Drugs, Health Technologies, Health Systems
Reimbursement Review
Finerenone (Kerendia)
Sponsor: Bayer Inc.
Therapeutic area: Heart failure
Summary
What Is Heart Failure?
Heart failure (HF) is a disorder in which the heart is unable to adequately pump blood through the body to maintain the metabolic needs of tissues and organs. Common symptoms of HF include dyspnea and fatigue, exercise intolerance, and fluid buildup, which in turn may lead to pulmonary congestion and peripheral edema (mainly of the feet, ankles, or legs) that substantially affects quality of life. Patients living with HF often deal with other comorbidities, including diabetes, hypertension, atrial fibrillation, kidney problems, and chronic obstructive pulmonary disease.
In Canada, HF is the third most common reason for hospitalization. It was estimated that more than 750,000 individuals in Canada were living with HF. The crude prevalence of HF among patients aged 40 years or older in Canada has been steadily increasing from 3.62% in 2012 to 4.01%% in 2024.
What Are the Treatment Goals and Current Treatment Options for HF?
Treatment goals for HF include reducing hospitalizations and emergency department visits; lowering the risk of death; increasing patients’ energy, strength, and ability to participate in daily activities; and improving symptom management, stability, and quality of life, as well as minimizing treatment side effects. In the patient group input, patients emphasized that new treatments should address goals beyond just symptom management.
Current treatment options for HF include sodium-glucose cotransporter-2 (SGLT2) inhibitors (empagliflozin and dapagliflozin) and off-label medications, such as angiotensin receptor neprilysin inhibitors, angiotensin II receptor blockers, angiotensin-converting enzyme inhibitors, steroidal mineralocorticoid receptor antagonists (spironolactone and eplerenone), diuretics, beta-blockers, cardiac glycosides, nitrates, potassium supplements, potassium-lowering drugs, alpha-blocking drugs, calcium-channel blockers, nonsteroidal anti-inflammatory drugs, statin, insulins and analogues, and other antidiabetic drugs.
What Is Kerendia and Why Did Canada’s Drug Agency Conduct This Review?
Kerendia is a drug containing finerenone, available as 10 mg, 20 mg, and 40 mg oral tablets. At the time of this review, Health Canada was reviewing Kerendia for the treatment of HF with a mildly reduced or preserved ejection fraction (a left ventricular ejection fraction [LVEF] ≥ 40%) to reduce the risk of cardiovascular (CV) death, hospitalization for HF, and urgent HF visits.
Canada’s Drug Agency (CDA-AMC) reviewed Kerendia to inform a recommendation as to whether the participating public drug programs should reimburse the drug for the indication under review by Health Canada.
How Did CDA-AMC Evaluate Kerendia?
CDA-AMC reviewed the clinical evidence on the beneficial and harmful effects, as well as the economic evidence, of Kerendia versus other treatments used in Canada as an adjunct to standard of care (SOC) therapy in adults for the treatment of HF with a mildly reduced or preserved ejection fraction (LVEF ≥ 40%) to reduce the risk of CV death, hospitalization for HF, and urgent HF visits. SGLT2 inhibitors and off-label treatments (e.g., angiotensin receptor neprilysin inhibitors, angiotensin II receptor blockers, angiotensin-converting enzyme inhibitors, steroidal mineralocorticoid receptor antagonists [i.e., spironolactone and eplerenone]) were considered relevant treatments to compare with Kerendia when reviewing the clinical evidence.
CDA-AMC identified equity and ethical considerations relevant to Kerendia and patients living with HF.
The review was informed by materials submitted by the sponsor and included clinical and economic evidence.
The review was also informed by 1 patient group submission, 3 clinician group submissions, input from a group of clinicians (consisting of family physicians, cardiologists, general internists, a clinical pharmacist, and a nurse practitioner) from Canada in support of Kerendia access for HF with preserved ejection fraction, a solo submission by a cardiologist in response to our call for input, and input from the participating public drug programs addressing issues that may affect their ability to implement a recommendation.
Two cardiologists (1 each from Alberta and Ontario) with expertise in the diagnosis and management of HF were consulted as part of the review process.
What Were the Findings?
Clinical Evidence
CDA-AMC reviewed the following clinical evidence:
1 randomized, controlled, double-blind, parallel-group, placebo-controlled, multicentre, phase III trial (FINEARTS-HF) comparing Kerendia with placebo (as an add-on therapy to SOC in both groups) in 6,001 patients with HF (New York Heart Association class II to IV and an LVEF ≥ 40%)
1 network meta-analysis of Kerendia versus SGLT2 inhibitors (empagliflozin and dapagliflozin).
For the comparison of Kerendia versus placebo, both in addition to SOC, in the FINEARTS-HF trial:
Evidence from the FINEARTS-HF trial demonstrated that after a median follow-up of 32 months, Kerendia likely results in a clinically important reduction in the composite outcome of CV death and total HF events (hospitalization for HF and urgent HF visits) compared to placebo. The reduction was mainly driven by the reduction in total HF events. Specifically:
Kerendia results in a reduction of hospitalizations for HF and urgent HF visits compared to placebo; however, the clinical importance of the reduction is uncertain.
Kerendia likely results in a reduction of CV deaths compared to placebo; however, the clinical importance of the reduction is uncertain.
Kerendia may result in an improvement in patient’s quality of life from baseline to 12 months as measured by the Kansas City Cardiomyopathy Questionnaire total symptom score compared with placebo, but the clinical importance of the effect is uncertain.
Kerendia results in an increase in events of hyperkalemia and worsening of renal function compared to placebo; however, the clinical importance of the increase is unknown.
For the comparison of Kerendia versus SGLT2 inhibitors (empagliflozin and dapagliflozin): Evidence from 1 network meta-analysis was insufficient to draw a firm conclusion regarding the efficacy (i.e., CV death and hospitalizations for HF and total HF events) and the safety of Kerendia versus empagliflozin and dapagliflozin in patients with HF (New York Heart Association class II to IV and an LVEF ≥ 40%) due to limitations such as significant heterogeneity and sparse network.
There was no clinical evidence to determine how Kerendia compares with spironolactone. The sponsor could not perform an indirect treatment comparison due to differences in trial patient selection, study design, and regional factors resulting in substantial heterogeneity that could substantially limit the interpretability of the results. CDA-AMC partially accepted the sponsor’s deviation request, and spironolactone was only included in the budget impact analysis.
Economic Evidence
Kerendia is available as oral tablets (10 mg, 20 mg, and 40 mg). At the submitted price of $3.26 per tablet, the annual cost of Kerendia is expected to be $1,189 per patient, based on the Health Canada–recommended dosage.
Key clinical efficacy data (CV deaths and total HF events) used to inform the economic model were derived from the FINEARTS-HF trial, which compared Kerendia plus SOC with placebo plus SOC. Evidence submitted by the sponsor indicates that Kerendia plus SOC resulted in a reduction of hospitalizations for HF and urgent HF visits; however, the clinical importance of the reduction is uncertain. Kerendia plus SOC likely results in a reduction of CV deaths compared to placebo plus SOC. However, the trial was not designed to detect a difference between groups in CV deaths, and the clinical meaningfulness of the reduction was considered uncertain. Patients in the placebo plus SOC group experienced numerically more deaths than patients in the Kerendia plus SOC group, but time to all-cause mortality was tested outside the testing hierarchy and did not reach the nominal significance.
The results of the CDA-AMC analysis compare Kerendia plus SOC to SOC alone over a 3-year time horizon, with SOC largely informed by the FINEARTS-HF trial. Because SGLT2 inhibitor use in the trial is only estimated to be 13%, the economic results are not generalizable to a population with higher SGLT2 inhibitor use.
Kerendia plus SOC is predicted to be associated with higher costs to health care systems than SOC alone (incremental costs = $2,538), primarily driven by increased costs associated with drug acquisition.
Kerendia plus SOC is predicted to be associated with a gain of 0.02 life-years compared to SOC alone and may result in a gain of 0.03 quality-adjusted life-years (QALYs) compared to SOC alone. The close alignment between incremental life-years and QALYs suggests that the incremental benefit was primarily driven by extended survival, with minimal changes in health-related quality of life.
The incremental cost-effectiveness ratio of Kerendia plus SOC compared to SOC alone was $77,195 per QALY gained in the CDA-AMC base case. The cost-effectiveness of Kerendia using a longer time horizon is highly uncertain due to data limitations. The potential value of Kerendia plus SOC is dependent on patients realizing the predicted survival benefit compared to SOC alone, which is uncertain due to limitations with the clinical data. If these health outcomes are not realized, the incremental cost-effectiveness ratio for Kerendia plus SOC compared to SOC alone will be higher than predicted in the CDA-AMC analysis, and greater price reductions may be required to achieve a given willingness-to-pay threshold.
CDA-AMC estimates that the budget impact of reimbursing Kerendia as an adjunct to SOC in adults for the treatment of HF with mildly reduced or preserved ejection fraction (LVEF ≥ 40%) to reduce the risk of CV death, hospitalization for HF, and urgent HF visits will be approximately $107 million over the first 3 years of reimbursement compared to the amount currently spent on SOC alone. Given Kerendia is an add-on to existing care, its estimated expenditure is identical to the estimate budget impact (i.e., $107 million). The actual budget impact of reimbursing Kerendia will depend on the expected market uptake of Kerendia.
Abbreviations
AE
adverse event
AF
atrial fibrillation
ARNI
angiotensin receptor neprilysin inhibitor
BIA
budget impact analysis
CCS
Canadian Cardiovascular Society
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
CV
cardiovascular
CSS
clinical summary score
DIC
deviance information criterion
eGFR
estimated glomerular filtration rate
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HF
heart failure
HFmrEF
heart failure with mildly reduced ejection fraction
HFpEF
heart failure with preserved ejection fraction
HR
hazard ratio
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
KCCQ
Kansas City Cardiomyopathy Questionnaire
LVEF
left ventricular ejection fraction
MID
minimal important difference
MRA
mineralocorticoid receptor antagonist
NMA
network meta-analysis
NT-proBNP
N-terminal prohormone B-type natriuretic peptide
NYHA
New York Heart Association
OR
odds ratio
QALY
quality-adjusted life-year
RCT
randomized controlled trial
RR
rate ratio
SAE
serious adverse event
SD
standard deviation
SGLT2
sodium-glucose cotransporter-2
SLR
systematic literature review
SOC
standard of care
TSS
total symptom score
Background
Introduction
Context for the Review
The objectives of this report are as follows:
Review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of finerenone in the form of 10 mg, 20 mg, and 40 mg oral tablets as an adjunct to standard of care (SOC) in adults for the treatment of heart failure (HF) with mildly reduced or preserved ejection fraction (a left ventricular ejection fraction [LVEF] ≥ 40%) to reduce the risk of cardiovascular (CV) death, hospitalization for HF, and urgent HF visits. The evidence will focus on comparing finerenone to relevant comparators in clinical practice in Canada, as outlined in Table 1, and identify gaps in the current evidence.
Review and critically appraise the economic information submitted by the sponsor, including a cost-effectiveness analysis and budget impact analysis (BIA). The focus of the economic review is aligned with the scope of the clinical review, unless otherwise stated. For most reviews, a CDA-AMC base case is developed, informed by clinical expert input, the available clinical evidence, and the best interpretation of the economic evidence based on the information provided by the sponsor.
The sponsor submitted a Reimbursement Review application before receiving a Notice of Compliance from Health Canada. This report reflects the anticipated indication and recommended dosage for finerenone as indicated in the sponsor’s submission.
Table 1: Information on the Application Submitted for Review and on the CDA-AMC Review
Item | Description |
|---|---|
Information on the application submitted for review | |
Drug | Finerenone (Kerendia): 10 mg, 20 mg, or 40 mg oral tablets |
Sponsor | Bayer Inc. |
Health Canada indication | Finerenone is indicated as an adjunct to standard of care in adults for the treatment of heart failure with left ventricular ejection fraction (LVEF) ≥ 40% to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits. |
Health Canada approval status | Under review |
Health Canada review pathway | Standard |
NOC date | January 19, 2026 |
Mechanism of action | Finerenone is a nonsteroidal selective antagonist of the mineralocorticoid receptor. It binds to the mineralocorticoid receptor and blocks the binding of aldosterone, a component of the renin-angiotensin-aldosterone system. The mechanism of action by which finerenone reduces renal and cardiovascular events is not completely understood. |
Recommended dosage | The starting dose of finerenone is:
For treatment of heart failure with mildly reduced or preserved ejection fraction (LVEF ≥ 40%), the recommended target dose of finerenone depends on renal function (eGFR) at initiation of finerenone treatment:
|
Submission type | Initial |
Sponsor’s reimbursement request | Finerenone is indicated as an adjunct to standard of care therapy in adults for the treatment of heart failure with a mildly reduced or preserved ejection fraction (LVEF ≥ 40%) to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits. |
Submitted price | $3.26 per 10 mg tablet $3.26 per 20 mg tablet $3.26 per 40 mg tablet |
Information on the CDA-AMC review | |
Review type | Standard |
Clinical review focusa | Population: As defined in the reimbursement request Subgroups: None Intervention: Per recommended dosage Comparators: Standard of care (includes SGLT2 inhibitors, dapagliflozin, and empagliflozin; and off-label treatmentsa) Outcomes: Composite of CV death and the first and recurrent heart failure events (heart failure hospitalizations, urgent heart failure visits), change in NYHA class, change in KCCQ-TSS, renal composite, all-cause mortality, standard harms (AEs, SAEs, WDAEs, deaths), and notable harms (hyperkalemia, worsening of renal function) |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; CV = cardiovascular; eGFR = estimated glomerular filtration rate; HFpEF = heart failure with preserved ejection fraction; HFrEF = heart failure with reduced ejection fraction; KCCQ = Kansas City Cardiomyopathy Questionnaire; MRA = mineralocorticoid receptor antagonist; LVEF = left ventricular ejection fraction; NYHA = New York Heart Association; NOC = Notice of Compliance; SAE = serious adverse event; SGLT2 = sodium-glucose cotransporter-2; TSS = total symptom score; WDAE = withdrawal due to adverse event.
aThe economic review aligns with the scope of the clinical review, unless otherwise stated. Off-label treatments include steroidal MRAs (spironolactone), angiotensin receptor neprilysin inhibitors, angiotensin II receptor blockers, angiotensin-converting enzyme inhibitors, beta-blockers, loop diuretics, cardiac glycosides, nitrates, potassium supplements, potassium-lowering drugs, alpha-blocking drugs, calcium-channel blockers, nonsteroidal anti-inflammatory drugs, statins, insulins and analogues, and other antidiabetic drugs. The clinical experts consulted for the review noted that calcium-channel blockers, nonsteroidal anti-inflammatory drugs, statins, insulins and analogues, and other antidiabetic drugs are ancillary and would not be used to treat HF (HFrEF or HFpEF), although they are commonly used for other conditions among patients with HF.
Submission History for the Drug Under Review
Canada’s Drug Agency (CDA-AMC) previously reviewed finerenone through the reimbursement review process as an adjunct to SOC therapy in adult patients with chronic kidney disease and type 2 diabetes to reduce the risk of end-stage kidney disease and a sustained decrease in estimated glomerular filtration rate (eGFR), CV death, nonfatal myocardial infarction, and hospitalization for HF. The drug was issued a recommendation to “Reimburse with clinical criteria and/or conditions” for that indication in March 2023.
Sources of Information
The contents of the Reimbursement Review report are informed by materials submitted by the sponsor, input received from interested parties (patient groups, clinician groups, and provincial drug programs), and input from clinical experts consulted for this review. Calls for patient group and clinician group input are issued for each Reimbursement Review.
We received 1 patient group submission from HeartLife Foundation. The patient group conducted an online national survey in July 2025 of residents of Canada diagnosed with HF with a mildly reduced or preserved ejection fraction. Patients living with HF and multiple comorbidities also provided written input.
We received submissions from 3 clinician groups: the University of Ottawa and Heart Institute Cardiology and Heart Failure; Regional Wellness Kidney Centre; and the Fraser Health Authority – Cardiology and Nephrology Divisions. There was also a submission from a group of clinicians (consisting of family physicians, cardiologists, general internists, a clinical pharmacist, and a nurse practitioner) from Canada in support of access to finerenone for HF with preserved ejection fraction (HFpEF). There was a solo submission from a cardiologist practising in Ontario. The clinicians provided input based on information from published studies and their clinical experiences. The full submissions received are available on the project landing page in the Supplemental Material document.
We considered input from patient and clinician groups throughout the review, including input on which outcomes to include in the report and the interpretation of the clinical and economic evidence. Relevant patient and clinician group input is summarized in the Disease Background, Current Management, and Unmet Needs and Existing Challenges sections.
The drug programs provide input on the reimbursement review process of each drug by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 in the Supplemental Material document.
Each review team includes at least 1 clinical expert with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process. Two cardiologists (1 each from Alberta and Ontario) with expertise in the diagnosis and management of HF participated as part of the review team.
Disease Background
HF is a disorder in which the heart is unable to adequately pump blood throughout the body to maintain the metabolic needs of tissues and organs. HF is a complex clinical condition characterized by symptoms (e.g., fatigue and dyspnea) resulting from structural or functional impairment of ventricular filling or ejection of blood.1,2 Common symptoms of HF include dyspnea and fatigue, exercise intolerance, and fluid buildup, which in turn may lead to pulmonary congestion and peripheral edema (mainly in the feet, ankles, or legs) that substantially affects quality of life. Patients living with HF often deal with other comorbidities, including diabetes, hypertension, atrial fibrillation (AF), kidney problems, and chronic obstructive pulmonary disease.
HF is classified based on the percentage of blood that is being pumped out of the left ventricle, otherwise known as the LVEF.2 Patients with an LVEF of up to 40% are classified as HF with reduced ejection fraction, patients with an LVEF from 41% to 49% are classified as having HF with mildly reduced ejection fraction (HFmrEF), and patients with an LVEF of 50% or greater are classified as having HFpEF.3,4 Patients with HFpEF are more likely to have chronic systemic comorbidities that secondarily cause myocardial dysfunction.5,6
The Heart and Stroke Foundation of Canada reported in 2022 that more than 750,000 individuals in Canada were living with HF.7 The crude prevalence of HF among patients aged 40 years or older in Canada has been steadily increasing from 3.63% in 2012 to 4.01% in 2024.8 Rates of HF are higher among Indigenous people in Canada compared to non-Indigenous people.7 HF imposes a substantial health care burden, resulting mainly from hospitalizations.9 In Canada, HF is the third most common reason for hospitalization.10 The all-cause mortality rate in people living in Canada aged 40 years and older with HF is 6 times higher than in those without an HF diagnosis.11
Patient Group Input
Patients living with HFmrEF and/or HFpEF noted swelling in the legs, ankles, and stomach; shortness of breath with light activity; fatigue and low energy; and difficulty sleeping flat and concentrating as some of the burdensome symptoms. They emphasized that small tasks such as climbing stairs can lead to fluid retention, resulting in emergency department visits or hospitalizations. The constant uncertainty and sudden worsening in health also affected patients, caregivers, family members, and communities. Patients stopped working, cooking, walking, travelling, or socializing. In addition, they noted that identifying the cause of a patient’s symptoms was difficult in patients with comorbidities such as kidney disease, pulmonary hypertension, chronic obstructive pulmonary disorder, or arthritis.
Current Management
Treatment Goals
Patient Group Input
Important outcomes identified by patients included fewer hospital stays and emergency department visits, lowering the risk of death, increasing energy and strength to participate in daily activities, and having milder side effects, better symptom management, and stability. They also emphasized the importance of affordability and a treatment that is easier to incorporate into their daily routines.
Clinical Experts Consulted for This Review
The clinical experts considered reduction in hospitalizations (first and recurrent) and outpatient urgent HF exacerbations as the most important goals. In addition, the experts noted that a reduction in CV deaths and/or all-cause mortality would be added benefits. Improvement in quality of life, symptom burdens, and functional capacity were also identified as treatment goals.
Clinician Group Input
The input from the clinician groups aligned with that of the clinical experts. A reduction in albuminuria or proteinuria was also identified as a treatment goal.
Current Treatment Options
According to the 2017 Canadian Cardiovascular Society (CCS) guidelines,2 the pharmacological management of HFpEF includes the identification and treatment of underlying etiological factors implicated in development of HFpEF; identification and treatment of comorbid conditions that may exacerbate HF syndrome; control of symptoms; and the realization of clinically meaningful reduction in CV end points, such as HF hospitalization and mortality. Nonpharmacological measures include lifestyle modifications such as fluid restriction, avoiding salt and alcohol, and regular exercise.2
The 2025 CCS updated guidelines12 on the pharmacological management of patients with an LVEF of 40% or greater identified these 4 therapies for improving outcomes based on evidence from contemporary clinical trials and meta-analyses:
sodium-glucose cotransporter-2 (SGLT2) inhibitors
mineralocorticoid receptors (MRAs)
angiotensin receptor neprilysin inhibitors (ARNIs)
evidence-based drugs with glucagon-like peptide-1 receptor agonist activity.
The updated guidelines strongly recommend the use of SGLT2 inhibitors (empagliflozin and dapagliflozin) for people with symptomatic HF and an LVEF of greater than 40% to reduce the risk of hospitalization for HF.12 Evidence from 2 phase III randomized controlled trials (RCTs), EMPEROR-Preserved and DELIVER, demonstrated a statistically significant reduction in the primary end point, which was driven primarily by a substantial reduction in hospitalizations for HF with an insignificant reduction in CV deaths. In addition, the safety profile of SGLT2 inhibitors was considered favourable in patients with HF across the spectrum of LVEF.
The 2017 CCS guidelines included a weak recommendation for spironolactone based on a post hoc geographic subgroup analysis of the TOPCAT trial conducted within North and South America. In contrast, the 2025 updated guidelines strongly recommend the use of MRAs (both steroidal and nonsteroidal) in people with symptomatic HF and an LVEF of greater than 40% to reduce the risk of hospitalization for HF. The guidelines are informed by a systematic review of 13 studies, including 9 RCTs in the meta-analysis. Eplerenone and spironolactone were the 2 steroidal MRAs evaluated, while finerenone was the only nonsteroidal MRA. Furthermore, the inclusion of results from the FINEARTS-HF trial, which comprised a large study population, improved precision and certainty in the evidence for reduction in hospitalizations for HF with MRAs compared with placebo or SOC. The updated guidelines provided a weak recommendation for the use of ARNIs in patients with symptomatic HF and an LVEF of greater than 40%, particularly those at low risk of symptomatic hypotension. A similarly weak recommendation was made for evidence-based glucagon-like peptide-1 receptor agonists in individuals with obesity and an LVEF of 45% or greater, based on emerging evidence supporting their role in improving clinical outcomes in this population.
Other pharmacological therapies with proven efficacy for improving outcomes include the use of candesartan (weak recommendation, moderate quality evidence) to reduce hospitalization for HF, particularly in patients who are ineligible for or unable to be treated with an ARNI. Additionally, the use of loop diuretics to control symptoms of congestion and peripheral edema is strongly recommended. These recommendations remain unchanged from the 2017 CCS guidelines.
Clinical Experts Consulted for This Review
The clinical experts noted that currently only SGLT2 inhibitors and diuretics offer clinical benefit in the population with an LVEF of 40% or greater. They noted that SGLT2 inhibitors reduce CV mortality and hospitalizations for HF. They added that, while diuretics may reduce congestive symptoms and risk of readmission to the hospital, there is no evidence indicating they reduce mortality.
Clinician Group Input
The clinician groups generally agreed with the clinical experts consulted for this review.
Key characteristics of finerenone are summarized with other treatments available for HF in the Key Characteristics of Finerenone and Its Comparators (by Drug Class) table in Appendix 1 in the Supplemental Material document.
Unmet Needs and Existing Challenges
Patient Group Input
Patients living with HF noted that current treatments help reduce symptoms such as swelling or shortness of breath; however, they struggled to keep their condition stable. The patient group emphasized that, while SGLT2 inhibitors have been beneficial for patients with HFpEF, many patients continue to experience fluid overload, fatigue, hospitalizations, and a decline in their ability to function. They also noted that the wait times for a cardiologist may be several months. Patients emphasized the burden of being on multiple medications, the need for frequent dose adjustments, the inability to access specialists, and treatment delays leading to emotional distress. The patient group noted that patients had to live with ongoing symptoms and frequent medication changes, and deal with side effects, fluid limits, and bloodwork.
Clinical Experts Consulted for This Review
The clinical experts noted several challenges with the use of SGLT2 inhibitors. First, they are contraindicated in patients with advanced kidney dysfunction (an eGFR < 25 mL/min/1.73 m2) and in those with type 1 diabetes. Second, 10% to 15% of patients discontinue using SGLT2 inhibitors due to side effects such as genitourinary infections and a decrease in renal function (usually transient).
Clinician Group Input
The clinicians noted that patients with HFpEF have limited treatment options and are particularly underserved (i.e., patients with eGFR < 60 mL/min/1.73 m2, which is consistent with chronic kidney disease stages 3 or 4). They noted that patients with HFpEF:
were largely without mortality-reducing therapies
were at substantial risk of repeated hospitalization
had tolerability and safety concerns with current therapies (e.g., use of spironolactone associated with gynecomastia)
had limited tools to match therapies for patients with heterogenous phenotypes (e.g., AF dominant, cardiorenal, hypertensive)
had comorbidities (e.g., experiencing frailty, chronic kidney disease) requiring polypharmacy.
Considerations for Using the Drug Under Review
Contents within this section have been informed by input from the clinical experts consulted for the purpose of this review and from clinician groups, as well as the reimbursement conditions proposed by the sponsor (refer to the Initiation, Renewal, Discontinuation, and Prescribing Conditions Proposed by the Sponsor [and Not Modified by CDA-AMC] table in Appendix 1 in the Supplemental Material document). The implementation questions from the public drug programs and corresponding responses from the clinical experts consulted for this review are summarized in the Summary of Drug Program Input and Clinical Expert Responses table in Appendix 1 in the Supplemental Material document. The following has been summarized by the review team.
Place in Therapy
Clinical Experts Consulted for This Review
The clinical experts indicated that finerenone would cause a shift in the current treatment paradigm as it addresses the underlying disease process. The experts noted that finerenone should be used as an add-on to SOC therapy, including SGLT2 inhibitors because they modify disease trajectory. The experts noted that treatment with finerenone would require monitoring of renal function and potassium after initiation.
Clinician Group Input
The clinician groups noted that patients would be treated with SGLT2 inhibitors in the first line and would be considered for treatment with finerenone if they had ongoing symptoms, mild renal dysfunction, recurrent risks of hospitalizations, and no contraindications. They added that finerenone should be considered as the first MRA option along with SGLT2 inhibitors in patients with HFpEF.
Patient Population
Clinical Experts Consulted for This Review
The clinical experts noted that patients with a symptomatic LVEF greater than or equal to 40% were most in need of an intervention. They also noted that the condition affects both men and women, with a higher prevalence among older individuals. It is commonly associated with diabetes mellitus, obesity, and systemic hypertension, but can also occur in people without any known risk factors. The experts also indicated that patients with advanced kidney dysfunction and hyperkalemia are not suitable for treatment with finerenone, according to the product monograph.
Regarding the initiation conditions proposed by the sponsor, the clinical experts identified no issues with the proposed conditions.
Clinician Group Input
Clinician input generally aligned with the opinions of the clinical experts consulted for this review.
Assessing the Response to Treatment
Clinical Experts Consulted for This Review
The clinical experts noted that a reduction in adverse outcomes related to the disease and symptomatic relief was the main concern in clinical practice. Therefore, primary considerations include reductions in hospitalizations and ambulatory visits, and reductions in mortality, which they indicated aligned with the pivotal trial.
Clinician Group Input
Clinician groups agreed with the opinion of clinical experts. They also noted that a clinically meaningful response would focus on function or exercise capacity (increase in walking distance and ability to do more activities without symptoms). One clinician group noted that response to treatment would be assessed every 1 month to 3 months by measuring the eGFR and levels of creatinine and serum potassium, symptoms, and hospitalizations.
Discontinuing Treatment
Clinical Experts Consulted for This Review
The clinical experts indicated that treatment with finerenone will be discontinued if a patient develops severe renal failure, persistent increase of serum potassium, or an intolerance or allergy to the drug.
Clinician Group Input
The clinician groups agreed with the clinical experts. They also noted that treatment with finerenone will be discontinued if a patient develops substantial hepatic insufficiency classified as Child-Pugh class C or started concomitant systemic therapy with potent cytochrome P450 isoenzyme 3A4 inhibitors or inducers (e.g., ritonavir, indinavir, cobicistat, clarithromycin, or itraconazole).
Prescribing Considerations
Clinical Experts Consulted for This Review
The clinical experts noted that a cardiologist may be required to make the initial diagnosis, but treatment could be initiated and monitored by a qualified clinician, including a nonspecialist. The experts acknowledged the disparities in access to care in Canada; however, they pointed out that these disparities would not affect the use of finerenone more than any other treatment that would require an initial specialist assessment.
The clinical experts did not identify any issues regarding the proposed prescribing conditions by the sponsor.
Clinician Group Input
The clinician group input generally aligned with the opinions of the clinical experts. They also noted that, as experience with finerenone increases over time, internal medicine and primary care providers would also be able to diagnose, treat, and monitor patients with HFpEF and HFmrEF.
Clinical Review
Methods
The review team considered the following evidence from the sponsor’s submission: a phase III RCT (FINEARTS-HF) identified in the sponsor’s systematic review and an indirect treatment comparison (ITC). Eligible studies for the systematic review included published and unpublished pivotal studies and phase III and IV RCTs. Relevant patients and interventions were defined by the reimbursement request and the recommended dosage in the product monograph. Relevant comparators were SGLT2 inhibitors and other medications used off-label in patients with HF (e.g., steroidal MRAs [spironolactone, eplerenone] and angiotensin-converting enzyme inhibitors, which are considered part of SOC in clinical practice in Canada). Because no direct comparison between finerenone and SGLT2 inhibitors exists and SGLT2 inhibitors are considered relevant comparators for this patient population in this review, CDA-AMC requested an indirect comparison. The drugs included in the ITC submitted by the sponsor were dapagliflozin and empagliflozin, which are SGLT2 inhibitors approved by Health Canada as adjuncts to SOC therapy for the treatment of HF in Canada.
The review team selected outcomes for review considering the sponsor’s Summary of Clinical Evidence and input from a patient group and clinician groups as well as from the 2 clinical experts consulted by CDA-AMC for this review. Included outcomes considered relevant to expert committee deliberations were selected in consultation with committee members. Evidence for the most important outcomes identified in the systematic review was evaluated using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach. These outcomes were selected for GRADE assessment because they reflect key treatment goals for HF and were deemed important based on input from patient groups, clinician groups, and clinical experts:
Primary end point: Composite of CV death and total (first and recurrent) HF events
CV death
Total HF events (hospitalizations for HF and urgent HF visits)
Kansas City Cardiomyopathy Questionnaire (KCCQ) total symptom score (TSS)
Safety: Hyperkalemia and worsening of renal function
Methods for data extraction, risk of bias appraisal, and certainty of evidence assessment are in Appendix 2 in the Supplemental Material document.
Clinical Evidence
The following sources of evidence submitted by the sponsor are reviewed and appraised in this report:
1 pivotal study included in the systematic review, FINEARTS-HF
1 ITC.
Systematic Review
Description of Studies
Study Characteristics
Characteristics of the included studies are summarized in Table 2. Details pertaining to the eligibility criteria, interventions and comparators, and relevant outcome measures are provided in Appendix 3 in the Supplemental Material document.
The FINEARTS-HF study was a phase III, double-blind, parallel-group, placebo-controlled, multicentre, event-driven RCT investigating the efficacy and safety of finerenone on morbidity and mortality in patients with HF (New York Heart Association [NYHA] class II to IV disease) with an LVEF of 40% or greater. The primary objective was to demonstrate the superiority of finerenone to placebo in reducing the rate of the composite CV end point. The primary composite end point comprised CV death and/or total HF events. Patients were eligible to enrol if they were aged 40 years and older with an LVEF of 40% or greater. A total of 7,463 patients were screened at 654 sites across 37 countries, including 21 sites in Canada. Of the 6,016 patients enrolled in the study, 116 were enrolled from sites in Canada.
Patients were randomly assigned in a 1:1 ratio to receive finerenone or placebo, both as an adjunct to SOC. Randomization was stratified by country and/or region and LVEF (< 60%, ≥ 60%). Two caps were applied to enrolment to ensure a representative study population: 20% for patients with an LVEF of 60% or greater and 50% for patients without an HF event in the 90 days before enrolment. There were 3 phases in the study design: a screening phase of 2 weeks, a treatment phase of 36 months, and a follow-up phase lasting 30 days (+ 5 days). Following visit 1, onsite assessments were performed at 1 month, 2 months, and every 3 months thereafter through month 12, then at month 16 and every 8 months thereafter (including month 20), until the end of the study.
All randomized patients remained in the study until the targeted number of primary efficacy events occurred, or the study was terminated early at the recommendation of the data monitoring committee. All patients attended the protocol-specified study visits to perform all remaining assessments. If a participant was unable to attend a scheduled study visit, all reasonable efforts were made, including contacting them by telephone or reviewing medical and public records, to determine whether any study end points had been reached during the intended visit time frame. Minimal assessments (e.g., central labs for eGFR) were performed for participants who permanently discontinued study intervention.
The study data remained blinded until database lock and authorization of data release according to standard operating procedures.
This report is based on the primary Clinical Study Report following the database lock on July 25, 2024.
Table 2: Characteristics of Studies Included in the Systematic Review
Characteristic | Details |
|---|---|
Study name, design, and sample size | FINEARTS-HF; phase III, multicentre, double-blind, placebo-controlled RCT Total N (patients who were randomized) = 6,016 (finerenone = 3,011; placebo = 3,005) |
Key inclusion criteria |
|
Key exclusion criteria |
|
Intervention and comparator | Intervention
Comparator
|
Relevant end points | Primary end point
Secondary end points
Safety
|
AE = adverse event; BNP = B-type natriuretic peptide; CV = cardiovascular; eGFR = estimated glomerular filtration rate; HF = heart failure; KCCQ = Kansas City Cardiomyopathy Questionnaire; LAA = left atrial area; LAD = left atrial diameter; LAVI = left atrial volume index; LVEF = left ventricular ejection fraction; LVMI = left ventricular mass index; NT-proBNP = N-terminal prohormone B-type natriuretic peptide; NYHA = New York Heart Association; SAE = serious adverse event; SBP = systolic blood pressure; TSS = total symptom score; WDAE = withdrawal due to adverse event.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.13
Statistical Testing and Analysis Populations
The sample size determination was based on a simulation study assuming a joint frailty model to account for the correlation between HF events and CV death, and participant heterogeneity was modelled with respect to baseline intensities and/or hazards. That is, given the participant-specific gamma-distributed frailties, a homogeneous Poisson process for HF events and an exponential distribution for the time to CV death were assumed. Furthermore, the frailty term was assumed to be the same for HF events and CV death.
As treatment effects, a hazard ratio (HR) for CV death of 0.8 and a rate ratio (RR) for HF events of 0.75 were assumed. With approximately 5,500 randomized participants, approximately 1,310 first events and 2,375 total events were expected, leading to a required power of 90% to show an effect at a 2-sided alpha level of 5%. Under these assumptions, a 19% decrease in the rate of the primary end point for finerenone was expected to be observed. An annual drug discontinuation rate of 5% was assumed, with patients receiving finerenone having the same risk of events as participants receiving placebo after discontinuation and no change in event rate for discontinuing participants receiving placebo. Patients who discontinued the study intervention were expected to remain under observation in the study. Due to blinded event rates being lower than those assumed in the sample-size calculation, the planned number of randomized patients was increased to approximately 6,000.
The efficacy analyses were performed using the full analysis set (FAS), which included all randomized patients (excluding 15 patients with critical violations of good clinical practice standards). The safety analysis was performed using the safety analysis set, which included all participants in the FAS who received at least 1 dose of the study intervention.
Patient Disposition
Patient disposition for the included study is summarized in the Patient Disposition in FINEARTS-HF table in Appendix 4 in the Supplemental Material document.
In the FINEARTS-HF trial, 7,463 patients were screened, and 6,016 patients were randomized to either the finerenone group (n = 3,011) or the placebo group (n = 3,005). A total of 7 patients in the finerenone group and 12 patients in the placebo group discontinued the study. Reasons for discontinuation were patients lost to follow-up (finerenone: 1, placebo: 5) and withdrawal of consent (finerenone: 6, placebo: 7). The FAS included 3,003 patients in the finerenone group and 2,998 patients in the placebo group.
From the patients in the FAS, 2,993 each in the finerenone and placebo groups started the treatment. Of these, 31.3% of patients in the finerenone group and 32.3% of patients in the placebo group did not complete treatment. Primary reasons for discontinuations were death (finerenone: 332 [11.0%], placebo: 358 [11.8%]), patient decision (finerenone: 208 [6.9%], placebo: 215 [7.2%]), and adverse events (AEs) (finerenone: 143 [4.7%], placebo: 127 [4.2%]). Of the patients who prematurely discontinued study treatment, 393 (13.1%) in the finerenone group and 387 (12.9%) in the placebo group completed the posttreatment follow-up. Of the patients who completed the study treatment, 2,050 (68.1%) in the finerenone group and 2,021 (67.3%) in the placebo group started posttreatment follow-up. A total of 1,987 patients (66.0%) and 1,947 patients (64.8%) in the finerenone and placebo groups, respectively, completed posttreatment follow-up.
Protocol deviations were similar across both treatment groups (finerenone: 63.8%, placebo: 63.1%) (refer to the Number of Patients With Important Protocol Deviations [All Randomized Patients] table in Appendix 4 in the Supplemental Material document). The most common reasons for protocol deviations were procedure deviations (finerenone: 34.4%, placebo: 32.7%) followed by time schedule deviations (finerenone: 25.2%, placebo: 24.0%), other deviations (finerenone: 18.9%, placebo: 19.4%), and treatment deviations (finerenone: 13.4%, placebo: 14.1%).
Baseline Characteristics
Baseline characteristics of patients in the FINEARTS-HF trial are presented in Table 3 and the Summary of Additional Baseline Characteristics From Studies Included in the Systematic Review (FAS) table in Appendix 4 in the Supplemental Material document. Across groups, the mean age of patients was 71.9 years and more than three-quarters of the cohort were aged 65 years or older. Overall, most of the patients were white (78.9%) and male (54.5%). Most patients (80.8%) had an LVEF of less than 60% and had NYHA class II disease (69.1%). There were fewer patients with diabetes (40.6%), NYHA class III (30.2%), and NYHA class IV (0.7%).
Table 3: Summary of Baseline Characteristics From Studies Included in the Systematic Review (FAS)
Characteristic | FINEARTS-HF trial | ||
|---|---|---|---|
Finerenone (N = 3,003) | Placebo (N = 2,998) | Overall (N = 6,001) | |
Female, n (%) | 1,355 (45.1) | 1,377 (45.9) | 2,732 (45.5) |
Male, n (%) | 1,648 (54.9) | 1,621 (54.1) | 3,269 (54.5) |
Race, n (%) | |||
American Indian or Alaskan Native | 75 (2.5) | 74 (2.5) | 149 (2.5) |
Asian | 497 (16.6) | 499 (16.6) | 996 (16.6) |
Black or African American | 49 (1.6) | 39 (1.3) | 88 (1.5) |
Multiple | 7 (0.2) | 8 (0.3) | 15 (0.2) |
Native Hawaiian or Other Pacific Islander | 1 (< 0.1) | 0 | 1 (< 0.1) |
White | 2,366 (78.8) | 2,369 (79.0) | 4,735 (78.9) |
Not reported | 8 (0.3) | 9 (0.3) | 17 (0.3) |
Age (years), mean (SD) | 71.94 (9.60) | 72.04 (9.69) | 71.99 (9.65) |
Body mass index (kg/m2), mean (SD) | 29.88 (6.10) | 30.00 (6.14) | 29.94 (6.12) |
Baseline LVEF category, n (%) | |||
< 60%, | 2,422 (80.7) | 2,424 (80.9) | 4,846 (80.8) |
≥ 60% | 575 (19.1) | 572 (19.1) | 1,147 (19.1) |
NYHA class, n (%) | |||
NYHA class II | 2,081 (69.3) | 2,065 (68.9) | 4,146 (69.1) |
NYHA class III | 903 (30.1) | 910 (30.4) | 1,813 (30.2) |
NYHA class IV | 18 (0.6) | 23 (0.8) | 41 (0.7) |
Missing | 1 (< 0.1) | 0 | 1 (< 0.1) |
Index HF event, n (%) | |||
≤ 7 days from randomization | 609 (20.3) | 610 (20.3) | 1,219 (20.3) |
> 7 days to ≤ 3 months | 1,030 (34.3) | 998 (33.3) | 2,028 (33.8) |
> 3 months or no index HF event | 1,364 (45.4) | 1,390 (46.4) | 2,754 (45.9) |
Atrial fibrillation present at baseline, n (%) | 1,165 (38.8) | 1,128 (37.6) | 2,293 (38.2) |
Baseline serum potassium (mmol/L), mean (SD) | 4.38 (0.48) | 4.37 (0.47) | 4.37 (0.47) |
Baseline eGFR (mL/min/1.73 m2) category, n (%) | |||
< 60 mL/min/1.73 m2 | 1,451 (48.3) | 1,437 (47.9) | 2,888 (48.1) |
≥ 60 mL/min/1.73 m2 | 1,552 (51.7) | 1,561 (52.1) | 3,113 (51.9) |
Presence of diabetes mellitus at baseline, n (%) | 1,217 (40.5) | 1,222 (40.8) | 2,439 (40.6) |
Use of ACEI, ARB, or ARNI at baseline, n (%) | 2,379 (79.2) | 2,380 (79.4) | 4,759 (79.3) |
Use of SGLT2 inhibitors at baseline, n (%) | 393 (13.1) | 424 (14.1) | 817 (13.6) |
ACEI = angiotensin-converting enzyme inhibitor; ARB = angiotensin receptor blocker; ARNI = angiotensin receptor neprilysin inhibitor; eGFR = estimated glomerular filtration rate; FAS = full analysis set; HF = heart failure; LVEF = left ventricular ejection fraction; NYHA = New York Heart Association; SGLT-2 = sodium-glucose cotransporter-2; SD = standard deviation.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.13
Treatment Exposure and Concomitant Medications
Details of patients’ treatment exposure, adherence, and use of concomitant medications are in the Summary of Patient Exposure From Studies Included in the Systematic Review and the Summary of Relevant Concomitant Medications After Start of Study Intervention (FAS) tables in Appendix 4 in the Supplemental Material document.
In the finerenone group, the mean treatment duration was 25.07 months (standard deviation [SD] = 12.48 months), and mean treatment compliance was 102.78 (SD = 98.59). In the placebo group, the mean treatment duration was 25.29 months (SD = 12.38 months), and the mean treatment compliance was 100.45 (SD = 27.11).
A total of 5,976 patients (99.6%) received at least 1 medication of interest before the start of the study (finerenone: 99.5%, placebo: 99.7%). Overall, most patients were taking loop diuretics (87.3%), beta-blockers (84.9%), an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker (79.3%), organic anion transporting polypeptides substrates (76.2%), and statins (67.5%). Other commonly used drugs were aspirin (33.7%), calcium-blocking channels (34.1%), alpha-blocking drugs (25.0%), and antidiabetic drugs (27.7%). A total of 817 patients (13.6%) were taking SGLT2 inhibitors at study baseline (finerenone: 13.1%, placebo: 14.1%).
The use of concomitant medications was generally well balanced after the start of the study. However, minor imbalances between the finerenone and placebo groups were observed in the proportion of patients using certain medications: SGLT2 inhibitors (19.8% versus 21.9%), calcium-channel blockers (17.4% versus 19.7%), potassium supplements (12.8% versus 18.6%), and potassium-lowering drugs (3.4% versus 1.4%). Before the end of the study intervention, 3.6% of the patients in the finerenone group and 4.8% in the placebo group started treatment with any MRA (mostly spironolactone).
Critical Appraisal
Internal Validity
Eligible patients were centrally randomized to the study intervention or control using an interactive response technology. Treatment allocation was adequately performed via a computer-generated randomization list. Blinding was maintained by ensuring tablets were identical in appearance (size, shape, and colour) for the treatment and placebo groups. Randomization was stratified by country and/or region and LVEF (< 60%, ≥ 60%). The baseline characteristics were generally well balanced between groups. The proportion of female participants was lower than that of male participants. However, the clinical experts consulted by CDA-AMC for this review did not consider this imbalance a major concern, citing challenges in recruiting female participants for trials in this population.
Premature emergency unblinding was recorded in 87 patients. Eighty-one patients had their randomization codes broken (finerenone: 51, placebo: 30) and 6 unblinding events were reported by the investigators (finerenone: 2, placebo: 4). About one-third of the participants in both groups discontinued study treatment. In both groups, the main reasons for discontinuing treatment was death (finerenone: 11.0%, placebo: 11.8%), followed by AEs (finerenone: 4.7%, placebo: 4.2%), and patient decision (finerenone: 6.9%, placebo: 7.2%). The potential impact of these unblinding events and discontinuations on efficacy results was not considered a major concern.
An independent committee blinded to the study intervention adjudicated all deaths and HF events. Over a median follow-up of 32 months, the between-group difference in CV deaths was insignificant. Clinical experts noted that the event numbers were too small to detect a difference in CV deaths between groups. In addition, they emphasized that, for outcomes that take longer to manifest, such as mortality, the follow-up duration of the trial may have been insufficient to adequately assess the magnitude of effect. The secondary outcomes of renal composite end point and change in NYHA class failed hierarchical testing, and all-cause mortality was tested outside the hierarchical testing. Therefore, the results of these assessments are inconclusive and are considered as supportive evidence only.
Evidence for the validity, reliability, and responsiveness of the KCCQ as a measure of health-related quality of life (HRQoL) in patients with HF is summarized in Appendix 3 in the Supplemental Material document. Based on the available literature and clinical experts consulted by CDA-AMC, a between-group minimal important difference (MID) of 5 points in the KCCQ-TSS was applied. All observed values, including for those who permanently discontinued treatment, were included in the analysis. However, at 12 months, the missing data rate was greater than 16% in both arms of the study and it was excluded from the analysis. Therefore, besides the risk of bias due to subjective patient-reported outcomes, there was an risk of bias due to attrition.
External Validity
Patients in the FINEARTS-HF trial were predominately white, which is not representative of the racially and ethnically diverse patient populations in Canada. However, how that affects the generalizability of the study findings in the context in Canada is unclear. The FINEARTS-HF trial excluded patients with high systolic blood pressure (i.e., ≥ 160 mm Hg not receiving treatment for lowering blood pressure). The clinical experts noted that these criteria would exclude a relatively small percentage of patients, who would not respond any differently to finerenone. Patients were also required to have structural heart and N-terminal prohormone B-type natriuretic peptide (NT-proBNP) assessments as specified by the inclusion criteria. The clinical experts noted that these criteria aimed to reduce the risk of misdiagnosis and ensure proper inclusion of patients with HF. The study design capped the proportion of patients with an LVEF of 60% or greater at 20%. However, the experts did not view this as a major concern and noted that the LVEF assessments were subjective and varied by physician. Overall, the clinical experts consulted by CDA-AMC indicated that the population in the FINEARTS-HF trial was reflective of the patients seen in clinical practice in Canada.
With regard to comparators, the clinical experts noted that, while SGLT2 inhibitors are now relevant comparators to finerenone, they were not indicated for HF at the time the FINEARTS-HF trial started, making placebo plus SOC an acceptable comparator for the trial. They pointed out that the use of older MRAs, such as spironolactone, was limited due to uncertainty regarding their clinical benefit in patients with HFpEF.
The outcomes evaluated in the trial and reported in this review are considered important by patients with HFmrEF and HFpEF and aligned with their treatment goals.
Results
The key efficacy and harms results, and findings from the GRADE assessment (Table 4) are presented in this section. Detailed efficacy and harms results can be found in Appendix 4 in the Supplemental Material document.
Efficacy
The following are results for the key efficacy outcomes of the FINEARTS-HF trial at database lock on July 25, 2024 (median follow-up duration of 32 months):
CV death and total HF events (first and recurrent): The incidence rates (per 100 patient-years) for the primary composite outcome were 14.88 in the finerenone group and 17.70 in the placebo group. The difference between groups was 2.81 (95% confidence interval [CI], █████ ██ █████), with an RR of 0.84 (95% CI, 0.74 to 0.95), in favour of finerenone. The results of the subgroup analyses were consistent with those of the primary analysis (refer to the Forest Plot for the Primary Composite End Point of CV Death and Total HF Events (First and Recurrent) (FAS) figure in Appendix 4 in the Supplemental Material document). For the supportive time to first event analyses, the RR was 0.84 (95% CI, 0.76 to 0.94) in favour of finerenone (refer to the Results for Adjudicated Composite End Point and Its Components (First Event – Full Analysis Set) table in Appendix 4 in the Supplemental Material document).
CV death: As 1 component of the primary composite end point, CV death was assessed in a supportive analysis. The incidence rates (per 100 patient-years) for CV death were 3.33 in the finerenone group and 3.59 in the placebo group, with a between-group difference of 0.6 (95% CI, ████ ██ ███) and an HR of 0.93 (95% CI, 0.78 to 1.11). Another supportive analysis indicated that the finerenone group experienced a lower number of CV death events due to total HF events in comparison to the placebo group, with an RR of 0.82 (95% CI, 0.71 to 0.94).
Total HF events: The total (first and recurrent) HF events comprising hospitalizations for HF and urgent HF visits was a key secondary end point. At the time of analysis, the incidence rates for the total numbers of HF events were 11.57 in the finerenone group and 14.12 in the placebo group. The difference between groups was 2.55 (95% CI, █████ ██ █████), with an RR of 0.82, (95% CI, 0.71 to 0.94). The results of the subgroup analysis were consistent with those in the overall analysis of HF events (refer to the Forest Plot of the Secondary End Point of Total HF Events (First and Recurrent) by Subgroups (FAS) figure in Appendix 4 in the Supplemental Material document).
For hospitalization for HF, the incidence rates were 10.22 in the finerenone group and 12.10 in the placebo group. The difference between groups was 1.87 (95% CI, █████ ██ █████), favouring finerenone. For urgent HF visits, the incidence rates were 1.35 in the finerenone and 2.03 in the placebo group. The difference between groups was 0.68 (95% CI, █████ ██ █████), favouring finerenone.
KCCQ-TSS: The least squares mean differences in KCCQ-TSS between groups were 1.41 points (95% CI, 0.50 points to 2.31 points) at 6 months, 1.75 points (95% CI, 0.81 points to 2.68 points) at 9 months, and 1.61 points (95% CI, 0.66 points to 2.55 points) at 12 months.
Summary of Other Findings
Change in NYHA class: The odds ratio (OR) for NYHA class was 1.01 (95% CI, 0.88 to 1.15), indicating no difference between treatment groups (refer to the Summary of Key Efficacy Results From Studies Included in the Systematic Review table in Appendix 4 in the Supplemental Material document).
Renal composite end point: A higher proportion of patients in the finerenone group (2.5%) reported renal composite events compared to patients in the placebo group (1.8%), with an HR of 1.33 (95% CI, 0.94 to 1.89) (refer to the Summary of Key Efficacy Results From Studies Included in the Systematic Review table in Appendix 4 in the Supplemental Material document).
All-cause mortality: Fewer deaths were reported in the finerenone group (n = 491, 16.4%) than in the placebo group (n = 522, 17.4%), corresponding to an HR of 0.93 (95% CI, 0.83 to 1.06) (refer to the Summary of Key Efficacy Results From Studies Included in the Systematic Review table in Appendix 4 in the Supplemental Material document).
Harms
Key results are presented in the Summary of Harms Results From Studies Included in the Systematic Review table in Appendix 4 in the Supplemental Material document and include the following:
Adverse events:
An AE was reported in 84.0% of patients in the finerenone group and 82.3% of patients in the placebo group.
The most common AEs (≥ 5% of patients) more frequently reported in the finerenone group compared to the placebo group were hyperkalemia (8.8% versus 3.8%, respectively), renal impairment (6.6% versus 3.9%), hypotension (7.3% versus 4.5%), dizziness (5.8% versus 4.7%), diarrhea (5.7% versus 4.4%), and decreased eGFR (5.2% versus 3.6%).
Other common AEs more frequently reported in the placebo group compared to the finerenone group included cardiac failure (6.1% versus 4.3%, respectively), hypertension (6.2% versus 3.6%), and hypokalemia (6.7% versus 3.0%).
Serious AEs (SAEs) were more commonly reported in patients receiving placebo (40.5%) than finerenone (38.7%). The most common SAEs were pneumonia (finerenone: 2.9%, placebo: 3.7%), AF (finerenone: 2.6%, placebo: 2.4%), and COVID-19 (finerenone: 2.3%, placebo: 2.4%). Serious acute kidney injury was frequently reported in patients in the finerenone group (1.8%) compared to patients in the placebo group (0.9%).
Withdrawals due to any AE (finerenone: 3.2%; placebo: 2.8%) and SAEs (1.2% for both groups) were similar between groups.
More deaths occurred in the placebo group (17.6%) than in the finerenone group (16.5%).
Notable AEs:
The proportion of patients reporting hyperkalemia and worsening of renal function was substantially higher in the finerenone group than the placebo group (Table 4).
The numbers of patients requiring hospitalizations for hyperkalemia were low but higher in the finerenone group (n = 16, 0.5%) compared to the placebo group (n = 6, 0.2%).
AEs related to worsening renal function included renal impairment, decreased eGFR, acute kidney injury, renal failure, and increased blood creatinine. Worsening of renal function was more frequently reported in patients receiving finerenone (17.7%) than in those receiving placebo (10.9%). The number of patients requiring hospitalization due to worsening of renal function was low in both groups (finerenone: 2.0%, placebo: 1.3%).
Summary of Findings and Certainty of the Evidence
Based on the available literature, a between-group MID threshold of 5 points was applied for the KCCQ‑TSS estimates (refer to the Summary of Outcome Measures and Minimal Important Differences table in Appendix 3 in the Supplemental Material document). In the absence of literature-based MID estimates, thresholds suggested by the clinical experts were used for the outcomes of composite end point of CV death and HF events (MID: 2 per 100 patient-years). In the absence of a known threshold, the certainty in the presented non-null effect was rated.
Table 4: Summary of Findings for Finerenone vs. Placebo for Patients With HF (LVEF of 40% or Greater)
Outcome and follow-up | Patients, N (studies) | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Finerenone | Difference | |||||
Composite end point — CV death and HF events | |||||||
CV death and total HF events (first and recurrent), incidence rate per 100 patient‑years (95% CI) Median study duration: 32 months | 6,001 (1 RCT) | RR: 0.84 (0.74 to 0.95) | 17.70 (16.22 to 19.31) | 14.88 (13.52 to 16.38) | −2.81 (███ ) | Moderatea | Finerenone likely results in a reduction of incident events compared to placebo. |
CV death | |||||||
CV death, % (95% CI) Median study duration: 32 months | 6,001 (1 RCT) | csHR: 0.93 (0.78 to 1.11) | 8.7 (NR) | 8.1 (NR) | −0.6 (████) | Moderateb | Finerenone likely results in a reduction of CV deaths compared to placebo. The clinical importance of the reduction is uncertain. |
HF events | |||||||
Total HF events (first and recurrent), incidence rate per 100 patient‑years (95% CI) Median study duration: 32 months | 6,001 (1 RCT) | RR: 0.82 (0.71 to 0.94) | 14.12 (12.79 to 15.60) | 11.57 (10.37 to 12.91) | −2.55 (████) | Highc | Finerenone results in a reduction of incident events compared to placebo. The clinical importance of the reduction is uncertain. |
Hospitalizations for HF, incidence rate per 100 patient‑years (95% CI) Median study duration: 32 months | 6,001 (1 RCT) | RR: 0.84 (0.73 to 0.98) | 12.10 (10.90 to 13.42) | 10.22 (9.18 to 11.39) | −1.87 (████) | Highc | Finerenone results in a reduction of incident events compared to placebo. The clinical importance of the reduction is uncertain. |
Urgent HF events, incidence rate per 100 patient‑years (95% CI) Median study duration: 32 months | 6,001 (1 RCT) | RR: 0.66 (0.47 to 0.91) | 2.03 (1.65 to 2.49) | 1.35 (1.04 to 1.75) | −0.68 (████) | Highc | Finerenone results in a reduction of incident events compared to placebo. The clinical importance of the reduction is uncertain. |
Health-related quality of life | |||||||
LSM mean change in KCCQ-TSS from baseline, (95% CI) Follow-up: 12 months | 5,004 (1 RCT) | NA | 6.54 (5.78 to 7.29) | 8.15 (7.42 to 8.87) | 1.61 (0.66 to 2.55) | Lowd | Finerenone may result in an improvement in the KCCQ‑TSS when compared to placebo. |
Harms | |||||||
Proportion of patients who reported hyperkalemia, % (95% CI) | 5,986 (1 RCT) | NA | 4.2 (NR) | 9.7 (NR) | 5.5 (███ █) | High | Finerenone results in an increase of hyperkalemia events compared to placebo. The clinical importance of the increase is unknown. |
Proportion of patients who reported worsening of renal function, % (95% CI) | 5,986 (1 RCT) | NA | 10.9 (NR) | 17.7 (NR) | 6.8 (███ █) | High | Finerenone results in an increase of events compared to placebo. The clinical importance of the increase is unknown. |
CDA-AMC = Canada’s Drug Agency; CI = confidence interval; csHR = cause-specific hazard ratio; CV = cardiovascular; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HF = heart failure; KCCQ = Kansas City Cardiomyopathy Questionnaire; LSM = least squares mean; LVEF = left ventricular ejection fraction; MID = minimal importance difference; NA = not applicable; NR = not reported; RCT = randomized controlled trial; RR = rate ratio; SOC = standard of care; TSS = total symptom score; vs. = versus.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes. Patients received finerenone and placebo as adjunct to SOC therapy.
CV deaths and HF events were adjudicated by an independent adjudication committee and considered from randomization up until the cut-off date. Hyperkalemia was defined as serum potassium ≥ 5.5 mmol/L. Worsening of renal function includes acute kidney injury, blood creatinine increased, glomerular filtration rate decreased, postrenal failure, prerenal failure, renal failure, and renal impairment. The difference in incidence rates and the risk differences with their corresponding 95% CIs were not a part of the statistical analysis plan and were requested from the sponsor by CDA-AMC to facilitate the GRADE assessment.
aRated down 1 level for serious imprecision. The 95% CI includes the threshold of clinical importance (i.e., MID of 2 per 100 patient-years between groups) suggested by the clinical experts.
bRated down 1 level for serious imprecision. In the absence of an established MID, the null was used. The CIs for the difference between groups included the null, suggesting a possibility of no difference.
cNot downgraded: No MID was available, and the effect was judged based on the null. The CDA-AMC review team acknowledges that the 95% CI for the point estimates excludes the null. However, in the absence of a known MID for the differences between groups, the clinical importance of the estimated between-group difference is uncertain.
dRated down1 level for risk of bias due to substantial missing data; 16.8% of patients in the finerenone group and 16.4% of patients in the placebo group had missing data at month 12. Rated down another level as the point estimate is below the between-group MID of 5 points for the KCCQ-TSS.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence13 and information from an additional information request (dated September 12, 2025)
Long-Term Extension Studies
The sponsor did not submit any long-term extension studies.
Indirect Evidence
No direct evidence exists to support the comparative efficacy and safety of finerenone versus other SGLT2 inhibitors. As such, the sponsor submitted a network meta-analysis (NMA) for the ITC evaluating finerenone versus 2 SGLT2 inhibitors (dapagliflozin and empagliflozin).
Description of Indirect Treatment Comparison
The objective of the ITC was to evaluate the efficacy and safety of finerenone compared to SGLT2 inhibitors (both as adjunct to SOC therapies) in a population with HF classified as NYHA II to IV and an LVEF of 40% or greater (HFmrEF or HFpEF). The NMA compared finerenone to dapagliflozin and empagliflozin, with SOC as background for each drug and using placebo plus SOC as the common comparator. Both dapagliflozin and empagliflozin are approved by Health Canada as an adjunct to SOC therapy for the treatment of HF in Canada. The base case used a Bayesian random-effects model with flat priors and restricted the inclusion to trials that were most comparable to the FINEARTS-HF trial. One study14 (DAPPER) was determined to be too heterogenous and was reserved for a sensitivity analysis. An additional sensitivity analysis was conducted using a frequentist NMA. Studies informing the NMA were identified through a systematic review (search cut-off: April 9, 2025).
Study Selection and Review Methods
Eligible studies enrolled adults with HFmrEF and/or HFpEF (NYHA class II to IV; LVEF ≥ 40%). Up to 10% of NYHA class I were allowed. Interventions were finerenone 10 mg, 20 mg, or 40 mg daily (dose by eGFR), dapagliflozin 5 mg or 10 mg daily, or empagliflozin 10 mg daily. Studies were not excluded due to comparators. Outcomes included composite CV death and/or total HF events, time to first HF event and mortality end points, recurrent HF events, KCCQ clinical summary score (CSS) and/or TSS at approximately 12 months, NYHA improvement, renal composite, and safety events. When data at multiple time points were reported, those most comparable to the FINEARTS-HF trial were chosen.
Only RCTs were included, with no limits on language, time, or geography. Beyond the broad eligibility of a systematic literature review (SLR), stricter criteria were applied for the quantitative analysis to enhance comparability (screening trials for clinical and/or methodologic similarity to the FINEARTS-HF trial).
Studies were identified via an SLR (original search on May 23, 2023; updates on March 5, 2024, and April 9, 2025) in Embase, MEDLINE, CENTRAL, and ClinicalTrials.gov, supplemented by manual citation searches. The sponsor’s summary referenced April 9, 2025, as the cut-off for the updated search.
Titles, abstracts, and full-text screening were performed independently by 2 reviewers with third-party resolution. Extraction used 1 reviewer with senior validation. The risk of bias for RCTs was assessed using the Cochrane Risk of Bias 2 tool by a single reviewer and with the overall results for each primary publication presented in the report. Judgments were summarized as low risk, some concerns, or high risk; outcome-level assessments were captured in the extraction tables.
Data extraction was handled by a single reviewer with a secondary reviewer conducting an accuracy check. Specific methods for quality control at this stage were not clear.
ITC Analysis Methods
For additional information on the analysis methods for the ITC, refer to Appendix 6 in the Supplemental Material document.
Random-effects models were initially preferred. Fixed-effects models were chosen only if heterogeneity was low (I2 ≤ 30%) and the deviance information criterion (DIC) was at least 5 points lower than the DIC value for the random-effects model. Inconsistency statistics were not produced given the absence of closed loops. Bayesian models were run in the gemtc package of JAGS software and frequentist analyses used netmeta in R.
Nodes were constructed by pooling different doses within each treatment. Dapagliflozin and empagliflozin were kept as separate SGLT2 inhibitor nodes. Heterogeneity was handled via random effects and judged with I2 (where calculable) and DIC for model selection. Convergence was assessed using trace plots. A sensitivity analyses (frequentist NMA; broadened study set) yielded overlapping estimates. No subgroup analyses were conducted.
Feasibility and robustness were addressed with 2 prespecified sensitivities: a Bayesian NMA with the addition of 1 excluded trial (DAPPER) and a frequentist NMA using the primary analysis set to test performance in a sparse, star-shaped network. Background therapy differences (SGLT2 inhibitors were prohibited in SGLT2 trials but permitted in the FINEARTS‑HF trial) as well as differences in the timing of the collection of end points were noted as context, but no specific analysis was conducted to explore their impact.
Summary of Included Studies
The sponsor’s SLR identified multiple SGLT2 inhibitor RCTs. Applying stricter, prespecified criteria for the ITC narrowed these to 3 comparator trials, which, together with the FINEARTS‑HF trial, formed a 4-study evidence set, with the primary NMA restricted to 3 double-blind, phase III, placebo-controlled RCTs: FINEARTS‑HF (finerenone; N = 6,001),14,15 DELIVER (dapagliflozin; N = 6,263),16-18 and EMPEROR‑Preserved (empagliflozin; N = 5,988).19-23 The DAPPER trial (open-label, N = 285)14 was excluded from the primary analysis but retained for sensitivity analyses to test the robustness of the results.
All interventions were oral, administered once daily at approved doses (finerenone titrated from 10 mg to 40 mg by eGFR; dapagliflozin from 5 mg to 10 mg; empagliflozin 10 mg). Trials differed in randomization and/or blinding (the DAPPER trial was open-label versus double-blind pivotal RCTs), event-driven versus fixed-time assessments, and handling of recurrent events.
The submission did not provide a study-by-study risk of bias table in the body text. The quality safeguards were implemented via design restrictions (phase III RCTs with adequate follow-up and full-text publication) and an explicit homogeneity appraisal before quantitative synthesis. The methods referenced health technology assessment good-practice guidance (Cochrane‑aligned Risk of Bias 2 framework) from the underlying SLR.
The evidence forms a star-shaped, anchored network with placebo plus SOC as the common comparator. The primary NMA includes 3 trials (1 of finerenone and 2 of SGLT2 inhibitors) informing all finerenone versus SGLT2 inhibitors that contrasts indirectly. As no closed loops are present, inconsistency testing is not informative; interpretation relies on the transitivity assumptions and the noted between-study differences in baseline therapy, populations, and follow-up timing.
Critical Appraisal of ITC
The evidence synthesis follows an appropriate pathway (systematic review with updates feeding an NMA) using standard methods. The sponsor reports using the Cochrane Risk of Bias 2 tool at the outcome level (although detailed ratings are not communicated), applying a reasonable fixed versus random-effects choice rule and including a frequentist NMA as a sensitivity analysis. The Bayesian implementation is conventional and competently executed, and the synthesis covers a broad set of clinically relevant efficacy and safety outcomes aligned to decision needs.
The certainty of the evidence is limited by a relatively sparse, star-shaped network. With 1 study per active node, I2 could not be estimated. Therefore, the assessment of heterogeneity between trials could only be performed qualitatively. The recognized clinical and methodological heterogeneity across the trials may have compromised the validity of the ITC analysis results. In the FINEARTS-HF trial, a higher proportion of patients (30%) with NYHA class III HF were enrolled, compared to the DELIVER trial (24%) and EMPEROR-Preserved trial (18%). Across the primary analysis set (the FINEARTS-HF, DELIVER, and EMPEROR-Preserved trials), baseline demographic and disease characteristics were broadly comparable. The mean age was approximately 72 years, the mean LVEF was 53% to 54%, the mean eGFR was 61 to 63 mL/min/1.73 m2, and hypertension was reported in 88% to 91% of patients. In the DELIVER and EMPEROR-Preserved trials, 45% and 49% of patients reported diabetes, respectively, compared to 40% of patients in the FINEARTS-HF trial. Regarding background therapy, the use of renin-angiotensin system inhibitors (angiotensin II receptor blockers and angiotensin-converting enzyme inhibitors) was reported in approximately 72% of patients, use of beta-blockers was high (≥ 83%) and near-universal use of diuretics was reported across the 3 trials. However, SGLT2 inhibitors were permitted in the FINEARTS-HF trial as SOC but prohibited in the DELIVER and EMPEROR-Preserved trials. In addition, NT-proBNP entry thresholds differed across the trials (≥ 300 pg/mL with AF or ≥ 600 pg/mL without AF for the DELIVER trial versus ≥ 300 pg/mL with AF or ≥ 900 pg/mL without AF for the FINEARTS-HF and EMPEROR-Preserved trials). Follow-up in the FINEARTS-HF trial was 32 months compared to 28 months in the DELIVER trial and 26 months in the EMPEROR-Preserved trial. The assessment time points of the KCCQ differed across the trials (12 months in the FINEARTS-HF and EMPEROR-Preserved trials, but 8 months in the DELIVER trial). The renal composite definitions were similar, but renal death was included in the DELIVER and EMPEROR-Preserved trials. The DAPPER trial was notably heterogeneous, with an age range of 20 to 85 years (versus ≥ 40 years in the FINEARTS-HF trial), included more males (71% versus 55% in the FINEARTS-HF trial), NYHA class I patients (10% versus 0), and patients with diabetes only, with a higher mean eGFR threshold (≥ 45 versus > 25), lower mean body mass index (25 kg/m2 versus 30 kg/m2) and hypertension rates (84% versus 88%), lower use of renin-angiotensin system inhibitors (64% versus 80%) and beta-blocker use (49% versus 85%), flexible dapagliflozin dosing (5 or 10 mg), lower placebo event rates, and an open-label design. Due to the heterogeneity, the DAPPER trial was excluded from the base case and used only to test robustness.
The proportional hazards assumption was not assessed for time to first end points. As recurrent-event outcomes were reported only in the DELIVER trials, those comparisons are limited to dapagliflozin. Therefore, the statistical estimates used in the assessment of treatment effect could have been affected. In base-case analysis results, all 95% credible intervals (CrIs) crossed the null. Model fit favoured random effects in the base case as determined by DIC thresholds; fixed effects produced similar point estimates where used.
Overall, the ITC is methodologically sound and transparently reported, but its inferential strength is curtailed by network sparsity, untested modelling assumptions, and cross-trial differences in baseline risk and background therapy. Given the wide CrIs and the described limitations, the current ITC does not support a conclusion on whether finerenone is superior or inferior to dapagliflozin or empagliflozin.
Efficacy and Harms Results of ITCs
Key results of the ITCs are in Table 5, Table 6, Table 7, and Table 8. A 95% CrI that includes 1 for the HR or RR estimates means that there is substantial uncertainty that precludes making a definitive conclusion that one drug is better or worse than the others in the comparisons.
Composite Outcome: CV Death and Total (First and Recurrent) HF Events
Compared to dapagliflozin, finerenone showed an RR of ████ ████ ████ ████ ██ █████ for the total primary composite (Table 5). Both CrIs cross the null (i.e., 1). No estimates versus empagliflozin were reported for this end point.
Total (First and Recurrent) HF Events
Compared to dapagliflozin, finerenone showed an RR of ████ ████ ████ ████ ██ █████ (Table 5). No estimates versus empagliflozin were reported for total HF events.
Time to CV Death or HF Events
Compared to dapagliflozin, finerenone yielded an HR of ████ ████ ████ ████ ██ █████ for CV death or first HF event, ████ ████ ████ ████ ██ █████ for first HF event, ████ ████ ████ ████ ██ █████ for hospitalization for HF, and ████ ████ ████ ████ ██ █████ for CV death. (Table 6). All CrIs included 1. Compared to empagliflozin, the HRs were ████ ████ ████ ████ ██ █████ for CV death or hospitalization for HF, ████ ████ ████ ████ ██ █████ for hospitalization for HF, ████ ████ ████ ████ ██ █████ for CV death (Table 6). All CrIs included 1.
Kansas City Cardiomyopathy Questionnaire
The mean differences for change from baseline in KCCQ-TSS or KCCQ-CSS at approximately 12 months were near 0, and CrIs spanned 0 (Table 7). The mean difference for finerenone versus dapagliflozin for KCCQ-TSS was █████ ████ ████ █████ ██ █████ and for KCCQ-CSS it was █████ ████ ████ █████ ██ █████. The mean difference for finerenone versus empagliflozin for KCCQ-TSS was █████ ████ ████ █████ ██ █████ and for KCCQ-CSS it was █████ ████ ████ █████ ██ █████.
NYHA Class Improvement
The ORs were ████ ████ ████ ████ ██ █████ for finerenone versus dapagliflozin and ████ ████ ████ ████ ██ ████ for finerenone versus empagliflozin.
Composite Renal Outcome
The RRs were ████ ████ ████ ████ ██ █████ for finerenone versus dapagliflozin and ████ ████ ████ ████ ██ █████ for finerenone versus empagliflozin (Table 6). All CrIs included 1.
All-Cause Mortality
The RRs were ████ ████ ████ ████ ██ █████ for finerenone versus dapagliflozin and ████ ████ ████ ████ ███████ for finerenone versus empagliflozin (Table 6). All CrIs included 1.
Harms
The safety analysis focused on several outcomes: any AEs, any SAEs, and discontinuation due to AEs. Acute renal failure and acute kidney injury were reported as SAEs. Hyperkalemia was reported as an AE and an SAE. For safety end points including any SAEs, specific SAEs (acute renal failure, acute kidney injury, and hyperkalemia), and discontinuations due to AEs, both SGLT2 inhibitors could be compared with finerenone.
Adverse Events
For any AEs and hyperkalemia, data were available only for empagliflozin (Table 8). For this outcome, the results of the random-effects NMA showed insufficient differences between finerenone and empagliflozin, and it could not be determined if 1 drug was more favourable that the other as the CrI included 1. The OR was ████ ████ ████ ████ ██ █████. For hyperkalemia reported as an AE, the results from the NMA suggested imprecise estimates, as indicated by the relatively wide CrIs when comparing finerenone to empagliflozin (OR = █████ ███ ████ ████ ██ █████).
Serious Adverse Events
For any SAE, the random-effects NMA showed insufficient differences between finerenone and dapagliflozin or empagliflozin, and it could not be determined if 1 drug was more favourable than the other, as the CrIs included 1 (OR = █████ ███ ████ ████ ██ ████) and dapagliflozin (OR = █████ ███ ████ ████ ██ ████). The random-effects NMA showed insufficient differences between groups to suggest 1 drug is more favourable than the other when comparing finerenone to empagliflozin and dapagliflozin for the following SAEs:
acute renal failure (empagliflozin: OR = █████ ███ ████ ████ ██ █████; dapagliflozin: OR = █████ ███ ████ ████ ██ █████)
acute kidney injury (empagliflozin: OR = █████ ███ ████ ████ ██ ████; dapagliflozin: OR = █████ ███ ████ ████ ██ ████)
hyperkalemia (empagliflozin: OR = █████ ███ ████ ████ ██ █████; dapagliflozin: OR = █████ ███ ████ ████ ██ █████).
Discontinuation Due to AEs
The random-effects NMA showed insufficient between-group differences to suggest 1 drug is more favourable than the other, as the CrIs included 1 for discontinuations due to AEs when comparing finerenone to empagliflozin (OR = █████ ███ ████ ████ ██ ████) and dapagliflozin (OR = █████ ███ ████ ████ ██ ████).
Table 5: ITC Data Table for Total (First and Recurrent) Outcomes — Finerenone vs. Comparators
Outcome | Rate ratio (95% CrI) | |
|---|---|---|
Dapagliflozin | Empagliflozin | |
Total (first and recurrent) primary composite outcomes | ████ █████ █ | ████ █████ █ |
Total (first and recurrent) HF events | ████ █████ █ | ████ █████ █ |
CrI = credible interval; HF = heart failure; ITC = indirect treatment comparison; NA = not applicable; vs. = versus.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the sponsor-submitted ITC technical report.13
Table 6: ITC Data Table for Time-to-Event Outcomes — Finerenone vs. Comparators
Outcome | Hazard ratio (95% CrI) | |
|---|---|---|
Dapagliflozin | Empagliflozin | |
CV death or first HF event | ████ ████ ██ ████ | ████ ████ ██ ████ |
CV death or HHF | ████ ████ ██ ████ | ████ ████ ██ ████ |
HF event | ████ ████ ██ ████ | ████ ████ ██ ████ |
Hospitalization for HF | ████ ████ ██ ████ | ████ ████ ██ ████ |
CV death | ████ ████ ██ ████ | ████ ████ ██ ████ |
All-cause mortality | ████ ████ ██ ████ | ████ ████ ██ ████ |
Composite renal outcome | ████ ████ ██ ████ | ████ ████ ██ ████ |
CrI = credible interval; CV = cardiovascular; HF = heart failure; HHF = hospitalization for HF; ITC = indirect treatment comparison; NA = not applicable; vs. = versus.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the sponsor-submitted ITC technical report.13
Table 7: ITC Data Table for Continuous Outcomes — Finerenone vs. Comparators
Outcome | Mean difference in change from baseline (95% CrI) | |
|---|---|---|
Dapagliflozin | Empagliflozin | |
KCCQ-TSS | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
KCCQ-CSS | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
CrI = credible confidence interval; CSS = clinical summary score; ITC = indirect treatment comparison; KCCQ = Kansas City Cardiomyopathy Questionnaire; TSS = total symptom score; vs. = versus.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the sponsor-submitted ITC technical report.13
Table 8: ITC Data Table for Harms — Finerenone vs. Comparators
Outcome | Odds ratio (95% CrI) | |
|---|---|---|
Dapagliflozin | Empagliflozin | |
Any AE | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Any SAE | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Discontinuation due to AEs | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Acute renal failure (SAE) | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Acute kidney injury (SAE) | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Hyperkalemia (AE) | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
Hyperkalemia (SAE) | █████ ██████ ██ █████ | █████ ██████ ██ █████ |
AE = adverse event; CrI = credible confidence interval; ITC = indirect treatment comparison; NA = not applicable; SAE = serious adverse event; vs. = versus.
Note: An odds ratio > 1 indicates “favoured comparator (dapagliflozin or empagliflozin),” while OR < 1 indicates “favoured finerenone.”
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence and the sponsor-submitted ITC technical report.13
Studies Addressing Gaps in the Systematic Review Evidence
The sponsor did not submit any studies to address evidence gaps.
Discussion
Efficacy
The key treatment goals identified by patients and clinicians include reducing the risk of hospitalization, lowering mortality, minimizing side effects, and improving quality of life. Patients living with HF also emphasized the need for treatments to go beyond symptom management and improve their ability to function. In the FINEARTS-HF trial, the primary end point was a composite of CV death and total HF events (i.e., first and recurrent urgent HF visits and hospitalizations for HF). After a median follow-up of 32 months, treatment with finerenone resulted in a 16% reduction in the relative risk in the primary outcome compared to placebo. The clinical experts assessed this difference between treatment groups as clinically meaningful, and a GRADE assessment rated the certainty of evidence as moderate, indicating that the reduction in the composite of CV death and total HF events was likely clinically important (Table 4). The reduction observed in the composite end point was largely driven by the reduction in HF events. Prespecified subgroup analyses conducted for several subgroups were considered exploratory, detailed in Appendix 4 in the Supplemental Material document, and revealed no major concerns aligning with the primary results. While the trial employed strict inclusion and exclusion criteria to ensure capturing a representative population, this may have led to an enriched study cohort. However, the clinical experts consulted by CDA‑AMC for this review did not consider the exclusions to be major concerns and noted that, overall, the characteristics of patients within the trial were generalizable to clinical practice in Canada. The results from the individual components (CV death and total HF events) of the primary composite were also reviewed. For CV death, the GRADE assessment of the absolute difference between groups demonstrated that finerenone likely resulted in a reduction of CV deaths compared to placebo. However, conclusions regarding this outcome were limited by the imprecision in the estimates. At the median follow-up of 32 months, the difference between the groups in CV deaths was statistically insignificant, with an HR of 0.93 (95% CI, 0.78 to 1.11). As the trial was not designed to detect a difference between groups in CV deaths, the clinical meaningfulness of the results was considered uncertain. For total HF events, the results of the trial demonstrated an 18% reduction in the relative risk of events in favour of finerenone. The clinical experts consulted by CDA-AMC considered this difference between treatment groups to be clinically meaningful. The GRADE assessment rated the certainty of evidence as high, indicating that treatment with finerenone resulted in a reduction in total HF events compared to placebo; however, the clinical importance of the reduction is uncertain. The clinical experts emphasized that total HF events constitute an important outcome because it affects the patients, treatment outcomes, caregivers, and the broader health care system.
Patient HRQoL was assessed using the KCCQ, which the clinical experts noted was a valid tool to assess HRQoL in patients with HF. The TSS domain of the KCCQ was a key secondary end point in the FINEARTS-HF trial. The difference between groups from baseline to 12 months suggested that treatment with finerenone may improve HRQoL. However, the certainty of evidence was considered low as the difference between groups was below the MID suggested by the clinical experts (i.e., 5 points between groups). Furthermore, there was uncertainty due to the subjectivity of patient-reported outcomes and a risk of attrition bias due to a substantial amount of missing data. The results demonstrated little to no difference between the finerenone and placebo groups for other key secondary end points, including change in NYHA class, composite renal end point, and all-cause mortality.
The clinical experts consulted by CDA-AMC noted that spironolactone was considered a relevant comparator; however, it is used off-label, and the potential for use is limited due to the uncertainty regarding its clinical benefit and safety in patients with HFpEF. The 2017 CCS guidelines provided a “weak” recommendation for use of spironolactone and candesartan in patients with HFpEF.2 Based on evidence from the EMPEROR-Preserved and DELIVER trials in patients with HF, the SGLT2 inhibitors (empagliflozin and dapagliflozin) were approved for use in patients with HF, which makes them relevant comparators for finerenone. The 2025 CCS guidelines strongly recommend the use of SGLT2 inhibitors in patients with HFmrEF and HFpEF. However, the clinical experts consulted by CDA-AMC indicated that, at the time the FINEARTS-HF trial started, SGLT2 inhibitors were not indicated in patients with HF and therefore would not be considered comparators for the trial.
One sponsor-submitted NMA compared finerenone, dapagliflozin, and empagliflozin using placebo plus SOC as the common comparator. The base case was a Bayesian random-effects model. The results from this model were also tested under a frequentist approach, and a sensitivity analysis was conducted adding another study (the DAPPER trial) to the base-case network. The DAPPER trial was excluded from the base case due to its deviating study design (e.g., open label) and patient characteristics (e.g., population with diabetes only); a sensitivity analysis was used to verify if its inclusion would affect the findings. The ITC had a number of limitations related to heterogeneities in the included studies. Most notably, other SGLT2 inhibitors (beyond the intervention) were permitted as part of SOC in the FINEARTS-HF trial (used by 13.6% of patients at baseline) but were prohibited as background therapy in the SGLT2 inhibitor trials (in which they were the active intervention). This creates a difference in the treatments received by the common comparator (placebo plus SOC) arms. The analysis was limited by a sparse, star-shaped network, which made statistical heterogeneity (I2) difficult or impossible to measure across most comparisons. Across key outcomes (HF events, mortality, KCCQ scores, and safety), all the point estimates were centred around 1, with wide 95% CrIs that crossed the null (i.e., 1). Therefore, in the comparison of finerenone versus the SGLT2 inhibitors, no comparative advantage was demonstrated in favour of any of the drugs evaluated.
Harms
The safety profile of finerenone was generally consistent with findings from previous studies24 and was considered acceptable by the clinical experts consulted for this review. Overall, the proportion of patients with AEs was comparable between finerenone (84.0%) and placebo plus SOC groups (82.3%). The proportion of SAEs was marginally higher in patients receiving placebo (40.5%) than in patients receiving finerenone (38.7%). Numerically, more deaths occurred in the placebo group. A higher proportion of patients in the finerenone group reported hyperkalemia compared to the placebo group (9.7% versus 4.2%, respectively). However, most cases were considered mild or moderate, and few resulted in hospitalization (finerenone: 0.5%; placebo: 0.2%). The clinical experts consulted by CDA-AMC noted that a higher risk of hyperkalemia was not concerning in itself as it was a part of the mechanism of action for finerenone and was clinically manageable. Worsening of renal function was also reported more frequently in the finerenone group (17.7%) than in the placebo group (10.9%). Renal impairment (finerenone: 6.6%; placebo: 3.9%) and decreased eGFR (finerenone: 5.2%, placebo: 3.6%) were most frequently reported. However, the majority of the events were nonserious and mild or moderate in intensity, and the percentage of hospitalizations due to renal impairment was low (finerenone: 2.0%, placebo: 1.3%). Hypotension was also more frequently reported in the finerenone group (7.3%) than in the placebo group (4.5%), with most cases being nonserious and mild or moderate in intensity. The product monograph of finerenone states that finerenone should be withheld in patients if serum potassium levels are 6.0 mmol/L or greater. In addition, the risk of hyperkalemia may increase with the intake of concomitant medications and other MRAs.
Results from the random-effects model from the ITC had insufficient between-group differences to suggest 1 drug is more favourable than the other when comparing finerenone to SGLT2 inhibitors (empagliflozin and dapagliflozin) for safety outcomes, including any SAE or discontinuation due to AEs. Treatment with finerenone was associated with higher odds of acute renal failure, acute kidney injury, or hyperkalemia; however, the estimates are considered imprecise due to the wide CrIs. No new safety signals were identified.
Ethics and Equity Considerations
The patient group noted that patients living with HFmrEF and HFpEF experience burdensome symptoms, and fear of a sudden decline in health can lead to reduced participation in daily activities, adding to emotional strain. The patient group also described treatment delays and an inability to access specialists, leading to emotional distress. The clinical experts consulted by CDA-AMC for this review agreed that there are disparities in access to care in Canada. They noted the inequality of diagnostic testing (NT-proBNP biomarkers) in Canada and acknowledged that patients may be living several hours away from a diagnostic centre or a cardiology clinic. They mentioned that, in real-world practice, decision-making of a diagnosis of HFpEF is not based exclusively on NT-proBNP levels, particularly in patients with obesity, for whom an NT-proBNP level may not be elevated. One clinician group noted that patients with HFpEF have limited treatment options and are particularly underserved (i.e., patients with an eGFR < 60 mL/min/1.73 m2, which is consistent with stages 3 or 4 of chronic kidney disease).
Although the trial enrolled a lower proportion of women and capped the patients with an LVEF of greater than 60%, the clinical experts consulted for this review suggested that the study results were generalizable to these patient groups in clinical practice. The fact that patients within the trial were predominately white raises some concerns regarding whether the magnitude of benefit observed in the FINEARTS-HF trial can be generalized to racially and ethnically diverse patient populations. HF disproportionately affects equity-deserving groups.7 The risk of HF is higher among Indigenous people and other marginalized communities (e.g., people experiencing low socioeconomic status, racially and ethnically diverse populations, and females).7,25,26 Therefore, inequities in accessing appropriate care and use of evidence-based therapies, and obstacles to navigating health care systems may contribute to disparate outcomes.25
Conclusion
One phase III, double-blind, placebo-controlled, multicentre RCT (FINEARTS-HF) provided evidence of the efficacy and safety of finerenone compared with placebo, both in addition to SOC therapies, in adults with HF (NYHA class II to IV and an LVEF ≥ 40%). The results of the FINEARTS-HF trial indicate that, in patients with HFmrEF and HFpEF, finerenone likely results in a clinically meaningful reduction in the risk of the composite of CV death and HF events compared to placebo. The reduction was mainly driven by a reduction in total HF events. Finerenone resulted in a reduction of hospitalizations for HF and urgent HF visits. In the absence of known thresholds for clinically meaningful effects, the clinical importance of the reductions is uncertain. Patient quality of life as measured by the KCCQ-TSS may be improved with finerenone treatment, but the clinical importance of the effect is uncertain. Overall, the proportion of patients reporting an AE was comparable between the treatment groups. However, finerenone resulted in an increase in events involving hyperkalemia and worsening of renal function compared to placebo. Patients in the placebo group experienced numerically more deaths than did patients in the finerenone group.
Evidence from an ITC submitted by the sponsor was insufficient to draw a firm conclusion regarding the efficacy and safety of finerenone versus empagliflozin or dapagliflozin in patients with HF (NYHA class II to IV and an LVEF ≥ 40%) due to study limitations, such as significant heterogeneity and a sparse network.
Economic Review
Methods
The review team appraised the submitted pharmacoeconomic evidence, which included an economic evaluation comparing the cost-effectiveness of finerenone as an adjunct therapy for those receiving SOC therapies to SOC therapies alone for the requested reimbursement population (i.e., as an adjunct to SOC therapies in adults for the treatment of HF with mildly reduced or preserved ejection fraction [an LVEF ≥ 40%] to reduce the risk of CV death, hospitalization for HF, and urgent HF visits). This aligns with the population studied in the FINEARTS-HF trial but is narrower than the proposed Health Canada indication. The sponsor also submitted a BIA assessing the budgetary impact of reimbursing finerenone for the requested reimbursement population.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of finerenone from the perspective of a public health care payer in Canada over a lifetime horizon of 28 years. The modelled population comprised adult patients receiving finerenone as an adjunct to SOC therapies, which is aligned with the reimbursement request. This is narrower than the proposed Health Canada population and was based on the participants in the FINEARTS-HF trial. The sponsor’s base-case analysis included costs related to drug acquisition, disease management, treatment monitoring, AEs, and end of life care.
In the sponsor’s base case, finerenone plus SOC was associated with incremental costs of $8,643 and 0.38 incremental quality-adjusted life-years (QALYs) relative to SOC alone. This resulted in an incremental cost-effectiveness ratio (ICER) of $22,712 per QALY gained. Of the incremental benefit compared to SOC alone (0.38 QALYs), approximately 92% of the benefit was predicted to accrue after the treatment duration of the FINEARTS-HF trial (mean treatment period = 25 months). Additional information about the sponsor’s submission is summarized in Appendix 10 in the Supplemental Material document.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 9; full details are provided in Appendix 11 in the Supplemental Material document).
Table 9: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The survival benefit associated with finerenone is likely overestimated. | The sponsor’s analysis suggests that patients treated with finerenone plus SOC would accrue an additional 0.38 LYs (i.e., 4.6 months) compared to patients treated with SOC alone. According to the CDA‑AMC Clinical Review report, finerenone likely results in a reduction of CV deaths; however, the clinical importance is uncertain. The clinical experts noted that there has been no survival benefit observed in any HFpEF trial. | This issue could not be addressed in the CDA‑AMC base case given the uncertainty of the evidence to support a survival benefit for patients receiving treatment for HF. If the survival benefit is expected to be less than modelled, the ICER would be expected to increase. | To explore uncertainty around this issue, CDA‑AMC conducted a scenario analysis in which the survival benefit associated with finerenone was removed. |
The rate of HF events does not replicate the trial. | The sponsor’s model overestimated event rates for patients receiving SOC alone and underestimated event rates for patients treated with finerenone plus SOC. As a result, the model produced a rate ratio between treatment arms (approximately 0.68) that was notably lower than that reported in the trial (0.82). The inconsistency of modelled outcomes during the trial period raises concerns with the validity of the modelled outcomes. | To model HF events for finerenone plus SOC, CDA‑AMC applied the rate ratio from the trial (0.82) to the placebo plus SOC event rates. Given that the CDA‑AMC reanalysis applies a fixed HR from the trial, any extrapolation of CV outcomes beyond 3 years is highly uncertain, and CDA‑AMC adopted a 3‑year time horizon in reanalysis. | No scenario analysis was conducted. |
The proportion of patients receiving treatment with SGLT2 inhibitors as SOC is likely underestimated. | The proportion of patients receiving treatment with SGLT2 inhibitors was derived from the FINEARTS-HF trial. Since the initiation of this trial, SOC has evolved, with SGLT2 inhibitors considered a standard treatment for patients with HF. As some SGLT2 inhibitors are now publicly reimbursed for this condition, a higher proportion of patients in real-world settings would be expected to be prescribed SGLT2 inhibitors, according to clinical expert feedback. The impact on the efficacy of finerenone as an adjunct to SOC, given a higher proportion of patients are receiving SGLT2 inhibitors, is uncertain. | CDA-AMC was unable to address this issue in the absence of comparative clinical data that reflects present SGLT2 inhibitor use in clinical practice in Canada. The increased cost associated with a larger proportion of patients receiving SGLT2 inhibitors as part of SOC is not expected to have an impact on the results given that the cost of SOC is the same for both treatment arms. | No scenario analysis was conducted. |
Hospitalization costs are likely double counted. | The sponsor’s analysis includes both the cost of HHF and the cost of CV deaths. As most CV deaths occur during a hospitalization event, the cost of CV deaths is likely double counted by adopting this costing approach. | CDA‑AMC removed the cost of CV mortality. | No scenario analysis was conducted. CDA‑AMC considered the revised base-case approach to be the most reasonable and conservative. |
CDA-AMC = Canada’s Drug Agency; CV = cardiovascular; HF = heart failure; HHF = hospitalization due to heart failure; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; LVEF = left ventricular ejection fraction; LY = life-year; SGLT2 = sodium-glucose cotransporter‑2; SOC = standard of care.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 11 in the Supplemental Material document.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions, in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 11 in the Supplemental Material document.
Impact on Health Care Costs
Finerenone is predicted to be associated with additional health care costs compared to SOC (incremental costs = $2,538). This increase in health care spending results from drug acquisition costs associated with finerenone, although it is partially offset by avoiding hospitalizations due to HF events (refer to Figure 1).
Figure 1: Impact of Finerenone Plus SOC vs. SOC on Health Care Costs
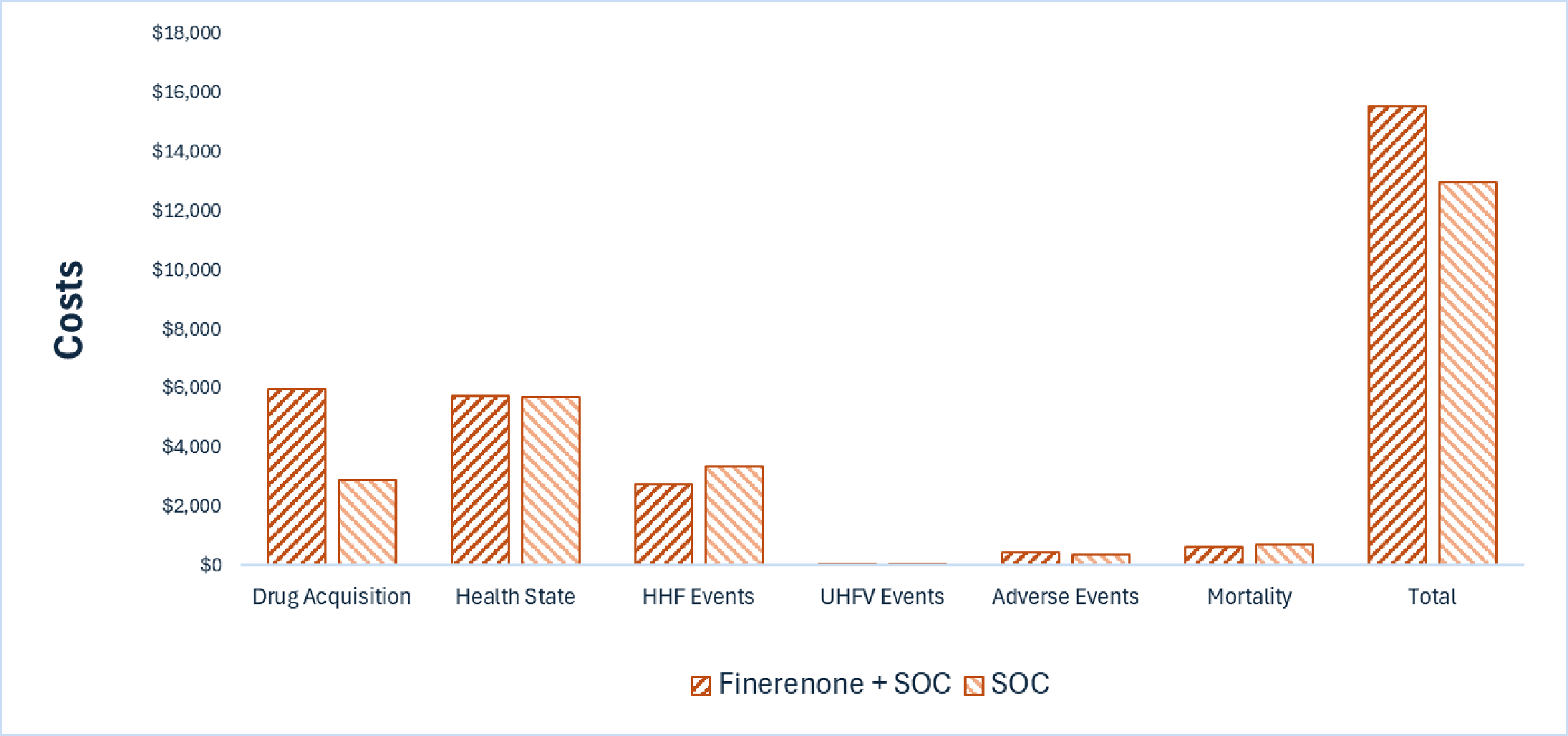
HHF = hospitalization due to heart failure; SOC = standard of care; UHFV = urgent heart failure visit; vs. = versus.
Note: Adverse event costs include costs associated with hyperkalemia.
Impact on Health
Relative to SOC alone, finerenone is predicted to increase the amount of time a patient remains in the NYHA class II health state by approximately 0.01 years and to extend overall survival by 0.02 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, finerenone is predicted to result in an additional 0.03 QALYs per patient compared to SOC alone, over a 3‑year time horizon. The close alignment between incremental life-years and QALYs suggests that the incremental benefit was primarily driven by extended survival, with minimal changes in HRQoL.
Figure 2: Impact of Finerenone Plus SOC vs. SOC on Patient Health
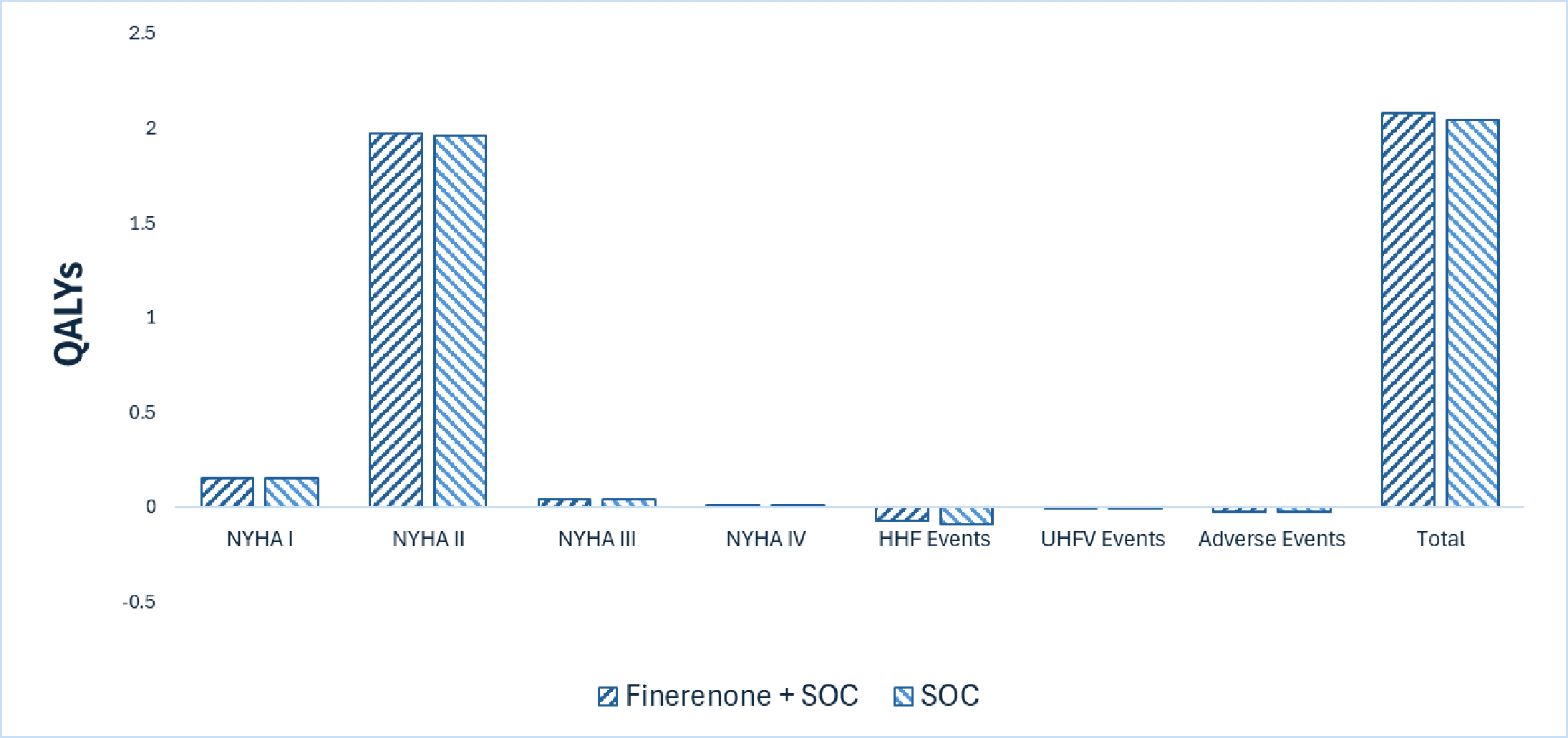
HHF = hospitalization due to heart failure; NYHA = New York Heart Association; QALY = quality-adjusted life-year; SOC = standard of care; UHFV = urgent heart failure visit; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $77,195 per QALY gained for finerenone compared to SOC alone (refer to Table 10). Additional details on the CDA-AMC base case are available in Appendix 11 in the Supplemental Material document.
Table 10: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|
SOC | 12,979 | 2.04 | Reference |
Finerenone plus SOC | 15,517 | 2.08 | 77,195 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
The CDA-AMC base case adopted a 3-year time horizon to apply the trial-based HR for HF events. The cost-effectiveness of finerenone plus SOC using a longer time horizon is highly uncertain due to a lack of long-term data to inform HF events beyond 3 years. If the treatment effect of finerenone wanes over time, the ICER would be higher (i.e., the incremental costs will be stable, but the incremental benefits will decrease), or, if there are additional benefits beyond what is captured in the 3-year period, the ICER will decrease (i.e., incremental costs will be stable, but the incremental benefits will increase).
Results of the CDA-AMC analysis suggest that the benefit of finerenone plus SOC compared to SOC alone is driven by the predicted survival benefit. In the submitted model, CV-related and all-cause mortality were estimated using parametric survival models fitted to patient data from the FINEARTS-HF trial assuming proportional hazards over time. CDA-AMC conducted a scenario analysis, assuming no difference in mortality for patients treated with finerenone plus SOC or SOC alone. In this analysis, the ICER for finerenone plus SOC was estimated to be $156,523 per QALY gained versus SOC.
Summary of the Budget Impact
The sponsor submitted a BIA to estimate the 3-year (2027 to 2029) impact of reimbursing finerenone as an adjunct to SOC therapies in adults for the treatment of HF with mildly reduced or preserved ejection fraction (an LVEF ≥ 40%) to reduce the risk of CV death, hospitalization for HF, and urgent HF visits. The sponsor assumed that the payer would be CDA‑AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The unit price of finerenone was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 12 in the Supplemental Material document.
CDA-AMC identified a number of issues with the sponsor’s submitted BIA that could not be addressed. The sponsor estimated that, by year 3 of reimbursement, 358,902 patients would be eligible for treatment with finerenone, 48,868 of whom would be expected to receive finerenone. The estimated incremental budget impact of reimbursing finerenone is predicted to be approximately $107 million over the first 3 years, with an expected expenditure of $107 million on finerenone. The actual budget impact of reimbursing finerenone as an adjunct to SOC for adults with HF will depend on the market share of finerenone.
Conclusion
Based on the CDA-AMC base case using a 3-year time horizon, finerenone would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $77,195 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Appendix 11 in the Supplemental Material document). The estimated ICER is uncertain as the model predicted a mortality improvement despite the uncertain clinical evidence. As noted in the CDA-AMC clinical review, finerenone likely resulted in a reduction of CV deaths compared to placebo. However, conclusions regarding this outcome were limited by the imprecision in the estimates. If the survival benefit is not realized, the ICER for finerenone plus SOC compared to SOC alone will be higher than predicted in the CDA-AMC base-case analysis, and greater price reductions may be required to achieve a given willingness-to-pay threshold. The cost-effectiveness of finerenone using a longer time horizon is highly uncertain due to data limitations. In addition, the impact on efficacy of finerenone as an adjunct to SOC is uncertain, as a higher proportion of patients are treated with SGLT2 inhibitors in practice compared to the proportion observed in the FINEARTS-HF trial.
The sponsor estimated that the budget impact of reimbursing finerenone as an adjunct to SOC therapies in adults for the treatment of HF with mildly reduced or preserved ejection fraction (an LVEF ≥ 40%) to reduce the risk of CV death, hospitalization for HF, and urgent HF visits would be approximately $107 million in the first 3 years. The 3‑year expenditure on finerenone (not accounting for current expenditure on comparators) is estimated to be $107 million.
The available evidence reviewed reflects the sponsor’s reimbursement request. The cost-effectiveness and budget impact of finerenone for the full Health Canada indication is unknown, although the clinical experts consulted by CDA‑AMC noted that finerenone is most likely to be prescribed as an adjunct to SOC in alignment with its reimbursement request.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
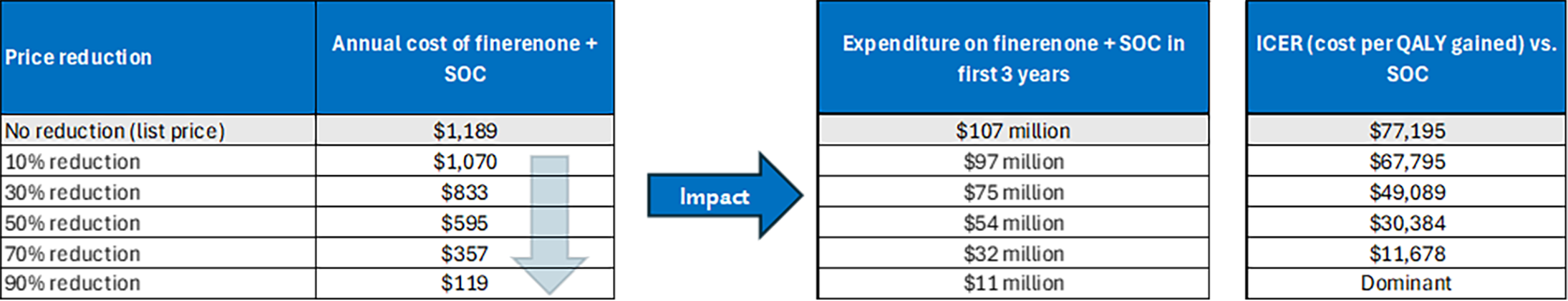
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs = versus.
Notes: Expenditure includes only the drug cost of finerenone. The term “dominant” indicates that a drug costs less and provides more QALYs than the comparator.
References
1.Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895-e1032. doi: 10.1161/CIR.0000000000001063 PubMed
2.Ezekowitz JA, O'Meara E, McDonald MA, et al. 2017 Comprehensive Update of the Canadian Cardiovascular Society Guidelines for the Management of Heart Failure. Can J Cardiol. 2017;33(11):1342-1433. doi: 10.1016/j.cjca.2017.08.022 PubMed
3.Bozkurt B, Coats AJ, Tsutsui H, et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J Card Fail. 2021;27(4):387-413. doi: 10.1016/j.cardfail.2021.01.022 PubMed
4.Savarese G, Stolfo D, Sinagra G, Lund LH. Heart failure with mid-range or mildly reduced ejection fraction. Nat Rev Cardiol. 2022;19(2):100-116. doi: 10.1038/s41569-021-00605-5 PubMed
5.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129-2200. doi: 10.1093/eurheartj/ehw128 PubMed
6.National Heart L, Blood I. Heart failure. Causes and risk factors [sponsor supplied reference]. 2022. Accessed Jun 21, 2024. https://www.nhlbi.nih.gov/health/heart-failure/causes
7.The Heart and Stroke Foundation of Canada. Heart failure in Canada: complex, incurable and on the rise. February 2022. Accessed at: https://www.heartandstroke.ca/what-we-do/media-centre/news-releases/heart-failure-in-canada-complex-incurable-and-on-the-rise#:~:text=In%20Canada%20there%20are%20750%2C000,family%20member%20or%20close%20friend [sponsor supplied reference].
8.Canadian Chronic Disease Surveillance System (CCDSS). 2025. Accessed October 28. https://health-infobase.canada.ca/ccdss/data-tool/index?G=00&V=11
9.Poon S, Leis B, Lambert L, et al. The State of Heart Failure Care in Canada: Minimal Improvement in Readmissions Over Time Despite an Increased Number of Evidence-Based Therapies. CJC Open. 2022;4(8):667-675. doi: 10.1016/j.cjco.2022.04.011 PubMed
10.Solomon Scott D, McMurray John JV, Anand Inder S, et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N Engl J Med. 2019;381(17):1609-1620. doi: 10.1056/NEJMoa1908655 PubMed
11.Canadian Chronic Disease Surveillance System data files provided by provinces and territories, as of July 2024. Public Health Agency of Canada, Health Infobase https://health-infobase.canada.ca [sponsor supplied reference].
12.Virani S, Zieroth S, Aleksova N, et al. Canadian Cardiovascular Society/Canadian Heart Failure Society 2025 Guideline Update for Pharmacologic Management of Heart Failure With Nonreduced Ejection Fraction (LVEF > 40%). Can J Cardiol. 2025;41(10):1857-1874. doi: 10.1016/j.cjca.2025.07.027 PubMed
13.Bayer Inc. Sponsor Summary of Clinical Evidence [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kerendia (finerenone), 10 mg, 20 mg and 40 mg, oral tablets. October 2025.
14.Yoshihara F, Imazu M, Sakuma I, et al. DAPagliflozin for the attenuation of albuminuria in Patients with hEaRt failure and type 2 diabetes (DAPPER study): a multicentre, randomised, open-label, parallel-group, standard treatment-controlled trial. EClinicalMedicine. 2023;66:102334. doi: 10.1016/j.eclinm.2023.102334 PubMed
15.Solomon SD, McMurray JJV, Vaduganathan M, et al. Finerenone in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med. 2024;391(16):1475-1485. doi: 10.1056/NEJMoa2407107 PubMed
16.Solomon SD, McMurray JJV, Claggett B, et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med. 2022;387(12):1089-1098. doi: 10.1056/NEJMoa2206286 PubMed
17.Ostrominski JW, Vaduganathan M, Claggett BL, et al. Dapagliflozin and New York Heart Association functional class in heart failure with mildly reduced or preserved ejection fraction: the DELIVER trial. Eur J Heart Fail. 2022;24(10):1892-1901. doi: 10.1002/ejhf.2652 PubMed
18.NCT03619213. Dapagliflozin Evaluation to Improve the LIVEs of Patients with Preserved Ejection Fraction Heart Failure. (DELIVER) [Available from: https://clinicaltrials.gov/study/NCT03619213 [sponsor supplied reference].
19.Anker SD, Butler J, Usman MS, et al. Efficacy of empagliflozin in heart failure with preserved versus mid-range ejection fraction: a pre-specified analysis of EMPEROR-Preserved. Nat Med. 2022;28(12):2512-2520. doi: 10.1038/s41591-022-02041-5 PubMed
20.Butler J, Packer M, Filippatos G, et al. Effect of empagliflozin in patients with heart failure across the spectrum of left ventricular ejection fraction. Eur Heart J. 2022;43(5):416-426. doi: 10.1093/eurheartj/ehab798 PubMed
21.NCT03057951. Empagliflozin outcome trial in Patients With chronic heart Failure with Preserved Ejection Fraction (EMPEROR-Preserved) [Available from: https://beta.clinicaltrials.gov/study/NCT03057951 [sponsor supplied reference].
22.Butler J, Usman MS, Filippatos G, et al. Safety and Efficacy of Empagliflozin and Diuretic Use in Patients with Heart Failure and Preserved Ejection Fraction: A Post Hoc Analysis of the EMPEROR-Preserved Trial. JAMA Cardiol. 2023;8(7):640-649. doi: 10.1001/jamacardio.2023.1090 PubMed
23.Anker SD, Butler J, Filippatos G, et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med. 2021;385(16):1451-1461. doi: 10.1056/NEJMoa2107038 PubMed
24.CADTH. Drug Reimbursement Review clinical guidance report: Finerenone (Kerendia) as an adjunct to standard of care therapy in adults with chronic kidney disease (CKD) and type 2 diabetes (T2D) to reduce the risk of: End-stage kidney disease and a sustained decrease in estimated glomerular filtration rate, Cardiovascular death, non-fatal myocardial infarction and hospitalization for heart failure. Standard of care includes: an angiotensin converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB), and a sodium-glucose cotransporter-2 inhibitor (SGLT2i), unless contraindicated or not tolerated. May 2023. Accessed September 1, 2025. https://www.cda-amc.ca/finerenone
25.Lewsey SC, Breathett K. Racial and ethnic disparities in heart failure: current state and future directions. Curr Opin Cardiol. 2021;36(3):320-328. doi: 10.1097/hco.0000000000000855 PubMed
26.Walia RS, Mankoff R. Impact of Socioeconomic Status on Heart Failure. J Community Hosp Intern Med Perspect. 2023;13(6):107-111. doi: 10.55729/2000-9666.1258 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.