CADTH Reimbursement Review
Alpelisib (Piqray)
Sponsor: Novartis Pharmaceuticals Inc.
Therapeutic area: Advanced or metastatic breast cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
AI
aromatase inhibitor
CBCN
Canadian Breast Cancer Network
CDK4/6
cyclin-dependent kinase 4 and 6
CI
confidence interval
CNS
central nervous system
ctDNA
circulating tumour DNA
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ-5D-5L
EQ-5D Five-Level
ESMO
European Society for Medical Oncology
ESO
European School of Oncology
FAS
full analysis set
HER2
human epidermal growth factor receptor 2
HRQoL
health-related quality of life
IM
intramuscular
LHRH
luteinizing hormone–releasing hormone
mFAS
modified full analysis set
NGS
next-generation sequencing
OS
overall survival
PCR
polymerase chain reaction
PFS
progression-free survival
PI3K
phosphatidylinositol 3-kinase
PP
per-protocol
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
SAE
serious adverse event
SOC
standard of care
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Alpelisib (Piqray), in 50 mg, 150 mg, and 200 mg oral tablets |
Indication | Alpelisib in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen |
Reimbursement request | Alpelisib in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 11, 2020 |
Sponsor | Novartis Pharmaceuticals Inc. |
CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; NOC = Notice of Compliance.
Introduction
Breast cancer is the most commonly diagnosed cancer in Canadian females and the most common subtype of breast cancer is hormone receptor–positive, human epidermal growth factor receptor 2 (HER2)–negative breast cancer. It was estimated that in 2020, there would be 27,200 new cases of breast cancer in Canada, with age-standardized incidence rates of 1.1 per 100,000 males and 128.2 per 100,000 females. It was also estimated that there would be 5,100 deaths from breast cancer, with age-standardized mortality rates of 0.3 per 100,000 males and 22.0 per 100,000 females. In an analysis of females diagnosed with breast cancer from 2012 to 2016 in the Ontario Cancer Registry, the percentages of patients with 5-year survival ranged from 24.0% to 94.7%, depending on disease stage at diagnosis.
There are no curative treatments for advanced or metastatic breast cancer. According to input from clinicians consulted by CADTH for the purpose of this review, standard first-line therapy for advanced or metastatic hormone receptor–positive HER2-negative breast cancer is a cyclin-dependent kinase 4 and 6 (CDK 4/6) inhibitor in combination with an aromatase inhibitor (AI). A luteinizing hormone–releasing hormone (LHRH) receptor agonist is also given for ovarian suppression, depending on menopausal status. Patients whose disease recurs while on or shortly after stopping adjuvant AI therapy are considered resistant and are frequently offered CDK4/6 inhibitor with fulvestrant instead of an AI. Upon disease progression, second-line therapy options include a different single-agent AI, a single-agent fulvestrant, an investigational therapy in a clinical trial, chemotherapy (commonly single-agent capecitabine or a taxane), combined everolimus and exemestane, and tamoxifen. Despite the number of options beyond first-line therapy, there is no high-quality evidence for prolongation of survival with these therapies. Coverage of fulvestrant and everolimus plus exemestane is inconsistent across jurisdictions in Canada.
Alpelisib is an oral phosphatidylinositol 3-kinase (PI3K) inhibitor that, in combination with fulvestrant, is indicated for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen. Alpelisib 300 mg is taken daily on a continuous basis along with intramuscular (IM) fulvestrant 500 mg on day 1, day 15, and day 29, and every 28 days thereafter. Dose reductions for alpelisib are permitted to 250 mg or 200 mg daily for the management of adverse drug reactions.
Alpelisib has not been previously reviewed by CADTH. The sponsor’s reimbursement request is for alpelisib in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor. The reimbursement request differs from the Health Canada indication in that it specifies that patients must have received CDK4/6 inhibitor with a previous endocrine-based regimen.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of 50 mg, 150 mg, and 200 mg alpelisib tablets in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups provided input for this review: the Canadian Breast Cancer Network (CBCN), Rethink Breast Cancer, and CanCertainty. The CBCN provided information gathered from patient and caregiver responses from 2 online surveys conducted in 2017 (90 patients) and 2012 (87 patients and caregivers) and a telephone interview with 1 patient. Rethink Breast Cancer’s submission was informed by an online survey in 2021 of 24 patients and telephone interviews with 6 of the survey respondents. CanCertainty developed its submission based on published reports relating to breast cancer and oral cancer drugs.
The CBCN reported that the physical impact of metastatic breast cancer is variable across individuals, with the vast majority of patients in the 2012 survey reporting a significant/debilitating or some/moderate impact on their quality of life due to the symptoms of fatigue, insomnia, and pain. Many negative impacts on patients and their families’ daily lives were identified, including restrictions in patients’ ability to remain employed, care for children and dependents, be social, exercise, pursue hobbies and interests, and spend time with loved ones. The patient groups identified the following measures of effectiveness as the most important: progression-free survival (PFS), overall survival (OS), quality of life, and adverse effects. Survey results indicated that patients are willing to tolerate side effects for drugs that can improve long-term health outcomes.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of breast cancer.
Current therapies for advanced or metastatic breast cancer beyond the first-line setting have low response rates and have not been shown to improve OS. Chemotherapy options are more poorly tolerated than endocrine therapy and many available chemotherapy options are administered intravenously, requiring more hospital visits and reliance on institutions. Alpelisib would be the first treatment available specifically for patients with PIK3CA-mutated cancer.
For patients harbouring a PIK3CA mutation, alpelisib would be added to an already established standard of care (SOC) option for the second-line treatment of advanced or metastatic hormone receptor–positive HER2-negative breast cancer (i.e., fulvestrant). Alpelisib would not be used as a first-line treatment given the strong evidence for the use of CDK4/6 inhibitors with endocrine therapy in that setting. Patients with advanced or metastatic hormone receptor–positive HER2-negative breast cancer, activating mutations in the PIK3CA gene (identified using liquid biopsy or tissue testing on archival or newly obtained tumour tissue), good performance status, expected survival of longer than 3 months, and no type 1 diabetes mellitus or uncontrolled type 2 diabetes mellitus would be best suited for treatment with alpelisib plus fulvestrant. Alpelisib with fulvestrant would not be reserved for patients who are intolerant of other treatments or for whom other treatments are contraindicated. In patients with life-threatening visceral organ metastases, chemotherapy would be recommended before considering treatment with alpelisib and fulvestrant. Patients would not be suited for treatment with alpelisib plus fulvestrant if they have poor performance status, have type 1 or uncontrolled type 2 diabetes mellitus, are unable to understand and manage potential toxicities and dosing and monitoring requirements, or are non-compliant with follow-up.
Treatment response is monitored using a combination of clinical examination, laboratory evaluation (markers of organ function with or without tumour markers), and radiographic evaluation. Treatment continues as long as the disease is stable or responding on radiographic scans according to the Response Evaluation Criteria in Solid Tumours (RECIST) criteria. Treatment with alpelisib and fulvestrant should be discontinued if there is disease progression, intolerable or dangerous toxicity (especially uncontrollable hyperglycemia), or an event or development of a comorbidity that adversely impacts performance status or survival (e.g., stroke).
Treatment with alpelisib and fulvestrant would be prescribed by medical oncologists or associated team physicians with expertise in cancer therapies and toxicity management. Patients would be treated on an outpatient basis under medical oncology supervision and fulvestrant injections would be administered in a hospital outpatient clinic or family doctor’s office.
Clinician Group Input
One clinician group submission was received from 6 clinicians with the Breast Medical Oncology group at the Ottawa Hospital Cancer Centre. Due to the small percentage of patients in the pivotal trial who had previously received the current first-line SOC with CDK4/6 inhibitors, opinions were divided on whether it would be appropriate to offer alpelisib to this patient population. While there was mention that patients had been treated recently through the sponsor’s access program, the submission did not describe the clinicians’ experience with alpelisib.
Drug Program Input
There were several questions from officials with the drug plans regarding patient populations that would be suitable for treatment with alpelisib plus fulvestrant, discontinuation of alpelisib or fulvestrant, and PIK3CA mutation testing. Patients were excluded from the pivotal trial for alpelisib if they had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 2 or more, were receiving LHRH agonist for the induction of ovarian suppression, had inflammatory breast cancer, had symptomatic visceral disease, had received prior chemotherapy in the metastatic setting, had received prior fulvestrant treatment, had uncontrolled central nervous system (CNS) metastases, or had type 1 diabetes or uncontrolled type 2 diabetes. According to the clinical experts consulted by CADTH, patients receiving LHRH agonist for the induction of ovarian suppression would be eligible, while patients in the other groups (aside from those with diabetes) could be considered for eligibility on a case-by-case basis or if they met certain other criteria.
The drug plan representatives also wanted to know if alpelisib could be continued as monotherapy if fulvestrant was discontinued or interrupted. The clinical experts indicated that alpelisib could be continued during an interruption but not after discontinuation. Conversely, the drug plans also wanted to know if patients who had to discontinue alpelisib due to intolerance could continue with single-agent fulvestrant. The clinical experts considered it appropriate to continue these patients on single-agent fulvestrant. In response to a related question, the experts also considered it appropriate to permanently discontinue alpelisib after it had been discontinued for more than 4 weeks due to unresolved toxicity. Another drug plan question was whether it would be appropriate to offer patients on chemotherapy with no evidence of progressive disease or intolerance alpelisib plus fulvestrant. The clinical experts did not consider this appropriate as patients doing well on chemotherapy would not be switched to a different therapy.
With regard to PIK3CA mutation testing, representatives for the drug plans asked which patients should be tested for the PIK3CA mutation and when in the course of treatment this testing should occur. According to the clinical experts consulted by CADTH, patients identified as best suited for alpelisib plus fulvestrant treatment minus the criterion of having an activating mutation in the PIK3CA gene should be tested. (Refer to the preceding section regarding input from clinical experts consulted by CADTH for patients identified as best suited for alpelisib plus fulvestrant treatment.) Testing should be performed at diagnosis of de novo metastatic breast cancer, relapse following treatment for early breast cancer, or progression on first-line therapy for advanced or metastatic breast cancer.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The CADTH systematic review identified 1 relevant study, the SOLAR-1 study. The SOLAR-1 study (N = 572) was a placebo-controlled, double-blind, parallel-group randomized controlled trial (RCT) that randomized patients 1:1 to alpelisib 300 mg daily or matching-administration placebo, in combination with fulvestrant 500 mg on day 1, day 15, and day 29, and every 28 days afterwards. Men and postmenopausal women with hormone receptor–positive, HER2-negative advanced or metastatic breast cancer and previous endocrine therapy were randomized within each of 2 cohorts based on PIK3CA mutation status: the PIK3CA mutant cohort and PIK3CA non-mutant cohort. The primary and key secondary outcomes were PFS and OS in the PIK3CA mutant cohort (N = 341). Endocrine therapy with a CDK4/6 inhibitor was not a part of the SOC at the time the study was conducted (enrolment was from 2015 to 2017) and only 20 patients in the PIK3CA mutant cohort had received prior CDK4/6 inhibitor treatment, meeting the reimbursement criteria requested by the sponsor.
Within the PIK3CA mutant cohort, 20 patients were identified as having prior CDK4/6 inhibitor treatment, according to the randomization stratum. Female patients were included only if they were postmenopausal and were not receiving LHRH agonist for the induction of ovarian suppression. In the subgroup with prior CDK4/6 inhibitor treatment, all patients had an ECOG PS of 0 or 1, most patients were White, and most had secondary endocrine resistance. In the entire cohort, most patients were White, had an ECOG PS of 0 (the remaining having a performance status of 1), had 1 or 2 metastatic sites, had 1 line of prior medication therapy, had no prior hormonal therapy in the metastatic setting, and had secondary endocrine resistance.
Efficacy Results
Efficacy results from the SOLAR-1 study PIK3CA mutant cohort in the subgroup of patients with prior CDK4/6 inhibitor treatment are presented in Table 2. At the final PFS analysis at the June 12, 2018, data cut-off date, median PFS was 5.5 months (95% confidence interval [CI], 1.58 months to 16.76 months) in the alpelisib group and 1.8 months (95% CI, 1.68 months to 3.58 months) in the placebo group. The hazard ratio for the alpelisib group versus the placebo group was 0.48 (95% CI, 0.17 to 1.36).
At the final OS analysis at the April 23, 2020, data cut-off date, median OS was 29.8 months (95% CI, 6.67 months to 38.21 months) in the alpelisib group and 12.9 months (95% CI, 2.46 months to 34.60 months) in the placebo group. The hazard ratio for the alpelisib group versus the placebo group was 0.67 (95% CI, 0.21 to 2.18).
Harms Results
Harms are presented for all patients in the PIK3CA mutant cohort in Table 2. Almost all patients reported at least 1 adverse event (AE) (99.4% in the alpelisib group and 90.6% in the placebo group). Most of the AEs occurring in at least 10% of patients of at least 1 treatment group were more common in the alpelisib group compared with the placebo group. All of the AEs reported by more than 20% of patients in the alpelisib group were also more common in the alpelisib group than in the placebo group: hyperglycemia, diarrhea, nausea, rash, decreased appetite, decreased weight, stomatitis, vomiting, fatigue, and alopecia.
Serious AEs (SAEs) were reported in 39.6% of the alpelisib group and 19.9% of the placebo group. The most common SAEs were hyperglycemia (10.1% in the alpelisib group and none in the placebo group), osteonecrosis of the jaw (3.6% in the alpelisib group and none in the placebo group), stomatitis, acute kidney injury, and rash (2.4% in the alpelisib group and none in the placebo group for the preceding 3 SAEs).
Withdrawals from treatment due to AEs were more common in the alpelisib group (27.2%) versus the placebo group (5.8%). The most common AEs leading to discontinuation were reported in the alpelisib group alone: hyperglycemia (6.5%), rash (4.7%), and diarrhea (3.6%).
On-treatment deaths up to 30 days after the last dose of study treatment occurred in 4.1% of the alpelisib group and 5.8% of the placebo group. The most common cause of on-treatment death was breast cancer (3.6% in the alpelisib group and 4.1% in the placebo group) and other causes of on-treatment death were reported for 1 patient each.
The following notable harms identified in the systematic review protocol occurred in more than 10% of at least 1 treatment group and were more common in the alpelisib group: hyperglycemia, diarrhea, nausea, rash, vomiting, and maculopapular rash.
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Outcome | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Patients with prior CDK4/6 inhibitor treatment, full analysis set | Alpelisib + fulvestrant N = 9 | Placebo + fulvestrant N = 11 |
PFS: June 12, 2018, data cut-off date (primary analysis) | ||
Median, months (95% CI)a | 5.5 (1.58 to 16.76) | 1.8 (1.68 to 3.58) |
HR (95% CI)b | 0.48 (0.17 to 1.36) | Reference group |
OS: April 23, 2020, data cut-off date (final analysis) | ||
Median, months (95% CI)a | 29.8 (6.67 to 38.21) | 12.9 (2.46 to 34.60) |
HR (95% CI)b | 0.67 (0.21 to 2.18) | Reference group |
All patients, safety analysis set | Alpelisib + fulvestrant N = 169 | Placebo + fulvestrant N = 171 |
Harms, n (%) | ||
AEs | 168 (99.4) | 155 (90.6) |
SAEs | 67 (39.6) | 34 (19.9) |
WDAEs (from study treatment) | 46 (27.2) | 10 (5.8) |
Deaths (up to 30 days after last dose of study treatment) | 7 (4.1) | 10 (5.8) |
Notable harms, n (%) | ||
Hyperglycemia | 113 (66.9) | 15 (8.8) |
Diarrhea | 97 (57.4) | 20 (11.7) |
Nausea | 82 (48.5) | 35 (20.5) |
Rash | 69 (40.8) | 12 (7.0) |
Vomiting | 46 (27.2) | 17 (9.9) |
Rash maculopapular | 25 (14.8) | 1 (0.6) |
Hypersensitivity | 6 (3.6) | 0 |
Glycated hemoglobin, increased | 5 (3.0) | 0 |
Blood glucose increased | 3 (1.8) | 1 (0.6) |
Pneumonitis | 2 (1.2) | 0 |
Erythema multiforme | 2 (1.2) | 0 |
Glucose urine present | 2 (1.2) | 0 |
Anaphylactic reaction | 1 (0.6) | 0 |
Drug hypersensitivity | 1 (0.6) | 0 |
Stevens-Johnson syndrome | 1 (0.6) | 0 |
Diabetes mellitus | 1 (0.6) | 1 (0.6) |
Ketoacidosis | 1 (0.6) | 0 |
Type 2 diabetes mellitus | 1 (0.6) | 0 |
AE = adverse event; CDK4/6 = cyclin-dependent kinase 4 and 6; CI = confidence interval; HR = hazard ratio; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aUsing Kaplan-Meier estimation.
bCox proportional hazards model, stratified by the presence of lung and/or liver metastases, performed in the full analysis set.
Source: Interim Clinical Study Report (2018)1 and final Clinical Study Report for the SOLAR-1 study (2020).2
Critical Appraisal
No relevant conclusions can be drawn regarding PFS and OS in patients treated with alpelisib and fulvestrant versus placebo and fulvestrant because the SOLAR-1 study was not designed to test hypotheses in the subgroup of patients with prior CDK4/6 inhibitor treatment and did not include outcomes in this subgroup in the statistical testing hierarchy. Only the results in this small subgroup can inform comparative efficacy in the patient population targeted by the sponsor’s reimbursement request, since the results in the entire PIK3CA mutant cohort cannot be generalized to the relevant patient population.
Other Relevant Evidence
Description of Studies
There were 2 additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review. The BYLieve study, a non-comparative cohort study, included 1 cohort of patients treated with alpelisib and fulvestrant that matched the relevant patient population. In a separate observational study, the relevant cohort of the BYLieve study was compared with a database-derived cohort treated with non-alpelisib SOC following propensity score weighting.
Non-Comparative Cohort Study
The BYLieve study assigned patients to 1 of 3 cohorts based on their most recent anticancer therapy. Of the 3 cohorts, cohort A (N = 127) was relevant to the review. Cohort A included patients with hormone receptor–positive, HER2-negative advanced or metastatic breast cancer and a confirmed PIK3CA mutation who had received any CDK4/6 inhibitor plus any AI as their immediate prior treatment. These patients were assigned to receive alpelisib plus fulvestrant at the same dosages as in the SOLAR-1 study. The primary end point in the BYLieve study was the proportion of patients who were alive without disease progression at 6 months by local investigator assessment using the criteria of RECIST Version 1.1 (RECIST 1.1). The outcomes of PFS and OS as well as safety data were also evaluated in the BYLieve study.
Progression and Survival Results
As of the data cut-off date, 61 of 121 (50.4%) patients in the modified full analysis set or mFAS (N = 121) were alive without progressive disease per investigator assessment at 6 months (95% CI, 41.2% to 59.6%). The study met the primary objective for cohort A because the lower bound of the 95% CI was greater than 30%. The median PFS by investigator assessment was 7.3 months (95% CI, 5.6 months to 8.3 months). The PFS rates by investigator assessment at 6 months and 12 months were 54.1% (95% CI, 44.3% to 62.9%) and 27.3% (95% CI, 17.6% to 37.8%), respectively.
The median OS was 17.3 months (95% CI, 17.2 months to 20.7 months). The OS rates at 6 months and 12 months were 91.9% (95% CI, 84.9% to 95.7%) and 75.2% (95% CI, 62.5% to 84.2%), respectively. The sponsor indicated in the BYLieve Clinical Study Report that OS data should be interpreted with caution due to the proportion of patients alive and to ongoing follow-up at the time of the data cut-off date.
Harms Results
Almost all (99.2%) of patients experienced at least 1 treatment-emergent AE. The most common AEs (≥ 20%) were diarrhea (59.8%), hyperglycemia (58.3%), nausea (45.7%), fatigue (29.1%), decreased appetite (28.3%), rash (28.3%), stomatitis (26.8%), and vomiting (23.6%). Overall, 26.0% of patients experienced an SAE. The most common SAEs were hyperglycemia (5.5%), maculopapular rash (3.1%), dyspnea (2.4%), pleural effusion (2.4%), abdominal pain (1.6%), and hematemesis (1.6%). The most common AEs leading to discontinuation of study treatment were rash (3.9%), colitis, hyperglycemia, urticaria, and vomiting (1.6% each). As of the data cut-off date, 7 (5.5%) patients had died during study treatment or within 30 days of the last dose of study drug, and 4 of these on-treatment deaths were attributed to breast cancer.
The following notable harms were reported: hyperglycemia (58.3%), hypersensitivity and anaphylactic reactions (10.2%), diarrhea (59.8%), nausea (45.7%), rash (28.3%), vomiting (23.6%), maculopapular rash (14.2%), pneumonitis (0.8%), and severe cutaneous skin reactions (0.8%).
Critical Appraisal
The BYLieve study is unable to inform the efficacy of alpelisib plus fulvestrant versus a relevant comparator due to its non-comparative study design. There was also no statistical hypothesis testing in the relevant outcomes of interest, PFS and OS.
Observational Study
The observational study compared cohort A from the BYLieve study with a real-world cohort derived from the Flatiron Clinico-Genomic Database. Cohort A from the BYLieve study (N = 120), whose patients received alpelisib plus fulvestrant following treatment with a CDK4/6 inhibitor plus AI, was compared to the Flatiron cohort (N = 95), whose patients received non-alpelisib SOC following treatment with a CDK4/6 inhibitor and non-fulvestrant endocrine therapy. The outcome PFS was compared between the cohorts following weighting of the Flatiron cohort based on propensity scores.
Efficacy Results
Following propensity score weighting to estimate the average treatment effect on those treated, median PFS was 3.7 months (95% CI, 3.1 months to 6.1 months) in the Flatiron cohort and 7.3 months (95% CI, 5.6 months to 8.3 months) in the BYLieve cohort with a P value of 0.040 for the log-rank test. The weighted hazard ratio for PFS in the BYLieve cohort versus the Flatiron cohort was 0.62 (95% CI, 0.44 to 0.85; P = 0.002). The observational study included sensitivity analyses assessing the sensitivity of the results to the form of confounding adjustment — namely, greedy matching and exact matching. The results of those analyses were not meaningfully different from the primary analysis results. No sensitivity analysis was performed on the assumption of no unmeasured confounding.
Harms Results
Harms were not assessed in the observational study.
Critical Appraisal
Overall, there remains a great deal of uncertainty regarding the efficacy of alpelisib in comparison to the SOC due to the inherent limitations of observational data. While the adjustment approaches taken in this study may have adequately balanced on observable prognostic factors categorized as they were, bias in the efficacy estimate due to selection bias, measurement error, unmeasured confounding, and residual confounding cannot be ruled out. No attempts were made to assess or estimate the possible magnitude of such bias.
Conclusions
No conclusions could be drawn from the SOLAR-1 and BYLieve studies regarding the comparative efficacy or effectiveness of alpelisib plus fulvestrant versus any relevant comparator in patients with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor. Neither study was designed to draw conclusions on comparative efficacy in this patient population. The sponsor-submitted observational study comparing a cohort from the BYLieve study that received alpelisib plus fulvestrant to a database-derived cohort that received a variety of non-alpelisib SOC therapies reported PFS results in favour of alpelisib. Methodological limitations in the observational study, including a high likelihood of residual confounding that may have led to bias in favour of alpelisib and differences in the methods used to determine PFS, contributed a substantial degree of uncertainty to the effect estimate. Considering the evidence in its entirety, the magnitude of any benefit associated with alpelisib plus fulvestrant in the relevant patient population remains unclear. In the SOLAR-1 and BYLieve studies, alpelisib treatment was associated with hyperglycemia, diarrhea, nausea, rash, decreased appetite, stomatitis, vomiting, and fatigue. Although the AEs reported in the studies can be medically managed, the percentages of patients who discontinued treatment due to AEs in the studies suggest that a large proportion of patients may not be able to remain on treatment with alpelisib due to AEs and/or subsequent side effects from treatments to manage these.
Introduction
Disease Background
Breast cancer is the most commonly diagnosed cancer in Canadian females. According to Canadian data from 2011 to 2015, more than 80% of breast cancers in Canadian females were diagnosed at an early stage (stage I or stage II).3 For the purposes of this review, advanced breast cancer refers to locoregionally advanced breast cancer not amenable to curative therapy. Metastatic breast cancer occurs when cancer spreads to other parts of the body. Most patients with metastatic breast cancer are those with relapse following treatment for early breast cancer as opposed to those with metastatic breast cancer at diagnosis (de novo cases).4 Independent of stage, there are 3 main subtypes of breast cancer, based on expression of hormone (estrogen and/or progesterone) receptors and overexpression of HER2. The most common subtype of breast cancer is hormone receptor–positive, HER2-negative breast cancer.
It was estimated that in 2020, there would be 27,200 new cases of breast cancer in Canada, with age-standardized incidence rates of 1.1 per 100,000 males and 128.2 per 100,000 females.5 It was also estimated that there would be 5,100 deaths from breast cancer, with age-standardized mortality rates of 0.3 per 100,000 males and 22.0 per 100,000 females.5 In an analysis of females diagnosed with breast cancer from 2012 to 2016 in the Ontario Cancer Registry, 64.8% of patients with a known subtype had hormone receptor–positive HER2-negative breast cancer, with an estimated annual incidence rate of 97 to 105 per 100,000 females.6 This estimate is in line with an estimate from registries in the US in patients diagnosed from 2010 to 2013 (66.6%).7 The percentages of Ontario patients with hormone receptor–positive, HER2-negative breast cancer who had stage III and stage IV disease at diagnosis were 12% and 3.7%, respectively.6 Patients with stage IV, hormone receptor–positive, HER2-negative breast cancer at diagnosis in the Ontario study had an estimated median survival of 35.2 months, though percentages of patients with 5-year survival ranged from 24.0% to 94.7%, depending on disease stage at diagnosis.6
Standards of Therapy
Although there are no curative treatments for advanced or metastatic breast cancer, there are multiple systemic therapies available for the disease. According to input from clinicians consulted by CADTH for the purpose of this review, standard first-line therapy for advanced or metastatic hormone receptor–positive, HER2-negative breast cancer is a CDK4/6 inhibitor with an AI. An LHRH receptor agonist is also given for ovarian suppression, depending on menopausal status. Exceptions to the use of a CDK4/6 inhibitor might include patients with a very low burden of disease, patients with significant comorbidities or contraindications to CDK4/6 inhibitors, and patients who prefer single-agent endocrine treatment for reasons such as desire for less frequent monitoring or concern about side effects. Patients whose disease recurs while on or shortly after stopping adjuvant AI therapy are considered resistant and are frequently offered CDK4/6 inhibitor with fulvestrant instead of an AI. Upon disease progression, second-line therapy options include a different single-agent AI, a single-agent fulvestrant, an investigational therapy in a clinical trial, chemotherapy (commonly single-agent capecitabine or a taxane), combined everolimus and exemestane, and tamoxifen. Coverage of fulvestrant and everolimus plus exemestane is inconsistent across jurisdictions in Canada. Aggressive disease progression (e.g., disease not responding to first-line therapy or significant visceral metastases) is treated with chemotherapy. Despite the number of options beyond first-line therapy, there is no high-quality evidence for prolongation of survival with these therapies. The SOC outlined by the clinicians consulted by CADTH is consistent with a recent international consensus guideline published by the European School of Oncology (ESO) and the European Society for Medical Oncology (ESMO) for the management of advanced breast cancer.8
The clinicians consulted by CADTH for this review described the following treatment goals: improving OS, delaying cancer progression, maintaining or improving quality of life, preventing or improving cancer-related symptoms (e.g., pain, dyspnea, fatigue), maintaining or improving organ function, maintaining patient independence, increasing ability to maintain employment, and delaying the initiation of chemotherapy. Input from patient groups for this review reported similar treatment goals. Patients indicated that they wanted treatments that delayed progression of their disease, prolonged life without sacrificing quality of life, reduced symptoms, and minimized the risk of side effects while stabilizing their disease. Patients were willing to tolerate side effects if treatments improved long-term health outcomes.
Drug
Alpelisib is an oral PI3K inhibitor that, in combination with fulvestrant, is indicated for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen. Alpelisib 300 mg is taken daily on a continuous basis along with IM fulvestrant 500 mg on day 1, day 15, and day 29, and every 28 days thereafter. Dose reductions for alpelisib are permitted to 250 mg or 200 mg daily for the management of adverse drug reactions.
The class IA isoforms of PI3K play a role in the control of cell growth, proliferation, metabolism, and migration via the PI3K/protein kinase B/mechanistic target of rapamycin pathway.9 There is some evidence to suggest that the presence of mutations in the gene encoding the p110 alpha catalytic subunit of PI3K (PIK3CA) is associated with worse OS and resistance to chemotherapy in patients with metastatic breast cancer.9 Alpelisib is the first approved therapy in Canada for patients with PIK3CA-mutated, hormone receptor–positive, HER2-negative advanced or metastatic breast cancer.
Alpelisib has not been previously reviewed by CADTH. The sponsor’s reimbursement request is for alpelisib in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor. The reimbursement request differs from the Health Canada indication in that it specifies that patients must have received CDK4/6 inhibitor with a previous endocrine-based regimen.
Table 3: Key Characteristics of Alpelisib, Fulvestrant, Capecitabine, and Combined Everolimus and Exemestane
Characteristic | Alpelisib | Fulvestrant | Capecitabine | Everolimus and exemestane |
|---|---|---|---|---|
Mechanism of action | Inhibits PI3K (predominantly against PI3K alpha) | ER antagonist | Prodrug metabolized to the cytotoxic moiety 5-FU | mTOR inhibitor (downstream of the PI3K/AKT pathway) Aromatase inactivator |
Indicationa | In combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen | Locally advanced or metastatic breast cancer in postmenopausal women, regardless of age, who have disease progression following prior antiestrogen therapy | Treatment of advanced or metastatic breast cancer after failure of standard therapy including a taxane, unless therapy with a taxane is clinically contraindicated | Everolimus: Treatment of postmenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer in combination with exemestane after recurrence or progression following treatment with letrozole or anastrozole Exemestane: Treatment of advanced breast cancer in women with natural or artificially induced postmenopausal status whose disease has progressed following antiestrogen therapy |
Route of administration | Oral | IM | Oral | Oral |
Recommended dosage | Alpelisib tablets 300 mg once daily on a continuous basis with fulvestrant 500 mg IM on day 1, day 15, and day 29, and every 28 days thereafter Dose reduction allowed to alpelisib 250 mg or 200 mg daily for the management of adverse drug reactions | 500 mg on day 1, day 15, and day 29, and then every 28 days thereafter | 1,250 mg/m2 twice daily (morning and evening) for 14 days, followed by a 7-day rest period Dose modifications recommended for management of AEs | Everolimus 10 mg once daily and exemestane 25 mg once daily Everolimus dose modifications recommended to manage AEs, in patients with hepatic impairment, and for drug-drug interactions (CYP3A4 and/or PgP inhibitors) Exemestane 25 mg once daily |
Serious adverse effects or safety issues | Hypersensitivity, severe cutaneous reactions, hyperglycemia, pneumonitis, severe diarrhea, and osteonecrosis of the jaw | Liver inflammation (elevated transaminase, bilirubin, and alkaline phosphatase) and hypersensitivity reactions, including angioedema and urticaria | Acute renal failure secondary to dehydration, sudden death due to cardiotoxicity, severe skin reactions, severe toxicity associated with 5-FU metabolite attributed to a deficiency of DPD activity | Everolimus Drug-drug interactions with strong inducers of CYP3A4 and/or PgP, concomitant ACE inhibitor therapy and increased risk for angioedema, stomatitis including mouth ulceration, coagulation or bleeding anomalies with concomitant use of drugs that affect platelet function or that can increase risk of hemorrhage, and in patients with history of bleeding disorders, increased risk of infection, non-infectious pneumonitis, hypersensitivity reactions, deep vein thrombosis, and pulmonary embolism events Exemestane Ischemic cardiovascular events, hypercholesterolemia, gastric ulcer, reduced bone mineral density, severe cutaneous reactions |
5-FU = 5-fluorouracil; ACE = angiotensin-converting enzyme; AE = adverse event; AKT = protein kinase B; DPD = dihydropyrimidine dehydrogenase; ER = estrogen receptor; HER2 = human epidermal growth factor receptor 2; IM = intramuscular; mTOR = mammalian target of rapamycin; PgP = P-glycoprotein; PI3K = phosphatidylinositol 3-kinase.
aHealth Canada–approved indication.
Source: Product monographs for Piqray (2020),10 Faslodex (2019),11 Xeloda (2021),12 Afinitor (2021),13 and Aromasin (2018).14
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
Three patient groups provided input for this review: the CBCN, Rethink Breast Cancer, and CanCertainty.
The CBCN is a patient-directed, national health charity that focuses on ensuring the best quality of care for all Canadians affected by breast cancer through the promotion of information, education, and advocacy activities. The CBCN provided information gathered from 2 online surveys conducted in 2017 and 2012. A total of 180 patients living with breast cancer responded to the 2017 survey. The submission only included data from a subset of 90 Canadian respondents with metastatic, hormone receptor–positive, HER2-negative breast cancer. A total of 71 patients and 16 caregivers responded to the 2012 survey. In addition, the CBCN conducted a telephone interview with 1 patient in May 2021.
Rethink Breast Cancer is a registered charity and the organization’s mission is to empower young people worldwide who are concerned about and affected by breast cancer through education, support, and advocacy. Rethink Breast Cancer conducted an online survey from March 31 to April 8, 2021, to inform its submission. A total of 24 postmenopausal women diagnosed with hormone receptor–positive, HER2-negative, advanced or metastatic breast cancer with a PIK3CA mutation completed the survey (4 of the women were from Canada). Six of these patients also participated in telephone interviews.
The CanCertainty Coalition consists of 30 Canadian patient groups, cancer health charities, and caregiver organizations from across the country. It works with oncologists and cancer care professionals to improve the affordability and accessibility of cancer treatment. CanCertainty developed its submission based on published reports relating to breast cancer and oral cancer drugs.
Disease Experience
The CBCN reported that the physical impact of metastatic breast cancer is variable across individuals and has a significant or debilitating impact on patients’ quality of life. In the 2012 survey, the vast majority of patients reported a significant/debilitating or some/moderate impact on their quality of life, due to the symptoms of fatigue, insomnia, and pain. The CBCN also reported that there are multiple social impacts of living with metastatic breast cancer. Metastatic breast cancer restricts an individual’s employment and career, their ability to care for children and dependents, and their ability to be social and meaningfully participate in their community. Most patient respondents also reported restrictions to their ability to exercise, pursue hobbies and interests, and spend time with loved ones. Other experiences identified by patients included guilt, the feeling of being a burden on caregivers, fear of death, poor body image, not knowing what functionality will be lost, fear of the impact of cancer and the loss of a parent on children, not knowing what will happen to children, the loss of support of loved ones, as well as marital stress/loss of fidelity and affection from their husband.
Experience With Treatment
Respondents to the Rethink Breast Cancer survey had experience with a variety of treatments, including fulvestrant, letrozole, palbociclib, exemestane, anastrozole, tamoxifen, and capecitabine. All 24 respondents had experience with alpelisib in combination with fulvestrant, and 20 respondents were treated with a CDK4/6 inhibitor before receiving alpelisib. Most respondents had undergone multiple lines of treatment and reported a wide range of outcomes and side effects. The most commonly reported side effects for breast cancer treatments overall were fatigue (100%), diarrhea (83%), loss of appetite (75%), nausea (54%), and headache (46%). Fatigue, diarrhea, and hyperglycemia were identified as the most difficult-to-tolerate side effects of previous treatments. Most respondents (86%) did not report any difficulty accessing treatment. An additional impact, identified in the CBCN submission, is difficulty associated with accessing quality child care during cancer treatment.
A total of 18 respondents to the Rethink Breast Cancer survey matched the requested reimbursement criteria for the present review and the responses from these patients with regard to experience with alpelisib were summarized in the submission. At the time of the survey, 12 of these patients were still receiving alpelisib, 5 stopped receiving it because it did not control their cancer, and 1 stopped receiving it because she could not tolerate the side effects. Compared to other treatments received, most patients indicated that the drug’s side effects were the same (33%) or worse (28%), though some patients (39%) indicated that the side effects were better. Most patients experienced diarrhea (89%), reduced appetite (78%), weight loss (72%), and alopecia (67%) while receiving alpelisib. While not reported as frequently, hyperglycemia was highlighted during patient interviews as being especially hard to manage. Several respondents reported that dose reductions made an important difference in helping them manage the side effects. Some comments from patients regarding the side effects included the following:
“I am tolerating, but it is difficult.”
“Important to find effective ways to manage SE [side effects] right away, especially in the first 4 months when there are so [many] SE that are pretty overwhelming.”
“Piqray worked for almost 18 months and was tough but manageable. I did not have any of the major side effects like blood sugar issues or the rash. I got itchy but that was controlled with antihistamines. I do lose my sense of taste and appetite but that was minor and manageable, although I did lose weight.”
The CBCN conducted a phone interview with a patient from the US who had direct experience with the treatment under review. This patient reported that she was personally satisfied with the treatment’s impact on her metastatic disease. The patient reported that she experienced side effects from alpelisib (hyperglycemia, nausea, and fatigue) but they were manageable. While the patient’s experience was in line with the experiences outlined in the Rethink Breast Cancer submission, the impact of fatigue on productivity and activity levels was highlighted. The patient also indicated that alpelisib was preferable compared to other treatments such as chemotherapy. Some comments from this patient included the following:
“Piqray is actually keeping the cancer under control even better than Ibrance did…. We looked at a couple of other things, my doctor and I, but because I had the mutation, the whole idea of precision medicine and focusing on the weak spots in the cancer specifically, that was why my doctor felt like it would be the best way to go.”
“When I was diagnosed, I had a super high disease load. So I went from so much disease to stability on Ibrance, but there was still a lot of active mets. And now I have one active mets. So it really was effective on the mets.”
“But outside of the hyperglycemia, pretty intense fatigue. For the hyperglycemia, I’m on Jardiance, and that’s kept the hyperglycemia under control. I do get a fair bit of nausea as well, and I’ve got a variety of medications that I take at different times of the day to keep the nausea under control. The fatigue: I drink a lot of coffee, and I’ve had to adjust my activity levels. The fatigue has been something that has been a side effect of every medication I’ve been on, so I feel that that’s a side effect that I’ve become a little bit more able to handle.”
Testing for the PIK3CA mutation was described in the Rethink Breast Cancer submission. Most respondents (79%) had received genomic testing and this was performed by blood test, tumour biopsy, or both. It should be noted that only 4 of the 24 respondents were from Canada. One of the Canadian patients interviewed described wait times of about 3 weeks for a biopsy and 4 weeks for the results, during which she experienced both excitement about having better information and anxiety over not having a treatment plan. The biopsy procedure was described as somewhat painful.
The patient groups reported significant financial challenges associated with treatments for metastatic breast cancer. The financial burden associated with living with advanced breast cancer includes loss of income and substantial costs associated with treatment and disease management. Patients indicated that the cost of medication, the cost of alternative treatments (e.g., massage, physiotherapy) to manage symptoms and side effects, and the time required to travel to treatment had a significant or debilitating impact on their quality of life. Other financial challenges were also reported, including not qualifying for insurance at work, the inability to change employers due to loss of insurance, and the prohibitive cost of new treatment options. CanCertainty noted that reimbursement of oral cancer drugs is not equal across jurisdictions in Canada. As a result, patients who do not have adequate insurance may have to pay out of pocket for medication and/or apply to funding assistance programs, which can take time and delay access to treatment. The groups also considered that for patients who do have private insurance, they may still have copayments, deductibles, and annual or lifetime caps that increase the financial burden on patients and families. Comments from patients included the following:
“Many of the next step treatments are very expensive [and not covered by government programs] and it is a HUGE struggle to get [coverage]. […] When dealing with an incurable disease the last thing you want to have to do is spend time on a letter writing campaign to argue about whether or not you should receive the drugs [recommended by your physician]. At about $1500.00 a week, I don't know many who can afford that.”
“When I turn 65 I will no longer have private insurance. I will not be able to afford the medication I currently take never mind any future medication that I may require.”
“I worry that in the future, a drug that may work for me won’t be accessible to me based on provincial formulary.”
Improved Outcomes
The patient groups indicated that the following measures of effectiveness were most important to respondents: PFS, OS, quality of life, and adverse effects. Reducing symptoms of the disease was also noted as an important outcome. Overall, patients indicated that controlling disease progression was most important to them. Patients indicated that they wanted treatments that delayed progression of their disease, prolonged life without sacrificing quality of life, and minimized the risk of side effects while stabilizing their disease.
The CBCN reported that it is very important for patients to have good quality of life when receiving treatment and the patients it speaks to acknowledge the importance of having the energy to attend their children’s activities and to spend time with family and friends. However, survey results indicated that patients are willing to tolerate side effects for drugs that can improve long-term health outcomes. Patients indicated that any treatment that gives people additional months or years of survival is beneficial. Many patient respondents indicated that treatment side effects such as fatigue, nausea, depression, problems with concentration, diarrhea, hair loss, and insomnia would be acceptable if the treatment extended PFS by approximately 6 months. Comments from patients included the following:
“Risks vs benefits. Some adverse side effects are worth the benefits for short term.”
“I can deal with pretty significant side effects if the outcome of treatment is optimistic.”
“I have a seven-year-old and nine-year-old. I’m not ready to leave them.”
“I will tolerate pretty much anything within reason in order to find and stay on a drug that keeps the tumour burden low.”
In addition to clinical outcomes, patients indicated that affordability and ease of access is important. Patients also expressed the need for personal choice regarding their treatment options. Patients want to be part of the decision-making process regarding which treatments they receive.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of breast cancer.
Unmet Needs
Current therapies for advanced or metastatic breast cancer beyond the first-line setting have low response rates and have not been shown to improve OS. Chemotherapy options are more poorly tolerated than endocrine therapy, causing nausea, vomiting, alopecia, fatigue, cytopenia, and sometimes dangerous adverse reactions. In addition, many available chemotherapy options are administered intravenously, requiring more hospital visits and reliance on institutions. The presence of PIK3CA gene mutations may be associated with poorer prognosis and resistance to endocrine therapy and alpelisib would be the first treatment available specifically for patients with PIK3CA-mutated cancer.
Place in Therapy
For patients harbouring a PIK3CA mutation, alpelisib would be added to an already established SOC option for the second-line treatment of advanced or metastatic hormone receptor–positive, HER2-negative breast cancer (i.e., fulvestrant). Alpelisib would not be used as a first-line treatment given the strong evidence for the use of CDK4/6 inhibitors with endocrine therapy in that setting. Additionally, alpelisib with fulvestrant would be a preferred treatment for patients with PIK3CA-mutated cancer and good performance status, as opposed to being reserved for patients who are intolerant of other treatments or for whom other treatments are contraindicated. Subsequent lines of therapy would include those previously used after fulvestrant monotherapy, including sequential single-agent chemotherapy drugs or investigational therapies in clinical trials.
If a patient received single-agent AI treatment in the first-line setting, it would be appropriate to recommend fulvestrant with a CDK4/6 inhibitor rather than fulvestrant with alpelisib. There is a lack of evidence from RCTs showing superiority of 1 approach over the other and the side-effect profiles of CDK4/6 inhibitors are favourable overall compared with alpelisib. In patients with life-threatening visceral organ metastases, chemotherapy would be recommended before considering treatment with alpelisib and fulvestrant.
Patient Population
Patients with advanced or metastatic hormone receptor–positive, HER2-negative breast cancer, activating mutations in the PIK3CA gene, good performance status (ECOG PS status of 0 or 1), expected survival of longer than 3 months, and no type 1 diabetes mellitus or uncontrolled type 2 diabetes mellitus would be best suited for treatment with the drug under review. As discussed earlier, patients should have progressed on first-line endocrine therapy and previously received treatment with a CDK4/6 inhibitor in the metastatic setting. Patients must be postmenopausal or rendered functionally postmenopausal. The presence of visceral metastases would not affect a patient’s eligibility.
A patient would not be suited for treatment with the drug under review if they have poor performance status, have type 1 diabetes mellitus or uncontrolled type 2 diabetes mellitus, are unable to understand and manage potential toxicities and dosing and monitoring requirements, or are non-compliant with follow-up. As mentioned before, patients with rapidly progressive visceral metastases may be better served by chemotherapy.
Patients with PIK3CA-mutated cancers are identified using liquid biopsy or tissue testing on archival or newly obtained tumour tissue. However, routine PIK3CA mutation testing is not part of the current SOC. PIK3CA mutation testing can be performed in commercial laboratories and academic hospitals in Canada, but it is not routinely funded or accessible. Ideally, testing would be offered to patients with advanced or metastatic hormone receptor–positive, HER2-negative breast cancer who are best suited for treatment with alpelisib (aside from the requirement for activating mutations in the PIK3CA gene). Since prevention of cancer symptoms is an important goal in the treatment of advanced or metastatic breast cancer, the presence of symptoms is not required to initiate treatment in this setting. In the absence of treatment, the disease will invariably progress and cause symptoms.
Some aspects of PIK3CA mutation testing have not been well characterized, such as concordance between methods (next-generation sequencing (NGS) versus polymerase chain reaction (PCR) methods; liquid biopsy versus tissue biopsy), concordance between tissue source (primary tumour versus metastasis), and stability of results over time. Evidence of concordance between liquid biopsy and tissue biopsy results has been summarized in a recent systematic review.15
Assessing Response to Treatment
Treatment response is monitored using a combination of clinical examination, laboratory evaluation (markers of organ function with or without tumour markers), and radiographic evaluation. Radiographic scans are initially performed at least every 3 months and toxicity or symptom assessments are initially performed every 2 weeks to 4 weeks or as needed. Clinical and laboratory evaluations are performed before each treatment cycle and may detect disease progression ahead of scheduled imaging. Treatment continues as long as the disease is stable or responding on radiographic scans according to the RECIST criteria.
Any of the following would also be considered a clinically meaningful response to treatment: improved survival (overall or progress-free), stabilization or reduction in the frequency or severity of cancer-related symptoms (e.g., pain, dyspnea), improvement in organ function (e.g., bone, liver, lung), maintenance or improvement of performance status and ability to perform activities of daily living, and delay in initiation of chemotherapy (especially IV chemotherapy).
Discontinuing Treatment
Treatment with alpelisib and fulvestrant should be discontinued if there is disease progression, intolerable or dangerous toxicity (especially uncontrollable hyperglycemia), or an event or development of a comorbidity that adversely impacts performance status or survival (e.g., stroke). Patient preference would also dictate treatment discontinuation.
Prescribing Conditions
Treatment with alpelisib and fulvestrant would be prescribed by medical oncologists or associated team physicians with expertise in cancer therapies and toxicity management. Patients would be treated on an outpatient basis under medical oncology supervision, which can include help from family physicians and/or nurse practitioners with additional training in oncology. Fulvestrant injections would be administered in a hospital outpatient clinic or family doctor’s office.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
One clinician group submission was received from 6 clinicians with the Breast Medical Oncology group at the Ottawa Hospital Cancer Centre, an academic teaching hospital centre in Ontario. The group offers routine SOC treatments and access to promising treatments in phase I to phase III clinical trials. The group serves a large referral base from the Champlain Local Health Integration Network in Ontario.
Unmet Needs
The most important treatment goals in this disease setting are to maintain or improve quality of life compared with currently available treatments, delay the progression of cancer, improve or maintain organ function, reduce cancer symptoms, and allow patients to be treated with oral therapy with a view to allowing patients to remain gainfully employed and independent, and preventing institutionalization.
Current therapies have the following shortcomings in terms of achieving treatment goals: disappointing responses to second-line therapy and later therapies and no improvement in OS, patients becoming more rapidly refractory to current therapies, and chemotherapy options that are poorly tolerated due to numerous side effects and sometimes dangerous adverse reactions.
Place in Therapy
Alpelisib with fulvestrant, the latter of which is an already established SOC second-line therapy, would be a paradigm-changing SOC option in the second line for patients with a PIK3CA mutation. It would replace fulvestrant monotherapy and lines of therapy following alpelisib with fulvestrant would be the same as those previously used following fulvestrant monotherapy. The improvement in PFS found in the SOLAR-1 study of 5 months would be considered worthwhile by patients and clinicians.
It would be appropriate to use standard first-line therapy, which includes CDK4/6 inhibitors, before using alpelisib plus fulvestrant. Chemotherapy would be recommended beforehand in patients with life-threatening visceral organ metastases.
Patient Population
The patients best suited for treatment with alpelisib and fulvestrant are those meeting the eligibility criteria for the SOLAR-1 study and with activating mutations of PIK3CA. Patients should also have ECOG PS of 0 to 1 with expected survival of more than 3 months, should not have diabetes mellitus type 1 or uncontrolled type 2 (glycated hemoglobin of greater than 6.4%), and should be able to understand and comply with the specific safety, monitoring, and side-effect management issues associated with the drug. Patients would be eligible with or without visceral metastases, with brain metastases if controlled or treated, and with ovarian function suppression to achieve postmenopausal state if not already postmenopausal. Patients with an ECOG PS of 2 would potentially be considered for treatment with alpelisib and fulvestrant if they were fit with aggressive disease but unfit for chemotherapy. Alpelisib would uniquely address the needs of patients with relevant PIK3CA mutations progressing after standard first-line therapy. Aside from activating PIK3CA mutations, patient subgroups and other clinical factors cannot be used to select patients who are likely to derive the greatest benefit from treatment with alpelisib and fulvestrant.
Patients least suitable for treatment with alpelisib and fulvestrant are those with ECOG PS of 2 to 4 and those with diabetes mellitus type 1 or uncontrolled type 2. One of the clinicians felt that, without evidence, patients who had first-line CDK4/6 inhibitor treatment would not be suitable for treatment with alpelisib, while most of the clinicians felt that access and funding for alpelisib should not be withheld. There was mention of further trials under way for assessing the efficacy of alpelisib after first-line treatment with CDK4/6 inhibitors.
Access to molecular testing for PIK3CA mutation status would be required to identify patients for treatment. Testing is not challenging but is not routinely funded or accessible currently in Canada. It can be done by commercial laboratories or in academic hospital laboratories if funded.
Assessing Response to Treatment
Response to treatment is determined based on symptoms, laboratory markers, and radiographic scans and tumour measurements. Scans are usually initially performed at least every 3 months and treatment is continued if disease is stable or responding radiographically according to RECIST criteria. Any of the following would be considered a clinically meaningful response to treatment: reduction in the frequency or severity of symptoms, improvement of organ function, stabilization of symptoms, and maintenance or improvement of performance status. Toxicity or symptom assessments would be performed every 2 weeks to 4 weeks early in treatment or as needed.
Discontinuing Treatment
The following factors should be considered when deciding to discontinue treatment: disease progression, intolerable or dangerous toxicity (especially uncontrolled grade 3 or grade 4 hyperglycemia, rash, or diarrhea), and patient preference or refusal.
Prescribing Conditions
Treatment should only be prescribed by certified medical oncologists or associated team physicians with expertise in cancer therapies and toxicity management. Treatment would take place in the community setting (for alpelisib), and in hospital outpatient clinics or family doctors’ offices (for fulvestrant).
Additional Considerations
Substantial discordance of opinion was noted regarding treatment of patients with prior first-line CDK4/6 inhibitor therapy. Some clinicians were of the opinion that the benefits of alpelisib in this population were uncertain and the toxicities with alpelisib were significant, with 1 clinician noting that capecitabine would likely yield a better therapeutic index. Other clinicians noted the lack of recent advances in this niche and were of the opinion that the benefits of alpelisib are commensurate with patient values and that the toxicities, though substantial, are predictable and manageable by medical oncologists.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Can the SOLAR-1 study results be generalized to patients with breast cancer who have received chemotherapy in the metastatic setting before treatment with CDK4/6 inhibitor and AI? | There is uncertainty as to whether the SOLAR-1 study results can be generalized to patients with previous chemotherapy in the metastatic setting. However, 1 previous line of chemotherapy in the metastatic setting is not expected to significantly alter the target population. |
If fulvestrant needs to be discontinued or interrupted, can alpelisib be continued as monotherapy? | Patients may continue on alpelisib monotherapy if fulvestrant therapy is interrupted. If fulvestrant is discontinued, alpelisib cannot be continued as monotherapy as there is no evidence to support this use. |
Can patients who are required to discontinue alpelisib due to intolerance continue treatment with single-agent fulvestrant? | Yes, these patients can continue on single-agent fulvestrant. |
If alpelisib is temporarily discontinued due to toxicity, what is the time frame in which it is appropriate to re-start (e.g., if discontinued for more than 4 weeks, should therapy be permanently discontinued)? | For discontinuation due to unresolved toxicity, it is appropriate to permanently discontinue alpelisib after it has been discontinued for more than 4 weeks. |
The following groups of patients were excluded from the SOLAR-1 study. Would they be considered eligible for treatment with alpelisib and fulvestrant? 1.ECOG PS ≥ 2 2.Patients receiving LHRH agonist for induction of ovarian suppression 3.Patients with inflammatory breast cancer 4.Patients with symptomatic visceral disease 5.Patients who have received prior chemotherapy in the metastatic setting 6.Patients who have received prior fulvestrant 7.Patients with CNS metastases 8.Patients with an established diagnosis of type 1 diabetes or uncontrolled type 2 diabetes | 1. Patients with good PS would be eligible. Generally, this entails a PS of 0 or 1. Sometimes, patients with a PS of 2 are suitable for treatment. 2. Yes, patients receiving LHRH agonist for the induction of ovarian suppression would be eligible. 3. Patients with inflammatory breast cancer that would be treated with curative intent are not eligible. However, if a patient has concurrent inflammatory and metastatic breast cancer, they would be considered eligible. 4. Symptomatic visceral disease would not be a reason to automatically exclude a patient from treatment. It would be considered on a case-by-case basis. 5. Patients with 1 prior line of chemotherapy in the metastatic setting can still be considered for eligibility. Chemotherapy may be initiated for reasons other than endocrine resistance, such as to reduce burden of disease or to start a patient on a more readily accessible therapy while waiting for access to a targeted agent. 6. If the patient has progressed on prior fulvestrant, they would not be eligible. Otherwise, alpelisib could be added to fulvestrant therapy, recognizing that patients may start fulvestrant therapy while waiting to find out PIK3CA mutation status. Also, it is appropriate to add alpelisib to fulvestrant as alpelisib would not be available for subsequent lines of treatment. 7. The CADTH review team noted that patients with CNS involvement were eligible for the SOLAR-1 study if they completed prior therapy for CNS metastases ≥ 28 days before the start of the study, if the CNS tumour was clinically stable at screening, and if they did not receive steroids and/or enzyme-inducing antiepileptic medications for brain metastases. The clinical experts agreed that to be eligible, patients would require local therapy to control CNS metastases. 8. No, patients with type 1 diabetes or uncontrolled type 2 diabetes would not be eligible. |
Is it appropriate to offer alpelisib and fulvestrant to patients eligible for this treatment who are currently receiving a chemotherapy option (e.g., capecitabine) with no evidence of progressive disease or intolerance? | No, it would not be appropriate to offer alpelisib and fulvestrant to this population. If patients are doing well on chemotherapy, they would not be switched to a different therapy. |
Which patients should be tested for PIK3CA mutation?a | Patients who are best suited for treatment with alpelisib and fulvestrant, as described in the summary of clinician input by the clinical experts consulted by CADTH, should be tested for PIK3CA mutation. This includes patients with advanced or metastatic hormone receptor–positive, HER2-negative breast cancer, good PS, expected survival of longer than 3 months, and no type 1 diabetes mellitus or uncontrolled type 2 diabetes mellitus who have progressed on first-line endocrine therapy and who previously received treatment with a CDK4/6 inhibitor in the metastatic setting. |
When in the course of treatment should PIK3CA mutation testing occur (e.g., at diagnosis or at point of relapse)?a | PIK3CA testing should be performed at diagnosis of de novo metastatic breast cancer, relapse following treatment for early breast cancer, or progression on first-line therapy for advanced or metastatic breast cancer. Although evidence is limited, it is generally thought that the mutation is stable in the metastatic setting. Testing for PIK3CA mutations is currently not routinely funded or accessible. If testing for mutations is accessible through a clinical trial or a special program, it is typically performed before first-line therapy in the metastatic setting. If testing is not available through these avenues, it is typically done after progression on first-line therapy in the metastatic setting. |
AI = aromatase inhibitor; CDK4/6 = cyclin-dependent kinase 4 and 6; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HER2 = human epidermal growth factor receptor 2; LHRH = luteinizing hormone–releasing hormone; PS = performance status.
aAdditional information regarding PIK3CA mutation testing is presented in Appendix 4.
Clinical Evidence
The clinical evidence included in the review of alpelisib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of 50 mg, 150 mg, and 200 mg alpelisib tablets in combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor |
Intervention | Alpelisib tablets 300 mg once daily on a continuous basis with fulvestrant 500 mg IM on day 1, day 15, and day 29, and every 28 days thereafter Dose reduction allowed to alpelisib 250 mg or 200 mg daily for the management of adverse drug reactions |
Comparator |
|
Outcomes | Efficacy outcomes
Harms outcomes
|
Study designs | Published and unpublished phase III and phase IV RCTs |
AE = adverse event; CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; HRQoL = health-related quality of life; IM = intramuscular; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.16
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid and Embase (1974‒) via Ovid. The search strategy comprised both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Piqray or alpelisib. Clinical trials registries were searched: the US National Institutes of Health’s ClinicalTrials.gov, the WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on May 19, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee on September 8, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.17 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses dealing with breast cancer was run in MEDLINE All (1946‒) on May 19, 2021. No limits were applied to the search.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
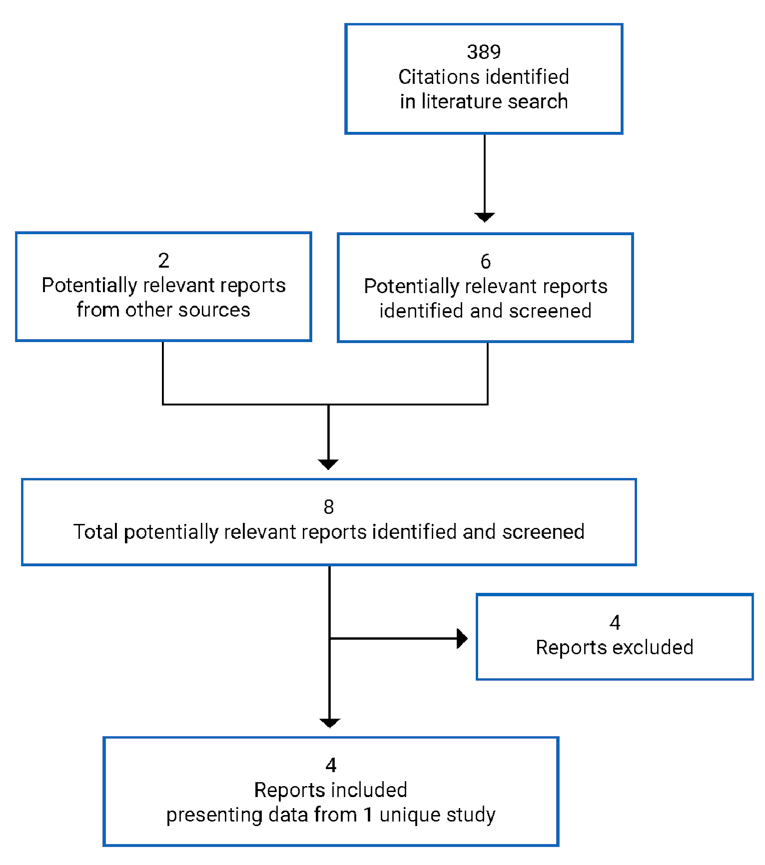
Table 6: Details of Included Studies
Characteristic | Description of SOLAR-1 Study |
|---|---|
Designs and populations | |
Study design | Double-blind, parallel-group, phase III RCT |
Locations | North America (including 9 sites in Canada), South America, Europe, Asia, Australia |
Patient enrolment dates | July 23, 2015, to July 21, 2017 |
Data cut-off dates |
|
Randomized (N) | 572, including:
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Alpelisib 300 mg p.o. q.d. and fulvestrant 500 mg IM on day 1 and day 15 of cycle 1 and on day 1 ± 3 days of subsequent cycles (each cycle being 28 days) |
Comparator(s) | Placebo 300 mg p.o. q.d. and fulvestrant 500 mg IM on day 1 and day 15 of cycle 1 and on day 1 ± 3 days of subsequent cycles (each cycle being 28 days) |
Duration | |
Phase | |
Screening | 35 days |
Treatment | Ongoing until disease progression, unacceptable toxicity, death, or discontinuation for any other reason |
Follow-up |
|
Outcomes | |
Primary end point | PFS in the PIK3CA mutant cohort |
Secondary and exploratory end points | Key secondary: OS in the PIK3CA mutant cohort Secondary
Exploratory
|
Notes | |
Publications | André et al. (2019)18 André et al. (2021)19 |
AI = aromatase inhibitor; AKT = protein kinase B; BPI-SF = Brief Pain Inventory–Short Form; CDK4/6 = cyclin-dependent kinase 4 and 6; CNS = central nervous system; ctDNA = circulating tumour DNA; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = EQ-5D Five-Level; EQ VAS = EuroQol Visual Analogue Scale; ER = estrogen receptor; ET = endocrine therapy; FPG = fasting plasma glucose; GI = gastrointestinal; IM = intramuscular; mTOR = mechanistic target of rapamycin; OS = overall survival; PFS = progression-free survival; PI3K = phosphatidylinositol 3-kinase; p.o. = orally; q.d. = every day; RCT = randomized controlled trial.
Note: Two additional reports were included from the FDA20 and European Medicines Agency.21
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and final Clinical Study Report (2020).2
Description of Studies
One relevant study, the SOLAR-1 study, was selected for inclusion in the CADTH systematic review. The SOLAR-1 study was a pivotal study provided in the sponsor’s submission to CADTH and Health Canada and was also identified in the CADTH systematic literature search. The SOLAR-1 study was a placebo-controlled, double-blind, parallel-group RCT with a primary objective of determining whether treatment with alpelisib and fulvestrant prolongs PFS compared with placebo and fulvestrant for patients with PIK3CA-mutated advanced breast cancer. The key secondary objective was to compare OS between the same groups as stated in the primary objective.
The SOLAR-1 study (N = 572; enrolment from 2015 to 2017) randomized patients 1:1 to alpelisib 300 mg daily or matching placebo, in combination with fulvestrant 500 mg on day 1, day 15, and day 29, and every 28 days afterwards. Patients were randomized within each of 2 cohorts based on the results of real-time PCR tumour tissue testing for mutations in exon 7, exon 9, and exon 20 of the PIK3CA gene: the PIK3CA mutant cohort and the PIK3CA non-mutant cohort. Randomization was stratified according to whether patients had received prior CDK4/6 inhibitor treatment (the total number of patients in the “yes” category was to be no more than 30% of the overall study population) and whether they had lung and/or liver metastases. The study was conducted in North America (including 9 sites in Canada), South America, Europe, Asia, and Australia. The data cut-off date for the analysis of the primary end point in the SOLAR-1 study was June 12, 2018. The final OS analysis was performed using a data cut-off date of April 23, 2020.
During the first 14 days of the 35-day screening phase, a tumour sample from archived tissue or a new biopsy was collected and sent to a sponsor-designated laboratory for PIK3CA mutation testing before randomization.
In the original study protocol, the primary and key secondary end points were to be evaluated in both the PIK3CA mutant and non-mutant cohorts. Following the first protocol amendment, PFS and OS in the PIK3CA non-mutant cohort became secondary end points.
At the sponsor’s request that the reimbursement criteria focus on patients with prior treatment with an endocrine-based regimen with a CDK4/6 inhibitor, the present systematic review reports efficacy results only for the stratum of patients in the PIK3CA mutant cohort who had received prior CDK4/6 inhibitor treatment (N = 20). The BYLieve study — a phase II, non-comparative trial with 1 cohort (N = 127) meeting the systematic review criteria for population, intervention, and outcomes — is summarized in the Other Relevant Evidence section of this report.
Populations
Inclusion and Exclusion Criteria
The main inclusion and exclusion criteria for the SOLAR-1 study are provided in Table 6. Women were eligible if they were postmenopausal, with the definition of postmenopausal excluding menopause induced by ovarian suppression. Patients had to have radiological or objective evidence of recurrence or progression during or after AI therapy, and it was not specified that this therapy had to be in the advanced or metastatic setting. While patients who were endocrine sensitive were initially enrolled, a protocol amendment later clarified that patients without previous treatment for metastatic disease had to have relapsed with progression while at or within 12 months of completion of adjuvant or neoadjuvant endocrine therapy. Patients who had relapsed with progression while at or within 12 months of completion of adjuvant endocrine therapy and then relapsed with progression after 1 line of endocrine therapy for metastatic disease were not included. Patients were excluded if they had received prior treatment with chemotherapy other than adjuvant or neoadjuvant chemotherapy, fulvestrant, or any PI3K, mammalian target of rapamycin, or protein kinase B inhibitor. Patients had to have an ECOG PS of 0 or 1. Patients with any of the following were excluded: patients with diabetes mellitus type 1 or uncontrolled diabetes mellitus type 2 or hyperglycemia, patients with inflammatory breast cancer, and patients with symptomatic visceral disease or any disease burden making the patient ineligible for endocrine therapy.
Baseline Characteristics
The primary end point in the SOLAR-1 study was PFS in the entire PIK3CA mutant cohort (N = 341) and results in patients in that cohort with previous CDK4/6 inhibitor treatment (N = 20) were only available in subgroup analyses. Since summaries of patient disposition, treatment exposure, and protocol deviations were not available for the relevant patient population for the present review, these data are presented for the entire PIK3CA mutant cohort. Baseline characteristics are presented for both the entire PIK3CA cohort and the subgroup with prior CDK4/6 inhibitor treatment; data for certain characteristics (disease stage at study entry, lines of prior medication therapy, and lines of prior hormonal therapy in metastatic setting) were not available for the subgroup.
Detailed information on key baseline characteristics for the full analysis set (FAS) in the PIK3CA mutant cohort in the SOLAR-1 study and in the subgroup with prior CDK4/6 inhibitor treatment are provided in Table 7. Overall, in the entire PIK3CA mutant cohort, baseline characteristics were balanced between the treatment groups, though there were some imbalances with regard to metastatic sites. Within the subgroup with prior CDK4/6 inhibitor treatment, there were imbalances in the alpelisib group versus the placebo group in ECOG PS (44.4% versus 72.7% with ECOG PS of 0), numbers of sites of metastases (44.5% versus 18.2% with 1 site and no patients versus 18.2% with 3 sites), and progesterone receptor status (55.6% versus 81.8% with positive status).
Within the entire PIK3CA mutant cohort, there was an approximately even split between patients who had received 1 line or no lines of endocrine therapy in the metastatic setting, with less than 3% having received 2 previous lines. This information was not reported for the subgroup with prior CDK4/6 inhibitor treatment and it was not clear how many of those patients had received 2 previous lines of endocrine therapy. In the subgroup, most patients (66.7% and 72.7% within each group) were categorized as having secondary endocrine resistance, while some were categorized under primary endocrine resistance (33.3% and 18.2%) or categorized as endocrine sensitive (11.8% and 11.0%).
The numbers of patients with prior CDK4/6 inhibitor treatment, as identified for randomization stratification with the interactive response technology, were 9 in the alpelisib group and 11 in the placebo group. These patients were the ones included in subgroup analyses for the prior CDK4/6 inhibitor treatment subgroup and it is these analyses that are presented in the current report. However, according to clinical data collected in case report forms and reported in summaries of prior antineoplastic therapy, the numbers of patients with prior CDK4/6 inhibitor therapy (those with prior abemaciclib, palbociclib, or ribociclib therapy) were 11 in the alpelisib group and 8 in the placebo group. Finally, a concordance table in the SOLAR-1 Interim Clinical Study Report comparing numbers of patients within each group according to each source of data showed 12 patients and 11 patients in the alpelisib and fulvestrant groups, respectively, according to the clinical data. According to the concordance table, all 9 patients in the alpelisib group in the randomization stratum were identified as having prior CDK4/6 inhibitor therapy in the clinical data. In contrast, 10 of 11 patients in the placebo groups were common between the randomization stratum and the clinical data. The reasons for these differences are unclear.
Table 7: Summary of Baseline Characteristics
Characteristic | SOLAR-1 study PIK3CA mutant cohort, FAS | SOLAR-1 study PIK3CA mutant cohort, patients with prior CDK4/6 inhibitor treatment, FAS | ||
|---|---|---|---|---|
Alpelisib + fulvestrant N = 169 | Placebo + fulvestrant N = 172 | Alpelisib + fulvestrant N = 9 | Placebo + fulvestrant N = 11 | |
Age in years, mean (SD) | 62.7 (10.22) | 64.0 (9.99) | 56.4 (12.18) | 62.9 (11.96) |
Age category in years, n (%) | ||||
18 to < 65 | 95 (56.2) | 89 (51.7) | NR | NR |
65 to < 85 | 73 (43.2) | 80 (46.5) | NR | NR |
≥ 85 | 1 (0.6) | 3 (1.7) | NR | NR |
Sex, n (%) | ||||
Female | 168 (99.4) | 172 (100) | 9 (100) | 11 (100) |
Male | 1 (0.6) | 0 | 0 | 0 |
Race, n (%) | ||||
White | 117 (69.2) | 109 (63.4) | 8 (88.9) | 8 (72.7) |
Asian | 34 (20.1) | 40 (23.3) | 1 (11.1) | 2 (18.2) |
Other | 8 (4.7) | 10 (5.8) | 0 | 1 (9.1) |
Unknown | 8 (4.7) | 8 (4.7) | 0 | 0 |
Black or African-American | 1 (0.6) | 3 (1.7) | 0 | 0 |
American Indian or Alaska Native | 1 (0.6) | 2 (1.2) | 0 | 0 |
ECOG PS, n (%) | ||||
0 | 112 (66.3) | 113 (65.7) | 4 (44.4) | 8 (72.7) |
1 | 56 (33.1) | 58 (33.7) | 5 (55.6) | 3 (27.3) |
Missing | 1 (0.6) | 1 (0.6) | 0 | 0 |
Disease stage at study entry, n (%) | ||||
III | 1 (0.6) | 7 (4.1) | NR | NR |
IV | 168 (99.4) | 165 (95.9) | NR | NR |
Sites of metastases, n (%) | ||||
CNS | 0 | 2 (1.2) | 0 | 0 |
Breast | 1 (0.6) | 3 (1.7) | 0 | 0 |
Bone | 131 (77.5) | 121 (70.3) | 8 (88.9) | 8 (72.7) |
Bone only | 42 (24.9) | 35 (20.3) | 3 (33.3) | 2 (18.2) |
Visceral | 93 (55.0) | 100 (58.1) | 4 (44.4) | 5 (45.5) |
Liver | 49 (29.0) | 54 (31.4) | 4 (44.4) | 4 (36.4) |
Lung | 57 (33.7) | 68 (39.5) | 1 (11.1) | 4 (36.4) |
Other visceral | 3 (1.8) | 1 (0.6) | 1 (11.1) | 1 (9.1) |
Skin | 4 (2.4) | 6 (3.5) | 1 (11.1) | 2 (18.2) |
Lymph nodes | 56 (33.1) | 65 (37.8) | 2 (22.2) | 5 (45.5) |
Other | 25 (14.8) | 18 (10.5) | 1 (11.1) | 1 (9.1) |
None | 0 | 1 (0.6) | 0 | 0 |
Metastatic sites, n (%) | ||||
0 | 0 | 1 (0.6) | 0 | 0 |
1 | 63 (37.3) | 52 (30.2) | 4 (44.4) | 2 (18.2) |
2 | 58 (34.3) | 60 (34.9) | 4 (44.4) | 5 (45.5) |
3 | 24 (14.2) | 42 (24.4) | 0 | 2 (18.2) |
4 | 19 (11.2) | 10 (5.8) | 0 | 1 (9.1) |
≥ 5 | 5 (3.0) | 7 (4.1) | 1 (11.1) | 1 (9.1) |
Hormone receptor status, n (%) | ||||
Estrogen receptor–positive | 167 (98.9) | 172 (100) | 9 (100) | 11 (100) |
Progesterone receptor–positive | 120 (71.0) | 132 (76.7) | 5 (55.6) | 9 (81.8) |
Lines of prior medication therapy, n (%) | ||||
1 | 118 (69.8) | 114 (66.3) | NR | NR |
2 | 45 (26.6) | 55 (32.0) | NR | NR |
3 | 6 (3.6) | 3 (1.7) | NR | NR |
Lines of prior hormonal therapy in metastatic setting, n (%) | ||||
0 | 88 (52.1) | 89 (51.7) | NR | NR |
1 | 77 (45.6) | 81 (47.1) | NR | NR |
2 | 4 (2.4) | 2 (1.2) | NR | NR |
Prior CDK4/6 inhibitor therapy, n (%) | ||||
According to entry in IRT for stratification factor | 9 (5.3)a | 11 (6.4)b | NA | NA |
According to clinical data for prior antineoplastic therapy (prior abemaciclib, palbociclib, or ribociclib therapy) | 11 (6.5) | 8 (4.7) | NA | NA |
According to clinical data from concordance table of IRT stratification factor vs. clinical data | 12 (7.1)a | 11 (6.4)b | NA | NA |
Endocrine resistance status, n (%) | ||||
Primary endocrine resistance | 23 (13.6) | 22 (12.8) | 3 (33.3) | 2 (18.2) |
Secondary endocrine resistance | 120 (71.0) | 127 (73.8) | 6 (66.7) | 8 (72.7) |
Endocrine sensitivity | 20 (11.8) | 19 (11.0) | NR | NR |
CDK4/6 = cyclin-dependent kinase 4 and 6; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FAS = full analysis set; IRT = interactive response technology; NA = not applicable; NR = not reported; SD = standard deviation; vs. = versus.
aAll 9 patients in the IRT stratum alpelisib group were also in the clinical data group.
bThere were 10 patients who were common to both groups.
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and sponsor response to June 23, 2021, additional information request.22
Interventions
Patients were assigned based on tissue testing to the PIK3CA mutant cohort or PIK3CA non-mutant cohort following confirmation of eligibility. Patients were randomized and allocated by an interactive response technology provider, with patients randomized in each cohort 1:1 to the alpelisib or placebo group using previous treatment with CDK4/6 inhibitor(s) (yes or no) and the presence of lung and/or liver metastasis (yes or no) as stratification factors. Patients and investigators were blinded to PIK3CA mutation status throughout the study. Patients, investigators, and local radiologists were blinded to treatment assignment throughout the study. The sponsor’s clinical trial team was blinded to treatment assignment until the database was locked for the main PFS analysis, subsequent to the June 12, 2018, data cut-off date. An independent data monitoring committee reviewed safety and efficacy data during the study and an independent statistical group, external to the sponsor and not involved in study conduct, prepared the data monitoring committee reports. Interim PFS analyses were performed by an independent statistician who was also not involved in study conduct.
Patients in the alpelisib group received alpelisib as 50 mg and 200 mg film-coated tablets and patients in the placebo group received placebo tablets that were identical to alpelisib medication in appearance and packaging. The starting dosage for alpelisib and placebo was 300 mg taken orally once daily, with instructions to take the medication within 1 hour after a meal at approximately the same time each day. Patients were given an adequate supply at each study visit, instructions for administration, and medication diaries. Treatment adherence was assessed using pill counts and information provided by the patients and/or caregiver. Fulvestrant 500 mg was administered intramuscularly on day 1 and day 15 of cycle 1 and on day 1 (± 3 days) of each subsequent 28-day cycle.
A maximum of 2 dose reductions of 50 mg each was permitted for alpelisib and placebo (with no re-escalation permitted) to manage intolerance of study medication, while no dose adjustments were permitted for fulvestrant. Guidance for dose modifications for alpelisib and placebo was outlined in the study protocol. Patients were required to permanently discontinue treatment with alpelisib or placebo if they required a dose delay of more than 28 days. Patients were required to permanently discontinue treatment with fulvestrant if it was withheld for more than 35 days since a planned injection. If 1 study medication was discontinued, patients could continue treatment with the other study medication.
Patients taking alpelisib or placebo in combination with fulvestrant were not permitted to take medications with a known risk for torsades de pointes, other investigational and antineoplastic therapies, or herbal preparation and/or medications and dietary supplements (except for vitamins). However, medications required to treat AEs, medications to manage cancer symptoms and concurrent diseases, and supportive care agents were permitted.
Patients could voluntarily withdraw from either or both study treatments, and an investigator could discontinue either or both study treatments if continuation was considered to be detrimental to the patient’s well-being. Patients taking only 1 of the 2 study medications continued with study visits. Patients who discontinued both medications were to attend an end-of-treatment visit within 14 days of study treatment discontinuation and were followed up for safety data until 30 days after the last dose.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the SOLAR-1 study is provided in Table 8. These end points are further summarized as follows. Although health-related quality of life (HRQoL) was assessed in the SOLAR-1 study using the EQ-5D Five-Level (EQ-5D-5L) questionnaire and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30), there were no analyses of those outcomes in the subgroup with prior CDK4/6 inhibitor treatment.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | End point in SOLAR-1 |
|---|---|
Progression-free survival | Primary |
Overall survival | Key secondary |
CDK4/6 = cyclin-dependent kinase 4 and 6.
Note: Only outcomes analyzed in the subgroup of patients with prior CDK4/6 inhibitor treatment are listed.
Source: SOLAR-1 study interim Clinical Study Report (2018).1
Efficacy
Progression-Free Survival
PFS was based on local radiology of tumour assessments according to the RECIST 1.1 guideline.23 Tumour assessments were also reviewed centrally by a blinded independent review committee using an audit-based approach to provide supportive analyses. Imaging assessments used the same imaging modality used at baseline and were performed every 8 weeks (± 7 days) during the first 18 months following randomization and every 12 weeks (± 7 days) thereafter until disease progression, death, withdrawal of consent, loss to follow-up, a patient or guardian decision, or end of treatment. Assessments continued after the start of new antineoplastic therapy. If there was clinical suspicion of disease progression, physical examination and imaging assessment were performed promptly. Baseline imaging assessments included CT or MRI of the chest, abdomen, and pelvis (and of the brain if clinically indicated), a whole body bone scan, localized bone CT, MRI, or X-ray for lesions identified on a whole body bone scan not visible on a chest, abdomen, or pelvis CT or MRI, colour photography for skin lesions, and CT or MRI of other metastatic sites if clinically indicated. Complete physical examinations were performed during screening (within 14 days of randomization), on day 8 and day 15 of cycle 1, on day 1 and day 15 of cycle 2, and on day 1 of each cycle thereafter.
Overall Survival
Following the discontinuation of study treatment and tumour assessments, survival follow-up continued until the final number of OS events was reached (178) or the study was stopped for other reasons. Survival follow-up was conducted through clinical visits or telephone calls every 12 weeks (± 1 week) until death, loss to follow-up, or withdrawal of consent for survival follow-up.
Adverse Events
AEs that began or worsened after patient consent were recorded at each study visit until at least 30 days following discontinuation of study treatment. AEs were detected by non-directive questioning at visits, when volunteered by patients between visits, or through physical examination, laboratory test, or other assessments.
Statistical Analysis
The primary outcome of the study was PFS in the FAS in the PIK3CA mutant cohort based on local radiology assessment. PFS was defined as the time from the date of randomization to the date of first documented disease progression or death due to any cause. Patients were censored at the last tumour assessment if they did not have an event or if they had an event that occurred after at least 2 missing tumour assessments. If a patient was missing the baseline tumour assessment, that patient was censored at the date of randomization for progressive disease events. The survival distribution was estimated using Kaplan-Meier analysis.
A 1-sided 2.0% significance level was used for rejecting the null hypothesis that the log-hazard ratio was 0 or more (or the hazard ratio was ≥ 1) for the alpelisib group versus the placebo group. A stratified log-rank test, with strata based on prior CDK4/6 inhibitor treatment status and the presence of lung and/or liver metastases, was used to compare PFS between treatment groups. The hazard ratio for PFS and its 95% CI were estimated using a stratified Cox proportional hazards model, using the aforementioned strata.
Haybittle-Peto boundaries were used to control for overall type I error rate in a 3-look group sequential design with an interim futility analysis planned after observing approximately 97 PFS events (40% of expected events) and an interim efficacy analysis for superiority after approximately 185 PFS events (76% of expected events). A final PFS analysis was to be performed after approximately 243 events. Based on the planned numbers of events, the significance levels for the interim and final PFS efficacy analyses were 0.0001 and 0.0199, respectively.
OS in the PIK3CA mutant cohort was the key secondary end point and was compared between treatment groups using the same methods as for PFS, though a separate Lan-DeMets (O’Brien-Fleming) alpha spending function was used to control type I error rate with an overall alpha of 2.5%. OS could be tested at the interim PFS efficacy analysis, but only if PFS was statistically significant. If OS was not significant at the interim analysis, a second OS analysis was performed at approximately 85% of expected deaths. If OS was not tested at the interim PFS efficacy analysis, it was tested at the final PFS analysis instead. A final OS analysis (if previously OS analyses were not significant) was planned at approximately 178 deaths if the final PFS was significant.
It was calculated that a total of 243 PFS events would be required for 83.8% power to reject the null hypothesis, assuming a true hazard ratio of 0.6 or an increase in median PFS from 7.0 months to 11.67 months. A total of 340 patients randomized in the PIK3CA mutant cohort would be needed, based on 40% of patients having a PIK3CA mutation; enrolment rates of 12 patients, 35 patients, and 59 patients per month during the first 6 months, the next 6 months, and the remainder of the study, respectively; and a 10% loss to PFS follow-up. With 340 patients randomized and a 5% loss to follow-up for OS, it was estimated that 178 deaths would occur by the OS final analysis and that with a true hazard ratio of 0.67, there would be 72% power to reject the null hypothesis for the key secondary end point.
Subgroup Analyses
Pre-specified subgroup analyses for the primary and key secondary end points were planned if statistical significance was reached. The same analysis methods that were used for the primary end point were applied to each subgroup. The subgroup analyses did not account for multiplicity and were not part of the sample size considerations for the study. In the analyses for the subgroup of patients with prior CDK4/6 inhibitor treatment, a Cox proportional hazards model was used and was stratified by the presence of lung and/or liver metastases.
Sensitivity Analyses
Sensitivity analyses for the primary end point were planned if statistical significance was reached. In the subgroup of patients with prior CDK4/6 inhibitor treatment, the primary end point analysis was to be repeated in the per-protocol (PP) set, using different censoring methods (an actual event approach, a backdating approach, and the approach of censoring patients after a new antineoplastic therapy), and using stratification factors based on the clinical database as opposed to those used in the interactive response technology. In the actual date censoring approach, events were included even if they occurred following 2 or more missing tumour assessments. In the backdating approach, events occurring after at least 1 missing assessment were assigned the date of the next scheduled assessment.
Table 9: Statistical Analysis of Efficacy End Points in SOLAR-1
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
PFS in the subgroup of patients with prior CDK4/6 inhibitor treatment in the PIK3CA mutant cohort | Stratified CPH model | Presence of lung/liver metastases |
|
OS in the subgroup of patients with prior CDK4/6 inhibitor treatment in the PIK3CA mutant cohort | Stratified CPH model | Presence of lung/liver metastases | NA |
CDK4/6 = cyclin-dependent kinase 4 and 6; CPH = Cox proportional hazards; NA = not applicable; OS = overall survival; PFS = progression-free survival; PP = per-protocol.
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and final Clinical Study Report (2020).2
Analysis Populations
All patients randomized to study treatment were included in the FAS and were analyzed according to the cohort, treatment, and strata assigned during randomization. Patients in the PP set were those from the FAS without certain protocol deviations that were specified in the statistical analysis plan. The safety analysis set included all patients who received any study treatment, with patients analyzed according to the study treatment received.
Results
Patient Disposition
Detailed patient disposition for the SOLAR-1 study in the entire PIK3CA mutant cohort for data cut-off dates representing the final PFS analysis (June 12, 2018, data cut-off date) and final OS analysis (April 23, 2020, data cut-off date) is presented in Table 10. The percentages of patients discontinuing from the study were 4.1% and 2.9% in the alpelisib and placebo groups, respectively, at the final PFS analysis and 5.9% and 2.9% at the final OS analysis. The most common reason for study discontinuation, progressive disease, was more common in the alpelisib group versus the placebo group (2.4% versus 0.6% at the final PFS analysis and 3.0% and 0.6% at the final OS analysis). Patient disposition information within the subgroup of interest — patients with prior CDK4/6 inhibitor treatment — was unavailable.
Disposition | SOLAR-1 study | |
|---|---|---|
Alpelisib + fulvestrant | Placebo + fulvestrant | |
Screened, N | 1,442 | |
Randomized, N (%) | 572 (39.7) | |
Screen failure, N (%) | 808 (56.0) | |
PIK3CA mutant cohort | ||
Randomized, N | 169 | 172 |
FAS, N | 169 | 172 |
PP set, N | 144 | 148 |
Safety analysis set, N | 169 | 171 |
PIK3CA mutant cohort: June 12, 2018, data cut-off date | ||
Discontinued from study, N (%) | 7 (4.1) | 5 (2.9) |
Reason for discontinuation, N (%) | ||
Progressive disease | 4 (2.4) | 1 (0.6) |
Patient/guardian decision | 2 (1.2) | 2 (1.2) |
Death | 0 | 2 (1.2) |
Physician decision | 1 (0.6) | 0 |
PIK3CA mutant cohort: April 23, 2020, data cut-off date | ||
Discontinued from study, N (%) | 10 (5.9) | 5 (2.9) |
Reason for discontinuation, N (%) | ||
Progressive disease | 5 (3.0) | 1 (0.6) |
Patient/guardian decision | 4. (2.4) | 2 (1.2) |
Death | 0 | 2 (1.2) |
Physician decision | 1 (0.6) | 0 |
PIK3CA mutant cohort, patients with prior CDK4/6 inhibitor treatment | ||
FAS, N | 9 | 11 |
PP set, N | 8 | 10 |
Safety analysis set, N | 9 | 11 |
CDK4/6 = cyclin-dependent kinase 4 and 6; FAS = full analysis set; PP = per-protocol.
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and final Clinical Study Report (2020).2
The most common protocol deviations at the final OS analysis in the SOLAR-1 study are presented in Table 11. The most common protocol deviation was use of prohibited concomitant medication (20.7% in the alpelisib group and 14.0% in the placebo group), though in most cases, the medication was 1 prohibited for safety reasons (i.e., those with known risk for torsades de pointes).
The reasons for the exclusion of patients from the PP set for the primary end point analysis in the PIK3CA mutant cohort were as follows: the patient relapsed at or within 12 months from completion of adjuvant or neoadjuvant endocrine therapy and subsequently progressed after 1 line of endocrine therapy or progressed on more than 1 line of endocrine therapy for metastatic disease, the patient was not postmenopausal, there was prohibited concomitant medication at baseline, the criteria for measurable disease or lytic bone lesion was not met, the criteria for prior antineoplastic therapy was not met, breast cancer type (HR status) was not met, or no study treatment was taken. Aside from the first reason, no more than 3.0% of patients in a group were excluded for each reason. In total, 14.8% and 14.0% of patients in the alpelisib and placebo groups were excluded from the PP set, respectively. Protocol deviation information within the subgroup of interest, patients with prior CDK4/6 inhibitor treatment, was unavailable.
Deviation | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant FAS N = 169 | Placebo + fulvestrant FAS N = 172 | |
Patients with any protocol deviation, N (%) | 92 (54.4) | 87 (50.6) |
Most common protocol deviationsa by category, N (%) | ||
Inclusion criteria deviation | ||
Patient relapsed while on or within 12 months from completion of adjuvant or neoadjuvant ET and subsequently progressed after 1 line of ET or progressed on > 1 line of ET for metastatic disease | 16 (9.5) | 15 (8.7) |
No ECG triplicate at screening | 13 (7.7) | 13 (7.6) |
Baseline creatinine clearance criterion missing | 5 (3.0) | 11 (6.4) |
Baseline fasting serum analysis criterion missing | 6 (3.6) | 10 (5.8) |
Prohibited concomitant medication | ||
Prohibited concomitant medication while on study | 35 (20.7) | 24 (14.0) |
Exclusion criteria deviation | ||
Prohibited concomitant medication at baseline | 7 (4.1) | 9 (5.2) |
Patient not withdrawn as per protocol | ||
Study treatment discontinued but patient not withdrawn from treatment phase | 4 (2.4) | 9 (5.2) |
Other deviation | ||
Patient has not provided consent for the optional biopsy sample collected | 4 (2.4) | 9 (5.2) |
ECG = electrocardiogram; ET = endocrine therapy; FAS = full analysis set.
Note: All patients at cut-off date of April 23, 2020.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Exposure to Study Treatments
Details on the duration of treatment exposure and study treatment discontinuations at the time of the final OS analysis (April 23, 2020, data cut-off date) are provided in Table 12. Most patients had discontinued alpelisib or placebo at the final PFS analysis (82.2% for alpelisib and 84.2% for placebo; these values are not shown in Table 12) and at the final OS analysis (91.7% for alpelisib and 97.7% for placebo). At the final OS analysis, treatment discontinuation due to progressive disease was less common in the alpelisib group compared with the placebo group (47.9% versus 78.9%) while discontinuation due to AE was more common in the alpelisib group than in the placebo group (26.6% versus 5.8%). The percentages of patients discontinuing fulvestrant were similar to those for alpelisib and placebo, though discontinuation due to AE was less common (3.6% for alpelisib and 1.8% for placebo). Treatment exposure information within the subgroup of interest, patients with prior CDK4/6 inhibitor treatment, was unavailable.
Exposure | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant Safety analysis set N = 169 | Placebo + fulvestrant Safety analysis set N = 171 | |
Duration of exposure to alpelisib or placebo, months | ||
Mean (SD) | 10.8 (12.87) | 9.7 (10.47) |
Median (IQR) | 5.5 (1.6 to 14.8) | 4.6 (1.9 to 13.8) |
Duration of exposure to fulvestrant, months | ||
Mean (SD) | 14.0 (13.45) | 10.4 (11.13) |
Median (IQR) | 8.3 (4.6 to 19.4) | 5.5 (2.2 to 14.7) |
Discontinued alpelisib or placebo, N (%) | 155 (91.7) | 167 (97.7) |
Reasons for alpelisib or placebo discontinuation, N (%) | ||
Progressive disease | 81 (47.9) | 135 (78.9) |
Adverse event | 45 (26.6) | 10 (5.8) |
Patient/guardian decision | 16 (9.5) | 7 (4.1) |
Physician decision | 8 (4.7) | 8 (4.7) |
Death | 2 (1.2) | 4 (2.3) |
Protocol deviation | 3 (1.8) | 3 (1.8) |
Discontinued fulvestrant, N (%) | 149 (88.2) | 164 (95.9) |
Reasons for fulvestrant discontinuation, N (%) | ||
Progressive disease | 111 (65.7) | 138 (80.7) |
Adverse event | 6 (3.6) | 3 (1.8) |
Patient/guardian decision | 17 (10.1) | 7 (4.1) |
Physician decision | 8 (4.7) | 9 (5.3) |
Death | 3 (1.8) | 4(2.3) |
Protocol deviation | 4 (2.4) | 3 (1.8) |
IQR = interquartile range; SD = standard deviation.
Note: All patients at cut-off date of April 23, 2020.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Follow-up duration and reasons for censoring patients for the primary end point analysis and final OS analysis are presented in Table 13. Median follow-up duration for PFS at the primary end point analysis (June 12, 2018, data cut-off date) was 20.2 months and 19.9 months in the alpelisib group and placebo group, respectively, in the overall PIK3CA mutant cohort. In the subgroup with prior CDK4/6 inhibitor treatment, the median follow-up duration was similar (19.0 months in the alpelisib group and 18.0 months in the placebo group). At the final OS analysis (April 23, 2020, data cut-off date), the mean follow-up duration was 42.6 months in the alpelisib group and 42.3 months in the placebo group, with similar median follow-up durations in the subgroup with prior CDK4/6 inhibitor treatment (41.4 months and 40.4 months).
Follow-up | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant FAS N = 169 | Placebo + fulvestrant FAS N = 172 | |
June 12, 2018, data cut-off date | ||
Median follow-up, months | 20.17 | 19.89 |
PFS events, n (%) | 103 (60.9) | 129 (75.0) |
Progressive disease | 99 (58.6) | 120 (69.8) |
Death | 4 (2.4) | 9 (5.2) |
Patients censored for PFS, n (%) | 66 (39.1) | 43 (25.0) |
Withdrew consent | 10 (5.9) | 5 (2.9) |
Event documented after ≥ 2 missing tumour assessments | 3 (1.8) | 1 (0.6) |
Adequate assessment no longer available | 8 (4.7) | 6 (3.5) |
April 23, 2020, data cut-off date | ||
Median follow-up, months | 42.55 | 42.27 |
Patients censored for OS, n (%) | 82 (48.5) | 78 (45.3) |
Lost to follow-up | 18 (10.7) | 17 (9.9) |
Patients with prior CDK4/6 inhibitor treatment | Alpelisib + fulvestrant FAS N = 9 | Placebo + fulvestrant FAS N = 11 |
June 12, 2018, data cut-off date | ||
Median follow-up, months | 19.02 | 18.00 |
PFS events, N (%) | 7 (77.8) | 10 (90.9) |
Progressive disease | 7 (77.8) | 8 (72.7) |
Death | 0 | 2 (18.2) |
April 23, 2020, data cut-off date | ||
Median follow-up for PFS and OS, months | 41.40 | 40.38 |
PFS events, N (%) | 7 (77.8) | 10 (90.9) |
OS events, N (%) | 7 (77.8) | 9 (81.8) |
CDK4/6 = cyclin-dependent kinase 4 and 6; FAS = full analysis set; OS = overall survival; PFS = progression-free survival.
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and final Clinical Study Report (2020).2
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported as follows.
Progression-Free Survival
The results for PFS in patients with prior CDK4/6 inhibitor treatment in the PIK3CA mutant cohort are presented in Table 14. At the primary PFS analysis at the June 12, 2018, data cut-off date, median PFS was 5.5 months (95% CI, 1.58 months to 16.76 months) in the alpelisib group and 1.8 months (95% CI, 1.68 months to 3.58 months) in the placebo group. The hazard ratio for the alpelisib group versus the placebo group was 0.48 (95% CI, 0.17 to 1.36). The results were identical between the primary PFS analysis (June 12, 2018, data cut-off) and the final PFS analysis (April 23, 2020, data cut-off date). Conducting sensitivity analyses in the PP set, using different censoring methods, and using stratification factors based on the clinical database yielded results consistent with the primary analysis.
Table 14: Progression-Free Survival
Outcome of patients with prior CDK4/6 inhibitor treatment | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant N = 9 | Placebo + fulvestrant N = 11 | |
Median, months (95% CI)a | 5.5 (1.58 to 16.76) | 1.8 (1.68 to 3.58) |
HR (95% CI)b | 0.48 (0.17 to 1.36) | Reference group |
CDK4/6 = cyclin-dependent kinase 4 and 6; CI = confidence interval; HR = hazard ratio; PFS = progression-free survival.
Note: PFS cut-off date of June 12, 2018.
aUsing Kaplan-Meier estimation.
bCox proportional hazards model, stratified by the presence of lung and/or liver metastases, performed in the full analysis set.
Source: SOLAR-1 study interim Clinical Study Report (2018).1
Kaplan-Meier curves for PFS in the 2 treatment groups in patients with prior CDK4/6 inhibitor treatment are shown in Figure 2.
Figure 2: Progression-Free Survival in Patients with Prior CDK4/6 Inhibitor Treatment
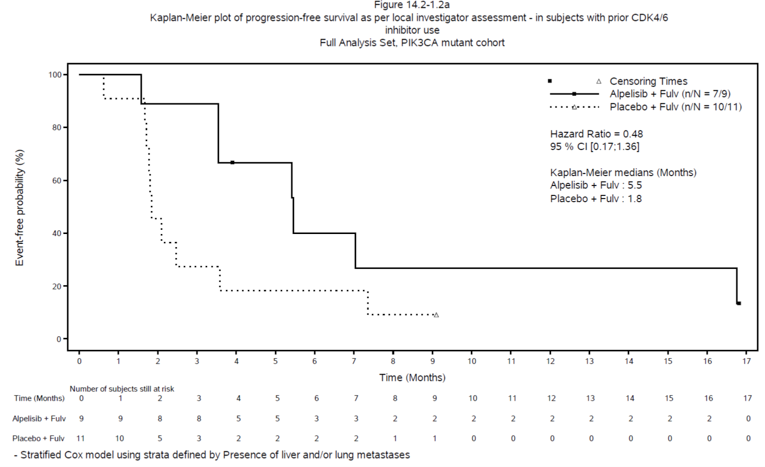
CDK4/6 = cyclin-dependent kinase 4 and 6; CI = confidence interval; fulv = fulvestrant.
Source: SOLAR-1 study interim Clinical Study Report (2018).1
Overall Survival
The results for OS in patients with prior CDK4/6 inhibitor treatment in the PIK3CA mutant cohort are presented in Table 15. At the final OS analysis at the April 23, 2020, data cut-off date, the median OS was 29.8 months (95% CI, 6.67 months to 38.21 months) in the alpelisib group and 12.9 months (95% CI, 2.46 months to 34.60 months) in the placebo group. The hazard ratio for the alpelisib group versus the placebo group was 0.67 (95% CI, 0.21 to 2.18).
Outcome of patients with prior CDK4/6 inhibitor treatment | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant N = 9 | Placebo + fulvestrant N = 11 | |
Median, months (95% CI)a | 29.8 (6.67 to 38.21) | 12.9 (2.46 to 34.60) |
HR (95% CI)b | 0.67 (0.21 to 2.18) | Reference group |
CDK4/6 = cyclin-dependent kinase 4 and 6; CI = confidence interval; HR = hazard ratio; OS = overall survival.
Note: All patients at cut-off date of April 23, 2020.
aUsing Kaplan-Meier estimation.
bCox proportional hazards model, stratified by presence of lung and/or liver metastases, performed in the full analysis set.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Kaplan-Meier curves for OS in the 2 treatment groups in patients with prior CDK4/6 inhibitor treatment are shown in Figure 3.
Figure 3: Overall Survival in Patients With Prior CDK4/6 Inhibitor Treatment
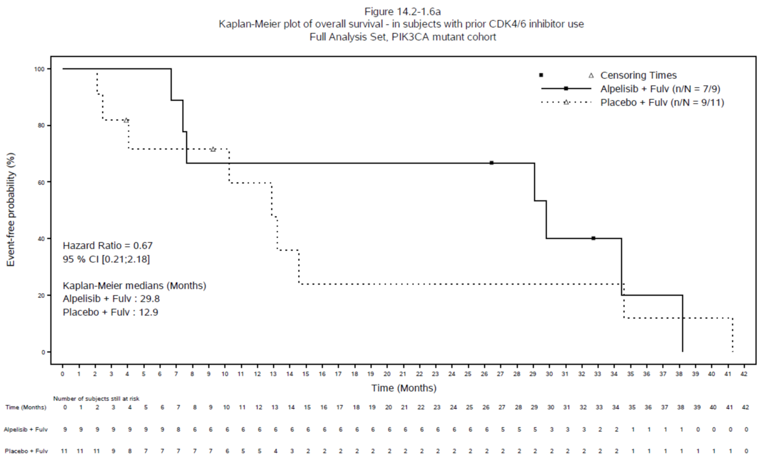
CDK4/6 = cyclin-dependent kinase 4 and 6; CI = confidence interval; fulv = fulvestrant.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Health-Related Quality of Life
Although HRQoL was assessed in the SOLAR-1 study using the EQ-5D-5L questionnaire and the EORTC QLQ-C30, analysis in the subgroup of patients with prior CDK4/6 inhibitor treatment was not available for HRQoL outcomes. Results for the index score of the EQ-5D-5L for the entire PIK3CA mutant cohort are presented in Appendix 3 to provide context for the CADTH Pharmacoeconomic Review Report.
Harms
Only those harms identified in the review protocol are reported as follows. Results for harms are presented for the entire PIK3CA mutant cohort (this aligns with the Health Canada–approved indication for alpelisib). The CADTH review team did not request data from the sponsor on AEs in the subgroup with prior CDK4/6 inhibitor treatment, since AE data were expected to be more informative in the entire PIK3CA mutant cohort than in the subgroup and results for harms were not expected by the clinicians consulted by CADTH to differ between patients who had or had not previously received CDK4/6 inhibitor treatment. See Table 16 for detailed harms data at the final OS analysis (April 23, 2020, data cut-off date).
Adverse Events
Almost all patients reported at least 1 AE (99.4% in the alpelisib group and 90.6% in the placebo group). Most of the AEs occurring in at least 10% of patients of at least 1 treatment group were more common in the alpelisib group compared with the placebo group. All of the AEs reported by more than 20% of patients in the alpelisib group were also more common in the alpelisib group than in the placebo group: hyperglycemia, diarrhea, nausea, rash, decreased appetite, decreased weight, stomatitis, vomiting, fatigue, and alopecia. The only AE reported by more than 20% of patients in the placebo group was nausea (20.5%).
Medications or therapies for AEs were required for 96.4% of patients in the alpelisib group and 63.8% of patients in the placebo group. All of the AEs requiring medications or therapies occurring in at least 10% of 1 treatment group were more common in the alpelisib group compared with the placebo group and were as follows: hyperglycemia, diarrhea, rash, nausea, stomatitis, maculopapular rash, urinary tract infection, and mucosal inflammation.
Serious Adverse Events
SAEs were reported in 39.6% of patients in the alpelisib group and 19.9% of patients in the placebo group. The most common SAEs were hyperglycemia (10.1% in the alpelisib group and none in the placebo group), osteonecrosis of the jaw (3.6% in the alpelisib group and none in the placebo group), stomatitis, acute kidney injury, and rash (2.4% in the alpelisib group and none in the placebo group for the preceding 3 SAEs).
Withdrawals Due to Adverse Events
Withdrawals from treatment due to AEs were more common in the alpelisib group (27.2%) versus the placebo group (5.8%). The most common AEs leading to discontinuation were reported in the alpelisib group alone: hyperglycemia (6.5%), rash (4.7%), and diarrhea (3.6%).
Mortality
On-treatment deaths were defined as those occurring within 30 days of the last dose of study treatment. The most common cause of on-treatment death was breast cancer (3.6% in the alpelisib group and 4.1% in the placebo group). Other causes of on-treatment death were reported for 1 patient each: cardiorespiratory arrest in the alpelisib group and gastrointestinal hemorrhage, pneumonia, and septic shock in the placebo group.
Notable Harms
The following notable harms identified in the systematic review protocol occurred in more than 10% of patients of at least 1 treatment group and were more common in the alpelisib group: hyperglycemia, diarrhea, nausea, rash, vomiting, and maculopapular rash. The following notable harms occurred in 3.6% or less of each treatment group: hypersensitivity, anaphylactic reaction, drug hypersensitivity, increased glycated hemoglobin, increased blood glucose, pneumonitis, erythema multiforme, glucose urine present, Stevens-Johnson syndrome, diabetes mellitus, ketoacidosis, and type 2 diabetes mellitus.
Harms | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant Safety analysis set N = 169 | Placebo + fulvestrant Safety analysis set N = 171 | |
Patients with ≥ 1 AE | ||
n (%) | 168 (99.4) | 155 (90.6) |
Most common events,a n (%) | ||
Hyperglycemiab | 113 (66.9) | 15 (8.8) |
Diarrheab | 97 (57.4) | 20 (11.7) |
Nauseab | 82 (48.5) | 35 (20.5) |
Rashb | 69 (40.8) | 12 (7.0) |
Decreased appetite | 58 (34.3) | 13 (7.6) |
Decreased weight | 47 (27.8) | 2 (1.2) |
Stomatitis | 46 (27.2) | 10 (5.8) |
Vomitingb | 46 (27.2) | 17 (9.9) |
Fatigue | 43 (25.4) | 28 (16.4) |
Alopecia | 36 (21.3) | 5 (2.9) |
Asthenia | 32 (18.9) | 23 (13.5) |
Headache | 32 (18.9) | 23 (13.5) |
Pruritus | 29 (17.2) | 7 (4.1) |
Mucosal inflammation | 28 (16.6) | 4 (2.3) |
Back pain | 27 (16.0) | 22 (12.9) |
Pyrexia | 26 (15.4) | 14 (8.2) |
Arthralgia | 25 (14.8) | 30 (17.5) |
Dry skin | 25 (14.8) | 5 (2.9) |
Rash, maculopapularb | 25 (14.8) | 1 (0.6) |
Dyspepsia | 24 (14.2) | 7 (4.1) |
Dysgeusia | 24 (14.2) | 4 (2.3) |
Edema, peripheral | 23 (13.6) | 9 (5.3) |
Blood creatinine, increased | 21 (12.4) | 1 (0.6) |
Anemia | 20 (11.8) | 10 (5.8) |
Abdominal pain | 20 (11.8) | 12 (7.0) |
Hypokalemia | 20 (11.8) | 4 (2.3) |
Cough | 20 (11.8) | 18 (10.5) |
Dry mouth | 19 (11.2) | 5 (2.9) |
Urinary tract infection | 19 (11.2) | 10 (5.8) |
Aspartate aminotransferase, increased | 19 (11.2) | 8 (4.7) |
Insomnia | 18 (10.7) | 3 (1.8) |
Alanine aminotransferase, increased | 17 (10.1) | 10 (5.8) |
Gamma-glutamyltransferase, increased | 17 (10.1) | 16 (9.4) |
Dyspnea | 17 (10.1) | 22 (12.9) |
Hypertension | 17 (10.1) | 9 (5.3) |
Patients with ≥ 1 SAE | ||
n (%) | 67 (39.6) | 34 (19.9) |
Most common events,c n (%) | ||
Hyperglycemiab | 17 (10.1) | 0 |
Osteonecrosis of jaw | 6 (3.6) | 0 |
Stomatitis | 4 (2.4) | 0 |
Acute kidney injury | 4 (2.4) | 0 |
Rashb | 4 (2.4) | 0 |
Abdominal pain | 3 (1.8) | 2 (1.2) |
Diarrheab | 3 (1.8) | 0 |
Pneumonia | 3 (1.8) | 5 (2.9) |
Dehydration | 3 (1.8) | 3 (1.8) |
Anemia | 2 (1.2) | 1 (0.6) |
Nauseab | 2 (1.2) | 0 |
General physical health deterioration | 2 (1.2) | 0 |
Pyrexia | 2 (1.2) | 0 |
Blood creatinine, increased | 2 (1.2) | 0 |
Hypokalemia | 2 (1.2) | 1 (0.6) |
Back pain | 2 (1.2) | 1 (0.6) |
Muscular weakness | 2 (1.2) | 0 |
Brain edema | 2 (1.2) | 0 |
Dyspnea | 2 (1.2) | 2 (1.2) |
Pleural effusion | 2 (1.2) | 4 (2.3) |
Pulmonary embolism | 2 (1.2) | 1 (0.6) |
Erythema multiforme | 2 (1.2) | 0 |
Rash, maculopapularb | 2 (1.2) | 0 |
Patients who stopped treatment due to AEs | ||
n (%) | 46 (27.2) | 10 (5.8) |
Most common events,c n (%) | ||
Hyperglycemiab | 11 (6.5) | 0 |
Rashb | 8 (4.7) | 0 |
Diarrheab | 6 (3.6) | 0 |
Nauseab | 3 (1.8) | 0 |
Stomatitis | 3 (1.8) | 1 (0.6) |
Fatigue | 3 (1.8) | 0 |
Lipase, increased | 3 (1.8) | 4 (2.3) |
Vomiting | 2(1.2) | 0 |
Mucosal inflammation | 2 (1.2) | 0 |
Decreased appetite | 2 (1.2) | 0 |
Rash, maculopapularb | 2 (1.2) | 0 |
Patients who required medications or therapies for AEs | ||
n (%) | 163 (96.4) | 109 (63.7) |
Most common events,a n (%) | ||
Hyperglycemiab | 101 (59.8) | 6 (3.5) |
Diarrheab | 53 (31.4) | 6 (3.5) |
Rashb | 52 (30.8) | 7 (4.1) |
Nauseab | 37 (21.9) | 13 (7.6) |
Stomatitis | 35 (20.7) | 5 (2.9) |
Rash, maculopapularb | 23 (13.6) | 1 (0.6) |
Urinary tract infection | 19 (11.2) | 8 (4.7) |
Mucosal inflammation | 17 (10.1) | 2 (1.2) |
On-treatment deaths (up to 30 days after last dose of study treatment) | ||
n (%) | 7 (4.1) | 10 (5.8) |
Breast cancer | 6 (3.6) | 7 (4.1) |
Cardiorespiratory arrest | 1 (0.6) | 0 |
Gastrointestinal hemorrhage | 0 | 1 (0.6) |
Pneumonia | 0 | 1 (0.6) |
Septic shock | 0 | 1 (0.6) |
Other notable harms, n (%) | ||
Pneumonitis | 2 (1.2) | 0 |
Hypersensitivity | 6 (3.6) | 0 |
Anaphylactic reaction | 1 (0.6) | 0 |
Drug hypersensitivity | 1 (0.6) | 0 |
Stevens-Johnson syndrome | 1 (0.6) | 0 |
Erythema multiforme | 2 (1.2) | 0 |
Glycated hemoglobin, increased | 5 (3.0) | 0 |
Blood glucose, increased | 3 (1.8) | 1 (0.6) |
Glucose urine present | 2 (1.2) | 0 |
Diabetes mellitus | 1 (0.6) | 1 (0.6) |
Ketoacidosis | 1 (0.6) | 0 |
Type 2 diabetes mellitus | 1 (0.6) | 0 |
AE = adverse event; SAE = serious adverse event.
Note: All patients at cut-off date of April 23, 2020.
aFrequency of more than 10%.
bFrequency of more than 1%.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Critical Appraisal
Internal Validity
The methods for randomization, treatment allocation, and maintenance of blinding to treatment assignment were appropriate in the SOLAR-1 study. However, the differences in side-effect profiles between the treatment groups may have resulted in treatment unblinding in both patients and investigators. For example, hyperglycemia was reported in 66.9% of patients in the alpelisib group and 8.8% of patients in the placebo group, while 26.6% of patients in the alpelisib group discontinued treatment due to AEs compared with 5.8% of patients in the placebo group. Since PFS was assessed objectively (with the radiologist remaining blinded to treatment assignment) and there was regular follow-up for PFS and OS, the risk of bias from potential treatment unblinding was low for determining these outcomes. However, the bias caused by potential differences in care, increased monitoring, or missing visits is unclear.
There were discrepancies among different approaches for identifying the subgroup of patients in the PIK3CA cohort with prior CDK4/6 inhibitor treatment, with the approach in the main analysis being the use of the randomization stratum (9 patients and 11 patients in the alpelisib group and placebo group, respectively). Although there was a sensitivity analysis of PFS performed in the subgroup of patients with prior CDK4/6 inhibitor treatment as identified by the clinical data, the numbers of patients in this sensitivity analysis (12 in the alpelisib group and 11 in the placebo group) did not match those reported in the summary of prior antineoplastic therapy (11 in the alpelisib group and 8 in the placebo group). It is uncertain if there were patients in the main analysis and sensitivity analysis who hadn’t received prior CDK4/6 inhibitor treatment and if this was more common in 1 group than the other. It is also possible that a number of patients who had received prior CDK4/6 inhibitor treatment were not included in the main analysis and sensitivity analysis. The potential impact on the results from differing approaches in identifying patients with prior CDK4/6 inhibitor treatment for analysis is unclear.
Within the whole PIK3CA mutant cohort, some imbalances in baseline characteristics were noted; however, the clinical experts consulted by CADTH for this review did not consider them likely to impact the efficacy and safety results. Within the subgroup with prior CDK4/6 inhibitor treatment, there was a lower percentage of patients in the alpelisib group with an ECOG PS of 0 (44.5% in the alpelisib group versus 72.7% in the placebo group), with potential bias in favour of the placebo group.
Despite the high percentages of patients discontinuing treatment in both groups, the percentages of patients discontinuing from the study were lower than 6% in each group. The percentages of patients discontinuing the study were higher in the alpelisib group versus the placebo group (4.1% versus 2.9% at the primary PFS analysis and 5.9% versus 2.9% at the final OS analysis). Similar trends were noted in percentages of patients discontinuing the study due to progressive disease. There was also a higher percentage of patients in the alpelisib group censored from the primary PFS analysis due to withdrawn consent than in the placebo group (5.9% versus 2.9%). The potential direction of bias is unclear for these imbalances in discontinuation and censoring, although the low percentages in each category (less than 6%) mean that any impact on the efficacy results is likely to be minor in the PIK3CA mutant cohort. However, patient disposition was not available for the subgroup with prior CDK4/6 inhibitor treatment and it is unclear what impact study discontinuations may have had on results in that subgroup.
There were some analytical issues of note in the OS analysis. The percentages of patients lost to follow-up were 10.7% and 9.9% in the alpelisib group and placebo group, respectively, and the potential impact of this missing data is uncertain. There were also no PP analyses conducted for OS in the subgroup with prior CDK4/6 inhibitor treatment. In addition, it is unclear from visual inspection of the OS Kaplan-Meier curves in Figure 3 whether the proportional hazards assumption was met. Additionally, no consideration was made for informative censoring via adjustment in the Cox model in the PFS and OS analyses, nor was the small sample size considered, as the well-known statistical properties of the Cox model likely do not hold in such a sample size.
Regardless of the aforementioned issues with internal validity in the SOLAR-1 study, the main limitation of the study that precludes any assessment of comparative efficacy in the population with prior CDK4/6 inhibitor treatment is that the study was not designed to test hypotheses in that population. The 2 outcomes in the testing hierarchy were PFS and OS in the entire PIK3CA mutant cohort and the study sample size considerations were based on those outcomes. As described in the SOLAR-1 Interim Clinical Study Report, “Analyses of efficacy in subgroups were intended to explore the intrinsic consistency of any treatment effect.” In the absence of pre-specified subgroup analyses within the statistical testing hierarchy, no conclusions can be drawn about efficacy in any of the subgroups.
External Validity
As with internal validity, the main limitation of the SOLAR-1 study in terms of external validity is that the objectives of the study do not align with the relevant population for this review. The clinical experts consulted by CADTH for this review indicated that the results for the entire PIK3CA cohort cannot be used to inform the comparative efficacy of alpelisib specifically in patients with prior CDK4/6 inhibitor treatment. This uncertainty, combined with the inability to draw conclusions in the subgroup with prior CDK4/6 inhibitor treatment, means that there is insufficient evidence to support the efficacy of alpelisib and fulvestrant compared with fulvestrant alone in the relevant population for this review. It is important to note that the population targeted by the sponsor’s reimbursement request largely represents the population that would be considered for alpelisib and fulvestrant therapy in Canadian clinical practice, since an endocrine regimen with a CDK4/6 inhibitor is the current SOC for first-line treatment for advanced and metastatic breast cancer. Another issue with the study population is that the SOLAR-1 study included patients with prior endocrine therapy, regardless of setting, and 52% of patients in the PIK3CA mutant cohort had not received endocrine therapy in the metastatic setting. In contrast, patients in Canada who are suited for treatment with alpelisib and fulvestrant will have received endocrine therapy in the metastatic setting, according to the clinical experts consulted by CADTH.
Female patients receiving an LHRH agonist for induction of ovarian suppression were excluded from the SOLAR-1 study, but the clinical experts consulted by CADTH considered it reasonable to generalize the results to this patient population. Although the PIK3CA mutant cohort had 1 male patient and the results were not considered to be generalizable to male patients, the clinical experts would not exclude male patients from treatment with alpelisib and fulvestrant in practice.
Aside from the aforementioned issues, the baseline characteristics, including ECOG PS, sites of metastases, and endocrine resistance status (primary resistance versus secondary resistance), appeared to be in line with patients treated in the Canadian setting, according to the clinical experts consulted by CADTH, although the clinical experts noted that patients with an ECOG PS of 2 may be considered for treatment with alpelisib plus fulvestrant in practice.
According to the study protocol, patients who discontinued treatment with 1 study drug (alpelisib/placebo or fulvestrant) could continue to receive the other study drug. The clinical experts consulted by CADTH for this review indicated that alpelisib monotherapy would not be an option upon discontinuation of fulvestrant.
The frequency of assessments of disease progression and patient follow-up in the study were similar to what would be found in Canadian clinical practice, according to the clinical experts consulted by CADTH, though in practice most patients would have imaging assessments every 12 weeks from the start.
Indirect Evidence
CADTH performed a literature review to identify any relevant indirect comparisons. A focused literature search for network meta-analyses dealing with breast cancer was run in MEDLINE All (1946–) on May 19, 2021. No limits were applied to the search. The search yielded 253 results whose abstracts were reviewed for relevance. Of those, 3 were identified as potentially relevant and, upon full-text review, were deemed not relevant because they did not perform any analyses restricted to the patient population of interest.24-26 An additional result27 from a clinical literature search alert was excluded upon full-text review for the same reason. Therefore, no indirect evidence was available for this review.
Other Relevant Evidence
This section includes 2 additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review. The patient population in the SOLAR-1 study did not align with the patient population relevant to this review, those with prior CDK4/6 inhibitor treatment, and conclusions on the comparative efficacy of alpelisib and fulvestrant versus fulvestrant alone could not be drawn in that population. The BYLieve study, a non-comparative cohort study, included 1 cohort of patients treated with alpelisib and fulvestrant that matched the relevant patient population. In a separate observational study, the relevant cohort of the BYLieve study was compared with a database-derived cohort treated with non-alpelisib SOC following propensity score weighting.
Non-Comparative Cohort Study
One sponsor-submitted, non-comparative cohort study is summarized to provide additional evidence regarding the efficacy and safety of alpelisib plus fulvestrant in patients with PIK3CA-mutated, hormone receptor–positive, HER2-negative advanced breast cancer who have been previously treated with a CDK4/6 inhibitor in combination with an endocrine-based therapy.
Table 17: Details of the BYLieve Study
Criteria | Description |
|---|---|
Design and population | |
Study design | Phase II, open-label, non-comparative cohort study |
Locations | 114 study locations in North America (including 3 sites in Canada), South America, Europe, and Asia |
Patient enrolment dates | Cohort A: August 14, 2017, to December 17, 2019 Cohort B: August 14, 2017, to ongoing Cohort C: May 9, 2019, to ongoing |
Enrolled as of DCO (n) | Cohort A: 127 Cohort B: 81 Cohort C: 1 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Cohorts | Cohort A: Alpelisib (300 mg p.o. q.d.) + fulvestrant (500 mg IM, day 1 and day 15 of cycle 1 and day 1 of each 28-day cycle thereafter) Cohort B: Alpelisib (300 mg p.o. q.d.) + letrozole Cohort C: Alpelisib (300 mg p.o. q.d.) + fulvestrant (500 mg IM, day 1 and day 15 of cycle 1 and day 1 of each 28-day cycle thereafter) |
Duration | |
Phase | |
Pre-screening and screening | 21 days |
Treatment | Ongoing until disease progression, intolerable toxicity, or 18 months after last patient’s first treatment. |
Follow-up | Ongoing until death, loss to follow-up, or withdrawal of consent for survival follow-up, or end of study |
Outcomes | |
Primary end point | Proportion of patients who are alive without disease progression at 6-month follow-up based on local investigator assessment using RECIST 1.1 |
Secondary and exploratory end points | Secondary
Exploratory
|
Notes | |
Publications | Rugo et al. (2021)28 – Primary analysis of Cohort A |
AI = aromatase inhibitor; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; CBR = clinical benefit rate; CDK4/6 = cyclin-dependent kinase 4 and 6; CNS = central nervous system; ctDNA = circulating tumour DNA; DCO = data cut-off; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ET = endocrine therapy; FPG = fasting plasma glucose; GI = gastrointestinal; HER2 = human epidermal growth factor receptor 2; IHC = immunohistochemistry; IM = intramuscular; INR = international normalized ratio; NGS = next-generation sequencing; ORR = objective response rate; OS = overall survival; PI3K = phosphatidylinositol-3-kinase; PFS = progression-free survival; PFS2 = progression-free survival after next line of treatment; p.o. = orally; q.d. = every day; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; ULN = upper limit of normal.
aPatients with PIK3CA mutation confirmed by a Novartis-designated laboratory, in which adequate formalin-fixed paraffin-embedded tissue sections with more than 10% tumour tissue had to be provided. It was recommended to provide a tumour sample collected after the most recent progression or recurrence, or patients with a pathology report confirming PIK3CA mutant status by a certified laboratory using a validated PIK3CA mutation assay (either from tissue or blood). It was also mandatory to provide a tumour sample (either archival or newly obtained) for a PIK3CA mutation confirmation by a Novartis-designated laboratory. It was recommended that the tumour sample be collected after the most recent progression or recurrence.
bANC of 1.5 × 109 per litre or more; platelets of 100 × 109 per litre or more (for patients with lesions involving the bone marrow, platelet count ≥ 75 × 109 per litre could be acceptable); hemoglobin of 9.0 g/dL or more; INR of 1.5 or less (INR ≤ 2.0 was allowed for those patients treated with vitamin K antagonist); in the absence of liver metastases, serum AST and ALT of 2.5 × ULN or less or of 5 × ULN or less if hepatic metastases were present; total serum bilirubin less than 2 × ULN (any elevated bilirubin should be asymptomatic at enrolment) except for patients with Gilbert syndrome, who could only be included if the total bilirubin was 3.0 × ULN or less or direct bilirubin of 1.5 × ULN or less; serum creatinine of 1.5 × ULN or less or creatinine clearance of 35 mL per minute or more using Cockcroft-Gault formula.
cPatients with CNS involvement were excluded unless they met the following criteria: (i) At least 4 weeks from prior therapy completion (including radiation and/or surgery) to starting the study treatment; (ii) Clinically stable CNS tumour at the time of screening untreated or without evidence of progression for at least 4 weeks after treatment as determined by clinical examination and brain imaging (MRI or CT) during the screening period; (iii) were on a stable low dose of steroids for 2 weeks before initiating study treatment.
dPatients were required to have FPG of 140 mg/dL (7.7 mmol/L) or less and hemoglobin A1C of 6.4% or less.
Source: Rugo et al. (2021),28 BYLieve Clinical Study Report,32 and BYLieve First Interpretable Results (2021).29
Methods
The BYLieve study is an ongoing phase II, multi-centre, open-label, 3-cohort, non-comparative study. The objective of the study, as stated in the BYLieve Clinical Study Report, is to assess the efficacy and safety of alpelisib plus endocrine therapy (either fulvestrant or letrozole) in premenopausal and postmenopausal women, and men, with PIK3CA-mutant, hormone receptor–positive, HER2-negative advanced breast cancer who have progressed on or after prior treatments. Gonadal suppression had to be achieved with either goserelin or leuprolide in men and premenopausal women. Patients were assigned to a cohort based on most recent previous therapy (AI or fulvestrant) as indicated in the following:
Cohort A: Patients who received any CDK4/6 inhibitor plus any AI as immediate prior treatment will receive alpelisib plus fulvestrant.
Cohort B: Patients who received any CDK4/6 inhibitor plus fulvestrant as immediate prior treatment will receive alpelisib plus letrozole.
Cohort C: Patients who received systemic chemotherapy or endocrine therapy (as monotherapy or in combination with targeted treatment, except CDK4/6 inhibitor plus AI) as immediate prior treatment will receive alpelisib plus fulvestrant. In this cohort, endocrine therapy included letrozole, fulvestrant, and CDK4/6 inhibitor plus fulvestrant.
The BYLieve study planned to enrol 112 patients in each cohort (total N = 336). As of the data cut-off date, a total of 209 patients were enrolled: 127 patients in cohort A, 81 patients in cohort B, and 1 patient in cohort C. The BYLieve study enrolled patients from 114 multinational study locations (including 3 sites in Canada).
The overall design of the BYLieve study is summarized in Figure 4. The BYLieve study included a pre-screening phase and a screening phase. In the pre-screening phase, patients were tested for eligible PIK3CA mutations. Patients who tested positive for the PIK3CA mutation were then offered the opportunity to sign the main study informed consent form to enter the screening phase. The pre-screening and screening phases were to be completed within 21 days. In the treatment phase, patients were treated until disease progression, intolerable toxicity, or until 18 months after the last enrolled patient’s first treatment. Treatment crossover between cohorts was not permitted. Patients who discontinued study treatment due to disease progression, death, loss to follow-up, or withdrawal of consent for efficacy follow-up were not followed for efficacy, though those who discontinued due to disease progression were followed for safety for 30 days. Patients who discontinued study treatment for reasons other than disease progression, death, loss to follow-up, or withdrawal of consent for efficacy follow-up entered the post-treatment follow-up phase, which included 30 days of safety follow-up and efficacy follow-up. For post-treatment efficacy follow-up, tumour response was assessed locally via CT or MRI per RECIST 1.1 every 12 weeks until disease progression, death, withdrawal of consent, loss to follow-up, a patient or guardian decision, or the end of the study. For the survival follow-up period following end-of-treatment follow-up, all patients were followed for survival status after progression every 12 weeks regardless of treatment discontinuation reason.
Figure 4: BYLieve Study Design — Overview
a All patients were followed for survival status (after progression) every 12 weeks regardless of the treatment discontinuation reason (except in cases of consent being withdrawn, death, or a patient being lost to follow-up) until death, loss to follow-up, or withdrawal of consent for survival follow-up or the end of the study.
Source: BYLieve Clinical Study Report.32
The primary outcome of the BYLieve study is the proportion of patients who were alive without disease progression at 6 months based on local investigator assessment using RECIST 1.1 in each cohort. Secondary outcomes included OS and PFS by local investigator assessment.
This report includes the primary end point, OS, PFS, and harms data for cohort A alone because this is the only cohort that aligns with the intervention under review and the reimbursement criteria requested by the sponsor. Cohort B does not align with the intervention under review because patients received alpelisib in combination with letrozole. Cohort C does not align with the reimbursement request because patients were not previously treated with an endocrine-based regimen with a CDK4/6 inhibitor.
Inclusion and Exclusion Criteria
The BYLieve study included men and premenopausal or postmenopausal women (≥ 18 years of age) with advanced (locoregionally recurrent or metastatic) hormone receptor–positive and HER2-negative breast cancer not amenable to curative therapy and a confirmed PIK3CA mutation determined by local or central laboratory testing of tumour tissue or plasma. The inclusion of premenopausal women who were receiving concomitant ovarian suppression with an LHRH agonist was allowed in the first protocol amendment. Patients were required to have measurable disease per RECIST 1.1 criteria or at least 1 predominantly lytic bone lesion present. To be enrolled in cohort A, patients must have had documented tumour progression on or after treatment with a CDK4/6 inhibitor in combination with an AI (the last treatment regimen before study entry), an ECOG PS of 2 or less, fasting plasma glucose of 140 mg/dL (7.7 mmol/L) or less, and glycated hemoglobin of 6.4% or less. Patients were excluded if they had an established diagnosis of diabetes mellitus type 1 or uncontrolled type 2 diabetes. Patients could have 2 or fewer previous anticancer therapies and no more than 1 previous chemotherapy regimen in the advanced or metastatic setting. Patients with CNS involvement were excluded unless they met the following criteria: they were at least 4 weeks from prior therapy completion (including radiation and/or surgery) to starting the study treatment; they had a clinically stable CNS tumour at the time of screening, untreated or without evidence of progression for at least 4 weeks after treatment as determined by clinical examination and brain imaging (MRI or CT) during the screening period; and they were on a stable low dose of steroids for 2 weeks before initiating study treatment.
Baseline Characteristics
The baseline characteristics of patients enrolled in cohort A of the BYLieve study are summarized in Table 18. The mean age of patients was 56.7 years. All patients (100%) were female. Most patients were postmenopausal (78.0%), were Caucasian (63.8%), had stage IV disease at study entry (97.6%), had invasive ductal carcinoma (68.5%), had secondary endocrine resistance (59.8%), and had an ECOG PS of 0 (62.2%). The mean time from most recent recurrence and/or relapse was 2.2 months.
Table 18: Summary of Baseline Characteristics — Cohort A of BYLieve Study
Baseline characteristic | Cohort A (N = 127) |
|---|---|
Age, mean (SD), years | 56.7 (10.7) |
Sex, n (%) | |
Female | 127 (100) |
Male | 0 |
Race | |
Caucasian | 81 (63.8) |
Asian | 12 (9.4) |
Black | 6 (4.7) |
Pacific Islander | 1 (0.8) |
Other | 3 (2.4) |
Unknown | 23 (18.1) |
Missing | 1 (0.8) |
Ethnicity | |
Hispanic or Latino | 20 (15.7) |
East Asian | 7 (5.5) |
South Asian | 3 (2.4) |
Southeast Asian | 3 (2.4) |
Mixed ethnicity | 1 (0.8) |
Other | 42 (33.1) |
Unknown | 19 (15.0) |
Not reported | 32 (25.2) |
BMI, mean (SD), kg/m2 a | 26.1 (5.5) |
Menopausal status, n (%) | |
Premenopausal | 28 (22.0) |
Postmenopausal | 99 (78.0) |
ECOG PS, n (%) | |
0 | 79 (62.2) |
1 | 41 (32.3) |
2 | 2 (1.6) |
Missing | 5 (3.9) |
Histology/cytology, n (%) | |
Invasive ductal carcinoma | 87 (68.5) |
Invasive lobular carcinoma | 18 (14.2) |
Adenocarcinoma | 12 (9.4) |
Lobular carcinoma in situ | 2 (1.6) |
Squamous cell carcinoma | 1 (0.8) |
Undifferentiated carcinoma | 1 (0.8) |
Other | 5 (3.9) |
Not applicable | 1 (0.8) |
Time since most recent recurrence/relapse, mean (SD), months | 2.2 (2.5) |
Stage at time of study entry, n (%) | |
III | 3 (2.4) |
IV | 124 (97.6) |
Lines of previous therapy in metastatic setting, n (%) | |
0 | 15 (11.8) |
1 | 89 (70.1) |
2 | 21 (16.5) |
3 | 2 (1.6) |
Lines of previous endocrine therapy in the metastatic setting, n (%) | |
0 | 15 (11.8) |
1 | 98 (77.2) |
2 | 14 (11.0) |
Endocrine status at study entry, n (%) | |
Primary endocrine resistance | 26 (20.5) |
Secondary endocrine resistance | 76 (59.8) |
Endocrine sensitivity | 1 (0.8) |
Current extent of disease, metastatic sites, n (%) | |
Bone | 108 (85.0) |
Bone only | 24 (18.9) |
Visceral | 85 (66.9) |
Liver | 59 (46.5) |
Lung | 43 (33.9) |
Other visceral | 8 (6.3) |
Lymph nodes | 37 (29.1) |
Breast | 5 (3.9) |
Skin | 4 (3.1) |
CNS | 2 (1.6) |
Other | 12 (9.4) |
BMI = body mass index; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; SD = standard deviation.
an = 117.
Source: Rugo et al. (2021)28 and BYLieve Clinical Study Report.32
Interventions
Patients were assigned to receive alpelisib plus fulvestrant (cohort A) if their immediate prior treatment was a CDK4/6 inhibitor plus any AI. Patients received 300 mg alpelisib orally once per day and 500 mg fulvestrant intramuscularly on day 1 of each 28-day cycle and on day 15 of cycle 1. Treatment continued until disease progression, unacceptable toxicity, death, or discontinuation from study treatment due to any other reason. For patients unable to tolerate alpelisib due to AEs, a maximum of 2 dose reductions of alpelisib were allowed (the first dose reduction was to 250 mg per day while the second dose reduction was to 200 mg per day). Men and premenopausal women also received goserelin (an injectable subcutaneous implant, 3.6 mg, every 28 days) or leuprolide (injectable IM depot, 7.5 mg, every 28 days) as study treatments.
Concomitant medications required to treat AEs, manage cancer symptoms, and treat concurrent diseases were allowed, as were supportive care agents (e.g., pain medications, antiemetics, antidiarrheals). Oral antidiabetics were permitted to treat patients who developed hyperglycemia during the study. Gastric protection agents, corticosteroids, and palliative radiotherapy (i.e., local radiotherapy for analgesic purposes or for lytic lesions at risk of fracture) were also permitted. Anticoagulants and bisphosphonates could be used with caution during study treatment. Stronger inducers of CYP3A4 were prohibited.
Outcomes
The outcomes identified in the CADTH systematic review protocol that were included in the BYLieve study design are summarized in Table 19.
Table 19: Summary of Outcomes of Interest Identified in the CADTH Review Protocol — BYLieve Study
Outcome measure | BYLieve study | Definition |
|---|---|---|
OS | Secondary | Time from the date of start of treatment to the date of death |
PFS | Secondary | Time from the date of start of treatment to the date of the first documented progression or death due to any cause. Disease progression was assessed by the local investigator using RECIST 1.1 criteria. |
HRQoL | Not reported | NA |
HRQoL = health-related quality of life; NA = not applicable; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source BYLieve Clinical Study Report.32
The primary end point of the BYLieve study was defined as the proportion of patients who were alive without disease progression at 6 months by local investigator assessment using RECIST 1.1 criteria. In the original study protocol, tumour response was assessed locally via CT or MRI per RECIST 1.1 at screening, every 8 weeks in the first 6 months, and every 12 weeks thereafter.28 After approval of a protocol amendment (January 30, 2019), assessments occurred every 12 weeks per SOC throughout the study until disease progression, death, withdrawal of consent, loss to follow-up, a patient or guardian decision, or the end of the study. If there was clinical suspicion of disease progression, physical examination and imaging assessment were performed promptly.
Safety was evaluated by collecting data on AEs at every visit as well as by physical examination, vital signs, performance status evaluation, electrocardiogram, cardiac imaging, and laboratory evaluations for hematology and biochemistry, including glucose monitoring.
Statistical Analysis
In the BYLieve study protocol, it was planned that the primary analysis for each cohort would be performed 6 months after the last patient enrolled in the cohort had started treatment. Two interim analyses were added as protocol amendments. The first interim analysis was planned to be performed after at least 20 patients receiving alpelisib plus fulvestrant (cohort A) had received at least 6 months of follow-up. The second interim analysis was planned to be performed when approximately 170 patients (50% of enrolled patients) had been treated in the study (regardless of cohort) and had received at least 6 months of follow-up. At the time of the interim analyses, preliminary disease progression, survival, and safety data were analyzed in a descriptive fashion. The primary end point was not analyzed, and no statistical hypothesis was tested. Another protocol amendment allowed analyses of the primary end point for each cohort independent of the other cohorts. Following this update, the primary analysis for each cohort was planned to be performed 6 months after the start of treatment by the last patient. The final analysis for cohort A with testing of the statistical hypothesis for the primary end point has been completed.
Analysis Sets
The FAS was defined as all patients to whom study treatment was assigned and who had received at least 1 dose of study treatment. Patients were analyzed according to the treatment to which they were assigned. The FAS was used for all baseline and demographic summaries, patient disposition, and biomarker analysis.
The mFAS included all patients of the FAS population who had PIK3CA mutation confirmed by a Novartis-designated laboratory; patients were analyzed according to the treatment to which they were assigned. The mFAS was designated as the primary population for the analysis of progression and survival end points.
The PP set included a subset of the patients in mFAS who were compliant with the protocol requirements. The PP set was used for sensitivity analyses of the primary end point.
The safety analysis set included patients who had received at least 1 dose of any component of the study treatment. Patients were analyzed according to the study treatment received.
Primary End Point Analysis
The proportion of patients alive without disease progression after 6 months was presented with a 95% CI that was determined using the Clopper and Pearson exact method.30 Per the statistical analysis plan, the null hypothesis would be rejected if the lower bound of the 95% CI was greater than 30%.
The study protocol defined 6 months as 24 weeks, plus or minus 1 week; therefore, tumour assessments between week 23 and week 25 were considered for the primary analysis. For the primary end point analysis, patients who progressed, died, or discontinued the study before 6 months were counted as a “failure.”
The mFAS is the primary analysis population for the primary end point. It was planned that analysis of the primary end point would also be performed on the FAS and possibly be repeated on the PP set as supportive analyses. As per protocol, the primary analysis for each cohort was to be performed 6 months after the last patient had started treatment or discontinued early in the respective cohort. The primary end point was planned to be analyzed only at the final analyses of cohort A; hence, the sponsor determined that the end point did not require adjustment for multiplicity.
Sample Size Calculation
The sample size was based on an exact binomial test for a single proportion to test the null hypothesis that the proportion of patients who are alive without progression at 6 months is less than or equal to 0.30. With a 1-sided 2.5% level of significance (2-sided 95% CI), a total sample size of 112 patients in each cohort was required to have a power of at least 90% when the true proportion of patients who are alive without progression at 6 months is greater than or equal to 0.45.
Secondary End Point Analysis
Overall Survival
OS was estimated using the Kaplan-Meier method. The median OS, the OS rate at 6 months, and the OS rate at 12 months with 95% CIs were reported. If a patient was not known to have died, OS was censored at the last known date that the patient was alive.
Progression-Free Survival
PFS was estimated using the Kaplan-Meier method. The median PFS, the PFS rate at 6 months, and the PFS rate at 12 months with 95% CIs were reported. If no PFS event was observed before the cut-off date, PFS was censored. The censoring date was the date of the last adequate tumour assessment before the data cut-off date. If a PFS event was observed after 2 or more missing or non-adequate tumour assessments, then PFS was censored at the date of the last adequate tumour assessment. If a PFS event was observed after a single missing or non-adequate tumour assessment, the actual date of the event was used.
Patient Disposition
A total of 127 patients were enrolled into cohort A of the BYLieve study and 100% of those patients were treated. As of the data cut-off date, 26.0% of patients had ongoing study treatment and 74.0% had discontinued treatment. The most common reasons for discontinuing study treatment were progressive disease (50.4%) and AEs (14.2%).
A total of 82 (64.6%) patients discontinued study treatment but did not enter the post-treatment efficacy follow-up phase of the study. These patients did not enter the efficacy follow-up phase per the study protocol because they discontinued study treatment due to documented disease progression, death, loss to follow-up, or withdrawal of consent.
Table 20: Patient Disposition — Cohort A of BYLieve Study, FAS
Disposition | Cohort A |
|---|---|
Enrolled, Na | 127 |
Treated, n (%) | 127 (100.0) |
Ongoing study treatment, n (%) | 33 (26.0) |
Completed study treatment, n (%) | 0 |
Discontinued study treatment, n (%) | 94 (74.0) |
Progressive disease | 64 (50.4) |
AE | 18 (14.2) |
Physician decision | 4 (3.1) |
Death | 3 (2.4) |
Patient/guardian decision | 3 (2.4) |
Protocol deviation | 1 (0.8) |
Technical problems | 1 (0.8) |
Post-treatment efficacy follow-up for patients who discontinued treatment, n (%) | NA |
Did not enter post-treatment efficacy follow-up | 82 (64.6) |
Ongoing | 4 (3.1) |
Discontinued due to progressive disease | 9 (7.1) |
Discontinued due to death | 1 (0.8) |
FAS, n (%) | 127 (100) |
mFAS, n (%)b | 121 (95.3) |
PP set, n (%)c | 106 (83.5) |
Safety analysis set, n (%) | 127 (100) |
AE = adverse event; FAS = full analysis set; mFAS = modified full analysis set; NA = not applicable; PP = per-protocol.
aEnrolled as of the December 17, 2019, data cut-off date.
bThe mFAS included all patients of the FAS population who had PIK3CA mutation confirmed by a Novartis-designated laboratory.
cThe PP set included patients in the mFAS who were compliant with the protocol requirements.
Source: Rugo et al. (2021),28 BYLieve Clinical Study Report,32 and BYLieve First Interpretable Results (2021).29
Protocol Deviations
Overall, 45.7% of patients in cohort A had protocol deviations. The most frequently reported protocol deviations were use of a prohibited concomitant medication (18.1%) and study treatment deviation (13.4%). The most common study treatment deviation was fulvestrant dose 500 mg not administered (10.2%).
Table 21: Summary of Protocol Deviations — Cohort A of BYLieve Study
Category of protocol deviation | Cohort A (N = 127) |
|---|---|
Any protocol deviation, n (%) | 58 (45.7) |
Use of prohibited concomitant medication | 23 (18.1) |
Study treatment deviation | 17 (13.4) |
Inclusion criteria deviation | 4 (3.1) |
Exclusion criteria deviation | 2 (1.6) |
Not discontinued after meeting withdrawal criteria | 2 (1.6) |
Other deviation | 10 (7.9) |
Source: BYLieve Clinical Study Report.32
Exposure to study treatments in cohort A of the BYLieve study is summarized in Table 22. The median duration of exposure to alpelisib was 5.1 months (N = 127); the median duration of exposure to fulvestrant was 6.5 months (N = 126). The median (interquartile range) relative dose intensity for alpelisib was 89.9% (75.0% to 100%).
PP premenopausal women were eligible to participate in the BYLieve study if they were receiving concomitant ovarian suppression with a LHRH agonist. A total of 16 patients were exposed to goserelin and the median duration of exposure was 8.3 months (range = 1 month to 17 months). Two patients were exposed to leuprolide: 1 for less than 2 months and 1 for 7 months.
Table 22: Summary of Exposure to Study Treatments — Cohort A of BYLieve Study, Safety Analysis Set
Parameter | Cohort A | |||
|---|---|---|---|---|
Alpelisib (N = 127) | Fulvestrant (N = 126) | Goserelin (N = 16) | Leuprolide (N = 2) | |
Average daily dose, mg | ||||
Mean (SD) | 281.5 (25.0) | 500.0 (0.0) | NR | NR |
Median (IQR) | 299.1 (262.1 to 300.0) | 500.0 (500.0 to 500.0) | NR | NR |
Dose intensity, mg/day | ||||
Mean (SD) | 253.5 (53.7) | 21.9 (6.2) | NR | NR |
Median (IQR) | 269.6 (225.0 to 300.0) | 20.6 (19.7 to 25.0) | NR | NR |
Relative dose intensity, % | ||||
Mean (SD) | 84.5 (17.9) | NR | NR | NR |
Median (IQR) | 89.9 (75.0 to 100.0) | NR | NR | NR |
Duration of exposure, months | ||||
Mean (SD) | 5.8 (4.7) | 6.7 (4.7) | 8.1 (4.5) | 4.6 (3.9) |
Median (IQR) | 5.1 (1.8 to 8.6) | 6.5 (2.3 to 9.0) | 8.3 (4.2 to 11.4) | 4.6 (1.9 to 7.4) |
IQR = interquartile range; NR = not reported; SD = standard deviation.
Note: Average dose does not consider drug-free days, whereas dose intensity and relative dose intensity include days of 0 doses in the calculation. Cumulative dose of a study treatment is defined as the total dose given during the study treatment exposure. Dose intensity for patients with non-zero duration of exposure is defined as dose intensity (mg/day) = actual cumulative dose (mg) ÷ duration of exposure to study treatment (day), where actual cumulative dose refers to the total actual dose administered over the duration for which the patient is on the study treatment. Relative dose intensity is defined as dose intensity ÷ planned dose intensity, where planned dose intensity is planned cumulative dose (mg) ÷ duration of exposure (day). The planned cumulative dose for a study treatment component refers to the total planned dose per the protocol up to the last date of study drug administration.
Source: BYLieve Clinical Study Report.32
Dose reductions and interruptions in cohort A of the BYLieve study are summarized in Table 23. Per the study protocol, a maximum of 2 dose reductions of alpelisib (first dose reduction to 250 mg per day; second dose reduction to 200 mg per day) were allowed for each patient, after which the patient was discontinued. Overall, 51.2% of patients required at least 1 dose reduction during treatment with alpelisib. The most common reason for a dose reduction was an AE (37.8%) and the median duration of dose reduction was 35 days. Overall, 63.8% of patients required at least 1 dose interruption during treatment with alpelisib. The most common reason for dose interruption was an AE (59.8%).
Table 23: Summary of Dose Reductions and Interruptions — Cohort A of BYLieve Study, Safety Analysis Set
Dose Parameter | Cohort A (N = 127) | |
|---|---|---|
Alpelisib | Fulvestrant | |
Dose reductions | ||
Required ≥ 1 dose reduction, n (%) | 65 (51.2) | NA |
1 | 48 (37.8) | NA |
≥ 2 | 17 (13.4) | NA |
Median duration of dose reduction, days (range) | 35 (1 to 527) | NA |
Reasons for dose reduction, n (%) | ||
AE | 48 (37.8) | NA |
Physician decision | 9 (7.1) | NA |
Per-protocol | 3 (2.4) | NA |
Dosing error | 2 (1.6) | NA |
Patient/guardian decision | 2 (1.6) | NA |
Dose interruptions | ||
Required ≥ 1 dose interruption, n (%) | 81 (63.8) | 4 (3.2) |
1 | 35 (27.6) | 4 (3.2) |
2 | 27 (21.3) | 0 |
≥ 3 | 19 (15.0) | 0 |
Median number of dose interruptions, n (range) | 1 (0 to 36) | NR |
Median duration of dose interruption, days (range) | 12 (1 to 60) | 1 (1 to 4) |
Reasons for dose interruption, n (%) | ||
AE | 76 (59.8) | 2 (1.6) |
Physician decision | 7 (5.5) | 0 |
Per-protocol | 0 | 1 (0.8) |
Dosing error | 12 (9.4) | 0 |
Patient/guardian decision | 5 (3.9) | 0 |
Dispensing error | 0 | 1 (0.8) |
AE = adverse event; NA = not applicable; NR = not reported.
Source: BYLieve Clinical Study Report.32
Progression and Survival Outcomes
Progression and survival outcomes for cohort A of the BYLieve study (alpelisib plus fulvestrant in patients who were previously treated with any CDK4/6 inhibitor plus any AI) are summarized in Table 24. At the time of the data cut-off date (December 17, 2019), the median duration of follow-up in cohort A was 11.7 months.
The CADTH review protocol identified PFS, OS, and HRQoL as the main efficacy outcomes of interest. Results for these, along with the primary outcome used in the BYLieve study, are the focus of this summary.
Primary Outcome
As of the data cut-off date, 61 of 121 (50.4%) patients in the mFAS were alive without progressive disease per investigator assessment at 6 months (95% CI, 41.2% to 59.6%). The study met the primary objective for cohort A because the lower bound of the 95% CI was greater than 30%.
A supportive analysis was done in the PP set, which showed 56 of 106 (52.8%) patients were alive without progressive disease at 6 months.
Progression-Free Survival
A total of 72 (59.5%) PFS events were observed as of the data cut-off date, comprising 66 (54.5%) progressive disease events and 6 (5.0%) deaths. The Kaplan-Meier plot of PFS by investigator assessment is depicted in Figure 5. The median PFS by investigator assessment was 7.3 months (95% CI, 5.6 months to 8.3 months). The PFS rates by investigator assessment at 6 months and 12 months were 54.1% (95% CI, 44.3% to 62.9%) and 27.3% (95% CI, 17.6% to 37.8%), respectively.
Overall, 40.5% of patients were censored in the analysis of PFS. The most common reason for censoring was that the follow-up of the patient was ongoing without an event (31.4%). No patients were censored due to loss to follow-up.
Figure 5: Kaplan-Meier Plot of Progression-Free Survival by Local Investigator Assessment — Cohort A of BYLieve Study, mFAS
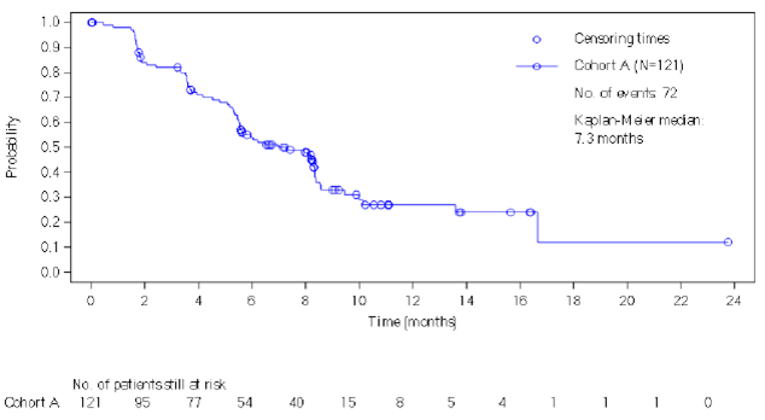
No. = number.
Source: BYLieve Clinical Study Report.32
Overall Survival
As of the data cut-off date, 25 (20.7%) deaths were reported in the mFAS. The Kaplan-Meier plot of OS is depicted in Figure 6. The median OS was 17.3 months (95% CI, 17.2 months to 20.7 months). The OS rates at 6 months and 12 months were 91.9% (95% CI, 84.9% to 95.7%) and 75.2% (95% CI, 62.5% to 84.2%), respectively.
Overall, 79.3% of patients were censored in the OS analysis. Of these, 55.4% were alive and 24.0% were lost to follow-up. The sponsor indicated in the BYLieve Clinical Study Report that OS data should be interpreted with caution due to the proportion of patients alive and ongoing follow-up at the time of the data cut-off date.
Figure 6: Kaplan-Meier Plot of Overall Survival — Cohort A of BYLieve Study, mFAS
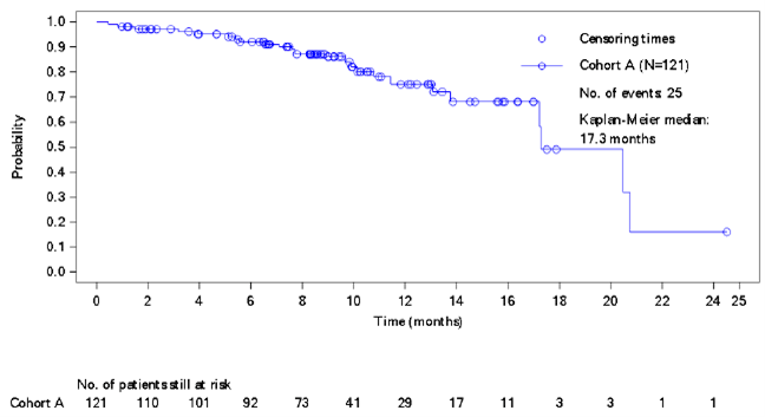
mFAS = modified full analysis set; no. = number.
Source: BYLieve Clinical Study Report.32
Health-Related Quality of Life
Data on HRQoL were not included in the BYLieve study.
Table 24: Summary of Progression and Survival Outcomes — Cohort A of BYLieve Study, mFAS
Outcome | Cohort A (N = 121) |
|---|---|
Primary outcome | |
Proportion of patients who are alive without disease progression per RECIST 1.1 at 6-month follow-up (95% CI)a | 50.4 (41.2 to 59.6) |
PFS | |
Patients with events, n (%) | 72 (59.5) |
Progressive disease | 66 (54.5) |
Death | 6 (5.0) |
Censored, n (%) | 49 (40.5) |
Ongoing follow-up without event | 38 (31.4) |
Lost to follow-up | 0 |
Withdrew consent | 6 (5.0) |
Adequate assessment no longer available | 5 (4.1) |
Median PFS, months (95% CI)b | 7.3 (5.6 to 8.3) |
PFS rate at 6 months, % (95% CI)c | 54.1 (44.3 to 62.9) |
PFS rate at 12 months, % (95% CI)c | 27.3 (17.6 to 37.8) |
OS | |
Patients with events, n (%) | 25 (20.7) |
Censored, n (%) | 96 (79.3) |
Alive | 67 (55.4) |
Lost to follow-up | 29 (24.0) |
Median OS, months (95% CI)b | 17.3 (17.2 to 20.7) |
OS rate at 6 months, % (95% CI)c | 91.9 (84.9 to 95.7) |
OS rate at 12 months, % (95% CI)c | 75.2 (62.5 to 84.2) |
CI = confidence interval; mFAS = modified full analysis set; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
aBased on local investigator assessment.
bCalculated from PROC LIFETEST output using the Brookmeyer and Crowley method.
cObtained from the Kaplan-Meier estimates; the Greenwood formula was used for CIs of Kaplan-Meier estimates.
Source: BYLieve Clinical Study Report.32
Harms
Only those harms identified in the CADTH review protocol are reported as follows. See Table 25 for detailed harms data for cohort A of the BYLieve study.
Adverse Events
Almost all patients (99.2%) experienced at least 1 treatment-emergent AE. The most common AEs (≥ 20%) were diarrhea (59.8%), hyperglycemia (58.3%), nausea (45.7%), fatigue (29.1%), decreased appetite (28.3%), rash (28.3%), stomatitis (26.8%), and vomiting (23.6%).
A total of 82 (64.6%) patients experienced an AE leading to dose adjustment or interruption. The most common AEs requiring a dose adjustment or interruption were hyperglycemia (29.1%), rash (12.6%), maculopapular rash (9.4%), and diarrhea (7.9%).
The majority of patients (94.5%) experienced an AE requiring additional therapy. The most common AEs requiring additional therapy were hyperglycemia (43.3%), diarrhea (26.0%), nausea (19.7%), rash (19.7%), stomatitis (18.1%), and maculopapular rash (11.8%).
Serious Adverse Events
Overall, 26.0% of patients experienced an SAE. The most common SAEs were hyperglycemia (5.5%), maculopapular rash (3.1%), dyspnea (2.4%), pleural effusion (2.4%), abdominal pain (1.6%), and hematemesis (1.6%).
Withdrawals Due to Adverse Events
The most common AEs leading to discontinuation of study treatment were rash (3.9%) and colitis, hyperglycemia, urticaria, and vomiting (1.6% each).
Deaths
As of the data cut-off date, 25 (20.7%) patients had died. Seven (5.5%) patients had died during study treatment or within 30 days of the last dose of study drug, and 4 of these on-treatment deaths were attributed to breast cancer.
Notable Harms
Overall, 74 (58.3%) patients experienced hyperglycemia. Thirteen (10.2%) patients experienced hypersensitivity and anaphylactic reactions. Diarrhea, nausea, and vomiting were reported in 59.8%, 45.7%, and 23.6% of patients, respectively. Pneumonitis and severe cutaneous skin reactions were experienced by 1 (0.8%) patient each. Overall, 36 (28.3%) patients experienced rash and 18 (14.2%) patients experienced maculopapular rash.
Table 25: Summary of Harms — Cohort A of BYLieve Study, Safety Analysis Set
Harms | Cohort A (N = 127) |
|---|---|
Patients with ≥ 1 AE | |
n (%) | 126 (99.2) |
Most common events,a n (%) | |
Diarrhea | 76 (59.8) |
Hyperglycemia | 74 (58.3) |
Nausea | 58 (45.7) |
Fatigue | 37 (29.1) |
Decreased appetite | 36 (28.3) |
Rash | 36 (28.3) |
Stomatitis | 34 (26.8) |
Vomiting | 30 (23.6) |
Patients with ≥ 1 SAE | |
n (%) | 33 (26.0) |
Most common events,b n (%) | |
Hyperglycemia | 7 (5.5) |
Rash maculopapular | 4 (3.1) |
Dyspnea | 3 (2.4) |
Pleural effusion | 3 (2.4) |
Abdominal pain | 2 (1.6) |
Hematemesis | 2 (1.6) |
Patients with ≥ 1 AE requiring dose adjustment or interruption | |
n (%) | 82 (64.6) |
Most common events,c n (%) | |
Hyperglycemia | 37 (29.1) |
Rash | 16 (12.6) |
Rash maculopapular | 12 (9.4) |
Diarrhea | 10 (7.9) |
Patients with ≥ 1 AE requiring additional therapy | |
n (%) | 120 (94.5) |
Most common events,d n (%) | |
Hyperglycemia | 55 (43.3) |
Diarrhea | 33 (26.0) |
Nausea | 25 (19.7) |
Rash | 25 (19.7) |
Stomatitis | 23 (18.1) |
Rash maculopapular | 15 (11.8) |
Patients who discontinued treatment due to AEs | |
n (%) | 26 (20.5) |
Most common events,b n (%) | |
Rash | 5 (3.9) |
Colitis | 2 (1.6) |
Hyperglycemia | 2 (1.6) |
Urticaria | 2 (1.6) |
Vomiting | 2 (1.6) |
Deaths | |
Deaths as of DCO of December 17, 2019 | 25 (20.7) |
On-treatment deaths,e n (%) | 7 (5.5) |
Primary reason for on-treatment death, n (%) | |
Breast cancer | 4 (3.1) |
Respiratory failure | 1 (0.8) |
Superior vena cava occlusion | 1 (0.8) |
Unspecified | 1 (0.8) |
Notable harms | |
Hyperglycemia, n (%) | 74 (58.3) |
Hypersensitivity and anaphylactic reaction, n (%) | 13 (10.2) |
Diarrhea, n (%) | 76 (59.8) |
Nausea, n (%) | 58 (45.7) |
Vomiting, n (%) | 30 (23.6) |
Severe cutaneous reactions, n (%) | 1 (0.8) |
Rash, n (%) | 36 (28.3) |
Rash maculopapular, n (%) | 18 (14.2) |
Pneumonitis, n (%) | 1 (0.8) |
AE = adverse event; DCO = data cut-off; SAE = serious adverse event.
aFrequency of 20% or more.
bReported in more than 1 patient.
cFrequency of 5% or more.
dFrequency of 10% or more.
eOn-treatment deaths, which included patients who died during treatment or within 30 days of the last dose.
Source: BYLieve Clinical Study Report.32
Critical Appraisal
Internal Validity
The BYLieve study is a prospective, non-comparative cohort study. Patients were assigned to cohorts based on their most recently received anticancer treatment (i.e., patients were assigned to cohort A and thus received alpelisib plus fulvestrant if their most recent treatment was any CDK4/6 inhibitor plus any AI). Due to the absence of a comparator group, no conclusions can be made regarding the efficacy and safety of alpelisib plus fulvestrant in the post-CDK4/6 inhibitor setting compared to other treatment options based on the BYLieve study data.
The BYLieve study was open label and, thus, patients and investigators were aware of the treatment status. Open-label studies are prone to various biases (e.g., patient selection bias, reporting bias). Furthermore, disease progression was assessed by the local investigator for the primary outcome and PFS. The open-label administration of alpelisib in combination with fulvestrant may have biased the reporting of these end points. Progressive disease was determined by the investigator using RECIST 1.1, which is commonly used in clinical trials.
The BYLieve study did not include all enrolled patients (i.e., the FAS) in the primary analyses. The primary analysis population was the mFAS, which included all enrolled patients who had PIK3CA mutation confirmed by a Novartis-designated laboratory (n = 121, 95.3%).
For the primary end point analysis, patients who progressed, died, or discontinued the study before 6 months had passed were counted as failures. The primary analysis for cohort A was performed as planned (i.e., 6 months after the last patient had started treatment or discontinued early). Since the primary end point was tested for statistical significance in 1 analysis, adjustment for multiplicity was not required. At the planned analysis, the study met its primary end point (i.e., the lower bound of the 95% CI for the proportion of patients alive without progression after 6 months was > 30%). The sponsor considered a proportion of 30% to be a clinically meaningful threshold for the primary end point of the study, but the rationale for this threshold is unclear. This is not a commonly used end point, or 1 that was considered of particular importance by the patient group or the clinical experts consulted by CADTH. Thus, the validity of the primary end point is unknown.
The secondary end points in the BYLieve study — OS and PFS — were estimated using the Kaplan-Meier method. The sponsor indicated that the OS data should be interpreted with caution due to the proportion of patients alive and having ongoing follow-up at the time of the data cut-off date. CADTH reviewers agree that the data are difficult to interpret and as a result, no conclusions can be made regarding the effect of alpelisib in combination with fulvestrant on OS. Furthermore, 24.0% of patients were censored due to being lost to follow-up in the analysis of OS. This loss to follow-up could lead to significant bias in the estimation of OS. In the analysis of PFS, 0 patients were lost to follow-up. Again, due to the lack of any comparator, the results of the PFS or OS analyses do not provide evidence of the effects of alpelisib in combination with fulvestrant on these outcomes.
The median duration of follow-up at the primary analysis of cohort A was less than 1 year and most (64.6%) patients did not enter the post-treatment follow-up phase per the study protocol (i.e., patients only entered the post-treatment follow-up phase if they did not discontinue study treatment due to documented disease progression, death, loss to follow-up, or withdrawal of consent for efficacy follow-up).
External Validity
The BYLieve study was an international, multi-centre study that included 3 sites in Canada. The majority of patients enrolled in cohort A of the BYLieve study were postmenopausal women, were Caucasian, had stage IV disease at study entry, had invasive ductal carcinoma, had secondary endocrine resistance, and had an ECOG PS of 0. The mean age of patients was 56.7 years and the mean time from most recent recurrence and/or relapse was 2.2 months. The clinical experts consulted by CADTH indicated that the baseline characteristics of the study patients were generally consistent with the Canadian patient population. The BYLieve study only enrolled women; thus, the study does not provide evidence on the use of alpelisib in combination with fulvestrant in men. However, the clinical experts consulted by CADTH considered it reasonable to generalize the results to the male population.
For the primary outcome analysis, the sponsor considered a proportion of 30% who had not progressed, died, or discontinued at 6 months to be a clinically meaningful threshold for the primary end point of the study. The clinical experts consulted by CADTH indicated that they would expect fewer than 30% to experience disease progression or death at 6 months after starting the second-line treatments that are currently used in standard care (e.g., fulvestrant alone). As a result, the 30% threshold used in the BYLieve study may not be clinically relevant in the Canadian treatment setting.
The intervention used in cohort A of the BYLieve study aligns with the reimbursement request and intended use of alpelisib in Canadian patients, per the clinical experts consulted by CADTH.
The patient groups that provided input for this review identified OS and PFS as important outcomes, which were secondary end points in the BYLieve study. Patients also identified HRQoL as an important outcome, but this was not captured in the BYLieve study.
The median duration of follow-up was less than 1 year as of the data cut-off date. As a result, the BYLieve study does not provide long-term data on alpelisib in combination with fulvestrant.
Observational Study
The sponsor-submitted observational study, which has been published,31 compared 1 of the cohorts of the BYLieve study to a set of patients who were identified from electronic medical records as provided by the Flatiron Clinico-Genomic Database. The study was not randomized, and propensity score weighting was used in an attempt to adjust for confounding.
Methods
Objectives
The primary objective of the study was to evaluate and compare PFS among patients who received alpelisib and fulvestrant in the BYLieve trial (cohort A) to patients who received a non-alpelisib SOC therapy in the Flatiron Clinico-Genomic Database, all of whom had hormone receptor–positive, HER2-negative, advanced breast cancer harbouring a PIK3CA mutation and had disease progression during or after treatment with CDK4/6 inhibitors with endocrine therapy. The secondary objective was to evaluate and compare the rate of patients who remained progression-free at 6 months as measured by a milestone PFS analysis between the same groups of patients.
Patient Selection Criteria and Selection Process
Key selection criteria of the 2 cohorts included in the study are summarized in Table 26, in addition to the treatments and outcomes. The index date was defined as the date when the treatment under consideration began. The Flatiron data included data collected between January 1, 2011, and June 30, 2019, and the index date was required to be on or before January 1, 2019, to allow for at least 6 months of potential follow-up. Most of the inclusion and exclusion criteria that were used in the BYLieve study were not possible to apply in the Flatiron database because the necessary information was not available in electronic medical records. The following patient characteristics were available for both cohorts: sex, age, race (White versus non-White), ECOG PS, cancer stage at diagnosis, cancer stage at the index date, site of metastasis (bone lesion only, lung and/or liver), number of sites of metastases (< 3 and ≥ 3), number of prior lines of treatment in the advanced setting, duration between diagnosis and the index date, and the index SOC regimen.
Table 26: Key Characteristics of Cohorts Included in the Observational Study
Characteristic | BYLieve study, cohort A | Flatiron database-derived cohort |
|---|---|---|
Patient inclusion criteria | See Table 17 for key inclusion criteria for cohort A in the BYLieve study | Key differences from cohort A of the BYLieve study were that:
|
Patient exclusion criteria | See Table 17 for key exclusion criteria for cohort A in the BYLieve study. In addition, patients who died within 14 days of treatment initiation were excluded. It is unclear whether progression events within 14 days would be excluded. | Key differences from cohort A of the BYLieve study were that:
|
Intervention | Alpelisib 300 mg p.o. q.d. and fulvestrant 500 mg IM on day 1 and day 15 of cycle 1 and on day 1 of subsequent cycles (each cycle being 28 days) | Standard of care, defined as any non-alpelisib treatment administered following treatment with a CDK4/6 inhibitor and endocrine therapy |
Outcomes |
|
|
AI = aromatase inhibitor; CDK4/6 = cyclin-dependent kinase 4 and 6; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IM = intramuscular; p.o. = orally; q.d. = every day; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: BYLieve interim Clinical Study Report (2020)32 and sponsor-submitted Observational Study Report (2020).33
The attrition table for patients in the Flatiron cohort is presented in Table 27.
Table 27: Attrition Table for the Flatiron Cohort
Step | Description | Number of patients | Percentage of patients remaining from previous step |
|---|---|---|---|
0 | CDK4/6 inhibitor + HT combination therapy at any time | 855 | 100% |
1 | Progressed to next line of therapy after CDK4/6 inhibitor + HT combination therapy (start of this next therapy is index date) | 637 | 74.5% |
2 | No more than 2 lines of prior anticancer regimens for advanced breast cancer | 370 | 58.1% |
3 | No more than 1 line of prior chemotherapy regimen in the advanced/metastatic setting | 368 | 99.5% |
4 | Patients with advanced breast cancer before the start of index treatment | 362 | 98.4% |
6 | Patients having a structured activity within 90 days of advanced diagnosis | 288 | 79.6% |
7 | Patients aged ≥ 18 at index | 288 | 100.0% |
8 | Patients having a confirmed PIK3CA mutation | 126 | 43.8% |
9 | Initiated treatment on or before January 31, 2019, to allow for a minimum of 6 months of observation time (data extraction end date in Flatiron was June 30, 2019) | 112 | 88.9% |
10 | Patients available for PFS analysis | 111 | 99.1% |
11a | Exclude patients with HER2+ drugs, clinical study drug, or alpelisib as part of the index regimen | 95 | 85.6% |
11b | Patients treated with a regimen that was not included for subgroup analysis | 65 | 58.6% |
CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; HT = hormonal therapy, PFS = progression-free survival.
Source: Sponsor-submitted Observational Study Report (2020).33
Propensity Score Development
The covariates in Table 28, described in the sponsor’s report33 as clinically relevant prognostic variables, summarize the patient characteristics that were used to balance the cohorts for effect estimation. In the primary analysis, these covariates were used as predictors in a logistic regression model with treatment with alpelisib plus fulvestrant as the binary outcome to estimate the conditional probability of receiving that treatment for each individual in the observational study.
Table 28: Covariates Used for Propensity Score Development and/or Matching
Characteristic | Propensity score matching covariates |
|---|---|
Site of metastasis | Bone lesion only, lung/liver |
Age group in years | < 50, 50 to < 65, ≥ 65 |
Time (months) between initial diagnosis of breast cancer and index date | < 27, 27 to < 60, 60 to < 128, ≥ 128 |
Number of sites of metastasis | < 3, ≥ 3 |
Source: Sponsor-submitted Observational Study Report (2020).33
Statistical Methods
Three methods of balancing were used: propensity score estimation34 followed by weighting by the odds of treatment with alpelisib plus fulvestrant,35 1:1 greedy nearest-neighbour matching based on the same propensity scores using a caliper of 0.2 times the standard deviation of the logit of the propensity scores, and 1:1 exact matching based on exact matches of the covariates (not propensity score based36). The first approach was considered the primary analysis while the latter 2 were considered sensitivity analyses. Missing data were not imputed.
Following weighting and/or matching, the median PFS, the PFS survival curves, and the 6-month probability of PFS were estimated using the Kaplan-Meier method with Hall-Wellner confidence bands.37 The survival curves were compared using the log-rank test, and Cox regression was used to estimate hazard ratios. Bootstrapping was used in the weighted analysis with 200 replicates to estimate the standard errors.38 It is unclear whether the weights were re-estimated at each bootstrap sample. Patients in the BYLieve cohort were given weights of 1 while patients in the Flatiron cohort were given weights of the odds of treatment propensity score divided by (1 minus propensity score), where propensity score is the propensity score of treatment. Balance was assessed by comparing standardized mean differences of the baseline variables.
Results
Patient Characteristics
Table 29 shows the characteristics of the cohorts before and after propensity score weighting. Patients in the BYLieve cohort received a weight of 1, so the study characteristics were the same before and after weighting. The clinical experts consulted by CADTH for the review agreed that before weighting, patients in the Flatiron cohort appeared to be older and sicker, on average, than those in the BYLieve cohort. The characteristics that were used in the propensity score development were balanced after weighting, but many other important measured characteristics were not balanced after weighting — notably, ECOG PS at baseline, and stage at initial diagnosis. In the Flatiron cohort, the SOC included a wide variety of treatments, the most common of which were capecitabine and fulvestrant.
Table 29: Patient Characteristics Before and After Weighting
Characteristic | Pre-weighted | Post-weighted | |||
|---|---|---|---|---|---|
Flatiron cohort N = 95 | Trial cohorta N = 120 | SMD, % | Flatiron cohort N = 116 | SMD, % | |
Sex, n (%) | |||||
Female | 94 (99.0) | 120 (100) | 14.5 | 112.3 (96.7) | 45.5 |
Male | 1 (1.1) | 0 (0) | 14.5 | 3.8 (3.3) | –45.5 |
Age in years at index: Continuous | |||||
Mean (SD) | 60.5 (10.76) | 57.0 (10.21) | 32.9 | 57.2 (12.8) | –1.9 |
Median (Q1 to Q3) | 61.0 (53.0 to 68.0) | 58.0 (48.0 to 65.0) | NR | 57.0 (49.0 to 65.0) | NR |
Min. (max.) | 38.00 (82.00) | 33.00 (83.00) | NR | 38 (82) | NR |
Age in years at index: Categories, n (%)b, c | |||||
< 50 | 13 (13.7) | 35 (29.2) | 38.2 | 30.1 (26.0) | 7.9 |
50 to < 65 | 49 (51.6) | 54 (45) | 13.1 | 55.8 (48.1) | –6.1 |
≥ 65 | 33 (34.7) | 31 (25.8) | 19.4 | 30.2 (26.0) | –0.3 |
Race, n (%) | |||||
Non-White | 22 (23.2) | 44 (36.7) | 29.7 | 28.7 (24.7) | 26.3 |
White | 73 (76.8) | 76 (63.3) | –29.7 | 87.4 (75.3) | –26.3 |
ECOG PS at baseline, n (%)b, d | |||||
0 | 35 (36.8) | 78 (65) | 58.4 | 44.3 (38.2) | 55.6 |
1 | 28 (29.5) | 38 (31.7) | 4.7 | 33.9 (29.2) | 5.4 |
2 | 8 (8.4) | 1 (0.8) | 36.5 | 11.1 (9.5) | –41.8 |
Missing | 24 (25.3) | 3 (2.5) | –69.4 | 26.8 (23.1) | –62.8 |
Stage at initial diagnosis, n (%) | |||||
0/I | 13 (13.7) | 22 (18.3) | 12.6 | 15 (13.0) | 14.6 |
II | 29 (30.5) | 42 (35) | 9.5 | 31.6 (27.2) | 16.6 |
III | 23 (24.2) | 17 (14.2) | –25.6 | 30.5 (26.2) | –30.8 |
IV | 22 (23.2) | 38 (31.7) | 19.1 | 30.6 (26.4) | 11.9 |
Missing | 8 (8.4) | 1 (0.8) | –36.5 | 8.4 (7.3) | –30.9 |
Stage at index, n (%) | |||||
III | 0 (0) | 3 (2.5) | 22.6 | 0 (0) | 22.6 |
IV | 95 (100) | 117 (97.5) | –22.6 | 116.1 (100) | –22.6 |
Pooled number of sites, n (%)b, e | |||||
< 3 | 57 (60) | 84 (70) | 21.0 | 79.2 (68.2) | 3.8 |
≥ 3 | 38 (40) | 36 (30) | –21.0 | 36.9 (31.8) | –3.8 |
Sites of metastases, n (%)b, e | |||||
Bone lesions only | 20 (21.1) | 22 (18.3) | –6.8 | 23.8 (20.5) | –5.4 |
Lung/liver | 56 (59) | 80 (66.7) | 15.9 | 73.1 (63.0) | 7.6 |
Number of prior lines of therapy in metastatic setting, n (%) | |||||
0 | 0 (0) | 14 (11.7) | 51.2 | 0 (0) | 51.2 |
1 | 61 (64.2) | 85 (70.8) | 14.1 | 76.6 (66.0) | 10.3 |
2 | 34 (35.8) | 20 (16.7) | –44.3 | 39.5 (34.0) | –40.2 |
3 | 0 (0) | 1 (0.8) | 12.9 | 0 (0) | 12.9 |
Time from initial diagnosis to index date in months: Continuousf | |||||
Mean (SD) | 84.69 (71.35) | 83.78 (73.92) | –1.3 | 76.8 (73.2) | 9.6 |
Median (Q1 to Q3) | 61.27 (30.9 to 131.8) | 56.61 (26.0 to 124.4) | NA | 58.2 (25.5 to 102.6) | NA |
Min. (max.) | 4.57 (331.20) | 6.21 (408.71) | NA | 4.6 (331.2) | NA |
Time from initial diagnosis to index date in months: Categories, n (%)b, f | |||||
< 27 | 22 (23.2) | 31 (25.8) | 6.2 | 30.5 (26.3) | –1.1 |
27 to < 60 | 24 (25.3) | 30 (25.0) | –0.6 | 29 (25.0) | 0.0 |
60 to < 128 | 24 (25.3) | 31 (25.8) | 1.3 | 31.3 (26.9) | –2.5 |
≥ 128 | 25 (26.3) | 28 (23.3) | –6.9 | 25.3 (21.8) | 3.6 |
Index treatment, n (%)g | |||||
Alpelisib, fulvestrant | NA | 120 (100) | NA | NA | NA |
Capecitabine | 14 (14.7) | NA | NA | 14 (12.1) | NA |
Fulvestrant | 14 (14.7) | NA | NA | 14 (12.4) | NA |
Fulvestrant, palbociclib | 13 (13.7) | NA | NA | 19 (16.0) | NA |
Everolimus, exemestane | 11 (11.6) | NA | NA | 12 (10.4) | NA |
Fulvestrant, letrozole, palbociclib | 5 (5.3) | NA | NA | 5 (4.0) | NA |
Fulvestrant, letrozole | 3 (3.2) | NA | NA | 4 (3.2) | NA |
Paclitaxel | 3 (3.2) | NA | NA | 5 (4.6) | NA |
Abemaciclib, fulvestrant | 2 (2.1) | NA | NA | 3 (2.2) | NA |
Carboplatin, gemcitabine | 2 (2.1) | NA | NA | 2 (1.7) | NA |
Eribulin | 2 (2.1) | NA | NA | 3 (2.5) | NA |
Exemestane | 2 (2.1) | NA | NA | 4 (3.6) | NA |
Exemestane, palbociclib | 2 (2.1) | NA | NA | 5 (3.9) | NA |
Paclitaxel protein-bound | 2 (2.1) | NA | NA | 4 (3.6) | NA |
Abemaciclib, fulvestrant, letrozole | 1 (1.1) | NA | NA | 1 (0.7) | NA |
Abemaciclib, letrozole | 1 (1.1) | NA | NA | 1 (0.6) | NA |
Anastrozole, palbociclib | 1 (1.1) | NA | NA | 1 (0.5) | NA |
Bevacizumab, paclitaxel protein-bound | 1 (1.1) | NA | NA | 2 (1.5) | NA |
Capecitabine, everolimus, exemestane | 1 (1.1) | NA | NA | 2 (1.5) | NA |
Docetaxel | 1 (1.1) | NA | NA | 1 (0.7) | NA |
Doxorubicin | 1 (1.1) | NA | NA | 1 (1.1) | NA |
Doxorubicin — pegylated liposomal | 1 (1.1) | NA | NA | 1 (0.6) | NA |
Everolimus, exemestane, fulvestrant | 1 (1.1) | NA | NA | 1 (0.8) | NA |
Everolimus, exemestane, goserelin | 1 (1.1) | NA | NA | 2 (2.0) | NA |
Everolimus, fulvestrant | 1 (1.1) | NA | NA | 1 (0.8) | NA |
Everolimus, letrozole | 1 (1.1) | NA | NA | 1 (0.8) | NA |
Everolimus, letrozole, ribociclib | 1 (1.1) | NA | NA | 1 (0.5) | NA |
Exemestane, fulvestrant, palbociclib | 1 (1.1) | NA | NA | 1 (0.6) | NA |
Fulvestrant, palbociclib, tamoxifen | 1 (1.1) | NA | NA | 2 (1.6) | NA |
Fulvestrant, ribociclib | 1 (1.1) | NA | NA | 1 (0.8) | NA |
Gemcitabine | 1 (1.1) | NA | NA | 1 (0.5) | NA |
Gemcitabine, letrozole, paclitaxel protein-bound, palbociclib | 1 (1.1) | NA | NA | 1 (1.2) | NA |
Letrozole, palbociclib | 1 (1.1) | NA | NA | 2 (1.5) | NA |
Letrozole, palbociclib, tamoxifen | 1 (1.1) | NA | NA | 2 (1.5) | NA |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; max. = maximum; min. = minimum; NA = not applicable; NR = not reported; Q1 = first quartile; Q3 = third quartile; SD = standard deviation; SMD = standardized mean difference.
Note: Age category, pooled number of metastatic sites, site of metastasis, and time since initial diagnosis (see Table 28) were used to generate the propensity scores used in weighting by odds. The weighting by odds method maintains the composition of trial patients (each of them having a weight of 1), while real-world patients were weighted by the odds to reflect the trial population. An absolute standardized difference of less than 25% between the 2 groups was considered to be balanced.
aOnly patients in the modified full analysis set without a death event within 14 days were included.
bThis was considered as a covariate in the weighting by odds analysis (see Table 28).
cAge at index is defined as the difference in years between year of birth and year of index treatment (year of index treatment minus year of birth) in the Flatiron database.
dECOG PS score is determined based on the record closest to the index date within a time window of 30 days before the index date in the Flatiron Clinico-Genomic Database to 7 days after the index date. This window of 30 days before the index date in the database to 7 days after the index date is recommended by Flatiron as an appropriate baseline period. ECOG PS was captured at screening in the BYLieve study.
eThe day on which the site of metastasis was observed was not available in Flatiron; only the month and year are available. Sites of metastases observed during the month of the index date in the Flatiron database or prior were considered. Sites of metastases were recorded at screening in the BYLieve study.
fThe difference in time was calculated using the following formula: (date 2 – date 1) divided by 30.4275. It was assumed that 1 year contains 365.25 days.
gThe numbers of patients for each index regimen was rounded to the nearest integer for the post-weighted cohort.
Source: Sponsor response to June 15, 2021, additional information request.39
Efficacy
Figure 7 shows the results of the primary analysis, the weighted Kaplan-Meier estimated survival curves of progression for the BYLieve cohort and the propensity score odds–weighted Flatiron cohort, along with the result of the adjusted log-rank test (P = 0.004).
Figure 7: Post-Weight Progression-Free Survival (Primary Analysis)
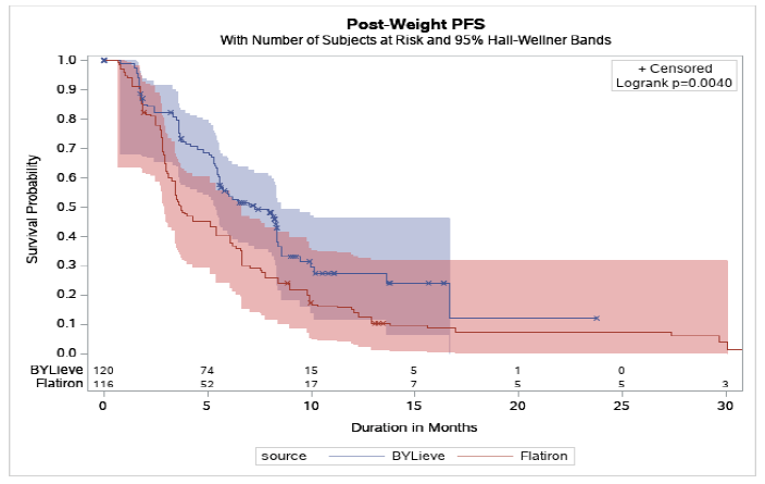
PFS = progression-free survival.
Source: Sponsor-submitted Observational Study Report (2020).33
Table 30 shows the summary statistics corresponding to the primary survival analysis. The median survival estimated by the weighted Kaplan-Meier method in the Flatiron cohort was 3.7 months (95% CI, 3.1 months to 6.1 months) compared to 7.3 months (95% CI, 5.6 months to 8.3 months) in the BYLieve cohort. The estimated hazard ratio estimated by weighted Cox regression comparing the BYLieve cohort to the Flatiron cohort was 0.62 (95% CI, 0.44 to 0.85; P = 0.002) where the P value was calculated by the Wald test using bootstrap standard errors. Due to the weighting method (propensity score odds), these represent estimates of the average treatment effect on those treated under the assumptions that the propensity score model was correctly specified, that there were no confounders other than the ones specified in Table 28, and that those covariates were measured without error.35
Table 30: Summary Statistics of Primary Analysis
Outcome | Pre-weighted | Post-weighted | ||
|---|---|---|---|---|
Flatiron cohort N = 95 | Trial cohorta N = 120 | Flatiron cohort N = 116 | Trial cohorta N = 120 | |
Summary of censored/failed patients | ||||
Progressions or deaths, n (%) | 88 (92.6) | 71 (59.2) | 107 (92.2) | 71 (59.2) |
Kaplan-Meier statistics | ||||
Median PFS, months (95% CI) | 3.6 (3.1 to 6.1) | 7.3 (5.6 to 8.3) | 3.7 (3.1 to 6.1) | 7.3 (5.6 to 8.3) |
P value | < 0.01 | NR | ||
Median PFS, months (95% CI based on bootstrap) | NR | NR | 3.7 (2.2 to 5.3) | 7.3 (5.3 to 9.2) |
P value | NR | < 0.01 | ||
PFS rates at specific time points | ||||
3-month rate, % | 60.8 | 82.3 | 62.1 | 82.3 |
6-month rate, % | 40.5 | 54.6 | 40.1 | 54.6 |
9-month rate, % | 24.4 | 33.3 | 21.8 | 33.3 |
12-month rate, % | 16.4 | 27.5 | 15.0 | 27.5 |
15-month rate, % | 10.0 | 24.1 | 9.8 | 24.1 |
18-month rate, % | 6.7 | 12.0 | 7.4 | 12.0 |
Hazard ratio statistics | ||||
Hazard ratio (95% CI) | Reference group | 0.64 (0.46 to 0.87) | Reference group | 0.62 (0.45 to 0.83) |
P value | 0.005 | NR | ||
Hazard ratio (95% CI based on bootstrap) | Reference group | NR | Reference group | 0.62 (0.44 to 0.85) |
P value | NR | 0.002 | ||
CI = confidence interval; NR = not reported; PFS = progression-free survival.
Note: The age category, pooled number of metastatic sites category, site of metastasis (bone lesion only, lung and/or liver) category, and time since initial diagnosis category are used to generate propensity score used in weighting by odds (see Table 28). The weighting by odds method maintains the composition of trial patients (each of them have a weight of 1), while real-world patients are weighted by the odds to reflect the trial population. The median survival and rates at specific time points were estimated using the weighted Kaplan-Meier method, with P values estimated using the adjusted log-rank test. Hazard ratios were estimated with weighted Cox regression, with confidence intervals and P values estimated using the Wald method with bootstrap standard errors based on 200 replicates.
aOnly patients in the modified full analysis set without a death event within 14 days were included.
Source: Sponsor response to June 15, 2021, additional information request.39
Sensitivity Analyses
The observational study included sensitivity analyses assessing the sensitivity of the results to the form of confounding adjustment — namely, greedy matching and exact matching. The results of those analyses were not meaningfully different from the primary analysis results. No sensitivity analysis to the assumption of no unmeasured confounding was done.
Critical Appraisal
Internal Validity
The sponsor submitted an observational study comparing patients exposed to alpelisib with patients exposed to non-alpelisib SOC using a cohort from the BYLieve study combined with data from the Flatiron health database. Differences between the cohorts were accounted for using a propensity score weighted analysis based on a set of clinically relevant prognostic variables. The main findings were not sensitive to the choice of adjustment method when compared to 2 types of matching methods. There are many weaknesses of this study that raise concerns regarding the reliability and validity of these estimates of efficacy between alpelisib and SOC. There was no apparent pre-specified and registered protocol for this study, and no power or sample size calculations were reported. Hence, the statistical inference obtained from this study has low reliability and validity.
The 2 cohorts are innately different, making them harder to compare and requiring confounding and selection bias adjustment. The BYLieve study was a multinational cohort study that recruited patients from all around the world, but the Flatiron database is based in the US. Data collection methods differ dramatically between carefully planned cohort studies and electronic health records that are not designed specifically for research purposes.
Confounding and selection bias adjustment is likely inadequate for several reasons. The set of covariates used for propensity score modelling was insufficient to control for known prognostic factors, including number and type of prior lines of therapy, ECOG PS, sex, and disease stage at index date. In the SOLAR-1 study, ECOG PS of 0 versus 1 was a highly significant prognostic variable, but was not included in the propensity score model due to the large amount of missing data in the Flatiron cohort.
There was a planned exclusion of patients from the Flatiron cohort who were treated with alpelisib. Due to this exclusion, the study treatment and the cohort definition were completely confounded. As such, it is unknown whether the differences observed in the study are attributable to the treatment or to the inherent differences in selection in the Flatiron database versus controlled clinical studies; this source of confounding cannot by accounted for by the weighting and/or matching approach. The patient disposition generally indicates that the Flatiron cohort is older and has more severe disease, on average. Hence, this source of confounding is a critical weakness of the study since it raises concerns for the validity of any causal inference from the study. No sensitivity analyses were done to address this limitation.
Finally, the justification provided by the sponsor for the exclusion of events that occurred within 14 days of starting treatment was given as follows: “This criterion was implemented based on Flatiron’s analytic guidance which suggests that progression or death observed within 14 days of treatment initiation may not be directly associated with the effectiveness of treatment.”33 No data to support this guidance were provided, but the clinical experts consulted by CADTH thought that this was reasonable. Furthermore, it is unclear whether patients with progression events that occurred within 14 days were excluded or if those events were simply not counted. The BYLieve study was a multinational cohort study that recruited patients from all around the world, but the Flatiron database is exclusively based in the US. Data collection methods differ dramatically between carefully planned cohort studies and electronic health records that are not designed specifically for research purposes.
Overall, there remains a great deal of uncertainty regarding the efficacy of alpelisib in comparison to SOC, due to the inherent limitations of observational data. While the adjustment approaches taken in this study may have adequately balanced on observable prognostic factors categorized as they were, bias in the efficacy estimate due to selection bias, measurement error, unmeasured confounding, and residual confounding cannot be ruled out. No attempts were made to assess or estimate the possible magnitude of such bias.
External Validity
The external validity of the results should be the same as that of the BYLieve cohort in that the results are weighted to match that study. However, the same limitations in the ability to weight properly given the covariates used and the innate differences in the cohorts remain. As the Flatiron cohort includes only US patients, it is likely that practices and patients are different from the Canadian population and are, in general, sicker and receiving a lower SOC.
Discussion
Summary of Available Evidence
The CADTH systematic review identified 1 relevant study, the SOLAR-1 study. The SOLAR-1 study (N = 572) was a placebo-controlled, double-blind, parallel-group RCT that randomized patients 1:1 to alpelisib 300 mg daily or matching placebo, in combination with fulvestrant 500 mg on day 1, day 15, and day 29, and every 28 days afterwards. Men and postmenopausal women with hormone receptor–positive, HER2-negative advanced or metastatic breast cancer and previous endocrine therapy were randomized within each of 2 cohorts based on PIK3CA mutation status: the PIK3CA mutant cohort and PIK3CA non-mutant cohort. The primary and key secondary outcomes were PFS and OS in the PIK3CA mutant cohort (N = 341). Endocrine therapy with a CDK4/6 inhibitor was not a part of the SOC at the time the study was conducted (enrolment was from 2015 to 2017) and only 20 patients in the PIK3CA mutant cohort were included in the subgroup analyses of patients who had received prior CDK4/6 inhibitor treatment and therefore met the reimbursement criteria requested by the sponsor.
Included in the sponsor’s submission was the non-comparative BYLieve study and an observational study comparing cohort A in the BYLieve study with a real-world, database-derived cohort. The BYLieve study assigned patients to 1 of 3 cohorts based on their most recent anticancer therapy. Of the 3 cohorts, cohort A (N = 127) was identified by the CADTH review team as relevant to the review; it included patients with hormone receptor–positive, HER2-negative advanced or metastatic breast cancer and a confirmed PIK3CA mutation and who had received any CDK4/6 inhibitor plus any AI as their immediate prior treatment. These patients were assigned to receive alpelisib plus fulvestrant at the same dosages as in the SOLAR-1 study. The primary end point in the BYLieve study was the proportion of patients who were alive without disease progression at 6 months by local investigator assessment using RECIST 1.1 criteria. The outcomes of PFS and OS as well as safety data were also evaluated in the BYLieve study. The observational study compared cohort A from the BYLieve study to a database-derived cohort that received non-alpelisib SOC immediately following treatment with a CDK4/6 inhibitor and non-fulvestrant endocrine therapy. The outcome PFS was compared between the cohorts following weighting of the database-derived cohort based on propensity scores.
Interpretation of Results
Efficacy
No relevant conclusions can be drawn regarding PFS and OS in patients treated with alpelisib and fulvestrant versus placebo and fulvestrant because the SOLAR-1 study was not designed to test hypotheses in the subgroup of patients with prior CDK4/6 inhibitor treatment and the results of the analyses were not statistically significant. The only outcomes in the testing hierarchy were PFS and OS in the entire PIK3CA mutant cohort and the study sample size considerations were based on those outcomes. Analyses of efficacy in subgroups, of which the subgroup of patients with prior CDK4/6 inhibitor treatment was 1, was for the purpose of examining the consistency of any treatment effect found in the entire cohort across the subgroups rather than demonstrating efficacy in any of the subgroups. In the absence of pre-specified subgroup analyses within the statistical testing hierarchy, no conclusions can be drawn about efficacy in any of the subgroups. As well, different approaches for identifying patients with prior CDK4/6 inhibitor therapy resulted in different numbers of patients being identified as such within the PIK3CA mutant cohort; it is unclear whether all the patients in the stratum with prior CDK4/6 inhibitor treatment did, in fact, receive that treatment.
The clinical experts consulted by CADTH for this review agreed that the results from the entire PIK3CA mutant cohort cannot be generalized to the patient population under review — namely, patients who have received a prior endocrine regimen with a CDK4/6 inhibitor. The design of the SOLAR-1 study reveals that efficacy results were not expected to be consistent between patients with and without prior CDK4/6 inhibitor treatment. Randomization was stratified according to previous CDK4/6 inhibitor usage, with the following rationale given in the SOLAR-1 study interim Clinical Study Report: “Prior CDK4/6 inhibitor use was selected since treatment with a CDK4/6 inhibitor in combination with letrozole has shown a strong PFS advantage over single-agent letrozole; therefore, prior treatment with a CDK4/6 inhibitor may impact the outcome of subsequent treatment with fulvestrant and/or PI3K inhibitors compared to CDK4/6 inhibitor naïve subjects.”1 Reviews of alpelisib from regulatory agencies did not contain any conclusions regarding the efficacy of alpelisib with fulvestrant in patients previously treated with a CDK4/6 inhibitor. Despite approving the use of alpelisib in patients with progression on or following an endocrine-based regimen, the FDA stated in its statistical and clinical evaluation of alpelisib that, based on the SOLAR-1 study, “no conclusions may be drawn regarding the efficacy of alpelisib plus fulvestrant in the CDK 4/6 inhibitor-pretreated population.”20 The European Medicines Agency made a similar statement in its review, based on the SOLAR-1 and BYLieve studies, and restricted the approved indication to patients previously treated with endocrine therapy as a monotherapy.21
Of note, the sponsor has indicated that a new phase III trial will be conducted for alpelisib plus fulvestrant in patients with prior CDK4/6 inhibitor treatment. The trial will be a double-blind, placebo-controlled RCT in men and postmenopausal women with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced breast cancer who have progressed on or after treatment with an AI and a CDK4/6 inhibitor. As stated by the sponsor, the planned study population aligns with the population targeted by the reimbursement request. The first patient visit was expected to occur in October of 2021, with the first interpretable results for the trial planned for the third quarter of 2024.
While the BYLieve study did include an entire cohort of patients from the patient population under review, as with the SOLAR-1 study, it is also unable to inform the efficacy of alpelisib plus fulvestrant versus a relevant comparator (i.e., single-agent fulvestrant, single-agent chemotherapy, or everolimus plus exemestane) due to its non-comparative study design. Patients were assigned to 1 of 3 cohorts depending on the prior anticancer therapy they had received, as opposed to being randomized to treatment groups. Although the study met its primary end point of the lower bound of the 95% CI for proportion of patients alive without progression after 6 months being greater than 0.30, the clinical experts consulted by CADTH indicated that they would expect less than 30% of the patients to experience disease progression or death within 6 months after starting the second-line treatments that are currently used in standard care (e.g., fulvestrant alone). Therefore, the 30% threshold used in the BYLieve study may not be clinically significant in the Canadian treatment setting. Aside from the primary end point, there was no statistical hypothesis testing in the BYLieve study and no conclusions could be drawn regarding the outcomes of interest in the CADTH systematic review protocol, PFS and OS.
The sponsor-submitted observational study comparing cohort A from the BYLieve study to the Flatiron database-derived, real-world cohort had several key limitations that are of concern for drawing conclusions about the comparative efficacy of alpelisib plus fulvestrant versus any relevant comparator. Following propensity score–based weighting of the cohorts, PFS was 3.7 months (95% CI, 3.1 months to 6.1 months) in the Flatiron cohort and 7.3 months (95% CI, 5.6 months to 8.3 months) in the BYLieve cohort. The hazard ratio for disease progression or death for the BYLieve cohort versus the Flatiron cohort was 0.62 (95% CI, 0.44 to 0.85). There remains a great deal of uncertainty regarding the efficacy of alpelisib in comparison to SOC due to the inherent limitations of observational data. While the adjustment approaches taken in this study may have adequately balanced on observable prognostic factors categorized as they were, bias in the efficacy estimate due to selection bias, measurement error, unmeasured confounding, and residual confounding cannot be ruled out. No attempts were made to assess or estimate the possible magnitude of such bias. Given these limitations, the comparative efficacy of alpelisib plus fulvestrant versus the SOC identified in the Flatiron cohort remains unclear.
Input from patient groups and clinicians emphasized that, in addition to OS and PFS, quality of life is a very important consideration for patients. Outcomes assessing HRQoL were not analyzed for the subgroup in the SOLAR-1 study and were not included in the BYLieve study. Even if analyses of HRQoL outcomes for the subgroup of patients with prior CDK4/6 inhibitor treatment in the SOLAR-1 study were available, they would be subject to the same limitations as PFS and OS in that subgroup. In addition, potential treatment unblinding due to the obvious differences in AE profiles between treatment groups could have led to bias in the EQ-5D-5L and EORTC QLQ-C30 outcomes.
Harms
The safety profile of alpelisib, as informed by the entire PIK3CA cohort in the SOLAR-1 study (corresponding with the Health Canada–approved indication for alpelisib) and cohort A in the BYLieve study, was consistent between studies. In the SOLAR-1 study, the most common AEs (those reported in at least 20% in either treatment group) were also more common in the alpelisib group versus the placebo group: hyperglycemia, diarrhea, nausea, rash, decreased appetite, decreased weight, stomatitis, vomiting, fatigue, and alopecia. The most commonly reported AEs in the BYLieve study in cohort A were similar, except for the absence of decreased weight and alopecia. According to the clinical experts consulted by CADTH for this review, all of the AEs identified in the clinical studies can be managed, aside from osteonecrosis of the jaw. The clinical experts identified hyperglycemia, rash, and diarrhea as the main AEs and indicated that rash can be prevented with prophylactic antihistamine, while hyperglycemia and diarrhea are more challenging to manage. However, the ability of patients to manage the AEs depends on their tolerance for side effects from additional treatments. Aside from osteonecrosis of the jaw, discontinuing treatment with alpelisib may be necessary due to medical reasons such as significant dehydration due to diarrhea or elevation of glucose levels to the point of requiring visits to the emergency department. The most common serious AE in both studies was hyperglycemia, with all other serious AEs occurring in less than 4% of any treatment group. Hyperglycemia, rash, and diarrhea were the most common reasons for discontinuing alpelisib due to an AE in the SOLAR-1 study, while rash was the most common reason in the BYLieve study. The percentage of patients who discontinued treatment due to an AE was notably higher in the alpelisib group than in the placebo group in the SOLAR-1 study; for the alpelisib groups, it was 27.2% in the SOLAR-1 study and 20.5% in the BYLieve study. The clinical experts consulted by CADTH did not have enough experience in treating patients with alpelisib to comment on which estimate was more reflective of clinical practice in Canada but indicated that both seemed reasonable. Safety outcomes were not assessed in the observational study.
According to input from patient groups, minimizing the risk of side effects was identified as being important to patients. The commonly reported AEs from the SOLAR-1 and BYLieve studies were in line with those reported by patients with experience with alpelisib treatment. The patient groups indicated that patients who had taken alpelisib experienced substantial difficulties with the side effects, including dealing with intense fatigue or being overwhelmed by the number of side effects early on. Hyperglycemia was commonly identified as being particularly hard to manage. Despite these difficulties, the patient group submissions also made it clear that overall, patients found the side effects manageable and were willing to tolerate them for the sake of a treatment that would improve long-term health outcomes.
Conclusions
No conclusions could be drawn from the SOLAR-1 and BYLieve studies regarding the comparative efficacy or effectiveness of alpelisib plus fulvestrant versus any relevant comparator in patients with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor. Neither study was designed to draw conclusions on comparative efficacy in this patient population. The sponsor-submitted observational study comparing a cohort from the BYLieve study that received alpelisib plus fulvestrant to a database-derived cohort that received a variety of non-alpelisib SOC therapies reported PFS results in favour of alpelisib. Methodological limitations in the observational study, including a high likelihood of residual confounding that may have led to bias in favour of alpelisib and differences in the methods used to determine PFS, contributed a substantial degree of uncertainty to the effect estimate. Considering the evidence in its entirety, the magnitude of any benefit associated with alpelisib plus fulvestrant in the relevant patient population remains unclear. In the SOLAR-1 and BYLieve studies, alpelisib treatment was associated with hyperglycemia, diarrhea, nausea, rash, decreased appetite, stomatitis, vomiting, and fatigue. Although the AEs reported in the studies can be medically managed, the percentages of patients who discontinued treatment due to an AE in the studies suggest that a large proportion of patients may not be able to remain on treatment with alpelisib due to AEs and/or subsequent side effects from treatments to manage these.
References
1.Interim Clinical Study Report: CBYL719C2301. A phase III randomized double-blind, placebo controlled study of alpelisib in combination with fulvestrant for men and postmenopausal women with hormone receptor positive, HER2-negative advanced breast cancer which progressed on or after aromatase inhibitor treatment [internal sponsor's report]. Basel (CH): Novartis; 2018.
2.Clinical Study Report: CBYL719C2301. SOLAR-1: A phase III randomized double-blind, placebo controlled study of alpelisib in combination with fulvestrant for men and postmenopausal women with hormone receptor positive, HER2-negative advanced breast cancer which progressed on or after aromatase inhibitor treatment. Clinical Study Report - final overall survival analysis and update of efficacy and safety data [internal additional sponsor's report]. Basel (CH): Novartis; 2020.
3.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2018. A special 2018 report on cancer incidence by stage. Toronto (ON): Canadian Cancer Society; 2018: www.cancer.ca/Canadian-Cancer-Statistics-2018-EN. Accessed 2021 Jun 11.
4.Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the number of women living with metastatic breast cancer in the United States. Cancer Epidemiol Biomarkers Prev. 2017;26(6):809-815. PubMed
5.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. Can Med Assoc J. 2020;192(9):E199-E205. PubMed
6.Seung SJ, Traore AN, Pourmirza B, Fathers KE, Coombes M, Jerzak KJ. A population-based analysis of breast cancer incidence and survival by subtype in Ontario women. Curr Oncol. 2020;27(2):e191-e198. PubMed
7.Howlader N, Cronin KA, Kurian AW, Andridge R. Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiol Biomarkers Prev. 2018;27(6):619-626. PubMed
8.Cardoso F, Paluch-Shimon S, Senkus E, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. 2020;31(12):1623-1649. PubMed
9.Mosele F, Stefanovska B, Lusque A, et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann Oncol. 2020;31(3):377-386. PubMed
10.PrPiqray® (alpelisib): 50 mg, 150 mg and 200 mg tablets [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada; 2020 Mar 11.
11.PrFaslodex® (fulvestrant): 50 mg/mL pre-filled syringe for intramuscular injection [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2019 Nov 15: https://pdf.hres.ca/dpd_pm/00053975.PDF. Accessed 2021 Jun 8.
12.PrXeloda® (capecitabine): 150 mg and 500 mg tablets [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 19: https://pdf.hres.ca/dpd_pm/00060777.PDF. Accessed 2021 Jun 8.
13.PrAfinitor® (everolimus): 2.5 mg, 5 mg and 10 mg, tablets [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 May 5: https://pdf.hres.ca/dpd_pm/00060902.PDF. Accessed 2021 Jun 8.
14.PrAromasin® (exemestane): 25 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2018 Mar 6: https://pdf.hres.ca/dpd_pm/00044156.PDF. Accessed 2021 Jun 8.
15.Anderson EJ, Mollon LE, Dean JL, et al. A systematic review of the prevalence and diagnostic workup of PIK3CA mutations in HR+/HER2- metastatic breast cancer. Int J Breast Cancer. 2020:3759179. PubMed
16.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
17.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 May 7.
18.Andre F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929-1940. PubMed
19.Andre F, Ciruelos EM, Juric D, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. 2021;32(2):208-217. PubMed
20.Center for Drug Evaluation Research. Multidiscipline review. Piqray (alpelisib) oral. Novartis Pharmaceuticals. Application No.:212526. Approval date: 05/24/2019 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2019: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212526Orig1s000MultidisciplineR.pdf. Accessed 2021 Jun 17.
21.Committee for Medicinal Products for Human Use. Assessment report: Piqray (alpelisib). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2020 May 28: https://www.ema.europa.eu/documents/assessment-report/piqray-epar-public-assessment-report_en.pdf. Accessed 2021 Jun 17.
22.Novartis Pharmaceuticals Inc. response to 2021 June 23 DRR request for additional information regarding Piqray DRR review. Additional SOLAR-1 study data in subgroup with prior CDK4/6 inhibitor use [internal additional sponsor's information]. Basel (CH): Novartis Pharmaceuticals; 2021.
23.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. PubMed
24.Giuliano M, Schettini F, Rognoni C, et al. Endocrine treatment versus chemotherapy in postmenopausal women with hormone receptor-positive, HER2-negative, metastatic breast cancer: a systematic review and network meta-analysis. Lancet Oncol. 2019;20(10):1360-1369. PubMed
25.Han Y, Wang J, Wang Z, Xu B. Comparative efficacy and safety of CDK4/6 and PI3K/AKT/mTOR inhibitors in women with hormone receptor-positive, HER2-negative metastatic breast cancer: a systematic review and network meta-analysis. Curr Probl Cancer. 2020;44(6):100606. PubMed
26.Wang S, Liu M, Lian S, et al. Which is the most appropriate PI3K inhibitor for breast cancer patients with or without PIK3CA status mutant? A systematic review and network meta-analysis. Biomed Res Int. 2020:7451576. PubMed
27.Leung JH, Leung HW, Wang SY, Huang SS, Chan AL. Efficacy and safety of CDK4/6 and PI3K/AKT/mTOR inhibitors as second-line treatment in postmenopausal patients with hormone receptor-positive, HER-2-negative metastatic breast cancer: a network meta-analysis. Expert Opin Drug Saf. 2021;28. PubMed
28.Rugo HS, Lerebours F, Ciruelos E, et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021;22(4):489-498. PubMed
29.Drug Reimbursement Review sponsor submission: Piqray (alpelisib), 50 mg, 150 mg and 200 mg tablets. [internal sponsor's package]. Dorval (QC): Novartis; 2021 Apr 20.
30.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26(4):404-413.
31.Turner S, Chia S, Kanakamedala H, et al. Effectiveness of alpelisib + fulvestrant compared with real-world standard treatment among patients with HR+, HER2-, PIK3CA-mutated breast cancer. Oncologist. 2021;26(7):e1133-e1142. PubMed
32.Clinical Study Report: CBYL719X2402. BYLieve: A phase II, multicenter, open-label, three-cohort, non-comparative study to assess the efficacy and sfaety of alpelisib plus fulvestrant or letrozole in patients with PIK3CA mutant, hormone receptor (HR) positive, HER2-negative advanced breast cancer (aBC), who have progressed on or after prior treatments. Interim Clinical study report (data cut-off date: 17-Dec-2019): primary endpoint analysis for Cohort A [internal sponsor's report]. Basel (CH): Novartis; 2020.
33.Non-interventional Final Study Report: CBYL719C2403. Clinical efficacy of alpelisib and fulvestrant from BYLieve compared with real-world effectiveness of standard treatment among patients with HR+ HER2– PIK3CA-mutated advanced breast cancer [internal sponsor's report]. Basel (CH): Novartis; 2020.
34.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41-55.
35.Imbens GW, Rubin DB. Causal inference for statistics, social, and biomedical sciences: an introduction. Cambridge (UK): Cambridge University Press; 2015.
36.Stuart EA. Matching methods for causal inference: a review and a look forward. Stat Sci. 2010;25(1):1-21. PubMed
37.Hall WJ, Wellner JA. Confidence bands for a survival curve from censored data. Biometrika. 1980;67(1):133-143.
38.Efron B, Tibshirani RJ. An introduction to the bootstrap. New York (NY): Chapman & Hall/CRC; 1994.
39.Novartis Pharmaceuticals Inc. response to 2021 June 15 DRR request for additional information regarding Piqray DRR review. Observational study supplemental materials [internal additional sponsor's information]. Basel (CH): Novartis Pharmaceuticals; 2021.
40.EuroQoL Research Foundation. EQ-5D-5L user guide. 2019: https://euroqol.org/publications/user-guides. Accessed 2021 Jul 6.
41.Breast cancer (version 4.2021). NCCN Clinical practice guideline in oncology. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2021 Apr 28: www.nccn.org. Accessed 2021 May 7.
42.Therascreen PIK3CA RGQ PCR kit; licence no. 104503. Medical Devices Active Licence Listing (MDALL): archived licence listing 2021; https://health-products.canada.ca/mdall-limh/information.do?deviceId_idInstrument=1014385&deviceName_nomInstrument=THERASCREEN+PIK3CA+RGQ+PCR+KIT&lang=eng&licenceId=104503. Accessed 2021 Jul 13.
43.Center for Drug Evaluation Research. Summary of Safety and Effectiveness Data (SSED). therascreen PIK3CA RGQ PCR Kit (real-time PCR test). QIAGEN GmbH. Application No.:P190001. Approval date: 05/24/2019. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2019: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190001B.pdf. Accessed 2021 Jun 18.
44.Coverage, billing, and financial assistance for Foundation Medicine patients. Cambridge (MA): Foundation Medicine; 2020: https://assets.ctfassets.net/w98cd481qyp0/PKWlcTAZ3RTtqdZcoyGnS/17bab00b0c408a62cd9a2ddf108646a2/Foundation_Medicine_Patient_Billing_Guidelines.pdf. Accessed 2021 Jul 6.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 19, 2021
Alerts: Bi-weekly search updates until project completion
Study types: No filters were applied to limit retrieval by study type.
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Piqray* or alpelisib* or BYL719 or BYL 719 or 08W5N2C97Q).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*Alpelisib/
(Piqray* or alpelisib* or BYL719 or BYL 719).ti,ab,kw,dq.
3 or 4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
Search -- Studies with results | Piqray/alpelisib AND breast neoplasms
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms -- Piqray/alpelisib AND breast cancer
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms -- Piqray/alpelisib AND breast cancer
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms -- Piqray/alpelisib AND breast cancer
Grey Literature
Search dates: May 7 to May 13, 2021
Keywords: Piqray, alpelisib, breast cancer
Limits: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Ciruelos EM, Rugo HS, Mayer IA, et al. Patient-reported outcomes in patients with PIK3CA-mutated hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer from SOLAR-1. J Clin Oncol. 2021:JCO2001139. | Study population |
Batalini F, Moulder SL, Winer EP, Rugo HS, Lin NU, Wulf GM. Response of brain metastases from PIK3CA-mutant breast cancer to alpelisib. JCO Precis Oncol. 2020;4:572-578. | Study design |
Rugo HS, Andre F, Yamashita T, et al. Time course and management of key adverse events during the randomized phase III SOLAR-1 study of PI3K inhibitor alpelisib plus fulvestrant in patients with HR-positive advanced breast cancer. Ann Oncol. 2020;31(8):1001-1010. | Study population |
Andre F, Ciruelos EM, Rubovszky G, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): Results of the phase III SOLAR-1 trial. Ann Oncol. 2018;29(Suppl 8):viii709. | Study population |
Note: This table has not been copy-edited.
Appendix 3: EQ-5D Five-Level Index Score Data From the SOLAR-1 Study
Note that this appendix has not been copy-edited.
Backgound
In the SOLAR-1 study, change from baseline in the EQ-5D-5L index score in the PIK3CA mutant cohort was an exploratory outcome. Although the index score was not analyzed in the subgroup with prior CDK4/6 inhibitor treatment, the results for the cohort are summarized in this appendix to support CADTH’s review of the sponsor’s economic evaluation.
EQ-5D is a generic quality-of-life instrument that can be applied to a wide range of health conditions and treatments.40 The EQ‑5D‑5L version was introduced in 2005 based on the earlier 3-Levels version (EQ-5D-3L).40 It consists of an EQ-5D descriptive system and the EQ visual analogue scale. The descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression, each with 5 levels: a level 1 response represents “no problems;” level 2 “slight problems;” level 3 “moderate problems;” level 4 “severe problems;” and level 5 “extreme problems” or “unable to perform,” which is the worst response in the dimension. Respondents are asked to choose the level that reflects their health state for each of the 5 dimensions. In total, there are 3,125 possible unique health states defined by the EQ-5D-5L, with 11111 and 55555 representing the best and worst health states, respectively. Results from the EQ-5D-5L descriptive system can be converted into a single index score using a scoring algorithm taking the local patient and population preferences into account. The range of index scores will differ according to the scoring algorithm used; however, in all scoring algorithms of the EQ-5D-5L, a score of 0 represents the health state “dead” and 1.0 reflects “perfect health.” Negative scores are also possible for those health states that society (not the individual patient) considers to be “worse than dead.”
Methods
In the SOLAR-1 study, the tablet version of the EQ-5D-5L was administered to patients in their local language during the screening period and every 8 weeks after randomization during the first 18 month and every 12 weeks thereafter (including at the end-of-treatment visit) until disease progression, death, withdrawal of consent, loss to follow-up, or patient/guardian decision. In patients who discontinued treatment for these reasons, EQ-5D-5L data were collected at an end-of-treatment visit. In patients who discontinued treatment for other reasons, such as AE, collection of EQ-5D-5L data continued until disease progression, death, withdrawal of consent, loss to follow-up, or patient/guardian decision. There was no indication in the SOLAR-1 Clinical Study Report that collection of EQ-5D-5L data in a patient was interrupted if their treatment was interrupted. The EQ-5D-5L was administered at each visit prior to any clinical assessment, drug dosing, or diagnostic testing.
In addition to descriptive statistics to summarize the index scores, a repeated measures model was used to compare change in baseline in the index score between the alpelisib and placebo groups. Statistical testing was not performed. The model included the following terms: treatment, terms for the randomization stratification factors, time, baseline value, and treatment-by-time. The percentage of patients completing the questionnaire out of patients remaining in the study at each visit was summarized.
Results
Baseline scores and percentage of patients completing the questionnaire are presented in Table 33. At least 80% of patients remaining in the study completed the questionnaire at each visit for the first 25 months. Mean change from baseline in index score and standard error of measurement estimated from the repeated measures model at each study visit are plotted in Figure 8. At each post-baseline time point, the estimated mean EQ-5D-5L index score from the model was numerically higher in the alpelisib group than in the placebo group.
Table 33: EQ-5D Five-Level Results From the SOLAR-1 Study
Characteristic | SOLAR-1 study PIK3CA mutant cohort | |
|---|---|---|
Alpelisib + fulvestrant FAS N = 169 | Placebo + fulvestrant FAS N = 172 | |
All patients: June 12, 2018, data cut-off date | ||
EQ-5D-5L index score at baseline, N | 155 | 157 |
Mean (SD) | 0.766 (0.1941) | 0.742 (0.2150) |
All patients: April 23, 2020, data cut-off date | ||
Patients with valid EQ-5D-5L questionnaire at selected time points, N (% of patients on study at scheduled day) | ||
Baseline | 161 (95.3) | 167 (97.1) |
Cycle 7, day 1 | 103 (90.4) | 82 (92.1) |
Cycle 13, day 1 | 69 (88.5) | 50 (86.2) |
Cycle 19, day 1 | 50 (92.6) | 36 (90.0) |
Cycle 25, day 1 | 36 (87.8) | 25 (80.6) |
Cycle 31, day 1 | 28 (90.3) | 15 (83.3) |
Cycle 37, day 1 | 17 (73.9) | 7 (70.0) |
Cycle 43, day 1 | 14 (77.8) | 5 (83.3) |
EQ-5D-5L = EQ-5D Five-Level; FAS = full analysis set; SD = standard deviation.
Source: SOLAR-1 study interim Clinical Study Report (2018)1 and final Clinical Study Report (2020).2
Figure 8: Change From Baseline in EQ-5D Five-Level Index Score in the PIK3CA Mutant Cohort
EQ-5D-5L = EQ-5D Five-Level; fulv = fulvestrant; LSMeans = least squares mean; SEM = standard error of the mean.
Source: SOLAR-1 study final Clinical Study Report (2020).2
Limitations of the Results
While the percentage of patients completing the EQ-5D-5L at each visit out of the patients being followed for efficacy remained above 80% for the first 25 months, this still means that there was some missing data and the resulting potential direction of bias is unclear. Also, the numbers of patients (and percentages of those still being followed for efficacy) steadily decreased over the study visits such that the proportion of patients in the FAS contributing EQ-5D-5L data was less than half by the end of the first year. Treatment discontinuations due to disease progression and patient/guardian decision were imbalanced between the alpelisib group versus the placebo group: 47.9% versus 78.9% due to disease progression and 9.5% versus 4.1% due to patient/guardian decision. The direction of any potential bias from this missing data is unclear. Although the approach of discontinuing HRQoL assessments following disease progression presumably allows for more focus on the effects of the treatments on HRQoL, it results in HRQoL data that is not representative of the entire study population.
The notable difference in AE profiles between the 2 treatment groups may have led to treatment unblinding of patients and investigators. While bias from treatment unblinding was likely mitigated for PFS and OS by the objective nature of those assessments, the EQ-5D-5L is a subjective assessment that is susceptible to bias from treatment unblinding. While the direction of bias is not certain, it remains a possibility that patients aware of being assigned to the alpelisib group could have had a more optimistic outlook, resulting in bias favouring alpelisib.
Appendix 4: Companion Diagnostic Testing
Note that this appendix has not been copy-edited.
PIK3CA Mutation Testing
Guidelines recommend that patients with hormone receptor–positive, HER2-negative advanced breast cancer be tested for PIK3CA mutations to determine eligibility for alpelisib.8,41 Specifically, ESO-ESMO guidelines state that alpelisib with fulvestrant is a treatment option for patients with PIK3CA-mutated tumours (in exons 9 or 20), previously exposed to an AI and with appropriate glycated hemoglobin levels.8 PIK3CA mutations can be detected by tumour tissue testing or liquid biopsy. Liquid biopsies can use circulating tumour DNA (ctDNA) or cell-free DNA.15 Multiple techniques for testing can be used including PCR, NGS, Sanger sequencing, and liquid chip technology.15
The ESO-ESMO guidelines recommend that patients should be tested for PIK3CA mutation in a tissue (metastasis or primary) and/or by ctDNA testing in blood if alpelisib is an available treatment option. Per National Comprehensive Cancer Network guidelines, if the liquid biopsy is negative, tumour tissue testing is recommended.41 Similarly, ESO-ESMO guidelines recommend that if patients do not have an available archival tissue sample and have an uninformative result using a liquid biopsy test, undergoing a tumour biopsy for PIK3CA mutation testing could be considered.8 Patients may undergo multiple tumour biopsies over time, particularly if the disease progresses during a specific treatment.15 Also, the tumour biopsy site may change over time.15 According to the clinical experts consulted by CADTH, testing is preferably performed on metastatic tumour tissue rather than primary tumour tissue.
In a systematic review of the prevalence and diagnosis of PIK3CA mutations in hormone receptor–positive, HER2-negative metastatic breast cancer, the authors found that the overall reported prevalence of PIK3CA mutations ranged from 13.3% to 61.5% (median, 36.4%; IQR: 31.2%, 45.6%).15 Among tissue biopsies, PIK3CA mutation prevalence ranged from 20.8% to 61.5%, compared to 43.4% to 46.8% among liquid biopsies. It is unclear whether the prevalence of PIK3CA mutations in patients who are best suited to treatment with alpelisib and fulvestrant differs from that in the overall patient population with hormone receptor–positive, HER2-negative metastatic breast cancer. Concordance of PIK3CA mutations between tissue and liquid biopsies ranged from 70.4% to 94%. Most hotspot mutations were located in H1047R in exon 20 and E545K in exon 9. Lastly, this systematic review found a lack of consistent approaches regarding if and how to re-test for PIK3CA mutations at different time points or between different testable materials. The authors reported that this lack of re-test protocol represents a significant gap in knowledge for the clinical applicability of PIK3CA testing for hormone receptor–positive, HER2-negative advanced breast cancer patients.
Health Canada42 and the FDA43 have approved the therascreen PIK3CA RGQ PCR Kit, which is a real-time PCR test for 11 mutations in the PIK3CA gene. The mutations tested include C420R in exon 7; E542K, E545A, E545D [1635G > T only], E545G, E545K, Q546E, and Q546R in exon 9; and H1047L, H1047R, and H1047Y in exon 20. This test uses genomic DNA extracted from breast tumour tissue or ctDNA from plasma. The test is intended to aid clinicians in identifying breast cancer patients who may be eligible for treatment with alpelisib. Per the FDA report, patients whose tissue or plasma specimen produce a positive therascreen test result for the presence of 1 or more PIK3CA mutations are eligible for treatment with alpelisib.43 The FDA recommends that patients whose plasma specimens produce a negative result using this test should be reflexed to testing with tumour tissue for PIK3CA mutations.43
Per the clinical experts consulted by CADTH, testing for PIK3CA mutations is challenging to access in Canada outside of special programs or research studies. Currently, private pay options for PIK3CA mutations are the most accessible option outside of clinical trials. The clinical experts reported that liquid biopsy tests (specifically the Canexia Health Follow It test) cost approximately CA$500 and that the FoundationOne CDx tumour biopsy test offered by Foundation Medicine costs approximately CA$7,000 per test. The current list price for the FoundationOne CDx tumour biopsy test in the US is US$5,800.44 Both the Canexia Follow It test and the FoundationOne CDx tumour biopsy test analyze a panel of genes rather than the PIK3CA gene alone. The clinical experts indicated that most Canadian clinicians currently access these tests after progression on first-line therapy given the limited accessibility and the cost to the patient. Based on the experiences of the clinical experts, NGS methods are preferred over the therascreen PCR test in hospital-based facilities as they can identify a wider variety of PIK3CA mutations; additionally, turnaround times for tests are approximately 1 to 2 weeks for liquid biopsy tests and up to 6 weeks for in-house tissue tests, depending on batching. The experts also indicated that in-house tissue testing may cost less than the FoundationOne CDx test as the cost of materials was estimated to be less than CA$1,000 per test.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CDK4/6
cyclin-dependent kinase 4 and 6
CI
confidence interval
EQ-5D-5L
EQ-5D Five-Level
HER2
human epidermal growth factor receptor 2
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
PFS
progression-free survival
PPS
post-progression survival
QALY
quality-adjusted life-year
RDI
relative dose intensity
SOC
standard of care
TTD
time to treatment discontinuation
WTP
willingness-to-pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Alpelisib (Piqray), in 50 mg, 150 mg, and 200 mg oral tablets |
Submitted price | Alpelisib: $95.23 per 50 mg tablet $190.46 per 150 mg tablet $95.23 per 200 mg tablet + 50 mg tablet |
Indication | In combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 11, 2020 |
Reimbursement request | In combination with fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor |
Sponsor | Novartis Pharmaceuticals Inc. |
Submission history | No |
CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-effectiveness analysis Semi-Markov cohort model |
Target population | Postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor (this aligns with the sponsor’s reimbursement request) |
Treatment | Alpelisib plus fulvestrant |
Comparators | SOC (everolimus plus exemestane) |
Perspective | Canadian publicly funded health care payer |
Outcomes | LYs, QALYs |
Time horizon | Lifetime (15 years) |
Key data source | BYLieve trial (alpelisib plus fulvestrant) and Flatiron data (SOC) |
Submitted results | The ICER for alpelisib plus fulvestrant compared to SOC was $69,674 per QALY (incremental costs = $25,184; incremental QALYS = 0.36). |
Key limitations |
|
CADTH reanalysis results |
|
AE = adverse event; CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; LY = life-year; PFS = progression-free survival; PPS = post-progression survival; QALY = quality-adjusted life-year; RDI = relative dose intensity; SOC = standard of care; TTD = time to treatment discontinuation; vs. = versus.
Conclusions
Based on the CADTH clinical review, the efficacy of alpelisib plus fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor remains unclear. Given the insufficient direct comparative clinical efficacy information for the relevant patient population, the sponsor submitted an observational study comparing progression-free survival (PFS) among patients who received alpelisib plus fulvestrant in cohort A of the BYLieve study (a single-arm, open-label trial) to patients who received a variety of non-alpelisib standard of care (SOC) therapies from the Flatiron Clinico-Genomic Database cohort (retrospective electronic health record data). The study used propensity score weighting methods that had several limitations, resulting in poor reliability and validity of the efficacy estimates. These limitations included the high likelihood of residual confounding, the potential for measurement error in the observed covariates due to the nature of the electronic health record data collection in the Flatiron data, the underlying differences in the patient populations before and after re-weighting, and the exclusion of patients from the Flatiron cohort who were treated with alpelisib. Due to the lack of sufficient comparative clinical efficacy data, there is a high degree of uncertainty in estimating the cost-effectiveness of alpelisib plus fulvestrant for the sponsor’s requested reimbursement population.
CADTH conducted exploratory reanalyses to assess the impact of alternative model assumptions on the cost-effectiveness estimates for alpelisib plus fulvestrant compared to SOC. These changes included a revised price for everolimus, an alternate parametric PFS curve, alternate estimates for the percentage of patients progressing due to death, the inclusion of PIK3CA retesting costs, a relative dose intensity (RDI) of 1.0 for oral drugs, the removal of treatment-specific health state utility estimates, the use of an alternate hazard ratio (HR) for the derivation of time to treatment discontinuation (TTD) curves from PFS, and the setting of adverse event (AE) incidence as being equal between treatments. Although CADTH conducted these exploratory analyses using alternate estimates in the model, the CADTH reanalyses were limited by the substantial uncertainty associated with the key efficacy estimates informing the model, including PFS and post-progression survival (PPS) (i.e., from the BYLieve versus Flatiron analysis). Due to the lack of alternative comparative clinical efficacy estimates for alpelisib plus fulvestrant and other relevant SOC agents (e.g., capecitabine and fulvestrant monotherapy), the results of any analysis remain highly uncertain.
In CADTH’s exploratory reanalyses, alpelisib plus fulvestrant compared to SOC had a 0% probability of being cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY, with an incremental cost-effectiveness ratio (ICER) of $319,592 per QALY gained. A price reduction of 99% is required for alpelisib plus fulvestrant to be cost-effective at a $50,000 per QALY threshold.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Three patient groups provided input for this review: the Canadian Breast Cancer Network, Rethink Breast Cancer, and CanCertainty. Input was gathered from multiple online surveys and from published reports relating to breast cancer and oral cancer drugs. Canadian respondents included 90 individuals with metastatic, hormone receptor–positive, HER2-negative breast cancer and 4 individuals with hormone receptor–positive, HER2-negative, advanced or metastatic breast cancer with a PIK3CA mutation. Patients reported having received multiple lines of treatment and reported a range of outcomes and side effects. The most commonly reported side effects for breast cancer treatments overall were fatigue (100%), diarrhea (83%), loss of appetite (75%), nausea (54%), and headache (46%). Fatigue, diarrhea, and hyperglycemia were identified as the most difficult to tolerate side effects of previous treatments. Most patients experienced diarrhea (89%), reduced appetite (78%), weight loss (72%), and alopecia (67%) while receiving alpelisib. While not reported as frequently, hyperglycemia was highlighted during patient interviews as being especially hard to manage. Compared to other treatments received, most patients indicated that the drug’s side effects were the same (33%) or worse (28%), though some patients (39%) indicated that the side effects were better. One of the Canadian patients interviewed described wait times of about 3 weeks for a biopsy and 4 weeks for the results, during which she experienced both excitement about having better information and anxiety over not having a treatment plan. The biopsy procedure was described as somewhat painful. The patient groups reported significant financial challenges associated with treatments for metastatic breast cancer and the differences in jurisdictional funding for oral cancer drugs across Canada. Paying out of pocket and/or applying to funding assistance programs can take time and delay access to treatment.
Input was received from 1 clinician group: the Breast Medical Oncology group at the Ottawa Hospital Cancer Centre. Clinician input indicated that in the first-line setting, patients are treated with a combination of an aromatase inhibitor and a CDK4/6 inhibitor (palbociclib, ribociclib, or abemaciclib). On disease progression, patients may receive chemotherapy if they have aggressive disease progression or significant visceral metastases (e.g., lung, liver), or patients may receive second-line endocrine therapy with fulvestrant or another single-agent aromatase inhibitor. Input indicated that response rates for second-line therapy with fulvestrant are less than 15% and response rates with aromatase inhibitors are less than 10%, and that alpelisib was likely to replace fulvestrant monotherapy in practice. It was mentioned that treatment is typically provided in a community setting by medical oncologists or team physicians with sufficient oncology experience. Toxicity and symptom assessment typically occur every 2 weeks to 4 weeks in early treatment and scans are initially performed every 3 months. PIK3CA mutations were stated to be common, and patients with PIK3CA are less sensitive to chemotherapy. Presently, there is no targeted therapy for patients with PIK3CA mutations and molecular testing is not currently funded in Canada, remaining challenging to access. Clinician opinion about alpelisib was mixed in the submission; some clinicians indicated that the evidence for use was too uncertain, while others noted that there has not been advances in treatment for years and the toxicities would be manageable. No particular subgroup was thought to benefit most from treatment with alpelisib, and there was no biologic rationale to believe that alpelisib would not be effective following a CDK4/6 inhibitor.
Drug plan input included questions about the relative efficacy of alpelisib plus fulvestrant compared to endocrine monotherapy and chemotherapy options, considerations for treatment discontinuation (e.g., can alpelisib be given as monotherapy), if cost-effectiveness is expected to differ between patients who receive prior treatment with a CDK4/6 inhibitor versus those who have not, and the appropriateness of switching patients from active treatment (e.g., fulvestrant or capecitabine) to alpelisib plus fulvestrant with no evidence of disease progression or intolerance. Drug plans also asked about the potential for wastage given that alpelisib is available as tablet packs based on dose, the appropriate monitoring and management of AEs (e.g., rash, hyperglycemia), and considerations related to PIK3CA mutation testing (e.g., who should be tested, when testing should occur, at what cost).
Several of these concerns were addressed in the sponsor’s model, including the inclusion of PIK3CA testing costs and some costs associated with AEs reported by patients (e.g., fatigue, diarrhea, hyperglycemia).
In addition, CADTH addressed some of these concerns regarding the inclusion of PIK3CA retesting costs.
CADTH was unable to address the following concerns raised from stakeholder input:
the lack of the inclusion of chemotherapy as a relevant comparator in the sponsor’s model
the impact of treatment discontinuation or switching from monotherapy (i.e., alpelisib or fulvestrant alone) to combined therapy (alpelisib plus fulvestrant).
Economic Review
The current review is for alpelisib (Piqray) plus fulvestrant for men and postmenopausal women with PIK3CA-mutant, hormone receptor–positive, HER2-negative advanced breast cancer who had received prior treatment with a CDK4/6 inhibitor plus an aromatase inhibitor.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-effectiveness analysis of alpelisib plus fulvestrant compared to SOC, which was assumed to reflect everolimus plus exemestane. The modelled population consisted of men and postmenopausal women with PIK3CA-mutant, hormone receptor–positive, HER2-negative advanced breast cancer who had received prior treatment with a CDK4/6 inhibitor plus an aromatase inhibitor, which aligns with the sponsor’s reimbursement request. Of note, the Health Canada–approved indication is for patients with disease progression following an endocrine-based regimen, not necessarily only those patients who have progressed on a CDK4/6 inhibitor plus aromatase inhibitor. Alpelisib is available in 50 mg, 150 mg, and 200 mg oral tablets, and the recommended dose is 300 mg once daily on a continuous basis. Dose reductions to 250 mg per day and 200 mg daily may be necessary for the management of adverse drug reactions. The submitted price of $95.23 per 150 mg tablet, $190.46 per 200 mg tablet, and $190.46 for both a 200 mg tablet plus 50 mg tablet results in a daily cost of $190.46 and the 28-day cycle cost is $5,333 for alpelisib. In addition to alpelisib, fulvestrant is given at a dose of 500 mg on day 1, day 15, and day 29, and then once every 28 days thereafter. Patients should discontinue alpelisib if more than 2 dose reductions are required, and may continue taking fulvestrant based on the clinical judgment of the treating physician.1 The daily cost of fulvestrant is $62.45 (first cycle) to $20.82 (after the first cycle). The total per cycle cost of alpelisib plus fulvestrant is $7,082 for the first cycle and $5,916 for subsequent cycles, per patient. Treatment wastage was accounted for on a per tablet or per vial basis by rounding up to the number of tablets or vials required to achieve the milligrams required per cycle. The comparator was SOC based on the Flatiron database, which is based on a US population receiving a range of treatments (see Figure 2, Appendix 3). For the purposes of calculating costs and resource utilization, the sponsor assumed that the SOC comparator was everolimus plus exemestane.
The clinical outcomes modelled included life-years and quality-adjusted life-years (QALYs). The model was undertaken from the perspective of the Canadian public health care system over a time horizon of 15 years and using a discount rate of 1.5% for life-years, QALYs, and costs.
Model Structure
The sponsor used a semi-Markov model with 3 health states (progression-free, post progression, and death). The model had a 28-day cycle length and included tunnel states, which allow for the probability of transitioning out of each state to be defined by the time since entering a state. An illustration of the model structure can be found in Figure 1 in Appendix 3.
Model Inputs
For the sponsor’s submitted base case, the modelled patient population had a mean age of 57 years and were 100% female, based on cohort A of the BYLieve trial. Patients were assumed to have a body surface area of 1.73 m2 and a weight of 68.63 kg based on data from the SOLAR-1 trial.2
The dosing that was used in the model for alpelisib (300 mg once daily) plus fulvestrant (500 mg on day 1, day 15, and day 29, and then once every 28 days thereafter) was consistent with the Health Canada–approved indication. Dosing for SOC was assumed to be that of everolimus plus exemestane, according to the dose used in the BOLERO-2 trial (10 mg per day for everolimus and 25 mg per day for exemestane continuously for 28 days). RDI was based on data from the SOLAR-1 trial (alpelisib = 0.837; fulvestrant = 1.0) and BOLERO-2 (everolimus = 0.86; exemestane = 1.0).
Comparative efficacy estimates were based on data from the BYLieve versus Flatiron analysis. Parametric survival curves for PFS were estimated separately for each treatment using patient-level data from BYLieve and the Flatiron cohort (restricted cubic spline 1 log-normal restricted for both treatments). The probability of a patient moving from the progression-free health state to the post-progression or death health state was estimated based on the distribution of PFS events (i.e., progression or death) experienced by patients from the SOLAR-1 trial, according to the treatment received (4.4% of patients who progressed were due to death in the alpelisib plus fulvestrant arm; 7.5% of patients who progressed were due to death in the fulvestrant monotherapy arm) and was assumed to be constant over time. For the sponsor’s base case, the distribution of the type of progression event for the historical control arm was assumed to be the same as that of fulvestrant monotherapy. The probability of a patient moving from the post-progression health state to the death health state was based on a single parametric survival curve for alpelisib plus fulvestrant and SOC using patient-level data from the BYLieve trial.
The probabilities of remaining on treatment (defined as time from randomization to either discontinuation of medication or death) were estimated using patient-level data from the BYLieve trial (alpelisib plus fulvestrant) and from the BOLERO-2 trial for SOC (everolimus plus exemestane). For alpelisib plus fulvestrant, TTD curves were estimated separately for each (an exponential curve for alpelisib and a log-normal curve for fulvestrant) and it was assumed that TTD would not exceed PFS. TTD curves for SOC were based on patient-level data from the BOLERO-2 trial and were assumed to be the same for both everolimus and exemestane. TTD curves were estimated by applying an HR (1.2; 95% confidence interval [CI], 1.04 to 1.39), which was estimated based on the relationship between PFS and TTD curves in the BOLERO-2 trial, to the PFS curve estimated in the sponsor’s submitted model (based on Flatiron data).
All-cause mortality and mortality due to metastatic breast cancer were included in the model. The sponsor assumed that the probability of death in a given cycle could not be lower than mortality estimates for the general population, based on Statistics Canada life tables.3
AEs considered in the model included all-cause grade 3-plus events with an incidence of at least 5% for any of the comparators of interest. Data from SOLAR-1 was used to estimate AEs for alpelisib plus fulvestrant, and data from BOLERO-2 was used for AEs for patients receiving SOC.
Health state utility values (PFS, PPS) were derived from EQ-5D Five-Level (EQ-5D-5L) data collected from the full population of SOLAR-1 (April 23, 2020, data cut-off date) using generalized estimating equation regression analyses. The regression model selected by the sponsor included covariates for baseline utility values and utility values from EQ-5D-5L assessments that occurred within 28 days of death. Treatment-specific utility values were used when patients were on treatment in the PFS health state. Predicted mean utility values based on the regression analysis were 0.8415 for alpelisib plus fulvestrant and 0.8286 for placebo plus fulvestrant. Utility data were not available from the Flatiron cohort, so utility values for SOC were derived by taking the utility values obtained for alpelisib plus fulvestrant and applying a disutility for treatment for SOC, which was assumed to be 0.03 less than that of the placebo plus fulvestrant arm. The 0.03 decrement in utility was based on the assumption that treatment with everolimus would result in a decrease in utility of 0.03 (based on data from the BOLERO-2 trial)4 and that there would be no difference in quality of life between treatment with exemestane and fulvestrant (based on data from the CONFIRM trial and the EFECT trial).5,6 The predicted mean utility while off treatment in the PFS health state was 0.7603 and post-progression utility was 0.7800 regardless of treatment, based on the results of the regression analysis. The sponsor included a disutility of –0.1508 to account for a reduction in quality of life within 3 months of death. The sponsor’s base case did not include disutilities due to AEs.
Costs included in the sponsor’s model consisted of PIK3CA mutation testing costs, medication costs (acquisition, administration, and dispensing costs), costs of AEs, post-progression treatment costs, follow-up and monitoring costs (upon initiation of treatment and while on treatment), and terminal care costs. PIK3CA mutation testing costs were estimated to be $500, assuming that testing would be completed using either real-time polymerase chain reaction, digital polymerase chain reaction, or next-generation sequencing. Medication costs were calculated based on the percentage of patients expected to be on therapy and drug costs sourced from Ontario data reported in IQVIA. Medication costs for SOC were based on estimated costs for everolimus and exemestane. The sponsor adjusted the cost of treatment using RDIs from the SOLAR-1 trial and the BOLERO-2 trial for SOC (assumed to be everolimus and exemestane). For oral drugs, the sponsor included dispensing fees and administration costs (physician visit), based on Ontario estimates ($29.33); for intramuscular injections (fulvestrant), the sponsor included dispensing, injection, and physician visit fees ($50.77). Costs for the management of AEs were calculated using the Ontario Case Costing Initiative’s costing analysis tool and the percentage of AEs anticipated to be managed on an outpatient basis and inpatient basis, according to the results of a physician survey coordinated by the sponsor. Post-progression treatment costs were based on data from the full population of the SOLAR-1 trial using post-progression treatments received by the fulvestrant plus placebo arm for SOC. The first, second, and third round of post-progression treatments were included in the model, based on the post-treatment anticancer therapy received after a PFS event for patients in the SOLAR-1 trial.
Summary of Sponsor’s Economic Evaluation Results
All of the sponsor’s analyses were run probabilistically using 1,000 iterations. The deterministic and probabilistic results were similar. The probabilistic findings are presented as follows. Comparator costs are based on publicly available list prices and may not reflect actual costs paid by public drug plans.
Base-Case Results
In the sponsor’s base case, alpelisib plus fulvestrant was associated with estimated costs of $114,730 and 1.78 QALYs over a 15-year time horizon. Compared to the historical control group, alpelisib plus fulvestrant was associated with an incremental cost of $25,184 and 0.36 additional QALYs. This resulted in an ICER of $69,674 per QALY gained for alpelisib plus fulvestrant compared to the historical control group. In the sponsor’s base case, alpelisib plus fulvestrant had a 20.5% probability of being cost-effective at a WTP threshold of $50,000 per QALY. At the end of the 15-year time horizon, approximately 0.1% of patients in the alpelisib plus fulvestrant arm and 0.01% of patients in the historical control arm were projected to be alive. Results were primarily driven by drug acquisition costs. Table 3 provides a summary of the sponsor’s economic evaluation results.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. historical control ($/QALY) |
|---|---|---|---|---|---|
Historical control | 89,546 | Reference | 1.42 | Reference | Reference |
Alpelisib + fulvestrant | 114,730 | 25,184 | 1.78 | 0.36 | 69,674 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Additional results from the sponsor’s submitted base-case economic evaluation are presented in Appendix 3 (disaggregated results and projected overall survival and PFS curves versus the Kaplan–Meier curve from the BYLieve trial). Also summarized in Appendix 3 is the sponsor’s secondary analysis, which included the use of data from the SOLAR-1 post-CDK4/6 inhibitor subgroup. However, CADTH did not critically appraise this analysis.
Sensitivity and Scenario Analysis Results
Scenario analyses conducted by the sponsor included alternate time horizons (10 years and 20 years), alternate discount rates (0% and 3%), an alternative utility value in the PPS state, and alternate assumptions for the PFS curve.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Unable to derive a CADTH base-case reanalysis due to the lack of sufficiently robust comparative efficacy data: The sponsor’s base-case economic analysis was informed by data from several sources, including the SOLAR-1 trial, the BYLieve trial, and an observational study comparing cohort A of the BYLieve trial to the Flatiron cohort using propensity score matching methods. These 3 studies were summarized in the CADTH Clinical Review Report, which concluded that the efficacy of alpelisib plus fulvestrant in the relevant patient population remains unclear based on the totality of the clinical evidence provided by the sponsor.
The sponsor’s base-case economic analysis was conducted using efficacy data from an observational study comparing alpelisib plus fulvestrant from cohort A of BYLieve to SOC based on a propensity matched real world cohort from the Flatiron database. The BYLieve trial was a non-comparative cohort study that included patients with PIK3CA mutation who had been previously treated with a CDK4/6 inhibitor plus an endocrine-based therapy. The study was also open-label and had a median duration of follow-up of less than 1 year. Comparative data were derived from the Flatiron database, which is a non-randomized, retrospective cohort of patients with information collected through electronic medical records from the US. As stated in the CADTH Clinical Review Report, there were several limitations of this analysis, including uncontrolled confounding (Eastern Cooperative Oncology Group Performance Status), differences between the nature of the Flatiron database compared to the BYLieve study (electronic data collection methods, the inclusion of older patients with more severe disease), and the exclusion of patients having received alpelisib in the Flatiron cohort. Based on these limitations, it is not possible to know whether the observed differences in PFS between alpelisib plus fulvestrant and SOC are due to the causal effect of treatment or due to biases from selection and uncontrolled confounding in the analysis.
The sponsor conducted an additional economic analysis using data from the SOLAR-1 trial. The SOLAR-1 study was a placebo-controlled, double-blind, parallel-group randomized controlled trial that was designed to assess PFS with alpelisib plus fulvestrant compared with placebo and fulvestrant for patients with PIK3CA-mutated advanced breast cancer. As stated in the CADTH Clinical Review Report, no conclusions can be drawn from this trial because the SOLAR-1 study was not designed to test hypotheses in the subgroup of patients with prior CDK4/6 inhibitor treatment.
Based on the CADTH clinical review, no robust comparative efficacy and safety data exists to appropriately assess the cost-effectiveness of alpelisib plus fulvestrant compared to relevant comparators in the patient population requested by the sponsor.
Due to the lack of sufficient comparative efficacy data, CADTH was unable to identify an appropriate base case to inform the derivation of price reduction estimates. CADTH conducted reanalyses to assess the impact of alternative model assumptions on the cost-effectiveness estimates for alpelisib plus fulvestrant; however, these analyses should only be considered exploratory.
Limited generalizability of modelled SOC comparator in Canadian clinical practice: The sponsor compared alpelisib plus fulvestrant to SOC based on the Flatiron database, which is a US population whose SOC regimens differ from those used in Canadian clinical practice. According to the clinical experts consulted by CADTH for this review, relevant comparators that are more commonly used in Canadian clinical practice include fulvestrant monotherapy and single-agent chemotherapy, which only made up approximately 30% of the index treatments used by patients in the unweighted Flatiron cohort, and was used by an even smaller percentage of patients (< 25%) in the weighted cohort (Figure 2, Appendix 3). The differences in comparator agents used between the modelled cohort and Canadian clinical practice limits the interpretability of the cost-effectiveness of alpelisib plus fulvestrant compared to relevant comparator agents in Canada.
For the purposes of calculating costs and resource use, the sponsor assumed that the SOC comparison for alpelisib in combination with fulvestrant would be equivalent to everolimus plus exemestane. Given that the costs of chemotherapy and fulvestrant monotherapy are substantially lower ($209 for capecitabine per 28-day cycle, and $1,749 in the first 28-day cycle and $583 in subsequent cycles for fulvestrant) than the costs of everolimus and exemestane ($2,874 per 28-day cycle), the incremental costs for alpelisib plus fulvestrant are likely to be underestimated, biasing the results in its favour under the assumption of equal efficacy and safety between everolimus and exemestane and chemotherapy or fulvestrant monotherapy.
Additionally, the assumption that AE incidence for SOC would be aligned with everolimus and exemestane is uncertain given the limited generalizability of this regimen in Canadian clinical practice and the sponsor’s use of a naive comparison between data from BOLERO-2 (SOC) and SOLAR-1 (alpelisib plus fulvestrant). The use of naive comparisons assumes that the patient populations and study methodology is sufficiently similar and that there is no confounding present, which is highly unlikely to hold in practice. The estimates derived from these studies for key parameters in the sponsor’s model compromise the validity and interpretability of the cost-effectiveness of the results presented by the sponsor and limit any CADTH exploratory reanalyses. The only direct evidence between alpelisib plus fulvestrant and a comparator agent (fulvestrant monotherapy) for the incidence of AEs is from the SOLAR-1 trial. As presented in the CADTH Clinical Review Report, AEs, serious AEs, withdrawals due to AEs, and notable harms (e.g., hyperglycemia, diarrhea, nausea, rash, vomiting, maculopapular rash) were more common in the alpelisib plus fulvestrant arm compared to the placebo with fulvestrant arm. However, AE incidence for alpelisib plus fulvestrant compared to other relevant SOC agents is unknown.
CADTH was unable to address the lack of cost-effectiveness results for alpelisib plus fulvestrant compared to SOC agents that are typically used in Canadian clinical practice. In the CADTH exploratory reanalyses, CADTH used the sponsor’s AE incidence rates from the SOLAR-1 trial for alpelisib plus fulvestrant and assumed that these would be the same for SOC. A scenario analysis was also conducted that assumed a price reduction for everolimus and exemestane of 80% (based on a weighted average of costs that assumed 60% of patients would receive capecitabine monotherapy and 40% would receive fulvestrant monotherapy).
Overestimate of PFS in patients receiving second-line treatment: The sponsor used data from the BYLieve versus the Flatiron study to estimate the PFS for patients receiving alpelisib plus fulvestrant and SOC. Study data (spanning approximately 2.5 years) were used to derive parametric PFS curves to extrapolate the study data over the model’s 15-year time horizon. The sponsor’s base case used a restricted cubic spline 1 log-normal model for both alpelisib plus fulvestrant and the historical control group, which predicted a 2-year PFS estimate of 11% of patients remaining progression-free for alpelisib plus fulvestrant and 5% of patients remaining progression-free in the historical control group. According to the clinical experts consulted by CADTH for this review, the sponsor’s estimates for alpelisib plus fulvestrant and SOC were thought to be an overestimate of the percentage of patients remaining progression-free at 2 years for someone who has progressed on first-line endocrine therapy. The experts anticipated that PFS would be less than 5% at 2 years, and by 4 years most patients would have progressed. This was more closely aligned to the PFS estimates predicted by a Gamma unrestricted model (i.e., at 2 years = 4% progression-free with alpelisib plus fulvestrant and 1.4% for historical control; at 4 years = < 0.1% progression-free in both treatment arms). Overestimating the percentage of patients remaining progression-free over the 15-year time horizon led to an overestimate of incremental QALYs gained for alpelisib plus fulvestrant compared to SOC.
The sponsor used data from the SOLAR-1 trial to inform the percentage of progression events that are due to death (4.4% for alpelisib plus fulvestrant and 7.5% for fulvestrant monotherapy) or due to progression. The sponsor assumed that the 7.5% of patients progressing due to death when receiving fulvestrant would also apply to patients receiving SOC (everolimus and exemestane). The clinical experts consulted by CADTH for this review indicated that they would anticipate that the percentage of patients who would progress due to death would not differ between alpelisib plus fulvestrant and SOC. The use of data from the SOLAR-1 trial to inform overall survival estimates was also applied to the BYLieve versus Flatiron analysis, and therefore relied on an additional naive comparison that is based on the assumption that the overall survival for patients in each of the 3 studies would have sufficiently similar underlying disease trajectories. As stated in the CADTH Clinical Review Report, the clinical experts consulted by CADTH for this review indicated that the efficacy results from the PIK3CA cohort of SOLAR-1 cannot be used to inform the efficacy of alpelisib plus fulvestrant specifically in patients with prior CDK4/6 inhibitor treatment. The lack of certainty and face validity of these estimates resulted in an overestimate of patients progressing to death in the SOC arm, and an overestimate of the incremental QALYs gained for alpelisib plus fulvestrant compared to SOC.
In CADTH’s exploratory reanalyses, an alternate PFS curve (i.e., Gamma unrestricted model) for alpelisib plus fulvestrant and SOC was used. CADTH’s exploratory reanalysis also assumed that the percentage of patients who are anticipated to progress due to death would be the same in both treatment arms (i.e., 4.4%).
Underestimate of PIK3CA mutation testing costs: The sponsor assumed that the cost of PIK3CA mutation testing would be CA$500 and the testing cost included in the model for alpelisib plus fulvestrant would be a 1-off mean cost of CA$1,368 based on a 36.4% test positivity rate. The sponsor accounted for the likelihood that not all patients tested for the PIK3CA mutation would receive alpelisib and used a cost of PIK3CA mutation testing that was in line with the current cost of liquid biopsy tests conducted at local centres in Canada. Retesting for patients who test negative on liquid biopsy was not accounted for in the sponsor’s model. According to the clinical experts consulted by CADTH for this review, it is recommended to universally retest patients who test negative for the PIK3CA mutation on the initial liquid biopsy. This is aligned with the National Comprehensive Cancer Network guidelines7 and the European School of Oncology-European Society for Medical Oncology guidelines8 outlined in the CADTH Clinical Review Report, which state that patients whose liquid biopsy is negative should be retested with tumour tissue for PIK3CA mutations. As stated in the CADTH Clinical Review Report, a FoundationOne CDx tumour biopsy test conducted by Foundation Medicine costs US$5,800 per test (CA$7,431.54 based on an average Bank of Canada exchange rate of 1.2813 from July 2020 to July 2021).9 PIK3CA mutation testing is challenging to access in Canada outside of special programs or research studies. Local in-house tissue testing for the PIK3CA mutation may be available in some centres at a lower cost; however, the total cost of local tests is unclear (e.g., 1 centre reported a cost of approximately CA$300 to CA$750 for materials, which would not include interpretation). Omitting the costs of PIK3CA retesting underestimated the incremental cost of treating with alpelisib plus fulvestrant relative to comparator agents.
In CADTH’s exploratory reanalyses, a retesting rate of 63.6% using tissue biopsy at a cost of CA$7,431.54 was included. CADTH also conducted a scenario analysis to assess the impact of a lower cost for tissue biopsy retesting of CA$500.
Uncertainty associated with RDI and TTD assumptions impact treatment costs: The sponsor adjusted the cost of alpelisib and the cost of everolimus using an RDI of 0.837 for alpelisib and 0.86 for everolimus. This practice underestimated the total expenditure associated with these agents. For oral treatments, Canadian pharmacies are likely to dispense the full quantity of medication for each treatment cycle and excess tablets are unlikely to be recuperated. The cost of medication is therefore independent of any dose reductions observed in the trial during the course of the treatment. The use of RDI for alpelisib and everolimus underestimated the incremental costs associated with alpelisib, biasing results in its favour.
TTD was estimated using parametric curves based on patient-level data from the BYLieve trial (alpelisib plus fulvestrant) and from the BOLERO-2 trial and Flatiron data for SOC (everolimus plus exemestane). TTD curves for SOC were estimated by applying an HR (1.2; 95% CI, 1.04 to 1.39), which was estimated based on the relationship between PFS and TTD curves in the BOLERO-2 trial, to the PFS curve estimated in the sponsor’s submitted model (based on Flatiron data). This assumption consequently implies a naive direct comparison, which assumes that the relationship between PFS and TTD curves found in the BOLERO-2 trial would be similarly found in the patient population of interest for this review, instead of explicit modelling of TTD data from patient-level data. According to the clinical experts consulted by CADTH for this review, the sponsor’s assumed relationship between TTD and PFS may be reasonable; however, the magnitude of this relationship is difficult to predict in practice. Given the uncertainty associated with the HR (95% CI, 1.04 to 1.39) and the differences in the patient populations between the BYLieve and BOLERO-2 trials (not all patients have a PIK3CA mutation or have previously received a CDK4/6 inhibitor), the incremental costs associated with alpelisib may be overestimated or underestimated, depending on the HR assumed.
In CADTH’s exploratory reanalyses, an RDI of 1.0 was used for alpelisib and everolimus, and a conservative estimate for the sponsor’s assumed relationship between TTD and PFS was used (i.e., upper 95% CI for the HR = 1.39).
Uncertainty and lack of face validity of the health state utility estimates used in the sponsor’s model: The sponsor used health-related quality of life data from a regression analysis of the full population of the SOLAR-1 trial to inform the health state utility values (i.e., PFS on treatment, PFS off treatment, and PPS) for alpelisib plus fulvestrant and SOC. Treatment-specific utilities were used while patients were on treatment in the PFS health state (i.e., 0.84 for alpelisib plus fulvestrant and 0.80 for SOC), and the same utility values were used for each treatment when patients were off treatment or had progressed (i.e., 0.76 for both treatments). As stated in the CADTH Clinical Review Report, the health-related quality of life data from the SOLAR-1 trial is limited by the low number of patients completing the questionnaire, especially at later time points; the potential risk of bias from treatment unblinding due to the frequencies of AEs; the differences in rates of discontinuation and reasons for discontinuation between the 2 treatment arms; and the lack of data specifically for patients post-CDK4/6 inhibitor. The impact of these limitations on the direction and/or magnitude of bias for alpelisib plus fulvestrant and SOC is uncertain.
Treatment-specific utility values used for the SOC arm (i.e., assumed to be everolimus plus exemestane) was estimated by the sponsor by means of several assumptions, including the fact that utility data for patients receiving fulvestrant 250 mg is equal to patients receiving fulvestrant 500 mg (based on data from the CONFIRM trial),5 utility data for patients receiving exemestane 25 mg is equal to patients receiving fulvestrant (based on data from the EFECT trial),6 and patients receiving everolimus 10 mg would experience a utility decrement of 0.03 compared to fulvestrant (based on data from the BOLERO-2 trial).4 These assumptions were derived from selected clinical trials (i.e., via a non-systematic review of the literature) in postmenopausal hormone receptor–positive advanced breast cancer. However, the trials were conducted in 3 different patient populations, recruited over different time frames, and used different quality-of-life instruments. Consequently, the utility data used for patients in the PFS on-treatment SOC health state may not appropriately reflect that which was observed in practice.
In CADTH’s exploratory reanalyses, the same on-treatment PFS health state utility value was used for alpelisib plus fulvestrant and for SOC.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (see Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Price of everolimus | Not appropriate. The sponsor used a price for everolimus of $172.2559 per tablet (IQVIA) that was substantially higher than the publicly available price of everolimus reported in Nova Scotia’s Exception Status Drugs Formulary list ($101.3270). There is variability in the price of everolimus between jurisdictions in Canada, so CADTH used the lower publicly available price for the exploratory reanalyses and conducted a scenario analysis using the higher price ($172.256 per tablet). |
Percentage of patients progressing due to death assumed to be constant over time | Uncertain. The sponsor assumed that the percentage of patients assumed to progress due to death in the alpelisib plus fulvestrant arm and the SOC arm would remain constant over time. According to the clinical experts consulted by CADTH, it is anticipated that the percentage of patients who progress due to death may get smaller over time because a longer time remaining progression-free likely signals that a patient is responding to treatment. However, the clinical experts indicated that a constant percentage may be a reasonable assumption. CADTH could not explore this assumption in the reanalyses. |
Post-progression survival was assumed to be the same for alpelisib plus fulvestrant and SOC | Reasonable. No data were available from the Flatiron database to inform post-progression survival for SOC, so the sponsor used BYLieve patient-level data to inform post-progression parametric survival curves. Post-progression survival was assumed to be the same regardless of index treatment received, which was deemed reasonable by the clinical experts consulted by CADTH for this review, as differences in post-progression survival benefits between alpelisib and comparators is not expected to occur. |
Post-progression treatment options | Not appropriate. The sponsor assumed that patients who progress on alpelisib plus fulvestrant or SOC would receive up to 3 lines of additional therapy and would receive a mix of treatment, such as additional CDK4/6 inhibitors, bevacizumab + chemotherapy, and that these agents would be different between treatment arms. The clinical experts consulted by CADTH for this review indicated that it is reasonable to assume that patients would receive an additional 3 lines of treatment post progression; however, the majority of those patients would be receiving chemotherapy or might be trying a third-line endocrine treatment (e.g., exemestane or tamoxifen). Bevacizumab, for example, is not used in Canada for the patient population of interest, and the clinical experts indicated that the post-progression treatment options are unlikely to differ between treatment arms. |
Post-progression health state utility values | Uncertain. The sponsor used a utility value of 0.780 from the regression analysis for the post-progression health state for both alpelisib plus fulvestrant and SOC, based on data from SOLAR-1. In SOLAR-1, EQ-5D-5L data were only collected until disease progression, death, withdrawal of consent, loss to follow-up, or end of treatment.2 If a patient discontinued due to an AE, EQ-5D-5L data collection continued until 1 of the aforementioned instances occurred (e.g., disease progression, withdrawal of consent). It is unclear based on the sponsor’s submission how these data were collected and if they represent data collected at an end-of-treatment visit or longer-term post-progression data. The higher utility value assumed for the post-progression health state compared to the progression-free off-treatment health state utility also lacked face validity, according to the clinical experts consulted by CADTH for this review. The sponsor acknowledged that a utility value of 0.780 post progression is higher than what has been found in other studies (e.g., a utility value of 0.505 from Lloyd and colleagues).10 |
Incidence of AEs | Uncertain. The sponsor included all-cause grade 3+ AEs with an incidence of at least 5% for any of the comparators of interest. Data from SOLAR-1 was used to estimate AEs for alpelisib plus fulvestrant, and data from BOLERO-2 was used for AEs for patients receiving SOC. CADTH requested that the sponsor clarify the source of these incidence rates as the incidence rates used in the model could not be confirmed by CADTH. The sponsor responded to CADTH’s request, indicating that the AEs were sourced from the BYLieve study; however, this source differed from the sponsor’s original reference to SOLAR-1 data, and it remained unclear what the sponsor intended to use for historical control AE incidence rates. Additionally, based on input provided by patient groups for this review, alopecia, weight loss, loss of appetite, and nausea were identified as important to patients. These events were not included in the sponsor’s model. |
Duration of treatment for AEs | Not appropriate. The sponsor assumed that all AEs would last 1 month and applied a 1-off cost to account for these events. The clinical experts consulted by CADTH for this review indicated that some AEs (e.g., diarrhea, rash) are likely to require treatment and management for the duration of time that patients are on treatment, while others may be resolved within the 1-month period (e.g., infection). The exclusion of ongoing costs for AE management is likely to underestimate the incremental costs associated with alpelisib if these events are more common compared to comparator agents. |
Parameter distributions used for PSA | Not appropriate. In the sponsor’s probabilistic analysis, normal distributions were specified for the probability of AEs, cost of AEs, drug acquisition costs, drug administration and dispensing costs, terminal care costs, and follow-up and monitoring. According to the CADTH guidelines for economic evaluations, the choice of the form of the distribution should reflect the nature of the input parameter and match the bounds of the parameter — for example, beta distributions are typically used for probabilities and gamma distributions for costs. The use of normal distributions in the sponsor’s model may not appropriately characterize the parameter uncertainty in the model. |
AE = adverse event; CDK4/6 = cyclin-dependent kinase 4 and 6; EQ-5D-5L = EQ-5D Five-Level; PSA = probabilistic sensitivity analysis; SOC = standard of care.
CADTH Reanalyses of the Economic Evaluation
CADTH was unable to determine a base case for the CADTH reanalyses and consequently conducted exploratory reanalyses of the sponsor’s submitted model. CADTH exploratory reanalyses were derived by making changes to the model parameter values and assumptions in consultation with clinical experts. Details of the exploratory reanalyses are presented in Appendix 4. Changes consist of correcting the price of everolimus, revising the assumption for the PFS curves used, changing the percentage of patients progressing due to death for SOC to be the same as that for alpelisib plus fulvestrant, including retesting costs for PIK3CA mutation, revising RDI assumptions for oral drugs to 1.0, removing treatment-specific utility values, using a revised estimate for the HR to derive the TTD curve from PFS for SOC, and the setting of AE incidence as being equal between treatments.
Exploratory and Scenario Reanalysis Results
The CADTH exploratory reanalyses suggested that alpelisib plus fulvestrant was associated with higher costs and higher QALYs than SOC over a 15-year time horizon: namely, ICER $319,592 per QALY gained (Table 5). There was a 0% probability that alpelisib plus fulvestrant would be cost-effective at a WTP threshold of $50,000 per QALY. An exploratory price reduction analysis suggests that the price of alpelisib plus fulvestrant would need to be reduced by 99% to be cost-effective at this threshold (Table 13, Appendix 4).
Table 5: Summary of CADTH’s Exploratory Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. historical control ($/QALY) |
|---|---|---|---|---|---|
Historical control | 79,119 | Reference | 1.42 | Reference | Reference |
Alpelisib + fulvestrant | 129,828 | 50,710 | 1.58 | 0.16 | 319,592 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; ref. = reference; vs. = versus.
Although CADTH has presented the results of this reanalysis, it is important to emphasize that these results should be considered in light of the lack of sufficient comparative clinical efficacy and safety data to inform the analysis. CADTH undertook this exploratory reanalysis based on input provided by the clinical experts consulted by CADTH for this review; however, CADTH was limited by the structure of the sponsor’s submitted model and the data available. The incremental effectiveness and costs identified in this analysis are associated with substantial uncertainty and could be underestimates of the true values.
CADTH also conducted scenario analyses on the exploratory reanalysis to assess the impact of an alternate assumption for tissue biopsy retesting costs (CA$500), and a price reduction for everolimus and exemestane of 80% to reflect lower cost comparator agents used in clinical practice. Details of the exploratory reanalyses and scenario analyses results are reported in Appendix 4.
Issues for Consideration
According to the clinical experts consulted by CADTH for this review, it is possible that, in practice, there may be a desire to continue treating patients with fulvestrant monotherapy for those patients who discontinue alpelisib due to intolerance. Of note, fulvestrant monotherapy is not publicly funded in all jurisdictions across Canada.
CADTH’s exploratory reanalyses assume that PIK3CA mutation testing will occur after progression on a first-line CDK4/6 inhibitor treatment (i.e., before initiating on alpelisib plus fulvestrant). As stated by the clinical experts consulted by CADTH for this review, the timing of testing is likely to occur following progression; however, if testing is done through patient participation in a trial or free of charge, it may take place before first-line metastatic treatment. Alternate assumptions used for the timing and cost of PIK3CA mutations are likely to change the cost-effectiveness of alpelisib plus fulvestrant compared to other relevant agents.
Overall Conclusions
Based on the CADTH clinical review, the efficacy of alpelisib plus fulvestrant for the treatment of postmenopausal women, and men, with hormone receptor–positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer after disease progression following an endocrine-based regimen with a CDK4/6 inhibitor remains unclear. Given the insufficient direct comparative clinical efficacy data, the sponsor submitted an observational study comparing PFS among patients who received alpelisib plus fulvestrant in cohort A of the BYLieve study (a single-arm, open-label trial) to patients who received a variety of non-alpelisib SOC therapies from the Flatiron Clinico-Genomic Database (retrospective electronic health record data). The study used propensity score weighting methods that had several limitations, resulting in poor reliability and validity of the efficacy estimates. These limitations included the high likelihood of residual confounding, the potential for measurement error in the observed covariates due to the nature of the electronic health record data collection in the Flatiron data, the underlying differences in the patient populations before and after re-weighting, and the exclusion of patients from the Flatiron cohort who were treated with alpelisib. Due to the lack of sufficient comparative clinical efficacy data, there is a high degree of uncertainty in estimating the cost-effectiveness of alpelisib plus fulvestrant for the sponsor’s requested reimbursement population.
CADTH conducted exploratory reanalyses to assess the impact of alternative model assumptions on the cost-effectiveness estimates for alpelisib plus fulvestrant compared to SOC. These changes included a revised price for everolimus, an alternate parametric PFS curve, alternate estimates for the percentage of patients progressing due to death, the inclusion of PIK3CA retesting costs, an RDI of 1.0 for oral drugs, the removal of treatment-specific health state utility estimates, the use of an alternate HR for the derivation of TTD curves from PFS, and the setting of AE incidence as being equal between treatments. Although CADTH conducted this exploratory analysis by using alternate estimates in the model, the CADTH reanalyses were limited by the substantial uncertainty associated with the key efficacy estimates informing the model, including PFS and PPS (i.e., from the BYLieve versus Flatiron analysis). Due to the lack of alternative comparative clinical efficacy estimates for alpelisib plus fulvestrant and other relevant SOC agents (e.g., capecitabine and fulvestrant monotherapy), the results of any analyses remain highly uncertain.
In CADTH’s exploratory reanalyses, alpelisib plus fulvestrant compared to SOC had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY with an ICER of $319,592 per QALY gained compared to SOC. A price reduction of 99% is required for alpelisib plus fulvestrant to be cost-effective at a $50,000 per QALY threshold. The cost of alpelisib and PIK3CA mutation testing costs were key drivers of the results.
References
1.PrTukysaTM (tucatinib): 50 mg and 150 mg oral tablets [product monograph]. Milton (ON): McKesson Speciality Distribution Inc; 2020 Jun 3.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Piqray (alpelisib), 50 mg, 150 mg and 200 mg tablets. Dorval (QC): Novartis; 2021 Apr 20.
3.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2021 Jun 28.
4.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2011;366(6):520-529. PubMed
5.Leo AD, Jerusalem G, Petruzelka L, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor–positive advanced breast cancer. J Clin Oncol. 2010;28(30):4594-4600. PubMed
6.Chia S, Gradishar W, Mauriac L, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor–positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008;26(10):1664-1670. PubMed
7.Breast cancer (version 4.2021). NCCN Clinical practice guideline in oncology. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2021 Apr 28: www.nccn.org. Accessed 2021 Jun 22.
8.Cardoso F, Paluch-Shimon S, Senkus E, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. 2020;31(12):1623-1649. PubMed
9.Bank of Canada. Currency converter. 2021; https://www.bankofcanada.ca/rates/exchange/currency-converter/. Accessed 2021 Jul 16.
10.Lloyd A, Nafees B, Narewska J, Dewilde S, Watkins J. Health state utilities for metastatic breast cancer. Br J Cancer. 2006;95(6):683-690. PubMed
11.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 May 14.
12.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 May 14.
13.Appendix III - Criteria for coverage of exception status drugs. Halifax (NS): Nova Scotia Department of Health and Wellness; 2021: https://novascotia.ca/dhw/pharmacare/documents/Criteria-for-Exception-Status-Coverage.pdf. Accessed 2021 May 14.
14.Turner S, Kanakamedala H, Hsu W-C. Clinical efficacy of alpelisib and fulvestrant from BYLieve compared with real-world effectiveness of standard treatment among patients with HR+ HER2– PIK3CA-mutated advanced breast cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Piqray (alpelisib), 50 mg, 150 mg and 200 mg tablets. Dorval (QC): Novartis; 2021 Apr 20. 2020.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CADTH Cost Comparison Table for Advanced or Metastatic Breast Cancer
Treatment | Strength/ concentration | Form | Price | Recommended dosage | Daily cost | 28-day cycle costa |
|---|---|---|---|---|---|---|
Alpelisib (Piqray) | 150 mg 200 mg 200 mg + 50 mg | Tab | 95.2300a 190.4600a 95.2300a | 300 mg once daily with fulvestrantb | 190.46 | 5,333 |
Fulvestrant (generic) | 50 mg/mL | Pre-filled syringe (1 dose) 2 × 5 mL (250 mg/5mL) | 58.2895c | 500 mg on day 1, day 15, and day 28, and every 28 days thereafter. | First cycle: 62.45 Thereafter: 20.82 | First cycle: 1,749 Thereafter: 583 |
Alpelisib plus fulvestrant (first cycle) | 252.91 | 7,082 | ||||
Alpelisib plus fulvestrant (thereafter) | 211.28 | 5,916 | ||||
Fulvestrant monotherapy | ||||||
Fulvestrant (generic) | 50 mg/mL | Pre-filled syringe (1 dose) 2 × 5 mL | 582.8950c | 500 mg on day 1, day 15, and day 28, and every 28 days thereafter | First cycle: 62.45 Thereafter: 20.82 | First cycle: 1,749 Thereafter: 583 |
Everolimus with exemestane | ||||||
Everolimus (generic) | 2.5 mg 5 mg 10 mg | Tab | 101.3270d | 10 mg once daily with exemestane | 101.33 | 2,837 |
Exemestane (generic) | 25 mg | Tab | 1.3263c | 25 mg once daily | 1.33 | 37 |
Everolimus with exemestane | 102.65 | 2,874 | ||||
Chemotherapy (as monotherapy) | ||||||
Capecitabine (generic) | 150 mg 500 mg | Tab | 0.4575c 1.5250c | 1,000 mg/m2 (twice daily, days 1 to 14 every 21 days) | 7.32 | 205 |
Paclitaxel (generic) | 6 mg/mL | Vial for IV infusion 5 mL 16 mL 50 mL | 300.00 1,196.80 3,740.00 | 80 mg/m2 (days 1, 8 and 15 over 28 days) e | 142.70 | 2,997 |
Docetaxel (generic) | 10 mg/mL 20 mg/mL | Vial for IV infusion 8 mL 16 mL 1 mL 4 mL 8 mL | 970.20 1,850.00 249.00 497.00 990.00 | 100 mg/m2 (once every 3 weeks) | 59.00 | 1,239 |
Chemotherapy (as dual therapy) | ||||||
Gemcitabine (generic) | 40 mg/mL | Vial for infusion 25 mL 50 mL | 270.00 540.00 | 600 or 750 mg/m2 (days 1 and 8 every 21 days) | 25.47 | 721 |
Cisplatin (generic) | 1 mg/mL | Vial for IV infusion 50 mL 100 mL | 135.00 270.00 | 30 mg/m2 (days 1 and 8 every 21 days) | 25.71 | 720 |
Carboplatin (generic) | 10 mg/mL | Vial for IV infusion 5 mL 15 mL 45 mL 60 mL | 70.000 210.0000 599.9985 775.002 | 300 mg (days 1 and 8 every 21 days) f | 40.00 | 1,120 |
Gemcitabine + cisplatin | 51.18 | 1,441 | ||||
Gemcitabine + carboplatin | 65.71 | 1,840 | ||||
Note: All prices are IQVIA Delta PA wholesale list prices (accessed May 2021), unless otherwise indicated, and do not include dispensing fees or markups.11 Costs assume a body weight of 75 kg or a body surface area of 1.8 m2 and include wastage of unused medication in vials.
aSponsor’s submitted price.2
bAlpelisib: Dose reductions may occur (first dose reduction: 250 mg/day; second dose reduction: 200 mg/day) to a maximum of 2 dosing reductions, after which it is recommended that the patient should be discontinued from treatment.
cOntario Drug Benefit Formulary list price (accessed May 2021).12
dNova Scotia’s Exception Status Drugs Formulary List (accessed May 2021).13
eRecommended dosage based on input from the clinical experts consulted for this review.
fAssuming a mean CrCl level of 150ml/min and a target AUC of 2 on days 1 and 8 every 21 days. According to the clinical experts consulted by CADTH for this review, dosing may be based on a target AUC of 5 on day 1 of a 21-day cycle (750 mg every 21 days). Dosing will vary depending on patient CrCl and target AUC.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | As highlighted in the key limitations section, relevant comparators (e.g., chemotherapy and fulvestrant monotherapy) were not included in the sponsor’s base-case analysis. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | The percentage of patients progressing due to death was based on a percentage from the SOLAR-1 trial applied to the PFS curves. This parameter lacked flexibility to test alternate assumptions over the model time horizon. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Normal distributions were specified for the probability of AEs, cost of AEs, drug acquisition costs, drug administration and dispensing costs, terminal care costs, and follow-up and monitoring. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The sponsor’s base-case analysis compared alpelisib plus fulvestrant to SOC and conducted a secondary analysis that used a separate dataset to compare alpelisib plus fulvestrant vs. fulvestrant monotherapy and everolimus and exemestane. The results of these 2 analyses differed and the sponsor did not provide an explanation for the differences between the results of these 2 analyses. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | In several instances (e.g., AE incidence, distributions specified for the PSA) there was a misalignment between the sponsor’s pharmacoeconomic report and economic model. |
AE = adverse events; PSA = probabilistic sensitivity analysis; SOC = standard of care.
Note: This table has not been copy-edited.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure2
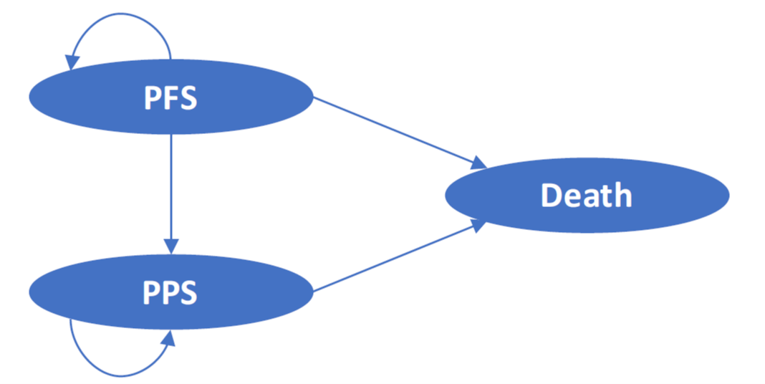
PFS = progression-free survival; PPS = post-progression survival.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Figure 2: Treatments Received by Patients in the Flatiron Database (Weighted and Unweighted Cohorts)2
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Detailed Results of the Sponsor’s Base Case
Figure 3: PFS and OS, 5 Years (Model Results for Alpelisib Plus Fulvestrant Versus Historical Control Versus Kaplan–Meier Curve)14
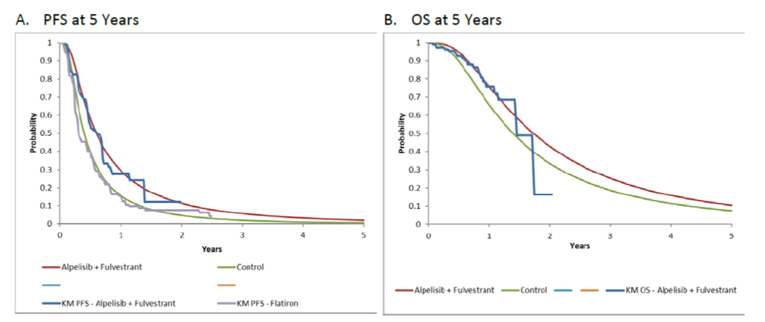
KM = Kaplan–Meier; OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Table 8: Disaggregated Summary of Sponsor’s Base-Case Economic Evaluation Results
Parameter | Alpelisib + fulvestrant | Historical control | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 2.28 | 1.87 | 0.41 |
By health state | |||
PFS | 0.99 | 0.61 | 0.38 |
PPS | 1.29 | 1.26 | 0.03 |
Discounted QALYs | |||
Total | 1.78 | 1.42 | 0.36 |
By health state | |||
PFS | 0.82 | 0.48 | 0.34 |
PPS | 0.96 | 0.93 | 0.02 |
Discounted costs ($) | |||
Total | 114,730 | 89,546 | |
Acquisition | 50,298 | 27,139 | 23,159 |
Administration and dispensing | 904 | 366 | 538 |
Adverse events | 402 | 576 | –174 |
Follow-up and monitoring | 6,835 | 5,354 | 1,481 |
Post-progression treatment | 33,387 | 34,384 | –997 |
Terminal costs | 21,536 | 21,728 | –191 |
Genetic testing and progression costs | 1,368 | 0 | 1,368 |
ICER ($/QALY) | 69,674 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; PFS = progression-free survival; PPS = post-progression survival; QALY = quality-adjusted life-year.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Results of the Sponsor’s Secondary Analyses Based on the SOLAR-1 Post-CDK4/6 Inhibitor Subgroup
The sponsor conducted a secondary analysis of alpelisib plus fulvestrant compared to fulvestrant alone and everolimus with exemestane based on data from the post-CDK4/6 inhibitor subgroup of the SOLAR-1 trial and an indirect treatment comparison. The results of this analysis found that alpelisib plus fulvestrant is associated with an incremental cost of $35,799 and incremental QALYs of 0.39 compared to fulvestrant monotherapy. Everolimus and exemestane was subject to extended dominance through fulvestrant monotherapy and alpelisib plus fulvestrant. Based on the sequential analysis, alpelisib plus fulvestrant had an ICER of $91,634 per QALY gained compared to fulvestrant monotherapy. The results of this analysis are presented in Table 9.
Table 9: Summary of the Sponsor’s Economic Evaluation Results for the Sponsor’s Secondary Analysis
Drug | Total costs ($) | Total QALYs | ICER vs. fulvestrant monotherapy ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Fulvestrant | 81,318 | 1.15 | Ref. | Ref. |
Everolimus + exemestane | 101,631 | 1.31 | $124,769 | Subject to extended dominance through fulvestrant and alpelisib + fulvestrant. |
Alpelisib + fulvestrant | 117,117 | 1.54 | $39,638 | $91,634 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; ref. = reference; vs. = versus.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Appendix 4: Details on the CADTH Exploratory Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the CADTH Exploratory Reanalysis
CADTH was unable to identify a base-case reanalysis. As an alternative, CADTH conducted exploratory reanalyses that consisted of a correction to the price of everolimus, a revised assumption for the PFS curve used, changing the percentage of patients progressing due to death for SOC to be the same as that for alpelisib plus fulvestrant, inclusion of retesting costs for PIK3CA mutation, revising RDI assumptions for oral drugs to 1.0, removing treatment-specific utility values, using a revised estimate for the HR to derive the TTD curve from PFS for SOC, and the setting of AE incidence as being equal between treatments. These changes are reported in Table 10.
Table 10: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
A. Price of everolimus | $172.2559 per tab (IQVIA) | $101.3270 per tab (NS Exceptional Access) |
Changes to derive the CADTH exploratory analysis | ||
1. Overestimate of PFS derived from selected parametric curve | Alpelisib + fulvestrant: RCS 1 log-normal model SOC: RCS 1 log-normal model | Alpelisib + fulvestrant: Gamma unrestricted model SOC: Gamma unrestricted model |
2. Percentage of patients progressing due to death | Alpelisib + fulvestrant: 4.4% SOC: 7.5% | Alpelisib + fulvestrant: 4.4% SOC: 4.4% |
3. PIK3CA mutation testing | Retesting rate: 0% | Retesting rate: 63.6% using tissue biopsy at a cost of CA$7,431.54 |
4. RDI for oral drugs | Alpelisib: 0837 Fulvestrant (loading dose): 1.0 Fulvestrant: 1.0 Everolimus: 0.86 Exemestane: 1.0 | Alpelisib: 1.0 Fulvestrant (loading dose): 1.0 Fulvestrant: 1.0 Everolimus: 1.0 Exemestane: 1.0 |
5. On-treatment utility estimates | Alpelisib + fulvestrant: 0.84 SOC: 0.80 | Alpelisib + fulvestrant: 0.84 SOC: 0.84 |
6. HR for derivation of TTD curves from PFS for SOC | HR: 1.202 | HR: 1.39 |
7. Equal AE incidence between alpelisib plus fulvestrant, and SOC | Alpelisib + fulvestrant: AE incidence from SOLAR-1 SOC: AE incidence from BOLERO-2 | Alpelisib + fulvestrant: AE incidence from SOLAR-1 SOC: AE incidence equal to alpelisib + fulvestrant |
CADTH exploratory reanalysis | 1 + 2 + 3 + 4 + 5 + 6 + 7 | |
AE = adverse event; HR = hazard ratio; NS = Nova Scotia; PFS = progression-free survival; RCS = restricted cubic spline; RDI = relative dose intensity; SOC = standard of care; TTD = time to treatment discontinuation.
Exploratory Reanalyses Results
As outlined in Table 11, CADTH’s exploratory reanalyses suggested that alpelisib plus fulvestrant was associated with higher costs and higher QALYs than SOC over a 15-year time horizon (ICER $319,592 per QALY gained). Disaggregated results are presented in Table 12. CADTH also conducted scenario analyses on the exploratory reanalysis to assess the impact of an alternate assumption for tissue biopsy retesting costs (CA$500), and a price reduction for everolimus and exemestane of 80% to reflect lower cost comparator agents used in clinical practice. Results of the price reduction analysis and scenario analyses are presented in Table 13 and Table 14, respectively. A price reduction of 99% is required for alpelisib plus fulvestrant to be cost-effective at a $50,000 per QALY threshold.
Table 11: Summary of the Stepped Analysis of the CADTH Reanalyses Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Historical control | 89,546 | 1.42 | Ref. |
Alpelisib + fulvestrant | 114,730 | 1.78 | 69,674 | |
Sponsor’s corrected base case | Historical control | 78,431 | 1.42 | Ref. |
Alpelisib + fulvestrant | 114,595 | 1.78 | 101,124 | |
CADTH reanalysis 1: PFS curve | Historical control | 77,736 | 1.38 | Ref. |
Alpelisib + fulvestrant | 109,766 | 1.59 | 155,072 | |
CADTH reanalysis 2: Percentage of patients progressing due to death | Historical control | 79,603 | 1.46 | Ref. |
Alpelisib + fulvestrant | 114,959 | 1.79 | 107,086 | |
CADTH reanalysis 3: PIK3CA mutation testing | Historical control | 78,548 | 1.43 | Ref. |
Alpelisib + fulvestrant | 127,917 | 1.79 | 136,878 | |
CADTH reanalysis 4: RDIs for oral drugs | Historical control | 81,046 | 1.43 | Ref. |
Alpelisib + fulvestrant | 122,146 | 1.79 | 119,945 | |
CADTH reanalysis 5: On-treatment utility estimates | Historical Control | 78,528 | 1.45 | Ref. |
Alpelisib + fulvestrant | 114,841 | 1.79 | 106,184 | |
CADTH reanalysis 6: HR for derivation of TTD curves from PFS for SOC | Historical control | 75,777 | 1.42 | Ref. |
Alpelisib + fulvestrant | 114,787 | 1.79 | 107,051 | |
CADTH reanalysis 7: Equal AE incidence | Historical control | 78,186 | 1.43 | Ref. |
Alpelisib + fulvestrant | 114,840 | 1.79 | 101,229 | |
CADTH exploratory reanalyses 1 + 2 + 3 + 4 + 5 + 6 + 7 | Historical control | 79,119 | 1.42 | Ref. |
Alpelisib + fulvestrant | 129,828 | 1.58 | 319,592 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; RDI = relative dose intensity; ref. = reference; SOC = standard of care; TTD = time to treatment discontinuation.
Table 12: Disaggregated Summary of CADTH’s Exploratory Reanalyses Results
Parameter | Alpelisib + fulvestrant | Historical control | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 2.04 | 1.85 | 0.18 |
By health state | |||
PFS | 0.74 | 0.55 | 0.18 |
PPS | 1.30 | 1.30 | 0.00 |
Discounted QALYs | |||
Total | 1.58 | 1.42 | 0.16 |
By health state | |||
PFS | 0.61 | 0.45 | 0.16 |
PPS | 0.96 | 0.97 | 0.00 |
Discounted costs ($) | |||
Total | 129,828 | 79,119 | 50,710 |
By health state | |||
PFS | 57,770 | 19,188 | 38,582 |
PPS | 36,218 | 38,186 | −1,968 |
By category | |||
Acquisition | 53,459 | 15,939 | 37,388 |
Administration and dispensing | 748 | 314 | 435 |
Adverse events | 402 | 402 | 0 |
Follow-up and monitoring | 5,830 | 5,073 | 758 |
Post-progression treatment | 33,590 | 35,555 | –1,966 |
Terminal costs | 21,680 | 21,744 | –65 |
Genetic testing and progression costs | 14,160 | 0 | 14,160 |
ICER ($/QALY) | 319,592 | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; PFS = progression-free survival; PPS = post-progression survival; QALY = quality-adjusted life-year; SOC = standard of care.
Table 13: CADTH Price Reduction Analyses (Deterministic Results)
Analysis | ICERs for alpelisib + fulvestrant vs. historical control | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | $75,815 | $324,068 |
10% | $65,107 | $296,347 |
20% | $54,400 | $268,627 |
30% | $43,692 | $240,907 |
40% | $32,985 | $213,187 |
50% | $22,277 | $185,466 |
60% | $11,569 | $157,746 |
70% | $862 | $130,026 |
80% | Alpelisib + fulvestrant dominant | $102,305 |
90% | Alpelisib + fulvestrant dominant | $74,585 |
98% | Alpelisib + fulvestrant dominant | $52,409 |
99% | Alpelisib + fulvestrant dominant | $49,637 |
100% | Alpelisib + fulvestrant dominant | $46,865 |
ICER = incremental cost-effectiveness ratio; SOC = standard of care; vs. = versus.
Scenario Analyses
CADTH conducted the following scenario analyses on CADTH’s exploratory reanalyses including: an alternate assumption for tissue biopsy retesting costs (CA$500), a price reduction for everolimus and exemestane of 80% to reflect lower cost comparator agents used in clinical practice, and a higher publicly available price of everolimus. Details of these analyses are reported in Table 14.
Table 14: Summary of the Scenario Analysis of the CADTH Exploratory Reanalyses Results
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH exploratory reanalysis | Historical control | 79,119 | 1.42 | Ref. |
Alpelisib + fulvestrant | 129,828 | 1.58 | 319,592 | |
Scenario 1: Lower PIK3CA mutation retesting costs | Historical control | 79,099 | 1.45 | Ref. |
Alpelisib + fulvestrant | 117,825 | 1.61 | 244,956 | |
Scenario 2: Alternate comparator cost assumptions | Historical control | 66,528 | 1.44 | Ref. |
Alpelisib + fulvestrant | 129,451 | 1.60 | 401,649 | |
Scenario 3: Alternate cost for everolimus | Historical control | 90,246 | 1.44 | Ref. |
Alpelisib + fulvestrant | 129,787 | 1.59 | 251,581 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; ref. = reference; SOC = standard of care.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 15: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) that compared the change in expenditure with the adoption of alpelisib plus fulvestrant compared to a reference scenario where alpelisib plus fulvestrant was not available. The BIA was modelled over a 3-year time period and a baseline year (current year). The population of interest was for men and postmenopausal women with hormone receptor–positive, /HER2-, PIK3CA mutant advanced breast cancer who have received prior CDK4/6i + aromatase inhibitor therapy, which is in line with the sponsor’s reimbursement request. The reference scenario included the availability of fulvestrant, everolimus and exemestane, exemestane, nonsteroidal aromatase inhibitors (anastrozole, letrozole), chemotherapy (paclitaxel, docetaxel, capecitabine), and tamoxifen, and the new drug scenario included the availability of alpelisib in addition to all treatments in the reference scenario. The BIA was undertaken from the public payer perspective for the Canadian setting (public drug plans and provincial cancer agencies, with the exception of Quebec).
Key assumptions of the sponsor’s BIA:
Expected annual costs are inclusive of medication acquisition costs, jurisdiction-specific markups, and dispensing fees. PIK3CA mutation testing costs were not included in the sponsor’s base-case results.
Exclusion of CDK4/6i as comparator agents given their unlikely use in the eligible population (i.e., prior receipt of CDK4/6 inhibitors).
Eligible population consists of 2 groups of patients according to early or advanced stage of breast cancer at time of diagnosis:
A. Patients with newly diagnosed advanced breast cancer (i.e., advanced disease at diagnosis) and progressed while receiving CDK4/6i + AI.
B. Patients previously diagnosed with early breast cancer in a prior year and experience locoregional or metastatic recurrence during each year of the BIA projection period.
The sponsor used an epidemiologic, incidence-based approach to estimate the eligible population size as illustrated in Table 16 and Table 17.
Table 16: Sponsor's Estimation of the Size of the Eligible Population
Parameter | Baseline year | Year 1 | Year 2 | Year 3 |
|---|---|---|---|---|
Canadian population (annual growth rate of 1.3%) | 30,177,413 | 30,565,955 | 30,959,499 | 31,358,111 |
A. Number of new cases of locally advanced or metastatic breast cancer progressed while receiving CDK4/6i + AI | ||||
Number of new cases of breast cancer (66.8 per 100,000 annual incidence of breast cancer) | 20,159 | 20,418 | 20,681 | 20,947 |
Number with locally advanced or metastatic breast cancer at diagnosis (8.6% of incident cases) | 1,738 | 1,760 | 1,783 | 1,806 |
Number with HR+/HER2- locally advanced or metastatic breast cancer (71%) | 1,234 | 1,250 | 1,266 | 1,282 |
Number of men and postmenopausal females (82% of women postmenopausal) | 1,015 | 1,029 | 1,042 | 1,055 |
Number receiving first-line CDK4/6i + AI (|) | || | || | || | || |
B. Number of previously diagnosed cases of early breast cancer that experience locoregional or metastatic recurrence | ||||
Number of prevalent cases of breast cancer (910 per 100,000) | 274,464 | 277,997 | 281,577 | 285,202 |
Number of cases of early breast cancer (94% of prevalent cases) | 258,270 | 261,596 | 264,964 | 268,375 |
Number of men and postmenopausal females with HR+/HER2- tumours (58%) | 150,930 | 152,873 | 154,842 | 156,835 |
Number with breast cancer recurrence (2.5% per year) | 3,751 | 3,800 | 3,849 | 3,898 |
Number receiving first-line CDK4/6i + AI (|) | |||| | |||| | |||| | |||| |
Total number of patients eligible for drug under review | ||||
Total number of newly diagnosed advanced breast cancer (A.) and previously diagnosed recurrent (B.) | 3,347 | 3,390 | 3,434 | 3,478 |
Total number tested for PIK3CA mutation (|||) | |||| | |||| | |||| | |||| |
Total number tested positive for PIK3CA mutation ( | ) | 368 | 373 | 377 | 382 |
AI = aromatase inhibitor; CDK4/6 = cyclin-dependent kinase 4 and 6; HER2 = human epidermal growth factor receptor 2.
Source: Sponsor’s Pharmacoeconomic Submission (2021).2
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Number of patients eligible for drug under review | 373 / 377 / 382 |
Market uptake (3 years) | |
Uptake (reference scenario) Everolimus + exemestane Capecitabine Paclitaxel Anastrozole Fulvestrant Exemestane Docetaxel Letrozole Tamoxifen | |||||||||||||||||||| |||||||||||||||||||| |||||||||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |
Uptake (new drug scenario) Alpelisib + fulvestrant Everolimus + exemestane Capecitabine Paclitaxel Anastrozole Fulvestrant Exemestane Docetaxel Letrozole Tamoxifen | |||||||||||||||||||| |||||||||||||| |||||||||||||||||| |||||||||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |||||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment per cyclea Alpelisib + fulvestrant Everolimus + exemestane Capecitabine Paclitaxel Anastrozole Fulvestrant Exemestane Docetaxel Letrozole Tamoxifen | $6,385; $5,425 $4,452 $267 $1,720 $49 $1,922; $961 $73 $1,821 $59 $11 |
RDI = relative dose intensity.
aSponsor’s estimate based on cost per dose (sourced from IQVIA, doses per cycle, and RDI). Depending on regimen, days per cycle may be 21, 28, or 30.
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor’s model estimated a net budget impact of $3,759,712 in year 1, $4,154,312 in year 2, and $4,762,549 in year 3 for a 3-year total of $12,676,573. The sponsor also conducted scenario analyses that included IV administration costs and PIK3CA mutation testing costs. The submitted analysis is based on the publicly available prices of the comparator treatments.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Limited generalizability of the modelled comparators: The sponsor included everolimus and exemestane, capecitabine, paclitaxel, anastrozole, fulvestrant, exemestane, docetaxel, letrozole, and tamoxifen as comparator agents in the new and reference scenario and assumed that everolimus and exemestane would comprise |% of the market share in the reference scenario. According to the clinical experts consulted by CADTH for this review, anastrazole and letrozole are not relevant comparator agents for the population of interest for this review, and chemotherapy (i.e., most commonly capecitabine) or an endocrine therapy is most used following progression on an endocrine-based regimen with a CDK4/6 inhibitor; everolimus plus exemestane is not funded by public drug programs in Canada in this setting. Given that everolimus and exemestane is the costliest comparator agent, its inclusion as a primary comparator overestimates the cost of treatment in the reference scenario and underestimates the incremental budget impact with the introduction of alpelisib plus fulvestrant.
In the CADTH reanalysis, anastrozole and letrozole were removed from the list of comparator agents, and market share assumptions in the reference scenario were revised to have chemotherapy and fulvestrant comprise the majority of the market share: chemotherapy (capecitabine: 45%; paclitaxel 10%; docetaxel 10%), fulvestrant (20%), everolimus and exemestane (5%), exemestane (5%), and tamoxifen (5%). These market share distributions were included in the same proportions for year 1, year 2, and year 3 in the new drug scenario.
Uncertainty in market share estimates for alpelisib plus fulvestrant and comparator agents in the new drug scenario: The sponsor assumed that alpelisib plus fulvestrant would take |% of the market share in year 1, |% market share in year 2, and |% market share in year 3 based on a survey of Canadian clinicians. The clinical experts consulted by CADTH for this review indicated that there could be a higher uptake of alpelisib plus fulvestrant (70 to 80%) especially if testing is available and there is an access program in place. An underestimate in market share estimates for alpelisib plus fulvestrant underestimates the cost of the new drug scenario and underestimates the incremental budget impact.
CADTH conducted a scenario analysis with revised market shares estimates for alpelisib plus fulvestrant, estimating 55% in year 1, 65% in year 2, and 75% in year 3. Market shares of comparator agents were reduced to maintain the distributions of the remaining market share available as used in the CADTH reanalysis (i.e., 65% chemotherapy, 20% fulvestrant, 15% everolimus and exemestane, exemestane, and tamoxifen).
Uncertainty in the estimates to derive the eligible patient population: The sponsor assumed that ||% of patients would be tested for the PIK3CA mutation in practice. According to the clinical experts consulted by CADTH for this review, the percentage of patients tested for PIK3CA mutation in practice is expected to be higher – i.e., approximately 50%. Underestimating the percentage of patients anticipated to be tested for PIK3CA mutation will underestimate the eligible patient population and consequently, underestimate the potential budget impact of alpelisib plus fulvestrant.
CADTH revised the percentage of patients likely to be tested for PIK3CA mutation (50%) of patients.
RDI less than 1.0 underestimated the cost of treatment: The sponsor adjusted the cost of alpelisib and the cost of treatment by applying an RDI of 0.837 for alpelisib, 0.86 for everolimus, 0.92 for paclitaxel, 0.92 for docetaxel, and 0.78 for capecitabine. This practice underestimated the total expenditure associated with these agents. For oral treatments, Canadian pharmacies are likely to dispense the full quantity of medication for each treatment cycle and excess tablets are unlikely to be recuperated. The use of RDIs less than 1.0 underestimated the total costs in the new drug scenario and underestimated the budget impact associated with the introduction of alpelisib plus fulvestrant.
CADTH revised the RDI for all oral drugs to 1.0 which aligns with the pharmacoeconomic evaluation.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
Underestimate of PIK3CA mutation testing costs: The sponsor’s base case excluded the costs of PIK3CA mutation testing and the sponsor undertook a scenario analysis that included these costs. The sponsor assumed that PIK3CA mutation testing would cost $500 and that no patients would require retesting (due to the lack of data). The clinical experts consulted by CADTH for this review indicated that it is recommended to universally retest patients who test negative for the PIK3CA mutation on the initial liquid biopsy. Based on a test positivity rate of 36.4%, it could then be inferred that 63.6% of patients would be retested with tissue biopsy. As stated in the CADTH Clinical Review Report, a FoundationOne CDx tumour biopsy test conducted by Foundation Medicine costs $5,800 US per test (CA$7,431.54 based on an average Bank of Canada exchange rate of 1.2813 from July 2020 to July 2021).9 Local in-house tissue testing for the PIK3CA mutation may be available in some centres at a lower cost; however, the total cost of local tests is unclear (e.g., 1 centre reported a cost of approximately CA$300 to CA$750 for materials, which would not include interpretation). Omitting the costs of PIK3CA retesting underestimated the costs of alpelisib plus fulvestrant and underestimated the incremental budget impact. CADTH undertook a scenario analysis to include testing and retesting costs in the new drug scenario assuming that 63.6% of patients would be retested assuming both higher costs of retesting ($7,431.54 CAD) and lower costs of retesting (CA$500).
Uncertainty in the percentage of patients covered by public drug plans: CADTH retained the sponsors assumptions regarding the percentage of the eligible population covered by public drug plans (i.e., 75.6% for patients < 65 years of age, and 100% for patients 65 years of age and older) in the CADTH base case. CADTH undertook a scenario analysis to assess the impact of 100% of patients younger than 65 years of age covered by public drug plans.
Discrepancy in results reported in the sponsor’s model: The sponsor’s model included 2 results tabs – 1 for “province” and 1 for “consolidate.” In the sponsor’s base case, the results reported on these 2 spreadsheets differed by $9,702. The source of this discrepancy is unclear. The CADTH reanalyses are based on the results reported in the provinces all costs spreadsheet which includes all Canadian public drug programs with the exception of Quebec.
CADTH Reanalyses of the Budget Impact Analysis
Table 18: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
A. Price of everolimus | 169.7193 per tab (IQVIA) | 101.3270 per tab (NS Exceptional Access) |
Changes to derive the CADTH base case | ||
1. Revised market share distributions of comparator agents. | Comparator (current, year 1, year 2, year 3) Fulvestrant: 6%, 3%, 3%, 3% Everolimus + Exemestane: 34%, 8%, 6%, 2% Anastrozole: 8%, 2%, 1%, 0% Letrozole: 4%, 1%, 1%, 0% Exemestane: 6%, 2%, 1%, 1% Paclitaxel: 10%, 10%, 10%, 10% Docetaxel: 6%, 5%, 5%, 5% Capecitabine: 23%, 11%, 10%, 9% Tamoxifen: 4%, 3%, 3%, 3% | Comparator (current, year 1, year 2, year 3) Fulvestrant: 20%, 9%, 8%, 6% Everolimus + Exemestane: 5%, 2%, 2%, 2% Anastrozole: 0%, 0%, 0%, 0% Letrozole: 0%, 0%, 0%, 0% Exemestane: 5%, 2%, 2%, 2% Paclitaxel: 10%, 5%, 4%, 3% Docetaxel: 10%, 5%, 4%, 3% Capecitabine: 45%, 20%, 18%, 14% Tamoxifen: 5%, 2%, 2%, 2% |
2. Percentage of patients tested for PIK3CA mutation | ||||% | 50% |
3. RDIs for oral drugs | Alpelisib: 0.84 Everolimus: 0.86 Paclitaxel: 0.92 Docetaxel: 0.92 Capecitabine: 0.78 | Alpelisib: 1.0 Everolimus: 1.0 Paclitaxel: 1.0 Docetaxel: 1.0 Capecitabine: 1.0 |
CADTH base case | Reanalysis 1 + 2 + 3 | |
NS = Nova Scotia; RDI = relative dose intensity.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20.
CADTH undertook scenario analyses that included:
Inclusion of PIK3CA mutation testing costs.
Inclusion of PIK3CA mutation retesting costs: Assuming higher cost ($7,431.54 CAD) of tissue testing.
Inclusion of PIK3CA mutation retesting costs: Assuming lower cost ($500 CAD) of tissue testing.
100% of patients regardless of age would be covered by the public payer.
99% price reduction for alpelisib.
Table 19: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $12,676,573 |
Corrected base case | $15,508,069 |
CADTH reanalysis 1: Alternative market share distributions for comparators | $17,839,083 |
CADTH reanalysis 2: Percentage of patients tested for PIK3CA mutation | $25,675,610 |
CADTH reanalysis 3: RDIs for oral drugs | $17,684,224 |
CADTH base case | $33,939,690 |
RDI = relative dose intensity.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 3,359,768 | 3,403,026 | 3,446,840 | 3,491,219 | 10,341,086 |
New drug | 3,359,768 | 7,162,737 | 7,601,152 | 8,253,769 | 23,017,658 | |
Budget impact | 0 | 3,759,712 | 4,154,312 | 4,762,549 | 12,676,573 | |
CADTH base case | Reference | 2,343,455 | 2,373,628 | 2,404,189 | 2,435,143 | 7,212,959 |
New drug | 2,343,455 | 12,439,712 | 13,526,757 | 15,186,180 | 41,152,650 | |
Budget impact | 0 | 10,066,084 | 11,122,569 | 12,751,037 | 33,939,690 | |
CADTH scenario analysis 1: Inclusion of PIK3CA testing costs | Reference | 2,343,455 | 2,373,628 | 2,404,189 | 2,435,143 | 7,212,959 |
New drug | 2,343,455 | 13,287,301 | 14,385,259 | 16,055,736 | 43,728,297 | |
Budget impact | 0 | 10,913,674 | 11,981,071 | 13,620,593 | 36,515,337 | |
CADTH scenario analysis 2: Inclusion of PIK3CA testing and retesting costs (high) | Reference | 2,343,455 | 2,373,628 | 2,404,189 | 2,435,143 | 7,212,959 |
New drug | 2,343,455 | 21,299,494 | 22,500,611 | 24,275,574 | 68,075,679 | |
Budget impact | 0 | 18,925,866 | 20,096,422 | 21,840,431 | 60,862,720 | |
CADTH scenario analysis 3: Inclusion of PIK3CA testing and retesting costs (low) | Reference | 2,343,455 | 2,373,628 | 2,404,189 | 2,435,143 | 7,212,959 |
New drug | 2,343,455 | 13,826,368 | 14,931,267 | 16,608,773 | 45,366,408 | |
Budget impact | 0 | 11,452,740 | 12,527,078 | 14,173,630 | 38,153,449 | |
CADTH scenario analysis 4: 99% price reduction | Reference | 3,359,768 | 3,403,026 | 3,446,840 | 3,491,219 | 10,341,086 |
New drug | 3,359,768 | 7,162,737 | 7,601,152 | 8,253,769 | 23,017,658 | |
Budget impact | 0 | 3,759,712 | 4,154,312 | 4,762,549 | 12,676,573 | |
CADTH scenario analysis 5: 100% of patients covered by public payer | Reference | 2,860,506 | 2,897,335 | 2,934,639 | 2,972,423 | 8,804,398 |
New drug | 2,860,506 | 15,184,361 | 16,511,247 | 18,536,798 | 50,232,406 | |
Budget impact | 0 | 12,287,025 | 13,576,608 | 15,564,375 | 41,428,008 |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.