CADTH Reimbursement Review
Budesonide (Jorveza)
Sponsor: Avir Pharma Inc.
Therapeutic area: Maintenance of eosinophilic esophagitis in adults
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ADR
adverse drug reaction
AE
adverse event
CDEC
Canadian Drug Expert Committee
CI
confidence interval
DB
double-blind
EEsAI
Eosinophilic Esophagitis Activity Index
EEsAI-PRO
Eosinophilic Esophagitis Activity Index Patient Reported Outcome
EoE
eosinophilic esophagitis
EoE-QoL-A
Adult Eosinophilic Esophagitis Quality of Life
eos
eosinophil
EOT
end of treatment
FAS
full analysis set
FDA
Food and Drug Administration
GI
gastrointestinal
HPF
high-power field
HRQoL
health-related quality of life
MID
minimal important difference
modSHS
modified Short Health Scale
NRS
numerical rating scale
OLE
open-label extension
OLI
open-label induction
OLRI
open-label re-induction
PatGA
Patient’s Global Assessment
PGA
Physician’s Global Assessment
PPI
proton pump inhibitor
PPI-REE
PPI-responsive esophageal eosinophilia
QoL
quality of life
SAE
serious adverse event
SAF
safety analysis set
SHS
Short Health Scale
TEAE
treatment-emergent adverse event
VDQ
Visual Dysphagia Question
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Budesonide orodispersible tablets (Jorveza), 0.5 mg and 1 mg, oral |
Indication | Induction and maintenance of clinico-pathological remission in adults with eosinophilic esophagitis (EoE) |
Reimbursement request | For the maintenance of clinico-pathological remission in adults with EoE |
Health Canada Approval Status | NOC |
Health Canada Review Pathway | Standard review |
NOC date | March 16, 2021 |
Sponsor | Avir Pharma Inc. |
EoE = eosinophilic esophagitis; NOC = Notice of Compliance.
Introduction
Eosinophilic esophagitis (EoE) is a chronic, local immune-mediated, esophageal disease characterized histologically by eosinophil-predominant inflammation and clinically by symptoms related to esophageal dysfunction.1,2 The most commonly reported symptoms in older children and adults are dysphagia (difficulty swallowing), food impactions (food getting stuck in the esophagus), and chest pain not associated with swallowing.1,2 EoE impairs patients’ social and psychological functioning and significantly affects their health-related quality of life (HRQoL).2 In Canada, the most recent estimates of EoE were published in a systematic review in 2018, which reported an incidence rate of 2.1 to 10.7 EoE cases per 100,000 per year.3 The diagnostic criteria of EoE include the following: presence of clinical symptoms indicative of esophageal dysfunction; eosinophil-predominant inflammation on esophageal biopsy, consisting of a peak value of at least 15 eosinophils per high-power field (HPF) (or 60 eosinophils/mm2 [eos/mm2]); and the exclusion of any non-EoE disorders that may be responsible for or may contribute to symptoms and esophageal eosinophilia.1,4
The management of EoE includes a variety of dietary, pharmacological, and endoscopic interventions.5 The aim of therapy is symptomatic relief, with histologic improvement in esophageal eosinophilia, and, in the case of children, restoration of normal growth and development.6 Dietary therapy is 1 of the first-line treatment options in children and adults and involves avoidance of certain foods, to minimize allergen exposure.2,6 Before budesonide orodispersible tablets were approved in Canada to induce remission in adults with EoE, there were no approved pharmacological drugs for the treatment of EoE. In addition, budesonide orodispersible tablets would be the first treatment approved in Canada for the maintenance of clinicopathologic remission in adults with EoE. Because of the lack of approved specific treatments, proton pump inhibitors (PPIs) and topical corticosteroids are being used off-label to treat the disease.2,7 Either might be offered as first-line anti-inflammatory pharmacological therapy.2,7 Topical corticosteroids fluticasone propionate and nebulized budesonide are generally prescribed. Fluticasone, per its instructions, is sprayed into the patient’s mouth and then swallowed, while, for budesonide, patients are instructed to mix budesonide with sucralose or another thickener to form an aqueous gel (slurry) for administration.2,6 Topical corticosteroids are associated with several limitations, preventing the development of an optimized formulation. Given that EoE is a chronic disease and symptoms commonly recur after discontinuing treatment, it is recommended that maintenance therapy be considered in certain patients. Patients who need maintenance therapy are those with a narrow-calibre esophagus, prior emergent endoscopy performed for esophageal food bolus impaction, prior esophageal stricture requiring repeated stretching (dilation), prior esophageal perforation, severe or ongoing symptoms, or prior Boerhaave syndrome, as well as those who prefer maintenance therapy.8
Budesonide orodispersible tablets are indicated for the induction and maintenance of clinicopathologic remission in adults with EoE.9 The recommended daily dosage of budesonide for the maintenance of remission is one 0.5 mg orodispersible tablet in the morning and another in the evening (a total dosage of 1 mg of budesonide daily). The duration of maintenance therapy is determined by the treating physician. It is recommended that budesonide be taken after a meal, and no food or liquid should be taken during or 30 minutes after administration.9
The objective of this report is to perform a systematic review of the beneficial and harmful effects of budesonide 0.5 mg orodispersible tablets for the maintenance of clinicopathologic remission in adults with EoE.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and by clinical experts consulted by CADTH for the purpose of this review.
Patient Input
A total of 3 patient group submissions were received for this review, from 2 Canadian patient organizations, the Gastrointestinal (GI) Society and Food Allergy Canada, and 1 international patient group, the EOS Network (formerly Families Affected by Eosinophilic Diseases [FABED]) from the UK.
The patient groups submitted input from a variety of sources, including results from published studies, a patient experience survey, telephone interviews with patients, experiences as a patient advocacy organization, social media commentary, and direct commentary and quotes from patients and caregivers.
According to the patient input received, the symptoms of EoE vary among individuals and can include difficulty swallowing, choking, regurgitation, nausea, vomiting, fatigue, reflux, and abdominal and/or chest pain, as well as malnutrition and failure to thrive in the case of young children.
Living with EoE greatly affects the daily lives of patients and their families — socially, mentally, and financially. Dietary restriction presents the biggest burden to the lives of patients and/or caregivers, negatively affecting activities such as holidays and family gatherings, social engagements, dining away from home, and travel.
The patient groups reported that corticosteroids generally reduce the number of eosinophils and improve symptoms; however, these are primarily asthma medications used beyond the Health Canada indication. They are swallowed from an inhaler or mixed, and the nonspecific nature of drug delivery makes the effectiveness varied and uncertain.
Patients expressed a desire for convenience in medication administration as well as clear instructions to maintain compliance. Patients expressed a need for a treatment that improves their day-to-day quality of life (i.e., eating, working, and socializing) and indicated that an effective therapy that resolves clinicopathologic symptoms, is easy to consume, and has minimal long-term complications is highly important.
Clinician Input
Input from Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH indicated that every treatment option currently available has some limitations. The dietary approach is challenging for patients to follow. Maintaining food avoidance over a long period is cumbersome for patients. Furthermore, several patients have no specific food trigger. PPI therapy is the most straightforward treatment and is well tolerated. PPIs are effective treatments in a subgroup of patients with mild symptoms; however, for patients who respond to PPI therapy, there is a risk of relapse upon discontinuation of the PPI. Response rates to topical corticosteroids are high; however, recurrence rates on withdrawal of the medication are high as well. In Canada, these agents are not commercially formulated for the EoE indication and are used off-label; hence, they must be compounded or administered via a different route than approved. Patients, physicians, and pharmacists are left to adapt these corticosteroid formulations for the patient. This can be confusing and cumbersome, leading to reduced patient compliance. Hence, other formulations of topical corticosteroids are needed to improve convenience and compliance.
The clinical experts indicated that budesonide orodispersible tablets would fulfill the same role as compounded topical corticosteroids; thus, introducing budesonide orodispersible tablets would not, by itself, represent a large treatment paradigm shift.
The clinical experts indicated that the patients best suited for maintenance treatment with budesonide orodispersible tablets are those who responded to the initial treatment with budesonide orodispersible tablets after failure to respond to PPI therapy and whose symptoms recur more than once per year or who have a history of severe disease, as manifested by food impactions or significant fibrosis; and patients with severe endoscopic disease who are also intolerant to or noncompliant with fluticasone or other formulations of budesonide. The clinical experts said the patients who are least likely to benefit from budesonide orodispersible tablets are those who respond to PPI treatment or other topical steroids (such as fluticasone propionate or budesonide slurry).
In practice, clinicians assess patients symptomatically. No scales are currently used in clinical practice to assess symptomatic response. Meaningful responses to treatment include the complete resolution of symptoms of dysphagia and food impaction. Other important assessments are an overall improvement in a patient’s symptoms, allowing them to consume solid food of all consistencies; reduced hospitalization; lack of need for dilation; absence of strictures; lack of need for endoscopy; and a decrease in the frequency and severity of dysphagia. The clinical experts indicated that treatment response should be assessed 3 months after initiating maintenance therapy, then every 6 months to 1 year thereafter.
The clinical experts indicated that patients who relapse while receiving 0.5 mg budesonide twice daily for maintenance of remission would have their dosage increased to 1 mg budesonide twice daily (for re-induction of remission). After achieving remission on the 1 mg budesonide twice daily dosage, patients would be switched back to the 0.5 mg budesonide twice daily dosage for maintenance of remission. Patients who relapse again while on 0.5 mg budesonide twice daily would have their dosage increased to 1 mg budesonide twice daily for re-induction of remission. After achieving remission on the 1 mg budesonide twice daily dosage, patients would remain on the 1 mg budesonide twice daily dosage for the maintenance of remission. Patients who relapse while receiving the 1 mg budesonide twice daily dosage as maintenance of remission need to be assessed for compliance and other factors associated with recurrence, and some patients might need to discontinue budesonide orodispersible tablets and try another treatment approach.
The clinical experts also indicated that, while maintenance therapy generally implies continuous treatment, in clinical practice, treatment might be titrated, intermittent, continuous, or stopped. The decision is affected by symptoms, complications, strictures, and persistent inflammation on endoscopy.
The clinical experts suggested that treatment discontinuation can be considered if unacceptable side effects are present (such as recurrent candidiasis, systemic side effects of topical corticosteroids, and hypersensitivity), or if patients are intolerant to the drug.
The clinical experts agreed that budesonide orodispersible tablets for the treatment of EoE should be prescribed by specialists in gastroenterology or allergy who have expertise in EoE.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
Input was obtained from the jurisdictions participating in CADTH reimbursement reviews. Key factors that could affect implementation include:
duration of maintenance therapy
use of 1 mg budesonide orodispersible tablets twice daily off-label for the maintenance of remission
use of budesonide orodispersible tablets in PPI-naive patients
use of 1 mg budesonide orodispersible tablets in children
time interval follow-up for assessment.
The clinical experts consulted by CADTH provided responses (Table 4).
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The BUL-2/EER trial (N = 204) was a pivotal phase III, double-blind (DB), randomized, multi-centre, placebo-controlled study that compared the efficacy and tolerability of a 48-week treatment with 2 different dosages of budesonide effervescent tablets (budesonide 0.5 mg twice daily and budesonide 1 mg twice daily) with placebo for the maintenance of clinicopathologic remission in adult patients with EoE. Patients enrolled in the trial were adults (18 to 75 years of age) with a confirmed clinicopathologic diagnosis of EoE, and clinicopathologic remission achieved either in the open-label induction (OLI) phase of BUL-2/EER or the induction trial BUL-1/EEA (reviewed by the CADTH Canadian Drug Expert Committee [CDEC] for the induction of remission indication), who must have undergone a documented trial with PPIs to exclude PPI-responsive esophageal eosinophilia (PPI-REE). Patients were assigned to 1 of 3 treatment groups via a central randomization procedure using a 1:1:1 allocation ratio to receive either budesonide 0.5 mg orodispersible tablet twice daily, budesonide 1 mg orodispersible tablet twice daily, or a placebo orodispersible tablet twice daily. The budesonide and placebo treatments were identical in physical appearance, which assured treatment blinding. Randomized treatment assignment was not stratified. The percentage of patients who had not had a treatment failure after 48 weeks of treatment was the primary end point. The percentage of patients with histologic relapse, change in the peak eos/mm2 HPF from baseline; the percentage of patients with a clinical relapse; the percentage of patients with a total weekly Eosinophilic Esophagitis Activity Index Patient Reported Outcome (EEsAI-PRO) score of 20 or less; and the percentage of patients in deep disease remission were key secondary end points. HRQoL was assessed using the modified Short Health Scale (modSHS) and the Adult Eosinophilic Esophagitis Quality of Life (EoE-QoL-A) questionnaire. modSHS and EoE-QoL-A were exploratory outcomes in the BUL-2/EER trial. Budesonide 1 mg twice daily is not an approved dosage for the maintenance of remission in Canada; therefore, this review focused on the budesonide 0.5 mg twice daily dosage only.
In the BUL-2/EER trial, the average age of the participants was 36 years, and the majority were men (84% and 81% for the budesonide 0.5 mg and placebo study arms, respectively). All baseline parameters of disease activity, including histologic results and endoscopic as well as patients’ and investigators’ assessments, showed low values for disease activity in all treatment groups, which is representative of EoE patients who are in remission. The disease duration since diagnosis and since first symptoms were shorter in the placebo group than in the budesonide 0.5 mg group. The mean time since an established EoE diagnosis was 4.3 years and 3.3 years for the budesonide 0.5 mg and placebo study groups, respectively, with a mean time since symptom onset of 12.6 years and 9.6 years, respectively. In addition, fewer patients in the placebo group (5.9%) had a previous esophageal dilation compared to the budesonide 0.5 mg group (19.1%).
Efficacy Results
The percentage of patients who had not had a treatment failure after 48 weeks of DB treatment was 73.5% in the budesonide 0.5 mg twice daily group and 4.4% in the placebo group. The difference between the budesonide 0.5 mg group and placebo group was 69.1 percentage points (97.5% confidence interval [CI], 55.89 to 82.34 percentage points; P < 0.0001), which was clinically relevant and statistically significant in favour of the budesonide 0.5 mg twice daily treatment group. The time to relapse was shorter for the placebo-treated group (86 days) than for the budesonide 0.5 mg treatment group (336 days). The clinical experts consulted by CADTH considered the definition of treatment failure comprehensive, given that it took into account both clinical and histologic aspects of deterioration of the disease, and almost any indication of lapse of control was noted as treatment failure.
The percentage of patients with histologic relapse was 13.2% in the budesonide 0.5 mg twice daily group and 89.7% in the placebo group. The difference between the budesonide 0.5 mg group and placebo group was −76.5 percentage points (97.5% CI, −88.8 to −64.1 percentage points; P < 0.0001), which was statistically significant in favour of the budesonide 0.5 mg twice daily treatment group.
Clinical relapse during the DB phase was observed in 10.3% of the patients in the budesonide 0.5 mg twice daily group, and in 60.3% of those in the placebo group. The difference between the budesonide 0.5 mg group and placebo group was −50.0 percentage points (97.5% CI, −65.7 to −34.3 percentage points; P < 0.0001), which was statistically significant in favour of the budesonide 0.5 mg twice daily treatment group.
None of the patients in the budesonide 0.5 mg treatment group and 1 patient in the placebo group experienced a food impaction requiring endoscopic intervention during the treatment phase. No patient needed an endoscopic dilation at any time during the DB treatment phase.
In the BUL-2/EER trial, HRQoL was assessed using EoE-QoL-A and modSHS. The difference between the budesonide 0.5 mg twice daily group and the placebo treatment group in mean absolute change in score from baseline to end of treatment (EOT) of the DB phase for EoE-QoL-A (30 items), EoE-QoL-A (24 items), EoE-QoL-A eating/diet impact (10 items), and EoE-QoL-A eating/diet impact (4 items) was 0.46 (95% CI, 0.27 to 0.66), 0.49 (95% CI, 0.30 to 0.68), 0.65 (95% CI, 0.39 to 0.92), and 0.75 (95% CI, 0.49 to 1.02), respectively. These between-group differences were in favour of the budesonide 0.5 mg twice daily group. The difference between the budesonide 0.5 mg twice daily group and the placebo treatment group in mean absolute change in score from baseline to EOT of the DB phase for symptom burden, social function, disease-related worry, and general well-being of the modSHS was −22 (95% CI, −30.5 to −13.9), −15 (95% CI, −23.6 to −7.3), −12 (95% CI, −19.4 to −3.7), and −12 (95% CI, −18.9 to −4.3), respectively. These between-group differences were in favour of the budesonide 0.5 mg twice daily group. A minimal important difference (MID) for the EoE-QoL-A and modSHS was not identified for patients with EoE. Also, the analysis of modSHS and EoE-QoL-A was not specifically tested for statistical significance with methods adjusted for multiplicity, despite reporting the 95% CI. It is likely, however, that budesonide 0.5 mg twice daily maintained the patients’ HRQoL, while the HRQoL deteriorated in patients who received placebo.
The percentage of patients with a total weekly EEsAI-PRO score of 20 or less (which indicates remission) at the end of the DB phase was 72.1% in the budesonide 0.5 mg twice daily group and 20.6% in the placebo group. The difference between the budesonide 0.5 mg group and the placebo group was 51.5 percentage points (95% CI, 35.1 to 67.9 percentage points; P < 0.0001), which was statistically significant in favour of the budesonide 0.5 mg twice daily treatment group.
The percentage of patients in deep disease remission (i.e., deep clinical, deep endoscopic, and histologic remission, based on the peak number of eosinophils per HPF) at EOT was 39.7% in the budesonide 0.5 mg twice daily group and 0% in the placebo group. The difference between the budesonide 0.5 mg group and placebo group was 39.7 percentage points (97.5% CI, 26.4 to 53.0; P < 0.0001), which was statistically significant in favour of the budesonide 0.5 mg twice daily treatment group.
Harms Results
In the BUL-2/EER trial, the majority of patients reported at least 1 treatment-emergent adverse event (AE). A total of 87 patients (83.8%) in the budesonide 0.5 mg twice daily group and 61 patients (89.7%) in the placebo group experienced at least 1 treatment-emergent AE.
No deaths occurred. During the DB phase, only for 3 patients (4.4%) in the budesonide 0.5 mg twice daily group and none of the patients in the placebo group reported serious AEs, none of which were related to the study medication, as assessed by the investigator. Moreover, only 10% of patients in the budesonide 0.5 mg twice daily group, in contrast to 62% of patients in the placebo group, experienced an AE leading to premature withdrawal of the investigational products, most often due to deterioration/relapse of EoE or to an esophageal food impaction. Bolus impaction leading to discontinuation of DB the investigational products was observed in 2 patients in the placebo group. No patient needed a dilation during the DB phase.
The most frequently reported treatment-emergent adverse drug reactions (ADRs) in the budesonide 0.5 mg twice daily treatment group were 17 suspected candidiasis ADRs, occurring in 12 patients (17.6%), versus no such events in the placebo group. Candidiasis is a known ADR caused by the immunosuppressive action of budesonide. Not all macroscopically suspected fungal infections were confirmed by Grocott silver staining. In 5 patients, the suspected candidiasis was histologically confirmed, and, finally, in 4 patients, the suspected candidiasis was both histologically confirmed and clinically manifested.
Table 2: Summary of Key Results From Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Patients who did not have a treatment failure after 48 weeks of treatment | ||
n (%) | 50 (73.5) | 3 (4.4) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | 69.1 (55.89 to 82.34) | Reference |
P valuea | < 0.0001 | Reference |
Percentage of patients with histologic relapse, defined as peak of ≥ 48 eos/mm2 HPF at end of treatment of the DB phase | ||
n (%) | 9 (13.2) | 61 (89.7) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | −76.5 (−88.8 to −64.1) | Reference |
P valuea | < 0.0001 | Reference |
Percentage of patients with a clinical relapse, having experienced a food impaction that needed endoscopic intervention, or needed an endoscopic dilation during the DB treatment phase | ||
n (%) | 7 (10.3) | 41 (60.3) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | −50.0 (−65.7 to −34.3) | Reference |
P valuea | < 0.0001 | Reference |
EoE-QoL-A 30 items overall score (weighted average)b | ||
N | 66 | 65 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.46 (0.27 to 0.66) | Reference |
P valuec | < 0.0001d | Reference |
EoE-QoL-A 24 items overall score (weighted average)b | ||
N | 66 | 65 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.49 (0.30 to 0.68) | Reference |
P valuec | < 0.0001d | Reference |
EoE-QoL-A eating/diet impact 10 items (weighted average)b | ||
N | 66 | 65 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.65 (0.39 to 0.92) | Reference |
P valuec | < 0.0001d | Reference |
EoE-QoL-A eating/diet impact 4 items (weighted average)b | ||
N | 66 | 65 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.75 (0.49 to 1.02) | Reference |
P valuec | < 0.0001d | Reference |
Modified Short Health Scales Symptom burdene | ||
N | 66 | 64 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −22 (−30.5 to −13.9) | Reference |
P valuec | < 0.0001f | Reference |
Modified Short Health Scales — Social functione | ||
N | 66 | 64 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −15 (−23.6 to −7.3) | Reference |
P valuec | 0.0003f | Reference |
Modified Short Health Scales — Disease-related worrye | ||
N | 65 | 64 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −12 (−19.4 to −3.7) | Reference |
P valuec | 0.0041f | Reference |
Modified Short Health Scales — General well-beinge | ||
N | 66 | 64 |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −12 (−18.9 to −4.3) | Reference |
P valuec | 0.0022f | Reference |
Percentage of patients with a total weekly EEsAI-PRO score of ≤ 20 at end of treatment of the DB phase | ||
n (%) | 49 (72.1) | 14 (20.6) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | 51.5 (35.1 to 67.9) | Reference |
P valuea | < 0.0001 | Reference |
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF, i.e., < 15 eos/HPF), at end of treatment of the DB phase | ||
n (%) | 27 (39.7) | 0 |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | 39.7 (26.4 to 53.0) | Reference |
P valuea | < 0.0001 | Reference |
Harms, n (%) | ||
AEs | 57 (83.8) | 61 (89.7) |
SAEs | 3 (4.4) | 0 |
WDAEs (from study treatment) | 7 (10.3) | 41 (60.3) |
Deaths | 0 | 0 |
Notable harms, n (%) | ||
Oropharyngeal candidiasis | 5 (7.4) | 0 |
Dysgeusia | 0 | 0 |
Cataract | 0 | 1 (1.5) |
Psychiatric disorders | ||
Anxiety | 3 (4.4) | 0 |
Depression | 0 | 1 (1.5) |
Insomnia | 2 (2.9) | 0 |
Mood swings | 1 (1.5) | 0 |
Sleep disorder | 0 | 1 (1.5) |
Sore throat (pharyngitis) | 3 (4.4) | 1 (1.5) |
AE = adverse effect; b.i.d. = twice a day; CI = confidence interval; DB = double-blind; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; HPF = high-power field; SAE = serious adverse effect; vs. = versus; WDAE = withdrawal due to adverse effect.
aTesting of null hypothesis by means of the 1-sided normal approximation test, Bonferroni adjusted alpha = 0.0125.
bThe EoE-QoL-A weighted average scores range from 0 to 4. Higher scores indicate better quality of life.
c2-sided t-test used for exploratory testing.
dEoE-QoL-A scores were outside the statistical testing hierarchy.
eRange of each score: 0 to 100. Lower numbers indicate higher quality of life.
fModified Short Health Scale was outside the statistical testing hierarchy.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Critical Appraisal
The patients’ baseline characteristics and prior treatment experience appeared to be roughly balanced at baseline between groups, although the disease duration since diagnosis and since first symptoms were shorter in the placebo group than in the budesonide 0.5 mg group. The mean time since an established EoE diagnosis was 4.3 years and 3.3 years in the budesonide 0.5 mg and placebo study groups, respectively, with a mean time since symptom onset of 12.6 years and 9.6 years, respectively. In addition, fewer patients in the placebo group (5.9%) had a previous esophageal dilation compared to those in the budesonide 0.5 mg group (19.1%). The impact of such imbalance on the treatment effect assessment is unknown. A large number of patients discontinued the trial, which could also have biased the results for patient-reported outcomes, HRQoL, and other exploratory outcomes. For example, only 23 out of 68 patients (33.8%) in the placebo group completed the 48-week DB phase. Also, MID in the EoE population is unavailable for any of the patient-reported outcomes assessed. Subjective recall biases in the assessment of clinical relapse would be highly likely, particularly when such recall differed between treatment groups, due perhaps to patients’ or the assessing physicians’ awareness of the treatment assignment as a result of drug-related side effects. For example, 19.1% of patients in the budesonide 0.5 mg group had suspected candidiasis, whereas no such events were reported in the placebo group. Moreover, the majority of patients in the placebo group had experienced aggravation of the disease (“condition aggravated”) (64.7% versus 16.2%) during the 48-week treatment period, which may have led to recall of more severe or worsening symptoms or pain among patients on placebo than their counterparts on active treatment.
Patients enrolled in the BUL-2/EER trial were deemed to be similar to patients with EoE in Canada, although no Canadian study site was included in this trial. Only patients with clinicopathologic remission (defined as fulfilling both histologic remission and resolution of symptoms criteria) after receiving budesonide orodispersible tablets were enrolled. Hence, results may not be generalizable to patients who achieved clinicopathologic remission using other treatments. Patients with cardiovascular disease, diabetes mellitus, osteoporosis, active peptic ulcer disease, glaucoma, cataracts, or infections who did not have careful medical monitoring were excluded from the trial, which limits the generalizability of the trial results for patients with comorbidities. The BUL-2/EER trial was designed to demonstrate superiority over placebo at week 48. It was unclear how long the patients would remain in remission while on treatment, or whether patients would relapse after they stopped treatment. Hence, the optimal duration of maintenance treatment was not explored. The BUL-2/EER trial excluded patients with severe strictures, which may limit the interpretations of the efficacy findings to patients who have strictures with a predominant inflammatory component.
Indirect Comparisons
No indirect comparisons were identified or submitted by the sponsor.
Conclusions
The BUL-2/EER trial provided evidence on the efficacy and safety of budesonide effervescent tablets 0.5 mg for the maintenance of clinicopathologic remission in adult patients with EoE. The DB phase of BUL-2/EER trial demonstrated that the majority of patients who have had a remission of EoE following a 6- or 12-week course of budesonide 1 mg twice daily orodispersible tablets can be maintained in clinical and histologic remission for 48 weeks with budesonide 0.5 mg twice daily. The time to relapse was shorter for the placebo-treated group than for the active treatment group. The effect of budesonide on HRQoL remains uncertain due to lack of MID, the high number of patients who discontinued placebo, and recall bias. During the BUL-2/EER trial, an effect on the long-term consequences of the disease could not be shown because the number of events, such as food impaction or esophageal dilation due to stricture formation, was too low. It is uncertain whether patients would relapse if they discontinued treatment or if they switched to a lower dosage. Safety data from the BUL-2/EER trial did not demonstrate any notable concern. Long-term safety, particularly in combination with other pharmacological therapies, remains unknown.
Introduction
Disease Background
Eosinophilic esophagitis (EoE) is a chronic, local immune-mediated, esophageal disease characterized histologically by eosinophil-predominant inflammation and clinically by symptoms related to esophageal dysfunction.1,2 The most commonly reported symptoms in older children and adults are dysphagia (difficulty swallowing), food impactions (food getting stuck in the esophagus), and chest pain not associated with swallowing.1,2 EoE is considered a progressive condition; patients do not “outgrow” it. Left untreated, EoE can progress to a fibrostenotic condition that is characterized by stricture formation and functional abnormalities, such as food bolus impaction (choking on food) requiring bolus removal by an emergency endoscopic procedure.2,11 Another serious and potentially life-threatening complication of EoE is esophageal perforation and/or rupture, termed spontaneous Boerhaave syndrome, which can occur following prolonged and severe vomiting during an endoscopy or as a complication of esophageal food bolus impaction.12 EoE impairs patients’ social and psychological functioning and significantly affects their health-related quality of life (HRQoL).2
EoE can occur at any age; as well, there is a male predominance, it is more common in White people, and there is a strong association with atopic diseases.13 Epidemiologic data on EoE are relatively sparse due to poor awareness and recognition of the disease in the past. Recent literature suggests that the prevalence of EoE is on the rise, in part due to increased recognition and improved diagnosis.14 In Canada, the most recent estimates of EoE were published in a systematic review in 2018, which reported an incidence rate of 2.1 to 10.7 EoE cases per 100,000 per year.3 Notably, separate estimates for pediatric and adult populations were not provided. The global incidence of EoE in adults is estimated at 7.7 per 100,000, with no significant differences among the results from different countries.15
The diagnosis of EoE is based on symptoms, histologic findings, and endoscopic appearance. In patients with chronic symptoms of esophageal dysfunction (e.g., food impaction, dysphagia, odynophagia [pain when swallowing], abdominal pain, heartburn, food refusal, regurgitation, or chest pain) EoE is suspected.1 The index of suspicion is raised if the patient has a history of atopic comorbidities (e.g., atopic dermatitis, asthma, or immediate food-type allergies) and a family history of dysphagia or EoE. A history of severe pain after dilation of a stricture or of esophageal perforation also raises suspicion of this disorder. The diagnosis is established by upper endoscopy with esophageal biopsies in addition to an evaluation to exclude other disorders that can cause esophageal eosinophilia.1 Because the symptoms of EoE are not specific, the diagnosis may be missed.1 The diagnostic criteria of EoE are based on an updated international consensus published by Dellon et al. (2018),4 and include (1) clinical symptoms indicative of esophageal dysfunction; (2) eosinophil-predominant inflammation on esophageal biopsy, consisting of a peak value of at least 15 eosinophils per high-power field (HPF) (or 60 eosinophils/mm2); and (3) the exclusion of any non-EoE disorders that may be responsible for or contributing to symptoms and esophageal eosinophilia.1,4
Standards of Therapy
The management of EoE includes a variety of dietary, pharmacological, and endoscopic interventions.5 The aim of therapy is symptomatic relief, with histologic improvement in esophageal eosinophilia and, in the case of children, restoration of normal growth and development.6 The clinicians consulted by CADTH for the purpose of this review indicated that there is no formal Canadian guideline for this condition. Clinicians generally consult evidence from literature and follow personal experience when prescribing treatments. They also indicated that the currently available treatment for EoE includes dietary, pharmacological, and endoscopic treatment. The endoscopic treatment is used to treat complications of persistent EoE. Current treatments for EoE mainly manage symptoms rather than modifying the underlying disease mechanisms. While a high proportion of patients treated with dietary and/or pharmacological treatment achieve histologic remission and resolution of symptoms, the recurrence of symptoms and inflammation is common after treatment is discontinued. Although not indicated for EoE, proton pump inhibitors (PPIs), which reduce esophageal exposure to acid, are the first line of pharmacological therapy for EoE. They can be used in conjunction with dietary eliminations. The second line of pharmacological treatment is topical corticosteroids. After the topical corticosteroids, biologics such as mepolizumab, benralizumab, or dupilumab drugs might be used. However, they are not approved by Health Canada or reimbursed for the treatment of EoE. Esophageal dilation might be used in symptomatic patients with strictures that persist despite dietary and pharmacological treatment. With any therapy, monitoring is difficult. Endoscopy is required for the initial diagnosis of EoE, but repeat endoscopy becomes unwieldy and difficult for individuals. The clinicians consulted by CADTH indicated that the ideal treatment in adults would prevent strictures, resolve symptoms (i.e., dysphagia and food impaction), as well as reverse the histologic changes in the esophagus. The clinicians consulted by CADTH also indicated that reversing the endoscopic changes in the esophagus is also an important goal, as it would prevent long-term complications and the need for repeat dilation.
The most recent treatment guidelines identified from the literature were developed by authors participating on behalf of United European Gastroenterology; the European Society for Paediatric Gastroenterology, Hepatology and Nutrition; the European Academy of Allergy and Clinical Immunology; and the European Society of Eosinophilic Esophagitis. The guidelines were published by Lucendo et al. (2017)2; they provided a treatment algorithm that was used in the updated international consensus guidelines4 and a recent Canadian article entitled “Practical Guide to Allergy and Immunology in Canada.”16 Commonly used treatments can be broadly classified into dietary therapy, pharmacotherapy, and surgical interventions. Dietary therapy is 1 of the first-line treatment options in children and adults and involves diets that avoid certain foods to minimize allergen exposure.2,6 The empirical 6- or 4-food elimination diet involves the avoidance of the most common allergy-triggering food groups (e.g., milk, eggs, wheat and gluten, soy and legumes, peanuts, tree nuts, and fish and shellfish) and is a common dietary management strategy for EoE. Before budesonide orodispersible tablets were approved in Canada for the induction of remission in adults with EoE, there were no approved drugs for the treatment of EoE. Budesonide orodispersible tablets would also be the first treatment approved in Canada for the maintenance of clinicopathologic remission in adults with EoE. Because of the lack of approved specific treatments, PPIs and topical corticosteroids are being used off-label to treat the disease.2,7 Either might be offered as first-line anti-inflammatory pharmacological therapy.2,7 If patients respond to PPI therapy, it is recommended that it be continued at the lowest dose needed to control symptoms; however, the best maintenance doses have yet to be defined.2,6
Patients who are nonresponsive to PPIs are treated with corticosteroids — in particular, drugs used for the treatment of asthma, given the pathological similarities between the 2 conditions.2 Two drugs, fluticasone propionate and nebulized budesonide, are generally prescribed. Fluticasone is sprayed into the patient’s mouth and then swallowed, while, for budesonide, patients are instructed to mix budesonide with sucralose or another thickener to form an aqueous gel (slurry) for administration.2,6 Topical corticosteroids are associated with several limitations, preventing the development of an optimized formulation. Their efficacy has been investigated in a limited number of studies and patients, but those studies have limited comparability, since the drugs, daily dosages, length of treatment, methods of administration, and definition of outcomes were not standardized.2 Systemic corticosteroids, such as prednisone, are not recommended for the treatment of EoE.2 A number of recent biologics show promising results; however, these are not yet approved in Canada.2 It is worth noting that the US FDA has granted an Orphan Drug Designation to benralizumab for the treatment of EoE.17
Esophageal dilation is a nonpharmacological treatment in which the narrowed area of the esophagus is dilated, or stretched, using either a bougie (cone-shaped tube) or a balloon as the dilator.18 This procedure is effective for relieving dysphagia but has no effect on underlying inflammation.6 Esophageal dilation is generally reserved for patients with strictures or rings who have not responded to medical therapy.6 Esophageal dilation should be performed carefully, since it sometimes leads to complications such as chest pain and life-threatening esophageal perforation.6,8
Given that EoE is a chronic disease and symptoms commonly recur after treatment is discontinued, it is recommended that maintenance therapy be considered in certain patients. The American College of Gastroenterology Clinical Guideline8 indicates that maintenance therapy is indicated in patients with narrow-calibre esophagus, prior emergent endoscopy performed for esophageal food bolus impaction, prior esophageal stricture requiring repeated dilations, prior esophageal perforation, severe or ongoing symptoms, and prior Boerhaave syndrome therapy, as well as those who prefer maintenance therapy.
Drug
Budesonide orodispersible tablets are indicated for the induction and maintenance of clinicopathologic remission in adults with EoE.9 The European Medicines Agency has approved budesonide orodispersible tablets for the treatment of EoE in adults.19
Budesonide is formulated as a 0.5 mg or 1 mg orodispersible tablet, which is designed to dissolve by effervescence in the mouth and mix with saliva before swallowing. It should be placed on the tip of the tongue and gently pressed against the top of the mouth, where it dissolves. This usually takes about 2 minutes. The effervescence process of the tablet starts after budesonide comes into contact with saliva and stimulates the production of further saliva. The dissolved material should be swallowed with saliva little by little while the orodispersible tablet disintegrates. The orodispersible tablet should not be chewed or swallowed undissolved.9
The recommended daily dosage of budesonide for the maintenance of remission is one 0.5 mg orodispersible tablet in the morning and another in the evening (total dosage of 1 mg of budesonide daily). The duration of maintenance therapy is determined by the treating physician.9 It is recommended that budesonide be taken after a meal and that no food or liquid be taken during or 30 minutes after administration.9 The Health Canada product monograph indicates that treatment with budesonide orodispersible tablets should be initiated by a physician experienced in the diagnosis and treatment of EoE.9
Budesonide is a nonhalogenated glucocorticosteroid, which acts primarily as an anti-inflammatory. Following its binding to the glucocorticoid receptor, budesonide inhibits the antigen-stimulated secretion of various pro-inflammatory signal molecules in the esophageal epithelium. The inhibition of these pro-inflammatory signals may significantly reduce the eosinophilic infiltration of the esophagus.9
CADTH Reimbursement Review has previously reviewed budesonide orodispersible tablets for the induction of clinicopathologic remission in adults with EoE,20 and the CADTH Canadian Drug Expert Committee (CDEC) recommended reimbursement of budesonide orodispersible tablets for the induction of clinicopathologic remission in adults with EoE.21 The sponsor has requested reimbursement of budesonide orodispersible tablets for the maintenance of clinicopathologic remission in adults with EoE.
Table 3 describes key characteristics of drugs commonly recommended for EoE.
Table 3: Key Characteristics of Budesonide Orodispersible Tablets, Proton Pump Inhibitors, Budesonide Nebules, and Topical Fluticasone
Characteristic | Budesonide orodispersible tablets (Jorveza) | PPIs | Budesonide nebules | Topical fluticasone |
|---|---|---|---|---|
Mechanism of action | Reduces the eosinophilic infiltration of the esophagus | Effectively block acid secretion | Anti-inflammatory corticosteroid | Anti-inflammatory corticosteroid |
Indicationa | Under review: for the maintenance of clinicopathologic remission in adults with EoE | Reflux esophagitis | Asthma | Asthma |
Route of administration | Oral | Oral | Can be administered using a nebulizer; patients are then instructed to swallow the accumulated liquid or take as an oral viscous slurry | Administered using a metered-dose inhaler without a spacer; sprayed into the patient’s mouth and then swallowed |
Recommended dosage | 1 mg as 1 tablet (0.5 mg) in the morning and 1 tablet (0.5 mg) in the evening | Varies by drug | Induction dosage (usually divided doses): 2 mg/day to 4 mg/day Maintenance dosage (usually divided doses): 2 mg/day Nebules available in concentrations of 0.125 mg/mL, 0.25 mg/mL, or 0.5 mg/mL | Induction dosage (usually divided doses): Maintenance dosage (usually divided doses): 880 mcg/day to 1,760 mcg/day |
Serious adverse effects or safety issues | Fungal infections (candidiasis) of the mouth, pharynx, and esophagus | Warnings and precautions:
| Systemic corticosteroid effects (especially high-dose inhaled corticosteroid) Localized candidiasis | Systemic corticosteroid effects (especially high-dose inhaled corticosteroid) Localized candidiasis |
Effects of long-term treatment include hypergastrinemia, possible enterochromaffin-like cell hyperplasia and carcinoid formation in the stomach, adenomas and carcinomas in the liver, and neoplastic changes in the thyroid |
CDAD = Clostridium difficile-associated diarrhea; CDI = Clostridium difficile infection; EoE = eosinophilic esophagitis; PPI = proton pump inhibitor.
aHealth Canada–approved indication.
Source: CADTH Common Drug Review clinical expert, e-CPS,22 Lucendo et al. (2017),2 and Jorveza product monograph.9
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
Two Canadian patient organizations, the Gastrointestinal (GI) Society and Food Allergy Canada, and 1 international patient group, the EOS Network (formerly Families Affected by Eosinophilic Diseases [FABED]) from the UK, supplied input for the review of budesonide for the maintenance treatment of clinicopathologic remission in adults with EoE. The GI Society in Canada is committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to health care, and promoting gastrointestinal and liver health. Food Allergy Canada is a national non-profit charity helping Canadians live safely and confidently with food allergy. The international EOS Network aims to provide information and support to patients with eosinophilic GI diseases and their families through advocacy, educational resources, and educational events for health care professionals.
The patient groups submitted input from a variety of sources, including results from published studies, a patient experience survey, telephone interviews with patients, experiences as a patient advocacy organization, social media commentary, and direct commentary and quotes from patients and caregivers. Food Allergy Canada conducted patient telephone interviews and data analysis with the help of an independent consultant. A total of 7 patients with EoE as well as 1 caregiver were interviewed as part of the Food Allergy Canada submission. The patient experience public survey in the EOS Network input was completed in conjunction with Guts UK Charity; 39 forms were completed during the survey.
Disease Experience
Symptoms of EoE are often similar to other well-known GI disorders such as gastroesophageal reflux disease and include difficulty swallowing, food obstruction, choking, regurgitation, nausea, vomiting, fatigue, reflux, and abdominal and/or chest pain. Left untreated, EoE may lead to malnutrition, poor growth, anemia, and increased severity of food obstructions that require medical intervention. In some patients, EoE is complicated by further narrowing of the esophagus (strictures) that compound swallowing and choking. In the EOS Network patient survey, 74% of respondents reported they had suffered food obstructions and 39% reported they had needed dilation (stretching of the esophagus) to treat strictures.
Given the generic symptoms and similarities to other GI diseases, many individuals can go years without a proper diagnosis. Patients visit multiple specialists, requiring a battery of tests, creating frustration and anxiety, with patients expressing concern over health care providers’ lack of knowledge about EoE. These visits are also time-consuming, requiring individuals to take time off work or school, which increases the burden of disease.
Living with EoE has a significant financial, social, and mental impact on an individual’s quality of life (QoL). Dietary restrictions associated with having EoE mean that individuals always need to be on high alert for possible food triggers. The inability to eat the same food as family, friends, and colleagues, to dine at restaurants, and to attend work or social events results in social isolation, fear, embarrassment, anxiety, and an overall decrease in QoL. Patients’ comments reflected their daily hardships due to EoE:
“I can’t enjoy communal meals. It’s difficult to socialize, difficult to go out with groups of people. I’m always worried that I’ll be embarrassed by my reactions to food.”
“It's very restricted and so most of the time you eat alone because you cannot eat what others are eating. Eating the same restricted meals all the time is not great.”
Experience with Currently Available Treatment
There is currently no cure for EoE, and the goal of therapy is to eliminate the eosinophils in the affected area, thereby reducing inflammation and alleviating symptoms. Corticosteroid medications are widely used off-label in Canada to reduce the number of eosinophils and improve symptoms. The 2 most frequently prescribed corticosteroids are budesonide and fluticasone. In both cases, these medications are intended to be mixed in a liquid or slurry and swallowed 30 minutes before each meal to coat the esophagus. Unfortunately, neither option provides a convenient, reliable method of administration to ensure a consistent dose of medication to manage the disease. Both options require patients/caregivers to disregard the patient leaflet instructions, and verbal instructions from the prescriber are open to misinterpretation, resulting in noncompliance. Patients noted the following about taking these treatments:
“This medication helped me as it improved my symptoms, but it was difficult to take and I was very unhappy taking 5 teaspoons of Splenda daily.”
“[Oral viscous budesonide] tasted terrible. It was hard to hold down.”
“I found it difficult to know whether I was swallowing enough to make any difference. It gave me oral thrush.”
Other pharmacological options include PPIs, including omeprazole, lansoprazole, and esomeprazole, to manage symptoms. However, the majority of survey participants felt that this treatment did not improve their QoL.
Patients with EoE often have high rates of atopic allergy-related disorders. EoE is known to be triggered by a delayed reaction to food. As this is not related to immunoglobulin E (IgE), there is no available testing to determine which foods may trigger symptoms. Dietary restriction therapy involves the elimination of the food(s) likely to be causing the reaction and resulting in the accumulation of eosinophils, with the most common foods being eggs, milk and dairy, wheat, soya, seafood and shellfish, and peanuts and tree nuts. Elimination diets are challenging for patients to follow, and the trial-and-error process of elimination is very time-consuming and burdensome. Such diets require access to timely and frequent endoscopies, which is a real and significant challenge in Canada. This situation puts an even greater emphasis on having an approved drug for maintenance of the disease.
If a patient’s symptoms do not improve with an altered diet or medication, physicians may recommend an elemental diet. Individuals are placed on a liquid diet consisting of a cocktail of amino acids, sugars, vitamins, minerals, and fats for approximately 4 to 6 weeks. If an individual is not able to consume enough calories, or does not tolerate the elemental diet, then a feeding tube is required. There are cases of EoE in which individuals are no longer able to tolerate food and are permanently on a feeding tube to survive, which has a huge impact on daily life for patients and caregivers and can require 24-hour support.
The Food Allergy Canada survey asked about how current therapies were able to manage EoE symptoms on a scale of 1 (strongly disagree) to 10 (strongly agree). The average score was 6.7, suggesting that respondents were partially satisfied with their current symptom management. All patients had made dietary changes, and most had received endoscopies. Three patients had received different forms of budesonide. Of the patients responding to the EOS Network survey, 36 (92%) had been prescribed PPIs to manage symptoms and 28 (72%) had tried corticosteroids.
Of the 10 patients who received Jorveza, 80% indicated it was an effective, simple, and convenient option that improved their symptoms and QoL:
“I have felt a big improvement in my symptoms since taking the drug Jorveza (budesonide), while it has not cured my disease it has made living with it easier. I feel this is due to the convenience and simplicity of taking the right dose of medication in a dispersible tablet, especially when away from home.”
“Taking Jorveza has much improved my quality of life in a positive way, in comparison to taking budesonide slurry with Splenda. Jorveza also fits in better with my lifestyle. It has transformed my life, I feel “normal” again.”
Generally, patients agree that the benefits of Jorveza outweigh the side effects of taking corticosteroids. However, there are some unconfirmed comments that patients have experienced brittle hair and nails as well as pancreatitis, alongside reports of side effects from all current treatments (i.e., PPIs and corticosteroids).
Improved Outcomes
Currently, there are few treatments available for EoE. For patients, this means there is little hope of improving and managing their disease state over the long-term. Patients would like treatments that are effective, that have no or minimal side effects, that are easy to consume, and for which long-term safety has been tested.
For patients, the improvement in symptom control and QoL is of paramount importance. Patients in the EOS Network survey noted that the most negatively affected areas of daily life include diet and eating, work, social life, and travel. Other EoE treatment needs noted by patients included:
“Something that gets the symptoms under control. Something that doesn’t require 9 packages of Splenda a day (oral viscous budesonide) — there’s no research on whether that’s harmful.”
“Pre-mixed budesonide — that would encourage me to use it more.”
Patient organizations noted that the benefits of new therapies such as Jorveza as maintenance treatment for EoE could reduce the need for restricted/elemental diets; physician, dietitian, and hospital/emergency visits and appointments; medical procedures to remove food or dilation (stretching) of the esophagus due to long-term strictures; and stress and anxiety caused by inconsistent care.
The harsh reality for individuals with EoE is that, after a considerable amount of time, they can have a short period of remission but frequently experience symptoms again once treatment or dietary restriction stops. Given that EoE is chronic, and symptoms resume when treatment is stopped, patient groups believe that treatment for the maintenance of remission should be funded. Otherwise, patients will have to revert to using off-label, inferior options. Jorveza would be the first on-label treatment for this indication.
Clinician Input
Input from Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of EoE.
Unmet Needs
EoE is a chronic disease; lasting remission is not frequently seen in patients. Currently, no treatment that can induce long-term remission in the majority of patients is available. Also, it is sometimes difficult to assess treatment efficacy, given that there is no noninvasive measure of the inflammation in the esophagus. Hence, patients could be subject to several endoscopies to confirm that they responded to treatment. Some patients who still have inflammation in their esophagus but who achieved symptomatic response stop taking any medication for their condition.
Every treatment option currently available has some limitations. The dietary approach is challenging for patients to follow; it has a significant impact on the patient’s QoL, and it often requires the assistance of a dietitian. Also, maintaining food avoidance over a long period is cumbersome for patients. Furthermore, some patients have no specific food trigger. Although PPI therapy is the most straightforward treatment and is well tolerated, PPIs seem to be effective in less than a third of patients with mild symptoms. A substantial proportion of patients are nonresponders to PPI therapy, and, even for patients who respond to PPI therapy, there is a risk of relapse upon discontinuation of the PPI. Corticosteroids with topical activity and low systemic bioavailability are the mainstay of treatment for patients with EoE. Response rates are high, but recurrence rates following withdrawal of the medication are high as well. Currently, in Canada, these drugs are not commercially formulated for EoE and are used off-label; hence, they must be compounded or administered via a different route than that approved. Patients, physicians, and pharmacists are left to adapt these corticosteroid formulations to EoE patients, which can be confusing and cumbersome, leading to reduced patient compliance. Hence, other formulations of topical corticosteroids are needed to improve convenience and compliance. Esophageal dilation is effective; however, it can lead to very significant complications, ranging from severe transient pain to life-threatening perforation. Some patients with long-standing disease with strictures and fibrosis may not respond to topical corticosteroids. However, it is unclear what is the best treatment option for these patients, and there is no good indicator of which patients will progress to strictures and fibrosis.
Patients who have the greatest unmet need for an intervention such as budesonide orodispersible tablets for the maintenance of clinicopathologic remission are those with complications (such as dysphagia, impaction, and stricture), those who did not respond to PPIs, those with persistent symptoms or inflammation, and those who responded to the initial treatment with budesonide orodispersible tablets but experienced symptom recurrence after discontinuing budesonide orodispersible tablets.
Place in Therapy
Currently, topical corticosteroids (which are often thickened with something like Splenda to be viscous enough to adhere to the esophagus) are used off-label (without a Health Canada indication) for the treatment of EoE in adults. However, the topical corticosteroids currently used are cumbersome. Also, the compliance rate in patients receiving topical corticosteroids is low. The drug under review, budesonide orodispersible tablets, will fulfill the same role as the compounded topical corticosteroids. It would mainly be used in patients who fail to achieve symptomatic or histologic response after a trial of a PPI; thus, introducing budesonide orodispersible tablets would not, by itself, represent a large treatment paradigm shift.
Patient Population
The clinical experts indicated that patients who would be best suited for maintenance treatment with budesonide orodispersible tablets are (1) those who responded to the initial treatment with budesonide orodispersible tablets after having failed to respond to PPI therapy and whose symptoms recur more than once per year or who have a history of severe disease as manifested by food impactions or significant fibrosis; and (2) patients with severe endoscopic disease, who are intolerant to or noncompliant with fluticasone or other formulation of budesonide. Patients who respond to PPI treatment or other topical steroids (such as fluticasone propionate or budesonide slurry) are the least suitable for maintenance treatment with budesonide orodispersible tablets.
Assessing Response to Treatment
In practice, clinicians assess patients symptomatically. No scales are currently used in clinical practice to assess symptomatic response. Meaningful responses to treatment include the complete resolution of symptoms of dysphagia and food impaction. Other important assessments include an overall improvement in patient symptoms, allowing them to consume solid food of all consistencies; reduced hospitalization; lack of need for dilation; absence of strictures; lack of need for endoscopy; and a decrease in the frequency and severity of dysphagia.
The clinical experts indicated that treatment response should be assessed 3 months after initiating maintenance therapy, then every 6 months to 1 year thereafter.
Discontinuing Treatment
The clinical experts indicated that patients who relapse while receiving 0.5 mg budesonide twice daily for maintenance of remission would have their dosage increased to 1 mg budesonide twice daily (for re-induction of remission). After achieving remission on the 1 mg budesonide twice daily dosage, patients would be switched back to 0.5 mg budesonide twice daily for maintenance of remission. Patients who relapse again while on the 0.5 mg budesonide twice daily would have their dosage increased to 1 mg budesonide twice daily for re-induction of remission. After achieving remission on the 1 mg budesonide twice daily dosage, patients would remain on the 1 mg budesonide twice daily for the maintenance of remission. Patients who relapse while receiving the 1 mg budesonide orodispersible tablets twice daily as maintenance of remission need to be assessed for compliance and other factors associated with recurrence, and some patients might need to discontinue budesonide orodispersible tablets and try another treatment approach. Patients would receive oral steroids after discontinuing budesonide orodispersible tablets, as well as endoscopic procedures and dilation of the esophagus, as needed.
Also, treatment discontinuation should be considered if there are unacceptable side effects (such as recurrent candidiasis, systemic side effects from topical corticosteroids, or hypersensitivity), or if patients are intolerant to the drug.
The clinical experts also indicated that, while maintenance therapy in general would imply continuous treatment, in clinical practice treatment might be titrated, intermittent, continuous, or stopped. The decision is affected by symptoms, complications, strictures, and persistent inflammation on endoscopy.
Prescribing Conditions
The clinical experts agreed that budesonide orodispersible tablets for the treatment of EoE should be prescribed by specialists in gastroenterology or allergy who have expertise in EoE. The expertise necessary to monitor response is available in outpatient clinics.
Clinician Group Input
No clinician group input was submitted for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug Program Implementation Questions | Clinical Expert Response |
|---|---|
Duration of maintenance therapy seems to be dependent on individual patient factors. What factors would you take into consideration to help determine an authorization period? | The clinical experts indicated that patients who discontinued budesonide orodispersible tablets after induction of remission and relapsed within 3 to 6 months of treatment discontinuation should be treated for at least 1 year using budesonide orodispersible tablets as maintenance therapy. After the first year of authorization, renewal should be individualized, with some patients stopping treatment with budesonide orodispersible tablets. If the symptoms recur, then they should restart therapy using budesonide orodispersible tablets. The clinical experts also indicated that the evidence from the BUL-2/EER trial is insufficient to provide criteria to decide which patients would be able to stop treatment after 48 weeks without risk of a relapse. However, from clinical experience, the clinical experts think that patients with a history of severe disease, as manifested by food impactions or significant fibrosis, need to stay on 0.5 mg budesonide orodispersible tablets twice daily for a long period. |
In what context, based on patient factors, would you envisage an off-label dosage regimen (e.g., increasing the dose to 1 mg budesonide twice daily) in the adult population for the maintenance of remission? | The clinical experts indicated that disease severity before remission would guide dosage decisions and that patients with more severe disease tend to need more aggressive therapy. However, the clinical experts indicated that they would try first to maintain the remission using the budesonide 0.5 mg twice daily dosage; if the patient relapsed, then re-induction of remission using the budesonide 1 mg twice daily dosage would be tried. After achieving remission again using the 1 mg budesonide twice daily dosage, patients would be switched back to the 0.5 mg budesonide twice daily for maintenance of remission. Patients who relapsed again while on the 0.5 mg budesonide twice daily would have their dosage increased to 1 mg budesonide twice daily for re-induction of remission. After achieving remission on the 1 mg budesonide twice daily dosage, patients would remain on the 1 mg budesonide twice daily dosage for the maintenance of remission. |
Which subgroup of PPI-naive patients with EoE benefit from maintenance treatment with budesonide orodispersible tablets? | The clinical experts indicated that PPI-naive patients would probably respond in a similar manner as patients who had failed to respond to PPI treatment. However, they also indicated that PPI will always be the first line of treatment unless a patient who is PPI-naive presents with food impaction and severe disease, in which case the treating physician would initiate treatment with budesonide orodispersible tablets. |
Would you be able to comment on whether budesonide orodispersible tablets can be used off-label in the pediatric population? | The clinical experts indicated that budesonide orodispersible tablets might be used off-label in children; however, there are some concerns about safety and what dosage to prescribe. They also mentioned that children are already prescribed off-label fluticasone propionate and budesonide slurry, and it would be much easier to teach children to use budesonide orodispersible tablets than how to swallow fluticasone propionate and budesonide slurries. In addition, the taste of fluticasone propionate and budesonide slurry is problematic in terms of adherence in this patient population. |
How would you follow up with these patients with regard to time intervals and tests? Safety and efficacy? Other? | The clinical experts indicated that treatment response should be assessed 3 months after initiating maintenance therapy, then every 6 months to 1 year thereafter. The clinical experts indicated that they would not provide endoscopy and histology for maintenance therapy to determine effectiveness during scheduled follow-up appointments, given that they try to limit the use of endoscopy to once every 1 to 2 years. |
EoE = eosinophilic esophagitis; PPI = proton pump inhibitor.
Clinical Evidence
The clinical evidence included in the review of budesonide is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor; however, no indirect evidence was submitted by the sponsor, nor was any indirect evidence that met the selection criteria specified in the review identified from the literature. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objective
To perform a systematic review of the beneficial and harmful effects of 0.5 mg budesonide orodispersible tablets for the maintenance of clinicopathologic remission in adults with EoE.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented below was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adults with clinicopathologic remission of EoE Subgroups:
|
Intervention | 1 mg budesonide orally, as 1 of the 0.5 mg tablet in the morning and 1 of the 0.5 mg tablet in the evening |
Comparators | PPI Topical budesonide Topical fluticasone Systemic steroids Montelukast Food elimination diets Placebo |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; EoE = eosinophilic esophagitis; PGA = Physician’s Global Assessment; PPI = proton pump inhibitor; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse events.
The literature search was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist (https://www.cadth.ca/resources/finding-evidence/press).23
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid and Embase (1974‒) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Jorveza (budesonide) and eosinophilic esophagitis. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, and Health Canada’s Clinical Trials Database.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on November 30, 2020. Regular alerts updated the search until the meeting of the CADTH CDEC on April 21, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist (https://www.cadth.ca/grey-matters).24 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings from the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Included Studies
Detail | Study BUL-2/EER |
|---|---|
Designs and populations | |
Study design | Phase III, DB, multi-centre, placebo-controlled RCT |
Locations | Belgium, Germany, Spain, Switzerland, the Netherlands, and the UK |
Patient enrolment dates | 29 January 2016 |
Randomized (N) | 204 |
Inclusion criteria |
|
Exclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 0.5 mg budesonide orodispersible tablet twice daily 1 mg budesonide orodispersible tablet twice daily |
Comparator(s) | Placebo orodispersible tablet twice daily |
Duration | |
Phase | |
Open-label induction phase | 6 weeks |
DB treatment | 48 weeks |
Open-label extension | Up to 96 weeks |
Follow-up | 4 weeks |
Outcomes | |
Primary end points | Percentage of patients who had not had a treatment failure after 48 weeks of treatment. Treatment failure after 48 weeks of treatment was “yes,” if at least 1 of the following criteria was met at any time during the DB treatment phase:
|
Secondary and exploratory end points | Secondary end points:
Exploratory end points:
|
Notes | |
Publications | Straumann et al. (2020)25 |
DB = double-blind; EEsAI = Eosinophilic Esophagitis Activity Index; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; EoE = eosinophilic esophagitis; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; eos = eosinophil; GERD = gastroesophageal reflux disease; HPF = high-power field; modSHS = modified Short Health Scale; NRS = numerical rating scale; OLI = open-label induction; PatGA = Patient’s Global Assessment; PGA = Physician’s Global Assessment; PPI = proton pump inhibitor; PPI-REE = PPI-responsive esophageal eosinophilia; RCT = randomized controlled trial.
aclinicopathologic response to PPIs was defined as having original symptoms of esophageal dysfunction, with marked improvement of symptoms and peak eosinophils of fewer than 15 per HPF after 4 to 8 weeks’ treatment with PPIs. The PPI dosage used for a minimum of 4 weeks should have been at least the standard dosage according to the authorized summary of product characteristics of the respective PPI (e.g., omeprazole at 20 mg/day, pantoprazole at 40 mg/day, esomeprazole at 40 mg/day, lansoprazole at 30 mg/day, or rabeprazole at 20 mg/day).
Note: 3 additional reports were included — Drug Reimbursement Review,26 European public assessment report,27 and the Clinical Study Report of the BUL-2/EEA trial.10
Source: Straumann et al. (2020)25 and the Clinical Study Report of the BUL-2/EEA trial.10
Description of Studies
One trial (BUL-2/EER) met the inclusion criteria. The BUL-2/EER trial (N = 204) was a double-blind (DB), randomized, placebo-controlled, phase III study that compared the efficacy and tolerability of a 48-week treatment with 2 different doses of budesonide effervescent tablets with placebo for maintenance of clinicopathologic remission in adult patients with EoE. The BUL-2/EER trial was conducted in 29 centres in 6 countries (Belgium, Germany, the Netherlands, Spain, Switzerland, and the UK).
Patients who achieved clinicopathologic remission during the DB or open-label induction (OLI) phases of the induction trial BUL-1/EEA were eligible to enrol in the BUL-2/EER trial. In addition, a 6-week OLI-treatment arm with budesonide 1 mg orodispersible tablet twice daily was opened in the BUL-2/EER study for sites participating in the BUL-1/EEA study after completion of enrolment of BUL-1/EEA. This OLI-treatment arm was also available at sites that did not participate in the BUL-1/EEA trial. To ensure that the patient population enrolled in the OLI phase of the BUL-2/EER trial remained comparable throughout BUL-2/EER, patients enrolled in the OLI phase of the BUL-2/EER trial were recruited according to the major inclusion and exclusion criteria of study BUL-1/EEA. Patients in clinicopathologic remission at the end-of-treatment (EOT) visit of either the OLI phase of BUL-2/EER or the induction trial BUL-1/EEA were offered to continue in the 48-week DB phase of the BUL-2/EER trial.
At start of the DB treatment phase of the BUL-2/EER trial, patients were assigned to 1 of the 3 treatment groups via a central randomization procedure using a 1:1:1 allocation ratio to receive a 48-week, DB treatment with either budesonide 0.5 mg orodispersible tablet, budesonide 1 mg, or placebo orodispersible tablet twice daily. The budesonide 0.5 mg, budesonide 1 mg, and placebo tablets were identical in appearance, shape, and taste, which assured treatment blinding. Randomized treatment assignment was not stratified.
Patients in the BUL-2/EER trial with a clinical or histologic relapse or a food impaction that needed endoscopic intervention during the DB treatment phase were offered an open-label re-induction (OLRI) of remission or response treatment with budesonide 1 mg for up to 6 weeks. Patients completing the DB treatment phase without treatment failure and patients with clinical improvement in the OLRI phase could receive open-label extension (OLE) treatment with budesonide 0.5 mg twice daily (or with an escalated dose of 2 times budesonide 0.5 mg twice daily) for up to 48 weeks, with a further optional continuation of the OLE phase up to 96 weeks of total OLE treatment, or until the date budesonide orodispersible tablets had received marketing authorization and were available in the market (whichever came first).
Patients were followed up to 4 weeks after their last treatment visit in the DB or open-label (OLI, OLRI, or OLE) phase, which primarily served to check safety parameters such as outcomes of adverse events (AEs) and follow-up of laboratory values. Patients without clinical symptoms at EOT/withdrawal DB, OLRI, or OLE visit remained untreated during this follow-up period. Patients who had clinical symptoms of EoE at EOT/withdrawal OLI, DB, OLRI, or OLE visit could be treated symptomatically during the follow-up period in accordance with treatment, as decided by the investigator.
Of note, the BUL-1/EEA trial was reviewed by CADTH and presented to CDEC in the drug reimbursement review of budesonide for the induction of clinicopathologic remission in adults with EoE.20 Hence, the BUL-1/EEA trial was not summarized in this report.
Data will not be presented for the budesonide 1 mg twice daily dosage because it is not aligned with the Health Canada–approved dosage.
Figure 2 is a schematic design of the BUL-2/EER trial.
Figure 2: Study Design for BUL-2/EER
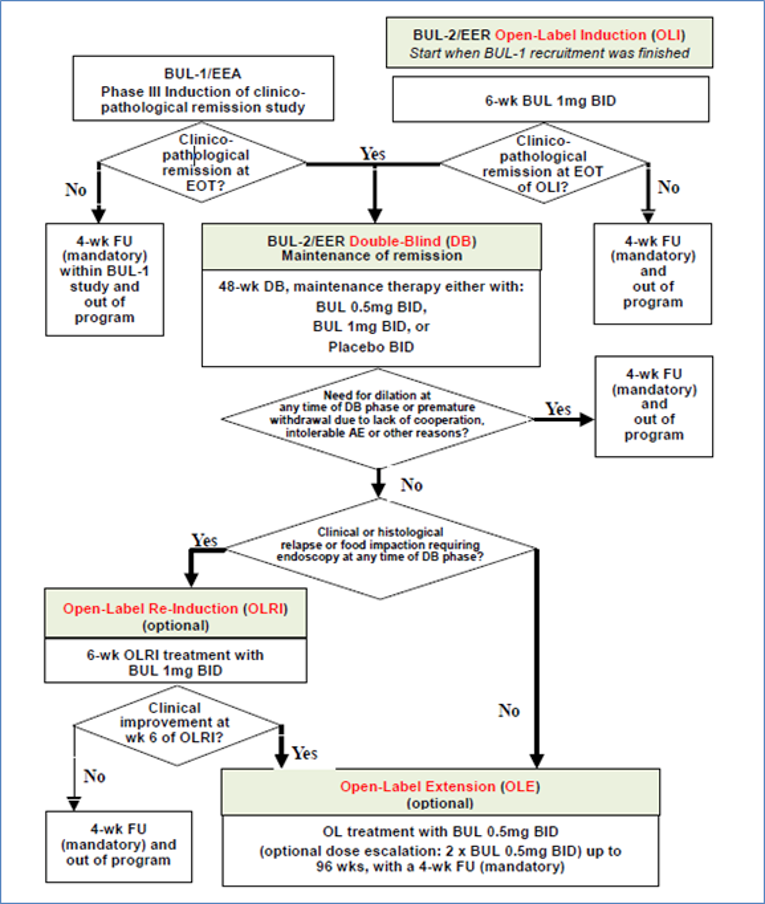
BID = twice a day; BUL = budesonide orodispersible tablet; DB = double-blind; EOT = end of treatment; FU = follow-up; OLE = open-label extension; OLI = open-label induction; OLRI = open-label re-induction.
Note: Withdrawal from the DB treatment phase due to lack of efficacy (without fulfilling either clinical or histologic relapse criteria or food impaction requiring endoscopy) was not a criterion to decide which study phase followed after the DB phase. The criteria displayed in the diagram were used to make the decision.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Populations
Inclusion and Exclusion Criteria
Patients enrolled in the BUL-2/EER trial were adults (18 to 75 years of age) with a confirmed clinicopathologic diagnosis of EoE and clinicopathologic remission, defined as fulfilling both histologic remission and resolution of symptoms criteria at EOT visit of either the OLI phase of BUL-2/EER or of the induction trial BUL-1/EEA. Histologic remission was defined as peak eosinophil count less than 16 eosinophils (eos)/mm2 HPF, and resolution of symptoms was defined as a severity of 2 points or less on a 0- to 10-point numerical rating scale (NRS) for dysphagia and a severity of 2 points or less on 0- to 10-point NRS for pain during swallowing on each day in the last week of induction treatment.
Patients were excluded from the BUL-2/EER trial if they:
were pregnant or breastfeeding
had PPI-responsive esophageal eosinophilia (PPI-REE)
were intolerant or hypersensitive to the study drug
had a history of abnormal pH monitoring of the distal esophagus
had clinical evidence of any causes other than EoE for eosinophilia of the esophagus
had signs or symptoms of gastroesophageal reflux disease, achalasia, scleroderma, abnormal renal or hepatic function, AIDS, active tuberculosis, a relevant gastrointestinal disease, or relevant systemic disease without proper medical monitoring (cardiovascular disease, diabetes mellitus, osteoporosis, active peptic ulcer disease, glaucoma, cataract, or infection)
had used topical glucocorticoids within 2 weeks of screening
had used systemic glucocorticoids, biologics, or immunosuppressants within 4 weeks of screening
had had esophageal surgery at any time
had undergone dietary restriction in the preceding 4 weeks
had experienced esophageal dilation or upper gastrointestinal bleeding in the preceding 8 weeks
had had cancer in the preceding 5 years.
Baseline Characteristics
As shown in Table 7, the patients’ baseline characteristics appeared to be roughly balanced, despite certain large variations between the treatment groups. Notably, the disease duration since diagnosis and since first symptoms were shorter in the placebo group than in the budesonide 0.5 mg group. The mean time since an established EoE diagnosis was 4.3 years and 3.3 years for the budesonide 0.5 mg and placebo groups, respectively, with a mean time since symptom onset of 12.6 years and 9.6 years, respectively. In addition, fewer patients in the placebo group (5.9%) had a previous esophageal dilation compared to the budesonide 0.5 mg group (19.1%). The average age of the participants was 36 years, and the majority were men (84% and 81% for the budesonide 0.5 mg and placebo study arms, respectively), which is representative of the EoE adult patient population. All baseline parameters of disease activity, including histologic results and endoscopic as well as patients’ and investigators’ assessments, showed similarly low values for disease activity in all treatment groups. Of note, all patients in all treatment groups were in deep histologic remission, defined as 0 eos/mm2 HPF, and two-thirds of the patients in all treatment groups were in deep histologic and endoscopic remission.
Table 7: Summary of Baseline Characteristics in Study BUL-2/EER, Full Analysis Set
Characteristic | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Sex, n (%) | ||
Male | 57 (83.8) | 55 (80.9) |
Female | 11 (16.2) | 13 (19.1) |
Age, years | ||
Mean (SD) | 36 (10.9) | 36 (9.9) |
Range | 19 to 69 | 18 to 64 |
Race, n (%) | ||
White | 68 (100) | 68 (100) |
Smoking habits, n (%) | ||
Current | 8 (11.8) | 2 (2.9) |
Former | 7 (10.3) | 8 (11.8) |
Never | 53 (77.9) | 58 (85.3) |
BMI, kg/m2 | ||
Mean (SD) | 24.1 (3.02) | 24.4 (4.12) |
Range | 18.0 to 30.4 | 17.6 to 41.5 |
Time since EoE diagnosis, years | ||
Mean (SD) | 4.3 (3.47) | 3.3 (2.85) |
Median (range) | 4.1 (0.2 to 15.7) | 2.1 (0.2 to 11.7) |
Time since first EoE symptoms, years | ||
Mean (SD) | 12.6 (8.50) | 9.6 (8.22) |
Median (range) | 10.4 (0.3 to 35.7) | 7.0 (1.0 to 37.6) |
Previous esophageal dilations, n (%) | 13 (19.1) | 4 (5.9) |
Previous esophageal surgeries, n (%) | 0 | 0 |
Conducted PPI trial, n (%) | 68 (100) | 68 (100) |
Clinical response to PPI, n (%) | 8 (11.8) | 5 (7.4) |
Pathological response to PPI, n (%) | 0 | 0 |
Previous EoE treatment, n (%)a | ||
PPI | 46 (67.6) | 46 (67.6) |
Topical budesonide | 11 (16.2) | 14 (20.6) |
Topical fluticasone | 29 (42.6) | 16 (23.5) |
Systemic steroids | 1 (1.5) | 0 (0) |
Other | 3 (4.4) | 1 (1.5) |
Endoscopic dilation | 13 (19.1) | 4 (5.9) |
Elemental diet | 0 (0) | 2 (2.9) |
Directed elimination diet (based on allergy test) | 3 (4.4) | 6 (8.8) |
Nondirected elimination diet | 28 (41.2) | 24 (35.3) |
History of allergic disease, n (%) | 54 (79.4) | 50 (73.5) |
Peak eos/mm2 HPF, mean (SD) | 0 (1.4) | 1 (3.6) |
Blood eos/mm3 (baseline), mean (SD) | 205 (141.2) | 170 (156.8) |
Modified EEsAI endoscopic instrument score | ||
Total (0 to 9), mean (SD) | 1 (1.1) | 1(1.0) |
Inflammatory signs (0 to 4), mean (SD) | 0 (0.6) | 0 (0.6) |
Fibrotic signs (0 to 4), mean (SD) | 1 (0.7) | 0 (0.6) |
All grade 0, n (%) | 34 (50.0) | 35 (51.5) |
Endoscopist’s overall assessment of EoE activity: No endoscopic findings, n (%) | 50 (73.5) | 43 (63.2) |
Dysphagia (NRS of 0 to 10 points), last 7 days, mean (SD) | 1 (0.9) | 1 (0.8) |
Pain during swallowing (NRS of 0 to 10 points), last 7 days, mean (SD) | 1 (0.9) | 0 (0.8) |
Patient’s Global Assessment of EoE activity (NRS of 0 to 10 points), mean (SD) | 1 (0.8) | 1 (0.9) |
Physician’s Global Assessment of EoE activity (NRS of 0 to 10 points), mean (SD) | 1 (0.8) | 1 (0.9) |
Weekly EEsAI-PRO, mean (SD) | 16 (14.1) | 18 (16.6) |
EEsAI-PRO ≤ 20 | 43 (63.2) | 38 (55.9) |
modSHS (VAS 0 to 100), mean (SD)b | ||
Symptom burden | 11 (9.6) | 11 (12.1) |
Social function | 12 (13.7) | 13 (17.3) |
Disease-related worry | 25 (20.9) | 27 (24.0) |
General well-being | 12 (13.0) | 15 (14.3) |
EoE-QoL-A eating/diet impact), mean (SD)c | 3.3 (0.62) | 3.2 (0.84) |
Histologic remission, n (%)d | 68 (100) | 68 (100) |
Deep histologic remission, n (%)e | 65 (95.6) | 64 (94.1) |
Deep endoscopic remission, n (%)f | 45 (66.2) | 47 (69.1) |
Deep endoscopic and histologic remission, n (%)e,f | 45 (66.2) | 47 (69.1) |
Deep clinical remission, n (%)g | 17 (25.0) | 19 (27.9) |
Deep disease remission, n (%)e,f,g | 9 (13.2) | 15 (22.1) |
Previous and current episodes of EoE | ||
EoE: Duration of last remission phase, months | ||
N | 30 | 29 |
Median (range) | 9 (0 to 72) | 5 (0 to 81) |
EoE: Time since end of last remission phase, months | ||
N | 30 | 31 |
Median (range) | 6 (2 to 55) | 4 (2 to 45) |
EoE: Duration of last acute episode, months | ||
N | 55 | 61 |
Median (range) | 5 (2 to 363) | 8 (2 to 449) |
EoE: Time since end of last acute episode, months | ||
N | 68 | 67 |
Median (range) | 0 (0 to 6) | 0 (0 to 6) |
EoE: Time since start of current remission phase, months | ||
N | 68 | 67 |
Median (range) | 0 (0 to 4) | 0 (0 to 2) |
b.i.d. = twice a day; BMI = body mass index; EEsAI = Eosinophilic Esophagitis Activity Index; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; EoE = eosinophilic esophagitis; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; eos = eosinophil; HPF = high-power field; modSHS = modified Short Health Scale; NRS = numerical rating scale; PPI = proton pump inhibitor; SD = standard deviation; VAS = visual analogue scale.
aTreatment options used in patient’s history before enrolment into the study program of BUL-1/EEA and BUL-2/EER.
bRange of each score: 0 to 100. Lower numbers indicate higher QoL.
cWeighted average of 10 items with each range from 0 to 4. Higher scores indicate better QoL.
dPeak eos less than 48 /mm2 HPF (corresponding to less than 15 eos/HPF).
ePeak eos of 0/mm2 HPF (corresponding to 0 eos/HPF).
fFixed rings = Grade 0: none or Grade 1: mild, exudates = Grade 0: none, furrows = Grade 0: absent, and edema = Grade 0: absent
gBoth NRS (24-hour recall period) scores for dysphagia and pain during swallowing are 0 on each day in the last week before baseline
Source: Clinical Study Report of the BUL-2/EEA trial.10
Interventions
In the BUL-2/EER trial, the patients received either budesonide 0.5 mg, budesonide 1 mg, or placebo orodispersible tablet twice daily. The placebo orodispersible tablets were indistinguishable in appearance, size, and taste from the budesonide orodispersible tablets. One orodispersible tablet was taken in the morning and another in the evening after the meal. The orodispersible tablet was placed on the tongue, which allowed it to dissolve rapidly and to be swallowed with saliva little by little. Patients were advised not to drink or eat for 30 minutes after study drug administration.
The use of systemic or topical glucocorticoids, biologics, or immunosuppressants as concomitant medication was not permitted during treatment phase. In addition, the initiation of dietary restrictions was also not permitted within 4 weeks before the screening visit or during treatment. Existing, permitted concomitant treatments were not changed during the DB treatment phase and the OLI-treatment phase of the BUL-2/EER trial.
Patients were prematurely withdrawn from the trial due to lack of efficacy, which was defined as no change or a deterioration in the weekly Patient’s Global Assessment (PatGA) concerning the severity of EoE symptoms after at least 4 weeks of treatment compared to start of treatment phase, a clinical relapse, a histologic relapse, a food impaction at any time that needed endoscopic intervention, or an endoscopic dilation. Patients were also prematurely withdrawn from the trial if they experienced intolerable AEs.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized below. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 5.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | BUL-2/EER |
|---|---|
Percentage of patients who had not had a treatment failure after 48 weeks of treatment. Treatment failure after 48 weeks of treatment was “yes,” if at least 1 of the following criteria was met at any time during the DB treatment phase:
| Primary efficacy end point |
Percentage of patients with histologic relapse, defined as peak of ≥ 48 eos/mm2 HPF (corresponding to ≥ 15 eos/HPF) at end of treatment of the DB phase | Key secondary end pointa |
Change in the peak eos/mm2 HPF from baseline of the DB phase to end of treatment of the DB phase | Key secondary end pointa |
Percentage of patients with a clinical relapse, having experienced a food impaction that needed endoscopic intervention or having needed an endoscopic dilation during the DB treatment phase | Key secondary end pointa |
Percentage of patients with a total weekly EEsAI-PRO score of ≤ 20 at end of treatment of the DB phase | Key secondary end pointa |
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF), at end of treatment of the DB phase | Key secondary end pointa |
Percentage of patients with histologic remission, defined as a peak of < 16 eos/mm2 HPF, at end of treatment of the DB phase | Exploratory end point |
Percentage of patients with deep histologic remission, defined as a peak of 0 eos/mm2 HPF, at end of treatment of the DB phase | Exploratory end point |
Percentage of patients maintaining deep histologic remission, defined as a peak of 0 eos/mm2 HPF, from baseline of the DB phase to end of treatment of the DB phase | Exploratory end point |
Change from baseline of the DB phase in total modified EEsAI endoscopic instrument score | Exploratory end point |
Change from baseline of the DB phase in the inflammatory signs of the modified EEsAI endoscopic instrument score | Exploratory end point |
Change from baseline of the DB phase in the fibrotic signs of the modified EEsAI endoscopic instrument score | Exploratory end point |
Percentage of patients with all grades 0 in modified EEsAI endoscopic instrument score at end of treatment of the DB phase | Exploratory end point |
Percentage of patients with no endoscopic findings (endoscopist’s overall assessment of EoE activity) at end of treatment of the DB phase | Exploratory end point |
Change from baseline of the DB phase in the PGA of EoE activity (0 to 10 NRS) | Exploratory end point |
Percentage of patients with increase of ≥ 3 points from baseline of the DB phase in the PatGA concerning the severity of EoE symptoms (0 to 10 NRS) at end of treatment of the DB phase | Exploratory end point |
Change from baseline of the DB phase in the PatGA concerning the severity of EoE symptoms (0 to 10 NRS) | Exploratory end point |
Percentage of patients with a clinical relapse during the DB phase | Exploratory end point |
Change from baseline of the DB phase in the EoE-QoL-A questionnaire | Exploratory end point |
Change from baseline of the DB phase in modSHS | Exploratory end point |
Time to treatment failure | Exploratory end point |
Time to first occurrence of clinical relapse | Exploratory end point |
DB = double-blind; EEsAI = Eosinophilic Esophagitis Activity Index; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; EoE = eosinophilic esophagitis; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; eos = eosinophil; HPF = high-power field; modSHS = modified Short Health Scale; NRS = numerical rating scale; PatGA = Patient’s Global Assessment; PGA = Physician’s Global Assessment.
aEfficacy significance testing continued in hierarchical fashion for the 5 key secondary end points until the first of these comparisons of budesonide 0.5 mg twice daily versus placebo or budesonide 0.5 mg twice daily versus placebo showed a 1-sided P value > 0.0125. Once a nonsignificant P value occurred, all subsequent significance tests were considered exploratory in nature. Both streams were tested independently from each other, such that key secondary efficacy variables were tested in a confirmatory fashion for each active treatment group only if the primary efficacy variable had shown significance for that treatment group. Conversely, nonsignificance in a key secondary efficacy variable for 1 of the active treatment groups did not imply stopping the hierarchical testing in the other treatment group. The sequence used was in the same order as the key secondary outcomes presented in Table 8.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Treatment failure: The primary outcome in the BUL-2/EER trial was percentage of patients who had not had a treatment failure after 48 weeks of treatment. Where patients were considered to be experiencing treatment failure after 48 weeks of treatment if at least 1 of the following criteria was met at any time during the DB treatment phase:
clinical relapse
histologic relapse, i.e., a peak of ≥ 48 eos/mm2 HPF (corresponding to ≥ 15 eos/HPF), at EOT in the DB phase
a food impaction that needed endoscopic intervention
need for an endoscopic dilation
premature withdrawal for any reason.
Clinical Relapse
In the BUL-2/EER study, clinical relapse was defined as dysphagia or pain during swallowing in the past 7 days (7-day recall period) of a severity of at least 4 points on a 0 to 10 NRS for dysphagia or pain during swallowing, respectively, confirmed by a severity of least 4 points on at least 1 day during the subsequent week on the respective 0 to 10 NRS for dysphagia or pain during swallowing (24-hours recall period).10 Evidence regarding the validity, reliability, and minimal important difference (MID) for clinical relapse for patients with EoE was not found in the literature.
The Dysphagia NRS is a 10-point rating scale in which patients assess the severity of dysphagia symptoms experienced in the past 24 hours or 7 days. The scale ranges from 0 to 10 (0 represents no trouble swallowing, 10 represents the most severe trouble swallowing). The Dysphagia NRS captures dysphagia symptoms associated with EoE only and not symptoms associated with cold, e.g., sore throat. Patients in the trial received the scale in the form of a diary, and daily ratings were used to calculate a weekly sum.10 No studies validating the Dysphagia NRS in patients with EoE were identified from the literature; neither was an MID found.
The Pain During Swallowing NRS is a 10-point rating scale in which patients assess the severity of pain during swallowing experienced in the past 24 hours or 7 days. The scale ranges from 0 to 10 (0 represents no pain during swallowing, 10 represents the most severe pain during swallowing).10 Evidence regarding the validity, reliability, and MID of the Pain During Swallowing NRS for patients with EoE was not found in the literature.
Patients received a diary for daily documentation of the Dysphagia NRS and Pain During Swallowing NRS. In case of a suspected clinical relapse during the DB treatment phase, the entries on the patient diary cards on each day between the DB visit at which clinical relapse was suspected and the subsequent DB extra visit were used. One patient diary card contains the data for 7 days. If the patient’s documentation was incomplete, the investigator asked the patient to give the information on the missing parameters retrospectively. If the patient could not remember, the corresponding fields remained empty. If a patient showed signs of a clinical relapse at 1 of the visits, an extra visit was scheduled, and the patient had to complete a daily diary for the next 7 days until this DB extra visit.
Percentage of patients with a clinical relapse, with a food impaction that needed endoscopic intervention, or needing an endoscopic dilation during the DB treatment phase was a key secondary end point in the BUL-2/EER trial. Percentage of patients with a clinical relapse during the DB phase and time to first occurrence of clinical relapse were exploratory end points in the BUL-2/EER trial.
Histologic Relapse
The peak number of eosinophils per mm2 HPF was derived from the evaluation of 6 HPFs derived from 6 esophageal biopsies. Two biopsy specimens each were taken from the proximal, mid-, and distal part of the esophagus for the assessment of 6 HPFs. In each biopsy specimen, the number of eosinophils per HPF were counted and transformed to the number of eosinophils per mm2 HPF. The highest number of eosinophils per mm2 HPF across the 6 biopsies was used to decide on histologic relapse. A patient was considered to have experienced a histologic relapse if the highest number of eosinophils per mm2 HPF across the 6 biopsies was ≥ 48. At least 1 evaluable biopsy had to be available for this purpose. Otherwise, histologic relapse was considered not evaluable.
There is no clear threshold of eosinophils per HPF universally established as an end point in EoE, but other literature reports have used this cut-off, and the diagnosis of EoE is commonly based on more than 15 eosinophils per HPF.28 The number of eosinophils per HPF is often less than the number of eosinophils per mm2 HPF. The FDA draft guidance for industry for developing drugs for the treatment of EoE states that the assessment of histologic response should be documented, defined as “peak esophageal eosinophil per HPF count of less than or equal to 6 across all available esophageal levels at the final treatment period evaluation.”29
Percentage of patients with histologic relapse, defined as peak of 48 eos/mm2 HPF or greater (corresponding to 15 eos/HPF or greater) at EOT of the DB phase, and change in the peak eos/mm2 HPF from baseline of the DB phase to EOT of the DB phase, were key secondary end points in the BUL-2/EER trial.
Percentage of patients with histologic remission, defined as a peak of less than 16 eos/mm2 HPF at EOT of the DB phase, percentage of patients with deep histologic remission, defined as a peak of 0 eos/mm2 HPF at EOT of the DB phase, and percentage of patients maintaining deep histologic remission, defined as a peak of 0 eos/mm2 HPF from baseline of the DB phase to EOT of the DB phase, were exploratory end points in the BUL-2/EER trial.
Food Impaction and Endoscopic Dilation
Food impaction that needed endoscopic intervention during the DB treatment phase was calculated as “yes” at the EOT/withdrawal visit of the DB phase if the question on occurrence of food impaction and need for endoscopic intervention since last visit was answered “yes” at any visit during the DB treatment phase.
Health-Related Quality of Life
In the BUL-2/EER trial, HRQoL was evaluated using the modified Short Health Scale (modSHS) and the Adult Eosinophilic Esophagitis Quality of Life Questionnaire (EoE-QoL-A). Change from baseline of the DB phase in the EoE-QoL-A questionnaire and change from baseline of the DB phase in modSHS were exploratory end points in the BUL-2/EER trial.
Adult Eosinophilic Esophagitis Quality of Life Questionnaire (EoE-QoL-A)
The EoE-QoL-A is a self-reported questionnaire. There is an original version and a refined version, which was used in the BUL-2/EER trial. The refined 30-item questionnaire (a 24-item scale with a 6-question addendum for those on elimination diet therapies) is categorized according to the following 5 dimensions: impact of the disease on eating patterns and diet, social impact, emotional impact, disease anxiety, and swallowing anxiety. Patients provide responses based on their life over the past week that best describe their experiences with living with EoE. Each question has 5 answers, ranging from 4, which corresponds to “does not describe their experiences at all,” to 0, which corresponds to “extremely describes their experiences.” Based on the responses, an overall score and 5 subscale scores are generated. Higher scores indicate better quality of life. Notably, there is a standard version (24 items) and a standard plus dietary restrictions version (30 items) of the EoE-QoL-A questionnaire. The latter is used for patients on elimination diet therapy. Since the dietary restrictions section is not applicable to all patients, a weighted average is calculated for the overall score and the 5 subscales by adding the value of the response for each item answered, then dividing by the total number of questions answered.10 Validity, reliability, and responsiveness were shown for the original version;30 however, only construct validity was assessed for the shorter version.31 An MID for the total score of the 5 domains was not reported by the authors or identified from the literature.
Modified Short Health Scale (modSHS)
The modSHS is a 4-item questionnaire, representing each of 4 health dimensions: symptom burden, social function, disease-related worry, and general well-being. The patient answers a total of 4 questions (health dimensions) that assess the effects of esophageal disease on the patient’s QoL.10
Patients respond to each of the following questions representing the 4 health dimensions, scored on a scale of 0 to 100: How severe are the symptoms from esophageal disease? (0 represents no symptoms, 100 represents very severe symptoms), Do the symptoms interfere with activities in daily life due to esophageal problems? (0 represents not at all, 100 represents that they interfere to a very high degree), Does the patient worry about esophageal disease? (0 represents no worry, 100 represents constant worry), and What is the patient’s general feeling of well-being? (0 represents very good, 100 represents dreadful)?
While the Short Health Scale has demonstrated discrimination validity, reliability (including internal consistency and test-retest reliability), and responsiveness in gastrointestinal conditions such as ulcerative colitis32 and Crohn disease,33 a psychometric analysis of the modSHS in EoE was not found in the literature. Additionally, an MID was not identified for any of these conditions.
EoE Activity
In the BUL-2/EER trial, EoE activity was evaluated using the Eosinophilic Esophagitis Activity Index Patient Reported Outcome (EEsAI-PRO), Physician’s Global Assessment (PGA) of EoE activity, and PatGA concerning the severity of EoE symptoms. The percentage of patients with a total weekly EEsAI-PRO score of 20 or less at EOT of the DB phase was a key secondary end point in the BUL-2/EER trial, while change from baseline of the DB phase in the PGA of EoE activity, percentage of patients with increase of 3 points or more from baseline of the DB phase in the PatGA concerning the severity of EoE symptoms at EOT of the DB phase, and change from baseline of the DB phase in the PatGA concerning the severity of EoE symptoms were exploratory end points in the BUL-2/EER trial.
Eosinophilic Esophagitis Activity Index Patient Reported Outcome
The EEsAI-PRO score is used to assess EoE activity in adult patients over a 7-day recall period; it consists of the following 5 items: frequency of trouble swallowing, duration of dysphagia episodes, pain during swallowing, Visual Dysphagia Questions (VDQs), and behavioural change strategies. The scores of each item are added to provide an overall score out of 100, with disease severity rated as remission (0 to 20), mild (21 to 40), moderate (41 to 65), and severe (66 to 100).10 Construct and content validity has been demonstrated34; no information on reliability and responsiveness were found in the literature.
Physician’s Global Assessment of EoE Activity
In this scale, physicians are asked to provide an overall assessment of the patients’ EoE activity and severity, taking into consideration the symptoms, endoscopy, histology, and laboratory markers. The EoE activity is rated on a 10-point scale, ranging from 0 (inactive EoE) to 10 (most active EoE).10 Evidence of validity and reliability as well as MID were not found in the literature.
Patient’s Global Assessment Concerning the Severity of EoE Symptoms
The PatGA scale evaluates the severity of EoE symptoms from a patient’s perspective. Patients were asked to rate the severity of their EoE symptoms in the past 7 days on a scale that ranges from 0 to 10 (0 represents no symptoms, 10 represents most severe symptoms).10 Evidence of validity and reliability as well as MID were not found in the literature.
Patients in Deep Disease Remission
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF), at EOT of the DB phase was a key secondary end point in the BUL-2/EER trial.
Deep disease remission consists of 3 components: deep clinical, deep endoscopic, and histologic remission, which are based on the peak number of eosinophils per HPF. Deep disease remission is fulfilled if all 3 components are “Yes” (based on the peak number of eosinophils at time of assessment). If at least 1 is “No,” deep disease remission is also “No.” Otherwise, deep disease remission is not evaluable.10 No studies validating deep disease remission and its components were identified from the literature, and an MID was not found.
Deep Clinical Remission
Deep clinical remission is “Yes” if the NRS (7-day recall period) for dysphagia and pain during swallowing is 0 at the respective visit. If the NRS (7-day recall period) exceeds 0 for dysphagia or for pain during swallowing, deep clinical remission at the visit is not fulfilled (“No”). Otherwise, deep clinical remission is not evaluable.10
Deep Endoscopic Remission
Deep endoscopic remission is fulfilled (“Yes”) if the following modified EEsAI endoscopic instrument subscores meet the following criteria: fixed rings = Grade 0: none or Grade 1: mild, exudates = Grade 0: none, furrows = Grade 0: absent, edema = Grade 0: absent.
If at least 1 of these features exceeds this grading, deep endoscopic remission at the respective visit is not fulfilled (“No”). Otherwise, deep endoscopic remission at the respective visit is not evaluable.10
The fibrotic signs subscore of the modified EEsAI endoscopic instrument (range 0 to 4) consisted of the assessment of the following subscores: Fixed rings and stricture; whereas, the inflammatory signs subscore (range 0 to 4) consisted of the assessment of the following subscores: Exudates, Furrows, and Edema.
Interobserver agreement/reliability was demonstrated for the modified endoscopic scale.35 No studies validating the modified EEsAI endoscopic instrument score were identified in the literature search; neither was an MID found.
Histologic Remission
Histologic remission for deriving deep disease remission is defined by a peak eosinophil count of less than 15 eos/HPF. If the peak eosinophil count is 15 eos/HPF or more, histologic remission is “No” at the respective visit. Otherwise, histologic remission for deriving deep disease remission is not evaluable.10
Deep Endoscopic and Histologic Remission
Deep endoscopic and histologic remission is fulfilled (“Yes”) if both deep endoscopic remission and histologic remission are “Yes.” If at least 1 of these is “No,” deep endoscopic and histologic remission is also “No.” Otherwise, deep endoscopic and histologic remission is not evaluable.10
Safety
Treatment-emergent adverse events (TEAEs) were defined as any event with an onset after the first administration of the investigational products or, if pre-existing, worsening after the first administration of investigational products, and occurring within the period of treatment with the investigational products.
An SAE was defined as any untoward medical occurrence that at any dose results in death, is life-threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability and/or incapacity, or is a congenital anomaly or birth defect.
Statistical Analysis
Power Calculation
The determination of sample size assumed that the percentage of patients with failure would be 50% for the placebo group, 30% for the budesonide 0.5 mg twice daily group, and 25% for the budesonide 1 mg twice daily group. In total, 192 patients were needed to detect a difference of 20% to 25% in percentage of treatment failure, at 1-sided alpha level of 0.025 and a statistical power of more than 80%. This sample size was increased to account for 5% of randomized patients who did not receive at least 1 dose of treatment therapy. In total, 204 patients were randomized in a 1:1:1 randomization ratio into 1 of the treatment groups.
Primary Outcome
The primary outcome was the percentage of patients who had not had a treatment failure after 48 weeks of treatment. As shown in more detail in Table 9, treatment failure primarily consisted of both clinical and histologic failure as part of the key secondary outcomes and was analyzed using the normal approximation tests. To adjust for multiplicity in the comparison of the 2 budesonide treatment groups with the placebo group, a Bonferroni correction was made; calculations were performed for each pairwise comparison with placebo separately, at a 1-sided level of significance of 0.0125.
Handling of Missing Data
Patients who withdraw prematurely for any reason or with unknown outcome due to missing data were treated as treatment failures.
Subgroup Analysis
The primary efficacy outcome was analyzed descriptively with respect to the following subgroups:
Path to remission (i.e., BUL-1/EEA or BUL-2/EER OLI phase)
Localization of the inflammation at screening for either BUL-1/EEA or BUL-2/EER:
Proximal (yes/no), median (yes/no), and distal (yes/no) esophagus
1, 2, or 3 esophageal segments affected
An esophageal segment was defined as affected by inflammation if the peak number of eos/mm2 HPF is 16 or more.
Concomitant use of PPIs (yes/no) during the DB treatment phase
History of allergic diseases (yes/no)
Time interval since first symptoms of EoE at DB baseline (years): less than median (years) and median or more (years)
No subgroup analysis was conducted by history of relapses, strictures, food impaction, or induction treatment used.
Sensitivity Analysis
A sensitivity analysis was conducted using a logistic regression model for the treatment failure (yes or no) with treatment group (0.5 mg and 1 mg versus placebo) adjusted with baseline characteristics as covariates, defined as follows:
Peak number of eos/mm2 HPF at screening (screening visit for BUL-1/EEA or BUL-2/EER study)
Weekly sum of Dysphagia NRS at baseline of induction phase (first visit of BUL-1/EEA or of OLI of BUL-2/EER)
Weekly sum of Pain During Swallowing NRS at baseline of induction phase (first visit of BUL-1/EEA or of OLI of BUL-2/EER)
In case the actual number of treatment failures did not allow a joint model containing these variables, separate sensitivity analyses were performed by entering each covariate separately.
Secondary Outcomes
To control the family-wise type I error at 0.0125 level, the statical significance testing was performed in hierarchical fashion in a pre-specified order for the 5 key secondary outcomes (Table 9) until the first of these comparisons of budesonide 0.5 mg or 1 mg twice daily versus placebo showed a 1-sided P > 0.0125; then the testing was stopped. Once a nonsignificant P value occurred, all subsequent significance tests were considered exploratory in nature. Below is the hierarchy of the key secondary end points:
Percentage of patients with histologic relapse, defined as peak of 48 eos/mm2 HPF or more at EOT of the DB phase
Change in the peak eos/mm2 HPF from baseline of the DB phase to EOT of the DB phase
Percentage of patients with a clinical relapse, a food impaction that needed endoscopic intervention, or an endoscopic dilation during the DB treatment phase
Percentage of patients with a total weekly EEsAI-PRO score of 20 or less at EOT of the DB phase
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF), at EOT of the DB phase
Dichotomous outcomes (key secondary outcomes number 1, 3, 4, and 5) were analyzed using the normal approximation test. It is not reported whether the normal approximation assumption was tested. Dichotomous key secondary outcomes with a corresponding baseline measurement (number 1 and 4) were also analyzed using logistic regression, including the screening/baseline value(s) of the preceding induction phase in addition to treatment group. For the first key secondary end point, the peak number of eos/mm2 HPF at screening was used as covariate; for the fourth key secondary end point, the weekly EEsAI-PRO score at baseline of the preceding induction phase was used as covariate.
Change in the peak eos/mm2 HPF was analyzed using the Wilcoxon rank sum test.
Time-to-event outcomes (time to treatment failure, and time to first occurrence of clinical relapse) were described using Kaplan-Meier methods.
For both EoE-QoL-A overall scores as well as for both eating/diet impact subscores, the differences between absolute changes from DB baseline to 48 weeks were tested in an exploratory way (t-test, 2-sided, type I error of 0.05) for both budesonide groups versus placebo. In addition, 2-sided 95% confidence intervals were calculated for the group differences in means. For all 4 dimensions of the modSHS, the differences between absolute changes from DB baseline to 48 weeks were tested (t-test, 2-sided, type I error of 0.05) for both budesonide groups versus placebo.
In case of missing diary values (i.e., NRS for dysphagia and pain during swallowing, respectively) within a week before the respective visit for 1 or 2 days, the weekly sum of these values was calculated for valid data only and the sum was divided by the number of days with valid data and multiplied with 7. If data were available for less than 5 days (more than 2 days missing) the weekly-based variables were “not evaluable” for the respective week.
If at least 1 of the 5 items relevant for the calculation of the EEsAI-PRO score was missing, the EEsAI-PRO score was missing as well. In case the only missing item was “duration of trouble swallowing,” and item “frequency of trouble swallowing” was answered with “never,” the EEsAI-PRO score was evaluable and the score for the item “duration of trouble swallowing” was set to 0 for the EEsAI-PRO score calculation.
The absolute change from DB baseline to any DB visit was calculated as the value at the visit minus the DB baseline value. If either of the 2 values was missing, the absolute change was missing.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Sensitivity analyses |
|---|---|---|
Primary outcome | ||
Percentage of patients who had not had a treatment failure after 48 weeks of treatment. Treatment failure after 48 weeks of treatment was “yes,” if at least 1 of the following criteria was met at any time during the DB treatment phase:
| Normal approximation tests | Logistic regression model |
Key secondary outcomes | ||
Percentage of patients with histologic relapse, defined as peak of ≥ 48 eos/mm2 HPF (corresponding to ≥ 15 eos/HPF) at end of treatment of the DB phase | Normal approximation tests | Logistic regression model |
Change in the peak eos/mm2 HPF from baseline of the DB phase to end of treatment of the DB phase | Wilcoxon rank sum test | None |
Percentage of patients with a clinical relapse, a food impaction that needed endoscopic intervention, or an endoscopic dilation during the DB treatment phase | Normal approximation tests | None |
Percentage of patients with a total weekly EEsAI-PRO score of ≤ 20 at end of treatment of the DB phase | Normal approximation tests | Logistic regression model |
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF), at end of treatment of the DB phase | Normal approximation tests | None |
Exploratory end point | ||
Change from baseline of the DB phase in the EoE-QoL-A questionnaire | t-test | None |
Change from baseline of the DB phase in modSHS | t-test | None |
DB = double-blind; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; eos = eosinophil; HPF = high-power field; modSHS = modified Short Health Scale; NRS = numerical rating scale.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Analysis Populations
The full analysis set (FAS) included all randomized patients (as randomized) who received at least 1 dose of therapy.
The safety analysis set (SAF) included all randomized patients (as treated) who received at least 1 dose of therapy. If the administration of any therapy was not certain, the patient was included in the SAF.
The evaluation of primary and secondary efficacy end points was performed for the FAS. The SAF was used for the evaluation of safety.
Results
Patient Disposition
In total, 297 patients were included in the BUL-2/EER trial, of which 66 patients were former participants in the BUL-1/EEA trial and were all randomized for the DB treatment phase and 231 patients were screened for the BUL-2/EER trial without previous participation in BUL-1/EEA. A total of 50 patients of the 231 screened patients were screening failures and were not included in the OLI phase of the BUL-2/EER trial. As a result, 181 patients were included in the OLI phase and received at least 1 dose of budesonide 1 mg twice daily. At OLI EOT, 43 patients (23.8%) of OLI patients were not considered for transition to the DB phase, mainly because they were not in clinicopathologic remission. Thus, 138 patients were considered for transition into the DB phase and were subsequently randomized.
A total of 204 patients were randomized for the DB treatment phase, and all were treated. Of these, 141 patients completed the DB phase and 63 patients were prematurely withdrawn, mainly due to lack of efficacy of the investigational products, with only 2 patients withdrawing from the study due to AEs. The dropout rate and lack of efficacy of the investigational products as the primary reason for premature discontinuation were higher in the placebo group (66.2%) than in the budesonide 0.5 mg twice daily group (13.2%).
After the DB phase, |||||||||||||||||||||||| with the OLRI phase, 105 patients continued with the OLE phase, 8 patients continued with the follow-up phase, and 9 patients did not participate in any further study phase. Complete details are presented in Table 10.
Table 10: Patient Disposition in Study BUL-2/EER
Patient disposition | Budesonide 0.5 mg b.i.d. | Placebo |
|---|---|---|
Randomized and treated, N (%) | 68 (100.0) | 68 (100.0) |
Full DB treatment phase completed, N (%) | 59 (86.8) | 23 (33.8) |
DB treatment phase prematurely discontinued, N (%) | 9 (13.2) | 45 (66.2) |
Reason for discontinuation, N (%) | ||
| 7 (10.3) | 42 (61.8) |
| 0 | 0 |
| 2 (2.9) | 3 (4.4) |
Study phases following the DB treatment phase, N (%) | ||
| | | | |
| 49 (72.1) | 5 (7.4) |
| 2 (2.9) | 2 (2.9) |
| 2 (2.9) | 4 (5.9) |
FAS-DB, N (%) | 68 | 68 |
SAF-DB, N (%) | 68 | 68 |
PP-DB, N (%) | 56 | 57 |
b.i.d. = twice a day; DB = double-blind; FAS = full analysis set; OLE = open-label extension; OLRI = open-label re-induction; PP = per-protocol; SAF = safety analysis set.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Exposure to Study Treatments
Budesonide 0.5 mg twice daily was administered on average on 308 days (SD = 87.0 days‚ median = 337 days), and placebo was administered on average on 164 days (SD = 132.9 days‚ median = 97 days).
Overall, compliance of the patients who received the treatments was high in all treatment groups. Compliance (%) was calculated as the number of used tablets divided by the total number of tablets prescribed during the treatment period, then multiplied by 100. Compliance was calculated based on drug accountability. Unreturned blisters were either counted as used medication (Method 1) or unused medication (Method 2). Mean values for the calculated compliance (Method 1) were 93.4% (SD = 10.62, median = 96.1%) in the budesonide 0.5 mg twice daily group, and 94.2% (SD = 8.99, median = 96.4%) in the placebo group. Mean values for the calculated compliance (Method 2) were 91.0% (SD = 13.10, median = 93.1%) in the budesonide 0.5 mg twice daily group, and 91.2% (SD = 10.72, median = 94.4%) in the placebo group.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below. See Appendix 3 for detailed efficacy data.
Treatment Failure
As per study design, treatment failure was defined as at least 1 of the following criteria being met at any time during the DB treatment phase:
clinical relapse
histologic relapse, i.e., a peak of 48 eos/mm2 HPF or more (corresponding to 15 eos/HPF or more) at EOT in the DB phase
food impaction that needed endoscopic intervention
need for an endoscopic dilation
premature withdrawal for any reason.
The percentage of patients who had not had a treatment failure after 48 weeks of DB treatment were 73.5% in the budesonide 0.5 mg twice daily group, and 4.4% in the placebo group. Patients experienced treatment failures primarily driven by either clinical and/or histologic relapse.
Histologic relapse was observed in 13.2% of the patients in the budesonide 0.5 mg twice daily group and in 89.7% of the patients in the placebo group. Clinical relapse was observed in 10.3% of the patients in the budesonide 0.5 mg twice daily group and in 60.3% of the patients in the placebo group. None of the patients in the budesonide 0.5 mg treatment group and 1 patient in the placebo group experienced a food impaction during the treatment phase that needed endoscopic intervention. No patient needed an endoscopic dilation at any time during the DB treatment phase. Of note, 9 out of 68 patients (13.2%) in the budesonide 0.5 mg group, in contrast to 45 of 68 patients (66.2%) in the placebo group, withdrew prematurely from the 48-week DB treatment phase (Table 11). This was consistent with the high percentage of treatment failure observed in the placebo arm.
For the comparison between the budesonide 0.5 mg twice daily group and the placebo group, the difference between percentages of patients who had not had a treatment failure was 69.1 percentage points (97.5% confidence interval [CI], 55.89 to 82.34 percentage points; P < 0.0001) in favour of the budesonide 0.5 mg group (Table 11).
The median time to treatment failure was 336 days in the budesonide 0.5 mg twice daily group and 86 days in the placebo group (Table 11).
A sensitivity analysis was performed using a logistic regression model for the budesonide 0.5 mg twice daily group, with a number of factors as covariates, such as baseline overall peak eos/mm2 HPF values, baseline weekly sum of Dysphagia NRS, and baseline weekly sum of Pain During Swallowing NRS of the preceding induction phase (either BUL-1/EEA or BUL-2/EER OLI phase). The sensitivity analysis supported the results of the primary analysis. However, the impact of notable imbalances at baseline on, for example, the duration since EoE diagnosis, duration since the first symptoms, and number of previous esophageal dilations was not assessed by using a multivariate adjustment approach.
Overall, all subgroup results showed a pattern similar to that observed for the total population. Percentage of patients who had not had a treatment failure after 48 weeks of treatment were higher in the budesonide 0.5 mg twice daily group compared to the placebo group for all subgroups assessed. Of note, in the budesonide 0.5 mg twice daily group, a higher percentage of patients who had not had a treatment failure was observed for patients who entered the DB phase after participation in the BUL-1/EEA trial (20/22 patients [90.9%] in the budesonide 0.5 mg group) than for patients who entered the DB phase after participation in the OLI phase of the BUL-2/EER trial (30/46 patients [65.2%] in the budesonide 0.5 mg group). The subgroup analyses by localization of the inflammation at screening (either proximal, mid-, or distal esophagus) as well as by the extent of inflammation (number of affected esophageal segments [1, 2, or 3 segments]) were also consistent with the results in the overall population. Results of the subgroup analysis are presented in Table 19.
Table 11: Lack of Treatment Failure after 48 Weeks of Treatment in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Patients who had not had a treatment failure after 48 weeks of treatment | ||
Number of patients contributing to the analysis | 68 | 68 |
Number (%) of patients who had not had a treatment failure after 48 weeks of treatment | 50 (73.5) | 3 (4.4) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI) | 69.1 (55.89 to 82.34) | Reference |
P valuea | < 0.0001 | Reference |
Time to treatment failure (days)b | ||
Median (IQR) | 336 (333 to 340) | 86 (29 to 333) |
Number (%) of patients with histologic relapse, defined as peak of ≥ 48 eos/mm2 HPF at the end of treatment in the DB phase | ||
Yes | 9 (13.2) | 61 (89.7) |
No | 57 (83.8) | 4 (5.9) |
Not evaluable | 2 (2.9) | 3 (4.4) |
Number (%) of patients with clinical relapse during the DB phase | ||
Not suspected | 60 (88.2) | 21 (30.9) |
Suspicion resolved | 1 (1.5) | 5 (7.4) |
Yes | 7 (10.3) | 41 (60.3) |
Suspected but not assessable | 0 (0.0) | 1 (1.5) |
Patients experiencing a food impaction that needed endoscopic intervention | ||
N (%) | 0 | 1 (1.5) |
Patients needing an endoscopic dilation | ||
N (%) | 0 | 0 |
Premature withdrawal for any reason | ||
N (%) | 9 (13.2) | 45 (66.2) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; eos = eosinophil; HPF = high-power field; IQR = interquartile range; vs. = versus.
aTesting of null hypothesis by means of the 1-sided normal approximation test, Bonferroni adjusted alpha = 0.0125.
bTime to treatment failure was calculated as the time interval in days from date of first intake of DB investigational medicinal product to date of the first occurrence of at least 1 of the following events during the DB phase: clinical relapse, histologic relapse (occurrence date = date of endoscopy), occurrence of a food impaction that needed endoscopic intervention (occurrence date = date of food impaction according to AE reporting), need for an endoscopic dilation (occurrence date = date of endoscopic dilation according to AE reporting), or premature withdrawal for any reason. The time to variable was calculated as the earliest of these occurrence dates – date of first intake of DB investigational medicinal product.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Histologic Response
Percentage of patients with histologic relapse, defined as peak of 48 eos/mm2 HPF or more (corresponding to 15 eos/HPF or more) at end of treatment of the DB phase, and change in the peak eos/mm2 HPF from baseline to EOT of the DB phase were key secondary end points in the BUL-2/EER trial. Percentage of patients with histologic remission, defined as a peak of less than 16 eos/mm2 HPF at EOT of the DB phase, percentage of patients with deep histologic remission, defined as a peak of 0 eos/mm2 HPF at EOT of the DB phase, and percentage of patients maintaining deep histologic remission, defined as a peak of 0 eos/mm2 HPF from baseline of the DB phase to EOT of the DB phase, were exploratory end points in the BUL-2/EER trial.
Nine patients (13.2%) in the budesonide 0.5 mg twice daily group and 61 patients (89.7%) in the placebo group experienced a histologic relapse. When comparing to placebo, the difference in percentages of patients with histologic relapse was −76.5 percentage points (97.5% CI, −88.8 to −64.1 percentage points; P < 0.0001) in favour of the budesonide 0.5 mg twice daily group (Table 12).
The mean change in the peak eosinophils per mm2 HPF from baseline to 48 weeks was 38 (SD = 112.6) in the budesonide 0.5 mg group compared to 262 (SD = 216.3) in the placebo group. The difference between the budesonide 0.5 mg twice daily group and the placebo group was statistically significant at P < 0.0001 (Table 12).
In the budesonide 0.5 mg twice daily group, the percentages of patients with deep histologic remission (i.e., peak of 0 eos/mm2 HPF) at DB EOT, as well as the proportion of patients with maintenance of deep histologic remission from DB baseline to DB EOT, was more than 75%, whereas the percentages in the placebo group were below 2% (Table 12).
Table 12: Histologic Outcomes in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Percentage of patients with histologic relapse, defined as peak of ≥ 48 eos/mm2 HPF at end of treatment of the DB phasea | ||
Number of patients contributing to the analysis | 68 | 68 |
Patients with histologic relapse, n (%) | 9 (13.2) | 61 (89.7) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI)b | −76.5 (−88.8 to −64.1) | Reference |
P valuec | < 0.0001 | Reference |
Change in the peak eos/mm2 HPF from baseline of the DB phase to end of treatment of the DB phased | ||
Number of patients contributing to the analysis | 66 | 65 |
Mean (SD) | 38 (112.6) | 262 (216.3) |
P valuee | < 0.0001 | Reference |
Percentage of patients with histologic remission, defined as a peak of < 16 eos/mm2 HPF at end of treatment of the DB phase | ||
n (%) | 53 (77.9) | 2 (2.9) |
Percentage of patients with deep histologic remission, defined as a peak of 0 eos/mm2 HPF at end of treatment of the DB phase | ||
n (%) | 52 (76.5) | 1 (1.5) |
Percentage of patients maintaining deep histologic remission, defined as a peak of 0 eos/mm2 HPF from baseline of the DB phase to end of treatment of the DB phasef | ||
N | 65 | 64 |
n (%) | 50 (76.9) | 1 (1.6) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; eos = eosinophil; HPF = high-power field; vs. = versus.
aFor this analysis, not evaluable results were set to “No.”
bBonferroni correction.
cNormal approximation test was used for testing.
dIn case the change at DB EOT visit could not be calculated because no valid DB EOT visit value was available, the change at DB EOT visit was missing and the patient was excluded from this analysis.
eWilcoxon rank sum test was used for testing.
fMaintaining deep histologic remission from DB baseline was only evaluated for the subset of patients who were in deep histologic remission at DB baseline.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Clinical Relapse
Percentage of patients with a clinical relapse during the DB treatment phase was a key secondary end point and time to first occurrence of clinical relapse was an exploratory end point in the BUL-2/EER trial.
Seven patients (10.3%) in the budesonide 0.5 mg twice daily group and 41 patients (60.3%) in the placebo group experienced clinical relapse. The difference from placebo group in percentages of patients with histologic relapse was −50.0 percentage points (97.5% CI, −65.7 to −34.3 percentage points; P < 0.0001) in favour of the budesonide 0.5 mg twice daily group (Table 13).
The median time to first clinical relapse was 336 days in the budesonide 0.5 mg twice daily group, and 86 days in the placebo group (Table 13).
Table 13: Clinical Relapse in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Percentage of patients with a clinical relapse, having experienced a food impaction that needed endoscopic intervention or having needed an endoscopic dilation during the DB treatment phasea | ||
Number of patients contributing to the analysis | 68 | 68 |
Patients with a clinical relapse, have experienced a food impaction that needed endoscopic intervention, or needed an endoscopic dilation during the DB treatment phase, n (%) | 7 (10.3) | 41 (60.3) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI)b | −50.0 (−65.7 to −34.3) | Reference |
P valuec | < 0.0001 | Reference |
Time to first occurrence of clinical relapse (days)d | ||
Median (IQR) | 336 (333 to 340) | 86 (29 to 333) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; IQR = interquartile range; vs. = versus.
aFor this analysis not evaluable results were set to “No.”
bBonferroni correction.
cNormal approximation test was used for testing.
dTime to first occurrence of clinical relapse was calculated as the time interval in days from the date of first intake of DB investigational medicinal product to the date of the first clinical relapse as follows: Date of the visit at which the first subsequently confirmed clinical relapse was suspected minus date of first intake of DB investigational medicinal product.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Health-Related Quality of Life
Change from baseline in the EoE-QoL-A questionnaire and change from baseline in modSHS were exploratory end points in the BUL-2/EER trial.
The difference between the budesonide 0.5 mg twice daily group and the placebo treatment group in mean absolute changes in score from baseline to 48 weeks of the DB phase for EoE-QoL-A (30 items), EoE-QoL-A (24 items), EoE-QoL-A eating/diet impact (10 items), and EoE-QoL-A eating/diet impact (4 items) were 0.46 (95% CI, 0.27 to 0.66), 0.49 (95% CI, 0.30 to 0.68), 0.65 (95% CI, 0.39 to 0.92), and 0.75 (95% CI, 0.49 to 1.02), respectively. These between-group differences were in favour of the budesonide 0.5 mg twice daily group (Table 14).
Table 14: Eosinophilic Esophagitis Quality of Life Scale for Adults Questionnaire in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
EoE-QoL-A 30 items overall score (weighted average)a | ||
Baseline | ||
N | 64 | 64 |
Mean (SD) | 3.2 (0.56) | 3.0 (0.70) |
End of treatment of the DB phase | ||
N | 66 | 65 |
Mean (SD) | 3.3 (0.46) | 2.8 (0.75) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.46 (0.27 to 0.66) | Reference |
P valueb | < 0.0001c | Reference |
EoE-QoL-A 24 items overall score (weighted average)a | ||
Baseline | ||
N | 64 | 64 |
Mean (SD) | 3.1 (0.57) | 3.0 (0.70) |
End of treatment of the DB phase | ||
N | 66 | 65 |
Mean (SD) | 3.3 (0.46) | 2.7 (0.75) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.49 (0.30 to 0.68) | Reference |
P valueb | < 0.0001c | Reference |
EoE-QoL-A eating/diet impact 10 items (weighted average)a | ||
Baseline | ||
N | 64 | 64 |
Mean (SD) | 3.3 (0.62) | 3.2 (0.84) |
End of treatment of the DB phase | ||
N | 66 | 65 |
Mean (SD) | 3.5 (0.48) | 2.8 (0.99) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.65 (0.39 to 0.92) | Reference |
P valueb | < 0.0001c | Reference |
EoE-QoL-A eating/diet impact 4 items (weighted average)a | ||
Baseline | ||
N | 64 | 64 |
Mean (SD) | 3.2 (0.69) | 3.2 (0.83) |
End of treatment of the DB phase | ||
N | 66 | 65 |
Mean (SD) | 3.5 (0.49) | 2.8 (0.97) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | 0.75 (0.49 to 1.02) | Reference |
P valueb | < 0.0001c | Reference |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; EoE-QoL-A = Adult Eosinophilic Esophagitis Quality of Life; SD = standard deviation; vs. = versus.
aThe EoE-QoL-A weighted average scores range from 0 to 4. Higher scores indicate better QoL.
b2-sided t-test was used for exploratory testing.
cEoE-QoL-A scores were outside the statistical testing hierarchy.
Source: Clinical Study Report of the BUL-2/EEA trial.10
The difference between the budesonide 0.5 mg twice daily group and the placebo treatment group in mean absolute changes in score (95% CI) from baseline to 48 weeks of the DB phase for symptom burden, social function, disease-related worry, and general well-being were −22 (−30.5 to −13.9), −15 (−23.6 to −7.3), −12 (−19.4 to −3.7), and −12 (−18.9 to −4.3), respectively. These between-group differences were in favour of the budesonide 0.5 mg twice daily group (Table 15).
Table 15: Modified Short Health Scales in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Symptom burdena | ||
Baseline | ||
N | 68 | 68 |
Mean (SD) | 11 (9.6) | 11 (12.1) |
End of treatment of the DB phase | ||
N | 66 | 64 |
Mean (SD) | 12 (18.7) | 34 (26.0) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −22 (−30.5 to −13.9) | Reference |
P valueb | < 0.0001c | Reference |
Social functiona | ||
Baseline | ||
N | 68 | 68 |
Mean (SD) | 12 (13.7) | 13 (17.3) |
End of treatment of the DB phase | ||
N | 66 | 64 |
Mean (SD) | 11 (17.3) | 27 (26.0) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −15 (−23.6 to −7.3) | Reference |
P valueb | 0.0003c | Reference |
Disease-related worrya | ||
Baseline | ||
N | 68 | 68 |
Mean (SD) | 25 (20.9) | 27 (24.0) |
End of treatment of the DB phase | ||
N | 65 | 64 |
Mean (SD) | 22 (23.0) | 36 (27.5) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −12 (−19.4 to −3.7) | Reference |
P valueb | 0.0041c | Reference |
General well-beinga | ||
Baseline | ||
N | 68 | 68 |
Mean (SD) | 12 (13.0) | 15 (14.3) |
End of treatment of the DB phase | ||
N | 66 | 64 |
Mean (SD) | 12 (15.5) | 26 (25.2) |
Difference between absolute change from DB baseline to end of treatment of the DB phase (budesonide vs. placebo) (95% CI) | −12 (−18.9 to −4.3) | Reference |
P valueb | 0.0022c | Reference |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; SD = standard deviation; vs. = versus.
aRange of each score: 0 to 100. Lower numbers indicate higher QoL.
b2-sided t-test was used for exploratory testing.
cModified Short Health Scale was outside the statistical testing hierarchy.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Eosinophilic Esophagitis Activity
Eosinophilic Esophagitis Activity Index Patient Reported Outcome
The percentage of patients with a total weekly EEsAI-PRO score of 20 or less at end of treatment of the DB phase was a key secondary end point in the BUL-2/EER trial.
Forty-nine patients (72.1%) in the budesonide 0.5 mg twice daily group and 14 patients (20.6%) in the placebo group achieved a total weekly EEsAI-PRO score of 20 or less at EOT of the DB phase. The difference between the budesonide 0.5 mg twice daily group and the placebo treatment group was 51.5 percentage points (95% CI, 35.1 to 67.9 percentage points; P < 0.0001) in favour of the budesonide 0.5 mg twice daily group (Table 16).
Table 16: Total Weekly EEsAI-PRO Score in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Percentage of patients with a total weekly EEsAI-PRO score of ≤ 20 at end of treatment of the DB phasea | ||
Number of patients contributing to the analysis | 68 | 68 |
Number (%) of patients who had not had a treatment failure after 48 weeks of treatment | 49 (72.1) | 14 (20.6) |
Difference in percentage: budesonide vs. placebo, percentage points (97.5% CI)b | 51.5 (35.1 to 67.9) | Reference |
P valuec | < 0.0001 | Reference |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; vs. = versus.
aFor this analysis not evaluable results were set to “No.”
bBonferroni correction.
cNormal approximation test was used for testing.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Physician’s Global Assessment
The change from baseline of the DB phase in the PGA of EoE activity (NRS of 0 to 10) was an exploratory end point in the BUL-2/EER trial.
The mean change in the PGA of EoE activity from baseline to EOT was 0 (SD = 1.8) in the budesonide 0.5 mg twice daily group, and 4 (SD = 2.4) the placebo group (Table 20 in Appendix 4). No statistical analysis for the between-group difference was conducted for this analysis.
Patient’s Global Assessment
The percentage of patients with an increase of 3 points or more from baseline in the PatGA regarding the severity of EoE symptoms (NRS scale of 0 to 10) at the end of 48 weeks’ DB phase and the mean change from baseline were exploratory end points in the BUL-2/EER trial.
The results showed a much higher percentage of patients with overall symptom resolution (PatGA 2 points or less) in the budesonide 0.5 mg twice daily group (88.2%) than in the placebo group (32.4%). The percentage of patients with increase of 3 points or more from baseline was 7.4% in the budesonide 0.5 mg twice daily group, and 50% the placebo group. The mean change in the PatGA regarding the severity of EoE symptoms from baseline to 48 weeks was 0 (SD = 2.0), in the budesonide 0.5 mg twice daily group and 3 (SD = 2.7) the placebo group (Table 21). No statistical analysis for the between-group difference was conducted for this analysis.
Endoscopy Outcomes
In the budesonide 0.5 mg twice daily group, the percentage of patients with all features graded as 0 (based on the modified EEsAI endoscopic instrument score) was more than 50% at 48 weeks, whereas the percentage in the placebo group was 5.9%. Furthermore, in the budesonide 0.5 mg twice daily group the percentage of patients with no or only mild endoscopic findings was more than 90%, whereas the percentage in the placebo group was 35.3% (Table 23). No statistical analysis for the between-group difference was conducted for these analyses.
Patients in Deep Disease Remission
The percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF), at end of 48 weeks was a key secondary end point in the BUL-2/EER trial.
Twenty-seven patients (39.7%) in the budesonide 0.5 mg twice daily group, and none of the patients in the placebo group achieved deep disease remission at EOT. The difference from placebo group in the percentage of patients with deep disease remission was 39.7 percentage points (97.5% CI, 26.4 to 53.0 percentage points; P < 0.0001) in favour of the budesonide 0.5 mg twice daily group (Table 24).
Results from the subgroup analyses results showed a pattern similar to that observed for the total population. The percentage of patients in deep disease remission after 48 weeks of treatment was higher in the budesonide 0.5 mg twice daily group compared to the placebo group. Of note, a higher percentage of patients with deep disease remission was observed for patients who entered the DB phase after participation in the BUL-1/EEA trial (13/22 patients [59.1%] in the budesonide 0.5 mg twice daily group) than for patients who entered the DB phase after participation in the OLI phase of the BUL-2/EER trial (14/46 patients [30.4%] in the budesonide 0.5 mg twice daily group). The subgroup analyses by localization of the inflammation at screening (either proximal, mid-, or distal esophagus) as well as by the extent of inflammation (number of affected esophageal segments [1, 2, or 3 segments]) at screening showed consistent results as in the overall population. Results of the subgroup analysis are presented in Table 25.
Harms
Only those harms identified in the review protocol are reported below. See Table 17 for detailed harms data.
Adverse Events
In the BUL-2/EER trial, the majority of patients reported at least 1 TEAE, including 57 patients (83.8%) in the budesonide 0.5 mg twice daily group and 61 patients (89.7%) in the placebo group (Table 17).
The most frequently reported TEAEs in the budesonide 0.5 mg treatment group were nasopharyngitis (in 25 patients [36.8%] in the budesonide 0.5 mg twice daily group and 19 patients [27.9%] in the placebo group), headache (in 14 patients [20.6%] in the budesonide 0.5 mg twice daily group and 5 patients [7.4%] in the placebo group), and suspected candidiasis AEs (in 13 patients [19.1%] in the budesonide 0.5 mg twice daily group and in none of the patients in the placebo group). AEs that occurred only in the budesonide 0.5 mg treatment group, but not in the placebo group, were diarrhea, bronchitis, suspected candidiasis AEs, urinary tract infection, anxiety, and asthma (Table 17).
Serious Adverse Events
Three patients (4.4%) in the budesonide 0.5 mg twice daily group, and none in the placebo group reported serious AE (SAE). All SAEs were treatment-emergent and assessed by the investigator as unrelated to the investigational products. The reason for classifying the AE as serious was hospitalization.
Withdrawals Due to Adverse Events
Seven patients (10.3%) in the budesonide 0.5 mg twice daily group and 42 patients (61.8%) in the placebo group had AEs that led to discontinuation of treatment, with the main reason being “condition aggravated” (Table 17). All 7 patients who withdrew from the treatment group did so due to “condition aggravated.”
Mortality
No deaths occurred during the DB phase of the BUL-2/EER trial.
Notable Harms
A total of 21 suspected candidiasis AEs occurred in 13 patients in the budesonide 0.5 mg twice daily group, and none occurred in the placebo group. Of the 21 treatment-emergent suspected candidiasis AEs, 17 were rated as adverse drug reaction (ADRs) in 12 patients (17.6%) in the budesonide 0.5 mg twice daily group. Fifteen of these ADRs (esophageal, oral, or oropharyngeal candidiasis) were clinically manifested; 5 ADRs were histologically confirmed, and 4 ADRs were both histologically confirmed and clinically manifested. All suspected candidiasis ADRs with either histologic confirmation or clinical manifestation were of mild or moderate severity. All of the ADRs in the budesonide 0.5 mg twice daily group were resolved and the patients recovered.
Dysgeusia was not reported in any patient in the budesonide 0.5 mg twice daily and the placebo groups. Cataract was reported in 1 patient (1.5%) in the placebo group versus none in the budesonide 0.5 mg twice daily treatment group. Depression and sleep disorder were each reported in 1 patient (1.5%) in the placebo group versus none in the budesonide 0.5 mg twice daily treatment group. Anxiety was reported in 3 patients (4.4%) in the budesonide 0.5 mg twice daily group versus none in the placebo group. Insomnia was reported in 2 patients (2.9%) and mood swings were reported in 1 patient (1.5%) in the budesonide 0.5 mg twice daily group versus none in the placebo groups. Symptoms of sore throat (pharyngitis) were reported in 3 patients (4.4%) in the budesonide 0.5 mg twice daily group and 1 patient (1.5%) in the placebo group.
Table 17: Summary of Harms in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Patients with ≥ 1 adverse event | ||
n (%) | 57 (83.8) | 61 (89.7) |
Most common events,a n (%) | ||
Blepharitis | 3 (4.4) | 1 (1.5) |
Diarrhea | 5 (7.4) | 0 |
Dyspepsia | 3 (4.4) | 3 (4.4) |
Dysphagia | 7 (10.3) | 2 (2.9) |
Esophageal food impaction | 0 | 2 (2.9) |
Chest pain | 4 (5.9) | 1 (1.5) |
Condition aggravated | 11 (16.2) | 44 (64.7) |
Bronchitis | 1 (1.5) | 0 |
Gastroenteritis | 3 (4.4) | 1 (1.5) |
Influenza | 3 (4.4) | 2 (2.9) |
Nasopharyngitis | 25 (36.8) | 19 (27.9) |
Suspected candidiasis adverse eventb | 13 (19.1) | 0 |
Balanitis candidiasis | 1 (1.5) | 0 |
Genital candidiasis | 1 (1.5) | 0 |
Esophageal candidiasis | 7 (10.3) | 0 |
Oral candidiasis | 7 (10.3) | 0 |
Oropharyngeal candidiasis | 3 (4.4) | 0 |
Vulvovaginal candidiasis | 0 | 0 |
Pharyngitis | 3 (4.4) | 1 (1.5) |
Urinary tract infection | 3 (4.4) | 0 |
Back pain | 3 (4.4) | 1 (1.5) |
Headache | 14 (20.6) | 5 (7.4) |
Anxiety | 3 (4.4) | 0 |
Asthma | 4 (5.9) | 0 |
Patients with ≥ 1 SAE | ||
n (%) | 3 (4.4) | 0 |
Inguinal hernia | 1 (1.5) | 0 |
Sinusitis | 1 (1.5) | 0 |
Cartilage injury | 1 (1.5) | 0 |
Skull fracture | 0 | 0 |
Upper limb fracture | 1 (1.5) | 0 |
Patients who stopped treatment due to adverse events | ||
n (%) | 7 (10.3) | 41 (60.3) |
Esophageal food impaction | 0 | 2 (2.9) |
Chest pain | 0 | 0 |
Condition aggravated | 7 (10.3) | 40 (58.8) |
Retinitis | 0 | 0 |
Oropharyngeal pain | 0 | 0 |
Dermatitis allergic | 0 | 0 |
Deaths | ||
n (%) | 0 | 0 |
Notable harms | ||
Suspected candidiasis | ||
Esophageal candidiasis | 6 (8.8) | 0 |
Oral candidiasis | 7 (10.3) | 0 |
Oropharyngeal candidiasis | 3 (4.4) | 0 |
Histologically confirmed candidiasis | ||
Oropharyngeal candidiasis | 5 (7.4) | 0 |
Dysgeusia | 0 | 0 |
Decreased bone mineral density | NR | NR |
Cataract | 0 | 1 (1.5) |
Glaucoma | 0 | 0 |
Psychiatric disorders | ||
Anxiety | 3 (4.4) | 0 |
Depression | 0 | 1 (1.5) |
Insomnia | 2 (2.9) | 0 |
Mood swings | 1 (1.5) | 0 |
Sleep disorder | 0 | 1 (1.5) |
Symptoms of sore throat (pharyngitis) | 3 (4.4) | 1 (1.5) |
Osteonecrosis of the hip | NR | NR |
b.i.d. = twice a day; NR = not reported; SAE = serious adverse event.
aOccurring in at least 3 patients in 1 treatment group.
bPatients with more than 1 candidiasis adverse event may appear several times in different subcategories but are counted only once in the “Suspected candidiasis adverse event” category.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Critical Appraisal
Internal Validity
The BUL-2/EER trial used accepted methods to conceal allocation and randomize patients to treatments; in addition, matched placebo was used to maintain blinding. Randomization was performed using randomly permuted blocks. The patients’ baseline characteristics and prior treatment experience appeared to be roughly balanced at baseline between groups, although the disease duration since diagnosis and since first symptoms were shorter in the placebo group than in the budesonide 0.5 mg group. The mean time since an established EoE diagnosis was 4.3 years and 3.3 years in the budesonide 0.5 mg and placebo groups, respectively, with a mean time since symptom onset of 12.6 years and 9.6 years, respectively. In addition, fewer patients in the placebo group (5.9%) had a previous esophageal dilation compared to the budesonide 0.5 mg group (19.1%). The impact of such imbalance on the treatment effect assessment is unknown.
In this study, clinical relapse was defined as patient’s experiencing dysphagia or pain during swallowing in the past 7 days (7-day recall period), with a severity of at least 4 points on a 0 to 10 NRS for dysphagia or pain during swallowing. The benefit of budesonide 0.5 twice daily, as measured by treatment failure, was primarily driven by histologic and clinical relapse. Subjective recall biases in the assessment of clinical relapse would be highly likely, particularly when such recall differed between treatment groups, due perhaps to patients’ or the assessing physicians’ awareness of the treatment assignment as a result of drug-related side effects. For example, 19.1% of patients in the budesonide 0.5 mg group had suspected candidiasis AEs, whereas no such events were reported in the placebo group. Moreover, the majority of patients in the placebo group had experienced aggravated conditions of the disease (64.7% in the placebo group versus 16.2% in the 0.5 mg budesonide group) during the 48-week treatment period, which may have led to recall of more severe or worsening experience of symptoms or pains among patients on placebo than their counterparts on active treatment.
The large number of patients who discontinued from the trial could also have biased the results for patient-reported outcomes, HRQoL, and other exploratory outcomes. For example, only 23 out of 68 patients (33.8%) in the placebo group completed the 48-week DB phase. It was not reported how missing values were handled for the analysis of EoE-QoL-A and modSHS. The number of patients included in these analyses was very similar to the number of patients randomized in the FAS; however, 66% of patients in the placebo group discontinued treatment before week 48. Hence, it is unclear how patients who discontinued were accounted for. Hence, there is uncertainty around the potential benefit of budesonide on HRQoL. Finally, the clinical expert consulted on this review indicated that, due to the absence of an estimated MID for the HRQoL measures used in the BUL-2/EER trial, it is impossible to draw any firm conclusions about the effects of budesonides on HRQoL. Of note, using different HRQoL outcome measures, patients receiving placebo experienced deterioration in their HRQoL, while patients who received budesonide did not.
Subgroup analysis by the path to remission (i.e., BUL-1/EEA or BUL-2/EER OLI phase) showed that a higher percentage of patients had not had a treatment failure and were in deep disease remission among those who entered the DB phase of the BUL-2/EER trial after participation in the BUL-1/EEA trial than among those who entered the DB phase after participation in the OLI phase of the BUL-2/EER trial. However, it is not clear why this difference in response occurred and whether the open-label design of the OLI phase of the BUL-2/EER and recall bias affected the results.
The validity, test-retest reliability, and responsiveness of the outcome measures (e.g., Dysphagia NRS, Pain During Swallowing NRS, PGA of EoE activity, and PatGA concerning the severity of EoE symptoms) used in the BUL-2/EER trial were not established. Also, the MID in the EoE population is not available for any of the patient-reported outcomes assessed. The clinical assessment of symptom resolution and patients’ HRQoL were based on patient-reported outcomes using a diary recording over a week or questionnaires.
External Validity
Patients enrolled in the BUL-2/EER trial were deemed to be similar to patients with EoE in Canada, although no Canadian study site was included in this trial. Only patients with clinicopathologic remission, defined as fulfilling both criteria for histologic remission and resolution of symptoms after receiving budesonide orodispersible tablets, were enrolled. Hence, results may not be generalizable to patients who achieved clinicopathologic remission using other treatments. If careful medical monitoring was not ensured, patients with cardiovascular disease, diabetes mellitus, osteoporosis, active peptic ulcer disease, glaucoma, cataract, or infection were excluded from the trial, which limits the generalizability of the trial results for patients with comorbidities.
The clinical experts indicated that using placebo as a control group is acceptable given that no other medicinal product is licensed for the long-term treatment of EoE.
EoE is a chronic condition in which patients experience recurrences of inflammation, requiring re-treatment or changes in therapy over time. The BUL-2/EER trial was designed to demonstrate superiority over placebo at week 48, and it was unclear how long the patients would remain on remission while on treatment. It was unclear whether patients would relapse after they stopped treatment. Hence, the optimal duration of maintenance treatment was not explored. Also, the clinical experts indicated that, in clinical practice, patients might receive treatment intermittently. However, the BUL-2/EER trial did not explore intermittent use of budesonide.
Patients enrolled in the BUL-2/EER trial after they achieved clinicopathologic remission in either the OLI phase of BUL-2/EER or the induction trial BUL-1/EEA. Some patients in the BUL-1/EEA trial received induction treatment for 12 weeks, while the majority of patients received budesonide for 6 weeks. CDEC’s final recommendation for budesonide orodispersible tablets for the induction of clinicopathologic remission in adults with EoE21 limits treatment duration for the induction of remission to a maximum period of 6 weeks, with no option for extending treatment with budesonide for the induction of remission to 12 weeks or for re-treating with budesonide in case of relapse. In the BUL-2/EER trial, no subgroup analysis by prior treatment duration with budesonide (6 weeks versus 12 weeks) was conducted. Hence, it is uncertain how the course of therapy during the induction of remission affects the response rate for the maintenance of remission.
The BUL-2/EER trial excluded patients with severe strictures, which may limit the interpretation of the efficacy findings to patients with severe strictures
Indirect Evidence
No indirect evidence was submitted by the sponsor or identified in our literature search that would match the inclusion and exclusion criteria of this review.
Other Relevant Evidence
There is currently 1 ongoing OLE study that includes patients from the BUL-2/EER; however, results were not available at the time of this review.
Discussion
Summary of Available Evidence
One pivotal phase III, DB, randomized, multi-centre, placebo-controlled study met the inclusion criteria. The BUL-2/EER trial (N = 204) compared the efficacy and tolerability of a 48-week treatment with 2 different doses of budesonide effervescent tablets (budesonide 0.5 mg twice daily and budesonide 1 mg twice daily) with placebo for the maintenance of clinicopathologic remission in adult patients with EoE. The percentage of patients who had not had a treatment failure after 48 weeks of treatment was the primary end point. The percentage of patients with histologic relapse, change in the peak eos/mm2 HPF from baseline, the percentage of patients with a clinical relapse, the percentage of patients with a total weekly EEsAI-PRO score of 20 or less, and the percentage of patients in deep disease remission were key secondary end points. HRQoL — evaluated using the modSHS and the EoE-QoL-A — was an exploratory outcome in the BUL-2/EER trial. All baseline parameters of disease activity, including histologic results and endoscopic as well as patients’ and investigators’ assessments, showed low values for disease activity in all treatment groups, which is representative of the EoE patients who are in remission.
The main limitations of the BUL-2/EER trial are the imbalances between treatment groups in some baseline characteristics. The disease duration since diagnosis and since first symptoms were shorter in the placebo group than in the budesonide 0.5 mg group. Fewer patients in the placebo group (5.9%) had a previous esophageal dilation compared to the budesonide 0.5 mg group (19.1%). However, the impact of such imbalance on the treatment effect assessment is unknown. In addition, a large number of patients in the placebo group (45 patients [66.2%]) discontinued from the trial, which could also have biased the results for patient-reported outcomes, HRQoL, and other exploratory outcomes.
Interpretation of Results
Efficacy
EoE is a chronic, immune-mediated esophageal disease characterized by an eosinophil-predominant inflammation of the esophageal mucosa causing symptoms of esophageal dysfunction, and thus requiring anti-inflammatory treatment to achieve clinicopathologic remission. Chronic inflammation with symptoms such as frequent episodes of food impaction have an unfavourable impact on patients’ QoL. Endoscopic procedures, including removal of impacted food and dilation, are often needed. These, coupled with the typical fragility of the mucosa in this disease, are associated with complications such as mucosal tears, significant pain, and even rare esophageal perforations, which could be serious and life-threatening. Aims of treatment for maintenance of remission are to control symptoms, normalize endoscopic appearance, maintain histologic remission, maximize QoL, and avoid complications, especially long-term tissue remodelling, leading to a fibrostenotic esophagus.36
The primary end point of the DB maintenance phase of the BUL-2/EER trial (percentage of patients who had not had a treatment failure after 48 weeks of treatment) took into account both clinical and histologic aspects of deterioration due to the disease, which the clinical experts consulted by CADTH considered comprehensive and set a high bar for demonstrating efficacy, given that almost any indication of lapse of control was noted as treatment failure. The percentage of patients who had not had a treatment failure after 48 weeks of treatment was 73.5% in the budesonide 0.5 mg twice daily group versus 4.4% in the placebo group, with P values for the comparisons between the budesonide 0.5 mg group and placebo significant at the < 0.0001 level. These results clearly demonstrate that a 48-week treatment with budesonide 0.5 mg twice daily is statistically significant and was considered clinically relevant by the clinical experts consulted by CADTH. It is worth noting that the time to relapse was shorter for the placebo-treated group (86 days) as compared to the budesonide 0.5 mg treatment group (336 days).
All pre-specified subgroup analyses of the primary end point (e.g., localization and extent of inflammation, concomitant PPI use, or time since first symptoms) confirmed the primary outcome and convincingly showed the robustness of the observed superiority of the budesonide 0.5 mg twice daily group over placebo. In the budesonide 0.5 mg twice daily group, a higher percentage of patients who had not had a treatment failure was observed for patients who entered the DB phase after participation in the BUL-1/EEA trial (20/22 patients [90.9%]) than for patients who entered the DB phase after participation in the OLI phase of the BUL-2/EER trial (30/46 patients [65.2%]). However, it is not clear why this difference in response occurred and whether the open-label design of the OLI phase of the BUL-2/EER and recall bias affected the results.
The first 4 of the 5 pre-specified key secondary end points, including the percentage of patients with histologic relapse (13.2% of the patients in the budesonide 0.5 mg twice daily group, and in 89.7% of the patients in the placebo group), change in the peak eosinophil count/mm2 HPF (mean 38 [SD = 112.6] in the budesonide 0.5 mg group, and 262 [SD = 216.3] in the placebo group), percentages of patients with clinical relapse, food impaction requiring endoscopic intervention, or need for dilation (10.3% in the budesonide 0.5 mg twice daily group, and 60.3% in the placebo group), and proportions of patients with a total weekly EEsAI-PRO score of 20 or less at DB EOT (72.1% in the budesonide 0.5 mg twice daily group and 20.6% in the placebo group) were all consistently and substantially higher (P < 0.0001) for the budesonide 0.5 mg twice daily group compared with placebo.
The fifth a priori ordered key secondary end point, deep disease remission (i.e., achieving a complete clinical, complete endoscopic, and histologic remission), which was the most stringent secondary end point, showed that the budesonide 0.5 mg twice daily group was also statistically significantly superior to placebo (39.7% in the budesonide 0.5 mg twice daily group and none of the patients in the placebo group achieved deep disease remission at EOT). Of the 9 patients in the budesonide 0.5 mg twice daily group and the 15 patients in the placebo group who were in deep disease remission at baseline, deep disease remission was maintained over a period of 48 weeks in 5 patients (55.6%) in the budesonide 0.5 mg twice daily group and none of the patients in the placebo group.
It was also clear from the patient group input received for this submission that patients consider improved QoL to be an important outcome of treatment. In the BUL-2/EER trial, HRQoL was assessed using the EoE-QoL-A and modSHS instruments. The differences between absolute changes from DB baseline to DB EOT for EoE-QoL-A overall scores revealed that the budesonide 0.5 mg twice daily was better at maintaining patients’ HRQoL than placebo. Also, for all 4 questions of the modSHS — symptom burden, social function, disease-related worry, and general well-being — the differences between the budesonide 0.5 mg twice daily group versus placebo in the absolute changes from DB baseline to DB EOT were in favour of the budesonide groups. However, due to the absence of an estimated MID for the HRQoL measures used in the BUL-2/EER trial, it is not possible to draw any firm conclusions about the effects of orodispersible tablets on HRQoL. In addition, change from baseline in the EoE-QoL-A questionnaire and change from baseline in modSHS were exploratory end points in the BUL-2/EER trial. Finally, a large number of patients discontinued from the trial, which could also have biased the results for patient-reported outcomes, HRQoL, and other exploratory outcomes. For example, only 23 out of 68 patients (33.8%) in the placebo group completed the 48-week DB phase. It was not reported how missing values were handled for the analysis of EoE-QoL-A and modSHS. The number of patients included in these analyses was very similar to the number of patients randomized in the FAS; however, 66% of patients in the placebo group discontinued treatment before week 48. Hence, it is unclear how patients who discontinued were accounted for and whether their response collected before discontinuation was carried forward or responses collected at the end of the DB phase were used. Hence, there is uncertainty concerning the potential benefit of budesonide for HRQoL.
The product monograph indicates that the duration of maintenance therapy is determined by the treating physician.9 The clinical experts consulted by CADTH for this review indicated that patients who discontinued budesonide orodispersible tablets after induction of remission and relapsed within 3 to 6 months of treatment discontinuation should be treated for at least 1 year using budesonide orodispersible tablets as maintenance therapy. After the first year of authorization, renewal should be individualized. Some patients might stop treatment with budesonide orodispersible tablets; if symptoms recur, then they should restart using budesonide orodispersible tablets. The clinical experts also indicated that the evidence from the BUL-2/EER trial is insufficient to provide a criterion for which patients would be able to stop treatment after 48 weeks without risking a relapse. However, from clinical experience, the clinical experts think that patients with a history of severe disease, as manifested by food impactions or significant fibrosis, need to stay on 0.5 mg budesonide orodispersible tablets twice daily for a long period.
The daily dose recommended by the European Medicines Agency is 1 mg budesonide as one 0.5-mg tablet in the morning and another in the evening or 2 mg budesonide as one 1 mg tablet in the morning and another in the evening, depending on the individual clinical requirement of the patient. A maintenance dose of 1 mg budesonide twice daily is recommended for patients with a long-standing disease history and/or high extent of esophageal inflammation in their acute disease state.19 Health Canada has approved only the 0.5 mg budesonide orodispersible tablets twice daily dosage for the maintenance of remission. The clinical experts consulted by CADTH for this review indicated that disease severity before remission would guide dosage decisions and that patients with more severe disease tend to need more aggressive therapy. However, the clinical experts indicated that they would try first to maintain the remission using the budesonide 0.5 mg twice daily dosage; if the patient relapsed, then re-induction of remission using the budesonide 1mg twice daily dosage would be tried. After achieving remission again using the 1 mg budesonide twice daily dosage, patients would be switched back to the 0.5 mg budesonide twice daily dosage for maintenance of remission. Patients who relapsed again while on the 0.5 mg budesonide twice daily dosage would have their dosage increased to 1 mg budesonide twice daily for re-induction of remission. After achieving remission on the 1 mg budesonide twice daily dosage, patients would remain on the 1 mg budesonide twice daily for the maintenance of remission.
It is worth noting that CDEC’s final recommendation for budesonide orodispersible tablets for the induction of clinicopathologic remission in adults with EoE21 limits treatment duration for the induction of remission to a maximum period of 6 weeks, with no option for extending treatment with budesonide for the induction of remission to 12 weeks or for re-treating with budesonide in case of relapse. The clinical experts indicated that, in clinical practice, patients would be prescribed budesonide 1 mg twice daily dosage for the re-induction of remission in case of relapse on the budesonide 0.5 mg twice daily dosage.
The clinical experts also indicated that, in clinical practice, patients might receive treatment intermittently. However, the BUL-2/EER trial did not explore dose switching and intermittent use of budesonide. The clinical experts also indicated that patients who relapsed while receiving the 1 mg budesonide orodispersible tablets twice daily for maintenance of remission needed to be assessed for compliance and other factors associated with recurrence. Some patients might need to discontinue budesonide orodispersible tablets, try another treatment approach and undergo endoscopy by a gastroenterologist.
The clinical experts consulted by CADTH for this review indicated that the aim of treatment is, first and foremost, the treatment and prevention of the inflammatory changes within the esophagus and the reduction (or cure) of the associated symptoms. Long-term treatment is aimed at keeping the esophagus inflammation-free and preventing symptoms from recurring. The long-term consequences of ongoing inflammation and symptoms are the development of fibrotic changes in the esophageal lining, narrowing of the esophageal lumen, and formation of strictures. The consequences for the patients are modification of eating behaviour, avoidance of certain types of food, and, finally, esophageal obstruction, and the need for endoscopic disimpaction and dilation. However, an effect on the long-term sequelae of EoE, namely, stricture formation (as assessed by endoscopy) or food impaction events, could not be demonstrated in the BUL-2/EER trial. However, this might be due to the longer time period needed for observation for these end points.
Patients enrolled in the BUL-2/EER trial after they achieved clinicopathologic remission in either the OLI phase of BUL-2/EER or the induction trial BUL-1/EEA. Some patients in the BUL-1/EEA trial received induction treatment for 12 weeks, while the majority of patients received budesonide for 6 weeks. CDEC’s final recommendation for budesonide orodispersible tablets for the induction of clinicopathologic remission in adults with EoE21 limits treatment duration for the induction of remission to a maximum period of 6 weeks, with no option for extending treatment with budesonide for the induction of remission to 12 weeks or for re-treating with budesonide in case of relapse. In the BUL-2/EER trial, no subgroup analysis by prior treatment duration with budesonide (6 weeks versus 12 weeks) was conducted. Hence, it is uncertain how the course of therapy during the induction of remission affects the response rate for the maintenance of remission. It is also uncertain whether patients who achieved remission using nonpharmacological or pharmacological treatment other than budesonide orodispersible tablets would respond in the same manner for maintenance of remission using budesonide orodispersible tablets as those who achieved induction of remission using budesonide orodispersible tablets. On the other hand, the clinical experts indicated that, if patients are responding well to therapies other than budesonide orodispersible tablets, they would not switch such patients to budesonide orodispersible tablets.
Patients in the BUL-2/EER study with a clinical or histologic relapse (as defined in the primary end point) or a food impaction that needed endoscopic intervention during the DB treatment phase were offered an open-label re-induction |||||| of remission or response treatment with budesonide 1 mg group twice daily for up to 6 weeks. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Harms
The clinical experts indicated that the nature and frequency of AEs observed in the budesonide 0.5 mg group were consistent with the known safety profile of topical budesonide.
No deaths occurred. During the DB phase, only for 3 patients (4.4%) in the budesonide 0.5 mg twice daily group and none of the patients in the placebo group reported SAEs, none of which were related to study medication, as assessed by the investigator. Moreover, only 10% to 12% of patients in the budesonide 0.5 mg treatment group, in contrast to 62% of patients in the placebo group, experienced an AE leading to premature withdrawal of the investigational products, most often due to deterioration/relapse of EoE. Bolus impaction leading to discontinuation of DB the investigational products was observed in 2 patients in the placebo group. No patient needed a dilation during the DB phase.
The most frequently reported TEAEs in the budesonide 0.5 mg treatment group were nasopharyngitis (in 36.8% of patients in the budesonide 0.5 mg twice daily group and 27.9% of patients in the placebo group) and headache (in 20.6% of patients in the budesonide 0.5 mg twice daily group and 7.4% of patients in the placebo group).
The most frequently reported treatment-emergent ADRs in the budesonide 0.5 mg twice daily treatment group were 17 suspected ADRs of candidiasis, occurring in 12 patients (17.6%), versus no such events in the placebo group. These are known ADRs caused by the immunosuppressive action of budesonide. It is noteworthy that not all macroscopically suspected fungal infections were confirmed by the Grocott silver staining. In 5 patients in the budesonide 0.5 mg twice daily group, the suspected candidiasis was histologically confirmed, and, in 4 patients in the budesonide 0.5 mg twice daily group, the suspected candidiasis was both histologically confirmed and clinically manifested.
Conclusions
The BUL-2/EER trial provided evidence on the efficacy and safety of budesonide effervescent tablets 0.5 mg for the maintenance of clinicopathologic remission in adult patients with EoE. The DB phase of BUL-2/EER trial demonstrated that the majority of patients who have had a remission of EoE following a 6- or 12-week course of budesonide 1 mg twice daily orodispersible tablets can be maintained in clinical and histologic remission for 48 weeks with budesonide 0.5 mg twice daily. The time to relapse was shorter for the placebo-treated group than for the active treatment group. The effect of budesonide on HRQoL remains uncertain due to lack of MID, the high number of patients who discontinued placebo, and recall bias. During the BUL-2/EER trial, an effect on the long-term consequences of the disease could not be shown because the number of events, such as food impaction or esophageal dilation due to stricture formation, was too low. It is uncertain whether patients would relapse if they discontinued treatment or if they switched to a lower dosage. Safety data from the BUL-2/EER trial did not demonstrate any notable concern. Long-term safety, particularly in combination with other pharmacological therapies, remains unknown.
References
1.Bonis PA. Clinical manifestations and diagnosis of eosinophilic esophagitis. In: Talley NJ, ed. UpToDate. Watham (MA): UpToDate; 2020: www.uptodate.com. Accessed 2020 Nov 24.
2.Lucendo AJ M-IJ, Arias A, et al. Guidelines on eosinophilic esophagitis: evidence-based statements and recommendations for diagnosis and management in children and adults. United European Gastroenterol J. 2017;5(3):335-358. PubMed
3.Shaheen NJ, Mukkada V, Eichinger CS, Schofield H, Todorova L, Falk GW. Natural history of eosinophilic esophagitis: a systematic review of epidemiology and disease course. Dis Esophagus. 2018;31(8). PubMed
4.Dellon ES, Liacouras CA, Molina-Infante J, et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology. 2018;155(4):1022-1033.e1010. PubMed
5.Furuta GT, Katzka DA. Eosinophilic Esophagitis. N Engl J Med. 2015;373(17):1640-1648. PubMed
6.Bonis PA. Treatment of eosinophilic esophagitis. In: Talley NJ, ed. UpToDate. Watham (MA): UpToDate; 2019: www.uptodate.com. Accessed 2020 Nov 25.
7.Lucendo AJ. Pharmacological treatments for eosinophilic esophagitis: current options and emerging therapies. Expert Rev Clin Immunol. 2020;16(1):63-77. PubMed
8.Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA. ACG clinical guideline: Evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol. 2013;108(5):679-692; quiz 693. PubMed
9.Jorveza (budesonide): 0.5mg and 1 mg orodispersible tablets [product monograph]. Blainville (QC): AVIR Pharma Inc.; 2021 March 16.
10.Clinical Study Report: BUL-2/EER. Double- blind, randomized, placebo-controlled, phase III study on the efficacy and tolerability of a 48-week treatment with two different doses of budesonide effervescent tablets vs. placebo for maintenance of clinico-pathological remission in adult patients with eosinophilic esophagitis [CONFIDENTIAL sponsor's report]. Freiburg (DE): Dr. Falk Pharma GmbH; 2019 May 15.
11.Dellon ES HI. Epidemiology and Natural History of Eosinophilic Esophagitis. Gastroenterology. Gastroenterology. 2018;154(2):319-332 e313. PubMed
12.Sengupta N TE, Corban C, Sommers T, Leffler DA, Lembo AJ. The clinical predictors of aetiology and complications among 173 patients presenting to the Emergency Department with oesophageal food bolus impaction from 2004-2014. Aliment Pharmacol Ther. 2015;42(1):91-98. PubMed
13.Dellon ES. Epidemiology of eosinophilic esophagitis. Gastroenterol Clin North Am. 2014;43(2):201-218. PubMed
14.Carr S, Chan ES, Watson W. Eosinophilic esophagitis. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):58. PubMed
15.Navarro P, Arias A, Arias-Gonzalez L, Laserna-Mendieta EJ, Ruiz-Ponce M, Lucendo AJ. Systematic review with meta-analysis: the growing incidence and prevalence of eosinophilic oesophagitis in children and adults in population-based studies. Aliment Pharmacol Ther. 2019;49(9):1116-1125. PubMed
16.Carr S, Chan ES, Watson W. Correction to: Eosinophilic esophagitis. Allergy, Asthma & Clinical Immunology. 2019;15(1):22. PubMed
17.Orphan Drug Designations and Approvals. Benralizumab. Silver Spring (MD): U.S. Food and Drug Administration; 2019 Aug 26: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=670018. Accessed 2021 Jan 29.
18.Dougherty M, Runge TM, Eluri S, Dellon ES. Esophageal dilation with either bougie or balloon technique as a treatment for eosinophilic esophagitis: a systematic review and meta-analysis. Gastrointest Endosc. 2017;86(4):581-591.e583. PubMed
19.Product information: Jorveza (budesonide) (European public asssessment report). London (GB): European Medicines Agency; 2020 Jun: https://www.ema.europa.eu/en/documents/product-information/jorveza-epar-product-information_en.pdf. Accessed 2020 Dec 14.
20.CADTH Drug Reimbursment Review: Jorveza (AVIR Pharma Inc.). Ottawa (ON): CADTH; 2020: https://cadth.ca/budesonide-0. Accessed 2021 Jan 29.
21.CADTH Drug Reimbursment Expert Review Committee final recommendation: Jorveza (AVIR Pharma Inc.). Ottawa (ON): CADTH; 2020: https://cadth.ca/sites/default/files/cdr/complete/SR0634%20Jorveza%20-%20CDEC%20Final%20Recommendation%20October%2030%2C%202020_for%20posting.pdf. Accessed 2021 Jan 29.
22.e-CPS. Ottawa (ON): Canadian Pharmacists Association; 2020: https://www.e-therapeutics.ca/. Accessed 2021 Jan 05.
23.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
24.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Feb 17.
25.Straumann A, Lucendo AJ, Miehlke S, et al. Budesonide Orodispersible Tablets Maintain Remission in a Randomized, Placebo-Controlled Trial of Patients With Eosinophilic Esophagitis. Gastroenterology. 2020;159(5):1672-1685.e1675.
26.Drug Reimbursement Review sponsor submission: Jorveza (budesonide), 0.5mg and 1 mg orodispersible tablets. Blainville (QC): AVIR Pharma Inc.; 2020 Nov 16.
27.Assessment report: Jorveza (budesonide) (European public asssessment report). Amsterdam (The Netherlands): European Medicines Agency; 2020 Mar: https://www.ema.europa.eu/en/documents/variation-report/jorveza-004655-x-0007-g-epar-assessment-report-variation_en.pdf. Accessed 2020 Dec 14.
28.Straumann A, Conus S, Degen L, et al. Budesonide is effective in adolescent and adult patients with active eosinophilic esophagitis. Gastroenterology. 2010;139(5):1526-1537, 1537.e1521.
29.Center for Drug Evaluation Research. Eosinophilic esophagitis: Developing drugs for treatment guidance for industry (FDA guidance document). Rockville (MD): U. S. Food and Drug Administration (FDA); 2019: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/eosinophilic-esophagitis-developing-drugs-treatment-guidance-industry. Accessed 2021 Feb 10.
30.Taft TH, Kern E, Kwiatek MA, Hirano I, Gonsalves N, Keefer L. The adult eosinophilic oesophagitis quality of life questionnaire: a new measure of health-related quality of life. Aliment Pharmacol Ther. 2011;34(7):790-798. PubMed
31.Bajaj S, Taft T, Keefer L, al. e. Validity, usability, and acceptability of the Eosinophilic Esophagitis Quality of Life Scale for Adults (EoE-QOL-A). Gastroenterol Hepatol (N Y). 2012;142(5 Suppl 1):S-434.
32.Hjortswang H, Järnerot G, Curman B, et al. The Short Health Scale: a valid measure of subjective health in ulcerative colitis. Scand J Gastroenterol. 2006;41(10):1196-1203. PubMed
33.Stjernman H, Grännö C, Järnerot G, et al. Short health scale: a valid, reliable, and responsive instrument for subjective health assessment in Crohn's disease. Inflamm Bowel Dis. 2008;14(1):47-52. PubMed
34.Schoepfer AM, Straumann A, Panczak R, et al. Development and validation of a symptom-based activity index for adults with eosinophilic esophagitis. Gastroenterology. 2014;147(6):1255-1266.e1221. PubMed
35.Hirano I, Moy N, Heckman MG, Thomas CS, Gonsalves N, Achem SR. Endoscopic assessment of the oesophageal features of eosinophilic oesophagitis: validation of a novel classification and grading system. Gut. 2013;62(4):489-495. PubMed
36.Philpott H, Dellon ES. The role of maintenance therapy in eosinophilic esophagitis: who, why, and how? J Gastroenterol. 2018;53(2):165-171. PubMed
37.Avir Pharma response to Dec 9 2020 CDR request for additional information regarding the Jorveza CDR review [CONFIDENTIAL additional sponsor's information]. Blainville (QC): Avir Pharma 2020 Dec 22.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of Search: November 30, 2020
Alerts: Weekly search updates until project completion
Study types: No filters were applied to limit the retrieval by study type.
Limits:
No date limit
Humans
Language limit: No language limits used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for 1 character |
? | Truncation symbol for 1 or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase); |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq = # | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
budesonide/
(jorveza* or budesonide* or map-0010 or map0010 or s-1320 or s1320 or Q3OKS62Q6X).ti,ab,rn,nm,kf,ot.
eosinophilic esophagitis/
(eosinophil* adj3 (esophagitis or esophagitis)).ti,ab,kf.
(eoe or oeoe).ti,ab,kf.
One or 2
Three or 4 or 5
Six and 7
Eight use medall
*budesonide/
(jorveza* or budesonide* jorveza* or budesonide* or map-0010 or map0010 or s-1320 or s1320).ti,ab,kw,dq.
eosinophilic esophagitis/
(eosinophil* adj3 (esophagitis or esophagitis)).ti,ab,kw,dq.
(eoe or oeoe).ti,ab,kw.
Ten or 11
Twelve or 13 or 14
Fifteen and 16
Seventeen use oemezd
conference abstract.pt.
conference review.pt.
Nineteen or 20
Eighteen not 21
Nine or 22
remove duplicates from 23
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search — Studies with results for: budesonide AND eosinophilic esophagitis]
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
[Search terms — budesonide AND eosinophilic esophagitis]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms — budesonide AND eosinophilic esophagitis]
EU Clinical Trials
Register European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms — budesonide AND eosinophilic esophagitis]
Grey Literature
Search dates: November 26, 2020
Keywords: [budesonide AND eosinophilic esophagitis]
Limits: No limits
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool For Searching Health-Related Grey Literature (https://www.cadth.ca/grey-matters) were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Table 19: Studies Excluded From Review
Reference | Reason for exclusion |
|---|---|
Straumann A, Conus S, Degen L, et al. Long-term budesonide maintenance treatment is partially effective for patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2011;9(5):400-409.e401. Dellon ES, Woosley JT, Arrington A, et al. Rapid Recurrence of Eosinophilic Esophagitis Activity After Successful Treatment in the Observation phase of a Randomized, Double-Blind, Double-Dummy Trial. Clin Gastroenterol Hepatol. 2020;18(7):1483-1492.e1482. | Intervention (inappropriate formulation) |
Miehlke S, Hruz P, Vieth M, et al. A randomized, double-blind trial comparing budesonide formulations and dosages for short-term treatment of eosinophilic esophagitis. Gut. 2016;65(3):390-399. | Study design (not a phase III or phase IV RCT) |
Lucendo AJ, Miehlke S, Schlag C, et al. Efficacy of budesonide Orodispersible Tablets as Induction Therapy for Eosinophilic Esophagitis in a Randomized Placebo-Controlled Trial. Gastroenterology. 2019;157(1):74-86.e15. | Study population |
De Heer J, Miehlke S, Rosch T, et al. Histologic and Clinical Effects of Different Topical Corticosteroids for Eosinophilic Esophagitis: Lessons from an Updated Meta-Analysis of Placebo-Controlled Randomized Trials. Digestion. 2020:1-9. | Systematic Review |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 20: Subgroup Analyses for the Freedom of Treatment Failure After 48 Weeks of Treatment in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Path to remission, n/N (%) | ||
BUL-1/EEA | 20/22 (90.9) | 0/22 (0.0) |
BUL-2/EER OLI phase | 30/46 (65.2) | 3/46 (6.5) |
Localization of inflammation at Screening of either BUL-1/EEA or BUL-2/EER, n/N (%) | ||
Proximal esophagus | ||
No | 11/11 (100.0) | 1/7 (14.3) |
Yes | 39/57 (68.4) | 2/61 (3.3) |
Median esophagus | ||
Not evaluable | 0/0 (0) | 0/1 (0.0) |
No | 10/11 (90.9) | 0/3 (0.0) |
Yes | 40/57 (70.2) | 3/64 (4.7) |
Distal esophagus | ||
Not evaluable | 0/0 (0) | 0/1 (0.0) |
No | 2/3 (66.7) | 0/1 (0.0) |
Yes | 48/65 (73.8) | 3/66 (4.5) |
Extent of inflammation at Screening: Number of esophageal segments affected, n/N (%) | ||
1 | 7/7 (100.0) | 0/3 (0.0) |
2 | 9/11 (81.8) | 1/7 (14.3) |
3 | 34/50 (68.0) | 2/58 (3.4) |
Concomitant use of PPIs during the DB phase, n/N (%) | ||
No | 37/52 (71.2) | 2/59 (3.4) |
Yes | 13/16 (81.3) | 1/9 (11.1) |
History of allergic diseases, n/N (%) | ||
No | 9/14 (64.3) | 1/18 (5.6) |
Yes | 41/54 (75.9) | 2/50 (4.0) |
Time since first symptoms of EoE at DB baseline, n/N (%)a | ||
Not evaluable | 0/0 (0) | 0/2 (0.0) |
< median | 21/27 (77.8) | 2/41 (4.9) |
> median | 29/41 (70.7) | 1/25 (4.0) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; PPI = proton pump inhibitor.
aMedian time since first symptoms of EoE was 9.2 years
Source: Clinical Study Report of the BUL-2/EEA trial.10
Table 21: Physician’s Global Assessment of EoE Activity in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Physician’s Global Assessment of EoE Activity (0-10)a | ||
Baseline, Mean (SD) | 1 (0.8) | 1 (0.9) |
End of treatment of the DB phase | ||
N | 66 | 66 |
Mean (SD) | 1 (1.8) | 5 (2.4) |
Absolute change from DB baseline to end of treatment of the DB phase | ||
N | 66 | 66 |
Mean (SD) | 0 (1.8) | 4 (2.4) |
b.i.d. = twice a day; CI = confidence interval; NRS = numerical rating scale.
aNRS 0 to 10: Lower values indicate more favourable outcomes.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Table 22: Patient’s Global Assessment of EoE Severity in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Patient’s Global Assessment of EoE Severity (0-10)a | ||
Baseline, Mean (SD) | 1 (0.8) | 1 (0.9) |
End of treatment of the DB phase | ||
N | 66 | 64 |
Mean (SD) | 1 (1.8) | 4 (2.6) |
Absolute change from DB baseline to end of treatment of the DB phase | ||
N | 66 | 63 |
Mean (SD) | 0 (2.0) | 3 (2.7) |
Percentage of patients with increase of ≥ 3 points from baseline of the DB phase in the PatGA concerning the severity of EoE symptoms (0-10 NRS) at end of treatment of the DB phase, n (%) | 5 (7.4) | 34 (50.0) |
Percentage of patients with PatGA NRS 2 points or less at end of treatment of the DB phase, n (%) | 60 (88.2) | 22 (32.4) |
b.i.d. = twice a day; CI = confidence interval; NRS = numerical rating scale.
aNRS 0 to 10: Lower values indicate more favourable outcomes.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Table 23: Endoscopy Outcomes in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Change from baseline of the DB phase in total modified EEsAI endoscopic instrument scorea | ||
Baseline, Mean (SD) | 1 (1.1) | 1 (1.0) |
End of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 1 (1.2) | 4 (1.8) |
Absolute change from DB baseline to end of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 0 (1.4) | 3 (1.9) |
Percentage of patients with all grades ‘0’ in modified EEsAI endoscopic instrument score at end of treatment of the DB phase, n (%)b | ||
Baseline | 34 (50.0) | 35 (51.5) |
End of treatment of the DB phase | 36 (52.9) | 4 (5.9) |
Change from baseline of the DB phase in the ‘inflammatory signs’ of the modified EEsAI endoscopic instrument scorec | ||
Baseline, Mean (SD) | 0 (0.6) | 0 (0.6) |
End of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 0 (0.9) | 3 (1.1) |
Absolute change from DB baseline to end of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 0 (1.0) | 2 (1.2) |
Change from baseline of the DB phase in the ‘fibrotic signs’ of the modified EEsAI endoscopic instrument scorec | ||
Baseline, Mean (SD) | 1 (0.7) | 0 (0.6) |
End of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 0 (0.6) | 1 (1.1) |
Absolute change from DB baseline to end of treatment of the DB phase | ||
N | 65 | 65 |
Mean (SD) | 0 (0.7) | 1 (1.0) |
Endoscopist's overall assessment of EoE activity: Percentage of patients with 'none or only mild' endoscopic findings | ||
Baseline | 67 (98.5) | 67 (98.5) |
End of treatment of the DB phase | 64 (94.1) | 24 (35.3) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; EEsAI = Eosinophilic Esophagitis Activity Index
aRange 0 to 9: Lower values indicate more favourable outcomes.
bAll features with grade = 0 is 'Yes' if the total modified EEsAI endoscopic instrument score is 0. It is 'No' if the total modified EEsAI endoscopic instrument score exceeds 0. Otherwise, it is 'not evaluable'.
cRange 0 to 4: Lower values indicate more favourable outcomes.
dThe endoscopist gave an overall assessment of EoE activity based on the general appearance of all endoscopic EoE findings by choosing 1 of the following responses: none, mild, moderate, severe.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Table 24: Deep Disease Remission in Study BUL-2/EER
End point | Budesonide 0.5 mg b.i.d. (N = 68) | Placebo (N = 68) |
|---|---|---|
Percentage of patients in deep disease remission, i.e., deep clinical, deep endoscopic, and histologic remission (based on the peak number of eos per HPF, i.e., <15 eos/HPF), at end of treatment of the DB phasea | ||
Number of patients contributing to the analysis | 68 | 68 |
Number (%) of patients who had not had a treatment failure after 48 weeks of treatment | 27 (39.7) | 0 |
Difference in proportions: Budesonide vs. placebo (97.5% CI)b | 39.7% (26.4 to 53.0) | Reference |
P valuec | < 0.0001 | Reference |
Subgroup analyses for deep disease remission | ||
Path to remission, n/N (%) | ||
BUL-1/EEA | 13/22 (59.1) | 0/22 (0.0) |
BUL-2/EER OLI phase | 14/46 (30.4) | 0/46 (0.0) |
Localization of inflammation at Screening of either BUL-1/EEA or BUL-2/EER, n/N (%) | ||
Proximal esophagus | — | — |
No | 7/11 (63.6) | 0/7 (0.0) |
Yes | 20/57 (35.1) | 0/61 (0.0) |
Median esophagus | — | — |
Not evaluable | 0/0 (0) | 0/1 (0.0) |
No | 5/11 (45.5) | 0/3 (0.0) |
Yes | 22/57 (38.6) | 0/64 (0.0) |
Distal esophagus | — | — |
Not evaluable | 0/0 (0) | 0/1 (0.0) |
No | 1/3 (33.3) | 0/1 (0.0) |
Yes | 26/65 (40.0) | 0/66 (0.0) |
Extent of inflammation at Screening: Number of esophageal segments affected, n/N (%) | ||
1 | 3/7 (42.9) | 0/3 (0.0) |
2 | 7/11 (63.6) | 0/7 (0.0) |
3 | 17/50 (34.0) | 0/58 (0.0) |
Concomitant use of PPIs during the DB phase, n/N (%) | ||
No | 17/52 (32.7) | 0/59 (0.0) |
Yes | 10/16 (62.5) | 0/9 (0.0) |
No | 4/14 (28.6) | 0/18 (0.0) |
Yes | 23/54 (42.6) | 0/50 (0.0) |
Time since first symptoms of EoE at DB baseline, n/N (%)d | ||
Not evaluable | 0/0 (0) | 0/2 (0.0) |
< median | 11/27 (40.7) | 0/41 (0.0) |
> median | 16/41 (39.0) | 0/25 (0.0) |
b.i.d. = twice a day; CI = confidence interval; DB = double-blind; EoE = eosinophilic esophagitis; PPI = proton pump inhibitor; vs. = versus.
aFor this analysis not evaluable results were set to 'No.'.
bBonferroni correction.
cNormal approximation test was used for testing.
dMedian time since first symptoms of EoE was 9.2 years.
Source: Clinical Study Report of the BUL-2/EEA trial.10
Appendix 4: Open-label Re-Induction BUL-2/EER
Note that this appendix has not been copy-edited.
Patients in the BUL-2/EER study with a clinical or histologic relapse (as defined in the primary end point) or experiencing a food impaction that needed endoscopic intervention during the double-blind treatment phase, were offered an open-label re-induction (OLRI) of remission or response treatment with budesonide 1 mg group twice daily for up to 6 weeks.
The proportion of patients presenting a resolution of symptoms (i.e., no or only minimal symptoms defined as NRS 2 or less for dysphagia and odynophagia, respectively) are presented in Table 25 below. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||| Patients who relapsed while receiving placebo had previously achieved clinicopathologic remission before being enrolled in study BUL-2/EER and assigned to the placebo group.37
Table 25: Percentage of patients with resolution of symptoms (i.e., no or minimal problems) in the week before OLRI Week 6 (LOCF)
End point | Budesonide 0.5 mg b.i.d. → Budesonide 1 mg b.i.d. | | Placebo → Budesonide 1 mg b.i.d. | |
|---|---|---|
Resolution of symptoms (i.e., no or minimal problems) in the week prior to analysis visit in the OLRI Week 6 (LOCF) | ||
Yes, n (%) | | | | |
No, n (%) | | | | |
b.i.d. = twice a day; NRS = Numerical Rating Scale (0 to 10 point scale, lower values indicate more favourable outcomes); OLRI = open-label re-induction.
Resolution of symptoms is defined as a severity of 2 points or less on 0- to 10-point NRS (7-day recall period) for dysphagia and a severity of 2 points or less on 0 to 10-point NRS (7-day recall period) for pain during swallowing in the week before visit.
Source: Sponsor provided additional information.37
Limitations
The main limitations of the OLRI phase of the BUL-2/EER trial arise from the open-label study design, lacking randomization. These may impact the subjective patient-reported outcomes such as clinical response could have been overestimated, given that patients were aware that they were receiving active treatment. Finally, it is worth noting that the sponsor provided these data and was not audited at the time these results were provided.
Appendix 5: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Dysphagia NRS
Pain During Swallowing NRS
EEsAI-PRO
PGA of EoE activity (NRS of 0 to 10)
PatGA concerning the severity of EoE symptoms
modSHS
EoE-QoL-A questionnaire
Modified EEsAI endoscopic instrument score (‘Inflammatory signs’ and ‘Fibrotic signs’)
Deep disease remission (Deep clinical remission, deep endoscopic, and histologic remission)
Findings
Table 26: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Dysphagia Numerical Rating Scale | 10-point Likert-type scale concerning the severity of dysphagia symptoms | No evidence found assessing the validity, reliability, and responsiveness of the scale | Not identified from the literature, although a score of 4 or more was used in the BUL-2/EER study to define relapse.10 |
Pain During Swallowing Numerical Rating Scale | 10-point Likert-type scale concerning the severity of pain when swallowing | No evidence found assessing the validity, reliability, and responsiveness of the scale | Not identified from the literature, although a score of 4 or more was used in the BUL-2/EER study to define relapse.10 |
EEsAI-PRO | 5-item scale to assess EoE activity | Construct and content validity was demonstrated34; no information on reliability and responsiveness | Not identified from the literature, although in the BUL-2/EER study, a ≥ 20-point decrease from baseline was used as a response to treatment and remission was defined as EEsAI-PRO score of 20 or less.10 |
PGA | 10-point Likert-type scale for global assessment of patients’ EoE activity | No evidence found assessing the validity, reliability, and responsiveness of the scale | Not identified from the literature |
PatGA | 10-point Likert-type scale for global assessment of EoE symptoms | No evidence found assessing the validity, reliability, and responsiveness of the scale | Not identified from the literature |
Modified Short Health Scale | Generic 4-item HRQoL questionnaire | Validity, reliability, and responsiveness demonstrated in ulcerative colitis32 and Crohn disease,33 but not in EoE | Not identified from the literature |
Adult Eosinophilic Esophagitis Quality of Life Questionnaire | 5-dimension and 37-item questionnaire, refined to a 30-item questionnaire (24-item scale with a 6-question addendum for those on elimination diet therapies) | Validity, reliability, and responsiveness shown for the original version30; only construct validity assessed for the shorter version31 | Not identified from the literature |
Modified EEsAI endoscopic instrument score | Modified EEsAI to assess endoscopic characteristics (both fibrotic and inflammatory) of EoE using major and minor subscores (0-9) of fixed rings, exudates, furrows, edema, and crêpe paper esophagus | Interobserver agreement/reliability was demonstrated for the modified endoscopic scale.35 No evidence found assessing validity and responsiveness of the scale. | Not identified from the literature |
Deep Disease Remission | Three component assessment of disease remission consisting of deep clinical remission, deep endoscopic remission, and histologic remission based on scores from the Dysphagia NRS, Pain During Swallowing NRS, EEsAI endoscopic instrument subscores, and peak number of eosinophils | No evidence found assessing the validity, reliability, and responsiveness of the scales | Not identified from the literature |
EoE = eosinophilic esophagitis; EEsAI-PRO = Eosinophilic Esophagitis Activity Index Patient Reported Outcome; HRQoL = health-related quality of life; MID = minimal important difference; PatGA = Patient’s Global Assessment; PGA = Physician’s Global Assessment.
Dysphagia Numerical Rating Scale
The Dysphagia NRS is a 10-point rating scale where patients provide an assessment of the severity of dysphagia symptoms experienced in the past 24 hours or 7 days. The scale ranges from 0 to 10 (0 represents no trouble swallowing, 10 represents the most severe trouble swallowing). The Dysphagia NRS captures dysphagia symptoms associated with EoE only and not symptoms associated with cold, e.g., sore throat. Patients in the trial received the scale in the form of a diary, and daily ratings were used to calculate a weekly sum.10
No studies validating the Dysphagia NRS in patients with EoE were identified from the literature; neither was an MID found.
Pain During Swallowing Numerical Rating Scale
The Pain During Swallowing NRS is a 10-point rating scale where patients provide an assessment of the severity of pain during swallowing experienced in the past 24 hours or 7 days. The scale ranges from 0 to 10 (0 represents no pain during swallowing, 10 represents the most severe pain during swallowing).10
Evidence regarding the validity, reliability, and MID of the Pain During Swallowing NRS were not found for patients with EoE in the literature.
Clinical Relapse
In the BUL-2/EER study, clinical relapse was defined as experiencing dysphagia or pain during swallowing in the past 7 days (7-day recall period) of a severity of at least 4 points on a 0 to 10 NRS for dysphagia or pain during swallowing, respectively, confirmed by a severity of least 4 points on at least 1 day during the subsequent week on the respective 0 to 10 NRS for dysphagia or pain during swallowing (24-hours recall period).10 Evidence regarding the validity, reliability, and MID for clinical relapse were not found for patients with EoE in the literature.
The Eosinophilic Esophagitis Activity Index Patient Reported Outcome
The EEsAI-PRO score is used to assess EoE activity over a 7-day recall period in adult patients that consists of 5 items:
Frequency of trouble swallowing — Patients are asked the number of times they had trouble swallowing in the past week, and the regularity is scored in 4 increments ranging from never to daily, with higher frequency associated with a higher score.
Duration of dysphagia episodes — The duration of dysphagia episodes in the past 7 days is scored based on a 5-minute cut-off, with longer duration associated with a higher score.
Pain during swallowing — Patients are asked if they experienced pain when swallowing and are scored higher when pain was present.
VDQ — The VDQ score measures the occurrence of dysphagia induced by the virtual ingestion of 8 reference foods, each at different consistencies, which are graded on a scale of 0 to 3 (described in detail as follows).
Behavioural change strategies — This item evaluates the change in patients’ behaviour in response to specific foods with 8 different consistencies; it also has 3 sub-items that are scored separately, i.e., avoidance, modification and slow eating (AMS) (described in detail as follows).
The scores of each item are added to provide an overall score out of 100, with disease severity rated as remission (0 to 20), mild (21 to 40), moderate (41 to 65), and severe (66 to 100).10
Assessment of Validity
The EEsAI-PRO score was developed and validated in a clinical trial setting by Schoepfer et al., conducted in 3 phases.34 In the first phase, items for the PRO instrument were generated by patient input from a mixed-methods approach using open-ended patient symptom surveys, focus groups, and semi-structured patient interviews. In total, the PRO instrument consisted of 45 items on symptoms severity and behavioural adaptations, which were grouped into 5 domains: a general domain to assess sociodemographic characteristics, 2 symptom domains to address symptoms dependent and independent of food intake, a comorbidities domain, and a medication domain. Three different time periods were assessed for the optimum recall period of the PRO instrument, e.g., 24 hours, 7 days, and 30 days. For each recall period, patients were asked to provide a PatGA of EoE severity on an 11-point Likert scale, as described previously (where a score of 0 is defined as “no symptoms” and a score of 10 is defined as “most severe symptoms”). This was used as the gold standard and the main outcome in the trial.34
During the second phase, the prototype of the PRO instrument was assessed in a test sample of 153 patients with EoE in Switzerland and the US who completed the PatGA at study entry. The data obtained from the VDQ and AMS items were used to create a composite score. Using multivariable linear regression analysis in which the PatGA was used as the outcome, and responses to specific items in the PRO instrument as predictors, 7 PRO factors used to assess characteristics of dysphagia, behavioural adaptations to living with dysphagia, and pain while swallowing accounted for 67% of the variation in patients’ assessment of disease severity. After grouping the 3 AMS items, 5 variables were selected for the final instrument. In terms of recall period, the majority of patients (> 70%) indicated that 7 days was the best recall period.34
Finally, the PRO instrument and PRO score were tested in an independent validation sample of 120 adult patients with EoE. By comparing the PRO score with the PatGA in the validation sample, it was shown that the EEsAI-PRO score for the 7-day recall period predicted 65% of the variability in PatGA, closely reflecting the results obtained in the test set. The EEsAI-PRO score showed construct validity with the PatGA based on high agreement between the scales. Patients required a median of 8 minutes to complete the questionnaire (range of 4 to 10 minutes), and rated a median difficulty of 1 on an 11-point Likert scale (where 0 stands for “no difficulties at all” and 10 stands for “very difficult”) in response to the question, “How difficult was it for you to complete this questionnaire?” Content validity was assessed by asking patients if the scale adequately measured the complaints they had or currently experienced due to EoE, with responses mapped on an 11-point Likert scale (where 10 stands for “perfectly” and 0 stands for “not at all”). The median response from patients was a score of 8 (range of 4 to 10). An assessment of reliability was not done.34
MID
A response to treatment was defined by the authors of the BUL-2EER trial as a 20-point or more decrease in the EEsAI-PRO score from baseline, however, it is not clear how this threshold was estimated.10
Physician’s Global Assessment of EoE Activity
In this scale, physicians are asked to provide an overall assessment of the patients’ EoE activity and severity taking into consideration the symptoms, endoscopy, histology, and laboratory markers. The EoE activity is rated on a 10-point scale, ranging from 0 (inactive EoE) to 10 (most active EoE).10
Evidence of validity and reliability as well as MID were not found from the literature.
Patient’s Global Assessment Concerning the Severity of EoE Symptoms
The PatGA scale evaluates the severity of EoE symptoms from a patient’s perspective. Patients were asked to rate the severity of their EoE symptoms in the past 7 days on a scale that ranges from 0 to 10 (0 represents no symptoms and 10 represents the most severe symptoms).10
Evidence of validity and reliability as well as MID were not found for the PatGA from the literature.
Patient’s Quality of Life: Modified Short Health Scale
The modSHS is a slightly modified form of the Short Health Scale (SHS). The SHS demonstrated discrimination validity, reliability (including internal consistency and test-retest reliability), and responsiveness in gastrointestinal conditions such as ulcerative colitis and Crohn disease.33 To be used in the BUL-1/EEA trial, the SHS was modified by replacing the terms with respect to the underlying disease in questions (1) to (3), i.e., “bowel” replaced by the term “esophageal.”10
The modSHS is a simplified 4-item questionnaire, representing each of 4 health dimensions: symptom burden, social function, disease-related worry, and general well-being. The patient answers 4 questions that assess the effects of esophageal disease on the patient’s QoL.10
Patients respond to each of the following questions representing the 4 health dimensions, which is scored on a scale of 0 to 100.
the severity of the symptoms from esophageal disease (0 represents no symptoms, 100 represents very severe symptoms)
interference with activities in daily life due to esophageal problems (0 represents not at all, 100 represents interference to a very high degree)
worry caused by esophageal disease (0 represents no worry, 100 represents constant worry)
a general feeling of well-being (0 represents very good, 100 represents dreadful).
While the SHS demonstrated discrimination validity, reliability (including internal consistency and test-retest reliability), and responsiveness in gastrointestinal conditions such as ulcerative colitis32 and Crohn disease,33 a psychometric analysis of the modified version of the SHS in EoE was not found from the literature. Additionally, an MID was not identified for any of these conditions.
Adult Eosinophilic Esophagitis Quality of Life Questionnaire
The EoE-QoL-A is a self-reported questionnaire that was originally developed as a 5-dimension and 37-item symptom inventory for adult patients with EoE, and included symptoms of esophageal dysfunction, disease impact, and anxiety.30 A refined version with 24 items and a 6-question addendum was later developed for those on elimination diet therapies. It met the recommended FDA guideline for PRO development31 and was used in the BUL2/EER trial. The refined 30-item questionnaire (24-item scale with a 6-question addendum for those on elimination diet therapies) is categorized according the original 5 dimensions, listed here:
impact of the disease on eating patterns and diet (10 items)
social impact (4 items)
emotional impact (8 items)
disease anxiety (5 items)
swallowing anxiety (3 items).
Patients provide responses based on their life over the past week by checking the responses that best describe their experiences living with EoE. Each question had 5 answers ranging from 4, which corresponds to “does not describe their experiences at all,” to 0, which corresponds to “extremely describes their experiences.” An overall score and 5 subscale scores are generated based on the responses. Weighted average scores range from 0 to 4; higher scores indicate better QoL. Notably, there is a standard version (24 items) and a standard plus dietary restrictions version (30 items) of the EoE-QoL-A questionnaire; the latter is used for patients on elimination diet therapy. Since the dietary restrictions section is not applicable to all patients, a weighted average is calculated for the overall score and the 5 subscales by adding the value of the response for each item answered, then dividing by the total number of questions answered.10
Assessment of Validity and Reliability
The original 37-item version of the EoE-QoL-A version was evaluated for scale reliability, internal consistency, factor structure, and concurrent and convergent validity in 201 adult patients with EoE in the US.30 Patients were assessed for their current EoE symptoms, illness perceptions, psychological distress, and HRQoL based on the Esophageal Symptoms Questionnaire; the Brief Illness Perception Questionnaire and the Perceived Health Competence Scale; the Brief Symptom Inventory-18; and the Medical Outcomes Study Short Form-12, version 2, and the Centers for Disease Control and Prevention (CDC) Healthy Days Measure, respectively — all previously validated scales for the respective measures. Results from analyses of principal components yielded the 37-item, 5-factor structure, which explained 70% of the variance and showed excellent internal consistency (Cronbach alpha = 0.96, Guttman Split-half = 0.88). Excellent test-retest reliability was shown for the individual items (range of r = 0.54 to 0.88) and for the total scale (r = 0.86). Concurrent validity was supported by a moderate negative relationship with the number of unhealthy days as reported by the CDC-HRQoL-4 (r = − 0.41) and moderate positive relationship with HRQoL as measured by the Short Form 12 item (version 2) Health Survey (range of r = 0.43 to 0.52). Participants who were in remission scored statistically significantly higher on the EoE-QoL-A scale than those who were not, supporting evidence for discrimination validity. Finally, convergent validity for the Adult Eosinophilic Esophagitis Quality of Life instrument was demonstrated by moderate negative relationships with psychological distress and esophageal symptoms as measured by the Esophageal Symptoms Questionnaire and Brief Symptom Inventory 18 (range of r = − 0.37 to −0.57) and moderate positive relationships with illness perception measures as measured by the Brief Illness Perception Questionnaire and the Perceived Health Competence Scale (range of r = 0.44 to 0.73).30
The shorter and more refined 30-item version of the EoE-QoL-A scale was assessed for validity, usability, and acceptability by the same group of researchers, although the information were published in a conference proceeding.31 Construct validity and acceptability were confirmed via qualitative cognitive interviews (to assess the clarity, understandability, length, rhetoric, and potential variability in interpretation for each question) of 10 patients and item refinement. Based on interview data, 7 questions were deleted due to nearly unanimous agreement in lack of clarity, relevance, and/or repetitiveness with other questions within the survey. Eight questions were rephrased to minimize leading rhetoric and/or ambiguity in wording that resulted in extensive variability in interpretation. Finally, 6 questions were only reserved for patients on elimination diet therapies as the majority (80%) of patients not on such therapies found these questions to be irrelevant or not applicable. Even though other psychometric properties were not assessed, the authors reported that the final measure met the recommended guidelines for PRO development, allowing for assessment of the impact of EoE across multiple domains in both research and clinical settings.31
MID
An MID for the total score or the 5 domains was not reported by the authors or identified from the literature.
Modified Eosinophilic Esophagitis Activity Index Endoscopic Instrument Score
Endoscopic signs of EoE present as a variety of individually unique characteristics. The overall occurrence of endoscopic abnormalities was classified using the modified EEsAI endoscopic instrument score. The modified EEsAI endoscopic instrument scope differs from the original EEsAI as some esophageal features (feline esophagus and narrow-calibre esophagus) were removed due to poor agreement from practising clinicians.35 The modified EEsAI endoscopic instrument score is calculated as the sum of the modified major and minor subscores (total score range: 0 to 9) based on the presence of the following features:
Major Features:
Fixed rings (also referred to as concentric rings, corrugated esophagus, corrugated rings, ringed esophagus, trachealization)
Grade 0: none
Grade 1: mild (subtle circumferential ridges)
Grade 2: moderate (distinct rings that do not impair passage of a standard diagnostic adult endoscope [outer diameter 8 to 9.5 mm])
Grade 3: severe (distinct rings that do not permit passage of a diagnostic endoscope)
Exudates (also referred to as white spots, plaques)
Grade 0: none
Grade 1: mild (lesions involving 10% or less of the esophageal surface area)
Grade 2: severe (lesions involving more than 10% of the esophageal surface area)
Furrows (also referred to as vertical lines, longitudinal furrows)
Grade 0: absent
Grade 1: present
Edema (also referred to as decreased vascular markings, mucosal pallor)
Grade 0: absent (distinct vascularity present)
Grade 1: loss of clarity or absence of vascular markings
Stricture
Grade 0: absent
Grade 1: present
Minor Features:
Crêpe paper esophagus (mucosal fragility or laceration upon passage of diagnostic endoscope but not after esophageal dilation).
Grade 0: absent
Grade 1: present
The ‘fibrotic signs’ subscore (range: 0 to 4) consisted of the assessment of the following subscores: ‘Fixed rings’ and ‘Stricture,’ whereas the ‘inflammatory signs’ subscore (range: 0 to 4) consisted of the assessment of the following subscores: ‘Exudates,’ ‘Furrows,’ and ‘Edema.’
Assessment of Reliability
The overall occurrence of endoscopic abnormalities was classified using the modified EEsAI endoscopic instrument score, which was reported to have good interobserver agreement. The proposed system incorporated the grading of 4 major esophageal features (rings, furrows, exudates, edema) and the presence of additional features of narrow-calibre esophagus, feline esophagus, stricture, and crêpe paper esophagus. Using a series of endoscopic videos, 21 gastroenterologists were blinded to the histologic status of patients, and surveys were provided to assess the proposed system. Interobserver agreement between reviewers regarding assessment of endoscopic abnormalities was assessed and interpreted based on a combination of estimates of multi-rater κ and the proportion of pairwise agreement of 21 gastroenterologists for each video (210 total pairwise comparisons per video for each of the 25 videos = 5,250 total pairwise comparisons).35
Using the original grading system, poor agreement between gastroenterologists was seen for esophageal features of edema (κ = 0.23, 51% agreement) and feline esophagus (κ = 0.15, 68% agreement), fair agreement for narrow-calibre esophagus (κ = 0.30, 74% agreement), and moderate agreement for rings, furrows and exudates (κ = 0.38 to 0.46, 56% to 65% agreement). The modified grading system consisted of removal of poorly performing features, after which, the 4 major features of EoE (fixed rings, exudates, furrows, and edema; κ = 0.40 to 0.54, 71% to 81% agreement), and additional minor features of stricture and crêpe paper esophagus (κ = 0.52 and 0.58, 79% and 92% agreement) demonstrated good agreement.35
No studies validating the modified EEsAI endoscopic instrument score was identified in the literature search; neither was an MID found.
Deep Disease Remission
Deep disease remission consists of 3 components including deep clinical remission, deep endoscopic remission, and histologic remission, which are based on the peak number of eosinophils per HPF. Deep disease remission is fulfilled if all, deep clinical remission, deep endoscopic remission, and histologic remission are ‘Yes’ (based on the peak number of eosinophils at time of assessment). If at least 1 is ‘No,’ deep disease remission is also ‘No.’ Otherwise, deep disease remission is not evaluable.10
No studies validating deep disease remission and its components were identified from the literature, and an MID was not found.
Deep Clinical Remission
Deep clinical remission is ‘Yes’ if the NRS (24-hour recall period) scores for dysphagia and pain during swallowing are ‘0’ on each day prior the visit, respectively. If the diary values are missing for at most 2 days, and all remaining 5 days have a NRS score of ‘0,’ the criterion is fulfilled (‘Yes’). If at least 1 NRS score exceeds ‘0,’ deep clinical remission is not fulfilled (‘No’) (independent of the number of days with valid values). Otherwise, deep clinical remission at OLI Week 6, OLI Week 6 (LOCF), or DB baseline is not evaluable.10
Deep clinical remission is ‘Yes’ if the NRS (7-day recall period) for dysphagia and pain during swallowing is ‘0’ at the respective visit. If the NRS (7-day recall period) exceeds ‘0’ for dysphagia or for pain during swallowing, deep clinical remission at the visit is not fulfilled (‘No’). Otherwise, deep clinical remission is not evaluable.10
Deep Endoscopic and Histologic Remission
Deep endoscopic and histologic remission is fulfilled (‘Yes’) if the patient fulfills the following 2 criteria: Deep endoscopic and histologic remission. Deep endoscopic and histologic remission is ‘Yes’ if both, deep endoscopic remission and histologic remission are ‘Yes’ (criteria outlined below). If at least 1 of these is ‘No,’ deep endoscopic and histologic remission is also ‘No.’ Otherwise, deep endoscopic and histologic remission is not evaluable.10
Deep Endoscopic Remission
Deep endoscopic remission is fulfilled (‘Yes’) if the following modified EEsAI endoscopic instrument subscores meet the following criteria:
fixed rings = 'Grade 0: none' or 'Grade 1: mild'
exudates = 'Grade 0: none'
furrows = 'Grade 0: absent'
edema = 'Grade 0: absent'
If at least 1 of the above features exceeds the grading as given above, deep endoscopic remission at the respective visit is not fulfilled (‘No’). Otherwise, deep endoscopic remission at the respective visit is not evaluable.10
Histologic Remission
Histologic remission for deriving deep disease remission is defined by a peak eosinophil count of < 15 eos/HPF (Note: this differs from the definition of histologic remission based on the peak number of eos per mm2 HPF). If the peak eosinophil count is ≥ 15 eos/HPF, histologic remission for deriving deep disease remission is ‘No’ at the respective visit. Otherwise, histologic remission for deriving deep disease remission is not evaluable.10
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDEC
Canadian Drug Expert Committee
EoE
eosinophilic esophagitis
GERD
gastroesophageal reflux disease
ICER
incremental cost-effectiveness ratio
NIHB
Non-Insured Health Benefits
OLRI
open-label re-induction
PPI
proton pump inhibitor
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Budesonide orodispersible (Jorveza), 0.5 mg tablets |
Submitted price | Budesonide 0.5 mg orodispersible tablet: $4.6750 |
Indication | For the maintenance of clinico-pathological remission in adults with eosinophilic esophagitis (EoE) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | March 16, 2021 |
Reimbursement request | As per indication |
Sponsor | Avir Pharma Inc. |
Submission history | Previously reviewed: Yes Indication: Indicated for the induction of clinicopathologic remission in adults with EoE Recommendation date: October 28, 2020 Recommendation: Reimburse with clinical criteria and/or conditions |
EoE = eosinophilic esophagitis; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults diagnosed with EoE who were refractory to treatment with a PPI, and who achieved clinicohistologic remission after a 6- or 12-week induction treatment with budesonide |
Treatment | Budesonide orodispersible tablets 0.5 mg twice dailya |
Comparator | No maintenance treatment with budesonidea |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | Lifetime (46 years) |
Key data source | BUL-2/EER maintenance trial |
Submitted results for base case | Base Case: ICER = $28,806 per QALY ($50,502 incremental costs, 1.75 incremental QALYs) |
Key limitations |
|
CADTH reanalysis results | Given the structure of the sponsor’s model and the absence of clinical information for treatment of subsequent recurrence, CADTH was unable to estimate a revised base case. CADTH did examine the cost-effectiveness of budesonide for a single maintenance period up to the first recurrence (i.e., no re-induction) aligning with the available clinical evidence. In doing so, CADTH also corrected the sponsor’s model to address limitations with the calculation of utility values. CADTH estimated that budesonide maintenance therapy was associated with an ICER of $26,645 ($6,478 incremental costs, 0.24 incremental QALYs) compared to no maintenance therapy over a lifetime time horizon, in patients who were refractory to or who had experienced a recurrence on PPI therapy, and who had responded to initial induction therapy with budesonide when only a single maintenance period up to thefirst recurrence (i.e., no re-induction) was considered. CADTH considered shorter time horizons to address concerns related to overestimating the predicted clinical benefits from a single maintenance course of budesonide therapy in the absence of additional subsequent treatment; the ICER increased with shorter time horizons. While the ICER is lower than reported by the sponsor for CADTH’s reanalyses, the results are uncertain due to the limitations associated with the available clinical data and do not reflect how budesonide will likely be used in practice over multiple inductions and maintenance treatments. As a result, the cost-effectiveness of budesonide beyond the initial maintenance treatment is uncertain. |
CDEC = CADTH Canadian Drug Expert Committee; EoE = eosinophilic esophagitis; ICER = incremental cost-effectiveness ratio; PPI = proton pump inhibitor; QALY = quality-adjusted life-year.
aRe-induction treatment with budesonide 1 mg twice daily was allowed in the sponsor’s base case.
Conclusions
The BUL-2/EER maintenance trial demonstrated that budesonide was effective in prolonging remission in patients with eosinophilic esophagitis (EoE) who were refractory to proton pump inhibitor treatment and who had responded to 6 or 12 weeks of budesonide 1 mg twice daily induction therapy. The trial found that 73.5% of patients in the budesonide 0.5 mg group remained in clinicopathologic remission at 48 weeks compared to 4.4% of patients in the placebo group.1,2 Patients receiving budesonide maintenance therapy maintained a symptom-free state at a high rate compared to patients in the placebo group. No clinical data are available on time to subsequent recurrence.
Due to an absence of data, CADTH was unable to estimate the cost-effectiveness of maintenance therapy with budesonide in the full population represented by the Health Canada indication or in comparison to other current treatments for the maintenance of remission in patients with EoE. Although the sponsor attempted to model multiple recurrences and re-inductions over a patient’s lifetime, CADTH considered the available evidence to be insufficient to determine the clinical efficacy or cost-effectiveness of re-inductions of budesonide in either patients who experienced a recurrence on budesonide therapy or while not receiving maintenance therapy. There was also a lack of available data on the rate of subsequent recurrence after a successful re-induction for patients who had previously experienced a recurrence while receiving maintenance therapy.
CADTH made corrections to the sponsor’s base case. However, given the clinical uncertainty regarding the long-term efficacy of budesonide and its use to re-induce remission in patients after they experienced a recurrence while on budesonide, CADTH could not determine an estimate of the cost-effectiveness of budesonide for maintenance treatment for patients with EoE. CADTH was able to provide an estimate of the cost-effectiveness for budesonide for initial maintenance treatment following response to budesonide induction. However, this analysis does not reflect how budesonide will likely be used in practice over multiple inductions and maintenance treatments; it should be viewed in the context of the limitations associated with the clinical evidence.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups that participated in the CADTH review process. Feedback from clinician groups was not received for this review.
Two Canadian and 1 international patient group submissions were received, from the Gastrointestinal (GI) Society, Food Allergy Canada, and the EOS Network of the UK. A total of 7 patients with EoE, as well as 1 caregiver, were interviewed as part of the Food Allergy Canada submission while a public survey used in the EOS Network input received 39 completed forms. The patient groups reported that key pharmacological treatments for the maintenance of remission in EoE include off-label corticosteroids mixed in a liquid or slurry. Patients reported the difficulties involved in using corticosteroids in this manner, including difficulties interpreting verbal administration instructions, inability to know if the dose was consistent, swallowing multiple packages of artificial sweeteners daily at unknown risk, terrible taste, and side effects such as oral thrush. Other pharmacological options include proton pump inhibitors (PPIs); however, the majority of survey participants did not feel PPIs improved their quality of life. As EoE patients have high rates of atopic allergy-related disorders, dietary restriction therapy may also be used, although patients found following such restrictive diets challenging. Respondents to Food Allergy Canada’s survey rated their current therapies’ ability to manage their symptoms at an average of 6.7 out of 10, suggesting patients were partially satisfied with their current symptom management. All of these patients had made dietary changes, and most had undergone endoscopies. Three patients had received different forms of budesonide. Of patients responding to the EOS Network survey, 92% had been prescribed PPIs to manage symptoms, and 72% had tried corticosteroids. Of the 10 patients who had received Jorveza, 80% indicated it was an effective, simple, and convenient option that improved their symptoms and quality of life. Patients generally agreed that the benefits of Jorveza outweighed the side effects, although there were some unconfirmed comments about experiencing brittle hair and nails as well as pancreatitis, alongside reports of side effects from other treatments. For patients, improvement in symptom control and quality of life were of paramount importance. Patients in the EOS Network survey indicated that the most negatively affected areas of daily life included diet, work, social life, and travel. Patient organizations noted that the benefits of new and on-label therapies such as Jorveza for maintenance treatment of EoE could reduce the need for restricted/elemental diets; physician, dietitian, and hospital visits; medical procedures to remove impacted food or dilatation of the esophagus due to long-term stricture damage; and stress and anxiety due to inconsistent care.
Several aspects of the patient input were addressed in the sponsor’s model:
Improved quality of life associated with improved symptoms was incorporated into the model, as were reduced numbers of physician visits, dilatations, and food impactions. Adverse events resulting from treatments were associated with costs but not quality-of-life decrements.
Off-label comparators mentioned by patients and reported as partially satisfactory were not included in the sponsor’s model.
CADTH was unable to address the following concerns raised from stakeholder feedback:
The incorporation of off-label comparators providing some level of satisfactory symptom management, or the level of improvement due to budesonide compared to more difficult-to-use steroid treatments.
The cost-effectiveness of budesonide when used consistently, even after recurrences.
Economic Review
The current review is for budesonide orodispersible tablets (Jorveza) for the maintenance of clinicopathologic remission of EoE in adults.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis3 in which budesonide 0.5 mg orodispersible tablets (hereafter referred to as budesonide) were compared with no treatment in adult patients (aged 18 to 75 years) in confirmed clinicopathologic EoE remission at baseline following an induction treatment with budesonide 1 mg twice daily for 6 or 12 weeks, who had been refractory to PPI treatment, consistent with patients enrolled in the BUL-2/EER maintenance trial (Straumann et al., 2020).2 This population is different than the indicated population and the reimbursement request, which do not restrict the population to patients refractory to PPIs, or limit maintenance treatment with budesonide to those who achieved remission with budesonide induction.4 No data were submitted for patients who are not refractory to PPIs or who achieved remission without budesonide induction therapy.
The recommended dose of budesonide is 0.5 mg twice daily. At the submitted price of $4.68 per 0.5 mg tablet, the annual cost of maintenance therapy with budesonide is $3,413 per patient (Appendix 1). The sponsor allowed re-induction of budesonide after recurrence in the model; re-induction involved a dosage of 1 mg twice daily for 6 weeks, identical to the recommended dosage of initial induction with budesonide.4,5
For the base case, the sponsor estimated costs and quality-adjusted life-years (QALYs) for each treatment regimen from the perspective of a Canadian health care payer, over a lifetime time horizon. The model used a 1-week cycle and incorporated a 1.5% annual discount rate for both costs and QALYs.
Model Structure
The sponsor submitted a Markov state transition model with 4 health states: “responder”, “recurrence”, “nonresponder,” and “death” (Figure 1 in Appendix 3). All patients entered the model in the responder state, consistent with being in confirmed clinicohistologic remission. Each week, patients had a risk of entering the recurrence state, and then transitioned to the nonresponder state, where they remained until and unless they responded to re-induction therapy with budesonide, whereupon they re-entered the responder state. Modelled patients could repeat transitions between recurrence, re-treatment, and response for the remainder of the modelled time horizon. Patients could also move to the death state from any other state in the model.
Model Inputs
Patients entered the model at 36 years of age, with 84% of patients being male, consistent with the population of the BUL-2/EER maintenance trial.2 As EoE and budesonide therapy were not expected to affect mortality, patients had an annual risk of death consistent with age- and gender-matched mortality rates of the Canadian population.6
Efficacy within the first year of the model was based on Kaplan-Meier time-to-event curves reported in the BUL-2/EER trial2 (Figure 2). The remaining time horizon was exponentially extrapolated based on recurrences from weeks 1 to 50 of the trial for the budesonide 0.5 mg group, and from weeks 13 through 50 of the placebo group for no treatment (Figure 3). Patients on budesonide maintenance therapy who experienced recurrence immediately stopped treatment and waited a median of 12 weeks before visiting their gastroenterologist, at which point re-induction therapy with 1 mg budesonide twice daily for 6 weeks was initiated. The efficacy of re-induction with budesonide for all groups in the model was based on clinical resolution observed among patients in the placebo arm from the BUL-2/EER maintenance trial who undertook budesonide re-induction after experiencing a recurrence.
Patients receiving budesonide maintenance therapy were assigned an adherence status, falling into 3 categories: “adherent” patients would take budesonide forever unless they had a recurrence (22.5%); “at risk of nonadherence” patients were still currently taking their budesonide maintenance therapy (77.5%) but would cease to adhere at a median of 36 weeks, based on an asthma adherence study by Kang et al. (2013)7; and “nonadherent” patients were no longer taking maintenance therapy and were thus assumed to be at a risk of recurrence equivalent to that of patients taking no maintenance treatment. The sponsor included adverse events if they occurred in at least 5% of patients in either active treatment arm of the BUL-2/EER maintenance trial.
Health-related utilities for patients in the response state were consistent with age- and gender-matched norms reported for the general Canadian population,8 with an overall population average utility value of 0.869. For the recurrence and nonresponse states, a utility consistent with moderate gastroesophageal reflux disease (GERD; 0.67) was used as a proxy for active EoE, from a German and Swedish study. This utility was then made age- and gender-dependent by adjusting the responder health states downward, based on the relative reduction in utility for GERD compared to the weighted average of the modelled population. Adverse events were assumed not to affect utility values within the model.
Costs included the drug acquisition cost of budesonide. Additionally, patients in remission were assumed to visit their gastroenterologist once a year. Patients experiencing a recurrence saw their doctor an average of 12 weeks after the recurrence began and every 17 weeks thereafter until and unless they re-entered the remission state. Patients in remission also had an endoscopy once a year, while those in the nonresponse state did so every 17 weeks. Costs were also applied to adverse events. However, as all adverse events in the trial were considered mild, only fungal infections and asthma were associated with resource use, which included an extra physician visit ($77.20) and a fluconazole ($2.29) or salbutamol ($5.00) prescription, respectively. Modelled patients in recurrence or nonresponse could also experience a food impaction based on the 10-year probability that a patient would have at least 1 food impaction, as reported by Dellon et al. (2014)9 and converted to a weekly probability by the sponsor (0.07%). Of modelled patients with a food impaction, 1.73% experienced a perforation of the esophagus, based on a retrospective analysis by Sengupta et al. (2015).10 Food impactions were associated with a 2.6-day hospital stay ($4,613), while a perforation of the esophagus was associated with a 13.3-day stay, based on Ontario case costing data.3
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted a probabilistic analysis of 5,000 iterations. The results of the deterministic analysis were very similar to those of the probabilistic analysis. The probabilistic findings are reported below.
Base-Case Results
For the maintenance of clinicohistologic remission in adults with EoE who were refractory to PPIs, when compared to no maintenance therapy over a lifetime time horizon, the sponsor concluded that budesonide was associated with $50,502 in increased costs, yielding an additional 1.75 QALYs, for an incremental cost-effectiveness ratio (ICER) of $28,806 per QALY (Table 3). More details can be found in Table 10.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no maintenance ($/QALY) |
|---|---|---|---|---|---|
No maintenance | 26,913 | Reference | 23.69 | Reference | Reference |
Budesonide maintenance | 77,414 | 50,502 | 25.44 | 1.75 | 28,806 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Adapted from sponsor’s pharmacoeconomic submission (Table 12).3
Sensitivity and Scenario Analysis Results
The sponsor conducted a series of 1-way sensitivity analyses, with results being most affected by varying the health utility for nonresponders or those experiencing a recurrence, varying the recurrence rate post-induction, and varying the rate at which patients responded to re-inductions with budesonide. The utility assumed for nonresponders had the largest impact on the ICER.
The sponsor also conducted a number of scenario analyses by shortening the time horizon, assuming all patients were adherent long-term, allowing for only 1 or 0 re-inductions, varying the waiting time after a recurrence, varying the utility for active EoE, varying the discount rate, and altering patient starting age to 20 or 65 years. The ICER for budesonide remained below $50,000 per QALY for most scenarios (range = $20,822 to $55,014 per QALY), with the exception of the 5-year time horizon and when Crohn disease was used as a proxy for active EoE.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
Modelled population differs from the Health Canada indication: To enroll in the BUL-1/EEA and BUL-2/EER trials, patients had to have been refractory to treatment with a PPI for 4 to 8 weeks. Patients with PPI-responsive esophageal eosinophilia (i.e., a typical EoE symptom presentation, where GERD is diagnostically excluded, and who demonstrated a clinicopathologic response to PPIs) were excluded from the trials. Health Canada approved budesonide without restriction on prior PPI use. There is an evidence gap between the population in which data exist and the broader, indicated population. No PPIs currently available in Canada have been approved by Health Canada for the treatment of EoE, and the positioning of PPIs in the treatment algorithm for EoE is not clear. Furthermore, the diagnosis of EoE has evolved since the trials began. Recent international clinical guidelines have classified PPIs as a treatment for EoE rather than as a diagnostic criterion and have not recommended that patients try a PPI first and then switch to topical corticosteroids, but rather that either PPIs or topical corticosteroids be first-line pharmacological treatment.
Given the lack of clinical data for the Health Canada–indicated population, CADTH was unable to conduct reanalyses to adjust for this limitation. An estimate of cost-effectiveness is available only for patients who received both budesonide induction and maintenance therapy after they were refractory to 4 to 8 weeks of PPI therapy. Furthermore, patients in the BUL-2/EER trial could receive up to 12 weeks’ treatment in the induction phase, which does not align with the criteria recommended for reimbursement by the CADTH Canadian Drug Expert Committee (CDEC) for budesonide for induction. The implications of these population differences on the cost-effectiveness are unknown.
Efficacy of budesonide re-induction unknown: At the time of review, the BUL-2/EER maintenance trial was not able to demonstrate the efficacy of re-induction with budesonide after a recurrence of EoE symptoms, and multiple inductions with budesonide are not the current standard of care in Canada. The sponsor assumed that modelled patients in both the budesonide maintenance and no-treatment groups who experience recurrence would undergo a re-induction with budesonide, in effect comparing budesonide maintenance and on-demand budesonide re-induction therapy with on-demand budesonide re-induction therapy alone. The response rate of this re-induction, regardless of the number of previous inductions, was assumed to be equivalent to that observed during the open-label re-induction (OLRI) phase of the BUL-2/EER maintenance trial, in which 94.7% of 57 placebo patients who had experienced a recurrence clinically responded to a 6-week open-label re-induction with budesonide. At the request of CADTH, the sponsor provided early, unaudited data from the OLRI phase of the BUL-2/EER maintenance trial suggesting that 22 of 25 patients who had received budesonide maintenance had responded to a 6-week re-induction with budesonide. While there is substantial uncertainty associated with these data, the clinical experts consulted by CADTH did not agree that patients who experienced a recurrence while on budesonide maintenance therapy would respond to re-induction with budesonide 1 mg tablets at rates equivalent to those not taking maintenance therapy. There are no data to inform re-induction response rates for subsequent relapses (i.e., more than 1 re-induction). As 98% of incremental QALYs in the sponsor’s base case come from the extrapolated period (i.e., not within the first year of the model), the sponsor’s results are highly uncertain.
CADTH excluded budesonide re-induction in reanalyses, as insufficient data exist to inform the rate at which patients on either budesonide or no maintenance therapy would respond to such re-induction.
CADTH assumed that 94.7% of patients receiving no maintenance treatment and 88% of budesonide patients would respond to re-induction therapy in scenario analyses.
Duration of remission after re-induction and number of subsequent recurrences are unknown: No data were available to inform the duration of remission before recurrence that would be experienced by patients who re-induced with budesonide after having at least once recurrence while taking budesonide maintenance therapy or no maintenance therapy. Furthermore, the number of subsequent recurrences over time and any changes in time to recurrence are unknown.
CADTH excluded budesonide re-induction in reanalyses, as insufficient data exist to inform the rate at which patients on either budesonide or no maintenance therapy would relapse after such re-induction.
Relevant comparators were not considered: The sponsor compared budesonide maintenance with no maintenance treatment using the placebo group of the BUL-2/EER trial as a proxy. However, there are therapies currently used in Canada for the maintenance treatment of EoE; the availability of budesonide may displace or supplement such therapies. These include dietary modifications, PPIs, and swallowed corticosteroids designed for inhalation, such as fluticasone and budesonide.
CADTH was unable to conduct reanalyses to account for this limitation.
Recurrence and nonresponder utility underestimated: The sponsor’s choice of health utility value for the recurrence and nonresponder states was from a population of German and Swedish patients studied in 1999 to 2000.11 While the experts consulted by CADTH agreed that moderate GERD is a reasonable proxy for estimating the utility of active EoE, CADTH reviewers did not consider it appropriate to directly compare the utility value for moderate GERD of 0.67 from Kartman et al. (2004)11 with the modelled utility for patients in the response state, who were assumed to have an equivalent utility to age- and sex-matched Canadian general population norms (modelled mean responder utility = 0.869)8 The sponsor calculated that being in a recurrence or non-responding state was associated with a 22.9% (1 to 0.67/0.869) reduction in utility. However, the mean utility of not having heartburn from Kartman et al. (2004) was 0.84, lower than that modelled by the sponsor for Canadian EoE patients in the response state.
CADTH calculated the relative utility of having moderate GERD compared with not having heartburn within Kartman et al. (2004)11 to be a 20.2% reduction (1 to 0.67/0.84) in health utility, and thus applied that reduction to estimate the utility of being in the recurrence or nonresponse states relative to the responder state for modelled EoE patients.
CADTH also identified limitations in the model logic, which occurred when changes to various parameters resulted in outcomes that did not meet face validity (Table 4, Appendix 2, and Appendix 4).
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined in Table 4, along with other key assumptions made by the sponsor and appraised by CADTH.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Key Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Recurrence rates are based on Kaplan-Meier time-to-recurrence data from BUL-2/EER trial in the first 50 weeks, with an exponential extrapolation based on weeks 0 to 50 for the budesonide maintenance groups and 13 to 50 of the placebo group for no maintenance. | Uncertain. Recurrence rates in the real world after 50 weeks may not match the sponsor’s extrapolated predictions; however, halving and doubling the assumed recurrence rates in each group did not have a large impact on the ICER. |
All patients enter the model at 36 years of age, the mean baseline age in the BUL-2/EER maintenance trial. | Inappropriate, as patients in clinical practice will be of various ages at induction and subsequent maintenance, with younger patients associated with higher incremental costs and QALYs over their lifetime. However, as this is expected to increase the variability but not the mean ICER, CADTH did not add a distribution for patient age. |
Quality of life of responders is equivalent to that of the general population. | Acceptable |
Mortality is equivalent to that of the general population. | Acceptable |
Patients begin budesonide maintenance therapy after successful induction with budesonide. | Acceptable. Some patients in clinical practice may start budesonide maintenance therapy after achieving remission with another therapy; however, the clinical experts consulted by CADTH believed this would be a small population who would mostly be switching from other forms of oral steroid maintenance therapy to the more convenient tablet form. |
Adverse events are not associated with quality-of-life decrements. | Inappropriate. Patients experiencing fungal infections, asthma exacerbations, endoscopies, food impactions, and perforation of the esophagus would all have lower quality of life for the duration of their adverse event or procedure. As these events are relatively fleeting compared to the lifetime time horizon, it is unlikely that including them would have a large impact on the ICER, and thus CADTH did not attempt to incorporate the QoL impact of adverse events. Including a QoL decrement for adverse events and endoscopies would slightly favour budesonide maintenance therapy (incremental QALYs would be slightly lower). |
77.5% of patients would become nonadherent after a median 36 months on treatment, based on an asthma study. | Uncertain. While the clinical experts consulted by CADTH agreed that a proportion of patients would become nonadherent when their symptoms resolved, they were not convinced that an asthma population would appropriately reflect the proportion and rate at which patients would become nonadherent in an EoE population. The sponsor tested a 100% adherence rate, which did not substantially change the ICER. CADTH tested both 50% and 100% adherence rates in scenario analyses and also determined the impact was minimal on the ICER. |
EoE = eosinophilic esophagitis; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; QoL = quality of life.
CADTH Reanalyses of the Economic Evaluation
Due the limitations with the submitted data and model, CADTH was unable to determine a base-case estimate for the cost-effectiveness of budesonide as maintenance therapy in the full population indicated by Health Canada, including patients who are not refractory to PPIs, nor to compare budesonide with other forms of maintenance therapy for EoE currently in use in Canada.
Corrected Base-Case Results
CADTH did note some base assumptions with the sponsor’s model that either did not align with best practice or did not align with CADTH’s CDEC’s prior recommendation.5 As a result, CADTH undertook 2 reanalyses to revise the sponsor’s base case to address these limitations (Table 5).
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Re-inductions | Unlimited; patients allowed to re-induce upon recurrence as needed for length of time horizon | As induction with budesonide in patients with active EoE was considered separately,5 re-induction with budesonide after a recurrence of EoE was not considered part of maintenance therapy (in line with the prior CDEC recommendation) |
2. Utility in recurrence and nonresponse | Mean 0.67, consistent with mean found by Kartman et al. (2004)11 | Mean 0.69, consistent with mean found by Kartman et al. (2004) adjusted for difference in sponsor’s modelled responder utility compared to the Kartman et al. (2004) responder utility |
Revised estimate | — | Reanalyses 1 and 2 |
The results of these reanalyses are presented in Table 6. CADTH’s corrections to the sponsor’s base case — considering only maintenance therapy (no re-inductions) and a slightly higher utility for patients in the recurrence and nonresponse states — indicated that the use of budesonide maintenance therapy was associated with an additional 0.24 QALYs at an incremental cost of $6,478 over the lifetime time horizon, resulting in an ICER of $26,645 per QALY. This represents the cost-effectiveness estimate when only a single maintenance period up to the first recurrence (i.e., no re-induction) was considered.
In the reanalysis, 74.5% of incremental QALYs are accrued beyond the trial observation period, i.e., beyond 1 year. At a willingness to pay of $50,000 per QALY, 99.1% of iterations would be considered cost-effective. CADTH reviewers found there were insufficient data to estimate the cost-effectiveness of multiple re-inductions of budesonide when recurrences occurred on maintenance therapy, should maintenance therapy and re-inductions be considered together. As a result of the limitations identified, these results should be viewed with caution.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | No maintenance | 26,913 | 23.69 | Reference |
Budesonide maintenance | 77,414 | 25.44 | 28,806 | |
Correction 1 | No maintenance | 29,859 | 21.72 | Reference |
Budesonide maintenance | 36,353 | 21.99 | 23,606 | |
Correction 2 | No maintenance | 26,863 | 24.17 | Reference |
Budesonide maintenance | 77,515 | 25.72 | 32,584 | |
Revised estimate (reanalyses 1 and 2) | No maintenance | 29,922 | 22.43 | Reference |
Budesonide maintenance | 36,400 | 22.67 | 26,645 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH conducted additional scenario analyses exploring the effect of shortening the time horizon; allowing a single re-induction, allowing a single or unlimited re-inductions, assuming the unaudited, open-label data available from the BUL-2/EER maintenance trial; and assuming either 100% or 50% adherence to budesonide. Similar to the sponsor’s scenario analyses, most CADTH scenarios reported ICERs under $50,000 per QALY (range = $21,966 to $67,085 per QALY; Table 12). Given the limitations previously identified, these results should be viewed with caution. An additional analysis was included to acknowledge the CDEC recommendation for budesonide for induction therapy, which recommended a price reduction. When the change was applied to the revised estimate, this ICER was reduced to $16,919 per QALY.
Issues for Consideration
Budesonide nebules: In addition to swallowed fluticasone powder for inhalation, swallowed budesonide suspension for inhalation, available in nebules, has been used to treat EoE in Canada, including for maintenance therapy. According to clinical experts consulted by CADTH, the suspension is typically mixed with a sweetener or other vehicle to make it more palatable and to increase viscosity to prolong contact with the esophagus. Health Canada stipulates that pharmaceuticals should be compounded only if a product is unavailable and not solely for economic reasons12; however, some off-label use of budesonide nebules may continue despite the availability of budesonide tablets. At the Ontario Drug Benefit Formulary list price, budesonide nebules used in this manner are less expensive than the submitted price of budesonide tablets (Table 8).
Budesonide tablet for induction: CDEC recommended that budesonide be reimbursed for the induction of clinicopathologic remission in adults with EoE with certain conditions and criteria, including a maximum duration of 6 weeks’ treatment. CADTH’s appraisal of the economic evaluation identified substantial uncertainty surrounding the cost-effectiveness of budesonide, noting that a price reduction of up to 35% may be required to achieve an ICER of $50,000 per QALY. As a result, CDEC recommended reimbursement on the condition of a reduced price. The current review did not include the initial induction component (out of scope); the sponsor did consider re-induction within its submission, although this was revised in CADTH reanalyses.
Potential for off-label use of 1 mg budesonide for maintenance therapy: Although Health Canada approved only the 0.5 mg twice daily dosage of budesonide for maintenance therapy, a 1 mg twice daily dosage was included in the pivotal trial.1 The 1 mg twice daily dosage has been approved for maintenance therapy in other jurisdictions.14 The clinical experts consulted by CADTH stated that, if a patient experienced a recurrence on maintenance therapy with the 0.5 mg twice daily dosage, they would consider escalating the patient to the 1 mg twice daily dosage, particularly if the patient had experienced a recurrence more than once or had experienced a food impaction. At a current price of $5.50 per tablet,5,15 the annual cost of maintenance therapy with 1 mg twice daily budesonide would be $4,015 per patient, compared to $3,413 for the 0.5 mg twice daily dosage. Additionally, the clinical experts consulted by CADTH indicated that patients using off-label budesonide slurries sometimes take them once daily rather than twice and may prefer to continue such a schedule after switching to budesonide tablets. Once daily dosage of 1 mg budesonide tablets was not studied in clinical trials1 and is not approved by Health Canada4 but would be less expensive than twice daily dosage at either available tablet strength.
Biologic therapies are under development: The clinical experts consulted by CADTH noted that biologic therapies are currently being studied for the treatment of EoE and are likely to become available within the next few years. Biologic therapies are more costly (at the current dosage regimens) than any currently available pharmacological therapy for EoE, including budesonide tablets.
Overall Conclusions
The BUL-2/EER maintenance trial demonstrated that budesonide was effective in prolonging remission in EoE patients who were refractory to PPI treatment and who had responded to 6 or 12 weeks of budesonide 1 mg twice daily induction therapy. In the trial, 73.5% of patients in the budesonide 0.5 mg group remained in clinicopathologic remission at 48 weeks compared to 4.4% of patients in the placebo group.1,2 Patients receiving budesonide maintenance therapy maintained a symptom-free state at a higher rate than patients in the placebo group. No clinical data are available on time to subsequent recurrence.
Due to an absence of data, CADTH was unable to estimate the cost-effectiveness of maintenance therapy with budesonide in the full population represented by the Health Canada indication or in comparison with other treatments currently in use for the maintenance of remission in patients with EoE. Although the sponsor attempted to model multiple recurrences and re-inductions over a patient’s lifetime, CADTH considered the issue of re-induction to be separate from maintenance therapy and considered the available evidence to be insufficient to determine the clinical efficacy or cost-effectiveness of re-inductions of budesonide in either patients who experienced a recurrence on budesonide therapy or while not receiving maintenance therapy. There was also a lack of available data to inform the rate at which patients who had previously experienced a recurrence on maintenance therapy would experience a recurrence again after a successful re-induction.
CADTH made corrections to the sponsor’s base case, which suggested that the use of budesonide maintenance therapy was associated with an ICER of $26,645 per QALY when compared with no maintenance therapy in patients who were refractory to PPI therapy or who had experienced a recurrence while receiving PPI therapy and who had responded to an induction with budesonide when considering only 1 maintenance period and no subsequent re-inductions. When shorter time horizons were considered in order to minimize the impact of removing re-inductions in the modelled clinical pathway, the ICER increased as the time horizon decreased. However, given the clinical uncertainty regarding the long-term efficacy of budesonide and its use to re-induce remission in patients after they experienced a recurrence while on budesonide, CADTH could not determine an estimate of the cost-effectiveness of budesonide for maintenance treatment for patients with EoE. CADTH is also cognizant that this analysis does not reflect how budesonide will likely be used in practice over multiple inductions and maintenance treatments. CADTH considers that these results should be interpreted in the context of the CADTH clinical review findings, which note that an effect on the long-term consequences of the disease could not be elucidated due to the low number of events observed in the BUL-2/EER trial, although patients receiving budesonide had a superior experience of their quality of life than those who did not receive budesonide. If the price reduction recommended by CDEC for budesonide in the induction phase is achieved, budesonide is more likely to be cost-effective in the maintenance phase.
Given the lack of comparative clinical data for treatments currently used for the induction and maintenance of EoE, the cost-effectiveness of budesonide compared with these treatments is unknown. At the submitted price, the drug acquisition cost of budesonide for maintenance therapy is $3,413 per patient per year, which is more expensive than other pharmacological therapies currently in use in Canada for the treatment of EoE.
References
1.Clinical Study Report: BUL-2/EER. Double- blind, randomized, placebo-controlled, phase III study on the efficacy and tolerability of a 48-week treatment with 2 different doses of budesonide effervescent tablets vs. placebo for maintenance of clinico-pathological remission in adult patients with eosinophilic esophagitis [CONFIDENTIAL sponsor's report]. Freiburg (DE): Dr. Falk Pharma GmbH; 2019 May 15.
2.Straumann A, Lucendo AJ, Miehlke S, et al. Budesonide Orodispersible Tablets Maintain Remission in a Randomized, Placebo-Controlled Trial of Patients With Eosinophilic Esophagitis. Gastroenterology. 2020;159(5):1672-1685.e1675.
3.Pharmacoeconomic evaluation. In: Drug Reimbursement Review sponsor submission: Jorveza (budesonide), 0.5mg and 1 mg orodispersible tablets.. Blainville (QC): AVIR Pharma Inc; 2020 Nov 16..
4.Jorveza (budesonide): 0.5mg and 1 mg orodispersible tablets [product monograph]. Blainville (QC): AVIR Pharma Inc.; 2019 May 15.
5.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: Jorveza Ottawa (ON): CADTH; 2020: https://cadth.ca/sites/default/files/cdr/complete/SR0634%20Jorveza%20-%20CDEC%20Final%20Recommendation%20October%2030%2C%202020_for%20posting.pdf. Accessed 2021 Feb 04.
6.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2020 Dec 29.
7.Kang MG, Kim JY, Jung JW, et al. Lost to follow-up in asthmatics does not mean treatment failure: causes and clinical outcomes of non-adherence to outpatient treatment in adult asthma. Allergy Asthma Immunol Res. 2013;5(6):357-364. PubMed
8.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
9.Dellon ES, Kim HP, Sperry SL, Rybnicek DA, Woosley JT, Shaheen NJ. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc. 2014;79(4):577-585 e574. PubMed
10.Sengupta N, Tapper EB, Corban C, et al. The clinical predictors of aetiology and complications among 173 patients presenting to the Emergency Department with oesophageal food bolus impaction from 2004-2014. Aliment Pharmacol Ther. 2015;42(1):91-98. PubMed
11.Kartman B, Gatz G, Johannesson M. Health state utilities in gastroesophageal reflux disease patients with heartburn: a study in Germany and Sweden. Med Decis Making. 2004;24(1):40-52. PubMed
12.Policy on manufacturing and compounding drug products in Canada (POL-0051). Ottawa (ON): Health Canada; 2009: https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/good-manufacturing-practices/guidance-documents/policy-manufacturing-compounding-drug-products.html#a51. Accessed 2021 Apr 20.
13.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2020; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2020 Dec 20.
14.Product Information: Jorveza (budesonide) (European Public Assessment Report). London (GB): European Medicines Agency; 2020 Jun 4: https://www.ema.europa.eu/en/medicines/human/EPAR/jorveza#product-information-section. Accessed 2021 Apr 20.
15.DeltaPA. [Ottawa (ON)]: IQVIA; 2020: https://www.iqvia.com/. Accessed 2020 Dec 20.
16.Budget Impact Analysis [CONFIDENTIAL sponsor's report]. In: Drug Reimbursement Review sponsor submission: Jorveza (budesonide), 0.5mg orodispersible tablets. Blainville (QC): AVIR Pharma Inc.; 2021.
17.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2018 to 2019. 2020: https://www.sac-isc.gc.ca/DAM/DAM-ISC-SAC/DAM-HLTH/STAGING/texte-text/nihb-Annual_Report_2018-19_1589921777815_eng.pdf. Accessed 2021 Feb 03.
18.Table: 17-10-0005-01 Population estimates on July 1st, by age and sex. Ottawa (ON): Statistics Canada; 2020: 10.25318/1710000501-eng. Accessed 2021 Feb 03.10.25318/1710000501-eng
19.Lucendo AJ, Miehlke S, Schlag C, et al. Efficacy of Budesonide Orodispersible Tablets as Induction Therapy for Eosinophilic Esophagitis in a Randomized Placebo-Controlled Trial. Gastroenterology. 2019;157(1):74-86.e15. PubMed
20.Saskatchewan Drug Plan: search formulary. 2020; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2020 Dec 20, 2020.
21.Gastro-oesophageal reflux disease and dyspepsia in adults: Investigation and management, Appendix A: Dosage information on proton pump inhibitors. [CG184]. 2014; https://www.nice.org.uk/guidance/cg184/chapter/Appendix-A-Dosage-information-on-proton-pump-inhibitors. Accessed 20 Dec 2020.
22.Navarro P, Arias A, Arias-Gonzalez L, Laserna-Mendieta EJ, Ruiz-Ponce M, Lucendo AJ. Systematic review with meta-analysis: the growing incidence and prevalence of eosinophilic oesophagitis in children and adults in population-based studies. Aliment Pharmacol Ther. 2019;49(9):1116-1125. PubMed
23.Lucendo AJ, Arias A, Molina-Infante J. Efficacy of Proton Pump Inhibitor Drugs for Inducing Clinical and Histologic Remission in Patients With Symptomatic Esophageal Eosinophilia: A Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol. 2016;14(1):13-22 e11. PubMed
24.Lucendo AJ, Molina-Infante J, Arias A, et al. Guidelines on eosinophilic esophagitis: evidence-based statements and recommendations for diagnosis and management in children and adults. United European Gastroenterol J. 2017;5(3):335-358. PubMed
25.Kapel RC, Miller JK, Torres C, Aksoy S, Lash R, Katzka DA. Eosinophilic esophagitis: a prevalent disease in the United States that affects all age groups. Gastroenterology. 2008;134(5):1316-1321. PubMed
26.Prescribed drug spending in Canada, 2019: A Focus on public drug programs. Ottawa, ON: Canadian Institute for Health Information (CIHI); 2019: https://www.cihi.ca/sites/default/files/document/pdex-report-2019-en-web.pdf. Accessed 2021 Jan 05.
Appendix 1: Cost Comparison Tables
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for Maintenance of Remission in Eosinophilic Esophagitis
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Budesonide (Jorveza) | 0.5 mg 1 mg | orodispersible tablet | 4.6750a 5.5000b | 1 mg daily, 0.5 mg in the morning and 0.5 mg in the evening | 9.35 | 3,413a |
Topical steroids | ||||||
Fluticasone propionate (Flovent, generic) | 50 mcg 125 mcg 250 mcg | HFA metered dose inhaler, 120 doses | 26.1000 45.0200 67.5300 | 500 to 1,000 mcg daily in divided doses, swallowed | 1.13 to 2.25 | 411 to 821 |
Proton pump inhibitors | ||||||
Dexlansoprazole (Dexilant) | 30 mg 60 mg | Delayed release cap | 2.2977b | 30 mg to 60 mg daily | 2.30 | 838 |
Esomepazole (generic) | 20 mg 40 mg | Delayed release tab or cap | 0.5500c | 20 mg to 40 mg dailyd | 0.55 | 201 |
Lansoprazole (generic) | 15 mg 30 mg | Delayed release cap | 0.5000 | 30 mg once or twice dailyd | 0.50 to 1.00 | 182 to 365 |
Omeprazole (generic) | 20 mg | tab, cap regular or delayed release | 0.2287 | 20 mg to 40 mg dailyd | 0.23 to 0.46 | 83 to 167 |
Pantoprazole (generic) | 20 mg 40 mg | Enteric coated tab | 0.1803c 0.1875 | 40 mg once or twice dailyd | 0.19 to 0.38 | 68 to 137 |
Rabeprazole (generic) | 10 mg 20 mg | Enteric coated tab | 0.0669 0.1338 | 20 mg once or twice dailyd | 0.13 to 0.27 | 47 to 95 |
Cap = capsule; mg = milligram; tab = tablet.
All prices are from the Ontario Drug Benefit Formulary (accessed Apr 2021),13 unless otherwise indicated, and do not include dispensing fees.
aSponsor’s submitted price.3 Re-induction with budesonide is as previously reported, 1 mg twice daily for 6 weeks, at a cost of $462.5
bIQVIA Delta PA wholesale price (accessed Apr 2021).15
cSaskatchewan Formulary list price (accessed Apr 2021).20
dStandard and double dose recommendations for the treatment of GERD or erosive esophagitis, as per individual product monographs and the 2014 NICE Clinical Guideline 184 Gastro-oesophageal reflux disease and dyspepsia in adults: investigation and management, Appendix A.21
Table 8: CADTH Cost Comparison Table for Eosinophilic Esophagitis, Other Budesonide Products
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Budesonide (Pulmicort Nebuamp, generic) | 0.125 mg/mL 0.250 mg/mL 0.500 mg/mL | Nebuamp suspension | 0.1714 0.4630 0.6839 | 2 mg daily, swallowed, in divided doses | 2.74 | 998 |
All prices are from the Ontario Drug Benefit Formulary (accessed Apr 2021),13 unless otherwise indicated, and do not include dispensing fees.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Submission is based on the best available data, but does not address the full Health Canada–indicated population, and thus does not fully address the reimbursement request “As per indication.” |
Model has been adequately programmed and has sufficient face validity | No | Depending on the analysis run, some model results did not meet face validity. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
![The figure denotes the model structure, including initial and subsequent induction and post-induction phases. Patients could respond or not. If they didn’t initially respond, they could not later respond. If they initially respond, patients can continue to respond until they experience a recurrence. After which, they can be retreated and respond or not, starting the process again.]](https://canjhealthtechnol.ca/index.php/cjht/article/download/sr0666r/version/175/342/1036/SR0666PE-fig01.png)
![The figure denotes the time to recurrence over the first year of the sponsor’s model, based on the clinical trial. It reports the proportion of patients in clinical remission at weekly timepoints for patients receiving budesonide 0.5 mg and placebo. The proportion of patients receiving budesonide 0.5 mg in clinical remission reduces steadily then remains stable at approximately 90% from week 11 onwards; the proportion of patients receiving placebo in clinical remission reduces quickly within the first 6 weeks, then slowly reduces until plateauing at approximately 40% at around 35 weeks.]](https://canjhealthtechnol.ca/index.php/cjht/article/download/sr0666r/version/175/342/1037/SR0666PE-fig02.png)
Figure 3: Time to Recurrence in Subsequent Years of the Sponsor’s Model
![The figure denotes the time to recurrence in subsequent years, as extrapolated within the sponsor’s model. It reports the proportion of patients in clinical remission at weekly timepoints for patients receiving budesonide 0.5 mg and placebo. The proportion of patients receiving budesonide 0.5 mg in clinical remission reduces steadily from approximately 90% at the end of Year 1 to approximately 20% at Year 10; the proportion of patients receiving placebo in clinical remission reduces steadily from approximately 40% at the end of Year 1 to 0% at Year 10.]](https://canjhealthtechnol.ca/index.php/cjht/article/download/sr0666r/version/175/342/1038/SR0666PE-fig03.png)
Source: Sponsor’s pharmacoeconomic submission, Figure 4.3
Detailed Results of the Sponsor’s Base Case
Table 10: Detailed Results of Sponsor’s Base-Case Analysis
Category | No maintenance | Budesonide | Incremental |
|---|---|---|---|
Total survival time (years) | 45.84 | 45.84 | 0 |
Years in remission | 13.66 | 26.60 | 12.94 |
Years in recurrence | 5.57 | 3.56 | −2.01 |
Years in nonresponder | 26.60 | 15.68 | −10.93 |
Number of recurrence/re-treatment loops | 16.26 | 10.46 | −5.81 |
Total costs ($) | 26,913 | 77,414 | 50,502 |
Drug costs ($) | 6,020 | 56,329 | 50,308 |
Adverse events ($) | 5,566 | 9,755 | 4,189 |
Resource costs ($) | 15,326 | 11,330 | −3,996 |
Total QALYs | 23.69 | 25.44 | 1.75 |
QALYs in remission | 9.60 | 17.26 | 7.66 |
QALYs in recurrence | 3.00 | 1.80 | −1.19 |
QALYs in nonresponder | 11.09 | 6.38 | −4.72 |
ICER | 28,806 | ||
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Reanalyses
Table 11: Detailed Results of Revised Estimate
Category | No maintenance | Budesonide | Incremental |
|---|---|---|---|
Total survival time (years) | 45.84 | 45.84 | 0 |
Years in remission | 1.14 | 2.59 | 1.46 |
Years in recurrence | 0 | 0 | 0 |
Years in nonresponder | 44.70 | 43.25 | −1.46 |
Number of recurrence/re-treatment loops | 0 | 0 | 0 |
Total costs ($) | 29,922 | 36,400 | 6,478 |
Drug costs ($) | 0 | 6,702a | 6,702 |
Adverse events ($) | 5,958 | 6,702 | 575 |
Resource costs ($) | 23,964 | 23,165 | −799 |
Total QALYs | 22.43 | 22.67 | 0.24 |
QALYs in remission | 1.00 | 2.20 | 1.20 |
QALYs in recurrence | 0 | 0 | 0 |
QALYs in nonresponder | 21.43 | 22.47 | −0.96 |
ICER | 26,645 | ||
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aCADTH noted that this cost may be less than expected, although the reanalysis accounts for discontinuation and assumes a continuation of effect after treatment stoppage in at least a proportion of patients.
Results of Scenario Analyses
Table 12: CADTH Scenario Analyses
Scenario | CADTH Revised Estimate | CADTH Scenario | ICER ($/QALY) |
|---|---|---|---|
Sponsor base case | 28,806 | ||
CADTH corrected base case | 26,645 | ||
Scenario Analyses | |||
Scenario A: 1-year time horizon | Time horizon: lifetime | Time horizon: 1 year | 35,939 |
Scenario B: 5-year time horizon | Time horizon: lifetime | Time horizon: 5 years | 30,553 |
Scenario C: 10-year time horizon | Time horizon: lifetime | Time horizon: 10 years | 28,262 |
Scenario D: Single re-induction allowed | No re-inductions | Single re-induction, efficacy consistent with placebo group of open-label BUL-2/EER OLRI phase data 94.7% | 23,630a |
Scenario E: Re-induction efficacy (single re-induction) | No re-inductions | Single re-induction consistent with open-label BUL-2/EER data for each group Budesonide: 88.0% No maintenance: 94.7% | 23,289a |
Scenario F: Re-induction efficacy (lifetime re-inductions) | No re-inductions | Lifetime re-inductions assuming open-label BUL-2/EER data Budesonide: 88.0% No maintenance: 94.7% | 67,085 |
Scenario G: 100% Adherence | 77.5% patients nonadherent | 0% patients nonadherent | 21,966 |
Scenario H: 50% Adherence | 77.5% patients nonadherent | 50% patients nonadherent | 23,601 |
Scenario I: Budesonide price | As submitted | Including 35% price reduction noted in CDEC recommendation for budesonide for induction | 16,919 |
ICER = incremental cost-effectiveness ratio; OLRI = open-label re-induction; QALY = quality-adjusted life-year.
aCADTH noted that there is uncertainty with the model logic. Lowering the re-induction efficacy in patients in the maintenance treatment group means that fewer people remain on budesonide maintenance therapy, making maintenance therapy appear more cost-effective.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
In the sponsor-submitted budget impact analysis (BIA),16 the sponsor assessed the inclusion of budesonide tablets for adults with EoE who are either refractory to treatment with a PPI, or who had relapsed on PPI maintenance therapy compared to no maintenance therapy. The BIA was conducted from a Canadian public drug payer perspective over a 3-year time horizon using an epidemiology-based model and included drug utilization costs. Key inputs to the BIA are documented in Table 15.
State the key assumptions:
Only patients who are refractory to treatment with a PPI, or who have a recurrence while on PPI maintenance therapy, will be eligible for budesonide tablets, therefore PPI maintenance therapy was not considered a comparator.
Only patients achieving clinic-histologic remission after budesonide tablet induction will be eligible for budesonide therapy.
The use of budesonide tablets for maintenance therapy does not alter the use of other medications (e.g., PPI therapy).
All patients are adherent, and claims are made every 30 days.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Canadian Population ≥ 18 years (excl. Quebec) | |
Annual population growth | |
Prevalence (Incidence) of EoE | 0.04% (0.01%)22 |
Percentage EoE patients refractory to treatment with PPI | 39.2%23 |
Percentage non-refractory to PPI patients who recur | 20.0%24 |
Percentage of eligible patients receiving budesonide tablet induction therapy | 100%, assumption |
Percentage of patients achieving clinic-histologic remission after induction with budesonide tablets | 57.6%19 |
Percentage eligible population under/over age 65 years | 87.9% (under 65), 12.1% (over 65)25 |
Percentage population covered by public drug plans under/over age 65 years | 31.4% (under 65), 91.5% (over 65)26 |
Number of patients eligible for drug under review | 1,410 / 1,634 / 1,862 |
Market Uptake (3 years) | |
Uptake (reference scenario) No maintenance therapy | 100% |
Uptake (new drug scenario) Budesonide tablets No maintenance therapy | 80% / 90% / 100% 20% / 10% / 0% |
Cost of treatment (per patient) | |
Cost of treatment per year, including weighted average markup and fees Budesonide tablets No maintenance | $3,793 $0 |
EoE = eosinophilic esophagitis; PPI = proton pump inhibitor.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base-case BIA revealed that the yearly incremental expenditures associated with the reimbursement of budesonide tablets, including dispensing fees and markups, for patients refractory to PPI treatment or who had relapsed on PPI maintenance therapy were expected to be $3,980,984 in Year 1, $5,072,966 in Year 2, and $6,421,948 in Year 3, for a 3-year cumulative total budget impact of $15,385,899. When dispensing fees and markups are excluded, the sponsor’s model reports an expected budgetary cost of $3,854,951 in year 1, $5,026,067 in year 2, and $6,362,703 in year 3, for a 3-year cumulative total of $15,243,721. The factors which had the greatest impact on the BIA included: the exclusion of patients not shown to be refractory to PPIs or who had not relapsed on PPIs, and the assumed market share for budesonide tablets.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Modelled population differs from the Health Canada indication: The sponsor’s BIA limits the population who could receive budesonide tablets to those who are refractory to or who recurred while on treatment with PPIs. Health Canada–approved budesonide tablets without restriction on prior PPI use4 (see CADTH Appraisal of the Sponsor’s Economic Evaluation, above). While the clinical experts consulted by CADTH agree it is likely that most patients who receive budesonide tablets will have tried a PPI and found it insufficient to control their symptoms, some patients would have severe symptoms and would start steroid therapy immediately, likely budesonide tablets. The potential remains for a broader patient population to receive budesonide induction and maintenance therapy than estimated by the sponsor.
CADTH conducted a scenario analysis eliminating the requirement for a patient to have been refractory to or have recurred while using a PPI. This analysis approximates the largest population that could receive maintenance therapy with budesonide tablets under the Health Canada indication rather than the most likely population to do so.
Relevant comparators omitted from analysis: The sponsor excluded off-label pharmacological treatments used in Canada for the maintenance of remission in EoE.
CADTH was unable to adjust the analysis to account for the use of other therapies such as swallowed fluticasone in the population of interest. However, should budesonide tablets displace some of the market share of these off-label therapies, the budget impact associated with reimbursing budesonide tablets would be lower than estimated due to the offset of their cost.
Non-Insured Health Benefits (NIHB) population double-counted and overestimated: In calculating the number of Canadians at risk of EoE, the sponsor summed the provincial populations (excluding Quebec), and then added the number of NIHB recipients. This method introduces a degree of double counting as the vast majority of NIHB recipients do not live in the Territories,17 and thus are already included in Statistics Canada’s provincial population estimates. Additionally, the sponsor included all NIHB recipients, not just those over the age of 18.
To estimate the total population at risk of EoE, CADTH summed the adult (≥ 18 years) populations of all provinces and territories,18 excluding Quebec, and then separated the number of adult NIHB recipients reported in each region,17 including them instead as NIHB recipients. This change decreases the total eligible population and thus decreases the estimated budget impact of budesonide.
Lack of discontinuation: While the sponsor’s cost-utility analysis included an estimate that 77.5% of patients will become nonadherent a median of 36 months into budesonide maintenance therapy,3 the sponsor’s BIA assumes all patients who start maintenance treatment remain on it for the full extent of the 3-year time horizon. This overestimates the incremental budgetary cost associated with the reimbursement of budesonide tablets compared to the assumptions made in the cost-effectiveness model. Discontinuing patients are not assumed to undergo a second induction within the time horizon under this analysis.
CADTH incorporated discontinuations due to nonadherence into its base-case reanalysis by calculating the probability of patients discontinuing treatment due to nonadherence from 1 year to another to be 15.1%, based on half of 77.5% of patients becoming nonadherent by 36 months (the median time to nonadherence in the sponsor’s cost-utility analysis). This change decreases the number of patients receiving budesonide tablets in years 2 and 3, and thus reduces the estimated budget impact.
Uptake of budesonide maintenance therapy may be overestimated: Feedback on the proportion of patients who were eligible for budesonide induction therapy who would choose to receive it varied among the clinical experts consulted by CADTH, ranging from 100% as assumed by the sponsor, to 50%, given that some patients may prefer therapies they’d used before such as fluticasone inhalers, while others may prefer not to use a steroid at all. The clinical experts did not agree with the sponsor that 100% of patients who responded to induction therapy would continue on maintenance therapy by year 3 of its reimbursement, or indeed ever, again predicting that some patients would prefer therapies they’d used before or would avoid long-term steroid use.
CADTH assumed in its base-case reanalysis that 40% of patients who responded to budesonide induction would continue to receive it as maintenance therapy in the first year, rising to 70% by year 3.
Based on a range of clinical expert input, CADTH conducted a scenario analysis assuming that of patients who had failed a PPI, approximately 50% would undergo induction with budesonide.
Population limited to successful 6-week budesonide induction: While the budesonide product monograph indicates that the usual duration of induction therapy is 6 weeks,4 and the CDEC recommendation for induction with budesonide recommended limiting duration to 6 weeks,5 patients were included in the BUL-2/EER maintenance trial if they had responded to a 6- or 12-week induction with budesonide. Patients in the BUL-1/EEA induction trial19 open-label extension who had not achieved remission by week 6 were allowed an additional 6 weeks of open-label budesonide. By the end of 6 or 12 weeks, 84.7% of patients had achieved clinicohistologic remission.
CADTH included a scenario analysis assuming that 84.7% of patients who are given a 6- or 12-week induction with budesonide respond and are eligible to continue with budesonide maintenance therapy.
CADTH Reanalyses of the BIA
CADTH revised the sponsor's base case by: ensuring NIHB beneficiaries were not double-counted in the overall population and that only adults were included, considering nonadherence to budesonide maintenance therapy, and reducing the proportion of patients who continue with budesonide maintenance therapy after achieving remission during the induction phase. Table 15 outlines the parameters used by the sponsor in comparison to those used by CADTH.
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. NIHB overestimate removed | Total adult population: 24,248,480 | Total adult population: 23,466,536 |
2. Nonadherence considered | Patients discontinuing from previous year: 0% | Patients discontinuing from previous year: 15.1% |
3. Lower proportion of successful inductions continue on maintenance in years 1, 2, and 3 | Proportion of budesonide induction patients in remission continuing on maintenance: 80% / 90% / 100% | Proportion of budesonide induction patients in remission continuing on maintenance: 40% / 55% / 70% |
CADTH base case | — | Reanalyses 1 + 2 + 3 |
NIHB = Non-Insured Health Benefits; PPI = proton pump inhibitor.
Applying these changes decreased the total 3-year budget impact to $8,319,438 when markups and dispensing fees are included, or $8,252,819 when excluded. The results of the CADTH step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $15,385,899 |
CADTH reanalysis 1: Excess NIHB population removed | $14,505,394 |
CADTH reanalysis 2: Nonadherence considered | $14,123,005 |
CADTH reanalysis 3: Lowered budesonide tablet maintenance uptake | $9,541,002 |
CADTH base case | $8,319,438 |
CADTH base case, without markups or dispensing fees | $8,252,819 |
BIA = budget impact analysis; NIHB = Non-Insured Health Benefits.
CADTH also conducted additional scenario analyses to explore areas of uncertainty:
Scenario A. Eligible population was not limited by being refractory to or recurring while using PPIs
Scenario B. 50% of patients who fail PPIs undergo induction therapy with budesonide
Scenario C. Patients who achieve remission on up to 12 weeks of budesonide induction therapy are eligible for continuing maintenance (84.7%).
While assuming that only 50% of patients who are refractory to PPIs or who have a recurrence while using PPIs try budesonide therapy decreased the estimated budget impact compared to the CADTH base case, both assuming budesonide patients did not need to be refractory to PPIs and including patients who achieved remission on 12 weeks of budesonide therapy expanded the eligible population and thus increased the estimated budget impact. Of these, the inclusion of patients who were not refractory to or who had not recurred while using PPI therapy, consistent with the full population represented by the Health Canada indication, had the largest impact on the estimated budget, see Table 17.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|
Submitted base case | $0 | $3,890,984 | $5,072,966 | $6,421,948 | $15,385,899 |
CADTH base case | $0 | $1,834,365 | $2,646,272 | $3,838,801 | $8,319,438 |
CADTH Scenario A: Not limited to PPI refractory | $0 | $3,571,582 | $5,152,398 | $7,474,302 | $16,198,282 |
CADTH Scenario B: 50% of patients failing PPIs try budesonide induction | $0 | $917,182 | $1,323,136 | $2,178,299 | $4,418,617 |
CADTH Scenario C: 12-week responders included | $0 | $2,697,408 | $3,891,306 | $5,644,904 | $12,233,617 |
BIA = budget impact analysis; PPI = proton pump inhibitor.
Note. The reference scenario in all analyses had a total cost of $0, thus only the cost of the new drug scenario/budget impact is presented.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.