CADTH Health Technology Review
Drugs for Rare Diseases: A Review of National and International Health Technology Assessment Agencies and Public Payers’ Decision-Making Processes
Environmental Scan
Abbreviations
AHS
Alberta Health Services
ATU
Temporary Authorizations for Use
CDEC
Canadian Drug Expert Committee
CDR
Common Drug Review
CEPAG
Comparative Effectiveness Public Advisory Council
CPEC
Canadian Plasma Protein Product Expert Committee
CSEMI
Comité scientifique d'évaluation des médicaments aux fins d'inscription
CTAF
California Technology Assessment Forum
DRDs
drugs for rare diseases
EDRD
Expensive Drugs for Rare Diseases
EDS
Exception Drug Status
EMA
European Medicines Agency
GBA
Gemeinsamer Bundesausschuss or Federal Joint Committee
HAS
Haute Autorité de Santé
HST
highly specialised technologies
HSTEC
Highly Specialised Technologies Evaluation Committee
HTA
health technology assessment
ICER
Institute for Clinical and Economic Review
INESSS
Institut national d’excellence en santé et en services sociaux
IQWiG
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen or Institute for Quality and Efficiency in Health Care
LSDP
Life Saving Drugs Program
MPS
mucopolysaccharidosis
NDC
New Drugs Committee
NHS
National Health Services
NICE
National Institute for Health and Care Excellence
NIHB
Non-Insured Health Benefits
NPAF
New Products Assessment Form
NPPA
Named Patient Pharmaceutical Assessment
QALY
quality-adjusted life-year
PACE
Patient and Clinician Engagement
PAS
Patient Access Scheme
PASAG
Patient Access Scheme Assessment Group
PBAC
Pharmaceutical Benefits Advisory Committee
PBS
Pharmaceutical Benefits Scheme
pERC
pan-Canadian Oncology Drug Review Expert Review Committee
PHARMAC
Pharmaceutical Management Agency
PTAC
Pharmacology and Therapeutics Advisory Committee
RAMQ
Régie de l'assurance maladie du Québec
RWE
real-world evidence
SMC
Scottish Medicines Consortium
STEDT
Short Term Exceptional Drug Therapy
Key Messages
Drugs for rare diseases can address significant unmet therapeutic needs for patients living with seriously debilitating and life-threatening conditions; however, the high costs of these drugs can pose challenges for public drug programs and health care systems.
There are challenges with the application of standard health technology assessment methods for the assessment of drugs for rare diseases, including uncertainty with the clinical and economic evidence due to small sample sizes, poorly characterized natural history of disease, uncertain epidemiology, absence of comparative studies, heterogenous phenotypes, and lack of diagnostic accuracy. In addition, there are challenges applying commonly accepted economic benchmarks due to the very high cost of these drugs.
To address these challenges, agencies and public payers have established separate or modified processes and programs to review and make reimbursement recommendations for drugs for rare diseases. There is a lack of consistency across agencies with respect to how drugs for rare diseases are defined and what aspects of the process are modified to address the challenges with these drugs; however, common features include greater acceptance of uncertainty with the clinical and economic evidence and a higher willingness-to-pay threshold.
The majority of health technology assessment agencies have highlighted that drugs for ultra-rare diseases are particularly challenging and warrant special consideration. This includes the creation of completely separate review processes for drugs indicated for use in the treatment of ultra-rare conditions in the UK (both England and Scotland).
The majority of processes for funding drugs for rare diseases by public drug programs included in this report manage the drugs through standard formulary processes involving the use of special authorization to ensure that patients meet the eligibility criteria for the drugs. Those that have specialized formularies have largely focused on providing access to ultra-rare conditions.
Abstract
CADTH conducted an Environmental Scan to identify, describe, and compare how health technology assessment (HTA) agencies in Canada and internationally make reimbursement recommendations on drugs for rare diseases (DRDs). The report provides a comparison of the review and decision-making processes for agencies in Canada (CADTH and Institut national d’excellence en santé et en services sociaux [INESSS]), the UK (National Institute for Health and Care Excellence [NICE] and Scottish Medicines Consortium [SMC]), Australia (Pharmaceutical Benefits Advisory Committee [PBAC]), New Zealand (Pharmaceutical Management Agency [PHARMAC]), Germany (Federal Joint Committee [G-BA] and Institute for Quality and Efficiency in Health Care [IQWiG]), France (Haute Autorité de Santé [HAS]), and the US (Institute for Clinical and Economic Review [ICER]). The report also presents information on how selected public drug programs in Canada and internationally evaluate and make funding decisions on DRD.
The HTA agencies included in this review have established separate or modified processes to review and make reimbursement recommendations for DRDs. The agencies differ with respect to how they identify which DRDs will be selected for consideration through the specialized or modified review processes. All agencies with clearly defined eligibility criteria state that the drug must be indicated for a serious disease for which there is unmet medical need. The prevalence threshold is the key characteristic that differs across the agencies. SMC, NICE, PHARMAC, and ICER have all adopted criteria that specifically isolate drugs indicated for ultra-rare diseases for special consideration (prevalence thresholds ranging from 1 in 50,000 to 3 in 100,000).
There is variation across the different agencies with respect to how their process has been modified to accommodate DRDs. Common features include greater acceptance of uncertainty with the clinical and economic evidence, a higher willingness-to-pay threshold, and greater engagement with patient groups and the clinical community. The newly launched SMC process for ultra-orphan drugs is the most unique HTA process identified in this Environmental Scan. This process consists of an initial HTA evaluation to identify gaps in the evidence, followed by an evidence generation phase when the drug is reimbursed for a period of up to 3 years, and concluding with a reassessment and a final decision issued regarding availability of the drug for use in National Health Service (NHS) Scotland.
The majority of public drug programs included in this report manage DRDs through standard formulary processes involving the use of special authorization to ensure that patients meet the eligibility criteria for the drugs. Those that have specialized formularies, such as the Australian Life Saving Drugs Program (LSDP), have largely focused on providing drug access for ultra-rare conditions.
Context
DRDs are medicinal products intended for the prevention or treatment of rare diseases or disease subtypes. There is a lack of consensus on how rare diseases are defined across international regulatory and HTA agencies.1 In addition to the prevalence of the diseases, other criteria such as disease severity, lack of alternative treatment, and the hereditary nature of the condition are also used to define these diseases.1 For the purposes of this Environmental Scan, the term drugs for rare diseases (DRDs) is used to identify these drugs in general terms.
Estimates for the number of Canadians living with rare diseases range from 1 million (approximately 2% to 3% of the population)2 to as high as 2.8 million.3 There are more than 7,000 identified rare diseases and this number has been increasing.3 DRDs are often very expensive, with costs often exceeding CA$100,000 per year for 1 patient.4 These high costs are typically attributed to the high cost of research and overall small market size for DRDs. The high cost of drugs, the increasing prevalence of rare diseases, and the severity of rare diseases pose a significant societal, clinical, and economic burden to patients and caregivers, as well as to the health care system.
Various factors make it challenging to apply standard HTA methodologies to assess these drugs, such as uncertainty of evidence and low prevalence of rare diseases, as well as poorly explored epidemiology, absence of comparable treatment alternatives on the market, and failure of DRDs to meet the set economic benchmarks because of their high cost.5 To address these challenges posed by standard HTA methodologies, some HTA agencies and public payers have established separate or modified processes and programs to review and make funding recommendations or decisions on DRDs.
This Environment Scan was conducted as an update to CADTH’s 2018 report Drugs for Rare Diseases: A Review of National and International Health Technology Assessment Agencies and Public Payers’ Decision-Making Processes.6
Objectives
The objective of this Environmental Scan is to identify, describe, and compare how HTA agencies in Canada and internationally make reimbursement recommendations on DRDs. The report will also present information on how selected public drug programs evaluate and make funding decisions on DRDs. This Environmental Scan will aim to answer the following key questions:
How do HTA agencies review and make reimbursement recommendations for DRDs?
How do public payers make funding decisions for DRDs?
Do any of Canada’s publicly funded drug plans use a DRD-specific evaluation framework to evaluate DRD funding?
This comparison of the review and decision-making processes, for both HTA organizations and public payers, will include (but is not limited to) definitions, program eligibility criteria, the submission process, and evaluation frameworks including clinical and economic assessments. Publicly available reports, guidelines, and evaluation frameworks from the HTA organizations in the countries listed in Table 1 were reviewed to gather the information. These countries were selected because of commonalities with the Canadian context, including geography and regulatory HTA or reimbursement processes. If available, this Environmental Scan will also present information on a separate funding program for DRDs in these countries. Information on Canada’s public drug plans, specific to their evaluation and decision-making process for DRDs, will also be presented. Some of these HTA agencies and public payers make a distinction between drugs for “rare” and drugs for “ultra-rare” conditions and have established separate or modified processes for each of these categories, whereas some categorize DRDs under lifesaving drugs or highly specialized technologies. This report includes information on the HTA process and funding program for all these categories if they were explicitly designed to address the unique needs of DRDs.
Table 1: Agencies and Programs Reviewed
Country | Agency | Primary committee(s) | Formularies informed |
Canada | CADTH | CADTH Canadian Drug Expert Committee CADTH pan-Canadian Oncology Drug Review Expert Review Committee Canadian Plasma Protein Product Expert Committee | Public drug programs and cancer agencies |
INESSS | Comité scientifique d'évaluation des médicaments aux fins d'inscription | Régie de l'assurance maladie du Québec | |
UK | NICE | Technology Appraisal Committees Cancer drugs fund rapid reconsideration committee Highly Specialised Technologies Evaluation Committee | NHS England |
SMC | New Drugs Committee SMC Committee | NHS Scotland | |
Australia | PBAC Secretariat | Pharmaceutical Benefits Advisory Committee | Pharmaceutical Benefits Scheme |
LSDP Secretariat | LSDP Expert Panel | LSDP | |
New Zealand | PHARMAC | Pharmacology and Therapeutics Advisory Committee Rare Disorders Subcommittee | Pharmaceutical Schedule |
PHARMAC board or by staff under delegated authority from the Board | Named Patient Pharmaceutical Assessment Policy | ||
Germany | IQWiG | G-BA | Social Health Insurance benefit catalogue |
France | HAS | Transparency Committee | National Health Insurance |
US | ICER | California Technology Assessment Forum Midwest Comparative Effectiveness Public Advisory Council New England Comparative Effectiveness Public Advisory Council | Non-specific |
G-BA = Gemeinsamer Bundesausschuss; HAS = Haute Autorité de Santé; ICER = Institute for Clinical and Economic Review; INESSS = Institut national d’excellence en santé et en services sociaux; IQWiG = Institute for Quality and Efficiency in Health Care; LSDP = Life Saving Drugs Program; NHS = National Health Service; NICE = National Institute for Health and Care Excellence; PBAC = Pharmaceutical Benefits Advisory Committee; PHARMAC = Pharmaceutical Management Agency.
Methods
This report used a literature search strategy developed for a previous CADTH report.6 For the current report, a limited literature search was conducted on key resources including MEDLINE, Canadian and major international health technology agencies, as well as a focused internet search. No filters were applied to limit the retrieval by study type. The initial search was run on January 25, 2018. For the current report, database searches were rerun on January 14, 2021, to capture any articles published since the initial search date. The search of major health technology agencies was also updated to include documents published since January 2018.
Findings
The following sections present information on how HTA agencies and public payers in Canada, Australia, the UK, New Zealand, France, Germany, and the US make reimbursement recommendations or decisions for DRDs. An overview of the evaluation process or programs in each of the countries is provided. In addition, a summary of specialized funding processes for DRDs are provided for the public drug programs that currently operate them.
Overview of HTA Processes for Drugs for Rare Diseases
Rare Disease Processes
Table 2 provides a summary of the different processes used by HTA agencies for the evaluation of DRDs. The majority of agencies review DRDs through modified versions of their standard HTA processes (CADTH, INESSS, PBAC, PHARMAC, HAS, and ICER). These agencies have introduced specialized accommodations for DRDs within their administrative, review, and/or deliberative and recommendation processes. NICE and SMC currently operate separate processes for the review of drugs for ultra-rare diseases (i.e., the highly specialised technologies [HST] process and ultra-orphan pathway, respectively). SMC has also introduced a modified process for the review of orphan drugs (i.e., those designated as orphan drugs by the European Medicines Agency [EMA], but that do not qualify for review through the ultra-orphan pathway). Germany has a separate review process for drugs designated as orphan drugs by EMA provided the costs of the drug are less than €50 million over 12 months but applies its standard review processes for orphan drugs if the costs exceed €50 million over 12 months.
Eligibility for Specialized or Modified Review Processes
There is considerable variation across regulatory and HTA agencies with respect to how DRDs are defined to be assessed through specialized or modified review pathways.1 The common characteristics for eligibility for specialized DRD processes are small patient populations, severe disease, and a lack of alternative treatments. As shown in the findings for this scan, the HTA agencies that have accommodations for DRDs will typically apply them for drugs that are designated as orphan drugs by a regulatory authority (e.g., G-BA/IQWiG),7 specifically for ultra-rare diseases (NICE, PHARMAC, and ICER),8-10 or have accommodations for both rare and ultra-rare diseases (SMC).11,12 CADTH, INESSS, and HAS have broader criteria that are not necessarily restricted to a particular prevalence threshold for when the accommodations are applied.13,14
The agencies that have specialized or modified processes for ultra-rare diseases have adopted prevalence thresholds of 1 in 50,000 (SMC, NICE, and PHARMAC) or approximately 3 in 100,000 (ICER).9-12 Both PHARMAC and ICER restrict the ultra-rare process based on the total patient populations that may be eligible for treatment with the drug in question (i.e., all potential indications are pooled when considering whether or not the drug has met the ultra-rare thresholds set by the agencies).9,10 PHARMAC has indicated that this approach ensures that the modified processes are limited to those manufacturers who are disadvantaged by the small patient population for their product(s).15
Both SMC and G-BA/IQWiG have aligned their orphan drug processes with the EMA’s orphan drug designation7,11; however, the G-BA/IQWiG process includes an economic criterion that expenditures for the drug must not exceed €50 million per 12 months.7,16 Orphan drugs with sales that exceed €50 million in a 12-month period must file a complete dossier for evaluation by IQWiG.7,16 This was the only agency included in the scan that specifically included an economic threshold for determining the review pathway of DRDs. NICE also includes an economic criterion for eligibility in the HST process (i.e., very high acquisition cost), but a threshold is not specified in the procedures.8
Administrative Processes
PHARMAC and HAS offer opportunities for earlier filing of applications for drugs that meet the criteria for the specialized processes (i.e., before regulatory approval). Both CADTH and INESSS currently allow parallel regulatory and HTA reviews for all drug types; therefore, no further modifications are made for DRDs. Topic selection for NICE and ICER occurs in the same manner for DRDs as with other drugs.17,18 The times for the review and recommendation phases for specialized DRD processes vary across agencies. CADTH, INESSS, PBAC, NICE, ICER, and PHARMAC all have the same timelines for DRDs as with other drugs. The SMC processes for orphan drugs and the reassessment phase of the ultra-orphan pathway is 22 weeks to 26 weeks in duration, which is longer than the timelines for other drugs (18 weeks).12,19 None of the agencies that charge application fees have a modified fee schedule for DRDs.
For all HTAs that initiate the process through applications filed by an industry sponsor (i.e., all except ICER), the application requirements for DRDs are generally similar with those of other drugs, including for the SMC ultra-orphan pathway. The G-BA/IQWiG process allows an abbreviated application for orphan drugs below the threshold of €50 million per 12 months; however, a complete application is required if and when the sales exceed this economic threshold. NICE and the SMC ultra-orphan pathway are the other processes that use completely specialized submission templates for DRDs.20,21 All the others use the same requirements or use them with slight adjustments (e.g., optional inclusion of information supporting rule of rescue in the PBAC process).22
Review Process
Enhanced opportunities for engagement with patient groups were noted in the processes used by SMC (Patient and Clinician Engagement [PACE] meetings), PBAC (Stakeholder Meetings), and INESSS (conducting interviews and focus groups).23-25 The other agencies apply their standard processes for patient engagement for DRDs and other products. Both CADTH and INESSS may convene panels of experts to advise on the review of DRDs and other complex drugs.26 As with patient groups, SMC and PBAC offer opportunities for enhanced engagement with the clinical community as part of the PACE and stakeholder meetings, respectively.23,24
The clinical review processes are largely similar across agencies for DRDs and other drugs, with the exception of the G-BA/IQWiG process for orphan drugs and the SMC ultra-orphan review pathway, for which the processes are significantly different than the standard HTA processes.7,12For the G-BA/IQWiG process for orphan drugs, with sales less than €50 million per 12 months, added benefit of the drug under review is assumed to be proven based on regulatory approval. This allows the sponsor to file an abbreviated submission package with IQWiG. The new ultra-orphan review pathway for SMC consists of an initial HTA review focused on the identification of gaps in the evidence, a period of market access of up to 3 years to generate evidence, and a reassessment and final recommendation from SMC.12 Most of the agencies note that there is greater allowance and consideration of evidence from non-randomized clinical studies in their evaluation processes for DRDs.
All the HTA agencies consider economic evidence as part of their review processes; however, there is typically greater acceptance of uncertainty in the economic evaluation due to the challenges in generating robust evidence and/or in the limited knowledge concerning the natural history of the disease. In addition, NICE, SMC, and PBAC have processes that may allow acceptance of a high cost per quality-adjusted life-year (QALY) for DRDs relative to other drugs, either through explicitly higher willingness-to-pay thresholds or simple acknowledgement that a recommendation in favour of reimbursement may be issued despite a high cost per QALY.11,22,27
Recommendation Processes
All agencies, with the exception of NICE, issue recommendations for DRDs using the same committees as their standard review processes. NICE has established a separate committee for the HST process (i.e., the Highly Specialised Technologies Evaluation Committee).28 PHARMAC has established a dedicated Rare Disease Subcommittee for the review of ultra-rare drugs. However, this subcommittee makes the initial recommendation; the final recommendation is issued by the standard Pharmacology and Therapeutics Advisory Committee.29 Most of the agencies have modified their deliberative processes to accommodate the unique challenges with DRDs. This includes greater allowances for uncertainty in the clinical and economic evidence due to challenges generating robust evidence (e.g., CADTH),13 acceptance of a higher willingness-to-pay threshold (e.g., NICE and SMC),11,27 and the potential for a recommendation that is conditional upon further evidence generation (e.g., NICE and INESSS).25,27
Table 2: Features of HTA Processes for DRDs
CADTH | INESSS | NICE | SMC | G-BA/IQWiG | PBAC/LSDP | PHARMAC | ICER |
Does the agency have specialized processes or modifications to accommodate DRDs? | |||||||
Modified review and deliberative process13 | Modified review and deliberative process | Ultra-rare drugs: Separate review and deliberative process8 | Ultra-orphan: Separate review process12 Orphan: Modified review and deliberative process (PACE meetings)11 | Separate process for orphan drugs if costs are < €50 million (added benefit is proven)7 | PBAC: Modified deliberative process (rule of rescue; stakeholder meetings)22 LSDP: Separate process for ultra-rare drugs30 | Modified application process (earlier filing)9 | Modified value assessment framework10 |
Are there criteria to be met for a drug to be considered under the DRD review process or modified review process? | |||||||
Specific criteria related to rarity of the condition, severity of the illness, and unmet need13 | Criteria not specified | Eligibility criteria are defined; all criteria have to be met to be eligible for HST8 | Ultra-orphan: Prevalence ≤ 1 in 50,00012 Orphan drug: EMA designation11 | EMA orphan drug definition provided the budget impact is < €50 million7 | PBAC: Severe disease, small population, unmet need, evidence drug provides improvement22 LSDP: Prevalence ≤ 1 in 50,000; rejected by PBAC for cost-effectiveness30 | Prevalence ≤ 1 in 50,0009 | Prevalence ≤ 10,000 (approximately 3 in 100,000)10 |
Is there a separate submission template or does the standard template require additional information for DRDs? | |||||||
No separate submission requirements13 | No separate submission requirements | Separate submission template20 | Ultra-orphan: Separate submission template21 Orphan: No separate template31 | Only requires information on the “extent of additional benefit” | PBAC: May include information to support rule of rescue22 LSDP: Separate submission templates and process30 | No separate submission requirements9 | Not applicable |
Does a special evaluation committee review and make recommendations on DRDs? | |||||||
No separate committee (CDEC, CPEC, pERC)13 | No separate committee (CSEMI)32 | HSTEC28 | No separate committee (NDC and SMC)33 | No separate committee (G-BA) | PBAC: No separate committee22 LSDP: LDSP expert panel30 | Rare Disease Subcommittee makes initial recommendation for PTAC to finalize29 | No separate committee (CTAF, Midwest CEPAG, New England CEPAG)10 |
Are any considerations made for economic evaluation? | |||||||
Greater allowance for uncertainty in economic evaluation13 | Criteria not specified | Consideration made in the benchmark for QALYs and discounting27 | Greater allowance for uncertainty in economic evaluation; PACE meeting may result in acceptance of a higher cost per QALY11 | Only the cost of treatment is considered | PBAC: Rule of rescue may result in listing irrespective of a high ICER22 LSDP: Must be rejected by PBAC on basis of cost-effectiveness30 | Criteria not specified | Additional context included; scenario analyses presented in tandem with base-case; mapping studies conducted10 |
Are there specialized processes for patient engagement? | |||||||
Standard processes for patient engagement13 | Opportunities for interviews and focus groups with patients and caregivers25 | Standard processes for patient engagement | Opportunities for PACE meetings24 | Standard processes for patient engagement | Opportunities for stakeholder meetings23 | Standard processes for patient engagement | Standard processes for patient engagement |
Is a financial risk-sharing arrangement a part of the proposal or recommendation for DRDs? | |||||||
Not considered by CADTH13 (occurs through negotiation) | Not considered by INESSS (occurs through negotiation) | Managed access arrangement could be a part of the HSTEC recommendation | Ultra-orphan: Must include a patient access scheme12 Orphan: May include a patient access scheme19 | Criteria not specified | Not considered by PBAC (occurs through negotiation) | Criteria not specified | Not applicable |
CDEC = CADTH Canadian Drug Expert Committee; CEPAG = Comparative Effectiveness Public Advisory Council; CPEC = Canadian Plasma Protein Product Expert Committee; CSEMI = Comité scientifique d'évaluation des médicaments aux fins d'inscription; CTAF = California Technology Assessment Forum; DRD = drugs for rare diseases; EMA = European Medicines Agency; G-BA = Gemeinsamer Bundesausschuss or Federal Joint Committee; HST = highly specialised technologies; HTA = health technology assessment; ICER = Institute for Clinical and Economic Review; INESSS = Institut national d’excellence en santé et en services sociaux; IQWiG = Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen or Institute for Quality and Efficiency in Health Care; LSDP = Life Saving Drugs Program Australia); NDC = New Drugs Committee; NICE = National Institute for Health and Care Excellence; PACE = Patient and Clinician Engagement; PBAC = Pharmaceutical Benefits Advisory Committee; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; PHARMAC = Pharmaceutical Management Agency; PTAC = Pharmacology and Therapeutics Advisory Committee; QALY = quality-adjusted life-year; SMC = Scottish Medicines Consortium.
Canada (With the Exception of Quebec)
Program Overview
CADTH conducts HTA and issues reimbursement recommendations to federal, provincial, and territorial drug programs, provincial cancer agencies, and Canadian Blood Services. CADTH has specialized accommodations for DRDs and other complex files throughout its review and recommendation processes (Figure 1).
Administrative Processes
CADTH administrative processes are the same for initial submissions of DRDs and other drugs (e.g., application requirements, fees, opportunities for meeting with CADTH, and timelines for filing and reviewing applications). Accommodations for DRDs can be made when determining resubmission eligibility with greater allowances for and consideration of new evidence derived from non-randomized studies (including real-world evidence). CADTH does not have a modified application fee structure specifically for DRDs, though a higher application fee applies if the drug under review is a cell or gene therapy (which are often indicated for use in the treatment of rare diseases).34
Review Process
In the review of DRDs and other complex drugs, CADTH applies a modified approach that includes greater consideration of real-world evidence and other forms of non-randomized studies and enhanced engagement with the clinical community. The clinician engagement process is augmented through the formation of panels of experts that are used to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the drug’s potential place in therapy (e.g., potential reimbursement conditions).13 In addition, these panels can be formed jointly with INESSS to establish a common pan-Canadian advisory panel for the drug under review.13 Drugs selected jointly by CADTH and INESSS typically involve the following characteristics: challenges in generating robust evidence due to the rarity of the condition, potential for challenging implementation issues, perceived ethical challenges for decision-makers, and high acquisition costs and/or substantial budget impact.13,26 As such, the process is most commonly applied during the review of DRDs.
Recommendation Process
CADTH does not have an expert committee specifically for the review of DRDs, and recommendations are issued by 1 of the 3 standing committees depending on the type of product and target drug formulary (i.e., non-oncology drugs, oncology drugs, or plasma protein products).13 These committees issue recommendations according to a framework, which has been modified to address some of the unique challenges associated with generating evidence for DRDs. The framework allows the committee to issue recommendations that are in favour of reimbursement, despite limitations with the available evidence, when a DRD has the potential to address significant unmet medical need for the target population.13
The modified deliberative process can be applied in situations where the drug under review is indicated for a condition with the following characteristics: (1) is life-threatening, seriously debilitating, or both serious and chronic in nature; (2) affects a relatively small number of patients (incidence of less than 5 in 10,000, but typically closer to 1 in 100,000); (3) is often genetically based, with onset at birth or early childhood, and leads to a shortened lifespan; (4) places a heavy burden on caregivers and the health care system; (5) is difficult to study because of the small patient population; (6) there is an absence of clinically effective drug or non-drug alternative treatments; (7) substantial morbidity and mortality exist despite the available drug or non-drug alternative treatments.13 For drugs and indications that meet these criteria the committee may issue a recommendation in favour of reimbursement despite limitations with the evidence, provided the available evidence reasonably suggests that the drug could substantially reduce morbidity and/or mortality associated with the disease and the condition can be identified with reasonable diagnostic precision.13
Figure 1: CADTH Process Enhancements in the Review and Recommendation Phases for DRDs and Other Complex Drugs
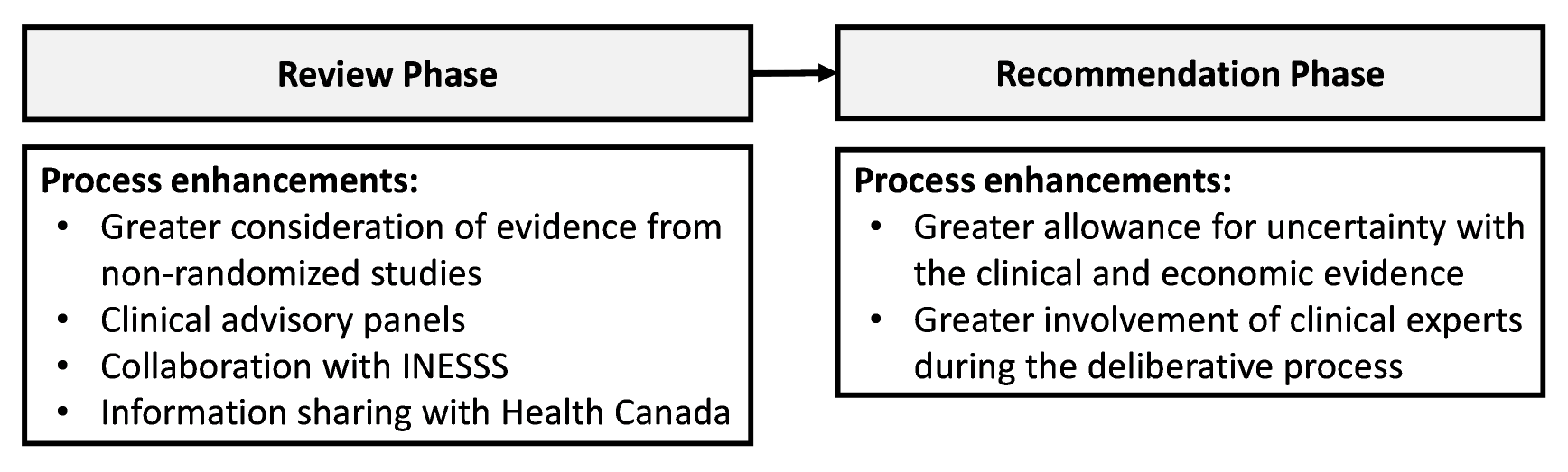
INSESS = Institut national d’excellence en santé et en services sociaux.
Canada (Quebec)
Program Overview
INESSS conducts HTA for the province of Quebec and issues recommendations to the Ministry of Health and Social Services regarding drug reimbursement.35 Similar to CADTH, INESSS has specialized accommodations for DRDs and other complex files throughout its review and recommendation processes.
Administrative Processes
INESSS currently applies the same application process, review timelines, and fees for DRDs and other drugs.36
Review Process
INESSS has demonstrated specialized accommodations for DRDs and other complex files with respect to patient and caregiver engagement,25 formation of clinical advisory panels in collaboration with CADTH,26 and greater allowances for alternative non-randomized trial evidence.37 With respect to patient engagement, INESSS has conducted interviews and focus groups with patients and caregivers as part of the review process for DRDs.25
Recommendation Process
There is no specialized DRD committee in the INESSS process, and all drugs are reviewed by the Comité scientifique d'évaluation des médicaments aux fins d'inscription (CSEMI).32,38 INESSS has issued recommendations for DRDs based on the promise of therapeutic value that are conditional upon additional evidence collection.25,39
UK (England)
Program Overview
NICE conducts HTA and advises the NHS in England on the clinical effectiveness, cost-effectiveness, and service impact of new and emerging as well as established health care technologies.27,40 NICE has a separate review process for drugs indicated for use in the treatment of “very rare conditions,” known as the HST process.8,27 Guidance for the HST process does not provide a definition for very rare8; however, the 13 drugs that have been reviewed through the HST process to date were indicated for diseases that would typically be considered ultra-rare diseases based on criteria used by other HTA agencies (i.e., familial chylomicronemia syndrome, Batten disease, inherited retinal dystrophies caused by RPE65 gene mutations, hereditary transthyretin amyloidosis, X-linked hypophosphatemia, adenosine deaminase deficiency, hypophosphatasia, Gaucher disease, Fabry disease, Duchenne muscular dystrophy with nonsense mutation, mucopolysaccharidosis (MPS) type IVA, atypical hemolytic uremic syndrome).41
Administrative Processes
The process for selecting drugs for review through the HST is similar to the process used by NICE for other technologies; however, the drugs need to demonstrate that they meet all of the eligibility criteria for the HST process8,18:
Target patient group for the technology in its licensed indication is so small that treatment will usually be concentrated in very few centres in the NHS
Target patient group is distinct for clinical reasons
Disease is chronic and severely disabling
Technology is expected to be used exclusively in the context of a highly specialized service
Technology is likely to have a very high acquisition cost
Technology has the potential for lifelong use
Need for national commissioning of the technology is significant8,27
The submission process for HST evaluation is similar to NICE standard technology appraisal, but with dedicated proforma and submission templates.8,42,43 The submission templates for the HST and single technology appraisal processes are similar, but with some modifications to the HST template to accommodate additional information (e.g., evidence on the burden of illness).27
Review Process
THE HST review process is similar to the standard single technology appraisal process used by NICE. A report on the comparative clinical and cost-effectiveness of the drug is prepared by an Evidence Review Group.27 Opportunities for stakeholder input and feedback are the same for the HST and standard single technology appraisal process. The guidance from NICE states that the preferred methods for evaluating the cost-effectiveness of drugs reviewed through the HST process are consistent with the standard technology appraisal process; however, some considerations are made for drugs reviewed through the HST that may allow for different weighting of QALYs.8,27
Recommendation Process
NICE recommendations for the drugs reviewed through the HST process are issued by the Highly Specialised Technologies Evaluation Committee (HSTEC), which is distinct from the Technology Appraisal Committees used for the standard NICE processes for single and multiple technology drug reviews.28 HSTEC members are appointed on a 3-year term, and members are drawn from the NHS, patient and carer organizations, academia, and pharmaceuticals and medical devices industries.28
The evaluation and decision-making processes for HST are largely similar to NICE’s standard technology appraisal. HSTEC therefore considers advice from NICE on the appropriate approach to making scientific and social value judgments. HSTEC considers the following factors in its deliberation: the nature of the condition, the clinical effectiveness, value for money, and the impact of the technology beyond direct health benefits.27 A key difference is the willingness-to-pay threshold for drugs reviewed the HST process (£100,000 to £300,000 per QALY) compared with those reviewed through the standard process (£20,000 to £30,000 per QALY).
UK (Scotland)
Program Overview
The Scottish Medicines Consortium (SMC) advises NHS Scotland regarding which medicines provide good value for patients.11,19 SMC has established additional processes within its evaluation framework for orphan drugs (prevalence less than 5 per 10,000) and a new process for ultra-orphan drugs (prevalence not exceeding 1 in 50,000 in Scotland) that was introduced in 2019.21,44
Administrative Processes
Orphan Drugs
For orphan drugs, companies have the option to indicate whether they wish their submission to be considered under the end of life or orphan process in the event of “not recommended” advice from the New Drugs Committee; that is, with the option for a PACE meeting and/or opportunity for new or revised Patient Access Scheme (PAS). The process for orphan drugs includes modifications to the standard HTA process, including additional patient and clinician engagement (i.e., PACE meetings) and extended timelines (22 to 26 weeks) relative to the standard review process (18 weeks).11,24,45
Ultra-Orphan Drugs
For ultra-orphan drugs, sponsors should confirm that the drug meets the criteria for consideration through the ultra-orphan review process by completing an ultra-orphan proforma before filing the application.12,46 For products that meet the ultra-orphan eligibility criteria, sponsors must meet the following conditions:
Make a full submission to the SMC for the initial assessment stage that meets SMC requirements for assessment under the ultra-orphan process
Offer a PAS that complies with the standard terms and conditions considered acceptable by the Patient Access Scheme Assessment Group (PASAG)
Support the data collection arrangements that meets the evidence generation requirements for assessment under the ultra-orphan pathway12
Applications for ultra-orphan drugs are filed using a dedicated submission template (i.e., New Product Assessment Form for Ultra-Orphan Medicines) for the initial assessment by SMC.21 The company will be required to generate additional evidence and file for reassessment with SMC after a period of 3 years.12,47
Review Process
Orphan Drugs
Companies opting for an assessment via the PACE process must provide additional information on the categorization of the medicine and supporting evidence and rationale for this categorization. This should include data on the prevalence of the condition in the fully licensed indication in NHS Scotland. All other sections of the New Products Assessment Form, including economic analysis, are the same for all full submissions or re-submissions of any new product.31,44
Ultra-Orphan Drugs
The ultra-orphan process involves 4 steps: (1) confirmation that the drug meets the criteria to be considered an ultra-orphan medicine; (2) submission to SMC for initial assessment of the clinical and cost-effectiveness, focusing on areas of uncertainty that should be addressed with additional evidence generation; (3) data collection by the manufacturer for a period of up to 3 years to address areas of uncertainty with the evidence; (4) submission to SMC for reassessment and decision on whether or not the drug should be available for routine usage in NHS Scotland.12,47,48
SMC conducts an initial assessment of the clinical and cost-effectiveness for the ultra-orphan drug. This assessment highlights uncertainties within the available evidence and will be used to inform the data collection and evidence generation stage of the review pathway. The initial assessment is completed in approximately 18 weeks.12 Patient groups have the opportunity to provide input during the initial assessment. PACE meetings are not offered during the initial assessment but may occur as part of the reassessment phase of the process. Sponsors must file a PAS for all ultra-orphan drugs. The PASAG, which operates separately from SMC, will review the PAS proposal and advise on the feasibility of implementation. Guidance for the ultra-orphan review pathway notes that companies are encouraged to offer a fair price in return for the flexibility of the ultra-orphan process, which provides market access for a period of 3 years and opportunities to address uncertainty in the evidence.12
The next phase of the process involves a period of up to 3 years for the sponsor to collect additional data for the drug to address any areas of uncertainty highlighted in the initial assessment conducted by SMC. Sponsors will need to develop a data collection plan to be accepted into the ultra-orphan pathway.47 The Scottish Government has established guidance for sponsors regarding the evidence generation phase of the process, with advice on planning, collection, governance, and reporting.
Within the 3-year period, sponsors will be required to submit an updated full submission for reassessment by SMC, including the additional evidence generated through the initial market access period. The reassessment is conducted by SMC within 22 weeks (e.g., similar to the timelines for the orphan drug process) and involves a reimbursement recommendation from the New Drugs Committee and the SMC Committee.
Recommendation Process
Both orphan drugs and ultra-orphan drugs are assessed by the New Drugs Committee and SMC Committee for a recommendation regarding whether or not they should be accepted for use by NHS Scotland and under what conditions reimbursement should be considered. The recommendation is issued as part of the initial assessment for orphan drugs and part of the reassessment for ultra-orphan drugs.11,12,19 The framework for the committees allows greater acceptance of uncertainty in the economic evaluation and may allow for the committee to accept a higher willingness-to-pay threshold for DRDs.
If the committee issues a draft recommendation that is not in favour of reimbursement, the sponsor may request that SMC convene a PACE meeting. The purpose of PACE is to gather detailed information on the drug that may not be fully captured within the conventional clinical and economic assessment process. This may allow for a more in-depth discussion on the potential benefits of the drug under review, including its impact on the quality of those living with the condition. The sponsor is given the opportunity to provide information as part of the PACE process, including commentary on unmet need, severity of the condition, added value of the drug under review for patients and caregivers, and details of any subgroups who may benefit most from treatment. The sponsor is also provided with the opportunity to file a new or revised PAS aimed at improving the cost-effectiveness of the drug.
Figure 2: SMC New Ultra-Orphan Review Process
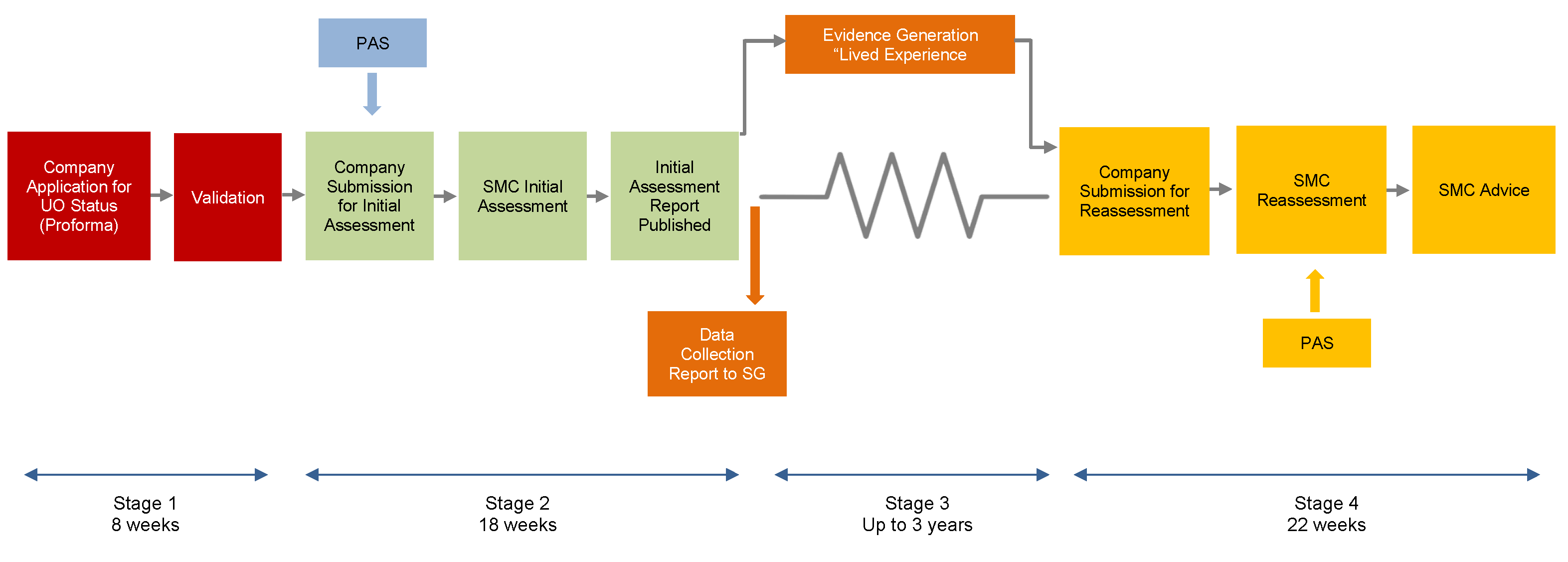
UO = ultra-orphan; PACE = Patient and Clinician Engagement; PAS = Patient Access Scheme; SG = Scottish Government.
Source: Figure from A Guide to the Ultra-Orphan Pathway.12 Contains public sector information licensed under the Open Government Licence v3.0. http://www.nationalarchives.gov.uk/doc/open-government-licence/version/3/
Germany
Program Overview
The G-BA issues directives for the benefits catalogue of the statutory health insurance funds in Germany.49 The G-BA commissions the IQWiG to conduct early assessments to examine the added benefits of a medicine and to make recommendations.5,7,49 Legislation in Germany grants orphan drugs a special status in the early benefit assessment of drugs, whereby its added benefit is already considered proven through market authorization, with IQWiG only assessing information provided by the manufacturers on the number of patients affected by the rare disease and the cost of treatment.5,7 This special provision only applies to orphan drugs with sales not exceeding €50 million over a 12-month period. Orphan drugs with sales that exceed €50 million over 12 months undergo the standard HTA evaluation process conducted by IQWiG, as commissioned by the G-BA, using the same method as for other drugs.5,7,16
Administrative Processes
For orphan drugs with revenues not exceeding €50 million in the past 12 months, sponsors do not need to submit proof of medical benefit and additional medical benefit over an appropriate comparator to IQWiG. Additional benefit of the drug is assumed to be proven once it has received market authorization. The G-BA evaluates the magnitude of therapeutic benefit for relevant patient groups to create the basis for price negotiations with the sponsor.7,16 For orphan drugs with revenues exceeding €50 million in the past 12 months, the sponsor must file a standard submission for evaluation of additional benefit by IQWiG.7,16
Review Process
For orphan drugs with revenues not exceeding €50 million in the past 12 months, IQWiG is commissioned to assess information provided by the manufacturers on the number of eligible patients (plausibility check of the epidemiological model) and the costs of the drug under review.7,16 For any orphan drugs with revenues exceeding €50 million in the past 12 months, IQWiG would be commissioned to conduct an evaluation of additional benefit using their standard methodology and based on a complete submission by the sponsor.7,16
Recommendation Process
The G-BA decides the extent of the additional benefit for all new drugs in Germany. As added benefit is already concluded by market authorization of orphan drugs, the G-BA will determine the magnitude of benefit (i.e., major, considerable, minor, or non-quantifiable) for those with revenues not exceeding €50 million in a 12-month period.7,16 For orphan drugs exceeding the €50 million threshold, the G-BA would make a decision based on IQWiG’s complete assessment and considering all available options.7,16
Australia
Program Overview
PBAC is an independent expert body appointed by the Australian Government to primarily recommend new medicines for listing on the Pharmaceutical Benefits Scheme.50,51 A subset of drugs for ultra-rare diseases that have been rejected by PBAC for economic reasons may be eligible for reimbursement through the Australian LSDP (described below in the section on funding mechanisms).
Administrative Process
PBAC administrative processes are the same for DRDs and other drugs, with the exception that the PBAC guidelines specify that sponsors may submit additional information to describe and justify any claims for why the rule of rescue should be considered for the drug under review and the opportunity to convene a stakeholder meeting (described in detail below).22
Review Process
PBAC applies the same review process for DRDs as with other drugs, with specialized accommodations introduced at the recommendation phase of the process.22
Recommendation Process
The PBAC does not apply a separate evaluation framework for DRDs; however, a provision within the standard PBAC process, called the “rule of rescue;” that is, the value for rescuing life regardless of the cost of the treatment.22 Criteria outlined in the rule of rescue, if met, allows flexibility in exceptional circumstances and may be particularly influential in favour of reimbursement. The criteria are focused on disease severity (e.g., likely to lead to premature death), small patient populations (threshold not defined), unmet medical need (absence of alternative treatment options), and evidence that the drug may provide clinical improvement (Table 3).22 A decision to invoke the rule of rescue is particularly relevant when the committee would reject reimbursement due to comparative cost-effectiveness. A recommendation in favour of reimbursement may be favoured under the rule of rescue, irrespective of a high incremental cost-effectiveness ratio.
Stakeholder Meetings may be convened when a drug has not been recommended by PBAC. These meetings are reserved for drugs that treat serious, disabling, or life-threatening conditions, where there are no other realistic treatment options for that condition but where insufficient cost-effectiveness prevents PBAC from recommending listing. Hence, they are applicable to DRDs that have not been recommended for reimbursement. The objective of the meeting is to gain additional insights from stakeholders regarding the drug under review.23
New Zealand
Program Overview
PHARMAC, a government agency, makes decisions on which drugs to publicly fund in New Zealand.29,52 Drugs indicated for the treatment of ultra-rare diseases (prevalence of less than 1 in 50,000 people in New Zealand) are reviewed through a modified process for rare disorders.9,15,52 The current process used by PHARMAC was developed following a 2014 pilot process that was initiated to stimulate competition across manufacturers of drugs for ultra-rare diseases. The pilot concluded in 2017 and led to the funding of 10 new drugs and a series of process improvements for how drugs for ultra-rare diseases are managed through the PHARMAC process.15,53,54 New Zealand has additional processes in place to provide case-by-case funding for patients with rare diseases (described in Funding Programs).15,55
Administrative Process
The modified DRDs process includes opportunities for earlier application filing and opportunities for pre-engagement with PHARMAC. These applications may be filed before approval by the New Zealand regulatory authority (Medsafe).9,52 The application requirements and review timelines are the same for DRDs and other drugs.29
Review Process
PHARMAC applies the same review process for DRDs and other drugs, with specialized accommodations introduced at the recommendation phase of the process.9
Recommendation Process
Submissions for drugs for ultra-rare diseases are considered by a dedicated Rare Disorders Subcommittee of the Pharmacology and Therapeutics Advisory Committee.9,52 The subcommittee reviews the evidence for health benefit and need, then issues a recommendation to the Pharmacology and Therapeutics Advisory Committee, who then makes a final determination. The factors considered by the committees for DRDs are the same as those considered for all drugs: (1) need, (2) health benefits, (3) costs and savings, and (4) suitability.56 Each of these factors is evaluated for 3 different aspects: the individual; the family, whānau, and society; and the health system.56 PHARMAC has noted that factors that are particularly relevant for DRDs are:
The health needs of the person (as rare diseases are often deliberating and severe illness)
The health needs of others (as caring for someone with a rare disease can have a significant impact on the health and well-being of those with that responsibility)
The availability and suitability of alternative treatments (as there are often few options for those living with rare diseases)9,15
US
Program Overview
ICER is an independent and non-partisan research organization that objectively evaluates the clinical and economic value of health care and health care delivery innovations, including prescription drugs.17,57,58 The key modification for DRDs in the ICER process involves the application of a modified version of its value assessment framework for drugs for ultra-rare diseases (prevalence not exceeding 10,000 Americans; approximately 3 in 100,000).10 These modifications were introduced following a multi-stakeholder consultation that was undertaken in 2017.59,60
Administrative Processes
Unlike the other HTA processes included in this report, the ICER process is not initiated by drug manufacturers filing an application and seeking reimbursement from a set of specific public payers. As a result, there are no administrative process adjustments for DRDs. The modified approach to value assessment is applied for treatments for ultra-rare diseases when:
An eligible patient population for the treatment indication(s) included in the scope of the ICER review is estimated at fewer than approximately 10,000 individuals
There are no ongoing or planned clinical trials of the treatment for a patient population greater than approximately 10,000 individuals.10
As part of the initial stakeholder consultation on a topic, ICER includes a draft recommendation its initial draft scoping document noting whether or not the drug(s) of interest will be assessed in accordance with the framework for ultra-rare diseases. Stakeholders have the opportunity to comment on the recommendation and ICER will make the final decision regarding application of the modified framework for ultra-rare diseases.10
Review Process
ICER uses its standard Evidence-Based Medicine matrix to assess the comparative clinical effectiveness of treatment of ultra-rare diseases (i.e., the instrument used to evaluate net health benefit and the level of certainty with the evidence).10,61 However, ICER provides specific commentary in its reports noting the challenges in generating robust evidence when studying ultra-rare diseases (e.g., need for alternative study designs, use of surrogate outcome measures, absence of long-term safety, and use of historical control groups).
For all drugs, including those that meet the criteria for ultra-orphan drugs, ICER provides willingness-to-pay threshold results from US$50,000 per QALY per equal value of life years gained (evLYG) to US$200,000 per QALY per evLYG. There is no specialized quantitative weighting system to different magnitudes of QALY gains. However, the following modifications are made in the reports: additional context is included noting that decision-makers often give special weighting to other benefits and to contextual considerations that may result in a drug for an ultra-rare disease being reimbursed at higher prices in comparison with treatments for more common conditions; presenting a scenario analysis with broader societal costs in tandem with the base-case analysis when such costs are substantial; and, when necessary, conducting a search for “mapping” studies to help translate surrogate outcomes into quality of life measures.10
Recommendation Process
ICER does not use a specialized committee for the evaluation of drugs reviewed through the ultra-rare process. Recommendations are issued by 1 of the 3 committees used by ICER (California Technology Assessment Forum, Midwest Comparative Effectiveness Public Advisory Council, New England Comparative Effectiveness Public Advisory Council).58 During their deliberations, the appraisal committee(s) vote on the long-term value for money of treatments for serious ultra-rare diseases using the same approach as other interventions by having members vote on the value regardless of the base-case results (i.e., even if results exceed US$200,000 per QALY per evLYG).10
France
Program Overview
HAS is the French HTA body that evaluates drugs and issues recommendations to the Ministère des Solidarités et de la Santé for inclusion on the list of reimbursable drugs.62
Administrative Processes
HAS does not have specialized review processes specifically for DRDs but offers 2 expedited review pathways (the procédure d’instruction anticipée and the procédure de pré-dépôt) that can be applied to DRDs (as well as other products that may fulfill unmet medical needs).14,62,63
Review Process
HAS applies the same review process for DRDs as with other drugs.
Recommendation Process
HAS does not use a specialized review committee for DRDs, and recommendations are issued by the Transparency Committee for all drugs. The committee is able to issue recommendations in favour of reimbursement that are conditional upon additional evidence generation to verify the clinical benefit of the drug within 3 years.63
Table 3: Eligibility for Specialized or Modified Processes for DRDs
HTA Agency | Eligibility | Implications |
CADTH | The condition for which the drug is indicated has the following characteristics:
There is an absence of clinically effective drug or non-drug alternative treatments. Substantial morbidity and mortality exist despite the available drug or non-drug alternative treatments.13 | Accommodations (as a result of modifications) in the deliberative process |
NICE | HSTs are selected using the following criteria, all of which have to apply:
| Separate review process |
SMC | Ultra-orphan drugs:To be considered as an ultra-orphan medicine, all these criteria should be met: | Separate review process |
Orphan drugs: A drug with EMA designated orphan status or a medicine to treat an equivalent size of population (< 5 per 10,000) irrespective of whether it has designated orphan status.31,44 | Modified review process | |
G-BA | Drug has been designated as an orphan medicinal product by the EMA, and the value of sales is < €50 million per 12 months.7 | Separate review process |
PBAC | The rule of rescue may be applied in exceptional circumstances when the 4 criteria are met:
| Accommodations in the deliberative process |
PHARMAC | Applications for drugs that meet all 3 policy principles:
| Modified application process |
ICER | Modified approach to value assessment for treatments for ultra-rare diseases when:
| Modified value assessment framework |
EMA = European Medicines Agency; G-BA = Gemeinsamer Bundesausschuss or Federal Joint Committee; HST = highly specialised technologies; ICER = Institute for Clinical and Economic Review; LSDP = Life Savings Drug Program; NICE = National Institute for Health and Care Excellence; PBAC = Pharmaceutical Benefits Advisory Committee; PHARMAC = Pharmaceutical Management Agency; SMC = Scottish Medicines Consortium.
Funding Programs for Drugs for Rare Diseases
Canada
As shown in Table 4, the public drug programs in Canada have established a variety of processes to evaluate claims and facilitate the reimbursement of DRDs. All the drug programs use special and/or exceptional authorization processes for DRDs that involve the completion of request forms that demonstrate the patient meets a series of criteria to be considered eligible for reimbursement. These criteria are typically informed through the recommendations issued by CADTH and INESSS (e.g., initiation criteria, renewal criteria, discontinuation criteria, and administration criteria). Details regarding DRD-focused reimbursement processes are provided in the following sections by jurisdiction. Many of the drug programs also offer compassionate or exceptional access for patients on a case-by-case basis for drugs that are not otherwise reimbursed through alternative mechanisms (e.g., not approved in Canada or failure of other treatment options).64
Table 4: Reimbursement Processes for DRDs by Canadian Jurisdiction
Jurisdiction | Reimbursement processes for DRDs |
British Columbia | BC Pharmacare (Special Authority)65 |
Alberta | Alberta Drug Benefit List (Special Authorization)66 |
Rare Diseases Drug Coverage Program67 | |
Short Term Exceptional Drug Therapy Program68 | |
Saskatchewan | Saskatchewan Formulary (Exception Drug Status Program)69 |
Inherited Metabolic Disease Benefits70 | |
Manitoba | Manitoba Pharmacare Drug Formulary (Exception Drug Status) |
Ontario | Ontario Drug Benefit Program (Exceptional Access Program)71 |
Inherited Metabolic Diseases Program72 | |
Special Drug Programs73 | |
Compassionate Review Policy74 | |
Quebec | RAMQ List of Medicines (Exceptional Medications)75 |
New Brunswick | New Brunswick Drug Plan (Special Authorization) |
New Brunswick Drugs for Rare Diseases Plan76 | |
Nova Scotia | Nova Scotia Pharmacare (Exception Status Drugs)77 |
Prince Edward Island | PEI Pharmacare Formulary (Special Authorization)78 |
Newfoundland and Labrador | NL Prescription Drug Program Formulary (Special Authorization)79 |
Exceptional Review Process80 | |
Yukon | Yukon Drug Formulary (Exception Drug Status)81 |
Northwest Territories | Specified Disease Conditions Program82 |
Nunavut | Extended Health Benefits72 |
Non-Insured Health Benefits | NIHB Drug Benefit List (limited use benefits)83 |
Exception drugs83 |
AHS = Alberta Health Services; NIHB = Non-Insured Health Benefits; PEI = Prince Edward Island; RAMQ = Régie de l'assurance maladie du Québec.
British Columbia6,64
British Columbia uses an Expensive Drugs for Rare Diseases (EDRD) program for making case-by-case reimbursement decisions about DRDs.
Patient eligibility: Residents of British Columbia who are actively enrolled in the BC Medical Services Plan.
Patient cost-sharing: None
Eligible drugs: Eligibility is determined on a case-by-case basis, but the EDRD process uses the following for guidance: drugs that have an annual cost of $100,000 or more per patient and are indicated for a non–cancer-related condition.
Funds SAP drugs: Unknown
Process for accessing drug coverage: Physicians submit reimbursement requests for specific patients. An expert advisory committee (comprised of pediatric and adult rare diseases specialists, a medical geneticist, pharmacists, health administrators, a health economist, and an ethicist) reviews the request and makes a recommendation on a case-by-case, exceptional, last-resort basis. The committee’s recommendations are also supported by an initial review and recommendation from clinical expert subcommittees.
Transparency of decisions: The decision and rationale are not made public.
Alberta6,64,66,84
In Alberta, access to DRDs occurs through the Alberta Drug Benefit List (ADBL). Additionally, access to some DRDs is also available through the Rare Diseases Drug Coverage Program and the Short Term Exceptional Drug Therapy (STEDT) program.
The Alberta Drug Benefit List (ADBL)
There are a number of DRDs on the ADBL listed with a special authorization status.
Patient eligibility: Patients must enroll into 1 of the community drug and health benefit programs sponsored by the Alberta government and administered by Alberta Blue Cross.
Patient cost-sharing: Varies based on the cost-sharing rules of the program the patient is enrolled in.
Eligible drugs: Confirmation of whether a DRD is listed on the ADBL can be checked by searching the ADBL.
Funds SAP drugs: No
Process for accessing drug coverage: The patient’s specialist physician must complete a Special Authorization request form specific to the drug and indication requested and send it to Alberta Blue Cross. The form specifies clinical criteria. The request may require a review by an expert clinician and a formal approval by Alberta Health. The patient and the requesting physician may be required to sign a consent form. Alberta Blue Cross notifies the patient’s specialist about the funding decision. Funding is provided for a finite period of time. Ongoing funding requires an application to renew coverage.
Transparency of decisions: Funding decisions for individual cases are not public record.
The Rare Diseases Drug Coverage Program
The Rare Diseases Drug Coverage Program enables access to a defined set of DRDs. A rare disease is defined as a genetic disorder that occurs in fewer than 1 in 50,000 Canadians or fewer than 50 Albertans.
Patient eligibility: Patients must be registered in the Alberta Health Care Insurance Plan and have no other drug or funding options available. Patients must meet the clinical criteria and must not have an additional significant illness that is likely to affect life expectancy.
Patient cost-sharing: None
Eligible drugs: Drug products must have a Notice of Compliance. Examples of diseases currently eligible for coverage consideration under the program include MPS type I and type VI, Hunter syndrome, Pompe disease, atypical hemolytic uremic syndrome, and Niemann-Pick disease type C.
Funds SAP drugs: No
Process for accessing drug coverage: A Rare Disease Specialist must complete a STEDT/Rare Diseases Drug Coverage Request form and forward it to STEDT. Each application is screened for completeness and then forwarded to Alberta Health for final approval. STEDT notifies the individual’s Rare Disease Specialist about the funding decision. Approvals are granted for an initial period of 6 months and renewals are granted for a maximum of 12 months.
Transparency of decisions: Funding decisions for individual cases are not public record.
The Short Term Exceptional Drug Therapy (STEDT) Program
The STEDT Program enables Albertans with life-, limb-, or organ -threatening medical conditions to access, on a case-by-case basis, high-cost drug therapies when no other treatment or funding options exist.
Patient eligibility: Patients must be registered in the Alberta Health Care Insurance Plan and have no other drug therapy or funding options available.
Patient cost-sharing: None
Eligible drugs: There is no defined list of eligible drugs. The drug requested must be for a condition that is directly or indirectly life-, limb-, or organ-threatening where conventional therapies have failed or are not tolerated, and the patient’s clinical circumstances are rare. The requesting specialist must reference studies to support the request. The total cost of the requested drug must be at least $1,500 per patient per year. Drugs used in the direct treatment of cancer are not eligible. Drugs under review by CADTH, Alberta’s Expert Committee on Drug Evaluation and Therapeutics, or under negotiations by the pan-Canadian Pharmaceutical Alliance are not eligible.
Funds SAP drugs: Yes
Process for accessing drug coverage: Same as the Rare Diseases Drug Program.
Transparency of decisions: Funding decisions for individual cases are not public record.
Saskatchewan64,69,85
In Saskatchewan, reimbursement requests for rare disease drugs, including expensive drugs for rare diseases, may be considered for coverage. Some of these drugs may be included in the EDS program (such as Myozyme, Kalydeco, Spinraza) or the Inherited Metabolic Disease Benefit List (such as Kuvan and medications for Fabry disease).
Patient eligibility: Patients must be enrolled in the Saskatchewan Drug Plan.
Patient cost-sharing: Coverage for listed or approved medications is subject to the patient’s deductible and co-payment for the majority of drugs.
Eligible drugs: In general, drugs may be listed with EDS in a variety of circumstances when there is a concern regarding the appropriate place in therapy or the potential cost impact of the medication. In addition to this, a limited number of rare disease medications are captured within the Inherited Metabolic Disease Benefit List. Requests for high-cost drugs, or for the use of other non-listed medications or indications, may be considered on a case-by-case basis.
Funds SAP drugs: SAP drugs may be considered on a case-by-case basis.
Process for accessing drug coverage: Prescribers and pharmacists may submit applications on behalf of their patients for drugs listed on the EDS program or the Inherited Metabolic Disease Benefit List. In addition, prescribers and pharmacists may initiate case-by-case requests for drugs not otherwise listed on the Drug Plan for a specific patient. Case-by-case requests will be assessed and may be taken to the Drug Advisory Committee of Saskatchewan for consideration and advice regarding coverage. Patients are notified by letter if coverage has been approved and for what time period. If a request has been denied, letters are sent to the patient and prescriber to notify them of the decision.
Transparency of decisions: Decisions regarding drug coverage applications for individual patients are not made public.
Manitoba64,86,87
Manitoba does not have a dedicated process for enabling access to DRDs. Reimbursement requests are considered case-by-case under the Exception Drug Status (EDS) Program.
Patient eligibility: Manitoba residents enrolled in the Manitoba Pharmacare Program.
Patient cost-sharing: There is an annual, income-based deductible in which the beneficiary must pay out-of-pocket for their prescription drugs until the deductible is reached, after which Pharmacare will pay 100% of eligible prescription costs for the remainder of the benefit year. There are no premiums or co-payments.
Eligible drugs: Drugs are approved for coverage under the EDS Program if they meet specific criteria and upon review and recommendation of the Manitoba Drug Standards and Therapeutics Committee (MDSTC). In general, the drugs fall into 1 of the following categories:
A drug that is ordinarily administered to hospital in-patients is administered outside of a hospital because of unusual circumstances
A drug that is required in the diagnosis and/or treatment of an illness, disability, or condition rarely found in Manitoba
The evidence provided to the minister, in accordance with established criteria, supports a specific treatment regime which includes use of the drug or other item.
Funds SAP drugs: SAP drugs may be considered on a case-by-case basis.
Process for accessing drug coverage: Duly licensed practitioners prescribing within their scope of practice may submit an application to the EDS Program. Applications for drugs for rare diseases with established EDS criteria are assessed by a pharmacist consultant with the EDS Program. Drugs without established EDS criteria may be considered case-by-case by the Provincial Drug Programs Review Process. Prescribers and patients are notified by letter on whether coverage has been approved or denied.
Transparency of decisions: Decisions or rationales are not made public.
Ontario6,71,73,88
Ontario does not have a dedicated process for enabling access to DRDs. Once the decision to fund a drug under Ontario’s public drug programs has been made — subsequent to completion of the national health technology assessment and negotiation processes — reimbursement requests are usually considered on a case-by-case basis under the Exceptional Access Program. In addition, the Special Drugs Program covers the full cost of a drug to treat Gaucher disease, and the Inherited Metabolic Diseases Program covers the full cost of listed treatments for certain inherited metabolic diseases.
Patient eligibility: To qualify for the Exceptional Access Program, patients must qualify for the Ontario Drug Benefit program, be prescribed the drug by an Ontario doctor or nurse practitioner (Quebec or Manitoba doctors may prescribe if the patient lives on the border with either of these provinces), and have the prescription filled at a pharmacy in Ontario that has an account with the Ontario Ministry of Health to dispense drugs under the Ontario Drug Benefit program.
To qualify for the Special Drugs Program, patients must live in Ontario, have a valid Ontario health card, be under the care of an Ontario doctor at a designated hospital, and meet the conditions for coverage as specified in the Health Insurance Act.
To qualify for the Inherited Metabolic Diseases Program, patients must live in Ontario, have a valid Ontario health card, be diagnosed with 1 of the disorders covered by the program, and be under the care of a doctor from a designated treatment centre. Patients do not qualify if they receive coverage through private insurance or employee benefits.
Patient cost-sharing: Reimbursement through the Ontario Drug Benefit Program, including the Exceptional Access Program, is subject to deductibles and/or co-payments. The Special Drugs Program and the Inherited Metabolic Diseases Program cover the full cost of reimbursed treatments.
Eligible drugs: The Exceptional Access Program facilitates patient access to drugs not funded on the Ontario Drug Benefit Formulary or if no listed alternative is available.
The Special Drugs Program covers imiglucerase for patients with Gaucher disease.
The Inherited Metabolic Diseases Program covers drugs identified in the Program’s List of Disorders, Covered Drugs, Supplements and Specialty Foods.
Funds SAP drugs: Yes, funding may be considered on a case-by-case basis.
Process for accessing drug coverage: Only a doctor or nurse practitioner (on behalf of an individual patient) can apply for coverage from the Exceptional Access Program, which includes the Compassionate Review Policy. Each application is considered by pharmacists who specialize in the Exceptional Access Program. Sometimes applications are sent out for medical expert review. These pharmacists and experts base their review on criteria approved through the established HTA process. The Executive Officer of the Ontario Public Drug Programs (OPDP) makes the final decision.
To access imiglucerase under the Special Drugs Program, it must be prescribed by a physician, the use of the medication must be recommended by the Gaucher’s Disease Review Committee, and the medication must be provided to a patient with Gaucher disease.
For the Inherited Metabolic Diseases Program, Ontario physicians can submit applications for new products to be listed. Completed applications are assigned to a primary reviewer who then submits a report to the Inherited Metabolic Diseases subcommittee. The subcommittee then makes funding recommendations to the Executive Officer of OPDP. For some drug products, the Executive Officer may ask the Committee to Evaluate Drugs to provide a separate review and recommendation. The Executive Officer makes the final funding decision.
Transparency of decisions: For new drugs or indications (including DRDs) reviewed through the CADTH reimbursement review process, broad funding decisions (i.e., the decision that a drug product will or will not be considered for coverage) are posted at the Ministry of Health’s Status For Single-Source Submissions webpage. Criteria for coverage, as applicable, are posted online for drugs reimbursed via the Exceptional Access Program and Inherited Metabolic Diseases program.
Quebec64,75,89,90
Quebec does not have a dedicated process for considering reimbursement requests for DRDs. Under the Régie de l’assurance maladie de Québec (RAMQ), most listed DRDs would be listed as medicament d’exception. DRDs not listed could be covered on a case-by-case basis via the Exceptional Patients program.
Patient eligibility: Everyone who is permanently settled in Quebec must have prescription drug insurance coverage at all times, either through the public plan or through a private plan. Only those persons who are not eligible for a private plan may register for the public plan.
Patient cost-sharing: Those covered by the public plan must pay an annual premium, with some exceptions.
Eligible drugs: Quebec has an “exception patient program” for the treatment of severe medical conditions. This public plan covers certain prescription drugs, notably those not on the list of medications for insured persons with an exceptional need. Exceptional medicines must be used for the therapeutic indications recognized by INESSS.
Funds SAP drugs: No
Process for accessing drug coverage: The health professional sends an authorization request to RAMQ, which ultimately decides whether the reimbursement is authorized or not.
Transparency of decisions: RAMQ informs prescribers and insured persons of its decisions. Funding decisions are not posted publicly.
New Brunswick64,76
The New Brunswick Drugs for Rare Diseases Plan (NBDRDP) provides assistance with the cost of certain drugs for specific rare diseases.
Patient eligibility: Patients must be permanent residents of New Brunswick with a valid Medicare card who meet the clinical criteria for the drug requested.
Patient cost-sharing: None
Eligible drugs: The NBDRDP considers requests for a defined set of drugs: Aldurazyme for the treatment of Hurler and Hurler Scheie forms of MPS type I; Elaprase for the treatment of Hunter syndrome; Ilaris for the treatment of cryopyrin-associated periodic syndrome; Myozyme for infantile, early-onset, and adult or late-onset Pompe disease; Naglazyme for the treatment of MPS type VI; and Zavesca for the treatment of Niemann-Pick type C. New DRDs follow the same drug review process as other drugs considered for reimbursement by the public drug plans.
Funds SAP drugs: No
Process for accessing drug coverage: A request form for a listed drug must be completed by a physician for each individual patient. Requests for coverage are assessed by the external medical experts.
Transparency of decisions: Funding decisions for individual cases are not public record.
Nova Scotia77
Nova Scotia does not have a dedicated process for enabling access to DRDs. In general, DRDs are listed and processed as per other benefits in the Nova Scotia Pharmacare Programs. Funding for some medications may be provided by the hospital system via an Exception Drug Fund.
Patient eligibility: Nova Scotia residents enrolled in a Nova Scotia Pharmacare Program and subject to usual parameters of each program.
Patient cost-sharing: The Seniors Pharmacare Program includes a premium and co-payment. The Family Pharmacare Program — which is open to all Nova Scotians with a valid Nova Scotia Health Care Card — includes a deductible and co-payment. Medications dispensed within the hospital system may either be fully insured or are subject to a dispensing fee.
Eligible drugs: DRDs may be eligible for coverage under the Pharmacare Programs when a patient meets the criteria for coverage developed by the Atlantic and/or Canadian Expert Advisory Committees.
Funds SAP drugs: SAP drugs may be funded on a case-by-case basis.
Process for accessing drug coverage: Coverage of exception status drugs will be approved according to specific criteria upon review of a prescriber’s written request.
Transparency of decisions: Criteria for drugs funded in the Nova Scotia Pharmacare Programs are published in the Nova Scotia Formulary.
Prince Edward Island64,78
Prince Edward Island does not have a dedicated process for enabling access to DRDs. DRDs may be considered case-by-case for Special Authorization coverage.
Patient eligibility: Patients must be enrolled in certain PEI Drug Programs that permit Special Authorization coverage and meet established clinical criteria.
Patient cost-sharing: Patients share in the cost depending on which drug program the medication is funded through.
Eligible drugs: DRDs may be considered for Special Authorization coverage under the following circumstances: therapeutic alternatives listed in the Formulary are contraindicated or have been found to be ineffective or drugs for which there are no alternatives listed in the Formulary. To be eligible for Special Authorization, drug products must be approved for sale in Canada and reviewed and approved for coverage by the Canadian Expert Drug Advisory Committee or the Atlantic Expert Advisory Committee. Special Authorization coverage will normally only be approved for the treatment of indications and dosages listed in the official product monograph approved by Health Canada and published in the most recent edition of the Compendium of Pharmaceuticals and Specialties. On a case-by-case basis, new drugs not yet approved for sale in Canada or not yet reviewed by CDR may be reviewed. Drugs not authorized for sale and use in Canada (e.g., drugs obtained through Health Canada’s Special Access Program, experimental or investigational drugs) are not eligible for consideration of coverage.
Funds SAP drugs: Drugs not authorized for sale and use in Canada (e.g., drugs obtained through Health Canada’s Special Access Program, experimental or investigational drugs) are not eligible for consideration of coverage.
Process for accessing drug coverage: Prescribers may apply for Special Authorization for individual patients. Special Authorization requests are reviewed by drug program staff. Patients and prescribers are notified by letter if coverage has been approved. Medications approved through this process are limited to a maximum 30-day supply per fill unless otherwise noted in the drug criteria. If additional information is required or if a request is denied, a letter is sent to the patient and physician notifying them of the need for more information or reason for denial.
Transparency of decisions: Decisions are made public via listing on the PEI Pharmacare Formulary.
Newfoundland and Labrador79
Newfoundland and Labrador does not have a dedicated process for enabling access to DRDs. Reimbursement requests are considered based on defined criteria as recommendations from CDR, CADTH’s pan-Canadian Oncology Drug Review, or Atlantic Common Drug Review in addition to the Newfoundland and Labrador Prescription Drug Program (NLPDP) benefit list.
Patient eligibility: Patients must be enrolled in the NLPDP and must meet certain defined criteria (including clinical criteria).
Patient cost-sharing: Depending on NLPDP plan eligibility, co-payments may vary.
Eligible drugs: Once a DRD is accepted for coverage under NLPDP, it is classified as a Special Authorization drug product.
Funds SAP drugs: No
Process for accessing drug coverage: A prescriber can request a Special Authorization drug on behalf of a patient. The request is considered in accordance with criteria developed by the CDR, pan-Canadian Oncology Drug Review, the Atlantic Common Drug Review, or other committees or processes as determined or adopted by the minister. If a Special Authorization request has been declined, a prescriber may request an internal review of the matter. If the request is again denied after consideration in an internal review, a prescriber may apply to the medical practitioner designated by the minister for a review of the decision. The decision of that medical practitioner is final.
Transparency of decisions: Special Authorization decisions are not made public; notification of decisions are made to the patient and health care provider only.
Yukon81
Yukon does not have a dedicated process for DRDs. Coverage of a drug, including DRDs, not on the Yukon Drug Formulary may be granted through the EDS process.
Patient eligibility: Yukon residents who are enrolled in eligible drug plans under the Yukon Health Care Insurance Plan (YHCIP). In all cases, the YHCIP is the insurer of last resort (Yukon residents enrolled in the NIHB Program may access DRDs through the NIHB process).
Patient cost-sharing: One drug plan (the Chronic Disease Program) requires clients to pay the first $250 per annum per individual to a maximum of $500 per family. The deductible may be reduced and is always waived for palliative patients as per program regulations.
Eligible drugs: Pharmaceutical products that qualify under the respective program regulation.
Funds SAP drugs: SAP drugs may be funded on a case-by-case basis.
Process for accessing drug coverage: Only Yukon physicians (on behalf of individual patients) may apply for EDS. The Formulary Working Group will review applications for EDS coverage on a monthly basis. The Working Group’s decision will be communicated to the physician and patient. Only the physician will be informed about the therapeutic reasons for non-approval.
Transparency of decisions: Decisions or rationales are not made public.
Northwest Territories64,91,92
The Northwest Territories does not have a dedicated process for enabling access to DRDs. Funding requests for drugs (including DRDs) not on the Non-Insured Health Benefit Drug Benefit (NIHB) List must be submitted to Alberta Blue Cross for prior authorization.
Patient eligibility: Residents of the Northwest Territories who are enrolled in the NWT Health Care Plan and are eligible for extended health benefits (residents enrolled in the NIHB Program may access DRDs through the NIHB process).
Patient cost-sharing: None
Eligible drugs: Drug products not listed in the NIHB Drug Benefit List.
Funds SAP drugs: SAP requests will be considered on a case-by-case basis.
Process for accessing drug coverage: Alberta Blue Cross administers benefits for a drug program on behalf of the Government of Northwest Territories. If a drug has been prescribed that is not on the NIHB Drug Benefit List, a request may be submitted by a health care professional or pharmacist to Alberta Blue Cross on the patient’s behalf for prior authorization. Final decisions are made by the Deputy Minister.
Transparency of decisions: Decisions or rationales are not made public.
Nunavut
Information from Nunavut is awaiting validation.
Non-Insured Health Benefits64,93-95
The NIHB program follows the same process as other public drug plans for enabling access to DRDs. This includes following the criteria established by CDR and pan-Canadian Pharmaceutical Alliance price negotiations. Reimbursement requests for DRDs are considered for coverage on a case-by-case basis to allow for appropriate consultation with prescribers. The program is working on developing criteria for DRDs published on the NIHB Drug Benefit List that will also be consistent with criteria for other public drug plans.
Patient eligibility: Residents of Canada who are any of the following:
a First Nations person who is registered under the Indian Act
an Inuk recognized by an Inuit land claim organization
a child less than 18 months old whose parent is a registered First Nations person or a recognized Inuk.
Patient cost-sharing: None
Eligible drugs: All drugs with a CADTH recommendation that meet the pricing requirements set out by CADTH recommendations.
Funds SAP drugs: Yes
Process for accessing drug coverage: Requests are submitted to NIHB’s Drug Exception Centre for Review. The prescriber will receive a questionnaire to be completed and sent back to the NIHB Program. The information from the questionnaire will be reviewed by an NIHB health care professional, and the submitting pharmacy and prescriber will be informed of the NIHB decision.
Transparency of decisions: NIHB is developing a list of DRDs with eligibility criteria. The criteria that are used are the same as other public drug plans who follow CDR recommendations.
Australia
The Australian Government has established the LSDP to provide subsidized access to expensive and lifesaving medicines for very rare and life-threatening medical conditions.50,96 Following a review of the LSDP, the Australian Government introduced a number of changes to the program in 2018.97,98 These included the formal adoption of a rare disease threshold (prevalence ≤ 1 per 50,000) and the establishment of the LSDP Expert Panel to advise and assist the Commonwealth Chief Medical Officer in matters related to the LSDP.30
To be eligible for reimbursement through the LSDP, the drug must be found to meet each of the following criteria:
The drug has regulatory approval for the treatment of a rare (prevalence ≤ 1 per 50,000 people) but clinically definable disease.
The disease is identifiable with reasonable diagnostic precision.
Epidemiological and other studies provide evidence that the disease causes a significant reduction in age-specific life expectancy.
There is evidence to predict that the drug will substantially extend the lifespan of patients.
The drug must be accepted as clinically effective by PBAC but rejected for Pharmaceutical Benefits Scheme listing because it fails to meet the required cost-effectiveness criteria.
There is no alternative drug listed on the Pharmaceutical Benefits Scheme or available for public hospital in-patients that can be used as lifesaving treatment of the disease. However, the availability of an alternative drug under the LSDP does not disqualify the proposed drug from consideration for the LSDP.
There is no alternative non-drug therapeutic modality that is a suitable and cost-effective treatment of the disease.
The cost of the drug would constitute an unreasonable financial burden on patients or their guardians.30,96
Funding for drugs included in the LSDP is separate from the Pharmaceutical Benefits Scheme and there are currently 16 drugs available through the program for the treatment of 10 ultra-rare diseases (Gaucher disease, Fabry disease, Pompe disease, Batten disease, paroxysmal nocturnal hemoglobinuria, hereditary tyrosinemia type I, and MPS types I, II, IVA, and VI).99
New Zealand
The majority of DRDs available in New Zealand are listed on the Pharmaceutical Schedule; however, an alternative mechanism exists to allow PHARMAC to consider reimbursement for patients based on exceptional circumstances.15 The Named Patient Pharmaceutical Assessment (NPPA) policy may allow patients to access drugs that are not listed on the Pharmaceutical Schedule.15,55 Any authorized medical prescriber is permitted to file an NPPA application provided it is within their scope of practice. The request is evaluated against 3 core principles for the NPPA policy to determine the following:
Does the person have exceptional clinical circumstances?
Has the person tried all existing funded alternative treatments?
Has PHARMAC considered the treatment for funding previously?55
The decision regarding reimbursement through the NPPA policy is made by the PHARMAC Board or by staff under delegated authority from the Board.55 PHARMAC reported that in 2017 to 2018, 176 patients received access to 76 medications specifically for the treatment of rare disorders.15
France
France has also established the Temporary Authorizations for Use (ATU) process to allow patients living with rare or serious diseases to access drugs before market authorization. This process is available for situations that meet the following criteria: the drug is intended to treat, prevent, or diagnose serious or rare diseases; there is no suitable alternative treatment available on the market; and the available evidence suggests that the drug has an acceptable risk/benefit profile.62 The ATU process has been cited as a mechanism to generate real-world evidence as part of the HAS Innovative Medicines Assessment Action Plan.63
Limitations
This report does not present information on the regulatory framework around orphan drugs or DRD designation or definition, the market approval process, or the various legislative frameworks and incentives regarding research and development of these products. CADTH has previously reviewed some of those aspects in the 2013 report Drugs for Rare Diseases: Evolving Trends in Regulatory and Health Technology Assessment Perspectives.100 This report only highlights information on separate or modified HTA processes or reimbursement programs specific to DRDs. There are many aspects of the standard review processes that apply to reviews of DRDs as well (e.g., patient input, reconsideration and/or appeal processes, RWE processes), which are not reviewed in this report. CADTH conducted a report in 2016 that provides insight into the standard HTA processes at national and international HTA agencies.101
This report is not a comprehensive review of all HTA agencies and was focused solely on a subset of countries including Canada, Australia, New Zealand, UK, France, Germany, and a single agency in the US (ICER). The Appraisal of Orphan Medicinal Products (OMPs) project through the IMPACT HTA initiative is currently compiling the DRD review processes for many additional countries in Europe.5,102
Conclusions and Implications for Decision- or Policy-Making
HTA Processes for DRDs
Canadian and international HTA agencies have established separate or modified processes to review and make reimbursement recommendations or decisions for DRDs. The agencies differ with respect to how DRDs are identified for special consideration (e.g., prevalence thresholds, presence of absence of economic criteria). As all the eligibility criteria tend to specify that the drug under review must be indicated for a serious disease for which there is unmet medical need, and nearly all DRDs can be considered to be costly medications (particularly in comparison with more common diseases), the prevalence threshold is a primary factor that differentiates the criteria used by the HTA agencies included in this report. SMC, NICE, PHARMAC, and ICER have all adopted criteria that specifically isolate drugs indicated for ultra-rare diseases for special consideration. A similar approach has been adopted by the Australian LSDP (classified as a funding process rather than an HTA process for the purposes of this scan).30
When selecting their threshold for ultra-orphan drugs, ICER noted that the ability to conduct clinical studies with adequate outcome measures, duration, and follow‐up is typically maintained until the size of the target patient population falls below a prevalence of approximately 3 in 100,000.59,60 Similar observations were made by CADTH when developing guidelines for application of the modified deliberative process for DRDs (e.g., typically applied for drugs with an incidence that is closer to 1 in 100,000 as opposed to the lower 5 in 10,000 threshold).13 These observations were subsequently verified through a retrospective review of CADTH reports and recommendations.103 The criteria specified by CADTH were adapted from the 2012 draft Canadian Orphan Drug Regulatory Framework, which specified an incidence threshold of less than 5 in 10,000 to be considered an orphan drug (similar to the definition used by the EMA), but with the caveat that specialized accommodations are more applicable for ultra-rare diseases for which there are significant challenges in generating high-quality evidence.13,104 This approach has been adopted to reflect the heterogeneity of DRDs and to ensure flexibility for the expert committee to determine how and when accommodations should be made based on the totality of information for the drug and the disease.
The majority of HTA agencies are consistent across their processes for DRDs and other drugs with respect to issuing a recommendation following the review of a complete submission; however, the G-BA/IQWiG process for orphan drugs and the SMC process for ultra-orphan drugs are different. The newly launched SMC process for ultra-orphan drugs is the most unique HTA process identified in this Environmental Scan and represents a significant change in how these drugs are reviewed and introduced into the NHS Scotland formulary. Rather than conducting a complete evaluation of the drug to determine if it should be reimbursed, the process begins with only an initial evaluation to identify gaps in the evidence. This is followed by an evidence generation phase when the drug is reimbursed for a period of up to 3 years before a reassessment is conducted by SMC and a final decision issued regarding availability of the drug for use in NHS Scotland.12,47 The G-BA/IQWiG process for orphan drugs is an abbreviated pathway where the drug is assumed to have proven added benefit at the time of regulatory approval; hence, traditional HTA is not conducted and IQWiG only assesses information provided by the manufacturers on the number of patients affected by the rare disease and the cost of treatment.
There are minor variations in the administrative processes for DRDs across some of the agencies. These include the use of different submission templates for NICE and SMC,9,20,21 allowances for earlier filing for PHARMAC, and additional opportunities for meeting with the agency (e.g., PACE and Stakeholder Meetings).23,24 SMC was the only agency that explicitly reported longer timelines for orphan drugs and ultra-orphan drugs (reassessment phase), which have timelines ranging from 22 weeks to 26 weeks as opposed to the 18 weeks applied for other drugs.12,19 PHARMAC currently offers opportunities for earlier filing and engagement for sponsors with pending applications for drugs indicated for use in the treatment of ultra-rare diseases.9 CADTH used to offer opportunities for earlier engagement with sponsors with pending applications for DRDs; however, these opportunities are now offered to all sponsors irrespective of the type of product. Similarly, CADTH allows any sponsor to file their application up to 180 calendar days before the anticipated date of approval by Health Canada. As CADTH is bound by its performance metrics to issue a recommendation to the sponsor within 180 calendar days, there is limited utility for any expedited review processes for DRDs (with the exception of re-submissions where CADTH may expedite the timelines for DRDs or other products that have been designated by Health Canada as priority reviews).13 The application and review timelines for INESSS are the same as those by CADTH.
All of the agencies with the exception of NICE use the same expert committees to issue recommendations for both DRDs and other drugs. Accommodations to the deliberative processes for committees typically include greater allowances for uncertainty in the clinical and economic evidence and higher willingness-to-pay thresholds. These are among the most common challenges when dealing with DRDs, particularly those indicated for use in the treatment of ultra-rare diseases for which there can be significant challenges in generating robust clinical evidence and the drugs have very high acquisition costs.5
Processes for Funding DRDs
Across national and international public payers, there are a variety of different approaches used to facilitate the reimbursement of DRDs. In Canada, the public drug programs have established a variety of processes to evaluate claims and facilitate the reimbursement of DRDs. Many of the drug programs offer compassionate or exceptional access for patients on a case-by-case basis for DRDs that are not otherwise reimbursed through alternative mechanisms.64 Similar mechanisms have been established in New Zealand under the NPPA policy, which provided access to 76 medicines for 176 patients in 2017 to 2018.15,55
The specialized formularies used in Canada are largely comprised of drugs that would be considered ultra-rare conditions. The Australian Government has also established a unique funding process for drugs for ultra-rare diseases. The LSDP provides subsidized access to expensive and lifesaving medicines for very rare and life-threatening medical conditions.50,96 Among other criteria related to disease severity, unmet need, and absence of alternatives, the LSDP has adopted a prevalence of 1 per 50,000 or less as the threshold for rare diseases. A key feature of this program is that it is only eligible for drugs for which PBAC has concluded that there is clinical benefit but rejected it for listing on the Pharmaceutical Benefits Scheme formulary due to concerns related to the cost-effectiveness. Hence, the LSDP provides a small formulary of drugs for ultra-rare diseases that is separate from the Pharmaceutical Benefits Scheme. There are currently 16 drugs available through the program for the treatment of 10 conditions (Gaucher disease, Fabry disease, Pompe disease, Batten disease, paroxysmal nocturnal hemoglobinuria, hereditary tyrosinemia type I, and MPS types I, II, IVA, and VI).99
Overall, the majority of DRDs are managed through standard formulary processes (e.g., special authorization) and are not listed on separate formularies. Those that have specialized formularies have largely focused on providing access to ultra-rare conditions.
References
1.Richter T, Nestler-Parr S, Babela R, et al. Rare disease terminology and definitions - a systematic global review: report of the ISPOR Rare Disease Special Interest Group. Value Health. 2015;18(6):906-914. https://www.valueinhealthjournal.com/article/S1098-3015(15)01979-8/pdf. Accessed 2021 Mar 1. PubMed
2.Standing Committee on Health. Canadians Affected by Rare Diseases and Disorders: Improving Access to Treatment. Report of the Standing Committee on Health: Government of Canada; 2019: https://www.ourcommons.ca/Content/Committee/421/HESA/Reports/RP10349306/hesarp22/hesarp22-e.pdf. Accessed 2021 Mar 1.
3.Canadian Organization for Rare Disorders. Now is the Time: A Strategy for Rare Diseases is a Strategy for all Canadians. Canada's Rare Disease Strategy. Toronto: Canadian Organization for Rare Disorders; 2015: https://www.raredisorders.ca/content/uploads/CORD_Canada_RD_Strategy_22May15.pdf. Accessed 2021 Mar 1.
4.Patented Medicine Prices Review Board. What is the “Expense” for Expensive Drugs for Rare Diseases? : Government of Canada; 2019: http://pmprb-cepmb.gc.ca/view.asp?ccid=1461&lang=en. Accessed 2021 Mar 1.
5.Nicod E, Whittal A, Drummond M, Facey K. Are supplemental appraisal/reimbursement processes needed for rare disease treatments? An international comparison of country approaches. Orphanet J Rare Dis. 2020;15(1):189. PubMed
6.Drugs for Rare Diseases: A Review of National and International Health Technology Assessment Agencies and Public Payers’ Decision-Making Processes CADTH environmental scan. Ottawa: CADTH; 2018: https://www.cadth.ca/sites/default/files/pdf/es0326_drugs_for_rare_diseases.pdf. Accessed 2021 Feb 26.
7.Impact HTA: Germany. 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_3ce53a1d83e84d6d83866c55195d1056.pdf. Accessed 2021 Jan 11.
8.National Institute for Health Care Excellence. Interim process and methods of the highly specialised technologies programme: updated to reflect 2017 changes. London: National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-highly-specialised-technologies-guidance/HST-interim-methods-process-guide-may-17.pdf. Accessed 2021 Mar 1.
9.PHARMAC. Funding for rare disorders. Wellington (NZ): Pharmaceutical Management Agency (PHARMAC); 2020: https://pharmac.govt.nz/medicine-funding-and-supply/the-funding-process/from-application-to-funded-medicine-how-we-fund-a-medicine/medicines-for-rare-disorders/. Accessed 2021 Mar 1.
10.Institute for Clinical and Economic Review. Modifications to the ICER value assessment framework for treatments for ultra-rare diseases. Boston (MA): Institute for Clinical and Economic Review; 2020: http://icerorg.wpengine.com/wp-content/uploads/2020/10/ICER_URD_Framework_Adapt_013120.pdf. Accessed 2021 Mar 1.
11.Impact HTA: United Kingdom (Scotland). 2019: https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_74a19c766e544af090a2d43229cb6295.pdf. Accessed 2021 Mar 1.
12.Scottish Government. Ultra-orphan medicines pathway guide. Scottish Government; 2019: https://www.gov.scot/binaries/content/documents/govscot/publications/advice-and-guidance/2019/05/ultra-orphan-medicine-pathways-guidance/documents/ultra-orphan-medicines-pathway-guide/ultra-orphan-medicines-pathway-guide/govscot%3Adocument/Generic%2BGuidance%2B-%2BUltra-orphan%2Bpathway_draft%2Bguidance%2B-%2Bfinal.pdf. Accessed 2021 Mar 1.
13.Procedures for CADTH Reimbursement Reviews. Ottawa: CADTH; 2021: https://cadth.ca/sites/default/files/Drug_Review_Process/CADTH_Drug_Reimbursement_Review_Procedures.pdf. Accessed 2021 Mar 1.
14.Haute Autorité de Santé. Procédures d’évaluation anticipée «Fast-Tracking» par la Commission de la Transparence. Haute Autorité de Santé; 2021: https://www.has-sante.fr/upload/docs/application/pdf/2019-07/fast_track_22072019.pdf. Accessed 2021 Mar 1.
15.PHARMAC. 2019 report: Funding medicines for rare disorders. Wellington, New Zealand: PHARMAC; 2019: https://pharmac.govt.nz/assets/2019-Report-Funding-Medicines-for-Rare-Disorders-PDF-version.pdf. Accessed 2021 Mar 1.
16.Wenzl MP, Valérie. Pharmaceutical Reimbursement and Pricing in Germany. OECD; 2018: https://www.oecd.org/health/health-systems/Pharmaceutical-Reimbursement-and-Pricing-in-Germany.pdf. Accessed 2021 Mar 1.
17.Institute for Clinical and Economic Review. Topic selection. Institute for Clinical and Economic Review (ICER); 2021: https://icer.org/our-approach/methods-process/value-assessment-framework/topic-selection/. Accessed 2021 Mar 1.
18.National Institute for Health Care Excellence. Proposed highly specialised technology evaluations. 2021: https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-highly-specialised-technologies-guidance/proposed-highly-specialised-technology-evaluations. Accessed 2021 Mar 1.
19.Scottish Medicines Consortium. A Guide to the Scottish Medicines Consortium. Glasgow: Scottish Medicines Consortium; 2018: https://www.scottishmedicines.org.uk/media/3574/20180712-a-guide-to-the-scottish-medicines-consortium.pdf. Accessed 2021 Mar 1.
20.National Institute for Health Care Excellence. Highly Specialised Technologies Evaluation Programme: Specification for company submission of evidence. London: National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-highly-specialised-technologies-guidance/hst-interim-evidence-submission-template.doc. Accessed 2021 Mar 1.
21.Scottish Medicines Consortium. New Product Assessment Form for Ultra-Orphan Medicines. Glasgow (GB): Scottish Medicines Consortium; 2019: https://www.scottishmedicines.org.uk/media/4467/uo-npaf-updated-100619.docx. Accessed 2021 Mar 1.
22.Department of Health. Guidelines for preparing a submission to the Pharmaceutical Benefits Advisory Committee. Version 5.0. Canberra (AU): Commonwealth of Australia; 2016: https://pbac.pbs.gov.au/content/information/files/pbac-guidelines-version-5.pdf. Accessed 2021 Mar 1.
23.Department of Health. PBAC Stakeholder Meetings. 2019; https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/stakeholder-meetings. Accessed 2021 Mar 1.
24.Scottish Medicines Consortium. Patient and Clinician Engagement (PACE) Meetings Overview. Glasgow (GB): Scottish Medicines Consortium; 2020: https://www.scottishmedicines.org.uk/media/5423/pace-overview-document-v32.pdf. Accessed 2021 Mar 1.
25.Sprinraza – Amyotrophie spinale 5q de type II, de type III et présymptomatique. Avis transmis à la ministre en décembre 2018: Institut national d’excellence en santé et en services sociaux 2018: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Janvier_2019/Spinraza_2018_12.pdf. Accessed 2021 Mar 1.
26.Pilot Process: Joint CADTH And INESSS Engagement with Clinical Specialists. CADTH; Institut national d'excellence en santé et services sociaux; 2018: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/ACMTS-INESSS_Annonce_projet_pilote__130918_EN.pdf. Accessed 2021 Mar 1.
27.Impact HTA: United Kingdom (England). 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_685ce2997dc94213adaee15e05e03077.pdf. Accessed 2021 Jan 11.
28.National Institute for Health Care Excellence. Highly Specialised Technologies Evaluation Committee. London: National Institute for Health and Care Excellence; 2021: https://www.nice.org.uk/Get-Involved/Meetings-in-public/Highly-Specialised-Technologies-Evaluation-Committee. Accessed 2021 Mar 1.
29.PHARMAC. Guidelines for funding applications to PHARMAC. Wellington (NZ): PHARMAC; 2020: https://pharmac.govt.nz/assets/supplier-guidelines.pdf. Accessed 2021 Mar 1.
30.Department of Health. Procedure guidance for medicines funded through the Life Saving Drugs Program (LSDP). Canberra (AU): Australian Government; 2018: https://www.health.gov.au/resources/publications/procedure-guidance-for-medicines-funded-through-the-life-saving-drugs-program-lsdp. Accessed 2021 Mar 1.
31.Scottish Medicines Consortium. New Product Assessment Form Glasgow (GB): Scottish Medicines Consortium; 2020: https://www.scottishmedicines.org.uk/media/5764/20210802-new-product-assessment-form-npaf.docx. Accessed 2021 Mar 1.
32.l’Institut national d’excellence en santé et en services sociaux. Standing Scientific Committee on Entry on the List of Medications. 2018; https://www.inesss.qc.ca/en/about-us/structure/standing-scientific-committees/standing-scientific-committee-on-entry-on-the-list-of-medications.html.
33.Scottish Medicines Consortium. Who is involved in the Scottish Medicines Consortium? 2021; https://www.scottishmedicines.org.uk/about-us/who-we-are/. Accessed 2021 Mar 1.
34.Fee Schedule for CADTH Pharmaceutical Reviews. Ottawa: CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/CADTH_Application_Fees.pdf. Accessed 2021 Mar 1.
35.Act Respecting the Institut National D’excellence en Santé et en Services Sociaux Gouvernement du Québec; 2020: http://www.legisquebec.gouv.qc.ca/en/pdf/cs/I-13.03.pdf. Accessed 2021 Mar 1.
36.Institut national d’excellenceven santé et en services sociaux. Evaluation of Drugs for Listing Purposes: A Change of Approach. 2018: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Processus/Evaluation_of_drugs_a_change_of_approach.pdf. Accessed 2021 Mar 1.
37.Impact HTA: Canada. 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_972b2cc8eef84a619de9d4218fbdd33b.pdf. Accessed 2021 Jan 11.
38.Règles de fonctionnement du Comité scientifique d’évaluation des médicaments aux fins d’inscription. l’Institut national d’excellence en santé et en services sociaux; 2018: https://www.inesss.qc.ca/fileadmin/doc/INESSS/DocuAdmin/Comites_scientifiques/Regles-CSEMI.pdf. Accessed 2021 Mar 1.
39.Galafold – Maladie de Fabry. Avis transmis au ministre le 23 octobre 2018: Institut national d’excellence en santé et en services sociaux 2018: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Novembre_2018/20181023_AvisMinistre_Web.pdf. Accessed 2021 Mar 1.
40.National Institute for Health Care Excellence. NICE Technology Appraisal Guidance. 2021; https://www.nice.org.uk/About/What-we-do/Our-Programmes/NICE-guidance/NICE-technology-appraisal-guidance. Accessed 2021 Mar 1.
41.National Institute for Health Care Excellence. Guidance and advice list. 2021; https://www.nice.org.uk/guidance/published?type=hst. Accessed 2021 Mar 1.
42.National Institute for Health Care Excellence. Interim Highly Specialised Technologies Evaluation Programme: Specification for company submission of evidence London: National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-highly-specialised-technologies-guidance/hst-interim-evidence-submission-template.doc. Accessed 2018.
43.National Institute for Health Care Excellence. Highly Specialised Technologies Evaluation Programme: Company Initial Decision Problem proforma. London: National Institute for Health and Care Excellence; 2017: https://www.nice.org.uk/Media/Default//About/what-we-do/NICE-guidance/NICE-highly-specialised-technologies-guidance/company-initial-decision-problem-pro-forma-hst.doc. Accessed 2021 Mar 1.
44.Scottish Medicines Consortium. Guidance to Submitting Companies for Completion of New Product Assessment Form (NPAF). Glasgow (GB): Scottish Medicines Consortium; 2020: https://www.scottishmedicines.org.uk/media/5599/20200611-guidance-on-npaf.pdf. Accessed 2021 Mar 1.
45.Scottish Medicines Consortium. Timelines & Submission Scheduling. 2021; https://www.scottishmedicines.org.uk/media/3574/20180712-a-guide-to-the-scottish-medicines-consortium.pdf.
46.Scottish Medicines Consortium. Proforma: evidence to support SMC ultra-orphan status. Glasgow (GB): Scottish Medicines Consortium; 2020: https://www.scottishmedicines.org.uk/media/4808/ultra-orphan-proforma.docx. Accessed 2021 Mar 1.
47.Scottish Government. Ultra-orphan medicines pathway evidence generation phase: guidance. Scottish Government; 2019: https://www.gov.scot/binaries/content/documents/govscot/publications/advice-and-guidance/2019/05/ultra-orphan-medicine-pathways-guidance/documents/ultra-orphan-medicines-pathway-evidence-generation-phase-guidance/ultra-orphan-medicines-pathway-evidence-generation-phase-guidance/govscot%3Adocument/Guidance%2Bfor%2Bthe%2Bdata%2Bcollection%2Belement%2Bof%2BUO%2BPathway%2B-%2Bfinal.pdf. Accessed 2021 Mar 1.
48.Scottish Medicines Consortium. Ultra-orphan medicines for extremely rare conditions. Glasgow: Scottish Medicines Consortium; 2019: https://www.scottishmedicines.org.uk/how-we-decide/ultra-orphan-medicines-for-extremely-rare-conditions/. Accessed 2021 Mar 1.
49.The Federal Joint Committee Decisions on Healthcare Benefits. Berlin, Germany: Gemeinsamer Bundesausschuss 2018: https://www.g-ba.de/downloads/17-98-3769/2018-12-12_G-BA_Infobrosch%C3%BCre_EN_bf.pdf. Accessed 2021 Mar 1.
50.Impact HTA: Australia. 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_344ab206492f4c84ba2774ff32fd1dc8.pdf. Accessed 2021 Jan 11.
51.Department of Health. Procedure guidance for listing medicines on the Pharmaceutical Benefits Scheme. Version 2.0. Canberra (AU): Commonwealth of Australia; 2020: https://www.pbs.gov.au/industry/listing/procedure-guidance/files/Procedure-guidance-for-listing-medicines-on-the-Pharmaceutical-Benefits-Scheme-v2.0.pdf. Accessed 2021 Mar 1.
52.Impact HTA: New Zealand. 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_e525dad657da4d19b2e629bca1dfbe4f.pdf. Accessed 2021 Jan 11.
53.PHARMAC. Consultation: Contestable fund for medicines for rare disorders. Wellington, New Zealand: PHARMAC; 2014: https://pharmac.govt.nz/assets/consultation-2014-07-08-rare-disorders.pdf. Accessed 2021 Mar 1.
54.Worth M, Town I, Hensen M. Evaluation of PHARMAC's commercial approach to fund medicines for rare disorders. Wellington (NZ): PHARMAC; 2017: https://www.pharmac.govt.nz/assets/2017-06-final-Grant-Thornton-evaluation.pdf. Accessed 2021 Mar 1.
55.PHARMAC. Exceptional Circumstances Framework (including the Named Patient Pharmaceutical Assessment Policy). Wellington, New Zealand: PHARMAC; 2019: https://pharmac.govt.nz/assets/exceptional-circumstances-framework.pdf. Accessed 2021 Mar 1.
56.PHARMAC. Factors for Consideration. Wellington (NZ): Pharmaceutical Management Agency (PHARMAC); 2020: https://pharmac.govt.nz/medicine-funding-and-supply/the-funding-process/policies-manuals-and-processes/factors-for-consideration/. Accessed 2021 Mar 1.
57.Institute for Clinical and Economic Review. Who We Are. Institute for Clinical and Economic Review (ICER); 2021: https://icer.org/who-we-are/. Accessed 2021 Mar 1.
58.Institute for Clinical and Economic Review. Methods & Process. Institute for Clinical and Economic Review (ICER); 2021: https://icer.org/our-approach/methods-process/. Accessed 2021 Mar 1.
59.Institute for Clinical and Economic Review. Assessing the Effectiveness and Value of Drugs for Rare Conditions. A Technical Brief for the ICER Orphan Drug Assessment & Pricing Summit. Boston (MA): Institute for Clinical and Economic Review; 2017: https://icer.org/wp-content/uploads/2020/10/ICER_Assessing-the-Value-of-Drugs-for-Rare-Conditions_051017.pdf. Accessed 2021 Mar 1.
60.Institute for Clinical and Economic Review. Methods Update: Treatments for Ultra-Rare Diseases. 2017; https://icer.org/explore-our-research/policy-papers/orphan-drug-summit-2017/. Accessed 2021 Mar 1.
61.Institute for Clinical and Economic Review. ICER Evidence Rating Matrix: A User's Guide. Boston (MA): Institute for Clinical and Economic Review; 2021: http://icerorg.wpengine.com/wp-content/uploads/2020/10/ICER_EBM_Matrix_User_Guide_013120.pdf. Accessed 2021 Mar 1.
62.Impact HTA: France. 2019; https://8c3e11d9-5f36-452f-abe3-c95befd6e85d.filesusr.com/ugd/e1a359_d034501772354317af6747881b5ef9f4.pdf. Accessed 2021 Jan 11.
63.Haute Autorité de Santé. Innovative medicines assessment action plan. Haute Autorité de Santé; 2020: https://www.has-sante.fr/upload/docs/application/pdf/2020-03/innovative_medicine_action_plan_27.01.20.pdf. Accessed 2021 Mar 1.
64.Menon D, Clark D, Stafinski T. Reimbursement of drugs for rare diseases through the public healthcare system in Canada: where are we now? Healthc Policy. 2015;11(1):15-32. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4748363/pdf/policy-11-015.pdf. Accessed 2021 Feb 26. PubMed
65.Government of British Columbia. Drugs Requiring Pre-approval (Special Authority). 2021: https://www2.gov.bc.ca/gov/content/health/health-drug-coverage/pharmacare-for-bc-residents/what-we-cover/drug-coverage/drugs-requiring-pre-approval. Accessed 2021 Mar 1.
66.Special Authorization Guidelines. Government of Alberta; 2020: https://www.ab.bluecross.ca/dbl/pdfs/dbl_sec1_sa.pdf. Accessed 2021 Mar 1.
67.Rare Diseases Drug Coverage Program, section 4. Alberta Drug Benefit List. Edmonton (AB): Alberta Blue Cross; 2018: https://www.ab.bluecross.ca/dbl/pdfs/dbl_sec4.pdf. Accessed 2021 Mar 1.
68.Medication Orders Policy and Procedures Support Document: Frequently Asked Questions (FAQ). Government of Alberta; 2017: https://www.albertahealthservices.ca/assets/info/hp/gen/if-hp-gen-medication-orders-frequently-asked-questions.pdf. Accessed 2021 Mar 1.
69.Exception Drug Status Program. Government of Saskatchewan; 2021: https://formulary.drugplan.ehealthsask.ca/PDFs/APPENDIXA.pdf. Accessed 2021 Mar 1.
70.Inherited Metabolic Disease Benefits. Government of Saskatchewan; 2021: https://formulary.drugplan.ehealthsask.ca/PDFs/InheritedMetabolicDiseaseBenefitList_Aug_2016.pdf. Accessed 2021 Mar 1.
71.Government of Ontario. Applying to the Exceptional Access Program. 2020 Jan 8; https://www.ontario.ca/page/applying-exceptional-access-program. Accessed 2021 Feb 26.
72.Ontario Ministry of Health and Long-Term Care. Inherited Metabolic Diseases Program. 2021; https://www.health.gov.on.ca/en/pro/programs/drugs/funded_drug/fund_inherited_drug.aspx. Accessed 2021 Mar 1.
73.Ontario Ministry of Health and Long-Term Care. Special Drug Programs. 2013; https://www.health.gov.on.ca/en/pro/programs/drugs/funded_drug/fund_special.aspx. Accessed 2021 Feb 26.
74.Compassionate Review Policy. Ontario Ministry of Health and Long-Term Care,: https://www.health.gov.on.ca/en/pro/programs/drugs/pdf/compassionate_review_policy.pdf. Accessed 2021 Mar 1.
75.Government of Quebec. List of Medications (Appendices IV and IV.1). 2020 Dec; https://www.ramq.gouv.qc.ca/sites/default/files/documents/liste-med-2020-12-16-en.pdf. Accessed 2021 Mar 1.
76.New Brunswick drugs for rare diseases plan. Fredericton (NB): New Brunswick Canada; 2018: https://www2.gnb.ca/content/gnb/en/services/services_renderer.201352.New_Brunswick_Drugs_for_Rare_Diseases_Plan.html. Accessed 2021 Mar 1.
77.Government of Nova Scotia. Nova Scotia Pharmacare: Exception Status Drugs. 2021; https://novascotia.ca/dhw/pharmacare/exception-status-drugs.asp. Accessed 2021 Feb 26.
78.Health PEI. P.E.I. Pharmacare Formulary. 2021 Feb; https://www.princeedwardisland.ca/sites/default/files/publications/pei_pharmacare_formulary.pdf. Accessed 2021 Mar 1.
79.Government of Newfoundland and Labrador. Special Authorization Drug Products. 2021; https://www.gov.nl.ca/hcs/prescription/covered-specialauthdrugs/. Accessed 2021 Mar 1.
80.Government of Newfoundland and Labrador. Exceptional Review Process. 2021; https://www.gov.nl.ca/hcs/prescription/erp/. Accessed 2021 Mar 1.
81.Government of Yukon. Find drug coverage information. 2021; https://yukon.ca/en/health-and-wellness/medical-professionals/find-drug-coverage-information. Accessed 2021 Feb 26.
82.Government of the Northwest Territories. Applying for the Extended Health Benefits for Specified Disease Conditions Program. 2021; https://www.hss.gov.nt.ca/en/services/applying-extended-health-benefits-specified-disease-conditions-program. Accessed 2021 Feb 26.
83.Indigenous Services Canada. Non-Insured Health Benefits Drug Benefit List. 2020: https://www.sac-isc.gc.ca/DAM/DAM-ISC-SAC/DAM-HLTH/STAGING/texte-text/nihb_benefits-services_drugs_dbl-index_1573154657223_eng.pdf. Accessed 2021 Feb 26.
84.Alberta Health. Welcome to the iDBL. 2021 Mar; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2021 Mar 29.
85.Government of Saskatchewan. Extended Benefits and Drug Plan. 2021; https://www.saskatchewan.ca/residents/health/prescription-drug-plans-and-health-coverage/extended-benefits-and-drug-plan. Accessed 2021 Mar 1.
86.Government of Manitoba. Part 3 exception drug status (EDS). 2021 Jan 21: https://www.gov.mb.ca/health/mdbif/docs/edsnotice.pdf. Accessed 2021 Feb 26.
87.Government of Manitoba. General Pharmacare Questions. 2021; https://www.gov.mb.ca/health/pharmacare/general.html. Accessed 2021 Feb 26.
88.Inherited Metabolic Diseases (IMD) Program: List of Disorders, Covered Drugs, Supplements and Specialty Foods. Ontario Ministry of Health; 2019 Dec: https://www.health.gov.on.ca/en/pro/programs/drugs/funded_drug/pdf/list_food.pdf. Accessed 2021 Mar 1.
89.Government of Quebec. Exceptional medications. 2021; https://www.inesss.qc.ca/en/themes/medicaments/evaluation-process-and-criteria/exceptional-medications.html. Accessed 2021 Feb 26.
90.Gouvernement du Québec. Know the conditions for coverage. 2020; https://www.ramq.gouv.qc.ca/en/citizens/prescription-drug-insurance/know-conditions-coverage. Accessed 2021 Mar 1.
91.Government of the Northwest Territories. Applying for the Extended Health Benefits for Seniors Program. 2021; https://www.hss.gov.nt.ca/en/services/supplementary-health-benefits/extended-health-benefits-seniors-program. Accessed 2021 Feb 26.
92.Government of the Northwest Territories. Applying for Métis Health Benefits Program. 2021; https://www.hss.gov.nt.ca/en/services/supplementary-health-benefits/metis-health-benefits. Accessed 2021 Feb 21.
93.Government of Canada. Who is eligible for the Non-Insured Health Benefits program. 2019; https://www.sac-isc.gc.ca/eng/1574187596083/1576511384063. Accessed 2021 Feb 26.
94.Indigenous Services Canada. Non-Insured Health Benefits Program. Drug Benefit List – Exceptions. 2020 Sep; https://www.sac-isc.gc.ca/eng/1572888328565/1572888420703#s4e. Accessed 2021 Feb 26.
95.Drug benefit list. Toronto (ON): Express Scripts Canada 2020: https://nihb.express-scripts.ca/NIHBProvider/benefits/pharmacy?page=drugbenefit-grid&benefit=pharmacy. Accessed 2021 Feb 26.
96.Department of Health. Life Saving Drugs Program (LSDP) frequently asked questions (FAQs). Canberra (ACT): Australian Government; 2018: https://www.pbs.gov.au/reviews/lsdp-report/lsdp-faq.pdf. Accessed 2021 Mar 1.
97.Australian Government response to the Post-market Review of the Life Saving Drugs Program. Canberra, Australia: Australian Government 2018: https://www.pbs.gov.au/reviews/lsdp-report/government-response-to-lsdp-review.pdf. Accessed 2021 Mar 1.
98.Department of Health. Post-market review of the Life Saving Drugs Programme Report to the Australian Government. Canberra, Australia: Australian Government 2018: https://www.pbs.gov.au/reviews/lsdp-report/lsdp-review-report.pdf. Accessed 2021 Mar 1.
99.Health Do. Life Saving Drugs Program - Information for patients, prescribers and pharmacists. 2020; https://www.health.gov.au/initiatives-and-programs/life-saving-drugs-program?utm_source=health.gov.au&utm_medium=callout-auto-custom&utm_campaign=digital_transformation. Accessed 2021 Mar 1.
100.Drugs for rare diseases: evolving trends in regulatory and health technology assessment perspectives. CADTH environmental scan No.42. Ottawa: CADTH; 2013: https://www.cadth.ca/sites/default/files/pdf/ES0300_Rare_Disease_Drugs_e.pdf. Accessed 2021 Mar 1.
101.Single drug technology assessment processes across health technology assessment organizations. Environmental scan; issue 55. Ottawa: CADTH; 2016: https://www.cadth.ca/dv/single-drug-technology-assessment-processes-across-health-technology-assessment-organizations. Accessed 2021 Mar 1.
102.Appraisal of Orphan Medicinal Products (OMPs). 2020; https://www.impact-hta.eu/work-package-10. Accessed 2021 Mar 1.
103.Richter T, Janoudi G, Amegatse W, Nester-Parr S. Characteristics of drugs for ultra-rare diseases versus drugs for other rare diseases in HTA submissions made to the CADTH CDR. Orphanet J Rare Dis. 2018;13(1):15. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5793441. Accessed 2021 Mar 1. PubMed
104.Office of Legislative and Regulatory Modernization. Initial Draft Discussion Document for a Canadian Orphan Drug Regulatory Framework. Ottawa (ON): Health Canada; 2012.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.