CADTH Reimbursement Review
Enfortumab Vedotin (Padcev)
Sponsor: Seagen Canada Inc.
Therapeutic area: Locally advanced or metastatic urothelial carcinoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ADC
antibody-drug conjugate
AE
adverse event
AESI
adverse event of special interest
BCC
Bladder Cancer Canada
CI
confidence interval
CPI
checkpoint inhibitor
CR
complete response
DAC
Drug Advisory Committee
DCR
disease control rate
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ-5D-5
EuroQol 5-Dimensions 5-Levels questionnaire
EQ VAS
EuroQol Visual Analogue Scale
FAS
full analysis set
FGFR
fibroblast growth-factor receptor gene
HCRU
health care resource utilization
HR
hazard ratio
HRQoL
health-related quality of life
IPCW
inverse probability of censoring weights
IRR
infusion-related reactions
MIBC
muscle-invasive bladder cancer
MMAE
monomethyl auristatin E
MMRM
mixed model for repeated measures
NMIBC
non–muscle-invasive bladder cancer
ORR
overall response rate
OS
overall survival
PAG
Provincial Advisory Group
PD-1
programmed death receptor 1
PD-L1
programmed death ligand 1
PFS
progression-free survival
PFS1
progression-free survival on study therapy
PFS2
progression-free survival on subsequent therapy
PR
partial response
PRO
patient-reported outcome
QoL
quality of life
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
RES
response evaluable set
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TURBT
transurethral resection of the bladder tumour
UC
urothelial carcinoma
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Enfortumab vedotin, 1.25 mg/kg, 20 mg and 30 mg single-use vials for IV infusion. |
Indication | For the treatment of adult patients with unresectable locally advanced or metastatic urothelial cancer who have previously received a platinum-containing chemotherapy and programmed death receptor 1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor therapy. |
Reimbursement request | For the treatment of patients with locally advanced or metastatic urothelial cancer who have previously received a PD-1 or PD-L1 inhibitor and who: Have received a platinum-containing chemotherapy in the neoadjuvant, adjuvant, locally advanced, or metastatic setting. |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | October 29, 2021 |
Sponsor | Seagen Canada Inc. |
NOC = Notice of Compliance; PD-1 = programmed death receptor 1; PD-L1 = programmed death ligand 1.
Introduction
Bladder cancer is the fifth most common cancer in Canada, with approximately 12,200 cases, resulting in an estimated 2,600 deaths in 2020. Urothelial carcinoma (UC) is the most common type of bladder cancer, accounting for 90% to 95% of cases.1-4 Urothelial cancer typically arises in the bladder but may develop in any location lined with urothelium, including the renal pelvis, ureter, urethra, and prostatic urethra.5 Age, tobacco use, chemical carcinogens, family history, arsenic exposure, and use of indwelling catheters are known risk factors for bladder cancer. Bladder cancer is more common in males, although the reason is unknown.6-9 The most common presentation of UC is visible or microscopic hematuria. Other symptoms of UC include painful urination, back or flank pain, fatigue, and unexplained weight loss.4 Approximately 15% of patients have locally advanced or metastatic UC at presentation.10 In Canada, the 5-year net survival rate for bladder cancer is 75%; however, there are no Canadian-specific survival statistics for locally advanced or metastatic UC patients. The estimated 5-year relative survival for patients with metastatic disease is approximately 6.4%,7 and up to 15% when treated with contemporary regimens.11,12
Standard of care for locally advanced or metastatic UC consists of platinum-based chemotherapy, mainly gemcitabine plus cisplatin. In cisplatin-ineligible patients, gemcitabine plus carboplatin is recommended.10 Recently, avelumab was granted a conditional listing by CADTH for the first-line maintenance treatment of locally advanced or metastatic UC in patients whose disease has not progressed following first-line platinum-based induction chemotherapy.13 In patients who progress following platinum-based chemotherapy, treatment with checkpoint inhibitor (CPI) immunotherapy, preferably pembrolizumab, is recommended as second-line systemic therapy.14,15 Following failure of platinum chemotherapy and CPIs, or when CPIs are unavailable, salvage chemotherapy with taxanes is recommended, with paclitaxel or docetaxel preferred for most patients.10
Enfortumab vedotin is an antibody-drug conjugate (ADC) consisting of a fully human immunoglobulin G1Κ antibody and the microtubule-disrupting drug monomethyl auristatin E (MMAE). It acts via a protease-cleavable linker directed against nectin-4,16 which is an adhesion protein located on the surface of UC cells. Non-clinical data suggest that enfortumab vedotin binds cells expressing nectin-4, resulting in internalization of the ADC–nectin-4 complex and intracellular release of MMAE via proteolytic cleavage, inducing apoptosis through a disrupted microtubule network.17 The MMAE released through cellular apoptosis can further diffuse into nearby cells expressing low levels of nectin-4, resulting in cytotoxic cell death.18
Enfortumab vedotin is currently under review by Health Canada for the treatment of patients with locally advanced or metastatic UC who have previously received a programmed death receptor 1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and who have received a platinum-containing chemotherapy in the neoadjuvant, adjuvant, locally advanced, or metastatic setting or who are not eligible for cisplatin-containing chemotherapy.18
The Health Canada Notice of Compliance was expected on November 2, 2021. Enfortumab vedotin has not been previously reviewed by CADTH.
The objective of the current review is to perform a systematic review of the beneficial and harmful effects of enfortumab vedotin in patients with locally advanced or metastatic UC who have previously received a platinum-containing chemotherapy and a PD-1 or PD-L1 inhibitor.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group, Bladder Cancer Canada (BCC) provided input for the review of enfortumab vedotin in patients with locally advanced or metastatic UC. Bladder Cancer Canada is a nationally registered Canadian charity and is the first and only Canadian patient advocacy organization dedicated to bladder cancer issues. Supported by a Medical Advisory Board and a Medical Research Board consisting of the top bladder cancer specialists across Canada, its mission is to help bladder cancer patients and their support teams address the day-to-day issues of this disease; increase awareness among the public and medical community; and fund research into the diagnosis, treatment, and elimination of the disease. The organization’s vision is patient support, awareness, and research to create a world where bladder cancer is “just a memory.”
The information provided by BCC was gathered through an online survey and telephone interviews conducted between May 27 and June 11, 2021. Most survey respondents were from Canada, with a small number from the US. Telephone interviews with 2 patients from Canada who had experience with enfortumab vedotin were also conducted in June 2021. In total, 38 patients diagnosed with stage II or higher muscle-invasive bladder cancer (MIBC), of whom one-third reported living with locally advanced or metastatic bladder cancer, and 6 caregivers completed the survey.
Many patients and caregivers reported that symptoms of bladder cancer including fatigue, lack of sleep, and loss of strength and stamina were problematic but manageable, while some patients indicated that having bladder cancer has had a minimal impact on their day-to-day lives. Additional symptoms including blood in the urine, pain in the abdomen and bones, decreased mobility and difficulty and/or pain when urinating were also commonly reported. Frequent need for urination and loss of control, urostomy and catheter management, and urinary tract infections were the most commonly reported issues related to continence that affect the day-to-day life of patients and result in the need for additional planning, discomfort, and time lost. Financial impacts related to the costs of catheters and urostomy supplies that are not covered by some provincial governments were reported to strain the already limited financial resources of patients and caregivers.
Patients reported experiencing a number of side effects with current treatments, including fatigue, constipation, low blood cell count, loss of appetite, neuropathy, nausea, vomiting, hair loss, insomnia, diarrhea, and mouth sores. While most patients identified minimal barriers to accessing treatment for their bladder cancer; some mentioned they did have difficulties due to travel distances, treatment costs, unavailability of treatment in Canada, no access to a physician, and the need for time off work to receive treatment. Two patients had experience with enfortumab vedotin through a clinical trial. Patients noted that side effects of treatment with enfortumab vedotin were temporary and manageable compared to previous treatments received. When asked what key benefits provided by enfortumab vedotin have been important to them as patients, they said that the treatment has given them their “life back again” — allowing them to resume activities that they enjoy. Patients with experience with enfortumab vedotin emphasized the importance of publicly funded access to this treatment.
Overall, patients and caregivers expressed a desire for fewer and less-severe side effects than those experienced with current bladder cancer treatments, as well as treatments that induce remission or are curative. Specifically, patients described an ideal treatment as one that would slow or stop disease progression, recurrence and spread; reduce pain, fatigue, and impaired sexual function; increase energy levels and strength; improve mental health, continence, and urination control; and result in fewer or no infections and avoidance of surgery.
Clinician Input
Input From Clinical Experts Consulted by CADTH
In patients with incurable locally advanced or metastatic UC, the clinical expert identified an unmet need for an effective third-line treatment option after progression with platinum chemotherapy and a PD-1 or PD-L1 inhibitor. The mainstay of treatment for incurable patients is cytotoxic platinum-based chemotherapy with gemcitabine with or without cisplatin. Maintenance avelumab was reported to show an overall survival (OS) benefit and is likely to become a funded standard of care. Pembrolizumab is now a funded second-line standard of care in Canada following demonstration of a survival benefit after progression despite first-line chemotherapy, displacing second-line taxane therapy. The only options following immunotherapy are paclitaxel and docetaxel, which are associated with modest response rates and treatment duration; enfortumab vedotin would therefore provide a new alternative to taxane therapy. The clinical expert noted that identifying patients who would respond to enfortumab could not be done, and that patients at this stage are typically under the care of expert medical oncologists, who would be able to identify progressive disease to initiate new treatment. Response to treatment would rely on improvement in symptoms, which would be assessed before treatment and/or evidence of objective tumour shrinkage on imaging. The clinical expert also stated that there are additional adverse events (AEs) with enfortumab vedotin that may require assessment by ophthalmologists or dermatologists.
Clinician Group Input
Two clinicians from the Ontario Health (Cancer Care Ontario) Genitourinary Cancer Drug Advisory Committee (DAC) and a group of 17 Canadian physicians who treat bladder cancer and who, with the support of BCC, provided input for this review. The DAC provides timely evidence-based clinical and health-system guidance on drug-related issues in support of Ontario Health’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. The group of Canadian physicians represents the specialty from across Canada in both academic and community settings and shares BCC’s goal of improving the management of bladder cancer.
The clinicians agreed that a standard of care for patients with advanced urothelial cancer post–platinum chemotherapy and post-immunotherapy is an unmet need in these patients. Enfortumab vedotin is indicated in a third-line setting. Alternative third-line options would be non-platinum chemotherapy, for which there is little evidence of efficacy and for which the toxicity rate is much higher, or fibroblast growth-factor receptor gene (FGFR)-targeted therapy, which would not be favoured due to the lack of FGFR testing in Canada. The experts agreed that enfortumab vedotin will redefine the current treatment paradigm as there are no other beneficial therapies in this setting other than taxanes, which are associated with significant toxicity, Offering enfortumab vedotin to all eligible patients would provide them with hope for improved life expectancy with tolerable side effects. The clinician group stated that patients would be assessed for toxicity and clinical progression every month throughout treatment, with imaging every 2 to 3 months. Blood work should be performed before each treatment cycle, and patients should be seen by their treating oncologist following each cycle. Patients with disease that has metastasized to the bones should also have a bone scan. The clinician group noted that decisions to discontinue treatment should be made in consultation with the patient and would include progressive disease, worsening symptoms, severe AEs, deterioration to end of life, dose-limiting toxicity resulting in intolerable adverse effects such as significant neuropathy, and patient wishes to discontinue treatment for any number of personal reasons.
Although no marked experience with enfortumab vedotin was mentioned, the clinicians consider this drug of great importance in the management of bladder cancer, filling an unmet need for patients requiring treatment following progression on platinum-based chemotherapy and immunotherapy. The approval of enfortumab vedotin would give medical oncologists an option to offer to patients with advanced urothelial cancer that has progressed on first- and second-line therapy. Enfortumab vedotin offers significant OS benefits compared to taxane chemotherapy, with tangible benefits for patients. Enfortumab vedotin would offer a longer life expectancy with preservation of quality of life (QoL) as the drug is generally well tolerated. For a patient population with such a poor prognosis, the inclusion of enfortumab vedotin in the treatment algorithm has the potential to significantly improve the outcomes associated with bladder cancer.
Drug Program Input
Input was obtained from the provinces (ministries of health and/or cancer agencies) participating in CADTH reimbursement reviews. The Provincial Advisory Group (PAG) noted that taxanes (paclitaxel and docetaxel) are the most relevant comparators in this setting and are funded and available in all provinces. They noted that an updated algorithm for metastatic UC would help jurisdictions navigate funding as there are now several therapies available for multiple lines of treatment, increasing the complexity of funding.
The PAG identified several factors that could affect implementation. The main concerns of the PAG involved indication creep; they were interested to know if patients who have not received previous platinum-based chemotherapy, or those who did but did not receive PD-1 or PD-L1 inhibitors, should be treated with enfortumab vedotin. The clinical expert explained that these patients would not generally be eligible; however, there would be instances in which these patients may receive enfortumab vedotin. Additionally, the PAG was interested in whether patients who discontinue immunotherapy due to toxicity should be offered enfortumab vedotin before disease progression, and if patients currently being treated with taxanes or alternative chemotherapies should be switched to enfortumab vedotin at the time of public funding or wait until their disease progresses. The clinical expert noted that initiation of enfortumab vedotin should follow the criteria of Study EV-301, in which patients who discontinued CPI treatment due to toxicity were eligible provided they had evidence of disease progression following discontinuation, and that if current treatment options are working, there would be no reason to switch to enfortumab vedotin until disease progression. However, this is up to the treating physician, as well as the patient.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Study EV-301 was a global, open-label, phase III randomized controlled trial (RCT) comparing enfortumab vedotin to standard salvage chemotherapy regimens in adults with locally advanced or metastatic UC who had received a platinum-containing chemotherapy and who had experienced disease progression or relapse during or following treatment with PD-1 or PD-L1 inhibitors. Patients were randomized 1:1 to receive enfortumab vedotin (n = 301) 1.25 mg/kg on days 1, 8, and 15 of every 28-day cycle, or standard chemotherapy consisting of paclitaxel, docetaxel, or vinflunine (n = 307) on day 1 of each 21-day cycle until disease progression. The primary end point of the EV-301 study was OS, with secondary end points of progression-free survival (PFS), overall response rate (ORR), disease control rate (DCR), and health-related quality of life (HRQoL).17
Baseline characteristics of the EV-301 trial were well balanced between treatment arms; however, the study may have enrolled a healthier group of patients with a younger median age and lower Eastern Cooperative Oncology Group Performance Status (ECOG PS) compared to the Canadian population. Study EV-301 patients were mostly White (51.6%) and male (77.3%), with a median age of 68 years. Most patients had an ECOG PS of 1 (59.9%) and metastatic disease (95.2%).17
Efficacy Results
In the final primary efficacy analysis of EV-301, the median OS was 12.88 months (95% confidence interval [CI], 10.58 to 15.21) in the enfortumab vedotin arm, and 8.97 months (95% CI, 8.05 to 10.74) in the chemotherapy arm. Enfortumab vedotin was associated with a statistically significantly prolonged OS compared to chemotherapy (hazard ratio [HR] = 0.702; 95% CI, 0.556 to 0.886; P = 0.00142).17 Results for all sensitivity and subgroup analyses were consistent with the primary analysis.
The secondary end point of PFS was in line with the primary end point. Enfortumab vedotin was associated with a statistically significantly prolonged PFS compared to chemotherapy (HR = 0.615; 95% CI, 0.505 to 0.748; P < 0.00001), with a median PFS of 5.55 months (95% CI, 5.32 to 5.82) in the enfortumab vedotin arm and 3.71 months (95% CI, 3.52 to 3.94) in the chemotherapy arm.17 Sensitivity and subgroup analyses for PFS were consistent with the overall analysis.
Health-related quality of life, a secondary outcome of EV-301, was assessed by the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and EuroQol 5-Dimensions 5-Levels questionnaire (EQ-5D-5L). In the enfortumab vedotin arm, change in scores from baseline to week 12 for functional scales of the EORTC QLQ-C30 ranged from 2.17 (standard deviation [SD] = 16.20) for emotional functioning to −5.12 (SD = 23.80) for social functioning. In the chemotherapy arm, change from baseline scores at week 12 ranged from 3.27 (SD = 18.06) in emotional functioning to −9.15 (SD = 26.29) in role functioning. For symptom scores in the enfortumab vedotin arm, the change from baseline at week 12 ranged from 5.77 (SD = 32.56) for appetite loss to −6.96 (SD = 26.26) for pain, while in the chemotherapy arm, scores ranged from −1.63 (SD = 27.90) for insomnia to 6.64 (SD = 22.56) for fatigue. For the EQ-5D-5L, the mean change from baseline to week 12 for the EuroQol Visual Analogue Scale (EQ VAS) was −1.8 (SD = 16.6) for enfortumab vedotin and −5.3 (14.5) for the chemotherapy arm.17
The ORR was a secondary outcome in EV-301. The confirmed ORR was 40.6% compared to 17.9% for the chemotherapy arm, which was statistically significantly in favour of enfortumab vedotin (P < 0.001). Totals of 4.9% and 35.8% of patients achieved a confirmed complete response (CR) and partial response (PR), respectively, in the enfortumab vedotin arm compared to 2.7% and 15.2%, respectively, in the chemotherapy arm. Results for sensitivity and subgroup analyses for ORR were comparable to those of the primary analysis.17
Harms Results
The overall incidence of treatment-emergent adverse events (TEAEs) was consistent between enfortumab vedotin (98.0%) and taxane chemotherapy arms (||||||); however, there were imbalances in the specific TEAEs experienced in each arm, with differences of 5% or greater for enfortumab vedotin in 15 preferred term TEAEs. The incidence of serious adverse events (SAEs) was higher in the enfortumab vedotin arm compared to taxane chemotherapy (46.6% versus ||||), with acute kidney injury occurring most frequently in the enfortumab vedotin arm (6.4% versus ||||||) and febrile neutropenia occurring most frequently with taxane chemotherapy (1.4% versus ||||). Withdrawals due to adverse events (WDAEs) and TEAEs resulting in death were similar between the enfortumab vedotin and taxane chemotherapy arms (17.2% versus ||||||, and 7.1% versus ||||, respectively). The most common reason for WDAEs was peripheral sensory neuropathy, which occurred in 2.4% and |||||| of patients in the enfortumab vedotin and taxane chemotherapy groups, respectively.
The incidence of notable harms, including infusion-related reactions (IRRs), ocular disorders, skin reactions, and peripheral neuropathy, was generally more frequent in the enfortumab vedotin arm than the chemotherapy arm. Infusion-related reactions were the least frequently occurring group of notable harms in 9.1% versus |||| of patients in the enfortumab vedotin and taxane chemotherapy arms, respectively. Drug eruption was the most common IRR with enfortumab vedotin (5.7% versus ||||||), while general systemic IRRs were most frequent in the taxane chemotherapy arm (1.4% versus ||||). Incidence of treatment-emergent ocular disorders was higher in the enfortumab vedotin arm compared to the taxane chemotherapy arm (28.0% versus ||||||, respectively), the most frequent being increased lacrimation (10.1% versus ||||, respectively), dry eye (6.4% versus ||||||, respectively), and conjunctivitis (6.4% versus ||||, respectively). Skin reactions were more frequent in the enfortumab vedotin arm (53.7%) compared to the taxane chemotherapy arm (||||). The most frequently occurring skin reactions were rash (16.9% versus ||||||), maculopapular rash (16.9% versus ||||||), stomatitis (9.1% versus ||||||), and drug eruption (8.8% versus ||||). Peripheral neuropathy events occurred in 50.3% and |||||| of patients in the enfortumab vedotin and taxane chemotherapy arms, respectively. The majority of notable harms were of mild to moderate severity.
Critical Appraisal
Study EV-301 was a phase III, open-label RCT. In general, patients in the 2 treatment arms did not differ with regard to baseline disease or treatment characteristics, indicating that randomization was successful. The reviewers and the clinical expert consulted by CADTH agreed that the open-label design was appropriate; however, they noted that this could increase the risk of bias in the reporting of outcomes such as response, HRQoL, and AEs, which are subjective in measurement and interpretation. The primary end point of OS is objective, and therefore unlikely to be affected by biases of open-label study designs. Secondary end points of PFS and ORR are subjective, and therefore subject to potential bias. Reporting of patient-rated outcomes, such as symptom reduction and HRQoL, as well as some of the harms outcomes, may have been biased or influenced by the patient or investigator’s knowledge of treatment assignment. All study outcomes were investigator-assessed and did not include full evaluation through an independent review committee to mitigate the biases associated with the open-label study design. Discontinuation rates were higher in the chemotherapy arm compared with the treatment arm (81.4% versus 92.8%, respectively) while the rate of discontinuation due to disease progression was nearly identical (58.8% versus 58.6%, respectively), which may reflect the open-label design, given that the proportion of discontinuations due to patient and physician decision was higher in the chemotherapy arm. The study was stopped early for efficacy based on a statistically significant OS result in favour of enfortumab vedotin. Trials that stop early for benefit may show a higher or better treatment-effect estimate in the intervention group; however, given that the primary end point of the study, OS, was not subjective, the review team’s concerns were minor. Still, the decision to conduct the primary analysis using an information fraction of only 68.6% raises the possibility of an increased and notable risk of overestimation.
In discussions with the clinical expert consulted by CADTH, the inclusion and exclusion criteria for EV-301 were generally as expected for patients with locally advanced or metastatic UC. However, the expert hypothesized that the patients included in the trial may reflect a “less sick” population than would be seen in the real world, and noted that the median age of 68 implies a younger study population than would be expected. Additionally, the clinical expert considered the ECOG PS of patients to be unreflective of patients at this stage of disease, as most patients would not be ECOG PS 0 or 1 (0: 40.1%, 1: 59.9%). The chosen comparator of standard chemotherapy generally aligns with the recommended standard-of-care guidelines in Canada; however, vinflunine is not available as a treatment option in Canadian clinical practice, and any aggregate results for the chemotherapy arm should therefore consider the proportion of patients that may have received this treatment. The clinical expert noted that this may not affect efficacy outcomes but would affect the results for safety. Given the known differences in safety profiles of enfortumab vedotin, taxanes, and vinflunine, any safety results must be interpreted with caution and may not be generalizable. The high rate of dropouts in completion of the patient-reported outcome (PRO) measures should also be taken into account when interpreting the results.
Indirect Comparisons
No indirect evidence that matched the inclusion and exclusion criteria of this review was included in the sponsor’s submission to CADTH or identified in the literature search.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
Enfortumab vedotin is a first-in-class treatment that has been studied in patients who have received a prior PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy and demonstrated a statistically significant improvement in OS and PFS compared to chemotherapy in this patient population. Enfortumab vedotin was also associated with a clinically meaningful ORR compared to chemotherapy for a single-drug therapy, which is an important consideration for this stage of disease. The ORR results are in line with the survival benefit seen for OS. Both measures are important to patients, but the effect of the clinically meaningful ORR on improvement in cancer symptoms is uncertain. Given the open-label design, the PFS and ORR results must be interpreted with caution. Enfortumab vedotin was not associated with any major improvement or deterioration in HRQoL in the pivotal study; however, because of high patient-attrition rates, the effect of enfortumab vedotin on HRQoL remains uncertain.
Overall, despite the similar rates of TEAEs between the enfortumab vedotin and chemotherapy arms, any interpretation of the comparative results must take into consideration the individual safety profiles of the study treatments, particularly for taxanes, as vinflunine is not available in Canada. Generally, the rate of specific TEAEs was higher for enfortumab vedotin compared to taxane therapies. Compared to taxane chemotherapy, enfortumab vedotin was also associated with more SAEs and symptomatic notable harms, including ocular disorders, skin reactions, and peripheral neuropathy. Despite the feedback from 2 patients in the patient group input who noted that side effects of treatment with enfortumab vedotin were temporary and manageable, enfortumab vedotin was hypothesized by the clinical expert to be more toxic than current therapeutic options, which may explain the lack of improvement in QoL.
Overall, enfortumab vedotin provides an effective third-line treatment option, extending survival and demonstrating good clinical response after progression with platinum chemotherapy and a PD-1 or PD-L1 inhibitor. However, there were several limitations in the generalizability of results given the stage of disease, as well as potential safety concerns.
Introduction
Disease Background
Bladder cancer is the fifth most common cancer in Canada, with approximately 12,200 cases, resulting in an estimated 2,600 deaths in 2020. Urothelial carcinoma is the most common type of bladder cancer, accounting for 90% to 95% of cases.1-4 Urothelial cancer typically arises in the bladder but may develop in any location lined with urothelium, including the renal pelvis, ureter, urethra, and prostatic urethra.5 Age, tobacco use, chemical carcinogens, family history, arsenic exposure, and use of indwelling catheters are known risk factors for bladder cancer. Bladder cancer is more common in males, although the reason is unknown.6-9
Urothelial carcinoma can be characterized as non–muscle-invasive bladder cancer (NMIBC), MIBC, or metastatic based on the extent of invasion into the wall of the bladder (Figure 1). Approximately 70% to 80% of newly diagnosed patients present with NMIBC.19 However, superficial bladder tumours often recur, and a subset of these patients progress to develop high-grade muscle-invasive urothelial cancer. Nearly 40% to 50% of patients with early-stage MIBC will relapse after initial treatment. Approximately 15% of patients present with locally advanced or metastatic disease.10 In Canada, the 5-year net survival for bladder cancer is 75%; however, there are no Canadian-specific survival statistics for locally advanced or metastatic UC patients. The estimated 5-year relative survival rate for patients with metastatic disease is approximately 6.4%,7 and up to 15% when treated with contemporary regimens.11,12
Figure 1: Stages of Urothelial Carcinoma
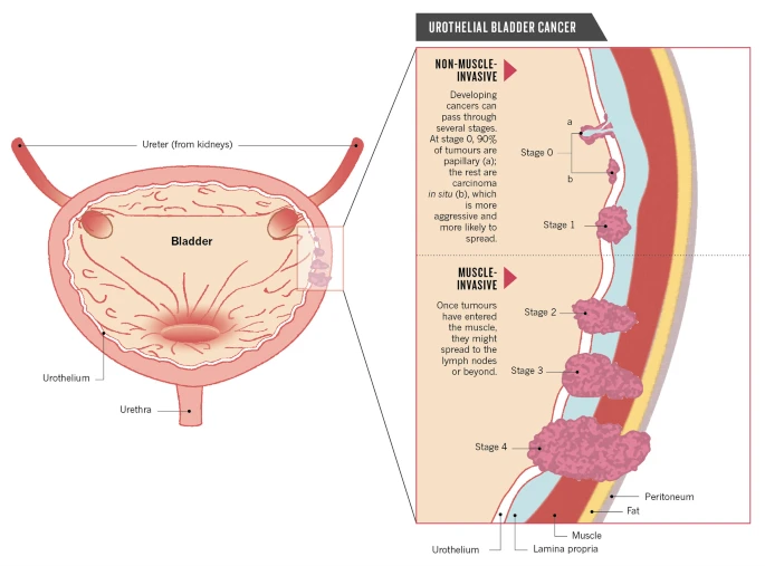
Source: Berdik (2017).20 Reprinted by permission from Springer Nature Limited: Springer Nature, Berdik, C., Unlocking bladder cancer, Nature, 551:S34-S35, 2017. https://www.nature.com/articles/551S34a.
The most common presentation of UC is visible or microscopic hematuria. Other symptoms of UC include painful urination, back or flank pain, fatigue, and unexplained weight loss.4 Coupled with numerous imaging and clinical tests, including urinalysis, cystoscopy, and urine cytology, diagnosis is confirmed with a high-quality transurethral resection of the bladder tumour (TURBT), which also confirms the pathology and extent of the disease. If the tumour is determined to be NMIBC, localized chemotherapy is initiated. If it is determined to be MIBC, additional laboratory tests (complete blood count, and blood chemistry), imaging (X-ray, CT, and MRI), and biomarker testing (PD-L1 and FGFR) are recommended.4,14
Standards of Therapy
Standard of care for locally advanced or metastatic UC consists of platinum-based chemotherapy. In patients who are eligible for cisplatin-based chemotherapy, the preferred regimen is gemcitabine plus cisplatin. In select cases in which more aggressive treatment is needed, dose-dense methotrexate, vinblastine, doxorubicin, and cisplatin is used. Routine assessment of cisplatin eligibility consists of creatinine clearance of greater than 60 mL/min, an ECOG PS of less than 1, absence of hearing loss greater than grade 2 (per Common Terminology Criteria for Adverse Events), absence of neuropathy of greater than grade 2, and absence of New York Heart Association grade III or IV heart failure. In these patients, gemcitabine plus carboplatin is recommended.10 Recently, CADTH granted a conditional listing to avelumab for first-line maintenance treatment of locally advanced or metastatic UC in patients whose disease has not progressed following first-line platinum-based induction chemotherapy.13 Although it did not receive a positive recommendation from CADTH,21 pembrolizumab may be used in a first-line setting in cisplatin-ineligible patients with advanced UC.10
In patients who progress following platinum-based chemotherapy, treatment with CPI immunotherapy, preferably pembrolizumab, is recommended as second-line systemic therapy.14,15 Alternative CPI regimens include nivolumab, avelumab, and erdafitinib, if available.14 Following failure of platinum chemotherapy and CPIs, or when CPIs are unavailable, salvage chemotherapy with taxanes; paclitaxel or docetaxel are preferred in most patients. Alternatively, re-treatment with previously unused chemotherapy and immunotherapy regimens can be used, following a prolonged response to initial chemotherapy.10
Drug
Enfortumab vedotin is an ADC consisting of a fully human immunoglobulin G1Κ antibody and a microtubule-disrupting drug, MMAE, that acts via a protease-cleavable linker directed against nectin-416 adhesion proteins on UC cell surfaces. Non-clinical data suggest that enfortumab vedotin binds nectin-4–expressing cells, resulting in internalization of the ADC–nectin-4 complex and intracellular release of MMAE via protolytic cleavage, and inducing apoptosis through a disrupted microtubule network.17 When released through cellular apoptosis, MMAE can further diffuse into nearby cells expressing low levels of nectin-4, resulting in cytotoxic cell death.18
Enfortumab vedotin is administered via IV infusion and must be reconstituted and diluted with 2.3 mL or 3.3 mL of sterile water for injection before administration from 20 mg and 30 mg vials, respectively. Enfortumab vedotin is administered over 30 minutes through an IV line, is not to be administered as an IV push or bolus, and is not to be co-administered with other drugs.18
Enfortumab vedotin is approved by Health Canada for the treatment of adult patients with unresectable locally advanced or metastatic UC who have previously received a platinum-containing chemotherapy and PD-1 or PD-L1 inhibitor therapy.18
A Health Canada Notice of Compliance was granted on October 29, 2021. Enfortumab vedotin has not been previously reviewed by CADTH.
In 2019, enfortumab vedotin was approved by the FDA under accelerated approval for treatment of adult patients with locally advanced or metastatic UC who have previously received a PD-1 or PD-L1 inhibitor and a platinum-containing chemotherapy.17 In 2021, the European Medicines Agency accepted an application for a marketing authorization for enfortumab vedotin as an accelerated assessment.22
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient advocacy group, BCC, provided input for the review of enfortumab vedotin in locally advanced or metastatic UC. A nationally registered Canadian charity, BCC and is the first and only Canadian patient advocacy organization dedicated to bladder cancer issues. Supported by a Medical Advisory Board and a Medical Research Board consisting of the top bladder cancer specialists across Canada, its mission is to help bladder cancer patients and their support teams address the day-to-day issues of this disease; increase awareness among the public and medical community; and fund research into the diagnosis, treatment, and elimination of the disease. The organization’s vision is patient support, awareness, and research to create a world where bladder cancer is “just a memory.”
The information provided by BCC was gathered through an online survey and telephone interviews conducted between May 27 and June 11, 2021. Most survey respondents were from Canada, with a small number from the US. Additionally, telephone interviews with 2 patients from Canada who had experience with enfortumab vedotin were conducted in June 2021. In total, 38 patients diagnosed with stage II or higher MIBC, of which one-third reported living with locally advanced or metastatic bladder cancer, and 6 caregivers completed the survey.
Many patients and caregivers reported that symptoms of bladder cancer, including fatigue, lack of sleep, and loss of strength and stamina were problematic but manageable, while some patients indicated that having bladder cancer has had a minimal impact on their day-to-day lives. Blood in the urine, pain in the abdomen and bones, decreased mobility and difficulty or pain when urinating were also commonly reported symptoms. Frequent need for urination and loss of control, urostomy and catheter management, and urinary tract infections were the most commonly reported issues related to continence that affect the day-to-day lives of patients and result in the need for additional planning, discomfort, and time lost. Financial impacts related to the costs of catheters and urostomy supplies that are not covered by some provincial governments were reported to strain the already limited financial resources of patients and caregivers.
Patients described experiencing a number of side effects with current treatments, including fatigue, constipation, low blood cell count, loss of appetite, neuropathy, nausea, vomiting, hair loss, insomnia, diarrhea, and mouth sores. Most patients reported encountering minimal barriers to treatment for their bladder cancer; however, some mentioned they did have difficulties due to travel distances, treatment costs, unavailability of treatment in Canada, no access to a physician, and the need to take time off work to receive treatment. Two patients had experience with enfortumab vedotin through a clinical trial. Patients noted that the side effects of treatment with enfortumab vedotin were temporary and manageable compared to previous treatments received. When asked what key benefits from enfortumab vedotin have been important to them as patients, they reported that the treatment has given them their “life back again” — allowing them to resume the activities that they enjoy. Patients with experience with enfortumab vedotin emphasized the importance to publicly funded access to this treatment.
Overall, patients and caregivers hoped for fewer and less-severe side effects than those experienced with current bladder cancer treatments, as well as treatments that induced remission or were curative. Specifically, patients described the ideal treatment as one that would slow or stop disease progression, recurrence and spread, reduce pain, fatigue, and impaired sexual function; increase energy levels and strength; improve mental health, continence, and urination control; and result in fewer or no infections and avoidance of surgery.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of urothelial cancer.
Unmet Needs
The clinical expert stated that there is a need for an effective third-line treatment option after progression with platinum chemotherapy and a PD-1 or PD-L1 inhibitor. Currently these patients are offered either a trial of monotherapy taxane chemotherapy, targeted therapy (if next-generation sequencing is available and actionable mutations are found), or best supportive care. The clinical expert highlighted that, at this point, survival is short (usually less than 12 months), and it is questionable whether these approaches influence the natural history of the disease. At this stage of disease, current treatment should reduce tumour burden, in turn reducing cancer symptoms and prolonging length of life. Ideally, treatment would improve both survival and QoL for these patients as well.
Place in Therapy
Initially, bladder cancer is treated superficially with intravesical therapies, but often it progresses to muscle-invasive disease that requires more definitive therapy with cystectomy (with or without perioperative cisplatin-based chemotherapy) or chemoradiation. The clinical expert noted that patients may then develop incurable local recurrence or, in most cases, distant metastases, while some patients present with de novo incurable metastatic disease.
The clinical expert noted that the mainstay of treatment for incurable patients is cytotoxic platinum-based chemotherapy with gemcitabine with or without cisplatin (or carboplatin), which improves OS but is not curative. For the small minority of patients with absolute contraindications to chemotherapy, the clinical expert stated that the safest alternative is immunotherapy, provided PD-L1 is overexpressed. Enfortumab could be considered, however, it has the potential for serious toxicities comparable to those associated with chemotherapy, and it is unclear why enfortumab would be preferred. Patients with true contraindications or severe immune-mediated toxicity due to PD-1or PD-L1 inhibitors could be considered for enfortumab therapy.
Second-line chemotherapy consists of single-drug paclitaxel, docetaxel, or vinflunine, and only has modest activity. However, vinflunine is not approved or available in Canada. Pembrolizumab is now a funded second-line standard of care in Canada following demonstration of a survival benefit after progression despite first-line chemotherapy, displacing second-line taxane therapy. In patients without evidence of progressive disease after first-line treatment with gemcitabine and platinum-based chemotherapy, maintenance therapy with avelumab was reported to show an OS benefit and is likely to become a funded standard of care. It was also suggested by the clinical expert that avelumab maintenance therapy may be considered a form of second-line therapy, as there is no evidence to support re-treatment with other PD-1 or PD-L1 inhibitors following progression. The clinical expert emphasized that primary chemotherapy should continue to be the standard of care until comparative data are available.
According to the clinical expert, there is a need for a more effective third-line treatment options given the use of pembrolizumab as second-line treatment. The clinical expert stated that patients are often in good health following treatment with immunotherapy and are good candidates for additional systemic therapy; however, the only options in these patients are paclitaxel and docetaxel, which have modest response rates and treatment durations. The clinical expert acknowledged that the targeted agent erdafitinib is available from the sponsor for patients whose tumours express aberrations in FGFR genes. It was also noted that only 25% of incurable, locally advanced, or metastatic UC patients have tumours with eligible FGFR aberrations, and next-generation sequencing of tumours is required for confirmation. The clinical expert also noted that erdafitinib is not currently funded for this indication.
The clinical expert noted that enfortumab vedotin has only been studied in patients with progressive cancer and appears to be associated with high objective tumour response rates in metastatic UC; current data would therefore not support its use in the absence of disease progression. The clinical expert also expressed a high level of interest in the use of enfortumab vedotin earlier in the natural history of metastatic UC, adding that the drug may eventually have a role in perioperative adjuvant therapy.
Patient Population
The clinical expert suggested that the population under consideration would be incurable locally advanced or metastatic urothelial cancer, and that misdiagnosis is highly unlikely. Virtually all incurable patients will have progressed despite platinum-based chemotherapy and PD-1 or PD-L1 inhibitor therapy. The clinical expert noted that there is no way to assess which patients will most likely respond to treatment, and that no special tests are required. Patients are typically under the care of expert medical oncologists, and that symptoms and cross-sectional imaging make it easy to identify disease progression. The expert did note that some patients on immunotherapy may experience pseudo-progression, but most oncologists would not switch treatment until unequivocal progression despite immunotherapy was observed.
The clinical expert noted that the patients least likely to respond to treatment are those with poor functional status.
Assessing Response to Treatment
The clinical expert stated that there are no differences in disease characteristic or predictors of response to enfortumab among urothelial cancer patients. Instead, those with signs or symptoms of disease progression despite prior platinum-based chemotherapy and PD-1 or PD-L1 inhibitors currently have the greatest need. Response to treatment would rely on improvement in symptoms, which would be assessed before treatment and/or evidence of objective tumour shrinkage on imaging (i.e., every 3 months).
Discontinuing Treatment
The clinical expert suggested that discontinuation of treatment would be warranted in the event of disease progression despite treatment, a severely reduced functional status, or intolerable side effects.
Prescribing Conditions
The clinical expert noted that patients with incurable locally advanced or metastatic UC are typically under the care of expert medical oncologists, and that enfortumab can be administered by trained staff in an outpatient clinic. The expert emphasized that enfortumab may cause adverse ocular effects such as keratitis, and assessment and monitoring by an ophthalmologist may be required. Additionally, rash due to treatment with enfortumab may require assessment by a dermatologist.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
Two clinicians from the Cancer Care Ontario Genitourinary Cancer DAC and a group of 17 Canadian physicians who treat bladder cancer, with the support of BCC, provided input for this review. The DAC provides timely evidence-based clinical and health-system guidance on drug-related issues in support of Cancer Care Ontario’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. The group of Canadian physicians represents the specialty from across Canada in both academic and community settings and shares BCC’s goal of improving the management of bladder cancer.
The clinicians agreed that there was no standard of care for patients with advanced urothelial cancer post–platinum chemotherapy and post-immunotherapy, representing an unmet need in these patients. Enfortumab vedotin is indicated in the third-line setting, and the experts agreed that enfortumab vedotin will redefine the current treatment paradigm, with alternative third-line treatment options consisting of non-platinum chemotherapy (i.e., taxanes, for which there is little evidence of efficacy and for which the toxicity rate is much higher) or FGFR-targeted therapy (which would not be favoured for the reasons of unavailability of FGFR testing in Canada), becoming fourth-line options. The clinician groups indicated that offering enfortumab vedotin to all eligible patients would provide them with hope for improved life expectancy with tolerable side effects. The clinician group stated that, throughout treatment, patients would be seen by their treating oncologist following each cycle and would be assessed for toxicity and clinical progression every month, with imaging every 2 to 3 months. Blood work should be performed before each treatment cycle. One clinician group noted that patients with disease that has metastasized to the bones should also have a bone scan. The clinician groups noted that decisions to discontinue treatment should be made in consultation with the patient, and generally be based on progressive disease, worsening symptoms, dose-limiting toxicity resulting in intolerable severe AEs such as significant neuropathy, patient wishes to discontinue treatment for personal reasons, and deterioration to end of life.
Although no marked experience with enfortumab vedotin was mentioned, the clinician groups consider this drug of great importance in the management of bladder cancer, filling an unmet need for patients requiring treatment following progression on platinum-based chemotherapy and immunotherapy and giving medical oncologists an additional option to offer patients with advanced urothelial cancer that has progressed on first- and second-line therapy. The clinician groups believe that enfortumab vedotin would offer a longer life expectancy with preservation of QoL, as the drug is generally well tolerated. The clinician groups also noted that, for a patient population with such a poor prognosis, the inclusion of enfortumab vedotin in the treatment algorithm has the potential to significantly improve the outcomes associated with bladder cancer.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 2.
The PAG noted that taxanes (paclitaxel and docetaxel) are the most relevant comparators in this setting and are funded and available in all provinces. The group noted that an updated algorithm for metastatic UC would help jurisdictions navigate funding as several therapies are now available for multiple lines of treatment, increasing the complexity of funding.
Table 2: Summary of Drug Plan Input and Clinical Expert Response
Additional implementation questions from the drug programs | Advice from CADTH |
|---|---|
Relevant comparators | |
Erdafitinib is approved by Health Canada for patients with metastatic UC whose tumours have FGFR genetic alterations, and who have disease progression during or following at least 1 line of prior chemotherapy. It may be available through a sponsor’s patient support program but has not been reviewed by CADTH yet and is not publicly funded. Erdafitinib could also be considered a relevant comparator in patients with FGFR genetic alterations for patients previously treated with PD-1 or PD-L1 inhibitors and chemotherapy. | For consideration by the expert review committee. |
Considerations for initiation of therapy | |
Some patients may not be candidates for platinum-based chemotherapy due to comorbidities and may have received alternate non-platinum–based or single-drug chemotherapy. Should patients who have not received previous platinum-based chemotherapy be eligible for enfortumab vedotin? | Generally, these patients would not be eligible for enfortumab vedotin; however, exceptions would be made for patients not eligible for platinum chemotherapy (i.e., elderly, frailty). In this case, immunotherapy should be given first, followed by enfortumab vedotin. |
Some patients may have a contraindication to or may not be candidates for immunotherapy or experience immune toxicity necessitating discontinuation of immunotherapy. Should patients who received platinum-based chemotherapy, but did not receive PD-1 or PD-L1 inhibitors be eligible for enfortumab vedotin? | Generally, these patients would not be eligible for enfortumab vedotin; however, exceptions would be made for patients with absolute contraindications to immunotherapy. |
Should patients who have immunotherapy permanently discontinued for toxicity reasons be eligible for enfortumab vedotin at the time of disease progression or could they be switched to enfortumab vedotin before disease progression? | Initiation of enfortumab vedotin should be in line with EV-301, in which patients who discontinued CPI treatment due to toxicity were eligible provided they had evidence of disease progression following discontinuation. |
Considerations for prescribing of therapy | |
The dosing of enfortumab vedotin is 1.25 mg/kg IV over 30 minutes on days 1, 8, and 15 every 28 days (maximum dose of 125 mg for patients > 100 kg) until disease progression or unacceptable toxicity. Weekly dosing is more labour-intensive and requires frequent patient visits for administration. | For consideration by the expert review committee. |
Skin and soft-tissue injury following administration has been observed when extravasation occurred. It is important to ensure good venous access before starting, and the infusion site should be monitored for extravasation during administration. If extravasation occurs, it is recommended to stop the infusion and monitor for adverse reactions. Enfortumab vedotin should only be administered by staff trained to manage extravasations of vesicants/irritants in appropriate facilities. | For consideration by the expert review committee. |
Generalizability | |
The eligibility criteria in the EV-301 study included patients with an ECOG PS of 0 or 1. Should patients with an ECOG PS > 1 be eligible for enfortumab vedotin? | Selected patients with an ECOG PS of 2 could be considered for treatment with enfortumab vedotin. |
Patients currently receiving taxanes or alternate chemotherapy would have a time-limited opportunity to switch to enfortumab vedotin. Should patients receiving these treatments be switched to enfortumab vedotin at the time of public funding, or would they be eligible after disease progression on these treatments? | Both options should be considered. If treatment with taxanes or alternative chemotherapy is working, there is no reason to switch therapies. However, after disease progression, and if eligible for enfortumab vedotin, the patient may be switched. |
Care provision issues | |
The PAG notes that enfortumab vedotin is available in single-use vials of 20 mg and 30 mg. Vial sharing is not expected due to the size of the patient population, and it is anticipated that drug wastage will occur, especially at the maximum dose of 125 mg. The vial sizes do not match the maximum dose at some dosing levels (1.25 mg/kg up to 125 mg; 1.0 mg/kg up to 100 mg; 0.75 mg/kg up to 75 mg; 0.5 mg/kg up to 50 mg), so wastage is expected with doses. Also, because the vial sizes are small relative to the usual starting dose, there is a resource impact (e.g., 125 mg dose requires 3 × 30 mg plus 2 × 20 mg to minimize wastage but requires 5 vials to reconstitute and dilute to final preparation), and an impact on pharmacy resources. The PAG also notes the chemical and physical stability of the final preparation is limited (16 hours, refrigerated), and treatment will likely need to occur at facilities where sterile compounding pharmacies are nearby or onsite. | For consideration by the expert review committee. |
The draft product monograph states that no dose adjustments are required for patients with mild hepatic impairment, mild to severe renal impairment, or with concomitant use of strong inhibitors of CYP3A4. However, drug-information databases note that strong inducers or inhibitors of CYP3A4 may decrease or increase the serum concentration of enfortumab vedotin. The PAG notes there is potential for clinically significant drug-drug interactions with strong CYP3A4 inducers and inhibitors, which may affect pharmacy resources for identification, monitoring and resolution of these drug-drug interactions. | For consideration by the expert review committee. |
System and economic Issues | |
The number of patients eligible for enfortumab vedotin in Canada (excluding Québec) was estimated by the manufacturer at 388 for year 1, 461 for year 2 and 534 for year 3, for a total of 1,382 patients over the 3-year period. The sponsor’s BIA estimates $5,950,573 in year 1, $12,707,014 in year 2, and $21,272,715 in year 3, for a total of $39,930,302 over the projection period. The BIA predicts that funding of enfortumab vedotin for the treatment of locally advanced or metastatic urothelial carcinoma would result in incremental costs of $4,804,551 in year 1, $11,347,174 in year 2, $19,696,563 in year 3, for a total incremental cost of $35,848,288 over the 3-year projection period. This is based on market share estimates of 15%, 30%, and 45% for years 1 through 3 in second line, and 25%, 40%, and 55% for years 1 through 3 in third line. PAG is concerned the market share and BIA may be underestimated, resulting in a substantially higher budget impact. | For consideration by the expert review committee. |
BIA = budget impact analysis; CPI = checkpoint inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FGFR = fibroblast growth-factor receptor gene; PAG = Provincial Advisory Group; PD-1 = programmed death receptor 1; PD-L1 = programmed death ligand 1.
Clinical Evidence
The clinical evidence included in the review of enfortumab vedotin is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. No indirect treatment comparisons were submitted to CADTH by the sponsor. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of enfortumab vedotin 1.25 mg/kg for the treatment of patients with locally advanced or metastatic urothelial cancer who have previously received a PD-1 or PD-L1 inhibitor and a platinum-containing chemotherapy in the neoadjuvant, adjuvant, locally advanced, or metastatic setting or who are not eligible for cisplatin-containing chemotherapy.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 3. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
The systematic review protocol was established before the granting of a Notice of Compliance from Health Canada.
Table 3: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients with locally advanced or metastatic urothelial cancer who have previously received a PD-1 or PD-L1 inhibitor and who:
Subgroups:
|
Intervention | Enfortumab vedotin 1.25 mg/kg, 20 mg and 30 mg single-use vials for IV injection |
Comparators | Chemotherapy:
Immunotherapy:
Targeted therapy:
|
Outcomesa | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV randomized controlled trials |
AE = adverse event; CR = complete response; DCR = disease control rate; DOR = duration of response; ECOG = Eastern Cooperative Oncology Group; FGFR2 = fibroblast growth-factor receptor 2; FGFR3 = fibroblast growth-factor receptor 3; GP = general practitioner; HCRU = health care resource utilization; ORR = overall response rate; PD-1 = programmed death receptor 1; PDL1 = programmed death ligand 1; PR = partial response; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aOutcomes in bold were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the Peer Review of Electronic Search Strategies checklist.23
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was enfortumab vedotin. Clinical trials registries searched included the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on July 22, 2021. Regular alerts updated the search until the meeting of CADTH pan-Canadian Oncology Drug Review Expert Committee on November 10, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.24 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies for inclusion, and differences were resolved through discussion. A focused literature search for network meta-analyses dealing with UC was run in MEDLINE All (1946–) on July 22, 2021. No limits were applied.
Findings From the Literature
A total of 105 studies were identified from the literature for inclusion in the systematic review (Figure 2). The included study is summarized in Table 4. A list of excluded studies is presented in Appendix 2.
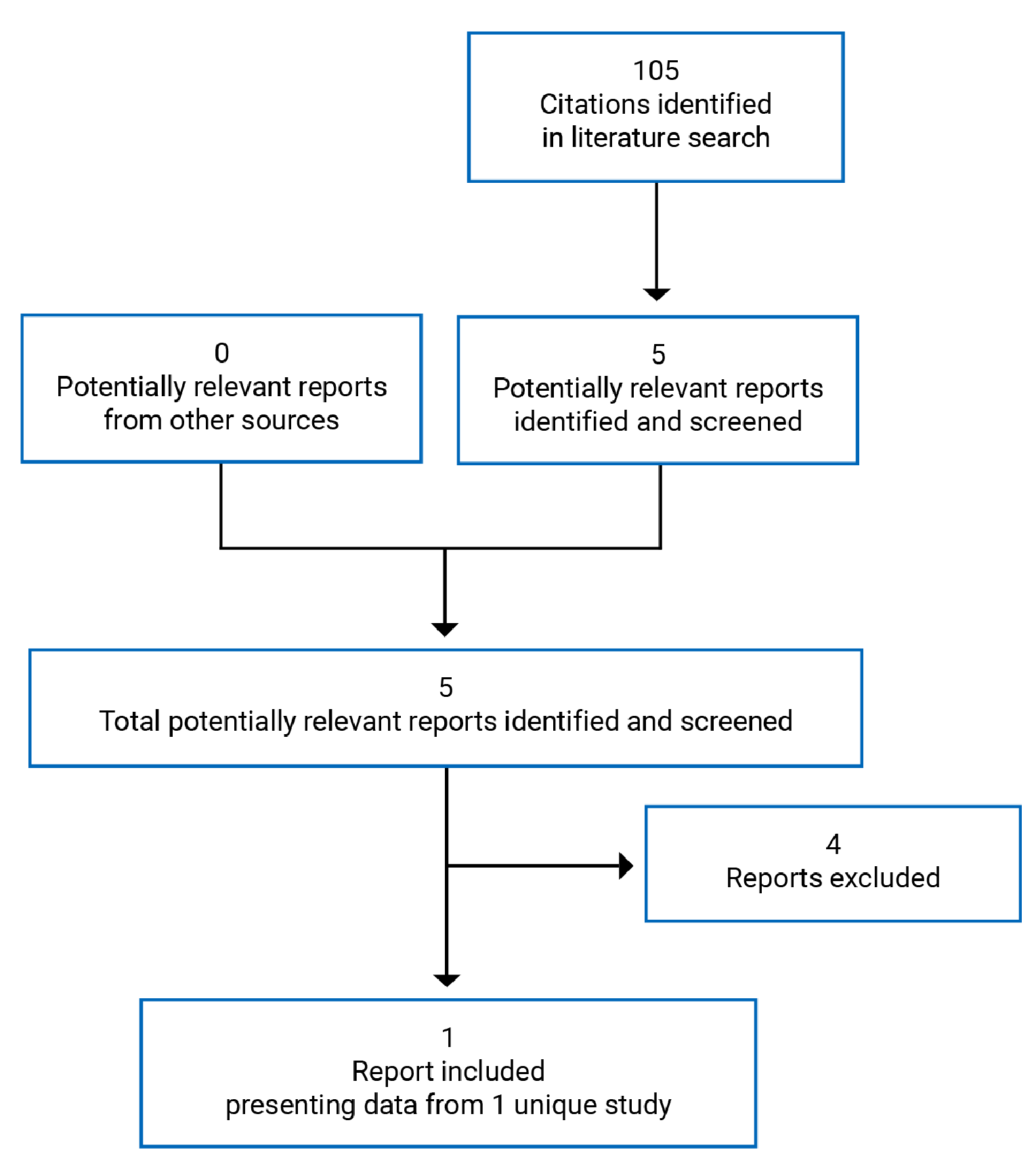
Table 4: Details of Included Studies
Study EV-301 | |
|---|---|
Designs and populations | |
Study design | Open-label, randomized, phase III study |
Locations | 19 countries, including Argentina, Australia, Austria, Belgium, Canada, Denmark, France, Germany, Italy, Japan, The Netherlands, Portugal, Republic of Korea, Russian Federation, Spain, Switzerland, Taiwan, UK, and the US |
Patient enrolment dates | July 2018 to January 2020 |
Randomized (N) | 608 |
Inclusion criteria | Legally an adult according to local regulation at the time of signing informed consent Histologically or cytologically confirmed urothelial carcinoma (i.e., cancer of the bladder, renal pelvis, ureter or urethra); patients with urothelial carcinoma (transitional cell) with squamous differentiation or mixed cell types were eligible Experienced radiographic progression or relapse during or after a CPI (anti–PD-1 or anti–PD-L1) for locally advanced or metastatic disease; patients who discontinued CPI treatment because of toxicity were eligible provided that they had evidence of disease progression following discontinuation; CPI need not have been the most recent therapy; patients for whom the most recent therapy had been a non-CPI based regimen were eligible if they had progressed or relapsed during or after their most recent therapy; locally advanced disease must not have been amenable to resection with curative intent per the treating physician Patients must have received a platinum-containing regimen (cisplatin or carboplatin) in the metastatic or locally advanced, neoadjuvant, or adjuvant setting; if platinum was administered in the adjuvant or neoadjuvant setting, the patient must have progressed within 12 months of completion Radiologically documented metastatic or locally advanced disease at baseline An archival tumour tissue sample was to be available for submission to the central laboratory before study treatment; if an archival tumour tissue sample was not available, a fresh tissue sample was to have been provided; if a fresh tissue sample could not be provided because of safety concerns, enrolment into the study was to have been discussed with the medical monitor ECOG PS of 0 or 1 The following baseline laboratory data: ANC ≥ 1,500/mm3, platelet count ≥ 100 × 109, hemoglobin ≥ 9 g/dL, serum total bilirubin ≤ 1.5 x ULN or ≤ 3 × ULN for patients with Gilbert disease, creatinine clearance ≥ 30 mL/min as estimated per institutional standards or as measured by 24 hour urine collection (GFR could have been used instead of creatinine clearance), ALT and AST ≤ 2.5 × ULN or ≤ 3 × ULN for patients with liver metastases (docetaxel was not to be chosen as a comparator for patients if total bilirubin > ULN, or if AST and/or ALT > 1.5 × ULN concomitant with ALP > 2.5 × ULN) |
Exclusion criteria | Pre-existing sensory or motor neuropathy grade ≥ 2 Active CNS metastases: patients with treated CNS metastases were permitted on study if all the following were true:
Ongoing clinically significant toxicity (≥ grade 2 with the exception of alopecia) associated with prior treatment (including systemic therapy, radiotherapy, or surgery); patients with ≤ grade 2 immunotherapy-related hypothyroidism or panhypopituitarism may have been enrolled when well-maintained or controlled on a stable dose of hormone replacement therapy (if indicated); patients with ongoing ≥ grade 3 immunotherapy-related hypothyroidism or panhypopituitarism were excluded; patients with ongoing immunotherapy-related colitis, uveitis, myocarditis, or pneumonitis or patients with other immunotherapy-related AEs requiring high doses of steroids (> 20 mg/day of prednisone or equivalent) were excluded Prior treatment with enfortumab vedotin or other MMAE-based ADCs Received prior chemotherapy for UC with all available study therapies in the control arm (i.e., both prior paclitaxel and docetaxel in regions where vinflunine is not an approved therapy, or prior paclitaxel, docetaxel and vinflunine in regions where vinflunine is an approved therapy) Received more than 1 prior chemotherapy regimen for locally advanced or metastatic UC, including chemotherapy for adjuvant or neo-adjuvant disease if recurrence had occurred within 12 months of completing therapy; substitution of carboplatin for cisplatin was not to constitute a new regimen provided no new chemotherapeutic agents had been added to the regimen Patient was currently receiving systemic antimicrobial treatment for viral, bacterial, or fungal infection at the time of first dose of enfortumab vedotin; routine antimicrobial prophylaxis was permitted Known active hepatitis B or active hepatitis C Known history of HIV infection Documented history of a cerebral vascular event (stroke or transient ischemic attack), unstable angina, myocardial infarction, or cardiac symptoms (including CHF) consistent with NYHA class III–IV within 6 months before the first dose of study drug Radiotherapy or major surgery within 4 weeks before first dose of study drug Chemotherapy, biologics, investigational drugs, and/or antitumour treatment with immunotherapy that was not completed 2 weeks before first dose of study drug History of uncontrolled diabetes mellitus within 3 months of the first dose of study drug; uncontrolled diabetes is defined as hemoglobin A1C ≥ 8% or between 7% and < 8% with associated diabetes symptoms (polyuria or polydipsia) that are not otherwise explained |
Drugs | |
Intervention | Enfortumab vedotin, 1.25 mg/kg on days 1, 8, and 15 of every 28-day cycle via IV infusion until disease progression. |
Comparator(s) | Chemotherapy: Docetaxel 75 mg/m2 on day 1 of every 21-day cycle via IV infusion until disease progression Paclitaxel 175 mg/m2 on day 1 of every 21-day cycle via IV infusion until disease progression Vinflunine 320 mg/m2 on day 1 of every 21-day cycle via IV infusion until disease progression |
Duration | |
Phase | |
Screening | 28 days before randomization |
Treatment | 28- and 21-day treatment cycles for enfortumab vedotin and chemotherapy regimens, respectively |
Follow-up | Until disease progression or treatment discontinuation |
Outcomes | |
Primary end point | Overall survival, defined as the time from randomization to the date of death |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Powles et al. (2021)25 |
ADC = antibody-drug conjugate; AE = adverse event; ALP = alkaline phosphatase; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; CHF = congestive heart failure; CNS = central nervous system; CPI = checkpoint inhibitor; CR = complete response; DCR = disease control rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GFR = glomerular filtration rate; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; MMAE = monomethyl auristatin E; NYHA = New York Heart Association; ORR = overall response rate; PD-1 = programmed cell death protein 1; PD-L1 = programmed cell death ligand 1; PFS1 = progression-free survival on study therapy; PFS2 = progression-free survival on subsequent therapy; PR = partial response; PRO = patient-reported outcome; QoL = quality of life; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; TTR = time to response; UC = urothelial carcinoma; ULN = upper limit of normal.
Source: EV-301 Clinical Study Report.17
Description of Studies
One study was included in the review. Study EV-301 was a global, open-label, phase III RCT comparing enfortumab vedotin to standard salvage chemotherapy regimens in adults with locally advanced or metastatic UC who had received a platinum-containing chemotherapy and who had experienced disease progression or relapse during or following treatment with PD-1 or PD-L1 inhibitors. The design of EV-301 is shown in Figure 3.
Figure 3: Schematic Overview of the EV-301 Study
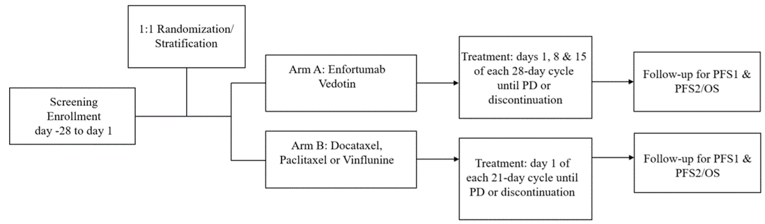
OS = overall survival; PD = progressive disease; PFS1 = progression-free survival on study therapy; PFS2 = progression-free survival on subsequent therapy.
Source: EV-301 Clinical Study Report.17
The primary objective of EV-301 was to compare the OS of patients with locally advanced or metastatic UC treated with enfortumab vedotin to those treated with chemotherapy. It was conducted in 19 countries, with 11 sites in Canada accounting for 52 patients.17
Key eligibility criteria for EV-301 are summarized in Table 4. Briefly, eligible patients included adults with histologically or cytologically confirmed urothelial carcinoma previously treated with a platinum-containing regimen (cisplatin or carboplatin) in the metastatic or locally advanced, neoadjuvant, or adjuvant setting and must have experienced radiographic progression or relapse during or after a CPI (anti–PD-1 or anti–PD-L1) for locally advanced or metastatic disease.17
Patients were randomized 1:1 via interactive response technology to either arm A, which consisted of enfortumab vedotin (n = 301), or arm B, which consisted of various chemotherapy regimens (n = 307). Prior to randomization, investigators selected docetaxel, vinflunine, or paclitaxel to be used in the event a patient was randomized to arm B, with a maximum of 35% of patients receiving vinflunine. Patients were stratified according to ECOG PS (0 versus 1), regions of the world (Western Europe versus US versus rest of world) and liver metastasis (yes versus no). To maintain trial integrity, aggregate analyses or summaries by randomized treatment assignment were limited and documented before the primary hard database lock. Interim analysis was conducted externally by an independent data analysis centre and the results were reviewed by an independent data monitoring committee. The data cut-off date for the primary analysis of EV-301 was July 15, 2020, and database lock took place on September 15, 2020.17
Any subgroup results for patients receiving vinflunine in arm B will not be discussed in this report as vinflunine is not available in Canada. Vinflunine was only offered as a choice of comparator in countries where it is approved for UC.17
Populations
Inclusion and Exclusion Criteria
Inclusion and exclusion criteria for the EV-301 trial are summarized in Table 4. Patients were required to have histologically or cytologically confirmed UC with locally advanced or metastatic UC at baseline and an ECOG PS of 0 or 1. Patients must have received a platinum-containing regimen (cisplatin or carboplatin) in the metastatic or locally advanced, neoadjuvant, or adjuvant setting. In line with current guidelines, if the platinum regimen was administered in the adjuvant or neoadjuvant setting, patients must have progressed within 12 months of completion. Finally, patients must have received treatment with, and experienced radiographic progression or relapse during or after, a CPI for locally advanced or metastatic disease. Patients were excluded if they had received prior chemotherapy with any study therapies in the control arm.17
Investigators were free to terminate a patient’s involvement in the study if they felt that the patient’s clinical condition warranted discontinuation, and each patient was free to discontinue or withdraw from the study at any time for any reason. All patients who discontinued study treatment were to remain in the study and continue to be followed for 30 days after their last dose for safety assessments, and every 3 months for survival status and progression on subsequent therapy or if the patient specifically withdrew consent for contact. If a patient was discontinued for ongoing AEs or unresolved laboratory results, the patient was followed up until the event was stabilized or no longer clinically significant.17
Baseline Characteristics
Baseline characteristics for the EV-301 study are summarized in Table 5. Baseline characteristics were well balanced between enfortumab vedotin and chemotherapy arms. Patients were mostly White (52.8% versus 50.5%, respectively) and male (79.1% versus 75.6%, respectively). The median age was 68 years in each treatment arm, with 64.1% and 63.8% aged 65 years or older in the enfortumab vedotin and chemotherapy arms, respectively. Patients were stratified by ECOG PS, geographic region, and presence of liver metastases. Geographic region varied, with most patients living in the rest of the world category (43.8%), followed by Western Europe (41.9%), and the US (14.3%). Most patients had an ECOG PS of 1 (60.1% versus 59.6% in the enfortumab vedotin and chemotherapy arms, respectively), and metastatic disease at baseline (96.3% versus 94.1%, respectively), of whom 30.9% of all patients had liver metastases, while most patients had metastases to the viscera (77.7% versus 81.7%, respectively). A total of 27 patients had mismatched stratification (19 on liver metastasis and 8 on ECOG PS), which was accounted for using sensitivity analyses.17
Totals of 277 patients (92.0%) and 284 patients (92.5%) in the respective enfortumab vedotin and chemotherapy arms underwent prior procedures for primary cancer; the most common being TURBT (n = 318, 52.3%). Prior radiation therapy was received by 32.7% of patients overall — 96 (31.9%) in the enfortumab vedotin arm and 103 (33.6%) in the chemotherapy arm). The majority of patients received 2 prior lines of therapy in the locally advanced or metastatic setting (74.1% versus 77.5%, respectively). Most patients received cisplatin-based platinum regimens (64.1% versus 61.9% in the enfortumab vedotin and chemotherapy arms, respectively), and pembrolizumab was the most commonly received PD-1 or PD-L1 inhibitor received (48.5% versus 46.9%, respectively). In patients with prior CPI therapy, progressive disease was the most common best overall response, occurring in 50.7% of patients overall.17
Table 5: Summary of Baseline Characteristics (Safety Analysis Set)
Characteristic | EV-301 | |
|---|---|---|
Enfortumab vedotin (N = 301) | Chemotherapy (N = 307) | |
Age (years) | ||
Mean (SD) | 66.52 (9.11) | 66.81 (9.93) |
Median (range) | 68.0 (34.0 to 85.0) | 68.0 (30.0 to 88.0) |
< 65 | 108 (35.9) | 111 (36.2) |
65 to < 75 | 141 (46.8) | 128 (41.7) |
≥ 75 | 52 (17.3) | 68 (22.1) |
Sex, n (%) | ||
Male | 238 (79.1) | 232 (75.6) |
Female | 63 (20.9) | 75 (24.4) |
Race, n (%) | ||
White | 159 (52.8) | 155 (50.5) |
Black or African-American | 2 (0.7) | 2 (0.7) |
Asian | 97 (32.2) | 103 (33.6) |
Native Hawaiian or other Pacific Islander | 0 | 1 (0.3) |
Not reported | 43 (14.3) | 46 (15.0) |
Weight (kg) | ||
Mean (SD) | 74.51 (16.75) | 73.25 (15.90) |
Median (range) | 74.20 (40.0 to 146.5) | 72.20 (37.3 to 148.3) |
Body mass index (kg/m2) | ||
Mean (SD) | 25.68 (4.49) | 25.56 (4.86) |
Median (range) | 25.41 (15.9 to 43.0) | 25.05 (14.5 to 47.9) |
Region | ||
Western Europe | 126 (41.9) | 129 (42.0) |
US | 43 (14.3) | 44 (14.3) |
Rest of world | 132 (43.9) | 134 (43.6) |
ECOG PS, n (%) | ||
0 | 120 (39.9) | 124 (40.4) |
1 | 181 (60.1) | 183 (59.6) |
Extent of disease, n (%) | ||
Metastatic | 290 (96.3) | 289 (94.1) |
Locally advanced | 11 (3.7) | 18 (5.9) |
Primary disease site origin, n (%)a | ||
Upper tract | 98 (32.6) | 107 (34.9) |
Bladder/other | 203 (67.4) | 200 (65.1) |
Visceral metastasis, n (%)b | ||
Yes | 234 (77.7) | 250 (81.7) |
No | 67 (22.3) | 56 (18.3) |
Prior lines of systemic therapy under locally advanced or metastatic setting, n (%) | ||
1 | 39 (13.0) | 32 (10.4) |
2 | 223 (74.1) | 238 (77.5) |
≥ 3 | 39 (13.0) | 37 (12.1) |
Type of prior platinum-based treatment received, n (%) | ||
Cisplatin-based only | 193 (64.1) | 190 (61.9) |
Carboplatin-based only | 74 (24.6) | 85 (27.7) |
Both cisplatin- and carboplatin-based | 34 (11.3) | 31 (10.1) |
Type of prior CPI received, n (%) | ||
Nivolumab | 21 (7.0) | 13 (4.2) |
Pembrolizumab | 146 (48.5) | 144 (46.9) |
Atezolizumab | 86 (28.6) | 89 (29.0) |
Avelumab | 16 (5.3) | 13 (4.2) |
Durvalumab | 35 (11.6) | 56 (18.2) |
Other | 11 (3.7) | 11 (3.6) |
Best overall response on prior CPI therapy, n (%) | ||
Complete response | 16 (5.3) | 9 (2.9) |
Partial response | 45 (15.0) | 41 (13.4) |
Stable disease | 51 (16.9) | 63 (20.5) |
Progressive disease | 156 (51.8) | 152 (49.5) |
Nonevaluable | 6 (2.0) | 4 (1.3) |
Unknown | 20 (6.6) | 36 (11.7) |
Not applicable | 6 (2.0) | 2 (0.7) |
CPI = checkpoint inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; SD = standard deviation.
aUpper tract included renal pelvis and ureter. Bladder/other included urethra, bladder and other.
bPatients had baseline tumour results at the locations of lung, liver, spleen, adrenal gland, kidney, heart, colon, bone, or prostate gland.
Source: EV-301 Clinical Study Report.17
Interventions
Enfortumab Vedotin
Enfortumab vedotin was administered via IV infusion at a dose of 1.25 mg/kg over 30 minutes on days 1, 8, and 15 of every 28-day cycle until radiological disease progression per investigator assessment, or other discontinuation criteria were met, or upon study termination or completion. Enfortumab vedotin was not administered as an IV push or bolus. Weight-based dosing was calculated using the patient’s actual body weight on day 1 of each cycle. Doses were based on 100 kg for individuals weighing > 100 kg. The maximum dose of enfortumab permitted was 125 mg. Enfortumab vedotin was supplied by Astellas in single-use glass vials containing white, lyophilized powder for reconstitution. Each vial contained 30 mg of the study drug.17
Patients were observed during administration and for at least 60 minutes following the infusion during the first 3 cycles for redness, swelling, pain, and infection. Patients experiencing enfortumab vedotin IRRs may have received premedication before subsequent infusion, including pain medication, antihistamines, and corticosteroids 30 to 60 minutes before each infusion. If anaphylaxis occurred, treatment was immediately and permanently discontinued. If toxicities or AEs occurred on day 1 of any cycle and enfortumab vedotin could not be administered, the start of the cycle may have been delayed. If toxicities occurred on days 8 or 15 of any cycle and required the dose to be held for 3 days or longer, the dose(s) were to have been eliminated, rather than delayed. If a patient only received enfortumab vedotin on day 1 and needed to skip days 8 and 15, the patient could resume the next cycle as early as day 22 (new day 1), if the toxicity had resolved by then.17
Dose reduction to 1 mg/kg and to 0.75 mg/kg was allowed depending on the type and severity of toxicity. Patients with a dose reduction may have been re-escalated, provided the toxicity did not require discontinuation of the study drug. If the toxicity recurred, re-escalation was not permitted. Patients with grade 2 or higher corneal AEs were not permitted to re-escalate the dose. Dose interruptions for other enfortumab vedotin-associated toxicity were permitted at the discretion of the site investigator.17
Dose interruptions may have lasted up to 8 weeks (2 cycles). Dose interruptions for patients who were deriving clinical benefit from treatment may have been extended beyond 8 weeks if toxicity did not require permanent discontinuation. The schedule for response assessments was not adjusted in the event of dose interruption.17
For patients with hyperglycemia, blood glucose was assessed before each dose of study drug. The study drug was withheld in patients with blood glucose levels exceeding 250 mg/dL (13.9 mmol/L). Treatment discontinuation was required if blood glucose levels exceeded 500 mg/dL (27.8 mmol/L) and was considered related to enfortumab vedotin.17
Docetaxel, Paclitaxel, or Vinflunine
Local product labels and institutional guidelines were followed for the administration of chemotherapy agents.17
Docetaxel was administered as an IV infusion on day 1 of every 21-day cycle after all procedures and assessments had been completed, including the required premedication according to the local standard of care before day 1. Docetaxel was administered at 75 mg/m2 over a 1-hour infusion, and patients were observed during administration and at least 30 minutes following the first 3 cycles. Patients were pre-medicated with corticosteroids, such as dexamethasone 16 mg per day orally for 3 days starting 1 day before docetaxel administration to reduce the incidence and severity of fluid retention and hypersensitivity reactions.17
Vinflunine was administered as an IV infusion on day 1 of every 21-day cycle at a dose of 320 mg/m2 after all procedures and assessments had been completed. Patients were observed during administration and for at least 30 minutes following the first 3 cycles.17
Paclitaxel was administered as an IV infusion on day 1 of every 21-day cycle at a dose of 175 mg/m2 after all procedures and assessments had been completed. Paclitaxel was administered over 3 hours and patients were observed for at least 30 minutes following the first 3 cycles. Patients treated with paclitaxel were pre-medicated to prevent severe hypersensitivity; this may have consisted of dexamethasone 20 mg orally approximately 12 and 6 hours before paclitaxel, diphenhydramine 50 mg intravenously 30 to 60 minutes before paclitaxel, and cimetidine (300 mg) or ranitidine (50 mg) intravenously 30 to 60 minutes before paclitaxel, as determined by the investigator.17
Switching between chemotherapies during study treatment was not permitted. Dose modifications for chemotherapy regimens were considered according to local product labels. Transfusions or growth factors were used for hematologic toxicities of grade 3 or higher with chemotherapy, following institutional guidelines.17
Prior and Concomitant Therapy
Prior medication was considered medication with at least 1 dose taken before the date of the first dose of the study drug.26 Concomitant drug therapies were those medications or therapies with at least 1 dose taken between the date of the first dose and the date of the last dose of the study drug plus 30 days.17,26 Overall, 99.3% of enfortumab-treated and 99.7% of chemotherapy-treated patients were using concomitant drug therapies.17
Patients in the enfortumab vedotin arm who were concomitantly receiving strong CYP3A4 inhibitors or P-glycoprotein inhibitors were to be closely monitored for adverse reactions.17
In the chemotherapy arm (treatment with docetaxel, vinflunine, or paclitaxel), concomitant use of drugs that strongly inhibit or induce CYP3A4 were to be avoided. Additionally, medicinal products that prolong the QT-QTc interval were to be avoided in patients receiving vinflunine, and caution was to be exercised when paclitaxel was administered with strong inhibitors or inducers of CYP2C8.17
Subsequent Anticancer Therapy
Subsequent anticancer therapies included all systemic therapies and radiation, palliative radiation, and other therapies that irradiate or affect either target or non-target lesions. A total of 108 enfortumab-treated patients (35.9%) and 118 chemotherapy-treated patients (38.4%) received subsequent anticancer therapy after discontinuation of study treatment. The most common subsequent anticancer therapy was paclitaxel for both treatment arms (19 patients [6.3%] in the enfortumab vedotin arm and 18 patients [5.9%] in the chemotherapy arm).17
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 6. These end points are further summarized in the following section. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 6: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study EV-301 |
|---|---|
Overall survival | Primary efficacy end point |
PFS1 by RECIST 1.1 | Secondary efficacy end point |
ORR (CR + PR) by RECIST 1.1 | Secondary efficacy end point |
DCR (CR + PR + stable disease) by RECIST 1.1 | Secondary efficacy end point |
DOR by RECIST 1.1 | Secondary efficacy end point |
HRQoL and PRO parameters (EORTC QLQ-C30 and EQ-5D-5L) | Secondary efficacy end point |
PFS2 | Exploratory efficacy end point |
Time to response | Exploratory efficacy end point |
Health care resource utilization | Exploratory efficacy end point |
CR = complete response; DCR = disease control rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = EuroQol 5-Dimensions 5-levels questionnaire; PFS1 = progression-free survival on study therapy; PFS2 = progression-free survival on subsequent therapy; PR = partial response; PRO = patient-reported outcome; HRQoL = health-related quality of life; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1.
The primary outcome of the EV-301 trial was OS, defined as the time from randomization to the date of death from any cause.17
The secondary outcomes of EV-301 included PFS, ORR, duration of response (DOR), DCR, and HRQoL via the EORTC QLQ-C30 and EQ-5D-5L. For response outcomes, patients were evaluated based on investigator assessment according to Response Evaluation Criteria in Solid Tumours Version 1 (RECIST 1.1). Secondary outcomes were defined as follows:17
Progression-free survival on study therapy (PFS1) was defined as the time from the date of randomization until the date of radiological disease progression (per RECIST 1.1), or until death from any cause.
ORR was defined as the proportion of patients with complete or partial objective response based on the RECIST 1.1 per the investigator.
DCR was defined as the proportion of patients with a complete or partial objective response, or a stable disease based on RECIST 1.1.
DOR was defined as the time from the date of the first response CR or PR according to RECIST 1.1 (whichever is recorded first) that is subsequently confirmed as assessed by the investigator to the date of radiological progression or date of death for patients who achieved CR or PR.
HRQoL and PRO parameters were measured by the EORTC QLQ-C30 and EQ-5D-5L instruments. Detailed information on the scoring and validity of the EORTC QLQ-C30 and EQ-5D-5L is provided in Appendix 4.
The EORTC QLQ-C30 is a generic tool developed to assess the QoL of cancer patients. The tool yields the following scales: Global Health Status/QoL, functional scales, symptom scales or items, and financial impact. Outcome scores are computed using a linear transformation of the raw score such that scores range from 0 to 100. A higher score represents a higher (“better”) level of functioning, or a higher (“worse”) level of symptom.
The EQ-5D-5L is a generic, preference-based measure that indirectly measures the utility for health and generates an index-based summary score based upon societal preference weights. The EQ-5D-5L consists of 6 items that cover 5 main domains (mobility, self-care, usual activities, pain and discomfort, and anxiety or depression) and the EuroQol Visual Analogue Scale (EQ VAS) for health status.
Exploratory end points in EV-301 of interest included:17
Progression-free survival on subsequent therapy (PFS2), defined as the time from randomization to subsequent disease progression after initiation of new anticancer therapy, or death from any cause, whichever occurred first. Following PFS1, patients entered the long-term follow-up period and were followed per institutional guidelines (but not less than every 3 months from the date of the follow-up visit) for survival and progression status on subsequent therapy.
A health care resource utilization (HCRU) questionnaire focused on unplanned use of health care resources related to clinical events or AEs in patients assigned to treatment arms. Data on the incidence of emergency room visits, hospitalizations, and outpatient (either primary care or specialist) office visits were collected via the HCRU tool. Date and length of each hospitalization were collected.
Safety end points included AEs, laboratory tests, vital signs, electrocardiograms, and ECOG PS. All AEs were coded using the Medical Dictionary for Regulatory Activities by system organ class and preferred term and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events. An AE was considered “serious” if, in the view of either the investigator or sponsor, it resulted in death; was life-threatening (resulted in persistent or significant disability or incapacity or substantial disruption of the ability to conduct normal life functions); resulted in a congenital anomaly or birth defect; required inpatient hospitalization (except for planned procedures as allowed per study); or led to prolongation of hospitalization (except if prolongation of planned hospitalization is not caused by an AE), hospitalization for treatment/observation/examination caused by an AE, or other medically important events.17
Statistical Analysis
Sample Size and Power Calculation
The sample size calculation for EV-301 was based on the primary end point of OS. Assuming a 10% dropout rate, the estimated sample size was 600 patients, resulting in 85% power to detect a statistically significant difference at an overall 1-sided type I error rate of 0.025. Assuming an HR of 0.75 (the median OS values in enfortumab vedotin arm and chemotherapy arm were 10.7 months and 8 months, respectively), the final analysis was planned for when 439 death events had occurred, and 1 interim analysis was conducted at 285 death events (65% of planned events).17
The planned sample size also provided more than 90% power to detect statistically significant differences on secondary end points including PFS1 (assuming median PFS1 values in the enfortumab vedotin and chemotherapy arms were 6 months and 4 months, respectively), and ORR and DCR (assuming a 15% treatment difference between enfortumab vedotin and chemotherapy for both ORR and DCR).17
Interim and Final Analyses
The study design was a sequential group design with 2 planned analyses: 1 interim analysis and 1 final (primary) analysis. The interim analysis was to be performed after 285 deaths (65%), while the final analysis was to be performed after 439 death events. The interim analysis was conducted externally by an independent data analysis centre and the results reviewed by an independent data monitoring committee. At the interim analysis, OS was tested at the 1-sided 0.00541 significance level for efficacy according to the O’Brien-Fleming boundary as implemented by a Lan-DeMets alpha spending function. Secondary end points of PFS1, ORR, and DCR were tested sequentially when primary OS was rejected using the hierarchical gatekeeping procedure. The familywise type I error rate for this study was strongly controlled at 0.025 (1-sided) according to the multiplicity adjustment rule. The ORR and DCR were tested following rejection of both OS and PFS1 and the significance level of both ORR and DCR was 0.025.17
Predetermined early termination based on statistically significant OS results favouring enfortumab vedotin was suggested by the independent data analysis centre at the interim analysis.17,26
Multiplicity Adjustment
Multiplicity adjustment was incorporated within the interim and final analysis of the primary end point (OS), and between the primary and the selected secondary end points (PFS1, ORR, and DCR) using the familywise type I error rate at 2.5% (1-sided). The primary end point of OS was to be tested at both the interim and final analysis according to the O’Brien-Fleming boundary according to the Lan-DeMets method via East v6.5. As stated above, secondary outcomes were tested hierarchically if OS was rejected. The significance level of PFS1 at the interim and final analysis were to be based on the Pocock boundary following the Lan-DeMets method.17
Boundaries for OS and PFS1 were updated based on actual observed information fractions at the interim. Because study enrolment was completed in January 2020, and based on the definition of the response evaluable set (RES), the ORR and DCR analyses were ready after enrolment completion. Following the pre-specified multiplicity procedure, the ORR was to be tested once on the interim data (i.e., data per interim analysis cut-off) and was only to be tested when both OS and PFS1 were rejected either at interim or final analysis. The ORR analysis was to apply the interim data cut-off date when it was performed at the final analysis. The ORR test P value was to be compared to the significance level 0.025. Testing on DCR was similar to the testing on ORR, but DCR was to be tested only when ORR was rejected.17
Efficacy Analyses
All randomized patients were analyzed according to the treatment to which they were randomized. Efficacy analyses were conducted on the full analysis set (FAS) and RES. Imaging for both arms was performed at baseline and every 56 ± 7 days from the first dose of study treatment throughout the study until PFS1 was documented by radiological disease progression or the patient was lost to follow-up, died, withdrew study consent, or started a subsequent anticancer therapy. Images were analyzed for radiologic disease progression by study site investigator. Independent review of all or a sample of scans may have been considered following completion of PFS1 analyses.17
Descriptive statistics including the number of patients, mean, SD, median, minimum, and maximum were used for continuous variables. Frequencies and percentages are displayed for categorical data based on the number of patients with no missing data. Kaplan–Meier survival curves are displayed for time-to-event variables and median survival time was estimated with 2-sided 95% CIs. All nominal comparisons were made using 2-sided test at an alpha = 0.05 significance level.17
In the event of missing or partial dates, if the imputed date was after minimum (death date, cut-off date), minimum (death date, cut-off date) was used as imputed date.26
Missing or partial start and stop dates of AEs and concomitant medication were imputed using the following algorithm:26
Imputation rules for partial or missing stop dates:
If the month and year are present, then impute as the last day of that month.
If only the year is present, impute as December 31 of that year.
If the stop date is entirely missing, assume the event or medication is ongoing.
Imputation rules for partial or missing start dates are summarized in Table 7.
Table 7: Imputation Rules for Missing Dates
Start date | Stop date | |||||||
|---|---|---|---|---|---|---|---|---|
Complete: yyyymmdd | Partial: yyyymm | Partial: yyyy | Missing | |||||
< First dose | ≥ First dose | < First dose | ≥ First dose | < First dose | ≥ First dose | — | ||
Partial: yyyymm | = First dose yyyymm | 2 | 1 | NA | 1 | NA | 1 | 1 |
≠ First dose yyyymm | 2 | 2 | 2 | 2 | 2 | 2 | ||
Partial: yyyy | = First dose yyyy | 3 | 1 | 3 | 1 | NA | 1 | 1 |
≠ First dose yyyy | 3 | 3 | 3 | 3 | 3 | |||
Missing | 4 | 1 | 4 | 1 | 4 | 1 | 1 | |
1= impute as the date of first dose; 2 = impute as the first of the month; 3 = impute as January 1 of the year; 4 = impute as January 1 of the stop year; NA = not applicable; yyyy = year; yyyymm = year and month; yyyymmdd = year, month, and day.
Source: Study EV-301 Statistical Analysis Plan (2021 Jul 09 #6).
The imputed dates were used to determine whether an AE is or is not treatment-emergent. Listings of AEs and concomitant medications presented the actual partial dates; imputed dates were not shown.26
In the case of a partial starting date of subsequent anticancer therapy, the date was imputed to the first day of the month but not earlier than the last dosing date of the study drug. A month and year must have been present, or the date remained missing.26
Patients with missing baseline variable were excluded from the analysis of change from baseline.26
Patients who did not satisfy the criteria to be counted as responders or for whom data were insufficient to determine or confirm a response under the RECIST 1.1 guidelines were to be considered nonresponders in the final analysis of response rates. No imputation of data was done to determine individual patient response.26
Primary Efficacy End Point Analyses
The analysis population for the primary end point of OS was the FAS. Overall survival was defined as the time from the date of randomization until the documented date of death from any cause. Patients who were still alive at the data cut-off date (July 15, 2020) were censored at the last known alive date or at the analysis cut-off date, whichever is earlier. Overall survival (in days) was calculated as (date of death or censored) – (date of randomization) + 1.17
The distribution of OS was estimated for each treatment arm using Kaplan–Meier methodology, and the primary analysis comparing arm A and arm B was conducted using the log-rank test stratified by ECOG PS (0 versus 1), region (US, Western Europe, or the rest of world) and liver metastasis status (yes versus no) per interactive response technology. The stratified Cox proportional hazards model was used to estimate the HR and the corresponding 95% CIs with the same stratification factors as used for stratified log-rank test.17
Sensitivity Analyses
Sensitivity analysis for the primary end point of OS included:17
OS analysis based on unstratified log-rank test.
Imbalance in the use of subsequent anticancer therapy (including subsequent use of enfortumab vedotin) may have potentially biased the inference in OS. The following methods were to be used to assess the impact of the use of subsequent anticancer therapy on OS:
Rank-preserving structural failure time method. This method was to assess the impact on patients in arm B who took enfortumab vedotin as a subsequent therapy by reconstructing the patients’ survival durations, as if they had never received enfortumab vedotin.
Inverse probability of censoring weights (IPCW) method. Patients who took subsequent therapy were censored at the time of sequent anticancer therapy but were weighted according to their probability of taking subsequent therapy.
To assess the potential impact of COVID-19, the following sensitivity analyses may have been conducted:17
same as OS primary analysis, except those patients who died from COVID-19 were censored at the death date
same as OS primary analysis, but patients who died from of COVID-19 were excluded from the analysis.
An ad hoc sensitivity analysis using the actual stratification factors was conducted to account for any mismatched stratification factors at baseline.17
Secondary Efficacy End Point Analyses
Secondary efficacy end points of PFS1, ORR, DOR, and DCR were assessed by the investigator. Response and progression were evaluated using RECIST 1.1.17 Progression-free survival on study therapy was assessed in the FAS and was a secondary end point of the EV-301 trial. Formal statistical comparison of arm A (enfortumab vedotin) and arm B (chemotherapy) was performed per the planned multiplicity adjustment rule. Values for PFS were estimated using the Kaplan–Meier method, and the stratified Cox proportional hazard model was used to estimate HRs and 95% CIs using the same stratification factors as those used for OS. Sensitivity analyses for PFS1 included analysis based on the unstratified log-rank test, sensitivity analysis on FAS when removing the censoring of progressive disease or death events occurring after missing 2 consecutive tumour assessments, and sensitivity analyses to assess the potential impact of COVID-19 as detailed for OS. Secondary end point analyses of ORR and DCR were conducted in the RES using the stratified Cochran-Mantel-Haenszel test. Both ORR and DCR including 95% CIs were estimated for each treatment arm. Formal statistical comparison of enfortumab vedotin and chemotherapy was conducted according to the planned multiplicity adjustment rule only. Additional sensitivity analysis for ORR and DCR included the comparison of ORR and DCR regardless of confirmation. For patients with confirmed CR or PR from the analysis of ORR, the distribution of DOR was estimated using the Kaplan–Meier method for each treatment arm.17
Health-related quality of life was a secondary end point of the EV-301 trial and was measured using the EORTC QLQ-C30 and EQ-5D-5L.17 For the EORTC QLQ-C30, instrument completion rate at each analysis was reported. Completion rate (i.e., unadjusted) at each analysis visit was calculated as the number of patients meeting the minimum requirements for scoring at least 1 domain of the instrument divided by the number of patients in the FAS population. The compliance rate (adjusted) at each analysis visit was calculated among patients who were expected to have PRO assessments. The following was provided:17
number and percent of patients with all EORTC QLQ-C30 questions completed
number and percent of patients for whom at least 1 subscale of EORTC QLQ-C30 can be calculated (minimum requirements for scoring of the instrument)
number and percent of patients with at least 1 question completed on the EORTC QLQ-C30.
Means and change from baseline at each scheduled assessment were reported for each of the EORTC QLQ-C30 subscales. The analyses included data from the baseline assessment through the last available data for all patients in the FAS. Change from baseline in the Global Health Status (a 2-item QoL subscale of the EORTC QLQ-C30) score for enfortumab vedotin and chemotherapy at week 12 was analyzed using a restricted maximum likelihood–based repeated measures approach (mixed model for repeated measures [MMRM]). The week 12 time point was selected to minimize the impact of missing data given that the median of PFS1 for the chemotherapy arm was 4 months. Analysis of the EORTC QLQ-C30 was based on observed data (i.e., data collected at each time point without carrying forward previous values). Only patients with a baseline and at least 1 post-baseline score were included in the analysis. Data from a limited number of PRO assessments may have been used in case of substantial dropout (i.e., analysis was to be limited to time points at which at least 10% of subjects had non-missing data in both treatment arms) by week 12.17
For the EQ-5D-5L, instrument completion and compliance rates at each analysis visit were reported. Completion rate (i.e., unadjusted) at each analysis visit was calculated as the number of patients for whom either the utility index or EQ VAS score could be calculated (minimum requirements for scoring of the instrument) divided by the number of patients in the FAS population. The compliance rate (adjusted) at each analysis visit was calculated among patients who were expected to have PRO assessments. The following were provided:17
the number and percent of patients with all questions completed (i.e., all 5 items and EQ VAS)
the number and percent of patients for whom either the utility index or the VAS score could be calculated (minimum requirements for scoring of the instrument)
the number and percent of patients with at least 1 question completed on the EQ-5D-5L or EQ VAS.
The completion and compliance rates by treatment arm at each analysis visit were also provided graphically by means of a line graph. Descriptive characteristics of the EQ-5D-5L were summarized from baseline assessment through the last available data. The frequency and the percentage of reported problems for each level for each dimension were provided. All time point data were included and summarized. Scores for the EQ VAS were summarized by treatment arm at each visit using descriptive statistics.17
Due to a potential data integrity issue for electronic reporting at site 39004, sensitivity analysis for the MMRM analysis for the EQ-5D-5L, as well as the EORTC QLQ-C30 and for the mean change from baseline for each of the domains for the EORTC QLQ-C30, was conducted by removing the data from site 39004.17,26
Changes to Planned Analyses
For PRO outcomes, the pre-specified analysis approach created a week-by-week cut-off that started in the midpoint of each week and carried forward into the midpoint of the following week, which unintentionally created a mismatch that artificially lowered the calculation of compliance rates, particularly at the week 12 time point. As a result, questionnaire completion and compliance rates and the analyses for EORTC QLQ-C30 and EQ-5D were also assessed in ad hoc analyses that aligned the windows for the start and end of each week.17
Subgroup Analyses
Pre-specified subgroup analyses for OS, PFS1, and ORR were conducted to determine if the treatment effect was consistent across the following subgroups: age (younger than 65 versus 65 or older and younger than 75 versus 75 or older), sex (male versus female), region (Western Europe versus US versus rest of world), ECOG PS (0 versus 1), liver metastasis (yes versus no), pre-selected control therapy (paclitaxel versus docetaxel versus vinflunine), primary tumour site (upper tract versus bladder or other), prior lines of systemic therapy (1 to 2 lines versus 3 or more lines), and best response to prior CPI (responder versus nonresponder). Treatment-effect estimates and 95% CIs were provided.17,26
Exploratory Efficacy Analyses
The FAS was used for the analysis of PFS2 for each treatment arm following the Kaplan–Meier methodology. Enfortumab vedotin and chemotherapy were compared using log-rank tests stratified by ECOG PS (0 versus 1), region (US, Europe, and the rest of world) and liver metastasis status (yes versus no) per interactive response technology. The stratified Cox proportional hazards model was used to estimate the HR and the corresponding 95% CI.17
Incidences of HCRU, including emergency room visits, hospitalizations, and outpatient (either primary care or specialist) office visits, were summarized by treatment arm by each visit. The duration of hospital stays was also summarized by treatment arm using descriptive statistics (mean, SD, median, minimum, and maximum).17
Treatment Discontinuation and Follow-Up
Following discontinuation, patients had a follow-up visit 30 days after their last dose of study drug for safety assessments. If a patient discontinued the study drug before PFS1, the patient entered the post-treatment follow-up period and continued imaging assessments every 56 days until PFS1 was documented or a subsequent anticancer treatment was initiated, whichever occurred first. Following PFS1, patients entered the long-term follow-up period and were to be followed at a minimum of every 3 months from the date of the follow-up visit for survival status and progression status on subsequent therapy (i.e., PFS2). Patients were followed until PFS2 was documented or the patient started another anticancer treatment, whichever occurred first. Following PFS2, patients entered the survival follow-up period and were followed for survival status every 3 months until death, loss to follow-up, withdrawal of study consent, or study termination by sponsor. Patients were eligible to continue receiving treatment until they met a discontinuation criterion or upon study termination or completion, whichever occurred first.17
Safety Analyses
All safety analyses were presented as the number and percentage of patients by treatment arm and overall, for the safety analysis set. All AEs, TEAEs, and SAEs recorded on treatment, including those within 30 days from the last study treatment, were summarized by number and percentage of patients reporting AEs. Summary tables include patient counts as opposed to AE counts.17
Adverse events of special interest (AESIs), such as IRRs, ocular toxicity, skin reactions, and peripheral neuropathy, were summarized by treatment arm and overall and classified by sponsor-specific query (SSQ), customized medical query, or standard Medical Dictionary for Regulatory Activities query (SMQ) and preferred term. Time to onset of a specific AESI was calculated as time from the first dose of study drug to the start of the first TEAE that met the respective search criteria.17
Analysis Populations
The following analysis populations were defined in Study EV-301:17
The FAS consisted of all randomized patients. This analysis set is in compliance with the intent-to-treat principle that includes all randomized patients. The FAS was the primary analysis set for efficacy analyses except for response-related efficacy end points. Demographic and baseline characteristics were summarized for the FAS.
The safety analysis set consisted of all patients who received any amount of study drug and was used for safety analyses. Patients were analyzed based on actual treatment received.
The RES was defined as all patients in the FAS who had measurable disease (according to RECIST 1.1) as determined by the investigator at baseline. The RES was used for primary efficacy analysis of response-related end points (e.g., ORR and DCR). Patients were analyzed by assigned treatment.
Protocol Amendments and Deviations
Three substantial amendments were made to the study protocol. A total of 128 patients were randomized under the original protocol, and 480 were randomized under the second version of the protocol incorporating Amendment 1. At the time of the interim analysis, no patients had been enrolled under the third or fourth versions of the protocol, which incorporated Amendments 2 and 3.17
The first amendment included some additional exclusion criteria for the EV-301 population that were not outlined in the original study protocol, many of which were related to contraindications or hypersensitivities on the product labels of the comparators. Additional changes included hematology, biochemistry, and pregnancy tests in women of childbearing potential, as well as concomitant medication assessments included at follow-up visits. Amendment 2 increased the number of death events in the final and interim analyses to increase the power of the study from 80% to 85%. Amendment 3 included a crossover extension phase that was added to the design of the study.17,25,26 Overall, none of the protocol amendments affected the conduct or integrity of the study, nor did they affect the patients’ safety.
As of the cut-off date, 33 patients — 21 (7.0%) in the enfortumab vedotin arm and 12 (3.9%) in the chemotherapy arm — had 1 or more major protocol deviations. The most frequent protocol deviation involved patients entering the study despite not satisfying the entry criteria. This occurred in 8 patients in the enfortumab vedotin arm (2.7%) and 9 patients in the chemotherapy arm (2.9%).17
Eight patients (2.7%) in the enfortumab vedotin arm compared to 2 patients (0.7%) in the chemotherapy arm developed withdrawal criteria during the study but were not withdrawn. Four patients (1.3%) in the enfortumab vedotin arm received the wrong treatment or incorrect dose; 1 received weight-based dosing rather than weight-capped dosing and 3 had a total dose ± 10% of the recommended dose. Last, 1 patient (0.3%) in each arm received excluded concomitant treatments.17
Results
Patient Disposition
Study EV-301 was a randomized, open-label, phase III clinical trial. Table 8 summarizes the disposition of enrolled patients. A total of 745 patients were screened for eligibility and 608 were randomized to receive enfortumab vedotin (n = 301) or chemotherapy (n = 307). As of the data cut-off date, 56 patients (18.6%) in the enfortumab vedotin arm and 22 patients (7.2%) in the chemotherapy arm remained on the study drug. A greater proportion of patients in the chemotherapy arm discontinued from the study compared to the enfortumab vedotin arm (92.8% versus 81.4%, respectively), most due to patient withdrawal (8.8% versus 5.0%, respectively) and physician decision (7.2% versus 2.3%, respectively). The most common reasons for treatment discontinuation in both treatment arms were disease progression (177 patients [58.8%] in the enfortumab vedotin arm and 180 patients [58.6%] in the chemotherapy arm) and AEs (42 patients [14.0%] in the enfortumab vedotin arm and 46 patients [15.0%] in the chemotherapy arm).17
Table 8: Patient Disposition (Safety Analysis Set)
Patient disposition | Study EV-301 | |
|---|---|---|
Enfortumab vedotin | Chemotherapy | |
Screened, N | 745 | |
Randomized, N (%) | 608 | |
Randomized to open-label drug | 301 | 307 |
Discontinued from study, N (%) | 245 (81.4) | 285 (92.8) |
Reason for discontinuation, N (%) | ||
Adverse events | 42 (14.0) | 46 (15.0) |
Death | 2 (0.7) | 2 (0.7) |
Lost to follow-up | 0 | 1 (0.3) |
Progressive disease | 177 (58.8) | 180 (58.6) |
Protocol deviation | 1 (0.3) | 1 (0.3) |
Withdrawal by patient | 15 (5.0) | 27 (8.8) |
Physician decision | 7 (2.3) | 22 (7.2) |
Othera | 1 (0.3) | 6 (2.0) |
Full analysis set, N (%) | 301 (100) | 307 (100) |
Safety analysis set, N (%) | 296 (98.3) | 291 (94.8) |
Response evaluable set, N (%) | 288 (95.7) | 296 (96.4) |
Note: Data cut-off date: July 15, 2020.
aReasons for other: enfortumab vedotin arm: coronavirus disease 2019 travel restrictions; chemotherapy arm: hospitalization; 2 patients received standard number of cycles of chemotherapy consistent with standard of care; chose weekly paclitaxel; patient had known sensitivity to alcohol confirmed after randomization; surgery.
Source: EV-301 Clinical Study Report.17
Exposure to Study Treatments
Exposure to study treatments is summarized in Table 9. The median duration of treatment with enfortumab vedotin was 4.99 months (range = 0.5 to 19.4), and 3.45 (range = 0.2 to 15.0) months for the chemotherapy arm, with individual median durations of treatment of 2.23 months, 3.68 months, and 3.91 months for patients treated with docetaxel, paclitaxel, and vinflunine, respectively.17
The median relative dose intensities were 80.73% for the enfortumab vedotin arm and 97.36% for the chemotherapy arm. The median relative dose intensities for docetaxel, paclitaxel, and vinflunine were 98.17%, 96.71%, and 92.54%, respectively.17
Table 9: Study Drug Exposure and Treatment Compliance (Safety Analysis Set)
Measure | Enfortumab vedotin (N = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Combined (N = 291) | Docetaxel (N = 109) | Paclitaxel (N = 107) | Vinflunine (N = 75) | ||
Duration of exposure (months)a | |||||
Mean (SD) | 5.36 (3.72) | 3.96 (2.95) | 3.62 (2.85) | 3.90 (2.61) | 4.55 (3.45) |
Median (range) | 4.99 (0.5 to 19.4) | 3.45 (0.2 to 15.0) | 2.23 (0.3 to 15.0) | 3.68 (0.3 to 12.3) | 3.91 (0.2 to 13.9) |
Relative dose intensity (%)b | |||||
Mean (SD) | 79.35 (17.52) | 91.76 (11.61) | 91.78 (12.29) | 92.25 (10.25) | 91.01 (12.53) |
Median (range) | 80.73 (30.6 to 104.9) | 97.36 (32.5 to 114.2) | 98.17 (32.5 to 105.0) | 96.71 (57.9 to 107.2) | 92.54 (37.6 to 114.2) |
SD = standard deviation.
Note: Data cut-off: July 15, 2020.
aCalculated as: last date of exposure – first dose date + 1. Enfortumab vedotin cycle length is 28 days, chemotherapy cycle length is 21 days.
bCalculated as: (dose intensity/planned dose intensity) × 100.
Source: EV-301 Clinical Study Report.17
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported.
Overall Survival
At the pre-planned interim analysis, a total of 299 death events had occurred. The independent data analysis centre then informed the sponsor that the efficacy boundary for OS had been crossed and recommended stopping the study for efficacy based on an August 20, 2020, data snapshot, with a July 15, 2020, data cut-off. For OS at the interim, the HR for enfortumab vedotin versus chemotherapy was 0.70 (95% CI, 0.56 to 0.89). The study database was subsequently locked on September 15, 2020 (data cut-off, July 15, 2020) for primary efficacy analysis, and the protocol was amended to allow for patients in the chemotherapy arm to cross over to receive enfortumab vedotin therapy (Protocol Amendment 3).17
In the final primary efficacy analysis (Table 10), a total of 301 (68.6%) of the planned 439 death events occurred in the FAS with 11.1 months of follow-up. The HR for the enfortumab vedotin versus chemotherapy was 0.702 (95% CI, 0.556 to 0.886) in favour of enfortumab vedotin. The median OS was 12.88 months (95% CI, 10.58 to 15.21) in the enfortumab vedotin arm and 8.97 months (95% CI, 8.05 to 10.74) in the chemotherapy arm (P = 0.00142). The OS rate was 77.9% versus 69.5% and 51.5% versus 39.2% for enfortumab vedotin and chemotherapy arms at 6 and 12 months, respectively.17 Figure 4 summarizes OS in a Kaplan–Meier plot.
Table 10: Primary Efficacy Analysis Based on Overall Survival (Full Analysis Set)
Measure | Enfortumab vedotin (n = 301) | Chemotherapy (n = 307) |
|---|---|---|
Overall survival | ||
Deaths, n (%) | 134 (44.5) | 167 (54.4) |
Median (95% CI) overall survival (months)a | 12.88 (10.58 to 15.21) | 8.97 (8.05 to 10.74) |
1-sided P valueb | 0.00142 | |
Stratified analysisc | ||
HR (95% CI)d | 0.702 (0.556 to 0.886) | |
Overall survival rate, % (95% CI)e | ||
6 months | 77.9 (72.74 to 82.25) | 69.5 (63.85 to 74.38) |
12 months | 51.5 (44.63 to 58.03) | 39.2 (32.60 to 45.64) |
CI = confidence interval.
aBased on Kaplan–Meier estimate.
bBased on log-rank test of the Kaplan–Meier curves. The P value of OS is less than or equal to the predetermined 1-sided significance level of 0.00679 based on the number of observed deaths.
cStratification factors were ECOG PS, geographic region, and liver metastasis per interactive response technology.
dBased on Cox proportional hazards model with treatment, ECOG PS, geographic region, and liver metastasis as the explanatory variables. Assuming proportional hazards, a hazard ratio of less than 1 indicates a reduction in hazard rate in favour of the treatment arm.
eSurvival rate and 95% CI were estimated using the Kaplan–Meier method and Greenwood formula.
Source: EV-301 Clinical Study Report.17
Figure 4: Kaplan–Meier Plot of Overall Survival (Full Analysis Set)
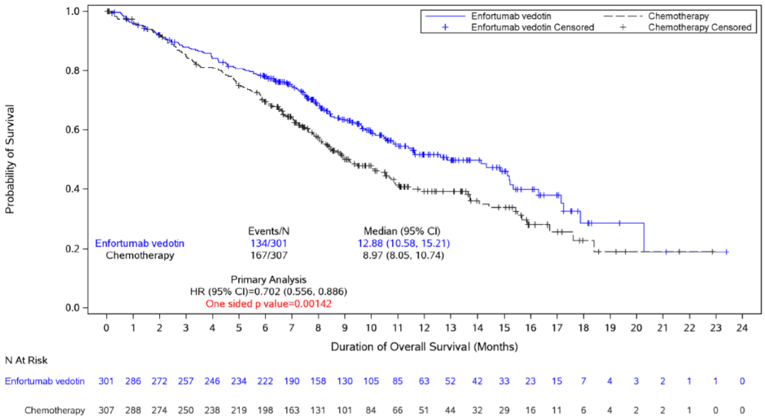
CI = confidence interval; HR = hazard ratio.
Source: EV-301 Clinical Study Report.17
Sensitivity Analysis
Pre-specified sensitivity analyses evaluating the effect of various censoring rules on OS are summarized in Table 11. Results for all sensitivity analyses (rank-preserving structural failure time method, IPCW method, and ad hoc sensitivity by stratification factors) were consistent with the primary analysis, with all methods demonstrating statistically significant improvements in OS compared to chemotherapy. For the rank-preserving structural failure time method, only 4 patients from the chemotherapy arm received subsequent therapy with enfortumab vedotin, and the results are similar. Results for the IPCW method were based on the 95 patients (31.6%) in the enfortumab vedotin arm the and 102 patients (33.2%) in the chemotherapy arm who received subsequent therapy as of the data cut-off date. Results for the IPCW method varied most from the primary analysis, with a HR of 0.630 (95% CI, 0.435 to 0.912) for enfortumab versus chemotherapy. The median OS was 17.87 months (95% CI, 11.63 to not evaluable) for enfortumab vedotin, and 10.32 months (95% CI, 8.31 to 15.67) for chemotherapy (P = 0.001). Results for sensitivity analyses based on actual stratification factors were also comparable to those of the primary analysis as there was only a total of 14 patients in the enfortumab vedotin arm and 13 patients in the chemotherapy arm with mismatched stratification at baseline.17
Table 11: Sensitivity Analysis — Overall Survival (Full Analysis Set)
Measure | Enfortumab vedotin (n = 301) | Chemotherapy (n = 307) |
|---|---|---|
Adjusting in chemotherapy arm based on the RPSFT method | ||
Median (95% CI) overall survival (months)a | 12.88 (10.58 to 15.21) | 8.94 (8.05 to NE) |
1-sided P valueb | 0.001 | |
HR (95% CI)cb | 0.705 (0.516 to 0.853) | |
Adjusting in chemotherapy arm based on the IPCW method | ||
Median (95% CI) overall survival (months)a | 17.87 (11.63 to NE) | 10.32 (8.31 to 15.67) |
1-sided P valued | 0.001 | |
HR (95% CI)e | 0.630 (0.435 to 0.912) | |
Adjusting in chemotherapy arm based on the actual stratification factors (ad hoc) | ||
Median (95% CI) overall survival (months)a | 12.88 (10.58 to 15.21) | 8.97 (8.05 to 10.74) |
1-sided P valuef | 0.002 | |
HR (95% CI)g | 0.712 (0.562 to 0.901) | |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; IPCW = inverse probability of censoring weights; NE = not evaluable; RPSFT = rank-preserving structural failure time.
aBased on Kaplan–Meier estimate.
bP value was calculated from 1,000 simulations.
cStratified Cox proportional hazards model was analyzed on 1,000 bootstrapping simulated datasets. The CI was from the 2.5 percentile and 97.5 percentile of 1,000 simulations.
dBased on weighted stratified log-rank test of the Kaplan–Meier curves.
eBased on weighted stratified Cox proportional hazards model with treatment as the explanatory variables. Stratification factors were ECOG PS, geographic region, and liver metastasis per interactive response technologies. Weight was calculated from 2 logistic models. One model included only baseline covariates (age, primary tumour site, and prior lines of therapy in locally advanced or metastatic settings. The other model included both baseline covariates and time-dependent variables (sum of diameter and ECOG assessments).
fBased on log-rank test of the Kaplan–Meier curves.
gBased on Cox proportional hazards model with treatment, baseline ECOG PS, region, and baseline liver metastasis as the explanatory variables. Assuming proportional hazards, a hazard ratio of less than 1 indicates a reduction in hazard rate in favour of the treatment arm.
Source: EV-301 Clinical Study Report.17
Subgroup Analysis
Subgroups of interest outlined in the review protocol (Table 3) for OS in the FAS are summarized in Table 12. Across subgroups of interest, results were mostly consistent with the overall population. The subgroups of patients with an ECOG PS of 0, primary tumour site of the upper tract, patients with 3 or more prior lines of systemic therapy, and responders to prior CPI were favoured over chemotherapy (HR = 0.810 [95% CI, 0.530 to 1.240], HR = 0.848 [95% CI, 0.567 to 1.269], HR = 0.875 [95% CI, 0.466 to 1.644], and HR = 0.630 [95% CI, 0.338 to 1.174], respectively);17 however, there was a small number of patients within these subgroups.
Table 12: Subgroup Analysis for Overall Survival, Investigator Assessment (Full Analysis Set)
Subgroup | Enfortumab vedotin event/N | Chemotherapy event/N | Hazard ratio (95% CI)a |
|---|---|---|---|
All subjectsb | 134/301 | 167/307 | 0.702 (0.556 to 0.886) |
ECOG PS per IRT | |||
0 | 40/120 | 46/124 | 0.810 (0.530 to 1.240) |
1 | 94/181 | 121/183 | 0.666 (0.508 to 0.873) |
Liver metastases per IRT | |||
Yes | 53/93 | 63/95 | 0.660 (0.456 to 0.957) |
No | 81/208 | 104/212 | 0.734 (0.549 to 0.981) |
Site of primary tumour | |||
Upper tract | 44/98 | 52/107 | 0.848 (0.567 to 1.269) |
Bladder/other | 90/203 | 115/200 | 0.666 (0.506 to 0.879) |
Prior lines of systemic therapy | |||
1 to 2 | 115/262 | 147/270 | 0.692 (0.542 to 0.883) |
≥ 3 | 19/39 | 20/37 | 0.875 (0.466 to 1.644) |
Best response to CPI | |||
Responder | 18/61 | 23/50 | 0.630 (0.338 to 1.174) |
Nonresponder | 100/207 | 120/215 | 0.757 (0.580 to 0.988) |
CPI = checkpoint inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; IRT = interactive response technology.
aIn each subgroup, the hazard ratio was estimated using an unstratified Cox proportional hazards model with treatment. Assuming proportional hazards, an HR of less than 1 indicates a reduction in hazard rate in favour of the treatment arm.
bThe HR reported for all subjects was based on stratified analysis. Stratification factors were ECOG PS, geographic region, and liver metastasis per interactive response technologies.
Source: EV-301 Clinical Study Report.17
Progression-Free Survival
Progression-Free Survival on Study Therapy
Progression-free survival was a secondary end point of EV-301; the results are summarized in Table 13 and by Kaplan–Meier plot in Figure 5. Enfortumab vedotin was associated with a statistically significantly prolonged PFS compared to chemotherapy (HR = 0.615; 95% CI, 0.505 to 0.748). The median PFS1 was 5.55 months (95% CI, 5.32 to 5.82) in the enfortumab vedotin arm, and 3.71 months (95% CI, 3.52 to 3.94) in the chemotherapy arm (P < 0.00001). The 6- and 12-month PFS rates for enfortumab vedotin and chemotherapy were 44.0% and 28.2%, and 21.7% and 8.3%, respectively.17
Table 13: PFS1 According to Investigator Assessment (Full Analysis Set)
Progression-free survival measure | Enfortumab vedotin (n = 301) | Chemotherapy (n = 307) |
|---|---|---|
PFS events, n (%) | 201 (66.8) | 231 (75.2) |
Radiographical progression | 172 (57.1) | 195 (63.5) |
Death without documented progression | 29 (9.6) | 36 (11.7) |
Censored, n (%) | 100 (33.2) | 76 (24.8) |
No PFS event | 89 (29.6) | 64 (20.8) |
PFS event after new anticancer therapy | 8 (2.7) | 9 (2.9) |
PFS event after missing ≥ 2 consecutive assessments | 3 (1.0) | 3 (1.0) |
Median (95% CI) PFS (months)a | 5.55 (5.32 to 5.82) | 3.71 (3.52 to 3.94) |
Stratified analysisb | ||
Hazard ratio (95% CI)c | 0.615 (0.505 to 0.748) | |
1-sided P valued | < 0.00001e | |
PFS rate, % (95% CI)f | ||
6 months | 44.0 (37.96 to 49.84) | 28.2 (22.85 to 33.76) |
12 months | 21.7 (16.26 to 27.71) | 8.3 (4.61 to 13.36) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; PFS = progression-free survival; PFS1 = progression-free survival on study therapy.
aBased on Kaplan–Meier estimate.
bStratification factors were ECOG PS, geographic region, and liver metastasis per interactive response technologies.
cBased on Cox proportional hazards model with treatment, ECOG PS, geographic region, and liver metastasis as the explanatory variables. Assuming proportional hazards, a hazard ratio of less than 1 indicates a reduction in the hazard rate in favour of the treatment arm. For unstratified analysis, treatment is the only explanatory variable.
dBased on log-rank test of the Kaplan–Meier curves.
eFor stratified analysis, the P value of overall survival is less than or equal to the predetermined 1-sided significance level of 0.02189 based on the number of observed PFS events.
fPFS rate and 95% CI were estimated using the Kaplan–Meier method and Greenwood formula.
Source: EV-301 Clinical Study Report.17
Figure 5: Kaplan–Meier Plot of PFS1, Investigator Assessment (Full Analysis Set)
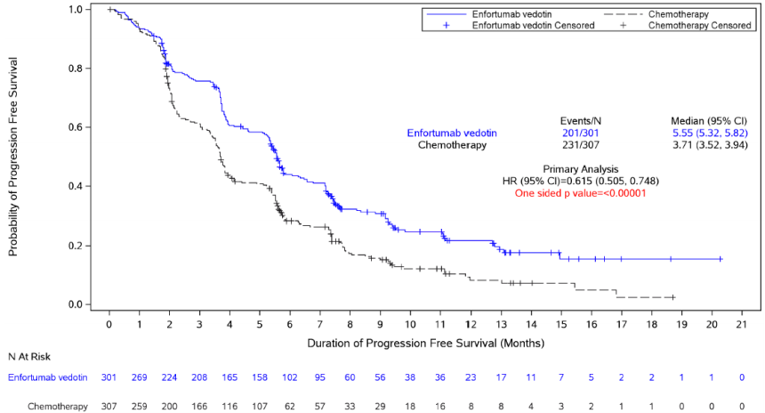
CI = confidence interval; HR = hazard ratio.
Source: EV-301 Clinical Study Report.17
Sensitivity Analysis
Pre-specified sensitivity analyses evaluating the effect of various censoring rules on PFS are summarized in Table 30. Sensitivity analyses were comparable to the primary analysis of PFS1, with an HR for PFS for enfortumab vedotin versus chemotherapy of 0.616 (95% CI, 0.507 to 0.749) in favour of enfortumab vedotin.17
Subgroup Analysis
Subgroups of interest outlined in the review protocol (Table 3) for PFS in the FAS are summarized in Table 31. Results of the subgroup analyses were consistent with the overall PFS1 observed, ranging from HR = 0.511 to HR = 0.716 for subgroups of interest, including ECOG PS, liver metastasis, primary tumour site, prior lines of systemic therapy, and best response to prior CPI.17
Progression-Free Survival on Subsequent Therapy
Progression-free survival on subsequent therapy was defined as the time from randomization to subsequent disease progression after initiation of new anticancer therapy, or death from any cause, and was an exploratory end point of EV-301. Results for PFS2 are summarized in Table 32 of Appendix 3.17
Health-Related Quality of Life
EORTC QLQ-C30
Health-related quality of life as assessed by the EORTC QLQ-C30 was a secondary outcome of EV-301. At baseline, 90.7% and 88.6% of patients completed the EORTC QLQ-C30 in the enfortumab vedotin and chemotherapy arms, respectively. Completion and compliance rates for the EORTC QLQ-C30 generally diminished with each visit (Figure 7, Appendix 3).17
Results of the EORTC QLQ-C30 functional and symptom scales from baseline to week 12 are summarized in Table 14. Following ad hoc adjustments (due to mismatched data entry and visit schedule), the mean baseline Global Health Status scores for the adjusted analysis were 63.83 (19.89) in the enfortumab vedotin arm and 64.58 (19.19) in the chemotherapy arm. The change from baseline to week 12 by MMRM in the FAS (n = 227 in enfortumab vedotin arm and n = 193 in chemotherapy arm) was −2.825 (standard error = 1.348) in the enfortumab vedotin arm, and −4.996 (standard error = 1.479) in the chemotherapy arm (P = 0.2429). Results of the sensitivity analysis excluding 1 study site were similar to the base-case MMRM.17
In the enfortumab vedotin arm, mean change from baseline scores from the EORTC QLQ-C30 ranged from −0.92 (SD = 15.76) in cognitive functioning to −5.12 (SD = 23.80) in social functioning, with an increase of 2.17 (SD = 16.20] in the emotional functioning score. In the chemotherapy arm, reduction in score from baseline to week 12 ranged from −0.49 (SD = 16.66) in cognitive functioning to −9.15 (SD = 26.29) in role functioning, with an increase of 3.27 (SD = 18.06) in emotional functioning.
For enfortumab vedotin, a decrease in symptom burden from baseline to week 12 was observed for nausea and vomiting (−0.39 [SD = 16.73]), insomnia (−3.67 [SD = 30.06]), constipation (−6.04 [SD = 27.99]), and financial difficulties (−0.26 [SD = 20.36]) scores, with the largest numerical improvement in reduction of self-reported pain (−6.96 [SD = 26.26]), while increases were demonstrated in other symptom scales. Conversely, improvements in insomnia (−1.63 [SD = 27.90]) and constipation (−0.98 [SD = 24.13]) scores occurred from baseline to week 12 in the chemotherapy arm, while the other symptom scores worsened.17 In an additional analysis provided to CADTH, enfortumab vedotin was favoured over chemotherapy in reduction of reported pain symptoms (−5.73; 95% CI, −10.80 to −0.66). Improvement in appetite loss favoured the chemotherapy arm (7.29; 95% CI, 0.90 to 13.69).27
Table 14: Summary of EORTC QLQ-C30 Functional and Symptom Scales, Result and Change From Baseline to Week 12 (Full Analysis Set)
EORTC QLQ-C30 domain | Enfortumab vedotin | Chemotherapy | ||||
|---|---|---|---|---|---|---|
Baseline (n = 273) | Week 12 (n = 133) | Change from baseline | Baseline (n = 272) | Week 12 (n = 104) | Change from baseline | |
Functional scales, mean (SD) | ||||||
Physical functioning score | 75.21 (22.34) | 76.74 (21.92) | −1.36 (19.38) | 75.05 (21.06) | 73.97 (20.62) | −6.21 (16.44) |
Global health status | 63.83 (19.89) | 64.35 (18.90) | −2.30 (18.02) | 64.58 (19.19) | 64.42 (18.55) | −5.72 (16.04) |
Role functioning score | 74.30 (28.40) | 74.56 (27.18) | −3.28 (25.29) | 72.06 (27.23) | 71.31 (26.42) | −9.15 (26.29) |
Emotional functioning score | 78.54 (20.65) | 82.64 (17.83) | 2.17 (16.20) | 76.53 (19.87) | 83.73 (18.48) | 3.27 (18.06) |
Cognitive functioning score | 85.53 (18.11) | 86.22 (17.47) | −0.92 (15.76) | 84.01 (19.80) | 87.18 (16.46) | −0.49 (16.66) |
Social functioning score | 80.10 (25.34) | 78.57 (24.92) | −5.12 (23.80) | 80.39 (23.52) | 79.33 (24.08) | −5.39 (23.12) |
Symptom scales, mean (SD) | ||||||
Fatigue symptom score | 33.58 (24.97) | 34.67 (22.93) | 3.94 (23.32) | 35.17 (24.14) | 33.87 (22.93) | 6.64 (22.56) |
Nausea and vomiting symptom score | 6.04 (14.19) | 4.76 (11.16) | −0.39 (16.73) | 6.56 (13.77) | 4.01 (11.02) | 0.16 (14.55) |
Pain symptom score | 30.89 (27.83) | 21.18 (22.76) | −6.96 (26.26) | 32.17 (27.01) | 25.96 (25.32) | 1.96 (24.07) |
Dyspnea symptom score | 16.97 (23.06) | 16.54 (20.36) | 4.20 (20.14) | 16.91 (23.44) | 19.87 (26.08) | 4.90 (20.66) |
Insomnia symptom score | 25.15 (27.16) | 20.80 (24.82) | −3.67 (30.06) | 24.88 (27.28) | 18.59 (24.06) | −1.63 (27.90) |
Appetite loss symptom score | 22.22 (27.61) | 26.32 (27.54) | 5.77 (32.56) | 24.02 (27.52) | 19.87 (28.07) | 3.92 (27.87) |
Constipation symptom score | 20.88 (27.26) | 14.29 (19.37) | −6.04 (27.99) | 21.94 (28.14) | 13.78 (21.59) | −0.98 (24.13) |
Diarrhea symptom score | 6.72 (15.65) | 10.53 (19.83) | 3.94 (24.35) | 7.23 (16.72) | 6.73 (14.96) | 3.27 (19.63) |
Financial difficulties symptom score | 11.84 (21.44) | 12.78 (22.37) | −0.26 (20.36) | 10.78 (20.40) | 11.86 (21.24) | 2.61 (16.04) |
EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; SD = standard deviation.
Source: EV-301 Clinical Study Report.17
EuroQol 5-Dimensions 5-Levels Questionnaire
At baseline, 91.0% and 89.9% of patients completed the EQ-5D-5L in the enfortumab vedotin and chemotherapy arms, respectively. Completion and compliance rates for the EQ-5D-5L decreased over time, with a notable decrease after week 12 (Figure 8, Appendix 3).17
Descriptive results from the EQ-5D-5L are summarized in Table 15. Mean EQ VAS scores at baseline were 68.2 (SD = 18.1) and 68.3 (SD = 18.8) in the enfortumab vedotin and chemotherapy arms, respectively. A reduction of −1.8 (SD = 16.6) in mean EQ VAS scores was reported from baseline to week 12 in the enfortumab vedotin arm compared to −5.3 (SD = 14.5) in the chemotherapy arm.17
Table 15: EQ-5D-5L Questionnaire Results, EQ VAS, and Change from Baseline (Full Analysis Set)
EQ 5-Dimensions 5-Levels questionnaire | Enfortumab vedotin (n = 301) | Chemotherapy (n = 307) | ||
|---|---|---|---|---|
Baseline | Week 12 | Baseline | Week 12 | |
Mobility, n (%) | ||||
No problems | 144 (47.8) | 71 (23.6) | 151 (49.2) | 50 (16.3) |
Slight problems | 78 (25.9) | 37 (12.3) | 73 (23.8) | 29 (9.4) |
Moderate problems | 35 (11.6) | 19 (6.3) | 35 (11.4) | 18 (5.9) |
Severe problems | 14 (4.7) | 3 (1.0) | 14 (4.6) | 7 (2.3) |
Extreme problems | 3 (1.0) | 3 (1.0) | 3 (1.0) | 0 |
Self-care, n (%) | ||||
No problems | 219 (72.8) | 102 (33.9) | 214 (69.7) | 79 (25.7) |
Slight problems | 34 (11.3) | 19 (6.3) | 43 (14.0) | 21 (6.8) |
Moderate problems | 14 (4.7) | 8 (2.7) | 10 (3.3) | 3 (1.0) |
Severe problems | 4 (1.3) | 1 (0.3) | 9 (2.9) | 1 (0.3) |
Extreme problems | 3 (1.0) | 3 (1.0) | 0 | 0 |
Usual activities | ||||
No problems | 138 (45.8) | 57 (18.9) | 131 (42.7) | 42 (13.7) |
Slight problems | 76 (25.2) | 50 (16.6) | 95 (30.9) | 41 (13.4) |
Moderate problems | 42 (14.0) | 21 (7.0) | 37 (12.1) | 13 (4.2) |
Severe problems | 14 (4.7) | 2 (0.7) | 12 (3.9) | 8 (2.6) |
Extreme problems | 4 (1.3) | 3 (1.0) | 1 (0.3) | 0 |
Pain or discomfort | ||||
No problems | 95 (31.6) | 51 (16.9) | 80 (26.1) | 39 (12.7) |
Slight problems | 91 (30.2) | 53 (17.6) | 114 (37.1) | 43 (14.0) |
Moderate problems | 65 (21.6) | 24 (8.0) | 63 (20.5) | 18 (5.9) |
Severe problems | 23 (7.6) | 4 (1.3) | 17 (5.5) | 4 (1.3) |
Extreme problems | 0 | 1 (0.3) | 2 (0.7) | 0 |
Anxiety or depression | ||||
No problems | 154 (51.2) | 81 (26.9) | 134 (43.6) | 59 (19.2) |
Slight problems | 85 (28.2) | 38 (12.6) | 98 (31.9) | 36 (11.7) |
Moderate problems | 31 (10.3) | 12 (4.0) | 35 (11.4) | 7 (2.3) |
Severe problems | 4 (1.3) | 2 (0.7) | 7 (2.3) | 1 (0.3) |
Extreme problems | 0 | 0 | 2 (0.7) | 1 (0.3) |
EQ VASa | ||||
n | 267 | 130 | 270 | 104 |
Mean (SD) | 68.2 (18.1) | 69.0 (19.2) | 68.3 (18.8) | 69.9 (19.5) |
Median (range) | 70.0 (0 to 100) | 75.0 (13 to 100) | 71.0 (0 to 100) | 72.0 (10 to 100) |
Mean (SD) change from baseline | −1.8 (16.6) | −5.3 (14.5) | ||
EQ VAS = EQ-5D Visual Analogue Scale; SD = standard deviation.
aEQ VAS data from 13 subjects from 4 sites (31001, 31002, 31003, and 31009) were removed from the analysis due to an issue on the EQ VAS item on the EQ-5D-5L.
Source: EV-301 Clinical Study Report.17
Clinical Response Outcomes
Overall Response Rate and Disease Control Rate
Results for the secondary end points of EV-301, ORR, DCR, and other clinical response outcomes per RECIST 1.1 according to the investigator are summarized in Table 16. In patients with measurable disease, the confirmed ORR was statistically significant in favour of enfortumab vedotin, with an ORR of 40.6% for the enfortumab vedotin arm and 17.9% for the chemotherapy arm (stratified 1-sided P < 0.001). Totals of 14 patients (4.9%) and 8 patients (2.7%) achieved a CR, and 103 (35.8%) versus 45 (15.2%) achieved a PR in the enfortumab and chemotherapy arms, respectively.17
The confirmed DCR was statistically significant in favour of enfortumab vedotin, with a DCR of 71.9% in the enfortumab arm and 53.4% in the chemotherapy arm (1-sided P value < 0.001).17
Table 16: Overall Response Rate and Disease Control Rate, Investigator Assessment (Response Evaluable Set)
Response | Enfortumab vedotin (n = 288) | Chemotherapy (n = 296) |
|---|---|---|
Best overall response, confirmed, n (%)a | ||
Confirmed complete response | 14 (4.9) | 8 (2.7) |
Confirmed partial response | 103 (35.8) | 45 (15.2) |
Stable disease | 90 (31.3) | 105 (35.5) |
Progressive disease | 44 (15.3) | 83 (28.0) |
Not evaluable | 37 (12.8) | 55 (18.6) |
Overall response rate, confirmed, n (%) | 117 (40.6) | 53 (17.9) |
95% CI (%)b | (34.90 to 46.54) | (13.71 to 22.76) |
Stratified 1-sided P valuec | < 0.001d | |
Disease control rate, confirmed, n (%)e | 207 (71.9) | 158 (53.4) |
95% CI (%)b | (66.30 to 76.99) | (47.52 to 59.17) |
Stratified 1-sided P valuec | < 0.001 d | |
CI = confidence interval; CR = complete response; PR = partial response.
aThe definition of best overall response followed RECIST 1.1. A CR and PR must have been confirmed by 2 scans a minimum of 4 weeks apart. The minimum duration for stable disease was 7 weeks.
bUsing exact method based on binomial distribution (Clopper-Pearson).
cBased on Cochran-Mantel-Haenszel test. Stratification factors were Eastern Cooperative Oncology Group Performance Status, geographic region, and liver metastasis.
dIndicates that the P values of end points are less than or equal to 0.025 (1-sided predetermined efficacy boundary per 100% information fraction).
eDCR was defined as the proportion of patients who had a best overall response of confirmed CR, confirmed PR, or stable disease (≥ 7 weeks).
Source: EV-301 Clinical Study Report.17
Of the 117 patients in the enfortumab vedotin arm who achieved a confirmed CR or PR, the time to response was 1.87 months, while in the 53 patients who received chemotherapy, the time to response was 1.91 months.17
Sensitivity Analysis
Results for the sensitivity analysis of ORR and DCR regardless of confirmation of best overall response are summarized in Table 33. While still in favour of enfortumab vedotin versus chemotherapy (50.7% versus 28.4%; 1-sided P value < 0.001), the proportion of patients with unconfirmed ORR were higher than the primary analysis for ORR. Sensitivity analysis results for DCR were comparable to those for the primary analysis.17
Subgroup Analysis – ORR
Subgroups of interest outlined in the review protocol (Table 3) for OS in the FAS are summarized in Table 34. Across subgroups of interest, the treatment effect was consistent with the overall analysis, with all subgroups favouring enfortumab vedotin.17
Duration of Response
The DOR for patients with confirmed CR or PR is summarized in Table 17 and Figure 6. Duration of response was similar between treatment arms, with a median DOR of 7.39 months (95% CI, 5.59 to 9.46) versus 8.11 months (95% CI, 5.65 to 9.56) for enfortumab vedotin and chemotherapy, respectively.17
Table 17: Duration of Response According to Investigator Assessment (All Patients With Confirmed Complete or Partial Response – Response Evaluable Set)
Duration of response measure | Enfortumab vedotin (n = 117) | Chemotherapy (n = 53) |
|---|---|---|
Events, n (%) | 63 (53.8) | 29 (54.7) |
Radiographical progression | 62 (53.0) | 28 (52.8) |
Death without documented progression | 1 (0.9) | 1 (1.9) |
Censored, n (%) | 54 (46.2) | 24 (45.3) |
No event | 52 (44.4) | 23 (43.4) |
Event after new anticancer therapy | 2 (1.7) | 1 (1.9) |
Median DOR (95% CI), monthsa | 7.39 (5.59 to 9.46) | 8.11 (5.65 to 9.56) |
Rate of patients with neither progressive disease nor death, % (95% CI)b | ||
6 months | 53.8 (43.67 to 62.97) | 56.0 (40.13 to 69.24) |
12 months | 27.7 (17.00 to 39.53) | 19.8 (7.04 to 37.18) |
CI = confidence interval; DOR = duration of response; RES = response evaluable set.
aBased on Kaplan–Meier estimate.
bRate of patients with neither PD nor death since they achieved the first confirmed CR or PR. Rates at other time points and the 95% CIs are estimated based on DOR using the Kaplan–Meier method and Greenwood formula.
Source: EV-301 Clinical Study Report.17
Figure 6: Kaplan–Meier Plot of Duration of Response, Investigator Assessment (All Patients With Confirmed Complete or Partial Response)
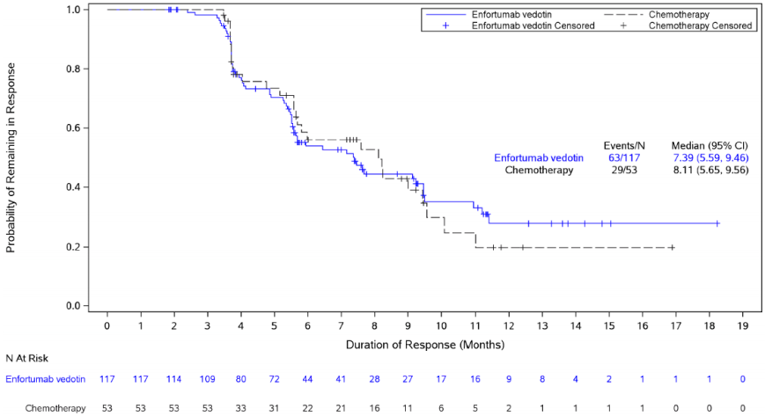
CI = confidence interval; CR = complete response; DOR = duration of response; PR = partial response.
Source: EV-301 Clinical Study Report.17
Health Care Resource Utilization
Measures of HCRU, an exploratory outcome in the EV-301 study, are summarized in Table 18. No notable differences in specific HCRU measures were observed between enfortumab vedotin and chemotherapy.17
Table 18: Summary of Health Care Resource Utilization Questionnaire (Full Analysis Set)
Health care resource utilization measure | Enfortumab vedotin (n = 288) | Chemotherapy (n = 296) |
|---|---|---|
ER visits (EOT) | ||
Number of ER visits | 14 | 24 |
Number of post–emergency room hospitalizations | 18 | 14 |
Duration of post–emergency room hospitalizations (days), mean (SD) | 5.7 (4.99) | 7.9 (5.93) |
Hospital admissions (EOT) | ||
Number of hospital admissions | 12 | 10 |
Duration of hospital admissions (days), mean (SD) | 10.5 (10.08) | 8.9 (6.84) |
GP visits (EOT) | ||
Number of general practitioner visits | 24 | 21 |
Specialist visits (EOT) | ||
Number of specialist visits | 31 | 22 |
EOT = end of treatment.
Source: EV-301 Clinical Study Report17
Harms
Only those harms identified in the review protocol are reported. Specific harms associated with vinflunine are not presented.
Adverse Events
Overall, 98.5% of patients experienced at least 1 TEAE — 290 (98.0%) in the enfortumab vedotin arm and 288 (99%) in the chemotherapy arm (Table 19). The most common TEAEs in the enfortumab vedotin were alopecia (47.0%), decreased appetite (40.9%), fatigue (36.1%), diarrhea (34.8%), peripheral sensory neuropathy (34.5%), and pruritus (34.5%). The most common TEAEs for taxane chemotherapies included alopecia (||||||), fatigue (||||||), peripheral sensory neuropathy (||||||), and anemia (||||).17
The incidence of TEAEs, including decreased appetite, diarrhea, pruritis, nausea, constipation, dysgeusia, pyrexia, dry skin, rash, maculopapular rash, decreased weight, asthenia, abdominal pain, increased aspartate aminotransferase, and hyperglycemia, were each at least 5% higher in the enfortumab vedotin than in the taxane chemotherapy arm, while anemia, decreased neutrophil count, peripheral edema, arthralgia, myalgia, and decreased white blood cell count were at least 5% higher with taxane chemotherapy than with enfortumab vedotin.17
The TEAEs resulting in dose reduction or dose interruption are summarized in Table 35 and Table 36 of Appendix 3. Of all patients with TEAEs, 101 (34.1%) and 81 (27.8%) in the enfortumab vedotin and chemotherapy arms (including vinflunine), respectively, experienced TEAEs that led to a dose reduction (Table 35), while 180 (60.8%) and 85 (29.2%) in the enfortumab vedotin and chemotherapy arms, respectively, experienced a TEAE leading to a dose interruption (Table 36). In the enfortumab vedotin arm, the most common TEAEs resulting in a dose reduction were peripheral sensory neuropathy (7.4%) and maculopapular rash (4.4%). In the chemotherapy arm, the most common TEAEs resulting in dose reduction were peripheral sensory neuropathy (6.2%) and fatigue (4.1%). In the enfortumab vedotin arm, the most common TEAEs leading to dose interruption were peripheral sensory neuropathy (15.5%) and fatigue (6.1%). In the chemotherapy arm, the most common TEAEs leading to dose interruption were decreased neutrophil count (3.8%) and anemia (3.4%).17
Table 19: Treatment-Emergent Adverse Events Reported in at Least 10% of Patients (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Overall | 290 (98.0) | |||||| | |||||||| | |||||||| | 288 (99.0) |
Alopecia | 139 (47.0) | |||||||| | |||||||| | |||||||| | 110 (37.8) |
Decreased appetite | 121 (40.9) | |||||||| | |||||||| | |||||||| | 78 (26.8) |
Fatigue | 107 (36.1) | |||||||| | |||||||| | |||||||| | 78 (26.8) |
Diarrhea | 103 (34.8) | |||||||| | |||||||| | |||||||| | 66 (22.7) |
Peripheral sensory neuropathy | 102 (34.5) | |||||||| | |||||||| | |||||||| | 66 (22.7) |
Pruritus | 102 (34.5) | |||||||| | |||||||| | |||||||| | 20 (6.9) |
Nausea | 89 (30.1) | |||||||| | |||||||| | |||||||| | 74 (25.4) |
Constipation | 82 (27.7) | |||||||| | |||||||| | |||||||| | 73 (25.1) |
Dysgeusia | 74 (25.0) | |||||||| | |||||||| | |||||||| | 23 (7.9) |
Pyrexia | 65 (22.0) | |||||||| | |||||||| | |||||||| | 41 (14.1) |
Anemia | 59 (19.9) | |||||||| | |||||||| | |||||||| | 87 (29.9) |
Dry skin | 50 (16.9) | |||||||| | |||||||| | |||||||| | 11 (3.8) |
Rash | 50 (16.9) | |||||||| | |||||||| | |||||||| | 16 (5.5) |
Maculopapular rash | 50 (16.9) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Decreased weight | 47 (15.9) | |||||||| | |||||||| | |||||||| | 20 (6.9) |
Asthenia | 46 (15.5) | |||||||| | |||||||| | |||||||| | 40 (13.7) |
Vomiting | 42 (14.2) | |||||||| | |||||||| | |||||||| | 44 (15.1) |
Abdominal pain | 39 (13.2) | |||||||| | |||||||| | |||||||| | 27 (9.3) |
Increased aspartate aminotransferase | 36 (12.2) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Hematuria | 33 (11.1) | |||||||| | |||||||| | |||||||| | 25 (8.6) |
Decreased neutrophil count | 33 (11.1) | |||||||| | |||||||| | |||||||| | 54 (18.6) |
Hyperglycemia | 31 (10.5) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Insomnia | 31 (10.5) | |||||||| | |||||||| | |||||||| | 23 (7.9) |
Increased lacrimation | 30 (10.1) | |||||||| | |||||||| | |||||||| | 12 (4.1) |
Peripheral edema | 27 (9.1) | |||||||| | |||||||| | |||||||| | 39 (13.4) |
Arthralgia | 19 (6.4) | |||||||| | |||||||| | |||||||| | 36 (12.4) |
Myalgia | 15 (5.1) | |||||||| | |||||||| | |||||||| | 32 (11.0) |
Decreased white blood cell count | 16 (5.4) | |||||||| | |||||||| | |||||||| | 32 (11.0) |
MedDRA = Medical Dictionary for Regulatory Activities.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
A least 1 grade 3 or higher TEAE was experienced by 403 patients (68.7%) — 210 (70.9%) with enfortumab vedotin, and |||||||||||| with taxane chemotherapy (|||||| for docetaxel, and |||| for paclitaxel) (Table 20). For enfortumab vedotin, the most common grade 3 or greater TEAEs (occurring in at least 5% of patients) were maculopapular rash (7.4%), hyperglycemia and decreased neutrophil count (7.1%, each), fatigue (6.8%), anemia (6.4%), and decreased appetite (5.4%). The most frequent grade 3 or higher TEAEs with docetaxel were decreased neutrophil count (||||), anemia (||||),decreased white blood cell count (||||), and febrile neutropenia (||||). The most frequent grade or higher 3 TEAEs with paclitaxel were decreased neutrophil count (||||), anemia (||||), and febrile neutropenia (||||||).17
Some imbalances across treatment arms were evident between the incidence rates of grade or greater 3 TEAEs. The taxanes were associated with a higher grade or greater decreased neutrophil counts (|||| versus 7.1%), anemia (|||| versus 6.4%), febrile neutropenia (|||| vs 1.4%) and decreased white blood cell counts (|||||| versus 1.4%) compared to enfortumab vedotin, while the incidence of grade or greater maculopapular rash and hyperglycemia was higher in the enfortumab vedotin arm compared to taxane chemotherapies (7.4% versus | ; and 7.1% versus ||||, respectively) (Table 20).17
Table 20: Grade 3 or Higher Treatment-Emergent Adverse Events Reported for at Least 2% of Patients (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Overall | 210 (70.9) | |||||||| | |||||||| | |||||||| | 193 (66.3) |
Maculopapular rash | 22 (7.4) | |||||||| | |||||||| | |||||||| | 0 |
Hyperglycemia | 21 (7.1) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Decreased neutrophil count | 21 (7.1) | |||||||| | |||||||| | |||||||| | 43 (14.8) |
Fatigue | 20 (6.8) | |||||||| | |||||||| | |||||||| | 14 (4.8) |
Anemia | 19 (6.4) | |||||||| | |||||||| | |||||||| | 34 (11.7) |
Decreased appetite | 16 (5.4) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Neutropenia | 14 (4.7) | |||||||| | |||||||| | |||||||| | 22 (7.6) |
Pneumonia | 13 (4.4) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Hyponatremia | 12 (4.1) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Malignant neoplasm progression | 12 (4.1) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Diarrhea | 11 (3.7) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Peripheral sensory neuropathy | 9 (3.0) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Urinary tract infection bacterial | 9 (3.0) | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Acute kidney injury | 8 (2.7) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Drug eruption | 8 (2.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Increased lipase | 8 (2.7) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Asthenia | 7 (2.4) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Decreased lymphocyte count | 7 (2.4) | |||||||| | |||||||| | |||||||| | 12 (4.1) |
Pyrexia | 7 (2.4) | |||||||| | |||||||| | |||||||| | 0 |
Hematuria | 6 (2.0) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Urinary tract infection | 6 (2.0) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Febrile neutropenia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 16 (5.5) |
Decreased white blood cell count | 4 (1.4) | |||||||| | |||||||| | |||||||| | 21 (7.2) |
Constipation | 3 (1.0) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
General physical health deterioration | 3 (1.0) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Abdominal pain | 2 (0.7) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
MedDRA = Medical Dictionary for Regulatory Activities.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Serious Adverse Events
The incidence of SAEs is summarized in Table 21. The overall incidence of treatment-emergent SAEs was similar between the enfortumab vedotin (46.6%) and taxane chemotherapy arms (||||). The most common SAEs in the enfortumab vedotin arm were acute kidney injury (6.4%), malignant neoplasm progression (4.1%), and pneumonia (4.1%). The most common SAEs in the taxane chemotherapy arm were febrile neutropenia (||||||) and pneumonia (||||).17
Table 21: Serious Treatment-Emergent Adverse Events in 1% or More of Patients (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | ||||
|---|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | |||
Overall | 138 (46.6) | |||||||| | |||||||| | |||||||| | 128 (44.0) | |
Acute kidney injury | 19 (6.4) | |||||||| | |||||||| | |||||||| | 7 (2.4) | |
Malignant neoplasm progression | 12 (4.1) | |||||||| | |||||||| | |||||||| | 7 (2.4) | |
Pneumonia | 12 (4.1) | |||||||| | |||||||| | |||||||| | 7 (2.4) | |
Urinary tract infection, bacterial | 9 (3.0) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Diarrhea | 7 (2.4) | |||||||| | |||||||| | |||||||| | 4 (1.4) | |
Urinary tract infection | 7 (2.4) | |||||||| | |||||||| | |||||||| | 6 (2.1) | |
Pyrexia | 6 (2.0) | |||||||| | |||||||| | |||||||| | 9 (3.1) | |
Atrial fibrillation | 5 (1.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Decreased appetite | 5 (1.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Hematuria | 5 (1.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Sepsis | 5 (1.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Vomiting | 5 (1.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Anemia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 6 (2.1) | |
Dyspnea | 4 (1.4) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Febrile neutropenia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 16 (5.5) | |
Hyperglycemia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Neutropenia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 8 (2.7) | |
Maculopapular rash | 4 (1.4) | |||||||| | |||||||| | |||||||| | 0 | |
Septic shock | 4 (1.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Abdominal pain | 3 (1.0) | |||||||| | |||||||| | |||||||| | 6 (2.1) | |
Asthenia | 3 (1.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Cellulitis | 3 (1.0) | |||||||| | |||||||| | |||||||| | 2 (0.7) | |
Urinary tract infection, Escherichia | 3 (1.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Fatigue | 3 (1.0) | |||||||| | |||||||| | |||||||| | 2 (0.7) | |
Hydronephrosis | 3 (1.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) | |
Multiple organ dysfunction syndrome | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 | |
Rash | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 | |
Back pain | 2 (0.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Constipation | 2 (0.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Dehydration | 2 (0.7) | |||||||| | |||||||| | |||||||| | 4 (1.4) | |
General physical health deterioration | 2 (0.7) | |||||||| | |||||||| | |||||||| | 5 (1.7) | |
Hyponatremia | 2 (0.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Malaise | 2 (0.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Decreased neutrophil count | 2 (0.7) | |||||||| | |||||||| | |||||||| | 5 (1.7) | |
Urosepsis | 2 (0.7) | |||||||| | |||||||| | |||||||| | 6 (2.1) | |
Delirium | 1 (0.3) | |||||||| | |||||||| | |||||||| | 3 (1.0) | |
Hyperkalemia | 1 (0.3) | |||||||| | |||||||| | |||||||| | 4 (1.4) | |
MedDRA = Medical Dictionary for Regulatory Activities; NR = not reported.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Withdrawals Due to Adverse Events
As of the July 15, 2020, data cut-off, a total of 51 patients (17.4%) in the enfortumab vedotin arm, and |||||||||| in the taxane chemotherapy arm withdrew from treatment due to TEAEs. The most common reason for a WDAE in both arms was peripheral sensory neuropathy, which occurred in 2.4% and |||| of patients in the enfortumab vedotin and taxane chemotherapy arms, respectively.17
Table 22: Treatment-Emergent Adverse Events Resulting in Drug Withdrawal for 2 or More Patients by Preferred Term (Safety Analysis Set)
System organ class preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Overall | 51 (17.2) | |||||||| | |||||||| | |||||||| | 51 (17.5) |
Peripheral sensory neuropathy | 7 (2.4) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Malignant neoplasm progression | 5 (1.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Peripheral motor neuropathy | 5 (1.7) | |||||||| | |||||||| | |||||||| | 0 |
Maculopapular rash | 4 (1.4) | |||||||| | |||||||| | |||||||| | 0 |
Acute kidney injury | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 |
Dermatitis bullous | 2 (0.7) | |||||||| | |||||||| | |||||||| | 0 |
Drug eruption | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Hepatic function abnormal | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Hyperglycemia | 2 (0.7) | |||||||| | |||||||| | |||||||| | 0 |
Peripheral neuropathy | 2 (0.7) | |||||||| | |||||||| | |||||||| | 0 |
Pneumonia | 2 (0.7) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Neutropenia | 1 (0.3) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Anemia | 0 | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Constipation | 0 | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Febrile neutropenia | 0 | |||||||| | |||||||| | |||||||| | 3 (1.0) |
General physical health deterioration | 0 | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Sepsis | 0 | |||||||| | |||||||| | |||||||| | 3 (1.0) |
MedDRA = Medical Dictionary for Regulatory Activities.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Mortality
Treatment-emergent AEs resulting in death during the study are summarized in Table 23. Overall, 21 patients (7.1%) in the enfortumab vedotin and |||||||||| patients in the taxane chemotherapy arms experienced a TEAE leading to death, with malignant neoplasm progression the most common TEAE leading to death in both arms.17
As of the July 15, 2020, data cut-off date, 130 patients (43.9%) in the enfortumab vedotin arm and 161 patients (55.3%) in the chemotherapy arm (including those who received vinflunine) died. The most common cause of death on study was disease progression.17
Table 23: Treatment-Emergent Adverse Events Resulting in Death (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Overall | 21 (7.1) | |||||||| | |||||||| | |||||||| | 16 (5.5) |
Malignant neoplasm progression | 10 (3.4) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Multiple organ dysfunction syndrome | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 |
Pneumonia | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Brain edema | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Dyspnea | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Hepatic function abnormal | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Hyperglycemia | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Pelvic abscess | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Septic shock | 1 (0.3) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Cardiac arrest | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Cardiogenic shock | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Death | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
General physical health deterioration | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Neutropenic sepsis | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Pancytopenia | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Pneumocystis jirovecii pneumonia | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Respiratory distress | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Sepsis | 0 | |||||||| | |||||||| | |||||||| | 2 (0.7) |
MedDRA = Medical Dictionary for Regulatory Activities.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Notable Harms
Infusion-Related Reactions
Infusion-related reactions by preferred term are summarized in Table 24. Overall, 27 patients (9.1%) in the enfortumab vedotin arm and |||||||||||||||||||| in the taxane chemotherapy arm experienced an IRR.
The majority of the IRRs were grades 1 or 2, with 4 grade 3 events, all in the enfortumab vedotin arm (1 event of drug hypersensitivity and 3 events of drug eruption), considered related to enfortumab vedotin. One case of drug eruption in the enfortumab vedotin arm was considered serious, while 2 events consisting of extravasation and infusion-related reaction were considered serious in the chemotherapy arm (including the vinflunine-treated patients).17
Table 24: Infusion-Related Reactions (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Any infusion-related reactions (SSQ/CMQ) | 27 (9.1) | |||||||| | |||||||| | |||||||| | 17 (5.8) |
Any local IRR events (SSQ/CMQ) | 4 (1.4) | |||||||| | |||||||| | |||||||| | 9 (3.1) |
Any infusion-site reactions (SSQ/CMQ) | 3 (1.0) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Infusion-site extravasation | 2 (0.7) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Infusion-site hemorrhage | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Infusion-site irritation | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Infusion-site joint swelling | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Any extravasation site reactions (SMQ) | 3 (1.0) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Administration-site extravasation | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Extravasation | 1 (0.3) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Infusion-site extravasation | 2 (0.7) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Any systemic IRR events (SSQ/CMQ) | 23 (7.8) | |||||||| | |||||||| | |||||||| | 11 (3.8) |
Drug eruption | 17 (5.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Drug hypersensitivity | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Face edema | 0 | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Fixed eruption | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
IRR | 4 (1.4) | |||||||| | |||||||| | |||||||| | 9 (3.1) |
CMQ = customized medical query; IRR = infusion-related reaction; MedDRA = Medical Dictionary for Regulatory Activities; SMQ = standard MedDRA query; SSQ = sponsor-specific query.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Ocular Disorders
Ocular disorders were considered AEs of special interest for this review. The incidence of treatment-emergent ocular disorders was higher in the enfortumab vedotin arm and is summarized in Table 25. Ocular disorders were experienced by 83 patients (28%) in the enfortumab vedotin arm and 21 patients (||||)in the chemotherapy arm (taxanes). In the enfortumab vedotin and taxane chemotherapy arms, 71 patients (24.0%), and |||||||||| patients experienced an event in the category of any dry eye, respectively. Blurred vision was experienced by 18 patients (6.1%) in the enfortumab vedotin arm and |||||||| of patients in the taxane chemotherapy arm. Three grade 3 events were reported by 3 patients; 2 in the enfortumab vedotin arm that were considered serious (1 event each of conjunctivitis and blepharitis) and 2 in the chemotherapy arm (lacrimation increased and visual impairment). One patient in the enfortumab vedotin arm withdrew from treatment because of conjunctivitis.17
In patients who experienced an event, the median time to onset was 1.9 months in the enfortumab vedotin arm and 0.5 months in the chemotherapy arm (including vinflunine).17
Table 25: Treatment-Emergent Ocular Disorders (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Any ocular disorders | 83 (28.0) | |||||||| | |||||||| | |||||||| | 23 (7.9) |
Any corneal disorders (SMQ) | 3 (1.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Keratitis | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Corneal epithelium defect | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Keratopathy | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Any dry eye (SSQ/CMQ) | 71 (24.0) | |||||||| | |||||||| | |||||||| | 17 (5.8) |
Lacrimation increased | 30 (10.1) | |||||||| | |||||||| | |||||||| | 12 (4.1) |
Dry eye | 19 (6.4) | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Conjunctivitis | 19 (6.4) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Blepharitis | 6 (2.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Keratitis | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Conjunctivitis allergic | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Eye irritation | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Keratopathy | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Meibomian gland dysfunction | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Ocular discomfort | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Any blurred vision (SSQ/CMQ) | 18 (6.1) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Vision blurred | 16 (5.4) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Visual impairment | 0 | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Visual acuity reduced | 2 (0.7) | |||||||| | |||||||| | |||||||| | 0 |
CMQ = customized medical query; MedDRA = Medical Dictionary for Regulatory Activities; SMQ = standard MedDRA query; SSQ = sponsor-specific query.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Skin Reactions
Table 26 summarizes skin reactions in the safety analysis set. In the enfortumab vedotin arm, 159 patients (53.7%) experienced any rash or serious cutaneous adverse reactions, compared to |||||||||| in the taxane chemotherapy arm. The most common rash per sponsor-specific or customized medical queries in the enfortumab vedotin compared to the taxane chemotherapy arm were rash (16.9% versus ||||, maculopapular rash (16.9% versus ||||), and drug eruption (8.8% versus ||||). The most frequent severe cutaneous adverse reactions (SMQ) were stomatitis (9.1% versus ||||), drug eruption (8.8% versus ||||), and conjunctivitis (6.4% versus ||||). Grade 1 or 2 skin reactions occurred in 112 (39%) patients in the enfortumab vedotin arm, and 56 (19%) patients in the chemotherapy arm (including vinflunine-treated patients).17
In the enfortumab vedotin arm, 42 patients reported grade 3 events, including stomatitis (0.7%), drug eruption (2.7%), and conjunctivitis, blister, dermatitis bullous, skin exfoliation, and toxic skin eruption (0.3% each), and 1 patient experienced grade 4 dermatitis bullous that was considered related to treatment. Fourteen (4.7%) patients in the enfortumab vedotin arm experienced skin reactions that were considered serious, while 12 patients experienced a skin reaction that led to withdrawal of treatment (4 rash maculopapular, 2 drug eruption, 2 dermatitis bullous, and 1 patient each with rash, rash erythematous, conjunctivitis, and toxic skin eruption). In the overall chemotherapy arm (including vinflunine-treated patients), 2 patients reported grade 3 events: drug eruption and mouth ulceration (0.3% each), and only 1 patient discontinued treatment due to drug eruption. Twenty-four patients (8.1%) in the enfortumab vedotin arm and 2 patients (0.7%) in the chemotherapy arm experienced skin reactions that led to a dose reduction, while 33 patients (11.1%) in the enfortumab vedotin arm and no subjects in the chemotherapy arm experienced events that led to a dose interruption.17
In patients who experienced an event, median time to onset was 0.5 months in the enfortumab vedotin arm and 0.7 months in the chemotherapy arm.17
Table 26: Treatment-Emergent Skin Reactions Including Severe Cutaneous Adverse Reactions (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Any rashes or severe cutaneous adverse reactions | 159 (53.7) | |||||||| | |||||||| | |||||||| | 58 (19.9) |
Any rash (SSQ/CMQ) | 142 (48.0) | |||||||| | |||||||| | |||||||| | 38 (13.1) |
Rash | 50 (16.9) | |||||||| | |||||||| | |||||||| | 16 (5.5) |
Maculopapular rash | 50 (16.9) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Drug eruption | 26 (8.8) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Erythema | 12 (4.1) | |||||||| | |||||||| | |||||||| | 5 (1.7) |
Erythematous rash | 10 (3.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Blister | 9 (3.0) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Dermatitis bullous | 7 (2.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Palmar-plantar erythrodysesthesia syndrome | 3 (1.0) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Eczema | 4 (1.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Macular rash | 2 (0.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Pruritic rash | 2 (0.7) | |||||||| | |||||||| | |||||||| | 0 |
Dermatitis | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Erythema multiforme | 1 (0.3) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Fixed eruption | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Pemphigus | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Perivascular dermatitis | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Papular rash | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Vesicular rash | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Skin irritation | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Toxic skin eruption | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Any severe cutaneous adverse reactions (SMQ) | 77 (26.0) | |||||||| | |||||||| | |||||||| | 0 |
Stomatitis | 27 (9.1) | |||||||| | |||||||| | |||||||| | 27 (9.3) |
Drug eruption | 26 (8.8) | |||||||| | |||||||| | |||||||| | 19 (6.5) |
Conjunctivitis | 19 (6.4) | |||||||| | |||||||| | |||||||| | 4 (1.4) |
Blister | 9 (3.0) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Dermatitis bullous | 7 (2.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Skin exfoliation | 8 (2.7) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Erythema multiforme | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Exfoliative rash | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Fixed eruption | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Mouth ulceration | 0 | |||||||| | |||||||| | |||||||| | 0 |
Pemphigus | 1 (0.3) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Toxic skin eruption | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
CMQ = customized medical query; MedDRA = Medical Dictionary for Regulatory Activities; SMQ = standard MedDRA query; SSQ = sponsor-specific query.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Peripheral Neuropathy
Table 27 summarizes the incidence of peripheral neuropathy in the safety analysis set. A total of 149 patients (50.3%) and |||||||||| patients in the enfortumab vedotin and taxane chemotherapy arms experienced an event of any peripheral neuropathy, of which 139 patients (47%) and |||||||||| patients in the enfortumab vedotin and chemotherapy arms, respectively, experienced any sensory peripheral neuropathy, and 33 patients (11.1%) and |||||||| experienced motor events.
A total of 23 peripheral neuropathy events were grade 3 (15 in the enfortumab vedotin arm and 8 in the overall chemotherapy arm), with 6 patients in the enfortumab vedotin arm and 2 patients in the chemotherapy arm experiencing peripheral neuropathy that was considered serious. Fourteen patients in the enfortumab vedotin arm and 8 patients in the chemotherapy arm reported peripheral neuropathy events that resulted in treatment withdrawal.17
In patients who experienced a neuropathy event, the median time to onset in the enfortumab vedotin arm was 2.5 months compared to 0.8 months in the chemotherapy arm. For grade 2 or higher events, the median time to onset was 4.3 months in the enfortumab vedotin arm and 1.8 months in the chemotherapy arm.17
Table 27: Treatment-Emergent Peripheral Neuropathy (Safety Analysis Set)
Preferred term (MedDRA v23.0) | Enfortumab vedotin (n = 296) | Chemotherapy | |||
|---|---|---|---|---|---|
Docetaxel (n = 109) | Paclitaxel (n = 107) | Total taxanes (n = 216) | Total chemotherapya (n = 291) | ||
Any peripheral neuropathy (SMQ) | 149 (50.3) | |||||||| | |||||||| | |||||||| | 100 (34.4) |
Any peripheral neuropathy motor events (SSQ/CMQ) | 33 (11.1) | |||||||| | |||||||| | |||||||| | 9 (3.1) |
Muscular weakness | 16 (5.4) | |||||||| | |||||||| | |||||||| | 7 (2.4) |
Peripheral motor neuropathy | 11 (3.7) | |||||||| | |||||||| | |||||||| | 0 |
Peripheral sensorimotor neuropathy | 8 (2.7) | |||||||| | |||||||| | |||||||| | 2 (0.7) |
Peroneal nerve palsy | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 |
Any peripheral neuropathy sensory events (SSQ/CMQ) | 139 (47.0) | |||||||| | |||||||| | |||||||| | 97 (33.3) |
Peripheral sensory neuropathy | 102 (34.5) | |||||||| | |||||||| | |||||||| | 66 (22.7) |
Peripheral neuropathy | 20 (6.8) | |||||||| | |||||||| | |||||||| | 16 (5.5) |
Paresthesia | 15 (5.1) | |||||||| | |||||||| | |||||||| | 8 (2.7) |
Polyneuropathy | 5 (1.7) | |||||||| | |||||||| | |||||||| | 6 (2.1) |
Gait disturbance | 8 (2.7) | |||||||| | |||||||| | |||||||| | 0 |
Hypoesthesia | 2 (0.7) | |||||||| | |||||||| | |||||||| | 3 (1.0) |
Neuralgia | 4 (1.4) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Neurotoxicity | 4 (1.4) | |||||||| | |||||||| | |||||||| | 0 |
Dysesthesia | 3 (1.0) | |||||||| | |||||||| | |||||||| | 0 |
Burning sensation | 1 (0.3) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Sensory loss | 1 (0.3) | |||||||| | |||||||| | |||||||| | 1 (0.3) |
Demyelinating polyneuropathy | 1 (0.3) | |||||||| | |||||||| | |||||||| | 0 |
Sensory disturbance | 0 | |||||||| | |||||||| | |||||||| | 1 (0.3) |
CMQ = customized medical query; MedDRA = Medical Dictionary for Regulatory Activities; SMQ = standard MedDRA query; SSQ = sponsor-specific query.
aIncludes vinflunine.
Source: EV-301 Clinical Study Report.17
Critical Appraisal
Internal Validity
Study EV-301 was a phase III, open-label RCT. Appropriate methods for randomization (via interactive response technologies) and treatment allocation were employed. Patients were randomized based on ECOG PS, presence of liver metastases, and geographic region (Western Europe versus US versus rest of world). The methods for randomization were considered by the CADTH review team to result in a low risk of bias, as patients were randomly assigned to a treatment arm using technology that ensured relatively equal proportions of patients would be randomized, not only to each treatment arm, but to each pre-specified stratification factor. In general, patients did not differ with regard to baseline disease or treatment characteristics, indicating that randomization was successful. No imbalances across treatment arms or analysis populations therefore could have resulted in important differences in outcomes. A total of 27 patients in the study had mismatched stratification from randomization and at baseline. It is unclear if the baseline characteristics were those of the patients with correct or incorrect stratification factors. Moreover, it was unclear what the cause for the misclassification was, as no reason was provided. Regardless, because the results of the ad hoc sensitivity analyses were similar to those of the primary analysis, the results were not likely influenced by this error.
An open-label design was used, potentially increasing the risk of bias in the reporting of outcomes that are subjective in measurement and interpretation, such as response, HRQoL, and AEs, particularly given the expected AEs of special interest, such as skin reactions and ocular disorders associated with enfortumab vedotin. The primary end point of OS is objective, and unlikely to be affected by biases of open-label study designs. Secondary end points of PFS and ORR are subjective, and therefore subject to potential bias. Reporting of patient-rated outcomes, such as symptom reduction and HRQoL, and some of the harms outcomes may have been biased or influenced by the patient or investigator’s knowledge of treatment assignment. Moreover, all study outcomes were investigator-assessed and did not include full evaluation via an independent review committee to mitigate the biases associated with the open-label study design. To maintain trial integrity and increase the credibility of study results, aggregate analyses or summaries by randomized treatment assignment or actual treatment assignment were limited and documented before the primary hard database lock. Given the differences in administration methods and dosing of the intervention and comparator arms, as well as the known treatment-related toxicities associated with enfortumab vedotin, the CADTH reviewers and clinical expert consulted by CADTH agreed that the open-label design was appropriate, and that a double-blind study would not be feasible in this setting.
The original study protocol was amended 3 times. None of the amendments were considered to negatively affect the conduct or integrity of the study outcomes or data. Major protocol deviations occurred slightly more frequently in the enfortumab vedotin arm. Important protocol deviations were due to adding study patients who did not satisfy entry criteria (n = 17), developed withdrawal criteria during the study but were not withdrawn (n = 10), received the wrong treatment or incorrect dose (n = 4), or received excluded concomitant treatments (n = 2). It was noted that deviations related to development of withdrawal criteria occurred primarily when a subject developed a withdrawal criterion but the investigator believed the patient was continuing to derive clinical benefit and therefore chose to keep them on treatment. As there were few important protocol deviations, which were generally similar between arms, it is unlikely that they had a significant impact on efficacy or safety analyses.
As of the data cut-off, study discontinuation rates were high, and the majority of patients in both the enfortumab and chemotherapy arms had discontinued the study (81.4% and 92.8%, respectively). Although the overall proportion of discontinuation was higher in the chemotherapy arm, the proportion of patients discontinuing treatment due to disease progression was nearly identical between treatment arms (58.8% and 58.6% for the enfortumab and chemotherapy arms, respectively). It is unclear why the proportion of patients discontinuing treatment due to progressive disease was similar, given the significantly improved OS and PFS with enfortumab vedotin, as fewer cases of progressive disease would occur in a more effective regimen. Moreover, patient withdrawals and physician-decision dropouts were higher in the chemotherapy arm, potentially reflecting the open-label design of the study and introducing bias.
Although the method of accounting for missing data was provided, the actual amount of missing data for each outcome was not provided, making it impossible to determine the impact of the missing data on the results. The EORTC QLQ-C30 and EQ-5D-5L reported the proportion of patients completing each measure at specified time points and, unsurprisingly, the number of patients completing HRQoL measures dropped throughout the study; results related to HRQoL should therefore be interpreted with caution. As stated by the sponsor, a 12-week time of assessment was used for the PRO measures, which was considered appropriate given the median follow-up duration, as well as the expected PFS for the chemotherapy arm. Appropriate measures were used to account for the issues with mismatched assessment windows for HRQoL, as well as the data integrity issues.
Acceptable methods to account for multiplicity, including a hierarchical gatekeeping procedure, were used in the EV-301 trial for primary and secondary outcomes. The primary efficacy outcome of OS was controlled for using the familywise error rate, followed by the secondary end points of PFS1, ORR, and DCR. Other secondary and exploratory outcomes, including HRQoL, were not controlled for multiplicity, and any interpretation should consider the possibility of type I error.
A Cox proportional hazards model was used for OS and PFS outcomes. It was assumed in all cases that the proportional hazards assumption was met; however, it was not specified or tested, and was therefore unclear.
Predefined subgroup analyses based on various disease characteristics were conducted to examine the consistency of the primary and secondary analyses results across subgroup levels. The results of subgroup analyses were generally aligned with the overall analysis for OS, PFS1, and ORR. Subgroups were not confirmatory in nature, and therefore were not adjusted for multiplicity or missing data, and should be considered with this limitation in mind. Although generally in favour of the overall analyses, the results must be interpreted with caution due to the small number of patients in some subgroups.
At the planned interim analysis of EV-301, the independent data analysis centre suggested that the sponsor stop the study early for efficacy based on a statistically significant result for OS in favour of enfortumab vedotin. Trials that stop early for benefit may typically show a higher or superior treatment-effect estimate in the intervention group. Given that the primary end point of the study, OS, was not subjective, the review team’s concerns were minor. However, the primary analysis was conducted early based on the information fraction (68.6%). suggesting an increased, and notable risk of overestimation.
External Validity
In discussions with the clinical expert consulted by CADTH, the inclusion and exclusion criteria for EV-301 were generally as expected for patients with locally advanced or metastatic UC. The clinical expert consulted by CADTH noted that, in clinical practice, patients with HIV would not be excluded, as they were in this trial. Additionally, patients with metastases in the central nervous system would be eligible for treatment if their disease was under control. It was considered that patients included in the study may be a less sick population for various reasons, including laboratory assessments at inclusion, as well as the baseline characteristics of the included patients given the stage of the disease. The median age of patients in this trial was 68 years, which the clinical expert noted represent a younger population, as the median age of diagnosis is 65 years. Moreover, the proportion of patients over the age of 75 (17.3% and 22.1% for the enfortumab and chemotherapy arms, respectively) was underrepresented according to the clinical expert. The clinical expert also considered the ECOG PS of patients to be unreflective of patients at this stage of disease, and most patients would not have an ECOG PS of 0 or 1, whereas approximately 40% of patients in this trial had an ECOG PS of 0. It was also noted that Asian patients were overrepresented in the EV-301 study at 32.9%, which is not likely what would be seen in the Canadian population. Together, the results may not be generalizable to a typical population of third-line patients with metastatic UC in Canada.
The chosen comparator of standard chemotherapy consisting of paclitaxel, docetaxel, or vinflunine in EV-301 generally aligns with the recommended standard-of-care guidelines in Canada for patients who have failed platinum-containing chemotherapy and a PD-1 or PD-L1 inhibitor. Vinflunine is not a treatment option available in Canadian clinical practice, and any aggregate results for the chemotherapy arm should consider the proportion of patients that may have received this treatment. In EV-301, vinflunine was limited to 35% of the overall population of patients in the chemotherapy arm as it is not available in all countries and was given to 75 patients (25.8%) in the safety analysis set of the chemotherapy arm.
Outcomes in the EV-301 trial were clinically relevant, and important to patients. The survival benefit in terms of OS was statistically significant in favour of enfortumab vedotin, and was considered clinically meaningful, albeit moderately, by the clinical expert. This was considered important to the review team, as EV-301 was stopped early for efficacy based on an information fraction of 68.6%, and the moderately meaningful OS benefit (despite the threat of a potentially overestimated treatment effect) should therefore be interpreted with caution. The clinical expert consulted by CADTH noted that the OS seen in the comparator chemotherapy arm was higher than anticipated; however, when coupled with the assumption that patients may be less sick, it was reasonable. Moreover, the observed PFS was as expected. As previously mentioned, the proportion of patients discontinuing due to progressive disease was the same in each treatment arm despite the significantly improved PFS with enfortumab vedotin, likely due to the open-label design, and PFS results should therefore be interpreted with caution. The trial had a median follow-up of 11.1 months, which was considered acceptable given the virulence of the disease at this stage.
Health-related quality of life was assessed by the EORTC QLQ-C30 and EQ-5D-5L measures. At the 12-week time of assessment chosen for the analysis, 140 and 168 patients in the enfortumab vedotin and chemotherapy arms did not complete the EORTC QLQ-C30, with similar decreases in completion and compliance rates seen for the EQ-5D-5L. As HRQoL was a secondary outcome, and not controlled for multiplicity, results for PROs should be interpreted with caution and may not be generalizable to the Canadian population.
Originally, safety results for the chemotherapy arm were not separated by individual treatment received (paclitaxel, docetaxel, or vinflunine). The CADTH review team requested the safety results of interest be separated by individual treatment to reflect Canadian clinical practice, given the known differences in safety profiles for these treatments (particularly for vinflunine, which is not available in Canada), compared to the taxanes, which were inflating the results of certain AEs for the chemotherapy arm. For example, the rate of constipation, which is not generally an issue when a patients is treated with taxanes but is a known AE with vinflunine use, between enfortumab vedotin and chemotherapy arms was similar at 27.7% versus 25.1%, respectively. Results by individual chemotherapy were provided by the sponsor.
Overall, generalizability of the study results to the Canadian patient population could be limited due to the possible overestimate of the treatment effect following early termination of the study, the inclusion of vinflunine as a comparator, and the inclusion of a potentially healthier population than would be found in the real world.
Indirect Evidence
A focused literature search for network meta-analyses dealing with urothelial carcinoma was run in MEDLINE All (1946–) on July 22, 2021. No limits were applied.
No indirect evidence was included in the sponsor’s submission to CADTH or identified in the literature search that matched the inclusion and exclusion criteria of this review.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One phase III, open-label RCT; Study EV-301, was included in this review. Study EV-301 consisted of 608 patients with locally advanced or metastatic UC who were randomized to receive either enfortumab vedotin (n = 301) 1.25 mg/kg on days 1, 8, and 15 of every 28-day cycle or standard chemotherapy consisting of paclitaxel, docetaxel, or vinflunine (n = 307) on day 1 of each 21-day cycle until disease progression or unacceptable toxicity. The primary end point of EV-301 was OS, with secondary end points of PFS, ORR, DCR, and HRQoL.17
Baseline characteristics of the EV-301 trial were well balanced between treatment arms, but a healthier group of patients with a younger median age and lower ECOG PS compared to the Canadian population may have been enrolled. Patients in EV-301 were mostly White (51.6%) and male (77.3%), with a median age of 68 years. Most patients had an ECOG PS of 1 (59.9%) and had metastatic disease (95.2%).17
Interpretation of Results
Efficacy
The primary efficacy end point of EV-301 was OS, which is a clinically relevant outcome to this review. Based on an interim analysis, EV-301 was terminated early for efficacy due to significant interim OS results. The results for OS at the primary analysis were statistically significantly in favour of enfortumab vedotin compared to chemotherapy (HR = 0.70; 95% CI, 0.556 to 0.886; P = 0.00142), although the median OS for the chemotherapy arm was higher than expected at 8.97 months, according to the clinical expert consulted by CADTH. The included population was considered to be less sick than typical of locally advanced or metastatic UC patients, overestimating the true OS. Sensitivity analyses and subgroup analyses of OS were consistent with the primary analysis, further enforcing the results. Results for the secondary end point of PFS were in line with OS, and median PFS was reflective of what would be expected in the chemotherapy arm (3.71 months).
Results for HRQoL as measured by the EORTC QLQ-C30, demonstrated a minimal change from baseline in QoL for functional scales, with reductions in QoL in both the enfortumab vedotin and chemotherapy arms at week 12. There was a marked decrease in respondents at week 12 compared to baseline, as evidenced by the large standard deviations for the change from baseline. For symptom scales, the enfortumab vedotin arm had more symptom improvements than the chemotherapy arm at week 12, particularly for pain, which was an outcome important to patients. However, given the known side effects of treatment with enfortumab vedotin and chemotherapy, it is difficult to differentiate between cancer-related symptoms, and treatment symptoms. Although enfortumab vedotin was favoured for reduction in self-reported pain (−5.73; 95% CI, −10.80 to −0.66), the results must be interpreted with caution given the open-label study design and the lack of adjustment for multiple comparisons for HRQoL outcomes.
For the EQ VAS, patients in the chemotherapy arm demonstrated a worse QoL at 12 weeks compared to baseline than did members of the enfortumab vedotin arm (−5.3 versus −1.8, respectively). Overall, while there was no marked deterioration or improvement in HRQoL, the results must be interpreted with caution as they were secondary outcomes that were not controlled for multiplicity and were subject to decreased completion rates over time. Improvement in QoL was described by patients as an important aspect of treatment. The clinical expert suspected that enfortumab vedotin may display more toxicities than chemotherapy, and this would likely have a negative impact on QoL.
The secondary outcome of ORR in EV-301 was statistically significant in favour of enfortumab vedotin and deemed clinically meaningful by the clinical expert consulted by CADTH. Typical response at this stage of disease in this population is 20%, which is consistent with the patients in the chemotherapy arm (17.9%). The ORR of 40.6% in the enfortumab vedotin arm was considered exceptional for a single-drug therapy. As per RECIST 1.1 guidelines,28 a PR is determined by a 30% decrease in the sum of diameters in target lesions. With 35.8% of patients in the enfortumab vedotin arm demonstrating a confirmed PR according to RECIST 1.1, the clinical expert consulted by CADTH expected that this would result in survival benefits, as well as an improvement in cancer-related symptoms. The results for ORR are in line with the survival benefit seen for OS; however, it remains uncertain what impact the clinically meaningful ORR has on improvement in cancer symptoms. Given the known toxicities associated with enfortumab vedotin, the improvement in symptoms may be difficult to determine. Regardless, halting progression of the disease, improved survival, and improvement in symptoms were outcomes important to patients.
Harms
The overall incidence of harms reported in EV-301 was well balanced, with the exception of the known AESIs with enfortumab vedotin. However, given the known safety profiles of both enfortumab vedotin and the individual chemotherapies, the comparable distribution of AEs between groups was questioned by the clinical expert consulted by CADTH. There were some imbalances in the specific incidences of TEAEs between patients receiving enfortumab vedotin and those receiving chemotherapy despite the similar overall incidence. Regarding grade 3 or higher TEAEs, the overall incidence was unbalanced between the treatment arms. In general, the incidence of grade 3 or higher treatment-emergent hematologic toxicities was higher with taxane chemotherapy, including decreased neutrophil count (7.1% versus ||||), neutropenia (4.7% versus ||||), febrile neutropenia (1.4% versus ||||), and severe anemia potentially requiring blood transfusions (6.4% versus ||||). According to the clinical expert, hematologic toxicities are more critical if they require treatment (i.e., resulting in febrile neutropenia or requiring blood transfusions). For example, decreased neutrophil counts can be considered asymptomatic, compared to the grade 3 or higher AEs that were more frequent for enfortumab vedotin (e.g., maculopapular rash), which are highly symptomatic. Despite higher rates of potentially symptomatic hematologic toxicity with chemotherapy, the clinical expert consulted by CADTH noted that the nature of toxicities must be considered when evaluating the comparative safety as the overall harms results suggest that the treatments have similar safety profiles. The clinical expert also noted that, although highlighted, skin reactions (dry skin, rash, maculopapular rash, and pruritis) were presented individually, making them appear less frequent in the enfortumab vedotin arm, which should be interpreted carefully as it is a known side effect.
A unique side effect of enfortumab vedotin is the development of related ocular disorders, such as increased lacrimation, dry eye, keratitis, or conjunctivitis, that require monitoring and examination by an ophthalmologist. This is an important consideration that may negatively affect patients and will add incremental costs and health care resources. Skin reactions including rash are also an AESI associated with the use of enfortumab vedotin. The incidence of skin reactions in EV-301 was higher in the enfortumab vedotin arm compared to taxane chemotherapy (53.7% versus ||||||). Rash (48% versus ||||) is a known side effect of treatment with enfortumab vedotin and, according to the clinical expert consulted by CADTH, it can be quite severe and debilitating for patients, affecting their QoL and imposing additional costs. The clinical expert consulted by CADTH suggested that enfortumab vedotin may be more toxic than current chemotherapies and hypothesized that the toxicities seen with enfortumab vedotin treatment explain why QoL did not improve greatly. However, the patient group input cites temporary and manageable toxicities experienced in the 2 patients who received enfortumab vedotin.
The proportions of patients discontinuing the study due to AEs were similar between treatment arms (14% versus 15% for enfortumab vedotin and chemotherapy, respectively). As previously mentioned, withdrawals by patient and physician decision were more common in the chemotherapy arm, which could be a result of the open-label design of the study as patients and investigators were aware of treatment, potentially biasing the reason for discontinuation, and affecting the interpretability and generalizability of results.
Conclusions
Enfortumab vedotin is a first-in-class treatment that has been studied in patients who have received a prior PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy and demonstrated a statistically significant improvement in OS and PFS compared to chemotherapy. Enfortumab vedotin was also associated with a clinically meaningful ORR compared to chemotherapy for a single-drug therapy, which is an important consideration for this stage of disease. The results for ORR are in line with the survival benefit seen for OS, both of which are important to patients; however, it remains uncertain what impact the clinically meaningful ORR has on improvement in cancer symptoms. Moreover, given the open-label design, the results for PFS and ORR must be interpreted with caution. Enfortumab vedotin was not associated with any major improvement or deterioration in HRQoL in the pivotal study; however, because of high patient-attrition rates, the effect of enfortumab vedotin on HRQoL remains uncertain.
Overall, despite the overall similar rates of TEAEs between the enfortumab vedotin and chemotherapy arms, interpretation of the comparative results should consider the individual safety profiles of the study treatments, particularly for taxanes, as vinflunine is not available in Canada. Generally, the rate of specific TEAEs was higher for enfortumab vedotin compared to taxane therapies. Enfortumab vedotin was also associated with more SAEs and symptomatic notable harms, including ocular disorders, skin reactions, and peripheral neuropathy, compared to taxane chemotherapy. Despite the feedback from 2 patients in the patient group input who noted that side effects of treatment with enfortumab vedotin were temporary and manageable, enfortumab vedotin was hypothesized by the clinical expert to be more toxic than current therapeutic options, which may explain the lack of improvement in QoL.
Overall, enfortumab vedotin provides an effective third-line treatment option, extending survival and demonstrating good clinical response after progression with platinum chemotherapy and a PD-1 or PD-L1 inhibitor. However, there were several limitations in the generalizability of results given the stage of disease, as well as potential safety concerns.
References
1.Flaig TW, Spiess PE, Agarwal N, et al. Bladder Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(3):329-354. PubMed
2.Fleshner NE, Herr HW, Stewart AK, Murphy GP, Mettlin C, Menck HR. The National Cancer Data Base report on bladder carcinoma. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1996;78(7):1505-1513. PubMed
3.Cancer Treatment Centers of America. Bladder cancer types. 2021; https://www.cancercenter.com/cancer-types/bladder-cancer/types. Accessed 2021 Oct 14.
4.Lotan Y, Choueiri TK. Clinical presentation, diagnosis, and staging of bladder cancer. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2020: www.uptodate.com. Accessed 2021 Jul 13.
5.Canadian Cancer Society. Bladder cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/bladder. Accessed 2021 Oct 14.
6.American Cancer Society. Cancer facts & figures 2021. Atlanta (GA): American Cancer Society; 2021: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2021/cancer-facts-and-figures-2021.pdf?bcs-agent-scanner=6d745fe2-1baa-7a44-a2b6-2f677a8a646c. Accessed 2021 Oct 14.
7.SEER cancer stat facts: bladder cancer. Bethesda (MD): National Cancer Institute; 2021: https://seer.cancer.gov/statfacts/html/urinb.html. Accessed 2021 Oct 14.
8.Daneshmand S. Epidemiology and risk factors of urothelial (transitional cell) carcinoma of the bladder. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Jul 13.
9.Malats N, Real FX. Epidemiology of bladder cancer. Hematol Oncol Clin North Am. 2015;29(2):177-189, vii. PubMed
10.Warren M, Kolinsky M, Canil CM, et al. Canadian Urological Association/Genitourinary Medical Oncologists of Canada consensus statement: management of unresectable locally advanced and metastatic urothelial carcinoma. Can Urol Assoc J. 2019;13(10):318-327. PubMed
11.Bellmunt J. Treatment of metastatic urothelial cancer of the bladder and urinary tract. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Jul 13.
12.von der Maase H, Sengelov L, Roberts JT, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. 2005;23(21):4602-4608. PubMed
13.CADTH pCODR Reimbursement Expert Review Committee (pERC) final recommendation: avelumab (Bavencio - Pfizer). Ottawa (ON): CADTH; 2021 Mar 23: https://cadth.ca/sites/default/files/pcodr/Reviews2021/10225AvelumabUC_FnRec_REDACT_EC23Mar2021_final.pdf?bcs-agent-scanner=4f537223-81fc-6a4b-b667-f4b3c17e7608. Accessed 2021 Oct 14.
14.NCCN Guidelines. Bladder Cancer version 3.2021. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2021 Apr 22: www.nccn.org. Accessed 2021 Jul 13.
15.CADTH pCODR Reimbursement Expert Review Committee (pERC) final recommendation: pembrolizumab (Keytruda - Merck Canada Inc). Ottawa (ON): CADTH; 2018: https://cadth.ca/sites/default/files/pcodr/pcodr_pembrolizumab_keytruda_muc_fn_rec.pdf. Accessed 2021 Oct 14.
16.Challita-Eid PM, Satpayev D, Yang P, et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016;76(10):3003-3013. PubMed
17.Clinical Study Report: 7465-CL-0301. An open-label, randomized phase 3 study to evaluate enfortumab vedotin vs chemotherapy in subjects with previously treated locally advanced or metastatic urothelial cancer (EV-301) [internal sponsor's report]. Northbrook (IL): Astellas Pharma Global Development, Inc.; 2021.
18.Padcev (enfortumab vedotin): 20 mg and 30 mg single-use vials of lyophilized powder for solution for intravenous infusion only [product monograph]. Mississauga (ON): Seagen Canada; 2021 Oct 29.
19.National Cancer Institute. Bladder cancer — health professional version. 2021; https://www.cancer.gov/types/bladder/hp. Accessed 2021 Oct 14.
20.Berdik C. Unlocking bladder cancer. Nature. 2017;551(7679):S34-S35. PubMed
21.CADTH pCODR Reimbursement Expert Review Committee (pERC) final recommendation: pembrolizumab (Keytruda - Merck Canada Inc). Ottawa (ON): CADTH; 2019 Oct 03: https://cadth.ca/sites/default/files/pcodr/Reviews2019/10177PembrolizumabMUC%28firstline%29_FnRec_REDACT_Post_03Oct2019_final.pdf. Accessed 2021 Oct 14.
22.European Medicines Agency accepts marketing authorization application for enfortumab vedotin Toyko (JP): Astellas Pharma Inc.; 2021: https://www.astellas.com/system/files/news/2021-03/20210326_en_5.pdf?bcs-agent-scanner=361d7c92-58e4-384b-9a14-e94faec6b24b. Accessed 2021 Oct 14.
23.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
24.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Jul 12.
25.Powles T, Rosenberg JE, Sonpavde GP, et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N Engl J Med. 2021;384(12):1125-1135. PubMed
26.Drug Reimbursement Review sponsor submission: Padcev (enfortumab vedotin), 20 mg and 30 mg single-use vials of lyophilized powder for solution for intravenous infusion only [internal sponsor's package]. Mississauga (ON): Seagen Canada; 2021 Jul 09.
27.Mamtani R, Rosenberg JE, Powles T, et al. Quality of life, functioning, and symptoms in patients with previously treated locally advanced or metastatic urothelial carcinoma from EV-301: a randomized phase 3 trial of enfortumab vedotin versus chemotherapy. J Clin Oncol. 2021;39(15_suppl):4539-4539.
28.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. PubMed
29.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-1807. PubMed
30.EQ-5D-5L user guide, 2019. Rotterdam (NL): EuroQol Research Foundation; 2019: https://euroqol.org/publications/user-guides/. Accessed 2021 Oct 14.
31.Pickard AS, De Leon MC, Kohlmann T, Cella D, Rosenbloom S. Psychometric comparison of the standard EQ-5D to a 5 level version in cancer patients. Med Care. 2007;45(3):259-263. PubMed
32.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-defined estimates of the minimally important difference for EQ-5D-5L index scores. Value Health. 2017;20(4):644-650. PubMed
33.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
34.Fayers PM. Interpreting quality of life data: population-based reference data for the EORTC QLQ-C30. Eur J Cancer. 2001;37(11):1331-1334. PubMed
35.Osoba D, Aaronson N, Zee B, Sprangers M, te Velde A. Modification of the EORTC QLQ-C30 (version 2.0) based on content validity and reliability testing in large samples of patients with cancer. The Study Group on Quality of Life of the EORTC and the Symptom Control and Quality of Life Committees of the NCI of Canada Clinical Trials Group. Qual Life Res. 1997;6(2):103-108. PubMed
36.Blazeby JM, Hall E, Aaronson NK, et al. Validation and reliability testing of the EORTC QLQ-NMIBC24 questionnaire module to assess patient-reported outcomes in non-muscle-invasive bladder cancer. Eur Urol. 2014;66(6):1148-1156. PubMed
37.Osoba D, Burchmore M. Health-related quality of life in women with metastatic breast cancer treated with trastuzumab (Herceptin). Semin Oncol. 1999;26(4 Suppl 12):84-88. PubMed
38.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 22, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Padcev* or enfortumab vedotin* or enfortumabvedotin* or AGS-22MSE or AGS22MSE or AGS-22CE or AGS22CE or AGS-22ME or AGS22ME or ASG-22ME or ASG22ME or AGS-22M6E or AGS22M6E or DLE8519RWM).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*enfortumab vedotin/
(Padcev* or enfortumab vedotin* or enfortumabvedotin* or AGS-22MSE or AGS22MSE or AGS-22CE or AGS22CE or AGS-22ME or AGS22ME or ASG-22ME or ASG22ME or AGS-22M6E or AGS22M6E or DLE8519RWM).ti,ab,kw,dq.
or/3-4
5 use oemezd
6 not conference abstract.pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Padcev OR Enfortumab vedotin OR enfortumab vedotin-ejfv OR enfortumab vedotin ejfv OR AGS-22MSE OR AGS22MSE OR AGS-22CE OR AGS22CE OR AGS-22ME OR AGS22ME OR AGS-22M6E OR AGS22M6E]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- Padcev OR Enfortumab vedotin OR enfortumab vedotin-ejfv OR enfortumab vedotin ejfv OR AGS-22MSE OR AGS22MSE OR AGS-22CE OR AGS22CE OR AGS-22ME OR AGS22ME OR AGS-22M6E OR AGS22M6E]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Enfortumab vedotin]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Enfortumab OR Padcev OR AGS-22MSE OR AGS22MSE OR AGS-22CE OR AGS22CE OR AGS-22ME OR AGS22ME OR AGS-22M6E OR AGS22M6E]
Grey Literature
Search dates: July 12, 2021 – July 16, 2021
Keywords: [Padcev OR Enfortumab vedotin OR urothelial carcinoma OR bladder cancer OR urinary cancer]
Limits: Publication years: none
Updated: Search updated prior to the meeting of CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC).
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Deininger S, Torzsok P, Oswald D, Lusuardi L. Current Systemic Treatment Options in Metastatic Urothelial Carcinoma after Progression on Checkpoint Inhibition Therapy-A Systemic Review Combined with Single-Group Meta-Analysis of Three Studies Testing Enfortumab Vedotin. Cancers (Basel). 2021 Jun 26;13(13):26. | Systematic Review |
Hirotsu KE, Rana J, Wang JY, et al. Clinicopathologic characterization of enfortumab vedotin-associated cutaneous toxicity in patients with urothelial carcinoma. J Am Acad Dermatol. 2020 Dec 07;07:07. | Review article |
Rosenberg J, Sridhar SS, Zhang J, et al. EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients With Nectin-4-Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. J Clin Oncol. 2020 04 01;38(10):1041-1049. | Study design (phase I) |
Rosenberg JE, O'Donnell PH, Balar AV, et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J Clin Oncol. 2019 10 10;37(29):2592-2600. | Study design (phase II) |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 30: Sensitivity Analysis — PFS1, Investigator Assessment (Full Analysis Set)
Outcome | Enfortumab Vedotin (n = 301) | Chemotherapy (n = 307) |
|---|---|---|
PFS Events, n (%) | 204 (67.8) | 234 (76.2) |
Radiographical Progression | 172 (57.1) | 195 (63.5) |
Death without Documented Progression | 32 (10.6) | 39 (12.7) |
Censored, n (%) | 97 (32.2) | 73 (23.8) |
No PFS Event | 89 (29.6) | 64 (20.8) |
PFS Event After New Anticancer Therapy | 8 (2.7) | 9 (2.9) |
Median (95% CI) PFS (months)a | 5.55 (5.32, 6.08) | 3.71 (3.52, 3.98) |
Stratified Analysisb | ||
HR (95% CI)c | 0.616 (0.507, 0.749) | |
1-sided P valued | < 0.001 | |
PFS Rate, % (95% CI)e | ||
6 Months | 44.2 (38.21, 50.03) | 28.6 (23.28, 34.18) |
12 Months | 21.3 (15.92, 27.23) | 8.2 (4.52, 13.14) |
HR = hazard ratio; PFS = progression-free survival.
aBased on Kaplan-Meier estimate.
bStratification factors were ECOG PS, geographic region and liver metastasis per IRT
cBased on Cox proportional hazards model with treatment, ECOG PS, geographic region and liver metastasis as the explanatory variables. Assuming proportional hazards, a hazard ratio < 1 indicates a reduction in hazard rate in favour of treatment arm.
dBased on log-rank test.
ePFS rate and 95% CI were estimated using Kaplan-Meier method and Greenwood formula.
Source: EV-301 Clinical Study Report.17
Table 31: Subgroup Analysis of PFS1, Investigator Assessment (Full Analysis Set)
Subgroup | Enfortumab Vedotin Event/N | Chemotherapy Event/N | HR (95% CI)a |
|---|---|---|---|
All Subjectsb | 201/301 | 231/307 | 0.615 (0.505, 0.748) |
ECOG PS per IRT | |||
0 | 71/120 | 86/124 | 0.617 (0.450, 0.846) |
1 | 130/181 | 145/183 | 0.659 (0.519, 0.837) |
Liver Metastases per IRT | |||
Yes | 71/93 | 75/95 | 0.597 (0.428, 0.833) |
No | 130/208 | 156/212 | 0.648 (0.512, 0.818) |
Site of Primary Tumour | |||
Upper Tract | 63/98 | 74/107 | 0.716 (0.511, 1.003) |
Bladder/Other | 138/203 | 157/200 | 0.606 (0.481, 0.763) |
Prior Lines of Systemic Therapy | |||
1-2 | 175/262 | 203/270 | 0.640 (0.523, 0.785) |
≥3 | 26/39 | 28/37 | 0.672 (0.393, 1.150) |
Best Response to CPI | |||
Responder | 32/61 | 36/50 | 0.511 (0.317, 0.826) |
Nonresponder | 146/207 | 160/215 | 0.697 (0.556, 0.873) |
CPI = checkpoint inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EV = enfortumab vedotin; HR = hazard ratio; IRT = interactive response technology.
[1]In each subgroup, the HR was estimated using unstratified Cox proportional hazards model with treatment. Assuming proportional hazards, HR < 1 indicates a reduction in hazard rate in favour of treatment arm.
[2]The HR reported for all patients was based on stratified analysis. Stratification factors were ECOG PS, geographic region and liver metastasis per IRT
Source: EV-301 Clinical Study Report.17
Table 32: Progression-Free Survival After Next-Line Therapy, Investigator Assessment (Full Analysis Set)
Outcome | Enfortumab Vedotin (n = 301) | Chemotherapy (n = 307) |
|---|---|---|
PFS2 Events, n (%) | 152 (50.5%) | 195 (63.5%) |
Progression of Disease | 36 (12.0%) | 54 (17.6%) |
Death from any Cause | 104 (34.6%) | 125 (40.7%) |
Other PFS2 Eventsa | 12 (4.0%) | 16 (5.2%) |
Censored, n (%) | 149 (49.5%) | 112 (36.5%) |
Median (95% CI) PFS2 (months)b | 9.63 (8.21, 10.58) | 7.00 (6.54, 8.05) |
Stratified Analysisc | ||
HR (95% CI)d | 0.619 (0.497, 0.771) | |
1-sided P valuee | < 0.001 | |
CI = confidence interval; HR = hazard ratio; PFS2 = progression-free survival after next-line therapy.
aIncludes start of a different treatment after the subsequent new anticancer therapy after end of study drug treatment.
bBased on Kaplan-Meier estimate.
cStratification factors were ECOG PS, geographic region and liver metastasis from the electronic Case Report Form.
dBased on Cox proportional hazards model with treatment, ECOG PS, geographic region and liver metastasis as the explanatory variables. Assuming proportional hazards, a hazard ratio < 1 indicates a reduction in hazard rate in favour of treatment arm.
eBased on log-rank test.
Source: EV-301 Clinical Study Report.17
Figure 7: EORTC QLQ-C30 Completion Rate by Visit (Full Analysis Set)
EOT = end of treatment; FUP = follow-up; Wk = week.
Source: EV-301 Clinical Study Report.17
Figure 8: EQ-5D-5L Completion Rate by Visit (Full Analysis Set)
EOT = end of treatment; FUP = follow-up; Wk = week.
Source: EV-301 Clinical Study Report.17
Table 33: Sensitivity Analysis — ORR and DCR, Investigator Assessment (Response Evaluable Set)
Outcome | Enfortumab Vedotin (n = 288) | Chemotherapy (n = 296) |
|---|---|---|
BOR, Unconfirmed, n (%)a | ||
Unconfirmed Complete Response | 20 (6.9) | 8 (2.7) |
Unconfirmed Partial Response | 126 (43.8) | 76 (25.7) |
Stable Disease | 62 (21.5) | 76 (25.7) |
Progressive Disease | 44 (15.3) | 81 (27.4) |
Not Evaluable | 36 (12.5) | 55 (18.6) |
Unconfirmed ORR, n (%) | 146 (50.7) | 84 (28.4) |
95% CI for ORR (%)b | (44.77, 56.61) | (23.31, 33.88) |
Stratified 1-sided P valuec | < 0.001 | |
Unconfirmed DCR, n (%)d | 208 (72.2) | 160 (54.1) |
95% CI for DCR (%)b | (66.66, 77.32) | (48.19, 59.83) |
Stratified 1-sided P valuec | < 0.001 | |
BOR = best overall response; CR = complete response; DCR: disease control rate; ORR = overall response rate; PR = partial response.
aThe definition of best overall response followed RECIST 1.1. The minimum duration for stable disease was 7 weeks.
bUsing exact method based on binomial distribution (Clopper-Pearson).
cBased on Cochran-Mantel-Haenszel test. Stratification factors were ECOG PS, geographic region and liver metastasis.
dDCR was defined as the proportion of patients who had a best overall response of confirmed CR, confirmed PR, or stable disease (≥ 7 weeks).
Source: EV-301 Clinical Study Report.17
Table 34: Subgroup Analysis of ORR, Investigator Assessment (Full Analysis Set)
Subgroup | Enfortumab Vedotin n/N | Chemotherapy n/N | Absolute Difference (95% CI) |
|---|---|---|---|
All Subjectsa | 117/288 | 53/296 | 22.7% (14.7, 30.6) |
ECOG PS per IRT | |||
0 | 49/115 | 30/121 | 17.8% (5.0, 30.2) |
1 | 68/173 | 23/175 | 26.2% (15.8, 36.1) |
Liver Metastases per IRT | |||
Yes | 33/93 | 10/93 | 24.7% (10.0, 38.7) |
No | 84/195 | 43/203 | 21.9% (12.1, 31.3) |
Site of Primary Tumour | |||
Upper Tract | 43/98 | 20/105 | 24.8% (11.1, 37.8) |
Bladder/Other | 74/190 | 33/191 | 21.7% (11.6, 31.1) |
Prior Lines of Systemic Therapy | |||
1-2 | 103/251 | 47/262 | 23.1% (14.5, 31.4) |
≥3 | 14/37 | 6/34 | 20.2% (-3.6, 41.7) |
Best Response to CPI | |||
Responder | 28/56 | 12/49 | 25.5% (6.3, 43.4) |
Nonresponder | 79/199 | 36/207 | 22.3% (12.7, 31.7) |
CI = confidence interval; CPI = checkpoint inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FAS = full analysis set; IRT = interactive response technology; ORR = overall response rate.
aBased on unstratified analysis.
Source: EV-301 Clinical Study Report.17
Table 35: TEAEs Resulting in Dose Reduction (Safety Analysis Set)
Preferred Term (MedDRA v23.0) | Enfortumab Vedotin (n = 296) | Chemotherapy (n = 291) |
|---|---|---|
Overall | 101 (34.1) | 81 (27.8) |
Peripheral sensory neuropathy | 22 (7.4) | 18 (6.2) |
Rash maculopapular | 13 (4.4) | 0 |
Decreased appetite | 10 (3.4) | 3 (1.0) |
Fatigue | 8 (2.7) | 12 (4.1) |
Neuropathy peripheral | 6 (2.0) | 2 (0.7) |
Neutrophil count decreased | 6 (2.0) | 8 (2.7) |
Neutropenia | 5 (1.7) | 7 (2.4) |
Diarrhea | 4 (1.4) | 2 (0.7) |
Drug eruption | 4 (1.4) | 0 |
Pruritus | 4 (1.4) | 0 |
Lipase increased | 3 (1.0) | 0 |
Rash | 3 (1.0) | 0 |
Vomiting | 3 (1.0) | 1 (0.3) |
Abdominal pain | 2 (0.7) | 1 (0.3) |
Aspartate aminotransferase increased | 2 (0.7) | 0 |
Hyperglycemia | 2 (0.7) | 0 |
Mucosal inflammation | 2 (0.7) | 1 (0.3) |
Nausea | 2 (0.7) | 2 (0.7) |
Abdominal pain upper | 1 (0.3) | 2 (0.7) |
Asthenia | 1 (0.3) | 5 (1.7) |
Constipation | 1 (0.3) | 6 (2.1) |
Thrombocytopenia | 1 (0.3) | 2 (0.7) |
White blood cell count decreased | 1 (0.3) | 2 (0.7) |
Anemia | 0 | 2 (0.7) |
Febrile neutropenia | 0 | 8 (2.7) |
Leukopenia | 0 | 2 (0.7) |
Malaise | 0 | 3 (1.0) |
Polyneuropathy | 0 | 2 (0.7) |
Pyrexia | 0 | 2 (0.7) |
Source: EV-301 Clinical Study Report.17
Table 36: TEAEs Resulting in Dose Interruption (Safety Analysis Set)
Preferred Term (MedDRA v23.0) | Enfortumab Vedotin (n = 296) | Chemotherapy (n = 291) |
|---|---|---|
Overall | 180 (60.8) | 85 (29.2) |
Peripheral sensory neuropathy | 46 (15.5) | 4 (1.4) |
Fatigue | 18 (6.1) | 4 (1.4) |
Neutrophil count decreased | 15 (5.1) | 11 (3.8) |
Rash maculopapular | 13 (4.4) | 0 |
Hyperglycemia | 11 (3.7) | 1 (0.3) |
Rash | 10 (3.4) | 0 |
Neuropathy peripheral | 9 (3.0) | 1 (0.3) |
Anemia | 8 (2.7) | 10 (3.4) |
Asthenia | 8 (2.7) | 1 (0.3) |
Pyrexia | 8 (2.7) | 4 (1.4) |
Acute kidney injury | 7 (2.4) | 0 |
Diarrhea | 7 (2.4) | 0 |
Drug eruption | 7 (2.4) | 0 |
Pneumonia | 7 (2.4) | 1 (0.3) |
Alanine aminotransferase increased | 6 (2.0) | 1 (0.3) |
Aspartate aminotransferase increased | 6 (2.0) | 2 (0.7) |
Decreased appetite | 6 (2.0) | 0 |
Neutropenia | 6 (2.0) | 5 (1.7) |
Blood creatinine increased | 5 (1.7) | 1 (0.3) |
Pruritus | 5 (1.7) | 0 |
Cellulitis | 4 (1.4) | 0 |
Dizziness | 4 (1.4) | 0 |
Dyspnea | 4 (1.4) | 0 |
Hematuria | 4 (1.4) | 3 (1.0) |
Peripheral sensorimotor neuropathy | 4 (1.4) | 0 |
Urinary tract infection | 4 (1.4) | 1 (0.3) |
White blood cell count decreased | 4 (1.4) | 1 (0.3) |
Dermatitis bullous | 3 (1.0) | 0 |
Hyponatremia | 3 (1.0) | 1 (0.3) |
Nausea | 3 (1.0) | 2 (0.7) |
Polyneuropathy | 3 (1.0) | 0 |
Thrombocytopenia | 3 (1.0) | 1 (0.3) |
Vomiting | 3 (1.0) | 0 |
Abdominal pain | 2 (0.7) | 1 (0.3) |
Amylase increased | 2 (0.7) | 0 |
Chronic kidney disease | 2 (0.7) | 0 |
Confusional state | 2 (0.7) | 1 (0.3) |
Dehydration | 2 (0.7) | 0 |
Dermatitis acneiform | 2 (0.7) | 0 |
Dysgeusia | 2 (0.7) | 0 |
Escherichia urinary tract infection | 2 (0.7) | 0 |
Eye pain | 2 (0.7) | 0 |
General physical health deterioration | 2 (0.7) | 2 (0.7) |
Hepatic function abnormal | 2 (0.7) | 2 (0.7) |
Herpes zoster | 2 (0.7) | 0 |
Hypokalemia | 2 (0.7) | 1 (0.3) |
Hypomagnesemia | 2 (0.7) | 0 |
Hypophosphatemia | 2 (0.7) | 0 |
Hypotension | 2 (0.7) | 0 |
Infusion-related reaction | 2 (0.7) | 4 (1.4) |
Lipase increased | 2 (0.7) | 1 (0.3) |
Lymphocyte count decreased | 2 (0.7) | 1 (0.3) |
Myalgia | 2 (0.7) | 1 (0.3) |
Nasopharyngitis | 2 (0.7) | 1 (0.3) |
Neurotoxicity | 2 (0.7) | 0 |
Oedema peripheral | 2 (0.7) | 3 (1.0) |
Paresthesia | 2 (0.7) | 0 |
Peripheral motor neuropathy | 2 (0.7) | 0 |
Sepsis | 2 (0.7) | 0 |
Urinary tract infection bacterial | 2 (0.7) | 1 (0.3) |
Urosepsis | 2 (0.7) | 2 (0.7) |
Vision blurred | 2 (0.7) | 0 |
Bone pain | 1 (0.3) | 2 (0.7) |
Malaise | 1 (0.3) | 5 (1.7) |
Blood alkaline phosphatase increased | 0 | 2 (0.7) |
Extravasation | 0 | 2 (0.7) |
Injection site reaction | 0 | 2 (0.7) |
Leukopenia | 0 | 2 (0.7) |
Pneumocystis jirovecii pneumonia | 0 | 2 (0.7) |
Urticaria | 0 | 2 (0.7) |
Source: EV-301 Clinical Study Report.17
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference):
EORTC QLQ-C30
EQ-5D-5L
Findings
Table 37: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about Measurement Properties | Minimal important difference |
|---|---|---|---|
EORTC QLQ-C30 | 30-item, patient-reported, cancer-specific, quality of life questionnaire using 4- and 7-point Likert scales | No evidence of validity, reliability, or responsiveness in patients with locally advanced or metastatic urothelial cancer. | Patients with cancer29: 5-10 points small change 10-20 points moderate change > 20 points large change No MID identified in patients with locally advanced or metastatic urothelial cancer |
EQ-5D-5L | Generic, preference-based measure of HRQoL.30 | No evidence of validity, reliability, or responsiveness in patients with locally advanced or metastatic urothelial cancer. | All cancers in the US: 0.07-0.09 (by ECOG), and 0.06-0.07 (by FACT-G).31 Canadian population: 0.037 for the health state index score.32 Patients with advanced cancer: 7 to 12 for the VAS.31 No MID identified in patients with locally advanced or metastatic urothelial cancer. |
EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; MID = minimal important difference.
EORTC QLQ-C30
Description and Scoring
The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) is one of the most used PRO measures in oncology clinical trials. It is a multidimensional, cancer-specific, measure of HRQoL.33
The EORTC QLQ-C30 is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), a Global Health Status/QoL scale, and 6 single items assessing additional symptoms commonly reported by cancer patients (dyspnea, loss of appetite, insomnia, constipation and diarrhea) and perceived financial impact of the disease.33
The EORTC QLQ-C30 uses a 1-week recall period to assess function and symptoms. All the scales and single-item measures range in score from 0 to 100. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global QoL scale, the response format is a 7-point Likert-type scale with anchors at 1 = “very poor” and 7 = “excellent.” Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Scale sum scores are transformed so that a high score on the functional scales represents a high/healthy level of functioning, a high score on the symptom scales represents a high level of symptomatology, and a high score on the Global Health Status/QoL represents a high QoL.34
According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least half of the items. It is assumed that the missing items have values equal to the average of those items for what the respondent completed.34
Assessment of Validity, and Reliability
In its initial development, the EORTC QLQ-C30 underwent an evaluation of its psychometric properties and demonstrated reliability and validity in cancer patients in multicultural clinical research settings.33 A revision of the EORTC QLQ-C30 was undertaken to improve low internal consistency estimates and content validity for the role functioning scale and emphasis on physical functioning in the global QoL scale.35 The original and new versions were in a total of 1,181 patients with cancer in Canada and the Netherlands. Internal consistency improved in role functioning scale in the new version (Cronbach alpha ranging from 0.78-0.88), and substitution of the new item for the previous did not alter internal consistency (Cronbach alpha ranging from 0.81-0.92.35
The EORTC QLQ-C30 (Version 3.0) is the version currently in use, which differed from the previous Version 2.0 in that the number of response options for the first 5 items of the questionnaire that comprise the Physical Function scale were increased from 2 response options (yes/no in Version 2.0) to 4 (not at all, a little, quite a bit, very much). Internal consistency reliability, construct validity, criterion validity, and responsiveness of the EORTC QLQ-C30 Version 3.0 was assessed in 622 head and neck cancer patients from 12 countries which demonstrated that version 3.0 was more reliable than previous versions.29 Internal consistency of the multi-item scales was assessed using Cronbach alpha, with a value of 0.70 being considered adequate. The internal consistency of the new Physical Function scale of the EORTC QLQ-C30 Version 3.0 was 0.84, compared with 0.66 in Version 1.0. The EORTC QLQ-C30 Version 3.0 was able to discriminate between head and neck cancer patients who were disease-free, who were newly diagnosed, and those with recurrent disease. As well, differences were noted between stages and according to Karnofsky performance status (KPS), as the new scale had a stronger association with KPS. Further, there was a high correlation observed between scores on the EORTC QLQ-C30 Version 3.0 and symptom/toxicity scores. Responsiveness to change was assessed using the standardized response mean (SRM), with an SRM of 0.20 being considered small, 0.50 being considered medium, and 0.80 being considered large. The changes in the scores of QLQ-C30 demonstrated a small to medium SRM in response to treatment over time with scores mostly deteriorating between 5 and 10 points.29
Evidence of validity, and reliability of the EORTC QLQ-C30 was not identified in the literature for patients with locally advanced or metastatic urothelial cancer. However, one study evaluated the reliability, validity, and responsiveness of the EORTC QLQ-C30 NMIBC24 for the population of patients with non-invasive muscle bladder cancer (NMIBC).36 The study recruited participants from the Bladder COX-2 Inhibition trial, which is a randomized placebo-controlled trial evaluating the addition of celecoxib to standard treatment (transurethral resection of bladder tumour, single-dose MMC, and BCG induction and maintenance for disease at high risk for recurrence or multiple MMC instillations for disease at intermediate risk for recurrence). The EORTC QLQ-NMIBC24 is a 24-item survey that evaluates HRQoL for patients with intermediate to high risk NMIBC. It has multi-item scales assessing urinary symptoms (items 1–7), intravesical treatment issues (items 10 and 11), future perspective (items 12–14), fever and feeling ill (items 8 and 9), and abdominal bloating and flatulence (items 15 and 16), along with single items addressing different aspects of sexual functioning (items 17–24). All responses are linearly transformed from 0 to 100, with a high score indicating more symptoms or problems or better function for the functional scales. Internal consistency was measured by the Cronbach alpha coefficient. The internal consistency of the scales at each time point were good (>0.70) for the urinary symptoms, future worries, sexual function, and sexual function in men scales. The fever and malaise scale had coefficients of 0.57 and 0.76; in the abdominal bloating scale, the coefficients ranged between 0.49 and 0.62. To measure criterion validity, the study correlated scales of the C30 with those of the BLS24. Most correlations were low (r < 0.4), indicating that the scales have very little overlap and can be co-administered. The module was responsive to changes over time.36
Minimal Important Difference
One study from 1998 conducted in patients with breast cancer and small-cell lung cancer estimated a clinically relevant change in score on any scale of the EORTC QLQ-C30 to be 10 points. The estimate was based on a study that used an anchor-based approach to estimate the minimal important difference (MID) in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire (SSQ) had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported being “very much” changed had corresponding changes of more than 20 points.37
No MID in patients with locally advanced or metastatic UC has been identified.
EQ-5D-5L
Description and Scoring
The EQ-5D is a generic, utility-based measure of HRQoL. The EQ-5D-5L is a 2-part questionnaire consisting of the EQ-5D descriptive system and the EQ visual analogue scale (VAS) with a recall period of one day. The descriptive system consists of 5 dimensions including mobility, self-care, usual activities, pain/discomfort and anxiety/depression. Each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems and extreme problems. Each decision corresponds to a 1-digit number that expresses the level selected for that dimension which are combined into a 5-digit number that describes the patient’s health state, for a total of 3125 possible health states. Health states can be summarized using the 5-digit code or represented by a single summary index value which reflects how good or bad a health state is according to the preferences of the general population of a country/region. The summary index is derived by applying weights to each level in each dimension. The index is calculated by deducting the appropriate weights from 1, the value for full health (i.e., state 11111). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states ‘dead’ and ‘perfect health,’ respectively.30
Valuation of the EQ-5D summary value sets for Canada was undertaken in 2012 based on composite time trade-off and traditional time trade-off techniques. Scores of −0.148 and 0.949 were reported as the worst and best EQ-5D-5L states, respectively.38
The EQ VAS records the patient’s self-rated health on a vertical visual analogue scale, where the end points are labelled ‘The best health you can imagine’ and ‘The worst health you can imagine.’ The VAS can be used as a quantitative measure of health outcome that reflect the patient’s own judgement at that specific time point.30
Assessment of Validity and Reliability
No evidence of validity and reliability of the EQ-5D-5L for patients with locally advanced or metastatic UC was identified.
Minimal Important Difference
Pickard et al. conducted a retrospective analysis of 534 patients with 11 types of cancer (including lymphoma) to estimate the MID using distribution-based (SEM, 1/2 SD, and 1/3 SD) and anchor-based (ECOG) methods.31 Using both anchor-based and distribution-based methods, estimates of the MID for the EQ-5D-5L ranged from 0.07 to 0.09 grouped by ECOG PS for all cancers, and 0.06 to 0.07 when based on FACT-G quintiles. Minimally important differences for the EQ-5D VAS ranged from 8 to 12 based on the ECOG performance status, and from 7 to 10 based on FACT QoL questionnaire quintiles.
McClure et al. (2017) obtained the minimal important difference (MID) for the EQ-5D-5L by calculating the average absolute difference between the index score of the baseline health state and the index score of all single-level transitions from the baseline state. Single-level transitions across all 3,125 health states were averaged to arrive at MIDs for various countries, by applying country-specific scoring algorithms. For Canada, transitions between levels 3 and 4 were excluded from the average to form a constant distribution of MID values across the range of baseline scores. This analysis resulted in a Canadian-specific MID of 0.037.32
No information on the MID of the EQ-5D-5L in patients with locally advanced or metastatic urothelial cancer was found.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
FGFR
fibroblast growth factor receptor gene
ICER
incremental cost-effectiveness ratio
mUC
locally advanced or metastatic urothelial cancer
ORR
overall response rate
OS
overall survival
PD-1
programmed death receptor 1
PD-L1
programmed death ligand 1
PFS
progression-free survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Enfortumab vedotin (Padcev), lyophilized powder for reconstitution for IV infusion |
Submitted price | Enfortumab vedotin, 20 mg vial: $1,181.00 Enfortumab vedotin, 30 mg vial: $1,772.00 |
Indication | For the treatment of adult patients with unresectable locally advanced or metastatic urothelial cancer who have previously received a platinum-containing chemotherapy and programmed death receptor 1 or programmed death ligand 1 inhibitors |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review (Project Orbis) |
NOC date | October 29, 2021 |
Reimbursement request | Same as indication |
Sponsor | Seagen Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adult patients with mUC previously treated with a platinum-based chemotherapy and a PD-1 or PD-L1 inhibitor, which is consistent with the reimbursement request |
Treatment | Enfortumab vedotin |
Comparator | A combined taxane comparator consisting of docetaxel and paclitaxel |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (10 years) |
Key data source | Clinical efficacy was modelled using the overall survival, progression-free survival, and duration of treatment observed in the EV-301 trial. This trial was also used to generate health-state utility values based on the EQ-5D and to estimate the incidence of relevant adverse events. |
Submitted results | ICER = $316,921 per QALY for enfortumab vedotin vs. docetaxel or paclitaxel (incremental QALYs: 0.31; incremental costs: $96,788) |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; mUC = locally advanced or metastatic urothelial cancer; PD-1 = programmed death receptor 1; PD-L1 = programmed death ligand 1; QALY = quality-adjusted life-year; RDI = relative dose intensity.
Conclusions
The CADTH Clinical Review noted that treatment with enfortumab vedotin resulted in a statistically significant survival advantage in terms of overall survival (OS) and progression-free survival (PFS) compared to chemotherapy in subjects who had received a prior programmed death receptor 1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and platinum-containing chemotherapy. As OS data were available up to 24 months, the extrapolated OS benefits beyond the trial duration in the sponsor’s base case remain uncertain. Enfortumab vedotin was also associated with a clinically meaningful overall response rate (ORR) compared to chemotherapy for a single-drug therapy, which is an important consideration for this stage of disease. The ORR results are in line with the benefit seen for OS, both of which are important to patients; however, the effect of ORR on improvement in cancer symptoms is uncertain. As a result of patient attrition, the effect of health-related quality of life also remains uncertain, although enfortumab vedotin was not associated with any major change in quality of life.
CADTH identified several limitations with the sponsor’s pharmacoeconomic model involving the OS extrapolation, treatment-specific utilities, and relative dose intensity (RDI). As part of the base case, CADTH used a Gompertz parametric function to estimate OS, shortened the time horizon to 5 years, used health-state utilities, and excluded consideration of RDI. Based on the CADTH base case, enfortumab vedotin was associated with an incremental cost-effectiveness ratio (ICER) of $506,439 per quality-adjusted life-year (QALY), and the probability of cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY was 0%. A price reduction of 93% would be required to achieve cost-effectiveness at this threshold. The CADTH results are aligned with those of the sponsor’s, which suggest that enfortumab vedotin is not cost-effective at conventional willingness-to-pay thresholds.
The pharmacoeconomic model is driven by the OS extrapolation assumptions and the drug acquisition cost of enfortumab vedotin and was sensitive to assumptions about the methods used to estimate OS and about RDI.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
As part of the call for patient input CADTH received feedback from Bladder Cancer Canada (BCC), a patient advocacy organization dedicated to bladder cancer issues that conducted an online survey of 38 patients and 6 caregivers, the majority of whom resided in Canada. The organization also conducted telephone interviews with 2 Canadian patients who had experience with enfortumab vedotin. Among the survey respondents, 39% had received platinum-based chemotherapy, with 22% reporting that their cancer progressed after treatment, and 44% reporting that it did not progress. Some patients also received immunotherapy, radiation, or surgical resection. Side effects of current treatments included fatigue, constipation, and low blood cell counts. Patients indicated that they hoped enfortumab vedotin would help slow disease progression and recurrence. Patients with experience using the drug noted that enfortumab vedotin was associated with a shorter treatment schedule, shorter time in hospital, and fewer side effects overall compared to other treatments. Side effects of note with enfortumab vedotin included neuropathy and skin irritation.
CADTH received clinician input from the Ontario Health (Cancer Care Ontario) Genitourinary Cancer Drug Advisory Committee. The clinicians stated that there is no standard of care for third-line treatment of locally advanced or metastatic urothelial cancer (mUC) and that patients are treated based on physician choice and expert opinion. Docetaxel or paclitaxel are sometimes used but are associated with significant toxicity. Clinicians would use enfortumab vedotin in any patient treated previously with platinum-based chemotherapy and immunotherapy. As there are no other beneficial therapies with any survival advantage in this setting, enfortumab vedotin would become the standard of care.
The drug plan input received by CADTH for this review noted that erdafitinib is approved by Health Canada for patients with mUC who experience disease progression during or following at least 1 line of prior chemotherapy. The plans were interested in whether patients who were ineligible or who had not received either platinum-based chemotherapy or immunotherapy would still be eligible for enfortumab vedotin. The plans noted that a weekly dosing schedule is more labour-intensive and that administration side effects such as extravasation are a concern. The plans cited implementation issues, such as high pharmacy resource utilization for drug reconstitution and a limited stability after reconstitution necessitating onsite compounding pharmacies. Finally, the plans noted that generic versions of docetaxel and paclitaxel are available.
The sponsor’s model addressed the concern that its model compared enfortumab vedotin to a combined taxane comparator (docetaxel or paclitaxel).
In addition, CADTH addressed some of these concerns as follows:
CADTH included the generic prices of paclitaxel and docetaxel as part of the cost comparison table (Table 8).
CADTH increased the market share of enfortumab vedotin in the budget impact analysis (BIA) to align with clinical input that suggested this drug would become standard of care.
CADTH was unable to address the concern that the sponsor did not explicitly model the disutilities associated with adverse events (AEs) due to treatment.
Economic Review
The current review is for enfortumab vedotin for adult patients with mUC previously treated with a platinum-based chemotherapy and a PD-1 or PD-L1 inhibitor.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing enfortumab vedotin to docetaxel or paclitaxel for the treatment of adult patients with mUC previously treated with a platinum-based chemotherapy and a PD-1 or PD-L1 inhibitor. The modelled population was derived from the EV-301 trial and aligns with the reimbursement request.1
Enfortumab vedotin is available in single-use vials containing lyophilized powder for reconstitution at 10 mg/mL for IV infusion. The recommended dose of enfortumab vedotin is 1.25 mg/kg (up to a maximum of 125 mg for patients weighing more than 100 kg) administered intravenously over 30 minutes on days 1, 8, and 15 of a 28-day cycle until disease progression or unacceptable toxicity.2 According to the Health Canada indication and clinician input, enfortumab vedotin is expected to be used as a third-line therapy in adult patients with mUC who have already received platinum-based chemotherapy and immunotherapy. The cost for enfortumab vedotin is $1,181 per 20 mg vial and $1,772 per 30 mg vial3; the 28-day cost is $17,718 per patient, as calculated by CADTH (Table 8), based on the mean patient weight of 74 kg in the EV-301 trial.1
This analysis used a combined taxane comparator consisting of docetaxel and paclitaxel, the costs and benefits of which were weighted according to the proportion of patients in the taxane arms of the EV-301 trial: 51% and 49%, respectively.1 The recommended doses of docetaxel and paclitaxel in the EV-301 trial were 75 mg/m2 and 175 mg/m2 every 3 weeks, respectively, which aligned with the product monographs.4,5 The 28-day cost for docetaxel was $2,196, and paclitaxel cost $4,433 over 28 days, as calculated by CADTH (Table 8).
The sponsor calculated a monthly cost of $15,131 for enfortumab vedotin based on an RDI of ||||%.1,3 The sponsor’s base case considered wastage for enfortumab vedotin on account of the small number of patients who would receive it. An administration cost of $504 per month was applied based on the administration costs of pembrolizumab.6 The monthly cost of the weighted docetaxel or paclitaxel comparator was calculated to be $568, based on an RDI of ||||||% and ||||||% for docetaxel and paclitaxel, respectively, with an administration cost of $422 per month also included. Wastage was not considered for docetaxel or paclitaxel as these drugs are routinely used in clinical practice.
Outcomes of the model included QALYs and life-years over a lifetime horizon of 10 years. The base-case analysis was conducted from the perspective of the Canadian public health care system, with a 1.5% annual discount applied to both costs and outcomes. The cycle length was monthly with a half-cycle correction applied.
Model Structure
The sponsor submitted a partitioned survival model consisting of 3 mutually exclusive health states: pre-progression, post-progression, and death. All patients entered the model with stable disease in the pre-progression state after receipt of treatment with enfortumab vedotin or either docetaxel or paclitaxel. The proportion of patients in the pre-progression health state followed the PFS curve of each treatment as observed in the EV-301 study, in which progression was defined according to radiological disease progression per Response Evaluation Criteria in Solid Tumours Version 1.1.1 The proportion of patients in the post-progression health state was equal to the difference between the OS and the PFS curves. Patients transitioning to the death state remained there until the end of the model time horizon. The sponsor’s model structure is available in Appendix 3 (Figure 1).
Model Inputs
The population used for this model was derived from the EV-301 trial (n = 608).7 Patients had a mean body weight of 74 kg and body surface area of 1.9 m2, and 77% were male. The starting age in the model was 67 years.7
The sponsor used parametric modelling to extrapolate the OS and PFS data from EV-301. For PFS, a log-logistic distribution was considered the best fit for both the enfortumab vedotin and docetaxel or paclitaxel treatment arms based on fit statistics and visual inspection. Furthermore, the proportional hazards assumption was considered satisfied; therefore, in the sponsor’s base case the PFS of docetaxel or paclitaxel was estimated based on the hazard ratio of PFS for docetaxel or paclitaxel versus enfortumab vedotin (1.84; 95% confidence interval, 1.48 to 2.29). For OS, a Weibull distribution was considered the best fit for both treatments. The sponsor’s submission claimed that the proportional hazards assumption was also satisfied for OS. Consequently, in the sponsor’s base case the OS of docetaxel or paclitaxel was estimated based on the hazard ratio of OS for docetaxel or paclitaxel versus enfortumab vedotin (1.54; 95% confidence interval, 1.19 to 1.99). The PFS rates over time were capped by the estimated OS rates, and OS was capped by the age-gender adjusted national mortality rates in Canada.8 Patients in the EV-301 study received the study treatment until the disease progression, a protocol-defined discontinuation criterion was met, study termination, or study completion. The duration of treatment for enfortumab vedotin and docetaxel or paclitaxel was based on Kaplan–Meier curves and was capped by the estimated PFS.
The dose of enfortumab vedotin used in the model, based on the EV-301 trial and product monograph2,7 was as described in the Overview section. The sponsor constructed a normal distribution of body weight in percentile form around the mean weight of EV-301 and calculated the number of 20 mg and 30 mg vials required for each | % percentile.
Health-related quality of life data were collected from EV-301 for both the pre- and post-progression health states as measured by the EuroQol 5-Dimensions 5-Levels questionnaire. Pre-progression utility was assumed to vary by treatment to account for the differences in safety profiles between the treatments. The pre-progression utility for enfortumab vedotin and docetaxel or paclitaxel was calculated to be |||||||| and ||||||||, with the post-progression utility calculated to be ||||||||.1 Disutilities due to AEs were not explicitly modelled in the sponsor’s base case.
All costs used in the model except drug prices were inflated to 2021 Canadian dollars. In addition to drug acquisition costs, the sponsor calculated administration costs for enfortumab vedotin, docetaxel, and paclitaxel as outlined in the Overview section, based on physician fees, nurse and pharmacy workload costs, and infusion-chair time costs.9-13 Costs associated with subsequent treatment were not considered as later-line therapies are limited in this population. Resource use costs were also included in the model and these costs were treatment-independent. Costs associated with medical care included $84 per urologist visit, $159 per oncologist visit, $154 per radiation oncologist visit, $85 per general practitioner visit, and $39 per nurse visit. A cost of $87 was applied per chest CT scan, and $109 was assumed per CT scan of the abdomen and pelvis. A cost of $155 was applied per bone scan. These costs were all obtained from the Ministry of Health of Ontario Schedule of Benefits.9 A cost of $187 was applied for each emergency department visit,14 and $7,888 for each inpatient admission based on the Ontario Case Costing Initiative for neoplasm of the bladder.15 Costs of managing grade 3 or 4 AEs occurring in at least 5% of patients in EV-301 were applied as a 1-time AE cost at the beginning of the model. These AEs included anemia, (febrile) neutropenia, maculopapular rash, decreased appetite, hyperglycemia, decreased neutrophil count, decreased white blood cell count, and fatigue. The unit costs per AE were derived from the Ontario Case Costing Initiative and ranged from $525 to $7,735 when weighted by inpatient versus outpatient management.15 Finally, patients were assumed to incur a 1-time terminal care cost before death of $43,270 based on a gender-weighted average terminal care cost for bladder cancer patients.16
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following section.
Base-Case Results
Enfortumab vedotin was associated with incremental costs of $96,788 and 0.31 QALYs in comparison with docetaxel or paclitaxel, resulting in an ICER of $316,921 per QALY (Table 3). CADTH noted that approximately 35% of the incremental QALYs in the sponsor’s base case were accrued after 24 months, which is the maximum time for which OS data are available from the EV-301 trial.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Total QALYs | Incremental QALYs | ICER vs. DP ($ per QALY) |
|---|---|---|---|---|---|---|
Docetaxel or paclitaxel | 64,082 | Reference | 0.94 | 0.69 | Reference | Reference |
Enfortumab vedotin | 160,870 | 96,788 | 1.31 | 1.00 | 0.31 | 316,921 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Note: Submitted analyses are based on the publicly available prices of comparators and may not reflect confidential, negotiated prices.
Source: Sponsor’s pharmacoeconomic submission.3
Sensitivity and Scenario Analysis Results
The sponsor conducted a number of sensitivity and scenario analysis involving the time horizon, discount rate, modelling assumptions for docetaxel or paclitaxel, and utility values. In these analyses, the ICER was most sensitive to extrapolation assumptions surrounding OS. Specifically, when OS for docetaxel or paclitaxel was extrapolated using a Weibull function rather than applying a hazard ratio, the resulting ICER was $373,608.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Extrapolation overestimates the survival benefit of enfortumab vedotin beyond the EV-301 trial period: The sponsor extrapolated the OS data observed in the EV-301 trial using a Weibull parametric function. This resulted in an assumed survival benefit for enfortumab vedotin beyond the trial period ( | % versus | % alive for enfortumab vedotin versus docetaxel or paclitaxel, respectively, after 3 years). This was determined to be clinically implausible according to the clinical expert consulted by CADTH and was not supported by the survival evidence in EV-301, which suggested that OS was equal between enfortumab vedotin and docetaxel or paclitaxel at the end of the trial.
In reanalysis, CADTH used a Gompertz parametric function to extrapolate OS, based on feedback from clinical experts. In addition, as this extrapolation resulted in 0% of patients being alive at 5 years, CADTH shortened the time horizon to 5 years as part of the base case. In scenario analyses, CADTH used independent Gompertz functions to estimate OS in the enfortumab and docetaxel or paclitaxel arms separately.
Use of treatment-specific utilities: As part of the base case, the sponsor used treatment-specific utilities of |||||||| for enfortumab vedotin and |||||| for docetaxel and paclitaxel in the pre-progression state, while the utility post-progression was |||||||| for both treatments. According to CADTH’s Guidelines for the Economic Evaluation of Health Technologies, health utilities should reflect the health states in the model rather than the treatment comparators.17 The use of treatment-specific utilities implies that a patient’s quality of life will differ as a result of the treatment received due to different safety profiles for the 2 treatments. However, this approach does not adequately describe the mechanism by which patients experience quality of life differences, and therefore there is meaningful uncertainty around the extent to which such differences are explained by AEs versus other potential explanatory factors. The sponsor justified the inclusion of treatment-specific utilities by suggesting that patients on enfortumab vedotin were expected to have a better quality of life based on the safety profiles of the 2 treatments, an assertion that did not align with feedback from the clinical expert consulted by CADTH. The clinical expert noted that enfortumab vedotin can be associated with corneal and ocular toxicity, anorexia, fatigue, neuropathy, diarrhea, and skin toxicity, all of which may affect quality of life. The clinical expert indicated that the quality of life of patients on enfortumab vedotin would not differ from those on docetaxel or paclitaxel.
As part of the base case, CADTH used the sponsor-provided health-state utility values for the pre- and post-progression states. Treatment-specific utilities were explored in scenario analysis.
Relative dose intensity does not correlate well with and may underestimate drug costs: The sponsor incorporated a RDI of ||||||% for enfortumab vedotin, ||||||||% for docetaxel, and ||||||% for paclitaxel, and then multiplied each value by the respective recommended doses for each comparator. These values were calculated from the EV-301 trial as the mean dose received per patient divided by the planned dose.1 Multiplying the RDI by the drug costs is problematic as RDI can be influenced by many different things. For example, the dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation. Each of these reasons has a different impact on drug costs. Furthermore, it is unclear how these assumptions interact with considerations about vial size and wastage, which were incorporated into the sponsor’s calculations of the per cycle drug costs. As enfortumab vedotin is supplied in single-use vials of 20 mg and 30 mg each, dose reductions due to AEs may not necessarily lead to fewer vials of the product being used. By multiplying the RDI by the calculated drug acquisition costs, the sponsor implicitly assumed that the relative vial distribution (20 mg versus 30 mg) and wastage considerations for enfortumab vedotin are identical for a patient with perfect dose adherence and a patient with dose delays and reductions. Overall, without explicitly modelling dose delays and reductions for the patient population, this method of multiplying RDI by drug acquisition costs is associated with uncertainty about the true drug cost incurred by payers.
As part of the base case, CADTH assumed a RDI of 100% for enfortumab vedotin and docetaxel or paclitaxel, while including the sponsor’s original RDI assumptions in a scenario analysis.
Discrepancy surrounding comparator prices: The sponsor assumed prices for docetaxel and paclitaxel of $1.52 and $2.00 per mg, respectively. Cost sources consulted by CADTH listed the prices for docetaxel and paclitaxel to be $11.56 and $10.00 per mg, respectively.18 This discrepancy was explored in a scenario analysis.
As part of a scenario analysis, CADTH used costs of $11.56 and $10.00 per mg of docetaxel and paclitaxel, respectively.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (See Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Disutilities due to AEs were not explicitly modelled. | Not appropriate. Clinical experts emphasized potential toxicities affecting quality of life for patients on enfortumab vedotin that were not explicitly modelled. |
Total management costs for AEs were applied as a 1-time cost during the first model cycle and estimated as the sum of the product of the AE incidence and associated unit costs. | Uncertain. This approach does not allow for discounting of AE costs as all are applied in the first cycle. |
The prevalence of subsequent treatment use in EV-301 was comparable between the enfortumab vedotin and the docetaxel or paclitaxel treatment groups and these costs were not considered. | Appropriate. Clinical experts noted that few patients would receive further treatment after progression on enfortumab vedotin. |
Probabilistic uncertainty around drug acquisition and administration costs was characterized by a gamma distribution. | Not appropriate. Drug acquisition and administration costs are not likely to vary and should not have an associated uncertainty. |
AE = adverse event.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived, in consultation with clinical experts, by making changes in model parameter values and assumptions. These changes are summarized in Table 5 and include changes to the OS modelling extrapolation, time horizon, utility values, and RDI.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Parametric overall survival modelling | Weibull function | Gompertz function |
2. Time horizon | 10 years | 5 years |
3. Utility values | Treatment-specific utility values: Pre-progression enfortumab: |||||| Pre-progression docetaxel or paclitaxel: |||||| Post-progression: |||||||| | Health-state–specific utility values: Pre-progression: 0.795 Post-progression: 0.697 |
4. Relative dose intensity | Enfortumab: ||||||||% Docetaxel: ||||||% Paclitaxel: ||||||||% | Enfortumab: 100% Docetaxel: 100% Paclitaxel: 100% |
CADTH base case | — | Reanalysis 1 + 2 + 3 + 4 |
In the CADTH base case, enfortumab vedotin was associated with estimated total costs of $180,440 and 0.89 QALYs, compared to total costs and QALYs of $63,398 and 0.66, respectively, for patients receiving docetaxel or paclitaxel. The ICER for enfortumab vedotin compared to docetaxel or paclitaxel was $506,439 per QALY, and the probability of cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY was 0%. A detailed breakdown of the disaggregate results is available in Appendix 4 (Table 11). Enfortumab vedotin is associated with higher drug acquisition costs compared with docetaxel or paclitaxel, due in part to the longer duration of therapy (i.e., 5 months versus 3 months for docetaxel or paclitaxel) and also the higher unit cost for the drug.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case | DP | 64,082 | 0.69 | Reference |
Enfortumab vedotin | 160,870 | 1.00 | 316,921 | |
CADTH reanalysis 1 – OS modelling | DP | 63,222 | 0.66 | Reference |
Enfortumab vedotin | 158,539 | 0.90 | 400,858 | |
CADTH reanalysis 2 – time horizon | DP | 63,994 | 0.69 | Reference |
Enfortumab vedotin | 160,267 | 0.99 | 321,971 | |
CADTH reanalysis 3 – utility values | DP | 64,073 | 0.69 | Reference |
Enfortumab vedotin | 160,826 | 0.99 | 323,602 | |
CADTH reanalysis 4 – relative dose intensity | DP | 64,256 | 0.69 | Reference |
Enfortumab vedotin | 182,769 | 1.00 | 387,612 | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) | DP | 63,398 | 0.66 | Reference |
Enfortumab vedotin | 180,440 | 0.89 | 506,439 |
DP = docetaxel or paclitaxel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s and CADTH’s base case. Based on the CADTH base case, a price reduction of 93% would be necessary to achieve cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY (Table 7).
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for enfortumab vedotin vs. docetaxel or paclitaxel ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 316,921 | 506,439 |
10% | 286,125 | 456,832 |
20% | 255,805 | 407,225 |
30% | 225,486 | 357,618 |
40% | 195,166 | 308,011 |
50% | 164,846 | 258,404 |
60% | 134,526 | 208,797 |
70% | 104,207 | 159,190 |
80% | 73,887 | 109,583 |
90% | 43,567 | 59,976 |
93% | 34,471 | 45,094 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CADTH undertook a series of exploratory analyses to determine the impact of alternative assumptions on the cost-effectiveness of enfortumab vedotin, which are outlined as follows:
Per-milligram costs of $11.56 and $10.00 were used for docetaxel and paclitaxel, respectively.
The sponsor’s original RDI assumptions were used.
The sponsor’s original treatment-specific utilities were used.
Gompertz curves were fit independently for the OS data for enfortumab vedotin and docetaxel or paclitaxel.
The results of these analyses are presented in Appendix 4 (Table 12). The scenario analysis in which individual Gompertz curves were fitted to the OS data had the largest effect on the ICER, which was calculated to be $687,056 per QALY. The scenario analysis involving the sponsor’s original RDI assumptions resulted in an ICER of $412,286 per QALY. The scenario analysis involving different costs for taxanes resulted in an ICER of $459,487. The scenario analysis involving treatment-specific utilities had a minimal impact on the ICER, which was calculated to be $493,126.
Issues for Consideration
Avelumab is a PD-L1 inhibitor that received a positive recommendation from the CADTH pan-Canadian Oncology Drug Review Expert Review Committee on March 23, 2021, for the first-line maintenance treatment of patients with mUC whose disease has not progressed with first-line platinum-based induction chemotherapy.19 The cost-effectiveness of enfortumab vedotin in a population that has already received avelumab is unknown.
Erdafitinib is an inhibitor of fibroblast growth factor receptor (FGFR), a gene that is aberrant in approximately 25% of patients with mUC. Erdafitinib has received a Notice of Compliance with conditions from Health Canada for the treatment of adult patients with mUC whose tumours have susceptible FGFR2 or FGFR3 genetic alterations and who have disease progression during or following at least 1 line of prior chemotherapy.20 Clinical experts noted that, in this subset of patients, erdafitinib may be used in a similar setting and may compete with enfortumab vedotin. The cost-effectiveness of enfortumab vedotin versus erdafitinib is unknown.
The clinical expert consulted by CADTH for this review noted that a minority of patients who progress on enfortumab vedotin may receive docetaxel or paclitaxel in a fourth-line setting. However, this likely represents a small proportion of the patient population, as the efficacy of monotherapy with taxanes at that point is limited and patients may prefer not to receive those drugs due to toxicity.
The clinical expert consulted by CADTH for this review further noted that the duration of treatment would likely be shorter in a real-world setting due to treatment toxicity. The effect that this probable higher rate of discontinuation has on the cost-effectiveness of enfortumab vedotin is unknown.
Overall Conclusions
The CADTH Clinical Review noted that treatment with enfortumab vedotin resulted in a statistically significant survival advantage in terms of OS and PFS compared to chemotherapy in subjects who had received a prior PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy. As OS data were available up to 24 months, the extrapolated OS benefits beyond the trial duration in the sponsor’s base case remain uncertain. Enfortumab vedotin was also associated with a clinically meaningful ORR compared to chemotherapy for a single agent therapy, which is an important consideration for this stage of disease. The results for ORR are in line with the survival benefit seen for OS. Both outcomes are important to patients; however, the impact ORR has on improvement in cancer symptoms remains uncertain. As a result of patient attrition, the effect of health-related quality of life remains uncertain, although enfortumab vedotin was not associated with any major improvement or deterioration in quality of life.
The CADTH review identified several limitations with the sponsor’s pharmacoeconomic model involving the OS extrapolation, treatment-specific utilities, and RDI. As part of the base case, CADTH used a Gompertz parametric function to estimate OS, shortened the time horizon to 5 years, used health-state utilities, and excluded consideration of RDI. Based on the CADTH base case, enfortumab vedotin was associated with an ICER of $506,439 per QALY, and the probability of cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY was 0%; these results are aligned with the sponsor’s base case. A price reduction of 93% would be required to achieve cost-effectiveness at this threshold.
The pharmacoeconomic model is driven by the OS extrapolation assumptions and the drug acquisition cost of enfortumab vedotin and was sensitive to assumptions about the methods used to estimate OS and about RDI.
References
1.Clinical Study Report: 7465-CL-0301. An open-label, randomized phase 3 study to evaluate enfortumab vedotin vs chemotherapy in subjects with previously treated locally advanced or metastatic urothelial cancer (EV-301) [internal sponsor's report]. Northbrook (IL): Astellas Pharma Global Development, Inc.; 2021 Jan.
2.Padcev (enfortumab vedotin): 20 mg and 30 mg single-use vials of lyophilized powder for solution for intravenous infusion only [product monograph]. Mississauga (ON): Seagen Canada; 2021 Oct 29.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Padcev (enfortumab vedotin), 20 mg and 30 mg single-use vials of lyophilized powder for solution for intravenous infusion only [internal sponsor's package]. Mississauga (ON): Seagen Canada; 2021 Jul 09.
4.Docetaxel: 10 mg/mL solution for intraveous infusion. Boucherville (QC): Sandoz Canada Inc; 2020 Nov 17: https://pdf.hres.ca/dpd_pm/00058887.PDF. Accessed 2021 Aug 23.
5.Paclitaxel: 6 mg/mL solution for injection [product monograph]. Kirkland (QC): Accord Healthcare Inc; 2020 Sep 14: https://pdf.hres.ca/dpd_pm/00058617.PDF. Accessed 2021 Aug 23.
6.Cancer Care Ontario. PEMB Regimen. 2020; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/65201. Accessed 2021 Oct 15.
7.Powles T, Rosenberg JE, Sonpavde GP, et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N Engl J Med. 2021;384(12):1125-1135. PubMed
8.Table 13-10-0710-01 Mortality rates, by age group. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1310071001. Accessed 2020 Dec 28.
9.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Aug 23.
10.Job Bank. Wages for Pharmacists. Ottawa (ON): Government of Canada; 2020: https://www.jobbank.gc.ca/wagereport/occupation/18196. Accessed 2021 Oct 15.
11.Job Bank. Wages for registered nurses and registered psychiatric nurses. Ottawa (ON): Government of Canada; 2020: https://www.jobbank.gc.ca/wagereport/occupation/993. Accessed 2021 Oct 15.
12.Table 14-10-0043-01 Average usual and actual hours worked in a reference week by type of work (full- and part-time), annual. Ottawa (ON): Statistics Canada; 2021: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410004301. Accessed 2021 Oct 15.
13.Pettigrew M, Kavan P, Surprenant L, Lim HJ. Comparative net cost impact of the utilization of panitumumab versus cetuximab for the treatment of patients with metastatic colorectal cancer in Canada. J Med Econ. 2016;19(2):135-147. PubMed
14.Dawson H, Zinck G. CIHI Survey: ED spending in Canada: a focus on the cost of patients waiting for access to an in-patient bed in Ontario. Healthc Q. 2009;12(1):25-28. PubMed
15.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Oct 15.
16.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
17.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/pdf/guidelines_for_the_economic_evaluation_of_health_technologies_canada_4th_ed.pdf. Accessed 2021 Oct 15.
18.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Oct 15.
19.CADTH pCODR Reimbursement Expert Review Committee (pERC) final recommendation: avelumab (Bavencio - Pfizer). Ottawa (ON): CADTH; 2021 Mar 23: https://cadth.ca/sites/default/files/pcodr/Reviews2021/10225AvelumabUC_FnRec_REDACT_EC23Mar2021_final.pdf. Accessed 2021 Oct 15.
20.Balversa - Notice of Compliance with Conditions - Qualifying Notice. Ottawa (ON): Therapeutic Products Directorate, Health Canada; 2019: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/notice-compliance/conditions/qualifying-notice-balversa-224529.html. Accessed 2021 Oct 15.
21.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Padcev (enfortumab vedotin), 20 mg and 30 mg single-use vials of lyophilized powder for solution for intravenous infusion only [internal sponsor's package]. Mississauga (ON): Seagen Canada; 2021 Jul 09.
22.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
23.Global Cancer Observatory. Population fact sheets: Canada 2020. Lyon (FR): International Agency for Research on Cancer (IARC); 2020: https://gco.iarc.fr/today/data/factsheets/populations/124-canada-fact-sheets.pdf. Accessed 2020 Dec 08.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison for Patients With mUC Who Have Previously Received Platinum Chemotherapy and Immunotherapy
Treatment | Strength/ concentration | Form (vial size if single-use) | Price | Recommended dosagea | Daily cost | 28-day cost |
|---|---|---|---|---|---|---|
Enfortumab vedotin | 10 mg/mL | 20 mg 30 mg Powder for IV infusion | $1,181.0000b $1,772.0000 | 1.25 mg/kg 3 times every 4 weeks | $632.79 | $17,718 |
Taxanes | ||||||
Docetaxel | 10 mg/mL | Vial for IV infusion | $115.6250 | 75 mg/m2 every 3 weeks | $78.46 | $2,196 |
Paclitaxel | 6 mg/mL | Vial for IV infusion | $60.0000 | 175 mg/m2 every 3 weeks | $158.33 | $4,433 |
mUC = locally advanced or metastatic urothelial cancer.
Note: All prices are from the IQVIA Delta PA database (accessed August 2021),18 unless otherwise indicated, and do not include dispensing fees. Costs are calculated based on a mean body weight of 74 kg and mean body surface area of 1.9 m2 as per the EV-301 trial.1
aThe recommended dosages are from the respective product monographs.2,4,5
bSponsor-submitted price.3
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | The submitted budget impact analysis relied on VBA code, making it difficult to validate assumptions about market shares and the proportion of patients receiving each drug. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Probabilistic uncertainties were applied to drug acquisition and administration costs, which are not typically associated with uncertainty. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Table 10: Disaggregated Results of the Sponsor’s Base-Case Analysis
Parameter | Enfortumab vedotin | Docetaxel or paclitaxel | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 1.31 | 0.94 | 0.38 |
Pre-progression | 0.76 | 0.40 | 0.36 |
Post-progression | 0.56 | 0.54 | 0.02 |
Discounted QALYs | |||
Total QALYs | 1.00 | 0.69 | 0.31 |
Pre-progression | 0.61 | 0.32 | 0.29 |
Post-progression | 0.39 | 0.38 | 0.01 |
Discounted costs ($) | |||
Total costs | 160,870 | 64,082 | 96,788 |
Pre-progression drug costs | 92,787 | 2,139 | 90,649 |
Pre-progression administration costs | 3,088 | 1,591 | 1,497 |
Pre-progression disease management costs | 10,300 | 5,396 | 4,904 |
Post-progression disease management costs | 11,186 | 10,830 | 357 |
AE cost | 598 | 979 | -381 |
Terminal care cost | 42,909 | 43,147 | -238 |
ICER ($ per QALY) | 316,921 | ||
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.3
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Enfortumab vedotin | Docetaxel or paclitaxel | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 1.18 | 0.89 | 0.29 |
Pre-progression | 0.70 | 0.39 | 0.31 |
Post-progression | 0.47 | 0.50 | −0.02 |
Discounted QALYs | |||
Total QALYs | 0.89 | 0.66 | 0.23 |
Pre-progression | 0.56 | 0.31 | 0.25 |
Post-progression | 0.33 | 0.35 | -0.02 |
Discounted costs ($) | |||
Total costs | 180,440 | 63,398 | 117,042 |
Pre-progression drug costs | 114,646 | 2,328 | 112,318 |
Pre-progression administration costs | 3,109 | 1,604 | 1,504 |
Pre-progression disease management costs | 9,642 | 5,374 | 4,269 |
Post-progression disease management costs | 9,540 | 10,019 | −479 |
AE cost | 599 | 967 | −368 |
Terminal care cost | 42,904 | 43,107 | −202 |
ICER ($ per QALY) | 506,439 | ||
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Summary of Scenario Analyses Conducted on CADTH Base Case
Scenario | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
CADTH base case | DP | 63,398 | 0.66 | Reference |
Enfortumab | 180,440 | 0.89 | 506,439 | |
1. Taxane prices | DP | 74,249 | 0.66 | Reference |
Enfortumab | 180,440 | 0.89 | 459,487 | |
2. Sponsor’s RDI assumptions used | DP | 63,215 | 0.66 | Reference |
Enfortumab | 158,497 | 0.89 | 412,286 | |
3. Treatment-specific utilities | DP | 63,398 | 0.66 | Reference |
Enfortumab | 180,440 | 0.90 | 493,126 | |
4. Gompertz curves fit independently for DP and enfortumab | DP | 65,124 | 0.72 | Reference |
Enfortumab | 180,440 | 0.89 | 687,056 |
DP = docetaxel or paclitaxel; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life year; RDI = relative dose intensity.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted BIA assessed the introduction of enfortumab vedotin for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) previously treated with a platinum-based chemotherapy and a PD-1/PD-L1 inhibitor. The analysis was taken from the perspective of the Canadian public drug plans using an epidemiology-based approach, with only drug acquisition costs included. A 3-year time horizon was used, from 2022 to 2024, with 2021 as a base year. The population size was estimated using the prevalence and incidence of mUC, followed by a series of stepwise attritions to specify the population size. A summary of the sponsor’s derivation of the eligible population size is presented in Figure 2 and Figure 3.
The reference case scenario included the comparators docetaxel or paclitaxel. The new drug scenario included enfortumab vedotin, docetaxel, and paclitaxel. Key inputs to the BIA are documented in Table 14.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population for Treatment Post-Avelumab and Post-Platinum and Pembrolizumab Treatment
1L = first-line; 2L = second-line; 3L = third-line; BC = bladder cancer; CR = complete response; EV = enfortumab vedotin; la/mUC = locally advanced or metastatic urothelial cancer; MIBC = muscle invasive bladder cancer; NMIBC = non-muscle invasive bladder cancer; PR = partial response; SD = stable disease; UC = urothelial cancer.
Note: The left side of the figure categorizing patients as “Eligible for 2L Treatment with EV – Post Avelumab” refers to patients who have progressed on avelumab maintenance therapy. The right side of the figure categorizing patients as “Eligible for 3L Treatment with EV” refers to patients who did not respond to platinum chemotherapy and have progressed on subsequent pembrolizumab.
Source: Sponsor’s budget impact submission.21
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population for Treatment After Progressing on Platinum Therapy Within 12 Months and Progressing on Subsequent Pembrolizumab
Note: The categorizing of patients as “Eligible for 2L Treatment with EV – Post Pembrolizumab” refers to patients who have progressed within 12 months of platinum chemotherapy and progressed on subsequent pembrolizumab.
Source: Sponsor’s budget impact submission.21
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Incidence of bladder cancer (2020) | 25 per 100,00022 |
Mortality rate of bladder cancer (2020) | 5.7 per 100,00022 |
Prevalence of bladder cancer (2020) | 31,29723 |
Number of patients eligible for treatment with EV after progressing on avelumab maintenance therapy | 276 / 328 / 380 |
Number of patients eligible for treatment with EV after not responding to platinum chemotherapy and progressing on pembrolizumab | 41 / 48 / 56 |
Number of patients eligible for treatment with EV after progressing within 12 months of platinum chemotherapy and further progressing on pembrolizumab | 71 / 85 / 98 |
Number of total patients eligible for enfortumab vedotin | 388 / 460 / 534 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
2L setting | — |
EV | 0% / 0% / 0% |
Paclitaxel | ||||||% / ||||||% / ||||||% |
Docetaxel | ||||||% / ||||||% / ||||||% |
3L setting | — |
EV | 0% / 0% / 0% |
Paclitaxel | ||||||% / ||||||% / ||||||% |
Docetaxel | ||||||% / ||||||% / ||||||% |
Uptake (new drug scenario) | |
2L setting | — |
EV | ||||||% / ||||||% / ||||||% |
Paclitaxel | ||||||% / ||||||% / ||||||% |
Docetaxel | ||||||% / ||||||% / ||||||% |
3L setting | — |
EV | ||||||% / ||||||% / ||||||% |
Paclitaxel | ||||||% / ||||||% / ||||||% |
Docetaxel | ||||||% / ||||||% / ||||||% |
Cost of treatment (per patient) | |
Cost of treatment over lifetime | — |
EV (5-month duration) | $83,952 |
Paclitaxel (3.5-month duration) | $3,039 |
Docetaxel (3.5-month duration) | $984 |
2L = second-line (patients progressed on avelumab maintenance therapy or progressed within 12 months of platinum chemotherapy and subsequent pembrolizumab); 3L = third-line (patients who did not respond to platinum chemotherapy and progressed on subsequent pembrolizumab); EV = enfortumab vedotin.
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor’s estimated budget impact of funding enfortumab vedotin for the treatment of adult patients with mUC previously treated with a platinum-based chemotherapy and a PD-1/PD-L1 inhibitor was $4,804,551 in year 1, $11,347,174 in year 2, and $19,696,563 in year 3, for a 3-year total of $35,848,288.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Market share for enfortumab vedotin likely underestimated: The sponsor used 2 different sets of market share assumptions for the patients depending on their prior treatment course (Table 14)..The clinical expert consulted by CADTH felt that the market share for enfortumab vedotin was underestimated given the specificity with which the sponsor derived the eligible patient population and clinicians’ anticipated preference for the drug. The clinical expert noted that most patients who have previously received platinum chemotherapy and immunotherapy would receive enfortumab vedotin, a position supported by the clinical input received for this review which also noted that enfortumab would become standard of care in this population. Furthermore, in response to a concern from the drug plans, the clinical expert noted that some patients currently receiving taxanes may choose to switch to enfortumab vedotin at the time of public funding, further supporting the assumption of a rapid uptake of this product if it became available. The clinical expert consulted by CADTH estimated the market share for enfortumab vedotin to be 50% in year 1, 65% in year 2, and 80% in year 3 in the full patient population.
As part of the base case, CADTH increased the market shares of enfortumab vedotin in each year of the BIA, while maintaining the relative market shares of docetaxel and paclitaxel.
Median treatment duration from EV-301 used: The sponsor’s BIA included estimates of how long patients would be on treatment for, and multiplied the duration of treatment by monthly drug costs to estimate drug acquisition costs per patient. In their base case the sponsor used the median treatment durations of 5.0 and 3.5 months for enfortumab vedotin and docetaxel or paclitaxel, respectively. However, it is more appropriate to use the mean treatment durations, which were 5.36 and 3.96 months for enfortumab vedotin and docetaxel or paclitaxel, respectively.
As part of the base case, CADTH used the mean treatment durations for all drugs included in the BIA.
Relative dose intensity: The sponsor incorporated the same RDIs from the pharmacoeconomic model into the BIA which were multiplied by the respective drug acquisition costs for each comparator. The limitations of this approach have been previously described in CADTH Appraisal of the Sponsor’s Economic Evaluation section.
To align with the pharmacoeconomic model CADTH used RDIs of 100% for all comparators in the BIA.
Additional limitations were identified but were not considered to be key limitations. The sponsor made a transcription error in their model regarding the prevalence of bladder cancer. The corrected 5-year prevalence estimate of patients with bladder cancer in Canada is 31,297.
CADTH Reanalyses of the Budget Impact Analysis
Based on the limitations identified, CADTH increased the market shares for enfortumab vedotin, used mean treatment duration, and assumed RDIs of 100%.
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Prevalence of bladder cancer | 31,927 | 31,297 |
Changes to derive the CADTH base case | ||
1. Market shares in the new drug scenario | 2L setting (Y1 / Y2 / Y3): EV = ||||||% / ||||||% / ||||||% Paclitaxel = ||||||% / ||||||% / ||||||% Docetaxel = ||||||% / ||||||% / ||||||% 3L setting (Y1 / Y2 / Y3): EV = ||||||% / ||||||% / ||||||% Paclitaxel = ||||||% / ||||||% / ||||||% Docetaxel = ||||||% / ||||||% / ||||||% | All lines of therapy (Y1 / Y2 / Y3): EV = 50% / 65% / 80% Docetaxel = 30% / 21% / 12% Paclitaxel = 20% / 14% / 8% |
2. Treatment duration | Median: Enfortumab vedotin – 5.0 months Docetaxel – 3.5 months Paclitaxel – 3.5 months | Mean: Enfortumab vedotin – 5.36 months Docetaxel – 3.96 months Paclitaxel – 3.96 months |
3. Relative dose intensity | Enfortumab: ||||||||||||% Docetaxel: ||||||||||||% Paclitaxel: ||||||||||||% | Enfortumab: 100% Docetaxel: 100% Paclitaxel: 100% |
CADTH base case | Reanalysis 1 + 2 + 3 | |
2L = second-line (patients progressed on avelumab maintenance therapy or progressed within 12 months of platinum chemotherapy and subsequent pembrolizumab); 3L = third-line (patients who did not respond to platinum chemotherapy and progressed on subsequent pembrolizumab); EV = enfortumab vedotin; Y1 = year 1; Y2 = year 2; Y3 = year 3.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. Based on the CADTH base case, the budget impact of the reimbursement of enfortumab vedotin for the treatment of adult patients with mUC previously treatment with platinum chemotherapy and immunotherapy is expected to be $20,806,133 in year 1, $32,299,559 in year 2, and $46,273,397 in year 3, with a 3-year total of $99,379,089. A scenario analysis using the sponsor’s original RDI assumptions resulted in a 3-year budget impact of $78,326,191.
Table 16: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Submitted base case (corrected) | $35,386,568 |
CADTH reanalysis 1 – market shares | $73,232,729 |
CADTH reanalysis 2 – mean treatment duration | $37,816,112 |
CADTH reanalysis 3 – relative dose intensity | $44,978,994 |
CADTH base case | $99,379,089 |
BIA = budget impact analysis.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case (corrected) | Reference | $916,262 | $1,127,578 | $1,341,396 | $1,557,708 | $4,026,681 |
New drug | $916,262 | $5,854,804 | $12,534,663 | $21,023,782 | $39,413,249 | |
Budget impact | $0 | $4,727,226 | $11,193,267 | $19,466,075 | $35,386,568 | |
CADTH base case | Reference | $1,123,254 | $1,382,309 | $1,644,430 | $1,909,609 | $4,936,347 |
New drug | $1,123,254 | $22,188,442 | $33,943,989 | $48,183,005 | $104,315,436 | |
Budget impact | $0 | $20,806,133 | $32,299,559 | $46,273,397 | $99,379,089 | |
CADTH scenario analysis: sponsor’s original RDI assumptions | Reference | $1,036,685 | $1,275,774 | $1,517,693 | $1,762,435 | $4,555,902 |
New drug | $1,036,685 | $17,662,491 | $26,973,424 | $38,246,178 | $82,882,094 | |
Budget impact | $0 | $16,386,717 | $25,455,731 | $36,483,743 | $78,326,191 |
BIA = budget impact analysis; RDI = relative dose intensity.
Stakeholder Input
Patient Group Input
Bladder Cancer Canada
About Bladder Cancer Canada
BLADDER CANCER CANADA (BCC) is the first and only Canadian patient advocacy organization dedicated to bladder cancer issues.
The story of Bladder Cancer Canada began with two people who have bladder cancer – David Guttman and Jack Moon. Their medical teams tended to the disease in their bodies, but they realized nourishment for their inner beings was missing – and was just as critical to their recovery. They needed other bladder cancer patients to talk to about their fears and uncertainty. Someone to help them find up-to-date information about diagnosis, treatment options and prognosis. At that time, there was no one to turn to. So, in 2009, they formed BCC to fill the void.
Today, BCC is a nationally registered Canadian charity. Supported by a Medical Advisory Board and a Medical Research Board consisting of the top bladder cancer specialists across Canada, its mission aims to help bladder cancer patients and their support teams address the day-to-day issues of this disease; increase awareness among the general public and medical community; and fund research into the diagnosis, treatment and elimination of the disease. BCC’s vision is patient support, awareness and research to create a world where bladder cancer is just a memory.
Connect with Bladder Cancer Canada at www.bladdercancercanada.org.
Information Gathering
The information was gathered through an online survey and telephone interviews. All of the data was contributed anonymously. Bladder Cancer Canada (BCC) developed and designed a 20-minute online survey that was disseminated in English only.
Recruitment was undertaken by BCC through social media, e-newsletters and other online platforms, as well as direct email to
physicians, patients and caregivers. The online survey was open between May 27 and June 11, 2021. All of the 38 patients and 6 caregivers who completed the survey reported being diagnosed or a caregiver to someone diagnosed with Stage II or higher muscle-invasive bladder cancer. One-third of patients said they were living with locally advanced or metastatic bladder cancer, and two-thirds of caregivers reported caring for someone with advanced or metastatic disease. The same percentage of patients (40%) reported either having or not having previously received platinum-based chemotherapy. Of those who had received platinum-based chemotherapy, 44% said their cancer did not progress (i.e., it stabilized or improved), while 22% said that their cancer did progress after receiving chemotherapy. The majority of survey respondents were from Canada, with a small number from the U.S.
Telephone interviews were conducted in June 2021 with two patients from Canada who had experience on enfortumab vedotin.
Disease Experience
While some survey respondents reported that having bladder cancer has had a minimal impact on their day-to-day lives, many patients and caregivers mentioned that fatigue, lack of sleep, and loss of strength and stamina were problematic, but manageable to a greater or lesser extent. The most commonly mentioned impacts on day-to-day life were related to continence issues, including frequent need for urination and loss of control,” urostomy and catheter management, and urinary tract infections. One caregiver said that due to frequent “accidents,” she and her husband have become less social. Some patients and caregivers also mentioned that day-to-day activities take more time and planning and can even be uncomfortable for some – such as walking, sitting, driving, sleeping, getting dressed and wearing clothing. Caregivers mentioned that they were constantly worried for their loved ones and were no longer able to plan anything ahead of time. One caregiver said they had become the sole earner, as her loved one was no longer able to work, and another provided in-home care to her husband, helping to manage his chronic pain caused by bladder cancer. Some patients reported that their mental health has been impacted by living with bladder cancer, leading to a loss of confidence, avoidance of leaving home/going out, loss of employment and fear of looking for another job, and reduced intimacy. It was also noted by some respondents that the COVID-19 pandemic has exacerbated the day-to-day impacts of living with bladder cancer. And finally, financial impacts were mentioned related to the costs of catheter and urostomy supplies that are not covered by some provincial governments – in some cases putting additional strain on already limited financial resources.
“My biggest peeve is that Pharmacare WILL NOT pay for my catheters. They will pay for all other ostomy supplies, BUT NOT CATHETERS!! No one should have to pay to pee!!”
“Urostomy supplies strain my already limited financial resources.”
“Everything takes more time and planning. Clothes are uncomfortable. Bending is uncomfortable.”
“I completely lost my confidence avoid going anywhere, lost my job and don't dare to apply for another as I need to go to empty my bag.”
“When my husband was first diagnosed, day-to-day tasks were hard. Between all the appointments and COVID restrictions it was terrible. Chemo was hard, and I wanted to be close by but was unable to. Now, he is on Immunotherapy every 6 weeks and doing well.”
When asked about the impact of bladder cancer on their quality of life, the responses ranged from “not much” to “I have no life.” One caregiver said their “life has been shattered.” Fatigue was the most commonly reported symptom of bladder cancer by both patients and caregivers, followed by blood in the urine, pain in the abdomen and bones, decreased mobility and difficulty/pain when urinating. Recovery from surgery was difficult for many respondents, having a major impact on quality of life for months to years following the surgery. Pain – extreme in some cases – was mentioned by patients and caregivers alike as being a very difficult aspect of bladder cancer to manage. Many people cited frequent urinary tract infections as having a significant and sustained impact on their quality of life, and in some cases life-threatening, when the infections led to hospitalization due to sepsis or kidney problems. One patient whose ureters were both blocked by tumours underwent surgery to insert bilateral nephrostomy tubes and bags through their back. Side effects from treatment – such as chemotherapy and immunotherapy – were also mentioned as impacting patients’ quality of life, leaving them to cope with nausea, fatigue, pain, a weakened immune system and reduced mobility –or causing them to discontinue treatment altogether.
“Were at a stop presently. We were going to retire and enjoy our grandkids. We feel numb now, our life has been shattered.”
“During 5 years, I was always having UTIs and had to rush to hospital every time. My quality of life was very difficult. I had to stay home just in case and I had problems with my kidney every time I had a UTI.”
“Fear and stress can get me down. Many doctors’ appointments and scans are a continual reminder the cancer could reappear elsewhere.”
Experiences With Currently Available Treatments
Regarding patients’ response to platinum-based chemotherapy, 39% of respondents said they had previously received this form of treatment and among them, 44% reported that their bladder cancer did not progress, while 22% said that it did progress after receiving platinum-based chemotherapy. Of the currently available pharmaceutical treatments for bladder cancer, survey respondents specifically mentioned having received one or more of the following: Platinol (cisplatin) – 47%; Gemzar (gemcitabine) – 18%; Keytruda (pembrolizumab) – 16%; and Paraplatin (carboplatin) – 7%. Imfinzi (durvalumab), Adriamycin/Rubex (doxorubicin), and Trexall (methotrexate) were each mentioned by 3% of patients. Other treatments mentioned by survey respondents include Bacillus Calmette-Guerin (BCG) and vinblastine.
In addition to pharmaceutical interventions, respondents also mentioned receiving radiation therapy and surgery, including TURBT (transurethral resection of bladder tumour) and full/partial cystectomy, and some reported that they were being monitored and had not yet received any treatment for their bladder cancer. The extent and duration of efficacy of the various bladder cancer treatment modalities and combinations varied widely among survey respondents. Some reported that their cancer was gone following chemotherapy alone, or with radiation and TURBT (tri-modal therapy), and others have found success with immunotherapy, including in reducing metastases. For some patients, bladder removal surgery (full/partial cystectomy) was successful in eliminating the cancer, while others required additional treatment following surgery. A few patients shared that a full cystectomy was not successful in stopping tumour growth and that other forms of treatment were not able to stop the cancer from metastasizing. Others expressed frustration with having to undergo various treatments with significant side effects, only to end up having to have their bladder removed after all. One caregiver felt that a standard of care for the treatment of bladder cancer was needed across the country.
When asked what side effects patients experienced from treatments for bladder cancer, the following were mentioned by patients/caregivers: fatigue (67%/100%), constipation (48%/67%), low blood cell count (39%/67%), loss of appetite (36%/50%), neuropathy (27%/33%), nausea (27%/67%), hair loss (24%/33%), insomnia (21%/17%), diarrhea (18%/17%), mouth sores (6%/17%), and vomiting (3%/17%), as well as skin problems, shortness of breath, hearing loss/tinnitus, and incontinence. Surgery was mentioned by many respondents as being extremely difficult and involving a lengthy and very painful recovery. People reported being able to manage side effects of treatment by resting, “waiting it out,” taking naps/sleeping more, taking medications for symptom management (including opioids for pain), leading a more sedentary lifestyle, taking time off work, and by having a positive and a supportive home life.
The majority of respondents said they have had no difficulties accessing treatment for bladder cancer, however, some mentioned they did have difficulties due to: the travel distance to access treatment (8%); the cost of treatment (5%); the treatment being unavailable in Canada (5%); not having access to a physician (5%); and requiring time off work to receive treatment (3%). One patient mentioned that if they hadn’t been able to take early retirement, they would have had to quit their job due to their treatment regimen. Additionally, the COVID-19 pandemic has made it difficult for some patients to reach their doctors and one caregiver reported that their loved one’s surgery was cancelled and then rescheduled a few weeks later due to COVID.
With respect to the financial impact of undergoing treatment for bladder cancer, just over two-thirds of patients and caregivers indicated they hadn’t experienced any financial challenges. Of those who said they had experienced financial challenges as a result of being treated for bladder cancer, costs related to reduced income due to work absence, travel, parking, accommodations, medications, and homecare were mentioned. One caregiver noted that having Short Term Disability benefits was helpful while her husband was receiving chemotherapy and unable to work. In addition to some patients in certain provinces having to cover some of the costs of urostomy and catheter supplies (as mentioned in Section 3), one patient added that paying out-of-pocket for a costly diagnostic CT scan because of long wait-times at public facilities in their province.
“Surgical bladder removal unsuccessful as the tumour had enveloped the left femoral artery and was too risky to try surgery. Next tried immunotherapy, but the drug used boosted my immune system, causing a flare up of my rheumatoid arthritis symptoms, so that was discontinued. I then had six radiation treatments which, as evidenced by two subsequent CT scans, has stopped the tumour growth, so far. Another treatment was explored but my tumour didn't have a certain mutation and so I didn't qualify for it.”
“My cancer is gone, but the residual loss of kidney function is not good. I am not a candidate for at home dialysis if my kidney function goes lower because of my Indiana pouch.”
“During radiation treatments, I had to urinate every hour or so. Lack of sleep is my main problem. During my two chemo cycles, I had absolutely no energy and could barely walk across the street. TURBTs were painful, and the catheter is very uncomfortable.”
Improved Outcomes
When asked what improvements patients and caregivers would like to see if enfortumab vedotin was accessible to them as a treatment for bladder cancer, they mentioned: slowing or stopping the disease from progressing, recurring and spreading; a reduction in pain; fewer/no infections; reduced fatigue, increased energy level/strength; improved mental health; reduced impairment of sexual functionality; avoidance of surgery (cystectomy); improved continence/urination control; to go into remission/be cured; and overall, patients and caregivers hoped for fewer/less severe side effects than experienced with bladder cancer treatments currently in use.
Assuming that the desired improvements were provided through treatment with enfortumab vedotin, respondents were asked how their day-to-day life and quality of life as patients/caregivers would be different. People mentioned they would be able to go for a walk and no longer fear leaving the house, and that they would be able to help around the house (i.e. cutting the grass, meal preparation) and participate in family activities. They also mentioned being able to get a longer and continuous night’s sleep, drive for longer periods of time, and travel to visit family. Additionally, respondents said that their outlook on life would be more positive and they would be able to plan for the future – with one person “life would improve immensely.” One patient also noted that “if [enfortumab vedotin] can help more patients avoid surgery, it's well worth the cost, as surgery (cystectomy) is quite life-changing.”
“I would expect an increase in physical strength and a decrease in fatigue and shortness of breath. I would hope to be able to go on walks with my wife, participate in family activities, and travel to visit family (assuming Covid is under control or eliminated).”
“I'd have more mobility and be able to do pretty much what I'd like, including the ability to drive for up to two-four hours at a time. Increased amount of continuous sleep would be nice (not having to urinate every 1-2 hours).”
“Any new drug or advance in treatment of bladder cancer will directly affect me as a caregiver. It would hopefully give me more time with my husband, affect his quality of life and ease my state of mind.”
Experience With Drug Under Review
Two patients were interviewed regarding their experiences with enfortumab vedotin. Both live in Canada and accessed the treatment through a clinical trial, and both have been on treatment for more than two years. One of the two patients first learned about the treatment from a family member in the U.S. and then contacted her physician to find out if there were any clinical trials in Canada that she might qualify for.
By way of comparison with other treatments, the patients interviewed said that past treatments they had received had left them feeling nauseous, tired and weak. One patient noted that while on other treatments, she often had “bad days” when all she wanted to do was sit all day, or stay in bed. And on two occasions, the fatigue and weakness from her previous treatments was so extreme that friends from England flew to Canada to help her husband take care of her, because he was overwhelmed. One patient also experienced vision problems from one treatment which temporarily prevented him from performing his job.
With respect to any disadvantages of treatment with enfortumab vedotin, one of the two patients interviewed said that the only side effect she could think of was itchy skin, described as “more of a nuisance than anything else,” which she manages with Benadryl, as needed. The other patient interviewed mentioned hair loss, alterations to sense of taste, nausea and skin sores which he noted were temporary and resolved over time. This same patient also said that he continues to experience numbness and tingling in his hands due to neuropathy and some gastrointestinal (GI) upset from time to time; however, he feels these side effects haven’t been as bad as those experienced on other treatments. He manages them by reducing his hours at work as needed for the neuropathy, and by taking an antacid to address any GI issues.
Also as compared with other treatments for bladder cancer, the benefits of enfortumab vedotin that were mentioned by the patients interviewed included a shorter treatment schedule, shorter time in hospital for each treatment, and fewer side effects. One patient commented that enfortumab vedotin is “very easy on the body,” and both remarked that their energy levels were much improved on enfortumab vedotin, as compared with the extreme fatigue they experienced on other treatments.
When asked what key values about enfortumab vedotin have been important to them as patients, they said that the treatment has made a “huge difference” in their life and has given them their “life back again” – allowing them to resume the activities that they enjoy, such as cooking, gardening, taking short road trips, and playing guitar in a rock band. One patient recalled that when he was first diagnosed, he had to make a choice to live or to die – and he chose to live. Of all of the treatments he’s been on, he says that enfortumab vedotin is “by far the best” – the treatment shrunk his tumour (he has had a complete response to therapy) and it has helped him survive bladder cancer thus far.
Companion Diagnostic Test
N/A
Anything Else?
Based on the interviews conducted and some of the survey responses, it is important to patients to have publicly funded access to enfortumab vedotin. One patient commented that all the other treatments he tried before enfortumab vedotin were a waste – both to him and to the healthcare system in terms of their expense. He suggested that the savings gained from not using an ineffective treatment on patients could go towards funding enfortumab vedotin. This patient also pointed out that because the treatment has allowed him to live longer and keep working, he is able to continue paying his taxes.
“My bladder has been removed. However, having lived with bladder cancer for 10 years, I would hope that all current and future Bladder cancer patients would have access to Padcev [enfortumab vedotin] and the cost be covered under the applicable government program. As a patient I was focussed on being positive. As a caregiver, my wife was under significant stress and she felt helpless.”
Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Advocacy Solutions, experts in healthcare advocacy, provided best practices for data collection, consultation and discussions with our patient group.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Advocacy Solutions collected the responses from our patient group and provided analysis on the results.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Name: Michelle Colero
Position: Executive Director, Bladder Cancer Canada
Patient Group: Bladder Cancer Patients & Caregivers
Date: July 12, 2021
Table 1: Conflict of Interest Declaration for Bladder Cancer Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
EMD Serono (Indirect) | X | — | — | — |
Pfizer Canada (Indirect) | X | — | — | — |
Verity Pharmaceuticals (Indirect) | X | — | — | — |
Merck Canada (Indirect) | — | — | X | — |
Ferring Canada (Indirect) | — | — | X | — |
Astra Zenica (Indirect) | — | — | X | — |
Bristol Myers Squibb (Indirect) | — | — | X | — |
Seagen (Indirect as the funding was from the pat-ed fund) | — | — | X | — |
Clinician Group Input
Ontario Health (Cancer Care Ontario) Genitourinary Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Genitourinary Cancer Drug Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Please describe how you gathered the information included in the submission.
Discussed jointly via emails.
Current treatments
Describe the current treatment paradigm for the disease
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Current treatment (3rd line chemo) are basically non-existent with respect to level 1 evidence. Patients are thus given a “dealer’s choice” of chemotherapy agents which are based on expert opinion.
No standard of care treatment for patients with advanced urothelial cancer post-platinum chemotherapy, post- immunotherapy. Taxanes (docetaxel and paclitaxel) are sometimes used in this setting, but more on the basis of expert opinion with little Level 1 evidence. These chemotherapies are associated with significant toxicity.
Treatment goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Improve overall survival, decrease the risk of progression, delay radiologic progression and onset of symptoms, and improve quality of life.
Treatment gaps (unmet needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatments needed to improve compliance. Formulations are needed to improve convenience.
Response: No standard of care treatment. There are many reasons current treatments are not ideal:
Not all patients respond to available treatments
Patients become refractory to current treatment options
No treatments are available to reverse the course of disease
No treatments are available to address key outcomes
Treatments are needed that are better tolerated
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Those who have failed the 2 lines of systemic therapy outlined in the indication for enfortumab do not have any other reasonable options.
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Enfortumab vedotin is a new therapy with a novel mechanism of action. It would be used in patients who have had prior treatment with platinum-based chemotherapy and immunotherapy. As phase III randomized clinical trial of enfortumab vedotin in this patient population (NEJM Powles et al, 2021) demonstrated improved overall survival and progression free survival compared to chemotherapy (Docetaxel, paclitaxel or vinflunine).
This will redefine the current treatment paradigm. There are no other beneficial therapies in this setting with any kind of survival advantage. Enfortumab fills an unmet need.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: No, not recommended or appropriate in the 3rd line setting.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: This drug would provide an effective 3rd line agent when the first 2 lines have failed. There would not be other opportunities to treat patients with enfortumab downstream as the longevity of these patients is brief.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Patient with advanced urothelial cancer who have progressed after platinum-based chemotherapy and immunotherapy.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Those who fail platinum-based chemo and immune checkpoint inhibitors as per indication for the drug. Patients who are pre-symptomatic should be treated.
Which patients would be least suitable for treatment with the drug under review?
Response: Those who have not had prior systemic therapy
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: Not aware of a biomarker that enriches for response.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Yes, the outcomes used in clinical practice align with the outcomes typically used in the clinical trial. This is based on serial imaging (CT scans) and patient reported outcomes.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Survival, response to therapy are both objective meaningful responses. Subjective meaningful responses include: improvements in disease symptoms, stabilization of symptoms, improvements in ADLs.
How often should treatment response be assessed?
Response: As per medical oncology standards of care. Patients would be assessed for toxicity and clinical progression monthly. Restaging CT scans every 2-3 months.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: Progressive disease while on treatment, worsening symptoms, severe adverse events and clear deterioration to end of life.
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic.
Response: Cancer centre infusion clinics.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: NA
Additional information
Is there any additional information you feel is pertinent to this review?
Response: This drug is really important and will revolutionize the management of bladder cancer as it fills an unmet need for these patients. There is no good therapy in the 3rd line setting.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Girish Kulkarni
Position: Ontario Cancer Lead
Date: 21 July 2021
Table 2: Conflict of Interest Declaration for OH-CCO’s DAC Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Christina Canil
Position: Medical oncologist
Date: 23 July 2021
Table 3: Conflict of Interest Declaration for OH-CCO’s DAC Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Canadian Physicians Who Treat Bladder Cancer
About the Clinician Group
Please describe the purpose of your organization. Include a link to your website (if applicable).
We are a group of Canadian physicians who treat bladder and who, with the support of Bladder Cancer Canada, a Canadian patient advocacy organization dedicated to bladder cancer issues (https://bladdercancercanada.org/en/), wish to make this submission to CADTH. We represent the specialty from across Canada in both academic and community settings and share Bladder Cancer Canada’s goal to improve the management of bladder cancer.
Information Gathering
Please describe how you gathered the information included in the submission.
Interviews based on the questions in this template were conducted with one Canadian urologic oncologist and one Canadian medical oncologist. The resulting submission was then circulated to Canadian physicians who treat bladder cancer. The distribution list was provided by Bladder Cancer Canada. Physicians who wished to support this Clinician Group Input submission signed the submission. In addition, the following articles were referenced for background information.
Peter C. Black, Nimira S. Alimohamed, David Berman, Normand Blais, Bernhard Eigl, Pierre I. Karakiewicz, et al. Optimizing management of advanced urothelial carcinoma: A review of emerging therapies and biomarker-driven patient selection. Can Urol Assoc J 2020;14(8):E373-82. http://dx.doi.org/10.5489/cuaj.6458
Mark Warren, Michael Kolinsky, Christina M. Canil, Piotr Czaykowski, Srikala S. Sridhar, Peter C. Black, et al, on behalf of GUMOC. Canadian Urological Association/Genitourinary Medical Oncologists of Canada consensus statement: Management of unresectable locally advanced and metastatic urothelial carcinoma. Can Urol Assoc J 2019;13(10):318-27. http://dx.doi.org/10.5489/cuaj.6015
Current treatments
Describe the current treatment paradigm for the disease
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: In Canada, bladder cancer is the fifth most common cancer, with an estimated 12,200 new cases and 2,600 deaths (2020). Approximately 15% of patients have locally advanced or metastatic disease at presentation. Of those patients with non-metastatic muscle-invasive disease, about half will relapse after initial treatment. These patients and the majority of patients with advanced-stage disease will die of their cancer. Urothelial carcinoma of the upper tract (kidney and ureter) is a relatively rare cancer that is very similar to bladder cancer and is treated in the same way. The current treatment paradigm for locally advanced or metastatic urothelial carcinoma of the bladder or upper tract can be summarized as follows (source: Warren et al. Can Urol Assoc J 2019;13(10):318-27).
First-line systemic therapy
Patient eligible for cisplatin-based chemotherapy (Cisplatin kills cancer cells by damaging their DNA and stopping them from dividing.)
Preferred routine regimen is gemcitabine/cisplatin (GC).
Dose-dense methotrexate, vinblastine, doxorubicin and cisplatin (DD-MVAC) with growth factor support may be considered in select cases where a more aggressive treatment approach is being considered.
Patients who have at least stable disease (i.e., no worsening on imaging studies) after platinum-based chemotherapy are treated with switch maintenance avelumab, an immunotherapy. This agent is provided through a special access program.
Patient ineligible for cisplatin-based chemotherapy:
Preferred regimen is gemcitabine/carboplatin (GCa).
In patients not suitable for combination chemotherapy, single-agent gemcitabine, paclitaxel, or docetaxel is recommended.
Immunotherapy has been approved for all platinum-ineligible patients and for cisplatin-ineligible patients whose tumours are positive for PD-L1 expression by immunohistochemistry, but there is no coverage for this treatment option in Canada and it is not provided through a special access program. It is therefore not used routinely
Second-line systemic therapy (Immunotherapy stimulates the body’s defenses to attack and kill cancer cells.) Patients who have progressive disease during or after platinum-based chemotherapy
Pembrolizumab is the preferred regimen (if available).
Where pembrolizumab is unavailable or a patient is ineligible, single-agent paclitaxel or docetaxel is preferred for the majority of patients.
Re-treatment with a platinum-based regimen is a reasonable option in a patient who has disease progression following a prolonged (>6- to 12-month) initial response to platinum-based chemotherapy.
Treatment with erdafitinib in eligible patients (mutations and fusions in fibroblast growth factor receptors (FGFR) 2 and 3 are found in approximately 20% of patients with invasive urothelial carcinoma. (Erdafitinib is a tyrosine kinase CADTH Clinician Group Input Template Page 3 of 14 June 2021 inhibitor that targets FGFR 1–4 in order to stop or slow the growth of cancer cells). Erdafitinib is provided through a special access program that is anticipated to continue until phase III trial results are available.
If a patient experiences progressive disease on any one of these treatments he/she can potentially be treated with the other agents listed in the third and fourth line if he/she is eligible.
If a patient experiences progressive disease during or after switch maintenance avelumab therapy, that patient could receive erdafinitib, if eligible, or more chemotherapy, but not pembrolizumab.
The role of aggressive surgical/radiotherapeutic management in oligometastatic disease
Routine metastasectomy or other localized treatment (e.g., stereotactic radiation) to metastatic sites in patients with oligometastatic or limited metastatic disease is not recommended. However, such treatment may be appropriate in selected cases. Sometimes lymph node metastases in the retroperitoneum (i.e., in the abdomen above the pelvis) are removed surgically after preceding chemotherapy.
The routine use of radical cystectomy (RC) or radiation to the primary tumour is not recommended in patients with metastatic urothelial carcinoma other than for palliation. However, such treatment may be appropriate in selected cases.
The decision to treat oligometastatic disease with local therapies should be made in a multidisciplinary context with involvement of an experienced medical oncologist, uro-oncologist, and radiation oncologist, where appropriate.
Treatment goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: The ideal treatment would: delay disease progression; prolong life while minimizing symptoms; improve health- related quality of life; increase the ability to maintain employment and maintain independence; and reduce burden on caregivers.
Treatment gaps (unmet needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatments needed to improve compliance. Formulations are needed to improve convenience
Response: Most patients will be offered platinum-based chemotherapy (usually cisplatin) as first-line option, and virtually all will eventually progress on this treatment. Some common side effects of chemotherapy include fatigue; increased risk of CADTH Clinician Group Input Template Page 4 of 14 June 2021 infection and bleeding; hearing problems; bowel changes; decreased kidney function; peripheral nerve damage (neuropathy); hair thinning or loss; skin rash; weight changes; anemia and thrombocytopenia.
Those who survive will be offered immunotherapy, but only ~20% of patients will respond to this therapy. Side effects of these drugs can include fatigue, loss of appetite, diarrhea, skin rashes, and itchy skin. These drugs can also trigger an immune response against any normal tissue, which can cause serious or even life-threatening problems in the lungs, intestines, liver, hormone-making glands, or other organs. The same side effects can be observed in patients receiving switch maintenance avelumab.
Currently, FGFR-targeted therapy is an option for those patients whose tumours harbour an FGFR alteration – only about 20-25% of patients. Furthermore, FGFR-alteration testing is not available in Canada, meaning FGFR-targeted therapy is not an option for the majority of patients. Some common side effects of FGFR-targeted therapy for bladder cancer include: sore mouth; fatigue; diarrhea; dry mouth; loss of appetite; taste changes; skin and nail problems (including redness, itching and dryness); muscle and joint pain; low blood cell counts; and vision changes.
A significant proportion of patients are not eligible for platinum-based chemotherapy due to other medical conditions, frailty and age. These patients are then also ineligible for subsequent lines of therapy.
The side effect profiles of existing treatments are so significant that they limit their usefulness in many patients. As such, a treatment that can be offered to all patients with diverse tumour histology with a demonstrable survival benefit and tolerable side effect profile is needed for this population.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: EV would meet an unmet need in virtually all patients with advanced urothelial carcinoma, as almost all patients will eventually progress on existing first- and second-line therapies, and such a small percentage (~25%) are eligible for the FGFR-targeted therapy. Offering EV to all eligible patients would provide them with hope for improved life expectancy with tolerable side effects.
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: EV targets a molecule (Nectin-4) that is expressed in >95% of bladder cancers, delivering a chemotherapy payload directly to the cancer cell with fewer ensuing systemic impacts. Because virtually all patient tumours express this molecule, there is no need for any biomarker testing and EV could be offered to almost all patients. EV would be offered as a third-line option to patients who progress on chemotherapy and immunotherapy. FGFR-targeted CADTH Clinician Group Input Template Page 5 of 14 June 2021 therapy is supported only by a single arm phase II trial, whereas EV is supported by a randomized phase III trial. As FGFR testing is not available in Canada and so few patient tumours harbour an FGFR alteration, EV will become the standard third-line treatment, pushing FGFR- targeted therapy to fourth-line in eligible patients.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: EV is indicated in the third-line setting. All patients must have previously been treated with platinum-based chemotherapy and immunotherapy. Alternative third-line options would be non-platinum chemotherapy, for which there is little evidence of efficacy and for which the toxicity rate is much higher, or FGFR-targeted therapy, which would not be favoured for the reasons outlined under 6.1.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: As outlined above, EV would push FGFR-targeted therapy to the fourth line in eligible patients. Non-platinum chemotherapy (especially with a taxane) could also follow EV therapy. This represents a major improvement in treatment options for these patients.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: All patients with locally advanced or metastatic urothelial carcinoma who have progressed on chemotherapy and immunotherapy would be considered for treatment with EV. Patients best suited to treatment with EV would include: those with a reasonable life expectancy (>3 months), adequate performance status (i.e., ECOG score of 0-1), adequate renal function, and without significant neuropathy or other toxicity concerns. A patient’s ability to access the treatment centre for the duration of treatment should also be considered.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Progression on first- and second-line treatment is determined by radiographic imaging. This is part of routine clinical practice and does not represent a challenge. Pseudoprogression on immunotherapy represents one potential for premature determination of progression, but this is very rarely observed in urothelial carcinoma. Urothelial carcinoma typically progresses rapidly and treatment decisions are not generally dependent on a patient developing symptoms, since delays in therapy can lead to a patient becoming unfit for treatment. Only patients for whom EV is indicated would be considered. Patient assessment should include: estimation of life expectancy; determination of performance status (i.e., ECOG score); renal function testing; determination of existing toxicities such as neuropathy; and level of frailty. Finally, any determination about whether to move to EV treatment would include the patient’s wishes. No biomarker testing is needed to determine eligibility.
Which patients would be least suitable for treatment with the drug under review?
Response: The following patients would be considered least suitable for treatment with EV:
Patients with a very short life expectancy (i.e., <3 months)
Patients with significant frailty • Patients with low functional status
Patients with insufficient renal function to tolerate treatment
Patients with significant existing peripheral neuropathy
Patients who could not access the treatment centre for weekly infusions over a 3-month period (typically 3 or 4 cycles of treatment consisting of weekly treatments for 3 weeks then 1 week off).
Patients who decline treatment for any personal reasons.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: There are currently no clinical or molecular biomarkers to predict response to EV.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: The outcomes used in clinical practice are aligned with outcomes typically used in clinical trials, i.e., duration of response, progression-free survival, safety, and tolerability. These endpoints are measured by clinical history and examination, as well as radiographic imaging.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: The following would be considered meaningful clinical responses:
Lack of progression of disease as determined by CT (or other imaging as required such a bone scan).
Stabilization or reduction in the frequency or severity of symptoms such as fatigue, pain, loss of appetite, or respiratory symptoms.
Attainment of major motor milestones due to reduction in pain and/or fatigue.
Ability to perform activities of daily living.
The magnitude of response to treatment would not be expected to vary across physicians. Variation would likely be more affected by disease and patient characteristics than physician or treating centre characteristics.
How often should treatment response be assessed?
Response: There are normally 3 or 4 treatment cycles consisting of 3 weekly infusions followed by 1 week off. Blood work should be performed prior to each treatment cycle, and patients should be seen by their treating oncologist following each cycle. Radiographic imaging should be performed at least every 3 months. Patients with disease that has metastasized to the bones should also have a bone scan.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Response: Decisions to discontinue treatment should be made in consultation with the patient and would include disease progression, dose-limiting toxicity resulting in intolerable adverse effects such as significant neuropathy, and patient wishes to discontinue treatment for any number of personal reasons.
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: The most appropriate treatment setting is one that has staff who are specialized and experienced in delivering chemotherapy treatments and a dedicated physical space for chemotherapy infusions. Therefore, treatments can be delivered in the community setting, hospital outpatient setting, or a specialty clinic (such as an infusion clinic) that have the appropriate staff and facilities.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: NA
Additional information
Is there any additional information you feel is pertinent to this review?
Response: The approval of EV would give medical oncologists an option to offer to patients with advanced urothelial cancer that has progressed on first- and second-line therapy. EV offers significant overall survival benefit compared to taxane chemotherapy, with tangible benefits for patients. EV would offer a longer life expectancy with preservation of quality of life, as the drug is generally well tolerated. For a patient population with such a poor prognosis, the inclusion of EV in the treatment algorithm has the potential to significantly improve the outcomes associated with bladder cancer.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Medical writer, Cynthia Lank (Cynthia N. Lank Editorial Services, Halifax, NS), provided logistical support in the form of conducting interviews, collating responses, and drafting submission for review and revision by participating physicians.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Medical writer, Cynthia Lank (Cynthia N. Lank Editorial Services, Halifax, NS), provided logistical support in the form of conducting interviews, collating responses, and drafting submission for review and revision by participating physicians.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review
Declaration for Clinician 1
Name: Dr. Peter Black
Position: Medical Oncologist, Chair, Bladder Cancer Canada, Medical Advisory and Research Boards; Senior Research Scientist, Vancouver Prostate Centre; Associate Director, Clinical Research, Vancouver Prostate Centre; Professor, Department of Urologic Sciences, University of British Columbia
Date: 06/07/2021
Table 4: Conflict of Interest Declaration for Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Francisco E. Vera Badillo
Position: Associate Professor, Department of Oncology, Queen’s University
Date: 06/07/2021
Table 5: Conflict of Interest Declaration for Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Seagen | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Naveen S. Basappa
Position: Medical Oncologist
Date: 11/07/2021
Table 6: Conflict of Interest Declaration for Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Nayyer Iqbal
Position: Medical Oncologist and Professor in Division of Oncology, College of Medicine, University of Saskatchewan
Date: 11/07/2021
Table 7: Conflict of Interest Declaration for Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 5
Name: Cristiano Ferrario
Position: Medical Oncologist, Jewish General Hospital and Assistant Professor, Oncology, McGill University
Date: 12/07/2021
Table 8: Conflict of Interest Declaration for Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 6
Name: Dr. Samantha Gray
Position: Medical Oncologist
Date: 12/07/2021
Table 9: Conflict of Interest Declaration for Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 7
Name: Dr. April Rose
Position: Medical Oncologist
Date: 12/07/2021
Table 10: Conflict of Interest Declaration for Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 8
Name: Dr. Normand Blais
Position: Medical Oncologist, CHUM
Date: 12/07/2021
Table 11: Conflict of Interest Declaration for Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Seagen | X | — | — | — |
Declaration for Clinician 9
Name: Dr. Scott North
Position: Medical Oncologist
Date: 12/07/2021
Table 12: Conflict of Interest Declaration for Clinician 9
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Seagen | X | — | — | — |
Declaration for Clinician 10
Name: Dr. Nimira Alimohamed
Position: Medical Oncologist, Tom Baker Cancer Centre, Alberta Health Services
Date: 12/07/2021
Table 13: Conflict of Interest Declaration for Clinician 10
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 11
Name: Dr. Daniel Heng
Position: Medical Oncologist
Date: 12/07/2021
Table 14: Conflict of Interest Declaration for Clinician 11
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | X | — | — | — |
Declaration for Clinician 12
Name: Dr. Tina Cheng
Position: Medical Oncologist
Date: 12/07/2021
Table 15: Conflict of Interest Declaration for Clinician 12
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 13
Name: Dr. Simon Yu
Position: Medical Oncologist
Date: 12/07/2021
Table 16: Conflict of Interest Declaration for Clinician 13
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 14
Name: Dr. Michel Pavic
Position: Medical Oncologist
Date: 12/07/2021
Table 17: Conflict of Interest Declaration for Clinician 14
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 15
Name: Dr. Srikala Sridhar
Position: Genitourinary Medical Oncologist
Date: 13/07/2021
Table 18: Conflict of Interest Declaration for Clinician 15
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Seagen Inc. | X | — | — | — |
Declaration for Clinician 16
Name: Dr. Lori Wood
Position: Medical Oncologist
Date: 13/07/2021
Table 19: Conflict of Interest Declaration for Clinician 16
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
Declaration for Clinician 17
Name: Dr. Aly-Khan Lalani
Position: Medical Oncologist and Assistant Professor, McMaster University
Date: 06/07/2021
Table 20: Conflict of Interest Declaration for Clinician 17
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.