CADTH Health Technology Review
Lenalidomide Plus Rituximab Chemotherapy for Relapsed or Refractory Indolent B-Cell Non-Hodgkin Lymphomas
Rapid Review
Authors: Sara D. Khangura, Andrea Ryce
Abbreviations
AE
adverse event(s)
ASCT
autologous stem cell therapy
CI
confidence interval
CR
complete response
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
ERG
Expert Review Group
FL
follicular lymphoma
HTA
health technology assessment
ICER
incremental cost-effectiveness ratio
IRC
independent review committee
ITT
intention to treat
MZL
marginal zone lymphoma
NHS
National Health Service
NICE
National Institutes for health Care Excellence
NIH
National Institutes of Health
ORR
overall response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PR
partial response
R2
lenalidomide + rituximab
R-CHOP
rituximab, cyclophosphamide, epirubicin, vincristine, and prednisone
RCT
randomized controlled trial
R-CVP
rituximab, cyclophosphamide, vincristine, and prednisone
R-LEN
rituximab and lenalidomide
R-mono
rituximab monotherapy
SD
stable disease
SR
systematic review
WTP
willingness to pay
Key Messages
Data from 2 randomized controlled trials indicated a statistically significant benefit in progression-free survival and overall survival for patients with follicular lymphoma who received R2 as compared to patients who received rituximab plus placebo or R-CHOP.
The frequency of all types of adverse events in patients receiving R2 as compared to rituximab plus placebo or R-CHOP was comparable, but patients receiving R2 experienced more severe adverse events.
Two economic analyses concluded that R2 was cost-effective for the treatment of patients with follicular lymphoma as compared to rituximab plus placebo (UK and Dutch contexts).
Evidence identified in this review was mostly limited to that describing patients with follicular lymphoma.
Most evidence identified in this review was generated with support and/or funding from a private industry pharmaceutical manufacturer.
Context and Policy Issues
Non-Hodgkin lymphomas (NHL) are a group of more than 30 diseases that affect the lymphatic system in the human body and are categorized as either indolent (low-grade) or aggressive (high-grade).1 In Canada, NHL is the fifth most common cancer diagnosed in adults, with an increasing incidence being observed over recent decades.2 Follicular lymphoma (FL) is the most common indolent B-cell NHL in North America,1 representing an estimated half of indolent NHLs, with another 15% being marginal zone lymphoma (MZL).3
Most indolent B-cell NHLs are identified in the advanced stages and are incurable4; however, due to their slow progression, many can be treated and often result in remission that can last for 10 years, or more.3 For some patients, however, there is no response, or an insufficient response, to initial therapy e.g., it is estimated that approximately 10% of patients living with FL are refractory to initial therapy.5 Treatment for indolent B-cell NHL varies based on the features of the disease and patient, but may include observation, radiotherapy, chemotherapy and/or stem cell transplant, according to the Canadian Cancer Society.6 In general, chemoimmunotherapy, combining rituximab and cytotoxic chemotherapy (i.e., cyclophosphamide, doxorubicin hydrochloride, vincristine and prednisone) — also known as R-CHOP — has become a commonly recommended treatment option in patients with relapsed, indolent B-cell NHL.4 However, R-CHOP may not be suitable for frail elderly patients, those with comorbidities or those who have several disease relapses.4
Lenalidomide is a novel immunomodulatory agent with unique and promising mechanisms of action in the context of B-cell NHL.7,8 Lenalidomide has demonstrated efficacy as monotherapy in patients with B-cell NHL, and preclinical data suggested a potential benefit of combining lenalidomide with rituximab.9 Rituximab has also been used as a monotherapy, as well as combined with other agents, demonstrating a benefit to patients with B-cell NHL.10,11 The combination of rituximab with lenalidomide (R2) has demonstrated favourable findings early on — in both previously untreated and previously treated indolent B-cell NHL patients — and has been suggested as a possible alternative to chemotherapy.9,11,12
A Health Canada Notice of Compliance does not exist for lenalidomide in patients with NHL, and CADTH’s reimbursement review process does not typically review generic drugs. With the recent approval of some generic lenalidomide products in Canada, the aim of this report is to summarize available evidence on the clinical and cost-effectiveness of lenalidomide plus rituximab (R2) for the treatment of relapsed or refractory indolent B-cell lymphomas.
Research Questions
What is the clinical effectiveness of lenalidomide plus rituximab combination chemotherapy (R2) for relapsed or refractory indolent B-cell non-Hodgkin lymphomas?
What is the cost-effectiveness of lenalidomide plus rituximab combination chemotherapy (R2) for relapsed or refractory indolent B-cell non-Hodgkin lymphomas?
Methods
Literature Search Methods
A limited literature search was conducted by an information specialist on key resources including MEDLINE and Embase, all via Ovid, the Cochrane Library, the University of York Centre for Reviews and Dissemination databases, the websites of Canadian and major international health technology agencies, as well as a focused Internet search. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were lenalidomide and rituximab and indolent B-cell non-Hodgkin lymphomas. Where possible, retrieval was limited to the human population. The search was also limited to English language documents published between January 1, 2016 and October 7, 2021.
Selection Criteria and Methods
One reviewer screened citations and selected studies. In the first level of screening, titles and abstracts were reviewed and potentially relevant articles were retrieved and assessed for inclusion. The final selection of full-text articles was based on the inclusion criteria presented in Table 1.
Criteria | Description |
|---|---|
Population | Adult patients with any grade of relapsed or refractory indolent B-cell non-Hodgkin lymphomas (i.e., relapsed/refractory follicular lymphoma, marginal zone lymphoma, lymphoplasmacytic lymphoma and Waldenström macroglobulinemia, MALT lymphoma) |
Intervention | Lenalidomide plus rituximab combination (R2) chemotherapy |
Comparator | Rituximab single agent therapy or combination chemotherapy other than R2 therapy (i.e., R-CHOP, R-CVP, R-FCM, R-CEOP, R-fludarabine, R-bendamustine) |
Outcomes | Q1: Clinical effectiveness (i.e., progression-free survival, overall survival, response rate, duration of response, quality of life); safety (i.e., adverse events of ≥ grade 3 and grade 4, serious adverse events, deaths) Q2: Cost-effectiveness (e.g., cost per quality-adjusted life-years) |
Study designs | Health technology assessments, systematic reviews,, randomized controlled trials, economic evaluations |
MALT = mucosa-associated lymphoid tissue; R-CEOP = rituximab, cyclophosphamide, etoposide, vincristine, prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine (Oncovin), and prednisone; R-CVP = rituximab, cyclophosphamide, vincristine, and prednisone; R-FCM = rituximab, fludarabine, cyclophosphamide, mitoxantrone.
Exclusion Criteria
Articles were excluded if they did not meet the selection criteria outlined in Table 1, they were duplicate publications, or were published before 2016. Systematic reviews in which all relevant studies were captured in other more recent or more comprehensive systematic reviews were excluded. Primary studies retrieved by the search were excluded if they were captured in 1 or more included health technology assessments or systematic reviews.
Critical Appraisal of Individual Studies
The included publications were critically appraised by 1 reviewer using the following tools as a guide: A MeaSurement Tool to Assess systematic Reviews 2 (AMSTAR 2)13 for systematic reviews, the Downs and Black checklist14 for randomized and non-randomized studies, and the Drummond checklist15 for economic evaluations. Summary scores were not calculated for the included studies; rather, the strengths and limitations of each included publication were described narratively.
Summary of Evidence
Quantity of Research Available
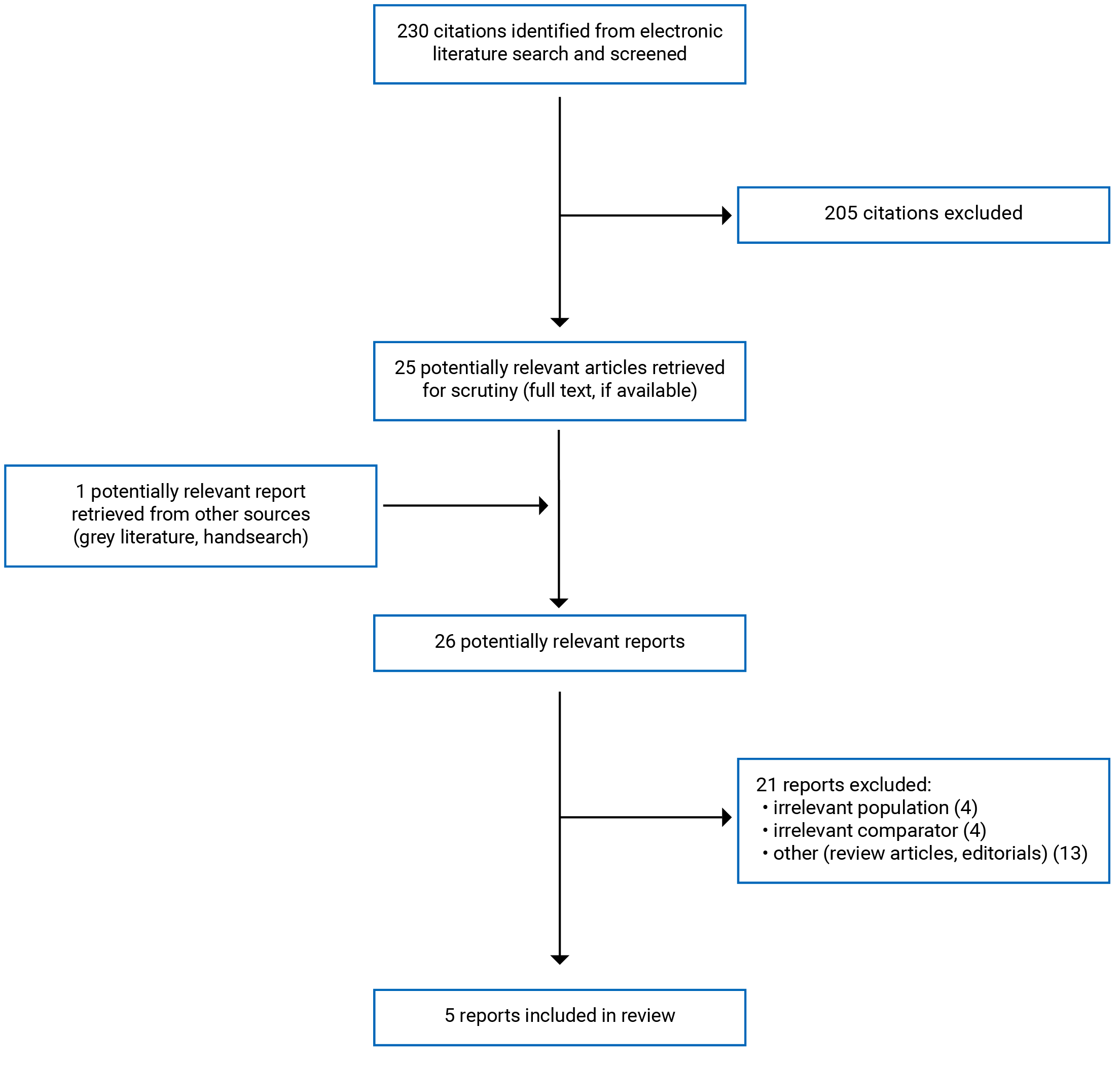
A total of 230 citations were identified in the literature search. Following screening of titles and abstracts, 205 citations were excluded and 25 potentially relevant reports from the electronic search were retrieved for full-text review. One potentially relevant publication was retrieved from the grey literature search for full-text review. Of these potentially relevant articles, 21 publications were excluded for various reasons, and 5 publications met the eligibility criteria and were included in this report. Eligible publications were 1 health technology assessment (HTA) (including both an eligible systematic review (SR) and an eligible economic evaluation), 3 randomized controlled trial (RCT) reports representing 2 unique studies, and 1 report of an economic evaluation. Appendix 1 presents the PRISMA16 flow chart of the study selection.
Additional references of potential interest are provided in Appendix 5.
Summary of Study Characteristics
Additional details regarding the characteristics of included publications are provided in Appendix 2.
Study Design
The HTA identified by this review was published in 2020 and incorporated a SR and an economic evaluation. Both the SR and economic evaluation were conducted by an industry sponsor, and submitted for review to an Expert Review Group (ERG) that was commissioned by the National Institute for Health and Clinical Excellence (NICE) in the UK.17
While the SR included in the HTA reports identifying 5 eligible studies, the analyses in the HTA focus primarily on data from the AUGMENT trial18 — including the ERG’s critique of the SR, which focuses only on the AUGMENT trial, as the other 4 studies did not contain data of relevance to the scope of the HTA, according to the ERG.17 Similarly, only the data from the AUGMENT study as summarized in the HTA met the eligibility criteria for this review, and so the summary of the SR is limited to a description of the findings from the AUGMENT trial only.
Of the 3 RCTs identified by this review,19-21 219,20 report on data from the AUGMENT trial.18 AUGMENT is described by the authors of the studies included in this review as a phase III, multi-centre, double-blind RCT, using intention-to-treat (ITT) analyses.19,20 One of these papers was published in 2019 and described the full study population of the AUGMENT RCT,20 while the other was published in 2020 and described a subset of the AUGMENT study patients.19 The third RCT was published in 2019 and based in a single centre.21
The 2 economic evaluations identified by this review conducted cost-effectiveness analyses and used data from the AUGMENT study18 One study considered 3 perspectives in the analyses (i.e., societal, health care and societal including future non-medical costs), and used a 3-state partitioned model, incorporating a lifetime time horizon and assuming a willingness-to-pay (WTP) threshold of €50,000 per quality-adjusted life-year (QALY).22 The other economic evaluation was incorporated within the HTA included in this report and described analyses conducted both by an industry sponsor and an ERG — the latter of which also prepared and reported on a critical assessment of the industry sponsor s cost-effectiveness analyses submission.17 The HTA-based cost-effectiveness analyses considered 2 perspectives (i.e., both National Health Service (NHS) and personal social services perspectives), and the economic model used a 3-state (including progression-free, post-progression and death) partitioned design that incorporated a lifetime time horizon and assumed a WTP threshold of £30,000 per QALY. Both cost-effectiveness analyses conducted sensitivity (probabilistic and deterministic) as well as scenario analyses.
Finally, despite the overlap between the HTA’s findings (i.e., clinical outcomes and cost-effectiveness data generated from the AUGMENT trial) and the search for this review (i.e., 3 primary study reports describing clinical outcomes and cost-effectiveness analyses using AUGMENT trial data19,20,22), we retained all 4 publications as included studies, because each of them reported on some unique findings not described in the other papers. Another important contributing factor in this decision was the large amount of redacted data in the HTA (where indicated as confidential by NICE),17 rendering a large amount of the data unavailable. Consequently, most of the findings describing clinical effectiveness from the AUGMENT trial were taken from the primary clinical report from AUGMENT that was included in this review.20
Country of Origin
The HTA was conducted for NICE in the UK; consequently, the industry sponsor’s submission (including both the SR and economic evaluation), ERG’s report and other associated documents within HTA report are specific to a UK context.17 The AUGMENT RCT is described as having been conducted across 97 centres representing 15 countries (not specified).20 The subset of patients from the AUGMENT RCT described in the paper reported by Izutsu and colleagues included only those participants recruited and followed up in Japan.19 The single-centre RCT was conducted in China.21 The economic evaluations were conducted in the Netherlands22 and the UK,17 respectively.
No evidence from a Canadian context was identified by this review.
Patient Population
Because the SR from the HTA reported relevant data from the AUGMENT RCT only,17 details describing the population were taken from the included report describing the primary study.20 Leonard and colleagues describe the AUGMENT RCT, which recruited patients with previously treated FL or MZL, including 358 participants with a median age of 63 years (range 26 to 88).20 The Japanese substudy of the AUGMENT trial reported data on 36 participants with a median age of 61 years (range 44 to 83).19 The single-centre RCT described 60 patients with FL and a mean age of 49.3 years (SD 10.4) in the intervention group and 51.1 years (SD 10.6) in the control group.21 The proportions of participants who were male in the RCT study populations were 50% in the AUGMENT trial,20 48% in the Japanese sub study of the AUGMENT RCT,19 and 58% in the single-centre RCT.21 Among the AUGMENT trial participants, 82% had FL and 18% had MZL20; whereas, in the Japanese substudy of AUGMENT, 97% of the patients had FL and 3% had MZL.19 In all 3 RCTs, baseline characteristics were otherwise described as being similar across the intervention and control groups.19-21
Both economic evaluations used patient data from the AUGMENT trial.18 to inform base-case, sensitivity and scenario analyses.17,22 The models used in the HTA pooled data across patients with FL and MZL,;7 whereas, the cost-effectiveness study used only the subset of patients with FL.22
No evidence was identified by this review describing the use of R2 in patients with lymphoplasmacytic lymphoma, Waldenström macroglobulinemia, or mucosa-associated lymphoid tissue (MALT) lymphoma.
Interventions and Comparators
Because the SR from the HTA reported relevant data from the AUGMENT RCT only,17 details describing the intervention and comparator from that study were taken from the other included reports describing the primary study.19,20 The intervention and comparators were the same in all papers describing clinical effectiveness data from the AUGMENT trial17,19,20 i.e., the active intervention arm was R2, comprised of lenalidomide (20 mg or 10 mg) administered once per day on day 1 to day 21 of a 28-day cycle for 12 cycles, combined with rituximab (375 mg/m2) administered intravenously once per week on days 1, 8, 15, and 22 of cycle 1, and day 1 of cycles 2 to 5.19,20 For the comparison arm, AUGMENT used rituximab plus placebo, with no other details described about its administration, other than it was “administered similarly” (p. 1189)20 as the R2 regimen.19,20 In the active comparator group, the single-centre RCT used lenalidomide (15 mg) administered orally with rituximab (375 mg/m2) on day 1; cyclophosphamide (750 mg/ m2) on day 2; epirubicin (60 mg/ m2) on day 2; vincristine (1.4 mg/m2) on day 2, and; prednisone (100 mg) on days 1 to 5, administered intravenously every 21 days for 6 cycles — described by the authors as R-CHOP.21 Control arm patients in the single-centre RCT received R-CHOP (without lenalidomide).21
The economic evaluations both drew from the AUGMENT trial as well, and so assessed R2 as the intervention of interest i.e., rituximab (375 mg/m2) administered on days 1, 8, 15, and 22 in cycle 1, and on day 1 of every subsequent 28-day cycle, until cycle 5; with lenalidomide (20 mg or 30 mg) per day, administered orally on days 1 to 21 of a 28-day cycle, for a total of up to 12 cycles.17,22 In the cost-effectiveness study, authors used rituximab monotherapy (R-mono) from the AUGMENT RCT as the comparison intervention i.e., rituximab 375 mg/m2 administered intravenously on days 1, 8, 15, and 22 in cycle 1, and on day 1 of every subsequent 28-day cycle until cycle 5.22 In the HTA’s economic evaluation, the industry sponsor’s economic model included R-CHOP and R-CVP as indirect comparators relevant to this review; that is R-CVP included rituximab 375 mg/m2, cyclophosphamide 750 mg/ m2, vincristine 1.4 mg/ m2 all on day 1; prednisolone 100 mg on day 1 to 5 of a 21-day cycle, for up to 8 cycles. The R-CHOP comparator comprised the same constituents and followed the same protocol, but also included the addition of doxorubicin (50 mg/m2) on day 1 of the 21-day cycle.17 Notably, as part of its critique of the industry sponsor’s economic model, the ERG requested additional cost-effectiveness analyses using R-mono as a comparator; while the findings of these analyses are provided in the HTA, the methods (including the constituents and treatment protocol for R-mono as included in the economic model), were not detailed in the HTA report.
No evidence was identified by this review describing rituximab, fludarabine, cyclophosphamide, mitoxantrone (R-FCM); rituximab, cyclophosphamide, etoposide, vincristine, prednisone (R-CEOP); rituximab with fludarabine (R-fludarabine); rituximab with bendamustine (R-bendamustine), or; rituximab, cyclophosphamide, etoposide, vincristine, prednisone (R-CEOP) as comparators.
Outcomes
The primary and secondary outcomes from the AUGMENT trial are described in detail below (and in Appendix 2) from the primary report of findings that was included in this review20; however, the HTA’s SR did provide some information on health-related quality of life (HRQoL) in addition to the primary and secondary outcomes described below,17 and the available data from that outcome are included in this report. The primary outcome of the AUGMENT trial, as described in both papers included in this review that reported data from it, was progression-free survival (PFS), measured in months.19,20 PFS was not otherwise defined in either paper,19,20 but is described in a clinical trials registry record for the study as the time at which either disease progression or death is observed (whichever comes first), beginning from randomization.18 The AUGMENT trial studies also described secondary outcomes data, including overall response rate (ORR), measured in months; complete response (CR), measured by counting numbers of patients (reported by Leonard and colleagues, only); duration of response (DOR), measured in months (reported by Leonard and colleagues, only); overall survival (OS), measured in months; event-free survival (EFS), measured in months, and; safety, measured by observations of adverse events (AEs).19,20 Data describing response and progression outcomes for AUGMENT were reviewed by an independent review committee (IRC) and findings were then reported both from study investigators and the IRC; IRC data were favoured over study investigator data for use in this review, as IRC review is recommended by WHO and has shown effectiveness in mitigating biased effect estimates.23 Patients in the AUGMENT trial are being followed for 5 years for disease progression, subsequent treatment and responses, as well as subsequent malignancies.20 The single-centre RCT measured ORR, defined as including CR, partial response (PR), stable disease (SD) and progressive disease (PD).21 Study investigators also measured overall PFS and OS at 1-, 2- and 3-year intervals.21 Data on safety outcomes were also collected by measuring and grading observed AE.21
The economic evaluations both reported on cost-effectiveness using costs and QALYs,17,22 and the cost-effectiveness study also reported on total average life-years (LY) per patient.22 Both studies presented their findings using incremental cost-effectiveness ratios (ICERs) reporting on the cost per unit of health benefit gained i.e., QALY or LY.17,22
Additional details regarding the characteristics of included studies are provided in Appendix 2
Summary of Critical Appraisal
SR
The SR described in the HTA included in this report demonstrated several strengths and limitations. The most notable limitation was the absence of a detailed description of the methods. For instance, while the SR portion of the HTA included in the industry sponsor’s submission makes reference to appendices describing important details abut the SR and its methods, these appendices are not included within the HTA document17 and are therefore unavailable to the reader. Nonetheless, the ERG report summarizes some of key pieces of information necessary for critically appraising the review methods.17 Using the available information, it was apparent that a comprehensive search of the literature was performed. In addition, duplicate data extraction was completed and risk of bias assessments were performed.17 The study characteristics were described and the approach to synthesis was explained and justified appropriately.17 Another strength was the comprehensive critique provided by the ERG of the SR (as well as the entire submission from the industry sponsor) as the third-party assessment provides the reader with helpful insights into the strengths and limitations of the review. Limitations to the SR were also noted, however; neither research question(s) is/are nor a link to a protocol are provided in the report.17 The study selection process is not described in detail, and the ERG confirms that duplicate study selection was not indicated by the authors of the SR.17 Because access to the appendix detailing the methods is unavailable, there are many important pieces of information that were absent such as the search strategies are referenced but not available; duplicate critical appraisal is not specified; a study flow (PRISMA) diagram is not provided, and; reasons for excluding ineligible studies are not detailed.17 Another important limitation of the SR report itself as included in the HTA is the large amount of data that are redacted from the report. Since the SR was conducted by an industry sponsor as part of an HTA, confidentiality requirements necessitated the redaction of some data that are then unavailable to the reader.
RCTs
Strengths and limitations were identified in all 3 RCT reports included in this review.19-21 The clarity of reporting was generally very good across all 3 papers, with only minor concerns identified. While the AUGMENT trial was double-blind (according to both Izutsu and colleagues19 as well as the AUGMENT clinical trials registration record18) this was not indicated in the paper reported by Leonard and colleagues.20 And while most details were clearly reported in the single-centre RCT report, there were no estimates of random variability included,21 which limits the reader’s ability to understand the impact of the small sample size of the trial on the effect estimates reported. In addition, no information on the representativeness of the study populations assessed was provided in any of the 3 reports, preventing the reader from considering whether this may pose a threat to external validity. Bias and confounding were generally well accounted for in the AUGMENT trial reports, with a randomized, double-blind design described, appropriate outcome measures and statistical tests, and both survival and ITT analyses performed.19,20 The limitations observed in the 2 AUGMENT reports included no clear description of whether the patients were recruited from the same population,19,20 as well as an apparent oversight in the paper by Leonard and colleagues, which did not describe all outcomes assessed in the methods section of the report.20 In the single-centre RCT, some features supporting the internal validity of the study were reported (e.g., patients were randomized to treatment groups, outcome measures and statistical tests were considered appropriate, and survival analyses were included).21 However, there were no blinding procedures described,21 leaving the reader uncertain as to whether the study may have been open-label; an open-label study design can threaten the internal validity of the study by introducing bias among both the patients, health care providers and investigators, who have awareness of which patient is receiving which treatment regimen. Further, the single-centre RCT did not describe the time period over which patients were recruited; the randomization procedure, or; any information about loss to follow-up.21 These oversights in reporting and/or study design can also threaten internal validity by introducing potential confounding, which may compromise the accuracy of the estimates of differences between patient groups in the study. Finally, no mention of sample size was reported, preventing the reader from considering whether the study was sufficiently powered to render valid and reliable results.
Economic Evaluations
In general, both economic evaluations studies demonstrated more strengths than limitations according to the Drummond checklist15 assessment, providing sufficient information from a robust source of clinical data (i.e., the AUGMENT study18) for their analyses.17,22 Both the study designs and data collection methods were well described in both papers with perspectives and approaches clearly stated and data sources (including justifications for their use) explicitly provided and cited.17,22 Similarly, the analysis and interpretation for both economic evaluations were generally clear and comprehensive, with time horizons stated, discount rates provided, sensitivity, scenario and incremental analyses conducted, and conclusions supported by the data generated.17,22 Despite the strengths, however, both studies also demonstrated some important limitations e.g., both studies were either funded or conducted by private industry sources, which constitutes a potential conflict of interest and can introduce important sources of bias.24,25 The economic evaluation incorporated into the HTA demonstrated some important limitations that were explicated by the ERG which assessed the industry sponsor’s model (e.g., the ERG stated its primary concern as the indirect comparison used to inform the model, which they suggest may have inflated the efficacy estimates used for R2).17 The ERG highlighted several additional concerns,17 which may be a function — at least to some extent — of the conflict of interest limitation that exists due to the source of funding from private industry.
Additional details regarding the strengths and limitations of included publications are provided in Appendix 3.
Summary of Findings
Clinical Effectiveness of Lenalidomide Plus Rituximab for Relapsed or Refractory Indolent Non-Hodgkin Lymphoma
As explained above, clinical effectiveness data describing primary and most secondary outcomes from the AUGMENT study as reported in the HTA’s SR17 have been taken from the primary study report included in this review,20 as they were described in more detail and unredacted in the latter. Only HRQoL as described in the HTA’s SR is reported here.
PFS
The paper reported by Leonard and colleagues described the median follow-up time of PFS in the AUGMENT RCT as 28.3 months for the overall study population; during which time, a total of 185 events were observed.20 Median PFS in the R2 arm was 39.4 months (95% CI, 22.9 to not reached), and in the R-placebo arm was 14.1 months (95% CI, 11.4 to 16.7), rendering a hazard ratio (HR) of 0.46 (95% CI, 0.34 to 0.62), which indicates a statistically significant benefit observed in the R2 arm (P < 0.0001).20 In the subset of patients participating in AUGMENT in Japan, median PFS in the R2 arm was not reached (95% CI, 19.7 to not estimable), and in the placebo arm, was 16.5 (95% CI, 11.3 to 30.6), resulting in a HR of 0.32 (95% CI, 0.11 to 0.96), also favouring the R2 group.19 Izutsu and colleagues also calculated the estimated probability of PFS at 2 years, rendering an estimate of 69% (95% CI, 40 to 86) for the R2 group and 33% (95% CI, 14 to 55) for the R-placebo group.19 In their assessment of PFS, Liu and colleagues found no statistical difference between the groups at 1-year of follow-up, observing a statistically significant benefit in favour of lenalidomide plus R-CHOP at both 2- and 3-years follow-up as 23 and 17 patients, respectively, achieved PFS at 2- and 3-years in the lenalidomide plus R-CHOP arm, and 17 and 10 patients at 2- and 3-years, respectively, in the R-CHOP-only arm (P = 0.031 at 2-years and P = 0.035 at 3-years follow-up).21 Overall PFS in the single-centre study was presented using a Kaplan–Meier curve indicating a significant benefit of lenalidomide plus R-CHOP (P = 0.032).21
OS
Median OS as observed in the overall AUGMENT RCT,20 as well as the subset of patients reported in the Japanese subanalysis19 was not reached in either the R2 or R-placebo arms. In the single-centre RCT, overall OS favoured the lenalidomide plus R-CHOP group (P = 0.024); though, at 1-year of follow-up, no statistical difference between the groups was observed (P = 0.313).21 Nonetheless, at 2 and 3-years of follow-up, significantly more patients experienced OS in the lenalidomide plus R-CHOP group (P = 0.021 at 2-years and P = 0.030 at 3-years follow-up).21
ORR
In the full study population of the AUGMENT trial, ORR was found to be statistically significantly superior in the R2 group i.e., 79% of patients (95% CI 73 to 85) as compared to the R-placebo group with 59% of patients (95% CI, 52 to 67) (P < 0.0001).20 The Japanese substudy reported higher ORR in the R2 group with 94% of patients (95% CI, 73 to 100) as compared to the R-placebo group with 56% of patients (95% CI, 31 to 79), but did not characterize the difference statistically.19 The single-centre RCT also found a statistically significant benefit for patients receiving lenalidomide plus R-CHOP, with 83.33% of patients achieving ORR as compared to 66.67% in the R-CHOP-only group (P = 0.027).21
EFS
Median EFS in the AUGMENT trial was 27.6 months (95% CI, 22.1 to not reached) in the R2 arm and 13.9 months (95% CI, 11.4 to 16.7) among patients receiving R-placebo, rendering a HR of 0.51 (95% CI, 0.38 to 0.67) and indicating a statistically significant benefit of treatment with R2 (P < 0.0001).20 Similarly, EFS was found to be higher in both the Japanese substudy of AUGMENT i.e., in the R2 arm, EFS was not reached (95% CI 17.2 months to not estimable) and in the R-placebo arm, EFS was 16.5 months (11.3 to 30.6) producing a HR or 0.35 (95% CI, 0.13 to 0.97).19
DOR
The DOR also favoured treatment with R2 in the AUGMENT trial, with 36.6 months observed (95% CI, 22.9 to not reached) as compared to 21.7 months (95% CI, 12.8 to 27.6) in the R-placebo group (HR 0.53 [95% CI, 0.36 to 0.79] P = 0.0015).20 The findings for this outcome in the Japanese sub study were less clear, with the DOR not reached in the R2 group (95% CI, 13.7 to not estimable) as compared to 19.0 months in the R-placebo group (95% CI 2.8 to not estimable) (HR 0.40 [95% CI, 0.13 to 1.25]); though, authors did highlight that this finding does indicate a benefit of R2 as compared to R-placebo.19
HRQoL
The HTA’s SR report explained that HRQoL between the R2 and R-mono groups in the AUGMENT study was comparable with no clinically meaningful difference identified.17 The report also makes reference to additional detail that was tabulated in an appendix; however, the appendices were not included in the report as published to the NICE website,17 and so, no additional detail was available.
Safety
Authors of both reports presenting primary data from the AUGMENT trial described a similar number of AEs overall in both patient groups, but a larger proportion of patients overall experienced more severe AEs (i.e., grade III/IV) in the R2 as compared to the R-placebo arm.19,20 The most common AEs observed in the R2 arm were neutropenia, diarrhea and constipation, with more variability in the proportions of patients experiencing AEs in the R-placebo arm.19,20 On the other hand, the single-centre study reported no statistically significant difference in the proportions of patients experiencing AEs between the lenalidomide plus R-CHOP and R-CHOP-only groups.21
Cost-Effectiveness of Lenalidomide Plus Rituximab for Relapsed or Refractory Indolent Non-Hodgkin Lymphoma
While the total costs estimated for R-LEN exceeded those of R-mono from all 3 perspectives considered in the cost-effectiveness study, at a WTP of €50,000, R-LEN was found to be cost-effective as compared to R-mono.22 From the societal perspective, the ICER for the base-case analysis was found to be €40,493; from the health care perspective, it was €37,951, and; from the societal perspective including future non-medical costs, it was €49,296.22 Sensitivity analyses indicated uncertainty; however, estimating that the probability of cost-effectiveness for R-LEN ranged from as low as 3% when including projected future non-medical costs, to as high as 82% when considering health care costs alone. Deterministic sensitivity analyses suggested that the ICERs were most sensitive to changes in the mean age of the patient, variations in the utility value for PFS and PD.22 The economic analysis presented in the HTA redacted much of the relevant comparative data on costs, incremental costs and QALYs due to confidentiality concerning negotiated drug costs for R2; nonetheless, the industry sponsor’s analysis resulted in their conclusion that R2 was cost-effective at a WTP threshold of £30,000 as compared to R-CHOP, R-CVP and R-mono with ICERs of £11,471, £16,814 and £22,580 reported, respectively.17 The HTA’s ERG conducted a follow-up analysis for the base case, reporting on ICERs of £15,505 when compared to R-CHOP; £21,759 compared to R-CVP and £27,372 as compared to R-mono. The ERG portion of the HTA report concludes that there remains significant uncertainty around the comparative cost-effectiveness of R2, given indirect comparative data used in the economic modelling, as well as uncertainty around cost estimates and other parameters.17
Appendix 4 presents the main study findings and authors’ conclusions.
Limitations
There was no evidence of particular relevance to Canada identified by this review, limiting the generalizability of the clinical and cost-effectiveness findings to the Canadian context. As well, no evidence was identified describing the use of R2 in patients with lymphoplasmacytic lymphoma, Waldenström macroglobulinemia or MALT lymphoma, limiting the extent to which the findings of this review may be generalized to a broader B-cell NHL patient population, outside of FL.
Of the 5 included publications,17,19-22 4 reported on data from the AUGMENT RCT,17,19,20,22 essentially limiting the findings of this review to data from 2 eligible study populations.18,21 Clinical effectiveness outcomes were reported from both of the eligible studies; whereas, cost-effectiveness data describing R2 were drawn solely from AUGMENT RCT data.17,22 While the AUGMENT trial is a large, international, multi-centre RCT, the limited number of studies identified by this review indicate that additional research may be needed that investigates the clinical and cost-effectiveness of R2 for patients with relapsed or refractory, indolent B-cell NHL.
All of the reports identified by this review demonstrated strengths and limitations in the critical appraisal; importantly however, the 4 publications reporting on data from the AUGMENT trial17,19,20,22 described research that was either conducted and/or funded by a private industry sponsor, which represents a potential conflict of interest and introduces a risk of bias. The single-centre RCT did not report on its source of funding.21
Lastly, the HTA was limited in its description of eligible data; that is, only data from the AUGMENT RCT were eligible for this review. In addition, large portions of the HTA describing key details were not available (e.g., appendices describing the methods of the SR; tabulated HRQoL data, etc). Further, the HTA redacted a large proportion of the data reported, rendering much of the report unusable.
Conclusions and Implications for Decision- or Policy-Making
This review identified 5 eligible publications, including 1 HTA (describing both an SR and an economic evaluation),17 3 RCT reports representing 2 unique studies.19-21 and 1 additional economic evaluation.22
Most of the clinical evidence identified favours the use of R2 as compared to other rituximab-containing regimens for the treatment of relapsed or refractory FL.17,19-21 While 2 of the studies reporting on clinical effectiveness outcomes were limited by small sample sizes,19,21 the data from the full AUGMENT RCT was generated from a larger sample across multiple centres and countries.17,20 Nonetheless, it is important to consider that the source of funding for the AUGMENT RCT comes from a private industry sponsor and may introduce the potential for bias from conflict of interest.
Similarly, the economic analyses identified by this review indicate R2 as being cost-effective when compared to other rituximab-containing regimens in both UK and Dutch contexts.17,22 Notably, there were multiple uncertainties highlighted in both publications as to factors that could affect the cost-effectiveness of R2 — particularly the analyses using indirect comparisons — and importantly, the generalizability of these findings to the Canadian context is unknown and potentially limited.
Notably, since the publication of the main findings from the AUGMENT trial,20 both NICE (UK) and FDA (US) have approved the use of R2 for previously treated FL (as well as MZL in the US).26,27
While the findings of this review are generally favourable toward the clinical and cost-effectiveness of R2 for the treatment of relapsed or refractory, indolent B-cell NHL, available evidence remains limited as most evidence describes patients living with FL; has been produced and/or funded by private industry; and none of the evidence is specific to Canadian health care settings, which makes it difficult to draw conclusions on the clinical and cost-effectiveness of R2 for the treatment of relapsed or refractory, indolent NHL. Additional research that considers evidence of the effects of R2 on additional subtypes of relapsed or refractory indolent NHL, as well as that which further mitigates potential conflicts of interest, and describes information of relevance to the Canadian context, will provide broader insights into the clinical and cost-effectiveness of R2 and help stakeholders in decision-making regarding the use of lenalidomide.
References
1.Canadian Cancer Society. What is non-Hodgkin lymphoma? 2014: https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/what-is-non-hodgkin-lymphoma. Accessed 1 Nov 2021.
2.Le M, Ghazawi FM, Alakel A, et al. Incidence and mortality trends and geographic patterns of follicular lymphoma in Canada. Curr Oncol. 2019;26(4):e473-e481. PubMed
3.Gaur R. Augment: Lenalidomide/rituximab vs placebo/rituximab in relapsed or refractory indolent lymphoma. Journal of Clinical Outcomes Management. 2019;26(5):200-203.
4.Yilmaz U, Salihoglu A, Soysal T. An Overview of Lenalidomide in Combination with Rituximab for the Treatment of Adult Patients with Follicular Lymphoma: The Evidence to Date. Drug Des Devel Ther. 2021;15:3809-3820. PubMed
5.Freedman AS, Friedberg JW. Treatment of relapsed or refractory follicular lymphoma. Waltham (MA): UpToDate; 2021 May 21.
6.Canadian Cancer Society. Treatments for indolent non-Hodgkin lymphoma. 2014: https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/treatment/indolent-lymphoma. Accessed 1 Nov 2021.
7.Blair HA. Lenalidomide: A Review in Previously Treated Follicular Lymphoma. Drugs. 2020;80(13):1337-1344. PubMed
8.Gordon LI. Lenalidomide and R-CHOP in follicular lymphoma: where do we go from here? The Lancet Haematology. 2018;5(9):e381-e382. PubMed
9.Bari A, Marcheselli R, Barbolini M, Ferri P, Sacchi S, Pozzi S. A concise review of lenalidomide therapy for follicular lymphoma. Expert Opinion on Orphan Drugs. 2017;5(3):269-275.
10.Sacchi S, Marcheselli R, Bari A, et al. Safety and efficacy of lenalidomide in combination with rituximab in recurrent indolent non-follicular lymphoma: final results of a phase II study conducted by the Fondazione Italiana Linfomi. Haematologica. 2016;101(5):e196-199. PubMed
11.Cheson BD, Morschhauser F, Martin P. Management of Adverse Events From the Combination of Rituximab and Lenalidomide in the Treatment of Patients With Follicular and Low-Grade Non-Hodgkin Lymphoma. Clin Lymphoma Myeloma Leuk. 2020;20(9):563-571. PubMed
12.Flowers CR, Leonard JP, Fowler NH. Lenalidomide in follicular lymphoma. Blood. 2020;135(24):2133-2136. PubMed
13.Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. PubMed
14.Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. PubMed
15.Higgins JPT, Green S, editors. Figure 15.5.a: Drummond checklist (Drummond 1996). Cochrane handbook for systematic reviews of interventions. London (GB): The Cochrane Collaboration; 2011: http://handbook-5-1.cochrane.org/chapter_15/figure_15_5_a_drummond_checklist_drummond_1996.htm. Accessed 13 Oct 2021.
16.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1-e34. PubMed
17.NICE. Technology appraisal guidance. Lenalidomide with rituximab for previously treated follicular lymphoma. 2020: https://www.nice.org.uk/guidance/ta627/evidence. Accessed 13 Oct 2021.
18.ClinicalTrials.gov. Rituximab Plus Lenalidomide for Patients with Relapsed/Refractory Indolent Non-Hodgkin’s Lymphoma (Follicular Lymphoma and Marginal Zone Lymphoma) (AUGMENT). Last updated 26 May 2021.: https://clinicaltrials.gov/ct2/show/NCT01938001 Accessed 28 Oct 2021.
19.Izutsu K, Minami Y, Fukuhara N, et al. Analysis of Japanese patients from the AUGMENT phase III study of lenalidomide + rituximab (R2) vs. rituximab + placebo in relapsed/refractory indolent non-Hodgkin lymphoma. Int J Hematol. 2020;111(3):409-416. PubMed
20.Leonard JP, Trneny M, Izutsu K, et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol. 2019;37(14):1188-1199. PubMed
21.Liu Z, Gao H, Peng Q, Yang Y. Efficacy of rituximab combined with lenalidomide in patients with recurrent follicular lymphoma. International Journal of Clinical and Experimental Medicine. 2019;12(9):11708-11715.
22.Thielen FW, Kersten MJ, Kuizenga P, et al. Cost-effectiveness of lenalidomide plus rituximab versus rituximab monotherapy in patients with previously treated follicular lymphoma: a societal view. Expert Rev Anticancer Ther. 2021:1-12. PubMed
23.Tang PA, Pond GR, Chen EX. Influence of an independent review committee on assessment of response rate and progression-free survival in phase III clinical trials. Annals of Oncology. 2010;21(1):19-26. PubMed
24.Lexchin J. Sponsorship bias in clinical research. Int J Risk Saf Med. 2012;24(4):233-242. PubMed
25.Yaphe J, Edman R, Knishkowy B, Herman J. The association between funding by commercial interests and study outcome in randomized controlled drug trials. Family Practice. 2001;18(6):565-568. PubMed
26.NICE News. NICE recommends new ‘chemotherapy-free’ treatment for lymphoma. 2020; https://www.nice.org.uk/news/article/nice-recommends-new-chemotherapy-free-treatment-for-lymphoma Accessed 31 Oct 2021.
27.US Food & Drug Administration. FDA approves lenalidomide for follicular and marginal zone lymphoma. 2019: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lenalidomide-follicular-and-marginal-zone-lymphoma. Accessed 31 Oct 2021.
Appendix 1: Selection of Included Studies
Appendix 2: Characteristics of Included Publications
Note that this appendix has not been copy-edited.
Table 2: Characteristics of Included Systematic Review
Study, country, funding source | Study designs, no. of primary studies included | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
NICE 201917 Country: UK Funding: NICE | PICOS: SRs, RCTs and NRS Eligible for inclusion in this review: 1 RCT included/assessed i.e., the AUGMENT trial20 – also included in this review due to the lack of available, relevant data in the HTA report | PICOS: Adults with relapsed or refractory FL or MZL Baseline characteristics from the eligible RCT: Randomized to treatment arm, n (%)
Disease, n (%)
Sex, n (%)
Age, n (%)
Baseline ECOG score, n (%)
Refractory to the last prior regimen, n (%)
POD24, n (%)
| PICOS interventions of interest: Systemic induction therapies (including R2 among others) PICOS comparator interventions: a list that includes 3 of those eligible for this review i.e., R-CVP, R-CHOP, R-mono Intervention from the eligible RCT: Lenalidomide (10 mg or 20 mg administered orally once per day from days 1 to 21 of a 28-day cycle) with rituximab (R2) (375 mg/m2, administered IV once per week beginning on day 1 of a 28-day cycle each week in Cycle 1 and on Day 1 of every 28-day Cycle from Cycles 2 through 5) Comparator from the eligible RCT: Rituximab plus placebo (R-mono) (not otherwise detailed/described) | PICOS outcomes of interest: survival, response, duration of treatment, duration of response, quality of life, time to next lymphoma treatment, adverse events Outcomes included in the eligible RCT*:
Follow-up:
*Outcome data were redacted throughout much of the HTA report17; consequently, most of the findings were summarized in this report from the published report of the AUGMENT study20 with the exception of health-related quality of life, which appeared only in the HTA report |
ECOG = Eastern Cooperative Oncology Group; FL = follicular lymphoma; HTA = health technology assessment; ITT = intention-to-treat; IV = IV; m2 = metres squared; mg = milligram(s); mo = months; MZL = marginal zone lymphoma; n = number; NICE = National Institutes for health Care Excellence; NR = not reported; NRS = non-randomized study; PICOS = population, intervention, comparator, outcomes, study design (eligibility criteria); POD24 = relapsed within 2 years of initial chemotherapy; R2 = lenalidomide + rituximab; R-CHOP = rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine (Oncovin), and prednisone; RCT = randomized controlled trial; R-CVP = rituximab, cyclophosphamide, vincristine, and prednisone; R-mono = rituximab monotherapy; SR = systematic review; yrs = years.
Table 3: Characteristics of Included Primary Clinical Studies
Study citation, country, funding source | Study design | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
|---|---|---|---|---|
Izutsu, 202019 Japan Funding: Celgene Corp. | Phase III, multi-centre placebo-controlled, double-blind RCT (AUGMENT trial) | Adults with previously treated FL or MZL (N = 36, ITT study subpopulation of Japanese patients from the AUGMENT trial) Randomized to treatment arm, n
Disease, n (%)
Sex, n (%)
Age, median (range)
Baseline ECOG score, n (%)
Number of prior systemic therapies, n (%)
Refractory to the last prior regimen, n (%)
| Intervention: R2: Lenalidomide 20 mg or 10 mg administered once per day on day 1 to day 21 of a 28-day cycle for 12 cycles with rituximab 375 mg/m2 administered IV once per week on days 1, 8, 15, and 22 of cycle 1, and day 1 of cycles 2 to 5 Comparator: R-placebo: administered on the same schedule as R2 (not otherwise described) | Primary outcome*:
Secondary outcomes*:
Follow-up: 12 treatment cycles or until discontinuation; patients subsequently are being followed up for 5 yrs for *Where reported in duplicate, study outcomes assessed by an IRC (as opposed to those assessed by the study investigators) are presented in this report |
Leonard, 201920 US (lead author) Funding: Celgene Corp. | Phase III, multi-centre (15 countries), placebo-controlled RCT (AUGMENT trial) | Adults with previously treated FL or MZL (N = 358, ITT study population) Disease, n (%)
Sex, n (%)
Age, median (range)
Baseline ECOG score, n (%)
Number of prior systemic antilymphoma therapies, n (%)
Prior rituximab therapy, n (%)
Refractory to the last regimen, n (%)
Randomized to treatment arm, n (%)
| Intervention: R2: Lenalidomide 20 mg or 10 mg administered orally once per day on day 1 to day 21 of a 28-day cycle for 12 cycles with rituximab 375 mg/m2 administered IV once per week on days 1, 8, 15, and 22 of cycle 1 and day 1 of cycles 2 to 5 Comparators: R-placebo: Rituximab plus placebo “administered similarly” (p. 1189) — no other detail described | Primary outcome*:
Secondary outcomes*:
Follow-up: 12 treatment cycles or until disease progression, unacceptable toxicity, patient withdrawal; patients subsequently are being followed up for 5 yrs for progression, subsequent therapy and response(s), as well as subsequent malignancy/ies *Where reported in duplicate, study outcomes assessed by an IRC (as opposed to those assessed by the study investigators) are presented in this report |
Liu, 201921 China Funding: NR | Single-centre RCT | Adults with recurrent FL (N = 60) Randomized to treatment arm, n (%)
Sex, n (%)
Age, mean (SD)
Grading, n (%)
| Intervention: Lenalidomide 15 mg administered orally with R-CHOP (as described by the study authors) i.e., rituximab 375 mg/m2 on day 1; cyclophosphamide 750 mg/m2 on day 2; epirubicin 60 mg/m2 on day 2; vincristine 1.4 mg/m2 on day 2; prednisone 100 mg on days 1-5, administered intravenously every 21 days for 6 cycles Comparator: R-CHOP (as described by the study authors) i.e., rituximab 375 mg/m2 on day 1; cyclophosphamide 750 mg/m2 on day 2; epirubicin 60 mg/m2 on day 2; vincristine 1.4 mg/m2 on day 2; prednisone 100 mg on days 1-5, administered intravenously every 21 days for 6 cycles | Outcomes:
Follow-up: For ORR, CR, PR, SD, PD: 6 cycles of treatment For PFS, OS: 1 yr, 2 yr, 3 yr |
AE = adverse event; CR = complete response; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; FL = follicular lymphoma; IRC = independent review committee; IV = IV; m2 = metres squared; mg = milligram(s); mo = month(s); MZL = marginal zone lymphoma; N/n = number; NIH = National Institutes of Health; NR = not reported; NA = not applicable; ORR = overall response rate; OS = overall survival; p. = page; PD = progressive disease; PFS = progression-free survival; POD24 = relapsed within 2 years of initial chemotherapy; PR = partial response; R2 = lenalidomide + rituximab; R-CHOP = rituximab + cyclophosphamide + epirubicin + vincristine + prednisone; R-mono = rituximab monotherapy; R-placebo = rituximab + placebo; R-CHOP = rituximab, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine (Oncovin), and prednisone; RCT = randomized controlled trial; SD = stable disease; yr = year(s).
Table 4: Characteristics of Included Economic Evaluations
Study citation country, funding source | Type of analysis, time horizon, perspective | Population characteristics | Intervention and comparator(s) | Approach | Source of clinical, cost, and utility data used in analysis | Main assumptions |
|---|---|---|---|---|---|---|
Thielen, 202122 Country: Netherlands Funding: Celgene Corp. | Type of analysis: Cost-effectiveness Time horizon: Lifetime Perspective: 3 perspectives considered: societal, health care and societal including future non-medical costs | Adults with previously treated FL (N = 295, data taken from the AUGMENT trial) Sex, % female
Age, mean (SD)
Body surface area, mean units NR (SD)
History of 1 prior systemic therapy, %
| R-LEN: Rituximab 375 mg/m2 administered intravenously/subcutaneously on days 1, 8, 15, and 22 in cycle 1 and on day 1 of every subsequent 28-day cycle until cycle 5 with lenalidomide 20 mg or 30 mg administered orally (frequency NR) R-mono: Rituximab 375 mg/m2 administered intravenously on days 1, 8, 15, and 22 in cycle 1 and on day 1 of every subsequent 28-day cycle until cycle 5. | Three-state partitioned survival model Outcomes:
Sensitivity analyses:
Scenario analyses:
| Clinical data: AUGMENT trial20 Cost data: Several published and cited sources (i.e., academic and government) informing estimates for travel, productivity loss and informal care Utility data: AUGMENT study20 and other published, cited academic sources | A 4-week treatment cycle length Potential for use of biosimilars instead of rituximab Subcutaneous administration of rituximab following the first IV dose All patient received 20 mg dose of lenalidomide That clinical effectiveness outcomes were similar/comparable across the R2 and R-CHOP/R-CVP patient populations |
NICE 202017 UK Funding: NICE | Cost-effectiveness analyses (conducted both by the industry sponsor and an Expert Review Group); lifetime time horizon; NHS and Personal Social Services perspective | Previously treated patients with FL and MZL (pooled, from the AUGMENT study) Sex, % female
Age, mean (range)
Baseline ECOG score, n (%)
Refractory to the last regimen, n (%)
POD24, n (%)
| Intervention R2: Lenalidomide 20 mg per day administered orally on days 1 to 21 of a 28-day cycle for up to 12 cycles; rituximab 375 mg/m2 per week on days 1, 8, 15, and 22 in cycle 1 and on day 1 of every subsequent 28-day cycle until cycle 5 Comparators R-CHOP: Rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2 all on day 1; prednisolone 100 mg on days 1 to 5 of a 21-day cycle, for up to 8 cycles R-CVP: Rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, vincristine 1.4 mg/m2 all on day 1; prednisolone 100 mg on day 1 to 5 of a 21-day cycle, for up to 8 cycles R-mono: rituximab plus placebo (not otherwise detailed/described) | Three-state partitioned survival model i.e., PF, PP, death Outcomes:
Costs were in British Pounds (£); a 3.5% discount rate was used for utilities and costs, and a WTP threshold of £30,000/QALY was assumed for the base-case analyses Sensitivity analyses:
Scenario analyses:
| Clinical data AUGMENT study data for R2 data, including PFS and OS; HMRN trial data for R-CHOP and R-CVP comparators Cost data NHS and Personal Social Services data sources Utility data AUGMENT EQ-5D-3L data; published literature for scenario analyses |
AE = adverse event; CR = complete response; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; EFS = event-free survival; EQ-5D-3L = 3-level EQ-5D questionnaire; FL = follicular lymphoma; HMRN = Haematological Malignancy Research Network; ICER = incremental cost-effectiveness ratio; IRC = independent review committee; IV = IV; LY = life-year(s); m2 = metres squared; mg = milligram(s); mo = month(s); MZL = marginal zone lymphoma; N/n = number; NHS = National Health Service (UK); NICE = National Institutes for health Care Excellence; NIH = National Institutes of Health; NR = not reported; NA = not applicable; ORR = overall response rate; OS = overall survival; PF = progression free; POD24 = defined as relapse within 2 years of initial chemoimmunotherapy; PP = post-progression; POD24 = relapsed within 2 years of initial chemotherapy; QALY = quality-adjusted life-year(s); R2 = lenalidomide + rituximab; QALY = quality-adjusted life-year(s); R-CHOP = rituximab + cyclophosphamide + epirubicin + vincristine + prednisone; R-mono = rituximab monotherapy; R-placebo = rituximab + placebo; RCT = randomized controlled trial; R-CVP = rituximab, cyclophosphamide, vincristine, and prednisone; SD = standard deviation; WTP = willingness-to-pay; yr = year(s).
Appendix 3: Critical Appraisal of Included Publications
Note that this appendix has not been copy-edited.
Table 5: Strengths and Limitations of Systematic Review using AMSTAR 213
Strengths | Limitations |
|---|---|
NICE 202017 | |
AMSTAR assessment Search dates and sources are summarized in the ERG report portion of the HTA Data extraction is described as being completed in duplicate and well done by the ERG report Risk of bias assessment was completed and corroborated by the ERG Study characteristics are reported Summary statistics were included in the available data Synthesis methods were appropriate i.e., no quantitative synthesis undertaken SR conducted by private industry (drug manufacturer) and underwent a thorough critique by a third party (i.e., ERG unaffiliated with either the drug manufacturer or NICE/NHS) | AMSTAR assessment Research questions are not explicitly stated/provided Some reference to a protocol is made, but no link to a registration record or protocol document is provided Study selection process is not described No details of methods are provided in the SR portion of the report e.g.,
Duplicate critical appraisal not indicated Study flow diagram not available (i.e., referenced as being included in an appendix that is not available in the HTA document) Excluded studies list not provided Other SR conducted by private industry (drug manufacturer) i.e., conflict of interest A great deal of data were redacted from the report and therefore not useful (though, this limitation does not significantly detract from the current review as the data for AUGMENT were taken from the primary study report that was also included in this review)20) |
AMSTAR 2 = A MeaSurement Tool to Assess systematic Reviews 2; ERG = Expert Review Group; HTA = health technology assessment; SR = systematic review.
Table 6: Strengths and Limitations of Clinical Studies Using the Downs and Black Checklist14
Strengths | Limitations |
|---|---|
Izutsu, 202019 | |
Reporting The objective of the study is stated Patient characteristics are clearly described Interventions are clearly described Main outcomes are described in the methods section Main findings are clearly described Estimates of random variability are provided Adverse events were described Actual P values were reported No loss to follow-up is reported Internal Validity (bias) Patients and health care workers were blinded to the interventions Main outcome measures were appropriate No evidence of data dredging was apparent Statistical tests used were appropriate Survival analyses were included Compliance with the interventions was described Internal Validity (confounding) Patients were recruited over the same time period Patients were randomized to treatment groups Randomization was concealed from patients, health care providers, investigators and outcome assessors18 ITT analyses were undertaken | External Validity No information reported on the representativeness of the patients or health care institutions Internal Validity (confounding) Unclear whether patients were recruited from the same population Power This substudy of the AUGMENT trial was explicitly stated as being underpowered to detect differences in outcomes |
Leonard 201920 | |
Reporting The objective of the study is stated Patient characteristics are clearly described Interventions are clearly described Main outcomes are described in the methods section Main findings are clearly described Estimates of random variability are provided Adverse events were described Actual P values were reported No loss to follow-up is reported Internal Validity (bias) Patients and health care workers were blinded to the interventions Main outcome measures were appropriate Statistical tests used were appropriate Survival analyses were included Compliance with the interventions was described Internal Validity (confounding) Patients were recruited over the same time period Patients were randomized to treatment groups Randomization was concealed from patients, health care providers, investigators and outcome assessors18 ITT analyses were undertaken | Reporting A double-blind method was used in the AUGMENT trial,18 but this was not reported in the paper by Leonard and colleagues External Validity No information reported on the representativeness of the patients or health care institutions Internal Validity (bias) No significant evidence of data dredging was apparent, but some outcomes (e.g., best response, PR, PD, SD) were reported in the results that were not pre-specified in the methods Internal Validity (confounding) Unclear whether patients were recruited from the same population Power Unclear whether the sample size was sufficient |
Liu 201921 | |
Reporting The objective of the study is stated Patient characteristics are clearly described Interventions are clearly described Main outcomes are described in the methods section Main findings are clearly described Adverse events were described Actual P values were reported No loss to follow-up is reported Internal Validity (bias) Main outcome measures were appropriate No evidence of data dredging was apparent Statistical tests used were appropriate Survival analyses were included Internal Validity (confounding) Patients were randomized to treatment groups | Reporting Estimates of random variability are not provided External Validity No information reported on the representativeness of the patients or health care institution Internal Validity (bias) No information on blinding is reported Compliance with the interventions was not described Internal Validity (confounding) The time period over which patients were recruited in not reported The randomization procedure is not described in any detail Loss to follow-up was not described Power No information on the impact of the sample size or the power of the study to detect a meaningful difference between groups is provided |
ITT = intention-to-treat; PD = progressive disease; PR = partial response; SD = stable disease.
Table 7: Strengths and Limitations of Economic Evaluations Using the Drummond Checklist15
Strengths | Limitations |
|---|---|
Thielen 202122 | |
Study design The research aim and its importance are clearly stated The perspectives and form of economic analysis used are clearly stated and justified Comparator is described Data collection Sources of effect estimates and details of the study design are explicitly provided Outcome measures are explicitly stated Methods and sources describing valuation are reported Productivity changes are described Currency and price data are reported Details and justification for the model used are described Analysis and interpretation Time horizon is stated Discount rate clearly reported Sensitivity and scenario analyses are reported and variables are described Incremental analysis included Findings address the research aim Conclusions follow from the data reported with appropriate caveats included | Study design Rationale for use of the comparator is not explicitly justified Data collection Relevance of productivity changes is not made explicitly clear Quantities of resource use are not reported separately from their costs Analysis and interpretation Choice of discount rate not explicitly justified (but is cited) Choice of variables for sensitivity analyses not explicitly justified Other Funding for the study provided by private industry (drug manufacturer) Absence of long-term data Uncertainty in several of the model parameters |
NICE 202017 | |
Study design The perspectives and form of economic analysis used are clearly stated and justified Some comparators are described and their selection is justified Data collection Sources of effect estimates and details of the study design are explicitly provided Outcome measures are explicitly stated Methods and sources describing valuation are reported Currency and price data are reported Details and justification for the model used are described Analysis and interpretation Time horizon is stated Discount rate clearly reported and cited Sensitivity and scenario analyses are reported and variables are described Incremental analysis included Findings address the research aim Conclusions follow from the data reported with appropriate caveats included Other Original economic model developed by private industry (drug manufacturer) underwent a thorough critique by a third party (i.e., ERG unaffiliated with either the drug manufacturer or NICE/NHS) | Study design The research question and its importance are not made explicit (but may arguably be implicit in the HTA method) One comparator (R-mono) is not described explicitly (as it was included, post hoc, in follow-up analyses only) Data collection Quantities of resource use are not reported separately from their costs Analysis and interpretation The time horizon is criticized by the ERG as having been stated incorrectly Choice of discount rate not explicitly justified (but is cited) Clarity of reporting Many of the findings were redacted from the report Multiple appendices are referred to throughout the report but are not available/accessible Rituximab control arm from the AUGMENT trial is described as R-mono; whereas, the AUGMENT trial control arm is actually R-placebo Additional limitations of relevance to this report identified by the HTA’s ERG Partitioned analysis with no state transition model Indirect comparisons may have introduced inaccuracy into the model (e.g., inflated efficacy of R2) Choice of curves for PFS and OS are not best fit and not justified Data describing AEs is inappropriate/missing Utility values likely inaccurate inflating ICERs in favour of R2 Some data describing resources and costs include overestimates that are likely inflating ICERs in favour of R2 Other Original economic model developed by drug manufacturer (potential conflict of interest) |
ERG = Expert Review Group; HTA = health technology assessment; NHS = National Health Service (UK); NICE = National Institutes for health Care Excellence; R2 = lenalidomide + rituximab; R-mono = rituximab monotherapy; R-placebo = rituximab + placebo.
Appendix 4: Main Study Findings and Authors’ Conclusions
Note that this appendix has not been copy-edited.
Table 8: Summary of Findings of Included Systematic Review
Main study findings | Authors’ conclusion |
|---|---|
NICE 202017 | |
Data describing PFS, OS, EFS, ORR and AEs were taken from the primary study paper describing the AUGMENT trial (see below) Health-related quality of life
| “Clinical-effectiveness evidence shows that, when people take lenalidomide with rituximab, their follicular lymphoma does not progress as quickly as when they take rituximab with chemotherapy. There is also evidence that lenalidomide with rituximab helps people live longer than rituximab with chemotherapy, although it is too early to tell for how much longer. Lenalidomide with rituximab costs more than rituximab with chemotherapy. However, its cost-effectiveness estimate is within the range that NICE normally considers an acceptable use of NHS resources. Therefore, lenalidomide with rituximab is recommended.” (https://www.nice.org.uk/guidance/TA627/chapter/1-Recommendations) |
AEs = adverse events; EFS = event-free survival; HTA = health technology assessment; PFS = progression-free survival; ORR = overall response.
Summary of Findings of Included Primary Clinical Studies
Izutsu 202019
Main study findings
Primary end point (N = 30)
PFS
Median PFS (IRC), mo (95% CI)
R2
NR (19.7 to not estimable [NE])
R-placebo
16.5 (11.3 to 30.6)
Statistical difference between groups
Hazard ratio (95% CI)
0.32 (0.11 to 0.96) (favours R2)
Probability of PFS at 2 years (IRC), % (95% CI)
R2
69 (40 to 86)
R-placebo
33 (14 to 55)
Statistical difference between groups
NR
Secondary end points
ORR, n (% [95% CI])
R2
17 (94 [73 to 100])
R-placebo
10 (56 [31 to 79])
CR, n (% [95% CI])
R2
3 (17 [4 to 41])
R-placebo
2 (11 [1 to 35])
DOR
Median DOR, mo (95% CI)
R2
NR (13.7 to NE)
R-placebo
19.0 (2.8 to NE)
Statistical difference between groups
Hazard ratio (95% CI)
0.40 (0.13 to 1.25)
EFS
Median EFS, month (95% CI)
R2
NR (17.2 to NE)
R-placebo
16.5 (11.3 to 30.6)
Statistical difference between groups
Hazard ratio (95% CI)
0.36 (0.13 to 0.97)
OS
Median OS, month (95% CI)
R2
NR (NE to NE)
R-placebo
NR (NE to NE)
Statistical difference between groups
Hazard ratio (95% CI)
NE
Safety
Adverse events observed in at least 20% of study patients (all grades), n (%)
Infusion-related reaction
All grades
R2: 13 (72)
R-placebo: 8 (44)
Grade 3 or 4
R2: 1 (6)
R-placebo: 0 (0)
Neutropenia
All grades
R2: 11 (61)
R-placebo: 6 (33)
Grade 3 or 4
R2: 9 (50)
R-placebo: 3 (17)
Constipation
All grades
R2: 10 (56)
R-placebo: 2 (11)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Nasopharyngitis
All grades
R2: 7 (39)
R-placebo: 5 (28)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Rash
All grades
R2: 7 (39)
R-placebo: 3 (17)
Grade 3 or 4
R2: 1 (6)
R-placebo: 0 (0)
Diarrhea
All grades
R2: 7 (39)
R-placebo: 0 (0)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Decreased lymphocyte count
All grades
R2: 5 (28)
R-placebo: 4 (22)
Grade 3 or 4
R2: 3 (17)
R-placebo: 2 (11)
Decreased white blood count
All grades
R2: 5 (28)
R-placebo: 5 (28)
Grade 3 or 4
R2: 1 (6)
R-placebo: 2 (11)
Leukopenia
All grades
R2: 5 (28)
R-placebo: 2 (11)
Grade 3 or 4
R2: 1 (6)
R-placebo: 0 (0)
Thrombocytopenia
All grades
R2: 5 (28)
R-placebo: 0 (0)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Alanine aminotransferase increased
All grades
R2: 5 (28)
R-placebo: 0 (0)
Grade 3 or 4
R2: 1 (6)
R-placebo: 0 (0)
Maculopapular rash
All grades
R2: 5 (28)
R-placebo: 0 (0)
Grade 3 or 4
R2: 1 (6)
R-placebo: 0 (0)
Peripheral sensory neuropathy
All grades
R2: 4 (22)
R-placebo: 3 (17)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Decreased blood immunoglobulin
All grades
R2: 4 (22)
R-placebo: 1 (6)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Authors’ conclusion
“R2 in Japanese patients from AUGMENT demonstrated superior efficacy compared with R-placebo and reduced the risk of progression by 68% (HR 0.32; 95% CI 0.11–0.96) compared with R-placebo. Median PFS was not reached in the R2 group compared with 16.5 months in the R-placebo group.” (p. 414)
Leonard 201920
Main study findings
Primary end point (N = 358 ITT analyses)
PFS (assessed by IRC)
Median PFS, month (95% CI)
R2
39.4 (22.9 to NR)
R-placebo
14.1 (11.4 to 16.7)
Difference between groups
Hazard ratio (95% CI)
0.46 (0.34 to 0.62) (favours R2)
P value
< 0.0001 (favours R2)
Median follow-up (all), month
28.3
Total events; that is progression or death assessed by IRC before censoring (all), n
185
Secondary end points (N = 358 ITT analyses)
OS
Median (95% CI), months
R2
NR (NR to NR)
R-placebo
NR (NR to NR)
Difference between groups
Hazard ratio (95% CI)
0.61 (0.33 to 1.13)
P value
Not reported
Probability of OS at 2 years, % (95% CI)
R2
93 (87 to 96)
R-placebo
87 (81 to 92)
Difference between groups
Not reported
EFS (assessed by IRC), months
Median (95% CI)
R2
27.6 (22.1 to NR)
R-placebo
13.9 (11.4 to 16.7)
Statistical difference between groups
Hazard ratio (95% CI)
0.51 (0.38 to 0.67)
P value
< 0.0001 (favours R2)
ORR, n patients (% [95% CI])
R2
141 (79 [73 to 85])
R-placebo
107 (59 [52 to 67])
Difference between groups
P value
< 0.0001 (favours R2)
DOR, month (95% CI)
Median (95% CI)
R2
36.6 (22.9 to NR)
R-placebo
21.7 (12.8 to 27.6)
Difference between groups
Hazard ratio (95% CI)
0.53 (0.36 to 0.79)
P value
0.0015 (favours R2)
CR, n patients (% [95% CI])
R2
57 (32 [25 to 39])
R-placebo
37 (21 [15 to 27])
Difference between groups
P value
0.0119 (favours R2)
Safety
Adverse events observed in at least 10% of study patients, n (%)
Neutropenia
All grades
R2: 102 (58)
R-placebo: 40 (22)
Grade 3 or 4
R2: 88 (50)
R-placebo: 23 (13)
Diarrhea
All grades
R2: 55 (31)
R-placebo: 41 (23)
Grade 3 or 4
R2: 5 (3)
R-placebo: 0 (0)
Constipation
All grades
R2: 46 (26)
R-placebo: 25 (14)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Cough
All grades
R2: 40 (23)
R-placebo: 31 (17)
Grade 3 or 4
R2: 1(1)
R-placebo: 0 (0)
Fatigue
All grades
R2: 38 (22)
R-placebo: 33 (18)
Grade 3 or 4
R2: 2 (1)
R-placebo: 1 (1)
Pyrexia
All grades
R2: 37 (21)
R-placebo: 27 (15)
Grade 3 or 4
R2: 1 (1)
R-placebo: 3 (2)
Leukopenia
All grades
R2: 36 (20)
R-placebo: 17 (9)
Grade 3 or 4
R2: 12 (7)
R-placebo: 3 (2)
Upper respiratory tract infection
All grades
R2: 32 (18)
R-placebo: 23 (13)
Grade 3 or 4
R2: 2 (1)
R-mono: 4 (2)
Anemia
All grades
R2: 28 (16)
R-placebo: 8 (4)
Grade 3 or 4
R2: 8 (5)
R-mono: 1 (1)
Headache
All grades
R2: 26 (15)
R-placebo: 17 (9)
Grade 3 or 4
R2: 1 (1)
R-placebo: 0 (0)
Infusion-related reaction
All grades
R2: 26 (15)
R-placebo: 24 (13)
Grade 3 or 4
R2: 4 (2)
R-placebo: 0 (0)
Thrombocytopenia
All grades
R2: 26 (15)
R-placebo: 8 (4)
Grade 3 or 4
R2: 4 (2)
R-placebo: 2 (1)
Asthenia
All grades
R2: 24 (14)
R-placebo: 19 (11)
Grade 3 or 4
R2: 2 (1)
R-placebo: 1 (1)
Decreased appetite
All grades
R2: 23 (13)
R-placebo: 11 (6)
Grade 3 or 4
R2: 2 (1)
R-placebo: 0 (0)
Muscle spasms
All grades
R2: 23 (13)
R-placebo: 9 (5)
Grade 3 or 4
R2: 1 (1)
R-placebo: 1 (1)
Peripheral edema
All grades
R2: 23 (13)
R-placebo: 16 (9)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Abdominal pain
All grades
R2: 22 (13)
R-placebo: 16 (9)
Grade 3 or 4
R2: 2 (1)
R-placebo: 0 (0)
Pruritis
All grades
R2: 21 (12)
R-placebo: 7 (4)
Grade 3 or 4
R2: 2 (1)
R-placebo: 0 (0)
Nausea
All grades
R2: 20 (11)
R-placebo: 23 (13)
Grade 3 or 4
R2: 0 (0)
R-placebo: 1 (1)
Dyspnea
All grades
R2: 19 (11)
R-placebo: 8 (4)
Grade 3 or 4
R2: 2 (1)
R-placebo: 1 (1)
Rash
All grades
R2: 19 (11)
R-placebo: 7 (4)
Grade 3 or 4
R2: 2 (1)
R-placebo: 1 (1)
Tumour flare
All grades
R2: 19 (11)
R-placebo: 1 (1)
Grade 3 or 4
R2: 1 (1)
R-placebo: 0 (0)
Alanine aminotransferase increased
All grades
R2: 18 (10)
R-placebo: 15 (8)
Grade 3 or 4
R2: 3 (2)
R-placebo: 1 (1)
Influenza
All grades
R2: 17 (10)
R-placebo: 8 (4)
Grade 3 or 4
R2: 1 (1)
R-placebo: 0 (0)
Vomiting
All grades
R2: 17 (10)
R-placebo: 13 (7)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Back pain
All grades
R2: 14 (8)
R-placebo: 18 (10)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
Nasopharyngitis
All grades
R2: 13 (7)
R-placebo: 18 (10)
Grade 3 or 4
R2: 0 (0)
R-placebo: 0 (0)
End points not described in the methods, but appearing in the results section
Best response (not otherwise described/defined), n (95% CI)
PR, n (%)
R2
84 (47)
R-placebo
70 (39)
Difference between groups
P value
Not reported
SD, n (%)
R2
22 (12)
R-placebo
56 (31)
Difference between groups
P value
Not reported
PD/death, n (%)
R2
6 (3)
R-placebo
15 (8)
Difference between groups
P value
Not reported
Missing, n (%)
R2
9 (5)
R-placebo
2 (1)
Difference between groups
P value
Not reported
Post-hoc subgroup analysis of PFS based on prior treatment
PFS, month (95% CI), prior exposure to rituximab + bendamustine
R2 (n = 19)
NR (20.2 to NR)
R -placebo (n = 14)
11.1 (3.0 to 30.6)
Difference between groups
Hazard ratio (95% CI)
0.23 (0.06 to 0.85)
PFS, month (95% CI), prior exposure to rituximab + cyclophosphamide + doxorubicin + vincristine + prednisolone
R2 (n = 66)
39.4 (22.1 to NR)
R-placebo (n = 69)
13.9 (8.7 to 28.0)
Statistical difference between groups
Hazard ratio (95% CI)
0.50 (0.30 to 0.82)
Authors’ conclusion
“The magnitude of efficacy differences between the two treatments is clinically meaningful and suggests that lenalidomide plus rituximab should replace rituximab monotherapy as a standard of care for patients with relapsed or refractory indolent NHL.” (p. 1197)
Liu 201921
Main study findings
ORR, % patients
Lenalidomide + R-CHOP
83.33
R-CHOP
66.67
Difference between groups
x2
6.193
P value
0.027
CR, n patients (%)
Lenalidomide + R-CHOP
15 (50)
R-CHOP
11 (36.67)
PR, n patients (%)
Lenalidomide + R-CHOP
10 (33.33)
R-CHOP
9 (30)
SD, n patients (%)
Lenalidomide + R-CHOP
6 (20)
R-CHOP
3 (10)
PD, n patients (%)
Lenalidomide + R-CHOP
4 (13.33)
R-CHOP
2 (6.67)
Overall PFS (presented using Kaplan–Meier curve)
Difference between groups
x2
5.183
P value
0.032
1-year PFS, n patients (%)
Lenalidomide + R-CHOP
28 (93.33)
R-CHOP
26 (86.67)
Difference between groups
x2
0.741
P value
0.389
2-year PFS, n patients (%)
Lenalidomide + R-CHOP
23 (76.67)
R-CHOP
17 (56.67)
Difference between groups
x2
5.279
P value
0.031 (favours intervention)
3-year PFS, n patients (%)
Lenalidomide + R-CHOP
17 (56.67)
R-CHOP
10 (33.33)
Difference between groups
x2
4.986
P value
0.035 (favours intervention)
Overall OS (presented using Kaplan–Meier curve)
Difference between groups
x2
6.576
P value
0.024
1-year OS, n patients (%)
Lenalidomide + R-CHOP
30 (100)
R-CHOP
29 (96.67)
Difference between groups
x2
1.017
P value
0.313
2-year OS, n patients (%)
Lenalidomide + R-CHOP
26 (86.67)
R-CHOP
19 (63.33)
Difference between groups
x2
6.830
P value
0.021 (favours intervention)
3-year OS, n patients (%)
Lenalidomide + R-CHOP
20 (66.67)
R-CHOP
14 (46.67)
Difference between groups
x2
5.407
P value
0.030 (favours intervention)
Adverse events, n (%)
Neutropenia (grade 3/4)
Lenalidomide + R-CHOP
6 (20)
R-CHOP
7 (23.33)
x2
0.600
P value
0.438
Thrombocytopenia (grade 3/4)
Lenalidomide + R-CHOP
7 (23.33)
R-CHOP
4 (13.33)
x2
1.200
P value
0.273
Rash
Lenalidomide + R-CHOP
12 (40)
R-CHOP
10 (33.33)
x2
0.073
P value
0.787
Thrombosis
Lenalidomide + R-CHOP
9 (30)
R-CHOP
7 (23.33)
x2
0.073
P value
0.787
Digestive tract reaction
Lenalidomide + R-CHOP
7 (23.33)
R-CHOP
5 (16.67)
x2
0.089
P value
0.766
Hepatic impairment
Lenalidomide + R-CHOP
6 (20)
R-CHOP
4 (13.33)
x2
0.111
P value
0.379
Authors’ conclusion
“In conclusion, combined therapy of rituximab and lenalidomide for patients with recurrent FL can not only improve short-term clinical benefit rate, but also effectively controls the progression of disease, prolongs the survival period of patients, and offers good safety. This therapy can be a new option of treatment for recurrent FL.” (p. 11713)
Summary of Findings of Included Economic Evaluations
Thielen 202122
Main study findings
Total Costs (average, lifetime, undiscounted, per patient), €
Societal perspective
R-LEN
200,355
R-mono
132,789
Health care perspective
R-LEN
165,547
R-mono
102,223
Societal perspective (including future non-medical costs)
R-LEN
299,943
R-mono
217,687
Life-Year
Societal perspective, total, average, per patient (PFS, PD)
R-LEN
12.9 (3.2, 0)
R-mono
10.9 (1.7, 9.1)
ICER (discounted, per life-year gained), €
33,681
Health care perspective, total, average, per patient (PFS, PD)
R-LEN
12.9 (3.2, 0)
R-mono
10.9 (1.7, 9.1)
ICER (discounted, discounted, per life-year gained), €
31,567
Societal perspective (including future non-medical costs), total, average, per patient (PFS, PD)
R-LEN
12.9 (3.2, 0)
R-mono
10.9 (1.7, 9.1)
ICER (discounted, per life-year gained), €
41,004
Quality-adjusted life-year
Societal perspective, total, average, per patient (PFS, PD)
R-LEN
10.8 (2.8, 8)
R-mono
9.1 (1.5, 7.6)
ICER (discounted, per quality-adjusted life-year gained [QALY]), €
40,493
Health care perspective, total, average, per patient (PFS, PD)
R-LEN
10.8 (2.8, 8)
R-mono
9.1 (1.5, 7.6)
ICER (discounted, per QALY gained), €
37,951
Societal perspective (including future non-medical costs), total, average, per patient (PFS, PD)
R-LEN
10.8 (2.8, 8)
R-mono
9.1 (1.5, 7.6)
ICER (discounted, per QALY gained), €
49,296
Sensitivity/scenario analyses
Deterministic (societal perspective)
ICERs, range (€/QALY)
37,116 to 44,816
Top 10 influential model parameters (in order of influence on ICER)
Age (mean age of patient)
Utility value of PFS (varied by 0.1)
Increase resulted in lowest ICER
Utility value of PD (varied by 0.1)
Informal care (price per hour)
Informal care (probability of receiving)
Increase to 60% from 22% assumption in base case resulted in highest ICER
Informal care (time per week)
Costs of adverse events (neutropenia)
Follow-up costs (outpatient clinic)
% adverse events (neutropenia with R-LEN)
Drug administration costs
ICERs per QALY
Lowest
37,116
Highest
44,816
Probabilistic, probability of cost-effectiveness of R-LEN, %
Base-case societal perspective (assuming a €50,000 WTP threshold)
67
Base-case health care costs perspective (assuming a €50,000 WTP threshold) – health care costs only
82
Base-case societal perspective (assuming a €50,000 WTP threshold) – including future non-medical costs
3
Scenario analyses
Societal perspective, n cost-effective scenarios/32
WTP threshold of €20 thousand
0/32
WTP threshold of €50 thousand
29/32
WTP threshold of €80 thousand
32/32
Health care perspective, n cost-effective scenarios/32
WTP threshold of €20 thousand
0/32
WTP threshold of €50 thousand
29/32
WTP threshold of €80 thousand
32/32
Societal perspective including future non-medical costs, n cost-effective scenarios/32
WTP threshold of €20 thousand
0/32
WTP threshold of €50 thousand
24/32
WTP threshold of €80 thousand
32/32
Authors’ conclusion
“Our results show that R-LEN can be considered cost-effective at a WTP threshold of 50,000 EUR/QALY gained from the base-case analysis. Nevertheless, this result is marked by some uncertainty. Long-term efficacy data could validate the model results and reduce this uncertainty. Although more recent and extensive data would be preferred, a further exploration of real-world evidence (e.g., from cancer registries) could be a first step.” (p. 11)
NICE 202017
Main study findings
Deterministic base-case analysis (manufacturer)
Total costs, £
NR for any comparisons (i.e., redacted from the HTA report)
Total costs of AEs per treatment, £
R2 (non-refractory to rituximab)
1,831.71
R2 (refractory to rituximab)
1773.94
R-CHOP
3,604.13
R-CVP
2,753.56
R-mono
462.41
Incremental costs, £
NR for any comparisons (i.e., redacted from the HTA report)
Total QALYs
NR for any comparisons (i.e., redacted from the HTA report)
Incremental QALYs
NR for any comparisons (i.e., redacted from the HTA report)
ICERs (£/QALY)
R2 vs. R-CHOP
11,471
R2 vs. R-CVP
16,814
R2 vs. R-mono
22,580
Deterministic base-case analysis (Expert Review Group)
Total costs, £
NR for any comparisons (i.e., redacted from the HTA report)
Incremental costs, £
NR for any comparisons (i.e., redacted from the HTA report)
Total QALYs
NR for any comparisons (i.e., redacted from the HTA report)
Incremental QALYs
NR for any comparisons (i.e., redacted from the HTA report)
ICERs (£/QALY)
R2 vs. R-CHOP
15,505
R2 vs. R-CVP
21,759
R2 vs. R-mono
27,372
Incremental and pairwise deterministic base-case analysis (Expert Review Group)
ICER (£/QALY) compared to next relevant alternative
R-CVP
ref
R-CHOP
dominated
R2
21,759
Sensitivity/scenario analyses
Probabilistic (manufacturer)
ICERs (£/QALY)
R2 vs. R-CHOP
13,443
R2 vs. R-CVP
20,896
R2 vs. R-mono
26,116
Probability of cost-effectiveness (assuming a £30,000 WTP threshold), %
R2 vs. R-CHOP
81.7
R2 vs. R-CVP
72.4
R2 vs. R-mono
69
Probabilistic (Expert Review Group)
ICERs (£/QALY)
R2 vs. R-CHOP
15,818
R2 vs. R-CVP
23,367
R2 vs. R-mono
29,010
Deterministic
R2 vs. R-CHOP
ICERs, range (£/QALY)
9,177 to 13,766
Top 3 influential model parameters (in order of influence on ICER)
Cost of ASCT
Subsequent treatment cost post-R-CHOP
Proportion of patients w/ASCT post-R-CHOP
R2 vs. R-CVP
ICERs, range (£/QALY)
NR to < £30,000
Top 3 influential model parameters (in order of influence on ICER)
Subsequent treatment cost (post-R-CVP)
Resource use cost
Administration cost of complex chemotherapy
R2 vs. R-mono
ICERs, range (£/QALY)
NR to < £30,000
Top 3 influential model parameters (in order of influence on ICER)
Subsequent treatment cost (post-R-CVP)
Resource use cost
Administration cost of complex chemotherapy
Scenario analyses
R2 vs. R-CHOP, n cost-effective scenarios/ > 80 (assuming a WTP threshold of £30,000)
All scenarios/ > 80
R2 vs. R-CVP, n cost-effective scenarios/ > 80 (assuming a WTP threshold of £30,000)
All but 1 scenario (5-year time horizon)/ > 80
Authors’ conclusion
“…the uncertainty around the cost effectiveness of R2 is substantial, mainly caused by the possible bias introduced by the indirect treatment comparison, which could not be accounted for in the ERG analyses. The ICER for R-CVP is higher and suffers from the same uncertainty. The R-mono analysis is based on a direct comparison, but is also surrounded by substantial uncertainty, as the ICER is rather sensitive to, for instance, the time-point at which treatment waning starts and utilities in the PP health state.” (p. 99 of the ERG report; p. 528/890 of the HTA document)
Appendix 5: References of Potential Interest
Note that this appendix has not been copy-edited.
Review Articles
Narrative reviews, evidence summaries and/or briefings
Shah H, Stephens D, Seymour J, Maddocks K. Incorporating novel targeted and immunotherapeutic agents in treatment of B-Cell lymphomas. American Society of Clinical Oncology Educational Book. 2021;41:1-18. PubMed
Witlox WJA, Grimm SE, Riemsma R, et al. Lenalidomide with Rituximab for Previously Treated Follicular Lymphoma and Marginal Zone Lymphoma: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. Pharmacoeconomics. 2021 02;39(2):171-180. PubMed
Blair HA. Lenalidomide: A Review in Previously Treated Follicular Lymphoma. Drugs. 2020 Sep;80(13):1337-1344. PubMed
Flowers CR, Leonard JP, Fowler NH. Lenalidomide in follicular lymphoma. Blood. 2020 06 11;135(24):2133-2136. PubMed
Healthcare Improvement Scotland. Scottish Medicines Consortium. lenalidomide 2.5mg, 5mg, 7.5mg, 10mg, 15mg, 20mg and 25mg hard capsules (Revlimid®) Celgene Ltd. 04 Sept 2020. https://www.scottishmedicines.org.uk/media/5465/lenalidomide-revlimid-fl-final-september-2020-for-website.pdf
NIHR Innovation Observatory. Lenalidomide (Revlimid) + rituximab for relapsed/refractory follicular lymphoma and marginal zone lymphoma. Oct 2017. https://www.io.nihr.ac.uk/wp-content/uploads/migrated_new/13719-Lenalidomide+rituximab-for-follicular-or-marginal-zone-lymphoma.pdf
Additional References
Relevant editorial, commentary, letter to the editor
Barr PM. Augmenting Indolent Lymphoma Treatment Options With the Combination of Lenalidomide and Rituximab. J Clin Oncol. 2019 05 10;37(14):1151-1153. PubMed
No eligible comparative data
Thanarajasingam G, Leonard JP, Witzig TE, et al. Longitudinal Toxicity over Time (ToxT) analysis to evaluate tolerability: a case study of lenalidomide in the CALGB 50401 (Alliance) trial. The Lancet Haematology. 2020 Jun;7(6):e490-e497. PubMed
Non-comparative study
Sacchi S, Marcheselli R, Bari A, et al. Safety and efficacy of lenalidomide in combination with rituximab in recurrent indolent non-follicular lymphoma: final results of a phase II study conducted by the Fondazione Italiana Linfomi. Haematologica. 2016 05;101(5):e196-199. PubMed
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up to date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
Questions or requests for information about this report can be directed to Requests@CADTH.ca