CADTH Reimbursement Review
Pertuzumab (Perjeta)
Sponsor: Hoffmann-La Roche Ltd.
Therapeutic area: Early-stage breast cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ASCO
American Society of Clinical Oncology
BCC-BTG
BC Cancer Breast Tumour Group
bpCR
pathologic complete response in the breast
CBCN
Canadian Breast Cancer Network
CI
confidence interval
CR
complete response
DFS
disease-free survival
ECOG
Eastern Cooperative Oncology Group
EFS
event-free survival
EMA
European Medicines Agency
FEC
5-fluorouracil and epirubicin plus cyclophosphamide
FISH
fluorescent in-situ hybridization
HER2
human epidermal growth factor receptor 2
IHC
immunohistochemistry
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention to treat
IxRS
interactive web and voice response system
LVEF
left ventricular ejection fraction
NYHA
New York Heart Association
OH-CCO BCDAC
Ontario Health (Cancer Care Ontario) Breast Cancer Drug Advisory Committee
OS
overall survival
PAG
Provincial Advisory Group
pCR
pathologic complete response
PD
progressive disease
PFS
progression-free survival
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumors
SD
standard deviation
T-DM1
trastuzumab emtansine
Tis
tumour in situ
tpCR
total pathologic complete response
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Pertuzumab (Perjeta), 420 mg/14 mL vial, IV infusion. |
Indication | In combination with trastuzumab and chemotherapy for neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive). |
Reimbursement request | Per indication. Patients should receive neoadjuvant treatment with pertuzumab in combination with trastuzumab and chemotherapy for 3 to 6 cycles, depending on the regimen chosen. Patients who start pertuzumab in combination with trastuzumab and chemotherapy in the neoadjuvant setting and do not have residual disease following surgery should continue to receive adjuvant trastuzumab to complete 1 year of HER2-directed therapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 25, 2021 |
Sponsor | Hoffmann-La Roche Ltd. |
HER2 = human epidermal growth factor receptor 2; NOC = Notice of Compliance.
Introduction
Breast cancer will strike 1 in 8 Canadian women during their lifetime and 1 in 33 will die of the disease. Due to advances in and widespread use of screening programs, breast cancer is typically diagnosed at an early stage, and approximately 95% of cases are diagnosed at stages I to III. Prior to the advent of anti–human epidermal growth factor receptor 2 (HER2) therapies, patients who had HER2-positive breast cancer had a poorer prognosis than those without HER2 overexpression. Patients undergoing neoadjuvant therapy can be subclassified into those with locally advanced breast cancers that are not operable, those with locally advanced cancers that are operable, and those with primary operable breast cancers. In locally advanced cancers, 1 purpose of neoadjuvant therapy is to convert the tumour from an inoperable to an operable state. In all cancers, the purpose of neoadjuvant therapy is to downstage the tumour to potentially avoid mastectomy in favour of breast-conserving surgery, to assess response to systemic therapy, to potentially escalate or de-escalate subsequent adjuvant therapy based on the response to neoadjuvant therapy, and to initiate systemic therapy early to try to limit systemic spread. Neoadjuvant therapy is the standard of care according to international and local guidelines for patients who are at stage II or III, and some patients with stage I disease. By treating with chemotherapy early, the risk of systemic recurrence is also decreased. A small number of patients with small tumours may have surgery first followed by adjuvant therapy, and this approach would be the norm for stage I disease. The standard regimen, according to the clinical experts consulted by CADTH for this review, for stages II and III HER2-positive breast cancer, would be doxorubicin, cyclophosphamide, and paclitaxel plus trastuzumab or docetaxel and carboplatin plus trastuzumab. The American Society of Clinical Oncology (ASCO) guidelines for neoadjuvant management of HER2-positive, node-positive, or high-risk node-negative breast cancer recommend an anthracycline and taxane or a non-anthracycline-based regimen in combination with trastuzumab; they also note that pertuzumab may be added to this regimen. In many regions, including the US, Europe, and the UK, pertuzumab would be added to this regimen.1 The European Society for Medical Oncology recommends a dual blockade with pertuzumab and trastuzumab in high-risk patients (with node-positive or estrogen receptor–negative disease), starting before or after surgery; for neoadjuvant therapy, the German Gynecological Oncology Group recommends adding pertuzumab when treating patients with node-positive disease.2,3 The goal of neoadjuvant treatment is curative.
Pertuzumab is an HER2 inhibitor. It is administered by IV infusion in combination with trastuzumab and chemotherapy. After an initial 840 mg loading dose, the maintenance dose is 420 mg every 3 weeks for 3 to 6 cycles in the neoadjuvant setting. It is indicated, in combination with trastuzumab and chemotherapy, for the neoadjuvant treatment of patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive). This drug was previously reviewed by CADTH for off-label use under the same indication.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of pertuzumab by IV infusion in combination with trastuzumab and chemotherapy for the neoadjuvant treatment of patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive).
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups provided input, the Canadian Breast Cancer Network (CBCN) and Rethink Breast Cancer. Information was gathered through phone interviews (11 interviewees) and 2 surveys, 1 distributed by CBCN (52 respondents, all Canadian) and 1 by Rethink Breast Cancer (62 respondents, 60% Canadian).
Patients described the emotional distress associated with being diagnosed with a type of breast cancer that is known to have a poor prognosis in the absence of HER2-directed therapy. Patients also noted the adverse effects associated with the disease and the treatments (cardiotoxicity, fever, cough, muscle pain, fatigue, diarrhea, and nausea) and noted that fatigue, pain, and nausea are most likely to impact their daily lives. Patients also noted the financial burden associated with lost income and treatment costs, with 17% of respondents in 1 survey reporting a very large financial impact and 38% reporting some financial impact.
The most important outcomes for patients were the elimination of cancer cells, prevention of recurrence, and preventing metastases. Maintaining quality of life was also rated by the majority of patients as very important or important, as was managing adverse effects. Patients were clear that they were very willing to tolerate new adverse effects from drugs to extend life expectancy.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Per indication, pertuzumab in the neoadjuvant setting would be used in combination with trastuzumab and chemotherapy. The shift in the treatment paradigm would simply be the addition of pertuzumab to the standard therapies already being used.
According to the clinical experts, the patients most likely to respond to the addition of pertuzumab would be those who have HER2-positive (or HER2-overexpressing) breast cancer. According to the clinical experts, all patients who are HER2-positive and are candidates for neoadjuvant therapy would be eligible for the addition of pertuzumab to their regimen, and those who are not candidates for either chemotherapy (due to being too ill) or neoadjuvant therapy (those with small stage I cancer) would not be eligible for pertuzumab. It was noted that it is very rare for a patient to be too ill to receive chemotherapy.
The clinical experts noted that, ultimately, response in the neoadjuvant setting is determined at the time of surgery, when assessment of pathologic complete response (pCR) is performed. Prior to surgery, patients would most likely be assessed every 2 to 3 weeks at the time they come in to receive their chemotherapy, typically by a physical exam, although sometimes this may be supplemented by imaging of the breast (ultrasound or MRI). If, during therapy, the tumour is growing or not responding, the chemotherapy protocol may be modified or the patient may be sent for surgery earlier than planned. A clinically meaningful response is a shrinkage of the tumour to facilitate surgical removal.
One of the clinical experts consulted by CADTH said they believe that increasing pCR rates would result in a reduced risk of relapse in this population.
With respect to deciding when to discontinue treatment, the clinical experts noted this may occur if the tumour is growing, in which case surgery may be performed earlier than planned or, in some cases, other chemotherapy protocols may be instituted. Patients with clear disease progression after receiving 1 to 2 cycles of optimized taxane-based chemotherapy should be considered for discontinuation.
One clinical expert noted that the addition of pertuzumab to the current treatment paradigm is important, given that this is a curable disease that often occurs in younger patients. The other clinical expert noted the importance of increased rates of tumour downstaging and pCR in reducing longer-term treatment-related morbidity.
Clinician Group Input
Two clinician groups provided input, the BC Cancer Breast Tumour Group (BCC-BTG) and the Ontario Health (Cancer Care Ontario) Breast Cancer Drug Advisory Committee (OH-CCO BCDAC).
One clinician group noted that the greatest need for pertuzumab is in patients with inflammatory breast cancer and inoperable stage IIIC breast cancers to downstage to get to primary surgery.
The groups did not specifically refer to their experiences with pertuzumab; however, 1 clinician group noted that combining pertuzumab with trastuzumab is the international standard of care in stage II to III HER2-positive breast cancer.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The Provincial Advisory Group (PAG) noted that in most provinces, the current standard of care for the neoadjuvant treatment of HER2-positive breast cancer is trastuzumab plus chemotherapy. Pertuzumab, being an IV drug, would be administered in an outpatient chemotherapy centre for appropriate administration and monitoring of infusion-related reactions. PAG highlighted several enablers of the implementation of pertuzumab in the neoadjuvant setting, including that the dose and frequency of pertuzumab in the neoadjuvant setting are the same as in the metastatic setting, that it is used as an add-on drug to existing treatment, and that drug wastage is not a concern since pertuzumab vials contain the amount of the fixed dose. PAG also identified barriers to implementation that include the high cost of pertuzumab and the additional preparation time and chair time needed for the infusion. Pertuzumab is administered for 4 to 6 cycles before surgery and PAG noted that given the high cost of pertuzumab, there is a significant difference in cost between 4 cycles and 6 cycles.
Clinical experts were consulted by CADTH for questions related to the implementation of pertuzumab in current provincial drug plans. Overall, most implementation questions related to the dosing schedule and administration, the eligible patient population, pCR as an end point, and re-treatment with pertuzumab in subsequent lines of treatment.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Four trials, all identified as pivotal by the sponsor, were included in the CADTH review. NEOSPHERE (N = 417, randomized 1:1:1:1 across 4 groups) was an open-label randomized controlled trial (RCT) that had an arm that contained pertuzumab and trastuzumab plus docetaxel and an arm that contained trastuzumab plus docetaxel. PEONY (N = 329, randomized 2:1 across 2 treatment arms) was a double-blind RCT that randomized patients to either pertuzumab and trastuzumab plus docetaxel or trastuzumab plus docetaxel. TRYPHAENA (N = 225, randomized 1:1:1 across 3 treatment arms) and BERENICE (N = 400, distributed 1:1 across 2 cohorts, non-RCT) were designed to compare different background regimens of chemotherapy combined with pertuzumab and trastuzumab. The primary focus of this review was the NEOSPHERE and PEONY trials, with TRYPHAENA and BERENICE providing supportive evidence, where available. All trials included patients with early breast cancer that was HER2-positive.
All trials featured a neoadjuvant treatment phase followed by surgery and then an adjuvant treatment phase.
In NEOSPHERE and PEONY, the neoadjuvant phase lasted 4 cycles and consisted of the treatments described previously. In the adjuvant phase of NEOSPHERE, treatment arms received 3 cycles of 5-fluorouracil and epirubicin plus cyclophosphamide (FEC) and trastuzumab for up to 1 year. The adjuvant phase of PEONY included 3 cycles of FEC followed by pertuzumab and trastuzumab for cycles 8 to 17 in the arm that received pertuzumab and trastuzumab plus docetaxel in the neoadjuvant phase, and placebo plus trastuzumab for cycles 8 to 17 in the arm that received trastuzumab plus docetaxel in the neoadjuvant phase.
In the neoadjuvant phase of TRYPHAENA, patients in arm A received pertuzumab and trastuzumab plus FEC for 3 cycles followed by pertuzumab and trastuzumab plus docetaxel for 3 cycles, patients in arm B received FEC for 3 cycles then pertuzumab and trastuzumab plus docetaxel for 3 cycles, and patients in arm C received pertuzumab plus docetaxel and carboplatin plus trastuzumab for 6 cycles. In the adjuvant phase, patients received trastuzumab from cycle 7 onward, up to 1 year.
In BERENICE, patients in arm A received doxorubicin plus cyclophosphamide for cycles 1 to 4, pertuzumab and trastuzumab plus paclitaxel for cycles 5 to 8, and patients in arm B received FEC for cycles 1 to 4, followed by pertuzumab and trastuzumab plus docetaxel for cycles 5 to 8. For the adjuvant phase, patients in both arms received pertuzumab and trastuzumab.
The primary outcome of NEOSPHERE was the pCR rate at the conclusion of the neoadjuvant treatment period, and the primary outcome of PEONY was the total pathologic complete response (tpCR) rate, also at the conclusion of the neoadjuvant treatment period. PEONY also reported on the pathologic complete response in the breast (bpCR) rate at the conclusion of the neoadjuvant period. Both trials were designed to report on various longer-term outcomes such as overall survival (OS), progression-free survival (PFS), event-free survival (EFS), and disease-free survival (DFS); however, these outcomes were assessed during or after the adjuvant treatment period. The primary objective of TRYPHAENA and BERENICE was to assess safety and tolerability. The primary safety outcomes in TRYPHAENA were incidence of symptomatic cardiac events and clinically significant left ventricular ejection fraction (LVEF) decline, and the primary safety outcomes in BERENICE were incidence of New York Heart Association (NYHA) class III and IV heart failure and incidence of LVEF decline.
Patients in the included trials were about 50 years old at baseline and the majority (approximately 70% to 80%) were White, except for PEONY, where all patients were Asian. Most patients (nearly 90%) had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 and the rest had a status of 1. Approximately half (47% in NEOSPHERE, 51% in PEONY) of patients were either estrogen or progesterone receptor–positive, except for BERENICE, in which approximately 2-thirds of patients were estrogen or progesterone receptor–positive. In terms of baseline disease category, the majority of patients in NEOSPHERE and TRYPHAENA were stage T2N0M0 (NEOSPHERE, |;|;|; TRYPHAENA, 31%) or stage T2N1M0 (NEOSPHERE, |;|; TRYPHAENA, 33%). In PEONY, most patients were stage T2 (67%), followed by T3 (22%), and had lymph node–positive disease (76%). In BERENICE, most patients were stage T2 (67%) followed by T3 (20%); 47% were N1, 8% were N2, and 2% were N3; 100% were M0.
Efficacy Results
The median overall time on study in NEOSPHERE was |||||||||||||||||||||||||||||||||||||||||| in the pertuzumab and trastuzumab plus docetaxel arm and |||||||||||||||||||||||||||||||||||||||||||| in the trastuzumab plus docetaxel arm. In PEONY, the median time on study was |||||||||||||||||||||||||||||||||||||||||||| in the pertuzumab and trastuzumab plus chemotherapy arm and |||||||||||||||||||||||||||||||||||||| in the trastuzumab plus chemotherapy arm. In TRYPHAENA, the median time on study ranged from ||||||||||||||||||||||||||||||||||||||||||||||||||||||||| in the 3 treatment arms. In BERENICE, the median time on study was |||||||||||||||||||||||||||||||||||||||||||||| in cohort A and |||||||||||||||||||||||||||||||||||||||||||||| in cohort B.
Assessment of longer-term outcomes, such as OS, EFS, DFS, and PFS, included treatment regimens received in both the neoadjuvant and adjuvant phases of treatment. OS was not assessed in NEOSPHERE; OS data are not yet mature from PEONY, according to the sponsor, and there are no comparative OS data available from TRYPHAENA or BERENICE. Data for invasive DFS or EFS were not available from the included trials, either because it was not assessed or because the data were reported as not yet mature by the sponsor. With respect to PFS, in NEOSPHERE, progression events occurred in 15.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 17.8% of patients in the trastuzumab plus docetaxel arm, for a hazard ratio (HR) of 0.69 (95% confidence interval [CI], 0.34 to 1.40). These results were consistent with the PFS data reported in TRYPHAENA, where the PFS event rates were 13.7% in arm A, 14.7% in arm B, and 18.2% in arm C. Data on PFS were not yet mature in PEONY, according to the sponsor, and PFS was not assessed in BERENICE. DFS events occurred in 14.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm in NEOSPHERE, and 17.5% of patients in the trastuzumab plus docetaxel arm, and these results were consistent with those reported in TRYPHAENA, where the PFS events were 14.5% in arm A, 11.9% in arm B, and 15.3% in arm C. The DFS data were not yet mature in PEONY, according to the sponsor, and DFS was not assessed in BERENICE. None of the trials were powered to assess between-group differences in these longer-term outcomes.
In NEOSPHERE, a pCR was achieved by 45.8% of patients in the pertuzumab and trastuzumab plus docetaxel arm, and 29.0% of patients in the trastuzumab plus docetaxel arm, for a difference in response rates between groups of 16.8% (95% CI, 3.5 to 30.1; P = 0.0094). In PEONY, the tpCR rate assessed by an independent review committee (IRC) was 39.3% in the pertuzumab and trastuzumab plus docetaxel arm and 21.8% in the trastuzumab plus docetaxel arm, for a difference in response rates of 17.45% (95% CI, 6.89 to 28.01; P = 0.0014). The difference in pCR rates between the 2 trials may reflect the different definitions of pCR used, as NEOSPHERE used only breast tissue to assess pCR, while PEONY used breast and nodes. Additionally, PEONY reported the bpCR rate as a secondary outcome, and the IRC-assessed bpCR rate was consistent with that of the tpCR rate (42.0% versus 23.6%), for a between-group difference of 18.37% (95% CI, 7.60 to 29.15). The pCR rates ranged from 57.3% to 66.2% across the 3 arms in TRYPHAENA and were 60.7% and 61.8% in the 2 cohorts in BERENICE.
In NEOSPHERE, a complete response (CR) was observed in 18.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 18.3% of patients in the trastuzumab plus docetaxel arm, and a partial response (PR) was observed in 49.1% of patients and 49.3% of patients, respectively, when assessed by X-ray or mammography. When assessed by clinical exam, a CR was observed in 25.0% versus 21.6% of patients, respectively, and a PR was observed in 63.0% versus 59.8% of patients, respectively. In PEONY, clinical response was assessed as a secondary outcome, and an objective response, defined as obtaining either a CR or PR) during cycles 1 to 4 occurred in 88.6% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 78.2% of patients in the trastuzumab plus docetaxel arm, for a difference in objective response rates between groups of 10.4% (95% CI, 1.12 to 19.69). A CR was observed in 11.0% versus 10.0% of patients, and a PR was observed in 77.6% versus 68.2% of patients, respectively.
Duration of response, health-related quality of life, and symptoms were not assessed in the included studies. Among patients in whom mastectomy was initially planned, breast-conserving surgery was achieved in 23.2% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 22.6% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE. This outcome was not assessed in PEONY. In TRYPHAENA, the percentage of patients undergoing breast-conserving surgery was consistent with that of NEOSPHERE, ranging between 16.7% and 27.0% of patients across arms in the subgroup of patients in whom mastectomy was initially planned. In the BERENICE study, 44.4% and 42.9% of patients with T2 or T3 tumours in the 2 cohorts had breast-conserving surgery.
Harms Results
The percentage of patients experiencing adverse events was similar between pertuzumab and trastuzumab plus docetaxel and trastuzumab plus docetaxel, occurring in 96% to 98% of patients across treatment arms in NEOSPHERE and PEONY. The most common adverse events in the trials for pertuzumab and trastuzumab plus docetaxel versus trastuzumab plus docetaxel, were alopecia (63.6% versus 65.4% in NEOSPHERE; 49.1% in PEONY for both treatments), neutropenia (50.5% versus 62.6% in NEOSPHERE; 48.2% versus 44.5% in PEONY), and diarrhea (45.8% versus 33.6% in NEOSPHERE; 38.5% versus 16.4% in PEONY). The most common grade 3 or greater adverse event was neutropenia (44.9% versus 57.0% in NEOSPHERE; 38.1% versus 32.7% in PEONY). Similar results were seen in TRYPHAENA and BERENICE, where approximately 99% of patients experienced an adverse event at some time during the study, and neutropenia was the most common grade 3 or greater adverse event.
Serious adverse events occurred in 10.3% of patients in the pertuzumab and trastuzumab plus docetaxel group and 16.8% of patients in the trastuzumab plus docetaxel group in NEOSPHERE, and in 10.1% versus 8.2% of patients, respectively, in PEONY. Febrile neutropenia was the most common serious adverse event in both NEOSPHERE groups, occurring in 5.6% of patients treated with pertuzumab and trastuzumab plus docetaxel and 6.5% of patients treated with trastuzumab plus docetaxel. In PEONY, febrile neutropenia occurred in 1.8% of patients treated with pertuzumab and trastuzumab plus docetaxel and in no patients treated with trastuzumab plus docetaxel. In TRYPHAENA, 28% of patients experienced a serious adverse event across the treatment arms, and, in BERENICE, 24% of patients experienced a serious adverse event. Febrile neutropenia was the most common serious adverse event in both studies, occurring in about 10% of patients.
Few patients across the trials stopped treatment due to an adverse event: 0.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm versus no patients in the trastuzumab plus docetaxel arm in NEOSPHERE, and 0.5% of patients in the pertuzumab and trastuzumab plus docetaxel arm and no patients in the trastuzumab plus docetaxel arm in PEONY. The number of patients withdrawing due to an adverse event was 7% across arms in TRYPHAENA and 3.5% across cohorts in BERENICE.
One patient died in each of the pertuzumab and trastuzumab plus docetaxel and trastuzumab plus docetaxel arms in NEOSPHERE; both deaths were considered to be due to complications of breast cancer. One patient died in the pertuzumab and trastuzumab plus docetaxel arm in PEONY, due to a suicide, and there were no deaths in the trastuzumab plus docetaxel arm.
Among notable harms, cardiac dysfunction occurred in 2.8% of patients in the pertuzumab and trastuzumab plus docetaxel arm, and 0.9% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE; no patients in PEONY had an LVEF decline to less than 40% or a primary or secondary cardiac event. Events of drug hypersensitivity or anaphylaxis occurred in NEOSPHERE in |||| of patients in the pertuzumab and trastuzumab plus docetaxel arm and in |||| of patients in the trastuzumab plus docetaxel arm, and in |||| versus |||| of patients in PEONY, respectively.
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Characteristic | NEOSPHERE | PEONY | TRYPHAENA | BERENICE | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Arm B pert + trast + doce N = 107 | Arm A trast + doce N = 107 | Arm C pert + trast N = 107 | Arm D pert + doce N = 96 | Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 | Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 | Cohort A ddAC then pacli + pert + trast N = 199 | Cohort B FEC then doce + pert + trast N = 201 | |
OSa | |||||||||||
Patients with event, n (%) | NR | NR | NR | NR | NM | NM | 5 (6.8) | 7 (9.3) | 10 (13.0) | NM | NM |
PFSa | |||||||||||
Patients with event, n (%) | 17 (15.9) | 19 (17.8) | 27 (25.2) | 24 (25.0) | NR | NR | 10 (13.7) | 11 (14.7) | 14 (18.2) | NR | NR |
HR (95% CI)b vs. trast + doce | 0.69 (0.34 to 1.40) | Reference | 1.25 (0.68 to 2.30) | NR | NA | NA | NA | NA | NA | NA | NA |
DFSa | |||||||||||
Patients with event, n (%) | 15 (14.9) | 18 (17.5) | 19 (19.8) | 22 (23.9) | NM | NM | 10/69 (14.5) | 8/67 (11.9) | 11/72 (15.3) | NR | NR |
HR (95% CI)b vs. trast + doce | 0.60 (0.28 to 1.27) | Reference | 0.83 (0.42 to 1.64) | NR | NA | NA | NA | NA | NA | NA | NA |
pCR | bpCR | tpCR | bpCR | tpCR | |||||||
Responders, n (%) | 49 (45.8) | 31 (29.0) | 18 (16.8) | 23 (24.0) | 86 (39.3) | 24 (21.8) | 45 (61.6) | 43 (57.3) | 51 (66.2) | 123 (61.8) | 122 (60.7) |
Difference in response rates vs. trast + doce (95% CI) | 16.82 (3.5 to 30.1) | Reference | −12.15 (−23.8 to −0.5) | NR | 17.45 (6.89 to 28.01)c | NA | NA | NA | NA | NA | |
P value from CMHd | 0.0094 | 0.0198 | 0.0010 | 0.0014e | NA | NA | NA | NA | NA | ||
P value (Simes correction for CMH test)f | 0.0141 | 0.0198 | 0.0030 | NA | NA | NA | NA | NA | NA | NA | |
Objective response | |||||||||||
By CBE | By CBE and/or MRI | As per local practice | By CBE and/or MRI | ||||||||
Responders, n (%) | 88 (88.0) | 79 (81.4) | 65 (66.3) | 65 (73.9) | 194 (88.6) | 86 (78.2) | 67 (91.8) | 71 (94.7) | 69 (89.6) | 134 (67.3) | 121 (60.2) |
Complete response | 25 (25.0) | 21 (21.6) | 11 (11.2) | 14 (15.9) | 24 (11.0) | 11 (10.0) | 37 (50.7) | 21 (28.0) | 31 (40.3) | 79 (39.7) | 48 (23.9) |
Partial response | 63 (63.0) | 58 (59.8) | 54 (55.1) | 51 (58.0) | 170 (77.6) | 75 (68.2) | 30 (41.1) | 50 (66.7) | 38 (49.4) | 55 (27.6) | 73 (36.3) |
Stable disease | 12 (12.0) | 17 (17.5) | 31 (31.6) | 23 (26.1) | 18 (8.2) | 21 (19.1) | 3 (4.1) | 1 (1.3) | 5 (6.5) | 14 (7.0) | 20 (10.0) |
Disease progression | 0 (0.0) | 1 (1.0) | 2 (2.0) | 0 (0.0) | 1 (0.5) | 2 (1.8) | 0 (0.0) | 1 (1.3) | 0 (0.0) | 1 (0.5) | 2 (1.0) |
BCS | |||||||||||
BCS, n (%), T2 or T3 patients only | 13/56 (23.2) | 14/62 (22.6) | 11/61 (18.0) | 19/60 (31.7) | NR | NR | 10/46 (21.7) | 6/36 (16.7) | 10/37 (27.0) | 76/171 (44.4) | 75/175 (42.9) |
Harms | |||||||||||
Total AE, n (%) | |||||| | |||||| | |||| | |||||| | |||||| | |||||||| | |||||| | |||||| | |||||| | 197 (99.0) | 198 (100.0) |
Total SAE, n (%) | 11 (10.3) | 18 (16.8) | 4 (3.7) | 16 (17.0) | 22 (10.1) | 9 (8.2) | 20 (27.8) | 15 (20.0) | 27 (35.5) | 45 (22.6) | 52 (26.3) |
WDAE, n (%) | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
AE = adverse event; BCS = breast-conserving surgery; bpCR = pathologic complete response in the breast; CBE = clinical breast exam; chemo = chemotherapy; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; ddAC = dose-dense doxorubicin and cyclophosphamide; DFS = disease-free survival; doce = docetaxel; EFS = event-free survival; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; HR = hazard ratio; NA = not applicable; NM = data not mature; NR = not reported; OS = overall survival; pCR = pathologic complete response; pert = pertuzumab; PFS = progression-free survival; SAE = serious adverse event; tpCR = total pathologic complete response; trast = trastuzumab; WDAE = withdrawal due to adverse event.
Note: Invasive DFS, EFS, health-related quality of life, and symptoms were not assessed in any of the included studies.
aIncludes both neoadjuvant and adjuvant treatment.
bHR is based on Cox proportional hazard regression stratified by breast cancer type and hormone positivity.
cApproximate 95% CI for difference of 2 rates using Hauck-Anderson method.
dCMH test stratified by breast cancer type (operable, locally advanced, or inflammatory) and estrogen and or progesterone positivity (either is positive vs. both are negative).
eCMH test stratified by disease category (early stage and locally advanced) and hormone receptor status (positive for estrogen and/or progesterone receptor or negative for both) from an interactive web and voice response system.
fP value from CMH test, with Simes multiplicity adjustment.
Source: Clinical Study Report for NEOSPHERE,4 PEONY,5 TRYPHAENA,6 and BERENICE.7
Critical Appraisal
NEOSPHERE was an open-label study, and no centralized blinded review of pathology was conducted when assessing pCR responses. Although pathology findings are unlikely to be biased by knowledge of treatment assignment, a blinded review of pathology is recommended by regulatory bodies. With respect to the primary outcome, pCR was defined differently between NEOSPHERE and PEONY. In NEOSPHERE, the primary outcome of pCR included only breast tissue (commonly described as bpCR) while, in PEONY, assessment of pCR for the primary outcome included breast and nodes, referred to as tpCR; the latter is the method recommended by the FDA. TRYPHAENA and BERENICE provide only limited supportive information regarding efficacy; as neither trial had a comparator, neither was designed to test hypotheses with respect to efficacy outcomes, and BERENICE was not a randomized trial. The alpha in NEOSPHERE was set at 0.2 instead of the traditional 0.05, and this might have increased the risk of finding a statistically significant difference in pCR rates between arms where none existed.
OS was not assessed as an efficacy outcome in NEOSPHERE and the OS data for PEONY were not yet mature, according to the sponsor; therefore, there is no information to determine whether the addition of pertuzumab to neoadjuvant treatment with trastuzumab and docetaxel improves this important outcome. Health-related quality of life and symptoms were also not assessed and, although these outcomes may not be as important in early breast cancer and in the neoadjuvant setting, assessment of health-related quality of life would help in assessing what impact the addition of pertuzumab has on adverse effects.
Indirect Treatment Comparisons
No indirect treatment comparisons (ITCs) were submitted by the sponsor, and none were found in the literature that would inform this review.
Other Relevant Evidence
There were no other studies that were found that would be relevant to this review.
Conclusions
Four trials that were identified as pivotal by the sponsor were included in this review. Rates of pCR were improved when pertuzumab was added to standard neoadjuvant regimens with trastuzumab plus chemotherapy in the 2 trials that featured a comparator, NEOSPHERE and PEONY. It is unclear whether these improvements in pCR translate into improved OS, as this outcome was not studied in NEOSPHERE, and the survival data from PEONY were not available at the time of review. The combination of pertuzumab with trastuzumab plus chemotherapy did not appear to improve invasive DFS, PFS, EFS, or DFS, either because these outcomes were not studied, the data were not yet available, or there was a lack of statistical significance when they were assessed. Health-related quality of life and symptoms were not assessed in any of the included studies. Based on the included studies, the addition of pertuzumab to trastuzumab plus chemotherapy did not appear to introduce significant safety or tolerability issues.
Introduction
Disease Background
It is estimated that 1 in 8 Canadian women will be diagnosed with breast cancer during their lifetime, and that 1 in 33 will die of the disease.8 Due to advances in and widespread use of screening programs, breast cancer is typically diagnosed at an early stage, and approximately 95% of cases are diagnosed at stages I to III. Generally speaking, primary operable breast cancers are categorized as stage I or II, and locally advanced breast cancers are categorized as stage III. The overall 5-year survival rate from breast cancer is 88%, though this will vary based on the stage and subtype of breast cancer.8 Prior to the use of anti-HER2 therapy, patients who had HER2-positive breast cancer tended to have a poorer prognosis than patients without HER2 overexpression.
Patients who are undergoing neoadjuvant therapy can be subclassified into those with locally advanced breast cancers that are not operable, those with locally advanced cancers that are operable, and those with primary operable breast cancers. In locally advanced cancers, 1 purpose of neoadjuvant therapy is to convert the tumour from an inoperable to an operable state. In all cancers, the purpose of neoadjuvant therapy is to downstage the tumour to potentially avoid mastectomy in favour of breast-conserving surgery, to assess response to systemic therapy, to potentially escalate or de-escalate subsequent adjuvant therapy based on the response to neoadjuvant therapy, and to initiate systemic therapy early to try to limit systemic spread.
Standards of Therapy
For the majority of patients with HER2-positive breast cancer who are either stage II or III, as well as some patients who are stage I, neoadjuvant systemic treatment is the current standard of care in Canada. According to the clinical experts consulted by CADTH, this represents current accepted international guidelines and is increasingly the standard therapy used in Canada, with some exceptions. By treating with chemotherapy early, the risk of systemic recurrence is also decreased. There are a small number of patients who may have surgery first and then have adjuvant therapy, particularly if they have small cancers, and this would be the norm for stage I disease.
According to the clinical experts, standard neoadjuvant chemotherapy is doxorubicin, cyclophosphamide, and paclitaxel plus trastuzumab or docetaxel and carboplatin plus trastuzumab. A small number of patients might get 5-fluoruracil, epirubicin, and docetaxel plus trastuzumab. These are all standard protocols used internationally, although pertuzumab is often given with trastuzumab in other countries, according to the clinical experts, and there are some patients who gain access to pertuzumab through private insurance. The ASCO guidelines for the neoadjuvant management of HER2-positive, node-positive, or high-risk node-negative breast cancer recommend an anthracycline and taxane or non-anthracycline-based regimen in combination with trastuzumab, and they also suggest that pertuzumab can be used with trastuzumab in the neoadjuvant setting.1 The European Society for Medical Oncology recommends a dual blockade with pertuzumab and trastuzumab in high-risk patients (node-positive or estrogen receptor–negative), starting before or after surgery and for neoadjuvant therapy; the German Gynecological Oncology Group recommends adding pertuzumab when treating node-positive disease.2,3 According to 1 clinical expert, patients who achieve a pCR after neoadjuvant treatment tend to have a better prognosis and are recommended to receive adjuvant trastuzumab for up to 14 cycles, while patients with residual invasive disease are recommended to receive trastuzumab emtansine (T-DM1) in the adjuvant setting. Patients with hormone receptor–positive and HER2-positive breast cancer are also recommended for adjuvant endocrine therapy. The goal of treatment in the neoadjuvant setting is curative, as this means that otherwise healthy patients can go on to live a normal life span and maintain employment. This also reduces caregiver burden and is generally beneficial to society.
Drug
Pertuzumab is a monoclonal antibody that targets the extracellular dimerization domain of the HER2 receptor protein and thus blocks ligand-dependent heterodimerization of HER2 with other members of the human epidermal growth factor receptor family. Pertuzumab therefore inhibits ligand-initiated intracellular signalling through 2 pathways, the mitogen-activated protein kinase pathway and the phosphoinositide-3 kinase pathway, causing cell growth arrest and apoptosis. Additionally, pertuzumab mediates antibody-dependent cell-mediated cytotoxicity.
On February 25, 2021, Health Canada issued a Notice of Compliance for the use of pertuzumab in the neoadjuvant setting.9 Pertuzumab is indicated, in combination with trastuzumab and chemotherapy, for the neoadjuvant treatment of patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive).9 Pertuzumab was previously reviewed by CADTH for off-label use under the same indication in 2015 and the CADTH pan-Canadian Oncology Drug Review (pCODR) Expert Review Committee (pERC) issued a negative recommendation for reimbursement. Pertuzumab in combination with trastuzumab is also indicated as adjuvant treatment in HER2-positive, early breast cancer with lymph node–positive and/or hormone receptor–negative disease, and with docetaxel for patients with HER2-positive metastatic breast cancer who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease. CADTH reviewed pertuzumab for both indications and pertuzumab received a negative recommendation for reimbursement for the adjuvant indication and a recommendation for reimbursement for the metastatic breast cancer indication.
Pertuzumab is administered by IV infusion. In the neoadjuvant setting, after an initial 840 mg loading dose, the maintenance dose is 420 mg every 3 weeks for 3 to 6 cycles. Pertuzumab is given in combination with trastuzumab, as part of 1 of the following regimens in early-stage breast cancer:
Four pre-operative cycles of pertuzumab in combination with trastuzumab and docetaxel (75 mg/m2 with the option to escalate to 100 mg/m2 at physician discretion if the initial dose is well tolerated) every 3 weeks, followed by 3 post-operative cycles of FEC (5-fluorouracil: 600 mg/m2; epirubicin: 90 mg/m2; cyclophosphamide: 600 mg/m2) every 3 weeks, as given in the NEOSPHERE trial.
Three or 4 pre-operative cycles of FEC (5-fluorouracil 500 mg/m2; epirubicin: 100 mg/m2; cyclophosphamide: 600 mg/m2) alone, every 3 weeks, followed by 3 or 4 pre-operative cycles of pertuzumab in combination with docetaxel (75 mg/m2 with the option to escalate to 100 mg/m2 at physician discretion if the initial dose is well tolerated) and trastuzumab every 3 weeks, as given in the TRYPHAENA and BERENICE trials, respectively.
Six pre-operative cycles of pertuzumab in combination with docetaxel and carboplatin plus trastuzumab: 75 mg/m2 docetaxel (escalation of docetaxel above 75 mg/m2 is not recommended), carboplatin (area under the plasma concentration versus time curve [AUC] 6) and trastuzumab every 3 weeks, as given in the TRYPHAENA trial.
Four pre-operative cycles of dose-dense doxorubicin and cyclophosphamide (doxorubicin: 60 mg/m2; cyclophosphamide: 600 mg/m2) alone every 2 weeks, followed by 4 pre-operative cycles of pertuzumab in combination with trastuzumab every 3 weeks, and paclitaxel (80 mg/m2) every week for 12 weeks, as given in the BERENICE trial.
The sponsor’s reimbursement request is the same as the Health Canada indication, in combination with trastuzumab and chemotherapy, for the neoadjuvant treatment of patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive). The sponsor also added that patients should receive neoadjuvant treatment with pertuzumab in combination with trastuzumab and chemotherapy for 3 to 6 cycles, depending on the regimen chosen. Patients who start pertuzumab in combination with trastuzumab and chemotherapy in the neoadjuvant setting and who do not have residual disease following surgery should continue to receive adjuvant trastuzumab to complete 1 year of HER2-directed therapy.
Table 3: Key Characteristics of Pertuzumab and Trastuzumab
Characteristic | Pertuzumab | Trastuzumab |
|---|---|---|
Mechanism of action | Monoclonal antibody that targets the extracellular dimerization domain (subdomain 2) of HER2 and blocks ligand-dependent heterodimerization of HER2 with other HER family members, including HER1 (EGFR), HER3, and HER4. This inhibits intracellular signalling through 2 major pathways, the MAP kinase pathway and the PI3K pathway | Monoclonal antibody that targets the extracellular domain of the HER2 receptor |
Indicationa | Early breast cancer: In combination with trastuzumab and chemotherapy, for:
Metastatic breast cancer:
| Early breast cancer: For patients with an ECOG status of 0 to 1 who overexpress HER2; it is administered:
Metastatic breast cancer:
|
Route of administration | IV | IV or SC |
Recommended dose | Metastatic and early breast cancer:
When administered with pertuzumab, the recommended dose of trastuzumab is either an IV infusion with an initial dose of 8 mg/kg followed every 3 weeks by a dose of 6 mg/kg, or a fixed dose of 600 mg SC initially and every 3 weeks thereafter | Early breast cancer:
|
Serious adverse effects or safety issues |
|
|
ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; HER1, HER2, HER3, HER4 = human epidermal growth factor receptor 1, 2, 3, or 4; MAP = mitogen-activated protein; PI3K = phosphoinositide-3 kinase; SC = subcutaneous.
aHealth Canada–approved indication.
Source: Product monographs for pertuzumab9 and trastuzumab.10
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
Two patient groups provided input for this submission, the CBCN and Rethink Breast Cancer. The CBCN is a patient-directed, national health charity committed to ensuring the best quality care for all Canadians affected by breast cancer through the promotion of information, education, and advocacy activities. Rethink Breast Cancer’s mission is to empower young people who are affected by or concerned about breast cancer through education, support, and advocacy. It represents the voice of young women to ensure their needs and values are considered in all aspects of breast cancer treatment and care.
For the preparation of this submission, the CBCN noted that Roche Canada, the sponsor, connected them with patients who had experience with pertuzumab. Rethink Breast Cancer requested information from Roche and its scientific advisory committee on the characteristics of the drug and its benefits and they contracted a freelance health technology assessment writer to help prepare the submission and to develop a survey and analyze the responses received.
Information for the submission was obtained from CBCN’s 2017 Lived Experience Breast Cancer Patient Survey, from which CBCN summarized the responses from 52 Canadians with early-stage, HER2-positive breast cancer (stage I to III). All respondents identified as female and from various regions: Alberta (4% of respondents), Ontario (23%), Saskatchewan (12%), Quebec (8%), British Columbia (8%), Manitoba (4%), and the Atlantic provinces (17%); 25% did not specify their location. The age at which respondents were diagnosed with breast cancer was categorized as follows: 30 to 39 years of age (19%), 40 to 49 years (40%), 50 to 59 years (29%), and 60 to 69 years (6%); the remaining 6% of respondents did not disclose their age. In addition, 4 patients who had direct experience with pertuzumab participated in a phone interview. The CBCN also conducted a literature review to identify issues and experiences shared among women with breast cancer.
Rethink Breast Cancer conducted an online survey from March 23 to April 19, 2021 that was circulated through the organization’s mailing list, partner organizations, and social media. Of the 62 respondents, 37 (60%) were from Canada (Alberta, British Columbia, New Brunswick, Ontario, Quebec, and Saskatchewan), 22 were from the US (35%), and 3 were from other countries or did not disclose their location. Seven respondents agreed to participate in telephone interviews with staff. All 62 respondents were diagnosed with HER2-positive breast cancer in stage I, II, or III. Among the respondents, 41 (66%) had treatment experience with pertuzumab including 35 (56%) who match the full indication for this review.
Disease Experience
A diagnosis of early-stage, HER2-positive breast cancer has a significant impact on the day-to-day life of the patient, particularly as this cancer subtype is traditionally associated with more aggressive cancer with a poor prognosis in the absence of HER2-directed therapy. It is also associated with a higher risk of recurrence or metastases. Both the diagnosis as well as the treatments that are used impact the emotional and physical well-being of a patient. Some of the adverse effects of HER2-positive breast cancer and the therapies used to manage this disease include cardiac toxicity, fever, cough, muscle pain, fatigue, diarrhea, and nausea. Many of these symptoms have the ability to impact daily life, primarily, fatigue, pain, and nausea.
The financial burden associated with living with breast cancer extends far beyond any loss of income during a temporary or permanent absence from employment. In addition to the loss of income during illness, breast cancer patients can incur substantial costs associated with treatment and disease management. In the CBCN survey, 17% of respondents had a very large financial impact, and 38% had some financial impact from their diagnosis. One patient stated:
“Very hard on my family...had to return to work still not feeling strong enough...very hard ...when you’re sick you don’t need this stress on top of everything else.”
Experience With Treatment
In the Rethink Breast Cancer survey, 56 of 62 patients had received trastuzumab and 12 had received T-DM1. The most commonly reported chemotherapy drugs received included carboplatin, docetaxel, paclitaxel, doxorubicin (Adriamycin), and cyclophosphamide. Fatigue was the most commonly reported adverse effect of these treatments (80%), followed by diarrhea (64%), nausea (44%), and insomnia (39%). Fatigue was most frequently cited as the hardest-to-tolerate adverse effect of these treatments. Diarrhea, nausea, neuropathy, and taste changes were also cited by at least 10% of respondents. Most respondents (73%) did not report any problems accessing treatment.
In the CBCN 2017 survey, most of the HER2-positive, early-stage breast cancer patients had been or were currently being treated with a combination of surgery, radiation, chemotherapy, and the HER2-directed therapy, trastuzumab. Most patients had undergone surgery (44 out of the 52 respondents), radiation therapy (33 respondents), and chemotherapy (35 respondents) as part of their overall breast cancer treatment.
In the CBCN survey, respondents reported barriers to treatment including lack of access to private insurance coverage and support medications. While 40 of the 52 patients surveyed reported having private insurance coverage, several (6 respondents) also reported challenges accessing medications not publicly reimbursed. Respondents stated they had to use private insurance (17 respondents) or pay out of pocket (11 respondents) to access medications they had been prescribed. One patient said:
“When I found out how expensive my treatment is, I was absolutely flabbergasted. I had to leave the pharmacy empty handed because a one month supply was over $1400 and I didn't have the money or amount available on credit. I was told about a form to fill out if my income was below a certain amount but I didn't qualify. So, we paid for it out of pocket and did get some reimbursed from work insurance plan.”
Rethink Breast Cancer reported the experience of 35 patients who received neoadjuvant pertuzumab in combination with trastuzumab and chemotherapy for locally advanced, inflammatory, or early-stage breast cancer. Twenty-one patients achieved a pCR within 1 year of their surgery. Of these 21 patients, 1 patient had since had a recurrence; the other 20 remained free of cancer. Most of the respondents felt that pertuzumab had improved their quality of life in every listed area, including activities such as the ability to work, sleep, drive, care for children, and perform household chores. Said 1 patient respondent:
“If you have a scan or you have another ultrasound and you see the reduction in the tumor so quickly, it had an impact on anxiety, on positivity, on quality of life.”
Diarrhea and fatigue were the most commonly reported adverse effects of pertuzumab (84% and 81%, respectively), followed by alopecia (38%), neutropenia (25%), and nausea (22%). However, respondents overwhelmingly described these adverse effects as tolerable. Moreover, respondents explicitly said they were willing to tolerate the adverse effects of pertuzumab for its medical benefits. A few respondents noted they had difficulties with the loading dose of pertuzumab but found the subsequent doses to be tolerable. Others were not able to distinguish the adverse effects of pertuzumab from the other drugs they were receiving concurrently.
The CBCN reported the experience of 4 patients who had received pertuzumab in addition to trastuzumab, chemotherapy, and surgery. It is not clear if pertuzumab was administered in the adjuvant or neoadjuvant setting. All patients were hopeful that this addition to their treatment regimen would help improve their survival and reduce the risk of recurrence. The patients had difficulty determining if the adverse effects they experienced were related to pertuzumab or other therapies, but all rated their quality of life as mid-range to high on a 10-point scale. All patients expressed concerns about the lack of access to new treatments and the potential financial burden of paying out of pocket. One patient respondent said:
“Having just that additional little bit of peace of mind that I’m doing everything that I can. I’m pretty young. I’ve got a young family. I’ve got a three-year-old. So I need to be able to say that I’ve done everything that I possibly can to beat it. So having that peace of mind that I’m getting the same care that others are getting elsewhere in the world, so I don’t have to look at going somewhere else and all the costs and finances involved. If there was a breakthrough treatment that was working in the U.S. but not available in Canada, having to somehow try to finance going there to go get that treatment.”
Improved Outcomes
Among respondents to the Rethink Breast Cancer survey, eliminating cancer, preventing recurrence, and preventing metastases were overwhelmingly rated as the most important outcomes for their breast cancer treatment with greater than 96% of respondents rating these goals as “very important.” Maintaining quality of life was also rated as “very important” or “important” for 63% and 18% of respondents, respectively, while managing adverse effects was “very important” for 46% and “important” for 23% of respondents. Respondents also indicated they were highly willing to tolerate new adverse effects from new drugs to extend life expectancy:
“…I just finished chemo and feel like there’s no way I’d ever do it again. Period. But at the same time how do you not do *whatever* it takes to stay alive? I’d undergo near death side effects in order to avoid death…”
“I will tolerate whatever symptoms I have to so that I can survive and take care of my children.”
Respondents of CBNC’s 2017 survey indicated that the following key factors influenced their decision-making around treatments:
Effectiveness of the treatment: How well the treatment stabilized their disease and delayed the progression of their cancer. Effectiveness was ranked as “very important” by 40 of 52 respondents (77%), with 73% of respondents stating effectiveness was the single most important factor in their treatment decisions. The majority of respondents indicated that reducing the risk of recurrence was “important” or “very important.”
“I was willing to do whatever was best to rid myself of the cancer. I could deal with the side effects and disruption in my life for the long term good.”
“I just wanted to make sure they did everything to get rid of the cancer.”
Reducing the risk of recurrence without sacrificing quality of life: Being able to maintain productive, active lives with minimal disruption to daily routines and avoiding relapse of their cancer. Approximately 2-thirds of respondents indicated maintaining quality of life and mobility was “important” or “very important,” while maintaining productivity was an important concern for 35% of respondents. Six respondents raised concerns regarding their ability to provide childcare, and this was an important factor in their treatment decisions.
“I am a mother to 3 children. I wanted to be aggressive in order to increase my chances of survival.”
“I only wanted to reduce my risk of recurrence as much as possible. Everything else was secondary.”
“My quality of life during and after treatment was the biggest issue for me.”
“If I had to do it over again, I would opt out of chemo.”
Adverse effect management: Minimizing risk while stabilizing their disease. Minimal adverse effects of treatment was an “important” or “very important” concern for half the respondents.
“Les effets de la chimio qui nous sont inconnus et qui fait peur car on en attends parler tellement négativement.” [Translation: The effects of chemo that are unknown and scary because we hear people talk about it so negatively.]
Cost and accessibility of treatments: Affordability and ease of accessing treatments. Having access to new treatments was important to patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of breast cancer.
Unmet Needs
One of the clinical experts consulted by CADTH said they believe that, with respect to pCR rates, there is room for improvement with current treatments, as lower rates of pCR correspond to higher relapse rates. The other clinical expert noted that increasing the rate of tumour downstaging and pCR should improve cosmesis and decrease survivorship morbidity through less invasive local-regional management approaches and post-operative T-DM1, a drug associated with a higher risk of toxicity than trastuzumab. These toxicities include peripheral neuropathy, cytopenia, abnormal liver enzymes, and, rarely, pneumonitis and hepatic nodular regenerative hyperplasia.
Place in Therapy
Per indication, pertuzumab would be used in combination with trastuzumab and taxane-based chemotherapy in the neoadjuvant setting. The clinical expert consulted by CADTH noted that, if pertuzumab were used in the neoadjuvant setting, this would likely reduce the number of relapses and thus the number of patients who would need to be treated for advanced disease. The shift in the treatment paradigm would simply be the addition of pertuzumab to the standard therapies already being used in the neoadjuvant setting.
Patient Population
The patients most likely to respond to the addition of pertuzumab would be those who are HER2-positive (or overexpressing HER2) with T2 or more advanced breast tumours and/or lymph node involvement, according to the clinical experts consulted by CADTH. One of the clinical experts noted that pCR occurs in patients with either estrogen receptor–positive or estrogen receptor–negative disease; however, the pCR rate is consistently higher in those who are estrogen receptor–negative. According to the clinical experts, all patients who are HER2-positive and considered candidates for neoadjuvant therapy would be eligible for the addition of pertuzumab to their neoadjuvant regimen. Patients who are not candidates for either chemotherapy (due to being too ill) or for neoadjuvant therapy (because they have a tiny stage I cancerous tumour) would not be eligible for neoadjuvant pertuzumab. It was also noted that it is very rare for a patient to be too ill to receive chemotherapy.
Assessing Response to Treatment
One clinical expert noted that, ultimately, response in the neoadjuvant setting is determined at the time of surgery, when assessment of pCR is performed. Prior to surgery, patients would most likely be assessed every 2 to 3 weeks depending on the regimen at the time they come in to receive their chemotherapy. Assessment is typically done by a physical exam, although sometimes this may be supplemented by imaging of the breast (ultrasound or MRI). A clinically meaningful response is a shrinkage of the tumour to facilitate surgical removal.
Discontinuing Treatment
With respect to deciding when to discontinue treatment, the clinical experts noted this may occur if the tumour is growing, in which case surgery may be performed earlier than planned or, in some cases, other chemotherapy protocols may be instituted. Patients with clear disease progression after receiving 1 to 2 cycles of optimized taxane-based chemotherapy should be considered for discontinuation. One of the clinical experts noted that hypersensitivity to pertuzumab may be a reason to discontinue therapy.
Prescribing Conditions
Pertuzumab would be administered wherever chemotherapy is given; in Canada, most chemotherapy is administered in cancer centres, hospitals with medical oncologists and chemotherapy units, hospitals with regional chemotherapy units or, occasionally, in hospitals with general practitioner oncologists who are being directed by medical oncologists at cancer centres. Chemotherapy in Canada is very protocol-oriented and is also funded on a provincial level, which requires that standard protocols be followed in specialized units. The other clinical expert noted that if there were no signs of infusion reactions, subsequent infusions could be administered in centres that are not accredited to give an advanced drug if it is appropriate to do so for the patient’s convenience.
Additional Considerations
One clinical expert noted that the addition of pertuzumab to the current treatment paradigm is important, given that this is a curable disease that often occurs in younger patients. The other clinical expert noted the importance of increased rates of tumour downstaging and pCR in reducing longer-term treatment-related morbidity.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
Input from 2 clinician groups was received on the reimbursement review of pertuzumab as neoadjuvant treatment: 1 from the BCC-BTG and the other from the OH-CCO BCDAC.
The BCC-BTG is a multidisciplinary group that operates within BC Cancer and sets the standards of care, treatment options, and pathways across the spectrum of breast cancer care in British Columbia. Information from a previous review for neoadjuvant pertuzumab in HER2-positive breast cancer was also used to inform this submission.
The OH-CCO BCDAC provides timely evidence-based clinical and health system guidance on drug-related issues in support of OH-CCO’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. Information for this submission was discussed jointly through email at a BCDAC meeting.
Unmet Needs
The BCC-BTG noted the intent of treatment is complete eradication of disease (both gross disease in breast and nodes and potential microscopic metastases) to improve cure rates. The group added that pertuzumab concurrent with taxane and trastuzumab is not publicly funded as a standard neoadjuvant regimen in eligible stage II to III HER2-positive breast cancer in Canada, though there are patient assistance programs. For example, Roche’s OnCare program provides assistance in insurance navigation and co-payments and coordination of infusion care. The program does not include compassionate or free pertuzumab; therefore, at present, there is significant inequity for access to pertuzumab in Canada.
With respect to the most important goals that an ideal treatment would address, the BCC-BTG noted that inducing a tumour response for enhanced breast cancer surgery (i.e., tumour downstaging for breast-conservation surgery) and pathological response (hopefully pCR) such that adjuvant treatment is not escalated to T-DM1) are important goals for treatment. The OH-CCO BCDAC noted that improving survival, preventing recurrence, acceptable toxicities, and improvement in health-related quality of life are the most important treatment goals for therapy.
The OH-CCO BCDAC noted that despite optimal systemic and local therapy, approximately 25% to 30% of HER2-positive patients still experience disease recurrence; thus, better and improved treatments are needed. They added that the patients who have the greatest unmet need for an intervention like pertuzumab are patients diagnosed at a higher stage of disease, since they have the greatest risk of disease recurrence
The BCC-BTG noted that the international standard of care for stage II to III HER2-positive breast cancer is neoadjuvant pertuzumab and trastuzumab concurrent with a taxane. The clinician group noted that the treatment gap in Canada is a significantly lower pCR rate (approximately 50% lower) without pertuzumab, which not only translates into a potentially lower long-term clinical outcome, but also exposes more patients to more toxic adjuvant treatments like T-DM1 rather than adjuvant trastuzumab alone. The clinicians from the BCC-BTG added that the greatest need for pertuzumab is in the inflammatory breast cancer population and for inoperable stage IIIC breast cancers to downstage the tumour for primary surgery.
Place in Therapy
The BCC-BTG noted that for HER2-positive stage II to III breast cancer, the current treatment pathway in Canada includes neoadjuvant chemotherapy (taxane ± anthracycline) concurrent with neoadjuvant trastuzumab (for the taxane component) for 6 to 8 cycles before surgery. The BCC-BTG added that the 2 main clinical reasons for this neoadjuvant approach is to downstage the primary tumour and axillary nodes to improve the surgical approach (lumpectomy rather than mastectomy and/or sentinel lymph node biopsy versus axillary lymph node dissection), and to use pathological response determination (pCR versus no pCR) to decide on the course of adjuvant treatment, where trastuzumab is given to patients who achieve a pCR and T-DM1 is given to patients who do not achieve a pCR.
The OH-CCO BCDAC noted that the current standard of care for T1c or greater (if N0) or any node-positive disease is neoadjuvant trastuzumab and chemotherapy. The group added that if there is residual disease post surgery, patients would be offered adjuvant T-DM1.
Both clinician groups agreed that pertuzumab would be added to neoadjuvant trastuzumab. The BCC-BTG noted that pertuzumab would fit into the current treatment pathways of neoadjuvant taxane and trastuzumab plus or minus anthracycline. Both clinician groups agreed it would not be appropriate for patients to try other treatment options before pertuzumab, as it is a neoadjuvant therapy with curative intent. The BCC-BTG clinicians noted that clinical and radiological imaging are not accurate in predicting pCR; thus, as long as the treatment is tolerated, the goal is to deliver chemotherapy and anti-HER2 therapy upfront before surgery.
Both clinician groups agreed that the use of pertuzumab in the neoadjuvant setting may impact the use of pertuzumab in the metastatic setting, as pertuzumab and trastuzumab are funded in the first-line metastatic setting. The BCC-BTG noted that it would be reasonable to consider pertuzumab, trastuzumab, and a taxane as first-line treatment (the current standard of care) for metastatic HER2-positive breast cancer if 12 months or longer had elapsed since the last dose of neoadjuvant pertuzumab.
Patient Population
The BCC-BTG noted that patients with stage II to III HER2-positive breast cancer who are medically fit to receive neoadjuvant chemotherapy and dual anti-HER2-directed therapies would be best suited to receive treatment with pertuzumab. The OH-CCO BCDAC noted that the sponsor’s funding request is for pertuzumab in combination with trastuzumab and chemotherapy for the neoadjuvant treatment of patients with HER2-positive locally advanced inflammatory or early-stage breast cancer (either 2 cm in diameter or node-positive). The OH-CCO BCDAC noted that when selecting patients who may be potential candidates for adjuvant treatment with T-DM1, patents with tumours 1 cm in diameter or node-positive disease were eligible in the KATHERINE trial. The group commented that in Ontario’s Evidence Building Program, approximately 100 patients per year are diagnosed with T1a to T1b N0 HER2-positive disease.
Both clinician groups agreed that testing for HER2 status is standard for all patients newly diagnosed with invasive breast cancer, as per the ASCO and College of American Pathologists (ASCO/CAP) guidelines. The BCC-BTG noted that most pathology laboratories participate in a quality-assurance program and patients are identified by the surgeon for referral to a medical oncologist for consideration of neoadjuvant systemic therapies. The BCC-BTG commented that the future direction is for greater neoadjuvant treatment in primary operable breast cancer and that breast cancer surgeons across the country are very aware and familiar with this approach and treatment strategy.
Both clinician groups agreed that patients who are not suited for neoadjuvant chemotherapy or HER2-directed therapy due to the presence of comorbidities or poor LVEF would be least suitable for treatment with pertuzumab.
With respect to patients who are the most likely to exhibit a response to neoadjuvant treatment with pertuzumab in practice, the BCC-BTG noted there is currently no predictive clinical factor or biomarker that has been validated as predictive of clinical benefit to neoadjuvant anti-HER2-directed therapy other than HER2 positivity (immunohistochemistry [IHC] score of 3 or greater and/or positive for fluorescent in situ hybridization [FISH], as per ASCO/CAP guidelines). The clinicians from the BCC-BTG added there are emerging data that stromal tumour-infiltrating lymphocytes are predictive of a higher pCR to both chemotherapy and to HER2-directed antibodies (pertuzumab and trastuzumab), though this biomarker is not standardly reported or used in clinical decision-making today. The OH-CCO BCDAC noted that the drug under review will be used only in breast cancer patients who are HER2-positive.
Assessing Response to Treatment
The BCC-BTG noted that a pCR, defined as no invasive disease in the breast and lymph nodes, where residual DCIS is permissible in the definition, can be used to determine whether a patient is responding to treatment in clinical practice. Both clinical groups agreed that a clinically meaningful response to treatment includes achievement of a pCR, which is associated with improved long-term clinical outcomes, including DFS and OS. The BCC-BTG added that enhanced breast cancer surgeries, such as downstaging from mastectomy to breast-conserving surgery or from axillary node dissection to sentinel node biopsy, are also clinically meaningful responses. The clinicians from BCC-BTG also added that achieving a pCR would abrogate the need for adjuvant T-DM1.
According to the clinician groups, treatment response should be assessed pathologically and patients should be monitored routinely during neoadjuvant therapy to evaluate response to treatment.
Discontinuing Treatment
As per the BCC-BTG input, factors to consider when deciding to discontinue treatment include clinical progression of disease (as documented by physical exam and/or radiological imaging) or significant adverse events (such as clinically significant cardiotoxicity or diarrhea). The OH-CCO BCDAC added disease progression on treatment (which is rare) and toxicities are important factors to consider when deciding whether to discontinue treatment.
Prescribing Conditions
Both clinician groups agreed that outpatient clinics are the most appropriate setting for treatment with pertuzumab. The BCC-BTG added that delivery is concurrent with taxane and trastuzumab; thus treatment should be in the outpatient chemotherapy unit.
Additional Considerations
The OH-CCO BCDAC noted that the treatment strategy proposed has not been specifically evaluated in any clinical trial.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. PAG noted that in most provinces, the current standard of care for the neoadjuvant treatment of HER2-positive breast cancer is trastuzumab plus chemotherapy. Pertuzumab, being an IV drug, would be administered in an outpatient chemotherapy centre for appropriate administration and monitoring of infusion-related reactions. PAG highlighted several enablers to the implementation of pertuzumab in the neoadjuvant setting: the dose and frequency of pertuzumab in the neoadjuvant setting is the same as in the metastatic setting, it is an add-on to existing treatment, and drug wastage is not a concern since pertuzumab vials contain the amount of the fixed dose. PAG also identified barriers to implementation that include: the high cost of pertuzumab and the additional preparation time and chair time needed for the infusion. Pertuzumab is administered for 4 to 6 cycles before surgery and PAG noted that given the high cost of pertuzumab, there is a significant difference in cost between 4 cycles and 6 cycles. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
PAG noted that the current standard of care in most provinces for neoadjuvant treatment of HER2-positive breast cancer is trastuzumab plus chemotherapy. However, it noted that different regimens using different numbers of cycles with trastuzumab are involved.
| Although the number of cycles of trastuzumab varies by regimen, trastuzumab is administered for 1 year regardless of the regimen given. The standard anthracycline chemotherapy regimens currently used include FEC docetaxel (6 cycles) or ddAC paclitaxel (8 cycles). The clinical experts noted that other anthracycline regimens would not typically be used in combination with trastuzumab. |
PAG noted the phase II NEOSPHERE trial shows an improvement in pCR when pertuzumab is added in the neoadjuvant setting. The outcomes of interest for the new phase III trial in this submission (PEONY) are also based on pCR and it is again using pertuzumab with trastuzumab and docetaxel, as did the phase II trial.
| Numerous trials in early breast cancer have demonstrated a correlation between pCR and improved survival outcomes.11 The Neo ALTTO trial evaluated the efficacy of neoadjuvant lapatinib, trastuzumab, and the combination of lapatinib plus trastuzumab.12 The trial showed the combination treatment significantly improved rates of pCR compared with the 2 single-drug treatment arms, but EFS and OS were not significantly different between the treatment groups. However, the trial demonstrated that regardless of the neoadjuvant received, survival outcomes were significantly improved in the patients who achieved a pCR. The ALTTO trial, which evaluated lapatinib plus trastuzumab in the adjuvant setting, also demonstrated no difference in survival outcomes between the combination compared with the sequential administration of lapatinib and trastuzumab or either single-drug therapy alone.13 Based on these results, lapatinib is not considered to add clinical benefit to trastuzumab in the adjuvant or neoadjuvant treatment settings. Lapatinib, a dual TKI, has a mechanism of action that is different from pertuzumab. In contrast, the combination of pertuzumab and trastuzumab has demonstrated improved treatment efficacy compared with trastuzumab alone in both treatment settings. |
PAG noted that HER2-positive, locally advanced, and inflammatory breast cancer is clearly indicated for neoadjuvant treatment; however, the early-stage breast cancer population encompasses a wider range of patients.
|
|
PAG noted that pertuzumab is administered for 4 to 6 cycles before surgery. Given the high cost of pertuzumab, there is a significant difference between 4 cycles and 6 cycles. In the most recent phase III study (PEONY), it was given for 4 cycles. PAG would like to confirm this would be the number of cycles (e.g., 4).
| The number of cycles of neoadjuvant pertuzumab is dependent on the regimen used:
|
Upon public listing, would clinical experts support adding pertuzumab to the treatment of patients currently undergoing neoadjuvant therapy with trastuzumab and chemotherapy? | Yes, for patients currently receiving neoadjuvant trastuzumab and chemotherapy, it would be reasonable to add pertuzumab. One clinical expert noted that if pertuzumab were to be added, it should be at the beginning of the taxane component of the regimen, not simply at the discretion of the treating physician and not halfway through the taxane regimen. |
This drug may change the place in therapy for drugs reimbursed in subsequent lines.
|
|
ddAC = dose-dense doxorubicin and cyclophosphamide; EFS = event-free survival; FEC = 5-fluorouracil and epirubicin plus cyclophosphamide; HER2 = human epidermal growth factor receptor 2; OS = overall survival; PAG = Provincial Advisory Group; pCR = pathologic complete response; TDM-1 = trastuzumab emtansine; TKI = tyrosine kinase inhibitor.
Clinical Evidence
The clinical evidence included in the review of pertuzumab is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those studies that were selected according to an a priori protocol. The second section normally includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review; however, no indirect evidence was submitted and none was found in the literature. The third section includes sponsor-submitted, long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review; however, none were considered relevant for this review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of pertuzumab by IV infusion in combination with trastuzumab and chemotherapy for the neoadjuvant treatment of patients with HER2-positive locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive).
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5.
Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Patients with HER2-positivea locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive) Subgroups:
|
Intervention | Pertuzumab 840 mg IV loading dose (over 60 minutes) followed by maintenance dose of 420 mg every 3 weeks by IV infusion (over 30 to 60 minutes), as part of a neoadjuvant chemotherapy treatment regimen outlined subsequentlyb |
Comparators | Trastuzumab plus chemotherapy |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; FEC = 5-fluorouracil and epirubicin plus cyclophosphamide; HER, HER2, HER3, HER4 = human epidermal growth factor receptor 2, 3, or 4; IHC = immunohistochemistry; pCR = pathologic complete response; RCT = randomized controlled trial; SAE = serious adverse event; TCH = docetaxel and carboplatin plus trastuzumab; WDAE = withdrawal due to adverse event.
Note: Patients who start pertuzumab and trastuzumab in the neoadjuvant setting may, at the discretion of the physician, continue to receive adjuvant pertuzumab and trastuzumab to complete 1 year of treatment.
aHER2-positive tumour status is defined as a score of 3 or higher by IHC or a ratio of ≥ 2.0 by in situ hybridization assessed by a validated test.
bBased on the Health Canada product monograph, pertuzumab should be administered every 3 weeks for 3 to 6 cycles as part of 1 of the following treatment regimens for early breast cancer:
Four pre-operative cycles of pertuzumab in combination with trastuzumab and docetaxel every 3 weeks (75 mg/m2 with the option to escalate to 100 mg/m2 at physician discretion if the initial dose is well tolerated), followed by 3 post-operative cycles of FEC (F: 600 mg/m2; E: 90 mg/m2; C: 600 mg/m2) every 3 weeks, as given in NEOSPHERE.
Three or 4 pre-operative cycles of FEC (F: 500 mg/m2; E: 100 mg/m2; C: 600 mg/m2) alone, every 3 weeks, followed by 3 or 4 pre-operative cycles of pertuzumab in combination with docetaxel (75 mg/m2 with the option to escalate to 100 mg/m2 at physician discretion if the initial dose is well tolerated) and trastuzumab every 3 weeks, as given in TRYPHAENA and BERENICE, respectively.
Six pre-operative cycles of pertuzumab in combination with docetaxel (75 mg/m2; escalation of docetaxel above 75 mg/m2 is not recommended), carboplatin (area under the curve 6), and trastuzumab (TCH) every 3 weeks, as given in TRYPHAENA.
Four pre-operative cycles of dose-dense doxorubicin and cyclophosphamide (60 mg/m2 doxorubicin, 600 mg/m2 cyclophosphamide) every 2 weeks followed by 4 pre-operative cycles of pertuzumab in combination with trastuzumab every 3 weeks, and paclitaxel (80 mg/m2) every week for 12 weeks, as given in BERENICE.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) through Ovid and Embase (1974—) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Perjeta (pertuzumab) and Herceptin (trastuzumab). Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
Methodological filters were applied to limit retrieval to RCTs or controlled clinical trials. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on May 12, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on Sep 8, 2021.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist. Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency [EMA]). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 4 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6 through to Table 9. A list of excluded studies is presented in Appendix 2.
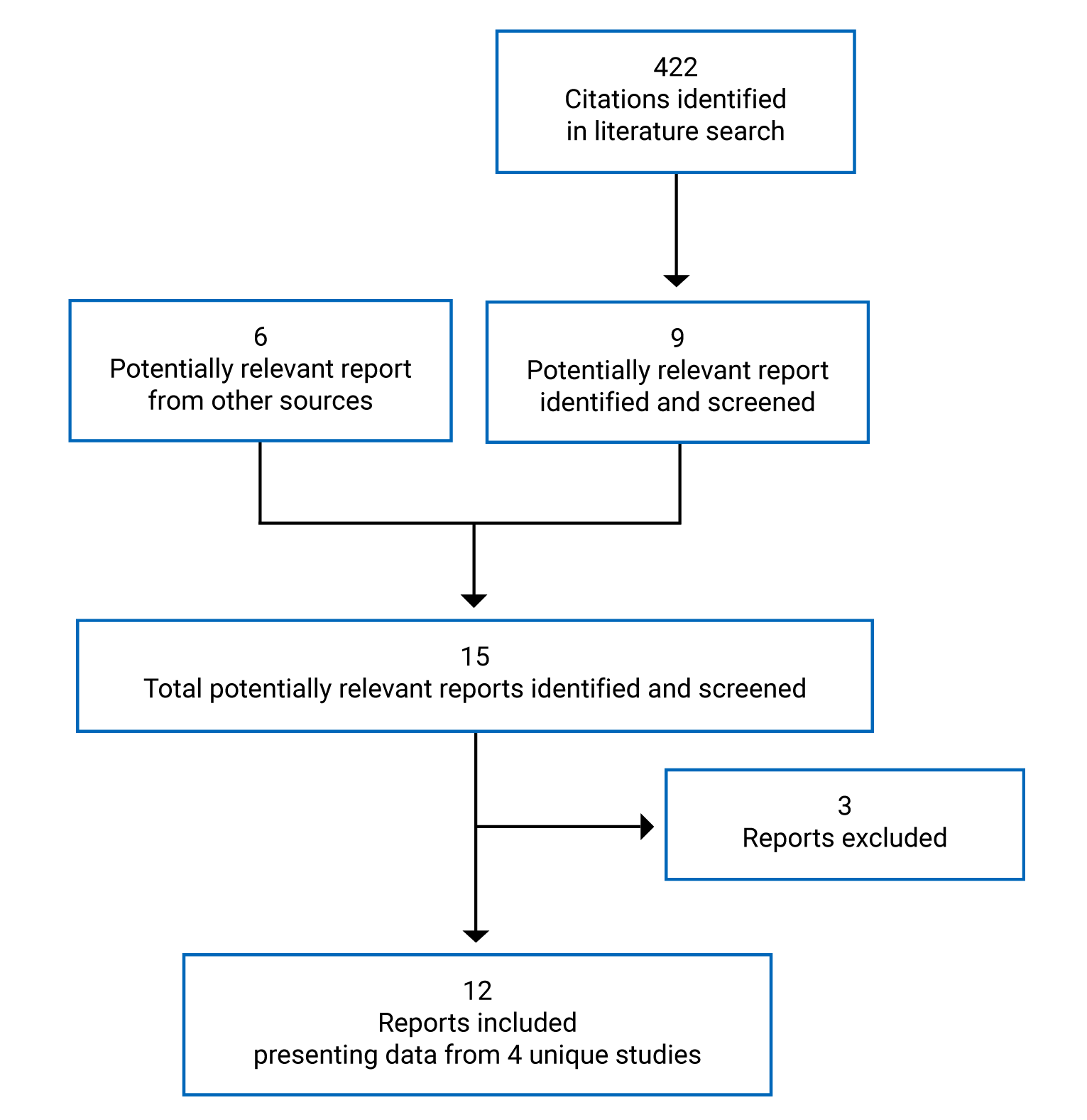
Table 6: Details of Included Studies — NEOSPHERE
Detail | Description |
|---|---|
Designs and populations | |
Study design | OL RCT (phase II) |
Locations | 59 sites; 16 countries in North America (Canada, Mexico), Europe, South America, and Asia |
Study period | First patient enrolled: December 17, 2007 Primary analysis data cut-off: December 22, 2009 |
Randomized (N) | N = 417 |
Inclusion criteria |
|
Exclusion criteria |
|
Intervention | Neoadjuvant phase:
Adjuvant phase:
|
Comparator(s) | Neoadjuvant phase:
Adjuvant phase:
|
Phase | |
Screening | 28 days |
Treatment |
|
Follow-up |
|
Outcomes | |
Primary end point | pCR rate |
Other end points | Secondary:
Safety:
|
Notes | |
Publications | |
Afib = atrial fibrillation; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; BC = breast cancer; BCC = basal cell carcinoma; CHF = congestive heart failure; DBP = diastolic blood pressure; DM = diabetes mellitus; ECOG = Eastern Cooperative Oncology Group; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; Hb = hemoglobin; HBV = hepatitis B virus; HCV = hepatitis C virus; HER2+ = human epidermal growth factor receptor 2–positive; LVEF = left ventricular ejection fraction; MI = myocardial infarction; MUGA = multiple-gated acquisition; NYHA = New York Heart Association; OL = open label; pCR = pathologic complete response; RCT = open-label randomized controlled trial; SBP = systolic blood pressure; SVT = supraventricular tachycardia; ULN = upper limit of normal.
Source: Clinical Study Report for NEOSPHERE.4
Table 7: Details of Included Studies — PEONY
Detail | Description |
|---|---|
Designs and populations | |
Study design | DB RCT (phase III) |
Locations | 23 sites (China, Korea, Thailand, Taiwan) |
Study period | First patient randomized: March 14, 2016 (ongoing) Primary analysis data cut-off: October 23, 2017 |
Randomized (N) | N = 329 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention |
|
Comparator(s) |
|
Duration | |
Phase | |
Screening | 28 days |
Treatment |
|
Follow-up | Adjuvant phase: Follow-up (every 3 months for the first year and every 6 months thereafter) continued until disease progression or recurrence or until 5 years after randomization of the last patient, whichever comes first |
Outcomes | |
Primary end point | tpCR rate (assessed by IRC) |
Other end points | Secondary:
Exploratory:
|
Notes | |
Publications | Shao (2020)16 |
AV = atrioventricular; BC = breast cancer; bpm = beats per minute; bpCR = breast pathologic complete response; DBP = diastolic blood pressure; DB = double blind; ECG = electrocardiogram; ECHO = echocardiography; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; HER2+ = human epidermal growth factor receptor 2–positive; HTN = hypertension; IHC = immunohistochemistry; IRC = independent review committee; ISH = in situ hybridization; LVEF = left ventricular ejection fraction; NMSC = non-melanoma skin cancer; PgR = progesterone receptor; RCT = randomized controlled trial; SBP = systolic blood pressure; tpCR = total pathologic complete response.
Source: Clinical Study Report for PEONY.5
Table 8: Details of Included Studies — TRYPHAENA
Detail | Description |
|---|---|
Designs and populations | |
Study design | Open-label RCT (phase II) |
Locations | North America (Canada, Mexico), Europe, South America, Asia, South Africa |
Study period | First patient enrolled: ||||||||||||||, 2009 Clinical cut-off: June 21, 2011 |
Randomized (N) | N = 225 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Neoadjuvant phase:
|
(continued) | Doses (by IV infusion):
Adjuvant phase:
|
Comparator(s) | No comparator |
Duration | |
Phase | |
Screening | 28 days |
Treatment |
|
Follow-up |
|
Outcomes | |
Primary end point |
|
Other end points | Secondary safety end points:
Key secondary efficacy end points:
Other secondary end points:
|
Notes | |
Publications | |
Afib = atrial fibrillation; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; BC = breast cancer; BCC = basal cell carcinoma; BM = bone marrow; CHF = congestive heart failure; CISH = chromogenic in situ hybridization; DBP = diastolic blood pressure; ECHO = echocardiogram; ECOG = Eastern Cooperative Oncology Group; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; FISH = fluorescent in situ hybridization; Hb = hemoglobin; HBV = hepatitis B virus; HCV = hepatitis C virus; HER2+ = human epidermal growth factor receptor 2–positive; HTN = hypertension; IHC = immunohistochemistry; LVEF = left ventricular ejection fraction; LVSD = left ventricular systolic dysfunction; MI = myocardial infarction; MUGA = multiple-gated acquisition; NYHA = New York Heart Association; pCR = pathologic complete response; SBP = systolic blood pressure; SCC = squamous cell carcinoma; SVT = supraventricular tachycardia; TCH = docetaxel + carboplatin + trastuzumab; ULN = upper limit of normal.
Source: Clinical Study Report for TRYPHAENA.6
Table 9: Details of Included Studies — BERENICE
Detail | Description |
|---|---|
Designs and populations | |
Study design | Non-RCT (phase II) |
Locations | 75 sites (Canada, Mexico, US, Europe) |
Study period | First patient enrolled: July | , 2014 Clinical cut-off: January 7, 2017 |
Randomized (N) | N = 401 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Neoadjuvant phase:
Doses (by IV infusion):
Adjuvant phase:
|
Comparator(s) | No comparator |
Duration | |
Phase | |
Screening | 28 days |
Treatment |
|
Follow-up |
|
Outcomes | |
Primary end point |
|
Other end points | Secondary safety:
Secondary efficacy:
|
(continued) | Exploratory:
|
Notes | |
Publications | Swain (2018)19 |
5-FU = 5-fluorouracil; AFib = atrial fibrillation; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; BC = breast cancer; BCC = basal cell carcinoma; BM = bone marrow; bpCR = breast pathologic complete response; CHF = congestive heart failure; CISH = chromogenic in situ hybridization; CR = complete response; DBP = diastolic blood pressure; ECOG = Eastern Cooperative Oncology Group; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; FISH = fluorescent in situ hybridization; HBV = hepatitis B virus; HCV = hepatitis C virus; HER2+ = human epidermal growth factor receptor 2 positive; HTN = hypertension; IHC = immunohistochemistry; ISH = in situ hybridization; LVEF = left ventricular ejection fraction; MI = myocardial infarction; MUGA = multiple-gated acquisition; NYHA = New York Heart Association; pCR = pathologic complete response; q.2.w. = every 2 weeks; q.3.w. = every 3 weeks; RCT = randomized controlled trial; SBP = systolic blood pressure; SCC = squamous cell carcinoma; SVT = supraventricular tachycardia; ULN = upper limit of normal.
Source: Clinical Study Report for BERENICE.7
Description of Studies
NEOSPHERE was an open-label multinational RCT (5 Canadian sites) whose objective was to make a preliminary assessment of the efficacy of neoadjuvant treatment with trastuzumab plus docetaxel (N = 107) compared with pertuzumab and trastuzumab plus docetaxel (N = 107), or compared with pertuzumab and trastuzumab (N = 107), and to compare pertuzumab plus docetaxel (N = 96) with pertuzumab and trastuzumab plus docetaxel in patients with T2 to T4d HER2-positive breast cancer, based on pCR (Figure 2). The treatment comparison relevant to this review is pertuzumab and trastuzumab plus docetaxel compared with trastuzumab plus docetaxel.
In NEOSPHERE, randomization was conducted 1:1:1:1 by an interactive web and voice response system (IxRS) and was stratified by breast cancer type (operable, locally advanced, or inflammatory) and by hormone receptor status. Arm D was added as a protocol amendment; therefore, there were fewer patients enrolled into this treatment group. The study consisted of a 12-week, 4-cycle neoadjuvant phase followed by surgery, and then an adjuvant phase where patients received FEC for 3 cycles and trastuzumab for 1 year. The treatment group that did not receive docetaxel in the neoadjuvant phase (Arm C) received docetaxel for 4 cycles, then 3 cycles of FEC, in addition to trastuzumab for 1 year. The primary outcome of the trial was evaluated when all patients had received 4 cycles of neoadjuvant treatment and had undergone surgery or had withdrawn from the study.
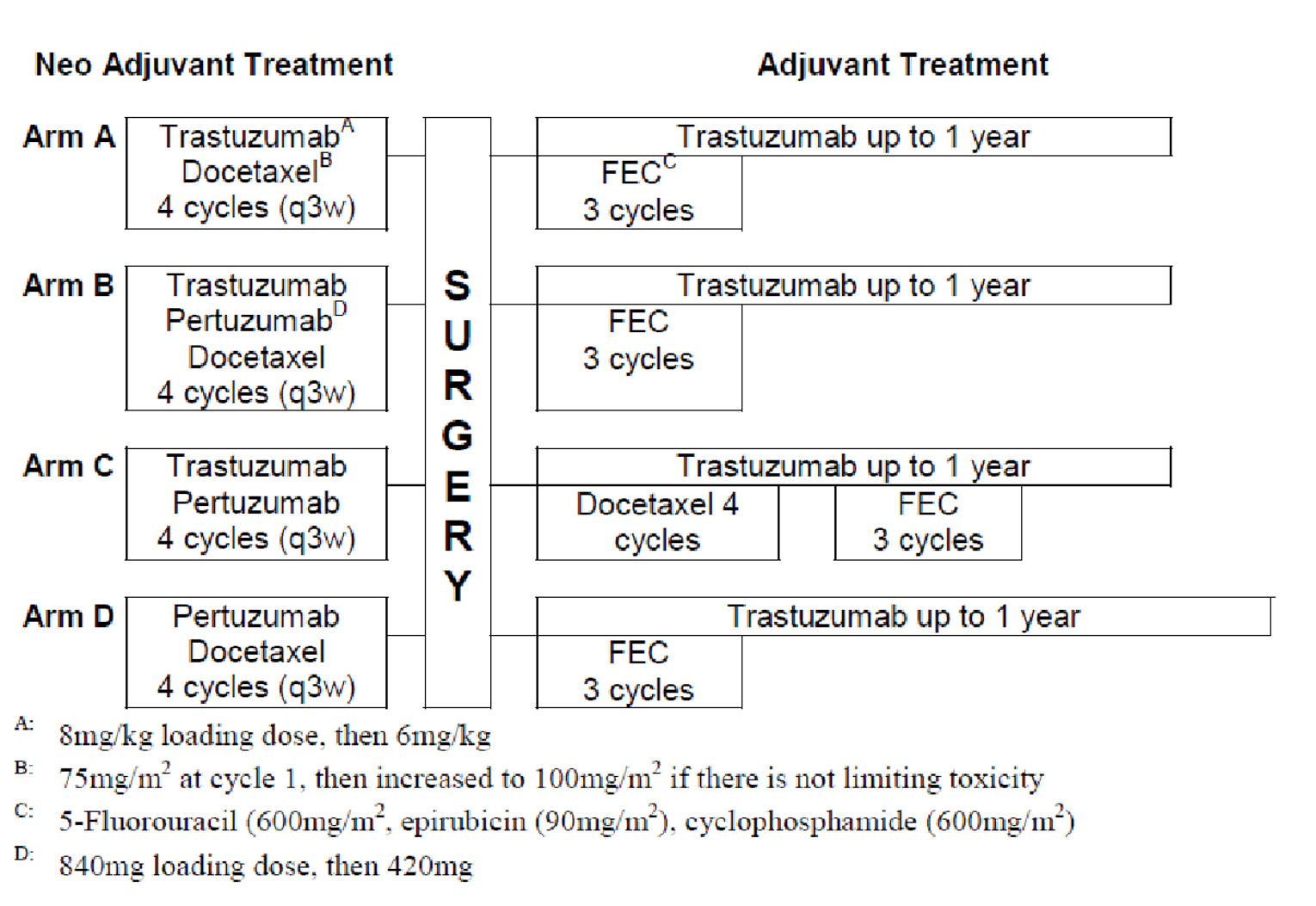
FEC = 5-fluorouracil and epirubicin plus cyclophosphamide; q3w = every 3 weeks.
Source: Clinical Study Report for NEOSPHERE.4
PEONY was a double-blind RCT conducted entirely in Asia whose primary objective was to evaluate the efficacy of pertuzumab and trastuzumab plus docetaxel (N = 219) compared with placebo plus trastuzumab plus docetaxel (N = 110), in a neoadjuvant setting before surgery, in patients with early-stage or locally advanced HER2-positive breast cancer. Other efficacy outcomes were to be assessed in the final analysis of the study 5 years after randomization of the last patient. Randomization was carried out 2:1 using IxRS, stratified by disease category (early-stage or locally advanced breast cancer) and hormone receptor status (estrogen receptor–positive and/or progesterone receptor–positive or negative for both). Patients had their pathologic response evaluated after completing 4 cycles of neoadjuvant therapy and surgery. Post surgery, patients received FEC for 3 cycles and then continued on HER2 therapy every 3 weeks until disease recurrence or unacceptable toxicity for up to 1 additional year.
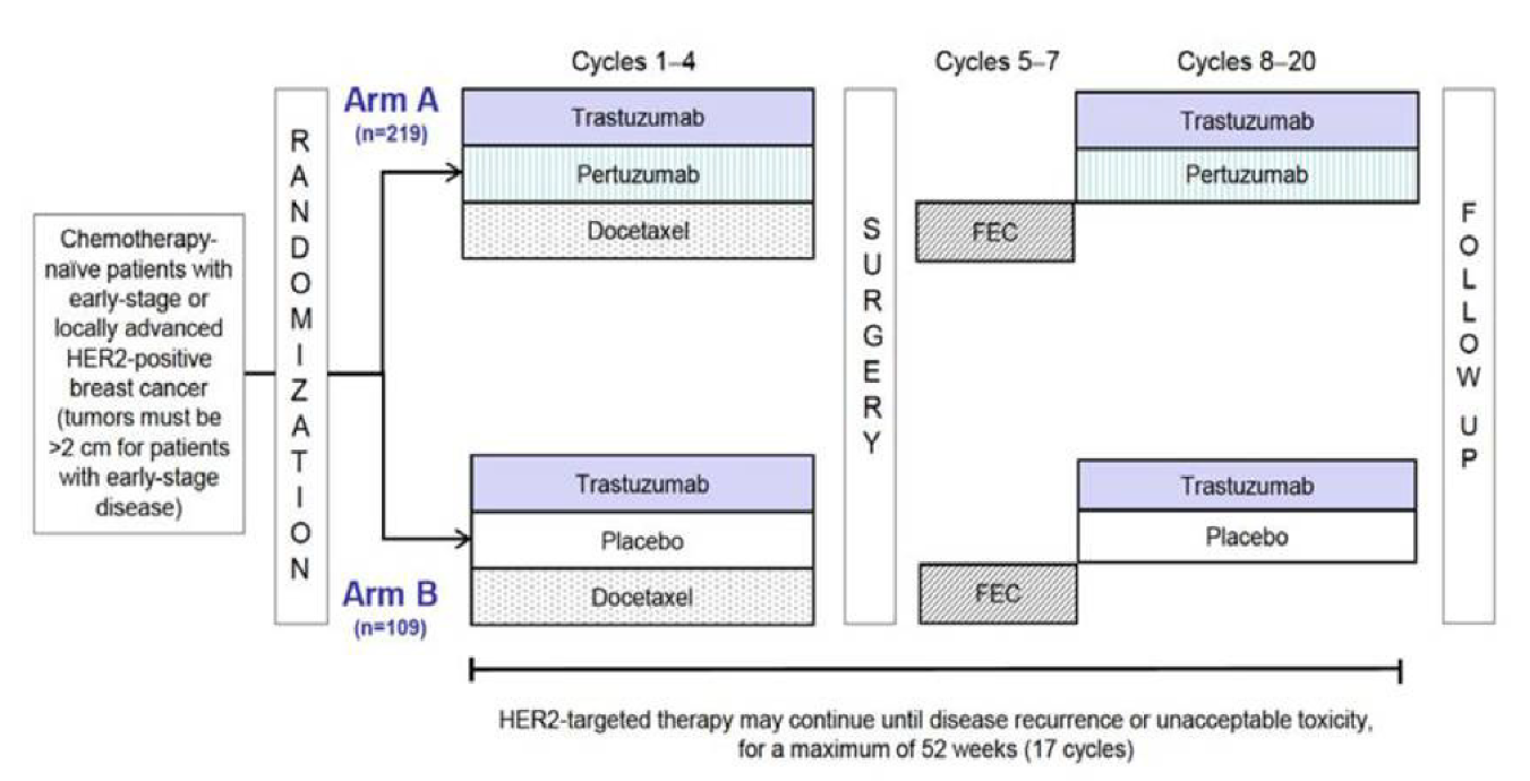
FEC = 5-fluorouracil and epirubicin plus cyclophosphamide; HER2 = human epidermal growth factor receptor 2.
Source: Clinical Study Report for PEONY.5
TRYPHAENA was an open-label, multinational study with 2 Canadian sites. TRYPHAENA (N = 225) and BERENICE (N = 400) were multinational studies with Canadian sites (3 sites in TRYPHAENA, 5 sites in BERENICE). In TRYPHAENA, randomization of 225 patients was carried out 1:1:1 by IxRS and was stratified by breast cancer type (operable, locally advanced, or inflammatory) and by hormone receptor status (hormone receptor–positive or –negative). In TRYPHAENA, patients received 6 cycles of neoadjuvant therapy with pertuzumab, trastuzumab, and an anthracycline or carboplatin-based chemotherapy, followed by surgery then trastuzumab for 1 year. TRYPHAENA did not have a comparator group, and the primary objective was to assess safety and tolerability of pertuzumab in combination with various regimens (Figure 4). After 5 years had elapsed since the randomization of the last patient, the study was completed on January 25, 2016.
BERENICE was an open-label, non-randomized study where patients were allocated 1:1 to 1 of 2 cohorts. The choice of neoadjuvant treatment was made by the investigator; however, only 1 cohort at a time was opened to enrolment at any given site; thus, investigators could not enrol patients into both cohorts simultaneously. In BERENICE, patients received 4 cycles (12 weeks) of neoadjuvant therapy, followed by surgery, then 13 cycles of adjuvant therapy with pertuzumab and trastuzumab, for a total of 17 cycles. BERENICE did not have a comparator group, and the primary objective was to assess the safety and tolerability of pertuzumab in combination with various regimens (Figure 5). BERENICE is ongoing, and the study is planned to end 5 years after enrolment of the last patient, or when all patients have died or the trial is terminated by the sponsor, whichever comes earlier. It is expected that the study will last approximately 6.5 years, including time on treatment of approximately 1 year and follow-up for cardiac safety and efficacy for an additional 4 years.
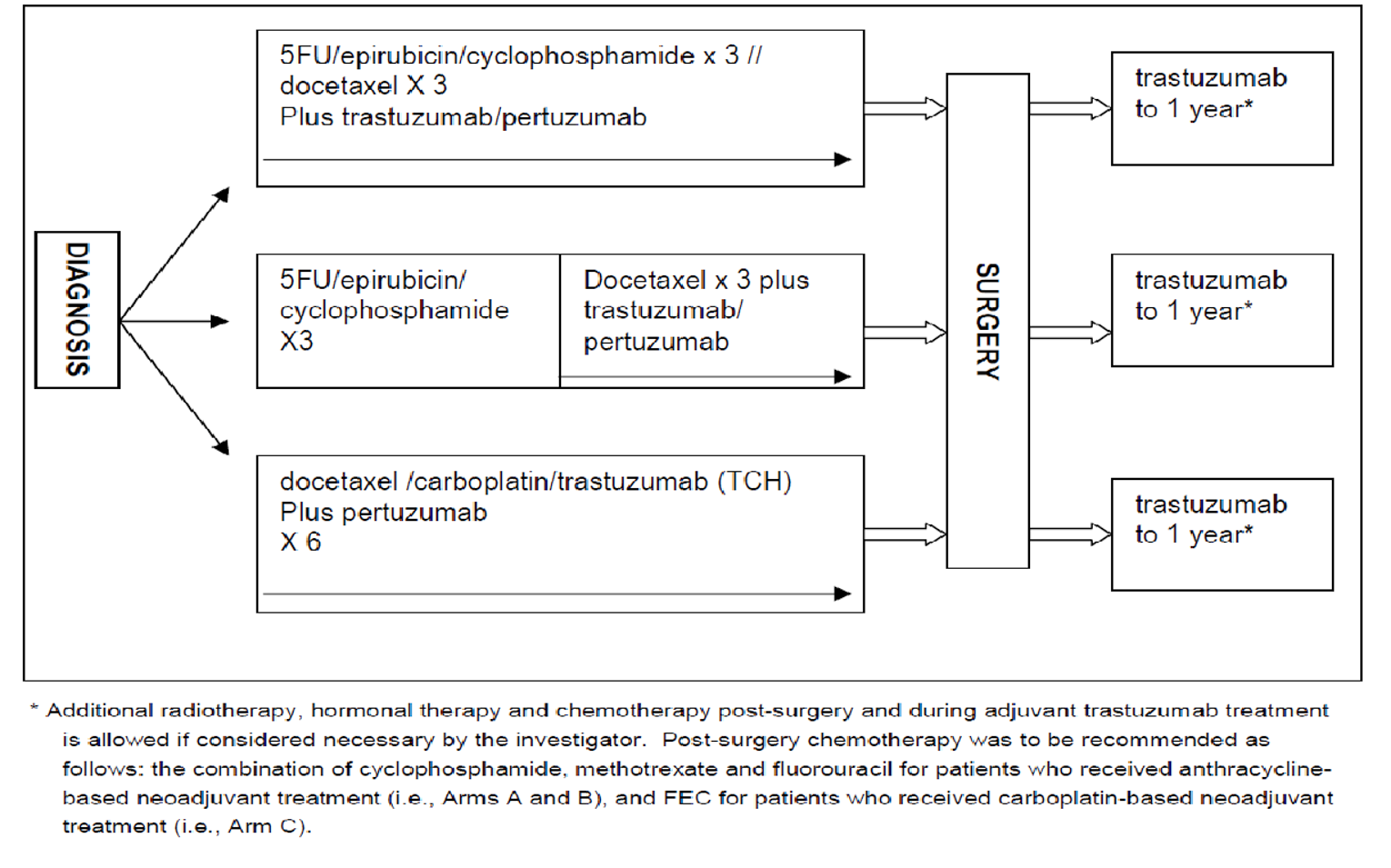
5FU = 5-fluorouracil; FEC = 5-fluorouracil and epirubicin plus cyclophosphamide; TCH = docetaxel and carboplatin plus trastuzumab.
Source: Clinical Study Report for TRYPHAENA.6
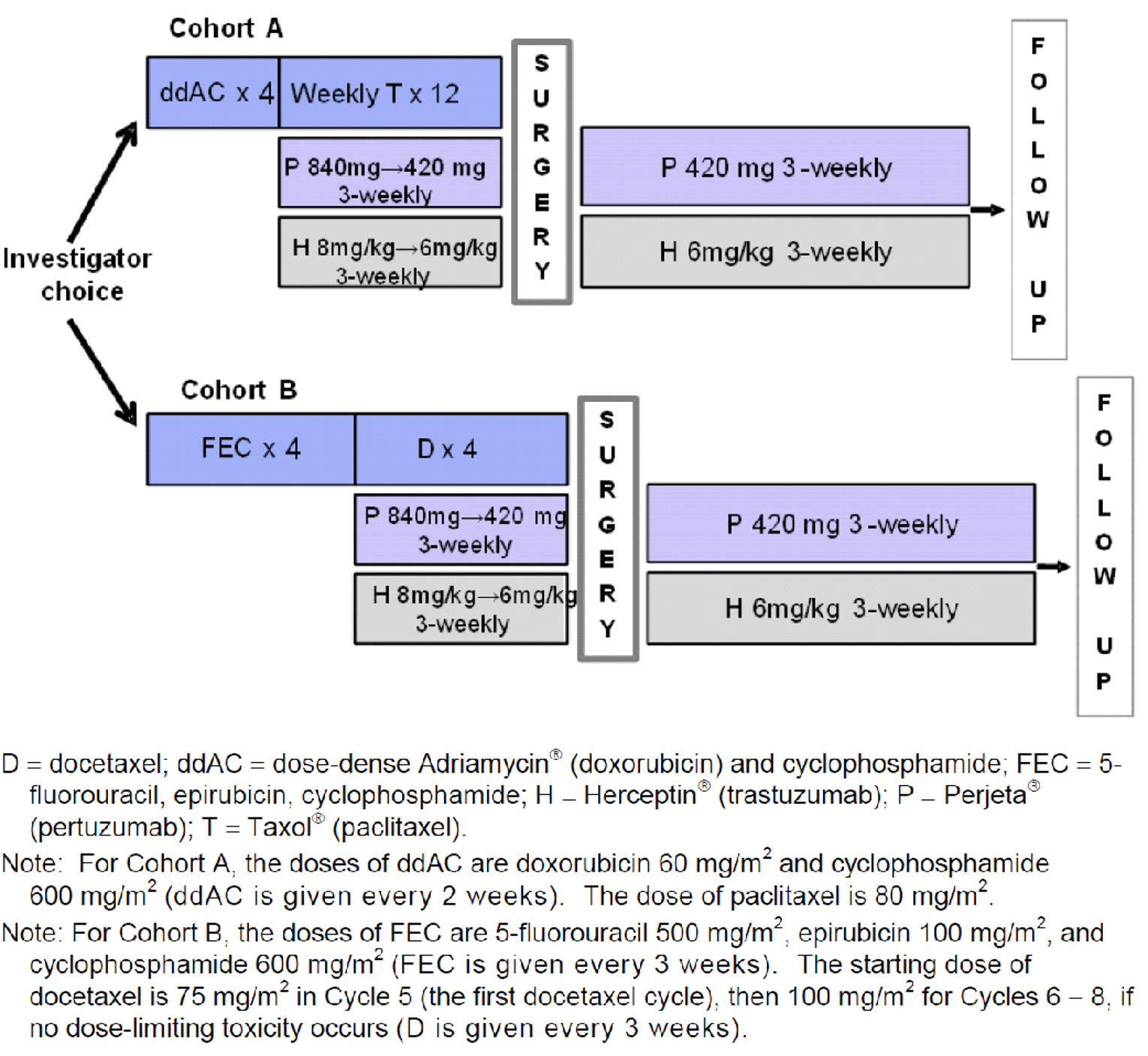
Populations
Inclusion and Exclusion Criteria
NEOSPHERE included females with locally advanced, inflammatory, or early-stage unilateral invasive breast cancer. The primary tumour had to be greater than 2 cm in diameter and confirmed to be HER2-positive, and patients were to have an ECOG performance status of 0 or 1. PEONY did not require patients to be female (but all were); otherwise, PEONY and NEOSPHERE had similar inclusion criteria, stipulating that patients had to have early-stage or locally advanced HER2-positive breast cancer with an ECOG performance status of 0 or 1. Both studies required an LVEF of 55% or greater at baseline.
Both studies excluded patients with stage IV metastatic disease, patients who had received prior anti-cancer therapy or radiotherapy for any malignancy, and patients with other malignancies (excluding carcinoma in situ of the cervix and basal cell carcinoma). PEONY excluded patients with inflammatory breast cancer, but NEOSPHERE did not. Patients with serious cardiac illness were also excluded from both studies.
The inclusion and exclusion criteria for TRYPHAENA and BERENICE did not differ markedly from that of NEOSPHERE and PEONY. BERENICE was the only study of the 4 included studies that included patients with a primary tumour that was less than 2 cm in diameter as long as it was greater than 5 mm and the cancer was node-positive.
Baseline Characteristics
Patients in NEOSPHERE and PEONY were about 50 years old at baseline (in NEOSPHERE, the mean age was 49.8 years, standard deviation [SD] 10.0 years; in PEONY, it was 48.8 years, SD 9.5 years). The majority of patients (approximately 70%) were White in NEOSPHERE, and all patients were Asian in PEONY. The mean age was similar in TRYPHAENA (50.2 years, SD 10.9) and BERENICE (49.6 years, SD 11.6), and a higher percentage of patients were White (approximately 75% to 85%) compared with NEOSPHERE. The majority of patients (nearly 90%) in NEOSPHERE, PEONY, and TRYPHAENA had an ECOG performance status of 0; the rest had a status of 1. The ECOG performance status of patients at baseline was not reported in BERENICE. Approximately half (47% in NEOSPHERE, 51% in PEONY, 51% in TRYPHAENA) of patients were either estrogen or progesterone receptor–positive while, in BERENICE, about 2-thirds of patients were estrogen or progesterone receptor–positive. The majority of patients in NEOSPHERE had breast cancer types that were either locally advanced (32%) or operable (61%) and the remainder had inflammatory breast cancer while, in PEONY, 70% were considered early stage and the remainder were considered locally advanced. Similar to NEOSPHERE, greater than 90% of patients in TRYPHAENA had breast cancer that was considered locally advanced or operable, and the remainder had inflammatory breast cancer. In BERENICE, baseline disease was not categorized as locally advanced and so forth; instead, disease category was reported using tumour, nodes, and metastases (TNM) staging. In BERENICE, the majority of patients had T2 (67%) tumours followed by T3 (20%); 47% had N1 disease, 8% had N2, 2% had N3, and 100% had disease classified as M0. The majority of patients in NEOSPHERE and TRYPHAENA had disease classified as T2N0M0 (NEOSPHERE: ||||||, TRYPHAENA: 31%) or T2N1M0 (NEOSPHERE: ||||||, TRYPHAENA: 33%). In PEONY, most patients had disease classified as T2 (67%), followed by T3 (22%), and most had lymph node–positive disease (76%).
In NEOSPHERE, there were some imbalances in baseline characteristics across treatment arms; the percentage of patients who were White in the pertuzumab plus docetaxel arm was 63.5% compared with 72.0% to 74.8% in the other 3 arms. ECOG performance status at baseline also appeared to be imbalanced across treatment arms in NEOSPHERE, as was histologic tumour grade (between 26.0% and 34.6% of patients had tumours that were classified as moderately differentiated). In PEONY, 70.8% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 64.5% in the trastuzumab plus docetaxel arm had T2 tumours, and most had positive lymph node status (73.1% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 80.9% of patients in the trastuzumab plus docetaxel arm). There were also imbalances between arms in TRYPHAENA for race (76.4% of patients were White in arm A versus 69.3% in arm B versus 84.2% in arm C, |||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, hormone receptor status (estrogen and/or progesterone receptor–positive: 53.4% versus 46.7% versus 51.9%, respectively), and disease category (20.5% had locally advanced disease versus 22.7% versus 31.2%, respectively). In BERENICE, there were imbalances between groups in hormone receptor status (44.2% of the patients in 1 group had disease that was estrogen and progesterone receptor–positive versus 36.8% in the other group).
Table 10: Summary of Baseline Characteristics in NEOSPHERE — ITT Population
Characteristic | Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 107 | Arm D pert + doce N = 96 |
|---|---|---|---|---|
Mean (SD) age, years | 49.6 (10.05) | 50.9 (8.94) | 49.7 (10.67) | 48.9 (10.50) |
Female gender, n (%) | 107 (100) | 107 (100) | 107 (100) | 96 (100) |
Race, n (%) | ||||
White | 77 (72.0) | 80 (74.8) | 79 (73.8) | 61 (63.5) |
Black or of African descent | 2 (1.9) | 0 | 1 (0.9) | 3 (3.1) |
Asian | 23 (21.5) | 25 (23.4) | 22 (20.6) | 25 (26.0) |
Other | 5 (4.7) | 2 (1.9) | 5 (4.7) | 7 (7.3) |
|||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||| | |||| | |||| | |||| | |||| |
ECOG performance status, n (%) | ||||
0 | 96 (89.7) | 100 (94.3) | 92 (86.0) | 80 (83.3) |
1 | 11 (10.3) | 6 (5.7) | 15 (14.0) | 16 (16.7) |
|||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||| | |||| | |||| | |||| | |||| |
Hormone receptor status, n (%) | ||||
ER and PgR negative | 57 (53.3) | 57 (53.3) | 55 (51.9) | 50 (52.1) |
ER and/or PgR positive | 50 (46.7) | 50 (46.7) | 51 (48.1) | 46 (47.9) |
|||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||| | |||| | |||| | |||| | |||| |
Breast cancer type, n (%) | ||||
Inflammatory | 10 (9.3) | 7 (6.5) | 7 (6.5) | 5 (5.2) |
Locally advanced | 32 (29.9) | 36 (33.6) | 35 (32.7) | 31 (32.3) |
Operable | 65 (60.7) | 64 (59.8) | 65 (60.7) | 60 (62.5) |
|||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||| | |||| | |||| | |||| | |||| |
|||||||||||| | |||| | |||| | |||| | |||| |
doce = docetaxel; ECOG = Eastern Cooperative Oncology Group; FISH = fluorescent in situ hybridization; HER2 = human epidermal growth factor receptor 2; IHC = immunohistochemistry; ITT = intention to treat; pert = pertuzumab; SD = standard deviation; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for NEOSPHERE.4
Table 11: Summary of Baseline Characteristics in PEONY — ITT Population
Characteristic | Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 |
|---|---|---|
Mean (SD) age, years | 48.4 (9.7) | 49.5 (9.1) |
Female gender, n (%) | 219 (100.0) | 110 (100.0) |
Race, n (%) | ||
Asian | 219 (100.0) | 110 (100.0) |
Menopausal status, n (%) | ||
Premenopausal | 132 (60.3) | 65 (59.1) |
Postmenopausal | 87 (39.7) | 45 (40.9) |
ECOG performance status, n (%) | ||
0 | 198 (90.4) | 97 (88.2) |
1 | 21 (9.6) | 13 (11.8) |
|||||||||||||||||||||||||||||||||||||||||| | ||
|||||||||||| | |||| | |||| |
Histologic subtype, n (%) | ||
Ductal | 203 (92.7) | 103 (93.6) |
Lobular | 4 (1.8) | 1 (0.9) |
Comedo | 0 | 1 (0.9) |
Other | 15 (6.8) | 7 (6.4) |
Hormone receptor status, n (%) | ||
ER and PgR negative | 105 (47.9) | 54 (49.1) |
ER and/or PgR positive | 114 (52.1) | 56 (50.9) |
Disease category, n (%) | ||
Early stage | 152 (69.4) | 77 (70.0) |
Locally advanced | 67 (30.6) | 33 (30.0) |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | ||
|||||||||||| | |||| | |||| |
|||||||||||| | |||| | |||| |
Primary tumour stage, n (%) | ||
T2 | 155 (70.8) | 71 (64.5) |
T3 | 45 (20.5) | 29 (26.4) |
T4 | 19 (8.7) | 10 (9.1) |
Lymph node status, n (%) | ||
Positive | 160 (73.1) | 89 (80.9) |
Negative | 59 (26.9) | 21 (19.1) |
chemo = chemotherapy; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; FISH = fluorescent in situ hybridization; HER2 = human epidermal growth factor receptor 2; IHC = immunohistochemistry; pert = pertuzumab; PgR = progesterone receptor; PLA = placebo; SD = standard deviation; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for PEONY.5
Table 12: Summary of Baseline Characteristics in TRYPHAENA — ITT Population
Characteristic | Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 |
|---|---|---|---|
Mean (SD) age, years | 49.4 (11.41) | 50.5 (10.70) | 50.5 (10.62) |
Female, n (%) | 72 (100) | 75 (100) | 76 (100) |
Race, n (%) | |||
White | 55 (76.4) | 52 (69.3) | 64 (84.2) |
Black or of African descent | 4 (5.6) | 3 (4.0) | 2 (2.6) |
Asian | 12 (16.7) | 18 (24.0) | 10 (13.2) |
Other | 1 (1.4) | 2 (2.7) | — |
|||||||||||||||||||||||||||||||| | |||
|||||||||||||||| | |||| | |||| | |||| |
ECOG performance status, n (%) | |||
0 | 65 (91.5) | 66 (88.0) | 67 (88.2) |
1 | 6 (8.5) | 9 (12.0) | 9 (11.8) |
|||||||||||||||||||||||||||||||| | |||
|||||| | |||| | |||| | |||| |
|||||| | |||| | |||| | |||| |
ER and/or PgR status, n (%) | |||
Negative | 34 (46.6) | 40 (53.3) | 37 (48.1) |
Positive | 39 (53.4) | 35 (46.7) | 40 (51.9) |
Disease category, n (%) | |||
Inflammatory | 5 (6.8) | 4 (5.3) | 4 (5.2) |
Locally advanced | 15 (20.5) | 17 (22.7) | 24 (31.2) |
Operable | 53 (72.6) | 54 (72.0) | 49 (63.6) |
Patients with operable BC, n (%) | |||
T2N0M0 | 9 (17.0) | 20 (37.0) | 19 (38.8) |
T2N1M0 | 18 (34.0) | 15 (27.8) | 19 (38.8) |
T3N0M0 | 6 (11.3) | 5 (9.3) | 4 (8.2) |
T3N1M0 | 20 (37.7) | 14 (25.9) | 7 (14.3) |
BC = breast cancer; doce = docetaxel; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; pert = pertuzumab; ITT = intention to treat; PgR = progesterone receptor; SD = standard deviation; TCH = docetaxel + carboplatin + trastuzumab; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for TRYPHAENA.6
Table 13: Summary of Baseline Characteristics in BERENICE — ITT Population
Characteristic | Cohort A ddAC then pacli + pert + trast N = 199 | Cohort B FEC then doce + pert + trast N = 201 |
|---|---|---|
Mean (SD) age, years | 49.8 (11.7) | 49.5 (11.5) |
Female, n (%) | 199 (100.0) | 200 (99.5) |
Race, n (%) | ||
White | 169 (84.9) | 163 (81.1) |
Black | 11 (5.5) | 0 |
Asian | 6 (3.0) | 4 (2.0) |
Other | 13 (6.5) | 34 (16.9) |
|||||||||||||||||||||||||||||||| | ||
|||||||||||||||||| | |||||| | |||||| |
|||||||||||||||||||||| | |||||| | |||||| |
Histological tumour grade, n (%) | ||
GX | 3 (1.5) | 9 (4.5) |
G1 | 4 (2.0) | 2 (1.0) |
G2 | 67 (33.8) | 56 (27.9) |
G3 | 108 (54.5) | 106 (52.7) |
Unknown | 16 (8.1) | 28 (13.9) |
Histologic subtype, n (%) | ||
Ductal | 171 (85.9) | 176 (87.6) |
Lobular | 9 (4.5) | 4 (2.0) |
Medullary | 0 | 0 |
Mucinous | 1 (0.5) | 0 |
Comedo | 4 (2.0) | 0 |
Tubular | 2 (1.0) | 0 |
NOS | 14 (7.0) | 19 (9.5) |
Other | 8 (4.0) | 8 (4.0) |
Hormone receptor status, n (%) | ||
Positive | 128 (64.3) | 124 (61.7) |
Negative | 65 (32.7) | 75 (37.3) |
Unknown | 6 (3.0) | 2 (1.0) |
|||||||||||||||||||||||| | ||
|||||||||||| | |||||| | |||||| |
|||||||||||| | |||||| | |||||| |
Primary tumour stage, n (%) | ||
TX | 0 | 1 (0.5) |
T0 | 1 (0.5) | 0 |
T1 | 18 (9.0) | 12 (6.0) |
T2 | 138 (69.3) | 130 (64.7) |
T3 | 33 (16.6) | 45 (22.4) |
T4 | 9 (4.5) | 13 (6.5) |
Regional lymph node stage, n (%) | ||
NX | 8 (4.0) | 9 (4.5) |
N0 | 80 (40.2) | 74 (36.8) |
N1 | 92 (46.2) | 98 (48.8) |
N2 | 16 (8.0) | 15 (7.5) |
N3 | 3 (1.5) | 5 (2.5) |
Distant metastasis, n (%) | ||
M0 | 199 (100.0) | 201 (100.0) |
ddAC = dose-dense doxorubicin and cyclophosphamide; doce = docetaxel; ECOG = Eastern Cooperative Oncology Group; ER = estrogen receptor; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; FISH = fluorescent in situ hybridization; HER2 = human epidermal growth factor receptor 2; IHC = immunohistochemistry; NOS = not otherwise specified; pacli = paclitaxel; pert = pertuzumab; PgR = progesterone receptor; PS = performance status; SD = standard deviation; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for BERENICE.7
Interventions
In NEOSPHERE and the other studies, pertuzumab was administered on day 1 of cycle 1 as an IV infusion at a loading dose of 840 mg. On day 22 (3 weeks after the first dose) and every 3 weeks thereafter, pertuzumab was administered as an IV infusion at a dose of 420 mg. The initial dose of pertuzumab was administered over 60 (± 10) minutes and patients were observed for a further 60 minutes. The infusion was slowed or interrupted if the patient experienced fever, chills, or other infusion-associated symptoms. If the infusion was well tolerated, subsequent doses were administered over 30 (± 10) minutes and patients observed for a further 60 minutes. All infusion-associated symptoms had to be resolved either before chemotherapy was given or the patient was discharged. Trastuzumab was administered on day 1 of cycle 1 as an IV infusion at a loading dose of 8 mg/kg. On day 22 (3 weeks after the first dose), and every 3 weeks thereafter, trastuzumab was administered as an IV infusion at 6 mg/kg. The initial dose of trastuzumab was administered over 90 (± 10) minutes and patients were observed for at least 30 minutes from the end of the infusion for infusion-associated symptoms. Interruption or slowing of the infusion was used to control symptoms, which could be resumed if symptoms abated. If the infusion was well tolerated, subsequent infusions were administered over 30 (± 10) minutes and patients were observed for a further 30 minutes. All infusion-associated symptoms had to be resolved either before further study treatment was given or the patient was discharged. Docetaxel was administered at 75 mg/m2 as an IV infusion over 60 (± 10) minutes, after the trastuzumab or pertuzumab infusion observation period. From day 22 onward (3 weeks after the first dose), docetaxel was escalated in the subsequent cycle(s) up to 100 mg/m2 if no limiting toxicity was experienced. In the adjuvant phase, patients exposed to docetaxel in the neoadjuvant phase received trastuzumab 6 mg/kg IV followed by FEC on day 1 and every 3 weeks thereafter for cycles 5 to 7, with each cycle lasting 21 days beginning 2 weeks after surgery. Trastuzumab monotherapy was administered for cycles 8 to 17, at the 6 mg/kg IV dose, every 3 weeks. In the arm that did not receive docetaxel in the neoadjuvant phase, trastuzumab 6 mg/kg IV was administered, followed by docetaxel 75 mg/m2 IV at cycle 5, and docetaxel was escalated to 100 mg/m2 for cycles 6 to 8 (as long as there were no dose-limiting toxicities). For cycles 9 to 11, 6 mg/kg IV of trastuzumab followed by FEC was administered on day 1 and every 3 weeks thereafter, and trastuzumab 6 mg/kg IV monotherapy was continued every 3 weeks for cycles 12 to 17.
In PEONY, the study treatment was administered in 3-week cycles. Patients were treated with trastuzumab, pertuzumab, and docetaxel (arm A) or trastuzumab, placebo, and docetaxel (arm B) for 4 cycles in the neoadjuvant setting. Matched placebo contained the formulation but not the antibody itself. After surgery, patients received 3 cycles of FEC chemotherapy. Patients then continued HER2-targeted therapy with trastuzumab and pertuzumab (arm A) or trastuzumab and placebo (arm B) for up to 1 year in total (17 cycles, including 4 cycles in the neoadjuvant setting) or until disease recurrence, as assessed by the investigator, or unacceptable toxicity.
For a summary of regimens used in TRYPHAENA, see Figure 4. This study also featured a neoadjuvant phase followed by surgery and then an adjuvant phase, and combined pertuzumab with various chemotherapy regimens at the doses described previously. In the neoadjuvant phase of TRYPHAENA, pertuzumab and trastuzumab were given either concomitantly or sequentially with anthracycline-based chemotherapy or concomitantly with non-anthracycline-based chemotherapy. Neoadjuvant treatment was for 6 cycles in TRYPHAENA, which was longer than the 4 cycles in NEOSPHERE and PEONY.
For a summary of regimens used in BERENICE, see Figure 5. In BERENICE, the patients in arm A received doxorubicin for 4 cycles before receiving 4 cycles of pertuzumab plus trastuzumab combined with paclitaxel, while patients in arm B received 4 cycles of FEC followed by 4 cycles of pertuzumab plus trastuzumab combined with docetaxel. Adjuvant therapy in TRYPHAENA consisted of trastuzumab 6 mg/kg IV every 3 weeks and continued for a maximum of 1 year, while adjuvant therapy in BERENICE consisted of 13 cycles of pertuzumab plus trastuzumab. Additional radiotherapy, hormonal therapy, and chemotherapy post surgery and during adjuvant trastuzumab was allowed at the discretion of the investigator in TRYPHAENA while, in BERENICE, additional radiotherapy and adjuvant hormonal therapy could be given as clinically indicated, according to the guidelines provided per protocol.
As for concomitant medications, in general, across the trials, all medications taken by the patient for concomitant diseases were allowed to continue during the study treatment period and were recorded on the electronic case report form.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 14. These end points are further summarized subsequently. A detailed discussion and critical appraisal of pCR as an outcome measure is provided in Appendix 4.
Table 14: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | NEOSPHERE | PEONY | TRYPHAENA | BERENICE |
|---|---|---|---|---|
Overall survivala | Not assessed | Secondary (includes adjuvant) | Exploratory | Exploratory |
Invasive disease–free survivala | Not assessed | Not assessed | Not assessed | Exploratory |
Event-free survivala | Not assessed | Secondary | Not assessed | Exploratory |
Progression-free survivala | Secondary | Secondary | Exploratory | Not assessed |
Disease-free survivala | Secondary | Secondary | Exploratory | Not assessed |
pCR | Primary | Primary | Exploratory | Exploratory |
Objective response | Secondary | Secondary | Exploratory | Exploratory |
Health-related QoL | Not assessed | Not assessed | Not assessed | Not assessed |
Symptoms | Not assessed | Not assessed | Not assessed | Not assessed |
Breast-conserving surgery rate | Secondary | Not assessed | Exploratory | Exploratory |
pCR = pathologic complete response; QoL = quality of life.
aThese outcomes are influenced by treatments received in the adjuvant phase.
Source: Clinical Study Report for NEOSPHERE,4 PEONY,5 TRYPHAENA,6 BERENICE.7
Overall Survival
OS was not assessed as an efficacy outcome in NEOSPHERE. In PEONY, patients were to be followed for survival every 3 months for the first year and every 6 months thereafter until disease progression or recurrence, or until 5 years after randomization of the last patient, whichever came first. OS was an exploratory outcome in TRYPHAENA and BERENICE.
Invasive Disease–Free Survival
Invasive DFS was not assessed in NEOSPHERE, PEONY, or TRYPHAENA; these data were not yet mature in BERENICE, according to the sponsor, and were therefore not reported.
Disease-Free Survival
DFS was a secondary outcome. It was defined in NEOSPHERE as the time from the first date of no disease (i.e., date of surgery) to the first documentation of progressive disease (PD) or death, while, in PEONY, it was until disease recurrence or death. In NEOSPHERE, PD was identified by the investigator in response to a question on the electronic case report form and was not based solely on Response Evaluation Criteria in Solid Tumors (RECIST) criteria, and additional clinical information could be considered in the assessment of PD. Any evidence of contralateral disease in situ was not considered PD. Patients who had surgery but did not achieve a pCR were censored at the date of surgery. Patients who withdrew from the study without documented progression and for whom there was evidence that evaluations were made were censored at the date of the last assessment when the patient was known to be disease-free. DFS was an exploratory outcome in TRYPHAENA and was defined as it was in NEOSPHERE and was not assessed in BERENICE. Across trials, DFS included the adjuvant phase of treatment.
Progression-Free Survival
PFS was a secondary outcome in NEOSPHERE and PEONY and was defined as the time from the date of randomization to the first documentation of PD or death. Any evidence of contralateral disease in situ was not considered as PD, although invasive contralateral disease was counted as PD in PEONY. Otherwise, PD in PEONY was defined as described previously. Patients who withdrew from the study without documented progression and for whom there was evidence that evaluations were made were censored at the date of the last assessment when the patient was known to be free from PD. Patients without post-baseline assessments but known to be alive were censored at the time of randomization. PFS was an exploratory outcome in TRYPHAENA, defined as the time from randomization to first documentation of PD or death, and PFS was not studied in BERENICE. Across the trials, PFS included the adjuvant phase of treatment.
Event-Free Survival
EFS was a secondary outcome in PEONY and was defined as the time from randomization to the first documentation of 1 of the following: death from any cause, disease recurrence, or disease progression (before surgery). PD was determined by the investigator using RECIST v1.1. Any evidence of contralateral disease in situ was not counted as PD.
Patients who did not have an event at the time of the analysis were censored as of the date they were last known to be alive and event-free. EFS was not studied in NEOSPHERE and was not assessed in TRYPHAENA. EFS was exploratory in BERENICE, defined as the time from enrolment to first occurrence of PD, relapse, or death. Across trials, EFS included the adjuvant phase of treatment.
Pathologic Complete Response
The primary outcome of NEOSPHERE and PEONY was pCR. In NEOSPHERE, pCR was assessed specifically in the breast; in PEONY, tpCR was assessed as the primary outcome and bpCR was assessed as a secondary outcome. In both studies, pCR was assessed post surgery, after patients had received 4 cycles of neoadjuvant therapy or after study withdrawal. In NEOSPHERE, pCR was defined as the absence of invasive neoplastic cells on microscopic examination of the surgical specimen (residual in situ disease was allowed). In PEONY, tpCR was defined as the absence of invasive neoplastic cells in the surgically resected breast specimen and all sampled ipsilateral nodes. In NEOSPHERE, all tumour responses were assessed locally and were not independently reviewed while, in PEONY, tumour responses were assessed both locally and centrally by an IRC. In PEONY, IRC assessments were used for the primary outcome and were conducted in a blinded manner consistent with FDA recommendations. In TRYPHAENA, pCR was assessed in the breast as a main efficacy outcome and was evaluated post surgery; in BERENICE, pCR was assessed in breast and nodes post surgery and was also a main efficacy outcome.
Objective Response
Clinical response, defined as achieving a CR or PR, was a secondary outcome in NEOSPHERE and PEONY. In NEOSPHERE, tumour burden was assessed at baseline using mammography and clinical breast exam after completion of all pre-operative treatment cycles. Any additional conventional assessment methods used by local practice were also collected (i.e., ultrasound, CT, MRI, X-ray). Clinical breast exam was also conducted after each treatment cycle in the neoadjuvant phase. In PEONY, the clinical response rate was defined as the proportion of patients who achieved a clinical response during cycles 1 to 4 (pre-surgery). If PD was suspected, then an unscheduled assessment was performed and, if confirmed, the patient was removed from study treatment and offered local standard of care, such as a second-line cytotoxic regimen, radiation, or surgery. For assessment of response, modifications to RECIST were employed in NEOSPHERE. For the primary lesion in the breast, the RECIST criteria were applied in terms of percentages, but the sum of lesions was not used; instead, only the size of the primary breast lesion was used to determine response. For overall response, the sizes would be summed only if the method of assessment was the same for all lesions (breast and nodes). For example, if a patient had a breast lesion assessed by mammogram and lymph nodes by ultrasound, then each would be summed only within that method of assessment. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| In PEONY, clinical response was required to be assessed by clinical breast exam, at each cycle between days 15 and 21, or on study day 1 of the next cycle, and by mammography at baseline and cycle 4. Clinical response was defined as CR, PR, stable disease, and PD, and was identified as per local practice based on RECIST criteria. Clinical response was an exploratory outcome in TRYPHAENA and BERENICE and was defined as patients achieving either a CR or PR at any time pre-surgery. In TRYPHAENA, clinical response was assessed as per local practice at each cycle, between days 15 and 21, or on study day 1 of the next cycle, and, in BERENICE, it was assessed before each new cycle by clinical exam, mammography, and/or other methods, as per local practice.
Breast-Conserving Surgery
The breast-conserving surgery rate was defined as the proportion of patients who achieved breast-conserving surgery out of the intention-to-treat (ITT) population without inflammatory breast cancer, as these patients received mastectomy irrespective of their response to neoadjuvant therapy. The breast-conserving surgery rate among patients in whom mastectomy was initially planned was a secondary outcome in NEOSPHERE and an exploratory outcome in TRYPHAENA. The breast-conserving surgery rate among all patients was an exploratory outcome in the BERENICE study and was not assessed in PEONY.
Symptoms and Health-Related Quality of Life
Symptoms and health-related quality of life were not assessed in any of the included studies.
Statistical Analysis
Primary Outcome(s) of the Studies
Power Calculation
With a sample size of 400 patients and a randomization ratio of 1:1:1:1, NEOSPHERE had 80% power to detect a 15% increase between each of the 3 primary comparisons between treatment arms at an overall alpha of 0.2. A pCR rate of 25% was expected in the trastuzumab plus docetaxel and the pertuzumab plus docetaxel arms, while a 40% pCR in the pertuzumab and trastuzumab plus docetaxel and pertuzumab and the trastuzumab arms was described as being “of clinical interest.” |||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||. PEONY assumed a tpCR of 20% in the trastuzumab plus docetaxel arm, an absolute increase of 15% in the pertuzumab and trastuzumab plus docetaxel group, and a 2-sided significance of 5% to arrive at 85% power for a sample of 328 patients and a 2:1 randomization ratio. The data source that informed the estimates for tpCR was not reported.
No formal hypothesis testing was planned in TRYPHAENA. The planned sample size was based on the primary safety end point (incidence of symptomatic cardiac events and clinically significant declines in LVEF during the neoadjuvant period), and approximately 75 patients were to be recruited into the study. The approximate expected pCR rates were 50% in arm A, 45% in arm B, and 40% in arm C. The data source informing the estimates for pCR was not reported. For the incidence of symptomatic left ventricular dysfunction, if the true underlying incidence was 3%, the probability of observing more than 5 events in a treatment arm was 0.025. No power calculation was provided in BERENICE.
Statistical Test or Model
The 3 comparisons between the treatment arms for the primary outcome in NEOSPHERE were considered to be of equal importance and were made using a Cochran-Mantel-Haenszel test, stratified by operable (T2 to T3, N0 to N1, M0), locally advanced (T2 to T3, N2 or N3, M0; T4a to T4c, any N, M0) and inflammatory (T4d, any N, M0) breast cancer and estrogen and/or progesterone positivity (either is positive versus both are negative) (Table 15). This primary analysis focused on the neoadjuvant phase. Secondary outcomes (best tumour response, clinical response rate, time to clinical response, proportion of patients with T2 to T3 tumours receiving breast-conserving surgery, PD) were calculated and summarized for descriptive purposes only.
In PEONY, for the primary outcome of tpCR rate (IRC assessed), a 2-sided Cochran-Mantel-Haenszel test was performed for assessment of tpCR, stratified by disease category (early stage, locally advanced) and hormone receptor status (estrogen and/or progesterone receptor–positive, or negative for both) (Table 15). An unadjusted Fisher exact test was also performed. Secondary outcomes were assessed in a manner similar to how the primary outcome was assessed; however, the P values were stated to be “descriptive only.”
All efficacy outcomes in TRYPHAENA and BERENICE were assessed descriptively; therefore, no formal statistical analyses were performed.
Data Imputation Methods
In both NEOSPHERE and PEONY, patients with missing or unevaluable pCR, tpCR, or bpCR assessments were considered nonresponders. Other outcomes were not assessed formally and, therefore, no sensitivity analyses appear to have been planned.
Subgroup Analyses
In NEOSPHERE, pre-specified subgroups of interest included age (< 65 versus ≥ 65), menopausal status (postmenopausal versus premenopausal), primary tumour stage (T2 versus T3 or larger), lymph node status (positive versus negative), histological subtype (ductal versus non-ductal, not including unknown), disease category at baseline (early stage versus locally advanced), hormone receptor status at baseline (positive for estrogen and/or progesterone receptor versus negative for both), HER2 subgroups defined as an IHC score of 3+ (regardless of FISH status), IHC score of 2+ and FISH-positive, or IHC score of 0 or 1+ and FISH-positive, and pathological tumour stage at surgery (T0 or tumour in situ [Tis] versus T1 or larger). Comparability between groups (tests for interaction) was not checked nor was multiplicity accounted for.
The pre-specified subgroups of interest in PEONY included age (< 65 versus ≥ 65), menopausal status, primary tumour stage at baseline (T2 versus T3 or larger), lymph node status at baseline (positive versus negative), histological subtype (ductal versus non-ductal, not including unknown values), disease stage (early stage versus locally advanced), hormone receptor status at baseline (estrogen and/or progesterone receptor–positive versus negative for both), HER2 subgroups defined as an IHC score of 3+ (regardless of FISH status), IHC score of 2+ and FISH-positive, and IHC score of 0 or 1+ and FISH-positive and pathologic tumour stage at surgery (T0 or Tis versus T1 or larger). In PEONY, subgroups were assessed for various biomarkers as a sensitivity analysis for the primary outcome and were tested using an unstratified instead of a stratified Cochran-Mantel-Haenszel test. No tests were performed for interactions, and no P values were reported.
Multiplicity
In NEOSPHERE, for the primary outcome, there were 3 between-group comparisons made during the study, and a Simes multiplicity adjustment was applied to the individual P values obtained at the end of the study to maintain the overall false-positive risk at 0.2. No multiplicity adjustment for the secondary outcomes assessed was described in PEONY, and the P values for secondary outcomes were noted as descriptive.
Sensitivity Analyses
In NEOSPHERE, a sensitivity analysis was performed where assessment of pCR was repeated, counting only patients who completed surgery and had a valid pCR assessment (Table 15). This was done to exclude early dropouts due to PD from the analysis. A sensitivity analysis was also performed to assess the impact of changes to the TNM codes that were made in protocol version C (see list of protocol amendments that follows). A cross tabulation of responses was performed for patients recruited under protocol B and protocol C, as well as a chi-square test to test whether the proportion of patients in each breast cancer type under protocol B was not significantly different under protocol B versus protocol C.
Protocol Amendments
There were 3 protocol amendments in NEOSPHERE:
Amendment 1 (December 4, 2007) added arm D to evaluate the efficacy of pertuzumab without trastuzumab and to update the hypothesis testing and analyses to reflect this change. Additionally, the number of patients in the study was increased from 180 to 400 and the number of study centres from between 45 to 55, to 100. Insulin-dependent diabetes mellitus was added as an exclusion criterion and the offset dosing schedule was clarified.
Amendment 2 (December 11, 2008) corrected the TNM categories used to classify patients’ disease (operable, locally advanced, or inflammatory cancer) for the stratification groups.
Amendment 3 (June 27, 2009) updated the definition of postmenopausal women, the contraceptive requirements for women of childbearing potential (as recommended by the Medicines and Health care products Regulatory Agency in accordance with the International Conference on Harmonization M3 guideline), and updated the pregnancy testing schedule. This amendment also clarified the clinical response definition.
There were 3 protocol amendments in PEONY:
Amendments 2 (August 5, 2014) and 3 (August 15, 2014) were made to provide clarity and consistency around protocol procedures, assessments, and analyses (e.g., clinical tumour assessments, safety reporting, contraception use).
Amendment 4 (December 6, 2016) changed the timing of the primary efficacy analysis. Originally, the analysis was to occur when all patients completed the treatment or discontinued visits (i.e., after completion of all adjuvant and neoadjuvant treatments). This was changed to occur when all patients who were eligible for surgery had completed surgery and been assessed for pathologic response (i.e., after surgical treatment following neoadjuvant therapy). This change is consistent with the globally accepted scientific conduct of neoadjuvant studies, according to the sponsor, and consistent with other pertuzumab neoadjuvant studies.
The total number of protocol amendments in TRYPHAENA was not reported but included changes to the timing of baseline mammography, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
|||||||||||||| protocol amendments in BERENICE, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| partners of male patients are not in scope), and discrepancies in the pathology manual and the schedule of assessments were corrected.
Table 15: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
NEOSPHERE | |||
pCR | CMH | Stratified by operable (T2 to T3, N0 to N1, M0), locally advanced (T2 to T3, N2 or N3, M0; T4a to T4c, any N, M0) and inflammatory (T4d, any N, M0) BC and estrogen and/or progesterone positivity (either is positive vs. both are negative) |
|
| Descriptive purposes only | NA | NA |
PEONY | |||
tpCR (IRC) | ● CMH, 2-sided ● Fisher exact test (unadjusted) | CMH stratified by disease category (early stage or locally advanced) and hormone receptor status (positive for ER and/or PgR or negative for both) |
|
bpCR rate (IRC) | Same as for primary | Same as primary, though P values were described as “descriptive only” | NA |
TRYPHAENA | |||
No formal hypothesis testing | NA | NA | NA |
BERENICE | |||
No formal hypothesis testing | NA | NA | NA |
BC = breast cancer; bpCR = breast pathologic complete response; CMH = Cochran-Mantel Haenszel; ER = estrogen receptor; IRC = independent review committee; ITT = intention to treat; NA = not applicable; pCR = pathologic complete response; PgR = progesterone receptor; tpCR = total pathologic complete response.
Source: Clinical Study Report for NEOSPHERE,4 PEONY,5 TRYPHAENA,6 BERENICE.7
Analysis Populations
In NEOSPHERE, PEONY, and TRYPHAENA, the ITT population included all randomized patients regardless whether they received study medication and, in BERENICE, the ITT included all enrolled patients regardless whether they received study medication. The analyses for all studies were conducted according to the randomized or assigned treatment group. The safety population in all studies included all patients who received at least 1 dose of the study drug and had at least 1 safety assessment performed at baseline. NEOSPHERE also identified a per-protocol population that excluded patients from the ITT population who had a major protocol violation before the adjuvant phase of the study. TRYPHAENA included a per-protocol population that had to fulfill all of the following criteria: received 3 cycles of study medication (neoadjuvant setting), received no other anti-cancer treatment (non-study medication or radiotherapy), and underwent surgery. This analysis occurred only in the per-protocol population and differed from the ITT population by 10% or greater.
Results
Patient Disposition
Study withdrawals were less than 10% across all treatment arms in each study. The percentage of patients in each study who withdrew from treatment was generally higher than those withdrawing from the study, except for PEONY. The percentage of screen failures was typically greater than 25%. Not all studies reported the reasons for screen failures; however, in the ones that did (TRYPHAENA and BERENICE), common reasons included HER2-negative disease, metastatic disease, and HER2 positivity not confirmed by central laboratory.
Table 16: Patient Disposition — NEOSPHERE
Disposition | Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 107 | Arm D pert + doce N = 96 |
|---|---|---|---|---|
Screened | 603 | |||
Randomized | 107 | 107 | 107 | 96 |
Randomized and treated | 106 | 106 | 107 | 94 |
Discontinued neoadjuvant treatment | 5 (4.7) | 4 (3.7) | 14 (13.0) | 6 (6.4) |
Adverse event | 0 | 0 | 2 (1.9) | 2 (2.1) |
Death | 1 (0.9) | 0 | 0 | 1 (1.0) |
Insufficient therapeutic response | 1 (0.9) | 0 | 7 (6.5) | 1 (1.0) |
Violation of selection criteria at entry | 2 (1.9) | 1 (0.9) | 1 (0.9) | 1 (1.0) |
Refused treatment | 1 (0.9) | 1 (0.9) | 4 (3.7) | 1 (1.0) |
Failure to return | 0 | 1 (0.9) | 0 | 0 |
Other | 0 | 1 (0.9) | 0 | 0 |
Withdrew from study during neoadjuvant period | 4 (3.7) | 2 (1.9) | 10 (9.3) | 3 (3.1) |
Surgery and valid pCR assessment | 102 (95.3) | 104 (97.2) | 96 (89.7) | 90 (93.8) |
Entered adjuvant treatment | 102 (95.3) | 103 (96.3) | 94 (87.9) | 88 (91.7) |
Withdrew from adjuvant treatment | 8 (7.5) | 5 (4.7) | 4 (3.7) | 14 (14.6) |
Withdrew from study during adjuvant phase | 2 (1.9) | 8 (7.5) | 1 (0.9) | 6 (6.3) |
Entered post-treatment follow-up phase | 102 (95.3) | 98 (91.6) | 98 (91.6) | 87 (91) |
ITT population | 107 (100) | 107 (100) | 107 (100) | 96 (100) |
Per protocol | 101 (94.4) | 105 (98.1) | 105 (98.1) | 91 (94.8) |
Safety | 107a | 107b | 108c | 94 |
Protocol deviations, n (%) | ||||
Number violating at least 1 inclusion criterion | 11 (10.3) | 11 (10.3) | 8 (7.5) | 8 (8.3) |
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
Number violating at least 1 exclusion criterion | 7 (6.5) | 14 (13.1) | 11 (10.3) | 7 (7.3) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
Number with at least 1 on-study violation | 11 (10.3) | 5 (4.7) | 7 (6.5) | 6 (6.3) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
doce = docetaxel; ITT = intention to treat; pCR = pathologic complete response; pert = pertuzumab; trast = trastuzumab.
aOne patient was randomized to the pert + doce arm but received treatment according to the trast + pert + doce arm.
bOne patient randomized to the trast + pert + doce arm actually received the treatment for the trast + doce arm; therefore, they are included in the safety population for the trast + doce arm.
cOne patient was randomized to the pert + doce arm but received treatment according to the trast + pert arm.
Note: Redacted rows were deleted.
Source: Clinical Study Report for NEOSPHERE.4
Table 17: Patient Disposition — PEONY
Disposition | Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 |
|---|---|---|
Screened | 383 | |
Randomized | 219 | 110 |
Randomized and treated | 218 (99.5) | 110 (100) |
Discontinued study (neoadjuvant period) | 13 (5.9) | 8 (7.3) |
Death | 1 (0.5) | 0 |
Withdrawal by patient | 11 (5.0) | 7 (6.4) |
Physician decision | 0 | 1 (0.9) |
Other | 1 (0.5) | 0 |
Discontinued study treatment (neoadjuvant period) | 4 (1.8) | 2 (1.8) |
Adverse event | 2 (0.9) | 0 |
Withdrawal by patient | 2 (0.9) | 1 (0.9) |
Progression of disease | 0 | 1 (0.9) |
Other | — | — |
Started adjuvant treatment, n (%) | 208 (95.0) | 103 (93.6) |
ITT population | 219 (100) | 110 (100) |
Safety | 218 (99.5) | 110 (100) |
Protocol deviations, n (%) | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; chemo = chemotherapy; doce = docetaxel; ITT = intention to treat; LVEF = left ventricular ejection fraction; pCR = pathologic complete response; pert = pertuzumab; PLA = placebo; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for PEONY.5
Table 18: Patient Disposition — TRYPHAENA
Disposition | Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 |
|---|---|---|---|
Screened | 300 | ||
Randomized | 73 | 75 | 77 |
Withdrew before neoadjuvant period | 1 | 0 | 1 |
Entered neoadjuvant period | 72 | 75 | 76 |
Withdrew from neoadjuvant treatment | 4 (5.5) | 10 (13.3) | 7 (9.1) |
Adverse event | 3 (4.1) | 4 (5.3) | 5 (6.5) |
Violation of selection criteria | 1 (1.4) | 1 (1.3) | 0 |
Refused treatment | 0 | 3 (4.0) | 0 |
Disease progression | 0 | 1 (1.3) | 0 |
Disease recurrence | 0 | 1 (1.3) | 0 |
Other | 0 | 0 | 2 (2.6) |
Surgery and valid pCR assessment | 67 (91.8) | 67 (89.3) | 71 (92.2) |
Entered adjuvant treatment | 68 (93.2) | 65 (86.7) | 67 (87.0) |
Completed adjuvant treatment, n (%) | 62 (84.9) | 60 (80.0) | 64 (83.1) |
ITT population | 73 | 75 | 77 |
Safety | 72 | 75 | 76 |
Protocol violations, n (%) | |||
Patients with at least 1 protocol violation, n (%) | 29 (39.7) | 25 (33.3) | 24 (31.2) |
Patients with at least 1 inclusion violation | 10 (13.7) | 4 (5.3) | 5 (6.5) |
|||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
Patients with at least 1 exclusion violation, n (%) | 9 (12.3) | 4 (5.3) | 6 (7.8) |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
Patients with at least 1 on-study violation, n (%) | 17 (23.3) | 20 (26.7) | 18 (23.4) |
|||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| |
doce = docetaxel; ECOG = Eastern Cooperative Oncology Group; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; HER2 = human epidermal growth factor receptor 2; ITT = intention to treat; LVEF = left ventricular ejection fraction; pCR = pathologic complete response; pert = pertuzumab; pla = placebo; TCH = docetaxel + carboplatin + trastuzumab; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for TRYPHAENA.6
Table 19: Patient Disposition — BERENICE
Disposition | COHORT A ddAC then pacli + pert + trast N = 199 | Cohort B FEC then doce + pert + trast N = 201 |
|---|---|---|
Screened | 523 | |
Enrolled | 199 | 202 |
Started neoadjuvant treatment | 199 | 198 |
Early surgery resulted in incomplete neoadjuvant treatment, n (%) | 4 (2.0) | 3 (1.5) |
Discontinued study treatment, n (%) | 13 (6.5) | 6 (3.0) |
Adverse event | 6 (3.0) | 3 (1.5) |
Withdrawal by patient | 1 (0.5) | 0 |
Progression of disease | 1 (0.5) | 1 (0.5) |
Physician decision | 2 (1.0) | 0 |
Lack of efficacy | 0 | 1 (0.5) |
Other | 1 (0.5) | 1 (0.5) |
Completed adjuvant treatment | 163 (81.9) | 176 (87.6) |
ITT population | 199 | 201 |
Safety | 199 | 198 |
Protocol deviations, n (%) | |||||| | |||||| |
|||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||||| | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||| | |||||| | |||||| |
AST = aspartate aminotransferase; ddAC = dose-dense doxorubicin and cyclophosphamide; doce = docetaxel; ECOG = Eastern Cooperative Oncology Group; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; HER2 = human epidermal growth factor receptor 2; ITT = intention to treat; LVEF = left ventricular ejection fraction; pacli = paclitaxel; pCR = pathologic complete response; pert = pertuzumab; TCH = docetaxel + carboplatin + trastuzumab; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for BERENICE.7
Exposure to Study Treatments (Neoadjuvant Period)
Treatment exposure data are summarized in Table 20 to Table 23.
Across studies, patients generally received 3.9 or 4.0 of the 4 planned cycles of neoadjuvant treatment, on average. The use of co-interventions was reported to varying extents across the studies. Dose delays, interruptions, and discontinuations with pertuzumab occurred in 7.4% of patients in NEOSPHERE, 3.2% of patients in PEONY, 10% of patients in TRYPHAENA, and 17% of patients in BERENICE.
The use of radiotherapy in NEOSPHERE was similar between the pertuzumab and trastuzumab plus docetaxel group and the trastuzumab plus docetaxel group. The most common co-interventions in PEONY were 5-hydroxytryptamine 3 (5-HT3) antagonists, corticosteroids, and colony-stimulating factors, and their use was similar between groups. The most common co-interventions in TRYPHAENA were anti-estrogens, anti-anemia drugs, and colony-stimulating factors; radiotherapy, 5-HT3 antagonists, and steroids were the most common co-interventions in BERENICE.
Table 20: Exposure to Study Treatments in Neoadjuvant Treatment Period — NEOSPHERE
Characteristic | Treatment arms | |||
|---|---|---|---|---|
Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 107 | Arm D pert + doce N = 96 | |
Number of pertuzumab cycles administered per patient, mean (SD) | 3.9 |||||| | NA | 3.9 |||||| | 3.9 | |
Total dose received, mg, mean (SD) | 2,059.6 ( | ) | NA | 2,047.7 (||||) | 2,051.0 (||||) |
Number (%) of patients completing at least: | ||||
1 cycle | |||||| | |||||| | |||||| | |||||| |
2 cycles | |||||| | |||||| | |||||| | |||||| |
3 cycles | |||||| | |||||| | |||||| | |||||| |
4 cycles | 102 (95) | NA | 100 (93) | 88 (94) |
Pertuzumab infusion administered, delayed, slowed down, interrupted, or discontinued, n (%) | 31 (7.4) | NA | 42 (10.0) | 28 (7.7) |
Due to adverse events, n (%) | 13 (3.1) | NA | 12 (2.9) | 2 (0.5) |
|||||||||||||||||||||||||||||||||||||||||| | |||||| | |||||| | |||||| | |||||| |
doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; NA = not applicable; pert = pertuzumab; SD = standard deviation; trast = trastuzumab.
Source: Clinical Study Report for NEOSPHERE.4
Table 21: Exposure to Study Treatments in Neoadjuvant Treatment Period — PEONY
Characteristic | Treatment arms | |
|---|---|---|
Arm A trast + pert + chemo | Arm B PLA + trast + chemo | |
Exposure to pertuzumab or placebo | ||
Treatment duration, weeks, mean (SD) | 12.0 (1.2) | 12.0 (0.7) |
Number of cycles, mean (SD) | 3.9 (0.4) | 4.0 (0.2) |
Cumulative dose, mg, mean (SD) | 2,082.7 (162.9) | 2,084.9 (96.6) |
Number of infusion modifications, n (%) | ||
0 | 211 (96.8) | 110 (100.0) |
1 | 7 (3.2) | 0 |
2 or more | 0 | 0 |
Number of infusion modifications due to an adverse event, n (%) | ||
0 | 212 (97.2) | 110 (100.0) |
1 | 6 (2.8) | 0 |
2 or more | 0 | 0 |
Concomitant treatments | ||
|||||||||||||||||||| | |||||| | |||||| |
|||||| | |||||| | |||||| |
chemo = chemotherapy; pert = pertuzumab; PLA = placebo; SD = standard deviation; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for PEONY.5
Table 22: Exposure to Study Treatments in Neoadjuvant Treatment Period — TRYPHAENA
Characteristic | Treatment arms | |||||
|---|---|---|---|---|---|---|
Arm A | Arm B | Arm C | ||||
Number of pert cycles administered, mean (SD) | |||||| | |||||| | |||||| | |||
Total dose received, mg, mean (SD) | |||||| | |||||| | |||||| | |||
Dose received per cycle, mg, mean (SD) | |||||| | |||||| | |||||| | |||
Number of cycles of pertuzumab delayed, modified, or discontinued | |||||| | |||||| | |||||| | |||
Number of cycles of pertuzumab delayed, modified, or discontinued due to adverse event | |||||| | |||||| | |||||| | |||
Number (%) of patients completing at least: | ||||||
1 cycle | |||||| | |||||| | |||||| | |||
2 cycles | |||||| | |||||| | |||||| | |||
3 cycles | |||||| | 66 (88.0) | |||||| | |||
4 cycles | |||||| | |||||| | |||||| | |||
5 cycles | |||||| | |||||| | |||||| | |||
6 cycles | 66 (91.7) | 0 | 70 (92.1) | |||
Patients taking previous concomitant and concomitant treatments during the adjuvant period, n (%) | ||||||
|||||||||||||||| | |||||| | |||||| | |||||| | |||
|||||||||||||||||||| | |||||| | |||||| | |||||| | |||
doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; pert = pertuzumab; SD = standard deviation; TCH = docetaxel + carboplatin + trastuzumab; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for TRYPHAENA.6
Table 23: Exposure to Study Treatments in Neoadjuvant Treatment Period — BERENICE
Characteristic | Treatment arms | |
|---|---|---|
COHORT A | Cohort B | |
Treatment duration, weeks, mean (SD) | |||||| | |||||| |
Number of cycles, mean (SD) | |||||| | |||||| |
Cumulative dose, mg, mean (SD) | |||||| | |||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||
|||||| | |||||| | |||||| |
Number of dose modifications or delays due to adverse event, n (%) | ||
0 | 163 (83.6) | 176 (89.8) |
1 | 30 (15.4) | 19 (9.7) |
2 | 2 (1.0) | 1 (0.5) |
> 2 | 0 | 0 |
Patients taking previous concomitant and concomitant treatments during the adjuvant period, n (%) | ||
|||||||||||||||||||| | |||||| | |||||| |
|||||||||||||||||||||| | |||||| | |||||| |
ddAC = dose-dense doxorubicin + cyclophosphamide; doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; pacli = paclitaxel; pert = pertuzumab; SD = standard deviation; trast = trastuzumab.
Note: Redacted rows were deleted.
Source: Clinical Study Report for BERENICE.7
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently. See Appendix 3 for detailed efficacy data. Note that data for longer-term survival outcomes reflect the treatment received in both the neoadjuvant and adjuvant phases. The overall median time on study in NEOSPHERE was |||||||||||||||||||||||||||||||||||||||||| in the pertuzumab and trastuzumab plus docetaxel arm and |||||||||||||||||||||||||||||||||||||||||||| in the trastuzumab plus docetaxel arm. In PEONY, the median time on study was |||||||||||||||||||||||||||||||||||||||||||| in the pertuzumab and trastuzumab plus chemotherapy arm and |||||||||||||||||||||||||||||||||||||||| in the trastuzumab plus chemotherapy arm. In TRYPHAENA, the median follow-up time was ||||||| in arm A |||||||||||||||||||||||||||||||||||||||| in arm B |||||||||||| and |||||||||||||| in arm C |||||||||||||| In BERENICE, median time on study was |||||||||||||||||||||||||||||||||||||||||||||| in cohort A and |||||||||||||||||||||||||||||||||||||||||||||| in cohort B.
Overall Survival
OS was only reported for the adjuvant period, where reported.
In NEOSPHERE, OS was not a pre-specified outcome; in PEONY, the data on OS were considered not mature by the sponsor and were therefore not reported. In TRYPHAENA, OS events ranged between 6.8% and 13.0% across arms (Table 26); in BERENICE, the data were not yet mature, according to the sponsor, and were therefore not reported.
Invasive Disease–Free Survival
This outcome was not assessed in NEOSPHERE, PEONY, or TRYPHAENA. In BERENICE, the data were not yet mature, according to the sponsor, and were therefore not reported.
Event-Free Survival
EFS was not assessed in NEOSPHERE. The data were not yet mature in PEONY and in BERENICE, according to the sponsor, and were therefore not reported. In TRYPHAENA, EFS was not assessed.
Progression-Free Survival
Assessment of PFS included the adjuvant phase. In NEOSPHERE (Table 24), progression events occurred in 15.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 17.8% of patients in the trastuzumab plus docetaxel arm, for an HR of 0.69 (95% CI, 0.34 to 1.40). These data were not yet mature in PEONY, according to the sponsor. In TRYPHAENA, the PFS event rates (Table 26) were 13.7% in arm A, 14.7% in arm B, and 18.2% in arm C. This outcome was not assessed in BERENICE.
Figure 6: Kaplan–Meier Curve of PFS in NEOSPHERE
ITT = intention to treat; PFS = progression-free survival; vs = versus.
Source: Clinical Study Report for NEOSPHERE.
Disease-Free Survival
Reporting of DFS events included the adjuvant treatment phase in NEOSPHERE. A DFS event was reported for 14.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 17.5% of patients in the trastuzumab plus docetaxel arm (Table 24). The DFS data in PEONY were not yet mature, according to the sponsor, and were therefore not reported. Results for DFS in TRYPHAENA were consistent with that of NEOSPHERE, with DFS events occurring in 14.5% of patients in arm A, 11.9% of patients in arm B, and 15.3% of patients in arm C (Table 26). DFS was not assessed in BERENICE.
Pathologic Complete Response
Of the 417 patients randomized into NEOSPHERE (Table 24), 392 underwent surgery and all of these patients had a valid assessment of pathologic response. In NEOSPHERE, the pCR rate was 45.8% in the pertuzumab and trastuzumab plus docetaxel arm and 29.0% in the trastuzumab plus docetaxel arm, for a difference in response rates between groups of 16.8% (95% CI, 3.5 to 30.1; P = 0.0094).
In PEONY (Table 25), the IRC-assessed tpCR rate was 39.3% in the pertuzumab and trastuzumab plus docetaxel arm and 21.8% in the trastuzumab plus docetaxel arm, for a difference in response rates of 17.45% (95% CI, 6.89 to 28.01; P = 0.0014). The tpCR rates in PEONY assessed by the local pathologist were 39.3% versus 20.9%, respectively (Table 25). Additionally, PEONY reported the bpCR rate as a secondary outcome, and the IRC-assessed bpCR rate was 42.0% versus 23.6%, for a between-group difference of 18.37% (95% CI, 7.60 to 29.15; P = 0.0010). The pCR rates in TRYPHAENA (Table 26) were 61.6% in arm A, 57.3% in arm B, and 66.2% in arm C, and 60.7% and 61.8% in the 2 cohorts in BERENICE (Table 27).
Subgroups
Complete subgroup data reported for the primary outcome in NEOSPHERE and in PEONY can be found in Table 34 and Table 35, respectively. In NEOSPHERE, in the subgroup of patients with locally advanced breast cancer, the pCR rate was 43.8% in the pertuzumab and trastuzumab plus docetaxel arm and 41.7% in the trastuzumab plus docetaxel arm. In the subgroup of patients with operable breast cancer, the rates were 47.7% versus 23.4%, respectively and, in those with inflammatory breast cancer, the rates were 40.0% versus 14.3%, respectively. The difference between the pertuzumab and trastuzumab plus docetaxel arm and the trastuzumab plus docetaxel arm for those patients who were estrogen and progesterone receptor–positive was 26.0% versus 20.0%, respectively; in those who were estrogen and progesterone receptor–negative, the rates were 63.2% versus 36.8%, respectively. The treatment effect appeared consistent across the various biomarkers assessed. In PEONY, the difference in tpCR rates in patients who were estrogen and/or progesterone receptor–positive was 33.3% versus 25.0%, respectively; in those who were estrogen and progesterone receptor–negative, the rates were 46.1% versus 18.5%, respectively.
Objective Response
In NEOSPHERE, when assessed by X-ray or mammography, a CR was observed in 18.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 18.3% of patients in the trastuzumab plus docetaxel arm, and a PR was observed in 49.1% of patients and 49.3% of patients, respectively (Table 24). When assessed by clinical exam, the rates of CR were 25.0% versus 21.6%, respectively and, for PR, were 63.0% versus 59.8%, respectively. In PEONY, clinical response was assessed as a secondary outcome, and an objective response (defined as obtaining either a CR or PR) during cycles 1 to 4 occurred in 88.6% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 78.2% of patients in the trastuzumab plus docetaxel arm, for a difference in objective response rates between groups of 10.4% (95% CI, 1.12 to 19.69). A CR was observed in 11.0% versus 10.0% of patients, and a PR was observed in 77.6% versus 68.2% of patients, respectively (Table 25).
Duration of Response
The duration of response was not reported in the included studies.
Health-Related Quality of Life
This outcome was not assessed in the included studies.
Symptoms
This outcome was not assessed in the included studies.
Breast-Conserving Surgery
Among patients with T2 or T3 tumours for whom mastectomy was initially planned, breast-conserving surgery was achieved in 23.2% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 22.6% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE (Table 24). This outcome was not assessed in PEONY. In TRYPHAENA, for the subgroup of patients with T2 or T3 tumours in whom mastectomy was initially planned, the percentage of patients undergoing breast-conserving surgery was 21.7% in arm A, 16.7% in arm B, and 27.0% in arm C (Table 26). In the BERENICE study, 44.4% and 42.9% of patients with T2 or T3 tumours in each of the groups received breast-conserving surgery (Table 27).
Table 24: Efficacy — NEOSPHERE (ITT Population)
Outcome | Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 107 | Arm D pert + doce N = 96 |
|---|---|---|---|---|
Progression-free survival (Results of final analysis (data cut-off October 20, 2014) | ||||
Patients with progression events, n (%) | 17 (15.9) | 19 (17.8) | 27 (25.2) | 24 (25.0) |
HR (95% CI)a vs. trast + doce | 0.69 (0.34 to 1.40) | Reference | 1.25 (0.68 to 2.30) | NR |
HR (95% CI)a vs. pert + trast + doce | Reference | NR | NR | 2.05 (1.07 to 3.93) |
Disease-free survival Results of final analysis (data cut-off October 20, 2014) | ||||
Patients with an event, n (%) | 15 (14.9) | 18 (17.5) | 19 (19.8) | 22 (23.9) |
HR (95% CI)a vs. trast + doce | 0.60 (0.28 to 1.27) | Reference | 0.83 (0.42 to 1.64) | NR |
HR (95% CI)a vs. pert + trast + doce | Reference | NR | NR | 2.16 (1.08 to 4.32) |
pCR | ||||
Responders, n (%) | 49 (45.8) | 31 (29.0) | 18 (16.8) | 23 (24.0) |
Difference in response rates vs. trast + doce (95% CI) | 16.82 (3.5 to 30.1) | Reference | −12.15 (−23.8 to −0.5) | NR |
Difference in response rates vs. pert + trast + doce (95% CI) | Reference | NR | NR | −21.84 (−35.1 to −8.5) |
P value from CMHb | P = 0.0094 | P = 0.0198 | P = 0.0010 | |
P value (Simes correction for CMH test)c | P = 0.0141 | P = 0.0198 | P = 0.0030 | |
Objective response | ||||
By X-ray and/or mammography | N = 53 | N = 71 | N = 55 | N = 43 |
Overall response, n (%) | ||||
Responders | 36 (67.9) | 48 (67.6) | 26 (47.3) | 28 (65.1) |
Complete response | 10 (18.9) | 13 (18.3) | 7 (12.7) | 8 (18.6) |
Partial response | 26 (49.1) | 35 (49.3) | 19 (34.5) | 20 (46.5) |
Stable disease | 16 (30.2) | 22 (31.0) | 25 (45.5) | 15 (34.9) |
Disease progression | 1 (1.9) | 1 (1.4) | 4 (7.3) | 0 (0.0) |
By clinical examination | N = 100 | N = 97 | N = 98 | N = 88 |
Overall response, n (%) | ||||
Responders | 88 (88.0) | 79 (81.4) | 65 (66.3) | 65 (73.9) |
Complete response | 25 (25.0) | 21 (21.6) | 11 (11.2) | 14 (15.9) |
Partial response | 63 (63.0) | 58 (59.8) | 54 (55.1) | 51 (58.0) |
Stable disease | 12 (12.0) | 17 (17.5) | 31 (31.6) | 23 (26.1) |
Disease progression | 0 (0.0) | 1 (1.0) | 2 (2.0) | 0 (0.0) |
Breast-conserving surgery | ||||
Breast-conserving surgery, n/N (%) T2 or T3 patients in whom mastectomy was planned | 13/56 (23.2) | 14/62 (22.6) | 11/61 (18.0) | 19/60 (31.7) |
chemo = chemotherapy; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; doce = docetaxel; HR = hazard ratio; ITT = intention to treat; NR = not reported; pCR = pathologic complete response; pert = pertuzumab; trast = trastuzumab.
aHR is based on Cox proportional hazard regression stratified by breast cancer type and hormone positivity.
bCMH test stratified by breast cancer type (operable, locally advanced, or inflammatory) and estrogen and or progesterone positivity (either was positive vs. both were negative).
cP value from Cochran-Mantel-Haenszel test with Simes multiplicity adjustment.
Source: Clinical Study Report for NEOSPHERE.4
Table 25: Efficacy — PEONY (ITT Population)
Outcome | Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 |
|---|---|---|
pCR | ||
tpCR rate (IRC assessed) | ||
Responders, n (%) | 86 (39.3) | 24 (21.8) |
Difference in response rates (95% CI)a | 17.45 (6.89 to 28.01) | reference |
P value, CMH testb | P = 0.0014 | reference |
P value, Fisher | P = 0.0019 | reference |
tpCR rate (local pathologist) | ||
Responders, n (%) | 86 (39.3) | 23 (20.9) |
Difference in response rates (95% CI)a | 18.36 (7.89 to 28.83) | reference |
P value, CMH testb | P = 0.0008 | reference |
bpCR rate (IRC assessed) | ||
Responders, n (%) | 92 (42.0) | 26 (23.6) |
Difference in response rates (95% CI)a | 18.37 (7.60 to 29.15) | reference |
P value, CMH testb | P = 0.0010 | reference |
Objective response | ||
Objective response during cycles 1 to 4 | ||
Responders, n (%) | 194 (88.6) | 86 (78.2) |
Difference in response rates (95% CI)a | 10.40 (1.12 to 19.69) | reference |
P value, CMH testb | P = 0.0125 | reference |
Complete response, n (%) | 24 (11.0) | 11 (10.0) |
Partial response, n (%) | 170 (77.6) | 75 (68.2) |
Stable disease, n (%) | 18 (8.2) | 21 (19.1) |
Progressive disease, n (%) | 1 (0.5) | 2 (1.8) |
Missing or unevaluable, n (%) | 6 (2.7) | 1 (0.9) |
bpCR = pathologic complete response in the breast; chemo = chemotherapy; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; HR = hazard ratio; IRC = independent review committee; ITT = intention to treat; pCR = pathologic complete response; pert = pertuzumab; PLA = placebo; tpCR = total pathologic complete response; trast = trastuzumab.
aApproximate 95% CI for difference of 2 rates using Hauck-Anderson method.
bCochran-Mantel-Haenszel test stratified by disease category (early stage and locally advanced) and hormone receptor status (positive for estrogen receptor and/or progesterone receptor or negative for both) from interactive voice and web response system. Note that statistical analyses performed beyond the primary outcome (IRC-assessed tpCR) were descriptive only.
Source: Clinical Study Report for PEONY.5
Table 26: Efficacy — TRYPHAENA (ITT Population)
Outcome | Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 |
|---|---|---|---|
Overall survival | |||
Patients with event, n (%) | 5 (6.8) | 7 (9.3) | 10 (13.0) |
Progression-free survival | |||
Patients with event, n (%) | 10/73 (13.7) | 11/75 (14.7) | 14/77 (18.2) |
Disease-free survival | |||
Patients with event, n (%) | 10/69 (14.5) | 8/67 (11.9) | 11/72 (15.3) |
pCR | |||
Patients with pCR, n (%) | 45 (61.6) | 43 (57.3) | 51 (66.2) |
(95% CI) | (49.5 to 72.8) | (45.4 to 68.7) | (54.6 to 76.6) |
Objective response | |||
Best tumour response, n (%) | |||
Clinical response (CR + PR) | 67 (91.8) | 71 (94.7) | 69 (89.6) |
CR | 37 (50.7) | 21 (28.0) | 31 (40.3) |
PR | 30 (41.1) | 50 (66.7) | 38 (49.4) |
Stable disease | 3 (4.1) | 1 (1.3) | 5 (6.5) |
Disease progression | 0 (0.0) | 1 (1.3) | 0 (0.0) |
Missing | 3 (4.1) | 2 (2.7) | 3 (3.9) |
Breast-conserving surgery, n/N (%) | |||
T2 or T3 patients in whom mastectomy was planned | 10/46 (21.7) | 6/36 (16.7) | 10/37 (27.0) |
CI = confidence interval; CR = complete response; doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; ITT = intention to treat; pCR = pathologic complete response; pert = pertuzumab; PR = partial response; TCH = docetaxel + carboplatin + trastuzumab; tpCR = total pathologic complete response; trast = trastuzumab.
Source: Clinical Study Report for TRYPHAENA.6
Table 27: Efficacy — BERENICE (ITT Population)
Outcome | COHORT A ddAC then pacli + pert + trast N = 199 | Cohort B FEC then doce + pert + trast N = 201 |
|---|---|---|
pCR | ||
Patients with pCR, n (%) | 123 (61.8) | 122 (60.7) |
95% CI for response rates using Pearson-Clopper method | (54.67 to 68.59) | (53.58 to 67.49) |
Missing or unevaluablea | 14 (7.0) | 8 (4.0) |
Patients with bpCR, n (%) | 134 (67.3) | 132 (65.7) |
Objective response, n (%) | ||
Clinical response rate, ipsilateral breast, ITT population | 134 (67.3) | 121 (60.2) |
Complete response | 79 (39.7) | 48 (23.9) |
Partial response | 55 (27.6) | 73 (36.3) |
Stable disease | 14 (7.0) | 20 (10.0) |
Disease progression | 1 (0.5) | 2 (1.0) |
Missing or unevaluable | 50 (25.1) | 58 (28.9) |
Breast-conserving surgery, n/N (%) | ||
T2 or T3 patients only | 76/171 (44.4) | 75/175 (42.9) |
bpCR = pathologic complete response in the breast; CI = confidence interval; ddAC = dose-dense doxorubicin and cyclophosphamide; doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; ITT = intention to treat; pacli = paclitaxel; pCR = pathologic complete response; pert = pertuzumab; tpCR = total pathologic complete response; trast = trastuzumab.
aPatients are classified as missing or unevaluable if they do not undergo surgery or do not have a valid tpCR assessment and are considered to be nonresponders.
Source: Clinical Study Report for BERENICE.7
Harms
Only those harms identified in the review protocol are reported subsequently. See Table 28 for detailed harms data.
Adverse Events
Adverse events occurred in 98.1% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in the trastuzumab plus docetaxel arm in NEOSPHERE, and in 97.7% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 96.4% of patients in the trastuzumab plus docetaxel arm in PEONY. The most common adverse events in the trials for pertuzumab and trastuzumab plus docetaxel versus trastuzumab plus docetaxel, were alopecia (63.6% versus 65.4% in NEOSPHERE; 49.1% in each arm in PEONY), neutropenia (50.5% versus 62.6% in NEOSPHERE; 48.2% versus 44.5% in PEONY), and diarrhea (45.8% versus 33.6% in NEOSPHERE; 38.5% versus 16.4% in PEONY). The most common grade 3 or greater adverse event was neutropenia (44.9% versus 57.0% in NEOSPHERE; 38.1% versus 32.7% in PEONY). Similar results were seen in TRYPHAENA and BERENICE, where approximately 99% of patients experienced an adverse event at some time during the study, and neutropenia was the most common grade 3 or greater adverse event.
Serious Adverse Events
Serious adverse events occurred in 10.3% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 16.8% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE, and in 10.1% versus 8.2% of patients, respectively, in PEONY. Febrile neutropenia was the most common serious adverse event in both NEOSPHERE arms, occurring in 5.6% of patients treated with pertuzumab and trastuzumab plus docetaxel, and 6.5% of patients treated with trastuzumab plus docetaxel and, in PEONY, occurring in 1.8% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in no patients in the trastuzumab plus docetaxel arm. In TRYPHAENA, 28% of patients experienced a serious adverse event across arms and, in BERENICE, 24% of patients experienced a serious adverse event. Febrile neutropenia was the most common serious adverse event in both studies, occurring in about 10% of patients.
Withdrawals Due to Adverse Events
Few patients across studies stopped treatment due to an adverse event, which occurred in 0.9% of patients in the pertuzumab and trastuzumab plus docetaxel arm versus in no patients in the trastuzumab plus docetaxel arm in NEOSPHERE, and in 0.5% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in no patients in the trastuzumab plus docetaxel arm in PEONY. The number of patients withdrawing due to an adverse event was 7% across arms in TRYPHAENA and 3.5% across cohorts in BERENICE.
Mortality
One patient died in each of the pertuzumab and trastuzumab plus docetaxel and trastuzumab plus docetaxel arms in NEOSPHERE, and both deaths were considered to be due to complications of breast cancer. One patient died in the pertuzumab and trastuzumab plus docetaxel arm in PEONY, due to a suicide, and there were no deaths in the trastuzumab plus docetaxel arm. There were no deaths in TRYPHAENA and no deaths in BERENICE.
Notable Harms
Notable harms of diarrhea were reported previously. Cardiac dysfunction occurred in 2.8% of patients in the pertuzumab and trastuzumab plus docetaxel arm and 0.9% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE, and no patients in PEONY had an LVEF decline to less than 40%, or a primary or secondary cardiac event. Events of drug hypersensitivity or anaphylaxis occurred in 5.6% of patients in the pertuzumab and trastuzumab plus docetaxel arm and in 1.9% of patients in the trastuzumab plus docetaxel arm in NEOSPHERE, and in 3.2% versus 1.8% of patients in PEONY, respectively.
Table 28: Summary of Harms — NEOSPHERE (Neoadjuvant Period; Safety Population)
Harms | All | Grade ≥ 3 | ||||||
|---|---|---|---|---|---|---|---|---|
Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 108 | Arm D pert + doce N = 94 | Arm B trast + pert + doce N = 107 | Arm A trast + doce N = 107 | Arm C trast + pert N = 108 | Arm D pert + doce N = 94 | |
Total, n (%) | |||||| | |||||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Most common events,a n (%) | ||||||||
Alopecia | 68 (63.6) | 70 (65.4) | 1 (0.9) | 63 (67.0) | 5 (4.7) | 1 (0.9) | 0 | 4 (4.3) |
Neutropenia | 54 (50.5) | 67 (62.6) | 1 (0.9) | 59 (62.8) | 48 (44.9) | 61 (57.0) | 1 (0.9) | 52 (55.3) |
Diarrhea | 49 (45.8) | 36 (33.6) | 30 (27.8) | 51 (54.3) | 6 (5.6) | 4 (3.7) | 0 | 4 (4.3) |
Nausea | 41 (38.3) | 39 (36.4) | 15 (13.9) | 34 (36.2) | NR | NR | NR | NR |
Fatigue | 28 (26.2) | 29 (27.1) | 13 (12.0) | 24 (25.5) | NR | NR | NR | NR |
Rash | 28 (26.2) | 23 (21.5) | 12 (11.1) | 27 (28.7) | 2 (1.9) | 2 (1.9) | 0 | 1 (1.1) |
Mucosal inflammation | 28 (26.2) | 23 (21.5) | 3 (2.8) | 24 (25.5) | 2 (1.9) | 0 | 0 | 0 |
Myalgia | 24 (22.4) | 24 (22.4) | 10 (9.3) | 19 (20.2) | NR | NR | NR | NR |
Asthenia | 22 (20.6) | 19 (17.8) | 3 (2.8) | 15 (16.0) | 2 (1.9) | 0 | 0 | 2 (2.1) |
Headache | 12 (11.2) | 12 (11.2) | 15 (13.9) | 12 (12.8) | NR | NR | NR | NR |
Vomiting | 14 (13.1) | 13 (12.1) | 5 (4.6) | 15 (16.0) | NR | NR | NR | NR |
Pyrexia | 18 (16.8) | 11 (10.3) | 9 (8.3) | 8 (8.5) | NR | NR | NR | NR |
Leukopenia | 10 (9.3) | 23 (21.5) | 0 | 12 (12.8) | 5 (4.7) | 13 (12.1) | 0 | 7 (7.4) |
Stomatitis | 19 (17.8) | 8 (7.5) | 5 (4.6) | 9 (9.6) | NR | NR | NR | NR |
Dysgeusia | 16 (15.0) | 11 (10.3) | 5 (4.6) | 7 (7.4) | NR | NR | NR | NR |
Decreased appetite | 15 (14.0) | 7 (6.5) | 2 (1.9) | 14 (14.9) | NR | NR | NR | NR |
Arthralgia | 11 (10.3) | 9 (8.4) | 5 (4.6) | 9 (9.6) | NR | NR | NR | NR |
Peripheral sensory neuropathy | 9 (8.4) | 13 (12.1) | 2 (1.9) | 10 (10.6) | NR | NR | NR | NR |
Insomnia | 9 (8.4) | 12 (11.2) | 4 (3.7) | 8 (8.5) | NR | NR | NR | NR |
Bone pain | 10 (9.3) | 11 (10.3) | 0 | 4 (4.3) | NR | NR | NR | NR |
Febrile neutropenia | 9 (8.4) | 8 (7.5) | 0 | 7 (7.4) | 9 (8.4) | 8 (7.5) | 0 | 7 (7.4) |
Patients with ≥ 1 SAE | ||||||||
n (%) | 11 (10.3) | 18 (16.8) | 4 (3.7) | 16 (17.0) | NA | NA | NA | NA |
Most common events, n (%) | ||||||||
Febrile neutropenia | 6 (5.6) | 7 (6.5) | 0 | 6 (6.4) | NA | NA | NA | NA |
Neutropenia | 4 (3.7) | 1 (0.9) | 0 | 6 (6.4) | NA | NA | NA | NA |
Patients who stopped treatment due to AEs | ||||||||
n (%) | 1 (0.9) | 0 | 2 (1.9) | 2 (2.1) | NA | NA | NA | NA |
Deaths | ||||||||
n (%) | 1 | 1 | 0 | 1 | NA | NA | NA | NA |
Cause | Fulminant hepatitis (BC) | BC metastatic | BC metastatic | NA | NA | NA | NA | |
Notable harms | ||||||||
Cardiac dysfunction, n (%) | 3 (2.8) | 1 (0.9) | 1 (0.9) | 1 (1.1) | NA | NA | NA | NA |
LV dysfunction | 3 (2.8) | 1 (0.9) | 0 | 1 (1.1) | NA | NA | NA | NA |
Hypersensitivity or anaphylaxis, n (%) | 6 (5.6) | 2 (1.9) | 6 (5.6) | 6 (6.4) | NA | NA | NA | NA |
Drug hypersensitivity | 6 (5.6) | 2 (1.9) | 6 (5.6) | 5 (5.3) | NA | NA | NA | NA |
AE = adverse event; BC = breast cancer; doce = docetaxel; LV = left ventricular; NA = not applicable; NR = not reported; pert = pertuzumab; SAE = serious adverse event; trast = trastuzumab.
aOverall adverse events reported in at least 7% of patients in any treatment group.
Source: Clinical Study Report for NEOSPHERE.4
Table 29: Summary of Harms — PEONY (Neoadjuvant Period; Safety Population)
Harms | All | Grade ≥ 3 | ||
|---|---|---|---|---|
Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 | Arm A trast + pert + chemo N = 219 | Arm B PLA + trast + chemo N = 110 | |
Patients with at least 1 AE, n (%) | |||| | |||| | |||| | ||| |
Most frequent AE, ≥ 10% in any group, n (%) | ||||
Alopecia | 107 (49.1) | 54 (49.1) | NR | NR |
Neutropenia | 105 (48.2) | 49 (44.5) | 83 (38.1) | 36 (32.7) |
Leukopenia | 92 (42.2) | 43 (39.1) | 45 (20.6) | 21 (19.1) |
Diarrhea | 84 (38.5) | 18 (16.4) | NR | NR |
Anemia | 53 (24.3) | 30 (27.3) | NR | NR |
ALT increased | 49 (22.5) | 26 (23.6) | NR | NR |
Nausea | 45 (20.6) | 21 (19.1) | NR | NR |
AST increased | 37 (17.0) | 22 (20.0) | NR | NR |
Pyrexia | 31 (14.2) | 11 (10.0) | NR | NR |
Upper respiratory tract infection | 33 (15.1) | 7 (6.4) | NR | NR |
Mouth ulceration | |||| | |||| | NR | NR |
Rash | |||| | |||| | NR | NR |
Insomnia | |||| | |||| | NR | NR |
Patients with at least 1 SAE, n (%) | 22 (10.1) | 9 (8.2) | — | — |
Most common SAE, occurring in 2% of patients | — | — | NA | NA |
Febrile neutropenia | 4 (1.8) | 1 (0.9) | NA | NA |
Patients who stopped treatment due to AEs | ||||
AE leading to withdrawal from treatment (pertuzumab or placebo), n (%) | |||| | |||| | NA | NA |
AE leading to withdrawal from any treatment, n (%) | |||| | |||| | NA | NA |
AE leading to dose interruption of treatment (pertuzumab or placebo), n (%) | |||| | |||| | NA | NA |
Deaths, n | 1 | 0 | — | — |
Cause | Suicide | — | NA | NA |
Notable harms | ||||
Primary or secondary cardiac event: Heart failure (NYHA class III or IV) and significant LVEF decline, n (%) | 0 | 0 | NA | NA |
LVEF decline to < 40%, n (%) | 0 | 0 | NA | NA |
Mean (SD) baseline LVEF % | 66.41 (4.93) | 66.03 (5.19) | NA | NA |
Mean (SD) change from baseline to worst value LVEF % | −5.06 (5.18) | −4.58 (5.34) | NA | NA |
Patients with grade ≥ 3 diarrhea, n (%) | 2 (0.9) | 0 | NA | NA |
Infusion reactions, n (%) | 48 (22.0) | 10 (9.1) | NA | NA |
Anaphylaxis or hypersensitivity, n (%) | |||| | |||| | NA | NA |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; chemo = chemotherapy; LVEF = left ventricular ejection fraction; NA = not applicable; NR = not reported; NYHA = New York Heart Association; pert = pertuzumab; PLA = placebo; SAE = serious adverse event; SD = standard deviation; trast = trastuzumab.
Source: Clinical Study Report for PEONY.5
Table 30: Summary of Harms — TRYPHAENA (Neoadjuvant Period, Safety Population)
Harms | All | Grade ≥ 3 | ||||
|---|---|---|---|---|---|---|
Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 | Arm A FEC + pert + trast × 3 then doce + pert + trast × 3 N = 73 | Arm B FEC × 3 then doce + pert + trast × 3 N = 75 | Arm C TCH + pert × 6 N = 77 | |
Patients with at least 1 AE, n (%) | |||| | |||| | |||| | |||| | |||||| | |||| |
Most frequent AE, ≥ 10% in any group, n (%) | ||||||
Diarrhea | 44 (61.1) | 46 (61.3) | 55 (72.4) | 3 (4.2) | 4 (5.3) | 9 (11.8) |
Alopecia | 35 (48.6) | 39 (52.0) | 41 (53.9) | NR | NR | NR |
Nausea | 38 (52.8) | 40 (53.3) | 34 (44.7) | 0 | 2 (2.7) | 0 |
Neutropenia | 37 (51.4) | 35 (46.7) | 37 (48.7) | 34 (47.2) | 32 (42.7) | 35 (46.1) |
Vomiting | 29 (40.3) | 27 (36.0) | 30 (39.5) | 0 | 2 (2.7) | 4 (5.3) |
Fatigue | 26 (36.1) | 27 (36.0) | 32 (42.1) | 0 | 0 | 3 (3.9) |
Anemia | 14 (19.4) | 6 (8.0) | 28 (36.8) | 1 (1.4) | 2 (2.7) | 13 (17.1) |
Mucosal inflammation | 17 (23.6) | 15 (20.0) | 13 (17.1) | 0 | 0 | 1 (1.3) |
Constipation | 13 (18.1) | 17 (22.7) | 12 (15.8) | NR | NR | NR |
Dyspepsia | 18 (25.0) | 6 (8.0) | 17 (22.4) | 1 (1.4) | 0 | 0 |
Leukopenia | 16 (22.2) | 12 (16.0) | 13 (17.1) | 14 (19.4) | 9 (12.0) | 9 (11.8) |
Decreased appetite | 15 (20.8) | 8 (10.7) | 16 (21.1) | NR | NR | NR |
Headache | 16 (22.2) | 11 (14.7) | 12 (15.8) | NR | NR | NR |
Rash | 14 (19.4) | 8 (10.7) | 16 (21.1) | 0 | 0 | 1 (1.3) |
Dysgeusia | 8 (11.1) | 10 (13.3) | 16 (21.1) | NR | NR | NR |
Insomnia | 8 (11.1) | 10 (13.3) | 16 (21.1) | NR | NR | NR |
Febrile neutropenia | 13 (18.1) | 7 (9.3) | 13 (17.1) | 13 (18.1) | 7 (9.3) | 13 (17.1) |
Stomatitis | 10 (13.9) | 13 (17.3) | 9 (11.8) | NR | NR | NR |
Pyrexia | 12 (16.7) | 7 (9.3) | 12 (15.8) | NR | NR | NR |
Myalgia | 12 (16.7) | 9 (12.0) | 8 (10.5) | 0 | 1 (1.3) | 0 |
Thrombocytopenia | 5 (6.9) | 1 (1.3) | 23 (30.3) | NR | NR | 9 (11.8) |
Asthenia | 7 (9.7) | 11 (14.7) | 10 (13.2) | 0 | 1 (1.3) | 1 (1.3) |
Epistaxis | 8 (11.1) | 8 (10.7) | 12 (15.8) | 0 | 0 | 1 (1.3) |
Dizziness | 6 (8.3) | 6 (8.0) | 12 (15.8) | 0 | 1 (1.3) | 0 |
Dyspnea | 9 (12.5) | 6 (8.0) | 8 (10.5) | 0 | 2 (2.7) | 1 (1.3) |
Arthralgia | 8 (11.1) | 9 (12.0) | 5 (6.6) | NR | NR | NR |
Cough | 7 (9.7) | 4 (5.3) | 9 (11.8) | NR | NR | NR |
Oropharyngeal pain | 6 (8.3) | 5 (6.7) | 9 (11.8) | NR | NR | NR |
Dry skin | 4 (5.6) | 7 (9.3) | 8 (10.5) | NR | NR | NR |
Lacrimation decreased | 9 (12.5) | 4 (5.3) | 6 (7.9) | NR | NR | NR |
Palmar-plantar erythrodysesthesia | 5 (6.9) | 8 (10.7) | 6 (7.9) | NR | NR | NR |
Peripheral edema | 8 (11.1) | 3 (4.0) | 7 (9.2) | NR | NR | NR |
ALT increased | 5 (6.9) | 2 (2.7) | 8 (10.5) | 0 | 0 | 3 (3.9) |
Peripheral neuropathy | 4 (5.6) | 1 (1.3) | 8 (10.5) | NR | NR | NR |
Patients with at least 1 SAE, n (%) | 20 (27.8) | 15 (20.0) | 27 (35.5) | NA | NA | NA |
Most common SAE, 4% of patients, n (%) | — | — | — | NA | NA | NA |
Febrile neutropenia | 10 (13.9) | 4 (5.3) | 11 (14.5) | NA | NA | NA |
Neutropenia | 2 (2.8) | 3 (4.0) | 1 (1.3) | NA | NA | NA |
Diarrhea | 1 (1.4) | 3 (4.0) | 4 (5.3) | NA | NA | NA |
AE leading to discontinuation of study medication, n (%) | 4 (5.6) | 5 (6.7) | 6 (7.9) | NA | NA | NA |
Left ventricular dysfunction | |||| | |||| | |||| | NA | NA | NA |
Drug hypersensitivity | |||| | |||| | |||| | NA | NA | NA |
Neutropenia | |||| | |||| | |||| | NA | NA | NA |
Deaths | 0 | 0 | 0 | NA | NA | NA |
Notable harms, n (%) | ||||||
Symptomatic LVEF assessed by investigator: NYHA class III or IV | 0 | 1 (1.3) | 0 | NR | NR | NR |
Left ventricular dysfunction | 4 (5.6) | 3 (4.0) | 2 (2.6) | NR | NR | NR |
Left ventricular dysfunction grade ≥ 3 | 0 | 2 (2.7) | 0 | NR | NR | NR |
SAE suggestive of CHF | 1 (1.4) | 2 (2.7) | 0 | NA | NA | NA |
Patients with grade ≥ 3 diarrhea | 3 (4.2) | 4 (5.3) | 9 (11.8) | NR | NR | NR |
Anaphylaxis or hypersensitivity | |||| | |||| | |||| | 2 (2.8) | 0 | 2 (2.6) |
Drug hypersensitivity | 7 (9.7) | 1 (1.3) | 8 (10.5) | NR | NR | NR |
AE = adverse event; ALT = alanine transaminase; CHF = congestive heart failure; doce = docetaxel; FEC = 5-fluorouracil + epirubicin + cyclophosphamide; LVEF = left ventricular ejection fraction; NA = not applicable; NR = not reported; NYHA = New York Heart Association; pert = pertuzumab; SAE = serious adverse event; TCH = docetaxel + carboplatin + trastuzumab; trast = trastuzumab.
Source: Clinical Study Report for TRYPHAENA.6
Table 31: Summary of Harms — BERENICE (Neoadjuvant Period, Safety Population)
Harms | All | Grade ≥ 3 | ||
|---|---|---|---|---|
Cohort A ddAC then pacli + pert + trast N = 199 | Cohort B FEC then doce + pert + trast N = 201 | Cohort A N = 199 | Cohort B N = 201 | |
Patients with at least 1 AE, n (%) | 197 (99.0) | 198 (100.0) | 99 (49.7) | 108 (54.5) |
Most frequent AE, ≥ 20%, any group, n (%) | ||||
Nausea | 141 (70.9) | 137 (69.2) | 5 (2.5) | 4 (2.0) |
Diarrhea | 133 (66.8) | 137 (69.2) | 6 (3.0) | 20 (10.1) |
Constipation | 69 (34.7) | 76 (38.4) | NR | NR |
Vomiting | 45 (22.6) | 69 (34.8) | 2 (1.0) | 8 (4.0) |
Stomatitis | 49 (24.6) | 54 (27.3) | 0 | 9 (5.1) |
Dyspepsia | 38 (19.1) | 32 (16.2) | NR | NR |
Abdominal pain upper | 12 (6.0) | 26 (13.1) | NR | NR |
Abdominal pain | 10 (5.0) | 20 (10.1) | NR | NR |
Gastroesophageal reflux | 23 (11.6) | 4 (2.0) | NR | NR |
Fatigue | 116 (58.3) | 76 (38.4) | 2 (1.0) | 9 (4.5) |
Asthenia | 37 (18.6) | 82 (41.4) | NR | NR |
Mucosal inflammation | 43 (21.6) | 74 (37.4) | 2 (1.0) | 7 (3.5) |
Alopecia | 124 (62.3) | 116 (58.6) | NR | NR |
Headache | 60 (30.2) | 28 (14.1) | NR | NR |
Peripheral neuropathy | 46 (23.1) | 26 (13.1) | 4 (2.0) | 0 |
Myalgia | 40 (20.1) | 66 (33.3) | NR | NR |
Arthralgia | 39 (19.6) | 42 (21.2) | NR | NR |
Epistaxis | 50 (25.1) | 37 (18.7) | NR | NR |
Cough | 40 (20.1) | 17 (8.6) | NR | NR |
Anemia | 54 (27.1) | 60 (30.3) | 6 (3.0) | 5 (2.5) |
Neutropenia | 44 (22.1) | 32 (16.2) | 24 (12.1) | 17 (8.6) |
Decreased appetite | 39 (19.6) | 45 (22.7) | NR | NR |
Febrile neutropenia | NR | NR | 14 (7.0) | 34 (17.2) |
Patients with at least 1 SAE, n (%) | 45 (22.6) | 52 (26.3) | — | — |
Febrile neutropenia | 12 (6.0) | 27 (13.6) | NA | NA |
Diarrhea | 1 (0.5) | 11 (5.6) | NA | NA |
Neutropenic sepsis | 0 | 7 (3.5) | NA | NA |
AE leading to discontinuation of pert or trast | 10 (5.0) | 4 (2.0) | — | — |
Cardiac failure | |||| | |||| | NA | NA |
Ejection fraction decreased | |||| | |||| | NA | NA |
Neutropenia | |||| | |||| | NA | NA |
AE leading to dose interruption or delay, n(%) | |||| | |||| | NA | NA |
Notable harms, n (%) | ||||
Patients who developed HF during neoadjuvant treatment | 3 (1.5) | 0 | NA | NA |
NYHA class III | 3 (1.5) | 0 | NA | NA |
NYHA class IV | 1 (0.5) | 0 | NA | NA |
Total number of patients with at least 1 LVEF significant decline event | 13 (6.5) | 4 (2.0) | NA | NA |
Onset during HER2 antibody neoadjuvant therapy | 13 (6.5) | 3 (1.5) | NA | NA |
Total number of patients with at least 1 confirmed LVEF significant decline | 2 (1.0) | 1 (0.5) | NA | NA |
Onset during HER2 antibody neoadjuvant therapy | 2 (1.0) | 1 (0.5) | NA | NA |
Patients with grade ≥ 3 diarrhea | 6 (3.0) | 20 (10.1) | NA | NA |
Anaphylaxis or hypersensitivity | |||| | |||| | |||| | |||| |
Infusion reactions related to pert or trast | |||| | |||| | NA | NA |
AE = adverse event; ddAC = dose-dense doxorubicin and cyclophosphamide; HER2 = human epidermal growth factor receptor 2; HF = heart failure; LVEF = left ventricular ejection fraction; NA = not applicable; NR = not reported; NYHA = New York Heart Association; pacli = paclitaxel; pert = pertuzumab; trast = trastuzumab.
Note: LVEF significant decline is the decline in LVEF of ≥ 10% points from baseline to an LVEF of < 50%.
Confirmed LVEF declines are defined as at least 2 consecutive readings of significant declines in LVEF and single LVEF declines are defined as only 1 reading of significant decline (no consecutive readings) in LVEF.
Source: Clinical Study Report for BERENICE.7
Critical Appraisal
Internal Validity
There was no blinding in the NEOSPHERE trial. Additionally, pCR rates in NEOSPHERE were not assessed by a centralized, blinded IRC, a design feature of this trial that has been criticized by Health Canada and does not follow FDA guidance for assessing pCR. This is less likely to have biased outcomes such as survival; however, the impact on outcomes such as pCR and clinical response, and the assessment of these outcomes, is unclear. PEONY was double-blinded, and blinding included patients, investigators, and study personnel, and there were steps taken to facilitate blinding, such as the use of a matching placebo.
NEOSPHERE, PEONY, and TRYPHAENA used randomization to assign patients to treatment arms, while BERENICE did not. In the randomized trials, randomization was stratified and steps were taken, namely, the use of an IxRS, to maintain allocation concealment in PEONY, the only blinded study. The fact that BERENICE was not randomized is a limitation, as this increases the potential for bias in the allocation of patients to the groups within the study. Additionally, randomization, when stratified, assists in maintaining equal distribution of important baseline characteristics between groups. Without randomization, there is potential for confounding of the treatment effect estimates for efficacy and in the assessment of harms. However, baseline characteristics in BERENICE were reasonably well balanced between groups.
The percentage of patients withdrawing during the neoadjuvant period was generally low (< 10%) across studies, and generally similar between treatment groups, with 1 exception being the pertuzumab and trastuzumab arm (9.3%) in NEOSPHERE. On average, patients received 3.9 cycles of neoadjuvant therapy, which is very close to the planned 4 cycles, and about 3% of patients in the pertuzumab and trastuzumab plus docetaxel arm had treatment delays or modifications to their regimens, mainly due to adverse events, and this was similar to what was seen in the trastuzumab plus docetaxel arm in both NEOSPHERE and PEONY.
Missing data for the primary dichotomous outcomes were accounted for by counting missing values as nonresponders. This is a conservative approach that may bias results in favour of 1 group if there are clear differences in withdrawals between treatment arms. This was not the case for the comparisons of interest in either NEOSPHERE or PEONY; therefore, this approach is unlikely to have biased results.
Neither TRYPHAENA nor BERENICE were designed to perform any hypothesis testing and, therefore, any of the reported efficacy data are for supportive purposes only.
The NEOSPHERE trial used a Simes method to control for multiplicity. There were numerous statistical tests conducted between the 4 groups in this trial, and Simes appears to be an acceptable method for controlling for type I error. However, in NEOSPHERE, the alpha was set at 0.2 rather than the traditional 0.05, as the trial was designed as a “proof of concept” phase II study rather than a confirmatory phase III study. This means that in NEOSPHERE, there was a greater chance of finding a statistically significant difference between treatment groups where none actually existed compared with what would be expected in a pivotal trial. The method used for controlling for multiple statistical testing was not described in the protocol for PEONY, and P values beyond the primary outcome were stated to be “descriptive only.” For this reason, statistical analyses and P values beyond the primary outcome should be interpreted with consideration of the risk of type I error.
The primary outcome of both NEOSPHERE and PEONY was pCR, although each trial defined pCR differently, with bpCR being used in NEOSPHERE and tpCR in PEONY. The validity of pCR as a surrogate outcome is reviewed in detail in Appendix 4. A number of meta-analyses were found that assess the evidence for pCR as a prognostic marker for OS and EFS in breast cancer. Based on these meta-analyses, there is evidence for pCR as a prognostic factor on an individual-patient basis, but not sufficient evidence to identify the magnitude of pCR improvement that predicts long-term prognosis on a trial level. Additionally, the FDA only recommends use of pCR as a primary outcome for drugs that are undergoing an accelerated review pathway. Both the EMA and the FDA recommend that a pCR be declared only when there is no evidence of invasive cancer from breast tissue and all sampled regional lymph nodes. This definition was used in PEONY for the primary outcome, described as tpCR while, in NEOSPHERE, a pCR appears to have been defined based on breast tissue only. Therefore, the definition of pCR used for the primary outcome in PEONY may more closely reflect contemporary guidance on the assessment of this outcome. The difference in the definition of pCR in these 2 studies reflects changes in guidance that occurred between the time of the design of NEOSPHERE and the more recent PEONY trial.
Breast-conserving surgery is likely an important outcome to some patients with early breast cancer; however, interpretation of results for this outcome is greatly complicated by patient (and physician) preferences. There are some patients who may wish to undergo a mastectomy regardless of the status of their tumour, as their priority is to eliminate the cancer at all costs, while others may place a greater emphasis on cosmesis. The patient’s physician may also influence this decision based on their biases, 1 way or the other; therefore, while this is an important outcome, it may not be possible to interpret these data.
External Validity
The clinical expert consulted by CADTH for this review stated they believed the populations in NEOSPHERE and PEONY reflect the populations that would be expected to be treated with pertuzumab in Canada. Although PEONY was based entirely in Asia, the clinical expert stated they believed this study was still generalizable to the Canadian population. Neither NEOSPHERE nor PEONY included males with breast cancer and, although breast cancer in men is relatively uncommon, this is still a gap in knowledge about pertuzumab for this indication, which does not restrict based on gender. There was a relatively high percentage of patients who were screened out of each of the studies (25% or more); however, in the studies that reported reasons for screen failures, the most common reasons (HER2-negative disease, metastatic disease, HER2 status unavailable) were unlikely to present significant generalizability issues. There was also a relatively large percentage of patients with protocol violations, including those involving the inclusion and exclusion criteria in NEOSPHERE. The exact nature of the violations involving inclusion or exclusion criteria is not known, nor is the impact of the inclusion of these patients on the study results.
The primary outcome of both NEOSPHERE and PEONY was pCR. This and objective response are relevant outcomes to assess in a neoadjuvant trial, as survival data are not generally assessable from the neoadjuvant phase alone because there are typically very few deaths in the neoadjuvant phase in a population such as this. As the clinical expert consulted by CADTH noted, the goal of neoadjuvant therapy is curative, and that is what pCR is intended to assess.
OS was not assessed as an outcome in NEOSPHERE. While OS was assessed as a secondary outcome in PEONY, the sponsor indicated the data are not yet mature to report on this or any other survival outcome. Once OS data are reported from PEONY, interpretation of it and other longer-term outcomes may be complicated by the fact that patients in the comparison groups received different adjuvant regimens; therefore, any differences seen in OS cannot necessarily be attributed to the addition of pertuzumab to trastuzumab and chemotherapy in the neoadjuvant phase.
Health-related quality of life and symptoms were not assessed in any of the included trials. The focus of the neoadjuvant treatment is curative, and it is clear from patient input to CADTH that patients are willing to trade adverse effects in exchange for a treatment that can alter their disease course. Many of the patients at this stage of their disease are largely asymptomatic or mildly symptomatic; therefore, improving their quality of life with drug therapy at this stage may be challenging, and patients may be concerned about a treatment that will reduce their quality of life due to adverse effects. Therefore, although health-related quality of life may not be as important a consideration in early-stage versus late-stage disease, it is still important to patients and should have been assessed as an outcome in the included studies.
The treatment regimens used in the neoadjuvant phase of the included studies appear to be consistent with what 1 would expect a patient to receive for this indication in Canada, according to the clinical experts consulted by CADTH. The adjuvant phase of all of the included trials included trastuzumab, which would be standard for this indication in Canada; however, the 2 recent trials, PEONY and BERENICE, also included pertuzumab in at least 1 treatment arm in the adjuvant phase. Pertuzumab is not publicly funded for adjuvant treatment of early breast cancer in Canada; therefore, this may be a generalizability issue, as its use is likely limited in this country.
Indirect Evidence
No ITCs were submitted by the sponsor.
CADTH conducted a literature search to determine if there was additional indirect evidence that evaluated the efficacy and safety of pertuzumab as a neoadjuvant treatment for patients with HER2-positive, locally advanced, inflammatory, or early breast cancer.
A focused literature search for ITCs dealing with HER2 and breast cancer was run in MEDLINE All (1946—) on May 12, 2021. No limits were applied to the search.
One researcher screened the search results for ITCs that met the population, intervention, comparator, and outcome criteria listed in the systematic review protocol (Table 5). Six potentially relevant ITCs were identified from the literature.20-25 Three reports were excluded because they did not include pertuzumab,23 were focused on adjuvant therapy,20 or were replaced by a more recent update by the same authors.21 Thus, there were 3 ITCs that included the population and intervention of interest.22,24,25 A further review of these reports was conducted to determine if the data available addressed gaps in the direct evidence.
Two reports22,24 did not include the most recent evidence for pertuzumab from the PEONY trial, and 1 ITC25 focused on the intensity of the chemotherapy regimens used with targeted therapies.
Considering that the systematic review included 2 head-to-head RCTs for the comparison of interest, it was determined that these ITCs did not address any gaps and, thus, these reports have not been summarized.
Other Relevant Evidence
This section is for the inclusion of submitted long-term extension studies and additional relevant studies included in the sponsor’s submission to CADTH that are considered to address important gaps in the evidence included in the systematic review. In this case, there were no additional studies found that would inform this review.
Discussion
Summary of Available Evidence
Four trials that were identified by the sponsor as pivotal were included in this review, all focusing on neoadjuvant pertuzumab in early breast cancer. Two of the trials, NEOSPHERE and PEONY, had an appropriate comparator group relevant to the Canadian context and, of these 2 trials, NEOSPHERE (n = 417) was open-label and PEONY (N = 329) was double-blinded, and both assessed pCR as the primary outcome. The objective of the other 2 trials, TRYPHAENA (N = 225) and BERENICE (N = 400), was to assess the safety and tolerability of the addition of pertuzumab to trastuzumab across a variety of different chemotherapy regimens and, thus, neither had a comparator of interest for this review. TRYPHAENA was an open-label RCT while, in BERENICE, investigators were responsible for allocating patients to groups.
The was no ITC submitted by the sponsor, and no ITCs were found in the literature that were thought to be potentially informative to this review. No studies were found for the “other relevant evidence” section.
With respect to baseline characteristics, patients were approximately 50 years of age, on average, and the majority (about 70% to 80%) were White, except in PEONY, where all patients were Asian. Most patients (90%) had an ECOG performance status of 0 and the remaining had an ECOG performance status of 1. Approximately half of the patients were either estrogen or progesterone receptor–positive, with the exception of BERENICE, where about 2-thirds of patients were estrogen or progesterone receptor–positive. The majority of study patients had operable (approximately 60% in NEOSPHERE) or early-stage disease (70% in PEONY), followed by locally advanced disease (approximately 30%). Approximately 7% of patients in NEOSPHERE had inflammatory breast cancer, while this subtype was excluded from PEONY. Similar proportions of patients were seen for these disease categories in TRYPHAENA, with the majority being operable (approximately 70%), followed by locally advanced (approximately 25%) and inflammatory breast cancer. BERENICE did not report disease category at baseline.
Interpretation of Results
Efficacy
NEOSPHERE (bpCR) and PEONY (tpCR) both assessed pCR as their primary outcome, and this is an appropriate outcome for the neoadjuvant setting, according to the clinical experts consulted by CADTH, as the goal of treatment in early-stage breast cancer is to cure the patient of their disease. Therefore, a pCR not only implies freedom from malignant cells at the time of pathologic assessment at surgery (pCR) but also a reduced risk of relapse. Although the former is readily assessed in the neoadjuvant phase of treatment, assessment of the latter requires a much longer follow-up period. Statistically significant improvements were observed with the addition of pertuzumab over trastuzumab plus docetaxel in both RCTs; in NEOSPHERE, the difference between groups for pCR was 16.8% (95% CI, 3.5 to 30.1) and, in PEONY, the difference between groups for tpCR was 17.5% (95% CI, 6.9 to 28.0). In the non-comparative trials, depending on chemotherapy regimen, the bpCR ranged from 57.3% to 66.2% across groups in TRYPHAENA, and tpCR was 61.8% and 60.7% across groups in BERENICE. NEOSPHERE was not a blinded study; assessment of pathology for pCR was carried out in an unblinded manner by pathologists and was not performed by an IRC, a design feature that has been criticized by Health Canada and which does not follow FDA guidance for such assessments. It is unclear what impact an unblinded assessment would have on this outcome. However, generally, across the 4 trials, the evidence suggests that the addition of pertuzumab to trastuzumab and chemotherapy provides additional benefit with respect to pCR, whether it is assessed as tpCR or bpCR, although the long-term benefits of achieving a pCR in the neoadjuvant phase are less clear.
OS was not assessed as an efficacy outcome in NEOSPHERE, and the data are not yet mature in PEONY, according to the sponsor, to provide an assessment of this outcome. Once survival data are mature enough to report on survival outcomes in PEONY, these findings may be confounded by the fact that the 2 groups in the study received different treatments in the adjuvant phase of treatment. To assess the impact of neoadjuvant treatment on survival outcomes, it is important that all groups receive the same treatment in the adjuvant phase, as this is the only way 1 can attribute differences in survival responses to different treatments in the neoadjuvant phase. PFS and DFS were not improved when pertuzumab was added to trastuzumab plus docetaxel; therefore, there is no clear evidence that the improvements in pCR reported in NEOSPHERE have a positive impact on longer-term efficacy, and there are no survival data from PEONY as of this report. The predictive value of pCR for these longer-term outcomes is reviewed in Appendix 4, and there is conflicting evidence for the value of pCR as a surrogate for outcomes such as OS, depending on whether the analysis is based on individual patient–level data (responder analyses) or trial-level data.
Health-related quality of life and symptoms were not assessed in any of the included trials. There is no question that breast cancer has a significant impact on a patient’s health-related quality of life, and this is well described in patient input to CADTH. However, 1 of the clinical experts consulted by CADTH noted that in the neoadjuvant setting, where patients have early-stage breast cancer and the goal is curative, neither of these outcomes are as relevant as they would be in more advanced stages of cancer. The clinical expert stated they believed the fact that there were relatively few treatment withdrawals in either study (in either the pertuzumab and trastuzumab plus docetaxel group or trastuzumab plus docetaxel group) suggests that the addition of pertuzumab to the standard regimen for these patients is not causing intolerable adverse effects, and that should be a consideration when assessing a drug intended for the neoadjuvant setting. In their input to CADTH, patients were clear they would be willing to accept additional adverse effects from the addition of pertuzumab if the trade-off were a clinically meaningful improvement in efficacy, although they also noted the importance of maintaining quality of life through treatment. As such, the lack of data on health-related quality of life and symptoms should still be considered an important gap in knowledge about pertuzumab for this indication.
Harms
The most common adverse effects from NEOSPHERE and PEONY were those either typically associated with chemotherapy (neutropenia) or those associated with epidermal growth factor receptor inhibitors (diarrhea). Given the relatively small number of patients who withdrew from treatment due to an adverse event, and given that, on average, patients completed 3.9 of a possible 4 cycles of neoadjuvant therapy in both the pertuzumab and trastuzumab plus docetaxel group and the trastuzumab plus docetaxel group, the addition of pertuzumab to trastuzumab plus docetaxel appeared to be well tolerated by patients.
Cardiotoxicity is identified as a serious potential harm associated with the combination of pertuzumab and trastuzumab plus docetaxel. Cardiotoxicity, most notably heart failure, is a side effect observed with a number of cytotoxic chemotherapy drugs as well as with trastuzumab. Therefore, adding pertuzumab, which may also cause cardiotoxicity as a class effect of HER2 inhibitors, is a valid concern, although data from NEOSPHERE and PEONY do not suggest a clear increased risk of reduced ejection fraction or any other indicators of cardiotoxicity from adding pertuzumab to trastuzumab plus docetaxel.
Conclusions
Four trials that were identified as pivotal by the sponsor were included in this review. Rates of pCR were improved when pertuzumab was added to standard neoadjuvant regimens with trastuzumab plus chemotherapy in the 2 trials that featured a comparator, NEOSPHERE and PEONY. It is unclear whether these improvements in pCR translate into improved OS, as this outcome was not studied in NEOSPHERE, and the survival data from PEONY were not available at the time of review. The combination of pertuzumab with trastuzumab plus chemotherapy did not appear to improve invasive DFS, PFS, EFS, or DFS, either because these outcomes were not studied, the data were not yet available, or there was a lack of statistical significance when they were assessed. Health-related quality of life and symptoms were not assessed in any of the included studies. Based on the included studies, the addition of pertuzumab to trastuzumab plus chemotherapy did not appear to introduce significant safety or tolerability issues.
References
1.Korde LA, Somerfield MR, Carey LA, et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J Clin Oncol. 2021;39(13):1485-1505. PubMed
2.Cardoso F, Kyriakides S, Ohno S, et al. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann Oncol. 2019;30(8):1194-1220. PubMed
3.Ditsch N, Kolberg-Liedtke C, Friedrich M, et al. AGO Recommendations for the Diagnosis and Treatment of Patients with Early Breast Cancer: Update 2021. Breast Care (Basel). 2021;16(3):214-227. PubMed
4.Clinical Study Report: 1032196. A randomized, multicenter, multinational Phase II study on trastuzumab plus docetaxel versus trastuzumab plus docetaxel plus pertuzumab versus trastuzumab plus pertuzumab versus pertuzumab and docetaxel in patients with locally advanced, inflammatory or early stage HER2 positive breast cancer. Report No. 1032196.Version 1: September 2010; Version 2: December 2010 (to include investigators’ CVs); Version 3: June 2011 (to correct one patient’s primary endpoint data and add biomarker results). [internal sponsor's report]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2011.
5.Clinical Study Report: PEONY. YO28762 A randomized, multicenter, double-blind, placebo-controlled, phase III study to evaluate pertuzumab in combination with docetaxel and trastuzumab as neoadjuvant therapy, and pertuzumab in combination with trastuzumab as adjuvant therapy after surgery and chemotherapy in patients with early-stage or locally advanced HER2-positive breast cancer. [internal sponsor's report]. Mississauga (ON): Hoffmann-La Roche Ltd; 2018.
6.Clinical Study Report: TRYPHAENA. BO22280 - A randomised, multicentre, multinational Phase II study to evaluate pertuzumab in combination with trastuzumab, given either concomitantly or sequentially with standard anthracycline-based chemotherapy or concomitantly with a non-anthracycline-based chemotherapy regimen, as neoadjuvant therapy for patients with locally advanced, inflammatory or early stage HER2-positive breast cancer. [internal sponsor's report]. Mississauga (ON): Hoffmann-La Roche Ltd; 2012.
7.Clinical Study Report: BERENICE. WO29217 – A Multicenter, Multinational, Phase II Study to Evaluate Perjeta in Combination with Herceptin and Standard Neoadjuvant Anthracycline-Based Chemotherapy in Patients with HER2-Positive, Locally Advanced, Inflammatory, or Early-Stage Breast Cancer. [internal sponsor's report]. Mississauga (ON): Hoffmann-La Roche, Ltd; 2017.
8.Canadian Cancer Society. Breast Cancer Statistics 2021; www.cancer.ca/en/cancer-information/cancer-type/breast/statistics/?region = on. Accessed 2021 Jun 17.
9.Perjeta® pertuzumab for injection Sterile Concentrate for Solution for Infusion, 420mg/14 mL vial [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Mar 11.
10.Herceptin (Trastuzumab) for injection [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 1999.
11.Spring LM, Fell G, Arfe A, et al. Pathologic Complete Response after Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive Meta-analysis. Clin Cancer Res. 2020;26(12):2838-2848. PubMed
12.de Azambuja E, Holmes AP, Piccart-Gebhart M, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): survival outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. The Lancet Oncology. 2014;15(10):1137-1146. PubMed
13.Moreno-Aspitia A, Holmes EM, Jackisch C, et al. Updated results from the phase III ALTTO trial (BIG 2-06; NCCTG (Alliance) N063D) comparing one year of anti-HER2 therapy with lapatinib alone (L), trastuzumab alone (T), their sequence (T→L) or their combination (L+T) in the adjuvant treatment of HER2-positive early breast cancer. Journal of Clinical Oncology. 2017;35(15_suppl):502-502.
14.Gianni L, Pienkowski T, Im YH, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25-32. PubMed
15.Gianni L, Pienkowski T, Im YH, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016;17(6):791-800. PubMed
16.Shao Z, Pang D, Yang H, et al. Efficacy, Safety, and Tolerability of Pertuzumab, Trastuzumab, and Docetaxel for Patients With Early or Locally Advanced ERBB2-Positive Breast Cancer in Asia: The PEONY Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6(3):e193692. PubMed
17.Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Annals of Oncology. 2013;24(9):2278-2284. PubMed
18.Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. European Journal of Cancer. 2018;89:27-35. PubMed
19.Swain SM, Ewer MS, Viale G, et al. Pertuzumab, trastuzumab, and standard anthracycline- and taxane-based chemotherapy for the neoadjuvant treatment of patients with HER2-positive localized breast cancer (BERENICE): a phase II, open-label, multicenter, multinational cardiac safety study. Annals of Oncology. 2018;29(3):646-653. PubMed
20.Debiasi M, Polanczyk CA, Ziegelmann P, et al. Efficacy of Anti-HER2 Agents in Combination With Adjuvant or Neoadjuvant Chemotherapy for Early and Locally Advanced HER2-Positive Breast Cancer Patients: A Network Meta-Analysis. Front. 2018;8:156. PubMed
21.Nagayama A, Hayashida T, Jinno H, et al. Comparative effectiveness of neoadjuvant therapy for HER2-positive breast cancer: a network meta-analysis. J Natl Cancer Inst. 2014;106(9). PubMed
22.Nakashoji A, Hayashida T, Yokoe T, et al. The updated network meta-analysis of neoadjuvant therapy for HER2-positive breast cancer. Cancer Treatment Reviews. 2018;62:9-17. PubMed
23.Pathak M, Deo SV, Dwivedi SN, Thakur B, Sreenivas V, Rath GK. Regimens of neo-adjuvant chemotherapy in the treatment of breast cancer: A systematic review & network meta-analysis with PRISMA-NMA compliance. Crit Rev Oncol Hematol. 2020;153:103015. PubMed
24.Wu D, Chen T, Jiang H, et al. Comparative Efficacy and Tolerability of Neoadjuvant Immunotherapy Regimens for Patients with HER2-Positive Breast Cancer: A Network Meta-Analysis. J. 2019;2019:3406972.
25.Zhang J, Yu Y, Lin Y, et al. Efficacy and safety of neoadjuvant therapy for HER2-positive early breast cancer: a network meta-analysis. Therapeutic Advances in Medical Oncology. 2021;13:17588359211006948. PubMed
26.Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet (London, England). 2014;384(9938):164-172. PubMed
27.Cortazar P, Zhang L, Untch M, et al. Abstract S1-11: Meta-analysis Results from the Collaborative Trials in Neoadjuvant Breast Cancer (CTNeoBC). Cancer Res. 2012;72(24 Supplement):S1-11-S11-11.
28.Berruti A, Amoroso V, Gallo F, et al. Pathologic complete response as a potential surrogate for the clinical outcome in patients with breast cancer after neoadjuvant therapy: a meta-regression of 29 randomized prospective studies. J Clin Oncol. 2014;32(34):3883-3891. PubMed
29.Broglio KR, Quintana M, Foster M, et al. Association of Pathologic Complete Response to Neoadjuvant Therapy in HER2-Positive Breast Cancer With Long-Term Outcomes: A Meta-Analysis. JAMA Oncol. 2016;2(6):751-760. PubMed
30.Huang M, O'Shaughnessy J, Zhao J, et al. Evaluation of Pathologic Complete Response as a Surrogate for Long-Term Survival Outcomes in Triple-Negative Breast Cancer. J. 2020;18(8):1096-1104. PubMed
31.Kong X, Moran MS, Zhang N, Haffty B, Yang Q. Meta-analysis confirms achieving pathological complete response after neoadjuvant chemotherapy predicts favourable prognosis for breast cancer patients. European Journal of Cancer. 2011;47(14):2084-2090. PubMed
32.Korn EL, Sachs MC, McShane LM. Statistical controversies in clinical research: assessing pathologic complete response as a trial-level surrogate end point for early-stage breast cancer. Ann Oncol. 2016;27(1):10-15. PubMed
33.Nekljudova V, Loibl S, von Minckwitz G, et al. Trial-level prediction of long-term outcome based on pathologic complete response (pCR) after neoadjuvant chemotherapy for early-stage breast cancer (EBC). Contemp Clin Trials. 2018;71:194-198. PubMed
34.European Medicines Agency. Appendix 4 to the guideline on the evaluation of anticancer medicinal products in man: condition specific guidance. 2015: https://www.ema.europa.eu/en/documents/scientific-guideline/evaluation-anticancer-medicinal-products-man-appendix-4-condition-specific-guidance-rev2_en.pdf. Accessed 2021 Aug 18.
35.U.S. Food and Drug Administration. Pathological Complete Response in Neoadjuvant Treatment of High-Risk Early-Stage Breast Cancer: Use as an Endpoint to Support Accelerated Approval Guidance for Industry. 2020: https://www.fda.gov/media/83507/download. Accessed 2021 Jun 1.
36.Ciani O, Buyse M, Drummond M, Rasi G, Saad ED, Taylor RS. Time to Review the Role of Surrogate End Points in Health Policy: State of the Art and the Way Forward. Value Health. 2017;20(3):487-495. PubMed
37.Validity of surrogate endpoints in oncology: Executive summary of rapid report A10-05, Version 1.1. 2011: https://www.ncbi.nlm.nih.gov/books/NBK198799/pdf/Bookshelf_NBK198799.pdf. Accessed 2015 Jun 15.
38.U.S. Food and Drug Administration. Table of Surrogate Endpoints That Were the Basis of Drug Approval or Licensure. 2021: https://www.fda.gov/drugs/development-resources/table-surrogate-endpoints-were-basis-drug-approval-or-licensure. Accessed 2021 Jul 14.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 12, 2021
Alerts: Monthly search updates until project completion
Study types: RCTs; controlled clinical trials
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
# Searches
(Perjeta* or pertuzumab* or 2C4 or omnitarg* or r 1273 or r1273 or rg 1273 or rg1273 or K16AIQ8CTM or HSDB 8141 or HSDB8141).ti,ab,kf,ot,hw,rn,nm.
exp Trastuzumab/
(trastuzumab* or Herceptin* or Trazimera* or ontruzant* or vivitra* or zedora* or P188ANX8CK or kanjinti* or ogivri* or herzuma* or herclon* or herticad* or hertraz* or herzemab* or trastunix*).ti,ab,kf,ot,hw,rn,nm.
2 or 3
1 and 4
5 use medall
*Pertuzumab/
(Perjeta* or pertuzumab* or 2C4 or omnitarg* or r 1273 or r1273 or rg 1273 or rg1273 or HSDB 8141 or HSDB8141).ti,ab,kw,dq.
7 or 8
exp *Trastuzumab/
(trastuzumab* or Herceptin* or Trazimera* or ontruzant* or vivitra* or zedora* or kanjinti* or ogivri* or herzuma* or herclon* or herticad* or hertraz* or herzemab* or trastunix*).ti,ab,kw,dq.
10 or 11
9 and 12
13 use oemezd
6 or 14
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
"Randomized Controlled Trial (topic)"/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
"Controlled Clinical Trial (topic)"/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf,kw.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf,kw.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf,kw.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf,kw.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(pragmatic study or pragmatic studies).ti,ab,hw,kf,kw.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf,kw.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(phase adj3 (III or "3") adj3 (study or studies or trial*)).ti,hw,kf,kw.
or/16-46
15 and 47
48 not (conference abstract or conference review).pt.
remove duplicates from 49
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- neoadjuvant | breast cancer | Pertuzumab AND trastuzumab]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- neoadjuvant | breast cancer | Pertuzumab AND trastuzumab]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- neoadjuvant | breast cancer | Pertuzumab AND trastuzumab]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- neoadjuvant | breast cancer | Pertuzumab AND trastuzumab]
Grey Literature
Search dates: April 30, 2021 to May 6, 2021
Keywords: breast cancer | pertuzumab
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
The complete search archive of sites consulted for this report is available on request.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Van Ramshorst, Lancet Oncology (2018) | Comparator |
Tan, Lancet Oncology (2021) | Comparator |
Bines, British Journal of Cancer (2021) | Population |
Note: This table has not been copy-edited.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 34: Subgroup Analyses for pCR From NEOSPHERE
Characteristic | NEOSPHERE | |||
|---|---|---|---|---|
Trast + Pert + Doce | Trast + Doce | Trast + Pert | Pert + Doce | |
pCR by breast cancer type | ||||
Operable breast cancer | (N = 65) | (N = 64) | (N = 65) | (N = 60) |
Responders | 31/65 (47.7) | 15/64 (23.4) | 11/65 (16.9) | 16/60 (26.7) |
Nonresponders | 34/65 (52.3) | 49/64 (76.6) | 54/65 (83.1) | 44/60 (73.3) |
95% CI for response rates | [35.1; 60.5] | [13.8; 35.7] | [8.8; 28.3] | [16.1; 39.7] |
Inflammatory breast cancer | (N = 10) | (N = 7) | (N = 7) | (N = 5) |
Responders | 4/10 (40.0) | 1/7 (14.3) | 2/7 (28.6) | 2/5 (40.0) |
Nonresponders | 6/10 (60.0) | 6/7 (85.7) | 5/7 (71.4) | 3/5 (60.0) |
95% CI for response rates | [12.2; 73.8] | [0.4; 57.9] | [3.7; 71.0] | [5.3; 85.3] |
Locally advanced breast cancer | (N = 32) | (N = 36) | (N = 35) | (N = 31) |
Responders | 14/32 (43.8) | 15/36 (41.7) | 5/35 (14.3) | 5/31 (16.1) |
Nonresponders | 18/32 (56.3) | 21/36 (58.3) | 30/35 (85.7) | 26/31 (83.9) |
95% CI for response rates | [26.4; 62.3] | [25.5; 59.2] | [4.8; 30.3] | [5.5; 33.7] |
By hormone receptor status | ||||
Estrogen and/or progesterone positive | (N = 50) | (N = 50) | (N = 51) | (N = 46) |
Responders | 13/50 (26.0) | 10/50 (20.0) | 3/51 (5.9) | 8/46 (17.4) |
Nonresponders | 37/50 (74.0) | 40/50 (80.0) | 48/51 (94.1) | 38/46 (82.6) |
95% CI for response rates | [14.6; 40.3] | [10.0; 33.7] | [1.2; 16.2] | [7.8; 31.4] |
Estrogen and progesterone negative | (N = 57) | (N = 57) | (N = 55) | (N = 50) |
Responders | 36/57 (63.2) | 21/57 (36.8) | 15/55 (27.3) | 15/50 (30.0) |
Nonresponders | 21/57 (36.8) | 36/57 (63.2) | 40/55 (72.7) | 35/50 (70.0) |
95% CI for response rates | [49.3; 75.6] | [24.4; 50.7] | [16.1; 41.0] | [17.9; 44.6] |
Receptor status not known | (N = 0) | (N = 0) | (N = 1) | (N = 0) |
Lymph node status | ||||
pCR achieved and neg. lymph nodes at surgery | 42 (39.3) | 23 (21.5) | 12 (11.2) | 17 (17.7) |
pCR achieved and pos. lymph nodes at surgery | 7 (6.5) | 8 (7.5) | 6 (5.6) | 6 (6.3) |
pCR by biomarkers | ||||
HER2 Mem H-score | ||||
High | 36/57 (63.16) | 17/54 (31.48) | 10/52 (19.23) | 13/51 (25.49) |
Low | 13/50 (26.00) | 14/52 (26.92) | 8/55 (14.55) | 10/45 (22.22) |
Chi-Sqa P value | 0.0010 | 0.8899 | 0.6437 | 0.9164 |
HER3 Mem | ||||
High | 25/55 (45.45) | 12/51 (23.53) | 7/57 (12.28) | 11/44 (25.00) |
Low | 21/41 (51.22) | 17/48 (35.42) | 10/42 (23.81) | 9/39 (23.08) |
Chi-Sqa P value | 0.5983 | 0.4079 | 0.4863 | 0.9418 |
IGF-1R Mem | ||||
High | 18/43 (41.86) | 15/46 (32.61) | 6/56 (10.71) | 11/41 (26.83) |
Low | 21/39 (53.85) | 13/42 (30.95) | 5/35 (14.29) | 11/37 (29.73) |
Chi-Sqa P value | 0.9932 | 0.3417 | 0.5085 | 0.7610 |
pAKT Cyt | ||||
High | 28/51 (54.90) | 14/47 (29.79) | 6/53 (11.32) | 14/46 (30.43) |
Low | 10/27 (37.04) | 5/25 (20.00) | 3/26 (11.54) | 6/24 (25.00) |
Chi-Sqa P value | 0.3008 | 0.3942 | 1.0000 | 0.6058 |
PTEN Cyt | ||||
High | 37/78 (47.44) | 20/70 (28.57) | 11/64 (17.19) | 14/57 (24.56) |
Low | 6/17 (35.29) | 9/25 (36.00) | 4/34 (11.76) | 9/28 (32.14) |
Chi-Sqa P value | 0.0996 | 0.6050 | 0.2080 | 0.8134 |
PTEN Nuc | ||||
High | 22/49 (44.90) | 15/53 (28.30) | 8/43 (18.60) | 12/42 (28.57) |
Low | 21/46 (45.65) | 14/42 (33.33) | 7/55 (12.73) | 11/43 (25.58) |
Chi-Sqa P value | 0.8855 | 0.4364 | 0.1565 | 0.5919 |
pAKT Nuc | ||||
High | 21/41 (51.22) | 8/31 (25.81) | 5/41 (12.20) | 12/39 (30.77) |
Low | 17/37 (45.95) | 11/41 (26.83) | 4/38 (10.53) | 8/31 (25.81) |
Chi-Sqa P value | 0.4140 | 0.7296 | 0.5823 | 0.5325 |
EGFR-CR | ||||
High | 24/51 (47.06) | 13/42 (30.95) | 11/54 (20.37) | 11/42 (26.19) |
Low | 20/46 (43.48) | 16/54 (29.63) | 5/45 (11.11) | 12/43 (27.91) |
Chi-Sqa P value | 0.5121 | 0.9919 | 0.3459 | 0.9318 |
HER2-CR | ||||
High | 28/50 (56.00) | 20/53 (37.74) | 11/47 (23.40) | 14/45 (31.11) |
Low | 17/49 (34.69) | 9/47 (19.15) | 6/55 (10.91) | 9/41 (21.95) |
Chi-Sqa P value | 0.1616 | 0.0774 | 0.2009 | 0.3937 |
HER2/HER3-CR | ||||
High | 29/55 (52.73) | 18/55 (32.73) | 12/41 (29.27) | 15/41 (36.59) |
Low | 16/43 (37.21) | 11/43 (25.58) | 5/61 (8.20) | 8/45 (17.78) |
Chi-Sqa P value | 0.5524 | 0.9616 | 0.0790 | 0.1765 |
HER3-CR | ||||
High | 19/47 (40.43) | 12/45 (26.67) | 7/53 (13.21) | 12/47 (25.53) |
Low | 26/51 (50.98) | 17/53 (32.08) | 10/49 (20.41) | 11/39 (28.21) |
Chi-Sqa P value | 0.5874 | 0.9141 | 0.8707 | 0.7987 |
Target/Cen. Ratio (C-Myc) | ||||
High | 12/24 (50.00) | 6/19 (31.58) | 2/23 (8.70) | 3/21 (14.29) |
Low | 19/44 (43.18) | 19/55 (34.55) | 6/44 (13.64) | 13/45 (28.89) |
Chi-Sqa P value | 0.4717 | 0.8257 | 0.4630 | 0.1566 |
Serum amphiregulin [pg/mL] | ||||
High | 21/47 (44.68) | 17/53 (32.08) | 9/49 (18.37) | 11/44 (25.00) |
Low | 25/52 (48.08) | 11/44 (25.00) | 9/52 (17.31) | 11/43 (25.58) |
Chi-Sqa P value | 0.7598 | 0.3249 | 0.8756 | 0.6483 |
Serum EGF [pg/mL] | ||||
High | 26/54 (48.15) | 16/47 (34.04) | 7/48 (14.58) | 9/43 (20.93) |
Low | 20/45 (44.44) | 12/50 (24.00) | 11/53 (20.75) | 13/44 (29.55) |
Chi-Sqa P value | 0.5762 | 0.1966 | 0.4493 | 0.5319 |
Serum TGF-alpha [pg/mL] | ||||
High | 23/45 (51.11) | 18/49 (36.73) | 7/56 (12.50) | 10/42 (23.81) |
Low | 23/54 (42.59) | 10/48 (20.83) | 11/45 (24.44) | 12/45 (26.67) |
Chi-Sqa P value | 0.4000 | 0.0642 | 0.0445 | 0.8828 |
Serum sHER2 [ng/mL] | ||||
High | 21/46 (45.65) | 14/46 (30.43) | 12/46 (26.09) | 13/53 (24.53) |
Low | 24/51 (47.06) | 14/51 (27.45) | 6/54 (11.11) | 9/34 (26.47) |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 340 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 360 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 365 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 370 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 380 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 391 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HER2 Mem H-score 394 | ||||
High | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Low | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Chi-Sqa P value | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
CI = confidence interval; doce = docetaxel; HER2 = human epidermal growth factor receptor 2; pCR = pathologic complete response; pert = pertuzumab; trast = trastuzumab.
a CMH Chi-square test based on biomarker subgroup × pCR status (2 × 2) stratified by hormone receptor status and breast cancer type.
Source: Clinical Study Report for NEOSPHERE.4
Table 35: Subgroup Analyses for tpCR From PEONY
PEONY | ||
|---|---|---|
Characteristic | Trast + Pert + Chemo | Trast + Chemo |
tpCR response, n/N (%) (95% CI) by: | ||
Disease category | ||
Early stage | 63/153 (41.2) (33.29 to 49.41) | 20/77 (26.0) (16.64 to 37.23) |
Difference in response rates (95% CI) | 15.20 (1.97,28.44) | |
Locally advanced | 23/66 (34.8%) (23.53 to 47.58) | 4/33 (12.1) (3.40 to 28.20) |
Difference in response rates (95% CI) | 22.73 (5.02 to 40.43) | |
Hormone receptor status | ||
ER and PgR negative | 47/102 (46.1) (36.16 to 56.23) | 10/54 (18.5) (9.25 to 31.43) |
Difference in response rates (95% CI) | 27.56 (12.36 to 42.76) | |
ER and/or PgR positive | 39/117 (33.3) (24.89 to 42.64) | 14/56 (25.0) (14.39 to 38.37) |
Difference in response rates (95% CI) | 8.33 (−6.86 to 23.53) | |
Age group (years) | ||
< 65 | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
>= 65 | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
Menopausal status at randomization | ||
Postmenopausal | 38//87 (43.7) (33.06 to 54.74) | 15/45 (33.3) (20.00 to 48.95) |
Difference in response rates (95% CI) | 10.34 (−8.20 to 28.89) | — |
Premenopausal | 48/132 (36.4) (28.17 to 45.18) | 9/65 (13.8) (6.53 to 24.66) |
Difference in response rates (95% CI) | 22.52 (9.94 to 35.10) | — |
Primary tumour stage at baseline | ||
T2 | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
T3 or larger | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
Lymph nodes status | ||
Negative | 31/59 (52.5) (39.12 to 65.70) | 8/21 (38.1) (18.11 to 61.56) |
Difference in response rates (95% CI) | 14.45 (−12.80 to 41.69) | — |
Positive | 55/160 (34.4) (27.06 to 42.28) | 16/89 (18.0) (10.64 to 27.55) |
Difference in response rates (95% CI) | 16.40 (4.93 to 27.86) | — |
Histological subtype at baseline | ||
Ductal | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
Non-Ductal | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
ER/PgR status | ||
ER negative, PgR negative | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
ER negative, PgR positive | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
ER positive, PgR negative | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
ER positive, PgR positive | |||||||||||||||| | |||||||||||||||| |
Difference in response rates (95% CI) | |||||||||||||||| | — |
HER2 IHC | ||
2+ | 23/65 (35.4) (23.92 to 48.23) | 6/22 (27.3) (10.73 to 50.22) |
Difference in response rates (95% CI) | 8.11 (−16.52 to 32.75) | — |
3+ | 63/152 (41.4) (33.52 to 49.71) | 18/88 (20.5) (12.60 to 30.39) |
Difference in response rates (95% CI) | 20.99 (8.87 to 33.12) | — |
chemo = chemotherapy; CI = confidence interval; ER = estrogen receptor; HER2 = human epidermal growth factor receptor 2; pCR = pathologic complete response; pert = pertuzumab; PgR = progesterone receptor; trast = trastuzumab.
Source: Clinical Study Report for PEONY.5
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To evaluate the validity of pCR as a surrogate outcome for longer-term survival in patients with early breast cancer. pCR was the primary outcome in the NEOSPHERE and PEONY trials.
Findings
The relationship between pCR and longer-term survival outcomes, such as EFS and OS, in patients who received neoadjuvant treatment for breast cancer has been explored in a number of meta-analyses.11,26-33 In addition, both the FDA and EMA have issued guidance on the role of pCR as an end point in neoadjuvant breast cancer studies.34,35
Meta-Analyses
The FDA-funded meta-analysis by Cortazar et al. 201426 was based on a systematic literature review with a search of OVID (MEDLINE, Embase) and PubMed conducted in 2011. Eligibility criteria included studies having at least 200 patients with primary breast cancer treated with pre-operative chemotherapy followed by surgery. Studies were required to have available data for pCR, EFS, and OS, and have a median follow-up of at least 3 years. Twelve studies were identified, and individual-patient data from these studies were pooled for analysis.
The authors conducted a patient-level analysis, that explored the relationship between pCR and survival at an individual level, as well as a trial-level association, that could be used to predict population treatment benefits. The responder analysis, that compared patients with and without pCR irrespective of neoadjuvant treatment received (N = 11,955), estimated the HR and log-rank test for EFS and OS for the pooled population and subgroups based on Cox regression models stratified by study. The trial-level analysis of the correlation between EFS or OS and pCR was conducted using a weighted linear regression model on a log scale (N = 9,440). Different definitions of pCR were used across studies and these were analyzed separately to determine the which definitions correlate best with long-term outcomes. The 3 definitions analyzed were: ypT0 ypN0 (i.e., absence of invasive cancer and in-situ cancer in the breast and axillary nodes), ypT0/is ypN0 (i.e., absence of invasive cancer in the breast and axillary nodes, irrespective of ductal carcinoma in situ), and ypT0/is (i.e., absence of invasive cancer in the breast irrespective of ductal carcinoma in situ or nodal involvement).
The responder analyses found that eradication of tumour from both the breast and axillary lymph nodes (ypT0 pN0 and ypT0/is ypN0) was better associated with improved EFS and OS than was eradication of invasive tumour from the breast alone. It was also found that the association between pCR and long-term survival outcomes was stronger in patients with high-grade tumours, than with those with low-grade tumours. In the HER2-positive subgroup, pCR was associated with long-term outcomes regardless of hormone receptor status (EFS: HR 0.39; 95% CI, 0.31 to 0.50; OS: HR 0.34; 95% CI, 0.24 to 0.47). The most favourable outcomes after pCR were recorded in patients with HER2-positive, hormone receptor–negative tumours who received trastuzumab (EFS: HR 0.15; 95% CI, 0.09 to 0.27; OS: HR 0.08; 95% CI, 0.03 to 0.22), and in the triple-negative subgroup (EFS: HR 0.24; 95% CI, 0.18 to 0.33; OS: HR 0.16; 5% CI, 0.11 to 0.25).26
At the trial level, little association was found between an increase in the frequency of pCR and the impact of treatment on EFS or OS. These results were reported based on the coefficient of determination (R2) which represents the proportion of the variation in the outcome (i.e., difference in EFS or OS) that can be explained by the regression on the difference in pCR. The analyses reported an R2 of 0.03 (95% CI, 0.00 to 0.25) between pCR and EFS, and R2 of 0.24 (95% CI, 0.00 to 0.70) between pCR and OS. Results were similar when analyzed according to breast cancer subtype.26 Cortazar et al. concluded that “our pooled analysis could not validate pCR as a surrogate end point for improved EFS or OS.”26
Other meta-analyses have reported an association between pCR and long-term survival outcomes at the individual patient level,11,29,31 but generally weak association when analyzed at the trial (population) level.28-30,32 Berruti et al. 201428 reported weak trial-level association between pCR and DFS or OS in their analysis of 29 RCTs of neoadjuvant breast cancer treatments (R2 0.08 and 0.09, respectively). The analysis by Broglio et al. 201629 reported weak trial-level association between pCR and EFS (R2 0.23) but not for OS (R2 0) based on their analysis of 38 randomized and non-randomized studies in HER2-positive breast cancer. Based on 8 RCTs in patients with triple-negative breast cancer, Huang et al. 2020,30 reported moderate correlation between the log OR of pCR and the log HR of EFS (R2 0.68; 95% CI, 0.41 to 0.95) but weaker association between pCR and OS (R2 0.24; 95% CI, 0.01 to 0.77).
None of the trial-level associations reported in the meta-analyses met the thresholds that have been reported in the literature to validate surrogates.36,37 The German HTA organization Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWIG) reviewed the methodological approaches to validate surrogate outcomes and reported that while there is no universally applicable measure for surrogate validation, correlation-based approaches, which include the estimation of study-level and individual-level correlation measures, are frequently used in current practice. IQWIG states that proof of validity of a surrogate may be inferred from high trial-level correlation between the surrogate and the patient-relevant outcome (if the lower limit of the 95% CI for R [correlation coefficient] exceeds 0.85), whereas low correlation (upper limit of the 95% CI for R ≤ 0.70) results in proof of the lack of validity of the surrogate.37 Validity of surrogate outcomes is unclear if R values fall between these 2 thresholds.37 A report by Ciani et al. 2017 suggests that trial-level correlation coefficients (r or ρ) of 0.8 or R2 of 0.65 may be required for surrogate validation.36 Both reports state that validation of surrogate outcomes should be based on a meta-analysis of several RCTs that measure both the surrogate and clinically-relevant outcomes.36,37 Further, Ciani et al.36 state that an individual patient–level meta-analysis of all RCTs for a treatment is the most reliable approach. These reports maintain that conclusions regarding the validity of a surrogate is specific to a given context and cannot be transferred between different diseases, disease severity, or interventions.36,37
The meta-analyses exploring the validity of pCR as a surrogate outcome in patients with breast cancer were not without limitations, which may have impacted the results of the individual and trial-level analyses. The studies included in the meta-analyses were heterogeneous, with some analyses pooling randomized and non-randomized trials, and with the trials reporting outcomes using different definitions of pCR and breast cancer recurrence. The analyses may also be confounded by the heterogeneity in patient and treatment characteristics, as the trials enrolled patients with different subtypes and grades of breast cancer who received a variety of treatment regimens before and after surgery. pCR rates, as well as the relationship between pCR and longer-term survival outcomes may vary by breast cancer subtype and grade, or treatment received, thus inclusion of heterogeneous studies my obscure the association between pCR and survival. Cortazar et al.26 also noted that their analysis included few trials of HER2 targeted therapy (i.e., trastuzumab), and so the generalizability to neoadjuvant pertuzumab may be limited. Other limitations of the clinical trials included in the meta-analyses may be lack of statistical power to detect longer-term outcomes, or insufficient follow-up time.
Overall, the meta-analyses provide evidence of pCR as a prognostic marker on an individual-patient basis, but the evidence was insufficient to identify the magnitude of pCR improvement that is able to predict long-term prognosis at a trial level.
Regulatory Organizations
The FDA has accepted pCR as a surrogate outcome for accelerated approval in patients with breast cancer.38 In 2020, the FDA published updated guidance on pCR as an end point for neoadjuvant treatment of high-risk early-stage breast cancer, which was informed by the meta-analysis by Cortazar et al.27 that included approximately 13,000 patients enrolled in neoadjuvant trials. Based on this analysis the FDA recognized 2 definitions of pCR for marketing approval:
pCR is defined as the absence of residual invasive cancer on hematoxylin and eosin evaluation of the complete resected breast specimen and all sampled regional lymph nodes following completion of neoadjuvant systemic therapy (i.e., ypT0 or Tis ypN0 in the current American Joint Committee on Cancer [AJCC] staging system) or
pCR is defined as the absence of residual invasive and in-situ cancer on hematoxylin and eosin evaluation of the complete resected breast specimen and all sampled regional lymph nodes following completion of neoadjuvant systemic therapy (i.e., ypT0 ypN0 in the current AJCC staging system) (page 5)35
Guidance from the EMA also recommends the absence of residual invasive cancer in the breast and all sampled ipsilateral lymph nodes (ypT0/Tis ypN0) as the preferred definition of pCR, as eradication of the tumour from the breast and lymph nodes is associated with better EFS and OS, compared with eradication from the breast only.34 The presence of ductal carcinoma in situ does not appear to affect long-term outcomes.34,35
In their guidance, the EMA notes that several studies have shown an association between pCR following primary systemic therapy, and better OS in patients with breast cancer. However, based on these data, it is not possible to separate the importance of the treatment factor from the patient factor, thus true surrogacy of pCR (i.e., to what extent a certain difference in pCR rate can predict a certain difference in EFS) has not been established.34 The EMA states “Currently available data do not allow a precise prediction of the magnitude of the EFS/DFS/OS effect from a certain pCR effect. Therefore, a substantial increase in pCR shown in sufficiently large randomized trials is required for there to be a reasonable likelihood that this will translate into a clinically meaningful improvement in long-term outcomes.”34 The EMA also states that approval based on pCR for an add-on neoadjuvant regimen in patients with high-risk breast cancer may be accepted provided the mechanism of action is well characterized, the results show a high increase in pCR with only minor changes in toxicity, and supportive evidence for safety and efficacy is available in the metastatic setting. These data may lead to approval provided that confirmatory study data showing EFS, DFS, or OS are to follow.34
The FDA cited the following conclusion supporting the use of pCR for accelerated drug approval:
“Given the substantial improvements in survival for individual patients who attain pCR, a novel agent administered with standard therapy that produces a marked absolute increase in pCR rate compared with standard therapy alone in the intent-to-treat population (i.e., all randomized patients) may be reasonably likely to result in long-term improvements in EFS or OS. Different breast cancer subtypes may require different magnitudes of improvement in pCR rate to achieve clinically meaningful improvement in EFS or OS. Therapies that modestly increase pCR rate are less likely to improve long-term outcomes in any subtype.”35
The FDA concluded “… that important regulatory questions persist regarding use of pCR to support accelerated approval in high-risk early-stage breast cancer. A trial-level relationship between improvement in pCR and improvement in long-term outcome has not been established. If such a relationship exists, it is unknown whether the necessary magnitude of improvement in pCR will differ according to breast cancer subtype or drug class.”35 Confirmatory trials that demonstrate clinically meaningful and statistically significant improvement in EFS, DFS, or OS are required to verify the clinical benefit a drug granted accelerated approval based on pCR.35
Pharmacoeconomic Review
Abbreviations
AE
adverse event
DFS
disease-free survival
eBC
early-stage breast cancer
EFS
event-free survival
HER2
human epidermal growth factor receptor 2
HR
hazard ratio
HT
trastuzumab plus taxane
ICER
incremental cost-effectiveness ratio
iDFS
invasive disease–free survival
mBC
metastatic breast cancer
OS
overall survival
pCODR
CADTH pan-Canadian Oncology Drug Review
pCR
pathologic complete response
PHT
pertuzumab plus trastuzumab plus taxane
QALY
quality-adjusted life-year
T-DM1
trastuzumab emtansine
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pertuzumab (Perjeta), IV infusion |
Submitted price | Pertuzumab, 840 mg loading dose and 420 mg maintenance dose, IV infusion: $8.05 per mg or $3,381.81 per pack (420 mg) |
Indication | Neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or early-stage breast cancer (tumour either > 2 cm in diameter or node-positive disease) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | February 25, 2021 |
Reimbursement request | As per indication |
Sponsor | Hoffmann-La Roche Limited |
Submission history | Previously reviewed: Yes Indication: Patients with HER2-positive, early breast cancer at high risk of recurrence. A high risk of recurrence is defined as either node-positive or hormone receptor–negative disease Recommendation date: November 29, 2018 Recommendation: Do not reimburse due to uncertain clinical net benefit Indication: Neoadjuvant HER2-positive locally advanced, inflammatory, or early breast cancer Recommendation date: July 16, 2015 Recommendation: Do not reimburse due to uncertain clinical net benefit Indication: Metastatic breast cancer Recommendation date: August 1, 2013 Recommendation: Reimburse if the cost-effectiveness is improved to an acceptable level |
HER2 = human epidermal growth factor receptor 2; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Patients with HER2-positive, locally advanced, inflammatory, or early-stage breast cancer (tumour either > 2 cm in diameter or node-positive disease); aligns with the reimbursement request |
Treatment | Pertuzumab (Perjeta) IV infusion in combination with trastuzumab and taxane (PHT) chemotherapy |
Comparator | Neoadjuvant IV trastuzumab (100% biosimilar) plus taxane (HT) chemotherapy |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs, LYs |
Time horizon | Lifetime (51 years) |
Key data source |
|
Submitted results | ICER for PHT compared with HT was $27,986 per QALY (0.29 incremental QALYs; $8,000 incremental costs) |
Key limitations |
|
CADTH reanalysis results |
|
EFS = event-free survival; HER2 = human epidermal growth factor receptor 2; HR = hazard ratio; HT = trastuzumab plus taxane; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; pCR = pathologic complete response; PHT = pertuzumab plus trastuzumab and taxane; QALY = quality-adjusted life-year; WTP = willingness to pay.
Conclusions
Adding pertuzumab to neoadjuvant regimens with trastuzumab and chemotherapy improved pathologic complete response (pCR) rates. However, it remains unclear whether improvements in pCR translate into better survival outcomes, particularly overall survival (OS), as this outcome was either not studied or not yet mature in the submitted pivotal studies. There was no evidence that the combination of pertuzumab with trastuzumab plus chemotherapy improved invasive disease–free survival (iDFS), disease-free survival (DFS), or event-free survival (EFS), either because these outcomes were not studied, the data were not yet mature, or there was a lack of statistical significance when they were assessed. The available evidence does not establish that a difference in pCR rates between treatment arms will predict a difference in long-term survival end points (DFS, EFS, or OS) at the trial or population level. The relationship between pCR status and EFS was a key driver in the economic analysis.
CADTH was unable to determine a base case due to the inconclusive evidence regarding the validity of the association between pCR status and long-term survival outcomes (i.e., EFS and OS). CADTH made corrections to the sponsor’s base case to address minor errors identified with the sponsor’s submission; however, these corrections did not have a notable impact on the cost-effectiveness results. Pertuzumab plus trastuzumab plus taxane chemotherapy (PHT) was more costly (incremental cost, $7,797) and more effective (incremental quality-adjusted life-years [QALYs], 0.288) than trastuzumab plus taxane chemotherapy (HT), generating an incremental cost-effective ratio (ICER) of $27,112 per QALY. These results are contingent on the acceptability of the assumption that higher pCR translates into better survival outcomes (i.e., the sponsor’s hazard ratio [HR] of 0.33 for improved EFS for patients with a pCR compared with no pCR), particularly as 99% of the incremental benefit was accrued in the post-trial period. CADTH explored scenarios that indicated that the cost-effectiveness results were highly sensitive to the extent to which pCR improvement could lead to improved EFS. The higher the EFS HR for patients achieving a pCR compared with patients not achieving a pCR, the less likely that PHT was cost-effective. If achieving a pCR does not translate into better EFS, PHT would be dominated by HT, as PHT incurred higher costs and generated fewer QALYs than HT in that scenario. If the HR is greater than 0.41, then PHT is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY compared with HT.
Additionally, the sponsor’s model rested on several assumptions that could not be tested in CADTH’s exploratory scenario analyses. Most importantly, the model did not account for the direct impact of neoadjuvant PHT on survival outcomes and health utility. Moreover, the characteristics of the modelled population were based on the adjuvant trastuzumab emtansine (T-DM1) (KATHERINE) trial, and pCR rates were obtained from the ongoing PEONY trial, which included a 100% Asian population. There is no published evidence supporting the similarity of characteristics of patients who received neoadjuvant and adjuvant therapies for early-stage breast cancer (eBC) and the comparability of pCR status across racial groups. As a result of these limitations, in addition to the uncertainty with the association between pCR and long-term survival outcomes, the cost-effectiveness of PHT relative to HT in patients with HER2-positive, locally advanced, inflammatory, or early breast cancer is highly uncertain.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process, specifically, the information that pertains to the economic submission.
Two patient groups provided input for this review: the Canadian Breast Cancer Network (CBCN) and Rethink Breast Cancer. CBCN noted that Roche Canada, the sponsor, connected the CBCN with patients who had experience with pertuzumab, while Rethink Breast Cancer requested information from Roche and its scientific advisory committee on the characteristics of pertuzumab and its benefits. Patient input was gathered through new or previous retrospective online surveys and phone interviews. The patient groups noted that HER2-positive eBC is traditionally associated with more aggressive cancer with a poor prognosis in the absence of HER2-directed therapy. In addition to loss of income during illness, breast cancer patients can incur substantial costs associated with treatment and disease management. In the Rethink Breast Cancer survey, 56 of 62 patients (60% of whom were from Canada) had received trastuzumab and 12 had received T-DM1. The most commonly reported chemotherapy drugs received included carboplatin, docetaxel, paclitaxel, doxorubicin (Adriamycin), and cyclophosphamide. Rethink Breast Cancer reported the experience of 35 patients who received neoadjuvant pertuzumab in combination with trastuzumab and chemotherapy for locally advanced, inflammatory, or eBC. Twenty-one patients (60%) achieved pCR within 1 year of their surgery. Of these, 1 patient had since had a recurrence while the other 20 remained free of cancer cells. The majority of respondents felt that pertuzumab had improved their quality of life in every listed area, including activities such as the ability to work, sleep, drive, care for children, and perform household chores. The CBCN reported the experience of 4 patients who had received pertuzumab. The patients had difficulty determining if the adverse effects they experienced were related to pertuzumab or other therapies, but all rated their quality of life as mid-range to high on a 10-point scale. All patients expressed concerns about the lack of access to new treatments and the potential financial burden of paying out of pocket.
Input was received from 2 clinician groups, the Ontario Health (Cancer Care Ontario) Breast Cancer Drug Advisory Committee (CCO-DAC) and the BC Cancer Breast Tumour Group. The CCO-DAC noted that while the sponsor defined eBC for the purpose of reimbursement as tumours that are either 2 cm or more in diameter or node-positive disease, patients with either a tumour that was 1 cm in diameter or who had node-positive disease were eligible for the KATHERINE trial. The CCO-DAC also noted that the standard of care in Canada for this group is neoadjuvant trastuzumab plus chemotherapy. The BC Cancer Breast Tumour Group noted that the international standard of care for stage II and III HER2-positive breast cancer is neoadjuvant pertuzumab and trastuzumab with a taxane, leading to a gap where pCR rates are significantly lower than when pertuzumab is used, and more patients are exposed to more toxic adjuvant T-DM1 rather than to trastuzumab. Important outcomes were noted to include pCR and improved DFS and OS, with the BC Cancer Breast Tumour Group stating that achievement of pCR has consistently been associated with improved clinical outcomes. This group also noted that the use of neoadjuvant pertuzumab may also impact the use of pertuzumab in the metastatic setting, although it may still be reasonable to use pertuzumab for metastatic cancer if it has not been used for 12 months or more since the last neoadjuvant pertuzumab dose.
Drug plans identified the following barriers for the implementation of pertuzumab as relevant to the economic analysis: the high cost associated with pertuzumab, the increased preparation and chair time associated with an add-on therapy such as pertuzumab, and the need for clarity on the number of cycles required for pertuzumab treatment. The drug plans also noted 2 advantages regarding implementation: the potential offsetting of T-DM1 in the post-surgical setting, and that wastage of pertuzumab is unlikely to occur due to its fixed dosing. The drug plans also noted that the doubling of pCR observed in patients treated with neoadjuvant lapatinib plus trastuzumab compared with neoadjuvant trastuzumab in the Neo ALTTO trial did not correspond to the combination therapy being associated with improved survival outcomes. This increases the uncertainty that improved pCR, as reported in the pertuzumab group of the PEONY trial, will lead to increased cure and survival rates and a decreased risk of recurrence.
Several of the following concerns were addressed in the sponsor‘s model:
Quality of life was incorporated in the sponsor’s model, and a societal perspective incorporating productivity loss was assessed within a scenario analysis.
The sponsor assessed the relative impact of pertuzumab in combination with trastuzumab and chemotherapy on pCR when compared with trastuzumab combined with chemotherapy. The impact of these neoadjuvant therapies on survival outcomes, including EFS, DFS, and death, was indirectly estimated from the assumed relationship between pCR status and EFS.
Administration costs covering preparation and chair time associated with IV therapies were included in the sponsor’s model.
Scenario analyses that incorporated drug wastage or the possibility of adjuvant pertuzumab use were included in the sponsor’s model.
In addition, CADTH addressed some of these concerns as follows:
Drug plans had concerns regarding the lack of association between pCR and improved survival outcomes, as shown in the Neo ALTTO trial. While the patients in the Neo ALTTO trial comprised a population that was slightly different from the specific indication under review, CADTH explored the impact of varying EFS HRs among patients achieving a pCR compared with those not achieving a pCR.
CADTH was unable to address the following concern raised from stakeholder input:
The impact of neoadjuvant pertuzumab on quality of life could not be addressed because the pivotal trials focusing on neoadjuvant pertuzumab did not measure health utility.
Economic Review
The current review is for pertuzumab (Perjeta) for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or eBC (tumour either > 2 cm in diameter or node-positive disease).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis1 comparing costs and outcomes for pertuzumab in combination with PHT versus HT chemotherapy for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or eBC (tumour either > 2 cm in diameter or node-positive disease). The sponsor clarified that it was not seeking use in the adjuvant setting; the model therefore assumed that no patients would continue pertuzumab after surgery although, per the product monograph,2 this is allowed for up to 1 year at the discretion of the physician.
Pertuzumab is available as a sterile concentrate for solution for infusion (420 mg/14 mL vial). The recommended initial dose is 840 mg administered as a 60-minute IV infusion, followed every 3 weeks thereafter by a dose of 420 mg administered over a period of 30 to 60 minutes.2 The costs per monthly cycle of pertuzumab for the loading and maintenance doses were $6,763.62 (840 mg) and $3,381.81 (420 mg), respectively. The sponsor assumed that branded trastuzumab (Herceptin) was used with neoadjuvant pertuzumab, with the costs per cycle of $3,732.08 for the loading cycle and $2,799.06 for the maintenance cycle. Biosimilar trastuzumab was assumed in the adjuvant setting; the costs per cycle were $1,840.29 and $1,380.22 for the loading and maintenance cycles, respectively. The cost of TDM-1 in the adjuvant setting per monthly cycle was $5,473.73. The average cost per monthly cycle for neoadjuvant chemotherapy drugs was $1,067.35 for the 6 months of chemotherapy received.
The clinical outcomes of interest were QALYs and life-years (LYs). The economic analysis was undertaken over a time horizon of 51 years (lifetime) using monthly cycles from the perspective of a Canadian publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum.
Model Structure
The sponsor submitted a Markov model with 6 health states (EFS on treatment before surgery or iDFS after surgery [EFS/iDFS], non-metastatic recurrence (locoregional recurrence and contralateral breast cancer), remission after a non-metastatic recurrence, first-line metastatic breast cancer (mBC), subsequent lines of treatment in mBC, and death) (Appendix 3; Figure 1). All patients entered the model in the EFS health state and were assumed to receive neoadjuvant treatment with PHT or HT. Following surgery, patients received various adjuvant treatments, depending on their pCR status.
Patients with a pCR continued with trastuzumab alone for a maximum of 18 cycles (including the neoadjuvant cycles). Patients who did not achieve a pCR received TDM-1 for a maximum of 14 cycles. Patients could discontinue adjuvant treatments due to intolerability or other reasons. In each model cycle, patients could remain in the EFS/iDFS health state or experience 1 of these events: non-metastatic recurrence (locoregional recurrence and contralateral breast cancer), metastatic recurrence, or death. The sponsor included a non-metastatic recurrence as a tunnel state where patients who remained alive could stay for 12 months and receive another episode of an adjuvant therapy. After the 12-month period, patients might transition to remission or death. In the remission health state, patients were off treatment and assumed to have no further sign of the disease. In this health state, patients might transition to death or the metastatic recurrence health state. The metastatic recurrence health state was divided based on whether patients were receiving first- or subsequent-line mBC treatment. The risk of disease progression and death in both first- and subsequent-line mBC depended on treatments received in the mBC setting.
Model Inputs
The modelled population reflected the baseline patient characteristics of the KATHERINE trial, a phase III, open-label trial involving patients with HER2-positive eBC who were found to have residual invasive disease in the breast or axilla at surgery after receiving a neoadjuvant therapy containing a taxane (with or without an anthracycline) and trastuzumab.3 The sponsor’s model assumed a mean age of 49 years, a mean body surface area of 1.77 m2, a body weight of 71.42 kg, and an average height of 163.10 cm. The sponsor indicated that the characteristics of patients participating in the KATHERINE trial were generalizable to the Canadian eBC population in previous CADTH pan-Canadian Oncology Drug Review (pCODR) and Institut national d’excellence en santé et en services sociaux (INESSS) submissions for TDM-1,4,5 and also in line with the reimbursement request6 and Health Canada–approved indication.2
Rates of pCR were obtained from the PEONY trial, which is an ongoing phase III trial designed to evaluate the efficacy of pertuzumab in combination with trastuzumab and chemotherapy (docetaxel) compared with placebo in combination with trastuzumab and docetaxel as a neoadjuvant treatment before surgery in patients who are chemotherapy-naive with non-metastatic HER2-positive breast cancer. The probability of remaining in the EFS/iDFS health state depends on whether a patient achieves a pCR. The risk of disease recurrence in patients not achieving a pCR was estimated from the iDFS data reported by the KATHERINE trial. To derive the iDFS in patients treated with trastuzumab who achieved a pCR, the sponsor noted that patients with a pCDR had a decreased risk of EFS compared to those with residual disease (i.e., those who did not achieve pCR) and applied an HR of 0.33 (95% CI, 0.25 to 0.43) to patients who did not achieve a pCR in the trastuzumab arm from the KATHERINE trial, creating a survival estimate (HR EFS) for patients achieving pCR with HT. The HR was derived from a pooled analysis of the NEOSPHERE, TRYPHAENA, BERENICE, HannaH, and KRISTINE HER2-positive breast cancer trials.7 After the end of follow-up for the KATHERINE trial (i.e., 62 months), the sponsor used parametric survival models with an exponential function and a log-normal function to extrapolate the iDFS data for the non-pCR TDM-1 arm and the non-pCR trastuzumab arm, respectively. The long-term iDFS data for patients with a pCR was extrapolated using a log-normal distribution. The extrapolated recurrence rate was adjusted to replicate the trend in the recurrence rate observed in previous trastuzumab studies. The sponsor also assumed that patients could be considered functionally cured at 36 months, and that the cure rate would reach the maximum rate of 95% at 120 months. From this point onward, patients had a very low risk of recurrence and were only at risk of death due to natural causes.
The risk of a second malignancy was based on the British Columbia Cancer Registry study that estimated the risk of a second malignancy after adjuvant therapy from a cohort of 12,836 Canadian patients with eBC.8 The sponsor derived survival estimates from the EMILIA study for patients experiencing a metastatic recurrence within the first 18 months of the initiation of adjuvant treatment.9 The risk of disease progression and death in the metastatic setting (recurrence observed at least 18 months after adjuvant treatment initiation) were derived from the CLEOPATRA trial10 (pertuzumab plus trastuzumab plus taxane chemotherapy versus trastuzumab plus taxane chemotherapy) for PHT and HT and from the M77001 trial11 (trastuzumab plus taxane versus taxane alone). Alternative data sources for the risk of disease recurrence were tested in scenario analyses.
The model accounted for grade 3 or higher adverse events (AEs), which were based on the AEs in the APHINITY trial12 observed in at least 1% of patients with node-positive disease receiving either PHT or HT as neoadjuvant treatments. In the adjuvant setting, the frequency of AEs for patients who achieved a pCR versus those who did not achieve a pCR was obtained from the trastuzumab and TDM-1 arms of the KATHERINE trial.
A health utility value for the EFS/iDFS health state was derived from EQ-5D 3-Levels questionnaire data using the Canadian utility weights collected as part of the KATHERINE trial,3 which focused on higher-risk patients with residual invasive breast cancer who completed prior neoadjuvant chemotherapy with trastuzumab-containing therapy. A health utility was assumed to be the same regardless of treatment arm and pCR status. Health utility values for mBC were obtained from a study by Lloyd et al.13 Utility scores were adjusted to reflect declining utility with age.
Costs included drug-related (acquisition, administration, subsequent treatments), monitoring, supportive care, surgery, AEs, and terminal care. The unit costs for paclitaxel and docetaxel were obtained from the Keytruda submission to pCODR for metastatic urothelial carcinoma.14 The unit costs for the oral chemotherapeutic drugs capecitabine and lapatinib were obtained from the Ontario Drug Benefit Formulary/Comparative Drug Index15 and the Exceptional Access Program16 formulary, respectively. All other treatment costs were based on IQVIA Delta PA Ontario wholesale costs.17 Treatment and administration costs were estimated using the doses published in the Cancer Care Ontario treatment protocols, supplemented with information from 4 clinical advisors. Administration costs included laboratory tests, pre-treatment medications, and the cost of administering each treatment (regimen preparation, chair time, pharmacist and chemotherapy nurse time, and cancer centre overhead). Unit costs for pre-medications and supportive care were based on the Ontario Drug Benefit Formulary/Comparative Drug Index,15 Ontario Schedule of Benefits for Physician Services,18 and the Ontario Schedule of Benefits for Laboratory Services.19 Surgery cost was obtained from the Canadian Institute for Health Information Patient Cost Estimator. A terminal care cost was applied to patients who transitioned to the death health state, based on a study by Walker et al.20 that included the direct medical costs of palliative and end-of-life care in the last 6 months of life.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 2,000 iterations with the deterministic and probabilistic results being similar. The probabilistic findings are presented subsequently. The submitted analyses were based on the publicly available prices of the comparator and subsequent treatments.
Base-Case Results
In the sponsor’s base-case analysis, PHT were associated with an ICER of $27,986 per QALY compared with HT over a 51-year time horizon (Table 3). At a WTP value of $50,000 per QALY, the probability of PHT being cost-effective was 72.9% compared with HT. The main cost drivers were the drug-acquisition cost of pertuzumab and subsequent costs associated with TDM-1, followed by supportive care and end-of-life care costs. At the end of the model time horizon (i.e., 51 years), 4.1% of patients in the model were still alive.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drugs | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. HT ($/QALY) |
|---|---|---|---|---|---|
HT | 191,897 | Reference | 18.305 | Reference | Reference |
PHT | 199,861 | 7,964 | 18.589 | 0.285 | 27,986 |
ICER = incremental cost-effectiveness ratio; HT = trastuzumab and taxane; PHT = pertuzumab plus trastuzumab and taxane; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.
Sensitivity and Scenario Analysis Results
The sponsor performed scenario analyses by changing a discount rate, reducing a time horizon, using alternative data sources for neoadjuvant EFS and iDFS and health utilities, changing treatment duration, using alternative parametric models to extrapolate iDFS, and changing the assumptions regarding market share of neoadjuvant sequential chemotherapy, treatment mix for mBC, and cure rate. The ICERs for PHT compared with HT remained lower than $50,000 per QALY in most scenarios, except when pertuzumab was continued in the adjuvant setting. Compared with HT, the estimated ICERs for PHT ranged from $15,989 per QALY (changing the market share of neoadjuvant sequential chemotherapy) to $58,434 per QALY (continuation of pertuzumab in the adjuvant setting). Key drivers of the cost-effectiveness results included treatment duration of pertuzumab, discount rate, and treatment mix for mBC.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications on the economic analysis:
Inconclusive evidence regarding the association between pCR status and long-term survival outcomes led to high uncertainty in the cost-effectiveness results. The sponsor’s model predicts that patients receiving PHT will live an additional 0.49 years over the 51-year time horizon compared with HT. As noted in the CADTH Clinical Review, while patient-level evidence suggests an association between pCR and improved survival outcomes based on responder analyses, this association has not been established at the trial or population level. Specifically, the available evidence does not establish that a difference in pCR rates between treatment arms will predict a difference in long-term survival end points (DFS, EFS, or OS) between 2 treatments. As such, the evidence linking pCR with survival outcomes was associated with uncertainty, particularly as it relates to OS.
CADTH noted that the sponsor indirectly estimated EFS according to pCR status by using an HR reported by Swain et al.,7 which pooled pCR and EFS data from the NEOSPHERE,21,22 TRYPHAENA,23,24 BERENICE,25 HannaH,26,27 and KRISTINE28,29 trials. CADTH was concerned about the validity of the HR reported in this study for several reasons. First, the study did not describe how relevant studies were identified, selected, and abstracted; thus, it is unknown whether the authors identified and considered all relevant studies. Second, the HR reported in this pooled analysis may be of limited use in the Canadian setting, given the imbalance in baseline characteristics and differences in treatment modalities considered in the included trials. Specifically, the BERENICE and KRISTINE trials had more patients with clinical stage II disease than did the NEOSPHERE, TRYPHAENA, and HannaH trials. Moreover, the HannaH trial focused on neoadjuvant and adjuvant HT treatment modalities, while TRYPHAENA focused on neoadjuvant PHT and adjuvant HT therapy, and BERENICE and KRISTINE focused on neoadjuvant and adjuvant PHT treatments.
CADTH attempted to address this limitation in its scenario analyses by varying the HRs and assuming no association between pCR status and EFS (i.e., EFS HR for pCR versus non-pCR = 1).
High uncertainty of cost-effectiveness findings due to a structural uncertainty. The sponsor’s model did not account for the direct impact of neoadjuvant PHT on survival outcomes (disease recurrence or death) and health utility. The model combined clinical events, such as disease recurrence and death, that might occur during the neoadjuvant and adjuvant phases of treatment, with an assumption that the probabilities of developing such clinical events depended on whether pCR was achieved after surgery. Similarly, the sponsor used the health utility values reported in the KATHERINE trial, which focused on higher-risk patients with residual invasive breast cancer after completion of neoadjuvant chemotherapy. The EFS and health utility values observed in the adjuvant setting may not accurately represent the effects of neoadjuvant PHT because these outcomes may be confounded by several treatment regimens after surgery.
In addition, CADTH identified inconsistency in how data sources were used to inform the model. In particular, the economic analysis focused on neoadjuvant therapies for HER2-postive breast cancer; however, characteristics of the modelled population and AE rates related to neoadjuvant PHT were based on the adjuvant breast cancer (KATHERINE and APHINITY) trials.
CADTH was unable to address this limitation given the submitted model structure. The sponsor’s model does not have the capacity to capture disease recurrence, death, and health utility for patients who received neoadjuvant therapies. Additionally, health utility and health-related quality of life were not assessed in any of the relevant trials. Therefore, it remains unclear how the cost-effectiveness of PHT may be changed if the impact of neoadjuvant PHT on EFS and health utility outcomes are considered.
The included first-line metastatic chemotherapy treatments do not align with current practice in Canada. The model assumed that patients who received first-line metastatic treatments might be prescribed vinorelbine and lapatinib in combination with capecitabine, in addition to other relevant comparators. However, the clinical expert consulted by CADTH advised that the aforementioned regimens are no longer used for the treatment of mBC in Canada.
CADTH addressed this limitation in its reanalyses by changing the proportions of patients who received vinorelbine and lapatinib in combination with capecitabine to zero.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations of the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The baseline demographic characteristics used in the sponsor’s model were based on the intention-to-treat population from the KATHERINE trial, but the key efficacy of PHT vs. HT (pCR) was based on the PEONY trial. | The clinical expert consulted by CADTH agreed the patients who participated in the KATHERINE trial were generalizable to eBC patients in Canada. CADTH noted that the PEONY trial included a 100% Asian population, but most participants (~73%) in the KATHERINE trial were White. An exploratory subgroup analysis of the KATHERINE trial showed a slight variation in the iDFS HRs between TDM-1 and trastuzumab by race in the adjuvant setting. Although the sponsor stated that the difference in demographic characteristics between PEONY and the Canadian population was unlikely to affect treatment efficacy, there was no published evidence to support this assumption. |
100% of patients achieving pCR were treated in the adjuvant setting with trastuzumab, and 100% of patients without a pCR were treated with TDM-1. | Acceptable. CADTH performed a scenario analysis by allowing pertuzumab to be used in the adjuvant setting for patients receiving neoadjuvant PHT who achieve pCR. |
The cure adjustment started at 36 months and reached a maximum cure of 95% at 10 years. | Acceptable. This assumption was supported by a study by Takeuchi et al.30 that showed only 1.08% of patients experienced recurrence after 10 years in the trastuzumab chemotherapy arm. This assumption was also consistent with a previous T-DM1 pCODR eBC submission.4 CADTH performed a scenario analysis by varying the duration at which most patients are considered cured of BC. |
The duration of the incremental treatment effect of T-DM1 over trastuzumab was assumed to be maintained for 7 years and then linearly decreased to null at 10 years. | The clinical expert consulted by CADTH suggested that this assumption was acceptable. CADTH performed a scenario analysis by varying the duration of the treatment effect of T-DM1. The longer treatment effect was expected to reduce ICERs. |
The type of neoadjuvant and adjuvant treatments and pCR status did not affect health utility. | The clinical expert consulted by CADTH advised that systemic therapies can have different impacts on quality of life due to their different toxicity profiles, which could have been addressed using disutilities due to adverse events. The sponsor’s model, however, did not account for the impact of neoadjuvant treatments on health utility. CADTH conducted a scenario analysis to assess how the variation in health utility values by adjuvant treatments might influence the cost-effectiveness results. |
Full (100%) vial sharing is assumed for all IV medications. | Acceptable. The sponsor’s model assumed no drug wastage for all IV medication. Although the clinical expert consulted by CADTH stated that this was likely appropriate, drug wastage is likely to occur in practice. CADTH performed a scenario analysis by considering drug wastage for all IV medications. |
Patients with the following AEs (anemia, fatigue, leukopenia, neutropenia, white blood cell count decreased, or neutrophil count decreased) did not require any additional health resources. | Acceptable. The clinical expert consulted by CADTH indicated that patients with anemia may need medical attention and transfusion; therefore, this assumption may be inappropriate. However, the incidence of anemia was very low; this assumption was therefore unlikely to influence the cost-effectiveness results. |
A dispensing fee of $8.83 per prescription was applied for oral medications. | CADTH removed a dispensing fee in its reanalysis to ensure consistency across drug reviews. |
A log-normal distribution was used to represent the variation in cost data. | Cost data are very highly skewed; therefore, a gamma distribution would better reflect the high uncertainty of cost data. CADTH revised this in its reanalysis. |
BC = breast cancer; eBC = early breast cancer; HT = trastuzumab plus taxane; ICER = incremental cost-effectiveness ratio; pCODR = CADTH pan-Canadian Oncology Drug Review; pCR = pathologic complete response; PHT = pertuzumab plus trastuzumab plus taxane; T-DM1 = trastuzumab emtansine; vs. = versus.
CADTH Reanalyses of the Economic Evaluation
CADTH-Corrected Analysis Results
Key limitations of the sponsor’s model could not be adequately addressed due to the lack of data and limitations with the model structure (i.e., the association between pCR and survival outcomes and inability to account for the potential impact of neoadjuvant treatments on EFS, death, and health utility).
CADTH corrected the sponsor’s analysis by making the following changes to the model to align with best economic practices and Canadian clinical practice: excluding dispensing fees for oral medication, using a gamma distribution to represent the uncertainty of cost data, and removing vinorelbine and lapatinib in combination with capecitabine from a list of first-line mBC treatments. CADTH also increased the number of simulations from 2,000 to 5,000 to increase the stability of cost-effectiveness results. Table 7 details the revisions for the CADTH-corrected analysis.
Given the high uncertainty surrounding the evidence on the relationship between pCR and survival outcomes, CADTH did not perform a base-case analysis. The summary results of the CADTH-corrected analysis are presented in Table 8, while the disaggregated results are presented in Table 9. Results from this corrected analysis were consistent with the sponsor’s base case, suggesting that PHT was associated with higher costs ($7,797) and an improved QALY (0.288), with an ICER of $27,112 per QALY. PHT had a 70.4% chance of being cost-effective at a WTP threshold of $50,000 per QALY. CADTH calculated that more than 99% of the incremental QALYs were accrued after the trial period (defined by CADTH as the median trial follow-up period of 11.6 months).
Results from the CADTH-corrected analysis were contingent on the acceptability of the assumption that improved pCR translates into better survival outcomes. Scenarios exploring alternate assumptions are provided in the section that follows.
Exploratory Scenario Analysis Results
Based on CADTH’s corrected analysis, a series of exploratory scenario analyses were conducted. These analyses explored the impact of the following model parameters and assumptions: changes to pCR rates, EFS source, EFS HRs for patients with a pCR versus without a pCR, alternative parametric survival models for iDFS prediction, varying the time at which most patients are cured, change to state-specific health utility, using PHT as adjuvant treatment, applying treatment waning among patients with a pCR, applying drug wastage, and adopting a societal perspective.
These analyses demonstrated that the ICERs for PHT, when compared with HT, ranged from $11,987 per QALY (scenario 2, using the pCR rates from the trial data pooled by the FDA Collaborative Trials in Neoadjuvant Breast Cancer working group) to PHT being dominated, i.e., more costly and fewer QALYs than HT (scenario 5, which assumed no association between pCR and improved EFS). Cost-effectiveness results were largely driven by the association between pCR status and EFS; the weaker the association between achieving pCR and better EFS, the larger the ICER for PHT relative to HT. PHT was no longer cost-effective at a WTP threshold of $50,000 per QALY if the EFS HR for pCR versus non-pCR was greater than 0.41. Other key drivers included the time at which patients with no pCR are considered cured and the continuation of pertuzumab as an adjuvant therapy (Table 10).
Price-reduction analyses were not conducted on either the sponsor’s or CADTH’s corrected analysis, given that PHT was already cost-effective at a WTP threshold of $50,000 per QALY at the submitted price. CADTH performed exploratory price-reduction analyses on scenarios 4 and 5. Based on these exploratory analyses and a WTP threshold of $50,000 per QALY, PHT would be considered cost-effective if the price of pertuzumab were reduced by at least 9% and the EFS HR for pCR and non-pCR was assumed to be 0.43, which was the upper bound of the 95% CI for the EFS HR from Swain et al.7). However, if achieving a pCR was not associated with improved EFS, PHT would not be cost-effective regardless of the magnitude of price reduction (Appendix 4, Table 11).
Issues for Consideration
In 2015, the pCODR Expert Review Committee (pERC) considered the evidence for pertuzumab combined with trastuzumab and a taxane chemotherapy as neoadjuvant treatments for patients with HER2-positive primary operable or locally advanced or inflammatory breast cancer and recommended that PHT not be reimbursed. The negative recommendation was due to concerns about the validity of pCR as a surrogate for survival outcomes and the high uncertainty around the clinical benefits considered in the economic model. Similar concerns have been identified in the current submission.
The CADTH Clinical Review highlighted the variation in pCR between PHT and HT among subgroups of interest, such as breast cancer type (locally advanced versus operable breast cancer) and hormone receptor status. CADTH was unable to assess whether these factors would affect the cost-effectiveness results, given the submitted model structure and clinical information.
The clinical expert consulted for this review advised that breast cancer does occur in men, and these patients may be eligible for neoadjuvant pertuzumab. However, efficacy data for this population was not available; thus, the cost-effectiveness for this population is not known.
The clinical experts consulted by CADTH noted that emerging new therapies for HER2-positive breast cancer, such as neratinib or trastuzumab deruxtecan, were not included in the economic submission. The impact of these therapies on the cost-effectiveness of neoadjuvant PHT requires further exploration.
While beyond the scope of this review, pertuzumab is also available in a combination pack with trastuzumab; the Perjeta-Herceptin Combo Pack contains 420 mg of pertuzumab (Perjeta) and 440 mg of trastuzumab (Herceptin) at a wholesale price of $6,256 per kit in most of Canada, although the wholesale price in Quebec is lower.17 Outside of Quebec, the cost of this kit is more than the Association québécoise des pharmaciens propriétaires (AQPP) price of $2,700 for 440 mg of Herceptin-brand trastuzumab IV (no wholesale price available)17 and the submitted price of $3,382 for pertuzumab1 ($6,082 combined). It is also more than the cost of the combination when biosimilar trastuzumab IV ($1,416 for 440 mg) is considered ($4,798 combined). The potential availability of confidentially negotiated prices for the individual products and/or the combination kit may alter the relative costs associated with each option.
Overall Conclusions
Based on the CADTH Clinical Review, adding pertuzumab to neoadjuvant regimens with trastuzumab and taxane chemotherapy improved pCR rates. However, it remains unclear whether improvements in pCR translate into better survival outcomes, particularly OS, as this outcome was either not studied or not yet mature in the submitted pivotal studies. There was no evidence that PHT improved iDFS, DFS, or EFS relative to HT, either because these outcomes were not studied, the data were not yet mature, or there was a lack of statistical significance when they were assessed. Available evidence does not establish that a difference in pCR rates between treatment arms will predict a difference in long-term survival end points (DFS, EFS, or OS). The relationship between pCR status and EFS was considered a key driver in the economic analysis.
CADTH was unable to determine a base case due to the inconclusive evidence on the validity of pCR as a surrogate end point for long-term survival outcomes (EFS, OS). CADTH undertook corrections to the sponsor’s base case to address errors identified with the sponsor’s submission: removing dispensing fees for oral medications, using a gamma distribution to represent the uncertainty of cost data, removing vinorelbine and lapatinib combined with capecitabine from a list of first-line mBC treatments, and increasing the number of simulations from 2,000 to 5,000. These corrections did not have a notable impact on the sponsor’s base-case cost-effectiveness results, but did have slightly greater uncertainty; PHT was more costly (incremental cost, $7,797) and more effective (incremental QALYs, 0.288) than HT, generating an ICER of $27,112 per QALY. However, this estimate is contingent on the acceptability of the assumption that improved pCR translates into better survival outcomes; cost-effectiveness results were mostly driven by the extent to which pCR improvement could lead to improved EFS. The greater the EFS HR for patients achieving a pCR compared with patients not achieving a pCR, the less likely that PHT was cost-effective. If achieving a pCR does not translate into better EFS, PHT would be dominated by HT, as PHT incurred higher costs and generated fewer QALYs. An EFS HR of 0.41 or lower is required for pCR compared with no pCR for PHT to be considered cost-effective at a WTP threshold of $50,000 per QALY compared with HT.
Furthermore, there is no direct evidence to inform the comparative impact of PHT and HT on survival outcomes. The sponsor’s model was based on the relationship between pCR and EFS reported in a pooled analysis of 5 clinical trials in patients with HER2-positive breast cancer who achieved a pCR after neoadjuvant systemic anti-HER2 therapy. The pooled results should be carefully interpreted, as limited descriptions of the sponsor’s search strategies were provided and the included trials considered patients with different clinical stages and hormone receptor statuses.
Finally, the sponsor’s model rested on several assumptions that could not be fully tested in CADTH’s scenario analyses. Most importantly, the model did not account for the direct impact of neoadjuvant PHT therapy on survival outcomes and health utility. Characteristics of the modelled population were based on the adjuvant T-DM1 (KATHERINE) trial, and pCR rates were obtained from the ongoing PEONY trial, which included a 100% Asian population. There is no published evidence supporting the similarity of characteristics of patients who received neoadjuvant and adjuvant therapies for eBC and the comparability of pCR status across racial groups. As a result of these limitations, in addition to the uncertainty with the association between pCR and long-term survival outcomes, the cost-effectiveness of PHT relative to HT in patients with HER2-positive, locally advanced, inflammatory, or eBC is highly uncertain.
References
1.Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Perjeta® (pertuzumab). Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 14.
2.Perjeta® pertuzumab for injection Sterile Concentrate for Solution for Infusion, 420mg/14 mL vial [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Mar 11.
3.von Minckwitz G, Huang CS, Mano MS, et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N Engl J Med. 2019;380(7):617-628. PubMed
4.Final Economic Guidance Report. Trastuzumab Emtansine (Kadcyla) for Early Breast Cancer. Ottawa (ON): CADTH; 2019: https://cadth.ca/sites/default/files/pcodr/Reviews2020/10182TrastuzumabEmtansineEBC_fnEGR_NOREDACT-ABBREV_EarlyConv_22Jan2020_final.pdf. Accessed 2021 Jul 3.
5.Kadcyla – Cancer du sein précoce. Avis transmis à la ministre en janvier 2020. Québec, QC: INESSS; 2020: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Fevrier_2020/Kadcyla_2020_01.pdf. Accessed 2021 Jul 3.
6.Drug Reimbursement Review sponsor submission: Perjeta® (pertuzumab) [internal sponsor’s package]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 14.
7.Swain SM MH, Cortes J, et al. Risk of Recurrence and Death in Patients with Early HER2-Positive Breast Cancer Who Achieve a Pathological Complete Response after Different Types of HER2-Targeted Therapy: A Pooled Analysis. . Poster 1151 presented at the 2019 San Antonio Breast Cancer Symposium. San Antonio, TX: San Antonio Breast Cancer Symposium; 2019: https://sabcs19.posterview.com/nosl/p/P1-18-01. Accessed 2021 Jul 3.
8.Hamilton SN, Tyldesley S, Li D, Olson R, McBride M. Second malignancies after adjuvant radiation therapy for early stage breast cancer: is there increased risk with addition of regional radiation to local radiation? Int J Radiat Oncol Biol Phys. 2015;91(5):977-985. PubMed
9.Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(6):732-742. PubMed
10.Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724-734. PubMed
11.Marty M, Cognetti F, Maraninchi D, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23(19):4265-4274. PubMed
12.von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med. 2017;377(2):122-131. PubMed
13.Lloyd A, Nafees B, Narewska J, Dewilde S, Watkins J. Health state utilities for metastatic breast cancer. Br J Cancer. 2006;95(6):683-690. PubMed
14.pCODR. Final Economic Guidance Report. Pembrolizumab (Keytruda) for Metastatic Urothelial Carcinoma. Ottawa ON: CADTH; 2018: https://cadth.ca/sites/default/files/pcodr/pcodr_pembrolizumab_keytruda_muc_fn_egr.pdf. Accessed 2021 Jul 3.
15.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Jul 3.
16.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Jul 3.
17.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Jul 3.
18.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Jul 3.
19.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Jul 3.
20.Walker H, Anderson M, Farahati F, et al. Resource use and costs of end-of-Life/palliative care: Ontario adult cancer patients dying during 2002 and 2003. J Palliat Care. 2011;27(2):79-88. PubMed
21.Gianni L, Pienkowski T, Im YH, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncology. 2012;13(1):25-32. PubMed
22.Gianni L, Pienkowski T, Im YH, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncology. 2016;17(6):791-800. PubMed
23.Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Annals of Oncology. 2013;24(9):2278-2284. PubMed
24.Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer. 2018;89:27-35. PubMed
25.Swain SM, Ewer MS, Viale G, et al. Pertuzumab, trastuzumab, and standard anthracycline- and taxane-based chemotherapy for the neoadjuvant treatment of patients with HER2-positive localized breast cancer (BERENICE): a phase II, open-label, multicenter, multinational cardiac safety study. Annals of Oncology. 2018;29(3):646-653. PubMed
26.Ismael G, Hegg R, Muehlbauer S, et al. Subcutaneous versus intravenous administration of (neo)adjuvant trastuzumab in patients with HER2-positive, clinical stage I-III breast cancer (HannaH study): a phase 3, open-label, multicentre, randomised trial. Lancet Oncol. 2012;13(9):869-878. PubMed
27.Jackisch C, Stroyakovskiy D, Pivot X, et al. Subcutaneous vs Intravenous Trastuzumab for Patients With ERBB2-Positive Early Breast Cancer: Final Analysis of the HannaH Phase 3 Randomized Clinical Trial. JAMA Oncol. 2019;5(5):e190339. PubMed
28.Hurvitz SA, Martin M, Jung KH, et al. Neoadjuvant Trastuzumab Emtansine and Pertuzumab in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: Three-Year Outcomes From the Phase III KRISTINE Study. J Clin Oncol. 2019;37(25):2206-2216. PubMed
29.Hurvitz SA, Martin M, Symmans WF, et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncology. 2018;19(1):115-126. PubMed
30.Takeuchi H, Muto Y, Tashiro H. Clinicopathological characteristics of recurrence more than 10 years after surgery in patients with breast carcinoma. Anticancer Res. 2009;29(8):3445-3448. PubMed
31.Cancer Care Ontario: funded evidence-informed regimens. 2021; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2021 Jun 1.
32.Launay-Vacher V, Oudard S, Janus N, et al. Prevalence of Renal Insufficiency in cancer patients and implications for anticancer drug management: the renal insufficiency and anticancer medications (IRMA) study. Cancer. 2007;110(6):1376-1384. PubMed
33.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41. PubMed
34.Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7(11):1748-1756. PubMed
35.Budget Impact Analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Perjeta® (pertuzumab). Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 14.
36.Shao Z, Pang D, Yang H, et al. Efficacy, Safety, and Tolerability of Pertuzumab, Trastuzumab, and Docetaxel for Patients With Early or Locally Advanced ERBB2-Positive Breast Cancer in Asia: The PEONY Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6(3):e193692. PubMed
37.Summit Strategy Group. 2020 HER2+ eBC and mBC Survey and Chart Review. Confidential Market Research Conducted for ROCHE Canada: Roche Canada; 2020. Accessed 28-June-2021.
38.Canadian Cancer Statistics Advisory C. Canadian cancer statistics 2019. Toronto (ON): Canadian Cancer Society; 2019: https://www.cancer.ca/~/media/cancer.ca/CW/cancer%20information/cancer%20101/Canadian%20cancer%20statistics/Canadian-Cancer-Statistics-2019-EN.pdf?la=en. Accessed 2021 Jul 3.
39.Canadian Partnership Against Cancer. Stage Distribution: Distribution of cases by stage at diagnosis for breast cancer (women only) – 2013 diagnosis year. https://www.systemperformance.ca/disease-sites/breast/stage-distribution/. Accessed 2021 Jun 28.
40.Table 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000) - using the M1 medium growth scenario. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2021 Jun 28.
41.First Nations and Inuit Health Branch. Non-Insured Health Benefits Program Annual Report 2018/2019. Indigenous Services Canada,; 2020: https://www.sac-isc.gc.ca/eng/1584392581890/1584393350542#sec2. Accessed 2021 Jun 28.
42.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177-182. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for Neoadjuvant Treatments for HER2-Positive Early-Stage Breast Cancer
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Cost per 28-day cyclea ($) |
|---|---|---|---|---|---|---|
Pertuzumab (Perjeta) | 30 mg/mL | 420 mg vial | 3,381.8106b | 840 mg IV on Day 1 for the first cycle, followed by 420 mg on Day 1 for subsequent cycles. Administer 3 to 6 21-day cycles as part of one of the regimens outlined below, when trastuzumab is administeredc,d | First cycle: 322.08 Subsequent cycles: 161.04 | First cycle: 7,891 Subsequent cycles: 4,509 |
Pertuzumab (4 cycles) plus AC-PACL (Dose Dense) plus trastuzumab IV regimen, per 28 days | First cycle: 20,135 Subsequent cycles: 16,247 | |||||
Pertuzumab (4 cycles) plus AC-PACL (Dose Dense) plus trastuzumab SC regimen, per 28 days | First cycle: 21,105 Subsequent cycles: 17,723 | |||||
Pertuzumab (4 cycles) plus AC-PACL (Weekly) plus trastuzumab IV regimen, per 28 days | First cycle: 18,698 Subsequent cycles: 14,810 | |||||
Pertuzumab (4 cycles) plus AC-PACL (Weekly) plus trastuzumab SC regimen, per 28 days | First cycle: 19,667 Subsequent cycles: 16,285 | |||||
Pertuzumab (4 cycles) plus AC-D plus trastuzumab IV regimen, per 28 days | First cycle: 13,168 Subsequent cycles: 9,280 | |||||
Pertuzumab (4 cycles) plus AC-D plus trastuzumab SC regimen, per 28 days | First cycle: 14,137 Subsequent cycles: 10,755 | |||||
Pertuzumab (3 or 4 cycles) plus FEC-D + Tras-IV regimen, per 28 days | First cycle: 13,693 Subsequent cycles: 9,804 | |||||
Pertuzumab (3 or 4 cycles) plus FEC-D + Tras SC regimen, per 28 days | First cycle: 14,662 Subsequent cycles: 11,280 | |||||
Pertuzumab (6 cycles) plus carboplatin, docetaxel, and trastuzumab IV regimen, per 28 days | First cycle: 13,300 Subsequent cycles: 9,412 | |||||
Pertuzumab (6 cycles) plus carboplatin, docetaxel, and trastuzumab SC regimen, per 28 days | First cycle: 14,270 Subsequent cycles: 10,888 | |||||
AC-PACL (DD) + TRAS | ||||||
Doxorubicin (generics) | 2 mg/mL | 10 mg vial 50 mg vial 200 mg vial | 50.0000 255.0000 770.0000 | 60 mg/m2 IV on Day 1 every 2 weeks for 4 cycles | 40 | 1,120 |
Cyclophosphamide (generics) | 20 mg/mL | 500 mg vial 1,000 mg vial 2,000 mg vial | 91.3100 165.5200 304.4000 | 600 mg/m2 IV on Day 1 every 2 weeks for 4 cycles | 18 | 514 |
Paclitaxel (Taxol) | 6 mg/mL | 30 mg vial 96 mg vial 300 mg vial | 300.0000 1,196.8000 3,740.0000 | After AC is complete: 175 mg/m2 on Day 1 every 2 weeks for 4 cycles | 289 | 8,080 |
Trastuzumab IV (biosimilar) | 21 mg/mL | 150 mg vial 440 mg vial | 506.1405 1,417.196 | After AC is complete: First cycle: 8 mg/kg IV on day 1; Thereafter: 6 mg/kg IV on day 1 every 3 weeks for another 3 cyclese | First cycle: 92 Subsequent cycles: 67 | First cycle: 2,531 Subsequent cycles: 2,024 |
Trastuzumab SC (Herceptin SC) | 120 mg/mL | 600 mg single-dose vial | 2,625.0000 | After AC is complete: 600 mg SC on day 1 every 3 weeks for 4 cyclese | 125 | 3,500 |
AC-PACL(DD) + Tras-IV regimen, per 28-days | First cycle: 12,244 Subsequent cycles: 11,738 | |||||
AC-PACL(DD) + Tras SC regimen, per 28-days | 13,214 | |||||
AC-PACL (W) + TRAS | ||||||
Doxorubicin (generics) | 2 mg/mL | 10 mg vial 50 mg vial 200 mg vial | 50.0000 255.0000 770.0000 | 60 mg/m2 IV on Day 1 every 3 weeks for 4 cycles | 27 | 747 |
Cyclophosphamide (generics) | 20 mg/mL | 500 mg vial 1,000 mg vial 2,000 mg vial | 91.3100 165.5200 304.4000 | 600 mg/m2 IV on Day 1 every 3 weeks for 4 cycles | 12 | 342 |
Paclitaxel (Taxol) | 6 mg/mL | 30 mg vial 96 mg vial 300 mg vial | 300.0000 1,196.8000 3,740.0000 | After AC is complete: 80 mg/m2 every week for 12 weeks | 257 | 7,187 |
Trastuzumab IV (biosimilar) | 21 mg/mL | 150 mg vial 440 mg vial | 506.1405 1,417.196 | After AC is complete: First cycle: 8 mg/kg IV on day 1; Thereafter: 6 mg/kg IV on day 1 every 3 weeks for another 3 cyclese | First cycle: 96 Subsequent cycles: 72 | First cycle: 2,530 Subsequent cycles: 2,025 |
Trastuzumab SC (Herceptin SC) | 120 mg/mL | 600 mg single-dose vial | 2,625.0000 | After AC is complete: 600 mg SC on day 1 every 3 weeks for 4 cyclese | 125 | 3,500 |
AC-PACL(W) + Tras-IV regimen, per 28-days | First cycle: 10,807 Subsequent cycles: 10,301 | |||||
AC-PACL(W) + Tras SC regimen, per 28-days | 11,776 | |||||
AC-D + TRAS | ||||||
Doxorubicin (generics) | 2 mg/mL | 10 mg vial 50 mg vial 200 mg vial | 50.0000 255.0000 770.0000 | 60 mg/m2 IV on Day 1 every 3 weeks for 4 cycles | 27 | 747 |
Cyclophosphamide (generics) | 20 mg/mL | 500 mg vial 1,000 mg vial 2,000 mg vial | 91.3100 165.5200 304.4000 | 600 mg/m2 IV on Day 1 every 3 weeks for 4 cycles | 12 | 342 |
Docetaxel (generics) | 10 mg/mL 10 mg/mL 20 mg/mL 20 mg/mL 20 mg/mL | 80 mg vial 160 mg vial 20 mg vial 80 mg vial 160 mg vial | 970.2000 1,850.0000 249.0000 497.0000 994.0000 | After AC is complete: 100 mg/m2 IV on Day 1 every 3 weeks for 4 cycles | 59 | 1,657 |
Trastuzumab IV (biosimilar) | 21 mg/mL | 150 mg vial 440 mg vial | 506.1405 1,417.196 | After AC is complete: First cycle: 8 mg/kg IV on day 1c Thereafter: 6 mg/kg IV on day 1 every 3 weeks for another 3 cyclese | First cycle: 96 Subsequent cycles: 72 | First cycle: 2,530 Subsequent cycles: 2,025 |
Trastuzumab SC (Herceptin SC) | 120 mg/mL | 600 mg single-dose vial | 2,625.0000 | After AC is complete: 600 mg SC on day 1 every 3 weeks for 4 cyclese | 125 | 3,500 |
AC-D + Tras-IV regimen, per 28-days | First cycle: 5,277 Subsequent cycles: 4,771 | |||||
AC-D + Tras SC regimen, per 28-days | 6,246 | |||||
FEC-D + TRAS | ||||||
Fluorouracil (generics) | 50 mg/mL | 500 mg vial | 160.9000 | 500 mg/m2 IV on Day 1 every 3 weeks for 3 cycles. | 107 | 429 |
Epirubicin | 2 mg/mL | 10 mg vial 50 mg vial 100 mg vial | 40.1200 200.9100 779.5400 | 100 mg/m2 IV on Day 1 every 3 weeks for 3 cycles | 241 | 964 |
Cyclophosphamide (generics) | 20 mg/mL | 500 mg vial 1,000 mg vial 2,000 mg vial | 91.3100 165.5200 304.4000 | 500 mg/m2 IV on Day 1 every 3 weeks for 3 cycles | 8 | 221 |
Docetaxel (generics) | 10 mg/mL 10 mg/mL 20 mg/mL 20 mg/mL 20 mg/mL | 80 mg vial 160 mg vial 20 mg vial 80 mg vial 160 mg vial | 970.2000 1,850.0000 249.0000 497.0000 994.0000 | After FEC is complete: 100 mg/m2 IV on Day 1 every 3 weeks for 3 or 4 cycles | 59 | 1,657 |
Trastuzumab IV (biosimilar) | 21 mg/mL | 150 mg vial 440 mg vial | 506.1405 1,417.196 | After AC is complete: First cycle: 8 mg/kg IV on day 1; Thereafter: 6 mg/kg IV on day 1 every 3 weeks for another 2 or 3 cyclese | First cycle: 96 Subsequent cycles: 72 | First cycle: 2,531 Subsequent cycles: 2,024 |
Trastuzumab SC (Herceptin SC) | 120 mg/mL | 600 mg single-dose vial | 2,625.0000 | After AC is complete: 600 mg SC on day 1 every 3 weeks for 3 or 4 cyclese | 125 | 3,500 |
FEC-D + Tras-IV regimen, per 28 days | First cycle: 5,802 Subsequent cycles: 5,296 | |||||
FEC-D + Tras SC regimen, per 28 days | 6,771 | |||||
CRBP-D + Tras | ||||||
Carboplatin (generics) | 10 mg/mL | 50 mg 150 mg 450 mg 600 mg | 70.0000 210.0000 600.0000 775.0000 | Target AUC 6 on Day 1 every 3 weeks for 6 cyclesf | 43.57 | 1,220 |
Docetaxel (generics) | 10 mg/mL 10 mg/mL 20 mg/mL 20 mg/mL 20 mg/mL | 80 mg vial 160 mg vial 20 mg vial 80 mg vial 160 mg vial | 970.2000 1,850.0000 249.0000 497.0000 994.0000 | 75 mg/m2 IV on Day 1 every 3 weeks for 6 cycles | 59 | 1,657 |
Trastuzumab IV (biosimilar) | 21 mg/mL | 150 mg vial 440 mg vial | 506.1405 1,417.196 | First cycle: 8 mg/kg IV on day 1; Thereafter: 6 mg/kg IV on day 1 every 3 weeks for another 5 cyclese | First cycle: 96 Subsequent cycles: 72 | First cycle: 2,531 Subsequent cycles: 2,025 |
Trastuzumab SC (Herceptin SC) | 120 mg/mL | 600 mg single-dose vial | 2,625.0000 | After AC is complete: 600 mg SC on day 1 every 3 weeks for 6 cyclese | 125 | 3,500 |
CRBP-D plus Tras-IV, per 28 days | First cycle: 5,409 Subsequent cycles: 4,903 | |||||
CRBP-D plus Tras SC, per 28 days | 6,379 | |||||
A = doxorubicin; AUC = product of serum concentration (mg/mL) and time (min); C = cyclophosphamide; CRBP = carboplatin; D = docetaxel; DD = dose dense; F = fluorouracil; E = epirubicin; GFR = glomerular filtration rate; PACL = paclitaxel; SC = subcutaneous; tras = trastuzumab.
Note: All prices are wholesale prices from the IQVIA Delta PA database (accessed June 2021), unless otherwise indicated, and do not include dispensing or administration fees but do assume wastage of excess medication in vials. Doses are from the Cancer Care Ontario Drug Formulary regimen database.31 Mean patient body weight was assumed to be 75 kg, while mean body surface area was 1.8 m2. For the purposes of calculating glomerular filtration rate, patient age was assumed to be 49,3 and serum creatinine was 0.8871.32
aCost standardized to 28-day cycles to allow for comparison among regimens of different cycle lengths.
bSponsor’s submitted price.
cThe number of cycles of pertuzumab is dependent upon the number of cycles of trastuzumab given in the neoadjuvant setting with each chemotherapy regimen.
dPertuzumab is indicated for continuing adjuvant therapy after neoadjuvant therapy in combination with trastuzumab at the discretion of the physician. The sponsor is not requesting reimbursement for this indication for this review.
eAfter the initial doses during neoadjuvant chemotherapy, trastuzumab is continued in 21-day cycles for a total of one year, or until progression or unacceptable toxicity occurs.
fDose calculated using the Cockcroft-Gault method33 where GFR for women = (((140 – age) x body weight) / (72 × serum creatinine)) × 0.85 and the Calvert method34 where dose is Target AUC * (GFR + 25).
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Characteristics of the modelled population were based on the KATHERINE trial which focused on adjuvant T-DM1. Moreover, the key treatment efficacy (pCR rates) was based on the PEONY trial, where all participants were Asian. Although this assumption was acceptable by the clinical expert consulted by CADTH, there is no published evidence supporting that patients receiving neoadjuvant and adjuvant treatments have similar baseline characteristics and that a pCR response is independent of racial groups. |
Model has been adequately programmed and has sufficient face validity | Yes | Acceptable. |
Model structure is adequate for decision problem | No | See CADTH appraisal section. Furthermore, the clinical expert consulted by CADTH noted that several health states definitions were inappropriate and not used in the treatment of this condition in practice (e.g., “non-metastatic recurrence”). |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | See CADTH appraisal section. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | See CADTH appraisal section. Due to the large number of input parameters and assumptions used in the sponsor’s model, CADTH was unable to assess the impact of all variables and assumptions on cost-effectiveness results. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The sponsor’s model was based on several input parameters drawn from many data sources. The economic report should have included a table summarizing the sources used to inform key parameters to improve transparency. |
HT = trastuzumab-taxane chemotherapy; ICER = incremental cost-effectiveness ratio; PHT = pertuzumab- trastuzumab-taxane chemotherapy.
Note: This table has not been copy-edited.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Reanalyses
Table 7: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1.Dispensing fee for oral medications | $8.83 | 0 |
2.Distribution for cost data in probabilistic analyses | Log-normal | Gamma |
3.Number of simulations for probabilistic analyses | 2,000 | 5,000 |
4.Distribution of first-line therapies for early recurrence (< 18 months) mBC (%): a) Non-pCR arm; b) pCR arm | Pertuzumab + trastuzumab + docetaxel: 50.8 Pertuzumab + trastuzumab + paclitaxel: 44.3 Trastuzumab + capecitabine: 1.3 Vinorelbine: 2.5 Lapatinib + capecitabine: 1.3 TDM-1: 56.3 Pertuzumab + trastuzumab + docetaxel: 15.0 Pertuzumab + trastuzumab + paclitaxel: 20.0 Trastuzumab + vinorelbine: 1.9 Trastuzumab + capecitabine: 3.1 Vinorelbine: 2.5 Lapatinib + capecitabine: 1.9 | Pertuzumab + trastuzumab + docetaxel: 52.7 Pertuzumab + trastuzumab + paclitaxel: 46.2 Trastuzumab + capecitabine: 1.3 Vinorelbine: 0 Lapatinib + capecitabine: 0 TDM-1: 58.4 Pertuzumab + trastuzumab + docetaxel: 15.6 Pertuzumab + trastuzumab + paclitaxel: 20.8 Trastuzumab + vinorelbine: 2.0 Trastuzumab + capecitabine: 3.3 Vinorelbine: 0 Lapatinib + capecitabine: 0 |
BC = breast cancer; TDM-1 = trastuzumab emtansine; H = trastuzumab; mBC = metastatic breast cancer; pCR = pathologic complete response.
Table 8: Summary of the Stepped Analysis of the CADTH-Corrected Analysis Results
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | HT | 191,897 | 18.589 | Reference |
PHT | 199,861 | 18.305 | 27,986 | |
CADTH-corrected analysis | HT | 192,001 | 18.309 | Reference |
PHT | 199,798 | 18.597 | 27,112 |
HT = trastuzumab-taxane chemotherapy; ICER = incremental cost-effectiveness ratio; PHT = pertuzumab- trastuzumab-taxane chemotherapy, QALY = quality-adjusted life-year.
Table 9: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | PHT | HT | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 23.566 | 23.218 | 0.348 |
By health state | |||
iDFS | 22.397 | 21.999 | 0.398 |
Non-metastatic recurrence | 0.033 | 0.031 | 0.002 |
Remission | 0.323 | 0.303 | 0.020 |
First-line mBC | 0.374 | 0.407 | −0.033 |
Subsequent treatment line of mBC | 0.438 | 0.477 | −0.039 |
Discounted QALYs | |||
Total | 18.597 | 18.309 | 0.288 |
By health state or data source | |||
iDFS | 17.814 | 17.500 | 0.314 |
Non-metastatic recurrence | 0.027 | 0.025 | 0.002 |
Remission | 0.256 | 0.240 | 0.016 |
First-line mBC | 0.276 | 0.301 | −0.025 |
Subsequent treatment line of mBC | 0.223 | 0.243 | −0.020 |
Discounted costs ($) | |||
Total | 199,798 | 192,001 | 7,797 |
Acquisition | 86,306 | 70,557 | 15,749 |
Administration | 7,867 | 7,415 | 452 |
AE management | 144 | 74 | 69 |
Surgery | 4,697 | 4,697 | 0 |
Supportive care | 47,066 | 50,425 | −3,359 |
End of life | 5,900 | 6,406 | −506 |
ICER ($/QALY) | 27,112 | ||
AE = adverse event; ICER = incremental cost-effectiveness ratio; iDFS = invasive disease–free survival; LY = life-year; mBC = metastatic breast cancer; QALY = quality-adjusted life-year.
Scenario Analyses
Based on CADTH’s corrected reanalyses, a series of scenario analyses were conducted (Table 10).
Table 10: Summary of CADTH Scenario Analyses
Treatment regimen | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case | |||
HT | 191,897 | 18.589 | Reference |
PHT | 199,861 | 18.305 | 27,986 |
CADTH’s corrected analysis | |||
HT | 192,001 | 18.309 | Reference |
PHT | 199,798 | 18.597 | 27,112 |
CADTH’s scenario analysis 1: Alternative source for pCR (NEOSPHERE trial) | |||
HT | 184,701 | 18.451 | Reference |
PHT | 193,187 | 18.730 | 30,390 |
CADTH’s scenario analysis 2: Alternative source for EFS data (CTneoBC) | |||
HT | 261,949 | 14.819 | Reference |
PHT | 269,830 | 15.476 | 11,987 |
CADTH’s scenario analysis 3: Lower Bound of HR EFS for pCR vs. non-pCR (HR = 0.25) | |||
HT | 188,793 | 18.457 | Reference |
PHT | 194,091 | 18.832 | 14,116 |
CADTH’s scenario analysis 4: Upper Bound of HR EFS for pCR vs. non-pCR (HR = 0.43) | |||
HT | 195,355 | 18.204 | Reference |
PHT | 206,013 | 18.393 | 56,253 |
CADTH’s scenario analysis 5: Assuming no difference in EFS between pCR vs. non-pCR (HR = 1) | |||
HT | 212,888 | 17.573 | Reference |
PHT | 237,288 | 17.269 | Dominated by HT |
CADTH’s scenario analysis 6: Alternative parametric survival models for iDFS, non-pCR, TDM-1 and H (log-normal) | |||
HT | 183,153 | 18.569 | Reference |
PHT | 193,021 | 18.805 | 41,869 |
CADTH’s scenario analysis 7: Alternative parametric survival models for iDFS, non-pCR, TDM-1 (exponential) and H (generalized Gamma) | |||
HT | 189,403 | 18.412 | Reference |
PHT | 197,049 | 18.708 | 25,857 |
CADTH’s scenario analysis 8: Alternative parametric survival models for iDFS, pCR, PHT (gompertz) and HT (log-normal) iDFS | |||
HT | 192,115 | 18.353 | Reference |
PHT | 199,909 | 18.642 | 26,965 |
CADTH’s scenario analysis 9: Reducing duration at which the cure proportion of patients with pCR reached its maximum (95%) to 96 months | |||
HT | 190,449 | 18.382 | Reference |
PHT | 197,136 | 18.704 | 20,768 |
CADTH’s scenario analysis 10: Increasing duration at which the cure proportion of patients with pCR reached its maximum (95%) to 144 months | |||
HT | 193,031 | 18.275 | Reference |
PHT | 201,887 | 18.531 | 34,521 |
CADTH’s scenario analysis 11: Reducing duration at which the cure proportion of non-pCR reached its maximum (95%) to 96 months | |||
HT | 180,594 | 18.662 | Reference |
PHT | 191,027 | 18.875 | 48,981 |
CADTH’s scenario analysis 12: Increasing duration at which the cure proportion of non-pCR reached its maximum (95%) to 144 months | |||
HT | 202,198 | 18.019 | Reference |
PHT | 207,669 | 18.374 | 15,427 |
CADTH’s scenario analysis 13: Using alternative utility source for eBC (APHYNITY trial) | |||
HT | 192,092 | 18.277 | Reference |
PHT | 199,945 | 18.564 | 27,315 |
CADTH’s scenario analysis 14: Allowing 50% of eBC patients to use pertuzumab in an adjuvant setting | |||
HT | 191,973 | 18.328 | Reference |
PHT | 208,674 | 18.690 | 46,193 |
CADTH’s scenario analysis 15: Allowing 100% of eBC patients to use pertuzumab in an adjuvant setting | |||
HT | 191,924 | 18.335 | Reference |
PHT | 217,463 | 18.776 | 57,965 |
CADTH’s scenario analysis 16: Assuming the treatment effect in patients with pCR is maintained over time | |||
HT | 194,641 | 18.302 | Reference |
PHT | 201,909 | 18.593 | 24,983 |
CADTH’s scenario analysis 17: Assuming drug wastage for IV medications | |||
HT | 194,404 | 18.359 | Reference |
PHT | 204,240 | 18.646 | 34,248 |
CADTH’s scenario analysis 18: Removing trastuzumab monotherapy from a list of second-line treatments for metastatic BC | |||
HT | 192,164 | 18.361 | Reference |
PHT | 200,023 | 18.648 | 27,419 |
CADTH’s scenario analysis 19: Adopting a societal perspective (human capital approach) | |||
HT | 275,864 | 18.312 | Reference |
PHT | 269,078 | 18.598 | 23,732 |
CADTH’s scenario analysis 20: Adopting a societal perspective (friction method) | |||
HT | 215,678 | 18.310 | Reference |
PHT | 223,190 | 18.596 | 26,212 |
BC = breast cancer; EFS = event-free survival; HR = hazard ratio; HT = , trastuzumab-taxane chemotherapy; PHT = pertuzumab- trastuzumab-taxane chemotherapy; ICER = incremental cost-effectiveness ratio; pCR = pathologic complete response.
Table 11: CADTH Price Reduction Exploratory Analyses
Price reduction analysis | ICERs for PHT vs. HTa | |
|---|---|---|
CADTH Scenario Analysis 4 HR EFS pCR vs. non-pCR = 0.43 | CADTH Scenario Analysis 5 HR EFS pCR vs. non-pCR = 1.00 | |
No price reduction | $56,253 | Dominated by HT |
9% | $49,628 | Dominated by HT |
20% | $40,011 | Dominated by HT |
40% | $22,527 | Dominated by HT |
60% | $5,042 | Dominated by HT |
80% | PHT was dominant | Dominated by HT |
CTneoBC = Collaborative Trials in Neoadjuvant Breast Cancer; EFS = event-free survival; HR = hazard ratio; HT = trastuzumab-taxane chemotherapy; PHT = pertuzumab- trastuzumab-taxane chemotherapy; ICER = incremental cost-effectiveness ratio; pCR = pathologic complete response.
aBased on deterministic analyses.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 12: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted an incidence-based (budget impact analysis) BIA,35 assessing the expected budgetary impact of the introduction of pertuzumab in combination with trastuzumab and chemotherapy for the neoadjuvant treatment of patients with HER2-positive, locally advanced, inflammatory, or eBC, over a 3-year time horizon from the perspective of a Canadian drug plan payer. The population considered in the model is consistent with the indicated population.
Data from the model were obtained from various sources including: the PEONY trial,36 sponsor-conducted internal chart review,37 Canadian Cancer Society statistics,38 the Canadian Partnership Against Cancer statistics,39 Statistics Canada projections,40 and clinical expert opinion. Only drug costs were included, and the duration of therapy with pertuzumab was consistent with the proportions of trastuzumab plus chemotherapy neoadjuvant regimens assigned as recommended in the product monograph.2 No wastage was assumed.
Key inputs to the BIA are documented in Table 13. Other assumptions made by the sponsor include:
Incident patients of any gender were included if they had stage IIA, IIB, IIIA, IIIB, or IIIC breast cancer.
Perfect vial sharing of all treatments (no wastage).
Subsequent treatments for recurrent or metastatic breast cancer were not considered.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Breast Cancer Incidence, Canada excluding Quebeca Proportion of patients covered by public plans Incident patients by stageb % Incident patients in stage IB % Incident patients in stage IIA % Incident patients in stage IIB % Incident patients in stage IIIA % Incident patients in stage IIIB % Incident patients in stage IIIC Proportion of patients tested for HER2 (stage IIA to IIIC)c Proportion of patients who are HER2+d Proportion of patients receiving neoadjuvant treatmente Proportion of patients going on to receive surgeryc Proportion of patients going on to receive adjuvant treatmentc | 21,247 / 21,500 / 21,754 |||| 0% included 19.1% 10.6% 5.9% 2.1% 2.1% |||| 18% |||||||||||||||||||| |||| |||| |
Number of patients eligible for drug under review | 820 / 845 / 871 |
Market Uptake (3 years) | |
Uptake (reference scenario) Trastuzumab-IV (biosimilar) Trastuzumab-SC | |||||||||||||||||||||||| |||||||||||||| |
Uptake (new drug scenario)f Pertuzumab + trastuzumab-IV (Herceptin) Pertuzumab + trastuzumab-SC (Herceptin) Trastuzumab-IV (biosimilar) Trastuzumab-SC (Herceptin) | |||||||||||||||||| |||||||||||||| |||||||||||||||||||| |||||||||||||| |
Background Chemo Regimens, regardless of scenarioe AC-PACL(DD) AC-PACL(W) AC-D FEC-D CRBP-D | |||| |||| |||| |||| |||| |
Proportion of patients achieving pCR+g Pertuzumab + Trastuzumab + Chemotherapy Trastuzumab + Chemotherapy | 39.3% 21.8% |
Adjuvant Continuation therapy, regardless of scenario Patients achieving pCR Trastuzumab-IV Trastuzumab-SC Patients not achieving pCR Trastuzumab-IV Trastuzumab-SC Trastuzumab emastine (TDM-1) | |||||||||||||||||||||||| |||||||||||||| |||||||||||||||||||| |||||||||||||| |||||||||||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment for full course of neoadjuvant treatment AC-PACL(DD) + TRAS-IV (Herceptin) + PERT AC-PACL(DD) + TRAS-IV (biosimilar) AC-PACL(W) + TRAS-IV (Herceptin) + PERT AC-PACL(W) + TRAS-IV (biosimilar) CRBP-D + TRAS-IV (Herceptin) + PERT CRBP-D + TRAS-IV (biosimilar) | $34,318 $10,741 $35,238 $11,662 $49,891h $16,504h |
A = doxorubicin; C = cyclophosphamide; CRBP = carboplatin; D = docetaxel; DD = dose dense; F = fluorouracil; E = epirubicin; PACL = paclitaxel; PERT = pertuzumab; SC = subcutaneous; TRAS = trastuzumab; W = weekly.
aIncident cases derived from projected new cases of breast cancer from the Canadian Cancer Society,38 or calculated based on Statistics Canada population projections40 or NIHB 2018/2019 annual report.41
bDerived from 2013 breast cancer stage distribution reported by Canadian Partnership Against Cancer, only included proportions are reported.39
cReported as expert opinion.35
dSlamon et al. 1987.42
eBased on a Roche internal chart review of patients who started neoadjuvant therapy since April 1, 2020.37
fSource not reported, presumably internal market research.
gPEONY trial.36
hThe sponsor’s carboplatin dose calculation contained an error within the model When this error was corrected, the cost of a full course of neoadjuvant treatment with CRBP-D+TRAS+PERT was $51,207, while the full course cost of CRBO-D+TRAS was $17,821.
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s base case suggest that the incremental budget impact of reimbursing pertuzumab, in combination with trastuzumab and chemotherapy, for the neoadjuvant treatment of HER2-positive, locally advanced, inflammatory, or early-stage breast cancer (either > 2 cm in diameter or node-positive disease) would be $1,198,120, $8,578,658, and $13,794,812 in Years 1, 2, and 3 respectively, for a 3-year cumulative total of $23,571,591.
Key scenario analyses included: assuming wastage of excess medication in vials, increasing the rate at which the use of neoadjuvant treatment is increasing, varying the absolute uptake of pertuzumab by 5% in all 3 years, varying the proportion of HER2-positive patients, assuming that patients using pertuzumab receive biosimilar trastuzumab, assuming that all patients use the FEC-D based chemotherapy regimen (i.e., only 3 cycles of pertuzumab), and assuming patients achieving pCR with neoadjuvant pertuzumab continued on adjuvant pertuzumab for the remainder of a year. Results of all scenarios over 3 years ranged from $14.5 million to $54.9 million.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Some stage II and III patients excluded: The sponsor included early breast cancer patients consistent with the proportions of patients having stages IIA, IIB, IIIA, IIIB, and IIIC cancer as reported by the Canadian Partnership Against Cancer in 2013.39 However, this source also reports 0.1% of patients having stage III cancer not otherwise specified, and that 1.8% of patients with an unknown stage of cancer. CADTH assumed that patients with missing stage information in the dataset had the same likelihood of being in stages II or III as the rest of the reported population.
CADTH corrected the sponsor’s base case to include patients with stage III NOS cancer, as well as 40.7% of the 1.8% of patients with an unknown/unreported stage, proportional to the percentage of stage II and III patients overall.
Proportion of patients receiving neoadjuvant therapy underestimated: The model assumes that only |% of patients who would otherwise qualify for neoadjuvant therapy (i.e., HER2+, stage II or III) actually receive it in the base year of the model, based on an internal chart review of Canadian HER2+ early breast cancer patients with tumours greater than 3 cm who started on adjuvant therapy since April 1, 2020.37 A total of | breast cancer charts were obtained through | oncologists who responded to a survey request from the sponsor. The number of oncologists who did not respond to the survey was not reported. Data from the same report indicated that only about |% of responding oncologists highly believe that all patients eligible to receive neoadjuvant treatment are appropriately triaged by surgeons to receive it. The sponsor then assumed that the proportion of patients who receive neoadjuvant treatment is increasing by | % a year, based on expert opinion. The clinical expert consulted by CADTH considered both the initial proportion of patients currently receiving neoadjuvant therapy and the rate at which that proportion was increasing to be too low to accurately reflect current Canadian practice.
CADTH assumed that 65% of eligible patients in the base year receiving a neoadjuvant therapy, increasing by 5% per year to 80% by Year 3. A scenario analysis was run where the sponsor’s initial estimate of |% was assumed in the base year, with that proportion increasing by 5% per year.
Branded trastuzumab in combination with pertuzumab: The sponsor’s model assumes that patients receiving pertuzumab will also receive branded trastuzumab (Herceptin) for the duration of time they receive pertuzumab, and receive biosimilar trastuzumab otherwise, i.e., if using non-pertuzumab neoadjuvant regimens or during the year of adjuvant trastuzumab therapy after the neoadjuvant pertuzumab regimen is complete. The sponsor is thus modelling the budgetary impact of using 2 of their products in combination for neoadjuvant therapy, rather than just the budgetary impact of adding pertuzumab. While Herceptin was the brand of trastuzumab used in the clinical trial, unless an alternate pricing arrangement for this combination of products is specifically negotiated, it is likely that pertuzumab patients in clinical practice will instead receive the trastuzumab product they would otherwise have received in the absence of pertuzumab for neoadjuvant therapy.
In the base case, CADTH assumed that all patients would receive biosimilar trastuzumab IV as part of their neoadjuvant regimens.
Uptake of pertuzumab underestimated: The sponsor’s analysis assumes that only | % of eligible patients would receive pertuzumab in the first year of its reimbursement, rising to ||% and |% in Years 2 and 3, respectively. In contrast, the clinical expert consulted by CADTH as well as clinician group input considered neoadjuvant treatment to be the international standard of care, and as such, predict uptake of pertuzumab in the neoadjuvant setting will be swift and near-universal for eligible patients.
CADTH’s base case assumes that 60% of eligible patients will receive pertuzumab in Year 1, rising to 80% and 100% in Years 2 and 3, respectively.
Subsequent therapies were not considered: While the sponsor’s BIA model considers differences in adjuvant continuation therapy choice (i.e., trastuzumab or TDM-1) based on the proportion of patients achieving or not achieving pCR after surgery, it does not consider the impact of neoadjuvant pertuzumab on downstream therapies for recurrent or metastatic disease. There is uncertainty in the impact of pCR rates on survival outcomes such as EFS (see CADTH Appraisal of the Sponsor’s Economic Evaluation section, above), and therefore these is uncertainty in the budgetary impact of neoadjuvant pertuzumab on later therapies for recurrent or metastatic cancers.
CADTH was unable to adjust for this limitation due to structural limitations in the BIA model
Confidential prices are unknown: Both the sponsor’s and CADTH’s BIA analyses are based on wholesale or otherwise publicly available list prices for trastuzumab, TDM-1, and all chemotherapy regimens. It is likely that confidentially negotiated prices for some or all comparators exist. As such, the actual cost of adding pertuzumab to trastuzumab plus chemotherapy and the subsequent offsetting of adjuvant therapy with TDM-1 for patients who are pCR- is unknown.
CADTH was unable to adjust for this limitation.
Additional limitations were identified but were not considered to be key limitations. These limitations included: uncertainty in the proportions of patients who receive neoadjuvant therapy who go on to receive surgery and then adjuvant therapy, uncertainty in the market shares of neoadjuvant chemotherapy regimens, uncertainty in the proportion of pCR- patients who will receive TDM-1 adjuvant therapy, uncertainty in whether pCR rates derived from the PEONY trial are most generalizable to Canadian clinical practice, and a miscalculation of the required dose of carboplatin. Additionally, while not part of the reimbursement request, the product monograph states that pertuzumab in the neoadjuvant setting may be continued at the physician’s discretion in the adjuvant setting to complete 1 year of treatment.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by: including patients with stage III not otherwise specified and a proportion of unknown stage patients with breast cancer, correcting the dose of carboplatin, increasing the proportion of patients who receive neoadjuvant therapy, assuming patients receiving pertuzumab will receive biosimilar trastuzumab, and increasing the assumed uptake of pertuzumab. Table 14 outlines the parameters used by the sponsor in comparison to those used by CADTH.
Table 14: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Stage III NOS and 41% of unknown stage cancers included | 40% of overall BC patients | 41% of overall BC patients |
2. Dose of carboplatin correctedb | Calculated dose per cycle: 558 mg | Calculated dose per cycle: 670 mg |
Changes to derive the CADTH base case | ||
1. Proportion receiving neoadjuvant therapy | | / | / | | 70% / 75% / 80% |
2. Biosimilar trastuzumab | Patients receive branded trastuzumab for the duration of their pertuzumab regimen, otherwise they receive biosimilar trastuzumab | Patients receive biosimilar trastuzumab in combination with pertuzumab |
3. Uptake of pertuzumab | | / | / | | 60% / 80% / 100% |
CADTH base case | Reanalysis 1 + 2 + 3 | |
BC = breast cancer.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses) that are not identified as limitations.
bThe sponsor’s calculation of carboplatin dosing had a misplaced bracket in the formula. CADTH corrected this error so that carboplatin dosing matched that assumed in the submitted economic evaluation. This error altered the total costs of the reference and new drug scenarios but did not alter the incremental budget impact as it applied to both scenarios equally.
Applying these changes increased the total 3-year budget impact to $31,190,490. The results of the CADTH step-wise reanalysis are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16.
Table 15: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Submitted base case | 23,571,591 |
Corrected base case | 24,160,880 |
CADTH reanalysis 1: increased neoadjuvant use | 33,184,677 |
CADTH reanalysis 2: biosimilar trastuzumab | 14,866,580 |
CADTH reanalysis 3: increased pertuzumab uptake | 37,439,060 |
CADTH base case | 31,190,490 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to explore other areas of interest or uncertainty:
Excess medication in vials is wasted.
IV administration costs are included.
TDM-1 use in pCR- patients increased to 100% to match CUA model assumption.
All patients who receive neoadjuvant therapy are assumed to receive surgery and adjuvant therapy.
pCR rates from the NEOSPHERE trial (45.8% for pertuzumab, 29.0% for non-pertuzumab regimens) used.
Proportion of eligible patients receiving neoadjuvant therapy is |% in the base year, increasing by 5% annually.
50% of pCR+ patients receiving neoadjuvant pertuzumab continue it for adjuvant therapy.
100% of pCR+ patients receiving neoadjuvant pertuzumab continue it for adjuvant therapy.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 46,377,113 | 51,108,448 | 54,359,306 | 55,982,872 | 207,827,740 |
New drug | 46,377,113 | 52,306,568 | 62,937,964 | 69,777,685 | 231,399,330 | |
Budget impact | 0 | 1,198,120 | 8,578,658 | 13,794,812 | 23,571,591 | |
Corrected base case | Reference | 47,804,965 | 52,662,863 | 56,003,386 | 57,676,056 | 214,147,270 |
New drug | 47,804,965 | 53,890,936 | 64,796,510 | 71,815,739 | 238,308,150 | |
Budget impact | 0 | 1,228,073 | 8,793,125 | 14,139,683 | 24,160,880 | |
CADTH base case | Reference | 57,543,013 | 67,025,462 | 75,004,534 | 80,948,851 | 280,521,861 |
New drug | 57,543,013 | 74,344,203 | 85,166,764 | 94,658,370 | 311,712,350 | |
Budget impact | 0 | 7,318,741 | 10,162,230 | 13,709,519 | 31,190,490 | |
CADTH scenario 1: wastage assumed | Reference | 77,975,288 | 88,789,119 | 98,326,418 | 106,119,059 | 371,209,883 |
New drug | 77,975,288 | 97,237,123 | 110,170,308 | 122,097,247 | 407,479,966 | |
Budget impact | — | 8,448,004 | 11,843,890 | 15,978,189 | 36,270,083 | |
CADTH scenario 2: Administration costs | Reference | 57,543,013 | 67,025,462 | 75,004,534 | 80,948,851 | 280,521,861 |
New drug | 57,543,013 | 74,711,155 | 85,697,239 | 95,374,015 | 313,325,423 | |
Budget impact | — | 7,685,693 | 10,692,705 | 14,425,164 | 32,803,562 | |
CADTH scenario 3: pCR- patients all receive TDM-1 | Reference | 69,394,997 | 75,637,564 | 82,007,579 | 88,506,906 | 315,547,046 |
New drug | 69,394,997 | 81,799,948 | 90,916,067 | 100,525,044 | 342,636,056 | |
Budget impact | — | 6,162,385 | 8,908,488 | 12,018,138 | 27,089,011 | |
CADTH scenario 4: All patients continue with surgery + adjuvant | Reference | 60,855,055 | 70,954,583 | 79,437,564 | 85,733,211 | 296,980,414 |
New drug | 60,855,055 | 77,930,516 | 89,073,250 | 98,732,386 | 326,591,208 | |
Budget impact | — | 6,975,933 | 9,635,685 | 12,999,175 | 29,610,793 | |
CADTH scenario 5: pCR rates from NEOSPHERE | Reference | 54,996,807 | 63,853,742 | 71,350,772 | 77,005,518 | 267,206,839 |
New drug | 54,996,807 | 71,357,500 | 81,797,183 | 91,098,416 | 299,249,906 | |
Budget impact | — | 7,503,758 | 10,446,412 | 14,092,898 | 32,043,068 | |
CADTH scenario 6: | % neoadjuvant therapy in base year | Reference | 47,804,965 | 56,492,889 | 64,003,869 | 69,818,384 | 238,120,108 |
New drug | 47,804,965 | 62,661,543 | 72,675,639 | 81,642,844 | 264,784,990 | |
Budget impact | — | 6,168,653 | 8,671,770 | 11,824,460 | 26,664,883 | |
CADTH scenario 7: 50% continue pert adjuvant | Reference | 57,543,013 | 67,025,462 | 75,004,534 | 80,948,851 | 280,521,861 |
New drug | 57,543,013 | 79,707,590 | 92,920,203 | 105,118,270 | 335,289,077 | |
Budget impact | — | 12,682,129 | 17,915,669 | 24,169,419 | 54,767,216 | |
CADTH scenario 8: 100% continue pert adjuvant | Reference | 57,543,013 | 67,025,462 | 75,004,534 | 80,948,851 | 280,521,861 |
New drug | 57,543,013 | 85,070,978 | 100,673,642 | 115,578,170 | 358,865,803 | |
Budget impact | — | 18,045,516 | 25,669,108 | 34,629,319 | 78,343,943 |
BIA = budget impact analysis; pCR = pathologic complete response; pert = pertuzumab; TDM-1 = trastuzumab emtansine.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.