Drugs, Health Technologies, Health Systems
Reimbursement Review
Efgartigimod Alfa Injection (Vyvgart SC)
Sponsor: argenx Canada Inc.
Therapeutic area: Chronic inflammatory demyelinating polyneuropathy
Summary
What Is Chronic Inflammatory Demyelinating Polyneuropathy?
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a rare and often progressive autoimmune neuropathy that causes muscle weakness, sensory disturbances, fatigue, pain, and significant disability, including impaired mobility and impaired daily functioning. It presents in both typical and variant forms, is difficult to diagnose, and imposes a substantial burden on patients’ quality of life, often leading to long-term disability and economic strain. CIDP has an estimated prevalence of 8.9 cases per 100,000 individuals based on US data (2021). Accurate estimates for Canada are unavailable due to diagnostic challenges and clinical heterogeneity.
What Are the Treatment Goals and Current Treatment Options for CIDP?
The treatment goals for CIDP focus on improving patients’ daily functioning, independence, and overall quality of life by addressing both symptom relief and disease progression.
Input from both patient and clinician groups highlights that the relapse-remission cycles and negative side effects associated with current treatments diminish some patients’ quality of life and well-being. Both groups also note the need for treatments that improve strength and mobility, while being less burdensome.
Current treatment options for CIDP in Canada include IV immunoglobulin and subcutaneous immunoglobulin as the main therapies, with corticosteroids used less frequently due to side effects. For refractory cases, off-label immunosuppressants may be considered, and nonpharmacologic interventions like physiotherapy are essential for maintaining function and quality of life.
What Is Efgartigimod Alfa Injection and Why Did Canada’s Drug Agency Conduct This Review?
Efgartigimod alfa is a drug that is administered by subcutaneous injection. At the time this review was initiated, efgartigimod alfa was under regulatory review. Health Canada approved efgartigimod alfa injection as a monotherapy for adult patients with active CIDP on November 3, 2025.
Canada’s Drug Agency (CDA-AMC) reviewed efgartigimod alfa injection to inform a recommendation to the participating public drug programs on whether it should be reimbursed for the Health Canada–approved indication.
How Did CDA-AMC Evaluate Efgartigimod Alfa Injection?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of efgartigimod alfa injection versus other treatments used in Canada for adult patients with CIDP. Immunoglobulin (Ig) treatments (including IV immunoglobulin and subcutaneous immunoglobulin), corticosteroids (e.g., dexamethasone, methylprednisolone, prednisone), immunosuppressive treatments (e.g., rituximab, cyclosporine, cyclophosphamide), and plasma exchange (PLEX) were considered as relevant treatments to compare with efgartigimod alfa when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to the use of efgartigimod alfa injection in the treatment of CIDP.
The review was informed by materials submitted by the sponsor, which included clinical and economic evidence.
The review was also informed by 1 patient group submission and 1 clinician group submission in response to the CDA-AMC call for input and by input from the participating public drug programs around issues that may impact their ability to implement a recommendation.
Two clinical specialists with expertise in the diagnosis and management of CIDP were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 randomized withdrawal phase II trial (the ADHERE study) comparing efgartigimod alfa injection with placebo in 221 adult patients with CIDP
1 indirect treatment comparison comprising 5 matching-adjusted indirect comparisons of efgartigimod alfa injection versus Ig therapy (i.e., Hizentra and HyQvia).
For the comparison of efgartigimod alfa injection versus placebo based on the ADHERE study:
In adults with CIDP, treatment with efgartigimod alfa injection demonstrated a clinically meaningful reduction in the risk of clinical deterioration (CIDP relapse) at both 24 and 48 weeks compared to placebo.
At the final assessment of the randomized withdrawal phase, patients receiving efgartigimod alfa showed likely improvements in disability and functional impairment, as measured by the adjusted Inflammatory Neuropathy Cause and Treatment score, the Inflammatory Rasch-Built Overall Disability Scale, and the Medical Research Council sum score.
Quality of life, assessed via the EQ visual analogue scale, may also be improved with efgartigimod alfa injection.
No notable safety concerns were identified during the study period.
No additional safety concerns were identified with longer-term efgartigimod alfa injection treatment in the ADHERE+ study.
For the comparison of efgartigimod alfa injection versus Ig treatments based on the indirect treatment comparison:
The results suggested that efgartigimod alfa injection likely results in a clinically meaningful difference in Inflammatory Rasch-Built Overall Disability Scale score and in little to no clinically meaningful difference in adjusted Inflammatory Neuropathy Cause and Treatment score or Medical Research Council sum score up to 32 weeks, but the uncertainty in the evidence means that CDA-AMC cannot draw a definitive conclusion on the comparative efficacy of efgartigimod alfa injection versus Igs.
There was no evidence to inform how efgartigimod alfa injection compares with corticosteroids (e.g., dexamethasone, methylprednisolone, prednisone), immunosuppressive treatments (e.g., cyclosporine, cyclophosphamide, rituximab), or PLEX.
Economic Evidence
Efgartigimod alfa is available as a single-dose prefilled syringe (200 mg/mL). At the submitted price of $19,750.00 per 5 mL syringe, the annual cost of efgartigimod alfa is expected to be $1,030,527 per patient, based on the Health Canada–recommended dosing.
Comparative clinical efficacy in the economic analysis was derived from a sponsor-submitted indirect treatment comparison, with the efficacy of efgartigimod alfa informed by the ADHERE trial, which compared efgartigimod alfa to Ig. Indirect evidence submitted by the sponsor suggests that there may be little to no clinically meaningful difference in efficacy between efgartigimod alfa and Ig among patients with CIDP. The safety of efgartigimod alfa versus Ig was not assessed in the sponsor-submitted indirect treatment comparison.
The results of the CDA-AMC base case suggest the following:
Efgartigimod alfa is predicted to be associated with higher costs to the health care system than Ig (incremental costs = $6,405,231 per patient), primarily driven by increased costs associated with drug acquisition.
Efgartigimod alfa is predicted to be associated with no life-year gains compared to Ig and may result in a gain of 3.42 quality-adjusted life-years (QALYs) compared to Ig.
The incremental cost-effectiveness ratio of efgartigimod alfa compared to Ig was $1,872,111 per QALY gained in the CDA-AMC base case. The proportion of QALYs derived during the extrapolated period (i.e., after the trial period of 24 months) is estimated to be 87%. QALY gains are therefore contingent on long-term treatment efficacy and no treatment waning over time.
The incremental cost is driven primarily by the drug acquisition cost of efgartigimod alfa. The majority of the benefit with efgartigimod alfa is the increased time spent in the less impaired health states, although this benefit is largely derived during the extrapolated period, as noted previously. There is remaining uncertainty in the analysis associated with lack of head-to-head evidence versus comparators, limitations with the model structure, and the unknown price of Ig; these uncertainties could not be resolved by CDA-AMC and may result in a higher price reduction being warranted. The cost-effectiveness of efgartigimod alfa versus off-label comparators such as corticosteroids, PLEX, immunosuppressives, and rituximab remains unknown.
CDA-AMC estimates that the budget impact of reimbursing efgartigimod alfa for the treatment of patients with CIDP will be approximately $691 million over the first 3 years of reimbursement compared to the amount currently spent on comparators, with an estimated expenditure of $692 million on efgartigimod alfa over this period. The actual budget impact of reimbursing efgartigimod alfa will depend on the consideration of nondrug costs (i.e., blood products) to the health system, public coverage rates, and uncertainty in market shares and market uptake. At the submitted price, the incremental budget impact of reimbursing efgartigimod alfa is predicted to be greater than $40 million in years 1, 2, and 3, and the economic feasibility of adoption must be addressed. Additionally, the magnitude of uncertainty in the budget impact must be addressed to ensure the feasibility of adoption, given the difference between the sponsor’s estimate and the CDA-AMC estimate from the drug plan perspective.
Abbreviations
AE
adverse event
AESI
adverse event of special interest
aINCAT
adjusted Inflammatory Neuropathy Cause and Treatment
CDA-AMC
Canada’s Drug Agency
CDAS
CIDP Disease Activity Status
CIDP
chronic inflammatory demyelinating polyneuropathy
ECI
evidence of clinical improvement
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IgG
immunoglobulin G
IMP
investigational medicinal product
INCAT
Inflammatory Neuropathy Cause and Treatment
I-RODS
Inflammatory Rasch-Built Overall Disability Scale
ITC
indirect treatment comparison
IVIg
IV immunoglobulin
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
MRC
Medical Research Council
OLE
open-label extension
PLEX
plasma exchange
QALY
quality-adjusted life-year
RCT
randomized controlled trial
SAE
serious adverse event
SAF-A
stage A safety analysis set
SAF-B
stage B safety analysis set
SC
subcutaneous
SCIg
subcutaneous immunoglobulin
SD
standard deviation
SE
standard error
SLR
systematic literature review
VAS
visual analogue scale
Background
Introduction
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of efgartigimod alfa 1,000 mg/5 mL (200 mg/mL) solution in single-dose prefilled syringe for subcutaneous (SC) use as a monotherapy for adult patients with active chronic inflammatory demyelinating polyneuropathy (CIDP). The focus will be placed on comparing efgartigimod alfa injection to relevant comparators in clinical practice in Canada and identifying gaps in the current evidence, and this focus is outlined in Table 1.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis. The focus of the Economic Review is aligned with the scope of the Clinical Review, unless otherwise stated. For most reviews, a Canada’s Drug Agency (CDA-AMC) base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Efgartigimod alfa injection (Vyvgart SC), 1,000 mg/5 mL (200 mg/mL) solution in single-dose prefilled syringe for SC use |
Sponsor | argenx Canada Inc. |
Health Canada indication | As a monotherapy for adult patients with active chronic inflammatory demyelinating polyneuropathy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 3, 2025 |
Mechanism of action | A human IgG1 antibody fragment that functions as an FcRn blocker, reducing circulating IgG and pathogenic autoantibodies |
Recommended dosage | 1,000 mg once weekly, by SC injection |
Submission type | Initial |
Sponsor’s reimbursement request | Per indication |
Submitted price | $19,750.00 per 5 mL single-dose prefilled syringe |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: as defined in the Health Canada indication Subgroups: none Intervention: per recommended dosage Comparators:
Outcomes: clinical deterioration, disability and functional impairment outcomes (aINCAT, I-RODS, MRC sum score), health-related quality of life (EQ VAS), patient satisfaction (TSQM-9), and standard harms outcomes (AEs, SAEs, WDAEs, deaths, AESIs) |
AE = adverse event; AESI = adverse event of special interest; aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment; CDA-AMC = Canada’s Drug Agency; Ig = immunoglobulin; IgG = immunoglobulin G; IgG1 = immunoglobulin G1; I-RODS = Inflammatory Rasch-Built Overall Disability Scale; IVIg = IV immunoglobulin; MRC = Medical Research Council; NOC = Notice of Compliance; SAE = serious adverse event; SC = subcutaneous; SCIg = subcutaneous immunoglobulin; TSQM-9 = 9-item Treatment Satisfaction Questionnaire for Medication; VAS = visual analogue scale; WDAE = withdrawal due to adverse event.
aThe Economic Review aligns with the scope of the Clinical Review, unless otherwise stated.
Submission History for the Drug Under Review
CDA-AMC previously reviewed efgartigimod alfa (IV) through the reimbursement review process for the treatment of adult patients with generalized myasthenia gravis who are anti–acetylcholine receptor antibody positive and issued a recommendation of reimburse with conditions.
Sources of Information
The contents of this Reimbursement Review Report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and drug programs), and input from the clinical experts consulted for this review.
Calls for patient group and clinician group input are issued for each reimbursement review. One patient group submission from Muscular Dystrophy Canada and 1 clinician group submission from the Neuromuscular Disease Network for Canada were received. In the patient group, 79 patients with CIDP and 2 caregivers or family members of individuals with CIDP participated in a health care experience survey as well as optional semistructured virtual interviews. Seventeen clinicians with experience treating CIDP contributed to the clinician group submission. The full submissions received are available on the project landing page in the Patient and Clinician Group Input document. The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation.
Input from patient and clinician groups is considered throughout the review, including in the selection of outcomes to include in the Clinical Review and in the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections of this report.
Each review team includes at least 1 clinical expert with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two clinical specialists with expertise in the diagnosis and management of CIDP participated in this review.
Disease Background
CIDP is a rare, debilitating, severe, often progressive neuropathy that causes muscle weakness and sensory disturbances. The disease results in fatigue, pain, and disability that impairs one’s ability to independently perform activities of daily living.1-4 The disease is characterized by immune-mediated peripheral nerve demyelination and is a heterogenous disorder in which most patients develop relatively symmetric motor and sensory symptoms in the proximal and distal upper and lower limbs (i.e., typical CIDP).5 However, distal predominant, asymmetric, pure motor, and pure sensory variants are also recognized as CIDP variants.6
CIDP has a prevalence of 8.9 per 100,000 individuals in Minnesota, US.7 Accurate estimates on the prevalence and incidence of CIDP in Canada are not available; however, the best estimate of the existing total number of individuals with CIDP across Canada (excluding Quebec) is 3,004. The annual incidence of CIDP is estimated to be 1.6 cases per 100,000 individuals in the US, with the international range between 1 and 2 per 100,000.7,8 CIDP onset typically occurs when individuals are aged between 48 and 60 years.9
CIDP prevalence is difficult to estimate due to the disease’s varied clinical presentation and the challenges associated with defining and diagnosing the condition, particularly in patients with CIDP variants. Fifteen sets of diagnostic criteria have been developed to reflect the wide clinical spectrum of CIDP. Accordingly, the Guillain-Barré Syndrome–Chronic Inflammatory Demyelinating Polyneuropathy Foundation of Canada reports that initial visits to physicians often lead to misdiagnosis or dismissal.10 As a result, 65% of patients in Canada face diagnostic delays of 1 to 3 years, and 5% wait more than 3 years.11 Until diagnosis, there is ongoing nerve damage, and up to 80% of patients experience motor difficulties that interfere with daily functioning. Ongoing demyelination and axonal damage can occur, which may lead to irreversible neurologic impairment if left untreated.11 In a real-world study of the experience of patients with CIDP, conducted in the US, 27% of 234 patients initially received a diagnosis other than CIDP.12 The 2021 European Academy of Neurology–Peripheral Nerve Society guidelines are globally recognized and currently applied for diagnosing CIDP.6 Diagnosis typically begins with a clinical assessment by a neurologist in an outpatient setting, followed by electrodiagnostic tests (i.e., nerve conduction studies) to determine whether a patient has “typical CIDP” or a “CIDP variant.” According to a survey of 11 clinical experts based in Canada, it is estimated that approximately 73% of individuals with CIDP living in Canada have typical CIDP and 27% have CIDP variants.12
Input from the 81 individuals included in the patient group submission — 79 individuals with CIDP and 2 caregivers or family members — identified that CIDP affects their daily life and quality of life through the following elements associated with the disease: loss of physical function and mobility, persistent fatigue, chronic pain, emotional and mental health challenges, loss of independence, difficulties with daily activities, disruptions to employment and financial stability, social withdrawal, reduced participation in recreational activities, and increased burden and strain in relationships.
Current Management
Treatment Goals
Patients highlighted that ideal therapies should be evaluated not only by symptom relief but also by their ability to improve daily functioning, restore independence, and support a better overall quality of life. They identified several symptom-specific and disease-related goals they want new therapies to address, including better mobility and walking stability, reduced neuropathic pain, improved motor control, and increased energy and relief from chronic neuropathic pain. In addition, patients stressed the importance of reducing the frequency and severity of relapses and unpredictable symptoms, as well as the need for fewer treatment-related side effects. Furthermore, patients emphasized that therapies should offer consistent symptom control, fewer hospital visits, and the possibility of self-administration at home. Overall, participants described how meaningful it would be to regain the ability to manage daily activities on their own; improve their mental and emotional health; return to consistent employment; and take part in hobbies, relationships, and community life. Some patients reported that they would accept minor side effects or closer monitoring if it meant avoiding frequent infusions or hospital trips. Others were less open to trade-offs if safety or long-term stability were affected.
Input from the clinician group highlighted that an ideal treatment should address both immune dysregulation and neurodegeneration, offering sustained efficacy, minimal side effects, and easier administration. Also, it should ideally enable patients to regain or maintain independence, reduce the health care system burden, and maintain employment and social participation. In the long-term, treatments that can delay progression, restore nerve function, or provide durable remissions would be transformative in CIDP care.
The clinical experts noted that the primary goal of CIDP treatment is to mitigate immune-mediated nerve damage early to preserve function and prevent irreversible axonal loss, with treatment priorities focused on enabling daily activities, improving quality of life, controlling disease quickly with minimal side effects, and achieving clinical remission.
Current Treatment Options
There are currently no Canadian clinical guidelines specific to the management of CIDP. Based on the 2021 European Academy of Neurology–Peripheral Nerve Society guidelines,6 which are widely used in Canada to guide the diagnosis and treatment of CIDP, IV immunoglobulin (IVIg) and SC immunoglobulin (SCIg) are the standard of care, with corticosteroids used less often due to side effects.6,13,14 The clinical experts consulted by CDA-AMC for this review noted that IVIg is generally preferred, particularly in certain variants such as multifocal CIDP and motor or motor-predominant CIDP, where corticosteroids may worsen symptoms. SCIg is commonly used for maintenance in patients stabilized on IVIg; PLEX is reserved for severe or refractory cases but is not feasible for long-term use due to its invasiveness and limited availability.6,13,14
For patients whose disease is refractory to first-line therapies, both the clinician group and the clinical experts identified the use of immunosuppressive drugs such as rituximab, cyclophosphamide, azathioprine, and mycophenolate mofetil as potential therapeutic options. However, these treatments are often used off-label, are associated with an increased risk of infection (particularly rituximab and cyclophosphamide), and are supported by limited clinical evidence. The clinician group noted that access to drugs like rituximab may be facilitated through Canada’s province-specific special access programs, which typically require documentation of prior treatment failure. Nonpharmacologic interventions, including physiotherapy and multidisciplinary care, are also considered essential for maintaining function and quality of life.
Access and reimbursement vary across provinces. IVIg and SCIg are funded through Canadian Blood Services, while corticosteroids are reimbursed without restriction. Immunosuppressants are generally covered but may require special authorization. Both the clinician group and the clinical experts emphasized the importance of early diagnosis and timely treatment to improve outcomes.6,13,14
Key characteristics of efgartigimod alfa injection are summarized, along with other treatments available for adults with CIDP, in the Supplemental Material document available on the project landing page, in the Key Characteristics table in Appendix 1.
Unmet Needs and Existing Challenges
In the patient submission, patients with CIDP described challenges related to their disease being refractory to current treatments or losing responsiveness over time. Moreover, they emphasized the need for therapies that result in fewer side effects — such as weight gain, osteoporosis or bone fractures, mood changes, insomnia, headaches, and nausea — that negatively impact their quality of life. They also noted that current treatments, particularly the immunoglobulin (Ig) regimens, require frequent hospital visits, long infusions, and rigid administration protocols that disrupt daily routines. As a result, patients noted that CIDP often placed strain on caregivers and family members, who frequently reported emotional stress, feelings of guilt, and logistical challenges including out-of-pocket costs and transportation. This burden may be exacerbated in patients and caregivers of low socioeconomic status. These concerns highlight the need for flexible, targeted, and home-based treatment options.
The clinician group identified several key gaps in CIDP management, including limited response or tolerance to first-line therapies in some patients, and the absence of disease-modifying therapies that reverse or halt progression. They noted that patients often face burdensome infusion regimens and highlight the need for more convenient formulations with better long-term safety profiles.
Both groups underscored that frequent visits to hospital-based infusion centres or home care services are required, which can be difficult to coordinate, particularly in rural or underserved regions. Overall, there is a need for timely, equitable, and flexible access to therapies that are effective and tolerable and that fit within the realities of everyday life.
Considerations for Using the Drug Under Review
The contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor table in the Supplemental Material document, Appendix 1). The implementation questions from the public drug programs and the corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in the Supplemental Material document, Appendix 1. The following has been summarized by the review team.
Place in Therapy
The clinical experts and clinician group anticipate that efgartigimod alfa injection could shift the current CIDP treatment paradigm due to its targeted mechanism, rapid onset, and generally well-tolerated safety profile. The clinical experts noted that efgartigimod alfa injection may be used as monotherapy or in combination with corticosteroids, either as a first-line option — particularly in patients who have not yet received treatment for the disease — or in those whose disease is refractory to existing therapies. The clinical experts pointed out that unlike current nontargeted treatments, such as IVIg and corticosteroids, efgartigimod directly inhibits the neonatal FcRn, reducing pathogenic immunoglobulin G (IgG) levels and addressing the underlying disease process. The clinical experts noted the drug’s potential to replace or complement current first-line therapies, especially given its convenience of administration and lower risk of adverse effects compared to steroids, rituximab, or cyclophosphamide. The clinical experts noted that while some clinicians support its early use regardless of prior treatment failure, others suggest its positioning in the treatment sequence may depend on cost considerations.
Patient Population
Both the clinical experts and the clinician group agreed that efgartigimod alfa injection may benefit patients across nearly all CIDP subtypes, particularly those whose disease is refractory to Igs, those who have limited access to IVIg treatment, those who experience intolerance to corticosteroids, or those who do not experience response to corticosteroids. The clinical experts noted that efgartigimod alfa’s targeted mechanism, rapid action, and favourable safety profile make it a valuable option, potentially as a first-line therapy or for those with contraindications to IVIg or steroids. However, cost may influence its positioning in the treatment sequence.
Both the clinician group and the clinical experts noted that patients with atypical variants, such as pure sensory CIDP, or patients lacking IgG-mediated pathology may be less suited to treatment with efgartigimod alfa. They also agreed that outcome scales like CIDP Disease Activity Status (CDAS) or Inflammatory Neuropathy Cause and Treatment (INCAT) are not routinely used in practice and should not be required for treatment initiation. Instead, clinical judgment and validated functional assessments should guide decisions. The experts emphasized that requiring prolonged discontinuation of Ig therapy before starting efgartigimod alfa would be impractical and clinically unjustified, particularly given that patients already had a confirmed diagnosis of CIDP.
Assessing the Response to Treatments
The clinical experts noted that clinicians typically assess treatment response in CIDP using bedside muscle strength testing, such as the Medical Research Council (MRC) sum score, along with patient-reported outcomes. When appropriate, electrodiagnostic improvements are also considered. The clinician group further highlighted the use of functional assessments — including gait performance, grip strength, and the ability to perform activities of daily living — as practical indicators of response. Both the clinician group and clinical experts agreed that improvements of 4 points in the MRC sum score, 1 point in the INCAT score, or 4 points in the centile Inflammatory Rasch-Built Overall Disability Scale (I-RODS) score are generally regarded as clinically meaningful.
The clinical experts indicated that treatment response is generally observed within 4 to 12 weeks. Both the clinician group and the clinical experts consulted for this review noted that clinical measures are typically reassessed every 8 to 12 weeks during the initiation phase of treatment. During the maintenance phase, reassessment intervals generally extend to every 3 to 6 months, depending on the stability of the patient’s disease, and to every 6 to 12 months for patients whose disease remains stable on therapy.
The clinical experts noted that there are no additional access limitations for patients — including those in rural or remote areas — to the type of care and specialist assessments required to evaluate treatment response when using efgartigimod alfa injection. The monitoring procedures involved, such as muscle strength testing and functional assessments, are part of routine clinical care for CIDP and are not necessitated by the addition of this drug specifically. Patients receiving other therapies for CIDP typically undergo similar, though not identical, monitoring and assessment of treatment response, and at comparable intervals.
Discontinuing Treatment
The clinical experts noted that discontinuation of efgartigimod alfa injection should be considered in patients who experience no clinical improvement or stabilization in strength, disability, or relapse frequency after an adequate treatment period (typically 3 to 6 months) or who experience disease worsening during maintenance, with adverse events (AEs) also being a valid reason to stop therapy. The clinician group also emphasized that, in the event of treatment discontinuation followed by clinical deterioration, prompt reinitiation of efgartigimod alfa or transition to an alternative immunotherapy should be considered to prevent potential irreversible nerve damage.
Prescribing Considerations
The clinical experts noted that while diagnosis, treatment initiation, and monitoring of patients receiving efgartigimod alfa injection should be conducted by a neurologist or neuromuscular specialist, the drug’s ease of self-administration is expected to improve access for patients in remote areas, especially compared to IVIg, which requires hospital-based infusions. This improved accessibility will help reduce the treatment burden, which is an ethically important consideration and could benefit both patients and caregivers. The clinical experts noted that there is currently no standardized approach to switching CIDP therapies and that clinical judgment should guide treatment transitions.
Clinical Review
Methods
The review team considered studies in the sponsor’s systematic review (pivotal studies and randomized controlled trials [RCTs]), sponsor-submitted long-term extensions, indirect treatment comparisons (ITCs), and studies addressing gaps in the evidence for inclusion. Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and IV RCTs, or other designs as relevant. Relevant patients and interventions were defined by the indication and the recommended dosing in the draft product monograph. CDA-AMC did not identify any subgroups as potentially important for informing the reimbursement recommendation. Relevant comparators were drugs and nondrug treatments used in clinical practice in Canada to treat patients described in the indication under review. These treatments included Ig (IVIg, SCIg), corticosteroids (e.g., dexamethasone, methylprednisolone, prednisone), immunosuppressives (e.g., cyclosporine, cyclophosphamide, rituximab), and PLEX. Long-term extensions of included pivotal studies and RCTs were included, regardless of whether there was a comparison group. ITCs and studies addressing gaps submitted by the sponsor were included when they filled an identified gap in the systematic review evidence (e.g., missing comparator, longer follow-up time). The sponsor-submitted ITC only included efgartigimod alfa injection versus Ig therapy (i.e., Hizentra and HyQvia), as it was not feasible to conduct ITCs to evaluate the comparative clinical effectiveness of efgartigimod alfa injection versus corticosteroids (e.g., dexamethasone, methylprednisolone, prednisone) or immunosuppressives (e.g., cyclosporine, cyclophosphamide, rituximab) according to the sponsor’s feasibility assessment. PLEX was excluded from the ITC as it represents a nondrug comparator that is also rarely used in clinical practice in Canada (less than 5% of patients) according to the clinical experts consulted by the sponsor and CDA-AMC.
The review team selected outcomes (and follow-up times) for review based on the sponsor’s summary of clinical evidence, the clinical expert input, and the patient and clinician group input. Included outcomes are those considered relevant to expert committee deliberations, and they were selected in consultation with committee members. Evidence from the systematic review for the most important outcomes was assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach.15,16 The following outcomes were assessed using GRADE because they address important treatment goals for CIDP and are considered important to patients according to the patient and clinician input:
clinical deterioration (CIDP relapse)
adjusted INCAT (aINCAT) score
I-RODS score
MRC sum score
health-related quality of life (HRQoL) assessed with the EQ visual analogue scale (VAS)
serious AEs (SAEs).
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are in the Supplemental Material document in Appendix 2.
Clinical Evidence
In this report, the following sources of evidence submitted by the sponsor are reviewed and appraised:
1 pivotal study included in the systematic review: the ADHERE study
1 long-term extension study: the ADHERE+ study
1 ITC comprising 5 matching-adjusted indirect comparisons (MAICs) of efgartigimod alfa injection versus Ig therapy (i.e., Hizentra and HyQvia).
Systematic Review
Description of Studies
Study Characteristics
The characteristics of the included study are summarized in Table 2. Details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are in the Supplemental Material document in Appendix 3.
The ADHERE study investigated the efficacy and safety of efgartigimod alfa 1,000 mg once weekly by SC injection compared with placebo in adults with CIDP. The study was conducted at 146 sites across 22 countries in North America, Europe, and Asia and included 2 treatment periods: an open-label stage A and a randomized withdrawal, double-blind, phase II, placebo-controlled stage B. In stage A (open label), all eligible patients received 1,000 mg of SC efgartigimod alfa weekly for up to 12 weeks. Patients who demonstrated confirmed evidence of clinical improvement (ECI) — based on disability and functional impairment measures — during stage A were randomized 1:1 in stage B (randomized withdrawal) to either continue efgartigimod alfa injection or receive placebo. Randomization was stratified by aINCAT score during stage A (no change versus decrease of 1 point or more) and by prior CIDP therapy (not previously treated, received recent corticosteroids [within 6 months before screening], or received recent IVIg or SCIg). Efficacy and safety assessments occurred during stage B visits every 4 weeks through week 48.
The main body of this report focuses on data from stage B (randomized withdrawal), with select stage A data included when appropriate. Additional stage B details and the majority of stage A data, derived from a single-arm design, are provided in the Supplemental Material document.
Table 2: Characteristics of Studies Included in the Systematic Review
Study name, design, and sample size | Key inclusion criteria | Key exclusion criteria | Intervention and comparator | Relevant end points |
|---|---|---|---|---|
ADHERE study Multicentre, phase II, double-blind, placebo-controlled, randomized withdrawal study Total N (randomized withdrawal period) = 221 |
|
| Intervention: once-weekly injections of efgartigimod alfa (1,000 mg/5 mL) for up to 48 weeks Comparator: once-weekly injections of matched placebo SC for up to 48 weeks | Primary:
Key secondary:
Exploratory:
Safety:
|
AE = adverse event; aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment; BPI-SF = Brief Pain Inventory–Short Form; CDAS = CIDP Disease Activity Score; CIDP = chronic inflammatory demyelinating polyneuropathy; ECI = evidence of clinical improvement; EFNS = European Federation of Neurologic Societies; HADS = Hospital Anxiety and Depression Scale; INCAT = Inflammatory Neuropathy Cause and Treatment; I-RODS = Inflammatory Rasch-Built Overall Disability Scale; IVIg = IV immunoglobulin; MRC = Medical Research Council; PGIC = Patient Global Impression of Change; PNS = Peripheral Nerve Society; RT-FSS = Rasch-Transformed Fatigue Severity Scale; SAE = serious adverse event; SC = subcutaneous; SCIg = subcutaneous immunoglobulin; TSQM-9 = 9-item Treatment Satisfaction Questionnaire for Medication; TUG = Timed Up and Go.
Notes: In stage A, 322 patients received efgartigimod alfa injection. Patients who experienced confirmed ECI — based on disability and functional impairment measures — during stage A were eligible for randomization in stage B. The definition of ECI is detailed in the Descriptions of Outcomes section in the Supplemental Material document, Appendix 3. In stage B, the randomized withdrawal period, 111 patients received efgartigimod alfa injection and 110 patients received placebo.
Sources: Clinical Study Report for ADHERE.18 Details included in the table are from the sponsor’s summary of clinical evidence.19
Statistical Testing and Analysis Populations
Approximately 360 adult patients were planned to be enrolled in stage A. The stage A primary end point was summarized using an exact Clopper-Pearson 2-sided 95% confidence interval (CI), also used to compare this study’s results with those in historical controls.20-23 Secondary end points were analyzed descriptively.
For stage B, it was hypothesized that treatment with efgartigimod alfa injection would reduce the hazard of the event by 50% compared to placebo (i.e., a hazard ratio of 0.50). Based on this hypothesis, 88 events were required to demonstrate a difference in the time to first occurrence of clinical deterioration between the efgartigimod alfa injection arm and the placebo arm, using a log-rank test at a 1-sided alpha level of 0.025. An event was defined as an aINCAT increase during stage B. An increase (worsening) of 1 point had to be confirmed at a consecutive visit 3 to 7 days after the first aINCAT score increase of 1 point. For patients with an increase of at least 2 points on the aINCAT score compared to the stage B baseline, no confirmation was required. Patients were randomized into stage B until 88 events occurred per the sample size calculation. The study ended when the 88th event occurred. A Cox proportional hazard model was fitted, with the time to the first occurrence of clinical deterioration compared with the stage B baseline as the dependent variable and treatment as the independent variable. The model was stratified by prior CIDP therapy and a decrease of aINCAT score during stage A (both as stratified). The summary measure of the estimand was the hazard ratio for efgartigimod alfa injection versus placebo, along with the corresponding Wald-type 95% CI and the 2-sided P value.
The stage A safety analysis set (SAF-A) was defined as patients who received at least 1 dose or part of a dose of efgartigimod alfa injection in stage A. The SAF-A was used for general characteristics, efficacy, and safety analyses in stage A. For stage B, the modified intention-to-treat analysis set — composed of patients who were randomized and who received at least 1 dose or part of a dose of the investigational medicinal product (IMP) (efgartigimod alfa injection or placebo) in stage B — was the population for primary efficacy analysis. Sensitivity analyses were performed on the per-protocol analysis set, which included patients from the modified intention-to-treat analysis set who did not have major protocol deviations. The stage B safety analysis set (SAF-B) — defined as patients from the all-screened patients analysis set who received at least 1 dose or part of a dose of efgartigimod alfa injection in stage B — was used for safety analysis and general characteristics.
Patient Disposition
Patient disposition for the ADHERE study is summarized in the Supplemental Material document, Appendix 4. A total of 629 patients were screened, and 322 entered stage A (36 entered stage A directly, and 306 started the run-in period, of whom 286 entered stage A). During stage A, 101 patients (31.4%) were withdrawn from the study, including 22 patients who continued into the open-label extension (OLE). In stage B, 221 patients (68.6% of the patients who entered stage A) were randomized 1:1, with 111 and 110 in the efgartigimod alfa injection and placebo groups, respectively. The proportion of patients who continued into the OLE after the 88th event in the efgartigimod alfa injection group was numerically higher (31.5%) than in the placebo group (23.6%). The proportion of patients who withdrew from the study was slightly higher in the efgartigimod alfa injection group (10.8%) than in the placebo group (9.0%), with withdrawal by patients being the most common reason in both groups (2.7%). In stage B, both the modified intention-to-treat and SAF-B populations included all randomized patients in the treatment groups.
In stage B, major protocol deviations occurred in 15 patients (13.5%) in the efgartigimod alfa injection group and 22 patients (20.0%) in the placebo group, most commonly due to treatment deviations (efgartigimod alfa: 8.1%; placebo: 14.5%).18
Baseline Characteristics
A summary of key baseline patient characteristics in the ADHERE study is presented in Table 3, with additional baseline characteristics provided in the Supplemental Material document, Appendix 4.
In stage B of the ADHERE study, baseline characteristics were generally balanced between treatment groups. Among the 221 patients in the SAF-B, the mean age was 53 years. The proportion of male patients (64%) was higher than that of female patients (36%). The majority of patients were white (65%); the next largest group by ethnicity was Asian (30%). The average time since CIDP diagnosis was approximately 4 years. Most patients had typical CIDP (87%), and 13% had atypical forms (6% asymmetric; 6% distal). Two patients (1%) — 1 in each treatment group — had pure motor CIDP.
Among the patients in stage B, a slightly higher proportion had relapsing CIDP in the placebo group (48%) than in the efgartigimod alfa injection group (44%), whereas progressive CIDP was more common in the efgartigimod alfa injection group (56%) than in the placebo group (52%).
The SAF-B population included patients whose disease was refractory to CIDP therapy (CDAS scores of 5B [24%] or 5C [22%]), as well as patients with active disease that was stable or improving on therapy (CDAS scores: 3A [2%], 3B [24%], 4A [0.5%], 4B [5%]). Patients exhibited a range of disability and functional impairment, with a mean total INCAT score of 3.2, a mean I-RODS score of 52, and a mean dominant hand grip strength of 56.5 kPa.
The baseline disease characteristics in the SAF-B population were generally representative of those in the SAF-A, with the following 2 differences. The time since CIDP diagnosis was longer in the SAF-A population, with a mean of 4.9 years (standard deviation [SD] = 6.1 years), compared to 3.7 years (SD = 4.4 years) and 3.8 years (SD = 4.7 years) in the efgartigimod alfa injection and placebo groups, respectively, in the SAF-B. Compared to stage A, where 51% of patients had received prior IVIg or SCIg therapy, a lower proportion of patients entering stage B had received such treatment: 44% in the efgartigimod alfa injection group and 43% in the placebo group. Consequently, the proportions of patients previously treated with corticosteroids or who were considered off treatment were higher in stage B. Corticosteroid use was reported in 24% of the efgartigimod alfa injection group and 22% of the placebo group in stage B, versus 20% in stage A. Patients who were considered off treatment composed 32% and 35.5% of the efgartigimod alfa injection and placebo groups, respectively, in the SAF-B, compared to 29% in the SAF-A.
Table 3: Summary of Baseline Characteristics From the ADHERE Study (Safety Analysis Set)
Characteristic | Stage A | Stage B | |
|---|---|---|---|
Efgartigimod alfa injection (N = 322) | Efgartigimod alfa injection (N = 111) | Placebo (N = 110) | |
Age (years), mean (SD) | 54.0 (13.9) | 54.5 (13.2) | 51.3 (14.5) |
Sex at birth, n (%) | |||
Male | 208 (64.6) | 73 (65.8) | 69 (62.7) |
Female | 114 (35.4) | 38 (34.2) | 41 (37.3) |
Race, n (%) | |||
Asian | 89 (27.6) | 33 (29.7) | 34 (30.9) |
Black or African American | 4 (1.2) | 1 (0.9) | 1 (0.9) |
Native Hawaiian or other Pacific Islander | 1 (0.3) | 0 | 0 |
White | 211 (65.5) | 73 (65.8) | 71 (64.5) |
Other | 6 (1.9) | 2 (1.8) | 1 (0.9) |
Not reported | 11 (3.4) | 2 (1.8) | 3 (2.7) |
Time since CIDP diagnosisa (years) | |||
Mean (SD) | 4.9 (6.1) | 3.7 (4.4) | 3.8 (4.7) |
Median (range) | 2.8 (0 to 46.0) | 2.1 (0 to 22.0) | 2.2 (0 to 29.0) |
CIDP diagnosis,b n (%) | |||
Typical CIDP | 268 (83.2) | 97 (87.4) | 95 (86.4) |
Atypical CIDP | 54 (16.8) | 14 (12.6) | 15 (13.6) |
Asymmetric | 29 (9.0) | 6 (5.4) | 7 (6.4) |
Distal | 20 (6.2) | 7 (6.3) | 7 (6.4) |
Pure motor | 5 (1.6) | 1 (0.9) | 1 (0.9) |
CIDP disease evolution, n (%) | |||
Relapsing | 147 (45.7) | 49 (44.1) | 53 (48.2) |
Progressive | 174 (54.0) | 62 (55.9) | 57 (51.8) |
Unknown | 1 (0.3) | 0 | 0 |
CIDP disease activity status (CDAS score) at study screening, n (%) | |||
2 to 4 | 125 (38.8)c | 37 (33.3) | 34 (30.9) |
5 | 197 (61.2) | 74 (66.7) | 76 (69.1) |
Prior CIDP therapy,d n (%) | |||
Corticosteroids | 63 (19.6) | 26 (23.4) | 24 (21.8) |
IVIg or SCIg | 165 (51.2) | 49 (44.1) | 47 (42.7) |
Off treatment | 94 (29.2) | 36 (32.4) | 39 (35.5) |
Baseline disability and functional impairment measures at stage A or stage B, mean (SD) | |||
Total INCAT score | 4.6 (1.7) | 3.1 (1.5) | 3.3 (1.6) |
I-RODS score | 40.1 (14.7) | 53.6 (17.9) | 51.2 (15.4) |
Mean grip strength, dominant hand (kPa) | 38.5 (24.2) | 54.9 (23.6) | 58.0 (25.1) |
Mean grip strength, nondominant hand (kPa) | 39.0 (24.7) | 55.4 (28.3) | 56.7 (24.8) |
CDAS = CIDP Disease Activity Status; CIDP = chronic inflammatory demyelinating polyneuropathy; DAC = diagnostic adjudication committee; INCAT = Inflammatory Neuropathy Cause and Treatment; I-RODS = Inflammatory Rasch-Built Overall Disability Scale; NA = not applicable; IVIg = IV immunoglobulin; SCIg = subcutaneous immunoglobulin; SD = standard deviation.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aTime since CIDP diagnosis (years) was calculated as follows: (date of informed consent form − date of diagnosis) / 365.25. For stage B, stratification factors show the categories as randomized. Patients were considered off treatment if they had no history of prior CIDP therapy or had discontinued their CIDP therapy (corticosteroids, IVIg, or SCIg therapy) for at least 6 months before screening. The denominator for the percentage calculations was the total number of patients in the safety analysis set, excluding missing values.
bThe CIDP diagnosis was determined by the investigator and verified by the CIDP DAC.
cAt stage A baseline, 6 patients (1.9%) had a CDAS score of 2, and 119 patients (36.9%) had a CDAS score of 3 or 4.
dAt screening for stage A.
Sources: Clinical Study Report for ADHERE.18 Details included in the table are from the sponsor’s summary of clinical evidence.19
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure and adherence in the ADHERE study are in the Supplemental Material document, Appendix 4. In stage A, the mean duration of treatment with efgartigimod alfa injection was 5.2 weeks (SD = 3.3 weeks). In stage B, the mean treatment duration was longer in the efgartigimod alfa injection arm than in the placebo arm (25.1 weeks [SD = 17.2 weeks] and 18.7 weeks [SD = 16.9 weeks], respectively). The median number of IMP administrations per patient was 22.0 (range, 2.0 to 48.0) and 12.0 (range, 2.0 to 48.0) in the efgartigimod alfa injection and placebo arms, respectively. Dose modifications were not permitted during the ADHERE study.
In stage B, the median compliance rate was comparable between the 2 groups (efgartigimod alfa: 100.0% [range, 72.9% to 133.3%]; placebo: 100.0% [range, 73.8% to 200.0%]).
Concomitant CIDP therapy was not allowed during the run-in period, stage A, or stage B. Rescue therapy was not allowed during the study.
Critical Appraisal
Internal Validity
The ADHERE study employed a randomized withdrawal design in stage B, meaning all patients had received efgartigimod alfa injection for 12 weeks (with an optional additional week) during stage A, and only those patients who experienced response to the study drug entered the stage B randomized phase to assess the efficacy and safety of the drug. With half of the patients having discontinued the active treatment, any differences in the treatment outcomes, including AEs, between the 2 arms were attributed to the study drug. The limitations of this trial design include carryover effects, difficulties assessing whether the underlying disease process is still active, and long lag times to AEs if the disease is in remission. This withdrawal trial design may be more appropriate for phase I or II trials, such as the phase II ADHERE trial.24 Therefore, this study design may increase the uncertainty in the efficacy and safety results: the efficacy of the active drug might be overestimated, and AEs may be underdetected and thus underestimated.24
At the stage B baseline of the ADHERE study, all eligible patients were centrally randomized to either the efgartigimod alfa injection or placebo group using interactive response technology, which ensured allocation concealment. Randomization was stratified based on factors relevant to CIDP prognosis, and this approach was considered appropriate by the clinical experts consulted by CDA-AMC.
Blinding of patients and study personnel (e.g., the investigator, investigational site staff) in stage B was effectively preserved through the use of identical IMPs, with placebo and active drug matched in volume and vial appearance and administered using amber-coloured, masked syringes. The 2-physician model further ensured blinding integrity: the treating physician managed patient care while remaining blinded, and the evaluating physician, fully unaware of treatment details, conducted all clinical assessments and oversaw the evaluations of patient-reported outcomes. Although the use of placebo as a control could have led patients to infer their treatment assignment based on perceived symptom changes, potentially introducing bias in favour of efgartigimod alfa injection for subjective efficacy outcomes (e.g., I-RODS, HRQoL, the 9-item Treatment Satisfaction Questionnaire for Medication) and in favour of placebo for harms outcomes, this risk was considered low in the ADHERE study. The clinical experts consulted for this review and the CDA-AMC review team assessed that blinding was well maintained, resulting in a low risk of bias for both objective and subjective outcome measures.
Baseline patient demographics and disease characteristics were generally well balanced between treatment groups. Patient withdrawal during stage B was slightly higher in the efgartigimod alfa injection group (11%) than in the placebo group (9%). The mean treatment duration also differed between groups, with patients receiving efgartigimod alfa injection treated for an average of 25.1 weeks, compared to 18.7 weeks in the placebo group. The clinical experts and the CDA-AMC team assessed that these imbalances were due to protocol-defined end points — as stage B concluded once a total of 88 clinical deterioration events occurred across randomized patients — and were not considered indicative of unblinding or bias.
Concomitant CIDP therapies, including IVIg, SCIg, and corticosteroids, were not allowed during the run-in period, stage A, or stage B. Only a small number of patients took prohibited medications during stage B — with 6 patients (2.7%) reported as having major protocol deviations — indicating minimal risk of bias from concomitant therapy and no impact on the interpretation of the study results.
The estimands, end points, and measures used in the ADHERE study were appropriate and relevant to the objectives outlined in the protocol. The definition of the primary end point during stage B (clinical deterioration) was considered by the clinical experts to be clinically relevant for assessing efficacy and has been used in other CIDP clinical studies.21,23,25
Ideally, HRQoL outcomes should be reported at week 12 and week 48 to capture both early and sustained treatment effects. However, there was an imbalance in missing data between treatment groups at these time points, which may introduce bias. EQ VAS data were available for 69 of 111 patients in the efgartigimod alfa injection group and 49 of 110 patients in the placebo group at week 12 and for 29 and 14 patients in the 2 groups, respectively, at week 48. While the decision to use the last assessment during stage B for GRADE evaluation presents limitations — given that the timing of this assessment varied across patients, resulting in inconsistent treatment durations — the clinical experts and the CDA-AMC review team, taking these limitations into account, considered this approach appropriate.
An adequate sample size was achieved in stage B based on a priori sample size calculations for the primary end point. Overall, the statistical methods used in the ADHERE study were appropriate. The subgroup analyses were likely underpowered to identify subgroup differences. Adjustments for multiplicity were not conducted as these were regarded as unnecessary by the sponsor because different populations were tested in stage A and stage B. Lack of adjustments for multiplicity may be associated with an increased risk of type I error — the chance of incorrectly rejecting a true null hypothesis (false-positive) — and may introduce uncertainty about the meaningfulness of statistically significant findings, particularly when multiple end points or subgroup analyses are tested without correction.26,27
External Validity
The clinical experts consulted for this review noted that the ADHERE study population, including its inclusion and exclusion criteria, baseline characteristics, CIDP phenotypes, and prior therapies, were representative of patients eligible for efgartigimod alfa injection treatment in Canada. The ethnic distribution observed in the study may be explained by the geographic locations in which it was conducted, with patients identifying as white or Asian representing 93% of the study population in stage A and 95% in stage B. However, the lack of data on populations outside of these groups may limit the generalizability of the ADHERE study findings to other ethnic groups. Although the study required CDAS and INCAT scores for enrolment, the clinical experts commented that these tools are not routinely used in clinical practice in Canada. Specifically, CDAS is primarily a tool in CIDP research and is not commonly used to guide diagnosis or treatment decisions, which are instead guided by clinical assessment and electrodiagnostic testing. Similarly, INCAT is not required for access to other CIDP therapies, such as IVIg or PLEX, and its sensitivity in detecting meaningful clinical changes is limited and may restrict the generalizability of the efficacy results to real-world clinical practice. In addition, the clinical experts noted that the exclusion of patients with pure sensory CIDP and those without IgG-mediated pathology in the ADHERE study limits the applicability of the results to these subgroups.
As per the FDA guidance “Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products,”24 the randomized withdrawal design is regarded as an enrichment strategy in trials. In the ADHERE study, only patients who demonstrated clinical response during the open-label stage A were eligible for randomization, which may limit the generalizability of the results from the randomized comparative stage B to patients with CIDP who do not exhibit an initial response to efgartigimod alfa injection. Notably, the 2-stage study design was specifically employed to identify patients with active CIDP and to support diagnostic confirmation, consistent with the 2021 European Academy of Neurology–Peripheral Nerve Society guidelines,6 which include treatment response to immunotherapy as a supportive criterion for diagnosis. This approach aligns with prior CIDP studies such as the PATH21 and PRIMA trials,22 which similarly used treatment response during an initial open-label phase to enrich the randomized population and strengthen diagnostic certainty.
The intervention used in the ADHERE study, consisting of weekly SC administration of efgartigimod alfa injection without concomitant therapies, was considered feasible and practical. The dosing regimen aligns with the draft product monograph, and self-administration is consistent with current practice for similar therapies. The clinical experts expressed no concerns with patients self-administering SC injections of efgartigimod alfa.
The outcomes assessed in the ADHERE study, including clinical deterioration, functional measures, and HRQoL, were considered clinically important. The use of validated tools such as the MRC sum score and the duration of the randomized withdrawal period, which extended up to 48 weeks, were regarded by the clinical experts as appropriate for evaluating treatment efficacy.
Overall, the trial’s population, intervention, and outcomes were considered reflective of real-world CIDP care, supporting the generalizability of the findings to clinical practice in Canada.
Results
The key efficacy and harms results and findings from the GRADE assessment are presented in this section. Detailed efficacy and harms results can be found in the Supplemental Material document, Appendix 4. The ADHERE study has been completed, and the study’s efficacy and harms outcomes reported in this review were based on the data cut-off date of May 11, 2023.
Efficacy
Key efficacy results during stage B of the ADHERE study include the following:
There was a reduction in the risk of clinical deterioration (CIDP relapse) over the 48-week treatment period, with a hazard ratio of 0.39 (95% CI, 0.25 to 0.61) compared with placebo. The median time to clinical deterioration (and the 95% CI) could not be calculated for the efgartigimod alfa injection group because less than 50% of the patients had clinically deteriorated. The median time to clinical deterioration in the placebo group was 140 days (95% CI, 75 days to not calculated). The estimated percentage of clinical deterioration at week 24 was 26% (95% CI, 19% to 36%) in the efgartigimod alfa injection group and 54% (45% to 64%) in the placebo group; at week 48, the rates were 34% (25% to 45%) and 60% (50% to 70%), respectively.
At the last assessment in stage B, the observed clinical deterioration rate was lower in the efgartigimod alfa injection group (28%; 95% CI, 20% to 36%) than in the placebo group (54%; 95% CI, 44% to 63%), with a between-group risk difference of −26% (95% CI, −38% to 11%).
The between-group difference in mean change from baseline to the last assessment during stage B was −0.8 (95% CI, −1.2 to −0.3) in aINCAT score, 7.8 (95% CI, 3.5 to 12.1) in I-RODS score, and 2.7 (95% CI, 0.9 to 4.6) in MRC sum score, respectively.
The between-group differences in mean change from baseline to the last assessment during stage B in EQ VAS score were ████ ████ ██ █████ in a total of ███ patients.
The 9-item Treatment Satisfaction Questionnaire for Medication global satisfaction score increased over time in both groups. Mean scores and standard errors (SEs) were ██ ███ in the efgartigimod alga injection group and ██ ███ in the placebo group, respectively, at the stage B baseline, and the actual assessment scores were ██ ███ ███ ██ ███ at the last stage B assessment.
Harms
Key harms results during stage B of the ADHERE study include the following:
The harms results were reported during the 48-week stage B study period. The efgartigimod alfa injection group had a longer treatment duration, with a median of 22 weeks for efgartigimod alfa injection compared to 11 weeks for placebo.
There was a higher proportion of patients with any AEs in the efgartigimod alfa injection group (64%) than in the placebo group (56%). The frequently reported common AEs were COVID-19 (efgartigimod alfa: 17%, versus placebo: 13%), upper respiratory tract infection (2% versus 10%), injection site bruising (5% versus 1%), and headache (4% versus 2%).
SAEs were reported in 5.4% of patients in the efgartigimod alfa injection group and 5.5% of patients in the placebo group, with similar proportions observed in both groups.
AEs leading to discontinuation of the IMP occurred in a higher proportion of patients in the efgartigimod alfa injection group (2.7%) than in the placebo group (0.9%).
No patients in the efgartigimod alfa injection group died, and 1 patient in the placebo group died due to an SAE of pneumonia.
The proportion of patients who experienced any AEs of special interest (AESIs) was generally comparable between the efgartigimod alfa injection and placebo groups (31.5% versus 33.6%, respectively), as was the proportion of IMP-related AESIs (5.4% versus 6.4%). A higher proportion of patients in the placebo group experienced grade 3 or higher AESIs (efgartigimod alfa: 0.9%; placebo: 3.6%).
In terms of localized injection site reactions, higher proportions of patients reported injection site reactions in the efgartigimod alfa injection group than in the placebo group (14% versus 6%, respectively), treatment-related reactions as assessed by the principle investigator (12% versus 5%), and procedure-related reactions (7% versus 3%). No patients in either group reported serious injection site reactions, grade 3 or higher reactions, or injection site reactions leading to discontinuation of the IMP.
Summary of Findings and Certainty of the Evidence
Literature-based minimal important difference (MID) estimates were used as the thresholds for the following outcomes: aINCAT (MID = 1 point), I-RODS (MID = 4 points), and MRC sum score (MID = 4 points). Refer to the summary of outcome measures in the Supplemental Material document, Appendix 3. In the absence of a known threshold, the certainty in the presence of a non-null effect was rated for EQ VAS.
Table 4: Summary of Findings for Efgartigimod Alfa Injection vs. Placebo for Adult Patients With CIDP
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Efgartigimod alfa injection | Difference | |||||
CIDP clinical deterioration (relapse) | |||||||
Estimated percentage of clinical deterioration Time point: at 24 weeks | 221 (1 RCT) | HR = 0.39 (0.25 to 0.61) | 54.3 (95% CI, 44.8 to 64.4) | 26.2 (95% CI, 95% CI, 18.6 to 36.2) | −28.1 (95% CI, −40.7 to −14.3)a | High | Efgartigimod alfa injection results in a clinically important reduction in the risk of clinical deterioration compared with placebo at 24 weeks. |
Estimated percentage of clinical deterioration Time point: at 48 weeks | 221 (1 RCT) | HR = 0.39 (0.25 to 0.61) | 59.8 (95% CI, 50.0 to 69.9) | 34.0 (95% CI, 24.9 to 45.2) | −25.8 (95% CI, −39.4 to −10.9)a | High | Efgartigimod alfa injection results in a clinically important reduction in the risk of clinical deterioration compared with placebo at 48 weeks. |
Observed clinical deterioration rate Follow-up: at the last stage B assessment | 221 (1 RCT) | NR | 53.6 (95% CI, 44.3 to 63.0) | 27.9 (95% CI, 19.6 to 36.3) | −25.7 (95% CI, −38.0 to −11.4) | High | Efgartigimod alfa injection results in a clinically important reduction in clinical deterioration compared with placebo at the last assessment of the randomized withdrawal phase. |
Disability and functional impairment scales | |||||||
Change from stage B baseline in aINCAT (0 [best] to 10 [worst]), mean (SD) Follow-up: at the last stage B assessment | 220 (1 RCT) | NR | 0.9 (SD = 2.0) | 0.1 (SD = 1.1) | −0.8 (95% CI, −1.2 to −0.3)a | Moderateb (serious imprecision) | Efgartigimod alfa injection likely results in an improvement in aINCAT score compared with placebo at the last assessment of the randomized withdrawal phase. The clinical importance of the improvement is unclear. |
Change from stage B baseline in I-RODS (0 [worst] to 100 [best]), mean (SD) Follow-up: at the last stage B assessment | 219 (1 RCT) | NR | −7.0 (SD = 19.1) | 0.8 (SD = 12.3) | 7.8 (95% CI, 3.5 to 12.1)a | Highc | Efgartigimod alfa injection results in a clinically important improvement in I-RODS score compared with placebo at the last assessment of the randomized withdrawal phase. |
Change from stage B baseline in MRC sum score (0 [worst] to 60 [best]), mean (SD) Follow-up: at the last stage B assessment | 220 (1 RCT) | NR | −3.0 (SD = 9.0) | −0.3 (SD = 4.5) | 2.7 (95% CI, 0.9 to 4.6)a | Moderated (serious imprecision) | Efgartigimod alfa injection likely results in an improvement in MRC sum score compared with placebo at the last assessment of the randomized withdrawal phase. The clinical importance of the improvement is unclear. |
HRQoL (EQ VAS) | |||||||
Change from stage B baseline in EQ VAS (0 [worst] to 100 [best]), mean (SE) Follow-up: at the last stage B assessment | 187 (1 RCT) | NR | −10.2 (SD = 2.5) | 0.5 (SD = 1.8) | 10.7 (95% CI, 4.7 to 16.7)a | Highe | Efgartigimod alfa injection results in a clinically important improvement in EQ VAS compared with placebo at the last assessment of the randomized withdrawal phase. |
Harms | |||||||
Proportion of patients with any SAEs Follow-up: 48 weeks | 221 (1 RCT) | NR | 5.5% (NR) | 5.4% (NR) | 0 (95% CI, −6.7 to 6.5)a | Moderatef (serious imprecision) | Efgartigimod alfa injection likely results in little to no difference in SAEs when compared with placebo. |
aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment; CI = confidence interval; CIDP = chronic inflammatory demyelinating polyneuropathy; HR = hazard ratio; HRQoL = health-related quality of life; I-RODS = Inflammatory Rasch-Built Overall Disability Scale; MID = minimal important difference; MRC = Medical Research Council; NR = not reported; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation; SE = standard error; VAS = visual analogue scale; vs. = versus.
Notes: The ADHERE study has been completed, and the data cut-off date (last participant, last visit) was May 11, 2023. Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThis analysis was not part of the statistical analysis plan and was requested from the sponsor by Canada’s Drug Agency to facilitate the Grading of Recommendations Assessment, Development and Evaluation assessment.
bThe level of evidence was rated down by 1 level for serious imprecision, as the 95% CI for the between-group difference crossed the MID identified in the literature (a change of 1 point or more).
cRisk of bias was not rated down. Although the outcome was patient reported (a subjective measure), patients were blinded to treatment allocation during the randomized withdrawal stage. Imprecision was not rated down. Based on the MID identified in the literature (a change of 4 points or more), the point estimate and higher bound of the 95% CI exceeded the MID threshold, while the lower bound was close to the threshold. The clinical experts consulted for this review assessed the point estimate and the 95% CI of the absolute effect to be a clinically important difference.
dThe level of evidence was rated down 1 level for serious imprecision as the 95% CI for the between-group difference crossed the MID identified in the literature (a change of 4 points or more in centile score).
eRisk of bias was not rated down. Although the outcome was patient reported (a subjective measure), patients were blinded to treatment allocation during the randomized withdrawal stage. The impact of missing outcome data (15% of the patients) was unclear; however, no notable between-group imbalances in missing data were identified. The review team was unable to identify an MID from the literature or from the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CI of the absolute effect did not include the null threshold of 0, and imprecision was not rated down.
fThe review team was unable to identify an MID from the literature or from the clinical experts consulted for this review; therefore, the null was used to assess certainty. The 95% CIs of the absolute effects included the null threshold of 0.
Sources: Clinical Study Report for ADHERE;18 sponsor’s submission.28 Details included in the table are from the sponsor’s summary of clinical evidence.19
Long-Term Extension Studies
Description of Studies
The ADHERE study design is described in the Systematic Review section of this report. The ADHERE+ study is a long-term, single-arm, open-label, multicentre, phase II follow-up study of patients who enrolled in the ADHERE study. A summary of the long-term extension study (ADHERE+) is presented in this section to provide evidence on the long-term safety and tolerability of efgartigimod alfa injection in adult patients with CIDP.
The ADHERE+ study was conducted at 125 sites. Patients in the ADHERE+ study were eligible to continue from the ADHERE study if they experienced clinical deterioration (i.e., an increase of ≥ 1 point in the aINCAT disability score) in stage B of the ADHERE study, if they completed the week 48 visit of stage B of the ADHERE study without experiencing clinical deterioration, if the required number of events for the primary end point analysis was reached and the study was stopped, or if they completed the week 48 visit of this OLE study. Patients were excluded if they were pregnant and lactating, if they intended to become pregnant during the trial or within 90 days after the last treatment administration, if they had clinical evidence of another significant serious disease, or if they had undergone a recent major surgery or had a planned major surgery.
The dosing regimen consisted of once-weekly injections up to 48 weeks, with the option to renew for an additional 48-week treatment period. ██ ███ ████████ ██████████ ████████ █████ ███████ ████████████ ████ █████████ ████ ██ ████ █████ ███ █████ ██ ████ █████████ ██ █████ ██ █████ ██ ███████████ ██████ ███ ███ █ ██████ ████████ █████████ ███ ██ █████ ██ ██████ ████████ █████ ███████ ██ ████████████ ████ █████████ ████ █████ █████ █████ ██ ████ ████ ██████ ███ ██ █████ ██ █████ █████ █████████ █████ ████ █████ ███ ██████
The primary end points were the incidence of AEs and SAEs. Secondary objectives of interest included evaluating the long-term treatment effect by assessing the change from baseline over time in aINCAT score, MRC sum score, I-RODS score, and mean grip strength. The percentage of patients who did not experience clinical deterioration over time, defined by an increase in aINCAT score of 1 or more points, was also assessed. Additional secondary end points of interest included the evaluation of patient-reported outcomes, such as patient-reported quality of life, by assessing the change from baseline over time in EQ VAS.
The results presented in this interim analysis reflect the data available as of February 16, 2024.
Patient Disposition
Patient disposition for the ADHERE+ study is summarized in the Supplemental Material document, Appendix 5.
As of the clinical data cut-off date, 229 patients out of 322 patients in the main study had enrolled in the ADHERE+ study: 29 from the run-in or stage A efgartigimod alfa injection group, 99 from the stage B efgartigimod alfa injection group, and 101 from the stage B placebo group.
In total, 228 patients received at least 1 dose or part of a dose of efgartigimod alfa injection. As of the clinical data cut-off date of February 16, 2024, 3 patients (1.3%) had completed the study, 48 (21.1%) had withdrawn from the study, and 177 (77.6%) were ongoing in the study.
A total of 208 patients had been in the study for 24 weeks or longer, 111 for 48 weeks or longer, 33 for 96 weeks or longer, and 6 for 144 weeks or longer. The remaining patients had not yet reached week 24 or had discontinued the study earlier. Additionally, 66 patients (28.9%), 15 patients (6.6%), and 5 patients (2.2%) had entered a second, third, and fourth 48-week treatment period, respectively.
Baseline Characteristics
The median age of the patients was 56.0 years (range, 21.0 to 83.0 years). There was a higher percentage of males (62.3%) than females (37.7%). Most patients were white (62.2%); the next largest group by ethnicity was Asian (29.8%). For the patients included in the ADHERE+ study, the mean total INCAT score, mean I-RODS score, and mean grip strength of the dominant hand at the stage A baseline of the ADHERE study were 4.5 (SD = 1.6), 41.2 (SD = 15.4), and 39.0 kPa (SD = 23.6 kPa), respectively. Most baseline disease characteristics of patients in the ADHERE+ study were balanced between those from the ADHERE stage B efgartigimod alfa injection and ADHERE stage B placebo groups. The median time since CIDP diagnosis was shorter among those from the ADHERE stage B efgartigimod alfa injection group (2.89 years; range, 0.3 to 23.3 years) than from the ADHERE stage B placebo group (3.2 years; range, 0.3 to 29.2 years).
Exposure to Study Treatments
Details of patients’ treatment exposure in the ADHERE+ study are included in the Supplemental Material document, Appendix 5.
As of the clinical data cut-off date of February 16, 2024, 51 patients (22.4%) had discontinued treatment. In the total group, the mean treatment duration was 58.0 weeks (SD = 33.8 weeks), and the mean number of administrations was 56.0 (SD = 32.3) per patient. Efgartigimod alfa injection exposure was variable because patients entered this study at different times. The mean duration of exposure to the efgartigimod alfa injection was 57.4 weeks (SD = 32.2 weeks) for patients in the stage B efgartigimod alfa injection group and 64.1 weeks (SD = 37.7 weeks) for patients in the stage B placebo group. Mean compliance for patients in the total group was 98.5% (SD = 5.0%). Sixty patients had enrolled in the substudy exploring less frequent dosing (once every 2 weeks and once every 3 weeks).
Critical Appraisal
Internal Validity
The ADHERE+ study is open label, which may influence the perception of improvement by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., subjective AEs, patient-reported outcomes). Other limitations as discussed for stage B in the critical appraisal of the Systematic Review section — for example, the potential bias due to varied timing in the assessment of outcomes — could also apply in this long-term study.
External Validity
Because the patients who took part in the open-label long-term safety extension phase were originally from the ADHERE trial, and the eligibility criteria remained the same, similar limitations to generalizability are relevant to the open-label long-term extension phase as well. Moreover, the extension phase excluded patients who had clinical evidence of any other significant serious disease or who had undergone a recent major surgery or had a planned major surgery. It also excluded patients who were pregnant and lactating and those intending to become pregnant during the trial or within 90 days after the last efgartigimod alfa injection. These exclusions may limit the generalizability of the findings to these populations.
Results
Efficacy
Detailed results for outcomes relevant to this review are in the Supplemental Material document, Appendix 5. Key efficacy results include the following:
The mean change from the OLE baseline to the last assessment in the aINCAT score was −0.3 (SE = 0.1) in the stage B efgartigimod alfa injection group and −1.2 (SE = 0.2) in the placebo group.
The mean change from the OLE baseline to the last assessment in the I-RODS score was 3.4 (SE = 1.3) in the stage B efgartigimod alfa injection group and 11.1 (SE = 1.8) in the placebo group.
The mean change from the OLE baseline to the last assessment in the MRC sum score was 1.5 (SE = 0.4) in the stage B efgartigimod alfa injection group and 3.0 (SE = 0.6) in the placebo group.
The mean change from the OLE baseline to week 48 in EQ VAS was 9.7 (SE = 3.8) in the stage B efgartigimod alfa injection group and 18.3 (SE = 4.1) in the placebo group.
Harms
Detailed results for harms are presented in Appendix 5 in the Supplemental Material document. Key results include the following:
In the total group, 75.0% of patients experienced 1 or more AEs.
The most frequently reported AEs were COVID-19 in 16% of patients, upper respiratory tract infections in 10% of patients, nasopharyngitis in 7% of patients, and headache in 6% of patients.
In the total group, 66 SAEs occurred in 15% of patients. The most frequently reported SAEs were CIDP in 3% of patients and COVID-19 in 1% of patients.
AEs leading to efgartigimod alfa injection discontinuation occurred in 8% of patients in the total group.
Two patients died: 1 patient died due to CIDP and 1 patient died due to cardiac arrest.
In the total group, 46% of patients had an AESI. The most reported AESI was COVID-19, which was reported in 16% of patients.
Injection site reactions occurred in 13% of patients.
Indirect Evidence
Direct comparative evidence exists between efgartigimod alfa injection and placebo from the ADHERE trial, but there is a gap in evidence comparing efgartigimod alfa injection with Ig treatments. Indirect comparisons were required to address this gap.
Description of ITC
The sponsor submitted 1 ITC comprising 20 MAICs comparing efgartigimod alfa injection with Ig treatments for CIDP on symptom-related outcomes in adult patients.
Study Selection and Review Methods
The ITC was informed by a systematic literature review (SLR) of evidence in patients with CIDP (including typical and/or atypical presentations). Studies with efgartigimod alfa injection, oral or IV corticosteroids (including oral dexamethasone, IV methylprednisolone, oral prednisone, or prednisolone), or Ig (IVIg, SCIg, or both) for induction and/or maintenance treatment as an intervention were eligible. Immunosuppressives such as rituximab, cyclosporine, or cyclophosphamide were not included as interventions of interest in the sponsor-commissioned SLR. Relevant outcomes for the SLR included INCAT and/or aINCAT score, I-RODS score, MRC sum score, mean grip strength, Timed Up and Go score, and safety. Eligible study designs included RCTs, single-arm trials, and observational studies (including cross-sectional, case-control, cohort, or retrospective cohort); case reports or case series with fewer than 5 patients and preclinical studies were excluded.
The database search included PubMed and the Cochrane Library. Clinical trial registries (i.e., Prospero International Prospective Register of Systematic Reviews and ClinicalTrials.gov), health technology assessment submissions, and government and international body websites were also searched. The search was limited to publications from 2003 onward. The sponsor previously completed a targeted literature review that identified 62 studies on Ig in patients with CIDP. The list of eligible articles from this targeted literature review was obtained so that these articles could be screened for eligibility in the SLR. Titles, abstracts, and full text were each reviewed by 2 independent reviewers. Conflicts were discussed to decide whether the record should proceed to the next stage of screening. Where a disagreement could not be resolved, a conservative approach was taken and the record proceeded to the next stage of screening. Data extraction was performed by 1 reviewer, and a random 10% of extracted papers were checked by a second reviewer, as the initial error rate was less than 10%. Quality assessments were not done at this stage of the SLR. Risk of bias in the studies included in the ITC analyses was assessed at the study level using the Clinical Appraisal Skills Programme RCT checklist29 by 2 independent reviewers, based solely on published information. Discrepancies were resolved through discussion without the need for a third reviewer, and each study received a subjective overall rating (good, fair, poor); individual scores were presented, but no studies were excluded based on this assessment.
ITC Analysis Methods
For additional information on the analysis methods of the sponsor-submitted ITC, refer to Appendix 6 in the Supplemental Material document.
In the sponsor-submitted ITC, Ig treatments were considered the most appropriate comparator for efgartigimod alfa injection in patients with CIDP, as it is the current standard of care according to the clinical experts consulted by the sponsor and CDA-AMC. PLEX was excluded from the ITC as it represents a nondrug comparator that is also rarely used in clinical practice in Canada (less than 5% of patients) according to the clinical experts consulted by the sponsor and CDA-AMC. Corticosteroids (e.g., dexamethasone, methylprednisolone, prednisone) and immunosuppressives (e.g., rituximab, cyclosporin, cyclophosphamide) were excluded as potential comparators because indirect comparisons were not feasible according to the sponsor’s feasibility assessment. Adjusted potential prognostic factors and/or treatment effect modifiers in the ITC analyses included age, sex, disease duration and/or time from diagnosis, prior treatment with Ig, prior treatment with corticosteroids, treatment exposure, total INCAT score, aINCAT score, I-RODS score, mean grip strength (dominant hand), and MRC sum score; these factors were identified based on feedback from 2 clinical experts consulted by the sponsor. CIDP type (typical versus atypical), symmetric versus asymmetric symptoms, and CDAS score were also identified as potential prognostic factors and/or treatment effect modifiers by at least 1 clinical expert consulted by the sponsor but were not reported in any of the comparator studies at baseline; thus, they were not adjusted for. In each analysis, adjustment for effect modifiers was limited to the factors listed in this paragraph that were consistently reported in the ADHERE study and the comparator study under evaluation.
The sponsor evaluated the feasibility of assessing outcomes at weeks 12, 25, 32, and 48. A connected network of evidence was available for some of the key end points in stage B of the ADHERE study: time to aINCAT deterioration and change from stage B baseline in aINCAT, I-RODS, and mean grip strength. However, given the differences between studies in the patient populations and the time points at which the end points were evaluated, the sponsor concluded that the targeted comparisons based on population adjustment using MAICs were considered more appropriate than a network meta-analysis. Thus, the sponsor-conducted ITC included:
6 unanchored MAICs evaluating 4 efficacy outcomes (change from stage A baseline in aINCAT score, change from stage A baseline in I-RODS score, change from stage A baseline in grip strength [dominant hand], and time to initial aINCAT response) at weeks 13 or 25
14 anchored MAICs evaluating 6 efficacy outcomes (time to first aINCAT deterioration, change from stage B baseline in aINCAT score, change from stage B baseline in I-RODS score, change from stage B baseline in MRC sum score, and change from stage B baseline in grip strength [dominant hand and nondominant hand]) at weeks 25, 32, or 48.
For end points with data from multiple Ig treatments, fixed-effects meta-analyses (with random effects as sensitivity checks yielding identical results) were used to compare efgartigimod alfa injection versus Ig treatments. This report focused on aINCAT score, I-RODS score, and MRC sum score, as they were considered potentially important for informing the reimbursement recommendation. The MAIC weights were calculated using the method of moments and adjusted for population characteristics to estimate the relative treatment effect of efgartigimod alfa injection versus placebo. Both the effective sample size and the relative weight distribution were calculated for each MAIC to evaluate whether very high weights were assigned to a small number of patients. An unadjusted comparison of the relative effect of efgartigimod alfa injection versus placebo and the relative effect of Ig versus placebo was conducted as a sensitivity analysis.
Summary of Included Studies
Overview: In total, the SLR identified 78 eligible studies. Only studies that included efgartigimod alfa injection or Ig treatments and that reported at least 1 outcome also reported in the ADHERE trial were included. The ITCs ultimately included 4 studies of patients with CIDP: 1 assessed HyQvia (facilitated SCIg 10%) versus placebo (ADVANCE-CIDP 1 study; N = 62); 1 assessed Hizentra (20% SCIg; low dose = 0.2 g/kg; high dose = 0.4 g/kg) versus placebo (PATH study; N = 57 for low dose; N = 58 for high dose), 1 assessed efgartigimod alfa injection versus placebo (ADHERE study; N = 322 for stage A; N = 111 for stage B), and 1 assessed Privigen (10% liquid human IVIg, stabilized with L-proline) (PRIMA study; N = 28).
Study characteristics: All 4 trials evaluated Ig-based or novel therapies in adults with CIDP, though they differed in design, treatment modality, and end points. The PATH and ADVANCE-CIDP 1 studies were phase III, randomized, double-blind, placebo-controlled trials assessing SCIg (Hizentra and HyQvia, respectively) as maintenance therapy following IVIg stabilization. The PRIMA study was a phase III, single-arm, open-label study evaluating IVIg (Privigen) alone, and the ADHERE study was a 2-stage phase II trial investigating efgartigimod alfa injection, a novel FcRn antagonist, with an open-label induction phase followed by a randomized withdrawal phase. Across trials, relapse or response was primarily measured using the aINCAT score, though definitions varied slightly (e.g., single versus consecutive assessments). The sample sizes ranged from 28 (PRIMA study) to more than 300 (ADHERE study), and all trials included safety end points. The ADHERE and ADVANCE-CIDP 1 studies incorporated patients who both had and had not received previous treatment, while the PATH and PRIMA studies focused on patients whose disease stabilized or was responsive to prior IVIg. Differences also emerged in treatment duration, with randomized phases ranging from 24 to 48 weeks, and in the use of adjunctive functional measures such as grip strength, MRC sum score, and I-RODS score. The overall study quality was rated “good” for the ADHERE, PATH, and ADVANCE-CIDP 1 trials and “fair” for the PRIMA trial due to the open-label design. The sponsor narratively summarized AEs in the included studies.
Patient characteristics: All 4 trials enrolled adult patients (aged ≥ 18 years) with a diagnosis of definite or probable CIDP, generally based on European Federation of Neurological Societies–Peripheral Nerve Society 2010 criteria. While all trials included patients who had received prior Ig or corticosteroid treatment, the ADHERE and PRIMA trials also included participants whose disease had not previously been treated. Differences emerged in baseline disease activity requirements: the ADHERE study required a CDAS of at least 2 and an INCAT score of at least 2, while the ADVANCE-CIDP 1 study allowed baseline INCAT scores of 0 to 7 but required patients with low baseline scores to have historical documentation of an INCAT score of at least 2. The PRIMA study required at least a 1-point deterioration in aINCAT score during washout, and the PATH study required IVIg responsiveness but did not specify a score threshold. The studies’ exclusion criteria were broadly similar, focusing on other causes of neuropathy or neurologic conditions, though the specifics on the criteria varied (e.g., the ADHERE study excluded patients with pure sensory CIDP; the PRIMA study excluded patients with multifocal motor neuropathy and distal acquired demyelinating symmetric neuropathy).
The patients’ baseline characteristics showed variability across the trials. The median age ranged from 54.5 to 58.9 years. The proportion of female patients ranged from 26.7% to 46.6%, while male patients accounted for 53.4% to 73.7% of the population across the trials. Functional disability and strength scores (aINCAT score, I-RODS score, MRC sum score, grip strength) were reported in all trials, with ADHERE applying statistical weighting to align its population with the PATH and ADVANCE-CIDP 1 studies. Corticosteroid pretreatment rates also varied, reflecting differences in prior treatment exposure across study populations. Additional information on the patient characteristics of the sponsor’s MAICs can be found in Appendix 6 in the Supplemental Material document.
Critical Appraisal of ITCs
The search for potentially eligible studies was reasonably comprehensive, and the methods used to select relevant studies, extract data, and assess the risk of bias of the included studies were adequate. The adjustment for confounders and effect modifiers included all variables reported in both the ADHERE study and comparator studies, with key prognostic or effect modifiers informed by clinical expert input. However, the residual bias due to potential unknown confounders cannot be ruled out. Although MAICs provide comparative efficacy estimates for efgartigimod alfa injection versus Igs, the lack of comparative efficacy of efgartigimod alfa injection versus other comparators that are relevant to clinical practice in Canada may limit the contextual interpretation and generalizability of the findings.
In the sponsor-submitted ITC, the analyses were limited to data from SCIg treatments, with no results reported for IVIg. The impact of this limitation may be minimal: the clinical experts consulted by CDA-AMC indicated that the treatment effect would be expected to be similar between SCIg and IVIg in clinical practice. Yet the ITC included only patients previously treated with Ig due to the limited data in comparator trials on patients whose disease had not yet been treated with Ig, leaving the comparative treatment effect of efgartigimod alfa injection in patients who had not previously been treated with Ig unknown. This ITC did not reflect the sponsor’s reimbursement request, which has no restrictions on Ig exposure and limits the generalizability of the findings to the broader patient population.
The patient baseline characteristics showed heterogeneity across trials, which may introduce bias and uncertainty. Adjustment for prognostic factors and/or effect modifiers was limited to those consistently reported in both the ADHERE study and the comparator studies. As a result, not all relevant effect modifiers were accounted for, and the potential for residual confounding remains; the magnitude and direction of bias introduced by these limitations is uncertain.
The use of fixed-effects meta-analyses in the MAICs to compare the treatments of efgartigimod alfa injection versus Igs was reasonable given the availability of data from multiple Ig treatments. However, this approach assumed homogeneity across studies, which may not hold in the presence of clinical or methodological heterogeneity across trials. Although random-effects models were used as sensitivity analyses and yielded identical results, this does not confirm the absence of heterogeneity, particularly because the sponsor did not report any formal assessment of between-study variability. The assumption of no effect modification across trials was not supported, and differences in patient populations, study designs, and end point definitions may still bias the results. Although the fixed-effects model may offer precision, the potential for residual confounding and unaccounted heterogeneity complicated the interpretation of the comparative efficacy estimates.
A significantly reduced effective sample size (approximately 8% to 49% reduction across various analyses), after matching, would have resulted in a highly selective patient population in the comparison, which further compromised the generalizability of the results. Moreover, the resulting sample size led to wide 95% CIs, indicating notable imprecision of the findings in this ITC. HRQoL was identified by patient groups as an important outcome. However, no indirect comparison was conducted for HRQoL.
Efficacy and Harms Results of ITCs
Key results of the ITCs are given in Table 5. The analyses using data from the PRIMA study compared to stage A of the ADHERE trial were not included in this review due to the small effective sample size (ranging from ███ ██ ███ ██████████ ███ ████ ██ ████ of the available population). In the sponsor-submitted MAICs, the following effect modifiers and/or prognostic factors were included in the adjusted model: age, sex, disease duration and/or time from diagnosis, prior treatment with Ig, prior treatment with corticosteroids, treatment exposure, total INCAT score, aINCAT score, I-RODS score, mean grip strength (dominant hand), and MRC sum score. The effective sample size was ████ ██ ████ for efgartigimod alfa injection versus low-dose Hizentra, ████ ██ ████ for efgartigimod alfa injection versus high-dose Hizentra, and ████ ██ ████ for efgartigimod alfa injection versus HyQvia.
Efficacy
Across outcomes, efgartigimod alfa injection showed favourable trends compared with both Hizentra and HyQvia. The results of all sensitivity analysis (using data only up to 25 and 32 weeks from the ADHERE trial to match the same time point as the PATH trial and ADVANCE-CIDP 1 trial, respectively) were consistent with the primary analysis.
Table 5: Summary of MAIC Results for Efgartigimod Alfa Injection vs. SCIg (Hizentra and HyQvia)
Outcome | ADHERE (Efgartigimod alfa) Number/ESS | PATH (Hizentra) | ADVANCE-CIDP 1 (HyQvia) (N = 132) | |
|---|---|---|---|---|
Low dosea (N = 114) | High dosea (N = 115) | |||
Change from baseline in aINCAT | ||||
Difference in means up to 25 weeks for efgartigimod alfa vs. SCIg (Hizentra), points (95% CI) | N = 91 ███ █ ████ ███ ████████ ████████ ███ ████ ███ █████████ ████████ | –1.13 (–2.05 to –0.21) | –0.58 (–1.45 to 0.29) | NA |
Difference in means up to 32 weeks for efgartigimod alfa vs. SCIg (HyQvia), points (95% CI) | N = 96 ███ █ ████ | NA | NA | –0.88 (–1.72 to –0.04) |
Meta-analysis of difference in means up to 32 weeks for efgartigimod alfa vs. SCIg,a points (95% CI) | NA | –0.85 (–1.36 to –0.35) | ||
Change from baseline in I-RODS | ||||
Difference in means up to 25 weeks for efgartigimod alfa vs. SCIg (Hizentra), points (95% CI) | N = 91 ███ █ ████ ███ ████████ ████████ ███ ████ ███ █████████ ████████ | 11.35 (3.00 to 19.71) | 9.35 (0.78 to 17.93) | NA |
Difference in means up to 32 weeks for efgartigimod alfa vs. SCIg (HyQvia), points (95% CI) | N = 96 ███ █ ████ | NA | NA | 12.16 (2.67 to 21.65) |
Meta-analysis of difference in means up to 32 weeks for efgartigimod alfa vs. SCIg,a points (95% CI) | NA | 10.89 (5.83 to 15.95) | ||
Change from baseline in MRC sum score | ||||
Difference in means up to 32 weeks for efgartigimod alfa vs. SCIg (HyQvia), points (95% CI) | N = 96 ███ █ ████ | NA | NA | 2.83 (–0.82 to 6.47) |
aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment; CI = confidence interval; ESS = effective sample size; I-RODS = Inflammatory Rasch-Built Overall Disability Scale; MAIC = matching-adjusted indirect comparison; MRC = Medical Research Council; NA = not applicable; SCIg = subcutaneous immunoglobulin; vs. = versus.
aFixed effects meta-analyses were used to compare efgartigimod alfa injection vs. Ig treatments (Hizentra [high and low doses] and HyQvia) based on results from the ADHERE, PATH, and ADVANCE-CIDP 1 studies. Data were available for up to 25 weeks in the PATH trial and 32 weeks in the ADVANCE-CIDP 1 trial.
Source: Sponsor’s indirect treatment comparison technical report.30
Harms
The sponsor performed a narrative synthesis to qualitatively summarize safety data at the treatment level, based on safety data from multiple studies for Igs. Due to substantial heterogeneity in the reporting of safety outcomes, particularly in time frames and grading of nonserious AEs, the sponsor did not conduct any formal statistical analysis and comparative analysis between treatments for the safety outcomes in the sponsor-submitted ITC.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the systematic review evidence were included in the review.
Discussion
This report summarizes the evidence for efgartigimod alfa injection for the treatment of adults with CIDP based on 1 phase II study, 1 OLE, and 1 ITC.
Efficacy
CIDP is a rare, immune-mediated neuropathy with significant clinical burden. Despite the availability of first-line therapies, many patients experience suboptimal responses or treatment-related challenges, underscoring the need for more effective and convenient options. Efgartigimod alfa injection helps reduce inflammation and nerve damage by inhibiting the FcRn, thereby accelerating the removal of pathogenic IgG autoantibodies associated with CIDP.
The pivotal ADHERE study enrolled a geographically diverse population of 342 patients and employed rigorous diagnostic confirmation by an independent adjudication committee, an important consideration given the high rates of misdiagnosis and diagnostic uncertainty commonly reported in CIDP.
In stage B, the primary outcome was time to first event of clinical deterioration, defined as a change from baseline of 1 or more points (the MID identified in previous CIDP studies31,32) in the aINCAT score.18,33,34 Although INCAT31,35 or aINCAT31,36 score is not routinely used in clinical practice, it is a validated measure of functional disability in CIDP20-22,25,37,38 and was considered a reasonable and clinically meaningful end point by the clinical experts consulted for this review.
During the 12-week stage A treatment period, 66.5% of patients achieved confirmed improvements, with a median time to initial confirmed improvements of 43 days (95% CI, 31 to 51 days). In stage B, efgartigimod alfa injection reduced the risk of clinical deterioration compared with placebo at both 24 weeks (between-group difference = −28%) and 48 weeks (between-group difference = −26%), with a hazard ratio of 0.39 (95% CI, 0.25 to 0.61), indicating a 60% risk reduction in the event of clinical deterioration. The treatment effect was maintained throughout the long-term extension phase. The clinical experts assessed the difference in risk between treatment groups to be clinically meaningful. Subgroup analyses were conducted based on prior therapies (in both stage A and stage B), aINCAT score changes during stage A (for stage B subgrouping), CIDP disease activity as measured by the CDAS score, and age. These analyses, detailed in the Supplemental Material document, revealed no major concerns of differential effect compared to the primary results. The clinical experts confirmed that efgartigimod alfa injection is suitable for patients who both have and have not received prior treatment for the disease. While the observed treatment effect was derived from a highly selected population largely pretreated with CIDP therapies, generalizability to typical clinical practice in Canada may still hold because most patients receive Ig or corticosteroids soon after diagnosis.
Improvements were observed across multiple validated efficacy measures compared to placebo, including aINCAT score, I-RODS score, grip strength, and Timed Up and Go scores, and were sustained in the treatment arm. The aINCAT score remained stable in the efgartigimod alfa group during stage B, with a mean change of 0.1 (SE = 0.1) from baseline to last assessment. In contrast, the placebo group experienced a mean deterioration of 0.9 (SE = 0.19). The I-RODS results supported the treatment effect of efgartigimod alfa injection compared to placebo. A higher proportion of patients achieved functional improvement (≥ 4-point increase in I-RODS score) in the efgartigimod alfa injection group than in the placebo group (45% versus 36%). The mean change in I-RODS score from baseline to last assessment during stage B was 0.8 points (SE = 1 point) in the efgartigimod alfa injection group versus –7.0 points (SE = 2 points) in the placebo group, suggesting preservation of function and prevention of decline with efgartigimod alfa treatment. Additional data on grip strength and Timed Up and Go scores are provided in Appendix 4 in the Supplemental Material document.
Muscle function, as measured by the objective measure of MRC sum score, showed relative stability in the efgartigimod alfa injection group (mean change from stage B baseline = –0.3; SE = 0.4), while the placebo group experienced a greater decline (–3.0; SE = 0.9), suggesting that efgartigimod alfa injection may help maintain neuromuscular strength in patients with CIDP.
The patient and clinician groups, along with the clinical experts, considered HRQoL an outcome of importance. The EQ VAS used in the ADHERE study is a validated, generic, self-reported instrument that assesses 5 dimensions: mobility, self-care, usual activities, pain and discomfort, and anxiety and depression. Based on this measure, an increase of 11 points (SE = 1 point) was observed from baseline to the last assessment during stage A. This improvement was sustained during stage B among patients receiving efgartigimod alfa injection. From the baseline of stage B to the last assessment, the treatment group demonstrated a mean increase of 0.5 points (SE = 2 points), while the placebo group showed a decline in mean score (–10 points; SE = 2.5 points). The between-group mean difference was 11 points (95% CI, 5 to 17 points) in favour of efgartigimod alfa injection. The clinical experts noted that this difference is clinically meaningful in terms of patients’ perceived health status.
Efficacy outcomes for aINCAT score, I-RODS score, MRC sum score, grip strength, and EQ VAS score during the OLE were consistent with those observed in the ADHERE study.
███ ████████ ███████ ████ ██████████ ███ ██████████ ██ ███████ ████████████ ██ ████████ ██ ███ ███████ ███ ██████ ██████ ████████████ █████ █████████ ████ █ ████████ ████ ████ ██ ████ ███ ██████ ██████ ███ ██████ ██████ ██ ████ ███ ██████ ██ ███ ████ ██████████ ██ █████ ██ ██ █████ ██ █████ ████████ ███ █████████ ██ ███ ████████████ ████ █████████ ██████ ███ ████ ████ █████ ████████ ███████ ████████ ████████ ████ ████ ███ ██ █████ █ ████████ ██ ████ ███ ██ ███ ████ ███████████ ██ █████████ ███ ███████ █████ ██████ █ ██████ ███████ ████ ████ ███ ██ ████ ███ ███████ █████ ████████ ███████ █████████ ███████ ██████ ██ ████████████ ████ ████████████ ████ █████████ ███ ███████ ██████
Treatment adherence is important and can impact the efficacy of a CIDP therapy.19 In the ADHERE study, only authorized site staff or a trained home nurse administered the IMP, and compliance might exceed 100% because the calculation assumed dosing every 7 days; however, some patients received treatment earlier within the allowed window, potentially leading to 1 less expected dose (Supplemental Material document, Appendix 3).18,34 Patient compliance with the IMP was high throughout both stage A (median = 100%; range, 55% to 200%) and stage B (efgartigimod alfa: median = 100%; range, 73% to 133%; placebo: median = 100%; range, 74% to 200%). Moreover, each patient received individualized training for self-administration in preparation for anticipated use in the OLE trial (ADHERE+ study). The median number of training sessions required before a patient was deemed capable of self-administration was 3 (range, 1 to 16). Mean compliance across all patients was high at 98.5% (SD = 5.0%) during the ADHERE+ study (N = 228). Both patient and clinician groups, along with the clinical experts, noted that the SC route of administration and the potential for home-based care are particularly relevant in the Canadian context, where access to infusion services may be limited in rural or underserved areas. This approach may help address existing unmet needs.
Evidence from the ADHERE study showed that efgartigimod alfa injection helps address several unmet needs in the management of CIDP by offering a targeted and potentially more convenient treatment option. The outcome data showed a clinically meaningful benefit compared to placebo in patients with CIDP who experienced response to efgartigimod alfa injection, particularly in reducing relapse rates and improving functional and HRQoL outcomes. The study population is broadly representative of the CIDP population in Canada, including individuals whose disease both has and has not been previously treated, and is characterized by a predominance of typical CIDP phenotypes. The inclusion of participants with varying degrees of disability, from mild to severe, supports the generalizability of the findings.
However, certain limitations of the ADHERE study should be acknowledged and taken into account when interpreting the results. The absence of an active comparator limits the ability to draw direct comparisons with standard therapies such as IVIg, SCIg, or corticosteroids. Additionally, while the randomized withdrawal design offers practical and ethical advantages, it may introduce potential carryover effects and challenges in assessing ongoing disease activity.
Given the lack of direct comparative evidence between efgartigimod alfa injection and relevant comparators, the sponsor-conducted ITCs using 6 MAICs to estimate the relative efficacy of efgartigimod alfa injection compared with Igs in patients with CIDP. Overall, the results of the MAICs suggested that efgartigimod alfa injection likely results in a clinically meaningful difference in I-RODS score and little to no clinically meaningful difference in aINCAT score and MRC sum score up to 32 weeks, when compared with Igs. However, the reduced effective sample size as shown by ██ ██ ███ ██ ███ and wide 95% CIs suggest the lack of precision of the treatment effect results. In addition, the MAICs were further limited by recognized heterogeneity as shown in baseline characteristics across trials, which potentially results in a biased estimate of comparative efficacy. In addition, the MAICs were only conducted for patients who had been pretreated with Ig due to limited data in comparator trials on patients who had not received prior Ig treatment, which may not fully reflect the reimbursement request (with no restrictions on Ig exposure) and may not be generalizable to all patients with CIDP. Therefore, the MAIC results should be interpreted with caution, taking into consideration the limitations due to the lack of precision and the key differences in the trial population.
Harms
The safety profile of efgartigimod alfa injection was generally consistent with findings from previous studies39-42 and was considered acceptable by the clinical experts. Over the ADHERE study period of up to 60 weeks, the treatment was well tolerated. SAEs occurred in 6.5% of the patients during stage A (12 weeks plus an optional additional week); in stage B (48 weeks), SAEs were reported in 5.4% of patients in the efgartigimod alfa injection group and 5.5% in the placebo group. These SAEs did not follow a specific pattern (Supplemental Material document, Appendix 4), and no new safety signals emerged. The proportions of patients who reported an AE classified under “infection and infestations” system organ class were comparable between treatment groups (efgartigimod alfa: 31.5%; placebo: 33.6%), with no opportunistic infections reported. The ongoing OLE study (ADHERE+ study), with up to 48 weeks of follow-up, has shown consistent safety outcomes. Although efgartigimod alfa injection reduces circulating IgG levels, it does not impair IgG production, potentially preserving immune function. While the reduction in IgG levels warrants consideration of infection risk, the overall safety data were deemed expected and not concerning by the clinical experts. Nevertheless, the product monograph advises caution with immunization practices and recommends monitoring for infections during treatment, as live or live-attenuated vaccines are not recommended.43
No formal statistical analysis was conducted for harms in the sponsor-submitted ITC.
Ethics and Equity Considerations
The ADHERE study was conducted across Asia, Europe, and North America, with most stage A patients enrolled from China (18%), the US (15%), and Japan (7.5%). No patients were from Canada, nor were any identified as belonging to an Indigenous population in Canada. There is limited evidence on the efficacy and safety of efgartigimod alfa injection in patients in Canada or Indigenous populations. While study findings may be broadly generalizable, the absence of specific outcomes for diverse groups presents limitations for clinical and health systems decision-making, raising important ethical considerations around equitable access and representation.
Qualitative insights gathered through patient group input from Muscular Dystrophy Canada highlight the lived experiences of individuals with CIDP in Canada who accessed efgartigimod alfa injection. Two participants reported rapid, meaningful improvements in mobility, strength, and daily functioning with efgartigimod alfa injection, highlighting greater independence in daily life and valuing the stable symptom relief and convenience of home administration, which reduced treatment burden and enhanced autonomy.
The clinical experts consulted for this review noted that while CIDP does not disproportionately affect systemically marginalized populations in terms of prevalence, patients in nonurban or remote areas may face inequities in access to timely diagnosis and treatment due to limited availability of neuromuscular specialists. The SC administration of efgartigimod alfa may help reduce geographic barriers to care by offering a more accessible and convenient treatment option that promotes patient autonomy and may lessen caregiver burden. It is important to recognize that access barriers extend beyond geography and include socioeconomic and other systemic factors. More accessible and convenient options like efgartigimod alfa injection can support home-based care, which some patients and caregivers may prefer, potentially improving adherence and addressing certain access challenges. However, as patient monitoring is required, some barriers to access may persist.
Conclusion
One double-blind, phase II, randomized withdrawal trial suggested that in adults with CIDP, efgartigimod alfa injection results in a clinically meaningful reduction in the risk of clinical deterioration (CIDP relapse) compared to placebo. At the last assessment of the randomized withdrawal phase, efgartigimod alfa injection is likely associated with an improvement in disability and functional impairment, based on aINCAT score, I-RODS score, and MRC sum score. Patients’ quality of life as measured by EQ VAS may be improved with efgartigimod alfa treatment, and patient satisfaction with the treatment remained unchanged. These beneficial effects were likely maintained or preserved over the long-term extension phase. No notable safety concerns were identified with efgartigimod alfa injection during the study period. Efgartigimod alfa injection also offers a more convenient alternative to existing therapies, as a weekly SC injection that may be self-administered at home, potentially reducing nonclinical treatment burden on patients and caregivers. Its short administration time may further contribute to reducing this burden.
The ITC suggests that when compared with Igs, efgartigimod alfa injection likely results in a clinically meaningful difference in I-RODS score and little to no clinically meaningful difference in aINCAT score and MRC sum score up to 32 weeks. However, interpretation of the results is compromised by the significant amount of clinical and methodological heterogeneity and the imprecisions of the results across all assessed outcomes. No conclusion can be drawn for harms due to the lack of formal statistical analysis and the absence of comparative evaluation across treatments.
Economic Review
Methods
The review team appraised the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of efgartigimod alfa compared to Ig, as either IVIg or SCIg, for the treatment of adult patients with CIDP.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of efgartigimod alfa from the perspective of a public health care payer in Canada over a lifetime horizon (55 years). The modelled population comprised patients aged 18 years and older with CIDP, which is aligned with the Health Canada indication and was based on the participants in the ADHERE trial. The sponsor’s base-case analysis included costs related to drug acquisition, administration, AEs, and health care resource use.
In the sponsor’s base case, efgartigimod alfa was associated with incremental costs of $6,542,660 and 4.40 incremental QALYs relative to Ig. This resulted in an incremental cost-effectiveness ratio (ICER) of $1,488,146 per QALY gained. Of the incremental benefit compared to Ig (4.40 incremental quality-adjusted life-years [QALYs]), approximately 88% of the benefit was predicted to be accrued after the treatment duration of the ADHERE trial (mean observation period = 24 months). Additional information about the sponsor’s submission is summarized in the Supplemental Material document, Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 6; full details are provided in the Supplemental Material document, Appendix 4).
Table 6: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The model structure, which was based on aINCAT score, may not adequately reflect the management of CIDP in clinical practice and does not represent homogenous health states. | The definitions of health states were based on arbitrary aINCAT score cut-offs, and how clinically meaningful they were is unknown. Clinicians also noted that patients within each health state may have very different levels of physical function due to the pooling of arm and leg scores to obtain combined aINCAT scores. The use of aINCAT-based health states also does not incorporate aspects of disease important to patients such as nerve pain and fatigue. | CDA-AMC could not address this issue in the base case due to limitations in the model structure. | No scenario analysis was conducted. |
The magnitude of the clinical effectiveness of efgartigimod alfa versus Ig is highly uncertain based on the indirect evidence submitted by the sponsor. | There is no head-to-head evidence comparing efgartigimod alfa versus Ig. The sponsor-conducted ITC indicates that there is likely little to no clinically meaningful difference in aINCAT score from baseline comparing efgartigimod alfa versus Ig. | Due to lack of direct evidence and limitations with the comparative evidence used by the sponsor in the pharmacoeconomic analysis, the cost-effectiveness of efgartigimod alfa versus Ig is highly uncertain. CDA-AMC was unable to address limitations with the clinical data. | No scenario analysis was conducted. |
Relevant comparators were excluded from the analysis. | The clinical experts consulted by CDA-AMC and input received for this review indicate that standard of care in Canada for CIDP also includes PLEX, corticosteroids, and immunosuppressive treatments (e.g., rituximab, cyclosporin, cyclophosphamide) and that efgartigimod alfa has the potential to replace or complement these comparators should it be reimbursed. | CDA-AMC was unable to address limitations with the available comparator data. The cost-effectiveness of efgartigimod alfa versus these comparators could therefore not be determined. | No scenario analysis was conducted. |
The impact on caregiver disutility is uncertain, and the utility value for the very severe impairment health state is uncertain. | No evidence was available on caregiver disutility in the indicated population, and the proxy values used were uncertain given the uncertainty in how health states were defined. Furthermore, the utility value for the very severe impairment health state implied a state of health worse than death, which may not meet face validity according to clinical expert feedback obtained for this review. | CDA-AMC removed caregiver disutility from the reanalysis. | CDA-AMC tested the impact of adopting a utility value of 0.00 for the “very severe impairment” health state but determined that this was minimally impactful on the estimated ICER. |
The cost of Ig is unknown. | The sponsor assumed the cost of $100.00 per mg based on an INESSS publication.44 The cost of Ig is confidential, and the true price remains unknown. | CDA-AMC could not address this limitation in reanalysis. | If the true price of Ig is lower or higher than that estimated by the sponsor, the ICER comparing efgartigimod alfa versus Ig would be underestimated or overestimated, respectively. |
aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment; CDA-AMC = Canada’s Drug Agency; CIDP = chronic inflammatory demyelinating polyneuropathy; ICER = incremental cost-effectiveness ratio; Ig = immunoglobulin; INESSS = L’Institut national d'excellence en santé et en services sociaux; ITC = indirect treatment comparison; PLEX = plasma exchange.
Note: Full details of the issues identified by CDA-AMC are provided in the Supplemental Material document, Appendix 3.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to the Supplemental Material document, Appendix 11, Table 23), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in the Supplemental Material document, Appendix 11.
Impact on Health Care Costs
Efgartigimod alfa is predicted to be associated with additional health care costs compared to Ig (incremental costs = $6,405,231). This increase in health care spending results from higher drug acquisition costs associated with efgartigimod alfa (an additional $6,525,798 per patient). There are reductions in nondrug costs associated with efgartigimod alfa (cost savings of $62,951 per patient through reduced subsequent therapy costs and $47,082 in cost savings per patient from reduced hospitalizations) (refer to Figure 1).
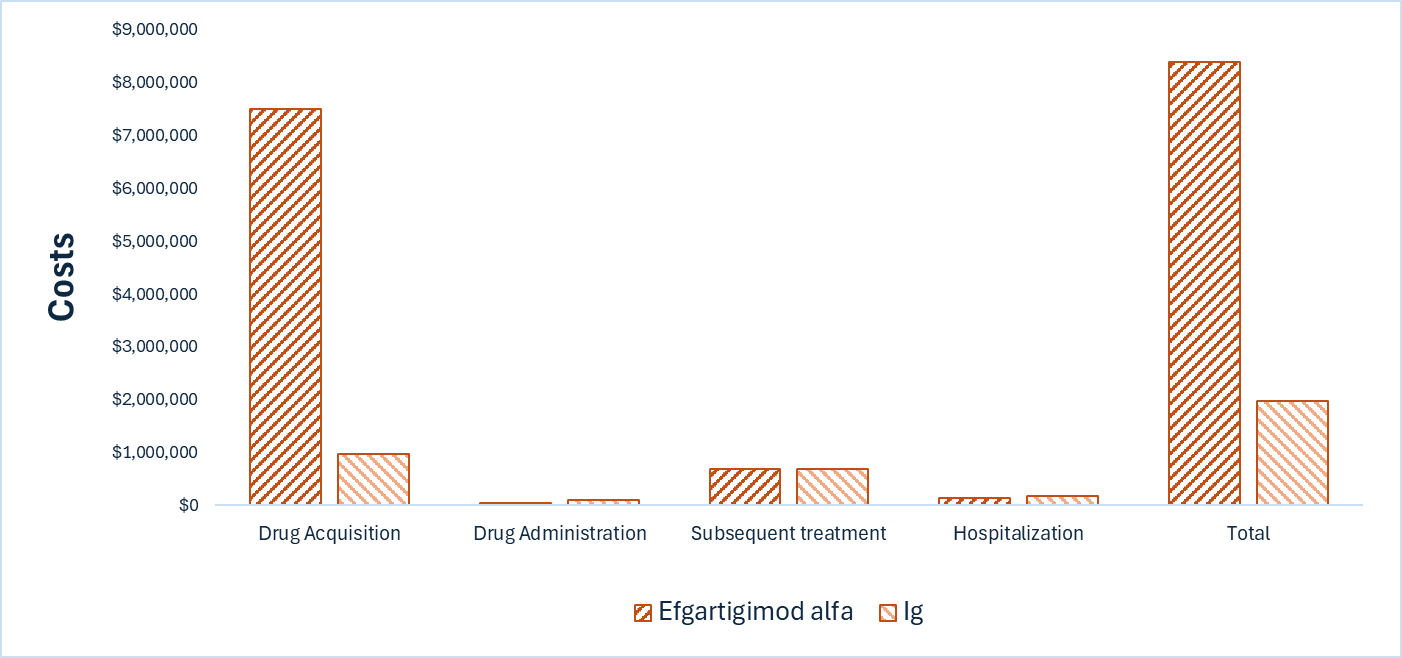
Impact on Health
Considering the impact of treatment on both quality and length of life, efgartigimod alfa is predicted to result in 3.42 additional QALYs per patient compared to Ig (refer to Figure 2). Approximately 87% of the predicted incremental benefit was accrued on the basis of extrapolation. Most incremental QALYs are generated in the very limited impairment and mild impairment health states. Negative QALYs are accrued during the very severe impairment health states, signifying a health state worse than death. Negative QALYs are also accrued due to disutility with drug administration. In both cases, a higher number of negative QALYs are accrued with Ig.
Figure 2: Impact of Efgartigimod Alfa vs. Ig on Patient Health
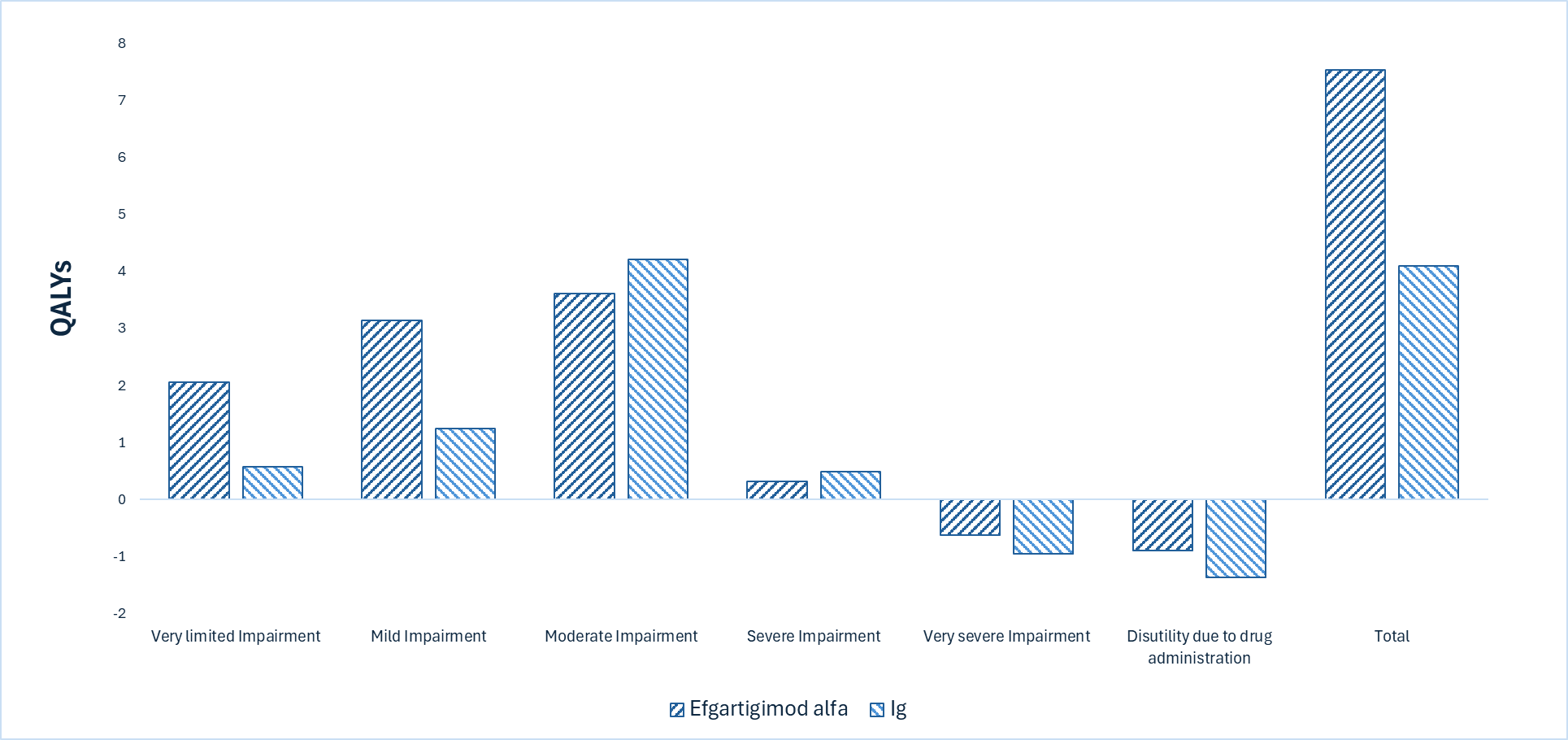
Ig = immunoglobulin; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $1,872,111 per QALY gained for efgartigimod alfa compared to Ig (refer to Table 7). Additional details on the CDA-AMC base case are available in the Supplemental Material document, Appendix 4.
Table 7: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. Ig ($/QALY) |
|---|---|---|---|
Ig | 1,981,029 | 4.10 | Reference |
Efgartigimod alfa | 8,386,260 | 7.53 | 1,872,111 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; Ig = immunoglobulin; QALY = quality-adjusted life-year; vs. = versus.
Note: The cost of Ig was sourced from an Institut national d’excellence en santé et en services sociaux (INESSS) publication on the optimal use of IV immunoglobulin.44
Uncertainty and Sensitivity
There is remaining uncertainty in the analysis associated with lack of head-to-head evidence versus comparators, model structure limitations, and the unknown price of Ig; this uncertainty could not be resolved by CDA-AMC and may result in a higher price reduction being warranted.
Due to lack of clinical evidence to support the disutility associated with the treatment administration of Ig, the extent of the impact of disutility associated with Ig administration was uncertain. The exclusion of disutility associated with treatment administration was explored in a scenario analysis (refer to the Supplemental Material document, Appendix 11, Table 27). Based on this scenario analysis, the ICER for efgartigimod alfa may increase to $2,338,527 per QALY gained compared with Ig.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis to estimate the 3-year (2027 to 2029) budget impact of reimbursing efgartigimod alfa for use in the Health Canada–indicated population. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach.45 The price of efgartigimod alfa was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices and published literature (for Igs).44,46 Additional information pertaining to the sponsor’s submission is provided in the Supplemental Material document, Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Supplemental Material document, Appendix 5). CDA-AMC estimated that by year 3 of reimbursement, 2,525 patients would be eligible for treatment with efgartigimod alfa, of whom 379 would be expected to receive efgartigimod alfa. The estimated incremental budget impact of reimbursing efgartigimod alfa is predicted to be approximately $691 million over the first 3 years, with an expected expenditure of $692 million on efgartigimod alfa. The actual budget impact will depend on the perspective applied (drug costs only or broader health care system) and the public reimbursement rate of certain treatments.
Conclusion
Based on the CDA-AMC base case, efgartigimod alfa would be considered cost-effective at the submitted price if the public health care system were willing to pay at least $1,872,111 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in the Supplemental Material document, Appendix 11, Table 26). The estimated cost-effectiveness of efgartigimod alfa compared to Ig is uncertain due to the lack of head-to-head evidence, limitations with the model structure, and the unknown price of Ig.
The budget impact of reimbursing efgartigimod alfa to the public drug plans in the first 3 years is estimated to be approximately $691 million. The 3-year expenditure on efgartigimod alfa (i.e., not accounting for current expenditure on comparators) is estimated to be $692 million. The estimated budget impact is uncertain due to the health system perspective applied, the public reimbursement rate of certain treatments, and the market share estimates.
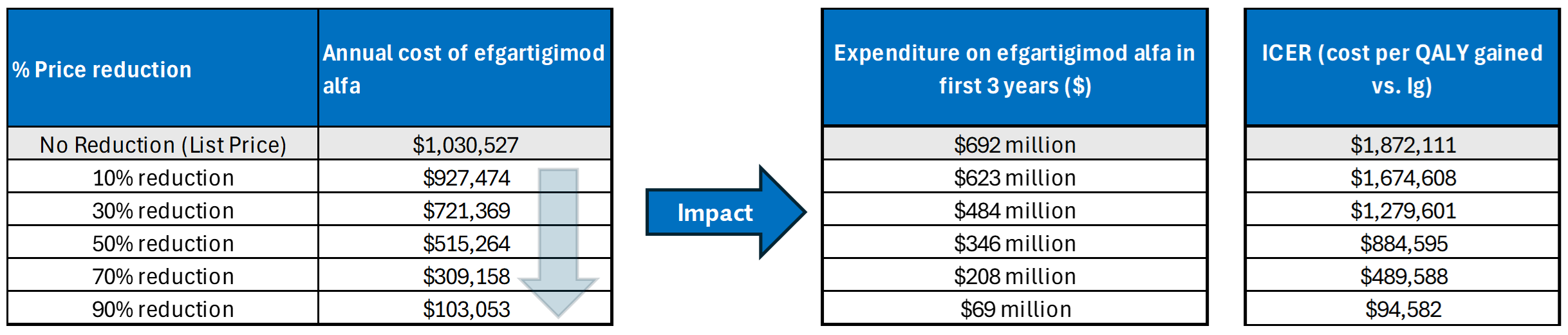
References
1.Bunschoten C, Jacobs BC, Van den Bergh PYK, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18(8):784–794. doi: 10.1016/S1474-4422(19)30144-9 PubMed
2.Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: a systematic review and meta-analysis. Neuroepidemiology. 2019;52(3-4):161–172. doi: 10.1159/000494291 PubMed
3.Lunn MP, Manji H, Choudhary PP, Hughes RA, Thomas PK. Chronic inflammatory demyelinating polyradiculoneuropathy: a prevalence study in south east England. J Neurol Neurosurg Psychiatry. 1999;66(5):677–680. doi: 10.1136/jnnp.66.5.677 PubMed
4.GBS/CIDP Foundation International. CIDP Chronic Inflammatory Demyelinating Polyneuropathy. Updated N/A. Accessed June 2025. https://www.gbs-cidp.org/wp-content/uploads/2012/05/CIDP-Booklet.pdf?utm_source
5.Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86(9):973–985. doi: 10.1136/jnnp-2014-309697 PubMed
6.Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force – second revision. Eur J Neurol. 2021;28(11):3556–3583. doi: 10.1111/ene.14959 PubMed
7.Laughlin RS, Dyck PJ, Melton LJ, 3rd, Leibson C, Ransom J, Dyck PJ. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology. 2009;73(1):39-45. doi: 10.1212/WNL.0b013e3181aaea47 PubMed
8.GBS/CIDP Foundation of Canada. Canadian CIDP patient Journey. Accessed March 24, 2025. https://www.gbscidp.ca/wp-content/uploads/2020/09/canadian-cidp-patient-journey-landscape-english.pdf
9.GBS CIDP Foundation of Canada. Voice of the Patient [sponsor supplied reference]. Accessed August 26, 2022. https://www.gbs-cidp.org/wp-content/uploads/2022/08/GBSCIDP-Voice-of-the-Patient-Report_Final.pdf
10.GBS CIDP Foundation of Canada. Canadian CIDP patient Journey [sponsor supplied reference]. https://www.gbscidp.ca/wp-content/uploads/2020/09/canadian-cidp-patient-journey-landscape-english.pdf
11.argenx. Illuminating the CIDP Journey [sponsor supplied reference]. 2025.
12.argenx Canada, Inc. VYVGART in CIDP - Clinical Questionnaire Summary Report [sponsor supplied reference]. 2025.
13.Allen JA, Lewis RA. Treatment of chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2022;66(5):552-557. doi: 10.1002/mus.27709 PubMed
14.Sivadasan A, Bril V. Therapies in Autoimmune Peripheral Neuropathies beyond Intravenous Immunoglobulin, Plasma Exchange and Corticosteroids: An Analytical Review. Transfus Med Rev. 2022;36(4):220-229. doi: 10.1016/j.tmrv.2022.05.002 PubMed
15.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
16.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
17.Van den Bergh PY, Hadden RD, Bouche P, et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. Eur J Neurol. 2010;17(3):356-63. doi: 10.1111/j.1468-1331.2009.02930.x PubMed
18.argenx BV. Clinical Study Report: ARGX-113-1802 (ADHERE). A Phase 2 Trial to Investigate the Efficacy, Safety, and Tolerability of Efgartigimod PH20 SC in Adult Patients With Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) [internal sponsor's report]. November 21, 2023.
19.argenx BV. VYVGART SC (efgartigimod alfa injection) for the treatment of adult patients with chronic inflammatory demyelinating polyneuropathy (CIDP): clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: VYVGART SC (efgartigimod alfa), 1000 mg/5 mL (200 mg/mL) solution in single-dose prefilled syringe, subcutaneous use. June 2025.
20.Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7(2):136-44. doi: 10.1016/S1474-4422(07)70329-0 PubMed
21.van Schaik IN, Bril V, van Geloven N, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2018;17(1):35-46. doi: 10.1016/s1474-4422(17)30378-2 PubMed
22.Léger J-M, De Bleecker J, Sommer C, et al. Efficacy and safety of Privigen® in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single-arm, open-label Phase III study (the PRIMA study). Journal of the peripheral nervous system: JPNS. 2013;18(2):130-140. doi: 10.1111/jns5.12017 PubMed
23.Cornblath DR, van Doorn PA, Hartung HP, et al. Randomized trial of three IVIg doses for treating chronic inflammatory demyelinating polyneuropathy. Brain. 2022;145(3):887-896. doi: 10.1093/brain/awab422 PubMed
24.FDA. Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products Guidance for Industry. U.S. Food and Drug Administration; 2019. https://www.fda.gov/media/121320/download
25.Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50(2):195-201. doi: 10.1002/ana.1088 PubMed
26.Li G, Taljaard M, Van den Heuvel ER, et al. An introduction to multiplicity issues in clinical trials: the what, why, when and how. Int J Epidemiol. 2016;46(2):746-755. doi: 10.1093/ije/dyw320 PubMed
27.Proschan MA, Waclawiw MA. Practical guidelines for multiplicity adjustment in clinical trials. Control Clin Trials. 2000;21(6):527-539. PubMed
28.argenx Canada Inc. argenx Canada Inc. response to Canada's Drug Agency request for additional information regarding Vyvgart SC review on July 10, 2025 [internal additional sponsor's information]. July 17, 2025.
29.Critical Appraisal Skills Programme. CASP Randomised Controlled Trial Checklist. 2023. Accessed August 11, 2025. https://casp-uk.net/casp-tools-checklists/randomised-controlled-trial-rct-checklist/
30.argenx B.V. Efgartigimod for CIDP: Systematic Literature Review and Indirect Treatment Comparison [internal sponsor's report]. argenx B.V.; 2024. Accessed June 5, 2025.
31.Wolfe GI, Nowak RJ, Lewis RA, Gelinas DF, Allen JA. Optimizing the Use of Outcome Measures in Chronic Inflammatory Demyelinating Polyneuropathy. US Neurology. 2017;13(01):26-34. doi: 10.17925/usn.2017.13.01.26
32.Allen JA, Merkies ISJ, Lewis RA. Monitoring Clinical Course and Treatment Response in Chronic Inflammatory Demyelinating Polyneuropathy During Routine Care: A Review of Clinical and Laboratory Assessment Measures. JAMA Neurol. 2020;77(9):1159-1166. doi: 10.1001/jamaneurol.2020.0781 PubMed
33.Allen JA, Lin J, Basta I, et al. Safety, tolerability, and efficacy of subcutaneous efgartigimod in patients with chronic inflammatory demyelinating polyradiculoneuropathy (ADHERE): a multicentre, randomised-withdrawal, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2024;23(10):1013-1024. doi: 10.1016/s1474-4422(24)00309-0 PubMed
34.argenx BV. ADHERE protocol version 5.0: A Phase 2 Trial to Investigate the Efficacy, Safety, and Tolerability of Efgartigimod PH20 SC in Adult Patients With Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) [internal additional sponsor's information]. October 12, 2022.
35.Breiner A, Barnett C, Bril V. INCAT disability score: a critical analysis of its measurement properties. Muscle Nerve. 2014;50(2):164–169. doi: 10.1002/mus.24207 PubMed
36.Khoo A, Frasca J, Schultz D. Measuring disease activity and predicting response to intravenous immunoglobulin in chronic inflammatory demyelinating polyneuropathy. Biomark Res. 2019;7:3. doi: 10.1186/s40364-019-0154-2 PubMed
37.Katzberg HD, Latov N, Walker FO. Measuring disease activity and clinical response during maintenance therapy in CIDP: from ICE trial outcome measures to future clinical biomarkers. Neurodegener Dis Manag. 2017;7(2):147–156. doi: 10.2217/nmt-2016-0058 PubMed
38.Vanhoutte EK, Latov N, Deng C, et al. Vigorimeter grip strength in CIDP: a responsive tool that rapidly measures the effect of IVIG—the ICE study. Eur J Neurol. 2013;20(5):748–755. doi: 10.1111/j.1468-1331.2012.03851.x PubMed
39.Howard JF, Jr., Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-536. doi: 10.1016/S1474-4422(21)00159-9 PubMed
40.Gwathmey KG, Broome CM, Goebeler M, et al. Safety profile of efgartigimod from global clinical trials across multiple immunoglobulin G-mediated autoimmune diseases. Expert Rev Clin Immunol. 2025;21(5):627-638. doi: 10.1080/1744666X.2025.2497840 PubMed
41.Broome CM, McDonald V, Miyakawa Y, et al. Efficacy and safety of the neonatal Fc receptor inhibitor efgartigimod in adults with primary immune thrombocytopenia (ADVANCE IV): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2023;402(10413):1648-1659. doi: 10.1016/s0140-6736(23)01460-5 PubMed
42.Canada's Drug Agency. Reimbursement recommendation: Efgartigimod Alfa (Vyvgart - argenx Canada Inc.) for the treatment of adult patients with generalized myasthenia gravis who are anti-acetylcholine receptor antibody positive. 2023. Accessed July 22, 2025.
43.argenx BV. VYVGART SC (efgartigimod alfa): 1000 mg/5 mL (200 mg/mL) solution in single-dose prefilled syringe, subcutaneous use [product monograph]. 2024. Updated 2025.
44.INESSS. Immunoglobulines Intraveineuses et sous-cutanées [sponsor supplied reference]. Accessed January 2025. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Rapports/Usage_optimal/Immunoglobulines/GUIDE_Immunoglobulines_intraveineuses_sous-cutanees.pdf
45.argenx BV. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: VYVGART SC (efgartigimod alfa), 1000 mg/5 mL (200 mg/mL) solution in single-dose prefilled syringe, subcutaneous use. June 5, 2025.
46.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed August 14, 2025. https://www.formulary.health.gov.on.ca/formulary/
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.