CADTH Reimbursement Review
Cemiplimab (Libtayo)
Sponsor: Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc.
Therapeutic area: Basal cell carcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
BCC
basal cell carcinoma
BICR
blinded independent central review
CI
confidence interval
CR
complete response
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
HHI
hedgehog pathway inhibitor
HRQoL
health-related quality of life
laBCC
locally advanced basal cell carcinoma
mBCC
metastatic basal cell carcinoma
MID
minimal important difference
MNC
Melanoma Network of Canada
NMSC
non-melanoma skin cancer
ORR
objective response rate
OS
overall survival
PD
progressive disease
PD-1
programmed cell death protein 1
PD-L1
programmed death-ligand 1
PD-L2
programmed death-ligand 2
PFS
progression-free survival
PR
partial response
QoL
quality of life
RECIST
Response Evaluation Criteria in Solid Tumors
SAE
serious adverse event
SD
standard deviation
SYSF
Save Your Skin Foundation
TEAE
treatment-emergent adverse event
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for cemiplimab is provided in Table 1.
Item | Description |
|---|---|
Drug product | Cemiplimab (Libtayo) 350 mg for IV use |
Indication | For the treatment of patients with locally advanced basal cell carcinoma (BCC) previously treated with a hedgehog pathway inhibitor |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review pathway |
NOC date | October 26, 2021 |
Sponsor | Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc. |
NOC = Notice of Compliance.
Introduction
Non-melanoma skin cancer accounts for approximately 28% of new cancer diagnoses in Canada,1 with basal cell carcinoma (BCC) accounting for 75% of all non-melanoma skin cancers.2 BCC generally develops on sun-exposed skin. Other risk factors include male sex, light hair, northern European ancestry, and the inability to tan. Seventy percent of cases occur on the head, frequently on the face, whereas 25% occur on the trunk and limbs and 5% in the perineal region.3 Most BCCs are diagnosed and treated early; however, some BCCs become extensive and infiltrative, posing a greater risk to patients.4
The objective of this report was to perform a systematic review of the beneficial and harmful effects of cemiplimab (IV injection, 350 mg) for the treatment of patients with locally advanced BCC (laBCC) previously treated with a hedgehog pathway inhibitor (HHI).
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Input was provided for this review by 2 patient groups, the Save Your Skin Foundation (SYSF) and the Melanoma Network of Canada (MNC). The SYSF gathered information from online surveys, virtual patient roundtables, and 1-on-one conversations from March 2021 to September 2021. All 23 patients consulted (20 of which were women) had been diagnosed with BCC; 5 patients had experience with cemiplimab. It was not reported whether patients had experience with HHI therapy before receiving cemiplimab. All but 4 patients were from Canada, with most of the Canadian responders being from Ontario. The MNC input was sourced from an online survey of 62 patients (44 of which were women) and 45 caregivers. All but 1 of the patients were from Canada, with 50% located in Ontario. Only 1 patient indicated they had experience with cemiplimab in metastatic disease and no patients had experience with HHI therapy.
In both surveys, patients highlighted the negative aspects of BCC and its treatment, including disfigurement, scaring, and the associated self-esteem difficulties. In both surveys, pain from the lesions and anxiety over finding recurrent disease were also mentioned by patients as key concerns. In the MNC survey, caregivers expressed that the disease caused much emotional stress from seeing their loved 1 in pain. Patients expressed a desire for less radiation and disfiguring surgery and greater access to treatments closer to their home and support network. Respondents from the SYSF submission with experience with cemiplimab indicated the side effects were manageable and the benefits would outweigh the side effects. Of the 5 patients with experience with cemiplimab, 2 had no side effects, 2 had fatigue, and 1 patient had skin rash. The 1 patient from the MNC submission who had experience with cemiplimab indicated that having the option for therapy was worth experiencing treatment side effects, which included difficulty with liver-related issues and flu-like symptoms.
Patients indicated there are no other options for treatment following progression on HHI therapy and the ability to access new treatments to eliminate disease and prevent recurrence is needed. Earlier diagnosis, access to specialists, and less invasive procedures were highlighted as important to patients and caregivers in the MNC survey.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical experts with expertise in the diagnosis and management of laBCC highlighted the lack of options available for patients with laBCC whose disease did not respond to HHI therapy, especially given the fact that response to HHI therapy is low and some patients cannot tolerate treatment side effects. As eligibility is based on whether the tumour is deemed unresectable or unsuitable for radiotherapy, this can be uncertain and, therefore, the clinical experts suggested that tumour eligibility should be determined by a multidisciplinary tumour board. The main goals of therapy are to shrink the tumour and increase the health-related quality of life (HRQoL) of patients with laBCC; the clinical experts highlighted the extreme importance of HRQoL in this patient population, given the disfiguring nature of the disease. Treatment would usually be discontinued upon disease progression (increase in size or extension of lesions), severe or intolerable side effects, or a lack of response after an adequate duration of treatment (identified as 4 to 6 months of treatment). According to the clinical experts, treatment with cemiplimab would be initiated by a medical oncologist or associated team physician with expertise in cancer therapies and toxicity management.
Clinician Group Input
One clinician group, Ontario Health (Cancer Care Ontario), provided input for this review. No major views that were contrary to those provided by the clinical experts consulted by CADTH were presented. Ontario Health echoed the lack of options for patients with laBCC whose disease has not responded to HHI therapy, as well as the importance of HRQoL outcomes specifically relating to disfiguring lesions and surgical scarring.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The drug plans identified implementation issues related to relevant comparators, consideration for initiation of therapy, consideration for prescribing of therapy, generalizability, care provision, system issues, and economic considerations. The clinical experts consulted by CADTH for this review weighed evidence from the included study and other clinical considerations to provide responses to the drug plans’ implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal and Protocol Selected Studies
Description of Study
One phase II, single-arm, non-randomized, open-label multi-centre trial, Study 1620,5,6 was included in the systematic review (Table 6). The primary objective of the study was to determine the efficacy of cemiplimab in achieving an objective tumour response in 2 cohorts: patients with laBCC and patients with metastatic basal cell carcinoma (mBCC).
The study enrolled patients with laBCC and mBCC who had previously received HHI therapy; however, the laBCC population (N = 84) was the focus of the CADTH review, since the Health Canada indication and requested reimbursement request were restricted to this patient population.
In Study 1620, patients were treated with cemiplimab for up to 93 weeks or until progressive disease (PD) or unacceptable toxicity. Tumour response was assessed using a composite of the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 for lesions with radiologically measurable components, and modified WHO clinical criteria for lesions with externally visible components, and responses were designated by blinded independent central review (BICR).
Most patients in the study with laBCC were male (66.7%) and White (67.9%). Infiltrative tumour histology accounted for 8.3% of laBCC lesions while the broad “other” category accounted for 66.7% of lesions, with most (89.3%) occurring in the head or neck region. The mean age of patients with laBCC was 69.1 (standard deviation [SD] = 12.8). The primary outcome was objective response rate (ORR) by BICR, and secondary outcomes included ORR by investigator assessment, duration of response (DOR), progression-free survival (PFS), overall survival (OS), time-to-tumour response, disease control rate, and HRQoL.
As Study 1620 was a single-arm non-comparative trial, the primary outcome was based on rejecting the null hypothesis of an ORR equal to a chosen non-clinically meaningful response rate. In the laBCC group, the null hypothesis was an ORR equal to 20% and would be rejected if the lower bound of the 2-sided 95% confidence interval (CI) excluded the value of 20%. This threshold was chosen to be consistent with what was determined to be clinically meaningful in previous trials for HHI therapy in advanced BCC though, notably, these trials were conducted in the first-line setting.7,8 The assessment of secondary outcomes was descriptive.
The primary analysis of Study 1620 was conducted based on a data cut-off date of February 17, 2020, at which time the mean duration of patient follow-up was 13.53 months and the mean duration of treatment with cemiplimab was 52.80 weeks. An updated analysis was performed |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, |||||||||||||||||||||||||||, at which time the mean duration of follow-up was ||||||||||||| and the mean duration of exposure was ||||||||||||.
Outcome Results
A summary of the results for key outcomes from Study 1620 is shown in Table 2.
The ORR at the time of the primary analysis was 28.6% (95% CI, 19.2% to 39.5%), which failed to meet the 20% pre-specified threshold based on the lower bound of the 95% CI. At the updated analysis, the pre-specified threshold was reached with an ORR (95% CI) of |||||||||||||||||||||||||||||||||||||||||||||. At the primary analysis, the median Kaplan–Meier estimation of DOR in the 24 patients who achieved either a complete response (CR) or partial response (PR) had not been reached. The observed DORs ranged from 2.1 months to greater than 21.4 months, with 79.2% of responders achieving a DOR greater than 6 months, and 45.8% of responders achieving a DOR greater than 12 months.
HRQoL was measured in Study 1620 using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the Skindex-16. Changes over time in the global health status (HRQoL) score for the EORTC QLQ-C30 were smaller than the minimal important difference (MID) estimate of 5 to 10 points at both the primary and updated analyses. An analysis of the EORTC QLQ-C30 functional and symptom scales showed scores consistent with the results for the global health status scale. Symptom scales remained stable over time with the exception of fatigue, which showed worsening in excess of the MID for the fatigue scale at cycles 7 and 9, though patient numbers were reduced at these time points. An improvement in excess of the MID of 10 points or greater was achieved in the emotion scale of the Skindex-16 at cycle 4 and maintained through the end of the study, while the symptom and functioning scales remained stable.
At the time of the primary analysis, 45.2% of patients in the laBCC group had experienced a PFS event, with 39.3% of patients experiencing disease progression and 6.0% experiencing death. The median PFS was 19.3 months (95% CI, 8.6 to not evaluable). At the updated analysis, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
At the time of the primary analysis, deaths had occurred in 11.9% of patients and the median (95% CI) OS had not been reached. At the updated analysis, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Harms Results
Treatment-emergent adverse events (TEAEs) occurred in almost all patients (97.6% and 98.8% at the primary and updated analyses, respectively). Serious adverse events (SAEs) occurred in 34.5% of patients and 36.9% at the primary and updated analyses, respectively, while TEAEs that led to treatment discontinuation occurred in 16.7% and 17.9% of patients, respectively. The most common TEAE that led to a dose delay was diarrhea in 4.8% of patients, followed by blood creatinine increased, fatigue, and urinary tract infection, each occurring in 3.6% of patients. Deaths due to TEAEs occurred in 3.6% of patients and 4.8% at the primary and updated analyses, respectively; these included 1 occurrence each of cachexia, malignant brain neoplasm, and acute kidney injury.
Immune-related adverse events (AEs) occurred in 56% and 58.3% of patients at the primary and updated analyses, respectively. This included 11.9% of patients who experienced grade 3 or greater TEAEs, 9.5% who experienced serious immune-related AEs, and 9.5% who experienced an immune-related AE leading to treatment discontinuation. Infusion reactions occurred at a much lower rate, with only 1.2% of patients experiencing any infusion-related reaction. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 2: Summary of Key Results From Study 1620
Outcomes | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
ORR | ||
ORR, n (%) | 24 (28.6)a | (32.1) |
95% CIb | 19.2 to 39.5 | 22.4 to 43.2 |
CRR,c n (%) | 5 (6.0) | ||||||||||||||| |
95% CIb | 2.0 to 13.3 | ||||||||||||||| |
Best overall response, n (%) | ||
CRc | 5 (6.0) | ||||||||||||||| |
PRc | 19 (22.6) | ||||||||||||||| |
Stable diseased | 43 (51.2) | ||||||||||||||| |
Non-CR and non-PDe | 0 | ||||||||||||||| |
PD | 9 (10.7) | ||||||||||||||| |
NEf | 8 (9.5) | ||||||||||||||| |
Global health status (HRQoL)g | ||
Baseline mean (SD); n | 64.30 (19.14); 74 | ||||||||||||||| |
Change from baseline, mean (SD); n | ||
Cycle 3 | −2.55 (19.82); 62 | ||||||||||||||| |
Cycle 5 | −1.91 (21.21); 48 | ||||||||||||||| |
Cycle 7 | −3.13 (19.72); 32 | ||||||||||||||| |
Cycle 9 | −6.37 (23.48); 17 | ||||||||||||||| |
Skindex-16 emotion scaleh | ||
Baseline mean (SD); n | 39.15 (30.53); 75 | ||||||||||||||| |
Change from baseline, mean (SD); n | ||
Cycle 3 | −8.60 (25.64); 63 | ||||||||||||||| |
Cycle 5 | −10.25 (24.65); 46 | ||||||||||||||| |
Cycle 7 | −13.65 (27.13); 30 | ||||||||||||||| |
Cycle 9 | −14.89 (36.84); 17 | ||||||||||||||| |
Skindex-16 symptoms scaleh | ||
Baseline mean (SD); n | 20.72 (23.04); 76 | ||||||||||||||| |
Change from baseline, mean (SD); n | ||
Cycle 3 | −0.26 (24.16); 64 | ||||||||||||||| |
Cycle 5 | −4.11 (18.06); 47 | ||||||||||||||| |
Cycle 7 | 0.69 (24.52); 30 | ||||||||||||||| |
Cycle 9 | −5.64 (26.76); 17 | ||||||||||||||| |
Skindex-16 functioning scaleh | ||
Baseline mean (SD); n | 25.64 (26.92); 7 | ||||||||||||||| |
Change from baseline, mean (SD); n | ||
Cycle 3 | −4.76 (20.20); 63 | ||||||||||||||| |
Cycle 5 | −3.76 (16.37); 47 | ||||||||||||||| |
Cycle 7 | −6.00 (15.77); 30 | ||||||||||||||| |
Cycle 9 | −4.31 (23.68); 17 | ||||||||||||||| |
PFS | ||
KM estimation of PFS | ||
Number of events, n (%) | 38 (45.2) | ||||||||||||||| |
PD, n (%) | 33 (39.3) | ||||||||||||||| |
Death, n (%) | 5 (6.0) | ||||||||||||||| |
Number of censored patients, n (%) | 46 (54.8) | ||||||||||||||| |
Median (95% CI), months | 19.3 (8.6 to NE) | ||||||||||||||| |
OS | ||
KM estimation of OS | ||
Number of deaths, n (%) | 10 (11.9%) | ||||||||||||||| |
Number of censored patients, n (%) | 74 (88.1%) | ||||||||||||||| |
Median (95% CI), months | NR (NE to NE) | ||||||||||||||| |
DOR | ||
KM estimation of DOR (CR or PR) | ||
Number of events,i n (%) | 6 (25.0) | ||||||||||||||| |
Number of censored patients,i n (%) | 18 (75.0) | ||||||||||||||| |
Median (95% CI), months | NR (15.0 to NE) | ||||||||||||||| |
TTR | ||
Observed time to response (CR or PR), months | ||
Mean (SD) | 5.17 (2.60) | ||||||||||||||| |
Harms, n (%) | ||
TEAEs | 82 (97.6) | ||||||||||||||| |
SAEs | 29 (34.5) | ||||||||||||||| |
WDAEs | 14 (16.7) | ||||||||||||||| |
TEAEs leading to death | 3 (3.6) | ||||||||||||||| |
Notable harms, n (%) | ||
Immune-related AEj | 47 (56.0) | ||||||||||||||| |
Grade 3, 4, or 5 immune-related AE | 10 (11.9) | ||||||||||||||| |
Serious immune-related AE | 8 (9.5) | ||||||||||||||| |
Immune-related AE leading to discontinuation | 8 (9.5) | ||||||||||||||| |
Immune-related AE leading to dose delay | 10 (11.9) | ||||||||||||||| |
Immune-related AE leading to drug interruption | 0 | ||||||||||||||| |
Immune-related AE leading to dose reduction | 0 | ||||||||||||||| |
Immune-related AE resulting in death | 0 | ||||||||||||||| |
Infusion-related reactions | 1 (1.2) | ||||||||||||||| |
Grade 3, 4, or 5 infusion-related reaction | 0 | ||||||||||||||| |
Serious infusion reaction | 0 | ||||||||||||||| |
Infusion reaction leading to discontinuation | 0 | ||||||||||||||| |
Infusion reaction leading to dose delay | 0 | ||||||||||||||| |
Infusion reaction leading to drug interruption | 1 (1.2) | ||||||||||||||| |
Infusion reaction leading to dose reduction | 0 | ||||||||||||||| |
Infusion reaction resulting in death | 0 | ||||||||||||||| |
AE = adverse event; CI = confidence interval; CR = complete response; CRR = complete response rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; KM = Kaplan–Meier; laBCC = locally advanced basal cell carcinoma; MedDRA = Medical Dictionary for Regulatory Activities; NA = not applicable; NE = not evaluable; NR = not reported; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SAE = serious adverse event; SD = standard deviation; TEAE = treatment-emergent adverse event; TTR = time to response; WDAE = withdrawal due to adverse event.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on |||||||||||||||||||||||||||||||||.
aAs per the protocol that requires confirmation of response to be considered a CR or PR, 2 patients who had initial responses that were not confirmed until after the data cut-off are not included.
bClopper-Person exact CI.
cCR and PR must be confirmed by repeated assessments no less than 4 weeks apart.
dStable disease criteria must be met at least once after a minimum duration of 39 days after the first dose date.
eNon-CR and non-PD categories apply to patients with non-measurable disease only.
fNE response includes missing and unknown tumour response.
gScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For global health status, a higher score signifies better HRQoL.
hItem scores are transformed to a linear scale of 0 to 100, with 0 representing “never bothered” and 100 representing “always bothered.”
iEvents include PD or deaths. Percentages are based on number of patients with confirmed CR or PR.
jAs there is currently no MedDRA-coded classification for immune-related AEs, the sponsor created a customized list of MedDRA-preferred terms for the identification of immune-related AEs.
Source: Study 1620 Clinical Study Report.5
Critical Appraisal
The most notable limitation of Study 1620 relates to its single-arm open-label design. Due to this, it is impossible to draw any conclusions about efficacy with any level of certainty. The clinical experts consulted by CADTH agreed with the clinically meaningful ORR threshold of 20%; it was also noted that this threshold is consistent with what was used in previous single-arm trials in patients with laBCC. Rejection of the null hypothesis (ORR = 20%) required the lower bound of the 95% CI to exclude 20%; this was not achieved at the time of the primary analysis (ORR = 28.6; 95% CI, 19.2% to 39.5%). Additionally, 2 patients did not meet the inclusion criterion requiring enrolled patients to have at least 1 measurable lesion but were enrolled in the study despite this. According to the clinical experts consulted for this review, this would likely bias the results by increasing the ORR. Important protocol deviations occurred in 23.8% of the patients in the laBCC group of Study 1620, though the observed protocol deviations were considered acceptable for a second-line oncology clinical trial. The most common important protocol deviations were related to enrolling patients despite deviations in the inclusion (15.5%) and exclusion (3.6%) criteria. A relatively high number of patients discontinued the study for reasons other than PD or death (19.0% at the primary analysis data cut-off); these reasons included AEs, lost to follow-up, non-compliance with the protocol, withdrawal of consent, patient decision, and sponsor decision. Specifically, in the case of non-compliance with the protocol and sponsor decision, the CADTH review team indicated that these are not valid reasons to discontinue the study and are likely to bias the results in favour of cemiplimab.
According to the study protocol, for a patient to have achieved a CR or PR, the response must have been confirmed at least 4 weeks following the initial documented response. If a response was not confirmed, the patient was reported as having stable disease. The sponsor presented an unplanned sensitivity analysis in which the pre-specified threshold to reject the null hypothesis was reached; this analysis includes initial responses from 2 patients that were unconfirmed at the time of the primary analysis. Both patients did ultimately have their responses confirmed; however, these results are based on an ad hoc redefinition of the primary outcome that differs from the study protocol. Since there was no adjustment for multiplicity in this analysis, there is an increased risk of type I error and, therefore, the results obtained should be interpreted with caution. The sponsor also provided the results of an unplanned updated analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| the reported ORR (95% CI) at this data cut-off was |||||||||||||||||||||||. The same limitations regarding no adjustment for multiplicity and increased risk of type I error apply to the updated analysis and results.
According to the clinical experts consulted by CADTH, the demographic and disease characteristics of the Study 1620 population were reflective of the Canadian population with laBCC. The dosage of cemiplimab in Study 1620 was aligned with the Health Canada–approved dosing and with clinical practice. In the study, treatment with cemiplimab was administered until PD or unacceptable toxicity up to 93 weeks. The protocol allowed for re-treatment of patients who had completed the full treatment course but experienced PD during the follow-up period. The sponsor confirmed that 1 patient had entered re-treatment with cemiplimab. The trial data may not be generalizable to treatment beyond the 93-week treatment course or within a re-treatment setting for patients who experience PD following discontinuation of cemiplimab, given the lack of data.
Indirect Comparisons
No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Conclusions
Study 1620 was a single-arm study of cemiplimab (350 mg every 3 weeks up to a maximum of 93 weeks) in patients with laBCC. The study did not meet the pre-specified threshold of a 20% ORR, which is considered clinically meaningful. At the updated analysis, the ORR ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In the opinion of the clinical experts consulted by CADTH, despite the limitations of Study 1620, the observed ORR in patients with laBCC previously treated with an HHI was considered clinically meaningful and of value in a high-burden disease for which there is a high unmet need for treatment options. The descriptive assessment of HRQoL in the study was limited by the low number of respondents contributing to assessments at later time points, but these assessments suggested multiple measures of HRQoL were stable over the course of the study and a clinically meaningful improvement was observed for emotional functioning. Data on PFS and OS were immature, but longer-term data for these outcomes will be challenging to interpret due to the non-comparative trial design. The notable harms that were observed with cemiplimab, specifically immune-related AEs, were consistent with the known safety profile of the drug and were considered manageable, with appropriate supportive care, by both the clinical experts consulted by CADTH and by patients, according to the patient input. The major limitation of Study 1620 is its open-label, single-group study design, which introduces bias in favour of cemiplimab and precludes the ability to evaluate the efficacy and magnitude of the clinical benefit of cemiplimab in this treatment setting.
Introduction
Disease Background
Non-melanoma skin cancer accounts for approximately 28% of new cancer diagnoses in Canada,1 with BCC accounting for 75% of all non-melanoma skin cancers.2 BCC is a malignancy derived from the non-keratinizing cells that form the basal layer of the epidermis. Tumour size can be quite variable, from a few millimetres to several centimetres; BCC tends to invade locally and rarely metastasizes distantly. BCC is principally a disease of the elderly but has been increasingly detected among younger adults.9 BCC generally develops on sun-exposed areas of the skin, and other risk factors include male sex, light hair, northern European ancestry, and the inability to tan. Seventy percent of cases of BCC occur on the head and frequently on the face, whereas 25% occur on the trunk and limbs and 5% in the perineal region.3
Most BCCs are generally diagnosed and treated early; however, some BCCs become extensive and infiltrative, posing a greater risk to patients.4 Generally, BCC is a slow-growing tumour with a doubling rate of between 6 months to 1 year but, left untreated, it may invade into the subcutaneous tissue, muscle, and bone. Direct extension into the central nervous system can also occur. Perineural invasion is uncommon in BCC but does imply a more aggressive phenotype, which is associated with more extensive invasion and more frequent recurrences.10 In BCCs occurring in the periocular region, perineural progression can lead to invasion of the orbital structures and result in pain, paresthesias, eye muscle weakness, and blindness.11 Metastasis of BCC is rare, with rates estimated to be less than 1%.12,13 The most common sites of metastatic spread are the lymph nodes and lungs.14 In those rare cases, squamous differentiation may be present in the primary or metastatic sites and may contribute to the aggressive phenotype.
Standards of Therapy
The principal modality of therapy for BCC is surgery. Curettage and electric dissection are commonly employed, with cure rates of up to 98%.15 However, for larger BCCs, surgical excision offers the most potential for margin control and often provides optimal cosmetic results. To achieve local control, adequate surgical margins are required. Clear surgical margins may be difficult to achieve while still maintaining acceptable cosmesis and can be particularly challenging for eradicating extensive BCCs involving the face.16
Radiotherapy is also commonly used. It has the advantage of sparing normal tissue and may reduce the need for reconstructive surgery. However, in some sites, such as the nose, ear, and periocular regions, collateral damage of normal tissue may occur. Radiotherapy remains an option for poor surgical candidates, but higher failure rates may occur in large, recurrent, and aggressive subtypes of BCCs. Radiotherapy can also be used in the palliation and debulking of tumours that are otherwise inoperable. Adjuvant post-operative radiotherapy may also be considered in cases when the risk of recurrence is high.17
Although laBCC and mBCC are relatively rare disease states, they lead to significant morbidity in patients. In patients with locally advanced and recurrent disease, the primary goal of therapy is local control and not OS. With respect to lesions on the face and distal extremities, an additional therapeutic goal is to maintain or optimize organ function. In some cases of laBCC, extensive surgical resection may not be technically possible. Furthermore, resection may involve removing vital structures such as the orbits or cranial bones, which would result in significant deformity and functional impairment. Moreover, in cases where recurrent disease occurs, further radiotherapy may not possible and the goal of obtaining clear surgical margins may be impossible to achieve.
Canadian clinical practice guidelines for laBCC and mBCC report that treatment involves any combination of surgery, radiation, and chemotherapy, and that there is a lack of evidence to inform treatment recommendations for a standard therapy.18 These guidelines were published in 2015 and are therefore out of date. The most recent guidelines from the National Comprehensive Cancer Network state that systemic HHI therapy is to be considered in patients with laBCC and mBCC if surgery or radiotherapy are unlikely to be curative19 and, for patients previously treated with HHI therapy or for whom HHI therapy is not appropriate, treatment with cemiplimab can be considered.19
At least 90% of BCCs appear to have an acquired aberrant activation of the hedgehog pathway. Linkage analyses have identified a locus on chromosome 9 that is deleted in sporadic BCC.20 The locus encodes for the patch 1 (PTCH1) gene, a transmembrane receptor that inhibits smoothened signalling and the downstream activation of cellular proliferation.21 Because abnormalities in the hedgehog signalling pathway are common in sporadic cases of BCC, routine testing to determine the precise nature of the signalling aberration is not recommended for clinical practice.
Currently, the only HHI therapy in Canada approved and publicly reimbursed for both laBCC and mBCC is vismodegib. The efficacy of vismodegib was evaluated in the multi-centre phase II ERIVANCE trial, which included 63 patients with laBCC and 33 patients with mBCC.8 Sekulic et al. reported a tumour response rate of 43% in patients with laBCC and 30% in patients with mBCC. AEs were common and generally mild and included muscle spasms, dysgeusia, weight loss, and fatigue. Sonidegib, another HHI, is also approved in Canada; however, CADTH did not recommend it for reimbursement when it was reviewed in 2021. The BOLT trial reported an ORR that ranged from 43% to 38% in patients with laBCC, depending on the dose of sonidegib received.7
Because there is currently no available therapy approved in Canada for patients who progress or become intolerant to HHI therapy, patients are treated according to best supportive care.
Drug
Cemiplimab is a recombinant human immunoglobulin G4 (IgG4) monoclonal antibody that binds to the programmed cell death 1 (PD-1) receptor, inhibiting the interaction with programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2). T-cells lose proliferation and function through increased expression of proteins like PD-1 and interaction with PD-L1 and PD-L2, down-modulating the antitumour response of T-cells.22 The inhibitory action of cemiplimab acting on PD-1 counteracts this inhibition of the immune response, including the antitumour immune response of T-cells.
On October 26, 2021, cemiplimab was issued market authorization without conditions for the treatment of patients with laBCC previously treated with an HHI. The sponsor’s reimbursement request for cemiplimab is aligned with the Health Canada–approved indication. Cemiplimab underwent review by Health Canada through a standard review pathway. Cemiplimab is also indicated for the treatment of patients with metastatic or locally advanced cutaneous squamous cell carcinoma and for first-line treatment of patients with non–small cell lung cancer expressing PD-L1, with no epidermal growth factor receptor, anaplastic lymphoma kinase, or ROS1 aberrations.23 Cemiplimab received a positive recommendation with conditions from CADTH in 2020 for the cutaneous squamous cell carcinoma indication.24
Cemiplimab was approved by the FDA for use in patients with laBCC previously treated with HHI therapy or for whom an HHI is inappropriate.25 FDA approval in patients with mBCC was granted under the accelerated approval process and may be dependent on verification of clinical benefit.25 The European Medicines Agency also approved cemiplimab in 2021 for the treatment of laBCC or mBCC following HHI therapy.26
Cemiplimab is administered as an IV infusion over 30 minutes every 3 weeks at a dose of 350 mg and is continued until symptomatic disease progression or unacceptable toxicity. Key characteristics of cemiplimab are shown in Table 3.
Table 3: Key Characteristics of Cemiplimab
Characteristic | Cemiplimab |
|---|---|
Mechanism of action | Cemiplimab is a recombinant human IgG4 monoclonal antibody that binds to the programmed cell death 1 receptor, inhibiting the interaction with PD-L1 and PD-L2. This counteracts the PD-L1-mediated inhibition of the immune response, including the antitumour immune response of T-cells. |
Indicationa | For the treatment of patients with locally advanced basal cell carcinoma previously treated with a hedgehog pathway inhibitor. |
Route of administration | IV infusion. |
Recommended dose | 350 mg every 3 weeks. |
Serious adverse effects or safety issues |
|
IgG4 = immunoglobulin G4; PD-L1 = programmed death-ligand 1; PD-L2 = programmed death-ligand 2.
aHealth Canada–approved indication.
Source: Cemiplimab product monograph.23
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Input was provided by 2 patient groups for this review, the SYSF and the MNC. The SYSF gathered information from online surveys, virtual patient roundtables, and 1-on-one conversations from March 2021 to September 2021. All 23 patients consulted (20 of whom were women) had been diagnosed with BCC, 5 of whom had experience with cemiplimab. It was not reported whether patients had experience with HHI therapy before receiving cemiplimab. All but 4 patients were from Canada, with most of the Canadian responders being from Ontario. The MNC input was sourced from an online survey of 62 patients (44 of which were women) and 45 caregivers. All but 1 of the patients were from Canada, with 50% located in Ontario. Only 1 patient indicated they had experience with cemiplimab in metastatic disease and no patients had experience with HHI therapy.
In both surveys, patients highlighted the negative aspects of BCC and its treatment, including disfigurement, scaring, and associated self-esteem difficulties. In both surveys, pain from the lesions and anxiety over finding recurrent disease were also mentioned by patients as key concerns. In the MNC survey, caregivers expressed that the disease caused much emotional stress due to seeing their loved 1 in pain. Patients expressed a desire for less radiation and disfiguring surgery and greater access to treatments closer to their home and support network. Respondents from the SYSF submission with experience with cemiplimab indicated that the side effects were manageable and the benefits would outweigh the side effects. Of the 5 patients with experience with cemiplimab, 2 had no side effects, 2 patients had fatigue, and 1 patient had skin rash. The 1 patient from the MNC submission who had experience with cemiplimab indicated that having the option for therapy was worth experiencing treatment side effects, which included difficulty with liver-related issues and flu-like symptoms.
Patients indicated there are no other options for treatment following progression on HHI therapy and the ability to access new treatments to eliminate disease and prevent recurrence is needed. Earlier diagnosis, access to specialists, and less invasive procedures were highlighted as important to patients and caregivers in the MNC survey.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adults with laBCC previously treated with HHI therapy.
Unmet Needs
The clinical experts consulted by CADTH highlighted the immense patient burden due to physical disfigurement from the external lesions that commonly present on the face of the patient. There are currently no treatment options for patients with laBCC who have been treated previously with HHI therapy, especially given that response to HHI is low and some patients cannot tolerate the side effects of this therapy.
Place in Therapy
The clinical experts noted that cemiplimab is indicated for patients with laBCC following HHI therapy and, therefore, cemiplimab would be used as a second-line therapy. There is currently no available treatment option for patients following HHI failure and, therefore, cemiplimab would not be displacing any currently prescribed second-line treatment. The clinical experts noted that in the future, combination therapy with cemiplimab and an HHI in the first-line setting could be explored; however, the associated phase II trial is still in the recruitment phase.27
Patient Population
According to the clinical experts, patients with laBCC and mBCC previously treated with HHI therapy would be best suited to receive cemiplimab. In the case of laBCC, lesions should be determined to be unresectable or unsuitable for radiation therapy by a multidisciplinary tumour board. As there were small numbers of patients in the pivotal trial, it is unclear if there are prognostic features that would determine response to treatment, and PD-L1 expression measurements do not appear to correlate with response in this setting. The patients most in need of intervention are those with laBCC that is disfiguring and invading vital structures.
Assessing Response to Treatment
Response to treatment is assessed by serial imaging demonstrating shrinking disease (objective responses) and clinical assessment, as well as more subjective measures such as maintained or improved HRQoL, cancer symptoms, and functional status. The outcomes used in clinical practice are the same as those used in clinical trials. Given the outward nature of locally advanced disease in these patients and the impact it has on HRQoL, patient-reported outcomes were identified by the clinical experts as extremely important in this setting. Increased survival is important if it comes with improvements in HRQoL, particularly in relation to disfigurement changes. Response to treatment should be assessed at each follow-up visit, with imaging performed as appropriate, typically every 3 months.
Discontinuing Treatment
Treatment would usually be discontinued upon disease progression (increase in size or extension of lesions), severe or intolerable side effects, or a lack of response after an adequate duration of treatment (identified as 4 to 6 months of treatment).
Prescribing Conditions
According to the clinical experts, treatment with cemiplimab would be initiated by a medical oncologist or associated team physician with expertise in cancer therapies and toxicity management. Cemiplimab would be administered in cancer centres or centres supervised by cancer centre–approved physicians with the expertise and staff (chemotherapy nurses, oncology pharmacists) to administer systemic therapies and manage treatment-related toxicities.
Additional Considerations
It was reiterated by the clinical experts that individual or community-based physicians may not be aware of the specific indications or contraindications of surgery and radiation for patients with laBCC. Decisions on which lesions are unresectable or not fit for radiation are therefore best determined by a multidisciplinary tumour board.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
One clinician group, Ontario Health (Cancer Care Ontario), provided input for this review. No major views contrary to those provided by the clinical experts consulted by CADTH were presented. Ontario Health echoed the lack of options for patients with laBCC whose disease did not respond to HHI therapy as well as the importance of HRQoL outcomes, specifically those related to disfiguring lesions and surgical scarring.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Questions | Clinical expert response |
|---|---|
Relevant comparators | |
Comments from the drug plans (response not required):
| For consideration by CADTH. |
Considerations for initiation of therapy | |
The treatment protocol includes re-treatment for an additional 4 cycles for patients who complete 9 cycles without disease progression. Should patients who completed 9 cycles but subsequently experience disease progression while off treatment be eligible for re-treatment? | The sponsor confirmed that 1 patient in Study 1620 received re-treatment with cemiplimab after they experienced progression while off treatment following completion of 9 cycles. The protocol allowed for re-treatment if recurrence occurred within the first 7 follow-up visits (visits every 28 days). The clinical experts consulted by CADTH indicated that experience with other immunotherapies suggests that patients with rapid recurrence within 6 months of completing treatment would be less likely to benefit from re-treatment than those experiencing recurrence that occurs 6 to 12 months after completing treatment, provided a significant response was observed during initial treatment. |
Comments from the drug plans (response not required):
| For consideration by CADTH. |
Considerations for prescribing of therapy | |
The usual starting dose of cemiplimab is 350 mg IV every 3 weeks. What is the maximum treatment duration? Should the maximum treatment duration be 93 weeks or until disease progression or unacceptable toxicity? | There are no data from Study 1620 for treating patients beyond the 93-week treatment schedule. Therefore, it is difficult to say whether patients should be treated beyond 93 weeks. The laBCC population that is felt suitable for second treatment is relatively fit, so it would be expected that they would be willing to be treated for the full 93 weeks. If patients with mBCC are treated off-label, it would be expected that they would be treated for as long as they are able to tolerate the treatment without progression. |
Comments from the drug plans (response not required):
| For consideration by CADTH. |
Generalizability | |
Study 1620 included patients who had mBCC, those with no better than stable disease for 9 months following HHI therapy, and an ECOG performance status of 0 and 1. Should treatment with cemiplimab be extended to the following patients:
| Metastatic patients were excluded from the Health Canada indication due to low patient numbers and immature interim results,28 however, there is no reason to believe that cemiplimab would not work in patients with mBCC. Most clinicians would wait 3 to 5 months for a response before exploring other treatment options; therefore, it is reasonable to expect that patients without a response after 9 months on an HHI would be offered cemiplimab. It is important to determine the acceptable criteria for intolerance. The criteria used in Study 1620 (any grade 3 or 4 AE deemed related to an HHI or grade 2 myalgia, dysgeusia, anorexia, nausea, or diarrhea in patients with at least 3 months of exposure to an HHI) would be reasonable for use in the Canadian setting. Patients with an ECOG performance status of ≥ 2 were excluded from Study 1620; however, given the lack of options for patients with this indication, it would be expected that these patients would be offered cemiplimab. |
Care provision issues | |
Serious immune-mediated reactions can be severe to fatal and usually occur during the treatment course. Early diagnosis and appropriate management are essential to minimize life-threatening complications. Should cemiplimab be reimbursed, is a statement needed ensuring access to a treatment centre with expertise to manage these side effects, should they occur? | The clinical experts noted that the oncology community is well accustomed to the use of immunotherapies and their associated side effects and risks. Cemiplimab does not appear to have any additional safety concerns beyond those that treatment clinics and prescribing clinicians are familiar with and able to manage should they arise. Therefore, there does not appear to be a need for a special safety statement for cemiplimab in this indication. |
Comments from the drug plans (response not required):
| For consideration by CADTH. |
System and economic issues | |
Comments from the drug plans (response not required):
| For consideration by CADTH. |
AE = adverse event; BCC = basal cell carcinoma; ECOG = Eastern Cooperative Oncology Group; HHI = hedgehog pathway inhibitor; LOI = letter of intent; mBCC = metastatic basal cell carcinoma; pCPA = pan-Canadian Pharmaceutical Alliance.
Clinical Evidence
The clinical evidence included in the review of cemiplimab is presented in the systematic review, which includes the pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those studies that were selected according to an a priori protocol. No indirect evidence was provided by the sponsor or met the selection criteria specified in the review. No additional relevant studies were identified that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of cemiplimab for the treatment of laBCC in patients previously treated with an HHI.
Methods
The studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults (age ≥ 18 years) with laBCC previously treated with an HHI Subgroups:
|
Intervention | Cemiplimab 350 mg administered as an IV infusion every 3 weeks |
Comparator | Best supportive care |
Outcomesa | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; BCC = basal cell carcinoma; CRR = complete response rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; HHI = hedgehog pathway inhibitor; HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PTCH1 = patched 1 gene; RCT = randomized controlled trial; SAE = serious adverse event; TP53 = tumour protein p53 gene; TTP = time to progression; TTR = time to response; WDAE = withdrawal due to adverse event; vs. = versus.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.29
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Libtayo (cemiplimab) and BCC. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on September 13, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on January 12, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist.30 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
One study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Study 1620
Characteristic | Study 1620 |
|---|---|
Study design | Phase II, open-label, non-randomized, 2-group, multi-centre study |
Locations | 49 sites in 10 countries in North America and Europe (1 site in Canadaa) |
Patient enrolment dates | First patient was enrolled on June 29, 2017; study is ongoing |
Primary analysis (February 17, 2020) enrolled and treated (N) | 132:
|
Updated analysis (||||||||||||) enrolled and treated (N) |
|
Inclusion criteria |
|
Exclusion criteria |
|
(continued) |
|
Intervention | Cemiplimab 350 mg IV infusion every 3 weeks |
Comparator(s) | Not applicable (non-comparative trial) |
Phase | NA |
Screening | Up to 28 days |
Open-label treatment period | Treatment q.3.w. up to a maximum of 93 weeks |
Follow-up | After 93 weeks of treatment, follow-up visits every 28 days for 7 visits and with extended follow-up of 1 year with quarterly assessments |
Primary end point | ORR as determined by BICR |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publications | Stratigos et al. (2021)6 |
AE = adverse event; BICR = blinded independent central review; CR = complete response; DCR = disease control rate; DB = double-blind; ECOG = Eastern Cooperative Oncology Group; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HHI = hedgehog pathway inhibitor; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NA = not applicable; ORR = objective response rate; OS = overall survival; PD-1 = programmed death-1; PD-L1 = programmed death-ligand 1; PFS = progression-free survival; PRO = patient-reported outcome; PS = performance status; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumors; TTR = time-to-tumour response.
aThere was 1 site in Canada that enrolled patients at the time of the primary analysis; a second site was reported at the updated data cut-off.
Source: Study 1620 Clinical Study Report.5
Description of Studies
Study 1620 (R2810-ONC-1620) is a phase II, single-arm, non-randomized, open-label study of cemiplimab in laBCC and patients with laBCC and mBCC following treatment with HHI therapy. The study was funded by Regeneron Pharmaceuticals and Sanofi. The primary objective of Study 1620 was to determine the efficacy of cemiplimab (350 mg every 3 weeks) in achieving an objective response in adult patients with laBCC or mBCC. The laBCC and mBCC populations were recruited and analyzed as 2 distinct groups and therefore the description of the study design includes references to the mBCC group. As the sponsor’s reimbursement request is limited to the laBCC population only, the results presented focus on the laBCC population. The results for the mBCC population are available in Appendix 3. Beginning on June 29, 2017, patients with laBCC or mBCC who had previously been treated with HHI therapy were enrolled in Study 1620 at 49 sites across North America (N = 3 patients in Canada) and Europe.
There were 4 amendments made to the trial protocol, 2 of which were made after patients had been enrolled. Notable amendments to mention include 1 made before the enrolment of patients where the protocol was amended to increase the dose from 250 mg to 350 mg every 3 weeks and to increase the length of the treatment period to 9 cycles. After patients had begun treatment, another protocol amendment was made to add an exclusion criterion for patients previously treatment with idelalisib. This amendment was made in response to safety findings from a separate trial of cemiplimab in patients with lymphoma where 2 patients previously treated with idelalisib experienced severe stomatitis and/or skin reactions, and a third patient experienced myositis and myasthenia gravis after treatment with cemiplimab. The second amendment was made after enrolment of patients began to extend the post-treatment follow-up for an additional year. How many patients had been enrolled before the implementation of these amendments was not reported.
Patients were screened for eligibility for up to 28 days before beginning study treatment. Patients were treated for up to 93 weeks or until progression, unacceptable toxicity, or confirmed response following a minimum of 48 weeks of treatment. Following the open-label treatment phase, patients entered the follow-up phase. This consisted of follow-up visits every 28 days for 7 visits and extended follow-up for 1 year with quarterly assessments. Patients who completed 9 cycles of treatment without progression but who progressed during the first 7 follow-up visits, without receiving any other systemic anti-cancer therapy, were permitted to enter re-treatment for an additional 48 weeks (maximum of 4 re-treatment cycles, 12 weeks per cycle).
The primary analysis was conducted based on a February 17, 2020, data cut-off date. An updated ||||||||||||||||||||||||||||||||||
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria for Study 1620 are summarized in Table 6. Adult patients (age ≥ 18 years) with a histologically confirmed diagnosis of laBCC or mBCC were eligible for inclusion if they were deemed unlikely to benefit from further HHI therapy due to prior progression on HHI therapy, intolerance to prior HHI therapy, or who achieved no better than stable disease after 9 months of HHI therapy. Eligible patients were required to have at least 1 measurable lesion with a longest diameter and a perpendicular diameter of 10 mm or greater if measured by digital medical photography (specifically for the laBCC group), and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Valid justification for unresectable disease included BCC that had recurred in the same location after 2 or more surgical resections, significant local invasion that precluded complete resection, as well as an anatomically difficult location for which surgery could result in severe disfigurement or disfunction. Similarly, patients were required to be deemed not fit for radiation therapy with acceptable justification being that a further dose would exceed the acceptable cumulative dose, the disease was unlikely to respond to therapy according to the judgment of the radiation oncologist, or an individualized risk-benefit assessment by a multidisciplinary team had deemed radiation to be contraindicated.
Patients were considered ineligible for enrolment in Study 1620 if they had received prior treatment with a PD-1 or PD-L1 pathway inhibitor or they had an ongoing or recent autoimmune disease that required treatment with systemic immunosuppressive treatments. Other exclusion criteria included untreated brain metastasis, active infection requiring therapy, or prior treatment with idelalisib. Patients were excluded if they received any anti-cancer treatment other than radiation therapy, investigational or standard care, within 30 days of the initial administration of cemiplimab.
Baseline Characteristics
The baseline characteristics of the patients in Study 1620 are shown in Table 7. Two-thirds of the patients in both the laBCC and mBCC groups were male, with a mean age of 69.1 (SD = 12.8) and 63.6 (SD = 11.4) years in the laBCC and mBCC groups, respectively. Most patients were White (67.9% of the laBCC group and 85.4% of the mBCC group), though a large proportion of the laBCC group (32.1%) reported missing data for race.
Table 7: Summary of Baseline Characteristics in Study 1620
Characteristic | mBCC (N = 48) | laBCC (N = 84) |
|---|---|---|
Sex, n (%) | ||
Male | 33 (68.8) | 56 (66.7) |
Female | 15 (31.3) | 28 (33.3) |
Age, years | ||
Mean (SD) | 63.6 (11.4) | 69.1 (12.8) |
Median (range) | 63.5 (38 to 90) | 70.0 (42 to 89) |
< 65, n, (%) | 24 (50.0) | 31 (36.9) |
≥ 65 to > 75, n (%) | 16 (33.3) | 19 (22.6) |
≥ 75, n (%) | 8 (16.7) | 34 (40.5) |
Race, n (%) | ||
White | 41 (85.4) | 57 (67.9) |
Not reported | 1 (2.1) | 0 |
Missinga | 6 (12.5) | 27 (32.1) |
Ethnicity, n (%) | ||
Not Hispanic or Latino | 41 (85.4) | 56 (66.7) |
Hispanic or Latino | 1 (2.1) | 1 (1.2) |
Missinga | 6 (12.5) | 27 (32.1) |
Height (cm)b | ||
Mean (SD) | 173.06 (8.48) | 170.13 (9.52) |
Median (range) | 173.00 (156.0 to 194.0) | 170.00 (147.0 to 192.0) |
Body weight | ||
Mean (SD) | 79.27 (21.87) | 75.70 (17.51) |
Median (range) | 74.25 (48.0 to 129.9) | 72.95 (44.6 to 134.8) |
BMI (kg/m2)b | ||
Mean (SD) | 26.15 (5.87) | 26.17 (5.47) |
Median (range) | 25.59 (16.81 to 42.91) | 24.49 (17.50 to 42.74) |
BMI = body mass index; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; SD = standard deviation.
aThis information was not reported for patients in countries that prohibit collection or reporting of patient race or ethnicity.
bData are missing for 1 patient.
Source: Study 1620 Clinical Study Report.5
The baseline disease characteristics of patients in Study 1620 are shown in Table 8. Most patients were classified as having an ECOG performance status of 0 at baseline in both the laBCC (60.7%) and mBCC (64.6%) groups. At baseline, the histological subtype as measured by central pathology review was mostly “other” (54.2%) or “unknown” (27.1%) in the mBCC group while, in the laBCC group, “other” accounted for 66.7% of participants’ histologic subtype. Of note, the classification of other could include morpheaform, metatypical, superficial, micronodular, mixed, basosquamous, keratotic, or desmoplastic subtypes. The proportion of tumours with infiltrative subtype was 8.3% in both the laBCC and mBCC groups. There were differences between laBCC and mBCC groups with regard to the primary site of tumour, with the majority of patients with laBCC with head and neck tumours (89.3%), while the mBCC group were more evenly distributed, with 41.7% head and neck tumours and 47.9% with trunk tumours. All patients had received prior HHI therapy. Vismodegib was the most common, received by 94.0% of patients in the laBCC group and 95.8% of patients in the mBCC group. Most patients (72.9% of patients with mBCC and 58.3% of patients with laBCC) received 1 prior HHI therapy, though some patients had received more than 1 prior HHI (27.1% of patients with mBCC and 41.7% of patients with laBCC). Progression of disease was reported as the most common reason for discontinuation of prior HHI therapy (71.4% of patients in the laBCC group and 81.3% of patients in the mBCC group), while 29.2% of patients with mBCC and 38.1% of patients with laBCC discontinued HHI therapy due to intolerance and only 8.3% and 12.5% of patients, respectively, indicated no better than stable disease for longer than 9 months as a reason for discontinuation. The proportion of patients with any number of prior cancer-related surgery was 83.3% in both the laBCC and mBCC groups, while prior radiotherapy was reported in 50.0% and 60.4%, respectively. For the laBCC population, the most common justification for surgical unresectability was an anatomically difficult location, with surgery likely to result in disfigurement or disfunction, which was cited for 40.5% of patients. The most common justification for further radiation being infeasible was that an individualized risk-benefit assessment by a multidisciplinary team deemed radiation to be contraindicated.
Table 8: Summary of Baseline Disease Characteristics in Study 1620
Characteristic | mBCC (N = 48) | laBCC (N = 84) |
|---|---|---|
ECOG performance status, n (%) | ||
0 | 31 (64.6) | 51 (60.7) |
1 | 17 (35.4) | 33 (39.3) |
Histological subtype by BICR | ||
Infiltrative | 4 (8.3) | 7 (8.3) |
Nodular | 5 (10.4) | 21 (25.0) |
Othera | 26 (54.2) | 56 (66.7) |
Unknown | 13 (27.1) | 0 |
Primary site of tumour, n (%) | ||
Head and neck | 20 (41.7) | 75 (89.3) |
Extremity | 4 (8.3) | 2 (2.4) |
Trunk | 23 (47.9) | 7 (8.3) |
Anogenital | 1 (2.1) | 0 |
Number of patients with prior HHI therapy, n (%) | 48 (100) | 84 (100) |
Sonidegib | 7 (14.6) | 14 (16.7) |
Vismodegib | 46 (95.8) | 79 (94.0) |
Both vismodegib and sonidegibb | 5 (10.4) | 9 (10.7) |
Number of HHI regimens at baseline, n (%) | ||
1 | 35 (72.9) | 49 (58.3) |
2 | 9 (18.8) | 27 (32.1) |
≥ 3 | 4 (8.3) | 8 (9.5) |
Reason for discontinuation of prior HHI therapy, n (%) | ||
Progression of disease | 39 (81.3) | 60 (71.4) |
Intolerance | 14 (29.2) | 32 (38.1) |
No better than stable disease after 9 months on HHI therapy | 6 (12.5) | 7 (8.3) |
Number of prior cancer-related surgeries, n (%) | ||
Any | 40 (83.3) | 70 (83.3) |
0 | 8 (16.7) | 14 (16.7) |
1 | 7 (14.6) | 22 (26.2) |
2 | 12 (25.0) | 10 (11.9) |
3 | 7 (14.6) | 9 (10.7) |
> 3 | 14 (29.2) | 29 (34.5) |
Number of prior cancer-related radiotherapies, n (%) | ||
Any | 29 (60.4) | 42 (50.0) |
0 | 19 (39.6) | 42 (50.0) |
1 | 22 (45.8) | 27 (32.1) |
2 | 6 (12.5) | 11 (13.1) |
3 | 0 | 2 (2.4) |
> 3 | 1 (2.1) | 2 (2.4) |
Primary reason for unresectability, n (%) | ||
BCC has recurred in the same location after 2 or more surgical procedures | NA | 22 (26.2) |
Anatomically challenging location that may result in severe disfigurement | NA | 34 (40.5) |
Significant local invasion that precludes complete resection | NA | 26 (31.0) |
Other conditions deemed to be contraindicating surgery | NA | 2 (2.4) |
Primary reason for not being a candidate for radiation, n (%) | NA | NA |
Further radiation would exceed acceptable cumulative dose | NA | 23 (27.4) |
Risk-benefit assessment deemed radiation to be contraindicated | NA | 36 (42.9) |
Judgment of radiation oncologist that tumour is unlikely to respond | NA | 25 (29.8) |
BCC = basal cell carcinoma; BICR = blinded independent central review; ECOG = Eastern Cooperative Oncology Group; HHI = hedgehog pathway inhibitor; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NA = not applicable.
a“Other” can include morpheaform, metatypical, superficial, micronodular, mixed, basosquamous, keratotic, or desmoplastic subtypes.
bPatients received both sonidegib and vismodegib as separate lines of therapy.
Source: Study 1620 Clinical Study Report.5
Interventions
All patients enrolled into Study 1620 were administered cemiplimab as an IV infusion every 3 weeks at a dose of 350 mg. The infusion was administered in an outpatient setting over approximately 30 minutes. As this was an open-label and single-arm trial, there was no blinding to treatment for patients or investigators.
Other than the study drug, all treatment administered from the time of signed consent to 30 days following the last administration of the study drug was considered concomitant medication. Focal palliative radiation was allowed for local control of a tumour if the patient had been on treatment for 24 weeks; the patient was considered to have experienced disease progression if radiation therapy was initiated. It was recommended that patients not receive systemic corticosteroids, except for a life-threatening emergency or to treat an immune-related AE. Physiologic replacement doses of systemic corticosteroids were permitted, along with any other medication considered in the investigator’s judgment to be necessary for the patient’s welfare and not expected to interfere with the study drug. Premedications for study treatments were permitted if deemed necessary by the investigator; however, no premedication was permitted for the first dose of the study drug.
Study rules for treatment-dose modifications and discontinuations are summarized in Table 9. Dose reductions of the first order reduced the dose to 120 mg every 3 weeks, and patients requiring a second dose reduction were reduced further to 60 mg every 3 weeks. Patients who experienced grade 3 or greater AEs were required to temporarily discontinue treatment with cemiplimab and could be considered for re-treatment when the toxicity resolved to grade 1 or baseline. Patients who required treatment to be discontinued for more than 84 consecutive days and patients with grade 3 or greater uveitis were permanently discontinued from cemiplimab. If a patient experienced an immune-related AE of grade 3 or greater, treatment was withheld and, if the corticosteroid (prednisone or equivalent) dose could not be brought down to less than 10 mg per day within 12 days of onset, treatment was discontinued.
Table 9: Study Treatment-Dose Modifications or Discontinuations
Toxicity | Grade | Hold treatment? | Restarting criteria | Restarting dose and/or schedule | Discontinuation criteria |
|---|---|---|---|---|---|
Hematological toxicity (other than grade 3 thrombocytopenia lasting longer than 7 days or associated with bleeding) | 1, 2, 3 | No | NA | NA | NA |
4 | Yes | Toxicity resolves to grade ≤ 1 or baseline | Decrease cemiplimab dosage to the next-lower dosing levela |
| |
Grade 3 thrombocytopenia lasting longer than 7 days or associated with bleeding | 3 | Yes | Toxicity resolves to grade ≤ 1 or baseline | Decrease cemiplimab dosage to the next-lower dosing levela |
|
Non-hematological toxicity | 1 | No | NA | NA | NA |
2 | Consider withholding for persistent symptoms | Toxicity resolves to grade 0 to 1 or baseline |
| Toxicity does not resolve within 84 days of last infusion | |
3 | Yes | Toxicity resolves to grade 0 to 1 or baseline | Decrease cemiplimab dosage to the next-lower dosing level | Toxicity does not resolve within 84 days of last infusion | |
4 | Yes | NA | NA | Patient must be discontinued |
AE = adverse event; NA = not applicable.
aFirst dose reduction = cemiplimab 120 mg every 3 weeks; second dose reduction = cemiplimab 60 mg every 3 weeks.
Source: Study 1620 Clinical Study Report.5
Outcomes
A list of end points identified in the CADTH review protocol that were assessed in the study included in this review is provided in Table 10. A detailed discussion and critical appraisal of the outcome measures of HRQoL used in Study 1620, EORTC QLQ-C30 and Skindex-16, is provided in Appendix 4. The EORTC QLQ-C30 has been used extensively in oncology trials and has demonstrated reliability, validity, and responsiveness to change in cancer patients, including patients with non-melanoma skin cancer (NMSC). The consensus of several studies was that the MID on any of the instrument’s scales was approximately 5 to 10 points. The Skindex-16 has been used extensively in dermatologic diseases and has demonstrated reliability, validity, and responsiveness to change in patient populations, including those with NMSC. The consensus of several studies was that the MID on any of the instrument’s scales was approximately 10 points.
Table 10: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 1620 |
|---|---|
ORR according to BICR was assessed separately for patients with laBCC and mBCC:
| Primary |
ORR by investigator assessment | Secondary |
DOR was measured as the time from criteria first met for CR or PR to the first date of recurrent disease or PD, or death from any cause | Secondary |
PFS measured (photographically or radiographically) from the start of treatment until the first date of recurrent disease or PD, or death due to any cause, by BICR and investigator assessment | Secondary |
OS measured from the start of treatment until death due to any cause | Secondary |
Time-to-tumour response was measured from the start of treatment to the first time measurement criteria were met for CR or PR | Secondary |
CR rate with tumour biopsy required for laBCC | Secondary |
Disease control rate was measured as the proportion of patients with a best overall response for CR, PR, or stable disease | Secondary |
HRQoL as measured by the EORTC QLQ-C30 and Skindex-16 | Secondary |
BICR = blinded independent central review; CR = complete response; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors.
Source: Study 1620 Clinical Study Report.5
The primary outcome of ORR was assessed separately for the mBCC and laBCC groups. Tumour response was assessed every 9 weeks for cycles 1 through 5 and every 12 weeks for cycles 6 through 9. Patients with radiologically measurable lesions were assessed using RECIST 1.1, where CR was predefined as disappearance of all target lesions, PR represented at least a 30% reduction in the sum diameters of the target lesion, and PD represented at least a 20% increase in the sum of diameters of the target lesion, while stable disease was achieved when there was neither sufficient growth nor shrinkage to qualify for PD or PR, respectively.
Specifically for the laBCC population, patients with only externally visible lesions were assessed according to digital medical photography and clinical response was scored according to a modified bi-dimensional WHO criteria, where CR was achieved with the disappearance of all target and non-target lesions, PR was achieved with a decrease of 50% or more in the sum products of the perpendicular longest dimensions, PD was represented by an increase of 25% or more in the sum products of perpendicular longest dimensions, and stable disease was achieved if there was neither sufficient shrinkage nor growth to qualify for PR or PD. Patients with visible external lesions were also considered to have PD if there was a new lesion with a longest diameter and a perpendicular diameter of 10 mm or greater that was clearly documented as not having been present previously. Of note, PR and CR responses were required to be maintained and confirmed 4 weeks following initial documentation of response before the patient could be considered to have achieved a PR, while a CR also required a confirmatory biopsy result.
For patients with laBCC with lesions that were both visibly measurable with digital medical photography and radiologically measurable according to RECIST 1.1, composite scoring criteria were used. Methodology for the composite scoring is shown in Table 11. Additionally, any previously inoperable lesion that was deemed to become operable following study treatment was considered a PR.
Table 11: Objective Response Composite Scoring Criteria
Clinical response (digital medical photography) | RECIST 1.1 response (radiology) | Composite (overall) |
|---|---|---|
CR | CR or NA | CR |
NA | CR | CR |
CR | PR or stable disease | PR |
PR | CR, PR, or stable disease or NA | PR |
NA | PR | PR |
Stable disease | CR or PR | PR |
Stable disease | Stable disease or NA | Stable disease |
NA | Stable disease | Stable disease |
PD | Any | PD |
Any | PD | PD |
CR = complete response; NA = not applicable; PD = progressive disease; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors.
Source: Study 1620 Clinical Study Report.5
All imaging data and response outcomes were reviewed by both the investigator and by BICR; however, these assessments were not completed in real time and, as such, clinical management decisions were made according to local investigator assessment. In the event of differing opinions between the investigator decision and the BICR, such that it would impact ongoing patient management, the situation would be discussed between the sponsor and the investigator.
HRQoL, as measured by the EORTC QLQ-C30 and Skindex-16 instruments, was measured on the first day of every cycle and at end of study, with the change in score from baseline measured from day 1 of the first treatment cycle. DOR was analyzed for patients who achieved a CR or PR from the time of meeting response criteria to the first date of recurrence, progression, or death due to any cause. Patients were censored at the last evaluable tumour assessment if they did not have documented tumour progression or death, or if they initiated new anti-cancer therapy without progression. PFS was analyzed from the start of treatment to the first date of recurrence, progression, or death due to any cause. Patients with no evaluable post-baseline tumour assessment were censored for PFS on the date of first study treatment. OS was measured from the start of treatment to death due to any cause; patients whose survival status was unknown were censored at the date at which they were last known to be alive. Time-to-tumour response was analyzed in patients with confirmed responses from the start of treatment to the date the response criteria were first met. All time-to-response variables were measured as date of event or censor minus the date of the first study treatment plus 1 day.
Harms outcomes included TEAEs, SAEs, and AEs requiring dose interruption or reduction; WDAEs; and AEs of special interest. AEs and SAEs were collected from the time of informed consent until 105 days following the last dose of the study drug, with AEs occurring before first dose recorded on the medical history page. AEs were coded to a preferred term and associated primary system organ class according to the Medical Dictionary for Regulatory Activities version 22.1. AEs of special interest for Study 1620 included grade 2 or greater infusion-related reactions, allergic or hypersensitivity reactions, grade 3 or greater immune-related toxicities, and any immune-related toxicity occurring in a patient previously treated with phosphoinositide 3-kinase inhibitors (PI3Ks). Laboratory safety variables, vital signs, electrocardiograms, and physical examination variables were also monitored.
Statistical Analysis
As Study 1620 was a single-arm non-comparative trial, the primary end point was based on rejecting the null hypothesis of an ORR equal to a chosen non-clinically meaningful response rate. For the laBCC group, the null hypothesis was an ORR equal to 20% and would be rejected if the lower bound of the 2-sided 95% CI excluded the value of 20%. For the mBCC group, the null hypothesis was an ORR equal to 15% and would be rejected if the lower bound of the 2-sided 95% CI excluded the value of 15%. These thresholds were chosen to be consistent with what was determined to be clinically meaningful for HHI therapy in advanced BCC.7,8
In the laBCC group, it was determined that a sample size of 80 was required to provide 85% power to reject the null hypothesis if the true ORR was 30% or more. In the mBCC group, it was determined that a sample size of 50 was required to provide 85% power to reject the null hypothesis if the true ORR was 28% or more. Target sample sizes were further increased by 5% to 53 patients in the mBCC group and 84 patients in the laBCC group.
The primary outcome of ORR, as measured by BICR in both groups, was tested according to the previously described hypothesis; all other secondary outcomes were summarized descriptively. ORR as well as CR rate and disease control rate were summarized by group, with the exact binomial 95% CI reported according to the Clopper-Pearson method. DOR, PFS, and OS distributions were estimated using the Kaplan–Meier method along with their medians and 95% CI. Kaplan–Meier estimates were produced for specific time points. Time-to-tumour response was summarized descriptively for specific time periods of interest. Table 12 provides a summary of the statistical analysis for each end point in Study 1620. All statistical analyses were conducted separately for the mBCC and laBCC groups.
Table 12: Statistical Analysis of Progression and Response End Points
Type | End point | Statistical model | Sensitivity analysis |
|---|---|---|---|
Primary | ORR as measured by BICR | Two-sided 95% exact binomial CIs were derived using the Clopper-Pearson method | The sponsor presented analysis of ORR based upon the inclusion of unconfirmed responses at the primary analysisa |
Secondary | ORR as measured by investigator assessment | Two-sided 95% exact binomial CIs were derived using the Clopper-Pearson method | NA |
Secondary | DOR as measured by BICR and investigator assessment | Distribution estimated using the Kaplan–Meier method, 2-sided 95% CI at specified time points | NA |
Secondary | PFS as measured by investigator assessment and BICR | Distribution estimated using the Kaplan–Meier method, 2-sided 95% CI at specified time points | Start of anti-cancer therapy considered as an event |
Secondary | OS | Distribution estimated using the Kaplan–Meier method, 2-sided 95% CI at specified time points | Patients censored at the start date of subsequent therapy |
Secondary | TTR as measured by BICR and investigator assessment | Summarized descriptively at specified time points | NA |
Secondary | CR rate as measured by BICR and investigator assessment | Two-sided 95% exact binomial CIs were derived using the Clopper-Pearson method | NA |
Secondary | DCR as measured by BICR and investigator assessment | Two-sided 95% exact binomial CIs were derived using the Clopper-Pearson method | NA |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; DCR = disease control rate; DOR = duration of response; NA = not applicable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TTR = time to response.
aSensitivity analysis to include unconfirmed responses in the ORR outcome was not pre-specified.
Source: Study 1620 Clinical Study Report.5
The data cut-off date for the primary analysis was chosen to allow the last patient enrolled to be followed for 27 weeks in order for an adequate time for response, plus an additional 30 weeks to allow for adequate follow-up for DOR, for a total of 57 weeks of follow-up. At the time of the primary analysis for the laBCC group, an interim analysis was conducted in the mBCC group for patients with sufficient follow-up. For tests where alpha spending was required, a 2-sided alpha of 0.0001 was applied to the interim analysis, with 0.0499 preserved for the final analysis. For the primary outcome of ORR in patients with mBCC at the interim analysis, both an adjusted 2-sided 99.99% CI and unadjusted 2-sided 95% CI were reported. The updated analysis |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Analysis Populations
The full analysis set included all patients who were enrolled in Study 1620 and deemed eligible for treatment by way of passing the screening criteria. All response and progression end points were analyzed in the full analysis set. The full analysis set for the interim analysis of the mBCC group included only patients with sufficient follow-up time, which was considered to be 6 months. The safety analysis set included all patients who received any study drug at the time of the data cut-off date.
Results
Patient Disposition
At the time of the primary analysis in the laBCC group, a total of 165 patients were screened, of which 132 were enrolled into Study 1620 and 33 were screening failures. The proportion of screen failures that comprised patients with laBCC or mBCC was not provided; therefore, Table 13 summarizes the reasons for screening failures for the 2 groups combined. Only the results for the laBCC group are presented in the following section, as these align with the reimbursement request; results for the mBCC group are presented in Appendix 3. The most common reason for screening failure was failure to meet the inclusion or exclusion criteria (72.7%), with the most commonly failed inclusion criteria being the requirement for a histologically confirmed diagnosis of invasive BCC and at least 1 measurable lesion, according to study criteria. Of the 33 total screening failures, 18.2% were classified as other.
Table 13: Screening Failures in Study 1620
Details | mBCC + laBCC groups combined |
|---|---|
Screened, N | 165 |
Enrolled, n | 132 |
Screened but not enrolled, n (%) | 33 (100) |
Reason for screen failure, n (%) | NA |
Withdrawal of consent | 2 (6.1) |
Death | 1 (3.0) |
Other | 6 (18.2) |
Did not meet inclusion or exclusion criteria, n (%) | 24 (72.7) |
Exclusion criteria 1: Ongoing or recent evidence of significant autoimmune disease that required treatment with systemic immunosuppressive treatments | 1 (3.0) |
Exclusion criteria 11: Concurrent malignancy other than BCC and/or history of malignancy other than BCC within 3 years of date of first planned dose of cemiplimab | 3 (9.1) |
Exclusion criteria 14: Any medical comorbidity, physical examination finding, or metabolic dysfunction or clinical laboratory abnormality that, in the opinion of the investigator, rendered the patient unsuitable for participation in a clinical trial | 2 (6.1) |
Exclusion criteria 15: Inability to undergo any contrast-enhanced radiologic response assessment | 1 (3.0) |
Exclusion criteria 6: Active infection requiring therapy, including positive tests for HIV-1 or HIV-2 serum antibody, hepatitis B virus, or hepatitis C virus | 1 (3.0) |
Exclusion criteria 7: History of pneumonitis within the past 5 years | 1 (3.0) |
Inclusion criteria 1: Histologically confirmed diagnosis of invasive BCC | 6 (18.2) |
Inclusion criteria 13: Patient willing and able to comply with clinic visits and study-related procedures | 1 (3.0) |
Inclusion criteria 3: At least 1 lesion that was measurable by study criteria | 6 (18.2) |
Inclusion criteria 4: ECOG performance status ≤ 1 | 1 (3.0) |
Inclusion criteria 7: Renal function — serum creatinine ≤ 2 × ULN or estimated creatinine clearance > 35 mL/min | 1 (3.0) |
BCC = basal cell carcinoma; ECOG = Eastern Cooperative Oncology Group; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NA = not applicable; ULN = upper limit of normal.
Source: Study 1620 Clinical Study Report.5
Patient disposition in the laBCC group of Study 1620 is summarized in Table 14. Of the 84 patients enrolled, 15.5% of patients had completed treatment at the time of the primary analysis, while 61.9% of patients had discontinued treatment. The most common reason for discontinuing treatment was disease progression (34.5%) followed by AEs as the second most common reason (15.5%). At the time of the primary analysis, 33.3% of patients were ongoing in the study, and 7.1% of patients had completed 93 weeks of treatment plus follow-up periods. With regard to study discontinuation, PD was still the most common reason (33.3%); however, 19.0% of patients discontinued study for reasons other than PD or death. These reasons included lost to follow-up, non-compliance with protocol, patient decision, sponsor decision, withdrawal of consent, AEs, or other. Of the 2 patients who discontinued the study for reasons classified as “other,” 1 was due to a general worsening of clinical condition and the other was due to pulmonary inflammation and worsening dyspnea. The full analysis set included all patients who had received the study treatment and had sufficient follow-up; this set was therefore equal to the safety analysis set. At the time of the primary analysis, 21.4% of patients had entered the follow-up period. The mean follow-up duration was 13.53 (SD = 6.54) months.
At the time of the updated analysis, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The proportion of patients who had completed study treatment increased to ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The full analysis and safety analysis sets ||||||||||||||||||||||||||||||||||||||||.
Table 14: Patient Disposition and Survival Follow-up in Study 1620
Disposition | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
Enrolled, N (%) | 84 (100) | |||||||| |
Treatment ongoing, n (%) | 19 (22.6) | |||||||| |
Off treatment, n (%) | 65 (77.4) | |||||||| |
Treatment completed, n (%) | 13 (15.5) | |||||||| |
Treatment discontinued, n (%) | 52 (61.9) | |||||||| |
Reason for treatment discontinuation, n (%) | NA | |||||||| |
AEs | 13 (15.5) | |||||||| |
Death | 1 (1.2) | |||||||| |
Lost to follow-up | 2 (2.4) | |||||||| |
Non-compliance with protocol | 1 (1.2) | |||||||| |
Patient decision | 5 (6.0) | |||||||| |
PD | 29 (34.5) | |||||||| |
Withdrawal of consent | 0 | |||||||| |
Confirmed CR per investigator assessment | 1 (1.2) | |||||||| |
Other | 0 | |||||||| |
Study ongoing, n (%) | 28 (33.3) | |||||||| |
Off study, n (%) | 56 (66.7) | |||||||| |
Study completed, n (%) | 6 (7.1) | |||||||| |
Study discontinued, n (%) | 50 (59.5) | |||||||| |
Reason for study discontinuation, n (%) | ||
AEs | 2 (2.4) | |||||||| |
Death | 6 (7.1) | |||||||| |
Lost to follow-up | 2 (2.4) | |||||||| |
Non-compliance with protocol | 1 (1.2) | |||||||| |
Patient decision | 5 (6.0) | |||||||| |
Sponsor decision | 1 (1.2) | |||||||| |
PD | 28 (33.3) | |||||||| |
Withdrawal of consent | 3 (3.6) | |||||||| |
Other | 2 (2.4) | |||||||| |
Entered follow-up, n (%) | 18 (21.4) | |||||||| |
Duration of study follow-up, months | ||
Mean (SD) | 13.53 (6.54) | |||||||| |
Median (range) | 15.06 (0.5 to 25.1) | |||||||| |
FAS, N | 84 (100) | |||||||| |
Safety, N | 84 (100) | |||||||| |
AE = adverse event; CR = complete response; FAS = full analysis set; laBCC = locally advanced basal cell carcinoma; NA = not applicable; PD = progressive disease; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on |||||||||||||||||||||||||.
Source: Study 1620 Clinical Study Report.5
Protocol deviations are shown in Table 15. Of the 23.8% patients with an important protocol deviation, the most common deviations related to meeting exclusion criteria but being enrolled (3.6%), or failing to meet inclusion criteria but being enrolled (15.5%). Most notably, from the patients who did not meet the inclusion criteria, there were 2 patients with no measurable lesions who were enrolled in the study. Other more common inclusion criteria that were not met were tumour material not confirmed by central pathology review before enrolment, and creatine phosphokinase testing not performed at screening.
Table 15: Important Protocol Deviations In Study 1620
Details | laBCC (N = 84) |
|---|---|
Number of important protocol deviations | 29 |
Patients with any important protocol deviation, n (%) | 20 (23.8) |
Exclusion criteria met but patient enrolled, n (%) | 3 (3.6) |
Exclusion criterion 8: Any anti-cancer treatment other than radiation therapy, investigational or standard of care, within 30 days of the initial administration | 2 (2.4) |
Exclusion criterion 15: Inability to undergo any contrast-enhanced radiologic response assessment | 1 (1.2) |
Inclusion criteria not met but patient enrolled, n (%) | 13 (15.5)a |
Inclusion criterion 3: No measurable lesion | 2 (2.4) |
Inclusion criterion 8: Creatine phosphokinase not performed at both screening and on C1D1 | 6 (7.1) |
Inclusion criterion 11: Archival or newly obtained tumour material for central pathology review for confirmation of BCC was not confirmed as received by central laboratory before enrolment | 7 (8.3) |
Inclusion criterion 6: Hepatic function not meeting protocol criteria for alkaline phosphatase levels that were higher than 2.5 × the upper limit of normal at both screening and on C1D1 | 1 (1.2) |
Inadequate administration of informed consent, n (%) | 1 (1.2) |
SAEs and AESIs not reported within 24 hours to PVRM, n (%) | 1 (1.2) |
Treatment deviation, n (%) | 2 (2.4) |
Other, n (%) | 2 (2.4) |
AESI = adverse event of special interest; BCC = basal cell carcinoma; C1D1 = cycle 1, day 1; CPK = creatine phosphokinase; laBCC = locally advanced basal cell carcinoma; PVRM = pharmacovigilance and risk management; SAE = serious adverse event.
aSome patients did not meet multiple inclusion requirements; therefore, the total specific inclusion criteria deviations sums to greater than the number of patients with an inclusion criteria deviation.
Source: Study 1620 Clinical Study Report.5
Exposure to Study Treatments
Treatment exposure in Study 1620 is shown in Table 16. At the time of the primary analysis, the mean duration of exposure for patients in the laBCC group was 52.80 (SD = 28.85) weeks, with a mean number of 16.7 (SD = 9.42) doses administered. Patients received a mean relative dose intensity of 0.95 (SD = 0.11), and 89.3% of patients had a treatment compliance rate of 80% or greater. At the updated analysis, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Table 16: Treatment Exposure in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
Duration of exposure, weeksa | ||
Mean (SD) | 52.80 (28.85) | |||||||||||||||| |
Median (range) | 47.15 (2.1 to 94.0) | |||||||||||||||| |
Number of doses administered | ||
Mean (SD) | 16.7 (9.42) | |||||||||||||||| |
Median (range) | 15 (1 to 31) | |||||||||||||||| |
Actual dose intensity (mg/week)b | ||
Mean (SD) | 110.65 (12.44) | |||||||||||||||| |
Median (range) | 115.50 (69.6 to 163.3) | |||||||||||||||| |
Relative dose intensityc | ||
Mean (SD) | 0.95 (0.11) | |||||||||||||||| |
Median (range) | 0.99 (0.6 to 1.4) | |||||||||||||||| |
Treatment compliance,d n (%) | ||
< 60% | 1 (1.2) | |||||||||||||||| |
≥ 60% < 80% | 8 (9.5) | |||||||||||||||| |
≥ 80% | 75 (89.3) | |||||||||||||||| |
laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||.
aDuration of exposure (weeks) = Minimum of (last dose date minus first dose date plus 21 days) divided by 7 AND (data cut-off date or death date minus first dose date plus 1 day) divided by 7 for 350 mg every 3 weeks..
bActual dose intensity (mg/week) = Total dose received (mg) divided by duration of exposure (weeks).
cRelative dose intensity = Actual dose intensity divided by planned dose intensity. Planned dose intensity (mg/week) = Planned dose (mg) divided by 3 (weeks).
dTreatment compliance = (Number of investigational product doses administered during the treatment period divided by the number of investigational product doses planned to be taken during treatment period) multiplied by 100%.
Source: Study 1620 Clinical Study Report.5
A summary of dose delays and infusion interruptions is shown in Table 17. At the time of the primary analysis, 45.2% of patients had experienced at least 1 dose delay or an infusion interruption. More patients experienced at least 1 dose delay (40.5%) compared with infusion interruptions (4.8%). Of the 4 patients who experienced an infusion interruption, 3 were interrupted due to an AE, while the reason for the interruption in the other patient was reported as “other.” Details on TEAEs and infusion interruptions are summarized in the Harms section.
Table 17: Summary of Dose Delays and Infusion Interruptions in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
Patients with at least 1 dose delay or infusion interruption, n (%) | 38 (45.2) | |||||||||||||||| |
Patients with at least 1 dose delay, n (%) | 34 (40.5) | |||||||||||||||| |
Number of dose delays, n (%) | ||
1 | 17 (20.2) | |||||||||||||||| |
2 | 10 (11.9) | |||||||||||||||| |
≥ 2 | 5 (6.0) | |||||||||||||||| |
Patients with at least 1 infusion interruption, n (%) | 4 (4.8) | |||||||||||||||| |
Number of infusion interruptions, n (%) | ||
1 | 4 (4.8) | |||||||||||||||| |
Reason for infusion interruption | ||
AE | 3 (3.6) | |||||||||||||||| |
Other | 1 (1.2) | |||||||||||||||| |
AE = adverse event; laBCC = locally advanced basal cell carcinoma; NA = not applicable.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||.
Source: Study 1620 Clinical Study Report.5
Concomitant medication use in Study 1620 is shown in Table 18. Overall, the use of any concomitant medication was high in patients with laBCC (97.6%). In the laBCC group, the most common categories of concomitant medications administered were analgesics (51.2%) and antibacterials for systemic use (52.4%).
Table 18: Concomitant Medications and Procedures in Study 1620
Details | laBCC (N = 84) |
|---|---|
Number of patients with any concomitant medications, n (%) | 82 (97.6) |
Analgesics | 43 (51.2) |
Antibacterial for systemic use | 44 (52.4) |
Antithrombotic drugs | 28 (33.3) |
Drugs acting on the renin-angiotensin system | 39 (46.4) |
Drugs for acid-related disorders | 31 (36.9) |
Beta-blocking drugs | 26 (31.0) |
Corticosteroid for systemic use | 26 (31.0) |
Anti-inflammatory and antirheumatic products | 17 (20.2) |
Lipid-modifying drugs | 17 (20.2) |
Psycholeptics | 19 (22.6) |
Diuretics | 21 (25.0) |
Ophthalmological | 24 (28.6) |
Vitamins | 14 (16.7) |
Corticosteroids, dermatological preparations | 21 (25.0) |
Antianemic preparations | 18 (21.4) |
Antidiarrheals, intestinal | 17 (20.2) |
Thyroid therapy | 20 (23.8) |
Blood substitutes and perfusion solution | 18 (21.4) |
Drugs used in diabetes | 20 (23.8) |
Psychoanaleptics | 15 (17.9) |
Calcium channel blockers | 14 (16.7) |
Number of patients with any concomitant procedures, n (%) | 61 (72.6) |
Investigations | 53 (63.1) |
Surgical and medical procedures | 32 (38.1) |
Uncoded | 1 (1.2) |
laBCC = locally advanced basal cell carcinoma.
Source: Study 1620 Clinical Study Report.5
Outcomes
Only those outcomes and analyses of subgroups identified in the review protocol are reported subsequently. See Appendix 3 for detailed results on other outcomes.
Objective Response Rate
ORR by BICR assessment was the primary end point in Study 1620 and the results are presented in Table 19. At the time of the primary analysis, the ORR in the laBCC group was 28.6% (95% CI, 19.2% to 39.5%), of which 6% achieved CR and 22.6% achieved PRs. The lower bound of the 95% CI did not exclude the pre-specified threshold of 20%.
Two pre-specified subgroup analyses of ORR were of interest to this review: reason for HHI discontinuation (progression or intolerance) and histologic subtype, as measured by central pathology review. In patients who discontinued HHI due to progression or lack of response (N = 63), the ORR was 28.6% (95% CI, 17.9% to 41.33%) and, in patients who discontinued due to intolerance (N = 21), the ORR was 28.6% (95% CI, 11.3% to 52.2%). The ORRs based on histologic subtypes were as follows: in patients with infiltrative histology (N = 7), the ORR was 42.9% (95% CI, 9.9% to 81.6%); in patients with nodular histology (N = 21), the ORR was 19.0% (95% CI, 5.4% to 41.9%); in patients with other histology (N = 56), the ORR was 30.4% (95% CI, 18.8% to 44.1%).
At the time of the primary analysis, it was noted that 2 patients with a PR were not included as responders in the analysis due to their responses not having been confirmed by BICR and, therefore, according to the protocol, were to be considered as having stable disease. These patients were included in a sensitivity analysis of the primary outcome because they ultimately were confirmed by BICR as having achieved a PR, albeit after the primary analysis data cut-off had passed. The results of this analysis show that the inclusion of these 2 patients pushes the lower bounds of the 95% CI above 20% (results not shown).
At the updated analysis, the ORR (95% CI) for the full analysis set was ||||||||||||||||||||||||||||||||||||||||||, of which |||||||| were CRs and |||||||| were PRs.
Table 19: ORR by BICR Assessment in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
ORR, n (%) | 24 (28.6)a | (32.1) |
95% CIb | 19.2 to 39.5 | 22.4 to 43.2 |
CRR,c n (%) | 5 (6.0) | |||||||||||| |
(95% CI)b | (2.0 to 13.3) | ||||||||||| |
DCR,d n (%) | 67 (79.8) | ||||||||||| |
95% CIb | 69.6 to 87.7 | ||||||||||| |
Best overall response, n (%) | ||
CRe | 5 (6.0) | ||||||||||| |
PRe | 19 (22.6) | ||||||||||| |
Stable diseasee | 43 (51.2) | ||||||||||| |
Non-CR and non-PDf | 0 | ||||||||||| |
PD | 9 (10.7) | ||||||||||| |
NEg | 8 (9.5) | ||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||.
aConfirmation of response is required to be considered a CR or PR; 2 patients who had initial responses but were not confirmed until after the data cut-off are not included.
bClopper-Person exact CI.
cCR and PR must be confirmed by repeated assessments no less than 4 weeks apart.
dCR + PR + stable disease.
eStable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
fNon-CR and non-PD is for patients with non-measurable disease only.
gNE response includes missing and unknown tumour responses.
Source: Study 1620 Clinical Study Report.5
Duration of Response
The results for DOR are summarized in Table 20. At the time of the primary analysis, the median Kaplan–Meier estimation of DOR in the 24 patients who achieved either a CR or PR had not been reached. The observed DORs ranged from 2.1 months to greater than 21.4 months, with 79.2% of responders achieving a DOR greater than 6 months and 45.8% of responders achieving a DOR greater than 12 months. At the updated analysis, the Kaplan–Meier estimate of median DOR ||||||||||||||||||||. The observed DOR ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| of responders achieving a DOR greater than 6 months and |||||| of responders achieving a DOR greater than 12 months.
Table 20: DOR by BICR in Study 1620
Details | Primary analysis laBCC (N = 24) | Updated analysis laBCC (|||||) |
|---|---|---|
KM estimation of DOR (CR or PR) | ||
Number of events,a n (%) | 6 (25.0) | |||||||| |
Number of censored patients,a n (%) | 18 (75.0) | |||||||| |
Median (95% CI), months | NR (15.0 to NE) | |||||||| |
Observed DOR (CR or PR)b | ||
Range, months | 2.1 to 21.4+ | |||||||| |
≥ 4 months, n (%) | 22 (91.7) | |||||||| |
≥ 6 months, n (%) | 19 (79.2) | |||||||| |
≥ 8 months, n (%) | 16 (66.7) | |||||||| |
≥ 12 months, n (%) | 11 (45.8) | |||||||| |
≥ 16 months, n (%) | 9 (37.5) | |||||||| |
≥ 20 months, n (%) | 2 (8.3) | |||||||| |
≥ 24 months, n (%) | 0 | |||||||| |
≥ 28 months, n (%) | NA | |||||||| |
CI = confidence interval; CR = complete response; DOR = duration of response; KM = Kaplan–Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; PR = partial response.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||.
aEvents include progressive disease and deaths. Percentages are based on number of patients with confirmed CR or PR.
bPercentages are based on number of patients with confirmed a CR or PR. The numerator includes the number of patients whose observed DOR reached at least the specified time. Patients who did not have the opportunity to reach the specified time point were included in the denominator only. Because responses for some patients are ongoing, the percentages at the specified time points may increase as data mature.
Source: Study 1620 Clinical Study Report.5
Health-Related Quality of Life
Results for HRQoL, measured using the global health status (HRQoL) scale of the EORTC QLQ-C30 at baseline and up to cycle 9, are shown in Table 21. Baseline completion of the questionnaire was 92.9% in patients with laBCC, with post-baseline completion rates greater than 80% through to cycle 7. Baseline global health status (on a scale of 0 to 100, with higher values signifying better HRQoL) was 64.30 (SD = 19.14) in the laBCC group. Changes in global health status (HRQoL) over time were smaller than the MID estimate of 5 to 10 points at |||||| the primary analysis ||||||||||||||||||||||. Analysis of the EORTC QLQ-C30 functional and symptom scales was consistent with the results for the global health status scale (Appendix 3). Symptom scales remained stable over time with the exception of fatigue, which showed worsening in excess of the MID for the fatigue scale at cycles 7 and 9, though patient numbers were reduced at these time points (Appendix 3).
Table 21: Global Health Status Scale of the EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) | ||||||
|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | |||||
n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 74 | 64.30 (19.14) | NA | NA | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 2 | 72 | 66.55 (20.15) | 72 | 2.55 (15.30) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 3 | 62 | 61.96 (21.95) | 62 | −2.55 (19.82) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 4 | 51 | 64.71 (20.38) | 51 | −0.49 (18.14) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 5 | 48 | 63.19 (22.53) | 48 | −1.91 (21.21) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 6 | 38 | 68.64 (20.45) | 38 | 4.17 (19.45) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 7 | 32 | 65.10 (19.68) | 32 | −3.13 (19.72) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 8 | 25 | 67.00 (20.90) | 25 | 3.00 (22.16) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 9 | 17 | 66.18 (19.20) | 17 | −6.37 (23.48) | |||||| | |||||||||||| | |||||| | |||||||||||| |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on |||||||||||||||||||||||||||||.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For global health status, a higher score signifies better HRQoL.
Source: Study 1620 Clinical Study Report.5
The results for HRQoL, as measured by the Skindex-16 emotional, symptom, and functioning scales at baseline and up to cycle 9 (on a linear scale of 0 to 100, with 0 representing never bothered and 100 representing always bothered), are summarized in Table 22, Table 23, and Table 24, respectively. Patients with laBCC had baseline completion rates for the questionnaire of 94.0%, 96.4%, and 95.2% for the emotions, symptoms, and functioning domains, respectively. Post-baseline completion rates were greater than 80% through cycle 6. The baseline mean emotional scale score was 39.15 (SD = 39.15). By cycle 4, and through to the end of treatment, there was a change of 10 points or greater, exceeding the MID threshold for improvement in this scale. The Skindex-16 symptom and functioning scales remained stable across time. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Table 22: Skindex-16 Emotion Scale in Study 1620
Cycle | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) | ||||||
|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | |||||
n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 75 | 39.15 (30.53) | NA | NA | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 2 | 69 | 30.49 (28.08) | 69 | −8.93 (27.77) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 3 | 63 | 29.54 (28.39) | 63 | −8.60 (25.64) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 4 | 51 | 23.65 (26.20) | 51 | −11.45 (24.22) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 5 | 46 | 23.96 (26.42) | 46 | −10.25 (24.65) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 6 | 35 | 17.55 (21.10) | 35 | −19.73 (27.30) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 7 | 30 | 24.60 (26.55) | 30 | −13.65 (27.13) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 8 | 24 | 23.81 (26.55) | 24 | −13.10 (26.98) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 9 | 17 | 25.72 (25.13) | 17 | −14.89 (36.84) | |||||| | |||||||||||| | |||||| | |||||||||||| |
laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||||||||||||.
aItem scores are transformed to a linear scale (0 to 100), with 0 representing never bothered and 100 representing always bothered.
Source: Study 1620 Clinical Study Report.5
Table 23: Skindex-16 Symptoms Scale in Study 1620
Cycle | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) | ||||||
|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | |||||
n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 76 | 20.72 (23.04) | NA | NA | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 2 | 71 | 18.58 (19.48) | 71 | −1.31 (21.61) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 3 | 64 | 19.86 (24.02) | 64 | −0.26 (24.16) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 4 | 52 | 12.05 (15.90) | 52 | −6.62 (23.92) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 5 | 47 | 12.83 (15.39) | 47 | −4.11 (18.06) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 6 | 36 | 15.74 (18.66) | 36 | −1.85 (21.42) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 7 | 30 | 18.61 (18.85) | 30 | 0.69 (24.52) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 8 | 25 | 18.33 (15.82) | 25 | −5.33 (26.15) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 9 | 17 | 15.93 (14.82) | 17 | −5.64 (26.76) | |||||| | |||||||||||| | |||||| | |||||||||||| |
laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||.
aItem scores are transformed to a linear scale of 0 to 100, with 0 representing never bothered and 100 representing always bothered.
Source: Study 1620 Clinical Study Report.5
Table 24: Skindex-16 Functioning Scale in Study 1620
Cycle | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) | ||||||
|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | |||||
n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 75 | 25.64 (26.92) | NA | NA | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 2 | 71 | 21.50 (28.18) | 71 | −4.98 (23.65) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 3 | 63 | 19.15 (24.58) | 63 | −4.76 (20.20) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 4 | 51 | 16.47 (23.78) | 51 | −5.82 (23.28) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 5 | 47 | 15.04 (20.17) | 47 | −3.76 (16.37) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 6 | 35 | 12.38 (18.60) | 35 | −11.14 (18.11) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 7 | 30 | 14.44 (20.48) | 30 | −6.00 (15.77) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 8 | 24 | 14.72 (21.65) | 24 | −7.22 (19.53) | |||||| | |||||||||||| | |||||| | |||||||||||| |
Cycle 9 | 17 | 13.53 (16.05) | 17 | −4.31 (23.68) | |||||| | |||||||||||| | |||||| | |||||||||||| |
laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||.
aItem scores are transformed to a linear scale of 0 to 100, with 0 representing never bothered and 100 representing always bothered.
Source: Study 1620 Clinical Study Report.5
Progression-Free Survival
The results for PFS by BICR assessment are shown in Table 25 and Figure 2. At the time of the primary analysis, 45.2% of patients in the laBCC group had experienced a PFS event, with 39.3% of patients experiencing disease progression and 6.0% experiencing death. The median PFS was 19.3 months (95% CI, 8.6 to not evaluable) with an estimate of event-free survival of 76.3% (95% CI, 65.1% to 84.4%) at 6 months, 56.5% (95% CI, 44.3% to 67.0%) at 12 months, and 35.3% (95% CI,19.1% to 52.0%) at 24 months. At the updated analysis, |||| of patients had experienced a PFS event, with |||||| of patients experiencing disease progression and ||||| experiencing death. The median (95% CI) PFS was ||||||||||||||||||||||||||||||||||, with an estimate of event-free survival (95% CI) at 6 months of ||||||||||||||||||||||||||||||||||||||||||||||||||||||||| at 12 months, and ||||||||||||||||||||||| at 24 months. A sensitivity analysis of PFS by BICR assessment was conducted to include patients starting anti-cancer therapy without documented progression as a PFS event. Results of this analysis were consistent with the primary analysis |||||||||||||||||||||||||||||||||||||||||||||| in the updated analysis (Appendix 3).
Table 25: PFS by BICR Assessment in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
KM estimation of PFS | ||
Number of events, n (%) | 38 (45.2) | |||||||||||| |
PD, n (%) | 33 (39.3) | |||||||||||| |
Death, n (%) | 5 (6.0) | |||||||||||| |
Number of censored patients, n (%) | 46 (54.8) | |||||||||||| |
Median (95% CI), months | 19.3 (8.6 to NE) | |||||||||||| |
Estimate of event-free probability, % (95% CI) | ||
4 months | 84.4 (74.1 to 90.8) | |||||||||||| |
6 months | 76.3 (65.1 to 84.4) | |||||||||||| |
8 months | 68.1 (56.3 to 77.4) | |||||||||||| |
12 months | 56.5 (44.3 to 67.0) | |||||||||||| |
16 months | 51.0 (38.6 to 62.1) | |||||||||||| |
20 months | 46.4 (32.2 to 59.4) | |||||||||||| |
24 months | 35.3 (19.1 to 52.0) | |||||||||||| |
28 months | NA | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; KM = Kaplan–Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; PD = progressive disease; PFS = progression-free survival.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||.
Source: Study 1620 Clinical Study Report.5
Figure 2: Kaplan–Meier of PFS by BICR Assessment in Study 1620
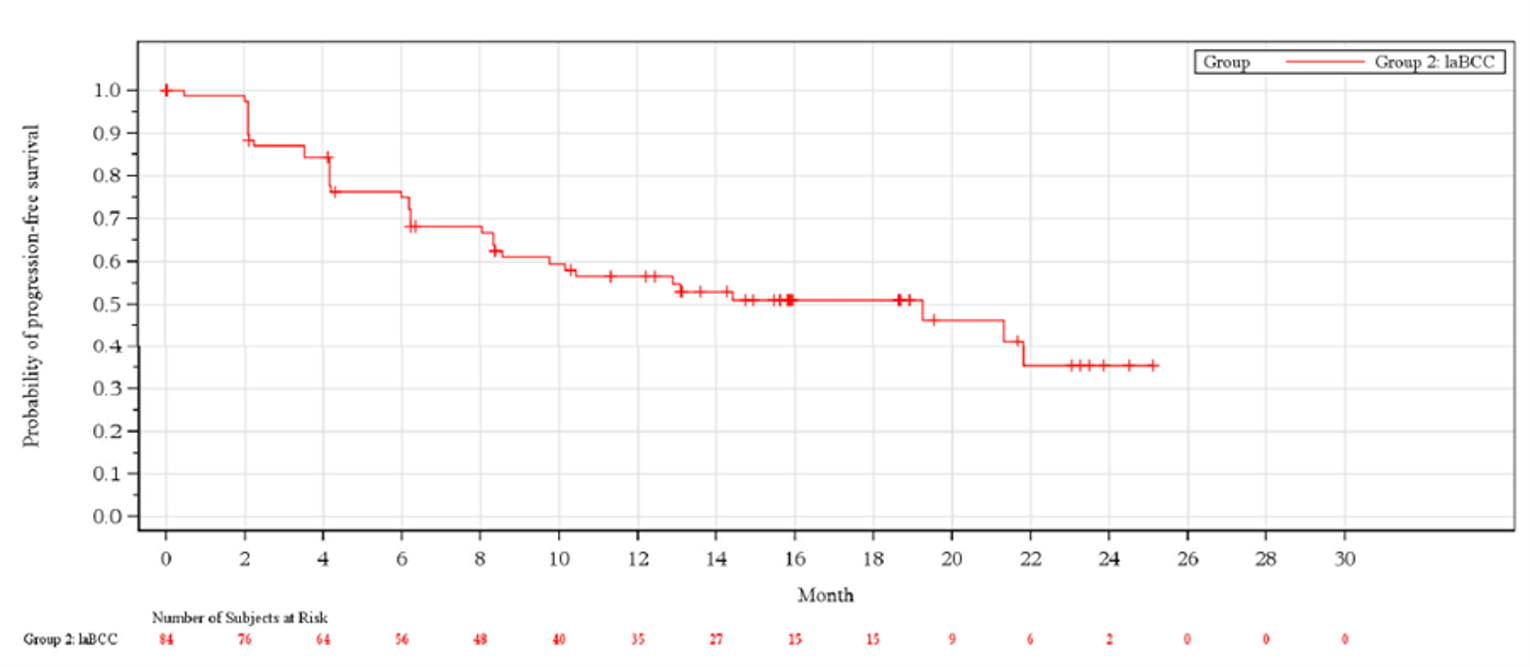
BICR = blinded independent central review; laBCC = locally advanced basal cell carcinoma; PFS = progression-free survival.
Source: Study 1620 Clinical Study Report.5
Overall Survival
The results for OS are summarized in Table 26 and Figure 3. At the time of the primary analysis, deaths had occurred in 11.9% of patients and the median OS had not been reached. The 6-month estimate of OS was 98.8% (95% CI, 91.8% to 99.8%) and, at 12 months, the estimate was 92.3% (95% CI, 83.6% to 96.5%). At 24 months, the estimate was 80.3% (95% CI, 62.6% to 90.3%). At the updated analysis, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The 6-month estimate of OS (95% CI) was |||||||||||||||||||||||||||||||||||||| and, at 12 months, the estimate was ||||||||||||||||||||||||||||||||||||||. At 24 months, the estimate was ||||||||||. ||||||||||||||||||||||. A sensitivity analysis was conducted censoring patients with subsequent therapy from the OS analysis. The results of this analysis were consistent with the primary analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| the analysis (Appendix 3).
Table 26: Summary of OS in Study 1620
Details | Primary analysis | Updated analysis |
|---|---|---|
KM estimation of OS | ||
Number of deaths, n (%) | 10 (11.9%) | ||||||||||| |
Number of censored patients, n (%) | 74 (88.1%) | ||||||||||| |
Median (95% CI), months | NR (NE to NE) | ||||||||||| |
Estimate of survival, % (95% CI) | ||
4 months | 98.8 (91.8 to 99.8) | ||||||||||| |
6 months | 98.8 (91.8 to 99.8) | ||||||||||| |
8 months | 96.3 (88.9 to 98.8) | ||||||||||| |
12 months | 92.3 (83.6 to 96.5) | ||||||||||| |
16 months | 90.8 (81.7 to 95.5) | ||||||||||| |
20 months | 85.7 (73.2 to 92.6) | ||||||||||| |
24 months | 80.3 (62.6 to 90.3) | ||||||||||| |
28 months | NA | ||||||||||| |
CI = confidence interval; KM = Kaplan–Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; OS = overall survival.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||||||||||||||.
Source: Study 1620 Clinical Study Report.5
Figure 3: Kaplan–Meier of OS in Study 1620
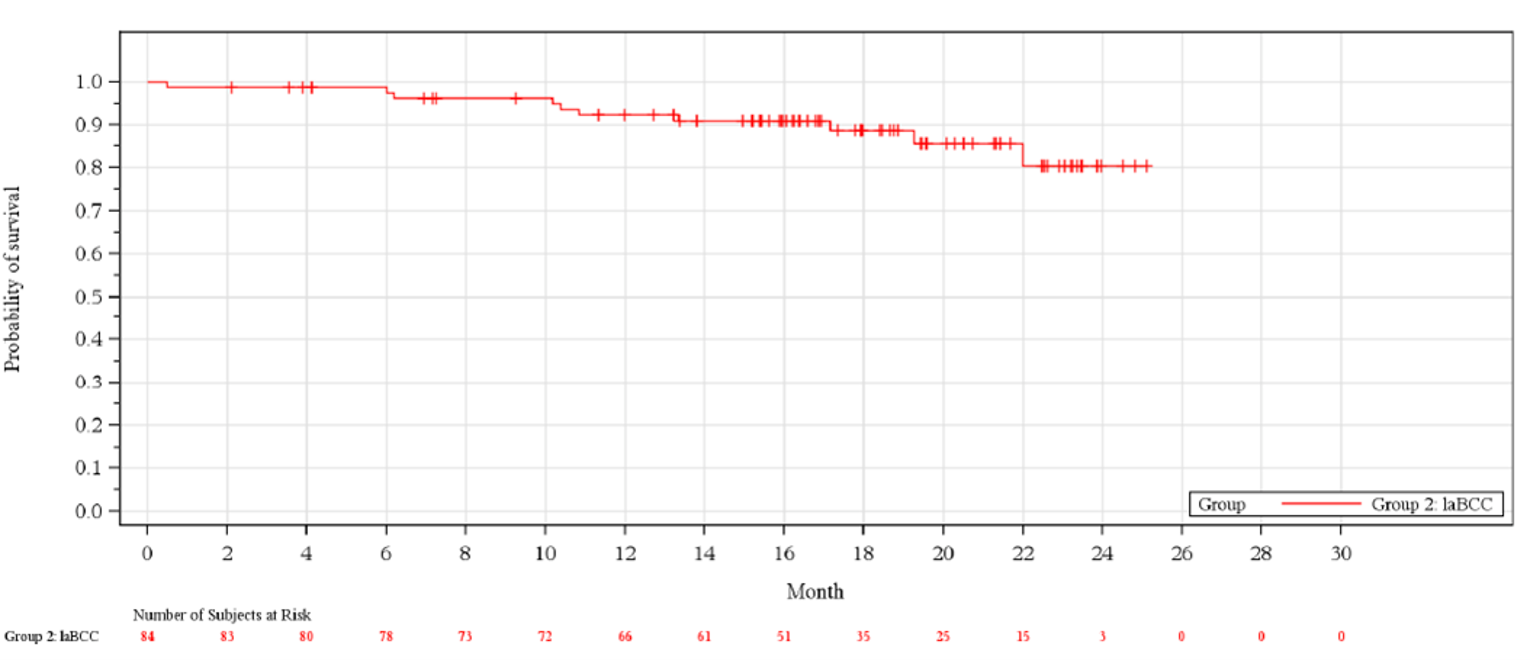
laBCC = locally advanced basal cell carcinoma; OS = overall survival.
Source: Study 1620 Clinical Study Report.5
Time to Response
The time to response according to BICR assessment is summarized in Table 27. At the time of the primary analysis, the mean time to response in patients with laBCC with a CR or PR (N = 24) was 5.17 (SD = 2.60) months and ranged from 2.1 to 13.4 months, with 50% of those responses occurring from 4 to 6 months from the start of treatment. At the updated data cut-off, the mean (SD) time to response in patients with laBCC with CR or PR ||||||||||||||||||||||||||||||||||||||||| and ranged from ||||||||||||||||||||||, with ||||| of responses occurring from 4 to 6 months and ||||| of responses occurring greater than 6 months from the start of treatment.
Table 27: Summary of Time to Response by BICR Assessment in Study 1620
Details | Primary analysis laBCC (N = 24) | Updated analysis laBCC (||||) |
|---|---|---|
Observed time to response (CR or PR), months | ||
Mean (SD) | 5.17 (2.60) | |||||||||||| |
Median (range) | 4.21 (2.1 to 13.4) | |||||||||||| |
Observed time to response (CR or PR), n (%)a | ||
< 2 months | 0 | |||||||||||| |
2 to 4 months | 5 (20.8) | |||||||||||| |
4 to 6 months | 12 (50.0) | |||||||||||| |
≥ 6 months | 7 (29.2) | |||||||||||| |
BICR = blinded independent central review; CR = complete response; laBCC = locally advanced basal cell carcinoma; NA = not applicable; PR = partial response; SD = standard deviation.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||||||||||||||||.
aPercentages are based on the number of patients with confirmed CR or PR.
Source: Study 1620 Clinical Study Report.5
Time to Progression
Results for time to progression were not assessed in Study 1620.
Harms
Only those harms identified in the review protocol are reported subsequently. Refer to Table 28 for detailed harms data.
Treatment-Emergent Adverse Events
At the time of the primary analysis, almost all patients (97.6%) treated with cemiplimab experienced at least 1 TEAE. The TEAEs (≥ 20%) that were common in patients receiving cemiplimab included fatigue (29.8%), diarrhea (23.8%), pruritis (21.4%), and asthenia (20.2%). At the updated analysis, 98.8% of patients reporting at least 1 TEAE, and the most frequently reported TEAEs ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| included fatigue |||||), diarrhea ||||||, pruritis ||||||, and asthenia |||||||.
Serious Adverse Events
At the time of the primary analysis, SAEs had occurred in 34.5% of patients, and the most commonly reported SAE was urinary tract infection (4.8% of patients). At the updated data cut-off, SAEs were reported in 36.9% of patients and |||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
TEAEs Leading to Interruption, Dose Delay, or Dose Reduction
At the primary analysis, TEAEs that led to a dose delay occurred in 36.9% of patients. The most common TEAE that led to a dose delay was diarrhea (4.8% of patients), followed by blood creatinine increased, fatigue, and urinary tract infection, each occurring in 3.6% of patients. Less common were TEAEs that led to study drug interruption (3.6%); these included 1 occurrence each of palpitations, extravasation, and flank pain. One patient experienced a TEAE leading to dose reduction (1.2%), specifically, a cutaneous soft tissue infection of BCC. No additional AEs leading to study drug interruption, dose delay, or dose reduction were reported at the updated analysis.
Withdrawals Due to TEAEs
TEAEs that led to discontinuation of cemiplimab occurred in 16.7% of patients at the primary analysis and 17.9% of patients at the updated analysis. The most common TEAE leading to treatment discontinuation was colitis, occurring in 2.4% of patients.
Mortality
At the time of the primary analysis, TEAEs resulting in death occurred in 3.6% of patients. These included 1 occurrence each of cachexia, malignant brain neoplasm, and acute kidney injury. At the updated analysis, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Notable Harms
Notable harms specified in the CADTH review protocol included immune-related AEs and infusion-related reactions. At the time of the primary analysis, 56.0% of patients had experienced an immune-related AE, 11.9% had experienced an immune-related AE of grade 3 or greater, and 9.5% discontinued treatment with cemiplimab due to an immune-related AE. These | |||||||||||||||||||||||||||||||||||| at the updated data analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||. Infusion-related reactions were less common, reported in ||||||||||||||||||| patients at |||| the primary analysis ||||||||||||||||||||||||||||||| |||||||||||||||||||.
Table 28: Summary of Harms in Study 1620
Harms | Primary analysis laBCC (N = 84) | Updated analysis |
|---|---|---|
Patients with ≥ 1 TEAE | ||
n (%) | 82 (97.6) | |||||||||||| |
Frequent TEAEs, n (%) | ||
Fatigue | 25 (29.8) | |||||||||||| |
Diarrhea | 20 (23.8) | |||||||||||| |
Pruritus | 18 (21.4) | |||||||||||| |
Asthenia | 17 (20.2) | |||||||||||| |
Anemia | 13 (15.5) | |||||||||||| |
Decreased appetite | 13 (15.5) | |||||||||||| |
Headache | 12 (14.3) | |||||||||||| |
Nausea | 12 (14.3) | |||||||||||| |
Urinary tract infection | 12 (14.3) | |||||||||||| |
Arthralgia | 11 (13.1) | |||||||||||| |
Dyspnea | 10 (11.9) | |||||||||||| |
Pyrexia | 5 (6.0) | |||||||||||| |
Constipation | 5 (6.0) | |||||||||||| |
Vomiting | 5 (6.0) | |||||||||||| |
Weight decreased | 7 (8.3) | |||||||||||| |
Weight increased | 2 (2.4) | |||||||||||| |
Dizziness | 8 (9.5) | |||||||||||| |
Hyperglycemia | 2 (2.4) | |||||||||||| |
Hypertension | 7 (8.3) | |||||||||||| |
Cough | 8 (9.5) | |||||||||||| |
Tumour hemorrhage | 8 (9.5) | |||||||||||| |
Patients with ≥ 1 SAE | ||
n (%) | 29 (34.5) | |||||||||||| |
Frequent SAEs, n (%) | ||
Urinary tract infection | 4 (4.8) | |||||||||||| |
Colitis | 2 (2.4) | |||||||||||| |
Myocardial infarction | 1 (1.2) | |||||||||||| |
Infected neoplasm | 2 (2.4) | |||||||||||| |
Anemia | 2 (2.4) | |||||||||||| |
Adrenal insufficiency | 2 (2.4) | |||||||||||| |
Acute kidney injury | 2 (2.4) | |||||||||||| |
TEAEs leading to study drug discontinuation | ||
n (%) | 14 (16.7) | |||||||||||| |
TEAEs leading to dose delay | ||
n (%) | 31 (36.9) | |||||||||||| |
TEAEs leading to study drug interruption | ||
n (%) | 3 (3.6) | |||||||||||| |
TEAEs leading to dose reduction | ||
n (%) | 1 (1.2) | |||||||||||| |
TEAEs leading to death | ||
n (%) | 3 (3.6) | |||||||||||| |
Notable harms | ||
AESIs, n (%) | NA | |||||||||||| |
Immune-related AEa | 47 (56.0) | |||||||||||| |
Grade 3, 4, or 5 immune-related AE | 10 (11.9) | |||||||||||| |
Serious immune-related AE | 8 (9.5) | |||||||||||| |
Immune-related AE leading to discontinuation | 8 (9.5) | |||||||||||| |
Immune-related AE leading to dose delay | 10 (11.9) | |||||||||||| |
Immune-related AE leading to drug interruption | 0 | |||||||||||| |
Immune-related AE leading to dose reduction | 0 | |||||||||||| |
Immune-related AE resulting in death | 0 | |||||||||||| |
Infusion-related reactions | 1 (1.2) | |||||||||||| |
Grade 3, 4, or 5 infusion-related reaction | 0 | |||||||||||| |
Serious infusion reaction | 0 | |||||||||||| |
Infusion reaction leading to discontinuation | 0 | |||||||||||| |
Infusion reaction leading to dose delay | 0 | |||||||||||| |
Infusion reaction leading to drug interruption | 1 (1.2) | |||||||||||| |
Infusion reaction leading to dose reduction | 0 | |||||||||||| |
Infusion reaction resulting in death | 0 | |||||||||||| |
AE = adverse event; AESI = adverse event of special interest; laBCC = locally advanced basal cell carcinoma; MedDRA = Medical Dictionary for Regulatory Activities; NA = not applicable; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: The primary analysis was based on a February 17, 2020 data cut-off; the updated analysis is based on ||||||||||||||||||||||||.
aAs there is currently no MedDRA-coded classification for immune-related AEs, the sponsor created a customized list of MedDRA-preferred terms for the identification of immune-related AEs.
Source: Study 1620 Clinical Study Report.5
Critical Appraisal
Internal Validity
Study 1620 was a phase II, single-arm, non-randomized, open-label, multi-centre study that evaluated cemiplimab in patients with laBCC and mBCC who had been previously treated with HHI therapy. Although non-comparative trial evidence is of lower quality, with an increased risk of bias compared with the preferred randomized controlled trial (RCT) design, it was acknowledged by the CADTH review team and the clinical experts consulted for this review that, due to the lack of a relevant active comparator and given the relative size of the patient population, conducting an RCT would be difficult. Nevertheless, it is impossible to draw conclusions of efficacy from a single-arm trial, as the causal relationship between outcomes and intervention cannot be ascertained without the inclusion of a comparator arm.
The outcomes assessed in Study 1620 (ORR, PFS, OS, HRQoL) are standard in oncology trials, and tumour responses were evaluated by BICR. As patients with laBCC had the potential for both external and internally measurable lesions, a composite scoring method was used that combined RECIST 1.1 and visual response criteria based on digital medical photography. The method of assessment was considered by the clinical experts consulted by CADTH to be valid for measuring response and consistent with previous trials conducted within the laBCC population.7,8 For the analysis of PFS and DOR, patients receiving a new anti-cancer therapy before an event were censored, and this outcome was not treated as an event. As per FDA guidance, this is considered a biased censoring rule, and starting another treatment before a documented event should be considered as an event in the analysis.31 Using this censoring rule biases the results in favour of cemiplimab; however, its impact was small in Study 1620, since the results of the sensitivity analysis that considered starting a new anti-cancer therapy included only 1 additional patient with a progression event.
The statistical analysis was appropriate, given the single-arm study design. The statistical analysis plan provided by the sponsor specified 20% as the threshold for a clinically meaning primary end point of ORR. The clinical experts consulted by CADTH agreed that response rates that exceed this level would be clinically meaningful, and it was also noted that this threshold is consistent with the ORR thresholds used in previous single-arm trials in BCC patients.7,8 Rejection of the null hypothesis (ORR = 20%) required the lower bound of the 95% CI to exclude 20%; however, this was not achieved at the time of primary analysis (ORR = 28.6; 95% CI, 19.2% to 39.5%).
According to the study protocol, for a patient to have achieved a CR or PR, a response must have been confirmed at least 4 weeks following the initial documented response. If the response was not confirmed, the patient was reported as having stable disease. The sponsor presented an unplanned sensitivity analysis in which the pre-specified threshold to reject the null hypothesis was reached; this includes the responses from 2 patients who had unconfirmed initial responses at the time of the primary analysis. Ultimately, both patients did have their responses confirmed; however, these results are based on an ad hoc redefinition of the primary outcome that differs from the study protocol. Since there was no adjustment for multiplicity in this analysis, there is an increased risk of type I error and, therefore, the results obtained should be interpreted with caution. The sponsor also provided the results of an ||||||||||||||||||||||| updated analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||; the reported ORR (95% CI) at this data cut-off was ||||||||||||||||||||||||||||||||||||||. The same limitations regarding no adjustment for multiplicity and increased risk of type I error apply to the updated analysis and results.
Important protocol deviations occurred in a relatively high number of patients in the laBCC group (23.8%), though the observed protocol deviations were considered acceptable for a second-line oncology clinical trial. The most common important protocol deviations were related to enrolling patients despite deviations in inclusion (15.5%) and exclusion (3.6%) criteria. Notably, 2 patients did not meet the inclusion criterion that required enrolled patients to have at least 1 measurable lesion, which would likely bias the results in favour of cemiplimab. Neither patient was recorded as achieving either a PR or CR; 1 achieved a best overall response of stable disease and the status of the second was recorded as not evaluable at all visits. Therefore, it is unlikely that including these patients biased the primary end point in favour of cemiplimab, though it is likely that secondary end points such as HRQoL, PFS, and OS could have been biased in favour of cemiplimab. The impact of the high number of other important protocol deviations on the characteristics of the study population and the direction of bias was unclear.
The blinding of patients was not possible in the context of a non-comparative trial and, therefore, the open-label nature of the design may have contributed to the introduction of several potential biases, although their overall impact is unclear. There was a relatively high number of patients who discontinued the study for reasons other than PD or death (19.0% at the primary analysis data cut-off); the other reasons for discontinuation included AEs, lost to follow-up, non-compliance with the protocol, withdrawal of consent, patient decision, and sponsor decision. Specifically in the case of non-compliance with the protocol and sponsor decision, the CADTH review team indicated these are not valid reasons to discontinue the study and are likely to bias the results in favour of cemiplimab. The high overall number of study dropouts may have introduced potential bias into the results; however, their overall impact on patient characteristics and study outcomes as well as the direction of bias is unclear. The decision to discontinue patients from therapy was made by investigators based on unblinded review of local imaging results and/or clinical assessments. These decisions may have altered treatment exposure to cemiplimab and thus influenced results. Investigators were less likely to classify PD than BICR (6.0% versus 10.7% at the primary analysis), which suggests patients were exposed to cemiplimab for longer than would have been the case if treatment decisions were based on blinded review.
The secondary outcomes of DOR, PFS, OS, and HRQoL did not have any formal hypothesis testing conducted. HRQoL outcomes had good completion rates at baseline (89%) and up to cycle 6 (> 80%); however, as none of these end points were part of a formal testing hierarchy, these results should be viewed as exploratory. The subgroup analyses of interest to this review (outcome of prior HHI therapy and histologic subtype) were specified a priori; however, as these analyses were not part of a formal testing hierarchy, these results must also be considered exploratory.
External Validity
According to the clinical experts consulted by CADTH, the demographic and disease characteristics of the Study 1620 population were reflective of the Canadian population with laBCC. The patients enrolled were required to have previously been treated with HHI therapy, 16.7% of whom were previously treated with sonidegib, an HHI that is Health Canada–approved but not publicly reimbursed in Canada. The clinical experts consulted by CADTH did not expect this to impact the generalizability of the study results to Canadian patients, who are unlikely to have received sonidegib. Similarly, 41.7% of patients received 2 or more lines of HHI therapy before receiving cemiplimab. As vismodegib is the only reimbursed HHI therapy in Canada, it is unlikely that the Canadian population would have received multiple lines of HHI therapy, suggesting a potential for better outcomes in Canadian patients.
The application of the inclusion and exclusion criteria to this patient population resulted in a relatively high proportion of screen failures (33 out of 165; 20%), though it was unclear what proportion of screen failures were in the laBCC group compared with the mBCC group. According to the clinical experts, the enrolment criteria, as in most oncology trials, likely selected for a healthier cross-section of the overall patient population who were better able to tolerate protocol therapy. The potential administration of cemiplimab outside of the Health Canada indication was identified as possible by the clinical experts consulted for this review. Study 1620 also included a small group of patients with mBCC who had previously received HHI therapy. The mBCC population was removed from the indication at the request of Health Canada, which cited the immaturity of the data at the time of the interim analysis for the mBCC group, with the ORR not reaching the pre-specified threshold for a clinically meaningful response, and uncertainty in the DOR due to limited follow-up and low patient numbers. Consequently, the generalizability of the trial data to the mBCC population is unclear. Study 1620 limited enrolment to patients with an ECOG performance status of 0 or 1 and, thus, the generalizability of the laBCC trial results to patients with a poorer ECOG performance status (score greater than 2) is unclear. According to the clinical experts, in clinical practice, the performance status of patients with mBCC or those with an ECOG performance status greater than 2 is expected to improve after initiating treatment with cemiplimab.
The dosage of cemiplimab in Study 1620 was aligned with the Health Canada–approved dosing and with clinical practice. In the study, treatment with cemiplimab was administered until PD or unacceptable toxicity, up to 93 weeks. The protocol allowed for the re-treatment of patients who had completed the full treatment course but who experienced PD during the follow-up period. The sponsor confirmed that 1 patient in the study had entered re-treatment with cemiplimab.32 The trial data may not be generalizable to treatment beyond the 93-week treatment course or within a re-treatment setting for patients who experience PD following discontinuation of cemiplimab, given the lack of data. All outcomes evaluated in the trial and considered in this review (ORR, DOR, HRQoL, PFS, OS) were clinically relevant, important to patients, and are used in clinical practice. The duration of follow-up was sufficient for assessment of the primary outcome of ORR, DOR, and HRQoL; however, longer-term outcomes of PFS and OS are difficult to interpret, given the single-group trial design. Subgroup analyses were not powered to detect treatment effect in patients who progressed or who achieved only stable disease after 9 months on HHI therapy, patients intolerant to HHI therapy, and patients based on their histologic subtype. Nevertheless, the clinical experts consulted for this review felt that the results of the trial were generalizable across strata for all of these subgroups.
Since the administration of cemiplimab would occur in a hospital or specialty clinic setting, background care (oncologist visits, imaging frequency, bloodwork, and so forth) would be expected to be similar for Canadian patients compared with those participating in Study 1620.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
No indirect evidence was submitted by the sponsor. A focused literature search for network meta-analyses dealing with BCC was run in MEDLINE All (1946–) on September 13, 2021. No limits were applied to the search. No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Discussion
Summary of Available Evidence
One phase II, single-arm, non-randomized, open-label multi-centre trial, Study 1620 (patients with laBCC; N = 84, primarily US and Europe)5,6 comprised the evidence for this CADTH review. The study enrolled patients with laBCC and mBCC who had previously received HHI therapy; however, the laBCC population was the focus of this review, as the Health Canada indication and reimbursement request were focused on this subgroup of patients. Patients were treated with cemiplimab until PD or unacceptable toxicity for up to 93 weeks. Tumour response was assessed using a composite of RECIST 1.1 for lesions with radiologically measurable components and modified WHO clinical criteria for lesions with externally visible components, and responses were designated by BICR. The primary outcome was ORR, while secondary outcomes included DOR, PFS, OS, and HRQoL.
According to the clinical experts consulted by CADTH, the baseline characteristics of Study 1620 were representative of patients with laBCC in Canada who would be candidates for cemiplimab. Most patients in the laBCC subgroup were male (66.7%) and White (67.9%). Infiltrative tumour histology accounted for 8.3% of laBCC lesions, while the broad “other” category accounted for 66.7% of lesions, with most (89.3%) occurring in the head or neck region. The mean age of the patients with laBCC was 69.1 (SD = 12.8) years. The limitations of the study included potential biases inherent to its single-arm, non-comparative design prohibiting the ability to draw causal conclusions between the intervention and outcomes.
Interpretation of Results
Outcomes
Administration of cemiplimab in Study 1620 resulted in an ORR of 28.6% (95% CI, 19.2% to 39.5%) at the primary analysis, and this increased to 32.1% (95% CI, 22.4% to 43.2%) at the updated analysis. At the primary analysis, the primary end point of ORR did not meet the pre-specified threshold (lower bound of the 95% CI, excluding the value of 20%) to reject the null hypothesis. The sponsor noted that with the inclusion of the 2 patients with unconfirmed initial responses (a deviation from the study protocol), the requirement for the rejection of the null hypothesis was met. The DOR at the primary analysis ranged from 2.1 months to 21.4 months, with 79.2% of responders achieving a DOR of greater than 6 months and 45.8% of responders achieving a DOR of greater than 12 months. The clinical experts consulted by CADTH considered the results to be clinically meaningful in this population, which had been previously treated with HHI therapy and had no alternative treatment options. Responses from the clinician and patient groups also specifically highlighted the lack of treatment options in this patient population. However, there are significant issues with analyzing the primary outcome in a manner that deviates from the study protocol to include additional responders.
As laBCC is a very disfiguring malignancy, there is a very large impact on the HRQoL of the patient, including reduced self-esteem due to their physical appearance, constant anxiety about finding new lesions, and pain. As such, HRQoL was identified in the patient group input as being very important to patients and was measured in Study 1620 using the EORTC QLQ-C30 and Skindex-16. The global health status or HRQoL score remained stable throughout the study, with no increase or decrease from baseline that met the MID estimate of 5 to 10 points. Functional and symptom scales were similarly stable throughout the study. An improvement in excess of the MID was achieved in the emotion scale of the Skindex-16 at cycle 4 and maintained through to the end of the study, while the symptom and functioning scales remained stable. The clinical experts consulted were hopeful, given the signal of improvement in some scales and the lack of deterioration in others, and commended the sponsor for using dermatology-specific HRQoL measures along with an oncology-specific instrument. However, the analyses of HRQoL outcomes were descriptive and potentially impacted by the low number of patients participating in later cycles of the study; there is also the potential for bias in favour of cemiplimab due to the open-label design of the study. At the primary analysis, the median PFS was 19.3 (95% CI, 8.6 to not evaluable) months, and the median OS was not reached. Given the low number of PFS and OS events in the study and the non-comparative study design, the effect of cemiplimab on long-term outcomes is unknown. The subgroup analyses were consistent with the primary analysis; however, as they were not part of a formal testing hierarchy, these results must be considered exploratory.
Harms
The safety profile of cemiplimab in Study 1620, which did not identify any new safety signals, was as expected, based on prior experience with the drug. Immune-related AEs occurred in more than half of patients and should be monitored, as is common practice with immunotherapies. The clinical experts consulted for this review felt the safety profile of cemiplimab in patients with laBCC is acceptable and can be managed with appropriate supportive care, and this aligned with the perspectives of patients gathered from patient groups indicating the side effects of cemiplimab were manageable and worth the potential benefit.
Other Considerations
No other considerations were identified for this review.
Conclusions
Study 1620 was a single-arm study of cemiplimab (350 mg every 3 weeks up to a maximum of 93 weeks) in patients with laBCC. The study did not meet the pre-specified threshold of a 20% ORR, which is considered clinically meaningful. At the updated analysis, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In the opinion of the clinical experts consulted by CADTH, despite the limitations of Study 1620, the observed ORR in patients with laBCC previously treated with an HHI was considered clinically meaningful and of value in a high-burden disease for which there is a high unmet need for treatment options. The descriptive assessment of HRQoL in the study was limited by the low number of respondents contributing to assessments at later time points, but suggested multiple measures of HRQoL were stable over the course of the study and a clinically meaningful improvement was observed for emotional functioning. Data on PFS and OS were immature, but longer-term data for these outcomes will be challenging to interpret due to the non-comparative study design. The notable harms that were observed with cemiplimab, specifically immune-related AEs, were consistent with the known safety profile of the drug and were considered by the clinical experts consulted by CADTH as well as by patients to be manageable with appropriate supportive care. The major limitation of Study 1620 is its open-label, single-group trial design, which introduces bias in favour of cemiplimab and precludes the ability to evaluate the efficacy and magnitude of the clinical benefit of cemiplimab in this treatment setting.
References
1.Non melanoma skin cancer. Ottawa (ON): Public Health Agency of Canada; 2019: https://www.canada.ca/en/public-health/services/chronic-diseases/cancer/non-melanoma-skin-cancer.html. Accessed 2021 Nov 15.
2.Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146(3):283-287. PubMed
3.Erba P, Farhadi J, Wettstein R, Arnold A, Harr T, Pierer G. Morphoeic basal cell carcinoma of the face. Scand J Plast Reconstr Surg Hand Surg. 2007;41(4):184-188. PubMed
4.Mohan SV, Chang AL. Advanced basal cell carcinoma: epidemiology and therapeutic innovations. Curr Dermatol Rep. 2014;3(1):40-45. PubMed
5.Clinical Study Report: R2810-ONC-1620. A phase 2 study of REGN2810, a fully human monoclonal antibody to programmed death-1, in patients with advanced basal cell carcinoma who experienced progression of disease on hedgehog pathway inhibitor therapy, or were intolerant of prior hedgehog pathway inhibitor therapy [internal sponsor's report]. Tarrytown (NY): Regeneron Pharmaceuticals, Inc.; 2020 Aug 21.
6.Stratigos AJ, Sekulic A, Peris K, et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: an open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021;22(6):848-857. PubMed
7.Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16(6):716-728. PubMed
8.Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171-2179. PubMed
9.Christenson LJ, Borrowman TA, Vachon CM, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294(6):681-690. PubMed
10.Niazi ZB, Lamberty BG. Perineural infiltration in basal cell carcinomas. Br J Plast Surg. 1993;46(2):156-157. PubMed
11.Leibovitch I, McNab A, Sullivan T, Davis G, Selva D. Orbital invasion by periocular basal cell carcinoma. Ophthalmology. 2005;112(4):717-723. PubMed
12.Goldenberg G, Karagiannis T, Palmer JB, et al. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: A retrospective cohort study. J Am Acad Dermatol. 2016;75(5):957-966.e952. PubMed
13.Stevens VW, Stenehjem DD, Patterson OV, et al. Characterization and survival of patients with metastatic basal cell carcinoma in the Department of Veterans Affairs: a retrospective electronic health record review. Arch Dermatol Res. 2018;310(6):505-513. PubMed
14.Ting PT, Kasper R, Arlette JP. Metastatic basal cell carcinoma: report of two cases and literature review. J Cutan Med Surg. 2005;9(1):10-15. PubMed
15.Spiller WF, Spiller RF. Treatment of basal cell epithelioma by curettage and electrodesiccation. J Am Acad Dermatol. 1984;11(5 Pt 1):808-814. PubMed
16.Bart RS, Schrager D, Kopf AW, Bromberg J, Dubin N. Scalpel excision of basal cell carcinomas. Arch Dermatol. 1978;114(5):739-742. PubMed
17.Thissen MR, Neumann MH, Schouten LJ. A systematic review of treatment modalities for primary basal cell carcinomas. Arch Dermatol. 1999;135(10):1177-1183. PubMed
18.Zloty D, Guenther LC, Sapijaszko M, et al. Non-melanoma Skin Cancer in Canada Chapter 4: Management of Basal Cell Carcinoma. J Cutan Med Surg. 2015;19(3):239-248. PubMed
19.NCCN Clinical Practice Guidelines. Basal cell skin cancer version 2.2021. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2021: https://www.nccn.org/. Accessed 2020 Nov 15.
20.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841-851. PubMed
21.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66(4):2081-2088. PubMed
22.Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537-1544. PubMed
23.Libtayo (cemiplimab for injection): 50mg/mL solution for infusion; Antineoplastic Agent, Monoclonal Antibody [product monograph]. Laval (QC): Sanofi-Aventis Canada Inc.; 2021 Nov 9.
24.CADTH pCODR Expert Review Committee (pERC) final recommendation: cemiplimab (Libtayo) for cutaneous squamous cell carcinoma drug name. Ottawa (ON): CADTH; 2020 Jan 22: https://cadth.ca/sites/default/files/pcodr/Reviews2020/10187CemiplimabCSCC_fnRec_REDACT_EarlyConv_22Jan2020_final.pdf. Accessed 2021 Nov 15.
25.Regeneron Pharmaceuticals Inc. Libtayo Prescribing information: Libtayo (cemiplimab-rwlc) injection, for intravenous use. Silver Sprin (MD): U.S. Food and Drug Administration (FDA); 2021: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761097s008lbl.pdf. Accessed 2021 Nov 03.
26.European Public Assessment Report (EPAR): Libtayo (cemiplimab). Amsterdam (NL): European Medicines Agency; 2021: https://www.ema.europa.eu/en/medicines/human/EPAR/libtayo. Accessed 2021 Nov 03.
27.Reinhard Dummer. NCT04679480: Anti-PD1-antibody and Pulsed HHI for Advanced BCC. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT04679480. Accessed 2021 Nov 15.
28.Drug Reimbursement Review sponsor submission: Libtayo (cemipliumab), 350 mg every three weeks as an intravenous (IV) infusion [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Aug 19.
29.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
30.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Sep 15.
31.Clinical trial endpoints for the approval of cancer drugs and biologics: guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018: https://www.fda.gov/media/71195/download. Accessed 2021 Nov 03.
32.Sanofi Genzyme response to October 20, 2021 DRR request for additional information regarding Libtayo (cemipliumab) DRR review [internal additional sponsor's information]. Mississauga (ON): Sanofi Genzyme; 2021 Nov 10.
33.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
34.Fayers P, Bottomley A, Group EQoL, Quality of Life U. Quality of life research within the EORTC-the EORTC QLQ-C30. European Organisation for Research and Treatment of Cancer. Eur J Cancer. 2002;38 Suppl 4:S125-133. PubMed
35.Osoba D, Aaronson N, Zee B, Sprangers M, te Velde A. Modification of the EORTC QLQ-C30 (version 2.0) based on content validity and reliability testing in large samples of patients with cancer. The Study Group on Quality of Life of the EORTC and the Symptom Control and Quality of Life Committees of the NCI of Canada Clinical Trials Group. Qual Life Res. 1997;6(2):103-108. PubMed
36.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-1807. PubMed
37.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
38.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-216. PubMed
39.Steinbauer J, Koller M, Kohl E, Karrer S, Landthaler M, Szeimies RM. Quality of life in health care of non-melanoma skin cancer - results of a pilot study. J Dtsch Dermatol Ges. 2011;9(2):129-135. PubMed
40.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
41.Muller K, Karrer S, Szeimies RM, et al. Quality of life assessment in patients with nonmelanoma skin cancer - psychometric validation of the EORTC QLQ-C30 questionnaire. J Dtsch Dermatol Ges. 2017;15(11):1090-1100. PubMed
42.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
43.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
44.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
45.Chren MM. The Skindex instruments to measure the effects of skin disease on quality of life. Dermatol Clin. 2012;30(2):231-236, xiii. PubMed
46.Chren MM, Lasek RJ, Sahay AP, Sands LP. Measurement properties of Skindex-16: a brief quality-of-life measure for patients with skin diseases. J Cutan Med Surg. 2001;5(2):105-110. PubMed
47.Hansson J, Bartley K, Karagiannis T, et al. Assessment of quality of life using Skindex-16 in patients with advanced basal cell carcinoma treated with vismodegib in the STEVIE study. Eur J Dermatol. 2018;28(6):775-783. PubMed
48.Nunnally JC, Bernstein IH. The assessment of reliability. Psychometric theory, 3rd ed. New York (NY): McGraw-Hill; 1994:248-249.
49.Lee EH, Klassen AF, Nehal KS, Cano SJ, Waters J, Pusic AL. A systematic review of patient-reported outcome instruments of nonmelanoma skin cancer in the dermatologic population. J Am Acad Dermatol. 2013;69(2):e59-67. PubMed
50.Chren MM, Sahay AP, Bertenthal DS, Sen S, Landefeld CS. Quality-of-life outcomes of treatments for cutaneous basal cell carcinoma and squamous cell carcinoma. J Invest Dermatol. 2007;127(6):1351-1357. PubMed
51.Guyatt GH, Feeny DH, Patrick DL. Measuring health-related quality of life. Ann Intern Med. 1993;118(8):622-629. PubMed
52.Guyatt GH, Osoba D, Wu AW, Wyrwich KW, Norman GR. Methods to explain the clinical significance of health status measures. Mayo Clin Proc. 2002;77(4):371-383. PubMed
53.Juniper EF, Guyatt GH, Willan A, Griffith LE. Determining a minimal important change in a disease-specific Quality of Life Questionnaire. J Clin Epidemiol. 1994;47(1):81-87. PubMed
54.Sprangers MA, Moinpour CM, Moynihan TJ, Patrick DL, Revicki DA. Assessing meaningful change in quality of life over time: a users' guide for clinicians. Mayo Clin Proc. 2002;77(6):561-571. PubMed
55.Rubin DB. Estimating causal effects from large data sets using propensity scores. Ann Intern Med. 1997;127(8 Pt 2):757-763. PubMed
56.D'Agostino RB, Jr. Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat Med. 1998;17(19):2265-2281. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946–present)
Embase (1974–present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: September 13, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Cemiplimab* or Libtayo* or regn2810 or regn 2810 or sar 439684 or sar439684 or 6QVL057INT).ti,ab,ot,kf,,hw,nm,rn.
1 use medall
*cemiplimab/
(Cemiplimab* or Libtayo* or regn2810 or regn 2810 or sar 439684 or sar439684).ti,ab,kw,dq.
3 or 4
5 use oemezd
(conference abstract or conference review).pt.
6 not 7
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search — Studies with results: Libtayo (cemiplimab) AND basal cell carcinoma (BCC)
WHO International Clinical Trials Registry Platform
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) AND basal cell carcinoma (BCC)
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) AND basal cell carcinoma (BCC)
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) AND basal cell carcinoma (BCC)
Grey Literature
Search dates: September 7, 2021 September 10, 2021
Keywords: Search terms: (Libtayo OR cemiplimab OR REGN-2810 ) AND basal cell carcinoma
Limits: No limits
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
No publications beyond the included pivotal trial were ordered for full-text review, therefore there are no excluded studies to report in this appendix.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Detailed Outcome Data in the laBCC Population
Main Outcome Analysis by Investigator Assessment
Table 30: ORR by Investigator Assessment in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
ORR, n (%) | 27 (32.1) | |||||||||||| |
95% CI | 22.4 to 43.2 | |||||||||||| |
CRRa, n (%) | 5 (6.0) | |||||||||||| |
95% CIb | 2.0 to 13.3 | |||||||||||| |
DCRc, n (%) | 73 (86.9) | |||||||||||| |
95% CIb | 77.8 to 93.3 | |||||||||||| |
Best overall response, n (%) | ||
CRa | 5 (6.0) | |||||||||||| |
PRa | 22 (26.2) | |||||||||||| |
Stable diseased | 46 (54.8) | |||||||||||| |
PD | 5 (6.0) | |||||||||||| |
NEe | 6 (7.1) | |||||||||||| |
CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; laBCC = locally advanced basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR+PR+Stable disease.
d Stable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
e Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
Table 31: DOR by Investigator Assessment in Study 1620
Details | Primary analysis laBCC (N = 27) | Updated analysis laBCC (|||||||||) |
|---|---|---|
KM estimation of DOR (CR or PR) | ||
Number of events,a n (%) | 6 (22.2) | |||||||||||| |
Number of censored patients,a n (%) | 21 (77.8) | |||||||||||| |
Median (95% CI), months | NR (19.6 to NE) | |||||||||||| |
Observed DOR (CR or PR)b | ||
Range, months | 2.8 to 21.4 | |||||||||||| |
≥ 4 months, n (%) | 26 (96.3) | |||||||||||| |
≥ 6 months, n (%) | 24 (88.9) | |||||||||||| |
≥ 8 months, n (%) | 22 (81.5) | |||||||||||| |
≥ 12 months, n (%) | 14 (51.9) | |||||||||||| |
≥ 16 months, n (%) | 8 (29.6) | |||||||||||| |
≥ 20 months, n (%) | 3 (11.1) | |||||||||||| |
≥ 24 months, n (%) | 0 | |||||||||||| |
≥ 28 months, n (%) | NA | |||||||||||| |
CI = confidence interval; CR = complete response; DOR = duration of response; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; PR = partial response.
a Events include progressive disease or deaths. Percentages are based on number of patients with confirmed CR or PR.
b Percentages are based on number of patients with confirmed CR or PR. The numerator includes the number of patients whose observed DOR reached at least the specified time. Patients who did not have the opportunity to reach the specified time point were included in the denominator only. Because responses for some patients are ongoing, the percentages at the specified time points may increase as data mature.
Source: Study 1620 Clinical Study Report.5
Table 32: PFS by Investigator Assessment in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
KM estimation of PFS | ||
Number of events, n (%) | 41 (48.8) | |||||||||||| |
PD, n (%) | 36 (42.9) | |||||||||||| |
Death, n (%) | 5 (6.0) | |||||||||||| |
Number of censored patients, n (%) | 43 (51.2) | |||||||||||| |
Median (95% CI), months | 17.1 (10.3 to 19.3) | |||||||||||| |
Estimate of event-free probability, % (95% CI) | ||
4 months | 91.1 (82.2 to 95.6) | |||||||||||| |
6 months | 84.5 (74.4 to 90.9) | |||||||||||| |
8 months | 76.4 (65.2 to 84.5) | |||||||||||| |
12 months | 59.4 (47.2 to 69.6) | |||||||||||| |
16 months | 50.3 (37.7 to 61.6) | |||||||||||| |
20 months | 35.9 (22.4 to 49.6) | |||||||||||| |
24 months | 30.8 (16.7 to 46.0) | |||||||||||| |
28 months | NA | |||||||||||| |
CI = confidence interval; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; PD = progressive disease; PFS = progression-free survival.
Source: Study 1620 Clinical Study Report.5
Figure 4: Kaplan-Meier of PFS by Investigator Assessment in Study 1620
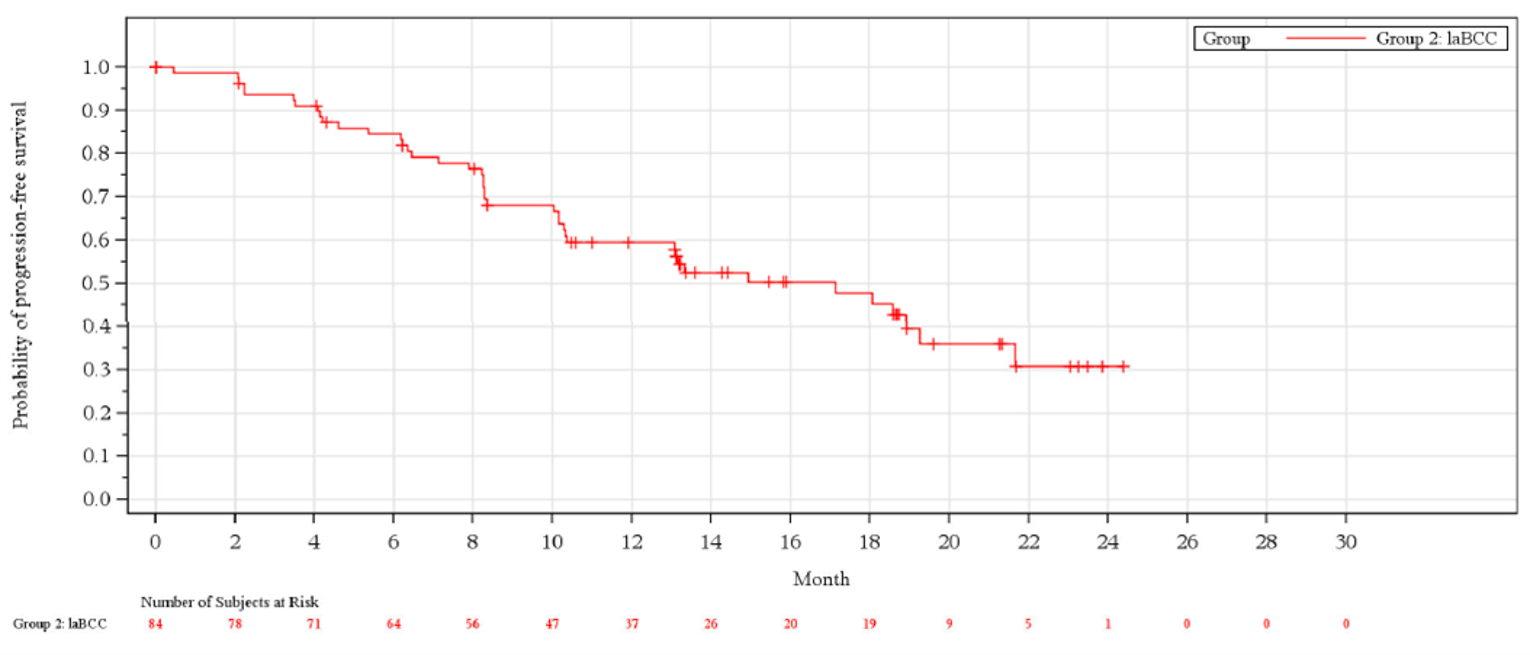
Source: Study 1620 Clinical Study Report.5
Table 33: Summary of Time to Response by Investigator Assessment in Study 1620
Details | Primary analysis laBCC (N = 27) | Updated analysis laBCC (|||||||||) |
|---|---|---|
Observed time to response (CR or PR), months | ||
Mean (SD) | 4.97 (3.562) | |||||||||||| |
Median (range) | 4.17 (2.0 to 13.9) | |||||||||||| |
Observed time to response (CR or PR), n (%)a | ||
< 2 months | 0 | |||||||||||| |
2 to 4 months | 12 (44.4) | |||||||||||| |
4 to 6 months | 6 (22.2) | |||||||||||| |
≥ 6 months | 9 (33.3) | |||||||||||| |
CR = complete response; laBCC = locally advanced basal cell carcinoma; PR = partial response; SD = standard deviation.
a Percentages are based on number of patients with confirmed CR or PR.
Source: Study 1620 Clinical Study Report.5
ORR Subgroup Analysis by Outcome of Prior HHI
Table 34: ORR by BICR in Patients Who Progressed/Lack of Response on HHI Therapy in Study 1620
Details | mBCC FAS (N = 26) | Primary analysis laBCC (N = 63) | Update analysis laBCC (|||||||||) |
|---|---|---|---|
ORR, n (%) | 6 (23.1) | 18 (28.6) | |||||||||||| |
95% CI | 9.0 to 43.6 | 17.9 to 41.3 | |||||||||||| |
CRRa, n (%) | 0 | 5 (7.9) | |||||||||||| |
(95% CI)b | (0 to 13.2) | (2.6 to 17.6) | |||||||||||| |
DCRc, n (%) | 18 (69.2) | 48 (76.2) | |||||||||||| |
(95% CI)b | (48.2 to 85.7) | (63.8 to 86.0) | |||||||||||| |
Best overall response, n (%) | |||
CRa | 0 | 5 (7.9) | |||||||||||| |
PRa | 6 (23.1) | 13 (20.6) | |||||||||||| |
Stable diseased | 9 (34.6) | 30 (47.6) | |||||||||||| |
Non-CR/non-PDe | 3 (11.5) | 0 | |||||||||||| |
PD | 6 (23.1) | 8 (12.7) | |||||||||||| |
NEf | 2 (7.7) | 7 (11.1) | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; HHI = hedgehog pathway inhibitor; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR+PR+Stable disease.
d Stable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
eNon-CR/Non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
Table 35: ORR by BICR in Patients Who Were Intolerant to HHI Therapy in Study 1620
Details | mBCC FAS (N = 2) | Primary analysis laBCC (N = 21) | Updated analysis laBCC (||||||||) |
|---|---|---|---|
ORR, n (%) | 0 | 6 (28.6) | |||||||||||| |
95% CI | 0 to 84.2 | 11.3 to 52.2 | |||||||||||| |
CRR,a n (%) | 0 | 0 | |||||||||||| |
(95% CI)b | (0 to 84.2) | (0 to 16.1) | |||||||||||| |
DCR,c n (%) | 1 (50.0) | 19 (90.5) | |||||||||||| |
(95% CI)b | (1.3 to 98.7) | (69.6 to 98.8) | |||||||||||| |
Best overall response, n (%) | |||
CRa | 0 | 0 | |||||||||||| |
PRa | 0 | 6 (28.6) | |||||||||||| |
Stable diseased | 1 (50.0) | 13 (61.9) | |||||||||||| |
Non-CR/Non-PDe | 0 | 0 | |||||||||||| |
PD | 1 (50.0) | 1 (4.8) | |||||||||||| |
NEf | 0 | 1 (4.8) | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; HHI = hedgehog pathway inhibitor; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR + PR + stable disease.
d Stable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
d Non-CR/Non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
ORR Subgroup Analysis by Histologic Subtype
Table 36: ORR by BICR in Patients With Infiltrative Histology in Study 1620
Details | mBCC FAS (N = 3) | Primary analysis laBCC (N = 7) | Updated analysis laBCC (||||||||) |
|---|---|---|---|
ORR, n (%) | 0 | 3 (42.9) | |||||||||||| |
95% CI | 0 to 70.8 | 9.9 to 81.6 | |||||||||||| |
CRRa, n (%) | 0 | 0 | |||||||||||| |
(95% CI)b | (0 to 70.8) | (0 to 41.0) | |||||||||||| |
DCRc, n (%) | 2 (66.7) | 6 (85.7) | |||||||||||| |
(95% CI)b | (9.4 to 99.2) | (42.1 to 99.6) | |||||||||||| |
Best overall response, n (%) | |||
CRa | 0 | 0 | |||||||||||| |
PRa | 0 | 3 (42.9) | |||||||||||| |
Stable diseased | 1 (33.3) | 3 (42.9) | |||||||||||| |
Non-CR/non-PDe | 1 (33.3) | 0 | |||||||||||| |
PD | 1 (33.3) | 1 (14.3) | |||||||||||| |
NEf | 0 | 0 | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; HHI = hedgehog pathway inhibitor; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR + PR + stable disease.
d Stable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
e Non-CR/Non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
Table 37: ORR by BICR in Patients With Nodular Histology in Study 1620
Details | mBCC FAS (N = 4) | Primary analysis laBCC (N = 21) | Updated analysis laBCC (||||||||) |
|---|---|---|---|
ORR, n (%) | 2 (50.0) | 4 (19.0) | |||||||||||| |
95% CI | 6.8 to 93.2 | 5.4 to 41.9 | |||||||||||| |
CRR,a n (%) | 0 | 2 (9.5) | |||||||||||| |
95% CIb | 0 to 60.2 | 1.2 to 30.4 | |||||||||||| |
DCR,c n (%) | 3 (75.0) | 17 (81.0) | |||||||||||| |
95% CIb | 19.4 to 99.4 | 58.1 to 94.6 | |||||||||||| |
Best overall response, n (%) | NA | NA | |||||||||||| |
CRa | 0 | 2 (9.5) | |||||||||||| |
PRa | 2 (50.0) | 2 (9.5) | |||||||||||| |
Stable diseased | 1 (25.0) | 13 (61.9) | |||||||||||| |
Non-CR/Non-PDe | 0 | 0 | |||||||||||| |
PD | 1 (25.0) | 0 | |||||||||||| |
NEf | 0 | 4 (19.0) | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR + PR + stable disease.
d Stable disease criteria must be met at least once after a minimum duration of 39 days after first dose date.
e Non-CR/non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
Table 38: ORR by BICR in Patients With Other Histology in Study 1620
Details | mBCC FAS (N = 20) | Primary analysis laBCC (N = 56) | Updated analysis laBCC (|||||||) |
|---|---|---|---|
ORR, n (%) | 3 (15.0) | 17 (30.4) | |||||||||||| |
95% CI | 3.2 to 37.9 | 18.8 to 44.1 | |||||||||||| |
CRR,a n (%) | 0 | 3 (5.4) | |||||||||||| |
(95% CI)b | (0 to 16.8) | (1.1 to 14.9) | |||||||||||| |
DCR,c n (%) | 13 (65.0) | 44 (78.6) | |||||||||||| |
(95% CI)b | (40.8 to 84.6) | (65.6 to 88.4) | |||||||||||| |
Best overall response, n (%) | |||
CRa | 0 | 3 (5.4) | |||||||||||| |
PRa | 3 (15.0) | 14 (25.0) | |||||||||||| |
Stable diseased | 8 (40.0) | 27 (48.2) | |||||||||||| |
Non-CR/non-PDe | 2 (10.0) | 0 | |||||||||||| |
PD | 5 (25.0) | 8 (14.3) | |||||||||||| |
NEf | 2 (10.0) | 4 (7.1) | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR+PR+Stable disease.
d SD criteria must be met at least once after a minimum duration of 39 days after first dose date.
e Non-CR/Non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
DOR Subgroup Analysis by Outcome of Prior HHI Therapy
Table 39: DOR by BICR in Patients Who Progressed/Lack of Response to HHI Therapy in Study 1620
Details | mBCC FAS (N = 6) | Updated analysis laBCC (N = 18) | Updated analysis laBCC (||||||||) |
|---|---|---|---|
KM estimation of DOR (CR or PR) | |||
Number of events,a n (%) | 2 (33.3) | 3 (16.7) | |||||||||||| |
Number of censored patients,a n (%) | 4 (66.7) | 15 (83.3) | |||||||||||| |
Median (95% CI), months | NR (9.0 to NE) | NR (15.5 to NE) | |||||||||||| |
Observed DOR (CR or PR)b | |||
Range, months | 9.0 to 23.0 | 2.1 to 21.4 | |||||||||||| |
≥ 4 months, n (%) | 6 (100) | 16 (88.9) | |||||||||||| |
≥ 6 months, n (%) | 6 (100) | 14 (77.8) | |||||||||||| |
≥ 8 months, n (%) | 6 (100) | 11 (61.1) | |||||||||||| |
≥ 12 months, n (%) | 2 (33.3) | 7 (38.9) | |||||||||||| |
≥ 16 months, n (%) | 1 (16.7) | 6 (33.3) | |||||||||||| |
≥ 20 months, n (%) | 1 (16.7) | 2 (11.1) | |||||||||||| |
≥ 24 months, n (%) | 0 | 0 | |||||||||||| |
≥ 28 months, n (%) | NA | NA | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; DOR = duration of response; HHI = hedgehog pathway inhibitor; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; PR = partial response.
a Events include progressive disease or deaths. Percentages are based on number of patients with confirmed CR or PR.
b Percentages are based on number of patients with confirmed CR or PR. The numerator includes the number of patients whose observed DOR reached at least the specified time. Patients who did not have the opportunity to reach the specified time point were included in the denominator only. Because responses for some patients are ongoing, the percentages at the specified time points may increase as data mature.
Source: Study 1620 Clinical Study Report.5
Table 40: DOR by BICR in Patients Who Were Intolerant to HHI Therapy in Study 1620
Details | mBCC FAS (N = 0) | Primary analysis laBCC (N = 6) | Updated analysis laBCC (|||||||) |
|---|---|---|---|
KM estimation of DOR (CR or PR) | |||
Number of events,a n (%) | NA | 3 (50.0) | |||||||||||| |
Number of censored patients,a n (%) | NA | 3 (50.0) | |||||||||||| |
Median (95% CI), months | NA | 19.0 (8.5 to NE) | |||||||||||| |
Observed DOR (CR or PR)b | |||
Range, months | NA | 4.9 to 19.1 | |||||||||||| |
≥ 4 months, n (%) | NA | 6 (100) | |||||||||||| |
≥ 6 months, n (%) | NA | 5 (83.3) | |||||||||||| |
≥ 8 months, n (%) | NA | 5 (83.3) | |||||||||||| |
≥ 12 months, n (%) | NA | 4 (66.7) | |||||||||||| |
≥ 16 months, n (%) | NA | 3 (50.0) | |||||||||||| |
≥ 20 months, n (%) | NA | 0 | |||||||||||| |
≥ 24 months, n (%) | NA | 0 | |||||||||||| |
≥ 28 months, n (%) | NA | NA | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; DOR = duration of response; HHI = hedgehog pathway inhibitor; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; mBCC = metastatic basal cell carcinoma; NA = not applicable; NE = not evaluable; PR = partial response.
a Events include progressive disease or deaths. Percentages are based on number of patients with confirmed CR or PR.
b Percentages are based on number of patients with confirmed CR or PR. The numerator includes the number of patients whose observed DOR reached at least the specified time. Patients who did not have the opportunity to reach the specified time point were included in the denominator only. Because responses for some patients are ongoing, the percentages at the specified time points may increase as data mature.
Source: Study 1620 Clinical Study Report.5
PFS Sensitivity Analysis: Including Start of Anti-Cancer Therapy as PFS Event
Table 41: PFS by BICR Including Start of Anti-Cancer Therapy as PFS Event in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
KM estimation of PFS | ||
Number of events, n (%) | 39 (46.4) | |||||||||||| |
PD, n (%) | 33 (39.3) | |||||||||||| |
Death, n (%) | 5 (6.0) | |||||||||||| |
Anti-cancer therapy, n (%) | 1 (1.2) | |||||||||||| |
Number of censored patients, n (%) | 45 (53.6) | |||||||||||| |
Median (95% CI), months | 14.4 (8.6 to NE) | |||||||||||| |
Estimate of event-free probability, % (95% CI) | ||
4 months | 84.4 (74.1 to 90.8) | |||||||||||| |
6 months | 76.3 (65.1 to 84.4) | |||||||||||| |
8 months | 68.1 (56.3 to 77.4) | |||||||||||| |
12 months | 55.0 (42.8 to 65.6) | |||||||||||| |
16 months | 49.6 (37.3 to 60.8) | |||||||||||| |
20 months | 45.1 (31.2 to 58.1) | |||||||||||| |
24 months | 34.4 (18.6 to 50.8) | |||||||||||| |
28 months | NA | |||||||||||| |
BICR = blinded independent central review; CI = confidence interval; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; PD = progressive disease; PFS = progression-free survival.
Source: Study 1620 Clinical Study Report.5
OS Sensitivity Analysis: Censoring Patients With Subsequent Therapy
Table 42: OS With Censoring of Patients With Subsequent Therapy in Study 1620
Details | Primary analysis laBCC (N = 84) | Updated analysis laBCC (N = 84) |
|---|---|---|
KM estimation of OS | ||
Number of deaths, n (%) | 9 (10.7) | |||||||||||| |
Number of censored patients, n (%) | 75 (89.3) | |||||||||||| |
Median (95% CI), months | NR (NE to NE) | |||||||||||| |
Estimate of probability of survival, % (95% CI) | ||
4 months | 98.8 (91.8 to 99.8) | |||||||||||| |
6 months | 98.8 (91.8 to 99.8) | |||||||||||| |
8 months | 96.3 (88.9 to 98.8) | |||||||||||| |
12 months | 92.2 (83.5 to 96.4) | |||||||||||| |
16 months | 92.2 (83.5 to 96.4) | |||||||||||| |
20 months | 86.8 (73.9 to 93.6) | |||||||||||| |
24 months | 81.0 (62.0 to 91.1) | |||||||||||| |
28 months | NA | |||||||||||| |
CI = confidence interval; KM = Kaplan-Meier; laBCC = locally advanced basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; OS = overall survival.
Source: Study 1620 Clinical Study Report.5
EORTC QLQ-C30 Functional and Symptom Scales
Table 43: Physical Functioning EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 81.25 (21.04) | NA | NA |
Cycle 2 | 75 | 79.38 (21.48) | 75 | −1.55 (12.10) |
Cycle 3 | 65 | 80.72 (18.93) | 65 | −0.56 (15.95) |
Cycle 4 | 55 | 78.97 (22.11) | 55 | −3.79 (17.81) |
Cycle 5 | 50 | 78.31 (21.51) | 50 | −2.86 (17.06) |
Cycle 6 | 40 | 80.83 (18.81) | 40 | 0.33 (12.07) |
Cycle 7 | 33 | 81.62 (17.64) | 33 | −0.20 (15.41) |
Cycle 8 | 27 | 76.79 (21.19) | 27 | −4.44 (14.32) |
Cycle 9 | 18 | 80.37 (21.02) | 18 | −3.33 (16.69) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For functioning scales, a higher score signifies higher functioning.
Source: Study 1620 Clinical Study Report.5
Table 44: Role Functioning EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 79.87 (24.53) | NA | NA |
Cycle 2 | 75 | 76.22 (27.56) | 75 | −3.11 (20.26) |
Cycle 3 | 65 | 75.13 (25.54) | 65 | −4.87 (25.30) |
Cycle 4 | 55 | 74.24 (27.74) | 55 | −6.06 (26.33) |
Cycle 5 | 50 | 75.33 (24.57) | 50 | −5.33 (26.39) |
Cycle 6 | 40 | 79.17 (24.09) | 40 | −3.33 (16.96) |
Cycle 7 | 33 | 75.76 (21.69) | 33 | −7.07 (16.68) |
Cycle 8 | 27 | 77.78 (24.02) | 27 | −2.47 (20.52) |
Cycle 9 | 18 | 75.00 (26.35) | 18 | −10.19 (26.90) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For functioning scales, a higher score signifies higher functioning.
Source: Study 1620 Clinical Study Report.5
Table 45: Emotional Functioning EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 81.36 (19.43) | NA | NA |
Cycle 2 | 74 | 83.03 (19.22) | 74 | 1.95 (20.48) |
Cycle 3 | 64 | 82.81 (21.30) | 64 | 1.56 (18.54) |
Cycle 4 | 54 | 82.41 (20.65) | 54 | 1.39 (21.28) |
Cycle 5 | 49 | 78.40 (21.78) | 49 | −4.59 (19.62) |
Cycle 6 | 40 | 85.63 (18.39) | 40 | 1.04 (19.81) |
Cycle 7 | 33 | 79.97 (19.31) | 33 | −4.63 (19.58) |
Cycle 8 | 27 | 84.67 (20.02) | 27 | 1.95 (18.34) |
Cycle 9 | 18 | 84.88 (19.58) | 18 | −1.23 (14.20) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For functioning scales, a higher score signifies higher functioning.
Source: Study 1620 Clinical Study Report.5
Table 46: Cognitive Functioning EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 85.75 (17.58) | NA | NA |
Cycle 2 | 74 | 82.88 (25.73) | 74 | −2.48 (26.99) |
Cycle 3 | 64 | 82.03 (23.44) | 64 | −4.17 (23.38) |
Cycle 4 | 54 | 80.86 (21.33) | 54 | −4.94 (21.39) |
Cycle 5 | 49 | 79.25 (26.69) | 49 | −6.46 (25.19) |
Cycle 6 | 40 | 82.50 (24.15) | 40 | −1.25 (19.02) |
Cycle 7 | 33 | 80.30 (21.43) | 33 | −5.56 (18.94) |
Cycle 8 | 27 | 85.19 (18.68) | 27 | 0.62 (18.19) |
Cycle 9 | 18 | 81.48 (24.18) | 18 | −5.56 (21.39) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For functioning scales, a higher score signifies higher functioning.
Source: Study 1620 Clinical Study Report.5
Table 47: Social Functioning EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 81.80 (24.52) | NA | NA |
Cycle 2 | 74 | 85.81 (22.19) | 74 | 4.50 (21.42) |
Cycle 3 | 64 | 84.38 (22.40) | 64 | 0.78 (21.09) |
Cycle 4 | 54 | 87.04 (21.15) | 54 | 1.85 (23.27) |
Cycle 5 | 49 | 87.07 (23.39) | 49 | 0.34 (23.45) |
Cycle 6 | 40 | 87.50 (20.59) | 40 | 0.00 (24.75) |
Cycle 7 | 33 | 91.41 (15.09) | 33 | 0.00 (18.16) |
Cycle 8 | 27 | 90.12 (19.75) | 27 | 3.70 (23.72) |
Cycle 9 | 18 | 87.96 (18.79) | 18 | −6.48 (25.01) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For functioning scales, a higher score signifies higher functioning.
Source: Study 1620 Clinical Study Report.5
Table 48: Fatigue Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 23.02 (24.78) | NA | NA |
Cycle 2 | 75 | 30.07 (26.58) | 75 | 6.44 (24.54) |
Cycle 3 | 65 | 28.72 (26.27) | 65 | 6.58 (24.90) |
Cycle 4 | 55 | 29.70 (25.31) | 55 | 7.58 (25.91) |
Cycle 5 | 50 | 31.56 (23.80) | 50 | 9.67 (23.40) |
Cycle 6 | 40 | 26.94 (24.90) | 40 | 7.78 (17.65) |
Cycle 7 | 33 | 30.30 (23.45) | 33 | 14.14 (20.84) |
Cycle 8 | 27 | 28.81 (21.63) | 27 | 9.47 (20.83) |
Cycle 9 | 18 | 27.78 (23.57) | 18 | 13.58 (21.75) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 49: Nausea/Vomiting Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 4.11 (12.14) | NA | NA |
Cycle 2 | 75 | 4.44 (11.73) | 75 | 0.22 (12.70) |
Cycle 3 | 65 | 2.82 (6.95) | 65 | −1.79 (12.88) |
Cycle 4 | 55 | 4.55 (12.61) | 55 | 0.30 (13.41) |
Cycle 5 | 50 | 6.00 (14.19) | 50 | 1.00 (13.64) |
Cycle 6 | 40 | 5.00 (10.13) | 40 | 1.67 (13.50) |
Cycle 7 | 33 | 5.05 (11.40) | 33 | 3.03 (13.47) |
Cycle 8 | 27 | 4.32 (10.93) | 27 | 1.23 (13.81) |
Cycle 9 | 18 | 0.00 (0.00) | 18 | −0.93 (3.93) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 50: Pain Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 22.94 (26.50) | NA | NA |
Cycle 2 | 75 | 23.33 (27.40) | 75 | −0.22 (26.35) |
Cycle 3 | 65 | 20.77 (27.01) | 65 | −1.54 (30.15) |
Cycle 4 | 55 | 20.30 (24.57) | 55 | −4.85 (25.80) |
Cycle 5 | 50 | 19.33 (24.60) | 50 | −4.00 (25.10) |
Cycle 6 | 40 | 15.42 (19.75) | 40 | −2.92 (24.43) |
Cycle 7 | 33 | 16.16 (21.03) | 33 | −4.04 (24.66) |
Cycle 8 | 27 | 13.58 (20.69) | 27 | −9.88 (25.43) |
Cycle 9 | 18 | 12.96 (18.57) | 18 | −8.33 (24.42) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; NA = not applicable; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 51: Dyspnea Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 14.29 (25.61) | NA | NA |
Cycle 2 | 75 | 16.00 (24.73) | 75 | 1.33 (24.16) |
Cycle 3 | 65 | 12.31 (21.71) | 65 | −0.51 (23.93) |
Cycle 4 | 55 | 13.94 (21.93) | 55 | 2.42 (24.72) |
Cycle 5 | 49 | 13.61 (20.32) | 49 | 0.00 (28.05) |
Cycle 6 | 40 | 12.50 (18.00) | 40 | −1.67 (19.90) |
Cycle 7 | 33 | 14.14 (18.69) | 33 | 2.02 (21.95) |
Cycle 8 | 27 | 14.81 (19.25) | 27 | 4.94 (20.05) |
Cycle 9 | 18 | 5.56 (12.78) | 18 | 0.00 (11.43) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0 For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 52: Insomnia Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 77 | 22.94 (26.08) | NA | NA |
Cycle 2 | 75 | 23.11 (27.93) | 75 | 0.44 (24.20) |
Cycle 3 | 65 | 22.56 (28.93) | 65 | −0.51 (26.02) |
Cycle 4 | 55 | 21.21 (24.31) | 55 | −1.82 (19.69) |
Cycle 5 | 50 | 24.00 (30.89) | 50 | 1.33 (30.09) |
Cycle 6 | 40 | 15.83 (25.02) | 40 | −4.17 (24.09) |
Cycle 7 | 33 | 21.21 (28.65) | 33 | 1.01 (28.24) |
Cycle 8 | 27 | 24.69 (28.63) | 27 | 2.47 (26.03) |
Cycle 9 | 18 | 25.93 (29.27) | 18 | 5.56 (17.15) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 53: Appetite Loss Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 14.47 (24.55) | NA | NA |
Cycle 2 | 74 | 12.61 (23.21) | 74 | −2.25 (27.22) |
Cycle 3 | 62 | 10.75 (18.87) | 62 | −4.30 (22.16) |
Cycle 4 | 54 | 13.58 (26.32) | 54 | −3.09 (26.12) |
Cycle 5 | 49 | 14.97 (31.23) | 49 | −1.36 (30.40) |
Cycle 6 | 39 | 9.40 (21.56) | 39 | −2.56 (29.00) |
Cycle 7 | 33 | 12.12 (23.30) | 33 | −1.01 (30.60) |
Cycle 8 | 26 | 11.54 (18.72) | 26 | −6.41 (28.31) |
Cycle 9 | 18 | 14.81 (28.52) | 18 | 0.00 (36.16) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 54: Constipation Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 7.02 (16.61) | NA | NA |
Cycle 2 | 74 | 9.46 (19.52) | 74 | 2.25 (21.60) |
Cycle 3 | 63 | 6.88 (16.02) | 63 | 1.06 (20.71) |
Cycle 4 | 54 | 8.02 (19.36) | 54 | 1.23 (21.44) |
Cycle 5 | 49 | 6.80 (19.22) | 49 | −0.68 (22.04) |
Cycle 6 | 40 | 4.17 (11.16) | 40 | −1.67 (18.41) |
Cycle 7 | 33 | 10.10 (22.80) | 33 | 4.04 (13.84) |
Cycle 8 | 27 | 6.17 (13.20) | 27 | −3.70 (16.88) |
Cycle 9 | 18 | 12.96 (20.26) | 18 | 5.56 (23.57) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Table 55: Diarrhea Symptom Scale EORTC QLQ-C30 in Study 1620
Cycle | Primary analysis laBCC (N = 84) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 76 | 9.21 (18.54) | NA | NA |
Cycle 2 | 74 | 9.46 (21.74) | 74 | 0.45 (24.36) |
Cycle 3 | 64 | 9.38 (20.97) | 64 | −1.04 (20.55) |
Cycle 4 | 54 | 7.41 (17.93) | 54 | −1.85 (19.87) |
Cycle 5 | 49 | 9.52 (22.57) | 49 | −0.68 (23.06) |
Cycle 6 | 40 | 6.67 (18.80) | 40 | −1.67 (18.41) |
Cycle 7 | 33 | 10.10 (25.67) | 33 | 1.01 (19.52) |
Cycle 8 | 27 | 2.47 (8.90) | 27 | −4.94 (15.20) |
Cycle 9 | 18 | 5.56 (12.78) | 18 | 1.85 (17.98) |
HRQoL = health-related quality of life; laBCC = locally advanced basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For symptom scales, a higher score signifies higher symptomatology.
Source: Study 1620 Clinical Study Report.5
Main Outcome Analysis in the mBCC Population
Table 56: Patient Disposition and Survival Follow-Up (mBCC Population)
Details | mBCC February 2020 Data Cut (N = 48) | mBCC |||||||||||| Data Cut (|||||||||) |
|---|---|---|
Enrolled, N (%) | 48 (100) | |||||||||||| |
Treatment ongoing, N (%) | 13 (27.1) | |||||||||||| |
Off treatment, N (%) | 35 (72.9) | |||||||||||| |
Treatment completed, N (%) | 3 (6.3) | |||||||||||| |
Treatment discontinued, N (%) | 32 (66.7) | |||||||||||| |
Reason for treatment discontinuation, N (%) | ||
Adverse events | 3 (6.3) | |||||||||||| |
Death | 1 (2.1) | |||||||||||| |
Lost to follow-up | 1 (2.1) | |||||||||||| |
Non-compliance with protocol | 0 | |||||||||||| |
Patient decision | 0 | |||||||||||| |
Progressive disease | 24 (50.0) | |||||||||||| |
Withdrawal of consent | 2 (4.2) | |||||||||||| |
Confirmed CR per investigator assessment | 1 (2.1) | |||||||||||| |
Other | 0 | |||||||||||| |
Study ongoing, N (%) | 18 (37.5) | |||||||||||| |
Off study, N (%) | 30 (62.5) | |||||||||||| |
Study completed, N (%) | 0 | |||||||||||| |
Study discontinued, N (%) | 30 (62.5) | |||||||||||| |
Reason for study discontinuation, N (%) | NA | |||||||||||| |
Adverse events | 0 | |||||||||||| |
Death | 2 (4.2) | |||||||||||| |
Lost to follow-up | 1 (2.1) | |||||||||||| |
Non-compliance with protocol | 1 (2.1) | |||||||||||| |
Patient decision | 0 | |||||||||||| |
Sponsor decision | 0 | |||||||||||| |
Progressive disease | 24 (50.0) | |||||||||||| |
Withdrawal of consent | 2 (4.2) | |||||||||||| |
Other | 0 | |||||||||||| |
Entered follow-up, N (%) | 4 (8.3) | |||||||||||| |
Duration of study follow-up, months | NA | |||||||||||| |
Mean (SD) | 8.47 (6.82) | |||||||||||| |
Median (range) | 6.21 (0 to 27.2) | |||||||||||| |
FAS, N | 28 (58.3) | |||||||||||| |
Safety, N | 48 (100) | |||||||||||| |
CR = complete response; FAS = full analysis set; mBCC = metastatic basal cell carcinoma; SD = standard deviation.
Source: Study 1620 Clinical Study Report.5
Table 57: Important Protocol Deviations (mBCC Population)
Details | mBCC (N = 48) |
|---|---|
Number of important protocol deviations | 5 |
Patients with any important protocol deviation, n (%) | 5 (10.4) |
Exclusion criteria met but patient enrolled, n (%) | 2 (4.2) |
Exclusion criterion 8: Any anti-cancer treatment other than radiation therapy investigational or standard of care, within 30 days of the initial administration | 2 (4.2) |
Exclusion criterion 15: Inability to undergo any contrast-enhanced radiologic response assessment | 0 |
Inclusion criteria not met but patient enrolled, n (%) | 2 (4.2) |
Inclusion criterion 3: No measurable lesion | 1 (2.1) |
Inclusion criterion 8: Creatine phosphokinase (CPK) not performed on both screening and C1D1 | 0 |
Inclusion criterion 11: Archival or newly obtained tumour material for central pathology review for confirmation of BCC was not confirmed as received by central laboratory prior to enrolment | 1 (2.1) |
Inclusion criterion 6: Hepatic function not meeting protocol criteria for alkaline phosphatase levels that were higher than 2.5 × the upper limit of normal (ULN) on both screening and C1D1 | 0 |
Inadequate informed consent administration, n (%) | 0 |
SAEs/AESIs not reported within 24 hours to PVRM, n (%) | 0 |
Treatment deviation, n (%) | 1 (2.1) |
Other, n (%) | 0 |
AESI = adverse event of special interest; BCC = basal cell carcinoma; C1D1 = cycle 1, day 1; CPK = Creatine phosphokinase; mBCC = metastatic basal cell carcinoma; PVRM = pharmacovigilance and risk management; SAE = serious adverse event; ULN = upper limit of normal.
a Some patients did not meet multiple inclusion requirements, therefore the total specific inclusion criteria deviations sums to greater than the number of patients with an inclusion criteria deviation.
Source: Study 1620 Clinical Study Report.5
Table 58: Treatment Exposure (mBCC Population)
Details | mBCC February 2020 Data Cut (N = 48) | mBCC |||||||||||| Data Cut (|||||||||) |
|---|---|---|
Duration of exposure, weeksa | NA | |||||||||||| |
Mean (SD) | 34.71 (28.37) | |||||||||||| |
Median (range) | 26.00 (2.7 to 93.4) | |||||||||||| |
Number of doses administered | NA | |||||||||||| |
Mean (SD) | 10.8 (8.5) | |||||||||||| |
Median (range) | 8.0 (1 to 30) | |||||||||||| |
Actual dose intensity (mg/week)b | NA | |||||||||||| |
Mean (SD) | 112.16 (10.72) | |||||||||||| |
Median (range) | 116.67 (66.5 to 128.9) | |||||||||||| |
Relative dose intensityc | NA | |||||||||||| |
Mean (SD) | 0.96 (0.09) | |||||||||||| |
Median (range) | 1.00 (0.6 to 1.1) | |||||||||||| |
Treatment compliance,d n (%) | NA | |||||||||||| |
< 60% | 1 (2.1) | |||||||||||| |
≥ 60% < 80% | 2 (4.2) | |||||||||||| |
≥ 80% | 45 (93.8) | |||||||||||| |
mBCC = metastatic basal cell carcinoma; SD = standard deviation; q.3.w. = every 3 weeks.
a Duration of exposure (weeks) = Minimum of (last dose date − first dose date + 21 days)/7 AND (data cut-off date or death date − first dose date + 1 day)/7 for 350 mg q.3.w.
b Actual dose intensity (mg/week) = Total dose received (mg) / Duration of exposure (weeks).
c Relative dose intensity = Actual dose intensity / Planned dose intensity. Planned dose intensity (mg/week) = Planned dose (mg) / 3 (weeks).
d Treatment Compliance = (Number of investigational product administered during treatment period/Number of investigational product planned to be taken during treatment period) × 100%.
Source: Study 1620 Clinical Study Report.5
Table 59: Concomitant Medications and Procedures (mBCC Population)
Details | mBCC (N = 48) |
|---|---|
Number of patients with any concomitant medications, n (%) | 46 (95.8) |
Analgesics | 34 (70.8) |
Antibacterial for systemic use | 26 (54.2) |
Antithrombotic agents | 23 (47.9) |
Agents acting on the renin-angiotensin system | 11 (22.9) |
Drugs for acid-related disorders | 19 (39.6) |
Beta-blocking agents | 12 (25.0) |
Corticosteroid for systemic use | 12 (25.0) |
Anti-inflammatory and antirheumatic products | 20 (41.7) |
Lipid-modifying agents | 13 (27.1) |
Psycholeptics | 11 (22.9) |
Diuretics | 8 (16.7) |
Ophthalmological | 4 (8.3) |
Vitamins | 14 (29.2) |
Corticosteroids, dermatological preparations | 6 (12.5) |
Antianemic preparations | 7 (14.6) |
Antidiarrheals, Intestinal | 8 (16.7) |
Thyroid therapy | 5 (10.4) |
Blood substitutes and perfusion solution | 5 (10.4) |
Drugs used in diabetes | 3 (6.3) |
Psychoanaleptics | 7 (14.6) |
Calcium channel blockers | 4 (8.3) |
Number of patients with any concomitant procedures, n (%) | 25 (52.1) |
Investigations | 23 (47.9) |
Surgical and medical procedures | 12 (25.0) |
Uncoded | 0 |
mBCC = metastatic basal cell carcinoma.
Source: Study 1620 Clinical Study Report.5
Table 60: ORR by BICR Assessment (mBCC Population)
Details | mBCC FAS (N = 28) |
|---|---|
ORR, n (%) | 6 (21.4) |
(95% CI)b | (8.3 to 41.0) |
CRR,a n (%) | 0 |
(95% CI)b | (0 to 12.3) |
DCR,c n (%) | 19 (67.9) |
(95% CI)b | (47.6 to 84.1) |
Best overall response, n (%) | |
CRa | 0 |
PRa | 6 (21.4) |
Stable diseased | 10 (35.7) |
Non-CR/Non-PDe | 3 (10.7) |
PD | 7 (25.0) |
NEf | 2 (7.1) |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; CR = complete response; CRR = complete response rate; DCR = disease control rate; FAS = full analysis set; mBCC = metastatic basal cell carcinoma; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response.
a CR/PR must be confirmed by repeated assessments no less than 4 weeks apart.
b Clopper-Person exact CI.
c CR+PR+Stable disease.
d SD criteria must be met at least once after a minimum duration of 39 days after first dose date.
e Non-CR/Non-PD is for patients with non-measurable disease only.
f Not evaluable response includes the missing and unknown tumour response.
Source: Study 1620 Clinical Study Report.5
Table 61: DOR by BICR (mBCC Population)
Details | mBCC FAS (N = 6) |
|---|---|
KM estimation of DOR (CR or PR) | |
Number of events,a n (%) | 2 (33.3) |
Number of censored patients,a n (%) | 4 (66.7) |
Median (95% CI), months | NR (9.0 to NE) |
Observed DOR (CR or PR)b | |
Range, months | 9.0 to 23.0+ |
≥ 4 months, n (%) | 6 (100) |
≥ 6 months, n (%) | 6 (100) |
≥ 8 months, n (%) | 6 (100) |
≥ 12 months, n (%) | 2 (33.3) |
≥ 16 months, n (%) | 1 (16.7) |
≥ 20 months, n (%) | 1 (16.7) |
≥ 24 months, n (%) | 0 |
≥ 28 months, n (%) | NA |
BICR = blinded independent central review; CI = confidence interval; CR = complete response; DOR = duration of response; FAS = full analysis set; KM = Kaplan-Meier; mBCC = metastatic basal cell carcinoma; NA = not applicable; NE = not evaluable; NR = not reached; PR = partial response.
a Events include progressive disease or deaths. Percentages are based on number of patients with confirmed CR or PR.
b Percentages are based on number of patients with confirmed CR or PR. The numerator includes the number of patients whose observed DOR reached at least the specified time. Patients who did not have the opportunity to reach the specified time point were included in the denominator only. Because responses for some patients are ongoing, the percentages at the specified time points may increase as data mature.
Source: Study 1620 Clinical Study Report.5
Table 62: PFS by BICR Assessment (mBCC Population)
Details | mBCC FAS (N = 28) |
|---|---|
KM estimation of PFS | |
Number of events, n (%) | 17 (60.7) |
PD, n (%) | 14 (50.0) |
Death, n (%) | 3 (10.7) |
Number of censored patients, n (%) | 11 (39.3) |
Median (95% CI), months | 8.3 (3.6 to 19.5) |
Estimate of event-free probability, % (95% CI) | |
4 months | 70.0 (48.8 to 83.7) |
6 months | 58.1 (37.1 to 74.3) |
8 months | 58.1 (37.1 to 74.3) |
12 months | 49.8 (29.5 to 67.1) |
16 months | 33.6 (15.2 to 53.2) |
20 months | 29.6 (10.0 to 47.3) |
24 months | 29.6 (10.0 to 47.3) |
28 months | NA |
BICR = blinded independent central review; CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; mBCC = metastatic basal cell carcinoma; NA = not applicable; PD = progressive disease; PFS = progression-free survival.
Source: Study 1620 Clinical Study Report.5
Table 63: Summary of OS (mBCC Population)
Details | mBCC FAS (N = 28) |
|---|---|
KM estimation of OS | |
Number of deaths, n (%) | 7 (25.0%) |
Number of censored patients, n (%) | 21 (75.0%) |
Median (95% CI), months | 25.7 (19.5 to NE) |
Estimate of survival, % (95% CI) | |
4 months | 96.4 (77.2 to 99.5) |
6 months | 96.4 (77.2 to 99.5) |
8 months | 92.6 (73.4 to 98.1) |
12 months | 92.6 (73.4 to 98.1) |
16 months | 78.3 (54.7 to 90.5) |
20 months | 71.2 (45.1 to 86.5) |
24 months | 71.2 (45.1 to 86.5) |
28 months | NA |
CI = confidence interval; FAS = full analysis set; KM = Kaplan-Meier; mBCC = metastatic basal cell carcinoma; NA = not applicable; NE = not evaluable; OS = overall survival.
Source: Study 1620 Clinical Study Report.5
Table 64: Summary of Time to Response by BICR Assessment (mBCC Population)
Details | mBCC FAS (N = 6) |
|---|---|
Observed time to response (CR or PR), months | |
Mean (SD) | 4.54 (3.34) |
Median (range) | 3.17 (2.1 to 10.5) |
Observed time to response (CR or PR), n (%)a | |
< 2 months | 0 |
2 to 4 months | 3 (50.0) |
4 to 6 months | 1 (16.7) |
≥ 6 months | 2 (33.3) |
BICR = blinded independent central review; CR = complete response; FAS = full analysis set; mBCC = metastatic basal cell carcinoma; PR = partial response; SD = standard deviation.
a Percentages are based on number of patients with confirmed CR or PR.
Source: Study 1620 Clinical Study Report.5
Table 65: Global Health Status EORTC QLQ-C30 (mBCC Population)
Cycle | mBCC FAS (N = 28) | |||
|---|---|---|---|---|
Scorea | Change from baseline | |||
n | Mean (SD) | n | Mean (SD) | |
Baseline | 22 | 61.74 (24.89) | NA | NA |
Cycle 2b | 21 | 58.73 (27.45) | 21 | −5.16 (23.20) |
Cycle 3b | 15 | 66.67 (25.59) | 15 | 6.11 (14.59) |
Cycle 4b | 14 | 59.52 (25.50) | 14 | 0.60 (14.42) |
Cycle 5b | 10 | 72.50 (24.86) | 10 | 12.50 (15.34) |
Cycle 6b | 12 | 67.36 (23.43) | 12 | 12.50 (11.51) |
Cycle 7 | 8 | 69.79 (29.53) | 8 | 21.88 (25.95) |
Cycle 8 | 6 | 75.00 (14.91) | 6 | 13.89 (11.39) |
Cycle 9 | 5 | 70.00 (19.19) | 5 | 3.33 (12.64) |
FAS = full analysis set; HRQoL = health-related quality of life; mBCC = metastatic basal cell carcinoma; SD = standard deviation.
a Scores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire version 3.0. For global health status, a higher score signifies better HRQoL.
Source: Study 1620 Clinical Study Report.5
Table 66: Skindex-16 Emotion Scale (mBCC Population)
Cycle | mBCC emotional scale (N = 28) | mBCC symptom scale (N = 28) | mBCC functioning scale (N = 28) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
Baseline | 22 | 33.08 (36.67) | NA | NA | 22 | 19.89 (27.93) | NA | NA | 22 | 27.73 (33.67) | NA | NA |
Cycle 2 | 22 | 23.59 (32.55) | 22 | −9.49 (25.47) | 22 | 16.10 (21.45) | 22 | −3.79 (19.54) | 22 | 20.15 (31.66) | 22 | −7.58 (19.31) |
Cycle 3 | 13 | 19.38 (26.98) | 13 | −6.93 (22.89) | 13 | 14.10 (18.68) | 13 | 0.64 (15.94) | 13 | 13.08 (27.37) | 13 | −8.46 (19.32) |
Cycle 4 | 13 | 30.04 (34.15) | 13 | −2.50 (20.00) | 13 | 20.19 (21.37) | 13 | 3.53 (16.48) | 13 | 16.92 (30.84) | 13 | −11.79 (26.06) |
Cycle 5 | 10 | 19.05 (27.65) | 10 | 1.67 (3.73) | 10 | 6.25 (8.62) | 10 | −1.67 (13.92) | 10 | 12.67 (29.01) | 10 | −5.00 (24.56) |
Cycle 6 | 11 | 19.70 (28.71) | 11 | −9.52 (27.42) | 11 | 15.53 (28.87) | 11 | −1.52 (18.09) | 11 | 15.76 (29.59) | 11 | −13.64 (31.39) |
Cycle 7 | 8 | 40.18 (35.37) | 8 | 8.33 (13.59) | 8 | 25.00 (27.55) | 8 | 4.17 (23.78) | 8 | 34.17 (41.85) | 8 | −5.00 (24.37) |
Cycle 8 | 6 | 6.35 (8.20) | 6 | 2.78 (6.10) | 6 | 13.89 (16.60) | 6 | 11.11 (18.19) | 6 | 0.00 (0.00) | 6 | −12.22 (29.94) |
Cycle 9 | 5 | 10.00 (13.92) | 5 | 6.67 (13.82) | 5 | 18.33 (18.31) | 5 | 16.67 (17.43) | 5 | 0.00 (0.00) | 5 | 0.00 (0.00) |
HRQoL = health-related quality of life; mBCC = metastatic basal cell carcinoma; SD = standard deviation.
a Item scores are transformed to a linear scale (0 to 100, with 0 representing never bothered and 100 representing always bothered).
Source: Study 1620 Clinical Study Report.5
Table 67: Summary of Harms (mBCC Population)
Harms | mBCC Safety analysis (N = 48) | mBCC |||||||||||| data cut-off (|||||||||) |
|---|---|---|
Patients with ≥ 1 TEAE | ||
n (%) | 43 (89.6) | |||||||||||| |
Frequent TEAEs, n (%) | ||
Fatigue | 19 (39.6) | |||||||||||| |
Diarrhea | 13 (27.1) | |||||||||||| |
Pruritus | 8 (16.7) | |||||||||||| |
Asthenia | 4 (8.3) | |||||||||||| |
Anemia | 4 (8.3) | |||||||||||| |
Decreased appetite | 6 (12.5) | |||||||||||| |
Headache | 4 (8.3) | |||||||||||| |
Nausea | 4 (8.3) | |||||||||||| |
Urinary tract infection | 4 (8.3) | |||||||||||| |
Arthralgia | 6 (12.5) | |||||||||||| |
Dyspnea | 3 (6.3) | |||||||||||| |
Pyrexia | 6 (12.5) | |||||||||||| |
Constipation | 9 (18.8) | |||||||||||| |
Vomiting | 5 (10.4) | |||||||||||| |
Weight decreased | 5 (10.4) | |||||||||||| |
Weight increased | 7 (14.6) | |||||||||||| |
Dizziness | 5 (10.4) | |||||||||||| |
Hyperglycemia | 5 (10.4) | |||||||||||| |
Hypertension | 6 (12.5) | |||||||||||| |
Cough | 3 (6.3) | |||||||||||| |
Tumour hemorrhage | 1 (2.1) | |||||||||||| |
Patients with ≥ 1 SAE | ||
n (%) | 13 (27.1) | |||||||||||| |
Frequent SAEs, n (%) | ||
Urinary tract infection | 1 (2.1) | |||||||||||| |
Colitis | 2 (4.2) | |||||||||||| |
Myocardial infarction | 0 | |||||||||||| |
Infected neoplasm | 0 | |||||||||||| |
Anemia | 0 | |||||||||||| |
Adrenal insufficiency | 0 | |||||||||||| |
Acute kidney injury | 0 | |||||||||||| |
TEAEs leading to study drug discontinuation | ||
n (%) | 3 (6.3) | |||||||||||| |
TEAEs leading to dose delay | ||
n (%) | 14 (29.2) | |||||||||||| |
TEAEs leading to study drug interruption | ||
n (%) | 4 (8.3) | |||||||||||| |
TEAEs leading to dose reduction | ||
n (%) | 0 | |||||||||||| |
TEAEs leading to death | ||
n (%) | 1 (2.1) | |||||||||||| |
Notable harms | ||
AESIs, n (%) | ||
Immune-related AEa | 25 (52.1) | |||||||||||| |
Grade 3/4/5 immune-related AE | 5 (10.4) | |||||||||||| |
Serious immune-related AE | 5 (10.4) | |||||||||||| |
Immune-related AE leading to discontinuation | 2 (4.2) | |||||||||||| |
Immune-related AE leading to dose delay | 7 (14.6) | |||||||||||| |
Immune-related AE leading to drug interruption | 0 | |||||||||||| |
Immune-related AE leading to dose reduction | 0 | |||||||||||| |
Immune-related AE resulting in death | 0 | |||||||||||| |
Infusion-related reactions | 5 (10.4) | |||||||||||| |
Grade 3, 4, or 5 infusion-related reaction | 0 | |||||||||||| |
Serious infusion reaction | 1 (2.1) | |||||||||||| |
Infusion reaction leading to discontinuation | 0 | |||||||||||| |
Infusion reaction leading to dose delay | 0 | |||||||||||| |
Infusion reaction leading to drug interruption | 4 (8.3) | |||||||||||| |
Infusion reaction leading to dose reduction | 0 | |||||||||||| |
Infusion reaction resulting in death | 0 | |||||||||||| |
AE = adverse event; AESI = adverse event of special interest; mBCC = metastatic basal cell carcinoma; SAE = serious adverse event; TEAE = treatment-emergent adverse events.
a As there is currently no MedDRA-coded classification for immune-related AEs, the sponsor created a customized list of MedDRA-preferred terms for the identification of immune-related AEs.
Source: Study 1620 Clinical Study Report.5
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-C30
Skindex-16
Findings
EORTC QLQ-C30
Description and Scoring
The EORTC QLQ-C30 is one of the most used patient-reported outcome measures in oncology clinical trials. It is a multi-dimensional, cancer-specific, self-administered, measure of HRQoL.33
The EORTC QLQ-C30 is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), a global health status/HRQoL scale, and 6 single items assessing additional symptoms commonly reported by cancer patients (dyspnea, loss of appetite, insomnia, constipation and diarrhea) as well as perceived financial impact of the disease.33
The EORTC QLQ-C30 uses a 1-week recall period to assess functional status and symptoms. All scales and single-item measures are scored from 0 to 100. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global HRQoL scale, the response format is a 7-point Likert-type scale with anchors at 1 (“very poor”) and 7 (“excellent”). Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Scale sum scores are transformed such that a high score on the functional scales represents a high/healthy level of functioning, a high score on the symptom scales represents a high level of symptomatology, and a high score on the global health status/HRQoL scale represents a high HRQoL.34
According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least half of the items. The values for missing items are interpolated with the average of the respondent-completed items.34
Assessment of Validity and Reliability
In its initial development, the EORTC QLQ-C30 underwent an evaluation of its psychometric properties and demonstrated reliability and validity in cancer patients in an international field trial of 305 patients in 13 multicultural clinical research settings.33 A revision of the EORTC QLQ-C30 was undertaken to improve low internal consistency, content validity for the role functional scale, and a conceptual difficulty (undue emphasis on physical function in the global HRQoL scale).35 The original and new versions were applied in a total of 1,181 patients with cancer in Canada and the Netherlands. Internal consistency improved for the role functional scale in the new version (Cronbach alpha ranging from 0.78 to 0.88), and substitution of the new item for the previous version did not alter internal consistency (Cronbach alpha ranging from 0.81 to 0.92.35
The EORTC QLQ-C30 (version 3.0) is the version currently in use and was used in Study 1620. Version 3.0 differs from the previous version 2.0 in that the number of response options for the first 5 items of the questionnaire comprising the physical function scale was increased from 2 options (yes/no in version 2.0) to 4 options (not at all, a little, quite a bit, very much). Internal consistency, reliability, construct validity, criterion validity, and responsiveness of the EORTC QLQ-C30 version 3.0 was assessed in 622 patients with head and neck cancer from 12 countries. Version 3.0 was more reliable than previous versions.36 Internal consistency of the multi-item scales was assessed using Cronbach alpha, with a value of 0.70 being considered adequate.37 The internal consistency of the new physical function scale of the EORTC QLQ-C30 version 3.0 was 0.84 compared with 0.66 in version 1.0. The EORTC QLQ-C30 version 3.0 was able to discriminate between head and neck cancer patients who were disease-free, who were newly diagnosed, and who had recurrent disease. As well, differences were noted between patients with different stages of disease and according to Karnofsky performance status (KPS); the new scale had a stronger association with KPS. Furthermore, there was a strong correlation observed between scores on the EORTC QLQ-C30 version 3.0 and symptom/treatment toxicity scores. Responsiveness to change was assessed using the standardized response mean (SRM), with an SRM of 0.20 considered small, 0.50 considered medium, and 0.80 considered large. The changes in the scores of the QLQ-C30 demonstrated a small to medium SRM in response to treatment over time with scores mostly changing between 5 and 10 points.36
In patients with NMSC, a prospective study conducted between May and December 2010 among 172 patients in Germany assessed the psychometric properties of the EORTC QLQ-C30 questionnaire. The reliability/internal consistency was assessed using Cronbach alpha coefficient. The validity of the EORTC QLQ-C30 scale was evaluated in 3 ways – by using item/scale correlations, by conducting inter-scale correlations within the EORTC QLQ-C30 scales as well as between the EORTC QLQ-C30 and the validated Dermatology Life Quality Index (DLQI) scale,38-40 and by using known-group comparisons.41
The Cronbach alpha coefficient for the multi-item EORTC QLQ-C30 scale was between 0.71 and 0.93, demonstrating an adequate reliability/internal consistency for all items except cognitive functioning, which contained a value of 0.68, just below the accepted cut-off value of 0.70. Convergent validity was obtained in 8 out of 9 multi-item scales (r > 0.40) with an exception for the physical functioning scale (r = 0.32). The scaling success was defined as the correlation between an item and its respective scale not being significantly lower than the correlation between that specific item and another scale. The scaling success rate for discriminant validity was demonstrated in 100% of the item/scale correlations, thus supporting the hypothesized scale structure of the EORTC QLQ-C30. One-third of inter-scale correlations within the EORTC QLQ-C30 were found substantial (r > 0.40), whereas the range of r varied from 0.01 to 0.70. Substantial correlations (r > 0.40) were observed between the DLQI total score and EORTC QLQ-C30 subscales (role, emotional, social functioning) as well as with global quality of life (QoL), the range of r being 0.16 to 0.49. Overall, the inter-scale correlations between DLQI and EORTC QLQ-C30 indicated a conceptually related but different aspects of the QoL construct. While evaluating the clinical validity of the EORTC QLQ-C30 scale using known-group comparisons, patients were divided into 3 subgroups: mild, moderate, and severe condition. To test the statistical significance of group differences, the Kruskal-Wallis H-test was used. Ten out of 15 scales showed patients with more severe conditions reported lower functioning and higher symptom scores compared with patients with less severe conditions. The EORTC QLQ-C30 significantly differentiated between clinically distinct patient groups, demonstrating that severe clinical conditions were associated with greater impairment in physical, role, and cognitive functioning (P ≤ 0.030). Overall, the clinical validity was demonstrated through the responsiveness of the EORTC QLQ-C30 scale to various health conditions. The results of this study demonstrated the EORTC QLQ-C30 scale as a reliable and valid tool for measurement of QoL among NMSC patients.41
Minimal Important Difference
No MID for BCC has been found for the EORTC QLQ-C30 scale.
For use in clinical trials, scores on the EORTC QLQ-C30 can be compared between groups of patients or within a group of patients over time. One study conducted in breast cancer and small-cell lung cancer patients in 1998 estimated a clinically relevant change in score on any scale of the EORTC QLQ-C30 to be 10 points.42 The estimate was based on a study that used an anchor-based approach to estimating the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported being “very much” changed had corresponding changes of more than 20 points.42
More recently in 2015, a Canadian study estimated the MIDs of EORTC QLQ-C30 scales using data from 193 newly diagnosed breast and colorectal cancer patients.43 The Supportive Care Needs Survey-Short Form-34 (SCNS-SF34) was used as an anchor; mean changes in EORTC QLQ-C30 scales associated with improvement, worsening, and no-change in supportive care based on the SCNS-SF34 was then calculated. MIDs were assessed for the following scales: Physical function, role function, emotional function, global health/QoL (i.e., GHS), pain, and fatigue. For improvement, MIDs associated with a statistically significantly improved supportive care needs ranged from 10 to 32 points. For worsening, MIDs associated with a statistically significantly worsening of supportive care needs ranged from 9 to 21 points. The range for unchanged supportive care needs was from 1-point worsening to 16-point improvement in EORTC QLQ-C30 score.43 Based on this, the authors suggested a 10-point change in EORTC QLQ-C30 score represented changes in supportive care needs, and therefore should be considered for clinical use.43
In 2014, another Canadian study estimated the MID for EORTC QLQ-C30 in 369 patients with advanced cancer, who completed the questionnaire at baseline and 1 month post radiation.44 The most common cancer type was breast cancer, followed by lung, prostate, gastrointestinal, renal cell, and others. The MID was estimated using both anchor and distribution-based methods for improvement and deterioration. Two anchors of overall health and overall QoL were used, both taken directly from the EORTC QLQ-C30 (questions 29 and 30) where patients rated their own overall health and QoL. Improvement and deterioration were categorized as an increase or decrease by 2 units to account for the natural fluctuation of patient scoring. Based on these anchors, the estimated MIDs across all EORTC QLQ-C30 scales ranged from 9.1 units to 23.5 units for improvement, and from 7.2 units to 13.5 units for deterioration. Distribution-based estimates were closest to 0.5 SD.44
Skindex-16
Description and Scoring
The Skindex-16 questionnaire is a generic HRQoL instrument that, according to the developers, can be used with skin diseases of any sort. The questionnaire is self-administered and is intended for an adult population. Skindex-16 consists of 16 items with a recall period of 4 weeks. Skindex-16 has 3 scales that address symptoms (4 items), emotions (7 items), and functioning (5 items). Using a continuous bipolar scale anchored by 7 boxes with the words “never bothered” and “always bothered” at each end, item scores are transformed to a linear scale (0 to 100, with 0 representing never bothered and 100 representing always bothered).45 Scale scores are calculated as the average of the transformed item scores. A total score is calculated as the average of all 16 items.
Skindex-16 was developed in 2001 by Chren et al. (2001).46 The Skindex-16 questionnaire is a refined version of the original 65-item Skindex and the 29-item Skindex-29.45 Skindex-16 includes additional items that were not included in Skindex-29 as well as the items that had the best performance in the previous versions.46
Assessment of Validity and Reliability
The validation study assessed the questionnaire among 692 patients waiting for dermatology appointments in clinics between 1996 and 1997. Reliability, reproducibility, validity, and responsiveness of the Skindex-16 questionnaire were determined. Internal-consistency reliability was assessed using Cronbach coefficient, alpha, whereas reproducibility was determined using Pearson's correlation coefficient. Construct validity was determined both clinically and psychometrically. Patients with inflammatory dermatoses (e.g., psoriasis or eczema) were hypothesized to show higher scale scores than patients with isolated lesions (e.g., benign growths or NMSC). Then Wilcoxon rank sum test was used to compare scale scores among these groups. Moreover, exploratory principal axes factor analysis was conducted along with an oblique rotation to identify the factor structure underlying patients' responses to the Skindex-16 scale. For content validity evaluation, responses of patients were examined using an open-ended question, "What is it about your skin problem that bothers you the most?" Lastly, to assess the responsiveness of patients to clinical change, paired t-tests was used to compare any change in scale scores among the 3 patient groups – patients who responded having improvement in their overall skin condition, patients whose condition remained unchanged, or patients whose condition deteriorated since last response time.46
While assessing the psychometric properties of Skindex-16 scales, a high level of internal consistency was achieved, with alpha corresponding to 0.86, 0.93, and 0.92 for the symptoms, emotions, and functioning scales, respectively. The Skindex-16 scale scores demonstrated high reproducibility after 72 hours, with a coefficient of 0.90, 0.89, and 0.88 for the symptoms, emotions, and functioning scales, respectively. Furthermore, the instrument demonstrated content and construct validity as items in the questionnaire captured information from the open-ended question. For patients who reported that their skin had improved or remained the same, mean scale sores were consistent with these changes.46
HRQoL outcomes had been reported in another study titled STEVIE (NCT01367665), a phase II, open-label study assessing safety of vismodegib among patients with mBCC or laBCC who showed unsuitability for surgery or radiotherapy. To assess the HRQoL, Skindex-16 questionnaire was completed by patients at baseline and at 3 subsequent visits.47 Good internal consistency (alpha = 0.775 to 0.936) was demonstrated while assessing the validity of the Skindex-16 through a pre-specified exploratory analysis during the STEVIE trial after 1 year of treatment.47,48 However, negligible effect of the indication on QoL was revealed in the STEVIE trial, with a lack of specificity and low baseline scores in the Skindex-16 scale.47 The limitation of the Skindex-16 scale to capture all BCC-specific effects among patients with NMSC or BCC were also evident in other studies.46,49
Minimal Important Difference
In a prospective cohort study of 633 patients with cutaneous BCC and squamous cell carcinoma (NMSC), diagnosed in 1999 and 2000 and followed for 2 years after treatment, tumour-related QoL outcomes of 3 therapies were measured and compared after 1 to 2 years of therapy using the Skindex-16. To determine the MID in 485 patients from the cohort, Skindex-16 scores were computed before and 1 week after corresponding treatment and were compared with patients’ responses to a global question with 7 response options showing how they were bothered from the skin cancer.50 The assumption to compute minimal clinically meaningful difference was that a difference in 1 response option to the global question would correspond to the MID.51-54 Fisher’s exact test was used to evaluate differences between treatment groups for dichotomous variables, whereas the chi-square test was used for categorical variables and analysis of variance was used for continuous variables. Change in tumour-related QoL after treatment was defined as the average difference between the baseline subscale score, and the subscale scores at 12, 18, or 24 months for each Skindex-16 subscale and each patient. Multivariate models were used to demonstrate changes in tumour-related QoL after corresponding treatment, adjusted for any prior treatment features; and propensity scores were used to adjust for differences between patients that would correspond to the choice of treatment.55,56 Propensity scores were derived from a logistic regression model. Lastly, changes in Skindex-16 scores were compared in the groups of matched pair patients created on the basis of the propensity scores. Based on this analysis, any changes for improvement or deterioration from the skin cancer were similar for different treatments, and the MID among NMSC patients for all Skindex-16 subscales was determined to be 10 points in this study. Clinically meaningful results were defined as a difference from baseline in any domain scores of 10 points or greater.50
Clinically meaningful difference also has been assessed in the phase II, open-label study STEVIE (NCT01367665).47 In this study the safety of vismodegib among patients with mBCC or laBCC who were unsuitable for surgery or radiotherapy was assessed. Based on the definition of clinically meaningful result,50 a clinically meaningful improvement was observed among 730 patients with laBCC in emotional well-being at each time point (median change from baseline measured: −11.9 at first visit; −21.4 at second visit; and −17.9 at last visit), but no clinically meaningful improvement or deterioration in symptom and functional scores was shown. No clinically meaningful changes were observed among 10 patients with mBCC in any domain scores of Skindex-16 at any time points. While comparing patients with and without clinically meaningful improvements during the study, grouped according to their tumour response, it was observed that there were more patients with partial or complete tumour responses among the clinically meaningful improvement group than those without a meaningful improvement, suggesting an association between treatment effectiveness and improvement in QoL.47
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BCC
basal cell carcinoma
BIA
budget impact analysis
BSC
best supportive care
ECOG
Eastern Cooperative Oncology Group
GP
general practitioner
HHI
hedgehog pathway inhibitor
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
laBCC
locally advanced basal cell carcinoma
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Cemiplimab (Libtayo), sterile solution for IV infusion |
Submitted price | Cemiplimab, 350 mg vial: $8,200 |
Indication | For the treatment of patients with locally advanced basal cell carcinoma previously treated with a hedgehog pathway inhibitor |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review pathway |
NOC date | October 26, 2021 |
Reimbursement request | As per indication |
Sponsor | Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc. |
Submission history |
|
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Adult patients with locally advanced BCC previously treated with a hedgehog pathway inhibitor, consistent with the reimbursement request |
Treatment | Cemiplimab |
Comparator | Best supportive care in the context of palliative care (no active therapy, palliative radiotherapy, wound management, and physician visits) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (35 years) |
Key data source |
|
Submitted results | ICER = $61,738 per QALY for cemiplimab vs. BSC (incremental costs: $207,123; incremental QALYs: 3.35) |
Key limitations |
|
CADTH reanalysis results |
|
AE = adverse event; BCC = basal cell carcinoma; BSC = best supportive care; GP = general practitioner; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; WTP = willingness to pay.
Conclusions
The CADTH Clinical Review noted that the potential benefit associated with receiving cemiplimab with regard to objective response, defined as lesion reduction, is highly uncertain. Similarly, CADTH notes that the sponsor’s economic model is based on progression-free survival (PFS) and overall survival (OS) data, and there is no existing evidence to inform the relative benefit of cemiplimab compared with best supportive care (BSC). Therefore, the extent of clinical benefit associated with cemiplimab is highly uncertain and the relative impact of cemiplimab compared with BSC is unknown.
CADTH identified several limitations with the sponsor’s economic evaluation that involved additional survival benefit, OS and PFS extrapolation, resource costs of progressive disease, wound dressings, and disutilities. CADTH was unable to derive a base case due to considerable uncertainty regarding comparative clinical benefit alongside a highly uncertain model structure. As an exploratory reanalysis, CADTH used a Weibull parametric function to extrapolate OS and assumed a similar OS for both arms, used a gamma parametric function to extrapolate PFS, reduced the frequency of post-progression health care visits, increased the frequency of wound dressings, and applied disutilities annually. Based on the CADTH exploratory reanalysis, cemiplimab was associated with an incremental cost-effectiveness ratio (ICER) of $2,259,421 per quality-adjusted life-year (QALY) and the probability of cost-effectiveness at a willingness-to-pay (WTP) threshold of $50,000 per QALY was 0%. A price reduction of 97% is necessary to achieve cost-effectiveness at this threshold.
The cost-effectiveness of cemiplimab is driven by assumptions around the extrapolation of OS, the utility benefit from tumour reduction, and the potential cost savings due to the reduced frequency of wound dressings. CADTH conducted a scenario analysis with alternate utilities for the post-progression state based on the objective response rate from Study 1620, which resulted in an increased ICER of $3,331,586 per QALY. CADTH was unable to evaluate the disutilities and costs associated with several immune-related and treatment-emergent adverse events (AEs) that were measured in Study 1620, as they were excluded by the sponsor in the economic model. Given that most patients do not experience a treatment response with cemiplimab, net benefit may be negative in the presence of treatment-related AEs and cemiplimab could be dominated (more costly, less effective than BSC). Consequently, a price reduction of even 100% would not make cemiplimab cost-effective. Crucially, the exploratory analyses conducted by CADTH still assume a net benefit of receiving cemiplimab relative to BSC; this is highly uncertain, given the clinical evidence presented. CADTH could not fully explore this uncertainty, given the model structure; therefore, the possibility that cemiplimab generates fewer QALYs at a higher cost than BSC at any price reduction should be seriously considered.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient input from the Melanoma Network of Canada (MNC) and the Save Your Skin Foundation (SYSF), 2 national-level advocacy and support organizations that are dedicated to skin cancer. The MNC conducted an online survey of 62 patients and 45 caregivers, with almost all respondents residing in Canada. One patient indicated they were currently being treated for metastatic disease with cemiplimab. A total of 77% of respondents underwent surgical excision, with others receiving Mohs surgery, cryosurgery, reconstructive surgery, radiation therapy, or topical creams. No patients reported undergoing chemotherapy or being treated with the hedgehog pathway inhibitor (HHI) vismodegib. SYSF conducted online surveys, patient roundtables, and individual interviews of 23 individuals with basal cell carcinoma (BCC), with the majority also from Canada. Five patients were undergoing treatment with cemiplimab. Respondents reported undergoing chemotherapy, surgery, radiation therapy, or Mohs surgery. Side effects of current treatments include inability to work, pain and scarring, nerve damage, and emotional distress. Patients noted they would like cemiplimab to delay disease progression and recurrence while reducing the associated pain, scarring, and disfigurement. Cemiplimab would address the lack of available, effective treatments for advanced BCC. Patient experience with the drug noted that side effects included liver problems, fatigue, skin rash, and flu-like symptoms.
CADTH received registered clinician input from the Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee. The clinicians stated there is currently no standard of care for the second-line treatment of advanced BCC and that patients may either reattempt HHI, if they had not progressed on HHI previously, or receive palliative treatment. Given the lack of other therapy options, cemiplimab would become the standard of care after HHI intolerance or failure.
CADTH received drug plan input expressing interest in patients ineligible for treatment with HHI or with an Eastern Cooperative Oncology Group (ECOG) performance status that is different from the ECOG scores in Study 1620 and whether they could also receive cemiplimab. They also highlighted that the IV infusion administration method would require adequate supplies and that temperature-controlled storage requirements might be of concern to pharmacies. Serious immune-mediated reactions due to treatment were also of concern to the drug plans, and ensuring access to a treatment centre to ensure early diagnosis and appropriate management should be further discussed. Finally, the plans noted there is an existing pan-Canadian Pharmaceutical Alliance letter of intent for the metastatic cutaneous squamous cell carcinoma indication.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model compared cemiplimab with BSC following HHI intolerance or failure.
In addition, CADTH addressed some of these concerns, as follows:
CADTH increased the market share of cemiplimab in the budget impact analysis (BIA) to reflect clinical expert feedback suggesting that cemiplimab would become standard of care.
CADTH was unable to address the following concerns raised in the stakeholder input:
Disutilities due to several immune-mediated or treatment-emergent reactions following administration of cemiplimab were not modelled.
The exploration of patients ineligible for HHI therapies and those with an ECOG performance status different from that in Study 1620 was not possible in the model.
Economic Review
The current review is for cemiplimab (Libtayo) for patients with locally advanced BCC (laBCC) previously treated with an HHI.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of cemiplimab compared with BSC for the treatment of patients with laBCC previously treated with an HHI. The model population comprised patients from the single-arm trial Study 1620 receiving cemiplimab and patients receiving BSC from the retrospective observational study, Cowey et al.1-3 The target population aligns with the Health Canada–indicated population and reimbursement request.
Cemiplimab is available in 7 mL single-use vials containing sterile solution at 50 mg/mL for IV infusion. The recommended dose of cemiplimab is 350 mg administered intravenously over 30 minutes on day 1 of a 21-day cycle until disease progression, unacceptable toxicity, or completion of planned treatment, up to 93 weeks.4 Cemiplimab is intended to be used as a second-line therapy for patients with laBCC who have previously received an HHI. The cost for cemiplimab is $8,200 per 350 mg vial and the 21-day cost is $8,200, as calculated by CADTH (Table 8).5
The comparator for this economic analysis is BSC, based on patients from Cowey et al. who discontinued first-line treatment with an HHI due to disease progression, toxicity, or lack of complete response.2,3 These patients received routine supportive care and no active therapy following HHI discontinuation.4 No drug acquisition or administration costs were included for BSC.
The cost per cycle is $8,200 for cemiplimab based on the treatment cycle of 21 days.5 An administration cost of $105 per cycle was also applied based on the total fixed IV administration costs of cemiplimab.4 Wastage costs were not incorporated because a flat dose is used for cemiplimab.
Outcomes modelled included QALYs and life-years over a lifetime time horizon of 35 years. The base-case analysis was conducted from the Canadian public health care system with costs and outcomes discounted at 1.5%. The cycle length was weekly with a half-cycle correction.
Model Structure
The sponsor submitted a partitioned survival model that consists of 3 mutually exclusive health states: pre-progression, post-progression, and death. Patients receiving cemiplimab enter the model in the pre-progression health state where they receive treatment and are either stable or responding to therapy. These patients can then transition directly to the death state or to the post-progression state where they remain until they transition to the death state. However, patients receiving BSC are assumed to enter the post-progression state where they remain until they transition to the death state. The proportion of patients in the pre-progression state is estimated by the PFS curve of the cemiplimab treatment arm from Study 1620, where progression is defined as recurrent or progressive disease, as per photographic or radiographic evidence noted by a clinician.1 The proportion of patients in the post-progression health state follows the OS curves from Study 1620 and Cowey et al. for patients receiving cemiplimab and BSC, respectively. A figure of the sponsor’s model structure can be found in Appendix 3 (Figure 1).
Model Inputs
The population used for this model comprised patients with laBCC and was derived from Study 1620 (n = 84) and Cowey et al. (n = 15).1-3 For Study 1620, the patients had a mean body mass index of 26.17 kg/m2 and 67% were male. The mean age was 69.1 For Cowey et al., 14 patients had laBCC and 1 had metastatic BCC. 80% of patients were male and the median age was 80.2,3 All patients included in the model discontinued prior treatment with HHI therapy.
The sponsor used parametric modelling to extrapolate the PFS and OS data from Study 1620 and OS data from Cowey et al. PFS was not included for the BSC arm. For PFS, a log-normal distribution was selected for the cemiplimab arm based on best statistical fit, visual inspection, and clinical expert advice. Exponential distributions were selected for both BSC and cemiplimab OS due to visual inspection and clinical expert advice. The sponsor claimed that the probability of death in those receiving cemiplimab was relatively constant over time. The PFS rate was capped by the OS rates and the extrapolation of OS was capped by the general mortality rate in Canada.6 Patients in Study 1620 received cemiplimab until disease progression, unacceptable toxicity, or completion of planned treatment, up to 93 weeks. Treatment duration for cemiplimab was based on Kaplan–Meier curves and was capped at 93 weeks, with the option to extrapolate data past 93 weeks using an exponential distribution.
The dose of cemiplimab used in the model is consistent with the description in the Overview section, being based on Study 1620 and the product monograph.1,5
Health-related quality of life (HRQoL) data were collected from Study 1620 for the pre-progression and post-progression health states using the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. These scores were then mapped to the EuroQol 5-Dimensions 5-Levels questionnaire. The pre-progression and post-progression utilities were calculated to be ||||||||||| and ||||||||||, respectively.1 Disutilities due to grade 3 and 4 AEs were incorporated as utility decrements ranging from −0.218 to −0.000 based on duration of the AE, which were derived from the Ontario Case Costing Initiative.4 These AEs included blood pressure increase, colitis, fatigue, hypertension, hypokalemia, urinary tract infection, visual impairment, and weight decrease.4
All costs used in the model, except drug costs and selected pre-progression routine costs, were inflated to 2021 Canadian dollars. Drug acquisition costs included the cost per 350 mg vial of cemiplimab, and IV administration costs included a fixed cost per administration of $105.7 Costs of subsequent treatments were not included due to lack of further options for the laBCC patient population. The model included resource use and costs associated with routine care, such as $157 per oncologist visit, $72 per dermatology visit, $84 per general practitioner (GP) visit, $53 for pre-progression blood test treatment monitoring, $215 for palliative radiotherapy, and $680 for complex palliative radiotherapy. These costs were all sourced from the Ontario Ministry of Health and Long-Term Care Schedule of Benefits.8 The cost of wound management was also included at $208, as sourced from the Registered Nurses’ Association of Ontario and assumptions.4 Costs for managing grade 3 and 4 AEs as described in the Overview section were sourced from the Ontario Case Costing Initiative, ranging from $703 to $1,193.9 Lastly, a 1-time terminal-care cost of $35,898 was applied based on an average expenditure per patient for end-of-life care for other cancer locations.10
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
Cemiplimab was associated with incremental costs of $208,043 and 3.39 QALYs compared with BSC, resulting in an ICER of $61,314 per QALY gained (Table 3). Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Treatment | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
BSC | 96,638 | Reference | 2.22 | Reference | Reference |
Cemiplimab | 304,681 | 208,043 | 5.61 | 3.39 | 61,314 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The submitted analysis is based on publicly available prices for the comparator treatments and may not reflect confidential prices.
Source: Sponsor’s pharmacoeconomic submission.4
Sensitivity and Scenario Analysis Results
The sponsor conducted various sensitivity and scenario analyses involving extrapolation assumptions for OS and PFS, utility values, time horizon, discount rate, and costs of routine care. In these analyses, the ICER was most sensitive to extrapolation and survival benefit assumptions applied to OS in the BSC and cemiplimab arms, leading to an ICER of $74,655 and $77,578 per QALY, respectively. The ICER values across all scenario analyses conducted by the sponsor varied from $49,405 to $78,809.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications on the economic analysis:
No direct or indirect evidence comparing cemiplimab with BSC: Effectiveness concerning OS and PFS for cemiplimab was derived from Study 1620. This was a single-arm trial and therefore does not address what the current outcomes are for patients who receive BSC. The sponsor assumes 100% of patients who receive BSC start in the disease-progressed state, thus excluding PFS from the BSC cohort. For OS, the sponsor derives estimates from Cowey et al., an unadjusted retrospective cohort of 15 patients that also includes 1 patient with metastatic laBCC. The use of assumptions and unadjusted external data to inform the relative effects of cemiplimab compared with BSC introduces a high degree of uncertainty into the sponsor’s analyses. There is a complete absence of trial data available for those receiving BSC, which makes all comparative conclusions between cemiplimab and BSC highly tenuous.
CADTH could not address this limitation, but notes that the assumptions and evidence used to inform PFS and OS in the economic model likely bias results in favour of cemiplimab.
Inappropriate model structure: In the sponsor’s model, 100% of patients who receive cemiplimab start “pre-progression” and 100% of patients who receive BSC start “post-progression.” Progression in the trial is defined as “recurrent or progressive disease,” which could still occur in an untreated cohort. Therefore, it does not make sense for 100% of patients who receive BSC to start in this state. This means that patients who receive BSC start the model with the same outcomes as those who receive cemiplimab and develop new lesions or who have lesions that increase in size. Second, this model structure assumes an immediate impact of cemiplimab as soon as the model starts. This assumes that patients who respond to cemiplimab have their tumours shrink on day 1 of treatment initiation. In the clinical trial, the average time to response was 6 months, with no patients responding sooner than 2 months. In the clinical trial, 32% of patients who received cemiplimab achieved an objective response, defined as either a partial response (50% or greater reduction of lesions) or complete response (complete reduction of all lesions). This indicates there was no noticeable reduction in the tumour site for most patients. Therefore, it seems implausible that 100% of patients who receive cemiplimab begin the model in a state with better utility and lower costs relative to BSC.
Overall, the sponsor’s modelling approach is not granular enough to capture the nuances of the decision problem. Patients who receive cemiplimab either respond, experiencing tumour shrinkage, or do not respond and their tumour remains the same size. Those who do respond likely do not do so immediately but experience tumour shrinkage after a period of time. For those who respond, tumour size may return to pre-treatment levels. Finally, tumour size may increase, meaning the patient moves to a state worse than pre-treatment levels. For patients receiving BSC, tumour size will likely only increase or remain the same, as they are not on active therapy. The rate at which cancer progresses may also be different between the 2 arms. The benefit of cemiplimab is therefore when it results in patients experiencing a tumour shrinkage that lasts until treatment response wanes and when it potentially slows the rate the cancer progresses. Given the sponsor has no comparative evidence on progression, it would have been more appropriate to build a model around treatment response, as tumour shrinkage is something that is unlikely to occur in the BSC arm, since they are not on active therapy. Any benefit regarding delaying cancer progression is speculative and the sponsor has attributed the maximum possible benefit with regard to cancer progression by assuming progression occurs over time in cemiplimab patients, but occurs straight away for patients receiving BSC.
CADTH could not address this limitation, but notes that the sponsor’s model overestimates the benefits of cemiplimab associated with progression. A sensitivity analysis was conducted that reduced the incremental utility benefit of cemiplimab in line with objective response to treatment.
Overestimation of survival benefit attributed to treatment with cemiplimab and inappropriate extrapolation of OS beyond the Study 1620 trial period: The sponsor assumed a substantial survival benefit associated with receiving cemiplimab compared with BSC based on immature survival data from Study 1620 and Cowey et al. The sponsor assumed that the 10-year survival for patients receiving cemiplimab was 30% compared with 3% for patients receiving BSC. However, there is no existing evidence that suggests treatment with cemiplimab leads to an increased patient lifespan. Although there may be a benefit relative to PFS, clinical expert feedback suggested that progression itself should not be affecting survival for most patients (> 90%). The sponsor uses an unadjusted retrospective cohort of 15 patients to determine survival in patients receiving BSC. However, mortality is likely higher in the Cowey et al. cohort due to a higher mean age, which represents the BSC arm in the model. The differing performance status scores across the cemiplimab and BSC cohorts also attributed an implausible survival benefit to those receiving cemiplimab, since patients in Study 1620 had ECOG scores of either 0 or 1, as per the study inclusion criteria, whereas some patients had a missing or higher ECOG score of 2 or 3 in the Cowey et al. cohort. An inappropriate survival advantage may also be attributed to patients in Study 1620 due to a high proportion of screen failures (33 out of 165) that likely selected a healthier patient population more capable of tolerating treatment. Based on clinical expert feedback, there is not expected to be a survival benefit associated with cemiplimab and the main purpose of treatment is to reduce tumours and increase HRQoL.
In reanalysis, CADTH assumed that patients receiving cemiplimab would have similar OS compared with patients receiving BSC, and that the main benefit derived from cemiplimab is on patient HRQoL. Based on feedback from clinical experts, CADTH also applied a Weibull parametric function to extrapolate OS.
Extrapolation overestimates the delay of progression attributable to cemiplimab beyond the Study 1620 trial period: The sponsor extrapolated the PFS data from Study 1620 using a log-normal parametric function. This resulted in an estimate of 14% and 5% of patients remaining progression-free after 5 and 10 years, respectively, which was deemed improbable based on expert opinion.
In reanalysis, CADTH used a gamma parametric function to extrapolate PFS, based on feedback from clinical experts.
Sponsor determination of costs of progressive disease overestimates costs for patients receiving BSC: The sponsor assumes that patients who receive BSC will require additional dermatology, oncology, and GP visits, relative to those who receive cemiplimab. These assumptions do not align with feedback from the clinical experts consulted by CADTH. The sponsor assumes that patients receiving BSC begin in the post-progression state and will visit a dermatologist every 12 weeks for disease monitoring and visit a GP monthly for a check-up and comorbidity management. The sponsor also assumes that patients in the post-progression health state will see an oncologist every 6 weeks, which is more frequent than expected, according to clinical expert feedback. Clinical experts suggested that dermatologist visits are not necessarily needed for disease monitoring, which is managed by a nurse practitioner visit or equivalent that is already included in resource costs. Clinical experts also suggested that the frequency of GP visits would not differ between patients receiving cemiplimab and BSC. Finally, experts suggested that all patients in the post-progression health state would likely see an oncologist every 3 months, as they are no longer on active therapy.
CADTH assumed that patients will not require dermatologist visits for disease-monitoring purposes, that receiving cemiplimab does not influence the frequency with which patients visit a GP, and that all patients in the post-progression health state will see an oncologist every 3 months.
Underestimating the frequency of wound dressings required for patients receiving cemiplimab deflates resource costs: The sponsor assumes that 25% of patients receiving cemiplimab will have 1 wound dressing per week, with the rest requiring no wound dressings. For patients receiving BSC, the sponsor assumed that 1.5 wound dressings a week would be required. The clinical experts consulted by CADTH suggested that patients on cemiplimab who have not experienced an objective response, defined as either a partial response or complete response, would not require a different number of wound dressings compared with patients receiving BSC. Expert feedback suggested that those experiencing a partial response to treatment would likely experience a decrease from 3 wound dressings per week to 2, and that those experiencing a complete response would not require any wound dressings per week.
CADTH assumed that patients receiving cemiplimab who are progression-free will require 2 wound dressings a week if experiencing a partial response, while patients not experiencing any objective response will continue to receive 3 wound dressings a week, similar to patients in the post-progression health state. Identical assumptions are applied to wound-management visits with a registered nurse. Patients experiencing a complete response to treatment will not require wound dressings or wound-management visits.
Impact of AEs following treatment with cemiplimab is not fully captured: Several sponsor-identified immune-related or treatment-emergent AEs measured in Study 1620 that met the sponsor’s threshold for inclusion (occurring in at least 2% of patients) were not included in the accompanying economic evaluation submitted to CADTH. These AEs included grade 3 adrenal insufficiency, grade 3 abdominal pain, grade 3 hyponatremia, grade 3 infected neoplasm, and grade 3 supraventricular tachycardia.1 The sponsor also applies utility decrements based on a length of inpatient admission for each AE drawn from an external data source instead of using the AE durations from Study 1620. Furthermore, disutilities were applied for select grade 3 and 4 AEs only in the first cycle of the model. This underestimates the disutility associated with cemiplimab, since patients continue treatment for up to 93 weeks and may experience AEs throughout the treatment duration.
In its reanalysis, CADTH used the sponsor-provided option to apply disutilities annually for patients receiving cemiplimab until treatment discontinuation. CADTH could not address the limitations regarding the duration of AEs and exclusion of the select AEs measured in Study 1620 from the economic evaluation.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (See Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations of the Submission)
Sponsor’s key assumption | CADTH comments |
|---|---|
Re-treatment is excluded from the economic evaluation. | Uncertain. Re-treatment is not modelled, despite being included in the study protocol. One patient was re-treated in Study 1620. The potential impact of re-treatment on cost-effectiveness is unknown and should be explored to reflect the high discontinuation rates observed in Study 1620 that may lead to patients entering re-treatment after disease progression or recurrence. |
A gamma distribution was used to represent probabilistic uncertainty for drug acquisition and administration costs. | Not appropriate. These costs are not likely to vary and should not be included in probabilistic sensitivity analyses. |
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with the clinical experts. Changes to the sponsor’s analyses are summarized in Table 4 and include alterations to the OS extrapolation and survival assumptions, PFS extrapolation, resource costs related to progressive disease, frequency and costs of wound dressings, and applied disutilities due to AEs.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH exploratory reanalysis | ||
1. Parametric OS modelling |
|
|
2. Parametric PFS modelling | Log-normal function | Gamma function |
3. Costs of progressive disease | Patients in the post-progression health state:
|
|
4. Costs of wound dressings |
|
|
5. Disutilities associated with AEs | Utility decrement and costs for AEs are applied for the first cycle of the model | Utility decrement and costs for AEs are applied annually until treatment discontinuation |
CADTH base case | — | Reanalysis 1 + 2 + 3 + 4 + 5 |
AE = adverse event; GP = general practitioner; OS = overall survival; PFS = progression-free survival.
In the CADTH base case, cemiplimab was associated with a total cost of $310,220 and 3.14 QALYs compared with $164,624 and 3.08 QALYs for patients receiving BSC. The ICER for cemiplimab compared with BSC was $2,259,421 per QALY, with a probability of being cost-effective at a WTP of $50,000 of 0%. Detailed information and disaggregated results are presented in Table 11 in Appendix 4.
Table 6: Summary of the Stepped Analysis of the CADTH Exploratory Reanalysis Results
Stepped analysisa | Treatment | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 96,547 | 2.22 | Reference |
Cemiplimab | 303,671 | 5.57 | 61,738 | |
CADTH reanalysis 1: OS modelling | BSC | 119,976 | 3.08 | Reference |
Cemiplimab | 238,894 | 3.16 | 1,412,093 | |
CADTH reanalysis 2: PFS modelling | BSC | 96,547 | 2.22 | Reference |
Cemiplimab | 316,032 | 5.54 | 65,988 | |
CADTH reanalysis 3: Resource costs | BSC | 90,694 | 2.22 | Reference |
Cemiplimab | 294,036 | 5.57 | 60,611 | |
CADTH reanalysis 4: Wound dressing costs | BSC | 134,819 | 2.22 | Reference |
Cemiplimab | 424,650 | 5.57 | 86,391 | |
CADTH reanalysis 5: Disutilities for AEs | BSC | 96,547 | 2.22 | Reference |
Cemiplimab | 303,811 | 5.57 | 61,783 | |
CADTH base case (reanalysis 1 + 2 + 3 + 4 + 5) | BSC | 164,624 | 3.08 | Reference |
Cemiplimab | 310,220 | 3.14 | 2,259,421 |
AE = adverse event; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH performed price-reduction analyses based on the sponsor base case and CADTH base-case reanalysis. Based on the CADTH base case, a price reduction of approximately 97% would be required to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY (Table 7).
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for cemiplimab vs. BSC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 61,314 | 2,259,421 |
10% | 56,686 | 2,030,699 |
20% | 52,058 | 1,801,978 |
30% | 47,430 | 1,573,256 |
40% | 42,802 | 1,344,535 |
50% | 38,175 | 1,115,813 |
60% | 33,547 | 887,092 |
70% | 28,919 | 658,371 |
80% | 24,291 | 429,649 |
90% | 19,663 | 200,928 |
97% | 16,424 | 40,823 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CADTH performed a scenario analysis to determine the impact of alternative assumptions on the cost-effectiveness of cemiplimab. A weighted average for the utility value for patients in the post-progression health state was applied based on the objective response rate from Study 1620 for patients with laBCC.
The results of these analyses are presented in Table 12 of Appendix 4. The scenario analysis using alternate utilities for patients in the post-progression health state, resulted in an ICER of $3,331,586 per QALY.
Issues for Consideration
First-line treatment with sonidegib, an HHI, is not recommended for reimbursement by public drug plans in Canada.11 Canadian patients with laBCC receiving HHI therapy likely are treated with vismodegib, the sole HHI therapy that is reimbursed in Canada.12 The cost-effectiveness of cemiplimab in populations that received multiple lines of HHI therapy in Canada is therefore unknown.
The clinical experts consulted by CADTH noted that cemiplimab could be administered to patients for periods longer than 93 weeks, depending on the prescribing physician. OS, PFS, and objective response rate are uncertain beyond the 93-week treatment course and for patients entering re-treatment that previously experienced recurrent or progressive disease following discontinuation of cemiplimab. The cost-effectiveness of cemiplimab beyond 93 weeks and in re-treatment is unknown.
The clinical experts consulted by CADTH suggested that select patients on HHI therapy may experience early intolerance and may switch to early second-line treatment with cemiplimab. The impact of early intolerance on the cost-effectiveness of cemiplimab is unknown.
Cemiplimab has been previously reviewed and recommended to be conditionally reimbursed in patients with metastatic or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation only if cost-effectiveness is improved to an acceptable level.13 The submitted price of $8,200 per 350 mg single-dose vial is identical to that of the laBCC indication.13
The cost-effectiveness of cemiplimab in the metastatic setting is unknown. Although the sponsor provided a scenario analysis to explore this, there was too much uncertainty to provide a meaningful analysis, given the model structure and assumptions. The sponsor predicts cost-effectiveness to be worse in the metastatic setting due to a shorter life expectancy and, therefore, less opportunity to benefit.
Overall Conclusions
The CADTH Clinical Review noted that the potential benefit associated with receiving cemiplimab with regard to objective response, defined as lesion reduction, is highly uncertain. Similarly, CADTH notes that the sponsor’s economic model is based on PFS and OS data, despite the lack of existing evidence to inform the relative benefit of cemiplimab compared with BSC. Therefore, the extent of the clinical benefit associated with cemiplimab is highly uncertain and the relative impact of cemiplimab compared with BSC is unknown.
CADTH identified several limitations with the sponsor’s economic evaluation that involved additional survival benefit, OS and PFS extrapolation, resource costs of progressive disease, wound dressings, and disutilities. CADTH was unable to derive a base case due to considerable uncertainty regarding comparative clinical benefit alongside a highly uncertain model structure. As an exploratory reanalysis, CADTH used a Weibull parametric function to extrapolate OS and assumed similar OS for both arms, used a gamma parametric function to extrapolate PFS, reduced the frequency of post-progression health care visits, increased the frequency of wound dressings, and applied disutilities annually. Based on the CADTH exploratory reanalysis, cemiplimab was associated with an ICER of $2,259,421 per QALY and the probability of cost-effectiveness at a WTP threshold of $50,000 per QALY was 0%. A price reduction of 97% is necessary to achieve cost-effectiveness at this threshold.
The sponsor’s economic evaluation is driven by the assumed survival benefit of cemiplimab, OS extrapolation assumptions, and the frequency of wound dressings. CADTH conducted a scenario analysis with alternate utilities for patients in the post-progression health state that resulted in an increased ICER of $3,331,586 per QALY. The alternate utilities were derived by adjusting the post-progression utility value with a weighted average, based on the objective response rate from Study 1620, such that only those who responded to treatment experienced an increase in quality of life. CADTH was unable to evaluate the disutilities and costs associated with several grade 3 immune-related and treatment-emergent AEs that were measured in Study 1620 but excluded by the sponsor in the economic model, such as adrenal insufficiency, abdominal pain, hyponatremia, infected neoplasm, and supraventricular tachycardia. CADTH also could not address the limitations with the sponsor’s model structure and assumptions about disease progression, such as the sponsor’s assumption that all patients receiving BSC begin in the disease-progressed state while all patients receiving cemiplimab begin in the progression-free state. Crucially, the exploratory analyses conducted by CADTH still assume a net benefit of receiving cemiplimab relative to BSC; this is highly uncertain, given the clinical evidence presented. CADTH could not fully explore this uncertainty, given the model structure and, therefore, the possibility that cemiplimab generates fewer QALYs at a higher cost than BSC at any price reduction should be seriously considered.
References
1.Clinical Study Report. Protocol No. R2810-ONC-1620. A phase 2 study of REGN2810, a fully human monoclonal antibody to programmed death-1, in patients with advanced basal cell carcinoma who experienced progression of disease on Hedgehog pathway inhibitor therapy, or were intolerant of prior Hedgehog pathway inhibitor therapy [internal sponsor's report]. Tarrytown (NY): Regeneron Pharmaceuticals, Inc.; 2020 Aug 21.
2.Cowey CL, Chen C-I, Aguilar KM, et al. Outcomes in patients (pts) with advanced basal cell carcinoma (aBCC) who discontinued hedgehog inhibitors (HHI) as first-line (1L) systemic treatment (Tx) in a U.S. community oncology setting: A retrospective observational study [meeting abstract]. J Clin Oncol. 2021;39(15_suppl):e18742-e18742.
3.Cowey CL, Chen C-I, Aguilar KM, et al. Frequency, characteristics, and subsequent treatment (Tx) of real-world patients (pts) who discontinue hedgehog inhibitors (HHIs) as first-line (1L) systemic Tx for advanced basal cell carcinoma (aBCC) [meeting abstract]. J Clin Oncol. 2021;39(15_suppl):e18740-e18740.
4.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: LIBTAYO (cemipliumab), 350 mg every three weeks as an intravenous (IV) infusion. Mississauga (ON): Sanofi Genzyme; 2021 Aug 19.
5.LIBTAYO (cemiplimab for injection): 50mg/mL solution for infusion; antineoplastic agent, monoclonal antibody [product monograph]. Laval (QC): Sanofi-Aventis Canada Inc.; 2021 Nov 9.
6.Statistics Canada. Deaths and mortality rates, by age group (Table: 13-10-0710-01). 2019; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310071001. Accessed 2021 Aug 28.
7.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/. Accessed 2021 Dec 15.
8.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Dec 15.
9.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Dec 15.
10.Walker H, Anderson M, Farahati F, et al. Resource use and costs of end-of-Life/palliative care: Ontario adult cancer patients dying during 2002 and 2003. J Palliat Care. 2011;27(2):79-88. PubMed
11.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: Sonidegib (odomzo) for basal cell carcinoma. Ottawa (ON): CADTH; 2021: https://cadth.ca/sonidegib-odomzo-basal-cell-carcinoma-details. Accessed 2021 Dec 15.
12.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: Vismodegib (erivedge) for advanced BCC. Ottawa (ON): CADTH; 2014: https://www.cadth.ca/vismodegib-erivedge-advanced-bcc-details. Accessed 2021 Dec 15.
13.pan-Canadian Oncology Drug Review Committee (pERC) final recommendation: cemiplimab (Libtayo) for cutaneous squamous cell carcinoma. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/cemiplimab-libtayo-cutaneous-squamous-cell-carcinoma-details. Accessed 2021 Dec 15.
14.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: LIBTAYO (cemipliumab), 350 mg every three weeks as an intravenous (IV) infusion. Mississauga (ON): Sanofi Genzyme; 2021 Aug 19.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Treatment of Patients With laBCC Previously Treated With an HHI
Treatment | Strength/ concentration | Form (vial size if single use) | Price | Recommended dosagea | Daily cost | 21-day cost |
|---|---|---|---|---|---|---|
Cemiplimab | 50 mg/mL | 350 mg vial / 7 mL sterile solution for IV infusion | 8,200.0000b | 350 mg every 21 days | 390.48 | 8,200 |
HHI = hedgehog inhibitor; laBCC = locally advanced basal cell carcinoma.
Note: All prices are from the sponsor’s pharmacoeconomic submission,4 unless otherwise indicated, and do not include dispensing fees.
a The recommended dosages are from the respective product monographs.5
b Sponsor-submitted price.4
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | CADTH identified inflexibilities in the sponsor’s model where certain calculations, such as the application of resource costs, which were not modifiable based on treatment arm. |
Model structure is adequate for decision problem | No | The model structure assumes 100% of patients benefit from receiving cemiplimab. There is a disconnect between trial evidence and how this relates to potential incremental benefit of cemiplimab versus BSC. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | CADTH identified limitations with the exclusion of PFS for all patients receiving BSC, the use of an external data source for parameters pertaining to BSC, and the exclusion of immune-related AEs due to treatment. See CADTH appraisal and assumptions section. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Probabilistic uncertainty was applied to costs of drug acquisition and administration, which are typically excluded. The model continued to assume OS benefit with cemiplimab when probabilistic iterations were run. See CADTH assumptions section. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Disaggregated results for discounted life-years pre-progression and post-progression are not available in the sponsor’s submitted economic model. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
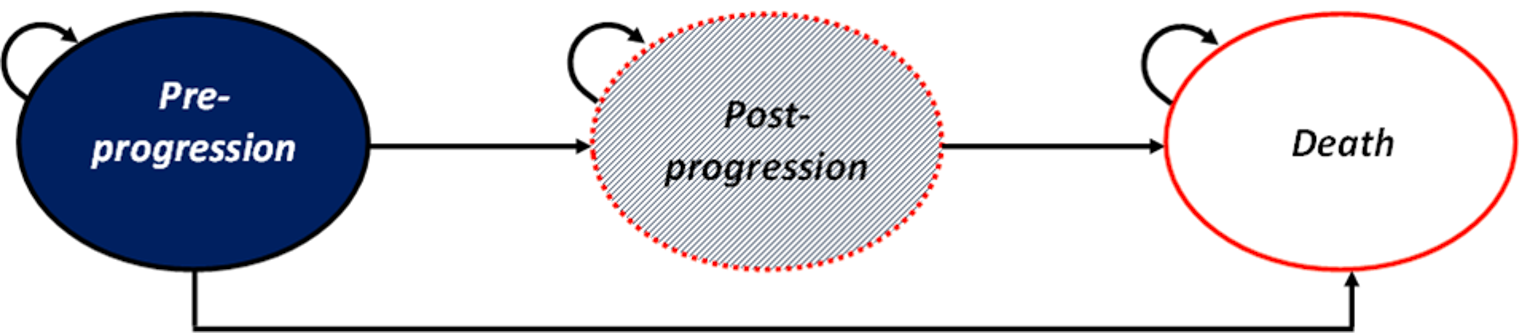
Figure 2: Sponsor Base Case PFS Extrapolation for Cemiplimab and BSC
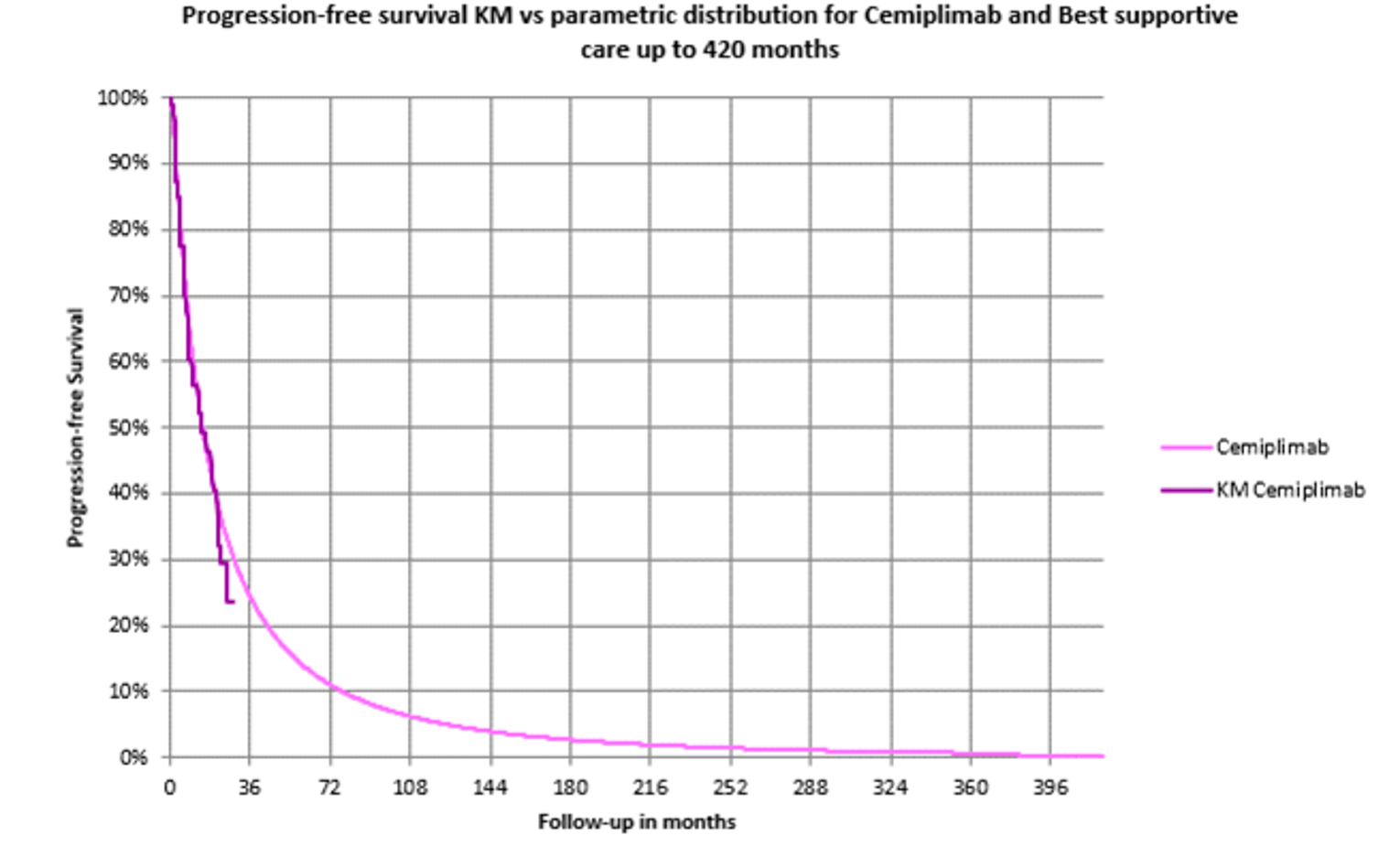
Source: Sponsor’s pharmacoeconomic submission.4
Figure 3: Sponsor Base Case OS Extrapolation for Cemiplimab and BSC
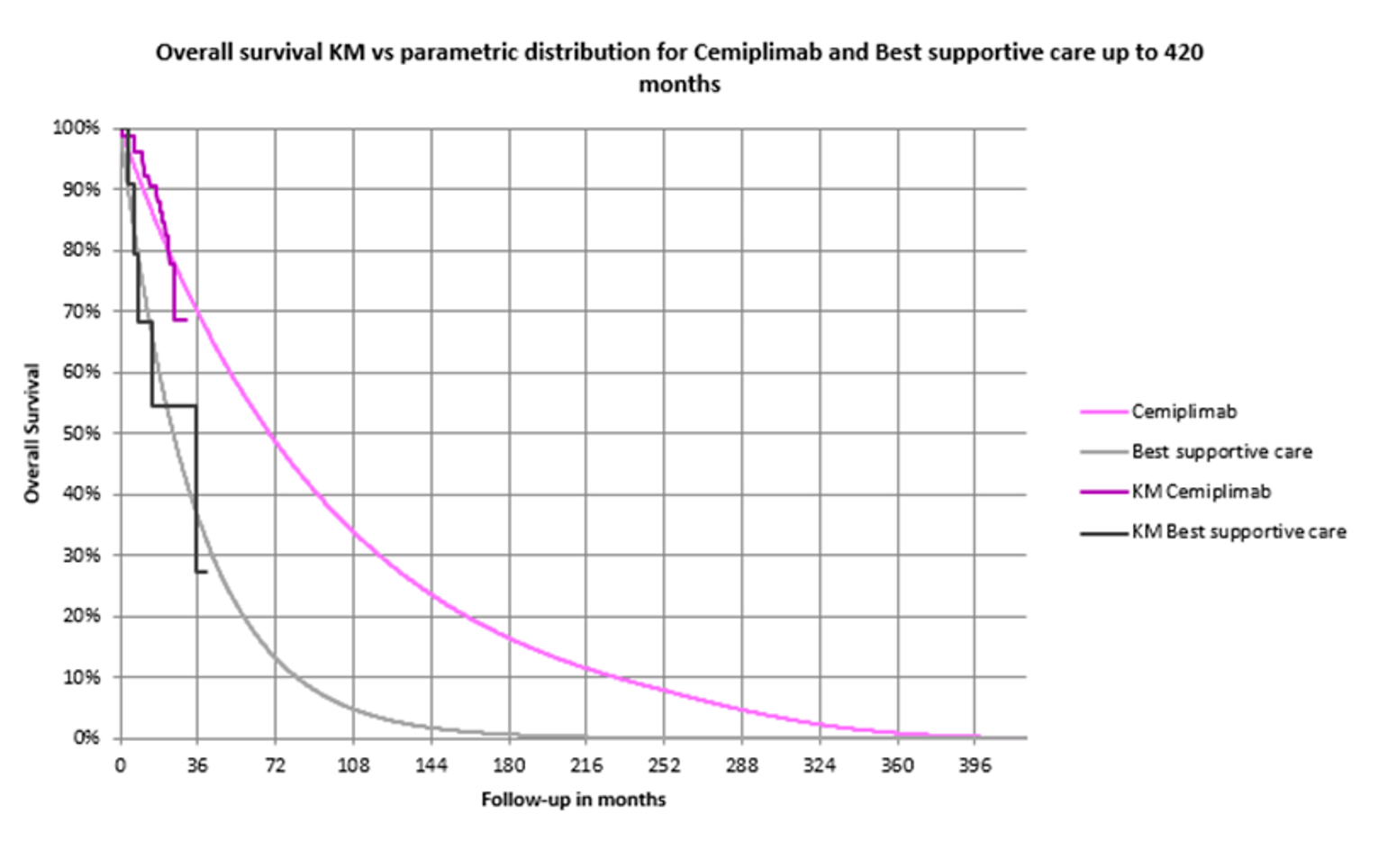
Source: Sponsor’s pharmacoeconomic submission.4
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Results of the Sponsor's Base-Case Analysis
Parameter | Cemiplimab | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 7.26 | 2.84 | 4.42 |
Pre-progressiona | NR | NR | NR |
Post-progressiona | NR | NR | NR |
Discounted QALYs | |||
Total QALYs | 5.61 | 2.22 | 3.39 |
Pre-progression | 2.08 | 0 | 2.08 |
Post-progression | 3.53 | 2.22 | 1.31 |
Discounted costs ($) | |||
Total costs | 304,681 | 96,638 | 208,043 |
Pre-progression drug costs | 151,445 | 0 | 151,445 |
Pre-progression administration costs | 1,952 | 0 | 1,952 |
Pre-progression disease management costs | 16,350 | 0 | 16,350 |
Post-progression disease management costs | 102,771 | 62,384 | 40,387 |
AE cost | 272 | 0 | 272 |
Terminal-care cost | 31,890 | 34,254 | -2,364 |
ICER ($/QALY) | 61,314 | ||
AE = adverse event; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; NR = not reported; QALY = quality-adjusted life-year.
a LY estimates disaggregated by health state were not available as discounted values in the economic model. Total LYs were available as discounted values.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 4: CADTH Exploratory Reanalysis PFS Extrapolation for Cemiplimab and BSC
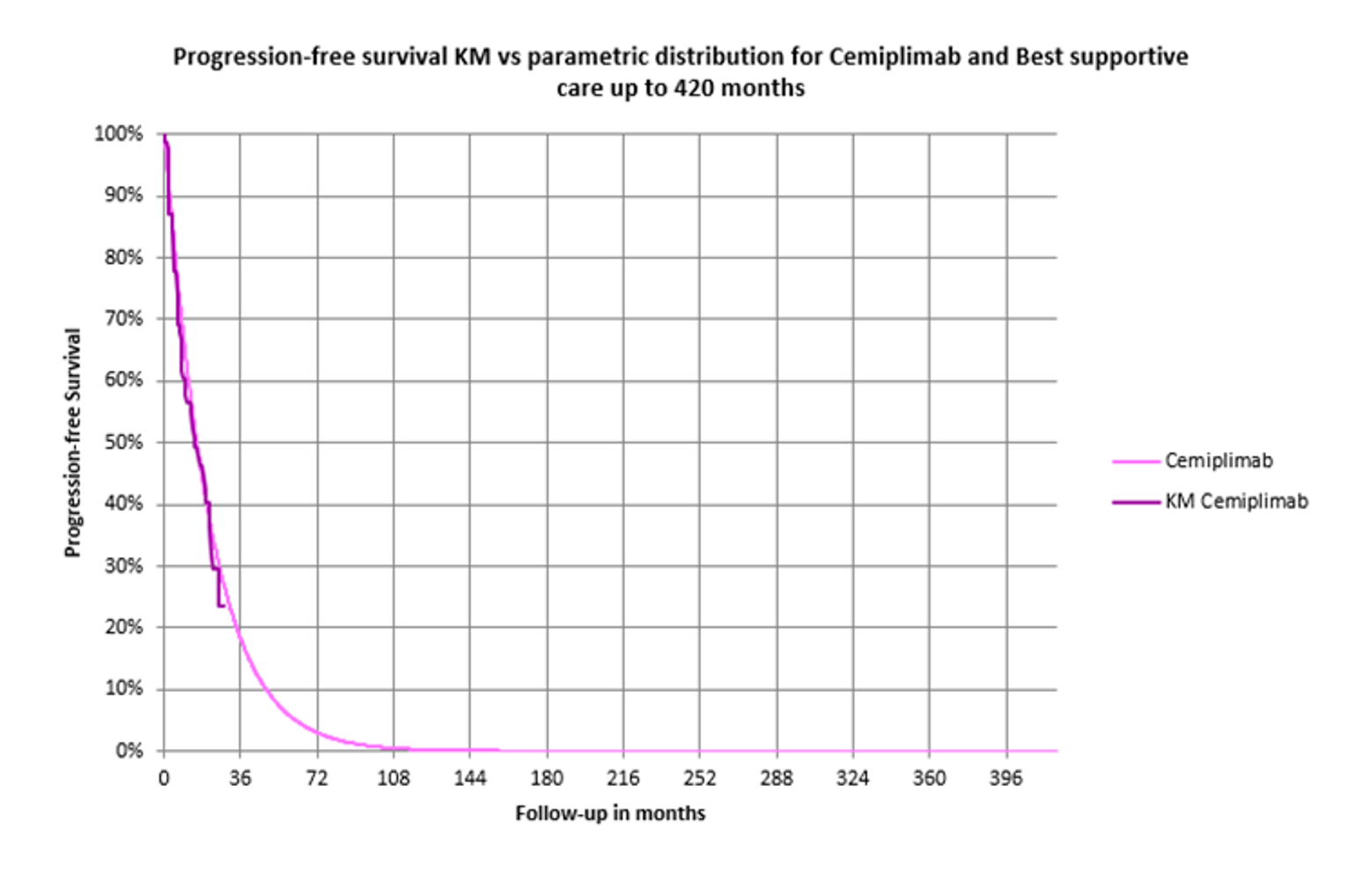
Source: Sponsor’s pharmacoeconomic submission, modified for CADTH’s exploratory analyses.4
Figure 5: CADTH Exploratory Reanalysis OS Extrapolation for Cemiplimab and BSC
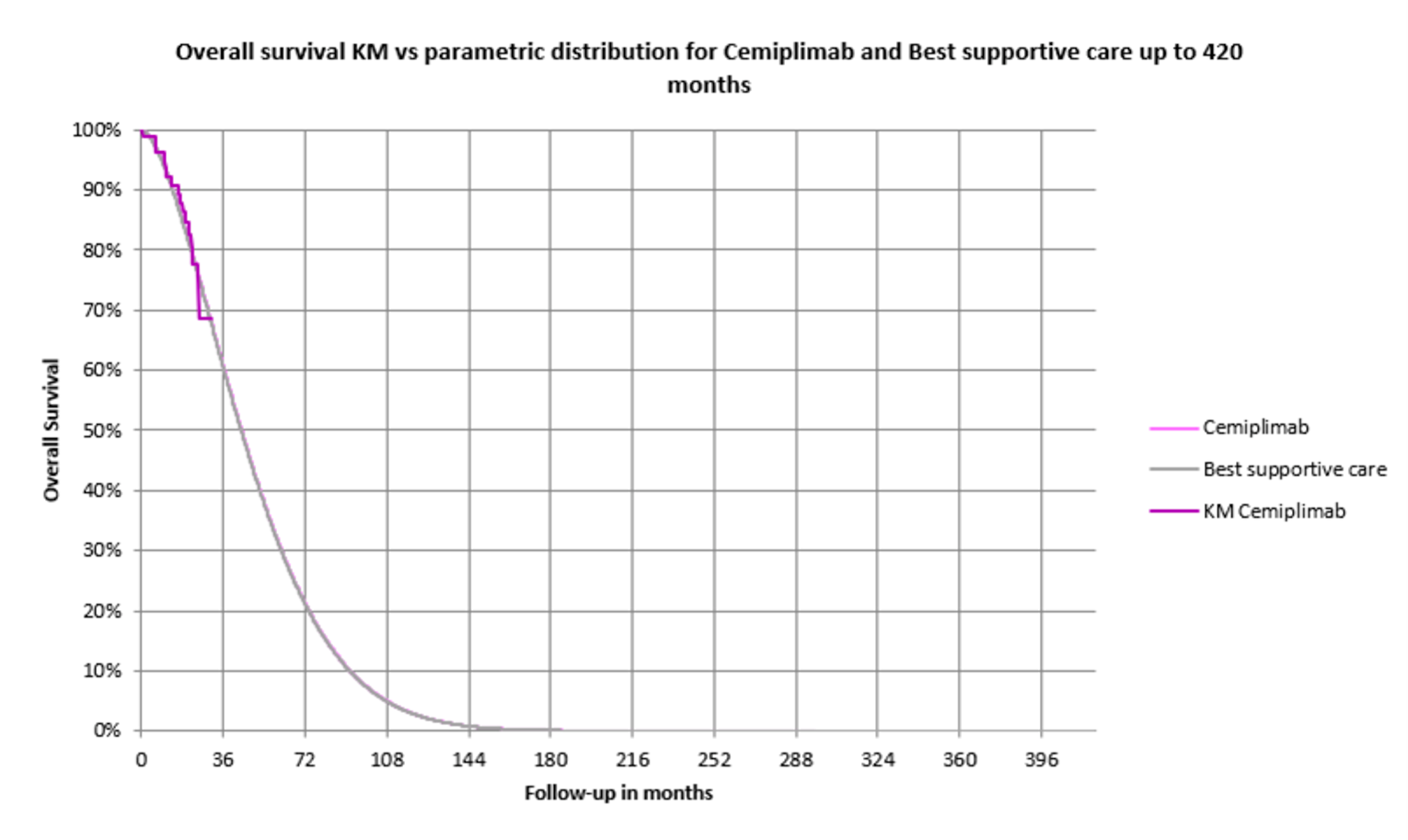
Source: Sponsor’s pharmacoeconomic submission, modified for CADTH’s exploratory analyses.4
Detailed Results of CADTH Base Case
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Cemiplimab | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 3.94 | 3.94 | 0 |
Pre-progressiona | NR | NR | NR |
Post-progressiona | NR | NR | NR |
Discounted QALYs | |||
Total QALYs | 3.14 | 3.08 | 0.06 |
Pre-progression | 1.45 | 0.00 | 1.45 |
Post-progression | 1.70 | 3.08 | 1.38 |
Discounted costs ($) | |||
Total costs | 310,220 | 164,624 | 145,596 |
Pre-progression drug costs | 150,238 | 0 | 150,238 |
Pre-progression administration costs | 1,926 | 0 | 1,926 |
Pre-progression disease management costs | 51,278 | 0 | 51,278 |
Post-progression disease management costs | 72,636 | 130,894 | -58,258 |
AE cost | 412 | 0 | 412 |
Terminal-care cost | 33,730 | 33,730 | 0 |
ICER ($/QALY) | 2,259,421 | ||
AE = adverse event; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; NR = not reported; QALY = quality-adjusted life-year.
a LY estimates disaggregated by health state were not available as discounted values in the economic model. Total LYs were available as discounted values.
Scenario Analyses
CADTH performed a scenario analysis to examine the impact of alternate utilities for the progressed disease health state on cost-effectiveness. The sponsor’s base case utility value of 0.793 was replaced with 0.805, which was calculated by applying a weighted average that gave a utility benefit only to those who experienced an objective response based on Study 1620 data. The alternate utility value was calculated by assuming that 32% of patients experienced the progression-free disease utility value and the remainder experienced the progressed disease utility value, based on the objective response rate from Study 1620 data.
Table 12: Summary of Scenario Analyses Conducted on CADTH Base Case
Scenario | Treatment | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH base case | BSC | 164,624 | 3.08 | Ref. |
Cemiplimab | 310,220 | 3.14 | 2,259,421 | |
1. Utility values | BSC | 165,038 | 3.13 | Ref. |
Cemiplimab | 310,932 | 3.17 | 3,331,586 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA estimated the introduction of cemiplimab for the treatment of patients with laBCC previously treated with an HHI.14 The analysis took the perspective of Canadian public drug plans using a top-down epidemiological approach and incorporating drug acquisition costs. A time horizon of 3 years between 2022 to 2024 was taken, with 2021 being the base year of the model. The target population size was estimated using the incidence of BCC and the proportion of patients with BCC whose disease is locally advanced, followed by further specifications of population size based on the proportion of patients receiving HHI, discontinuation rates of HHI, and medical eligibility. The reference case scenario included BSC (i.e., non-active therapy). The new drug scenario included cemiplimab and BSC. Key inputs to the BIA and the sponsor’s methodology in calculating target population are documented in Table 14.
The sponsors assume that 100% of patients were assumed to be receiving BSC (non-active therapy) in the reference scenario.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
CADTH-participating Pan-Canadian Population | 30,699,986 |
Incidence of BCCa | 0.1659% |
Proportion of patients with laBCC | 0.8% |
Proportion of patients with laBCC receiving HHI | 80% |
Proportion of patients with laBCC receiving HHI and requiring further therapy | 72% |
Proportion of patients requiring further therapy that are medically eligible for cemiplimab | 80% |
Number of total patients eligible for cemiplimab | 187 / 189 / 192 |
Market uptake (3 years) | |
Uptake (reference scenario) Cemiplimab Best supportive care | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Cemiplimab Best supportive care | ||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment over lifetime Cemiplimab Best supportive care | $152,213 $0 |
HHI = hedgehog pathway inhibitor.
a The incidence rate applied by the sponsor is prevalence data from 2014 Canadian Cancer Society surveillance estimates.14
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding cemiplimab for the treatment of laBCC for patients previously treated with a HHI was $5,715,066 in Year 1, $13,040,979 in Year 2, and $14,695,234 in Year 3, for a 3-year total of $33,451,279.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Market shares for cemiplimab are likely underestimated: The sponsor anticipated a gradual uptake of cemiplimab. Given that there is no second-line treatment available for laBCC, clinical experts consulted by CADTH noted that the market share for cemiplimab was likely underestimated, given clinicians’ anticipated preference for the drug when considering the high failure rates for HHI therapy. Both clinician and drug plan inputs indicated that cemiplimab would replace BSC as the new standard of care. Therefore, rapid uptake of this product is anticipated if it were to be made available. The clinical experts consulted by CADTH estimated the market share of cemiplimab to be 70% after 3 years on market.
CADTH increased the market shares of cemiplimab in each year included in the BIA linearly up to 70% and proportionately reduced the market share of BSC. An additional scenario analysis was performed to examine the budget impact of cemiplimab when increasing market shares up to 90%.
Median treatment discontinuation from Study 1620 was used to calculate drug acquisition costs: The sponsor used median time to discontinuation derived from Study 1620 to calculate drug acquisition costs per patient based on the recommended 3-week dosing schedule for cemiplimab. In the base case, the median treatment discontinuation of |||||||||| months is used for patients receiving cemiplimab. It is more appropriate to use mean treatment duration, which was |||||||||| months as reported in Study 1620.
In reanalysis, CADTH used the mean treatment duration for cemiplimab from Study 1620 to calculate drug acquisition costs per patient.
Proportion of patients that are medically eligible for cemiplimab is likely overestimated: Clinical experts advised that the number of patients medically eligible for cemiplimab per year is overestimated in certain jurisdictions in Canada. The sponsor assumed that 20% of patients with laBCC discontinuing treatment with HHI would be immunocompromised and therefore medically ineligible for treatment with cemiplimab. The clinical experts consulted by CADTH suggested that the proportion of patients ineligible for cemiplimab due to pre-existing comorbidities and limitations associated with IV administration was likely underestimated by the sponsor.
In a scenario analysis to explore the impact of medical eligibility on the budget impact of cemiplimab, CADTH reduced by 50% the proportion of patients with laBCC who are medically eligible for cemiplimab.
CADTH Reanalyses of the BIA
Based on the key limitations identified in the sponsor’s analysis, CADTH increased the market shares for cemiplimab and used mean treatment duration to calculate drug acquisition costs per patient.
Table 15: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market shares underestimated | Cemiplimab = |||||||||||||||||||||||||||||| BSC = |||||||||||||||||||||||||||||||||||||||| | Cemiplimab = 23% / 47% / 70% BSC = 77% / 53% / 30% |
2. Mean treatment duration | Median treatment discontinuation: |||||||||| months | Mean treatment duration: |||||||||| months |
CADTH base case | Reanalysis 1 + 2 | |
BIA = budget impact analysis; BSC = best supportive care; HHI = hedgehog pathway inhibitor.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. Based on the CADTH base case, the budget impact of the reimbursement of cemiplimab for the treatment of adult patients with laBCC previously treated with an HHI is expected to be $6,481,980 in year 1, $13,433,342 in year 2, and $20,290,516 in year 3. The 3-year total budget impact for cemiplimab is $40,205,838. A scenario analysis assessing the budget impact if the price of the drug under review reflected the price in which the ICER would be under the threshold of $50,000 per QALY resulted in a 3-year budget impact of $1,206,175. An additional scenario analysis applying a 50% reduction in the number of medically eligible patients led to a 3-year budget impact of $25,128,648. Lastly, the scenario analysis increasing market shares up to 90% led to a 3-year budget impact of $51,691,509.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $33,451,279 |
CADTH reanalysis 1 – market shares | $40,766,232 |
CADTH reanalysis 2 – mean treatment duration | $32,991,440 |
CADTH base case | $40,205,838 |
BIA = budget impact analysis.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | BSC | $0 | $0 | $0 | $0 | $0 |
Cemiplimab | $0 | $5,715,066 | $13,040,979 | $14,695,234 | $33,451,279 | |
Budget impact | $0 | $5,715,066 | $13,040,979 | $14,695,234 | $33,451,279 | |
CADTH base case | BSC | $0 | $0 | $0 | $0 | $0 |
Cemiplimab | $0 | $6,481,980 | $13,433,342 | $20,290,516 | $40,205,838 | |
Budget impact | $0 | $6,481,980 | $13,433,342 | $20,290,516 | $40,205,838 | |
CADTH scenario analysis: 97% price reduction | BSC | $0 | $0 | $0 | $0 | $0 |
Cemiplimab | $0 | $194,459 | $403,000 | $608,715 | $1,206,175 | |
Budget impact | $0 | $194,459 | $403,000 | $608,715 | $1,206,175 | |
CADTH scenario analysis: 50% reduction of medically eligible patients | BSC | $0 | $0 | $0 | $0 | $0 |
Cemiplimab | $0 | $4,051,237 | $8,395,839 | $12,681,572 | $25,128,648 | |
Budget impact | $0 | $4,051,237 | $8,395,839 | $12,681,572 | $25,128,648 | |
CADTH scenario analysis: market shares increased to 90% | BSC | $0 | $0 | $0 | $0 | $0 |
Cemiplimab | $0 | $8,454,756 | $17,148,947 | $26,087,806 | $51,691,509 | |
Budget impact | $0 | $4,051,237 | $8,395,839 | $12,681,572 | $51,691,509 |
BIA = budget impact analysis; BSC = best supportive care.
Stakeholder Input
Patient Group Input
Note that this appendix has not been copy-edited.
Save Your Skin Foundation
About Save Your Skin Foundation
SYSF is a national patient-led not-for-profit group dedicated to the fight against non-melanoma skin cancers, melanoma and ocular melanoma through nationwide education, advocacy, and awareness initiatives. SYSF provides a community of oncology patient and caregiver support throughout the entire continuum of care, from prevention and diagnosis to survivorship. www.saveyourskin.ca
Information Gathering
Information was obtained through online surveys, virtual patient roundtables and one-on-one conversations. Information collected for section 3, 4, and 5 included all BCC patients (23, inclusive of 5 on treatment under review), and was gathered over the past 6 months, while section 6 was information from patients treated by drug under review collected over the past 2 weeks.
There were 20 females and 3 males ranging between the age of 30 – 80+, the majority of respondents (6) being between 30-49, (7) 60 – 69 and (7) 70-79. 15 respondents were retired, 6 working fulltime, 1 looking for work and 1 disabled and unable to work.
There were 5 respondents from BC, 4 from Alberta, 7 from Ontario, 1 from NS. 2 from QC, and 4 from outside of Canada (Netherlands, Algeria and 2 from France)
Disease Experience
SYSF asked patients to describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
Fear and/or anxiety (all respondents)
Scarring and disfigurement (all respondents)
Fatigue (all respondents)
Fear of reoccurrence (all respondents)
Long, scary, roller coaster - surgeries, immunotherapy treatments, happy to be NED at this
always fearful of the future.
Difficult and scarry
Horrific. Horrific does not begin to describe the experience
Bleeding and sensitivity
Scars itchy and not healing
Bad scarring
Fever
Post-surgical nerve damage in face. Constantly looking for new signs of disease
Experiences With Currently Available Treatments
SYSF asked patients to describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
Chemo
Surgery
Chemo and surgery
Radiation
Mohs surgery
Patients in remote areas of Canada have problems getting to specialist if needed. Travel costs and time off from work puts extra stress on patients and caregivers. Fear and anxiety of reoccurrence. Disfigurement in patients with BCC of the face cause lack of confidence, nerve damage, some patients became isolated from friends and family. See above (3) for side effects to surgery and radiation. One patient had to travel to the US to get treatment as it was not available in Canada. Huge expenses and increased stress to her and her family and the added concern being treated outside her Country if anything was to go wrong. For patients with rare skin cancer the patient has to advocate for treatments as they are not on formulary or sometimes not offered by their physicians.
Improved Outcomes
SYSF asked patients what improvements would patients and caregivers like to see in a new treatment that is not achieved in currently available treatments? How might daily life and quality of life for patients, caregivers, and families be different if the new treatment provided those desired improvements? What trade-offs do patients, families, and caregivers consider when choosing therapy?
More new treatment options will minimal side effects
Less surgery
Less radiation
Treatments or procedures closer to home and to their support network
Experience With Drug Under Review
SYSF asked how did patients have access to the drug under review (for example, clinical trials, private insurance)? Compared to any previous therapies patients have used, what were the benefits experienced? What were the disadvantages? How did the benefits and disadvantages impact the lives of patients, caregivers, and families? Consider side effects and if they were tolerated or how they were managed. Was the drug easier to use than previous therapies? If so, how? Are there subgroups of patients within this disease state for whom this drug is particularly helpful? In what ways?
2 patients received treatment on clinical trial
3 patients did not know how they received treatment
3 patients are still on treatment
2 have completed treatment
2 patients had no side effect
2 patient had fatigue
1 patient had skin rash
The 2 patients that completed treatment both said that the side effects were manageable and the benefits outweighed the experience of the side effect. The 3 patients that are currently still on treatment said the side effects were manageable and they hoped the benefit would outweigh the experience of them. All 5 patients expressed knowledge that if they had had access to the treatment upon diagnosis of metastatic disease the disease experience would have been different, reducing scarring, disfigurement, and psychosocial side effect.
Companion Diagnostic Test
SYSF asked What are patient and caregiver experiences with the biomarker testing (companion diagnostic) associated with regarding the drug under review?
4 of the 5 patients knew nothing about testing, it was not discussed with them. 1 patients comment below (8).
Anything Else?
“Receiving this treatment was extremely important as nothing was available in Canada. As a patient you need to know how to access these drugs, if they are compatible with genetic mutations of tumour. In my experience any information I gathered was because of the persistence of us asking the questions and doing the ground work. Options did not come to us, we had to find them.”
“Grateful for the Support of SYS. Had to travel frequently (every 3 weeks) for 10 months to the States”
Patients with rare skin cancers feel alone and isolated as they can find very little information or resources and feel left behind by the system.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for Save Your Skin Foundation
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi | — | — | — | $75,000 |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Name: Kathleen Barnard
Position: President
Patient Group: Save Your Skin Foundation
Date: Sept 10th 2021
Melanoma Network of Canada
About Melanoma Network of Canada
The MNC was founded in 2009 by Annette Cyr, a patient and three-time survivor of melanoma, in conjunction with support and assistance from Jo Anne Adams, a patient survivor, and Terra Deeth, caregiver to a husband with melanoma. The network was founded to respond to the need for patients in Canada to have a nationally-based organization to coordinate educational and prevention efforts, provide a strong voice for advocacy, and assist in efforts to target funding for melanoma research. https://www.melanomanetwork.ca/
Information Gathering
While there are estimates of over 80,000 cases annually in Canada of basal cell carcinoma (BCC), it is estimated that only approximately 1% are advanced or metastatic. There were very few patients in Canada that have been prescribed this new therapy (we believe at the time of our survey, less than 50), which made data gathering difficult. Data was gathered for this submission by way of an on-line survey. A letter to physicians along with a link to the survey and purpose of the survey to mailed health care providers, including oncologists, surgeons, plastic surgeons and dermatologists. We posted the survey on line for patients and caregivers, regardless of stage. Reaching patients and physicians during Covid made it particularly challenging. We also used social media via Facebook, Twitter to promote the survey. The survey was made available August Demographics: We received a total of 62 individual patient responses and a further 45 caregiver responses. Of the total responses for patients, 44 were female and 18 were male. The survey was open to all patients, regardless of stage. The many of respondents were early stage or did not know their staging. @ were stage 2, 2 were stage 3 and 4 were stage 4. We had 1 respondent from the US, and the remainder from Canada. Only 1 patient indicated they had been on treatment for metastatic disease with cemiplimab. The majority of patients were over 50 years of age. The locations of respondents are:
Table 2: Location of Survey Respondents
Answer Choices | Responses | |
|---|---|---|
British Columbia | 17.74% | 11 |
Alberta | 12.90% | 8 |
Saskatchewan | 1.61% | 1 |
Manitoba | 1.61% | 1 |
Ontario | 50.00% | 31 |
Quebec | 6.45% | 4 |
Nova Scotia | 4.84% | 3 |
PEI | 1.61% | 1 |
New Brunswick | 1.61% | 1 |
Newfoundland & Labrador | 0.00% | 0 |
Nunavut, Yukon or Northwest Territories | 0.00% | 0 |
N/A Live Outside Canada | 1.61% | 1 |
Answered 62
Disease Experience
Most often, patients and family members indicate surviving is critical, which of course it is. With advanced BCC however, patients go through significant agony and distress from pain associated with the cancer itself and treatment. This cancer most often occurs on the head and neck and is very visible, disfiguring, and horrific for the average person to look at. The advanced age of many of the patients adds to the distress from challenges with travel, home support and care and other health issues. Side effects of these treatments (surgery and radiation) can be debilitating and traumatizing to the patient and their family. With advanced or metastatic disease, the scaring, disfigurement, pain, social isolation and depression due to the cancer and treatment are really impossible to fully describe. Patients would like less pain, less scarring and disfigurement, less debilitating surgery and effects from treatment.
Table 3: Side Effects Reported by Patients From the Disease Itself
Answer Choices | Responses | |
|---|---|---|
Pain | 13.04% | 6 |
Scarring or disfigurement | 47.83% | 22 |
Edema or fluid retention | 4.35% | 2 |
Peripheral neuropathy (nerve pain or damage) | 6.52% | 3 |
Disrupted sleep | 4.35% | 2 |
Fear or anxiety | 41.30% | 19 |
Fatigue | 15.22% | 7 |
Depression | 8.70% | 4 |
Negative impact to self-image, family or social life | 10.87% | 5 |
Financial loss or job loss | 2.17% | 1 |
Impact on sexuality | 6.52% | 3 |
None - there has been no impact | 32.61% | 15 |
Other (please explain) | — | 8 |
Answered 46
Some of the specific comments from patients on their experience with the cancer and treatment:
I have had my left eye start to go blind, I have had to take Apheresis treatments and have had multiple visits to eye doctors.
Have a lot of inflammation, and lots of severe bone pain. I have had Basal cell removed from leg and eye lid, and also removed ankle leg.
Paranoid of finding more. Constant stress and worry.
A lot of anxiety and depression as a result of how I look. People stare.
I had part of my nose skin grafted. I am happy with the results of the surgery, but I still feel that people look at my face.
After surgery couldn’t golf and couldn’t go to our boat slip no swimming! Depressed scared to go outside scared of the sun! Very moody and sad!
I have to get steroids shots to shrink my scar, makes it painful and difficult to make a tight fist. It is also hard to pick up my children.
Experiences With Currently Available Treatments
The majority of locally advanced or metastatic BCC is treated with surgery and/or topical treatments. The disease occurs most commonly on the head and neck, which is very challenging and difficult to treat. Current therapies often leave advanced patients in pain, often with side effects from topical treatments and significant physical and emotional scarring. Most often patients are treated with surgery, cryotherapy or topical drug treatments. Patients indicated significant issues with surgical procedures having a negative impact on quality of life.
Table 4: Treatments Received for Your BCC
Answer Choices | Responses | |
|---|---|---|
Surgical excision - removes the cancer and some normal tissue surrounding | 77.78% | 35 |
Mohs Surgery (surgical removal layer by layer) | 24.44% | 11 |
Curettage and Electrodesiccation (scraping and use of electrical current on scraped area) | 8.89% | 4 |
Cryosurgery (freezing) | 17.78% | 8 |
Reconstructive Surgery | 8.89% | 4 |
Lymph node dissection | 11.11% | 5 |
Photodynamic Therapy (PDT) | 2.22% | 1 |
Radiation Therapy | 6.67% | 3 |
Topical creams or gels - example: 5-fluorouracil (5-FU, Efudex) or imiquimod (Aldara, Zyclara) | 15.56% | 7 |
Chemotherapy | 0.00% | 0 |
Hedgehog pathway inhibitor (HHI)- vismodegib (Erivedge) | 0.00% | 0 |
Other (please specify) | 13.33% | 6 |
Answered 45
Patients comments on existing treatments:
Severe reaction to chemotherapy "cream".
Emotional,because living with pain everyday is hard to cope with
Mental health/emotional-it really takes a toll on you each time you need more excisions/treatment.
No longer work. On long-term disability.. money tighter for family
While healing I can’t see my grandchildren because of the appearance of surgery site on face. Swelling, stitches, redness
Just a bit of pain and scarring. The cream burns
Caregivers indicated worries and challenges, trying to support their loved ones. As this cancer is largely found in the 60+ population, many spouses already have their own health issues or are no longer alive to support the patient. Family members are often tasked with support, which drains them emotionally and financially with the amount of time and resources required to care for their loved one. There were challenges getting in to see a dermatologist or surgeon, particularly during covid times. Frequent travel expenses and time commitment to travel distances for treatment. There was additional stress for caregivers not being able to attend appointments with their loved one. About 30% of patients reported having no caregiver, so were dealing with treatment on their own.
Feedback from the caregivers on the experience included:
Stress is real and many worried nights and very scared to go to an appointment and possible worse news
Mostly emotional. She realizes that it was important to remove the cancer from the affected area, but, will never understand why they left so many scars.
Has caused a lot of worry.
Ongoing concern about potential impacts of treatment. I don’t like to see her in pain.
Improved Outcomes
When asked, both patients and caregivers had similar responses about what they would like to see in a new treatment. First and foremost, earlier diagnosis and access to specialist. Second, less invasive procedures. And third includes topping progression. Patient’s comments included:
Yes anything to eliminate the disease Educating family doctors for a quicker diagnosis as my family dr did not know what it was! I made my own appointment at my dermatologist ! I should have gone sooner!!!
Better screening from dermatologists it seems that they are to busy with other parts of their business.
I would like to a drug therapy that would eliminate the disease - no recurrences. Right now, I am a little anxious about it reoccurring, don’t feel comfortable about going out in the sun, always checking for new spots.
The very negative impact of successive and disfiguring surgeries and quality of life impacts after some of the surgeries and topical treatments leads to other health issues for many of the patients. Better treatments for advanced disease could also lessen the amount of surgery and lessening the burden of care for caregivers and the pain and disfigurement and associated issues for patients. Unfortunately, there are not many effective treatments for when BCC is metastatic.
Experience With Drug Under Review
Only one patient responded that they had been treated with the drug therapy under review. He had no trouble accessing the treatment. He is currently on therapy and hoping for a positive outcome, or regression of disease. He felt the option of having another therapy that might work was worth the treatment side effects. He indicated some difficulty with the liver and some flu like symptoms. There was very small numbers of patients offered the therapy in the country, so it made reaching them for comment very difficult.
The advantages for advanced or metastatic patients is the therapy is one other where options have run out completely and they are facing a horrible and likely outcome of death from this cancer. In a reasonable number of patients, this therapy makes the difference between life and death. Of course patients and caregivers would like access to any therapy that gives them an improved chance of survival along with a reasonable quality of life. Patients and caregivers are more than willing to accept some of the side effects for the trade-off of survival and/or disease control.
Companion Diagnostic Test
There is no companion diagnostic test with this therapy.
Anything Else?
This is a needed therapy for the small number of patients that have failed the existing option for treatment for advanced or metastatic basal cell carcinoma. At advanced stages, this disease is horribly painful, disfiguring and creates an enormous burden on the quality of life and mental health of the patient and family. It is a much needed therapy to provide an easily tolerable option that may result in delay of progression or potentially curative. The need for options at this stage of disease is imperative.
Conflict of Interest Declaration for Melanoma Network of Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
None received.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
N/A
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 5: Conflict of Interest Declaration for Melanoma Network of Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi | — | X | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Name: Annette Cyr
Position: Chair of the Board
Patient Group: Melanoma Network of Canada
Date: September 13, 2021
Clinician Group Input
Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug- related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Please describe how you gathered the information included in the submission.
This input was jointly discussed at a DAC meeting.
Current Treatments
Describe the current treatment paradigm for the disease
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: laBBC or metastatic disease – patient may retry HHI (if not progressed on HHI previously) or receive palliative treatment Currently there is no standard of care treatment for 2nd line treatment. This is an area of unmet need.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Prolong life, delay disease progression, maintain independence, reduce the severity of symptoms, improve health-related QoL, avoidance of disfiguring surgical procedures (e.g., removal of ears, enucleation of eye)
Treatment Gaps (unmet needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatments needed to improve compliance. Formulations are needed to improve convenience.
Response: There are no standard second-line treatment for this population. Cemiplimab is generally well tolerated, including the older population.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Patients who are not eligible for HHI, intolerant of HHI, or progressed on HHI
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Cemiplimab will be 2nd line treatment after HHI intolerance or failure.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: Patients would have been treated with HHI previously (either intolerance or failure).
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Cemiplimab will be a second-line therapy
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Intolerance or failure to HHI
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice?
Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.). Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: No companion diagnostic required.
Which patients would be least suitable for treatment with the drug under review?
Response: No specific restrictions
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: No
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Clinical response
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms, Stabilization (no deterioration) of symptoms.
Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response:
Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth)
Ability to perform activities of daily living
Improvement in symptoms
Stabilization (no deterioration) of symptoms
Avoidance of disfiguring surgical procedures
How often should treatment response be assessed?
Response: Patients are seen usually every 6-8 weeks. In the trial, the time to response was approximately 4 months.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: Disease progression and adverse events
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Outpatient at chemo suits
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: N/A
Additional information
Is there any additional information you feel is pertinent to this review?
Response: This is an unmet need for patients who are intolerant or progressed on HHI. Cemiplimab is well tolerated, including the elderly population who presents with laBCC or mBCC.
The primary analysis for Study 1620 is reported only for the laBBC group.
Conflict of Interest Declarations for Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Frances Wright
Position: Surgeon – Sunnybrook Health Sciences Centre
Date: 13 August 2021
Table 6: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Tara Baetz
Position: Medical oncologist
Date: 13 August 2021
Table 7: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Marcus Butler
Position: Medical oncologist
Date: 13 August 2021
Table 8: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | X | — | — | — |
Declaration for Clinician 4
Name: Dr. Elaine McWhirter
Position: Medical oncologist
Date: 13 August 2021
Table 9: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Teresa Petrella
Position: Medical oncologist
Date: 13 August 2021
Table 10: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Xinni Song
Position: Medical oncologist
Date: 13 August 2021
Table 11: Declaration for OH-CCO Skin Cancer Drug Advisory Committee Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None Declared | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.