CADTH Reimbursement Review
Normal Immunoglobulin (Human) 10% and Recombinant Human Hyaluronidase (HyQvia)
Sponsor: Takeda Canada Inc.
Therapeutic area: Humoral immunodeficiency
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
BW
body weight
CI
confidence interval
cSCIg
conventional subcutaneous immunoglobulin
FAS
full analysis set
fSCIg
facilitated subcutaneous immunoglobulin
HRQoL
health-related quality of life
IgA
immunoglobulin A
IgG
immunoglobulin G
IgHy10
normal immunoglobulin (human) 10% and recombinant human hyaluronidase
IgRT
immunoglobulin replacement therapy
IQR
interquartile range
IVIg
intravenous immunoglobulin
MedDRA
Medical Dictionary for Regulatory Activities
PASS
post-authorization safety study
PedsQL
Pediatric Quality of Life Inventory
PID
primary immunodeficiency disorder
PPS
per-protocol analysis set
rHuPH20
recombinant human hyaluronidase
SAE
serious adverse event
SAS
safety analysis set
SCIg
subcutaneous immunoglobulin
SD
standard deviation
SF-36
Short Form (36) Health Survey
SID
secondary immunodeficiency disorder
VASBI
validated acute serious bacterial infection
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Immunoglobulin human and recombinant human hyaluronidase (HyQvia) as a solution for subcutaneous infusion, available in the following strengths:
|
Indication | As replacement therapy for primary humoral immunodeficiency and secondary humoral immunodeficiency in adult patients |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 14, 2022 |
Sponsor | Takeda Canada Inc. |
NOC = Notice of Compliance.
Introduction
Immunodeficiencies are characterized by the inability to produce an adequate immune response because the components of the immune system are either absent or functionally inadequate. Primary immunodeficiency disorders (PIDs) or inborn errors of immunity encompass a heterogeneous group of disorders that are genetically determined, resulting from inherited defects in the development and/or function of the immune system.1 An estimated 1 in 1,200 people living in Canada live with a PID, with more than 70% of patients remaining undiagnosed.2 The drug under review is indicated for the treatment of humoral immunodeficiencies, which result from B-cell defects that lead to antibody deficiencies and account for 50% to 60% of PIDs.3 Living with a PID predisposes affected people to an increase in the frequency and severity of infections, autoimmunity, and aberrant inflammation and malignancy.1 The presentation of PIDs can occur at any age, and patients with B-cell (antibody deficiency) disorders typically present after 6 months of age with recurrent and often severe sinopulmonary infections such as otitis media, sinusitis, pneumonia, and gastrointestinal infections. Diarrhea, fatigue, autoimmune manifestations (such as autoimmune cytopenia), and hearing loss are also common.1,4 Consultation with a clinical immunologist or other specialist experienced in managing PIDs is recommended for diagnosis,5 which is based on a combination of clinical presentation and laboratory tests. Typically, these tests include an evaluation of serum antibody levels and a measurement of serum-specific antibody titres in response to vaccine antigens.1,6
Secondary immunodeficiency disorders (SIDs) are acquired and much more common than PIDs. They may result from systemic disorders, immunosuppressive treatments, or prolonged serious illness.3,7 Additionally, patients who are critically ill, older, or hospitalized are susceptible to an acquired immunodeficiency or SID.3,7 Secondary humoral immunodeficiency is a type of SID that occurs across a wide spectrum of diseases with a range in the level of susceptibility to infection and can sometimes be reversed.7 The diagnosis of secondary humoral immunodeficiency relies on screening and monitoring patients who are at risk of developing a secondary antibody deficiency.7 The laboratory tests used to confirm a diagnosis are consistent with those used for a PID. An estimation of the prevalence and incidence of SIDs or secondary humoral immunodeficiency in Canada was not identified; however, a study by Patel et al. (2019)7 reported that secondary humoral deficiencies are estimated to be 30-fold more common than primary humoral immunodeficiencies.
Human immunoglobulin preparations for IV or subcutaneous administration, or immunoglobulin replacement therapy (IgRT), are the cornerstone of treatment in patients with immunodeficiencies affecting the humoral immune system. The clinical experts consulted by CADTH indicated it is important to note that patients with predominantly antibody deficiencies and those with other forms of PIDs, such as combined immunodeficiencies or inborn errors of immunity, also might require long-term or even lifelong IgRT. Both IV immunoglobulin (IVIg) and subcutaneous immunoglobulin (SCIg) therapies have limitations, 1 of which is that both IVIg and SCIg are associated with adverse events (AEs). AEs associated with SCIg tend to be local reactions, whereas systemic AEs are more commonly reported with IVIg.8 Other reasons for using SCIg include improved consistency of immunoglobulin G (IgG) serum levels (less of a difference between peak and trough levels), that administration may be performed at home, and that there is no requirement for venous access. Disadvantages of SCIg compared with IVIg include a higher frequency of infusions and a requirement for multiple injection sites, as well as reduced compliance by some patients.1 Treatment with IVIg allows for the administration of larger volumes of IgG compared with SCIg, more frequent and direct contact with health care professionals, and less frequent administration.8 In addition to IgRT, early and aggressive treatment of infections with antimicrobial drugs is essential in patients with several forms of inborn errors of immunity, including primary humoral immunodeficiency.1,9 Non-drug approaches for the treatment of humoral immunodeficiencies include avoidance of some live vaccines for selected diseases and close monitoring and co-treatment for comorbidities, including autoimmune diseases (cytopenias, enteropathies, celiac disease, thyroid disease, granulomas), respiratory status and function, and malignancies.9
The product under review at CADTH is normal immunoglobulin (human) 10% and recombinant human hyaluronidase (IgHy10) (HyQvia) used in combination, which is a replacement therapy for immunodeficiencies. The sponsor has requested reimbursement of IgHy10 as per the indication, which is as replacement therapy for primary and secondary humoral immunodeficiency in adult patients.10
Both components of IgHy10 are unique drug products provided as individual solutions. They are administered by subcutaneous infusion and must be infused sequentially, beginning with hyaluronidase. The full therapeutic dose can be administered in 1 to 2 sites up to every 4 weeks. The frequency and number of infusion sites used to administer IgHy10 may be adjusted for volume, total infusion time, and tolerability to ensure the patient receives the same weekly dose equivalent of the required therapeutic dose.
The recommended dose of IgHy10 varies by previous treatment experience, as follows:
For patients naive to IgG treatment, administer IgHy10 gradually from a weekly equivalent dose to a 3- or 4-week interval at 300 mg/kg to 800 mg/kg. Adjust the dosage and treatment interval as necessary based on serum IgG trough levels and infection rates.
For patients switching directly from IV administration of immunoglobulin, or who have had a previous IV dose of immunoglobulin that can be referenced, IgHy10 should be administered at the same dose and at the same frequency as their previous IVIg treatment. When switching from IV treatment, begin IgHy10 1 to 2 weeks after the last IV dose. If patients were previously on a 3-week dosing regimen, increasing the interval to 4 weeks can be accomplished by administering the same weekly equivalents.
For patients currently being administered immunoglobulin subcutaneously, the initial dose of IgHy10 is the same as for subcutaneous treatment but may be adjusted to a 3- or 4-week interval based on the weekly equivalents. The first infusion of IgHy10 should be given 1 week after the last treatment with the previous immunoglobulin.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of IgHy10 for the treatment of primary and secondary humoral immunodeficiency in adult and pediatric patients 2 years of age and older. Of note, the systematic review protocol was established before the granting of a Notice of Compliance from Health Canada.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient group submitted input for the review of IgHy10, the Canadian Immunodeficiencies Patient Organization (CIPO). Information for the patient group submission was collected through an online survey of patients and caregivers (N = 246) and semi-structured telephone interviews with patients (N = 8) living with a PID. A total of 233 (95%) of the survey respondents were patients living with a PID, and 13 (5%) were caregivers responding on behalf of patients. The telephone interviews were with patients currently receiving IgRT for PID. None of the patients captured in the patient group submission had experience with IgHy10.
The patient group submission described living with a PID as being prone to a wide range of infections that are often severe, chronic, and debilitating. According to CIPO, the frequency of infections is dramatically reduced with IgRT. As many patients with a PID require lifelong therapy, the method of administration and setting where the treatment is administered are important factors that can significantly affect health-related quality of life (HRQoL). Patient preference is strongly considered when selecting SCIg or IVIg therapy, along with the efficacy of the specific therapy.
According to CIPO, patients want a treatment that minimizes disruptions to their career or personal life. It was noted that administering therapy at a lower frequency and reducing the need to travel to an infusion clinic would help minimize disruption to everyday life as well as reduce the risk of hospital-acquired infections. Telephone interview respondents indicated curiosity about trying IgHy10, but also described a desire for the same result from treatment, voiced concerns about having a negative experience due to switching therapies and wondered whether IgHy10 would be an appropriate treatment option for an individual patient seeking treatment.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PID and SID in pediatric and adult patients.
The clinical experts stated that treatments for primary humoral immunodeficiency that reverse the course of disease are limited. The experts also noted that treatments for secondary humoral immunodeficiency are limited, but that some etiologies can be reversed. Current options for IgRT are effective, but the clinical experts noted there is a need for treatment options that improve adherence and convenience. The clinical experts indicated that current treatments are time-consuming and can negatively affect daily function due to the duration and/or frequency of administration, as well as the need to administer IVIg in a hospital setting. The clinical experts also expressed a need for treatments that are better tolerated, noting IVIg is associated with side effects, including headaches, myalgia, arthralgia, and nausea, and requires pre-medication with antipyretics and anti-nausea drugs. Some patients were also reported as having experienced transient acute kidney injury, aseptic meningitis, thrombotic- and hyperviscosity-related side effects, and fever. Lastly, the experts reported that not all patients respond to currently available IVIg and conventional SCIg (cSCIg) treatments, and that it is difficult to achieve target levels of immunoglobulin in selected patients with IVIg due to its pharmacokinetics (greater variation in peaks and troughs between infusions), which can translate to suboptimal clinical control of disease.
According to the clinical experts consulted by CADTH for this review, IgHy10 would present an additional treatment option to patients, either as first line for patients who were expected to benefit from SCIg or as second line for those who do not tolerate IVIg or SCIg. Both experts felt IgHy10 had the potential to cause a shift in the current treatment paradigm, particularly in terms of SCIg therapies, as it would expand options for IgRT for patients with primary and secondary humoral immunodeficiency.
The clinical experts indicated the patients who would be expected to benefit from SCIg or IVIg would also be expected to benefit from treatment with IgHy10. The clinical experts also indicated that patients who would be expected to benefit most from switching from IVIg to treatment with IgHy10 include patients with certain comorbidities as well as those with limited access to health care facilities, who have had severe adverse effects to IVIg, who have difficult venous access, or who prefer not to miss school or work to receive treatment. The clinical experts also noted that the patients who are likely to benefit from switching from cSCIg to IgHy10 include those who require large doses or volumes due to higher body weight (BW) or who require immunomodulation, those who have a needle phobia and want to avoid using multiple injection sites, those who want to infuse SCIg less frequently, and those who are unable to adhere to weekly SCIg infusions.
Patients identified by the clinical experts as being least suitable for treatment included those who have little to no subcutaneous tissue (very low body fat) or who have severe skin conditions preventing the subcutaneous administration. The clinical experts also felt that patients with complex conditions who require frequent clinical and immunoglobulin monitoring and regular in-person reassessment (potentially coordinated with other IV treatments) would be less suitable for treatment with IgHy10. Lastly, the clinical experts noted that patients with a history of poor adherence to treatment, inappropriate compliance, or inappropriate support from a caregiver (particularly for pediatric patients) may also be less suitable for treatment with IgHy10.
The clinical experts indicated that patients with primary or secondary humoral immunodeficiency who are starting treatment with IgHy10 and who also have chronic or active infections would be expected to respond to treatment with an improvement in clinical state and normalized blood IgG levels (i.e., IgG target levels achieved). The clinical experts described long-term expectations for response to treatment as a reduction in the frequency and severity of infections, which should result in fewer visits to outpatient clinics, a reduced rate of emergency visits and/or hospitalizations due to infections, a reduced need for antimicrobial treatments or prophylactic use, fewer missed days from school or work, and improved overall health and HRQoL. The experts would also expect the overall burden experienced by caregivers and by the health care system to be reduced.
The experts described a clinically meaningful response to treatment as one that would include maintenance of target steady-state serum IgG trough levels (at least 7 g/L), reduced infection rate (no serious infections or significantly less severe infections per year), no emergency room visits or hospital admissions due to infections, significant reduction in days missed from work or school, improved survival and HRQoL, and remission of autoimmune and inflammatory diseases associated with inborn error of immunity, if present. Further, the experts noted that all of these outcomes are clinically meaningful when achieved without serious adverse events (SAEs).
The clinical experts suggested that patients treated with IgHy10 should be assessed for response to treatment every 3 to 6 months, depending on the disease severity, which is consistent with current clinical practice for patients with a PID or SID.
Feedback from the clinical experts indicated that discontinuation of treatment with IgRT would be considered if there is a lack of improvement in immunoglobulin replacement levels, or if response to treatment (as described previously) is not achieved despite increasing doses of IgRT. The experts also noted that issues with adherence (identified by an inability to maintain therapeutic serum IgG levels) may result in discontinuation from IgHy10, as these patients may be better served by IVIg. Discontinuation from IgHy10 may also be considered based on patient preference, or for patients who do not exhibit an improvement in HRQoL, as described by the clinical experts.
According to the clinical experts, IgHy10 can be administered at home after appropriate training by a patient support program, which is similar to current practice for other SCIg preparations. The experts also noted that select cases may warrant infusion of IgHy10 in a health care facility (infusion clinic, outpatient clinic, in-hospital medical day treatment unit), such as when the caregiver/patient is unable to administer the infusion. The experts indicated that a specialist with appropriate knowledge and training in PIDs and SIDs should be involved in the diagnosis and initiation of therapy. This may include immunologists, rheumatologists, and hematologists with training in IgRT.
Clinician Group Input
Seven clinicians authored 2 clinician group input submissions on behalf of 2 clinician groups: the Clinical Immunology Network-Canada (CINC) and the CIPO Medical and Scientific Advisory Committee. The clinician group input was aligned with the input provided by the clinical experts consulted by CADTH, with the exception of place in therapy and assessment of treatment response, which differed slightly. The clinician groups felt that IgHy10 would likely be used as a second-line treatment (following IVIg or cSCIg) for primary or secondary humoral immunodeficiency in general but could become a first-line treatment option in certain subpopulations of patients with the greatest unmet need. The clinician group input recommended that treatment response be assessed at least every 6 to 12 months, which is longer than the frequency of 3 to 6 months suggested by the clinical experts consulted by CADTH. The clinician groups also noted there are considerable benefits associated with the ability to treat patients at home, particularly in terms of rising capacity issues for both inpatient and outpatient beds and the increasing costs of medicines.
Drug Program Input
The drug program implementation questions were aimed at gaining insight from the clinical experts consulted by CADTH about how IgHy10 compares with other available treatments. The clinical experts consulted by CADTH did not have direct experience using IgHy10 in their clinical practices, but they generally described their expectations for IgHy10 as performing similarly to cSCIg with less frequent administration, which is aligned with the draft product monograph. The drug plans were also interested in how the implementation of IgHy10 would differ from cSCIg. The clinical experts consulted by CADTH expected that patients who were able to administer cSCIg at home would also be able to self-administer IgHy10 if patient education and training specific to IgHy10 were added to the specialized training program that is currently used for cSCIg. The clinical experts estimated this would involve an extra 15 to 30 minutes of training on top of the currently available program. Additionally, the drug plans were interested in whether patients could switch to other brands of immunoglobulin during the course of treatment with IgHy10. The clinical experts consulted by CADTH did not expect there would be any issues with switching from IgHy10 to other options for IgRT and, further, they did not express any safety concerns with the use of IgHy10 that would require additional monitoring of safety issues.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Two sponsor-submitted, phase III, open-label, non-randomized, single-group, prospective studies were included in this review. The pivotal study, Study 160603 (N = 89), evaluated the efficacy and tolerability of IgHy10 in patients aged 2 years and older with a PID that required antibody replacement therapy. Patients were also required to have a serum IgG trough level greater than 4.5 g/L and to have received regular IgRT for at least 3 months before enrolment. Study 160902 (N = 66) was an extension of Study 160603 that evaluated the long-term tolerability and safety of IgHy10. The safety extension also followed patients after discontinuation of hyaluronidase to monitor for delayed adverse reactions.
Study 160603 consisted of 2 epochs. During epoch 1, patients received IVIg 10% for 12 weeks. Of the 89 patients who were enrolled in epoch 1, 87 (98%) received IVIg treatment. Subsequently, 83 (93%) of the patients from epoch 1 continued to epoch 2, in which patients received IgHy10 (75 U/g recombinant human hyaluronidase [rHuPH20] followed by SCIg 10%), administered at 108% of the monthly total IVIg dose every 3 or 4 weeks for 14 to 18 months. The primary end point in Study 160603 was the rate of acute serious bacterial infections, defined as the mean number of validated acute serious bacterial infections (VASBIs) per patient per year. The annual rate of all infections, IgG levels, antibiotic use, health care utilization, productivity, HRQoL, tolerability, and safety were also evaluated.
Patients in Study 160603 who were exposed to treatment had a median age of 35.0 years (range = 4 to 78), were primarily White (91%) and non-Hispanic or non-Latino (91%), and 51% were male. The most commonly diagnosed PID among patients was common variable immune deficiency (56%), followed by hypogammaglobulinemia (20%) and X-linked agammaglobulinemia (7%). All patients reported a medical history that included disorders of the hematopoietic or lymphatic system, and the majority also reported medical conditions relating to the eyes, ears, nose, and throat (98%) and respiratory system (87%). The median serum IgG trough level assessed up to 6 months before enrolment was 10.34 g/L (range = 4.05 to 32.00). There were no notable differences in the demographic characteristics of patients who continued into Study 160902.
Efficacy Results
The efficacy results have been summarized with a focus on the results reported during epoch 2 of Study 160603, when patients were treated with IgHy10. It is important to note that in Study 160603, treatment with IgHy10 (epoch 2) cannot be compared with IVIg treatment (epoch 1) because the study was not designed to assess this.
Infections
The primary end point of Study 160603 was analyzed based on the null hypothesis of 1 or more VASBIs per patient per year tested against the alternate hypothesis of fewer than 1 VASBI per patient per year, as per the regulatory guidance from the FDA.11 No comparisons of IgHy10 with other IgRTs were available. A total of 2 VASBIs were reported during treatment with IgHy10 in epoch 2; both cases were episodes of bacterial pneumonia that were treated with oral antibiotics without hospitalization.12 This corresponded to a rate of 0.025 VASBIs per year (upper limit of the 99% confidence interval [CI] = 0.046). The clinical experts for this review agreed that the reported rate of VASBIs was aligned with what would be expected from an IgRT treatment. The rate of VASBIs was not reported during treatment with IVIg in epoch 1. The rate of VASBIs per year before the safety follow-up period was similar among the 66 patients who continued to Extension Study 160902 (0.020; upper limit of the 99% CI = 0.045).
In Study 160603, the rate of all infections reported during treatment with IVIg in epoch 1 was 4.51 infections per year (95% CI, 3.50 to 5.69). During treatment with IgHy10 in epoch 2, the rate of all infections was 2.97 infections per year (95% CI, 2.51 to 3.47). In Extension Study 160902, the rate of all infections during treatment with IgHy10 was 2.86 infections per year (95% CI, 2.36 to 3.43).
Additionally, a post hoc analysis by Wasserman et al. (2016)13 evaluated patients from Study 160603 and Study 160902 from the first administration of IgHy10 through the end of treatment. The annual rate of infections and annual rate of VASBIs during IgHy10 treatment were reported by age group (< 18 and ≥ 18 years). For patients who were at least 18 years of age (n = 59), the rate of infections was 2.98 per year (95% CI, 2.56 to 3.44) and the rate of VASBIs was 0.01 per year (upper limit of the 99% CI = 0.02). The rate of infections and VASBIs were consistent with the results reported for each of the individual studies.
Immunoglobulin Levels
The pivotal study and extension study evaluated IgG trough levels. The clinical experts consulted by CADTH indicated that IgG trough levels were routinely assessed in clinical practice and used as an indicator for the ability to prevent infection and disease-related comorbidities. The published recommendations for treatment with IgRT suggest that IgG trough levels should exceed 5 g/L and, ideally, be greater than 7 g/L.5 At baseline in Study 160603, the median serum IgG trough level was approximately 10 g/L. While receiving IgHy10 during epoch 2, the median IgG trough level in patients younger than 12 years old was 9.95 g/L (95% CI, 7.87 to 15.00). In patients who were at least 12 years old, the median IgG trough level was 10.70 g/L (95% CI, 9.46 to 11.80). The IgG trough levels while patients were receiving IgHy10 during epoch 2 appeared similar to the IgG trough levels while patients were receiving IVIg in epoch 1.
In Extension Study 160902, patients were asked to increase the frequency of IgHy10 administration to a 2- or 3-week interval for a minimum of 4 months to evaluate the effect of varying dose intervals on IgG levels. In summary, the steady-state IgG trough levels were maintained at a median of at least 10 g/L at a dose administration interval of 2, 3, or 4 weeks. After 4 months, patients had the option of returning to their initial dose interval or staying on the 2- or 3-week interval (17 of 66 [25.8%] patients stayed on a 2-week interval for more than 4 months). The ratio of IgG trough levels measured at the end of the safety follow-up period compared with the IgG trough levels measured at the end of IgHy10 treatment was also reported, which indicated that IgG levels were maintained or increased following discontinuation of hyaluronidase over a period of approximately 1.5 years (mean duration of treatment was 565.9 days; standard deviation [SD] = 211.8). Although the clinical experts consulted by CADTH indicated that IgG levels are routinely assessed and useful as a reference point, they also noted that treatment decisions are based predominantly on clinical assessments of the patient as opposed to relying solely on laboratory values.
Antibiotic Use
Outcomes related to antibiotic use were identified as important to clinicians and patients. Antibiotic use was reported as a point estimate of the number of days per month on antibiotics. Based on this, it was estimated that during treatment with IgHy10 in Study 160603, patients were on antibiotics for 1.68 days per month (95% CI, 1.29 to 2.16). In the extension study (Study 160902), the point estimate for days on antibiotics was reported annually. While receiving treatment with IgHy10 before the safety follow-up period, patients were on antibiotics for 64.03 days per year (95% CI, 45.16 to 87.54). The clinical experts consulted by CADTH indicated that the rate of antibiotic use was higher than expected, noting it is atypical for patients to use antibiotics on a monthly basis. Patients who could not discontinue prophylactic antibiotic use were excluded from the trials and concomitant prophylactic antibiotic use was not permitted during the trials. This is likely to overestimate the use of antibiotics in the trials; however, the magnitude of overestimation is difficult to determine, as the proportion of patients who were using antibiotics prophylactically before enrolment was not reported. Thus, there is significant uncertainty associated with the results for antibiotic use.
Health-Related Quality of Life
HRQoL was evaluated in Study 160603 and the extension study, Study 160902, using the Pediatric Quality of Life Inventory (PedsQL) and Short Form (36) Health Survey (SF-36) and reported by age group (2 to 7, 7 to 14, and at least 14 years old). Evidence of validity, reliability, and responsiveness was identified for both the PedsQL and SF-36; however, this did not include patients with immunodeficiencies. In Study 160603, using the SF-36 for patients aged at least 14 years, the mental and physical summary score was a median of 52.2 (range = 21.5 to 70.8) and 44.8 (range = 13.1 to 61.1), respectively. The HRQoL results were similar in the extension study, as the mental and physical summary score was a median of 51.0 (range = 25.3 to 56.9) and 48.7 (range = 11.6 to 52.9), respectively. The data for patients younger than 14 years of age were limited by sample size, and different outcome measures were used for patients between the age of 8 and 13 in the 2 studies. Although limited by methods of analysis and sample size, the evidence suggests there was no change in HRQoL following treatment with IgHy10.
Tolerability and Adherence
In Study 160603, 77 out of 87 patients (89%) and 350 out of 365 infusions (96%) did not require a reduction, interruption, or stoppage of infusion due to tolerability concerns while receiving IVIg in epoch 1. During treatment with IgHy10 in epoch 2, 68 out of 81 patients (84%) and 1,103 out of 1,129 infusions (98%) did not require a reduction, interruption, or stoppage of infusion due to tolerability concerns. The findings for patients receiving treatment with IgHy10 during Extension Study 160902 were similar to what was observed in Study 160603.
One of the key advantages that was anticipated for IgHy10 was the ability to administer treatment at home and less frequently than cSCIg, thereby improving the convenience and minimizing the impact of treatment on a patient’s life. The clinical experts consulted by CADTH indicated they expected the majority of patients (more than 90%) would be able to self-administer IgHy10 at home, similar to what they had observed with cSCIg in clinical practice. In epoch 2 of Study 160603, 282 out of 1,129 infusions (25%) were administered at home. Of these, 231 (82% of the infusions administered at home or 20.5% of all infusions) were administered without nurse intervention, and the majority of IgHy10 infusions (847 out of 1,129 or 75.0%) were administered at the investigational site. Infusions were required to occur at the investigational site until at least 2 infusions at the maximum infusion interval were tolerated, but the proportion of infusions to which this applies is unknown, based on the data available. While receiving IgHy10 (before the safety follow-up period) in Extension Study 160902, 64% of patients and 52% of infusions required assistance with self-administration at home. The reasons given for being unable to continue self-administration of infusions at home were medical reason (9 of 63 or 14.3% of patients), because of a family member (10 of 63 or 15.9% patients), or other reasons (37 of 63 or 58.7% of patients). Additional information about the reasons for being unable to self-administer infusions at home was not provided. Further, whether the ability to self-administer IgHy10 improves over time is unknown.
Health Care Utilization and Productivity
Outcomes related to health care utilization and productivity were of importance to clinicians and patients. A point estimate of the number of acute care physician visits, days in hospital, and days in hospital due to infection were reported per month in Study 160603 and per year in Study 160902.
In Study 160603, patients reported a rate of acute care visits to a physician of approximately 0.40 visits per month (95% CI, 0.32 to 0.49) while receiving treatment with IgHy10. There was no substantial difference in the number of acute care physician visits reported during IgHy10 (after the ramp-up) and during epoch 1 while receiving IVIg (0.33 visits per month; 95% CI, 0.23 to 0.45). The monthly rate of days in hospital and days in hospital due to infection were similar in epoch 1 and epoch 2, with overlapping CIs for the reported point estimates. The rate of days spent in hospital per month was 0.06 (95% CI, 0.03 to 0.10) while receiving IVIg and 0.02 (95% CI, 0.01 to 0.03) while receiving IgHy10. Whether the days spent in hospital per month included time spent in hospital due to infusions is unknown. The monthly rate of days spent in hospital due to infection was 0.03 (95% CI, 0.01 to 0.05) while receiving IVIg and zero days per month while receiving treatment with IgHy10. No days spent in hospital is a treatment goal for patients with humoral immunodeficiency, and these results were considered a clinically meaningful result by the clinical experts consulted by CADTH. Overall, these results describing health care utilization and productivity suggest there was minimal disruption to the everyday life of patients, as the number of acute care physician visits, days in hospital, and days in hospital due to infection occurred at a rate of less than 1 day (or 1 visit) per month per patient. Similar assessments conducted in the extension study yielded comparable results, reported annually.
In Study 160603, the point estimate for days off school or work during treatment with IgHy10 (epoch 2 after the ramp-up period) and treatment with IVIg (during epoch 1) was less than 1 day per month, or approximately 0.28 days per month (95% CI, 0.20 to 0.37) and 0.23 days per month (95% CI, 0.15 to 0.34), respectively. This suggests that patients experienced minimal disruption during IgRT in the trial, based on days of missed work or school.
Harms Results
The overall rate of AEs while receiving IgHy10 was 13.40 per patient during epoch 2 of Study 160603 and 19.75 per patient before the safety follow-up of Extension Study 160902. For reference, exposure to IgHy10 in Study 160603 was a mean of 38 days (SD = 10) during the ramp-up period, plus 368 days (SD = 104) following the ramp-up period. In Study 160902, patients were exposed to IgHy10 for a mean of 566 days (SD = 212). The rate of AEs was 4.45 per patient during treatment with IVIg in Study 160603, and 7.78 per patient during the safety follow-up with IVIg or SCIg without hyaluronidase during the extension study. The most frequently reported AEs while on treatment with IgHy10 was infusion site pain, which occurred at a rate of 1.14 events per patient, as well as the following AEs that occurred at a rate of less than 1 event per patient: headache, sinusitis, upper respiratory tract infection, asthma, nausea, fatigue, myalgia, infusion site pruritus, and viral upper respiratory tract infection. During the safety extension study, Study 160902, the overall rate of AEs was 19.75 per patient while receiving IgHy10. The rate of local AEs was 2.62 per patient, and the rate of systemic AEs was 17.13 per patient (including infections) or 12.71 per patient (excluding infections).
Additionally, AEs were reported by patients during epoch 2 following the ramp-up period. Overall, 53.1% of patients reported a local AE. The most frequently reported local AEs were infusion site pain, infusion site discomfort, infusion site erythema, and infusion site pruritus. From the available safety evidence, it is unknown whether the local AEs were specifically related to hyaluronidase or SCIg. The most frequently reported systemic AEs were headache (30% of patients), asthma (17%), nausea (15%), pyrexia (15%), and fatigue (15%), and the following AEs were reported in less than 15% of patients: myalgia, vomiting, arthralgia, dizziness, and diarrhea. Additionally, a post hoc integrated analysis of safety outcomes from Study 160603 and Extension Study 160902 reported the overall rate of systemic and local AEs (both excluding infections) by age group (< 18 and ≥ 18 years).14 Among adult patients (≥ 18 years), 1,200 systemic AEs were reported, corresponding to a rate of 8.63 AEs per patient-year. For local AEs, a total of 429 AEs were reported among adult patients, corresponding to a rate of 3.08 AEs per patient-year.
A total of 2 deaths were reported between the pivotal and extension study; both occurred during Extension Study 160902. The deaths were caused by toxicity to various drugs (n = 1) and cardiac arrest (n = 1), and neither death was considered related to the study treatments. Serious AEs were reported infrequently; a combined total of 22 patients reported an SAE in Study 160603 and Study 160902. Two SAEs due to chronic obstructive pulmonary disease were reported and were the only SAEs reported more than once.
The majority of notable harms identified in the systematic review protocol were captured in the standard reporting of safety results. While receiving IgHy10 in Study 160603, the systemic effects included as notable harms occurred at a rate of less than 1 AE per patient, as previously described where overall AEs are summarized. Infusion site pain was the most frequently occurring infusion-related AE (1.14 AEs per patient) and other infusion-related events occurred at a rate of less than 1 AE per patient. For local reactions, swelling or edema, and contact dermatitis were reported at a rate of less than 1 AE per patient, as were infusion site hypersensitivity and thrombotic events. No cases of hypersensitivity, anaphylaxis, thrombocytopenia, acute kidney injury, or aseptic meningitis were reported for patients receiving IgHy10 during epoch 2.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Results | Study 160603 | Extension Study 160902 | ||
|---|---|---|---|---|
Epoch 1 IVIg N = 87 | Epoch 2 IgHy10 N = 83 | Before safety follow-up IgHy10 N = 66 | Safety follow-up IgHy10 by IV or SC N = 66 | |
Rate of VASBIsa,b | ||||
N analyzed | NR | 81 | 66 | NR |
Rate per patient per year (upper limit of 99% CI) | NR | 0.025 (0.046) | 0.020 (0.045) | NR |
P value | NR | < 0.0001 | < 0.0001 | NR |
Rate of all infectionsa,b | ||||
N analyzed | 81 | 81 | 66 | NR |
Rate per patient per year (upper limit of 99% CI) | 4.51 (3.50 to 5.69) | 2.97 (2.51 to 3.47) | 2.86 (2.36 to 3.43) | NR |
P value | NR | NR | NR | NR |
IgG trough levels (g/L) by age and by infusion intervalc | ||||
N analyzed | 68 | 60 | 66 | NR |
By age, median (95% CI) | ||||
IgG trough levels in patients < 12 years (n = 11) | 9.63 (8.29 to 13.60) | 9.95 (7.87 to 15.00) | NR | NR |
IgG trough levels in patients ≥ 12 years (n = 70) | 10.40 (9.63 to 11.40) | 10.70 (9.46 to 11.80) | NR | NR |
By infusion interval, median (95% CI) | ||||
2-week interval (n = 17) | NA | NA | 10.90 (9.39 to 13.30) | NR |
3-week interval (n = 9) | NR | NR | 12.30 (11.50 to 15.30) | NR |
4-week interval (n = 47) | NR | NR | 9.67 (9.26 to 10.70) | NR |
Antibiotic use: Number of days on antibiotics per month (Study 160603) or per year (Study 160902)b,c | ||||
N analyzed | 81 | 81 | 66 | NR |
Days on antibiotics (95% CI) | 3.15 (2.19 to 4.35) | 1.69 (1.29 to 2.16) | 64.03 (45.16 to 87.54) | NR |
HRQoL via SF-36 in patients at least 14 years of agec (SAS) | ||||
N analyzed | 46 | 58 | 49 | NR |
Mental component summary, median (range) | 51.2 (20.5 to 66.9) | 52.2 (21.5 to 70.8) | 51.0 (25.3 to 56.9) | NR |
Physical component summary, median (range) | 44.7 (15.2 to 64.0) | 44.8 (13.1 to 61.1) | 48.7 (11.6 to 52.9) | NR |
Health care utilization: Number of days in hospital and number of acute care physician visits per month (Study 160603) or per year (Study 160902)b,c | ||||
N analyzed | — | 81 | 66 | NR |
Days off from school or work, rate (95% CI) | 0.23 (0.15 to 0.34) | 0.28 (0.20 to 0.37) | 7.70 (5.33 to 10.69) | NR |
Acute care physician visits, rate (95% CI) | 0.33 (0.23 to 0.45) | 0.40 (0.32 to 0.49) | 4.19 (3.14 to 5.45) | NR |
Days in hospital, rate (95% CI) | 0.06 (0.03 to 0.10) | 0.02 (0.01 to 0.03) | 0.83 (0.42 to 1.45) | NR |
Days in hospital due to infection, rate (95% CI) | 0.03 (0.01 to 0.05) | 0.00 (0.00 to 0.01) | 0.11 (0.06 to 0.18) | NR |
Productivity: Number of days off school or work per month (Study 160603) or per year (Study 160902)b,c | ||||
N analyzed | — | 81 | 66 | NR |
Days off school/work, rate (95% CI) | 0.23 (0.15 to 0.34) | 0.28 (0.20 to 0.37) | 7.70 (5.33 to 10.69) | NR |
Tolerability and adherencec (SAS) | ||||
N analyzed (number of patients) | 87 | 81 | 63 | 51 |
No reduction, interruption, or stop, n (%) | 77 (88.5) | 68 (84.0) | 54 (85.7) | 48 (94.1) |
Patients requiring assistance with self-administration of infusions at home, n (%) | NR | NR | 40 (63.5) | NR |
Harms (SAS) | ||||
Patients with ≥ 1 AE, n (%) | NR | NR | 63 (100.0) | 51 (77.3) |
Number of AEs, n (rate per patient) | 387 (4.45) | 1,085 (13.40) | 1,244 (19.75) | 407 (7.98) |
Patients with ≥ 1 SAE, n (%) | 3 (3.4) | 11 (13.6)d | 11 (16.7)e | |
WDAE (from study treatment) | NR | NR | NR | NR |
Deaths | 0 | 0 | 2f | |
Notable harms by number of events (rate of AEs per patient)g | ||||
Systemic effects | ||||
Headache | 53 (0.61) | 56 (0.69) | 47 (0.75) | 41 (0.80) |
Fatigue | 10 (0.12) | 21 (0.26) | 27 (0.43) | 23 (0.45) |
Nausea | 13 (0.15) | 25 (0.31) | 46 (0.73) | 25 (0.49) |
Vomiting | 11 (0.13) | 18 (0.22) | 17 (0.27) | 4 (0.08) |
Pyrexia | 11 (0.13) | 22 (0.27) | 16 (0.25) | 5 (0.10) |
Arthralgia | 1 (0.01) | 14 (0.17) | 8 (0.13) | 7 (0.14) |
Myalgia | 5 (0.06) | 20 (0.25) | 28 (0.44) | 8 (0.16) |
Infusion-related AEs | ||||
Infusion site discomfort | 0 | 30 (0.37) | 8 (0.13) | 2 (0.04) |
Infusion site pain | 1 (0.01) | 92 (1.14) | 84 (1.33) | 19 (0.37) |
Infusion site erythema | 0 | 28 (0.35) | 23 (0.37) | 0 |
Infusion site pruritus | 0 | 17 (0.21) | 31 (0.49) | 0 |
Local reactions | ||||
Swelling/edema | 0 | 9 (0.11) | 1 (0.02) | 0 |
Contact dermatitis | 2 (0.02) | 4 (0.05) | 4 (0.06) | 0 |
Infusion site hypersensitivity | 0 | 2 (0.03) | 0 | 0 |
Drug hypersensitivity | 0 | 0 | 3 (0.05) | 0 |
Hypersensitivity | 0 | 0 | 2 (0.03) | 0 |
Thrombotic events (thrombosis) | 0 | 2 (0.03) | 0 | 0 |
Thrombocytopenia | 0 | 0 | 1 (0.02) | 0 |
AE = adverse event; CI = confidence interval; HRQoL = health-related quality of life; IgG = immunoglobulin G; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; NA = not applicable; NR = not reported; rHuPH20 = recombinant human hyaluronidase; SAE = serious adverse event; SAS = safety analysis set; SC = subcutaneous; SF-36 = Short Form (36) Health Survey; VASBI = validated acute serious bacterial infection; WDAE = withdrawal due to adverse event.
Note: The safety follow-up period for Study 160902 was reported to provide evidence of long-term safety following discontinuation of hyaluronidase.
aVASBIs: The mean rate of infections per patient per year and its variance were calculated using a Poisson model accounting for the length of the observation period for each patient. To handle over-dispersion, the exponential distribution dispersion parameter was assumed to be given by the deviance divided by the degrees of freedom, and all statistics were adjusted accordingly. All infections: Point estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including allowance for over-dispersion, as described for the primary end points.
bResults for Study 160603 were analyzed using the full analysis set; results for Study 160902 were analyzed using the SAS.
cOutcomes were summarized using descriptive statistics.
dIncludes SAEs that occurred during the ramp-up period. In total, 11 patients experienced an SAE while receiving IgHy10; 3 of these patients experienced SAEs that occurred during the ramp-up period.
eSAEs were reported using the IgHy10 plus safety follow-up dataset (n = 66).
fDeaths were reported using the IgHy10 plus safety follow-up dataset (n = 66). One of the deaths occurred 4 weeks after completion of the study.
gThe following were included as notable harms but were not reported/observed in any of the included studies: anaphylaxis, acute kidney injury, aseptic meningitis.
Critical Appraisal
The evidence informing the safety and efficacy of IgHy10 is based on 2 single-group, open-label studies. Neither a historical control or concurrent comparator group were used and, consequently, there was no control for potential confounding variables. Additionally, it is not possible to infer causality for the reported outcomes that were assessed in the trials, such as the annual rate of infections, which lacks both context and a reference point for evaluation. The single-group study design was also a particular issue in the assessment of safety, in addition to the outcomes that were reported as a rate per patient or per infusion. While this may partly account for the varying duration of treatment in each of the reported observation periods, the proportion of patients experiencing a particular AE is unknown, with the exception of the AEs commonly reported during treatment with IgHy10 in Study 160603. Most of the outcomes of interest for this review, including the primary end point of the pivotal trial, were not expected to have been impacted by an open-label design; however, patient-reported outcomes, such as those related to HRQoL, tolerability and adherence, and safety outcomes, may have been impacted based on patient beliefs about IgHy10. The risk of attrition bias is a concern due to the overall discontinuation rate in Study 160603, where 21% of patients discontinued from the study primarily due to patient request for withdrawal or AEs. Further, 43% of the patients aged 2 to younger than 12 years (n = 14) in Study 160603 discontinued from the study either due to a requested withdrawal (29%) or an AE (14%). As a result, the reported study results are likely biased in favour of IgHy10, as the data analyzed are largely based on patients who did not discontinue from the study. Although the outcome measures selected for reporting in the studies were considered clinically relevant, the methods of analysis hindered the ability to meaningfully interpret the outcomes. Statistical testing was used only for the primary end point in Study 160603; therefore, multiplicity was not an issue. Within-group changes were generally not reported, as results were analyzed as a rate or summarized using descriptive statistics.
The clinical experts consulted by CADTH did not have any concerns regarding the generalizability of the evidence to the groups of patients excluded from the trials (such as pregnant females, patients with immunoglobulin A [IgA] deficiency, patients required to remain on prophylactic systemic antibacterial antibiotics, or patients with certain pre-existing conditions), or regarding patients living in Canada specifically, despite only 1 Canadian study site being included. There were also no concerns regarding the generalizability to patients with secondary humoral immunodeficiency, patients who were IgRT-naive, or patients with lower serum levels of IgG. Concomitant use of prophylactic antibacterial antibiotics and pre-medications used before the administration of IVIg or SCIg were avoided if possible during the studies, but their use is common in clinical practice. The clinical experts consulted for this review suggested that while an at-home training/support system would be implemented to facilitate administration of IgHy10 at home, the level of supervision in the trials was more intensive than what would be used in clinical practice. Lastly, IgHy10 was not studied in patients with secondary humoral immunodeficiency, despite a proposed indication and reimbursement request for this patient population. However, the clinical experts consulted by CADTH were comfortable with extrapolating evidence of efficacy from patients with primary humoral immunodeficiency to patients with secondary humoral immunodeficiency.
Indirect Evidence
Indirect evidence matching the inclusion and exclusion criteria of this review was not submitted by the sponsor or identified in the CADTH literature review.
Other Relevant Evidence
Sponsor-Submitted Study in Adult Patients Switching From cSCIg to IgHy10 (NCT02881437)
Description of Study
NCT02881437 was a phase IV, open-label, non-randomized, single-group prospective study17 that filled an important gap in the comparison of cSCIg with IgHy10. The primary objective was to examine the difference in steady-state IgG trough levels among adults (18 years or older) with a PID requiring IgRT during cSCIg treatment (primarily once weekly) compared with steady-state IgG trough levels after switching to IgHy10 administration every other week at equivalent doses. Among the 22 enrolled patients, the median age was 45.0 (interquartile range [IQR] = 32.0 to 54.0) years and 68.2% were female.
The study began with a 1-week ramp-up period (subcutaneous IgHy10 provided at 1-quarter of the usual monthly cSCIg dose) that started 1 week after the last cSCIg infusion before enrolment. Subcutaneous IgHy10 infusions then occurred every 2 weeks at 1-half of the initial monthly cSCIg dose, with follow-up measurements at 3 and 6 months.
Results
The mean change in steady-state IgG trough level when switching from cSCIg to IgHy10 was −0.30 g/L (SD = 1.54) after 3 months, and −0.29 g/L (SD = 1.35) after 6 months (n = 16). A total of 11 out of 19 patients (57.9%) had at least 1 infection in the first 3 months of follow-up and 8 out of 17 (47.1%) had at least 1 infection at between 3 and 6 months of follow-up. The mean change in the physical component summary of the SF-36 was −0.90 (SD = 4.37) from baseline to 3 months and −2.67 (SD = 5.17) from 3 to 6 months (n = 12); the mean change in the mental component summary was −2.67 (SD = 5.17) from baseline to 3 months and 1.33 (SD = 5.13) from 3 to 6 months (n = 14).
A total of 21 of 22 patients (95.5%) reported at least 1 local AE, and all reported at least 1 systemic AE between baseline and follow-up at 3 months; 12 of 22 (66.7%) reported at least 1 local AE and 16 of 22 patients (88.9%) reported at least 1 systemic AE between the 3-month and 6-month follow-ups. Commonly reported AEs were similar to those in the pivotal studies.
Critical Appraisal
There are several internal validity concerns that limit the certainty of the conclusions that can be drawn. The primary concern is that there is no control group and confounders are unaccounted for; thus, causal relationships cannot be established. Though the inclusion and exclusion criteria are clear, some details of the participant disposition are limited (i.e., number screened versus randomized). The open-label design is likely to have biased the subjective end points (direction unclear). A large number of patients were lost to follow-up (6 of 22; 37%), which reduces the reliability of the findings for HRQoL. The statistical analyses were not adjusted for multiplicity.
Despite some differences in setting (all of the study sites were in France) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to adult patients in Canada; the applicability of the findings to children is less clear. The exposure to study treatments appeared to match the product monograph, aside from the ramp-up phase (not described in the monograph). Outcomes were clinically relevant, though the clinical experts indicated they would not rely on IgG trough levels alone for clinical decision-making. The sample size and length of follow-up may have been inadequate to capture rare and/or long-term harms.
Sponsor-Submitted Study of Safety, Tolerability, and Immunogenicity of IgHy10 in Pediatric Patients (Study 161504, Interim Analysis)
Description of Study
Clinical Study 161504 was a phase IV, open-label, non-randomized, single-group prospective study18 that provided further post-authorization safety, tolerability, and immunogenicity data on IgHy10 among 42 pediatric patients (2 to < 18 years old) with primary humoral immunodeficiency requiring IgRT. The findings are based on a planned interim analysis after 75% of patients had completed 1 year of study in epoch 2.19 Those naive to facilitated SCIg (fSCIg) (n = 23, 54.8%) had a mean age of 10.3 years (SD = 3.8); the group was 78% male and 96% White. The characteristics of patients with prior fSCIg exposure (pre-treated) were similar.
Patients naive to fSCIg were enrolled in a maximum 6-week ramp-up period of introduction to IgHy10 infusions (epoch 1). In epoch 2, IgHy10 treatment continued for up to 3 years. After 1 year in epoch 2, patients with an anti-rHuPH20 titre of 160 or lower at any time proceeded to study completion; others continued on treatment for another 2 years. Patients with an anti-rHuPH20 titre of 160 or higher who experienced an SAE or severe AE continued in a safety follow-up (epoch 3) in which IgHy10 was discontinued and patients received IVIg or cSCIg. The exact details of the treatment regimens are not available. At the time of the interim analysis, 22 patients (52.3%) had completed the study, 17 (40.5%) were ongoing, and 3 (7.1%) had discontinued prematurely.
Results
The mean IgG trough level was 9.6 mg/dL (SD = 2.1) at enrolment and 8.2 mg/dL (SD = 2.9) at 12 months. A total of 31 out of 42 patients (73.8%) experienced at least 1 treatment-emergent infection. One patient (2.4%) had a serious acute bacterial infection.
Of 42 enrolled patients, 27 (64.3%) reported at least 1 AE. Eleven patients (26.2%) experienced at least 1 local AE, 24 (57.1%) experienced at least 1 systemic AE, and 4 (9.5%) experienced at least 1 SAE. Across all treatments (n not reported), 7.4% of infusions were stopped, interrupted, or adjusted due to an AE. No patients developed an anti-rHuPH20 antibody titre of 160 or greater.
Critical Appraisal
There are several internal validity concerns that limit the certainty of conclusions that can be drawn. The primary concern is that there is no control group and confounders are unaccounted for; thus, causal relationships cannot be established. No statistical hypothesis testing was conducted. Interim findings of safety and efficacy should be interpreted cautiously, as these findings could overestimate the benefits and/or underestimate the harms of a treatment.20,21 Though the inclusion and exclusion criteria are clear, some details of the participant disposition are limited (i.e., number screened versus randomized). The open-label design is likely to have biased the subjective end points; however, the direction of the bias is unclear. It is not fully clear how outcomes were defined and collected, and “treatment-emergent infections” were not pre-specified in the study protocol. The small sample size may have negatively impacted the reliability of the findings.
Despite some differences in setting (all of the study sites were in Europe) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to pediatric patients in Canada; the applicability of the evidence to adults is less clear. Dosing information was not provided. Outcomes were clinically relevant, though the clinical experts indicated they would not rely on IgG trough levels alone for clinical decision-making. The sample size was likely too small to capture rare harms.
Sponsor-Submitted Study on the Safety of IgHy10 in Pregnant Women and Their Infants (Registry Study 161301)
Description of Study
Study 161301 was a registry study providing safety data on women who had previously been treated with IgHy10 and their infants — a population excluded from the pivotal studies. All pregnant women who had ever been treated with IgHy10 were eligible in 1 of 2 study arms: the IgHy10 arm, which continued IgHy10 during pregnancy, and the alternative program arm, which switched patients to another IgRT or an alternative treatment. Nine mothers were enrolled; they had a median age of 34.0 years (IQR = 32.0 to 36.0), were primarily non-Hispanic or non-Latino (8 out of 9; 88.9%), and all were White. Seven of the mothers’ infants were enrolled. Patients visited their own physicians and were treated according to routine medical practice. Data on IgHy10 treatment were available for 6 (85.7%) mothers in the IgHy10 arm. Among these, the median number of infusions was 4 (IQR = 1.5 to 5.75); infusions were received on a 3- or 4-week interval.
Results
Among the 9 mothers, 4 (44.4%) reported at least 1 AE, 3 (42.9%) mothers in the IgHy10 arm and 1 (50.0%) in the alternative product arm. There were no local or immunologic AEs. One (11.1%) mother in the IgHy10 arm reported SAEs. There were no AEs leading to death or withdrawals due to adverse events (WDAEs). Four (44.4%) mothers (2 in each arm) were assessed for anti-rHuPH20 antibodies, and all were negative (titre less than 160). Eight (88.9%) mothers provided data on pregnancy outcomes; all were live births. One birth in the IgHy10 arm was considered abnormal because it was a Caesarean section.
Infants were born at a median of 38.0 (IQR = 37.0 to 40.0) weeks of gestation; weight, length, and head circumference were normal for all. Two (40.0%) infants in the IgHy10 arm had congenital malformations. During follow-up, 6 infants (85.7%) experienced at least 1 AE: 5 (100%) in the IgHy10 arm and 1 (50.0%) in the alternative product arm; 2 (40.0%) infants in the IgHy10 arm experienced an SAE. There were no AEs leading to death or WDAEs.
Critical Appraisal
There are several internal validity concerns that limit the certainty of the conclusions that can be drawn. The primary concern is that there was no control group and confounders are unaccounted for; thus, causal relationships cannot be established. There was no statistical hypothesis testing. Selection bias is possible because very few (n = 9) mothers were enrolled, and it is not clear whether women from various centres would differ systematically. As this is an open-label study, the subjective end points may be biased; however, the direction of the bias is unclear. Most of the data were collected retrospectively, which may have negatively affected quality and completeness. In the IgHy10 arm, 29% of mothers were lost to follow-up, which may have biased AE data in favour of IgHy10.
Despite some differences in setting (all of the study sites were in Europe or the US) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to patients in Canada. The dosage and administration of IgHy10 appear to align with the Health Canada–approved dosing; however, pregnant women are not an indicated population in the product monograph. The harm outcomes seem to be clinically important, but no efficacy outcomes were collected. The sample size and length of follow-up were likely inadequate to capture rare and long-term AEs.
Post-Authorization Safety Studies and Patient Registry Study
Results from 2 post-authorization safety studies (PASSes), Global PASS (NCT02593188)22 and EU PASS (EUPAS5812),23,24 and a patient registry study, FIGARO (NCT03054181),25-27 were submitted by the sponsor as supportive evidence. Both the Global PASS (N = 264) and EU PASS (N = 106) were non-interventional, prospective, uncontrolled, open-label, multi-centre PASSes that evaluated the long-term safety of IgHy10 under clinical routine conditions in the US and Europe, respectively. The Global PASS study was conducted between 2015 and 2021 and enrolled patients with a PID. The EU PASS study was conducted between 2014 and 2021 and enrolled patients who had been prescribed treatment for a PID or SID. The Global PASS reported that 56% of 909 infusions were self-administered at home. The EU PASS reported the proportion of treatments that were administered at a clinical site and at home by year since the first fSCIg treatment. During the first, second, and third year, and after the third year, 91.2% (n = 83 patients; 909 infusions), 93.2% (n = 556 patients; 600 infusions), 93.2% (n = 28 patients; 237 infusions), and 85.2% (n = 12 patients; 54 infusions) of treatments were administered at home, respectively. FIGARO was a long-term observational study on the utilization and outcomes of IgHy10 under everyday clinical practice conditions. FIGARO was conducted in Europe between 2016 and 2021 and enrolled 156 patients with a PID or SID. Data were available for 154 patients, of which 13 were pediatric (younger than 18 years), 120 were adults (18 to 64 years), and 21 were older adults (at least 65 years); results were analyzed by patient age. FIGARO reported that 81.7% of adults and 57.1% of older adults infused at home.
The results provided by the sponsor for infusions administered at home in Global PASS, EU PASS, and FIGARO suggest that the ability to administer treatment at home was more successful in a real-world setting than in the clinical trial setting; however, the generalizability of this evidence to patients treated in Canadian clinical practice is unknown. Additionally, the interpretation of the additional evidence should take into consideration the limitations associated with real-world evidence studies.
Post Hoc Analysis
The sponsor submitted a post hoc analysis by Wasserman et al. (2021)28 as supportive evidence. The analysis examined 3 consecutive, open-label, uncontrolled clinical studies of IgG therapy. Each of the studies included a subset of patients with primary immunodeficiency. Two of the 3 studies, Study 160603 and Study 160902, informed CADTH’s systematic review of HyQvia. The retrospective post hoc analysis included 30 patients who had received at least 1 infusion of each type of therapy, i.e., IVIg, cSCIg, and fSCIg, and was designed to evaluate the efficacy (rates of infection) and tolerability of the 3 routes of IgG administration. The duration of exposure, total number of infusions, and mean IgG dose received during a 4-week period differed between the 3 treatments. As noted in the publication, the limitations of the study include a small sample size; selection bias, as study participation was voluntary; and year-to-year variations in community infections and other factors that change over time, which cannot be accounted for in a sequential study design. The post hoc analysis concluded that across the 3 treatment modalities (IVIg, cSCIg, and fSCIg), the annualized rates of VASBIs (0, 0.09, and 0.04, respectively) and all infections (4.17, 3.68, and 2.42, respectively) were similarly low.
Conclusions
The pivotal trial (Study 160603) and related extension study (Study 160902) provided evidence on the efficacy and safety of IgHy10 as replacement therapy in patients with a PID requiring antibody replacement therapy. The rate of VASBIs was less than 1.0 infections per patient per year (P < 0.0001), which was aligned with expectations for treatment with IgRT in patients with a PID, according to the experts consulted by CADTH. The rate of all infections per patient per year was less than 3 during treatment with IgHy10 in both Study 160603 and Study 160902. Additionally, the monthly rate of days spent in hospital due to infection was minimal (0.03 days per month or less). This was considered clinically important, as per the feedback from the clinical experts consulted by CADTH. HRQoL is an outcome of importance to patients; however, the results provided in the included studies do not allow for conclusions regarding a change in HRQoL during IgHy10 treatment. Regarding the safety of IgHy10, 2 deaths deemed unrelated to treatment were reported during the extension study and SAEs were infrequent overall. Significant safety concerns were not identified for IgHy10.
The lack of comparative evidence (direct or indirect) for IgHy10 versus other IgRTs represents a major limitation in the context of this review. Conclusions cannot be drawn about how IgHy10 compares with other IgRTs, and the single-group study design hinders the ability to appropriately interpret the reported outcomes. Also of note, the available evidence excluded patients with secondary humoral immunodeficiency. The clinical experts consulted by CADTH did not express any issues with using evidence in patients with primary humoral immunodeficiency to inform treatment decisions for patients with secondary humoral immunodeficiency, although this still represents a gap in the evidence. In summary, there are many other SCIg and IVIg therapies available in Canada. Although IgHy10 offers a unique therapy because it comprises 2 drug products (IgG facilitated by rHuPH20), how IgHy10 differs from other immunoglobulin products in terms of safety and efficacy is currently unknown. Therefore, IgHy10 was viewed as another option for SCIg treatment.
Introduction
Disease Background
Immunodeficiencies are characterized by the inability to produce an adequate immune response because the components of the immune system are either absent or functionally inadequate. PIDs or inborn errors of immunity encompass a heterogeneous group of disorders that are genetically determined, resulting from inherited defects in the development and/or function of the immune system.1 More than 450 different PIDs have been identified and the clinical presentation of PIDs is variable; however, most patients with a PID present as having an increased susceptibility to infection.1 Living with a PID predisposes affected patients to an increase in the frequency and severity of infections, autoimmunity, and aberrant inflammation and malignancy.1
PIDs are often categorized by mechanistic and clinical characteristics.4 The drug under review is indicated for primary and secondary humoral immunodeficiencies. Humoral immunodeficiencies result from B-cell defects that lead to antibody deficiencies and account for 50% to 60% of PIDs.3 The most common B-cell disorders include X-linked agammaglobulinemia, common variable immunodeficiency, and selective IgA deficiency.1,3 The presentation of PIDs can occur at any age, and patients with B-cell (antibody deficiency) disorders typically present after 6 months of age with recurrent and often severe sinopulmonary infections such as otitis media, sinusitis and pneumonia, and gastrointestinal infections. Diarrhea, fatigue, autoimmune manifestations (such as autoimmune cytopenia), and hearing loss are also common.1,4
Early diagnosis and treatment are essential for the prevention of significant disease-associated morbidity and for improving patient outcomes.1 Despite this, patients living with a PID often go undiagnosed, as they present with “routine” infections of the sinuses, ears, and lungs, which require a high degree of suspicion and specialized testing to arrive at a diagnosis of PID.1 Immunodeficiency Canada estimates that 1 in 1,200 people living in Canada live with a PID, noting that more than 70% remain undiagnosed.2 Consultation with a clinical immunologist or other specialist experienced in managing PIDs is recommended for diagnosis.5 A diagnosis is made based on a combination of clinical presentation and screening/laboratory tests. Typically, these tests include an evaluation of serum antibody levels and a measurement of serum-specific antibody titres in response to vaccine antigens.1,6 Serum antibody levels that are lower than reference values by age and/or a weak or absent antibody response to antigens are indicative of B-cell immunodeficiencies.1,6 Genetic testing and flow cytometry may also be conducted to identify a specific cause of primary humoral immunodeficiencies6; however, this is not required to initiate treatment, according to the clinical experts consulted by CADTH.
Separate from the genetically determined PIDs are SIDs, which are acquired and much more common. Secondary immunodeficiencies are a consequence of extrinsic factors and may result from systemic disorders, immunosuppressive treatments, or prolonged serious illness. Additionally, patients who are critically ill, older, or hospitalized are susceptible to acquired or secondary immunodeficiency.3,7 Secondary humoral immunodeficiency is a type of SID that occurs across a wide spectrum of diseases with a range in the level of susceptibility to infection.7 Hematological malignancies, such as chronic lymphocytic leukemia, lymphoma, and multiple myeloma, are commonly associated with secondary antibody deficiency, either due to the conditions themselves or the therapies used to treat them. The diagnosis of secondary antibody deficiency relies on the screening and monitoring of patients who are at risk of developing secondary antibody deficiency.7 The laboratory tests used to confirm a diagnosis are consistent with those used for PID. An estimation of the prevalence and incidence of SIDs was not identified.
Standards of Therapy
According to the clinical experts consulted by CADTH, in Canada, the treatment of primary and secondary humoral immunodeficiency is complex. Human immunoglobulin preparations for IV or subcutaneous administration are the cornerstone of treatment in patients with primary immunodeficiency diseases affecting the humoral immune system.1 IgRT helps fight against infections and is typically a lifelong necessity for patients. The clinical experts also noted that IgRT may also have significant anti-inflammatory and immunomodulating effects. According to the clinical experts, the appropriate use of immunoglobulin can be life-saving and cannot be replaced by any other drug or non-drug treatment. The National Immunoglobulin Replacement Expert Committee recommends that IgRT use and associated changes in dosage, or changes in product or its mode of delivery, should be initiated directly by or in consultation with an immunologist with expertise in the field of PID.5
Dosing for IgRT is weight-based, with a recommended dose of 400 mg/kg to 600 mg/kg administered once every 3 to 4 weeks for IVIg, or 100 mg/kg to 150 mg/kg per week for SCIg.5 Further, the dose or frequency should be adjusted according to desired trough levels and catered to the needs of the individual patient. The clinical experts consulted for this review stated the administration schedule for SCIg can be safely adjusted and the interval shortened, but not more frequently than every 2 weeks. It is also recommended that serum IgG trough levels should be higher than 5 g/L and, ideally, more than 7 g/L, and they should be maintained as per the age-normalized range for optimal clinical response.5 Both IVIg and SCIg therapies have limitations, 1 of which is associated with AEs. The most common AEs include headache, flushing, chills, myalgia, wheezing, tachycardia, lower back pain, nausea, and hypotension.1 AEs associated with SCIg tend to be local reactions, whereas systemic AEs are more commonly reported with IVIg.8 In addition to fewer systemic AEs, SCIg offers advantages such as more consistent serum levels of IgG and administration at home, and does not require venous access. Disadvantages of SCIg include a higher frequency of infusions, requirement for multiple injection sites, and compliance, in certain situations.8 Treatment with IVIg allows for the administration of larger volumes of IgG compared with SCIg, more frequent and direct contact with health care professionals, and less frequent administration.8
In addition to IgRT, early and aggressive treatment of infections with antimicrobial drugs is essential in patients with primary or secondary humoral immunodeficiency.1,9 Antimicrobial or antifungal prophylaxis may be required as an additive treatment in select patients with chronic/recurrent severe infections not fully controlled with IgRT.1 The clinical experts consulted by CADTH noted that disease-modifying therapy is available only for selected immunodeficiencies and entails the use of immunoglobulin for immunomodulating therapy, which requires a much higher dose than IgRT.1
Non-drug approaches for the treatment of humoral immunodeficiencies include avoidance of some live vaccines for selected diseases, and close monitoring and co-treatment for comorbidities, including autoimmune diseases (inflammatory bowel disease, celiac disease, thyroid disease), respiratory status and function, and malignancies.9
IgRT is indicated also for the prevention of bacterial infections in patients with hypogammaglobulinemia and/or recurrent bacterial infections associated with B-cell malignancies and other secondary humoral immunodeficiencies affecting B cells, such as after a hematopoietic stem cell transplant or use of B cell–depleting biologics.4 In summary, the IgRT treatment approach for secondary humoral immunodeficiency is similar to the IgRT treatment approach for primary humoral immunodeficiency, as noted by the clinical experts consulted for this review.
The clinical experts consulted by CADTH described the treatment goals for patients living with primary or secondary humoral immunodeficiencies as similar to the goals prioritized by patients, such as an improvement in HRQoL, preserved function, and prevention of hospitalizations through the prevention of infections. The experts also identified treatment goals from the treating specialist’s perspective, which included a reduction in morbidity and mortality related to disease, such as organ damage. Additionally, the experts described an ideal treatment as 1 that is well tolerated by patients and has options for administration in terms of home and hospital settings. Reducing the number of missed days of school or work, reducing the time required for infusions, and reducing the caregiver and health care system burden were also identified as goals of treatment.
Drug
IgHy10 (HyQvia), a combination of normal immunoglobulin (human) 10% and rHuPH20, is the subject of this CADTH review. The sponsor has requested reimbursement of IgHy10 as per the indication, which is as replacement therapy for primary humoral immunodeficiency and secondary humoral immunodeficiency in adult patients.10
Both components of IgHy10 are provided as solutions and are administered by subcutaneous infusion. The immunoglobulin is provided as a 10% solution and hyaluronidase is provided as a solution containing 160 U/mL. The 2 components must be infused sequentially, beginning with hyaluronidase. The full therapeutic dose can be administered in 1 to 2 sites up to every 4 weeks. The frequency and number of infusion sites used to administer IgHy10 may be adjusted for volume, total infusion time, and tolerability to ensure the patient receives the same weekly dose equivalent of the required therapeutic dose. The recommended dose of IgHy10 varies by previous treatment experience, as follows:
For patients naive to IgG treatment, administer IgHy10 gradually from a weekly equivalent dose to a 3 or 4 week interval at 300 mg/kg to 800 mg/kg. Adjust the dosage and treatment interval as necessary based on serum IgG trough levels and infection rates.
For patients switching directly from IVIg, or who have had a previous IV dose of immunoglobulin that can be referenced, IgHy10 should be administered at the same dose and at the same frequency as their previous IVIg treatment. When switching from IV treatment, begin IgHy10 at 1 to 2 weeks after the last IV dose. If patients were previously on a 3-week dosing regimen, increasing the interval to 4 weeks can be accomplished by administering the same weekly equivalents.
For patients currently being administered SCIg, the initial dose of IgHy10 is the same as for subcutaneous treatment but may be adjusted to a 3- or 4-week interval based on the weekly equivalents. The first infusion of IgHy10 should be given 1 week after the last treatment with the previous immunoglobulin.
Hyaluronidase is administered using a minimum of 75 U/g of IgG.14 As per the draft product monograph,10 the recommended infusion rates for immunoglobulin (10%) vary by BW. For patients with a BW under 40 kg, infusion rates should begin at 5 mL/hour per infusion site and can be increased to a maximum of 160 mL per hour per infusion site. For patients with a BW of 40 kg or more, infusion rates should begin at 10 mL/hour per infusion site and can be increased to a maximum of 300 mL/hour per infusion site. Hyaluronidase may be infused manually or using a pump at a rate of 1 mL/minute to 2 mL/minute per infusion site, or as tolerated.
The immunoglobulin (10%) component of IgHy10 provides the therapeutic effect, while hyaluronidase facilitates the dispersion and absorption of the immunoglobulin (10%).10 IgG is the most common antibody in the human immune system and protects against a wide variety of bacterial and viral infections. The role of these antibodies and the mechanism of action of IgG in the immunoglobulin (10%) solution component of IgHy10 have not been fully elucidated. The other component is a soluble recombinant form of human hyaluronidase that modifies the permeability of connective tissues through the hydrolysis of hyaluronan.10
Table 3: Key Characteristics of fSCIg, cSCIg, and IVIg
Characteristic | fSCIg | cSCIg | IVIg |
|---|---|---|---|
Brand names of products | HyQvia | Cutaquig, Cuvitru, Hizentra, Xembify | Gamunex, IGIVnex, Gammagard Liquid, Gammagard S/D, Octagam, Panzyga, Privigen |
Mechanism of action |
| Provides passive immunity by administering exogenous IgG; restores abnormally low IgG levels to normal range, helping to prevent infection | Same as cSCIg |
Indicationa | Replacement therapy for primary and secondary humoral immunodeficiency in adult and patients |
| PID, SID, and neurologic and/or autoimmune conditionsb |
Route of administration | SC infusion (hyaluronidase followed by immunoglobulin) | SC infusion | IV infusion |
Infusion rate |
| Varies by product up to a maximum of 60 mL/hour per infusion site | Varies by product: 0.14 mL/kg/minute to 3.3 mL/kg/minute |
Immunoglobulin concentration | HyQvia = 10% |
|
|
Recommended dose |
| Weight-based; varies by product | Weight-based; varies by product |
Serious adverse effects or safety issues | Contraindicated in patients with: • a history of anaphylactic or severe systemic hypersensitivity reactions to IgG • known systemic hypersensitivity to hyaluronidase | • Thromboembolic events • Thrombosis • Fall in blood pressure or anaphylactic reaction | • Thromboembolic events • Renal dysfunction, acute renal failure, osmotic nephrosis, death • Hemolytic anemia, hemolysis, and hemolytic reaction |
Advantages | Same advantages as cSCIg, in addition to:
|
|
|
Disadvantages |
|
|
|
AE = adverse event; cSCIg = conventional subcutaneous immunoglobulin; fSCIg = facilitated subcutaneous immunoglobulin; IgG = immunoglobulin G; IgRT = immunoglobulin replacement therapy; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency; SC = subcutaneous; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency.
aHealth Canada–approved indication.
bIndication for specific conditions (PID, SID, or neurologic and/or autoimmune) vary by product.
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The original and complete patient input submission can be found in the Stakeholder Feedback section.
One patient group, CIPO, submitted input for the review of IgHy10. Information for the patient group submission was collected through an online survey of patients and caregivers (N = 246) and semi-structured telephone interviews with patients (N = 8) living with a PID. A total of 233 (95%) of the survey respondents were patients living with a PID and 13 (5%) were caregivers responding on behalf of patients. The telephone interviews were with patients currently receiving IgRT for a PID. None of the patients captured in the patient group submission had experience with IgHy10.
The patient group submission described living with a PID as being prone to a wide range of infections that are often severe, chronic, and debilitating. According to CIPO, the frequency of infections is dramatically reduced with IgRT. As many patients with a PID require lifelong therapy, the method of administration and setting where the treatment is administered are important factors that can significantly affect HRQoL. The patient preference is strongly considered when selecting SCIg or IVIg therapy, along with the efficacy of the specific therapy.
According to CIPO, patients want a treatment that minimizes disruptions to their career or personal life. It was noted that administering therapy at a lower frequency and reducing the need to travel to an infusion clinic would help minimize disruption to everyday life as well as reduce the risk of hospital-acquired infections. Telephone interview respondents indicated curiosity about trying IgHy10, but also described a desire for the same result from treatment, voiced concerns about having a negative experience due to switching therapies and wondered whether IgHy10 would be an appropriate treatment option for an individual patient seeking treatment.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PID and SID in pediatric and adult patients.
Unmet Needs
The clinical experts stated that treatments for primary humoral immunodeficiency that reverse the course of disease are limited. The experts also noted that treatments for secondary humoral immunodeficiency are limited, but some etiologies can be reversed. Current options for IgRT are effective, but the clinical experts noted that other treatments are needed to improve adherence and new formulations are needed to improve convenience. Current treatments were described as time-consuming and having an adverse effect on daily function, as IVIg requires monthly hospital visits lasting several hours and SCIg infusions are administered weekly or every 2 weeks. The clinical experts also expressed the need for treatments that are better tolerated, noting IVIg is associated with systemic side effects, including headaches, fever, myalgia, arthralgia, nausea, and allergic reactions, and requires pre-medication with antipyretics and anti-nausea drugs as well as antihistamines or corticosteroids. Some patients were also reported as having experienced transient acute kidney injury, aseptic meningitis, and thrombotic- and hyperviscosity-related side effects with IVIg, according to the experts. Lastly, the experts reported that not all patients respond to currently available treatments. Further, it was noted that it is difficult to achieve target levels of immunoglobulin in selected patients with IVIg due to its pharmacokinetics (greater fluxes in peaks and troughs between infusions), which can translate to suboptimal clinical control of disease.
Place in Therapy
According to the clinical experts consulted for this review, IgHy10 would present an additional option to patients who are already on IgRT (either IVIg or cSCIg), either as first line for patients who were expected to benefit from SCIg or as second line (following IVIg or cSCIg) for those who do not tolerate their current IgRT. The clinical experts reported that when determining which treatment to use, both the availability and different types of IgRT are discussed with the patient. The experts noted that the decision to use IgHy10 would be similar to the decision to use other available products. It was noted that this is because IgHy10 is very similar and comparable to other SCIg products in terms of providing the same anti-infective, immunomodulatory IgG molecules. Both experts felt IgHy10 had the potential to cause a shift in the current treatment paradigm, particularly in terms of SCIg therapies, as it would expand the available treatment options for subcutaneous IgRT for patients with primary or secondary humoral immunodeficiency.
Patient Population
The clinical experts indicated that patients who need IgRT and who would be expected to benefit more from SCIg compared with IVIg would also be expected to benefit from treatment with IgHy10. This would include patients with certain comorbidities (e.g., renal failure, thrombophilia, hyperviscosity disorders or older patients), those who live far from or have limited access to health care facilities, those who had severe side effects to IVIg, have difficult venous access, or those who prefer not to miss school or work or are unable to adhere to weekly SCIg infusions. The clinical experts also noted that patients who require large doses or large volumes due to higher BW or the need for immunomodulation (such as patients who suffer from additional autoimmune or hyperinflammatory diseases similar to the immune dysregulation in PID), or those who have a needle phobia and want to avoid using multiple injection sites, or those who want to infuse SCIg less frequently, would especially benefit from IgHy10. Both of the experts stated there are clear guidelines for diagnosing and treating primary and secondary humoral immunodeficiency; therefore, the identification of patients who require IgRT is not a challenge. More specifically, the experts stated a diagnosis is made using laboratory tests that demonstrate a quantitative and/or qualitative defect in innate immunoglobulin manifesting in clinical sequelae, such as recurrent infections, autoimmune disease, and/or malignancy.
Patients identified by the clinical experts as being least suitable for treatment with IgHy10 included those who have little to no subcutaneous tissue (very low body fat) or who have severe skin conditions preventing the subcutaneous administration. The clinical experts also felt that patients who require frequent clinical and immunoglobulin monitoring, such as patients with complex conditions with severe disease manifestation that require regular in-person reassessment (potentially coordinated with other IV treatments) would be less suitable for treatment with IgHy10. This is because it is more appropriate to follow those patients with monthly hospital visits when IVIg can be administered, and potentially coordinated with, other IV treatments. Lastly, the clinical experts noted patients with a history of poor adherence to treatment, inappropriate compliance, or inappropriate support from a caregiver (particularly for pediatric patients) may also be less suitable for treatment with IgHy10.
Assessing Response to Treatment
The clinical experts indicated that patients with primary or secondary humoral immunodeficiency who are starting treatment with IgHy10 and who also have chronic or active infections would be expected to respond to treatment with an improvement in clinical state and normalized blood IgG levels (i.e., IgG target levels achieved). In terms of long-term outcomes to assess response to treatment, the clinical experts would expect patients to present with fewer infections (and a reduction in frequency and severity), which should result in fewer visits to outpatient clinics, a reduced rate of emergency visits and/or hospitalizations due to infections, a reduced need for antimicrobial treatments or prophylactic use, fewer missed days from school or work, improved overall health and HRQoL, and a reduced need to access health care facilities for the treatment of infections. The experts also noted that in some cases, primary humoral immunodeficiency–associated organ dysfunction (such as lung function deterioration or malignancies) and autoimmune/hyperinflammatory diseases may go into remission. The experts would also expect the overall burden on caregivers and the health care system to be reduced.
The clinical experts reported that the outcomes used to assess the response to treatment with IgRT have a high degree of objectivity and therefore are not expected to vary between physicians. The experts described a clinically meaningful response to treatment as 1 that would include maintenance of target steady-state serum IgG trough levels (at least 7g/L), reduced infection rate (i.e., no serious infections or a notable reduction in severe infections per year), no emergency room visits/hospital admissions due to infections, a significant reduction in days missed from work or school, improved survival and HRQoL, and improvement or remission of autoimmune and inflammatory diseases associated with inborn error of immunity, if present.
The clinical experts suggested that patients treated with IgHy10 should be assessed for response to treatment every 3 to 6 months depending on disease severity, which is consistent with current clinical practice for patients with primary or secondary humoral immunodeficiency.
Discontinuing Treatment
Feedback from the clinical experts indicated that discontinuation of treatment with IgRT would be considered if there is a lack of improvement in immunoglobulin replacement levels, or response to treatment is not observed despite increasing doses of IgRT. The experts also noted that a patient who is not able to adhere to therapy (identified by an inability to maintain therapeutic serum IgG levels) and who therefore requires frequent monitoring, may be discontinued from IgHy10, as they may be better served by IVIg replacement. Discontinuation from IgHy10 may also be considered based on patient preference or because the patient does not exhibit an improvement in HRQoL, as described by the clinical experts.
Prescribing Conditions
According to the clinical experts, IgHy10 can be administered at home after appropriate training by a patient support program, which is similar to current practice for other SCIg preparations. The clinical experts also noted that selected cases may warrant infusion of IgHy10 in a health care facility (infusion clinic, outpatient clinic, in-hospital medical day treatment unit), such as when the caregiver or patient is unable to administer the infusion.
The experts also felt that a specialist with appropriate knowledge and training in primary or secondary humoral immunodeficiency should be involved in the diagnosis and initiation of therapy. This may include immunologists, rheumatologists, and hematologists with training in IgRT.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Input for the review of IgHy10 was received from 2 clinician groups, the CINC and the Medical and Scientific Advisory Committee of the CIPO. The CINC is a group of clinicians and researchers that was created to enhance clinical and research collaborations and promote clinical immunology and associated research in Canada. The CIPO Medical and Scientific Advisory Committee works with patient groups to ensure activities and health information are relevant and useful for patients and caregivers.
Unmet Needs
The unmet needs of patients living with primary and secondary humoral immunodeficiency described by clinician groups were consistent with the input from the clinical experts consulted by CADTH.
Place in Therapy
Similar to the input received from the clinical experts consulted by CADTH, the clinician group input described IgHy10 as an intervention that would complement other available treatment options due to its unique mechanism of action compared with the currently available immunoglobulin therapies. The clinician groups felt that IgHy10 would likely be used as a second-line treatment (following IVIg or cSCIg) for primary or secondary humoral immunodeficiency in general but could become a first-line treatment in certain subpopulations of patients with the greatest unmet need. That includes patients requiring significantly large volumes of immunoglobulin that require a high frequency of cSCIg infusions, those who have difficulty tolerating their required volume of cSCIg, those receiving IVIg that is associated with frequent and bothersome systemic symptoms, those receiving IVIg who would greatly benefit from not missing school/work on infusion days, and those patients living in remote areas without easy access to health care facilities.
Patient Population
The feedback from the clinician groups regarding the patient population best suited for treatment with IgHy10 was consistent with the input from the clinical experts consulted by CADTH.
Assessing Response to Treatment
In general, the feedback from the clinician groups in terms of assessing response to treatment with IgHy10 was consistent with the input from the clinical experts consulted by CADTH. Regarding a clinically meaningful response to treatment, the clinician group input further described the target for infection rate as fewer than 4 infections per year.
The clinician group input recommended that treatment response be assessed at least every 6 to 12 months, which is longer than the frequency of 3 to 6 months suggested by the clinical experts consulted by CADTH.
Discontinuing Treatment
The information submitted by the clinician groups regarding considerations for the discontinuation of treatment with IgHy10 was consistent with the input from the clinical experts consulted by CADTH.
Prescribing Conditions
In general, the clinician group input on the prescribing conditions for IgHy10 was consistent with the input from the clinical experts consulted by CADTH. The clinician group input also noted that the administration of IgHy10 in the outpatient setting would be an important improvement, as the infusions can be given very quickly without significant risk of the systemic side effects seen with IVIg or the local side effects seen with traditional SCIg. The clinician groups also mentioned allergists and oncologists as specialists qualified for care of patients with primary or secondary humoral immunodeficiency, in addition to those listed by the experts consulted by CADTH.
Additional Considerations
According to the clinical groups, SCIg has been an important development in the treatment of immunodeficiency, freeing patients from outpatient units and freeing outpatient units to treat other patients. They noted there are considerable benefits associated with the ability to treat patients at home, particularly in terms of rising capacity issues for both inpatient and outpatient beds and the increasing costs of medicines.
Drug Program Input
The drug programs provide input on every drug reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. Their implementation questions and the corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Does IgHy10 allow a similar dose as IVIg and a potentially lower dose per patient than cSCIg? If a patient is switched from IVIg/cSCIg to IgHy10, how much would the dose be expected to change? | Similar to the other cSCIg products, the IgHy10 dose is calculated the same way it would be for IVIg dosing and administered similarly every 4 weeks. A change in the dose or dosing interval would not be expected for a switch from IVIg to IgHy10. When switching from SCIg to IgHy10, the monthly dose would be the same and the dosing interval would be extended to every 3 or 4 weeks. For example, a patient on a dosage of 40 g per month of IVIg would be given 10 g of cSCIg every 7 days or 40 g of IgHy10 every 28 days. Therefore, a switch from SCIg and IgHy10 would not be expected to result in a change in monthly dose, but the dosing interval would be adjusted as described. Of note, IgHy10 dosing in Study 160603 and 160902 was 108% of the dose of IVIg received during epoch 1 of Study 160603. |
How does IgHy10 compare with IVIg and cSCIg in terms of safety?
| At the time of this review, no evidence comparing treatment with IgHy10 with other forms of IgRT (IVIg or cSCIg) was identified. Based on the results presented in Study 160603 and Study 160902, the clinical experts felt that, similarly to other cSCIg products, IgHy10 appears to translate into more local but fewer systemic adverse events than what is observed with IVIg treatment in clinical practice. |
What is the average dosing interval for IgHy10 and how does it compare with IVIg and cSCIg? | The clinical experts consulted by CADTH do not have experience using IgHy10 in clinical practice; however, cSCIg can be administered at intervals of up to every 2 weeks. Based on the literature, the average dosing interval of IgHy10 is expected to be greater than cSCIg, reaching up to every 3 or 4 weeks. This is consistent with the dosing interval used for IVIg in clinical practice. |
How does IgHy10 compare with other Ig brands in terms of overall infusion time? | Individual IgHy10 infusion times are expected to be shorter compared with the same dose of IVIg and longer compared with cSCIg. This is because, with cSCIg, patients infuse smaller volumes administered more frequently. For reference, in Study 160603, the median duration of IVIg infusion was 2.33 hours (range = 0.92 to 6.33) and the median duration of IgHy10 infusion was 2.08 hours (range = 0.83 to 4.68).12 |
If IgHy10 is found to be more costly than other Ig brands for not much benefit, we would need clear criteria on when IgHy10 should be considered over other Ig brands; for example, should patients with treatment compliance issues with cSCIg or who are unable to tolerate it be eligible for IgHy10? Should patients with poor venous access who are unable to use IVIg be eligible for IgHy10? | The clinical experts indicated they would consider the use of IgHy10 for patients they would consider for cSCIg. This would include patients with poor venous access as well as patients with tolerability or compliance issues with cSCIg. |
What percentage of the patient population would be able to manage their condition at home with monthly frequency? | Again, the clinical experts consulted by CADTH do not have experience with IgHy10; however, they would expect the proportion of patients who are able to manage their condition with cSCIg at home would be similar to IgHy10, i.e., more than 90% of patients. Of note, the proportion of patients who were able to independently self-administer IgHy10 at home was much lower. In Study 160603, 20.5% of all infusions were administered at home without nurse intervention. In Study 160902, 64% of patients and 52% of infusions required assistance with self-infusion (home infusion). |
IgHy10 is a dual vial unit. It has the same route but a different administration method than cSCIg. What level of special training would a patient/caregiver need to be comfortable administering IgHy10? | A specialized training program (such as the OnePath program in the case of another cSCIg product) would be essential to help orient patients to the administration of IgHy10 as well as educate patients on possible adverse events and how to treat them. The support program could also help navigate access issues. It was recommended that education around the mechanism of action for IgHy10 be included. Additionally, the program should be continuously available to support patients on IgHy10. The current capacity of training programs for SCIg products would likely not need to be increased; however, the duration of training sessions would likely need to be increased by approximately 15 to 30 minutes. IgHy10 infusion would require slightly more training (not more sessions but a longer session) than cSCIg products. The clinical experts felt that more than 90% of patients would be able to successfully complete training to be able to comfortably administer IgHy10 at home. |
If a patient starts IgHy10, can they switch to other Ig brands during the course of treatment? | The clinical experts indicated they would not expect to have any issues with switching patients on IgHy10 to other brands of IgRT. |
Is any additional monitoring required or are there any safety concerns due to the administration of recombinant hyaluronidase? | The clinical experts did not express any safety concerns or the need for any additional monitoring for the administration of IgHy10. |
cSCIg = conventional subcutaneous immunoglobulin; Ig = immunoglobulin; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IgRT = immunoglobulin replacement therapy; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency.
Clinical Evidence
The clinical evidence included in this part of the review of IgHy10 is presented in 3 sections. The first section, the systematic review, includes the pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review; however, indirect evidence was not provided or identified in the literature. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of IgHy10 for the treatment of primary and secondary humoral immunodeficiency in adult and pediatric patients 2 years of age and older.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented subsequently was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult and pediatric patients (2 years of age and older) with primary and secondary humoral immunodeficiency. Subgroups:
|
Intervention | Normal human immunoglobulin (10%)a and recombinant human hyaluronidase (160 units/mL)b as a solution for subcutaneous infusion, according to the following dosing recommendations:
|
Comparator | Ig replacement therapy (IVIg or cSCIg) |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; BMI = body mass index; cSCIg = conventional subcutaneous immunoglobulin; HRQoL = health-related quality of life; Ig = immunoglobulin; IgG = immunoglobin G; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = IV immunoglobulin; RCT = randomized controlled trial; SAE = serious adverse event; SCIg = subcutaneous immunoglobin; SF-36 = Short Form (36) Health Survey; WDAE = withdrawal due to adverse event.
aNormal immunoglobulin (human) 10% is available as 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL.
bRecombinant human hyaluronidase 160 units/mL is available as 200 units/1.25 mL, 400 units/2.5 mL, 800 units/5 mL, 1,600 units/10 mL, and 2,400 units/15 mL.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies tool.40
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) through Ovid and Embase (1974‒) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were human immunoglobulin and recombinant human hyaluronidase. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on July 22, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Plasma Protein Product Expert Committee on November 24, 2021.
A focused literature search for network meta-analyses dealing with immunoglobulins or immunodeficiencies was run in MEDLINE All (1946–) on July 21, 2021. No filters were applied to limit the retrieval by study type.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature tool.41 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
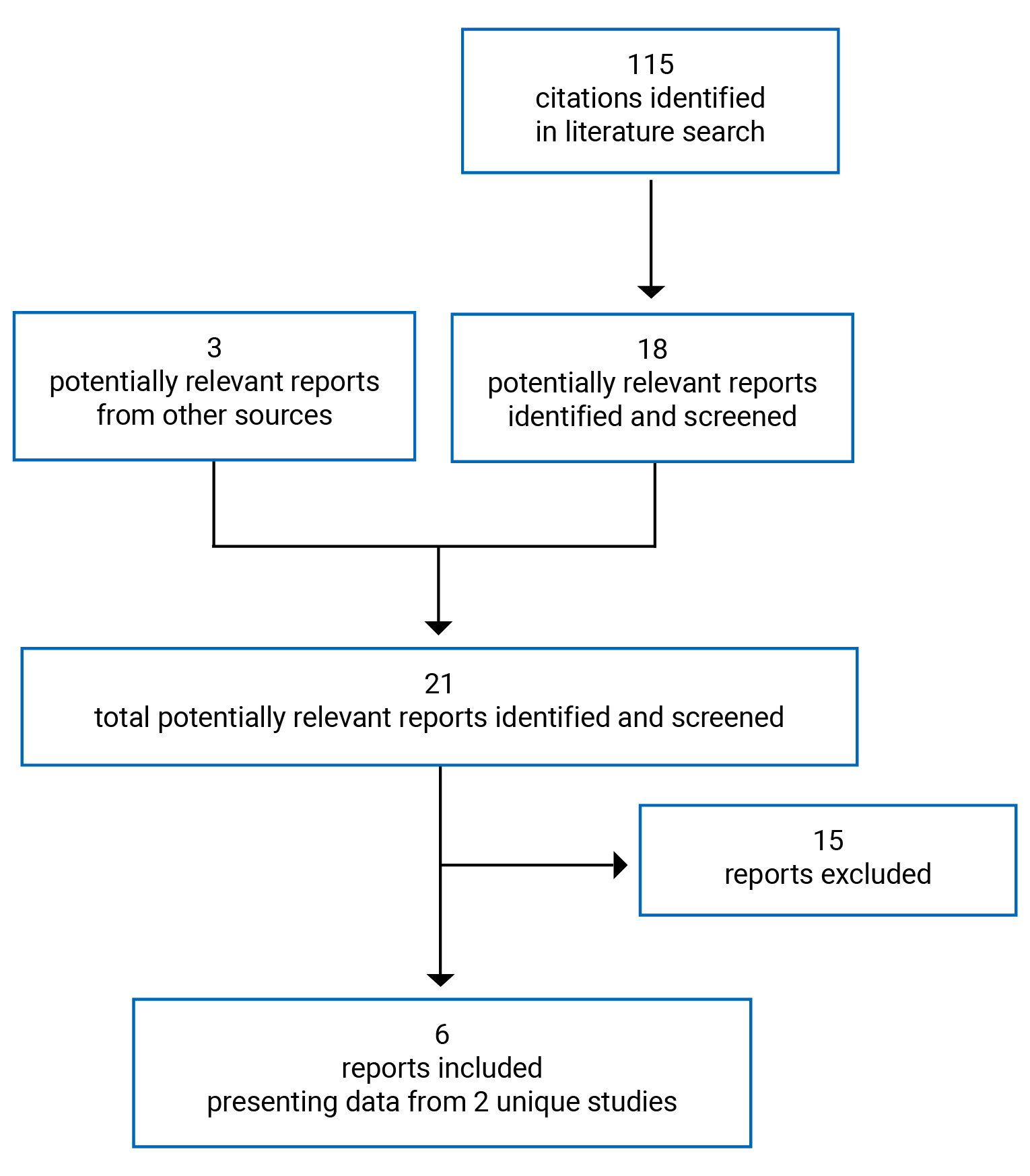
Table 6: Details of Included Studies
Detail | Study 160603 | Study 160902 |
|---|---|---|
Designs and populations | ||
Study design | Open-label, non-randomized, single-group prospective study | Open-label, non-randomized, single-group prospective study |
Locations | 14 study sites in the US and 1 in Canada | 11 study sites in the US |
Patient enrolment dates | December 18, 2008, to November 11, 2010 | Patients in Study 160603 proceeded directly into the open-label extension; safety follow-up began July 31, 2012. The study end date was August 6, 2013 |
Enrolled (N) | 89 | 66 |
Inclusion criteria |
| Completion of Baxter Clinical Study protocol 160603 |
Exclusion criteria |
| Refer to Study 160603 |
Drugs | ||
Interventions |
| The treatment dose and infusion interval from Study 160603 were maintained for the first 3 infusions. Thereafter, patients had the option to switch from a 3- or 4-week interval to a 2-week treatment interval using one-half the calculated 4-week dosage for a maximum of 4 months. In a final safety follow-up, patients changed to IVIg or SCIg alone (without hyaluronidase). |
Duration | ||
Phase | ||
Study 160601 (pre-study) | 12 months | Not applicable |
Open label |
| Not applicable |
Open-label extension | Not applicable | Minimum 4 months after switching to a 2-week treatment interval |
Safety follow-up | Not applicable | Up to 48 weeks (up to 3.5 years from study initiation) |
Outcomes | ||
Primary end point |
| Efficacy (not during safety follow-up):
|
Secondary and exploratory end points | Secondary Efficacy end points:
Safety end points:
Exploratory
| Secondary Long-term safety and tolerabilityc of IgHy10, evaluated by:
Exploratory
|
Notes | ||
Publications | Wasserman et al. (2012)12 | |
AE = adverse event; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; AUC = area under the concentration vs. time curve; BW = body weight; IgA = immunoglobulin A; IgG = immunoglobulin G; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IP = investigational product; ISG = immune serum globulin; IVIg = intravenous immunoglobulin; PedsQL = Pediatric Quality of Life Inventory; PID = primary immunodeficiency disorder; SC = subcutaneous; SCIg = subcutaneous immunoglobulin; SF-36 = Short Form (36) Health Survey; VASBI = validated acute serious bacterial infection.
Note: 3 additional reports were included, the CADTH submission6 and the Clinical Study Reports for Study 16060315 and Study 160902.16
aSerious conditions included: hepatitis B or C, HIV type 1 or 2, ALT or AST > 2.5 × the upper limit of normal, persistent severe neutropenia (ANC ≤ 500/mm3), a creatinine clearance < 60% of normal, malignancy in the past 12 months (other than adequately treated basal cell or squamous cell carcinoma of the skin or carcinoma in situ of the cervix), a thrombotic episode in the past 12 months, abnormal protein loss, anemia, significant risk of bleeding or bruising, an active infection, and severe dermatitis that precluded safe administration of SC treatment.
bDiagnostic criteria were according to the US Department of Health and Human Services, FDA Guidance for Industry, June 2008.11 Essential diagnostic features: Bacteremia required a positive blood culture. Sepsis was defined as 2 of the following in adults: temperature > 38°C oral or > 39°C rectal, or < 36°C oral or < 37°C rectal; heart rate > 90 beats/minute; respiratory rate > 20 breaths/minute or partial pressure of carbon dioxide (PaCO2) < 32 mm Hg; white blood cell count > 12,000/mm3, < 4,000/mm3, or > 10%. In children, sepsis was based on the criteria of the International Consensus Conference on Pediatric Sepsis. Blood-borne infections related to an indwelling catheter or vascular access device were not included. Bacterial meningitis required a positive cerebrospinal fluid Gram stain and/or culture and/or positive cerebrospinal fluid bacterial antigen assay. Osteomyelitis and septic arthritis required a positive X-ray, nuclear medicine bone scan, MRI, or CT scan showing bony destruction with radiolucent areas. Bacterial pneumonia required pulmonary infiltrate with consolidation on a chest X-ray. A visceral abscess required typical findings on ultrasound, CT scan, MRI, or radionuclide scan. Other features of the diagnoses are elaborated in the study protocol.
cSafety and tolerability end points were not defined as primary end points in the Clinical Study Report; however, they were aligned with the primary objectives of Study 160902.
Sources: Clinical Study Reports,15,16 Wasserman et al. (2012),12 Wasserman et al. (2016a),44 and Wasserman et al. (2016b).13
Description of Studies
The details of the included studies are summarized in Table 6. Two sponsor-provided Clinical Study Reports, funded by Baxter Health care Corporation, were included. Study 160603 (N = 89) was a phase III, open-label, non-randomized, single-group prospective study with enrolment from December 2008 to November 2010. A total of 15 study sites were included, with 14 located in the US and 1 in Canada. Study 160603 aimed to evaluate the efficacy and tolerability of IgHy10 in patients with a PID. The study also aimed to assess HRQoL for IgHy10, cSCIg, and IVIg.
An overview of the design of Study 160603 is presented in Figure 2. Patients were enrolled in 1 of 2 study arms (cohorts) in Study 160603 based on whether they had previously participated in Study 160601. Patients who participated in Study 160601 were enrolled in study arm 1. Patients in study arm 1 were not required to receive treatment with IVIg during epoch 1 of Study 160603 because data regarding the bioavailability and exposure to IVIg had already been obtained in Study 160601, which evaluated the pharmacokinetics, efficacy, and safety of IVIg and was considered equivalent to epoch 1 of Study 160603. Patients enrolled in study arm 2 included all other patients enrolled in Study 160603 and were entered into epoch 1. During epoch 1, patients in study arm 2 received treatment with IVIg (10%) for 3 months. All patients (in study arms 1 and 2) proceeded to epoch 2, where they received IgHy10 for 14 months to 18 months. The overall study duration was planned to be 17 months for each patient but could extend to a maximum of 21 months, pending approval of Study 160902.
Extension Study 16090216 (N = 66) aimed to assess the long-term safety, tolerability, and practicability of IgHy10. Patients who had previously completed Study 160603 proceeded directly into the open-label, non-randomized, single-group extension study. Patients maintained the same dose and infusion interval for IgHy10 for the first 3 infusions, after which they had the option of switching to a 2-week treatment interval using one-half of the 4-week dose for up to 4 months. The purpose of the switch to a shorter treatment interval was to evaluate whether this would result in improved immunoglobulin trough levels. On July 31, 2021, following a discussion between the sponsor and the FDA, all study patients entered the safety follow-up period where they stopped treatment with rHuPH20. This decision was based on the theoretical risk of exposure to anti-rHuPH20 antibodies. Patients were therefore treated with immunoglobulin (10%) alone via IV or subcutaneously for 24 to 48 weeks, depending on the levels of rHuPH20-binding antibody titre, to ensure a safe transfer off of the study drug. A total of 50 patients completed the safety follow-up; 3 of these had previously switched directly to the safety follow-up phase during epoch 2 of Study 160603.
Figure 2: Overview of Study 160603 Study Design

max = maximum; PK = pharmacokinetic; rHuPH20 = recombinant human hyaluronidase; SC = subcutaneous; w = weeks.
Source: Clinical Study Report.15
Populations
Inclusion and Exclusion Criteria
Study 160603 enrolled patients aged 2 years and older with a PID requiring antibody replacement as defined by WHO criteria.42,43 Patients were eligible if they had completed or were about to complete Study 160601, or had been receiving regular IVIg treatment at mean intervals of 21 (SD = 3) days or subcutaneous treatment at mean intervals of 5 to 16 days for at least 3 months before enrolment at a minimum dose of 300 mg/kg every 4 weeks. The main criteria for exclusion were serious medical conditions or hypersensitivity to immunoglobulin, enrolment in another non-Baxter clinical study, pregnancy, or not employing birth control measures. A prerequisite to inclusion in Extension Study 160902 was completion of Study 160603.
Baseline Characteristics
Table 7 and Table 8 show a summary of the patient baseline characteristics. In Study 160603, 2 patients withdrew before treatment, leaving 87 patients in the safety population for whom baseline characteristics were described. Of these, 87 patients (31 from Study 160601 and 56 from epoch 1 of Study 160603) were included among those who received IVIg (10%), and 83 patients received IgHy10 in epoch 2. Patients were distributed approximately equally by sex (51% male), had a median age of 35.0 years (range = 4 to 78), and were primarily White, and non-Hispanic and non-Latino (91%).
The most commonly diagnosed PID disease was common variable immune deficiency (56%), followed by hypogammaglobinemia (20%), and X-linked agammaglobulinemia (7%). All patients reported a medical history that included disorders of the hematopoietic and lymphatic systems, and the majority also reported medical conditions relating to the eyes, ears, nose, and throat (98%) and respiratory system (n = 76 of 87; 87%). The median serum IgG trough level up to 6 months before enrolment (before Study 160601 for study arm 1 and before Study 160603 for study arm 2) was 10.34 g/L (range = 4.05 to 32.00). Also, 64 (74%) of patients were receiving IVIg every 28 days, 17 (20%) every 21 days, 3 (3%) every 30 days, and 3 (3%) every 10 days or less before enrolment in Study 160603. It is unclear whether patients who had experience with IVIg administered every 10 days also had experience with cSCIg. Additionally, 33 patients (38%) were naive to cSCIg before enrolment.
There were no notable differences in the demographic characteristics of patients who continued into Extension Study 160902 versus patients in the main study. The most commonly diagnosed PID among those in the extension was common variable immune deficiency (59%), followed by humoral immune deficiency (9%) and hypogammaglobulinemia (9%). Patient medical history was generally similar to that of the patients who participated in Study 160603.
Table 7: Summary of Baseline Demographic Characteristics in Study 160603 and Study 160902 (SAS)
Characteristic | Study 160603 (N = 87)a | Study 160902 (N = 66) | ||||||
|---|---|---|---|---|---|---|---|---|
2 to < 12 years (n = 14) | ≥ 12 years (n = 73) | All | 2 to < 12 years (n = 4) | 12 to < 16 years (n = 7) | 16 to < 65 years (n = 47) | ≥ 65 years (n = 8) | All | |
Sex, n (%) | ||||||||
Male | 8 (57.1) | 36 (49.3) | 44 (50.6) | 3 (75.0) | 6 (85.7) | 24 (51.1) | 1 (12.5) | 34 (51.5) |
Female | 6 (42.9) | 37 (50.7) | 43 (49.4) | 1 (25.0) | 1 (14.3) | 23 (48.9) | 7 (87.5) | 32 (48.5) |
Age (years), median (range) | 8.0 (4 to 11) | 44.0 (12 to 78) | 35.0 (4 to 78) | 10.5 (9 to 11) | 15.0 (13 to 15) | 47.0 (16 to 64) | 71.0 (65 to 80) | 43.0 (9 to 80) |
Race, n (%) | ||||||||
White | 14 (100.0) | 65 (89.0) | 79 (90.8) | 4 (100.0) | 6 (85.7) | 41 (87.2) | 8 (100.0) | 59 (89.4) |
Other | 0 (0.0) | 8 (11.0) | 8 (9.2) | 0 (0.0) | 1 (14.3) | 4 (8.5) | 0 (0.0) | 5 (7.5) |
Ethnicity, n (%) | ||||||||
Hispanic or Latino | 1 (7.1) | 7 (9.6) | 8 (9.2) | 0 (0.0) | 0 (0.0) | 2 (4.3) | 0 (0.0) | 5 (7.6) |
Non-Hispanic or non-Latino | 13 (92.9) | 66 (90.4) | 79 (90.8) | 4 (100.0) | 7 (100.0) | 42 (89.4) | 8 (100.0) | 61 (92.4) |
Height (cm), median (range) | 130.0 (94 to 153) | 166.0 (108 to 193) | 165.0 (94.0 to 193.0) | 148.5 (127 to 154) | 156.0 (150 to 181) | 167.6 (132 to 188) | 162.3 (154 to 184) | 165.1 (127 to 188) |
Weight (kg), median (range) | 27.5 (15.0 to 59.4) | 69.0 (34.5 to 135.9) | 63.8 (15.0 to 135.9) | 42.0 (30.5 to 51.2) | 55.7 (42.7 to 86.7) | 70.9 (40.2 to 141.3) | 67.8 (60.7 to 81.8) | 67.2 (30.5 to 141.3) |
SAS = safety analysis set.
Note: Baseline refers to the start of epoch 1 in Study 160603.
aIncludes patients who received at least 1 dose of the study treatment.
Sources: Wasserman et al. (2012),12 Wasserman et al. (2016a),44 Wasserman et al. (2016b),13and Clinical Study Reports.15,16
Table 8: Summary of Baseline Disease Characteristics in Study 160603 and Study 160902 (SAS)
Characteristic | Study 160603 (N = 87)a | Study 160902 (N = 66) |
|---|---|---|
Primary immunodeficiency disease diagnosis, n (%) | ||
Common variable immunodeficiency disease | 49 (56.3) | 39 (59.1) |
Hypogammaglobulinemia | 17 (19.5) | 6 (9.1) |
X-linked agammaglobulinemia | 6 (6.9) | 3 (4.5) |
IgG subclass deficiency | 4 (4.6) | 4 (6.1) |
Specific antibody deficiency | 4 (4.6) | 1 (1.5) |
Hyper IgM syndrome | 2 (2.3) | 2 (3.0) |
Common variable immunodeficiency disease + hyper IgE syndrome | 1 (1.1) | NR |
Dysgammaglobulinemia | 1 (1.1) | 1 (1.5) |
Hyper IgE syndrome | 1 (1.1) | 1 (1.5) |
Severe combined immunodeficiency | 1 (1.1) | 2 (3.0) |
Severe combined immunodeficiency plus hypogammaglobulinemia | 1 (1.1) | NR |
Combined immune deficiency | NR | 1 (1.5) |
Humoral immune deficiency | NR | 6 (9.1) |
Medical history (disorders/conditions by system), n (%) | ||
Eyes, ears, nose, throat | 85 (97.7) | 63 (95.5) |
Respiratory | 76 (87.4) | 56 (84.8) |
Cardiovascular | 37 (42.5) | 34 (51.5) |
Gastrointestinal | 64 (73.6) | 52 (78.8) |
Musculoskeletal | 54 (62.1) | 49 (74.2) |
Neurologic | 73 (83.9) | 55 (83.3) |
Endocrine | 34 (39.1) | 25 (37.9) |
Hematopoietic/lymphatic | 87 (100.0) | 63 (95.5) |
Dermatological | 58 (66.7) | 47 (71.2) |
Genitourinary | 41 (47.1) | 36 (54.5) |
Serum IgG trough levels (g/L) up to 6 months before enrolment, median (range) | 10.335 (4.05 to 32.00) | NA |
IgE = immunoglobulin E; IgG = immunoglobulin G; IgM = immunoglobulin M; NA = not applicable; NR = not reported; SAS = safety analysis set.
Note: Baseline refers to the start of epoch 1 in Study 160603.
aTwo patients withdrew before treatment and are excluded.
Sources: Wasserman et al. (2012),12 Wasserman et al. (2016a),44 Wasserman et al. (2016b),13 Clinical Study Reports.15,16
Interventions
Study 160603
During epoch 1, all patients were treated with IV infusion of normal human immunoglobulin (10%) for 12 weeks, at their usual dose and frequency (≥ 300 mg/kg BW every 4 weeks with a 3- or 4-week interval). All infusions were administered at the study site. Infusions started at a rate of 0.5 mL/kg BW per hour and increased as tolerated up to a maximum of 5.0 mL/kg BW per hour. For patients who had previously received subcutaneous treatment, IV treatment in epoch 1 was administered at a 3- or 4-week interval, with a dose based on the weekly equivalent of subcutaneous treatment. If a patient experienced an AE of at least moderate severity, the infusion rate was reduced to the rate immediately below where the AE occurred. In the case of hypersensitivity reactions or AEs that continued after a reduction of the infusion rate, the infusion was stopped and treated according to the standard of care.
In epoch 2, patients were treated with IgHy10 for 14 to 18 months, which was delivered by portable IV pump or syringe pump at an infusion site chosen by the patient. Initial infusions were administered at the study site until at least 2 infusions at the maximum infusion interval were tolerated. Thereafter, home treatment was allowed following adequate training and in the presence of a home nurse. Patients first received rHuPH20 by subcutaneous infusion at a rate of 1 mL to 2 mL per minute and a dose of at least 75 U/g immunoglobulin. The maximum volume for the majority of patients was approximately 20 mL and was infused at a single site, though 2 sites could be used. This was followed, within 10 minutes, by infusion of subcutaneous normal human immunoglobulin (10%) at 108% of the weekly equivalent dose of IVIg.
Treatment intervals and doses for the initial subcutaneous infusions were gradually increased during the first weeks of treatment, referred to as the ramp-up period. The first dose was a single dose for a 1-week interval based on the pre-study IVIg dose. A dose was considered to be tolerated if there were no SAEs, no non-serious moderate or severe local AEs preventing completion of the infusion, and no non-serious moderate or severe systemic AEs during or within 60 minutes of the infusion. If tolerated, each week, the interval was increased by 1 week, with the dose of immunoglobulin calculated based on the weekly equivalent of the dose that should be received on a 4-week interval until the treatment interval was the same as the pre-study treatment interval for IVIg (every 3 or 4 weeks). The dose then remained constant throughout epoch 2 and was calculated based on immunoglobulin in grams per BW in kg. At that time, IgG trough levels were reviewed and, if the trough level ratio was not between 93% and 123%, the dose was corrected for the next infusion and further reviewed until the ratio fell within the desired range. The infusion rate varied by week and patient weight (Table 9) up to a maximum of 300 mL per hour. Generally, a single infusion site was used, but a second was allowed when the volume infused was above 600 mL or if a single site was not tolerated. If an AE occurred, the infusion rate could be adjusted, as described for epoch 1. Serum trough levels of IgG greater than 4.5 g/L had to be maintained throughout the study; if levels fell to 4.5 g/L or less, the dose was to be adjusted and trough levels re-evaluated at the next infusion.
Prior and concomitant therapies taken for up to 2 weeks before the start of Study 160603 were allowed and were recorded; prophylactic treatment with antibacterial antibiotics, other immunoglobulin products, and hyperimmune serum was not allowed. Pre-medication before IV infusions with antihistamines, antipyretics, and/or steroids was to be avoided unless the same AE had been observed during or following at least 2 infusions either before or during the study. Pre-medication before subcutaneous infusions was not allowed unless an AE of at least moderate severity that did not resolve with a reduction of the infusion rate occurred during or after at least 2 previous infusions.
Extension Study 160902
Treatment from epoch 2 of Study 160603 was continued (at the same dose as the last infusion) in Extension Study 160902, but patients had the option to switch from a 3- or 4-week to a 2-week treatment interval using 1-half of the calculated 4-week dose for a maximum of 4 months. Patients were requested to change their drug administration interval to evaluate whether a more frequent administration of IgHy10 improved IgG trough levels. In the subsequent safety follow-up period, patients discontinued use of hyaluronidase and were switched to either IVIg or SCIg alone for up to 48 weeks. Those who were switched to IV infusion received a weekly dose equivalent to 100% (± 5%) of the most recent IV dose at a 3- or 4-week interval; those continuing with subcutaneous infusions received a weekly dose equivalent to the most recent IV dose multiplied by 1.37 (± 5%) at 1-week intervals.
Table 9: Infusion Rates for Subcutaneous Normal Immunoglobulin (Human) 10%
Time | Infusion rate by body weight (mL/hour per site) | |
|---|---|---|
< 40 kg | ≥ 40 kg | |
First 2 SC infusions | ||
15 ± 10 minutes | 5 | 10 |
15 ± 10 minutes | 10 | 30 |
15 ± 10 minutes | 20 | 60 |
15 ± 10 minutes | 40 | 120 |
Until end of infusion | 80 | 240 |
Subsequent SC infusions | ||
15 ± 10 minutes | 10 | 10 |
15 ± 10 minutes | 20 | 30 |
15 ± 10 minutes | 40 | 120 |
15 ± 10 minutes | 80 | 240 |
Until end of infusion | 160 | 300 |
SC = subcutaneous.
Sources: Wasserman et al. (2012),12 Clinical Study Report.15
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 10. These end points are further summarized subsequently. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 10: Summary of Efficacy Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 160603 | Study 160902 |
|---|---|---|
Infections | ||
Acute serious bacterial infections | Primary (epoch 2 only) | Primary |
All infections, by organ system | Secondary | Secondary |
Immunoglobulin levels | ||
Trough levels of immunoglobulin | Secondary | Secondary (in relation to dose frequency) |
Antibiotic use | ||
Days on antibiotics | Secondary | Secondary |
HRQoL | ||
HRQoL (PedsQL, SF-36) | Exploratory | Secondary |
Tolerability | ||
Infusion rate reduced, interrupted, or stopped due to tolerability or AE (proportion of patients and infusions) | Secondary | Secondary |
Number of patients/caregivers unable to continue with home infusion | Not reported | Secondary |
Health care utilization | ||
Number of hospitalizations | Secondary | Secondary |
Days hospitalized | Secondary | Secondary |
Number of acute care physician visits due to infection or illness | Secondary | Secondary |
Productivity | ||
Days of school/work missed | Secondary | Secondary |
AE = adverse event; HRQoL = health-related quality of life; PedsQL = Pediatric Quality of Life Inventory; SF-36 = Short Form (36) Health Survey.
aTemporally associated events were those that occurred during or within 72 hours following the infusion.
Sources: Wasserman et al. (2012),12 Wasserman et al. (2016a),13 Wasserman et al. (2016b),44 Clinical Study Reports.15,16
Infections
In Study 160603 and during the treatment with IgHy10 in Extension Study 160902 (i.e., before the safety follow-up period), the primary efficacy end point was the rate of VASBIs per patient per year throughout the study period. These were reported as AEs and the number and type were determined. Serious acute bacterial infections included bacteremia/sepsis, bacterial meningitis, osteomyelitis/septic arthritis, bacterial pneumonia, and visceral abscesses caused by a recognized bacterial pathogen. Diagnostic criteria were according to the US Department of Health and Human Services, FDA Guidance for Industry, June 2008,11,45 and included an evaluation of symptoms and physical and laboratory findings. Essential diagnostic features were as follows:
Bacteremia required a positive blood culture.
Sepsis was defined as 2 of the following in adults: temperature greater than 38°C oral or 39°C rectal or less than 36°C oral or less than 37°C rectal, heart rate greater than 90 beats per minute, respiratory rate greater than 20 breaths per minute, or partial pressure of carbon dioxide (PaCO2) less than 32 mm Hg, white blood cell count greater than 12,000/mm3, less than 4,000/mm3, or greater than 10%. In children, sepsis was based on the criteria of the International Consensus Conference on Pediatric Sepsis.46 Blood-borne infections related to an indwelling catheter or vascular access device were not included.
Bacterial meningitis required a positive cerebrospinal fluid Gram stain and/or culture and/or positive cerebrospinal fluid bacterial antigen assay.
Osteomyelitis or septic arthritis required a positive X-ray, nuclear medicine bone scan, MRI, or CT scan showing bony destruction with radiolucent areas.
Bacterial pneumonia required pulmonary infiltrate with consolidation on chest X-ray.
Visceral abscess required typical findings on ultrasound, CT scan, MRI, or radionuclide scan.
The annual rate of all infections per patient during treatment with IgHy10 was also calculated.
Immunoglobulin Levels
Immunoglobulin trough levels were determined using standard assay methods for the determination of IgG, performed at a central laboratory. In Study 160603, samples were collected at baseline, on the day of each infusion, and at the end-of-study visit. In Extension Study 160902, assessments occurred every 12 weeks, monthly during the first 4 months of 2-week interval infusions, and at the end-of-study visit.
Antibiotic Use
In Study 160603 and before the safety follow-up of Extension Study 160902, days on antibiotics were collected throughout the studies using patient diaries that were transcribed to case report forms. The frequency of assessment was not reported.
Health-Related Quality of Life
Assessments of quality of life occurred at the time of the first subcutaneous infusion in epoch 2 of Study 160603 and at the end-of-study visit for all patients. In the extension study (Study 160902), assessments occurred at the start of the safety follow-up period. Quality of life among patients aged 2 to 13 years was assessed using the PedsQL.47 Scores were self-reported by patients aged 8 to 13 years and reported by a parent or primary caregiver for children aged 2 to 7 years. The PedsQL 4.0 (most recent version) consists of 23 items in 4 multi-item scales: physical functioning (8 items), emotional functioning (5 items), social functioning (5 items), and school functioning (5 items). Each item asks how much of a problem the item has been in the past month, with responses ranging from 0 (never a problem) to 4 (almost always a problem). Scale scores are then a sum of the items divided by the total number of questions answered. Two summary scores can then be computed: the physical health summary (8 items) is the same as the physical functioning scale, while the psychological summary score (15 items) is the sum of all items in the remaining scales divided by the number of items for which a response was provided.
Quality of life among patients 14 years and older was self-reported using the SF-36. The tool consists of 3 domains (functional status, well-being, and overall evaluation of health) comprising a total of 8 health dimensions. Functional status includes questions related to physical functioning (10 questions), social functioning (2 questions), physical role limitations (4 questions), and emotional role limitations (3 questions); well-being includes questions related to mental health (5 questions), vitality (4 questions), and pain (2 questions); and overall evaluation of health includes questions related to general health perception (5 questions). Response options to each question include Likert-like scales and yes/no options; responses within each domain are summed to obtain total scores. The developer’s recommended minimal important difference threshold for improved quality of life is 3 points,48 but a minimal important difference specific to patients with immunodeficiencies was not identified.
All scores on both the PedsQL and SF-36 were transformed to a scale of 0 to 100, where higher scores indicate better quality of life.
Tolerability and Adherence
In Study 160603, infusions were deemed tolerated if no serious adverse drug reactions, no non-serious moderate or severe local adverse drug reactions preventing completion of the infusion, and no non-serious moderate or severe systemic adverse drug reactions occurred within 60 minutes of completion of the infusion. In Study 160902, the number of patients and caregivers who were unable to continue with patient- or caregiver-administered home infusions and the reasons why they could not continue.
Health Care Utilization
In Study 160603 and during treatment with IgHy10 in Extension Study 160902 (before the safety follow-up), the number of acute care physician visits, number of days in hospital, and number of days in hospital due to infection were reported throughout each of the studies. Patient diaries that were transcribed to case report forms were used to obtain data informing health care utilization.
Productivity
In Study 160603 and during treatment with IgHy10 in Extension Study 160902 (before the safety follow-up), days off school or work were reported throughout each of the studies. Patient diaries that were transcribed to case report forms were used to obtain data informing productivity.
Safety
Data on AEs were assessed from the time consent forms were signed for Study 160603 to the end-of-study visit in Extension Study 160902. Observable swelling following SCIg infusion that did not cause discomfort was not considered an AE.
For each infusion scheduled to be given at the study site, vital signs (sitting blood pressure, pulse, respiratory rate, body temperature) were monitored and recorded pre-infusion (within 30 minutes before immunoglobulin infusion), 30 minutes after initiation of the infusion, hourly during the infusion, and post infusion (within 30 minutes of completion of the infusion). For infusions given at home or at an infusion centre, vital signs were to be monitored and recorded pre-infusion (within 30 minutes before immunoglobulin infusion) and post infusion (within 30 minutes of completion of the infusion). Any vital sign changes occurring during the infusion that were considered clinically significant by the investigator were recorded as an AE.
Statistical Analysis
Power Calculation and Data Handling
The sample size for Study 160603 was determined based on collecting safety data on a sufficient number of patients (approximately 30) who were naive to subcutaneous infusion, in addition to patients rolling over from Study 160601 (approximately 45). For the primary end point, a sample size of 80 was planned to provide 81% power to reject the null hypothesis of 1 or more VASBIs per year at the 1% level of statistical significance.
Primary Outcomes of the Studies
The primary end point in Study 160603 was serious acute bacterial infection rate, defined as the mean number of VASBIs per patient per year. Serious acute bacterial infections included: bacteremia/sepsis, bacterial meningitis, osteomyelitis/septic arthritis, bacterial pneumonia, and visceral abscesses caused by a recognized bacterial pathogen (as per the diagnostic criteria defined by the FDA).11 The primary end point was analyzed in the full analysis set (FAS).
The mean rate of VASBIs per patient per year and its 99% upper confidence limit were calculated using a Poisson model accounting for the length of the observation period for each patient. The null hypothesis of 1 or more VASBIs per patient per year was to be tested against the alternate hypothesis of less than 1 VASBI per patient per year. The observation period started after the ramp-up period and ended on the day of the end-of-study visit. To handle over-dispersion, the exponential distribution dispersion parameter was assumed to be given by the deviance divided by the degrees of freedom, and all statistics were adjusted accordingly. To assess efficacy, the null hypothesis of 1 or more infections per patient per year was tested using a generalized linear model assuming the Poisson distribution against the 1-sided alternative hypothesis of less than 1 infection per patient per year at the 1% level of statistical significance.
The primary end point was also assessed using the per-protocol analysis set (PPS).
There was no adjustment for covariates in any of the analyses. Statistical techniques were not used to identify or exclude any observations as outliers. If data were considered spurious, the reasoning and analysis from which the data were excluded were documented. For the primary end point, imputation techniques were not required, as the statistical method used accounted for the length of the observation period for each patient.
Since the number of patients at each study centre was small, analyses of centre effects were not performed. There were no adjustments made for multiple comparisons, as only 1 outcome was tested statistically. In Study 160603, in addition to the FAS and PPS, the primary end point and some secondary end points were assessed in the subcutaneous-naive population.
Interim Analyses
In Study 160603, a planned interim analysis was conducted that included data from patients in study arm 1 who had completed epoch 2 by the end of May 2010. The purpose of the interim analysis was to monitor the safety and tolerability of the treatments and to share updates about the study externally. No changes to the study were made as a result of the interim analysis.
In the extension study (Study 160902), a planned interim analysis occurred when the last patient reached 48 weeks of therapy. The analysis included serious AEs, all infections per patient, occurrence of neutralizing antibodies to rHuPH20, percentage of infusions requiring adjustment, all AEs by severity and classified by Medical Dictionary for Regulatory Activities (MedDRA) terms and IgG trough levels.
Sensitivity Analyses
Two sensitivity analyses of the rate of infections were performed to address the potential effects of patients not completing the full year of subcutaneous treatment:
Use multiple imputation for the infection rate in unobserved time periods:
For patients who terminated the study for increased frequency or severity of infections: Use twice the highest infection rate observed in patients who completed more than 2 months in the study in the same season to address concerns that these patients may have done worse than others in the study.
For patients who did not leave the study due to increased frequency or severity of infections: Use the patient’s infection rate in a season; if more than 2 months were observed, then use the rate observed in patients who completed 2 or more months in the study in the same season. Rubin multiple imputations were used, where the number of infections in an unobserved time period were imputed by random numbers from the Poisson distribution, with the Poisson parameter set differently by season and by reason for study termination. Five or more datasets were imputed and analyzed, then the point estimate and CI were calculated using the Rubin method.
Analyze a full year in the subset of patients who completed a full year. To avoid ambiguity, the first 365 days following the first subcutaneous infusion was selected.
Secondary Outcomes of the Studies
Infections
The annual rate of VASBIs was evaluated during the observation phase of Extension Study 160902 (before the safety follow-up). The analyses were similar to the analysis of the primary outcome in Study 160603. The annual rate of all infections was included as a secondary outcome in both studies. The rate of VASBIs was calculated per patient using the same Poisson model, including the allowance for over-dispersion. Additionally, in Study 160603, a post hoc exploratory analysis was conducted to assess the apparent difference in infection rates for IVIg versus IgHy10. Because the length of treatment and season of administration differed, the role of seasonal imbalance was investigated. Therefore, the infection rates were analyzed using a general linear Poisson model for the days per patient where the treatments were on the same calendar days. This analysis eliminated seasonal effects but not the possibility of year-to-year variation.
Immunoglobulin Levels
In Study 160603, IgG trough levels were summarized by medians and their 95% non-parametric CIs.
In Extension Study 160902, IgG trough levels over time were calculated for 2-, 3-, and 4-week infusion intervals and for the following age groups: 2 to 11 years, 12 to 15 years, 16 to 64 years, and 65 years and older. Descriptive statistics were used for the change in IgG trough levels from the end of the study period before the safety follow-up, to the end of the safety follow-up, stratifying for mode of administration (IV versus subcutaneous) and treatment interval.
Antibiotic Use, Health Care Utilization, and Productivity
In Study 160603 and during the observation phase of Extension Study 160902 (before the safety follow-up), point estimates and 95% CIs for the monthly rates of days off school/work, days on antibiotics, number of acute care physician visits, and days in hospital per patient were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end point in Study 160603.
Safety and Tolerability
For Study 160603, analyses of the safety end points were provided for both the IV and subcutaneous administrations at the dose used in epoch 2 after the ramp-up phase. For the ramp-up phase in epoch 2, tolerability was summarized descriptively by the number of patients who tolerated the dose of the first infusion, the dose of the second infusion, and the number of patients who did not tolerate the dose. The individual patient’s rate for each of the secondary safety end point events (e.g., AEs) was calculated by dividing the total number of events by the total number of IV or subcutaneous infusions, respectively.
In Extension Study 160902, the analyses of safety end points were stratified by age: 2 to 11 years, 12 to 15 years, 16 to 64 years, and 65 years and older. Analyses were conducted for the period of IgHy10 treatment, for the safety follow-up period, and for all periods combined. AEs were reported descriptively by category (MedDRA preferred term and seriousness) as a rate per patient and as a rate per infusion. Temporally associated AEs, defined as occurring during or within 72 hours of an infusion, were also reported descriptively by rate per patient and rate per infusion.
Exploratory Outcomes of the Studies
Health-Related Quality of Life
HRQoL was analyzed using descriptive statistics. In Study 160603, HRQoL was analyzed separately for the 2 to 7 years, 8 to 13 years, and 14 years of age and older groups. Patients younger than 14 years old were assessed using the PedsQL and patients who were 14 years of age and older were assessed using the SF-36.
Analysis Populations
The analysis sets included in Study 160603 are described subsequently.
The FAS included all patients who had been exposed to either or both study drugs and who provided data for the primary end point for any period of time.
The PPS was a subset of the FAS that included only patients who had completed at least 6 months of subcutaneous treatment after the ramp-up.
The safety analysis set (SAS) included all patients who were exposed to either or both study drugs.
The primary dataset used for the analysis of primary and secondary efficacy outcomes was the FAS. The SAS was used to analyze safety and tolerability outcomes as well as HRQoL outcomes.
In Extension Study 160902, the SAS included data obtained in Study 160902 on all patients who received at least 1 dose of the study drug in the context of Study 160902. All analyses were conducted using the SAS.
Results
Patient Disposition
Study 160603
Full details of the patient disposition in Study 160603 are shown in Table 11. Of the 89 potential patients who were screened, 2 (2%) failed screening and withdrew before treatment. The remaining 87 patients began IV treatment in epoch 1 or in Study 160601. Of the 87 patients treated, 31 (36%) were in arm 1 (those who had participated in Study 160601) and the remaining 56 were in arm 2. One patient in the 2 years to younger than 12 years group asked to withdraw during epoch 1, and 2 patients aged 12 years or older were withdrawn because they missed 2 consecutive treatments. Of the 87 patients who started treatment, 84 (97%) completed epoch 1.
One patient who completed epoch 1 did not receive any infusions in epoch 2; therefore, 83 (93%) patients began treatment in epoch 2. One patient began treatment in epoch 2 despite failing to meet an inclusion criterion (pre-study dose was 16mg/kg, which was too low due to weight gain); this was documented as a protocol deviation. Of those who started epoch 2, 5 patients aged from 2 years to younger than 12 years and 11 patients aged 12 years or older withdrew (reasons in Table 11). Of those who started treatment, 68 (78%) completed epoch 2.
There were 15 major protocol deviations detected during the study, the most common being the administration of an incorrect dose (n = 7) and failure to conduct pharmacokinetic assessments (n = 4). No patients were excluded from the per-protocol analysis due to deviations. An audit at 1 of the study sites detected significant departures from good clinical practice; however, the site was not censored, as it was judged that this would introduce bias in Baxter’s favour.
Table 11: Patient Disposition for Study 160603
Detail | Patient group by age | ||
|---|---|---|---|
2 to < 12 years | ≥ 12 years | All ages | |
Screened, N | 14 | 75 | 89 |
Withdrawn before treatment, N (%) | 0 (0.0) | 2 (2.7) | 2 (2.2) |
Discontinued from study,a N (%) | 6 (42.9) | 13 (17.3) | 19 (21.3) |
Requested withdrawal | 4 (28.6) | 2 (2.7) | 6 (6.7) |
Missed 2 consecutive administrations of IP | 0 (0.0) | 2 (2.7) | 2 (2.2) |
Adverse event | 2 (14.3) | 4 (5.3) | 6 (6.9) |
Increased frequency of infections | 0 (0.0) | 1 (1.3) | 1 (1.1) |
Switched to safety follow-up | 0 (0.0) | 3 (4.0) | 3 (3.4) |
Lost to follow-up | 0 (0.0) | 1 (1.3) | 1 (1.1) |
Full analysis set, N | 11 | 70 | 81 |
Per-protocol set,b N | 9 | 65 | 74 |
Safety analysis set,c N | 14 | 73 | 87 |
aIncludes patients who withdrew or discontinued from the study. A total of 16 patients discontinued from study and 3 switched to the safety follow-up period in Study 160902.
bSubset of the FAS that included only patients who completed at least 6 months of subcutaneous treatment after the ramp-up.
cAll patients exposed to either or both study drugs.
Source: Clinical Study Report.15
Extension Study 160902
Full details of the patient disposition in Extension Study 160902 are shown in Table 12. Out of the 66 patients who rolled over from Study 160603 into 160902, 63 (95%) were treated with SCIg (10%) plus rHuPH20 (IgHy10); 3 (5%) who had switched to the safety follow-up received IVIg (10%) alone. Of the 63 patients who were receiving IgHy10, 15 (23%) withdrew or were discontinued from the study (reasons in Table 12); 48 (91%) switched to the safety follow-up when protocol amendment 5 went into effect. Therefore, of those who started treatment in Study 160603, 48 (55%) completed the safety follow-up in Extension Study 160902.
There were 31 major protocol deviations during the study; the most common were described as a procedure not being done (n = 16) or other causes (n = 12). No patients were excluded from the analyses due to protocol deviations.
Table 12: Patient Disposition for Study 160902
Detail | Patient group by age | ||||
|---|---|---|---|---|---|
2 to < 12 years | 12 to < 16 years | 16 to < 65 years | ≥ 65 years | All ages | |
Rolled over from Study 160603, N | 4 | 7 | 47 | 8 | 66 |
Received IVIg 10% only without hyaluronidase, N (%) | 0 (0.0) | 0 (0.0) | 3 (6.4) | 0 (0.0) | 3 (4.5) |
Discontinued from IgHy10, N (%) | 0 (0.0) | 0 (0.0) | 14 (29.8) | 1 (12.5) | 15 (22.7) |
Patient withdrew | 0 (0.0) | 0 (0.0) | 3 (6.4) | 1 (12.5) | 4 (6.1) |
Patient died | 0 (0.0) | 0 (0.0) | 1 (2.1) | 0 (0.0) | 1 (1.5) |
Patient received bone marrow transplant | 0 (0.0) | 0 (0.0) | 1 (2.1) | 0 (0.0) | 1 (1.5) |
Site was closed out by sponsor | 0 (0.0) | 0 (0.0) | 6 (12.8) | 0 (0.0) | 6 (9.1) |
Site elected to exit study | 0 (0.0) | 0 (0.0) | 3 (6.4) | 0 (0.0) | 3 (4.5) |
Switched to safety follow-up, N (%) | 4 (100.0) | 7 (100.0) | 30 (63.8) | 7 (87.5) | 48 (72.7) |
Discontinued safety follow-up, N (%) | 0 (0.0) | 0 (0.0) | 1 (2.1) | 0 (0.0) | 1 (1.5) |
Adverse event | 0 (0.0) | 0 (0.0) | 1 (2.1) | 0 (0.0) | 1 (1.5) |
Completed safety follow-up, N (%) | 4 (100.0) | 7 (100.0) | 32 (68.1)a | 7 (87.5) | 50 (75.8) |
SAS, Nb | 4 | 7 | 47 | 8 | 66 |
IgHy10 treatment dataset, Nc | 4 | 7 | 47 | 8 | 66 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; SAS = safety analysis set.
a32 patients total: 29 patients who had completed the safety follow-up plus 3 patients who had completed IVIg (normal immunoglobulin [human] 10%) without hyaluronidase.
bAll patients who received at least 1 dose of the study drug in the context of Study 160902.
cThe data obtained in Study 160603 started with patients’ first exposure; for Study 160902, the data were for all patients who participated.
Source: Clinical Study Report.16
Exposure to Study Treatments
Study 160603
A total of 365 IV infusions of immunoglobulin and 1,359 subcutaneous infusions of IgHy10 (90% in the abdomen, 9% in the thighs, < 1% in both abdomen and thigh, and < 1% in another site) were administered during the study. The median duration of IVIg treatment in epoch 1 was 91.0 days for patients younger than 12 years of age and those aged 12 years and older. Patients received IgHy10 for a median of approximately 1 year in epoch 2 (excluding the ramp-up) in both the SAS and FAS. The median duration of exposure in epoch 2 (excluding the ramp-up) was slightly higher in the PPS (423.0 days and 378.0 days for patients aged < 12 years and ≥ 12 years, respectively). In the ramp-up period, patients were exposed to IgHy10 for a median of 42 days in both age groups and in all datasets.
In 78 of the 83 patients (94.0%) who began treatment in epoch 2, the dosing interval that was used for IV infusions was reached for subcutaneous infusions with rHuPH20 during the ramp-up. In 15 patients (18.1%), this was a 3-week interval and in 63 patients (75.9%), this was a 4-week interval.
Infusions of IVIg were initiated at a median flow rate of 13.3 mL/hour (range = 4.0 to 32.0) in patients younger than 12 years of age and 36.5 mL/hour (range = 10.0 to 240.0) in those aged 12 years and older. IgHy10 was given at a median initial infusion rate of 10.0 mL/hour in both age groups assessed in epoch 2 (excluding the ramp-up); this result was identical in all datasets. The initial infusion rate ranged from 5.0 mL/hour to 20.0 mL/hour in patients younger than 12 years of age and 5.0 mL/hour to 300.0 mL/hour in those aged 12 years and older in all datasets except the SAS, in which the initial infusion rate was always 10.0 mL/hour in patients younger than 12 years of age. In the ramp-up, the median initial infusion rate was 5.0 mL/hour (range = 5.0 to 10.0) in patients younger than 12 years of age in all datasets except the SAS (median = 10.0; range = 5.0 to 10.0 mL/hour) and 10.0 (range = 5.0 to 10.0) mL/hour in patients aged 12 years and older.
The median maximum infusion rate used in patients younger than 12 years old was 62.5 (range = 25.0 to 316.0) mL/hour for IVIg infusions (epoch 1), 160.0 (range = 80.0 to 300.0) mL/hour for IgHy10 infusions in epoch 2 (excluding the ramp-up), and 80.0 (range = 40.0 to 300.0) mL/hour for IgHy10 infusions during the ramp-up. Comparable results were obtained for the SAS.
Table 13: Exposure in Study 160603 (SAS)
Population | N | Duration of treatment (days), mean (SD) | Duration of treatment (days), median (range) | Duration of infusion (hours), median (range) | Total dose per kg body weight per week (g/kg/week), mean (SD) |
|---|---|---|---|---|---|
All patients | |||||
Epoch 1: IVIg | 87 | 93.0 (10.6) | 91.0 (54 to 148) | 2.33 (0.92 to 6.33) | 0.14 (0.06) |
Ramp-up: IgHy10 | 83 | 38.4 (9.5) | 42.0 (20 to 78) | 1.58 (0.63 to 5.38) | 0.14 (0.05) |
Epoch 2: IgHy10 | 81 | 367.7 (103.9) | 366.0 (42 to 507) | 2.08 (0.83 to 4.68) | 0.16 (0.05) |
By age | |||||
Epoch 1: IVIg | |||||
2 to < 12 years | 14 | 88.7 (7.7) | 91.0 (63 to 93) | 2.49 (1.40 to 5.03) | 0.14 (0.04) |
≥ 12 years | 73 | 93.8 (10.9) | 91.0 (54 to 148) | 2.33 (0.92 to 6.33) | 0.14 (0.06) |
Ramp-up: IgHy10 | |||||
2 to < 12 years | 13 | 39.7 (14.4) | 42.0 (21 to 78) | 1.51 (0.73 to 2.28) | 0.15 (0.04) |
≥ 12 years | 70 | 38.1 (8.4) | 42.0 (20 to 49) | 1.61(0.63 to 5.38) | 0.14 (0.05) |
Epoch 2: IgHy10 | |||||
2 to < 12 years | 11 | 360.3 (120.5) | 366.0 (156 to 505) | 1.73 (1.15 to 3.28) | 0.16 (0.04) |
≥ 12 years | 70 | 368.8 (102.0) | 365.5 (42 to 507) | 2.13 (0.83 to 4.68) | 0.15 (0.05) |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; SAS = safety analysis set; SD = standard deviation.
Source: Clinical Study Report.15
Extension Study 160902
Prior to the safety follow-up period in Study 160902, 63 patients received IgHy10 for a median treatment duration of 669 days (range = 60 to 729). The mean dose received was 0.156 g/kg per week (SD = 0.051). The median duration of IgHy10 infusion was 1.42 hours (range = 0.3 to 44.6). The high maximum infusion time was due to 1 patient being unable to complete an infusion at home due to technical problems with the infusion pump. The treatment therefore had to be completed at the clinic and the duration of the infusion was reported to be 2 days.
Across all age groups, the median initial rate of IgHy10 infusion was 10 mL/hour (range = 5 to 300) (Table 14) and the median maximum rate of infusion achieved was 300 mL/hour (range = 10 to 350). Across all age groups and infusion intervals, a median of 1.09 infusions per month (range = 0.3 to 2.1) was administered. Patients following the 3-week infusion interval received a median of 1.45 infusions per month (range = 0.8 to 1.6), and those following a 4-week infusion interval received a median of 1.09 infusions per month (range = 0.3 to 1.3). During the 2-week infusion interval period, patients received a median of 2.18 infusions per month (range = 2.0 to 2.5). Treatment with IgHy10 required a median of 1.58 infusion sites per month (range = 0.3 to 2.4) across all age groups and infusion intervals. Patients following the 3-week infusion interval used a median number of 2.40 infusion sites per month (range = 0.8 to 3.0); those following a 4-week infusion interval used a median of 1.09 infusion sites per month (range = 0.3 to 2.6). During the 2-week infusion interval period, patients used a median of 2.18 infusion sites per month (range = 2.0 to 5.1).
At the first infusion of the study, 10 of 66 patients (15%) received IgHy10 on a 3-week interval basis and 53 of 66 patients (80%) on a 4-week interval basis. Three out of 66 patients (4.5%) did not receive any IgHy10 in Study 160902.
Of the patients initially treated with IgHy10, 15 of 66 patients (22.7%) terminated the study while on IgHy10 and 51 of 66 patients (77.3%) were switched to the safety follow-up period. Of the patients on a baseline infusion interval of 3 weeks, 3 of 66 (4.5%) switched to a 2-week infusion interval and none returned to a 3-week infusion interval. Of the patients starting on a 4-week interval, 15 of 66 (22.7%) switched to a 2-week infusion interval and 3 of 66 (4.5%) returned to a 4-week infusion interval. Of a total of 18 patients who were switched to the 2-week infusion interval, 1 of 66 (1.5%) stayed on this treatment interval for less than 4 months, and 17 of 66 (25.8%) stayed on this interval for more than 4 months.
Table 14: Exposure to IgHy10 Before the Safety Follow-Up in Study 160902 (SAS)
Population | N | Duration of treatment (days), mean (SD) | Duration of treatment (days), median (range) | Duration of infusion (hours), median (range) | Total dose per kg body weight per week (g/kg/week), mean (SD) |
|---|---|---|---|---|---|
All patients | 63 | 565.9 (211.8) | 669 (60 to 729) | 1.42 (0.3 to 44.6) | 0.16 (0.05) |
By age | |||||
2 to < 12 years | 4 | 681.3 (9.3) | 679.5 (672 to 694) | 1.38 (0.8 to 3.2) | 0.17 (0.04) |
12 to < 16 years | 7 | 679.7 (34.1) | 694 (645 to 729) | 1.57 (0.6 to 3.0) | 0.14 (0.03) |
16 to < 65 years | 44 | 524.4 (238.8) | 664 (60 to 728) | 1.50 (0.3 to 44.6) | 0.17 (0.05) |
≥ 65 years | 8 | 636.6 (93.8) | 652.5 (414 to 715) | 1.28 (0.3 to 44.6) | 0.12 (0.03) |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; SAS = safety analysis set; SD = standard deviation.
Source: Clinical Study Report.16
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently. See Appendix 4 for detailed efficacy data.
Infections
Annual Rate of VASBIs
In Study 160603, the mean annual rate of VASBIs during IgHy10 treatment in epoch 2 was 0.025 per patient per year (upper limit of the 99% CI = 0.046) in the FAS, which was below 1.0 (P < 0.0001), the threshold suggested by FDA guidance. Two VASBIs were reported, occurring on the day of the first subcutaneous infusion at the final infusion interval after the ramp-up. Rates of VASBI in the PPS and SAS in Extension Study 160902 also fell below 1.0 VASBI per patient per year (P < 0.0001).
Annual Rate of All Infections
In Study 160603, the mean annual rate of all infections during treatment with IVIg (epoch 1) and IgHy10 (epoch 2) was 2.97 infections per patient per year (95% CI, 2.51 to 3.47) and 4.51 infections per patient per year (95% CI, 3.50 to 5.69), respectively. The annual rate of infections occurring in more than 5% of patients was also reported (Table 15).12 The most frequently reported infections during both epoch 1 and epoch 2 included sinusitis, upper respiratory tract infection, and bronchitis.
Two sensitivity analyses for the rate of infections were performed to address the potential effects of patients not completing the full year of IgHy10 in epoch 2. When Rubin multiple imputation methods were employed for any patients who terminated the study after less than 1 year, the annualized rate of all infections was 3.24 infections per patient per year (95% CI, 2.65 to 3.96). The rate of all infections per year in the 41 patients in the FAS who had completed a full year of treatment was 2.75 infections per patient per year (95% CI, 2.09 to 3.54).
The rate of infections in Extension Study 160902 was similar to the FAS in Study 160603.
In a post hoc analysis, Wasserman et al. (2016)13 evaluated patients from Study 160603 and Study 160902 from the first administration of IgHy10 through the end of treatment. Exposure to IgHy10 exceeded 30 months for 48 of the 83 patients included in the analysis. The annual rate of infections and annual rate of VASBIs during IgHy10 treatment were reported by age group (< 18 and ≥ 18 years). For patients who were at least 18 years of age (n = 59), the rate of infections was 2.98 per year (95% CI, 2.56 to 3.44) and the rate of VASBIs was 0.01 per year (upper limit of the 99% CI = 0.02). The rate of infections and VASBIs were consistent with the results reported for each of the individual studies. The rate of infections during treatment with IgHy10 was also reported for 1-year periods by age group. The rate of infections per 1-year period ranged from 2.52 (95% CI, 1.69 to 3.59) to 3.61 (95% CI, 2.84 to 4.50).
Table 15: Rate of Infections in Study 160603
Detail | Epoch 1: IVIg (N = 87) | Epoch 2: IgHY10 (N = 83) |
|---|---|---|
Full analysis set | ||
Validated acute serious bacterial infectionsa | ||
N analyzed | NR | 81 |
Rate per patient per year (upper limit of 99% CI) | NR | 0.025 (0.046) |
P valueb | NR | < 0.0001 |
All infectionsa | ||
N analyzed | 81 | |
Rate per patient per year (95% CI)b | 4.51 (3.50 to 5.69) | 2.97 (2.51 to 3.47) |
Infections occurring in > 5% of patients, n (95% CI) | ||
Sinusitis | 0.86 (0.55 to 1.26) | 0.65 (0.47 to 0.87) |
Upper respiratory tract infection | 0.41 (0.25 to 0.63) | 0.54 (0.39 to 0.73) |
Bronchitis | 0.32 (0.18 to 0.50) | 0.12 (0.07 to 0.19) |
Urinary tract infection | 0.27 (0.16 to 0.42) | 0.07 (0.04 to 0.12) |
Gastroenteritis (viral) | NR | 0.15 (0.09 to 0.22) |
Viral upper respiratory tract infection | NR | 0.11 (0.06 to 0.18) |
Viral infection | NR | 0.12 (0.07 to 0.19) |
Post-procedural infection | NR | 0.09 (0.05 to 0.13) |
Gastroenteritis | NR | 0.07 (0.04 to 0.12) |
Cellulitis | NR | 0.06 (0.04 to 0.10) |
Influenza | 0.23 (0.13 to 0.36) | NR |
Per-protocol analysis set | ||
Validated acute serious bacterial infectionsa | ||
N analyzed | NR | 74 |
Rate per patient per year (upper limit of 99% CI) | NR | 0.025 (0.048) |
P valuec | NR | < 0.0001 |
All infectionsa | ||
N analyzed | 74 | |
Rate per year (95% CI) | 4.44 (3.38 to 5.72) | 2.95 (2.48 to 3.48) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; NR = not reported; rHuPH20 = recombinant human hyaluronidase; SC = subcutaneous; SCIg = subcutaneous immunoglobulin.
aThe mean rate of infections per patient per year and its variance were calculated using a Poisson model accounting for the length of the observation period for each patient. To handle over-dispersion, the exponential distribution dispersion parameter was assumed to be given by the deviance divided by the degrees of freedom and all statistics were adjusted accordingly.
bIn an exploratory analysis using the same calendar days per patient for the IVIg only and SCIg plus rHuPH20 treatment periods, the infection rate of 4.3 per year for IVIg was higher than the rate of 2.3 per year for SCIg plus rHuPH20 (P = 0.0021). This analysis accounted for potential seasonal effects but could not account for year-to-year variations in weather conditions. Sensitivity analyses addressing the potential effects of patients who did not complete the full year of treatment, using Rubin imputation methods, resulted in an annualized rate of infections of 3.24 (95% CI, 2.65 to 3.96). The point estimate of the rate of all infections per year in the 41 patients in the full analysis set who had completed a full year of treatment was 2.75 (95% CI, 2.09 to 3.54).
cThere was no adjustment for multiple testing.
Source: Clinical Study Report15 and Wasserman et al. (2012).12
Table 16: Rate of Infections Before the Safety Follow-Up in Study 160902 (SAS)
Detaila,b | IgHy10 (N = 66) |
|---|---|
Validated acute serious bacterial infections | |
N analyzed | 66 |
Rate per year (upper limit of 99% CI) | 0.020 (0.045) |
P valuec | < 0.0001 |
All infections | |
N analyzed | 66 |
Rate per year (95% CI) | 2.86 (2.36 to 3.43) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; SAS = safety analysis set.
aThe mean rate of infections per patient per year and its variance were calculated using a Poisson model accounting for the length of the observation period for each patient. To handle over-dispersion, the exponential distribution dispersion parameter was assumed to be given by the deviance divided by the degrees of freedom, and all statistics were adjusted accordingly.
bThere was no adjustment for multiple testing.
Source: Clinical Study Report.16
Immunoglobulin Levels
While receiving IgHy10 in Study 160603 (Table 17), the median IgG trough level in patients younger than 12 years was 9.95 g/L (95% CI, 7.87 to 15.00). In patients who were at least 12 years old, the median IgG trough level was 10.70 g/L (95% CI, 9.46 to 11.80). Patients’ IgG trough levels while receiving IgHy10 (epoch 2) were similar to patients’ IgG trough levels while receiving IVIg (epoch 1). Results for IgG trough levels reported in the FAS were consistent with those reported using the PPS.
In Study 160902 (Table 18), the median IgG trough level for patients who received IgHy10 administered at a 2-week interval was 10.90 g/L (95% CI, 9.39 to 13.30). For patients who received IgHy10 administered at a 3-week interval and a 4-week interval, the median IgG trough level was 12.30 g/L (95% CI, 11.50 to 15.30) and 9.67 g/L (9.26 to 10.70), respectively. Study 160902 also reported IgG trough levels as a percent change for patients whose IgHy10 treatment was switched from a 3- or 4-week interval to a 2-week interval (Table 18). When receiving IgHy10 at a 2-week interval, the IgG trough level was 105.90% (SD = 0.03), and 113.23% (SD = 13.02) for patients who were previously on a 3- or 4-week interval.
Lastly, the ratio of IgG trough levels at the end of the safety follow-up period (treatment with IVIg or SCIg without hyaluronidase) compared with the end of the IgHy10 treatment period was analyzed in Study 160902 (Table 18). The IgG trough level in patients who received SCIg at the end of the safety follow-up period was 117.8% (95% CI, 104.4 to 145.7) of their IgG trough level while receiving IgHy10. The IgG trough level in patients who received IVIg at the end of the safety follow-up period was 102.5% (95% CI, 97.7 to 109.8) of their IgG trough level while receiving IgHy10. The ratio of IgG trough levels is also summarized by administration interval.
Table 17: IgG Trough Levels by Age in Study 160603
Detail | Epoch 1: IVIg (N = 87) | Epoch 2: IgHy10 (N = 83) |
|---|---|---|
Full analysis set, median (95% CI) | ||
N analyzed | 68 | 60 |
IgG trough level (g/L) in patients < 12 years (n = 11) | 9.63 (8.29 to 13.60) | 9.95 (7.87 to 15.00) |
IgG trough level (g/L) in patients ≥ 12 years (n = 70) | 10.40 (9.63 to 11.40) | 10.70 (9.46 to 11.80) |
Per-protocol analysis set, median (95% CI) | ||
N analyzed | 63 | 60 |
IgG trough level (g/L) in patients < 12 years (n = 9) | 9.63 (7.42 to 11.20) | 9.14 (7.83 to 11.40) |
IgG trough level (g/L) in patients ≥ 12 years (n = 65) | 10.40 (9.58 to 11.30) | 10.60 (9.42 to 11.70) |
CI = confidence interval; IgG = immunoglobulin G; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin.
Source: Clinical Study Report.15
Table 18: IgG Trough Levels by Dose Frequency in Extension Study 160902 (SAS)
Detail | IgHy10 (N = 66) |
|---|---|
Steady-state trough levels (g/L) maintained under IgHy10 treatment by infusion interval | |
2-week interval | |
N analyzed | 17 |
Median (95% CI) | 10.90 (9.39 to 13.30) |
3-week interval | |
N analyzed | 9 |
Median (95% CI) | 12.30 (11.50 to 15.30) |
4-week interval | |
N analyzed | 47 |
Median (95% CI) | 9.67 (9.26 to 10.70) |
Percent change of steady-state trough levels for patients who changed to a 2-week interval under IgHy10 treatment, % | |
3-week (previous interval) | |
N analyzed | 2 |
Mean (SD) | 105.90 (0.03) |
Median (95% CI) | 105.90 (NA) |
4-week (previous interval) | |
N analyzed | 9 |
Mean (SD) | 113.23 (13.02) |
Median (95% CI) | 112.44 (99.31 to 129.13) |
Ratio of IgG trough levels (g/L) at the end of safety follow-up vs. at end of IgHy10 treatment | |
SCIg at end of safety follow-up (total) | |
N analyzed | 9 |
Median, % (95% CI) | 117.8 (104.4 to 145.7) |
IVIg at end of safety follow-up (total) | |
N analyzed | 38 |
Median, % (95% CI) | 102.5 (97.7 to 109.8) |
SCIg at end of safety follow-up (2-week interval) | |
N analyzed | 6 |
Median, % (95% CI) | 108.5 (92.7 to 145.7) |
IVIg at end of safety follow-up (2-week interval) | |
N analyzed | 6 |
Median, % (95% CI) | 94.6 (90.3 to 96.5) |
SCIg at end of safety follow-up (3-week interval) | |
N analyzed | NA |
Median, % (95% CI) | NA |
IVIg at end of safety follow-up (3-week interval) | |
N analyzed | 5 |
Median, % (95% CI) | 116.7 (NA) |
SCIg at end of safety follow-up (4-week interval) | |
N analyzed | 3 |
Median, % (95% CI) | 142.7 (NA) |
IVIg at end of safety follow-up (4-week interval) | |
N analyzed | 38 |
Median, % (95% CI) | 103.1 (99.3 to 111.5) |
CI = confidence interval; IgG = immunoglobulin G; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; NA = not applicable; SAS = safety analysis set; SCIg = subcutaneous immunoglobulin; SD = standard deviation.
Source: Clinical Study Report.16
Antibiotic Use
Days on antibiotics were reported in Study 160603 and Study 160902 (before the safety follow-up period) and are summarized in Table 19 and Table 20, respectively.
While receiving treatment with IgHy10, patients were on antibiotics at a rate of 1.68 days per month (95% CI, 1.29 to 2.16). While on treatment with IVIg, patients were on antibiotics at a rate of 3.15 days per month (95% CI, 2.19 to 4.35). In Extension Study 160902, patients had to take antibiotics at an annual rate of 64.03 days per year (95% CI, 45.16 to 87.54).
Table 19: Days on Antibiotics in Study 160603
Detaila | Epoch 1: IVIg N = 87 | Epoch 2: IgHy10 N = 83 |
|---|---|---|
Full analysis set | ||
N analyzed | 81 | 81 |
Days on antibiotics per month (95% CI) | 3.15 (2.19 to 4.35) | 1.69 (1.29 to 2.16) |
Per-protocol analysis set (n = 74) | ||
N analyzed | 74 | 74 |
Days on antibiotics per month (95% CI) | 3.13 (2.12 to 4.42) | 1.67 (1.26 to 2.17) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
Source: Clinical Study Report.15
Table 20: Days on Antibiotics Before the Safety Follow-Up in Extension Study 160902 (SAS)
Detail | IgHy10 (N = 66) |
|---|---|
N analyzed | 66 |
Days on antibiotics per year (95% CI)a | 64.03 (45.16 to 87.54) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; SAS = safety analysis set.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
Source: Clinical Study Report.16
Health-Related Quality of Life
HRQoL was evaluated in Study 160603 and Extension Study 160902 using the PedsQL and SF-36 (Table 21 and Table 22). The scores from the SF-36 and PedsQL were combined and summarized for all patients. The median mental/psychosocial health summary score was 56.2 (range = 20.0 to 86.7) while receiving SCIg (patients enrolled from Study 160601), 53.1 (range = 20.5 to 90.0) while receiving IVIg, and 53.6 (range = 21.5 to 96.7) during treatment with IgHy10. The median physical component and health summary score was 47.0 (range = 13.1 to 100.0) while receiving SCIg (patients enrolled from Study 160601), 51.0 (range = 18.8 to 84.4) while receiving IVIg, and 56.0 (range = 55.2 to 56.6) during treatment with IgHy10.
HRQoL was also reported by age group (2 to 7 years, 8 to 13 years, and at least 14 years old). For IgHy10, among patients who were between 2 and 7 years old, the psychosocial health summary score was a median of 88.3 (range = 59.6 to 96.7), and the physical health summary score was a median of 88.0 (range = 66.7 to 97.8). For patients aged 8 to 13 years, the psychosocial health summary score was a median of 66.7 (range = 36.7 to 95.0), and the physical health summary score was a median of 90.6 (range = 28.1 to 100.0). Using the SF-36 for patients who were at least 14 years old and received IgHy10, the mental component and physical component summary scores were a median of 52.2 (range = 21.5 to 70.8) and 44.8 (range = 13.1 to 61.1), respectively.
Table 21: Health-Related Quality of Life in Study 160603 (SAS)
Detail | Patients enrolled from Study 160601, SCIg alone | Epoch 1: IVIg | Epoch 2: IgHy10 |
|---|---|---|---|
All patients (N = 87)a | |||
SF-36/PedsQL (all ages combined) | |||
N analyzed | 28 | 53 | 72 |
Mental/psychosocial health summary, median (range) | 56.2 (20.0 to 86.7) | 53.1 (20.5 to 90.0) | 53.6 (21.5 to 96.7) |
Physical component/health summary, median (range) | 47.0 (13.1 to 100.0) | 51.0 (18.8 to 84.4) | 56.0 (55.2 to 56.6) |
Total score, median (range) | NR | NR | NR |
HRQoL by age group | |||
PedsQL total score (age 2 to 7 years) | |||
N analyzed | 2 | 3 | 5 |
Psychosocial health summary, median (range) | 65.4 (65.4 to 65.4) | 70.0 (66.7 to 82.7) | 88.3 (59.6 to 96.7) |
Physical health summary, median (range) | 78.1 (75.0 to 81.3) | 96.9 (87.5 to 100.0) | 81.3 (78.1 to 100.0) |
Total score, median (range) | 71.4 (71.4 to 71.4) | 78.3 (76.1 to 88.1) | 88.0 (66.7 to 97.8) |
PedsQL total score (age 8 to 13 years) | |||
N analyzed | 6 | 4 | 9 |
Psychosocial health summary, median (range) | 67.5 (20.0 to 86.7) | 79.2 (51.7 to 90.0) | 66.7 (36.7 to 95.0) |
Physical health summary, median (range) | 78.1 (18.8 to 84.4) | 90.6 (84.4 to 100.0) | 90.6 (28.1 to 100.0) |
Total score, median (range) | 70.7 (19.6 to 83.7) | 83.2 (63.0 to 93.5) | 78.3 (33.7 to 96.7) |
SF-36 (age ≥ 14 years) | |||
N analyzed | 20 | 46 | 58 |
Mental component summary, median (range) | 53.7 (25.6 to 61.6) | 51.2 (20.5 to 66.9) | 52.2 (21.5 to 70.8) |
Physical component summary, median (range) | 47.5 (24.5 to 66.3) | 44.7 (15.2 to 64.0) | 44.8 (13.1 to 61.1) |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; NR = not reported; PedsQL = Pediatric Quality of Life Inventory; SAS = safety analysis set; SCIg = subcutaneous immunoglobulin; SF-36 = Short Form (36) Health Survey.
aThe range of both scales is 0 to 100, where higher scores indicate a better quality of life.
Source: Clinical Study Report.15
Table 22: Health-Related Quality of Life in Extension Study 160902 Assessed Before Safety Follow-Up (SAS)
Detail | IgHy10 (N = 66) |
|---|---|
PedsQLa total score (age 2 to 7 years) | |
N analyzed | 6 |
Psychosocial health summary, median (range) | 76.7 (61.7 to 93.3) |
Physical health summary, median (range) | 90.6 (53.1 to 100.0) |
Total score, median (range) | 79.9 (58.7 to 95.7) |
SF-36a (age 8 to 13 years) | |
N analyzed | 2 |
Mental component summary, median (range) | 53.3 (51.9 to 54.7) |
Physical component summary, median (range) | 50.8 (47.3 to 54.3) |
SF-36a (age ≥ 14 years) | |
N analyzed | 49 |
Mental component summary, median (range) | 51.0 (25.3 to 56.9) |
Physical component summary, median (range) | 48.7 (11.6 to 52.9) |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; PedsQL = Pediatric Quality of Life Inventory; SAS = safety analysis set; SF-36 = Short Form (36) Health Survey.
aThe range of both scales is 0 to 100, where higher scores indicate a better quality of life.
Source: Clinical Study Reports.15,16
Tolerability and Adherence
In Study 160603, all but 3 (3.7%) subcutaneous infusions of IgHy10 during the ramp-up were tolerated according to the predefined criteria. One of the 3 infusions that were not tolerated was in a patient younger than 12 years old, and the remaining 2 were in patients aged12 years or older. Two of the 3 infusions that were not tolerated were the first subcutaneous IgHy10 infusions administered to the respective patient. The median rate of IgHy10 infusions tolerated at the dose used in epoch 2 (excluding the ramp-up) was 100% (95% CI, 100 to 100) for both IVIg administration and subcutaneous infusions. This outcome was not reported in Extension Study 160902.
The rate of patients who had no infusions that required a reduction in flow rate or that had to be interrupted or stopped due to tolerability concerns or AEs, was similar for subcutaneous IgHy10 (84.0%) and IVIg administration (88.5%) (Table 23). Most (97.7%) subcutaneous infusions with IgHy10 were tolerated, as were most (95.9%) IVIg infusions. The findings for subcutaneous IgHy10 in the Extension Study 160902 were similar.
No patients required a dose correction due to IgG trough levels below 4.5 g/L in Study 160603. This outcome was not reported in Study 160902.
Table 23: Infusions Where the Rate Was Reduced or the Infusion Interrupted or Stopped Due to Tolerability Concerns or AEs in Study 160603 and Study 160902 (SAS)
Detaila | Study 160603 (N = 87) | Study 160902 (N = 66) | ||
|---|---|---|---|---|
Epoch 1: IVIg (n = 87) | Epoch 2: IgHy10 (n = 83) | IgHy10 (n = 63) | Safety follow-up (n = 51) | |
Tolerability by number of patients | ||||
N analyzed (number of patients) | 87 | 81 | 63 | 51 |
Infusion rate reduced, n (%) | 6 (6.9) | 8 (9.9) | 4 (6.3) | 1 (2.0) |
Infusion interrupted, n (%) | 4 (4.6) | 4 (4.9) | 4 (6.3) | 1 (2.0) |
Infusion stopped, n (%) | 0 (0.0) | 1 (1.2) | 1 (1.6) | 1 (2.0) |
No reduction, interruption, or stoppage, n (%) | 77 (88.5) | 68 (84.0) | 54 (85.7) | 48 (94.1) |
Tolerability by number of infusions | ||||
N analyzed (number of infusions) | 365 | 1,129 | 1,600 | 598 |
Infusion rate reduced, n (%) | 10 (2.7) | 19 (1.7) | 20 (1.3) | 2 (0.3) |
Infusion interrupted, n (%) | 5 (1.4) | 5 (0.4) | 13 (0.8) | 1 (0.2) |
Infusion stopped, n (%) | 0 (0.0) | 2 (0.2) | 1 (0.1) | 1 (0.2) |
No reduction, interruption, or stoppage, n (%) | 350 (95.9) | 1,103 (97.7) | 1,566 (97.9) | 594 (99.3) |
AE = adverse event; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; SAS = safety analysis set.
aOutcomes calculated using descriptive statistics.
Source: Clinical Study Reports.15,16
The location where IgHy10 was infused in Study 160603 is summarized in Table 24. Briefly, 847 (75.0%) infusions were administered at the investigational site and 282 (25.0%) were administered at home. Of those administered at home, 51 (4.5% of all infusions) required intervention by a nurse and 231 (20.5% of all infusions) were administered independently. Similarly, the reasons for not being able to continue home infusions in Extension Study 160902 have been summarized in Table 25. Overall, 63.5% of patients were unable to continue self-administration of IgHy10 at home. This was due to medical reasons (14.3%), a family member (15.9%), or other reasons (58.7%). Overall, 51.8% of infusions required assistance for self-administration of IgHy10 at home, with 14.1% due to a medical reason, 5.1% due to a family member, and 32.6% due to other reasons. A summary of patients and infusions requiring assistance for administration at home was also summarized by age (Table 25). Specific reasons for not being able to self-administer IgHy10 at home were not reported in Study 160603.
Table 24: Summary of IgHy10 Infusions Administered at Home in Study 160603 (SAS)
Location of IgHy10 infusion, n (%) | Study 160603 (Epoch 2 (N = 1,129)) |
|---|---|
Investigational site | 847 (75.0) |
At home (all infusions) | 282 (25.0) |
At home with intervention by nurse | 51 (4.5) |
At home without intervention by nurse | 231 (20.5) |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; SAS = safety analysis set.
Source: Clinical Study Reports.15
Table 25: Reasons for Being Unable to Continue Self-Administration of Infusions at Home in Study 160902 (SAS)
Detaila | Study 160902 (N = 66) | ||||
|---|---|---|---|---|---|
< 12 years (n = 4) | 12 to < 16 years (n = 7) | 16 to < 65 years (n = 47) | ≥ 65 years (n = 8) | All (n = 66) | |
Patients requiring assistance, with reasons | |||||
Total number of patients | 4 | 7 | 44 | 8 | 63 |
Medical reason, n (%) | 1 (25.0) | 0 (0.0) | 6 (13.6) | 2 (25.0) | 9 (14.3) |
Family member, n (%) | 4 (100.0) | 3 (42.9) | 2 (45.5) | 1 (12.5) | 10 (15.9) |
Other reason, n (%) | 4 (100.0) | 4 (57.1) | 22 (50.0) | 7 (87.5) | 37 (58.7) |
Total requiring assistance, n (%) | 4 (100.0) | 5 (71.4) | 24 (54.5) | 7 (87.5) | 40 (63.5) |
Infusions requiring assistance, with reasons | |||||
Total number of infusions | 127 | 189 | 1,046 | 238 | 1,600 |
Medical reason, n (%) | 26 (20.5) | 0 (0.0) | 136 (13.0) | 63 (36.5) | 225 (14.1) |
Family member, n (%) | 44 (34.6) | 23 (12.2) | 12 (1.1) | 2 (1.3) | 82 (5.1) |
Other reason, n (%) | 37 (29.1) | 36 (19.0) | 355 (33.9) | 93 (39.1) | 521 (32.6) |
Total requiring assistance, n (%) | 107 (84.3) | 59 (31.2) | 503 (48.1) | 159 (66.8) | 828 (51.8) |
SAS = safety analysis set.
aOutcomes calculated using descriptive statistics.
Source: Clinical Study Reports.15,16
Health Care Utilization
Days in Hospital and Number of Acute Care Physician Visits
The monthly rate of days in hospital and acute care physician visits reported in Study 160603 and Study 160902 (before the safety follow-up period) are summarized in Table 26 and Table 27, respectively.
While receiving treatment with IgHy10, patients reported acute care physician visits at a rate of 0.40 visits per month (95% CI, 0.32 to 0.49). While on treatment with IVIg, patients reported acute care physician visits at a rate of 0.33 visits per month (95% CI, 0.23 to 0.45). The rate of days spent in hospital per month was 0.06 (95% CI, 0.03 to 0.10) while receiving IVIg and 0.02 (95% CI, 0.01 to 0.03) while receiving IgHy10. The monthly rate of days spent in hospital due to infection was 0.03 days per month (95% CI, 0.01 to 0.05) while receiving IVIg, and zero days per month while receiving treatment with IgHy10.
In Extension Study 160902, patients reported acute care physician visits at a rate of 4.19 visits per year (95% CI, 3.14 to 5.45). Also in Study 160902, the rate of days in hospital was 0.83 days/year (95% CI, 0.42 to 1.45) and the rate of days in hospital due to infection was 0.11 days/year (95% CI, 0.06 to 0.18).
Table 26: Number of Days in Hospital and Acute Care Physician Visits in Study 160603
Detaila | Epoch 1: IVIg (N = 87) | Epoch 2: IgHy10 (N = 83) |
|---|---|---|
Full analysis set | ||
N analyzed | 81 | 81 |
Acute care physician visits per month (95% CI) | 0.33 (0.23 to 0.45) | 0.40 (0.32 to 0.49) |
Days in hospital per month (95% CI) | 0.06 (0.03 to 0.10) | 0.02 (0.01 to 0.03) |
Days in hospital due to infection per month (95% CI) | 0.03 (0.01 to 0.05) | 0.00 (0.00 to 0.01) |
Per-protocol analysis set | ||
N analyzed | 74 | 74 |
Acute care physician visits per month (95% CI) | 0.32 (0.22 to 0.45) | 0.39 (0.31 to 0.48) |
Days in hospital per month (95% CI) | 0.06 (0.03 to 0.11) | 0.02 (0.01 to 0.03) |
Days in hospital due to infection per month (95% CI) | 0.03 (0.02 to 0.06) | 0.00 (0.00 to 0.01) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
Source: Clinical Study Report.15
Table 27: Days in Hospital and Acute Care Physician Visits per Year Before the Safety Follow-Up in Extension Study 160902 (SAS)
Detaila | IgHy10 (N = 66) |
|---|---|
N analyzed | 66 |
Acute care physician visits per year (95% CI)b | 4.19 (3.14 to 5.45) |
Days in hospital per year (95% CI) | 0.83 (0.42 to 1.45) |
Days in hospital due to infection per year (95% CI) | 0.11 (0.06 to 0.18) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
bDefined as non-study outpatient visits.
Source: Clinical Study Report.16
Productivity
Days off school or work were reported in Study 160603 and Study 160902 (before the safety follow-up period) and are summarized in Table 28 and Table 29, respectively.
While receiving treatment with IgHy10, patients had to miss school or work at a rate of 0.28 days per month (95% CI, 0.20 to 0.37). While on treatment with IVIg, patients had to miss school or work at a rate of 0.23 days per month (95% CI, 0.15 to 0.34). In Extension Study 160902, patients missed school or work at an annual rate of 7.70 days per year (95% CI, 5.33 to 10.69).
Table 28: Days Off School or Work in Study 160603
Detaila | Epoch 1: IVIg (N = 87) | Epoch 2: IgHy10 (N = 83) |
|---|---|---|
Full analysis set (Study 160603) | ||
N analyzed | 81 | 81 |
Days off school or work per month, rate (95% CI) | 0.23 (0.15 to 0.34) | 0.28 (0.20 to 0.37) |
Per-protocol analysis set (Study 160603) | ||
N analyzed | 74 | 74 |
Days off school or work per month, rate (95% CI) | 0.25 (0.16 to 0.37) | 0.27 (0.19 to 0.37) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = IV immunoglobulin; SCIg = subcutaneous immunoglobulin.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
Source: Clinical Study Report.15
Table 29: Days Off School or Work Before the Safety Follow-Up in Extension Study 160902 (SAS)
Detaila | IgHy10 (N = 66) |
|---|---|
N analyzed | 66 |
Days off school or work per year (95% CI)b | 7.70 (5.33 to 10.69) |
CI = confidence interval; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; SAS = safety analysis set.
aPoint estimates and 95% CIs for the monthly and yearly rates were calculated using a Poisson model and the same methodology, including the allowance for over-dispersion, as described for the primary end points.
bDays off school or work due to fever was 0.72 (95% CI, 0.45 to 1.07) per year, due to infection was 2.81 (95% CI, 1.78 to 4.17) per year, and due to other illness was 4.18 (95% CI, 2.72 to 6.08) per year.
Source: Clinical Study Report.16
Functional Status
Outcomes related to functional status were not reported in the studies included in this review.
Harms
Only those harms identified in the review protocol are reported subsequently. The focus of the summary of harms is on harms that occurred while patients were receiving IgHy10 (during epoch 2 of Study 160603 and during the safety extension phase of Study 160902). See Table 30 and Table 31 for detailed harms data, which includes safety data corresponding to epoch 1 of Study 160603 (treatment with IVIg) and the safety follow-up phase of Study 160902 (treatment with SCIg or IVIg without hyaluronidase). Additional harms data have been reported in Table 35, Table 36, and Table 37. Note that safety data reported during epoch 2 exclude safety events that occurred during the ramp-up period (unless otherwise indicated), as safety outcomes evaluated during the ramp-up period were analyzed separately.
Adverse Events
The rate of AEs in Study 160603 and Study 160902 are summarized in Table 30. In Study 160603, the overall rate of AEs while receiving IgHy10 was 13.40 AEs per patient. The rate of local AEs was 2.90 AEs per patient and the rate of systemic AEs was 10.49 AEs per patient. The most frequently reported AEs while on treatment with IgHy10 was infusion site pain, which occurred at a rate of 1.14 events per patient, as well as the following AEs that occurred at a rate of less than 1 event per patient: headache, sinusitis, upper respiratory tract infection, asthma, nausea, fatigue, myalgia, infusion site pruritus, and viral upper respiratory tract infection. During the safety extension study (Study 160902), the overall rate of AEs was 19.75 per patient while receiving IgHy10. The rate of local AEs was 2.62 per patient and the rate of systemic AEs was 17.13 per patient (including infections) or 12.71 per patient (excluding infections).
In addition, a summary of the most commonly reported AEs (by proportion of patients reporting an AE) that occurred following the ramp-up period during epoch 2 (Study 160603) is provided in Table 31. Briefly, 53.1% of patients reported a local AE. The most frequently reported local AEs were infusion site pain, infusion site discomfort, infusion site erythema, and infusion site pruritus. The most frequently reported systemic AEs were headache (30% of patients), asthma (17%), nausea (15%), pyrexia (15%), and fatigue (15%), and the following AEs were reported in less than 15% of patients: myalgia, vomiting, arthralgia, dizziness, and diarrhea. The rate of AEs per infusion was also summarized in Table 31.
Additionally, a post hoc integrated analysis of safety outcomes from Study 160603 and Study 160902 reported the overall rate of systemic AEs and local AEs (both excluding infections) by age group (< 18 and ≥ 18 years).14 Among adult patients (≥ 18 years), 1,200 systemic AEs were reported, corresponding to a rate of 8.63 AEs per patient-year. For local AEs, a total of 429 AEs were reported among adult patients, corresponding to a rate of 3.08 AEs per patient-year.
A summary of the most commonly reported harms by rate of AEs per infusion in Study 160902 is available in Table 35. Additional harms data for Study 160603 and Study 160902, such as temporally associated AEs, are available in Table 36 and Table 37.
Serious Adverse Events
The SAEs reported in studies 160603 and 160902 are summarized in Table 30. In epoch 2 of Study 160603, a total of 14 SAEs occurred among 11 patients treated with IgHy10, including the ramp-up period. This included cervical dysplasia, grand mal convulsion, lobar pneumonia, status epilepticus with respiratory failure, back injury, acute adrenocortical insufficiency, headache, myocardial infarction, gastroenteritis, intervertebral disc degeneration, petit mal epilepsy, tonsillar hypertrophy, oral leukoplakia, and thrombosis. Three of the 11 patients reporting SAEs experienced the SAE during the ramp-up period. The SAEs reported for that period included intervertebral disc degeneration, lobar pneumonia, and oral leukoplakia.
In Study 160902, there were a total of 18 SAEs reported among 11 patients. Of the SAEs, 13 (72%) occurred during treatment with IgHy10, and 3 occurred during the safety follow-up. SAEs reported while receiving IgHy10 included: chronic obstructive pulmonary disease (2 events), hypertension, aortic valve incompetence, atrial fibrillation, adhesiolysis, appendicitis, cardiac failure congestive, cryptosporidiosis infection, cystocele, mental status change, pneumonia Pseudomonas aeruginosa, spinal meningeal cyst, toxicity to various drugs, and vaginal prolapse. During the safety follow-up, cellulitis, femur fracture, and transient ischemic attack were reported as SAEs.
The rate of SAEs per patient is also available in Table 30.
Withdrawals Due to Adverse Events
Withdrawals from treatment due to AEs were not reported. The number of patients who required that their infusion be stopped due to tolerability concerns or an AE was 2 (0.2%) during epoch 2 of Study 160603, 1 (0.1%) during the safety extension (Study 160902), and 1 (0.2%) during the safety follow-up of Study 160902 (Table 30). As described in the Results section of this report (under Patient Disposition), 6 (6.9%) patients withdrew from the study due to AEs during epoch 2 of Study 160603, including 2 of 14 patients (14.3%) who were younger than 12 years old and 4 of 73 patients (5.5%) who were at least 12 years old. In Study 160902, 1 patient (1.5%) withdrew from the study due to an AE; the patient was between 16 and 65 years old and experienced a severe anaphylactic reaction, which was considered to be related to the immunoglobulin (10%) treatment.
Mortality
No deaths were reported during Study 160603. Two deaths occurred during the Extension Study 160902, 1 patient died from toxicity to various drugs on study day 135, and 1 had a cardiac arrest approximately 4 weeks after they completed the study. None of the deaths were considered to be related to the study treatments.
Notable Harms
Notable harms for this review are summarized in Table 30 as the number of AEs and rate of AEs per patient. While receiving IgHy10 in Study 160603 (excluding the ramp-up period), systemic effects (including headache, fatigue, nausea, vomiting, pyrexia, arthralgia, and myalgia) occurred at a rate of less than 1 AE per patient, as previously described under AEs. Infusion site pain was the most frequently occurring infusion-related AE (1.14 AEs per patient). Infusion site discomfort, infusion site erythema, and infusion site pruritus occurred at a rate of less than 1 AE per patient. For local reactions, swelling/edema and contact dermatitis were reported at a rate of less than 1 AE per patient, as were infusion site hypersensitivity and thrombotic events. No cases of hypersensitivity, anaphylaxis, thrombocytopenia, acute kidney injury, or aseptic meningitis were reported for patients receiving IgHy10 during epoch 2.
While receiving IgHy10 during the safety extension Study 160902, the notable harms related to systemic effects, local reactions, drug hypersensitivity, hypersensitivity, and thrombocytopenia were reported at a rate of less than 1 AE per patient. Infusion-related AEs were also reported at a rate of less than 1 AE per patient, with the exception of infusion site pain, which occurred at a rate of 1.33 AEs per patient. No cases of infusion site hypersensitivity, anaphylaxis, thrombotic events, acute kidney injury, or aseptic meningitis were reported for patients receiving IgHy10 during the safety extension Study 160902.
Table 30: Summary of Harms in Study 160603 and Study 160902 (SAS)
Detail | Study 160603 | Study 160902 | ||
|---|---|---|---|---|
Epoch 1: IVIg (n = 87) | Epoch 2: IgHy10 (n = 83) | Study 160603 extension: IgHy10 (n = 63) | Safety follow-up: IgHy10 by IV or SC (n = 66) | |
N analyzed | 87 | 81 | 63 | 51 |
Patients with ≥ 1 adverse event | ||||
Number of patients, n (%) | NR | NR | 63 (100.0) | 51 (77.3) |
Total number of AEs, n (rate of AEs per patient) | ||||
All AEs | 387 (4.45) | 1,085 (13.40) | 1,244 (19.75) | 407 (7.98) |
Local AEs | 5 (0.06) | 235 (2.90) | 165 (2.62) | 25 (0.49) |
Systemic AEs | 382 (4.39) | 850 (10.49) | NR | NR |
Systemic AEs, including infections | NR | NR | 1,079 (17.13) | 382 (7.49) |
Systemic AEs, excluding infections | NR | NR | 801 (12.71) | 300 (5.88) |
Most common AEs,a n (rate of AEs per patient) | ||||
Infusion site pain | 1 (0.01) | 92 (1.14) | 84 (1.33) | 19 (0.37) |
Headache | 53 (0.61) | 56 (0.69) | 47 (0.75) | 41 (0.80) |
Sinusitis | 19 (0.22) | 53 (0.65) | 63 (1.00) | 16 (0.31) |
Upper respiratory tract infection | 9 (0.10) | 44 (0.54) | 29 (0.46) | 10 (0.20) |
Asthma | 8 (0.09) | 26 (0.32) | 32 (0.51) | 6 (0.12) |
Nausea | 13 (0.15) | 25 (0.31) | 46 (0.73) | 25 (0.49) |
Fatigue | 10 (0.12) | 21 (0.26) | 27 (0.43) | 23 (0.45) |
Myalgia | 5 (0.06) | 20 (0.25) | 28 (0.44) | 8 (0.16) |
Infusion site pruritus | 0 | 17 (0.21) | 31 (0.49) | 0 |
Viral upper respiratory tract infection | 2 (0.02) | 9 (0.11) | 30 (0.48) | 6 (0.12) |
Patients with ≥ 1 SAE | ||||
Number of patients, n (%) | 3 (3.4) | 11 (13.6)b | 11 (16.7)c | |
Number of SAEs, n (% rate per patient) | ||||
All SAEs | 4 (0.05) | 11 (0.14) | 15 (0.24) | 3 (0.06) |
Local AE | 0 | 0 | 0 | 0 |
Systemic AE | 4 (0.05) | 11 (0.14) | NR | NR |
Systemic AE, including infections | NR | NR | 15 (0.24) | 3 (0.06) |
Systemic AE, excluding infections | NR | NR | 12 (0.19) | 2 (0.04) |
Withdrawal from (stopped) treatment due to AEs | ||||
Total number of infusions | 365 | 1,129 | 1,600 | 598 |
Infusion stopped due to tolerability concerns or AE, n (%) | 0 | 2 (0.2) | 1 (0.1) | 1 (0.2) |
Deaths | ||||
n (%) | 0 | 0 | 2 (3.0)d | |
Toxicity to various drugs | NA | NA | 1 | |
Cardiac arrest | NA | NA | 1 | |
Notable harms, n (rate of AEs per patient) | ||||
Systemic effects | ||||
Headache | 53 (0.61) | 56 (0.69) | 47 (0.75) | 41 (0.80) |
Fatigue | 10 (0.12) | 21 (0.26) | 27 (0.43) | 23 (0.45) |
Nausea | 13 (0.15) | 25 (0.31) | 46 (0.73) | 25 (0.49) |
Vomiting | 11 (0.13) | 18 (0.22) | 17 (0.27) | 4 (0.08) |
Pyrexia | 11 (0.13) | 22 (0.27) | 16 (0.25) | 5 (0.10) |
Arthralgia | 1 (0.01) | 14 (0.17) | 8 (0.13) | 7 (0.14) |
Myalgia | 5 (0.06) | 20 (0.25) | 28 (0.44) | 8 (0.16) |
Infusion-related AEs | ||||
Infusion site discomfort | 0 | 30 (0.37) | 8 (0.13) | 2 (0.04) |
Infusion site pain | 1 (0.01) | 92 (1.14) | 84 (1.33) | 19 (0.37) |
Infusion site erythema | 0 | 28 (0.35) | 23 (0.37) | 0 |
Infusion site pruritus | 0 | 17 (0.21) | 31 (0.49) | 0 |
Local reactions | ||||
Swelling/edema | 0 | 9 (0.11) | 1 (0.02) | 0 |
Contact dermatitis | 2 (0.02) | 4 (0.05) | 4 (0.06) | 0 |
Infusion site hypersensitivity | 0 | 2 (0.03) | 0 | 0 |
Drug hypersensitivity | 0 | 0 | 3 (0.05) | 0 |
Hypersensitivity | 0 | 0 | 2 (0.03) | 0 |
Anaphylaxis | 0 | 0 | 0 | 0 |
Thrombotic events (thrombosis) | 0 | 2 (0.03) | 0 | 0 |
Thrombocytopenia | 0 | 0 | 1 (0.02) | 0 |
Acute kidney injury | 0 | 0 | 0 | 0 |
Aseptic meningitis | 0 | 0 | 0 | 0 |
AE = adverse event; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; NA = not applicable; NR = not reported; SAE = serious adverse event; SC = subcutaneous; SCIg = subcutaneous immunoglobulin.
aAE reported at a rate ≥ 0.40 per patient in any treatment group.
bIncludes SAEs that occurred during the ramp-up period. In total, 11 patients experienced an SAE while receiving IgHy10; 3 of these SAEs occurred during the ramp-up period.
cSAEs were reported using the IgHy10 plus safety follow-up (n = 66) dataset.
dDeaths were reported using the IgHy10 plus safety follow-up (n = 66) dataset. One of the deaths occurred 4 weeks after completion of the study.
Source: Clinical Study Reports.15,16
Table 31: Summary of the Most Commonly Reported Harms That Occurred After the Ramp-Up in Epoch 2 by Percentage of Patients and Rate of AEs per Infusion in Study 160603 (SAS)
Most common AEsa (excluding infections) | Epoch 2: IgHy10 (n = 83) | ||
|---|---|---|---|
Patients with AEs, n (%) | Infusions with AEs, number of AEs (rate per infusion) | ||
N analyzed | 81 | NR | |
Local AEs | 43 (53.1) | 235 (0.208) | |
Infusion site pain | 27 (33.3) | 92 (0.081) | |
Infusion site discomfort | 8 (9.9) | 30 (0.027) | |
Infusion site erythema | 10 (12.3) | 28 (0.025) | |
Infusion site pruritus | 6 (7.4) or < 10% | 17 (0.015) | |
Systemic AEs | NR | NR | |
Headache | 24 (29.6) | 56 (0.050) | |
Asthma | 14 (17.3) | 26 (0.023) | |
Nausea | 12 (14.8) | 25 (0.022) | |
Pyrexia | 12 (14.8) | 22 (0.019) | |
Fatigue | 12 (14.8) | 21 (0.019) | |
Myalgia | 10 (12.3) | 20 (0.018) | |
Vomiting | 11 (13.6) | 18 (0.016) | |
Arthralgia | 11 (13.6) | 14 (0.012) | |
Dizziness | 9 (11.1) | 12 (0.011) | |
Diarrhea | 8 (9.9) | 11 (0.010) | |
AE = adverse event; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; NR = not reported; SAS = safety analysis set.
aFrequency ≥ 10% of patients or ≥ 0.01 AEs per infusion.
Source: Clinical Study Report.15
Critical Appraisal
Internal Validity
The IgHy10 pivotal trial was a non-randomized, open-label, single-group, prospective study design. Due to the uncontrolled study design, randomization was not possible. Patients in Study 160603 were enrolled in 1 of 2 study arms based on participation in a prior pharmacokinetic study (Study 160601). Of the 87 patients included in the study, 31 had previously received IVIg and cSCIg through participation in Study 160601 and the data collected during that study were used for epoch 1 of Study 160603. The remaining 56 patients proceeded with IVIg treatment as part of epoch 1. Whether there are inherent differences between the patients included in study arms 1 and 2 or the care they received is unknown. Most of the outcomes of interest for this review, including the primary end point of Study 160603, were not expected to suffer from its open-label design; however, patient-reported outcomes such as those related to HRQoL, tolerability and adherence, and safety outcomes, may have been impacted based on patient beliefs about IgHy10. It is unknown whether knowledge of treatment would have caused bias in favour of or against IgHy10.
As a single-group study design, neither a historical control nor a concurrent comparator group was used in either Study 160603 or the subsequent Extension Study 160902. Consequently, there is substantial uncertainty associated with inferring causality associated with the outcomes reported in the trials. For example, it is unknown whether the number of VASBIs reported, which informed the primary end point, were solely the result of treatment with IgHy10, as potential confounding factors were not accounted for. Prior to receiving IgHy10 (epoch 2), patients had received treatment with IVIg in epoch 1, during which safety and efficacy data were collected; however, Study 160603 was not designed to compare IVIg with IgHy10 treatment, except for pharmacokinetics (IgG trough levels). Further, the duration of treatment with IVIg was much shorter (3 months) than with IgHy10 (14 to 18 months), which would have subjected any comparisons to limitations related to seasonality or year-to-year variation.
Missing data were not accounted for in either the pivotal study or the extension study. The statistical techniques employed in the 2 studies did not require imputation of missing data, as the methods used accounted for the length of time each patient spent in the study. Despite this, the risk of attrition bias is still a concern due to the overall discontinuation rate in Study 160603, where 21% of patients discontinued from the study. The impact of discontinuation rates was greater for patients in the 2 years to younger than 12 years group (N = 14) in Study 160603, where 43% of patients discontinued from the study either due to a requested withdrawal (29%) or an AE (14%). As a result, the reported study results are likely biased in favour of IgHy10, as the data analyzed is largely based on patients who did not discontinue from the study.
In general, the efficacy outcomes assessed in the pivotal study and extension study were considered appropriate based on feedback from the clinical experts consulted for this review. A validated definition of acute serious bacterial infections was used for the primary end point in Study 160603. In terms of clinical context, reporting of serious bacterial infections was restricted to VASBIs. This was seen as a limitation by the clinical experts, as non-acute infections were not specifically reported. Chronic infections that were considered clinically relevant, such as sinusitis, recurrent ear infections, and gastroenteritis, were likely captured under the secondary end point, annual rate of all infections; however, infections were not reported with that level of detail (acute versus chronic, or duration of infection). The clinical experts noted that the chronic infections described can be serious and require urgent intervention or a long course of antibiotics.
The other efficacy outcomes were objective/clinical (e.g., IgG trough levels, days on antibiotics) or based on validated measures, such as the HRQoL outcomes assessed using the PedsQL and SF-36. Evidence of validity, reliability, and responsiveness was identified for both the PedsQL and SF-36, but not specifically in patients with immunodeficiency.
Although the selection of outcome measures reported in the studies was considered appropriate, the methods of analysis used were associated with limitations that hindered the ability to meaningfully interpret the outcomes. The analysis of the primary end point, annual rate of VASBI, is an exception, as the analysis was aligned with FDA guidance for industry11 that states “a statistical demonstration of a serious infection rate per person-year less than 1.0 is adequate to provide substantial evidence of efficacy.” Statistical testing was used only for the primary end point in Study 160603; therefore, multiplicity was not an issue. Within-group changes were generally not reported, as results were analyzed as a rate or descriptively summarized for the observation period.
Both the patient and clinician input received for this review highlighted HRQoL as an important consideration for the treatment of primary and secondary humoral immunodeficiencies. The HRQoL outcomes included in the 2 studies were exploratory outcomes reported as a median (range) for each treatment period that did not offer the ability to draw any conclusions about a change in HRQoL with IgHy10. The HRQoL outcomes were reported by subgroups based on age, although they were limited by a small sample size, particularly for patients younger than 14 years old (N < 10). Additionally, HRQoL was reported using different outcomes for patients aged 8 to 13 years; in Study 160603, the PedsQL was used, whereas the SF-36 was used in the extension study.
The assessment of safety in the 2 studies was also challenging to interpret in a clinically meaningful way. The proportion of patients who experienced at least 1 AE was not reported in Study 160603, but the proportion of patients experiencing a local AE was available. The proportion of patients experiencing an AE (as defined by MedDRA) was reported as well, but only for patients receiving IgHy10 after the ramp-up period in Study 160603. Also, the majority of the safety outcomes were reported as a rate per patient or per infusion. While this may account for varying duration of treatment in each of the reported observation periods, the proportion of patients experiencing a particular AE is unknown, with the exception of the AEs commonly reported during treatment with IgHy10 in Study 160603.
External Validity
The pivotal trial (Study 160603) and extension study (Study 160902) were primarily located in the US, with 1 study site located in Canada, and were conducted between 2008 and 2013. The clinical experts consulted by CADTH felt the studies conducted in the US were generalizable to patients in Canada and that the age of the study was not a concern. They did note, however, that there has been a recent shift in the use of IVIg compared with SCIg, with the latter being used more commonly.
The patients included in Study 160603 and 160902 were required to have been diagnosed with a PID that required antibody replacement therapy and to have been receiving regular IgRT for at least 3 months before enrolment, and their last documented serum IgG trough level had to be greater than 4.5 g/L. The clinical experts consulted by CADTH did not have any concerns regarding the safety and efficacy of IgHy10 in terms of generalizing the evidence to patients with secondary humoral immunodeficiency, IgRT-naive patients, or patients with lower serum IgG levels. The sponsor also noted the guidance from Health Canada regarding the regulatory requirements for IVIg states that indications for SID can be accepted based on the evidence for a PID therapy49; however it is unclear whether this policy would apply to a novel, subcutaneous, combination product such as IgHy10.
Certain patient groups were excluded from the included trials, such as pregnant or lactating females, patients with IgA deficiency, patients who were required to remain on prophylactic systemic antibacterial antibiotics, and patients with various pre-existing conditions. The clinical experts indicated a significant proportion of patients with primary or secondary humoral immunodeficiency plus pre-existing conditions would be treated prophylactically with antibiotics; thus, the patient groups that were excluded from the trials would not be excluded from treatment with IgHy10. The clinical experts consulted by CADTH did not express concern with applying the results of the included studies to these patients.
Patients received IgHy10 (75 U/g rHuPH20 followed by SCIg 10%), administered at 108% of the monthly total IVIg dose used in epoch 1 every 3 or 4 weeks.10 The setting of the dose of SCIg to 108% of the dose of IVIg was based on a phase II pharmacokinetic study.14 This differs from the recommended dosing in the Health Canada product monograph, which is to follow the previous dose of IgRT administered. However, dose adjustments are made at the discretion of the treating physician in clinical practice; therefore, whether this is a major concern regarding the generalizability of the evidence is unknown. Prior to administration of IVIg or SCIg, concomitant use of prophylactic antibacterial antibiotics and pre-medications was avoided during the studies, if possible. However, because concomitant use of these medications is consistent with clinical practice, such avoidance may have exaggerated the reported tolerability and safety outcomes compared with what is expected in clinical practice. Administration of IgHy10 was either performed at home or an infusion centre, or under permanent medical supervision at the clinical study site. Patients who administered IgHy10 at home or at an infusion centre were observed by a health care professional until at least the fifth or sixth infusion, after which the ability to administer treatment without observation was evaluated. As per feedback from the clinical experts, an at-home training/support system would be implemented to facilitate the administration of IgHy10 at home, but the level of supervision in the trials exceeds expectations for clinical practice. The impact of this on the performance of IgHy10 and in relation to what would be expected in clinical practice is unknown.
The duration of treatment with IgHy10 in Study 160603 was 14 to 18 months, with a mean duration of treatment of approximately 1 year (368 days; SD = 104). This was sufficient to evaluate the primary outcome and most of the other outcomes assessed in the study. However, because IgRT is a lifelong treatment, there may not have been enough time to capture long-term outcomes, such as risk of malignancies. Study 160902 was designed as a long-term safety and tolerability study for IgHy10. Patients in the study received IgHy10 for a mean of 566 days (SD = 212), which was an acceptable duration of treatment to generate sufficient data to supplement the safety outcomes data reported in the pivotal trial.
Indirect Evidence
Indirect evidence matching the inclusion and exclusion criteria of this review was not submitted by the sponsor or identified in the CADTH literature review.
Other Relevant Evidence
This section includes 2 submitted phase IV single-arm studies and 1 pregnancy registry study. These were provided within the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review. The single-arm phase IV trials provide further evidence on the safety and tolerability of IgHy10 in children, and an assessment of IgG trough levels among patients switching from SCIg to subcutaneous IgHy10 every other week. The pregnancy registry study provides information on the potential harms of IgHy10 in pregnant women and their infants.
Sponsor-Submitted Study in Adult Patients Switching From CSCIg to IgHy10 (NCT02881437)
Description of Study
NCT02881437 was a phase IV, open-label, non-randomized, single-group prospective study,17 which filled an important gap in the comparison of cSCIg to IgHy10. The primary objective was to examine the difference in steady-state IgG trough levels among adults (18 years or older) with a PID requiring IgRT during cSCIg treatment (primarily once weekly) compared with the steady-state IgG trough levels after switching to IgHy10 administration every other week at equivalent doses. Full details of the study are in Appendix 5, Table 39. Among the 22 enrolled patients, the median age was 45.0 years (IQR = 32.0 to 54.0), and 68.2% were female. Appendix 5 shows the full baseline characteristics (Table 40) and patient disposition (Table 41).
The study began with a 1-week ramp-up period (subcutaneous IgHy10 provided at 1-quarter of the usual monthly cSCIg dose) which started 1 week after the last cSCIg infusion before enrolment. Subcutaneous IgHy10 infusions then occurred every 2 weeks at 1-half of the initial monthly SCIg dosage, with follow-up measurements at 3 and 6 months. Full details of prior treatment with cSCIg and exposure to IgHy10 are shown in Appendix 6, Table 42 and Table 43.
Outcomes of interest were change in IgG trough level, HRQoL (SF-36), and AEs.
Results
Table 44 in Appendix 5 shows the full details of the change in steady-state IgG trough level when switching from cSCIg to IgHy10. The mean change in steady-state IgG trough level when switching from cSCIg to IgHy10 was –0.30 g/L (SD = 1.54) after 3 months, and –0.29 g/L (SD = 1.35) after 6 months (n = 16).
A total of 11 of 19 patients (57.9%) had at least 1 infection in the first 3 months of follow-up, corresponding to an incidence of 0.34 (95% CI, 0.22 to 0.54) infections per month. Infection was reported among 8 of 17 patients (47.1%) at between 3 and 6 months of follow-up, corresponding to an incidence of 0.22 (95% CI, 0.10 to 0.48) infections per month.
The mean change in the physical component summary of the SF-36 was −0.90 (SD = 4.37) from baseline to 3 months and −2.67 (SD = 5.17) from 3 to 6 months (n = 12); the mean change in the mental component summary was −2.67 (SD = 5.17) from baseline to 3 months and 1.33 (SD = 5.13) from 3 to 6 months (n = 14).
A detailed record of the reported AEs is provided in Table 45. A total of 21 of 22 patients (95.5%) reported at least 1 local AE, and all reported at least 1 systemic AE between baseline and 3 months of follow-up; 12 of 22 patients (66.7%) reported at least 1 local AE and 16 of 22 (88.9%) reported at least 1 systemic AE between 3 and 6 months follow-up. Commonly reported AEs were similar to those in the pivotal studies.
Critical Appraisal
There are several internal validity concerns that limit the certainty of the conclusions that can be drawn. The primary concern is that there is no control group and confounders are unaccounted for; thus, causal relationships cannot be established. Though the inclusion and exclusion criteria are clear, some details of the disposition of the participants are limited (i.e., number screened versus randomized). The open-label design is likely to have biased the subjective end points; however, the direction of bias is unclear. There were large losses to follow-up (6 of 22; 37%), which reduces the reliability of the findings for HRQoL. Statistical analyses were not adjusted for multiplicity.
Despite some differences in setting (all study centres were located in France) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to adult patients in Canada; the applicability of the evidence to children is less clear. Exposure to study treatments appeared to match the product monograph, aside from the ramp-up phase (not described in the monograph). Outcomes were clinically relevant, though the clinical experts indicated they would not rely on IgG trough levels alone for clinical decision-making. The sample size and length of follow-up may have been inadequate to capture rare or long-term harms.
Sponsor-Submitted Study of Safety, Tolerability, and Immunogenicity of IgHy10 in Pediatric Patients (Study 161504, Interim Analysis)
Description of Study
Clinical Study 161504 was a phase IV, open-label, non-randomized, single-group prospective study18 that provided further post-authorization safety, tolerability, and immunogenicity data on IgHy10 among 42 pediatric patients (2 to < 18 years old) with primary humoral immunodeficiency requiring IgRT. The findings are based on a planned interim analysis after 75% of patients had completed 1 year of study in epoch 2.19 Full details of the study are in Appendix 6, Table 46. Patients who were naive to SCIg (n = 23; 54.8%) had a mean age of 10.3 years (SD = 3.8); the group was 78% male and 96% White. The characteristics of patients with prior SCIg exposure (pre-treated) were similar.
Patients naive to fSCIg were enrolled in a maximum 6-week ramp-up period where they were introduced to IgHy10 infusions (epoch 1). In epoch 2, IgHy10 treatment continued for up to 3 years. After 1 year in epoch 2, patients with an anti-rHuPH20 titre of 160 or lower at any time proceeded to study completion; others continued on treatment for another 2 years. Patients with an anti-rHuPH20 titre of 160 or higher who experienced an SAE or severe AE continued in a safety follow-up (epoch 3), where IgHy10 was discontinued and patients continued with IVIg or cSCIg. The exact details of the treatment regimens are not available. At the time of the interim analysis, 22 patients (52.3%) had completed the study, 17 (40.5%) were ongoing, and 3 (7.1%) had discontinued prematurely. At the time of the interim analysis, patients had received a mean of 12.5 infusions (SD = 6.9), with a median of 1.2 infusions per month (range = 0.5 to 7.6). The median infusion volume was 95.0 mL per site and the maximum infusion rate was 200.0 mL per hour per site.
Outcomes of interest were change in IgG trough level, immunogenicity, and AEs. Serious acute bacterial infections were not pre-specified but were reported. Details of the statistical analyses were unavailable.
Results
The mean IgG trough level was 9.6 mg/dL (SD = 2.1) at enrolment and 8.2 mg/dL (SD = 2.9) at 12 months. Full details of treatment-emergent infections are in Table 47 in Appendix 6. In total, 31 out of 42 patients (73.8%) experienced at least 1 treatment-emergent infection. One patient (2.4%) had a serious acute bacterial infection.
A detailed account of recorded AEs is provided in Table 48. Of 42 enrolled patients, 27 (64.3%) reported at least 1 AE. Eleven patients (26.2%) experienced at least 1 local AE, 24 (57.1%) experienced at least 1 systemic AE, and 4 (9.5%) experienced at least 1 SAE. Across all treatments (n not reported), 7.4% of infusions were stopped, interrupted, or adjusted due to an AE. No patients developed an anti-rHuPH20 antibody titre of 160 or greater.
Critical Appraisal
There are several internal validity concerns that limit the certainty of the conclusions that can be drawn. The primary concern is that there is no control group and confounders are unaccounted for; thus, causal relationships cannot be established. No statistical hypothesis testing was conducted. Interim findings of safety and efficacy should be interpreted cautiously, as these findings could overestimate the benefits and/or underestimate the harms of a treatment.20,21 Though the inclusion and exclusion criteria are clear, some details of the participant disposition are limited (i.e., number screened versus randomized). The open-label design is likely to have biased the subjective end points; however, the direction of the bias is unclear. It is not fully clear how outcomes were defined and collected, and “treatment-emergent infections” were not pre-specified in the study protocol. The small sample size may negatively impact the reliability of the findings.
Despite some differences in setting (all study sites were located in Europe) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to pediatric patients in Canada; the applicability of the evidence to adults is less clear. Dosing information was not provided. Outcomes were clinically relevant, though the clinical experts indicated they would not rely on IgG trough levels alone for clinical decision-making. The sample size was likely too small to capture rare harms.
Sponsor-Submitted Study on Safety of IgHy10 in Pregnant Women and Their Infants (Registry Study 161301)
Description of Study
Study 161301 was a registry study providing safety data on women who had previously received treatment with IgHy10 and their infants50 — a population excluded from the pivotal studies. All pregnant women who had ever been treated with IgHy10 were eligible in 1 of 2 study arms: continued IgHy10 during pregnancy (IgHy10 arm) and switched to another IgRT or alternative treatment (alternative product arm). Full details of the study are in Table 49. Nine mothers were enrolled; they had a median age of 34.0 years (IQR = 32.0 to 36.0), were primarily non-Hispanic and non-Latino (8 of 9; 88.9%), and all were White/Caucasian. Seven of the mothers’ infants were enrolled. The detailed characteristics of the mothers and their infants are in Appendix 7 (Table 50 and Table 51; patient disposition is in Table 52). Patients visited their own physicians and were treated according to routine medical practice. Data on IgHy10 treatment were available for 6 mothers (85.7%) in the IgHy10 arm. Among these, the median number of infusions was 4 (IQR = 1.5 to 5.75); infusions were received on a 3- or 4-week interval. Full details of exposure to IgHy10 are in Appendix 7 (Table 53 and Table 54).
Outcomes of interest were AEs (mothers and infants) and the development of anti-rHuPH20 antibodies (mothers). In addition, outcomes related to pregnancy and infant development were assessed. Full details of the statistical analysis are in Appendix 7.
Results
A full description of the AEs reported during the study is in Appendix 7 (Table 55). Among the 9 mothers, 4 (44.4%) reported at least 1 AE; 3 (42.9%) mothers in the IgHy10 arm and 1 (50.0%) in the alternative product arm. There were no local or immunologic AEs. One (11.1%) mother in the IgHy10 arm reported SAEs. There were no AEs leading to death or WDAEs. Four (44.4%) mothers (2 in each arm) were assessed for anti-rHuPH20 antibody titres, and all were negative (titre of less than 160). Eight (88.9%) mothers provided data on pregnancy outcomes; all were live births. One birth in the IgHy10 arm was considered abnormal because it was a Caesarean section.
Infants were born at a median gestation of 38.0 weeks (IQR = 37.0 to 40.0); weight, length, and head circumference were normal for all. Two (40.0%) infants in the IgHy10 arm had congenital malformations. During follow-up, 6 infants (85.7%) experienced at least 1 AE: 5 (100%) in the IgHy10 arm and 1 (50.0%) in the alternative product arm. Two (40.0%) infants in the IgHy10 arm experienced an SAE. There were no AEs leading to death or a WDAE.
Critical Appraisal
There are several internal validity concerns that limit the certainty of the conclusions that can be drawn. The primary concern is that there is no control group and confounders are unaccounted for; thus, causal relationships cannot be established. There was no statistical hypothesis testing. Selection bias is possible, as very few mothers (n = 9) were enrolled, and it is not clear whether women from various centres would differ systematically. As this is an open-label study, subjective end points may be biased; however, the direction of the bias is unclear. Most of the data were collected retrospectively, which may have negatively affected quality and completeness. In the IgHy10 arm, 29% of mothers were lost to follow-up, which may have biased AE data in favour of IgHy10.
Despite some differences in setting (study sites were in Europe and the US) and a narrower eligible population than would be seen in clinical practice, the clinical experts consulted by CADTH did not express concern in generalizing the evidence to patients in Canada. The dosage and administration of IgHy10 appear to align with the Health Canada–approved dosing; however, pregnant women are not an indicated population in the product monograph. The harm outcomes seem to be clinically important, but no efficacy outcomes were collected. The sample size and length of follow-up were likely inadequate to capture rare and/or long-term AEs.
Post-Authorization Safety Studies and Patient Registry Study
Results from 2 PASSes, Global PASS (NCT02593188)22 and EU PASS (EUPAS5812),23,24 and a patient registry study, FIGARO (NCT03054181),25-27 were submitted by the sponsor as supportive evidence. Both the Global PASS (N = 264) and EU PASS (N = 106) were non-interventional, prospective, uncontrolled, open-label, multi-centre PASSes that evaluated the long-term safety of IgHy10 under clinical routine conditions in the US and Europe, respectively. The Global PASS study was conducted between 2015 and 2021 and enrolled patients with a PID. The EU PASS study was conducted between 2014 and 2021 and enrolled patients who had been prescribed treatment for a PID or SID. The Global PASS reported that 56% of 909 infusions were self-administered at home. The EU PASS reported the proportion of treatments that were administered at a clinical site and at home by year since the first fSCIg treatment. During the first, second, and third year, and after the third year, 91.2% (n = 83 patients; 909 infusions), 93.2% (n = 556 patients; 600 infusions), 93.2% (n = 28 patients, 237 infusions), and 85.2% (n = 12 patients; 54 infusions) of treatments were administered at home, respectively. FIGARO was a long-term observational study on the utilization and outcomes of IgHy10 under everyday clinical practice conditions. FIGARO was conducted in Europe between 2016 and 2021 and enrolled 156 patients with a PID or SID. Data were available for 154 patients, of which 13 were pediatric (younger than 18 years), 120 were adults (18 to 64 years), and 21 were older adults (at least 65 years); results were analyzed by patient age. FIGARO reported that 81.7% of adults and 57.1% of older adults infused at home.
The results provided by the sponsor for infusions administered at home in Global PASS, EU PASS, and FIGARO suggest that the ability to administer treatment at home was more successful in a real-world setting than in the clinical trial setting. However, the generalizability of this evidence to patients treated in Canadian clinical practice is unknown. Additionally, the interpretation of the additional evidence should take into consideration the limitations associated with real-world evidence studies.
Post Hoc Analysis
The sponsor submitted a post hoc analysis by Wasserman et al., 2021,28 as supportive evidence. The analysis examined 3 consecutive, open-label, uncontrolled clinical studies of IgG therapy. Each of the studies included a subset of patients with primary immunodeficiency. Two of the 3 studies, Study 160603 and Study 160902, informed CADTH’s systematic review of HyQvia. The retrospective post hoc analysis included 30 patients who had received at least 1 infusion of each type of therapy, i.e., IVIg, cSCIg, and fSCIg, and was designed to evaluate the efficacy (rates of infection) and tolerability of the 3 routes of IgG administration. The duration of exposure, total number of infusions, and mean IgG dose received during a 4-week period differed between the 3 treatments. As noted in the publication, the limitations of the study include a small sample size; selection bias, as study participation was voluntary; and year-to-year variations in community infections and other factors that change over time, which cannot be accounted for in a sequential study design. The post hoc analysis concluded that across the 3 treatment modalities (IVIg, cSCIg, and fSCIg), annualized rates of VASBI (0, 0.09, and 0.04, respectively) and all infections (4.17, 3.68, and 2.42, respectively) were similarly low.
Discussion
Summary of Available Evidence
Two sponsor-submitted, phase III, open-label, non-randomized, single-group, prospective studies were included in this review. Study 160603 (N = 89) evaluated the efficacy and tolerability of IgHy10 in patients who were at least 2 years of age with a PID that required antibody replacement therapy. The patients who were enrolled in Study 160603 were between the age of 4 and 78 years, and the patients who continued to Study 160902 were between the ages of 9 and 80 years. All patients were also required to have a serum IgG trough level greater than 4.5 g/L and to have been receiving regular IgRT for at least 3 months before enrolment. Study 160902 (N = 66) was an extension of Study 160603 that evaluated the long-term tolerability and safety of IgHy10. The safety extension also followed patients for delayed adverse reactions following discontinuation of hyaluronidase.
Study 160603 consisted of 2 epochs. During epoch 1, patients received IVIg 10% for 12 weeks. In epoch 2, patients received IgHy10 (75 U/g rHuPH20 followed by SCIg 10%), administered at 108% of the monthly total IVIg dose every 3 or 4 weeks, for 14 to 18 months. The primary end point in Study 160603 was serious acute bacterial infection rate, defined as the mean number of VASBIs per patient per year. The annual rate of all infections, immunoglobulin levels, antibiotic use, health care utilization, productivity, HRQoL, tolerability, and safety were also evaluated. Safety outcomes, including those related to tolerability concerns and AEs, were evaluated in Study 160902; the efficacy outcomes that were evaluated in Study 160603 were also evaluated.
In addition to the 2 sponsor-submitted studies previously described, 2 phase IV single-arm studies and 1 pregnancy registry study were reviewed and critically appraised. The single-arm phase IV trials provided further evidence on the safety and tolerability of IgHy10 in children, and an assessment of immunoglobulin trough levels among patients switching from SCIg to subcutaneous IgHy10 every other week. The pregnancy registry study provided information on the potential harms of IgHy10 in pregnant women and their infants. The results are summarized in the Clinical Evidence section (under Other Relevant Evidence) of this report.
Interpretation of Results
Efficacy
Patients living with primary and secondary humoral immunodeficiency are unable to produce an adequate immune response because the components of the immune system, specifically the antibodies, are either absent or functionally inadequate. As a result, primary and secondary humoral immunodeficiency predispose affected patients to an increase in the frequency and severity of infections, autoimmunity, and malignancy. The standard of therapy for the treatment of these patients is the use of IgRT, with the goal of treatment being the prevention of infections leading to an improvement in HRQoL, preserved function, and prevention of hospitalizations. The goal of treatment is also to reduce morbidity and mortality related to disease and minimize disruptions to the patient’s life; the latter was identified as an outcome of strong importance to patients through the patient input received for this review.
Of note, the evidence summarized in this review does not include patients with secondary humoral immunodeficiency. The clinical experts consulted by CADTH did not express any issues with using evidence in patients with primary humoral immunodeficiency to inform treatment decisions for patients with secondary humoral immunodeficiency. Additionally, there was no comparative evidence for IgHy10 versus other available IgRTs.
In Study 160603, 2 VASBIs were reported during 14 to 18 months of treatment with IgHy10. Both cases were episodes of bacterial pneumonia that were treated with oral antibiotics without hospitalization.12 This corresponded to a rate of VASBIs per year of 0.025 (upper limit of the 99% CI, 0.046), thus meeting the primary end point of the study. The clinical experts for this review agreed that the reported rate of VASBIs in patients with a PID was aligned with what would be expected for patients receiving IgRT treatment. The rate of VASBIs per year before the safety follow-up period was similar among the 66 patients who continued to extension Study 160902 (rate of 0.020; upper limit of the 99% CI = 0.045). The rate of VASBIs was not analyzed during treatment with IVIg, but the rate of all infections was reported. The rate of all infections per patient per year was 2.97 (95% CI, 2.51 to 3.47) and 2.86 (95% CI, 2.36 to 3.43) during treatment with IgHy10 in Study 160603 and 160902, respectively. This was lower than the rate of all infections reported during treatment with IVIg in the pivotal trial, which was 4.51 (95% CI, 3.50 to 5.69). While we cannot statistically compare the infection rates reported during treatment with IgHy10 and treatment with IVIg, as the study was not designed to assess this, the rates appear to be lower when patients were treated with IgHy10. It is unknown whether this is an artifact of the different time periods or the different characteristics of the population as confounders such as temporal factors were not accounted for. Additionally, the results of a post hoc analysis of the annual rate of infections and annual rate of VASBIs during IgHy10 treatment were reported by age group (< 18 years and ≥ 18 years old).13 The annual rate of infections and annual rate of VASBIs in adult patients (≥ 18 years old) were consistent with the results reported for each of the individual studies.
The pivotal study and extension study included assessments of IgG trough levels. The clinical experts consulted by CADTH indicated that IgG levels are routinely assessed and useful as a reference point; however, treatment decisions are predominantly based on clinical assessments of the patient as opposed to relying solely on laboratory values. The median serum IgG trough level at baseline was approximately 10 g/L, which is aligned with published recommendations for treatment with IgRT.5 In Study 160603, the median IgG trough levels were maintained in both pediatric patients and patients who were at least 12 years of age or older over approximately 1 year of treatment. In the extension study, IgG trough levels were analyzed by infusion interval. Of note, patients were asked to increase the frequency of IgHy10 administration to a 2-week or 3-week interval to assess IgG levels following a change in administration frequency. In summary, the steady-state IgG trough levels were maintained for approximately 4 months at a median of at least 10 g/L despite changes to the administration frequency, although this is based on a small sample size of patients. Further, the ratio of IgG trough levels measured at the end of the safety follow-up period compared with the IgG trough levels measured at the end of IgHy10 treatment indicated that, following discontinuation of hyaluronidase, IgG levels were maintained or increased, with the exception of a switch to IVIg following administration of IgHy10 at a 2-week interval, where IgG levels decreased. Most of the assessments were based on a small sample size (between 3 and 38 patients) and statistical testing was not conducted, preventing any further conclusions from being drawn.
Outcomes related to antibiotic use, health care utilization, and productivity were of importance to clinicians and patients and were included in studies 160603 and 160902. While receiving treatment with IgHy10 in Study 160603, the rate of days off per month from either work or school was less than 1 day per month; no patients were hospitalized during treatment with IgHy10, and the rate of acute care physician visits was less than 1 visit per month. These outcomes suggest minimal disruption to the everyday life of patients and few days spent in hospital, which were key outcomes of interest for this review; however, the lack of historical data for the patients enrolled in the studies and the lack of a comparator group make it difficult to appropriately interpret these outcomes. Similar assessments conducted in the extension study yielded comparable results. The rate of days on antibiotics while on treatment with IgHy10 was also reported, which was less than 2 days per month in Study 160603 and 64 days per year (approximately 5 days per month) during the extension study. The clinical experts consulted by CADTH indicated that the rate of antibiotic use during treatment with IgHy10 in both studies was higher than expected, noting that it is atypical for patients to use antibiotics every month.
Another outcome that was of importance to patients was HRQoL. Two age-appropriate, validated outcomes, the PedsQL and SF-36, were used to assess HRQoL at the end of each epoch or observation period in both of the included studies. A significant limitation of the HRQoL data are the failure to evaluate outcomes as a change over time (i.e., at the start and end of treatment with IgHy10), especially considering the study was not designed to assess a comparison between epoch 1 (treatment with IVIg) and epoch 2 (treatment with IgHy10), and there was no other comparison group. As a result, no conclusions regarding HRQoL can be drawn from the available evidence.
The evidence that has been summarized so far suggests that in a clinical trial setting, IgHy10 seems to result in a rate of VASBIs that is similar to what might be expected with any IgRT; IgHy10 therapy also seems to maintain trough IgG levels and results in few visits to health care and disruptions to patient lives. However, the absence of comparative evidence for IgHy10 is a major limitation in the context of this review, as other SCIg and IVIg products are currently available in Canada. There is significant uncertainty associated with inferring causality of the reported results in studies 160603 and 160902, and there is no evidence on how IgHy10 performs compared with other available IgRTs. One of the key advantages that was anticipated for IgHy10 was the ability to administer treatment at home and less frequently than cSCIg, thereby improving the convenience and minimizing the impact of treatment on a patient’s life. The clinical experts consulted by CADTH indicated they expected that the majority of patients (more than 90%) would be able to self-administer IgHy10 at home, a level similar to what they had observed with cSCIg in clinical practice. The results from the extension, Study 160902, did not indicate this, as 64% of patients and 52% of infusions required assistance and were unable to continue self-administration of infusions at home. Also of note, 75.0% of all infusions in Study 160603 were administered at an investigational site. Of the 25% of infusions that were administered at home, 82% (20.5% of all infusions) were administered without nurse intervention (as opposed to 4.5% that were administered at home with intervention by a nurse). Some of the infusions were required to be administered at the investigational site, but the proportion of infusions this applies to is unknown, based on the data available.
From an implementation perspective, clinicians and decision-makers were interested in understanding how the efficacy of IgHy10 compares with the other available forms of IgRT, and cSCIg in particular, to determine the place in therapy. Again, conclusions cannot be drawn based on the evidence identified for this review that does not include comparative studies. This is a major limitation of the evidence identified for this review. There are many other SCIg and IVIg drugs available, and IgHy10 was viewed as another SCIg option by the clinical experts.
Three other studies did not meet the inclusion criteria of the CADTH review but were considered relevant in terms of addressing gaps in the evidence. In 1 study, patients did not experience a clinically meaningful change in IgG levels or HRQoL (measured by the SF-36) over a 6-month period. The other 2 studies provided additional safety evidence and have been described in the following section of the report.
Harms
One of the putative features of IgHy10 was that it possesses the benefits of both cSCIg (i.e., a favourable safety profile, particularly in terms of minimal systemic AEs) and IVIg (i.e., less frequent administration schedule). A total of 2 deaths were reported across the pivotal and the extension study; both occurred during Study 160902. One death was caused by toxicity to various drugs and the other by cardiac arrest; neither death was considered related to the study treatments. SAEs were reported infrequently. A total of 22 of 89 patients reported an SAE in studies 160603 and 160902. Two SAEs due to chronic obstructive pulmonary disease were reported, which were the only SAEs that were reported more than once.
The overall rate of AEs while receiving IgHy10 was 13.40 per patient during Study 160603 and 19.75 per patient during the extension study, where the mean duration of treatment was approximately 1 year and 1.5 years, respectively. In contrast, the rate of AEs was 4.45 per patient during treatment with IVIg in Study 160603, and 7.78 per patient during the safety follow-up with IVIg or SCIg without hyaluronidase during the extension study. The overall rate of AEs was driven by the rate of systemic AEs during treatment with IgHy10, which was not aligned with the expectations of the clinical experts consulted by CADTH. A notable limitation of this evidence is that the proportion of patients reporting an AE is limited to epoch 2 of Study 160603. For patients receiving IgHy10 following the ramp-up period in Study 160603, 53% of patients experienced at least 1 local AE. The most common AEs were infusion site pain (33% of patients), infusion site erythema (12%), and infusion site discomfort (10%). The proportion of patients with at least 1 systemic AE was not reported, but headache (30% of patients), asthma (17%), nausea (14%), pyrexia (14%), and fatigue (14%) were the most frequently reported individual systemic AEs. Again, the clinical experts indicated that the rate and proportion of patients reporting systemic AEs with IgHy10 was much higher than what is observed in clinical practice with cSCIg treatment. This suggests that IgHy10 may not offer a safety profile similar to cSCIg, as was anticipated; however, this is unknown without comparative evidence. Additionally, a post hoc integrated analysis of safety outcomes from Study 160603 and Study 160902 reported systemic AEs and local AEs among adult patients (aged ≥ 18 years) specifically. The rate of AEs in adult patients was similar to what was reported for the overall population in the individual studies, which included patients younger than 18 years of age, as well.
It should also be noted that temporally associated AEs (summarized in Appendix 4) were aligned with the results that have been discussed, but limited to data for overall AEs, local AEs, and systemic AEs. As noted by the clinical experts, the majority of patients treated with cSCIg administer treatment at home or without supervision from a health care professional, which was not the case in the included studies. The increased level of supervision may have impacted the frequency of AEs reported during the trial; however, whether AEs were overestimated in the trial or the AEs were underreported in clinical practice is unknown.
Two of the 3 other relevant studies summarized in this review provided additional safety data in pediatric patients and pregnant patients and their infants. Study 161504 (N = 42) was a single-arm phase IV trial that was conducted to collect post-authorization safety, tolerability, and immunogenicity data on the use of IgHy10 in pediatric patients. The most frequently reported AEs were cough (29% of patients), infusion site pain (17%), and pyrexia (14%). Evidence of immunogenicity (anti-rHuPH20 antibodies) was not observed in any patients. Additionally, only 1 patient experienced a serious acute bacterial infection. Study 161301 was a post-marketing pregnancy registry study designed to collect clinical safety data on the course and outcome of pregnancy among women who had ever been treated with IgHy10, and their infants. Overall, no major safety signals were observed; however, this is based on a very small sample size (fewer than 20 patients). Lastly, NCT02881437 (previously described) did not suggest any new safety signals associated with IgHy10 following a switch from cSCIg, based on physician record data.
The 2014 FDA assessment of IgHy10 summarized the safety profile of IgHy10 as being acceptable.51 Further, the FDA described the short-term risks as acceptable, but the long-term risks associated with antibodies to rHuPH20 remain unknown. The FDA report noted that Study 160603, Study 160902, and Study 160601 included a total of 26 pediatric patients (defined as patients from 2 years old to younger than 12 years), but that safety had not been established in pediatric patients. The European Medicines Agency originally restricted the use of IgHy10 to adults with safety concerns similar to those of pediatric patients, but has since authorized the use of IgHy10 as a replacement therapy in adults, children, and adolescents (from 0 to 18 years old).52,53 Specifically, IgHy10 is indicated for use in patients with PID syndromes with impaired antibody production and in specific populations with hypogammaglobulinemia due to an SID. In the US, HyQvia is currently restricted to the treatment of adults with a PID.
Conclusions
The pivotal trial (Study 160603) and related extension study (Study 160902) provided evidence on the efficacy and safety of IgHy10 as replacement therapy in patients with a PID requiring antibody replacement therapy. The rate of VASBIs was less than 1.0 infections per patient per year (P < 0.0001), which was aligned with expectations for treatment with IgRT in patients with a PID, according to the experts consulted by CADTH. The rate of all infections per patient per year was less than 3 during treatment with IgHy10 in Study 160603 and Study 160902. Additionally, the monthly rate of days spent in hospital due to infection was minimal (0.03 days per month or less), which was considered clinically important as per feedback from the clinical experts consulted by CADTH. HRQoL is an outcome of importance to patients; however, the results provided in the included studies do not allow for conclusions regarding a change in HRQoL during IgHy10 treatment. Regarding the safety of IgHy10, 2 deaths deemed unrelated to treatment were reported during the extension study, and SAEs were infrequent overall. Significant safety concerns were not identified for IgHy10.
The lack of comparative evidence (direct or indirect) for IgHy10 versus other IgRT represents a major limitation in the context of this review. Conclusions cannot be drawn about how IgHy10 compares with other IgRTs, and the single-group study design hinders the ability to appropriately interpret the reported outcomes. Also of note, the available evidence excluded patients with secondary humoral immunodeficiency. The clinical experts consulted by CADTH did not express any issues with using evidence in patients with primary humoral immunodeficiency to inform treatment decisions for patients with secondary humoral immunodeficiency, although this still represents a gap in the evidence. In summary, there are many other SCIg and IVIg therapies available in Canada. Although IgHy10 offers a unique therapy because it comprises 2 drug products (IgG facilitated by rHuPH20), how IgHy10 differs from other immunoglobulin products in terms of safety and efficacy is currently unknown. Therefore, IgHy10 was viewed as another option for SCIg treatment.
References
1.McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):61. PubMed
2.Immunodeficiency Canada. Primary immunodeficiency (PI). 2021; https://immunodeficiency.ca/primary-immunodeficiency/primary-immunodeficiency-pi/. Accessed 2021 Sep 10.
3.Fernandez J. Overview of immunodeficiency disorders. Merck Manual 2021; https://www.merckmanuals.com/en-ca/professional/immunology-allergic-disorders/immunodeficiency-disorders/overview-of-immunodeficiency-disorders#v27389893. Accessed 2021 Sep 10.
4.Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186-1205.e1178. PubMed
5.Betschel S, Dent P, Haddad E, et al. National Immunoglobulin replacement Expert Committee recommendations. LymphoSign J. 2017;4(3):117-118.
6.Chinn I. Primary humoral immunodeficiency. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2021 Sep 10.
7.Patel SY, Carbone J, Jolles S. The expanding field of secondary antibody deficiency: causes, diagnosis, and management. Front Immunol. 2019;10:33. PubMed
8.Jolles S. Subcutaneous and intramuscular immune globulin therapy. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2021 Sep 10.
9.Bundy V, Barbieri K, Keller M. Primary immunodeficiency: overview of management. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2021 Sep 10.
10.HyQvia® (normal immunoglobulin [human]) 10% 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL and recombinant human hyaluronidase 200 units/1.25 mL, 400 units/2.5 mL, 800 units/5 mL, 1600 units/10 mL and 2400 units/15 mL) solution for subcutaneous infusion [product monograph]. Toronto (ON): Takeda Canada Inc.; 2022 Jan.
11.U.S. Food and Drug Administration and Center for Biologics Evaluation and Research. Guidance for industry: safety, efficacy, and pharmacokinetic studies to support marketing of immune globulin intravenous (human) as replacement therapy for primary humoral immunodeficiency. Rockville (MD): U.S. Department of Health and Human Services, Food and Drug Administration; 2008: https://www.fda.gov/media/124333/download. Accessed 2021 Sep 10.
12.Wasserman RL, Melamed I, Stein MR, et al. Recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulins for primary immunodeficiency. J Allergy Clin Immunol. 2012;130(4):951-957.e911. PubMed
13.Wasserman RL, Melamed I, Stein MR, et al. Long-term tolerability, safety, and efficacy of recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulin for primary immunodeficiency. J Clin Immunol. 2016;36(6):571-582. PubMed
14.Drug Reimbursement Review sponsor submission: HyQvia® (normal immunoglobulin (human) 10% 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL and recombinant human hyaluronidase 200 units/1.25 mL, 400 units/2.5 mL, 800 units/5 mL, 1600 units/10 mL and 2400 units/15 mL) solution for subcutaneous infusion [internal sponsor's package]. Toronto (ON): Takeda Canada Inc.; 2021 Jun 24.
15.Clinical Study Report: 160603. Efficacy, tolerability and pharmacokinetic comparison of Immune Globulin Intravenous (Human), 10% (GAMMAGARD LIQUID/KIOVIG) administered intravenously or subcutaneously following administration of recombinant human hyaluronidase (rHuPH20) in subjects with primary immunodeficiency diseases [internal sponsor's report]. Westlake Village (CA): Baxter Healthcare Corporation; 2011 May 16.
16.Clinical Study Report: 160902. Long-term tolerability and safety of immune globulin subcutaneous solution (IGSC) administered subcutaneously following administration of recombinant human hyaluronidase (rHuPH20) in subjects with primary immunodeficiency diseases [internal sponsor's report]. Westlake Village (CA): Baxter Healthcare Corporation; 2014 Feb 3.
17.Clinical Study Report: 2015_31. Assessment of the IgG trough level in subjects with primary immunodeficiency switching from standard subcutaneous immunoglobulin (SCIG) to HyQvia every other week [internal sponsor's report]. Lille (FR): CHRU de Lille; 2020 Apr 27.
18.Picard C, Al-Herz W, Bousfiha A, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35(8):696-726. PubMed
19.Ciznar P, Roderick M, Schneiderova H, et al. Real-world safety and tolerability of facilitated subcutaneous immunoglobulin in pediatric patients with primary immunodeficiency diseases: interim analysis from a post-authorization study in Europe. J Clin Immunol. 2021;41(Suppl 1):S97-S98.
20.Montori VM, Devereaux PJ, Adhikari NK, et al. Randomized trials stopped early for benefit: a systematic review. JAMA. 2005;294(17):2203-2209. PubMed
21.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-1187. PubMed
22.Rubinstein A, Bridges T, Wedner HJ, et al. Infusion characteristics, adverse events, and patient preference during facilitated subcutaneous immunoglobulin treatment for primary immunodeficiency diseases: interim analysis of a global postauthorization safety study [internal sponsor's package]. Poster session presented at: Latin American Society for Immunodeficiencies meeting; October 9–12, 2019; Cancun, Mexico.
23.Ellerbroek PM, van Paassen P, Hanitsch LG, et al. Interim analysis of a postauthorization safety study on the long-term safety of hyaluronidase-facilitated subcutaneous immunoglobulin 10% in patients with primary immunodeficiency diseases in Europe. Poster session presented at: European Society for Immunodeficiencies Annual Meeting; September 18–21, 2019; Brussels, Belgium.
24.Baxalta Innovations GmbH. 161302: Non-interventional post-authorization safety study on the long-term safety of HyQvia in subjects treated with HyQvia. Amsterdam (NL): European Network of Centres for Pharmacoepidemiology and Pharmacovigilance; 2021: https://www.encepp.eu/encepp/viewResource.htm?id=26136. Accessed 2022 Jun 7.
25.Borte M, Hanitsch LG, Mahlaoui N, et al. Facilitated Immunoglobulin Administration Registry and Outcomes Study (FIGARO): interim results. Poster session presented at: European Society for Immunodeficiencies Annual Meeting; September 18–21, 2019; Brussels, Belgium.
26.Dimou M, Speletas M, Fasshauer M, et al. Facilitated immunoglobulin administration registry and outcomes study (FIGARO): interim results in pediatric, adult, and older-adult patients. Poster session presented at: European Society for Immunodeficiencies 19th Biennial Meeting, Online Meeting; October 14–17, 2020.
27.Dimou M, Speletas M, Quinti I, et al. Facilitated immunoglobulin administration registry and outcomes study (FIGARO): interim results in patients with primary or secondary immunodeficiency diseases. Poster session presented at: European Society for Immunodeficiencies 19th Biennial Meeting, Online Meeting; October 14–17, 2020.
28.Wasserman RL, Gupta S, Stein M, et al. Infection rates and tolerability of three different immunoglobulin administration modalities in patients with primary immunodeficiency diseases. Immunotherapy. 2022;14(4):215-224. PubMed
29.Gammagard S/D (immune globulin intravenous (human) [IGIV], solvent/detergent-treated (freeze-dried concentrate): 5 g/vial & 10 g/vial [product monograph]. Toronto (ON): Shire Pharma Canada ULC; 2018: https://pdf.hres.ca/dpd_pm/00046330.PDF. Accessed 2021 Sep 10.
30.Gammagard Liquid (immune globulin intravenous (human), [IGIV] 10%): solution for infusion and 1 g/10 mL, 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300mL [product monograph]. Toronto (ON): Shire Pharma Canada ULC; 2018: https://pdf.hres.ca/dpd_pm/00045116.PDF. Accessed 2021 Sep 10.
31.IGIVnex® (immune globulin intravenous (human), 10%): injectable solution [product monograph]. Mississauga (ON): Grifols Canada Ltd.; 2019: https://www.grifols.com/documents/89713601/0/IGIVnex+-+English+PM+-+2017-07-12.pdf/98eb0376-0d61-491a-9fda-713505c61379. Accessed 2021 Sep 14.
32.Gamunex (immune globulin intravenous (human), 10%): injectable solution [product monograph]. Mississauga (ON): Grifols Canada Ltd.; 2016: https://pdf.hres.ca/dpd_pm/00033486.PDF. Accessed 2021 Sep 10.
33.Cuvitru (normal immune globulin (human)): 200 mg/mL (20%) solution for subcutaneous infusion [product monograph]. Toronto (ON): Shire Pharma Canada ULC; 2018: https://pdf.hres.ca/dpd_pm/00044942.PDF. Accessed 2021 Sep 10.
34.Hizentra® (subcutaneous immunoglobulin (human)): 20% solution for injection (200mg/mL) [product monograph]. Ottawa (ON): CSL Behring Canada, Inc.; 2020: https://labeling.cslbehring.ca/PM/CA/Hizentra/EN/Hizentra-Product-Monograph.pdf. Accessed 2021 Sep 10.
35.Cutaquig® (Immunoglobulin (human) subcutaneous 16.5% solution for injection (165 mg/mL), subcutaneous use): 6 mL, 10 mL, 12 mL, 20 mL, 24 mL, 48 mL [product monograph]. Toronto (ON): Octapharma Canada Inc.; 2021: https://pdf.hres.ca/dpd_pm/00061820.PDF. Accessed 2021 Sep 10.
36.Privigen® (intravenous immunoglobulin (human) 10%): solution for infusion and intravenous [product monograph]. Ottawa (ON): CSL Behring Canada, Inc.; 2021: https://labeling.cslbehring.ca/pm/ca/privigen/en/privigen-product-monograph.pdf. Accessed 2021 Sep 10.
37.Panzyga® (immune globulin intravenous (human) solution for infusion, 100 mg/mL): 10 mL, 25 mL, 50 mL, 100 mL, 200 mL and 300 mL [product monograph]. Toronto (ON): Octapharma Canada Inc.; 2017: https://pdf.hres.ca/dpd_pm/00041520.PDF. Accessed 2021 Sep 10.
38.Octagam® (5% imunoglobulin intravenous (human)): solution for infusion, 50 mg/mL [product monograph]. Toronto (ON): Octapharma Canada Inc.; 2018: https://pdf.hres.ca/dpd_pm/00048166.PDF. Accessed 2021 Sep 10.
39.Xembify (subcutaneous immunoglobulin (human): 20% solution for subcutaneous use [product monograph]. Grifols Canada Ltd.: Mississauga (ON); 2019: https://pdf.hres.ca/dpd_pm/00054232.PDF. Accessed 2021 Sep 23.
40.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
41.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Jul 19.
42.Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120(4):776-794. PubMed
43.IUIS Scientific Committee. Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol. 1999;118 Suppl 1(Suppl 1):1-28.
44.Wasserman RL, Melamed I, Kobrynski L, et al. Recombinant human hyaluronidase facilitated subcutaneous immunoglobulin treatment in pediatric patients with primary immunodeficiencies: long-term efficacy, safety and tolerability. Immunotherapy. 2016;8(10):1175-1186. PubMed
45.Committee for Medicinal Products for Human Use. Guideline on the clinical investigation of human normal immunoglobulin for intravenous administration (IVIg). London (GB): European Medicines Agency; 2010: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-human-normal-immunoglobulin-intravenous-administration-ivig_en.pdf. Accessed 2021 Sep 10.
46.Glodstein B, Giroir B, Randolph A. International Pediatric Sepsis Consensus Conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6(1):2-8. PubMed
47.Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med Care. 1999;37(2):126-139. PubMed
48.Ware J, Kosinski M, Bjorner J, Turner-Bowker D, Gandek B, Maruish M. Determining important differences in scores. User’s Manual for the SF-36v2 Health Survey. Lincoln (RI): QualityMetric Incorporated; 2007.
49.Guidance Document: regulatory requirements for intravenous immunoglobulin products in Canada. Ottawa (ON): Health Canada; 2018: https://www.canada.ca/content/dam/phac-aspc/documents/services/drug-health-medication/Blood-Guidance-IVIG%20English-20180827.pdf. Accessed 2021 Nov 1.
50.Clinical Study Report: 161301. Pregnancy registry to collect long-term safety data from women treated with HyQvia [internal sponsor's report]. Lexington (MA): Baxalta US Inc.; 2021 Jan 13.
51.Center for Drug Evaluation Research. Multidiscipline review(s). HyQvia® (Normal Immunoglobulin (Human) 10% 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL and Recombinant Human Hyaluronidase 200 Units/1.25 mL, 400 Units/2.5 mL, 800 Units/5 mL, 1600 Units/10 mL and 2400 Units/15 mL) Solution for Subcutaneous Infusion. Company: Baxter Healthcare Corporation. Application No.: BL 125402/0. Approval date: 09/12/2014 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2014: https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/hyqvia. Accessed 2021 Sep 10.
52.Committee for Medicinal Products for Human Use. HyQvia (human normal immunoglobulin): Summary of opinion (initial authorisation). Amsterdam (NL): European Medicines Agency; 2013 Mar 21: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-hyqvia_en.pdf. Accessed 2021 Sep 10.
53.Committee for Medicinal Products for Human Use. HyQvia (human normal immunoglobulin): Summary of opinion (post authorisation). Amsterdam (NL): European Medicines Agency; 2016 Apr 28: https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-hyqvia_en.pdf. Accessed 2021 Sep 10.
54.McHorney CA, Ware JE, Jr., Raczek AE. The MOS 36-Item Short-Form Health Survey (SF-36): II. Psychometric and clinical tests of validity in measuring physical and mental health constructs. Med Care. 1993;31(3):247-263. PubMed
55.Brazier JE, Harper R, Jones NM, et al. Validating the SF-36 health survey questionnaire: new outcome measure for primary care. BMJ. 1992;305(6846):160-164. PubMed
56.McHorney CA, Ware JE, Jr., Lu JF, Sherbourne CD. The MOS 36-item Short-Form Health Survey (SF-36): III. Tests of data quality, scaling assumptions, and reliability across diverse patient groups. Med Care. 1994;32(1):40-66. PubMed
57.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3(6):329-341. PubMed
58.Varni JW, Seid M, Kurtin PS. PedsQL 4.0: reliability and validity of the Pediatric Quality of Life Inventory version 4.0 generic core scales in healthy and patient populations. Med Care. 2001;39(8):800-812. PubMed
59.Varni JW, Seid M, Knight TS, Uzark K, Szer IS. The PedsQL 4.0 Generic Core Scales: sensitivity, responsiveness, and impact on clinical decision-making. J Behav Med. 2002;25(2):175-193. PubMed
60.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
61.Desai AD, Zhou C, Stanford S, Haaland W, Varni JW, Mangione-Smith RM. Validity and responsiveness of the pediatric quality of life inventory (PedsQL) 4.0 generic core scales in the pediatric inpatient setting. JAMA Pediatr. 2014;168(12):1114-1121. PubMed
62.Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30(6):473-483. PubMed
63.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1(8476):307-310. PubMed
64.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
65.Tcheurekdjian H, Palermo T, Hostoffer R. Quality of life in common variable immunodeficiency requiring intravenous immunoglobulin therapy. Ann Allergy Asthma Immunol. 2004;93(2):160-165. PubMed
66.Frendl DM, Ware JE, Jr. Patient-reported functional health and well-being outcomes with drug therapy: a systematic review of randomized trials using the SF-36 health survey. Med Care. 2014;52(5):439-445. PubMed
67.Baxalta. NCT03116347: Post-authorization safety, tolerability and immunogenicity evaluation of HyQvia in pediatric PIDD subjects. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT03116347. Accessed 2021 Sep 10.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 22, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
“ | Surrounding a word or phrase, searches as an exact phrase; used to treat reserved words (words normally used as operators) as regular search terms |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(IgHy10* or IGHy* or fSCIG* or f-SCIG*).ti,ab,kf,ot,hw,rn,nm.
(rHuPH20 or rHuPH 20 or Enhanze* or 743QUY4VD8 or ((recombinant* or human* or facilitated) adj3 hyaluronidase)).ti,ab,kf,ot,hw,rn,nm.
Immunoglobulins/ or Immunoglobulins, Intravenous/ or Immunoglobulin G/ or gamma globulins/
(Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV or SCIG or SCIGG or IGSC or 66Y330CJHS).ti,ab,kf,ot,hw,rn,nm.
3 or 4
2 and 5
facilitated.ti,ab,kf,ot,hw,rn,nm.
(infusions, subcutaneous/ or injections, subcutaneous/ or subcutaneous tissue/) and (Immunoglobulins/ or Immunoglobulins, Intravenous/ or Immunoglobulin G/ or gamma globulins/)
(infusions, subcutaneous/ or injections, subcutaneous/ or subcutaneous tissue/) and (Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV or 66Y330CJHS).ti,ab,kf,ot,hw,rn,nm.
(((subcutaneous* or “sub cutaneous*” or subQ) adj3 (Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV or 66Y330CJHS)) or SCIG or SCIGG or SC IG or SC IGG or IGSC).ti,ab,kf,ot,hw,rn,nm.
8 or 9 or 10
7 and 11
1 or 6 or 12
13 use medall
(IgHy10* or IGHy* or fSCIG* or f-SCIG*).ti,ab,kw,dq.
recombinant hyaluronidase/
(rHuPH20 or rHuPH 20 or Enhanze* or ((recombinant* or human* or facilitated) adj3 hyaluronidase)).ti,ab,kw,dq.
16 or 17
immunoglobulin/ or human immunoglobulin/ or immunoglobulin G/ or immunoglobulin G1/ or immunoglobulin G2/ or immunoglobulin G2a/ or immunoglobulin G2b/ or immunoglobulin G3/ or immunoglobulin G4/
(Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV or SCIG or SCIGG or IGSC).ti,ab,kw,dq.
19 or 20
18 and 21
facilitated.ti,ab,kw,dq.
subcutaneous immunotherapy/ or immunoglobulin/sc or human immunoglobulin/sc
subcutaneous drug administration/ and (immunoglobulin/ or human immunoglobulin/ or immunoglobulin G/ or immunoglobulin G1/ or immunoglobulin G2/ or immunoglobulin G2a/ or immunoglobulin G2b/ or immunoglobulin G3/ or immunoglobulin G4/)
subcutaneous drug administration/ and (Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV).ti,ab,kw,dq.
(((subcutaneous* or “sub cutaneous*” or subQ) adj3 (Immunoglobulin* or immune globulin* or gammaglobulin* or gamma globulin* or IgG* or IG or NHIG or HNIG or IVIG or IGIV)) or SCIG or SCIGG or SC IG or SC IGG or IGSC).ti,ab,kw,dq.
24 or 25 or 26 or 27
23 and 28
15 or 22 or 29
30 not conference abstract.pt.
31 use oemezd
14 or 32
remove duplicates from 33
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search – IgHy10, IgHy, fSCIG, (facilitated OR hyaluronidase OR rHuPH20 OR Enhanze) AND (immunoglobulin OR immunoglobulins OR “immune globulin” OR “immune globulins” OR IVIG OR SCIG OR SCIGG OR IGSC OR IGIV)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms – IgHy10, IgHy, fSCIG, (facilitated, hyaluronidase, rHuPH20, Enhanze) AND (immunoglobulin*, “immune globulin,” “immune globulins,” SCIG)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – IgHy10, ighy, immunoglobulin, immunoglobulins, immune globulin, immune globulins, scig, fscig]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – IgHy10, IgHy, fSCIG, (facilitated, hyaluronidase, rHuPH20, Enhanze) AND (immunoglobulin, immunoglobulins, “immune globulin,” “immune globulins,” IVIG, SCIG, SCIGG, IGIV, IGSC)]
Grey Literature
Search dates: July 14 to 22, 2021
Keywords: IgHy10, IgHy, fSCIG, (facilitated subcutaneous OR hyaluronidase facilitated OR recombinant human hyaluronidase) AND (immunoglobulin OR immunoglobulins OR immune globulin OR immune globulins OR IVIG OR SCIG)
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Al-Zuhairy A, Jakobsen J, Andersen H, Sindrup SH, Markvardsen LK. Randomized trial of facilitated subcutaneous immunoglobulin in multifocal motor neuropathy. Eur J Neurol. 2019 10;26(10):1289-e1282. PubMed: PM31021036 | Study population |
Angelotti F, Capecchi R, Giannini D, Mazzarella O, Rocchi V, Migliorini P. Long-term efficacy, safety, and tolerability of recombinant human hyaluronidase-facilitated subcutaneous infusion of immunoglobulin (Ig) (fSCIg; IgHy10(R) in immunodeficiency diseases: real-life data from a monocentric experience. Clin Exp Med. 2020 Aug;20(3):387-392. PubMed: PM32385734 | Study design |
Blau IW, Conlon N, Petermann R, Nikolov N, Plesner T. Facilitated subcutaneous immunoglobulin administration (fSCIg): a new treatment option for patients with secondary immune deficiencies. Expert Rev Clin Immunol. 2016;12(7):705-711. PubMed: PM27156362 | Study design |
Danieli MG, Pulvirenti F, Rocchi V, Morariu R, Quinti I. Self-administered hyaluronidase-facilitated subcutaneous immunoglobulin therapy in complicated primary antibody deficiencies. Immunotherapy. 2016;8(9):995-1002. PubMed: PM27485073 | Study population |
Hustad NB, Degerud HM, Hjelmerud I, et al. Real-world experiences with facilitated subcutaneous immunoglobulin substitution in patients with hypogammaglobulinemia, using a three-step ramp-up schedule. Front Immunol. 2021;12:670547. PubMed: PM34012453 | Study design |
Jolles S. Hyaluronidase facilitated subcutaneous immunoglobulin in primary immunodeficiency. ImmunoTargets Ther. 2013;2:125-133. PubMed: PM27471693 | Review article |
Paassen PV, Pittrow D, Scheidegger C, Klotsche J, Ellerbroek PM. Use of recombinant human hyaluronidase-facilitated subcutaneous immunoglobulin in elderly patients. Immunotherapy. 2020 02;12(2):131-139. PubMed: PM32066296 | Study design |
Petersson C, Fust R, Hagstedt C, Wagstrom P, Nilsdotter-Augustinsson A. “Experiences of the burden of treatment”-Patient reports of facilitated subcutaneous immunoglobulin treatment in adults with immunodeficiency. J Clin Nurs. 2018 Dec;27(23-24):4270-4278. PubMed: PM29917296 | Study design |
Rosengren S, Dychter SS, Printz MA, et al. Clinical immunogenicity of rHuPH20, a hyaluronidase enabling subcutaneous drug administration. AAPS J. 2015;17(5):1144-1156. PubMed: PM25967925 | Study design |
Sanford M. Human immunoglobulin 10% with recombinant human hyaluronidase: replacement therapy in patients with primary immunodeficiency disorders. Biodrugs. 2014;28(4):411-420. PubMed: PM24925799 | Review article |
Wasserman RL, IgHy10 Experience Study Group. Clinical practice experience with IgHy10 in adults using alternative dosing regimens and pediatric patients: a retrospective study. Adv Ther. 2020 04;37(4):1536-1549. PubMed: PM32124273 | Study design |
Wasserman RL, Melamed I, Stein MR, et al. Recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulins for primary immunodeficiency. J Allergy Clin Immunol. 2012 October;130(4):951-957.e911. PubMed: PM22846381 | Duplicate study |
Wasserman RL. Overview of recombinant human hyaluronidase-facilitated subcutaneous infusion of IgG in primary immunodeficiencies. Immunotherapy. 2014;6(5):553-567. PubMed: PM24896624 | Review article |
Wasserman RL. Recombinant human hyaluronidase-facilitated subcutaneous immunoglobulin infusion in primary immunodeficiency diseases. Immunotherapy. 2017 09;9(12):1035-1050. PubMed: PM28871852 | Study design |
Wasserman RL. Subcutaneous immunoglobulin: facilitated infusion and advances in administration. Clin Exp Immunol. 2014;178 Suppl 1:75-77. PubMed: PM25546770 | Editorial |
fSCIg = facilitated subcutaneous immunoglobulin; IgG = immunoglobulin; rHuPH20 = recombinant human hyaluronidase.
Appendix 3: Detailed Outcome Data
Additional Efficacy Data
Table 34: Pharmacokinetic Parameters for Immunoglobulin in Relation to Dose Frequency in Extension Study 160902 (SAS)
Detail | Study 160902 (N = 66) |
|---|---|
Steady-state trough levels (g/L) maintained under SC with rHuPH20 treatment by infusion interval | |
2-week interval | |
N analyzed | 17 |
Median (95% CI) | 10.900 (9.390 to 13.300) |
3-week interval | |
N analyzed | 9 |
Median (95% CI) | 12.300 (11.500 to 15.300) |
4-week interval | |
N analyzed | 47 |
Median (95% CI) | 9.670 (9.260 to 10.700) |
Percent change of steady-state trough levels for patients who changed to a 2-week interval under SC with rHuPH20 treatment | |
3-week (previous interval) | |
N analyzed | 2 |
Mean (SD) | 105.90 (0.03)% |
Median (95% CI) | 105.90 (NA)% |
4-week (previous interval) | |
N analyzed | 9 |
Mean (SD) | 113.23 (13.02)% |
Median (95% CI) | 112.44 (99.31 to 129.13)% |
Ratio of IgG trough levels (g/L) at the end of safety follow-up vs. at end of SC with rHuPH20 treatment | |
SC at end of safety follow-up (total) | |
N analyzed | 9 |
Median (95% CI) | 117.8 (104.4 to 145.7)% |
IV at end of safety follow-up (total) | |
N analyzed | 38 |
Median (95% CI) | 102.5 (97.7 to 109.8)% |
SC at end of safety follow-up (2-week interval) | |
N analyzed | 6 |
Median (95% CI) | 108.5 (92.7 to 145.7)% |
IV at end of safety follow-up (2-week interval) | |
N analyzed | 6 |
Median (95% CI) | 94.6 (90.3 to 96.5)% |
SC at end of safety follow-up (3-week interval) | |
N analyzed | NA |
Median (95% CI) | NA |
IV at end of safety follow-up (3-week interval) | |
N analyzed | 5 |
Median (95% CI) | 11.7 (NA)% |
SC at end of safety follow-up (4-week interval) | |
N analyzed | 3 |
Median (95% CI) | 142.7 (NA)% |
IV at end of safety follow-up (4-week interval) | |
N analyzed | 38 |
Median (95% CI) | 103.1 (99.3 to 111.5)% |
CI = confidence interval; IgG = immunoglobulin; rHuPH20 = recombinant human hyaluronidase; SC = subcutaneous; SD = standard deviation.
Source: Clinical Study Report.16
Additional Harms Data
Table 35: Summary of the Most Commonly Reported Harms by Rate of AEs per Infusion in Study 160902 (SAS)
Detail | Study 160902 | |
|---|---|---|
160603 ext: IgHy10 (n = 63) | Safety follow-up: IgHy10 by IV or SC (n = 66) | |
Most common AEsa by percentage of patients | ||
Patients with AEs, n (%) | 63 (100.0) | 51 (77.3) |
Most common AEsa by rate per infusion | ||
Total number of infusions | 1,600 | 598 |
Infusions with AEs, number of AEs (rate per infusion) | ||
All AEs | 1,244 (0.778) | 407 (0.681) |
Local AEs | 603 (0.377) | 25 (0.042) |
Systemic AEs including infections | 1,079 (0.674) | 382 (0.639) |
Systemic AEs excluding infections | 801 (0.501) | 300 (0.502) |
Infusion site pain | 84 (0.053) | 19 (0.032) |
Sinusitis | 63 (0.039) | 16 (0.027) |
Headache | 47 (0.029) | 41 (0.069) |
Nausea | 46 (0.029) | 25 (0.042) |
Asthma | 32 (0.020) | 6 (0.010) |
Infusion site pruritus | 31 (0.019) | 0 |
Upper respiratory tract infection | 29 (0.018) | 10 (0.017) |
Myalgia | 28 (0.018) | 8 (0.013) |
Fatigue | 27 (0.017) | 23 (0.038) |
Infusion site erythema | 23 (0.014) | 0 |
Bronchitis | 23 (0.014) | 7 (0.012) |
Diarrhea | 19 (0.012) | rate < 0.010 |
Vomiting | 17 (0.011) | rate < 0.010 |
Asthenia | 16 (0.010) | rate < 0.010 |
Pyrexia | 16 (0.010) | rate < 0.010 |
Decreased appetite | rate < 0.010 | 8 (0.013) |
Migraine | rate < 0.010 | 8 (0.013) |
Arthralgia | rate < 0.010 | 7 (0.012) |
Peripheral edema | rate < 0.010 | 6 (0.010) |
Pruritus | rate < 0.010 | 6 (0.010) |
Urinary tract infection | rate < 0.010 | 6 (0.010) |
Viral upper respiratory tract infection | rate < 0.010 | 6 (0.010) |
aFrequency ≥ 10% of patients or ≥ 0.01 AEs per infusion.
Source: Clinical Study Report.16
Table 36: Summary of Harms — Temporally Associated AEs and All AEsa in Study 160603 (SAS)
Detail | Study 160603 | |
|---|---|---|
Epoch 1: IVIg (n = 87) | Epoch 2: IgHy10 (n = 83) | |
Temporally associated AEsb | ||
Patients with AEs | ||
N analyzed | 87 | 81 |
≥ 1 AE, including infections, n (%) | 58 (66.7) | 70 (86.4) |
≥ 1 AE, excluding infections, n (%) | 55 (63.2) | 67 (82.7) |
≥ 1 systemic AE, including infections, n (%) | 56 (64.4) | 61 (75.3) |
≥ 1 systemic AE, excluding infections, n (%) | 53 (60.9) | 55 (67.9) |
≥ 1 local AE, including infections, n (%) | 4 (4.6) | 42 (51.9) |
≥ 1 local AE, excluding infections, n (%) | 4 (4.6) | 42 (51.9) |
Infusions with AEs | ||
N analyzed | 365 | 1,129 |
≥ 1 AE, including infections, n (%) | 110 (30.1) | 277 (24.5) |
≥ 1 AE, excluding infections, n (%) | 105 (28.8) | 257 (22.8) |
≥ 1 moderate or severe AE, including infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 5.9 (0.0 to 7.1) |
≥ 1 moderate or severe AE, excluding infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 6.3) |
≥ 1 systemic AE, including infections, median rate (95% CI) | 25.0 (20.0 to 25.0) | 8.3 (7.7 to 12.5) |
≥ 1 systemic AE, excluding infections, median rate (95% CI) | 25.0 (16.7 to 25.0) | 8.3 (6.3 to 10.0) |
≥ 1 local AE, including infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 5.9 (0.0 to 8.3) |
≥ 1 local AE, excluding infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 5.9 (0.0 to 8.3) |
AEs at any time during the study | ||
Patients with AEs | ||
N analyzed | 87 | 81 |
≥ 1 local AE, including infections, n (%) | 5 (5.7) | 43 (53.1) |
≥ 1 local AE, excluding infections, n (%) | 5 (5.7) | 43 (53.1) |
Infusions with AEs | ||
N analyzed | 365 | 1,129 |
≥ 1 local AE, including infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 5.9 (0.0 to 8.3) |
≥ 1 local AE, excluding infections, median rate (95% CI) | 0.0 (0.0 to 0.0) | 5.9 (0.0 to 8.3) |
AE = adverse event; IVIg = intravenous immunoglobulin; rHuPH20 = recombinant human hyaluronidase; SCIg = subcutaneous immunoglobulin
aOutcomes calculated using descriptive statistics.
bTemporally associated events were those that occurred during or within 72 hours following the infusion.
Source: Clinical Study Report.15
Table 37: Summary of Harms in Study 160902 (SAS)
Detail | Study 160902a | ||
|---|---|---|---|
160603 ext: IgHy10 (n = 63) | Safety follow-up: IgHy10 by IV or SC (n = 66) | IgHy10 + safety follow-up (n = 66) | |
Temporally associated AEsb | |||
All AEs | |||
N patients analyzed | 63 | 66 | 66 |
N infusions analyzed | 1,600 | 598 | 2,198 |
Number of all AEs (rate per patient) [rate per infusion] | 477 (7.095) [0.279] | NR | NR |
Number of local AEs (rate per patient) [rate per infusion] | 165 (2.619) [0.103] | NR | NR |
Number of systemic AEs including infections (rate per patient) [rate per infusion] | 282 (4.476) [0.176] | NR | NR |
Number of systemic AEs excluding infections (rate per patient) [rate per infusion]) | 254 (4.032) [0.159] | NR | NR |
AEs at any time during the study | |||
Patients with AEs | |||
N analyzed | 63 | 66 | 66 |
≥ 1 AE, n (%) | 63 (100.0) | 51 (77.3) | NR |
Rate per patient | 19.75 | 7.980 | NR |
Infusions with AEs | |||
N analyzed | 1,600 | 598 | 2,198 |
Number of AEs (rate per infusion) | 1,244 (0.7775) | 407 (0.6806) | 1,651 (0.7511) |
≥ local AEs, n (%) | 140 (8.8) | 22 (3.7) | 162 (7.4) |
Serious AEs | |||
Patients with serious AEs | |||
Total number of patient-years c | 97.61 | 31.66 | 129.27 |
Number of SAEs | 15 | 3 | 18 |
Annual rate of SAEs c | 0.154 | 0.095 | 0.139 |
Infusions with serious AEs | |||
Total number of infusions | 1,600 | 598 | 2,198 |
Number of serious AEs (rate per infusiond) | 15 (0.0094) | 3 (0.0050) | 18 (0.0082) |
AE = adverse event; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; NR = not reported; rHuPH20 = recombinant human hyaluronidase; SC = subcutaneous; SCIg = subcutaneous immunoglobulin.
aOutcomes calculated using descriptive statistics.
bTemporally associated events were those that occurred during or within 72 hours following the infusion.
cPatient-years under respective treatment = Interval between the patient’s first SC+rHuPH20 treatment in 160902 and either a disposition event OR the date of the first treatment without rHuPH20 for the SC with rHuPH20 phase versus interval between first treatment in the safety follow-up and a disposition event.
cTotal number of SAEs divided by the total number of patient-years in the study phase
dTotal number of SAEs divided by the total number of infusions in the study phase
Source: Clinical Study Report.16
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference [MID]):
SF-36
PedsQL
Findings
Table 38: Summary of Outcome Measures and Their Measurement Properties
Type | Conclusions about measurement properties | MID |
|---|---|---|
Short Form (36) Health Survey (SF-36): Exploratory end point | ||
36-item self-reported generic health-related quality of health measure for adults | No data specific to immunodeficiency were identified. Validity: Construct validity demonstrated by the known group method. Patients with serious medical conditions scored significantly (P < 0.01) lower on all scales compared with those with minor conditions.54 People with chronic physical health problems have shown poorer GH scores than those without (GH score 53 vs. 66).55 Reliability: Acceptable internal consistency reliability (Cronbach alpha ≥ 0.70) for all scales and ≥ 0.90 for the MH and PF domains.55,56 Good test-retest reliability; < 1 point difference in scores after 1 week, non-clinically significant.55 Responsiveness:Has been tested among several distinct patient populations but not among people with immunodeficiency. | The tool’s developers propose the following48:
No MID specific to immunodeficiency was identified. |
Pediatric Quality of Life Inventory (PedsQL): Exploratory end point | ||
23-item self- or proxy-reported generic health-related quality of life measure for children and adolescents aged 2 to 18 years | No data specific to immunodeficiency were identified. Validity: Construct validity demonstrated by the known group method. All scales showed significantly (P < 0.001) higher scores for healthy versus acutely ill or chronically ill children.57-59 Scores show correlations of variable strength with markers of morbidity and illness burden.57,58 | 4.4 points on the total score for self-reports and 4.5 points for proxy reports in a general population of children in the US.57 No MID specific to immunodeficiency was identified. |
(continued) | Reliability: Acceptable internal consistency reliability (Cronbach alpha ≥ 0.70) for all subscales; 0.88 to 0.89 for self-reported, 0.90 to 0.92 for proxy-reported total score.57,58 Test-retest reliability is not reported. Intercorrelations between child and parent scores are strong (> 0.50) for most scales, and strengthen across age categories (5 to 7 years versus 8 to 13 years versus 13 to 18 years).57 Responsiveness: Among hospitalized children, Cohen effect size estimates for improvement in scores between admission and 2 to 8 weeks post-discharge were large60 for total scores (0.84 to 1.02 depending on age and caregiver versus child report), and moderate-to-large for physical (0.73 to 0.99) and psychosocial (0.71 to 0.85) subscales.61 | |
BP = bodily pain; GH = general health; MID = minimal important difference; MH = mental health; PF = physical functioning; RP = role physical; VT = vitality.
The Short Form (36) Health Survey
The SF-3662 was used in Study 160603 to measure HRQoL among children and adults ≥ 14 years, and in Study 160902 for children and adults ≥ 8 years. All scores were self-reported. HRQoL in adults was an exploratory end point.
The SF-36 is a 36-item, self-reported general health status instrument that has been used extensively in clinical trials across many disease areas.48 The SF-36 consists of 8 health domains: physical functioning (PF), role physical (RP), bodily pain (BP), general health (GH), vitality (VT), social functioning (SF), role emotional (RE), and mental health (MH).48 For each of the 8 domains, a subscale score can be calculated. The SF-36 also provides 2 component summaries, the physical component summaries (PCS) and the mental component summary (MCS), derived from aggregating the 8 domains according to a scoring algorithm. The PCS and MCS and 8 dimensions are each measured on a scale of 0 to 100, which are t scores (mean of 50 and SD of 10) that have been standardized to the US general population.48 Thus, a score of 50 on any scale would be at the average or norm of the general US population and a score 10 points lower (i.e., 40) would be 1 SD below the norm.48 On any of the scales, an increase in score indicates improvement in health status.48
McHorney et al. (1993)54 assessed the validity of the SF-36 among people with minor and serious medical conditions (n = 1,014). To test relative construct validity, the scores of groups with minor or serious medical conditions were compared (based on clinical criteria), showing that patients with serious medical conditions scored significantly (P < 0.01) lower on all scales compared with those with minor conditions. The physical functioning scale was considered most valid (mean difference −23.18 points, relative validity 1.00, P < 0.001) in detecting differences between groups. Brazier et al. (1992)55 further tested the tool’s validity and reliability in a sample of 1,980 patients aged 16 to 74 years in the UK. Test-retest reliability was assessed using the Bland and Altman method,63 with a 2-week interval between tests. The mean difference of scores on all scales was < 1 point and considered to be clinically insignificant. Construct validity was tested by comparing the scores of patients with and without health problems. Those who had chronic physical health problems had poorer general health scores than those without (GH score 53 versus 66), as did those who had visited a general practitioner in the past 2 weeks or had attended an outpatient visit in the past 3 months compared with those without visits. McHorney et al. (1994)56 later examined internal consistency reliability among a 3,445 patients in the US. The Cronbach alpha was found to be acceptable (> 0.70)64 for all scales and was ≥ 0.90 for the MH and PF domains. Some floor and ceiling effects were observed; floor effects were most prominent for the RP (24% of the sample) and RE (18%) domains, while ceiling effects were most prominent for the RP (37%), SF (46%), and RE (56%) domains. The SF-36 has not been validated specifically in people with primary or secondary immunodeficiency but has been used in this population previously.65
Based on anchor data, the developer of the SF-36 proposed the following minimally important mean group differences (MID) for the individual domain scores: PF, 3; RP, 3; BP, 3; GH, 2; VT, 2; SF, 3; RE, 4; and MH, 3.48 It should be noted that these MID values were determined as appropriate for groups with mean t score ranges of 30 to 40. For higher t score ranges, MID values may be higher.48 As these MID values were based on clinical and other non-patient-reported outcomes, they do not necessarily identify the smallest difference that patients would consider important. MIDs for a variety of conditions have been proposed, but none specific to immunodeficiency have been identified.66
Pediatric Quality of Life Inventory 4.0
The PedsQL47 was used in Study 160603 to assess HRQoL during the past month among children aged 2 to 13 years, and in Study 160902 for children aged 2 to 7 years. Assessment for children aged 2 to 7 years was by parent or caregiver report, and for 8 to 13 years was by self-report. HRQoL in children was an exploratory end point. The Clinical Study Reports did not indicate which version of the tool was used.
The PedsQL 4.0 is a brief (< 5 minutes to complete) modular approach to measuring HRQoL which is designed to be used among children with diverse acute or chronic health conditions.47 The tool is available in 2 forms: self-report for children and adolescents aged 5 to 18 years, and a parent proxy report form which is available for ages 2 to 18 years.58 The PedsQL includes 23 items on 4 scales: physical functioning (8 items), emotional functioning (5 items), social functioning (5 items), and school functioning (5 items).58 Questions ask how much of a problem the item has been in the past month.58 Possible responses differ by age category; a 5-point scale is used for ages 8 to 18 years, ranging from 0 (never a problem) to 4 (almost always a problem).58 For younger children (age 5 to 7 years), a simplified 3-point scale anchored by happy and sad faces is used, including 0 (not at all a problem), 2 (sometimes a problem), and 4 (a lot of a problem).58 The parent proxy report also includes a toddler (2 to 4 years) scale, with a reduced number of items related to school functioning (n = 3).58 Items are reverse-scored and linearly transformed to a scale of 0 to 100, where higher scores indicate better HRQoL.58 Scale scores are a sum of the component items divided by the total number of questions answered, to account for non-response.58 Two summary scores can then be computed: the Physical Health Summary (8 items) is the same as the physical functioning scale, while the psychological summary score (15 items) is the sum of all items in the remaining scales, divided by the number of items for which a response was provided.58
In order to validate the PedsQL Generic Core Scales, a sample of chronically ill (n = 683), acutely ill (n = 207), and healthy children (n = 730) aged 2 to 18 years and their parents were included in a study by Varni et al. (2001).58 No floor effects were observed but there were minimal (7.2% and 10.2% for the self- and proxy report of total score) to moderate (47.1 to 58.1% for the self- and proxy report of the social functioning subscale) ceiling effects. Construct validity was ascertained using the known-groups method, where 1-way analysis of variance comparing the scores of chronically ill, acutely ill, and healthy children revealed significant differences (P < 0.001), with healthy children having higher scores. Self- and proxy reports were correlated to a variable degree with indicators of morbidity and illness burden. Scale internal consistency reliability was shown to be acceptable (Cronbach alpha ≥ 0.70)64 for all scales except for self-reported school functioning (0.68). The total scores showed good reliability, with Cronbach alpha of 0.88 for self-reports and 0.90 for parent reports. Test-retest reliability was not examined. Heterotrait-monomethod correlations were used to estimate parent-child concordance on the same subscales, showing that all correlations were in the medium (r = 0.30) to large (r = 0.50) effect size range.
A later study by the same authors, Varni et al. (2003)57 performed further reliability and validity testing on a large sample 10,241 parents of children aged 2 to 16 years and 5,991 children aged 5 to 16 years. The findings of this study were similar, and additionally found that child-parent concordance in scores were large (r > 0.50) for most scales and tended to strengthen across categories of age (5 to 7 years versus 8 to 13 years versus 13 to 18 years). Weaker correlations (> 0.30) were found for physical health in children aged 5 to 13 years and social function in children aged 5 to 7 years. Varni et al. (2002)59 further assessed the sensitivity of the PedsQL 4.0 among 115 children presenting at a cardiology clinic and 730 healthy children. Statistically significant (P = 0.001) differences in PedsQL score were observed between children with varied severity of cardiovascular illness, as well as between children with cardiovascular conditions and those who were healthy. Methodological concerns that may limit the generalizability of the findings of the aforementioned reports include lack of information about non-respondents and overrepresentation of people with lower socioeconomic status.57,58
Desai et al. (2014)61 examined the responsiveness among hospitalized children at admission (n = 4,637) versus 2 to 8 weeks post-discharge (n = 2,694). Cohen effect size estimates for improvement in scores were large60 for total scores (0.84 to 1.02 depending on age and caregiver versus child report), and moderate-to-large for physical (0.73 to 0.99) and psychosocial (0.71 to 0.85) subscales. Construct validity was further demonstrated as patients with no chronic illness (and their parents) scored significantly (P < 0.05) higher on the total score, physical domain, and psychosocial domain when compared with patients with either complex or non-complex chronic illness.61 Finally, Varni et al. (2003)57 estimated the MID by calculating the standard error of measurement, suggesting a MID of 4.4 points on the total score for self-reports and 4.5 points for proxy reports. An MID specific to immunodeficiency was not identified.
Appendix 5: Details of Sponsor-Submitted Study in Adult Patients Switching From cSCIg to IgHy10 (NCT02881437)
Note that this appendix has not been copy-edited.
Table 39: Details of Study NCT02881437
Detail | NCT02881437 |
|---|---|
Designs and populations | |
Study design | Open-label single-group prospective study |
Locations | 7 study sites in France |
Patient enrolment dates | Started November 30, 2016; end date not reported. |
Enrolled (N) | 22 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Interventions | Treatment with subcutaneous IgHy10 for 6 months. After a 1-week ramp-up phase, subcutaneous infusions in the abdomen or thighs occurred biweekly at one-half of the initial (prior to enrolment) cSCIg dosage for 3 months. IgHy10 was administered using a 24G subcutaneous needle labelled for high flow rates; rHuPH20 was administered first at an initial rate of 1 or 2 mL/minute, followed 10 minutes later by infusion of the full dose of Ig (10%) at a rate of 300 mL/hour in patients weighing ≥ 40 kg and 160 mL/hour in patients weighting < 40 kg. |
Duration | |
Phase | |
Screening | Not reported |
Enrolment | Not reported |
Open-label | ~6 months |
Ramp-up | 1 week |
First follow-up | 3 months |
Second follow-up | 3 months |
Outcomes | |
Primary end point | Change in IgG trough level between visit 1 and visit 3 (baseline to 3 months follow-up) |
Secondary and exploratory end points | Secondary
|
Notes | |
Publications | None |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; cSCIg = conventional subcutaneous immunoglobulin; IgG = immunoglobulin G; IgHy10: normal human immunoglobulin (10%) with recombinant human hyaluronidase; IgRT = immunoglobulin replacement therapy; PID = primary immunodeficiency; rHuPH20 = recombinant human hyaluronidase; SF-36 = Short Form (36) Health Survey; TSQM-9 = treatment satisfaction questionnaire for medication.
Source: Clinical Study Report.17
Table 40: Baseline Characteristics of Patients in NCT02881437 (SAS)
Characteristic | NCT02881437 (n = 22) |
|---|---|
Age, years | |
N analyzed | 22 |
Median (IQR) | 45.0 (32.0, 54.0) |
Gender, n (%) | |
N analyzed | 22 |
Male | 7 (31.8) |
Female | 15 (68.2) |
Weight, kg | |
N analyzed | 20 |
Median (IQR) | 64.5 (57.5, 78.0) |
PID diagnosis, n (%) | |
N analyzed | 22 |
Common variable immunodeficiency | 14 (63.6) |
Deficit in IgG subclasses | 3 (13.6) |
X-linked gammaglobulinemia | 2 (9.1) |
Primary agammaglobulinemia | 2 (9.1) |
Other | 1 (4.6) |
Past disease history, n (%) a | |
N analyzed | 22 |
Hysterectomy | 4 (18.2) |
Appendicitis | 3 (13.6) |
Adenoidectomy, gastric bypass, herpes zoster, viral meningitis, lung disorders | 2 (9.1) each |
Concomitant diseases, n (%) | |
N analyzed | 22 |
Bronchiectasis | 2 (9.1) |
Interstitial lung disease, pulmonary fibrosis, chronic sinusitis, portal hypertension | 1 (4.5) each |
Vaccinations, n (%); mean (SD) years prior to entry | |
N analyzed | 22 |
Diphtheria, tetanus, and poliomyelitis | 8 (36.4); 10.9 (6.3) |
Hepatitis B | 5 (22.7); 22.6 (6.0) |
Pneumococcus | 10 (45.5); 4.4 (4.0) |
Meningococcus | 1 (4.5); 1 (NA) |
Influenza | 8 (36.4); 1.5 (2.7) |
Hemophilus influenzae | 3 (13.6); 2.0 (2.0) |
IQR = interquartile range; IgG = immunoglobulin G; NA = not applicable; PID = primary immunodeficiency disease.
aThe most common past diseases are listed, occurring in ≥ 2 patients.
Source: Clinical Study Report.50
Table 41: Patient Disposition for NCT02881437
Disposition | NCT02881437 |
|---|---|
Screened, n | NR |
Enrolled, n | 22 |
Completed, n (%) | 16 (72.7) |
Withdrawn, n (%) | 6 (27.3) |
Adverse event | 4 (18.2) |
Lost to follow-up a | 2 (9.0) |
Efficacy set, N b | 22 |
Safety set, N b | 22 |
Per-protocol set, N b | 22 |
IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase.
aReasons for losses to follow-up not reported.
bThe efficacy set and safety set included all enrolled patients who received at least 1 dose of IgHy10. Since there were no major protocol deviations, the per-protocol set was the same as the efficacy set.
Source: Clinical Study Report.17
Table 42: Prior and Ongoing Treatment With cSCIg Among Patients in NCT02881437 (SAS)
Treatment details | NCT02881437 (n = 22) |
|---|---|
Previous treatment | |
Age at start of IgRT, years | |
N analyzed | 22 |
Median (IQR) | 34.5 (18.0 to 46.0) |
Age at start of cSCIg, years | |
N analyzed | 20 |
Median (IQR) | 35.5 (22.5 to 48.5) |
Current treatment | |
Product, n (%) | |
N analyzed | 22 |
Gammanorm | 7 (31.8) |
Hizentra | 15 (68.2) |
Place of administration, n (%) | |
N analyzed | 22 |
Exclusively at home | 17 (81.0) |
At hospital then at home | 3 (14.3) |
At hospital and at home alternatively | 1 (4.8) |
Not determined | 1 (4.8) |
Average dose of Ig infused, g/treatment | |
N analyzed | 22 |
Median (IQR) | 7.5 (5.0 to 8.0) |
Average dose of Ig infused, g/kg/treatment | |
N analyzed | 20 |
Median (IQR) | 0.11 (0.08 to 0.14) |
Frequency of treatments, n (%) | |
N analyzed | 22 |
1 per week | 21 (95.5) |
Other | 1 (4.6) |
cSCIg = conventional subcutaneous immunoglobulin; Ig = immunoglobulin; IgRT = immunoglobulin replacement therapy; IQR = interquartile range.
Source: Clinical Study Report.50
Table 43: Exposure to IgHy10 Among Patients in NCT02881437 (Safety Set)
Detail | Timing of exposure measured in NCT02881437 (n = 22) | ||
|---|---|---|---|
Initial infusion | Initial infusion to 3 months follow-up | 3 months to 6 months follow-up b | |
Number of infusions | |||
N analyzed | 22 | 20 | 15 |
Total number of infusions | 22 | 108 | 62 |
Median (IQR) per patient | 1 (NA) | 7.00 (6.50 to 7.00) | 4.00 (4.00 to 5.00) |
Dose Ig prescribed, g | |||
Infusions analyzed | NR | 94 | 59 |
Median (IQR) | NR | 15.00 (10.00 to 20.00) | 30.00 (20.00 to 30.00) |
Dose Ig prescribed, g/kg | |||
Infusions analyzed | 19 | 85 | 55 |
Median (IQR) | 0.12 (0.08 to 0.14) | 0.23 (0.16 to 0.30) | 0.42 (0.33 to 0.51) |
Delay between infusions, days | |||
Infusions analyzed | NA | 88 | 47 |
Median (IQR) | NA | 14.00 (14.00 to 14.00) | 27.00 (21.00 to 29.00) |
Number of infusion sites | |||
Infusions analyzed | 20 | 100 | 61 |
One site | 19 (95.0) | 94 (94.0) | 53 (86.7) |
Two sites | 1 (5.0) | 6 (6.0) | 8 (13.1) |
Administered volume of rHuPH20, mL | |||
Infusions analyzed | 16 | 93 | 53 |
Median (IQR) | 3.75 (3.13 to 5.00) | 7.50 (5.00 to 10.00) | 10.00 (10.00 to 15.00) |
Duration of rHuPH20 infusion, minutes | |||
Infusions analyzed | 18 | 89 | 49 |
Median (IQR) | 4.00 (1.50 to 5.00) | 5.00 (5.00 to 8.00) | 10.00 (6.00 to 10.00) |
Time between rHuPH20 and Ig infusion, minutes | |||
Infusions analyzed | 16 | 76 | 42 |
Median (IQR) | 2.50 (1.00 to 4.82) | 4.00 (2.00 to 5.00) | 3.00 (2.00 to 5.00) |
Administered volume of Ig, mL | |||
Infusions analyzed | 17 | 89 | 56 |
Median (IQR) | 75.0 (74.7 to 80.0) | 150.00 (100.00, 150.00) | 300.00 (200.00, 300.00) |
Flow rate of Ig infusion, mL/minute | |||
Infusions analyzed | 16 | 82 | 52 |
Median (IQR) | 1.44 (1.16 to 1.70) | 1.89 (1.67 to 2.46) | 2.50 (2.00 to 3.33) |
Duration of Ig infusion, minutes | |||
Infusions analyzed | 16 | 87 | 54 |
Median (IQR) | 48.00 (42.50 to 55.00) | 60.00 (56.00 to 72.00) | 90.00 (75.00 to 120.00) |
Location of infusion | |||
Infusions analyzed | 21 | 99 | 61 |
Hospital | 21 (100.0) | 19 (19.2) | 14 (23.0) |
Home | 0 (0.0) | 80 (80.8) | 47 (77.1) |
Person responsible for infusion | |||
Infusions analyzed | 20 | 99 | 60 |
Nurse | 13 (65.0) | 24 (24.2) | 8 (13.3) |
Patient | 7 (35.0) | 71 (71.7) | 50 (83.3) |
Other | 0 (0.0) | 4 (4.0) | 2 (3.3) |
Ig = immunoglobulin; IQR = interquartile range; NA = not applicable; NR = not reported; rHuPH20 = recombinant human hyaluronidase.
Source: Clinical Study Report.17
Statistical Analysis in NCT02881437
The sample size was calculated using pharmacokinetic model simulations, which showed that an increase in the median IgG trough level of 15% could be expected when comparing cSCIg (20%) to IgHy10. The calculation assumed a mean difference in IgG trough level of ≥ 1.15 g/L, a common SD of 1.44 g/L, and 40% correlation between measures. A sample size of 22 pairs was determined to provide 90% power to show a significant change > 1.15 g/L, and > 80% power to show a significant change > 1 g/L at an alpha level of 0.05.
Change in IgG trough level between the first and last visits was calculated and compared with the null hypothesis of no change between visits using a student’s t-test for paired samples. Missing data at any time point were replaced with the last observed measurement. Additional subgroup analyses were performed on those who had their last IgHy10 injection 19 to 23 days prior to the last visit and those with the last injection 26 to 30 days prior to the last visit.
The number and proportion of patients having experienced at least 1 infection were described for each period and the incidence of infections was estimated using a Poisson regression model including the duration of exposure as an offset term. Missing data were not replaced. Change in HRQoL between visits was calculated and tested and compared with the null hypothesis of no change using a student’s t-test for paired samples.
The incidence of patients with at least 1 AE reported by the physician were analyzed at the patient level for local and systemic AEs.
Table 44: Change in Steady-State IgG Trough Levels in NCT02881437 (Efficacy Set)
Change measurement time | IgG trough level | |
|---|---|---|
With imputed values | Without imputed values | |
From baseline to 3 months (visit 3) | ||
N analyzed | 22 | 16 |
Baseline, mean (SD) | 9.36 (2.45) | 8.95 (2.11) |
3 months, mean (SD) | 9.14 (2.14)a | 8.64 (1.45) |
Change from baseline to 3 months, mean (SD) | −0.22 (1.31) | −0.30 (1.54) |
P value | 0.4372 | 0.4422 |
From 3 months (visit 3) to 6 months (visit 4) | ||
N analyzed | 22 | 15 |
3 months, mean (SD) | 9.14 (2.14)a | 8.40 (2.11) |
6 months, mean (SD) | 9.01 (2.05)b | 8.58 (1.88) |
Change from 3 to 6 months, mean (SD) | −0.13 (1.02) | 0.17 (0.80) |
P value | 0.5497 | 0.6904 |
From baseline to 6 months (visit 4) | ||
N analyzed | 22 | 16 |
Baseline, mean (SD) | 9.36 (2.45) | 8.92 (2.05) |
6 months, mean (SD) | 9.01 (2.05)b | 8.63 (1.51) |
Change from baseline to 6 months, mean (SD) | −0.35 (1.30) | −0.29 (1.35) |
P value | 0.2169 | 0.4011 |
IgG = immunoglobulin G; NA = not applicable.
aMissing values at first follow-up (visit 3) for 6 patients were replaced by the baseline value.
bMissing values at second follow-up (visit 4) were replaced by the last observed value (baseline for 5 patients and visit 3 for 1 patient).
Analysis: change has been calculated and null hypothesis of no change between visits has been tested by a student’s t-test for paired samples
Source: Clinical Study Report.17
Table 45: Physician Records of AEs and Most Commonly Reported AEs in NCT02881437 (Safety Set)
Adverse event | Timing of assessment in NCT02881437 (n = 22) | |
|---|---|---|
Baseline to 3 months | 3 to 6 months | |
Local AEs, n (%) | ||
N analyzed | 22 | 22 |
Any local AE | 21 (95.5) | 12 (66.7) |
Most common local AEsa | ||
Erythema | 18 (81.8) | 12 (66.7) |
Swelling | 17 (72.3) | 12 (66.7) |
Induration | 9 (40.9) | 3 (16.7) |
Injection site erythema | 6 (27.3) | 0 (0.0) |
Injection site pain | 3 (13.6) | 1 (5.6) |
Injection site paresthesia | 3 (13.6) | 0 (0.0) |
Other notable local AEs b | ||
Injection site pruritus | 1 (4.6) | 0 (0.0) |
Systemic AEs, n (%) | ||
N analyzed | 22 | 22 |
Any systemic AE | 22 (100.0) | 16 (88.9) |
Most common systemic AEs a | ||
Pain | 8 (36.4) | 9 (40.9) |
Fatigue | 6 (27.3) | 3 (13.6) |
Pyrexia | 4 (18.2) | 2 (9.1) |
Chills | 3 (13.6) | 4 (18.2) |
Feeling cold | 3 (13.6) | 2 (9.1) |
Abdominal pain | 9 (40.9) | 5 (22.7) |
Nausea | 6 (27.3) | 1 (4.6) |
Bronchitis | 3 (13.6) | 2 (9.1) |
Sinusitis | 3 (13.6) | 2 (9.1) |
Headache | 9 (40.9) | 8 (36.4) |
Pruritis | 8 (36.4) | 5 (22.7) |
Rash | 1 (4.6) | 3 (13.6) |
Back pain | 4 (18.2) | 2 (9.1) |
Arthralgia | 3 (13.6) | 3 (13.6) |
Myalgia | 3 (13.6) | 3 (13.6) |
Hot flush | 4 (18.2) | 1 (4.6) |
Vertigo | 4 (18.2) | 2 (9.1) |
Anxiety | 2 (9.1) | 3 (13.6) |
Other notable systemic AEs b | ||
Vomiting | 2 (9.1) | 0 (0.0) |
AE = adverse event; NA = not applicable; SF-36 = Short Form (36) Health Survey.
Note: Analysis: Descriptive statistics.
aAEs occurring in ≥ 10% of patients.
bOther notable harms are included here if they were not previously reported as occurring in ≥ 10% of patients.
Source: Clinical Study Report.17
Appendix 6: Details of Sponsor-Submitted Study on the Safety, Tolerability, and Immunogenicity of IgHy10 in Pediatric Patients (Study 161504, Interim Analysis)
Note that this appendix has not been copy-edited.
Table 46: Details of Clinical Study 161504
Detail | Clinical Study 161504 |
|---|---|
Designs and populations | |
Study design | Open-label non-randomized single-group prospective study |
Locations | 16 study sites across Europe (Czechia, Denmark, France, Greece, Slovakia, Sweden, UK) |
Patient enrolment dates | Started 13 June 2017; end date not reported. |
Enrolled (N) | 42 |
inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Exposures | Epoch 1 (ramp-up): IgHy10 (specific dosing schedule not reported) Epoch 2: IgHy10 continued for 1 year Epoch 3: Safety follow-up for patients with an anti-rHuPH20 antibody titer ≥ 160 during epoch 1 or 2 and who experienced either an SAE or severe AE. Patients received IVIg 100mg/mL solution (Kiovig) or cSCIg 200 mg/mL solution (Cuvitru) |
Duration | |
Phase | |
Screening | Not reported |
Enrolment | Not reported |
Open label | Epoch 1 (ramp-up): Up to 6 weeks Epoch 2: 1 to 3 years (depended on anti-rHuPH20 antibodies) Epoch 3 (safety follow-up): 1 year |
Outcomes | |
Primary end point |
|
Secondary and exploratory end points | Secondary
|
Notes | |
Publications | Ciznar et al.(2021) (poster)19 |
AE = adverse event; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; cSCIg = conventional subcutaneous immunoglobulin; EQ-5D = EuroQol 5D; HRQoL = health-related quality of life; IgG = immunoglobulin G; IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase; IgRT = immunoglobulin replacement therapy; IVIg = intravenous immunoglobulin; PedsQL = Pediatric Quality of Life Inventory; rHuPH20 = recombinant human hyaluronidase; SAE = serious adverse event.
aAs defined by the International Union of Immunological Societies Scientific Committee 2015.18
Source: NCT0311634767; Ciznar et al. (2021).19
Table 47: Treatment-Emergent Infections in Clinical Study 161504 (Interim Analysis, Enrolled Set)
Infection | cSCIg treatment group | ||
|---|---|---|---|
cSCIg naive (n = 23) | cSCIg pre-treated (n = 19) | Total (N = 42) | |
Patients with ≥ 1 infection, n (%) | |||
N analyzed | 23 | 19 | 42 |
Serious acute bacterial infection | NR | NR | 1 |
Any infectiona | 17 (73.9) | 14 (73.7) | 31 (73.8) |
Rhinitis | 2 (8.7) | 6 (31.6) | 8 (19.0) |
Gastroenteritis | 5 (21.7) | 0 (0.0) | 5 (11.9) |
Nasopharyngitis | 5 (21.7) | 0 (0.0) | 5 (11.9) |
Viral infection | 3 (13.0) | 0 (0.0) | 3 (7.1) |
Pneumonia | 1 (4.3) | 2 (10.5) | 3 (7.1) |
Upper respiratory tract infection | 0 (0.0) | 3 (15.8) | 3 (7.1) |
NR = not reported; cSCIg = conventional subcutaneous immunoglobulin.
Note: Analysis: descriptive statistics.
aReported as the preferred term below.
Source: Ciznar et al. (2021).19
Table 48: AEs in Clinical Study 161504 (Interim Analysis, Enrolled Set)
Adverse event | cSCIg treatment group | ||
|---|---|---|---|
cSCIg naive (n = 23) | cSCIg pre-treated (n = 19) | Total (n = 42) | |
Any AEs, n (%) | |||
N analyzed | 23 | 19 | 42 |
Patients with ≥ 1 AE | 16 (69.6) | 11 (57.9) | 27 (64.3) |
Patients with ≥ 1 local AE | 9 (39.1) | 2 (10.5) | 11 (26.2) |
Patients with ≥ 1 systemic AE | 13 (56.5) | 11 (57.9) | 24 (57.1) |
Patients with ≥ 1 SAE | 1 (4.3) | 3 (15.8) | 4 (9.5) |
Most common AEs, n (%) a | |||
N analyzed | 23 | 19 | 42 |
Local AEs | |||
Infusion site pain | 6 (26.1) | 1 (5.3) | 7 (16.7) |
Infusion site pruritis | 4 (17.4) | 0 (0.0) | 4 (9.5) |
Systemic AEs | |||
Cough | 7 (30.4) | 5 (26.3) | 12 (28.6) |
Pyrexia | 3 (13.0) | 3 (15.8) | 6 (14.3) |
Epistaxis | 2 (8.7) | 1 (5.3) | 3 (7.1) |
Diarrhea | 1 (4.3) | 1 (5.3) | 2 (4.8) |
Vomiting | 2 (8.7) | 1 (5.3) | 3 (7.1) |
Fatigue | 3 (13.0) | 1 (5.3) | 4 (9.5) |
Oropharyngeal pain | 1 (4.3) | 2 (10.5) | 3 (7.1) |
Uncoded | 1 (4.3) | 2 (10.5) | 3 (7.1) |
Treatment stopped, interrupted, or adjusted due to AE, n (%) | |||
N analyzed | 23 | 19 | 42 |
Number of infusions | 2 (9.3) | 1 (4.9) | 3 (7.4) |
AE = adverse event; cSCIg = conventional subcutaneous immunoglobulin.
Note: Analysis: descriptive statistics.
aReported in > 2 patients overall.
Source: Ciznar et al. (2021).19
Appendix 7: Details of Sponsor-Submitted Study on Safety of IgHy10 in Pregnant Women and Their Infants (Registry Study 161301)
Note that this appendix has not been copy-edited.
Table 49: Details of Registry Study 161301
Detail | Registry Study 161301 |
|---|---|
Designs and populations | |
Study design | Observational, uncontrolled, open-label, 2-arm post-marketing pregnancy registry study. Data were collected prospectively, if possible, but could be collected retrospectively if a physician became aware of eligibility at an advanced stage of pregnancy. |
Locations | 8 sites in Czech Republic, Germany, Poland, Slovakia, and the US |
Patient enrolment dates | Mothers: 4 December 2015 to 29 December 2017 Infants: 20 January 2017 to 17 December 2019 |
Enrolled (N) | 16 (9 mothers and 7 infants) |
Inclusion criteria | Women who became pregnant during or after treatment with IgHy10 |
Exclusion criteria | None |
Drugs | |
Exposures | There were 2 arms based on whether or not IgHy10 treatment was continued during pregnancy. Arm 1: Patients who previously received IgHy10, who then received a licensed human normal immunoglobulin other than IgHy10 (intravenous or subcutaneous) or an alternative treatment during pregnancy, as determined by their treating physician. Arm 2: Patients who previously received IgHy10 and continued with this treatment during pregnancy. |
Duration | |
Phase | |
Enrolment | 3.5 years |
Participation period for pregnant women | Enrolment to 6 months after delivery/end of pregnancy |
Participation period for infants | Enrolment until age of 2 years |
Total duration | Approximately 6 years from initiation |
Outcomes | |
Primary end point | • Incidence of all SAEs in expectant mothers and infants |
Secondary and exploratory end points | Secondary
|
Notes | |
Publications | None |
AE = adverse event; IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; rHuPH20 = recombinant human hyaluronidase.
Source: Clinical Study Report.50
Table 50: Baseline Characteristics of Expectant Mothers in Registry Study 161301 (Enrolled Set)
Detail | Registry Study 161301 arm | ||
|---|---|---|---|
IgHy10 arm (n = 7) | Alternative product arm (n = 2) | Total (n = 9) | |
Age (years), median (IQR) | 34.0 (32.0 to 36.0) | 33.0 (30.0 to 36.0) | 34.0 (32.0 to 36.0) |
Ethnicity, n (%) | |||
Hispanic/Latino | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Non-Hispanic/Latino | 6 (85.7) | 2 (100.0) | 8 (88.9) |
Race, n (%) | |||
White/Caucasian | 7 (100.0) | 2 (100.0) | 9 (100.0) |
Pre-pregnancy weight, kg | |||
N analyzed | 4 | 2 | 6 |
Median (IQR) | 64.5 (61.0 to 77.5) | 60.0 (58.0 to 62.0) | 63.0 (58.0 to 65.0) |
PID diagnosis, n (%) | |||
Common variable immunodeficiency | 6 (85.7) | 2 (100.0) | 8 (88.9) |
Hyper IgM Syndrome | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Medical history, n (%) | |||
Eyes, ears, nose, throat | 3 (42.9) | 1 (50.0) | 4 (44.4) |
Respiratory | 6 (85.7) | 1 (50.0) | 7 (77.8) |
Cardiovascular | 2 (28.6) | 0 (0.0) | 2 (22.2) |
Gastrointestinal | 4 (57.1) | 1 (50.0) | 5 (55.6) |
Musculoskeletal | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Neurological / Psychiatric | 1 (14.3) | 1 (50.0) | 2 (22.2) |
Endocrine | 2 (28.6) | 2 (100.0) | 4 (44.4) |
Hematopoietic / Lymphatic | 5 (71.4) | 2 (100.0) | 7 (77.8) |
Dermatological | 2 (28.6) | 1 (50.0) | 3 (33.3) |
Genitourinary / Obstetrical | 4 (57.1) | 1 (50.0) | 5 (55.6) |
Family history, n (%) | |||
Congenital abnormalities / Birth defects | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Adverse fetal outcome | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Psychomotor retardation | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Consanguinity between parents | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Pregnancy history, n (%) | |||
Any previous pregnancy | 4 (57.1) | 2 (100.0) | 6 (66.7) |
Number of previous pregnancies | 4 | 2 | 6 |
Number of normal live births | 3 (75.0) | 1 (50.0) | 4 (66.7) |
Number of abnormal live births | 0 (0.0) | 1 (50.0) | 1 (16.7) |
Number of fetal deaths | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Number of induced terminations | 1 (25.0) | 0 (0.0) | 1 (16.7) |
Number of spontaneous terminations | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Any obstetrical complications | 3 (75.0) | 0 (0.0) | 3 (50.0) |
Any fetal/neonatal abnormality | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Current pregnancy | |||
Time from date of last menstrual period to enrolment, median (IQR) days | 244.5 (168.0 to 279.5) | 265.5 (232.0 to 299.0) | 246.5 (228.0 to 298.0) |
Age at conception, median (IQR) years | 36.0 (30.0 to 37.0) | 30.0 (30.0 to 30.0) | 33.0 (30.0 to 36.5) |
History of subfertility/infertility, n (%) | 1 (14.3) | 0 (0.0) | 1 (11.1) |
Treatment for subfertility/infertility, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Any exposure to tobacco, (%) | 2 (28.6) | 1 (50.0) | 3 (33.3) |
Any exposure to alcohol, n (%) | 1 (14.3) | 1 (50.0) | 2 (22.2) |
Any exposure to recreational drug, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase; IQR = interquartile range; PID = primary immunodeficiency disease.
Source: Clinical Study Report.50
Table 51: Baseline Characteristics of Infants in Registry Study 161301 (Enrolled Set)
Detail | Registry Study 161301 arm | ||
|---|---|---|---|
IgHy10 arm (n = 5) | Alternative product arm (n = 2) | Total (n = 7) | |
Sex, n (%) | |||
Male | 2 (40.0) | 1 (50.0) | 3 (42.9) |
Female | 3 (60.0) | 1 (50.0) | 4 (57.1) |
Ethnicity, n (%) | |||
Non-Hispanic/Latino | 5 (100.0) | 2 (100.0) | 7 (100.0) |
Race, n (%) | |||
White/Caucasian | 5 (100.0) | 2 (100.0) | 7 (100.0) |
IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase.
Source: Clinical Study Report.50
Table 52: Patient Disposition for Registry Study 161301
Detail | Registry Study 161301 cohort | ||
|---|---|---|---|
Retrospective cohort | Prospective cohort | Total | |
Expectant mothers | |||
Screened | 7 | 2 | 9 |
Enrolled, n (%) | 7 (100.0) | 2 (100.0) | 9 (100.0) |
Completed, n (%) | 5 (71.4) | 2 (100.0) | 7 (77.8) |
Not completed, n (%) | 2 (28.6) | 0 (0.0) | 2 (22.2) |
Withdrawal by patient | 1 (50.0) in the IgHy10 arm | NA | 1 (50.0) in the IgHy10 arm |
Lost to follow-upa | 1 (50.0) in the IgHy10 arm | NA | 1 (50.0) in the IgHy10 arm |
Enrolled set, Nb | 7 | 2 | 9 |
Safety set, Nc | 7 | 2 | 9 |
Infants | |||
Screened | 5 | 2 | 7 |
Enrolled, n (%) | 5 (100.0) | 2 (100.0) | 7 (100.0) |
Completed, n (%) | 4 (80.0) | 2 (100.0) | 6 (85.7) |
Not completed, n (%) | 1 (20.0) | 0 (0.0) | 1 (14.3) |
Withdrawal by parent/guardian | 1 (100.0) | NA | 1 (14.3) |
Enrolled set, Nb | 5 | 2 | 7 |
Safety set, Nc | 5 | 2 | 7 |
IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase.
aReasons for losses to follow-up not reported.
bThe enrolled set consisted of all patients who had signed informed consent forms and met all inclusion/exclusion criteria at the time of enrolment.
cThe safety set consisted of all patients in the enrolled set.
Source: Clinical Study Report.50
Table 53: History of Treatment With IgHy10 in Registry Study 161301 (Enrolled Set)
Detail | Registry Study 161301 arm | ||
|---|---|---|---|
IgHy10 arm (n = 7) | Alternative product arm (n = 2) | Total (N = 9) | |
Treatment regimen (rHuPH20 volume, mL) per month | |||
N with data available | 5 | 2 | 7 |
Median (IQR) | 10.0 (10.0, 25.0) | 11.3 (7.5, 15.0) | 10.0 (7.5, 25.0) |
Total Ig dose (grams/month) | |||
N with data available | 4 | 2 | 6 |
Median (IQR) | 35.0 (20.0, 50.0) | 22.5 (15.0, 30.0) | 25.0 (20.0, 50.0) |
Treatment interval, n (%) | |||
N with data available | 6 | 2 | 8 |
Weekly | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Every 2 weeks | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Every 3 weeks | 1 (16.7) | 0 (0.0) | 1 (12.5) |
Every 4 weeks | 3 (50.0) | 2 (100.0) | 5 (62.5) |
Other | 2 (33.3) | 0 (0.0) | 2 (25.0) |
Treatment duration, days | |||
N with data available | 4 | 1 | 5 |
Median (IQR) | 613.0 (369.5, 710.5) | 139.0 (139.0, 139.0) | 579.0 (160.0, 647.0) |
IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase; IQR = interquartile range; rHuPH20 = recombinant human hyaluronidase.
Source: Clinical Study Report.50
Table 54: Actual IgHy10 Treatment and Changes to Treatment During Registry Study 161301 (Enrolled Set)
Detail | Registry Study 161301 arm | ||
|---|---|---|---|
IgHy10 arm (n = 7) | Alternative product arm (n = 2) | Total (N = 9) | |
Actual IgHy10 treatment during the study | |||
IgHy10 infusion characteristics | |||
N with data available | 6 | NA | NA |
Total number of infusions | 26 | NA | NA |
Infusions per mother, median (IQR) | 4 (1.5, 5.75) | NA | NA |
Infusions administered at home, n (%) | 25 (96.2) | NA | NA |
Infusions administered at study site, n (%) | 1 (3.8) | NA | NA |
Infusion interval, n (%) | |||
3 weeks | 3 (50.0) | NA | NA |
4 weeks | 3 (50.0) | NA | NA |
Number of infusion sites, mean (range) | 1.85 (1 to 2) | NA | NA |
Ig infusion characteristics | |||
Maximum Ig infusion rate, mean (range) mL/hour, n = 3 | 273.3 (200 to 300) | NA | NA |
Ig infusion volume, mean (range) mL, n = 5 | 305.0 (100 to 500) | NA | NA |
rHuPH20 infusion characteristics | |||
Infusion volume, mean (range), mL, n = 3 | 15.0 (5 to 20) | NA | NA |
Changes to IgHy10 treatment during the study | |||
Type of regimen change, n (%) | |||
N with data available | 2 | 1 | 3 |
Ongoing treatment revised | 2 (100.0) | 0 (0.0) | 2 (66.7) |
Treatment stopped after enrolment | 0 (0.0) | NA | 0 (0.0) |
Treatment restarted after enrolment | NA | 1 (100.0) | 1 (33.3) |
Reason for dose change, n (%) | |||
N with data available | 2 | 1 | 3 |
Low IgG trough level | 1 (50.0) | 0 (0.0) | 1 (33.3) |
Adverse event | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Other | 1 (50.0) | 1 (50.0)a | 2 (66.7) |
Ig = immunoglobulin; IgG = immunoglobulin G; IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase; rHuPH20 = recombinant human hyaluronidase.
aIgHy10 treatment restarted after delivery for an unspecified reason.
Source: Clinical Study Report.50
Table 55: AEs, SAEs, Mortality, and WDAEs in Mothers and Infants in Registry Study 161301 (Enrolled Set)
Detail | Registry Study 161301 arm | ||
|---|---|---|---|
IgHy10 arm (n = 7) | Alternative product arm (n = 2) | Total (n = 9) | |
Mothers, n (%) | |||
N analyzed | 7 | 2 | 9 |
Patients with ≥ 1 AE | 3 (42.9) | 1 (50.0) | 4 (44.4) |
Patients with ≥ 1 local AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Patients with ≥ 1 immunologic AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Patients with ≥ 1 SAE | 1 (14.3) | 0 (0.0) | 1 (1.1) |
Blood and lymphatic system disorders | 1 (14.3) | 0 (0.0) | 1 (1.1) |
Thrombocytopenia | 1 (14.3) | 0 (0.0) | 1 (1.1) |
Pregnancy, puerperium, and perinatal conditions | 1 (14.3) | 0 (0.0) | 1 (1.1) |
Pre-eclampsia | 1 (14.3) | 0 (0.0) | 1 (1.1) |
SAE leading to death | 0 (0.0) | 0 (0.0) | 0 (0.0) |
WDAE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Infants, n (%) | |||
N analyzed | 5 | 2 | 7 |
Patients with ≥ 1 AE | 5 (100.0) | 1 (50.0) a | 6 (85.7) |
Patients with ≥ 1 SAE | 2 (40.0) | 0 (0.0) | 2 (28.6) |
Congenital, familial, and genetic disorders | 2 (40.0) | 0 (0.0) | 2 (28.6) |
Cleft lip without cleft palate | 1 (20.0) | 0 (0.0) | 1 (14.3) |
Talipes calcaneovalgus | 1 (20.0) | 0 (0.0) | 1 (14.3) |
SAE leading to death | 0 (0.0) | 0 (0.0) | 0 (0.0) |
WDAE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
AE = adverse event; IgHy10 = normal human immunoglobulin (10%) with recombinant human hyaluronidase; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Note: Analysis: descriptive statistics.
aOne AE was not included in the summary count (because these are reported as related or unrelated to IgHy10), which was a tear duct blockage in 1 infant.
Source: Clinical Study Report.50
Statistical Analysis in Registry Study 161301
There was no pre-specified minimum sample size for the registry. All mothers were analyzed according to their initially assigned study arm (regardless of changes in treatment during the study) and together. Infants were analyzed in the study arm of their mother. Patients were analyzed according to the type of enrolment. Those where the condition of the fetus was assessed prior to enrolment or the outcome of pregnancy was known prior to enrolment were considered the retrospective cohort, whereas those without this information prior to enrolment were considered the prospective cohort. There were no stopping rules established for the study. Outcomes were summarized using descriptive statistics for patients in each arm.
All data were collected and entered directly into the electronic data capture system, and all sites were fully trained in its use. The clinical report forms were completed by trained personnel. All AEs and SAEs were coded using MedDRA version 19.0 or newer. An overall summary is provided on the number and proportion of patients with AEs and SAEs and AEs causing dose reduction, interruptions, or withdrawal for expectant mothers and infants. The Wilson score method was used to calculate a point estimate (95% CI) for the proportion of patients with any AE. Separate analyses were conducted for local/immunologic AEs, which were considered to be AEs of special interest. AEs were classified by organ system and preferred term.
In general, there was no imputation of missing data for outcomes of interest. Statistical techniques were not used to identify and exclude any observations as outliers. If any data were continued spurious, it was excluded and documented. No sensitivity analyses were performed.
Pharmacoeconomic Review
Abbreviations
CBS
Canadian Blood Services
FSS
Federal Supply Schedule
IgHy10
normal immunoglobulin (human) 10% and recombinant human hyaluronidase
IgRT
immunoglobulin replacement therapy
IVIg
intravenous immunoglobulin
PID
primary immunodeficiency disorder
SCIg
subcutaneous immunoglobulin
SID
secondary immunodeficiency disorder
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Normal immunoglobulin (human) and recombinant human hyaluronidase (HyQvia) as a solution for subcutaneous infusion, available in the following strengths:
|
Submitted price | $91.8751 per gram of immunoglobulin, 10% Available as:
|
Proposed indication | As replacement therapy for primary humoral immunodeficiency and secondary humoral immunodeficiency in adult patients |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | January 14, 2022 |
Reimbursement request | As per indication |
Sponsor | Takeda Canada Inc. |
Submission history | Previously reviewed: No |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IgRT = immunoglobulin replacement therapy; NOC = Notice of Compliance.
Note: Submitted price listed in Canadian dollars.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Adult Canadian patients with primary immunodeficiency disorder (PID) or secondary immunodeficiency disorder (SID), aligned to the anticipated Health Canada indication |
Treatment | Human immunoglobulin and recombinant human hyaluronidase (IgHy10) |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Time horizon | One year |
Key data source | Study 160603 (pivotal study) and Study 160902 (extension study): phase III, open-label, non-randomized, single-group studies |
Costs considered | Drug acquisition costs, hospital costs, infusion supply costs, and CBS service costs |
Submitted results | IgHy10 has annual cost savings of $1,334 for patients with a PID and savings of $530 for patients with an SID compared with a weighted average of the overall distribution of IVIg and SCIg products |
Key limitations |
|
CADTH reanalysis results |
|
CBS = Canadian Blood Services; CMA = cost-minimization analysis; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency disorder; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency disorder.
Conclusions
Based on the CADTH Clinical Review, no direct comparison between treatment with normal immunoglobulin (human) 10% and recombinant human hyaluronidase (IgHy10) and intravenous immunoglobulin (IVIg) or subcutaneous immunoglobulin (SCIg) was identified. The clinical evidence informing the efficacy and safety of IgHy10 was based on the pivotal and corresponding safety extension study (Study 160603 and Study 160902, respectively), which are single-group, non-randomized open-label studies. Importantly, neither study provided any comparative evidence on the efficacy and safety of IgHy10 with IVIg and SCIg therapies; therefore, the comparative efficacy and safety of IgHy10 to IVIg and SCIg therapies is unknown.
Assuming equal efficacy and safety for IgHy10 and IVIg and SCIg therapies, the sponsor conducted a cost-minimization analysis over a 1-year time horizon comparing drug costs and health care resource use costs. At the submitted price, the annual cost of IgHy10 is estimated to be $54,888 per patient with either a primary immunodeficiency disorder (PID) or a secondary immunodeficiency disorder (SID), which is more expensive than all comparators. In the CADTH reanalysis, in adult patients with a PID or SID, IgHy10 is estimated to have per-patient incremental costs ranging from $14,731 to $35,250 compared with IVIg products, and per-patient incremental costs ranging from $16,061 to $22,821 compared with SCIg products, over a 1-year time horizon. For IgHy10 to be equal in cost to the least expensive comparator for both PID and SID, a 73% price reduction is required.
This cost comparison assumes clinical similarity between IgHy10 and IVIg and SCIg therapies. Evidence from the pivotal study (Study 160603) and the extension study (Study 160902) appraised by CADTH did not include comparative evidence for the efficacy and safety of IgHy10 relative to IVIg or SCIg. Given this lack of comparative data, the use of a cost-minimization analysis is inappropriate to explore the clinical uncertainty. Therefore, if clinical similarity cannot be assumed, the cost-effectiveness of IgHy10 is unknown.
Economic Review
The current review is for IgHy10 for adult patients in Canada with a PID or SID.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a cost-minimization analysis for IgHy10 compared with IVIg and SCIg therapies. The target population of the analysis was adult and pediatric patients in Canada older than 2 years of age with a PID or SID, as per the anticipated Notice of Compliance at submission. The target population for this review is aligned with the sponsor’s reimbursement request. The recommended dosage of IgHy10 is 300 mg/kg to 800 mg/kg at 3- to 4-week intervals after the initial ramp-up, as per the product monograph. The recommended dosing for the individual IVIg and SCIg therapies is variable and weight-dependent for each product, based on their respective product monographs. In the cost-minimization analysis, drug cost calculations are weight-dependent based on an average adult weight of 69 kg and an average pediatric weight of 27.5 kg. The total amount of drug (number of grams) is further weighted by the distribution of adult and pediatric patients assumed in the target population. Patients with a PID who received IVIg or IgHy10 were assumed to receive a dose of 0.5275 g/kg every 4 weeks, while patients with a PID receiving SCIg are estimated to receive a dose of 0.1319 g/kg every week. Patients with an SID receiving IVIg or IgHy10 are estimated to receive a dose of 0.5101 g/kg every 4 weeks, while patients with an SID receiving SCIg are estimated to receive a dose of 0.1275 g/kg every week.
The sponsor assumed no differences in efficacy or safety between IgHy10 and IVIg and SCIg therapies. As a result, all clinical benefits were assumed to be equivalent. The sponsor’s base case considered drug acquisition costs, health care resource utilization costs (e.g., physician fees and nurse time), infusion pumps and materials costs, and costs associated with Canadian Blood Services (CBS) and blood tests. The analysis was conducted from the perspective of the public health care payer over a 1-year time horizon to capture differences in costs in the first year due to differences in the total costs of treatment, including direct medical costs. Costs were weighted by the market share distribution of individual therapies comprising IVIg and SCIg. Based on these aggregated and averaged costs, IgHy10 was associated with savings of $1,344 among patients with a PID and $530 among patients with an SID in its first year of use.
Table 3: Summary of the Sponsor’s Economic Evaluation Results for PID
Drug | Total drug costs ($) | Incremental drug costsa ($) | Total costsb ($) | Incremental costsc ($) |
|---|---|---|---|---|
IgHy10 | 39,427 | Reference | 40,563 | Reference |
IVIg therapies | 32,652 | 6,775 | 37,976 | 2,587 |
SCIg therapies | 50,026 | −10,599 | 51,046 | −10,483 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency disorder; SCIg = subcutaneous immunoglobulin.
aIncremental drug costs are total drug costs of IgHy10 minus the total drug costs of the comparator. Negative incremental costs indicated that IgHy10 is less costly.
bFor IVIg and SCIg, total costs are a weighted average of the individual therapies that incorporates the market shares of the comparators.
cIncremental costs are the total direct costs of IgHy10 minus the total direct costs of the comparator. Negative incremental costs indicated IgHy10 is associated with savings.
Source: Sponsor’s economic submission.1
Table 4: Summary of the Sponsor’s Economic Evaluation Results for SID
Drug | Total drug costs ($) | Incremental drug costsa ($) | Total costsb ($) | Incremental costsc ($) |
|---|---|---|---|---|
IgHy10 | 41,780 | Reference | 42,917 | Reference |
IVIg | 34,601 | 7,179 | 39,926 | 2,991 |
SCIg | 53,013 | −11,232 | 54,013 | −11,096 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency disorder.
aIncremental drug costs are total drug costs of IgHy10 minus the total drug costs of the comparator. Negative incremental costs indicated that IgHy10 is less costly.
bFor IVIg and SCIg therapies, total costs are a weighted average of the individual therapies that incorporates the market shares of the comparators.
cIncremental costs are the total direct costs of IgHy10 minus the total direct costs of the comparator. Negative incremental costs indicated IgHy10 is associated with savings.
Source: Sponsor’s economic submission.1
The sponsor also conducted scenario analyses testing alternative assumptions over the 1-year time horizon, which included the following:
changing the average adult and pediatric weights to 70 kg and 40 kg, respectively
reducing IgHy10 infusion materials costs, whereby 100% of pump costs and 90% of infusion supply costs are covered by the sponsor
changing the total immunoglobulin dosage per month such that an adult patient would receive 28 g in 4 weeks (based on the average patient weights in the submitted cost-minimization analysis)
assuming 100% of IgHy10 and SCIg training-associated nursing costs would be covered by the sponsor
assuming, specific to SID only, that the treatment duration is 3 months
adjusting costs to account for inflation for hospital costs and infusion supplies based on Ho et al. (2008)2
changing the patient weight and dosing based on Ho et al. (2008).1,2
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations of the sponsor’s analysis that have notable implications on the economic analysis:
The assumption of similar clinical efficacy and safety for IgHy10, IVIg, and SCIg is uncertain: The sponsor submitted a cost-minimization analysis based on the assumption of similar clinical efficacy and safety for IgHy10, IVIg, and SCIg. The clinical evidence informing the efficacy and safety of IgHy10 was based on the pivotal and corresponding safety extension studies (Study 160603 and Study 160902, respectively), which are single-group, non-randomized open-label studies. Evidence based on the primary end point in Study 160603 (i.e., validated acute serious bacterial infection [VASBI] rate) demonstrated that IgHy10 was able to prevent VASBIs based on a threshold of fewer than 1.0 VASBI per year, which was considered substantial evidence of efficacy by the FDA Guidance to Industry and was clinically meaningful, according to the clinical experts consulted by CADTH. Patient-reported outcomes, such as those related to health-related quality of life, tolerability and adherence, and safety outcomes, were also reported but likely subject to bias due to the open-label study design. Whether this would bias the results in favour of or against IgHy10 is unknown. Importantly, neither study provided any comparative evidence on the efficacy and safety of IgHy10 versus IVIg and SCIg therapies; therefore, the comparative efficacy and safety of IgHy10 compared with IVIg and SCIg is unknown.
CADTH is unable to address this limitation.
The sponsor’s analysis is based on an indicated population that is outdated (i.e., pre-Notice of Compliance): The sponsor’s submission included a population of adult and pediatric patients (2 years of age and older) with primary and secondary humoral immunodeficiency, which was aligned with the indication that was anticipated at submission. However, the CADTH reanalysis is specific to an adult population and the pediatric population is excluded; this change aligns with the new indication, which was updated post-submission.
CADTH addressed this limitation by excluding the pediatric population in its reanalysis.
Pricing of IVIg and SCIg comparators is uncertain due to the confidential nature of plasma products: As IVIg and SCIg prices in Canada are confidential, drug costs for this review were derived from the US Federal Supply Schedule (FSS), based on the average price per gram across available sizes for each IVIg and SCIg product. Because acquisition costs for IVIg and SCIg therapies are confidential, it is plausible that Canadian costs are not reflective of the US prices provided and may result in a bias favouring IgHy10. To obtain the pricing of plasma products that are potentially more reflective of the Canadian setting, CADTH calculated prices for IVIg and SCIg comparators using available publicly listed prices from the Australian National Blood Authority for Privigen and Hizentra, respectively. The Australian and US costs for Privigen and Hizentra were converted to Canadian dollars and divided to create a cost multiplier assumed to be reflective of the reduced cost of IVIg (Privigen) and SCIg (Hizentra) products in Canada. This multiplier was then applied to all other IVIg and SCIg costs, excluding IgHy10, to derive Canadian costs (see Appendix 1 for a full list of the CADTH-calculated Canadian prices). CADTH consulted with CBS to determine which costs (i.e., US- or Australian-derived Canadian prices) best represent Canadian practice and, based on the CBS feedback, the Australian-derived Canadian costs were used in the CADTH base-case analysis.
CADTH addressed this limitation by replacing the sponsor’s submitted pricing for IVIg and SCIg using CADTH-calculated prices for IVIg and SCIg that were more reflective of the Canadian setting.
The sponsor inappropriately compared IgHy10 with weighted-average IVIg and SCIg costs rather than with individual comparators: In the submitted cost-minimization analysis, the sponsor compared the costs of IgHy10 with IVIg and SCIg therapies by calculating aggregate drug costs from the individual IVIg and SCIg treatments comprised within each drug class and weighted their costs according to their market share distribution. There are 2 key issues with the sponsor’s methodological approach for calculating IVIg and SCIg drug costs. First, a direct head-to-head comparison of costs should have been facilitated between IgHy10 and each individual SCIg and IVIg product rather than with the entire class of SCIg and IVIg therapies; this would ensure generalizability and transparency, as the relative use of each product can differ between jurisdictions. Further, as a cost-minimization analysis inherently assumes no clinical differences between IgHy10 and comparator treatments, there is no justification for any price premium between individual IVIg and SCIg therapies in each drug class. Second, there is uncertainty around the market share distribution that was assumed for each blood product. The sponsor’s estimates are not indication-specific, whereas the clinical experts consulted by CADTH noted there are likely to be differences between the distribution of brands of IVIg and SCIg for adult and pediatric patients with a PID or SID in comparison with the overall IVIg and SCIg market for all indications.
CADTH addressed this limitation by omitting weights for each comparator within the IVIg and SCIg groups and calculating incremental costs for each comparator against IgHy10.
The sponsor’s selected dose may have underestimated drug costs: In the submitted cost-minimization analysis, drug costs are calculated according to weight-based dosing assumptions.1 Specifically, the sponsor assumed that IgHy10 would be administered at a dose of 0.5275 g/kg every 4 weeks and 0.5101 g/kg every 4 weeks for patients with a PID or an SID, respectively. Further, this dose was assumed to be equivalent to the dose of all IVIg therapies. For all SCIg therapies, an identical dose of 0.1319 g/kg every week and 0.1275 g/kg weekly was assumed for patients with a PID or SID, respectively. The clinical experts consulted by CADTH affirmed that the estimated doses selected by the sponsor fell within the recommended ranges for IVIg (0.3 g/kg to 0.7 g/kg per month) and SCIg (IVIg dose divided by 4) used in Canadian clinical practice. However, apart from individual differences in dosing based on patient weight, the clinical experts indicated patients with a PID or SID are expected to receive the same dose. The CADTH’s clinical experts further indicated that the sponsor’s dose is lower than the dose used in Canadian clinical practice, as most patients with a PID or SID treated with IVIg would receive a dose between 0.6 g/kg and 0.7 g/kg every 4 weeks.
CADTH addressed these limitations by first assuming patients with a PID or SID would receive an equal dose. Second, CADTH used the midpoint of the anticipated dose range (i.e., 0.65 g/kg) that was provided by CADTH’s clinical experts for all IVIg products and further divided this midpoint dose by 4 to obtain a dose for all SCIg products.
Additional limitations were identified but were not considered to be key limitations. These limitations include the following:
The exclusion of training costs for IgHy10 likely underestimated costs: In the sponsor-submitted base case, the sponsor did not incorporate the first year’s training time to administer IgHy10, which was estimated to require the same amount of training time as SCIg (3 hours per training visit and a total of 5 home visits). Rather, the sponsor incorporated only training time for IgHy10 and SCIg in a scenario analysis. The clinical experts consulted by CADTH indicated that additional training time of approximately 15 to 30 minutes might be required by physicians and nurses to learn how to administer the IgHy10 subcutaneous infusion due to the fact an extra vial is needed during administration.
CADTH was unable to address this limitation.
CADTH Reanalyses of the Economic Information
The CADTH reanalyses comprised the following changes to the sponsor’s base case:
excluding pediatric patients with PID or SID
applying CADTH-calculated pricing for all IVIg and SCIg comparators
omitting weighted average costs of IVIg and SCIg comparators
revising dosing assumptions to calculate drug costs.
The results of the reanalyses are available in Table 5 and Table 6 for patients with a PID or SID, respectively. Over a 1-year time horizon, IgHy10 was associated with additional costs per person ranging from $14,731 to $35,250 across all IVIg and SCIg comparators in either a PID or SID population.
Table 5: Summary of the CADTH Reanalysis Results for Patients With PID
Drug | Drug costs ($) | Incremental drug costs ($)a | Total costs ($) | Incremental costs ($)b |
|---|---|---|---|---|
IgHy10 | 53,752 | Reference | 54,888 | Reference |
IVIg | ||||
Gamunex | 15,957 | 37,795 | 21,281 | 33,607 |
IGIVnex | 15,957 | 37,795 | 21,281 | 33,607 |
Gammagard Liquid | 15,752 | 37,999 | 21,077 | 33,811 |
Gammagard S/D | 23,890 | 29,862 | 29,215 | 25,674 |
Octagam | 14,314 | 39,437 | 19,639 | 35,250 |
Panzyga | 34,833 | 18,919 | 40,157 | 14,731 |
Privigen | 24,556 | 29,196 | 29,880 | 25,008 |
SCIg | ||||
Cutaquig | 35,131 | 18,621 | 36,129 | 18,759 |
Cuvitru | 31,069 | 22,683 | 32,067 | 22,821 |
Hizentra | 32,277 | 21,475 | 33,275 | 21,613 |
Xembify | 37,830 | 15,922 | 38,828 | 16,061 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency disorder; SCIg = subcutaneous immunoglobulin.
Note: Reanalyses are based on CADTH-calculated prices of the comparator treatments.
aIncremental drug costs are total drug costs for IgHy10 minus the total drug costs of the comparator, where negative incremental costs indicated IgHy10 is associated with savings.
bIncremental costs are the total direct costs of IgHy10 minus the total direct costs of the comparator, where negative incremental costs indicated IgHy10 is associated with savings.
Table 6: Summary of the CADTH Reanalysis Results for Patients With an SID
Drug | Drug costs ($) | Incremental drug costs ($)a | Total costs ($) | Incremental costs ($)b |
|---|---|---|---|---|
IgHy10 | 53,752 | Reference | 54,888 | Reference |
IVIg | ||||
Gamunex | 15,957 | 37,795 | 21,281 | 33,607 |
IGIVnex | 15,957 | 37,795 | 21,281 | 33,607 |
Gammagard Liquid | 15,752 | 37,999 | 21,077 | 33,811 |
Gammagard S/D | 23,890 | 29,862 | 29,215 | 25,674 |
Octagam | 14,314 | 39,437 | 19,639 | 35,250 |
Panzyga | 34,833 | 18,919 | 40,157 | 14,731 |
Privigen | 24,556 | 29,196 | 29,880 | 25,008 |
SCIg | ||||
Cutaquig | 35,131 | 18,621 | 36,129 | 18,759 |
Cuvitru | 31,069 | 22,683 | 32,067 | 22,821 |
Hizentra | 32,277 | 21,475 | 33,275 | 21,613 |
Xembify | 37,830 | 15,922 | 38,828 | 16,061 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; SCIg = subcutaneous immunoglobulin therapy; SID = secondary immunodeficiency disorder.
Note: Reanalyses are based on CADTH-calculated prices of the comparator treatments.
aIncremental drug costs are total drug costs of IgHy10 minus the total drug costs of the comparator, where positive values indicate savings with IgHy10.
bIncremental costs are the total direct costs of IgHy10 minus the total direct costs of the comparator, where positive values indicate savings with IgHy10.
CADTH conducted price reduction analyses to determine the price point at which IgHy10 would be equal to the least costly comparator (IVIg or SCIg), in addition to determining the price point at which IgHy10 would be equal to the least costly IVIg and SCIg therapy (Table 7). A 73% price reduction was required to equal the least costly comparator.
Table 7: CADTH Price Reduction Analyses
Scenario | Sponsor’s submitted price ($) | Reduction neededa (%) | Reduced price ($) | Savings relative to submitted pricea,b ($) |
|---|---|---|---|---|
Price reduction required to equal least costly comparatorc | 91.88 | 73% | 24.46 | 67.41 |
Price reduction required to equal least costly IVIg comparator | 91.88 | 73% | 24.46 | 67.41 |
Price reduction required to equal least costly SCIg | 91.88 | 42% | 53.10 | 38.77 |
IVIg = intravenous immunoglobulin; SCIg = subcutaneous immunoglobulin.
aRelative to publicly available list prices of comparators.
bSavings from the sponsor list price per patient for 1 year.
cLeast costly comparator among all comparators (IVIg and SCIg therapies).
Issues for Consideration
Access to IVIg and SCIg therapies across jurisdictions: The IVIg and SCIg therapies considered in the sponsor’s submitted cost-minimization analysis is a comprehensive list of the treatments relevant to the Canadian clinical setting, according to the clinical experts consulted by CADTH. However, access to individual therapies comprising IVIg and SCIg therapies is based on available supply from the local CBS location. The clinical experts consulted by CADTH noted the commonly used IVIg therapies in their jurisdictions included Gamunex, IGIVnex, Gammagard Liquid, Gammagard S/D, and Octagam and, for SCIg, the majority of patients were treated with Cuvitru, followed by Hizentra and Cutaquig.
Off-label use of IgHy10: Immunoglobulin therapy is used for many off-label conditions, such as immunologic autoimmune/neurologic conditions (which require higher doses). It is likely that IgHy10 will also be used off-label and require higher doses for several indications. The introduction of IgHy10 could increase the use of immunoglobulin therapies in these conditions; however, the concentration of immunoglobulins is lower in IgHy10 (10%) compared with current IVIg formulations (20%). It remains uncertain how many additional patients could use IgHy10 for off-label use.
Conclusions
Based on the CADTH Clinical Review report, evidence of a direct comparison between treatment with IgHy10 and IVIg or SCIg was not identified. The clinical evidence informing the efficacy and safety of IgHy10 was based on the pivotal and corresponding safety extension study (Study 160603 and Study 160902, respectively), which are single-group, non-randomized open-label studies. Importantly, neither study provided any comparative evidence on the efficacy and safety of IgHy10 with IVIg and SCIg, and therefore, the comparative efficacy and safety of IgHy10 to IVIg and SCIg therapies is unknown.
Assuming equal efficacy and safety for IgHy10 and IVIg and SCIg, the sponsor conducted a cost-minimization analysis over a 1-year time horizon comparing drug costs and health care resource use costs. At the submitted price, the annual cost of IgHy10 is estimated to be $54,888 per patient with an SID or PID, which is more expensive than all comparators. In the CADTH reanalysis, in adult patients with a PID or SID, IgHy10 is estimated to have per-patient incremental costs ranging from $14,731 to $35,250 compared with IVIg products and per-patient incremental costs ranging from $16,061 to $22,821 compared with SCIg products, over a 1-year time horizon. For IgHy10 to be equal in cost to the least expensive comparator for both PID and SID, a 73% price reduction is required.
This cost comparison assumes clinical similarity between IgHy10 and IVIg and SCIg therapies. Evidence from the pivotal study (Study 160603) and the extension study (Study 160902) appraised by CADTH did not include comparative evidence for the efficacy and safety of IgHy10 relative to IVIg or SCIg therapies. Given this lack of comparative data, the use of a cost-minimization analysis is likely inappropriate. Therefore, if clinical similarity cannot be assumed, the cost-effectiveness of IgHy10 is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: HyQvia® normal immunoglobulin (human) 10% 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL and recombinant human hyaluronidase 200 units/1.25 mL, 400 units/2.5 mL, 800 units/5 mL, 1600 units/10 mL and 2400 units/15 mL solution for subcutaneous infusion. Toronto (ON): Takeda Canada Inc.; 2021 Jun 24.
2.Ho C, Membe S, Cimon K, Roifman C, Kanani A, Morrison A. Overview of subcutaneous versus intravenous immunoglobulin for primary immunodeficiencies: systematic review and economic evaluation. (Technology overview number 36). Ottawa (ON): CADTH; 2008.
3.National Blood Authority annual report 2019-20: Appendix 3. Plasma and recombinant products supplied under contract in 2019-20. Lyneham (AU): National Blood Authority Australia; 2020: https://www.transparency.gov.au/annual-reports/national-blood-authority/reporting-year/2019-20-16. Accessed 2021 Sep 27.
4.IGIVnex® (immune globulin intravenous (human), 10%): injectable solution [product monograph]. Mississauga (ON): Grifols Canada Ltd.; 2019: https://www.grifols.com/documents/89713601/0/IGIVnex+-+English+PM+-+2017-07-12.pdf/98eb0376-0d61-491a-9fda-713505c61379. Accessed 2021 Sep 14.
5.Gammagard S/D (immune globulin intravenous (human) [IGIV], solvent/detergent-treated (freeze-dried concentrate)): 5 g/vial & 10 g/vial [product monograph]. Toronto (ON): Takeda Canada Inc.; 2021: https://pdf.hres.ca/dpd_pm/00046330.PDF. Accessed 2021 Sep 27.
6.Octagam® 5% (imunoglobulin intravenous (human)): solution for infusion, 50 mg/mL [product monograph]. Toronto (ON): Octapharma Canada Inc.; 2018: https://pdf.hres.ca/dpd_pm/00048166.PDF. Accessed 2021 Sep 14.
7.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: HyQvia® normal immunoglobulin (human) 10% 2.5 g/25 mL, 5 g/50 mL, 10 g/100 mL, 20 g/200 mL, 30 g/300 mL and recombinant human hyaluronidase 200 units/1.25 mL, 400 units/2.5 mL, 800 units/5 mL, 1600 units/10 mL and 2400 units/15 mL solution for subcutaneous infusion. Toronto (ON): Takeda Canada Inc.; 2021 Jun 24.
8.Immunodeficiency Canada. Primary immunodeficiency (PI). 2021; https://immunodeficiency.ca/primary-immunodeficiency/primary-immunodeficiency-pi/. Accessed 2021 Sep 14.
9.Lindegren ML, Kobrynski L, Rasmussen SA, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Recomm Rep. 2004;53(RR-1):1-29. PubMed
Appendix 1: Additional Economic Information
Cost Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Primary and Secondary Immunodeficiency
Treatment | Strength and concentration | Form | Price per gram | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
IgHy10 | Normal Immunoglobulin (Human) 10%: 2.5 g/25 mL 5 g /50 mL 10 g/100 mL 20 g/200 mL 30 g/ 300 mL Recombinant human hyaluronidase (160 U/mL): 200 U/ 1.25 mL 400 U/2.5 mL 800 U/5 mL 1,600 U/10 mL 2,400 U/15 mL | Dual vial unit | $91.8751a | 0.3 to 0.8 g/kg every 3 to 4 weeks | Adult: 74.00 to 253.73 Pediatric: 31.72 to 99.68 | Adult: 27,011 to 92,610 Pediatric: 11,576 to 36,383 |
Intravenous Immunoglobulins (Human), 10% | ||||||
Gamunex b | 5 g/50 mL 10 g/ 100mL 20 g/ 100mL | Injectable solution | $27.2743 | 0.1 and 0.6 g/kg every 3 or 4 weeks | Adult: 7.32 to 56.49 Pediatric: 3.14 to 22.87 | Adult: 2,673 to 20,619 Pediatric: 1,146 to 8,346 |
IGIVnex c | 10% solution | Injectable solution | $27.2743 | 0.1 and 0.6 g/kg every 3 or 4 weeks | Adult: 7.32 to 56.49 Pediatric: 3.14 to 22.87 | Adult: 2,673 to 20,619 Pediatric: 1,146 to 8,346 |
Gammagard Liquid b | 1 g/10 mL 2.5 g/25 mL 5 g/50 mL 10 g/100 mL 20 g/200 mL 30 g/300mL | Solution for infusion | $26.9247 | Starting dose: 0.4 to 0.8 g/kg Thereafter: 0.2 to 0.8 g/kg | Initial: Adult: 28.92 to 74.36 Pediatric: 11.36 to 29.21 Subsequent: Adult: 14.46 to 111.53 Pediatric: 6.20 to 43.82 | Initial: Adult: 10,554 to 27,140 Pediatric: 4,146 to 10,662 Subsequent: Adult: 5,277 to 40,710 Pediatric: 2,262 to 15,993 |
Gammagard S/Dd | 5 g 10 g | Vial | $40.8342 | At least 0.1 mg/kg, monthly recommended. (Initially, patients may receive 0.2 to 0.4 g/kg) | Adult: 9.40 Pediatric: 4.03 | Adult: 3,430 Pediatric: 1,470 |
Octagamb,e | 5 g/50 mL 10 g/100 mL 20 g/100 mL 50 mg/mL | Solution for infusion | $24.4666 | Replacement therapy in PID: Monthly doses of at least 0.1 g/kg recommended. Replacement therapy in SIDs: 0.2 to 0.4 g/kg body weight every 3 to 4 weeks | Adult: 16.89 to 26.28 Pediatric: 7.24 to 10.32 | Adult: 6,166 to 9,591 Pediatric: 2,642 to 3,964 |
Panzyga | 100 mg/mL | Solution for infusion | $59.5375 | 0.2 to 0.8 g/kg, every 3 to 4 weeks | Adult: 31.97 to 164.42 Pediatric: 13.70 to 64.59 | Adult: 11,669 to 60,013 Pediatric: 5,001 to 23,577 |
Privigen b | 5 g/50 mL 10 g/ 100 mL 20 g/200 mL 40 g/ 400 mL | Solution for infusion and Intravenous | $41.9715 | 0.2 to 0.8 g/kg, every 3 to 4 weeks | Adult: 22.54 to 115.91 Pediatric: 9.66 to 45.54 | Adult: 8,226 to 42,307 Pediatric: 3,526 to 16,621 |
Subcutaneous immunoglobulins | ||||||
Cutaquig | 165 mg/mL Presentation sizes: 6 mL, 10 mL, 12 mL, 20 mL, 24 mL, 48 mL | 16.5% solution for injection | $60.0474 | Loading dose: at least 0.2 to 0.5 g/kg | Adult: 61.03 to 122.07 Pediatric: 26.16 to 52.32 | Adult: 22,278 to 44,555 Pediatric: 9,548 to 19,095 |
Cuvitru | 1 g/5 mL 2 g/10 mL 4 g/20 mL 10 g/50 mL 200 mg/mL (20%) | 20% solution for subcutaneous infusion | $53.1045 | At least 5 to 6 g/L A loading dose of at least 0.2 to 0.5 g/kg (1 to 2.5 mL/kg) body weight may be required. This may need to be divided over several days, with a maximal daily dose of 0.1 to 0.15 g/kg | Adult: 371.73 to 584.15 Pediatric: 159.31 to 265.52 | Adult: 135,682 to 213,215 Pediatric: 58,149 to 96,916 |
Hizentra b | 1 g/5 mL 2 g/10 mL 4 g/20 mL 10 g/50 mL 20% solution | 20% solution for subcutaneous injection | $55.1692 | 0.1 to 0.2 g/kg weekly | Adult: 56.08 to 112.15 Pediatric: 24.03 to 48.07 | Adult: 20,468 to 40,936 Pediatric: 8,772 to 17,544 |
Xembify | 20% solution | 20% solution for subcutaneous use | $64.6602 | 0.1 to 0.2 g/kg | Adult: 65.72 to 131.45 Pediatric: 28.17 to 56.33 | Adult: 23,989 to 47,978 Pediatric:10,281 to 20,562 |
IVIg = intravenous immunoglobulin; SCIg = subcutaneous immunoglobulin.
Note: Prices calculated by CADTH, unless otherwise indicated, and do not include dispensing fees. Assumes average adult weight of 69 kg and average pediatric weight of 27.5 kg. Strength and concentration for IVIg and SCIg products obtained from the Australian National Blood Authority price list.3 The pediatric costs presented reflect the outdated anticipated NOC at sponsor submission.
aSponsor’s submitted price.1
bNational Blood Authority Annual Report 2019-20.3
cObtained from the product monograph for IVIGnex.4
dObtained from the product monograph of Gammagard S/D.5
eObtained from the product monograph of Octagam 5%.6
Appendix 2: Submitted BIA and CADTH Appraisal
Table 9: Summary of Key Take-Aways
Key Take-Aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor assessed the budget impact of the introduction of IgHy10 for patients with PID and SID, from the perspective of the public drug plans in Canada (excluding Quebec), over a 3-year time horizon.7 The sponsor included drug acquisition costs in their base case, while costs of infusion pumps and infusion materials were included as part of a scenario analysis undertaken from the perspective of the public health care payer. Using the same perspective, the sponsor further included a scenario that examined the incremental budget impact of including the cost of physician and nurses and CBS and blood test costs. In the reference scenario, the sponsor assumed that patients receive IVIg and SCIg, which are comprised of a mix of individual treatments. In the new drug scenario, IgHy10 was assumed to capture market share from IVIg and SCIg therapies disproportionally. Drug costs of IgHy10 were calculated based on the average patient weights for adults and children, respectively, based on the pivotal study.7
The sponsor used an epidemiological approach to identify the total population eligible for treatment with IgHy10. The sponsor estimated the number of PID patients eligible (1 in 1,200 people living in Canada or 0.083%) for immunoglobulin treatment based on the pan-Canadian population.8 Among patients with PID, the sponsor assumed that approximately 30% of patients are diagnosed, and all diagnosed PID patients are eligible for treatment with immunoglobulin.8 The sponsor further applied an incidence rate of 0.01% to estimate the increase in the number of PID patients eligible for treatment with immunoglobulin over time, and assumed that this annual rate remained constant.9 A discontinuation rate of 10% was applied to this estimate to account for the proportion of PID patients who were assumed to discontinue immunoglobulin replacement therapy (IgRT) annually. Finally, the sponsor estimated that approximately 35% of PID and SID patients would have SID.7
The sponsor’s BIA also included the following key assumptions:
Contract agreements with CBS for the purchases of plasma protein products, including, Igs, are confidential. In the absence of Canadian pricing for immunoglobulin therapies, the prices per gram of the comparators were referenced from the US Department of Veteran Affairs FSS, as a proxy. Given the uncertainty of usage across the available sizes for each comparator, the average costs per gram was used as the price per gram for each comparator in the cost calculations for the BIA. In the absence of data, the price per gram of IGIVnex was assumed to be the same as Gamunex since both consist of immune globulin intravenous (human), 10% manufactured by Grifols Therapeutics Inc.7
The monthly amount of immunoglobulin a patient receives is assumed to be the same regardless of the route of administration (i.e., IVIg or SCIg).7
Dosing for IVIg and SCIg therapies were calculated based on an average adult weight of 69.0 kg and an average pediatric patient weight of 27.5 kg, derived from the pivotal Study 160603.7
The price per gram of IgHy10, as submitted by the sponsor, is US$75.7670 ($91.8751), and the dose for IgHy10 (e.g., grams per 4 weeks) is assumed to be equivalent to current immunoglobulin therapies.7
PID patients receiving IVIg or IgHy10 are estimated to receive a dose of 0.5275 g/kg every 4 weeks, while PID patients receiving SCIg therapies are estimated to receive a dose of 0.1319 g/kg every week (i.e., for SCIg therapies the dosage is administered weekly but assumed to be the same dose as IVIg therapies over a 4-week period).
SID patients receiving IVIg or IgHy10 are estimated to receive a dose of 0.5101 g/kg every 4 weeks, while SID patients receiving SCIg therapies are estimated to receive a dose of 0.1275 g/kg every week.
Key inputs to the BIA are documented in Table 10.
Table 10: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as baseline / year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Prevalence of PID | 0.083% |
Incidence of PID | 0.01% |
Diagnosis rate | 34% / 35% / 36% / 36% |
Treated with immunoglobulin | 100% |
Discontinuation rate | 10% |
Eligible PID patients | 7,996 / 8,258 / 8,522 / 8,776 |
Eligible SID patients | 4,306 / 4,446 / 4,589 / 4,726 |
Total number of eligible patients for drug under review | 12,302 / 12,704 / 13,112 / 13,502 |
Market uptake (3 years) | |
Uptake in the PID population (reference scenario) IVIg Gamunex IGIVnex Gammagard Liquid Gammagard S/D | 64.5% / 61.0% / 55.0% 14.2% / 13.4% / 12.1% 15.5% / 14.6% / 13.2% 13.5% / 12.8% / 11.6% 0.0% / 0.0% / 0.0% |
Octagam Panzyga Privigen SCIg Cutaquig Cuvitru Hizentra Xembify | 5.8% / 5.5% / 5.0% 3.2% / 3.1% / 2.8% 12.3% / 11.6% / 10.5% 35.5% / 39.0% / 45.0% 1.8% / 2.7% / 5.4% 30.2% / 31.6% / 28.3% 3.6% / 4.7% / 6.8% 0.0% / 0.0% / 4.5% |
Uptake in the SID population (reference scenario) IVIg Gamunex IGIVnex Gammagard Liquid Gammagard S/D Octagam Panzyga Privigen SCIg Cutaquig Cuvitru Hizentra Xembify | 69.5% / 66.0% / 60.0% 15.3% / 14.5% / 13.2% 16.7% / 15.8% / 14.4% 14.6% / 13.9% / 12.6% 0.0% / 0.0% / 0.0% 6.3% / 5.9% / 5.4% 3.5% / 3.3% / 3.0% 13.2% / 12.5% / 11.4% 30.5% / 34.0% / 40.0% 1.5% / 2.4% / 4.8% 25.9% / 27.5% / 25.2% 3.1% / 4.1% / 6.0% 0.0% / 0.0% / 4.0% |
Uptake in the PID population (new drug scenario) IVIg Gamunex IGIVnex Gammagard Liquid Gammagard S/D Octagam Panzyga Privigen | 62.2% / 56.6% / 49.1% 13.7% / 12.5% / 10.8% 14.9% / 13.6% / 11.8% 13.1% / 11.9% / 10.3% 0.0% / 0.0% / 0.0% 5.6% / 5.1% / 4.4% 3.1% / 2.8% / 2.5% 11.8% / 10.8% / 9.3% |
SCIg Cutaquig Cuvitru Hizentra Xembify | 34.4% / 36.9% / 41.9% 1.7% / 2.6% / 5.1% 29.2% / 29.9% / 26.4% 3.4% / 4.4% / 6.3% 0.0% / 0.0% / 4.2% |
Uptake in the SID population (new drug scenario) IVIg Gamunex IGIVnex Gammagard Liquid Gammagard S/D Octagam Panzyga Privigen SCIg Cutaquig Cuvitru Hizentra Xembify | 67.2% / 61.6% / 54.0% 14.8% / 13.5% / 11.9% 16.1% / 14.8% / 13.0% 14.1% / 12.9% / 11.3% 0.0% / 0.0% / 0.0% 6.0% / 5.5% / 4.9% 3.4% / 3.1% / 2.7% 12.8% / 11.7% / 10.3% 29.4% / 31.9% / 37.0% 1.5% / 2.2% / 4.5% 25.0% / 25.8% / 23.3% 2.9% / 3.8% / 5.6% 0.0% / 0.0% / 3.7% |
Cost of treatment (per patient, per year) ($) | |
Cost of treatment over 1 year ($); PID IgHy10 | 39,427 |
IVIg Gamunex IGIVnex Gammagard Liquid Gammagard S/D Octagam Panzyga Privigen | 28,959 28,959 28,588 43,357 25,978 63,216 44,564 |
SCIG Cutaquig Cuvitru Hizentra Xembify | 55,983 49,510 51,435 60,283 |
Cost of treatment over 1 year ($); SID IgHy10 | 41,780 |
IVIG Gamunex IGIVnex Gammagard Liquid Gammagard S/D Octagam Panzyga Privigen | 30,688 30,688 30,295 45,945 27,529 66,990 47,225 |
SCIG Cutaquig Cuvitru Hizentra Xembify | 59,325 52,466 54,505 63,882 |
IgHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency disorder; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency disorder.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s submitted base case suggested that the introduction of IgHy10 for the treatment of PID in adult and pediatric patients would result in an incremental budget of $200,278 in year 1, $307,131 in year 2, and a savings of $99,643 in year 3, for a total incremental budget impact of $407,766 over the 3-year time horizon.7
Results of the sponsor’s submitted base case suggested that the introduction of IgHy10 for the treatment of SID in adult and pediatric patients would result in an incremental budget of $138,344 in year 1, $223,342 in year 2, and $17,366 in year 3, for a total incremental budget impact of $379,053 over the 3-year time horizon.7
CADTH Appraisal of the Sponsor’s BIA
CADTH identified the following key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The sponsor’s submitted base case is based on an outdated pre-NOC indicated population: The sponsor’s submission included a population with primary humoral immunodeficiency and secondary humoral immunodeficiency in adult and pediatric patients (2 years of age and older), which was aligned with the anticipated indication at submission. However, the CADTH base case is specific to an adult population and the pediatric population is excluded; this change aligns with the new anticipated indication, which has been updated post-submission.
CADTH addressed this limitation by excluding the pediatric population in its reanalysis. CADTH notes that given the small proportion of pediatric patients, this change has a minor impact on the overall budget impact estimates.
Pricing of IVIg and SCIg comparators is uncertain in the Canadian setting: In the sponsor-submitted budget impact analysis, drug costs were derived from the US FSS based on the average price per gram across available sizes for each individual IVIg and SCIg, aligned with the sponsor’s submitted pharmacoeconomic analysis (see CADTH’s Appraisal of the Sponsor’s Economic Information). Due to the confidential nature of blood acquisition products in Canada and sources for IVIg and SCIg prices in contexts comparable to the Canadian setting, drug cost calculations using US pricing may overestimate the costs associated with comparator treatments in the Canadian setting.
CADTH addressed this limitation by revising all the prices of IVIg and SCIg comparators using known Australian prices, to align with CADTH’s base-case pharmacoeconomic analysis.
The anticipated market uptake of IgHy10 in the new drug scenario is likely underestimated: The sponsor assumed that IgHy10 would capture 3.4%, 6.5%, and 9.0% of the market share from IVIg and SCIg comparator treatments in year 1, year 2, and year 3, respectively, in the new drug scenario. The clinical experts consulted by CADTH indicated that this appeared to underestimate the anticipated uptake of IgHy10. Specifically, CADTH’s clinical experts noted that IgHy10 would likely comprise a larger share of the SCIg market (i.e., 30 to 40% of the SCIg market) as IgHy10 is most likely to compete with SCIg products due to the appeal of monthly infusions to patients. The experts further noted that the uptake of IgHy10 may be higher among adults rather than in children due to their smaller body size, which may result in fewer injection sites; however, the sponsor did not account for this in the submitted BIA
CADTH addressed the anticipated uptake of IgHy10 in the new drug scenario based on expert feedback, such that IgHy10 would capture 30%, 35% and 40% of the overall market share captured by SCIgs. In a scenario analysis, CADTH explored the impact of the same higher uptake (i.e., 30%, 35%, 40%) across all IVIgs and SCIgs over the 3-year time horizon.
The sponsor’s selected dose for IVIg and SCIg comparator treatments likely underestimates drug costs: In the submitted BIA, drug costs were calculated using weight-based dosing for adults and pediatric patients based on their average weight. The sponsor assumed an identical dose for all IVIgs, and for all SCIgs (i.e., provided weekly at one-quarter of the dose administered for IVIg). This was deemed reasonable according to CADTH’s clinical experts. However, the dose selected by the sponsor was noted to be slightly lower than the dose used in Canadian clinical practice, as patients with PID or SID treated with IVIg likely receive a dose between 0.6 and 0.7 g/kg every 4 weeks (see CADTH’s Appraisal of the Sponsor’s Economic Information).
CADTH addressed this limitation by first assuming patients with PID and SID would receive an equal dose, and second, CADTH changed the doses assigned to all IVIgs and SCIgs to 0.65 g/kg every 4 weeks and 0.1625 g/kg every week, respectively, to align with the CADTH pharmacoeconomic analysis.
Discontinuation rates may differ between the adult and pediatric populations, and do not align with clinical expectations: The sponsor assumed that the proportion of patients who discontinue treatment with IgRT therapy is the same in adult and pediatric populations, however, clinical experts consulted by CADTH indicated that this may differ between the 2 populations. Further, the discontinuation rate assumed by the sponsor (10%) was thought to be higher than expected according to CADTH’s clinical experts, who suggested that a discontinuation rate less than 5% was more appropriate since a very small proportion of patients are likely to discontinue in Canadian clinical practice. As such, there remains uncertainty with the sponsor’s assumed discontinuation rate in the BIA, and by overestimating the discontinuation rate, it is likely to bias the results by underestimating total costs.
CADTH addressed this limitation by changing the discontinuation rate to 4%. In scenario analyses, CADTH explored the assumption that all patients remained on treatment (i.e., no discontinuation rate, or 0%).
Uncertainty regarding the number of patients eligible to receive IgHy10: The sponsor used an epidemiological approach to identify the patient population eligible to receive IgHy10. The clinical experts consulted by CADTH noted several areas of uncertainty with the estimates and assumptions used to derive the target population size. First, the clinical experts consulted by CADTH highlighted uncertainty in the estimated number of patients with PID eligible for treatment. Specifically, CADTH’s clinical experts noted that it was unclear whether the target population truly reflected patients with primary humoral immunodeficiency within the broader population of patients with primary immunodeficiency, based on the sponsor’s assumption that all diagnosed patients with PID would be treated with immunoglobulin. CADTH’s clinical experts noted that patients with a selective immunoglobulin A (IgA) deficiency have a less severe defect and likely would not be treated with immunoglobulin. Second, among patients with PID, the sponsor assumed an annual incidence rate of 0.01%, which CADTH’s clinical experts indicated was lower than expected. Altogether, the inputs and assumptions used to estimate the eligible population size resulted in uncertainty.
CADTH was unable to address this limitation.
The market share distribution of IVIg and SCIg comparators in the reference scenario are uncertain and do not align with clinical expectations: In the submitted BIA, the sponsor assumed market share estimates for each comparator were based on internal data, such that for patients with PID, IVIgs were assumed to have the majority (70%) of the market share and SCIgs were assumed to have the remaining (30%) market share. For patients with SID, IVIgs were assumed to have a slightly higher market share (75%), and SCIgs were assumed to have the remaining (25%) market share. The clinical experts consulted by CADTH indicated that the sponsor’s market shares did not align with their clinical expectations. Specifically, clinical experts indicated there would be a higher use of SCIgs and lower use of IVIgs among the PID and SID populations. CADTH’s clinical experts affirmed that it is reasonable to expect an increased utilization of SCIg products over time for both the PID and SID populations, and that the market uptake would be similar for adults and children. The experts noted that the sponsor’s assumed uptake rates of each of the individual therapies within the IVIg and SCIg groups were uncertain.
CADTH was unable to address this limitation due to structural constraints imposed by the sponsor’s submitted model and therefore, was unable to explore an alternate market share distribution for the reference scenario in a scenario analysis.
The sponsor’s submitted model for the budget impact analysis is not user-friendly and unnecessarily complicated: Several of the model inputs and assumptions in the sponsor’s submitted budget impact analysis were difficult to test or modify with alternate inputs and assumptions due to the unnecessary complexity (e.g., illogical formulas, disconnected worksheets, non-functioning cells) and structural constraints.
CADTH was unable to address this key limitation.
CADTH Reanalyses of the BIA
A table noting the changes made to the sponsor’s BIA as part of the CADTH reanalysis is available in Table 11.
Table 11: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH base case | ||
1. Submission based on a pre-NOC indicated population | Includes adult and pediatric patients with PID and SID. | Excludes pediatric patients with PID and SID from eligible population, based on the anticipated post-NOC indicated population. |
2. Pricing | US pricing. | Cost multiplier to derive prices more likely to be reflective of Canadian setting. |
3. Market share estimates in the new drug scenario | Assumes that IgHy10 will capture the following market share uptake from IVIgs and SCIgs over the 3-year time horizon: 3.4% / 6.5% / 9.0% | Assumes that IgHy10 will capture the following market share uptake from IVIgs and SCIgs over the 3-year time horizon: 11.38% / 15.05% / 18.30% |
4. Dosing | Different doses assumed for PID and SID patients PID patients:
SID patients:
| Identical dose assumed for PID and SID patients for IVIgs and SCIgs
|
5. Discontinuation rate | 10% | 4% |
CADTH base case | Reanalyses 1 + 2 + 3 + 4 + 5 | |
gHy10 = normal immunoglobulin (human) 10% and recombinant human hyaluronidase; IVIg = intravenous immunoglobulin; PID = primary immunodeficiency; SCIg = subcutaneous immunoglobulin; SID = secondary immunodeficiency.
Applying the changes in Table 11 resulted in an increase in the estimated budget impact of $43,636,227 in year 1, $62,188,520 in year 2, and $80,037,821 in year 3, for a 3-year total budget impact of $185,862,568 for adult patients with PID and SID. The results of the CADTH stepwise reanalyses are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. For adult SID and PID patients, the 3-year total budget impact was $113,798,853 and $72,063,715, respectively.
Table 12: Summary of the CADTH Reanalyses of the BIA (Patients With PID and SID)
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $786,819 |
CADTH reanalysis 1 | $755,607 |
CADTH reanalysis 2 | $56,021,873 |
CADTH reanalysis 3 | $2,196,722 |
CADTH reanalysis 4 | $985,395 |
CADTH reanalysis 5 | $873,484 |
CADTH base case (Reanalysis 1 + 2 + 3 + 4 + 5) | $185,862,568 |
BIA = budget impact analysis; PID = primary immunodeficiency; SID = secondary immunodeficiency.
Table 13: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base casea | Reference | $471,575,148 | $504,871,345 | $529,742,395 | $568,181,274 | $1,602,795,014 |
New drug | $471,575,148 | $505,209,967 | $530,272,868 | $568,098,997 | $1,603,581,833 | |
Budget impact | $0 | $338,622 | $530,473 | –$82,277 | $786,819 | |
CADTH base case | Reference | $245,310,037 | $279,032,860 | $308,406,161 | $348,136,637 | $935,575,658 |
New drug | $245,310,037 | $322,669,088 | $370,594,680 | $428,174,459 | $1,121,438,227 | |
Budget impact | $0 | $43,636,227 | $62,188,520 | $80,037,821 | $185,862,568 |
BIA = budget impact analysis.
aThe sponsor-submitted base case was based on the anticipated pre-NOC indication at submission, and therefore includes both an adult and pediatric population. The new anticipated pre-NOC indication is specific to an adult population.
CADTH conducted the following additional scenario analyses from the drug plan perspective (Scenarios 1 and 2, Table 14):
Assumed a higher market share uptake of IgHy10 (30%, 35%, and 40% in years 1-3)
Assumed that no patients would discontinue treatment (discontinuation rate = 0%)
The scenario analysis assuming a higher uptake of IgHy10 resulted in an increase budget impact compared to the CADTH base case, from $185,862,568 over 3 years to $434,604,513. When assuming no patients discontinued IgRT therapy, the expected budget impact increased to $203,130,379 over 3 years.
Table 14: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1 | Reference | $245,310,037 | $279,032,860 | $308,406,161 | $348,136,637 | $935,575,658 |
New drug | $245,310,037 | $394,066,851 | $453,030,625 | $523,082,695 | $1,370,180,171 | |
Budget impact | $0 | $115,033,991 | $144,624,464 | $174,946,058 | $434,604,513 | |
CADTH scenario analysis 2 | Reference | $245,000,558 | $294,183,168 | $335,291,853 | $389,359,092 | $1,018,834,113 |
New drug | $250,000,558 | $340,188,659 | $402,901,735 | $478,874,099 | $1,221,964,492 | |
Budget impact | $0 | $46,005,490 | $67,609,881 | $89,515,007 | $203,130,379 |
Note: All scenario analyses are conducted based on the CADTH base case undertaken from the drug program plan perspective.
BIA = budget impact analysis.
Stakeholder Input
Patient Group Input
The Canadian Immunodeficiencies Patient Organization
About The Canadian Immunodeficiencies Patient Organization
The Canadian Immunodeficiencies Patient Organization (CIPO) is committed to creating awareness about primary immunodeficiency (PI), to advocate for new treatments, to help speed the diagnosis of patients, and to enable patients to become champions for their own quality of life. We equip patients, caregivers, family members and health care providers with the information, tools and resources they need to ensure that those affected by PI can live healthy and productive lives. http://www.cipo.ca/
Information Gathering
CIPO conducted an online survey of patients and caregivers from June 8, 2021 to June 27, 2021, to assess the challenges patients and caregivers face as a result of Primary Immunodeficiency (PI) and their experience with current treatments for PI.
A total of 246 individuals responded to the survey (n=246). 244 are from Canada, and 2 patients are from the U.S. 233 (95%) were individuals living with primary immunodeficiency, and 13 (5%) were caregivers answering the survey on behalf of patients. The survey contained the use of free-form commentary, scoring options and limited closed questions.
Additionally, semi-structured telephone interviews were conducted with eight (8) patients who are currently using either intravenous (IVIG) or subcutaneous (SCIG) immunoglobulin replacement therapy (IG) treatments for their PI.
This report reflects the results of the survey and patient interviews, as well intelligence and insights CIPO has garnered from more than two decades of experience in patient support and advocacy related to Primary Immunodeficiency.
Note: CIPO had hoped to identify patients that had experience with the treatment under review but there has been no recent experience in Canada with HyQvia. And, we were unable to contact the few patients in Vancouver that participated in the Phase III trial for this treatment that ran from December 2008 to November 2010. (ClinicalTrials.gov Identifier: NCT00814320).
Disease Experience
Primary Immunodeficiencies (PI) are a group of inherited and genetic defects of the immune system, resulting in an immune system which is partly or totally missing or does not function properly. These deficiencies make people with PI more prone to a wide range of infections (viral, bacterial and fungal) which can include: skin infections, sinopulmonary infections, and infections of the intestines. These infections are often chronic and debilitating.
We asked patients: Prior to starting immunoglobulin therapy (IVIG or SCIG) to treat your PI, approximately how many infections were you experiencing per year?
There was a wide range of reported infections (per year) prior to starting IG therapy, with the majority ranging from 2-12 infections per year. We plotted the infections per year of 184 patients who reported between 0 – 20 infections per year. However, 7 additional patients reported 30+ annual infections (not included in the plot below). The average # of infections per year was 8.12.
Below are excerpts from patient commentary regarding their experience with infections prior to starting IG therapy:
3 months of continuous illnesses that ended with shingles. In 6 months, every 10 days had no energy & in July had my first summer cold. Oct, had a near-death experience. Had encephalitis & pneumonia due to the severity Had severe coughs for years & had IBS.
“Previous to the year I was diagnosed, I had 1 or 2 (infections) per year. In the year leading up to my diagnosis I had recurrent sinus infections and extreme fatigue for 5-6 months that culminated in a severe pneumonia which I developed while on extended vacation in overseas.”
“Became ongoing mesenteric vasculitis with no relief. On top of consistent skin infections, mucous membrane ulcers.”
“Too many (infections) to count. Before being diagnosed late in elementary school I would miss sometimes a month of school. I was always sick with something and had constant ear and throat infections. Most colds or flues would leave me delirious it would get so bad.”
“I was experiencing sinus infections requiring antibiotics every 4-6 weeks for many years”.
We also asked patients: After starting immunoglobulin therapy (IVIG or SCIG) to treat your PI approximately how many infections are you now experiencing per year?
The frequency of infections changed dramatically. We plotted the infections per year of 207 patients who reported between 0 – 20 infections per year. (There were no patients reporting infection rates of over 20 per year). The average # of infections per year, for patients who had commenced immunoglobulin therapy, went down to 2.05 infections per year (from an average of 8.12 infections per year for patients prior to commencing therapy), representing almost a quartering of infections for patients who were properly diagnosed and prescribed treatment.
Because PI often presents in the form of infections, with a variety of symptoms and clinical manifestations, clinicians often treat the infections while missing the underlying cause. This frequently leads to reoccurrence of infections and persistence of the condition, leaving the patient vulnerable to health deterioration, physical disability, and possibly permanent organ damage, or even death. Patients may develop an autoimmune disorder or certain types of cancer. Left untreated, PI may result in unusually severe infections, organ damage, chronic disease and early morbidity.
Diagnostic delays are very common, and can lead to the steady deterioration of a patient’s health. These delays also have a negative impact on healthcare systems due to inappropriate use of health resources caused by avoidable visits to a variety of different specialists for recurring infections.
Because many patients with PI require therapy indefinitely, the method of administration, and setting where the treatment is administered, are important factors that can significantly affect health-related quality of life (HRQOL). For this submission we will be addressing topics related to HRQOL, specifically with regards to the satisfaction of patients with specific treatment regimens, and the performance of specific treatment regimens.
Experiences With Currently Available Treatments
Treatment of PI includes managing current infections and preventing future infections. Antibiotics are frequently prescribed to prevent or clear bacterial infections, and antivirals can be prescribed to help patients recover from infections caused by viruses.
Immunoglobulin G (IgG) replacement is the standard of care in many of these patients and may be given intravenously (IVIG) or subcutaneously (SCIG), to replace immune system components.
In our survey, we asked patients: What types of treatments do you have experience with? (select all that apply).
Table 1: Survey Results of Two Types of Treatments Experienced
Answer Choices | Answered (n=215) |
|---|---|
Intravenously administered IG (IVIG) therapy | 67.44% (n=146) |
Subcutaneously administered IG (SCIG) therapy | 84.19% (n=181) |
In our online survey and telephone interviews, we also asked patients to assess the advantages and disadvantages of IVIG and SCIG treatments.
Excerpts from Telephone Interviews:
Of the 3 patients interviewed by phone who were on SCIG treatment, two had previous experience with IVIG treatment (Patient 01 and Patient 02).
Patient 01 commented on his experience with IVIG treatment which he required every two weeks: “After treatment I would experience muscle spasms, headaches, nausea and chills etc., and while it started to get better over time, there was always a day after the bi-monthly infusion that I needed a rest day. I needed one day for the infusion and one day for recovery. This was very disruptive. 48 days per year allocated to treatment and recovery was not sustainable. I also experienced fatigue...IG levels would be high...then down. For me the subcutaneous method is much better than IV, even though with IV there was reduced frequency of infusions.”
Patient 02 commented that: “I am a nurse, and find the subcutaneous method much easier and much more convenient than going to the hospital. When I was going to the hospital for intravenous treatment, which was once every 4 weeks, treatment could run 5 or 6 hours, but it went faster when my body grew accustomed to treatment. Now I do subcutaneous treatment at home once a week, and it takes me about 1.5 hours, including prep time. I did not have any reaction with subcutaneous....only a small welt at injection site. I do not need to rest. I do it before going to sleep - on Monday night. No dips with subcutaneous but with IVIG, in the final week before my next treatment I would feel sluggish with subcutaneous I don’t feel that fatigue.”
With regards to SCIG, we asked in our online survey: If you are currently using a SCIG treatment at home, tell us what you think are the advantages are of this treatment. (select all that apply)
Table 2: Advantages of the SCIG Treatment
Answer Choices | Answered (n=168) |
|---|---|
Independence/Freedom | 90.48% (n=152) |
Ability to decide infusion place/time | 95.24% (n=160) |
Convenience | 89.29% (n=150) |
Avoidance of travel to infusion clinic/hospital | 88.1% (n=148) |
Other (please specify) | 26.19% (n=44) |
43 patients suggested other advantages of SCIG treatment including: 9 patients who mentioned that SCIG affords them the ability to travel, 9 patients who reported that SCIG is more tolerable with fewer side effects, 9 patients who reported that SCIG results in more stable IG levels (with no peaks and troughs). 6 patients who highlighted that SCIG enables them to avoid the risk of hospital-based infection (from attending IV clinics). 4 patients said that SCIG treatment is less disruptive or takes less time, with some patients focusing on how SCIG specifically enables them to pursue work/career with minimal disruption.
One patient (through the quantitative survey) mentioned the importance of being able to avoid hospitals in the context of COVID19 risk. Of the 8 patients interviewed by phone (of which 3 were on SCIG treatment) one patient specifically switched from IVIG treatment to SCIG due to risk of COVID19 infection (from the requirement to receive therapy in a hospital environment).
With regards to SCIG, we also asked: If you are currently using a SCIG treatment, tell us what you think the disadvantages are of this treatment. (select all that apply)
Table 3: Disadvantages of the SCIG Treatment
Answer Choices | Answered (n=154) |
|---|---|
Requires more frequent infusions (than IVIG) | 67.53% (n=104) |
Limited fluid volume that can be administered into a single site in a single infusion | 29.22% (n=45) |
Requires more infusion sites on my body (than IVIG) | 51.95% (n=80) |
Other (please specify) | 29.87% (n=48) |
35 patients provided additional comments regarding disadvantages of SCIG treatment including: 13 patients who used the opportunity to comment to expressly reiterate they thought there were NO disadvantages with SCIG treatment. 8 patients commented on the inconvenience of having to order, pick-up and store SCIG supplies, with one patient mentioning the risk of exposure to hospital-based infections when picking up treatment. 5 patients mentioned that self-infusing at home took a significant amount of time.
8 patients offered unique experiences or observations with regards to what they considered to be “disadvantages” with SCIG, including: risk of clots, appearance of “bulge” at injection site, the need to have a nurse present due to injection site bleeding, anaphylaxis due to the plastic and rubber components in the SCIG kit, and anxiety from self-injecting.
With regards to IVIG, we asked: If you are currently using an IVIG treatment, tell us what you think the advantages are of this treatment. (select all that apply)
Table 4: Advantages of the IVIG Treatment
Answer Choices | Answered (n=70) |
|---|---|
Less frequent dosing frequency (than SCIG) | 42.86% (n=30) |
Enables high-dose infusions | 35.71% (n=25) |
I have become familiar/comfortable with this method of treatment | 64.29% (n=45) |
Other (please specify) | 37.14% (n=26) |
19 patients offered comments regarding the advantages of IVIG treatment including: 5 patients who expressed how they value the monthly access to specialists (immunology residents, nurses etc) while at the infusion clinic. This theme (of valuing routine access to PI specialists) was also echoed in the qualitative interviews by patients who were receiving IVIG at a hospital infusion clinic.
One patient also commented on the “social aspect” of receiving IVIG at a clinic. She stated: “…the social aspect of having conversations and long-standing relationships with other PI patients and nurses in the specialty clinic cannot be understated. This is a rare disease and can be very isolating. …”
Excerpts from Telephone Interviews:
Patient 03, a 45 year-old woman in the Greater Toronto Area, employed full time, goes once a month to an infusion clinic in downtown Toronto. She reports that the option to switch to subcutaneous (at-home) treatment was discussed with her “but, I like the social support down at the clinic, along with the routine access to (PI) disease specialists. And, I didn’t like the idea of having to administer multiple times per week.” She further stated: “the medical support and the social support you get at the clinic is very important to me. They do blood screening two times per year and abdominal ultrasounds and pulmonary function tests. And a chest X-ray once per year. And I can routinely access an expert in PI. I don’t know if this would be available if I was receiving at-home treatment.”
Patient 04, a retired 59 year-old woman who also lives in the GTA goes every 4 weeks to an infusion clinic. With regards to the option of switching to SCIG, she states: “A few of my clinic buddies are on SubQ (SCIG). The major thing for them was to avoid loss time at work. I stuck with IV (IVIG). I liked that it was ‘my treatment day’ I would have tried SubQ perhaps, but I prefer to go to a clinic because there is a healthcare team in case of complications.”
With regards to IVIG, we also asked: If you are currently using an IVIG treatment, tell us what you think the disadvantages are of this treatment. (select all that apply)
Table 5: Disadvantages of the IVIG Treatment
Answer Choices | Answered (n=63) |
|---|---|
Invasive method of administration | 34.92% (n=22) |
Intolerable side effects | 17.46% (n=11) |
Requires travel to infusion clinic/hospital | 47.62% (n=30) |
Other (please specify) | 52.38% (n=33) |
21 patients offered comments regarding the disadvantages of IVIG treatment including: 7 patients who discussed the significant time commitment required to travel to the clinic, receive infusion, and return home and recuperate. Often these patients, when discussing this time commitment, commented on how IVIG received in a clinic was disruptive to their work or career, often requiring special dispensation from employers with regards to sick time/sick leave. One patient mentioned “…it has prevented me from taking on more leadership positions in my employee group”. 3 patients mentioned that IVIG resulted in damage to their veins and/or scarring at the regular infusion sites.
We also asked patients: If you have switched from an intravenous IG (IVIG) treatment to a subcutaneous IG (SCIG) treatment, please explain why you made that switch. 107 patients offered comments:
58 patients highlighted the convenience, freedom and flexibility as the incentives to switch from IVIG to SCIG. There were many references to the reduced time commitment, and elimination of the need to travel, as being key motivations to switch. Two patients related experiences of long distance travel (in one case 180 kilometers each way) required to attend an infusion clinic.
Many patients mentioned that switching to SCIG enabled them to pursue work and career without undue interruption.
21 patients switched to SCIG because it eliminated the peaks and troughs, they experienced on IVIG and enabled better overall maintenance of IG levels.
24 patients informed us that severe reactions or intolerable side effects from IVIG motivated their switch to SCIG. Notably two reported getting aseptic meningitis due to treatment with IVIG.
Safety issues, with respect to the risk of infection from hospital environments, were cited by 11 patients. 5 of those patients specifically made the switch to SCIG due to the risks of contracting COVID19 in a hospital/clinic environment.
9 patients reported the switch to SCIG was needed because of problems with venous access required for IVIG, mostly due to scarring.
And, 14 patients switched to SCIG based on the recommendation of their physician.
We also asked: If you have switched from a subcutaneous IG (SCIG) treatment to an intravenous IG (IVIG) treatment, please explain why you made that switch. 19 patients offered comments:
6 patients switched to IVIG (or in many cases “switched back” to IVIG) due to adverse reactions to SCIG treatment.
7 patients did not like the frequency of infusions and the total time commitment required for at- home SCIG treatment, and preferred the once monthly schedule of IVIG treatment.
3 patients reported that SCIG treatment did not work for them, citing either treatment failure or inadequate maintenance of IG levels.
2 patients valued the access to specialist care that was available with IVIG treatment at infusion clinics.
1 patient reported lacking trust in their ability to properly self-administer treatments.
We asked: Please rate on a scale of 1 – 5 how important it is to you to have access to new treatments for Primary Immunodeficiency (PI)? 1 is “not important” and 5 is “very important”?
Table 6: Importance of Access to New Treatments for Primary Immunodeficiency
N=219 | ||||||
|---|---|---|---|---|---|---|
1 (not important) | 2 | 3 | 4 | 5 (very important) | N/A | Weighted Average (WA) |
10pts 4.57%) | 7pts (3.2%) | 28pts (12.79%) | 41pts (18.72%) | 132pts (60.27%) | 1pts (0.46%) | 4.28 |
Conclusions
Experience with currently available treatments is varied with patients ascribing different values to the currently available (in Canada) treatments. Efficacy along with personal preference (with respect to lifestyle and HRQOL), along with treatment tolerability, are clearly the dominating factors that contribute to the treatment selection process. However, availability, familiarity, physician-recommendation and associated out-of-pocket costs all contribute to treatment selection decision-making. Lifestyle considerations are clearly important and consequential.
Recognizing that the distribution of patients with PI have a similar distribution as the rest of the population with respect to age and employment status, the ability to work without major impedance or disruption is considered to be of high importance to many PI patients. Patients requiring IVIG therapy, either due to lack of other treatment options or due to failure on SCIG therapy, typically require one day per month to be allocated to treatment and recovery, as treatment requires travel to a designated infusion clinic, significant time for the infusion and significant time for recovery (as fatigue is a common side-effect of IVIG). Many individuals with PI have difficulty accommodating a full day per month for IVIG treatment while meeting the requirements of their employment.
Patients with the option of selecting SCIG therapy also have trade-offs. While SCIG affords a measure of independence and flexibility because the treatment can be administered at-home at a scheduled largely determined by the patient, SCIG therapy requires a higher dosing frequency, as currently available SCIG treatments do not enable high-dose infusions. This higher dosing frequency is seen by many as burdensome.
There is clearly a population of PI patients that would derive great benefit from a therapy that can be administered at home while affording the same dosing frequency as intravenous immunoglobulin (IVIG) treatments (enabling high-dose infusions every 3–4 week) along with the benefit of reducing the number of infusion sites and adverse events (compared to IVIG therapies).
Improved Outcomes
Many patients with PI are anxious for new treatment options. While many patients hope for a treatment that could be, for instance, orally administered, the demand for a treatment that minimizes disruption in areas of career and personal life would represent a significant advancement in treatment for PI and would come closer to meeting the needs of a significant subset of PI patients.
Excerpts from Telephone Interviews:
In our telephone interviews with patients, we asked: If HyQvia (Immune globulin human and recombinant human hyaluronidase) was available to you as a treatment option, would you try it?
Patient 02 commented: “I am glad that there are other options coming through. While I am comfortable with my (SC) treatment, I would be interested in trying HyQvia.”
Patient 01 commented: “Having had a negative experience with a treatment change in the past, I have some concerns with switching, but I would consider it and I would discuss it with my doctor. I would ask: ‘Is there an adjustment period?’ If there was little risk I would consider it.”
Patient 05 commented: “If it gives same result...less often (treatment administration) is GOOD. If available I would ask Dr. about it. Is this a viable option for me? Would it have the same efficacy? Should I use it?”
Patient 06 commented: “HyQvia sounds cool. I would ask my doctor about. I did enjoy going to the hospital to chat with people (for her IVIG monthly treatment), but it is different now. I have a fear of going to the hospital because of risk of infection. If it (HyQvia) could reduce SubQ (SCIG) to once a month that would be a big reduction in personal burden. SubQ is an ever present chore. Anything to reduce the frequency would be welcome”
Patient 07 commented: “We need the ability to do IVIG at home. But that would require a nurse, so HyQvia might be good for some patients. I am not sure if it (HyQvia) is indicated for patients with autoimmune (disease), and PI patients are often autoimmune, so IVIG might be better. We are all very different. For working people, this new treatment option might be better for them. And younger people might prefer it.”
Conclusions
Treatment for PI requires lifelong therapy for most patients and there are many who may derive great benefit from Immune globulin human and recombinant human hyaluronidase (HyQvia).
These patients include:
Those that have difficulties accommodating the schedule of available (IVIG) infusion clinics due to career, academic pursuits etc.
Those that do not have convenient geographic proximity to an infusion clinic, or have other encumbrances on the ability to travel
Those that find the frequency of dosing of currently available SCIG therapies to be onerous
Those that are seeking to avoid the risk of hospital-acquired infections (including COVID19)
Every attempt should be made to optimize treatment outcomes and QoL for patients, and Immune globulin human and recombinant human hyaluronidase (HyQvia) would, for many patients improve their QoL significantly.
Experience With Drug Under Review
CIPO had hoped to identify patients that had experience with the treatment under review but there has been no recent experience in Canada with HyQvia. And, we were unable to contact the few patients in Vancouver that participated in the Phase III trial for this treatment that ran from December 2008 to November 2010. (ClinicalTrials.gov Identifier: NCT00814320).
However, HyQvia is a facilitated SCIG treatment that affords some of the best features of both intravenous and subcutaneous treatment modalities. HyQvia offers:
the same dosing frequency as intravenous immunoglobulin (IVIG) treatments: enabling high-dose infusions every 3–4 weeks with minimum infusion sites and fewer adverse events compared to IVIG therapies.
the opportunity to self-administer at home like other SCIG treatments, while also requiring fewer infusions compared to conventional SCIG therapies: enabling the flexibility to manage one’s own infusions, with no requirement for travel or disruption of work-week routines.
The content provided above identifies how patients in many cases prefer (and benefit from) the key features that are available through the treatment under review: HyQvia.
Companion Diagnostic Test
N/A
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
Canadian Blood Services (CBS) buys IVIG from several different manufacturers. In late 2020, CBS informed stakeholders that some manufacturers could not provide their contracted volumes. Based on current demand, some IVIG brands and vial sizes are in short supply or no longer available, at least until spring 2022. (FACT SHEET: Canadian Immunoglobulin (Ig) Supply and Intravenous Immunoglobulin (IVIg) Brand Switching. National Advisory Committee on Blood and Blood Products. (COMMON BRIEFING NOTE (nacblood.ca))
CBS has been asking all clinics to switch as many patients as possible to Subcutaneous IG therapies in light of the anticipated IVIG shortages. Recognizing the anticipated shortage of IVIG products, this new treatment (Immune globulin human and recombinant human hyaluronidase/HyQvia) may offer an opportunity to reduce demand of IVIG products while offering patients a treatment that affords some of the best features of both intravenous and subcutaneous treatment modalities.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
CIPO used regular and contracted employee assistance to conduct research and complete this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The collection and analysis of data was accomplished through the use of CIPO’s SurveyMonkey subscription which includes an online survey platform along with tools for data analysis, sample selection, bias elimination, and data representation. Both regular and contracted staff participated in data collection and analysis.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 7: Conflict of Interest Declaration for The Canadian Immunodeficiencies Patient Organization (CIPO)
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Takeda | — | — | — | x |
CSL Behring Canada | — | x | — | — |
Grifols Canada | — | — | — | x |
Octapharma Canada | — | x | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Clinician Group Input
Clinical Immunology Network-Canada
About Clinical Immunology Network-Canada
Please describe the purpose of your organization. Include a link to your website (if applicable).
The Clinical Immunology Network-Canada (CINC) is a newly formed organization with the mission of enhancing clinical and research collaborations and to promote clinical immunology and clinical immunology research in Canada. Our nearly 70 members are both clinicians and researchers with an interest and expertise in pediatric and adult clinical immunology as well as in rheumatology, and all major Canadian health centres and universities are represented in our network. Our website is under construction.
Information Gathering
Please describe how you gathered the information included in the submission.
a) Published information on:
Morbidity/mortality in immunodeficiency
Resnick ES, et al. Blood. 2012 Feb 16; 119(7): 1650–1657.
Historical use of hyaluronidase in hypodermoclysis
Hechter O, et al. Pediatrics 1947;30(6):645-656.
Phase III Trial on this product:
Wasserman RL, et al., J Allergy Clin Immunol. 2012;130:951-7.
Use of this product in primary immunodeficiencies:
Wasserman RL, Immunotherapy 2017;9(12):1035-1050.
Wiesik-Szewczyk E, et al., Front. Immunol. 2020 May;11:981.
Use of this product in secondary immunodeficiencies:
Dimou M, et al., Anticancer Res. 2018;38:4187-4191.
Practical considerations in the use of this product:
Ponsford M, et al., Clin Exp Immunol. 2015;182:302-313.
Patient experiences with the use of this product:
Petersson C, et al., J Clin Nurs. 2018;27:4270-4278.
b) Consultancy meeting.
c) Presentations on the topic during conferences.
d) Product monograph.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: The treatment for primary and secondary humoral immunodeficiency is immunoglobulin replacement therapy. This is supported by all clinical practice guidelines on treatment for immunodeficiencies. Various options of intravenous (IV) and subcutaneous (SC) immunoglobulin (IG) products are available in Canada, all with Health Canada approval; however, there are not any facilitated (hyaluronidase- containing) subcutaneous immunoglobulin (fSCIG) options.
The underlying disease mechanism in humoral immunodeficiencies is the primary or secondary inability to produce protective antibodies leading to a significantly increased risk of infections and the associated high morbidity and mortality in these patients, if left untreated. Immunoglobulin replacement therapy targets this issue and provides protection against infectious complications.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Prevent infections, prevent tissue damage (e.g. bronchiectasis), maintain overall health, prolong life, minimize adverse events, reduce need to access health care facilities for admission or infusion administration, improve quality of life, maintain independence, decrease missed school/work hours, decrease time of infusions.
Treatment Gaps (Unmet Needs)
Considering the treatment goals (see under Treatment Goal section), please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: Intravenous immunoglobulin (IVIG) requires that the patient attends a health care facility every 3 to 4 weeks, where they typically spend at least half a day, and depend on health care professionals for the intravenous access and administration of the infusion over a number of hours, leading to missed school and work hours. Systemic side effects are frequent and unpredictable with IVIg, associated with the large intravenous volumes required, such as headaches, aseptic meningitis, fever, chills, flu- like muscle and joint pains, nausea, vomiting, rashes, and blood clots.
The caregiver or the patient itself usually administers subcutaneous immunoglobulin (SCIG) with far less systemic side-effects but more frequent local reactions at the site of infusion; however, depending on the patient’s age, weight and body constitution, it requires daily to once weekly infusions due to the limited volume that can be tolerated per infusion. Given the frequency of infusions, compliance can be an issue. In addition, given the volume needed, it often requires the use of multiple infusion sites and prolonged infusion times.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Patients that would benefit from a hyaluronidase-containing fSCIG represent a subpopulation of those with primary/secondary humoral immunodeficiencies. These patients would be 1) those requiring significantly large volumes of SCIG in relation to their weight/body constitution or the nature of their underlying disorder, and therefore also requiring multiple injection sites and prolonged infusion times, these would also include patients on high immunomodulatory doses for associated autoimmune/inflammatory manifestations of their primary immunodeficiency, 2) those in whom a frequent number of SC infusions are needed per week to maintain their IgG levels and/or because the volume required per week cannot be tolerated if less infusions per week were given, 3) those receiving IVIG with frequent and bothersome associated systemic symptoms, 4) those receiving IVIG who would greatly benefit from not missing school/work on infusion days, 5) those patients living in remote areas without easy or near access to a health care facility for IV infusions or frequent pick-up of regular SCIG product and supplies.
Because SCIG can be given at home, there is an important improvement in capacity at Medical Outpatient units. Facilitated SCIG would increase the number of patients who are able to do this, unloading hospital outpatient units – a significant cost saving.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Hyaluronidase contained in this drug has been proven to lead to local subcutaneous hyaluronan depolymerization, causing a transient and reversible increase in tissue permeability for increased immunoglobulin diffusion and absorption, and also shortened time of infusion. Hyaluronidase has been used for subcutaneous infusions, known as clysis, for many decades, particularly in palliative treatments. The novelty of fSCIG is the combination with SCIG. This mechanism of action is not present in any of the currently available immunoglobulin therapies, therefore it would complement and would be added to these other treatments. This drug would represent a valuable option to the available immunoglobulin preparations and it would likely be used as a second-line treatment for primary/secondary humoral deficiencies in general, but could become a first-line treatment in the subpopulation of patients mentioned above. If the cost and availability of the drug are reasonable, it could cause a shift in the current treatment paradigm of SCIG replacement treatment. It has been reported to have the same bioavailability with longer infusion intervals than traditional SCIG and fewer systemic adverse reactions than seen with IVIG or local adverse reactions seen with traditional SCIG.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: If we do not take into account the potential cost and availability for this drug, there should not be any reason why patients should try any of the other options (IVIG or SCIG) first before switching to or starting this product. In that subpopulation of patients described above it could be started as first-line.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: In selected patients, IVIG is used to “load” a patient, before switching to SCIG. Alternatively, multiple SC infusions are used in the first 1-2 weeks, fSCIG could lead to more effective and efficient loading by the SC route.
If, for whatever reason, this product is either not tolerated or does not provide a stable protective IgG level, the other available immunoglobulin replacement options would be used and that would not represent a departure at all from the sequence employed in current practice. Currently, patients are frequently switched between IV and SC products for a variety of reasons and without any issues. And, yes, there would be an opportunity to treat patients in a subsequent line of therapy; this drug could be added and switched with others depending on what is deemed to be best for that patient at different points in their management.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: All Ig replacement patients could benefit from this treatment. As mentioned above, the patients most likely to have the greater benefit from this treatment option are: 1) those currently requiring large SCIG volumes usually associated with higher frequency of local reactions and the need to use multiple injection sites, 2) those currently requiring very frequent subcutaneous infusions given the volumes needed and/or their body constitution, 3) those experiencing frequent systemic reactions to the intravenous preparations, 4) those on IVIG with difficult IV access or frequent systemic side-effect, 5) those in whom absence from work or school would be problematic, 6) those living in remote areas with difficult access to health centers for either intravenous infusions or for frequent pick-up of SCIG product and supplies.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify) Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Clinician judgement and discussion with the caregiver/patient on the available treatment options, including this product. As long as a specialist trained and knowledgeable on primary/secondary humoral immunodeficiencies makes the diagnosis, there should not be any challenges or misdiagnosis. When the diagnosis is made, the initiation of immunoglobulin replacement therapy may be done even if patients are pre-symptomatic to prevent serious infections. The decision to use the drug under review in this setting would not be any different from the other available products.
Which patients would be least suitable for treatment with the drug under review?
Response: Those with skin conditions preventing the use of the subcutaneous route. Those in whom proper training on administration cannot be done. This would also apply to caregivers of pediatric patients. Those in whom compliance could be expected as an issue. Those benefiting from a more regular health check, which can usually happen during their monthly IVIG infusion visits.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: Yes, using clinical judgement based on the description of the subpopulation of patients that would benefit from this treatment mentioned above, i.e. patients with large volume infusions, frequent infusions, little subcutaneous fat or local reactions to traditional SCIG.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: The outcomes used in clinical practice are aligned to those typically used in clinical trials with immunoglobulin products and include: 1) rate of infections, 2) rate of emergency visits and/or hospitalizations due to infections, 3) IgG levels, 4) safety, 5) days missed from work/school, 6) quality of life.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Meaningful responses to treatment would include: 1) No infections or improved infection rate, aiming for less than 4 infections per year, 2) No or less frequent visits to the emergency department and no or less frequent hospitalizations due to infections, 3) maintenance of therapeutic IgG levels, 4) No serious adverse reactions, 5) Significant reduction in days missed from work/school, 6) Patient/caregiver improved quality of life, 7) remission of autoimmune/inflammatory manifestations associated with their primary immunodeficiency.
How often should treatment response be assessed?
Response: At least every 6-12 months, as it is usually done for other immunoglobulin replacement therapies.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Response: 1)Serious adverse reactions, 2) Inability to maintain therapeutic IgG levels, 3) no improvement in the quality of life or sense of benefit reported from the patient/caregiver as compared to the other available treatments, 4) Patient/caregiver decision to switch to any of the other options.
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: The best setting would be at home, after proper administration training. Alternatively, it could also happen in an outpatient clinic setting, particularly for those patients unable to do it at home. In the outpatient setting, facilitated SC infusion will be an important improvement, as the infusions can be given very quickly without significant risks of systemic side-effects seen with IVIG or local side-effects seen with traditional SCIG.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: Yes, a specialist trained and knowledgeable in the diagnosis and treatment of patients with primary/secondary humoral immunodeficiencies would be required, that is a Clinical Immunologist/Allergist, Clinical Immunologist, Rheumatologist, Hematologist, Oncologist, to name the most relevant.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: SCIG has been an important development in the treatment of immunodeficiency, freeing patients from outpatient units and freeing outpatient units to treat other patients. This saving in dollars and throughput should not be underestimated, since capacity issues are becoming increasingly acute for both inpatient and outpatient beds and the costs of medicine rising.
Conflict of Interest Declarations — Clinical Immunology Network-Canada (CINC)
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (see under Place in Therapy section) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
The Clinical Immunology Network-Canada (CINC) has not received any financial payment from any company or organization. We are presenting this input on behalf of and with the authorization of all our members.
Declaration for Clinician 1
Name: Luis Murguia-Favela
Position: Clinical Associate Professor. Pediatric Immunology. CINC-Chair
Date: 08-07-2021
Table 8: Declaration for CINC Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
SOBI | X | — | — | — |
Takeda | X | — | — | — |
Medexus | X | — | — | — |
Declaration for Clinician 2
Name: Beata Derfalvi
Position: Associate Professor, Pediatric Immunology, CINC- Executive Board Member and Inborn Errors of Immunity Registry Working Group Chair
Date: 09-07-2021
Table 9: Declaration for CINC Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
SOBI | X | — | — | — |
TAKEDA | X | — | — | — |
Sanofi Genzyme | X | — | — | — |
CSL Behring | — | — | — | X -- Investigator initiated research grant |
Declaration for Clinician 3
Name: Hugues Allard-Chamard
Position: Assistant Professor. Rheumatology. CINC-Executive Board Member and Education Committee Chair
Date: 14-07-2021
Table 10: Declaration for CINC Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Pfizer | X | — | — | — |
Sanofi | X | — | — | — |
Novartis | X | — | — | — |
Hoffmann-La Roche | X | — | — | — |
Eli Lilly | X | — | — | — |
Merk | X | — | — | — |
Janssen | X | — | — | — |
Amgen | X | — | — | — |
Abbvie | X | — | — | — |
Declaration for Clinician 4
Name: Bruce Ritchie
Position: Professor of Medicine, University of Alberta. CINC-Executive Board Member
Date: 15-07-2021
Table 11: Declaration for CINC Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
CSL Behring research grant | — | — | X | — |
CSL Honoraria donated to the University of Alberta | — | X | — | — |
Takeda Honoraria donated to the University of Alberta | — | X | — | — |
Octapharma Honoraria donated to the University of Alberta | — | X | — | — |
Medical and Scientific Advisory Committee of the Canadian Immunodeficiencies Patient Organization and Other PI-Treating Physicians
About the Medical and Scientific Advisory Committee of the Canadian Immunodeficiencies Patient Organization and Other PI-Treating Physicians
The Medical and Scientific Advisory Committee of the Canadian Immunodeficiencies Patient Organization (CIPO) works alongside the patient group to ensure its activities and health information are relevant and useful for patients and caregivers.
Information Gathering
This submission was informed by expert experience, acquired in Canadian clinical practice, of treating PI patients with IVIG and cSCIG therapies, and on expert review (by Canadian PI-treating physicians) of relevant data from the pivotal trial in support of HyQvia for the treatment of PI: Study 160603 (NCT00814320), a prospective, open-label, non-controlled, multi-center, phase III study.
Current Treatments
Describe the current treatment paradigm for the disease.
Response: Currently IgG replacement is the standard of care in many patients with PI and has been the therapeutic cornerstone for antibody-deficient patients who have recurrent infections and are deficient in total or specific IgG. Ig does not modify the underlying disease mechanism but is an essential life-saving replacement therapy which generally needs to be continued lifelong.
Ig replacement therapy is generally administered either intravenously (IVIG), or subcutaneously (SCIG) and SCIG can be given in two ways: conventional or facilitated. The facilitated method uses an additional enzyme to increase the amount of Ig that can be delivered during each subcutaneous infusion.
IVIG is done q4 weeks (usually), gives high peak levels and can cause systemic side effects. It needs IV access and medical utilization.
SCIG creates a stable trough level, lower peak, less systemic side effects and patients can do the treatments themselves. However this method of administration needs multiple sites of injections to reach the needed volume.
There are many different Ig preparations available worldwide. In Canada the products licensed to treat immunodeficiency include (or have included):
Gammagard Liquid (IVIG)
IGIVnex/Gamunex (IVIG)
Octagam (IVIG)
Panzyga (IVIG)
Cutaquig (SCIG)
Cuvitru (SCIG)
Gamunex (SCIG)
Hizentra (SCIG)
Treatment Goals
What are the most important goals that an ideal treatment would address?
Treatments for primary immunodeficiency is intended to reduce frequency and severity of infections and the chronic health effects of recurrent infections. IgG replacement therapy enhances the immune system, and treats the immunoglobulin deficiency which is vital for preventing infections.
Health-related quality of life (HRQOL) is becoming increasingly recognized as a factor that affects the well-being and treatment preferences of patients with PI. Patients with PI (in comparison with healthy children and adults) experience measurably lower general health with higher hospitalization rates and increased limitations on: physical activity, social participation, and school and work attendance and performance. A long delay in diagnosis and a large number of infectious episodes can further negatively impact HQROL. These infections, which can include pneumonia, meningitis, sepsis and severe sinusitis, are serious and can be life threatening. Treatment also prevents complications such as bronchiectasis, respiratory issues, and intestinal overgrowth. Early diagnosis and rapid treatment initiation is extremely important.
However, assuming a patient has been diagnosed and treatment (life-long IgG replacement therapy) has been initiated, the route of treatment administration has a significant impact on HRQOL.
An ideal treatment would provide a patient with the required antibody proteins needed for the immune system to fight infections, while affording minimal disruption to their life.
Multiple studies have found that the ability to manage replacement therapy by oneself at home increases patient/family empowerment, makes it easier to work or study, improves family relations, contributes to a sense of self control among patients and families, and improves the daily living situation (Jiang, F., Torgerson, T.R., & Ayars, A.G. (2015). Health-related quality of life in patients with primary immunodeficiency disease. Allergy, asthma, and clinical immunology: official journal of the Canadian Society of Allergy and Clinical Immunology, 11, 27. https://doi.org/10.1186/s13223-015-0092-y)
Subcutaneous delivery of IgG also offers more consistent IgG levels and does so safely.
Treatment Gaps (Unmet Needs)
Considering the treatment goals (see under Treatment Goals section), please describe goals (needs) that are not being met by currently available treatments.
Response: Current IVIG treatments enable high-dose infusions every 3–4 weeks with minimum infusion sites. However, IVIG treatment is administered by a healthcare professional at a hospital or infusion clinic scheduled typically on Mondays to Fridays during regular business hours. For many patients, this can lead to significant lost time at work or school, along with out-of-pocket costs related to travel. Of note, some infusion clinics closed during COVID. Hospital sites are problematic during pandemic and access can be a problem even in non pandemic situations
Current SCIG treatments offer the convenience of being able to self-administer at home. However limited subcutaneous infusion volumes and reduced bioavailability require more frequent treatment, multiple infusion sites, and dose adjustment to achieve pharmacokinetic equivalence to intravenous treatments. It is also not easy for many as it is time-consuming and can interfere with work and normal social pursuits.
Many patients would benefit from a subcutaneous treatment that enables the self-administration of treatment at home, while also requiring fewer infusions – and reduced infusion sites. In Canada, currently, there is an unmet need for such treatments.
Another unmet need is for those patients who cannot infuse at home and have bad veins. A subcutaneous treatment that requires fewer infusions – administered in a clinic setting (without needed a central line which puts them at significantly increase risk of life threatening infections) would meet the needs of these patients also.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: Patients who may benefit the most from Immune globulin human and recombinant human hyaluronidase (HyQvia) include patients that have difficulties accommodating the schedule of available infusion clinics (for IVIG therapies) due to work, school, distance from an infusion clinic, or inability to travel AND that find the frequency of dosing of currently available SCIG therapies to be onerous.
However, any patient with immunodeficiency could be a candidate for this new treatment including such obvious candidates as patients who require high doses (requiring high frequency of injections), those who have technical issues, younger/pediatric, and older patients.
Patients with primary immunodeficiency encounter a unique conundrum with respect to receiving (IVIG) treatment at a hospital. As their disease makes them uniquely susceptible to pathogens, the requirement to attend an infusion clinic at a hospital brings with it the risk of hospital-acquired infections.
The current COVID19 pandemic has created a unique situation whereby many patients, who preferred the dosing schedule afforded by IVIG therapies, have deemed the risks of exposure to COVID19 (to receive their IVIG treatment) to be too high. These patients would also benefit from a treatment that enabled the self-administration of treatment at home, while also requiring fewer infusions – and reduced infusion sites.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: SCIG can be given in two ways: conventional or facilitated (fSCIG). The facilitated method uses an additional enzyme to increase SC tissue permeability and allow for the increased amount of Ig that can be delivered during each subcutaneous infusion.
This method of delivering IgG brings together the advantages IVIG and SCIG, while providing comparable efficacy to currently used IVIG and SCIG treatments.
Hyaluronidase allows for subcutaneous IgG to be Administered at 10X or more the volume of traditional subcutaneous preparations per site: max 600 mL per site rather than the usual max of 20- 60 ml (depending on other subcutaneous preparations). This high volume in one site translates to only one or two subcutaneous sites monthly for the patient.
This preparation allows for the doing frequency of the intravenous route of treatment administration (once a month) while maintaining the benefits of the subcutaneous route of administration: less systemic side effects, convenience, and medical cost savings due to at-home treatment delivery.
While the fSCIG route of administration will not address the underlying disease process, this treatment will provide an important new treatment choice for patients, and has the strong potential to also improve compliance, given the less frequent dosing and lower burden of treatment.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: There are numerous considerations for choosing a route of administration for Ig replacement therapy including: frequency of dosing, IgG level concentration maintenance, # of needles sticks, time of infusion, side effects, and convenience and quality of life.
Ideally, the selection of treatment would be made, with the patient, weighing all the above considerations, with patient preference playing important role.
How would this drug affect the sequencing of therapies for the target condition?
Response: Persons with antibody deficiency require lifelong IgG replacement therapy. The rationale to change therapies would be based on side-effects and issues related to quality-of-life and the ability to administer a therapy where venous access (required for IVIG) is challenging.
Which patients would be best suited for treatment with the drug under review?
Response: See under Treatment Gaps (Unmet Needs). (Patients who may benefit the most from Immune globulin human and recombinant human hyaluronidase (HyQvia) include patients that have difficulties accommodating the schedule of available infusion clinics (for IVIG therapies) due to work, school, distance from an infusion clinic, or inability to travel AND that find the frequency of dosing of currently available SCIG therapies to be onerous.
However, any patient with immunodeficiency could be a candidate for this new treatment including such obvious candidates as patients who require high doses (requiring high frequency of injections), those who have technical issues, younger/pediatric, and older patients.
Patients with primary immunodeficiency encounter a unique conundrum with respect to receiving (IVIG) treatment at a hospital. As their disease makes them uniquely susceptible to pathogens, the requirement to attend an infusion clinic at a hospital brings with it the risk of hospital-acquired infections.
The current COVID19 pandemic has created a unique situation whereby many patients, who preferred the dosing schedule afforded by IVIG therapies, have deemed the risks of exposure to COVID19 (to receive their IVIG treatment) to be too high. These patients would also benefit from a treatment that enabled the self-administration of treatment at home, while also requiring fewer infusions – and reduced infusion sites.)
How would patients best suited for treatment with the drug under review be identified?
Response: These patients are under the care of specialists who identify them and recommend therapy. These would be specialists with expertise in this area-immunology, hematology etc. Though misdiagnosis or missed diagnosis can happen with any condition, specialist multidisciplinary care minimizes this.
Which patients would be least suitable for treatment with the drug under review?
Response: There may be certain patients where compliance is better managed through treatment administration in a clinical setting and who have no venous access issues.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: These patients will be assessed like any other immunodeficiency patient on replacement IgG with the most important criteria being frequency and severity of infection, with nothing needed to identify responders in advance.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: The outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials with reductions in frequency and severity of infections, and improved Quality of Life being the most important outcomes.
What would be considered a clinically meaningful response to treatment?
Response:
As above. A clinically meaningful response to treatment would be:
no serious infections
minimal infections and need for antibiotics
Normal participation in work and/or school
Normal quality of life
No infectious complications.
How often should treatment response be assessed?
Response: Assessment every 6 months is typical for newly diagnosed and new treated patients. Once patients are clinically stable, then annual reassessment is likely reasonable.
What factors should be considered when deciding to discontinue treatment?
Response: For PID treatment is generally lifelong. For Secondary Immunodeficiency (SID), if the underlying condition causing SID is treated and reversed then treatment discontinuation can be considered. Again, this would be a decision made by both the treating physician and the patient.
What settings are appropriate for treatment with the drug under review?
Response: For the therapy under review, home-based (self-administered) treatment after training for SCIG is appropriate. For patients who are not able to infuse at home, the treatment can also be administered in a facility setting (i.e. hospital, specialty clinic, etc.).
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: For optimal diagnosis management and treatment, a specialist is required to diagnose, treat and monitor patients who receive IgG replacement therapy, including HyQvia, the treatment under review.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: Recognizing that treatment for PI requires lifelong therapy for most, every attempt should be made to optimize treatment outcomes and QoL for patients.
Also, in light of Canadian Blood Services (CBS) request of clinics to switch as many patients as possible to Subcutaneous IG therapies (in light of the anticipated IVIG shortages, Immune globulin human and recombinant human hyaluronidase (HyQvia) could provide CBS with a new treatment option that serves to reduce demand of IVIG products.
Conflict of Interest Declarations — Medical and Scientific Advisory Committee of the Canadian Immunodeficiencies Patient Organization and Other PI-Treating Physicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (see under Place in Therapy section) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Administrative support (such as collecting conflict of interest declarations) was provided by The Canadian Immunodeficiencies Patient Organization (CIPO).
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
N/A
Declaration for Clinician 1
Name: Stephen Betschel
Position: Clinical Immunologist and Allergist, Unity Health - St. Michael's Hospital.
Date: 8-07-2021
Table 12: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
CSL | — | X | — | — |
Takeda | — | — | X | — |
Declaration for Clinician 2
Name: Amin Kanani
Position: Clinical Associate Professor, Division Head, Allergy and Immunology, University of British Columbia
Date: 05/07/2021
Table 13: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Takeda | — | X | — | — |
CSL Behring | — | X | — | — |
Declaration for Clinician 3
Name: Amanda Jagdis
Position: Clinical lmmunology & Allergy, Clinical instructor, UBC
Date: 13-06-2021
Table 14: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Astra Zeneca | X | — | — | — |
CSL Behring | X | — | — | — |
Takeda | X | — | — | — |
Miravo | X | — | — | — |
UBC Continuing professional development | X | — | — | — |
Declaration for Clinician 4
Name: Julia Upton
Position: Assoc prof dept Pediatrics, staff physician hospital for sick children
Date: 18-jun-2021
Table 15: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Astra Zeneca Bausch Health Kaleo Pfizer (Ad boards or non bureau talk) | X | — | — | — |
Dbv, Regeneron, alk (grants for industry sponsor trials) Novartis (donation of medication for a trial) | — | — | — | X |
Declaration for Clinician 5
Name: Persia Pourshahnazari
Position: Physician
Date: 04-07-2021
Table 16: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr Susan Waserman
Position: Professor of Medicine Division Director Allergy and Clinical Immunology, McMaster University
Date: 3 July 2021
Table 17: Declaration for the Medical and Scientific Advisory Committee of the CIPO and Other PI-Treating Physicians – Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Takeda | — | X | — | — |
CSL Behring | — | X | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.