated with luspatercept: In the sponsor’s base case, overall survival was estimated by fitting a Gompertz distribution to the overall survival data observed in MEDALIST (median follow-up of 26.25 months) and using that to estimate overall survival rates after year 1.3 Curves were fit to both the luspatercept + BSC arm and BSC alone, leading to an extrapolation of overall survival data for the lifetime model horizon. This approach is problematic because it assumes that the overall survival benefits observed during the trial period will be maintained throughout a patient’s lifetime, while the sponsor’s model estimates that after 1.5 years only 11.6% of patients will still be on luspatercept. Treatment benefits will, by their very nature, be highest during the trial, when the highest proportion of patients are on therapy, and it is not reasonable to expect benefits in overall survival to be sustained after the majority of patients have discontinued the drug. Clinical experts consulted by CADTH were asked to validate this assumption and they did not expect to see any residual overall survival benefit in patients who had discontinued luspatercept.
The sponsor provided several alternative methods for estimating overall survival in the model, including basing it on the TI, TD, and HR MDS states, IPSS-R levels, or WHO Prognostic Scoring System levels. Based on discussion with the clinical experts consulted by CADTH for this review, CADTH chose the option that modelled overall survival based on TI, TD, and HR MDS states, with hazard ratios for TD versus TI and HR MDS versus TI derived from the literature.7,8 This option appeared to be the most conservative with regard to overall survival benefits beyond the trial period.
CADTH assumed overall survival to be dependent on TI, TD, and HR MDS states as part of the base case. The estimate of overall survival based on IPSS-R level was explored in scenario analysis.
Different data cut-offs used in the clinical and pharmacoeconomic submissions: The sponsor used a data cut-off of July 2019 for its pharmacoeconomic analysis, while the clinical review was based on a May 2018 data cut-off. Upon request, the sponsor provided a pharmacoeconomic analysis that allowed the user to choose different scenarios based on the data cut-off. Both the sponsor’s and CADTH’s base case described in this report are based on the May 2018 data cut-off to align with the clinical review. The model inputs that were affected by the alternative data cut-off scenarios included discontinuation and loss of response parametric fits, probability of death in year 1, destination after loss of response, all transition matrices, overall survival data, and adverse event rates. The following inputs remained similar regardless of the data cut-off selected: the percent of TD patients receiving each type and dose of ICT (only the July 2019 data cut-off was available), and the number of luspatercept vials, transfusion units and visits (only the January 2019 data cut-off was available). It is unclear why inputs derived from the May 2018 data cut-off were not used exclusively in the May 2018 scenario, which increases the uncertainty associated with this analysis.
CADTH used the May 2018 data cut-off option for the base case. The original July 2019 data cut-off was tested in a scenario analysis.
Uncertainty regarding utility values in the HR MDS and AML health states: The sponsor assumed an equivalent utility value of 0.65 in patients with HTB, HR MDS, and AML. While unsure of the value of the HR MDS state, the clinical experts consulted by CADTH suspected that the quality of life of a patient with AML would be lower than that of a patient in the HTB state. The utility for AML was derived from a systematic review of health state values for AML,11 and CADTH chose an alternate value based on the included studies.
CADTH used a value of 0.524 for the AML health state for the base case but did not notice a substantial difference in the resulting ICER.
Uncertainty regarding incorporation of patient quality of life: The clinical experts consulted by CADTH noted that an increase in hemoglobin and decrease in RBC transfusion–dependence cannot necessarily be assumed to improve patients’ symptoms or quality of life. Although health-related quality-of-life data were collected in MEDALIST, they were not used to inform the pharmacoeconomic submission. As the sponsor has not demonstrated that luspatercept directly affects patient-important outcomes, these gains in QALYs may be considered speculative.
CADTH was unable to address this limitation in reanalysis.
The following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Response to luspatercept was defined as achieving TI for ≥ 8 weeks during weeks 1 to 24. | Uncertain. The sponsor notes in its report that 16 weeks has been proposed as a more relevant clinical end point,18 and that clinical expert feedback suggested a period of 8 weeks was not sufficient to capture quality-of-life changes. Clinical experts consulted by CADTH agreed with the assessment that the assessed response period should be longer than 8 weeks. It is unclear how this affects the cost-effectiveness estimate. |
Patients not achieving TI for ≥ 8 weeks during weeks 1 through 24 were assumed to discontinue luspatercept from week 25 onward. | Appropriate. Clinical experts confirmed that there is no reason to continue the drug if it is not effective. |
Patients discontinuing and losing response to luspatercept were assumed to experience the same transitions between health states as those in the BSC-alone arm after year 1. | Appropriate. Due to a lack of long-term follow-up of patients after discontinuing the drug and the expectation that they should no longer receive benefits; it is appropriate to apply BSC transitions. |
Patients discontinuing and losing response to luspatercept experienced differential transition probabilities during year 1 (weeks 0 to 52). | Uncertain. The sponsor based these transitions on MEDALIST results, which included small differences in transitions between the 2 arms, even after losing response. However, this may not be reflective of real-world practice, in which patients are not followed as closely as a clinical trial. Also, there is no reason to expect the clinical pathway following loss of response to differ based on initial treatment (luspatercept vs. BSC). A more conservative assumption applies equal transition probabilities to all discontinuers and nonresponders. This was tested in a scenario analysis but did not affect the results. |
BSC = best supportive care; TI = transfusion-independent.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts. The changes were made to address some of the uncertainty in the model and included modelling overall survival based on IPSS-R levels. These changes are summarized in Table 5.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Different data cut-off from clinical review | July 2019 | May 2018 – to align with clinical review |
Changes to derive the CADTH base case | ||
1. Lack of long-term OS data led to extrapolation in the base case | Fit a Gompertz directly to OS data from MEDALIST | OS benefits based on TI, TD, and HR MDS state |
2. Lower utility in the AML health state | 0.65 | 0.524 |
CADTH base case | — | Reanalysis 1 + 2 |
HR MDS = high-risk myelodysplastic syndrome; OS = overall survival; TD = transfusion-dependent; TI = transfusion-independent.
In the CADTH base case, luspatercept was associated with estimated total costs of $270,123 and total QALYs of 2.29, compared to $149,569 and 2.09, respectively, for patients receiving BSC. The ICER for luspatercept compared to BSC was $623,219 per QALY, with a 0% chance of being below $50,000 per QALY. A detailed breakdown of the disaggregate results is available in Appendix 4, Table 12.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case | BSC | 167,209 | 2.42 | Reference |
Luspatercept | 324,596 | 3.24 | 198,741 | |
Sponsor’s corrected base case: May 2018 data cut-off | BSC | 157,068 | 2.19 | Reference |
Luspatercept | 319,264 | 2.98 | 206,439 | |
CADTH reanalysis 1: OS assumptions based on TI, TD, HR MDS | BSC | 149,569 | 2.10 | Reference. |
Luspatercept | 270,123 | 2.29 | 622,796 | |
CADTH reanalysis 2: lower utility in AML | BSC | 157,002 | 2.19 | Reference |
Luspatercept | 320,075 | 2.98 | 204,223 | |
CADTH base case (reanalysis 1 + 2) | BSC | 149,569 | 2.09 | Reference |
Luspatercept | 270,123 | 2.29 | 623,219 |
AML = acute myeloid leukemia; HR MDS = high-risk myelodysplastic syndrome; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TD = transfusion-dependent; TI = transfusion-independent.
Scenario Analysis Results
CADTH undertook a price-reduction analysis based on the sponsor’s and CADTH’s base cases. Based on the CADTH base case, a price reduction of 85% would be necessary to achieve cost-effectiveness at a threshold of $50,000 per QALY. CADTH found that, for the sponsor’s base case, the price reduction required to achieve cost-effectiveness at a threshold of $50,000 per QALY was higher, at 94%. This was due in part to the cost of ICT, which is more influential in the sponsor’s base case as long-term overall survival benefits caused patients to remain on ICT longer while drug acquisition costs for luspatercept remained constant.
Table 7: CADTH Price-Reduction Analyses (Probabilistic)
Analysis | ICERs for luspatercept vs. BSC ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base case (corrected) | CADTH reanalysis |
No price reduction | 206,439 | 623,219 |
10% | 187,491 | 555,177 |
20% | 171,030 | 487,134 |
30% | 154,569 | 419,092 |
40% | 138,107 | 351,049 |
50% | 121,646 | 283,007 |
60% | 105,184 | 214,964 |
70% | 88,723 | 146,921 |
80% | 72,262 | 78,879 |
85% | 64,031 | 44,858 |
90% | 55,800 | 10,836 |
94% | 49,216 | Dominant |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CADTH undertook a series of exploratory analyses to determine the impact of alternative assumptions on the cost-effectiveness of luspatercept:
Overall survival assumptions based on IPSS-R level
Used July 2019 data cut-off
Assumed 100% of patients were LTB at baseline
Assumed 100% of patients were ITB at baseline
Assumed 100% of patients were HTB at baseline
The results of these analyses are presented in Appendix 4, Table 13. The scenarios that had the largest influence on the ICER were those that involved alternative assumptions surrounding baseline transfusion status. When all patients were assumed to start in the HTB state, the ICER was $1,170,786 per QALY. When overall survival was calculated based on the IPSS-R level, the ICER was $540,188 per QALY, emphasizing the impact of clinical uncertainty around overall survival on the cost-effectiveness of luspatercept.
Issues for Consideration
Health Canada recently approved a combination therapy, Inqovi (decitabine plus cedazuridine), for the treatment of intermediate-risk MDS with anemia and ring sideroblasts.19 Clinical experts consulted by CADTH noted that this drug is not yet available in clinical practice. The cost-effectiveness of luspatercept compared to this combination product is unknown; the budget impact of luspatercept could also be affected if this new product comes to market.
CADTH has previously reviewed luspatercept for the treatment of anemia associated with beta-thalassemia.20 The submitted price in that review was the same, at $2,189 per 25 mg vial and $6,567 per 75 mg vial. (This is currently under review.)
Overall Conclusions
The CADTH Clinical Review based on MEDALIST found that luspatercept was superior to placebo in terms of achieving RBC-TI for at least 8 weeks from week 1 to week 24, which was the primary end point. The study also demonstrated the superiority of luspatercept to placebo with respect to the secondary end points, achieving RBC-TI for at least 12 weeks between weeks 1 and 24 or between weeks 1 and 48. The other end points, including other hematologic responses, overall survival, iron accumulation, ICT usage, and progression to AML, were reported descriptively; however, these end points were not included in the statistical hierarchy and may be subject to type I error.
The CADTH review identified several key limitations with the sponsor’s pharmacoeconomic submission surrounding overall survival data assumptions, different data cut-offs in the clinical and pharmacoeconomic submissions, and utility in the AML state. CADTH reanalyses included an alternate assumption around overall survival extrapolation, use of a May 2018 data cut-off, and a reduced utility value in the AML state. Based on CADTH reanalyses, the ICER of luspatercept versus BSC for patients with MDS-associated anemia was $623,219 per QALY, with a 0% chance of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY. To achieve cost-effectiveness of luspatercept at a threshold of $50,000 per QALY, a price reduction of 85% would be required.
The scenario analyses considering baseline transfusion burden resulted in ICERs of $502,394, $502,951, and $1,170,786 per QALY for patients in the LTB, ITB, and HTB baseline states, respectively, indicating that luspatercept is least cost-effective in patients in the HTB state. Key drivers of the analysis are the transition probabilities between TI and TD health states during and after luspatercept treatment as indicated in MEDALIST results and the cost of luspatercept. Clinical experts consulted by CADTH indicated that transfusion burden alone is not necessarily an important outcome for patients and does not correlate closely with patients’ quality of life.
References
1.Reblozyl (luspatercept): 25 mg, 75 mg/vial of lyophilized powder for solution for subcutaneous injection [product monograph]. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2020 Sep 25.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Reblozyl (luspatercept) for myelodysplastic syndromes (MDS)-associated anemia, 25 mg, 75 mg/vial of lyophilized powder for solution for subcutaneous injection. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2021 Jan 28.
3.Clinical Study Report: ACE-536-MDS-001 (MEDALIST). A phase 3, double-blind, randomized study to compare the efficacy and safety of luspatercept (ACE-536) versus placebo for the treatment of anemia due to IPSS-R very low, low, or intermediate risk myelodysplastic syndromes in subjects with ring sideroblasts who require red blood cell transfusions [internal sponsor's report]. Summit (NJ): Celgene Corporation; 2019 Mar 13.
4.Delea T, Ouagari K, Sofrygin O. Cost of current iron chelation infusion therapy and cost-effectiveness of once-daily oral deferasirox in transfusion-dependent thalassemia patients in Canada. Blood. 2006;108:3349.
5.Greenberg P, Tuechler H, Schanz J. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454-2465. PubMed
6.Falantes J, Calderon C, Malaver F, et al. Clinical prognostic factors for survival and risk of progression to acute myeloid leukemia in patients with myelodysplastic syndromes with 10% marrow blasts and non-unfavorable cytogenetic categories. Clin Lymphoma Myeloma Leuk. 2013;13(2):144-152. PubMed
7.Harnan S, Ren S, Gomersall T. Associations between transfusion status and overall survival in patients with myelodysplastic syndrome - a systematic literature review and meta-analysis. Acta Haematol. 2016;136:13-42. PubMed
8.Voso M, Fenu S, Latagliata R. Revised International Prognostic Scoring System (IPSS) predicts survival and leukemic evolution of myelodysplastic syndromes significantly better than IPSS and WHO prognostic scoring system: validation by the Gruppo Romano Mielodisplasie Italian Regional Database. J Clin Oncol. 2013;31(21):2671-2677. PubMed
9.Oran B, Weisdorf D. Survival for older patients with acute myeloid leukemia: a population-based study. Haematologica. 2012;97:1916-1924. PubMed
10.Szende A, Schaefer C, Goss T, et al. Valuation of transfusion-free living in MDS: results of health utility interviews with patients. Health Qual Life Outcomes. 2009;7:81. PubMed
11.Forsythe A, Brandt P, Dolph M, Patel S, Rabe A, Tremblay G. Systematic review of health state utility values for acute myeloid leukemia. Clinicoecon Outcomes Res. 2018;10:83-92. PubMed
12.Osborne R, De Abreu L, Dalton A. Quality of life related to oral versus subcutaneous iron chelation: a time trade-off study. Value Health. 2007;10:451-456. PubMed
13.Lagerquist O, Poseluzny D, Westiuk G. The cost of transfusing a unit of red blood cells: a costing model for Canadian hospital use. ISBT Sci Ser. 2017;12:375-380.
14.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Apr 6.
15.Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2021 Apr 6.
16.Vidaza (azacitidine): 100 mg injection [product monograph]. Mississauga (ON): Celgene Inc; 2018 Mar 22: https://media.celgene.com/content/uploads/sites/23/Vidaza-Product_Monograph_-_English_Version.pdf. Accessed 2021 Jan 28.
17.Patient cost estimator. 2021; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2021 Jan 28.
18.Platzbecker U, Fenaux P, Ades L. Proposals for revised IWG 2018 hematological response criteria in patients with MDS included in clinical trials. Blood. 2019;133:1020-1030. PubMed
19.Inqovi (decitabine and cedazuridine): 35 mg decitabine / 100 mg cedazuridine tablets [product monograph]. Oakville (ON): Taiho Pharma Canada Inc.; 2020 Jul 7: https://pdf.hres.ca/dpd_pm/00057427.PDF. Accessed 2021 Jun 3.
20.Luspatercept (Reblozyl) for beta-thalassemia associated anemia. CADTH reimbursement review pharmacoeconomic report. Ottawa (ON): CADTH; 2021. Accessed Unpublished.
21.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Apr 6.
22.Apo-deferasirox: 125 mg, 250 mg, 500 mg dispersible tablets for oral suspension [product monograph]. Toronto (ON): Apotex Inc; 2020 May 4: https://pdf.hres.ca/dpd_pm/00056345.PDF. Accessed 2021 Apr 6.
23.Deferoxamine mesylate for injection: 500 mg deferoxamine mesylate /vial, 2 g deferoxamine mesylate /vial lyophilized powder [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2017 Jul 12: https://pdf.hres.ca/dpd_pm/00040145.PDF. Accessed 2021 Apr 6.
24.Ferriprox (deferiprone): 500 mg and 1000 mg tablets, 100 mg/mL oral solution [product monograph]. Toronto (ON): Chiesi Canada Corp; 2020 Mar 11: https://pdf.hres.ca/dpd_pm/00055386.PDF. Accessed 2021 Apr 6.
25.Budge Impact Analysis. Reblozyl (luspatercept) for myelodysplastic syndromes [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Reblozyl (luspatercept) for myelodysplastic syndromes (MDS)-associated anemia, 25 mg, 75 mg/vial of lyophilized powder for solution for subcutaneous injection. Saint-Laurent (QC): Celgene Inc., a Bristol-Myers Squibb company; 2021 Jan 28.
26.Leitch H, Parmar A, Wells R, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol. 2017;179:83-97. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison for Adult Patients With MDS-Associated Anemia
Treatment | Strength / concentration | Form (vial size if single-use) | Price | Recommended dosage | Daily costa | Annual cost |
|---|---|---|---|---|---|---|
Luspatercept (Reblozyl) | 50 mg/mL | 25 mg powder for SC injection 75 mg powder for SC injection | $2,189.0000b $6,567.0000b | 1.0 to 1.75 mg/kg every 3 weeks | $416.95 to $625.43 | $152,188 to $228,281 |
SC = subcutaneous.
Note: Annual costs are based on 365 days per year.
aBased on a mean weight of 76.0 kg in the MEDALIST trial.3
bSponsor submitted price.2
Table 9: CADTH Cost Comparison for the Treatment of Chronic Iron Overload
Treatment | Strength / concentration | Form (vial size if single-use) | Price | Recommended dosagea | Daily costb | Annual cost |
|---|---|---|---|---|---|---|
Deferasirox | 125 mg 250 mg 500 mg | Tablet | $9.2228 $18.4453 $36.8909 | 10 to 30 mg/kg daily | $64.56 to $175.23 | $23,564 to $63,960 |
Deferiprone | 1,000 mg 100 mg/mL | Tablet Oral solution | $33.4740 $3.3495 | 25 to 33 mg/kg 3 times daily | $200.84 to $301.27 | $73,308 to $109,962 |
SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary14 or Ontario Exceptional Access Program Formulary21 (accessed April 2021), unless otherwise indicated, and do not include mark-up or dispensing fees. Annual costs are based on 365 days per year.
aRecommended dosages are from the respective product’s monograph.22-24
bBased on a mean weight of 76.0 kg from patients in the MEDALIST trial.3
cSaskatchewan drug benefit formulary (accessed April 2021).15
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | Yes | No comment |
Model has been adequately programmed and has sufficient face validity . | No | There existed technical glitches with several of the radio and check boxes provided by the sponsor, making it difficult to identify the different scenarios presented in the pharmacoeconomic report. In addition, there were some discrepancies between the pharmacoeconomic report and the model that made validating sponsor assumptions around OS and AML challenging. |
Model structure is adequate for decision problem. | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | The probabilistic analysis was difficult to validate on account of there being almost 800 parameters in the model, a portion of which had the ability to be varied probabilistically. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | The submitted congruence test used an average of ICERs instead of the average incremental costs and QALYs. This led to unexpected results when incremental QALYs were close to 0. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details). | No | The submitted model included multiple (redundant) locations where parameter requirements were listed (e.g., percent of patients responding). Furthermore, the probabilistic model results were reported in multiple places and, in some cases, were inappropriately calculated (e.g., average of ICERs). This made validating the model unnecessarily complex and extra work was required to identify which cell values were driving the model. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Table 11: Detailed Results of the Sponsor’s Base Case
Category | Luspatercept + BSC | BSC | Incremental |
|---|---|---|---|
Costs | |||
MDS costs | $318,656 | $156,547 | $162,109 |
Luspatercept acquisition | $131,295 | $0 | $131,295 |
Luspatercept administration | $696 | $0 | $696 |
HR MDS drug acquisition | $1,897 | $1,694 | $204 |
HR MDS drug administration | $107 | $95 | $12 |
HR MDS allo-HSCT | $238 | $212 | $26 |
ICT acquisition | $60,983 | $50,100 | $10,883 |
ICT administration | $2,255 | $1,837 | $418 |
Treatment-related AEs | $23,911 | $17,272 | $6,640 |
Transfusions | $78,257 | $69,556 | $8,701 |
Physician visits | $7,052 | $5,882 | $1,170 |
Iron overload monitoring | $884 | $766 | $119 |
MDS complications monitoring | $2,102 | $1,844 | $258 |
Anemia-related complications | $8,799 | $7,143 | $1,655 |
ICT AE required monitoring | $179 | $146 | $33 |
AML costs | $608 | $522 | $87 |
Consolidation acquisition | $81 | $69 | $12 |
Physician visits | $259 | $222 | $36 |
Iron overload monitoring | $10 | $8 | $1 |
Complications monitoring | $64 | $55 | $9 |
Anemia-related complications | $195 | $167 | $28 |
Total | $319,264 | $157,068 | $162,196 |
Life-years (by health state) | |||
TI | 0.45 | 0.14 | 0.30 |
LTB | 0.67 | 0.38 | 0.30 |
ITB | 1.09 | 0.72 | 0.37 |
HTB | 1.82 | 1.83 | 0.00 |
HR MDS | 0.22 | 0.15 | 0.07 |
AML | 0.08 | 0.07 | 0.01 |
Total | 4.32 | 3.28 | 1.05 |
QALYs (by health state) | |||
TI | 0.38 | 0.12 | 0.26 |
LTB | 0.50 | 0.28 | 0.22 |
ITB | 0.75 | 0.50 | 0.25 |
HTB | 1.15 | 1.15 | 0.00 |
HR MDS | 0.14 | 0.10 | 0.04 |
AML | 0.05 | 0.04 | 0.01 |
Total | 2.98 | 2.19 | 0.79 |
ICER ($ per QALY) | $206,439 | ||
AML = acute myeloid leukemia; HR MDS = high-risk myelodysplastic syndrome; HTB = high transfusion burden; ICER = incremental cost-effectiveness ratio; ICT = iron chelation therapy; ITB = intermediate transfusion burden; LTB = low transfusion burden; QALYs = quality-adjusted life-years; TI = transfusion-independent.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Luspatercept | BSC | Incremental |
|---|---|---|---|
Discounted life-years | |||
Total | 3.29 | 3.13 | 0.16 |
TI | 0.44 | 0.14 | 0.30 |
LTB | 0.53 | 0.37 | 0.16 |
ITB | 0.82 | 0.70 | 0.12 |
HTB | 1.34 | 1.73 | −0.39 |
HR MDS | 0.11 | 0.13 | −0.02 |
AML | 0.06 | 0.06 | 0.00 |
Discounted QALYs | |||
Total | 2.29 | 2.09 | 0.19 |
TI | 0.37 | 0.12 | 0.25 |
LTB | 0.40 | 0.28 | 0.12 |
ITB | 0.57 | 0.48 | 0.08 |
HTB | 0.85 | 1.09 | −0.25 |
HR MDS | 0.07 | 0.09 | −0.02 |
AML | 0.03 | 0.03 | 0.00 |
Discounted costs ($) | |||
Total | 270,123 | 149,569 | 120,553 |
MDS costs | 269,629 | 149,080 | 120,549 |
Luspatercept acquisition | 131,619 | 0 | 131,619 |
Luspatercept administration | 718 | 0 | 718 |
HR MDS drug acquisition | 1,279 | 1,538 | −259 |
HR MDS drug administration | 72 | 87 | −15 |
HR MDS allo-HSCT | 161 | 194 | −32 |
ICT acquisition | 45,473 | 47,776 | −2,303 |
ICT administration | 1,691 | 1,761 | −71 |
Treatment-related AEs | 17,295 | 16,363 | 933 |
Transfusions | 57,273 | 66,341 | −9,068 |
Physician visits | 5,219 | 5,617 | −398 |
Iron overload monitoring | 630 | 722 | −93 |
MDS complications monitoring | 1,521 | 1,744 | −224 |
Anemia-related complications | 6,547 | 6,801 | −254 |
ICT AE required monitoring | 130 | 136 | −6 |
AML costs | 494 | 490 | 5 |
Consolidation acquisition | 58 | 61 | −3 |
Physician visits | 215 | 211 | 4 |
Iron overload monitoring | 8 | 8 | 0 |
Complications monitoring | 52 | 51 | 1 |
Anemia-related complications | 161 | 158 | 3 |
ICER ($/QALY) | 623,219 | ||
AE = adverse event; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TD = transfusion-dependent; TI = transfusion-independent.
Scenario Analyses
Table 13: Summary of Scenario Analyses Conducted on CADTH Base Case
Scenario | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
1. OS assumptions based on IPSS-R levels | BSC | 237,372 | 3.23 | Reference |
Luspatercept | 353,774 | 3.45 | 540,188 | |
2. Used July 2019 data cut | BSC | 158,073 | 2.31 | Reference |
Luspatercept | 272,782 | 2.48 | 658,849 | |
3. Assumed 100% of patients were LTB at baseline | BSC | 135,074 | 2.20 | Reference |
Luspatercept | 294,470 | 2.52 | 502,394 | |
4. Assumed 100% of patients were ITB at baseline | BSC | 153,477 | 2.07 | Reference |
Luspatercept | 274,297 | 2.31 | 502,951 | |
5. Assumed 100% of patients were HTB at baseline | BSC | 156,731 | 2.03 | Reference |
Luspatercept | 251,230 | 2.11 | 1,170,786 |
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the introduction of luspatercept for the treatment of adult patients with very low- to intermediate-risk MDS-associated anemia who have ring sideroblasts. The analysis was taken from the perspective of the Canadian public drug plans using an epidemiology-based approach, with only drug acquisition costs, mark-up, and dispensing fees included. A 3-year time horizon was used, from 2022 to 2024, with 2021 as a base year. The population size was estimated using the prevalence and incidence of MDS, followed by a series of stepwise attritions to specify the population size. A summary of the sponsor’s derivation of the eligible population size is presented in Figure 2.
In Canada, there are currently no medications specifically indicated for the treatment of TD anemia associated with very low- to intermediate-risk MDS, and thus no comparators. The reference case scenario consisted of BSC which comprised RBC transfusions and ICTs. The new drug scenario included luspatercept given in conjunction with BSC, and BSC alone. As the costs for RBC units are not reimbursed via Canadian public drug plans the costs associated with BSC only included ICT acquisition costs in both arms. Key inputs to the BIA are documented in Table 15.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
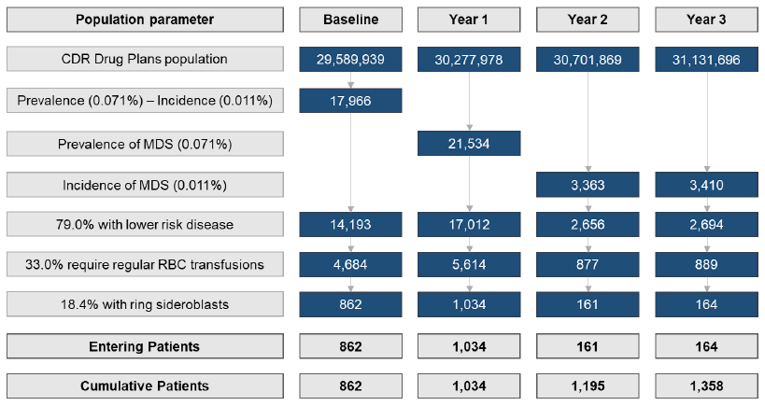
CDR = CADTH Common Drug Review; MDS = myelodysplastic syndromes; RBC = red blood cell.
Source: Sponsor’s budget impact submission.25
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Number of patients eligible for drug under review | 1,034 / 1,195 / 1,359 |
Market uptake (3 years) | |
Uptake (reference scenario) Luspatercept + BSC BSC alone | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Luspatercept + BSC BSC alone | ||||||% / ||||||% / ||||||% ||||||% / ||||||% / ||||||% |
Cost of treatment (per patient) | |
Cost of treatment annually (acquisition costs only) Luspatercept Deferoxamine mesylate Deferasirox | $195,591 $9,054 to $18,108 $23,580 to $94,321 |
BSC = best supportive care.
Summary of the Sponsor’s BIA Results
The estimated budget impact of funding luspatercept for the treatment of adult patients with very low- to intermediate-risk MDS-associated anemia who have ring sideroblasts was $25,947,853 in year 1, $28,395,584 in year 2, and $21,982,237 in year 3, for a 3-year total of $76,325,673.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Inclusion of patients who were not refractory to ESAs: The sponsor’s base case included all patients with lower-risk MDS who were TD with ring sideroblasts. However, the Health Canada indication further specifies that patients must have failed or are not suitable for erythropoietin-based therapy.1 This also aligns with the MEDALIST trial, which only included patients who were refractory to ESAs.3 The sponsor included a scenario in the BIA which restricted the population to those who were refractory to ESAs and would thus be eligible for MEDALIST, an estimate of ||||||%. This estimate was based on the MDS-CAN trial, but CADTH did not receive the clinical study report as part of the evidence package. Therefore, CADTH used data from a published paper of MDS-CAN to estimate the proportion of patients who were refractory to ESAs.26
CADTH only included patients who were refractory to ESAs in the base case, estimated to be 54% of the sponsor’s included base case population based on Leitch et al.26 The full population was explored in scenario analysis.
Market uptake of luspatercept is likely underestimated: The sponsor assumed market share uptake of luspatercept to be ||||||%, ||||||%, and ||||||% of the market in year 1, 2, and 3, respectively. Clinical experts consulted by CADTH felt that represented an underestimate of the rate at which this drug is expected to be prescribed, especially in year 1. Given that luspatercept is the first therapy to be indicated for patients with MDS-associated anemia with ring sideroblasts, the market uptake in year 1 is expected to be higher. Clinical experts suggested that patients and clinicians have been waiting for a treatment, and that if this drug were approved a high rate of use would be seen immediately. The sponsor provided an option in the analysis for a ‘fast uptake’ of the drug, where market share is equal to ||||% in all years.
CADTH assumed market shares of 44.3% in all 3 years of the BIA, as part of the base case, and included the sponsor-provided “optimistic” market share assumptions (+||% to each year) as part of scenario analysis.
CADTH Reanalyses of the BIA
Based on the limitations identified, CADTH’s base case included only patients who were ESA-refractory and increased the market shares of luspatercept in years 1 and 2.
Table 16: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Consideration of patients who are ESA-refractory | ||||||% (though not included in the base case) | 54.4% |
2. Market shares of luspatercept are underestimated | ||||||% / ||||% / ||||||% | 44.3% in all years |
CADTH base case | Reanalysis 1 + 2 | |
ESA = erythropoietin-stimulating agent.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. Based on the CADTH base case, the budget impact of the reimbursement of luspatercept for the treatment of MDS-associated anemia in expected to be $49,237,991 in year 1, $39,292,172 in year 2, and $12,948,948 in year 3, with a 3-year budget impact of $101,479,111. A scenario analysis involving the full population regardless of ESA eligibility resulted in a 3-year budget impact of $186,542,484. If an 85% price reduction were applied to luspatercept as per the pharmacoeconomic model appraisal, the resulting 3-year budget impact is $15,304,087.
Table 17: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $76,325,673 |
CADTH reanalysis 1 – only ESA-refractory patients | $41,521,166 |
CADTH reanalysis 2 – increased market shares in year 1 and 2 | $186,542,484 |
CADTH base case | $101,479,111 |
ESA = erythropoiesis-stimulating agent.
Table 18: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $14,424,657 | $17,289,128 | $17,962,892 | $17,822,167 | $67,498,845 |
New drug | $14,424,657 | $43,236,981 | $46,358,476 | $39,804,404 | $143,824,517 | |
Budget impact | $0 | $25,947,853 | $28,395,584 | $21,982,237 | $76,325,673 | |
CADTH base case | Reference | $7,847,014 | $9,405,286 | $9,771,813 | $9,695,259 | $36,719,371 |
New drug | $7,847,014 | $58,643,277 | $49,063,985 | $22,644,207 | $138,198,483 | |
Budget impact | $0 | $49,237,991 | $39,292,172 | $12,948,948 | $101,479,111 | |
CADTH scenario analysis 1: full population (including ESA eligible) | Reference | $14,424,657 | $17,289,128 | $17,962,892 | $17,822,167 | $67,498,845 |
New drug | $14,424,657 | $107,800,142 | $90,191,149 | $41,625,380 | $254,041,328 | |
Budget impact | $0 | $90,511,013 | $72,228,257 | $23,803,213 | $186,542,484 | |
CADTH scenario analysis 2: ‘optimistic’ market shares ( + 50% each year) | Reference | $7,847,014 | $9,405,286 | $9,771,813 | $9,695,259 | $36,719,371 |
New drug | $7,847,014 | $30,634,307 | $32,978,279 | $27,641,809 | $99,101,409 | |
Budget impact | $0 | $21,229,021 | $23,206,466 | $17,946,550 | $62,382,037 | |
CADTH scenario analysis 3: 85% price reduction | Reference | $7,847,014 | $9,405,286 | $9,771,813 | $9,695,259 | $36,719,371 |
New drug | $7,847,014 | $16,685,397 | $15,680,451 | $11,810,596 | $52,023,458 | |
Budget impact | $0 | $7,280,112 | $5,908,638 | $2,115,337 | $15,304,087 |
ESA = erythropoiesis-stimulating agent.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.