Drugs, Health Technologies, Health Systems
Reimbursement Review
Vanzacaftor-Tezacaftor-Deutivacaftor (Alyftrek)
Sponsor: Vertex Pharmaceuticals (Canada) Incorporated
Therapeutic area: Cystic fibrosis, F508del or responsive CFTR mutation, in patients aged 6 years and older
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
ALT
alanine aminotransferase
AST
aspartate aminotransferase
BMI
body mass index
CDA-AMC
Canada’s Drug Agency
CF
cystic fibrosis
CFQ-R
Cystic Fibrosis Questionnaire-Revised
CFTRm
CFTR modulator
CI
confidence interval
ELX-TEZ-IVA
elexacaftor-tezacaftor-ivacaftor and ivacaftor
FAS
full analysis set
FEV1
forced expiratory volume in 1 second
FRT
Fischer rat thyroid
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
LS
least squares
MID
minimal important difference
MMRM
mixed model for repeated measures
NOC
Notice of Compliance
ppFEV1
percent predicted forced expiratory volume in 1 second
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SE
standard error
SwCl
sweat chloride
VNZ–TEZ–D-IVA
vanzacaftor-tezacaftor-deutivacaftor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Vanzacaftor-tezacaftor-deutivacaftor (Alyftrek):
|
Sponsor | Vertex Pharmaceuticals (Canada) Incorporated |
Indication | For the treatment of cystic fibrosis in patients aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 21, 2025 |
Recommended dose | Once-daily dosing, orally administered:
|
NOC = Notice of Compliance.
Introduction
Cystic fibrosis (CF), an autosomal recessive condition, is the most common fatal genetic disease affecting children and young adults in Canada. It is caused by mutations in the CFTR gene, which is located on chromosome 7. A deletion of phenylalanine 508 in the first nucleotide binding domain (NBD1) (F508del) is the most common CFTR mutation that results in CF.1 The Canadian Cystic Fibrosis Registry reported there were 4,513 people in Canada living with CF in 2023. Of these, 87.8% carried 1 or more F508del mutations.2 CF results in airway obstruction, chronic endobronchial infection, and inflammation, which ultimately lead to destruction of lung tissue through the development of bronchiectasis and loss of lung function.3 Chronic endobronchial infection of the airways with bacterial pathogens, such as Pseudomonas aeruginosa, is associated with a more rapid loss of lung function.4 Acute or chronic endobronchial infections result in further destruction of lung tissue and are associated with respiratory morbidity. Lung disease accounts for the vast majority (> 80%) of deaths in patients with CF.1,5 Gastrointestinal and pancreatic involvement results in pancreatic exocrine insufficiency in most individuals with CF, causing malabsorption of fats and fat-soluble vitamins, which leads to malnutrition.6 Almost all of these patients, approximately 90%, according to the clinical experts, will have pancreatic insufficiency and will need to take lifelong pancreatic enzyme replacement with every meal as well as fat-soluble vitamin therapy. With increasing age, these patients may develop CF-related diabetes and require treatment with insulin. In 2023, CF-related diabetes was reported in 32.5% of adults and 2.4% of children living with CF in Canada.2
CFTR genetic testing is standard of care and part of the routine diagnostic framework to confirm CF. No implementation barriers due to testing are anticipated from testing done as part of establishing treatment eligibility for vanzacaftor-tezacaftor-deutivacaftor (VNZ–TEZ–D-IVA).
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the CDA-AMC call for input and from the clinical experts consulted for the purpose of this review.
Patient Input
One patient group, Cystic Fibrosis Canada, submitted input for this review. Information was gathered through focus groups with people in Canada with CF and their caregivers. Other sources of data included surveys (including a 2021 survey with more than 1,200 responses), data from the Canadian Cystic Fibrosis Registry, and a multiphase burden of disease study and other relevant publications.
The patient group highlighted that patients not on a CFTR modulator (CFTRm) frequently have periods of infection and acute inflammation called exacerbations that require a hospital stay of at least 2 weeks and that may last up to 4 weeks. Some of these patients could benefit from a CFTRm, but there may not be any CFTRm therapies currently that have a Health Canada indication for their mutation. The group noted that approximately 33% of all adult patients with CF in Canada have CF-related diabetes. Any of the other drugs that patients need to take on a regular basis can also have negative side effects and long-term risks, such as chronic use of antibiotics that may lead to resistance. Moreover, as patients age, they may need to try multiple antibiotics to find one that works, making management of drug-to-drug interactions difficult. The patient group reiterated that, currently, only CFTRm therapies target the underlying pathophysiology of CF by improving the function of the CFTR gene, while the other therapies used to treat CF are for the management of complications and control of symptoms.
The patient group highlighted that too many patients lack access to any CFTRm, including those patients who have ultra-rare mutations that are not on the narrow list of 152 mutations indicated for elexacaftor-tezacaftor-ivacaftor and ivacaftor (ELX-TEZ-IVA) or who cannot tolerate it, or those who have experienced a response to only VNZ–TEZ–D-IVA. The patient group noted that the FDA has approved the use of ELX-TEZ-IVA in 272 mutations, compared with 152 mutations in Canada; the group suggested there are approximately 23 patients in Canada aged 2 years or older with 1 or more of the 119 mutations that are not approved in Canada for treatment with ELX-TEZ-IVA. Thus, they could access ELX-TEZ-IVA in the US but cannot access it in Canada. Additionally, the FDA approved VNZ–TEZ–D-IVA for an additional 31 unique mutations for which ELX-TEZ-IVA is not approved in Canada; this represents approximately 13 additional people in Canada without access to ELX-TEZ-IVA. The group pointed out that, if VNZ–TEZ–D-IVA gets approved in Canada, up to 36 individuals aged 6 or older could access it for the first time. The group specifically requested that the committee recommend access to VNZ–TEZ–D-IVA for the 152 CFTR mutations currently indicated in Canada for ELX-TEZ-IVA. The group further asked that the committee recommend access to VNZ–TEZ–D-IVA for patients in Canada with any of the 151 rare mutations or mutation combinations the FDA lists as indications for VNZ–TEZ–D-IVA, and to recommend the broadest possible access to VNZ–TEZ–D-IVA for individuals with other CFTR mutations when there is evidence, and when more evidence is generated over time (including in vitro laboratory evidence or clinical evidence), that shows these patients will or may experience a response to this therapy. The group highlighted that CF has significant financial implications for patients and their caregivers, health systems, and society.
No patients reported experience with VNZ–TEZ–D-IVA treatment. The patient group highlighted that VNZ–TEZ–D-IVA may improve quality of life and adherence as a 1-pill-a-day treatment, which would provide an option for parents who may have difficulty administering medications to their children, who already take several medicines a day to maintain their health.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts described that ELX-TEZ-IVA is the current standard of care in patients with CF aged 6 years and older with eligible CF-causing mutations. However, there is an unmet need for more effective therapy that prolongs life, improves quality of life, and reduces the treatment burden of supportive medications in patients who have CF-causing mutations that are not eligible for treatment with ELX-TEZ-IVA, which represents approximately 10% of patients with CF in Canada and disproportionately represents racialized people. Additionally, even among patients eligible for treatment with ELX-TEZ-IVA, the clinical experts noted that most patients still have sweat chloride (SwCl) values in an abnormal range. Doses of ELX-TEZ-IVA are administered twice daily and require fat-containing meals for absorption; ergo, the experts noted that less frequent dosing is an unmet need that might improve adherence. Some patients may experience adverse events (AEs) or intolerance to ELX-TEZ-IVA requiring cessation of treatment, and there is an unmet need for alternative triple-combination CFTRm therapies in those circumstances. Finally, overall pill and treatment burden remains high in patients with CF, despite reductions after the advent of triple-combination CFTRm therapies (i.e., ELX-TEZ-IVA).
The clinical experts consulted by Canada’s Drug Agency (CDA-AMC) stated that VNZ–TEZ–D-IVA would occupy a place in therapy similar to that of ELX-TEZ-IVA, with the caveat that it may be applicable to a wider range of mutations, and that the current Health Canada indications begin at different ages (i.e., ELX-TEZ-IVA is currently indicated for patients aged at least 2 years, while VNZ–TEZ–D-IVA is indicated in patients aged at least 6 years). The experts noted that, given the once-daily dosing of VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA, improvements in treatment adherence may be expected. However, patients with CF are typically taking numerous pills daily in addition to CFTRm therapies; ergo, this may not represent a substantial difference in overall treatment or pill burden. Nonetheless, the expert specialized in pediatric care said that a reduction in treatment or pill burden may be particularly significant for the treatment of pediatric patients with CF. The experts noted that it may be premature for every patient currently stable on ELX-TEZ-IVA to be switched to VNZ–TEZ–D-IVA due to the longer clinical safety record of ELX-TEZ-IVA; however, VNZ–TEZ–D-IVA is generally expected to become the new standard of care for eligible patients. Additionally, if there are clinical signals that achieving a lower SwCl than previously seen with ELX-TEZ-IVA results in less end-organ damage, it may further shift treatment paradigms and reduce the need for other supportive therapies.
The clinical experts indicated that the patient population appropriate for treatment with VNZ–TEZ–D-IVA is similar to that for ELX-TEZ-IVA, with the exception that there may be additional CF-causing mutations eligible for treatment with VNZ–TEZ–D-IVA. Aside from genotype, the patient population is expected to be similar.
The clinical experts noted that the assessment of CF-related treatment is based on lung function spirometry, number of days treated with oral and IV antibiotics for pulmonary exacerbations, number of CF-related hospitalizations, and monitoring of body mass index (BMI) or BMI z score (as appropriate by age) for signs of malnutrition. The clinical experts informed CDA-AMC that the assessments related to VNZ–TEZ–D-IVA should be similar to those expected for ELX-TEZ-IVA, although, in any reimbursement criteria, it is also important to consider that assessments for initiation, continuation, or discontinuation should differ for patients newly starting triple-combination CFTRm therapies (i.e., patients who are generally expected to experience greater improvements with therapy) versus switching between triple-combination CFTRm therapies (i.e., the new treatment would be expected to maintain the benefit previously observed with the other therapy). Assessment of response in the previously listed metrics should generally be performed after a year of treatment because, as the experts highlighted, some metrics (especially pulmonary exacerbations and BMI or BMI z score) may have fluctuations with seasonal illness and short-term instability in pediatric patients that may not be reflective of the effect of the drug.
The clinical experts noted that discontinuation should be considered in the case of intolerable AEs that cannot be sufficiently managed or, in the case of a lung transplant, similar to current practice with ELX-TEZ-IVA (although this does not reflect a contraindication specified by Health Canada).
The clinical experts noted that a physician practising within or under the directed supervision of a Canadian CF clinic should manage the prescription and renewal of VNZ–TEZ–D-IVA.
Clinician Group Input
One clinician group, the Cystic Fibrosis Canada Health Care Advisory Council, provided input for this review. Information for this submission was gathered through the following: experience gained by working with and delivering medical services to people with CF; consultations with people with CF not eligible for ELX-TEZ-IVA; a review of medical and scientific literature, including clinical trial results and real-world experience; and the Canadian Cystic Fibrosis Registry, a collection of patient data and other information regarding CF care and outcomes. A total of 2 clinicians contributed to the input submitted for this review.
The input from the clinician group was generally consistent with that of the clinical experts consulted for this review, emphasizing that about 10% of patients have mutations not currently approved for treatment with available CFTRm therapies in Canada, and that there are no alternative triple-combination CFTRm treatments for patients with intolerance to ELX-TEZ-IVA. The group agreed that the once-daily regimen of VNZ–TEZ–D-IVA may improve adherence and convenience compared with the twice-daily regimen of ELX-TEZ-IVA.
The group noted that rare mutations for which there is currently no CFTRm indication are disproportionately found in racialized communities, so expanded access to this therapy may improve equitable access to disease-modifying treatments in Canada.
The clinician group noted that, according to recent Canadian clinical guidelines, all patients with CF who have at least 1 CFTRm-responsive CFTR variant should be treated with a CFTRm. The clinician group noted that patients with CF best suited for the drug under review include patients already on ELX-TEZ-IVA who are experiencing challenges due to side effects or related to adherence to a twice-daily treatment regimen, and patients with rare mutations not approved for treatment with ELX-TEZ-IVA but who may experience a response to VNZ–TEZ–D-IVA. The group agreed with the clinical experts regarding the metrics and yearly time frame of reassessments for treatment efficacy, as well as factors to consider for treatment discontinuation and prescribing.
Drug Program Input
The following were identified as key factors that could potentially impact the implementation of a recommendation for VNZ–TEZ–D-IVA:
Considerations for:
initiation of therapy
continuation or renewal of therapy
discontinuation of therapy
prescribing of therapy
care provision issues
system and economic issues.
The clinical experts provided advice on the potential implementation issues raised by the drug programs. Refer to Table 7 for more details.
Clinical Evidence
Systematic Review
Description of Studies
Two double-blind phase III randomized controlled trials (RCTs), SKYLINE 102 (VX21-121-102; N = 405) and SKYLINE 103 (VX21-121-103; N = 574), were included. One single-arm phase III study, RIDGELINE (VX21-121-105), was also included, specifically, cohort A1 (N = 17) and cohort B1 (N = 78).
The RCTs were randomized studies with a 52-week treatment phase and were similar in study design. Both SKYLINE studies compared VNZ–TEZ–D-IVA with ELX-TEZ-IVA in patients with CF aged at least 12 years with ELX-TEZ-IVA–responsive mutations. Both included patients with a percent predicted forced expiratory volume in 1 second (ppFEV1) value between 40% and 90% of the predicted mean for age, sex, and height for patients currently receiving ELX-TEZ-IVA, or a ppFEV1 of between 40% and 80% for patients not currently receiving ELX-TEZ-IVA. In terms of CF-related genotype, patients recruited to the SKYLINE 102 trial had to be heterozygous for F508del and a minimal function mutation, while patients recruited to the SKYLINE 103 trial had to have either homozygous F508del mutations, heterozygous F508del and a gating or residual function mutation, or at least 1 mutation identified as responsive to ELX-TEZ-IVA with no F508del mutation. All patients entered a 28-day run-in period receiving ELX-TEZ-IVA before being randomized 1:1 in a double-blind fashion to treatment with VNZ–TEZ–D-IVA or ELX-TEZ-IVA for 52 weeks, followed by an additional 28-day open-label follow-up for additional safety data.
In the RIDGELINE trial, patients aged between 1 and 11 years with a mutation responsive to ELX-TEZ-IVA were recruited. The RIDGELINE study included a part A, with the objective of evaluating the pharmacokinetics of VNZ–TEZ–D-IVA and relevant metabolites when dosed in therapeutic concentrations, and to evaluate the safety and tolerability of VNZ–TEZ–D-IVA. Part A preceded part B and informed the doses and weight-related dose thresholds for part B, in which the objectives were primarily to evaluate the safety and tolerability of VNZ–TEZ–D-IVA through week 24, and secondarily to evaluate the efficacy through week 24 as well as the pharmacokinetics. Parts A and B were each divided into 3 cohorts by age range, where the first cohort (A1 and B1) included patients aged 6 to 11 years (inclusive), the second (A2 and B2) included patients aged 2 to 5 years (inclusive), and the third (A3 and B3) included patients aged 1 year to younger than 2 years. Therefore, only cohorts A1 (N = 17) and B1 (N = 78) are relevant to the Health Canada indication considered here. The efficacy data presented in this report are informed by cohort B1, and the safety data are informed by both cohorts A1 and B1.
In the SKYLINE 102 trial, slightly more than 40% of patients were female and just under 60% of patients were male, whereas the sex distribution was closer to approximately 50% each in the SKYLINE 103 study. The mean age by treatment group in the 2 RCTs at baseline was 31 to 34 years across the SKYLINE studies, and the treatment groups were similar for these categories in each study. In cohorts A1 and B1 in the RIDGELINE study, 41.2% and 43.6% were female, 58.8% and 56.4% of patients were male, and the mean age was 9.5 years and 9.3 years, respectively. The baseline patient characteristics for ppFEV1 demonstrated more progressed disease in the studies that recruited older patients (the SKYLINE 102 and SKYLINE 103 trials recruited patients aged 12 years or older) compared with the RIDGELINE study, which recruited patients aged 6 to 11 years for cohorts A1 and B1; this reflects the natural course of CF, and there were no concerning within-study differences between treatment groups. In all studies, most patients (range, 84.6% to 94.1%) had immediate prior experience with a CFTRm, most commonly, ELX-TEZ-IVA. The mean duration of prior ELX-TEZ-IVA use was approximately 2 years in the SKYLINE 102 and SKYLINE 103 RCTs, and approximately 1 year in cohort B1 of the RIDGELINE trial. The SKYLINE 102 study recruited patients with a genotype that was heterozygous for F508del and a minimal function mutation; ergo, this genotype was represented in 100% of patients, whereas in the other studies, the most common genotype was homozygous for F508del (range, 47.4% to 78.2%). The majority of patients in every study were white (range, 90.7% to 97.5%), whereas a minority were Asian (range, 0% to 0.5%), Black or African American (range, 0% to 2.0%), more than 1 race (range, 0% to 1.5%), or other (range, 0.% to 0.4%), or information about race was not collected due to local regulations.
Efficacy Results
Number of Pulmonary Exacerbations
In the SKYLINE 102 trial full analysis set (FAS), the estimated event rate per year was 0.42 in the ELX-TEZ-IVA group (n = 50 patients with events) versus 0.32 in the VNZ–TEZ–D-IVA group (n = 60 patients with events). The pulmonary exacerbation rate difference was −0.10 events (95% confidence interval [CI], −0.24 to 0.04) per year.
In the SKYLINE 103 trial FAS, the annual event rate was 0.29 in the VNZ–TEZ–D-IVA group (n = 61 patients with events) and 0.26 in the ELX-TEZ-IVA group (n = 59 patients with events). The rate difference was 0.03 events per year (95% CI, −0.07 to 0.13).
In the RIDGELINE trial, there were 6 patients in the cohort B1 FAS who experienced an event, contributing to an annual event rate of 0.15.
No subgroup or sensitivity analyses were conducted for this end point in the included studies.
Time to First Pulmonary Exacerbation
In the SKYLINE 102 trial, based on a secondary analysis in the FAS of the time to first pulmonary exacerbation during the analysis period, the probability of event-free survival (Kaplan-Meier estimate) at 24 weeks was 0.820 (95% CI, 0.758 to 0.867) in the VNZ–TEZ–D-IVA group and 0.819 (95% CI, 0.758 to 0.866) in the ELX-TEZ-IVA group; at 52 weeks, it was 0.741 (95% CI, 0.673 to 0.797) and 0.695 (95% CI, 0.626 to 0.755), respectively.
In the SKYLINE 103 trial, based on a secondary analysis in the FAS of the time to first pulmonary exacerbation during the analysis period, the probability of event-free survival (Kaplan-Meier estimate) at 24 weeks was 0.857 (95% CI, 0.810 to 0.893) in the VNZ–TEZ–D-IVA group and 0.882 (0.838 to 0.914) in the ELX-TEZ-IVA group; at 52 weeks, it was 0.783 (95% CI, 0.729 to 0.827) and 0.792 (95% CI, 0.739 to 0.835), respectively.
Time to first pulmonary exacerbation was not reported in the RIDGELINE trial.
Pulmonary Exacerbations Requiring IV Antibiotics or Hospitalization
In the SKYLINE 102 trial FAS, 10 patients (5.1%) in the VNZ–TEZ–D-IVA group and 26 patients (12.9%) in the ELX-TEZ-IVA group had pulmonary exacerbations requiring hospitalization or IV antibiotic therapy during the analysis period. The probability of event-free survival (Kaplan-Meier estimate) in the 2 groups at 24 weeks was 0.969 (95% CI, 0.933 to 0.986) and 0.935 (95% CI, 0.890 to 0.962), respectively; at 52 weeks, it was 0.948 (95% CI, 0.906 to 0.972) and 0.868 (95% CI, 0.813 to 0.908), respectively. All patients with pulmonary exacerbations required IV antibiotic therapy; additionally, 8 patients in the VNZ–TEZ–D-IVA group and 17 patients in the ELX-TEZ-IVA group required hospitalization.
In the SKYLINE 103 trial FAS, 19 patients (6.7%) and 17 patients (5.9%) in the VNZ–TEZ–D-IVA group and ELX-TEZ-IVA group, respectively, had pulmonary exacerbations requiring hospitalization or IV antibiotic therapy. The probability of event-free survival (Kaplan-Meier estimate) in the 2 groups at 24 weeks was 0.961 (95% CI, 0.930 to 0.978) and 0.962 (95% CI, 0.932 to 0.979), respectively; at 52 weeks, it was 0.931 (95% CI, 0.894 to 0.956) and 0.940 (95% CI, 0.906 to 0.962), respectively. All patients with pulmonary exacerbations required IV antibiotic therapy; additionally, 14 patients in the VNZ–TEZ–D-IVA group and 9 patients in the ELX-TEZ-IVA group required hospitalization.
In cohort B1 of the RIDGELINE trial, 1 patient (1.3%) required both hospitalization and IV antibiotic therapy, for an observed event rate per year of 0.03.
Percent Predicted Forced Expiratory Volume in 1 Second
The primary outcome of ppFEV1 was evaluated in the FAS through week 24 in the SKYLINE 102 (n = 196 in the VNZ–TEZ–D-IVA group and n = 202 in the ELX-TEZ-IVA group) and SKYLINE 103 trials (n = 284 in the VNZ–TEZ–D-IVA group and n = 289 in the ELX-TEZ-IVA group). In the mixed model for repeated measures (MMRM) analysis of noninferiority comparing VNZ–TEZ–D-IVA with ELX-TEZ-IVA, the least squares (LS) mean treatment difference was 0.2% (95% CI, −0.7% to 1.1%; 1-sided P < 0.0001) in the SKYLINE 102 trial and 0.2% (95% CI, −0.5% to 0.9%; 1-sided P for noninferiority < 0.0001) in the SKYLINE 103 trial. The prespecified noninferiority margin was −3.0%; ergo, the results of both studies met the preplanned primary analysis of noninferiority for ppFEV1 because neither 95% CI crossed the noninferiority margin.
In cohort B1 of the RIDGELINE trial (n = 78), patients treated with VNZ–TEZ–D-IVA showed similar ppFEV1 values compared with baseline use of ELX-TEZ-IVA, based on a within-group LS mean absolute change from baseline to 24 weeks of 0% (95% CI, −2.0% to 1.9%).
The secondary end point of ppFEV1 through 52 weeks showed similar results in both studies, consistent with the 24-week primary analyses. In the SKYLINE 102 trial, the LS mean difference in absolute change from baseline was 0.1% (95% CI, −0.8% to 1.0%); in the SKYLINE 103 trial, it was 0.3% (95% CI, −0.4 to 1.0).
In the SKYLINE 102 and SKYLINE 103 trials, a supplementary sensitivity analysis was conducted using an alternative estimand where intercurrent events were addressed using the hypothetical strategy for the 24-week outcome of ppFEV1, and the results were consistent with the primary analyses.
Subgroup analyses were conducted on the primary outcome at 24 weeks in the SKYLINE 102 and SKYLINE 103 trials based on age (aged younger than 18 years versus at least 18 years at screening) and ppFEV1 at baseline (less than 70% versus at least 70%). Most of the results fell within the noninferiority margin of −3.0% and were similar to the primary analysis results. However, in the SKYLINE 102 trial in the subgroup of patients younger than age 18 years at screening, the 95% CI crossed this threshold, with an LS mean difference of −1.3% (95% CI, −3.9% to 1.3%). In the SKYLINE 103 trial, the value for this subgroup was 0.9% (95% CI, −1.2% to 3.0%).
No subgroup or sensitivity analyses were conducted in the RIDGELINE trial or for the week 52 outcomes in the SKYLINE studies.
Body Mass Index
In the RIDGELINE trial, the absolute change in BMI at week 24 was an LS mean of 0.22 kg/m2 (95% CI, 0.05 kg/m2 to 0.38 kg/m2).
In the SKYLINE 102 trial, the LS mean change from baseline in BMI in the VNZ–TEZ–D-IVA group (n = 179) and ELX-TEZ-IVA group (n = 187) at week 52 was 0.30 kg/m2 (standard error [SE] = 0.10 kg/m2) and 0.08 kg/m2 (SE = 0.10 kg/m2), respectively, with an LS mean difference of 0.22 kg/m2 (95% CI, −0.05 kg/m2 to 0.49 kg/m2).
In the SKYLINE 103 trial, the LS mean change from baseline in BMI in the VNZ–TEZ–D-IVA group (n = 248) and ELX-TEZ-IVA group (n = 266) at week 52 was 0.37 kg/m2 (SE = 0.08 kg/m2) and 0.22 kg/m2 (SE = 0.07 kg/m2), respectively, with an LS mean difference of 0.16 kg/m2 (95% CI, −0.05 kg/m2 to 0.36 kg/m2).
No subgroup or sensitivity analyses were reported.
BMI Z Score
In the RIDGELINE trial, the absolute change in BMI z score at week 24 was an LS mean of −0.05 (SE = 0.03; 95% CI, −0.12 to 0.02).
In the SKYLINE 102 trial at 52 weeks, the LS mean change from baseline in BMI z score among patients aged 20 years or younger at baseline was 0.25 (SE = 0.10) in the VNZ–TEZ–D-IVA group (n = 27) and −0.09 (SE = 0.09) in the ELX-TEZ-IVA group (n = 32), with an LS mean difference of 0.34 (95% CI, 0.07 to 0.61).
In the SKYLINE 103 trial, the LS mean change from baseline in BMI z score among patients aged 20 years or younger at baseline was 0.11 (SE = 0.06) in the VNZ–TEZ–D-IVA group (n = 42) and −0.13 (SE = 0.07) in the ELX-TEZ-IVA group (n = 35), with an LS mean difference of 0.24 (0.06 to 0.42) at 52 weeks.
No subgroup or sensitivity analyses were reported.
Cystic Fibrosis Questionnaire-Revised Respiratory Domain
Health-related quality of life (HRQoL) was measured using the Cystic Fibrosis Questionnaire-Revised (CFQ-R) respiratory domain.
In the SKYLINE 102 trial, the LS mean change in the CFQ-R respiratory domain score through week 24 was 0.5 points (SE = 1.1) in the VNZ–TEZ–D-IVA group (n = 186) and −1.7 points (SE = 1.0) in the ELX-TEZ-IVA group (n = 192). The LS mean difference was 2.3 points (95% CI, −0.6 to 5.2).
In the SKYLINE 103 trial, the LS mean change from baseline in the CFQ-R respiratory domain was −1.2 points (SE = 0.8) in the VNZ–TEZ–D-IVA group (n = 268) and the same value, i.e., −1.2 points (SE = 0.8) in the ELX-TEZ-IVA group (n = 270). The LS mean difference was −0.1 points (95% CI, −2.3 to 2.1).
Patients treated with VNZ–TEZ–D-IVA in cohort B1 of the RIDGELINE trial had a within-group LS mean absolute change from baseline through week 24 of 3.9 points (95% CI, 1.5 to 6.3).
Through week 52, the LS mean change in the SKYLINE 103 trial in the VNZ–TEZ–D-IVA group (n = 186) and ELX-TEZ-IVA group (n = 192) was 0.8 points (SE = 1.0) and −1.6 points (SE = 1.0), respectively, with an LS mean difference of 2.4 points (95% CI, −0.3 to 5.1).
In the SKYLINE 103 trial, the LS mean change in the VNZ–TEZ–D-IVA group (n = 268) and ELX-TEZ-IVA group (n = 270) through 52 weeks was −0.4 points (SE = 0.7) and −1.0 point (SE = 0.7), respectively, with an LS mean difference of 0.7 points (95% CI, −1.4 to 2.7).
No subgroup or sensitivity analyses were reported.
Sweat Chloride
In the SKYLINE 102 trial, the LS mean absolute change from baseline for SwCl through week 24 in the FAS was −7.5 mmol/L (standard deviation [SD] = 0.8 mmol/L) in the VNZ–TEZ–D-IVA group (n = 196) compared with 0.9 mmol/L (SD = 0.8 mmol/L) in the ELX-TEZ-IVA group (n = 202). The MMRM analysis of this secondary end point reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment compared with ELX-TEZ-IVA treatment, with an LS mean difference of −8.4 mmol/L (95% CI, −10.5 mmol/L to −6.3 mmol/L; P < 0.0001).
In the SKYLINE 103 trial, the LS mean absolute change from baseline in SwCl through week 24 (FAS; n = 294 and 289, respectively) was −5.1 mmol/L (SD = 0.7 mmol/L) in the VNZ–TEZ–D-IVA group compared with −2.3 mmol/L (SD = 0.7 mmol/L) in the ELX-TEZ-IVA group. The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment compared with ELX-TEZ-IVA treatment, with an LS mean difference of −2.8 mmol/L (95% CI, −4.7 mmol/L to −0.9 mmol/L; P = 0.0034).
In the single-arm study, RIDGELINE, the within-group LS mean absolute change from baseline through week 24 (averaging weeks 16 and 24) was −8.6 mmol/L (95% CI, −11.0 mmol/L to −6.3 mmol/L) in the cohort B1 FAS (n = 77).
In the SKYLINE 102 trial, the LS mean absolute change from baseline in SwCl through week 52 in the FAS was −7.5 mmol/L (SD = 0.7 mmol/L) in the VNZ–TEZ–D-IVA group (n = 196) compared with 0.5 mmol/L (SD = 0.7 mmol/L) in the ELX-TEZ-IVA group (n = 202). The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment with an LS mean difference of −8.0 mmol/L (95% CI, −9.9 mmol/L to −6.1 mmol/L).
In the SKYLINE 103 trial, the LS mean change in the VNZ–TEZ–D-IVA group (n = 284) was −5.0 mmol/L (SD = 0.6 mmol/L) and, in the ELX-TEZ-IVA group, it was −2.2 mmol/L (SD = 0.6 mmol/L). The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment, with an LS mean difference of −2.8 mmol/L (95% CI, −4.6 mmol/L to −1.0 mmol/L).
No subgroup or sensitivity analyses were reported.
Proportion of Patients With an SwCl Value of 30 mmol/L or Lower
The 24-week pooled outcome, including data from both the SKYLINE 102 and SKYLINE 103 studies, reported an odds ratio of 2.87 (95% CI, 2.00 to 4.12; P < 0.0001), indicating patients treated with VNZ–TEZ–D-IVA were more likely to have an SwCl of 30 mmol/L or lower at week 24 than patients treated with ELX-TEX-IVA.
In the RIDGELINE trial, 41 out of 78 patients (52.6%) had an SwCl of 30 mmol/L or lower at week 24 (95% CI, 40.9% to 64.0%).
The 52-week result in the SKYLINE 102 trial was an odds ratio of 5.77 (95% CI, 3.33 to 9.99) and in the SKYLINE 103 trial was 1.98 (95% CI, 1.36 to 2.88). While the magnitude of difference between the treatment arms appears to vary between the 2 studies, the trend was in the same direction and aligned with the 24-week outcome results reported in these studies.
No subgroup or sensitivity analyses were reported.
Harms Results
To contextualize the safety data from both the SKYLINE 102 and SKYLINE 103 studies, it is important to note that the majority of patients had received commercial ELX-TEZ-IVA before study enrolment, with a median exposure of approximately 2 years, and all patients received ELX-TEZ-IVA for 4 weeks during the run-in period. Additionally, patients who had a prior history of intolerance to ELX-TEZ-IVA were not eligible to enrol, and patients who developed intolerance to ELX-TEZ-IVA during the run-in period were discontinued from the study. Therefore, the safety data from the ELX-TEZ-IVA group are reflective of the experience in patients who have tolerated ELX-TEZ-IVA and who have continued on an existing treatment regimen, and the safety data from the VNZ–TEZ–D-IVA group are reflective of patients who initiated a new treatment regimen.
Similarly, in the RIDGELINE trial, all patients either had prior experience on ELX-TEZ-IVA or participated in a run-in period in which they received ELX-TEZ-IVA and then switched to VNZ–TEZ–D-IVA during the study period.
Overall Adverse Events
In the SKYLINE 102 and SKYLINE 103 trials, 94.4% and 96.5% of patients, respectively, who were treated with VNZ–TEZ–D-IVA experienced an AE, while 97% and 94.5% of patients, respectively, who were treated with ELX-TEZ-IVA experienced an AE. In both studies, the most common AEs were infective pulmonary exacerbations related to CF (range, 26.8% to 34.7% across the treatment groups in both studies), COVID-19 (range, 20.4% to 26.7%), cough (range, 20.3% to 23.0%), and nasopharyngitis (range, 17.3% to 23.0%).
In the RIDGELINE trial, the proportion of patients with at least 1 AE was 70.6% and 96.2% in cohorts A1 and B1, respectively.
Serious Adverse Events
In the SKYLINE 102 and SKYLINE 103 trials, among patients treated with VNZ–TEZ–D-IVA, 14.3% and 14.1%, respectively, experienced a serious adverse event (SAE), while 20.3% and 13.8% of patients, respectively, who were treated with ELX-TEZ-IVA experienced an SAE. The proportion of patients with any SAE appears elevated in the ELX-TEZ-IVA group of the SKYLINE 102 trial relative to the VNZ–TEZ–D-IVA group, but this was not the case in the SKYLINE 103 trial, where the values were similar across the 2 treatment groups. One life-threatening AE occurred, which was an infective pulmonary exacerbation of CF in a patient in the ELX-TEZ-IVA treatment group of the SKYLINE 103 trial.
In the 2 RCTs, the most common SAE was an infective pulmonary exacerbation of CF. In the SKYLINE 102 trial, this occurred in 5.6% of patients treated with VNZ–TEZ–D-IVA and 11.4% treated with ELX-TEZ-IVA. In the SKYLINE 103 trial, this occurred in 6.3% and 4.2% of patients, respectively. Other SAEs were individually uncommon (from 0 to approximately 2% of patients for each SAE) but included influenza, hemoptysis, pneumonia, suicidal ideation, syncope, COVID-19, alanine aminotransferase (ALT) increased, ALT or aspartate aminotransferase (AST) increased, constipation, distal intestinal obstruction syndrome, gamma-glutamyl transferase increased, cholelithiasis, and nephrolithiasis.
In the RIDGELINE trial, no patients in cohort A1 had an SAE, and 6 patients (7.7%) in cohort B1 had an SAE. In cohort B1, the SAEs included infective pulmonary exacerbations (n = 2); among these 2 patients, 1 also had an SAE of failure to thrive. Other SAEs that each occurred in 1 patient (1.3%) were adenovirus infection, constipation, pulmonary function test decreased, and cough. No life-threatening AEs occurred.
Withdrawals Due to Adverse Events
In the SKYLINE 102 trial, 2.0% of patients treated with VNZ–TEZ–D-IVA and 4.5% of patients treated with ELX-TEZ-IVA had an AE leading to treatment discontinuation. In the SKYLINE 103 trial, this occurred in 4.9% and 3.1% of patients, respectively. The AEs leading to treatment discontinuation were each individually uncommon (0% to approximately 2% of patients for each AE) and included a variety of types of AEs.
In the RIDGELINE trial, 1 patient (1.3%) had treatment interrupted due to a seizure, and 1 patient stopped treatment due to AEs of fatigue and coughing. Both patients belonged to cohort B1.
Mortality
No deaths occurred in the SKYLINE 102, SKYLINE 103, or RIDGELINE trials.
Notable Harms
Identified AEs of special interest included elevated aminotransferase levels, rash, elevated creatine kinase levels, cataracts, hypoglycemia, and neuropsychiatric events.
In the VNZ–TEZ–D-IVA and ELX-TEZ-IVA groups of the SKYLINE 102 trial, the proportion of patients with AEs of elevated aminotransferase levels was 8.2% and 6.4%, respectively. Rash occurred in 8.7% and 7.4%, elevated creatinine occurred in 9.2% and 11.9%, hypoglycemia occurred in 2.6% and 3.5%, and neuropsychiatric AEs occurred in 8.7% and 13.9%, respectively. No patients in either treatment group had AEs of █████████. No patients treated with VNZ–TEZ–D-IVA had serious ████████ ████████████, rash, creatine kinase elevation, ████████, or hypoglycemia events. Serious neuropsychiatric events occurred in 3 patients (1.5%) treated with VNZ–TEZ–D-IVA and in 2 patients (1.0%) treated with ELX-TEZ-IVA.
In the VNZ–TEZ–D-IVA and ELX-TEZ-IVA groups of the SKYLINE 103 trial, the proportion of patients with AEs of elevated aminotransferase levels was 9.5% and 7.6%, respectively. Rash occurred in 12.7% and 8.0%, elevated creatine kinase levels occurred in 8.8% and 5.9%, cataracts occurred in ████ ███ ████; hypoglycemia occurred in 1.1% and 3.8%, and neuropsychiatric AEs occurred in 13.4% and 10.7%, respectively. Serious neuropsychiatric events occurred in 1 patient (0.4%) treated with VNZ–TEZ–D-IVA and 2 patients (0.7%) treated with ELX-TEZ-IVA. Serious elevated aminotransferase events occurred in ███ ██████ patients treated with VNZ–TEZ–D-IVA and ████ treated with ELX-TEZ-IVA. Events of serious elevated creatine kinase occurred in 1 patient (0.4%) in the VNZ–TEZ–D-IVA group and in no patients in the ELX-TEZ-IVA group. No patients had serious rash, ████████, or hypoglycemia events.
In cohort A1 of the RIDGELINE trial, there were no events of ALT increased, AST increased, elevated creatine kinase levels, █████████, hypoglycemia, or neuropsychiatric AEs, while 3 patients (17.6%) had rash. In cohort B1, ALT increase occurred in 5.1% of patients, AST increase occurred in 2.6%, rash occurred in 5.1%, elevated creatine kinase levels occurred in 2.6%, cataracts occurred in ████, and neuropsychiatric events occurred in 5.1%; no patients in cohort B1 had hypoglycemia. No patients in either cohort had serious ████████ ████████████, rash, creatine kinase, ████████, hypoglycemia, or neuropsychiatric events.
Critical Appraisal
The SKYLINE 102 and SKYLINE 103 trials were double-blind, parallel-group, randomized studies with appropriate randomization, allocation concealment, blinding, and methodological approaches. Patient disposition and baseline characteristics were well balanced between the study groups and there were relatively few major protocol violations or discontinuations of treatment or study. The primary end point of change in ppFEV1 at 24 weeks was a formal noninferiority assessment tested at a 1-sided alpha level of 0.025, which was met according to the prespecified noninferiority margin of −3%. This margin is consistent with the statistical approach selected and has been used in previous studies of CF.7,8 It was considered to be appropriate by the clinical experts consulted by CDA-AMC, who noted that a between-group difference of approximately 5% in ppFEV1 may be the minimal important difference (MID), although there is no published consensus. Other outcomes were assessed using 2-sided alpha levels of 0.05. All missing data were assumed to be missing at random and were not imputed, and there was a generally low number of patients (less than 10%) with missing data, and this was well balanced between the treatment groups for each end point as of the 52-week outcomes in both studies. Given the expected similarity in the treatment efficacy and safety profiles, this aligned with expectations, albeit there was a slight imbalance in discontinuations due to AEs in the SKYLINE 102 trial. The 52-week end points assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) were not included in the hierarchical testing procedure and were not adjusted for multiplicity. This may raise the risk of bias, but was not considered to be particularly significant in the context of these noninferiority studies.
The RIDGELINE study was a single-arm, noncomparative, nonrandomized, open-label study that assessed outcomes similar to the SKYLINE 102 and SKYLINE 103 studies in terms of within-group change from baseline after 24 weeks of therapy with VNZ–TEZ–D-IVA. Because this is a noncomparative, nonrandomized study, the risk of bias is high and the degree of certainty in the results in comparison with ELX-TEZ-IVA is very low. This is especially the case for the self-reported (or caregiver-reported) HRQoL metric of the CFQ-R respiratory domain.
The SKYLINE 102 and SKYLINE 103 trials reflect the patient population aged 12 years and older with CF, while patients aged 6 to 11 years (inclusive) were recruited for cohorts A1 and B1 of the RIDGELINE study. The eligibility criteria and baseline characteristics were considered appropriate and reflective of the patient population with CF in Canada within those age groups, and appropriate and reflective of the patients expected to be eligible for treatment with VNZ–TEZ–D-IVA. The comparator, treatment setting, concomitant medications, and patient eligibility criteria were relevant and generalizable to the treatment setting in Canada. Because all of the included patients had treatment experience with ELX-TEZ-IVA and/or participated in run-in periods with ELX-TEZ-IVA treatment, and patients intolerant to or ineligible for ELX-TEZ-IVA were not included, the efficacy and safety data during the treatment period of the studies reflect the experience of patients switching from one CFTRm to another (if assigned to VNZ–TEZ–D-IVA), or reflect the experience of patients continuing on a therapy to which they have demonstrated tolerance (if assigned to ELX-TEZ-IVA in the 2 RCTs). This represents a gap in the generalizability of these studies because no data were captured on patients naive to CFTRm therapy who were beginning treatment with VNZ–TEZ–D-IVA. However, this is expected to be a minority of patients.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
number of pulmonary exacerbations per year
change from baseline to 52 weeks in:
lung function, as measured by ppFEV1
BMI
BMI z score in pediatric patients
HRQoL, as measured by CFQ-R respiratory domain score
SwCl
safety outcomes:
number of patients with AEs of elevated aminotransferase levels
number of patients with SAEs.
Table 2: Summary of Findings for VNZ–TEZ–D-IVA Versus ELX-TEZ-IVA for Patients Aged 12 Years or Older With CF
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Pulmonary exacerbations | ||||
Number of pulmonary exacerbations, event rate per year Follow-up: 52 weeks | 971 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderatea | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the annual event rate of pulmonary exacerbations when compared with ELX-TEZ-IVA. |
Lung function | ||||
Change from baseline in ppFEV1, LS mean percentage points (95% CI) Follow-up: 52 weeks | 933 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Highb | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older results in little to no difference (i.e., a noninferior effect) in the change in ppFEV1 when compared with ELX-TEZ-IVA. |
Body weight | ||||
Change from baseline in BMI, LS mean kg/m2 (95% CI) Follow-up: 52 weeks | 880 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderatec | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the change in BMI when compared with ELX-TEZ-IVA. |
Change from baseline in BMI z score among patients aged 20 years or younger, LS mean (95% CI) Follow-up: 52 weeks | 136 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderated | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the change in BMI z score when compared with ELX-TEZ-IVA. |
Health-related quality of life | ||||
Absolute change from baseline in CFQ-R respiratory domain, LS mean points (95% CI) Follow-up: 52 weeks | 916 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderatee | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the change in CFQ-R respiratory domain score compared with ELX-TEZ-IVA. |
SwCl | ||||
Absolute change from baseline in SwCl, LS mean mmol/L (95% CI) Follow-up: 52 weeks | 931 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderatef | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely reduces SwCl levels compared with ELX-TEZ-IVA. The clinical importance of a reduction in SwCl is unclear. The clinical experts judged the magnitude of the observed reduction to not be clinically meaningful.f |
Harms | ||||
Number of patients with elevated aminotransferase AEs, n (%) Follow-up: 52 weeks | 981 (2 RCTs) | SKYLINE 102 trial:
SKYLINE 103 trial:
| Moderateg | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the number of patients with AEs of elevated aminotransferase levels when compared with ELX-TEZ-IVA. |
Number of patients with SAEs, n (%) Follow-up: 52 weeks | 981 (2 RCTs) |
SKYLINE 103 trial:
| Moderateg | Treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the number of patients with SAEs compared with ELX-TEZ-IVA. |
AE = adverse event; BMI = body mass index; CDA-AMC = Canada’s Drug Agency; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LS = least squares; MID = minimal important difference; NR = not reported; ppFEV1 = percent predicted forced expiratory volume in 1 second; RCT = randomized controlled trial; SAE = serious adverse event; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for imprecision due to the small number of events. The null was used as the threshold for potential benefit or harms in the absence of a literature- or expert-informed MID, but this end point was not rated down an additional level because the narrow 95% CIs for the absolute between-group differences were judged by CDA-AMC to be unlikely to include important benefits or harms.
bThe prespecified noninferiority margin for ppFEV1 was 3%.
cRated down 1 level for imprecision. The null was used as the threshold for potential benefit or harms in the absence of a literature- or expert-informed MID. This efficacy outcome was also exploratory and not included in the primary or secondary estimand, increasing uncertainty in interpretation.
dRated down 1 level for imprecision. The null was used as the threshold for potential benefit or harms in the absence of a literature- or expert-informed MID. This efficacy outcome was also exploratory and not included in the primary or secondary estimand, increasing uncertainty in interpretation. Additionally, the sample size was small in this subgroup.
eRated down 1 level for imprecision. The 95% CIs for the LS mean differences in both trials include values below the published MID threshold of 4 points for the CFQ-R respiratory domain. Although both the point estimates are below the MID, the upper 95% CI of the SKYLINE 102 trial is greater than the MID, which was not observed in the SKYLINE 103 trial. The outcome was rated down by only 1 level because the observed effects are consistent with the hypothesis of noninferiority between the treatments and do not suggest a meaningful difference in harm or benefit.
fRated down 1 level for imprecision due to the lack of an established MID. Notably, the clinical importance of further reduction in SwCl (beyond that seen with ELX-TEZ-IVA) is unclear because the surrogacy of SwCl for clinical end points has not been established and was described by the clinical experts as being unreliable, the magnitude of reduction in SwCl was not considered to be meaningful by clinicians because a patient may experience greater changes in SwCl day to day, and the reduction in SwCl observed in these studies does not appear to correlate with significant improvement in any other outcomes on this timescale. If further reduction in SwCl is clinically important, it may take much longer studies to elucidate this relationship.
gRated down 1 level for imprecision. The null was used as the threshold for potential benefit or harms in the absence of a literature- or expert-informed MID. Additionally, the small number of events increases uncertainty in the interpretation.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the submitted Clinical Study Reports.9,10
Table 3: Summary of Findings for VNZ–TEZ–D-IVA Versus ELX-TEZ-IVA for Patients Aged 6 to 11 Years With CF
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Pulmonary exacerbations | ||||
Number of pulmonary exacerbations Follow-up: 24 weeks | 78 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa,b | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on the pulmonary exacerbation rate in patients with CF aged 6 to 11 years compared with any comparator. |
Lung function | ||||
Change from baseline in ppFEV1, LS mean percentage points (95% CI) Follow-up: 24 weeks | 74 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on ppFEV1 in patients with CF aged 6 to 11 years compared with any comparator. |
Body weight | ||||
Change from baseline in BMI, LS mean kg/m2 (95% CI) Follow-up: 24 weeks | 78 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on BMI in patients with CF aged 6 to 11 years compared with any comparator. |
Change from baseline in BMI z score among patients aged 20 years or younger, LS mean (95% CI) Follow-up: 24 weeks | 78 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on BMI z score in patients with CF aged 6 to 11 years compared with any comparator. |
Health-related quality of life | ||||
Change from baseline in CFQ-R respiratory domain, LS mean points (95% CI) Follow-up: 24 weeks | 75 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa,c | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on CFQ-R respiratory domain score in patients with CF aged 6 to 11 years compared with any comparator. |
SwCl | ||||
Change from baseline in SwCl, LS mean mmol/L (95% CI) Follow-up: 24 weeks | 75 (1 single-arm study) | RIDGELINE study cohort B1:
| Very lowa,d | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on SwCl in patients with CF aged 6 to 11 years compared with any comparator. |
Safety and tolerability | ||||
Number of patients with elevated aminotransferase–related AEs, n (%) Follow-up: 24 weeks | 78 (1 single-arm study) | RIDGELINE study:
| Very lowa,b | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on events of elevated aminotransferase levels in patients with CF aged 6 to 11 years compared with any comparator. |
Number of patients with SAEs, n (%) Follow-up: 24 weeks | 78 (1 single-arm study) | RIDGELINE study:
| Very lowa,b | The evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on SAEs in patients with CF aged 6 to 11 years compared with any comparator. |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LS = least squares; MID = minimal important difference; NR = not reported; ppFEV1 = percent predicted forced expiratory volume in 1 second; RCT = randomized controlled trial; SAE = serious adverse event; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated as very low certainty due to serious concerns about study design and imprecision. The evidence is derived from a single-arm noncomparative study, which limits the ability to attribute observed changes to the intervention and introduces a high risk of bias.
bNo change from baseline was reported.
cThe LS mean change from baseline was 3.9 points (95% CI, 1.5 to 6.3), just below the published MID target threshold of 4 points. Although the upper bound of the CI exceeds the MID, the lack of a control group and the proximity of the point estimate to the threshold preclude rating up for magnitude of effect. Additionally, the CFQ-R is a self-reported (or caregiver-reported) outcome measure, which contributes to uncertainty and potential bias in the context of an unblinded, single-arm study.
dThe clinical importance of further reduction in SwCl (beyond that seen with ELX-TEZ-IVA) is unclear for several reasons; the surrogacy of SwCl for clinical end points has not been established and was described by the clinical experts as being unreliable, the magnitude of reduction in SwCl was not considered by the clinicians to be meaningful because a patient may experience greater changes in SwCl day to day, and the reduction in SwCl observed in these studies does not appear to correlate with significant improvement in any other outcomes on this timescale. If further reduction in SwCl is clinically important, it may take much longer studies to elucidate this relationship.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the submitted Clinical Study Report.11
Long-Term Extension Studies
No long-term extension studies (with results) were submitted, although there are some presently ongoing for which results data are not yet available.
Indirect Comparisons
The sponsor submitted indirect comparisons between VNZ–TEZ–D-IVA and best supportive care; given the presence of direct evidence against ELX-TEZ-IVA, which is the standard of care, as previously discussed, this indirect comparison with supportive care was considered by CDA-AMC to have limited relevance; ergo, it was not summarized or critically appraised for the purpose of this review.
Studies Addressing Gaps in the Evidence
Four nonclinical studies evaluating the effect of VNZ–TEZ–D-IVA on cells in an in vitro setting were provided. For CFTR mutations with low prevalence, a conventional clinical trial approach to demonstrate efficacy is not always feasible.2,12 To evaluate the potential clinical benefit of VNZ–TEZ–D-IVA in these circumstances, additional mutations were identified using the following:
the in vitro Fischer rat thyroid (FRT) system (with a threshold of a 10% increase in chloride transport over baseline when expressed as a percentage of normal)
the in vitro human bronchial epithelial primary cell culture model
mechanistic data.13
There is precedent for using in vitro data in the approval of other CFTRm therapies in Canada (namely, ELX-TEZ-IVA) and other countries. A total of 303 CFTR mutations were proposed by the sponsor to be responsive to VNZ–TEZ–D-IVA. Based on the Health Canada Notice of Compliance (NOC) for VNZ–TEZ–D-IVA issued on July 21, 2025, 266 CFTR mutations were approved as part of the indication, of which 23 were based on clinical data, 227 were based on in vitro data, and 16 were based on extrapolation. The remaining CFTR mutations that were submitted by the sponsor were not included in the product monograph because they were excluded by Health Canada. Mutations were excluded by Health Canada if they were listed as not causing CF in both the CFTR2 and CFTR-France databases or if they were listed as not causing CF in 1 database and listed as variants of unknown significance or only causing CFTR-related disease in the other database and/or other sources.
Conclusions
Two noninferiority RCTs demonstrated with high-certainty evidence that VNZ–TEZ–D-IVA results in little to no difference in the change in lung function (measured using ppFEV1) when compared with ELX-TEZ-IVA through 52 weeks of treatment in patients aged at least 12 years with CF who had at least 1 F508del mutation or another responsive CFTR mutation. Moderate- to high-certainty evidence was also demonstrated for little to no difference between the treatment groups for outcomes related to the rate of pulmonary exacerbations, BMI, BMI z score in patients aged 20 years or younger, HRQoL measured using the CFQ-R respiratory domain, the number of patients who experienced events of elevated aminotransferase levels, and the number of patients with SAEs.
One single-arm study of VNZ–TEZ–D-IVA in patients aged 6 to 11 years with CF and ELX-TEX-IVA–responsive CFTR mutation(s) reported little change from baseline in the outcomes of ppFEV1, BMI, BMI z score, and CFQ-R respiratory domain, although the certainty of the evidence compared with ELX-TEZ-IVA or any comparator was very low owing to the noncomparative design of the study. Nonetheless, the efficacy and safety profile appeared to be consistent with that observed in the 2 RCTs and it was supportive of the extrapolation of the RCT results in this age group, in combination with the well-established precedent of ELX-TEZ-IVA treatment benefit in this age group and the shared mechanism of action between VNZ–TEZ–D-IVA and ELX-TEZ-IVA. The side effect profile was consistent with what was expected for triple-combination CFTRm therapies, and the rate of discontinuation due to AEs was very low.
There is precedent and support from the clinical experts for the practice of using in vitro studies to address gaps in the clinical study evidence related to rare CF-causing mutations for which it is infeasible to conduct standard clinical trials. Four nonclinical studies were provided evaluating the effect of VNZ–TEZ–D-IVA on cells in an in vitro setting. Based on the Health Canada NOC for VNZ–TEZ–D-IVA, 266 CFTR mutations were approved as part of the indication based on clinical data, in vitro data, and extrapolation. This represents an expansion from the 153 CFTR mutations approved for ELX-TEZ-IVA in Canada.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of VNZ–TEZ–D-IVA (Alyftrek) in the treatment of patients with CF who have a F508del mutation or another responsive CFTR mutation who are aged 6 years and older.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
CF, an autosomal recessive condition, is the most common fatal genetic disease affecting children and young adults in Canada. It is caused by mutations in the CFTR gene, which is located on chromosome 7. The CFTR gene encodes a chloride channel that regulates ion and fluid transport across cell membranes. When the CFTR gene is dysfunctional, secretions become tenacious and sticky, resulting in pathology in multiple organs, including the lungs, large and small intestines, pancreatic and bile ducts, and the vas deferens. F508del is the most common CFTR mutation that results in CF.1 The Canadian Cystic Fibrosis Registry reported there were 4,513 people in Canada living with CF in 2023. Of these, 87.8% carried 1 or more F508del mutations.2
More than 2,000 CFTR mutations have been identified among patients with CF.2 The mutations are classified as impaired biosynthesis (class I), defective protein maturation and accelerated degradation (class II), defective regulation of the CFTR gene at the plasma membrane (class III), defective chloride conductance (class IV), diminished transcription of the CFTR gene (class V), and accelerated turnover at the cell surface (class VI).14 CFTR mutations within classes I to III are associated with severe CF because they are considered nonfunctional, while mutations in classes IV to VI may retain CFTR function.14,15 Genotyping for mutations in the CFTR gene is performed routinely on almost all patients with CF in Canada and is also part of the newborn screening process.1
CF results in airway obstruction, chronic endobronchial infection, and inflammation, which ultimately lead to destruction of lung tissue through the development of bronchiectasis and loss of lung function.3 Although chronic pulmonary therapies instituted early in the disease have reduced the decline in lung function over time, patients with a genotype that is homozygous for the F508del mutation will develop chronic infection with Pseudomonas and progressive bronchiectasis and airway obstruction. In a cohort of approximately 1,000 healthy young children with CF who did not have a Pseudomonas infection at enrolment, there was a greater annual decline in forced expiratory volume in 1 second (FEV1) over the following 4 years in those patients with a genotype that was homozygous for the F508del mutation.16 Chronic endobronchial infection of the airways with bacterial pathogens, such as Pseudomonas aeruginosa,2 is associated with a more rapid loss of lung function.4 Acute or chronic endobronchial infections result in further destruction of lung tissue and are associated with respiratory morbidity. Lung disease accounts for the vast majority (> 80%) of deaths in patients with CF.1,5
Pulmonary exacerbations are associated with lung function decline and mortality and may require treatment with IV antibiotics and hospitalization. The Cystic Fibrosis Foundation has reported that approximately one-third of patients with CF will have at least 1 pulmonary exacerbation per year requiring IV antibiotics.17
Maintenance of pulmonary function (higher FEV1) and fewer respiratory exacerbations are associated with increased survival.18 Pulmonary management of CF therefore aims to clear the airways of secretions and treat lung pathogens to minimize inflammation.
Patients typically have pancreatic, gastrointestinal, and nutritional disease as well as progressive pulmonary damage. Gastrointestinal and pancreatic involvement results in pancreatic exocrine insufficiency in most individuals with CF, causing malabsorption of fats and fat-soluble vitamins, which leads to malnutrition. Maintaining adequate nutrition is associated with an improved clinical outcome and longevity for patients with CF.6 Almost all of these patients, approximately 90% according to the clinical experts, will have pancreatic insufficiency and will need to take lifelong pancreatic enzyme replacement with every meal as well as fat-soluble vitamin therapy. With increasing age, these patients may develop CF-related diabetes and require therapy with insulin. In 2023, CF-related diabetes was reported in 32.5% of adults and 2.4% of children living with CF in Canada.2
The median age of survival in Canada for a child born with CF was estimated to be 62.3 years in 2023.2 The Canadian Cystic Fibrosis Registry has reported an increase in the median age of death for patients with CF in Canada since the year 2000;1 The median age of death was 41.2 years in 20232 compared with 38.7 years in 2021 and 26.6 years in 2002.19
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Before the advent of CFTRm therapies, the key goals of CF management were to treat the complications of CF and relieve symptoms because there were no therapies available that could treat the fundamental CF-causing defects.20 Since CFTRm therapies became available, they have become the standard of care for eligible patients,21,22 and the treatment goals now include maintaining normal lung function, reducing or possibly eliminating CF-related symptoms, reducing or eliminating hospitalizations or pulmonary exacerbations, and maintaining adequate nutrition; additionally, in the long term, the clinical experts consulted by CDA-AMC noted they expect a reduction in future lung transplants related to CF, and that the goal is to push closer to normal life expectancy and better HRQoL in people with CF. Although CFTRm therapies are not curative, they are the first and only class of therapy developed to target the underlying pathophysiology of CF by working to improve the function of the CFTR protein.22 The first CFTRm available commercially was ivacaftor (Kalydeco), which works to target defects in the “gating” of CFTR channels, and is currently indicated by Health Canada for 10 CF-causative mutations in patients aged as young as 2 months.22 Two combination therapies with ivacaftor, lumacaftor-ivacaftor (Orkambi) and tezacaftor-ivacaftor (Symdeko), were later developed for the treatment of patients with 2 copies of the most common CF-causing variant, F508del, which represents approximately 50% of patients.22 In contrast, the relatively newer triple-combination therapy, ELX-TEZ-IVA (Trikafta),23-27 is indicated for patients aged 2 years and older with at least 1 F508del mutation or a mutation in 1 of the 152 non-F508del rare and responsive mutations, and it has become the current standard of care for most patients with CF in Canada.
There is an unmet need for patients with CF-causing mutations that are not eligible for treatment with ELX-TEZ-IVA, and for those who discontinue therapy due to intolerable side effects. Additionally, ELX-TEZ-IVA is administered twice daily and requires a fat-containing meal for absorption at each dose, which creates challenges for some patients around adherence. The clinical experts consulted by CDA-AMC noted that many patients while on therapy with ELX-TEZ-IVA will still have an SwCl level that is in an abnormal range, and that this represents an unmet need, i.e., for the improvement of SwCl levels into a normal range, although the long-term implications of this are still unclear. The experts also noted that non-F508del mutations are more likely to be diagnosed in racial minorities who may already be encountering systemic disadvantages within the health care system and who may be underrepresented in clinical trials; moreover, many non-F508del mutations are rare enough to preclude generating robust clinical evidence through a typical clinical trial. As a result, there is a disproportionate unmet need among racial minorities because a higher proportion of patients with CF who are racial minorities currently have no access to an approved CFTRm.
Patients receiving a CFTRm still receive symptom-relieving supportive care as needed, although the clinical experts consulted by CDA-AMC noted that treatment with a CFTRm may generally be expected to reduce (but not eliminate) the amount of other supportive care required.
Drug Under Review
VNZ–TEZ–D-IVA (Alyftrek) is a triple-combination CFTRm indicated for the treatment of CF in patients aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the CFTR gene; refer to Table 23 for the list of mutations acknowledged by Health Canada as responsive to VNZ–TEZ–D-IVA. The reimbursement request aligns with the Health Canada indication.
The sponsor proposed that VNZ–TEZ–D-IVA be reimbursed based on the criteria outlined in Table 4, which were judged by the clinical experts consulted by CDA-AMC to be reasonable and similar to those for ELX-TEZ-IVA.
Table 4: Sponsor’s Requested Reimbursement Criteria for VNZ–TEZ–D-IVA
Category | Criteria |
|---|---|
In patients newly initiating CFTRm therapy with VNZ–TEZ–D-IVA | |
Initiation criteria | Patient with a confirmed diagnosis of CF with at least 1 F508del mutation, or a non-F508del mutation in the CFTR gene that is found to be responsive based on clinical and/or in vitro evidence, and who is aged 6 years or older |
Renewal criteria (initial) | The sponsor requests that jurisdictions allow for any 1 of the following to demonstrate benefit after 6 months of treatment with VNZ–TEZ–D-IVA:
|
Renewal criteria (subsequent renewals annually) | Continued clinical benefit as determined by a physician specializing in the treatment of CF |
In patients switching from another CFTRm therapy to VNZ–TEZ–D-IVA | |
Initiation criteria | Patient with a confirmed diagnosis of CF with at least 1 F508del mutation or a non-F508del mutation in the CFTR gene that is found to be responsive based on clinical and/or in vitro evidence, and who is aged 6 years or older |
Renewal criteria (initial) | The sponsor requests that jurisdictions allow for any 1 of the following to demonstrate benefit after 6 months of treatment with VNZ–TEZ–D-IVA:
|
Renewal criteria (subsequent renewals annually) | Continued clinical benefit as determined by a physician specializing in the treatment of CF |
BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CFTRm = CFTR modulator; ppFEV1 = percent predicted forced expiratory volume in 1 second; VNZ–TEZ–D-IVA vanzacaftor-tezacaftor-deutivacaftor.
Sources: Sponsor submission materials.
VNZ–TEZ–D-IVA is administered by mouth once daily. It is available in 2 different doses. Patients who are less than 40 kg in body weight take 3 tablets per day of vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg. Patients who are at least 40 kg in body weight take 2 tablets per day of vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg. This treatment must be taken with a fat-containing meal to facilitate absorption.28
Vanzacaftor and tezacaftor are CFTR correctors that bind to different sites on the CFTR protein and have an additive effect in facilitating the cellular processing and trafficking of select mutant forms of the CFTR gene (including F508del) to increase the amount of CFTR protein delivered to the cell surface compared with either molecule alone. Deutivacaftor is a CFTR potentiator that increases the channel-open probability (or gating) of the CFTR protein at the cell surface. The half-life of deutivacaftor allows for once-daily dosing, in contrast with the twice-daily dosing of ivacaftor-containing therapies such as the other existing triple-combination CFTRm, ELX-TEZ-IVA (Trikafta). As with ELX-TEZ-IVA, the combination of multiple CFTR correctors and a CFTR potentiator in VNZ–TEZ–D-IVA is expected to be complementary and to increase both the quantity and function of CFTR protein and CFTR activity relative to monotherapies or 2-drug combination therapies.29,30
Key characteristics of VNZ–TEZ–D-IVA (Alyftrek) are summarized in Table 5 with other CFTRm-based treatments currently reimbursed in Canada for the treatment of CF.
Table 5: Key Characteristics of VNZ–TEZ–D-IVA (Alyftrek), ELX-TEZ-IVA (Trikafta), and IVA (Kalydeco)
Characteristic | VNZ–TEZ–D-IVA (Alyftrek) | ELX-TEZ-IVA (Trikafta) | IVA (Kalydeco) |
|---|---|---|---|
Mechanism of action | CFTR potentiator (IVA) and correctors (VNZ and TEZ) | CFTR potentiator (IVA) and correctors (ELX and TEZ) | CFTR potentiator |
Indicationa | For the treatment of CF in patients aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the CFTR gene | Patients aged 2 years and older who have 1 or more F508del mutations in the CFTR gene or a mutation in the CFTR gene that is responsive based on in vitro and/or clinical dataa | Granules (13.4 mg, 25 mg, 50 mg, and 75 mg): Indicated for the treatment of children with CF aged 2 months and older and weighing from 3 kg to less than 25 kg who have 1 of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, or R117H Tablets (150 mg): Indicated for the treatment of CF in patients aged 6 years and older and weighing 25 kg or more who have 1 of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, or R117H |
Route of administration | Oral tablets | Oral tablets and granules | Oral tablets and granules |
Recommended dose | Aged 6 years and < 40 kg: 3 tablets of D-IVA 50 mg, TEZ 20 mg, and VNZ 4 mg once daily Aged ≥ 6 years: ≥ 40 kg: 2 tablets of D-IVA 125 mg, TEZ 50 mg, and VNZ 10 mg once daily | Granules: Aged 2 to < 6 years and: < 14 kg: ELX 80 mg, TEZ 40 mg, and IVA 60 mg (morning); IVA 59.5 mg (evening) ≥ 14 kg: ELX 100 mg, TEZ 50 mg, and IVA 75 mg (morning); IVA 75 mg (evening) Tablets: Aged 6 to < 12 years and < 30 kg: ELX 100 mg, TEZ 50 mg, and IVA 75 mg (morning); IVA 75 mg (evening) Aged 6 to < 12 years and ≥ 30 kg or aged ≥ 12 years: ELX 200 mg, TEZ 100 mg, and IVA 150 mg (morning); IVA 150 mg (evening) | Tablets: IVA 150 mg every 12 hours Granules: 7 kg to < 14 kg: IVA 50 mg every 12 hours 14 kg to < 25 kg: IVA 75 mg every 12 hours |
Serious adverse effects or safety issues | Each of the product monographs include a warning about the risk of elevated aminotransferase (ALT and AST) levels, and monitoring of liver function is recommended before initiating treatment. The recommended frequency of monitoring for ELX-TEZ-IVA is every month during the first 6 months of treatment, every 3 months for the next 6 months, then at least annually thereafter. For VNZ–TEZ–D-IVA and IVA, the recommended frequency is every 3 months during the first year of treatment and annually thereafter.27,28,31 The product monograph recommends that these drugs not be used in patients with severe hepatic impairment.27,32-34 | ||
ALT = alanine aminotransferase; AST = aspartate aminotransferase; D-IVA = deutivacaftor; ELX = elexacaftor; IVA = ivacaftor; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; TEZ = tezacaftor; VNZ = vanzacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aHealth Canada–approved indication.
Sources: Product monographs for Alyftrek,28 Trikafta,27 and Kalydeco.31
Testing Procedure Considerations
CFTR genetic testing is the standard of care and part of the routine diagnostic work-up to confirm a CF diagnosis. An individual is diagnosed with CF when there is clinical presentation of disease and evidence of CFTR gene dysfunction through genetic testing for CFTR mutations.35 CFTR genetic testing is a staged process. Patients who present with clinical features or a family history indicative of CF or are identified through newborn screening will receive CFTR genetic assay testing.36 Initial genetic testing is used to identify the presence of 2 CF-causing mutations in the CFTR gene (1 inherited from each parent) to confirm a diagnosis of CF.37 The CFTR genetic assay test result is assessed against a panel of the most common CF-causing mutations, including F508del, to determine a potential CFTR mutation match. According to the clinical experts, CFTR mutation panels may vary between jurisdictions but are typically based on mutations that have been identified by the Clinical and Functional Translation of CFTR (CFTR2) project.38 In cases where none of the most common CFTR mutations are identified, only 1 mutation is identified, or no mutations are identified by the initial panel assay, follow-up genetic testing using an extended panel of CFTR mutations or full gene sequencing of the CFTR coding region can be used to confirm the mutation status of an individual.35
The clinical experts noted that standard CFTR gene panels, such as those based on the CFTR2 project, are often optimized for common mutations that are likely to be identified in white individuals, while rare or unclassified CFTR mutations may be more prevalent in racialized individuals.39 Based on the Cystic Fibrosis Canada Patient Registry, 91.7% of the currently identified population in Canada with CF is white, and 87.8% of the total number of patients with CF in Canada possess at least 1 F508del mutation.40 This raises important equity considerations about the mutations included in CFTR gene panels, and the potential impacts of the underrepresentation of racialized individuals. The patient group input highlighted that individuals with rare CFTR mutations may face significant challenges related to being diagnosed late with more advanced disease, which may also impact access to standard of care treatments. Initial CFTR genetic testing may be inconclusive if an individual has a rare mutation that has not yet been identified;36 however, according to the clinical experts, additional follow-up testing using full gene sequencing will likely capture patients who may have mutations not included in the panel. As previously mentioned, the Health Canada NOC approved 266 CFTR mutations as part of the indication for VNZ–TEZ–D-IVA, which may not capture individuals with rare or ultra-rare CFTR mutations.
CDA-AMC considered the potential impacts of CFTR genetic testing to confirm eligibility for VNZ–TEZ–D-IVA, including impacts to health systems, patients (including families and caregivers), and costs. No implementation barriers or only minimal barriers are anticipated because CFTR genetic testing is currently performed as the standard of care for a CF diagnosis across jurisdictions in Canada. The review team validated key considerations and the relevant information available from the materials submitted by the sponsor, input from the clinical experts consulted by the review team, patient group input, sources from the literature, and a scan of jurisdictional testing availability, when possible, which are summarized in Table 6.
Table 6: Considerations for CFTR Genetic Testing for Establishing Treatment Eligibility With VNZ–TEZ–D-IVA in CF
Consideration | Criterion | Available Information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | According to the clinical experts, all patients presenting with clinical features indicative of CF will receive CFTR genetic testing. Using data from the Cystic Fibrosis Canada Patient Registry, the sponsors estimated a baseline number of 3,810 patients with CF for 2025 with an annual increase of 1.68% per year. This would mean that 3,874 patients in year 1, 3,939 patients in year 2, and 4,005 patients in year 3 would be expected to require the test.a |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | According to the clinical experts and information from Cystic Fibrosis Canada, CFTR genetic testing is widely available and reimbursed across jurisdictions in Canada when patients meet the required indications and are referred by a treating physician for a CF diagnosis.36,b | |
Testing procedure as part of routine care | CFTR genetic testing is part of the standard of care and a routine diagnostic work-up to confirm a CF diagnosis in both NBS programs and patients receiving a CF diagnosis later in life. | |
Repeat testing requirements | According to the clinical experts, repeat testing is not required if a patient is diagnosed with 2 CFTR mutations through an initial panel test; however, if 1 or no mutations are identified using the panel test, then a patient will undergo full sequencing of the CFTR coding region to confirm mutation status. | |
Impacts on human and other health care resources caused by provision of the testing procedure | Because CFTR genetic testing is the standard of care for a CF diagnosis, no additional impacts are anticipated on human and other health care resources if VNZ–TEZ–D-IVA were to be reimbursed. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | According to the clinical experts and the sponsor, patients undergoing a CF diagnosis can access CFTR genetic testing across jurisdictions in Canada through treating physicians as part of the routine diagnostic work-up for CF. No barriers to CFTR genetic testing are anticipated if VNZ–TEZ–D-IVA is approved for reimbursement. |
Expected turnaround times for the testing procedure | According to the clinical experts and a scan of jurisdictional testing information, the turnaround time for CFTR genetic testing targeting common mutations varies depending on jurisdiction, but ranges from 1 to 2 weeks for urgent testing and 3 to 8 weeks for routine testing. Additionally, they reported that if full CFTR sequencing is needed to confirm mutation status, the turnaround time for results may take an additional 8 to 12 weeks. The turnaround time for testing is not anticipated to increase or pose additional barriers or delays for a patient receiving VNZ–TEZ–D-IVA, if they are eligible. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | Because CFTR genetic testing is currently part of the standard of care for a CF diagnosis, no additional burden to patients, families, or caregivers is anticipated from testing done as part of establishing treatment eligibility for VNZ–TEZ–D-IVA. | |
Clinical | Clinical utility and validity of the testing procedure | According to the clinical experts, CFTR genetic testing is accurate for identifying all known CF-causing mutations. Evidence from the Division of Genome Diagnostics in British Columbia provided the analytical sensitivity and specificity for the standard CFTR mutation panel assay, expanded mutation panel assay, and full gene sequencing assay used in its jurisdiction. The standard CFTR mutation panel has a sensitivity and specificity of 100% (95% CI, 97.9% to 100% and 99.9% to 100%, respectively).41 The expanded mutation panel assay has a sensitivity for single nucleotide variants of 100% (95% CI, 93.2% to 100%), a sensitivity for insertions and deletions of 100% (95% CI, 95.3% to 100%), and a specificity of 100% (95% CI, 99.9% to 100%).42 The full gene sequencing assay has a sensitivity for single nucleotide variants of 100% (95% CI, 95.0% to 100%), a sensitivity for insertions and deletions of 100% (95% CI, 95.6% to 100%), and a specificity of 100% (95% CI, 99.9% to 100%).43,c |
Risks of harm associated with the testing procedure | CFTR genetic testing can be done using blood samples drawn, typically through a heel-prick method, as part of an initial immunoreactive trypsinogen assessment.35 Because CFTR genetic testing is the standard of care for a CF diagnosis, no additional risk of harms associated with testing done as part of establishing treatment eligibility for VNZ–TEZ–D-IVA was identified. | |
Cost | Projected cost of the testing procedure | Evidence from the Molecular Genetics Laboratory in British Columbia and the clinical experts indicated that CF-related genetic testing costs approximately $300 for a basic CF panel test.44 There would be additional costs associated with full gene sequencing if needed; however, the estimated additional cost is unclear. Because testing is already part of routine care, there is no additional cost-associated impact from CFTR genetic testing done as part of establishing treatment eligibility for VNZ–TEZ–D-IVA. |
CF = cystic fibrosis; CI = confidence interval; NBS = newborn screening; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aSource: Sponsor-submitted budget impact assessment.
bCystic Fibrosis Canada did not provide testing information for Nunavut.
cCanada’s Drug Agency has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received for this review are available in the consolidated patient and clinician group input document on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by a patient group.
One patient group, Cystic Fibrosis Canada, submitted input for this review. Information was gathered through focus groups with people in Canada with CF and their caregivers. Other sources of data included surveys (including a 2021 survey with more than 1,200 responses), data from the Canadian Cystic Fibrosis Registry, and a multiphase burden of disease study and other relevant publications.
The patient group highlighted that patients not on a CFTRm frequently have periods of infection and acute inflammation called exacerbations that require a hospital stay of at least 2 weeks and that may last up to 4 weeks. Some of these patients could benefit from a CFTRm, but there may not be any CFTRm therapies currently that have a Health Canada indication for their mutation. The group noted that approximately 33% of all adult patients with CF in Canada have CF-related diabetes. Any of the other drugs that patients need to take on a regular basis can also have negative side effects and long-term risks, such as chronic use of antibiotics that may lead to resistance. Moreover, as patients age, they may need to try multiple antibiotics to find 1 that works, making management of drug-to-drug interactions difficult. The patient group reiterated that currently, only CFTRm therapies target the underlying pathophysiology of CF by improving the function of the CFTR gene, while other therapies used to treat CF are for the management of complications and control of symptoms.
The patient group highlighted that too many patients lack access to any CFTRm, including those patients who have ultra-rare mutations that are not on the narrow list of 152 mutations indicated for ELX-TEZ-IVA or who cannot tolerate it, or those who have experienced a response only to VNZ–TEZ–D-IVA. The patient group noted the FDA has approved the use of ELX-TEZ-IVA in 272 mutations, compared with 152 mutations in Canada; the group suggested there are approximately 23 patients in Canada aged 2 years or older with 1 or more of the 119 mutations that are not approved in Canada for treatment with ELX-TEZ-IVA. Thus, they could access ELX-TEZ-IVA in the US, but cannot access it in Canada. Additionally, the FDA approved VNZ–TEZ–D-IVA for an additional 31 unique mutations for which ELX-TEZ-IVA is not approved in Canada; this represents approximately 13 additional people in Canada without access to ELX-TEZ-IVA. The group pointed out that, if VNZ–TEZ–D-IVA gets approved in Canada, up to 36 individuals aged 6 years or older could access it for the first time. The group specifically requested that the committee recommend access to VNZ–TEZ–D-IVA for the 152 CFTR mutations currently indicated in Canada for ELX-TEZ-IVA. The group further asked that the committee recommend access to VNZ–TEZ–D-IVA for patients in Canada with any of the 151 rare mutations or mutation combinations the FDA lists as indications for VNZ–TEZ–D-IVA that currently are not indicated in Canada for any CFTRm, and to recommend the broadest possible access to VNZ–TEZ–D-IVA for individuals with other CFTR mutations when there is evidence, and when more evidence is generated over time (including in vitro laboratory evidence or clinical evidence), that shows these patients will or may experience a response to this therapy. The group highlighted that CF has significant financial implications for patients and their caregivers, health systems, and society.
No patients reported experience with VNZ–TEZ–D-IVA treatment. The patient group highlighted that VNZ–TEZ–D-IVA may improve quality of life and adherence as a 1-pill-a-day treatment, which would provide an option for parents who may have difficulty administering medications to their children, who already take several medicines a day to maintain their health.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of CF.
Unmet Needs
The clinical experts described that ELX-TEZ-IVA is the current standard of care in patients with CF aged 6 years and older with eligible CF-causing mutations, but there is an unmet need for more effective therapy that prolongs life, improves quality of life, and reduces the treatment burden of supportive medications in patients who have CF-causing mutations that are not eligible for treatment with ELX-TEZ-IVA, which represents approximately 10% of patients with CF in Canada, and disproportionately represents racialized people. Additionally, even among patients eligible for treatment with ELX-TEZ-IVA, the clinical experts noted that most patients still have SwCl levels in an abnormal range. Doses of ELX-TEZ-IVA are administered twice daily and require fat-containing meals for absorption; ergo, the experts noted that less frequent dosing is an unmet need that might improve adherence. Some patients may experience AEs or intolerance to ELX-TEZ-IVA, requiring dose adjustments or cessation, and there is an unmet need for alternative triple-combination CFTRm therapies in those circumstances. Finally, overall pill and treatment burden remains high in patients with CF, despite reductions after the advent of triple-combination CFTRm therapies (i.e., ELX-TEZ-IVA).
Place in Therapy
The clinical experts consulted by CDA-AMC stated that VNZ–TEZ–D-IVA would occupy a place in therapy similar to that of ELX-TEZ-IVA, with the caveat that it may be applicable to a wider range of mutations, and that the current Health Canada indications begin at different ages (i.e., ELX-TEZ-IVA is currently indicated for patients aged at least 2 years, while VNZ–TEZ–D-IVA is indicated in patients aged at least 6 years). The experts noted that, given the once-daily dosing of VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA, improvements in treatment adherence may be expected. However, patients with CF are typically taking numerous pills daily in addition to CFTRm therapies; ergo, this may not represent a substantial difference in overall treatment or pill burden. Nonetheless, the expert specialized in pediatric care said that a reduction in treatment or pill burden may be particularly significant for the treatment of pediatric patients with CF. The experts noted that it may be premature for every patient currently stable on ELX-TEZ-IVA to be switched to VNZ–TEZ–D-IVA due to the longer clinical safety record of ELX-TEZ-IVA; however, VNZ–TEZ–D-IVA is generally expected to become the new standard of care for eligible patients.
Additionally, if there are clinical signals that achieving a lower SwCl than previously seen with ELX-TEZ-IVA results in less end-organ damage, it may further shift treatment paradigms and reduce the need for other supportive therapies.
Patient Population
The patient population appropriate for treatment with VNZ–TEZ–D-IVA is similar to that for ELX-TEZ-IVA, with the exception that there may be additional CF-causing mutations eligible for treatment with VNZ–TEZ–D-IVA. Aside from genotype, the patient population is expected to be similar.
Assessing the Response to Treatment
The clinical experts noted that the assessment of CF-related treatment is based on lung function spirometry, number of days treated with oral and IV antibiotics for pulmonary exacerbations, number of CF-related hospitalizations, and monitoring of BMI or BMI z score (as appropriate by age) for signs of malnutrition. The clinical experts informed CDA-AMC that the assessments related to VNZ–TEZ–D-IVA should be similar to those expected for ELX-TEZ-IVA, although, in any reimbursement criteria, it is also important to consider that assessments for initiation, continuation, or discontinuation should differ for patients newly starting triple-combination CFTRm therapies (i.e., patients who are generally expected to experience greater improvements with therapy) versus switching between triple-combination CFTRm therapies (i.e., the new treatment would be expected to maintain the benefit previously observed with the other therapy). Assessment of response in the previously listed metrics should generally be performed after a year of treatment because, as the experts highlighted, some metrics (especially pulmonary exacerbations and BMI or BMI z score) may have fluctuations with seasonal illness and short-term instability in pediatric patients that may not be reflective of the effect of the drug.
Discontinuing Treatment
The clinical experts noted that discontinuation of VNZ–TEZ–D-IVA should be considered in the case of intolerable AEs that cannot be sufficiently managed or, in the case of a lung transplant, similar to current practice with ELX-TEZ-IVA (although this does not reflect a contraindication specified by Health Canada).
Prescribing Considerations
The clinical experts noted that a physician practising within or under the directed supervision of a Canadian CF clinic should manage the prescription and renewal of VNZ–TEZ–D-IVA.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One clinician group, the Cystic Fibrosis Canada Health Care Advisory Council, provided input for this review. Information for this submission was gathered through the following: experience gained by working with and delivering medical services to people with CF; consultations with people with CF not eligible for ELX-TEZ-IVA; a review of medical and scientific literature, including clinical trial results and real-world experience; and the Canadian Cystic Fibrosis Registry, a collection of patient data and other information regarding CF care and outcomes. A total of 2 clinicians contributed to the input submitted for this review.
The input from the clinician group was generally consistent with that of the clinical experts consulted for this review, emphasizing that about 10% of patients have mutations not currently approved for treatment with available CFTRm therapies in Canada, and that there are no alternative triple-combination CFTRm treatments for patients with AEs or intolerance to ELX-TEZ-IVA. The group agreed that the once-daily regimen of VNZ–TEZ–D-IVA may improve adherence and convenience compared with the twice-daily regimen of ELX-TEZ-IVA.
The group noted that rare mutations for which there is currently no CFTRm indication are disproportionately found in racialized communities, so expanded access to this therapy may improve equitable access to disease-modifying treatments in Canada.
The clinician group noted that, according to recent Canadian clinical guidelines, all patients with CF who have at least 1 CFTRm-responsive CFTR variant should be treated with a CFTRm. The clinician group noted that patients with CF best suited for the drug under review include patients already on ELX-TEZ-IVA who are experiencing challenges due to side effects or related to adherence to a twice-daily treatment regimen, and patients with rare mutations not approved for treatment with ELX-TEZ-IVA but who may experience a response to VNZ–TEZ–D-IVA. The group agreed with the clinical experts regarding the metrics and yearly time frame of reassessments for treatment efficacy, as well as factors to consider for treatment discontinuation and prescribing.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 7.
Table 7: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Consider alignment with the most recent reimbursement criteria for Trikafta (ELX-TEZ-IVA), as appropriate, for patients aged 6 years and older. | The clinical experts agreed that these criteria should be aligned with those for Trikafta for patients aged 6 years and older. |
Considerations for continuation or renewal of therapy | |
Consider alignment with the most recent reimbursement criteria for Trikafta, as appropriate, for patients aged 6 years and older. | The clinical experts agreed that these criteria should be aligned with those for Trikafta for patients aged 6 years and older, with the caveat that patients switching from 1 CFTRm to another should not be expected to have further improvements in outcomes at the same magnitude as patients who are first starting a CFTRm, but instead should be expected to have approximately equal or stable clinical status in CF-related outcomes and lung function. |
Considerations for discontinuation of therapy | |
Consider alignment with the most recent reimbursement criteria for Trikafta, as appropriate, for patients aged 6 years and older. | The clinical experts agreed that these criteria should be aligned with those for Trikafta for patients aged 6 years and older. |
Considerations for prescribing of therapy | |
Is there evidence for the use of Alyftrek (VNZ–TEZ–D-IVA) in combination with other CFTRm therapies? | The clinical experts stated there is no evidence for the use of VNZ–TEZ–D-IVA in combination with other CFTRm therapies. |
Consider alignment with the most recent reimbursement criteria for Trikafta, as appropriate, for patients 6 years and older. | The clinical experts agreed that the reimbursement criteria for Alyftrek should be aligned with the criteria for Trikafta for patients aged 6 years and older. |
Care provision issues | |
The clinical experts consulted for previous reviews (such as the review of Trikafta in November 2023) have noted that SwCl testing should not be used to evaluate response to treatment for the purposes of drug reimbursement because it is not clearly predictive of clinically important outcomes and reflects only the mechanism of action of CFTRm therapies. The clinical experts have also noted that access to SwCl testing can be challenging in some jurisdictions and that the timelines for receiving the test results can fluctuate. They also expressed concerns about the capacity of the health system to accommodate repeated SwCl testing. Should SwCl be used to assess response to treatment for the purposes of reimbursement? If so, under what circumstances? | The clinical experts agreed that SwCl is not appropriate for evaluating response to treatment for the purposes of drug reimbursement due to the issues listed. SwCl is an important metric for diagnosing CF, and a decrease in SwCl reflects the mechanism of action of initiated CFTRm therapies, but it is too heterogeneous between patients (and may be misleading in some CF-causing mutations) and has not been established as a clear surrogate for the prediction of important clinical outcomes such as ppFEV1. The clinical benefit and meaningfulness of an additional reduction in SwCl beyond that previously seen with ELX-TEZ-IVA has also not been established. Moreover, repeat SwCl testing in clinical practice may present a burden to the health system. However, the experts did consider SwCl to be an appropriate and sensitive end point for assessing whether a rare CF mutation, without supporting clinical evidence, responds to CFTRm therapies, and noted that repeat sweat testing may be warranted in the occasional patient with a rare genotype to demonstrate mechanistic response. |
System and economic issues | |
The sponsor has indicated that 98.7% of patients eligible for Alyftrek are also eligible for Trikafta. It is anticipated that Alyftrek will replace Trikafta for both new patients initiating treatment and existing patients currently receiving Trikafta, which has a budget impact. Do you agree Alyftrek is likely to replace Trikafta for patients initiating CFTRm therapy and patients currently receiving Trikafta? | The clinical experts agreed that a majority of patients with CF receiving ELX-TEZ-IVA would switch to VNZ–TEZ–D-IVA. Out of all patients with CF in Canada, the experts predicted that approximately 60% to 70% of patients would be interested in initiating VNZ–TEZ–D-IVA. |
CF = cystic fibrosis; CFTRm = CFTR modulator; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ppFEV1 = percent predicted forced expiratory volume in 1 second; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of VNZ–TEZ–D-IVA (Alyftrek) in the treatment of patients with CF who have a F508del mutation or another responsive CFTR mutation and are aged 6 years and older. The focus will be placed on comparing VNZ–TEZ–D-IVA with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of VNZ–TEZ–D-IVA is presented in 4 sections, with the review team’s critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The review team’s assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section typically includes sponsor-submitted long-term extension studies, although there are no completed long-term extension studies at this time. The third section typically includes indirect evidence from the sponsor, but there was no indirect treatment comparison relevant to the review. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence, which in this case pertain to in vitro assays of potentially responsive non-F508del mutations that may not have (and, due to rarity, may never have) robust clinical trial evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 RCTs and 1 single-arm pivotal study identified in the systematic review
4 additional studies addressing potentially responsive non-F508del mutations in an in vitro nonclinical setting.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 8. Two double-blind phase III RCTs, SKYLINE 102 (VX21-121-102, N = 405) and SKYLINE 103 (VX21-121-103; N = 574), and 1 single-arm phase III study, RIDGELINE (VX21-121-105), were included.
Both RCTs compared VNZ–TEZ–D-IVA with ELX-TEZ-IVA in patients with CF aged at least 12 years with ELX-TEZ-IVA–responsive mutations. These were randomized (1:1) studies with a 52-week treatment phase and were similar in study design. Randomization was done using an interactive web response system and was stratified by the patient’s age at screening visit (aged younger than 18 years versus at least 18 years), ppFEV1 during the run-in period (less than 70% versus at least 70%), SwCl during the run-in period (less than 30 mmol/L versus at least 30 mmol/L), and prior CFTRm use (yes versus no). All patients entered a 28-day run-in period receiving ELX-TEZ-IVA before being randomized in a double-blind fashion to treatment with VNZ–TEZ–D-IVA or ELX-TEZ-IVA for 52 weeks, followed by an additional 28-day open-label follow-up for additional safety data. In the SKYLINE 102 trial, there were no study sites in Canada, and no patients were located in Canada. In the SKYLINE 103 trial, there were 29 patients (4.9%) in Canada across 8 sites in various provinces. In both studies, the majority of patients were located in the US (44.7% and 33.8%, respectively), while the remainder were located in Australia, Europe, Israel, and New Zealand.
In the RIDGELINE trial, patients aged between 1 and 11 years with a mutation responsive to ELX-TEZ-IVA were recruited. The RIDGELINE study included a part A with the objective of evaluating the pharmacokinetics of VNZ–TEZ–D-IVA and relevant metabolites when dosed in therapeutic concentrations, and to evaluate the safety and tolerability of VNZ–TEZ–D-IVA. Part A preceded part B and informed the doses and weight-related dose thresholds for part B, in which the objectives were primarily to evaluate the safety and tolerability of VNZ–TEZ–D-IVA through week 24, and secondarily to evaluate the efficacy through week 24 as well as the pharmacokinetics. Parts A and B were each divided into 3 cohorts by age range, where the first cohort (A1 and B1) included patients aged 6 to 11 years (inclusive), the second (A2 and B2) included patients aged 2 to 5 years (inclusive), and the third (A3 and B3) included patients aged 1 year to younger than 2 years. Therefore, only cohorts A1 (N = 17) and B1 (N = 78) are relevant to the Health Canada indication considered here, and only cohort B1 is associated with results data for efficacy outcomes that are considered in this review, although both cohort A1 and cohort B1 contributed to the safety data that will be assessed.
Patients in cohort A1 of the RIDGELINE trial received VNZ–TEZ–D-IVA once daily for 22 days and were permitted to participate in cohort B1 if they met eligibility criteria. Patients in cohort B1 who were receiving stable ELX-TEZ-IVA treatment had the run-in period waived and entered the treatment period (VNZ–TEZ–D-IVA), which was 24 days, followed by a safety follow-up visit 28 days after the last dose. Patients entering cohort B1 who were not already receiving stable ELX-TEZ-IVA underwent a 28-day run-in period in which they received ELX-TEZ-IVA before the VNZ–TEZ–D-IVA treatment period. Dosing of ELX-TEZ-IVA during the run-in period and dosing of VNZ–TEZ–D-IVA during the treatment period were based on weight categories, as described in more detail in the subsequent Interventions section of this report. No sites or patients in Canada were included. The majority of sites and patients were in the US (100% of cohort A1 and 60.3% of cohort B1), and the remainder were located in Australia or Europe.
Table 8: Details of Studies Included in the Systematic Review
Details | SKYLINE 102 trial | SKYLINE 103 trial | RIDGELINE trial |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, randomized, double-blind, ELX-TEZ-IVA–controlled, parallel-group, multicentre study | Phase III, randomized, double-blind, ELX-TEZ-IVA–controlled, parallel-group, multicentre study | Phase III, 2-part (parts A and B), multicohort, multicentre study |
Locations | Australia, Canada, Europe, Israel, New Zealand, the US | Australia, Canada, Europe, Israel, New Zealand, the US | Australia, Europe, the US |
Study period | September 14, 2021, through November 21, 2023 | October 27, 2021, through November 30, 2023 | June 21, 2022, through November 27, 2023 |
Randomized (N) | 405 | 574 | NA (nonrandomized):
|
Inclusion criteriab |
|
|
|
Exclusion criteriaa |
|
| |
Drugs | |||
Intervention | VNZ–TEZ–D-IVA (N = 200) | VNZ–TEZ–D-IVA (N = 285) |
|
Comparator | ELX-TEZ-IVA (N = 205) | ELX-TEZ-IVA (N = 289) | NA |
Study duration | |||
Screening phase | Day −56 through day −29 | Day −56 through day −29 |
|
Treatment phase | 52 weeks | 52 weeks |
|
Follow-up phase | 4 weeks (28 days) | 4 weeks (28 days) |
|
Outcomes | |||
Primary end point | Absolute CFB in ppFEV1 through week 24 | Safety and tolerability | |
Secondary and exploratory end points | Key secondary
Other secondary
Other end points
| Cohort A1
Cohort B1: Secondary
Cohort B1: Other
| |
Publication status | |||
Publications | Keating et al. (2025) (includes both studies) plus supplementary materials and errata45 | Hoppe et al. (2025) plus supplementary materials and errata46 | |
NCT | NCT05033080 | NCT05076149 | NCT05422222 |
AE = adverse event; BMI = body mass index; CF = cystic fibrosis; CFB = change from baseline; CFQ-R = Cystic Fibrosis Questionnaire-Revised; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; CFTRm = CFTR modulator; F/F = homozygous for F508del; F/G = heterozygous for F508del and a gating mutation; F/MF = heterozygous for F508del and a minimal function mutation; F/other = heterozygous for F508del and a non-F508del mutation that does not fit into any of the other specified mutation categories (i.e., gating, residual function, minimal function, triple combination–responsive); F/RF = heterozygous for F508del and a residual function mutation; GLI = Global Lung Function Initiative; NA = not applicable; ppFEV1 = percent predicted forced expiratory volume in 1 second; RD = respiratory domain; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aThe trial was conducted in patients aged 1 to 11 years and included several cohorts (A1 and B1, A2 and B2, A3 and B3). The part B cohorts will not initiate until results are available from the corresponding part A cohort for the same age group. Cohorts A1 and B1 evaluated patients aged 6 years through 11 years, inclusive, while cohorts A2 and B2 included patients aged 2 to 5 years, and cohorts A3 and B3 included patients aged 1 year to younger than 2 years. Of the cohorts, only A1 and B1 were conducted in the age group evaluated in this study; ergo, the age group 2 and group 3 cohorts are not further described here. The purpose of cohort A1 was to determine doses and weight cut-offs for cohort B1; therefore, for efficacy outcomes, the focus is on cohort B1, while cohort A1 is also included in safety analyses.
bExclusion and inclusion criteria sourced from clinicaltrials.gov records.
Sources: Clinical Study Reports.9-11
Populations
Inclusion and Exclusion Criteria
The RCTs, SKYLINE 102 and SKYLINE 103, both included patients aged at least 12 years with a ppFEV1 value between 40% and 90% of the predicted mean for age, sex, and height for patients receiving ELX-TEZ-IVA, or a ppFEV1 between 40% and 80% for patients not receiving ELX-TEZ-IVA therapy. Patients recruited to the SKYLINE 102 trial had to be heterozygous for F508del and a minimal function mutation, while patients recruited to the SKYLINE 103 trial had to have a genotype that was either homozygous for F508del mutations, heterozygous for F508del and a gating or residual function mutation, or with at least 1 mutation identified as responsive to ELX-TEZ-IVA with no F508del mutation. Patients had to have stable CF, as determined by the investigator. Inadequate organ function or a recent (within 28 days of the run-in period) pulmonary exacerbation, respiratory infection, or changes in therapy were reasons for exclusion. Prohibited medications included moderate and strong CYP3A inducers or inhibitors (except ciprofloxacin) within 14 days of the first dose in the run-in period and during the study, and CFTRm therapies produced by other drug manufacturers taken within 28 days or 5 terminal half-lives (whichever is longer) before screening and during the study.
The single-arm RIDGELINE study included patients aged 1 to 11 years with stable CF and at least 1 triple combination–responsive mutation (including F508del) in the CFTR gene and ppFEV1 of at least 60% at screening. Cohorts A1 and B1 captured patients aged 6 to 11 years, inclusive, which is the focus of this review; the younger age groups were evaluated in cohorts A2, B2, A3, and B3, which will not be discussed in depth. Prohibited medications were the same as those listed for the RCTs described previously.
Key exclusion criteria for all trials were patients with a history of solid organ or hematological transplant, hepatic cirrhosis with portal hypertension, moderate hepatic impairment (Child-Pugh score of 7 to 9), severe hepatic impairment (Child-Pugh score of 10 to 15), lung infection with organisms associated with a more rapid decline in pulmonary status, or who were pregnant or breastfeeding.
Interventions
Intervention and Comparators
VNZ–TEZ–D-IVA was orally administered once daily as a fixed-dose combination tablet with a fat-containing meal or snack.
In the double-blind RCTs (SKYLINE 102 and SKYLINE 103), VNZ–TEZ–D-IVA, ELX-TEZ-IVA, ivacaftor, and identical placebos were supplied as tablets of similar size and appearance, and all patients received the same number of tablets per day to maintain blinding; i.e., the ELX-TEZ-IVA regimen included a second dose later in the day of ivacaftor only, so patients assigned to VNZ–TEZ–D-IVA received a matched placebo for that second dose to maintain blinding. The daily dose of VNZ–TEZ–D-IVA was 20 mg of vanzacaftor, 100 mg of tezacaftor, and 250 mg of deutivacaftor in the combination tablet. In the ELX-TEZ-IVA groups, the dose of elexacaftor was 200 mg daily, tezacaftor was 100 mg daily, and ivacaftor was 150 mg every 12 hours (i.e., twice daily for a total of 300 mg per day). There was a run-in period from day −28 to day −1 (after which the treatment period began), during which patients received ELX-TEZ-IVA at the same dose as previously described. Dose modifications were not allowed, although treatment interruptions were allowed or required to resolve toxicity.
In the single-arm open-label RIDGELINE study, because patients were younger (aged 6 to 11 years in cohorts A1 and B1), the dose was weight-dependent. In patients in cohort B1 who were under 30 kg in body weight at screening, the dosage for ELX-TEZ-IVA during the 28-day run-in period was 100 mg of elexacaftor once daily, 50 mg of tezacaftor once daily, and 75 mg of ivacaftor twice daily (i.e., a total of 150 mg of ivacaftor per day); in patients in this cohort who were at least 30 kg in body weight or greater, the dosage of ELX-TEZ-IVA in the 28-day run-in period was 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor twice daily (i.e., 300 mg daily over 2 doses). During the treatment period, the dosage of VNZ–TEZ–D-IVA in patients under 40 kg was 12 mg of vanzacaftor once daily, 60 mg of tezacaftor once daily, and 150 mg of deutivacaftor once daily. The dosage of VNZ–TEZ–D-IVA in patients 40 kg or greater was 20 mg of vanzacaftor once daily, 100 mg of tezacaftor once daily, and 250 mg of deutivacaftor once daily.
Concomitant Medications and Cointerventions
Patients with CF are typically managed with a variety of treatments to improve their symptoms, quality of life, and longevity. Patients could receive other medications during the studies, including CF medications consistent with standards of care (e.g., pancreatic enzymes), although some concomitant medications with potential interactions (such as products that are sensitive substrates of p-glycoprotein, products that are CYP2C9 substrates, or products that are substrates of OATP1B1 or OATP1B3, e.g., statins, among others) required additional caution and monitoring. As described in the inclusion and exclusion criteria listed previously, some medications were prohibited immediately preceding and during the studies if there was potential for them to affect the exposure to study therapies or to confound the study results, such as moderate and strong CYP31 inducers and inhibitors (except ciprofloxacin) or other CFTRm therapies.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 9, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review, and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 9: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | SKYLINE 102 trial | SKYLINE 103 trial | RIDGELINE triala |
|---|---|---|---|---|
Efficacy | ||||
Number of PEx (rate)b | Week 24 Week 52c | Secondary (52 weeks) | Secondary (52 weeks) | Secondary (annualized rate based on 24 weeks) |
Absolute change from baseline in ppFEV1 (noninferiority assessment) | Week 24 Week 52c | Primary (24 weeks)d Secondary (52 weeks) | Primary (24 weeks)d Secondary (52 weeks) | Secondary (24 weeks) |
Absolute change in BMIe | Week 24 Week 52c | Other | Other | Secondary (24 weeks) |
Absolute change in BMI z scoree | Week 24 Week 52c | Other | Other | Secondary (24 weeks) |
Absolute change from baseline in CFQ-R respiratory domain | Week 24 Week 52c | Secondary | Secondary | Secondary (24 weeks) |
Absolute change in SwCl | Week 24 Week 52c | Key secondary (24 weeks)d Secondary (52 weeks) | Key secondary (24 weeks)d Secondary (52 weeks) | Secondary (24 weeks) |
AEs | ||||
Number of patients with events of elevated aminotransferase levelsf | Week 24 Week 52c | Safety | Safety | Safety (24 weeks) |
Number of patients with SAEsf | Week 24 Week 52c | Safety | Safety | Safety (24 weeks) |
AE = adverse event; BMI = body mass index; CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire-Revised; FAS = full analysis set; GRADE = Grading of Recommendations Assessment, Development and Evaluation; PEx = pulmonary exacerbation(s); ppFEV1 = percent predicted forced expiratory volume in 1 second; SAE = serious adverse event; SwCl = sweat chloride.
aIn the RIDGELINE trial, which has no comparator arm, all outcomes were reported as within-group change only and reflect patients aged 6 to 11 years in cohort B1. Additionally, no 52-week outcomes were captured because this was a shorter study, so all outcomes from the RIDGELINE trial are approximately 24-week outcomes, unless otherwise specified.
bThe PEx events requiring IV treatment and/or hospitalization are also discussed in this Clinical Review, as well as hospitalizations overall, but have not been assessed using GRADE.
cThe 52-week end point has been preferentially assessed for this outcome, where available, based on clinical expert input that the longer duration is appropriate for assessment. End points of shorter duration (e.g., 24 weeks) for this outcome have been assessed using GRADE when they provide the only data available, which is the case for the RIDGELINE trial (which is the only source of data for patients aged 6 to 11 years) due to the shorter duration of the study.
dStatistical testing for this end point was adjusted for multiple comparisons (e.g., hierarchical testing) in the SKYLINE 102 and SKYLINE 103 trials (when the hierarchy included, in order, absolute change in ppFEV1 at 24 weeks, absolute change in SwCl through week 24, proportion of patients with an SwCl level of ≤ 60 mmol/L at 24 weeks, and proportion of patients with an SwCl level of ≤ 30 mmol/L at 24 weeks). No hierarchical testing or multiplicity adjustment was conducted for other outcomes, nor for any outcomes in the RIDGELINE trial.
eIn the SKYLINE 102 and SKYLINE 103 trials, the absolute change in BMI was assessed in the FAS (model adjusted for baseline BMI), while the absolute change in BMI z score was assessed in patients aged younger than 20 years. In the RIDGELINE trial, because all patients were aged 11 years at most, BMI and BMI z score were both assessed in the FAS.
fAll safety-related outcomes (including overall AEs, withdrawals due to AE, deaths, and so forth) have been reported, as is standard; however, per clinical expert input, only these 2 outcome measures (number of patients with SAEs and number with events of elevated aminotransferase levels) were assessed using GRADE.
Sources: Clinical Study Reports.9-11
Number of Pulmonary Exacerbations
A pulmonary exacerbation was defined as a new or change in antibiotic therapy (IV, inhaled, or oral) for at least 4 sinopulmonary signs or symptoms. Pulmonary exacerbation is a critical contributor to the clinical and economic burden experienced by people with CF. Not only do pulmonary exacerbations have an acute negative effect on patient health and HRQoL, they are also associated with several long-term consequences, including a faster rate of lung function decline, increased risk of future exacerbations and hospitalizations, increased likelihood of a future lung transplant, and increased risk of mortality.47-50 An empirically derived MID for pulmonary exacerbation rate has yet to be established, and the clinical experts consulted by CDA-AMC agreed there is no clear threshold. The threshold used in the GRADE assessments was therefore null.
The SKYLINE 102 and SKYLINE 103 trials both measured the change in the number of pulmonary exacerbations from baseline to the week 52 evaluation period, whereas the RIDGELINE trial measured from baseline to week 24.
Although not assessed using GRADE, related outcomes such as the time to first pulmonary exacerbation and the number of pulmonary exacerbation events requiring hospitalization or IV antibiotic therapies have also been summarized.
Percent Predicted Forced Expiratory Volume in 1 Second
Lung function in people with CF is typically reported using ppFEV1, which determines the maximum volume of air exhaled during the first second of a forced breath. The ppFEV1 value is calculated by dividing the patient’s FEV1 score by the normal FEV1 Global Lung Initiative reference value for healthy, nonsmoking individuals based on age, height, and sex and multiplying by 100.2,51 For every 1% decline in ppFEV1, 5-year mortality risk increases by 4%, highlighting the clinical importance of maintaining lung function at the highest possible ppFEV1 level.18,52 Although no empirically validated MID was identified, and there is no clinician consensus on an MID for ppFEV1, a 5% threshold was previously accepted by CDA-AMC and Cystic Fibrosis Canada as a meaningful improvement.22,24
The primary outcome of the 2 included RCTs (SKYLINE 102 and SKYLINE 103) was the absolute change from baseline in ppFEV1 at 24 weeks, which was also a secondary outcome in the single-arm study, RIDGELINE. The 2 RCTs also reported the 52-week outcome for ppFEV1 as a secondary outcome. In the SKYLINE 102 and SKYLINE 103 trials, the between-group findings were assessed based on the prespecified noninferiority margin of 3%.
BMI and BMI Z Score
BMI is a measure of a patient’s nutritional status and is based on their weight (in kilograms) and height (in metres), and BMI z score is a standardized measure of BMI that takes into account age group and sex.2 Malnutrition leads to a low BMI and failure to thrive (i.e., insufficient weight gain and growth), which increases susceptibility to lung infections.53,54 BMI has also been established as a predictor of survival in patients with CF.55 All 3 included studies measured weight-related parameters, including BMI and BMI z score as a change from baseline to 24 weeks and, in the 2 RCTs, 52 weeks. In the SKYLINE 102 and SKYLINE 103 studies, which included both adult and pediatric patients older than 12 years, the change in BMI z score was measured only in patients who were aged 20 years or younger, and the change in BMI score was measured in all patients. No MID was identified through the literature or clinical expert input for these end points, so the threshold used in the GRADE assessment was null.
Sweat Chloride
The SwCl test is a standard diagnostic tool for CF because elevated concentrations of chloride in sweat indicate abnormality of CFTR protein activity.22,35 The CFTR protein normally functions as a chloride ion transport channel. SwCl alone is not always sufficient to diagnose or exclude a diagnosis of CF because some patients with CF may have only mildly elevated SwCl at assessment, which is impacted in part by the specific CF-causing mutations carried by the patient.22,35 A normal concentration level of SwCl is typically considered to be less than 30 mmol/L, while values from 30 mmol/L to 59 mmol/L are diagnostically unclear, and values greater than 60 mmol/L indicate CF.22,35 Continued monitoring of SwCl is not common in real-world clinical practice after diagnosis.22,35
Despite some heterogeneity between patients and the relationship between specific CF-causing mutations, changes in SwCl shortly after treatment initiation are an indicator of the restoration of some CFTR ion channel function; as such, it may be a useful mechanistic biomarker of response to a CFTRm.22,35,56 The clinical experts consulted by CDA-AMC noted that SwCl can be a more sensitive indicator of mechanistic response to treatment initiation than clinical outcomes such as ppFEV1 in patients whose lung function is still within the normal range at baseline (wherein improvements beyond baseline are unlikely), as is typically the case in most children. Also, changes in SwCl can be evaluated sooner than clinical outcomes such as lung function, which will typically require a longer duration of treatment to show meaningful changes. Patients commonly continue to have elevated SwCl concentrations after treatment with CFTRm therapies, even if there have been significant reductions compared with their before-treatment SwCl concentration results.57 The clinical experts consulted by CDA-AMC also highlighted that there can be differences (5 mmol/L to 10 mmol/L) in SwCl levels in the same patient, depending on which arm is sampled (even when taken at the same time), or between different testing dates.
The experts consulted by CDA-AMC noted that, in their experience, SwCl is not strongly correlated with clinical outcomes owing to other sources of between-patient heterogeneity that more greatly impact clinical outcomes. They highlighted that even siblings with the same CF-causing genotype may appear to have a different relationship between changes in SwCl and clinical outcomes. The experts described that SwCl may not always be an appropriate biomarker, depending on the specific CF-causing mutation carried by a patient, because some CFTR mutations are associated with higher or lower SwCl elevations than others and, in some cases, such as the N1303K CFTR mutation, SwCl is not expected to substantially reduce with CFTRm therapy, despite objective improvement in clinical symptoms, indicating that the typical mechanistic reduction in SwCl may not always be directly relevant to the clinical improvements seen with a CFTRm. They also indicated there is currently no well-established MID for the end point of SwCl when comparing between groups treated with CFTRm therapies, and that it remains to be seen whether there is long-term clinical value with regard to reductions in SwCl beyond those seen with ELX-TEZ-IVA treatment.
In concordance with the clinical expert opinion presented to CDA-AMC, the magnitude and reliability of the correlation between SwCl and specific clinical outcomes has not been thoroughly established in published literature. Modern clinical studies of treatments for CF often include the assessment of SwCl, and patients treated with CFTRm therapies (compared with patients not treated with CFTRm therapies or compared with within-patient changes before and after CFTRm therapy) typically demonstrate reduced SwCl as well as improved clinical outcomes;56,58-60 however, a causal relationship or reliable prediction of clinical benefit of SwCl as a surrogate end point has not been reported.
Several studies were submitted by the sponsor as evidence of support for this relationship. This review will examine several of them here briefly, excluding those studies that did not conduct any analysis of correlation or causality.
In a phase II study of ivacaftor (NCT00457821) in patients with CF and at least 1 G551D mutation,56 within-patient SwCl was evaluated as a biomarker for changes in CFTR activity. There was a large amount of variability seen between patients in the ivacaftor treatment groups, which the study authors suggested may be due to differences in CFTR activity correction between individuals, variability in exposure to ivacaftor, differences in the mutation of the other allele, the relative quantity of CFTR channels at the cell surface, and/or the role of modifier genes impacting ion balance. Although this study demonstrated that CFTR activity increases after therapy with ivacaftor and that SwCl was also generally reduced, thereby demonstrating the effect of ivacaftor as a CFTRm that partially restores CFTR function, a clear connection or quantification of any potential correlation between changes in SwCl and specific clinical outcomes was not evaluated.56
In an analysis of the US Cystic Fibrosis Foundation Patient Registry data that used Cox regression to evaluate the correlation of SwCl with clinical end points, there was no independent association of SwCl with survival, lung function, height, or BMI in patients with a known CFTR genotype functional class. However, among patients with an unclassified functional class of CFTR genotype, SwCl was an independent predictor of survival.61
In a study by Zemanick et al. (2025),59 6 phase III placebo-controlled studies (and their open-label extensions) of CFTRm in patients with CF with a homozygous F508del mutation or a heterozygous F508del and a minimal function mutation genotype were pooled and evaluated to examine the relationship between attained values of SwCl and several clinical end points: lung function (ppFEV1), BMI, patient-reported outcomes, pulmonary exacerbations, and change in lung function over time. The studies included EVOLVE/EXTEND, AURORA F/MF, AURORA F/F, KEPLER, NCT03447249 (parent study) and NCT03447262 (open-label extension of NCT03447249), and NCT03460990 (parent study) and NCT03447262 (open-label extension of NCT03460990). The CFTR modulators used as interventions in these studies included tezacaftor-ivacaftor, ELX-TEZ-IVA, and VX-659-TEZ-IVA, and the median duration of the studies ranged from 0.5 years to 4.0 years. In total, the studies included 1,314 patients treated with a CFTRm and 642 patients treated with placebo. The change from baseline outcomes was analyzed using an analysis of covariance model; the number of pulmonary exacerbations through week 24 was analyzed using a negative binomial regression model, and the rate of change in ppFEV1 was analyzed using a linear random coefficients model. End points were analyzed for the association between SwCl and the stated clinical outcomes using 4 SwCl-based groupings: less than 30 mmol/L, at least 30 mmol/L to less than 60 mmol/L, at least 60 mmol/L to less than 80 mmol/L, and at least 80 mmol/L. The results of the study suggest a correlation between attainment of the lower categories of SwCl (< 30 mmol/L or ≥ 30 mmol/L to < 60 mmol/L) and numerically better clinical outcomes. However, the 95% CIs of the results for most end points across the SwCl-based subgroups were overlapping to a large degree, suggesting these differences were generally not statistically significant.
The results of the Zemanick et al. (2025)59 study provide support for the biologic plausibility of a correlation between SwCl and clinical end points, and it pools data across several trials in populations that are mostly relevant to the target population. However, there are limitations related to temporal ambiguity and causality, magnitude and clinical relevance of differences, and transferability across CFTR-causing mutation variants. These limitations will be discussed further subsequently.
Temporal ambiguity and causality: The Zemanick et al. (2025)59 study adjusted baseline values for patients who had run-in periods with Trikafta, using expected untreated baselines derived from the phase III trials. While this helps standardize comparisons, it introduces model-based assumptions rather than direct observation, which weakens causal inference. These adjustments are necessary for harmonizing data but inherently limit the ability to draw causal conclusions. The primary analyses assessed change from baseline in both SwCl and clinical outcomes over the same time period. This concurrent measurement means that temporal precedence is not established. It is difficult to determine whether changes in SwCl preceded, followed, or occurred simultaneously with clinical improvements. The analysis of covariance, negative binomial regression, and linear random coefficients models are appropriate for assessing associations and adjusting for covariates; however, they do not account for unmeasured confounding, nor do they establish a directional pathway from SwCl to clinical outcomes.
Magnitude and clinical relevance of differences: While lower SwCl levels were associated with greater mean improvements in ppFEV1, CFQ-R respiratory domain, BMI, and exacerbation rates, the 95% CIs overlapped across the 3 lower SwCl groups (< 80 mmol/L) for several outcomes. For example, the LS mean change in ppFEV1 was:
15.65% in the less than 30 mmol/L group
14.01% in the 30 mmol/L to less than 60 mmol/L group
12.74% in the 60 mmol/L to less than 80 mmol/L group.
Using an MID of 5% for ppFEV1, as mentioned previously, these differences (each under 2%) are unlikely to be clinically meaningful. This undermines the strength of the association and further limits the ability to infer causality. The article by Zemanick et al. (2025)59 notes this point as a limitation.
Transferability across CFTR variants: The Zemanick et al. (2025)59 article notes that clinical benefit has been observed in patients with the N1303K variant despite no consistent improvements in SwCl, suggesting that SwCl may not be informative across all genotypes. This limits its generalizability and further challenges its use as a surrogate end point because, mechanistically, it is apparent that these patients are experiencing a clinical benefit with CFTRm without necessarily also experiencing the reductions in SwCl that are typically seen in patients with CF caused by non-N1303K mutations.
While the Zemanick et al. study (2025)59 is well conducted and provides important data, it is not definitive in establishing a causative correlation between SwCl as a surrogate for clinical end points and is insufficient to be relied on alone for decision-making in the context of health technology assessment. Moreover, regulatory agencies have not accepted SwCl as a surrogate for clinical benefit, reinforcing the need for caution in its interpretation within a health technology assessment framework.
In summary, despite the uncertainty about its prognostic utility, SwCl is an important biomarker that provides information about the mechanistic effect of CFTRm therapies and could be supportive in assessing their comparability. Nonetheless, the clinical importance or surrogacy of SwCl reductions beyond that previously seen in studies of ELX-TEZ-IVA has not been clearly established to predict clinical outcomes and, accordingly, there is no established MID for SwCl for comparisons between treatment groups; therefore, the GRADE assessment was conducted using null as the threshold of interest.
In the SKYLINE 102, SKYLINE 103, and RIDGELINE trials, SwCl was collected before study drug dosing at each time point (2 samples, 1 from each arm) and sent to a central laboratory for testing and interpretation of results. In addition to the absolute change from baseline in SwCl, this review will also report (although not assessed with GRADE) the proportion of patients with an SwCl level of 30 mmol/L or lower at week 24 and week 52.
CFQ-R Respiratory Domain
The CFQ-R is the most widely used and validated patient-reported outcome measure in CF that assesses the clinical benefit of a given treatment from the patient or caregiver’s perspective.62 It includes 12 domains, each of which is scored from 0 to 100, with higher numbers representing better HRQoL or fewer symptoms. The respiratory domain includes items that measure patient- (or caregiver-) reported improvements in the following respiratory symptoms: waking up from coughing, wheezing, coughing, congestion, difficulty breathing, and mucus production.29,63,64 The MID, which corresponds to the smallest clinically important change a patient can detect, is estimated to be a 4-point change in CFQ-R respiratory domain scores.62 The SKYLINE 102, SKYLINE 103, and RIDGELINE trials measured the absolute change in CFQ-R respiratory domain score from baseline to the week 24 evaluation period. The measurement properties of the CFQ-R respiratory domain are detailed in Table 10.
Elevated Aminotransferase Levels
Elevated aminotransferase is an AE associated with the treatment of CF using CFTRm therapies that can indicate issues with the liver.22 This AE was identified by the clinical experts consulted by CDA-AMC as one that is particularly important to monitor. The magnitude of elevation can indicate the need to interrupt, reduce, or discontinue therapy.22
Serious Adverse Events
In the included studies, SAEs are defined as any AE that meets the following criteria:
fatal (death, regardless of cause, that occurs during participation in the study or occurs after participation and is suspected of being a delayed toxicity due to the administration of the study drug)
life-threatening, such that the patient was at immediate risk of death from the reaction as it occurred
inpatient hospitalization or prolongation of hospitalization
persistent or significant disability or incapacity (disability is defined as a substantial disruption of a person’s ability to conduct normal life functions)
congenital anomaly or birth defect
important medical event that, based on appropriate medical judgment, may jeopardize the patient or may require medical or surgical intervention to prevent 1 of the outcomes listed previously (e.g., an allergic bronchospasm requiring intensive treatment in an emergency department or at home).
Table 10: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
CFQ-R respiratory domain | The CFQ-R is a disease-specific HRQoL instrument designed for patients with CF that is available in age-appropriate versions for children aged 6 to 13 years (child self-report version), parents who serve as a proxy for their child (parent observer-reported version), and an adolescent and adult version for individuals aged 14 years and older.65 The domains in the adolescent and adult version have an HRQoL module that includes physical functioning, vitality, emotional functioning, social or school functioning, role functioning, body image, eating problems, and treatment burden domains; a symptoms module that includes respiratory symptoms, digestive symptoms, and weight; and a health perception module. A 4-point Likert scale is used to measure frequency (always, often, sometimes, or never), intensity (a great deal, somewhat, a little, or not at all), and true–false scales (very true, somewhat true, somewhat false, or very false). Items within domains are summed up and standardized. Individual domain scores range from 0 to 100, with higher scores indicating better HRQoL. The scales are designed to measure symptoms and function during the 2-week period before the administration of the questionnaire.65,66 | Validity: In 1 study conducted in the US with 212 patients with CF aged from 14 to 53 years, construct validity was assessed by examining the associations between the CFQ-R, age, disease severity, and nutritional status. The CFQ-R was inversely correlated with age (rs = −0.17 to −0.36; P < 0.05). Associations were found between the CFQ-R weight-related domains and nutritional status, with positive correlations between body image and weight domains, and body mass index scores (rs = 0.38 to 0.47; P < 0.01). The CFQ-R was able to differentiate between those with mild, moderate, and severe disease (Hotelling T2 = 0.44; F [22,384] = 3.84; P < 0.001), thus establishing construct validity.66 Convergent validity was evaluated by examining correlations between similar domains on the CFQ-R and SF-36, a generic HRQoL measure, with strong associations found between the following domains: physical (r = 0.81; P < 0.01), health perceptions or general health (r = 0.79; P < 0.01), vitality (r = 0.84; P < 0.01), role or role-physical (r = 0.73; P < 0.01), emotional functioning or mental health (r = 0.74; P < 0.01), and social (r = 0.57; P < 0.01).66 Another study conducted among 84 children with CF, ranging in age from 7 to 13 years, and their parents demonstrated convergent validity between the child self-reported and parent-proxy reports on several scales of the CFQ-R.67 Discriminant validity was assessed by evaluating the correlations between scales on the CFQ-R and SF-36 that do not measure similar constructs, but the CFQ-R was found to be moderately correlated with the SF-36 in terms of general health and mental health (rs = 0.19 to 0.42).66 Reliability: In the study conducted with patients with CF in the US, item-level analyses were assessed by examining item-to-scale correlations, internal consistencies, and test–retest reliability. Internal consistency coefficients indicated acceptable reliability (Cronbach alpha = 0.67 to 0.94), with acceptable test–retest stability (ICC = 0.45 to 0.90) in this study.66 Another study demonstrated acceptable reliability for all scales (Cronbach alpha = 0.60 to 0.76) except treatment burden (Cronbach alpha = 0.44).67 Responsiveness: In a study consisting of 24 adult patients with clinically improved CF, an acceptable level of responsiveness was found in terms of physical functioning, emotions, body image, and treatment burden, with effect sizes ranging from 0.25 to 0.38. Among the caregivers of 17 patients with CF who responded to the CFQ-R parent-proxy version, responsiveness was demonstrated for 3 dimensions: physical functioning, energy, and respiratory symptoms.65 | A difference of at least 4 points in the respiratory domain score of the CFQ-R is commonly cited as the MID for patients with CF.62 |
CF = cystic fibrosis; CFQ-R = Cystic Fibrosis Questionnaire-Revised; F = F statistic; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MID = minimal important difference; r = correlation coefficient; rs = Spearman correlation; SF-36 = Short Form (36) Health Survey.
Statistical Analysis
Sample Size and Power Calculation
For the SKYLINE 102 trial, assuming a within-group SD of 8, a 10% dropout rate at week 24, and a treatment difference of zero between VNZ–TEZ–D-IVA and ELX-TEZ-IVA, a sample size of 200 patients in each group for a total of 400 patients provided more than 90% power to test the primary hypothesis for the primary end point, based on a 1-sided, 2-sample t test at a significance level of 0.025.
For the SKYLINE 103 trial, assuming a within-group SD of 8, a 10% dropout rate at week 24, and a treatment difference of zero between VNZ–TEZ–D-IVA and ELX-TEZ-IVA, a sample size of 275 patients in each group for a total of 550 patients provided more than 95% power to test the primary hypothesis for the primary end point, based on a 1-sided, 2-sample t test at a significance level of 0.025.
For the RIDGELINE trial, between 12 and 20 patients were planned to be enrolled in cohort A1. Sample size calculations were determined based on VNZ–TEZ–D-IVA pharmacokinetic parameters, such as clearance and volume of distribution. Based on the variability observed in adults, data from 12 patients allowed 80% power to target a 95% CI within 60% and 140% of the geometric mean estimate of clearance for VNZ–TEZ–D-IVA. No formal power calculation was performed for cohort B1. Approximately 65 patients were planned for enrolment in cohort B1, which was deemed adequate to meet the primary safety objective. The occurrence of treatment-emergent AEs was a safety end point. With approximately 55 patients expected to complete cohort B1, the study had a 94% chance of observing an AE in at least 1 patient if the true incidence rate was 5%, and a greater than 99% chance of observing an AE in at least 1 patient if the true incidence rate was 10%. The probabilities were calculated by assuming a binomial distribution for the number of AEs.
End Points
Primary End Point Analysis (ppFEV1)
In the SKYLINE 102 and SKYLINE 103 trials, the primary noninferiority analysis was performed using an MMRM with change from baseline at day 15, week 4, week 8, week 16, and week 24 as the dependent variable. The model included treatment group, visit, and treatment-by-visit interaction as fixed effects, with continuous baseline ppFEV1, age at screening (aged < 18 years versus ≥ 18 years) and mutation group as covariates. The MMRM included data from all available visits up to week 24. A Kenward-Roger approximation was used for denominator degrees of freedom. Treatment effect through week 24 was estimated by averaging weeks 16 and 24. Intercurrent events of prohibited medication and treatment discontinuation were addressed using the treatment policy strategy (observed data following the event were included in the model). A hierarchical testing procedure was used to control the overall type I error at an alpha of 0.05. The primary end point of absolute change from baseline in ppFEV1 through week 24 was tested for noninferiority at a 1-sided alpha level of 0.025. Subgroups assessed for ppFEV1 included age (younger than 18 years, 18 years or older), ppFEV1 at baseline (less than 70%, at least 70% and greater), SwCl at baseline (less than 30 mmol/L, at least 30 mmol/L), sex (female, male), and region (North America, the rest of the world). A supplementary analysis was conducted in which intercurrent events were handled using the hypothetical strategy (observed data after the event were set to missing) instead of the treatment policy strategy; no other sensitivity analyses were described.
The noninferiority margin selected in the SKYLINE 102 and SKYLINE 103 trials was 3 percentage points. A statistical approach using the Rothmann method recommends that the noninferiority margin preserve at least 50% of the treatment effect of the active control (ELX-TEZ-IVA) compared with placebo, where the treatment effect is estimated by the lower bound of the 95% CI.68,69 The selected noninferiority margin of 3 percentage points is consistent with this statistical method and the noninferiority margin for ppFEV1 that is used in clinical studies evaluating symptomatic CF treatments.7,8
The analysis of the 52-week end point for change in ppFEV1 was conducted in a manner similar to that of the 24-week end point described previously, with the end points estimated by averaging weeks 16, 24, 36, and 52.
In the RIDGELINE trial, no primary efficacy analysis was reported, but the ppFEV1 analysis used an MMRM to estimate the within-group mean absolute change through week 24 (estimated by averaging weeks 16 and 24). The end point was the dependent variable, visit was a fixed-effect variable, and baseline value and genotype group were covariates. If there was nonconvergence due to there being only a small number of patients in the genotype group, then it was removed from the model. An unstructured covariance structure was used to model the within-patient errors. If model estimation did not converge, a compound symmetry covariance structure was used instead. No multiplicity adjustments, sensitivity analyses, or subgroup analyses were conducted in the RIDGELINE trial.
Secondary Continuous End Point Analysis (SwCl, CFQ-R Respiratory Domain, BMI, and Other Weight Metrics)
In all 3 trials, the analysis of absolute change from baseline in SwCl, CFQ-R respiratory domain, BMI, and BMI z score was based on an MMRM that was similar to the one described previously for ppFEV1, with absolute change from baseline at outcome assessment (e.g., week 24) as the dependent variable. For SwCl and the CFQ-R respiratory domain, the primary result obtained from the model was the estimated treatment difference through week 24, estimated by averaging weeks 16 and 24. No subgroup or sensitivity analyses were conducted for secondary outcomes in any of the 3 trials.
For the RCTs, a hierarchical testing procedure was used to control the overall type I error at an alpha of 0.05 for the primary outcome and the key secondary end points (the absolute change from baseline in SwCl through week 24, the proportion of patients with an SwCl level of 60 mmol/L or less at week 24, and the proportion of patients with an SwCl level of 30 mmol/L or less, pooled between the SKYLINE 102 and SKYLINE 103 trials). The primary end point of absolute change from baseline in ppFEV1 through week 24 was tested for noninferiority at a 1-sided alpha level of 0.025. The key secondary end points were formally tested at an alpha level of 0.05 only if the primary analysis of absolute change from baseline in ppFEV1 through week 24 was statistically significant, i.e., if the null hypothesis of inferiority was rejected. For a test at any step to be considered statistically significant within the testing hierarchy, it must have been statistically significant, and all previous tests (if any) within the hierarchy must have been statistically significant at the 0.05 level (1-sided 0.025 level for the primary end point). In the 2 RCTs, the order of the hierarchy after the primary outcome was as follows: absolute change from baseline in SwCl through week 24, proportion of patients with an SwCl level of less than 60 mmol/L through week 24 (pooled FAS in the SKYLINE 102 and SKYLINE 103 trials), and proportion of patients with an SwCl level of less than 30 mmol/L through week 24 (pooled FAS).
The analyses of the 52-week ppFEV1- and SwCl-related end points were conducted in a manner similar to that of the 24-week end points previously described, but these end points were not included in a hierarchical testing procedure and were estimated by averaging weeks 16, 24, 36, and 52.
No hierarchical testing or multiplicity adjustments were conducted in the RIDGELINE trial.
Secondary End Point Analysis: Number of Pulmonary Exacerbations (Rate)
In the SKYLINE 102 and SKYLINE 103 trials, pulmonary exacerbations through 52 weeks were analyzed by comparing differences in the annual exacerbation rates between treatment groups, with 95% CIs providing statistical context.
In the RIDGELINE study, the number of pulmonary exacerbations during the 24-week analysis period was assessed using descriptive statistics, including annualized event rate and duration of events. The annualized duration of events for each patient was the total number of days with the event multiplied by 48 weeks and divided by the total number of weeks in the study up to the end of the analysis period. The annualized duration was descriptively summarized.
No other details on the methodology of assessing this outcome were reported in any of the included studies, such as how missing data were handled, although the RIDGELINE study stated that, in general, missing data were not imputed unless otherwise specified.
No subgroup or sensitivity analyses were conducted for the secondary outcomes in any of the included studies. No hierarchical testing or multiplicity adjustment was conducted in the RIDGELINE study, and pulmonary exacerbations were not included in the hierarchical testing structure for either the SKYLINE 102 or SKYLINE 103 trial.
BMI and BMI Z Scores
Across all 3 trials, the BMI and BMI z scores were calculated by using Centers for Disease Control and Prevention growth charts.70
In the SKYLINE 102 and SKYLINE 103 RCTs, BMI and BMI z score were assessed as an absolute change from baseline at week 52 using an MMRM similar to the one described for ppFEV1. BMI was assessed in the FAS, while BMI z score was assessed in patients aged 20 years or younger at baseline, and the covariate of age at screening was not included in the BMI z score model for this reason. Data obtained from the day 15, week 4, week 8, week 16, and week 24 visits were included in the model.
In the RIDGELINE trial, because all patients in the study were aged 11 years at most, both BMI and BMI z score were assessed in the FAS (within the cohort in question, in this case, cohort B1). This end point was also assessed through an MMRM similar to the one described for SwCl.
No subgroup or sensitivity analyses were conducted for secondary outcomes in any of the 3 trials, and no BMI or BMI z scores were included in the hierarchical testing structure of either the SKYLINE 102 or SKYLINE 103 trials. No hierarchical testing or multiplicity adjustment was conducted in the RIDGELINE trial.
Safety Analyses
In all 3 trials, safety analyses were assessed during the treatment-emergent period in the safety analysis set, including follow-up time after the last day of the treatment period (approximately 1 month). Patients were analyzed according to the actual treatment received during the treatment period. Patients who received the study drug while in more than 1 treatment group were allocated to the VNZ–TEZ–D-IVA group.
For the safety analyses, baseline was defined as the most recent nonmissing measurement (scheduled or unscheduled) collected before the first dose of the study drug in the treatment period (i.e., the day 1 visit).
Subgroup Analyses
In both the SKYLINE 102 and SKYLINE 103 trials, the MMRM used for the primary analysis was used for the subgroup analysis of ppFEV1, where a similar model was applied to each category of the subgroup: age at screening (aged < 18 years, ≥ 18 years), ppFEV1 (< 70%, ≥ 70%), and sex (female, male). Model-based estimates for a given category are displayed if the analysis converges in that category. For subgroup analyses based on age, the term “age at screening” was removed from the MMRM model. The adjusted means with 2-sided 95% CIs were provided.
The clinical experts consulted by CDA-AMC suggested there may be differences in expected treatment outcomes in pediatric patients and patients with highly preserved lung function compared with others, so these subgroup results have been summarized in the ppFEV1 section of the efficacy results. However, sex is not expected to have an influence on prognosis or treatment response in CF, according to the clinical experts, so it will not be addressed. The studies are not powered to analyze treatment differences in the smaller sample sizes of subgroup analyses, so the findings may be influenced by variability and may lack precision, increasing the risk of false-positive or false-negative results. These analyses are exploratory and should be considered hypothesis-generating rather than confirmatory.
In the RIDGELINE trial, no subgroup analyses were performed due to the small sample size.
Table 11: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
RCTs: SKYLINE 102 and SKYLINE 103 | ||||
Number of pulmonary exacerbations through week 24 and week 52 (FAS) | MMRM based on the design used for the primary end point (absolute change in ppFEV1 at 24 weeks) | Refer to ppFEV1; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Absolute change in ppFEV1 through week 24 and week 52 (FAS) |
| Fixed categorical effects for treatment, visit, age at screening (aged < 18 years vs. ≥ 18 years), and treatment-by-visit interaction, with baseline ppFEV1 and baseline SwCl as continuous covariates | All data were assumed to be missing at random and were not imputed | Alternative estimand defined similarly to the primary estimand with the exception that intercurrent events were handled using the hypothetical strategy (observed data after the event were set to missing) |
BMI and BMI z score through week 52 (FAS) |
| Refer to ppFEV1; the same adjustment factors apply, with the exception that age was not adjusted for the BMI z score model because it included only patients aged 20 years or younger | All data were assumed to be missing at random and were not imputed | None |
CFQ-R RD scores through week 24 and week 52 (FAS) | MMRM similar to the one described for the primary outcome of ppFEV1 | Refer to ppFEV1; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Absolute change in baseline SwCl through week 24 and week 52 (mmol/L) (FAS) |
| Refer to ppFEV1; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Elevated aminotransferase through week 52 (SAS) | Safety analysis | None | All data were assumed to be missing at random and were not imputed | None |
SAEs through week 52 (SAS) | Safety analysis | None | All data were assumed to be missing at random and were not imputed | None |
Single-arm study: RIDGELINE | ||||
Number of pulmonary exacerbations through week 24 (cohort B1 FAS) |
| None | All data were assumed to be missing at random and were not imputed | None |
Absolute change in ppFEV1 from baseline through week 24 (cohort B1 FAS) |
| Refer to SwCl; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Absolute change in CFQ-R RD score (child version) from baseline through week 24 (cohort B1 FAS) |
| Refer to SwCl; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Absolute change in BMI and BMI z score from baseline at week 24 (cohort B1 FAS) |
| Refer to SwCl; the same adjustment factors apply | All data were assumed to be missing at random and were not imputed | None |
Absolute change in SwCl from baseline through week 24 (cohort B1 FAS) |
| Fixed categorical effects for genotype group (F/MF; F/F; other) and visit, and baseline SwCl as a continuous covariate | All data were assumed to be missing at random and were not imputed | None |
Elevated aminotransferase levels (SAS, reported for both cohorts A1 and B1) | Safety analysis | None | All data were assumed to be missing at random and were not imputed | None |
SAEs (SAS, reported for both cohorts A1 and B1) | Safety analysis | None | All data were assumed to be missing at random and were not imputed | None |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CI = confidence interval; F/F = homozygous for F508del; F/MF = heterozygous for F508del and a minimal function mutation; FAS = full analysis set; LS = least squares; MMRM = mixed model for repeated measures; ppFEV1 = percent predicted forced expiratory volume in 1 second; RCT = randomized controlled trial; RD = respiratory domain; SAE = serious adverse event; SAS = safety analysis set; SwCl = sweat chloride; vs. = versus.
Note: Only ppFEV1 at 24 weeks in the SKYLINE 102 and SKYLINE 103 trials, and key secondary outcomes (change from baseline through week 24 in SwCl, proportion of patients with an SwCl level of 60 mmol/L or less at week 24 and proportion of patients with an SwCl level of 30 mmol/L or less at week 24) were part of the hierarchical testing procedure. No 52-week outcomes were controlled for multiplicity. No outcomes from the RIDGELINE trial were controlled for multiplicity.
Sources: Clinical Study Reports.9-11
Analysis Populations
The analysis populations of the SKYLINE 102, SKYLINE 103, and RIDGELINE trials are described in Table 12.
Table 12: Analysis Populations of the Included Studies
Studies | Population | Definition | Application |
|---|---|---|---|
SKYLINE 102, SKYLINE 103, and RIDGELINE | Set of all patients | Included all patients who were randomized or received at least 1 dose of the study drug | This analysis set was used for all individual patient data listings and disposition summary tables, unless otherwise specified. |
FAS | Included all randomized patients who carried the intended CFTR mutation(s) and received at least 1 dose of the study drug during the treatment period | The FAS was used to summarize patient demographics and baseline characteristics, and for the analyses of all efficacy end points in which patient data were analyzed according to the patient’s randomized treatment group, unless otherwise specified. | |
Safety set for the treatment period | Included all patients who received at least 1 dose of the study drug during the treatment period | This safety set was used for all safety analyses during the treatment period in which patient data were analyzed according to the treatment the patient received, unless otherwise specified. |
FAS = full analysis set.
Sources: Clinical Study Reports.9-11
Results
Patient Disposition
Patient disposition in the SKYLINE 102, SKYLINE 103, and RIDGELINE trials is summarized in Table 13.
In the SKYLINE 102 trial, 200 patients in the VNZ–TEZ–D-IVA group and 205 patients in the ELX-TEZ-IVA group were randomized, although 4 and 3 patients in each group, respectively, did not receive any dose. Of the patients who received any dose, 12 (6.1%) and 11 (5.4%) patients, respectively, discontinued from the study, while 15 (7.7%) and 16 (7.9%) patients, respectively, discontinued from treatment (but not necessarily the study). The most common reasons for discontinuation from the study were AEs (2% in each group) and withdrawal of consent not due to AEs (2.6% and 2.5%, respectively). Discontinuation from treatment was also due most commonly to AEs (2% in the VNZ–TEZ–D-IVA group and 5% in the ELX-TEZ-IVA group), pregnancy (1.5% and 0.5%, respectively), or refusal of further dosing not due to AEs (2.6% and 1.0%, respectively). Patient disposition was generally well balanced between the groups, with some relatively small differences.
In the SKYLINE 103 trial, 285 patients were randomized to VNZ–TEZ–D-IVA and 289 were randomized to ELX-TEZ-IVA. One patient randomized to VNZ–TEZ–D-IVA did not receive any dose of the study drug. Discontinuations from the study occurred in 10 (3.5%) and 20 (7.0%) patients, respectively, and discontinuations from treatment occurred in 25 (8.8%) and 16 (5.5%) patients, respectively. The most common reasons for study discontinuation were again AEs (3.2% and 1.0%, respectively), followed by withdrawal of consent not due to AEs (1.1% and 0.3%, respectively). The most common reasons for discontinuation from treatment were AEs (4.9% and 3.1%, respectively), pregnancy (1.4% and 1.0%, respectively), and refusal of further dosing not related to AEs (0.7% and 0.3%, respectively). Patient disposition was generally well balanced between the groups, with some relatively small differences.
In the RIDGELINE trial, 17 patients were recruited for cohort A1, and 78 patients were recruited for cohort B1. No patients discontinued from cohort A1, and only 1 patient discontinued treatment (but not the study) in cohort B1, which was due to an AE.
Table 13: Summary of Patient Disposition From Studies Included in the Systematic Review (Set of All Patients)
Patient disposition | SKYLINE 102 trial | SKYLINE 103 trial | RIDGELINE trial | |||
|---|---|---|---|---|---|---|
VNZ–TEZ–D-IVA | ELX-TEZ-IVA | VNZ–TEZ–D-IVA | ELX-TEZ-IVA | Cohort A1 VNZ–TEZ–D-IVA | Cohort B1 VNZ–TEZ–D-IVA | |
Screened, N | NR | NR | NR | NR | NR | NR |
Randomized, N | 200 | 205 | 285 | 289 | NA | NA |
Randomized but not dosed, n | 4 | 3 | 1 | 0 | NA | NA |
FAS, N | 196 | 202 | 284 | 289 | 17 | 78 |
Safety, N | 196 | 202 | 284 | 289 | 17 | 78 |
Discontinued from study, n (% of safety set) | 12 (6.1) | 11 (5.4) | 20 (7.0) | 10 (3.5) | 0 (0) | 0 (0) |
Reason for discontinuation of study, n (%): | ||||||
Adverse events | 4 (2.0) | 4 (2.0) | 9 (3.2) | 3 (1.0) | NA | NA |
Withdrawal of consent (not due to AE) | 5 (2.6) | 5 (2.5) | 3 (1.1) | 1 (0.3) | NA | NA |
Lost to follow-up | 0 (0.0) | 1 (0.5) | 1 (0.4) | 0 (0) | NA | NA |
Other noncompliance | 1 (0.5) | 0 (0.0) | 1 (0.4) | 1 (0.3) | NA | NA |
Physician decision | 1 (0.5) | 0 (0.0) | 1 (0.4) | 0 (0) | NA | NA |
Death | 0 (0) | 0 (0) | 0 | 0 | NA | NA |
Commercial drug is available for patient | 0 | 0 | 1 (0.4) | 0 (0) | NA | NA |
Other | 1 (0.5) | 1 (0.5) | 4 (1.4) | 5 (1.7) | NA | NA |
Discontinued treatment, n (%) | 15 (7.7) | 16 (7.9) | 25 (8.8) | 16 (5.5) | 0 (0) | 1 (1.3) |
Reason for discontinuation of treatment, n (%): | ||||||
AEs | 4 (2.0) | 10 (5.0) | 14 (4.9) | 9 (3.1) | NA | 1 (1.3) |
Patient refused further dosing (not due to AE) | 5 (2.6) | 2 (1.0) | 2 (0.7) | 1 (0.3) | NA | 0 (0) |
Lost to follow-up | 0 (0) | 1 (0.5) | 1 (0.4) | 0 (0) | NA | 0 (0) |
Noncompliance with study drug | 2 (1.0) | 0 (0) | 1 (0.4) | 1 (0.3) | NA | 0 (0) |
Physician decision | 1 (0.5) | 0 (0) | 0 | 0 | NA | 0 (0) |
Pregnancy (self or partner) | 3 (1.5) | 1 (0.5) | 4 (1.4) | 3 (1.0) | NA | 0 (0) |
Commercial drug is available for patient | 0 | 0 | 1 (0.4) | 0 (0) | NA | 0 (0) |
Other | 0 (0) | 2 (1.0) | 3 (1.1) | 2 (0.7) | NA | 0 (0) |
AE = adverse event; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FAS = full analysis set; NA = not applicable; NR = not reported; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Sources: Clinical Study Reports.9-11
Important Protocol Deviations
In the SKYLINE 102 trial, 20 patients (5.0%) had important protocol deviations, which were defined as any protocol deviation that may have significantly impacted the completeness, accuracy, and/or reliability of key study data or that may have significantly affected a patient’s rights, safety, or well-being. Of these, 8 patients (5 in the VNZ–TEZ–D-IVA group and 3 in the ELX-TEZ-IVA group) had events related to investigational products, such as study drug temperature excursions, study drug dispensation errors, and study drug dosing outside of the relevant treatment period. The deviation was related to prohibited concomitant medications in 7 patients (3 in the VNZ–TEZ–D-IVA group and 4 in the ELX-TEZ-IVA group), including solumedrol, commercial ELX-TEZ-IVA (Trikafta), cimetidine, fluconazole, and dexamethasone. Deviations related to eligibility criteria occurred in 3 patients (2 in the VNZ–TEZ–D-IVA group and 1 in the ELX-TEZ-IVA group) due to acute respiratory infection or antibiotics received during the run-in period. Two patients had deviations related to study conduct or procedures: 1 in the VNZ–TEZ–D-IVA group (a subinvestigator performed a physical examination without documented training) and 1 in the ELX-TEZ-IVA group (morning and evening doses were exchanged at the week 36 visit). Finally, 1 patient had a deviation due to delay in SAE reporting in the VNZ–TEZ–D-IVA group.
In the SKYLINE 103 trial, 27 patients (4.7%) had important protocol deviations, which were defined in the same manner as in the SKYLINE 102 trial. Of these, 16 had deviations related to investigation products (11 in the VNZ–TEZ–D-IVA group and 5 in the ELX-TEZ-IVA group) such as study drug dispensation errors, compliance, study drug kits not being returned, and incorrect study drug dosing. Five had deviations related to eligibility criteria (1 in the VNZ–TEZ–D-IVA group and 4 in the ELX-TEZ-IVA group) due to medical history, acute illness, or antibiotics received during the run-in period. Three patients had a deviation due to delay in SAE reporting, 2 in the VNZ–TEZ–D-IVA group and 1 in the ELX-TEZ-IVA group. Two patients had deviations related to study conduct: 1 patient in the VNZ–TEZ–D-IVA group had delayed the interruption of the study drug and resumed the study drug without meeting resumption criteria, and 1 patient in the ELX-TEZ-IVA group performed day 1 assessments after study drug dosing. Finally, 1 patient’s parent signed the incorrect version of the consent form, and 1 patient took commercial CFTRm during an interruption in the study drug.
In the RIDGELINE trial, 2 patients in cohort A1 had protocol deviations (defined in the same manner as stated previously): 1 enrolled without a documented review of the screening laboratory results, and 1 did not have a baseline hemoglobin value confirmed before study drug dosing. In cohort B1, 2 patients had deviations related to incorrect study drug dispensing (the dose for higher-weight patients) on day 1 and were dispensed the correct kits (the dose for lower-weight patients) on day 15 and thereafter.
Baseline Characteristics
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The SKYLINE 102 (N = 398) and SKYLINE 103 (N = 573) trials were similarly designed RCTs. In the SKYLINE 102 trial, slightly more than 40% of patients were female and just under 60% of patients were male, whereas the sex distribution was closer to approximately 50% each in the SKYLINE 103 trial. The mean age by treatment group in the 2 RCTs at baseline was 31 to 34 years across the SKYLINE studies, and the treatment groups were similar for these categories in each study. In general, the characteristics appeared to be well balanced between the treatment groups in each study.
In cohorts A1 and B1 of the RIDGELINE trial, 41.2% and 43.6% were female, 58.8% and 56.4% of patients were male, and the mean age was 9.5 years and 9.3 years, respectively.
The majority of patients in every study were white (range, 90.7% to 97.5%), while a minority were Asian (range, 0% to 0.5%), Black or African American (range, 0% to 2.0%), more than 1 race (range, 0% to 1.5%), or other (range, 0.% to 0.4%), or information about race was not collected due to local regulations. The baseline data for ppFEV1 demonstrated more progressed disease in the studies that recruited older patients (the SKYLINE 102 and SKYLINE 103 trials recruited patients aged 12 years or older) compared with the RIDGELINE trial, which recruited patients aged 6 to 11 years for cohorts A1 and B1; this reflects the natural course of CF, and there were no concerning within-study differences between treatment groups. In all studies, most patients (range, 84.6% to 94.1%) had prior experience with a CFTRm, most commonly, ELX-TEZ-IVA. The mean duration of prior ELX-TEZ-IVA use was approximately 2 years in the SKYLINE 102 and SKYLINE 103 RCTs, and approximately 1 year in cohort B1 of the RIDGELINE trial. The SKYLINE 102 trial recruited patients with a genotype that was heterozygous for F508del and a minimal function mutation; ergo, this genotype was represented in 100% of patients while, in the other studies, the most common genotype was homozygous for F508del (range, 47.4% to 78.2%).
Table 14: Summary of Baseline Characteristics From the Studies Included in the Systematic Review (Full Analysis Sets)
Characteristic | SKYLINE 102 trial | SKYLINE 103 trial | RIDGELINE trial | |||
|---|---|---|---|---|---|---|
VNZ–TEZ–D-IVA (N = 196) | ELX-TEZ-IVA (N = 202) | VNZ–TEZ–D-IVA (N = 284) | ELX-TEZ-IVA (N = 289) | Cohort A1 VNZ–TEZ–D-IVA (N = 17) | Cohort B1 VNZ–TEZ–D-IV (N = 78) | |
Sex, n (%) | ||||||
Female | 80.0 (40.8) | 83 (41.1) | 135 (47.5) | 145 (50.2) | 7.0 (41.2) | 34.0 (43.6) |
Male | 116.0 (59.2) | 119 (58.9) | 149 (52.5) | 144 (49.8) | 10.0 (58.8) | 44.0 (56.4) |
Age | ||||||
Age, mean years (SD) | 30.8 (10.5) | 30.9 (11.4) | 33.3 (12.6) | 34.0 (12.4) | 9.5 (1.3) | 9.1 (1.7) |
Age, median years (range) | 30.3 (12.4 to 61.7) | 31.3 (12.2 to 71.6) | 32.6 (12.2 to 71.2) | 33.8 (12.7 to 63.4) | 9.6 (6.4 to 11.0) | 9.3 (6.2 to 12.0) |
Ethnicity, n (%) | ||||||
Hispanic or Latino | 13 (6.6) | 11 (5.4) | 4 (1.4) | 5 (1.7) | 3.0 (17.6) | 9.0 (11.5) |
Not Hispanic or Latino | 183 (93.4) | 190 (94.1) | 265 (93.3) | 261 (90.3) | 14.0 (82.4) | 62.0 (79.5) |
Information about race not collected per local regulations | 0 (0) | 1 (0.5) | 15 (5.3) | 23 (8.0) | NR | 7.0 (9.0) |
Race, n (%) | ||||||
American Indian or Alaska Native | NR | NR | 0 (0) | 1 (0.3) | NR | NR |
Asian | 1 (0.5) | 0 (0) | 1 (0.4) | 1 (0.3) | NR | NR |
Southeast Asian | NR | NR | 1 (0.4) | 0 (0) | NR | NR |
Other Asian | 1 (0.5) | 0 (0) | 0 (0) | 1 (0.3) | NR | NR |
Black or African American | 4 (2.0) | 1 (0.5) | 0 (0) | 0 (0) | NR | 1.0 (1.3) |
White | 191 (97.4) | 197 (97.5) | 270 (95.1) | 262 (90.7) | 17.0 (100.0) | 71.0 (91.0) |
Information about race not collected per local regulations | NR | NR | 10 (3.5) | 23 (8.0) | NR | 5 (6.4) |
More than 1 race | 0 (0) | 3 (1.5) | 2 (0.7) | 1 (0.3) | NR | 1.0 (1.3) |
Other | 0 (0) | 1 (0.5) | 1 (0.4) | 1 (0.3) | NR | NR |
Geographic region, n (%) | ||||||
North America | 87 (44.4) | 91 (45.0) | 114 (40.1) | 103 (35.6) | 17 (100.0) | 47.0 (60.3) |
Rest of the worlda | 109 (55.6) | 111 (55.0) | 170 (59.9) | 186 (64.4) | NA | 31.0 (39.7) |
ppFEV1 category, n (%) | ||||||
< 40 | 6 (3.1) | 3 (1.5) | 5 (1.8) | 7 (2.4) | 0 (0) | 1.0 (1.3) |
≥ 40 to < 70 | 95 (48.5) | 111 (55.0) | 149 (52.5) | 160 (55.4) | 0 (0) | 1.0 (1.3) |
≥ 70 to ≤ 90 | 85 (43.4) | 79 (39.1) | 112 (39.4) | 107 (37.0) | 2 (11.8) | 15 (19.2) |
> 90 | 7 (3.6) | 8 (4.0) | 13 (4.6) | 12 (4.2) | 15 (88.2) | 60 (76.9) |
Missing | 3 (1.5) | 1 (0.5) | 5 (1.8) | 3 (1.0) | NR | 1 (1.3) |
Mean (SD) | 67.0 (15.3) | 67.2 (14.6) | 67.2 (14.6) | 66.4 (14.9) | 106.8 (9.3) | 99.7 (15.1) |
Median (range) | 67.7 (28.0 to 108.6) | 68.1 (31.0 to 100.1) | 66.9 (38.3 to 112.5) | 66.9 (36.4 to 104.6) | 105.8 (88.2 to 123.0) | 100.5 (29.3 to 146.0) |
SwCI (mmol/L) | ||||||
Mean (SD) | 53.6 (17.0) | 54.3 (18.2) | 43.4 (18.5) | 42.1 (17.9) | 46.2 (22.7) | 40.4 (20.9) |
Median (range) | 52.0 (20.5 to 106.8) | 54.3 (10.0 to 113.5) | 43.0 (10.0 to 113.3) | 41.6 (10.0 to 109.3) | 43.5 (12.0 to 80.5) | 39.0 (11.5 to 109.5) |
CFQ-R RD score | ||||||
Mean (SD) | 85.8 (14.7) | 82.9 (15.7) | 85.7 (13.2) | 85.6 (13.2) | NR | 84.8 (16.1)b |
Median (range) | 88.9 (27.8 to 100.0) | 83.3 (22.2 to 100.0) | 88.9 (33.3 to 100.0) | 88.9 (27.8 to 100.0) | NR | 91.7 (16.7 to 100.0)b |
BMI (kg/m2) | ||||||
Mean (SD) | 22.71 (3.40) | 23.03 (3.85) | 23.27 (4.02) | 22.92 (3.27) | 17.75 (1.46) | 16.83 (2.13) |
Median (range) | 22.31 (14.28 to 35.31) | 22.40 (15.81 to 44.72) | 22.48 (15.56 to 44.98) | 22.83 (15.43 to 35.42) | 17.36 (14.72 to 20.47) | 16.33 (13.40 to 23.43) |
BMI z score (patients aged ≤ 20 years at baseline) | ||||||
Mean (SD) | −0.36 (1.09) | −0.14 (0.88) | −0.17 (0.95) | −0.30 (0.98) | 0.45 (0.51) | 0.07 (0.87) |
Median (range) | −0.31 (−4.15 to 1.92) | −0.29 (−2.06 to 1.98) | −0.13 (−2.62 to 2.12) | −0.26 (−3.07 to 1.21) | 0.30 (−0.57 to 1.32) | 0.08 (−1.75 to 1.84) |
Weight (kg) | ||||||
Mean (SD) | 65.08 (13.32) | 64.54 (13.75) | 66.58 (13.98) | 65.05 (13.35) | 32.84 (6.32) | 30.21 (7.48) |
Median (range) | 63.00 (33.00 to 130.18) | 63.00 (33.00 to 116.57) | 65.0 (34.00 to 122.47) | 63.0 (32.00 to 106.00) | 34.0 (18.60 to 43.70) | 28.90 (19.30 to 54.00) |
Height (cm) | ||||||
Mean (SD) | 168.8 (9.5) | 166.9 (9.5) | 168.7 (9.4) | 167.8 (10.1) | 135.3 (10.1) | 133.0 (10.5) |
Median (range) | 169.0 (147.0 to 195.0) | 167.0 (137.2 to 192.0) | 169.0 (139.7 to 191.0) | 167.0 (140.0 to 200.0) | 137.7 (112.4 to 149.2) | 133.1 (111.5 to 153.7) |
CFTR genotype group, n (%) | ||||||
F/F | 0 | 0 | 222 (78.2) | 224 (77.5) | 9 (52.9) | 37 (47.4) |
F/MF | 196 (100)c | 202 (100)c | 0 | 0 | 3 (17.6) | 24 (30.8) |
F/G | 0 | 0 | 19 (6.7) | 20 (6.9) | 1 (5.9) | 3 (3.8) |
F/RF | 0 | 0 | 23 (8.1) | 23 (8.0) | NR | 1 (1.3) |
F/other | 0 | 0 | 0 | 0 | NR | 2 (2.6) |
Triple combination–responsive, any mutation | 0 | 0 | NR | NR | 4 (23.5) | 11 (14.1) |
Triple combination–responsive, F508del mutation | 0 | 0 | 0 | 0 | 1 (5.9) | 5 (6.4) |
Triple combination–responsive, non-F508del mutation | 0 | 0 | 20 (7.0) | 22 (7.6) | 3 (17.6) | 6 (7.7) |
Prior CFTRm use, n (%) | ||||||
Yes (any) | 170 (86.7) | 177 (87.6) | 241 (84.9) | 250 (86.5) | 16 (94.1) | 66 (84.6) |
Kalydeco | 1 (0.5) | 0 | 11 (3.9) | 5 (1.7) | 0 | 2 (2.6) |
Orkambi | 0 | 0 | 19 (6.7) | 17 (5.9) | 0 | 2 (2.6) |
Symdeko | 1 (0.5) | 0 | 26 (9.2) | 24 (8.3) | 0 | 0 |
Trikafta | 168 (85.7) | 177 (87.6) | 185 (65.1) | 204 (70.6) | 16 (94.1) | 62 (79.5) |
No (none) | 26 (13.3) | 25 (12.4) | 43 (15.1) | 39 (13.5) | 1 (5.9) | 12 (15.4) |
Duration of ELX-TEZ-IVA use as a prior medication, years | ||||||
Number of patients contributing to the analysis, n | 168 | 177 | 185 | 204 | NR | 62 |
Mean (SD) | 2.09 (1.05) | 2.17 (1.16) | 2.03 (0.95) | 1.78 (1.00) | NR | 1.25 (0.73) |
Median | 2.27 | 2.26 | 2.17 | 1.92 | NR | 1.14 |
Range | 0.13 to 5.07 | 0.14 to 5.05 | 0.05 to 4.45 | 0.13 to 4.42 | NR | 0.13 to 2.90 |
BMI = body mass index; CFQ-R RD = Cystic Fibrosis Questionnaire-Revised respiratory domain; CFTRm = CFTR modulator; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del; F/G = heterozygous for F508del and a gating mutation; F/MF = heterozygous for F508del and a minimal function mutation; F/other = heterozygous for F508del and a non-F508del mutation that does not fit into any of the other specified mutation categories (i.e., gating, residual function, minimal function, triple combination–responsive); F/RF = heterozygous for F508del and a residual function mutation; NR = not reported; ppFEV1 = percent predicted forced expiratory volume in 1 second; SD = standard deviation; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aIn the SKYLINE 102 trial, North America includes patients from the US, and rest of the world includes patients from Australia, Europe, Israel, and New Zealand. In the SKYLINE 103 trial, North America includes patients from the US and Canada, and rest of the world includes patients from Australia, Europe, Israel, and New Zealand. In the RIDGELINE trial, North America includes patients from the US, and rest of the world includes patients from Australia and Europe.
bIn the RIDGELINE trial, the CFQ-R RD score is based on the child version and was assessed in 75 of the 78 patients in cohort B1.
cThe SKYLINE 102 trial included this genotype only, based on eligibility criteria.
Exposure to Study Treatments
The exposure to study treatments is summarized in Table 15.
Table 15: Summary of Patient Exposure in Studies Included in the Systematic Review (FAS)
Exposure | SKYLINE 102 trial | SKYLINE 103 trial | RIDGELINE trial | |||
|---|---|---|---|---|---|---|
VNZ–TEZ–D-IVA (N = 196) | ELX-TEZ-IVA (N = 202) | VNZ–TEZ–D-IVA (N = 284) | ELX-TEZ-IVA (N = 289) | Cohort A1 (N = 17) | Cohort B1 (N = 78) | |
Total patient-weeks (patient-years) | 9,809.7 (204.4) | 9,985.7 (208.0) | 13,973.1 (291.1) | 14,538.9 (302.9) | 53.3 (NR) | 1,855.3 (38.7) |
Duration, mean weeks (SD) | 50.0 (7.8) | 49.4 (10.0) | 49.2 (10.0) | 50.3 (7.6) | 3.1 (0.1) | 23.8 (2.1) |
Duration, median weeks (range) | 52.0 (1.3 to 54.1) | 52.0 (0.4 to 53.7) | 52.0 (0.4 to 53.7) | 52.0 (0.1 to 53.3) | 3.1 (3.0 to 3.3) | 24.0 (6.3 to 25.4) |
Exposure duration by interval, n (%) | ||||||
≤ 1 week | 0 (0) | 1 (0.5) | 1 (0.4) | 1 (0.3) | NR | 0 (0) |
> 1 to ≤ 2 weeks | 1 (0.5) | 2 (1.0) | 0 (0) | 0 (0) | NR | 0 (0) |
> 2 to ≤ 4 weeks | 0 (0) | 2 (1.0) | 3 (1.1) | 0 (0) | NR | 0 (0) |
> 4 to ≤ 12 weeks | 0 (0) | 3 (1.5) | 5 (1.8) | 2 (0.7) | NR | 1 (1.3) |
> 12 to ≤ 24 weeks | 6 (3.1) | 0 (0) | 8 (2.8) | 7 (2.4) | NR | 48 (61.5) |
> 24 to ≤ 36 weeks | 5 (2.6) | 4 (2.0) | 2 (0.7) | 3 (1.0) | NR | 29 (37.2)a |
> 36 to ≤ 52 weeks | 115 (58.7) | 129 (63.9) | 182 (64.1) | 186 (64.4) | NA | NA |
> 52 weeks | 69 (35.2) | 61 (30.2) | 83 (29.2) | 90 (31.1) | NA | NA |
> 15 to ≤ 22 days | NR | NR | NR | NR | 15 (88.2) | NR |
> 22 days | NR | NR | NR | NR | 2 (11.8) | NR |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FAS = full analysis set; NA = not applicable; NR = not reported; SD = standard deviation; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aFor the RIDGELINE trial only, this row represents an exposure of > 24 weeks instead of > 24 weeks to ≤ 36 weeks.
Sources: Clinical Study Reports.9-11
Concomitant Medications
A summary of concomitant medications in at least 20% of patients is presented in Table 16.
Table 16: Summary of Concomitant Medications in at Least 20% of Patients in Any Group During the Treatment Phase of the Included Studies (FAS)
Medication, N (%) | SKYLINE 102 | SKYLINE 103 | RIDGELINE | |||
|---|---|---|---|---|---|---|
VNZ–TEZ–D-IVA (N = 196) | ELX-TEZ-IVA (N = 202) | VNZ–TEZ–D-IVA (N = 284) | ELX-TEZ-IVA (N = 289) | Cohort A1 (N = 17) | Cohort B1 (N = 78) | |
Patients with any concomitant medication | 196 (100.0) | 202 (100.0) | 283 (99.6) | 289 (100.0) | 17.0 (100.0) | 78.0 (100.0) |
Sodium chloride | 138 (70.4) | 145 (71.8) | 169 (59.5) | 189 (65.4) | 13 (76.5) | 64 (82.1) |
Pancreatin | 139 (70.9) | 141 (69.8) | 195 (68.7) | 192 (66.4) | 5 (29.4) | 44 (56.4) |
Dornase alfa | 124 (63.3) | 139 (68.8) | 184 (64.8) | 184 (63.7) | 15 (88.2) | 49 (62.8) |
Salbutamol | 126 (64.3) | 122 (60.4) | 164 (57.7) | 170 (58.8) | 14 (82.4) | 53 (67.9) |
Azithromycin | 97 (49.5) | 97 (48.0) | 128 (45.1) | 118 (40.8) | 1 (5.9) | 14 (17.9) |
Colecalciferol | 77 (39.3) | 83 (41.1) | 137 (48.2) | 123 (42.6) | 2 (11.8) | 15 (19.2) |
Ibuprofen | 74 (37.8) | 74 (36.6) | 82 (28.9) | 102 (35.3) | 5 (29.4) | 26 (33.3) |
Paracetamol | 71 (36.2) | 76 (37.6) | 129 (45.4) | 110 (38.1) | 5 (29.4) | 25 (32.1) |
Tobramycin | 59 (30.1) | 59 (29.2) | 70 (24.6) | 70 (24.2) | NR | 5 (6.4) |
Ciprofloxacin | 47 (24.0) | 60 (29.7) | 56 (19.7) | 61 (21.1) | NR | 3 (3.8) |
Ursodeoxycholic acid | 58 (29.6) | 46 (22.8) | 48 (16.9) | 72 (24.9) | NR | 11 (14.1) |
Pancrelipase | 50 (25.5) | 49 (24.3) | 55 (19.4) | 65 (22.5) | 9 (52.9) | 23 (29.5) |
Vitamins | 50 (25.5) | 41 (20.3) | 56 (19.7) | 47 (16.3) | 6 (35.3) | 17 (21.8) |
Omeprazole | 46 (23.5) | 44 (21.8) | 61 (21.5) | 57 (19.7) | 2 (11.8) | 7 (9.0) |
Cetirizine hydrochloride | 16 (8.2) | 21 (10.4) | 27 (9.5) | 37 (12.8) | 7 (41.2) | 18 (23.1) |
Fluticasone propionate | 38 (19.4) | 40 (19.8) | 48 (16.9) | 42 (14.5) | 8 (47.1) | 26 (33.3) |
Macrogol 3350 | 16 (8.2) | 22 (10.9) | 24 (8.5) | 23 (8.0) | 7 (41.2) | 17 (21.8) |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FAS = full analysis set; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Sources: Clinical Study Reports.9-11
Efficacy
The results for key end points from the SKYLINE 102 and SKYLINE 103 studies are presented in Table 17, while results from the single-arm study, RIDGELINE, are presented in Table 18. These tables include the longest-duration end points available for the included studies and capture the results of any end points chosen to be assessed using GRADE.
Number of Pulmonary Exacerbations
Annual Event Rate
In the SKYLINE 102 trial FAS, the estimated event rate per year was 0.42 in the ELX-TEZ-IVA group (n = 50 patients with events) versus 0.32 in the VNZ–TEZ–D-IVA group (n = 60 patients with events). The pulmonary exacerbation rate difference was −0.10 events (95% CI, −0.24 to 0.04) per year.
In the SKYLINE 103 trial FAS, the annual event rate was 0.29 in the VNZ–TEZ–D-IVA group (n = 61 patients with events) and 0.26 in the ELX-TEZ-IVA group (n = 59 patients with events). The rate difference was 0.03 events per year (95% CI, −0.07 to 0.13).
In the RIDGELINE trial, there were 6 patients in the cohort B1 FAS who experienced an event, contributing to an annual event rate of 0.15.
No subgroup or sensitivity analyses were conducted for this end point in the included studies.
Time to First Pulmonary Exacerbation
In the SKYLINE 102 trial, based on a secondary analysis in the FAS of the time to first pulmonary exacerbation during the analysis period, the probability of event-free survival (Kaplan-Meier estimate) at 24 weeks was 0.820 (95% CI, 0.758 to 0.867) in the VNZ–TEZ–D-IVA group and 0.819 (95% CI, 0.758 to 0.866) in the ELX-TEZ-IVA group; at 52 weeks, it was 0.741 (95% CI, 0.673 to 0.797) and 0.695 (95% CI, 0.626 to 0.755), respectively.
In the SKYLINE 103 trial, based on a secondary analysis in the FAS of the time to first pulmonary exacerbation during the analysis period, the probability of event-free survival (Kaplan-Meier estimate) at 24 weeks was 0.857 (95% CI, 0.810 to 0.893) in the VNZ–TEZ–D-IVA group and 0.882 (0.838 to 0.914) in the ELX-TEZ-IVA group; at 52 weeks, it was 0.783 (95% CI, 0.729 to 0.827) and 0.792 (95% CI, 0.739 to 0.835), respectively.
Time to first pulmonary exacerbation was not reported in the RIDGELINE trial.
Pulmonary Exacerbations Requiring IV Antibiotics or Hospitalization
In the SKYLINE 102 trial FAS, 10 patients (5.1%) in the VNZ–TEZ–D-IVA group and 26 patients (12.9%) in the ELX-TEZ-IVA group had pulmonary exacerbations requiring hospitalization or IV antibiotic therapy during the analysis period. The probability of event-free survival (Kaplan-Meier estimate) in the 2 groups at 24 weeks was 0.969 (95% CI, 0.933 to 0.986) and 0.935 (95% CI, 0.890 to 0.962), respectively; at 52 weeks, it was 0.948 (95% CI, 0.906 to 0.972) and 0.868 (95% CI, 0.813 to 0.908), respectively. All patients with events required IV antibiotic therapy; additionally, 8 patients in the VNZ–TEZ–D-IVA group and 17 patients in the ELX-TEZ-IVA group required hospitalization.
In the SKYLINE 103 trial FAS, 19 patients (6.7%) and 17 patients (5.9%) in the VNZ–TEZ–D-IVA group and ELX-TEZ-IVA group, respectively, had pulmonary exacerbations requiring hospitalization or IV antibiotic therapy. The probability of event-free survival (Kaplan-Meier estimate) at 24 weeks was 0.961 (95% CI, 0.930 to 0.978) and 0.962 (95% CI, 0.932 to 0.979); at 52 weeks, it was 0.931 (95% CI, 0.894 to 0.956) and 0.940 (95% CI, 0.906 to 0.962), respectively. All patients with pulmonary exacerbations required IV antibiotic therapy; additionally, 14 patients in the VNZ–TEZ–D-IVA group and 9 patients in the ELX-TEZ-IVA group required hospitalization.
In cohort B1 of the RIDGELINE trial, 1 patient (1.3%) required both hospitalization and IV antibiotic therapy, for an observed event rate per year of 0.03.
Percent Predicted Forced Expiratory Volume in 1 Second
Twenty-Four Weeks
The primary outcome of ppFEV1 was evaluated in the FAS through week 24 in both the SKYLINE 102 trial (n = 196 in the VNZ–TEZ–D-IVA group and n = 202 in the ELX-TEZ-IVA group) and SKYLINE 103 trial (n = 284 and 289, respectively). In the MMRM analysis of noninferiority comparing VNZ–TEZ–D-IVA with ELX-TEZ-IVA, the LS mean treatment difference was 0.2% (1-sided P < 0.0001; 95% CI, −0.7% to 1.1%) in the SKYLINE 102 trial and was 0.2% (95% CI, −0.5% to 0.9%; 1-sided P for noninferiority < 0.0001) in the SKYLINE 103 trial. The prespecified noninferiority margin was −3.0%; ergo, the results of both studies met the preplanned primary analysis of noninferiority for ppFEV1 because neither 95% CI crossed the noninferiority margin.
In cohort B1 of the RIDGELINE trial (n = 78), patients treated with VNZ–TEZ–D-IVA showed similar ppFEV1 values compared with baseline use of ELX-TEZ-IVA, based on a within-group LS mean absolute change from baseline to week 24 of 0% (95% CI, −2.0% to 1.9%).
Fifty-Two Weeks
The secondary end point of ppFEV1 through 52 weeks showed similar results in both studies, consistent with the 24-week primary analyses. In the SKYLINE 102 trial, the LS mean difference in absolute change from baseline was 0.1% (95% CI, −0.8% to 1.0%); in the SKYLINE 103 trial, it was 0.3% (95% CI, −0.4 to 1.0).
Subgroup and Sensitivity Analyses
In the SKYLINE 102 and SKYLINE 103 trials, a supplementary sensitivity analysis was conducted using an alternative estimand where intercurrent events were addressed using the hypothetical strategy for the 24-week outcome of ppFEV1, and the results were consistent with the primary analyses.
Subgroup analyses were conducted on the primary outcome at 24 weeks in the SKYLINE 102 and SKYLINE 103 trials based on age (aged younger than 18 years versus at least 18 years at screening) and ppFEV1 at baseline (less than 70% versus at least 70%). Most of the results fell within the noninferiority margin of −3.0% and were similar to the primary analysis results. However, in the SKYLINE 102 trial in the subgroup of patients younger than age 18 years at screening, the 95% CI crossed this threshold, with an LS mean difference of −1.3% (95% CI, −3.9% to 1.3%). In the SKYLINE 103 trial, the value for this subgroup was 0.9% (95% CI, −1.2% to 3.0%).
No subgroup or sensitivity analyses were conducted in the RIDGELINE trial or for the week 52 outcomes in the SKYLINE studies.
Body Mass Index
Twenty-Four Weeks
In the RIDGELINE trial, the absolute change in BMI at week 24 was an LS mean of 0.22 kg/m2 (95% CI, 0.05 kg/m2 to 0.38 kg/m2).
Fifty-Two Weeks
In the SKYLINE 102 trial, the LS mean change from baseline in BMI in the VNZ–TEZ–D-IVA group (n = 179) and ELX-TEZ-IVA group (n = 187) at week 52 was 0.30 kg/m2 (SE = 0.10 kg/m2) and 0.08 kg/m2 (SE = 0.10 kg/m2), respectively, with an LS mean difference of 0.22 kg/m2 (95% CI, −0.05 kg/m2 to 0.49 kg/m2).
In the SKYLINE 103 trial, the LS mean change from baseline in BMI in the VNZ–TEZ–D-IVA group (n = 248) and ELX-TEZ-IVA group (n = 266) at week 52 was 0.37 kg/m2 (SE = 0.08 kg/m2) and 0.22 kg/m2 (SE = 0.07 kg/m2), respectively, with an LS mean difference of 0.16 kg/m2 (95% CI, −0.05 kg/m2 to 0.36 kg/m2).
Subgroup and Sensitivity Analyses
No subgroup or sensitivity analyses were reported.
BMI Z Score
Twenty-Four Weeks
In the RIDGELINE trial, the absolute change in BMI z score at week 24 was an LS mean of −0.05 (95% CI, −0.12 to 0.02).
Fifty-Two Weeks
In the SKYLINE 102 trial at 52 weeks, the LS mean change from baseline in BMI z score among patients aged 20 years or younger at baseline was 0.25 (SE = 0.10) in the VNZ–TEZ–D-IVA group (n = 27) and −0.09 (SE = 0.09) in the ELX-TEZ-IVA group (n = 32), with an LS mean difference of 0.34 (95% CI, 0.07 to 0.61).
In the SKYLINE 103 trial, the LS mean change from baseline in BMI z score among patients aged 20 years or younger at baseline was 0.11 (SE = 0.06) in the VNZ–TEZ–D-IVA group (n = 42) and −0.13 (SE = 0.07) in the ELX-TEZ-IVA group (n = 35), with an LS mean difference of 0.24 (0.06 to 0.42) at 52 weeks.
Subgroup and Sensitivity Analyses
No subgroup or sensitivity analyses were reported.
CFQ-R Respiratory Domain
Twenty-Four Weeks
In the SKYLINE 102 trial, the LS mean change in the CFQ-R respiratory domain score through week 24 was 0.5 points (SE = 1.1) in the VNZ–TEZ–D-IVA group (n = 186) and −1.7 points (SE = 1.0) in the ELX-TEZ-IVA group (n = 192). The LS mean difference was 2.3 points (95% CI, −0.6 to 5.2).
In the SKYLINE 103 trial, the LS mean change from baseline in the CFQ-R respiratory domain was −1.2 points (SE = 0.8) in the VNZ–TEZ–D-IVA group (n = 268) and the same value, i.e., −1.2 points (SE = 0.8) in the ELX-TEZ-IVA group (n = 270). The LS mean difference was −0.1 points (95% CI, −2.3 to 2.1).
Patients treated with VNZ–TEZ–D-IVA in cohort B1 of the RIDGELINE study had a within-group LS mean absolute change from baseline through week 24 of 3.9 points (95% CI, 1.5 to 6.3).
Fifty-Two Weeks
In the SKYLINE 102 trial, the LS mean change in the VNZ–TEZ–D-IVA group (n = 186) and ELX-TEZ-IVA group (n = 192) at 52 weeks was 0.8 points (SE = 1.0) and −1.6 points (SE = 1.0), respectively, with an LS mean difference of 2.4 points (95% CI, −0.3 to 5.1).
In the SKYLINE 103 trial, the LS mean change in the VNZ–TEZ–D-IVA group (n = 268) and ELX-TEZ-IVA group (n = 270) at 52 weeks was −0.4 points (SE = 0.7) and −1.0 points (SE = 0.7), respectively, with an LS mean difference of 0.7 points (95% CI, −1.4 to 2.7).
Subgroup and Sensitivity Analyses
No subgroup or sensitivity analyses were reported.
Sweat Chloride
Twenty-Four Weeks
In the SKYLINE 102 trial, the LS mean absolute change from baseline for SwCl through week 24 in the FAS was −7.5 mmol/L (SD = 0.8 mmol/L) in the VNZ–TEZ–D-IVA group (n = 196) compared with 0.9 mmol/L (SD = 0.8 mmol/L) in the ELX-TEZ-IVA group (n = 202). The MMRM analysis of this secondary end point reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment compared with ELX-TEZ-IVA treatment, with an LS mean difference of −8.4 mmol/L (95% CI, −10.5 to −6.3 mmol/L; P < 0.0001).
In the SKYLINE 103 trial, the LS mean absolute change from baseline in SwCl through week 24 in the FAS was −5.1 mmol/L (SD = 0.7 mmol/L) in the VNZ–TEZ–D-IVA group (n = 294) compared with −2.3 mmol/L (SD = 0.7 mmol/L) in the ELX-TEZ-IVA group (n = 289). The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment compared with ELX-TEZ-IVA treatment, with an LS mean difference of −2.8 mmol/L (95% CI, −4.7 mmol/L to −0.9 mmol/L; P = 0.0034).
In the single-arm study, RIDGELINE, the within-group LS mean absolute change from baseline through week 24 (averaging weeks 16 and 24) was −8.6 mmol/L (95% CI, −11.0 mmol/L to −6.3 mmol/L) in the cohort B1 FAS (n = 77).
Fifty-Two Weeks
In the SKYLINE 102 trial, the LS mean absolute change from baseline in SwCl through week 52 in the FAS was −7.5 mmol/L (SD = 0.7 mmol/L) in the VNZ–TEZ–D-IVA group (n = 196) compared with 0.5 mmol/L (SD = 0.7 mmol/L) in the ELX-TEZ-IVA group (n = 202). The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment, with an LS mean difference of −8.0 mmol/L (95% CI, −9.9 mmol/L to −6.1 mmol/L).
In the SKYLINE 103 trial, the LS mean change in the VNZ–TEZ–D-IVA group (n = 284) was −5.0 mmol/L (SD = 0.6 mmol/L) and, in the ELX-TEZ-IVA group, it was −2.2 mmol/L (SD = 0.6 mmol/L). The MMRM analysis reported a reduction (i.e., improvement) in SwCl associated with VNZ–TEZ–D-IVA treatment, with an LS mean difference of −2.8 mmol/L (95% CI, −4.6 mmol/L to −1.0 mmol/L).
Proportion of Patients With an SwCl of 30 mmol/L or Lower
The 24-week pooled outcome, including data from both the SKYLINE 102 and SKYLINE 103 studies, reported an odds ratio of 2.87 (95% CI, 2.00 to 4.12; P < 0.0001), indicating patients treated with VNZ–TEZ–D-IVA were more likely to have an SwCl of 30 mmol/L or lower at week 24 than patients treated with ELX-TEZ-IVA.
In the RIDGELINE trial, 41 out of 78 patients (52.6%) had an SwCl of 30 mmol/L or lower at week 24 (95% CI, 40.9% to 64.0%).
The 52-week result in the SKYLINE 102 trial was an odds ratio of 5.77 (95% CI, 3.33 to 9.99), and in the SKYLINE 103 trial was 1.98 (95% CI, 1.36 to 2.88). While the magnitude of difference between the treatment arms appears to vary between the 2 studies, the trend was in the same direction and aligned with the 24-week outcome results reported in these studies.
Subgroup and Sensitivity Analyses
No subgroup or sensitivity analyses were reported.
Table 17: Summary of Key Efficacy Results From the 2 RCTs, SKYLINE 102 and SKYLINE 103 (Full Analysis Sets)
Outcome | SKYLINE 102 trial | SKYLINE 103 trial | ||
|---|---|---|---|---|
VNZ–TEZ–D-IVA (N = 196) | ELX-TEZ-IVA (N = 202) | VNZ–TEZ–D-IVA (N = 284) | ELX-TEZ-IVA (N = 289) | |
Number of pulmonary exacerbations through week 52 | ||||
Total number of days (years) in the pulmonary exacerbation analysis period | 69,839 (207.9) | 71,832 (213.8) | 100,057 (297.8) | 103,480 (308.0) |
Number of patients with events, n (%) | 50 (25.5) | 60 (29.7) | 61 (21.5) | 59 (20.4) |
Total number of events | 67 | 90 | 86 | 79 |
Event rate per year | 0.32 | 0.42 | 0.29 | 0.26 |
Rate difference vs. ELX-TEZ-IVA (95% CI) | −0.10 (−0.24 to 0.04) | 0.03 (−0.07 to 0.13) | ||
Absolute change from baseline in ppFEV1 through week 52 | ||||
Number of patients contributing to the baseline evaluation | 193 | 201 | 279 | 286 |
Baseline mean, % (SD) | 67.0 (15.3) | 67.2 (14.6) | 67.2 (14.6) | 66.4 (14.9) |
Number of patients contributing to the analysis, n | 189 | 196 | 271 | 277 |
LS mean, % (SE) | 0.5 (0.3) | 0.4 (0.3) | 0.3 (0.2) | 0.0 (0.2) |
95% CI of LS mean, % | −0.1 to 1.1 | −0.3 to 1.0 | −0.2 to 0.8 | −0.5 to 0.5 |
LS mean difference, % (95% CI) | 0.1 (−0.8 to 1.0) | 0.3 (−0.4 to 1.0) | ||
Absolute change from baseline in BMI (kg/m2) at week 52 | ||||
Number of patients contributing to the baseline evaluation | 195 | 202 | 280 | 289 |
Baseline mean, kg/m2 (SD) | 22.71 (3.40) | 23.03 (3.85) | 23.27 (4.02) | 22.92 (3.27) |
Number of patients contributing to the analysis, n | 179 | 187 | 248 | 266 |
LS mean, kg/m2 (SE) | 0.30 (0.10) | 0.08 (0.10) | 0.37 (0.08) | 0.22 (0.07) |
95% CI of LS mean, kg/m2 | 0.11 to 0.49 | −0.11 to 0.27 | 0.22 to 0.52 | 0.07 to 0.36 |
LS mean difference, kg/m2 (95% CI) | 0.22 (−0.05 to 0.49) | 0.16 (−0.05 to 0.36) | ||
Absolute change from baseline in BMI z score at week 52 (patients aged ≤ 20 years at baseline) | ||||
Number of patients contributing to the baseline evaluation | 32 | 40 | 53 | 42 |
Baseline mean (SD) | −0.36 (1.09) | −0.14 (0.88) | −0.17 (0.95) | −0.30 (0.98) |
Number of patients contributing to the analysis, n | 27 | 32 | 42 | 35 |
LS mean change (SE) | 0.25 (0.10) | −0.09 (0.09) | 0.11 (0.06) | −0.13 (0.07) |
95% CI of LS mean | 0.05 to 0.45 | −0.27 to 0.09 | −0.01 to 0.23 | −0.27 to 0.01 |
LS mean difference (95% CI) | 0.34 (0.07 to 0.61) | 0.24 (0.06 to 0.42) | ||
Absolute change from baseline in CFQ-R RD score through week 52 | ||||
Number of patients contributing to the baseline evaluation | 192 | 197 | 280 | 282 |
Baseline mean (SD) | 85.8 (14.7) | 82.9 (15.7) | 85.7 (13.2) | 85.6 (13.2) |
Number of patients contributing to the analysis, n | 186 | 192 | 268 | 270 |
LS mean change (SE) | 0.8 (1.0) | −1.6 (1.0) | −0.4 (0.7) | −1.0 (0.7) |
95% CI of LS mean | −1.1 to 2.7 | −3.5 to 0.3 | −1.8 to 1.1 | −2.5 to 0.4 |
LS mean difference (95% CI) | 2.4 95% CI, −0.3 to 5.1 | 0.7 − 1.4 to 2.7 | ||
Absolute change from baseline in SwCl (mmol/L) through week 52 | ||||
Number of patients contributing to the baseline evaluation | 194 | 201 | 282 | 282 |
Baseline mean, mmol/L (SD) | 53.6 (17.0) | 54.3 (18.2) | 43.4 (18.5) | 42.1 (17.9) |
Number of patients contributing to the analysis, n | 188 | 195 | 271 | 277 |
LS mean change, mmol/L (SE) | −7.5 (0.7) | 0.5 (0.7) | −5.0 (0.6) | −2.2 (0.6) |
95% CI of LS mean, mmol/L | −8.9 to −6.2 | −0.8 to 1.8 | −6.2 to −3.7 | −3.5 to −1.0 |
LS mean difference, mmol/L (95% CI) | −8.0 (−9.9 to −6.1) | −2.8 (−4.6 to −1.0) | ||
P value | < 0.0001 | 0.0024 | ||
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FAS = full analysis set; LS = least squares; PFAS = pooled full analysis set; ppFEV1 = percent predicted forced expiratory volume in 1 second; RCT = randomized controlled trial; RD = respiratory domain; SD = standard deviation; SE = standard error; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: Analyses were based on the FAS unless noted otherwise. FAS was defined as all randomized patients who carry the intended CFTR mutation(s) and received at least 1 dose of the study drug during the treatment period. PFAS was defined as the pooled FAS from the SKYLINE 102 and SKYLINE 103 studies.
Baseline was defined as the predose day 1 value. If the predose day 1 value was missing, the most recent predose nonmissing value on or after the day −14 visit, including unscheduled visits, was used as baseline.
Sources: Clinical Study Reports.9,10
Table 18: Summary of Key Efficacy Results From the Single-Arm Study, RIDGELINE 103
Outcome | VNZ–TEZ–D-IVA (N = 78) (cohort B1 FAS) |
|---|---|
Number of pulmonary exacerbations through week 24 | |
Total number of days (years) in the pulmonary exacerbation analysis period | 13,188 (39.3) |
Number of patients with events, n (%) | 6 (7.7) |
Total number of events | 6 |
Event rate per year | 0.15 |
Absolute change from baseline in ppFEV1 through week 24 | |
Number of patients contributing to the baseline evaluation | 77 |
Baseline to mean, % (SD) | 99.7 (15.1) |
Number of patients contributing to the analysis, n | 74 |
LS mean, % (SE) | 0.0 (1.0) |
95% CI of LS mean, % | −2.0 to 1.9 |
Absolute change from baseline in BMI (kg/m2) at week 24 | |
Number of patients contributing to the baseline evaluation | 78 |
Baseline mean, kg/m2 (SD) | 16.83 (2.13) |
Number of patients contributing to the analysis, n | 78 |
LS mean, kg/m2 (SE) | 0.22 (0.08) |
95% CI of LS mean, kg/m2 | 0.05 to 0.38 |
Absolute change from baseline in BMI z score (kg/m2) at week 24 | |
Number of patients contributing to the baseline evaluation | 78 |
Baseline mean (SD) | 0.07 (0.87) |
Number of patients contributing to the analysis, n | 78 |
LS mean (SE) | −0.05 (0.03) |
95% CI of LS mean | −0.12 to 0.02 |
Absolute change in CFQ−R RD score through week 24 | |
Number of patients contributing to the baseline evaluation | 75 |
Baseline mean (SD) | 84.8 (16.1) |
Number of patients contributing to the analysis, n | 75 |
LS mean (SE) | 3.9 (1.2) |
95% CI of LS mean | 1.5 to 6.3 |
Absolute change from baseline in SwCl (mmol/L) through week 24 | |
Number of patients contributing to the baseline evaluation | 77 |
Baseline to mean, mmol/L (SD) | 40.4 (20.9) |
Number of patients contributing to the analysis, n | 77 |
LS mean, mmol/L (SE) | −8.6 (1.2) |
95% CI of LS mean, mmol/L | −11.0 to −6.3 |
BMI = body mass index; CFQ-R = Cystic Fibrosis Questionnaire-Revised; CI = confidence interval; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FAS = full analysis set; LS = least squares; PFAS = pooled full analysis set; ppFEV1 = percent predicted forced expiratory volume in 1 second; RD = respiratory domain; SD = standard deviation; SE = standard error; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Notes: Baseline was defined as the predose day 1 value. For patients who were on stable ELX-TEZ-IVA, if the predose day 1 value was missing, the most recent available predose value, including the screening assessment, was used as baseline.
Source: Clinical Study Report.11
Harms
Harms data are presented for the SKYLINE 102 and SKYLINE 103 trials in Table 19 and in Table 20 for the single-arm RIDGELINE trial.
To contextualize the safety data from both the SKYLINE 102 and SKYLINE 103 trials, it is important to note that the majority of patients had received commercial ELX-TEZ-IVA before study enrolment, with a median exposure of approximately 2 years, and all patients received ELX-TEZ-IVA for 4 weeks during the run-in period. Additionally, patients who had a prior history of intolerance to ELX-TEZ-IVA were not eligible to enrol, and patients who developed intolerance to ELX-TEZ-IVA during the run-in period were discontinued from the study. Therefore, the safety data from the ELX-TEZ-IVA group are reflective of the experience in patients who have tolerated ELX-TEZ-IVA and continued on an existing treatment regimen, and the safety data from the VNZ–TEZ–D-IVA group are reflective of patients who were switched to a new treatment regimen. Similarly, in the RIDGELINE trial, all patients either had prior experience on ELX-TEZ-IVA or participated in a run-in period in which they received ELX-TEZ-IVA and then switched to VNZ–TEZ–D-IVA during the study period.
Adverse Events
In the SKYLINE 102 and SKYLINE 103 trials, 94.4% and 96.5% of patients, respectively, who were treated with VNZ–TEZ–D-IVA experienced an AE, while 97% and 94.5% of patients, respectively, who were treated with ELX-TEZ-IVA experienced an AE. In both studies, the most common AEs were infective pulmonary exacerbations related to CF (range, 26.8% to 34.7% across the treatment groups in both studies), COVID-19 (range, 20.4% to 26.7%), cough (range, 20.3% to 23.0%), and nasopharyngitis (range, 17.3% to 23.0%).
In the RIDGELINE trial, the proportion of patients with at least 1 AE was 70.6% and 96.2% in cohorts A1 and B1, respectively.
Serious Adverse Events
In the SKYLINE 102 and SKYLINE 103 trials, among patients treated with VNZ–TEZ–D-IVA, 14.3% and 14.1%, respectively, experienced an SAE, while 20.3% and 13.8% of patients, respectively, who were treated with ELX-TEZ-IVA experienced an SAE. The proportion of patients with any SAE appears elevated in the ELX-TEZ-IVA group of the SKYLINE 102 trial relative to the VNZ–TEZ–D-IVA group, but this was not the case in the SKYLINE 103 trial, where the values were similar across the 2 treatment groups. One life-threatening AE occurred, which was an infective pulmonary exacerbation of CF in a patient in the ELX-TEZ-IVA treatment group of the SKYLINE 103 trial.
In the 2 RCTs, the most common SAE was an infective pulmonary exacerbation of CF. In the SKYLINE 102 trial, this occurred in 5.6% of patients treated with VNZ–TEZ–D-IVA and 11.4% treated with ELX-TEZ-IVA. In the SKYLINE 103 trial, this occurred in 6.3% and 4.2% of patients, respectively. Other SAEs were individually uncommon (from 0 to approximately 2% of patients for each SAE) but included influenza, hemoptysis, pneumonia, suicidal ideation, syncope, COVID-19, ALT increased, ALT or AST increased, constipation, distal intestinal obstruction syndrome, gamma-glutamyl transferase increased, cholelithiasis, and nephrolithiasis.
In the RIDGELINE trial, no patients in cohort A1 had an SAE, and 6 patients (7.7%) in cohort B1 had an SAE. In cohort B1, the SAEs included infective pulmonary exacerbations (n = 2); among these 2 patients, 1 also had an SAE of failure to thrive. Other SAEs that each occurred in 1 patient (1.3%) were adenovirus infection, constipation, pulmonary function test decreased, and cough. No life-threatening AEs occurred.
Withdrawals Due to Adverse Events
In the SKYLINE 102 trial, 2.0% of patients treated with VNZ–TEZ–D-IVA and 4.5% of patients treated with ELX-TEZ-IVA had an AE leading to treatment discontinuation. In the SKYLINE 103 trial, this occurred in 4.9% and 3.1% of patients, respectively. The AEs leading to treatment discontinuation were each individually uncommon (0% to approximately 2% of patients for each AE) and included a variety of types of AEs.
In the RIDGELINE trial, 1 patient (1.3%) had treatment interrupted due to a seizure, and 1 patient stopped treatment due to AEs of fatigue and coughing. Both patients belonged to cohort B1.
Mortality
No deaths occurred in the SKYLINE 102, SKYLINE 103, or RIDGELINE trials.
Notable Harms
Identified AEs of special interest included elevated aminotransferase levels, rash, elevated creatine kinase levels, cataracts, hypoglycemia, and neuropsychiatric events.
In the VNZ–TEZ–D-IVA and ELX-TEZ-IVA groups of the SKYLINE 102 trial, the proportion of patients with AEs of elevated aminotransferase levels was 8.2% and 6.4%, respectively. Rash occurred in 8.7% and 7.4%, elevated creatinine occurred in 9.2% and 11.9%, hypoglycemia occurred in 2.6% and 3.5%, and neuropsychiatric AEs occurred in 8.7% and 13.9%, respectively. No patients in either treatment group had AEs of █████████. No patients treated with VNZ–TEZ–D-IVA had serious ████████ ████████████, rash, creatine kinase elevation, ████████, or hypoglycemia events. Serious neuropsychiatric events occurred in 3 patients (1.5%) treated with VNZ–TEZ–D-IVA and in 2 patients (1.0%) treated with ELX-TEZ-IVA.
In the VNZ–TEZ–D-IVA and ELX-TEZ-IVA groups of the SKYLINE 103 trial, the proportion of patients with AEs of elevated aminotransferase levels was 9.5% and 7.6%, respectively. Rash occurred in 12.7% and 8.0%, elevated creatine kinase levels occurred in 8.8% and 5.9%, cataracts occurred in ████ ███ ████, hypoglycemia occurred in 1.1% and 3.8%, and neuropsychiatric AEs occurred in 13.4% and 10.7%, respectively. Serious neuropsychiatric events occurred in 1 patient (0.4%) treated with VNZ–TEZ–D-IVA and 2 patients (0.7%) treated with ELX-TEZ-IVA. Serious elevated aminotransferase–related events occurred in ███ ██████ patients treated with VNZ–TEZ–D-IVA and ████ treated with ELX-TEZ-IVA. Events of serious elevated creatine kinase occurred in 1 patient (0.4%) in the VNZ–TEZ–D-IVA group and in no patients in the ELX-TEZ-IVA group. No patients had serious rash, ████████, or hypoglycemia events.
In cohort A1 of the RIDGELINE trial, there were no events of ALT increased, AST increased, elevated creatine kinase levels, █████████, hypoglycemia, or neuropsychiatric AEs, while 3 patients (17.6%) had rash. In cohort B1, ALT increase occurred in 5.1% of patients, AST increase occurred in 2.6%, rash occurred in 5.1%, elevated creatine kinase levels occurred in 2.6%, cataracts occurred in ████, and neuropsychiatric events occurred in 5.1%; no patients in cohort B1 had hypoglycemia. No patients in either cohort had serious ████████ ████████████, rash, creatine kinase, ████████, hypoglycemia, or neuropsychiatric events.
Table 19: Summary of Harms Results in the SKYLINE 102 and SKYLINE 103 Trials (Safety Analysis Set)
Adverse event | SKYLINE 102 trial | SKYLINE 103 trial | ||
|---|---|---|---|---|
VNZ–TEZ–D-IVA N = 196 | ELX-TEZ-IVA N = 202 | VNZ–TEZ–D-IVA N = 284 | ELX-TEZ-IVA N = 289 | |
AEs occurring in ≥ 10% of patients in any treatment group by PT, n (%) | ||||
Patients with any AEs | 185 (94.4) | 196 (97.0) | 274 (96.5) | 273 (94.5) |
Infective pulmonary exacerbation of CF | 57 (29.1) | 70 (34.7) | 76 (26.8) | 88 (30.4) |
COVID-19 | 49 (25.0) | 54 (26.7) | 58 (20.4) | 73 (25.3) |
Cough | 45 (23.0) | 41 (20.3) | 63 (22.2) | 60 (20.8) |
Nasopharyngitis | 45 (23.0) | 35 (17.3) | 57 (20.1) | 60 (20.8) |
Headache | 25 (12.8) | 22 (10.9) | 51 (18.0) | 41 (14.2) |
Oropharyngeal pain | 24 (12.2) | 23 (11.4) | 45 (15.8) | 37 (12.8) |
Pyrexia | 24 (12.2) | 21 (10.4) | 28 (9.9) | 29 (10.0) |
Diarrhea | 21 (10.7) | 15 (7.4) | 37 (13.0) | 44 (15.2) |
Nasal congestion | 19 (9.7) | 24 (11.9) | 29 (10.2) | 23 (8.0) |
Blood creatine phosphokinase increased | 18 (9.2) | 24 (11.9) | 25 (8.8) | 17 (5.9) |
Sputum increased | 18 (9.2) | 21 (10.4) | 27 (9.5) | 29 (10.0) |
Upper respiratory tract infection | 17 (8.7) | 27 (13.4) | 55 (19.4) | 40 (13.8) |
Influenza | 19 (9.7) | 10 (5.0) | 33 (11.6) | 16 (5.5) |
Fatigue | 18 (9.2) | 16 (7.9) | 33 (11.6) | 30 (10.4) |
SAEs in ≥ 2 patients, n (%) | ||||
Patients with SAEs | 28 (14.3) | 41 (20.3) | 40 (14.1) | 40 (13.8) |
Infective pulmonary exacerbation of CF | 11 (5.6) | 23 (11.4) | 18 (6.3) | 12 (4.2) |
Influenza | 3 (1.5) | 1 (0.5) | 4 (1.4) | 2 (0.7) |
Hemoptysis | 2 (1.0) | 2 (1.0) | 1 (0.4) | 1 (0.3) |
Pneumonia | 2 (1.0) | 0 | 2 (0.7) | 6 (2.1) |
Suicidal ideation | 2 (1.0) | 1 (0.5) | 0 | 1 (0.3) |
Syncope | 2 (1.0) | 0 | NR | NR |
COVID-19 | 1 (0.5) | 3 (1.5) | 1 (0.4) | 1 (0.3) |
ALT increased | || | | █████ | | █████ | || |
AST increased | || | | █████ | | █████ | || |
Constipation | 0 | 2 (1.0) | 0 | 1 (0.3) |
GGT increased | 0 | 1 (0.5) | 2 (0.7) | 0 |
DIOS | 1 (0.5) | 1 (0.5) | 1 (0.4) | 2 (0.7) |
Cholelithiasis | 0 | 1 (0.5) | 0 | 2 (0.7) |
Nephrolithiasis | NR | NR | 0 | 2 (0.7) |
Patients who stopped treatment due to TEAEs n (%) | ||||
Patients with any AEs leading to treatment discontinuation | 4 (2.0) | 9 (4.5) | 14 (4.9) | 9 (3.1) |
Reasons for stopping treatment due to TEAEs that occurred in at least 3 patients in any group, n (%) | ||||
ALT increasedb | 1 (0.5) | 1 (0.5) | 6 (2.1) | 2 (0.7) |
AST increased | 1 (0.5) | 1 (0.5) | 5 (1.8) | 2 (0.7) |
Respiratory, thoracic, and mediastinal disorder | 1 (0.5) | 1 (0.5) | 3 (1.1) | 1 (0.3) |
Gastrointestinal disorder | 0 | 3 (1.5) | 0 | 1 (0.3) |
General disorders | 0 | 1 (0.5) | 3 (1.1) | 1 (0.3) |
AEs of special interest, n (%) | ||||
Elevated aminotransferase levels | 16 (8.2) | 13 (6.4) | 27 (9.5) | 22 (7.6) |
Rash | 17 (8.7) | 15 (7.4) | 36 (12.7) | 23 (8.0) |
Elevated creatine kinase levels | 18 (9.2) | 24 (11.9) | 25 (8.8) | 17 (5.9) |
Cataracts | || | || | | █████ | | █████ |
Hypoglycemia | 5 (2.6) | 7 (3.5) | 3 (1.1) | 11 (3.8) |
Neuropsychiatric | 17 (8.7) | 28 (13.9) | 38 (13.4) | 31 (10.7) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CF = cystic fibrosis; DIOS = Distal Intestinal Obstruction Syndrome; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; GGT = gamma-glutamyl transferase PT = preferred term; SAE = serious adverse event; TEAE = treatment-emergent adverse event; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Sources: Clinical Study Reports.9,10
Table 20: Summary of Harms Results in the RIDGELINE Trial (Safety Analysis Set)
AEs | Cohort A1: VNZ–TEZ–D-IVA (N = 17) | Cohort B1: VNZ–TEZ–D-IVA (N = 78) |
|---|---|---|
AEs occurring in ≥ 5% of patients (treatment period) n (%)a | ||
Patient with any AEs | 12 (70.6) | 75 (96.2) |
Cough | 3 (17.6) | 36 (46.2) |
Pyrexia | 1 (5.9) | 16 (20.5) |
Headache | NR | 14 (17.9) |
Infective PEx of CF | NR | 13 (16.7) |
Oropharyngeal pain | NR | 13 (16.7) |
Abdominal pain | 2 (11.8) | 9 (11.5) |
Nasal congestion | 1 (5.9) | 9 (11.5) |
Rhinorrhea | 1 (5.9) | 9 (11.5) |
Vomiting | 1 (5.9) | 8 (10.3) |
Fatigue | NR | 7 (9.0) |
Pharyngitis streptococcal | NR | 7 (9.0) |
Rhinitis | NR | 7 (9.0) |
Abdominal pain (upper) | NR | 6 (7.7) |
Diarrhea | NR | 6 (7.7) |
Neutrophil count decreased | NR | 6 (7.7) |
Productive cough | NR | 6 (7.7) |
Upper respiratory tract infection | NR | 6 (7.7) |
ALT increased | 0 | 4 (5.1) |
Ear infection | NR | 4 (5.1) |
Rash | 2 (11.8) | 4 (5.1) |
White blood cell count decreased | NR | 4 (5.1) |
SAEs in ≥ 2 patients, n (%) | ||
Patients with any SAEs | 0 | 6 (7.7) |
Infective PEx of CF | 0 | 2 (2.6) |
Patients who stopped treatment due to TEAEs n (%) | ||
Patients with any AEs leading to treatment discontinuation | 0 | 1 (1.3) |
Fatigue | 0 | 1 (1.3) |
Cough | 0 | 1 (1.3) |
AEs of special interest, n (%) | ||
ALT increased | 0 | 4 (5.1) |
AST increased | 0 | 2 (2.6) |
Rash | 3 (17.6) | 4 (5.1) |
Elevated creatine kinase levels | 0 | 2 (2.6) |
Cataracts | || | | █████ |
Hypoglycemia | 0 | 0 |
Neuropsychiatric | 0 | 4 (5.1) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CF = cystic fibrosis; COVID-19 = coronavirus disease 2019; NR = not reported; PEx = pulmonary exacerbation; PT = preferred term SAE = serious adverse event; TEAE = treatment-emergent adverse event; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aThe AEs displayed here are those experienced by at least 5% of the patients in cohort B1. Due to the small sample size of cohort A1, a single patient represents greater than 5% of the cohort’s population.
Source: Clinical Study Report.11
Critical Appraisal
Internal Validity
SKYLINE 102 and SKYLINE 103 Trials
The SKYLINE 102 and SKYLINE 103 trials were double-blind, parallel-group, randomized active-controlled trials. The treatments were administered in a double-blind manner, including the use of identical placebo formulations to prevent unblinding due to the twice-daily dosing schedule of ELX-TEZ-IVA versus the once-daily dosing schedule of VNZ–TEZ–D-IVA. The similarity in mechanism of action, expected efficacy, and expected AEs between the 2 therapies further reduced the likelihood of unintentional unblinding. Moreover, all patients received ELX-TEZ-IVA during the run-in period, and the majority had prior experience with ELX-TEZ-IVA or other CFTRm therapy, minimizing the potential for treatment inference based on subjective experience.
Randomization and allocation concealment were conducted in an appropriate manner, and randomization was stratified by relevant factors. Patient disposition was also generally well balanced between the arms. A few patients were randomized but not dosed: in the SKYLINE 102 trial, 4 in the VNZ–TEZ–D-IVA group and 3 in the ELX-TEZ-IVA group; in the SKYLINE 103 trial, 1 in the VNZ–TEZ–D-IVA group and in no patients in the ELX-TEZ-IVA group, indicating a low risk of bias due to postrandomization exclusions.
Discontinuation of treatment or study was under 10% in all groups across the 2 RCTs and the reasons were generally balanced. However, in the SKYLINE 102 trial, more patients discontinued treatment due to AEs in the ELX-TEZ-IVA group than the VNZ–TEZ–D-IVA group. This may just be attributable to random variation rather than a systematic issue, especially as it was not observed in the SKYLINE 103 trial.
Baseline demographic and disease-related characteristics were generally well balanced across treatment groups in both the SKYLINE 102 and SKYLINE 103 trials. Important protocol deviations were observed in 5% or fewer patients in each study and were not differentially distributed between the treatment groups or apparently driven by any systemic bias. The efficacy and safety results were generally consistent between the SKYLINE 102 and SKYLINE 103 trials, with minor differences that may be due to chance, such as the previously mentioned higher number of AE-related discontinuations in the ELX-TEZ-IVA group in the SKYLINE 102 trial.
Analyses of the efficacy outcomes of interest (which includes lung function [ppFEV1], body weight [BMI and BMI z score], HRQoL [CFQ-R respiratory domain], and SwCl at weeks 24 and 52) were conducted using MMRM using restricted maximum likelihood, and adjustment factors included fixed categorical effects for treatment, visit, age at screening (except for in the assessment of BMI z score which included only patients aged 20 years and younger), and treatment-by-visit interaction, with baseline ppFEV1 and baseline SwCl as continuous covariates. Notably, the sample size for BMI z score was very small (n = 32 to 53 in each treatment group) owing to the inclusion of only patients aged 20 years or younger, and this age category does not reflect the randomization stratification factors in the study design (which was divided by age 18 rather than 20 years), which suggests it may break randomization.
The primary end point of ppFEV1 at 24 weeks, and the key secondary end points of absolute change from baseline in SwCl through week 24, the proportion of patients with an SwCl level of 60 mmol/L or less at week 24, and the proportion of patients with an SwCl level of 30 mmol/L or less at week 24, were controlled for type I error using a hierarchical testing procedure. However, this does not appear to apply to the 52-week outcomes for the change in ppFEV1 or change in SwCl, which were the related end points that CDA-AMC assessed using GRADE, nor does it apply to any of the other outcomes assessed in this report. Nonetheless, the consistency between the 24-week and 52-week end point for the change in ppFEV1 bolsters confidence in the results.
The primary end point of change in ppFEV1 at 24 weeks (and secondary at 52 weeks) was a noninferiority assessment tested at a 1-sided alpha level of 0.025, which was met according to the prespecified noninferiority margin of −3%. The selection of a 3% noninferiority margin, based on the approach used, is consistent with FDA and European Medicines Agency guidance and has been used in previous noninferiority studies of treatments for CF.7,8 As mentioned previously, a 5% improvement in ppFEV1 has been recognized by CDA-AMC and other health technology assessment bodies and Cystic Fibrosis Canada as an MID, indicating that the 3% margin remains within the bounds of clinical relevance and supports the appropriateness of the noninferiority margin. The clinical experts consulted by CDA-AMC agreed that the noninferiority margin used in the trials was appropriate. The statistical plan for the primary end point was structured in the estimand framework, which was appropriate. The treatment policy strategy was used to handle the use of a nonstudy drug CFTRm for greater than 3 days during the run-in or treatment periods before week 24, as well as treatment discontinuation before week 24, which means that observed ppFEV1 values were used if available after the event. This approach was considered appropriate because it better reflects the effectiveness in a real-world setting where treatment discontinuation and modifying therapies are a part of practice. A sensitivity analysis was conducted for the primary end point in which intercurrent events were instead addressed using the hypothetical strategy, meaning that values were set to ‘missing’ after the intercurrent event, and the results were aligned. Only 2 intercurrent events were defined; ergo, it is hypothetically possible there could be overlooked sources of potential bias; however, the intercurrent events described were expected by CDA-AMC to be the most common and impactful events because deaths did not occur during these studies and changes in therapies other than CFTRm treatments are expected to be less impactful on the efficacy results due to the nature of the disease and the typical roster of concomitant medications used in CF. As such, this did not raise major concerns regarding increased risk of bias.
Other outcomes were assessed using 1-sided alpha levels of 0.05. The end point of pulmonary exacerbation rate did not appear to be calculated using a similar MMRM as described previously, which may suggest it was not adjusted for similar factors and may raise the risk of bias for that end point.
All missing data were assumed to be missing at random and were not imputed, and there was a generally low number of patients (less than 10%) with missing data, and this was well balanced between the treatment groups for each end point as of the 52-week outcomes in both studies. Given the expected similarity in the treatment efficacy and safety profiles, this aligned with expectations, albeit there was a slight imbalance in discontinuations due to AEs in 1 study, as previously mentioned.
The 52-week end points assessed using GRADE were not included in the hierarchical testing procedure and were not adjusted for multiplicity. This may raise concerns for an increased risk of type I error for these end points, but it was not considered to be particularly significant in the context of these noninferiority studies.
There were no concerns regarding elevated risk of bias with regard to the measurement of outcomes or selection of reported results for any of the study end points. The ppFEV1 is standardized and repeatable, and all outcomes assessed herein, other than the CFQ-R respiratory domain, are objective. The CFQ-R is a self-reported (or caregiver-reported) metric that is relatively well validated in assessing the HRQoL of patients with CF. Although these data are generated by either a child or caregiver, the double-blind randomized nature of the study mitigates the elevated risk of bias in this type of end point when comparing between treatment groups, except in the event of accidental unblinding, which was not considered to be likely in general.
Overall, the risk of bias for the SKYLINE 102 and SKYLINE 103 trials was considered low to moderate.
RIDGELINE Trial
The RIDGELINE study was a single-arm, noncomparative, nonrandomized, open-label study that assessed outcomes similar to the SKYLINE 102 and SKYLINE 103 studies in terms of within-group change from baseline after 24 weeks of therapy with VNZ–TEZ–D-IVA. There was no formal primary end point, no adjustment for multiplicity or hierarchical testing procedure, and no sensitivity or subgroup analyses were conducted. The MMRM approach was similar to that previously described, but with fixed categorical effects for genotype group and visit, and baseline SwCl as a continuous covariate. All patients either had prior treatment experience with ELX-TEZ-IVA or participated in a run-in period with it before the treatment period, during which they then received VNZ–TEZ–D-IVA. There was a very low rate of missing data (which were assumed to be missing at random and were not imputed) and a very low rate of discontinuations.
Because this is a noncomparative, nonrandomized study, it is impossible to conclude whether the observed effects can be attributable to the effects of VNZ–TEZ–D-IVA. As such, the risk of bias is high and the degree of certainty in the results in comparison with ELX-TEZ-IVA is very low. This is especially the case for the self-reported (or caregiver-reported) HRQoL metric of the CFQ-R respiratory domain.
External Validity
The SKYLINE 102 and SKYLINE 103 trials reflect the patient population aged 12 years and older with CF, while patients aged 6 to 11 years (inclusive) were recruited for cohorts A1 and B1 of the RIDGELINE study. The eligibility criteria and baseline characteristics were considered appropriate and reflective of the patient population with CF in Canada within those age groups and those expected to be eligible for treatment with VNZ–TEZ–D-IVA, based on clinical expert input. The intervention, comparator, and treatment setting were relevant to the study question and to real-world clinical practice because the active comparator, ELX-TEZ-IVA, is the standard of care for patients with CF who have eligible CF-causing genotypes and is received by the majority of patients with CF in Canada, in the setting of specialty CF clinics. Concomitant medications were evaluated by the clinical experts to be reflective of their experience in treating patients with CF in Canada, and the evaluated end points captured clinically relevant data, albeit some end points are not routinely assessed in clinical practice (e.g., CFQ-R respiratory domain and testing for SwCl on an ongoing basis after diagnosis), although they are still informative and useful in the context of a clinical trial setting for regulatory and reimbursement decision-making. The end point of SwCl is typically assessed in clinical studies of CF, but is not regularly assessed in clinical practice after diagnosis, and the importance of further reductions in SwCl with VNZ–TEZ–D-IVA relative to that seen with ELX-TEZ-IVA has not been established; moreover, the magnitude of difference in SwCl observed in these studies was not considered to be clinically important by the CF experts consulted by CDA-AMC, based on the data available to date.
Because all of the included patients had treatment experience with ELX-TEZ-IVA and/or participated in run-in periods with ELX-TEZ-IVA treatment, and patients intolerant to or ineligible for ELX-TEZ-IVA were not included, the efficacy and safety data during the treatment period of the studies reflect the experience of patients switching from 1 CFTRm to another (if assigned to VNZ–TEZ–D-IVA), or reflects the experience of patients continuing on a CFTRm therapy to which they have demonstrated tolerance (if assigned to ELX-TEZ-IVA in the 2 RCTs). This represents a gap in the generalizability of these studies because no data were captured on patients naive to CFTRm therapy who were beginning treatment with VNZ–TEZ–D-IVA. However, this is expected to be a minority of patients, as per clinical expert input. Some proportion of these patients in the real world would be those whose genotypes are rare and not captured in any phase III clinical trial of CF, which is a generalizability issue that was also present in the clinical trial program of ELX-TEZ-IVA. Data for rarer genotypes will be discussed in the next section, wherein in vitro evidence was considered in lieu of clinical data.
For RIDGELINE, the 24-week single-arm study conducted in patients aged 6 to 11 years, the generalizability of results such as ppFEV1 and weight-related outcomes may be potentially confounded by the shorter study duration due to the propensity for pediatric patients to catch seasonal illnesses.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.71,72
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention likely results in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
For single-arm trials: Although GRADE guidance is not available for noncomparative studies, the CDA-AMC review team assessed pivotal single-arm trials for study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias to present these important considerations. Because the lack of a comparator arm does not allow for a conclusion to be drawn on the effect of the intervention versus any comparator, the certainty of evidence for single-arm trials started at very low certainty, with no opportunity for rating up.
When possible, certainty is typically rated in the context of the presence of an important (nontrivial) treatment effect; if this is not possible, certainty is rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. In the context of the current assessment, the objective of the studies was to establish noninferiority, so unless signalled otherwise, the certainty was generally rated in the context of suggesting there may be little to no difference between treatments for a given outcome, where applicable. For most outcomes, the target-of-certainty assessment was therefore null. As described in Table 10, an MID of 4 points was identified for the CFQ-R respiratory domain.
Findings from the SKYLINE 102 and SKYLINE 103 trials, the RCTs that recruited patients aged 12 years or older, were considered together and summarized narratively in Table 2 because these studies were similar in populations, interventions, design, and outcome measures. The RIDGELINE trial was considered separately in Table 3, given the different patient population (aged 6 to 11 years), different end points (24 weeks rather than 52 weeks), and single-arm design.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
No long-term extension studies with results were submitted, although there are some presently ongoing for which results data are not yet available.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
The sponsor submitted indirect comparisons between VNZ–TEZ–D-IVA and best supportive care. Given the presence of direct evidence against ELX-TEZ-IVA, which is the standard of care, as previously discussed, this indirect comparison with supportive care was considered by CDA-AMC to have limited relevance; ergo, it was not summarized or critically appraised for the purpose of this review.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The sponsor submitted 4 nonclinical studies evaluating the effect of VNZ–TEZ–D-IVA on cells in an in vitro setting. For CFTR mutations with low prevalence, the submission suggested that a conventional clinical trial approach to demonstrate efficacy is not always feasible.2,12 To evaluate the potential clinical benefit of VNZ–TEZ–D-IVA in these circumstances, additional mutations were identified using 3 methods: the in vitro FRT system, the in vitro human bronchial epithelial primary cell culture model, and mechanistic data.13
The FRT assay was used to identify additional CFTR mutations for which it is predicted that VNZ–TEZ–D-IVA may offer clinical benefit. The FRT assay was developed to systematically characterize the responsiveness of individual CFTR mutations to CFTRm therapies.73 This in vitro FRT cell model has been used previously in the approval of ELX-TEZ-IVA in Canada and in other countries.
Using the FRT assay, the function of the CFTR at the cell surface was assessed in Ussing chambers, which quantify the CFTR-mediated transport in FRT cells expressing each mutant CFTR form as a fraction of the chloride transport in FRT cells expressing normal CFTR (percentage of normal). In vitro responsiveness in the FRT cell model is based on an increase of 10% or more over baseline in chloride transport as a percentage of normal CFTR chloride transport. This threshold was selected because analyses of CFTR activity and disease phenotype showed that a 10% increase in CFTR activity is typically associated with improved clinical status,56 and the clinical experts consulted by CDA-AMC concurred that this is an appropriate threshold; it was also the threshold applied in previous reviews of ELX-TEZ-IVA. The sponsor stated that this is a conservative threshold, and some mutations that did not meet this threshold were further studied.74
The N1303K CFTR mutation previously did not meet the 10% threshold for responsiveness in in vitro assessments of ELX-TEZ-IVA submitted in previous reviews, but real-world observational evidence suggested some potential for responsiveness in clinical outcomes for patients with the N1303K genotype when treated with ELX-TEZ-IVA. To further validate predicted efficacy for the N1303K CFTR mutation in the context of treatment with VNZ–TEZ–D-IVA, it was tested using in vitro human bronchial epithelial cells from 3 CF donors, 1 with a homozygous N1303K mutation, and 2 with a heterozygous N1303K and a minimal function mutation, in addition to clinical benefit demonstrated in published observational studies.
Results
Based on all data available, there were 303 CFTR mutations that were considered by the sponsor to be responsive to VNZ–TEZ–D-IVA; of these, 26 were confirmed as responsive by the clinical data from the phase III clinical studies. An overview of the study designs and key results is presented in Table 21, and a listing of all mutations considered by the sponsor to be responsive to VNZ–TEZ–D-IVA based on the in vitro assessments is presented in Table 22.
Table 21: Summary of Nonclinical Studies Submitted With Key Results
Study report number | Study name (long title) | Date | Study description | Summary of key results |
|---|---|---|---|---|
U020 | Effect of Vanzacaftor (VNZ; VX-121) in Combination With Tezacaftor (TEZ; VX-661) and Deuterated Ivacaftor (DIVA; VX-561) on Cl- Transport in Bronchial Epithelial Cells Isolated From CF Donors Heterozygous and Homozygous for the N1303K-CFTR Mutation | Date of report: November 17, 2023 Date of study: December 2023 | The effect of clinically relevant concentrations of VNZ–TEZ–D-IVA on N1303K CFTR mutation–mediated CI transport in N1303K/N1303K-HBE cells and N1303K/MF-HBE cells was measured using Ussing chamber electrophysiology techniques. CF-HBE cells were derived from the lungs of CF donors. The activity of VNZ–TEZ–D-IVA was benchmarked against that of ELX-TEZ-IVA at the clinically relevant concentrations for each component in each respective donor. | VNZ–TEZ–D-IVA caused an increase in CI transport in both types of cells. Treatment with VNZ–TEZ–D-IVA resulted in superior responses compared with treatment with TEZ-IVA and similar (or slightly improved) responses compared with ELX-TEZ-IVA in all donors tested. |
U015 | In Vitro Pharmacological Profiling of CFTR Mutations in FRT Cells Using Vanzacaftor (VNZ; VX-121), Tezacaftor (TEZ; VX-661), and Deutivacaftor (D-IVA; VX-561): Effects on CFTR Processing and Trafficking and Cl-Transport | Date of report: January 18, 2024 Date of study: April 2023 to December 2023 | CFTR mutations that produce full-length CFTR protein and meet 1 of the following criteria were selected for study:
FRT cells expressing CFTR mutations were generated and analyzed using western blot (assessing delivery of mutant CFTR protein to cell surface) and Ussing chamber (assessing function). | Based on western blot studies, most mutations responded to VNZ–TEZ–D-IVA. Based on Ussing chamber studies, VNZ–TEZ–D-IVA increased CI transport by ≥ 10 pp of normal (wild type) over baseline in F508del and 134 additional CFTR mutant forms, including 29 mutations previously shown to be responsive to ELX-TEZ-IVA but not responsive to IVA or TEZ-IVA in the FRT cell model, 27 mutations previously shown to be not responsive to ELX-TEZ-IVA in the FRT cell model, and 78 mutations not previously tested in the FRT cell model using Vertex Pharmaceuticals standard operating procedures. 26 of the 161 mutations were not responsive to VNZ–TEZ–D-IVA. |
M378 | Effect of VX-445 on Chloride Transport In Bronchial Epithelial Cells Isolated From CF Donors Homozygous or heterozygous for the F508del CFTR mutation | Date of report: May 25, 2019 Date of study: May 2016 to August 2016 | Ussing chamber electrophysiology was used to determine the potency (EC90) and efficacy (response at EC90 and clinically relevant Cave) of VNZ alone and in combination with TEZ and/or D-IVA on F508del-CFTR-mediated CI transport in primary HBE cells isolated from CF donors with either a heterozygous F508del mutation, where the mutation on the other allele was a minimal function mutation not expected to respond to a CFTRm, or with a homozygous for F508del mutation. | Under the experimental conditions tested, the triple-combination therapy increased CI transport more than dual combinations (VNZ-TEZ, VNZ-IVA, TEZ-IVA), and the potency of VNZ was improved in the presence of TEZ. The potency was similar in F/MF-HBE and F/F-HBE cells, although the concentration of VNZ required to reach the EC90 was 3- to 8-fold lower when used in triple-combination therapy than when used alone or in double-combination with TEZ or IVA. |
P289 | In Vitro Pharmacological Profiling of CFTR Mutations in FRT Cells Using VX-445, TEZ, and IVA: Effects on Processing and Trafficking, and Chloride Transport | Date of report: November 6, 2020 Date of study: July 2017 to September 2019 | A nonexhaustive set of 235 CF-causing mutations with in-silico translation consistent with production of full-length CFTR protein (e.g., missense mutations, small insertion and/or deletion mutations that are in frame). FRT cells were engineered to express 1 of the rare mutations, and control FRT cell lines were included as comparators.a Mutations causing truncated or deleted CFTR protein, or splice mutations, were excluded. Study methods used western blot (assessing delivery of mutant CFTR protein to cell surface) and Ussing chamber (assessing function). | Of the 235 CFTR mutations selected for testing, 219 (plus wild type and F508del) cell lines had the correct sequence and met the inclusion criteria for CFTR expression (others were excluded for having insufficient mRNA levels or displaying the incorrect sequence). Based on western blot, treatment with ELX-TEZ-IVA improved CFTR processing compared with vehicle for F508del and the majority of other CFTR mutations tested (n = 194 of 219). Treatment with IVA alone had little to no effect. ELX-TEZ-IVA increased CI transport in FRT cells expressing F508del to a level ≥ 10% over baseline (i.e., responsive), while the increase observed with TEZ-IVA was below 10% of normal (i.e., not responsive). The negative controls were not responsive to IVA and TEZ-IVA. In addition to F508del-CFTR, 80% of the mutant CFTR forms accepted in the study (175 of 219 mutations) were responsive to ELX-TEZ-IVA. 44 of the mutations were not responsive to any of the CFTR regimens tested using the threshold of 10% of normal CFTR function. |
CF = cystic fibrosis; CI = chloride; DIVA = deuterated ivacaftor (deutivacaftor); EC = effective concentration; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; F/F = homozygous for F508del; F/G = heterozygous for F508del and a gating mutation; F/MF = heterozygous for F508del and a minimal function mutation; F/other = heterozygous for F508del and a non-F508del mutation that does not fit into 1 of the other specified mutation categories (i.e., gating, residual function, minimal function, triple combination–responsive); FRT = Fischer rat thyroid; HBE = human bronchial epithelial; IVA = ivacaftor; LUM-IVA = lumacaftor-ivacaftor; SwCl = sweat chloride; TEZ = tezacaftor; TEZ-IVA = tezacaftor-ivacaftor; VNZ = vanzacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aFRT cells were each engineered to express 1 of the rare mutations, and the following control FRT cell lines were included as comparators for the western blot and Ussing chamber studies: untransfected (parental) FRT cells (used to establish a baseline and noise threshold for the system); wild-type (normal) CFTR (used to normalize CI transport to normal [% normal]), and F508del CFTR mutation (the most common CF-causing mutation and the subject of FDA approvals for LUM-IVA, TEZ-IVA, and ELX-TEZ-IVA therapies). The positive controls were 2 CFTR mutations (G551D and R117H) that were responsive in previous FRT studies and have been demonstrated in clinical trials to be IVA-responsive (studies 770-102, 770-103, and 770-110), and the negative controls were 3 CFTR mutations (G1061R, R1066C, and N1303K) that were not responsive to IVA or TEZ-IVA in previous in vitro studies.
Sources: Details included in the table are from the sponsor’s Summary of Clinical Evidence and Nonclinical Study Reports.75-78
Table 22: Summary of Mutations Suggested by the Sponsor as Being Responsive to VNZ–TEZ–D-IVA Based on the Sponsor’s Interpretation of the Submitted Nonclinical Studies
Mutations | ||||
|---|---|---|---|---|
ELX-TEZ-IVA label mutations (153 mutations, including F508del) — all responsive to VNZ–TEZ–D-IVA | ||||
3141del9 | E588V | H139R | P574H | S341P |
546insCTA | E822K | H199Y | Q98R | S364P |
711+3A→G | F191V | H1054D | Q237E | S492F |
2789+5G→A | F311del | H1085P | Q237H | S549N |
3272-26A→G | F311L | H1085R | Q359R | S549R |
3849+10kbC→T | F508C;S1251Na | H1375P | Q1291R | S737F |
A46D | F508del | I336K | R74Q | S912L |
A120T | F575Y | I502T | R74W | S945L |
A234D | F1016S | I601F | R74W; D1270Na | S977F |
A349V | F1052V | I618T | R74W; V201Ma | S1159F |
A455E | F1074L | I980K | R74W;V201M;D1270Na | S1159P |
A554E | F1099L | I1269N | R117C | S1251N |
A1006E | G27R | I1366N | R117G | S1255P |
A1067T | G85E | L15P | R117H | T338I |
D110E | G126D | L165S | R117L | T1036N |
D110H | G178R | L206W | R117P | V201M |
D192G | G194R | L346P | R258G | V232D |
D443Y | G194V | L453S | R334L | V456A |
D443Y; G576A; R668Ca | G314E | L967S | R334Q | V456F |
D579G | G463V | L1077P | R347H | V1153E |
D614G | G480C | L1324P | R347L | V1240G |
D924N | G551D | L1335P | R347P | W361R |
D979V | G551S | L1480P | R352Q | W1098C |
D1152H | G622D | M265R | R352W | W1282R |
D1270N | G628R | M952I | R933G | Y109N |
E56K | G970D | M952T | R1066H | Y161D |
E60K | G1061R | M1101K | R1070Q | Y161S |
E92K | G1069R | N1303K | R1070W | Y563N |
E116K | G1244E | P5L | R1283M | Y1032C |
E193K | G1249R | P67L | R1283S | — |
E474K | G1349D | P205S | S13F | — |
Additional mutations supported by in vitro FRT data (150 mutations) | ||||
Mutations previously rejected by Health Canada for ELX-TEZ-IVA; supported by clinical evidence for VNZ–TEZ–D-IVA (3 mutations) | ||||
R75Q | V562I | V754M | — | — |
CFTR mutations responsive to VNZ–TEZ–D-IVA that were not responsive to ELX-TEZ-IVA (31 mutations) | ||||
3195del6 | D513G | I506T | R1066C | V520F |
3199del6 | G149R | L102R | R1066L | Y569C |
A559T | G91R | L1065P | R1066M | Y913C |
A559V | H199R | M1101R | R516G | — |
A561E | H609R | P99L | R560S | — |
A613T | I1234Vdel6aa | Q1100P | R560T | — |
A72D | I1398S | Q452P | T604I | — |
CFTR mutations responsive to VNZ–TEZ–D-IVA that were not previously tested with ELX-TEZ-IVA (74 mutations) | ||||
1507_1515del9 | G1047R | I506L | N187K | R709Q |
2183A→G | G1123R | I556V | N418S | R75L |
A1067P | G1247R | K162E | P140S | S1045Y |
A107G | G27E | K464E | P499A | S108F |
A309D | G424S | L1011S | P750L | S1118F |
A62P | G480S | L137P | Q1313K | S1235R |
C491R | G551A | L333F | Q372H | S549I |
D1445N | G970S | L333H | Q493R | T1086I |
D565G | H620P | L441P | Q552P | T1246I |
D993Y | H620Q | L619S | R1048G | T1299I |
E116Q | H939R;H949La | M1137V | R117C;G576A;R668Ca | T351I |
E292K | I105N | M150K | R297Q | V392G |
F1107L | I125T | N1088D | R31C | V603F |
F200I | I148N | N1303I | R516S | Y301C |
F587I | I331N | N186K | R555G | — |
CFTR mutations responsive to either IVA and/or TEZ-IVA, and assumed responsive to VNZ–TEZ–D-IVA (27 mutations) | ||||
D836Y | G576A | I807M | R31L | T1053I |
G576A;R668Ca | K1060T | R553Q | V1293G | E403D |
H939R | L320V | R668C | Y1014C | I1027T |
L997F | R751L | F508C | I1139V | M152V |
R792G | G178E | I175V | R170H | S589N |
I148T | R1162L | — | — | — |
Noncanonical splice mutations (15 mutations) | ||||
1341G→A | 2789+2insA | 3849+40A→G | 4005+2T→C | 621+3A→G |
1898+3A→G | 3041-15T→G | 3849+4A→G | 5T;TG12a | 296+28A→G |
2752-26A→G | 3600G→A | 3850-3T→G | 5T;TG13a | E831X |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; FRT = Fischer rat thyroid; IVA = ivacaftor; TEZ-IVA = tezacaftor-ivacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aComplex/compound mutations in which a single allele of the CFTR gene has multiple mutations; these exist independent of the presence of mutations on the other allele.
Source: Sponsor-submitted Summary of Clinical Evidence.79
Perspective of the Clinical Experts
The clinical experts consulted for this review noted that responsiveness to VNZ–TEZ–D-IVA using the in vitro model applied by the sponsor in the clinical development program for patients with rare CFTR mutations is sufficient evidence to support initial prescribing to these patients. In addition to regulatory approval based on this information (e.g., FDA approvals), the clinical experts cited the following considerations that would support the use of VNZ–TEZ–D-IVA in clinical practice:
There is a lack of alternative treatment options for some of these patients, particularly those who are not eligible for or cannot tolerate ELX-TEZ-IVA but may potentially experience a response to VNZ–TEZ–D-IVA based on their CF-causing genotype.
The nature of CF is severe, progressive, and life-limiting.
In vitro data demonstrating activity on the CFTR channel for patients with these rare mutations support extrapolation of the clinical benefit demonstrated in studies involving more common CF-causing mutations in the CFTR gene.
There are concerns about equity for those living with CFTR mutations for which the incidence is sufficiently low to preclude the generation of robust clinical evidence. The clinical experts noted that CF with a non-F508del mutation is more likely to be diagnosed in racialized persons with CF who may already be encountering systemic disadvantages within the health care system. Based on the rarity of these mutations, it would be impossible to enrol patients with each mutation into clinical trials. The current focus on clinical trial data alone for drug approval is resulting in ethnic and racial inequity in medication access. It is well known that these patient groups are underrepresented in clinical trials for a multitude of complex reasons.
Based on prior experience with ELX-TEZ-IVA for the treatment of CF in Canada and the rest of the world, there is a mounting body of evidence from case reports, case series, and clinical experience that ELX-TEZ-IVA can have a clinically meaningful impact on patients with rare CFTR mutations shown to be responsive based on in vitro data, which supports the use of this in vitro data in this manner in the context of VNZ–TEZ–D-IVA. The clinical experts consulted by CDA-AMC noted that, in this small minority of patients, it may be appropriate to conduct repeat sweat testing to build evidence for responsiveness to CFTRm therapy, although that is not expected to be standard clinical practice for the majority of patients.
Health Canada Conclusions
The 266 CFTR-responsive mutations listed in the product monograph for Alyftrek (VNZ–TEZ–D-IVA) are provided in Table 23.
Table 23: CFTR Mutations Included in the Health Canada Indication and Listed in the Current Product Monograph as Responsive to VNZ–TEZ–D-IVAa
CFTR mutations | |||||||
|---|---|---|---|---|---|---|---|
Based on clinical datab | |||||||
A455E | G1244E | H1054D | L1077P | R1066H | S1159F | S549R | W1282R |
D1152H | G551D | I336K | L206W | R347P | S1251N | S945L | Y563N |
F508del | G85E | I502T | M1101K | R352Q | S549N | W1098C | — |
Based on in vitro datac | |||||||
1507_1515del9 | D443Y;G576A; | F587I | H1085R | L137P | Q1313K | R347L | T1086I |
2183A→G | D513G | G1047R | H1375P | L1480P | Q237E | R352W | T1246I |
3141del9 | D565G | G1061R | H139R | L15P | Q237H | R516G | T1299I |
3195del6 | D579G | G1069R | H199R | L165S | Q359R | R516S | T338I |
3199del6 | D614G | G1123R | H199Y | L333F | Q372H | R555G | T351I |
546insCTA | D924N | G1247R | H609R | L333H | Q452P | R560S | T604I |
A1006E | D979V | G1249R | H620P | L346P | Q493R | R560T | V1153E |
A1067P | D993Y | G126D | H939R;H949Ld | L441P | Q552P | R74Q | V1240G |
A1067T | E116K | G1349D | I105N | L453S | Q98R | R74W | V1293G |
A107G | E116Q | G149R | I1139V | L619S | R1048G | R74W;D1 | V201M |
A120T | E193K | G178R | I1234Vdel | M1101R | R1066C | R74W;V201M;D1270Nd | V232D |
A234D | E292K | G194R | I1269N | M1137V | R1066L | R74W;V2 | V392G |
A309D | E474K | G194V | I1366N | M150K | R1066M | R751L | V456A |
A349V | E56K | G27E | I1398S | M265R | R1070Q | R75L | V456F |
A46D | E588V | G27R | I148N | M952I | R1070W | R933G | V520F |
A554E | E60K | G314E | I331N | M952T | R117C | S1045Y | V603F |
A559T | E822K | G424S | I506L | N1088D | R117C;G576A;R668Cd | S108F | W361R |
A559V | E92K | G463V | I506T | N1303I | R117G | S1118F | Y1014C |
A561E | F1016S | G480C | I601F | N1303Ke | R117H | S1159P | Y1032C |
A613T | F1052V | G480S | I618T | N186K | R117L | S1255P | Y109N |
A62P | F1074L | G551A | I980K | N187K | R117P | S13F | Y161D |
A72D | F1099L | G551S | K1060T | P205S | R1283M | S341P | Y161S |
C491R | F1107L | G576A;R668Cd | K162E | P574H | R1283S | S364P | Y569C |
D110E | F191V | G622D | K464E | P5L | R258G | S492F | Y913C |
D110H | F200I | G628R | L1011S | P67L | R297Q | S549I | — |
D1270N | F311del | G91R | L102R | P750L | R31L | S737F | — |
D1445N | F311L | G970D | L1065P | P99L | R334L | S912L | — |
D192G | F508C;S1251Nd | G970S | L1324P | Q1100P | R334Q | S977F | — |
D443Y | F575Y | H1085P | L1335P | Q1291R | R347H | T1036N | — |
Based on extrapolationf | |||||||
1898+3A→G | 2789+5G→A | 3272-26A→G | 3849+10k | 3849+4A | 4005+2T | 5T;TG13 | 711+3A→G |
2789+2insA | 3041-15T→G | 3600G→A | 3849+40A | 3850-3T→G | 5T;TG12 | 621+3A→G | E831X |
FRT = Fischer rat thyroid; HBE = human bronchial epithelial; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aBased on the mechanism of action of VNZ–TEZ–D-IVA, CFTR mutations that do not allow for the production of CFTR protein are not expected to respond to VNZ–TEZ–D-IVA.
bClinical data obtained from the SKYLINE 102 and SKYLINE 103 trials.
cCFTR mutations were responsive to VNZ–TEZ–D-IVA or to ivacaftor and/or tezacaftor-ivacaftor in vitro in the FRT cell lines; therefore, response to VNZ–TEZ–D-IVA is expected.
dComplex or compound mutations in which a single allele of the CFTR gene has multiple mutations; these exist independent of the presence of mutations on the other allele.
eThe N1303K mutation is predicted to be responsive only by HBE assay. All other mutations predicted to be responsive with in vitro data are supported by FRT assay.
fEfficacy is extrapolated to certain noncanonical splice mutations because having clinical trials in all mutations in this subgroup is infeasible, and these mutations are not amenable to interrogation by FRT system.
Source: Product monograph for VNZ–TEZ–D-IVA.28
Health Canada excluded 37 mutations from the indication. They were excluded if they were listed as not causing CF in both the CFTR2 and CFTR-France databases or if they were listed as not causing CF in 1 database and listed as variants of unknown significance or only causing CFTR-related disease in the other database and/or other sources.
The excluded mutations are as follows, in no particular order:
D836Y
F508C
G576A
H620Q
I1027T
I125T
I148T
I556V
I807M
L967S
L997F
R1162L
R170H
R31C
R553Q
R668C
R75Q
R792G
S1235R
Y301C
2752-26A>G
1341G>A
296+28A>G
E403D
G178E
H939R
I175V
L320V
M152V
N418S
P140S
P499A
R709Q
S589N
T1053I
V562I
V754M.
Discussion
Summary of Available Evidence
The clinical evidence for VNZ–TEZ–D-IVA in patients with CF was derived from 2 double-blind phase III RCTs, SKYLINE 102 (VX21-121-102, N = 405) and SKYLINE 103 (VX21-121-103, N = 574), and cohorts A1 (N = 17) and B1 (N = 78) of the single-arm phase III study, RIDGELINE (VX21-121-105). The RCTs provided evidence in patients aged 12 years and older and had a 52-week treatment phase, while the single-arm study cohorts provided data about patients aged 6 to 11 years, inclusive, and had a 24-week treatment phase; all 3 studies included run-in periods with ELX-TEZ-IVA before the treatment phases. The studies assessed the annual rate of pulmonary exacerbations, change in ppFEV1, change in BMI and BMI z score, change in HRQoL as measured using the CFQ-R respiratory domain, change in SwCl, and safety and tolerability. In the 2 RCTs, the primary end point was the change from baseline in ppFEV1 through 24 weeks.
The baseline characteristics of patients were generally balanced between the treatment groups in the 2 RCTs, and all 3 studies were considered by the clinical experts to be applicable to the setting in Canada. The SKYLINE 102 trial recruited patients with a genotype that was heterozygous for F508del and a minimal function mutation; ergo, this genotype was represented in 100% of patients while, in the other studies, the most common genotype was homozygous for F508del (range, 47.4% to 78.2%). The majority of patients in every study were white (range, 90.7% to 97.5%), while a minority were Asian (range, 0% to 0.5%), Black or African American (range, 0% to 2.0%), or other (range, 0.% to 0.4%), or information about race was not collected due to local regulations. In all studies, most patients (range, 84.6% to 94.1%) had prior treatment experience with a CFTRm, most commonly, ELX-TEZ-IVA. The mean duration of prior ELX-TEZ-IVA use was approximately 2 years in the SKYLINE 102 and SKYLINE 103 RCTs, and approximately 1 year in cohort B1 of the RIDGELINE trial.
Nonclinical study reports were provided for 4 in vitro studies as supportive evidence to inform the potential responsiveness of CF-causing mutations to VNZ–TEZ–D-IVA. For CFTR mutations with low prevalence, a conventional clinical trial approach to demonstrate efficacy is not always feasible.2,12 To evaluate the potential clinical benefit of VNZ–TEZ–D-IVA in these circumstances, additional mutations were identified using the following:
the in vitro FRT system (with a threshold of a 10% increase in chloride transport over baseline when expressed as a percentage of normal)
the in vitro human bronchial epithelial primary cell culture model
mechanistic data.13
There is precedent for using in vitro data in the approval of other CFTRm therapies in Canada (namely, ELX-TEZ-IVA) and other countries.
Interpretation of Results
Efficacy
SKYLINE 102 and SKYLINE 103 Trials
The pivotal RCTs, SKYLINE 102 and SKYLINE 103, enrolled patients aged at least 12 years and assessed the key treatment goals of CF, including the reduction or elimination of pulmonary exacerbations, maintenance of normal lung function, maintenance of adequate nutrition, and maintenance or improvement of HRQoL.
The difference between the VNZ–TEZ–D-IVA group and the ELX-TEZ-IVA group in the annual event rate of pulmonary exacerbations studies was −0.10 (95% CI, −0.24 to 0.04) in the SKYLINE 102 trial and 0.03 (95% CI, −0.07 to 0.13). No MID was identified through the literature or clinical expert input for this end point, so the threshold used in the GRADE assessment was null. Per the GRADE assessment, there is moderate certainty based on the evidence that treatment with VNZ–TEZ–D-IVA through 52 weeks likely results in little to no difference in the annual event rate of pulmonary exacerbations compared with ELX-TEZ-IVA. The small number of events in the treatment groups reduced the certainty of the evidence.
Lung function was assessed based on the change in ppFEV1 from baseline, which is a standard, objective, and repeatable metric commonly used in studies of patients with CF. The studies formally demonstrated noninferiority between VNZ–TEZ–D-IVA and ELX-TEZ-IVA based on the primary end point of change in ppFEV1 from baseline at week 24 and the prespecified noninferiority margin of −3%. Although there is no formally established MID in the literature for ppFEV1, a 5% improvement has been previously accepted by CDA-AMC and Cystic Fibrosis Canada as a threshold for a clinically meaningful difference; for this reason, the 3% noninferiority margin was considered appropriate and conservative by the CDA-AMC review team, in consultation with clinical experts. Using the 3% prespecified noninferiority margin as the threshold for the GRADE assessment of change in ppFEV1 through 52 weeks, it was demonstrated with high certainty that treatment with VNZ–TEZ–D-IVA results in little to no difference in ppFEV1 compared with ELX-TEZ-IVA, based on a between-group difference of 0.1% (−0.8% to 1.0%) in the SKYLINE 102 trial and 0.3% (−0.4% to 1.0%) in the SKYLINE 103 trial. The results were consistent between the 24-week and 52-week end points of change in ppFEV1, as well as in the subgroup and sensitivity analyses conducted on the primary end point at 24 weeks.
In terms of body weight, no MID was identified through the literature or clinical expert input for BMI and BMI z score, so the threshold used in the GRADE assessment was null for both end points. There is moderate-certainty evidence for little to no difference in the change in BMI and BMI z score at week 52 between VNZ–TEZ–D-IVA and ELX-TEZ-IVA. The change in BMI z score was assessed in a subgroup of patients 20 years or younger; there is a concern for risk of bias due to the small sample size of the subgroup assessed (n = 72 in the SKYLINE 102 trial and n = 95 in the SKYLINE 103 trial), which was not concordant with the stratification factors applied during randomization.
HRQoL was assessed using the CFQ-R respiratory domain score, a validated disease-specific scale. Results of moderate certainty showed that VNZ–TEZ–D-IVA likely results in little to no difference in the change in CFQ-R respiratory domain score compared with ELX-TEZ-IVA. The certainty was lowered slightly by imprecision. The 95% CIs for the LS mean differences in both trials include values below the published MID threshold of 4 points for the CFQ-R respiratory domain. Although both the point estimates and the upper 95% CI for the SKYLINE 102 trial fall below the MID, the upper 95% CI of the SKYLINE 103 trial is greater than the MID. However, the outcome was rated down only 1 level rather than 2 because the observed effects are consistent with the hypothesis of noninferiority between the treatments and do not suggest meaningful harm or benefit.
Finally, CDA-AMC also assessed the reported SwCl results from the SKYLINE 102 and SKYLINE 103 trials. The generalizability and clinical relevance of the SwCl end point warrant some additional consideration. The CDA-AMC review team did not identify high-quality evidence in the literature or in the materials supporting the submission that confirms the causality and clinical utility and of measuring SwCl as a surrogate for patient-important outcomes. Several supporting materials were provided by the sponsor for the purpose of highlighting the utility of SwCl to this end. However, these materials, in line with the product monograph for VNZ–TEZ–D-IVA and previously for ELX-TEZ-IVA, highlight the utility of SwCl only in a pharmacodynamic context because it provides mechanistic information of the effect of a CFTRm on the partial restoration of CFTR protein function. SwCl is also important (in combination with other factors) in the diagnosis of CF but generally not in the ongoing treatment of CF. Neither the submitted supplementary evidence nor the VNZ–TEZ–D-IVA product monograph includes SwCl as evidence of similar or improved efficacy, nor is it included as a surrogate for patient-important outcomes such as the frequency of pulmonary exacerbations, the maintenance or improvement of lung function, healthy body weight, or HRQoL. Moreover, to the knowledge of the CDA-AMC review team, there are no data with regard to any correlation, causality, or surrogacy between the magnitude of an SwCl reduction and important long-term outcomes in patients with CF outside the timescale of a typical clinical study duration, such as downstream organ function later in life and the overall life expectancy of patients with CF.
In the SKYLINE 102 trial at 52 weeks, the LS mean difference in SwCl through 52 weeks was −8.0 mmol/L (95% CI, −9.9 mmol/L to −6.1 mmol/L), and in the SKYLINE 103 trial, it was −2.8 mmol/L (95% CI, −4.6 mmol/L to −1.0 mmol/L). The clinical significance of the absolute difference of up to approximately 8 mmol/L in favour of VNZ–TEZ–D-IVA was unclear, given the lack of an empirically validated MID and, moreover, the consulted clinical experts indicated this difference was unlikely to be clinically significant due to its objectively small magnitude. The clinical experts consulted by CDA-AMC noted that SwCl levels can be impacted by intrapatient variability in SwCl measurements and missed doses of CFTRm medication that may exceed the observed between-group differences. The experts consulted by CDA-AMC noted that, in their experience, SwCl is not strongly correlated with clinical outcomes owing to other sources of between-patient heterogeneity that more greatly impact clinical outcomes. The experts described that SwCl also may not always be an appropriate biomarker for clinical end points, depending on the specific CF-causing mutation carried by a patient, because some CFTR mutations are associated with higher or lower SwCl elevations than others and, in some cases, such as the N1303K CFTR mutation, SwCl is not expected to substantially reduce with CFTRm therapy, despite objective improvement in clinical symptoms. They also indicated there is currently no well-established MID for the end point of SwCl when comparing between groups treated with CFTRm therapies, and that it remains to be seen whether there is long-term clinical value with regard to reductions in SwCl beyond those seen with ELX-TEZ-IVA treatment. Ergo, while the result for change in SwCl is statistically significant, it was not considered to be clinically significant. This is also borne out in the observation that there was no apparent benefit in any clinical end points associated with VNZ–TEZ–D-IVA therapy for up to 1 year, despite the apparent reduction in SwCl relative to ELX-TEZ-IVA. The clinical experts consulted by CDA-AMC also stated that repeat sweat testing is not likely to be useful in a clinical context for the majority of cases, except perhaps in a limited fashion to demonstrate a mechanistic response to a CFTRm in cases of rare mutations for which there is no clinical evidence; however, it was expressed by the experts that the health care system would be burdened by an expectation to conduct repeat sweat testing in all or most patients with CF and that it would have very little if any clinical utility.
In summary, SwCl is an important biomarker in CF and has utility in providing mechanistic and pharmacodynamic information such as for diagnosis, and whether a patient is experiencing a response to CFTRm therapy or an in vitro analysis of a mutation model shows a response to such therapy (although even this is not universal), and it is included as an end point of interest in this review as a result. However, the clinical importance of SwCl reductions beyond that previously seen in studies of ELX-TEZ-IVA has not been clearly established, and there is currently no robust evidence that additional reduction in SwCl is associated with additional clinical benefits compared with ELX-TEZ-IVA, nor that further reduction in SwCl is an appropriate biomarker or specifically correlated with any patient-relevant clinical end points at this time.
The patient populations enrolled in the SKYLINE 102 and SKYLINE 103 trials were generally reflective of clinical practice in Canada, and the study design and characteristics of the patients included were considered to be generalizable based on consultation with the clinical experts. However, there is a gap in the evidence regarding patients who do not qualify for or do not tolerate treatment with ELX-TEZ-IVA because all of the patients recruited to either RCT had to both be eligible for ELX-TEZ-IVA and demonstrate tolerance to it. Most patients had recent treatment experience with ELX-TEZ-IVA, and all patients were included in an approximately 1-month run-in phase during which they received ELX-TEZ-IVA.
RIDGELINE Trial
The single-arm study, RIDGELINE, provided evidence of the safety and efficacy of VNZ–TEZ–D-IVA in patients aged 6 to 11 years, inclusive. Like the SKYLINE 102 and SKYLINE 103 studies, the RIDGELINE trial required patients to be eligible for treatment with ELX-TEZ-IVA and to demonstrate tolerance to it; patients who were not on a stable regimen of ELX-TEZ-IVA at screening participated in a run-in period during which they received it. The results of the study reported there was little to no difference in the change from baseline to week 24 for several of the key outcomes (including ppFEV1, BMI, BMI z score, and CFQ-R respiratory domain score). The annualized rate of pulmonary exacerbations was calculated based on 6 patients experiencing an event through 24 weeks of the treatment period, contributing to an annualized rate of 0.15, although no changes from baseline or comparisons with prestudy rates were provided for this end point, and the small number of events further reduces the certainty of the end point. Due to the noncomparative study design of the RIDGELINE trial, the GRADE assessment of the certainty of evidence was very low for all end points. However, in the totality of evidence, including the evidence reported in the SKYLINE 102 and SKYLINE 103 trials in patients aged 12 years and older, the shared mechanism of action with ELX-TEZ-IVA, and regulatory acceptance, there is a biologically and clinically plausible rationale for the extrapolation of the results of the SKYLINE 102 and SKYLINE 103 trials to this age group that is supported by the single-arm change-from-baseline results presented in the RIDGELINE trial. Younger patients, due to the nature of CF, tend to have more preserved lung function, which can result in less obvious “benefit” in ppFEV1 compared with patients with more accumulated CF-related lung damage — although, notably, the treatment goal in this circumstance is the maintenance of ppFEV1 rather than an improvement because it is not possible to improve ppFEV1 beyond a healthy control using a CFTRm. Nonetheless, in consultation with the clinical experts consulted by CDA-AMC, the age group of 6 to 11 years is not expected to have a different treatment response, side effects, or disease prognosis than older patients. In other words, although treatment goals may differ due to less accumulated CF-related damage at younger ages, there is no concern that the lower certainty evidence reflects a large evidence gap in the generalizability to these younger patients. Similar to the SKYLINE studies previously described, the RIDGELINE study also reported a decrease in SwCl from baseline to 24 weeks, but all of the same nuance and caveats regarding the interpretation of this outcome apply.
In summary, given the unmet need in this rare and severe disease population and the well-established precedent of ELX-TEZ-IVA’s use and profound clinical benefit in this age group, there is reasonable clinical rationale to expect the efficacy of VNZ–TEZ–D-IVA to be generally similar to ELX-TEZ-IVA in patients with CF aged 6 to 11 years for clinically important outcomes. Like with the SKYLINE studies, there is a gap in the evidence regarding patients who are not eligible for or do not tolerate treatment with ELX-TEZ-IVA; however, there is also clinical rationale to extrapolate the results to that population (as supported by the content in the Other Considerations subsection).
Harms
Based on evidence from 2 double-blind RCTs, SKYLINE 102 and SKYLINE 103, the treatment of CF with VNZ–TEZ–D-IVA in patients aged 12 years or older likely results in little to no difference in the number of patients with AEs of elevated aminotransferase levels or SAEs when compared with ELX-TEZ-IVA after 52 weeks of treatment. The side effect profile was consistent with what was expected for triple-combination CFTRm therapies, and the rate of discontinuation due to AEs was very low. No patients died during any of the included studies.
Notably, patients had to be eligible and demonstrate tolerance to treatment with ELX-TEZ-IVA at baseline. The incidence of AEs — particularly those associated with the initiation of CFTRm therapy, which tend to decline as a patient continues — may be generally underestimated (across both treatment groups) during the randomized treatment phase, which was corroborated by the clinical experts consulted by CDA-AMC, who stated that many patients will initially have side effects at treatment start-up that decline as they get used to CFTRm therapy.
Other Considerations
Based on a combination of clinical and in vitro evidence, there were 303 CFTR mutations that were considered by the sponsor to be responsive to VNZ–TEZ–D-IVA. Based on the Health Canada NOC for VNZ–TEZ–D-IVA issued on July 21, 2025, 266 CFTR mutations were approved as part of the indication, of which 23 were based on clinical data, 227 were based on in vitro data, and 16 were based on extrapolation. The remaining CFTR mutations that were submitted by the sponsor were not included in the product monograph. Mutations were excluded by Health Canada if they were listed as not causing CF in both the CFTR2 and CFTR-France databases or if they were listed as not causing CF in 1 database and listed as variants of unknown significance or only causing CFTR-related disease in the other database and/or other sources. In comparison, there are 153 CF-causing mutations (including F508del) approved for treatment with ELX-TEZ-IVA, all of which except 1 (L967S) are included in the list of 266 mutations approved for the indication of VNZ–TEZ–D-IVA.
Conclusion
Two noninferiority RCTs demonstrated with high-certainty evidence that VNZ–TEZ–D-IVA results in little to no difference in the change in lung function (measured using ppFEV1) when compared with ELX-TEZ-IVA through 52 weeks of treatment in patients aged at least 12 years with CF who had at least 1 F508del mutation or another responsive CFTR mutation. Moderate- to high-certainty evidence was also demonstrated for little to no difference between the treatment groups for outcomes related to the rate of pulmonary exacerbations, BMI, BMI z score in patients aged 20 years or younger, HRQoL measured using the CFQ-R respiratory domain, the number of patients who experienced events of elevated aminotransferase levels, and the number of patients with SAEs.
One single-arm study of VNZ–TEZ–D-IVA in patients aged 6 to 11 years with CF and ELX-TEX-IVA–responsive CFTR mutation(s) reported little change from baseline in the outcomes of ppFEV1, BMI, BMI z score, and CFQ-R respiratory domain, although the certainty of the evidence compared with ELX-TEZ-IVA or any comparator was very low owing to the noncomparative design of the study. Nonetheless, the efficacy and safety profile appeared to be consistent with that observed in the 2 RCTs and it was supportive of the extrapolation of the RCT results in this age group, in combination with the well-established precedent of ELX-TEZ-IVA treatment benefit in this age group and the shared mechanism of action between VNZ–TEZ–D-IVA and ELX-TEZ-IVA. The side effect profile was consistent with what was expected for triple-combination CFTRm therapies, and the rate of discontinuation due to AEs was very low.
There is precedent and support from the clinical experts for the practice of using in vitro studies to address gaps in the clinical study evidence related to rare CF-causing mutations for which it is infeasible to conduct standard clinical trials. Four nonclinical studies were provided evaluating the effect of VNZ–TEZ–D-IVA on cells in an in vitro setting. Based on the Health Canada NOC for VNZ–TEZ–D-IVA, 266 CFTR mutations were approved as part of the indication based on clinical data, in vitro data, and extrapolation. This represents an expansion from the 153 CFTR mutations approved for ELX-TEZ-IVA in Canada.
References
1.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis registry: 2019 annual data report. 2020. Accessed November 15, 2023. https://www.cysticfibrosis.ca/registry/2019AnnualDataReport.pdf.
2.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry: 2023 annual data report. 2024. Accessed July 6, 2025. https://cystic-fibrosis.cdn.prismic.io/cystic-fibrosis/Z9sI9jiBA97GirFZ_2023-Annual-Data-ReportWEB-2-.pdf
3.Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176(10):957-69. doi:10.1164/rccm.200705-664OC PubMed
4.Emerson J, Rosenfeld M, McNamara S, et al. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34(2):91-100. doi:10.1002/ppul.10127 PubMed
5.Cystic Fibrosis Canada. Canadian consensus statement on aerosolized antibiotic use in cystic fibrosis. 2020. Accessed November 15, 2923. https://www.cysticfibrosis.ca/uploads/About%20Us/CFC005%20AEROSOLIZED%20ANTIBIOTIC%20_englishFinal.pdf
6.Corey M, McLaughlin FJ, Williams M, et al. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol. 1988;41(6):583-91. doi:10.1016/0895-4356(88)90063-7 PubMed
7.Konstan MW, Flume PA, Kappler M, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J Cyst Fibros. 2011;10(1):54-61. doi:10.1016/j.jcf.2010.10.003 PubMed
8.Mayer-Hamblett N, Nichols DP, Odem-Davis K, et al. Evaluating the impact of stopping chronic therapies after modulator drug therapy in cystic fibrosis: the SIMPLIFY clinical trial study design. Ann Am Thorac Soc. 2021;18(8):1397-1405. doi:10.1513/AnnalsATS.202010-1336SD PubMed
9.Vertex Pharmaceuticals Incorporated. Clinical Study Report: VX20-121-102. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VNZ combination therapy in subjects with cystic fibrosis who are heterozygous for F508del and a Minimal Function mutation (F/MF) [internal sponsor's report]. April 5, 2024.
10.Vertex Pharmaceuticals Incorporated. Clinical Study Report: VX20-121-103. A phase 3, randomized, double-blind, controlled study evaluating the efficacy and safety of VNZ combination therapy in subjects with cystic fibrosis who are homozygous for F508del, Heterozygous for F508del and a Gating (F/G) or Residual Function (F/RF) mutation, or have at least 1 other triple combination responsive CFTR Mutation and no F508del mutation [internal sponsor's report]. April 5, 2024.
11.Vertex Pharmaceuticals Incorporated. Clinical Study Report: VX21-121-105. A phase 3 study evaluating the pharmacokinetics, safety, and tolerability of vanzacaftor/tezacaftor/deutivacaftor triple combination therapy in cystic fibrosis subjects 1 through 11 years of age. Final analysis for subjects 6 through 11 years of age (cohorts A1 and B1) [internal sponsor's report]. April 3, 2024.
12.European Cystic Fibrosis Society. ECFSPR patient registry 2021 annual data report [sponsor supplied reference]. 2023. https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf
13.Vertex. 2.5 clinical overview vanzacaftor/tezacaftor/d-ivacaftor (VNZ TC) for cystic fibrosis [sponsor supplied reference]. 2024.
14.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992-2001. doi:10.1056/NEJMra043184 PubMed
15.Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424-433. doi:10.1091/mbc.E14-04-0935 PubMed
16.Cogen J, Emerson J, Sanders DB, et al. Risk factors for lung function decline in a large cohort of young cystic fibrosis patients. Pediatr Pulmonol. 2015;50(8):763-70. doi:10.1002/ppul.23217 PubMed
17.Committee for Medicinal Products for Human Use. Assessment report: Kaftrio (ivacaftor/tezacaftor/elexacaftor). European Medicines Agency; 2020. Accessed 2020 Dec 17. https://www.ema.europa.eu/en/documents/assessment-report/kaftrio-epar-public-assessment-report_en.pdf
18.Liou TG, Adler FR, Fitzsimmons SC, et al. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol. 2001;153(4):345-52. doi:10.1093/aje/153.4.345 PubMed
19.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry: 2021 annual data report [sponsor supplied reference]. 2023:1-56.
20.Paranjape SM, Mogayzel PJ, Jr. Cystic fibrosis in the era of precision medicine. Paediatr Respir Rev. 2018;25:64-72. doi:10.1016/j.prrv.2017.03.001 PubMed
21.Southern KW, Castellani C, Lammertyn E, et al. Standards of care for CFTR variant-specific therapy (including modulators) for people with cystic fibrosis. J Cyst Fibros. 2022;doi:10.1016/j.jcf.2022.10.002 PubMed
22.Cystic Fibrosis Canada. Canadian clinical consensus guideline for initiation, monitoring and discontinuations of CFTR modulator therapies for patients with cystic fibrosis [sponsor supplied reference]. 2022. https://www.cysticfibrosis.ca/uploads/Consensus%20Guideline%20-%20CFTR%20Modulators%20June%202022%20(004)%20FINAL-ua.pdf
23.CADTH. Reimbursement recommendation: Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta). For the treatment of cystic fibrosis in patients aged 12 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Can J Health Technol. 2021;1(9)doi:10.51731/cjht.2021.153
24.CADTH. Reimbursement recommendation: Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta). For the treatment of cystic fibrosis in patients aged 6 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Can J Health Technol. 2022;2(7)doi:10.51731/cjht.2022.383
25.CADTH. Reimbursement recommendation: Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta). For the treatment of cystic fibrosis in patients aged 2 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Can J Health Technol. 2023;3(12):19. doi:10.51731/cjht.2023.799
26.Canada’s Drug Agency. Reimbursement recommendation: Elexacaftor-tezacaftor-ivacaftor and ivacaftor (Trikafta) for the treatment of cystic fibrosis in patients aged 2 years and older who have at least one mutation in the CFTR gene that is responsive based on clinical and/or in vitro data. Can J Health Technol. 2024;4(11)doi:10.51731/cjht.2024.101
27.Vertex Pharmaceuticals (Canada) Incorporated. Trikafta (elexacaftor/tezacaftor/ivacaftor and ivacaftor): elexacaftor 100 mg / tezacaftor 50 mg / ivacaftor 75 mg tablets and ivacaftor 150 mg tablets; elexacaftor 50 mg / tezacaftor 25 mg / ivacaftor 37.5 mg tablets and ivacaftor 75 mg tablets for oral use; elexacaftor 100 mg / tezacaftor 50 mg / ivacaftor 75 mg granules and ivacaftor 75 mg granules; elexacaftor 80 mg / tezacaftor 40 mg / ivacaftor 60 mg granules and ivacaftor 59.5 mg granules for oral use [product monograph]. May 13, 2025.
28.Vertex Pharmaceuticals (Canada) Incorporated. Alyftrek (vanzacaftor/tezacaftor/deutivacaftor): vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg film-coated tablets for oral use [product monograph]. July 21, 2025.
29.Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809-1819. doi:10.1056/NEJMoa1908639 PubMed
30.Clancy JP. Rapid therapeutic advances in CFTR modulator science. Pediatr Pulmonol. 2018;53(S3):S4-S11. doi:10.1002/ppul.24157 PubMed
31.Vertex Pharmaceuticals (Canada) Incorporated. Kalydeco (ivacaftor tablets): 150 mg, oral; ivacaftor granules 25 mg per packet, 50 mg per packet, 75 mg per packet, oral [product monograph]. May 23, 2022.
32.Vertex Pharmaceuticals (Canada) Incorporated. Orkambi (lumacaftor/ivacaftor tablets): 100 mg/125 mg, 200 mg/125 mg, lumacaftor/ivacaftor granules 100 mg/125 mg,150 mg/188 mg [product monograph]. December 11, 2018.
33.Vertex Pharmaceuticals (Canada) Incorporated. Kalydeco (ivacaftor tablets): 150 mg; ivacaftor granules 50 mg per packet, 75 mg per packetl [product monograph]. January 25, 2019.
34.Vertex. In vitro Responsive CFTR Mutations. VXMA-US-20-2000703 (v1.0) December 2020 [sponsor supplied reference]. 2020.
35.Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181s:S4-S15.e1. doi:10.1016/j.jpeds.2016.09.064
36.Cystic Fibrosis Canada. Genetic testing and sweat tests. Accessed July 4, 2025. https://cysticfibrosis.ca/resource/genetic-testing-and-sweat-tests
37.SickKids. Cystic Fibrosis. Accessed July 4, 2025. https://www.sickkids.ca/en/care-services/for-health-care-providers/lab-tests/244-Cystic-Fibrosis/
38.US CF Foundation, Johns Hopkins University, The Hospital for Sick Children. The Clinical and Functional Translation of CFTR (CFTR2) 2011.
39.Shemie G, Nguyen MT, Wallenburg J, et al. The equitable implementation of cystic fibrosis personalized medicines in Canada. J Pers Med. 2021;11(5)doi:10.3390/jpm11050382 PubMed
40.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry: 2022 annual data report [sponsor supplied reference]. 2023. https://www.cysticfibrosis.ca/our-programs/cf-registry
41.Division of Genome Diagnostics at BC Children's Hospital and BC Women Hospital. Cystic fibrosis targeted 130 variant assay. Accessed July 30, 2025. https://genebc.ca/uploads/CF%20Variant%20Lists/CWMG_EXT_0125JA3v1.3_CF_Targeted_130_variant_assay.pdf
42.Division of Genome Diagnostics at BC Children's Hospital and BC Women Hospital. Cystic fibrosis targeted expanded panel (EXP) v5.0. Accessed July 30, 2025. https://genebc.ca/uploads/CF%20Variant%20Lists/CWMG_EXT_0153JA1v1.7.1_CF_Targeted_expanded_panel.pdf
43.Division of Genome Diagnostics at BC Children's Hospital and BC Women Hospital. Cystic fibrosis comprehensive gene sequencing. Accessed July 30, 2025. https://genebc.ca/uploads/CF%20Variant%20Lists/CWMG_EXT_0153JA2v1.3_CF_Comprehensive_full_gene_sequencing.pdf
44.BC Children's Hospital & BC Women's Hospital. Molecular Genetics Laboratory. Test pricing. Accessed July 4, 2025. https://genebc.ca/uploads/CWMG_REQ_0310_Test_Pricing.pdf
45.Keating C, Yonker LM, Vermeulen F, et al. Vanzacaftor-tezacaftor-deutivacaftor versus elexacaftor-tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years and older (SKYLINE Trials VX20-121-102 and VX20-121-103): results from two randomised, active-controlled, phase 3 trials. Lancet Respir Med. 2025;13(3):256-271. doi:10.1016/s2213-2600(24)00411-9 PubMed
46.Hoppe JE, Kasi AS, Pittman JE, et al. Vanzacaftor-tezacaftor-deutivacaftor for children aged 6-11 years with cystic fibrosis (RIDGELINE Trial VX21-121-105): an analysis from a single-arm, phase 3 trial. Lancet Respir Med. 2025;13(3):244-255. doi:10.1016/s2213-2600(24)00407-7 PubMed
47.Waters V, Stanojevic S, Atenafu EG, et al. Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur Respir J. 2012;40(1):61. doi:10.1183/09031936.00159111 PubMed
48.Rubin JL, Thayer S, Watkins A, et al. Frequency and costs of pulmonary exacerbations in patients with cystic fibrosis in the United States. Curr Med Res Opin. 2017;33(4):667-674. doi:10.1080/03007995.2016.1277196 PubMed
49.de Boer K, Vandemheen KL, Tullis E, et al. Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis. Thorax. 2011;66(8):680. doi:10.1136/thx.2011.161117 PubMed
50.Stephenson AL, Tom M, Berthiaume Y, et al. A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur Respir J. 2015;45(3):670-679. doi:10.1183/09031936.00119714 PubMed
51.Cooper BG, Stocks J, Hall GL, et al. The Global Lung Function Initiative (GLI) Network: bringing the world’s respiratory reference values together. Breathe. 2017;13(3):e56-e64. doi:10.1183/20734735.012717 PubMed
52.National Institute for Health and Care Excellence. Lumacaftor-ivacaftor for treating cystic fibrosis homozygous for the F508del mutation. NICE Guideline TA398 [sponsor supplied reference]. 2016. https://www.nice.org.uk/guidance/ng78
53.Culhane S, George C, Pearo B, et al. Malnutrition in cystic fibrosis: a review. Nutr Clin Pract. 2013;28(6):676-83. doi:10.1177/0884533613507086 PubMed
54.Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22(127):58-65. doi:10.1183/09059180.00008412 PubMed
55.Nkam L, Lambert J, Latouche A, et al. A 3-year prognostic score for adults with cystic fibrosis. J Cyst Fibros. 2017;16(6):702-708. doi:10.1016/j.jcf.2017.03.004 PubMed
56.Accurso FJ, Van Goor F, Zha J, et al. Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J Cyst Fibros. 2014;13(2):139-147. PubMed
57.Zemanick ET, et al. Heterogeneity of CFTR modulator-induced sweat chloride concentrations in people with cystic fibrosis. J Cyst Fibros. 2024;23(4):676-684. PubMed
58.Zemanick ET, Taylor-Cousar J, Davies JC, et al. A phase 3 open-label study of ELX/TEZ/IVA in children 6 through 11 years of age with CF and at least one F508del allele. Am J Respir Crit Care Med. 2021;
59.Zemanick ET, Ramsey B, Sands D, et al. Sweat chloride reflects CFTR function and correlates with clinical outcomes following CFTR modulator treatment. J Cyst Fibros. 2025;doi:10.1016/j.jcf.2024.12.006 PubMed
60.Olivier M, Kavvalou A, Welsner M, et al. Real-life impact of highly effective CFTR modulator therapy in children with cystic fibrosis. Front Pharmacol. 2023;doi:10.3389/fphar.2023.1176815 PubMed
61.McKone EF, Velentgas P, Swenson AJ, et al. Association of sweat chloride concentration at time of diagnosis and CFTR genotype with mortality and cystic fibrosis phenotype. J Cyst Fibros. 2015;14(5):580-586. doi:10.1016/j.jcf.2015.01.005 PubMed
62.Quittner AL, Modi AC, Wainwright C, et al. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic pseudomonas aeruginosa airway infection. Chest. 2009;135(6):1610-1618. doi:10.1378/chest.08-1190 PubMed
63.U.S. Food and Drug Administration. Division of Risk Management Review of Patient Labelling for Cayaston (aztreonam for inhalation solution) [sponsor supplied reference]. 2010. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/050814Orig1s000Other%20R.pdf
64.Goss CH, Quittner AL. Patient-reported outcomes in cystic fibrosis. Proc Am Thorac Soc. 2007;4(4):378-386. doi:10.1513/pats.200703-039BR PubMed
65.Henry B, Aussage P, Grosskopf C, et al. Development of the Cystic Fibrosis Questionnaire (CFQ) for assessing quality of life in pediatric and adult patients. Qual Life Res. 2003;12(1):63-76. doi:10.1023/a:1022037320039 PubMed
66.Quittner AL, Buu A, Messer MA, et al. Development and validation of The Cystic Fibrosis Questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest. 2005;128(4):2347-54. doi:10.1378/chest.128.4.2347 PubMed
67.Modi AC, Quittner AL. Validation of a disease-specific measure of health-related quality of life for children with cystic fibrosis. J Pediatr Psychol. 2003;28(8):535-45. doi:10.1093/jpepsy/jsg044 PubMed
68.Schumi J, Wittes JT. Through the looking glass: understanding non-inferiority. Trials. 2011;12:106. doi:10.1186/1745-6215-12-106 PubMed
69.VanDevanter DR, Heltshe SL, Spahr J, et al. Rationalizing endpoints for prospective studies of pulmonary exacerbation treatment response in cystic fibrosis. J Cyst Fibros. 2017;16(5):607-615. doi:10.1016/j.jcf.2017.04.004 PubMed
70.Centers for Disease Control Prevention. CDC growth charts: percentile data files with LMS values [sponsor supplied reference]. https://www.cdc.gov/growthcharts/percentile_data_files.htm
71.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
72.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
73.Sheppard DN, Carson MR, Ostedgaard LS, et al. Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol. 1994;266(4 Pt 1):L405-13. doi:10.1152/ajplung.1994.266.4.L405 PubMed
74.Durmowicz AG, Lim R, Rogers H, et al. The U.S. Food and Drug Administration's experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. Ann Am Thorac Soc. 2018;15(1):1-2. doi:10.1513/AnnalsATS.201708-668PS PubMed
75.Vertex Pharmaceuticals Incorporated. Nonclinical Study Report: U020. Effect of vanzacaftor (VNZ; VX-121) in combination with tezacaftor (TEZ; VX-661) and deuterated ivacaftor (DIVA; VX-561) on Cl- transport in bronchial epithelial cells isolated from CF donors heterozygous and homozygous for the N1303K-CFTR mutation [internal sponsor's report]. December, 2023.
76.Vertex Pharmaceuticals Incorporated. Nonclinical Study Report: M378. Effect of VX-445 on chloride transport in bronchial epithelial cells isolated from CF donors homozygous or heterozygous for the F508del-CFTR mutation [internal sponsor's report]. May 25, 2019.
77.Vertex Pharmaceuticals Incorporated. Nonclinical Study Report: P289. In vitro pharmacological profiling of CFTR mutations in FRT cells using VX-445, TEZ, and IVA: effects on processing and trafficking, and chloride transport [internal sponsor's report]. November 6, 2020.
78.Vertex Pharmaceuticals Incorporated. Nonclinical Study Report: U015. In vitro pharmacological profiling of CFTR mutations in FRT cells using vanzacaftor (VNZ; VX-121), tezacaftor (TEZ; VX-661), and deutivacaftor (D-IVA; VX-561): effects on CFTR processing and trafficking and Cl- transport [internal sponsor's report]. January 18, 2024.
79.Vertex Pharmaceuticals (Canada) Incorporated. Summary of Clinical Evidence. Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) for the treatment of people with cystic fibrosis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg oral tablets. May 9, 2025.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
CF
cystic fibrosis
ELX-TEZ-IVA
elexacaftor-tezacaftor-ivacaftor and ivacaftor
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
PEx
pulmonary exacerbations
ppFEV1
percent predicted forced expiratory volume in 1 second
QALY
quality-adjusted life-year
RR
rate ratio
SwCl
sweat chloride
VNZ–TEZ–D-IVA
vanzacaftor-tezacaftor-deutivacaftor
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of vanzacaftor-tezacaftor-deutivacaftor (VNZ–TEZ–D-IVA) compared with elexacaftor-tezacaftor-ivacaftor and ivacaftor (ELX-TEZ-IVA) and best supportive care (BSC) for the treatment of cystic fibrosis (CF) in people aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the CFTR gene.
Item | Description |
|---|---|
Drug product | Vanzacaftor-tezacaftor-deutivacaftor (Alyftrek):
|
Indication | For the treatment of cystic fibrosis in patients aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene |
Submitted price | $24,696.00 per pack of 84 tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg ($294.00 per tablet) or per pack of 56 tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg ($441.00 per tablet)a |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 21, 2025 |
Reimbursement request | As per indication |
Sponsor | Vertex Pharmaceuticals (Canada) Incorporated |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
aThe sponsor has stated that vanzacaftor-tezacaftor-deutivacaftor is dispensed on a per-package basis.
Summary
VNZ–TEZ–D-IVA is available as a combination tablet for oral administration in 2 strengths (vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg; vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg). The sponsor has stated that the lower-strength tablet of VNZ–TEZ–D-IVA (vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg) is dispensed in packages of 84 tablets, while the higher-strength tablet (vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg) is available in packages of 56 tablets. The submitted price of both package sizes is $24,696.00, which equates to a price of $294.00 per lower-strength tablet (vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg) and $441.00 per higher-dose tablet (vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg). For both strengths, the annual cost of VNZ–TEZ–D-IVA is expected to be $322,151 per patient, based on the Health Canada–recommended dosage.
To inform the economic model, the sponsor used estimates of relative efficacy (change in percent predicted forced expiratory volume in 1 second [ppFEV1]) for VNZ–TEZ–D-IVA and ELX-TEZ-IVA from the SKYLINE and RIDGELINE trials. The CDA-AMC Clinical Review found that, based on observations from the head-to-head noninferiority SKYLINE trial, VNZ–TEZ–D-IVA likely results in little to no difference in the annual rate of pulmonary exacerbations (PEx) or change from baseline in ppFEV1 compared with ELX-TEZ-IVA after 52 weeks of treatment among patients aged 12 years and older. Among patients aged 6 to 11 years, the evidence is very uncertain about the efficacy of VNZ–TEZ–D-IVA compared with any other treatment because of the single-arm design of the RIDGELINE trial. For PEx, the sponsor used data from an indirect treatment comparison (ITC), the findings of which were inconsistent with the direct evidence submitted by the sponsor for PEx. This ITC was not critically appraised by CDA-AMC due to the availability of head-to-head evidence.
The results of the CDA-AMC base case suggest the following:
VNZ–TEZ–D-IVA is predicted to be associated with higher costs to the health care system than ELZ-TEZ-IVA (incremental costs = $494,835), primarily driven by increased costs associated with drug acquisition.
VNZ–TEZ–D-IVA is predicted to be associated with a gain of 0.07 life-years and a gain of 0.13 quality-adjusted life-years (QALYs) compared with ELX-TEZ-IVA.
The incremental cost-effectiveness ratio (ICER) of VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA was $3,727,216 per QALY gained in the CDA-AMC base case. More than 99% of the incremental QALYs were gained in the extrapolated period (i.e., after 52 weeks). In the absence of long-term data to support a difference in clinical outcomes, it is highly uncertain whether this difference will be realized in clinical practice. Further price reductions in addition to those presented in this report may therefore be required to achieve cost-effectiveness at a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing VNZ–TEZ–D-IVA for the indicated population will be approximately $68 million over the first 3 years of reimbursement compared with the amount currently spent on ELX-TEZ-IVA, with an estimated expenditure of $1 billion on VNZ–TEZ–D-IVA over this period. The actual budget impact of reimbursing VNZ–TEZ–D-IVA will depend on the uptake of VNZ–TEZ–D-IVA, the confidential price of ELX-TEZ-IVA, and the proportion of patients with public drug plan coverage. The magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption, given the difference between the sponsor’s estimate and the CDA-AMC estimate.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of VNZ–TEZ–D-IVA plus BSC versus ELX-TEZ-IVA plus BSC and BSC alone (referred to hereafter as VNZ–TEZ–D-IVA, ELX-TEZ-IVA, and BSC, respectively) from the perspective of a public health care payer in Canada over a lifetime horizon (70 years). The modelled population comprised patients aged 6 years and older with at least 1 F508del mutation or another responsive mutation in the CFTR gene, which is aligned with the Health Canada indication. The characteristics of patients in the model were derived by the sponsor from participants in various clinical trials (refer to Appendix 3). The sponsor’s base-case analysis included costs related to drug acquisition, disease management, PEx, lung transplant, adverse events, and monitoring. No acquisition costs were included for BSC.
In the sponsor’s pairwise analysis, VNZ–TEZ–D-IVA was associated with incremental costs of $527,982 and a gain of 1.15 incremental QALYs relative to ELX-TEZ-IVA (ICER = $457,263 per QALY gained). Compared with BSC, VNZ–TEZ–D-IVA was associated with an incremental cost of $7,852,391 and a gain of 14.42 incremental QALYs (ICER = $544,482). Of the incremental benefit compared with ELX-TEZ-IVA (1.20 incremental QALYs), approximately 98% of the benefit was predicted to be accrued after the treatment duration of the SKYLINE trials (52 weeks). In the sponsor’s sequential analysis, ELX-TEZ-IVA was extendedly dominated by BSC and VNZ–TEZ–D-IVA (ICER for VNZ–TEZ–D-IVA = $544,482 versus BSC). Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The long-term term benefits of VNZ–TEZ–D-IVA on lung function are uncertain. | The sponsor’s model predicts a gain of 1.15 QALYs for VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA, with 98% of the incremental benefit accrued after the trial duration. The sponsor assumed that VNZ–TEZ–D-IVA will be associated with a slower rate of ppFEV1 decline than ELX-TEZ-IVA, based on a reduction in SwCl levels with VNZ–TEZ–D-IVA. Head-to-head trial data submitted by the sponsor suggest there will be little to no difference in ppFEV1 with VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA with up to 52 weeks of treatment, and no long-term data were submitted by the sponsor to support a long-term difference in effectiveness. | CDA-AMC assumed that the rate of ppFEV1 decline would be equivalent for VNZ–TEZ–D-IVA and ELX-TEZ-IVA.a | CDA-AMC explored the impact of uncertainty in the long-term relative effectiveness of VNZ–TEZ–D-IVA and ELX-TEZ-IVA in scenario analyses. |
Whether VNZ–TEZ–D-IVA will be associated with a lower rate of PEx vs. ELX-TEZ-IVA is highly uncertain. | The sponsor used equations from the literature to estimate the annual rate of PEx for BSC based on patient ppFEV1 and age, and multiplied this by rate ratios for VNZ–TEZ–D-IVA and ELX-TEZ-IVA derived from sponsor-conducted ITCs. This approach predicted a lower annual PEx rate for VNZ–TEZ–D-IVA vs. ELX-TEZ-IVA, which is inconsistent with the submitted data from head-to-head clinical trials. | CDA-AMC assumed that the annual rate of PEx will be equivalent for VNZ–TEZ–D-IVA and ELX-TEZ-IVA. | No scenario analysis was conducted. |
The impact of VNZ–TEZ–D-IVA on survival is highly uncertain. | The sponsor’s model predicts an incremental gain of 0.44 LYs with VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA. Survival was not an outcome in the SKYLINE or RIDGELINE trials, and no long-term data were provided by the sponsor to support a difference in survival between treatments. | CDA-AMC was unable to directly modify the survival inputs owing to the structure of the sponsor’s model; however, the other changes implemented in the CDA-AMC base case resulted in a smaller incremental gain in LYs (0.07). | No scenario analysis was conducted. |
The inclusion of a treatment-specific utility benefit for VNZ–TEZ–D-IVA is inappropriate. | The sponsor applied treatment-specific utility benefits (in addition to health state utility values) for VNZ–TEZ–D-IVA (0.117) and ELX-TEZ-IVA (0.085), which were calculated by the sponsor by use of an ITC of CFQ-R-8D observations from the SKYLINE 102 trial (VX20-121-102). As noted in the Clinical Review, data from these trials suggest that VNZ–TEZ–D-IVA likely results in little to no difference in CFQ-R respiratory domain scores compared with ELX-TEZ-IVA. | Treatment-specific utility increments for VNZ–TEZ–D-IVA and ELX-TEZ-IVA were removed in the CDA-AMC base case. | No scenario analysis was conducted. |
Adjustment of drug costs by patient adherence underestimates acquisition costs. | The sponsor adjusted the price of VNZ–TEZ–D-IVA and ELX-TEZ-IVA by estimated adherence to treatment, assuming that patients will be equally adherent to both treatments. No data were submitted by the sponsor to support this assumption, and input from clinician and patient groups noted that adherence to VNZ–TEZ–D-IVA may be higher due to its once-daily administration. | CDA-AMC assumed 100% adherence for both VNZ–TEZ–D-IVA and ELX-TEZ-IVA in the base case. | No scenario analysis was conducted. |
The relevance of BSC as a comparator is limited. | The sponsor included BSCb as a comparator against VNZ–TEZ–D-IVA and ELX-TEZ-IVA. The clinical experts consulted by CDA-AMC indicated that BSC would be relevant as a comparator only among patients who are not eligible for ELX-TEZ-IVA based on their genetic mutation; this is estimated to be approximately 1.3% of patients with CF in clinical practice in Canada. There was no evidence submitted by the sponsor to support the comparative efficacy of VNZ–TEZ–D-IVA vs. BSC for any outcome other than PEx. | Due to uncertainty regarding the relevance of BSC as a comparator and minimal comparative efficacy data for VNZ–TEZ–D-IVA vs. BSC, BSC was removed as a comparator in the CDA-AMC reanalysis. | The cost-effectiveness of VNZ–TEZ–D-IVA vs. BSC was explored in a scenario analysis. |
Health care costs are underestimated for VNZ–TEZ–D-IVA and ELX-TEZ-IVA. | The sponsor assumed there will be no cost associated with CF-related disease management for patients receiving VNZ–TEZ–D-IVA or ELX-TEZ-IVA after the time of the expected death of a patient who receives only BSC. This underestimates costs associated with VNZ–TEZ–D-IVA and ELX-TEZ-IVA. | CDA-AMC could not address this limitation directly; however, the incremental gain in LYs between VNZ–TEZ–D-IVA and ELX-TEZ-IVA in the CDA-AMC base case are minimal (0.07). Therefore, the impact this issue has on costs in the CDA-AMC base case is small. | In the scenario analysis that compares VNZ–TEZ–D-IVA with BSC, costs to the public health care system are underestimated for VNZ–TEZ–D-IVA; as such, the ICER is likely higher than this scenario suggests. |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; CF = cystic fibrosis; CFQ-R-8D = Cystic Fibrosis Questionnaire-Revised 8 dimensions; CFTRm = CFTR modulator; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; PEx = pulmonary exacerbations; ppFEV1 = percent predicted forced expiratory volume in 1 second; QALY = quality-adjusted life-year; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
aAs noted in the Clinical Review, the surrogacy of SwCl for clinical end points has not been established and was described by the clinical experts to be unreliable and highly heterogeneous, and the reduction of SwCl observed in the studies submitted by the sponsor was not correlated with improvements in clinical end points at either 24 or 52 weeks compared with ELX-TEZ-IVA. As such, whether the observed differences in SwCl translate to a clinical benefit in lung function beyond the trial duration is highly uncertain.
bBSC was assumed by the sponsor to comprise mucolytics, inhaled and oral antibiotics, inhaled hypertonic saline, nutritional supplements, enteral tube feeding, pancreatic enzymes, antifungal drugs, corticosteroids, and physiotherapy.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
VNZ–TEZ–D-IVA is predicted to be associated with additional health care costs compared with ELX-TEZ-IVA (incremental costs = $494,835). This increase in health care spending results primarily from drug acquisition costs associated with VNZ–TEZ–D-IVA (refer to Figure 1). There are anticipated to be minimal cost differences between the 2 treatments in relation to nondrug health care costs (refer to Figure 1).
Figure 1: Impact of VNZ–TEZ–D-IVA Versus ELX-TEZ-IVA on Health Care Costs
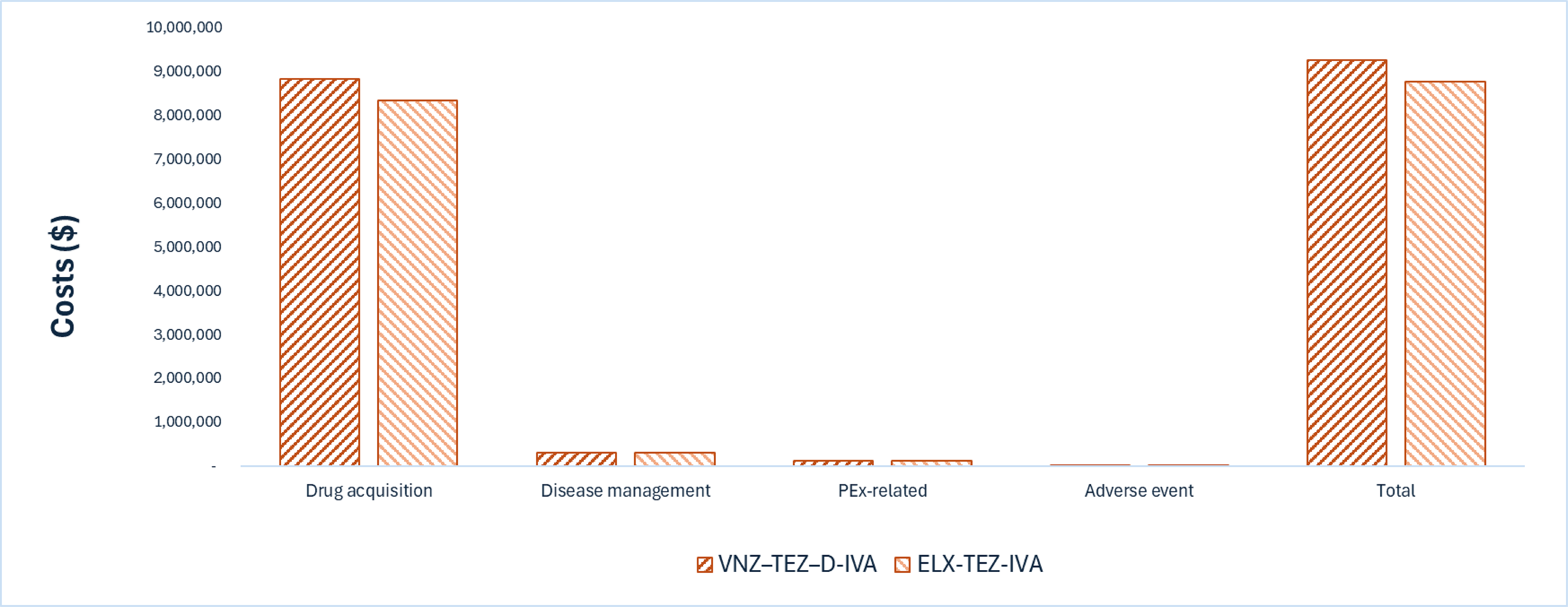
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; PEx = pulmonary exacerbation; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Impact on Health
Relative to ELX-TEZ-IVA, VNZ–TEZ–D-IVA is expected to result in similar survival outcomes (refer to Appendix 4,Table 8). Considering the impact of treatment on both quality and length of life, VNZ–TEZ–D-IVA is predicted to result in 0.13 additional QALYs per patient compared with ELX-TEZ-IVA over a lifetime horizon, primarily owing to differences in treatment discontinuation. More than 99% of the predicted incremental benefit was accrued on the basis of extrapolation.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $3,727,216 per QALY gained for VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. ELX-TEZ-IVA ($/QALY) |
|---|---|---|---|
ELX-TEZ-IVA | 8,766,740 | 21.24 | Reference |
VNZ–TEZ–D-IVA | 9,261,575 | 21.37 | 3,727,216 |
CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: Publicly available list prices were used for ELX-TEZ-IVA.
Uncertainty and Sensitivity
Uncertainty around the long-term impact of VNZ–TEZ–D-IVA on ppFEV1 relative to ELX-TEZ-IVA was explored in a scenario analysis. Based on the results of a scenario that adopted a 5% slower rate of ppFEV1 decline compared with ELX/TEZ/IVA (as assumed by the sponsor), the decision as to whether VNZ–TEZ–D-IVA is cost-effective is unlikely to be affected (ICER = $1,617,898 per QALY gained) (refer to Table 10).
The sponsor justified their its assumption of a 5% slower rate of ppFEV1 decline with VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA on the basis of sweat chloride (SwCl) findings from the SKYLINE trials. However, as noted in the Clinical Review, the surrogacy of SwCl for clinical end points has not been established, and the magnitude of reduction in SwCl was not considered to be meaningful by clinicians. Further, the reduction in SwCl observed in these studies was not correlated with significant improvement in any other outcomes during the trial period. In the absence of long-term data and established surrogacy, it is unknown whether a difference in SwCl will translate to a slower rate of ppFEV1 decline over time.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing VNZ–TEZ–D-IVA for use in the Health Canada–indicated population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of VNZ–TEZ–D-IVA was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that by year 3 of reimbursement, 2,936 patients will be eligible for VNZ–TEZ–D-IVA; of these, 2,210 patients are expected to receive VNZ–TEZ–D-IVA. The estimated incremental budget impact of reimbursing VNZ–TEZ–D-IVA is predicted to be approximately $67.9 million over the first 3 years, with an expected expenditure of $1.1 billion on VNZ–TEZ–D-IVA. The actual budget impact will depend on the uptake of VNZ–TEZ–D-IVA.
Conclusion
Based on the CDA-AMC base case, VNZ–TEZ–D-IVA would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $3,727,216 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 2; full details of the impact of price reductions on cost-effectiveness are presented in Table 9). Given that the acquisition costs of VNZ–TEZ–D-IVA and ELX-TEZ-IVA are similar (annual costs per patient of $322,151 and $306,810, respectively), a small (< 10%) price reduction is required for VNZ–TEZ–D-IVA to have a lower cost than ELX-TEZ-IVA, thus making it cost-effective at any willingness-to-pay threshold, i.e., more effective (incremental QALYs = 0.13) and less costly. Any negotiated price arrangements for ELX-TEZ-IVA will require further price reductions in addition to those presented in this report.
The difference in QALYs in the CDA-AMC base case (0.13 QALYs) was primarily due to differences in treatment discontinuation. However, findings from the submitted head-to-head trials suggest that VNZ–TEZ–D-IVA likely results in little to no difference in the annual event rate of PEx, change in ppFEV1, or health-related quality of life compared with ELX-TEZ-IVA. Therefore, the finding of any difference in QALYs between VNZ–TEZ–D-IVA versus ELX-TEZ-IVA is highly uncertain. If there are no differences in health between VNZ–TEZ–D-IVA and ELX-TEZ-IVA, then the cost of VNZ–TEZ–D-IVA should not exceed that of ELX-TEZ-IVA to ensure cost-effectiveness.
The budget impact on the public drug plans of reimbursing VNZ–TEZ–D-IVA in the first 3 years is estimated to be approximately $68 million. The 3-year expenditure on VNZ–TEZ–D-IVA (i.e., not accounting for current expenditure on comparators) is estimated to be $1.1 billion.
Figure 2: Summary of the CDA-AMC Economic Analysis and Price Reduction

CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: Expenditure includes only the drug cost of VNZ–TEZ–D-IVA. The term dominant indicates that a drug costs less and provides more QALYs than the comparator. In the CDA-AMC base case, VNZ–TEZ–D-IVA was associated with 0.13 higher QALYs versus ELX-TEZ-IVA.
Model Structure
The sponsor submitted a patient-level microsimulation model to track CF progression and treatment benefits for a typical patient profile informed by individual patient-level characteristics at baseline (Figure 3).2 In the sponsor’s probabilistic base case, 250 average patients were simulated across 600 iterations, and the expected costs and clinical effects of VNZ–TEZ–D-IVA, ELX-TEZ-IVA, and BSC alone were calculated.
At the beginning of each cycle, the model calculated a patient’s mortality risk based on a Cox proportional hazard model that linked an individual patient’s survival to 9 risk factors: age, sex, ppFEV1, annual number of PEx, Staphylococcus aureus infection, Burkholderia cepacia infection, CF-related diabetes (CFRD), weight-for-age z score, and pancreatic sufficiency status. In each cycle a patient remained alive, patient characteristics (age, ppFEV1, weight-for-age z score, PEx rate, eligibility for and occurrence of lung transplant, development of CFRD, and treatment discontinuation) were updated. Long-term health outcomes were predicted using clinical outcomes such as median predicted survival, mean time spent in ppFEV1 states, cumulative change in ppFEV1, annual and lifetime PEx rates, and proportion of patients receiving a lung transplant. Patients accrued life-years and QALYs in each treatment cycle, whereas costs were applied at the end of each run of 250 patients.
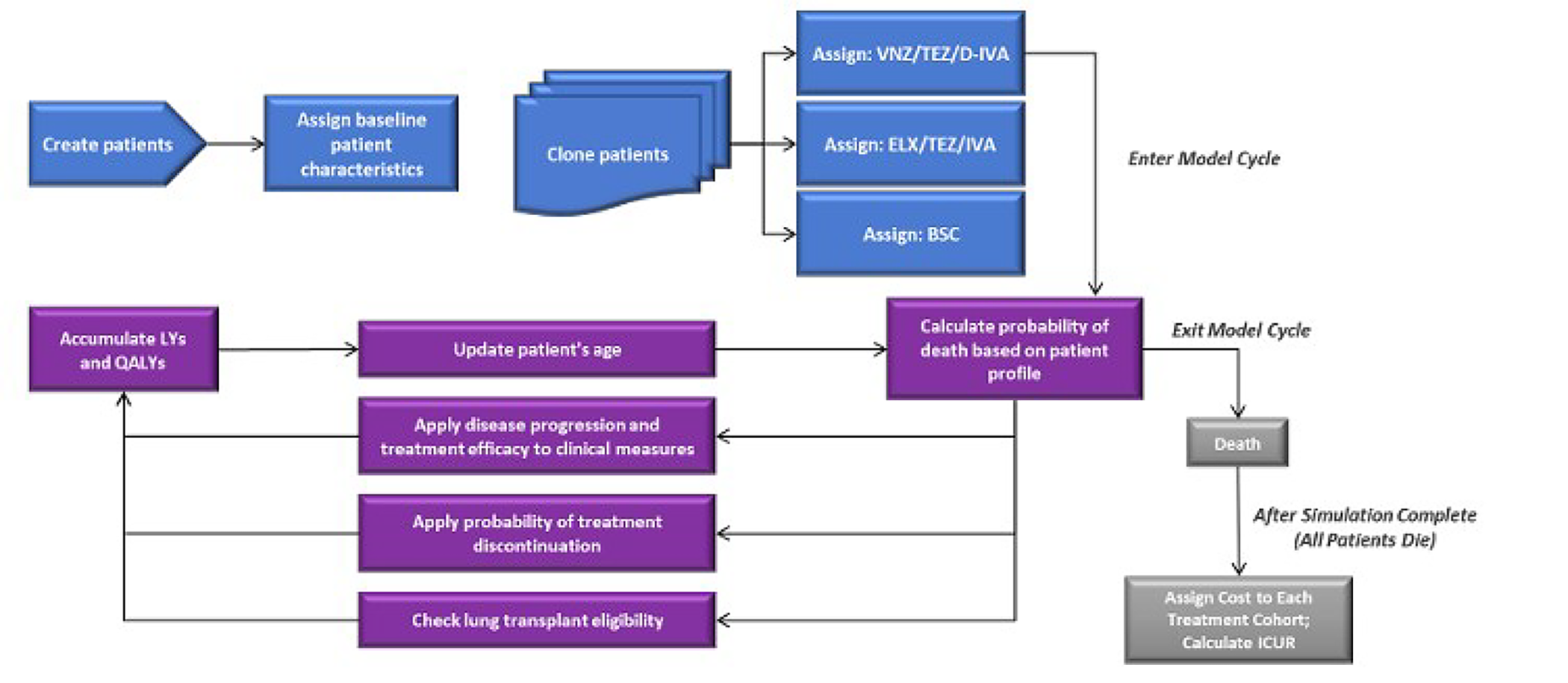
BSC = best supportive care; ELX/TEZ/IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICUR = incremental cost-utility ratio; LY = life-year; QALY = quality-adjusted life-year; VNZ/TEZ/D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Source: Sponsor’s pharmacoeconomic submission.2
References
1.Ontario Ministry of Health. Exceptional Access Program (EAP). Accessed July 9, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
2.Vertex Pharmaceuticals (Canada) Incorporated. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg oral tablets. May 9, 2025.
3.Vertex Pharmaceuticals (Canada) Incorporated. Alyftrek (vanzacaftor/tezacaftor/deutivacaftor): vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg film-coated tablets for oral use [product monograph]. July 21, 2025.
4.A phase 3 double-blind, randomized, placebo-controlled study evaluating the efficacy and safety of ELX/TEZ/IVA in cystic fibrosis subjects 6 years of age and older with a non-F508del ELX/TEZ/IVA-responsive CFTR mutation [sponsor supplied reference]. 2023.
5.Vertex Pharmaceuticals (Canada) Incorporated. DOF – HEOR – PEx Indirect Treatment Comparison of Study 121-102: VNZ/TEZ/D-IVA vs. Placebo [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg oral tablets. August 1, 2024.
6.Goss CH, Burns JL. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax. 2007;62(4):360-7. doi:10.1136/thx.2006.060889 PubMed
7.Konstan MW, Morgan WJ, Butler SM, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151(2):134-9, 139 e1. doi:10.1016/j.jpeds.2007.03.006
8.Konstan MW, Wagener JS, Vandevanter DR, et al. Risk factors for rate of decline in FEV1 in adults with cystic fibrosis. J Cyst Fibros. 2012;11(5):405-11. doi:10.1016/j.jcf.2012.03.009 PubMed
9.Whiting P, Al M, Burgers L, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014;18(18):1-106. doi:10.3310/hta18180 PubMed
10.Adler AI, Shine BS, Chamnan P, et al. Genetic determinants and epidemiology of cystic fibrosis-related diabetes: results from a British cohort of children and adults. Diabetes Care. 2008;31(9):1789-94. doi:10.2337/dc08-0466 PubMed
11.Canada’s Drug Agency. Reimbursement Review: Elexacaftor-Tezacaftor-Ivacaftor and Ivacaftor (Trikafta). Can J Health Technol. 2024;4(2)doi:10.51731/cjht.2024.826
12.Acaster S, Pinder B, Mukuria C, et al. Mapping the EQ-5D index from the cystic fibrosis questionnaire-revised using multiple modelling approaches. Health Qual Life Outcomes. 2015;13:33. doi:10.1186/s12955-015-0224-6 PubMed
13.Vertex Pharmaceuticals (Canada) Incorporated. DOF – HEOR – CFQ‐R‐8D treatment‐specific utility increment indirect treatment comparison for VNZ/TEZ/D-IVA (Study 121-102) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg oral tablets. August 1, 2024.
14.Solem C, Vera-Llonch M, Tai M, et al. Pulmonary exacerbations, lung dysfunction, and EQ-5D measures in adolescents and adults with cystic fibrosis homozygous for the F508del-CFTR mutation. Poster PRS40 presented at: ISPOR 21st International Meeting; 2016;
15.Solem CT, Vera-Llonch M, Liu S, et al. Impact of pulmonary exacerbations and lung function on generic health-related quality of life in patients with cystic fibrosis. Health Qual Life Outcomes. 2016;14(1):63. doi:10.1186/s12955-016-0465-z PubMed
16.Ontario Ministry of Health. Schedule of Benefits for Laboratory Services [sponsor supplied reference]. 2025. https://www.ontario.ca/files/2025-03/moh-ohip-schedule-of-benefits-laboratory-services-2025-03-03.pdf
17.Ontario Ministry of Health. Schedule of Benefits for Physician Services [sponsor supplied reference]. 2025. https://www.ontario.ca/files/2025-03/moh-schedule-benefit-2024-03-04.pdf
18.Vertex. Cystic Fibrosis Canada Registry Data (2014). Request for information from Vertex Pharmaceuticals. Release Date: December 2016 [sponsor supplied reference]. 2014.
19.Canadian Institute for Health Information. Patient cost estimator. Updated 2024. Accessed March 3, 2025. https://www.cihi.ca/en/patient-cost-estimator
20.Feng LB, Grosse SD, Green RF, et al. Precision medicine in action: the impact of ivacaftor on cystic fibrosis-related hospitalizations. Health Aff (Millwood). 2018;37(5):773-779. doi:10.1377/hlthaff.2017.1554 PubMed
21.Hassan M, Bonafede M, Limone B, et al. Reduction in pulmonary exacerbations (PEx) after initiation of ivacaftor: a retrospective cohort study among patients with cystic fibrosis (CF) treated in real-world settings. Journal of Cystic Fibrosis. 2016;15:S58.
22.Laflamme OD, Johnson N, Steele K, et al. Socioeconomic burden of cystic fibrosis in Canada. BMJ Open Respir Res. 2024;11(1)doi:10.1136/bmjresp-2024-002309 PubMed
23.Vasiliadis HM, Collet JP, Penrod JR, et al. A cost-effectiveness and cost-utility study of lung transplantation. J Heart Lung Transplant. 2005;24(9):1275-83. doi:10.1016/j.healun.2004.10.012 PubMed
24.Daines CL, Tullis E, Costa S, et al. Long-term safety and efficacy of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis and at least one F508del allele: 144-week interim results from a 192-week open-label extension study. Eur Respir J. 2023;62(6)doi:10.1183/13993003.02029-2022 PubMed
25.pan-Canadian Pharmaceutical Alliance. Trikafta (elexacaftor/tezacaftor/ivacaftor and ivacaftor). 2024. Accessed July 22, 2025. https://www.pcpacanada.ca/negotiation/22896
26.Vertex Pharmaceuticals (Canada) Incorporated. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Alyftrek (vanzacaftor/tezacaftor/deutivacaftor) vanzacaftor 4 mg / tezacaftor 20 mg / deutivacaftor 50 mg; vanzacaftor 10 mg / tezacaftor 50 mg / deutivacaftor 125 mg oral tablets. May 9, 2025.
27.Vertex Pharmaceuticals Incorporated. Data on file. Unpublished Number of patients with CF in Canada by age and genotype [sponsor supplied reference]. 2024.
28.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry 2012 annual data report [sponsor supplied reference]. 2014. Accessed September, 2021. https://www.cysticfibrosis.ca/our-programs/cf-registry
29.Cystic Fibrosis Canada. The Canadian Cystic Fibrosis Registry 2017 annual data report [sponsor supplied reference]. 2018. Accessed September, 2021. https://www.cysticfibrosis.ca/registry/2017AnnualDataReport.pdf
30.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed July 29, 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for CFTR Modulator Therapies for Cystic Fibrosis for Patients Aged 6 Years and Older With at Least 1 F508del Mutation or Another Responsive Mutation in the CFTR Gene
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Vanzacaftor-tezacaftor- deutivacaftor (Alyftrek) |
| Tablet | 294.0000a 441.0000a | < 40 kg: 3 tablets, each containing vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg once daily ≥ 40 kg: 2 tablets, each containing vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg once daily | 882.0000 | 322,151 |
CFTR modulator therapies | ||||||
Elexacaftor-tezacaftor-ivacaftor plus ivacaftor (Trikafta) |
| Tablet | 280.0000 | < 30 kg: 2 tablets, each containing elexacaftor 100 mg, tezacaftor 50 mg, and ivacaftor 75 mg, plus 1 tablet of ivacaftor 150 mg daily ≥ 30 kg: 2 tablets, each containing elexacaftor 50 mg, tezacaftor 25 mg, and ivacaftor 37.5 mg, plus 1 tablet of ivacaftor 75 mg daily | 840.00 | 306,810 |
Ivacaftor (Kalydeco) |
| Granules packet | 420.0000 | ≥ 14 kg to < 25 kg: 2 granules packets (ivacaftor 75 mg) daily (1 packet every 12 hours) | 840.00 | 306,810 |
150 mg | Tablet | 420.0000 | ≥ 25 kg: 2 tablets daily | 840.00 | 306,810 | |
ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: All prices are from the Ontario Drug Benefit Formulary Exceptional Access Program (accessed July 2025),1 unless otherwise indicated, and do not include dispensing fees.
aThe sponsor has stated that VNZ–TEZ–D-IVA is dispensed as packages of 84 combination tablets of vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg or as packages of 56 combination tablets of vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg. The submitted price of both package sizes is $24,696.00, which equates to a price of $294.00 per tablet of vanzacaftor 4 mg, tezacaftor 20 mg, and deutivacaftor 50 mg, and $441.00 per tablet of vanzacaftor 10 mg, tezacaftor 50 mg, and deutivacaftor 125 mg. For both strengths, the annual cost of VNZ–TEZ–D-IVA is expected to be $322,151 per patient, based on the Health Canada–recommended dosage.2
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient group input was received from Cystic Fibrosis Canada, based on data collected via focus groups including patients and caregivers in Canada. Participants included patients with and without ELX-TEZ-IVA experience. The patient group input was also informed by data collected by Cystic Fibrosis Canada in a 2021 survey of patients and caregivers (1,200 responses) on ELX-TEZ-IVA, data from the Canadian Cystic Fibrosis Registry, and findings from a burden of disease study conducted by Dalhousie University and Cystic Fibrosis Canada. Patients and caregivers reported that CF affects the physical, psychological, social, and financial aspects of their lives. Patients noted that managing CF requires a demanding treatment routine and that frequent clinic visits and hospital stays are needed to manage the progressive and debilitating symptoms of CF. Information from the CF Registry provided as part of this input suggests that, in addition to the 152 mutations covered by the Health Canada indication for ELX-TEZ-IVA, there are an additional 150 mutations for which there is currently no indicated CFTR modulator, which corresponds to approximately 36 patients in Canada without access to a CFTR modulator. Input indicated that desired goals of a new treatment include one that is better tolerated or is able to treat CF in patients with rare and ultra-rare mutations that respond minimally or not at all to ELX-TEZ-IVA or that are not indicated for ELX-TEZ-IVA.
Clinician group input was received from the Cystic Fibrosis Canada Healthcare Advisory Council. The clinician input noted that treatment for CF is lifelong and includes many nonmodulator treatments and medications, many of which are initiated at diagnosis. Clinician input noted that CFTR modulators are the first available therapies for CF that aim to correct the basic defect in the CF gene; however, they are not curative. Input noted that ELX-TEZ-IVA has been effective (e.g., reduced PEx and hospitalizations, improved and stabilized lung function, improved growth and well-being) for most patients with a single F508del mutation or other responsive mutation. Clinicians noted, however, that about 10% of patients with CF have mutations that are not currently approved for treatment with available CFTRm therapies. Clinician input noted that VNZ–TEZ–D-IVA is a once-daily treatment compared with the twice-daily regimen of ELX-TEZ-IVA, and that patients best suited for VNZ–TEZ–D-IVA would be those already receiving ELX-TEZ-IVA who have challenges with adherence on the twice-daily regimen, those with rare mutations not approved for treatment with ELX-TEZ-IVA, and those who experienced side effects with ELX-TEZ-IVA.
Input from CDA-AMC–participating drug plans noted that previous reviews have highlighted issues pertaining to sweat chloride testing, including whether it is predictive of clinically important outcomes, as well as access issues and health system capacity for testing. The plans additionally noted concerns about the anticipated budget impact of reimbursing VNZ–TEZ–D-IVA and indicated that confidential negotiated pricing exists for ELX-TEZ-IVA.
Several of these concerns were addressed in the sponsor’s model:
ELX-TEZ-IVA was included as a comparator.
VNZ–TEZ–D-IVA and ELX-TEZ-IVA were assumed to be received in addition to standard of care.
CDA-AMC was unable to address the following concerns:
Treatment switching from ELX-TEZ-IVA to VNZ–TEZ–D-IVA in the cost-utility analysis.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the Clinical Review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of VNZ–TEZ–D-IVA, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | VNZ–TEZ–D-IVA (Alyftrek), oral tablet, (4 mg/20 mg/50 mg; 10 mg/50 mg/125 mg) |
Submitted price of drug under review |
|
Regimen | |
Annual cost of drug under review |
|
Model information | |
Type of economic evaluation |
|
Treatment | VNZ–TEZ–D-IVA plus BSCa (referred to as VNZ–TEZ–D-IVA) |
Included comparators |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (70 years) |
Cycle length | Four-week cycles for the first 2 years, followed by annual cycles for the remainder of the model horizon |
Modelled population | Patients aged 6 years and older with at least 1 F508del mutation or another responsive mutation in the CFTR gene |
Characteristics of modelled population | Derived from clinical trials conducted by the sponsor for ELX-TEZ-IVA, TEZ/IVA, LUM/IVA, and IVA (ELX-TEZ-IVA: VX18-445-106, VX17-445-102, VX19-445-116, VX21-445-124; TEZ-IVA trials: VX14-661-106, VX15-661-113, VX16-661-115, VX14-661-108, VX15-661-113, VX15-661-115; LUM/IVA: VX12-809-103, VX12-809-104, VX13-809-011, VX14-809-109), IVA: VX08-770-102, VX08-770-103, VX12-770-111 and VX11-770-110) in which patients were CFTRm-naive at baseline (mean age: 29.8 years; 45.8% women, 54.2% men)2 |
Model health states | Refer to model structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs |
|
Summary of the submitted results | |
Base case results |
|
Scenario analysis results |
|
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; CF = cystic fibrosis; CFTRm = CFTR modulator; CFQ-R-8D = Cystic Fibrosis Questionnaire-Revised 8 dimensions; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in 1 second; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
aBSC was assumed by the sponsor to include mucolytics, inhaled and oral antibiotics, inhaled hypertonic saline, nutritional supplements, enteral tube feeding, pancreatic enzymes, antifungal agents, corticosteroids, and physiotherapy.
bIn sequential analyses, VNZ–TEZ–D-IVA was associated with an ICER of $544,482 vs. BSC alone. In this analysis, ELX-TEZ-IVA was extendedly dominated by BSC alone and VNZ–TEZ–D-IVA.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC Clinical Review found that, based on observations from the head-to-head noninferiority SKYLINE trial, VNZ–TEZ–D-IVA likely results in little to no difference in the annual rate of PEx or change from baseline in ppFEV1, compared with ELX-TEZ-IVA after 52 weeks of treatment among patients aged 12 years and older. Among patients aged 6 to 11 years, the evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on the rate of PEx or change in ppFEV1 compared with any other treatment because of the single-arm design of the RIDGELINE trial; however, the efficacy and safety profile appeared to be consistent with that observed in the randomized controlled trials for older patients.
To inform the economic model, the sponsor used estimates of relative efficacy (change in ppFEV1) for VNZ–TEZ–D-IVA and ELX-TEZ-IVA from the SKYLINE and RIDGELINE trials for the observed period. For PEx, the sponsor used a rate ratio (RR) for VNZ–TEZ–D-IVA and ELX-TEZ-IVA (versus BSC) from a sponsor-conducted ITC. The findings of this ITC were inconsistent with the direct evidence submitted by the sponsor for PEx. This ITC was not critically appraised by CDA-AMC, due to the availability of head-to-head evidence
In the sponsor’s economic model, treatment effect was extrapolated beyond the trial period over a 70-year lifetime time horizon, with an assumed a 5% incremental benefit in ppFEV1 for VNZ–TEZ–D-IVA versus ELX-TEZ-IVA (i.e., a 5% slower rate of ppFEV1 decline with VNZ–TEZ–D-IVA and ELX-TEZ-IVA). Of the total incremental QALYs gained over the model horizon (1.48) versus ELX-TEZ-IVA in the sponsor’s analysis, 98% were accrued in the extrapolated period. Several additional sources of uncertainty related to the clinical efficacy inputs in the economic model were noted (Refer to Key Issues of the Submitted Economic Evaluation). As such, the estimated incremental gain in life-years and QALYs predicted by the sponsor’s model for VNZ–TEZ–D-IVA versus ELX-TEZ-IVA are highly uncertain.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The long-term benefits of VNZ–TEZ–D-IVA are uncertain. In the sponsor’s base case, 98% of the incremental QALYs (1.20) for VNZ–TEZ–D-IVA versus ELX-TEZ-IVA were accrued in the extrapolated period of the model (i.e., after the trial period for which there are available data). This difference was based on the extrapolation of trial data (up to 1 year duration) over a 70-year horizon. No long-term data were submitted by the sponsor.
The sponsor’s model included the potential for treatment effectiveness waning, in that ppFEV1 will decline over time beginning after the trial duration. Relative to BSC, the sponsor assumed that treatment with either VNZ–TEZ–D-IVA or ELX-TEZ-IVA will slow the rate of ppFEV1 decline; however, a different rate of decline was modelled for each CFTR modulator. For ELX-TEZ-IVA, the model assumes that patients who receive this treatment will experience a 90% reduction in the rate of the lung function decline, based on a comparison of the rate of change in lung function for those who received ELX-TEZ-IVA in the open-label extension Study VX17-445-105 compared with untreated matched controls from the US Cystic Fibrosis Foundation Patient Registry.24 For VNZ–TEZ–D-IVA, the sponsor assumed a 95% reduction in rate of lung function decline versus BSC. As noted in the Clinical Review, results of the noninferiority SKYLINE trials suggest that VNZ–TEZ–D-IVA results in little to no difference in the change in ppFEV1 when compared with ELX-TEZ-IVA over the first 52 weeks of treatment, and no long-term evidence was submitted by the sponsor to support a difference in ppFEV1 between VNZ–TEZ–D-IVA and ELX-TEZ-IVA after 52 weeks.
The sponsor justified assuming a 5% incremental difference in rate of ppFEV1 decline between VNZ–TEZ–D-IVA and ELX-TEZ-IVA on the basis of differences in sweat chloride (SwCl) levels observed in the SKYLINE trial. However, as noted in the Clinical Review, although there was a statistically significant difference in SwCl in favour of VNZ–TEZ–D-IVA in the trial, the clinical importance of a reduction in SwCl is unclear, and the magnitude of difference between treatments was not considered to be clinically relevant. Further, surrogacy of SwCl for clinical end points has not been established and was described by clinical experts to be unreliable and highly heterogeneous, and the reduction of SwCl observed in the studies submitted by the sponsor was not correlated with improvements in clinical end points at either 24 or 52 weeks compared with ELX-TEZ-IVA.
Thus, it is highly uncertain whether there will be a difference in long-term lung function between VNZ–TEZ–D-IVA and ELX-TEZ-IVA. As such, the incremental costs and QALYs predicted in the sponsor’s base case are highly uncertain.
In the base case, CDA-AMC assumed that the rate of ppFEV1 decline would be equivalent for VNZ–TEZ–D-IVA and ELX-TEZ-IVA after the trial period. The sponsor’s assumption of a slower rate of ppFEV1 decline with VNZ–TEZ–D-IVA relative to ELX-TEZ-IVA was explored in a scenario analysis.
Whether VNZ–TEZ–D-IVA will be associated with a lower rate of PEx versus ELX-TEZ-IVA is highly uncertain. The sponsor’s model tracks PEx requiring treatment with IV antibiotics and/or hospitalizations, with the probability of PEx being dependent on a patient’s ppFEV1 and age. To inform the economic model, the sponsor used a relationship reported in the literature between ppFEV1 and age to estimate the annual rate of PEx for each patient, with separate equations used for patients aged up to 17 years and for those aged 18 years and older.6 In each model cycle, the model estimates that PEx rate for BSC alone using these equations and then multiplies the rate for BSC by RRs for VNZ–TEZ–D-IVA and ELX-TEZ-IVA derived from sponsor-conducted ITCs “to reflect the benefit of treatment on this outcome.” CDA-AMC notes that the incremental benefit in the annual PEx rate for VNZ–TEZ–D-IVA versus ELX-TEZ-IVA derived from this ITC is not supported by the submitted clinical evidence from the head-to-head SKYLINE trials. As noted in the Clinical Review, evidence from the SKYLINE trials suggests that, in patients aged 12 years or older, VNZ–TEZ–D-IVA results in little to no difference in the annual rate of PEx compared with ELX-TEZ-IVA. Additionally, the evidence is very uncertain about the effect of VNZ–TEZ–D-IVA on the rate of PEx or change in ppFEV1 compared to any other treatment among patients aged 6 to 11 years because of the single-arm design of the RIDGELINE trial.
Clinical expert input received by CDA-AMC suggests that it is plausible that there may be an impact of CFTRm treatment on PEx beyond the impact of treatment on ppFEV1; however, whether there is a difference between VNZ–TEZ–D-IVA and ELX-TEZ-IVA, the magnitude of any difference, and the duration of any benefit is highly uncertain. Because the PEx RRs derived from the sponsor’s ITC were lower for VNZ–TEZ–D-IVA (RR = 0.17) than for ELX-TEZ-IVA (RR = 0.33) despite trial evidence suggesting no difference in the annual rate of PEx, the results are likely biased in favour of VNZ–TEZ–D-IVA, especially considering that the sponsor assumed this additional benefit would be applicable over the entire model horizon (70 years). Treatment effectiveness waning for this input was not explored by the sponsor.
In the CDA-AMC base case, the RR for PEx was assumed to be equivalent for VNZ–TEZ–D-IVA and ELX-TEZ-IVA.
The impact of VNZ–TEZ–D-IVA on survival is highly uncertain. The sponsor’s base case predicts an incremental gain of 0.4 life-years with VNZ–TEZ–D-IVA compared to ELX-TEZ-IVA. Survival was not an outcome in the SKYLINE or RIDGELINE trials, and no long-term data were provided to support a difference in survival between treatments.
CDA-AMC notes that the sponsor’s model predicts a median survival of 73 to 75 years for patients who receive a CFTR modulator. Clinical experts consulted by CDA-AMC indicated that the model overestimates the survival of such patients in clinical practice in Canada. That is, experts noted that the life expectancy of patients with CF in Canadian clinical practice (the majority of whom receive a CFTR modulator) is approximately 62 years. As such, it is highly uncertain whether the sponsor’s model accurately reflects the survival of patients with CF in practice.
CDA-AMC was unable to directly modify the survival assumptions in the sponsor’s model for CFTRm therapies owing to the structure of the sponsor’s model. Although survival is likely still overestimated in the CDA-AMC base case; however, the changes in other parameters made to derive the CDA-AMC base case resulted a smaller incremental gain in life-years for VNZ–TEZ–D-IVA and ELX-TEZ-IVA, which better reflects clinical expert opinion obtained by CDA-AMC for this review.
The inclusion of a treatment-specific utility benefit for VNZ–TEZ–D-IVA is inappropriate. Health state utility values for the economic model were obtained by the sponsor from the literature (EQ-5D values) and stratified by ppFEV1 (< 40, 40 to 70,3 70). The sponsor included an additional treatment-specific utility benefit for VNZ–TEZ–D-IVA (0.117) and ELX-TEZ-IVA (0.085), which were derived from a sponsor-conducted ITC of the Cystic Fibrosis Questionnaire-Revised 8 dimensions observations from the SKYLINE 102 trial (VX20-121-102).13 The sponsor indicated that these values were included because utility values based on ppFEV1 and PEx does not “capture the extrapulmonary benefits of CFTRm therapies, including benefits to other organ systems and general improvements in functioning, well-being, and quality of life unrelated to respiratory outcomes.”2 The inclusion such treatment-specific utility benefits is contradictory to the CDA-AMC recommendation that utilities should reflect the health states in the economic model. That is, all outcomes associated with treatment, along with their impact on patient utility, should be explicitly modelled, rather than captured using a treatment-specific utility value. Including treatment-specific utility benefits that are intended to capture differences in consequences between treatments that have not been modelled is therefore inappropriate.
The treatment-specific utility benefits for VNZ–TEZ–D-IVA and ELX-TEZ-IVA were excluded from the CDA-AMC base case.
Adjustment of drug costs by patient adherence underestimates acquisition costs. In the sponsor’s base case, the sponsor adjusted the price of the VNZ–TEZ–D-IVA and ELX-TEZ-IVA to account for estimated adherence to treatment (96% in the observed period, 92% in the extrapolated period [the period for which there was no observed data]). The sponsor notes that there are currently no adherence data for patients aged 6 years or older in the target population beyond the trial period, and as such, long-term adherence was based on observations from patients aged 6 to 11 years who received lumacaftor-ivacaftor and who were enrolled in the sponsor’s Canadian Patient Support Program for more than 4 years.2 The appropriateness of this assumption is uncertain, given the input received from clinician and patient groups noting that VNZ–TEZ–D-IVA may have improved adherence due to once-daily administration. Additionally, because the drugs would be dispensed regardless of whether the patients were adherent, the public health care payer would bear the full costs of drug acquisition. This adjustment resulted in an underestimate of the total drug acquisition costs associated with CFTR modulators, biasing results in favour of VNZ–TEZ–D-IVA when compared to ELX-TEZ-IVA.
In the CDA-AMC base case, adherence was assumed to be 100% for all treatments.
The relevance of BSC as a comparator is limited. In the sponsor-submitted pharmacoeconomic analysis, BSC was included as a comparator against VNZ–TEZ–D-IVA and ELX-TEZ-IVA, with BSC was assumed to comprise mucolytics, inhaled and oral antibiotics, inhaled hypertonic saline, nutritional supplements, enteral tube feeding, pancreatic enzymes, antifungal agents, corticosteroids, and physiotherapy. In the sponsor’s analysis, no costs were associated with BSC, and usage was assumed to not vary between CFTR modulators or if used alone. Clinical expert input received by CDA-AMC for this review indicated that the proportion of people in the Health-Canada–indicated population for VNZ–TEZ–D-IVA who are not eligible for ELX-TEZ-IVA (and would thus receive BSC) is very low (< 10%). That is, BSC would only be a relevant comparator for the small proportion of patients who are currently not eligible for ELX-TEZ-IVA (i.e., currently receiving BSC alone) but who would be eligible for VNZ–TEZ–D-IVA based on the draft Health Canada indication for VNZ–TEZ–D-IVA.
Further, as noted in a prior Key Issue, there is a lack of head-to-head evidence to estimate the relative efficacy of VNZ–TEZ–D-IVA versus BSC alone. In the model, the sponsor used data from the placebo arm from the ELX-TEZ-IVA VX17-445-102 trial to inform the efficacy of BSC alone. This is inappropriate given that the VX17-445-102 trial included patients who are eligible for ELX-TEZ-IVA, and according to expert input received by CDA-AMC, these patients are highly unlikely to receive BSC alone.
Due to uncertainty regarding the relevance of BSC alone as a comparator in the pharmacoeconomic evaluation and the lack of comparative efficacy data to inform relative effects between VNZ–TEZ–D-IVA and BSC alone, BSC alone was removed as a comparator in the CDA-AMC reanalysis. BSC alone was included as a comparator in a scenario analysis.
Health care costs are underestimated for VNZ–TEZ–D-IVA and ELX-TEZ-IVA. The sponsor excluded costs associated with CF care for patients who receive either VNZ–TEZ–D-IVA or ELX-TEZ-IVA after the time of death of a similar modelled patient who received BSC alone. The sponsor justifies this exclusion by stating “Given that CF is associated with substantial disease management costs, increasing survival in CF is costly, independent of CFTRm therapy costs. As a result, counting these supportive care costs during the life-years gained by patients treated with a CFTRm makes life-extending therapies appear less cost-effective despite the extended survival.”2 Considering that the submitted economic evaluation is intended to assess the cost-effectiveness of VNZ–TEZ–D-IVA compared with ELX-TEZ-IVA and BSC from the perspective of a publicly funded health care payer, exclusion of disease management costs for periods of extended survival underestimates total costs to the public health care payer for both VNZ–TEZ–D-IVA and ELX-TEZ-IVA.
CDA-AMC could not address this limitation; however, in the CDA-AMC base case, the incremental gain in life-years between VNZ–TEZ–D-IVA and ELX-TEZ-IVA are minimal (0.07). In the scenario analysis that compares VNZ–TEZ–D-IVA with BSC, costs to the public health care system are underestimated for VNZ–TEZ–D-IVA; as such, the ICER is likely higher than this scenario suggests.
Model lacked transparency and its programming prevented CDA-AMC from fully exploring the associated uncertainties. The sponsor’s submitted model was programmed with limited transparency, with many inputs and outputs being the result of Visual Basic for Applications coding rather than formula-based operations. CDA-AMC was unable to fully explore the uncertainty with parameters in the model, although results of the deterministic stepwise analysis met face validity.
CDA-AMC was unable to address this limitation in reanalysis.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 6). The impact of these changes, individually and collectively, is presented in Table 7. Note that, in addition to the changes described in Table 6, BSC was excluded from CDA-AMC base case as noted in the Key Issues section.
Table 6: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Rate of ppFEV1 decline in the extrapolation perioda | VNZ–TEZ–D-IVA = 95.0% vs. BSC ELX-TEZ-IVA = 90.0% vs. BSC | 90.0% vs. BSC for both VNZ–TEZ–D-IVA and ELX-TEZ-IVA |
2. Rate ratio for PExb | VNZ–TEZ–D-IVA = 0.17 vs. BSC ELX-TEZ-IVA = 0.33 vs. BSC | 0.33 vs. BSC for both VNZ–TEZ–D-IVA and ELX-TEZ-IVA |
3. Treatment-specific utility benefit | Included (0.117 for VNZ–TEZ–D-IVA; 0.085 for ELX-TEZ-IVA) | Treatment-specific utility benefits were excluded |
4. Treatment adherence | Observed period: 96% Extrapolation period: 92% | 100% |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 + 3 + 4 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in 1 second; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: CDA-AMC was unable to resolve the issues with the model inflexibility and exclusion of CF-related resource utilization after the death of a similar BSC patient.
aThe observed period reflects the trial duration (24 weeks for patients aged 6 to 11 years in the VX21-121-105 trial; 52 weeks for patients3 aged 12 years in the VX20-121-102 and VX20-121-103 trial), while the extrapolation period reflects the remainder of the model horizon beyond the trial durations.
bApplied in the model for patients aged 12 years and older, consistent with the sponsor’s submission.
Table 7: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | ELX-TEZ-IVA | 8,080,740 | 23.7 | Reference |
VNZ–TEZ–D-IVA | 8,679,558 | 25.2 | 403,444 | |
Sponsor’s base case (probabilistic) | ELX-TEZ-IVA | 8,112,327 | 23.5 | Reference |
VNZ–TEZ–D-IVA | 8,640,309 | 24.6 | 457,263 | |
CDA-AMC reanalysis 1 | ELX-TEZ-IVA | 8,080,740 | 23.7 | Reference |
VNZ–TEZ–D-IVA | 8,587,984 | 24.9 | 428,637 | |
CDA-AMC reanalysis 2 | ELX-TEZ-IVA | 8,080,740 | 23.7 | Reference |
VNZ–TEZ–D-IVA | 8,657,864 | 25.1 | 421,357 | |
CDA-AMC reanalysis 3 | ELX-TEZ-IVA | 8,080,740 | 21.4 | Reference |
VNZ–TEZ–D-IVA | 8,679,558 | 21.9 | 1,148,002 | |
CDA-AMC reanalysis 4 | ELX-TEZ-IVA | 8,735,423 | 23.7 | Reference |
VNZ–TEZ–D-IVA | 9,387,365 | 25.2 | 439,235 | |
CDA-AMC base case: (Reanalysis 1 + 2 + 3 + 4) (deterministic) | ELX-TEZ-IVA | 8,735,423 | 21.4 | Reference |
VNZ–TEZ–D-IVA | 9,248,038 | 21.5 | 4,363,392 | |
CDA-AMC base case (Reanalysis 1 + 2 + 3 + 4) (probabilistic) | ELX-TEZ-IVA | 8,766,740 | 21.2 | Reference |
VNZ–TEZ–D-IVA | 9,261,575 | 21.4 | 3,727,216 |
CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; PEx = pulmonary exacerbation; ppFEV1 = percent predicted forced expiratory volume in 1 second; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 8: Disaggregated Results of the CDA-AMC Base Case
Parameter | VNZ–TEZ–D-IVA | ELX-TEZ-IVA |
|---|---|---|
Discounted LYs | ||
Total | 29.24 | 29.17 |
Discounted QALYsa | ||
Total | 21.37 | 21.24 |
Discounted costs ($) | ||
Total | 9,261,575 | 8,766,740 |
Drug acquisition | 8,831,154 | 8,336,748 |
Non-PEx–related disease management | 306,739 | 307,013 |
PEx-related | 110,875 | 109,173 |
Lung transplant | 319 | 305 |
Adverse event | 12,278 | 13,292 |
Monitoring | 209 | 209 |
CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; LY = life-year; PEx = pulmonary exacerbation; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aFurther breakdown in discounted QALYs were not available in the sponsor’s submitted model.
Note: Probabilistic results. The CDA-AMC base case is based on publicly available prices of comparators.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 9). Given that the sponsor’s submitted price for VNZ–TEZ–D-IVA is approximately 5% higher than the publicly available price for ELX-TEZ-IVA, once price reductions exceed this level VNZ–TEZ–D-IVA becomes the lower cost option, becoming the “dominant” option as both drugs are expected to produce at least similar health outcomes.
Table 9: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Annual cost ($) | ICERs for VNZ–TEZ–D-IVA vs. ELX-TEZ-IVA ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 24,696a | 322,151 | 457,263 | 3,727,216 |
10% | 22,226 | 289,936 | Dominant | Dominant |
20% | 19,757 | 257,721 | Dominant | Dominant |
30% | 17,287 | 225,506 | Dominant | Dominant |
40% | 14,818 | 193,291 | Dominant | Dominant |
50% | 12,348 | 161,076 | Dominant | Dominant |
60% | 9,878 | 128,860 | Dominant | Dominant |
70% | 7,409 | 96,645 | Dominant | Dominant |
80% | 4,939 | 64,430 | Dominant | Dominant |
90% | 2,470 | 32,215 | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: The term dominant indicates that a drug costs less and provides more QALYs than the comparator. In the CDA-AMC base case, VNZ–TEZ–D-IVA was associated with 0.13 higher QALYs vs. ELX-TEZ-IVA.
aSponsor’s submitted price for VNZ–TEZ–D-IVA, per package of 84 tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg or as packages of 56 tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg.2
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 10.
Assuming an improvement of 5% in long-term ppFEV1 decline for VNZ–TEZ–D-IVA versus ELX-TEZ-IVA, which is aligned with the sponsor’s submission.
Table 10: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | ELX-TEZ-IVA | 8,766,740 | 21.24 | Reference |
VNZ–TEZ–D-IVA | 9,261,575 | 21.37 | 3,727,216 | |
CDA-AMC scenario 1b | ELX-TEZ-IVA | 8,766,740 | 21.24 | Reference |
VNZ–TEZ–D-IVA | 9,321,001 | 21.58 | 1,617,898 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SwCl = sweat chloride; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aProbabilistic analyses.
bAs noted in the Clinical Review, VNZ–TEZ–D-IVA likely reduces SwCl compared with ELX-TEZ-IVA in patients aged 12 years or older. However, the clinical importance of a further reduction in SwCl (beyond that seen with ELX-TEZ-IVA) is unclear, and the magnitude of reduction in SwCl was not considered to be clinically meaningful by clinicians. No long-term evidence was submitted by the sponsor to support a long-term difference in ppFEV1 between VNZ–TEZ–D-IVA and ELX-TEZ-IVA. As such, the results of this scenario should be considered highly uncertain.
CDA-AMC additionally conducted a scenario analysis in which VNZ–TEZ–D-IVA was compared to BSC (Table 11). As noted in the Key Issues section, BSC would be considered a comparator primarily among patients who are not eligible for ELX-TEZ-IVA (approximately 10% of patients; based on mutations included in the Health Canada indication). Comparative data were submitted by the sponsor only for the relative impact of VNZ–TEZ–D-IVA versus BSC on annual PEx rate; as such, the findings of this scenario should be considered highly uncertain.
Table 11: Results of CDA-AMC Scenario Analyses — VNZ–TEZ–D-IVA vs. BSC Alone
Drug | Total costs ($) | Total QALYs | ICER vs. BSC ($/QALYs) |
|---|---|---|---|
BSC | 787,918 | 10.2 | Reference |
VNZ–TEZ–D-IVA | 9,261,575 | 21.4 | 759,359 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor; vs. = versus.
Note: Probabilistic analyses are presented.
Issues for Consideration
Based on the sponsor’s analysis of data from the CF patient registry for Canada, approximately 99% of patients eligible for VNZ–TEZ–D-IVA are also eligible for ELX-TEZ-IVA. The sponsor has indicated that VNZ–TEZ–D-IVA is intended to displace ELX-TEZ-IVA over time in this group of patients.
ELX-TEZ-IVA has successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance for patients aged 2 years and older who have at least 1 F508del mutation or another responsive mutation in the CFTR gene and are listed on public formularies.25 It is therefore likely that ELX-TEZ-IVA is reimbursed by jurisdictional drug plans at confidential prices that are less than the publicly available list price.
Clinical expert input received by CDA-AMC indicated that ELX-TEZ-IVA is the current standard of care for patients aged 6 years and older in Canada with CF who have an eligible. Ivacaftor monotherapy, although indicated for the target population, was considered by the experts to be of little clinical relevance in the Canadian context. As such, the exclusion of ivacaftor monotherapy as a comparator from the sponsor’s economic and BIA is expected to have limited impact. However, CDA-AMC notes that the cost-effectiveness of VNZ–TEZ–D-IVA versus IVA monotherapy is unknown.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing VNZ–TEZ–D-IVA for the treatment of CF in people aged 6 years and older who have at least 1 F508del mutation or another responsive mutation in the CFTR gene.26
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec). The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs for VNZ–TEZ–D-IVA and ELX-TEZ-IVA; no cost was included for BSC. The market uptake for VNZ–TEZ–D-IVA was estimated using the sponsor’s internal market share estimates. The sponsor assumed that 98.7% of patients in the Health Canada–indicated population for VNZ–TEX–D-IVA will be eligible for ELX-TEZ-IVA, with the remainder assumed to be eligible for only BSC.27 As such, the majority of displacement is expected by the sponsor to primarily be from ELX-TEZ-IVA. The key inputs to the BIA are documented in Table 12.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing VNZ–TEZ–D-IVA would be $29,924,808 (year 1 = $6,377,992; year 2 = $10,353,916; year 3 = $13,192,900).
Table 12: Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Number of patients with eligible CFTR mutations (excluding Quebec) | 3,81027 |
Annual growth rate of CF patient population | |
Proportion of patients eligible for ELX-TEZ-IVA | 98.7%27 |
Percentage of patients covered by public drug plan | 55%a |
Number of patients eligible for drug under review | 2,840 / 2,888 / 2,936 |
Market shares (reference scenario) | |
VNZ–TEZ–D-IVA | 0% / 0% / 0% |
ELX-TEZ-IVA | 95.8% / 95.8% / 95.8% / 95.8% |
BSC alone | 4.2% / 4.2% / 4.2% / 4.2% |
Market shares (new drug scenario) | |
VNZ–TEZ–D-IVA | 12.6% / 24.8% / 35.7% |
ELX-TEZ-IVA | 95.8% / 83.9% / 72.1% / 61.2% |
BSC alone | 4.2% / 3.5% / 3.2% / 3.1% |
Cost of treatment (per patient per year)b | |
VNZ–TEX–D-IVA | $309,264 / $296,378 / $289,935 |
ELX-TEZ-IVA | $294,538 / $282,265 / $276,129 |
BSC alone | $0 |
BSC = best supportive care; CF = cystic fibrosis; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aBased on sponsor assumption.
bDrug acquisition costs for VNZ–TEZ–D-IVA and ELX-TEZ-IVA were adjusted for estimated adherence to treatment (year 1 = 96%; year 2 = 92%; year 3 = 90%).26
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for VEN–TEZ–D-IVA is uncertain: The sponsor assumed 55% of patients eligible for VNZ–TEZ–D-IVA have drug plan public coverage, based on internal data not provided to CDA-AMC. A number of CDA-AMC–participating jurisdictions have universal drug programs that may provide 100% coverage for VNZ–TEZ–D-IVA.30 Underestimation of the proportion of patients eligible for public drug plan coverage would underestimate the budget impact of reimbursing VNZ–TEZ–D-IVA.
CDA-AMC explored uncertainty in the proportion of patients with public drug plan coverage in scenario analyses.
The market share of ELX-TEZ-IVA in the reference scenario is uncertain: The sponsor assumed that, in the base year (2025), 97% of eligible patients would receive ELX-TEZ-IVA and that the remainder would receive BSC alone. Expert input received by CDA-AMC for this review indicated that that usage of ELX-TEZ-IVA is lower than estimated by the sponsor (approximately 90%), with the remainder opting to receive BSC alone, due to ineligibility, patient preference, or intolerability.
In the CDA-AMC base case, the market share of ELX-TEZ-IVA was assumed to be 90% in the baseline year to align with clinical expert input.
The uptake of VNZ–TEZ–D-IVA is likely underestimated: The sponsor assumed an uptake of VNZ–TEZ–D-IVA of 12% in year 1, 24% in year 2, and 35% in year 3 among patients who are currently eligible for ELX-TEZ-IVA and 60% in year 1, 85% in year 2, and 93% in year 3 for patients who are ineligible for ELX-TEZ-IVA (i.e., those who currently receive BSC alone). Clinical expert feedback obtained by CDA-AMC noted that the uptake of VNZ–TEZ–D-IVA is likely to be considerably higher in the ELX-TEZ-IVA–eligible subgroup and that uptake will be more rapid overall than estimated by the sponsor. This input also indicated that a considerable number of patients will likely switch from ELX-TEZ-IVA to VNZ–TEZ–D-IVA because of the once-daily dosing and concerns with mental health impacts with ELX-TEZ-IVA. Among patients who are not eligible for ELX-TEZ-IVA, clinical expert input indicated that uptake of VNZ–TEZ–D-IVA is expected to be very rapid owing to the lack of alternative treatments for this subgroup of patients.
In the CDA-AMC base case, the uptake of VNZ–TEZ–D-IVA was increased to 65% in year 1, 70% in year 2, and 75% in year 3 among patients eligible for ELX-TEZ-IVA. Among patients who currently receive BSC, the market share of VNZ–TEZ–D-IVA was increased to 85% in year 1, 90% in year 2, and 95% in year 3.
The adjustment of drug acquisition costs by patient adherence underestimates costs to the public drug plans. The sponsor adjusted the price of VNZ–TEZ–D-IVA and ELX-TEZ-IVA by adherence (year 1 = 96%; year 2 = 92%; year 3 = 90%). Adjustment of drug costs by adherence is likely to underestimate costs to the public drug plans because the cost of the drug is borne at the time of dispensation, resulting in the full costs of treatment being incurred by the public drug payer.
CDA-AMC additionally notes that no data were submitted by the sponsor to support the adherence rates for VNZ–TEZ–D-IVA, and that the sponsor assumed that adherence to VNZ–TEZ–D-IVA would be equivalent to that of ELX-TEZ-IVA. The appropriateness of this assumption is uncertain, given the input received from clinician and patient groups noting that VNZ–TEZ–D-IVA may have improved adherence due to once-daily administration.
In the CDA-AMC base case, adherence was assumed to be 100% for all treatments.
The price of drugs paid by public drug plans is uncertain. The analyses by both the sponsor and CDA-AMC are based on publicly available list prices for all comparators. Because ELX-TEZ-IVA has gone through negotiations with the pan-Canadian Pharmaceutical Alliance, the prices paid by public drug plans are not known.25
CDA-AMC was unable to address this limitation in reanalysis.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 12.
Table 13: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Market shares in the reference scenario (ELX-TEX-IVA–eligible subgroup) | ELX-TEZ-IVA:
BSC:
| ELX-TEZ-IVA:
BSC:
|
2. Market shares in the new-drug scenario | ELX-TEZ-IVA–eligible subgroup: VNZ–TEZ–D-IVA:
ELX-TEZ-IVA:
BSC:
ELX-TEZ-IVA-ineligible subgroup: VNZ–TEZ–D-IVA:
BSC:
| ELX-TEZ-IVA–eligible subgroup: VNZ–TEZ–D-IVA:
ELX-TEZ-IVA:
BSC:
ELX-TEZ-IVA-ineligible subgroup: VNZ–TEZ–D-IVA:
BSC:
|
3. Adherence | Year 1: 96% Year 2: 92% Year 3: 90% | 100% in all years |
CDA-AMC base casea | ― | Reanalysis 1 + 2 + 3 |
BIA = budget impact analysis; BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
aThe final adjusted market shares for ELX-TEZ-IVA in the CDA-AMC new-drug scenario base case were 90% at baseline, 25% in year 1, 20% in year 2, and 15% in year 3.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 14 and a more detailed breakdown is presented in Table 15. In the CDA-AMC base case, the 3-year budget impact of reimbursing VNZ–TEZ–D-IVA for the treatment of CF in Health Canada–indicated population was $67,988,215 (year 1 = $20,769,361; year 2 = $22,643,695; year 3 = $24,575,159).
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing VNZ–TEZ–D-IVA. The results are provided in Table 15.
Assuming that 100% of eligible patients will receive public coverage for VNZ–TEZ–D-IVA.
Table 14: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 29,924,808 |
CDA-AMC reanalysis 1 | 123,536,929 |
CDA-AMC reanalysis 2 | 62,888,429 |
CDA-AMC reanalysis 3 | 32,556,776 |
CDA-AMC base case: Reanalysis 1 + 2 + 3 | 67,988,215 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of comparators.
Table 15: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 433,383,846 | 440,666,875 | 429,402,617 | 427,127,030 | 1,297,196,521.84 |
VNZ–TEZ–D-IVA | 0 | 0 | 0 | 0 | 0 | |
All comparatorsa | 433,383,846 | 440,666,875 | 429,402,617 | 427,127,030 | 1,297,196,521.84 | |
New drug total | 433,383,846 | 447,044,867 | 439,756,533 | 440,319,930 | 1,327,121,330.22 | |
VNZ–TEZ–D-IVA | 0 | 60,893,482 | 116,597,862 | 167,310,901 | 344,802,245 | |
All comparatorsa | 433,383,846 | 386,151,386 | 323,158,670 | 273,009,029 | 982,319,085 | |
Budget impact | 0 | 6,377,992 | 10,353,916 | 13,192,900 | 29,924,808 | |
CDA-AMC base case | Reference total | 418,863,253 | 425,902,263 | 433,059,564 | 440,337,144 | 1,299,298,972 |
VNZ–TEZ–D-IVA | 0 | 0 | 0 | 0 | 0 | |
All comparatorsa | 418,863,253 | 425,902,263 | 433,059,564 | 440,337,144 | 1,299,298,972 | |
New drug total | 418,863,253 | 446,671,625 | 455,703,259 | 464,912,303 | 1,367,287,187 | |
VNZ–TEZ–D-IVA | 0 | 328,365,440 | 359,467,800 | 391,522,779 | 1,079,356,020 | |
All comparatorsa | 418,863,253 | 118,306,184 | 96,235,459 | 73,389,524 | 287,931,167 | |
Budget impact | 0 | 20,769,361 | 22,643,695 | 24,575,159 | 67,988,215 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: 80% public coverage | Reference total | 609,255,641 | 619,494,201 | 629,904,821 | 640,490,391 | 1,889,889,414 |
New drug total | 609,255,641 | 649,704,181 | 662,841,104 | 676,236,077 | 1,988,781,363 | |
Budget Impact | 0 | 30,209,980 | 32,936,283 | 35,745,686 | 98,891,949 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; ELX-TEZ-IVA = elexacaftor-tezacaftor-ivacaftor and ivacaftor; VNZ–TEZ–D-IVA = vanzacaftor-tezacaftor-deutivacaftor.
Note: The CDA-AMC reanalysis is based on the publicly available prices of comparators.
aThe comparators include ELX-TEZ-IVA and BSC; however, no drug costs were included by the sponsor for BSC; thus, the values for all comparators reflect costs associated with ELX-TEZ-IVA only.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.