CADTH Health Technology Review
Rituximab for the Treatment of Myasthenia Gravis: A 2021 Update
Rapid Review
Authors: Calvin Young, Sarah C. McGill
Abbreviations
AChR
acetylcholine receptors
ADL
activities of daily living
AMSTAR 2
A MeaSurement Tool to Assess systematic Reviews
CI
confidence interval
IgG
immunoglobulin G
LRP4
lipoprotein receptor-related protein 4
MGFA
Myasthenia Gravis Foundation of America
MG-QOL 15
15-item Myasthenia Gravis-specific Quality of Life
MuSK
muscle-specific tyrosine kinase
QMG
quantitative myasthenia gravis
RCT
randomized controlled trial
SD
standard deviation
Key Messages
Low-quality evidence suggests that treatment with rituximab may be associated with improvements in clinical status, use of concurrent immunomodulatory therapies, quality of life, and various laboratory parameters in patients with myasthenia gravis, compared to before treatment. However, substantial methodological limitations of the included literature limit the use of these findings for informing clinical and policy decisions.
Adverse events associated with the use of rituximab were relatively common, occurring in approximately 25% to 45% of patients treated with rituximab. The adverse events experienced by patients were not considered serious by primary study authors.
No studies were identified that compared the effectiveness of rituximab to other therapies for the treatment of myasthenia gravis. Summarized studies lacked control groups, meaning that any outcomes observed in study participants should not be attributed to rituximab alone.
There is a lack of evidence on the cost-effectiveness of rituximab for the treatment of myasthenia gravis. Additionally, no evidence-based guidelines were identified.
Context and Policy Issues
Myasthenia gravis is a chronic autoimmune disease of the neuromuscular junction in which antibodies produced by the immune system target various components of the postsynaptic membrane and impair neuromuscular transmission, causing weakness and fatigue of skeletal muscle.1 Approximately 80% of patients with myasthenia gravis have antibodies against acetylcholine receptors (AChRs).2 The remaining 20% of patients typically have antibodies against muscle-specific tyrosine kinase (MuSK), antibodies against related proteins such as agrin and low-density lipoprotein receptor-related protein 4 (LRP4), or are seronegative (i.e., do not have any detectable antibodies associated with myasthenia gravis).3 This condition can be localized to specific muscle groups (e.g., extrinsic ocular muscles) or it can be generalized, affecting many regions of the body.4 Myasthenia gravis may become life-threatening when it involves the bulbar or respiratory muscles, resulting in respiratory failure that requires intubation and mechanical ventilation (known as a myasthenic crisis).5
Sex and age are important factors that appear to influence the occurrence of myasthenia gravis. In patients younger than 40 years, females have disproportionately high rates of myasthenia gravis. Conversely, in populations older than 50 years, myasthenia gravis is more common in males. Between the ages of 40 years and 50 years, and in adolescent populations, myasthenia gravis affects male and females approximately equally.1 In Ontario, myasthenia gravis has been estimated to have a crude prevalence rate of approximately 32.0 per 100,000 population, a number which has been increasing over time.6
Conventional therapies for the treatment of myasthenia gravis include cholinesterase inhibitors (e.g., pyridostigmine), which increase the amount of acetylcholine available at the neuromuscular junction, corticosteroids (e.g., prednisone), which are immunosuppressants, thymectomy, where the thymus gland is removed to stop the production of autoantibodies, and other immunomodulatory therapies (e.g., azathioprine, cyclosporine, IV immunoglobulin, and plasma exchange).7-9 The goal of these therapies is to achieve stable disease where patients experience limited symptoms (i.e., minimal manifestation status).10 Although many patients experience success with these approaches, approximately 10% of those with generalized myasthenia gravis are refractory to or are unable to tolerate conventional therapies.8 In these cases, individualized treatment strategies involving other immunomodulatory therapies, such as eculizumab or rituximab, may be considered.8,11 In October 2020, the CADTH Canadian Drug Expert Committee assessed the use of eculizumab for the treatment of adult patients with refractory generalized myasthenia gravis and gave a conditional recommendation in favour of reimbursement.12 However, the place of rituximab therapy in the treatment pathway for patients with refractory myasthenia gravis is unclear, and an assessment of the available literature could help inform clinicians and decision-makers on its appropriate use.
The objective of this report is to review the clinical effectiveness and cost-effectiveness of rituximab therapy for the treatment of myasthenia gravis in those who are refractory to standard therapy. Additionally, this report aims to summarize the evidence-based guidelines regarding the use of rituximab for the treatment of myasthenia gravis. This report updates a 2018 CADTH report13 that concluded that rituximab therapy was associated with improvement in patients with myasthenia gravis; however, definitive conclusions were not possible at that time due to limitations of the clinical literature.
Research Questions
What is the clinical effectiveness of rituximab induction therapy for the treatment of myasthenia gravis for those who are refractory to standard therapy?
What is the clinical effectiveness of rituximab re-treatment for the treatment of myasthenia gravis?
What is the clinical effectiveness of rituximab maintenance therapy for the treatment of myasthenia gravis?
What is the cost-effectiveness of rituximab therapy for the treatment of myasthenia gravis?
What are the evidence-based guidelines regarding the use of rituximab for the treatment of myasthenia gravis?
Methods
Literature Search Methods
The literature search strategy used in this report is an update of one developed for a previous CADTH report.13 For the current report, a limited literature search was conducted on key resources including MEDLINE and Embase via Ovid, the Cochrane Database of Systematic Reviews, the international HTA database, Canadian and major international health technology agencies, as well as a focused internet search. The main search concepts were rituximab and myasthenia gravis. No filters were applied to limit the retrieval by study type. The initial search was limited to English-language documents published between January 1, 1998, and July 11, 2018. For the current report, database searches were rerun on February 4, 2021 to capture any articles published since the initial search date. Conference abstracts, comments, newspaper articles, editorials, and letters were excluded. Where possible, retrieval was limited to the human population. The search of major health technology agencies was also updated to include documents published since July 2018.
Selection Criteria and Methods
The systematic review management software DistillerSR (Evidence Partners, Ottawa, Canada) was used for study selection.14 One reviewer screened citations and selected studies. In the first level of screening, titles and abstracts were reviewed and potentially relevant articles were retrieved and assessed for inclusion. The final selection of full-text articles was based on the inclusion criteria presented in Table 1.
Criteria | Description |
Population | Patients with myasthenia gravis (regardless of clinical or autoantibody subtype) who are/were refractory to standard therapy or who are unable to tolerate standard therapy |
Intervention | Q1: Rituximab induction therapy (i.e., an initial course of rituximab) Q2: Rituximab re-treatment in case of flares Q3: Rituximab, given as regularly scheduled treatment (i.e., maintenance therapy) irrespective of initial response Q4: Rituximab as induction therapy, re-treatment, or maintenance therapy Q5: Rituximab (any regimen) |
Comparator | Q1 and Q2: Standard therapy (e.g., plasma exchange, corticosteroids, IV immunoglobulin, cholinesterase inhibitors, steroid-sparing agents such as azathioprine, thymectomy, methotrexate, cyclophosphamide, cyclosporine, tacrolimus, mycophenolate); no treatment; no comparator Q3: Re-treatment with rituximab upon disease flare/relapse; standard therapy upon disease flare/relapse; no treatment; no comparator Q4: Any comparator for the treatment of myasthenia gravis Q5: Not applicable |
Outcomes | Q1 to Q3: Clinical effectiveness (e.g., remission, clinical response, need for steroids, plasmapheresis, or immunotherapy, quality of life, laboratory parameters, safety [e.g., adverse events]) Q4: Cost-effectiveness (e.g., cost per quality-adjusted life-year gained) Q5: Recommendations regarding best practices (e.g., appropriate patient populations, treatment algorithms) |
Study designs | Health technology assessments, systematic reviews, RCTs, non-randomized studies, economic evaluations, and evidence-based guidelines |
RCT = randomized controlled trial.
Exclusion Criteria
Articles were excluded if they did not meet the selection criteria outlined in Table 1, were duplicate publications, had been included in the 2018 CADTH report13 on rituximab for myasthenia gravis, or were published before 2018. Systematic reviews in which all relevant primary studies were captured in other more recent or more comprehensive systematic reviews were excluded. Primary studies retrieved by the search were excluded if they were captured in 1 or more included systematic review and their entire participant population was included in the review. In cases where only a portion of the participant population from a primary study was captured in a systematic review, the primary study was also included, and the degree of participant overlap from the systematic review and the primary study was described. Systematic reviews that had incomplete information to ascertain eligibility and did not include any information that was relevant to the current report were excluded. Guidelines with unclear methodology (i.e., if it was unclear whether a systematic search of the literature was undertaken to inform the recommendations) were excluded.
Critical Appraisal of Individual Studies
The included publications were critically appraised by 1 reviewer using the following tools as a guide: A MeaSurement Tool to Assess systematic Reviews 2 (AMSTAR 2)15 for systematic reviews and the Downs and Black checklist16 for non-randomized studies. Summary scores were not calculated for the included studies; rather, the strengths and limitations of each included publication were described narratively.
Summary of Evidence
Quantity of Research Available
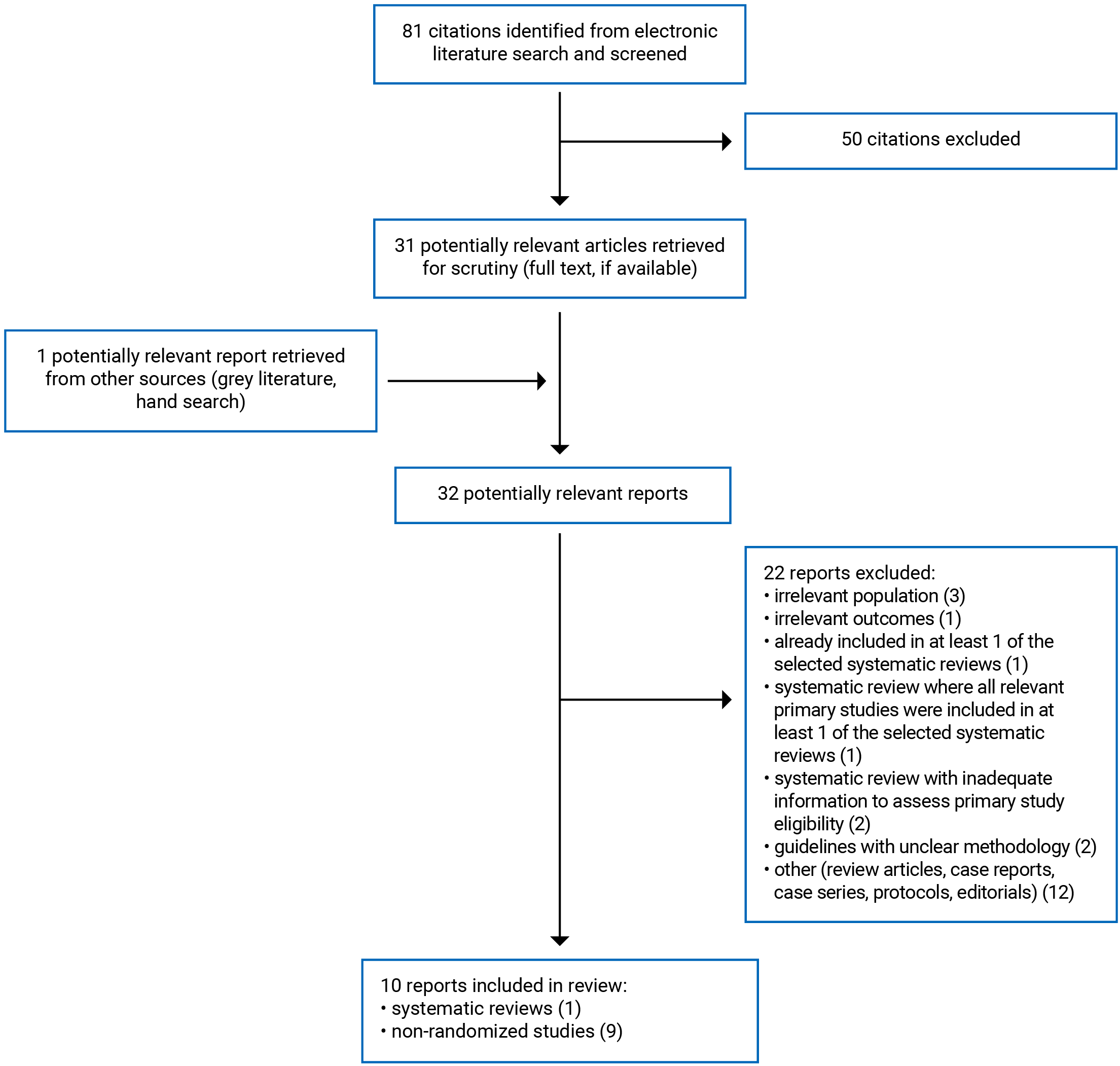
A total of 81 citations were identified in the literature search. Following screening of titles and abstracts, 50 citations were excluded and 31 potentially relevant reports from the electronic search were retrieved for full-text review. One potentially relevant publication was retrieved from the grey literature search for full-text review. Of these 32 potentially relevant articles, 22 publications were excluded for various reasons, and 10 publications met the inclusion criteria and were included in this report. These comprised 1 systematic review with meta-analysis17 and 9 non-randomized studies.18-26 Appendix 1 presents the PRISMA27 flow chart of the study selection. Additional references of potential interest are provided in Appendix 5.
Summary of Study Characteristics
One relevant systematic review with meta-analysis17 and 9 non-randomized studies18-26 were identified for inclusion in this review. No relevant health technology assessments, randomized controlled trials (RCTs), economic evaluations, or evidence-based guidelines were identified. Detailed study characteristics are available in Appendix 2 (Table 2 and Table 3).
The systematic review with meta-analysis17 had more specific inclusion criteria than the current report (i.e., narrower in scope). Specifically, the review17 assessed the effectiveness of rituximab for the treatment of patients with anti-AChR antibody–positive myasthenia gravis. The authors of the review17 included primary studies that enrolled patients with myasthenia gravis of any antibody type, but only extracted data pertaining to patients with anti-AChR antibody–positive myasthenia gravis. To ensure data from patients who had myasthenia gravis with other antibody types (e.g., anti-MuSK antibody positive, seronegative) were retained in the current report, primary studies retrieved from the electronic literature search were not excluded if they were also included in the Li et al. (2021)17 systematic review unless data from all patients relevant to the current report were summarized in the review. Of the 21 studies included in the Li et al. (2021)17 review, 519,23-26 were identified in the literature search conducted for the current report and were included separately from the systematic review as they provided information on additional patients. As a result, there is some overlap between data included in the meta-analysis from the Li et al. (2021)17 review and the findings from these 5 primary studies19,23-26 that were described narratively.
Study Design
The authors of the systematic review with meta-analysis17 searched for any human studies or clinical trials (e.g., RCTs, uncontrolled observational studies) published between January 2008 and January 2020 in MEDLINE, PubMed, and clinicaltrials.gov. There were 21 eligible studies included in the systematic review, all of which were relevant to the current report. All included primary studies were uncontrolled, single-arm observational studies.
Of the 9 non-randomized studies,18-26 2 studies21,24 were prospective, single-arm cohort studies, and 7 studies18-20,22,23,25,26 were retrospective, single-arm cohort studies. All 9 non-randomized studies18-26 did not include a control group. These studies made formal or informal comparisons from before treatment to after treatment and described outcomes in cohorts of patients who all received rituximab. Three studies18,23,26 were multi-centre, and 6 studies19-22,24,25 were conducted at a single centre.
Country of Origin
The included systematic review17 was by authors in China. The countries in which the primary studies included in the systematic review17 were conducted were not reported.
The non-randomized studies were conducted in Austria,26 China,21,24 France,18 India,25 Italy,20 the Republic of Korea,23 and the US.19,22
Patient Population
The systematic review with meta-analysis17 included studies of participants with refractory anti-AChR antibody–positive myasthenia gravis. A description of how refractory was defined was not provided in the review. A total of 260 patients were included in the review. The mean age of patients at the time of treatment was 43.7 years (median = 44.1 years), the proportion of female patients was 63.5%, and the mean disease duration at the time of treatment was 45.35 months.
All 9 non-randomized studies18-26 included patients with myasthenia gravis who were refractory, steroid-dependent, or who had unacceptable adverse reactions to conventional treatment. The number of relevant patients included in each study ranged between 5 and 56, and the total number of relevant patients included in all non-randomized studies18-26 was 184 (33 of these patients were also included in the systematic review with meta-analysis17). Seven studies18,20,21,23-26 were specific to adult populations, and 1 study22 was specific to children and adolescents. In 1 study,19 it was unclear if both adults and children were included or if the patient population only included adults. The mean or median ages of patients included in the non-randomized studies18-26 ranged between 11.6 years and 51.0 years. The proportion of female patients varied between 12.5% and 93.3%. Mean or median disease duration at the time of treatment ranged between 15.1 months and 184 months.
Interventions and Comparators
The eligible systematic review17 investigated rituximab. Studies included in the review17 were classified as having provided participants with low-dose rituximab or the routine dose of rituximab, but specific definitions of low dose and routine dose were not provided. The exact dosing of rituximab infusions varied across studies. In most instances, patients were given rituximab at 375 mg/m2. Other doses included 750 mg/m2 or fixed doses of 500 mg, 600 mg, or 1,000 mg. Most studies provided patients with maintenance treatments or re-treatments based on clinical course. The frequency of doses and the protocols for maintenance treatments and re-treatments was variable. None of the studies included in the systematic review17 included a control group (i.e., all studies were uncontrolled and only included patients who received rituximab).
All patients included in the non-randomized studies18-26 were treated with rituximab. Patients received different doses of rituximab across studies and within studies, including 375 mg/m2,18,23,25,26 750 mg/m2,22 or fixed doses of 500 mg,26 600 mg,21,24 1,000 mg.18-20,26 Dosing was often described as being at the discretion of the treating physician. The frequency of induction doses, and the protocols used for maintenance and re-treatment were variable across studies and within studies, and was typically informed by clinical status (e.g., patients who relapsed or who experienced CD19+ B cell repopulation may have been provided with additional treatments). Detailed descriptions of treatment protocols for each non-randomized study are provided in Appendix 2 (Table 3). None of the included non-randomized studies18-26 included a control group.
Outcomes
All included studies17-26 reported on outcomes relating to the clinical effectiveness of rituximab for the treatment of myasthenia gravis, including measures of clinical response, concurrent immunomodulatory therapies, myasthenia exacerbations, quality of life, laboratory parameters, and adverse events.
Measures of clinical response included the proportion of participants who achieved various improvements in Myasthenia Gravis Foundation of America (MGFA) post-intervention status,17-21,23,25,26 mean MGFA class,22 mean manual muscle test scores,18,21 the proportion of participants who experienced remission,19 mean number of days to achieve clinical remission,19 the proportion of participants who experienced relapse,19 mean number of days to relapse,19 and mean MGFA quantitative myasthenia gravis (QMG) scores.24 The MGFA post-intervention status is a scale that classifies patients in groups based on disease severity and the localization of the symptoms. Classes range from complete stable remission (healthiest class) to death from myasthenia gravis (lowest class).28 The manual muscle test examines the strength in 12 bilateral muscle groups and 6 ocular or axial muscles that are typically symptomatic in patients with myasthenia gravis. Each muscle is scored from 0 (normal strength) to 4 (paralysis). Total scores are the sum of each muscle score and can range between 0 and 72, where lower scores indicate more muscle strength (i.e., less disease severity).29 The QMG score is a 13-item scale used to quantify disease severity in myasthenia gravis. The scale assesses functionality in various muscles (e.g., ocular, bulbar, respiratory, limb) and grades each finding. Total scores range from 0 (no myasthenic findings) to 39 (maximal myasthenic deficits).30
Changes to concurrent immunomodulatory therapies (e.g., prednisone, steroids, oral steroid-sparing agents) were reported in the systematic review17 and 8 non-randomized studies.18-20,22-26 Specific outcomes included the proportion of patients who achieved the ability to reduce the daily dose of prednisone to 10 mg or less or to 50% or less,17-19,26 the proportion of patients who discontinued prednisone or other immunomodulatory therapies,17,18,20,25,26 the mean dose of immunomodulatory therapies,19,23,24 the mean number of concurrent immunomodulatory therapies,22 and the proportion of patients who used IV immunoglobulin, plasma exchange, or oral steroid-sparing agents.19,25,26
Myasthenia exacerbations were reported in 4 non-randomized studies.18,19,22,25 Reported outcomes were the proportion of patients who experienced myasthenia gravis exacerbation or myasthenic crisis,18,25 the mean number of disease exacerbations,19 the proportion of patients hospitalized for disease exacerbations,19 and the mean number of myasthenia gravis–related hospitalizations.22
Quality of life was reported in 2 non-randomized studies21,24 and was assessed using mean Myasthenia Gravis–related Activities of Daily Living (MG-ADL) scores21,24 and mean 15-item Myasthenia Gravis-specific Quality of Life (MG-QOL 15) scores.21,24 The MG-ADL scale is an 8-item patient-reported scale that assesses the individual’s ability to speak, chew, swallow, breathe, perform self-care activities, perform physical activities, and vision-related parameters (i.e., double vision and eyelid dropping). Each parameter is scored between 0 and 3 (total scores range between 0 and 24), with higher scores indicating increased limitations in daily activities caused by myasthenia gravis.31 The MG-QOL 15 is a short, self-administered questionnaire that evaluates 15 items categorized into 4 dimensions (i.e., mobility, symptoms, general contentment, and emotional well-being). Each item is scored between 0 (no impairment) and 4 (very much impaired). Total scores are the sum of scores from each item and can range between 0 (highest quality of life) and 60 (maximal impairment).32
Several laboratory parameters were reported in 5 non-randomized studies,20-22,24,26 including autoantibody levels (e.g., anti-AChR, anti-MuSK),20,22,24,26 MuSK-specific immunoglobulin G (IgG) and IgG4 levels, levels of various microRNAs (i.e., miR-150-5, miR-146a-5p),21 and various lymphocyte counts (e.g., B cells, T cells, natural killer cells).21,24
One systematic review17 and 7 non-randomized studies17-19,22-26 reported on outcomes related to the safety of rituximab. These outcomes included the proportion of patients who experienced any adverse event17-19,22,24 and the proportion of patients who experienced specific adverse events (e.g., cytopenia, arrythmia, infection, infusion reaction, and death).17,18,23,25,26
Summary of Critical Appraisal
Additional details regarding the strengths and limitations of the included publications are provided in Appendix 3 (Table 4 and Table 5).
Systematic Reviews
The included systematic review with meta-analysis17 had clearly defined objectives and eligibility criteria, searched multiple databases (i.e., MEDLINE, PubMed, and clinicaltrials.gov), and provided a description of key search terms (i.e., “myasthenia gravis” and “rituximab”) and search restrictions (e.g., only studies published in English were eligible, the search was restricted between January 2008 and January 2020), increasing the reproducibility of the literature searches. The methods for article selection and data extraction were well-documented and were conducted in duplicate or were conducted by 1 reviewer and verified by additional reviewers, decreasing the likelihood for inconsistency in these processes. In addition, the review17 included a figure that illustrated the article selection process, reasons for article exclusion were provided, and the authors described the included primary studies in adequate detail. Statistical heterogeneity and publication bias were assessed by the I2 test and the classic fail-safe N test (no indicators of publication bias were identified), respectively. Review authors disclosed their sources of funding (which were considered unlikely to have influenced the findings of the review) and stated that they had no related conflicts of interest.
As for methodological limitations, the review authors did not provide justification for their selection of eligible study designs (i.e., any human studies or clinical trials), a list of studies excluded after full-text review was not provided, sources of funding for the included primary studies were not reported, and the literature searching did not include a grey literature search, increasing the risk that relevant, non-indexed articles were not captured. Additionally, it was unclear whether the review methods were established before conducting the review (there was no mention of a protocol), increasing the risk for selective reporting. A significant methodological limitation to this systematic review17 was that the quality or risk of bias of included primary studies was not assessed. As a result, the quality or risk of bias among included primary clinical studies was not considered by the review authors when interpreting and discussing the results of the review and the potential impact of risk of bias in primary studies on the results of the meta-analyses was not examined. While not formally investigated, the quality of studies included in the systematic review was expected to be low due to their study design (i.e., uncontrolled single-arm cohort studies). Meta-analyses were conducted using fixed-effect models, which the authors justified as I2 values were less than 50%; however, this is not appropriate as primary study participants were sampled from different populations and there was a high degree of clinical heterogeneity across primary studies (e.g., there were differences in patient age, rituximab treatment regimen, and follow-up duration). Random-effect models would have been more appropriate.
Non-Randomized Studies
The 9 non-randomized studies18-26 were considered to be of low methodological quality based on the assessments using the Downs and Black checklist.16 There were several strengths common to all 9 non-randomized studies,18-26 including clearly described objectives, interventions, patient eligibility criteria, and main outcomes; participant characteristics, such as age, sex, antibody type, MGFA class, current and previous treatment, and disease duration, were provided; compliance with the intervention was reliable; main findings were clearly reported; any potential conflicts of interest were declared by the authors (in 8 studies there were no potential conflicts; the authors of 1 study declared some potential conflicts but they were considered unlikely to have influenced the findings of the study); and care providers and care settings appeared to be representative of the settings of interest, increasing external validity. The authors of 7 non-randomized studies19-24,26 reported their sources of funding, and none of these studies were industry-funded.
There were significant methodological limitations identified for each of the 9 non-randomized studies.18-26 In all cases, studies did not compare outcomes in patients treated with rituximab versus a control group of patients who did not receive rituximab (i.e., the studies were non-comparative and uncontrolled). As a result, the findings of these studies18-26 are susceptible to numerous forms of bias that threaten both internal and external validity. Any outcomes observed in study participants should not be attributed to rituximab alone, as there are many uncontrolled factors that may have contributed to the findings of these studies. Additionally, changes to concurrent medication provided to patients throughout the study periods, including immunomodulatory therapies (e.g., prednisone, azathioprine, IV immunoglobulin, mycophenolate, cyclosporine, plasma exchange), were permitted and were not controlled for. Patients who deteriorated or who experienced a myasthenic crisis while on rituximab during study periods were eligible for rescue therapy, and any additional treatments provided would have a confounding therapeutic effect (i.e., outcomes cannot be attributed to rituximab alone). Unlike in RCTs, participants in each of the included studies were not randomized to receive treatment with rituximab but were provided rituximab at the discretion of treating physicians. It is expected that treating physicians would have selected patients who were perceived as being more likely to succeed on rituximab than the average patient with refractory myasthenia gravis (i.e., there is a risk for selection bias, and it is unclear how findings may extend to patients in general practice). Similarly, Topakian and colleagues26 identified their patient population by inviting physicians via email to voluntarily submit anonymized data from all of their adult patients with myasthenia gravis who were treated with rituximab for inclusion in their study; physicians who had a positive clinical experience treating patients with myasthenia gravis with rituximab may have been more likely to submit data, and the findings may therefore not be representative of all patients who received rituximab in Austria. In the study by Marino et al. (2020),20 it was unclear if the patient population represented a true cohort or if patients were selected on the basis of an exposure and an outcome (i.e., there may have been other eligible patients treated with rituximab that were not included in this study). It was judged to likely be a cohort study; however, it cannot be ruled out that this study was a case series (and would then be ineligible for inclusion in the current review). There were some additional concerns relating to the generalizability of the findings. In particular, none of the included non-randomized studies were conducted in Canada, 6 studies19-22,24,25 were conducted at a single centre, and 3 studies20,22,25 included data from 10 or fewer patients. As a result, the generalizability of findings to Canadian settings was unclear. Finally, the authors of 2 studies18,25 did not report their sources of funding.
Summary of Findings
The overall findings of the included studies are highlighted below. Detailed summaries of the main findings and authors’ conclusions are available in Appendix 4.
Clinical Effectiveness of Rituximab
This report included 3 research questions regarding the effectiveness of rituximab induction therapy, rituximab re-treatment, and rituximab maintenance therapy for the treatment of myasthenia gravis. However, identified literature did not examine the effectiveness of rituximab used exclusively in 1 of these 3 manners. Rather, the studies provided patients with an initial course of rituximab and then decided whether subsequent maintenance or re-treatments were appropriate given the patients response to treatment. The dose and frequency of treatments was variable (as previously described) and was not applied consistently to all participants within the same study. Therefore, it is not possible to discuss results by research question but to provide a summary of the findings (by outcome) for patients who received any form of rituximab therapy to treat myasthenia gravis.
Clinical Response
Outcomes related to clinical response were reported in the systematic review with meta-analysis17 and 9 non-randomized studies.18-26 The pooled estimates from the Li et al. (2021)17 systematic review contain some of the same data from 85 patients in 5 non-randomized studies19,23-26 for which results are described separately.
Meta-analytic findings from the Li et al. (2021)17 systematic review, which included data from 260 patients reported in 21 primary studies, indicated that 77.0% (95% CI, 70.1% to 82.6%; P = 0.000) of participants achieved improved clinical status from baseline to follow-up (median duration after rituximab treatment was 37.5 months), which was defined as a MGFA post-intervention status of complete stable remission, minimal manifestations, and/or improved. Additionally, 50.8% (95% CI was not reported) achieved minimal manifestations or better status at follow-up; however, this result was not statistically significant (P = 0.921).
Within the non-randomized study by Dos Santos et al. (2020),18 which included 29 patients, the proportion of patients who achieved MGFA post-intervention status of improved or better was 86.2% at 6 months post-rituximab and 90.5% at 12 months post-rituximab. Compared to baseline (i.e., pre-rituximab), the mean myasthenic muscle score of participants statistically significantly improved at both 6 months (P < 0.0001) and 12 months (P = 0.006) after initiating rituximab. Of the 33 patients included in the study by Litchman and colleagues,19 63.6% had a MGFA post-intervention status of minimal manifestations or better at 12 months follow-up, 69.7% had a MGFA post-intervention status of minimal manifestations or better at final follow-up, and 63.6% achieved clinical remission. The proportion of patients who relapsed during the follow-up period, which had a mean duration of 1,861 days, was 48.5%.
The authors of the Marino et al. (2020)20 study, which included 9 patients, noted that 6 patients (66.6%) achieved optimal response (defined as the achievement and maintenance of the status of minimal manifestations or better together with a 50% or greater reduction in steroid dose, withdrawal of immunosuppressants, and no need for plasma exchange or IV immunoglobulin), 2 patients (22.2%) achieved partial response (defined as clinical improvements while failing to achieve the status of minimal manifestations or better or when prednisone reduction was less than 50% of pre-treatment dosage, with or without immunosuppressant withdrawal and no need for plasma exchange or IV immunoglobulin), and 1 patient (11.1%) had no response.
Participants in the Zhong et al. (2020)21 study (N = 12) had statistically significant decreases in mean MGFA QMG scores (27.8% decrease; P = 0.019) and mean myasthenia gravis–specific manual muscle test scores (67.4% decrease; P = 0.019) at 6 months after initiating rituximab compared to baseline values. Zingariello et al. (2020)22 reported a decrease in mean MGFA class of participants (N = 5) from 2.6 to 1.2 following treatment with rituximab; however, the statistical significance of this decrease was not assessed. Of the 17 participants included in the study by Choi et al. (2019),23 11 (65%) achieved MGFA post-intervention status of minimal manifestations or better with low-dose prednisolone (≤ 5 mg per day) during the follow-up period (median follow-up was 24 months). The median time to achieve this outcome was 7.6 months. The study by Jing et al. (2019)24 measured mean MGFA QMG scores and mean manual muscle test scores at baseline and 6 months post-rituximab in 15 study participants. For MGFA QMG scores, mean values decreased significantly from 15.7 (standard deviation [SD] = 4.9) to 11.2 (SD = 4.4; P = 0.013). Similarly, mean manual muscle test scores significantly decreased from 22.7 (SD = 18.1) to 6.9 (SD = 6.5; P = 0.004). Singh and Goyal (2019)25 stated that all 8 participants included in their study had improved MGFA post-intervention status following treatment with rituximab.
Topakian and colleagues26 categorized patients (N = 56) by MGFA post-intervention status at various follow-up periods. Three months after starting rituximab, 0% of patients died, 1.9% were worse, 11.3% were unchanged, 41.5% were improved, 18.9% had minimal manifestation status, and 26.4% were in complete stable remission. At final follow-up (median 20 months post-rituximab), 1.8% of patients died, 0% were worse, 1.8% were unchanged, 28.6% were improved, 25.0% had minimal manifestation status, and 42.9% were in complete stable remission. The distribution of patients into each category was also reported 6 months post-rituximab and 12 months post-rituximab (these findings are summarized in Appendix 4). The statistical significance of these findings was not assessed.
Concurrent Immunomodulatory Therapies
Information on the effect of rituximab treatment on concurrent immunomodulatory therapies was available in the systematic review with meta-analysis17 and 8 non-randomized studies.18-20,22-26
Findings from the meta-analysis,17 which included data from a total of 260 patients from 21 primary studies, indicated that 70.6% (95% CI, 57.8% to 80.7%; P = 0.002) of patients achieved the ability to reduce the daily dose of prednisone to 10 mg or less and 66.6% (95% CI, 50.1% to 79.8%; P = 0.048) of patients discontinued immunosuppressants after treatment with rituximab (at variable lengths of follow-up).
In the non-randomized study by Dos Santos et al. (2020),18 the mean dose of concurrent steroid treatment significantly decreased from 20.0 mg/day (N = 29) to 7.0 mg/day (N = 21) after 6 months of rituximab (P = 0.0005). Of the 19 patients with follow-up data, 36.8% and 57.9% of patients achieved the ability to reduce the daily dose of steroid to 10 mg or less at the 6-month and 12-month follow-ups, respectively. Additionally, 21.1% and 36.8% of patients discontinued steroid treatment 6 months and 12 months after initiating rituximab, respectively.
Litchman and colleagues19 reported that patients who were anti-AChR antibody positive (n = 17) had significantly decreased mean prednisone dose at 12 months (P < 0.01) and at final follow-up (mean follow-up of 20.06 months; P < 0.01), compared to baseline (i.e., before rituximab). Similarly, patients who were anti-MuSK antibody positive (n = 16) had significant decreases in mean prednisone dose at 12 months after initiating rituximab (P < 0.01) and at final follow-up (P < 0.01) compared to baseline. In the total study cohort (N = 33), 81.8% of patients had reduced the daily dose of prednisone to 10 mg or less and 63.3% had tapered prednisone completely at final follow-up. The number of patients on rescue therapy with IV immunoglobulin or plasma exchange decreased significantly between baseline and final follow-up in both patients who were anti-AChR antibody positive (n = 17; P < 0.01) and those who were anti-MuSK antibody positive (n = 16; P < 0.01). There were no significant differences in the number of patients on maintenance therapy with IV immunoglobulin or plasma exchange at baseline and at final follow-up.
Of the 9 participants included in the study by Marino et al. (2020),20 2 patients (22.2%) were tapered off prednisone completely before final follow-up (mean duration of 51.9 months). Zingariello and colleagues22 stated that the mean number of concurrent immunomodulatory medications used by their study participants (N = 5) decreased from 2.8 before rituximab to 1.2 at mean follow-up of 11.6 months; however, the statistical significance of this decrease was not assessed. In the Choi et al. (2019) study,23 mean prednisolone dose significantly decreased from 28 mg/day at the initiation of rituximab to 7.5 mg/day between weeks 20 and 24 post-rituximab (P < 0.001). Similarly, the mean dose of prednisone was significantly reduced by 40% (P = 0.001) from baseline after 6 months of rituximab in the study by Jing et al. (2019),24 which included 15 participants. Eight patients were included in the study by Singh and Goyal (2019)25: 6 (75%) were able to taper prednisone completely and 0 required treatment with IV immunoglobulin or plasma exchange after treatment with rituximab during the follow-up period (median duration of 18 months). The authors of the Topakian et al. (2019)26 study noted that during the follow-up period (median duration of 20 months), 59% of patients discontinued steroid treatment, 25.6% of patients reduced the daily dose of steroid treatment to 50% or less but were unable to discontinue altogether, and 23.2% of study patients required treatment (with IV immunoglobulin, subcutaneous immunoglobulin, immunoadsorption, eculizumab, or plasma exchange) after rituximab.
Myasthenia Exacerbations
Information on the effectiveness of rituximab with respect to myasthenia exacerbations was available in 4 non-randomized studies.18,19,22,25
The authors of the Dos Santos et al. (2020) study 18 noted that the proportion of patients from their study population (N = 29) who experienced myasthenia gravis exacerbation or myasthenic crisis during the follow-up period (mean follow-up of 20.06 months) was 20.6%. Participants in the Litchman et al. (2020) study 19 who were anti-AChR antibody positive (n = 17) experienced an average of 1.7 (SD = 1.2) disease exacerbations, while those who were anti-MusK antibody positive (n = 16) experienced a mean 1.4 (SD = 1.1) disease exacerbations during variable lengths of follow-up (mean follow-up duration after rituximab treatment was 1,861 days). Overall, 15.2% of study participants were hospitalized due to disease exacerbations. Zingariello and colleagues22 noted that at mean follow-up of 11.6 months after receiving rituximab, none of their study participants (N = 5) had been hospitalized due to juvenile myasthenia gravis exacerbations. The authors of the Singh and Goyal (2019) study 25 reported that none of their study participants (N = 8) experienced myasthenic crisis during the follow-up period, which had a median duration of 18 months. The statistical significance of these findings was not assessed by the authors of any of these studies18,19,22,25 because these findings were not compared to outcomes in a different treatment or control group.
Quality of Life
Two non-randomized studies21,24 reported on measures of quality of life. Six months after initiating rituximab, participants in the Zhong et al. (2020)21 study (N = 12) reported significant improvements in mean MG-ADL scores compared to baseline (before receiving rituximab) (P = 0.022) but no statistically significant differences in mean MG-QOL 15 scores (P = 0.13). Participants in the Jing et al. (2019) study24 (N = 15) reported statistically significant improvements at 6 months follow-up in mean myasthenia gravis–related ADL scores (P = 0.002) and mean MG-QOL 15 scores (P = 0.018) compared to baseline values.
Laboratory Parameters
Five non-randomized studies20-22,24,26 measured various laboratory parameters before and after treatment with rituximab. Marino and colleagues20 demonstrated that patients (N = 8) had significantly reduced MuSK-specific IgG and IgG4 levels at 2 months to 7 months and at 12 months to 30 months post-rituximab (IgG: P < 0.02; IgG4: P < 0.01). Additionally, patients had statistically significantly decreased MuSK-specific IgG/IgG4 ratios at 2 months to 7 months post-rituximab (P < 0.05). Participants in the study by Zhong et al. (2020)21 (N = 12) had significantly decreased mean levels of miR-150 to 5 (P = 0.006), CD19+ B cell counts (P = 0.006), and CD27+ memory B cell counts (P = 0.006) at 6 months after initiating rituximab compared to baseline (i.e., before treatment with rituximab). There were no significant differences in mean levels of miR-146a-5p, mean CD3+ T cell counts, mean CD4+ T cell counts, mean CD8+ T cell counts, and mean natural killer cell counts. The authors of the Zingariello et al. (2020)22 study stated that all study participants (N = 5) had decreased anti-AChR or anti-MuSK antibody titres following rituximab (at an unspecified length of follow-up); however, the magnitude and statistical significance of the decrease was not reported. Compared to baseline before treatment with rituximab, participants in the Jing et al. (2019)24 study had statistically significantly decreased counts of CD19+ B cells (P < 0.0001), CD27+ memory B cells (P < 0.0001), BAFF-R+ B cells (P < 0.0001), regulatory T cells (P < 0.001), and mean levels of anti-AChR antibodies (P = 0.016) after 6 months of treatment. There were no significant changes to CD3+ T cells, CD4+ T cells, or CD8+ T cells. The authors of the Topakian et al. (2019)26 study noted that 35.8% of their study population (20 of 56) had antibodies tested before and after treatment with rituximab. Although 13 of these patients had a decrease in antibody levels, the authors noted that interpretation of the data was compromised because follow-up durations were variable. The statistical significance and magnitude of the decrease were not reported.
Adverse Events
One systematic review17 and 7 non-randomized studies18,19,22-26 reported adverse events after treatment with rituximab. The systematic review and meta-analysis by Li et al. (2021)17 reported rates of adverse events in their patient population that included pooled data from 19 primary studies. Of the 260 patients included in the meta-analysis, 26.4% experienced any adverse event, 5.3% experienced cytopenia, 4.5% experienced arrhythmia, 12.9% experienced infection, 19.2% experienced infusion reaction, and 7.5% died at variable lengths of follow-up.
The authors of the non-randomized study by Dos Santos et al. (2020)18 noted that of the 28 patients included in their study, 42% experienced any adverse event, 7% experienced infusion reaction, 21.4% experienced infection, 7% experienced hematological disorders (i.e., thrombopenia or hypogammaglobulinemia), 3.7% experienced cardiologic disorders (i.e., bradycardia), and 3.7% experienced psychiatric disorders (i.e., suicide) at a mean follow-up of 20.06 months. Of the 17 participants in the study by Choi et al. (2019),23 2 patients experienced infusion reactions (11.8%), 1 patient was affected by herpes zoster (5.9%), and 1 patient died due to complications of invasive thymoma (5.9%) during the follow-up period (median duration of 24 months). The study by Singh and Goyal (2019)25 included 8 patients treated with rituximab. During the follow-up period (median duration of 18 months), 3 patients developed transaminitis, 1 patient developed osteonecrosis, and 1 patient tested positive for hepatitis B infection. None of the patients had infusion-associated reactions or cytopenia post-rituximab infusion. The authors of the Topakian et al. (2019)26 study stated that rituximab was generally well-tolerated by the 56 patients included in their study; however, several side effects and complications potentially related to rituximab were observed during follow-up (median duration of 20 months), including infusion reactions (n = 3), respiratory tract infections (n = 3), chronic pain syndromes (n = 2), enteritis (n = 2), herpes zoster (n = 1), erysipelas (n = 1), cholecystitis (n = 1), an unspecified mental disorder (n = 1), and alopecia areata (n = 1). One 59-year-old patient died 4.5 months after starting rituximab. The cause of death was assumed to be related to cardiac issues, although no autopsy was performed. The authors of 3 studies19,22,24 reported that patients in their study tolerated rituximab treatment without severe adverse events but did not elaborate any further.
Cost-Effectiveness of Rituximab
No relevant evidence regarding the cost-effectiveness of rituximab therapy for the treatment of myasthenia gravis was identified; therefore, no summary can be provided.
Guidelines
No relevant evidence-based guidelines regarding the use of rituximab for the treatment of myasthenia gravis were identified; therefore, no summary can be provided.
Limitations
There was a lack of high-quality studies and studies that compared the clinical effectiveness of rituximab to other standard therapies or to no treatment in patients with myasthenia gravis. The systematic review17 included data from 21 single-arm observational studies and did not evaluate the effectiveness of rituximab versus alternative treatment options. Similarly, the 9 non-randomized studies18-26 identified for inclusion in this report were single-arm cohort studies (i.e., they did not include a control group), and the majority were retrospective in design.18-20,22,23,25,26 The number of patients included in any single primary study (including studies summarized in the systematic review) ranged from 5 to 56, and the total number of relevant patients included in all 9 non-randomized studies18-26 was 184. Furthermore, the systematic review17 lacked methodical rigour and did not assess the quality of its included primary studies. As a result, the risk of bias is high, and the results summarized in this review should be interpreted with caution.
There was a high degree of clinical heterogeneity across studies, including differences in rituximab treatment protocols (i.e., the frequency and dosing of rituximab infusions), patient characteristics, reported outcomes, and length of follow-up. Further work is required to determine which patient characteristics (e.g., antibody type, age, sex, disease duration or severity) are most associated with benefit from rituximab treatment and the optimal dose and frequency of rituximab treatment.
None of the included studies17-26 discussed minimal clinically important difference values for any of the outcomes measured using continuous scales (e.g., clinical response measured with manual muscle test or QMG scores, quality of life measured with MG-QOL 15 scores). It is unclear if any of the reported changes in mean scores on these scales translate into clinically meaningful differences.
Only 1 included non-randomized study,22 which recruited a total of 5 patients younger than 18 years of age, was specific to children and adolescents; thus, the effectiveness of rituximab for the treatment of pediatric myasthenia gravis is unclear.
No evidence regarding the cost-effectiveness of rituximab for the treatment of patients with myasthenia gravis was identified. Additionally, no evidence-based guidelines regarding the use of rituximab for the treatment of patients with myasthenia gravis were identified.
Based on the information presented in the systematic review17 and in the non-randomized studies,18-26 none of the included primary clinical studies were conducted in Canada. Therefore, the clinical findings summarized in this report have unclear generalizability to Canadian settings.
Conclusions and Implications for Decision- or Policy-Making
This review comprised 1 systematic review with meta-analysis17 and 9 non-randomized studies18-26 regarding the clinical effectiveness of rituximab for the treatment of myasthenia gravis. All studies compared clinical outcomes from before treatment with rituximab to after treatment with rituximab within groups of patients. No studies compared the clinical effectiveness of rituximab with alternative therapies or no treatment with rituximab. No relevant cost-effectiveness literature or evidence-based guidelines were identified.
The findings of the systematic review with meta-analysis17 and the non-randomized studies18-26 suggested that rituximab may provide some clinical benefit to patients with myasthenia gravis. Treatment with rituximab was associated with improved clinical status,17-26 decreased usage of concurrent immunomodulatory therapies,17-20,22-26 improved quality of life,21,24 and improvements to various laboratory parameters (e.g., lymphocyte counts and levels of anti-AChR or anti-MuSK antibodies) compared to before treatment20-22,24,26; however, it was unclear whether the magnitude of improvement reported for these outcomes was clinically meaningful. Generally, the adverse events and side effects experienced by those treated with rituximab were not considered serious by study authors.17-19,22-26 When reported, the proportion of patients who experienced any adverse event following treatment with rituximab ranged between 26.4%17 and 42.8%.18
The findings of the current report are similar to the findings summarized in the 2018 CADTH report on this topic,13 which concluded that while rituximab appeared to be associated with improvement in patients with myasthenia gravis, definitive conclusions were not possible and that the findings needed to be interpreted with caution.
The limitations of the included literature (e.g., lack of studies with between-group comparisons, small sample sizes, high risk of bias, clinical heterogeneity between included studies, and unclear generalizability to Canadian settings) make it difficult to draw conclusions regarding the clinical effectiveness of rituximab for the treatment of myasthenia gravis. Future studies that directly compare the clinical effectiveness of rituximab with other treatment options (e.g., plasma exchange, corticosteroids, IV immunoglobulin, azathioprine, eculizumab) or with placebo-treated control groups will help to isolate the impact of rituximab and better inform clinical and policy decisions regarding its place in care.
References
1.Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J. Myasthenia gravis: a review. Autoimmune Dis. 2012;2012:874680-874680. PubMed
2.Rivner MH, Pasnoor M, Dimachkie MM, Barohn RJ, Mei L. Muscle-specific tyrosine kinase and myasthenia gravis owing to other antibodies. Neurol Clin. 2018;36(2):293-310. PubMed
3.Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Research. 2016;5:F1000 Faculty Rev-1513.
4.Sieb JP. Myasthenia gravis: an update for the clinician. Clin Exp Immunol. 2014;175(3):408-418. PubMed
5.Wendell LC, Levine JM. Myasthenic crisis. Neurohospitalist. 2011;1(1):16-22. PubMed
6.Breiner A, Widdifield J, Katzberg HD, Barnett C, Bril V, Tu K. Epidemiology of myasthenia gravis in Ontario, Canada. Neuromuscul Disord. 2016;26(1):41-46. PubMed
7.Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36(2):311-337. PubMed
8.Bird S. Overview of the treatment of myasthenia gravis. In: Shefner J, ed. UpToDate. Waltham (MA): UpToDate; 2020: https://www.uptodate.com/contents/overview-of-the-treatment-of-myasthenia-gravis/print. Accessed 2021 Feb 11.
9.Menon D, Barnett C, Bril V. Novel treatments in myasthenia gravis. Front Neurol. 2020;11:538-538. PubMed
10.Mantegazza R, Bonanno S, Camera G, Antozzi C. Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiatr Dis Treat. 2011;7:151-160. PubMed
11.Hoffmann S, Meisel A. Escalation strategies in the treatment of refractory myasthenia gravis. Neurology International Open. 2018;02(01):E56-E59.
12.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: eculzimab (Soliris - Alexium Pharma Canada Corp.). Ottawa (ON): CADTH; 2020 Oct: https://cadth.ca/sites/default/files/cdr/complete/SR0605%20Soliris%20MG%20-%20CDEC%20Final%20Recommendation%20October%2021%2C%202020_for%20posting.pdf. Accessed 2021 Mar 9.
13.Banerjee S, Adcock L. Rituximab for the treatment of myasthenia gravis: a review of clinical effectiveness, cost-effectiveness, and guidelines. (CADTH rapid response report: summary with critical appraisal). Ottawa (ON): CADTH; 2018: https://www.cadth.ca/rituximab-treatment-myasthenia-gravis-review-clinical-effectiveness-cost-effectiveness-and-0. Accessed 2021 Feb 4.
14.Evidence Partners. DistillerSR [computer program]. Ottawa (ON): Evidence Partners; 2011-.
15.Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. PubMed
16.Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377-384. PubMed
17.Li T, Zhang GQ, Li Y, et al. Efficacy and safety of different dosages of rituximab for refractory generalized AChR myasthenia gravis: A meta-analysis. J Clin Neurosci. 2021;85:6-12. PubMed
18.Dos Santos A, Noury JB, Genestet S, et al. Efficacy and safety of rituximab in myasthenia gravis: a French multicentre real-life study. Eur J Neurol. 2020;27(11):2277-2285. PubMed
19.Litchman T, Roy B, Kumar A, Sharma A, Njike V, Nowak RJ. Differential response to rituximab in anti-AChR and anti-MuSK positive myasthenia gravis patients: a single-center retrospective study. J Neurol Sci. 2020;411:116690. PubMed
20.Marino M, Basile U, Spagni G, et al. Long-lasting rituximab-induced reduction of specific-but not total-IgG4 in MuSK-positive myasthenia gravis. Front Immunol. 2020;11:613. PubMed
21.Zhong H, Lu J, Jing S, et al. Low-dose rituximab lowers serum Exosomal miR-150-5p in AChR-positive refractory myasthenia gravis patients. J Neuroimmunol. 2020;348:577383. PubMed
22.Zingariello CD, Elder ME, Kang PB. Rituximab as adjunct maintenance therapy for refractory juvenile myasthenia gravis. Pediatr Neurol. 2020;111:40-43. PubMed
23.Choi K, Hong YH, Ahn SH, et al. Repeated low-dose rituximab treatment based on the assessment of circulating B cells in patients with refractory myasthenia gravis. Ther Adv Neurol Disord. 2019;12:1756286419871187. PubMed
24.Jing S, Lu J, Song J, et al. Effect of low-dose rituximab treatment on T- and B-cell lymphocyte imbalance in refractory myasthenia gravis. J Neuroimmunol. 2019;332:216-223. PubMed
25.Singh N, Goyal V. Rituximab as induction therapy in refractory myasthenia gravis: 18 month follow-up study. J Neurol. 2019;266(7):1596-1600. PubMed
26.Topakian R, Zimprich F, Iglseder S, et al. High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol. 2019;266(3):699-706. PubMed
27.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. 2009;62(10):e1-e34. PubMed
28.Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis. Recommendations for clinical research standards. 2000;55(1):16-23.
29.Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring clinical treatment response in myasthenia gravis. Neurol Clin. 2018;36(2):339-353. PubMed
30.Barnett C, Katzberg H, Nabavi M, Bril V. The Quantitative Myasthenia Gravis Score: comparison with clinical, electrophysiological, and laboratory markers. J Clin Neuromuscul Dis. 2012;13(4):201-205. PubMed
31.Rozmilowska IM, Adamczyk-Sowa MH, Czyzewski D. The Myasthenia Gravis-specific Activities of Daily Living scale as a useful outcome measure and in routine clinical management in Polish patients. Neurol Neurochir Pol. 2018;52(3):368-373. PubMed
32.Ostovan VR, Fatehi F, Davoudi F, Nafissi S. Validation of the 15-item myasthenia gravis quality of life questionnaire (MG-QOL15) Persian version. Muscle Nerve. 2016;54(1):65-70. PubMed
Appendix 1: Selection of Included Studies
Appendix 2: Characteristics of Included Publications
Table 2: Characteristics of Included Systematic Reviews and Meta-Analyses
Study citation, country, funding source | Objectives, study designs, and numbers of primary studies included | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
Li et al. (2021)17 China Funding source: Youth Incubation Fund of Tianjin Medical University General Hospital and the National Key Clinical Specialty Construction Project of China | Objective: To evaluate the effectiveness and safety of rituximab at varying doses for the treatment of patients with refractory anti-AChR antibody–positive myasthenia gravis. Study design: Systematic review and meta-analysis of any human studies or clinical trials (e.g., RCTs, uncontrolled observational studies). Number of included studies: A total of 21 studies were included in the systematic review and meta-analysis (all were relevant to the current report). Quality assessment tool: The quality of included primary studies was not assessed by the authors of the systematic review. | Studies of people with refractory generalized anti-AChR antibody–positive myasthenia gravis were included Total number of included individuals: 260 Mean age, years (range): 43.7 (11 to 79) at the time of treatment initiation Sex: 63.5% female; sex of 49 individuals was not reported Antibody type: 260 patients were anti-AChR antibody positive Mean disease duration: 45.35 months (range = NR) | Intervention: Rituximab; treatment protocols varied by primary study. Of the 260 participants included in the meta-analyses, 171 patients (65.8%) received a routine regimen and 89 patients (34.2%) received a low-dose regimen (as described by the authors of the systematic review). Comparators: The included primary studies did not include control groups. All comparisons were from before treatment to after treatment with rituximab. | Clinical outcomes:
Length of follow-up: Median follow-up duration after rituximab treatment was 37.5 months |
AChR = acetylcholine receptor; MGFA = Myasthenia Gravis Foundation of America; NR = not reported; RCT = randomized controlled trial.
Table 3: Characteristics of Included Non-Randomized Studies
Study citation, country, funding source | Objective, study design, setting | Population characteristics | Intervention and comparator(s) | Clinical outcomes, length of follow-up |
Dos Santos et al. (2020)18 France Funding source: NR | Objective: To assess the efficacy and safety of rituximab for the treatment of adults with refractory or steroid-dependent myasthenia gravis Study design: Multi-centre, single-arm, retrospective cohort study Setting: Data from patients treated at 6 Departments of Neurology from the west of France (Nantes, Rennes, Brest, Angers, Poitiers, and Vannes) between 2011 and 2019 were reviewed | Inclusion criteria: Adults (≥ 18 years) diagnosed with refractory or steroid-dependent myasthenia gravis (MGFA class > II) with anti-AChR antibodies, anti-MuSK antibodies, or significant decrement after repetitive nerve stimulation (i.e., seronegative) who were treated with rituximab Number of participants: 29 Mean age: 49.6 (SD = 16.3; range = 18 to 82) years Sex: 58.6% female Antibody type: 20 patients were anti-AChR antibody positive, 5 were anti-MuSK antibody positive, and 4 patients were seronegative Mean disease duration: 8.8 (range = 0.41 to 33) years | Intervention: At the discretion of the treating neurologist, patients received 1 of the following rituximab treatment protocols:
Comparator: This study did not include a control group; within-group comparisons were made from before treatment to after treatment with rituximab | Clinical outcomes:
Length of follow-up: Mean follow-up duration after rituximab treatment was 20.06 (range = 0.17 to 68.93) months |
Litchman et al. (2020)19 US Funding source: No funding was received for this study | Objective: To compare the response to rituximab between patients with anti-AChR antibody–positive and anti-MuSK antibody–positive myasthenia gravis Study design: Single-centre, single-arm, retrospective cohort study Setting: Patients treated at the Yale Myasthenia Gravis Clinic, New Haven, Connecticut, between May 2003 and May 2017 were included | Inclusion criteria: People with refractory generalized myasthenia gravis with anti-AChR antibodies or anti-MuSK antibodies and who were followed for a minimum of 12 months after completion of the initial set of rituximab treatment cycles Patients whose immunotherapy dosage could not be lowered without clinical relapse, who could not achieve adequate clinical control, and/or who experienced severe adverse effects from their current immunosuppressive therapy were started on rituximab Number of participants: 33 (17 of these participants were included in the Li et al. 17 systematic review) Mean age: 35.9 (SD = 15.9; range = NR) years Gender: 72.7% female Antibody type: 17 patients were anti-AChR antibody positive and 16 patients were anti-MuSK antibody positive Mean disease duration: 1,579.4 (SD = 2,423.8) days | Intervention: Patients were treated with an initial 1 to 4 cycles of rituximab. Each cycle included 4 weekly infusions of 375 mg/m2 for 4 consecutive weeks. The interval between cycles was 6 months. The number of cycles was based on clinical improvement and patient toleration of tapering or withdrawal of other immunotherapies (i.e., corticosteroids).a Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab. | Clinical outcomes:
Length of follow-up: Mean follow-up duration after rituximab treatment was 1,861 (SD = 953.4) days |
Marino et al. (2020)20 Italy Funding source: Financial support was received from Università Cattolica del Sacro Cuore Fondazione Policlinico Universitario Agostino Gemelli IRCCS as a part of its programs on promotion and dissemination of scientific research | Objective: To investigate the effect of rituximab treatment in patients with refractory anti-MuSK antibody–positive myasthenia gravis. Study design: Single-centre, single-arm, retrospective cohort study Setting: Patients treated at Policlinico Gemelli, Università Cattolica between July 2006 and October 2018 were included | Inclusion criteria: People with refractory anti-MuSK antibody–positive myasthenia gravis. Patients were considered refractory if they met 1 of the following:
Number of participants: 9 Mean age: 50.4 (SD = 12.8; range = 38 to 73) years Gender: 88.9% female Antibody type: All 9 patients were anti-MuSK antibody positive Mean disease duration: NR | Intervention: Rituximab (375 mg/m2 once a week for 4 consecutive weeks followed by a single dose of 375 mg/m2 after 3 months). Infusions were preceded by IV methylprednisolone and oral antihistamine medication. Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab. | Clinical outcomes:
Length of follow-up: Mean follow-up duration after rituximab treatment was 51.9 (range = 20 to 144) months |
Zhong et al. (2020)21 China Funding source: Financial grants from the National Natural Science Foundation of China, the National Key Research and Development Program of China, and the Shanghai Municipal Science and Technology Major Project | Objective: To evaluate the effect of low-dose rituximab treatment on 2 potentially related microRNAs (miR-150-5p and miR-146a-5p) in patients with refractory anti-AChR antibody–positive myasthenia gravis. Study design: Single-centre, single-arm, prospective cohort study Setting: Patients who were treated at the Huashan Hospital between 2015 and 2018 were included | Inclusion criteria: Adults (≥ 18 years) with a confirmed diagnosis of refractory generalized myasthenia gravis who had not previously been treated with rituximab. Patients who failed to respond to multiple immunosuppressive therapies, had unacceptable adverse reactions to conventional treatments, who needed repeated treatment with IVIg or plasma exchange, or who experienced frequent myasthenic crises were considered refractory. Number of participants: 12b Mean age: 30.8 (SD = 13.5) years Sex: 83.3% female Antibody type: All 12 patients were anti-AChR antibody positive Mean disease duration: 54.8 (SD = 40.8) months | Intervention: Low-dose rituximab, given as a single dose of 600 mg Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab. | Clinical outcomes:
Length of follow-up: 6 months |
Zingariello et al. (2020)22 US Funding source: A grant from the Children’s Miracle Network. | Objective: To evaluate the efficacy and safety of rituximab for the treatment of children with refractory juvenile myasthenia gravis Study design: Single-centre, single-arm, retrospective cohort study Setting: Data from patients meeting inclusion criteria treated at a centre in Florida between 2014 and 2019 were included in the study | Inclusion criteria: Children (≤ 18 years) diagnosed with refractory myasthenia gravis. Patients were considered refractory if they had persistent symptoms despite therapy with pyridostigmine and at least 1 immunomodulatory medication. Number of participants: 5 Mean age: 11.6 (range = 6 to 16) years Sex: 60.0% female Antibody type: 4 patients were anti-AChR antibody positive and 1 patient was anti-MuSK antibody positive Mean disease duration: 15.1 (range = 4.5 to 27.5) months | Intervention: Rituximab; all participants were given 2 induction doses of 750 mg/m2 separated by 2 weeks to 3 weeks, followed by at least 1 maintenance dose. Maintenance dosing was 375 mg/m2 every 12 weeks. Each dose was followed by recue IVIg (1 g/kg). Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab (not compared statistically). | Clinical outcomes:
Length of follow-up: Mean follow-up duration after rituximab treatment was 11.6 (range = 4 to 24) months |
Choi et al. (2019)23 Republic of Korea Funding source: Grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea | Objective: To evaluate the long-term efficacy and safety of repeated low-dose rituximab for the treatment of patients with refractory myasthenia gravis. Additionally, the authors aimed to assess the correlation between circulating B cell repopulation and clinical relapse. Study design: Multi-centre, single-arm, retrospective cohort study Setting: Patients who were treated at 2 university-affiliated teaching hospitals (Seoul National University Hospital, and Seoul Metropolitan Government Boramae Medical Center) between September 2013 and January 2017 were included in this study | Inclusion criteria: Patients diagnosed with refractory myasthenia gravis. Patients were considered refractory if they had moderate-to-severe weakness, the MGFA clinical classification class III or worse despite conventional immunosuppressive treatment with prednisolone, plus 1 or more immunosuppressive agents over at least 1 year. Number of participants: 17 (9 of these participants were included in the Li et al.17 systematic review) Mean age: 51 years Sex: 64.7% female Antibody type: 9 patients were anti-AChR antibody positive, 6 patients were anti-MuSK antibody positive, and 2 patients were seronegative Median disease duration: 10 (range = 3 to 26) years | Intervention: Low-dose rituximab (375 mg/m2 given twice at 2-week intervals) was administered as an induction therapy. Re-treatment with 375 mg/m2 was provided as needed to patients who experienced CD19+ repopulation (i.e., an increase of the circulating CD19+ B cell proportion to > 1% of total lymphocytes) or clinical relapse (i.e., significant clinical worsening, as judged by treating neurologists). Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab. | Clinical outcomes:
Length of follow-up: Median follow-up duration after rituximab treatment was 24 (range = 7 to 49) months. |
Jing et al. (2019)24 China Funding source: Financial grants from the National Key Research and Development Program of China and the National Natural Science Foundation of China | Objective: To explore the effects of low-dose rituximab on circulating T and B cell lymphocytes and on clinical status in refractory and non-refractory myasthenia gravis patients Study design: Single-centre, single-arm, prospective cohort study Setting: Patients who were treated at the Huashan Hospital between 2014 and 2017 were included | Inclusion criteria: Adults (≥ 18 years) who were diagnosed with generalized myasthenia gravis. Patients were considered refractory if they had unsatisfactory response to 2 immunomodulatory agents (at least 1 of which was prednisone/prednisolone), unacceptable adverse reactions to conventional treatments, a requirement for repeated treatment with IVIg or plasma exchange, or experienced frequent myasthenic crises. Number of participants: 33 (15 were refractory and were considered relevant to the current report).c Characteristics of the refractory patients are summarized below; 14 of these participants were included in the Li et al. (2021)17 systematic review. Mean age: 34.4 (SD = 13.1) years Sex: 93.3% female Antibody type: Of those who were refractory, 14 patients were anti-AChR antibody positive and 1 patient was anti-MuSK antibody positive Mean disease duration: 57.3 (SD = 32.8) months | Intervention: Low-dose rituximab, given as a dose of 600 mg (100 mg on day 1 followed by 500 mg on day 2) Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab | Clinical outcomes:
Length of follow-up: 6 months |
Singh and Goyal (2019)25 India Funding source: NR | Objective: To investigate the efficacy of rituximab for the treatment of refractory myasthenia gravis Study design: Single-centre, single-arm, retrospective cohort study Setting: Data from patients meeting inclusion criteria treated at the All India Institute of Medical Sciences between January 2012 and December 2017 were included in the study | Patients diagnosed with refractory (as per the international consensus criteria by MGFA) myasthenia gravis Number of participants: 8 (6 of these participants were included in the systematic review by Li et al.17) Median age: 36 (range = 24 to 52) years Gender: 12.5% female Antibody type: 6 patients were anti-AChR antibody positive and 2 patients were anti-MuSK antibody positive Median disease duration: 184 (range = 10 to 264) months | Intervention: Rituximab, given as 4 weekly infusions at a dose of 375 mg/m2 for 4 consecutive weeks. Repeat cycles were provided at 6-month intervals, if required. Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab (not compared statistically). | Clinical outcomes:
Length of follow-up: Median follow-up duration after rituximab treatment was 18 (range = 14 to 29) months |
Topakian et al. (2019)26 Austria Funding source: No financial support was received for this work | Objective: To evaluate the real-world efficacy and safety of rituximab for the treatment of adults with refractory myasthenia gravis. Study design: Multi-centre, single-arm, retrospective cohort study Setting: All departments of neurology in Austrian hospitals and all members of the Austrian Neurological Society were invited in March 2017 to submit data from patients with myasthenia gravis treated with rituximab to be included in the study | Inclusion criteria: Adults (≥ 18 years of age) who were diagnosed with myasthenia gravis according to the guideline of the Austrian Neurological Society and who had a minimum length of follow-up of 3 months after initiating rituximab. Number of participants: 56 (39 of these participants were included in the systematic review by Li et al.17) Median age: 47.5 (IQR, 33 to 71) years Gender: 60.7% female Antibody type: 39 patients were anti-AChR antibody positive, 14 patients were anti-MuSK antibody positive, and 3 patients were seronegative Median disease duration: 4 (IQR, 1.3 to 10.8) years | Intervention: Treatment protocols varied across centres and between patients treated in the same centre. Participants received 1 of the following rituximab induction protocols:
Protocols for maintenance rituximab therapy were variable as well. Repeat rituximab infusions may have been given at the reappearance of B cells in peripheral blood, when clinical deterioration was observed, or according to a fixed-time/fixed dose protocol. At the time of the analysis, 21.4% of participants had only received induction therapy. Comparator: This study did not include a control group (i.e., no comparator); within-group comparisons were made from before to after treatment with rituximab (not compared statistically). | Clinical outcomes: • MGFA post-intervention status
• Adverse events Length of follow-up: Median follow-up duration after rituximab treatment was 20 (IQR, 10 to 53.5) months |
AChR = acetylcholine receptor; IgG = immunoglobulin G; IQR = interquartile range; IVIg = IV immunoglobulin; MGFA = Myasthenia Gravis Foundation of America; MuSK = muscle-specific kinase; NR = not reported; RNA = ribonucleic acid; SD = standard deviation.
aThe intention of this study was to compare outcomes between patients who were anti-AChR antibody positive and those who were anti-MuSK antibody positive, so while the study compared outcomes between 2 groups, this comparison does not align with the objective of the current report.
bWhile not explicitly stated in either publication, the patient population from this study may include some of the same participants as those in the Jing et al. (2019)24 study included in this report.
cWhile not explicitly stated in either publication, the patient population from this study may include some of the same participants as those in the Zhong et al. (2020)21 study included in this report.
Appendix 3: Critical Appraisal of Included Publications
Table 4: Strengths and Limitations of the Systematic Review and Meta-Analysis Using AMSTAR 215
Strengths | Limitations |
Li et al. (2021)17 | |
|
|
AMSTAR 2 = A MeaSurement Tool to Assess systematic Reviews 2; RCT = randomized controlled trial.
Table 5: Strengths and Limitations of Clinical Studies Using the Downs and Black Checklist16
Strengths | Limitations |
Dos Santos et al. (2020)18 | |
|
|
Litchman et al. (2020)19 | |
|
|
Marino et al. (2020)20 | |
|
|
Zhong et al. (2020)21 | |
|
|
Zingariello et al. (2020)22 | |
|
|
Choi et al. (2019)23 | |
|
|
Jing et al. (2019)24 | |
|
|
Singh and Goyal (2019)25 | |
|
|
Topakian et al. (2019)26 | |
|
|
MGFA = Myasthenia Gravis Foundation of America.
Appendix 4: Main Study Findings and Authors’ Conclusions
Summary of Findings of Included Systematic Review and Meta-Analysis
Li et al. (2021)17
Main Study Findings
Systematic review with meta-analysis that evaluated the effectiveness and safety of rituximab at varying doses for the treatment of patients with refractory anti-AChR antibody–positive myasthenia gravis (N = 260).
Relevant primary studies: All 21 primary studies included in the systematic review were relevant to the current report.
Summary of relevant findings:
Clinical response
Proportion of patients who achieved improved clinical status (defined as complete stable remission, minimal manifestations, and/or improved according to MGFA post-intervention status) from before to after treatment with rituximab: 77.0% (95% CI, 70.1% to 82.6%; P = 0.000)
Proportion of patients who achieved minimal manifestations or better status (according to MGFA post-intervention status) from before to after treatment with rituximab: 50.8% (95% CI, not reported [NR]; P = 0.921)
Concurrent immunomodulatory therapies
Proportion of patients able to reduce their daily dose of prednisone to ≤ 10 mg from before treatment to after treatment with rituximab: 70.6% (95% CI, 57.8% to 80.7%; P = 0.002)
Proportion of patients who discontinued immunosuppressants from before treatment to after treatment with rituximab: 66.6% (95% CI, 50.1% to 79.8%; P = 0.048)
Adverse events
Note: The rates experienced by participants were reported inconsistently for some outcomes in the Results and Discussion sections of the review; both values are provided as values from Results (Discussion).
Proportion of patients who experienced any adverse event: 26.4% (26.1%)
Proportion of patients who experienced cytopenia: 5.3% (1.6%)
Proportion of patients who experienced arrhythmia: 4.5% (1.2%)
Proportion of patients who experienced infection: 12.9% (8.6%)
Proportion of patients who experienced infusion reaction: 19.2%
Proportion of patients who experienced death: 7.5% (3.3%)
Authors’ Conclusion
“Our findings indicated that most of patients with refractory AChR-myasthenia gravis were responsive and well tolerated to rituximab. Repeated lower dose of rituximab might be effective for patients with refractory AChR-myasthenia gravis. A multi-center randomized controlled clinical trial using rituximab at different dosages and regimens in a larger population of patients with myasthenia gravis is necessary to assess the efficacy of rituximab in refractory AChR-myasthenia gravis (p. 11).”17
Summary of Findings of Included Primary Clinical Studies
Dos Santos et al. (2020)18
Main Study Findings
Multi-centre, single-arm, retrospective cohort study that included data from 29 patients who received rituximab to treat refractory or steroid-dependent myasthenia gravis.
Summary of relevant findings:
Clinical response
Proportion of patients who achieved MGFA post-intervention status of improved or better 6 months after initiating rituximab: 86.2% (25 of 29)
Proportion of patients who achieved MGFA post-intervention status of pharmacological remission 6 months after initiating rituximab: 14.3% (4 of 29)
Proportion of patients who achieved MGFA post-intervention status of improved or better 12 months after initiating rituximab: 90.5% (19 of 21)
Proportion of patients who achieved MGFA post-intervention status of complete stable remission or pharmacological remission 12 months after initiating rituximab: 38.1% (8 of 21)
Mean myasthenic muscle score 6 months after initiating rituximab
Prior to rituximab (N = 29): 68.8 (SD = 16.4)
After rituximab (N = 21): 83.1 (SD = 14.8)
P < 0.0001
Mean myasthenic muscle score 12 months after initiating rituximab
Prior to rituximab (N = 29): 68.8 (SD = 16.4)
After rituximab (N = 14): 85.0 (SD = 12.8)
P = 0.006
Myasthenia exacerbations
Proportion of patients who experienced myasthenia gravis exacerbation or myasthenic crisis after initiating rituximab during the follow-up period: 20.6% (6 of 29)
Concurrent immunomodulatory therapies
Mean dose of concurrent steroid treatment 6 months after initiating rituximab
Prior to rituximab (N = 29): 20.0 mg/day
After rituximab (N = 21): 7.0 mg/day
P = 0.0005
Proportion of patients who reduced the daily dose of steroid to ≤ 10 mg 6 months after initiating rituximab: 36.8% (7 of 19)
Proportion of patients who reduced the daily dose of steroid to ≤ 10 mg 12 months after initiating rituximab: 57.9% (11 of 19)
Proportion of patients who discontinued steroid treatment 6 months after initiating rituximab: 21.1% (4 of 19)
Proportion of patients who discontinued steroid treatment 12 months after initiating rituximab: 36.8% (7 of 19)
Adverse events – proportion of patients who experienced the following after initiating rituximab
Any adverse event: 42.8% (12 of 28)
Infusion reaction: 7% (2 of 28)
Infection: 21.4% (6 of 28)
Hematological disorders: 7% (2 of 28)
Cardiologic disorders: 3.7% (1 of 28)
Psychiatric disorders: 3.7% (1 of 28)
Authors’ Conclusion
“In conclusion, the present study corroborates the efficacy and the safety of rituximab in myasthenia gravis. Complementary studies are still necessary to define the best infusion protocol and the exact role of rituximab treatment in myasthenia gravis (p. 2284).”18
Litchman et al. (2020)19
Main Study Findings
Single-centre, single-arm, retrospective cohort study that compared the response to rituximab between patients with anti-AChR antibody–positive (n = 17) and anti-MuSK antibody–positive (n = 16) myasthenia gravis. Results of statistical tests comparing anti-AChR antibody–positive patients and anti-MuSK antibody–positive patients were not extracted as these were not relevant to the current report.
Summary of relevant findings:
Clinical response
Proportion of patients who achieved MGFA post-intervention status of minimal manifestations or better 12 months after initiating rituximab
Anti-AChR antibody positive (n = 17): 58.8% (10 of 17)
Anti-MuSK antibody positive (n = 16): 68.8% (11 of 16)
Total cohort (N = 33): 63.6% (21 of 33)
Proportion of patients who achieved MGFA post-intervention status of minimal manifestations or better at last follow-up visit
Anti-AChR antibody positive (n = 17): 65% (11 of 17)
Anti-MuSK antibody positive (n = 16): 75% (12 of 16)
Total cohort (N = 33): 69.7% (23 of 33)
Proportion of patients who achieved clinical remission, defined as when the patient achieved “asymptomatic” status based on the MGFA classification and had not relapsed for 12 months
Anti-AChR antibody positive (n = 17): 70.6% (12 of 17)
Anti-MuSK antibody positive (n = 16): 56.3% (9 of 16)
Total cohort (N = 33): 63.6% (21 of 33)
Mean days to clinical remission
Anti-AChR antibody positive (n = 17): 441 (SD = 336.6) days
Anti-MuSK antibody positive (n = 16): 230 (SD = 180.8) days
Total cohort (N = 33): NR
Proportion of patients with relapse
Anti-AChR antibody positive (n = 17): 58.8% (10 of 17)
Anti-MuSK antibody positive (n = 16): 37.5% (6 of 16)
Total cohort (N = 33): 48.5% (16 of 33)
Mean days to clinical relapse
Anti-AChR antibody positive (n = 17): 824.1 (SD = 479.3) days
Anti-MuSK antibody positive (n = 16): 1,529 (SD = 1,338.4) days
Total cohort (N = 33): NR
Concurrent immunomodulatory therapies
Mean prednisone dose 12 months after initiating rituximab
Anti-AChR antibody positive at baseline (n = 17): 46.8 (SD = 20.8) mg/day
Anti-AChR antibody positive at 12 months (n = 17): 11.8 (SD = 15.3) mg/day
Within-group P value: < 0.01
Anti-MuSK antibody positive at baseline (n = 16): 30.9 (SD = 24.9) mg/day
Anti-MuSK antibody positive at 12 months (n = 16): 9.5 (SD = 8.7) mg/day
Within-group P value: < 0.01
Mean prednisone dose at last follow-up (mg/day)
Anti-AChR antibody positive at baseline (n = 17): 46.8 (SD = 20.8) mg/day
Anti-AChR antibody positive last follow-up (n = 17): 17.9 (SD = 15.6) mg/day
Within-group P value: < 0.01
Anti-MuSK antibody positive at baseline (n = 16): 30.9 (SD = 24.9) mg/day
Anti-MuSK antibody positive at 12 months (n = 16): 2.8 (SD = 3.8) mg/day
Within-group P value: < 0.01
Proportion of patients who reduced the daily dose of prednisone to ≤ 10 mg
Anti-AChR antibody positive (n = 17): 82% (14 of 17)
Anti-MuSK antibody positive (n = 16): 81% (13 of 16)
Total cohort (N = 33): 81.8% (27 of 33)
Mean days to reduce the daily dose of prednisone to ≤ 10 mg
Anti-AChR antibody positive (n = 17): 361.9 (SD = 232.8) days
Anti-MuSK antibody positive (n = 16): 219.1 (SD = 152.0) days
Total cohort (N = 33): NR
Proportion of patients who tapered prednisone completely
Anti-AChR antibody positive (n = 17): 70% (12 of 17)
Anti-MuSK antibody positive (n = 16): 56% (9 of 16)
Total cohort (N = 33): 63.6% (21 of 33)
Mean days to taper prednisone completely
Anti-AChR antibody positive (n = 17): 811.4 (SD = 395.6) days
Anti-MuSK antibody positive (n = 16): 712.4 (SD = 395.6) days
Total cohort (N = 33): NR
Proportion of patients who used oral steroid-sparing agents
Anti-AChR antibody positive (n = 17): 18% (3 of 17)
Anti-MuSK antibody positive (n = 16): 12.5% (2 of 16)
Total cohort (N = 33): 15.2% (5 of 33)
Number of patients on maintenance therapy with IV immunoglobulin or plasma exchange
Anti-AChR antibody positive at baseline (n = 17): 4
Anti-AChR antibody positive post-rituximab (n = 17): 6
Within-group P value: 0.71
Anti-MuSK antibody positive at baseline (n = 16): 11
Anti-MuSK antibody positive post-rituximab (n = 16): 6
Within-group P value: 0.16
Mean total number of maintenance IV immunoglobulin and plasma exchange treatment cycles
Anti-AChR antibody positive (n = 17): 9.8 (SD = 31.8)
Anti-MuSK antibody positive (n = 16): 9.1 (SD = 19.8)
Total cohort (N = 33): NR
Number of patients on rescue therapy with IV immunoglobulin or plasma exchange
Anti-AChR antibody positive at baseline (n = 17): 16
Anti-AChR antibody positive post-rituximab (n = 17): 7
Within-group P value: < 0.01
Anti-MuSK antibody positive at baseline (n = 16): 13
Anti-MuSK antibody positive post-rituximab (n = 16): 3
Within-group P value: < 0.01
Mean total number of rescue IV immunoglobulin and plasma exchange treatment cycles
Anti-AChR antibody positive (n = 17): 0.7 (SD = 1.1)
Anti-MuSK antibody positive (n = 16): 0.5 (SD = 1.1)
Total cohort (N = 33): NR
Myasthenia exacerbations
Mean number of disease exacerbations
Anti-AChR antibody positive (n = 17): 1.7 (SD = 1.2)
Anti-MuSK antibody positive (n = 16): 1.4 (SD = 1.1)
Total cohort (N = 33): NR
Proportion of patients hospitalized for disease exacerbations
Anti-AChR antibody positive (n = 17): 29% (5 of 17)
Anti-MuSK antibody positive (n = 16): 0% (0 of 16)
Total cohort (N = 33): 15.2% (5 of 33)
Adverse events: “All patients in the study tolerated rituximab treatment without severe adverse side effects identified per chart review (p. 4).”19
Authors’ Conclusion
“Rituximab therapy is a reasonable option for both AChR+ and MuSK+ generalized MG patients, especially those with difficult to control disease. While MuSK+ MG patients may experience greater benefits, a significant differential response between groups was not observed in our retrospective study. B-cell targeted therapy continues to be a rational strategy in the MG treatment paradigm. Further investigations are warranted to explore how B-cell depletion therapy is best applied to achieve improved outcomes which are both meaningful and sustained in our patients (p. 5).”19
Marino et al. (2020)20
Main Study Findings
Single-centre, single-arm, cohort study that investigated the effect of rituximab treatment in patients (N = 9) with refractory anti-MuSK antibody–positive myasthenia gravis.
Summary of relevant findings:
Clinical response
Proportion of patients (N = 9) who achieved optimal response (defined as the achievement and maintenance of the status of minimal manifestations or better together with a ≥ 50% reduction of steroid dose, withdrawal of immunosuppressants, and no need for plasma exchange or IV immunoglobulin), partial response (defined as clinical improvements while failing to achieve the status of minimal manifestations or better or when prednisone reduction was < 50% of pre-treatment dosage, with or without immunosuppressant withdrawal and no need for plasma exchange or IV immunoglobulin), or no response
Optimal response: 66.6% (6 of 9)
Partial response: 22.2% (2 of 9)
No response: 11.1% (1 of 9)
Concurrent immunomodulatory therapies
Proportion of patients (N = 9) who achieved the ability to taper prednisone completely: 22.2% (2 of 9)
Laboratory parameters
“For statistical analysis, we compared MuSK-IgG and MuSK-IgG4 baseline levels with those at 2–7 months and at 12–30 months after rituximab in eight patients (samples from patient #5 were not available). We found a significant reduction at both time points (p < 0.02 for MuSK-IgG and p < 0.01 for MuSK-IgG4, by ANOVA), while the MuSK-IgG4/MuSK-IgG ratio was reduced significantly only at 2–7 months after rituximab (p < 0.05, by Student’s t test) (p. 4).”20
Authors’ Conclusion
“In line with previous reports, our data confirm that, in patients with MuSK- myasthenia gravis, rituximab is safe and induces long-term benefit associated with a strong steroid- and immunosuppressant-sparing effect. As MuSK- myasthenia gravis is often a life-threatening disease with a high proportion of patients refractory to conventional therapy, rituximab has been proposed as an early therapeutic option in patients unresponsive to first-line immunosuppression (p. 4).”20
Zhong et al. (2020)21
Single-centre, single-arm, prospective cohort study that evaluated the effect of low-dose rituximab treatment on 2 potentially related microRNAs (miR-150-5p and miR-146a-5p) in 12 patients with refractory anti-AChR antibody–positive myasthenia gravis.
Summary of relevant findings:
Clinical response
Mean MGFA QMG scores: Compared to baseline (before receiving rituximab), mean MGFA QMG scores 6 months after initiating rituximab decreased by 27.8% (P = 0.019)
Mean myasthenia gravis–specific manual muscle testing scores: Compared to baseline (before receiving rituximab), mean manual muscle testing scores 6 months after initiating rituximab decreased by 67.4% (P = 0.019)
Quality of life
Mean myasthenia gravis–related ADL scores: Compared to baseline (before receiving rituximab), mean myasthenia gravis–related ADL scores 6 months after initiating rituximab decreased by 43.9% (P = 0.022)
Mean MG-QOL15 scores: Compared to baseline (before receiving rituximab), mean MG-QOL15 scores 6 months after initiating rituximab were not significantly different (P = 0.13)
Laboratory parameters
Mean levels of miR-150-5: Compared to baseline (before receiving rituximab), mean miR-150-5 levels 6 months after initiating rituximab decreased by 47.9% (P = 0.006)
Mean levels of miR-146a-5p: Compared to baseline (before receiving rituximab), mean miR-146a-5p levels 6 months after initiating rituximab were not significantly different (P = 0.570)
Mean CD19+ B cell counts: Compared to baseline (before receiving rituximab), mean CD19+ B cell counts 6 months after initiating rituximab decreased by 85.4% (P = 0.006)
Mean CD27+ memory B cell counts: Compared to baseline (before receiving rituximab), mean CD27+ memory B cell counts 6 months after initiating rituximab decreased by 91.2% (P = 0.006)
Mean CD3+ T cell counts: Compared to baseline (before receiving rituximab), mean CD3+ T cell counts 6 months after initiating rituximab were not significantly different (P = 0.560)
Mean CD4+ T cell counts: Compared to baseline (before receiving rituximab), mean CD4+ T cell counts 6 months after initiating rituximab were not significantly different (P = 0.560)
Mean CD8+ T cell counts: Compared to baseline (before receiving rituximab), mean CD8+ T cell counts 6 months after initiating rituximab were not significantly different (P = 0.440)
Mean natural killer cell counts: Compared to baseline (before receiving rituximab), mean natural killer cell counts 6 months after initiating rituximab did not change significantly (P = 0.190)
Authors’ Conclusion
“In conclusion, as a self-controlled pilot trial, the current study shows the effectiveness of low-dose rituximab in alleviating symptoms of myasthenia gravis and decreasing the myasthenia gravis biomarker miR-150-5p. Furthermore, these results suggest that the relationship may be related to miR-150-5p interactions with CD19+ and CD27+ B cells (p. 5).”21
Zingariello et al. (2020)22
Main Study Findings
Single-centre, single-arm, retrospective cohort study that evaluated the efficacy and safety of rituximab for the treatment of children with refractory juvenile myasthenia gravis (N = 5). No statistical comparisons from before treatment to after treatment were conducted.
Clinical response
Mean MGFA class
Pre-rituximab (N = 5): 2.6
Post-rituximab (N = 5): 1.2
Concurrent immunomodulatory therapies
Mean number of concurrent immunomodulatory medications
Pre-rituximab (N = 5): 2.8
Post-rituximab (N = 5): 1.6
Myasthenia exacerbations
Mean number of juvenile myasthenia gravis–related hospitalizations
Pre-rituximab (N = 5): 2.8
Post-rituximab (N = 5): 0
Laboratory parameters: “All subjects had decreased AChR or MuSK antibody titers following rituximab, with antibodies becoming undetectable in three of the five cases (p. 41).”22
Adverse events: “No significant adverse events were recorded for any of the participants (p. 40).”22
Authors’ Conclusion
“Based on our cohort analysis, rituximab appears to be well-tolerated and potentially efficacious for children with juvenile myasthenia gravis, including those who have already had thymectomy. With respect to its safety profile, rituximab does not affect B-cell recovery, plasma cells, or antibody production. Rituximab has the potential to fill a significant therapeutic gap for refractory juvenile myasthenia gravis, but should be studied more rigorously in a larger cohort before more definitive recommendations can be made. Such an investigation will require the assembly of a multicenter consortium of pediatric neuromuscular clinics, perhaps modeled after ones that have already been established for inherited pediatric neuromuscular diseases such as spinal muscular atrophy and Duchenne muscular dystrophy (p. 43).”22
Choi et al. (2019)23
Main Study Findings
Multi-centre, single-arm, retrospective cohort study that evaluated the long-term efficacy and safety of repeated low-dose rituximab for the treatment of patients with refractory myasthenia gravis (N = 17).
Summary of relevant findings:
Clinical response
Proportion of patients (N = 17) who achieved MGFA post-intervention status of Minimal Manifestations or better with low-dose prednisolone (≤ 5 mg per day): 65%
Median time to achieve MGFA post-intervention status of Minimal Manifestations or better with low-dose prednisolone (≤ 5 mg per day) (N = 11): 7.6 (range = 2 to 17) months
Concurrent immunomodulatory therapies
Mean dose of concurrent prednisolone: Compared to during the first 4 weeks after rituximab induction treatment (i.e., weeks 0 to 4 after initiating rituximab), the time-weighted mean prednisolone dose was significantly decreased during weeks 20 to 24 after rituximab induction treatment (mean = 28 mg/day; SD = 17 mg/day versus mean = 7.5 mg/day; SD = 6.8 mg/day; P < 0.001)
Adverse events
“Two patients experienced infusion reactions, chest discomfort in one patient, and skin rash in the other. During follow up, one patient was affected by herpes zoster, and one patient died due to complications of invasive thymoma. Otherwise, there was no case with serious adverse events including severe infections other drug-related and laboratory abnormalities (p. 9).”23
Authors’ Conclusion
“This study provides support to the efficacy of low-dose rituximab in refractory MG for improving clinical outcomes and reducing the need for corticosteroid. Our results also suggest that repeated treatment based on the assessment of B-cell depletion in the peripheral blood could help to maintain clinical efficacy of rituximab with acceptable long-term safety profiles (p. 9-10).”23
Jing et al. (2019)24
Main Study Findings
Single-centre, single-arm, prospective cohort study that explored the effects of low-dose rituximab on circulating T cell and B cell lymphocytes and on clinical status in refractory and non-refractory myasthenia gravis patients. Only findings from refractory myasthenia gravis patients were extracted and summarized.
Summary of relevant findings:
Clinical response
Mean MGFA QMG scores
Baseline (N = 15): 15.7 (SD = 4.9)
6 months post-rituximab (N = 15): 11.2 (SD = 4.4)
P = 0.013
Mean manual muscle testing scores
Baseline (N = 15): 22.7 (SD = 18.1)
6 months post-rituximab (N = 15): 6.9 (SD = 6.5)
P = 0.004
Concurrent immunomodulatory therapies
Mean dose of concurrent prednisolone: Compared to baseline, mean prednisone dose was significantly decreased by 40% at 6 months after rituximab (P = 0.001)
Quality of life
Mean myasthenia gravis–related ADL scores
Baseline (N = 15): 8 (SD = 3.9)
6 months post-rituximab (N = 15): 3.6 (SD = 3.0)
P = 0.002
Mean MG-QOL15 scores
Baseline (N = 15): 35.1 (SD = 12.7)
6 months post-rituximab (N = 15): 23.2 (SD = 13.1)
P = 0.018
Laboratory parameters
Mean percentage of CD19+ B cells out of total lymphocytes
Baseline (N = 15): 10.9% (SD = 6.1%)
6 months post-rituximab (N = 15): 0.1% (SD = 0.3%)
P < 0.0001
Mean percentage of CD27+ memory B cells out of total B cells
Baseline (N = 15): 38.8% (SD = 14.6%)
6 months post-rituximab (N = 15): 3.2% (SD = 9.4%)
P < 0.0001
Mean percentage of BAFF-R+ B cells out of the total CD19+ B cells
Baseline (N = 15): 97.5% (SD = 2.5%)
6 months post-rituximab (N = 15): 8.7% (SD = 24.0%)
P < 0.0001
Mean percentage of CD19+ CD5+ CD1d+ regulatory B cells out of the total CD19+ B cells
Baseline (N = 15): 20.3% (SD = 2.5%)
6 months post-rituximab (N = 15): 1.4% (SD = 5.4%)
P < 0.0001
Mean percentage of CD19+ CD24+ CD38+ regulatory B cells out of the total CD19+ B cells
Baseline (N = 15): 0.2% (SD = 0.6%)
6 months post-rituximab (N = 15): 0%
P = 0.134
Mean percentage of regulatory T cells out of the CD4+ T cells
Baseline (N = 15): 1.9% (SD = 1.0%)
6 months post-rituximab (N = 15): 3.4% (SD = 1.1%)
P < 0.001
Mean percentage of CD3+ T cells
Baseline (N = 15): 77.5% (SD = 8.8%)
6 months post-rituximab (N = 15): 43.5% (SD = 8.0%)
P = 0.801
Mean percentage of CD4+ T cells
Baseline (N = 15): 43.5% (SD = 8.0%)
6 months post-rituximab (N = 15): 43.3% (SD = 10.5%)
P = 0.960
Mean percentage of CD8+ T cells
Baseline (N = 15): 29.8% (SD = 10.7%)
6 months post-rituximab (N = 15): 30.5% (SD = 10.2%)
P = 0.856
Mean levels of anti-AChR antibodies
Baseline (N = 15): 8.3 (SD = 3.5) nmol/L
6 months post-rituximab (N = 15): 6.9 (SD = 3.7) nmol/L
P = 0.016
Adverse events
“No allergic reactions or other serious side effects occurred during the follow-up period. All of the 15 patients tolerated the treatment well (p. 220).”24
Authors’ Conclusion
“Our investigation disclosed impacts of low-dose rituximab on clinical responses in refractory myasthenia gravis patients for up to six months following treatment. rituximab exerted its effect through B-cell elimination and subsequent B-cell and T-cell repopulation. The present study elucidated that there is indeed an immune imbalance of circulating T and B cells in refractory myasthenia gravis patients compared to that in non-refractory myasthenia gravis patients. It seems that, in refractory myasthenia gravis, the proportion of Bregs and Tregs with immunosuppressive effects is reduced and the expression of BAFF-R with survival and development is greater than in non-refractory myasthenia gravis. The increase of Tregs% in refractory myasthenia gravis was significantly correlated to the improvement of MGFA-QMG scores. Further studies are needed to investigate the clinical effect and mechanism of rituximab-treatment regimen in patients with myasthenia gravis (p. 222).”24
Singh and Goyal (2019)25
Main Study Findings
Single-centre, single-arm, retrospective cohort study that investigated the efficacy of rituximab for the treatment of patients with refractory myasthenia gravis (N = 8).
Summary of relevant findings:
Clinical response: Proportion of patients who had improved MGFA post-intervention status was 100% (8 of 8)
Myasthenia exacerbations: Proportion of patients who experienced myasthenic crisis during the follow-up period was 0% (0 of 8)
Concurrent immunomodulatory therapies
Proportion of patients who achieved the ability to taper prednisone completely was 75% (6 of 8)
Proportion of patients who required treatment with IV immunoglobulin or plasma exchange after rituximab was 0% (0 of 8)
Adverse events
“Three out of eight patients developed transaminitis which led to dose reduction of azathioprine in two patients and one patient was switched to mycophenolate mofetil due to recurrent episodes of transaminitis even at low doses of azathioprine and positive TPMT assay (p. 1598).”25
“One patient developed avascular necrosis of femur secondary to steroid use. One patient was later found to be HBsAg positive, probably due to prior history of receiving multiple cycles of plasma exchange for treating myasthenic crisis (p. 1598).”25
“None of the patients had infusion associated reactions or cytopenia post-rituximab infusion (p. 1596).”25
Authors’ Conclusion
“In conclusion, the marked effect of rituximab as induction therapy in patients with refractory myasthenia gravis in our clinic as well as its response in similar studies is promising, and suggests that further investigation of this agent in myasthenia gravis is warranted (p. 1599).”25
Topakian et al. (2019)26
Main Study Findings
Multi-centre, single-arm, retrospective cohort study that evaluated the real-world efficacy and safety of rituximab for the treatment of adults with refractory myasthenia gravis (N = 56).
Summary of relevant findings:
Clinical response
Proportion of patients by MGFA post-intervention status 3 months after starting rituximab (N = 53)
Death: 0%
Worse: 1.9%
Unchanged: 11.3%
Improved: 41.5%
Minimal manifestation: 18.9%
Complete stable remission: 26.4%
Proportion of patients by MGFA post-intervention status 6 months after starting rituximab (N = 52)
Death: 1.9%
Worse: 1.9%
Unchanged: 3.8%
Improved: 34.6%
Minimal manifestation: 23.1%
Complete stable remission: 34.6%
Proportion of patients by MGFA post-intervention status 12 months after starting rituximab (N = 41)
Death: 2.4%
Worse: 0%
Unchanged: 2.4%
Improved: 36.6%
Minimal manifestation: 19.5%
Complete stable remission: 39.0%
Proportion of patients by MGFA post-intervention status at final follow-up (median 20 months) after starting rituximab (N = 56)
Death: 1.8%
Worse: 0%
Unchanged: 1.8%
Improved: 28.6%
Minimal manifestation: 25.0%
Complete stable remission: 42.9%
Concurrent immunomodulatory therapies
Proportion of patients (N = 56) who required treatment with IV immunoglobulin, subcutaneous immunoglobulin, immunoadsorption, eculizumab, or plasma exchange after rituximab: 23.2%
Proportion of patients (N = 39) who discontinued steroid treatment: 59%
Proportion of patients (N = 39) who achieved the ability to reduce the daily dose of steroid to 50% or less: 25.6%
Laboratory parameters
“20 (35.8%) patients had antibody levels re-tested after start of rituximab. 13 (65%) of these patients showed some decrease of antibody levels, but this seemed unrelated to outcome. Interpretation of data was further compromised by the widely varying time points of antibody level testing (p. 702).”26
Adverse events
“Rituximab was generally well tolerated. Reported side effects and complications potentially related to rituximab therapy included infusion reactions (n = 3), respiratory tract infections (n = 3), chronic pain syndromes (n = 2), enteritis (n = 2), herpes zoster (n = 1), erysipelas (n = 1), cholecystitis (n = 1), unspecified mental disorder (n = 1), and alopecia areata (n = 1). There was one death during follow-up (p. 701).”26
Authors’ Conclusion
“In conclusion, this study found that rituximab is a well tolerated, safe, efficacious, and relatively fast-acting treatment for patients with myasthenia gravis. Benefit from rituximab was greatest in MuSK antibody-positive myasthenia gravis. Placebo-controlled, randomized trials are needed to further clarify the role of rituximab in myasthenia gravis (p. 705).”26
Appendix 5: References of Potential Interest
Previous CADTH Reports
CADTH Canadian Drug Expert Committee (CDEC) final recommendation: eculzimab (Soliris - Alexium Pharma Canada Corp.). Ottawa (ON): CADTH; 2020 Oct, last updated 2020 Dec 9: https://www.cadth.ca/eculizumab-16 Accessed 2021 Mar 9. Soliris is indicated in adult patients with generalized Myasthenia Gravis (gMG)
Chao Y, Argáez C. Video-oculography for the diagnosis of ocular myasthenia gravis: a review of diagnostic accuracy, cost-effectiveness, and guidelines. (CADTH rapid response report: summary with critical appraisal.) Ottawa (ON): CADTH; 2019: https://www.cadth.ca/video-oculography-diagnosis-ocular-myasthenia-gravis-review-diagnostic-accuracy-cost-effectiveness Accessed 2021 Mar 9.
Sutton D, Visintini S. Off-label use of intravenous immunoglobulin for neurological conditions: a review of clinical effectiveness. (CADTH rapid response: summary with critical appraisal.) Ottawa (ON): CADTH; 2018: https://www.cadth.ca/label-use-intravenous-immunoglobulin-neurological-conditions-review-clinical-effectiveness Accessed 2021 Mar 9.
Guidelines With Unclear Methodology
Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021 01 19;96(3):114-122. PubMed
Guidance for the management of Myasthenia Gravis (MG) and Lambert-Eaton Myasthenic Syndrome (LEMS) during the COVID-19 pandemic. Edmonton (AB): Alberta Health Services; 2020. https://www.albertahealthservices.ca/assets/info/ppih/if-ppih-covid-19-mg-lems-guidelines.pdf Accessed 2021 Mar 9.
Evoli A, Antonini G, Antozzi C, et al. Italian recommendations for the diagnosis and treatment of myasthenia gravis. Neurol Sci. 2019 Jun;40(6):1111-1124. PubMed
Review Articles
Farrugia ME, Goodfellow JA. A practical approach to managing patients with myasthenia gravis - opinions and a review of the literature. Front Neurol. 2020 Jul 7;11:604. PubMed
O'Connell K, Ramdas S, Palace J. Management of juvenile myasthenia gravis. Front Neurol. 2020 Jul 24;11:743. PubMed
Souto EB, Lima B, Campos JR, Martins-Gomes C, Souto SB, Silva AM. Myasthenia gravis: State of the art and new therapeutic strategies. J Neuroimmunol. 2019 12 15;337:577080. PubMed
Book Chapter
Punga AR, Kaminski HJ, Guptill JT. Emerging therapeutics for myasthenia gravis. In: Kaminski H, Kusner L, eds. Myasthenia Gravis and Related Disorders. Current Clinical Neurology. Totowa (NJ): Springer, Humana Press, Cham. 2018.
Additional References
NHS England. Clinical Commissioning Policy Statement: Rituximab bio-similar for the treatment of myasthenia gravis (adults). (NHS England Reference: 170084P). 2018; https://www.england.nhs.uk/wp-content/uploads/2018/10/Rituximab-biosimilar-for-the-treatment-of-myasthenia-gravis-adults-1.pdf Accessed 2021 Mar 9.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
Questions or requests for information about this report can be directed to Requests@CADTH.ca