CADTH Reimbursement Review
Sacituzumab Govitecan (Trodelvy)
Sponsor: Gilead Sciences Canada, Inc.
Therapeutic area: Locally advanced or metastatic triple-negative breast cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
BM
brain metastasis
BM-Neg
brain metastasis–negative
BM-Pos
brain metastasis–positive
CBCN
Canadian Breast Cancer Network
CI
confidence interval
CNS
central nervous system
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
ER
estrogen receptor
G-CSF
granulocyte colony–stimulating factor
HER2
human epidermal growth factor receptor 2
HR
hazard ratio
HRQoL
health-related quality of life
IRC
independent review committee
ITT
intention to treat
KM
Kaplan-Meier
MID
minimally important difference
mTNBC
metastatic triple-negative breast cancer
OL
open label
ORR
objective response rate
OS
overall survival
PARP
poly-(ADP-ribose) polymerase
PD
progressive disease
PD-1
programmed cell death protein 1
PD-L1
programmed death ligand 1
PFS
progression-free survival
PR
progesterone receptor
RECIST 1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
SAE
serious adverse event
SD
standard deviation
TNBC
triple-negative breast cancer
TPC
treatment of physician’s choice
Trop-2
trophoblast cell-surface marker 2
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for sacituzumab govitecan is provided in Table 1.
Item | Description |
|---|---|
Drug product | Sacituzumab govitecan (Trodelvy) 180 mg lyophilized powder for solution for injection, for IV use |
Indication | For the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received 2 or more prior therapies, at least 1 of them for metastatic disease |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review, Project Orbis |
NOC date | September 24, 2021 |
Sponsor | Gilead Sciences Canada, Inc. |
NOC = Notice of Compliance.
Introduction
Triple-negative breast cancer (TNBC), defined by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), is an aggressive cancer with poor prognosis.1,2 The disease is more common among younger women and racialized women.1,3 Of the approximately 27,000 Canadians diagnosed with breast cancer each year,4 15% will have TNBC,1,2 which carries higher risks of early recurrence and presentation as stage 4 metastatic disease. The exact prevalence and incidence of adult patients with unresectable locally advanced TNBC or metastatic TNBC (mTNBC) who have received 2 or more prior therapies, at least 1 of them for metastatic disease, is uncertain. As noted in the CADTH pharmacoeconomic report, the sponsor estimated that the total number of patients with locally advanced TNBC or mTNBC in Canada, including incident cases of de novo mTNBC as well as prevalent cases that have recurred or spread, was 633 in 2020, and the vast majority of patients would be candidates for sacituzumab govitecan.5,6 Median survival of locally advanced TNBC or mTNBC is approximately 12 months to 14 months.7 Symptoms including pain, fatigue, and insomnia impose limitations on patients’ ability to work, caregiving responsibilities, and physical activity, as well as significant financial burdens.
According to clinician and patient input, the main goals of systemic therapy for mTNBC are to extend survival and delay progression with minimal toxicity while maintaining or improving health-related quality of life (HRQoL). Immunotherapy and poly-(ADP-ribose) polymerase (PARP) inhibitors such as olaparib may induce responses in some patients, although the survival benefit is not clear, and these therapies are not currently funded. For recurrent locally advanced TNBC and mTNBC, sequential single-agent chemotherapy remains the standard of care.8 According to clinical experts consulted for this review, each line of therapy has diminishing response rates. The standard chemotherapies for locally advanced TNBC or mTNBC are taxanes, platinum agents, capecitabine, gemcitabine (with or without cisplatin or carboplatin), anthracyclines, eribulin, and vinorelbine. An optimal treatment sequence has not been determined, and treatment sequence varies across jurisdictions in Canada. According to clinical experts, in pre-treated patients with mTNBC, single-agent chemotherapies have low objective response rates (ORRs) (5% to 10%), as determined by tumour imaging, and low progression-free survival (PFS) (2 months to 3 months).9,10
Sacituzumab govitecan is an antibody-drug conjugate directed against human trophoblast cell-surface marker 2 (Trop-2), a transmembrane protein involved in calcium signal transduction that is overexpressed in many epithelial cancers, including TNBC. Sacituzumab govitecan is administered at a dose of 10 mg/kg as an IV infusion on day 1 and day 8 of a 21-day treatment cycle. Sacituzumab govitecan is indicated for the treatment of adult patients with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. According to the sponsor and clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who have been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication.
The objective of this report was to perform a systematic review of the beneficial and harmful effects of sacituzumab govitecan (IV injection, 10 mg/kg) for the treatment of adult patients with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Input was provided by 2 patient groups for this review: Rethink Breast Cancer and the Canadian Breast Cancer Network (CBCN). Rethink Breast Cancer conducted an online patient survey in June and July 2021. Of the 30 respondents with mTNBC (22 from the US and 6 from Canada), 4 with direct experience with sacituzumab govitecan participated in telephone interviews. The Canadian Breast Cancer Network distributed online surveys in 2012 and 2017 to patients living with breast cancer registered in the membership databases of CBCN and other patient organizations. Of the 157 respondents, data from 14 patients with mTNBC were captured in the input, and 1 Canadian patient with direct experience with sacituzumab govitecan participated in a telephone interview.
Patients highlighted the negative impacts of mTNBC, including spread to the bone, liver, lungs, and brain. Symptoms frequently included pain, fatigue, and insomnia and imposed significant financial burdens and limitations on patients’ ability to work, caregiving responsibilities, physical activity, and ability to spend time with loved ones. Patients highlighted the limited treatment options for mTNBC and their experiences with prior therapies (chemotherapy and immunotherapy), including their limited effectiveness in delaying progression, managing symptoms, and maintaining HRQoL, as well as their side effects (e.g., nausea and/or vomiting, fatigue, hand-foot syndrome). Twenty patients contacted by Rethink Breast Cancer and 1 patient contacted by CBCN had direct experience with sacituzumab govitecan. Patients felt that the drug was effective in controlling disease, extending survival, maintaining HRQoL, and reducing mTNBC symptoms (e.g., Jacksonian marches, bone pain, neuropathy) and noted that side effects (e.g., fatigue, alopecia, diarrhea, neutropenia) were manageable, although not all patients were currently able to access the drug.
Patients with mTNBC identified an important unmet need for treatments that control disease progression, prevent recurrence, and extend survival. Other needs identified by patients were treatments that maintain HRQoL, reduce symptom severity, and have a manageable safety profile.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical experts with expertise in the diagnosis and management of mTNBC highlighted the poor outcomes in patients with mTNBC and the limited effective treatment options for later-line therapy (single-agent chemotherapy, optimal sequencing undefined). ORRs by objective tumour imaging are low for lines of therapy beyond anthracyclines and taxanes, many patients become refractory to treatment, and many treatments are poorly tolerated. The main goals of therapy are to extend survival and delay progression with minimal toxicity while maintaining or improving HRQoL. Sacituzumab govitecan would be administered for later-line treatment after at least 2 prior lines of chemotherapy (at least 1 taxane in the adjuvant, neoadjuvant, or advanced setting and at least 1 therapy in the metastatic setting), where it may cause a shift in the current treatment paradigm by providing an option for targeted therapy. Any patient with mTNBC who has received at least 2 prior lines of systemic therapy with adequate performance status, adequate hematological and organ function, and stable central nervous system (CNS) disease would be a candidate for sacituzumab govitecan. Treatment would be initiated in suitable patients by the primary treating physician based on pathology or biomarker assessment and imaging or biopsy results. Response to treatment is assessed by serial imaging showing stable or shrinking disease (objective responses), laboratory markers, clinical assessment, and maintained or improved HRQoL and cancer symptoms. Treatment would be discontinued in patients with progressive disease (PD) or with significant worsening of symptoms, performance status, or HRQoL, as well as in patients with significant and persistent side effects (especially diarrhea).
Clinician Group Input
Three clinician groups provided input for this review: the Ontario Health–Cancer Care Ontario Breast Cancer Drug Advisory Committee (3 medical oncologists and 1 pharmacist), the Ottawa Hospital Cancer Centre Breast Medical Oncology group (4 medical oncologists), and the Rethink Breast Cancer Scientific Advisory Committee (6 medical oncologists from across Canada). No major contrary views were presented: clinical groups echoed the lack of effective options for later-line therapy of mTNBC. Sacituzumab govitecan may change the treatment paradigm and be used before vinorelbine or gemcitabine or reuse of doxorubicin.
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for discontinuation of therapy, generalizability, care provision, system issues, and economic considerations. The clinical experts consulted by CADTH for this review weighed evidence from the included study and other clinical considerations to provide responses to the Provincial Advisory Group’s drug program implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Study and Protocol-Selected Study
Description of Study
One phase III, randomized, open-label (OL), multi-centre study (ASCENT, N = 529)11,12 was included in the systematic review (Table 6). The primary objective of the study was to compare the efficacy of sacituzumab govitecan (10 mg/kg) with chemotherapy (the treatment of physician’s choice [TPC] selected from the following options: eribulin, capecitabine, gemcitabine, or vinorelbine) in prolonging PFS among adult patients with locally advanced TNBC or mTNBC previously treated with at least 2 systemic chemotherapy regimens (including 1 taxane in the adjuvant, neoadjuvant, or advanced setting). Of note, the study enrolled a small number of patients (approximately 3%) with locally advanced mTNBC who had not received prior therapy in the metastatic setting; according to the sponsor and clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who had been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication.
Patients with acceptable performance status (Eastern Cooperative Oncology Group [ECOG] 0 or 1) and organ function were randomized 1:1 to receive sacituzumab govitecan or TPC; randomization was stratified by number of prior therapies (2 to 3 versus > 3), baseline brain metastasis (BM) status, and region (North America versus rest of world). Most patients (88.5%) were negative for BM (BM-Neg), and 11.5% were positive for BM (BM-Pos). The mean age of participants was 54.0 (standard deviation [SD] = 11.5) years, and most were White (79.0%), not Hispanic or Latino (87.0%), and from North America (65.6%; nearly all from the US). The mean number of prior systemic therapies was 4.5 (SD = 2.1); nearly all patients had received prior breast cancer–related surgery (94.9%), most had received prior non-brain radiotherapy (81.1%), and approximately one-quarter had received prior programmed cell death protein 1 (PD-1) or programmed death ligand 1 (PD-L1) therapy (28.9%).
Patients were treated until PD or unacceptable toxicity and subsequently entered survival follow-up. Crossover was not permitted. The primary outcome was PFS by blinded independent review committee (IRC) assessment in the BM-Neg population, while PFS in the intention-to-treat (ITT) set, overall survival (OS) in the BM-Neg population and ITT set, ORR in the BM-Neg population and the ITT set, and HRQoL were secondary outcomes.
Efficacy Results
A summary of key results from the ASCENT trial is shown in Table 2. The primary outcome was PFS in the BM-Neg population. Median OS was longer in patients treated with sacituzumab govitecan than in those treated with TPC (BM-Neg population: median = 12.1; 95% confidence interval [CI], 10.7 to 14.0 months versus median = 6.7; 95% CI, 5.8 to 7.7 months; P < 0.0001; ITT set: median = 11.8; 95% CI, 10.5 to 13.8 months versus median 6.9; 95% CI, 5.9 to 7.7 months; P < 0.0001). The hazard ratio (HR) for death among patients treated with sacituzumab govitecan relative to TPC was 0.476 (95% CI, 0.383 to 0.592) in the BM-Neg population and 0.508 (95% CI, 0.414 to 0.624) in the ITT set. Median (95% CI) PFS by blinded IRC assessment was statistically significantly longer in patients treated with sacituzumab govitecan than in those treated with TPC (median = 5.6; 95% CI, 4.3 to 6.3 months versus median = 1.7; 95% CI, 1.5 to 2.6 months in the BM-Neg population; median = 4.8; 95% CI, 4.1 to 5.8 months versus median = 1.7; 95% CI, 1.5 to 2.5 months in the ITT set; P < 0.0001 in both the BM-Neg population and the ITT set). The HR for progression or death among patients treated with sacituzumab govitecan relative to TPC was 0.409 (95% CI, 0.323 to 0.519) in the BM-Neg population and 0.433 (95% CI, 0.347 to 0.541) in the ITT set. Median time to progression in the sacituzumab govitecan arm compared with the TPC arm was 5.8 (95% CI, 4.8 to 6.9) months versus 2.1 (95% CI, 1.5 to 2.7) months in the BM-Neg population and 5.6 (95% CI, 4.3 to 6.2) months versus 2.1 (95% CI, 1.5 to 2.8) months in the ITT set. The ORR in the sacituzumab govitecan arm compared with the TPC arm was 34.9% (95% CI, 28.8% to 41.4%) versus 4.7% (95% CI, 2.4% to 8.3%) in the BM-Neg population and 31.1% (95% CI, 25.6% to 37.0%) versus 4.2% (95% CI, 2.1% to 7.4%) in the ITT set. Mean time to response for patients receiving sacituzumab govitecan and TPC was 2.67 (SD = 1.91) months versus 1.86 (SD = 0.92) months in the BM-Neg population and 2.66 (SD = 1.91) months versus 1.86 (SD = 0.92) months in the ITT set. Subgroup analyses were not adjusted for multiplicity and were not powered to evaluate differences in the treatment effects of sacituzumab govitecan in patients with or without BRCA1 or BRCA2 mutations, patients who had received 2 to 3 or more than 3 prior lines of therapy, or BM-Pos and BM-Neg patients. Nevertheless, the clinical experts consulted for this review felt that the results of the trial were generalizable across strata for all these subgroups.
Harms Results
Adverse events (AEs) occurred in almost all patients treated with both sacituzumab govitecan and TPC (99.6% versus 97.8%). Serious AEs (SAEs) and withdrawals due to AEs (WDAEs) occurred in similar proportions of sacituzumab govitecan– and TPC-treated patients (26.7% versus 28.1% for SAEs and 4.7% versus 5.4% for WDAEs, in sacituzumab govitecan– and TPC-treated patients, respectively). Deaths due to AEs occurred in 1 sacituzumab govitecan–treated patient (0.4%) and 3 TPC-treated patients (1.3%).
Neutropenia or febrile neutropenia occurred more frequently in patients treated with sacituzumab govitecan than with TPC (65.1% versus 44.2%), including Grade 3 neutropenia (48.4% versus 29.0%), Grade 4 neutropenia (17.8% versus 13.4%), and serious neutropenia (7.4% versus 2.7%). Diarrhea occurred more frequently in patients treated with sacituzumab govitecan than with TPC (65.1% versus 17.0%), including Grade 3 diarrhea (11.2% versus 0.9%) and serious diarrhea (3.5% versus 0%). Only 1 patient discontinued sacituzumab govitecan due to a notable harm (diarrhea), although 10.9% and 4.7% of patients required sacituzumab govitecan dose reduction due to neutropenia and diarrhea, respectively.
Table 2: Summary of Key Results From the ASCENT Trial
Result | Sacituzumab govitecan | TPC |
|---|---|---|
OS | ||
BM-Neg | ||
Median (95% CI), monthsa | 12.1 (10.7 to 14.0) | 6.7 (5.8 to 7.7) |
Log-rank P value (stratified)b | < 0.0001 | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.476 (0.383 to 0.592) | |
ITT | ||
Median (95% CI), monthsa | 11.8 (10.5 to 13.8) | 6.9 (5.9 to 7.7) |
Log-rank P value (stratified)b | < 0.0001 | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.508 (0.414 to 0.624) | |
Global health status and HRQoL, SPc | ||
Baseline, mean (SD) [n] | 61.9 (21.3) [247] | 56.4 (22.2) [217] |
Change from baseline, mean (SD) [n] | ||
Cycle 2 | –3.8 (21.9) [216] | –1.4 (21.6) [157] |
Cycle 3 | 3.7 (22.6) [186] | –0.7 (23.2) [92] |
Cycle 4 | 3.6 (21.4) [177] | 1.1 (23.9) [71] |
Cycle 5 | 2.5 (23.5) [144] | 0 (21.1) [48] |
Cycle 6 | 3.9 (20.0) [141] | –1.6 (21.2) [36] |
End of treatment | –6.5 (23.1) [164] | –9.4 (20.5) [147] |
PFS | ||
BM-Neg | ||
Median (95% CI), monthsa | 5.6 (4.3 to 6.3) | 1.7 (1.5 to 2.6) |
Log-rank P value (stratified)b | < 0.0001 | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.409 (0.323 to 0.519) | |
ITT | ||
Median (95% CI), monthsa | 4.8 (4.1 to 5.8) | 1.7 (1.5 to 2.5) |
Log-rank P value (stratified)b | < 0.0001 | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.433 (0.347 to 0.541) | |
Time to progression | ||
BM-Neg | ||
Median (95% CI), monthsa | 5.8 (4.8 to 6.9) | 2.1 (1.5 to 2.7) |
Log-rank P value (stratified)b | < 0.0001d | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.406 (0.315 to 0.525) | |
ITT | ||
Median (95% CI), monthsa | 5.6 (4.3 to 6.2) | 2.1 (1.5 to 2.8) |
Log-rank P value (stratified)b | < 0.0001d | |
Stratified Cox regression HR relative to TPC (95% CI)b | 0.429 (0.338 to 0.545) | |
ORR | ||
BM-Neg | ||
n (%) [95% CI] | 82 (34.9) [28.8 to 41.4] | 11 (4.7) [2.4 to 8.3] |
OR (95% CI)e | 10.859 (5.590 to 21.095) | |
P valuef | < 0.0001d | |
ITT | ||
n (%) [95% CI] | 83 (31.1) [25.6 to 37.0] | 11 (4.2) [2.1 to 7.4] |
OR (95% CI)e | 10.994 (5.659 to 21.358) | |
P valuef | < 0.0001d | |
Time to response | ||
BM-Neg | ||
Mean (SD) [n], months | 2.67 (1.91) [82] | 1.86 (0.92) [11] |
ITT | ||
Mean (SD) [n], months | 2.66 (1.91) [83] | 1.86 (0.92) [11] |
Harms, n (%), SP | ||
AEs | 257 (99.6) | 219 (97.8) |
SAEs | 69 (26.7) | 63 (28.1) |
WDAEs | 12 (4.7) | 12 (5.4) |
Deaths | 1 (0.4) | 3 (1.3) |
Notable harms, n (%), SP | ||
Neutropeniag | 168 (65.1) | 99 (44.2) |
Grade 3 neutropenia | 125 (48.4) | 65 (29.0) |
Grade 4 neutropenia | 46 (17.8) | 30 (13.4) |
Serious neutropenia | 19 (7.4) | 6 (2.7) |
Neutropenia leading to study drug discontinuation | 0 | 3 (1.3) |
Neutropenia leading to dose interruption | 120 (46.5) | 48 (21.4) |
Neutropenia leading to dose reduction | 28 (10.9) | 43 (19.2) |
Hypersensitivityh | 88 (34.1) | 46 (20.5) |
Diarrhea | 168 (65.1) | 38 (17.0) |
Grade 3 diarrhea | 29 (11.2) | 2 (0.9) |
Grade 4 diarrhea | 0 | 0 |
Serious diarrhea | 9 (3.5) | 0 |
Diarrhea leading to study drug discontinuation | 1 (0.4) | 0 |
Diarrhea leading to dose interruption | 14 (5.4) | 1 (0.4) |
Diarrhea leading to dose reduction | 12 (4.7) | 99 (44.2) |
AE = adverse event; BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; HRQoL = health-related quality of life; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice; WDAE = withdrawal due to adverse event.
aMedian OS, PFS, and time to progression are from Kaplan-Meier estimates. CI for the median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cMeasured using European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30. Data at baseline and cycles 2 to 6 are presented while n was greater than 25 in both arms, as well as at the end of treatment.
dOutside statistical hierarchy; P values not adjusted for multiplicity.
eExact binomial CI for proportion is based on the beta distribution.
fP value is based on the Cochran-Mantel-Haenszel test.
gIncludes preferred terms neutropenia, febrile neutropenia, and neutrophil count decreased.
hIncludes “hypersensitivity” standardized Medical Dictionary for Regulatory Activities query (broad) and “anaphylactic reactions” standardized Medical Dictionary for Regulatory Activities query (broad); only events whose onset dates were on the day of or 1 day after an infusion were included. Includes preferred terms cough, dyspnea, rash, pruritus, stomatitis, hypotension, rash maculo-papular, rhinitis allergic, erythema, hypersensitivity, conjunctivitis, flushing, chest discomfort, dermatitis acneiform, rash pustular, rash macular, rash pruritic, bronchospasm, dermatitis contact, eye pruritis, mouth ulceration, edema, seasonal allergy, skin exfoliation, swollen tongue, urticaria, wheezing, choking, and localized edema.
Source: ASCENT Clinical Study Report.13
Critical Appraisal
Most of the notable limitations of the ASCENT trial were tied to its OL design. Although outcome assessment of PFS and OS was conducted by a blinded IRC, patient-reported HRQoL and harms outcomes may have been influenced to some degree by knowledge of treatment allocation. The decision to discontinue patients from therapy was made by investigators based on unblinded review of local imaging results and/or clinical assessments, and biased decision-making could have altered exposure to sacituzumab govitecan and/or TPC. Higher proportions of patients randomized to the TPC arm discontinued the study before receiving protocol therapy, during treatment, and during survival follow-up, and more PFS events were censored in the TPC arm due to initiation of other anticancer therapy and missed assessments. In addition, the magnitude of bias due to screening failures of unknown cause could not be evaluated as these data were not provided on a per-patient basis. The absence of formal statistical comparison and high amounts of missing HRQoL data (due to deaths and dropouts) limited interpretation of potentially important changes in this end point.
The demographic and disease characteristics of the ASCENT study population were broadly reflective of the Canadian population with mTNBC. Of note, the study enrolled small numbers of patients with locally advanced disease who had not received prior therapies in the metastatic setting (approximately 3%). According to the sponsor and the clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who had been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication. Thus, enrolment of these patients in the trial would not impact generalizability. A potentially important issue limiting generalizability to Canadian patients with mTNBC was the use of granulocyte colony–stimulating factor (G-CSF) to counteract neutropenia in approximately half of patients in the sacituzumab govitecan arm. According to the clinical experts, limited access to G-CSF in Canada may mean that dose reductions would be required in higher numbers of patients; the potential impact on efficacy is unclear. Generalizability to patients not included in the study (e.g., patients with ECOG Performance Status 2, patients who have not previously received taxanes, patients receiving sacituzumab govitecan in earlier lines of therapy) could not be evaluated.
Indirect Comparisons
No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Conclusions
Evidence from the ASCENT trial suggested that compared with TPC, administration of sacituzumab govitecan (10 mg/kg on days 1 and 8 of a 21-day treatment cycle) contributed to statistically significant and clinically meaningful prolongation of PFS and OS among patients with locally advanced TNBC or mTNBC who had received at least 2 prior therapies. ORRs were higher, and time to progression was longer, in patients treated with sacituzumab govitecan than in those treated with standard chemotherapy. Analyses of European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) data could not be interpreted due to absence of formal statistical testing and high rates of missing data resulting from deaths and withdrawals. The magnitude of the observed survival benefits and the potential impact on cancer symptoms are important outcomes to patients with mTNBC. Notable harms associated with sacituzumab govitecan (including neutropenia and diarrhea) were not insignificant but manageable with appropriate supportive care (including G-CSF) and dose modification and rarely required withdrawal of treatment. Minor limitations of the available evidence included bias in favour of sacituzumab govitecan on the part of patients and investigators due to the OL design of the ASCENT trial, which may have decreased exposure to TPC relative to sacituzumab govitecan, as well as potential for higher dose reduction and discontinuation rates for Canadian patients in the absence of G-CSF.
Introduction
Disease Background
TNBC, defined by the absence of cell-surface ER, PR, and HER2, is associated with an aggressive phenotype and poor prognosis.1,2 The disease is more common among younger women and Black women and often results in visceral metastasis.1,3 TNBC is characterized by heterogeneous molecular and immunological characteristics14 and has worse rates of PFS and OS than other breast cancer subtypes.1,2
More than 27,000 Canadians are diagnosed with breast cancer each year.4 Although the majority will be cured of their disease, up to 15% have TNBC (approximately 4,000 patients per year),1,2 which carries a higher risk of early recurrence and presentation as stage 4 metastatic disease. The exact prevalence and incidence of adult patients with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease, is uncertain. As noted in the CADTH Pharmacoeconomic Review, the sponsor estimated that the total number of patients with unresectable locally advanced TNBC or mTNBC was 633 in 2020. This estimation included incident cases of de novo mTNBC (calculated by applying the incidence of breast cancer in Canada according to the Canadian Cancer Statistical Advisory Committee to the population of Canada, then multiplying by the proportion with metastasis at presentation and the proportion with TNBC based on a study of Ontario women)15 as well as prevalent cases from prior years that had recurred or spread (calculated by multiplying the prevalence of breast cancer in Canada according to Statistics Canada by the annual probability of recurrence according to the Institut national d’excellence en santé et en services sociaux [INESSS] and the proportion of recurrent patients with TNBC based on a study of Ontario women). The vast majority (600) of the estimated 633 patients would be candidates for sacituzumab govitecan.5,6 Five-year OS of TNBC following neoadjuvant therapy is approximately 64%.16 Compared with other breast cancer subtypes, TNBC is highly invasive and frequently results in locally advanced disease (spread to nearby tissues and lymph nodes) or metastatic (distant spread) disease.1,2 Diagnosis of locally advanced TNBC or mTNBC is made by a medical oncologist based on pathological or biopsy and imaging results. Symptoms including pain, fatigue, and insomnia impose significant financial burdens as well as limitations on patients’ ability to work, caregiving responsibilities, physical activity, and ability to spend time with loved ones. Despite advances in understanding of the disease and development of new treatments, the median OS of advanced stage 4 TNBC is approximately 12 months to 14 months, which is lower than other breast cancer subtypes.7 Disease recurrence is often associated with metastasis to the bones, lungs, liver, and brain with significant symptomology.17 According to members of the Rethink Breast Cancer Scientific Advisory Committee (Appendix 2), this is often a debilitating and symptomatic death that occurs in younger women, and a diagnosis of mTNBC is devastating to patients and their families.
Standards of Therapy
Over the last several decades, hormone and targeted therapies have dramatically altered the landscape of other breast cancer subtypes, and multiple treatments are now available.10,18-20 Although immunotherapies (e.g., atezolizumab, pembrolizumab) have shown great promise in other tumour types, results in TNBC have been modest and/or conflicting.21 In patients with BRCA1 or BRCA2 mutations, PARP inhibitors such as olaparib may induce responses, although the survival benefit is not clear. According to clinical experts consulted by CADTH for this review, immunotherapies, PARP inhibitors, and nab-paclitaxel are not available and not funded in Canada, and special access programs are now closed. Thus, for recurrent or advanced TNBC, sequential single-agent chemotherapy until all options have been exhausted remains the standard of care.8 Each line of therapy has diminishing response; although initial lines of therapy may provide a few months of PFS, this decreases substantially with later lines.
Neoadjuvant chemotherapy, surgery and radiation, and adjuvant chemotherapy with curative intent continue to be mainstays for treatment of more limited TNBC. However, distant recurrence within several years of treatment will occur in many patients.
According to clinical experts, standard chemotherapies for locally advanced TNBC or mTNBC include taxanes (e.g., paclitaxel, docetaxel), platinum agents, capecitabine, gemcitabine (with or without cisplatin or carboplatin), anthracyclines (doxorubicin), eribulin, and occasionally vinorelbine.22 There is jurisdictional variation in treatment sequence across Canada. In the neoadjuvant and adjuvant setting, taxanes, anthracyclines, cyclophosphamide, and/or fluorouracil with or without platinum agents are given. Patients with de novo metastasis who are treatment naive would likely receive taxanes as first-line therapy. For patients who have previously received (neo)adjuvant therapy, first-line treatment in the metastatic setting can include taxane reuse, capecitabine, or cisplatin plus gemcitabine. Most patients develop PD after first-line therapy, then receive other standard chemotherapy options in variable order (e.g., taxane or cisplatin plus gemcitabine second line; anthracycline third line). Chemotherapy regimens are usually administered until PD, unacceptable toxicity, decline in performance status, or patient discontinuation. According to the clinical experts, these treatments have disappointingly low response rates (5% to 10%), and benefit beyond the first or second line (typically anthracyclines and taxanes) has not been convincingly demonstrated. Many patients quickly become refractory to treatment. Chemotherapy does not modify the underlying disease mechanism of mTNBC but can temporarily delay progression in some patients with pre-treated mTNBC,9,10 but often at the cost of side effects that impair HRQoL. OS in this population has not changed in 20 years.10
According to the clinical experts, the main goals of systemic therapy for mTNBC are to extend survival and delay progression with minimal toxicity while maintaining or improving HRQoL (compared with existing treatments). Additional goals of therapy are to maintain or improve organ function, reduce cancer symptoms, maintain patient independence, reduce caregiver burden, and minimize financial burdens and inconvenience for the patient. Better tolerated and more convenient therapies may help improve patient compliance.
Drug
Key characteristics of sacituzumab govitecan are shown in Table 3. Sacituzumab govitecan is administered at a dose of 10 mg/kg as an IV infusion on day 1 and day 8 of a 21-day treatment cycle. Sacituzumab govitecan is indicated for the treatment of adult patients with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. According to the sponsor and clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who had been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication. Sacituzumab govitecan was approved by the FDA in 2020 with the same indication and is undergoing accelerated review by the European Medicines Agency for the indication: “for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received at least two prior therapies, including at least one prior therapy for locally advanced or metastatic disease.”
Sacituzumab govitecan is an antibody-drug conjugate based on a humanized IgG1:k monoclonal antibody against Trop-2, a transmembrane protein involved in calcium signal transduction that is overexpressed in many epithelial cancers, including TNBC. The camptothecin-derived topoisomerase I inhibitor SN-38 is an active metabolite of irinotecan and is linked to the antibody by a hydrolyzable linker. The mechanism of action of sacituzumab govitecan is binding to Trop-2-expressing cancer cells followed by internalization23,24 and subsequent release of SN-38 via hydrolysis of the linker. SN-38 interacts with topoisomerase I and prevents religation of topoisomerase I–induced single strand breaks; the resulting DNA damage leads to tumour cell death. Sacituzumab govitecan releases its SN-38 payload both intra- and extracellularly in the tumour microenvironment.25,26 A higher amount of SN-38 is delivered to Trop-2-expressing tumours compared with conventional irinotecan therapy.27 Extracellular release of SN-38 from sacituzumab govitecan also results in bystander killing of Trop-2–negative tumour cells.28-30
Table 3: Key Characteristics of Sacituzumab Govitecan and Systemic Chemotherapy for mTNBC
Characteristic | Sacituzumab govitecan | Single-agent chemotherapy (e.g., eribulin) |
|---|---|---|
Mechanism of action | Binding and internalization of sacituzumab govitecan into Trop-2-expressing tumour cells followed by release of SN-38, DNA damage, and cell death | Microtubule inhibition |
Indicationa | For the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received 2 or more prior therapies, at least 1 of them for metastatic disease | For the treatment of patients with metastatic breast cancer who have previously received at least 2 chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane administered in either the adjuvant or metastatic setting. |
Route of administration | IV | IV |
Recommended dose | 10 mg/kg on day 1 and day 8 of a 21-day treatment cycle | 1.4 mg/m2 on day 1 and day 8 of a 21-day treatment cycle |
Serious adverse effects or safety issues | Severe neutropenia, diarrhea | Neutropenia, QT/QTc interval prolongation |
mTNBC = metastatic triple-negative breast cancer; Trop-2 = trophoblast cell-surface marker 2.
aHealth Canada–approved indication.
Source: CADTH review submission;5 draft product monograph for sacituzumab govitecan;31 product monograph for eribulin (Halaven).32
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. Raw patient group input can be found in Appendix 1.
Input was provided by 2 patient groups for this review: Rethink Breast Cancer and CBCN. Rethink Breast Cancer conducted online patient surveys in June and July 2021; of the 30 respondents with mTNBC (22 from the US and 6 from Canada), 4 with direct experience with sacituzumab govitecan participated in telephone interviews. CBCN distributed online surveys in 2012 and 2017 to patients living with breast cancer registered in the membership databases of CBCN and other patient organizations; of the 157 respondents, data from 14 patients with mTNBC were captured in the input, and 1 Canadian patient with direct experience with sacituzumab govitecan participated in a telephone interview.
Patients highlighted the negative impacts of mTNBC, including spread to the bone, liver, lungs, and brain. Symptoms frequently included pain, fatigue, and insomnia and imposed significant financial burdens and limitations on patients’ ability to work, caregiving responsibilities, physical activity, and ability to spend time with loved ones. Patients highlighted the limited treatment options for mTNBC and their experiences with prior therapies (chemotherapy and immunotherapy), including their limited effectiveness in delaying progression, managing symptoms, and maintaining HRQoL, as well as their sometimes-severe side effects (e.g., nausea and/or vomiting, fatigue, hand-foot syndrome). Twenty patients contacted by RBC and 1 patient contacted by CBCN had direct experience with sacituzumab govitecan. The patients felt that the drug was effective in controlling disease, extending survival, maintaining HRQoL, and reducing mTNBC symptoms (e.g., Jacksonian marches, bone pain, neuropathy) and noted that side effects (e.g., fatigue, alopecia, diarrhea, neutropenia) were manageable but that not all patients were currently able to access the drug.
Patients with mTNBC identified an important unmet need for treatments that control disease progression, prevent recurrence, and extend survival. Other needs identified by patients were treatments that maintain HRQoL, reduce symptom severity, and have a manageable safety profile.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adults with unresectable locally advanced TNBC or mTNBC who had received 2 or more prior therapies, at least 1 of them for metastatic disease.
Unmet Needs
Clinical experts highlighted the aggressive nature of mTNBC and its poor survival outcomes compared with other breast cancer subtypes, as well as the limited efficacy of current treatment options for later-line therapy. Unlike other subtypes of breast cancer, hormone and targeted therapies are not available to patients with mTNBC.
Place in Therapy
According to clinical experts, sacituzumab govitecan would serve as an additional line of treatment and would serve the same purpose as existing therapies (e.g., chemotherapy, immunotherapy, olaparib). Sacituzumab govitecan would not be expected to modify the underlying disease process of mTNBC but instead target Trop-2, which is present in more than 90% of tumours. Sacituzumab govitecan would be administered for later-line treatment after at least 2 prior lines of chemotherapy (at least 1 taxane in the adjuvant, neoadjuvant, or advanced setting and at least 1 therapy in the metastatic setting). However, 1 clinician suggested that for earlier-line therapy of patients who are not candidates for chemotherapy, sacituzumab govitecan might be considered. Sacituzumab govitecan would not be reserved for patients who are intolerant to other treatments or in whom other treatments are contraindicated. Clinicians expected that sacituzumab govitecan may cause a shift in the current mTNBC treatment paradigm by providing an option for targeted therapy, which is often better tolerated, after at least 2 lines of chemotherapy. Standard chemotherapies could then be considered following progression on sacituzumab govitecan.
Patient Population
Clinical experts stated that any patient with mTNBC who had received at least 2 prior lines of systemic therapy (1 taxane in the adjuvant, neoadjuvant, or advanced setting and 1 systemic therapy in the metastatic setting) with adequate performance status (ECOG 0 to 2 according to 1 expert), adequate hematological and organ function, and stable CNS disease (for patients with BM) would be a candidate for sacituzumab govitecan. Patients best suited for treatment would be identified by the primary treating physician based on pathology or biomarker assessment of ER, PR, and HER2 status and imaging or biopsy to confirm metastatic disease. Diagnosis is not challenging in routine clinical practice, and misdiagnosis or underdiagnosis is not a concern. Pre-symptomatic patients with evidence of PD following 2 lines of prior chemotherapy would also be candidates for sacituzumab govitecan. Patients who do not have mTNBC (who could receive hormone or other targeted therapies), patients with poor performance status, patients with inadequate hematological and organ function, patients with unstable CNS disease, and patients who have not yet tried taxanes would be least suited for treatment with sacituzumab govitecan. No biomarkers of response to sacituzumab govitecan are available.
Assessing Response to Treatment
Response to treatment is assessed by serial imaging demonstrating stable or shrinking disease (objective responses), laboratory markers, and clinical assessment, as well as more subjective measures such as maintained or improved HRQoL, cancer symptoms, and functional status. The outcomes used in clinical practice are the same as those used in trials. Clinically meaningful responses to treatment could be manifested by radiographic tumour response, improved survival, symptomatic stabilization, improvement or reduction in symptom frequency or severity (e.g., pain, dyspnea), stabilization of performance status, and prolonged independent ability to perform activities of daily living. Response to treatment should be assessed at each follow-up visit, with serial imaging typically performed at an 8-week to 12-week interval, with toxicity and safety assessments more often during early treatment (every 2 weeks to 4 weeks) or as needed.
Discontinuing Treatment
Treatment is usually continued in patients who have stable disease or objective responses based on tumour imaging. Treatment would be discontinued in patients with radiographically documented PD; in patients with significant worsening of symptoms, performance status, or HRQoL; and in patients with significant and persistent side effects from treatment (especially uncontrolled Grade 3 or Grade 4 diarrhea).
Prescribing Conditions
According to clinical experts, treatment with sacituzumab govitecan would be initiated by a medical oncologist or associated team physician with expertise in cancer therapies and toxicity management. Sacituzumab govitecan would be administered in a hospital setting or a specialty clinic with the expertise and staffing (chemotherapy nurses, oncology pharmacists) to administer systemic therapies and manage treatment-related toxicities.
Additional Considerations
Clinical experts emphasized that the benefits of sacituzumab govitecan demonstrated in the ASCENT trial11,12 are meaningful and aligned with patient values. The toxicities are predictable and manageable by medical oncologists. The drug would be a valuable addition for use in heavily pre-treated, treatment-resistant patients with mTNBC.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. Raw clinician group input can be found in Appendix 2.
Three clinician groups provided input for this review: the Ontario Health–Cancer Care Ontario Breast Cancer Drug Advisory Committee (3 medical oncologists and 1 pharmacist), the Ottawa Hospital Cancer Centre Breast Medical Oncology group (4 medical oncologists), and the Rethink Breast Cancer Scientific Advisory Committee (6 medical oncologists from across Canada). No major contrary views were presented: Clinical groups echoed the lack of effective options for later-line therapy of mTNBC. Sacituzumab govitecan could change the treatment paradigm and be used before drugs such as vinorelbine and gemcitabine or before reuse of agents such as doxorubicin.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact the ability of drug programs to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Implementation issues | Clinical experts’ response |
|---|---|
Relevant comparators | |
How does sacituzumab govitecan compare to other chemotherapy agents used in triple-negative breast cancer regimens (other than eribulin, gemcitabine, capecitabine, and vinorelbine)? | Treatments that are not represented in the ASCENT trial were likely used in an earlier line of therapy. Comparison of sacituzumab govitecan with other chemotherapies would likely show similar results. |
Considerations for initiation of therapy | |
Should patients with ECOG Performance Status of 2 or greater be eligible for sacituzumab govitecan? | No, as this would deviate from the study population. In the real world there would likely be some indication creep, but this would probably not have a large impact given the disease condition and survival expectations among patients with ECOG Performance Status of 2 or greater. |
In the ASCENT trial, inclusion criteria included previous exposure to taxanes. If a patient did not receive taxanes previously due to contraindications or intolerance, is that patient eligible for treatment with sacituzumab govitecan? | Yes. In the real world, patients would likely still be offered sacituzumab govitecan, although this situation would be rare for third- or further-line therapy. |
Considerations for continuation or renewal of therapy | |
None identified. | Not applicable. |
Considerations for discontinuation of therapy | |
None identified. | Not applicable. |
Considerations for prescribing of therapy | |
In comparison with currently available treatments, would you expect that sacituzumab govitecan will require more nursing resources and chair time? | Yes. Although the dosing schedules for sacituzumab govitecan and chemotherapy are similar, the first infusion of sacituzumab govitecan is approximately 3 hours, with subsequent infusions being 1 hour to 2 hours. This is due to concerns regarding infusion reactions that are mitigated by premedication (e.g., antihistamines, steroids). Comparator chemotherapies require much shorter chair times than sacituzumab govitecan. In rural oncology satellite sites, sacituzumab govitecan may not be initially accessible due to human resource limitations, monitoring difficulties, potential for adverse reactions, and drug wastage concerns. However, additional sites are likely to be added over time and as additional experience with the drug is gained. |
Generalizability | |
Should patients with ECOG Performance Status of 2 or greater be eligible for sacituzumab govitecan? | No, as this would deviate from the study population. In the real world there would likely be some indication creep, but this would probably not have a large impact given the disease condition and survival expectations among patients with ECOG Performance Status of 2 or greater. |
Funding algorithm (oncology only) | |
Do you expect that sacituzumab govitecan would impact the treatment paradigm such that administration of comparator chemotherapy regimens, previous lines of therapy, and subsequent lines of therapy will be impacted? Is there a certain subpopulation that would be mainly impacted? | Yes, potentially. Sacituzumab govitecan is a new treatment and appears to be tolerated with manageable side effects. If it is available, most patients will use it in the second- or third-line setting. If eligible, patients will likely use it as early as possible according to the indication. The impact on the treatment paradigm is not clear yet. |
Care provision issues | |
Sacituzumab govitecan is supplied as a 180 mg vial of lyophilized powder. The dosage of sacituzumab govitecan is 10 mg/kg on day 1 and day 8 of a 21-day treatment cycle. Do you expect drug wastage to occur? | This would depend on whether vial sharing is permitted. If a vial expires after 1 day, centres will schedule all patients on the same day to reduce spoilage, but this may be challenging due to the extended chair time. |
The preparation of sacituzumab govitecan requires a sterile compounding pharmacy, and the final product stability is also very short (4-hour storage at 4°C to 8°C, with administration within 4 hours, including infusion time, according to the FDA; 4-hour storage at 2°C to 8°C, with administration within 6 hours, including infusion time, according to Health Canada product monograph). In your opinion, which settings in Canada would be able to administer sacituzumab govitecan successfully? | Any site in Canada with the capacity to mix and administer IV chemotherapy, such as major cancer centres, would be able to successfully administer sacituzumab govitecan. The situation for rural and satellite sites is less certain. Administration by smaller sites could exacerbate drug wastage. Procedural modifications for administration may be needed depending on how long sacituzumab govitecan takes to prepare, local human resource constraints, and potential for vial sharing. For example, some centres may be able to arrange for administration of all sacituzumab govitecan on specific days. |
Hormone receptor status and HER2 are standard tests done in jurisdictions for metastatic breast cancer. | No response. For pERC consideration. |
System and economic issues | |
The sponsor estimates a 3-year pan-Canadian budget of $44 million, based on a market uptake of ||, ||, and || in years 1 to 3, respectively. PAG is concerned that market uptake may be underestimated since sacituzumab govitecan may represent the new standard of care for patients who meet the ASCENT trial criteria. | Refer to CADTH Pharmacoeconomic Review.6 |
Chair time and additional pharmacy and nursing resources will be required for administration and preparation of sacituzumab govitecan. | Refer to CADTH Pharmacoeconomic Review.6 |
Comparators used in the ASCENT trial are rather generic or have confidential prices. | Refer to CADTH Pharmacoeconomic Review.6 |
ECOG = Eastern Cooperative Oncology Group; HER2 = human epidermal growth factor receptor 2; PAG = Provincial Advisory Group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Committee.
Clinical Evidence
The clinical evidence included in the review of sacituzumab govitecan is presented in the Systematic Review section and includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. No indirect evidence was provided by the sponsor or met the selection criteria specified in the review. No additional relevant studies were identified that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review: Pivotal and Protocol-Selected Studies
Objectives
To perform a systematic review of the beneficial and harmful effects of sacituzumab govitecan (IV injection, 10 mg/kg) for the treatment of adult patients with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented below was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults (age ≥ 18 years) with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease Subgroups:
|
Intervention | Sacituzumab govitecan (10 mg/kg, administered as an IV infusion on day 1 and day 8 of a 21-day treatment cycle) |
Comparator(s) | Single- or multi-agent chemotherapy (e.g., eribulin, vinorelbine, gemcitabine, capecitabine, carboplatin, cisplatin-gemcitabine, carboplatin-gemcitabine, doxorubicin) |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; HRQoL = health-related quality of life; mTNBC = metastatic triple-negative breast cancer; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; TNBC = triple-negative breast cancer; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS (Peer Review of Electronic Search Strategies) checklist.33
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid and Embase (1974‒) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Trodelvy (sacituzumab govitecan). Clinical trial registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 3 for the detailed search strategies.
The initial search was completed on July 28, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee on December 1, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature tool.34 Included in this search were the websites of regulatory agencies (the FDA and the European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 3 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
Only 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 4.
Table 6: Details of the ASCENT Study
Detail | Description |
|---|---|
Designs and populations | |
Study design | Phase III, OL, multi-centre RCT |
Locations | 82 sites in Europe (Belgium, France, Germany, Spain, and the UK) and North America (US and Canada) |
Patient enrolment dates | November 7, 2017, to September 2019a |
Data cut-off | March 11, 2020 |
Randomized (N) | 529 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Sacituzumab govitecan (10 mg/kg, administered as an IV infusion on day 1 and day 8 of a 21-day treatment cycle) |
Comparators | TPC selected from one of:
|
Duration | |
Phase | |
Screening | 4 weeks |
OL treatment | Until progression requiring discontinuation of further treatment, unacceptable toxicity, study withdrawal, or death, whichever came first |
Survival follow-up | Until study withdrawal, death, or data cut-off, whichever came first |
Outcomes | |
Primary end points | PFS (BM-Neg) |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publicationsc | Bardia et al. (2021a)11 Bardia et al. (2021b)12 |
ADA = anti-drug antibody; AE = adverse event; ALT = alanine aminotransferase; ASCO = American Association for Clinical Oncology; AST = asparagine aminotransferase; BM = brain metastasis; BM-Neg = brain metastasis–negative; CAP = College of American Pathologists; CNS = central nervous system; COPD = chronic obstructive pulmonary disease; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; HBV = hepatitis B virus; HCV = hepatitis C virus; HER2 = human epidermal growth factor receptor 2; HRQoL = health-related quality of life; ITT = intention to treat; IULN = institutional upper limit of normal; OL = open label; ORR = objective response rate; OS = overall survival; PARP = poly-(ADP-ribose) polymerase; PFS = progression-free survival; PR = progesterone receptor; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; TNBC = triple-negative breast cancer; TPC = treatment of physician’s choice; Trop-2 = trophoblast cell-surface marker 2.
aThe calendar date in September 2019 on which enrolment ended was not stated.
bFor weekly vinorelbine injection, a cycle was defined as 3 weeks.
cOne additional report was included (ASCENT Clinical Study Report).
Source: ASCENT Clinical Study Report.13
Description of Study
ASCENT (IMMU-132-05, N = 529)11,12 was a phase III, randomized, OL, multi-centre study funded by Immunomedics (acquired in 2020 by the sponsor, Gilead Sciences). The primary objective of the study was to compare the efficacy of sacituzumab govitecan (10 mg/kg) with chemotherapy (TPC) in prolonging PFS among adult patients with locally advanced TNBC or mTNBC previously treated with at least 2 systemic chemotherapy regimens for unresectable locally advanced TNBC or mTNBC. Of note, the study enrolled a small number of patients with locally advanced disease who had not received prior therapy in the metastatic setting. According to the sponsor and clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who had been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication. Patients (adults aged ≥ 18 years with unresectable locally advanced TNBC or mTNBC who had received at least 2 prior therapies) were enrolled from November 7, 2017, until September 2019 at 82 sites in Europe and North America (3 sites; n = 5 patients in Canada). Patients were screened for eligibility within 4 weeks of initiating protocol therapy to assess eligibility.
Patients were randomized 1:1, using an interactive web response system, to receive sacituzumab govitecan (10 mg/kg IV infusion) or TPC (1 of eribulin, capecitabine, gemcitabine, or vinorelbine). Patients with Grade 2 neuropathy were ineligible to receive vinorelbine. The randomization procedure was not explicitly stated. Randomization was stratified by number of prior therapies (2 to 3 versus > 3), BM status (BM-Pos versus BM-Neg), and region (North America versus rest of world). Baseline assessments (tumour imaging, HRQoL) were conducted within 4 weeks of starting protocol therapy. Patients were treated until progression requiring discontinuation of further treatment, unacceptable toxicity, study withdrawal, or death, whichever came first. Treatment was also discontinued due to treatment delay of more than 3 weeks for any reason, pregnancy, physician decision, or withdrawal of consent. The first determination of PD did not require treatment discontinuation if the patient still derived benefit from therapy in the opinion of the investigator (and if agreed by the sponsor). However, treatment was discontinued if subsequent imaging documented PD. Following treatment discontinuation, patients entered survival follow-up until study withdrawal, death, or data cut-off, whichever came first. Crossover was not permitted. The database was closed on March 11, 2020.
Populations
Inclusion and Exclusion Criteria
Key inclusion and exclusion criteria for the ASCENT study are summarized in Table 6. Adult patients (age ≥ 18 years) with unresectable locally advanced or metastatic (CT or MRI confirmed) TNBC (histologically or cytogenetically confirmed as per American Association for Clinical Oncology or College of American Pathologists criteria) were eligible if disease was either refractory or relapsed after at least 2 prior standard of care systemic chemotherapy regimens for unresectable locally advanced TNBC or mTNBC. Poly-(ADP-ribose) polymerase inhibitors qualified as 1 prior therapy for patients with BRCA mutations, and chemotherapy in the (neo)adjuvant setting for more limited disease qualified as 1 prior therapy if development of unresectable locally advanced TNBC or mTNBC occurred within 1 year of completing chemotherapy. Patients had to have previously received taxanes in the adjuvant, neoadjuvant, or advanced setting. Only patients with CT or MRI measurable disease according to Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) and with ECOG Performance Status 0 or 1 were eligible, and for patients with baseline BM, brain MRI was required to show stable CNS disease. Other inclusion criteria were cessation of high-dose corticosteroids and prior cancer therapies, recovery from previous toxicities, eligibility for at least 1 TPC regimen, adequate hematological and organ function, and life expectancy of ≥ 3 months as judged by the investigator. Pregnant or lactating women and patients unwilling to use effective contraception were excluded, as were patients with Gilbert disease, HIV, hepatitis B virus, hepatitis C virus, active inflammatory bowel disease, and recent infections requiring antibiotic use. Patients with a history (≤ 6 months) of gastrointestinal disorders (bleeding, obstruction, or perforation), chronic obstructive pulmonary disease or other respiratory illness, or cardiac problems (unstable angina, myocardial infarction, congestive heart failure, or cardiac arrhythmia other than stable atrial fibrillation) were excluded. Patients had to be disease-free from prior malignancies — except for non-melanoma skin cancer and carcinoma of the cervix — for at least 3 years.
Baseline Characteristics
The baseline characteristics of participants in the ASCENT study are shown in Table 7. Nearly all patients were women. A total of 468 patients (88.5%) were BM-Neg, while 61 patients (11.5%) were BM-Pos. The mean (SD) age among the BM-Neg and the ITT populations was 54.1 (11.4) years and 54.0 (11.5) years, respectively. Most participants (63.7% of the BM-Neg population and 65.6% of the ITT set) were enrolled at sites in North America (US). Most participants were White (78.8% of the BM-Neg population and 79.0% of the ITT set) and not Hispanic or Latino (86.5% of the BM-Neg population and 87.0% of the ITT set). Baseline demographic characteristics were generally well balanced between study arms.
Table 7: Summary of Baseline Demographic Characteristics of Patients in the ASCENT Trial
Characteristic | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Total (N = 468) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) | |
Sex, n (%) | ||||||
Male | 2 (0.9) | 0 | 2 (0.4) | 2 (0.7) | 0 | 2 (0.4) |
Female | 233 (99.1) | 233 (100.0) | 466 (99.6) | 265 (99.3) | 262 (100.0) | 527 (99.6) |
If female, childbearing potential, n (%) | ||||||
n | 233 | 233 | 466 | 265 | 262 | 527 |
Yes | 59 (25.3) | 54 (23.2) | 113 (24.2) | 70 (26.4) | 60 (22.9) | 130 (24.7) |
No | 174 (74.7) | 179 (76.8) | 353 (75.8) | 195 (73.6) | 202 (77.1) | 397 (75.3) |
Age, years | ||||||
Mean (SD) | 54.2 (11.3) | 54.1 (11.6) | 54.1 (11.4) | 54.0 (11.3) | 54.0 (11.7) | 54.0 (11.5) |
Median (range) | 54 (29 to 82) | 53 (27 to 81) | 54 (27 to 82) | 54 (27 to 82) | 53 (27 to 81) | 54 (27 to 82) |
Age group, n (%) | ||||||
< 50 years | 84 (35.7) | 78 (33.5) | 162 (34.6) | 96 (36.0) | 89 (34.0) | 185 (35.0) |
50 to 64 years | 107 (45.5) | 109 (46.8) | 216 (46.2) | 122 (45.7) | 121 (46.2) | 243 (45.9) |
≥ 65 years | 44 (18.7) | 46 (19.7) | 90 (19.2) | 49 (18.4) | 52 (19.8) | 101 (19.1) |
Region, n (%) | ||||||
North America | 149 (63.4) | 149 (63.9) | 298 (63.7) | 175 (65.5) | 172 (65.6) | 347 (65.6) |
Rest of world | 86 (36.6) | 84 (36.1) | 170 (36.3) | 92 (34.5) | 90 (34.4) | 182 (34.4) |
Race, n (%) | ||||||
Asian | 9 (3.8) | 9 (3.9) | 18 (3.8) | 13 (4.9) | 9 (3.4) | 22 (4.2) |
Black | 28 (11.9) | 28 (12.0) | 56 (12.0) | 28 (10.5) | 34 (13.0) | 62 (11.7) |
White | 188 (80.0) | 181 (77.7) | 369 (78.8) | 215 (80.5) | 203 (77.5) | 418 (79.0) |
Other | 10 (4.3) | 15 (6.4) | 25 (5.3) | 11 (4.1) | 16 (6.1) | 27 (5.1) |
Ethnicity, n (%) | ||||||
Hispanic or Latino | 17 (7.2) | 22 (9.4) | 39 (8.3) | 20 (7.5) | 25 (9.5) | 45 (8.5) |
Not Hispanic or Latino | 205 (87.2) | 200 (85.8) | 405 (86.5) | 234 (87.6) | 226 (86.3) | 460 (87.0) |
Not reported | 7 (3.0) | 5 (2.1) | 12 (2.6) | 7 (2.6) | 5 (1.9) | 12 (2.3) |
Unknown | 6 (2.6) | 6 (2.6) | 12 (12.6) | 6 (2.2) | 6 (2.3) | 12 (2.3) |
Weight, kg | ||||||
Mean (SD) | 72.29 (18.20) | 70.33 (15.80) | 71.31 (17.06) | 71.74 (17.94) | 70.43 (15.74) | 71.09 (16.88) |
Median (range) | 69.45 (37.2 to 132.2) | 68.00 (41.4 to 118.0) | 68.70 (37.2 to 132.2) | 69.10 (37.2 to 132.2) | 68.35 (41.4 to 118.0) | 68.55 (37.2 to 132.2) |
Height, cm | ||||||
Mean (SD) | 163.54 (6.57) | 162.56 (7.20) | 163.05 (6.90) | 163.47 (6.64) | 162.54 (7.54) | 163.01 (7.11) |
Median (range) | 164.00 (142.0 to 185.0) | 163.00 (139.7 to 182.0) | 163.00 (139.7 to 185.0) | 163.70 (142.0 to 185.0) | 162.70 (129.5 to 185.4) | 163.00 (129.5 to 185.4) |
BMI, kg/m2 | ||||||
Mean (SD) | 27.00 (6.59) | 26.67 (6.10) | 26.83 (6.35) | 26.82 (6.48) | 26.74 (6.20) | 26.78 (6.34) |
Median (range) | 25.74 (15.0 to 49.3) | 25.88 (14.6 to 48.2) | 25.88 (14.6 to 49.3) | 25.41 (15.0 to 49.3) | 25.97 (14.6 to 48.2) | 25.82 (14.6 to 49.3) |
Body surface area, m2 | ||||||
Mean (SD) | 1.80 (0.23) | 1.77 (0.21) | 1.79 (0.22) | 1.79 (0.23) | 1.77 (0.21) | 1.78 (0.22) |
Median (range) | 1.76 (1.3 to 2.5) | 1.75 (1.3 to 2.4) | 1.76 (1.3 to 2.5) | 1.76 (1.3 to 2.5) | 1.75 (1.3 to 2.4) | 1.75 (1.3 to 2.5) |
BMI = body mass index; BM-Neg = brain metastasis–negative; ITT = intention to treat; SD = standard deviation; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
The baseline disease characteristics of participants in the ASCENT study are shown in Table 8. Approximately two-thirds of patients (68.8% of the BM-Neg population and 70.3% of the ITT set) were originally diagnosed with TNBC. The mean (SD) time from diagnosis of stage 4 TNBC to study entry was 21.44 (20.92) months in the BM-Neg population and 22.04 (20.768) months in the ITT set. Only 7.3% of the BM-Neg population and 8.1% of the ITT set were positive for either BRCA1 or BRCA2 mutations. Patients were roughly equally divided between ECOG Performance Status 0 (44.0% of the BM-Neg population and 43.3% of the ITT set) and ECOG Performance Status 1 (56.0% of the BM-Neg population and 56.7% of the ITT set). Approximately two-thirds of patients (70.5% of the BM-Neg population and 69.0% of the ITT set) had received 2 to 3 prior systemic chemotherapies, while approximately one-third had received more than 3 prior systemic chemotherapies. The mean (SD) number of prior systemic therapies received was 4.5 (2.1) in both the BM-Neg and the ITT populations. In addition to taxanes, most patients (82.1% of the BM-Neg population and 82.6% of the ITT set) had received cyclophosphamide, approximately two-thirds had received carboplatin (65.6% of the BM-Neg population and 64.8% of the ITT set), approximately two-thirds had received capecitabine (65.4% of the BM-Neg population and 66.9% of the ITT set), approximately half had received doxorubicin (51.9% of the BM-Neg population and 53.5% of the ITT set), approximately one-third had received eribulin (31.8% of the BM-Neg population and 32.7% of the ITT set), and approximately one-third had received gemcitabine (36.1% of both the BM-Neg and the ITT populations). Approximately one-fifth of patients had previously received investigational drugs (20.1% of the BM-Neg population and 20.4% of the ITT set). Approximately half of patients had received previous systemic chemotherapies in the adjuvant setting (57.5% of the BM-Neg population and 58.4% of the ITT set), approximately half had received previous systemic chemotherapies in the neoadjuvant setting (47.9% of the BM-Neg population and 47.1% of the ITT set), while nearly all had received previous systemic chemotherapies in the metastatic setting (97.6% of the BM-Neg population and 97.9% of the ITT set). A small number of patients (n = 12; 2.6% of the BM-Neg population and n = 15; 2.8% of the ITT set) had received previous systemic chemotherapies for locally advanced disease. Nearly all patients had received prior breast cancer–related surgery (94.9% of both the BM-Neg and ITT populations), most had received prior non-brain radiotherapy (81.4% of the BM-Neg population and 81.1% of the ITT set), and approximately one-quarter had received prior PD-1 or PD-L1 therapy (27.1% of the BM-Neg population and 28.9% of the ITT set).
Baseline disease characteristics were generally well balanced between study arms. Slightly higher proportions of patients in the TPC arm had previously received capecitabine (68.2% in the BM-Neg population and 69.8% in the ITT set) and gemcitabine (42.5% in the BM-Neg population and 40.5% in the ITT set) compared with patients in the sacituzumab govitecan arm (capecitabine: 62.6% in the BM-Neg population and 64.0% in the ITT set; gemcitabine: 29.8% in the BM-Neg population and 31.8% in the ITT set).
Table 8: Summary of Baseline Disease Characteristics of Patients in the ASCENT Trial
Characteristic | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Total (N = 468) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) | |
Original diagnosis TNBC, n (%) | ||||||
Yes | 165 (70.2) | 157 (67.4) | 322 (68.8) | 192 (71.9) | 180 (68.7) | 372 (70.3) |
No | 70 (29.8) | 76 (32.60) | 146 (31.2) | 75 (28.1) | 82 (31.3) | 157 (29.7) |
Time from diagnosis of stage 4 TNBC to study entry, months | ||||||
Mean (SD) | 21.53 (22.03) | 21.35 (19.79) | 21.44 (20.92) | 21.74 (21.20) | 22.35 (20.35) | 22.04 (20.77) |
Median (range) | 15.8 (0.1 to 202.9) | 15.15 (–0.4 to 140.1) | 15.29 (–0.4 to 202.9) | 16.82 (0.1 to 202.9) | 15.82 (–0.4 to 140.1) | 16.23 (–0.4 to 202.9) |
UGT1A1 genotype, n (%) | ||||||
*1/*1 | 99 (42.1) | NR | NR | 113 (42.3) | NR | NR |
*1/*28 | 84 (35.7) | NR | NR | 96 (36.0) | NR | NR |
*28/*28 | 30 (12.8) | NR | NR | 34 (12.7) | NR | NR |
Other | 7 (3.0) | NR | NR | 7 (2.6) | NR | NR |
Missing | 15 (6.4) | NR | NR | 17 (6.4) | NR | NR |
BRCA1 or BRCA2 mutational status, n (%)a | ||||||
Negative | 133 (56.6) | 125 (53.6) | 258 (55.1) | 150 (56.2) | 146 (55.7) | 296 (56.0) |
Positive | 16 (6.8) | 18 (7.7) | 34 (7.3) | 20 (7.5) | 23 (8.8) | 43 (8.1) |
ECOG PS at screening, n (%) | ||||||
0: Normal activity | 108 (46.0) | 98 (42.1) | 206 (44.0) | 121 (45.3) | 108 (41.2) | 229 (43.3) |
1: Symptoms but ambulatory | 127 (54.0) | 135 (57.9) | 262 (56.0) | 146 (54.7) | 154 (58.8) | 300 (56.7) |
Baseline serum bilirubin, n (%) | ||||||
≤ ULN | 224 (95.3) | 196 (84.1) | 420 (89.7) | 253 (94.8) | 218 (83.2) | 471 (89.0) |
> 1 and ≤ 1.5 × ULN | 4 (1.7) | 3 (1.3) | 7 (1.5) | 5 (1.9) | 4 (1.5) | 9 (1.7) |
> 1.5 × ULN | 0 | 1 (0.4) | 1 (0.2) | 0 | 1 (0.4) | 1 (0.2) |
Baseline creatinine clearance, mL/min | ||||||
Mean (SD) | 110.72 (38.76) | 109.12 (37.70) | 109.93 (38.20) | 110.95 (38.21) | 110.21 (38.33) | 110.58 (38.24) |
Median (range) | 100.81 (60.17 to 255.50) | 105.00 (53.00 to 253.32) | 102.34 (53.00 to 255.50) | 101.00 (60.17 to 255.50) | 106.58 (53.00 to 260.00) | 104.00 (53.00 to 260.00) |
Number of prior chemotherapies, n (%) | ||||||
2 to 3 | 166 (70.6) | 164 (70.4) | 330 (70.5) | 184 (68.9) | 181 (69.1) | 365 (69.0) |
> 3 | 69 (29.4) | 69 (29.6) | 138 (29.5) | 83 (31.1) | 81 (30.9) | 164 (31.0) |
Number of prior systemic therapies, n (%) | ||||||
Mean (SD) | 4.4 (2.1) | 4.5 (2.1) | 4.5 (2.1) | 4.5 (2.1) | 4.6 (2.1) | 4.5 (2.1) |
Median (range) | 4 (2 to 17) | 4 (2 to 14) | 4 (2 to 17) | 4 (2 to 17) | 4 (2 to 14) | 4 (2 to 17) |
Prior systemic therapies, n (%)b | ||||||
Plant alkaloids and other natural products | 235 (100.0) | 233 (100.0) | 468 (100.0) | 267 (100.0) | 262 (100.0) | 529 (100.0) |
Paclitaxel | 177 (75.3) | 187 (80.3) | 364 (77.8) | 204 (76.4) | 210 (80.2) | 414 (78.3) |
Docetaxel | 88 (37.4) | 75 (32.2) | 163 (34.8) | 101 (37.8) | 83 (31.7) | 184 (34.8) |
Paclitaxel albumin | 42 (17.9) | 37 (15.9) | 79 (16.9) | 49 (18.4) | 51 (19.5) | 100 (18.9) |
Vinorelbine | 14 (6.0) | 7 (3.0) | 21 (4.5) | 17 (6.4) | 7 (2.7) | 24 (4.5) |
Vinorelbine tartrate | 13 (5.5) | 10 (4.3) | 23 (4.9) | 13 (4.9) | 11 (4.2) | 24 (4.5) |
Other antineoplastic agents | 214 (91.1) | 215 (2.3) | 429 (91.7) | 242 (90.6) | 243 (92.7) | 485 (91.7) |
Carboplatin | 147 (62.6) | 160 (68.7) | 307 (65.6) | 164 (61.4) | 179 (68.3) | 343 (64.8) |
Eribulin | 77 (32.8) | 72 (30.9) | 149 (31.8) | 88 (33.0) | 85 (32.4) | 173 (32.7) |
Pembrolizumab | 42 (17.9) | 33 (14.2) | 75 (16.0) | 49 (18.4) | 41 (15.6) | 90 (17.0) |
Bevacizumab | 31 (13.2) | 26 (11.2) | 57 (12.2) | 34 (12.7) | 30 (11.5) | 64 (12.1) |
Cisplatin | 25 (10.6) | 25 (10.7) | 50 (10.7) | 32 (12.0) | 28 (10.7) | 60 (11.3) |
Trastuzumab | 19 (8.1) | 20 (8.6) | 39 (8.3) | 22 (8.2) | 22 (8.4) | 44 (8.3) |
Palbociclib | 18 (7.7) | 22 (9.4) | 40 (8.5) | 18 (6.7) | 25 (9.5) | 43 (8.1) |
Olaparib | 15 (6.4) | 13 (5.6) | 28 (6.0) | 18 (6.7) | 16 (6.1) | 34 (6.4) |
Nivolumab | 13 (5.5) | 12 (5.2) | 25 (5.3) | 16 (6.0) | 12 (4.6) | 28 (5.3) |
Atezolizumab | 11 (4.7) | 16 (6.9) | 27 (5.8) | 14 (5.2) | 22 (8.4) | 36 (6.8) |
Antimetabolites | 196 (83.4) | 205 (88.0) | 401 (85.7) | 226 (84.6) | 231 (88.2) | 457 (86.4) |
Capecitabine | 147 (62.6) | 159 (68.2) | 306 (65.4) | 171 (64.0) | 183 (69.8) | 354 (66.9) |
Gemcitabine | 70 (29.8) | 99 (42.5) | 169 (36.1) | 85 (31.8) | 106 (40.5) | 191 (36.1) |
Fluorouracil | 39 (16.6) | 42 (18.0) | 81 (17.3) | 44 (16.5) | 47 (17.9) | 91 (17.2) |
Gemcitabine hydrochloride | 20 (8.5) | 28 (12.0) | 48 (10.3) | 22 (8.2) | 31 (11.8) | 53 (10.0) |
Alkylating agents | 192 (81.7) | 192 (82.4) | 384 (82.1) | 221 (82.8) | 216 (82.4) | 437 (82.6) |
Cyclophosphamide | 192 (81.7) | 192 (82.4) | 384 (82.1) | 221 (82.8) | 216 (82.4) | 437 (82.6) |
Cytotoxic antibiotics and related substances | 188 (80.0) | 192 (82.4) | 380 (81.2) | 217 (81.3) | 218 (83.2) | 435 (82.2) |
Doxorubicin | 121 (51.5) | 122 (52.4) | 243 (51.9) | 142 (53.2) | 141 (53.8) | 283 (53.5) |
Epirubicin | 50 (21.3) | 55 (23.6) | 105 (22.4) | 55 (20.6) | 59 (22.5) | 114 (21.6) |
Pegylated liposomal doxorubicin hydrochloride | 14 (6.0) | 12 (5.2) | 26 (5.6) | 16 (6.0) | 15 (5.7) | 31 (5.9) |
Hormone antagonists and related agents | 61 (26.0) | 67 (28.8) | 128 (27.4) | 67 (25.1) | 76 (29.0) | 143 (27.0) |
Letrozole | 32 (13.6) | 30 (12.9) | 62 (13.2) | 36 (13.5) | 33 (12.6) | 69 (13.0) |
Tamoxifen | 29 (12.3) | 29 (12.4) | 58 (12.4) | 33 (12.4) | 37 (14.1) | 70 (13.2) |
Fulvestrant | 13 (5.5) | 18 (7.7) | 31 (6.6) | 13 (4.9) | 20 (7.6) | 33 (6.2) |
Anastrozole | 12 (5.1) | 21 (9.0) | 33 (7.1) | 14 (5.2) | 22 (8.4) | 36 (6.8) |
Exemestane | 9 (3.8) | 20 (8.6) | 29 (6.2) | 11 (4.1) | 21 (8.0) | 32 (6.0) |
Immunosuppressants | 16 (6.8) | 20 (8.6) | 36 (7.7) | 16 (6.0) | 23 (8.8) | 39 (7.4) |
Methotrexate | 11 (4.7) | 14 (6.0) | 25 (5.3) | 11 (4.1) | 17 (6.5) | 28 (5.3) |
Uncoded | 50 (21.3) | 44 (18.9) | 94 (20.1) | 56 (21.0) | 52 (19.8) | 108 (20.4) |
Investigational antineoplastic drugs | 43 (18.3) | 34 (14.6) | 77 (16.5) | 48 (18.0) | 42 (16.0) | 90 (17.0) |
Setting of prior systemic therapies, n (%) | ||||||
Adjuvant | 140 (59.6) | 129 (55.4) | 269 (57.5) | 161 (60.3) | 148 (56.5) | 309 (58.4) |
Neoadjuvant | 113 (48.1) | 111 (47.6) | 224 (47.9) | 124 (46.4) | 125 (47.7) | 249 (47.1) |
Metastatic | 226 (96.2) | 231 (99.1) | 457 (97.6) | 258 (96.6) | 260 (99.2) | 518 (97.9) |
Locally advanced disease | 8 (3.4) | 4 (1.7) | 12 (2.6) | 10 (3.7) | 5 (1.9) | 15 (2.8) |
Prior breast cancer–related surgery, n (%) | ||||||
Yes | 222 (94.5) | 222 (95.3) | 444 (94.9) | 252 (94.4) | 250 (95.4) | 502 (94.9) |
No | 13 (5.5) | 11 (4.7) | 24 (5.1) | 15 (5.6) | 12 (4.6) | 27 (5.1) |
Prior non-brain radiotherapy, n (%) | ||||||
Yes | 196 (83.4) | 185 (79.4) | 381 (81.4) | 223 (83.5) | 206 (78.6) | 429 (81.1) |
No | 39 (16.6) | 48 (20.6) | 87 (18.6) | 44 (16.5) | 56 (21.4) | 100 (18.9) |
Prior PD-1 or PD-L1 therapy, n (%) | ||||||
Yes | 67 (28.5) | 60 (25.8) | 127 (27.1) | 79 (29.6) | 74 (28.2) | 153 (28.9) |
No | 168 (71.5) | 173 (74.2) | 341 (72.9) | 188 (70.4) | 188 (71.8) | 376 (71.1) |
TPC, n (%)c | ||||||
Eribulin | 105 (44.7) | 126 (54.1) | 231 (49.4) | 115 (43.1) | 139 (53.1) | 254 (48.0) |
Capecitabine | 44 (18.7) | 31 (13.3) | 75 (16.0) | 48 (18.0) | 33 (12.6) | 81 (15.3) |
Gemcitabine | 41 (17.4) | 29 (12.4) | 70 (15.0) | 46 (17.2) | 38 (14.5) | 84 (15.9) |
Vinorelbine | 45 (19.1) | 47 (20.2) | 92 (19.7) | 58 (21.7) | 52 (19.8) | 110 (20.8) |
BM-Neg = brain metastasis–negative; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention to treat; NR = not reported; PD-1 = programmed cell death protein 1; PD-L1 = programmed death ligand 1; SD = standard deviation; TNBC = triple-negative breast cancer; TPC = treatment of physician’s choice; ULN = upper limit of normal.
aPositive denotes patient was either BRCA1-positive or BRCA2-positive. Negative denotes patient was both BRCA1-negative and BRCA2-negative.
bPrior systemic therapies used in ≥ 5% of either study arm are listed.
cSpecified by investigator before randomization.
Source: ASCENT Clinical Study Report.13
Interventions
Patients were randomized 1:1 to receive either sacituzumab govitecan or TPC. Because of the mixture of oral and IV therapies included in TPC, blinding was not possible. Both sacituzumab govitecan and TPC were administered by study personnel onsite; although not explicitly stated, capecitabine (orally twice daily for 2 weeks) was presumably self-administered. Sacituzumab govitecan was administered at a dose of 10 mg/kg based on data from the phase I/II IMMU-132-01 basket trial, suggesting that this dose had a similar safety profile to 8 mg/kg but was potentially associated with higher ORR.35 Sacituzumab govitecan was administered as an IV infusion on day 1 and day 8 of a 21-day treatment cycle. For the initial sacituzumab govitecan infusion, the initial rate over the first 15 minutes was 50 mg/hour or less; this rate was increased every 15 minutes to 30 minutes by 50 mg/hour, to a maximum of 500 mg/hour. For subsequent sacituzumab govitecan infusions, the initial rate over the first 15 minutes was 100 mg/hour to 200 mg/hour; this rate was increased every 15 minutes to 30 minutes by 100 mg/hour to 200 mg/hour, to a maximum of 1,000 mg/hour.
Eribulin was administered by IV infusion over 2 minutes to 5 minutes at a dose of 1.4 mg/m2 at North American sites and 1.23 mg/m2 at European sites on day 1 and day 8 of a 21-day cycle. Lower doses were administered to patients with moderate hepatic impairment (Child-Pugh score, B; 0.7 mg/m2 and 0.67 mg/m2 for North American and European sites, respectively). Capecitabine (1,000 mg/m2 to 1,250 mg/m2) was administered orally twice daily for 2 weeks, followed by a 1-week rest period, in a 21-day cycle. Gemcitabine (800 mg/m2 to 1,250 mg/m2) was administered by IV infusion over 30 minutes on day 1, day 8, and day 15 of a 28-day cycle. Vinorelbine (25 mg/m2) was administered via weekly IV injections over 6 minutes to 10 minutes. For weekly vinorelbine administration, a cycle was defined as 3 weeks.
Disallowed concomitant medications included other anticancer therapy or chemotherapeutic agents, high-dose corticosteroids, and strong inhibitors or inducers of CYP3A4 (because of a known interaction with irinotecan). Palliative and/or supportive medications, such as bone-modifying medications (bisphosphonates or denosumab), and/or radiation and surgery were allowed at the investigator’s discretion. Hematopoietic growth factors and blood transfusions were permitted. Low-dose, stable doses of corticosteroids (≤ 20 mg prednisone or equivalent daily) were permitted for treatment of BM, as were topical steroids and corticosteroid inhalers.
Premedications administered as primary prophylaxis to prevent infusion reactions to sacituzumab govitecan included antipyretics as well as H1 and H2 blockers (antihistamines). For anaphylactic reactions, appropriate medical measures (e.g., epinephrine, antihistamines, hydrocortisone, IV fluids) were taken. Premedication with a combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated) as primary prophylaxis to prevent as well as to treat chemotherapy- and sacituzumab govitecan–induced nausea and vomiting was strongly recommended. Premedications administered as secondary prophylaxis to prevent infusion reactions to sacituzumab govitecan included corticosteroids (50 mg hydrocortisone or equivalent). Appropriate premedication (e.g., atropine) was administered as secondary prophylaxis to patients who had an excessive cholinergic response to sacituzumab govitecan (e.g., abdominal cramping, diarrhea, salivation). Additional antiemetics, sedatives, and other supportive measures were administered as secondary prophylaxis if clinically indicated. Patients who experienced neutropenia were administered G-CSF for treatment and for secondary prophylaxis in subsequent infusions under the following conditions: Grade 4 neutropenia for at least 7 days; Grade 3 febrile neutropenia (absolute neutrophil count < 1,000/mm3; fever ≥ 38.5°C); or at time of scheduled treatment, Grade 3 or higher neutropenia which delays dose by 2 or 3 weeks for recovery to Grade 1 or lower neutropenia. Patients with diarrhea were treated with loperamide; additional supportive measures (fluid and electrolyte substitution) were used as clinically indicated.
The rules for sacituzumab govitecan therapy interruption, dose reduction, and discontinuation are shown in Table 9. Dose reductions for hematologic toxicity were implemented only after G-CSF was administered for treatment and as secondary prophylaxis in subsequent cycles. Patients on TPC could receive G-CSF per physician discretion, but this was not part of the recommended study protocol.
Table 9: Rules for Sacituzumab Govitecan Dose Interruption, Dose Reduction, and Discontinuation in the ASCENT Trial
Action | Event | Criteria |
|---|---|---|
Dose reduction | Infusion reaction | Grade 2: stopped for 15 minutes or until resolution, then resumed at a slower rate. Grade 1: slow infusion rate. Any infusion reaction must have resolved to < Grade 1 before the next scheduled infusion. |
Hematologic toxicity (used growth factors at any time as clinically indicated, including prophylactically) | If a patient had ≥ Grade 2 neutropenia on cycle 1 day 1, treatment was withheld until the neutropenia reduced to ≤ Grade 1. If treatment was delayed more than 3 weeks, the patient was discontinued from the study. If a patient had ≥ Grade 3 neutropenia in subsequent treatment cycles, treatment was withheld until neutropenia reduced to ≤ Grade 1, and growth factors were administered as clinically indicated. Patients were assessed weekly for Grade 3 and biweekly for Grade 4 neutropenia. If treatment was delayed more than 3 weeks, the patient was discontinued from the study. In the event of ≥ Grade 3 neutropenia on the scheduled treatment day:
Dose reduction scheme Grade 4 neutropenia ≥ 7 days, Grade 3 febrile neutropenia, ≥ Grade 3 neutropenia at time of treatment that delays dose by 2 or 3 weeks for recovery to ≤ Grade 1 neutropenia Occurrence:
| |
GI toxicity | If a patient experienced ≥ Grade 2 GI toxicity on cycle 1 day 1, treatment was withheld until GI toxicity reduced to ≤ Grade 1. Patient was assessed weekly for Grade 3 and biweekly for Grade 4 GI toxicity. If treatment was delayed more than 3 weeks, the patient was discontinued from the study. If a patient experienced ≥ Grade 3 GI toxicity in subsequent treatment cycles, treatment was withheld until GI toxicity reduced to ≤ Grade 1. Patient was assessed weekly for Grade 3 and biweekly for Grade 4 GI toxicity. If treatment was delayed more than 3 weeks, the patient was discontinued from the study. In the event of GI toxicity > Grade 3 on the scheduled treatment day:
| |
Non-hematologic toxicity | Dose reduction scheme
Occurrence:
| |
Discontinuation | Grade 3 to Grade 4 infusion reaction and any delay more than 3 weeks | |
ANC = absolute neutrophil count; G-CSF = granulocyte colony–stimulating factor; GI = gastrointestinal; Hgb = hemoglobin.
Note: Hematologic toxicity was defined as follows: Grade 1 or lower (ANC ≥ 1,500/mm3, platelets ≥ 75,000/mm3, or Hgb ≥ 10.0 g/dL); Grade 3 or higher (ANC < 1,000/mm3, platelets < 50,000/mm3, or Hgb < 8.0 g/dL); and Grade 4 (ANC < 500/mm3, platelets < 25,000/mm3, or Hgb < 6.5 g/dL).
aGranulocyte colony–stimulating factor was administered per National Comprehensive Cancer Network guidelines at a daily dose of 5 mcg/kg (rounding to the nearest vial size) until post-nadir ANC recovery to normal or near-normal levels by laboratory standards.
Source: ASCENT Clinical Study Report.13
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the ASCENT study is provided in Table 10. These end points are further summarized below. A detailed discussion and critical appraisal of the HRQoL outcome measure used in the ASCENT study, EORTC QLQ-C30, is provided in Appendix 6. The EORTC QLQ-C30 has been extensively used in oncology trials and has demonstrated reliability, validity, and responsiveness to change in cancer patients, including patients with mTNBC. The consensus of several studies, including studies of patients with locally advanced TNBC and mTNBC, was that the minimally important difference (MID) on any of the instrument’s scales was approximately 5 points to 10 points.
Table 10: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | ASCENT |
|---|---|
PFS in the BM-Neg population by IRC assessment: time from randomization until tumour progression as defined by RECIST 1.1 (20% increase in sum of target lesions or appearance of new non-target lesions) or death, whichever came first | Primary |
PFS in the ITT set | Secondary |
Time to progression in the BM-Neg population and the ITT set by IRC assessment: time from randomization until tumour progression among patients who progressed | Secondary |
HRQoL in the BM-Neg population and the ITT set assessed using the EORTC QLQ-C30 | Secondary |
OS in the BM-Neg population and the ITT set: time from start of study treatment to death from any cause | Secondary |
ORR in the BM-Neg population and the ITT set by IRC assessment: percentage of patients with confirmed partial responses and complete responses as defined by RECIST 1.1 | Secondary |
Time to response in the BM-Neg population and the ITT set by IRC assessment: time from randomization until the first partial response or complete response as defined by RECIST 1.1 | Secondary |
BM-Neg = brain metastasis–negative; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; IRC = independent review committee; ITT = intention to treat; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
Source: ASCENT Clinical Study Report.13
During the treatment period (until PD or unacceptable toxicity), tumour response was assessed by CT or MRI at baseline, every 6 weeks for the first 9 months, and then every 9 weeks. BM-Pos patients underwent brain MRI at the same time points. For each patient, the same imaging modality was used to assess tumour response throughout the study. Confirmatory scans for responses observed before week 36 were obtained at the next assessment, while confirmatory scans for responses after week 36 were confirmed 36 weeks after the initial response. Patients who discontinued treatment due to toxicity continued with radiological response assessments at the protocol-required schedule until progression of disease or initiation of new therapy. Additional scans to assess disease status were performed at the discretion of the treating physician.
Designation of response was based on the response of target and non-target lesions and the appearance of any new lesions according to RECIST 1.1. PD was defined as a predefined increase (+ 20%) in the sum of target lesions or the appearance of new non-target lesions, taking as reference the smallest sum of target lesions for each patient during the study period. Partial response was defined as at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum of diameters of target lesions. Complete response was defined as the disappearance of all target lesions with reduction of the short axis of any pathological lymph nodes to less than 10 mm. Stable disease was defined as either sufficient shrinkage (compared with baseline) to qualify as partial response but not sufficient increase (taking as reference the smallest sum of diameters of target lesions while on study) to qualify as PD.
All imaging data were reviewed both by the investigator and by a blinded IRC consisting of 2 board-certified radiologists, with discrepancies evaluated by a third board-certified radiologist. Decisions to discontinue protocol therapy due to PD were made by the investigator based on local imaging scans; the decision could also be made to discontinue protocol therapy due to clinical progression, based on the judgment of the investigator, in the absence of radiological evidence. Clinical progression leading to patient discontinuation was also documented by CT or MRI scans of target lesions if possible.
The primary outcome was PFS by IRC assessment in the BM-Neg population, defined as time from randomization until PD or death, whichever came first. For patients who started other anticancer therapies before PD or death, PFS was censored at the last assessment before initiation of new therapy. PFS by IRC assessment in the ITT set was a secondary outcome, as was time to progression in both the BM-Neg population and the ITT set, defined as time from randomization until progression among patients with PD. OS was assessed as a secondary outcome in both the BM-Neg population and the ITT set and was defined as time from start of study treatment until death from any cause. Patients without documentation of death were censored at the last date known alive; OS was not censored for patients who initiated other anticancer therapies. ORR response rate (the proportion of patients with partial or complete responses according to RECIST 1.1) by IRC assessment was evaluated in both the BM-Neg population and the ITT set as a secondary outcome, as was time to response, defined as time from randomization until the first partial or complete response.
During the treatment period, HRQoL was assessed using the EORTC QLQ-C30 every 3 weeks (4 weeks for gemcitabine) on day 1 of each cycle. Following treatment discontinuation, survival follow-up occurred every 4 weeks (in person or by phone) to assess survival status and to document any subsequent anticancer therapy. Survival status was also documented from public databases if allowed by local regulations.
Harms outcomes included treatment-emergent AEs, SAEs, AEs requiring dose interruption or reduction, WDAEs, and AEs of special interest. All AEs that began or worsened on or after the start of protocol therapy until 30 days after the last dose of the study drug were captured. AE were defined as any untoward medical occurrence and were coded according to the Medical Dictionary for Regulatory Activities version 22.1 and graded according to Common Terminology Criteria for Adverse Events 4.03. Incidence was calculated at the system organ class and preferred term levels. SAEs were defined as an untoward medical occurrence that was life threatening, required inpatient hospitalization or prolongation of hospitalization, resulted in persistent or significant disability or incapacity, resulted in death, or was a congenital abnormality, or as another important medical event that required medical or surgical intervention to prevent these outcomes. Adverse events of special interest were defined as shown in Table 11 to capture all AE preferred terms considered to be potentially associated with sacituzumab govitecan treatment based on the results of the phase I/II IMMU-132-01 basket trial.35 Clinical laboratory parameters, vital signs, and electrocardiograms were also monitored.
Table 11: Definition of Adverse Events of Special Interest in the ASCENT Trial
Adverse event | Preferred termsa |
|---|---|
Diarrhea | Diarrhea |
Nausea | Nausea |
Vomiting | Vomiting |
Neutropenia | Neutropenia, neutrophil count decreased, febrile neutropenia |
Febrile neutropenia | Febrile neutropenia |
Infections | SOC: infections and infestations |
Anemia | Anemia, hemoglobin decreased |
Thrombocytopenia | Thrombocytopenia, platelet count decreased |
Fatigue | Fatigue, asthenia |
Neuropathy | Gait disturbance, hypoesthesia, muscular weakness, neuropathy peripheral, paresthesia, and peripheral sensory neuropathy |
Hypersensitivityb | Hypersensitivity SMQ (broad), anaphylactic reactions SMQ (broad) |
Pulmonary events | Interstitial lung disease SMQ (narrow) |
MedDRA = Medical Dictionary for Regulatory Activities; SMQ = standardized MedDRA query; SOC = system organ class.
aWording of AESI preferred terms is according to MedDRA version 22.1.
bFor hypersensitivity, only events whose onset dates were on the day of or 1 day after an infusion were included.
Source: ASCENT Clinical Study Report.13
Statistical Analysis
Statistical analysis of efficacy outcomes in the ASCENT trial is summarized in Table 12. Type I error was controlled using a hierarchical testing strategy. In the 4-part hierarchy, differences in PFS by IRC assessment were tested first in the BM-Neg population. If significant, OS was tested in the BM-Neg population, followed by PFS by IRC assessment in the ITT set and then by OS in the ITT set. If fewer than 30 BM-Pos patients had been recruited, analyses in the ITT population would have been removed from the hierarchy. ORR and time to response were outside the statistical hierarchy and were evaluated in exploratory fashion.
Table 12: Statistical Analysis of Efficacy End Points in the ASCENT Trial
Type | End point | Position in statistical hierarchy | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|---|---|
Primary | PFS in the BM-Neg population | 1 | Log-rank test stratified by randomization factors; KM analysis with median PFS and 95% CIs calculated using the Brookmeyer and Crowley method with log-log transformation; HRs and 95% CIs calculated using stratified Cox proportional hazards model | Randomization variables only |
|
Secondary | PFS in the ITT set | 3 | As per primary analysis | Randomization variables only | As per primary analysis |
Secondary | Time to progression | Not included | As per primary analysis | Randomization variables only | None |
Secondary | HRQoL in the BM-Neg population and the ITT set | Not included | Descriptive statistics and summaries | None | None |
Secondary | OS in the BM-Neg population | 2 | As per primary analysis | Randomization variables only | None |
Secondary | OS in the ITT set | 4 | As per primary analysis | Randomization variables only | None |
Secondary | ORR in the BM-Neg population and the ITT set | Not included | Differences between groups in odds ratios were assessed using the Cochran-Mantel-Haenszel method stratified by factors used in randomization; 2-sided 95% CIs were calculated using the Clopper-Pearson exact method | Randomization variables only | Analysis of ORR in the “efficacy analyzable population,” defined as randomized and treated patients who received at least 1 cycle of sacituzumab govitecan or TPC and at least 2 post-baseline radiological assessments, or only 1 post-baseline radiological assessment that was assessed as PD |
Secondary | Time to response in the BM-Neg population and the ITT set | Not included | Descriptive statistics and summaries | None | None |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; HRQoL = health-related quality of life; ITT = intention to treat; KM = Kaplan-Meier; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
For the primary PFS analysis, sample size was based on an HR of 0.667, corresponding to a 50% improvement in PFS, which was considered to represent a clinically meaningful improvement in this patient population. A planned interim analysis of PFS for futility was cancelled in communication with the FDA. Anticipated enrolment was 488 patients, with BM-Pos patients capped at 15% (n = 74). The primary PFS analysis would be performed when investigator-assessed PFS in 425 patients occurred in the ITT set, as long as IRC-assessed PFS had occurred in 315 or more patients in the BM-Neg population. It was assumed that a maximum of 15% of patients would be BM-Pos and that there would be 13% fewer IRC-assessed PFS events compared with investigator-assessed PFS. Given these parameters, if the true HR was 0.667 in the BM-Neg population, the study would have 95% power to detect a statistically significant improvement in PFS with a 2-sided type I error rate of 5%. Based on an average PFS estimate in this patient population of approximately 3 months in the TPC arm, and assuming a 24-month enrolment period, the primary PFS analysis would be performed after a minimum follow-up of approximately 4 months.
OS was planned to be analyzed at the time of the PFS analysis as well as after 330 deaths had occurred in the BM-Neg population. At the time of the interim OS analysis, this would yield approximately 89.5% power to detect improved OS in the BM-Neg population with a 2-sided type I error rate of 5%, assuming that 72% of the pre-specified 330 deaths (i.e., 238 deaths) had occurred and that there was a true HR of 0.7. A Lan-DeMets spending function that approximates O’Brien-Fleming stopping boundaries would be applied to the interim OS analysis.
The data monitoring committee reviewed available data when the first 95 patients were randomized and every 6 months thereafter. Following each review, the data monitoring committee made recommendations to either continue the study unchanged, to modify the study, or to terminate the study because of safety concerns. Following the meeting of March 27, 2020, the data monitoring committee recommended that the final data analysis be conducted. At this time, 302 of the 315 pre-specified PFS events and 316 of the 330 pre-specified OS events had occurred (96% for both outcomes in the BM-Neg population). The statistical analysis plan was amended to specify the number of PFS events that would be used for the final analysis and to adjust the 2-sided alpha level for the primary PFS analysis. During database cleaning, additional PFS and OS events occurred, and the originally targeted numbers of events were reached for both outcomes. Therefore, the alpha adjustment was not applicable.
For the primary analysis, PFS in the BM-Neg population was compared between the sacituzumab govitecan and TPC arms using a log-rank test stratified by randomization factors. HR and its 95% CI were based on a stratified Cox proportional hazards model, with “group” as the only covariate stratified by randomization factors. PFS was analyzed using the Kaplan-Meier (KM) method, with median PFS and its 95% CI determined by the Brookmeyer and Crowley method with log-log transformation. Milestone PFS rates at 6, 9, and 12 months were derived from KM estimates. PFS in the ITT set was analyzed using the same method.
Five sensitivity analyses of PFS in the BM-Neg and ITT population were conducted by modification of censoring rules (Table 13). In sensitivity analysis 1, objectively documented progression or death were not censored, regardless of the timing of events. In sensitivity analysis 2, the dates for censoring events were only at scheduled assessment dates. In sensitivity analysis 3, the dates of discontinuation, change of treatment, or second missed scheduled assessment were assigned as event dates. In sensitivity analysis 4, clinical progression without radiographic evidence was considered an event. In sensitivity analysis 5, the censoring rules for the primary PFS analysis were applied to the safety population.
Table 13: Sensitivity Analysis of PFS and Censoring Rules in the ASCENT Trial
Event | Primary analysis | Sensitivity analysis 1 | Sensitivity analysis 2 | Sensitivity analysis 3 | Sensitivity analysis 4 (investigator-assessed PFS) |
|---|---|---|---|---|---|
No adequate response assessment after randomization | |||||
Died before second scheduled assessment | Date of death | Date of death | Date of death | Date of death | Date of death |
Did not die or died after missing 2 or more scheduled assessments | Censored at randomization | Progressed at date of death if died, or censored at date of randomization if did not die | Censored at randomization | Censored at randomization if did not die; progressed on the date of second missed scheduled assessment | Censored at randomization |
Continued scheduled response assessments until objective PD or death | |||||
PD at scheduled assessment or before missing 2 scheduled successive assessments | Not applicable | Date of PD | Date of PD if at scheduled assessment; date of next scheduled assessment if PD occurred between scheduled assessments or before missing 2 scheduled successive assessments (including PD that occurred at end of treatment or early withdrawal visits) | Date of PD | Date of PD if scheduled assessment; date of next scheduled assessment if PD occurred between scheduled assessments or before missing 2 scheduled successive assessments |
Clinical PD indicated between scheduled assessments or before missing 2 scheduled successive assessments | Not applicable | Not applicable | Not applicable | Not applicable | Date of next scheduled assessment |
Death between scheduled assessments or before missing 2 scheduled successive assessments | Date of death | Date of death | Date of death | Date of death | Date of death |
PD or death after missing 2 or more scheduled assessments | Censored at date of last adequate response assessment before missed assessments | Date of PD or death | Censored at date of last adequate response assessment before missed assessments | Progressed at second missed scheduled assessment | Censored at date of last adequate response assessment before missed assessments |
Treatment discontinuation for undocumented progression, toxicity, or other reason | Included in another scenario | Included in another scenario | Included in another scenario | Progressed at time of discontinuation | Included in another scenario |
Continued scheduled response assessments without objective PD or death | |||||
Initiated other anticancer treatment | Censored at date of last adequate response assessment with documented non-progression before starting other anticancer treatment | Date of documented progression or death, if occurred | Censored at date of last adequate response assessment with documented non-progression before starting anticancer treatment | Progressed on the date of start of anticancer treatment | Censored at date of last adequate response assessment with documented non-progression before starting other anticancer treatment |
No objective PD or death | Censored at date of last adequate response assessment | Censored at date of last adequate response assessment | Censored at date of last adequate response assessment | Censored at date of last adequate response assessment | Censored at date of last adequate response assessment |
PD = progressive disease; PFS = progression-free survival.
Source: ASCENT Clinical Study Report.13
OS and time to progression were analyzed in the BM-Neg population, and the ITT set as per the primary PFS analysis. Milestone OS rates at 12, 18, and 24 months were derived from KM estimates.
Subgroup analyses of PFS and OS were conducted for pre-specified subgroups (age < 65 versus ≥ 65 years; race; 2 to 3 versus > 3 prior therapies; region; original diagnosis of TNBC; prior breast cancer surgery; prior cancer radiotherapy; BRCA1 status; BRCA1 or BRCA2 status; prior PD-1 or PD-L1 use; Trop-2 status; baseline liver metastasis; UGT1A1 status), as per the primary analysis, but in exploratory fashion. Subgroup analyses of ORR were conducted for the same pre-specified subgroups. The study was not specifically powered to evaluate each stratum separately.
ORR was analyzed and compared between treatment arms using the Cochran-Mantel-Haenszel method stratified by randomization factors. Two-sided 95% CIs were calculated using the Clopper-Pearson exact method. A sensitivity analysis of ORR was conducted in the efficacy analyzable population, defined as randomized and treated patients who received at least 1 cycle of sacituzumab govitecan or TPC and had at least 2 post-baseline radiological assessments or only 1 post-baseline radiological assessment that was assessed as PD.
Time to response and HRQoL (EORTC QLQ-C30 scales and scores) were summarized using descriptive and summary statistics.
Analysis Populations
The screened population was defined as all patients who provided informed consent and participated in screening procedures to assess eligibility. The BM-Neg population was defined as patients without BM who were randomized to the stratum of no baseline BM. The ITT set was defined as all patients who were randomized (including BM-Neg and BM-Pos patients). The safety population was defined as all patients who received at least 1 dose of sacituzumab govitecan or TPC.
Results
Patient Disposition
In total, 730 patients were screened, of which 529 (72.5%) were randomized and 201 (27.5%) were screened but not randomized (Table 14). The proportion of patients who were screened but not randomized who were BM-Neg and BM-Pos was not provided. Reasons for screen failure were analyzed for the ITT set only and not in the BM-Neg population. The most frequent reasons for screen failure were lack of stable CNS disease for at least 4 weeks (26 of 201, 12.9%), inadequate renal and hepatic function (25 of 201, 12.4%), and absence of histologically or cytologically confirmed TNBC (24 of 210, 11.9%). The proportion of screen failures in which the reason for failure was unknown was at minimum 16.4% (33 of 201) but may have been much higher since reasons for screen failure were not provided on a per-patient basis.
Table 14: Summary of Screen Failures in the ASCENT Trial
Patients | Number |
|---|---|
Screened, N | 730 |
Randomized, n | 529 |
Screened but not randomized, n (%) | 201 (100.0) |
Reason for screen failure, n (%) | |
Inclusion criterion 1 (female or male patients, ≥ 18 years of age, able to understand and give written informed consent) | 6 (3.0) |
Inclusion criterion 2 (histologically or cytologically confirmed TNBC per ASCO or CAP criteria, based on the most recent analyzed biopsy or other pathology specimen) | 24 (11.9) |
Inclusion criterion 3 (metastatic disease documented by CT or MRI imaging) | 1 (0.5) |
Inclusion criterion 4 (measurable disease documented by CT or MRI imaging, as per RECIST 1.1; bone-only disease is not permitted) | 16 (8.0) |
Inclusion criterion 5 (brain MRI must be done for patients with brain metastasis, and patient must have had stable CNS disease for ≥ 4 weeks) | 26 (12.9) |
Inclusion criterion 6 (≥ 2 weeks beyond high-dose systemic corticosteroids; however, low-dose corticosteroids ≤ 20 mg prednisone or equivalent daily are permitted provided the dose is stable for 4 weeks) | 1 (0.5) |
Inclusion criterion 7 (refractory to or relapsed after ≥ 2 prior standard of care chemotherapy regimens for unresectable locally advanced or metastatic breast cancer) | 5 (2.5) |
Inclusion criterion 8 (all patients must have been previously treated with a taxane regardless of disease stage [adjuvant, neoadjuvant, or advanced] when it was given) | 1 (0.5) |
Inclusion criterion 10 (ECOG performance score of 0 or 1) | 10 (5.0) |
Inclusion criterion 10A (adequate hematology without transfusional support [hemoglobin > 9 g/dL, ANC > 1,500/mm3, platelets > 100,000/mm3]) | 1 (0.5) |
Inclusion criterion 11 (adequate hematology without ongoing transfusional support [hemoglobin > 9 g/dL, ANC > 1,500/mm3, platelets > 100,000/mm3]) | 11 (5.5) |
Inclusion criterion 11A (adequate renal and hepatic function [creatinine clearance > 60 mL/min, Cockcroft-Gault equation; bilirubin ≤ 1.5 × IULN; AST and ALT ≤ 2.5 × IULN or ≤ 5 × IULN; known liver metastases]) | 1 (0.5) |
Inclusion criterion 12 (adequate renal and hepatic function [creatinine clearance of > 60 mL/min, may be calculated using Cockcroft-Gault equation; bilirubin ≤ 1.5 × IULN, AST and ALT ≤ 3.0 × IULN or ≤ 5 × IULN]) | 25 (12.4) |
Inclusion criterion 12A (recovered from all toxicities to Grade 1 or less by NCI CTCAE version 4.03 [except that alopecia or peripheral neuropathy may be Grade 2 or less]) at randomization | 4 (2.0) |
Inclusion criterion 13 (recovered from all toxicities to Grade 1 or less by NCI CTCAE version 4.00 [except that alopecia or peripheral neuropathy may be Grade 2 or less]) at the time of randomization | 2 (1.0) |
Inclusion criterion 13A (patients must have completed all prior cancer treatments ≥ 2 weeks before randomization, including chemotherapy [includes endocrine treatment], radiotherapy, and major surgery) | 1 (0.5) |
Inclusion criterion 14 (patients must have completed all prior cancer treatments ≥ 2 weeks before randomization, including chemotherapy [includes endocrine treatment], radiotherapy, and major surgery) | 2 (1.0) |
Exclusion criterion 4 (presence of bulky disease, defined as any single mass > 7 cm in its greatest dimension) | 6 (3.0) |
Exclusion criterion 5 (patients with non-melanoma skin cancer or carcinoma in situ of the cervix are eligible, while patients with other prior malignancies must have had at least a 3-year disease-free interval) | 5 (2.5) |
Exclusion criterion 6 (patients known to be HIV-positive, hepatitis B–positive, or hepatitis C–positive) | 2 (1.0) |
Exclusion criterion 9 (prior history of clinically significant bleeding, intestinal obstruction, or GI perforation within 6 months of randomization) | 2 (1.0) |
Exclusion criterion 10 (infection requiring antibiotic use within 1 week of randomization) | 5 (2.5) |
Exclusion criterion 12 (other concurrent medical or psychiatric conditions that, in the investigator’s opinion, may be likely to confound study interpretation or prevent completion of study procedures and follow-up) | 6 (3.0) |
Exclusion criterion 14A (rapid deterioration during screening before randomization [e.g., significant change in performance status, ≥ 20% decrease in serum albumin levels, requiring modifications in analgesic management]) | 2 (1.0) |
Exclusion criterion 15A (other concurrent medical or psychiatric conditions that, in the investigator’s opinion, are likely to confound study interpretation or prevent completion of study procedures and follow-up examinations) | 3 (1.5) |
ALT = alanine aminotransferase; ANC = absolute neutrophil count; ASCO = American Association for Clinical Oncology; AST = asparagine aminotransferase; CAP = College of American Pathologists; CNS = central nervous system; ECOG = Eastern Cooperative Oncology Group; GI = gastrointestinal; IULN = institutional upper limit of normal; NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; TNBC = triple-negative breast cancer.
Note: If a patient has multiple reasons for screening failure, the patient is counted under each reason.
Source: ASCENT Clinical Study Report.13
Patient disposition in the ASCENT trial is summarized in Table 15. Among randomized patients, higher proportions received at least 1 dose of protocol therapy in the sacituzumab govitecan arm (97.0% in the BM-Neg population and 96.6% in the ITT set) than in the TPC arm (86.3% in the BM-Neg population and 85.5% in the ITT set), potentially because some patients in the TPC arm elected not to participate following randomization. Most patients randomized and treated eventually discontinued protocol therapy (88.5% of the BM-Neg population and 87.9% of the ITT set). The most common reason for treatment discontinuation was PD (78.0% of the BM-Neg population and 76.7% of the ITT set). Among patients who discontinued protocol therapy due to PD, a slightly higher proportion in the TPC arm were discontinued for clinical progression in the absence of radiological evidence (26 of 166 in the BM-Neg population and 28 out of 184 in the ITT set) than in the sacituzumab govitecan arm (24 out of 199 in the BM-Neg population and 24 out of 222 in the ITT set). Higher proportions of patients in the TPC arm discontinued treatment (7.3% in the BM-Neg population and 6.9% in the ITT set) and discontinued the study due to withdrawal of consent (9.9% in the BM-Neg population and 10.3% in the ITT set) compared with those in the sacituzumab govitecan arm (discontinued treatment: 1.7% of the BM-Neg population and 1.9% of the ITT set; discontinued study: 3.0% of the BM-Neg population and 3.0% of the ITT set). Only 1 patient (in the TPC arm) discontinued treatment due to unacceptable toxicity, and during the treatment phase, no patients were lost to follow-up.
Similar proportions of patients in the sacituzumab govitecan and TPC arms entered survival follow-up (78.7% and 81.1%, respectively, in the BM-Neg population; 79.8% and 80.2%, respectively, in the ITT set). The mean (SD) follow-up duration was longer in the sacituzumab govitecan arm (11.43 [5.87] months in the BM-Neg population and 11.06 [5.90] months in the ITT set) than in the TPC arm (7.26 [5.39] months in the BM-Neg population and 7.40 [5.51] months in the ITT set). Similar proportions of patients (approximately 5%) in both arms were lost to follow-up during survival follow-up. At the time of data cut-off, higher proportions of patients in the TPC arm had survival follow-up data collected at least 121 days prior (8.2% in the BM-Neg population and 8.0% in the ITT set) than those in the sacituzumab govitecan arm (2.6% in the BM-Neg population and 2.2% in the ITT set).
Table 15: Patient Disposition and Survival Follow-Up in the ASCENT Trial
Characteristic | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Total (N = 468) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) | |
Screened, N | NRa | 730 | ||||
Randomized, N (%) | 235 (100.0) | 233 (100.0) | 468 (100.0) | 267 (100.0) | 262 (100.0) | 529 (100.0) |
Treated, n (%) | 228 (97.0) | 201 (86.3) | 429 (91.7) | 258 (96.6) | 224 (85.5) | 482 (91.1) |
Discontinued from treatment, n (%) | 213 (90.6) | 201 (86.3) | 414 (88.5) | 241 (90.3) | 224 (85.5) | 465 (87.9) |
Primary reason for treatment discontinuation, n (%) | ||||||
PD | 199 (84.7) | 166 (71.2) | 365 (78.0) | 222 (83.1) | 184 (70.2) | 406 (76.7) |
Clinical progression | 24 (10.2) | 26 (11.2) | 50 (10.7) | 24 (9.0) | 28 (10.7) | 52 (9.8) |
Radiological progression | 175 (74.5) | 140 (60.1) | 315 (67.3) | 198 (74.2) | 156 (59.5) | 354 (66.9) |
Death | 1 (0.4) | 4 (1.7) | 5 (1.1) | 1 (0.4) | 4 (1.5) | 5 (0.9) |
Treatment delay > 3 weeks | 0 | 2 (0.9) | 2 (0.4) | 0 | 4 (1.5) | 4 (0.8) |
Withdrawal of consent | 4 (1.7) | 17 (7.3) | 21 (4.5) | 5 (1.9) | 18 (6.9) | 23 (4.3) |
Treatment only | 4 (1.7) | 11 (4.7) | 15 (3.2) | 5 (1.9) | 12 (4.6) | 17 (3.2) |
No survival follow-up | 0 | 6 (2.6) | 6 (1.3) | 0 | 6 (2.3) | 6 (1.1) |
AE | 6 (2.6) | 7 (3.0) | 13 (2.8) | 10 (3.7) | 8 (3.1) | 18 (3.4) |
Physician decision | 3 (1.3) | 4 (1.7) | 7 (1.5) | 3 (1.1) | 5 (1.9) | 8 (1.5) |
Pregnancy | 0 | 0 | 0 | 0 | 0 | 0 |
Lost to follow-up | 0 | 0 | 0 | 0 | 0 | 0 |
Unacceptable toxicity | 0 | 1 (0.4) | 1 (0.2) | 0 | 1 (0.4) | 1 (0.2) |
Discontinued from study, n (%) | 161 (68.5) | 203 (87.1) | 364 (77.8) | 185 (69.3) | 228 (87.0) | 413 (78.1) |
Primary reason for study discontinuation, n (%) | ||||||
Death | 151 (64.3) | 177 (76.0) | 328 (70.1) | 174 (65.2) | 197 (75.2) | 371 (70.1) |
Withdrawal of consent | 7 (3.0) | 23 (9.9) | 30 (6.4) | 8 (3.0) | 27 (10.3) | 35 (6.6) |
Lost to follow-up | 3 (1.3) | 3 (1.3) | 6 (1.3) | 3 (1.1) | 4 (1.5) | 7 (1.3) |
Survival follow-up, n (%) | ||||||
Entered survival follow-up | 185 (78.7) | 189 (81.1) | 374 (79.9) | 213 (79.8) | 210 (80.2) | 423 (80.0) |
Discontinued survival follow-up | 133 (56.6) | 161 (69.1) | 294 (62.8) | 155 (58.1) | 178 (67.9) | 333 (62.9) |
Ongoing with survival follow-up | 50 (21.3) | 28 (12.0) | 78 (16.7) | 56 (21.0) | 32 (12.2) | 88 (16.6) |
Survival status, n (%) | ||||||
Dead | 155 (66.0) | 185 (79.4) | 340 (72.6) | 179 (67.0) | 206 (78.6) | 385 (72.8) |
Alive, continuing in treatment | 15 (6.4) | 0 | 15 (3.2) | 17 (6.4) | 0 | 17 (3.2) |
Alive, continuing in follow-up | 54 (23.0) | 33 (14.2) | 87 (18.6) | 59 (22.1) | 39 (14.9) | 98 (18.5) |
Unknown or lost to follow-up | 11 (4.7) | 15 (6.4) | 26 (5.6) | 12 (4.5) | 17 (6.5) | 29 (5.5) |
Follow-up length, monthsb | ||||||
Mean (SD) | 11.43 (5.87) | 7.26 (5.39) | 9.35 (6.01) | 11.06 (5.90) | 7.40 (5.51) | 9.25 (5.99) |
Median (range) | 11.17 (0.3 to 23.8) | 6.21 (0.0 to 24.2) | 8.71 (0.0 to 24.2) | 10.55 (0.3 to 23.8) | 6.28 (0.0 to 24.2) | 8.38 (0.0 to 24.2) |
Currentness of survival follow-up, days, n (%)c | ||||||
0d | 228 (97.0) | 214 (91.8) | 442 (94.4) | 260 (97.4) | 241 (92.0) | 501 (94.7) |
> 1 to ≤ 30 | 0 | 0 | 0 | 0 | 0 | 0 |
≥ 31 to ≤ 60 | 1 (0.4) | 0 | 1 (0.2) | 1 (0.4) | 0 | 1 (0.2) |
≥ 61 to ≤ 90 | 0 | 0 | 0 | 0 | 0 | 0 |
≥ 91 to ≤ 120 | 0 | 0 | 0 | 0 | 0 | 0 |
≥ 121 | 6 (2.6) | 19 (8.2) | 25 (5.3) | 6 (2.2) | 21 (8.0) | 27 (5.1) |
Deaths during long-term follow-up, n (%) | 133 (56.6) | 158 (67.8) | 291 (62.2) | 155 (58.1) | 175 (66.8) | 330 (62.4) |
ITT, n (%) | — | — | — | 267 (100.0) | 262 (100.0) | 529 (100.0) |
BM-Neg, n (%) | 235 (100.0) | 233 (100.0) | 468 (100.0) | — | — | — |
SP, n (%) | — | — | — | 258 (96.6) | 224 (85.5) | 482 (91.1) |
AE = adverse event; BM-Neg = brain metastasis–negative; ITT = intention to treat; NR = not reported; PD = progressive disease; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aThe total number of BM-Neg patients screened was not reported. Reasons for screen failure were provided for the ITT set but not for the BM-Neg population.
bFollow-up length is the time from randomization to death or the last date known alive.
cTime from last date known alive to data cut-off date.
dPatients who died, who withdrew consent for study participation, or whose last date known alive was on or after the data cut-off date were classified as having current follow-up (0 days).
Source: ASCENT Clinical Study Report.13
Protocol deviations in the ASCENT trial are shown in Table 16. Approximately one-third of patients in the ITT set had at least 1 important protocol deviation (34.5% in the sacituzumab govitecan arm and 39.7% in the TPC arm). Important protocol deviations related to informed consent were more common in the TPC arm (21.0%) than in the sacituzumab govitecan arm (10.9%). Important protocol deviations related to investigational products (primarily handling, storage, and retention of the study drug) were more common in the sacituzumab govitecan arm (6.0%) than in the TPC arm (0.4%).
Table 16: Protocol Deviations in the ASCENT Trial — ITT Population
Characteristic | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) |
|---|---|---|---|
≥ 1 protocol deviation, n (%) | 225 (84.3) | 207 (79.0) | 432 (81.7) |
≥ 1 important protocol deviation, n (%) | 92 (34.5) | 104 (39.7) | 196 (37.1) |
Type of important protocol deviation, n (%)a | |||
Informed consent | 29 (10.9) | 55 (21.0) | 84 (15.9) |
Process | 9 (3.4) | 38 (14.5) | 47 (8.9) |
Study conduct or procedures | 55 (20.6) | 51 (19.5) | 106 (20.0) |
Inclusion and exclusion criteria | 23 (8.6) | 27 (10.3) | 50 (9.5) |
Dose formulation and dose administration | 29 (10.9) | 19 (7.3) | 48 (9.1) |
Investigational product | 16 (6.0) | 1 (0.4) | 17 (3.2) |
ITT = intention to treat; TPC = treatment of physician’s choice.
aTypes of important protocol deviations with frequencies of 10% or higher in any treatment arm are shown.
Source: ASCENT Clinical Study Report.13
Exposure to Study Treatments
Treatment exposure in the ASCENT trial safety population is shown in Table 17. Patients receiving sacituzumab govitecan had more than twice the exposure (mean = 5.77 [SD = 4.90] months, 8.5 [SD = 6.70] cycles) than those receiving eribulin (mean = 2.27 [SD = 2.18] months, 3.8 [SD = 2.97] cycles), capecitabine (mean = 2.16 [SD = 2.56] months, 3.3 [SD = 3.62] cycles), gemcitabine (mean = 2.25 [SD = 2.01] months, 3.0 [SD = 2.06] cycles), and vinorelbine (mean = 1.73 [SD = 2.31] months, 2.9 [SD = 2.97] cycles). Most protocol therapy was administered by study personnel at site visits, and thus adherence was not applicable. Although not explicitly stated, capecitabine was presumably self-administered, but no information on adherence was provided.
Table 17: Treatment Exposure in the ASCENT Trial — Safety Population
Exposure measure | Sacituzumab govitecan (N = 258) | Eribulin (N = 122) | Capecitabine (N = 22) | Gemcitabine (N = 31) | Vinorelbine (N = 43) |
|---|---|---|---|---|---|
Treatment duration, months | |||||
Mean (SD) | 5.77 (4.90) | 2.27 (2.18) | 2.16 (2.56) | 2.25 (2.01) | 1.73 (2.31) |
Median (range) | 4.39 (0.03 to 22.87) | 1.64 (0.03 to 15.34) | 1.18 (0.33 to 10.58) | 1.41 (0.23 to 8.08) | 0.95 (0.03 to 11.53) |
Treatment duration, cyclesa | |||||
Mean (SD) | 8.5 (6.70) | 3.8 (2.97) | 3.3 (3.62) | 3.0 (2.06) | 2.9 (2.97) |
Median (range) | 7 (1 to 33) | 3 (1 to 21) | 2 (1 to 15) | 2 (1 to 9) | 2 (1 to 15) |
SD = standard deviation.
aFor weekly injections with vinorelbine, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Concomitant medication use in the ASCENT trial is shown in Table 18. Higher proportions of patients in the sacituzumab govitecan arm received 1 or more concomitant medications (97.0% in the BM-Neg population and 96.6% in the ITT set) compared with those in the TPC arm (88.0% of the BM-Neg population and 87.0% of the ITT set). Medications used more often in the sacituzumab govitecan arm versus the TPC arm included (a) agents to prevent and treat infusion reactions (analgesics and antipyretics [76.2% versus 57.1% in the BM-Neg population; 74.5% versus 56.1% in the ITT set], corticosteroids [65.1% versus 35.2% in the BM-Neg population; 63.7% versus 35.1% in the ITT set], drugs for peptic ulcer and gastroesophageal reflux disease [67.7% versus 38.2% in the BM-Neg population; 67.0% versus 40.5% in the ITT set], and antihistamines [64.7% versus 15.9% in the BM-Neg population; 63.7% versus 16.4% in the ITT set]) and (b) agents to prevent and treat gastrointestinal symptoms (antiemetics and antinauseants [84.3% versus 53.2% in the BM-Neg population; 83.1% versus 53.8% in the ITT set] and antipropulsives [53.6% versus 9.9% in the BM-Neg population; 53.6% versus 8.8% in the ITT set]). Immunostimulants for secondary prophylaxis and treatment of myelosuppression were administered more frequently in the sacituzumab govitecan arm (46.4% of the BM-Neg population and 47.2% of the ITT set) than in the TPC arm (19.7% of the BM-Neg population and 19.8% of the ITT set).
Table 18: Concomitant Medications in the ASCENT Trial
Concomitant medication | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Total (N = 468) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) | |
≥ 1 Concomitant medication, n (%)a | 228 (97.0) | 205 (88.0) | 433 (92.5) | 258 (96.6) | 228 (87.0) | 486 (91.9) |
Antiemetics and antinauseants | 198 (84.3) | 124 (53.2) | 322 (68.8) | 222 (83.1) | 141 (53.8) | 363 (68.6) |
Ondansetron | 87 (37.0) | 56 (24.0) | 143 (30.6) | 96 (36.0) | 61 (23.3) | 157 (29.7) |
Ondansetron hydrochloride | 77 (32.8) | 46 (19.7) | 123 (26.3) | 84 (31.5) | 53 (20.2) | 137 (25.9) |
Palonosetron | 39 (16.6) | 2 (0.9) | 41 (8.8) | 40 (15.0) | 4 (1.5) | 44 (8.3) |
Prochlorperazine edisylate | 35 (14.9) | 22 (9.4) | 57 (12.2) | 41 (15.4) | 24 (9.2) | 65 (12.3) |
Prochlorperazine | 33 (14.0) | 12 (5.2) | 45 (9.6) | 37 (13.9) | 15 (5.7) | 52 (9.8) |
Fosaprepitant | 25 (10.6) | 0 | 25 (5.3) | 29 (10.9) | 0 | 29 (5.5) |
Analgesics and antipyretics | 179 (76.2) | 133 (57.1) | 312 (66.7) | 199 (74.5) | 147 (56.1) | 346 (65.4) |
Paracetamol | 148 (63.0) | 104 (44.6) | 252 (53.8) | 167 (62.5) | 112 (42.7) | 279 (52.7) |
Gabapentin | 36 (15.3) | 31 (13.3) | 67 (14.3) | 44 (16.5) | 37 (14.1) | 81 (15.3) |
Drugs for peptic ulcer and gastroesophageal reflux disease | 159 (67.7) | 89 (38.2) | 248 (53.0) | 179 (67.0) | 106 (40.5) | 285 (53.9) |
Famotidine | 75 (31.9) | 9 (3.9) | 84 (17.9) | 85 (31.8) | 12 (4.6) | 97 (18.3) |
Omeprazole | 39 (16.6) | 37 (15.9) | 76 (16.2) | 44 (16.5) | 41 (15.6) | 85 (16.1) |
Ranitidine hydrochloride | 36 (15.3) | 5 (2.1) | 41 (8.8) | 37 (13.9) | 6 (2.3) | 43 (8.1) |
Corticosteroids for systemic use | 153 (65.1) | 82 (35.2) | 235 (50.2) | 170 (63.7) | 92 (35.1) | 262 (49.5) |
Dexamethasone | 102 (43.4) | 47 (20.2) | 149 (31.8) | 116 (43.4) | 55 (21.0) | 171 (32.3) |
Antihistamines for systemic use | 152 (64.7) | 37 (15.9) | 189 (40.4) | 170 (63.7) | 43 (16.4) | 213 (40.3) |
Diphenhydramine | 59 (25.1) | 4 (1.7) | 63 (13.5) | 66 (24.7) | 5 (1.9) | 71 (13.4) |
Diphenhydramine hydrochloride | 33 (14.0) | 9 (3.9) | 42 (9.0) | 40 (15.0) | 10 (3.8) | 50 (9.5) |
Loratadine | 27 (11.5) | 9 (3.9) | 36 (7.7) | 28 (10.5) | 10 (3.8) | 38 (7.2) |
Dexchlorpheniramine maleate | 25 (10.6) | 1 (0.4) | 26 (5.6) | 25 (9.4) | 1 (0.4) | 26 (4.9) |
Opioids | 136 (57.9) | 128 (54.9) | 264 (56.4) | 154 (57.7) | 143 (54.6) | 297 (56.1) |
Oxycodone | 31 (13.2) | 33 (14.2) | 64 (13.7) | 43 (16.1) | 40 (15.3) | 83 (15.7) |
Oxycodone hydrochloride | 28 (11.9) | 35 (15.0) | 63 (13.5) | 29 (10.9) | 39 (14.9) | 68 (12.9) |
Fentanyl | 27 (11.5) | 31 (13.3) | 58 (12.4) | 32 (12.0) | 32 (12.2) | 64 (12.1) |
Antipropulsives | 126 (53.6) | 23 (9.9) | 149 (31.8) | 143 (53.6) | 23 (8.8) | 166 (31.4) |
Loperamide hydrochloride | 74 (31.5) | 11 (4.7) | 85 (18.2) | 83 (31.1) | 11 (4.2) | 94 (17.8) |
Loperamide | 54 (23.0) | 8 (3.4) | 62 (13.2) | 61 (22.8) | 8 (3.1) | 69 (13.0) |
Immunostimulants | 109 (46.4) | 46 (19.7) | 155 (33.1) | 126 (47.2) | 52 (19.8) | 178 (33.6) |
Filgrastim | 68 (28.9) | 34 (14.6) | 102 (21.8) | 75 (28.1) | 40 (15.3) | 115 (21.7) |
Pegfilgrastim | 42 (17.9) | 5 (2.1) | 47 (10.0) | 44 (16.5) | 5 (1.9) | 49 (9.3) |
BM-Neg = brain metastasis–negative; ITT = intention to treat; TPC = treatment of physician’s choice.
aConcomitant therapies used in ≥ 10% of either study arm are listed.
Source: ASCENT Clinical Study Report.13
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below. See Appendix 5 for detailed efficacy data.
Patients in the ASCENT trial were treated until PD or unacceptable toxicity. In the primary PFS analysis, censoring occurred at the time of data cut-off for patients who were still alive without PD or earlier for those who had no post-baseline tumour assessment, those who were lost to follow-up, those who withdrew consent, those who died or progressed after missing more than 1 assessment visit, or those who initiated other anticancer therapy before PD. In the OS analysis, censoring occurred at the time of data cut-off for patients who were still alive or earlier for those who were lost to follow-up or withdrew consent; however, OS was not censored at the time of initiating other anticancer therapy. Subsequent therapies were received following discontinuation of ASCENT trial protocol therapy in 47.6% of patients in the sacituzumab govitecan arm and 38.2% of patients in the TPC arm (Table 19). Specific therapies administered were similar for patients in both arms, except that more patients in the sacituzumab govitecan arm received eribulin (17.6% versus 3.8%).
Table 19: Subsequent Anticancer Therapies in the ASCENT Trial — ITT Population
Therapy | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Total (N = 529) | |||
|---|---|---|---|---|---|---|
n (%) | Mean duration, weeks | n (%) | Mean duration, weeks | n (%) | Mean duration, weeks | |
≥ 1 Subsequent medication, n (%)a | 127 (47.6) | NA | 100 (38.2) | NA | 227 (42.9) | NA |
Eribulin | 47 (17.6) | 11.0 | 10 (3.8) | 12.3 | 57 (10.8) | 11.3 |
Carboplatin | 22 (8.2) | 14.0 | 26 (9.9) | 12.0 | 48 (9.1) | 12.9 |
Capecitabine | 21 (7.9) | 9.9 | 14 (5.3) | 11.7 | 35 (6.6) | 10.6 |
Gemcitabine | 18 (6.7) | 14.7 | 16 (6.1) | 15.9 | 34 (6.4) | 15.3 |
Investigational drugs | 21 (7.9) | 10.9 | 13 (5.0) | 9.0 | 34 (6.4) | 10.1 |
Pegylated liposomal doxorubicin hydrochloride | 12 (4.5) | 9.0 | 19 (7.3) | 12.9 | 31 (5.9) | 11.4 |
Radiotherapy | 15 (5.6) | 3.4 | 14 (5.3) | 3.1 | 29 (5.5) | 3.3 |
Paclitaxel | 13 (4.9) | 9.0 | 15 (5.7) | 11.1 | 28 (5.3) | 10.1 |
Cyclophosphamide | 12 (4.5) | 7.4 | 15 (5.7) | 7.7 | 27 (5.1) | 7.6 |
Paclitaxel albumin | 10 (3.8) | 14.6 | 14 (5.3) | 21.3 | 24 (4.5) | 18.4 |
Vinorelbine | 6 (2.2) | 10.0 | 15 (5.7) | 9.9 | 21 (4.0) | 9.9 |
ITT = intention to treat; NA = not applicable; TPC = treatment of physician’s choice.
Note: Standardized medication names rather than raw reported names were used in the analysis.
aTreatments received by 5% of patients or more in either study arm are listed.
Source: CADTH review submission for sacituzumab govitecan.5
OS: BM-Neg Population and ITT Set
OS by IRC assessment in the BM-Neg population and the ITT set is shown in Table 20, Figure 2, and Figure 3. Approximately two-thirds of patients in the sacituzumab govitecan arm (66.0% in the BM-Neg population and 67.0% in the ITT set) died during follow-up, while approximately 80% of patients in the TPC arm died (79.4% in the BM-Neg population and 78.6% in the ITT set). Median (95% CI) OS was 12.1 (10.7 to 14.0) months in the sacituzumab govitecan arm, compared with 6.7 (5.8 to 7.7) months in the TPC arm in the BM-Neg population, and 11.8 (10.5 to 13.8) months in the sacituzumab govitecan arm compared with 6.9 (5.9 to 7.7) months in the TPC arm in the ITT set. This difference in median OS based on tests of the KM curves was statistically significant by stratified log-rank test (P < 0.0001) in both the BM-Neg population and the ITT set. The statistical test for differences in OS for both the BM-Neg population and the ITT set were included as part of the statistical testing hierarchy in the ASCENT trial. The HR (95% CI) for death by stratified Cox regression analysis was 0.476 (0.383 to 0.592) in the BM-Neg population and 0.508 (0.414 to 0.624) in the ITT set, comparing the sacituzumab govitecan arm with the TPC arm. The percentage of patients alive at 6, 12, 18, and 24 months was higher in the sacituzumab govitecan arm (BM-Neg population: 82.4%, 50.7%, 30.5%, and not calculable, respectively; ITT set: 79.3%, 48.8%, 28.6%, and not calculable, respectively) than in the TPC arm (BM-Neg population: 54.9%, 22.2%, 12.3%, and 6.6%, respectively; ITT set: 55.4%, 23.0%, 12.9%, and 6.8%, respectively).
Table 20: Overall Survival by IRC Assessment in the ASCENT Trial — BM-Neg and ITT Populations
Characteristic | BM-Neg | ITT | ||
|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | |
Patients with events, n (%) | 155 (66.0) | 185 (79.4) | 179 (67.0) | 206 (78.6) |
Patients without events (censored), n (%) | 80 (34.0) | 48 (20.6) | 88 (33.0) | 56 (21.4) |
Median (95% CI) OS, monthsa | 12.1 (10.7 to 14.0) | 6.7 (5.8 to 7.7) | 11.8 (10.5 to 13.8) | 6.9 (5.9 to 7.7) |
Log-rank P value (stratified)b | < 0.0001 | < 0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | 0.476 (0.383 to 0.592) | 0.508 (0.414 to 0.624) | ||
OS rate (95% CI) at 6 months, %c,d | 82.4 (76.9 to 86.7) | 54.9 (48.0 to 61.2) | 79.3 (73.9 to 83.7) | 55.4 (48.9 to 61.4) |
OS rate (95% CI) at 12 months, %c,d | 50.7 (43.9 to 57.0) | 22.2 (16.8 to 28.0) | 48.8 (42.5 to 54.8) | 23.0 (17.8 to 28.5) |
OS rate (95% CI) at 18 months, %c,d | 30.5 (24.1 to 37.1) | 12.3 (7.9 to 17.7) | 28.6 (22.6 to 34.8) | 12.9 (8.7 to 18.0) |
OS rate (95% CI) at 24 months, %c,d | NC | 6.6 (2.4 to 13.6) | NC | 6.8 (2.8 to 13.1) |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; NC = not calculable; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. The CI for the median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for OS rate at the specified time points are from Kaplan-Meier estimates.
dMilestone OS rate at 6 months was not pre-specified in the statistical analysis plan. Milestone time points for OS rates were mislabelled as 3, 6, 9, and 12 months in the Clinical Study Report.
Source: ASCENT Clinical Study Report.13
Figure 2: Overall Survival by IRC Assessment in the ASCENT Trial — BM-Neg Population
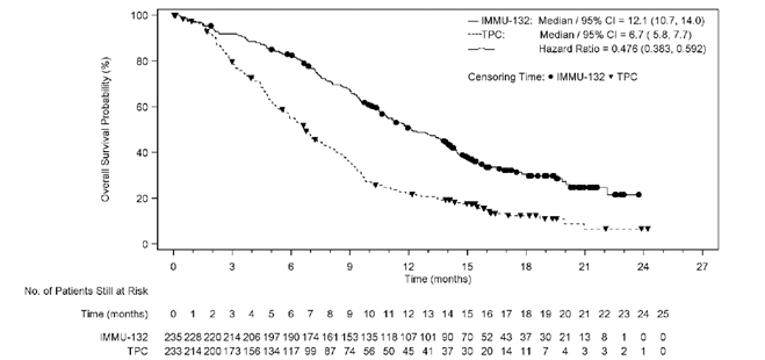
BM-Neg = brain metastasis–negative; CI = confidence interval; IMMU-132 = sacituzumab govitecan; IRC = independent review committee; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
Figure 3: Overall Survival by IRC Assessment in the ASCENT Trial — ITT Population
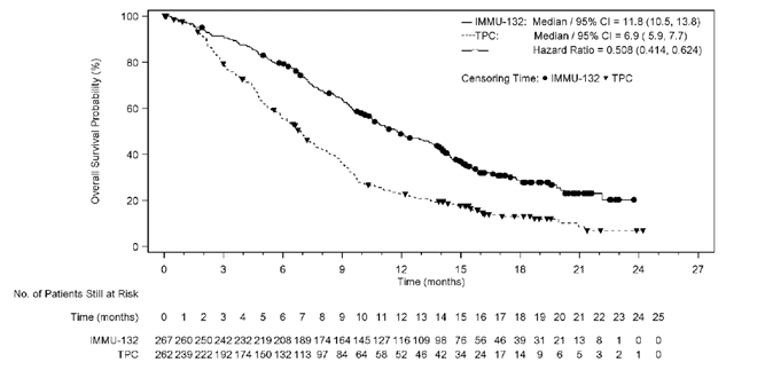
CI = confidence interval; IMMU-132 = sacituzumab govitecan; IRC = independent review committee; ITT = intention to treat; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
Three pre-specified subgroup analyses of OS in the ASCENT trial were of interest to this review (Appendix 5): BM at baseline (yes or no), prior therapies received (2 to 3 versus > 3) and BRCA1 or BRCA2 status. Among BM-Pos patients (n = 61), the median OS was 6.8 months in the sacituzumab govitecan arm and 7.5 months in the TPC arm (HR = 0.947; 95% CI, 0.523 to 1.716). Among patients in the ITT set who had received 2 to 3 prior therapies, the median OS was 12.1 months in the sacituzumab govitecan arm and 6.8 months in the TPC arm (HR = 0.442; 95% CI, 0.346 to 0.566). Among patients in the ITT set who had received more than 3 prior therapies, the median OS was 10.5 months in the sacituzumab govitecan arm and 7.6 months in the TPC arm (HR = 0.716; 95% CI, 0.501 to 1.022). Among BRCA1- or BRCA2-positive patients in the ITT set, the median OS was 15.6 months in the sacituzumab govitecan arm and 4.4 months in the TPC arm (HR = 0.411; 95% CI, 0.186 to 0.907); the number of BRCA1- or BRCA2-positive patients was small. Among BRCA1- or BRCA2-negative patients in the ITT set, the median OS was 10.5 months in the sacituzumab govitecan arm and 7.1 months in the TPC arm (HR = 0.595; 95% CI, 0.457 to 0.775).
HRQoL: Safety Population
HRQoL measured using the global health status or HRQoL scale of the EORTC QLQ-C30 at baseline and up to cycle 6 (n > 25 in both arms), as well as at the end of treatment, is shown in Table 21. The baseline mean global health status (on a scale of 0 to 100, with higher values signifying better HRQoL) was 61.9 (SD = 21.3) in patients treated with sacituzumab govitecan and 56.4 (SD = 22.2) in patients treated with TPC. Changes in global health status or HRQoL over time were smaller than the MID estimate of 5 points to 10 points.
Table 21: Global Health Status and HRQoL in the ASCENT Trial — Safety Population
Time point | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
Baseline | 247 | 61.9 (21.3) | — | — | 217 | 56.4 (22.2) | — | — | 464 | 59.4 (21.9) | — | — |
Cycle 2b | 223 | 59.4 (22.9) | 216 | –3.8 (21.9) | 162 | 57.7 (20.7) | 157 | –1.4 (21.6) | 385 | 58.7 (22.0) | 373 | –2.8 (21.8) |
Cycle 3b | 192 | 67.0 (18.8) | 186 | 3.7 (22.6) | 94 | 58.8 (19.4) | 92 | –0.7 (23.2) | 286 | 64.3 (19.4) | 278 | 2.2 (22.9) |
Cycle 4b | 183 | 67.6 (18.1) | 177 | 3.6 (21.4) | 74 | 59.7 (19.8) | 71 | 1.1 (23.9) | 257 | 65.3 (18.9) | 248 | 2.9 (22.1) |
Cycle 5b | 149 | 67.8 (19.3) | 144 | 2.5 (23.5) | 50 | 58.8 (21.2) | 48 | 0 (21.1) | 199 | 65.5 (20.1) | 192 | 1.9 (22.9) |
Cycle 6b | 147 | 69.7 (18.1) | 141 | 3.9 (20.0) | 37 | 60.8 (18.6) | 36 | –1.6 (21.2) | 184 | 67.9 (18.5) | 177 | 2.8 (20.3) |
End of treatment | 169 | 56.7 (23.4) | 164 | –6.5 (23.1) | 151 | 50.6 (22.4) | 147 | –9.4 (20.5) | 320 | 53.8 (23.1) | 311 | –7.9 (21.9) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; SD = standard deviation; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on the EORTC QLQ-C30 questionnaire, Version 3.0. For global health status, a higher score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Analysis of EORTC QLQ-C30 functional and symptom scales was consistent with the global health status scale (Appendix 5). Baseline values were generally slightly higher for functional scales and lower for symptom scales (indicating higher HRQoL) in the sacituzumab govitecan arm. Changes from baseline post-treatment suggested trends toward small improvements (or lesser deterioration compared with TPC) in physical functioning and role functioning, fatigue, pain, dyspnea, and insomnia. However, changes from baseline were also suggestive of worsening nausea and/or vomiting, as well as diarrhea, in patients treated with sacituzumab govitecan.
PFS: BM-Neg Population and ITT Set
PFS by IRC assessment in the BM-Neg population and the ITT set were the primary and key secondary outcomes in the ASCENT trial and are shown in Table 22, Figure 4, and Figure 5. Approximately 2-thirds of patients experienced events in both the sacituzumab govitecan arm (70.6% in the BM-Neg population and 71.2% in the ITT set) and the TPC arm (64.4% in the BM-Neg population and 65.3% in the ITT set). Among the approximately one-third of patients without events (censored), the most common reasons in the sacituzumab govitecan arm were alive without PD (15.7% in the BM-Neg population and 15.0% in the ITT set) and death after starting new anticancer therapy (11.5% in the BM-Neg population and 11.2% in the ITT set). In the TPC arm, the most common reasons for censoring were death after starting new anticancer therapy (20.2% in the BM-Neg population and 19.1% in the ITT set), lack of post-baseline tumour assessments (6.9% in the BM-Neg population and 7.6% in the ITT set), and death after missing more than 1 assessment visit (4.7% in the BM-Neg population and 4.2% in the ITT), while patients alive without PD were few (3.4% in both the BM-Neg population and the ITT set). Censoring of patients who died after starting new anticancer therapy applied to patients who were incorrectly deemed progressive by investigators and taken off protocol therapy in the absence of objective PD.
The median PFS was 5.6 (95% CI, 4.3 to 6.3) months in the sacituzumab govitecan arm, compared with 1.7 (95% CI,1.5 to 2.6) months in the TPC arm, in the BM-Neg population and 4.8 (95% CI, 4.1 to 5.8) months in the sacituzumab govitecan arm, compared with 1.7 (95% CI, 1.5 to 2.5) months in the TPC arm, in the ITT set. The differences in median PFS based on testing of the KM curves was statistically significant by stratified log-rank test (P < 0.0001) in both the BM-Neg population and the ITT set, and both statistical tests were adjusted for multiple comparisons under the hierarchical testing procedure. The HR (95% CI) for disease progression or death by stratified Cox regression analysis was 0.409 (95% CI, 0.323 to 0.519) in the BM-Neg population and 0.433 (95% CI, 0.347 to 0.541) in the ITT set, comparing the sacituzumab govitecan arm with the TPC arm. The percentage of patients alive and progression-free at 3, 6, 9, and 12 months was higher in the sacituzumab govitecan arm (BM-Neg population: 64.6%, 44.2%, 24.6%, and 17.2%, respectively; ITT set: 61.9%, 40.6%, 22.8%, and 16.2%, respectively) than in the TPC arm (BM-Neg population: 27.0%, 11.0%, 8.0%, and 6.7%, respectively; ITT set: 27.1%, 10.7%, 7.2%, and 6.0%, respectively).
Table 22: Progression-Free Survival by IRC Assessment in the ASCENT Trial — BM-Neg and ITT Populations
Characteristic | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with events, n (%) | 166 (70.6) | 150 (64.4) | — | 190 (71.2) | 171 (65.3) | — |
Deaths, n (%) | 16 (6.8) | 27 (11.6) | — | 19 (7.1) | 29 (11.1) | — |
Radiographic PD, n (%) | 150 (63.8) | 123 (52.8) | — | 171 (64.0) | 142 (54.2) | — |
Patients without events (censored), n (%) | 69 (29.4) | 83 (35.6) | — | 77 (28.8) | 91 (34.7) | — |
Alive without PD | 37 (15.7) | 8 (3.4) | — | 40 (15.0) | 9 (3.4) | — |
Died after missing ≥ 1 visit of assessment interval | 2 (0.9) | 11 (4.7) | — | 4 (1.5) | 11 (4.2) | — |
Died after starting new anticancer therapy | 27 (11.5) | 47 (20.2) | — | 30 (11.2) | 50 (19.1) | — |
Lost to follow-up | 1 (0.4) | 0 | — | 1 (0.4) | 0 | — |
No post-baseline evaluable tumour assessment | 1 (0.4) | 16 (6.9) | — | 1 (0.4) | 20 (7.6) | — |
PD after missing ≥ 1 visit of assessment interval | 1 (0.4) | 0 | — | 1 (0.4) | 0 | — |
Withdrawal of consent | 0 | 1 (0.4) | — | 0 | 1 (0.4) | — |
Median (95% CI) PFS, monthsa | 5.6 (4.3 to 6.3) | 1.7 (1.5 to 2.6) | — | 4.8 (4.1 to 5.8) | 1.7 (1.5 to 2.5) | — |
Log-rank P value (stratified)b | — | < 0.0001 | — | < 0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.409 (0.323 to 0.519) | — | 0.433 (0.347 to 0.541) | ||
PFS rate (95% CI) at 3 months, %c,d | 64.6 (57.9 to 70.5) | 27.0 (20.3 to 34.1) | — | 61.9 (55.5 to 67.6) | 27.1 (20.9 to 33.8) | — |
PFS rate (95% CI) at 6 months, %c | 44.2 (37.3 to 50.9) | 11.0 (6.4 to 17.1) | — | 40.6 (34.2 to 46.9) | 10.7 (6.4 to 16.3) | — |
PFS rate (95% CI) at 9 months, %c | 24.6 (18.5 to 31.2) | 8.0 (4.0 to 13.8) | — | 22.8 (17.2 to 28.9) | 7.2 (3.6 to 12.4) | — |
PFS rate (95% CI) at 12 months, %c | 17.2 (11.8 to 23.5) | 6.7 (3.0 to 12.5) | — | 16.2 (11.2 to 22.0) | 6.0 (2.7 to 11.2) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PD = progressive disease; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimates.
dMilestone PFS rate at 3 months was not pre-specified in the statistical analysis plan.
Source: ASCENT Clinical Study Report.13
Figure 4: Progression-Free Survival by IRC Assessment in the ASCENT Trial — BM-Neg Population
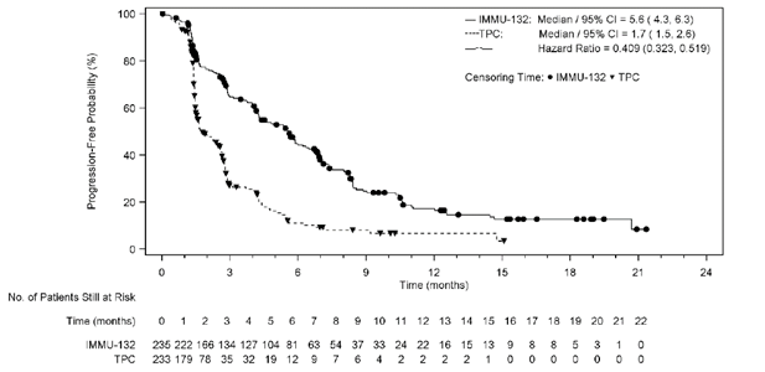
BM-Neg = brain metastasis–negative; CI = confidence interval; IMMU-132 = sacituzumab govitecan; IRC = independent review committee; PFS = progression-free survival; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
Figure 5: Progression-Free Survival by IRC Assessment in the ASCENT Trial — ITT Population
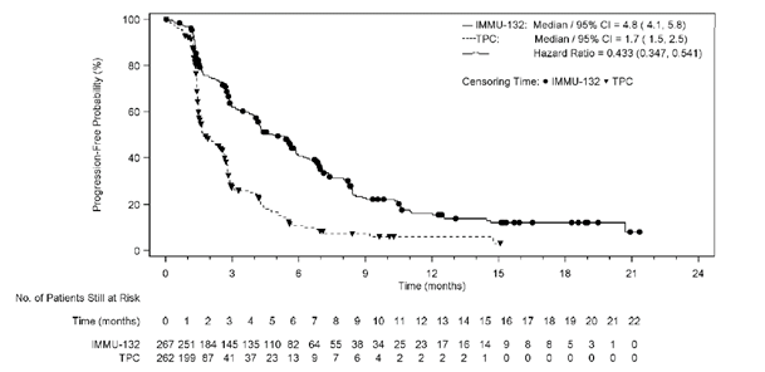
CI: confidence interval; IMMU-132 = sacituzumab govitecan; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
Sensitivity analyses of PFS by IRC assessment were consistent with the primary analysis (Appendix 5). Three pre-specified subgroup analyses of PFS in the ASCENT trial were of interest to this review: BM at baseline (yes or no), prior therapies received (2 to 3 versus > 3) and BRCA1 or BRCA2 status. Among BM-Pos patients (n = 61), the median PFS was 2.8 months in the sacituzumab govitecan arm and 1.6 months in the TPC arm (HR = 0.682; 95% CI, 0.379 to 1.228). Among patients in the ITT set who had received 2 to 3 prior therapies, the median PFS was 5.4 months in the sacituzumab govitecan arm and 1.6 months in the TPC arm (HR = 0.393; 95% CI, 0.300 to 0.515). Among patients in the ITT set who had received more than 3 prior therapies, the median PFS was 4.2 months in the sacituzumab govitecan arm and 2.2 months in the TPC arm (HR = 0.533; 95% CI, 0.369 to 0.771). Among BRCA1- or BRCA2-positive patients in the ITT set, the median PFS was 7.4 months in the sacituzumab govitecan arm and 2.5 months in the TPC arm (HR = 0.421; 95% CI, 0.181 to 0.980); however, the number of BRCA1- or BRCA2-positive patients was small. Among BRCA1- or BRCA2-negative patients in the ITT set, the median PFS was 4.3 months in the sacituzumab govitecan arm and 1.6 months in the TPC arm (HR = 0.454; 95% CI, 0.341 to 0.605).
Time to Progression: BM-Neg Population and ITT Set
Time to progression among patients with PD by IRC assessment was a secondary end point in the ASCENT trial (not adjusted for multiple comparisons in the hierarchical testing structure) and is shown in Table 23. The median time to progression was 5.8 (95% CI, 4.8 to 6.9) months in the sacituzumab govitecan arm, compared with 2.1 (95% CI, 1.5 to 2.7) months in the TPC arm in the BM-Neg population and 5.6 (95% CI, 4.3 to 6.2) months in the sacituzumab govitecan arm, compared with 2.1 (95% CI, 1.5 to 2.8) months in the TPC arm, in the ITT set. The HR for progression by stratified Cox regression analysis was 0.406 (95% CI, 0.315 to 0.525) in the BM-Neg population and 0.429 (95% CI, 0.338 to 0.545) in the ITT set, comparing the sacituzumab govitecan arm with the TPC arm.
Table 23: Time to Progression by IRC Assessment in the ASCENT Trial — BM-Neg and ITT Populations
Measure of progression | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with progression, n (%) | 150 (63.8) | 123 (52.8) | — | 171 (64.0) | 148 (54.2) | — |
Patients without progression (censored), n (%) | 85 (36.2) | 110 (47.2) | — | 96 (36.0) | 120 (45.8) | — |
Median (95% CI) time to progression, monthsa | 5.8 (4.8 to 6.9) | 2.1 (1.5 to 2.7) | — | 5.6 (4.3 to 6.2) | 2.1 (1.5 to 2.8) | — |
Log-rank P value (stratified)b,c | — | < 0.0001 | — | < 0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.406 (0.315 to 0.525) | — | 0.429 (0.338 to 0.545) | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; TPC = treatment of physician’s choice.
aMedian time to progression is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cP values not adjusted for multiple comparisons.
Source: ASCENT Clinical Study Report.13
ORR: BM-Neg Population and ITT Set
ORR by IRC assessment was a secondary end point in the ASCENT trial (not adjusted for multiple comparisons in the hierarchical testing structure) and is shown in Table 24. The ORR was 34.9% (95% CI, 28.8% to 41.4%) in the sacituzumab govitecan arm versus 4.7% (95% CI,2.4% to 8.3%) in the TPC arm among the BM-Neg population and 31.1% (95% CI, 25.6% to 37.0%) versus 4.2% (95% CI, 2.1% to 7.4%) in the TPC arm among the ITT set. The odds ratio for response was 10.859 (95% CI, 5.590 to 21.095) in the BM-Neg population and 10.994 (95% CI, 5.659 to 21.358) in the ITT set, comparing sacituzumab govitecan with TPC. The proportion of patients in the sacituzumab govitecan arm achieving partial responses (BM-Neg population: 30.6%; ITT: 27.3%) was higher than that of patients in the TPC arm (BM-Neg population: 3.9%; ITT: 3.4%). The proportion of patients with stable disease was similar in the sacituzumab govitecan and TPC arms. Very few patients in either arm had complete responses.
Table 24: ORR by IRC Assessment in the ASCENT Trial — BM-Neg and ITT Populations
ORR measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 230 | 230 | — | 261 | 257 | — |
ORR, n (%) [95% CI]a | 82 (34.9) [28.8 to 41.4] | 11 (4.7) [2.4 to 8.3] | — | 83 (31.1) [25.6 to 37.0] | 11 (4.2) [2.1 to 7.4] | — |
OR (95% CI)a | — | 10.859 (5.590 to 21.095) | — | 10.994 (5.659 to 21.358) | ||
P valueb | — | < 0.0001 | — | < 0.0001 | ||
Best overall response, n (%) | ||||||
Complete response | 10 (4.3) | 2 (0.9) | — | 10 (3.7) | 2 (0.8) | — |
Partial response | 72 (30.6) | 9 (3.9) | — | 73 (27.3) | 9 (3.4) | — |
Stable disease | 81 (34.5) | 62 (26.6) | — | 96 (36.0) | 71 (27.1) | — |
Stable disease > 6 months | 23 (9.8) | 9 (3.9) | — | 25 (9.4) | 10 (3.8) | — |
Progressive disease | 54 (23.0) | 89 (38.2) | — | 65 (24.3) | 100 (38.2) | — |
Not evaluable | 18 (7.7) | 71 (30.5) | — | 23 (8.6) | 80 (30.5) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the beta distribution.
bP value is based on Cochran-Mantel-Haenszel test.
Source: ASCENT Clinical Study Report.13
Sensitivity analysis of ORR in the efficacy analyzable population was consistent with the primary analysis (data not shown). Three pre-specified subgroup analyses of ORR in the ASCENT trial were of interest to this review: BM at baseline (yes or no), prior therapies received (2 to 3 versus > 3) and BRCA1 or BRCA2 status. These analyses were uninformative due to the low number of responders in any of the strata.
Time to Response: BM-Neg Population and ITT Set
Time to response among patients with partial responses or complete responses by IRC assessment is shown in Table 25. Mean time to response was 2.67 (SD = 1.91) months in the sacituzumab govitecan arm, compared with 1.86 (SD = 0.92) months in the TPC arm, in the BM-Neg population and 2.66 (SD = 1.91) months in the sacituzumab govitecan arm, compared with 1.86 (SD = 0.92) months in the TPC arm, in the ITT set.
Table 25: Time to Response by IRC Assessment in the ASCENT Trial — BM-Neg and ITT Populations
Measure of time to response | BM-Neg | ITT | ||
|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Sacituzumab govitecan (N = 267) | TPC (N = 262) | |
Patients with responses, n | 82 | 11 | 83 | 11 |
Mean (SD), months | 2.67 (1.91) | 1.86 (0.92) | 2.66 (1.91) | 1.86 (0.92) |
Median (range), months | 1.54 (0.7 to 10.6) | 1.45 (1.3 to 4.2) | 1.54 (0.7 to 10.6) | 1.45 (1.3 to 4.2) |
BM-Neg = brain metastasis–negative; IRC = independent review committee; ITT = intention to treat; SD = standard deviation; TPC = treatment of physician’s choice.
Source: ASCENT Clinical Study Report.13
Harms
Only those harms identified in the review protocol are reported below. Refer to Table 26 for detailed harms data.
Adverse Events
Almost all patients treated with sacituzumab govitecan (99.6%) and TPC (97.8%) experienced at least 1 AE. Frequent AEs (≥ 30% in either arm) that were more common in patients receiving sacituzumab govitecan than in patients receiving TPC included diarrhea (sacituzumab govitecan: 65.1%; TPC: 17.0%), nausea (sacituzumab govitecan: 62.4%; TPC: 30.4%), fatigue (sacituzumab govitecan: 51.6%; TPC: 39.7%), alopecia (sacituzumab govitecan: 46.9%; TPC: 16.1%); neutropenia (sacituzumab govitecan: 42.6%; TPC: 25.4%), anemia (sacituzumab govitecan: 39.1%; TPC: 27.2%), constipation (sacituzumab govitecan: 37.2%; TPC: 23.2%), and vomiting (sacituzumab govitecan: 33.3%; TPC: 16.1%).
Serious Adverse Events
SAEs occurred in similar proportions of patients receiving sacituzumab govitecan (26.7%) and TPC (28.1%). Frequent SAEs (≥ 2% in either arm) that were more common in patients receiving sacituzumab govitecan than in patients receiving TPC included febrile neutropenia (sacituzumab govitecan: 5.0%; TPC: 1.8%), diarrhea (sacituzumab govitecan: 3.5%; TPC: 0%), and pneumonia (sacituzumab govitecan: 2.7%; TPC: 1.8%). Frequent SAEs that were more common in patients receiving TPC than in patients receiving sacituzumab govitecan included pyrexia (sacituzumab govitecan: 1.2%; TPC: 2.2%), dyspnea (sacituzumab govitecan: 0.8%; TPC: 3.1%), and pleural effusion (sacituzumab govitecan: 0.8%; TPC: 2.7%).
AEs Leading to Dose Interruption or Dose Reduction
AEs leading to study drug interruption were more common in the sacituzumab govitecan arm (62.8%) than in the TPC arm (38.8%). AEs leading to dose reduction occurred less frequently in the sacituzumab govitecan arm (21.7%) than in the TPC arm (26.3%).
Withdrawals Due to Adverse Events
AEs leading to discontinuation of protocol therapy occurred in similar proportions of patients receiving sacituzumab govitecan (4.7%) and TPC (5.4%).
Mortality
Most patients died due to PD either during protocol therapy or during survival follow-up. Deaths occurring within 30 days of last study drug treatment were considered deaths due to AEs. Deaths due to AEs occurred in 1 patient (0.4%) in the sacituzumab govitecan arm, who died from respiratory failure, and 3 patients (1.3%) in the TPC arm, who died from neutropenic sepsis, sepsis, and general physical health deterioration related to PD, respectively.
AEs of Special Interest
AEs of special interest occurred in almost all sacituzumab govitecan–treated patients (98.8%) and most (89.7%) TPC-treated patients.
Notable Harms
Several notable harms specified in the CADTH review protocol were more frequent in patients treated with sacituzumab govitecan than in patients treated with TPC. These included neutropenia (neutropenia, febrile neutropenia, and neutrophil count decreased) (sacituzumab govitecan: 65.1%; TPC: 44.2%); anemia (anemia and hemoglobin decreased) (sacituzumab govitecan: 39.1%; TPC: 27.7%); hypersensitivity (sacituzumab govitecan: 34.1%; TPC, 20.5%); and diarrhea (sacituzumab govitecan: 65.1%; TPC: 17.0%). Thrombocytopenia (thrombocytopenia, platelet count decreased) was more frequent in patients receiving TPC than in patients receiving sacituzumab govitecan (sacituzumab govitecan: 6.2%; TPC: 12.5%).
Among notable harms, Grade 3 neutropenia, Grade 4 neutropenia, and Grade 3 diarrhea occurred in more than 10% of patients treated with sacituzumab govitecan (48.4%, 17.8%, and 11.2% of patients, respectively) but lower proportions of patients treated with TPC. Among patients receiving sacituzumab govitecan, neutropenia and diarrhea were frequent causes of dose interruption (46.5% and 5.4% of patients, respectively) and dose reduction (10.9% and 4.7% of patients, respectively). Only 2 patients discontinued sacituzumab govitecan due to notable harms, 1 for diarrhea and 1 for thrombocytopenia.
Table 26: Summary of Harms in the ASCENT Trial — Safety Population
Measure of harms | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) |
|---|---|---|---|
Patients with ≥ 1 AEa | |||
n (%) | 257 (99.6) | 219 (97.8) | 476 (98.8) |
Frequent AEs, n (%)b | |||
Diarrhea | 168 (65.1) | 38 (17.0) | 206 (42.7) |
Nausea | 161 (62.4) | 68 (30.4) | 229 (47.5) |
Fatigue | 133 (51.6) | 89 (39.7) | 222 (46.1) |
Alopecia | 121 (46.9) | 36 (16.1) | 157 (32.6) |
Neutropenia | 110 (42.6) | 57 (25.4) | 167 (34.6) |
Anemia | 101 (39.1) | 61 (27.2) | 162 (33.6) |
Constipation | 96 (37.2) | 52 (23.2) | 148 (30.7) |
Vomiting | 86 (33.3) | 36 (16.1) | 122 (25.3) |
Patients with ≥ 1 SAEc | |||
n (%) | 69 (26.7) | 63 (28.1) | 132 (27.4) |
Frequent SAEs, n (%)d | |||
Febrile neutropenia | 13 (5.0) | 4 (1.8) | 17 (3.5) |
Diarrhea | 9 (3.5) | 0 | 9 (1.9) |
Pneumonia | 7 (2.7) | 4 (1.8) | 11 (2.3) |
Pyrexia | 3 (1.2) | 5 (2.2) | 8 (1.7) |
Dyspnea | 2 (0.8) | 7 (3.1) | 9 (1.9) |
Pleural effusion | 2 (0.8) | 6 (2.7) | 8 (1.7) |
AEs leading to dose reduction | |||
n (%) | 56 (21.7) | 59 (26.3) | 115 (23.9) |
AEs leading to study drug interruption | |||
n (%) | 162 (62.8) | 87 (38.8) | 249 (51.7) |
AEs leading to study drug discontinuation | |||
n (%) | 12 (4.7) | 12 (5.4) | 24 (5.0) |
AEs leading to death | |||
n (%) | 1 (0.4) | 3 (1.3) | 4 (0.8) |
Notable harms, n (%) | |||
AESIs | 255 (98.8) | 201 (89.7) | 456 (94.6) |
Myelosuppression | |||
Neutropeniae | 168 (65.1) | 99 (44.2) | 267 (55.4) |
Grade 3 neutropenia | 125 (48.4) | 65 (29.0) | 190 (39.4) |
Grade 4 neutropenia | 46 (17.8) | 30 (13.4) | 76 (15.8) |
Serious neutropenia | 19 (7.4) | 6 (2.7) | 25 (5.2) |
Neutropenia leading to study drug discontinuation | 0 | 3 (1.3) | 3 (0.6) |
Neutropenia leading to dose interruption | 120 (46.5) | 48 (21.4) | 168 (34.9) |
Neutropenia leading to dose reduction | 28 (10.9) | 43 (19.2) | 71 (4.7) |
Anemiaf | 101 (39.1) | 62 (27.7) | 163 (33.8) |
Grade 3 anemia | 24 (9.3) | 13 (5.8) | 37 (7.7) |
Grade 4 anemia | 0 | 0 | 0 |
Serious anemia | 3 (1.2) | 2 (0.9) | 5 (1.0) |
Anemia leading to study drug discontinuation | 0 | 0 | 0 |
Anemia leading to dose interruption | 11 (4.3) | 6 (2.7) | 17 (3.5) |
Anemia leading to dose reduction | 3 (1.2) | 1 (0.4) | 4 (0.8) |
Thrombocytopeniag | 16 (6.2) | 28 (12.5) | 44 (9.1) |
Grade 3 thrombocytopenia | 3 (1.2) | 5 (2.2) | 8 (1.7) |
Grade 4 thrombocytopenia | 2 (0.8) | 0 | 2 (0.4) |
Serious thrombocytopenia | 2 (0.8) | 0 | 2 (0.4) |
Thrombocytopenia leading to study drug discontinuation | 1 (0.4) | 0 | 1 (0.2) |
Thrombocytopenia leading to dose interruption | 3 (1.2) | 6 (2.7) | 9 (1.9) |
Thrombocytopenia leading to dose reduction | 1 (0.4) | 2 (0.9) | 3 (0.6) |
Infusion reactions | |||
Hypersensitivityh | 88 (34.1) | 46 (20.5) | 134 (27.8) |
Grade 3 hypersensitivity | 3 (1.7) | 3 (1.3) | 6 (1.2) |
Grade 4 hypersensitivity | 0 | 0 | 0 |
Serious hypersensitivity | 1 (0.4) | 3 (1.3) | 4 (0.8) |
Hypersensitivity leading to study drug discontinuation | 0 | 0 | 0 |
Hypersensitivity leading to dose interruption | 3 (1.2) | 1 (0.4) | 4 (0.8) |
Hypersensitivity leading to dose reduction | 0 | 0 | 0 |
Diarrheai | 168 (65.1) | 38 (17.0) | 206 (42.7) |
Grade 3 diarrhea | 29 (11.2) | 2 (0.9) | 31 (6.4) |
Grade 4 diarrhea | 0 | 0 | 0 |
Serious diarrhea | 9 (3.5) | 0 | 9 (1.9) |
Diarrhea leading to study drug discontinuation | 1 (0.4) | 0 | 1 (0.2) |
Diarrhea leading to dose interruption | 14 (5.4) | 1 (0.4) | 15 (3.1) |
Diarrhea leading to dose reduction | 12 (4.7) | 1 (0.4) | 13 (2.7) |
AE = adverse event; AESI = adverse event of special interest; SAE = serious adverse event; SD = standard deviation; TPC = treatment of physician’s choice.
aAll AEs in this table were defined as treatment emergent (start date on or after the date of first dose of study treatment and up to 30 days after date of last dose of study treatment).
bSpecific AEs with a frequency greater than 30% in any group.
cDefined as AEs that were fatal, life threatening, or disabling and/or incapacitating, or that resulted in hospitalization or prolonged a hospital stay, or that resulted in congenital abnormalities.
dSpecific SAEs with a frequency greater than 2% in any group.
eIncludes preferred terms neutropenia, febrile neutropenia, and neutrophil count decreased.
fIncludes preferred terms anemia and hemoglobin decreased.
gIncludes preferred terms thrombocytopenia and platelet count decreased.
hIncludes hypersensitivity standardized Medical Dictionary for Regulatory Activities query (broad) and anaphylactic reactions standardized Medical Dictionary for Regulatory Activities query (broad); only events whose onset dates were on the day of or 1 day after an infusion were included. Includes preferred terms cough, dyspnea, rash, pruritus, stomatitis, hypotension, rash maculo-papular, rhinitis allergic, erythema, hypersensitivity, conjunctivitis, flushing, chest discomfort, dermatitis acneiform, rash pustular, rash macular, rash pruritic, bronchospasm, dermatitis contact, eye pruritis, mouth ulceration, edema, seasonal allergy, skin exfoliation, swollen tongue, urticaria, wheezing, choking, and localized edema.
iIncludes preferred term diarrhea.
Source: ASCENT Clinical Study Report.13
Critical Appraisal
Internal Validity
ASCENT was a phase III, randomized, OL, multi-centre study of heavily pre-treated patients with locally advanced TNBC or mTNBC (N = 529). The study was rigorously designed, and randomization appeared adequate in balancing baseline demographic and disease characteristics (including prior therapies) between the sacituzumab govitecan and TPC arms. There were no baseline imbalances in demographic or disease characteristics of prognostic importance according to the clinical experts consulted for this review. Use of an interactive web response system provided adequate allocation concealment. Application of the inclusion and exclusion criteria to this patient population resulted in a relatively high proportion of screen failures (201 out of 730; 27.5%), some of which were due to unstable CNS disease or inadequate liver or kidney function. However, reasons for screen failure were unknown in at least 16.4% (33 out of 201) patients, and possibly a much higher proportion, as these data were not provided on a per-patient basis. The impact of the potential bias resulting from screen failures is unclear. According to clinical experts consulted for this review, and as in most oncology trials, the enrolment criteria likely selected a healthier cross-section of the overall patient population with mTNBC who were better able to tolerate protocol therapy.
The interventions administered as part of TPC in the ASCENT trial were judged by the clinical experts for this review as appropriate in this patient population. The TPC did not include carboplatin, potentially because this agent is preferred for earlier lines of therapy and had been previously used in approximately two-thirds of the study population. However, a similar proportion of patients had previously received capecitabine, which was allowed as a TPC. According to the clinical experts, exclusion of carboplatin from TPC would be unlikely to have had a major impact on the study results. The outcomes used in the study (PFS, OS, and ORR) are standard in oncology trials, and tumour responses were objectively evaluated using RECIST 1.1 by a blinded IRC. However, patient-reported HRQoL and harms outcomes may have been influenced to some degree by knowledge of treatment allocation. This could explain higher baseline HRQoL scores in the sacituzumab govitecan arm, as HRQoL could have been evaluated post-randomization. Notably, OS was not censored for patients who initiated other anticancer therapies following ASCENT protocol therapy discontinuation; according to clinical experts consulted by CADTH for this review, this would be unlikely to have had a major impact on the study results since similar proportions of patients in both arms received additional therapy and the specific therapies administered were also largely similar for both arms.
Important protocol deviations occurred in 37.1% of all participants in the ASCENT trial. The most common important protocol deviations were related to the informed consent process (15.9%), study inclusion and exclusion criteria (9.5%), and dose formulation and administration (9.1%). The impact of important protocol deviations on the characteristics of the study population and on the administration of protocol therapy was unclear.
Blinding of patients and study personnel was not possible in the ASCENT study due to the mixture of IV and oral agents and variation in dosing schedules. As such, the OL nature of the study design may have contributed to the introduction of several potential biases. Although the overall impact of biases was unclear, several factors suggest that some biases may have been directional in favour of sacituzumab govitecan. Early dropouts and withdrawals from the study occurred more often in the TPC arm than in the sacituzumab govitecan arm. A lower proportion of patients randomized to receive TPC received protocol therapy (86.3% in the BM-Neg population and 85.5% in the ITT set) than did patients randomized to receive sacituzumab govitecan (97.0% in the BM-Neg population and 96.6% in the ITT set), potentially because some patients declined to participate in the study following randomization to the TPC arm. In addition, higher proportions of patients in the TPC arm discontinued treatment (approximately 7%) and discontinued the study (approximately 10%) than did patients in the sacituzumab govitecan arm (discontinued treatment: approximately 2%; discontinued study: approximately 3%). The impact of imbalanced early dropouts and withdrawals would be to decrease exposure to TPC relative to sacituzumab govitecan. Most critically, the decision to discontinue patients from therapy was made by investigators based on unblinded review of local imaging results and/or clinical assessments. These decisions could have altered exposure to sacituzumab govitecan and/or TPC and thus treatment efficacy. Higher proportions of patients in the TPC arm (approximately 19% to 20%) than in the sacituzumab govitecan arm (approximately 11% to 12%) were censored from PFS analyses due to initiation of new anticancer therapy before PD. These patients were incorrectly deemed progressive and withdrawn from protocol therapy by study investigators in the absence of objective PD; the increased frequency of such events in the TPC arm suggested potential bias in favour of sacituzumab govitecan on the part of the investigators. Many, but not all, patients withdrawn from protocol therapy would have subsequently been exposed to another chemotherapy regimen. The number of patients who continued protocol therapy inappropriately despite objective PD was unknown. According to clinical experts consulted for this review, these factors could have biased the PFS analyses in favour of sacituzumab govitecan to some degree, but not sufficiently to call the major findings of the study into question. Losses to follow-up were low and similar (approximately 5%) in both arms. At the time of data cut-off, a higher proportion of patients in the TPC arm (approximately 8%) than in the sacituzumab govitecan arm (approximately 2%) had survival data that were not current by at least 121 days, presumably due to missed survival follow-up assessments. Given the relatively poor survival outcomes in this population, this bias would likely be against sacituzumab govitecan due to missed deaths in the TPC arm.
There were no major issues with statistical analysis of ASCENT trial data that limited confidence in interpretation of the data. Although an interim analysis of OS was planned at the time of the final PFS analysis and could have led to early termination of the study, both analyses reached the pre-specified number of events in the statistical analysis plan and thus were considered final. The study was suitably powered, statistical tests were appropriate, and a strict hierarchical strategy was applied for multiplicity control of PFS and OS in both the BM-Neg and ITT population. The primary PFS analysis was robust to an array of sensitivity analyses conducted by varying censoring rules. ORR and HRQoL, as secondary outcomes, were both outside the statistical hierarchy and were not controlled for multiplicity. The absence of formal statistical comparison and high amounts of missing HRQoL data (due to deaths and dropouts) limited interpretation of potentially important changes in this end point. Subgroup analyses of interest to this review were specified a priori, and 2 of 3 (number of prior therapies and BM status) were based on stratification variables (BRCA mutational status was not a stratification variable). The study was not specifically powered to evaluate strata among subgroups, and the study included relatively few BM-Pos patients (n = 61) and BRCA1- or BRCA2-positive patients (n = 43); subgroup analyses were not controlled for multiplicity.
External Validity
According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics of the ASCENT study population were reflective of the Canadian population with mTNBC, despite only 5 patients having participated in the study at Canadian centres. Although the study population was primarily White (78.8% of the BM-Neg population and 79.0% of the ITT set) and not Hispanic or Latino (86.5% of the BM-Neg population and 87.0% of the ITT set), the clinical experts expected that this would not limit generalizability to other patients. Men can also develop mTNBC, and 2 men (0.4%) were included in the ASCENT trial; generalizability in this small patient subgroup is unclear. Of note, the ASCENT trial enrolled small numbers of patients with locally advanced disease who had not received prior therapies in the metastatic setting (n = 15; 2.8% in the ITT set). According to the sponsor as well as clinical experts consulted for this review, the treatment approach for unresectable locally advanced TNBC and mTNBC is the same, and patients with unresectable locally advanced TNBC who had been treated previously in the same manner as a metastatic patient and received at least 2 lines of systemic therapy would be eligible to receive sacituzumab govitecan in accordance with the Health Canada indication.
Potential administration of sacituzumab govitecan outside the Health Canada indication was identified as a possibility by the clinical experts consulted for this review. The ASCENT trial enrolled patients with ECOG Performance Status 0 or 1 who had previously received at least 2 lines of therapy (including a taxane in the adjuvant, neoadjuvant, or advanced setting). Thus, generalizability of the trial results to patients with ECOG Performance Status 2, to patients who have not yet tried taxanes, or to earlier-line therapy of patients who are not candidates for chemotherapy is unclear. According to the experts, in the real world at least some of these patients would likely be offered sacituzumab govitecan.
The doses of sacituzumab govitecan and TPC were aligned with Health Canada–approved dosing and were in line with clinical practice. In the ASCENT study, treatment with sacituzumab govitecan or TPC was administered until PD or unacceptable toxicity. Although this was in line with clinical practice according to clinical experts, the trial data may not be generalizable to other treatment durations (e.g., time-limited treatment or treatment until best response), especially since objective responses to sacituzumab govitecan occurred later than those to TPC (mean [SD], 2.67 [1.91] months versus 1.86 [0.92] months) and time to response was right skewed. The number of patients who continued sacituzumab govitecan beyond objective PD in the ASCENT study was unknown; however, clinical experts consulted for this review did not feel that this would limit generalizability to the strategy of treatment until PD or unacceptable toxicity. The clinical experts stated that the breakdown of TPC regimens used in the study was reflective of the third-line (and post-third-line) treatment setting in Canada. All the outcomes evaluated in the trial and considered in this review (PFS, HRQoL, symptoms, OS, and ORR) were clinically relevant, important to patients, and used in clinical practice. The duration of follow-up (7 months to 12 months) was sufficient for assessment of these outcomes in this population.
A potentially important issue limiting generalizability of the ASCENT study findings to Canadian patients was the high rate of immunostimulant (G-CSF) use among patients receiving sacituzumab govitecan (46.4% of the BM-Neg population and 47.2% of the ITT set). G-CSF was administered for treatment of neutropenia as necessary as well as for secondary prophylaxis to avoid dose reductions; in addition to Grade 4 neutropenia and Grade 3 or higher febrile neutropenia, G-CSF was administered in patients with Grade 3 neutropenia (not febrile) that delayed dosing by 2 or 3 weeks for recovery to Grade 1 or less. The clinical experts consulted by CADTH for this review emphasized that G-CSF cannot generally be accessed in Canada in the metastatic setting and that typical clinical practice would be dose reduction and, if necessary, discontinuation of treatment in patients with severe or persistent neutropenia or febrile neutropenia. Thus, more frequent sacituzumab govitecan dose reduction and/or discontinuation might occur for Canadian patients, leading to lower efficacy. The clinical experts did not expect the safety profile of sacituzumab govitecan to differ in the absence of G-CSF therapy: Although infections can be a consequence of myelosuppression, close monitoring of neutropenia and appropriate dose reductions are sufficient to ensure safe administration of sacituzumab govitecan.
Subgroup analyses were not adjusted for multiplicity and were not powered to evaluate differences in the treatment effects of sacituzumab govitecan in patients with and without BRCA1 or BRCA2 mutations, patients who had received 2 to 3 or more than 3 prior lines of therapy, or BM-Pos and BM-Neg patients. Nevertheless, the clinical experts consulted for this review felt that the results of the trial were generalizable across strata for all these subgroups. The clinical experts highlighted that the overall prognosis of BM-Pos patients is poor, and as a result, the BM-Pos patients who received sacituzumab govitecan or TPC derived less benefit than BM-Neg patients. The clinical experts acknowledged that, as with many oncologic drugs, there are some concerns that antibody drugs such as sacituzumab govitecan may not adequately penetrate the brain to elicit as strong an effect as in visceral tissues, thereby compromising response rates; however, the clinical experts noted that drugs are not withheld from patients with BM, as a clinical benefit may still be elicited. Ultimately, the clinical experts agreed that results are generalizable to BM-Pos patients and expressed that they would offer sacituzumab govitecan to BM-Pos patients. However, the clinical experts also acknowledged that the efficacy of all therapies for mTNBC decreases as line of therapy increases. Since the study included patients who had received from 2 to 17 prior systemic therapies, the generalizability of the trial evidence to any specific line or combination of prior therapies is unclear.
Since administration of sacituzumab govitecan would occur in a hospital or specialty clinic setting, background care (oncologist visits, imaging frequency, bloodwork, and so on) would be expected to be similar for Canadian patients compared with those participating in the ASCENT trial. Some patients in the ASCENT study receiving TPC (especially oral capecitabine) could have received additional background care via their participation in the trial. However, this would be expected to result in bias against sacituzumab govitecan.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
No indirect evidence was submitted by the sponsor. A focused literature search for network meta-analyses dealing with triple-negative breast neoplasms was run in MEDLINE All (1946–) on July 28, 2021. No limits were applied to the search. No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Discussion
Summary of Available Evidence
One phase III, randomized, OL, multi-centre study (ASCENT, N = 529, primarily US and Europe)11,12 contributed evidence to this report. The study enrolled patients with unresectable locally advanced TNBC or mTNBC (ECOG Performance Status 0 or 1 with adequate organ function) who had received 2 prior lines of therapy including 1 taxane in the adjuvant, neoadjuvant, or advanced setting. Patients were randomized 1:1 to receive either sacituzumab govitecan or TPC until PD or unacceptable toxicity. Tumour response was assessed using RECIST 1.1, and responses were designated by a blinded IRC. The primary outcome was PFS in the BM-Neg population, while secondary outcomes included PFS in the ITT set, OS in the BM-Neg population and ITT set, ORR, and HRQoL.
According to the clinical experts consulted by CADTH for this review, the baseline characteristics of the ASCENT study population were representative of Canadian patients with mTNBC who would be candidates for sacituzumab govitecan. Most patients (88.5%) were BM-Neg, White (79.0%), not Hispanic or Latino (87.0%), and BRCA1- or BRCA2-negative (91.9%). Most had received prior surgery (94.9%), non-brain radiotherapy (81.1%), and systemic therapies in the metastatic setting (97.9%), while a subset had received prior PD-1 or PD-L1 therapy (28.9%). The mean (SD) age of participants was 54.0 (11.5) years, and the mean (SD) number of prior systemic therapies received was 4.5 (2.1). Baseline demographic and disease characteristics were generally well balanced between study arms. There were no major methodological limitations of the study, apart from potential biases inherent to its OL design, although generalizability to administration in the Canadian context without G-CSF was an area of concern.
Interpretation of Results
Efficacy
Administration of sacituzumab govitecan in the ASCENT trial resulted in statistically significant prolongation of PFS (4.8 months to 5.6 months versus 1.7 months) and OS (11.8 months to 12.1 months versus 6.7 months to 6.9 months) compared with TPC in both the BM-Neg population and the ITT set. Although outside the statistical hierarchy, ORR was observed to be notably higher in the sacituzumab govitecan arm (31.1% to 34.9%) than in the TPC arm (4.2% to 4.7%) in both the BM-Neg population and the ITT set. According to the clinical experts consulted for this review, these results are highly clinically meaningful for later-line treatment of patients with mTNBC, for whom effective therapies are currently lacking. There were also potential signals of slight improvements in EORTC QLQ-C30 global health status, physical and role functioning, and cancer symptoms in patients receiving sacituzumab govitecan, although nausea and/or vomiting, as well as diarrhea, were potentially aggravated. However, because the MIDs for EORTC QLQ-C30 scores were not clearly achieved, HRQoL analyses were descriptive and limited by high rates of missing data, and EORTC QLQ-C30 was patient administered in a study with OL design, the clinical experts consulted for this review felt that changes in HRQoL in the ASCENT trial were hopeful but extremely uncertain. PFS, OS, HRQoL, and symptom relief were identified by patient groups as the most important outcomes to patients with mTNBC.
Although biases related to the OL design of the ASCENT trial, especially higher dropout rates in the TPC arm and unblinded decisions to discontinue patients from therapy, may have affected the study results to some degree, these were not considered by the clinical experts consulted for this review as likely to significantly influence interpretation. Although higher dropout rates in the TPC arm and higher rates of censoring due to initiation of other anticancer therapy without objective PD would have decreased exposure to TPC, many patients who discontinued ASCENT protocol therapy would have subsequently been treated with another chemotherapy regimen. Even if such biases contributed to slightly poorer outcomes in the TPC arm, these differences would be unlikely to alter the major conclusions of the study. Clinical experts speculated that based on the biology of mTNBC, variability in patient performance status (ECOG 2), previous taxane treatment, or line of therapy would have only minor impact on the efficacy of sacituzumab govitecan. Although administration of sacituzumab govitecan in the absence of G-CSF may require additional dose reductions, this would be required only in a subset of patients.
Harms
The safety profile of sacituzumab govitecan in the ASCENT trial was as expected based on prior experience with the drug, and AEs were considered manageable by patients and clinicians with appropriate supportive care. SAEs and WDAEs occurred at similar frequencies in patients treated with sacituzumab govitecan and TPC. Although neutropenia and diarrhea were associated with sacituzumab govitecan administration, with supportive care and dose reductions these notable harms did not generally require discontinuation of therapy. However, many neutropenic patients in the ASCENT trial received G-CSF, and the frequencies of dose reductions and discontinuations in the absence of G-CSF are unclear.
Other Considerations
No other considerations were identified for this review.
Conclusions
Evidence from the ASCENT trial suggested that compared with TPC, administration of sacituzumab govitecan (10 mg/kg on day 1 and day 8 of a 21-day treatment cycle) contributed to statistically significant and clinically meaningful prolongation of PFS and OS among patients with locally advanced TNBC or mTNBC who had received at least 2 prior therapies. ORRs were higher and time to progression was longer in patients treated with sacituzumab govitecan compared with standard chemotherapy. Analyses of EORTC QLQ-C30 data could not be interpreted due to absence of formal statistical testing and high rates of missing data resulting from deaths and withdrawals. The magnitude of the observed survival benefits and potential impact on cancer symptoms are important outcomes for patients with mTNBC. Notable harms associated with sacituzumab govitecan (including neutropenia and diarrhea) were not insignificant but manageable with appropriate supportive care (including G-CSF) and dose modification and rarely required withdrawal of treatment. Minor limitations of the available evidence included bias in favour of sacituzumab govitecan on the part of patients and investigators due to the OL design of the ASCENT trial, which may have decreased exposure to TPC relative to sacituzumab govitecan, as well as potential for higher dose reduction and discontinuation rates for Canadian patients in the absence of G-CSF.
References
1.Plasilova ML, Hayse B, Killelea BK, Horowitz NR, Chagpar AB, Lannin DR. Features of triple-negative breast cancer: Analysis of 38,813 cases from the national cancer database. Medicine (Baltimore). 2016;95(35):e4614. PubMed
2.DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66(1):31-42. PubMed
3.Trivers KF, Lund MJ, Porter PL, et al. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control. 2009;20(7):1071-1082. PubMed
4.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
5.Drug Reimbursement Review sponsor submission: Trodelvy® (sacituzumab govitecan),180 mg lyophilized powder for solution for injection, for intravenous use [internal sponsor's package]. Mississauga (ON): Gilead Sciences Canada, Inc; 2021.
6.CADTH Reimbursement Review pharmacoeconomic report: Sacituzumab govitecan (Trodelvy). Ottawa (Canada): CADTH; 2021.
7.Kassam F, Enright K, Dent R, et al. Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer. 2009;9(1):29-33. PubMed
8.Gradishar WJ, Anderson BO, Abraham J, et al. Breast Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(4):452-478. PubMed
9.Li CH, Karantza V, Aktan G, Lala M. Current treatment landscape for patients with locally recurrent inoperable or metastatic triple-negative breast cancer: a systematic literature review. Breast Cancer Res. 2019;21(1):143. PubMed
10.Zeichner SB, Terawaki H, Gogineni K. A Review of Systemic Treatment in Metastatic Triple-Negative Breast Cancer. Breast Cancer (Auckl). 2016;10:25-36. PubMed
11.Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2021;384(16):1529-1541. PubMed
12.Bardia A, Tolaney SM, Punie K, et al. Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Ann Oncol. 2021;08:08.
13.Clinical Study Report: IMMU-132-05. An international, multi-center, open-label, randomized, phase 3 trial of sacituzumab govitecan versus treatment of physician choice in patients with metastatic triple-negative breast cancer who received at least two prior treatments [internal sponsor's report]. Morris Plains (NJ): Immunomedics,Inc; 2020 Oct 26.
14.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674-690. PubMed
15.Seung SJ, Traore AN, Pourmirza B, Fathers KE, Coombes M, Jerzak KJ. A population-based analysis of breast cancer incidence and survival by subtype in Ontario women. Curr Oncol. 2020;27(2):e191-e198. PubMed
16.Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275-1281. PubMed
17.Tseng LM, Hsu NC, Chen SC, et al. Distant metastasis in triple-negative breast cancer. Neoplasma. 2013;60(3):290-294. PubMed
18.Cardoso F, Costa A, Senkus E, et al. 3rd ESO-ESMO international consensus guidelines for Advanced Breast Cancer (ABC 3). Breast. 2017;31:244-259. PubMed
19.Finn RS, Martin M, Rugo HS, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med. 2016;375(20):1925-1936. PubMed
20.Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783-1791. PubMed
21.Nagayama A, Vidula N, Bardia A. Novel Therapies for Metastatic Triple-Negative Breast Cancer: Spotlight on Immunotherapy and Antibody-Drug Conjugates. Oncology (Williston). 2021;35(5):249-254. PubMed
22.Moy B, Rumble RB, Come SE, et al. Chemotherapy and Targeted Therapy for Patients With Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer That is Either Endocrine-Pretreated or Hormone Receptor-Negative: ASCO Guideline Update. J Clin Oncol. 2021:JCO2101374. PubMed
23.Shih LB, Thorpe SR, Griffiths GL, et al. The processing and fate of antibodies and their radiolabels bound to the surface of tumor cells in vitro: a comparison of nine radiolabels. J Nucl Med. 1994;35(5):899-908. PubMed
24.Stein R, Goldenberg DM, Thorpe SR, Basu A, Mattes MJ. Effects of radiolabeling monoclonal antibodies with a residualizing iodine radiolabel on the accretion of radioisotope in tumors. Cancer Res. 1995;55(14):3132-3139. PubMed
25.Govindan SV, Cardillo TM, Sharkey RM, Tat F, Gold DV, Goldenberg DM. Milatuzumab-SN-38 conjugates for the treatment of CD74+ cancers. Mol Cancer Ther. 2013;12(6):968-978. PubMed
26.Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA, Sharkey RM. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget. 2015;6(26):22496-22512. PubMed
27.Sharkey RM, McBride WJ, Cardillo TM, et al. Enhanced Delivery of SN-38 to Human Tumor Xenografts with an Anti-Trop-2-SN-38 Antibody Conjugate (Sacituzumab Govitecan). Clin Cancer Res. 2015;21(22):5131-5138. PubMed
28.Lopez S, Perrone E, Bellone S, et al. Preclinical activity of sacituzumab govitecan (IMMU-132) in uterine and ovarian carcinosarcomas. Oncotarget. 2020;11(5):560-570. PubMed
29.Perrone E, Lopez S, Zeybek B, et al. Preclinical Activity of Sacituzumab Govitecan, an Antibody-Drug Conjugate Targeting Trophoblast Cell-Surface Antigen 2 (Trop-2) Linked to the Active Metabolite of Irinotecan (SN-38), in Ovarian Cancer. Front Oncol. 2020;10:118. PubMed
30.Zeybek B, Manzano A, Bianchi A, et al. Cervical carcinomas that overexpress human trophoblast cell-surface marker (Trop-2) are highly sensitive to the antibody-drug conjugate sacituzumab govitecan. Sci Rep. 2020;10(1):973. PubMed
31.Trodelvy®sacituzumab govitecan180 mg lyophilized powder for solutionfor injection, for intravenous use [draft product monograph]. Morris Plains (NJ): Immunomedics, Inc.; [date unknown].
32.Product monograph for Halaven®(eribulin mesylate) injection, 0.5 mg/mL. Mississauga (Canada): Eisai, Ltd.; 2017.
33.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. Journal of clinical epidemiology. 2016;75:40-46. PubMed
34.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2020 Nov 30.
35.Bardia A, Mayer IA, Vahdat LT, et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2019;380(8):741-751. PubMed
36.Bardia A, Messersmith WA, Kio EA, et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021;32(6):746-756. PubMed
37.Kalinsky K, Diamond JR, Vahdat LT, et al. Sacituzumab govitecan in previously treated hormone receptor-positive/HER2-negative metastatic breast cancer: final results from a phase I/II, single-arm, basket trial. Ann Oncol. 2020;31(12):1709-1718. PubMed
38.Bardia A, Mayer IA, Diamond JR, et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J Clin Oncol. 2017;35(19):2141-2148. PubMed
39.Ocean AJ, Starodub AN, Bardia A, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer. 2017;123(19):3843-3854. PubMed
40.Starodub AN, Ocean AJ, Shah MA, et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clinical Cancer Research. 2015;21(17):3870-3878. PubMed
41.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
42.Fayers P, Bottomley A, Group EQoL, Quality of Life U. Quality of life research within the EORTC-the EORTC QLQ-C30. European Organisation for Research and Treatment of Cancer. Eur J Cancer. 2002;38 Suppl 4:S125-133. PubMed
43.Osoba D, Aaronson N, Zee B, Sprangers M, te Velde A. Modification of the EORTC QLQ-C30 (version 2.0) based on content validity and reliability testing in large samples of patients with cancer. The Study Group on Quality of Life of the EORTC and the Symptom Control and Quality of Life Committees of the NCI of Canada Clinical Trials Group. Qual Life Res. 1997;6(2):103-108. PubMed
44.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-1807. PubMed
45.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
46.Groenvold M, Klee MC, Sprangers MA, Aaronson NK. Validation of the EORTC QLQ-C30 quality of life questionnaire through combined qualitative and quantitative assessment of patient-observer agreement. Journal of clinical epidemiology. 1997;50(4):441-450. PubMed
47.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159-174. PubMed
48.McLachlan SA, Devins GM, Goodwin PJ. Validation of the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (QLQ-C30) as a measure of psychosocial function in breast cancer patients. Eur J Cancer. 1998;34(4):510-517. PubMed
49.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
50.Musoro JZ, Coens C, Fiteni F, et al. Minimally Important Differences for Interpreting EORTC QLQ-C30 Scores in Patients With Advanced Breast Cancer. JNCI Cancer Spectr. 2019;3(3):pkz037. PubMed
51.Ousmen A, Conroy T, Guillemin F, et al. Impact of the occurrence of a response shift on the determination of the minimal important difference in a health-related quality of life score over time. Health Qual Life Outcomes. 2016;14(1):167. PubMed
52.Cocks K, King MT, Velikova G, Martyn St-James M, Fayers PM, Brown JM. Evidence-based guidelines for determination of sample size and interpretation of the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30. J Clin Oncol. 2011;29(1):89-96. PubMed
Appendix 1: Patient Group Input
Note this appendix has not been copy-edited.
Rethink Breast Cancer
About Rethink Breast Cancer (RBC)
Rethink Breast Canada’s mission is to empower young people worldwide who are concerned about and affected by breast cancer through education, support and advocacy. Since 2001, we have been building community for young women with breast cancer and providing support and resources to help them live the best quality of life. Because up to 30% of all breast cancers become metastatic, Rethink Breast Cancer has always worked closely with young MBC patients—who, sadly, leave our community far soon. We represent the voice of young people with breast cancer and strive to ensure their needs and values are heard and considered in all aspects of breast cancer treatment and care at all stages of their breast cancer experience. www.rethinkbreastcancer.com
Information Gathering
Online patient surveys were conducted between June 19 and July 10, 2021. The surveys asked questions about the impact of breast cancer on the lives of patients, the effect of current treatments and their willingness to accept side effects for improved health outcomes. The survey also included questions directed to patients with Trodelvy treatment experience. Potential respondents were identified through messages posted to Rethink’s Young Women’s Network and Instagram channel as well as through Facebook and Twitter. Messages were also posted on the Cancer Connection, BreastCancer.org and Cancer Survivors Network online discussion forums.
A total of 30 people completed the patient survey. Of these respondents, 6 are from Canada (representing Alberta, British Columbia, Manitoba and Ontario), 22 are from the United States, 1 is from the United Kingdom and 1 is from Antigua and Barbuda.
Disease Experience
All 30 respondents have been diagnosed with metastatic triple-negative breast cancer (mTNBC).
4 respondents were diagnosed in 2020, 9 were diagnosed in 2019, 4 were diagnosed in 2018, 6 were diagnosed in 2017, 3 were diagnosed between 2016, and 4 were diagnosed in 2015 or earlier.
9 respondents were originally diagnosed with mTNBC, while 21 had disease progression following their initial diagnosis.
10 respondents have brain metastases.
22 respondents are currently receiving third-line treatment or higher, 3 are receiving second-line treatment, 2 are receiving first-line treatment, 2 are receiving treatment after recurrence and 1 has had no evidence of disease for between six months and two years.
Experiences With Currently Available Treatments
All 30 respondents provided information about the treatments they have received since their diagnosis. Over half of respondents were treated with paclitaxel, capecitabine, doxorubicin, nabpaclitaxel and atezolizumab.
Treatments Received, n
Taxol (paclitaxel), 20
Xeloda (capecitabine), 20
Adriamycin (doxorubicin), 19
Abraxane (nab-paclitaxel), 17
Tecentriq (atezolizumab), 16
Gemzar (gemcitabine), 13
Paraplatin (carboplatin), 11
Cytoxan (cyclophosphamide), 10
Halaven (eribulin), 7
Keytruda (pembrolizumab), 7
Taxotere (docetaxel), 6
Lynparza (olaparib), 4
Radiation, 2
Cisplatin, 2
Epirubicin, 2
Navelbine (vinorelbine), 1
Opdivo (nivolumab), 1
Kadcyla (trastuzumab emtansine), 1
Herceptin (trastuzumab), 1
Kisqali (ribociclib), 1
Most respondents have undergone multiple lines of treatment and reported a wide range of outcomes and side effects. Their description of the side effects of previous treatments tended to be more severe than those reported in other surveys conducted by Rethink Breast Cancer for previous submissions. Many respondents reported hospitalizations due to the side effects of previous therapies. Xeloda was often identified as especially difficult to tolerate.
Fatigue was the most commonly reported side effect of previous treatments (97%, n=30), followed by loss of appetite (77%), nausea (70%), constipation (67%), diarrhea (60%) and headache (57%).
Hand and foot syndrome, nausea and fatigue were identified as the most difficult to tolerate side effects of these treatments.
Improved Outcomes
Rethink Breast Cancer asked patients to evaluate the importance of different outcomes for their breast cancer treatment on a scale of 1 (not important) to 5 (very important). All outcomes were rated over 4.4, but controlling disease progression, preventing recurrence and overall survival were considered the most important patient values. Preventing recurrence was rated higher by these respondents than respondents to surveys for previous submissions, likely reflecting their longer treatment history.
Table 27: Patients’ Rated Importance of Outcome From RBC Patient Group
Importance of outcome | 1 - not important | 2 | 3 | 4 | 5 – very important | Average |
|---|---|---|---|---|---|---|
Controlling disease progression | 0.00% 0 | 0.00% 0 | 0.00% 0 | 3.33% 1 | 96.67% 29 | 4.97 30 |
Reducing symptoms | 3.45% 1 | 6.90% 2 | 6.90% 2 | 10.34% 3 | 72.41% 21 | 4.41 29 |
Maintaining quality of life | 0.00% 0 | 0.00% 0 | 6.67% 2 | 10.00% 3 | 83.33% 25 | 4.77 30 |
Managing side effects | 0.00% 0 | 3.33% 1 | 3.33% 1 | 23.33% 7 | 70.00% 21 | 4.60 30 |
Preventing recurrence | 0.00% 0 | 0.00% 0 | 0.00% 0 | 3.33% 1 | 96.67% 29 | 4.97 30 |
Overall survival | 0.00% 0 | 0.00% 0 | 0.00% 0 | 3.33% 1 | 96.67% 29 | 4.97 30 |
Comments:
I am in treatment to LIVE; therefore I have to take a few side effects with a grain of salt sometimes.
I want to be around for my husband and my 2 kids. It breaks my heart to think of them experiencing milestones without me there to cheer them on.
Experience With Drug Under Review
Twenty respondents match the full indication for this review – they were treated as a breast cancer patient with Trodelvy, they received at least two lines of treatment for breast cancer before Trodelvy, and they received at least one line of treatment for metastatic breast cancer before receiving Trodelvy. 1 of these respondents is from Canada; the other 19 are from the United States. 4 of the respondents in this group agreed to participate in telephone interviews with staff members to discuss their treatment experience and elaborate on their feedback.
Patient Experience
5 respondents had received Trodelvy for less than 3 months, 8 respondents had received it for 3-6 months, and 7 respondents had received it for 6-12 months.
15 respondents were still receiving Trodelvy at the time of the survey, while 5 stopped receiving it because it did not control their cancer.
Quality of Life
Patients were asked to rate the change to their quality of life on Trodelvy compared to other treatments they had received on a scale of 1 (much worse) to 5 (much better). Patients indicated improvements in every area except for the ability to work where the effect was neutral. Stronger positive changes were noted for metastatic cancer symptoms, controlling disease, overall survival and preventing recurrence. It should be noted that the latter three categories were rated as the most important patient values in section 5.
Table 28: Patients’ Rated Change to Quality of Life From RBC Patient Group
Change to quality of life on Trodelvy | 1 – much worse | 2 | 3 | 4 | 5 – much better | n/a | Average |
|---|---|---|---|---|---|---|---|
Controlling disease | 5.00% 1 | 0.00% 0 | 15.00% 3 | 20.00% 4 | 45.00% 9 | 15.00% 3 | 4.20 17 |
Metastatic cancer symptoms | 0.00% 0 | 0.00% 0 | 20.00% 4 | 40.00% 8 | 40.00% 8 | 0.00% 0 | 4.20 20 |
Drug side effects | 10.53% 2 | 10.53% 2 | 31.58% 6 | 26.32% 5 | 15.79% 3 | 5.26% 1 | 3.28 19 |
Maintaining quality of life | 0.00% 0 | 5.00% 1 | 30.00% 6 | 35.00% 7 | 30.00% 6 | 0.00% 0 | 3.90 20 |
Preventing recurrence | 5.00% 1 | 0.00% 0 | 5.00% 1 | 25.00% 5 | 25.00% 5 | 40.00% 8 | 4.08 12 |
Overall survival | 5.00% 1 | 0.00% 0 | 15.00% 3 | 20.00% 4 | 40.00% 8 | 20.00% 4 | 4.13 16 |
Ability to work | 0.00% 0 | 10.00% 2 | 10.00% 2 | 10.00% 2 | 0.00% 0 | 70.00% 14 | 3.00 6 |
Ability to sleep | 0.00% 0 | 15.00% 3 | 30.00% 6 | 30.00% 6 | 15.00% 3 | 10.00% 2 | 3.50 18 |
Ability to drive | 0.00% 0 | 5.00% 1 | 30.00% 6 | 30.00% 6 | 15.00% 3 | 20.00% 4 | 3.69 16 |
Ability to perform household chores | 5.00% 1 | 0.00% 0 | 35.00% 7 | 30.00% 6 | 25.00% 5 | 5.00% 1 | 3.74 19 |
Ability to care for children | 0.00% 0 | 5.00% 1 | 10.00% 2 | 10.00% 2 | 20.00% 4 | 55.00% 11 | 4.00 9 |
Comments:
Some days I just have to sleep; some days I can’t really leave because of my stomach, and then other days, I’m moving around; I have grandkids and they spend time with me, and I just keep going like nothing else is going on in my life
Most days I feel normal, whereas before I wasn’t feeling normal
I remember it was crazy how Trodelvy worked immediately
Symptom Relief
7 respondents indicated that Trodelvy had helped to relieve some of the symptoms associated with mTNBC. Jacksonian marches, bone pain and neuropathy were all identified as specific cancer symptoms that improved during treatment with Trodelvy.
Comments include:
I haven’t had any brain episodes since starting Trodelvy which is huge because those were affecting my day-to-day life because if it happened the right side, then I couldn’t speak, on the left side, I couldn’t walk
I knew pretty much from the start back in November that it was helping because my bone pain … it disappeared - I had no pain
Because Trodelvy is really working, my pain kind of went away, so it really helped my quality of life
I definitely think its decreasing [my brain mets] which has given me less symptoms and allowed me to have a better quality of life
Side Effects
A majority of patients experienced fatigue (79%, n=19), alopecia (74%), diarrhea (68%) and neutropenia (59%) as side effects from Trodelvy.
When asked how much they could tolerate the side effects associated with Trodelvy on a scale of 1 (completely intolerable) to 10 (completely tolerable), the average score was 8.05. Only two respondents gave a score lower than 5.
Table 29: Patients’ Rated Tolerability of Side Effects From RBC Patient Group
Rating | Responses, n (%) |
|---|---|
1 | 0 (0.00) |
2 | 0 (0.00) |
3 | 2 (10.53) |
4 | 0 (0.00) |
5 | 2 (10.53) |
6 | 0 (0.00) |
7 | 1 (5.26) |
8 | 4 (21.05) |
9 | 3 (15.79) |
10 | 7 (36.84) |
Comments:
The only serious side effect was the neutropenia. All the others are tolerable or manageable with medication.
The diarrhea gets annoying, but is it continues to extend my life, I’ll take it.
All had their own challenges, but Trodelvy was the easiest by far
Trodelvy was the easiest for side effects.
Patients also emphasized that they were willing and able to tolerate these side effects for the medical benefits provided:
It’s not easy but cancer is rough
I can deal with an occasional day of not feeling well in my tummy for keeping my cancer at bay
Many respondents also noted that they were able to manage the side effects with the use of other drugs.
Anything Else?
When asked if they would recommend Trodelvy to other patients with breast cancer, all 20 respondents said that they would.
Asked to elaborate, comments included:
It was great! Very tolerable and I felt “normal”
I have made steady improvement. Less fatigue, more energy, regained appetite.
I would absolutely recommended this drug to other patients with breast cancer. Everyone is different when it comes to what drugs they respond to, but I feel this drug is especially important for those who have failed multiple treatments prior to trying this
I feel it is a great drug, especially for those with brain mets. As tolerable or more tolerable as other chemos I have been on. Neuropathy hit quick though and fatigue/insomnia is tough.
It’s working! Mets in lungs have disappeared, mets in liver and bones are shrinking.
It is an absolute must
This was the first medicine that got me clear – to NED – after just a couple of months, so it was really a blessing
I'm in USA getting Trodelvy, it is working for me and I hope every Canadian who is diagnosed with mTNBC has a chance to get this treatment.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
We asked Gilead to provide us with information about the general characteristics of the drug and its benefits. We asked our Scientific Advisory Committee (medical oncologists) about this drug and its benefits and whether it addressed an unmet need. Adam Waiser is a freelance health technology assessment writer who we contracted to help us with writing this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
We contracted Adam Waiser to help us develop the survey we used to collect the data used in this submission. All interviews were conducted by Rethink Breast Cancer staff. Adam Waiser helped us analyze the findings of our survey and interviews.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 30: Conflict of Interest Declaration for RBC
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
Gilead Sciences | X | — | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Rethink Breast Cancer
Date: July 21, 2021
Canadian Breast Cancer Network
About the Canadian Breast Cancer Network (CBCN)
The Canadian Breast Cancer Network (CBCN) is a leading, patient-directed, national health charity committed to ensuring the best quality of care for all Canadians affected by breast cancer through the promotion of information, education and advocacy activities. www.cbcn.ca
As a member of the Canadian Cancer Action Network, the Canadian Breast Cancer Network is committed to adhering to the Code of Conduct Governing Corporate Funding.
Information Gathering
Information for this submission was collected via:
CBCN’s 2017 Lived Experience Breast Cancer Patient Survey: An online survey was distributed in English and French to patients living with breast cancer. No patients surveyed had direct experience with the treatment under review. Survey questions comprised of a combination of scoring options and free form commentary. Patients were contacted through the membership databases of CBCN and other patient organizations.
Patient Respondents Profile:
157 Canadian metastatic patients participated in the survey. In this submission, CBCN specifically utilizes the data provided by the 14 patients who identified as having metastatic triple negative breast cancer (mTNBC).
The respondents all identified as female and all spoke English as a first-language. The majority of respondents were from Ontario (6) and British Columbia (2). The rest of the respondents were from New Brunswick (1), Alberta (1), Quebec (1), Nova Scotia (1). Saskatchewan (1) and Newfoundland and Labrador (1).
Most of the respondents (5) were between the ages of 50-59 when they were diagnosed with metastatic breast cancer, 4 respondents were in the 40-49 age range, 3 were between 30-39 years, and 2 were between 60-69 years of age.
All but 1 respondent was in a relationship. 13 of the mTNBC patients had children, with the majority (7) having children 20 years or older. 4 had children between the ages of 13-19, 3 had children 2-5 years of age, and 3 had children between 6-12 years old.
CBCN’s 2012 Lived Experience of Metastatic Breast Cancer Patients and Caregivers Survey Report: An online survey, conducted in collaboration with ReThink Breast Cancer, was distributed to patients living with metastatic breast cancer (mBC) and their caregivers. No patients surveyed had experience with the treatment under review. Survey questions comprised of a combination of scoring options and free form commentary. Patients were contacted through the membership databases of CBCN and other patient organizations.
Key informant interviews: A phone interview were conducted in June 2021 with a Canadian metastatic breast cancer patients living with metastatic triple negative breast cancer that had direct experience with the treatment under review.
Printed sources: A review was conducted of current studies and grey literature to identify issues and experiences that are commonly shared among many women living with breast cancer.
Disease Experience
Metastatic breast cancer is the spread of cancerous cell growth to areas of the body other than where the cancer first formed, and is often more severe. It is most commonly spread to the bones, but can include the lungs, liver, brain and skin. Current treatment options for metastatic breast cancer are only effective at prolonging progression-free disease, and most cases of advanced disease will progress and symptoms will worsen. Patients with a diagnosis of metastatic breast cancer understand the limitations of current treatment options and seek to live their remaining months and years with the best possible quality of life that they can achieve.
Triple negative breast cancer (TNBC) is an aggressive form of breast cancer whose growth is not driven by estrogen, progesterone, or by the overexpression of HER2 (human proteins epidermal growth factor receptor). While anyone can be diagnosed with triple negative breast cancer, this subtype of breast cancer has been found to be higher in young people, Black and Hispanic women, and those with a BRCA1 mutation (Triple-Negative Breast Cancer. Breastcancer.org. Accessed June 28, 2021. https://www.breastcancer.org/symptoms/types/triple-negative).Individuals in Canada, and in general, who are diagnosed with TNBC have a poor prognosis and poor survival outcomes. According to the American Cancer Society, the 5-year survival rate is 65% for regional mTNBC and 12% for distant mTNBC. This is compared to the 5-year survival rate for localized TNBC (Triple-negative Breast Cancer. American Cancer Society. Accessed June 28, 2021. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/types-of-breast-cancer/triple-negative.html).
In our 2017 Survey, the majority of respondents experienced metastases to their bones, liver and lungs.12% of metastatic patients reported metastases to their brain while 20% reported metastases to other body parts. Of the 14 patients who indicated that they are living with mTNBC, the majority of respondents (10) experienced metastases to their lungs. This was followed by metastases to other parts of their bodies (6), their bones (5), their liver (3) and their brain (2).
The Physical Impact of Metastatic Breast Cancer
How the disease presents itself through symptoms, how it progresses, and how it is experienced varies by patient, but many effects of metastatic breast cancer represent a significant or debilitating impact on their quality of life. In our 2012 Lived Experience of Metastatic Breast Cancer Patients and Caregivers Survey Report (2012 Survey), patients were asked what impact cancer-related symptoms had on their quality of life:
54% of patients reported that fatigue resulted in a significant or debilitating impact, and 40% reported some or moderate impact;
39% of patients reported that insomnia resulted in a significant or debilitating impact, and 46% reported some or moderate impact;
37% of patients reported that pain resulted in a significant or debilitating impact, and 44% reported some or moderate impact.
These results were further reinforced in our 2017 Lived Experience Breast Cancer Patient Survey (2017 Survey).
The Social Impact of Metastatic Breast Cancer
The impact of this disease spreads across all aspects of a patient’s life, restricting an individual’s employment and career, ability to care for children and dependents, and their ability to be social and meaningfully participate in their community. When asked in the 2012 Survey what kind of impact living with metastatic breast cancer has had on their quality of life:
Among those who were employed, 71% of patients identified significant restrictions to their ability to work;
Among those with children or dependents, 21% identified significant restrictions and 53% reported some or moderate restrictions to their caregiving responsibilities;
49% of patients identified significant restrictions and 38% identified some or moderate restrictions to their ability to exercise;
42% of patients identified significant restrictions and 42% identified some or moderate restrictions to their ability to pursue hobbies and personal interests;
41% of patients identified significant restrictions and 41% identified some or moderate restrictions to their ability to participate in social events and activities;
22% of patients identified significant restrictions and 52% identified some or moderate restrictions to their ability to spend time with loved ones.
Other experiences identified by patients included: guilt, the feeling of being a burden on caregivers, fear of death, poor body image, not knowing what functionality will be lost, fear of the impact of cancer and the loss of a parent on children, not knowing what will happen to children, the loss of support of loved ones, as well as marital stress/loss of fidelity and affection from husband.
Experiences With Currently Available Treatments
The Goals of Current Therapy
As with all treatment for metastatic breast caner, the goal of treatment for metastatic triple negative breast cancer is to control disease progression (extending life) and to manage cancer-related symptoms (extending or stabilizing quality of life). Treatment options for mBC and their effectiveness vary among type of cancer, location of cancer, and how symptoms are experienced.
Patients diagnosed with mTNBC have very limited treatment options. Targeted therapies that treat HR-positive and HER2-positive breast cancers are usually ineffective in treating TNBC. Because of the lack of effective treatment options for mTNBC, patients with this subtype of metastatic breast cancer face much lower overall survival (OS) rates than patients with other subtypes of metastatic breast cancer.
Currently, treatment for TNBC is very limited and usually involves chemotherapy, surgery and radiation. In the case of mTNBC however, the standard of care is single-agent chemotherapy. Unfortunately, as the disease continues to progress and treatment stops responding, individuals must move to second- and third-line treatments, making their treatment options even more limited as they require newer lines of treatment.
While immunotherapy can be helpful as a first-line treatment, single-agent chemotherapy is the standard treatment beyond first-line therapies, but it is associated with low response rates (<20%) and short median progression-free survival 2-3 months) (Bardia, A., et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 384, 1529-41 (2021). (2016). https://doi.org/10.1056/NEJMoa2028485). Eribulin is usually used for previously treated mTNBC but its PFS is low (>3 months).3 In addition to this, chemotherapy in general has a very high toxicity profile and is often associated with significant adverse events.
All of the14 mTNBC patients had been or were currently being treated with chemotherapy, 11 patients previously had surgery, 12 patients had or were receiving radiation therapy and 2 patients had or were currently receiving hormone therapy.
Key Factors for Decision-Making Around Treatment
Respondents in our 2017 Survey indicated that the following key factors influenced their decision-making around treatments:
Effectiveness of the treatment – how well the treatment stabilized their disease and delayed progression of their disease.
Prolonging life without sacrificing quality of life – being able to maintain productive, active lives with minimal disruption to daily routines.
Side effect management – minimizing risk while stabilizing their disease.
Cost and accessibility of treatments – affordability and ease of accessing treatments.
Treatment Efficacy
When asked how important progression-free survival was in considering treatments, the mTNBC patients in our 2017 Survey revealed that efficacy of the treatment is an important consideration to their decision-making. 69% of the 13 mTNBC who responded to the question indicated that progression-free survival of less than 3 months was important or very important. 86% of the 13 mTNBC who responded to the question indicated that progression-free survival of 3-5 months was important or very important. Of all of the 14 mTNBC in our 2017 Survey, 85% indicated that progression-free survival of 6 months or longer very important. When asked about overall survival, 85% of all mTNBC patients indicated that overall survival was very important when considering treatment options.
Metastatic patients in our 2017 Study also spoke on the importance of treatment effectiveness in their decision-making anecdotally:
“The most important factors for me are progression free survival and quality of life.” – mBC patient respondent
“Quality of life, efficacy of the drug to stabilize my TNBC” – mTNBC patient respondent
“Anything to prolong my survival and maintain quality of life.” – mBC patient respondent
“Survival is of upmost importance to me.” – mBC patient respondent
Quality of Life
Quality of life was routinely cited by patients as a key factor in making treatment decisions. In our 2017 Survey, 93% of the mTNBC patients revealed that quality of life was important or very important to them when considering treatment options. More specifically, 50%, 93% and 57% of mTNBC patients indicated that minimal side effects, mobility, and productivity, respectively, were important or very important considerations when making decisions regarding treatment options.
This concern was reiterated anecdotally:
“Making sure I have some quality of life so I can [spend] as much time with my kids and family I don't want them to watch me suffer” – mTNBC patient respondent
“Trying to balance the most effective treatment regime with the least impact on my day to day living/quality of life. Maintaining a certain level of independence is important to me.” – mTNBC patient respondent
“Definitely the balance of quality of life vs side effects with the [effectiveness].” – mTNBC patient respondent
Patient Willingness to Tolerate Treatment Side Effects
In our 2012 Metastatic Patient and Caregiver Survey, the responses to what level of side effects and how much impact on one’s quality of life would be worth extending progression-free disease by six months was shown to be determined at the personal level.
When asked to rate how much impact different symptoms of cancer and cancer treatment would be considered tolerable:
Almost two-thirds of patients indicated that when it comes to fatigue, nausea, depression, problems with concentration, memory loss, diarrhea and insomnia, some or a moderate impact on one’s quality of life would be considered acceptable, and approximately one quarter of patients indicated that a strong or debilitating impact would be considered acceptable.
70% of patients indicated that when it comes to pain, some or a moderate impact on one’s quality of life would be considered acceptable, and 27% of patients indicated that a strong or debilitating impact would be considered acceptable.
Similar responses were also found in our 2017 Survey. The majority of mTNBC respondents who responded to the question on acceptable side effects (13) indicated that pain, fatigue, nausea, insomnia, lack of concentration, memory loss, diarrhea, and hair loss were somewhat acceptable or very acceptable symptoms in exchange for 6 months or less of benefits from breast cancer treatment. The majority of mTNBC respondents indicated that depression as a symptom in exchange of 6 months or less of benefits from breast cancer treatment was somewhat acceptable (9 respondents). When it came to the symptom of vomiting, 10 mTNBC (the majority) indicated that it would not be acceptable (2 said it would be somewhat acceptable).
The Financial Burden of Treating and Managing Breast Cancer
The financial burden associated with living with advanced breast cancer extends far beyond any loss of income during a temporary or permanent absence from employment. In addition to the loss of income during illness, metastatic breast cancer patients can incur substantial costs associated with treatment and disease management. Research on the financial impact of breast cancer on patients identified the following: (Janet Dunbrack, Breast Cancer: Economic Impact and Labour Force Re-entry. Canadian Breast Cancer Network, 2010).
80% of breast cancer patients report a financial impact due to their illness.
44% of patients have used their savings, and 27% have taken on debt to cover costs.
These findings were consistent with the responses in our 2012 Survey:
Nearly one-third of patients indicated that the cost of medication, the cost of alternative treatments (i.e. massage, physiotherapy, etc.) to manage symptoms and side effects, and the time required to travel to treatment had a significant or debilitating impact on their quality of life.
24% of patients indicated that the costs associated with travel had a significant or debilitating impact on their quality of life, and 41% of patients indicated that it had some or moderate impact on their quality of life.
In our 2017 Survey, mTNBC patients reported that their diagnosis had some (57%) or a very large (43%) impact on their finances. In addition to this, 79% of mTNBC indicated that the time required to travel to treatment had some or a significant impact of their quality of life. 71% reported the same in regard to the cost of other treatments (i.e. massage, physiotherapy, etc.) and 79% reported the same in regard to costs associated with travel.
The financial impacts of a metastatic breast cancer diagnosed was also reiterated anecdotally:
“Many of the next step treatments are very expensive [and not covered by government programs] and it is a HUGE struggle to get [coverage]. […] When dealing with an incurable disease the last thing you want to have to do is spend time on a letter writing campaign to argue about whether or not you should receive the drugs [recommended by your physician]. At about $1500.00 a week, I don't know many who can afford that.” – mBC patient respondent
“Always a concern as you never know if the next drug will be covered or how long it takes to get approval from private coverage. Many times it delays treatment and this weighs on one's mind.” – mTNBC patient respondent
“I wanted to try [immunotherapy], but it is [$]7500.00 every 3 weeks not covered by private insurance, now will probably have to go on chemo again, and the last ones were very hard on me causing toxicity and having to get blood transfusions.” – mTNBC patient respondent
“Just because I am not in the lowest income bracket does not mean I don't need assistance. I am excluded from all programs I have tried to access.” – mTNBC patient respondent
Other financial barriers that metastatic breast cancer patients mentioned include: not qualifying for insurance at work, inability to change employers due to loss of insurance, and the prohibitive cost of new treatment options.
Patient Access to Local Resources and Supports During Treatment
When living with cancer, many patients experience significant barriers and challenges around availability of health care services and quality childcare in their community. In response to the 2012 Survey questions about the availability of supports such as childcare, transportation and alternative treatments in their community:
Among patients with children or other dependents, 53% indicated that there is minimal or no access to appropriate care for their loved ones when they are experiencing debilitating symptoms related to their cancer, and 40% identified barriers to accessing quality care during cancer treatment.
Similar results were found in our 2017 Survey among mTNBC with children at the time of their diagnosis:
7 patients reported that finding appropriate care for their children/dependents when experiencing side effects of cancer treatments was not accessible
7 patients indicated that finding appropriate care for their children/dependents during cancer treatment was not accessible
Among all mTNBC patients from our 2017 Survey, 43% indicated that finding symptom management options in or around their community was not accessible and 36% indicated that it was somewhat accessible.
Patient Willingness to Tolerate Risk
When asked in the 2012 Survey about their willingness to tolerate risk with a new treatment:
34% of respondents were willing to accept serious risk with treatment if it would control the disease
45% of respondents were willing to accept some risk with treatment
21% of respondents were very concerned and felt less comfortable with serious risks with treatment
Need for Personal Choice
The open ended question and the key informant interviews showed is that it is imperative that women with metastatic breast cancer have access to, and the option of what drugs they take. Most patients are well aware of the adverse effects of treatment up front and they want to make a personal choice that works for them. 57% percent of mTNBC patients in our 2017 Survey expressed being very comfortable in treatment decisions. Metastatic breast cancer patients expressed the need for personal choice and autonomy in our 2012 Survey as well as in the 2017 Survey:
“I think patients (ESPECIALLY young patients) should be given more decision making power in terms of access to radical treatments to control disease. […] With two small [children] I am determined to access any treatment that can extend my life and I hate struggling with doctors for this access.” – 2012 Survey
“I believe that I would prefer to tolerate severe restrictions in the quality of my life, if it meant that I would be able to have a longer period without progression.” – 2012 Survey mBC patient respondent
“It would be nice to have more choices and more information about them. I was lucky to get on a clinical trial perhaps because my oncologist was a research oncologist and involved in many. While I knew friend and acquaintances that had Stage IV BC and never informed of clinical trials, and sadly several did not survive the disease.” – 2017 Survey mBC patient respondent
“I am frustrated that ALL the treatment choices aren't given to me... I am told what I am taking next with no option or discussion on other options. My oncologist has assured me there are many treatments available, but have never shared which, so I have to turn to Facebook groups for guidance.” – 2017 Survey mTNBC patient respondent
Improved Outcomes
For metastatic patients, extension of progression-free survival (PFS) is of critical concern. Like any other treatment for metastatic breast cancer, patients have an expectation that sacituzumab govitecan (Trodelvy) will extend their progression-free survival with good quality of life when first- and second-line therapies stop working.
The phase 3 ASCENT trial evaluated and compared sacituzumab govitecan with the treatment of physician’s choice of a single-agent chemotherapy (eribulin, vinorelbine, capecitabine, or gemcitabine) in patients with relapsed or refractory metastatic triple-negative breast cancer.3
For patients without brain metastases, Phase 3 of the ASCENT trial showed a median PFS of 5.6 months for sacituzumab govitecan and a median PFS of 1.7 months for the comparison group. Median overall survival (OS) was 12.1 months with sacituzumab govitecan and 6.7 months with chemotherapy. For the full study population (those with or without brain metastases), median PFS was 4.8 months with sacituzumab govitecan compared to a median PFS of 1.7 months with chemotherapy. Median overall survival (OS) for sacituzumab govitecan was 11.8 months and 6.9 months with chemotherapy.
Around 20% of patients who are diagnosed with TNBC are 65 years and old (Kalinsky, K,. et al. Outcomes in Patients Aged ≥65 Years in the Phase 3 ASCENT Study of Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. J Clin Onc. 39 (2021). https://doi.org/10.1200/JCO.2021.39.15_suppl.1011.) These individuals are also less fit for chemotherapy because of higher comorbidity rates, higher use of medications, and pre-existing frailty or functional loss.5 As a result, older patients are more likely to receive less aggressive treatment for TNBC. To assess safety and efficacy outcomes of patients 65 years and older, a post-hoc subgroup analysis of phase 3 of the ASCENT data looked at patients 65 years and older without known brain metastases at baseline.5 This subgroup analysis found that patients 65 years and older who received sacituzumab govitecan had significant survival and clinical benefits compared to those in the control group who received the treatment of the physicians choice. For those 65 years and older, median PFS was 7.1 months compared to 2.4 months and median OS was 15.3 months compared to 8.2 months.
Adverse Effects
The phase 3 data from the ASCENT trial showed a few adverse effects in patients. Commonly reported side effects of any grade were: neutropenia, diarrhea, nausea, alopecia, fatigue, and anemia. Commonly reported side effects of grade 3 or higher were: neutropenia, leukopenia, diarrhea, anemia, and febrile neutropenia. 39 patients (15%) treated with sacituzumab govitecan reported serious adverse events.
In the subgroup analysis of the ASCENT trial, the safety profile of sacituzumab govitecan in patients 65 years and older was found to be comparable and as manageable as that among patients younger than 65 years old.5 Treatment discontinuation due to adverse events was 2% in both groups and there were no treatment-related deaths for either groups. While dose reductions happened more within patients 65 years and older compared to patients younger than 65, these rates were similar between sacituzumab govitecan and the treatment of physician's choice in the control group. Despite dose reductions, there was no considerable impact on efficacy of sacituzumab govitecan.
Impact of Treatment Options to Patients
By delaying the progression of the disease, this treatment can relieve cancer-related symptoms, and improve a patient’s quality of life. When living with no or with minimal cancer-related symptoms, and with minimal side effects from treatment, patients are able to reduce the impact of cancer on their ability to care for children and dependents, continue with their employment and earn income, spend time with loved ones and participate in their life in a meaningful way by engaging in social activities, travelling, maintaining friendships, and pursuing personal interests.
Value to Patients
The value to patients of extending the time that their cancer is progression-free cannot be overestimated. Patients living with metastatic breast cancer are aware that their advanced disease will progress with worsening symptoms until death, and embrace opportunities to try new treatments, even if benefits may be as little as a six month extension of progression-free disease. It is also very important for patients to have good quality of life when receiving treatment for metastatic disease. Patients that we speak to on a regular basis acknowledge the importance to have the energy to attend their children’s activities and to spend time with family and friends.
Experience With Drug Under Review
Patient Profile:
CBCN connected with the only Canadian patient who has experience with the treatment.
The patient is 37 years old and was diagnosed in November 2019. She was first diagnosed with stage III triple negative breast cancer which is now stage IV, triple negative breast cancer. She is able to access this treatment because of her oncologist. She has been previously been treated with an immunotherapy, AC chemotherapy, taxol, cisplatin, capecitabine, atezolizumab, and abraxane. She also had a double mastectomy and underwent 25 rounds of radiation.
The Impact of the Treatment on the Disease
With having been on a variety of treatments, the patient we interviewed expressed personal satisfaction with the treatment. She expressed that Trodelvy has been both impactful in treating her cancer and has been the treatment with the most manageable side effects.
“It’s the first thing that’s made any difference in my cancer at all since I was on AC chemo. The AC chemo shrunk my cancer, but then when I switched over to Taxol and cisplatin, my tumour grew back, and they continued chemotherapy and I went right into surgery to remove the tumour, because it was no longer responding. And then after the removal, I had a clean PET scan, and then I did radiation. And then while I was on capecitabine, it came back. And atezo and abraxane did nothing. And when I went on to Trodelvy, I had lymph nodes in my neck and chest that had cancer in them. I had a spot on my lung and my liver, and I had bone metastases. And after doing three cycles of Trodelvy, all of my lymph node involvement was gone. All of my bone metastases were gone. My lung spot was gone. The only thing left was my liver spot.”
Not only was Trodelvy helpful in treating her cancer, but after initial treatment with Trodelvy, the pain the patient felt in association to her bone metastases and her difficulty breathing due to the cancer on her lungs subsided.
“I was in an incredible amount of pain from the bone metastases specifically, and I was having trouble breathing from the lung spot when I started Trodelvy. And during the first cycle—I can’t remember if it was the first treatment or the second treatment, but I’m pretty sure it was the first treatment—my bone pain and my breathing difficulties were gone.”
Assessing Risks Associated With the Treatment
The patient that we interviewed shared that the side effects that she experienced from Trodelvy were very minor. The side effects from Trodelvy included hair loss, nausea, and headaches, which she emphasized were very minimal and more than manageable.
“They’re all fine. They’re all acceptable. I think they’re all within a realm of normal, manageable side effects.”
To address the headaches and nausea, she was able to take over-the-counter medications and take them on an as-needed basis. The patient found the side effects of Trodelvy to be much more tolerable than those she experienced while on other treatments.
“I take Tylenol for headaches if I need to. And I have some anti-nauseas that I take if I get nauseous. But it’s really on an as-needed basis, whereas with other therapies, I was taking them daily to make sure the side effects didn’t start, whereas with Trodelvy, the side effects I find are quite minor. So if I feel a little bit nauseous or a little bit headachy, I can take an over-the-counter medication and it’s enough to cut down the side effect, whereas with the other ones I was taking very heavy prescription medications daily.”
Alternatives to the Treatment
The patient was able to access Trodelvy through her oncologist and she acknowledged that most other metastatic triple negative breast cancer patients do not have access to this treatment.
“I feel incredibly lucky. I speak to a lot of other women, especially young women like me, some even younger, that have the same Stage IV triple negative diagnosis as I do, and they were unable to get this treatment.”
She sought out this treatment because everything else she had tried up to this point had not been working and was advised by her oncologist that this would give her the best chance. Prior to her oncologist getting involved, she had already begun seeing an oncologist in the United States to start therapy there due to a lack of alternatives in Canada.
“Everything else I tried wasn’t working. And it was what my oncologist said would give me the best chance. There were other therapies available in the United States, but really there weren’t any other good therapies available to me in Canada.”
Without access to this treatment, the patient we interviewed stated that she would have looked to get therapy in the United States and paid out-of-pocket. While she had this potential alternative, she acknowledged that she is privileged to have the financial means to do so and recognized that many other patients do not have the same means or access as she does.
“There’s actually not many options left available in Canada. I would probably be paying out-of-pocket for something In the United States. And I’m only really lucky enough to be able to do that because I have the financial means to do it, and I live close enough to the United States border that I can drive down for treatment. I talked to another patient who has the same cancer as me and that lives in Edmonton, and she can’t drive down to the States. So she had to fly down and is living there at great expense to her.”
In terms of how Trodelvy compared to other treatments that she had been on, the patient we interviewed found it to be the most preferable option, especially due to the difficult side effects from the alternatives.
“It’s actually one of the most manageable ones. I actually found I got the most life-impacting side effects when I went on capecitabine. And when I was on AC chemo, obviously that’s quite a difficult chemotherapy. There was a lot of nausea and that sort of thing. This one, it’s not so bad. I don’t have to take as many other drugs to manage the side effects.”
“Infusions are every couple of weeks so it’s not hard to do the treatment.”
The Social and Financial Impact of the Treatment
In terms of the social impacts of Trodelvy, the patient that we interviewed discussed that being on this treatment has allowed her to live a well-rounded life and uphold a good quality of life, rating her qualify of life while taking this treatment as an “eight out of ten”.
“I’ve been able to do a lot more while on Trodelvy than I have with other therapies.”
It has also had a positive impact on not just her life, but her family’s as well.
“It has had a hugely positive impact on our family. It’s been a huge relief to everyone in my family to have a treatment that actually works and to be able to have me functional and to be able to be happy while on treatment instead of in [bed] and in pain.”
While her mother lives with her and her husband to assist with childcare responsibilities, she has been able to actively participate in this as compared to other treatments.
“My husband and my mother live with us to help with my son. But I definitely have more ability and more energy to do things than I did with other therapies.”
In terms of the financial impacts of this treatment, the patient that we interviewed expressed that without being able to access this treatment, she would have been paying out-of-pocket and travelling to the United States to get treated. With this treatment being the only one that seems to be really helping her, the patient that we interviewed expressed the need for patients like her to have access to Trodelvy in Canada.
“I think that absolutely it should be acceptable here in Canada and that it should be funded by the government so that people who have Stage IV triple negative breast cancer have a chance at prolonging their lives. There’s a lot of additional therapies available for people who are hormone positive or HER2 positive. But there isn’t really a lot that is available that is effective for people that have triple negative.
Overall, the patient we interviewed expressed that she is really happy to be able to access a treatment that is effective in treating her cancer while allowing her a good quality of life.
“It’s saving my life. It’s saving my life. It’s giving me more time with my son, who’s only three. It’s the only thing that made any difference in my cancer.”
CBCN Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
CBCN did connect with the manufacturer, Gilead, to identify clinicians that could connect us with patients with experience on the treatment.
All other research, interviews and outreach to patients was conducted independently by the Canadian Breast Cancer Network, as was the compilation of information and data for the writing of this submission.
As a member of the Canadian Cancer Action Network, the Canadian Breast Cancer Network is committed to adhering to the Code of Conduct Governing Corporate Funding.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No. The Canadian Breast Cancer Network compiled and wrote this submission independently.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 31: Conflict of Interest Declaration for CBCN
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
Gilead | — | — | X | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Canadian Breast Cancer Network (CBCN)
Date: June 22, 2021
Appendix 2: Clinician Group Input
Note this appendix has not been copy-edited.
Ontario Health (Cancer Care Ontario) Breast Cancer Drug Advisory Committee (DAC)
Author of submission: Dr. Andrea Eisen, Dr. Phillip Blanchette, Dr. Orit Freedman, Annie Ngan (pharmacist)
About OH-CCO Drug Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Please describe how you gathered the information included in the submission.
Response: Discussed jointly at a DAC meeting.
Current Treatment
Describe the current treatment paradigm for the disease
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Current treatment options include single agent chemotherapy such as the ones included in ASCENT – e.g., eribulin, vinorelbine, gemcitabine, capecitabine
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Substantial improvement in overall survival in a difficult-to-treat population, delay disease progression, manageable toxicities
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples:
Not all patients respond to available treatments
Patients become refractory to current treatment options
No treatments are available to reverse the course of disease
No treatments are available to address key outcomes
Treatments are needed that are better tolerated
Treatment are needed to improve compliance
Formulations are needed to improve convenience
Response: This study highlights the current standard treatments are not effective for relapsed or refractory TNBC patients.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: All triple-negative breast cancer patients who are candidates for systemic therapy.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Previously treated population (at least 2 prior lines, and one prior line in the metastatic setting) and will replace current available therapy.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: No. It would not be appropriate – the current standard treatments are ineffective and associated with toxicities.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Depending on patient fitness as this is a late line treatment. If the patients are well enough to receive additional treatment, current standard treatment (e.g., single agent chemotherapy) may be considered.
Subsequent therapies were not reported in the trial.
Survival in TNBC is poor compared to other breast cancer subtypes.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Relapsed/refractory TNBC patients as per ASCENT protocol.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: The measurement of biomarkers is standard in breast cancer diagnosis.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients who are not well enough to receive systemic therapy.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: No predictive biomarkers to identify which patients may respond more.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Standard radiographic imaging and staging to assess response.
What would be considered a clinically meaningful response to treatment?
Examples:
Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth)
Attainment of major motor milestones
Ability to perform activities of daily living
Improvement in symptoms
Stabilization (no deterioration) of symptoms
Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: The large improvement in OS, PFS, and response rate seen with sacituzumab govitecan are clinically meaningful and previously unseen in this clinical setting. There also appears to be a delay in brain metastases.
How often should treatment response be assessed?
Response: As per current clinical standard – every 12 weeks for clinical assessment and staging
What factors should be considered when deciding to discontinue treatment?
Examples:
Disease progression (specify; e.g., loss of lower limb mobility)
Certain adverse events occur (specify type, frequency, and severity)
Additional treatment becomes necessary (specify)
Response: Disease progression or toxicities
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Outpatient treatment in cancer clinics
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: NA
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: This is a substantial result in advanced TNBC, a population with a huge unmet need.
Conflict of Interest Declarations for OH-CCO Breast Cancer DAC Clinicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Andrea Eisen
Position: Ontario Cancer Lead; Medical oncologist
Date: 23 June 2021
Table 32: Conflict of Interest Declaration for OH-CCO Breast Cancer DAC Clinician 1
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Phillip Blanchette
Position: Medical oncologist
Date: 23 June 2021
Table 33: Conflict of Interest Declaration for OH-CCO Breast Cancer DAC Clinician 2
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Orit Freedman
Position: Medical oncologist
Date: 23 June 2021
Table 34: Conflict of Interest Declaration for OH-CCO Breast Cancer DAC Clinician 3
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 4
Name: Annie Ngan
Position: pharmacist
Date: 23 June 2021
Table 35: Conflict of Interest Declaration for OH-CCO Breast Cancer DAC Clinician 4
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |
None declared | — | — | — | — |
The Ottawa Hospital Cancer Centre: Breast Medical Oncology Group
Author of the submission: Dr. Terry L. Ng
About the Ottawa Hospital Cancer Centre: Breast Medical Oncology Group
Please describe the purpose of your organization. Include a link to your website (if applicable).
We are members of the group of medical oncologists at the Ottawa Hospital Cancer Centre, affiliated with University of Ottawa, treating breast cancer. We are in an academic teaching hospital centre, and we are all involved in the care of breast cancer patients. We offer routine standard of care treatments and access to promising treatments in the context of phase 1 to 3 clinical trials and serve a large referral base from the Champlain LHIN in Ontario.
Information Gathering
Please describe how you gathered the information included in the submission.
Our members were canvassed electronically and in person for input and opinion, using this CADTH template, and the recommendations were condensed and coalesced into summary statements reflecting the breadth of opinions expressed. Our opinions were based on literature review, data from recent international congresses and publications.
Current Treatments
Describe the current treatment paradigm for the disease
Triple negative breast cancer (TNBC) is an aggressive disease characterized by heterogeneous molecular and immunological characteristics that portends worse overall survival compared to other breast cancer subtypes. Despite decades of research, chemotherapy has remained the mainstay of treatment in this population. Standard chemotherapy options include platinum agents, anthracyclines, taxanes and capecitabine but optimal sequencing is not well defined and meaningful benefits beyond 1st or 2nd line treatments have not been shown. For unresectable, locally-advanced and metastatic TNBC (mTNBC), recent data on immunotherapy using atezolizumab with nab-paclitaxel, and pembrolizumab with chemotherapy have demonstrated potential survival benefit in certain subgroups (PD-L1 ≥ 1% for atezolizumab combination and CPS ≥ 10% for pembrolizumab combination, progression-free survival (PFS) benefit only, overall survival (OS) not yet mature), but they are not approved for use in Canada. Median progression free survival on each line of chemotherapy is only 4-6 months, with diminishing return from each subsequent line of therapy. The median OS from time of diagnosis of mTNBC is only 14 months. There is a clear unmet need for this population. Current treatments improve symptoms, induce responses and improve survival modestly.
For selected patients with underlying germline mutations in brca1 or brca2, olaparib has demonstrated improved outcomes compared with standard chemotherapy options, however routine testing for brca germline mutations are not widely available across much of Canada, and testing for somatic tumoral mutations is not routinely available either. Despite demonstrated benefits, olaparib is not yet publicly funded.
Treatment Goals
What are the most important goals that an ideal treatment would address?
The most important goals of treatment would include: improved overall survival; maintained or improved quality of life compared with currently available treatments; delay of progression of cancer, improvement or maintenance of organ function (eg. liver, bone, lungs); and reduction of cancer symptoms.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Overall response and duration of response to currently available therapies after anthracyclines and taxanes are disappointing. Patients are quick to develop treatment resistance. There is greater propensity for TNBC patients to develop visceral crises in heavily pretreated patients. While eribulin has demonstrated slight OS improvement in mTNBC, the improvements were modest and long term survival remains quite poor. There is no defined optimal standard of care after past exposure to anthracycline and taxane chemotherapies.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: There is a great need for more effective treatments, especially for those that have become resistant to first and second line systemic therapy (e.g, taxanes and anthracyclines). The response rate after failure of two prior lines of systemic therapy for mTNBC is 5%. In brca mutated patients, platinum chemo drugs or olaparib might replace one of those first two lines of therapy but, again, olaparib is not publicly funded and the need beyond 2nd line remains.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Any patient who had at least two prior lines of systemic therapy for mTNBC, with one of the treatments being a taxane (neo- / adjuvant or palliative setting) with reasonable performance status (ECOG 0-2) and good organ function would be candidates for Sacituzumab govitecan-hzil. This treatment is already approved by the FDA in the US and would greatly benefit Canadian patients that fit these criteria.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
See response to 6.1. Patients need to have relapsed or progressed after two or more prior lines of mTNBC with at least one line of systemic therapy being a taxane, though those could have been in the early disease setting (neoadjuvant or adjuvant).
How would this drug affect the sequencing of therapies for the target condition?
Sacituzumab-govitecan (Trodelvy™) would be used in the second or later line setting for mTNBC. It would be used in lieu of other second or later line systemic therapies considered current standards in this population. Other standard systemic options (i.e., other chemotherapies) could then be considered after failure of Sacituzumab-govitecan (Trodelvy™).
Which patients would be best suited for treatment with the drug under review?
Patients meeting the eligibility criteria for the ASCENT trial. Patients would be eligible with or without visceral metastases, and in real world practice should be of ECOG PS 0-2 with expected survival of > 3 months. Patients with brain metastases should have stable brain lesions for ≥ 4 weeks.
How would patients best suited for treatment with the drug under review be identified?
Patients suitable for treatment with Sacituzumab-govitecan (Trodelvy™) would be identified by the primary treating physician based on diagnosis of mTNBC, clinical examination (performance status), and physician judgement about suitability of patient, and the confirmation of clinical and/or radiographic disease progression after the above defined preceding lines of therapy.
Which patients would be least suitable for treatment with the drug under review?
ECOG 3-4, patients who have not yet tried taxane treatment, those with active, symptomatic untreated brain metastases, and patients unable to understand the dosing and monitoring requirements and potential toxicities, or those non-compliant with required follow up.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
There are currently no clinical or molecular subgroups that can help us predict those most likely to respond to treatment (aside from having triple negative disease by standard definitions: ER negative, PR negative and Her2 negative).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Responses are determined based on symptoms, laboratory markers, and radiographic scans and tumour measurements, with scans usually performed at least every 3 months initially. Treatment is continued if disease is either stable or responding by RECIST criteria radiographically.
What would be considered a clinically meaningful response to treatment?
Reduction in the frequency or severity of symptoms (eg pain, dyspnea…)
Improvement of organ function (bone, liver, lung)
Stabilization of symptoms
Maintenance or improvement of performance status
Tumour radiographic response with either stabilization of disease or response by RECIST criteria
How often should treatment response be assessed?
At least every 3 months, with toxicity or symptom assessments more often early in treatment (every 2-4 weeks) or as needed
What factors should be considered when deciding to discontinue treatment?
Disease progression
Intolerable or dangerous toxicity, esp uncontrolled grade 3-4 rash or diarrhea
Patient preference or refusal
What settings are appropriate for treatment with the drug under review?
Hospital setting or specialty clinic that has expertise and staffing (chemotherapy nurses, oncology pharmacists) to administer chemotherapy and monitor / manage treatment-related toxicities
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Treatment should only be prescribed by certified medical oncologists or associated team physicians with expertise in cancer therapies and toxicity management.
Additional Information
Is there any additional information you feel is pertinent to this review?
The benefits seen in the ASCENT study are meaningful and valuable. There has not been a therapy demonstrating this magnitude of survival benefit in this heavily pre-treated, treatment-resistant population. The documented benefits are commensurate with patient values and the toxicities are predictable and manageable by medical oncologists.
Conflict of Interest Declarations for The Ottawa Hospital Cancer Centre: Breast Medical Oncology Group Clinicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No assistance was received in the completion of this report.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No assistance was received in collection or analysis of data to support this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Clinician Information
Name: Dr. Terry L. Ng
Position: Medical Oncologist, The Ottawa Hospital Cancer Centre
Date: 15-06-2021
Table 36: Conflict of Interest Declaration for The Ottawa Hospital Cancer Centre Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No financial conflicts to disclose from last 2 years | — | — | — | — |
Declaration for Clinician 2
Clinician Information
Name: Dr. Sandeep Sehdev
Position: Medical Oncologist, The Ottawa Hospital Cancer Centre
Date: 01-JULY-2021
Table 37: Conflict of Interest Declaration for The Ottawa Hospital Cancer Centre Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Gilead | X | — | — | — |
AstraZeneca | X | — | — | — |
Declaration for Clinician 3
Clinician Information
Name: Dr. Amirrtha Srikanthan
Position: Medical Oncologist, The Ottawa Hospital Cancer Centre
Date: July 2 2021
Table 38: Conflict of Interest Declaration for The Ottawa Hospital Cancer Centre Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None | — | — | — | — |
Declaration for Clinician 4
Clinician Information
Name: Dr. Moira Rushton-Marovac
Position: Medical Oncologist, The Ottawa Hospital Cancer Centre
Date: July 5 2021
Table 39: Conflict of Interest Declaration for The Ottawa Hospital Cancer Centre Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None | — | — | — | — |
Rethink Breast Cancer Scientific Advisory Committee
Author of the submission: KA Gelmon (lead author)
About Rethink Breast Cancer Scientific Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
This submission is from a group of six medical oncologists from across Canada. These clinicians voluntarily wished to advocate for metastatic triple negative breast cancer (TNBC) patients as this group of patients has significantly more limited lines of therapy in the metastatic setting than other subtypes of breast cancer and of all subtypes of metastatic breast cancer have the poorest survival. New and improved treatments for this population of metastatic breast cancer patients truly are an unmet need. The oncologists who have contributed have extensive clinical experience treating persons with breast cancer, including those with advanced triple negative cancer and also have experience with new drug development and approval.
Information Gathering
Please describe how you gathered the information included in the submission.
The information presented here is from the literature and has been collected by the authors. The content of the submission has been circulated to all the persons signing and they have contributed. The material presented has been approved by all of the authors. As well, in addition to the literature the experience of the authors has been included. The number of persons treated with Sacituzumab govitecan in Canada has been limited as participation in the pivotal clinical trial was very restricted but one of the authors has had direct experience with a patient treated with compassionate access and others have had patients treated in the US with the drug. All of the authors have read extensively on the drug and have heard in presentations about the agent in addition to dialogue with the overall study PI of the pivotal ASCENT trial.
Current Treatments
Describe the current treatment paradigm for the disease
Over 4000 persons will be diagnosed with breast cancer this year in Canada and the majority will be cured of their disease. However up to 15% of them will have a subtype known as triple negative breast cancer (TNBC) that is a more aggressive form, often occurring in young persons, and has a high risk of recurrence or presentation as Stage IV at diagnosis. Advanced TNBC has a median survival of only 12-14 months as compared to other types which now have prolonged survival for Stage IV disease. As well, when this disease recurs it commonly affects bones, lungs, brain and liver with significant symptoms. The diagnosis of advanced TNBC is devastating for the patient, the family, and the community.
Despite advances in the understanding of the heterogeneity of TNBC and the development of new treatments, survival remains very limited with most new treatments reporting limited or minimal impact on survival. Patients die quite rapidly from their cancer with this aggressive subtype. With the high incidence of CNS metastases this is often a very debilitating and symptomatic death. Moreover, this is often in young persons.
The standard therapies for recurrent or advanced TNBC remain chemotherapy with taxanes, platinum agents, capecitabine, gemcitabine, anthracyclines, eribulin, and occasionally vinorelbine. These treatments are given sequentially usually with diminishing responses with each line of therapy. Although initial lines of therapy may provide a few months of progression free survival this decreases substantially with later lines.
Immunotherapy which has shown great promise in other tumour types has modest and conflicting results in TNBC. After reports of an initial possible overall survival benefit for atezolizumab and abraxane in a PDL1 positive group, the sister study of paclitaxel and atezolizumab did not show benefit (in either PFS or OS) and neither treatment is available in Canada for persons with TNBC. Another immunotherapy, pembrolizumab has shown benefit in TNBC with PDL-1 positivity as defined as a CPS score of >10% in combination with either paclitaxel, abraxane, or gemcitabine/carboplatin but this is not yet available in Canada and although a progression free survival has been reported we do not yet have evidence of overall survival improvement.
A minority of persons have germline mutations of BRCA 1 or BRCA 2 or occasionally PALB2. The overall risk of a TNBC having a germline BRCA mutation is about 15% although this is higher in persons younger than 40 at diagnosis. These mutations have shown responses to PARP inhibitors in Phase I, II and III studies but no overall survival benefit. As well, these drugs are not publicly funded in Canada for advanced germline mutated breast cancer despite the Phase III data.
Thus, there is an unmet need for new agents with durable responses and moderate toxicity for advanced TNBC. The ASCENT trial compared Sacituzumab govitecan to chemotherapy (physician’s choice of eribulin, capecitabine, vinorelbine or gemcitabine as these are the commonly used agents for later lines of treatment) and showed a consistent benefit for the experimental arm with an improvement in progression free survival from 1.7 months for the chemotherapy of physicians choice arm to 5.6 months for the Sacituzumab govitecan arm, a doubling of overall survival from 6.7 months to 12.1 months and a response rate of 5% for the chemotherapy compared to 35% for the Sacituzumab govitecan. This was in heavily pretreated persons with a median of 3 prior treatments and prior exposure to taxanes. This data is the best data we have seen for this group of later stage TNBC and is extremely encouraging for patients and for the oncology community.
Treatment Goals
What are the most important goals that an ideal treatment would address?
The ideal treatment for triple negative breast cancer (TNBC) would be more effective treatments for early breast cancer to decrease the number of persons with advanced disease. For those with advanced cancer ‘cure’ would be the ultimate goal, but due to dissemination of cancer cells throughout the body in the setting of metastatic TNBC, this is not truly possible. In the metastatic setting, ideal treatment would provide long and durable overall survival benefit with minimal toxicity and modest impact on quality of life. As well, with TNBC at this time, a treatment that can be used in a large number of TNBC rather than a limited subset would be a goal of an ideal treatment. Delaying death and maintaining good quality of life are the current goals of our treatment of advanced TNBC. Patients consistently ask for stabilization of disease, suggesting a decrease in symptoms, no new cancer events and a durable treatment that can be maintained over a long period of time.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Current therapies for TNBC have shown only a minimal or no improvement in survival. Eribulin which was compared to other chemotherapies did have a modest improvement in overall survival in a population of pretreated patients. When 2 studies were pooled for an unplanned subset analysis, a 5-month difference was seen in a heterogenous population of pretreated TNBC. There are concerns however about this unplanned analysis. Older chemotherapies such as taxanes, capecitabine, anthracyclines, platinum agents, gemcitabine and vinorelbine were all developed prior to our clear understanding of TNBC and all have some efficacy but this is often weeks or a very few months of progression free survival, no major impact on overall survival and all have significant toxicities. Other agents such as pembrolizumab and chemotherapy or atezolizumab and abraxane are not approved or publicly funded, and the benefit of these agents in this setting has been inconsistent in trials. PARP inhibitors are limited to those with BRCA germline mutations which is a limited number of patients and have not shown an overall survival benefit for the intent to treat populations in their studies and are not funded in Canada for the treatment of advanced TNBC. Thus, there truly is an unmet need for patients with advanced TNBC in later lines of therapy. The results with Sacituzumab govitecan are therefore very important to meet this unmet need regardless of BRCA status or expression of PDL-1. The outcomes reported are part of the primary and secondary analysis of the phase III study and are statistically strong.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population.
This drug has been tested in later lines of therapy and there are not standard drugs for this group that have been shown to have efficacy. Many persons with advanced TNBC are young and relapse early after their initial treatment which often includes anthracyclines, cyclophosphamide, paclitaxel, carboplatin and capecitabine. With an early relapse the cancer is likely resistant to all of these agents and yet as the persons are often young, they are often quite well physically. Although immunotherapy with chemotherapy (although not yet approved) or eribulin may be prescribed, the options for treatment are very limited. Later lines of therapy are needed and Sacituzumab govitecan has been shown to have clinical efficacy in this group. Thus, this is a limited population without good therapeutic options at this time.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The criteria for treatment would be for any person with advanced TNBC who had received at least 2 prior therapies for systemic treatment for advanced TNBC with one of these treatments being a taxane which would have been given in the neo/adjuvant or advanced setting. As well, the person would have to be reasonably well, with good organ function and a good performance status (ECOG 0 -2). Thus, this drug would be for later lines of therapy (generally 2nd line or beyond).
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
The current data for this drug are for later lines with at least two prior lines of therapy for advanced TNBC and exposure to prior taxane at some point in their treatment. The rationale for this is the current published data on Sacituzumab govitecan and its efficacy as seen in the ASCENT trial. This is also consistent with the FDA approval for the drug.
How would this drug affect the sequencing of therapies for the target condition?
At this time, the drug would be used after 2 or more prior therapies and therefore would likely be given before drugs such as vinorelbine, gemcitabine or the reuse of agents such as Adriamycin. Patients would be eligible for those therapies after this drug, if they were progressing and well enough to take further lines of therapy. Some patients at academic centres at this stage of their treatment are offered experimental therapy on a clinical trial and these options would still be available or would be delayed until after Sacituzumab govitecan.
Which patients would be best suited for treatment with the drug under review?
The patients best suited for the treatment would be those that are similar to the subjects on the ASCENT trial as this is the population that benefited from this treatment. Eligible patients for the ASCENT trial had advanced TNBC, a good ECOG performance status, adequate organ function, were over 18 years of age, had no brain metastases or brain metastases that were stable for 4 or more weeks and a life expectancy of at least 3 months. Triple negative was defined as <1% expression for estrogen receptor and progesterone receptor, and negative for human epidermal growth factor receptor 2 by IHC or in-situ hybridization. All patients have prior treatment with a taxane. No limit to the number of prior treatments was stipulated.
In the Canadian context, the best suited patients would generally mirror these eligibility criteria as adult persons with prior exposure to taxanes, TNBC, adequate organ function, a good Performance status, and a life expectancy of > 3 months.
How would patients best suited for treatment with the drug under review be identified?
Persons with advanced breast cancer are treated in Canada by medical oncologists who are aware of guidelines and treatment policies as defined by regulatory approval and funding criteria. These persons would be seen by their treating health care provider and assessed for their suitability for treatment. If they progressed on prior therapies and fit eligibility they would be identified. Persons with advanced cancer are usually getting close follow-up and especially when they are on later lines of therapy.
Which patients would be least suitable for treatment with the drug under review?
The persons least suitable would be those with subtypes other than TNBC, with poor performance status, a short survival or uncontrolled brain metastases. As well, persons with earlier disease who had not yet had exposure to taxanes would not be suitable. If persons had poor organ function or were not compliant with follow-up they may be less suitable as there would be concerns about toxicity in a non-curative setting.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
One of the positive features of this treatment is that there is no marker other than TNBC that is associated with a response to treatment. That is different compared to a number of other new oncology treatments that may affect a small subset of the population. This treatment seems to be effective in TNBC as a whole which may reflect its mechanism of action. Although the marker for TROP-2 has been found in a large number of TNBC there is no evidence at this time that there is the need to measure it or any other factor other than TNBC (which is standardly assessed) to determine who may benefit.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
The most important outcome for a patient is the improvement of symptoms as this is meaningful to their quality of life. A decrease in pain, dyspnea, nausea or other symptoms related to advanced cancer is important. Stabilization of disease often suggests response; as well as an improvement in symptoms no growth in the cancer and a prolonged time without new events occurring suggests a response. Other outcomes are imaging to determine if the cancer is shrinking and to assess any new disease growth. Imaging is commonly done with CT scans, PET/CT scans, MRIs and ultrasounds. Blood work may also have utility in looking at improvement of liver enzymes or in some cases tumour markers.
What would be considered a clinically meaningful response to treatment?
The most clinically meaningful response would be an improvement in symptoms and the general well-being of the patient as most persons have symptoms related to the site of their metastases. Other meaningful responses are a shrinkage of the sites of tumour as assessed on imaging or sometimes physical examination and a lack of new lesions appearing. Tumours shrinking are often also associated with an improvement in symptoms. When there is efficacy of a treatment this universally improves patients’ mental health and quality of life. A prolonged duration of response is very meaningful to the patient especially when the treatment is well tolerated as stable or non-progressing disease is clinically meaningful. Finally, a lack of toxicity or well controlled side effects associated with a treatment that is keeping the disease stable is clinically meaningful. This all translates into longer good quality of life in the setting of advanced TNBC. Finally the ability to determine improvements in overall survival from real world evidence post implementation of public funding for this regimen will be a population based clinically meaningful response.
How often should treatment response be assessed?
Most persons with advanced cancer are seen every cycle which would be 3 weekly for this treatment and at that time they are assessed for toxicity, a history and a physical examination. There may be an ability to assess response through these maneuvers. In usual practice however, the best way to assess response is with imaging with CT, MRI, PET/CT or ultrasound. Most treatments are initially assessed for response with imaging after 3 cycles but this may range from 1 -4 cycles. This is usually followed by assessments every 3 – 4 cycles for persons with a response. If there are new symptoms scans to assess response may be required sooner.
What factors should be considered when deciding to discontinue treatment?
The most common reason to discontinue treatment in the advanced setting is progression of the disease, that is the cancer growing suggesting that the treatment is not working. This is usually assessed after 2 to 4 cycles of the treatment (a cycle being 3 weeks for Sacituzumab govitecan). Other factors would be severe toxicity that is not controlled, other health reasons or patient choice. Occasionally there may be a decision that there are other treatments that may be more optimal but in this situation with very few options this would be rare without progression of disease.
What settings are appropriate for treatment with the drug under review?
This treatment would be given in chemotherapy units within a cancer centre, hospital or specialty clinic similar to all other intravenous chemotherapy. As well as oncologists, these settings have special oncology trained physicians, nurses, and pharmacists. Many also have residents and fellows with expertise in oncology.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
This is an oncology drug. This treatment would be prescribed by medical oncologists or their delegates within the framework of the guidelines which would include prescribing details including dosing, toxicity treatment and monitoring.
Additional Information
Is there any additional information you feel is pertinent to this review?
Sacitizumab govitecan is a new agent that is exciting as it appears to have efficacy in a broad group of triple negative metastatic cancers. Despite being tested in later lines of therapy and the median number of treatments for persons in the trial was 3 with a range of 1 – 16 for the entire group significant efficacy was seen. To repeat, the progression free survival was 5.6 months compared to 1.7 for the standard chemotherapy, the overall survival almost doubled to 12. 1 months compared to 6.7 months and the response rate was 35% compared to 5%. There have not been as significant results for any other drug or intervention in this aggressive form of breast cancer in this heavily pretreated population. TNBC is a devastating disease for the person, their family, their friends and for society. Although we need still better treatments to avoid recurrences, this drug does have significant benefit for persons with advanced TNBC without undo toxicity. Canadian persons with aggressive recurrent TNBC would benefit from access to this drug as some may have significant benefit and be able to enjoy good quality lives for longer than if they were unable to be treated. We have not seen this impressive activity with any of the more targeted or untargeted treatments for TNBC in the advanced setting.
Conflict of Interest Declarations for Rethink Breast Cancer Scientific Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside funding or help was received to write this submission.
Rethink Breast Cancer provided some administrative project management report and is uploading the submission on behalf of our group.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside funding or help was received to write this submission.
Rethink Breast Cancer provided some administrative project management report and is uploading the submission on behalf of our group.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Clinician Information
Name: Karen A Gelmon
Position: Professor of Medicine, University of British Columbia, Chair UBC/BC Cancer Research Ethics Board, Medical Oncologist
Date: 12/07/2021
Table 40: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AstraZeneca | — | X | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Eli Lilly | X | — | — | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Seattle Genetics | X | — | — | — |
Ayala | X | — | — | — |
Gilead | X | — | — | — |
Genomic Health | X | — | — | — |
Nanostring | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 2
Clinician Information
Name: Christine Simmons
Position: Medical Oncologist
Date: 2021/07/14
Table 41: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Pfizer | X | — | — | — |
Merck | X | — | — | — |
Bayer | X | — | — | — |
Novartis | X | — | — | — |
Gilead | X | — | — | — |
Lilly | X | — | — | — |
Declaration for Clinician 3
Clinician Information
Name: Dr. Stephen K.L. Chia, MD, FRCPC
Position : Professor of Medicine, Medical Oncology, British Columbia Cancer Agency; Co-chair CCTG Breast Disease Site, Vancouver (BC)
Date: 22-07-2021
Table 42: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Gilead Sciences | X | — | — | — |
Novartis | — | X | — | — |
Pfizer | X | — | — | — |
Merck | X | — | — | — |
Hoffmann LaRoche | — | X | — | — |
Eli Lilly | X | — | — | — |
AstraZeneca | X | — | — | — |
Declaration for Clinician 4
Clinician Information
Name: Dr. Wendie Den Brok
Position: Medical Oncologist, BC Cancer
Date: 22-07-2021
Table 43: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | X | — | — | — |
Helsinn | X | — | — | — |
Declaration for Clinician 5
Clinician Information
Name: Dr. Dan Le, MD, MHA, FRCPC
Position: Medical Oncologist, BC Cancer – Surrey; Clinical Assistant Professor, Faculty of Medicine, University of British Columbia
Date: 22-07-2021
Table 44: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Eisai | X | — | — | — |
Declaration for Clinician 6
Clinician Information
Name: Dr. Christine Brezden-Masley, MD PhD FRCPC
Position: Medical Oncologist, Mount Sinai Hospital, Associate Professor, University of Toronto; Medical Director, Cancer Program at Sinai Health; Director, Marvelle Koffler Breast Centre
Date: 12-07-2021
Table 45: Conflict of Interest Declaration for Rethink Breast Cancer Scientific Advisory Committee Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Gilead Sciences | X | — | — | — |
Appendix 3: Literature Search Strategy
Note this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946–present)
Embase (1974–present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 28, 2021
Alerts: Biweekly search updates until project completion
Search filters applied: None
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase); |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
# Searches
(sacituzumab govitecan* or trodelvy* or hRS7-SN38 or hRS7-SN-38 or hRS7SN38 or hRS7SN-38 or hRS 7SN38 or IMMU-132 or IMMU132 or EX-A4354 or EXA4354 or M9BYU8XDQ6).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*sacituzumab govitecan/
*govitecan/
*sacituzumab/
(sacituzumab govitecan* or saci-tuzumab govitecan* or trodelvy* or hRS7-SN38 or hRS7-SN-38 or hRS7SN38 or hRS7SN-38 or hRS 7SN38 or IMMU-132 or IMMU132 or EX-A4354 or EXA4354).ti,ab,kw,dq.
or/3-6
7 use oemezd
8 not (conference abstract or conference review).pt.
2 or 9
remove duplicates from 10
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- "sacituzumab govitecan" OR trodelvy OR "hRS7-SN38" OR "hRS7-SN-38" OR hRS7SN38 OR "hRS7SN-38" OR "hRS 7SN38" OR "IMMU-132" OR IMMU132 OR "EX-A4354" OR EXA4354 | breast OR TNBC]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- "sacituzumab govitecan" OR trodelvy OR "hRS7-SN38" OR "hRS7-SN-38" OR hRS7SN38 OR "hRS7SN-38" OR "hRS 7SN38" OR "IMMU-132" OR IMMU132 OR "EX-A4354" OR EXA4354 | breast OR TNBC]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- "sacituzumab govitecan" OR trodelvy OR "hRS7-SN38" OR "hRS7-SN-38" OR hRS7SN38 OR "hRS7SN-38" OR "hRS 7SN38" OR "IMMU-132" OR IMMU132 OR "EX-A4354" OR EXA4354 | breast OR TNBC]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- "sacituzumab govitecan" OR trodelvy OR "hRS7-SN38" OR "hRS7-SN-38" OR hRS7SN38 OR "hRS7SN-38" OR "hRS 7SN38" OR "IMMU-132" OR IMMU132 OR "EX-A4354" OR EXA4354 | breast OR TNBC]
Grey Literature
Search dates: July 12, 2021 to July 16, 2021
Keywords: "sacituzumab govitecan" OR trodelvy OR "hRS7-SN38" OR "hRS7-SN-38" OR hRS7SN38 OR "hRS7SN-38" OR "hRS 7SN38" OR "IMMU-132" OR IMMU132 OR "EX-A4354" OR EXA4354
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 4: Excluded Studies
Note this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Bardia et al., 202136 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Kalinsky et al., 202037 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Bardia et al., 201935 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Bardia et al., 201738 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Ocean et al., 201739 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Starodub et al., 201540 | Study design (non-randomized phase I/II IMMU-132-01 basket trial) |
Appendix 5: Detailed Outcome Data
Note this appendix has not been copy-edited.
OS Subgroup Analysis by Primary Brain Metastasis
Table 48: Subgroup Analysis of OS for BM-Pos Versus BM-Neg in the ASCENT Trial — ITT Population
Outcome measure | BM-Pos | BM-Neg | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 32) | TPC (N = 29) | Treatment comparison | Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | |
Patients with events, n (%) | 24 (75.0) | 21 (72.4) | — | 155 (66.0) | 185 (79.4) | — |
Patients without events (censored), n (%) | 8 (25.0) | 8 (27.6) | — | 80 (34.0) | 48 (20.6) | — |
Median (95% CI) OS, monthsa | 6.8 (4.7 to 14.1) | 7.5 (4.7 to 11.1) | — | 12.1 (10.7 to 14.0) | 6.7 (5.8 to 7.7) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.947 (0.523 to 1.716) | — | 0.478 (0.385 to 0.593) | ||
P value for HR = 1 | — | 0.8576 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; BM-Pos = brain metastasis–positive; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
OS Subgroup Analysis by Prior Therapies
Table 49: Subgroup Analysis of OS for Patients With 2 to 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 166) | TPC (N = 164) | Treatment comparison | Sacituzumab govitecan (N = 184) | TPC (N = 181) | Treatment comparison | |
Patients with events, n (%) | 105 (63.3) | 134 (81.7) | — | 117 (63.6) | 146 (80.7) | — |
Patients without events (censored), n (%) | 61 (36.7) | 30 (18.3) | — | 67 (36.4) | 35 (19.3) | — |
Median (95% CI) OS, monthsa | 12.2 (10.6 to 14.5) | 6.7 (5.4 to 7.7) | — | 12.1 (10.5 to 14.4) | 6.8 (5.6 to 7.5) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.435 (0.336 to 0.564) | — | 0.442 (0.346 to 0.566) | ||
P value for HR = 1 | — | <0.0001 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
Table 50: Subgroup Analysis of OS for Patients With More Than 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 69) | TPC (N = 69) | Treatment comparison | Sacituzumab govitecan (N = 83) | TPC (N = 81) | Treatment comparison | |
Patients with events, n (%) | 50 (72.5) | 51 (73.9) | — | 62 (74.7) | 60 (74.1) | — |
Patients without events (censored), n (%) | 19 (27.5) | 18 (26.1) | — | 21 (25.3) | 21 (25.9) | — |
Median (95% CI) OS, monthsa | 12.1 (7.8 to 14.3) | 7.1 (4.6 to 9.1) | — | 10.5 (7.1 to 13.8) | 7.6 (5.2 to 9.2) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.600 (0.404 to 0.891) | — | 0.716 (0.501 to 1.022) | ||
P value for HR = 1 | — | 0.0113 | — | 0.0658 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
OS Subgroup Analysis by BRCA1/BRCA2 Mutational Status
Table 51: Subgroup Analysis of OS for Patients Who Were BRCA1- or BRCA2-Positive in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 16) | TPC (N = 18) | Treatment comparison | Sacituzumab govitecan (N = 20) | TPC (N = 23) | Treatment comparison | |
Patients with events, n (%) | 9 (56.3) | 14 (77.8) | — | 11 (55.0) | 17 (73.9) | — |
Patients without events (censored), n (%) | 7 (43.8) | 4 (22.2) | — | 9 (45.0) | 6 (26.1) | — |
Median (95% CI) OS, monthsa | 15.6 (6.2, -) | 4.4 (3.6 to 9.7) | — | 15.6 (7.1, -) | 4.4 (2.4 to 9.7) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.379 (0.156 to 0.921) | — | 0.411 (0.186 to 0.907) | ||
P value for HR = 1 | — | 0.0321 | — | 0.0278 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
Table 52: Subgroup Analysis of OS for Patients Who Were BRCA1- or BRCA2-Negative in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 133) | TPC (N = 125) | Treatment comparison | Sacituzumab govitecan (N = 150) | TPC (N = 146) | Treatment comparison | |
Patients with events, n (%) | 95 (71.4) | 99 (79.2) | — | 109 (72.7) | 115 (78.8) | — |
Patients without events (censored), n (%) | 38 (28.6) | 26 (20.8) | — | 41 (27.3) | 31 (21.2) | — |
Median (95% CI) OS, monthsa | 10.9 (9.6 to 13.4) | 7.0 (5.6 to 8.2) | — | 10.5 (9.2 to 12.2) | 7.1 (5.9 to 8.2) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.519 (0.390 to 0.690) | — | 0.595 (0.457 to 0.775) | ||
P value for HR = 1 | — | <0.0001 | — | 0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; OS = overall survival; TPC = treatment of physician’s choice.
aMedian OS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
EORTC QLQ-C30 Functional and Symptom Scales
Table 53: EORTC QLQ-C30 Physical Functioning Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 73.2 (21.69) | — | — | 217 | 71.2 (21.24) | — | — | 465 | 72.3 (21.48) | — | — |
Cycle 2b | 225 | 73.4 (21.24) | 219 | -1.0 (15.50) | 163 | 69.0 (23.48) | 158 | -4.4 (17.23) | 388 | 71.6 (22.28) | 377 | -2.4 (16.32) |
Cycle 3b | 194 | 78.3 (17.81) | 189 | 3.7 (16.70) | 96 | 71.0 (22.54) | 94 | -2.8 (19.56) | 290 | 75.9 (19.76) | 283 | 1.5 (17.93) |
Cycle 4b | 183 | 78.2 (17.16) | 178 | 3.6 (16.8) | 74 | 70.0 (20.59) | 71 | -2.9 (16.34) | 257 | 75.8 (18.55) | 149 | 1.8 (16.90) |
Cycle 5b | 149 | 78.5 (18.17) | 145 | 3.4 (19.17) | 50 | 73.5 (19.49) | 48 | -1.1 (17.52) | 199 | 77.2 (18.5) | 193 | 2.3 (18.83) |
Cycle 6b | 148 | 77.4 (19.28) | 143 | 0.9 (18.14) | 37 | 74.2 (19.97) | 36 | -0.6 (12.49) | 185 | 76.8 (19.41) | 179 | 0.6 (17.13) |
End of treatment | 170 | 72.5 (24.13) | 166 | -4.3 (21.49) | 151 | 61.0 (26.46) | 147 | -13.5 (20.54) | 321 | 67.1 (25.86) | 313 | -8.7 (21.51) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 54: EORTC QLQ-C30 Role Functioning Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 68.1 (30.35) | — | — | 217 | 65.1 (30.31) | — | — | 465 | 66.7 (30.33) | — | — |
Cycle 2b | 225 | 64.9 (28.90) | 219 | -4.2 (29.85) | 162 | 60.5 (30.23) | 157 | -9.0 (28.34) | 387 | 63.0 (29.51) | 376 | -6.2 (29.29) |
Cycle 3b | 194 | 74.1 (25.50) | 189 | 4.1 (29.65) | 96 | 63.9 (30.56) | 94 | -5.1 (34.39) | 290 | 70.7 (27.64) | 283 | 1.0 (31.54) |
Cycle 4b | 183 | 72.6 (26.20) | 178 | 1.3 (29.62) | 74 | 64.4 (28.98) | 71 | -4.5 (30.73) | 257 | 70.2 (27.23) | 249 | -0.3 (29.99) |
Cycle 5b | 149 | 72.9 (22.88) | 145 | 0.1 (29.89) | 50 | 66.0 (27.96) | 48 | -2.8 (25.80) | 199 | 71.2 (24.37) | 193 | -0.6 (28.8) |
Cycle 6b | 148 | 71.1 (25.89) | 143 | -3.7 (28.62) | 37 | 63.1 (30.72) | 36 | -6.5 (33.16) | 185 | 69.5 (27.03) | 179 | -4.3 (29.51) |
End of treatment | 170 | 62.1 (30.34) | 166 | -8.9 (32.34) | 150 | 51.7 (32.35) | 146 | -18.8 (29.83) | 320 | 57.2 (31.68) | 312 | -13.6 (31.53) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 55: EORTC QLQ-C30 Fatigue Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 39.4 (25.72) | — | — | 217 | 42.1 (25.99) | — | — | 465 | 40.7 (25.85) | — | — |
Cycle 2b | 225 | 44.9 (24.71) | 219 | 5.9 (24.48) | 162 | 47.4 (26.65) | 157 | 8.5 (18.80) | 387 | 45.9 (25.53) | 376 | 7.0 (22.30) |
Cycle 3b | 193 | 37.5 (21.41) | 188 | 0.8 (24.40) | 96 | 45.7 (24.66) | 94 | 7.2 (24.77) | 289 | 40.2 (22.83) | 282 | 2.9 (24.66) |
Cycle 4b | 183 | 38.0 (21.73) | 178 | 1.3 (23.91) | 74 | 45.8 (25.48) | 71 | 5.6 (24.91) | 257 | 40.2 (23.10) | 249 | 2.5 (24.23) |
Cycle 5b | 149 | 36.2 (21.23) | 145 | -0.3 (25.38) | 50 | 43.3 (24.82) | 48 | 1.6 (17.90) | 199 | 38.0 (22.34) | 193 | 0.2 (23.71) |
Cycle 6b | 148 | 37.0 (21.18) | 143 | 2.3 (23.56) | 37 | 44.0 (25.40) | 36 | 6.6 (24.95) | 185 | 38.4 (22.20) | 179 | 3.2 (23.84) |
End of treatment | 170 | 42.2 (25.05) | 166 | 5.5 (26.62) | 151 | 52.1 (27.49) | 147 | 14.0 (23.05) | 321 | 46.9 (26.64) | 313 | 9.5 (25.33) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 56: EORTC QLQ-C30 Pain Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 37.9 (30.54) | — | — | 217 | 42.5 (30.38) | — | — | 465 | 40.1 (30.52) | — | — |
Cycle 2b | 225 | 30.4 (27.05) | 219 | -7.0 (26.98) | 163 | 42.7 (31.48) | 158 | 2.4 (27.76) | 388 | 35.6 (29.59) | 377 | -3.1 (27.67) |
Cycle 3b | 194 | 23.0 (23.56) | 189 | -14.3 (27.86) | 96 | 30.6 (28.48) | 94 | -7.6 (29.40) | 290 | 25.5 (25.49) | 283 | -12.1 (28.50) |
Cycle 4b | 183 | 25.1 (24.51) | 178 | -11.9 (27.23) | 74 | 32.0 (28.37) | 71 | -5.2 (21.75) | 257 | 27.1 (25.81) | 249 | -10.0 (25.92) |
Cycle 5b | 149 | 25.6 (26.32) | 145 | -11.0 (29.48) | 50 | 30.7 (31.29) | 48 | -5.9 (27.39) | 199 | 26.9 (27.66) | 193 | -9.8 (28.99) |
Cycle 6b | 148 | 25.3 (24.47) | 143 | -7.8 (26.60) | 37 | 32.4 (29.90) | 36 | -4.2 (19.67) | 185 | 26.8 (25.72) | 179 | -7.1 (25.35) |
End of treatment | 170 | 38.6 (30.74) | 166 | 2.0 (27.69) | 151 | 45.1 (31.66) | 147 | 6.8 (30.33) | 321 | 41.7 (31.30) | 313 | 4.3 (29.01) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 57: EORTC QLQ-C30 Diarrhea Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 247 | 7.2 (17.73) | — | — | 217 | 6.5 (15.69) | — | — | 464 | 6.8 (16.80) | — | — |
Cycle 2b | 225 | 27.0 (33.98) | 218 | 19.4 (34.39) | 162 | 10.9 (20.63) | 157 | 3.6 (22.83) | 387 | 20.2 (30.17) | 375 | 12.8 (31.06) |
Cycle 3b | 194 | 24.1 (30.25) | 188 | 16.3 (31.87) | 95 | 12.6 (23.91) | 93 | 5.4 (23.72) | 289 | 20.3 (28.79) | 281 | 12.7 (29.83) |
Cycle 4b | 183 | 21.5 (27.50) | 177 | 14.9 (31.56) | 74 | 5.9 (12.77) | 71 | -0.5 (18.25) | 257 | 17.0 (25.19) | 248 | 10.5 (29.19) |
Cycle 5b | 149 | 23.5 (27.81) | 144 | 16.0 (31.28) | 50 | 8.0 (15.88) | 48 | 2.1 (19.94) | 199 | 19.6 (26.19) | 192 | 12.5 (29.44) |
Cycle 6b | 148 | 22.7 (28.05) | 142 | 15.7 (33.14) | 37 | 6.3 (13.24) | 36 | 0.9 (12.56) | 185 | 19.5 (26.57) | 178 | 12.7 (30.69) |
End of treatment | 170 | 17.8 (25.93) | 165 | 11.5 (28.19) | 151 | 9.9 (21.36) | 147 | 3.6 (22.46) | 321 | 14.1 (24.18) | 312 | 7.8 (25.91) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 58: EORTC QLQ-C30 Nausea or Vomiting Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 8.3 (16.36) | — | — | 217 | 10.3 (18.26) | — | — | 465 | 9.2 (17.28) | — | — |
Cycle 2b | 225 | 14.1 (19.76) | 219 | 6.5 (21.00) | 163 | 14.4 (20.16) | 158 | 4.3 (17.46) | 388 | 14.3 (19.90) | 377 | 5.6 (19.60) |
Cycle 3b | 194 | 11.3 (17.82) | 189 | 4.3 (21.75) | 96 | 12.2 (18.81) | 94 | 2.5 (21.86) | 290 | 11.6 (18.12) | 283 | 3.7 (21.77) |
Cycle 4b | 183 | 11.8 (16.09) | 178 | 5.0 (20.25) | 74 | 11.3 (15.39) | 71 | 0.7 (18.78) | 257 | 11.7 (15.86) | 249 | 3.7 (19.90) |
Cycle 5b | 149 | 10.1 (13.40) | 145 | 4.4 (16.55) | 50 | 10.0 (14.68) | 48 | 0.3 (15.56) | 199 | 10.1 (13.69) | 193 | 3.4 (16.37) |
Cycle 6b | 148 | 11.1 (14.07) | 143 | 5.0 (16.79) | 37 | 10.4 (16.36) | 36 | 0.0 (15.94) | 185 | 11.0 (14.51) | 179 | 4.0 (16.70) |
End of treatment | 170 | 13.4 (22.08) | 166 | 5.9 (24.76) | 151 | 16.6 (23.45) | 147 | 7.3 (23.33) | 321 | 14.9 (22.75) | 313 | 6.5 (24.07) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 59: EORTC QLQ-C30 Dyspnea Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 25.4 (30.36) | — | — | 217 | 25.0 (29.09) | — | — | 465 | 25.2 (29.75) | — | — |
Cycle 2b | 225 | 22.4 (28.14) | 219 | -3.0 (24.95) | 163 | 28.2 (29.30) | 158 | 4.0 (27.22) | 388 | 24.8 (28.74) | 377 | -0.1 (26.13) |
Cycle 3b | 192 | 17.0 (23.63) | 187 | -6.8 (26.13) | 96 | 28.8 (29.66) | 94 | 3.2 (27.25) | 288 | 20.9 (26.34) | 281 | -3.4 (26.87) |
Cycle 4b | 183 | 17.9 (24.40) | 178 | -6.6 (27.69) | 74 | 25.7 (27.89) | 71 | 0.0 (28.17) | 257 | 20.1 (25.64) | 249 | -4.7 (27.93) |
Cycle 5b | 149 | 17.4 (24.38) | 145 | -7.8 (31.43) | 50 | 25.3 (30.54) | 48 | -2.1 (35.33) | 199 | 19.4 (26.21) | 193 | -6.4 (32.45) |
Cycle 6b | 148 | 18.2 (23.76) | 143 | -6.3 (29.32) | 37 | 27.9 (30.95) | 36 | 0.9 (28.16) | 185 | 20.2 (25.56) | 179 | -4.8 (29.16) |
End of treatment | 169 | 25.2 (29.89) | 165 | 1.6 (30.09) | 150 | 30.9 (31.40) | 146 | 5.9 (28.95) | 319 | 27.9 (30.69) | 311 | 3.6 (29.59) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
Table 60: EORTC QLQ-C30 Insomnia Symptom Scale in the ASCENT Trial — Safety Population
Time | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Total (N = 482) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scorea | Change from baseline | Scorea | Change from baseline | Scorea | Change from baseline | |||||||
n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean score (SD) | n | Mean (SD) | |
Baseline | 248 | 33.2 (30.95) | — | — | 217 | 35.6 (31.42) | — | — | 465 | 34.3 (31.16) | — | — |
Cycle 2b | 225 | 29.8 (30.33) | 219 | -2.9 (29.55) | 162 | 35.2 (34.30) | 157 | 0.0 (31.80) | 387 | 32.0 (32.12) | 376 | -1.7 (30.50) |
Cycle 3b | 194 | 27.0 (26.71) | 189 | -3.4 (29.68) | 96 | 31.3 (30.13) | 94 | -3.5 (33.32) | 290 | 28.4 (27.91) | 283 | -3.4 (30.88) |
Cycle 4b | 182 | 25.8 (26.89) | 177 | -4.5 (30.23) | 74 | 34.7 (27.83) | 71 | 0.0 (30.86) | 256 | 28.4 (27.40) | 248 | -3.2 (30.42) |
Cycle 5b | 149 | 27.5 (30.69) | 145 | -2.5 (32.41) | 50 | 34.0 (26.50) | 48 | -1.4 (27.47) | 199 | 29.1 (29.76) | 193 | -2.2 (31.19) |
Cycle 6b | 148 | 23.6 (28.37) | 143 | -4.7 (31.55) | 37 | 35.1 (30.37) | 36 | 4.6 (24.11) | 185 | 25.9 (29.07) | 179 | -2.8 (30.37) |
End of treatment | 170 | 39.0 (31.41) | 166 | 6.0 (35.26) | 151 | 32.2 (28.65) | 147 | -4.3 (32.24) | 321 | 35.8 (30.29) | 313 | 1.2 (34.21) |
HRQoL = health-related quality of life; SD = standard deviation; SP = safety population; TPC = treatment of physician’s choice.
aScores range from 0 to 100 and are based on EORTC QLQ-C30 questionnaire, Version 3.0. For global health status and functional scales, a higher score signifies better HRQoL. For symptom scales, a lower score signifies better HRQoL.
bFor weekly vinorelbine injections, a cycle was defined as 3 weeks.
Source: ASCENT Clinical Study Report.13
PFS Sensitivity Analysis 1
Table 61: PFS by IRC Assessment Sensitivity Analysis 1 for the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with events, n (%) | 196 (83.4) | 208 (89.3) | — | 225 (84.3) | 232 (88.5) | — |
Patients without events (censored), n (%) | 39 (16.6) | 25 (10.7) | — | 42 (15.7) | 30 (11.5) | — |
Median (95% CI) PFS, monthsa | 5.6 (4.3 to 6.5) | 2.7 (2.0 to 2.9) | — | 5.1 (4.2 to 5.8) | 2.7 (1.8 to 2.8) | — |
Log-rank p value (stratified)b | — | <0.0001 | — | <0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.535 (0.437 to 0.654) | — | 0.552 (0.456 to 0.668) | ||
PFS rate (95% CI) at 3 months, %c | 66.0 (59.5 to 71.7) | 39.5 (32.9 to 46.0) | — | 63.7 (57.6 to 69.2) | 39.1 (33.0 to 45.3) | — |
PFS rate (95% CI) at 6 months, %c | 45.1 (38.6 to 51.4) | 22.1 (16.8 to 27.8) | — | 41.8 (35.7 to 47.7) | 21.4 (16.4 to 26.8) | — |
PFS rate (95% CI) at 9 months, %c | 27.8 (22.0 to 33.9) | 12.8 (8.7 to 17.7) | — | 26.0 (20.7 to 31.6) | 12.5 (8.6 to 17.1) | — |
PFS rate (95% CI) at 12 months, %c | 17.5 (12.6 to 23.1) | 5.9 (3.1 to 9.8) | — | 16.9 (12.3 to 22.0) | 6.3 (3.6 to 10.0) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimate.
Source: ASCENT Clinical Study Report.13
PFS Sensitivity Analysis 2
Table 62: PFS by IRC Assessment Sensitivity Analysis 2 for the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with events, n (%) | 166 (70.6) | 150 (64.4) | — | 190 (71.2) | 171 (65.3) | — |
Patients without events (censored), n (%) | 69 (29.4) | 83 (35.6) | — | 77 (28.8) | 91 (34.7) | — |
Median (95% CI) PFS, monthsa | 5.5 (4.1 to 6.9) | 2.2 (1.4 to 2.8) | — | 5.5 (4.1 to 6.5) | 2.2 (1.4 to 2.8) | — |
Log-rank p value (stratified)b | — | <0.0001 | — | <0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.402 (0.317 to 0.509) | — | 0.425 (0.341 to 0.530) | ||
PFS rate (95% CI) at 3 months, %c | 64.8 (58.1 to 70.7) | 26.9 (20.3 to 34.0) | — | 62.1 (55.7 to 67.8) | 27.0 (20.8 to 33.6) | — |
PFS rate (95% CI) at 6 months, %c | 47.6 (40.7 to 54.2) | 11.2 (6.5 to 17.3) | — | 44.1 (37.7 to 50.4)) | 10.9 (6.6 to 16.4) | — |
PFS rate (95% CI) at 9 months, %c | 25.9 (19.7 to 32.5) | 8.4 (4.3 to 14.1) | — | 24.0 (18.3 to 30.1) | 7.5 (3.9 to 12.7) | — |
PFS rate (95% CI) at 12 months, %c | 20.2 (14.4 to 26.6) | 7.0 (3.2 to 12.8) | — | 18.8 (13.6 to 24.8) | 6.3 (2.9 to 11.5) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimate.
Source: ASCENT Clinical Study Report.13
PFS Sensitivity Analysis 3
Table 63: PFS by IRC Assessment Sensitivity Analysis 3 for the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with events, n (%) | 222 (94.5) | 233 (100.0) | — | 252 (94.4) | 262 (100.0) | — |
Patients without events (censored), n (%) | 13 (5.5) | 0 | — | 15 (5.6) | 0 | — |
Median (95% CI) PFS, monthsa | 4.3 (3.5 to 5.4) | 1.5 (1.4 to 1.6) | — | 4.1 (3.2 to 4.5) | 1.5 (1.4 to 1.6) | — |
Log-rank p value (stratified)b | — | <0.00001 | — | <0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.385 (0.316 to 0.469) | — | 0.402 (0.333 to 0.484) | ||
PFS rate (95% CI) at 3 months, %c | 58.3 (51.7 to 64.3) | 21.9 (16.8 to 27.4) | — | 56.6 (50.4 to 62.3) | 21.8 (17.0 to 26.9) | — |
PFS rate (95% CI) at 6 months, %c | 36.6 (30.5 to 42.7) | 6.4 (3.8 to 10.1) | — | 32.9 (27.3 to 38.6) | 6.1 (3.6 to 9.5) | — |
PFS rate (95% CI) at 9 months, %c | 16.2 (11.8 to 21.2) | 2.6 (1.1 to 5.2) | — | 14.7 (10.8 to 19.3) | 2.3 (1.0 to 4.7) | — |
PFS rate (95% CI) at 12 months, %c | 10.1 (6.6 to 14.3) | 0.9 (0.2 to 2.8) | — | 9.3 (6.2 to 13.2) | 0.8 (0.2 to 2.5) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimate.
Source: ASCENT Clinical Study Report.13
PFS Sensitivity Analysis 4
Table 64: PFS by IRC Assessment Sensitivity Analysis 4 for the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with events, n (%) | 202 (86.0) | 189 (81.1) | — | 229 (85.8) | 211 (80.5) | — |
Patients without events (censored), n (%) | 33 (14.0) | 44 (18.9) | — | 38 (14.2) | 51 (19.5) | — |
Median (95% CI) PFS, monthsa | 5.5 (4.1 to 5.5) | 1.8 (1.4 to 2.8) | — | 5.5 (4.1 to 5.5) | 1.9 (1.4 to 2.8) | — |
Log-rank p value (stratified)b | — | <0.0001 | — | <0.0001 | ||
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.329 (0.265 to 0.409) | — | 0.361 (0.294 to 0.443) | ||
PFS rate (95% CI) at 3 months, %c | 68.1 (61.7 to 73.7) | 23.3 (17.6 to 29.6) | — | 65.5 (59.4 to 71.0) | 25.2 (19.5 to 31.2) | — |
PFS rate (95% CI) at 6 months, %c | 42.9 (36.4 to 49.2) | 8.1 (4.8 to 12.6) | — | 39.5 (33.5 to 45.5) | 8.2 (5.0 to 12.5) | — |
PFS rate (95% CI) at 9 months, %c | 26.1 (20.5 to 32.1) | 2.7 (1.0 to 5.8) | — | 24.1 (19.0 to 29.6) | 2.9 (1.2 to 5.9) | — |
PFS rate (95% CI) at 12 months, %c | 16.8 (12.1 to 22.1) | 0.5 (0.1 to 2.8) | — | 15.4 (11.1 to 20.2) | 1.0 (0.2 to 3.2) | — |
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimate.
Source: ASCENT Clinical Study Report.13
PFS Sensitivity Analysis 5
Table 65: PFS by IRC Assessment Sensitivity Analysis 5 for the ASCENT Trial — Safety Population
Outcome measure | Sacituzumab govitecan (N = 258) | TPC (N = 224) | Treatment comparison |
|---|---|---|---|
Patients with events, n (%) | 186 (72.1) | 162 (72.3) | — |
Patients without events (censored), n (%) | 72 (27.9) | 62 (27.7) | — |
Median (95% CI) PFS, monthsa | 5.4 (4.2 to 5.9) | 1.8 (1.5 to 2.7) | — |
Log-rank p value (stratified)b | — | <0.0001 | |
Stratified Cox regression analysis HR relative to TPC (95% CI) | — | 0.432 (0.344 to 0.541) | |
PFS rate (95% CI) at 3 months, %c | 62.8 (56.4 to 68.6) | 28.6 (22.1 to 35.5) | — |
PFS rate (95% CI) at 6 months, %c | 41.3 (34.8 to 47.6) | 11.3 (6.7 to 17.1) | — |
PFS rate (95% CI) at 9 months, %c | 23.2 (17.5 to 29.4) | 7.6 (3.8 to 13.1) | — |
PFS rate (95% CI) at 12 months, %c | 16.4 (11.3 to 22.3) | 6.3 (2.8 to 11.8) | — |
CI = confidence interval; HR = hazard ratio; IRC = independent review committee; PFS = progression-free survival; SP = safety population; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bStratified log-rank test and stratified Cox regression adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region.
cEstimate and CI for PFS rate at the specified time points are from Kaplan-Meier estimate.
Source: ASCENT Clinical Study Report.13
PFS Subgroup Analysis by Primary Brain Metastasis
Table 66: Subgroup Analysis of PFS by IRC Assessment for BM-Pos Versus BM-Neg in the ASCENT Trial — ITT Population
Outcome measure | BM-Pos | BM-Neg | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 32) | TPC (N = 29) | Treatment comparison | Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | |
Patients with events, n (%) | 24 (75.0) | 21 (72.4) | — | 166 (70.6) | 150 (64.4) | — |
Patients without events (censored), n (%) | 8 (25.0) | 8 (27.6) | — | 69 (29.4) | 83 (35.6) | — |
Median (95% CI) PFS, monthsa | 2.8 (1.5 to 3.9) | 1.6 (1.3 to 2.9) | — | 5.6 (4.3 to 6.3) | 1.7 (1.5 to 2.6) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.682 (0.379 to 1.228) | — | 0.411 (0.325 to 0.519) | ||
P value for HR = 1 | — | 0.2023 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; BM-Pos = brain metastasis–positive; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
PFS Subgroup Analysis by Prior Therapies
Table 67: Subgroup Analysis of PFS by IRC Assessment for Patients With 2 to 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 166) | TPC (N = 164) | Treatment comparison | Sacituzumab govitecan (N = 184) | TPC (N = 181) | Treatment comparison | |
Patients with events, n (%) | 109 (65.7) | 103 (62.8) | — | 122 (66.3) | 116 (64.1) | — |
Patients without events (censored), n (%) | 57 (34.3) | 61 (37.2) | — | 62 (33.7) | 65 (35.9) | — |
Median (95% CI) PFS, monthsa | 5.8 (4.2 to 7.1) | 1.6 (1.5 to 2.5) | — | 5.4 (4.1 to 6.8) | 1.6 (1.5 to 2.5) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.388 (0.291 to 0.517) | — | 0.393 (0.300 to 0.515) | ||
P value for HR = 1 | — | <0.0001 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
Table 68: Subgroup Analysis of PFS by IRC Assessment for Patients With More Than 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 69) | TPC (N = 69) | Treatment comparison | Sacituzumab govitecan (N = 83) | TPC (N = 81) | Treatment comparison | |
Patients with events, n (%) | 57 (82.6) | 47 (68.1) | — | 68 (81.9) | 55 (67.9) | — |
Patients without events (censored), n (%) | 12 (17.4) | 22 (31.9) | — | 15 (18.1) | 26 (32.1) | — |
Median (95% CI) PFS, monthsa | 5.6 (3.0 to 6.5) | 2.5 (1.5 to 2.8) | — | 4.2 (2.8 to 5.7) | 2.2 (1.5 to 2.8) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.483 (0.322 to 0.723) | — | 0.533 (0.369 to 0.771) | ||
P value for HR = 1 | — | 0.0004 | — | 0.0008 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
PFS Subgroup Analysis by BRCA1 or BRCA2 Mutational Status
Table 69: Subgroup Analysis of PFS by IRC Assessment for Patients Who Were BRCA1- or BRCA2-Positive in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 16) | TPC (N = 18) | Treatment comparison | Sacituzumab govitecan (N = 20) | TPC (N = 23) | Treatment comparison | |
Patients with events, n (%) | 11 (68.8) | 9 (50.0) | — | 12 (60.0) | 13 (56.5) | — |
Patients without events (censored), n (%) | 5 (31.3) | 9 (50.0) | — | 8 (40.0) | 10 (43.5) | — |
Median (95% CI) PFS, monthsa | 4.6 (1.3 to 10.3) | 2.5 (0.8 to 5.5) | — | 7.4 (1.5 to 14.5) | 2.5 (0.8 to 3.0) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.611 (0.237 to 1.571) | — | 0.421 (0.181 to 0.980) | ||
P value for HR = 1 | — | 0.3063 | — | 0.0447 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
Table 70: Subgroup Analysis of PFS by IRC Assessment for Patients Who Were BRCA1- or BRCA2-Negative in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 133) | TPC (N = 125) | Treatment comparison | Sacituzumab govitecan (N = 150) | TPC (N = 146) | Treatment comparison | |
Patients with events, n (%) | 100 (75.2) | 80 (64.0) | — | 115 (76.7) | 94 (64.4) | — |
Patients without events (censored), n (%) | 33 (24.8) | 45 (36.0) | — | 35 (23.3) | 52 (36.6) | — |
Median (95% CI) PFS, monthsa | 4.9 (3.8 to 5.9) | 1.6 (1.5 to 2.5) | — | 4.3 (3.2 to 5.6) | 1.6 (1.5 to 2.3) | — |
Stratified Cox regression analysis HR relative to TPC (95% CI)b | — | 0.419 (0.307 to 0.572) | — | 0.454 (0.341 to 0.605) | ||
P value for HR = 1 | — | <0.0001 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; PFS = progression-free survival; TPC = treatment of physician’s choice.
aMedian PFS is from Kaplan-Meier estimate. CI for median is computed using the Brookmeyer-Crowley method.
bHR and P value are from an unstratified Cox regression analysis.
Source: ASCENT Clinical Study Report.13
ORR Subgroup Analysis by Primary Brain Metastasis
Table 71: Subgroup Analysis of ORR for BM-Pos Versus BM-Neg in the ASCENT Trial — ITT Population
Outcome measure | BM-Pos | BM-Neg | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 32 | 29 | — | 235 | 233 | — |
ORR, n (%) [95% CI]a | 1 (3.1) [0.08 to 16.22] | 0 | — | 82 (34.9) [28.81 to 41.36] | 11 (4.7) [2.38 to 8.29] | — |
OR (95% CI)a | — | NC | — | 10.82 (5.58 to 20.97) | ||
P valueb | — | 0.3411 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; BM-Pos = brain metastasis–positive; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the Beta distribution.
bP value is based on Cochran-Mantel-Haenszel test
Source: ASCENT Clinical Study Report.13
ORR Subgroup Analysis by Prior Therapies
Table 72: Subgroup Analysis of ORR for Patients With 2 or 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 166 | 164 | — | 184 | 181 | — |
ORR, n (%) [95% CI]a | 66 (39.8) [32.26 to 47.63] | 7 (4.3) [1.73 to 8.60] | — | 67 (36.4) [29.46 to 43.81] | 7 (3.9) [1.57 to 7.81] | — |
OR (95% CI)a | — | 14.80 (6.53 to 33.56) | — | 14.23 (6.31 to 32.09) | ||
P valueb | — | <0.0001 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the Beta distribution.
bP value is based on Cochran-Mantel-Haenszel test
Source: ASCENT Clinical Study Report.13
Table 73: Subgroup Analysis of ORR for Patients With More Than 3 Prior Therapies in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 69 | 69 | — | 83 | 81 | — |
ORR, n (%) [95% CI]a | 16 (23.2) [13.87 to 34.91] | 4 (5.8) [1.60 to 14.18] | — | 16 (19.3) [11.44 to 29.41] | 4 (4.9) [1.36, 12.16] | — |
OR (95% CI)a | — | 4.91 (1.55 to 15.56) | — | 4.60 (1.47 to 14.42) | ||
P valueb | — | 0.0038 | — | 0.0052 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the Beta distribution.
bP value is based on Cochran-Mantel-Haenszel test
Source: ASCENT Clinical Study Report.13
ORR Subgroup Analysis by BRCA1 or BRCA2 Mutational Status
Table 74: Subgroup Analysis of ORR for Patients Who Were BRCA1- or BRCA2-Positive in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 16 | 18 | — | 20 | 23 | — |
ORR, n (%) [95% CI]a | 3 (18.8) [4.05 to 45.65] | 1 (5.6) [0.14 to 27.29] | — | 3 (15.0) [3.21 to 37.89] | 1 (4.3) [0.11, 21.95] | — |
OR (95% CI)a | — | 3.92 (0.36 to 42.20) | — | 3.88 (0.37 to 40.71) | ||
P valueb | — | 0.2403 | — | 0.2358 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the Beta distribution.
bP value is based on Cochran-Mantel-Haenszel test
Source: ASCENT Clinical Study Report.13
Table 75: Subgroup Analysis of ORR for Patients Who Were BRCA1- or BRCA2-Negative in the ASCENT Trial — BM-Neg and ITT Populations
Outcome measure | BM-Neg | ITT | ||||
|---|---|---|---|---|---|---|
Sacituzumab govitecan (N = 235) | TPC (N = 233) | Treatment comparison | Sacituzumab govitecan (N = 267) | TPC (N = 262) | Treatment comparison | |
Patients with measurable disease at baseline, n | 133 | 125 | — | 150 | 146 | — |
ORR, n (%) [95% CI]a | 44 (33.1) [25.17 to 41.77] | 7 (5.6) [2.28 to 11.20] | — | 45 (30.0) [22.80 to 38.01] | 7 (4.8) [1.95 to 9.63] | — |
OR (95% CI)a | — | 8.33 (3.58 to 19.38) | — | 8.51 (3.69 to 19.63) | ||
P valueb | — | <0.0001 | — | <0.0001 | ||
BM-Neg = brain metastasis–negative; CI = confidence interval; IRC = independent review committee; ITT = intention to treat; OR = odds ratio; ORR = objective response rate; TPC = treatment of physician’s choice.
aExact binomial CI for proportion is based on the Beta distribution.
bP value is based on Cochran-Mantel-Haenszel test
Source: ASCENT Clinical Study Report.13
Appendix 6: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-C30
Findings
Description and Scoring
The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) is one of the most used patient-reported outcome measures in oncology clinical trials. It is a multidimensional, cancer-specific, self-administered, measure of HRQoL.41
The EORTC QLQ-C30 is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), a global health status/HRQoL scale, and 6 single items assessing additional symptoms commonly reported by cancer patients (dyspnea, loss of appetite, insomnia, constipation and diarrhea) as well as perceived financial impact of the disease.41
The EORTC QLQ-C30 uses a 1-week recall period to assess functional status and symptoms. All scales and single-item measures are scored from 0 to 100. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from one to 4. For the 2 items that form the global HRQoL scale, the response format is a 7-point Likert-type scale with anchors at 1 = “very poor” and 7 = “excellent.” Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Scale sum scores are transformed such that a high score on the functional scales represents a high/healthy level of functioning, a high score on the symptom scales represents a high level of symptomatology, and a high score on the global health status/HRQoL scale represents a high HRQoL.42
According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least half of the items. The values for missing items are interpolated with the average of the respondent-completed items.42
Assessment of Validity and Reliability
In its initial development, the EORTC QLQ-C30 underwent an evaluation of its psychometric properties and demonstrated reliability and validity in cancer patients in an international field trial of 305 patients in 13 multicultural clinical research settings.41 A revision of the EORTC QLQ-C30 was undertaken to improve low internal consistency, content validity for the role functional scale, and a conceptual difficulty (undue emphasis on physical function in the global HRQoL scale).43 The original and new versions were applied in a total of 1,181 patients with cancer in Canada and the Netherlands. Internal consistency improved for the role functional scale in the new version (Cronbach alpha ranging from 0.78-0.88), and substitution of the new item for the previous version did not alter internal consistency (Cronbach alpha ranging from 0.81-0.92.43
The EORTC QLQ-C30 (Version 3.0) is the version currently in use and was used in the ASCENT trial. Version 3.0 differs from the previous Version 2.0 in that the number of response options for the first 5 items of the questionnaire comprising the physical function scale was increased from 2 options (yes/no in Version 2.0) to 4 options (not at all, a little, quite a bit, very much). Internal consistency, reliability, construct validity, criterion validity, and responsiveness of the EORTC QLQ-C30 Version 3.0 was assessed in 622 patients with head and neck cancer from 12 countries. Version 3.0 was more reliable than previous versions.44 Internal consistency of the multi-item scales was assessed using Cronbach alpha, with a value of 0.70 being considered adequate.45 The internal consistency of the new physical function scale of the EORTC QLQ-C30 Version 3.0 was 0.84 compared with 0.66 in Version 1.0. The EORTC QLQ-C30 Version 3.0 was able to discriminate between head and neck cancer patients who were disease-free, who were newly diagnosed, and who had recurrent disease. As well, differences were noted between patients with different stages of disease and according to Karnofsky Performance Status (KPS): the new scale had a stronger association with KPS. Furthermore, there was a strong correlation observed between scores on the EORTC QLQ-C30 Version 3.0 and symptom/treatment toxicity scores. Responsiveness to change was assessed using the standardized response mean (SRM), with an SRM of 0.20 considered small, 0.50 considered medium, and 0.80 considered large. The changes in the scores of QLQ-C30 demonstrated a small to medium SRM in response to treatment over time with scores mostly changing between 5 and 10 points.44
Specifically in patients with metastatic breast cancer, a 1997 studied the validity of the EORTC QLQ-C30 questionnaire through an analysis of patient-observer agreement. The median kappa coefficient for agreement across the 30 items in the EORTC QLQ-C30 was 0.86 with a range of 0.48 to 1.00,46 representing substantial or near-perfect agreement for most items.47 A further study of discriminative and convergent validity of the psychosocial subscales of EORTC QLQ-C30 in patients with breast cancer was conducted in 1998. The study found acceptable discriminative validity represented by correlation with external parameters such as ECOG Performance Status (Spearman’s rank correlation values ranging from 0.02 to 0.56). A correlation of 0.2 represented significance at the 0.01 level. The convergent validity, as represented by correlation with scores on the Profile of Mood States and Psychosocial Adjustment to Illness Scale, was also deemed to be acceptable.48
Minimal Important Difference
One study from 1998, conducted in patients with breast cancer and small-cell lung cancer, estimated that a change in score on any scale of the EORTC QLQ-C30 of 10 points would be clinically significant. This estimate was based on an anchor-based approach to estimate the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported “very much” change had corresponding changes in the EORTC QLQ-C30 of more than 20 points.49
A more recent study from 2019 aimed to describe the MID for the EORTC QLQ-C30 in patients with advanced breast cancer. This study used an anchor-based approach utilizing performance status and selected AEs as the anchor variables. MIDs for within-group changes ranged from 5 to 14 points for improvements and from -14 to -4 points for deterioration across the individual scales. For between-group differences, MIDs ranged from 4 to 11 points for improvements and from -18 to -4 points for deterioration across the individual scales.50
The impact of the response shift effect on EORTC QLQ-C30 MID in patients with breast cancer was assessed in a 2016 study.51 The response shift effect refers to the patient’s tendency to recalibrate, reprioritize, or reconceptualize the meaning of HRQoL over time. The authors found that, upon correcting for the response shift effect, a reliable and significant MID was in line with the commonly used value of 5 to 10 points on each scale established earlier by Osoba et al. (1998).49 A 2011 study combined systematic review, expert opinions, and meta-analysis to estimate large, medium and small differences for QLQ-C30 scores and recommended that small and medium differences corresponded with changes from 3 to 6 and 9 to 19 points, respectively, depending on the subscale.52
Pharmacoeconomic Review
Abbreviations
AE
adverse event
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
G-CSF
granulocyte colony–stimulating factor
ICER
incremental cost-effectiveness ratio
KM
Kaplan-Meier
mTNBC
metastatic triple-negative breast cancer
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
RDI
relative dosing intensity
TNBC
triple-negative breast cancer
TPC
treatment of physician’s choice
TTD
time to treatment discontinuation
Executive Summary
The executive summary comprises 2 tables, Table 1 and Table 2, and a conclusion.
Item | Description |
|---|---|
Drug product | Sacituzumab govitecan (Trodelvy) 180 mg lyophilized powder for solution for injection, for IV use |
Submitted price | Sacituzumab govitecan, 180 mg, vial for injection: $1,478.00 per vial |
Indication | For the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received 2 or more prior therapies, at least 1 of them for metastatic disease |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | September 24, 2021 |
Reimbursement request | As per indication |
Sponsor | Gilead Sciences Canada, Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adults with either locally advanced TNBC or mTNBC who were either refractory or had relapsed after at least 2 prior standard of care chemotherapy regimens |
Treatment | Sacituzumab govitecan |
Comparators | TPC comprised weighted single-agent chemotherapy regimens:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 5 years |
Key data source | The ASCENT trial, a phase III, multi-centre, randomized trial |
Submitted results | ICER = $347,009 per QALY gained vs. TPC (0.40 incremental QALYs and $137,427 incremental costs) |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; KM = Kaplan-Meier; LY = life-year; mTNBC = metastatic triple-negative breast cancer; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; TNBC = triple-negative breast cancer; TPC = treatment of physician’s choice; TTD = time to treatment discontinuation; vs. = versus.
Conclusions
The evidence identified in the CADTH clinical review suggests that when compared with treatment of physician’s choice (TPC), administration of sacituzumab govitecan (10 mg/kg on day 1 and day 8 of a 21-day treatment cycle) contributed to statistically significant and clinically meaningful prolongation of progression-free survival (PFS) and overall survival (OS) among patients with locally advanced triple-negative breast cancer (TNBC) or metastatic TNBC (mTNBC) who had received at least 2 prior therapies. Health-related quality of life data could not be interpreted due to absence of formal statistical testing and high rates of missing data resulting from deaths and withdrawals. The clinical efficacy of sacituzumab govitecan beyond the median follow-up in the ASCENT trial (approximately 12 months) is uncertain.
CADTH’s base-case reanalysis included selecting the gamma and log-logistic distributions for the PFS of sacituzumab govitecan and TPC, selecting the Weibull distribution for the OS of sacituzumab govitecan and TPC, selecting the gamma and Weibull distributions for the time to treatment discontinuation (TTD) of sacituzumab govitecan and TPC, applying the same overall utility value for patients with progressed disease, modifying the relative use of individual treatments among the TPC basket to align with the distribution expected in Canadian clinical practice, and increasing the relative dose intensity for patients who received sacituzumab govitecan. In the CADTH base case, sacituzumab govitecan was associated with an incremental cost-effectiveness ratio (ICER) of $375,333 per quality-adjusted life-year (QALY) gained (incremental costs of $116,613 and an incremental benefit of 0.31 QALYs) compared with TPC. CADTH’s findings are aligned with those of the sponsor. Based on the CADTH base case, a price reduction of at least 87% would be required for sacituzumab govitecan to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained.
The cost-effectiveness of sacituzumab govitecan is primarily driven by its drug acquisition costs and the magnitude of benefit attributed to sacituzumab govitecan. The cost-effectiveness of sacituzumab govitecan compared with the individual treatments that comprised the TPC basket remains unknown at this time given the lack of comparative effectiveness evidence.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Two patient groups, the Canadian Breast Cancer Network and Rethink Breast Cancer, provided input for the review of sacituzumab govitecan for the treatment of mTNBC via an online survey created by their respective organizations. Importantly, patient input indicated that they expected sacituzumab govitecan to extend PFS with a good quality of life when first- and second-line therapies stopped working. These patients further reported that improvements in quality of life attributed to disease control, OS, and prevention of recurrence were areas of greatest value to them. Patients reported that fatigue, nausea, constipation, diarrhea, and headache were the symptoms experienced most often with currently available treatments, while hand-foot syndrome, nausea, and fatigue were reported as the side effects most difficult to tolerate. None of the patients from the Canadian Breast Cancer Network group reported having lived experience with sacituzumab govitecan, while two-thirds of respondents from the Rethink Breast Cancer group had direct experience with sacituzumab govitecan. Commonly reported side effects of sacituzumab govitecan among patients included fatigue, alopecia, diarrhea, and neutropenia, although patients indicated that side effects from sacituzumab govitecan were much more tolerable than those from other treatments.
Registered clinician input reiterated the most important treatment goals for patients with mTNBC, which were outlined by patient input, such as a substantial improvement in OS and a delay in disease progression. These important clinical outcomes were recognized in the ASCENT trial and noted to be both clinically meaningful and previously unseen in patients with advanced TNBC, a population with a huge unmet need. Improved or maintained quality of life and an improvement of symptoms was also noted to be an important treatment goal according to the clinician group. Although single-agent chemotherapies such as eribulin, vinorelbine, gemcitabine, and capecitabine were recognized as currently available treatment options, registered clinicians indicated that these treatments are ineffective because they have no major impact on OS, have limited efficacy with respect to PFS, and are associated with toxicities. Sacituzumab govitecan addresses an unmet need for all TNBC patients who are candidates for systemic therapy since no standard treatments have shown improved efficacy for later lines. Registered clinicians stated that additional treatment with a single-agent chemotherapy may be helpful because survival outcomes are poorest for TNBC relative to other breast cancer subtypes and relapsed or refractory TNBC patients are the best candidates for this treatment. Registered clinicians noted that disease progression or toxicities were factors guiding decision-making related to treatment discontinuation and that treatment response for patients on sacituzumab govitecan should be assessed every 12 weeks along with staging. Lastly, the clinician group indicated that sacituzumab govitecan is most likely to be administered as an outpatient treatment in cancer clinics.
Feedback from the drug plans identified several items for CADTH to take into consideration for the review. First, drug plans noted that sacituzumab govitecan may change the place in therapy of comparator drugs (e.g., eribulin, gemcitabine, capecitabine, vinorelbine) or drugs reimbursed in previous lines and subsequent lines due to the complex therapeutic space, with multiple lines of therapy, subpopulations, and competing products. Drug plans raised concerns about patients’ eligibility for sacituzumab govitecan for those who have had no prior exposure to taxanes for reasons of intolerance or other contraindications, as per the ASCENT trial inclusion criteria. However, drug plans require clarity about whether patients with an Eastern Cooperative Oncology Group (ECOG) Performance Status of 2 or greater are eligible for sacituzumab govitecan. Drug plans anticipate that sacituzumab govitecan will require more chair time than other comparators and further noted that additional nursing and pharmacy resources will be required for administration and preparation of sacituzumab govitecan. Drug plans indicated that hormone receptor status and human epidermal growth factor receptor 2 (HER2) are standard tests done in jurisdictions for metastatic breast cancer. Drug plans noted that drug wastage is likely because dosing is weight dependent. Drug plans also noted that there are some negotiated prices for comparators because comparators in the ASCENT trial are rather generic or have confidential prices. Drug plans are concerned about the anticipated budget impact of sacituzumab govitecan because the market share uptake assumed by the sponsor over the 3-year time horizon ( | %, | %, and | % in years 1 to 3, respectively) may be underestimated if sacituzumab govitecan is the new standard of care for patients who meet the ASCENT trial criteria. Lastly, drug plans indicated that the comparators included in the ASCENT trial are generic treatments or have confidential prices, which are negotiated.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s base-case analysis compared sacituzumab govitecan with a treatment basket of single-agent chemotherapies based on the ASCENT trial, including eribulin, capecitabine, gemcitabine, and vinorelbine.
The sponsor captured disease management and monitoring costs that would be relevant to progression-free and progressive disease stages, which include the cost of community nurse visits.
The sponsor captured drug wastage that may be associated with each dose amount because dosing is dependent on patient weight or body surface area.
Several treatment-related adverse events (AEs) were captured, including fatigue, diarrhea, and neutropenia.
CADTH also addressed some of these concerns as follows:
increased market uptake of sacituzumab govitecan in the budget impact analysis.
CADTH was unable to address the following areas of concern raised in stakeholder input:
comparative clinical efficacy of sacituzumab govitecan versus the individual TPCs in the economic model
costs associated with pharmacy resources used to administer and prepare sacituzumab govitecan.
Economic Review
The current review is for sacituzumab govitecan (Trodelvy) for the treatment of adult patients with unresectable locally advanced TNBC or mTNBC who have received at least 2 prior therapies, including at least 1 prior therapy for locally advanced or metastatic disease.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing sacituzumab govitecan compared with TPC (i.e., a mix of single-agent chemotherapy treatments, including eribulin, capecitabine, gemcitabine, and vinorelbine) for adult mTNBC or unresectable locally advanced TNBC patients with at least 2 prior chemotherapies, including at least 1 prior therapy for locally advanced or metastatic disease.1 The modelled population was aligned with the pivotal clinical trial (ASCENT). The sponsor received a final Notice of Compliance from Health Canada (1 prior therapy for metastatic disease), which slightly differed from the initially submitted population referenced previously, although feedback indicated this would not impact the assessment of sacituzumab govitecan in this population. The sponsor conducted 2 subgroup analyses to explore the cost-effectiveness of sacituzumab govitecan for patients who are brain metastases–negative or for fast-relapsing, second-line patients.
Sacituzumab govitecan is available as a 180 mg vial for injection. The recommended total daily dose of sacituzumab govitecan is 10 mg/kg administered as an IV infusion on day 1 and day 8 of a 21-day treatment cycle and continued until progression of the underlying disease or unacceptable toxicity.1,2 At the sponsor-submitted price of $1,478 per vial, the cost per 21-day cycle was estimated to be $12,478 based on the sponsor’s assumption of a 94% dose intensity. The sponsor modelled 4 comparator single-agent chemotherapy treatments as part of the TPC basket — eribulin, capecitabine, gemcitabine, and vinorelbine — based on treatments received in the ASCENT trial.1 The TPC basket of comparators was modelled based on the sponsor’s assumed dosing regimens per administration, which were noted to differ from the Cancer Care Ontario–approved product monographs. Drug costs for eribulin, vinorelbine, gemcitabine, and capecitabine per respective treatment cycles were weighted according to the distribution of their relative use among patients, which was assumed to be 53%, 20%, 14%, and 13%, respectively, based on clinical expert feedback. The weighted cost of TPC based on this distribution was $1,649 per 21-day treatment cycle.1 Drug administration costs for eribulin, vinorelbine, gemcitabine, and capecitabine were further weighted, such that the weighted drug administration cost was $95 per 21-day treatment cycle. Drug wastage was assumed in the sponsor’s base case for all treatments.1
The economic analysis used a 5-year time horizon from the perspective of the publicly funded health care payer. Costs and clinical outcomes (i.e., QALYs and life-years) were discounted at a rate of 1.5% per annum.1
Model Structure
A partitioned survival model was submitted to capture the long-term costs and effects associated with the natural history of mTNBC over the model time horizon.1 The model consisted of 3 primary health states (PFS, progressed disease, and death), and the proportion of patients who were progression-free, experienced progressive disease, or were dead at any time over the model time horizon was derived from non–mutually exclusive survival curves. Overall survival and PFS curves were derived from the ASCENT trial for sacituzumab govitecan and TPC, and were used to determine the proportion of patients in each health state (Appendix 3, Figure 1).1 Specifically, the proportion of progression-free patients was derived as the area under the PFS curves, while the proportion of patients with progressed disease was derived by partitioning the OS curve (i.e., the difference in the area under the curve between the OS and PFS curves). Progression was defined according to objective PFS criteria, assessed by a blinded independent committee review using Response Evaluation Criteria in Solid Tumors Version 1.1. Time to treatment discontinuation was calculated using the ASCENT trial to identify the proportion of patients who were alive and who remained on treatment at any given point in time. Time to treatment discontinuation accounted for treatment discontinuation due to any cause.1
Model Inputs
The patient cohort comprised patients with mTNBC whose baseline characteristics were similar to the brain metastasis–negative patients in the ASCENT trial in age and the proportion of adult women. The modelled population mainly comprised adult women (99.6%), with an average age of 54 years, weight of 71.1 kg, and body surface area of 1.78 m2 based on the intention-to-treat population from the ASCENT trial.
Key clinical efficacy inputs (i.e., OS and PFS) and treatment duration (i.e., TTD) for sacituzumab govitecan and TPC were based on the results of the ASCENT trial (i.e., data cut-off March 11, 2020). Kaplan-Meier (KM) estimates of PFS, OS, and TTD from the trial period were used to fit parametric survival curves to extrapolate the treatment effect beyond the observed trial data (maximum follow-up: 23.8 months and 24.2 months for sacituzumab govitecan and TPC, respectively, at the data cut-off [i.e., date of final analysis]) over the entire model time horizon (5 years). Several parametric survival functions were fitted to the PFS, OS, and TTD data to determine the best-fitting distribution based on diagnostic plots, goodness-of-fit statistics, visual inspection, and clinical expectations regarding long-term progression risk and survival. The chosen parametric survival distribution of PFS for sacituzumab govitecan was the KM + log-normal distribution. For TPC, the KM + log-logistic distribution was used. The chosen parametric survival distribution for OS for both sacituzumab govitecan and TPC was the KM + log-logistic distribution. Individual TTD curves for sacituzumab govitecan and TPC were obtained from the ASCENT trial to identify the proportion of patients who were alive and remained on initial treatment. The chosen parametric survival distribution for TTD for sacituzumab govitecan was the KM + gamma distribution. For TPC, the KM + exponential distribution was used.
Health state utility values applied in the economic model were based on the ASCENT trial population using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30), administered to patients to assess their quality of life, and from which patients’ scores were mapped onto the EQ-5D-3L algorithm and adjusted for patient’s age, sex, and EORTC QLQ-C30 score. The sponsor applied treatment-specific utility values in the PFS state, whereas a single utility value was applied for patients in the progressed disease state. Utility decrements for treatment-related AEs were further applied to each health state and were sourced from a cost-effectiveness study of new treatments for advanced breast cancer in Canada.3
Costs are incorporated into the model as monthly costs and include drug acquisition costs derived from previous CADTH reimbursement review reports (eribulin, gemcitabine, capecitabine), with the exception of vinorelbine and sacituzumab govitecan, which were based on the sponsor’s internal pricing data. The model further incorporated concomitant medication costs, which were sourced from the Ontario Drug Benefit e-formulary,4 while the proportion of patients on concomitant medication was derived from the ASCENT trial. Subsequent treatment costs were also included, while subsequent treatment duration was informed by the ASCENT trial. Drug administration costs per chemotherapy agent were further included and sourced from the Ontario Schedule of Benefits of Physician Services.5 One-time AE costs were included on the assumption that patients who experienced AEs received inpatient care. Drug doses were weight dependent or calculated based on body surface area; thus, drug wastage was accounted for in the model.1 Patients who were on treatment in the PFS state accrued drug acquisition and administration costs, costs of concomitant medication, 1-time costs associated with AEs of treatment, costs associated with health care resource utilization, and monitoring costs. Patients who had progressed disease incurred costs of subsequent treatment, costs associated with health care resource utilization, and monitoring costs.
Summary of Sponsor’s Economic Evaluation Results
The sponsor’s cost-effectiveness analysis was based on 1,000 probabilistic iterations, for which findings are presented subsequently. All the sponsor’s scenario analyses were based on 1,000 iterations. Deterministic results were consistent with probabilistic findings.
Base-Case Results
In the sponsor’s base case, sacituzumab govitecan was associated with an incremental cost of $137,427 and 0.40 QALYs over a 5-year time horizon (see probabilistic results in Table 3). In the sponsor’s base case, 4.3% of sacituzumab govitecan patients and 1.3% of TPC patients were alive at the 5-year time horizon.
The sponsor did not present the proportion of incremental benefit derived within the trial compared with the extrapolation based on the sponsor’s model; therefore, CADTH undertook proxy analyses and determined that 39% of the incremental benefit (in terms of QALYs) associated with sacituzumab govitecan was accrued within the first year and 69% of the incremental benefit associated with sacituzumab govitecan was accrued within the first 2 years. The time points are similar to the mean and maximum follow-up in the ASCENT trial.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
TPC | 52,186 | 0.54 | Reference |
Sacituzumab govitecan | 189,614 | 0.94 | 347,009 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TPC = treatment of physician’s choice.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity and scenario analyses. These included varying the discount rate to 0% and 3% over the time horizon, varying the time horizon to 2 years and to 10 years, removing disutilities associated with AEs, setting the treatment discontinuation curve to the PFS curve (i.e., assuming that patients are treated until progression), setting granulocyte colony–stimulating factor (G-CSF) usage to 0%, and selecting alternate parametric distributions for the OS of sacituzumab govitecan and TPC.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations in the sponsor’s analysis that have notable implications for the economic analysis:
Long-term extrapolations of the comparative clinical efficacy (OS and PFS) are uncertain: The sponsor fitted several parametric survival curves to extrapolate OS and PFS for patients who received sacituzumab govitecan and TPC over the model’s time horizon (5 years) based on the observed period of the ASCENT trial (duration of follow-up: 24.2 months [maximum] and 11.43 months [mean]).1,6 The OS data informing both sacituzumab govitecan and TPC in the model were considered mature, as the median OS of 11.8 months (intention-to-treat population) was reached at the time of the available data cut-off from the trial. Specifically, the sponsor’s selected extrapolations for sacituzumab govitecan projected that, beyond the trial observed period, approximately 20% and 11% of patients would remain alive at year 2 and year 3, respectively, whereas for patients who received TPC, approximately 6% and 3% of patients were projected to remain alive at year 2 and year 3, respectively. CADTH’s clinical experts indicated that based on the natural history of disease in the indicated population, it is unlikely that patients on TPC would remain alive at year 3 because their life expectancy would be unlikely to extend beyond 1 year. Further, the sponsor’s extrapolations were based on KM data from the ASCENT trial over the maximum length of follow-up combined with the best-fitting parametric distribution from 24 months onward. When the KM data for survival outcomes were examined by CADTH’s clinical experts, despite the clinically meaningful improvements in survival shown in the ASCENT trial, the residual survival benefit of sacituzumab govitecan past progression is uncertain beyond the mean duration of follow-up because very few patients remained at risk and a high proportion of patients were censored (33.0% in sacituzumab govitecan and 21.4% in TPC). As such, feedback from the clinical experts consulted by CADTH indicated that the KM + log-normal distribution for the OS extrapolation of sacituzumab govitecan and the KM + log-logistic distribution for TPC selected in the sponsor’s economic base case were unlikely to be clinically plausible over the extrapolated period and overestimated the proportion of patients who would remain alive beyond the mean duration of follow-up for the remainder of the time horizon. CADTH selected alternate parametric distributions for the OS of sacituzumab govitecan and TPC that were likely clinically plausible based on expert feedback, visual fit, and best-fit statistics.
CADTH addressed this limitation by changing the OS extrapolated curves to the Weibull distribution for sacituzumab govitecan and TPC. In 1 scenario, CADTH changed the parametric distribution of OS for sacituzumab govitecan to the Gompertz distribution to explore the uncertainty of OS over the extrapolated period.
Although clinically meaningful improvements in PFS were observed in the ASCENT trial according to CADTH’s clinical experts and the median PFS was reached in both treatment arms (4.8 months for sacituzumab govitecan and 1.7 months for TPC), there were no patients remaining at risk in PFS at the end of follow-up. Although the median PFS was short, the sponsor selected survival distributions based on KM data over the maximum length of follow-up in the ASCENT trial (15 months for TPC and 21 months for sacituzumab govitecan, per the submitted model) combined with the best-fitting parametric distribution from these time points onward. CADTH’s clinical review noted that the KM data for PFS in the ASCENT trial show similar proportions of patients in the sacituzumab govitecan and TPC arms (28.8% and 34.7%) were censored and, importantly, very few patients in the TPC arm remained alive without having progressed and a higher number of patients were censored due to loss of follow-up or having initiated an alternate treatment. Despite the limitations noted with the available data to inform long-term PFS extrapolations, CADTH’s clinical experts indicated that the sponsor’s PFS extrapolation for sacituzumab govitecan may be clinically plausible up until year 3 but likely overestimates benefit thereafter; in comparison, based on the natural history of disease in the indicated population, the sponsor’s selected distribution for patients who received TPC may be considered optimistic. Additionally, CADTH’s clinical experts indicated that once a patient had progressed, it was reasonable to assume that the prognosis of patients who received sacituzumab govitecan could be expected to revert to patients who received TPC, in the absence of clinical evidence showing any long-term residual clinical benefit of treatment with sacituzumab govitecan. Given the limitations of the KM data for PFS in the ASCENT trial, and feedback from CADTH’s clinical experts pertaining to the clinical plausibility of the long-term extrapolations for PFS in the sponsor’s base case, CADTH selected alternate survival distributions for PFS based on clinical plausibility, visual fit, and best-fit statistics.
CADTH revised the parametric distributions of PFS for sacituzumab govitecan and TPC to the log-logistic and gamma parametric survival distributions, respectively. In a scenario analysis, CADTH changed the parametric distribution of PFS for sacituzumab govitecan to the log-normal and gamma distributions to explore the uncertainty of PFS over the extrapolated period.
Long-term extrapolation of TTD curves for sacituzumab govitecan and TPC are uncertain: The sponsor’s parametric distributions of TTD for sacituzumab govitecan and TPC did not align with clinical expectations according to the clinical experts consulted by CADTH. The main concern was the relationship between time on treatment and PFS. The clinical experts consulted by CADTH indicated that patients are unlikely to remain on the same treatment after having progressed and are more likely to receive a different treatment. CADTH’s clinical experts further noted that the TTD for both sacituzumab govitecan and TPC was likely to be similar to PFS due to the disease prognosis at this stage and stated that it was reasonable to expect that some patients would discontinue for reasons other than progression. In addition, in the ASCENT trial, patients received G-CSF to counteract neutropenia (which is a main side effect of sacituzumab govitecan). Publicly funded access to G-CSF is very limited in this patient population in Canada; therefore, it is possible that discontinuation may differ in the Canadian setting. Additionally, the differential dropout between treatment arms was of particular concern in the ASCENT trial and likely not generalizable to any real-world setting. Whether this may in turn impact PFS and OS is not known. Notably, CADTH was unable to validate the TTD data in the economic model with the sponsor’s submitted clinical study report because these data were neither reported nor provided to the CADTH clinical reviewers when requested.
CADTH revised the parametric survival distribution of TTD for sacituzumab govitecan and TPC to the gamma and Weibull distributions, respectively. In 1 scenario analysis, CADTH changed the parametric distribution of TTD for sacituzumab govitecan to the gamma distribution to explore the uncertainty of TTD over the extrapolated period.
Use of treatment-specific health state utility values is inappropriate: The sponsor assumed health state utilities for patients in PFS differed by treatment such that a lower utility value (0.626) was applied to patients who received TPC, and a higher utility value (0.710) was applied to patients who received sacituzumab govitecan. There are 2 main issues with the use of treatment-specific utility values. First, use of treatment-specific utility values is contradictory to CADTH’s Guidelines for the Economic Evaluation of Health Technologies: Canada, which recommends that utilities should reflect the health states in the economic model. Any differences by treatment should be transparently modelled and justified. Furthermore, the sponsor’s base case already incorporated disutilities due to AEs (which is the appropriate approach); therefore, there is limited justification for applying treatment-specific utilities. Second, there were no clinically meaningful differences in health-related quality of life between treatment arms for patients in PFS reported in the ASCENT trial; therefore, there is limited justification for the inclusion of these data in the economic model.
CADTH addressed this limitation by revising treatment-specific health state utility values for TPC to 0.710. In a scenario analysis, CADTH assessed the impact of lower health state utility values for patients in PFS for both treatment groups.
Relative use of each treatment in the TPC basket does not reflect usage in Canadian clinical practice: In the economic model, the sponsor assumed the relative use of each treatment comprising the TPC basket as follows: 53% on eribulin, 20% on vinorelbine, 14% on gemcitabine, and 13% on capecitabine. The distribution assumed by the sponsor overestimated drug acquisition costs for patients who received TPC. The clinical experts consulted by CADTH indicated that contrary to the sponsor’s estimates, eribulin is used less frequently among patients (estimated range between 25% and 40%) and capecitabine would be used at a higher frequency than assumed (approximately 25% to 40%).
CADTH revised the breakdown of the relative use of the individual treatments comprising the TPC basket to the following: 40% on eribulin, 20% on vinorelbine, 15% on gemcitabine, and 25% on capecitabine. In a scenario analysis, CADTH explored the impact of an alternate breakdown of the relative use of the individual treatments comprising the TPC basket by revising the proportion of patients on eribulin to 25% and capecitabine to 40%.
Relative dosing intensity (RDI) for patients who received sacituzumab govitecan was likely overestimated and did not align with clinical expectations: In the sponsor’s base-case analysis, both sacituzumab govitecan and TPC were administered at RDIs based on the ASCENT trial (94% and 100%, respectively), which were higher than expected in Canadian clinical practice, according to the clinical experts consulted by CADTH. The clinical experts consulted by CADTH noted that it is unlikely for either sacituzumab govitecan or TPC to be administered at a full dose and patients who receive TPC in clinical practice are unlikely to maintain such a high dose for the full duration of their therapy. The experts indicated that it is reasonable to assume both treatments would be administered at the same RDI (i.e., 90%) and they anticipated that patients on sacituzumab govitecan would maintain that dose. For patients who receive TPC, it is likely for patients to receive a dose reduction of approximately 50%. However, this approach of multiplying the RDI by the drug costs is problematic because RDI can be influenced by many factors. For instance, the dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation. Each of these reasons have differing impacts on drug costs. Furthermore, it is unclear how these assumptions interact with considerations about vial size and wastage, which were incorporated into the sponsor’s calculations of the per-cycle drug costs. Without explicitly modelling dose delays and reductions for the patient population, this method of multiplying RDI by drug acquisition costs contributes to uncertainty in the true drug costs incurred by payers.
CADTH revised the RDI for sacituzumab govitecan and TPC to reflect an RDI of 100%. In a scenario analysis, CADTH assumed a 90% RDI for sacituzumab govitecan and a 50% RDI for TPC, to align with expert feedback.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently (or in Appendix 4).
Relative use of concomitant medication for patients who received sacituzumab govitecan and TPC did not reflect concomitant medications used in Canadian clinical practice: In the economic model, the sponsor assumed that 47.2% and 19.8% of patients would receive concomitant medication with sacituzumab govitecan and TPC, respectively. Clinical experts consulted by CADTH indicated that immunostimulants are not widely used in Canadian clinical practice nor readily available. As such, a more reflective estimate for the proportion of patients receiving immunostimulants should be closer to 0%.
CADTH addressed this limitation by changing the proportion of patients assumed to receive immunostimulants concomitantly in both treatment arms to 0%.
Pricing of comparators (i.e., vinorelbine and gemcitabine) does not reflect current Canadian prices: In the economic model, the unit price of vinorelbine was $31.73 per mL for a 10 mg/mL vial and the unit price of gemcitabine was $1.14 per mL for a 38 mL vial; however, the sponsor’s assigned unit costs did not reflect Canadian pricing or the size of the product in the DeltaPA database7 and were underestimated.
CADTH reassigned unit costs to vinorelbine and gemcitabine based on current Canadian pricing from the DeltaPA database7 to $68.00 per mL in a 1 mL vial for vinorelbine (10 mg/mL) and $10.8120 per mL for gemcitabine (40 mg/mL), as per CADTH’s cost comparison table in Appendix 1.
Resource use associated with treatment administration and monitoring of sacituzumab govitecan is underestimated: The sponsor assumed that per-cycle administration costs for sacituzumab govitecan would be only slightly higher than TPC and that per-cycle monitoring costs would be reduced for sacituzumab govitecan relative to TPC. Feedback from the clinical experts consulted by CADTH and the CADTH participating drug plans indicated that these assumptions were unlikely to be aligned with clinical practice in Canada and that relevant costs related to sacituzumab govitecan monitoring and administration were not incorporated for sacituzumab govitecan.
CADTH could not assess the impact of this limitation given the sponsor’s model structure.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation Not Noted As Limitations to the Submission
Sponsor’s key assumption | CADTH comment |
|---|---|
The patient population in the model reflects the baseline characteristics of patients from the ASCENT trial expected to be treated in Canadian clinical practice. | Appropriate according to the clinical experts consulted for this review. |
A lifetime time horizon of 5 years. | Appropriate as this time horizon is adequate to capture all lifetime associated costs and outcomes for the indicated population. |
Utility decrements due to adverse events were captured separately as a 1-time occurrence for patients who are on treatment. | It is unclear whether incorporating this as a 1-time impact is appropriate. In the trial, patients experiencing adverse events could receive treatments to mitigate the adverse event. The same treatments to mitigate the adverse event may not be available in Canada. Furthermore, patients may experience more than 1 adverse event. As such, the utility decrements associated with sacituzumab govitecan may be underestimated. This is not expected to have a large impact on the overall QALYs. |
Distribution of subsequent treatments and proportion of patients receiving subsequent treatments. | Appropriate according to the clinical experts consulted for this review as this distribution aligned with that reported in the ASCENT clinical trial. |
Treatment duration of sacituzumab govitecan and individual treatments comprising TPC. | According to the clinical experts consulted by CADTH, if patients discontinue TPC due to disease progression, no further long-term benefit is expected, and patients are likely to switch to an alternate treatment. |
Mortality is assumed to be the same for patients in the PFS and PD health states to estimate newly progressed patients. | Uncertain. |
Subsequent treatments are assumed to begin upon progression and continue for fixed treatment durations independent of prior therapy received. Patients are assumed to be placed on subsequent treatments for palliative care only. | Likely appropriate. |
PD = progressive disease; PFS = progression-free survival; QALY = quality-adjusted life-year; TPC = treatment of physician’s choice.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook the reanalyses outlined in Table 5 to address, when possible, the limitations within the sponsor’s submitted economic model. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Overall survival for sacituzumab govitecan and TPC | Sacituzumab govitecan: KM + log-logistic TPC: KM + log-logistic | Sacituzumab govitecan: Weibull TPC: Weibull |
2. Progression-free survival for sacituzumab govitecan and TPC | Sacituzumab govitecan: KM + log-normal TPC: KM + log-normal | Sacituzumab govitecan: gamma TPC: log-logistic |
3. Time to treatment discontinuation for sacituzumab govitecan and TPC | Sacituzumab govitecan: KM + gamma TPC: exponential | Sacituzumab govitecan: gamma TPC: Weibull |
4. Health state utility values | Sacituzumab govitecan: 0.710 TPC: 0.626 | Sacituzumab govitecan: 0.710 TPC: 0.710 |
5. Relative use of each treatment among treatments of the TPC basket | Eribulin = 53% Vinorelbine = 20% Gemcitabine = 14% Capecitabine = 13% | Eribulin = 40% Vinorelbine = 20% Gemcitabine = 15% Capecitabine = 25% |
6. Relative dose intensity | Sacituzumab govitecan: 94% TPC: 100% | Sacituzumab govitecan: 100% TPC: 100% |
7. Concomitant medications | The proportion of patients assumed to receive immunostimulants concomitantly with sacituzumab govitecan and TPC were 47.2% and 19.8%, respectively | The proportion of patients assumed to receive immunostimulants concomitantly in both treatment arms was revised to 0% |
8. Pricing of comparators | Vinorelbine: $31.73 (for 10 mg/mL) Gemcitabine: $1.14 (for 40 mg/mL) | Vinorelbine: $68.00 (for 10 mg/mL) Gemcitabine: $10.8120 (for 40 mg/mL) |
CADTH base case | Reanalyses 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8 | |
KM = Kaplan-Meier; TPC = treatment of physician’s choice.
The results of these stepwise analyses can be found in Table 6. Results from the probabilistic analysis of the CADTH base case found that sacituzumab govitecan was associated with an incremental benefit of 0.31 QALYs and incremental costs of $116,613 compared with TPC. The ICER for sacituzumab govitecan versus TPC was $375,333 per QALY gained. Based on a willingness-to-pay threshold of $50,000, there is a 0% probability that sacituzumab govitecan would be the most cost-effective strategy.
The results were primarily driven by the drug acquisition cost of sacituzumab govitecan and the shortened incremental life-years gained based on the revised OS curves (Appendix 4, Table 10). This suggests that uncertainties in the extrapolation period remain key model drivers.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | TPCa | 52,186 | 0.54 | Reference |
Sacituzumab govitecan | 189,614 | 0.94 | 347,009 | |
CADTH reanalysis 1 | TPCa | 51,927 | 0.46 | Reference |
Sacituzumab govitecan | 189,752 | 0.81 | 398,100 | |
CADTH reanalysis 2 | TPCa | 52,227 | 0.54 | Reference |
Sacituzumab govitecan | 189,709 | 0.92 | 360,764 | |
CADTH reanalysis 3 | TPCa | 51,303 | 0.54 | Reference |
Sacituzumab govitecan | 174,509 | 0.94 | 311,100 | |
CADTH reanalysis 4 | TPCa | 52,186 | 0.57 | Reference |
Sacituzumab govitecan | 189,614 | 0.94 | 375,750 | |
CADTH reanalysis 5 | TPCa | 50,809 | 0.54 | Reference |
Sacituzumab govitecan | 189,614 | 0.94 | 350,488 | |
CADTH reanalysis 6 | TPCa | 52,186 | 0.54 | Reference |
Sacituzumab govitecan | 191,130 | 0.94 | 350,839 | |
CADTH reanalysis 7 | TPCa | 50,375 | 0.54 | Reference |
Sacituzumab govitecan | 177,832 | 0.94 | 321,835 | |
CADTH reanalysis 8 | TPCa | 53,386 | 0.54 | Reference |
Sacituzumab govitecan | 189,943 | 0.94 | 344,810 | |
CADTH base case | TPCa | 49,320 | 0.48 | Reference |
Sacituzumab govitecan | 165,933 | 0.79 | 375,333 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TPC = treatment of physician’s choice.
aReference product is the least costly alternative.
Scenario Analysis Results
CADTH undertook several scenario analyses on the CADTH base case to determine the impact of alternative assumptions on the cost-effectiveness of sacituzumab govitecan compared with TPC. These analyses included:
changing the parametric distribution of OS for sacituzumab govitecan to the Gompertz distribution
changing the parametric distribution of PFS for sacituzumab govitecan and TPC to the log-normal and gamma distributions, respectively
changing the parametric distribution of TTD for TPC to the gamma distribution
exploring an alternate distribution for the relative use of individual treatments within the TPC basket (i.e., 25% eribulin and 40% capecitabine)
exploring the impact of different RDI values on drug acquisition costs for sacituzumab govitecan and TPC: 90% and 50%, respectively
reducing the time horizon to 2 years (i.e., in line with the maximum duration of follow-up from the ASCENT trial).
The results of these analyses are presented in Appendix 4, Table 11. The ICER was most sensitive to the alternate parametric distributions of OS for sacituzumab govitecan (ICER = $420,046 per QALY gained) and to the time horizon (ICER = $454,335 per QALY gained); the ICER remained robust to scenarios 2 to 6.
CADTH undertook a series of price reduction analyses on the price of sacituzumab govitecan based on the sponsor’s submitted base case and CADTH’s base-case reanalyses (Table 7). The analyses indicate that a price reduction of 87% to 92% is required for sacituzumab govitecan to be considered cost-effective at $50,000 per QALY.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for sacituzumab govitecan vs. TPC ($/QALY gained) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 347,009 | 375,333 |
10% | 314,468 | 337,786 |
20% | 281,928 | 300,240 |
30% | 249,387 | 262,693 |
40% | 216,847 | 225,146 |
50% | 184,306 | 187,600 |
60% | 151,766 | 150,053 |
70% | 119,225 | 112,507 |
80% | 86,517 | 74,960 |
87% | 63,907 | 48,677 |
90% | 54,144 | 37,073 |
92% | 46,682 | 29,904 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TPC = treatment of physician’s choice; vs. = versus.
Issues for Consideration
The preparation of sacituzumab govitecan is labour intensive for pharmacy staff as multiple vial reconstitutions are required for a single dose. The final product stability is also very short, which will restrict the locations that can administer sacituzumab govitecan, as there must be a sterile compounding pharmacy onsite.
Treatment administration of sacituzumab govitecan is highly resource intensive. Administration occurs over 3 hours; patients must be under observation during this time and for at least 30 minutes following the initial dose for signs or symptoms of infusion-related reactions. If prior infusions were tolerated, the time to administer infusion may be reduced to a minimum of 1 hour; patients continue to need to be observed post-infusion.
Overall Conclusions
The evidence identified in the CADTH clinical review suggested that when compared with TPC, administration of sacituzumab govitecan (10 mg/kg on day 1 and day 8 of a 21-day treatment cycle) contributed to statistically significant and clinically meaningful prolongation of PFS and OS among patients with locally advanced TNBC or mTNBC who had received at least 2 prior therapies. Health-related quality of life data could not be interpreted due to the absence of formal statistical testing and high rates of missing data resulting from deaths and withdrawals. The clinical efficacy of sacituzumab govitecan beyond the median follow-up in the ASCENT trial (approximately 12 months) is uncertain.
The economic evaluation assessed the cost-effectiveness of sacituzumab govitecan compared with TPC for the treatment of adults with unresectable locally advanced TNBC or mTNBC who have received 2 or more prior therapies, at least 1 of them for metastatic disease. To address the identified limitations with the submitted model, CADTH revised the parametric distributions for OS, PFS, and TTD for both sacituzumab govitecan and TPC to reflect more clinically plausible values, revised the health state utility estimates for patients in PFS, revised the proportion of patients who received individual treatments among the TPC basket, and revised the relative dosing intensities of sacituzumab govitecan and TPC with values expected in Canadian clinical practice. Additionally, CADTH incorporated 2 minor changes to align the relative use of concomitant medications for patients who received sacituzumab govitecan and TPC in Canadian clinical practice and updated the pricing of comparators (i.e., vinorelbine and gemcitabine) to reflect the current Canadian prices.
CADTH’s reanalysis of the sponsor’s economic model estimated that sacituzumab govitecan was associated with 0.31 incremental QALYs and $116,613 incremental costs. The ICER for sacituzumab govitecan compared with TPC was $375,333 per QALY gained. CADTH’s findings are aligned with the sponsor’s findings. Based on CADTH’s reanalysis, the probability that sacituzumab govitecan is cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained is 0%. A price reduction of 87% for sacituzumab govitecan is required for it to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
The results were primarily driven by the treatment acquisition cost of sacituzumab govitecan and the incremental clinical benefit expected with sacituzumab govitecan over the model’s time horizon compared with TPC. Many of the uncertainties impacting the extrapolation could not be adequately addressed by CADTH (e.g., optimistic extrapolated OS and PFS distributions that are not consistent with clinical experts’ expectations) given the model structure and distributions available. The CADTH reanalyses suggested a small survival benefit of sacituzumab govitecan relative to TPC (i.e., 0.45 additional life-years), which supports the results of the ASCENT trial, which demonstrated a statistically significant difference in mortality between sacituzumab govitecan and TPC. The cost-effectiveness of sacituzumab govitecan compared with the individual treatments that comprised the TPC basket remains unknown at this time given the lack of comparative effectiveness evidence.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Trodelvy® (sacituzumab govitecan), 180 mg vials. Mississauga (ON): Gilead Sciences Canada, Inc.; 2021 Jun 30.
2.Trodelvy®sacituzumab govitecan, 180 mg lyophilized powder for solution for injection, for intravenous use [product monograph]. Morris Plains (NJ): Immunomedics, Inc.; 2021 Sep 24.
3.Beauchemin C, Letarte N, Mathurin K, Yelle L, Lachaine J. A global economic model to assess the cost-effectiveness of new treatments for advanced breast cancer in Canada. Journal of medical economics. 2016;19(6):619-629. PubMed
4.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Oct 6.
5.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Oct 6.
6.Clinical Study Report: IMMU-132-05. Sacituzumab govitecan, protocol IMMU-132-05. An international, multi-center, open-label, randomized, phase 3 trial of sacituzumab govitecan versus treatment of physician choice in patients with metastatic triple-negative breast cancer who received at least two prior treatments [internal sponsor's report]. Morris Plains (NJ): Immunomedics, Inc; 2020 Oct 26.
7.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Oct 6.
8.Paclitaxel. (BC Cancer Drug Manual). Vancouver (BC): BC Cancer Agency; 2012, revised 2019: http://www.bccancer.bc.ca/drug-database-site/Drug%20Index/Paclitaxel_monograph.pdf. Accessed 2021 Oct 6.
9.BC Cancer Protocol Summary for Neoadjuvant or Adjuvant Therapy for Breast Cancer using DOCEtaxel and Cyclophosphamide. Vancouver (BC): BC Cancer Agency; 2021: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Breast/BRAJDC_Protocol.pdf. Accessed 2021 Oct 6.
10.Cancer Care Ontario. Regimen Monograph: FEC-D, FEC-D+TRAS. FEC-D Regimen, Fluorouracil-EPIrubicin-Cyclophosphamide then DOCEtaxel; FEC-D+TRAS Regimen, Fluorouracil-EPIrubicin-Cyclophosphamide then DOCEtaxel-Trastuzumab. CCO Formulary 2021 Aug; https://www.cancercareontario.ca/sites/ccocancercare/files/FEC-DTRAS_BR_NADJ.pdf. Accessed 2021 Oct 6.
11.BC Cancer Protocol Summary for Palliative Therapy for Metastatic Breast Cancer using CISplatin and Gemcitabine. Vancouver (BC): BC Cancer Agency; 2007: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Breast/BRAVGEMP_Protocol.pdf. Accessed 2021 Oct 6.
12.Cancer Care Ontario. Regimen Monograph: CRBPDOCE Regimen, DOCEtaxel-CARBOplatin. CCO Formulary 2021 Mar; https://www.cancercareontario.ca/en/system/files_force/CRBPDOCE_GY_OV_A.pdf. Accessed 2021 Oct 6.
13.Cancer Care Ontario. Carboplatin - Provider Monograph. Drug Formulary [2019]; https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/43796. Accessed 2021 Oct 6.
14.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Trodelvy® (sacituzumab govitecan), 180 mg vials. Mississauga (ON): Gilead Sciences Canada, Inc.; 2021 Jun 30.
Appendix 1: Cost Comparison Table
Table 8: CADTH Cost Comparison Table for Metastatic Triple-Negative Breast Cancer
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | 28-day cost ($)a |
|---|---|---|---|---|---|---|
Monoclonal antibody | ||||||
Sacituzumab govitecan | 180 mg | Vial | $1,478.0000 per viala | 10 mg/kg once weekly on days 1 and 8 of continuous 21-day treatment cycles | 563.05 | 15,765 |
Single-agent chemotherapies | ||||||
Vinorelbineb | 10 mg/mL (in 1 mL) 10 mg/mL (in 5 mL) | Vial | 68.0000 | Continuous: 30 mg/m2 weekly Q3 weekly: 30 mg/m2 given on days 1 and 8 Q4 weekly: 30 mg/m2 given on days 1, 8, and 15 | 12.95 to 19.43 | 362 to 544 |
10 mg/mL (in 5 mL) | 80.0000 | |||||
Gemcitabine | 1,000 mg 2,000 mg 40 mg/mL (25 mL in 25 mL vial, or 50 mL in 50 mL vial) | Vial | 270.0000 540.0000 10.8120c | Q4W: 1,000 mg/m2 weekly for 3 weeks ± cisplatin 100 mg/m2 after infusion on Day 1 only Q3W: 1,250 mg/m2 weekly for 2 weeks ± cisplatin 100 mg/m2 after infusion on Day 1 only | 32.40 to 57.61 | 907 to 1,613 |
Capecitabine | 150 mg | Tablet | 0.4575 1.9478 | 1,250 mg/m2 twice daily for 14 days (Total daily dose 2,500 mg/m2) | 13.73 | 384 to 1,636 |
500 mg | 1.5250 6.4933 | |||||
Doxorubicind | 2 mg/mL (5 mL) 2 mg/mL (25 mL) 2 mg/mL (25 mL) 2 mg/mL (100 mL) 50 mg 150 mg | Vial | 10.0000 10.2000 11.4000 7.7000 360.3700 1,081.1000 | Q1W: 10-20 mg/m2 bolus Q3W: 60-75 mg/m2 bolus (40-60mg/m2 when used in combination) Q4W: 20-30 mg/m2/day bolus for 3 consecutive days | 10.68 to 64.82 | 300 to 1,815 |
Paclitaxel8 | 6 mg/mL (5 mL in 5 mL) 6 mg/mL (50 mL in 50 mL) | Vial Vial | 60.0000 74.8000 | 200 mg/m2 for one dose on day 1, every 3 weeks | 9.52 | 267 |
Carboplatine | 50 mg 150 mg 450 mg 600 mg | IV for solution | 70.0000 210.0000 599.9985 840.0000 | 750 to 900 mg | 50.00 to 60.00 | 1,400 to 1,680 |
Multi-agent chemotherapy regimens | ||||||
Docetaxel + Cyclophosphamide9 | ||||||
Docetaxel | 20 mg 80 mg 160 mg | Vial | 249.0000 497.0000 925.000 990.0000 1940.4000 1,850.0000 | 75 mg/m2 | 59.24 | 1,659 |
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 600 mg/m2 | 1.05 | 29 |
Docetaxel + Cyclophosphamidef | Every 21 days, for 4 cycles | 60.29 | 1,688 | |||
Docetaxel + Carboplatin | ||||||
Docetaxel | 20 mg 80 mg 160 mg | Vial | 249.0000 497.0000 925.000 990.0000 1940.4000 1,850.0000 | 75 mg/m2 | 59.24 | 1,659 |
Carboplatin | 50 mg 150 mg 450 mg 600 mg | Vial | 70.0000 210.0000 599.9985 840.0000 | 750 to 900 mg | 50.00 to 60.00 | 1,400 to 1,600 |
Docetaxel + Carboplatin | Every 21 days, for 6 cycles | 109.24 to 119.24 | 3,058 to 3,338 | |||
Fluorouracil-Epirubicin-Cyclophosphamide then Docetaxel (FEC-D) | ||||||
Fluorouracil | 50 mg/mL | Vial | 1.6090 1.9500 | 500 mg/m2 Day 1 | 28.96 | 39 |
5,000 mg/100 mL | Vial | 2.0000 | ||||
Epirubicin | 2 mg/mL (5mL) [10 mg] | Vial | 8.0240 9.5000 | 100 mg/m2 Day 1 | 7.80 | 10 |
2 mg/mL (25 mL) [50 mg] | 3.0000 8.0364 8.2800 | |||||
2 mg/mL (100 mL) [200 mg] | 8.2700 | |||||
50 mg | 4.9922 | |||||
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 500 mg/m2 Day 1 | 8.59 | 11 |
Docetaxel | 20 mg 80 mg 160 mg | Vial | 249.0000 497.0000 925.000 990.0000 1940.4000 1,850.0000 | 100 mg/m2 Day 1 (3 cycles) | 1,284.35 | 1,712 |
Fluorouracil + Epirubicin + Cyclophosphamide then Docetaxel (FEC-D)10 | FEC100 for 3 cycles, then docetaxel for 3 cycles | 1,254.47 (Total Day 1 Cost) | 1,773 | |||
Adriamycin + Cyclophosphamide, then Paclitaxel (ACP) | ||||||
Doxorubicin (Adriamycin) | 2 mg/mL (5 mL) [10 mg] 2 mg/mL (25 mL) [50 mg] 2 mg/mL (50 mL in 1x50mL) [100 mg] 2 mg/mL (100 mL) [200 mg] 150 mg | Vial | 10.0000 10.0900 11.4661 10.2000 11.4000 11.9245 26.0840 7.7000 9.7300 8.2898 | 60 mg/m2 Day 1 (first 4 cycles) | 560.00 | 747 |
Cyclophosphamide | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 600 mg/m2 Day 1 (first 4 cycles) | 10.50 | 14 |
Paclitaxel | 6 mg/mL | Vial | 60.0000 74.8000 | 175 mg/m2 Day 1 (last 4 cycles) | 3,120.00 | 4,160 |
Adriamycin + Cyclophosphamide then Paclitaxel (ACP) | AC for 4 cycles, then Paclitaxel for 4 cycles | 3,690.50 | 4,920 | |||
Cisplatin + Gemcitabine11 | ||||||
Cisplatin | 1 mg/mL (50mL) 1 mg/mL (100 mL) | Vial | 6.4600 2.7000 | 30 mg /m2 days 1 and 8, every 28 days | 0.62 | 17 |
Gemcitabine | 1,000 mg 2,000 mg 40 mg/mL (25 mL in 25 mL vial, or 50 mL in 50 mL vial) | Vial | 270.0000 540.0000 10.8120b | 750 mg/m2 days 1 and 8 | 17.49 | 490 |
Cisplatin + Gemcitabine | Every 21 days until disease progression. | 18.11 | 506 | |||
Carboplatin + Gemcitabine12 | ||||||
Carboplatin | 50 mg | Vial | 70.0000 | AUC 5 | 50.00 | 1,400 |
150 mg | 210.0000 | |||||
450 mg | 599.9985 | |||||
600 mg | 840.0000 | |||||
Gemcitabine | 1,000 mg 2,000 mg 40 mg/mL (25 mL in 25 mL vial) 40 mg/mL (50 mL in 50 mL vial) | Vial | 270.0000 540.0000 10.8120b | 1,000 to 1,250 mg /m2 | 23.15 to 28.80 | 648 to 806 |
Carboplatin + Gemcitabine | Every 21 days. For a usual total of 4 to 6 cycles | 73.15 to 78.80 | 2,048 to 2,206 | |||
AUC = area under the free carboplatin plasma concentration versus time curve; BCCA = British Columbia Cancer Agency; CCO = Cancer Care Ontario; GFR = glomerular filtration rate; Q1W = every week; Q3W = every 3 weeks; Q4W = every 4 weeks.
Note: The comparators presented in this table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans. All prices are from the IQVia DeltaPA database (accessed August 23, 2021), unless otherwise indicated, and do not include dispensing fees. Assumes patient weight of 71.09 kg and BSA = 1.78 m2 as per sponsor’s submission. All dosing regimens are based on regimen monographs from Cancer Care Ontario (CCO) unless otherwise stated. Eribulin was not included in the cost comparison table given no list price was available for public drug formularies (the list price for eribulin in the sponsor’s submitted model was reflective of AQPP [Quebec wholesale] pricing).
aSponsor-submitted price.
bRange of drug costs calculated based on continuous regimen (minimum) and Q3 (maximum). Dosing regimen is dependent on the protocol, which is not specified.
cPrice per mL.
dRange of drug costs calculated based on minimum and maximum doses (i.e., low is Q4W and high is Q1W).
eAs per CCO product monograph for Carboplatin13: Target AUC is 5 to 6. Carboplatin is dosed according to the following formula: Maximum carboplatin dose (mg) = target AUC (mg/mL per min) × (125 + 25); maximum dose is based on a capped GFR estimate at 125 mL/min for patients with normal renal function.
fEvery 21 days.9
gBCCA protocol.11
Note this appendix has not been copy-edited.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Transparency was limited by the number of hidden sheets and cells throughout the model, increasing time required to validate the model. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The probabilistic analysis was coded such that the model would report the same ICER run after run. CADTH was able to revise the parameters sheet to allow the model to run probabilistically without recoding the VBA, but for ease of reproduction, presented the results based on the sponsors model after ensuring the stability of the model. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Sacituzumab govitecan | TPC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 1.204 | 0.746 | 0.457 |
LYs in PFS | 0.585 | 0.263 | 0.322 |
LYs in PD | 0.618 | 0.483 | 0.135 |
Discounted QALYs | |||
Total QALYs | 0.793 | 0.482 | 0.311 |
QALYs in PFS | 0.415 | 0.187 | 0.228 |
QALYs in PD | 0.383 | 0.299 | 0.084 |
QALYs loss: AE disutility | –0.005 | –0.003 | –0.002 |
Discounted costs ($) | |||
Total | $165,933 | $49,320 | $116,613 |
Drug Acquisition | $116,655 | $5,111 | $111,544 |
Drug Administration | $1,390 | $345 | $1,045 |
Concomitant Medication | $1,079 | $225 | $854 |
Subsequent Treatment | $2,599 | $3,211 | -$611 |
Disease Management | $35,757 | $34,622 | $1,135 |
PFS | $1,788 | $795 | $993 |
PD | $1,929 | $1,505 | $424 |
Terminal Care | $32,039 | $32,321 | -$282 |
Monitoring | $1,353 | $1,223 | $130 |
PFS | $336 | $430 | -$94 |
PD | $1,017 | $793 | $224 |
AE management | $7,100 | $4,583 | $2,518 |
ICER ($/QALY) | 375,333 | ||
AE = adverse events; ICER = incremental cost-effectiveness ratio; LY= life-year; PF = progressed disease; PFS = progression-free survival; QALY = quality-adjusted life-year; SG = sacituzumab govitecan; TPC = treatment of physician’s choice.
Scenario Analyses
Table 11: Scenario Analyses for Sacituzumab Govitecan Versus TPC
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Scenario 1: OS parametric distribution for sacituzumab govitecan: Gompertz | TPC | 49,320 | 0.482 | Reference |
Sacituzumab govitecan | 165,691 | 0.759 | 420,046 | |
Scenario 2: PFS parametric distributions: Sacituzumab govitecan: Log-Normal TPC: Gamma | TPC | 49,340 | 0.482 | Reference |
Sacituzumab govitecan | 165,559 | 0.799 | 366,570 | |
Scenario 3: TTD parametric distributions for TPC: Gamma | TPC | 49,321 | 0.482 | Reference |
Sacituzumab govitecan | 165,933 | 0.793 | 375,328 | |
Scenario 4: Relative use of individual treatments within the TPC basket: 25% Eribulin and 40% Capecitabine | TPC | 47,814 | 0.482 | Reference |
Sacituzumab govitecan | 165,933 | 0.793 | 380,177 | |
Scenario 5: Relative dosing intensities: Sacituzumab govitecan: 90% TPC: 50% | TPC | 46,462 | 0.482 | Reference |
Sacituzumab govitecan | 159,480 | 0.793 | 363,759 | |
Scenario 6: Time horizon revised to 2 years | TPC | 48,266 | 0.474 | Reference |
Sacituzumab govitecan | 158,425 | 0.717 | 454,335 |
SG = sacituzumab govitecan; TPC = treatment of physician’s choice.
Appendix 5: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 12: Summary of Key Take-Aways
Key Take-Aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor assessed the budget impact of the introduction of sacituzumab govitecan compared with TPC (capecitabine, eribulin, vinorelbine, or gemcitabine) for adult patients with mTNBC, from the perspective of the public drug plan in the Canadian setting (excluding Quebec) over a 3-year time horizon.14 The sponsor’s submission only considered drug acquisition costs. In the reference scenario, the sponsor assumed that patients would be eligible to receive single-agent chemotherapies. In the new drug scenario, sacituzumab govitecan was assumed to proportionally displace market shares of the various chemotherapy treatments.14
The sponsor estimated the eligible population size using an epidemiological which was derived via several assumptions and inputs to first estimate the incident population (i.e., de novo metastatic population) and the prevalent population (i.e., those who progressed to mTNBC from earlier disease stages), respectively.14
The sponsor’s BIA also included the following key assumptions:
Key inputs to the BIA are documented in Table 13.
In the reference scenario, the sponsor assumed that 12.5% of patients were assumed to be enrolled in clinical trials.
In the new drug scenario, sacituzumab govitecan is expected to capture market share proportionately from all treatments in the reference scenario, with no capture from clinical trials. The introduction of sacituzumab govitecan is not expected to expand the market.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Annual incidence of breast cancer | 0.073% |
Proportion of patients with de novo metastases | 4.9% |
Proportion of patients with mTNBC among those with de novo metastases | 11.4% |
5-year breast cancer prevalence | 0.35% |
Annual probability of distant recurrence | 2.9% |
Proportion with mTNBC | 17.1% |
Proportion with systemic therapy prior to metastasis | 100% |
Stratification by prior systemic therapy | 633 |
Systemic therapy prior to metastasis (i.e., prevalent population) | 514 |
No systemic therapy prior to metastasis (i.e., incident population) | 119 |
Attrition rates by line of therapy Proportion receiving 1L treatment Proportion receiving 2L treatment Proportion receiving 3L treatment | 80% 74% 77% |
Proportion receiving full covered (i.e., drug plan eligible) | 100% |
Number of patients eligible for drug under review 2L mTNBC 3L mTNBC | 605 / 613 / 622 308 / 312 / 316 298 / 302 / 306 |
Market uptake (3 years) | |
Uptake (reference scenario) Capecitabine Eribulin Vinorelbine Gemcitabine Carboplatin Clinical trials | 11.0% / 11.0% /11.0% 46.5% / 46.5% / 46.5% 17.3% / 17.3% / 17.3% 12.7% / 12.7% / 12.7% 0% / 0% / 0% 12.5% / 12.5% / 12.5% |
Uptake (new drug scenario) Sacituzumab govitecan Capecitabine Eribulin Vinorelbine Gemcitabine Carboplatin Clinical trials | |||||| |||||| |||||| |||||| |||||| |||||| |||||| |
Cost of treatment (per patient) | |
Cost of treatment over year 1 Sacituzumab govitecan Capecitabine Eribulin Vinorelbine Gemcitabine Carboplatin | $75,406.15a $362.05 $5,087.41 $1,076.16 $197.62 $1,296.87 |
mTNBC = metastatic triple-negative breast cancer; 1L = first-line; 2L = second-line; 3L = third-line.
aSponsor’s calculated annual treatment cost for sacituzumab govitecan based on a 21-day treatment cycle and considered a treatment duration of 4.4 months, which differs from CADTH’s calculated annual treatment cost of sacituzumab govitecan.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base-case analysis under the drug plan perspective estimated that the introduction of sacituzumab govitecan in patients with mTNBC would result in an incremental budget impact of $6.6M in Year 1, $15.5M in Year 2 and $20.3M in Year 3, for a total budget impact of $42.4 over the 3-year time horizon.14
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The estimated eligible population for treatment with sacituzumab govitecan is uncertain: The sponsor undertook an epidemiological approach to estimate the size of the population eligible for sacituzumab govitecan. This required assessing the published literature and applying several assumptions to derive estimates for the incident and prevalent populations in a multi-step approach. The clinical experts consulted by CADTH indicated that the estimate of the target population derived from the sponsor’s assumptions and inputs may be associated with some uncertainty. First, the approximated total prevalent population (i.e., a total of 514 TNBC patients who progress to metastatic disease annually) was likely underestimated. Second, CADTH’s clinical experts noted uncertainty in several of the sponsor’s assumptions of attrition rates by line of therapy. The experts noted that among those who received first-line treatment, the proportion of patients who were assumed to progress and receive second-line treatment (74%), was likely underestimated, and among those who received second-line treatment, the proportion of patients who were assumed to progress and receive third-line treatment (77%) was likely overestimated. Based on the above estimates, CADTH’s clinical experts felt that the final population size was uncertain and likely underestimated with that in Canadian clinical practice.
CADTH did not address this limitation. In a scenario analysis, CADTH arbitrarily explored the impact of (i) an increase in the prevalent population by 25%; (ii) changing the proportion of patients who were assumed to progress and receive second-line treatment to 80%; and (iii) changing the proportion of patients who were assumed to progress and receive third-line treatment to 70%.
The anticipated uptake of sacituzumab govitecan in the new drug scenario is uncertain: The sponsor anticipated that sacituzumab govitecan would capture | ||%, || |% and || |% of the market share distribution in years 1, 2, and 3. The clinical experts consulted by CADTH described that the sponsor’s anticipated uptake is likely underestimated over the 3-year time horizon and that the market uptake would likely be higher across all years, particularly in years 2 and 3. CADTH revised the market share uptake of sacituzumab govitecan across years 1, 2, and 3 to 25%, 50%, and 85%, to align with experts’ feedback.
CADTH addressed this limitation by revising the market shares in the new drug scenario to 25%, 50%, and 85% in years 1, 2, and 3.
Pricing of comparators do not reflect Canadian prices, and dosing-related inputs (weight and BSA) are misaligned with the sponsor’s submitted pharmacoeconomic analysis: In the sponsor’s submitted budget impact analysis, the unit price of vinorelbine was $31.73 per mL for a 10 mg/mL vial and the unit price of gemcitabine was $1.14 per mL for a 38 mL vial, however, the sponsor’s assigned unit costs did not reflect Canadian pricing or available product in the DeltaPA database7 and were underestimated. Additionally, the sponsor assumed a patient weight of 69 kg and a BSA of 1.72 m2 which did not align with these inputs in the pharmacoeconomic analysis.
CADTH addressed this limitation by re-assigning unit costs to vinorelbine and gemcitabine based on current Canadian pricing from the DeltaPA database7 to $68.00 per mL in a 1 mL or 5 mL vial for vinorelbine (10 mg/mL) and $10.8120 per mL for gemcitabine (40 mg/mL as per CADTH’s cost comparison table in Appendix 1). CADTH further adjusted the units of drug per dose to align with the dosing in the pivotal trial; and revised the patient weight and BSA to align with the values used in the pharmacoeconomic analysis (71.09 kg and 1.78 m2).
CADTH Reanalyses of the BIA
A table noting the changes made to the sponsor’s BIA as part of the CADTH reanalysis is available in Table 14.
Table 14: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market share estimate in the new drug scenario in years 1, 2, and 3 | ||||||% / ||||||% / ||||||% | 25% / 50% / 85% |
2. Pricing of comparator treatments and medication dosing inputs | Vinorelbine: $31.73 per mL (for 10 mg/mL vial); 6 units per dose Gemcitabine: $1.14 (for 38 mg/mL); 46 units per dose Patient weight = 69 kg BSA = 1.72 m2 | Vinorelbine: $68.00 (for 10 mg/mL vial); 2 units per dose Gemcitabine: $10.8120 (for 40 mg/mL); 45 units per dose Patient weight = 71.09 kg BSA = 1.78 m2 |
3. Market share estimates in the reference scenario. | Capecitabine = 11.0% Eribulin = 46.5% Vinorelbine = 17.3% Gemcitabine = 12.7% Carboplatin = 0% Clinical trials = 12.5% | Capecitabine = 25% Eribulin = 40% Vinorelbine = 20% Gemcitabine = 15% Carboplatin = 0% Clinical trials = 0% |
CADTH base case | Reanalyses 1 + 2 + 3 | |
BSA = body surface area.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16.
Table 15: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $42,386,936 |
CADTH reanalysis 1 | $72,645,328 |
CADTH reanalysis 2 | $42,264,270 |
CADTH reanalysis 3 | $42,748,903 |
CADTH base case | $72,879,531 |
BIA = budget impact analysis.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $1,561,364 | $1,582,673 | $1,604,287 | $1,626,212 | $4,813,173 |
New drug | $1,561,364 | $8,156,603 | $17,152,948 | $21,890,558 | $47,200,109 | |
Budget impact | $0 | $6,573,931 | $15,548,660 | $20,264,345 | $42,386,936 | |
CADTH base case | Reference | $1,545,520 | $1,566,613 | $1,588,008 | $1,609,711 | $4,764,332 |
New drug | $1,545,520 | $12,740,364 | $24,161,313 | $40,742,186 | $77,643,864 | |
Budget impact | $0 | $11,173,751 | $22,573,305 | $39,132,475 | $72,879,531 |
BIA = budget impact analysis.
CADTH conducted the following additional scenario analyses from the drug plan perspective (Scenarios 1 to 5, Table 17):
Arbitrarily increasing the total proportion of patients who were assumed to progress and receive second-line treatment to 80% (i.e., 82% of those who received systemic therapy prior to metastasis and 72% of those who had no systemic therapy prior to metastasis).
Arbitrarily decreasing the proportion of patients who were assumed to progress and receive third-line treatment to 70% (i.e., 70% of those who received systemic therapy prior to metastasis and 70% of those who had no systemic therapy prior to metastasis).
Exploring the impact of an increase in the prevalent population by 25%.
Exploring the impact of a decrease in the prevalent population by 25%.
Mean duration of treatment with sacituzumab govitecan is increased from 4.4 months to 4.6 months.
Applied an 87% reduction in the price of sacituzumab govitecan to align with the point at which the ICER is within the willingness-to-pay threshold of $50,000 per QALY in the CADTH economic base case.
The model results were most sensitive to changes in the population size.
Table 17: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1 | Reference | $1,646,186 | $1,668,653 | $1,691,442 | $1,714,558 | $5,074,652 |
New drug | $1,646,186 | $13,570,195 | $25,735,036 | $43,395,888 | $82,701,119 | |
Budget impact | $0 | $11,901,542 | $24,043,595 | $41,681,330 | $77,626,467 | |
CADTH scenario analysis 2 | Reference | $1,477,315 | $1,497,476 | $1,517,928 | $1,538,673 | $4,554,077 |
New drug | $1,477,315 | $12,178,117 | $23,095,046 | $38,944,186 | $74,217,349 | |
Budget impact | $0 | $10,680,640 | $21,577,119 | $37,405,513 | $69,663,272 | |
CADTH scenario analysis 3 | Reference | $1,892,852 | $1,918,685 | $1,944,888 | $1,971,468 | $5,835,041 |
New drug | $1,892,852 | $15,603,561 | $29,591,189 | $49,898,353 | $95,093,104 | |
Budget impact | $0 | $13,684,876 | $27,646,301 | $47,926,885 | $89,258,063 | |
CADTH scenario analysis 4 | Reference | $1,198,189 | $1,214,541 | $1,231,128 | $1,247,954 | $3,693,623 |
New drug | $1,198,189 | $9,877,167 | $18,731,437 | $31,586,019 | $60,194,624 | |
Budget impact | $0 | $8,662,626 | $17,500,309 | $30,338,065 | $56,501,000 | |
CADTH scenario analysis 5 | Reference | $1,545,520 | $1,566,613 | $1,588,008 | $1,609,711 | $4,764,332 |
New drug | $1,545,520 | $13,258,943 | $25,212,636 | $42,553,861 | $81,025,441 | |
Budget impact | $0 | $11,692,330 | $23,624,628 | $40,944,150 | $76,261,109 | |
CADTH scenario analysis 6 | Reference | $1,545,520 | $1,566,613 | $1,588,008 | $1,609,711 | $4,764,332 |
New drug | $1,545,520 | $2,814,758 | $4,038,989 | $6,066,731 | $12,920,478 | |
Budget impact | $0 | $1,248,145 | $2,450,981 | $4,457,020 | $8,156,146 |
BIA = budget impact analysis.
Note: All scenario analyses are conducted based on the CADTH base case undertaken from the drug program plan perspective.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.