CADTH Health Technology Review
Platform, Basket, and Umbrella Trial Designs: Issues Around Health Technology Assessment of Novel Therapeutics
Discussion Paper
Authors: Jean-Eric Tarride, Matthew Cheung, Timothy P. Hanna, Lauren E. Cipriano, Dean A. Regier, Spencer Phillips Hey, Kelvin K.W. Chan, Nicole Mittmann
Author Information
Authors
Jean-Eric Tarride,1-3 Matthew Cheung,4 Timothy P. Hanna,5 Lauren E. Cipriano,6 Dean A. Regier,7 Spencer Phillips Hey,8 Kelvin K.W. Chan,4,9,10 Nicole Mittmann.4,9,11
Affiliations
1. Department of Health Research Methods, Evidence and Impact, McMaster University
2. Center for Health Economic and Policy Analysis (CHEPA), McMaster University
3. Programs for Assessment of Technology in Health (PATH), The Research Institute of St. Joe’s Hamilton, St. Joseph’s Healthcare Hamilton
4. Department of Medicine, University of Toronto
5. Division of Cancer Care and Epidemiology, Queen’s University Cancer Research Institute
6. Ivey Business School and Department of Epidemiology and Biostatistics, Schulich School of Medicine and Dentistry, Western University
7. Cancer Control Research, BC Cancer, School of Population and Public Health, University of British Columbia
8. Harvard Medical School, Harvard University
9. Sunnybrook Health Sciences Centre
10. Canadian Centre for Applied Research in Cancer Control
11. Canadian Agency for Drugs and Technologies in Health (CADTH)
Acknowledgements
We would like to thank Case Market Access Consulting Inc. for transcribing the webinar and creating a transcript and outline for the manuscript.
Key Messages
Advancements in genomic and precision medicine have changed the way oncology clinical trials are designed. Compared to the traditional single tumour-based trial, master protocols, which are often classified as basket, umbrella, or platform trials, conduct studies on multiple subgroups under a single overarching protocol.
This paper summarizes the discussion held during CADTH’s second webinar, which examined issues around health technology assessment of evidence generated using these trial designs.
In this workshop, several experts were invited to present on the clinical, health economic, patient value, and ethical aspects associated with these new study designs. The discussion section of this paper summarizes the major points raised by the presenters, as well as questions from the audience and answers from the presenters.
Introduction
Advancements in genomic and precision medicine have changed the way oncology clinical trials are designed. Compared to the traditional single tumour-based trial, master protocols, which are often classified as basket, umbrella or platform trials, conduct studies on multiple subgroups under a single overarching protocol. Basket trials enrol patients who share a specific genetic marker to test 1 or several targeted therapies independent of the anatomic location of a tumour. Umbrella trials simultaneously test multiple targeted therapies among patients who share a single tumour type but who are stratified into subgroups by molecular alteration. In platform trials, several interventions are tested against a common control group, but patients can enter and exit the trial based on efficacy and futility rules under a Bayesian framework. A 2019 systematic review identified 49 basket trials, 18 umbrella trials, and 16 platform trials.1 The majority of the basket (96%) and umbrella (89%) trials were exploratory (e.g., phase I or II) while approximately half (47%) of platform trials were phase III design. Most of these trials were conducted in the field of oncology (92%) and were ongoing (82%) at the time of the review. The number of therapeutics tested using these novel trial designs is expected to continue to increase rapidly over the coming years. Regulatory bodies, health technology assessment (HTA) agencies, and decision-makers are faced with numerous challenges when evaluating the evidence generated from these novel study designs.
To provide a forum to present broad perspectives and considerations when assessing the evidence generated by these new study designs from an HTA perspective, CADTH organized a series of 3 webinars in November and December 2020. The first webinar discussed the past, present, and future of economic evaluations of precision medicine at the Committee for Economic Analyses of the Canadian Cancer Trials Group.2 This paper summarizes the discussion held during our second webinar, which examined issues around HTA of evidence generated using these trial designs. In this workshop, several experts were invited to present on the clinical, health economic, patient value, and ethical aspects associated with these new study designs. The content of each presentation is summarized, starting with the clinical aspects. The discussion section of this paper summarizes the major points raised by questions from the audience and answers from the presenters. Our third paper presents the perspectives of patients, clinicians, public payers, private payers, and industry,3 which were discussed during the third webinar.
Clinical Methods and Outcomes for Health Technology Assessment in the Precision Medicine Era
Presenter: Dr. Timothy P. Hanna
There are several considerations for precision medicine clinical evidence generation given the master protocol designs, starting with the difficulty of effectively differentiating prognostic and predictive biomarkers when there is no comparison group. For example, if drug X is only given to patients with biomarker A and results in excellent treatment response, it remains unclear if patients with biomarker A would respond well to any treatment. It also remains unclear whether drug X would be effective for patients without biomarker A. With noncomparative designs it is also challenging to rigorously evaluate side effects because it may be unclear which symptoms are, and which are not, related to the drug.
Data for HTA is a consideration that is being addressed by the CanREValue initiative.4 The rapidly changing drug and treatment landscape requires timely data access, data sharing, data quality, data security, evidence relevance, and a functional learning health ecosystem, with appropriate ethical, legal, and social frameworks. The precision medicine evidence ecosystem exists along a continuum from drug pipeline through clinical trials (with associated subtypes and phenotypes), regulatory processes, and finally real-world data (RWD). Figure 1 illustrates this continuum and the interrelationships along the continuum.
Figure 1: The Precision Medicine Evidence Ecosystem
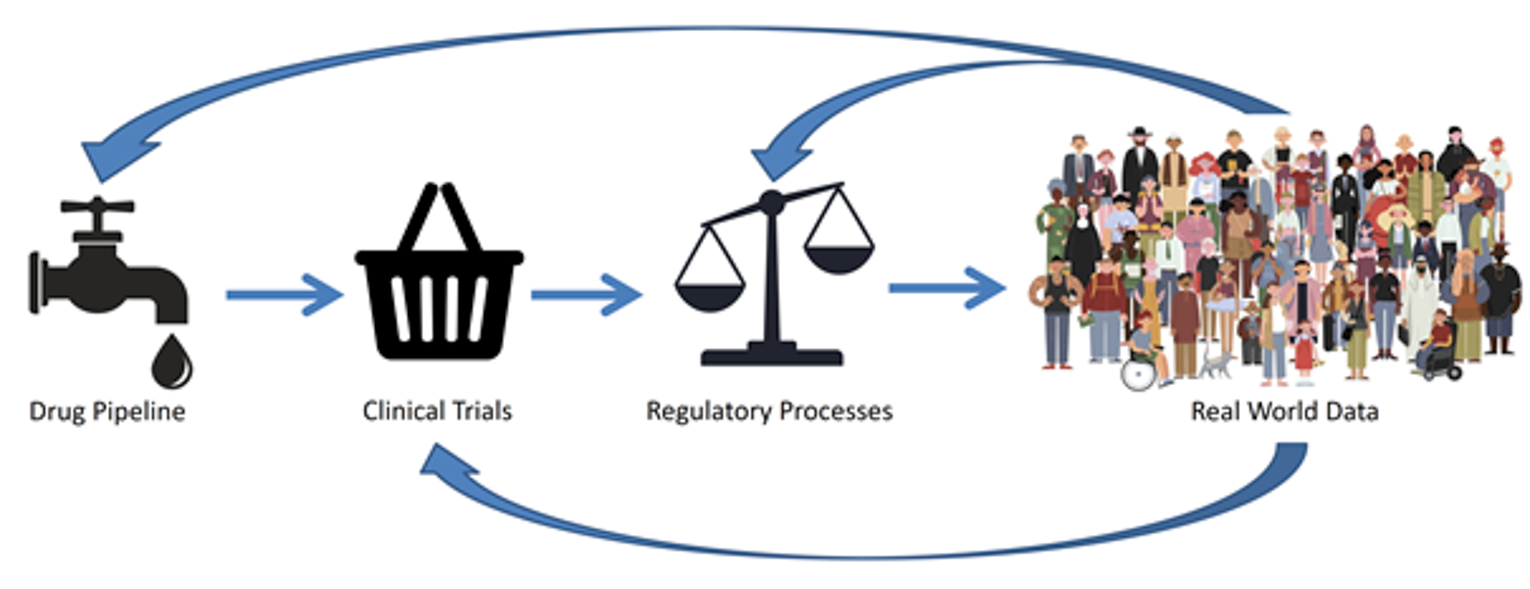
Note: This continuum illustrates the flow of information from the drug pipeline through clinical trials, regulatory processes, and ultimately to adoption into routine practice, where real-world data are generated. This real-world data can further inform the precision medicine evidence ecosystem; for example, informing re-evaluation of funding decisions, clinical trial design, and drug development.
Source: iStock/igorshi;smart;bagrovskam;vector_s.
There are many important outcomes in precision medicine, which can be broadly divided into health outcomes (e.g., overall survival, progression-free survival, quality-adjusted life-years [QALYs], time on treatment [TOT]) and non-health (e.g., timely access to information) or personal utility outcomes (e.g., knowledge of secondary findings).5 Common precision medicine questions relate to biomarker test performance characteristics, prognostic characteristics of the biomarker, and comparative effectiveness of treatment in patients with the biomarker.
Measuring outcomes for precision medicine is challenging for several reasons, including (1) small subgroups leading directly to imprecision in estimating benefit; (2) lack of a comparison group; (3) lack of clarity that a mutation itself is prognostic of observed outcomes (irrespective of the treatment) or that the treatment is uniquely effective in patients with the mutation; and (4) rapidly changing test and treatment landscape. RWD show promise in at least partly addressing these challenges. The population size available through administrative datasets is often large enough to ensure statistical power, even of small groups. There are also benefits to RWD in terms of causal inference and face validity, including:
data on untreated comparison groups
information of key health utilizations required to undertake real-world costing
RWD on patient and physician preferences in clinical practice.
Moreover, there are economic and procedural efficiencies, particularly for use of administrative data, including lower data collection costs6 compared to registries or trials, centralized data collection in the setting of universal health system, collection of key data elements (e.g., medical encounters, hospital encounters), and highly complete vital status information (i.e., mortality).
An assessment of RWD capture capability in Canada was carried out with a pilot study in collaboration with the Committee for Economic Analyses of the Canadian Cancer Trials Group to determine whether administrative data can improve the performance of cancer clinical trial economic analyses, specifically the CO.17 trial.7 Trial data from patients from Ontario was linked to the Ontario administrative data sources housed at ICES (formerly the Institute for Clinical Evaluative Sciences) where strong concordance was demonstrated between the 2 databases. Cost differences from a health ministry payer’s perspective were driven by aspects of utilization that could be collected with administrative data (e.g., drug costs, hospitalizations, and health care encounters). Additionally, there was longer follow-up time with more complete and potentially more accurate utilization information within the administrative data.7
However, there are several challenges with RWD. The most important relates to issues of confounding when RWD is used for estimating comparative effectiveness. Some data are not collected optimally; for example, socioeconomic status is typically an area-level measure. Performance status is rarely collected. In terms of costing, out of pocket costs, such as private-pay supportive care services or aspects of mental health services, are not covered publicly and therefore not collected routinely. In addition, quality of life is not collected, and non-health outcomes may be poorly represented. However, it can be possible, for example, to combine health utility data, such as EQ-5D questionnaires, collected longitudinally as part of trials or registries, with RWD at a later date for further analysis. In the same way, other variables not collected within RWD, such as performance status or recurrence, could be linked. Lastly, while there is the issue of accessing data in a reasonable, rapid time frame, RWD reflects reality and can be a rich source of data for modelling.
In addition, the gold standard for measuring antitumour activity is phase II clinical trials. It is not possible to directly measure activity with administrative data as it generally lacks response data. Possible useful proxies to direct measurements of antitumour activity include measuring TOT, difference in differences, or measuring TOT and looking at the ratio of TOT for a subsequent line of therapy (TOT2) to a preceding line of therapy (TOT1).8 The idea behind the use of this ratio (TOT2:TOT1) is that patients serve as their own control and that TOT or progression-free survival tend to be shorter on subsequent lines of therapy. If there is a sufficient threshold of patients where the TOT2:TOT1 ratio is beyond a certain number, then perhaps that can serve as a signal of treatment activity.9,10 However, these methods require further characterization and validation.
Recent computational advances such as machine learning can assist in some elements of precision medicine HTA. For example, with data cleaning and curation, natural language processing may be valuable for extracting information from patient charts. Causal inference requires an established outcome framework to understand the relationship between an exposure and an outcome, as well as to better map out what might be considered a mediator, a confounder, or an effect modifier. There are also machine learning methods that might be helpful to guide variable selection for matching.11,12
In summary, there is a crucial role for both clinical trial design and RWD in the precision medicine evidence ecosystem. RWD cannot replace properly conducted clinical trials but can provide complimentary information. When using RWD, causal inference must minimize the risk of bias; methods exist to do this but must be optimized for use in this context.
Health Economics Applied to Novel Therapies
Presenter: Dr. Lauren E. Cipriano
The goal of health economics is to identify the set of health technologies that will maximize population health given a constrained budget and health care infrastructure. Selecting technologies that provide poor value for money removes resources from the limited budget meaning other patients being cared for within the same system will have less access to technologies that would have provided them greater health. To ensure fairness across all individuals cared for within the health system, all interventions, including tumour agnostic strategies or other new precision medicine technologies, face the same health economic evaluation framework.
Technology Evaluation Must Be Comparative, and All Possible Alternatives Must Be Considered
In health economics, it is essential to identify the incremental costs and incremental benefits of a technology compared to its next best alternative, often the status quo. Comparative analysis is required because the choice is between the available technologies. Focused on a specific technology for a specific indication, the question health economics addresses is: What is the incremental cost that would need to be incurred to achieve the incremental benefits expected from using the new technology relative to current practice?
As is required in all other contexts, in the context of platform, basket, and umbrella trials, economic evaluation requires a separate analysis for each potential treatment and indication. This is, in part, because each indication has its own set of comparison treatment alternatives, each with their own prognosis, adverse event rates, and costs. As a result, even if the response rate, prognosis, cost, and adverse event rate of the new treatment is the same across all indications, each indication will have a unique incremental cost and incremental benefit when compared to the next best alternative for that indication.
This can be difficult in basket and umbrella trials without a comparator arm. The lack of a comparator arm leads to challenges in identifying meaningful treatment effect. Existing study populations may not have patient features in common with the population under study, most notably, the gene mutation or biomarker status. Without a comparison study arm or existing population with the same gene mutation or biomarker status, the treatment benefit cannot be reasonably separated from differences in prognosis attributable to the biomarker or gene itself.
Biostatistical Estimation of Progression-Free and Overall Survival Is Influenced by the Composition of the Heterogeneous Population
A core assumption of statistical survival models is that the sample is taken from a common population. This assumption ensures that all individuals in the study population remaining at risk at a specific point in time have the same probability of progression. A corollary of this assumption requires that the likelihood of event occurrence needs to be the same for participants enrolled both early and late in the study — that the study population cannot drift in features that may affect their survival trajectory such as age, disease severity, and prognosis. This assumption ensures that the estimated survival curve is representative of the population’s survival trajectory over the entire time frame estimated. Survival analysis on basket trials violates these core assumptions in numerous ways. The sample enrolled is heterogeneous in terms of primary cancer site, age, prognosis, number of prior lines of therapy, and time since previous treatment failure. Randomized controlled trials are able to overcome variation in time since previous treatment failure through the process of randomization, but single-armed trials are unable to overcome this limitation without stringent enrolment criteria. The remaining major sources of variation within the sample population — cancer site, age, and prognosis — cannot be overcome. There is too much heterogeneity in the study population when the sample population includes individuals with very poor survival prognosis (e.g., metastatic lung cancer or advanced glioma) and with better survival prognosis (e.g., thyroid cancer) as well as patients of different ages (e.g., adult patients with colon cancer in the same study as patients with pediatric soft tissue sarcoma or infantile fibrosarcoma). It is not sufficient that the entire population has 1 feature in common (e.g., a common tumour biomarker) when there are many other known sources of variation. At each point in time, the survival curve generated through this analysis can, at best, only represent the probability of survival over the next time interval of observation for the specific mixture sample population at risk at that point in time. This results in an analysis that is unable to be generalized into a continuous survival curve of any interpretability. It is also worth noting that the evaluation of hypothesis tests, including determining whether the median survival or survival to a specific time point is different than a pre-set clinically meaningful threshold, cannot also not be legitimately performed on such a heterogeneous population.
Survival analysis can be stratified by major sources of heterogeneity, such as tumour site, patient age, and number of lines of prior therapy. However, the very small sample sizes of individual cancer sites in basket trials published to date would result in highly uncertain survival trajectories, insufficient sample sizes to generate reliable confidence intervals, and inability to perform hypothesis tests, especially because of the lack of a randomized comparator arm. The remaining uncertainty makes meaningful inferences about treatment effectiveness difficult because bootstrapped confidence intervals around the estimated survival curves can include nearly the entire range of possible outcomes.
Modelling the Disease Progression as Well as Health and Economic Consequences Requires the Population to be Divided Into Homogeneous Health States
Mathematical models of disease progression generally divide the stages of disease progression into discrete health states. The definition of these health states needs to represent a homogeneous subpopulation in terms of the probability of departing the state, the cost of being in the state, and the health reward, often measured in QALYs, of being in the state. It has long been known that ignoring heterogeneity in the composition of a modelled health state’s population can lead to over- or underestimation of the cost-effectiveness of treatments.13
Specifically, if a health state, such as “progression-free,” comprises a heterogeneous population, then the definition of the state, the probability of leaving the state, and the costs and QALYs for being in the state all need to change over time. For example, if a “progression-free” health state is initially comprised of 50% patients with metastatic lung cancer and 50% patients with advanced thyroid cancer, because the patients with lung cancer are more likely to progress sooner, an analyst must keep track of the ratio of each patient type to correctly calculate the overall probability of state departure, weighted average cost, and weighted average quality of life at each time step in the model analysis. Similarly, differential rates of state departure influence the definition of subsequent states. Continuing with the example, initially the “progressed” health state will disproportionately represent patients with lung cancer, but as time goes on and the patients with lung cancer progress into the “dead” state, the “progressed” state will disproportionately represent patients with thyroid cancer. When upwards of 12 to 20 tumour types are being modelled simultaneously, keeping track of the precise composition of each health state at each time step is mathematically cumbersome, but necessary for ensuring an unbiased calculation of the expected costs and QALYs in each health state.
It is mathematically equivalent, but also more transparent, operationally simpler, and consistent with all best-practice guidelines related to the structure of mathematical models of disease to divide the population into subpopulations that are more homogeneous in state definition. As patients across subpopulations are not interacting and changing each other’s progression, as would occur in an infectious disease model or a queuing model, each subpopulation can be modelled separately and the results of the different subgroups can be combined post hoc, if combination is desired.
Decisions Regarding Technology Adoption Should Be Made for Individual Technology-Indication Pairs
Many health technologies are potential treatments for numerous clinical indications, but the decision about whether that technology is good value for money in a system with limited resources is specific to each clinical indication being considered. Generally, each of these decisions can be made independently of each other, with 2 main exceptions. First, when the subpopulations for a clinical indication cannot be treated differently for ethical reasons. Second, when a large capital infrastructure investment is required to acquire the technology and use across multiple indications is necessary for efficient use of the technology.
There are numerous examples of subpopulations that, generally, cannot or should not be provided differential access to health technologies even though the technology may not have the same effectiveness or cost-effectiveness in all subpopulations. This is particularly relevant when the reason a technology is less effective or cost-effective in 1 population is a structural marginalization of that subpopulation. However, in many cases, it is important and meaningful to know the effectiveness and cost-effectiveness in each subpopulation because it enables targeting of preventive and clinical care to populations that are historically marginalized that may not be cost-effective when made universally available.
When it is desirable to calculate an overall weighted average incremental cost-effectiveness ratio (ICER) across subpopulations or across indications, doing so can be performed after the fact. An overall cost-effectiveness ratio will be highly sensitive to the distribution over indications. Calculation of a weighted average ICER should use weights calculated using the relative size of each subpopulation or the incidence of each indication, representing the most likely distribution of use if approved. Combining economic evaluations using the relative number of patients in a clinical trial is generally not informative unless it represents the true relative incidence of each subpopulation or across each indication, which is unlikely given the challenges of recruiting clinical trial patients who vary across social and demographic characteristics as well as indications. Weighted averages of costs and benefits individually can be calculated to inform an overall weighted average ICER. ICERs themselves should not be averaged across subpopulations or indications as it is mathematically incorrect to do so.
Using the average ICER to inform decision-making should be done with extreme caution. The average ICER may be below the willingness-to-pay threshold, which may lead to adoption of the technology for some indications in which it is not effective or not cost-effective. Adoption for those indications will lead to an overall reduction in population health as resources are removed from other patients receiving other types of care to provide this treatment to these patients. Conversely, the average ICER may be above the willingness-to-pay threshold, which will prevent access for all indications, including those for which the new technology is effective and cost-effective. Again, this will result in lower population health than what is possible if the technology could be made available to the indications in which it is cost-effective. Any time the bundling of indications is considered, it is at the cost of economic efficiency in our health care system and that cost is generally born by decreasing the health investment into other patients, specifically members of communities that are historically marginalized, where underinvestment of health care resources is already substantial.
To illustrate this point, Figure 2 outlines the implications of adding a new technology to the current status quo treatment paradigm for a variety of indications, denoted as indications A, B, C, D, and E. In this example, introducing the new technology for just indication B (green text), results in an incremental cost of $20,000 per QALY gained compared to the status quo for all indications. Then, with the addition of the new technology for additional indications (in order or D, C, A, and then E), each subsequent ICER relates to the incremental costs and incremental benefits of adding that indication to the list of indications for which the technology is selected over the status quo for the indication. The black line connecting alternatives indicates the efficiency frontier. If there were an indication for which the technology was less effective than the comparator for that technology, including that indication in any bundle of interventions would be dominated and would not appear on the efficient frontier.
If a decision must be made to reimburse this technology for all or no indications, the dashed blue line is the resulting overall incremental cost of $500,000 per QALY gained. In this example, the technology would be deemed not cost-effective in the Canadian context, resulting in no reimbursement for any indication. However, it would be both health improving and a cost-effective policy in a Canadian context to provide reimbursement for indications B, C, and D because the ICERs for those indications are below the commonly cited willingness-to-pay threshold of $50,000 per QALY gained. In this case, as is typical, the costs and effectiveness of treatment for 1 group is fully independent of the other groups; therefore, the optimal reimbursement decision can be made by evaluating the cost-effectiveness of each treatment-indication pair separately. This is the typical case and is illustrated through numerous real-world examples where covered indications expanded with the development of indication-specific evidence (e.g., bevacizumab).
There are some technologies that require a substantial fixed cost for capital equipment, such as a proton beam synchrotron. While it may not be cost-effective to build the synchrotron for any 1 particular cancer type, it may be cost-effective if it were to be used for 10 to 15 indications. In this case, the costs and benefits of using the treatment in multiple indications is not independent because the marginal cost of the technology depends on its overall utilization rate. As a result, the evaluation of all potential combinations of interventions must be considered simultaneously to identify if there is a combination of indications together that warrant the capital investment.
Figure 2: Incremental Cost and Incremental QALY for the Addition of a New Technology, Successively, for Each Possible Indication Compared to the Status Quo Technology for That Indication
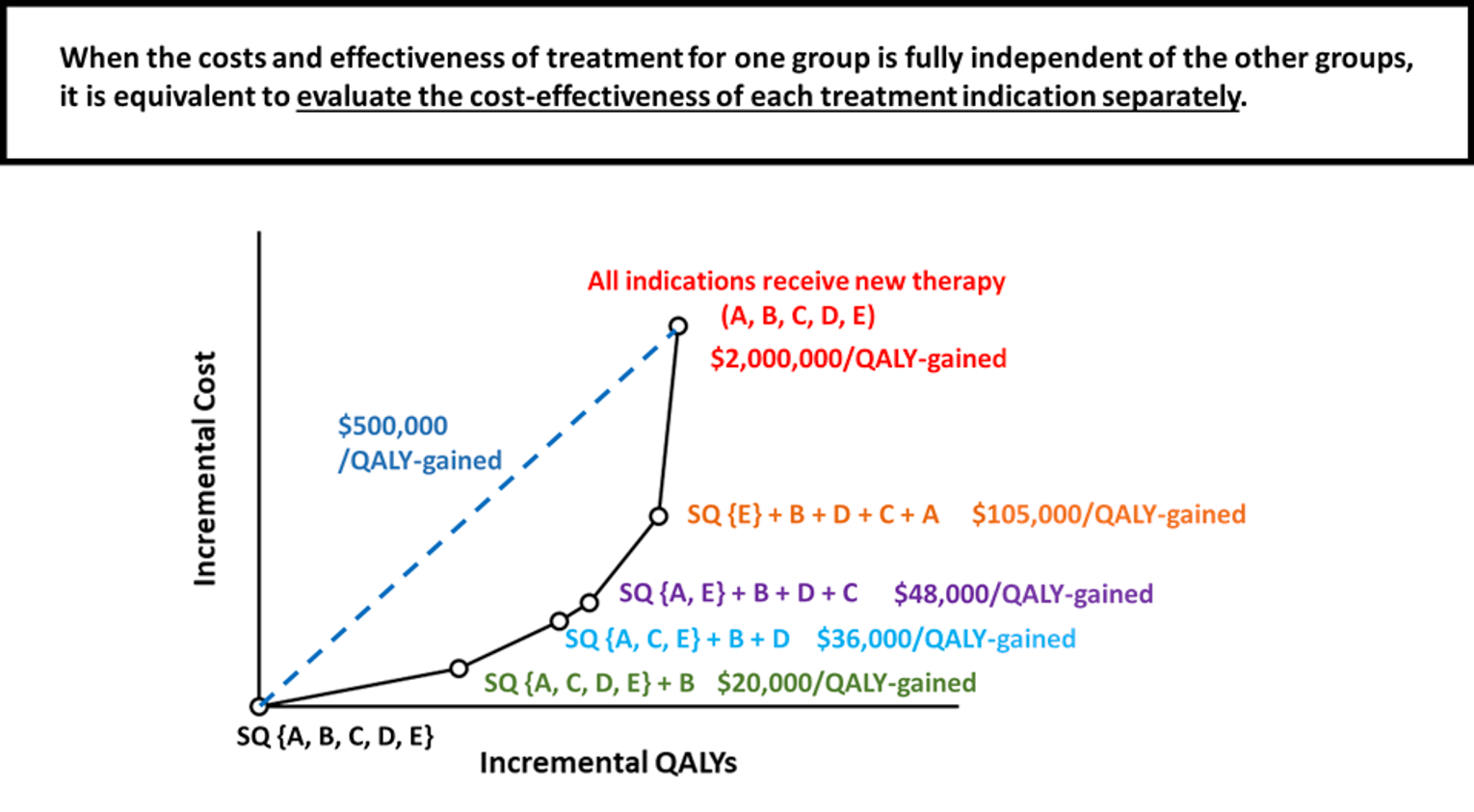
QALY = quality-adjusted life-year; SQ = status quo.
In the example presented in Figure 3, the fixed cost of the capital equipment results in an overwhelming marginal cost for a single indication. However, reimbursement for indications W, X, and Y is cost-effective. It is notable that this exception only exists when there is also a decision about whether to embark on the large capital investment. Once the decision has been made to make this investment, any subsequent health economic studies of whether to expand the use of the technology to more indications can be made independently. This is the case for technologies Z and V, for which the incremental cost per QALY gained on the efficient frontier is consistent regardless of whether the investment decision is considered independently or within a combined analysis.
In summary, as basket and umbrella trials increasingly become a source of clinical evidence, it is important to recall that the framework with which health economic analysis is carried out will not change. Each indication-technology pair requires its own effectiveness evidence, which necessitates reliable survival analysis, and each indication-technology pair has its own comparator technologies with its own prognosis, costs, and quality of life implications, which must be considered when evaluating whether or not a new technology offers incremental benefits compared to existing options. Accurate and unbiased mathematical modelling of disease processes requires division of the population into homogeneously defined health states that require fully stratified health economic analysis. Finally, decisions for each indication-technology pair need to be made independently to ensure that all uses of a technology contribute to maximizing population health. When the value of a technology is averaged over a wide variety of uses, such as a range of cancer tumour sites, there is a substantial risk that the technology is not made available to any populations because of the limited or highly uncertain evidence of efficacy across indications. Bundling indications in this manner imposes a harm to patients in whom the technology is effective and cost-effective. If, alternatively, it is funded for all indications, including those cancer sites for which it has not been shown to be effective or cost-effective, this decision takes valuable health-producing resources from other patients in a shared health care system.
Figure 3: Incremental Cost and Incremental QALY for the Addition of a New Technology, Successively, for Each Possible Indication Compared to the Status Quo Technology for That Indication When There Is a Large Capital Cost of Equipment
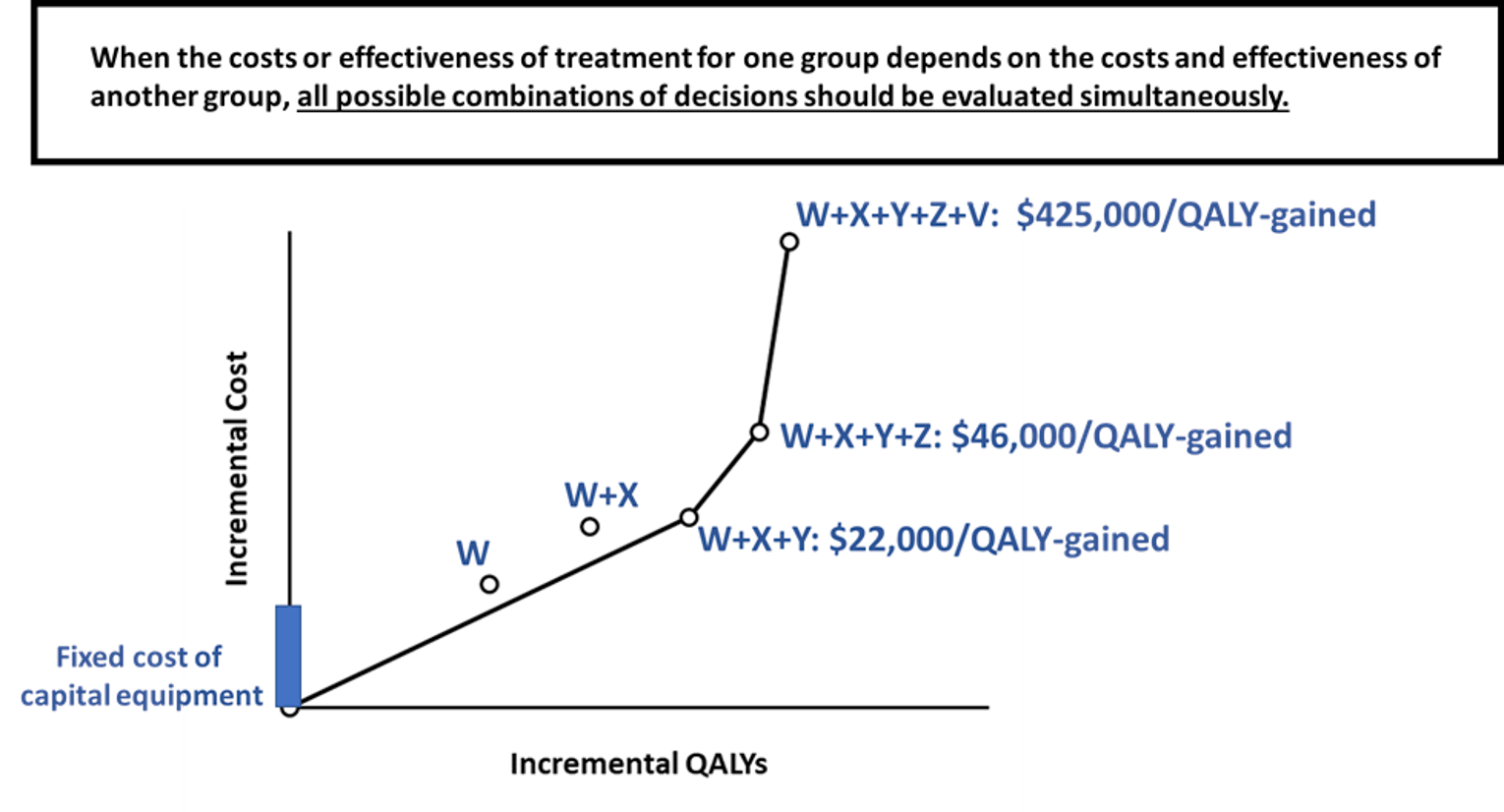
QALY = quality-adjusted life-year.
Patient-Centric Value
Presenter: Dr. Dean A. Regier
The objective of HTA deliberation at CADTH is to maximize health status, subject to budget constraint, from the perspective of a publicly funded health care system. To carry this out, trade-offs between costs and QALYs are examined and expressed as the ICER. The ICER is only 1 aspect of the deliberative framework used by the pan-Canadian Oncology Drug Review (pCODR) Expert Review Committee (pERC) or the Health Technology Expert Review Panel (HTERP) to assess drug and non-drug technologies, respectively. Patient values or preferences are used as part of both the pERC and HTERP deliberative frameworks. However, they are not captured in a way that is methodologically oriented. The quantitative or qualitative expression of value (i.e., preference-based utility) is lost due to lack of methodological rigour, which creates a missed opportunity to accurately characterize and report on patient values and patient preferences related to what it means to receive valued health care.
Recent research has been published to highlight a methodologically rigorous way to capture patient preferences in precision medicine through the elicitation of stated preferences for information on expected health and non-health outcomes.14 Estimates of demand and willingness to pay for information were examined both in the real world and through a discrete choice experiment, with external validity examined for chemotherapy treatment in estrogen receptor–positive and human epidermal growth factor receptor 2–negative breast cancer from 2005 to 2014. In the discrete choice experiment, a series of choice questions presenting 3 alternatives were given and participants were asked to make trade-offs between not only quality of life or potential health outcomes, but also the probability of having the genetic marker. Questions posed to participants are presented in Figure 4.14
Figure 4: Discrete Choice Experiment Question for Patients Undergoing Precision Medicine Interventions
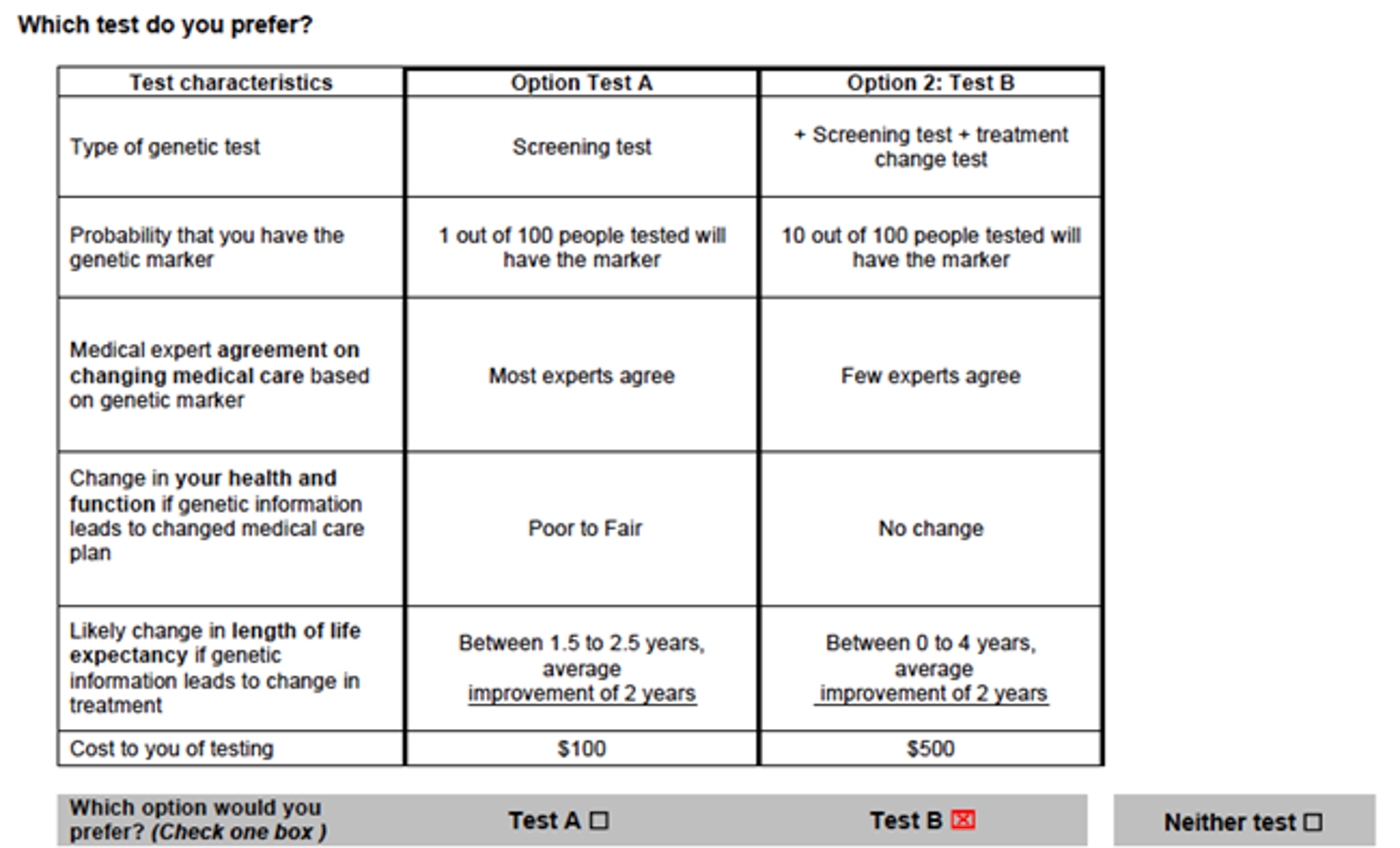
Note: Each choice task differed on 6 attributes. Patients were asked to choose between 2 scenarios for receiving precision medicine information. A neither alternative allowed for the possibility the respondents did not want to receive any information obtained from a precision medicine test.
Source: Reprinted by permission from Springer Nature: PharmacoEconomics. Demand precision medicine: a discrete-choice experiment and external validation study, Regier et al., 2020.14
The study revealed that, for patients, the most important aspects of precision medicine testing were survival gains with statistical uncertainty, cost of testing, and medical expert agreement on changing care based on test results. The value of a precision medicine test where most experts agree to treatment change based on the genomic test results compared to few was $1,100, reflecting a strong preference for consensus among experts around treatment change. Patients also demonstrated a willingness to pay for reduced uncertainty in health outcomes. For example, the survey estimated that a patient would pay an additional $265 for a test that could result in greater certainty around life expectancy gains.
The predicted demand (or uptake) of the 21-gene recurrence score assay for guiding adjuvant chemotherapy in women with estrogen receptor–positive and human epidermal growth factor receptor 2–negative breast cancer over a 9-year time horizon is in Figure 5. These estimates are compared to RWD (uptake) in the US over the same period. A strong relationship between the predictions and real-world estimates was found, with correlation coefficients of greater than 0.90 and a root-mean-squared error of 0.11. Predicted and real-world demand coincided when the evidence base was underdeveloped and uncertain (2005 to 2006) and also when uncertainty was eventually addressed through randomized controlled trials and subsequent increasing medical expert agreement (2012 to 2014).
Figure 5: Demand for the 21-Gene Recurrence Score Assay 2005 to 2014
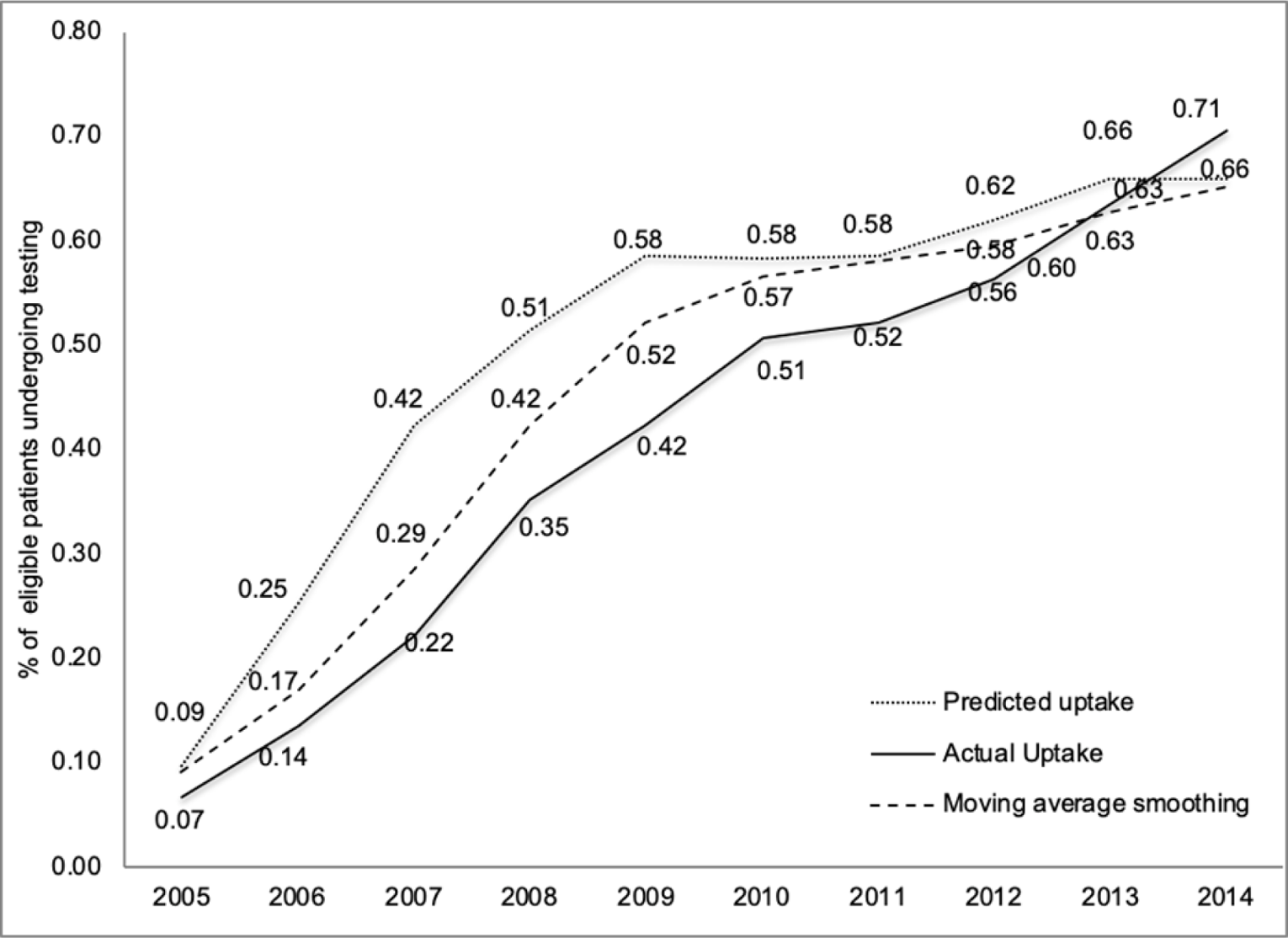
Note: Presented is the actual uptake (percentage of the eligible population) who underwent testing over the time period. Predicted uptake was derived from the discrete choice experiment. The moving average smoothing estimates were estimated by taking an average of the nearby uptake estimates (2 lagged values and the current value) and thus accounted for a lag in demand as the evidence base evolved.
Source: Reprinted by permission from Springer Nature: PharmacoEconomics. Demand precision medicine: a discrete-choice experiment and external validation study, Regier et al., 2020.14
In summary, deliberative frameworks require incorporating patients’ values and preferences; however, improvements could be made when applying and implementing the science of understanding patients’ preferences. There is a large amount of literature related to understanding patient preferences, qualitatively and quantitatively, and the literature converges on the idea that respondents can make complex decisions and trade-offs despite our skepticism of this.
Ethical and Social Issues
Presenter: Dr. Spencer Hey
Although master protocol frameworks are undoubtedly powerful and innovative, they raise some new ethical questions that require careful analysis. However, the goal of an ethical analysis is to illuminate the advantages, uncertainties, and challenges that stem from the specifics of these new designs and protocols, rather than classifying designs as more or less ethical. This paper will focus the ethical analysis around the so-called “big 3” principles15 of autonomy, beneficence, and justice (as illustrated in Figure 6).
In clinical trials, the principle of autonomy is most commonly operationalized through the practice of informed consent, whereby participants must understand the risks and burdens of the study and freely consent to taking part in research. However, patient engagement — the idea that participants have a right to be treated as equal research partners and should have input into the study’s design — is another, complementary way of operationalizing autonomy.
To satisfy the principle of beneficence, a study should be designed to maximize its validity and reliability of inferences to effectiveness in the real-world setting. Equally important is the idea of optimizing value, where research questions should be socially important, transformative to practice, and include appropriate populations and comparators. Trial costs and burdens on research participants should also be minimized, in keeping with sound scientific design.
The principle of justice includes the idea that the populations that participate in research should be able to benefit from knowledge gained. Recruitment should also aim to include a representative population. Indeed, a long-recognized problem with trials is that populations most likely to benefit from the intervention are not the same as those that are recruited and participate in trials. To ensure the equitable distribution of benefits, dissemination and implementation plans must be designed to reach diverse settings and populations, including those in both low- and high-resource settings. Lastly, to uphold scientific integrity and support future research initiatives, all research practices and communications about research should strive to safeguard public trust in the research enterprise.
It should be noted that these 3 ethical principles, and their implications for ethical trial design and conduct, will intersect and overlap. Indeed, part of the challenge in ethical analysis is navigating tensions between the principles (e.g., where considerations of justice might need to be traded off against beneficence, or vice versa). So let us examine how we might balance the application of these principles in the context of master protocol frameworks.
Beginning with basket and umbrella trials, the potential scientific and social value is high for these trials because they are intended to illuminate our biologic understanding of the disease. In basket trials, for example, diseases initially presumed to be discrete and distinct may be treated with the same molecular target — and this may reveal that a disease is better characterized by its molecular target, rather than its anatomic site.
Basket and umbrella designs may also reduce trial costs and burdens on participants by minimizing patient exposure to ineffective, toxic interventions. However, due to the importance of biomarker testing in classifying participants into different treatment subgroups, the validity of the research should be considered more uncertain in circumstances where the reliability or accuracy of a test is unclear.16
Justice issues may also arise for recruitment into basket trials, if, for example, patients without the relevant biomarkers are excluded. In situations where alternative treatments for patients without a specific biomarker are poor, there may be value in allowing enrolment in targeted therapy studies even if they do not possess the specific target as it may provide information about the utility of the biomarker test as well as if the therapy works despite the absence of the biomarker. However, this must be weighed against the risk and possible harm caused by exposing a participant to an ineffective therapy.17
The biggest ethical challenges regarding basket and umbrella designs relate to informed consent and the need to safeguard scientific integrity. Given the uncertainty in the relationships between biomarkers and treatments, there is the potential for harm to patient autonomy and public trust if that uncertainty is not made explicit in communications with the potential participants or the public more broadly.
Turning now to platform trials, these trial designs can offer large potential gains from a cost and burden perspective, as the research infrastructure that incorporates adding and dropping interventions during the trial reduces the cost per trial hypothesis. However, because of design complexity, there are new challenges related to informed consent, such as communicating a participant’s pathway through the study as treatment arms are added or dropped, as well as reconsenting and management of participant requests to enrol in a newly added treatment arm. Given the larger upfront costs with setting up a platform trial, there is the possibility of unintentionally selecting for higher resource settings and limiting equitable participant recruitment. From a validity perspective, it is also unclear how platform trial results can be folded into meta-analyses and contribute to the broader knowledge ecosystem given the dynamic nature of evidence evolving and the addition of intervention arms over time.
Finally, informed consent is often more challenging with elaborate designs, and demands thoughtful communication approaches to ensure a valid, truly informed consent process. While master protocol frameworks are an exciting frontier for clinical trials, ethical analysis should focus on the advantages, uncertainties, and challenges that stem from the specifics of designs and protocols.
Discussion
In precision medicine in oncology, at market entry there is limited evidence on outcomes, effectiveness and safety, and cost-effectiveness, yet expert agreement may be adopted into clinical practice by some clinicians. This creates tension among clinicians regarding approaches to care based on available and limited evidence. With large uncertainties in clinical outcomes, there is also lower confidence in the economic value associated with these technologies at the time of their submission. A suggested approach to deal with the uncertainty challenge in the context of basket trials is to conduct a basket trial with a large number of indications as part of a phase II trial, and subsequently narrow down this list to a few indications based on where the precision medicine has demonstrated the greatest treatment activity and where evaluations can then be conducted against comparator technologies to measure the magnitude of benefit and enable more accurate economic analysis. The incremental effectiveness is more easily observed when there is a strong clinical evidence base for the technology within the smaller list of indications where the greatest benefit is observed. Since this webinar, CADTH published (in June 2021) Guidance for Economic Evaluations of Tumour-Agnostic Products.18
Patient voices and preferences in design and outcomes of these novel trials should not be forgotten while at the same time there is a need for improvement on the systematic evaluation of quantitative and qualitative patient outcomes. Personalized medicine trials are not without their ethical concerns, most notably related to informed consent. Reducing the complexity of informed consent with conventional trials has been debated for some time and novel study designs add further complexity to this issue. However, there is opportunity to ensure patient awareness and understanding in a meaningful and credible way. For example, Genome Canada has introduced the concept “talking consent” when engaging those from Indigenous communities in gene profiling work, which has been effective to explain the process and requirements of consent.
Reimbursement with limited access, combined with a real-world evidence (RWE) generation plan to build on the evidence base may be a suitable mechanism to decrease the uncertainty associated with the evidence at product launch. Such managed access agreements that incorporate RWE may eventually contribute to the evidence available at market entry, further inform clinical trials, and ultimately inform clinical practice guidelines. From a process perspective in Canada, standards regarding data elements and the type of data that would be important for RWE generation are in development through the CanREValue initiative. These standards will ultimately be useful for individual jurisdictions should they develop resource capacity and data infrastructure. It is also critical to take advantage of RWE to measure patient-related outcomes (e.g., quality of life) and where possible to conduct economic evaluations in real conditions.
The precision medicine ecosystem requires consideration of the role of optimal clinical trial design and the best use of RWE to support HTA decision-making. New technologies will bring a level of evidence uncertainty; however, transparency around that uncertainty is necessary throughout the life cycle of evidence generation. The decision to invest in a particular technology results in an opportunity cost, and in the context of the publicly funded Canadian health care system, that opportunity cost is a reality.
References
1.Park JJH, Siden E, Zoratti MJ, et al. Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials. 2019;20(1):572. PubMed
2.Chan KKW, Cheung MC, Regier DA, et al. The past, present, and future of economic evaluations of precision medicine at the Committee for Economic Analyses of the Canadian Cancer Trials Group. Curr Oncol. 2021;28(5):3649-3658. PubMed
3.Mittmann N, Chan KKW, Cheung MC, et al. Platform, basket and umbrella trail designs: stakeholder perspectives of novel therapeutics [in press]. Can J Health Technol. 2022;2.
4.Chan K, Nam S, Evans B, et al. Developing a framework to incorporate real-world evidence in cancer drug funding decisions: the Canadian Real-world Evidence for Value of Cancer Drugs (CanREValue) collaboration. BMJ Open. 2020;10(1):e032884. PubMed
5.Regier DA, Weymann D, Buchanan J, Marshall DA, Wordsworth S. Valuation of health and nonhealth outcomes from next-generation sequencing: approaches, challenges, and solutions. Value Health. 2018;21(9):1043-1047. PubMed
6.Williams JG, Cheung WY, Cohen DR, Hutchings HA, Longo MF, Russell IT. Can randomised trials rely on existing electronic data? A feasibility study to explore the value of routine data in health technology assessment. Health Technol Assess. 2003;7(26):iii, v-x, 1-117.
7.Hanna TP, Nguyen P, Pater J, et al. Can administrative data improve the performance of cancer clinical trial economic analyses? J Oncol Pract. 2019;15(9):e807-e824. PubMed
8.Weymann D, Pollard S, Chan B, et al. Clinical and cost outcomes following genomics-informed treatment for advanced cancers. Cancer Med. 2021;10(15):5131-5140. PubMed
9.Buyse M, Quinaux E, Hendlisz A, Golfinopoulos V, Tournigand C, Mick R. Progression-free survival ratio as end point for phase II trials in advanced solid tumors. J Clin Oncol. 2011;29(15):e451-452; author reply e453. PubMed
10.Von Hoff DD, Stephenson JJ, Jr., Rosen P, et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28(33):4877-4883. PubMed
11.Drysdale E, Peng Y, Nguyen P, Baetz T, Hanna TP. A population-based study of the treatment effect of first-line ipilimumab for metastatic or unresectable melanoma. Melanoma Res. 2019;29(6):635-642. PubMed
12.Weymann D, Laskin J, Jones SJM, et al. Matching methods in precision oncology: An introduction and illustrative example. Mol Genet Genomic Med. 2021;9(1):e1554. PubMed
13.Zaric GS. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-396. PubMed
14.Regier DA, Veenstra DL, Basu A, Carlson JJ. Demand for precision medicine: a discrete-choice experiment and external validation study. PharmacoEconomics. 2020;38(1):57-68. PubMed
15.Office for Human Research Protections (OHRP). The Belmont report: ethical principles and guidelines for the protection of human subjects of research. Washington (DC): U.S. Department of Health and Human Services; 1979: https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html. Accessed 2022 Apr 28.
16.Hey SP, Weijer C, Taljaard M, Kesselheim AS. Research ethics for emerging trial designs: does equipoise need to adapt? BMJ. 2018;360:k226. PubMed
17.Hey SP, Gerlach CV, Dunlap G, Prasad V, Kesselheim AS. The evidence landscape in precision medicine. Sci Transl Med. 2020;12(540). PubMed
18.Haines A, LaPlante S, Lee K. Guidance for economic evaluations of tumour-agnostic products. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/pdf/mh0016-cadth-economic-guidance-for-tumor-agnostic-products-rev-june22.pdf. Accessed 2022 Apr 28.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up to date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third-party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.
Questions or requests for information about this report can be directed to Requests@CADTH.ca.