CADTH Health Technology Review
Rituximab for the Treatment of Primary Membranous Nephropathy
PROSPERO Registration Number: CRD42020182641
Health Technology Assessment
Authors: Saadul Islam, Ghayath Janoudi, Matthew Bryan, Christine Perras, Mohammed Jabr, Alex Haines, Nazila Assasi, Julie Boucher, Deba Hafizi, Phil Lafleur, Cody Black, Tessa Cornelissen,, Caitlyn Ford
Authorship
Project Development
Christine Perras contributed to the conceptualization of the project, the development of the topic brief and the scoping plan, and the writing of the protocol and sections of the draft reports. She ensured that all comments received during reviews were addressed. She liaised with external contractors, drug plan members, and clinical experts. She was instrumental in establishing and managing the implementation advice panel and in finalizing the final implementation advice report.
Clinical Review
Saadul Islam contributed to the conceptualization of the study, development of the protocol, collection of data, study quality assessment, statistical analysis, and interpretation of the results. He also wrote sections of the first draft of the report.
Ghayath Janoudi contributed to the protocol development, collection of data, statistical analysis, and interpretation of the results, and revised the report for important intellectual content.
Matthew Bryan contributed to the protocol development, statistical analysis, and interpretation of the results, and revised the report for important intellectual content. He provided oversight and guidance for conducting the statistical analysis and wrote key portions of the methods and results sections related to the network meta-analysis.
Mohammed Jabr was involved in the early part of the research. As project owner, he contributed to the protocol development, and oversaw data extraction, study quality assessment, and analysis of the data. He also led the research team meetings.
Nazila Assasi was project owner for the later part of the research. She reviewed drafts, provided project oversight, managed the implementation advice panel, and revised the report for important intellectual content.
Deba Hafizi contributed to the report’s editing, data auditing, and data updates.
Economic Review
Alex Haines oversaw the health economic protocol development and assessment of the feasibility of an economic evaluation, and provided oversight on the creation of economic tools to help inform decision-makers.
Cody Black contributed to the development of the health economic protocol.
Tessa Cornelissen developed economic tools to help inform decision-makers.
Patient Perspectives Review
Julie Boucher contributed to this review by gathering patient observations and experiences shared on online sources and drafting the summary of patient perspectives.
Implementation Advice Review
Philip Lafleur summarized the implementation advice panel input, provided assistance at the panel meeting, and finalized the panel advice.
Research Information Science
Caitlyn Ford contributed to this review by designing the database search strategies for all sections of the report, executing the search strategies, completing grey literature searches, maintaining search alerts, preparing the search methods and appendix, and providing final approval to the version of the report submitted for publication.
Acknowledgements
CADTH would like to acknowledge the following individuals for their contributions:
Clinical Expert Review
Dr. Marcello Tonelli provided clinical expertise throughout the course of the project. He provided comments on an earlier draft of the clinical report.
Jurisdictional Representation
Brent Ruddock contributed to the conceptualization of the project and reviewed the scoping plan, the protocol, and a draft report.
Implementation Advice Panel Members
Chair
Marcello Tonelli, MD, SM, MSc
Alberta Kidney Disease Network
University of Calgary
Calgary, AB
Members
Penelope Poyah, MD, FRCPC
Nephrologist and Associate Professor, Department of Medicine, Dalhousie University
Division of Nephrology, Nova Scotia Health Authority
Halifax, NS
Heather N. Reich, MD, CM, PhD, FRCPC
Gabor Zellerman Chair in Nephrology Research
Nephrologist, Clinician Scientist, University Health Network
Associate Professor, University of Toronto
Toronto, ON
Emily Reynen, MD, CM, PharmD, FRCPC
Department of Critical Care Medicine, Queen's University
Kingston, ON
Conflicts of Interest
Marcello Tonelli disclosed the following:
A speaking engagement for AstraZeneca that was not related to any technology or drug (“The future of nephrology”), 2021.
A research funding or grant for Daichi Sankyo that was not related to any technology or drug (“patient-centered kidney care”), 2017. Funds paid to institution.
Heather Reich disclosed the following:
Travel Funding or Payment
International society of nephrology guideline development for treatment of glomerulonephritis (GN), 2019 (International Society of Nephrology-Kidney Disease: Improving Global Outcomes meeting).
Speaking Engagements
Consultation: Complement inhibition in immunoglobulin A nephropathy (IgAN), 2021 (Novartis).
Invited lecture: New Kidney Disease: Improving Global Outcomes guidelines for treatment of membranous nephropathy, 2020 (Asian Pacific Congress in Nephrology 2020).
Educational Lectures
Consultation: Complement inhibition in nephropathy, 2021 (Novartis).
Lecture: Annual GN Continuing Medical Education conference, 2021 (University of Pennsylvania).
Lecture: IgAN annual Continuing Medical Education course, 2016 to present (American Society of Nephrology).
Lecture: IgAN annual meeting, 2021 (Saudi Society of Nephrology and Transplantation).
Lecture: IgAN annual meeting, 2021 (Pakistani Society of Nephrology).
Other
Calliditas, Omeros: IgAN: Personal fee for academic leadership committee advisory, National Coordinating Investigator, presentation, less than 5000$ each for 2020 to 2022. Clinical trial study site (no personal compensation) accounts for the remainder of the balance.
Chinook: National coordinating investigator role.
Alnylam: Clinical trial site sub-investigator, no personal fee.
Pfizer: Clinical trial site investigator. No personal compensation. One advisory meeting.
Louise Fast Foundation: Funder of the Toronto General Hospital GN fellowship. No personal compensation.
Payment as Advisor or Consultant
Nefecon: IgAN, 2018 to present (Calliditas).
Narsoplimab: IgAN, 2018 to present (Omeros).
Atrasentan: Align study in IgAN, 2021 to present (Chinook).
Consultation: Complement inhibition in IgAN, 2021 (Novartis).
ROBO study in focal segmental glomerulosclerosis: 2019 to present (Pfizer).
Sparsentan in IgAN: academic advisory, 2021 (Retrophin).
Research Funding or Grants
Nefecon: IgAN, 2018 to present (Calliditas).
Narsoplimab: IgAN, 2018 to present (Omeros).
Payment for Academic Appointments (Endowed Chairs)
University of Toronto, 2016 to present: Gabor Zellerman Chair in Nephrology Research.
No other authors declared conflicts of interest.
Abbreviations
AE
adverse event
BSC
best supportive care
CI
confidence interval
CR
complete remission
CTX
cyclophosphamide
CYC
cyclosporine
eGFR
estimated glomerular filtration rate
ESRD
end-stage renal disease
GRIPP2 SF
Guidance for Reporting Involvement of Patients and the Public 2 Short Form
HRQoL
health-related quality of life
HTA
health technology assessment
IMN
idiopathic membranous nephropathy
ITT
intention to treat
KDIGO
Kidney Disease: Improving Global Outcomes
KDQOL-SF
Kidney Disease and Quality of Life-Short Form
MN
membranous nephropathy
NR
no remission
PMN
primary membranous nephropathy
NMA
network meta-analysis
NS
nephrotic syndrome
NSAID
non-steroidal anti-inflammatory drug
OR
odds ratio
PLA2R
phospholipase A2 receptor
PR
partial remission
PRISMA
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
RCT
randomized controlled trial
SR
systematic review
TAC
tacrolimus
TR
total remission
Protocol Amendments
Section | Amendment | Page | Rationale |
|---|---|---|---|
Policy questions | Revisions to the second policy question. Amended policy questions were determined by the jurisdictional clients. 1. Based on the available evidence, what is the place in therapy of rituximab to treat primary membranous nephropathy? 2. What specific characteristics should patients possess or satisfy for access to rituximab? 3. What is the most appropriate rituximab dosing regimen? 4. What objective measures or outcomes, and at what time points, should be used to determine treatment success or failure in primary membranous nephropathy? 5. Are there special populations that require other considerations? | Page 7 in Protocol | An Implementation Advice Panel was convened to assist the jurisdictional clients in determining policy options for rituximab in primary membranous nephropathy. |
Key Messages
Membranous nephropathy is an autoimmune disease and one of the most common causes of nephrotic syndrome in adults. The incidence of membranous nephropathy is 1.2 in 100,000 persons per year worldwide. Approximately 80% of patients with membranous nephropathy have a classification of primary (or idiopathic) membranous nephropathy. The treatment goal of patients with primary membranous nephropathy is to induce remission. Current treatment options include calcineurin inhibitors (cyclosporine and tacrolimus), cyclophosphamide, and rituximab.
Rituximab is not approved for the indication of primary membranous nephropathy in Canada. This review aimed to evaluate the evidence on the use of rituximab compared to cyclophosphamide, tacrolimus, and cyclosporine in adult patients with primary membranous nephropathy.
A systematic review of the efficacy and safety of rituximab versus cyclosporine, tacrolimus, or cyclophosphamide was conducted with 18 included randomized controlled trials. A network meta-analysis of 11 of the 18 included randomized controlled trials was uninformative due to the small number of studies, the heterogeneity in the studies, and unreliable point estimates and wide credible intervals obtained with the network meta-analysis.
Due to the uninformative nature of the network meta-analysis, a narrative analysis was conducted of the head-to-head trials of rituximab. Two randomized controlled trials showed no evidence of a difference between rituximab and cyclophosphamide, whereas rituximab resulted in a better response rate (complete remission and the composite outcome of partial or complete remission) at 24 months compared with cyclosporine. There were no head-to-head trials comparing rituximab with tacrolimus.
Given the small network of studies, the heterogeneity in the included studies, and the limited information provided by the network meta-analysis and the pairwise comparisons of the MENTOR and RI-CYCLO trials, CADTH was unable to conduct an informative economic evaluation. Further, in addition to the clinical evidence gaps, there were also issues identifying information to inform key parameters to address the policy question of interest to decision-makers. Given the limitations associated with the clinical evidence and absence of evidence to inform key model parameters, an economic evaluation would not be able to quantify all relevant incremental costs and effects of using rituximab over the currently used alternatives.
Abstract
Background and Policy Context
Membranous nephropathy (MN) is an autoimmune disease and one of the most common causes of nephrotic syndrome in adults. The incidence of MN is 1.2 in 100,000 persons per year worldwide. Nephrotic syndrome is characterized by proteinuria, hypoalbuminemia, hyperlipidemia, and peripheral edema, and may lead to end-stage renal disease. Approximately 80% of patients with MN have a classification of primary MN (PMN) (or idiopathic MN [IMN]).
No patient groups or individual patients responded to CADTH’s call for feedback on the project scope. Thus, excerpts from patients’ experiences and perspectives shared on social media and other online sources were considered instead. As such, patients indicated that preventing or delaying end-stage renal disease and dialysis were important treatment goals.
There is evidence to show that immunosuppressive therapies such as cyclophosphamide (CTX) and the calcineurin inhibitors reduce proteinuria, all-cause mortality, and progression to end-stage renal disease. However, the use of these medications may be associated with serious adverse events (AEs) such as malignancy, infertility, and infection.
Rituximab is another treatment option for which there is evidence of efficacy. Rituximab is not approved for the indication of PMN in Canada, and as a such is used off-label. The review aims to evaluate the evidence on the use of rituximab compared to existing treatments in adult patients with PMN.
Clinical Evidence
A total of 18 randomized controlled trials (RCTs) were included in the systematic review (SR), of which 11 were included in the network meta-analysis (NMA). Due to the small network of studies and inherent heterogeneity in the included studies, primarily from differences in study interventions, population characteristics, and definition of outcomes, no conclusions could be drawn from the available data. The limitations of the NMA for drawing conclusions was evident from the extremely wide 95% credible intervals estimated by both the fixed effect and random effects models; therefore, reported estimates from these models were considered unreliable for drawing conclusions.
The aim of the project was to determine the place of rituximab in the treatment of PMN. Given the uninformative nature of the NMA results, a narrative synthesis of the head-to-head trials of rituximab was conducted. There were no head-to-head trials comparing rituximab to tacrolimus (TAC). Relevant evidence was taken from the MENTOR study (rituximab versus cyclosporine [CYC]) and the RI-CYCLO study (rituximab versus CTX). While both trials were generally well-conducted, the findings from RI-CYCLO were inconclusive with regards to the comparative efficacy and safety between rituximab and CTX for any of the outcomes measured at any of the time points, given that the study was not powered to detect a clinically meaningful difference. In MENTOR, rituximab was shown to be superior to CYC in terms of 2 efficacy outcomes at 24 months: complete remission [CR] and the composite outcome of partial remission [PR] or CR, this last one being the primary end point of the RCT. In addition, conclusions could not be drawn regarding the difference in time to remission and the difference in health-related quality of life (HRQoL) among patients treated with rituximab versus CYC. The lower response among patients who received CYC could have been due to the inclusion of patients with more severe proteinuria.
Rituximab showed a similar safety profile compared to both CTX and CYC; AEs occurred in similar frequency between treatment groups in both trials. With respect to notable harms, gastrointestinal events and infections were less common with rituximab relative to the respective comparator group, although 2 cases of end-stage renal disease were reported with rituximab.
Economic Evidence
Given the small network of studies, the heterogeneity in the included studies, and the limited information provided by the NMA and the pairwise comparisons of the MENTOR and RI-CYCLO trials, CADTH was unable to conduct an informative economic evaluation. Further, in addition to the clinical evidence gaps, there were also issues identifying information to inform key parameters to address the policy question of interest to decision-makers. Given the limitations associated with the clinical evidence and absence of evidence to inform key model parameters, an economic evaluation would not be able to quantify all relevant incremental costs and effects of using rituximab over the currently used alternatives. CADTH will work with the public drug plans to assist with tools to support policy decisions.
Conclusions and Implications for Decision- or Policy-Making
The evidence is inconclusive with regards to the comparative efficacy and safety of rituximab compared with CTX. Limited evidence from 1 RCT suggested that rituximab may result in better treatment response outcomes than CYC. There were no head-to-head trials comparing rituximab to TAC.
The evidence did not lend itself to conducting an economic evaluation; as such, the cost-effectiveness of rituximab to treat Canadians with PMN is unknown. Patients have indicated their willingness to try different treatment options while taking into consideration the safety profile of each drug.
CADTH convened a panel of experts who provided guidance on initiation, discontinuation, and prescribing criteria. These will be used by the public drug plans to inform local decisions.
Introduction
Background and Rationale
MN is an autoimmune disease and one of the most common causes of nephrotic syndrome (NS) in adults.1,2 NS is characterized by proteinuria (> 3.5 g per 24 hours), hypoalbuminemia (< 30 g/dL), hyperlipidemia, and peripheral edema. Patients are also at risk of thromboembolism.3 NS may lead to end-stage renal disease (ESRD).4
The incidence of MN is 1.2 per 100,000 persons per year worldwide.2 Approximately 80% of patients with MN have anti-phospholipase A2 receptor (anti-PLA2R) antibodies and are classified as PMN (or IMN); whereas 20% of patients have secondary MN due to a malignancy, an infection (e.g., hepatitis B or C), drugs (e.g., penicillamine, non-steroidal anti-inflammatory drugs [NSAIDs]), an autoimmune disease (e.g., systemic lupus erythematosus), or a non-identified autoantibody.1,2
Spontaneous remission of PMN is seen in approximately 30% of patients by 14 months and in approximately 60% of patients by 5 years,1,2,5 and 30% to 40% of patients will progress to ESRD within 10 years.5 The occurrence of remission is more common in patients with low antibody levels.1,2 Those with high levels of antibodies have higher risks of relapses, lower responses to therapy, and longer time to remission.1,2
The treatment goal of patients with PMN is to induce remission to reduce proteinuria and prevent kidney function loss.2 Treatments include supportive therapies for hypertension, hyperlipidemia, edema, and preventing thromboembolism.1,2 There is evidence to show that immunosuppressive therapy reduces proteinuria, all-cause mortality, and progression to ESRD. Alkylating drugs (CTX or chlorambucil) and calcineurin inhibitors (CYC or TAC) are immunosuppressive therapies recommended to treat patients with PMN.2The use of these medications is associated with serious AEs. Patients administered CTX are at risk of malignancy, infertility, infection, bone marrow suppression, liver toxicity, and cardiovascular events.1,2 Serious AEs seen in patients on calcineurin inhibitors include hypertension and nephrotoxicity.
The recently updated Kidney Disease: Improving Global Outcomes (KDIGO) guideline, KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases, makes recommendations for the use of rituximab, CTX, and calcineurin inhibitors in patients at moderate, high, and very high risk of progressive kidney injury, as well as for patients who relapse after therapy or who are treatment-resistant.6
Rituximab is a monoclonal antibody directed against the CD20 receptor. It induces the depletion of CD20 positive B cells. Its use in PMN was first reported in a case series in 2002 and subsequently in 3 single arm trials and 1 RCT that compared rituximab with supportive therapies.2 Rituximab does not have a Health Canada approval for the indication of PMN, and as a such is used off-label. Recently, 2 phase III RCTs have been conducted to compare rituximab with other immunosuppressive treatments in PMN: CYC in the MENTOR study and CTX in the RI-CYCLO study.7,8 A critical appraisal of the MENTOR study is posted on the CADTH website.9 In PMN, KDIGO recommends the administration of rituximab 1 g by IV twice within 2 weeks or 375 mg/m2 1 to 4 times at weekly intervals.6
CTX, a nitrogen mustard drug, is an antineoplastic and an immunosuppressant.10 In Canada, it is indicated for various cancers.11 Its used in PMN was first described in the 1970s.12 To treat PMN, KDIGO recommends a dose of 2.5 mg/kg per day in months 2, 4, and 6 in combination with methylprednisolone 1 g IV for 3 consecutive days at the start of month 1, 3, and 5 and prednisone 0.5 mg/kg per day in months 2, 4, and 6.6
CYC and TAC are calcineurin inhibitors that inhibit phosphatase calcineurin, which leads to a reduction in T-cell activation. CYC is indicated for the treatment of adults and children with NS, including MN.13 KDIGO recommends its administration at a dose of 3.5mg/kg per day to achieve serum trough level of 125 ng/mL to 225 ng/mL (101 nmol/L to 187 nmol/L).6 TAC, a drug approved for use in organ transplantation and in rheumatoid arthritis, is administered at a dose of 0.05 mg/kg per day to 0.1 mg/kg per day with a target trough level of 125 ng/mL to 225 ng/mL (104 nmol/L to 187 nmol/L) for 12 months for PMN.6,14 The calcineurin inhibitors may be given in combination with prednisone 10 mg per day, which can be withdrawn after 4 months in case of nonresponse or tapered after 12 months to a lower dose in responders.6
With the publication of 2 key studies, MENTOR and RI-CYCLO, the government-sponsored drug plans requested a review of the use of rituximab in adult patients with PMN.
Objective
CADTH undertook a health technology assessment (HTA) to review the available evidence on the use of rituximab for PMN to determine its effectiveness and cost-effectiveness relative to other treatments.
Policy Questions
The following policy questions were addressed with this project:
Is there evidence to support the use of rituximab in adult patients with PMN?
If so, what are the policy options for providing access to rituximab?
In a protocol amendment, the second policy question was replaced with these questions:
Based on the available evidence, what is the place in therapy of rituximab to treat PMN?
What specific characteristics should patients possess or satisfy for access to rituximab?
What is the most appropriate rituximab dosing regimen?
What objective measures or outcomes, and at what time points, should be used to determine treatment success or failure in PMN?
Are there special populations that require other considerations?
Research Questions
The project addressed the following research questions. Details on the specific interventions and outcomes are included in Table 2.
What are the efficacy and safety of rituximab compared with current treatments in patients with PMN?
What is the cost-effectiveness of rituximab compared with current treatments in patients with PMN?
Opportunities for Stakeholder Feedback
Stakeholders were given the opportunity to comment on the proposed project scope that informed this report. Stakeholders were also given the opportunity to provide feedback on the list of included studies and the draft report.
Summary of Patient Perspectives
CADTH involves patients, families, and patient groups to improve the quality and relevance of our assessments. When we are unable to speak to people with lived experience, we take steps to consider patient perspectives by including views from online patient groups. CADTH has adopted a Framework for Patient Engagement in Health Technology Assessment.15 The Framework includes Standards for Patient Involvement in Individual Health Technology Assessments and is used to support and guide CADTH’s activities involving patients and patient groups. For this HTA, understanding that patients have knowledge, perspectives, and experiences that are unique and contribute to essential evidence, guided CADTH’s approach for finding and collating patient insights.
CADTH wanted to speak with a patient with PMN to better understand the challenges of the disease and hear their firsthand experiences with rituximab. Though relevant patient groups were contacted and a public call for patient involvement posted, CADTH was unable to find and interview an individual with personal experience with PMN who had tried rituximab. As an alternative, excerpts from patient observations and experiences that had been shared on social media and other online sources were gathered. Appendix 1 follows the Guidance for Reporting Involvement of Patients and the Public 2 Short Form (GRIPP2 SF) checklist16 to outline the process of collecting patient perspectives and where and how that information was used in the review.
Patient age at diagnosis varied, but most patients reported being diagnosed before the age of 40. Disease symptoms included, but were not limited to, swelling and pain in the legs, weight gain, foamy urine, and fatigue. Some patients achieved proteinuria remission with corticosteroids and immunosuppressive therapies but reported that these medications had significant side effects. The risk of infertility associated with CTX was a major concern for 1 young female patient. Overall, patients expressed wanting to try a treatment if it meant preventing or delaying disease progression. It was not uncommon for them to have tried immunosuppressive therapies before rituximab. Effectiveness of rituximab, like immunosuppressive therapies, varied from one patient to the next. The consensus among patients seemed to be that they were willing to endure treatment side effects if it meant there was a chance to go into remission and delay progression to end-stage kidney disease.
The collection of patient perspectives enabled the research team to consider the evidence found in the literature alongside an understanding of the wider experiences of patients and family caregivers.
Methods
To inform the conduct of this HTA, a preliminary scoping review of the existing literature was conducted. A protocol was written a priori, using appropriate reporting guidelines (e.g., the Preferred Reporting Items for Systematic Reviews and Meta-Analyses Protocols ([PRISMA-P]) for guidance on clarity and completeness and they were followed throughout the study process.
Study Design
This clinical evaluation was designed as an SR and an NMA to answer the first research question. The SR and NMA of the primary studies focused on the clinical effectiveness, safety, and impact of rituximab treatment on PMN. The SR was conducted following the core methods and review steps, including screening, data extraction, and risk of bias assessment. The NMA was conducted in accordance with ISPOR guidelines for NMAs.17
To answer the second research question, an economic analysis was planned to evaluate the cost-utility of rituximab using a de novo decision analytic model to assess the costs and health outcomes associated with interventions for the treatment of biopsy-proven PMN in adult patients who have NS and have not achieved spontaneous remission within 6 months of diagnosis. The interventions were those in the clinical review.
Literature Search Methods
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.18 The search strategy is presented in Appendix 2.
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid, Embase (1974‒) via Ovid, the Cochrane Central Register of Controlled Trials (CENTRAL) via Ovid, and PubMed. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was membranous nephropathy. Clinical trial registries were also searched: the US National Institutes of Health’s clinicaltrials.gov and WHO’s International Clinical Trials Registry Platform (ICTRP) search portal.
Search filters were applied to limit retrieval to RCTs or controlled clinical trials. Retrieval was not limited by publication date and was limited to the English or French language. Conference abstracts were excluded from the search results.
The initial search was completed on April 22, 2020, with regular updates until the publication of the final report. The clinical trial registries search was updated before the completion of the stakeholder feedback period. Studies meeting the selection criteria of the review and identified in the alerts before the completion of the stakeholder feedback period were incorporated into the analysis of the final report.
Grey literature (literature that is not commercially published) was identified by searching sources listed in relevant sections of the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature resource,19 which included the websites of regulatory agencies, HTA agencies, clinical guideline repositories, SR repositories, patient-related groups, and professional associations. Google was also searched for additional internet-based materials. These searches were supplemented by reviewing bibliographies of key papers and through contacts with experts and industry, as appropriate. The grey literature search was updated before the completion of the stakeholder feedback period.
Selection and Eligibility Criteria
Studies were included if they met the eligibility criteria, including the specific population, intervention, comparators, outcomes, and study design, presented in Table 2. The inclusion criteria were informed by the informal scoping review of the existing literature, patient engagement, stakeholder feedback, and consultation with the clinical expert.
Table 2: Selection Criteria for Clinical Review
Criteria | Description |
|---|---|
Population and subgroups | Adults with biopsy-proven primary membranous nephropathy with nephrotic syndrome Subgroups:
|
Intervention and comparatorsa |
|
Outcomes | Clinical effectiveness:
Safety:
|
Study design | Published phase III randomized controlled trials |
eGFR = estimated glomerular filtration rate; PLA2R = phospholipase A2 receptor; Scr = serum creatinine.
aBest supportive care may be given on an as-needed basis with all treatment regimens, which can include angiotensin converting-enzyme inhibitors or angiotensin II receptor blockers; a diet low in salt and protein; and statins, chlorambucil, adrenocorticotropic hormone, azathioprine, mizoribine, and leflunomide.
Population and Subgroups
For this review, adult patients with biopsy-proven PMN with NS were included, although studies were not excluded if their definition of NS was ambiguous or largely captured with PMN. Subgroups relevant for this review were determined based on risk factors for disease progression, relapse, resistance to disease, and prior treatment history. The clinical expert indicated that there may be differences in treatment effectiveness between the subgroups and that risk factors for progression are essential for the decision of whether or not to treat.
Interventions and Comparators
All currently available treatments for PMN were considered potentially relevant; however, the clinical expert noted that mycophenolate mofetil was not a recommended intervention in this group of patients. The following interventions were therefore selected: rituximab, CTX, calcineurin inhibitors (CYC or TAC), and placebo. Recognizing that corticosteroids are sometimes given in combination with these drugs, no distinction was made between monotherapy or combination therapy for any of the included treatments.
It should be noted that best supportive care (BSC) was included as a background treatment, or as an add-on treatment to any of the included regimens as the clinical expert noted that BSC is generally given to all patients. Antihypertensive medications (angiotensin converting-enzyme inhibitors or angiotensin receptor blockers) constituted the most common classes of BSC.
Outcomes
Several potentially important outcomes were discussed to assess the clinical effectiveness and safety of the studied drugs. These included outcomes assessed clinical response, kidney function, and HRQoL. Outcomes that were noted less important from a clinical standpoint were excluded (e.g., biomarkers). Likewise, several safety end points were considered important for clinical decision-making and were therefore included in the protocol.
Study Design
Published phase III RCTs that met the previously described population; intervention and comparator criteria were eligible for inclusion.
Study Selection
Two reviewers independently screened titles and abstracts of all retrieved citations (i.e., literature searches of academic databases, grey literature searches, and clinical trial database) against the eligibility criteria (Table 2). Exclusion by both reviewers was required for a record to be excluded at the title and abstract level. Full-text articles that were judged to be potentially relevant by at least 1 reviewer were retrieved for the second level of screening. The same 2 reviewers independently examined all full-text articles against the eligibility criteria, and consensus was required for inclusion in the review. Discrepancies between reviewers were resolved by discussion.
Studies identified via monthly database search alerts and semi-annual grey literature search alerts meeting the selection criteria of the review were incorporated into the analysis.
Quality Assessment
The risk of bias of the primary studies was systematically evaluated using the methods described in the Cochrane Risk of Bias 2 assessment tool for RCTs.20
The Cochrane Risk of Bias 2 assessment tool20 allowed for the assessment of 5 sources of bias or “domains” — bias arising from the randomization process, bias due to deviations from intended interventions, bias due to missing outcome data, bias in measurement of the outcome, and bias in selection of the reported result. Each question within each domain was answered with a yes, probably yes, probably no, no, or no information. Afterward, a judgment of “low risk of bias,” “high risk of bias,” or “some concerns” was assigned for each domain, with rationale for each decision included in the comments box field.
The risk of bias assessments of the included studies was performed by 1 reviewer. The tools were used as a guide to evaluate the risk of bias in the included studies, and additional insight beyond the items on the instruments have been provided, when applicable. Summary scores were not calculated; rather, the strengths and limitations of each included study and how they affect the study findings were described narratively. Results of the risk of bias assessment were not used to exclude studies from this review.
Data Extraction
The original, primary publication for each included RCT was used for data extraction. In situations where multiple publications for a unique RCT were available (e.g., supplemental online appendices, companion publications of specific outcomes, or populations from the original study), the most recently adjudicated data for each outcome were extracted, with preference given to published records.
Reviewers used Microsoft Excel to document and tabulate all relevant information from the included studies. Data were extracted by the lead author, with data checking done by a secondary reviewer. Discrepancies were resolved through discussion until consensus was reached; a third reviewer was involved when necessary. The following relevant information were extracted, where available:
Study level: description of publication (e.g., first author last name, title, publication year), study characteristics (e.g., clinical trial registry identification number, trial acronym, study design, year of study conduct, sample size, study setting, country of study conduct, randomization ratio, blinding status, superiority or noninferiority design, eligibility criteria, study duration)
Patient level: number of patients, age, sex (as reported by study authors), clinical situation of the diagnosis, duration of disease, baseline characteristics
Intervention and comparator level: type, dose, total duration of treatment, dose frequency, route of administration, and concurrent and previous relevant therapies
Outcome level: description of outcomes (e.g., method of measurement, unit of measurement, length of follow-up)
Type of analysis: intention-to-treat (ITT) or safety population (data from figures were extracted if explicit numerical data were reported)
Data Analyses and Synthesis
After the conclusion of data extraction, a feasibility assessment was conducted for addressing the posed research questions. This included evaluating sources of methodological and clinical heterogeneity between the included studies. Study design, patients baseline characteristics, treatment characteristics, and outcomes definition were compared between studies. A qualitative assessment of feasibility was determined through close collaboration between the reviewers, methodologists, and clinical experts working on the HTA.
When feasible, the efficacy and safety of rituximab versus other relevant comparators outlined in the Table 2 were evaluated through an indirect treatment comparison using NMA.
All NMAs were conducted under a Bayesian framework. The modelling approach was suitably chosen for each outcome (i.e., binomial likelihood models for dichotomous outcomes and normal likelihood models for continuous data). Random effects models were identified a priori as the primary approach when feasible to account for anticipated clinical and methodological heterogeneity across studies. Fixed effects models were considered when the available network for a given outcome was insufficient for estimating a random-effects model. When possible, regardless of the primary analysis, both fixed effect and random effect models were reported along with diagnostic information criterion. Vague priors were used for all parameters in the model. Each NMA was estimated in a Markov Chain Monte Carlo simulation using 3 chains, and their convergence were assessed by examining the history, trace and Gelman-Rubin plots. A minimum of 10,000 burn-ins and 20,000 iterations were performed in the simulation. In addition, statistical heterogeneity were assessed through comparing the residual deviance between the fixed and random-effects model. An assessment of the consistency assumption was conducted through an inconsistency model. Additional sensitivity analysis were conducted by excluding studies with high risk of bias or studies with missing data to a relevant outcome.
Results of Clinical Evaluation
Quantity of Research Available
A total of 934 citations were identified in the literature search. Following screening of titles and abstracts, 14 studies were identified as potentially relevant and retrieved for full-text review. A total of 24 reports were retrieved from other sources (i.e., grey literature, hand search, and search alerts). Of these 38 potentially eligible reports, 19 reports presenting data from 18 unique studies met the inclusion criteria and were included for review. The report selection process is outlined in Appendix 3 using a PRISMA diagram. A list of included and excluded citations, with details describing the rationale for those excluded, are presented in appendices 4 and 5, respectively.
Study Characteristics
Study Design
A total of 19 publications met the inclusion criteria of this review, of which 2 publications reported findings from the same study, resulting in a total of 18 included trials. The study characteristics are shown in Appendix 6. The trials by Cattran et al.21 and Omrani et al.22 were single blinded and double blinded, respectively; 11 RCTs used an open-label design; and the remaining 5 trials did not specify the type of blinding. The sample sizes ranged from 26 patients to 130 patients across the studies; 10 studies had a sample size between 40 and 80 patients; 3 studies had a sample size of fewer than 40 patients; and 5 studies had a sample size greater than 80 patients. The duration of the studies (including follow-up) ranged from 6 months to 10 years, with 12 studies having a duration between 10 months and 25 months. Eight trials were multi centre, and 10 trials were single centre. Two studies were conducted in North America.
Inclusion Criteria
All trials were conducted in adults, although the definition of adults varied, with a minimum age of 15 years. Overall, the trials included patients with PMN, diagnosed primarily based on proteinuria and serum albumin and, to a lesser extent, creatinine clearance. Of the trials that reported a threshold for proteinuria, most studies used a threshold of 3.5 g per day or more for inclusion of patients, although studies consisting of patients with severe proteinuria and/or NS used a higher threshold (ranging from ≥ 5g per day to > 8 g per day). Serum albumin was generally used in combination with daily proteinuria as a criterion for defining PMN, with threshold ranging from less than 20 g/L to 35 g/L or more across studies. Creatinine clearance was reported as a function of estimated glomerular filtration rate (eGFR) or serum (or plasma) creatinine. For eGFR, the threshold ranged from 30 mL/min/1.73 m2 or greater to less than 60 mL/min/1.73 m2 across studies, whereas the threshold for serum creatinine ranged from lower than 221 µmol/L to lower than 133 µmol/L (or plasma creatinine concentration of less than 300 μmol/L). IMN stage as an eligibility criterion was reported in 5 trials. Three of these trials included IMN stage I to III, and 2 trials included IMN stage I to IV. IMN was generally diagnosed using renal biopsy and other pathological tests.
Exclusion Criteria
All trials excluded patients with IMN from a secondary source (systemic lupus erythematosus), or drug-associated nephropathy (phenytoin and gold salts). Patients with characteristics indicative of significant comorbidities were excluded, including any patients with any serious systemic infection or associated disorders requiring NSAIDS, liver function test abnormalities, severe renal diseases, diabetes mellitus, malignancy, infections (including malaria, HIV, tuberculosis, hepatitis B, and hepatitis C), severe cardiovascular conditions (cardiac dysfunction, uncontrolled hypertension, thromboembolism, unstable angina pectoris, renal vein thrombosis), and gastrointestinal diseases. Several trials restricted recruiting patients on immunosuppressive drugs, steroids, plasma exchange therapy, anti-lymphocyte products, gold, penicillamine, or NSAIDs between 1 month and 2 years before study, and rituximab (if the intervention of interest was rituximab). Pregnant patients, those using inadequate contraception, and those with hypersensitivity to the study drugs were also excluded.
Interventions and Comparators
Of the included studies, 2 trials included rituximab as a treatment group (MENTOR7 and RI-CYCLO8). In both trials, rituximab was administered in 2 separate IV doses (administered 14 days apart) of 1,000 mg over a period of 6 months, without concomitant or subsequent drug therapies, although the MENTOR study allowed subsequent re-treatment at the same dosage in the absence of remission. Three main treatment regimens were used across the studies: TAC, CTX, and CYC. Each of the 3 major treatment regimens were administered in different dosages across trials, usually given in 2 stages: a loading dose, to reach a certain plasma level, followed by a continuation dose, usually at a lower dose than the loading dose, continued through the end of the treatment period. Most trials administered corticosteroids (prednisone, prednisolone, or methylprednisolone) in combination with the primary treatment regimen, and these were also administered in 2 stages (i.e., a loading dose to reach a certain plasma level, and a continuation or tapering dose administered through the end of the treatment period). Details of the treatment regimen in each trial are provided in Appendix 7.
TAC was used as the treatment or comparator group in 10 of the included trials, and was compared with CTX, CYC, or other standards of care, including the Ponticelli regimen. TAC was generally administered at a dose of 0.5 mg/kg to 0.1 mg/kg per day, for a period ranging from 6 months to 24 months across studies. TAC was administered in combination with corticosteroids in all but 1 trial. Two trials compared TAC administered for different lengths of time: a short course (6 to 12 months) and a long-course (24 months).
CTX was used in 8 trials as an intervention or comparator (compared with TAC, CYC, rituximab, or BSC), in both IV and oral formulations, and administered for a period ranging from 3 to 12 months across trials. IV doses were administered once a month, at a dose of 500 mg/m2 to 750 mg/m2. Oral CTX doses ranged from 1 g to 2 g per day. In all cases, IV or oral corticosteroids were given in combination or alternatively with CTX.
CYC was used in 7 trials as an intervention or comparator (against placebo, rituximab, CTX, chlorambucil, or TAC), and administered for a period of 6 to 12 months. CYC was administered orally, at a dose of 1.5 mg/kg to 3.5 mg/kg per day across studies, with low doses for longer treatment periods, and higher doses for shorter treatment periods. CYC was given in combination with corticosteroids in all but 1 trial.
In addition to the study drugs, various medications were allowed in most trials. Antihypertensives were the most common medication, most notably angiotensin converting-enzyme inhibitors or angiotensin receptor blockers, administered as needed to maintain a target or stable blood pressure level (120 mm Hg to140 mm Hg for systolic blood pressure, 75 mm Hg to 90 mm Hg for diastolic blood pressure). Other medications included dietary modifications, cholesterol lowering drugs, and anticoagulants.
Outcomes
All included trials measured CR or PR as a primary or major outcome, or outcomes based on CR and PR, such as remission rates, total remission (TR), no remission (NR), relapse, or recurrence. Both CR and PR were based on proteinuria; however, the definition of CR and PR varied across studies (Table 3). For CR, proteinuria thresholds ranged from 0.3 g or more to 0.5 g per day for most trials. In addition, normal serum albumin level and stable or normal renal function were included in CR definition in a few trials. For PR, a wide range of definitions were used across the trials, including a proteinuria level 0.2 g per day or less to 3.5 g per day or less, 50% proteinuria reduction, and stable renal function. TR, sometimes referred to as any remission, consisted of those achieving either CR or PR, and NR consisted of those who achieved neither. Relapse and recurrence were outcomes based on a similar definition, most notably proteinuria level reaching outside of the range that was designated as PR after achieving CR and/or PR in the respective trials.
A number of outcomes assessing the kidney function were reported based on baseline creatinine or creatinine clearance. Most notable of these outcomes include doubling of baseline creatinine, referred to as renal survival in some trials. The incidence of the following outcomes were assessed less commonly: ESRD (defined as creatinine clearance of lower than 12 mL/min to 15 mL/minute or lower, the initiation of dialysis, or renal transplant), NS, or nephrotic proteinuria (severe proteinuria).
HRQoL was assessed in 2 trials, using the modified Kidney Disease Quality of Life-Short Form (KDQOL-SF) version 1.3, and visual analogue scale (score ranging from 0 to 10, with 10 indicating the best quality of life).
Table 3: Remission Definitions in the Included Studies
Remission | Definitions | Studies |
|---|---|---|
Complete remission | Proteinuria ≤ 0.3 g per day plus stable renal function | Cattran et al. (2001), Chen et al. (2010), He et al. (2013) |
Proteinuria < 0.3 g per day, normal serum albumin level, and stable renal function | Di et al. (2018), Li et al. (2017), Peng et al. (2015) | |
Proteinuria ≤ 0.3 g per day and serum albumin level of ≥ 3.5 g/dl | Fervenza et al. (2019) | |
Protein–creatinine index ≤ 0.2 g/10 mmol creatinine, and improved or stabilized renal function | Hofstra et al. (2010) | |
Proteinuria < 0.2g per day | Jha et al. (2007) | |
Proteinuria ≤ 0.3 g per day | Kosmadakis et al. (2010), Saito et al. (2014), Scolari et al. (2021) | |
Proteinuria < 0.5 g per day and stable per normal renal function | Praga et al. (2007), Xu et al. (2013) | |
Proteinuria < 0.5 g per day, normal serum albumin, and normal serum creatinine | Ramachandran et al. (2016 and 2017) | |
Proteinuria < 0.4 g per day | Yuan et al. (2013) | |
Proteinuria ≤ 0.3 g per day at 1 year | Scolari et al. (2021) | |
Partial remission | 50% proteinuria reduction, proteinuria < 3.5 g per day, and stable renal function | Cattran et al. (2001), Chen et al. (2010) |
Proteinuria 0.3 to 3.0 g per day; or 50% proteinuria reduction, serum albumin ≥ 30 g/L, and stable renal function | Di et al. (2018), Li et al. (2017) | |
50% proteinuria reduction, proteinuria < 3.5 g per day, and normal serum creatinine | He et al. (2013) | |
Protein–creatinine index < 2.0 g/10 mmol creatinine, and improved or stabilized renal function | Hofstra et al. (2010) | |
Proteinuria 0.2 g to 2.0 g per day; or 50% proteinuria reduction and stable renal function | Jha et al. (2007) | |
50% proteinuria reduction and proteinuria < 3.5 g per day | Kosmadakis et al. (2010), Peng et al. (2015), Praga et al. (2007) | |
Proteinuria 0.5 g to 1.9 g per day or 50% proteinuria reduction from baseline and stable renal function | Ramachandran et al. (2016 and 2017) | |
Proteinuria 0.3 g to 1.0 g per day | Saito et al. (2014) | |
Proteinuria 0.5 g to 3.5 g per day | Xu et al. (2013) | |
(continued) | 50% proteinuria reduction from baseline, proteinuria 0.4 g to 2.9 g per day, and serum albumin ≥ 30 g/L | Yuan et al. (2013) |
50% proteinuria reduction from baseline and final proteinuria between 0.31 and 3.5 g per day | Fervenza et al. (2019) | |
50% proteinuria reduction from baseline and proteinuria < 3.5 g per day | Scolari et al. (2021) |
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Scolari et al.,8 Xu et al.,36 Yuan et al.37
Baseline Patient Characteristics
The baseline patient characteristics are shown in Appendix 8. The mean age of the participants ranged from 37 to 58 years; for most trials, the patients were between 40 and 60 years old (15 studies). The trials consisted of 42.7% to 93.5% males, with 12 trials consisting of more than 60% males. Of the studies with available baseline data, the trials had a mean eGFR of 60 mL/min/1.73 m2 to 100 mL/min/1.73 m2 (15 studies), a mean systolic blood pressure of 100 mm Hg to 130 mm Hg (10 studies), and mean or median serum albumin of 2 g/dL to 3 g/dL (12 studies).
The following baseline characteristics were reported in no more than 9 studies: disease duration, total cholesterol, and urine protein. Of the studies that reported disease duration, 5 studies had patients who have had the disease for less than 15 months, whereas 1 trial included patients with the disease for more than 50 months. Six trials had patients with a mean total cholesterol greater than 250 mg/dL, whereas 1 trial had patients with a mean total cholesterol of less than 150 mg/dL. Mean or median urine protein levels ranged between 6 g and 12 g per day in all but 2 studies, which had protein levels of less than 4 g per day.
Data Analysis and Synthesis
Findings of the Network Meta-Analysis
Among the studies identified for inclusion in the NMA, 11 studies reported results for outcomes of interest for this review. The outcomes for which a viable network was available for analysis were CR and the composite outcome of CR or PR. A total of 10, 11, and 6 studies reported results for CR measured at 6, 12, and 18 months, respectively. A total of 11, 11, and 6 studies reported results for the composite outcome of PR or CR at 6, 12, and 18 months, respectively. The network diagrams and model results for both fixed effects and random effects models for each of these end points are reported in Appendix 9. All models successfully converged; however, upon examining the results of each model, several of the estimated credible intervals were found to be extremely wide. These results suggest a high level of instability in these models, likely a result of small networks of available studies and excessive heterogeneity across the network. Thus, the estimated results were considered to be unreliable for drawing conclusions for how rituximab compares to the available comparators in the network.
Narrative Synthesis
Given the uninformative results of the NMA, a narrative synthesis was conducted of the head-to-head trials of rituximab. Relevant evidence was taken from the MENTOR study (rituximab versus CYC) and the RI-CYCLO study (rituximab versus CTX). Unless otherwise specified, all efficacy results were based on the ITT population of the respective studies. There were no head-to-head trials comparing rituximab to TAC.
Summary of Critical Appraisal
The MENTOR and RI-CYCLO studies were considered well-conducted. These trials had a low risk of bias in each domain, with most signalling questions assessed adequately based on the level of detail provided in the publications. The critical appraisal of all the included trials is available in Appendix 10.
A complete critical appraisal of the MENTOR study is posted on the CADTH website.9 In summary, the CADTH critical appraisal of the MENTOR trial reported the key limitations of this study being small sample size, which limited power, and the open-label design of the study, which could introduce bias for subjectively measured end points. It was also noted that, due to the limited evidence, conclusions could not be made for the end points of HRQoL, creatinine clearance, and anti-PLA2R. While CADTH did not publish an in-depth peer-reviewed critical appraisal of the RI-CYCLO trial, based on the assessment conducted as part of this broader review, the study has similar limitations of low sample size, which limited power to detect difference, and an open-label design, which could introduce bias for subjectively measured end points.
Outcomes Assessing Clinical Response
Complete Remission
In the MENTOR study, CR was a secondary outcome that was not adjusted for multiple comparisons (Table 4). Thus, results must be interpreted cautiously with consideration to the increased risk of type I error. The risk difference comparing rituximab to CYC estimated a 2% decrease in CR at 6 months (95% confidence interval [CI], −5 to 2), a 9% increase in CR at 12 months (95% CI,−1 to 19), a 26% increase in CR at 18 months (95% CI,15 to 37), and a 35% increase in CR at 24 months (95% CI, 24 to 47).7
In the RI-CYCLO study, CR at 12 months was the primary efficacy outcome, although the study was conducted as a pilot study that was not powered to detect a clinically meaningful difference. As a result, the estimated 95% CI for each point estimate were extremely wide, and thus, do not provide sufficient evidence to support differences between rituximab and CTX for any end points. At 12 months, the odds ratio (OR) comparing rituximab to CTX estimated a numerically decreased odds of CR for rituximab compared to CTX with an estimated OR of 0.40 (95% CI, 0.13 to 1.23). Results from the RI-CYCLO trial were not adjusted for multiple comparisons; however, there is no increased risk of type I error from these results as none of the estimated 95% CIs exclude the null hypothesis.8
Complete or Partial Remission (Composite Outcome)
PR was reported as a composite outcome with CR (Table 4). In the MENTOR trial, the composite outcome of CR or PR at 24 months was the primary outcome. A gatekeeping approach was used to control the family-wise type I error by first testing the primary outcome for the noninferiority of rituximab compared to CYC and only testing for superiority if the test for noninferiority was successful. The results supported the conclusion that rituximab was noninferior to CYC at 24 months at a noninferiority margin of 15%. The noninferiority margin of 15% was based on the assumption that 55% of the patients in the rituximab group and 45% of those in the CYC group had a CR or PR at 24 months; however, only 20% patients in the CYC group achieved CR or PR through 24 months. The authors for the MENTOR study suggested that the lower response among patients receiving CYC could have been due to the inclusion of patients with more severe proteinuria. The risk difference comparing rituximab to CYC was 40% (95% CI, 25 to 55; 1-sided P < 0.001 for noninferiority) at 24 months. The effect estimate was consistent regardless of age (≤ 50 years and > 50 years), anti-PLA2R (≤ 40 u/mL and > 40 u/mL), and history of immunosuppressive therapy at baseline. Superiority of rituximab over CYC for CR or PR at 24 months was also supported by the results (2-sided P < 0.001). The secondary outcomes of CR or PR at 24 months based on the per-protocol population and at 12 months based on the ITT population were also included in the gatekeeping procedure for multiple testing and were simultaneously tested using a Bonferroni correction for multiple comparison, resulting in an alpha threshold of 0.0125 for the 1-sided P value testing noninferiority for these end points. Results for the per-protocol population have not been presented in this report. For the end point of CR or PR at 12 months, the results supported conclusions of noninferiority of rituximab compared to CYC at a noninferiority margin of 15%. The estimated risk difference showed a numerical increase of 8% (95% CI, −9 to 25) for rituximab compared to CYC. This end point was not tested for superiority as part of the pre-specified analysis nor was any other end point formally tested. Thus, any further conclusions drawn based on results from this study must consider the risk of increased type I error for such conclusions.7
In the RI-CYCLO trial, the composite outcome of CR or PR was a secondary outcome (Table 4). At month 6, 12, and 18, numerically fewer percentage of patients in the rituximab group achieved CR or PR compared to the CTX regimen group (OR = 0.57 [95% CI, 0.22 to 1.45]; OR = 0.61 [95% CI, 0.23 to 1.63]; OR = 0.49 [95% CI, 0.16 to 1.49], respectively), whereas more patient achieved this end point in the rituximab group at 24 and 36 months (OR = 1.32 [95% CI, 0.33 to 5.29]; OR = 2.12 [95% CI, 0.45 to 9.96], respectively). Of the reported subgroups, the composite CR or PR probability remained consistent across age (≤ 55 years and > 55 years) and serum albumin levels (≤ 2.5 g/dL and > 2.5 g/dL). As previously mentioned, the precision around the results from this study were generally insufficient to support any conclusions that the efficacy of rituximab differed from CTX.8
Table 4: Remission in the MENTOR and RI-CYCLO Studies (ITT)
Time point (months) | MENTOR | RI-CYCLO | ||||
|---|---|---|---|---|---|---|
Rituximab, n (%) (N = 65) | CYC, n (%) (N = 65) | Risk difference (95% CI) | Rituximab, n/N (%) (N = 37) | CTX, n/N (%) (N = 37) | Odds ratio (95% CI) | |
Complete remission | ||||||
6 | 0 (0) | 1 (2) | −2 (−5 to 2) | 3/37 (8) | 2/37 (5) | 1.54 (0.24 to 9.8) |
12 | 9 (14) | 3 (5) | 9 (−1 to 19) | 6/37 (16) | 12/37 (32) | 0.40 (0.13 to 1.23)a |
18 | 18 (28) | 1 (2) | 26 (15 to 37) | 10/32 (31) | 7/34 (21) | 1.75 (0.57 to 5.36) |
24 | 23 (35) | 0 (0) | 35 (24 to 47) | 11/26 (42) | 11/31 (35) | 1.33 (0.46 to 3.89) |
36 | ND | ND | ND | 6/20 (30) | 7/22 (32) | 0.92 (0.25 to 3.41) |
Complete or partial remission (composite outcome) | ||||||
6 | 23/65 (35) | 32/65 (49) | −14 (−31 to 3) | 19/37 (51) | 24/37 (65) | 0.57 (0.22 to 1.45) |
12 | 39/65 (60) | 34/65 (52) | 8 (−9 to 25) NI margin: 15 percentage points P value: 0.004b | 23/37 (62) | 27/37 (73) | 0.61 (0.23 to 1.63) |
18 | 40/65 (62) | 15/65 (23) | 38 (23 to 54) | 21/32 (66) | 27/34 (79) | 0.49 (0.16 to 1.49) |
24 | 39/65 (60) | 13/65 (20) | 40 (25 to 55) Inferiority result: NI margin: 15 percentage points P value: < 0.001a,b | 22/26 (85) | 25/31 (81) | 1.32 (0.33 to 5.29) |
Superiority result: P value: < 0.001a,b | ||||||
36 | ND | ND | ND | 17/20 (85) | 16/22 (73) | 2.12 (0.45 to 9.96) |
CI = confidence interval; CYC = cyclosporine; CTX = cyclophosphamide; ITT = intention to treat; ND = not done; NI = noninferiority.
Note: Data in bold represents primary outcome of the respective trial.
aPrimary outcome for the trial.
bIndicates analysis was controlled for family-wise type I error, with a stepwise approach (first testing the noninferiority of rituximab and then testing the superiority of rituximab if the noninferiority test was significant) or Bonferroni correction in the MENTOR study.
Time to Remission
Time-to-event curves for CR or PR during the 12-month treatment period was provided in the MENTOR trial only. Patients in the CYC group tended to have remission earlier, with a later catch-up in patients in the rituximab group (hazard ratio for response at 12 months = 0.85; 95% CI, 0.55 to 1.32) (data not presented).7
Relapse
Relapse was reported in the RI-CYCLO trial only. A total of 9 participants relapsed after they had achieved remission at 12 months: 3 of 23 patients in the rituximab group (13%) and 6 participants of 27 patients in the comparator group (22%).8
Health-Related Quality of Life
HRQoL was assessed in the MENTOR study only, using selected subscales of the KDQOL-SF in patients with CR or PR at months 6, 12, and 24. Results were only reported for patients that achieved CR or PR at a given time point; therefore, these results cannot be used to support conclusions regarding difference in HRQoL among patients treated with rituximab versus CYC (data not presented).7
Outcomes Assessing Harms
Adverse Events
Overall, both trials showed a similar AE profile for rituximab and the respective comparator group (Table 5). A higher proportion of AEs were reported in the MENTOR trial (71% and 78% in the rituximab and CYC groups, respectively) than in the RI-CYCLO trial (43% in both the rituximab and CTX groups). Subsequently, the number and rate of AEs per 100 patients were higher in the MENTOR study than in the RI-CYCLO study, with 275 AEs per 100 patients and 335 AEs per 100 patients in the rituximab and CYC groups, respectively, in the MENTOR trial, and 54 AEs per 100 patients and 47 AEs per 100 patients in the rituximab and CTX groups, respectively, in the RI-CYCLO trial. Only the MENTOR study reported AEs by grade; the proportion of patients with grade 3 or higher AEs was 35% in the CYC group compared to 17% in the rituximab group.7,8
Withdrawals Due to Adverse Events
Patients in the rituximab group in both trials had fewer treatment discontinuations than the respective comparator group: 2 with rituximab versus 11 with CYC in the MENTOR study, and 1 with rituximab versus 4 with CTX in the RI-CYCLO study.7,8
Serious Adverse Events
Both trials had a comparable number of serious AEs between the treatment groups (Table 5). One patient receiving rituximab in the RI-CYCLO study had a fatal serious AE due to lung cancer. The other serious AEs were nonfatal.7,8
Notable Harms
Of the notable harms listed in the selection criteria outlined in Table 2, patients in the rituximab group generally had a lower rate of gastrointestinal events and infections relative to the respective comparator group (Table 5). No cancers or deaths occurred during the MENTOR trial, whereas 3 patients developed cancer in the RI-CYCLO trial: 2 in the rituximab group (lung and breast carcinoma; the patient with lung cancer died), and 1 in the CTX group (prostate carcinoma). ESRD was reported in 1 patient in the CYC group in the MENTOR study, and 2 patients in the rituximab group in the RI-CYCLO study.7,8
Table 5: Harms in the MENTOR and RI-CYCLO Studies
Event | MENTOR | RI-CYCLO | ||||||
|---|---|---|---|---|---|---|---|---|
Rituximab (N = 65) | CYC (N = 65) | Rituximab (N = 37) | CTX (N = 37) | |||||
Patients, n (%) | Number of events (rate per 100 patients) | Patients, n (%) | Number of events (rate per 100 patients) | Patients, n (%) | Number of events (rate per 100 patients) | Patients, n (%) | Number of events (rate per 100 patients) | |
AE | 46 (71) | 179 (275) | 51 (78) | 218 (335) | 16 (43) | 25 (47) | 16 (43) | 30 (54) |
SAE | 11 (17) | 13 (20) | 20 (31) | 22 (34) | 7 (19) | 8 (11) | 5 (14) | 6 (7) |
Fatal | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (3) | 1 (1) | 0 (0) | 0 (0) |
Nonfatal | 11 (17) | 13 (20) | 20 (31) | 22 (34) | 7 (19) | 7 (9) | 5 (14) | 6 (7) |
Notable AEsa | ||||||||
GI pain | 1 (2) | 2 (3) | 9 (14) | 9 (14) | — | — | — | — |
GI infection | 4 (6) | 4 (6) | 4 (6) | 4 (6) | — | — | — | — |
Infections | 17 (26.2) | 28 (43.1) | 20 (30.8) | 26 (40.0) | — | — | — | — |
Influenza-like symptoms | 6 (9) | 8 (12) | 3 (5) | 3 (5) | — | — | — | — |
Pneumonia | 1 (2) | 1 (2) | 6 (9) | 6 (9) | 0 (0) | 0 (0) | 3 (8) | 7 (8) |
Other respiratory tract infection | 9 (14) | 12 (18) | 9 (14) | 10 (15) | — | — | — | — |
Skin infection | 4 (6) | 5 (8) | 0 | 0 | — | — | — | — |
Other infectious events | — | — | — | — | 5 (14) | 6 (8) | 6 (16) | 8 (10) |
Cancer | 0 | 0 | 0 | 0 | — | — | — | — |
Lung | — | — | — | — | 1 (3) | 1 (1) | 0 (0) | 0 (0) |
Prostate | — | — | — | — | 0 (0) | 0 (0) | 1 (3) | 1 (1) |
Breast | — | — | — | — | 1 (3) | 1 (1) | 0 (0) | 0 (0) |
AE = adverse event; CTX = cyclophosphamide; CYC = cyclosporine; GI = gastrointestinal; SAE = serious adverse event.
aNotable harms were only reported if they occurred in 4 or more patients in the MENTOR trial and 5 or more patients in the RI-CYCLO trial.
Results of Economic Evaluation
Given the small network of studies, the heterogeneity in the included studies, and the limited information provided by the NMA and the pairwise comparisons of the MENTOR and RI-CYCLO studies, CADTH was unable to conduct an informative economic evaluation. Furthermore, in addition to the clinical evidence gaps, there were also issues identifying information to inform key parameters to address the policy question of interest to decision-makers. Given the limitations associated with the clinical evidence and absence of evidence to inform key model parameters, an economic evaluation would not be able to quantify all relevant incremental costs and effects of using rituximab over the currently used alternatives. CADTH will work with the public drug plans to assist with tools to support policy decisions.
Discussion
Summary of Evidence
The aim of the review was to determine the place of rituximab in the treatment of PMN. To inform the project scope, CADTH searched online social platforms for firsthand experiences from individuals with MN, to get a better understanding of the challenges associated with the disease and its treatment. The selection criteria of the protocol were then adjusted to reflect these patient perspectives. With that, a total of 19 publications met the inclusion criteria for this review, with a total of 18 included trials.
The included RCTs enrolled patients with PMN, diagnosed primarily based on proteinuria and serum albumin. The sample sizes ranged from 26 patients to 130 patients across the trials. The mean age of the participants ranged from 37 years to 58 years and the participants were mostly male (range = 42.7% to 93.5%). The duration of the studies (including follow-up) ranged from 6 months to 10 years. Two studies were conducted in North America. All trials excluded patients if their disease was due to a secondary source such as systemic lupus erythematosus or drug-associated nephropathy. Various outcomes were reported, including CR or PR, remission rates, TR, NR, relapse, or recurrence measured at various time points.
A total of 11 studies reported the results for CR or the composite outcome of PR or CR that were used in the NMA. The fixed effects and random effects models of the NMA successfully converged; however, upon examining the results of each model, several of the estimated credible intervals were found to be extremely wide. These results suggested a high level of instability in these models likely a result of small networks of available studies and excessive heterogeneity across the network. Thus, the estimated results were unreliable for drawing conclusions.
Because of the noninformative results from the NMA, a narrative synthesis of the head-to-head trials of rituximab was performed. Relevant evidence was taken from the MENTOR study (rituximab versus CYC) and the RI-CYCLO study (rituximab versus CTX). These studies were considered well-conducted and the findings well reported. These trials had a low risk of bias in each domain, with most signalling questions assessed adequately based on the level of detail provided in the publications. There were no head-to-head trials comparing rituximab to TAC.
Rituximab Versus Cyclosporine (MENTOR Study Results)
In the MENTOR trial, the composite outcome of CR or PR at 24 months was the primary outcome. Rituximab was found to be noninferior to CYC at 24 months at a noninferiority margin of 15%, with a risk difference of 40%. Superiority of rituximab over CYC for CR or PR at 24 months was also supported by the results.
CR was a secondary outcome that was not adjusted for multiple comparisons. There was a decrease in the number of patients with CR (2%) at 6 months and an increase in the number of patients with CR of 9%, 26%, and 35% at 12 months, 18 months, and 24 months, respectively, with rituximab compared to CYC. Though the risk of increased type I error must be considered when interpreting these results, the magnitude of the effect observed, particularly at later time points, is unlikely to be explained by random chance. Patients in the CYC group tended to have remission earlier, with a later catch-up in patients in the rituximab group.
Results using selected subscales of KDQOL-SF in patients with CR or PR at months 6, 12, and 24 could not be used to support conclusions regarding differences in HRQoL among patients treated with rituximab versus CYC.
The percentage of AEs reported in the MENTOR trial was 71% and 78% in the rituximab and CYC groups, respectively. The number of AEs per 100 patients were 275 in the rituximab group and 335 in the CYC group. Patients in the rituximab group had fewer treatment discontinuations than those in the CYC group (2 with rituximab versus 11 with CYC).
Rituximab Versus Cyclophosphamide (RI-CYCLO Study Results)
In the RI-CYCLO trial, CR at 12 months was the primary efficacy outcome, although the study was conducted as a pilot study that was not powered to detect a clinically meaningful difference. As a result, the estimated 95% CI for each point estimate were extremely wide, and thus, do not provide sufficient evidence to support differences between rituximab and CTX at any time points.
The composite outcome of CR or PR was a secondary outcome in the RI-CYCLO trial. The results were insufficient to support any conclusions regarding the efficacy of rituximab versus CTX for this outcome.
More patients relapse with CTX compared with rituximab at 12 months: 3 of the 23 patients in the rituximab group and 6 of the 27 patients in the CTX group.
The percentage of AEs reported in the RI-CYCLO study was 43% with rituximab and 43% with CTX. The number of AEs per 100 patients were 54 with rituximab and 47 with CTX. Patients in the rituximab group had fewer treatment discontinuations with CTX (1 with rituximab versus 4 with CTX).
Economic Evaluation
Given the small network of studies, the heterogeneity in the included studies, and the limited information provided by the NMA and the pairwise comparisons of the MENTOR and RI-CYCLO trials, CADTH was unable to conduct an informative economic evaluation. Further, in addition to the clinical evidence gaps, there were also issues identifying information to inform key parameters to address the policy question of interest to decision-makers. Given the limitations associated with the clinical evidence and absence of evidence to inform key model parameters, an economic evaluation would not be able to quantify all relevant incremental costs and effects of using rituximab over the currently used alternatives. CADTH will work with the plans to assist with tools to support policy decisions.
Interpretation of Clinical Results
The SR was undertaken at the request of the government-sponsored drug plans. While 18 RCTs were found, only the findings of 2 RCTs were pertinent to the review.
The evidence from the RI-CYLCO trial was inconclusive with regards to the comparative efficacy and safety between rituximab and CTX for any of the outcomes measured at any of the time points, given that the study was not powered to detect a clinically meaningful difference. The MENTOR trial showed that rituximab may be superior to CYC in terms of the following efficacy outcomes at 24 months: CR and the composite outcome of PR or CR. In this same trial, conclusions could not be drawn regarding the difference in time to remission and the difference in HRQoL among patients treated with rituximab versus CYC. In the 2 RCTs, rituximab showed a similar safety profile as CTX or CYC.
The clinical expert consulted for this review indicated that equity-related issues need to be taken into consideration when determining the place in therapy of rituximab:
Besides clinical efficacy, the decision to fund rituximab or not may also pose issues for equity. Patient surveys as well as clinical experience suggest that some younger female patients with MN may wish to become pregnant. Since cyclophosphamide has an age-dependent effect on fertility in females whereas rituximab does not,38 a decision against funding rituximab could be seen as disproportionately affecting women.
In addition, if cyclophosphamide is selected and used for treatment of MN in a woman who desires children, provincial drug plans may pay for costly medications like gonadotropin-releasing hormone (GnRH) agonists and the health ministry will likely bear the costs of other treatments and/or specialist visits to preserve fertility and/or allow assisted reproduction. It might be less costly to pay for rituximab in such cases, although this possibility was not addressed in the economic analysis.
On the other hand, if publicly funded drug plans do not pay for fertility preservation or assisted reproduction following cyclophosphamide treatment, then a decision not to fund rituximab may be inequitable in that individuals with private drug coverage may be able to mitigate the consequences of cyclophosphamide-induced infertility, whereas those with only public coverage will not.
Strengths and Limitations of the Systematic Review
Strengths
The SR was developed using robust methodology and a protocol was developed a priori and registered with the PROPSERO database. The scoping plan was posted for stakeholder feedback. While no patient groups provided comments on the scoping plan, a search of various social platforms was undertaken to gather patient perspectives, which were used to inform the protocol. All available RCTs were included, and this list was posted for stakeholder feedback.
Evidence collection, data extraction, and evaluation of the quality of the studies were done in duplicate, with conflicts adjudicated through discussion. Heterogeneity across trials were carefully assessed. The analytical approach for the NMA was aligned with ISPOR guidelines and employed a standard methodology.
Limitations
The number of trials that contributed to the NMA was limited and reported results that were highly heterogeneous across the network. Due to the limited size of the network, it was not possible to adequately account for the level of heterogeneity, and as a result, the variation around estimated effects were extremely wide and generally not informative. Thus, CADTH was unable to use the findings of the NMA to draw conclusions for the report.
Implementation Advice
To support local policy decisions, CADTH convened a panel of experts who provided guidance on initiation, discontinuation, and prescribing criteria. The comprehensive implementation advice is available in Appendix 11.
Conclusions and Implications for Decision- or Policy-Making
MN is an autoimmune disease and one of the most common causes of NS in adults. Approximately 80% of patients with MN are classified as PMN (or IMN). While no patient groups or individual patients responded to the call for feedback to better understand the challenges of PMN, excerpts from patients’ experiences and perspectives shared on social media and other online sources were considered. Patients indicated that preventing or delaying ESRD and dialysis were important treatment goals.
Current treatment options for PMN include rituximab and immunosuppressive therapies such as CYC, TAC, and CTX. Most patients indicated that they had tried immunosuppressive therapies before rituximab. Rituximab is not approved for the indication of PMN in Canada; hence, the government-sponsored drug plans requested a review to determine the place in therapy of rituximab to treat adult patients with PMN.
To answer the policy questions, an SR was conducted and 18 RCTs that met the inclusion criteria were identified; of these, 11 trials were included in the NMA. Due to various factors, the estimates obtained from the NMA models were considered unreliable for drawing conclusions. Therefore, a narrative synthesis of 2 head-to-head trials, MENTOR and RI-CYCLO, was conducted. The limited evidence from the available studies did not provide conclusive evidence on the comparative efficacy of rituximab versus CTX; however, the findings from the MENTOR study suggest that rituximab may be superior to CYC in terms of CR and the composite outcome of PR or CR. No conclusions could be drawn from the available evidence regarding the difference in time to remission and the difference in HRQoL among patients treated with rituximab versus CYC. Finally, in the 2 RCTs, rituximab showed a similar safety profile compared to CTX and CYC. Of note, the conclusions drawn from the results of the SR are limited as they are only based on 2 RCTs as no other head-to-head trials of rituximab were retrieved.
Given the small network of studies, the heterogeneity in the included studies, and the limited information provided by the NMA and the pairwise comparisons of the MENTOR and RI-CYCLO studies, CADTH was unable to conduct an informative economic evaluation. Further, in addition to the clinical evidence gaps, there were also issues identifying information to inform key parameters to address the policy question of interest to decision-makers. Given the limitations associated with the clinical evidence and absence of evidence to inform key model parameters, an economic evaluation would not be able to quantify all relevant incremental costs and effects of using rituximab over the currently used alternatives. CADTH will work with the plans to assist with tools to support policy decisions.
Patients whose views were obtained from social media and other online platforms expressed their willingness to try a treatment if it meant preventing or delaying disease progression. As well, the AE profile of the drugs needs to be taken into consideration when choosing a treatment. The clinical expert consulted for this review indicated that women of child-bearing age should be given special consideration for funding rituximab in light of the adverse fertility profile of CTX.
To support local policy decisions, CADTH convened a panel of experts who provided guidance on initiation, discontinuation, and prescribing criteria. These will be used by the public drug plans to make local funding decisions.
References
1.Angioi A, Lepori N, Lopez AC, Sethi S, Fervenza FC, Pani A. Treatment of primary membranous nephropathy: where are we now? J Nephrol. 2018;31(4):489-502. PubMed
2.Bomback AS, Fervenza FC. Membranous Nephropathy: Approaches to Treatment. Am J Nephrol. 2018;47 Suppl 1:30-42. PubMed
3.Wang CS, Greenbaum LA. Nephrotic Syndrome. Pediatr Clin North Am. 2019;66(1):73-85. PubMed
4.Rojas-Rivera JE, Carriazo S, Ortiz A. Treatment of idiopathic membranous nephropathy in adults: KDIGO 2012, cyclophosphamide and cyclosporine A are out, rituximab is the new normal. Clinical kidney journal. 2019;12(5):629-638. PubMed
5.Couser WG. Primary Membranous Nephropathy. Clin J Am Soc Nephrol. 2017;12(6):983-997. PubMed
6.KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Journal of the International Society of Nephrology. 2021;100(4S):281.
7.Fervenza FC, Appel GB, Barbour SJ, et al. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. New Engl J Med. 2019;381(1):36-46. PubMed
8.Scolari F, Delbarba E, Santoro D, et al. Rituximab or Cyclophosphamide in the Treatment of Membranous Nephropathy: The RI-CYCLO Randomized Trial. JASN. 2021;01.
9.Murphy G. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy: A Critical Appraisal of the MENTOR Study. 2020: https://cadth.ca/sites/default/files/hta-he/ha0001-mentor-study-focused-crititcal-appraisal.pdf. Accessed 2021 Sep 18.
10.Ogino MH, Tadi P. Cyclophosphamide. StatPearls. Treasure Island (FL): StatPearls Publishing; 2021: https://www.ncbi.nlm.nih.gov/books/NBK553087/. Accessed 2021 Nov 3.
11.PrProcytox, Cyclophosphamide tablets USP: 25mg, 50mg, Cyclophsphamide for injection: 200mg, 500mg, 1000mg, 2000mg (powder for injection) per vial [product monograph]. Mississauga (ON): Baxter Corporation; 2012: https://pdf.hres.ca/dpd_pm/00017604.PDF. Accessed 2021 Nov 3.
12.Donadio JV, Jr., Holley KE, Anderson CF, Taylor WF. Controlled trial of cyclophosphamide in idiopathic membranous nephropathy. Kidney Int. 1974;6(6):431-439. PubMed
13.PrNeoral (cyclosporine capsules) (cyclosporine oral solution) for rmicroemulsion and Pr Sandimmune I.V. (cyclosporiine for injection). Neoral (solution 100 mg/mL), Neoral (soft gelatin capsules, 10, 25, 50 and 100 mg), Sandimmune I.V. (50 mg/mL for injection) [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2015: https://pdf.hres.ca/dpd_pm/00028894.PDF. Accessed 2021 Nov 3.
14.PrPrograf, Tacrolimus for injection: 5mg/ml, tacrolimus immediate release capsules, USP: 0.5mg, 1mg and 5mg [product monograph]. Markham (ON): Astellas Pharma Canada, Inc.; 2020: https://pdf.hres.ca/dpd_pm/00059061.PDF. Accessed 2021 Nov 3.
15.CADTH Framework for Patient Engagement in Health Technology Assessment. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/cadth-framework-patient-engagement-health-technology-assessment. Accessed 2021 Nov 12.
16.Staniszewska S, Brett J, Simera I, et al. GRIPP2 reporting checklists: tools to improve reporting of patient and public involvement in research. BMJ. 2017;358. PubMed
17.Hoaglin DC, Hawkins N, Jansen JP, et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices—part 2. . Value Health. 2011;14(4):429-437. PubMed
18.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
19.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2020 Apr 28.
20.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019(366: l4898.).
21.Cattran DC, Appel GB, Hebert LA, et al. Cyclosporine in patients with steroid-resistant membranous nephropathy: a randomized trial. Kidney Int. 2001;59(4):1484-1490. PubMed
22.Omrani H, Golmohamadi S, Hichi F, Sadeghi M. Comparison of the efficacy of tacrolimus versus cyclosporine in the treatment of idiopathic membranous nephropathy. Nephro-Urology Monthly. 2017;9(1):e42473.
23.Chen M, Li H, Li XY, et al. Tacrolimus combined with corticosteroids in treatment of nephrotic idiopathic membranous nephropathy: a multicenter randomized controlled trial. Am J Med Sci. 2010;339(3):233-238. PubMed
24.Di J, Qian Q, Yang M, et al. Efficacy and safety of long-course tacrolimus treatment for idiopathic membranous nephropathy. Exp Ther Med. 2018;16(2):979-984. PubMed
25.He L, Peng Y, Liu H, et al. Treatment of idiopathic membranous nephropathy with combination of low-dose tacrolimus and corticosteroids. J Nephrol. 2013;26(3):564-571. PubMed
26.Hofstra JM, Branten A J, Wirtz JJ, et al. Early versus late start of immunosuppressive therapy in idiopathic membranous nephropathy: a randomized controlled trial. Nephrology Dialysis Transplantation. 2010;25(1):129-136. PubMed
27.Howman A, Chapman TL, Langdon MM, et al. Immunosuppression for progressive membranous nephropathy: a UK randomised controlled trial. Lancet. 2013;381(9868):744-751. PubMed
28.Jha V, Ganguli A, Saha TK, et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol. 2007;18(6):1899-1904. PubMed
29.Kosmadakis G, Filiopoulos V, Smirloglou D, et al. Comparison of immunosuppressive therapeutic regimens in patients with nephrotic syndrome due to idiopathic membranous nephropathy. Ren Fail. 2010;32(5):566-571. PubMed
30.Li QH, Yang ZJ, Li L, et al. Comparison of efficacy and safety between tacrolimus and cyclosporine combined with corticosteroids in patients with idiopathic membranous nephropathy: A randomized controlled trial. Int J Clin Exp Med. 2017;10(6):9764-9770.
31.Peng L, Wei SY, Li LT, He YX, Li B. Comparison of different therapies in high-risk patients with idiopathic membranous nephropathy. J Formosan Med Assoc. 2016;115(1):11-18. PubMed
32.Praga M, Barrio V, Juarez GF, Luno J, Grupo Espanol de Estudio de la Nefropatia M. Tacrolimus monotherapy in membranous nephropathy: a randomized controlled trial. Kidney Int. 2007;71(9):924-930. PubMed
33.Ramachandran R, Hn HK, Kumar V, et al. Tacrolimus combined with corticosteroids versus Modified Ponticelli regimen in treatment of idiopathic membranous nephropathy: Randomized control trial. Nephrology. 2016;21(2):139-146. PubMed
34.Ramachandran R, Yadav AK, Kumar V, et al. Two-Year Follow-up Study of Membranous Nephropathy Treated With Tacrolimus and Corticosteroids Versus Cyclical Corticosteroids and Cyclophosphamide. KI Rep. 2017;2(4):610-616. PubMed
35.Saito T, Iwano M, Matsumoto K, et al. Significance of combined cyclosporine-prednisolone therapy and cyclosporine blood concentration monitoring for idiopathic membranous nephropathy with steroid-resistant nephrotic syndrome: a randomized controlled multicenter trial. Clin Exp Nephrol. 2014;18(5):784-794. PubMed
36.Xu J, Zhang W, Xu Y, et al. Tacrolimus combined with corticosteroids in idiopathic membranous nephropathy: a randomized, prospective, controlled trial. Contrib Nephrol. 2013;181:152-162. PubMed
37.Yuan H, Liu N, Sun GD, Jia Y, Luo P, Miao LN. Effect of prolonged tacrolimus treatment in idiopathic membranous nephropathy with nephrotic syndrome. Pharmacology. 2013;91(5-6):259-266. PubMed
38.Pendergraft WF, McGrath MM, Murphy AP, et al. Fetal outcomes after rituximab exposure in women with autoimmune vasculitis. Ann Rheum Dis. 2013;72(12):2051-2053. PubMed
39.Austin HA, Illei GG, Braun MJ, Balow JE. Randomized, controlled trial of prednisone, cyclophosphamide, and cyclosporine in lupus membranous nephropathy. J Am Soc Nephrol. 2009;20(4):901-911. PubMed
40.Cameron JS, Healy MJR, Adu D. The Medical Research Council Trial of short-term high-dose alternate day prednisolone in idiopathic membranous nephropathy with nephrotic syndrome in adults. Q J Med. 1990;74(274):133-156. PubMed
41.Cattran DC, Delmore T, Roscoe J, et al. A randomized controlled trial of prednisone in patients with idiopathic membranous nephropathy. New Engl J Med. 1989;320(4):210-215. PubMed
42.Choi JY, Kim DK, Kim YW, et al. The Effect of Mycophenolate Mofetil versus Cyclosporine as Combination Therapy with Low Dose Corticosteroids in High-risk Patients with Idiopathic Membranous Nephropathy: a Multicenter Randomized Trial. J Korean Med Sci. 2018;33(9):e74. PubMed
43.Coggins CH. A controlled study of short-term prednisone treatment in adults with membranous nephropathy. Collaborative study of the adult idiopathic nephrotic syndrome. New Engl J Med. 1979;301(24):1301-1306. PubMed
44.D'Amico G, Remuzzi G, Maschio G, et al. Effect of dietary proteins and lipids in patients with membranous nephropathy and nephrotic syndrome. Clin Nephrol. 1991;35(6):237-242. PubMed
45.Fernandez-Juarez G, Rojas-Rivera J, Logt AV, et al. The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy. Kidney Int. 2021;99(4):986-998. PubMed
46.Guo Y, Wu X, Liu L, Zhang H, Yang L, Chen W. Efficacy of leflunomide combined with prednisone for the treatment of PLA2R-associated primary membranous nephropathy. Ren Fail. 2020;42(1):122-130. PubMed
47.Liang Q, Li H, Xie X, Qu F, Li X, Chen J. The efficacy and safety of tacrolimus monotherapy in adult-onset nephrotic syndrome caused by idiopathic membranous nephropathy. Ren Fail. 2017;39(1):512-518. PubMed
48.Nikolopoulou A, Condon M, Turner-Stokes T, et al. Mycophenolate mofetil and tacrolimus versus tacrolimus alone for the treatment of idiopathic membranous glomerulonephritis: a randomised controlled trial. BMC Nephrol. 2019;20(1):352. PubMed
49.Ponticelli C, Zucchelli P, Imbasciati E, et al. Controlled trial of monthly alternated courses of steroid and chlorambucil for idiopathic membranous nephropathy. Proc Eur Dial Transplant Assoc. 1983;19:717-723. PubMed
50.Ponticelli C, Zucchelli P, Imbasciati E, et al. Controlled trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. New Engl J Med. 1984;310(15):946-950. PubMed
51.Ponticelli C, Zucchelli P, Passerini P, et al. A randomized trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. New Engl J Med. 1989;320(1):8-13. PubMed
52.Ponticelli C, Zucchelli P, Passerini P, Cesana B. Methylprednisolone plus chlorambucil as compared with methylprednisolone alone for the treatment of idiopathic membranous nephropathy. The Italian Idiopathic Membranous Nephropathy Treatment Study Group. New Engl J Med. 1992;327(9):599-603. PubMed
53.Ponticelli C, Zucchelli P, Passerini P, et al. A 10-year follow-up of a randomized study with methylprednisolone and chlorambucil in membranous nephropathy. Kidney Int. 1995;48(5):1600-1604. PubMed
54.Ponticelli C, Altieri P, Scolari F, et al. A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol. 1998;9(3):444-450. PubMed
55.Reichert LJ, Huysmans FT, Assmann K, Koene RA, Wetzels JF. Preserving renal function in patients with membranous nephropathy: daily oral chlorambucil compared with intermittent monthly pulses of cyclophosphamide. Ann Intern Med. 1994;121(5):328-333. PubMed
56.Saito T, Iwano M, Matsumoto K, et al. Mizoribine therapy combined with steroids and mizoribine blood concentration monitoring for idiopathic membranous nephropathy with steroid-resistant nephrotic syndrome. Clin Exp Nephrol. 2017;21(6):961-970. PubMed
57.West ML, Jindal KK, Bear RA, Goldstein M. A controlled trial of cyclophosphamide in patients with membranous glomerulonephritis. Kidney Int. 1987;32(4):579-584. PubMed
Appendix 1: Patient Perspectives
Note that this appendix has not been copy-edited.
Table 6: Results based on the Guidance for Reporting Involvement of Patients and the Public short form (GRIPP2 SF)
Section and topic | Item | Reported on page |
|---|---|---|
Aim | As part of this health technology assessment, CADTH collected patient perspectives through a summary of findings from online sources, to better understand all aspects of the disease and experiences with rituximab, and to identify concepts and assumptions to be explored further by CADTH. | 11-12, 24-25 |
Methods | CADTH contacted the Kidney Foundation of Canada and the Canadian Organization for Rare Disorders to help identify interested individual patients with personal experience with taking rituximab as a treatment for PMN. The call for participants was also shared on CADTH’s and the Kidney Foundation of Canada’s Facebook pages. Despite our best efforts, we were unable to identify a patient who met those criteria. Alternatively, we opted to report patient experiences and perspectives through a summary of findings from various online sources. We searched for and gathered excerpts from patients’ experiences and perspectives shared publicly on social media and other online sources. A total of 37 quotations were found, from 27 individuals [Facebook (n = 13), YouTube (n = 4), Inspire social network for health (n = 4), Reddit (n = 3), RareRenal (n = 2), and patient blog (n = 1)]. Quotations were anonymized; no identifying information was gathered. | 11-12, 24-25 |
Study results | The researchers were made aware of several outcomes and themes, in particular: Disease experience: Patient age at diagnosis varied from 11 years to 64 years, but most reported being diagnosed before the age of 40. Patients experienced symptoms such as swollen and aching legs, shortness of breath, frothy urine, raised blood pressure, fatigue, and weight gain, which typically led to a series of tests and confirmation of diagnosis via biopsy. Variability of treatment experiences: Efficacy of rituximab, like calcineurin inhibitors and alkylating drugs, varied considerably from one patient to the next. Some patients were in remission, some saw minor improvements, some were unsuccessful with rituximab, and others were starting rituximab as a last resort after being unsuccessful with other treatments. Most patients appeared to have tried other treatments prior to rituximab. Willingness to endure side effects: Most patients who shared their experiences on online social platforms were willing to endure side effects if it meant there was a chance of remission. Overall, patients reported wanting to try a treatment that offered the possibility of preventing or delaying end-stage renal disease and dialysis. | 8-9, 11, 26 |
Discussion and conclusions | Finding a Canadian patient with experience taking rituximab for PMN was the main challenge for this project. In terms of finding and collating perspectives shared publicly online, there are limitations to this approach, mainly, finding (and confirming) patients’ diagnoses and finding (and confirming) demographic information, both of which would help provide context to the experiences being used to in this health technology assessment. | 25 |
Reflections/critical perspective | The value in this approach to capture patient insights allowed us to learn about patients’ experiences, albeit, in an unconventional way. We were also able to include multiple perspectives, to reflect a diversity of needs rather than a singular patient perspective. However, engaging with an individual patient would have allowed us to learn more about barriers to access and use of treatment, as well as patient-borne costs, to better understand the challenges that patients in Canada specifically, must face. Additionally, people with limited or no access to computers or the Internet, such as elderly patients, racial/ethnic minorities, and those of lower socioeconomic status, may be underrepresented. It is possible that the patients who shared their experiences on online platforms skewed toward a younger and more digitally connected population; therefore, this sample may not reflect the broad range of patients with PMN. | 25 |
Appendix 2: Literature Search Strategy
Note that this appendix has not been copy-edited.
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Cochrane Central Register of Controlled Trials (CCTR)
Note: Subject headings will be customized for each database. Duplicates between databases will be removed in Ovid.
Date of Search: April 22, 2020
Alerts: Monthly search updates will be run until project completion
Study Types: Randomized controlled trials; controlled clinical trials
Limits:
Language limit: English and French-language
Conference abstracts excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ab | Abstract |
.dq | Candidate term word |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.pt | Publication type |
.mp | Mapped term |
.jw | Journal word title |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
glomerulonephritis, membranous/ or Heymann Nephritis Antigenic Complex/
((extra?membranous or membranous) adj5 (nephropath* or Glomerulo* or nephritis)).ti,ab,kf.
((glomerular basement membrane* or glomerular membrane basement or GMB or GBM) adj5 thick*).ti,ab,kf.
((PMN or MGN) adj5 (nephropath* or kidney* or glomerul*)).ti,ab,kf.
(Heymann* adj2 Nephritis).ti,ab,kf.
1 or 2 or 3 or 4 or 5
6 use medall
membranous glomerulonephritis/ or Heymann nephritis/
((extra?membranous or membranous) adj5 (nephropath* or Glomerulo* or nephritis)).ti,ab,kw,dq.
((glomerular basement membrane* or glomerular membrane basement or GMB or GBM) adj5 thick*).ti,ab,kw,dq.
((PMN or MGN) adj5 (nephropath* or kidney* or glomerul*)).ti,ab,kw,dq.
(Heymann* adj2 Nephritis).ti,ab,kw,dq.
8 or 9 or 10 or 11 or 12
13 use oemezd
7 or 14
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
"Randomized Controlled Trial (topic)"/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
"Controlled Clinical Trial (topic)"/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf,kw.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf,kw.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf,kw.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf,kw.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf,kw.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(pragmatic study or pragmatic studies).ti,ab,hw,kf,kw.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf,kw.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf,kw.
(phase adj3 (III or "3") adj3 (study or studies or trial*)).ti,hw,kf,kw.
or/16-46
15 and 47
48 and (english or french).lg.
exp animals/
exp animal experimentation/ or exp animal experiment/
exp models animal/
nonhuman/
exp vertebrate/ or exp vertebrates/
or/50-54
exp humans/
exp human experimentation/ or exp human experiment/
or/56-57
55 not 58
49 not 59
glomerulonephritis, membranous/ or Heymann Nephritis Antigenic Complex/
((extra?membranous or membranous) adj5 (nephropath* or Glomerulo* or nephritis)).ti,ab,kw.
((glomerular basement membrane* or glomerular membrane basement or GMB or GBM) adj5 thick*).ti,ab,kw.
((PMN or MGN) adj5 (nephropath* or kidney* or glomerul*)).ti,ab,kw.
(Heymann* adj2 Nephritis).ti,ab,kw.
61 or 62 or 63 or 64 or 65
66 use cctr
60 or 67
68 not conference abstract.pt.
remove duplicates from 69
Clinical Trial Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
Search updated prior to the completion of stakeholder feedback period.
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search updated prior to the completion of stakeholder feedback period.
Grey Literature
Search dates: May 2020
Keywords: Membranous nephropathy, glomerular basement membrane, GBM thickening, extramembranous nephropathy, Glomerular disease
Limits: English and French-language only documents
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool For Searching Health-Related Grey Literature (https://www.cadth.ca/grey-matters) will be searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug Class Reviews
Clinical Trial Registries
Databases (free)
Internet Search
Open Access Journals
Appendix 3: PRISMA
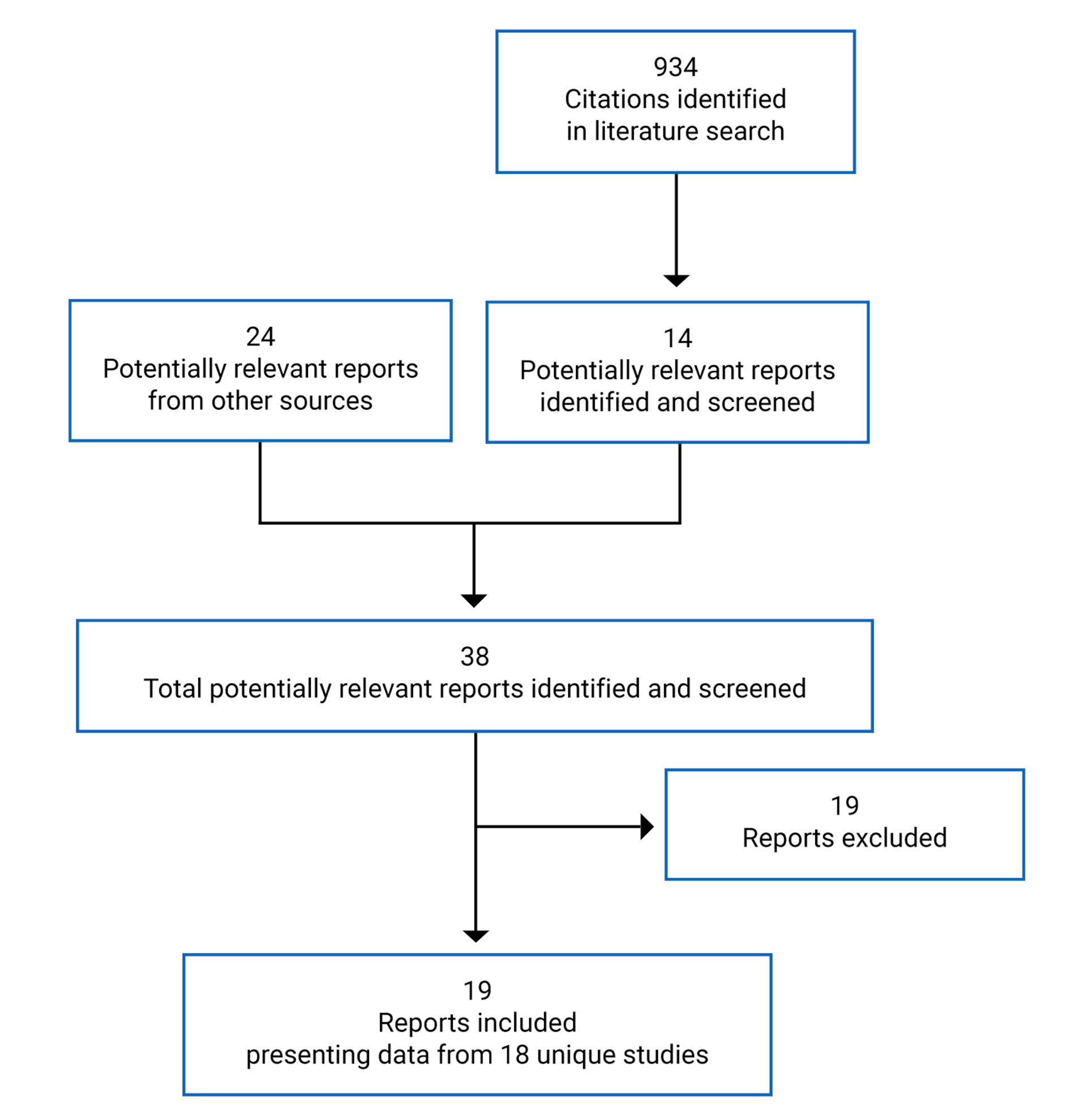
Appendix 4: List of Included Studies
Note that this appendix has not been copy-edited.
1.Cattran DC, Appel GB, Hebert LA, et al. Cyclosporine in patients with steroid-resistant membranous nephropathy: a randomized trial. Kidney Int. 2001;59(4):1484‐1490. PubMed
2.Chen M, Li H, Li XY, et al. Tacrolimus combined with corticosteroids in treatment of nephrotic idiopathic membranous nephropathy: a multicenter randomized controlled trial. Am J Med Sci. 2010;339(3):233‐238. PubMed
3.Di J, Qian Q, Yang M, et al. Efficacy and safety of long-course tacrolimus treatment for idiopathic membranous nephropathy. Exp Ther Med. 2018;16(2):979‐984. PubMed
4.Fervenza FC, Appel GB, Barbour SJ, et al. Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med. 2019;381(1):36‐46. PubMed
5.He L, Peng Y, Liu H, et al. Treatment of idiopathic membranous nephropathy with combination of low-dose tacrolimus and corticosteroids. J Nephrol. 2013 May-Jun;26(3):564-71. PubMed
6.Hofstra JM, Branten AJW, Joris JJM, et al. Early versus late start of immunosuppressive therapy in idiopathic membranous nephropathy: a randomized controlled trial. Nephrol Dial Transplant. 2010 Jan;25(1):129-36. PubMed
7.Howman A, Chapman TL, Langdon MM, et al. Immunosuppression for progressive membranous nephropathy: a UK randomised controlled trial. Lancet. 2013 Mar 2;381(9868):744-51. PubMed
8.Jha V, Ganguli A, Saha TK, et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol. 2007 Jun;18(6):1899-904. PubMed
9.Kosmadakis G, Filiopoulos V, Smirloglou D, et al. Comparison of immunosuppressive therapeutic regimens in patients with nephrotic syndrome due to idiopathic membranous nephropathy. Ren Fail. 2010 Jun;32(5):566-71. PubMed
10.Li Q, Yang Z, Li L, et al. Comparison of efficacy and safety between tacrolimus and cyclosporine combined with corticosteroids in patients with idiopathic membranous nephropathy: a randomized controlled trial. Int J Clin Exp Med. 2017;10(6):9764-9770.
11.Omrani H, Golmohamadi S, Hichi F, et al. Comparison of the efficacy of tacrolimus versus cyclosporine in the treatment of idiopathic membranous nephropathy. Nephrourol Mon. 2016; 9(1):e42473.
12.Peng L, Wei S, Li L, et al. Comparison of different therapies in high-risk patients with idiopathic membranous nephropathy. J Formos Med Assoc. 2016 Jan;115(1):11-8. PubMed
13.Praga M, Barrio V, Juárez GF, et al. Tacrolimus monotherapy in membranous nephropathy: a randomized controlled trial. Kidney Int. 2007 May;71(9):924-30. PubMed
14.Ramachandran R, Hn HK, Kumar V, et al. Tacrolimus combined with corticosteroids versus Modified Ponticelli regimen in treatment of idiopathic membranous nephropathy: Randomized control trial. Nephrology (Carlton). 2016 Feb;21(2):139-46. PubMed
15.Ramachandran R, Yadav AK, Kumar V, et al. Two-year follow-up study of membranous nephropathy treated with tacrolimus and corticosteroids versus cyclical corticosteroids and cyclophosphamide. Kidney Int Rep. 2017 Feb 9;2(4):610-616. PubMed
16.Saito T, Iwano M, Matsumoto K, et al. Significance of combined cyclosporine-prednisolone therapy and cyclosporine blood concentration monitoring for idiopathic membranous nephropathy with steroid-resistant nephrotic syndrome: a randomized controlled multicenter trial. Clin Exp Nephrol. 2014 Oct;18(5):784-94. PubMed
17.Xu J, Zhang W, Xu Y, et al. Tacrolimus combined with corticosteroids in idiopathic membranous nephropathy: a randomized, prospective, controlled trial. Contrib Nephrol. 2013;181:152-62. PubMed
18.Yuan H, Liu N, Sun GD, et al. Effect of prolonged tacrolimus treatment in idiopathic membranous nephropathy with nephrotic syndrome. Pharmacology. 2013;91(5-6):259-66. PubMed
19.Scolari F, Delbarba E, Santoro D, et al.; RI-CYCLO Investigators. Rituximab or Cyclophosphamide in the Treatment of Membranous Nephropathy: The RI-CYCLO Randomized Trial. J Am Soc Nephrol. 2021 Mar 1
Appendix 5: List of Excluded Studies
Note that this appendix has not been copy-edited.
Table 8: List of Excluded Studies and Reasons for Exclusion
Excluded studies | Reasons for exclusion |
|---|---|
Austin et al. 200939 | Patient population not relevant |
Cameron et al.200940 | Intervention/comparator not relevant |
Cattran et al.198941 | Intervention/comparator not relevant |
Choi et al. 201842 | Intervention/comparator not relevant |
Coggins et al. 197943 | Intervention/comparator not relevant |
D'Amico et al. 199144 | Intervention/comparator not relevant |
Fernandez-Juarez et al. 202145 | Intervention/comparator not relevant |
Guo et al. 202046 | Intervention/comparator not relevant |
Liang et al. 201747 | Study design not relevant |
Nikolopoulou et al. 201948 | Intervention/comparator not relevant |
Ponticelli et al. 198249 | Intervention/comparator not relevant |
Ponticelli et al. 198450 | Intervention/comparator not relevant |
Ponticelli et al. 198951 | Intervention/comparator not relevant |
Ponticelli et al. 199252 | Intervention/comparator not relevant |
Ponticelli et al. 199553 | Intervention/comparator not relevant |
Ponticelli et al. 199854 | Intervention/comparator not relevant |
Reichert et al. 199455 | Intervention/comparator not relevant |
Saito et al. 200756 | Intervention/comparator not relevant |
West et al. 198757 | Study design not relevant |
Appendix 6: Study Characteristics
Note that this appendix has not been copy-edited.
Table 9: Characteristics of the Included Randomized Controlled Trials
Study | Blinding | Number of centres (countries) | Total sample size | Follow-up |
|---|---|---|---|---|
Cattran et al. 2001 | single-blind | 11 (North America) | 51 | 78 weeks |
Chen et al. 2010 | open label | 6 (China) | 73 | 12 months |
Di et al. 2018 | not specified | 1 (China) | 76 | 24 months |
Fervenza et al. 2019 | open label | 22 (North America) | 130 | 24 months |
He et al. 2013 | open label | 1 (China) | 56 | 12 months |
Hofstra et al. 2010 | open label | multi centre (Netherlands) | 26 | 12 months |
Howman et al. 2013 | open label | 37 (UK) | 103 | 3 years |
Jha et al. 2007 | open label | 1 (India) | 93 | 10 years |
Kosmadakis et al. 2010 | open label | 1 (Greece) | 28 | 9 months |
Li et al. 2017 | not specified | 1 (China) | 31 | 6 months |
Omrani et al. 2017 | double-blind | 1 (Iran) | 68 | 6 months |
Peng et al. 2016 | not specified | 1 (China) | 90 | 9 months |
Praga et al. 2007 | open label | 13 (Spain) | 48 | 18 months |
Ramachandran et al. 2016 /17 | open label | 1 (India) | 70 | 24 months |
Saito et al. 2014 | open label | 30 (Japan) | 48 | 48 weeks |
Scolari et al. 2021 | open label | 11 (10 in Italy, 1 in Switzerland) | 74 | 6 months |
Xu et al. 2013 | not specified | 1 (China) | 100 | 18 months |
Yuan et al. 2013 | not specified | 1 (China) | 42 | 24 months |
Appendix 7: Interventions and Comparators
Table 10: Treatment Regimens in Included Studies
Study | Treatment regimen | Comparator regimen | Concomitant and background therapy for BP | |
|---|---|---|---|---|
Cattran et al. 2001 | Oral CYC 3.5 mg/kg/day [q12h] for 26 weeks then tapered off over 4 weeks period Target levels of 125 to 225 µg/L | Placebo 0.035 mL/kg/day [q12h] for 26 weeks then tapered off over 4 weeks period | Concomitant: Prednisone 0.15 mg/kg/day for 26 weeks (maximum dose 15 mg) then tapered off by thirds at 4-week intervals and stopped after 8 weeks. Background: Continuation of ACEI/ARB regimen as per usual – patients previously taking ACEI/ARB dose maintained their dose, patients not taking ACEI/ARB prescribed other antihypertensives, introduction of ACEI/ARB was forbidden, other dietary modifications made | |
Chen et al. 2010 | TAC (0.1 mg/kg/day [q12h] for 9 months 1st 6 months – plasma concentration 5‑10 μg/l Last 3 months – plasma concentration 2-5 μg/l | Oral CTX 100 mg/d for 4 months (accumulated dose 12 g) Dosage reduced by 50 mg/d if WBC < 4000/μl (dosage increased when WBC returned to the normal range) | Concomitant: Oral prednisone 1 mg/kg/d for 4 weeks, tapered gradually, and discontinued by 8 months Background: Continuation of ACEI/ARB regimen as per usual – patients previously taking ACEI/ARB dose maintained their dose, patients not taking ACEI/ARB prescribed other antihypertensives to maintain target BP 125/75 mm Hg | |
Di et al. 2018 | Short-course: Oral TAC (0.1 mg/kg/day) [q12h] for 12 months 1st 6 months – plasma concentration 5‑10 μg/l After 6 months – plasma concentration 2-4 μg/l | Long-course: Oral TAC (0.1 mg/kg/day) for 24 months 1st 6 months - plasma concentration 5‑10 μg/l After 6 months – plasma concentration 2-4 μg/l | Concomitant: Prednisone (0.5 mg/kg/day), dose reduced by 5 mg every 4 weeks, then maintained at a total of 10 mg/day | |
Fervenza et al. 2019 | Rituximab 1,000 mg IV on days 1 and 15, no second dose if complete remission achieved, second course administered if proteinuria reduced from baseline by ≥ 25% at 6 months | Oral CYC 3.5 mg/kg/day [q12h] for 6 months then tapered off over 2 months period (applicable for complete remission or failure, partial remission repeated treatment for 6 months); plus Prednisone 0.15 mg/kg/day for 26 weeks then tapered off for 8 weeks Target CYC levels of 125 to 175 ng/L | No concomitant corticosteroids Background: Renin–angiotensin system blockers, BP management targeting < 130/80 mm Hg, dietary sodium restriction to < 4 g/day, and dietary protein restriction to 0.8 – 1 g of protein/kg/d 3 months prior treatment | |
He et al. 2013 | Oral TAC 1 mg/day for 1 week then alternating 1 mg and 2 mg per day [q12h] for 12 months + Prednisone (1 mg/kg/day for 4 weeks tapered) Plasma concentration at 2-4 ng/l | IV CTX 750 mg/m2 once every 4 weeks for 24 weeks + Prednisone (1 mg/kg/day for 4 weeks tapered) | Concomitant: Oral prednisone 1 mg/kg/day for 4 weeks (max 60 mg/day) Prednisone tapered gradually by 5 mg/2 weeks till 30 mg/day, then further tapered at 5 mg/month rate till 10 mg/d for the rest of the 12 months treatment period Background: ACEI/ARB or other antihypertensives as needed to meet target BP of 125/75 mm Hg | |
Hofstra et al. 2010 | Early start (immediately after randomization): Oral CTX 1.5 mg/kg/day for 12 months | Late start (when renal function deteriorated): Same as early start group | Concomitant: methylprednisolone 1 g intravenously on Days 1, 2, 3, 60, 61, 62, 120, 121 and 122, and oral prednisone 0.5 mg/kg/day for 6 months, prednisone tapered at 5 mg/week rate Background: ACEI/ARB as needed to maintain target BP of 130/80 mmHg, 3-Hydroxy-3- methylglutaryl coenzyme A reductase inhibitors to decrease serum cholesterol levels, moderately salt-restricted diet, anticoagulant drugs (not routinely), famotidine for GI symptoms, trimethoprim–sulfamethoxazole for pneumocystis jiroveci pneumonia | |
Howman et al. 2013 | CYC 5 mg/kg/day for 12 months Target levels of 100 to 200 µg/L, dose reduced if toxicity developed | Prednisolone: IV methyl prednisolone 1 g/day for 3 consecutive days then oral prednisolone 0.5 mg/kg/day for 28 days during months 1, 3, and 5; plus Oral chlorambucil: 0.15 mg/kg/day during months 2, 4, and 6, dose reduced if leucopenia developed | Concomitant: renin-angiotensin blockade, statins, and anticoagulants as needed Background: ACEI, ARB, and other BP control medications as needed. | |
Jha et al. 2007 | 6-month course of alternate months of steroid and CTX: IV methylprednisolone 1 g/d for 3 consecutive days followed by oral prednisolone 0.5 mg/kg/d for 27 d in month 1, 3, and 5 and oral CTX at 2 mg/kg/d in month 2, 4, and 6 | Supportive therapy: Dietary sodium restriction, diuretics, and antihypertensive agents | Background: ACEI, ARB were not allowed in the first year | |
Kosmadakis et al. 2010 | Oral CYC 3–3.5 mg/kg/day plus oral methylprednisolone 12.5 mg/day for 9 months Target levels of 100 to 120 μg/L ACEI, ARB not allowed | Oral CTX 2 mg/kg/day plus oral methylprednisolone 1.5 mg/kg/48 hour for 9 months (dose adjusted per WBC count) ACEI, ARB not allowed | ACEI: lisinopril for 9 months | Background: low-sodium diet (5 g/day), doses of loop diuretics if indicated, antihypertensive agents (beta-blockers and/or dihydropyridine calcium-blockers), to maintain BP of 140/90 mmHg |
Li et al. 2017 | TAC 0.05-0.1 mg/kg/day [q12h]) for 6 months Plasma concentration 5‑10 ng/mL | CYC 3–5 mg/kg/day [q12h] for 6 months Target levels of 100 to 200 ng/mL | Concomitant: Oral Prednisone 0.5 mg/ kg/d, tapered at 5 mg/month rate till 10 mg/d, maintained through 6 month | |
Omrani et al. 2017 | TAC 0.05 mg/kg/d for 6 months. | CYC 3 - 6 mg/kg/d for 6 months | Concomitant: low dose of prednisolone for 6 months | |
Peng et al. 2016 | TAC 0.05 mg/kg/day [q12h] for 9 months + corticosteroid (0.5 mg/kg/d [tapered down after 2 months] 1st 6 months - plasma concentration 4–8 ng/l Last 3 months – plasma concentration 2-4 ng/l | IV CTX 750 mg/m2 once a month for 6 months, then reduced to every 3 months | Oral MMF 1.5 to 2.0 g/d | Concomitant: Oral corticosteroid 0.5 mg/kg/d (TAC) or 1 mg/kg/d (CTX and MMF) for 2 months Corticosteroids tapered by 5 mg/d every 2 weeks until 20 mg/d then tapered to zero based on patient’s condition Background: Antihypertensive agents to maintain target BP (130/80 mm Hg). ACEI/ARB not initiated, but continued among previous users. Anticoagulant drugs and simvastatin as needed. |
Praga et al. 2007 | TAC 0.05 mg/kg/d [q12h] for over 12 months with a 6-month taper by 25% Plasma concentration 3–5 ng/mL, or 5–8 ng/mL if remission not achieved by 2 months | Control group (no information available) | Background: Antihypertensive agents to maintain target BP (130/80 mm Hg). ACEI/ARB dose continued among previous users. Statins as indicated. | |
Ramachandran et al. 2016 /17 | Oral TAC 0.1 mg/kg/day [q12h] for 12 months plus prednisolone 0.5 mg/kg/day for 6 months, tapered by 0.1 mg/kg/week 1st 6 months – plasma concentration 5–10 ng/mL Last 6 months – plasma concentration 4–8 ng/mL | Modified Ponticelli regimen: IV methylprednisolone 1 g/d for 3 consecutive days followed by oral prednisolone 0.5 mg/kg/d for 27 d in month 1, 3, 5 and oral CTX at 2 mg/kg/d in month 2, 4, 6. | Background: All received max ACEI or ARBs and statins | |
Saito et al. 2014 | CYC 2 - 3 mg/kg/d [once daily] for 48 weeks | CYC 1.5 mg/kg/12h for 48 weeks | Concomitant: Prednisolone 40 mg/day, tapered until <10 mg/day by 48 weeks | |
Scolari et al. 2021 | Rituximab 1,000 mg IV on days 1 and 15 | Cyclical corticosteroid/CTX therapy: 3 consecutive cycles of 2-month duration each (for a total of 6 months) Month 1, 3, 5: 1 g IV methylprednisolone daily for 3 days, thereafter oral methylprednisolone (0.4 mg/kg/day) or prednisone (0.5 mg/ kg/day) for the remainder of the month. Month 2, 4, 6: Oral CTX daily (2.0 mg/kg/day) | Any medications not in the list of exclusion, at the discretion of the investigator | |
Xu et al. 2013 | TAC (0.1 mg/kg/day) for 10 months plus prednisolone (0.5 mg/kg per day [then tapered] | IV CTX 0.5–0.75 g/m2/month (maximum dosage 1.0 g/month) once a month for 6 months, then reduced to every 3 months Oral prednisone 0.5-1 mg/kg/d for 2 months | Background: ACEI, ARB allowed for BP control | |
Yuan et al. 2013 | Short-term: TAC 0.05-0.08 mg/kg/day [q12h] for 6 months Plasma concentration 5–8 ng/ml (maximum 0.15 mg/kg/day) | Long-term: TAC 0.05-0.08 mg/kg/day [q12h] for 24 months, tapered by 2 mg/day during months 6–12, then 0.5 mg BID for months 12–24 | Concomitant: Oral prednisone 30 mg/day for 8 weeks, tapered by 5 mg every 4 weeks till 10 mg/day, maintained throughout Background: NSAIDS, ACEI, ARB use prohibited, except for prior user of ACEI or ARB | |
ACEI = Angiotensin-converting enzyme; ARB = Angiotensin II receptor blockers; CYC = cyclosporine; CR= complete remission; CTX = cyclophosphamide; IV = intravenous; MMF = mycophenolate mofetil; NSAID= nonsteroidal anti inflammatory drug; q12h = every 12 hours; TAC = tacrolimus; WBC = white blood cell
Note that this appendix has not been copy-edited.
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Scolari et al.8 Xu et al.,36 Yuan et al.37
Appendix 8: Baseline Patient Characteristics
Note that this appendix has not been copy-edited.
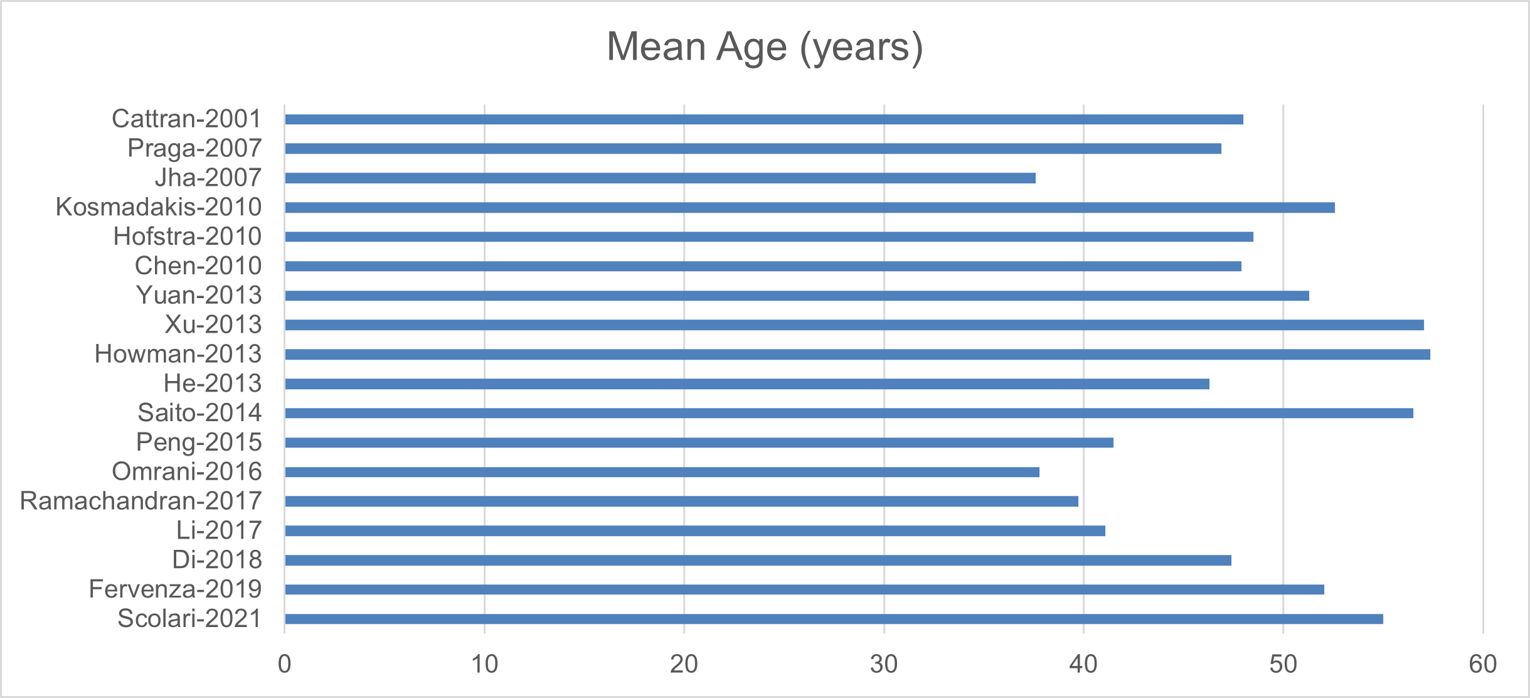
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
Figure 3: Estimated Glomerular Filtration Rate
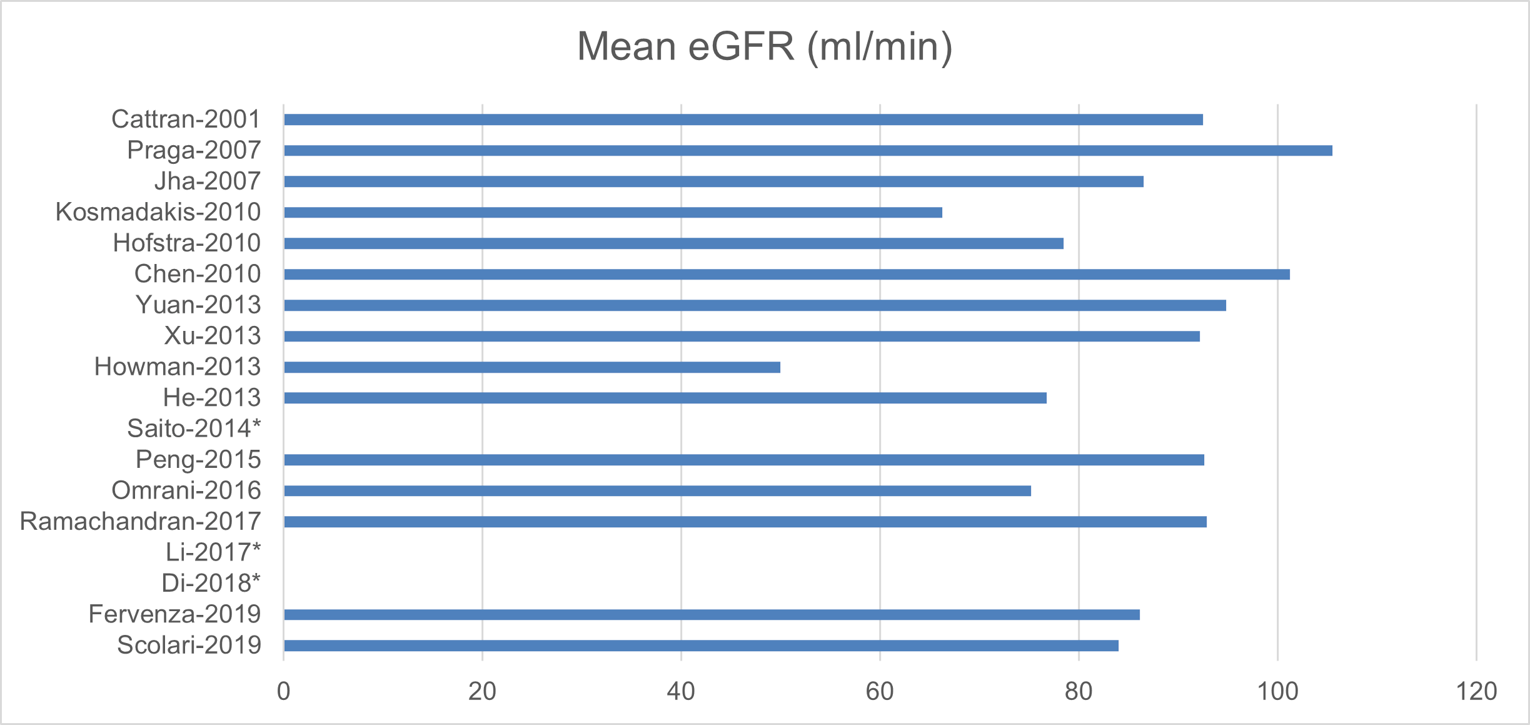
*Data not reported
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
Figure 4: Systolic Blood Pressure
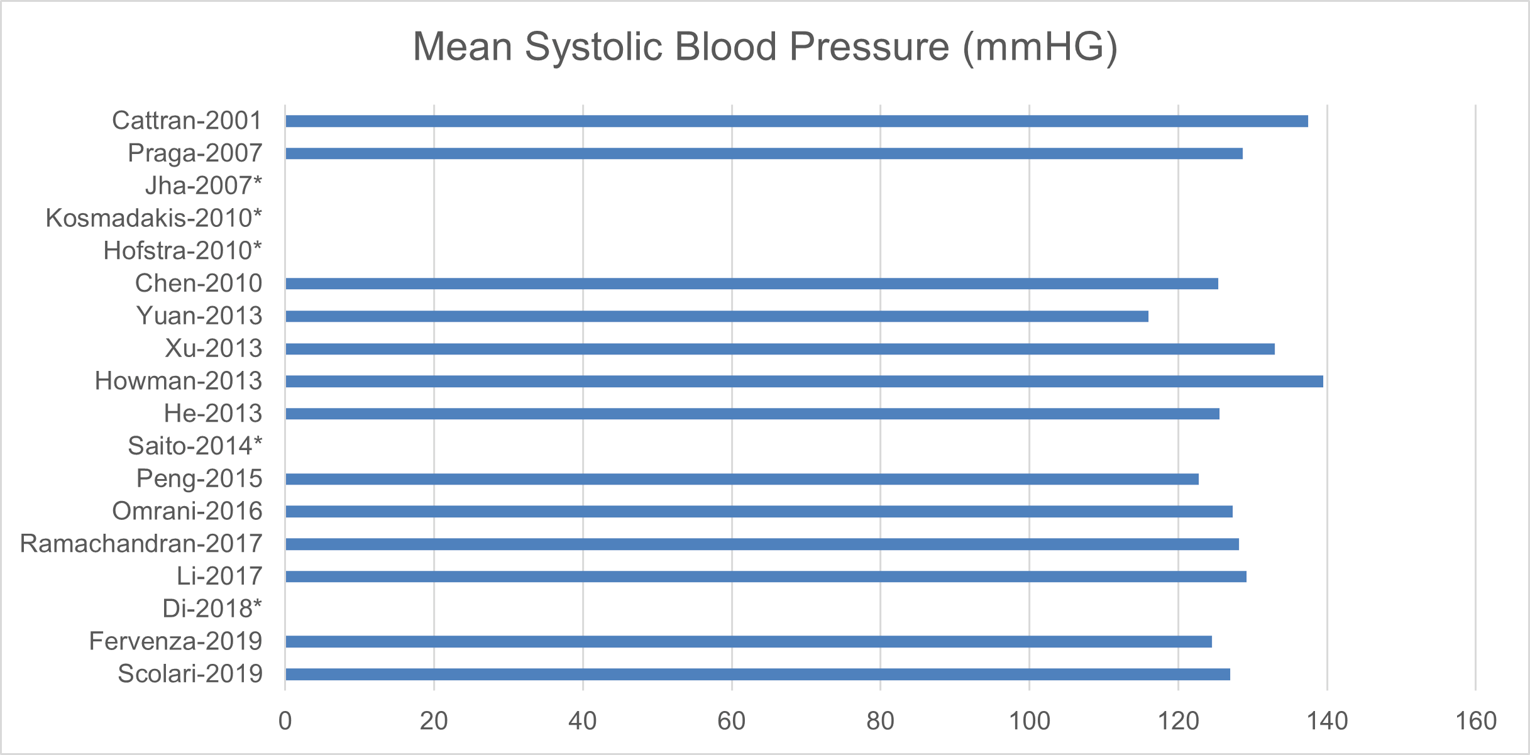
*Data not reported
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
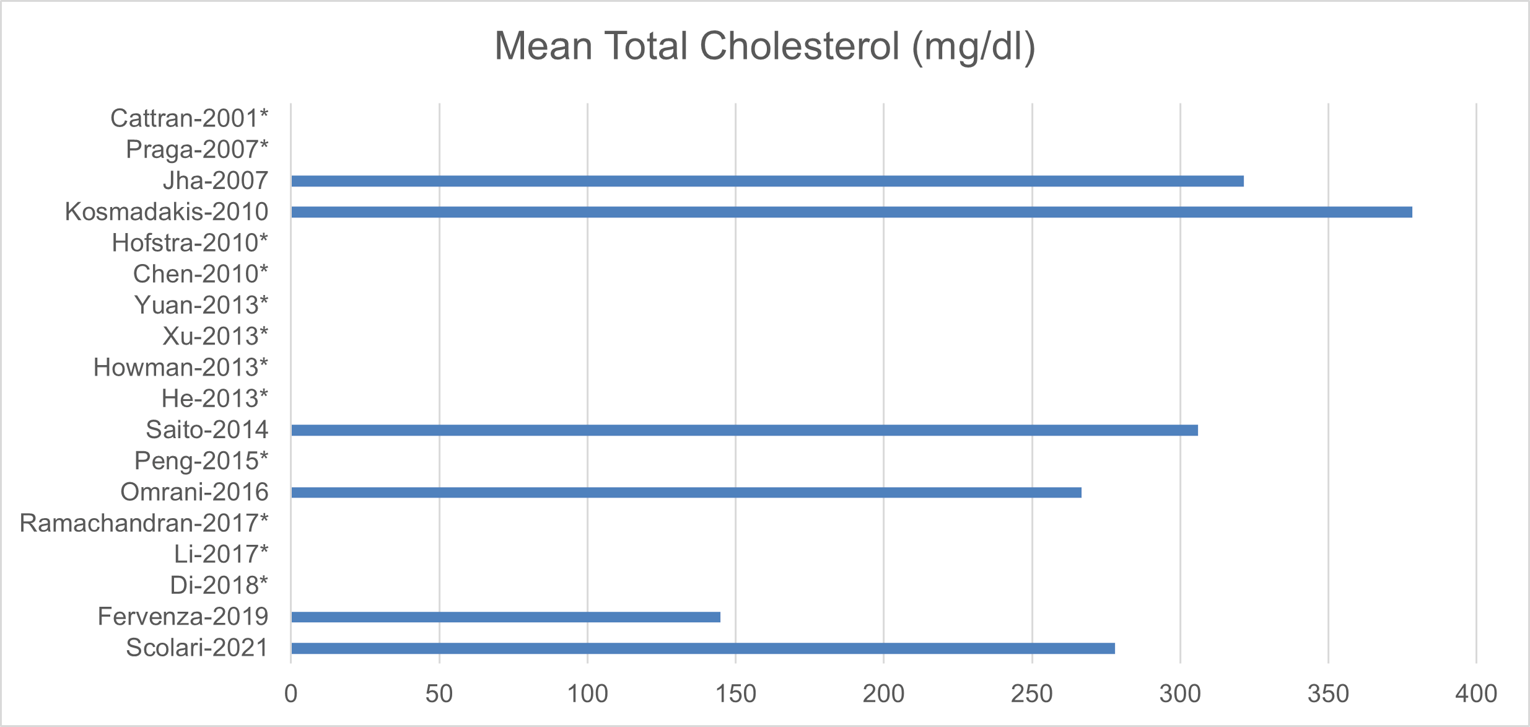
*Data not reported
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
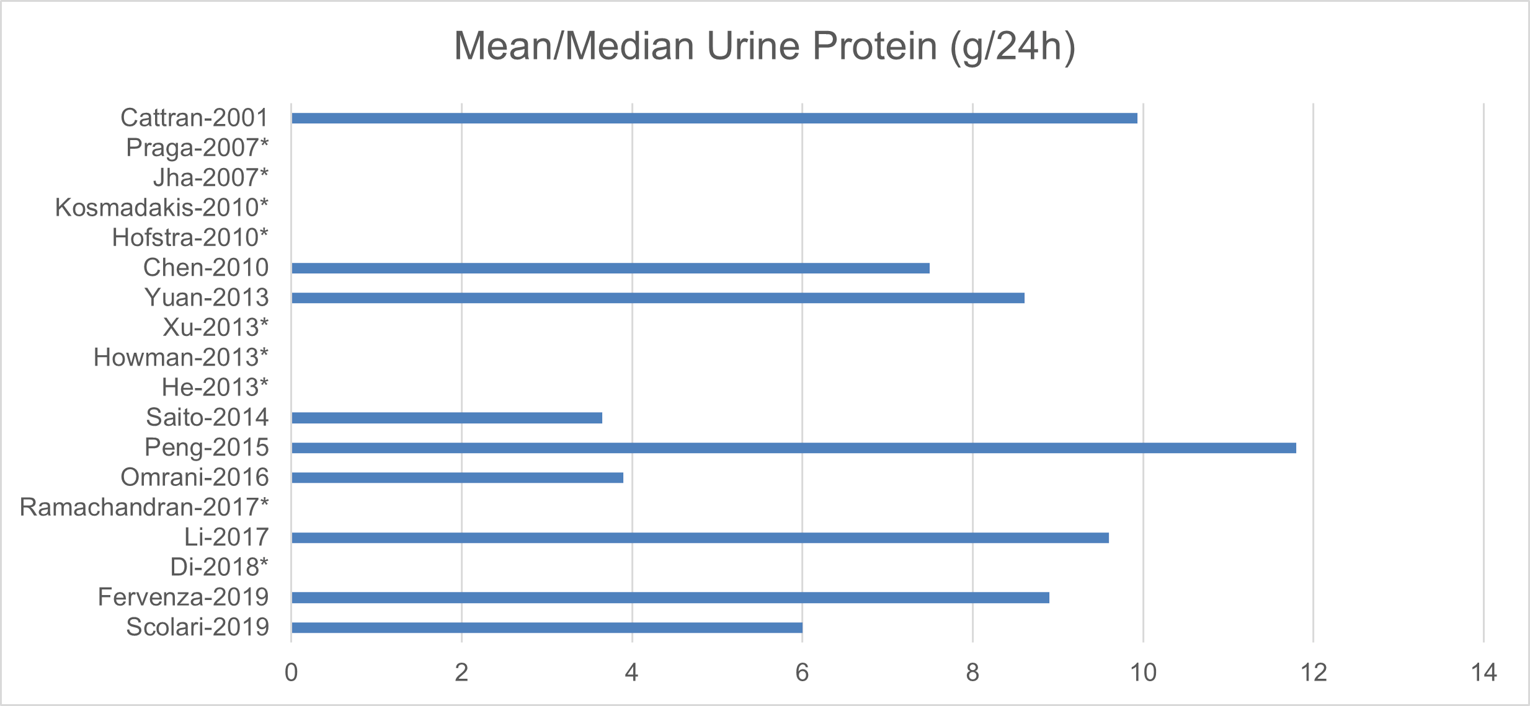
*Data not reported
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
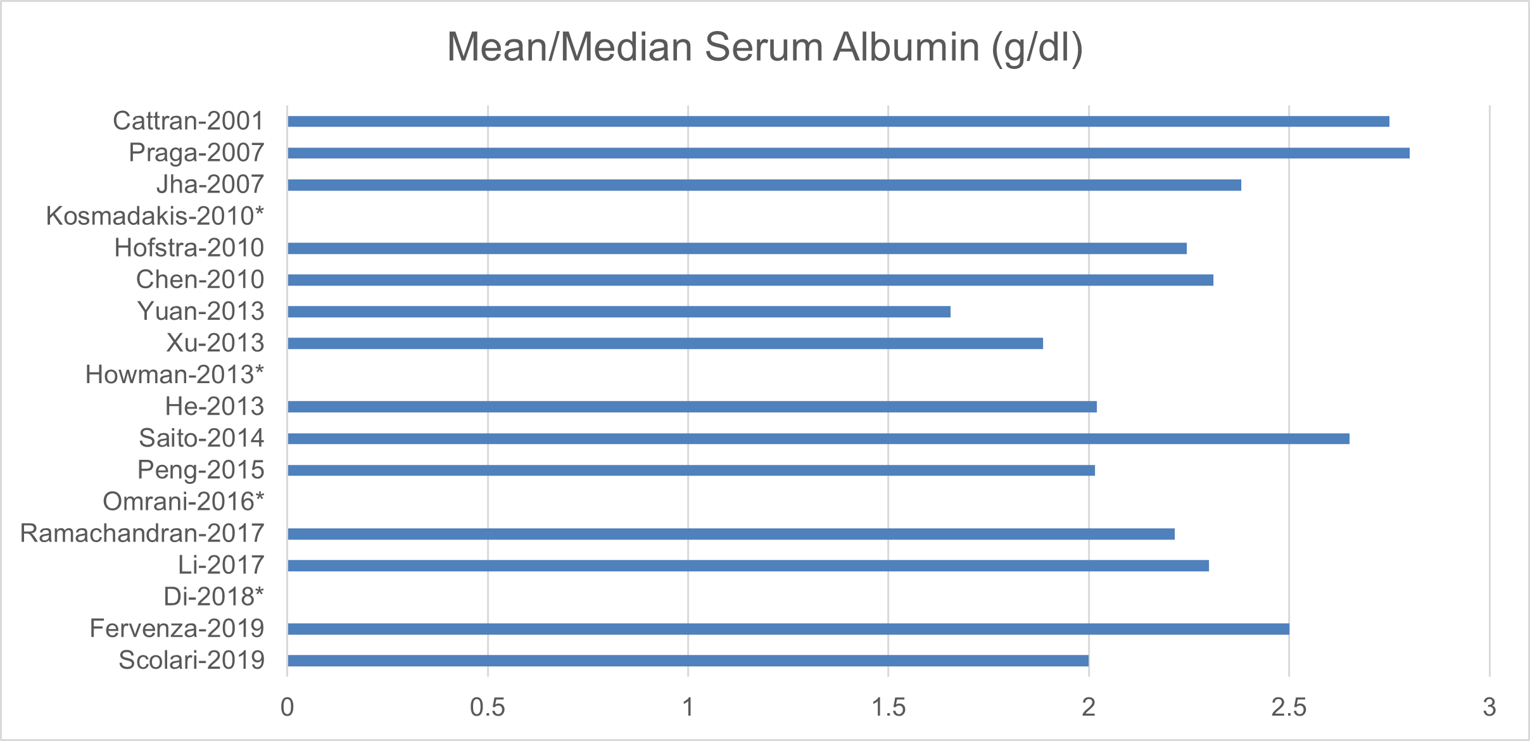
*Data not reported
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37 Scolari et al.8
Appendix 9: Network Meta-Analysis
Note that this appendix has not been copy-edited.
The NMA was based on comparisons between rituximab and TAC, CYC, CTX, and supportive care. Odds ratios were calculated for each rituximab treatment comparison. It should be noted that the results of the NMA cannot support any conclusions due to the high degree of uncertainty as demonstrated by the wide credible intervals (CrI).
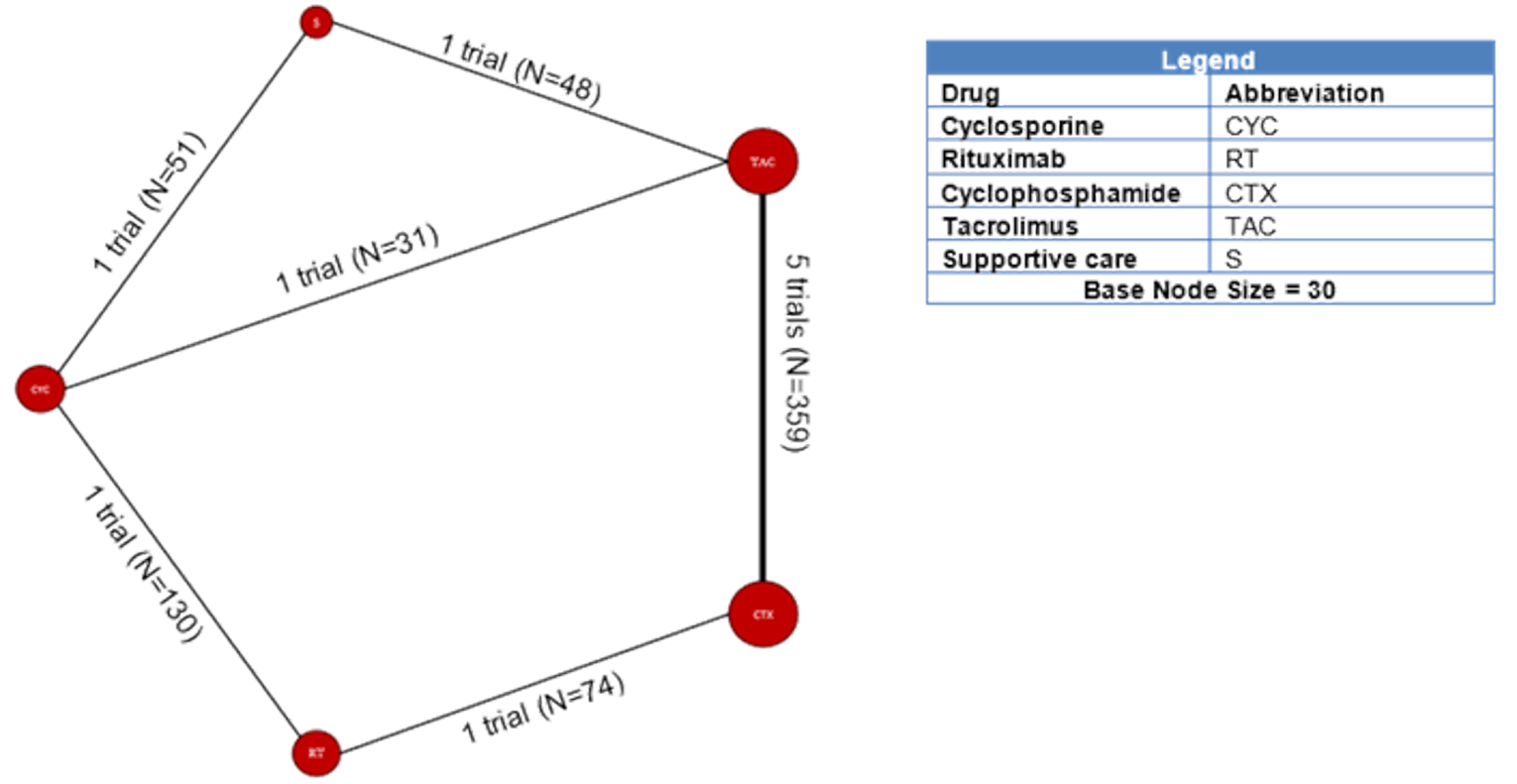
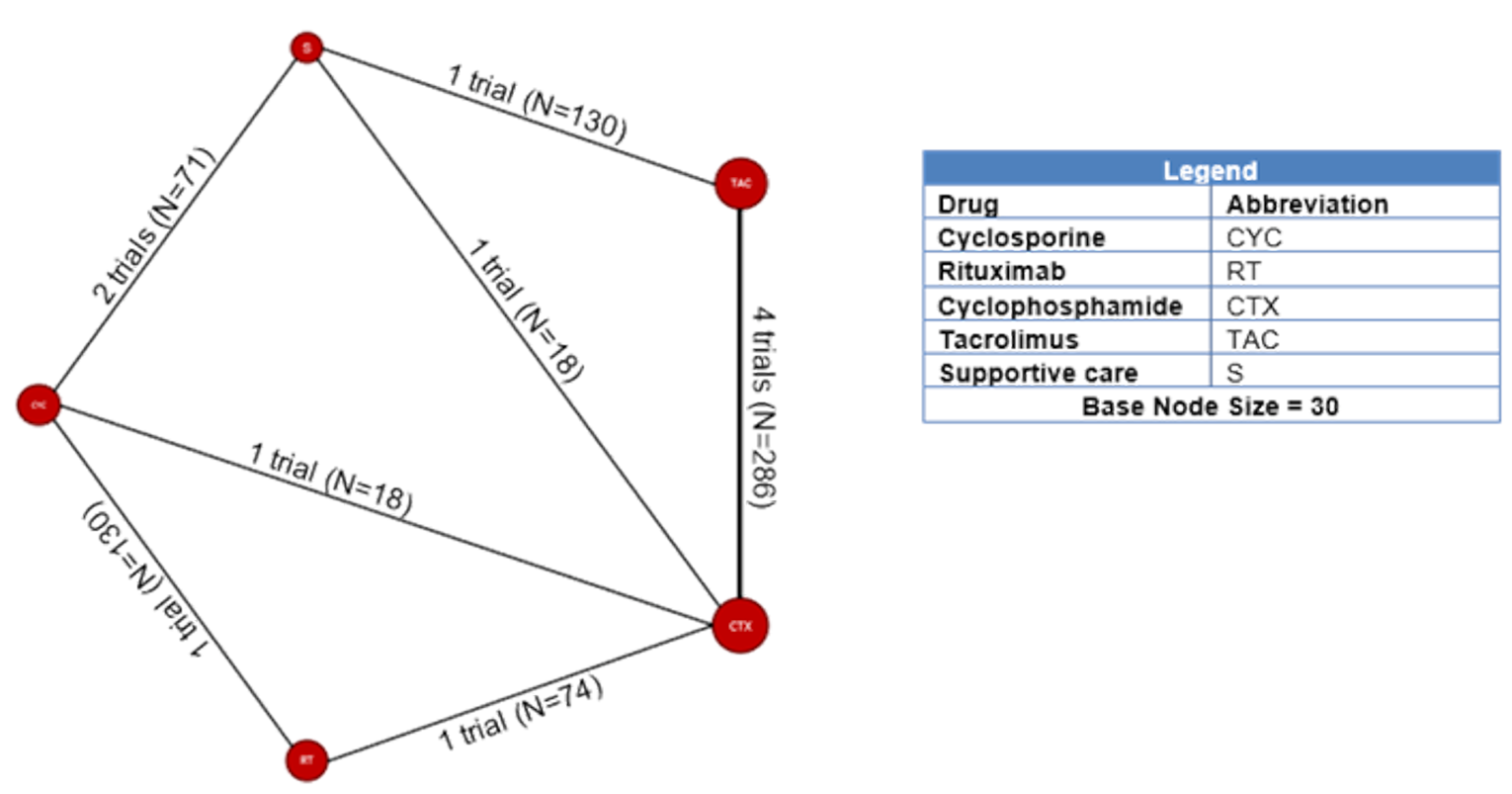
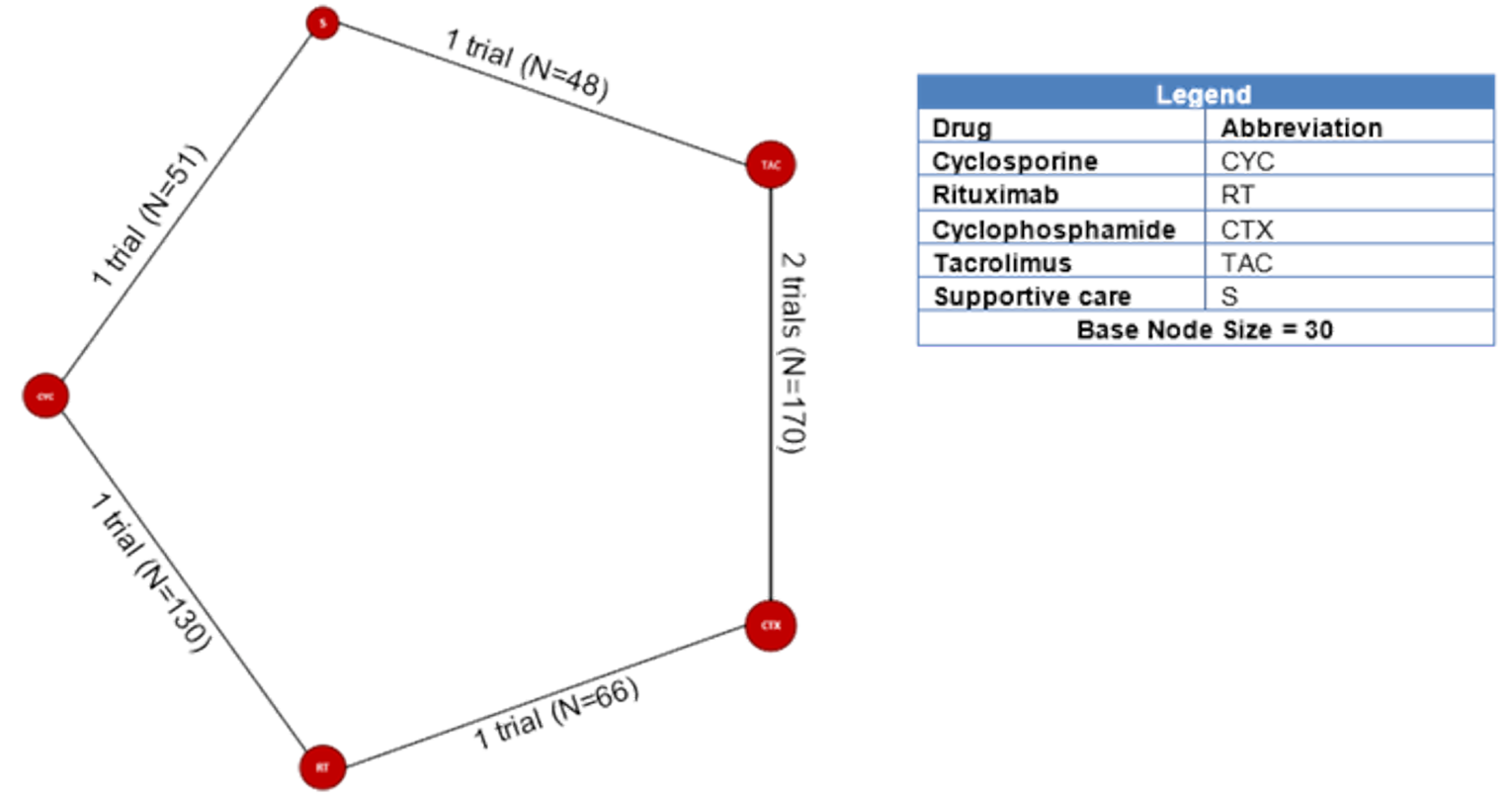
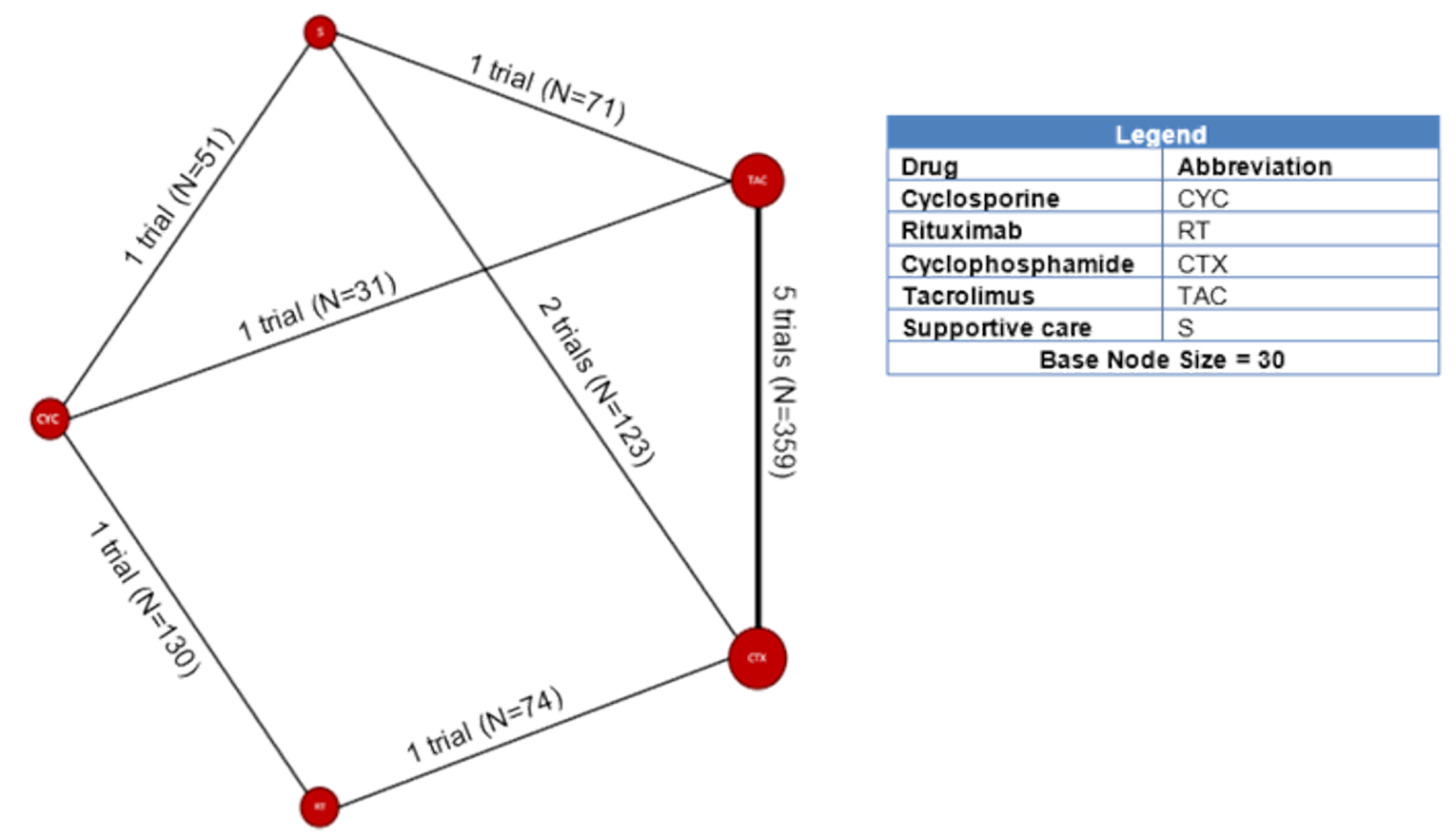
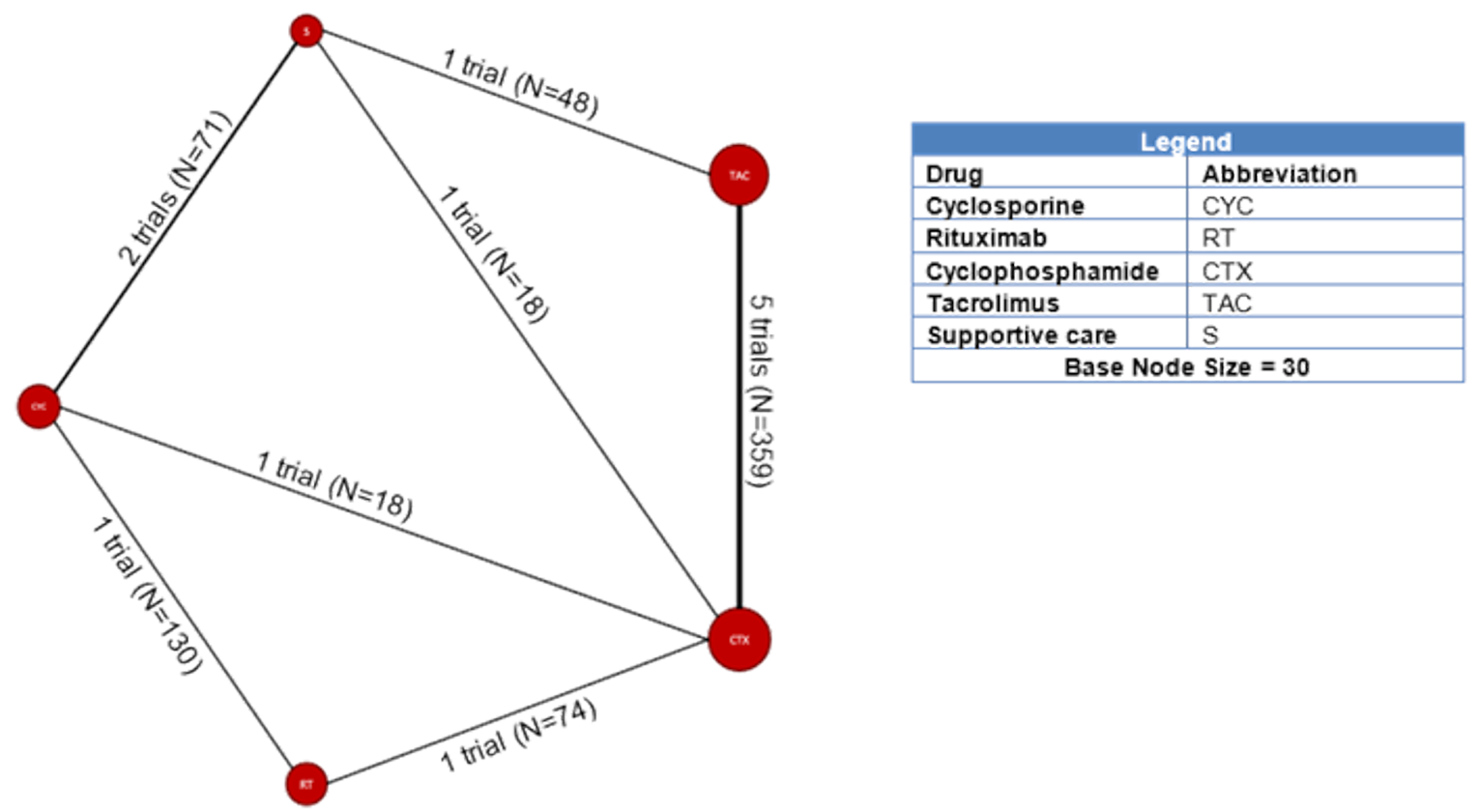
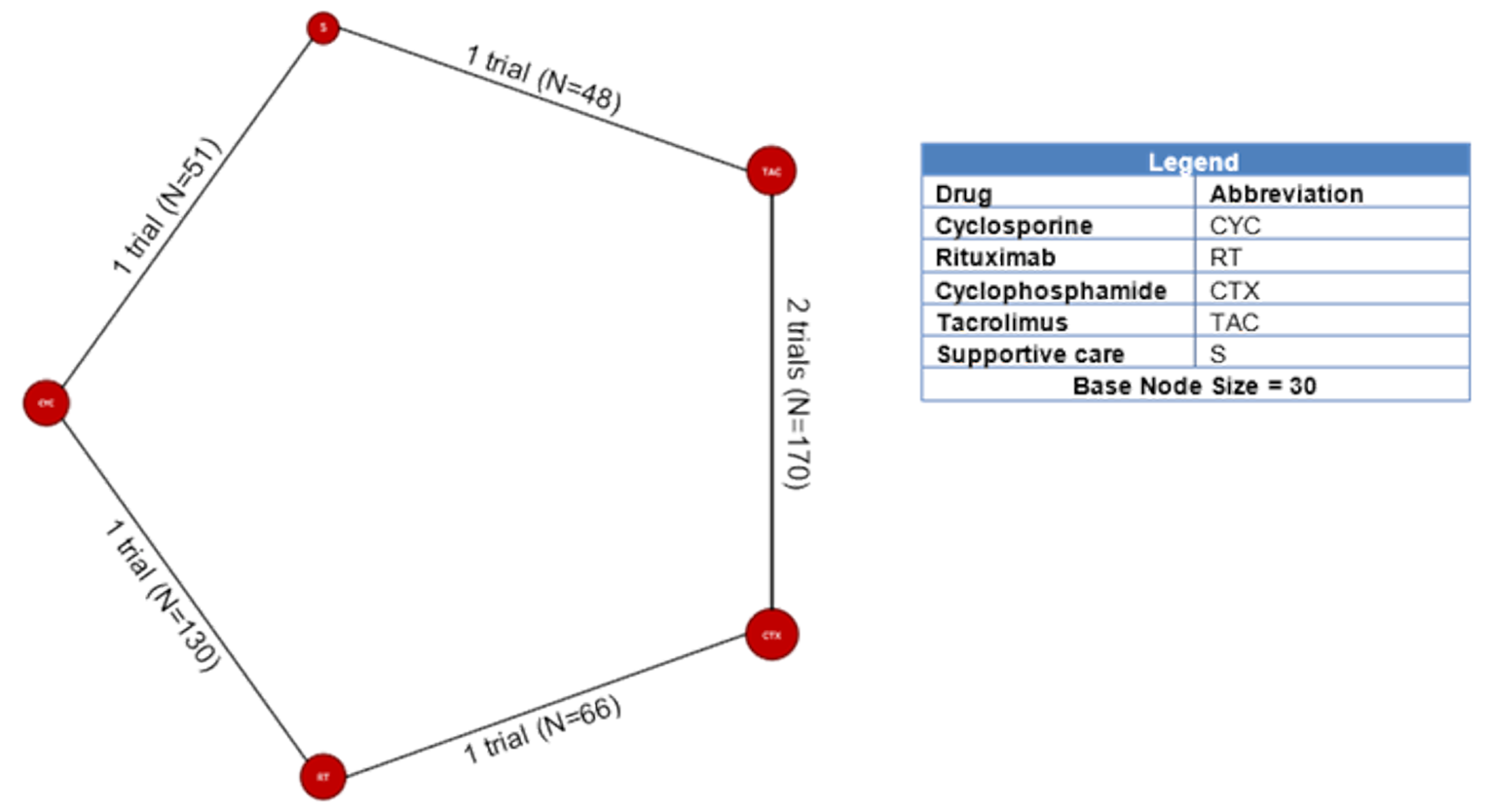
Table 11: Fixed and Random Effects Model Results for Complete Remission at 6, 12, and 18 months
Treatment Comparison | Odds Ratio (CrI) from Fixed Effects Model | Odds Ratio (CrI) from Random Effects Model | Fixed Effects Model DIC | Random Effects Model DIC |
|---|---|---|---|---|
6-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 1.08 (0.17 – 6.76) | 1.00 (0.09 – 9.23) | 99 | 99 |
Rituximab vs. cyclosporine | 0.68 (0.08-5.88) | 0.54 (0.03 – 7.25) | ||
Rituximab vs. supportive care | 1.03 (0.09 – 12.72) | 0.87 (0.04 – 16.78) | ||
Rituximab vs. tacrolimus | 0.59 (0.09-3.85) | 0.51 (0.04 – 4.96) | ||
12-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 0.48 (0.17-1.28) | 0.51 (0.06 – 4.60) | 105 | 99 |
Rituximab vs. cyclosporine | 2.62 (0.88 – 8.81) | 2.70 (2.91 – 26.60) | ||
Rituximab vs. supportive care | 1.81 (0.42 – 8.49) | 0.86 (0.02 – 4.54) | ||
Rituximab vs. tacrolimus | 0.36 (0.12-1.05) | 0.39 (0.04 – 4.82) | ||
18-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 1.99 (0.67 – 6.11) | 2.13 (0.29 - 18.84) | 63 | 64 |
Rituximab vs. cyclosporine | 22.20 (5.24 – 145.80) | 23.33 (2.39 - 356.70) | ||
Rituximab vs. supportive care | 25.07 (4.63 – 155.50) | 28.13 (2.01 - 583.09) | ||
Rituximab vs. tacrolimus | 5.97 (1.76 – 20.99) | 6.42 (0.65 - 81.23) | ||
CrI = credible interval; DIC = deviance information criterion
Table 12: Fixed and Random Effects Model Results for Any Remission at 6, 12, and 18 months
Treatment Comparison | Odds Ratio (CrI) from Fixed Effects Model | Odds Ratio (CrI) from Random Effects Model | Fixed Effects Model DIC | Random Effects Model DIC |
|---|---|---|---|---|
6-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 0.49 (0.22-1.09) | 0.48 (0.11 – 2.13) | 115 | 114 |
Rituximab vs. cyclosporine | 0.61 (0.32-1.15) | 0.63 (0.15 - 2.76) | ||
Rituximab vs. supportive care | 8.75 (3.13 – 26.10) | 9.12 (1.61 – 57.50) | ||
Rituximab vs. tacrolimus | 0.27 (0.11-0.65) | 0.24 (0.05 - 1.12) | ||
12-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 0.49 (0.20-1.18) | 0.35 (0.03 - 3.54) | 124 | 112 |
Rituximab vs. cyclosporine | 1.52 (0.79 – 2.95) | 2.25 (0.23 – 29.53) | ||
Rituximab vs. supportive care | 3.71 (1.31 – 10.84) | 2.54 (0.13 – 39.23) | ||
Rituximab vs. tacrolimus | 0.45 (0.57-1.47) | 0.27 (0.02 – 3.30) | ||
18-month Study Time Point | ||||
Rituximab vs. cyclophosphamide | 0.52 (0.19-1.39) | 0.51 (0.09 – 2.89) | 65 | 67 |
Rituximab vs. cyclosporine | 5.31 (2.60 – 11.17) | 5.29 (1.04 – 25.78) | ||
Rituximab vs. supportive care | 22.60 (6.80 – 81.70) | 22.59 (2.98 – 187.83) | ||
Rituximab vs. tacrolimus | 2.12 (0.69 – 6.51) | 2.13 (0.31 – 15.43) | ||
CrI = credible interval; DIC = deviance information criterion
Appendix 10: Critical Appraisal
Note that this appendix has not been copy-edited.
The included studies had a broad range of reporting quality, despite being published within the last two decades. Several domains in the Cochrane Risk of Bias tool could not be assessed as intended due to the context of the study. Firstly, not all studies reported if the analysis (at least for the primary outcomes) was done in the ITT population; however, the review team’s aim was to assess the ITT effect (assignment to intervention), as opposed to the per-protocol effect (adhering to intervention). Secondly, all but 2 trials were open-label, therefore the possibility remains that the assignment of treatment (including dosage determination or adjustment and adherence to treatment) and the assessment of outcomes were biased to a certain degree. However, all trials had an active comparator, and the dosage of the treatment and comparator arm could, in theory, be adjusted independently. Therefore, any bias resulting from the knowledge of treatment assignment is likely minimal. The outcomes in the trials were defined based on cutoffs from laboratory parameters, generally measured using appropriate procedure and instrument. Therefore, biases resulting from differential assessment of outcomes based on treatment knowledge is less likely, irrespective of whether the outcome assessors were blinded or not. Finally, the signaling questions in the “selection of reported result” domain were not applicable for most trials (refer to the Cochrane Risk of Bias tool for signaling questions). Even though results were analyzed in accordance with a pre-specified analysis plan for most trials, the analysis plan was not finalized before unblinded outcome data became available since most trials were open-label. The numerical results were neither selected on the basis of results from multiple eligible outcome measurements (e.g., scales, definitions, timepoints), nor from multiple eligible analyses of the data. Figure 14 provides a risk of bias plot showing an assessment of risk of bias in the individual domains as well as overall bias for each of the included trials.
Six trials were considered well conducted and reported, including Cattran et al., Chen et al., Fervenza et al., Praga et al., Scolari et al., and Ramachandran et al. These trials had a low risk of bias in each domain, with most signaling questions assessed adequately based on the level of detail provided in the publications. Seven trials had moderate risk of bias, including He et al., Hofstra t al., Howman et al., Jha et al., Kosmadakis et al., Saito et al., and Yuan et al. The trials had some concerns primarily with respect to randomization and treatment allocation as the publications did not provide adequate detail to assess the appropriateness of these processes. In addition, most of these trials provided no information if the outcome assessors were blinded to treatment assignment, therefore, the possibility that the results could be biased due to the knowledge of treatment assignment cannot be discounted. Table 11 provides additional details of the strengths and limitations of each trial based on risk of bias assessment.
Overall, 5 trials had a high risk of bias, including Di et al., Li et al., Omrani et al., Peng et al., and Xu et al. The high risk of bias primarily resulted from poor reporting of study methodology, specifically within the domains of “intended interventions” and “outcome measurement”. Except for Omrani et al., it was not clear if the other trials were open-label or not, as this information was not available, and could not be assessed from the respective publications. In addition, aspects of randomization, blinding, and allocation concealment were described with little information, or not at all. Additionally, it was not clear if the trial data were pre-specified to be analyzed in ITT population; however, the efficacy analyses were adjudicated to be conducted in the ITT population since data were available for all or almost all patients.
Figure 14: Risk of Bias Assessment Plot
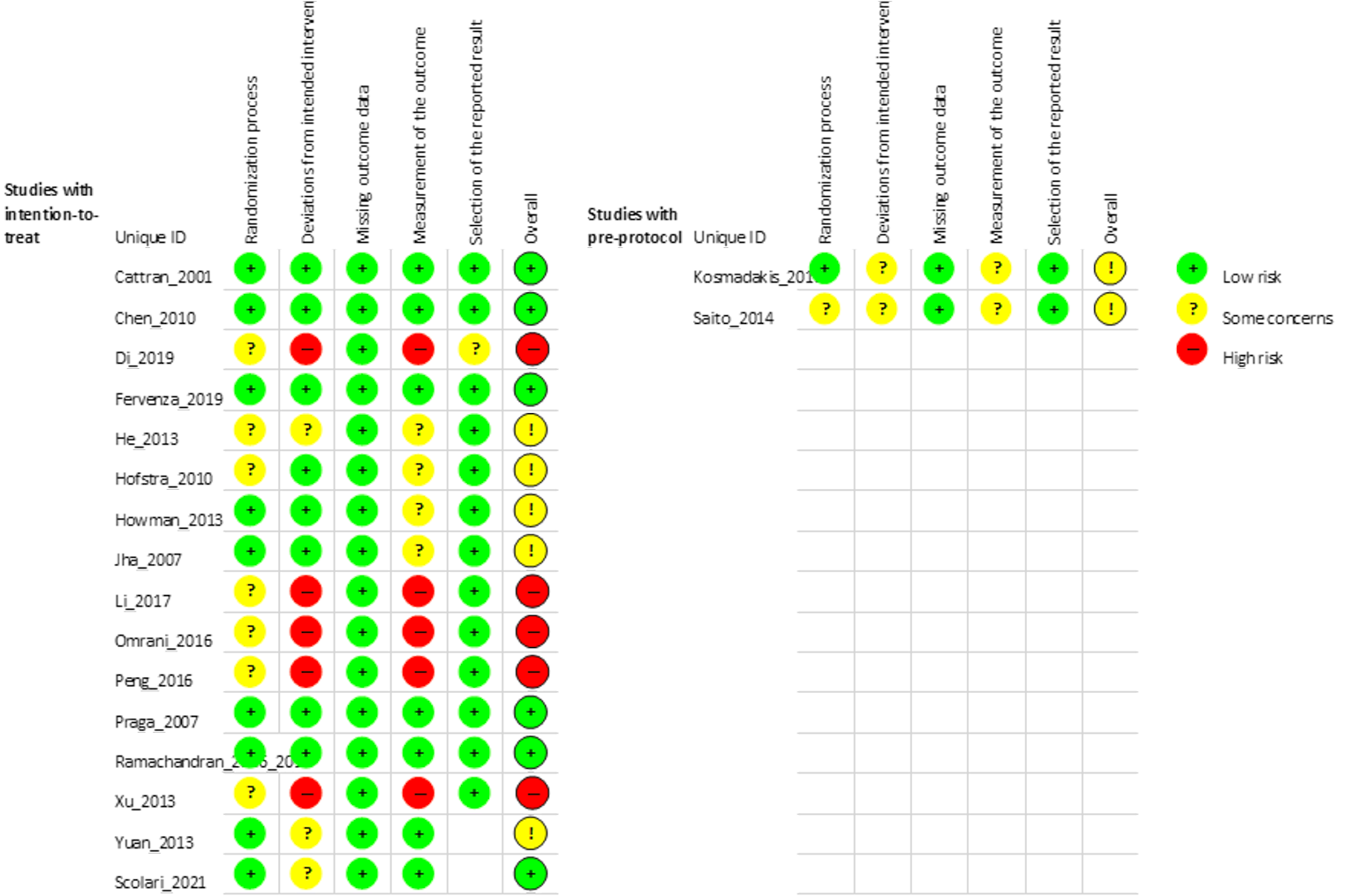
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.37
Table 13: Strengths and Limitations of Included Studies
First Author, Publication Year | Strengths | Limitations |
|---|---|---|
Cattran 2001 |
| |
Chen 2010 |
|
|
Di 2018 |
|
|
Fervenza 2019 |
|
|
He 2013 |
|
|
Hofstra 2010 |
|
|
Howman 2013 |
|
|
Jha 2007 |
|
|
Kosmadakis 2010 |
|
|
Li 2017 |
|
|
Omrani 2016 |
|
|
Peng 2016 |
|
|
Praga 2007 |
|
|
Ramachandran 2016 and 2017 |
|
|
Saito 2014 |
|
|
Xu 2013 |
|
|
Yuan 2013 |
|
|
Scolari 2021 |
|
|
Source: Cattran et al.,21 Chen et al.,23 Di et al.,24 Fervenza et al.,7 He et al.,25 Hofstra et al.,26 Howman et al.,27 Jha et al.,28 Kosmadakis et al.,29 Li et al.,30 Omrani et al.,22 Peng et al.,31 Praga et al.,32 Ramachandran et al.,33,34 Saito et al.,35 Xu et al.,36 Yuan et al.,37 Scolari et al.8
Appendix 11: Implementation Advice
Background
Representatives from public drug programs requested that a panel of clinical experts be convened to elaborate on the place of rituximab in the treatment of PMN to inform appropriate usage of this drug in the Canadian context. The panel was asked to focus their discussion on the following questions:
Based on the available evidence, what is the place in therapy of rituximab to treat primary membranous nephropathy?
What specific characteristics should patients possess or satisfy for access to rituximab?
What is the most appropriate rituximab dosing regimen?
What objective measures or outcomes, and at what time points, should be used to determine treatment success or failure in primary membranous nephropathy?
Are there special populations that require other considerations?
Consultation Process and Objectives
The clinical expert panel was comprised of 3 Canadian specialists with expertise in the diagnosis and management of patients with diseases of the kidneys and one Canadian specialist with expertise in critical care medicine. The objective of the panel was to provide advice to the participating drug programs. A consensus-based approach was used, and input was sought using a questionnaire and a panel meeting. In addition to the clinical panellists and CADTH staff, representatives from public drug programs were invited to participate in the discussion and to provide input in advance of the meeting on the topics up for discussion.
There was consensus among the panel members that there is considerable unmet need in the discussed patient population, and the panel agreed that rituximab has the potential to meet some of these unmet needs. The criteria specified in this appendix reflect the panel members’ conclusions based on the best available evidence and their clinical expertise in the diagnosis and management of primary membranous nephropathy.
The advice presented in this report is based on the evidence presented in the clinical review and the Kidney Disease: Improving Global Outcomes (KDIGO) 2021 guidelines, as well as the experience and expertise of the implementation advice panel members.
Implementation Advice
Initiation, discontinuation, and administration or prescribing criteria and conditions for rituximab for treatment of PMN are shown in Table 14.
Table 14: Summary of Initiation, Discontinuation, and Administration Criteria for Rituximab
Criteria | Description |
|---|---|
Definitions | Complete remission: Proteinuria < 0.3 g/day or protein-creatinine ratio < 30mg/mmol Partial remission: Reduction in proteinuria of at least 50% from baseline and final proteinuria between 0.3 g and 3.5 g per day Relapse: Recurrence of proteinuria (as per the initiation criteria) accompanied by a decrease in serum albumin to less than 30g/L in patients who have achieved a complete or partial remission following prior rituximab treatment Nonresponse: Lower than 25% reduction in proteinuria by 6 months after initial course |
Initiation criteria | Eligibility for treatment with rituximab is based on criteria for moderate to high risk of developing progressive kidney injury or complications of nephrotic syndrome. Initiation criteria Rituximab may be used in patients 18 years of age or older if one of the following criteria are met:
Initial treatment
Additional courses An additional course of rituximab may be administered using the same dosage and regimen at 6 months:
|
Discontinuation criteria |
|
Prescribing conditions | Populations that require special consideration:
|
eGFR = estimated glomerular filtration rate; PLA2R = phospholipase A2 receptor.
Clinical Panel Input Summary
Treatment Goals
The panel affirmed that the goals for treatment with rituximab and other therapies in primary membranous nephropathy are to achieve remission of proteinuria, maintain quality of life, prevent progressive loss of kidney function, prevent the need for renal replacement, and potentially prolong survival.
Place in Therapy
The panel discussed that there is uncertainty around the relative efficacy and harms of rituximab compared to other therapies for primary membranous nephropathy. There was consensus that the use of rituximab is associated with a lower risk of specific harms, relative to other drugs, due to a more favourable toxicity profile. The panel noted specific examples of toxicities associated with other treatment protocols, such as the use of higher doses of corticosteroids, higher risk of nephrotoxicity with calcineurin inhibitors, and cumulative toxicities related to cyclophosphamide protocols. The panel also acknowledged that there are risks to using rituximab, including serious infections; therefore, it should only be prescribed by clinicians with appropriate expertise.
The panel agreed that if rituximab is to be used as a first-line treatment, it may be used in patients newly diagnosed with primary membranous nephropathy, and is an alternative to CYC, calcineurin inhibitors, and cyclophosphamide in the first-line context. Rituximab is also a treatment option for patients who have had inadequate response to CYC, calcineurin inhibitors, or cyclophosphamide, or for patients in which continued treatment with those medications is not appropriate. Re-treatment with rituximab is an option in patients who had PR or CR and subsequently relapsed. Patients with recurrence of membranous nephropathy in a transplant kidney may also be considered for rituximab as first-line therapy; these patients are at risk of graft loss and are already immunosuppressed because of calcineurin inhibitor therapy.
Initiation Criteria
The panel agreed that the use of rituximab in patients with low risk of disease progression is not appropriate. Treatment with rituximab would be appropriate for patients (≥ 18 years of age) with moderate to high risk of progressive kidney disease based on proteinuria and estimated glomerular filtration rate.
The panel noted that the KDIGO guidelines recommend also using serum albumin and anti-phospholipase A2 receptor (PLA2R) titres to guide risk assessment, initiation, and continuation of treatment. While there is evidence that suggests a correlation between anti-PLA2R titres and resistance to treatment, laboratory tests for anti-PLA2R are not accessible in many Canadian centres and there is no consensus on how to use titre thresholds for treatment initiation. Additionally, the data that are available regarding anti-PLA2R titres in patients with primary membranous nephropathy do not provide sufficient evidence for informing treatment selection.
Discontinuation Criteria
The panel agreed that treatment with rituximab may be stopped when complete remission is achieved. Only one course of rituximab is required to determine whether or not a patient responds to therapy. In case of a nonresponse, re-treatment is not recommended.
Prescribing Conditions
The panel noted that the warnings and risks stated in the product monograph should be taken into account when prescribing rituximab. Rituximab may be used by those who are lactating, but careful consideration of risks to the fetus should be taken if prescribing for those who are pregnant. Special consideration should be given to risk of reactivation in patients with latent or active hepatitis B.
Acknowledgments: Implementation Advice Panel: Dr. Marcello Tonelli, Dr. Penelope Poyah, Dr. Heather Reich, and Dr. Emily Reynen. FPT Jurisdictional Representative: Brent Ruddock.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.