Drugs, Health Technologies, Health Systems
Reimbursement Review
Olezarsen (Tryngolza)
Sponsor: Theratechnologies Inc.
Therapeutic area: Familial chylomicronemia syndrome
Summary
What Is Familial Chylomicronemia Syndrome?
Familial chylomicronemia syndrome (FCS) is a rare, serious, recessively inherited metabolic disorder in which the body’s metabolism does not break down fats correctly due to a deficiency in lipoprotein lipase function. Patients with FCS have severely elevated levels of triglycerides in their blood, causing a high risk of acute pancreatitis episodes, which can be painful and potentially life-threatening.
FCS is a rare disease with an estimated global prevalence of approximately 1 to 10 per million people, although it is more common in some regions of Quebec due to a genetic founder effect.
Genetic testing for FCS involves assessing mutations in the LPL gene and other genes involved in triglyceride metabolism. However, due to limited availability of testing and the possibility of inconclusive testing results, genetic testing is not standard of care (SOC) as part of diagnosis across Canada.
What Are the Treatment Goals and Current Treatment Options for FCS?
An ideal treatment for FCS would safely and effectively reduce triglyceride levels, prevent pancreatitis, reduce FCS-related symptoms (such as abdominal pain), improve quality of life, ease dietary restrictions, and reduce caregiver and health care burden.
There are currently no treatments marketed in Canada for the treatment or management of FCS. Patients must eat a strict diet containing extremely low levels of fat, but most will still experience impactful symptoms. Adherence to the diet is also very difficult. Fibrates and omega-3 fatty acids (including icosapent ethyl) are frequently used off-label but are not supported by strong evidence in FCS and may be generally ineffective. Plasmapheresis may be used in acute settings but is not a viable long-term or preventive solution.
Therapies commonly used to lower low-density lipoprotein levels in the general population, such as statins, are not effective in patients with FCS due to the underlying cause of the disease.
What Is Tryngolza and Why Did Canada’s Drug Agency Conduct This Review?
Tryngolza is a drug that is administered as a subcutaneous injection. At the time this review was conducted, Health Canada was reviewing Tryngolza as an adjunct to diet in adult patients for the treatment of FCS.
Canada’s Drug Agency (CDA-AMC) reviewed Tryngolza to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the treatment of FCS in adult patients.
How Did CDA-AMC Evaluate Tryngolza?
The clinical evidence was identified through systematic searches for available studies.
CDA-AMC reviewed the clinical evidence on the benefits and harms, as well as the economic evidence, of Tryngolza relative to other treatments used in Canada for FCS. Because there are no other treatments indicated and marketed for FCS in Canada, SOC (e.g., restricted low-fat diet and management of symptoms) was the only comparator considered relevant when reviewing the evidence.
CDA-AMC identified equity and ethical considerations relevant to Tryngolza and FCS.
CDA-AMC considered the potential impacts of clinical diagnosis and/or genetic testing to determine eligibility for Tryngolza and FCS, including those related to health systems, patients (including families and caregivers), and costs.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission and 1 clinician group submission in response to our call for input, and by input from the participating public drug programs around issues that may impact their ability to implement a recommendation.
Four clinicians with specialties in endocrinology and metabolism, and/or internal medicine were consulted as part of the review process (2 as experts and 2 as panellists).
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
one randomized controlled phase III trial (the Balance trial) comparing Tryngolza with placebo in 66 patients with FCS
one single-group long-term extension of the Balance study
one single-group study (the Switch study) addressing gaps in the evidence (i.e., evidence for patients without genetic confirmation of FCS).
For the comparison of Tryngolza versus placebo (in combination with a low-fat diet):
Treatment with Tryngolza 80 mg likely reduces fasting triglyceride levels to a clinically meaningful degree relative to placebo as measured at month 6 and month 12 of treatment, but there was some uncertainty due to imprecision.
Treatment with Tryngolza 80 mg likely results in a reduction in the rate of acute pancreatitis events when compared to placebo as measured through weeks 1 to 53, but there was uncertainty due to imprecision.
Treatment with Tryngolza 80 mg likely has little to no impact on health-related quality of life as measured by the FCS Symptom and Impacts Scale at 12 months when compared to placebo, although there was some uncertainty due to imprecision.
Treatment with Tryngolza 80 mg may result in a lower proportion of patients who experience at least 1 serious adverse event when compared to placebo over a 12-month treatment duration, although there was uncertainty due to imprecision.
Based on evidence without a comparator:
Long-term follow-up data from the long-term extension study was limited due to the small sample size and interim data cut available at the time of this review. Based on the interim data, no new safety signals were identified.
The evidence was uncertain about the treatment effect in patients without genetic confirmation of FCS due to the limited number of patients in the Switch study, which provided interim data in 4 patients (16.7% of the study population) who had clinically diagnosed FCS. However, clinical experts consulted for this review did not expect differences in disease phenotype, prognosis, or treatment effect among patients without genetic confirmation of FCS.
Economic Evidence
Tryngolza is available as a prefilled autoinjector containing 80 mg Tryngolza in a 0.8 mL solution for subcutaneous use. At the submitted price of $41,000.00 per 0.8 mL autoinjector, the annual cost of Tryngolza is expected to be $492,000 per patient, based on the Health Canada–recommended dosage.
Key clinical efficacy data used to inform the economic model were derived from the Balance trial, which compared Tryngolza plus SOC with placebo plus SOC (defined as a low-fat diet, with concomitant use of fibrates, omega-3 fatty acids, medium-chain triglycerides oil, or other lipid-lowering medications). Evidence submitted by the sponsor indicates that Tryngolza plus SOC likely results in a clinically important reduction in fasting triglycerides and may result in a reduction in the rate of acute pancreatitis events compared with placebo plus SOC among adult patients with FCS.
The results of the CDA-AMC base case suggest:
Tryngolza plus SOC is predicted to be associated with higher costs to the health care system than SOC (incremental costs = $4,307,982), primarily driven by the drug acquisition cost of Tryngolza.
Tryngolza plus SOC is predicted to be associated with a gain of 1.15 life-years compared to SOC and may result in a gain of 1.42 quality-adjusted life-years (QALYs) compared to SOC. The survival benefit is predominantly driven by number of acute pancreatitis events and their impact on mortality, which is uncertain.
The incremental cost-effectiveness ratio of Tryngolza plus SOC compared to SOC was $3,041,706 per QALY gained in the CDA-AMC base case. The estimated cost-effectiveness ratio was highly sensitive to the drug acquisition cost of Tryngolza and the risk of acute pancreatitis events. Of the incremental benefit compared to SOC (1.42 incremental QALYs), approximately 93% of benefit was predicted to be accrued after the treatment duration of the Balance trial (observation period = 12 months).
CDA-AMC could not address the limitations related to productivity loss and caregiver health-related quality of life in the sponsor’s base case adopting a societal perspective because of constraints in the model structure and the lack of supporting evidence. As a result, CDA-AMC did not present a base case adopting the societal perspective.
CDA-AMC estimates that the budget impact of reimbursing Tryngolza plus SOC for the treatment in adult patients with FCS will be approximately $50 million over the first 3 years of reimbursement compared to the amount currently spent on SOC. Because Tryngolza is used as an add-on to SOC, the entire incremental budget impact reflects expenditure on Tryngolza (i.e., $50 million). The actual budget impact of reimbursing Tryngolza will depend on the market uptake of Tryngolza.
Abbreviations
AE
adverse event
AESI
adverse event of special interest
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
FAS
full analysis set
FCS
familial chylomicronemia syndrome
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ITT
intention to treat
LPL
lipoprotein lipase
LSM
least squares mean
LTE
long-term extension
MID
minimal important difference
NAFCS
North American Familial Chylomicronemia Score
QALY
quality-adjusted life-year
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SOC
standard of care
TEAE
treatment-emergent adverse event
Background
Introduction
Context for the Review
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of olezarsen 80 mg per 0.8 mL in a single-dose autoinjector for subcutaneous injection in the treatment of familial chylomicronemia syndrome (FCS) in adults. The focus is on comparing olezarsen with relevant comparators used in clinical practice in Canada and identifying gaps in the current evidence, as outlined in Table 1.
Consider the potential impacts of clinical diagnosis and/or genetic testing to ascertain eligibility for olezarsen and FCS, including the impacts on health systems, patients living with FCS (including families and caregivers), and costs.
Review and critically appraises the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Identify and describe ethical considerations associated with the use olezarsen in the treatment of FCS in adults.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Olezarsen (brand name to be confirmed), 80 mg per 0.8 mL solution in a single-dose autoinjector for subcutaneous use |
Sponsor | Theratechnologies Inc. |
Health Canada indication | As an adjunct to diet in adult patients for the treatment of FCS |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | December 16, 2025 |
Mechanism of action | Olezarsen is an antisense oligonucleotide–GalNAc conjugate that binds to apoC-III mRNA, leading to ribonuclease H1-mediated mRNA degradation and a reduction in triglycerides through a reduction of serum apoC-III protein. |
Recommended dosage | 80 mg administered by SC injection once monthly |
Submission type | Initial |
Sponsor’s reimbursement request | As per Health Canada indication |
Submitted price | $41,000.00 per 0.8 mL subcutaneous autoinjector |
Information on the CDA-AMC review | |
Review type | Complex |
Clinical review focusa | Population: As defined in the Health Canada indication Intervention: Per recommended dosage, as an add-on to standard of care (low-fat diet [≤ 20 g daily]) Comparators: Placebo, as an add-on to standard of care (low-fat diet [≤ 20 g daily]) Outcomes:
|
CDA-AMC = Canada’s Drug Agency; FCS = familial chylomicronemia syndrome; GalNAc = N-acetyl-D-galactosamine; mRNA = messenger ribonucleic acid; NOC = Notice of Compliance; SAE = serious adverse event; SC = subcutaneous.
aThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC has not previously reviewed olezarsen through the Reimbursement Review process.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each Reimbursement Review. One patient group submission from the Canadian Organization for Rare Disorders and 1 clinician group submission from the Lipid Clinic at Hamilton General Hospital were received. The patient group input gathered information through interviews with 9 patients and 1 caregiver living in Canada, in Quebec, Ontario, and British Columbia. The clinician group input was collaboratively authored by 6 physicians. The full submissions received are available on the project landing page in the consolidated input document. The drug programs provide input on each drug being reviewed through the Reimbursement Review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
Each review team includes at least 1 clinical expert with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two clinical experts with specialties in endocrinology, metabolism, and/or internal medicine participated as part of the review team.
In addition, a panel of 2 clinical experts from across Canada was convened to characterize unmet therapeutic needs, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with the condition, and explore the potential place in therapy of the drug.
Disease Background
FCS, also sometimes described as “monogenic chylomicronemia,” is a rare, genetic, autosomal recessive metabolic disease characterized by deficiency of lipoprotein lipase (LPL) function that causes severely elevated circulating triglycerides (hypertriglyceridemia) and chylomicrons (hyperchylomicronemia).3 Chylomicrons are large lipid-rich molecules that transport dietary fat from the intestines to other areas of the body.3 Normally, the function of LPL is to hydrolyze triglycerides in chylomicrons and very low–density lipoproteins, promoting clearance of triglycerides. Symptomatic presentation of FCS can be heterogeneous, nonspecific, and variable in severity.3 Classic symptoms of FCS include eruptive xanthomas (fatty deposits under the skin), fatigue, a variety of gastrointestinal complaints (nausea, vomiting, failure to thrive, abdominal pain), transient brain fog, enlargement of the spleen and liver (hepatosplenomegaly) or spleen alone (splenomegaly), and fatty discoloration of the retinal blood vessels (lipaemia retinalis).3 Patients with FCS are also at a high risk of relapsing episodes of acute pancreatitis, which require hospitalization, can be life-threatening, and may progress to chronic pancreatitis, pancreatic insufficiency, or pancreatic diabetes.3
The majority of patients (80% to 90%) with FCS have a homozygous mutation in the LPL gene, while the remainder have mutations in other causal genes that encode cofactors and proteins related to the activity of LPL, including APOC2, GPIHBP1, APOA5, and LMF1.3 FCS-causing mutations can be homozygous, compound heterozygous, or double heterozygous, and can be loss-of-function, null mutations, or nonsense mutations.4-6 Genetic tests exist for the 5 canonical FCS-causing mutations previously listed,3 but expert input informed CDA-AMC that access is limited in Canada. There are also rare cases (approximately 3%) of patients with autoimmune FCS-causing autoantibodies against LPL or GPIHBP1 who test negative on the genetic panel for FCS,3 and there are likely yet-uncharacterized mutations that are not captured by existing genetic tests.3 The clinical experts stated that diagnosis is typically based on clinical presentation and the exclusion of other, more common pathologies. The clinician group input highlighted that, although all mutations lead to severe hypertriglyceridemia, the severity of symptoms, age at onset, and residual enzyme activity can be influenced by the specific genes involved.
The prevalence of FCS is estimated to be between 1 to 10 per million people globally, equating to approximately 40 to 400 people across Canada. Due to a genetic founder effect, the prevalence of FCS is higher in Quebec and estimated to be approximately 20 to 40 per million people.
Patient group input: Interviews with patients with FCS and their caregivers highlighted that severe dietary restrictions, life-threatening complications, and acute pancreatitis, along with the associated severe pain and hospitalizations, all contribute to a profound burden of illness and reduced quality of life. Some patients expressed feeling very isolated because they were not able to share meals with family, friends, or work colleagues, as well as experiencing complications in their home and work lives as a result of disease-related flare-ups and highly restrictive lifestyle-based management. Some patients interviewed who reside in Quebec were diagnosed at birth or in infancy (due to the higher prevalence of FCS and greater awareness of the condition there), but the age at diagnosis for patients outside Quebec extended into adulthood and middle age for several patients interviewed. Patients with late diagnoses described dismissal by medical professionals of nonspecific symptoms (e.g., fatigue and stomach aches) throughout life, ineffective treatments, and stigma from doctors due to being mistaken for a person with alcohol dependency and/or due to a lack of knowledge about this rare condition.
Current Management
Treatment Goals
Patient group input: Based on the interviews with patients and caregivers, the patient group input indicated that all patients living with FCS would like a therapy that maintains their triglyceride levels at a very low (normal) level, has no or few tolerable side effects, and prevents future pancreatitis episodes. They also expressed the need for a therapy with a minimally disruptive administration schedule and one that does not require travel to treatment centres, which may place a disproportionate burden on patients who live far from specialty centres. Ideally, patients would like a therapy that would allow them to relax the restricted diet such that they could participate in social events without anxiety about severe medical consequences.
Clinician input: The clinician group input highlighted that the primary goal is reduction in fasting serum triglycerides, although there is no consensus on the specific magnitude of reduction that would be clinically meaningful. The second most important goal described by the group input was reduction in episodes of acute pancreatitis. Other secondary goals described include reduction of abdominal pain and other symptoms of FCS, although the group noted that these may be more challenging to quantify objectively. The group noted that patients with FCS typically have a fasting serum triglyceride concentration greater than 10 mmol/L despite adhering to intensive dietary restrictions. They also noted that the risk of acute pancreatitis is much lower in people with serum triglyceride levels less than 5 mmol/L to 10 mmol/L, and that they consider normal serum triglyceride levels to be less than 1.7 mmol/L.
Current Treatment Options
There are currently no treatments marketed in Canada for the treatment or management of FCS. The patient and clinician input described that patients living with FCS must follow a strict diet containing extremely low fat (up to 20 g of dietary fat per day, which is equivalent to 1 tablespoon of olive oil), but most will still experience impactful symptoms and triglyceride levels greater than 10 mmol/L despite the diet. Moreover, adherence to the diet is extremely difficult and limits the patient’s ability to participate in social activities of daily living. This may be particularly challenging for some patients whose cultural dietary practices include a higher fat content. The clinician group input described that fibrates and omega-3 fatty acids are frequently used off-label but are not supported by strong evidence in FCS and are generally ineffective. Moreover, the patient and clinician group inputs noted that pharmacotherapies (such as statins) or lifestyle changes commonly used to manage elevated triglycerides in the general population are ineffective in reducing triglyceride concentrations in patients with FCS because of the underlying LPL deficiency. The clinician group input noted that plasmapheresis may be used in acute settings but is not a viable long-term or preventive solution.
There are therefore no comparators aside from standard of care (SOC) (low-fat diet) for the context of this review. However, for context regarding the development of olezarsen, another pharmacotherapy — volanesorsen — will be discussed here briefly. Volanesorsen was developed to treat patients with FCS and has a similar mechanism of action as olezarsen, but due to safety concerns, it received a Notice of Non-Compliance from Health Canada and is not marketed in Canada or the US. For the purpose of this review, it is not considered a relevant comparator. In the patient group input, 3 patients had experience with volanesorsen, and all reported that it was difficult to tolerate or was intolerable.
Olezarsen, like volanesorsen, is a short, synthetic, single-stranded nucleotide molecule that targets and modifies the genetic material that produces apoC-III. This reduces serum apoC-III protein, which inhibits the breakdown of serum triglycerides; serum apoC-III levels are positively correlated with serum triglyceride levels. Due to differences in the biochemistry of olezarsen, the recommended dosage is 80 mg once every 4 weeks, which is less frequent than the weekly 285 mg dosage of volanesorsen.1 As a result of the lower dosage required, olezarsen is expected to have an improved safety profile relative to its parent drug.7,8
Unmet Needs and Existing Challenges
Patient group input: Patients interviewed described a lack of effective therapy for their disease, life-limiting lifestyle restrictions due to the difficult low-fat diet required, and continuing pain, fatigue, and emergency medical events (i.e., acute pancreatitis, which requires hospitalization and may be life-threatening) despite adherence to the diet. Additionally, many patients experienced delayed diagnosis and stigma or lack of understanding from friends, family, employers, and medical professionals, in addition to disruptions to their activities of daily living. Patients with experience with volanesorsen described the weekly administration schedule as onerous, in addition to substantial side effects, and the input reflected that there are challenges related to geographical access to specialty centres.
Clinician group input: The clinician group input concurred with the patient group input. The clinician group input also described that, because of the rarity of the disease, many patients are diagnosed late, misdiagnosed with more common conditions that share some clinical features, and/or dismissed by medical professionals regarding nonspecific symptoms (e.g., fatigue, stomach aches). The input highlighted that there are currently no therapies available in Canada for treating FCS that are safe and effective for lowering triglycerides and preventing acute pancreatitis.
Considerations for Using the Drug Under Review
The contents of this section have been informed by input from the clinical experts and panellists consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor table in Appendix 1 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 in the Supplemental Material document. The following information has been summarized by the review team.
Place in Therapy
Due to the rare nature of the disease and the nonspecific symptoms, all sources of input indicated that many patients living with FCS experience delayed diagnosis, misdiagnosis with similar but distinct conditions that are more common (e.g., multifactorial familial chylomicronemia, hypertriglyceridemia), and inappropriate treatment.
According to the patient and clinical expert input, olezarsen would represent a paradigm shift in the treatment of FCS given that there are no therapies indicated for this disease in Canada, and typical management strategies for elevated triglycerides (e.g., statins, diet, exercise) are largely ineffective at improving symptoms in patients with FCS because of the underlying pathophysiology of the condition. Olezarsen would therefore be the only therapy indicated for this patient population, and the clinician group input noted that it would be taken in combination with SOC, which is a restrictive low-fat diet (up to 20 g per day).
The clinical experts consulted by CDA-AMC noted that patients whose FCS is being treated with olezarsen may also receive comedications such as fibrates, omega-3 fatty acids, icosapent ethyl, and/or statins to help manage their FCS, although this is not an exhaustive list. Moreover, those who have developed exocrine or endocrine pancreatic dysfunction from prior pancreatitis episodes may be receiving pancreatic enzymes and/or insulin treatment, and patients with diabetes would be receiving antihyperglycemic therapy. Events of acute pancreatitis may require hospitalization.
Patient Population
FCS is a rare disease with an estimated global prevalence of approximately 1 to 10 per million people,9 although it is more common in some regions of Quebec due to a genetic founder effect.10 The clinician group input stated that patients most likely to benefit from olezarsen are any who are diagnosed with FCS, whether clinically diagnosed or confirmed by genetic diagnosis. They also mention that the genetic diagnostic tools are not exhaustive (i.e., not all patients with FCS will have mutations captured by existing genetic tests) and that there are barriers to accessing genetic testing in many parts of Canada. Clinical experts estimated that approximately 15% to 20% of patients with FCS may have mutations that are not included in current genetic testing panels for FCS, including FCS-causing mutations that have not yet been characterized.
Diagnosis of FCS, according to the clinician group input, is typically based on clinical presentation of patients and laboratory parameters, including persistently elevated triglyceride levels that are refractory to medication (i.e., triglyceride levels of at least 10 mmol/L) and clinical features that are typical of FCS (which may include symptoms such as recurrent abdominal pain, a history of pancreatitis, and young age at symptom onset, among others). Other, more common causes of elevated serum triglycerides must be ruled out, most importantly multifactorial familial chylomicronemia, which is not a monogenic condition but shares similar clinical phenotypes with FCS. Underdiagnosis, late diagnosis, and misdiagnosis with more common conditions are common issues faced by patients with FCS, especially outside of Quebec.
Validated scales for diagnosing FCS have been recently created, such as the North American Familial Chylomicronemia Score (NAFCS). A recent post hoc analysis of the Balance trial (presented in Section 2) reported that 95.5% of patients had a NAFCS of 45 or greater (“definite or likely” FCS), which suggests strong concordance between a NAFCS of 45 or greater and genetically diagnosed FCS.11 The clinical experts noted that the NAFCS is not commonly used in clinical practice at this time, but it aligns with the typical diagnostic approach for FCS.
Olezarsen is indicated for the treatment of adult patients. As this is a genetic disease and symptoms typically begin early in life, pediatric patients will continue to have an unmet need because there are no pharmacotherapies available to manage their condition.
The clinical experts noted that patients living with FCS who have a history of acute pancreatitis are at greater risk of repeated episodes; therefore, they have the highest unmet need. However, due to the potential severity, morbidity, mortality, and long-term sequelae associated with episodes of acute pancreatitis, prevention of even a first episode, and/or any reduction in the rate or risk of episodes, is of high clinical importance.
The sponsor-proposed initiation criteria were considered reasonable by the clinical experts and panel consulted by CDA-AMC. The proposed criteria suggested that olezarsen should be initiated as an adjunct to diet in adult patients with clinically or genetically confirmed FCS, with a maximum duration of 6 months for the initial recommendation (refer to Appendix 1 in the Supplemental Material document for additional details). The method of clinical diagnosis of FCS is not standardized across Canada, as diagnosis is typically made based on clinical gestalt according to the clinical experts consulted for this review. While the NAFCS algorithm has been newly developed and validated, it is not commonly used in clinical practice at this time. One clinical expert proposed that for the purpose of making the criteria more objective and uniform across providers while not excluding patients with a severe unmet need who could benefit from the therapy, the initiation criteria for olezarsen could require that patients meet any one of the following:
genetic confirmation of FCS
NAFCS greater than 45
combination of persistent triglyceride level of at least 10 mmol/L despite control of risk factors (alcohol abstinence, hemoglobin A1c < 8.5%), use of fibrate and/or other therapies to attempt to manage triglyceride levels, and any past episode of triglyceride-associated pancreatitis.
Testing Procedure Considerations
Genetic testing for FCS comprises testing for mutations in the LPL gene and a minimum of 4 other genes (GPIHBP1, APOA5, APOC2, and LMF1) involved in triglyceride metabolism.12 Clinical experts consulted for this review reported that genetic testing is not required for diagnosis and that clinicians instead primarily use clinical assessment and scoring algorithms to confirm a diagnosis.12 Input from the clinical experts and information submitted by the sponsor indicated that access to genetic testing is limited and is available in Canada through research programs or as out-of-country testing paid for out of pocket or through ministerial requests on a case-by-case basis. Clinicians ordering genetic testing through a commercial laboratory would submit samples to accredited labs to be sequenced with gene panels, and the costs could vary based on the laboratory and the specific genes being assessed. According to the information provided by the sponsor, genetic testing is publicly reimbursed in Quebec for patients and potential carriers. Clinical experts noted that, should olezarsen be reimbursed by public drug plans, there may be an increase in testing demand from clinicians and patients. However, it is unlikely to have major health system impacts because the number of potential patients living with FCS who would be considered for testing would remain small, and genetic testing would not be required to access the therapy.
We considered the potential impacts of genetic testing to inform eligibility for olezarsen in FCS, including impacts on health systems, patients living with FCS (including families and caregivers), and costs. No major impacts are anticipated because genetic testing would not be required for diagnosis or treatment. Key considerations and relevant information available from materials submitted by the sponsor, input from the clinical experts consulted by the review team, and sources from the literature were validated by the review team when possible and are summarized in Appendix 1 of the Supplemental Material document.
Assessing the Response to Treatment
The clinical experts consulted by CDA-AMC stated that, in FCS, the primary goal of treatment is reduction in triglyceride levels given the clear association of this biomarker with pancreatitis and other complications. They also indicated that there should be no arbitrary triglyceride thresholds in the reimbursement criteria.
The clinical experts highlighted that episodes of acute pancreatitis are the leading cause of morbidity and mortality in patients with FCS, and repeated pancreatic episodes are prognostic of a higher risk of FCS‑related sequelae (such as pancreatic insufficiency and pancreatic insufficiency–related diabetes). In light of this, the experts noted that any reduction in the event rate of pancreatitis is also clinically significant.
The sponsor-proposed renewal criteria were considered reasonable by the clinical experts and suggested that any reduction in triglyceride levels is sufficient to document beneficial clinical effects. The proposed criteria suggested that the initial renewal should be assessed at 6 months and subsequent renewals should be assessed annually (refer to Appendix 1 in the Supplemental Material document). The clinical experts communicated that in the absence of a reduction in triglycerides, other factors should be assessed before determining that olezarsen has been ineffective, such as adherence to the low-fat diet, abstinence from alcohol, and adequate management of other factors such as diabetes.
Discontinuing Treatment
The clinical experts and sponsor did not propose any specific discontinuation criteria aside from unmanageable side effects or a decision by the patient or clinician. FCS is a lifelong condition, and the beneficial effect of olezarsen is not curative; therefore, continued active treatment is expected to be required to maintain the benefits of therapy.
Prescribing Considerations
The clinical experts consulted by CDA-AMC concurred with the sponsor’s suggestion that olezarsen should be prescribed by specialists with qualifications and experience in the diagnosis and management of FCS (e.g., endocrinologists, cardiologists, lipidologists, or specialists in medical biochemistry or internal medicine).
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions (LTEs), indirect treatment comparisons, and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished phase III RCTs. Relevant patients and interventions were defined by the indication and the recommended dosage in the product monograph. Placebo as an adjunct to diet was the only relevant comparator. LTEs of included pivotal studies and RCTs were included, regardless of whether there was a comparison group. Studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (i.e., small sample size, limited follow-up data, and patients without genetic confirmation of FCS). There were no indirect treatment comparisons evaluated because of the lack of comparators available in Canada.
The review team selected outcomes (and follow-up times) for review considering the sponsor’s Summary of Clinical Evidence, clinical expert input, and patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations and were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. The following outcomes were included in the GRADE assessment because they are considered important to patients and/or clinicians who provided input to CDA-AMC:
change from baseline in fasting triglyceride levels (at month 6 and month 12)
adjudicated acute pancreatitis episode rate (through 12 months)
change from baseline in health-related quality of life (HRQoL) as measured by the FCS Symptoms and Impacts Scale at 12 months
the number of patients who experienced any serious adverse event (SAE) through 12 months.
In addition to the outcomes assessed using GRADE, other metrics related to triglyceride concentration, adjudicated acute pancreatitis events, and safety outcomes are discussed in the Results section to provide additional context as needed.
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are in in Appendix 2 in the Supplemental Material document. Results pertaining to health care resource utilization (hospitalizations, inpatient days, and emergency department visits) are summarized in Appendix 4 in the Supplemental Material document.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
one RCT included in the systematic review, the Balance study (NCT04568434)
one ongoing LTE study of the Balance study (NCT05130450; interim results)
one study addressing gaps in the evidence (inclusion of patients without genetic confirmation of FCS), the Switch study (NCT05185843).
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the Balance study are summarized in Table 2. Details of the full eligibility criteria and the relevant outcome measures are in in Appendix 3 in the Supplemental Material document.
The Balance study was a phase III, multicentre, double-blind, placebo-controlled, randomized study evaluating olezarsen 50 mg (cohort A) or olezarsen 80 mg (cohort B), administered once every 4 weeks as a subcutaneous injection in patients with FCS, compared to matched placebo, in conjunction with a low-fat diet (up to 20 g of dietary fat daily) over a 53-week treatment period. A total of 29 sites enrolled at least 1 patient in the study, including 3 sites in Canada, 16 sites in countries in the European Union, 1 site in the UK, and 9 sites in the US. In line with the Health Canada indication and the reimbursement request by the sponsor, this report focuses on the 80 mg olezarsen group; the 50 mg dose of olezarsen is mentioned only to provide context for the study design. The study included patients with a genetically confirmed FCS diagnosis and was designed to include mostly patients with a history of pancreatitis in the past 10 years. History of pancreatitis was defined as a recorded diagnosis of acute pancreatitis or hospitalization for severe abdominal pain consistent with acute pancreatitis and for which no alternative diagnosis was made. Patients without a recorded history of pancreatitis, or with no recorded history within 10 years before screening, were eligible for enrolment, but their enrolment was capped at 35% of the planned population size (i.e., up to 21 of the 60 planned patients). To qualify for enrolment, patients also had to be willing to follow a diet comprising up to a maximum of 20 g fat per day during the study and have elevated serum triglycerides (≥ 10 mmol/L or 880 mg/dL) at screening. Patients were randomized 1:1 using interactive response technology to receive olezarsen 50 mg or olezarsen 80 mg, and then 2:1 to receive olezarsen 50 mg, olezarsen 80 mg, or matching volume of placebo. Randomization was stratified by 2 factors: a prior history of pancreatitis within 10 years before screening (yes versus no), and previous treatment with volanesorsen (yes versus no).
The study consisted of the following periods:
screening period: 4 to 8 weeks, including at least a 2-week diet stabilization and run-in period for patients not already on a stable low-fat diet (≤ 20 g fat per day), and an approximately 2-week qualification period where trial eligibility was determined
treatment period: 53-week double-blind treatment period; dietary counselling commenced at the start of the diet stabilization period and was reinforced at intervals of approximately 4 to 8 weeks throughout the treatment and follow-up periods
posttreatment evaluation period: 13-week posttreatment evaluation or enrolment into the open-label extension study.
Table 2: Characteristics of Studies Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
Balance study, phase III, multicentre, randomized, double-blind, placebo-controlled Total N = 66 |
|
| Interventionb:
Comparatorb:
|
|
AE = adverse event; AESI = adverse event of special interest; FCS = familial chylomicronemia syndrome; HRQoL = health-related quality of life; SAE = serious adverse event; SC = subcutaneous.
aIf the fasting triglyceride level was < 10 mmol/L up to 2 additional tests may have been performed with any single test used to qualify.
bThe intervention received in cohort A (i.e., olezarsen 50 mg) is not relevant to the Health Canada indication and so will not be discussed further. It is included here for context given the matched placebo groups for cohort A and cohort B. Data from the pooled placebo groups informs the results.
Sources: Clinical Study Report (Balance)13 and the Sponsor’s Summary of Clinical Evidence.14
Statistical Testing and Analysis Populations
The Balance study was powered for the primary end point (change from baseline in triglyceride concentration at 6 months), assuming that the standard deviation (SD) in percent change in triglycerides was approximately 46% based on prior clinical trial experience in patients with FCS. With 14 patients in each olezarsen treatment group and 14 in the pooled placebo group, the study would have 90% power to detect a 60% difference between each treatment group and the pooled placebo group at a 2-sided alpha level of 0.05, assuming a 60% reduction in the treatment groups and no change in the placebo group. The planned sample size was 60 patients to account for potential early dropouts and to facilitate safety evaluation. The primary efficacy analysis was conducted after the last patient completed the treatment period (i.e., week 53).
All statistical tests were conducted using 2-sided tests with a 5% type I error rates unless otherwise stated. Efficacy results were summarized according to the treatment to which patients were randomized, and safety and pharmacokinetic results were summarized according to the treatment the patient actually received. The 6-month time point was defined as the average of weeks 23, 25, and 27; however, if 1 or 2 of the assessments were missing, only the nonmissing assessments were used. The 12-month time point was defined as the average of weeks 51 and 53.
To control the overall type I error rate at 0.05 across the primary and secondary end points, a hierarchical testing procedure was used in the following order: primary end point statistical significance in the 80 mg treatment group compared to pooled placebo; primary end point statistical significance in the 50 mg treatment group compared to pooled placebo; and thereafter, the secondary efficacy end points. The order of the multiple testing procedure is detailed in Appendix 3 in the Supplemental Material document. If the primary end point for either the 80 mg or 50 mg treatment group did not reach statistical significance, all subsequent end points in the hierarchy were considered exploratory.
Prespecified exploratory subgroup analyses were conducted on the primary efficacy end point by gender, age group, prior history of pancreatitis within 10 years of screening, previous treatment with volanesorsen, study sites location of (US versus non-US), region, race, ethnicity, diabetes status, and baseline triglyceride levels (less than the median versus greater than the median).
The analysis sets included the full analysis set (FAS), which included all patients who were randomized and received any amount of study drug, representing a modified intention-to-treat (ITT) population; the per-protocol set, which was a subset of the FAS that received at least 5 monthly doses of the study drug within the first 6 months, with no significant protocol violations; and the safety set, which included all randomized patients who received any amount of study drug.
Adjudicated acute pancreatitis event rates were compared between each treatment group and the pooled placebo group over several time periods: during the treatment period in the FAS; from week 13 to week 53 in the subset of the FAS with pancreatitis within the previous 10 years; from week 13 to week 53 in the FAS; during the treatment period (week 1 through week 53) in the subset of the FAS with at least 2 adjudicated pancreatitis events in the previous 5 years; and from week 13 to week 53 in the subset of the FAS with at least 2 adjudicated pancreatitis events in the previous 5 years.
Patient Disposition
A total of 144 patients were screened. There were 78 (54.2%) screening failures, primarily due to the eligibility criteria (n = 69; 88.5%). Other reasons included withdrawal of consent (n = 1; 1.3%), investigator decision (n = 2; 2.6%), and “other” (n = 6; 7.7%).
A total of 66 patients were randomly assigned to treatment; 43 patients were treated with at least 1 dose of olezarsen (22 patients in the olezarsen 80 mg group, 21 patients in the olezarsen 50 mg group) and 23 patients were treated with placebo.
Six patients (9.1%) discontinued the study drug (3 patients [13.6%] in the olezarsen 80 mg group; 2 patients [9.5%] in the olezarsen 50 mg group; 1 patient [4.3%] in the placebo group); no patients discontinued due to COVID-19 during the study. Two patients (9.1%) in the olezarsen 80 mg group and 1 patient (4.8%) in the olezarsen 50 mg group discontinued the study drug due to an adverse event (AE); no patients in the placebo group discontinued treatment due to an AE. Most patients terminated study follow-up to enrol in the LTE study, including 72.7% of patients in the olezarsen 80 mg group, 81.0% of patients in the olezarsen 50 mg group, and 82.6% in the placebo group.
Overall, 31 patients (47%) had a major protocol deviation (12 patients [54.5%] in the olezarsen 80 mg group; 9 patients [42.9%] in the olezarsen 50 mg group; 10 patients [43.5%] in the placebo group). In the olezarsen 80 mg group, the most common (> 15% of patients) major protocol deviations were related to improper informed consent procedures (31.8%) and missed visits (18.2%). In the placebo group, the most common major protocol deviations were related to improper informed consent procedures, study procedure, and missed visits (17.4% each). Other less common protocol deviations included study drug errors, use of restricted medications, and eligibility criteria not met.
Baseline Characteristics
A summary of the baseline characteristics in the Balance study is presented in Table 3.
Table 3: Summary of Baseline Characteristics in the Balance Study
Characteristic | Balance (FAS) | |
|---|---|---|
Olezarsen 80 mg (N = 22) | Placebo (N = 23) | |
Demographic characteristics | ||
Age (years), mean (SD) | 47.7 (13.30) | 44.0 (14.67) |
Sex, n (%) | ||
Female, n (%) | 11 (50.0) | 12 (52.2) |
Male, n (%) | 11 (50.0) | 11 (47.8) |
Race, n (%) | ||
Asian | 3 (13.6) | 0 |
White | 17 (77.3) | 22 (95.7) |
Other | 2 (9.1) | 1 (4.3) |
Diabetes status, n (%) | ||
No | 15 (68.2) | 17 (73.9) |
Type 1 or 2 | 7 (31.8) | 6 (26.1) |
Region, n (%) | ||
Europe | 14 (63.6) | 11 (47.8) |
North Americaa | 8 (36.4) | 12 (52.2) |
FCS genotype | ||
Genetic test outcome, n (%)b | 22 (100) | 23 (100) |
APOA5 | ||
Homozygote | 1 (4.5) | 1 (4.3) |
Double heterozygote | 1 (4.5) | 0 |
APOC2 | ||
Homozygote | 2 (9.1) | 0 |
GPIHBP1 | ||
Homozygote | 1 (4.5) | 1 (4.3) |
LMF1 | ||
Compound heterozygote | 1 (4.5) | 0 |
LPL | ||
Homozygote | 10 (45.5) | 16 (69.6) |
Hemizygote | 1 (4.5) | 0 |
Compound heterozygote | 5 (22.7) | 5 (21.7) |
Double heterozygote | 1 (4.5) | 0 |
Lipid and lipoprotein parameters | ||
Fasting triglycerides (mmol/L), mean (SD) | 29.5 (16.9) | 29.3 (14.9) |
Fasting apoC-III (mmol/L), mean (SD) | 0.3 (0.6) | 0.3 (0.1) |
Fasting apoB-48 (mg/dL), mean (SD) | 11.7 (8.1) | 14.2 (14.2) |
Fasting non–HDL-C (mmol/L), mean (SD) | 6.8 (2.6) | 7.0 (2.5) |
Pancreatitis history | ||
Patients with a history of acute pancreatitis or of a severe abdominal pain episode or other symptoms suggestive of pancreatitis requiring hospitalization, n (%) | 15 (68.2) | 19 (82.6) |
Number of episodes in the past 10 years | ||
Mean (SD) | 3.4 (6.0) | 6.3 (16.3) |
Median (range) | 1.0 (0 to 25) | 2.0 (0 to 78) |
Number of episodes in the past 5 years | ||
Mean (SD) | 1.5 (3.1) | 3.6 (8.2) |
Median (range) | 1.0 (0 to 15) | 1.0 (0 to 38) |
Patients with past documented diagnosis of acute pain or severe abdominal pain episode or other symptoms suggestive of pancreatitis, with or without hospitalization (but requiring an emergency department or urgent care visit), n (%) | 18 (81.8) | 21 (91.3) |
Number of episodes in the past 10 years | ||
Mean (SD) | 4.8 (7.5) | 6.6 (16.5) |
Median (range) | 1.5 (0 to 25) | 3.0 (0 to 79) |
Number of episodes in the past 5 years | ||
Mean (SD) | 1.7 (3.2) | 3.8 (8.4) |
Median (range) | 1.0 (0 to 15) | 1.0 (0 to 39) |
Patients with active pancreatitis within 4 weeks before informed consent, n (%) | 0 | 0 |
Patients with known chronic pancreatitis, n (%) | 8 (36.4) | 6 (26.1) |
Randomization stratification factors | ||
Prior history of pancreatitis within 10 years before screening, n (%) | 17 (77.3) | 15 (65.2) |
Prior treatment with volanesorsen, n (%) | 8 (36.4) | 10 (43.5) |
FAS = full analysis set; FCS = familial chylomicronemia syndrome; HDL-C = high density lipoprotein cholesterol; SD = standard deviation.
aFive patients (21.7%) assigned to the placebo group and 1 patient (4.5%) in the olezarsen 80 mg group were enrolled at sites in Canada.
bGenetic confirmation occurred either as a positive test outcome from the central genetics laboratory or, if the test outcome by the central genetics laboratory was indeterminate, a subject matter expert assessed that the results were consistent with FCS.
Sources: Clinical Study Report (Balance)13 and the Sponsor’s Summary of Clinical Evidence.14
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure and compliance, and use of concomitant medications in the Balance study, are provided in Appendix 4 in the Supplemental Material document.
The mean number of doses administered was 11.0 in the olezarsen 80 mg group and 12.1 in the placebo group. The median duration of treatment exposure was 364.0 days in both the olezarsen 80 mg and placebo groups (range, 29 to 400 days in the olezarsen group; range, 199 to 368 days in the placebo group). The treatment compliance was 96% in both the olezarsen 80 mg and placebo groups. In each of the 2 groups, 3 patients (13.6% and 13.0%, respectively) had dose interruptions due to AEs.
Most patients received at least 1 concomitant medication (95.5% of patients in the olezarsen 80 mg group and 100% of patients in the placebo group). The most common concomitant medications included paracetamol (13.6% and 17.4%, respectively), fenofibrate (31.8% and 34.8%, respectively), fish oil (27.3% and 26.1%, respectively), metformin (22.7% and 21.7%, respectively), acetylsalicylic acid (9.1% and 26.1%, respectively), ibuprofen (9.1% and 26.1%, respectively), pantoprazole (13.6% and 26.1%, respectively), and ondansetron (13.6% and 21.7%, respectively). Background medications included lipid-lowering medications, antidiabetic medications, antihypertensives, oral anticoagulants, tamoxifen, estrogen, or progestin to which patients were exposed before the first dose and which were continued after the first dose of study drug. The proportion of patients who received background medication was similar across treatment groups, including 90.9% in the olezarsen 80 mg group and 100.0% in the placebo group.
Critical Appraisal
Internal Validity
In the Balance study, the methods of randomization, stratification, treatment allocation, allocation concealment, and blinding were all considered appropriate.
The baseline characteristics of patients were generally balanced between treatment groups, with some exceptions. There were some between-group differences observed in geographical location and race (the majority of patients in all groups were white, with a higher proportion in the placebo group). There were slightly more patients with diabetes in the olezarsen 80 mg group (31.8%) than in the placebo group (26.1%), and subgroup results by diabetes status were noted by the clinical experts to be of potential interest. However, based on consultation with the clinical experts, these differences were not expected to impact the interpretation of the study results. There were also small differences in the proportions of patients with a history of pancreatitis in the past 10 years and prior treatment with volanesorsen, the 2 stratification factors. However, these differences correspond to only 2 patients between treatment groups in each comparison, which appear larger as percentages because of the small overall number of patients. Comparing the placebo group and olezarsen 80 mg group, there was a higher mean and median number of acute pancreatitis episodes in the past 10 years and a higher mean number, but similar median number, in the past 5 years in the placebo group. Given that a history of acute pancreatitis is prognostic of a higher risk of future acute pancreatitis episodes, this could potentially bias the pancreatitis-related outcomes in favour of olezarsen because patients assigned to the placebo group had a higher baseline risk of acute pancreatitis on average. Notably, the between-group difference was larger for the mean than the median, and the maximum number of episodes in the past 10 years was higher in the placebo group (78 episodes in the placebo group versus 25 in the olezarsen 80 mg group), suggesting that a small number of patients in the placebo group may have had an extremely high rate of acute pancreatitis, thereby inflating the mean value.
There were between-group differences observed in concomitant medications taken by patients in the placebo and olezarsen 80 mg groups. Of the 8 concomitant medications previously listed as the most common (≥ 20% of patients in any treatment group), the proportion of patients receiving the medication was notably higher in the placebo group for 5 of them: paracetamol, acetylsalicylic acid, ibuprofen, pantoprazole, and ondansetron. For the remaining 3 common concomitant medications (fenofibrate, fish oil, and metformin), the proportions differed by only a few percentage points between treatment groups. The concomitant medications used more frequently in the placebo group were medications for pain and/or inflammation relief (paracetamol, acetylsalicylic acid, and ibuprofen), acid reflux (pantoprazole), or nausea and vomiting (ondansetron). The higher use of these concomitant medications in the placebo group may also dampen any apparent between-group differences in acute pancreatitis and in patients self-assessing their symptoms via the FCS Symptoms and Impacts Scale.
The screening period included at least a 2-week diet stabilization or run-in period for patients not already on a stable diet, and patients received diet and alcohol counselling at regular intervals approximately 4 to 8 weeks apart during the study regardless of assignment to placebo or olezarsen 80 mg. However, it was unclear from the reported data whether and how adherence with the dietary restriction was measured. Further, differences in adherence to the diet between groups could introduce bias, but there is insufficient evidence to assess this.
Patients randomized to placebo could have received placebo matched to either the 50 mg or 80 mg olezarsen dose, but the placebo groups were pooled for all end points. This was not considered to increase risk of bias despite minor differences in injection volume between the 50 mg matched or 80 mg matched dosages. For end points evaluating the rate ratio of acute pancreatitis episodes, the olezarsen 50 mg and 80 mg groups were also pooled, which was considered appropriate given the challenges of evaluating an uncommon event in a rare disease population.
Because results for the 50 mg olezarsen dose did not reach statistical significance for the primary end point family, according to the hierarchical testing procedure, all secondary outcomes (for both doses of olezarsen compared to placebo) were nominal, exploratory, and supportive only, and were not controlled for multiplicity; therefore, they have an inflated type I error risk. For the comparisons of olezarsen 80 mg to placebo, only the percent change in triglycerides from baseline to month 6 was controlled for multiple comparisons, while the other end points evaluated in this report (triglycerides at month 12, acute pancreatitis, HRQoL, and safety end points) were not.
The study duration of 12 months was considered adequate for assessing the change in fasting triglyceride levels. A potential between-group difference was observed in the rate of acute pancreatitis in favour of olezarsen, but a longer duration of study would likely improve certainty in the outcome.
Triglyceride levels are an appropriate primary end point given that lowering triglyceride levels is the primary treatment goal for clinicians in real-world practice. However, this is a biomarker of elevated risk of pancreatitis (among other sequelae) rather than a clinical end point in itself, and the trial was not powered to detect differences in pancreatitis directly. The study capped the proportion of patients without a history of pancreatitis at 35%, which was considered appropriate by the clinical experts consulted for the review to enrich the study population for pancreatitis risk because a history of pancreatitis is a known prognostic factor for future events. Patients with a history of pancreatitis are also those with the greatest unmet need and the most likely to seek treatment, according to the clinical experts. This was not expected to increase the risk of internal bias because it applied to all treatment groups.
The number of patients recruited was appropriate to provide power for the primary end point, although the analyses were unlikely powered for secondary and exploratory end points. The statistical analysis of the primary end point, percent change from baseline in fasting triglycerides, was considered appropriate to account for the small sample size per stratum and heterogeneity in variance, using an analysis of covariance model. The 95% confidence intervals (CIs) were calculated using the robust variance estimator based on the Bell and McCaffrey method. The results were robust to 6 sensitivity analyses, which assessed the primary end point using different analysis sets, different methods for adjustment for missing data, and different methods of analysis.
All patients who were randomly assigned to treatment and received at least 1 dose of olezarsen or placebo were included in the FAS. Because all patients appeared to receive at least 1 dose, the FAS was the same as the ITT population. The modified ITT estimator is unbiased for the principle stratum estimand under the assumption that the intercurrent event is not affected by the assigned treatment arm, and there did not appear to be any imbalance between groups with regard to noninitiation of therapy (all patients initiated therapy) or major protocol deviations (there were many deviations, but they were similar between groups).
No patients had missing baseline values for triglycerides, apoC-III, apoB-48, and non–high-density lipoprotein levels. For months 3, 6, and 12, the missing values in the olezarsen treatment groups were imputed using the washout approach due to an insufficient number of retrieved dropouts. In the placebo group, missing values were imputed based on the missing-at-random assumption. For triglyceride-related end points, at month 6, data were missing for 1 patient in the placebo group and 3 patients in the olezarsen 80 mg group; at 12 months, data were missing for 2 and 3 patients, respectively. In the olezarsen 80 mg group, 2 patients discontinued due to AEs, whereas no patients in the placebo group did so, which may not be random. The multiple imputation strategy of “jump to reference” was considered appropriate and assumes that after a patient discontinues treatment, the missing outcome data would be similar to outcome data in the placebo group. This method assumes that there is no lasting effect of the drug. However, given the mechanism of action of this therapy, this assumption was expected to be reasonable; the clinical experts consulted by CDA-AMC indicated that the drug’s effect in reducing triglyceride levels would be expected to occur relatively quickly and remain stable during treatment. A sensitivity analysis was also conducted using the “copy increment from reference” multiple imputation approach, which assumes that after discontinuation, patients would follow the trajectory of the placebo group plus the difference observed up to the point of dropout; results were overall consistent.
Missing data were noted in the HRQoL assessments but did not appear to be especially high (e.g., 3 to 4 patients per treatment group were missing data for the FCS Symptoms and Impacts Scale), nor were they unbalanced between groups. Data were considered missing if patients completed fewer than 4 daily assessments in a week; otherwise, the average of the nonmissing scores was calculated. The method of imputation for missing data was considered appropriate. Although some validation studies have been performed for the FCS Symptoms and Impacts Scale (refer to Appendix 3 in the Supplemental Material document), there is no established minimal important difference (MID) in the literature.
Major protocol deviations occurred in 43.5% of patients in the placebo group and 54.5% of patients in the olezarsen 80 mg group. Most deviations were related to improper informed consent procedures, and the actions taken were to re-educate the site staff. Some individual patients had numerous associated major protocol deviations (e.g., 1 patient in the placebo group and 1 patient in the olezarsen 80 mg group each had more than 10 major protocol deviations), all of which were addressed by retraining site staff. The reason for such high deviations related to improper procedures is unknown, but this was not imbalanced between the study groups.
External Validity
The Balance study was generally appropriately designed to reflect the treatment landscape and the population of patients with FCS in Canada. According to the clinical experts consulted by CDA-AMC, the characteristics of the patients enrolled in the Balance study were mostly reflective of those in clinical practice in Canada, although the proportion of white patients was high. The limited inclusion of racialized patients reduces confidence in the generalizability of the study results to these populations. There is a lack of data regarding whether patients of different races or other demographic factors have differences in treatment response or prognosis, although the clinical experts consulted for this review stated that they expected no significant differences.
Patients were limited to those aged 18 years and older, which reflects the Health Canada indication. However, there remains an unmet need for patients aged younger than 18 years, and the clinical experts consulted for this review indicated that parents or guardians of pediatric patients may advocate for off-label access for their child or ward with FCS. In the clinical trial setting, there may be a higher degree of monitoring, greater access to counselling (e.g., genetic counselling and dietary counselling), and more frequent in-person appointments than in typical real-world practice. The comparator (i.e., placebo plus low-fat diet), concomitant medications, and end points were consistent with the SOC for FCS, although the quantified HRQoL scales are not typically used in clinical practice. The study duration and the primary and key secondary end points were considered reasonable because patients with lipid disorders may be followed up every 6 months to every 12 months.
There is a gap in the evidence regarding patients who do not have genetic confirmation of FCS, given that all patients in the Balance study were required to have genetic confirmation, whereas patients living in Canada may not always have access to genetic testing and, moreover, genetic testing is not comprehensive of all causes of FCS. However, the clinical experts consulted by CDA-AMC did not expect this to influence the results.
Adherence to the restrictive diet was described by the clinical experts as critical to achieving the benefits of treatment with olezarsen. The experts noted that they did not expect a large difference in dietary adherence in real-world practice compared with the clinical trial setting because of the severe potential consequences of dietary deviations (i.e., episodes of pancreatitis). Nonetheless, the greater degree of monitoring and access to dietary counselling may represent a difference between the trial setting versus nontrial clinical practice.
The FCS Symptoms and Impacts Scale is not commonly used in clinical practice, but it was considered appropriate for the purpose of a clinical trial to quantify HRQoL.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
Key results include the following:
Percent change in fasting triglycerides from baseline
From baseline to 6 months, the between-group difference in the least squares mean (LSM) change in fasting triglycerides was –43.50% (95% CI, –69.09% to –17.92%; P = 0.0009), favouring olezarsen.
From baseline to 12 months, the between-group difference in the LSM change in fasting triglycerides was –59.39% (95% CI, –90.66% to –28.12%), favouring olezarsen.
From baseline to 6 months, 9 patients (40.9%) in the olezarsen 80 mg group and 1 patient (4.3%) in the placebo group achieved at least a 40% reduction in fasting triglycerides; 2 patients (9.1%) in the olezarsen 80 mg group achieved at least a 70% reduction, compared with no patients in the placebo group.
Adjudicated acute pancreatitis episode rate through the treatment period (week 1 to week 53)
In the olezarsen 80 mg group, 1 patient (4.5%) experienced 1 adjudicated acute pancreatitis event, whereas in the placebo group, 7 patients (30.4%) experienced 11 such events.
Although the 50 mg group is not relevant for the indication, it is mentioned here to provide context and to support discussion of outcomes pooled across the 2 doses. One patient (4.8%) in the olezarsen 50 mg group experienced 1 event, and when the olezarsen 80 mg and 50 mg groups were pooled, there was an 88% lower rate of adjudicated pancreatitis events in the pooled olezarsen group compared with the placebo group (mean pancreatitis rate ratio = 0.12; 95% CI, 0.02 to 0.66).
For the subgroup of patients with a history of pancreatitis in the previous 10 years, the total olezarsen group (including both dose groups) had a 90% lower event rate compared with placebo (mean pancreatitis event rate ratio = 0.10; 95% CI, 0.02 to 0.66).
For the subgroup of patients with at least 2 adjudicated acute pancreatitis events in the 5 years before enrolment, the total olezarsen group had an 86% lower adjudicated pancreatitis event rate compared with placebo (mean pancreatitis event rate ratio = 0.14; 95% CI, 0.03 to 0.67).
Change from baseline for HRQoL
From baseline to month 12, the between-group difference in the LSM change in FCS Symptoms and Impacts Scale was ████████████████████.
Harms
Key results include the following.
Treatment-Emergent Adverse Events
The proportion of patients who experienced any AE was 86.4% (19 of 22) in the olezarsen 80 mg group and 95.7% (22 of 23) in the placebo group.
The most commonly reported treatment-emergent adverse events (TEAEs) (> 5% in olezarsen 80 mg group or placebo group) included the following for the olezarsen and placebo groups, respectively:
COVID-19 (13.6% and 34.8%)
abdominal pain (18.2% and 34.8%)
diarrhea (9.1% and 26.1%)
pancreatitis (4.5% and 17.4%)
fatigue (4.5% and 17.4%)
cellulitis (0% and 13.0%)
acute pancreatitis (0% and 13.0%)
headache (4.5% and 13.0%)
dental caries (0% and 8.7%)
gastroesophageal reflux disease (0% and 8.7%)
hypernatremia (0% and 8.7%)
back pain (4.5% and 8.7%)
migraine (0% and 8.7%)
proteinuria (0% and 8.7%)
cough (9.1% and 8.7%)
Serious Adverse Events
The proportion of patients who experienced serious TEAEs was lower in the olezarsen 80 mg group (13.6%) compared with the placebo group (39.1%).
The most common SAE was pancreatitis, which was experienced by 1 patient (4.5%) in the olezarsen group and 4 patients (17.4%) in the placebo group.
In the olezarsen 80 mg group, other SAEs included intestinal perforation and hepatic cirrhosis, each occurring in 1 patient. In the placebo group, other SAEs included gastric varices, gastric variceal hemorrhage, gastritis, cholangitis, cellulitis, hypoglycemia, hyponatremia, pancreatogenous diabetes, and squamous cell carcinoma of the oral cavity, each occurring in 1 patient.
Withdrawals Due to Adverse Events
Two patients in the olezarsen 80 mg group discontinued the study drug because of TEAEs. In 1 patient, the TEAEs were chills (moderate), trismus (moderate), and myalgia (moderate). In the other patient, the TEAEs were vomiting (moderate), diarrhea (moderate), flushing (moderate), and chest discomfort (moderate).
No patients in the placebo group discontinued due to AEs.
Mortality
No deaths occurred in the olezarsen 80 mg or the placebo group.
One patient in the olezarsen 50 mg group died of sudden death (the cause remains unknown because no TEAEs or significant laboratory or electrocardiogram abnormalities were detected).
Adverse Events of Special Interest
No patients in the olezarsen 80 mg or the placebo group experienced an adverse event of special interest (AESI) during the study.
Summary of Findings and Certainty of the Evidence
In the absence of literature-based MID estimates, a threshold of 30% suggested by the clinical experts was used in the GRADE assessment to assess the change in fasting triglyceride levels. In the absence of a known threshold, for all other end points, the review team, in consultation with the clinical experts, judged whether the point estimates and relevant bounds of the CI represented clinically important effects.
Table 4: Summary of Findings for Olezarsen (80 mg) Plus Low-Fat Diet (≤ 20 g of Fat per Day) Versus Placebo Plus Low-Fat Diet for Patients With FCS
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Olezarsen | Difference | |||||
Triglycerides | |||||||
Percent change from baseline in fasting triglyceride levels, LSM (95% CI) Follow-up: 6 months | 45 (1 RCT) | NA | 11.5% (−5.32% to 28.36%) | −31.99% (−49.03% to −14.94%) | −43.50% (−69.09% to −17.92%) | Moderate (serious imprecision)a | In patients with FCS, treatment with olezarsen plus low-fat diet likely results in a clinically important reduction in fasting triglyceride levels when compared to placebo plus low-fat diet. |
Percent change from baseline in fasting triglyceride levels, LSM (95% CI) Follow-up: 12 months | 45 (1 RCT) | NA | 20.89% (1.02% to 40.77%) | −38.50% (−58.19% to −18.82%) | −59.39% (−90.66% to −28.12%) | Moderate (serious imprecision)a | In patients with FCS, treatment with olezarsen plus low-fat diet likely results in a clinically important reduction in fasting triglyceride levels when compared to placebo plus low-fat diet. |
Acute pancreatitis | |||||||
Acute pancreatitis (adjudicated), mean event rate per 100 patient-years (95% CI) Follow-up: week 1 to week 53 | 66 (1 RCT)b | Rate ratio = 0.12 (0.02 to 0.66) | 36.31 (14.70 to 89.69) | 4.37 (0.94 to 20.30)b | ██████ | Moderate (serious imprecision)c | In patients with FCS, treatment with olezarsen plus low-fat diet likely results in a reduction in the rate of acute pancreatitis events when compared to placebo plus low-fat diet. |
Health-related quality of life | |||||||
FCS Symptoms and Impacts Scale, mean change from baseline in score (SD) Follow-up: 12 months | 45 (1 RCT) | NA | ███ ██ | ███ ██ | ███ ██ | Moderate (serious imprecision)d | In patients with FCS, treatment with olezarsen plus low-fat diet likely results in little to no difference in the change from baseline in FCS Symptoms and Impacts Scale scores when compared to placebo plus low-fat diet. |
Harms | |||||||
Number of patients with at least 1 SAE, n (%) Follow-up: 12 months | 45 (1 RCT) | NA | 9 (39.1 per 100) | 3 (13.6 per 100) | █████ | Low (very serious imprecision)e | In patients with FCS, treatment with olezarsen plus low-fat diet may result in a reduction in the proportion of patients with at least 1 SAE when compared to placebo plus low-fat diet. |
CI = confidence interval; FCS = familial chylomicronemia syndrome; HRQoL = health-related quality of life; LSM = least squares mean; NA = not applicable; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to rating down the level of certainty of the evidence are documented in the table footnotes. All mentions of “low-fat diet” in this table refer to the prescribed diet of up to 20 g of fat per day.
aRated down 1 level for serious imprecision. The clinical experts consulted by CDA-AMC considered a 30% between-group difference to be clinically meaningful. The point estimate suggests a clinically meaningful effect, while the upper bound of the 95% CI includes the potential for little to no difference. The small sample size raises concern about potential overestimation of the true effect.
bFor the end point of acute pancreatitis event rate, because the event is uncommon, the analysis population for olezarsen included the pooled 50 mg (N = 21) and 80 mg (N = 22) olezarsen groups. Although the 50 mg dose of olezarsen is not relevant to the Health Canada indication or reimbursement criteria, this was not expected to introduce bias because results suggested the 50 mg group had lower efficacy relative to the 80 mg group compared to placebo, and any expected bias would therefore have favoured the placebo group.
cThe level of certainty of the evidence was rated down 1 level for serious imprecision. A very small number of events were captured. Analysis of this outcome was not adjusted for multiplicity. There was no established MID, so the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect.
dThe level of certainty of the evidence was rated down 1 level for serious imprecision. There was no established MID, so the target of the certainty of evidence assessment was the presence or absence of any (non-null) effect. The 95% CI around the point estimate contains the potential for a positive impact on HRQoL, no impact on HRQoL, or a negative impact on HRQoL. The small sample size raises concern about potential overestimation of the true effect. Analysis of this outcome was not adjusted for multiplicity.
eConfidence intervals were not reported for this end point. The level of certainty of the evidence was rated down 2 levels for very serious imprecision. A very small number of events were captured. Between-group comparison was not available for this end point.
Sources: Clinical Study Report (Balance)13 and the Sponsor’s Summary of Clinical Evidence.14 Response to additional information requested by CDA-AMC.15
Long-Term Extension Studies
Description of Studies
ISIS 678354-CS13 (NCT05130450) is an ongoing, phase III, multicentre, open-label extension study designed to allow patients with FCS who participated in the pivotal Balance study (ISIS 678354-CS3, index study) to receive treatment with olezarsen (for patients who received placebo in the index study, referred to herein as the index-placebo group) or to continue treatment with olezarsen (for patients who received active treatment in the index study, referred to herein as the index-olezarsen [50 mg or 80 mg] group). Results presented in this section are based on data collected as of the database cut-off date of August 5, 2023.
The primary objective was to evaluate the safety and tolerability of olezarsen. Safety was evaluated through the collection and measurement of AEs, clinical laboratory parameters (including hematology, serum chemistry, and urinalysis), concomitant medication and procedure information, vital signs, and electrocardiograms. Secondary objectives were to evaluate the effect of olezarsen on reduction in fasting triglycerides, apoB-48, and the event rate of acute pancreatitis. Exploratory objectives were to evaluate the effect of olezarsen on health care utilization and to support content validity and evaluate patient-perceived meaningful change in the FCS Symptoms and Impacts Scale with olezarsen.
To be included in the extension study, patients needed to have completed the Balance study (i.e., received the last dose as scheduled) with an acceptable safety profile, in the judgment of the investigator, and to be willing to follow the restricted fat diet (up to 20 g per day) during the study. Other eligibility criteria were consistent with the Balance study. All patients in the extension study were to receive olezarsen 80 mg regardless of their randomly assigned treatment group in the Balance study. The study is planned to be 157 weeks, followed by a 13-week posttreatment follow-up period, with regular dietary counselling. However, at the time of the data cut-off, end points had been evaluated only up to 12 months of treatment in the open-label extension.
Patient Disposition
Approximately 60 patients were planned to be enrolled in this study, and 54 patients had been enrolled at the time of the data cut-off (August 5, 2023). Of these, 20 patients had received placebo, 17 patients had received olezarsen 50 mg, and 17 patients had received olezarsen 80 mg during the Balance study (which represents the FAS for the extension study). At the time of the data cut-off, 5 patients (9.3%) had discontinued the study drug: 4 patients (7.4%) due to AEs (2 patients in the group formerly assigned placebo and 2 patients in the group formerly assigned olezarsen 80 mg), and 1 patient (1.9%) because of voluntary withdrawal (in the group who had formerly received placebo). No patient had discontinued the study drug due to COVID-19 during the study, and no patients had completed the extension study by the cut-off date.
Baseline Characteristics
Demographic and baseline characteristics were similar across treatment groups and were generally similar to those in the Balance study, although mean baseline triglyceride and apoC-III levels were lower at baseline in the extension study among patients who had previously received olezarsen (50 mg or 80 mg) in the index study, as expected given treatment experience with olezarsen.
Exposure to Study Treatments
All patients enrolled in the extension study were receiving olezarsen 80 mg during the extension. At the time of the data cut-off, the mean duration of exposure during the extension study was lower in the index-placebo group (151.5 days) than in the index-olezarsen groups (235.0 days for the 50 mg group and 248.0 days for the 80 mg group). The median exposure was 151.5 days in the index-placebo group (range, 39 to 510 days), 583 days in the index-olezarsen 50 mg group (range, 57 to 962 days), and 613.5 days in the index-olezarsen 80 mg group (range, 28 to 943 days). Concomitant medication use was similar across treatment groups and similar to that in the index study.
Critical Appraisal
The extension study is a noncomparative, single-group, open-label safety study for which no randomization, blinding, or statistical inferences were conducted, and the available data are interim. The lack of a control group precluded causal statements about benefit and harm versus any comparator. The open-label nature of the study may increase the risk of bias in the assessment of subjective outcomes (such as self-reported AEs), because it may influence patient expectations of treatment. The external validity of the study is otherwise similar to that of the index study previously described.
Results
Efficacy
Detailed results for outcomes relevant to this review are in Appendix 5 in the Supplemental Material document.
Key results include the following:
The mean percent change in fasting triglycerides from baseline of the extension study to month 6 was ████ (SD = █████) in the index-olezarsen 80 mg group and ███████ ███ █ in the index-placebo group. The mean percent change in fasting triglycerides from baseline of the extension study to month 12 was ███████ ███ in the index-olezarsen 80 mg group and ████ (SD = ████) in the index-placebo group.
In patients with at least 2 acute pancreatitis events in the past 5 years before treatment in the index study, the number of patients with adjudicated acute pancreatitis events during the extension study was 1 (20.0%) in the index-olezarsen 80 mg group (N = 17) and 0 in the index-placebo group (N = 20).
In patients with any acute pancreatitis events in the 10 years before screening in the index study, the number of patients with adjudicated acute pancreatitis events was 1 (7.7%) in the index-olezarsen 80 mg group (N = 13) and 1 (7.7%) in the index-placebo group (N = 13).
Harms
Detailed results for harms are presented in Appendix 5 in the Supplemental Material document. Key results include the following:
The proportion of patients who experienced at least 1 AE was 64.7% in the index-olezarsen 80 mg group and 75% in the index-placebo group. The types of reported AEs were similar to those observed in the index study.
The proportion of patients who experienced SAEs during the extension study was greater in the index-olezarsen 80 mg group (23.5%) than in the index-placebo group (10.0%).
Four patients experienced AEs that led to permanent study drug discontinuation, including 2 patients in the index-olezarsen 80 mg group and 2 patients in the index-placebo group.
One AESI, requiring pretreatment to avoid a hypersensitivity reaction, occurred in 1 patient (5.9%) in the index-olezarsen 80 mg group.
No deaths occurred during the study.
Indirect Evidence
No indirect evidence was submitted for this review.
Studies Addressing Gaps in the Systematic Review Evidence
The sponsor identified 3 gaps in the evidence: small sample size, limited long-term safety and effectiveness data, and the requirement for genetic confirmation of FCS among all patients in the Balance study. To address these gaps, the sponsor submitted the Switch study. The Switch study (NCT05185843) is an ongoing, phase III, multicentre, open-label safety study of olezarsen 80 mg administered once every 4 weeks in patients with FCS who were previously treated with volanesorsen. The primary objective of the Switch study was to evaluate the safety and tolerability of olezarsen in patients with FCS who were previously treated with volanesorsen.
Description of Studies
Characteristics of the Switch study are described in Table 5.
The Switch study included 6 sites in Canada (representing 75% of enrolled patients) and is planned to include a 157-week treatment period, followed by a 13-week posttreatment follow-up period. Statistical analyses were descriptive in nature, and no inferential analyses were performed. The data reported herein are based on interim analyses from data cut-offs specified in each subsection.
The interim results of the Switch study were available for 24 patients with a median follow-up of 540 days (range, 54 to 616 days).
Table 5: Characteristics of Studies Addressing Gaps in Systematic Review Evidence
Study name, design, and sample size | Patient population | Intervention and comparator | Relevant end points |
|---|---|---|---|
Switch, phase III, multicentre, open-label, 157-week safety study Total N = 24 | Patients with FCS (clinically or genetically diagnosed) previously treated with volanesorsen | Olezarsen 80 mg every 4 weeks |
|
FCS = familial chylomicronemia syndrome.
Sources: Clinical Study Report (Switch)16 and the Sponsor’s Summary of Clinical Evidence.14
Patient Disposition
As of the data cut-off of the December 1, 2023, interim analysis, 24 patients were screened, all 24 were enrolled, and 1 patient (4.2%) discontinued study treatment due to an AE or SAE, while 23 patients remained ongoing in the study.
Baseline Characteristics
In the Switch study, 24 patients were enrolled, of which 4 (16.7%) had clinical diagnosis only. The mean age was 49.1 years (SD = 13.4 years) and 54.2% of patients were female and 45.8% were male. Of the enrolled patients, 18 (75.0%) were located in Canada. Race was reported as 8.3% Asian, 87.5% white, and 4.2% “other.” All patients had a history of pancreatitis in the past 10 years and had previously been treated with volanesorsen.
Exposure to Study Treatments
As of the data cut-off of the December 1, 2023, interim analysis, the mean duration of exposure was 497.8 days (SD = 132.98 days), with a median of 540 days (range, 54 to 616 days). Three patients (12.5%) experienced TEAEs that led to dose interruption. No patient experienced a dose reduction for any reason during the study.
Critical Appraisal
Internal Validity
The open-label nature of the study increases the risk of bias in the assessment of subjective outcomes. The primary objective of the study was to evaluate safety. Pharmacodynamic end points and change in fasting triglyceride levels were also assessed. No HRQoL results were reported.
There was a high degree of imprecision in the study results. For the end point of fasting triglyceride levels at week 53, only 11 patients had data available as of the cut-off date, and the SDs were very large relative to the mean change from baseline. The absolute mean change in fasting triglyceride levels was a reduction, whereas the mean percent change in fasting triglyceride levels was an increase. This discrepancy was caused by the small number of patients with data available at the time of analysis and a few patients with extremely high increases in triglyceride levels that skewed the results. In contrast, the median absolute change and median percent change were both reductions in triglyceride levels relative to baseline.
External Validity
The eligibility criteria and patient population appropriately reflect the target population of this assessment, with the caveat that the study population was disproportionately white. However, the clinical experts consulted for this review noted that this was not expected to impact the generalizability of the study results. The Switch study only included patients who were switching from volanesorsen, which may limit generalizability to patients starting this class of medications with olezarsen. However, because this is a study addressing gaps in the evidence, it was considered appropriate for the purpose for which it was submitted in this report.
The clinical experts estimated that approximately 15% to 20% of patients may have FCS due to causes that are not captured by existing genetic testing procedures. The Switch study did include patients with clinically diagnosed FCS only (i.e., without genetic confirmation of FCS), but because of the very small sample size (n = 4 patients without genetic confirmation), the noncomparative study design, and the lack of subgroup analysis by diagnosis type at the time of this review, the ability to draw conclusions from this study is limited.
Nonetheless, the clinical experts consulted for this review stated that no differences in prognosis, treatment effect, disease characteristics, or phenotype are expected when comparing patients with or without genetic confirmation of FCS, regardless of whether the absence of genetic confirmation is due to lack of access or to a negative result (i.e., because genetic testing is not comprehensive of all causes of FCS and that false-negative results may occur).
Results
Efficacy
Change and Percent Change From Baseline in Fasting Triglyceride Levels at Week 53
From baseline to week 53, based on a sample size of 11 patients as of the May 31, 2023, data cut-off, the mean change from baseline in fasting triglyceride levels was ████ mg/dL (███ ███), and the mean percent change was █ █████████.. The median change was ████████ and median percent change was █████ .
Acute Pancreatitis
As of the May 31, 2023, data cut-off, 1 patient had 1 adjudicated acute pancreatitis event, which occurred during the year 1 on-treatment period. Updated results were not available for the December 1, 2023, data cut-off.
Harms
Safety outcomes are available up to the December 1, 2023, data cut-off.
The most frequently experienced treatment-emergent AEs (TEAEs) (≥ 10% overall patients) were abdominal pain (8 patients; [33.3%]), COVID-19 (7 patients [29.2%]), injection site erythema (7 patients [29.2%]), oropharyngeal pain (6 patients [25.0%]), vomiting (5 patients [20.8%]), myalgia (4 patients [16.7%]), influenza (4 patients [16.7%]), fatigue (4 patients [16.7%]), nausea (4 patients [16.7%]), injection site swelling (3 patients [12.5%]), pancreatitis (3 patients [12.5%]), and back pain (3 patients [12.5%]).
No fatal TEAEs or deaths were reported during the study. Serious TEAEs were experienced by 3 patients (12.5%). No patients experienced AESIs. Injection site reactions were experienced by 4 patients (16.7%), hypersensitivity reactions were experienced by 3 patients (12.5%), and flu-like reactions by 4 patients (16.7%). Flu-like reactions were defined as AEs with preferred terms including influenza-like illness, pyrexia, feeling hot, body temperature increased, chills, myalgia, or arthralgia, beginning on the day of injection or the next day. Three patients experienced AEs with the preferred term pancreatitis, of which 1 had an event adjudicated as acute pancreatitis.
One patient withdrew from the study because of a serious TEAE of hypersensitivity, which was considered related to study drug. Two patients (8.3%) experienced TEAEs that led to dose interruption. No patient experienced a dose reduction for any reason while on study.
Key Takeaways
The Switch study provides data for an additional 24 patients with FCS for 12-month end points related to acute pancreatitis and safety. Although the ability to draw conclusions is limited because of the noncomparative design, the data are supportive of the pivotal study for the purpose of this review.
The Switch study does not address the gap of limited follow-up data, given that the outcome time points available at the current data cut-off are not longer than those in the pivotal study.
Only 4 patients (16.7%) had a clinical-only diagnosis of FCS. Additionally, the study is noncomparative, so conclusions cannot be drawn against placebo or SOC alone, and no subgroup analyses were conducted at the time of this review to evaluate differences between patients with clinical-only diagnoses and those with genetically confirmed FCS. This limits the ability to draw conclusions about the efficacy and safety of olezarsen in patients without genetic confirmation of FCS. There therefore remains a gap in the evidence pertaining to patients without genetic confirmation of FCS. However, the clinical experts consulted by CDA-AMC indicated that no differences are expected between this population and patients with genetic confirmation of FCS in terms of disease presentation, demographic characteristics, prognosis, or treatment effect.
Discussion
This report summarizes the evidence for olezarsen compared with placebo, in combination with a low-fat diet, for the treatment of FCS in adult patients. The evidence appraisal was based primarily on the results from 1 phase III, double-blind, placebo-controlled trial (the Balance study, N = 66). Interim results from the LTE study following the Balance study were also reported, and interim results from the ongoing Switch study were included to provide evidence in patients without genetic confirmation of FCS.
Efficacy
FCS is a rare, life-limiting genetic disease associated with substantial morbidity and mortality for which there are no safe and effective therapies available in Canada. Patients living with FCS have highly elevated serum triglyceride levels that are typically not responsive to standard lipid-lowering therapies, diet, or lifestyle adjustments due to the underlying deficiency in LPL activity that characterizes the disease. Chronically elevated triglyceride levels put patients with FCS at a high risk of episodes acute pancreatitis, which can be painful and life-threatening. Patients also face long-term risks associated with recurrent pancreatic episodes, including pancreatic insufficiency and pancreatic insufficiency–related diabetes. Patient and clinician groups indicated that there is a high unmet need for safe and effective therapies that can lower triglyceride levels and reduce the related risks for patients with FCS in Canada. The drug volanesorsen, which is not approved in Canada but is accessible in some countries for patients with FCS, is associated with a high burden of side effects and potentially life-threatening AEs.
The primary treatment goals in managing FCS are to reduce triglyceride levels and to reduce or prevent the occurrence of acute pancreatitis episodes, which are the largest source of morbidity and mortality in patients with FCS. In the Balance study, patients assigned to the placebo group experienced a mean increase in fasting triglyceride levels at months 6 and 12 compared with baseline, compared with a mean reduction associated with assignment to olezarsen 80 mg at both time points. Moreover, the between-group difference between olezarsen 80 mg and placebo exceeded the minimum clinically important difference of 30% suggested by clinical experts consulted by CDA-AMC, supporting that treatment with olezarsen likely results in a reduction in fasting triglyceride levels at months 6 and 12 compared with placebo, although there was imprecision because the 95% CI also included the potential for clinically unimportant benefit. This 30% threshold was suggested for the purpose of assessing certainty in health technology assessment, but in clinical practice, the experts noted that any reduction in triglyceride levels is considered a meaningful clinical effect. The clinical experts highlighted that treatment decisions regarding evaluating efficacy should be at the discretion of the treating clinician.
Because the 50 mg olezarsen dose group was included in the hierarchical testing procedure and did not meet statistical significance for the primary end point, all subsequent end points (i.e., all secondary end points in the study) are considered supportive and exploratory in nature with an elevated risk of type I error.
Over the duration of the study, the rate of acute pancreatitis was lower among patients receiving olezarsen than among those receiving placebo in an analysis that pooled the 50 mg and 80 mg olezarsen groups. There is uncertainty about the generalizability of this analysis because the 50 mg dose is not the recommended dose according to the product monograph. However, because the 50 mg dose was observed to be less effective relative to placebo than the 80 mg dose in the Balance study across the clinically important end points that were analyzed, the potential bias from pooling the 2 treatment groups was not expected to inflate the reported effect of the intervention relative to placebo. It was concluded that treatment with olezarsen may result in a reduction in the rate of acute pancreatitis events compared to placebo, with limitations due to very serious imprecision. Given that episodes of acute pancreatitis are generally uncommon, precision in measurement of the episode rate would likely benefit from longer-term follow-up.
Results of the patient-reported HRQoL assessments reported in the Balance study were more heterogeneous. For the purpose of this report, CDA-AMC applied the GRADE assessment to the FCS Symptoms and Impacts Scale and concluded that there is likely little to no difference between groups for this measure. Several other exploratory HRQoL outcomes were reported in the Balance study, including the Patient Global Impression of Change (PGIC), which did detect a difference in favour of the olezarsen group. The PGIC was not selected for assessment in this report because of the availability of the disease-specific measure, the FCS Symptoms and Impacts Scale. Standardized quantification of HRQoL is an important end point in clinical trials but is particularly difficult in the measurement of HRQoL impacts of diseases characterized by intermittent severe events, such as acute pancreatitis. In consultation with clinical experts, CDA-AMC understood that the effects of FCS on HRQoL are disproportionately associated with episodes of acute pancreatitis, whereas day-to-day HRQoL impacts are commonly related to the highly restrictive diet required for management of FCS, which severely limits typical social activities involving meals, food, and alcohol. Additionally, the rarity of and lack of knowledge about FCS in both the general population and with medical professionals contribute to stigma and dismissal experienced by patients in home, work, social, and medical settings. Based on current evidence, olezarsen is expected to be prescribed in conjunction with maintenance of the restrictive low-fat diet, and so the impact of this diet on patients’ activities of daily living is not expected to be alleviated. Overall, the clinical experts indicated that although HRQoL is important, improvements in triglyceride levels and acute pancreatitis are prioritized outcomes due to the high morbidity and mortality associated with them.
Health care resource utilization outcomes were also assessed in the Balance study as exploratory end points but were not selected for assessment using GRADE because the direct disease-related outcomes were considered higher priority; these outcomes are summarized in Appendix 4 in the Supplemental Material document. Briefly, the numbers of hospitalizations, inpatient days, and emergency department visits were nominally lower in the olezarsen 80 mg group than in the placebo group.
Interim results from the LTE study of the Balance study did not reveal any additional safety concerns as of the currently available data cut-off. Because this is a small, ongoing, noncomparative study with limited interim data available, no further conclusions related to this extension study could not be drawn at this time.
The sponsor identified 3 gaps in the pivotal evidence: small sample size, limited follow-up time, and that all patients in the Balance study were required to have genetic confirmation of FCS. As previously described, the experts indicated that genetic confirmation is not required to diagnose FCS. To address these gaps, the sponsor submitted interim results from the ongoing Switch study, which recruited 24 patients with clinically or genetically confirmed FCS who previously received volanesorsen and who received olezarsen 80 mg every 4 weeks for the duration of the study. The Switch study provides data for an additional 24 patients (which is objectively a small sample size, but the context of this rare disease must be noted); however, the data are noncomparative, which precludes comparisons with placebo or SOC. Only 4 of the 24 enrolled patients (16.7%) were clinically diagnosed without genetic confirmation. Because the Switch study was noncomparative and the sample size is insufficient for subgroup analyses, quantitative conclusions regarding the relative efficacy and safety of olezarsen in clinically diagnosed versus genetically confirmed patients could not be drawn, and there remains an evidence gap regarding whether outcomes differ between these patient subgroups when treated with olezarsen in comparison to placebo or SOC. Nonetheless, the clinical experts consulted by CDA-AMC expressed that patients with clinically and genetically confirmed FCS have no observable phenotypical differences and based on current data using the older drug volanesorsen (which has the same mechanism of action), there is no reason to suspect any difference in their prognosis or treatment response with olezarsen.
Harms
In general, AEs were very common across the treatment groups in the Balance study, and most AEs were mild to moderate in severity. More AEs and more SAEs were observed in the placebo group than the olezarsen 80 mg group, and the clinical experts consulted by CDA-AMC noted that most of the AEs observed in a higher proportion of patients in the placebo group were likely related to mild or subclinical pancreatitis, such as abdominal pain, diarrhea, and gastroesophageal reflux disease. More patients in the olezarsen 80 mg group experienced injection site AEs and decreases in platelet count, although none in any treatment group experienced the AESI of severe reductions in platelet count, which was identified as an AE of interest based on prior experience with volanesorsen. The other prespecified AESI, the requirement for pretreatment medication to avoid a hypersensitivity reaction, was not observed during the Balance study but did occur in 1 patient in the interim LTE data cut-off. Withdrawals due to AEs were generally uncommon, but there were more in the olezarsen 80 mg group than in the placebo group (2 versus 0). It was concluded that treatment with olezarsen may reduce the proportion of patients with at least 1 SAE when compared with placebo.
The safety of olezarsen has not been directly or indirectly quantified with respect to volanesorsen, which was the first drug in this class for the treatment of FCS. Given that volanesorsen was issued a Notice of Non-Compliance by Health Canada due to unpredictable severe thrombocytopenia events, volanesorsen is not considered an appropriate comparator because it is not used in clinical practice. However, it is of interest in the context of the development of olezarsen, which differs from volanesorsen in that it is conjugated to GaINAc. This increases uptake of the active component into hepatocytes and substantially reduces the required effective dose for therapeutic concentrations relative to volanesorsen. In this context, the results of the Balance study regarding AESIs are encouraging. However, because olezarsen is expected to be a lifelong treatment, additional long-term evidence and real-world data will help further elucidate its safety profile.
The clinical experts consulted by CDA-AMC noted that severe worsening of diabetes is a potential long-term AESI that warrants monitoring in the context of treating FCS in real-world practice. No such events were observed during the Balance study or in the interim results of the supportive studies, but the experts noted that this may be due to the small sample sizes, the short duration of follow-up relative to a patient’s lifetime, and/or the low proportion of patients with diabetes included in the pivotal study.
Ethics and Equity Considerations
FCS is a rare, genetic, incurable, life-limiting disease associated with increased morbidity and mortality. There are currently no therapies indicated for the treatment of FCS in Canada, leaving patients and their caregivers with few options beyond maintaining a strict low-fat diet. Clinical experts indicated that the requisite diet is difficult to adhere to and can limit an individual’s participation in some social activities. Although this is true for all patients with FCS, the experts added that some diets may be more difficult to modify due to higher fat content in cultural dietary practices. Input from clinicians and patient groups indicated that olezarsen is anticipated to help address the high burden of unmet need by effectively and safely lowering serum triglyceride levels and reducing the rate of acute pancreatitis in patients.
The diagnosis of FCS is typically based on clinical gestalt, although validated diagnostic algorithms such as the NAFCS have been recently developed. Patient and clinician group input highlighted that many patients experience late diagnosis, misdiagnosis, and stigma from medical professionals due to their lack of familiarity with this rare condition. It is possible to confirm a diagnosis of FCS through molecular testing; however, access to genetic testing is neither consistent nor equitable across Canada, and the genetic testing currently available is not comprehensive of all possible causes of FCS. Additionally, some patients may be uncomfortable with and may not consent to genetic testing for personal, privacy-related, practical (e.g., life insurance concerns), or family-related reasons. Although some patients may undergo molecular testing before treatment initiation, the clinical experts and clinician group input indicated that it would not be appropriate to require genetic confirmation of FCS to access olezarsen because they did not expect treatment effect differences based on genetic subtype. Given the rarity of FCS, initiation and management of treatment may place a disproportionate burden or impact on those who face challenges accessing specialist treatment centres that may be limited in some geographical areas of Canada. The clinical experts consulted for this review indicated that knowledge of FCS among medical providers is not uniform across the country. For example, because of the founder effect in Quebec, more medical professionals there are aware of FCS, and patients are more likely to be appropriately diagnosed at a younger age (e.g., because of known family history, newborn screening, or symptoms). There are no known disparities in FCS prevalence or disproportionate diagnostic barriers related to other demographic factors.
The indication and body of evidence apply only for patients aged 18 years or older. Given that FCS is a genetic, lifelong disease that typically becomes symptomatic at a young age relative to other causes of high triglyceride levels, pediatric patients with FCS may still have an unmet need because there are no Health Canada–approved therapies that can safely and effectively lower triglyceride levels and manage symptoms in this population. In the absence of other treatment options, caregivers may ask providers to prescribe olezarsen off-label for children. This could pose a challenge for providers, who may experience moral distress when trying to balance the uncertain risk-benefit profile in this population against the potential harm of waiting to treat in the context of this early-onset, progressive disease.
Conclusion
One phase III, double-blind, placebo-controlled, multicentre RCT (the Balance study) provided evidence on the efficacy and safety of olezarsen 80 mg administered subcutaneously once every 4 weeks for the treatment of FCS in adult patients, in addition to a low-fat diet of up to 20 g of fat per day. This evidence was supplemented by interim results for the ongoing LTE study of the Balance study and by interim results from the ongoing single-group Switch study, in which patients with clinically or genetically confirmed FCS who had previously received volanesorsen were switched to olezarsen. Patient and clinician groups highlighted an unmet need for any therapy that effectively reduces triglyceride levels in patients with FCS, given that standard lipid-lowering therapies and lifestyle changes are ineffective in the management of this disease. High serum triglyceride levels in FCS are an established risk factor for episodes of acute pancreatitis, which are the primary source of morbidity and mortality in patients with FCS. The results of the Balance study demonstrated that treatment with olezarsen 80 mg likely results in a clinically important reduction in fasting triglyceride levels at month 6 and month 12 compared to placebo. In addition, treatment with olezarsen 80 mg may reduce the rate of adjudicated acute pancreatitis events compared to placebo at 12 months; due to limitations inherent to clinical trials in rare diseases (small sample size and short duration of follow-up for rare events), this conclusion is of moderate certainty. Longer-term follow-up data from the LTE study were limited due to the small sample size and interim data cut-off available at the time of this review. Evidence was uncertain regarding the treatment effect in patients without genetic confirmation of FCS due to the limited number of patients in the Switch study. However, the clinical experts consulted for this review noted that no differences are expected between patients with and without genetic confirmation of FCS in terms of disease presentation, demographic characteristics, prognosis, or treatment effect. The harms results suggested that patients treated with olezarsen 80 mg experienced fewer AEs and SAEs than patients treated with placebo, and no patients experienced AESIs in the Balance study. In the interim results of the LTE, only 1 patient experienced an AESI requiring premedication to prevent hypersensitivity reaction, and no patients had experienced severe platelet count reductions to date. Although these AESIs and worsening of diabetes warrant careful monitoring in patients treated with olezarsen, the side effect profile of olezarsen was considered tolerable and manageable according to the clinical experts consulted by CDA-AMC and are encouraging in contrast to prior clinical experience with volanesorsen.
Economic Review
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of olezarsen as an adjunct to SOC (defined as a low-fat diet, with the addition of agents such as medium-chain triglyceride oil or fibrates) compared to SOC alone for the treatment of FCS in adults. Olezarsen is being reviewed by CDA-AMC through the complex review pathway; as such, CDA-AMC has appraised 2 cost-effectiveness analyses submitted by the sponsor, 1 adopting a publicly funded health care payer perspective and 1 adopting a societal perspective.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of olezarsen from the perspective of a public health care payer in Canada and from a societal perspective over a lifetime horizon (55 years). The modelled population comprised adults with FCS, which is aligned with the proposed Health Canada indication and was based on participants in the Balance trial. The sponsor’s base-case analysis included costs related to drug acquisition, administration, and health care resource use. The sponsor’s societal base case included additional costs associated with productivity loss.
In the sponsor’s base case, which adopted a health care payer perspective, olezarsen was associated with incremental costs of $4,103,329 and 1.97 incremental quality-adjusted life-years (QALYs) relative to SOC. This resulted in an incremental cost-effectiveness ratio (ICER) of $2,087,142 per QALY gained. Of the incremental benefit compared to SOC (1.97 incremental QALYs), approximately 94% of the benefit was predicted to be accrued after the treatment duration of the Balance trial (observation period = 12 months). From a societal perspective, the ICER was $1,888,773 per QALY gained. Additional information about the sponsor’s submission is summarized in Appendix 10 in the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 6; full details are provided in Appendix 11 in the Supplemental Material document).
Table 6: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
Long-term treatment effects of olezarsen are uncertain. | The sponsor assumed patients would maintain their 12-month response over the 55-year model horizon, without supporting long-term evidence. Because approximately 94% of incremental QALYs were accrued beyond the trial period, this assumption adds uncertainty to the estimated long-term benefits of olezarsen. | CDA-AMC was unable to address this limitation due to the lack of long-term clinical evidence. | No scenario analysis was conducted. |
The approach to model the effect of olezarsen on acute pancreatitis risk is not appropriate. | To model acute pancreatitis risk for olezarsen, calibration factors were applied to align with acute pancreatitis rate ratios from the Balance trial. Because the treatment effect on acute pancreatitis risk is already captured by triglyceride reduction, applying additional calibration factors may double count the effect and overestimate the clinical benefit of olezarsen. | CDA-AMC was unable to address this limitation due to the lack of direct clinical evidence. | CDA-AMC conducted a scenario analysis that removed the calibration factors for acute pancreatitis risk in the olezarsen arm. |
The mortality risk of acute pancreatitis is uncertain. | The sponsor informed the estimate of mortality per acute pancreatitis event using a conference abstract reporting results of a clinician survey with unclear event data, introducing substantial uncertainty in the mortality risk. | CDA-AMC conducted a reanalysis using acute pancreatitis mortality data from a large population-based study. | CDA-AMC conducted a scenario analysis maintaining the sponsor’s mortality risk. |
Drug acquisition costs of olezarsen are uncertain. | The sponsor applied an RDI of 96.16% for olezarsen over the model horizon, based on 12-month compliance data from the Balance trial. As long-term adherence and adverse event data are lacking, this assumption adds uncertainty to the estimated drug acquisition costs. | CDA-AMC conducted a reanalysis assuming 100% RDI for olezarsen. | No scenario analysis was conducted. |
The estimation of informal caregiver health-related quality of life is uncertain. | The sponsor applied a disutility intended to reflect the burden on informal caregivers as a treatment-specific utility decrement for patients receiving SOC, based on data on the impact of volanesorsen rather than olezarsen. The impact of olezarsen on caregiver quality of life is unknown. | CDA-AMC was unable to address these issues and did not present a base-case analysis from the societal perspective. | No scenario analysis was conducted. |
The estimation of productivity loss is uncertain. | First, productivity loss was applied by treatment status rather than health state, which is inappropriate because productivity is driven by changes in health and functional status rather than by whether a patient is receiving treatment. Second, productivity changes were based on data from volanesorsen rather than olezarsen, which may not reflect the potential effect of olezarsen on productivity. | CDA-AMC was unable to address these issues and did not present a base-case analysis from the societal perspective. | No scenario analysis was conducted. |
CDA-AMC = Canada’s Drug Agency; QALY = quality-adjusted life-year; RDI = relative dose intensity; SOC = standard of care.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 in the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 11 in the Supplemental Material document.
Impact on Health Care Costs
Olezarsen plus SOC is predicted to be associated with additional health care costs compared to SOC (incremental costs = $4,307,982). This increase in health care spending results from drug acquisition costs associated with olezarsen (refer to Figure 1).
Figure 1: Impact of Olezarsen Plus SOC vs. SOC on Health Care Costs
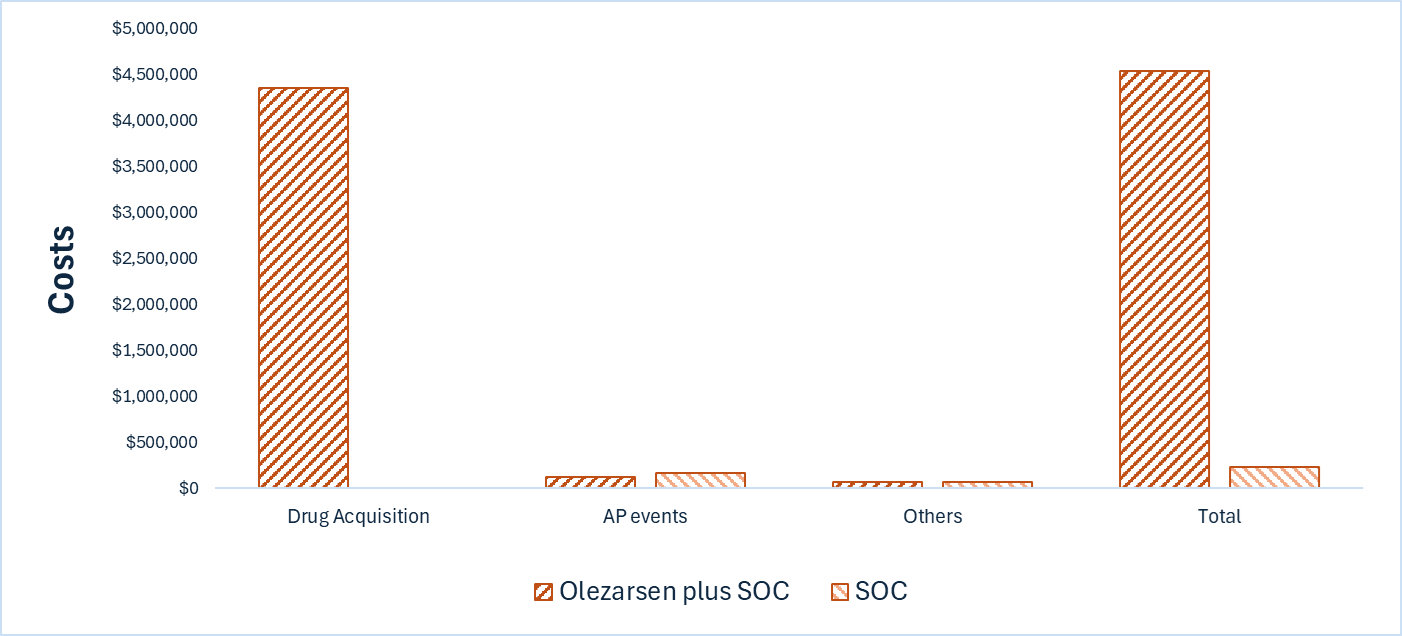
AP = acute pancreatitis; SOC = standard of care; vs. = versus.
Impact on Health
In patients with baseline triglyceride levels greater 10 mmol/L, olezarsen plus SOC is predicted to increase the proportion remaining in the triglyceride 10 mmol/L to less than 22.6 mmol/L health state compared with SOC and to extend overall survival by 1.15 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, olezarsen plus SOC is predicted to result in 1.42 additional QALYs per patient compared to SOC. Approximately 93% of the predicted incremental benefit was accrued on the basis of extrapolation.
Figure 2: Impact of Olezarsen Plus SOC vs. SOC on Patient Health
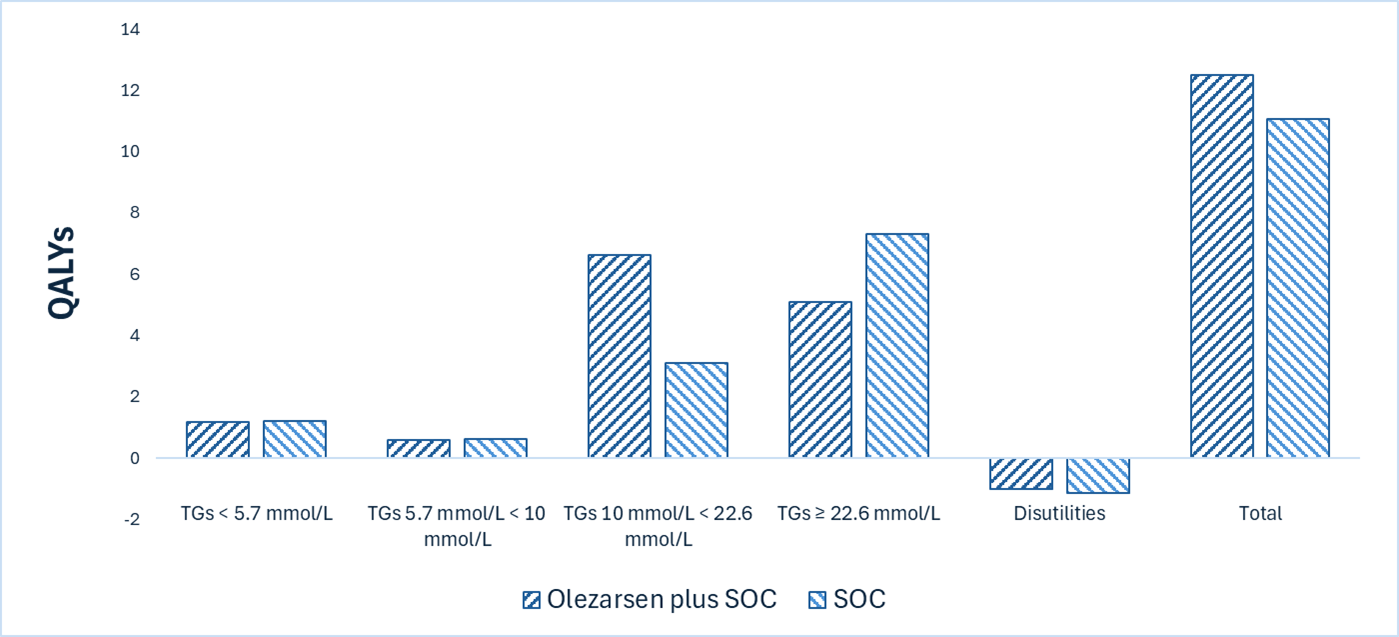
QALY = quality-adjusted life-year; SOC = standard of care; TG = triglyceride; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $3,041,706 per QALY gained for olezarsen plus SOC compared to SOC from the health care payer perspective (refer to Table 7). Additional details on the CDA-AMC base case are available in Appendix 11 in the Supplemental Material document.
Table 7: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|
SOC | 235,214 | 11.08 | Reference |
Olezarsen plus SOC | 4,543,196 | 12.50 | 3,041,706 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Uncertainty and Sensitivity
CDA-AMC explored several areas of uncertainty through scenario analysis. First, the calibration factor applied to estimate acute pancreatitis risk in the olezarsen plus SOC arm may have double-counted the treatment effect on acute pancreatitis risk reduction. Second, there was also uncertainty associated to the mortality risk of acute pancreatitis. These uncertainties were explored through scenario analyses, resulting in ICERs ranging from $2,145,693 and $3,582,871 per QALY gained, compared with SOC (refer to Appendix 11 in the Supplemental Material document).
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2026 to 2028) budget impact of reimbursing olezarsen for use in adults living with FCS. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of olezarsen was aligned with the price included in the sponsor’s economic evaluation. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 in the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (refer to Appendix 12 in the Supplemental Material document). CDA-AMC estimated that by year 3 of reimbursement, 48 patients would be eligible for olezarsen plus SOC; of these, 38 patients are expected to receive olezarsen plus SOC. The estimated incremental budget impact of reimbursing olezarsen plus SOC is predicted to be approximately $50 million over the first 3 years. Because SOC costs are assumed to be $0, the entire incremental budget impact reflects expenditures on olezarsen (i.e., $50 million). The actual budget impact will depend on the market uptake of olezarsen.
Conclusion
Based on the CDA-AMC base case, olezarsen plus SOC would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $3,041,706 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Appendix 11 in the Supplemental Material document). The estimated cost-effectiveness of olezarsen plus SOC compared with SOC alone is uncertain due to a lack of long-term data and uncertainty regarding the impact of treatment on the frequency and mortality risk of acute pancreatitis, which is a key driver of the survival benefit (i.e., 1.15 years). If the survival benefit is not realized, the ICER for olezarsen plus SOC compared with SOC alone will be higher than predicted in the CDA-AMC analysis.
The budget impact of reimbursing olezarsen plus SOC to public drug plans over the first 3 years is estimated to be approximately $50 million. The 3-year expenditure on olezarsen (i.e., not accounting for current expenditure on comparators) is estimated to be $50 million, because SOC costs are assumed to be $0.
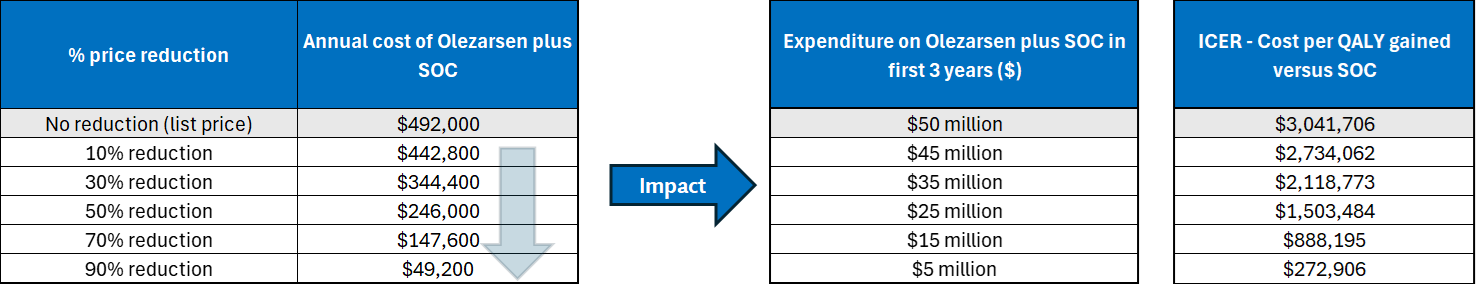
References
1.Theratechnologies, Inc. TRYNGOLZA™ (olezarsen) solution for subcutaneous injection 80 mg/0.8 mL [product monograph] [sponsor supplied reference]. 2025.
2.Stroes E AVK-PE, et al. A Randomized, Placebo-Controlled Phase 3 Study of Olezarsen In Patients With Familial Chylomicronemia Syndrome: The Balance Trial [sponsor supplied reference]. 2024:
3.Baass A, Paquette M, Bernard S, Hegele RA. Familial chylomicronemia syndrome: an under-recognized cause of severe hypertriglyceridaemia. J Intern Med. 2020;287(4):340-348. doi:10.1111/joim.13016 PubMed
4.Blom DJ, O'Dea L, Digenio A, et al. Characterizing familial chylomicronemia syndrome: Baseline data of the APPROACH study. J Clin Lipidol. 2018;12(5):1234-1243 e5. doi:10.1016/j.jacl.2018.05.013 PubMed
5.Hegele RA, Boren J, Ginsberg HN, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol. 2020;8(1):50-67. doi:10.1016/S2213-8587(19)30264-5 PubMed
6.Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272(2):185-96. doi:10.1111/j.1365-2796.2012.02516.x PubMed
7.Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42(13):8796-807. doi:10.1093/nar/gku531 PubMed
8.Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40(33):2785-2796. doi:10.1093/eurheartj/ehz209 PubMed
9.Dron JS, Hegele RA. Genetics of Hypertriglyceridemia. Front Endocrinol (Lausanne). 2020;11:455. doi:10.3389/fendo.2020.00455 PubMed
10.Bchetnia M, Bouchard L, Mathieu J, et al. Genetic burden linked to founder effects in Saguenay-Lac-Saint-Jean illustrates the importance of genetic screening test availability. J Med Genet. 2021;58(10):653-665. doi:10.1136/jmedgenet-2021-107809 PubMed
11.Brown A MP, Dibble A, et al. Brief Communication: Strong Concordance of the North American Familial Chylomicronemia Syndrome Score with a Positive Genetic Diagnosis in Patients from the Balance Study. Journal of Clinical Lipidology. 2025; PubMed
12.Javed F, Hegele RA, Garg A, et al. Familial chylomicronemia syndrome: An expert clinical review from the National Lipid Association. J Clin Lipidol. 2025;19(3):382-403. doi:10.1016/j.jacl.2025.03.013 PubMed
13.Theratechnologies Inc. Clinical Study Report: ISIS 678354-CS3. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of AKCEA-APOCIII-LRX Administered Subcutaneously to Patients with Familial Chylomicronemia Syndrome (FCS) [internal sponsor's report]. March 15, 2024.
14.Theratechnologies Inc. Sponsor Summary of Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TRYNGOLZA (olezarsen), solution for subcutaneous use, 80 mg/0.8 mL in a single dose autoinjector. July 29, 2025.
15.Theratechnologies. Theratechnologies response to Canada's Drug Agency request for additional information regarding olezarsen review on October 15, 2025: Absolute differences [internal additional sponsor's information]. October 17, 2025.
16.Theratechnologies Inc. Clinical Study Report: ISIS 678354-CS7. An Open-Label Safety Study of AKCEA-APOCIII-LRX Administered Subcutaneously to Patients with Familial Chylomicronemia Syndrome (FCS) Previously Treated with Volanesorsen (ISIS 304801)[internal sponsor's report]. March 15, 2024.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.