Drugs, Health Technologies, Health Systems
Reimbursement Review
Relugolix-Estradiol–Norethindrone Acetate (Myfembree)
Sponsor: Knight Therapeutics Inc.
Therapeutic area: Management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AUB
abnormal uterine bleeding
BMD
bone mineral density
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
DXA
dual-energy X-ray absorptiometry
E2
estradiol
E2-NETA
estradiol and norethindrone acetate
FIGO
International Federation of Gynecology and Obstetrics
GnRH
gonadotropin-releasing hormone
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HMB
heavy menstrual bleeding
HRQoL
health-related quality of life
ITC
indirect treatment comparison
LA
leuprolide acetate
LSM
least squares mean
MBL
menstrual blood loss
MD
mean difference
MFP
mifepristone
MID
minimal important difference
mITT
modified intention to treat
NETA
norethindrone acetate
NMA
network meta-analysis
NRS
numerical rating scale
OLE
open-label extension
RCT
randomized controlled trial
RGX
relugolix
RGX-E2-NETA
relugolix-estradiol–norethindrone acetate
RR
relative risk
RWS
randomized withdrawal study
SAE
serious adverse event
SD
standard deviation
SE
standard error
SLR
systematic literature review
SSS
Symptom Severity Scale
TEAE
treatment-emergent adverse event
UF
uterine fibroid
UFS-QoL
Uterine Fibroid Symptom Quality of Life questionnaire
UPA
ulipristal acetate
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Relugolix-estradiol–norethindrone acetate (Myfembree) 40 mg/1 mg/0.5 mg oral tablets |
Sponsor | Knight Therapeutics Inc. |
Indication | In premenopausal women for the management of heavy menstrual bleeding associated with uterine fibroids and for the management of moderate to severe pain associated with endometriosis |
Reimbursement request | For the management of heavy menstrual bleeding associated with uterine fibroids in women who are premenopausal |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 22, 2023 |
Recommended dose | Relugolix 40 mg, estradiol 1 mg, and norethindrone acetate 0.5 mg, once daily |
NOC = Notice of Compliance.
Introduction
Uterine fibroids (UFs), also known as myomas or leiomyomas, are benign neoplasms of uterine smooth muscle and are the most common tumours in females.1 UFs are typically diagnosed in people between menarche and menopause. The main risk factors are younger age at menarche, late onset of menopause, African ancestry, and a child-bearing history of few or no children.1 A common symptom is abnormal uterine bleeding (AUB), particularly heavy menstrual bleeding (HMB), which is reported by 50% to 80% of patients with symptomatic UFs in surveys and trials.2,3 Infertility, abdominal bloating, constipation, increased urinary frequency, incontinence, dyspareunia, anemia, and fatigue are among the other symptoms. These can affect health-related quality of life (HRQoL).1 For research purposes, HMB is defined as more than 80 mL of blood loss during each menstrual cycle, based on the direct measurement of menstrual blood loss (MBL) using the alkaline hematin method.4 However, from a clinical practice point of view, it is defined as a volume that interferes with a patient's HRQoL, including physical, social, and emotional aspects. Prevalence estimates of UFs vary widely, but have been estimated to range from 4.1% to 5.5%, based on females aged 15 to 49 years in Canada who self-report having physician-diagnosed UFs.5
According to the literature and the clinical expert consulted on this review, the overall goals of treatment are to reduce menstrual bleeding, allow for activities of daily living, increase hemoglobin levels, address pain and bulk symptoms, and improve HRQoL.6,7 The clinical expert also noted that reducing the size of UFs can allow for less invasive surgery, delay or reduce the need for surgery, manage symptoms until menopause (when symptoms tend to subside), and reduce the impact of UFs on fertility. Treatment can include surgical, procedural, and pharmacological options. The choice of treatment depends on the clinical presentation (i.e., symptoms, severity, size, and location of UFs), the patient’s age, and the patient’s goals (i.e., to preserve fertility and/or the uterus). First-line pharmacological options include combined estrogen-progestin contraceptives, progestin-releasing intrauterine systems, and tranexamic acid.6 However, the use of hormonal contraceptives for treating UFs is off-label, and HMB is often inadequately controlled with these drugs alone.8 For patients whose disease does not respond adequately to first-line treatments, gonadotropin-releasing hormone (GոRΗ) antagonists and agonists may be used with or without hormonal add-back therapy to mitigate hypoestrogenic side effects and protect the endometrium.9 GnRH agonists (e.g., leuprolide acetate [LA]) are often used as short-term preoperative therapies (i.e., for 3 months to 6 months) with concomitant iron therapy to reduce the size of UFs and improve anemia before surgery.10 A caveat with the use of GnRH agonists is the initial flare effect (i.e., symptom exacerbation), which is not seen with GոRΗ antagonists; in addition, once the GnRH agonist is stopped, UF regrowth often occurs within 3 months.7 Uterine artery embolization is a procedure used to address HMB and bulk symptoms, but it does not remove UFs, and it is associated with ovarian dysfunction and the need for reintervention.11 Hysteroscopic resection or myomectomy of submucosal UFs are also options; however, these have been associated with high blood loss and UF recurrence.6,7 Hysterectomy is the only definitive treatment for UFs, and it is associated with significant morbidity and mortality; therefore, it is a last-line option for patients who have persistent symptoms despite other treatments.7
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of oral relugolix-estradiol–norethindrone acetate (RGX-E2-NETA) (40 mg relugolix [RGX], 1 mg estradiol [E2], and 0.5 mg norethindrone acetate [NETA]) for the treatment of HMB associated with UFs in patients in the premenopausal stage. RGX-E2-NETA has not been reviewed before by Canada’s Drug Agency (CDA-AMC).
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient groups that responded to our call for input and from the clinical expert consulted for the purpose of this review.
Patient Input
CDA-AMC received 1 patient group submission from Canadian Women with Fibroids (CANFib), which had conducted a survey of 315 respondents with UFs in 2023. The submission included comments from the incoming Society of Obstetricians and Gynecologists of Canada president and an endorsement from the Women’s Health Coalition. CANFib noted wait times to consult a health care provider, new symptoms, and the ineffectiveness of current medications as constituting important unmet needs for patients. The group reported that most people with UFs experienced heavy bleeding and pain, absences from work, inability to function within their family unit, deficits in their social or professional lives, and eventual depression. Considering the ineffectiveness of the medications that patients have already tried and the long wait times to consult specialists, the respondents felt that more options and earlier treatment would benefit patients and reduce the need for follow-up visits and surgeries. Regarding the experience with currently available treatments, CANFib noted that numerous treatments and medications are available to treat UFs, but there is no cure other than surgical hysterectomy. According to the group, desirable outcomes for patients include access to an effective treatment that does not require a 3-month dosage commitment, regaining confidence, and being able to have meaningful family, social, and professional lives.
CANFib also conducted group discussions with 32 females who had experience with RGX-E2-NETA. Of these participants, 31 reported being “very happy” and 1 reported feeling “indifferent” with respect to how the drug improved symptoms. CANFib added that RGX-E2-NETA enabled those in the discussion group to lead full lives, be with their families, care for their children and/or parents, show up at work, and function without debilitating pain, heavy blood loss, or ongoing depression.
Clinician Input
Input From the Clinical Expert Consulted for This Review
According to the clinical expert consulted for this review, patients want new therapies that adequately and rapidly address symptoms without adverse effects, improve HRQoL, and are simple to administer, with quick resolution upon discontinuation.
Based on input from the clinical expert, first-line treatment of AUB or HMB due to UFs can include nonsteroidal anti-inflammatory drugs, hormonal contraceptives, tranexamic acid, and iron repletion. Once a diagnosis of UF-associated HMB has been confirmed, RGX-E2-NETA could be a treatment option in the absence of endometrial pathology, depending on the patient’s symptom severity, the location of UFs, and the patient’s goals. The clinical expert expects that RGX-E2-NETA will be used in practice to complement and facilitate less invasive medical procedures and surgical approaches, such as hysteroscopic procedures and fertility-preserving surgical approaches. As per the expert, RGX-E2-NETA may be used as a bridge treatment in patients who plan to have gynecological surgery within 3 months (to elevate hemoglobin and/or enable less invasive surgical methods); want to delay surgery for fertility; or anticipate menopause within a couple of years.
The clinical expert indicated that the patients most likely to respond to treatment with RGX-E2-NETA include adults in the premenopausal stage with HMB associated with UFs and bulk symptomatology secondary to UFs. It was also noted that patients with anemia secondary to severe AUB, severe pain, and ureteric obstruction by UFs are most in need of treatment. The patients best suited for treatment with RGX-E2-NETA would be identified based on clinical history, physical examination, and investigations. As part of confirming a diagnosis, endometrial and cervical pathologies must be ruled out based on the patient’s age and risk factors.
As per the clinical expert, the main outcomes used to assess treatment response in practice include qualitative clinical assessments for a reduction in primary symptomatology (i.e., reduced HMB), reduction in pain and bulk symptoms, return to normal function (e.g., activities of daily living and work), and fertility success, if this is the patient’s goal. Clinicians also focus on the patient’s satisfaction with the treatment, rather than quantitative measures, such as MBL volume. However, physicians may measure hemoglobin levels with the goal of increasing them to normal values in patients experiencing anemia. The expert explained that although the outcomes used in clinical trials may be informative, these are generally not used in practice (aside from measuring and improving hemoglobin levels).
The clinical expert stated that RGX-E2-NETA should be discontinued upon successful surgery or procedure, plans for pregnancy, arrival of menopause, a meaningful decline in bone mineral density (BMD), or intolerable adverse effects. Although the Health Canada product monograph states that the use of RGX-E2-NETA should be limited to 24 months, due to the risk of continued and irreversible bone loss, the expert suggested that treatment beyond 2 years may be considered if the patient is maintaining their goals for symptom improvement and has a healthy BMD. However, if RGX-E2-NETA is used to treat HMB and anemia in anticipation of a planned surgery or medical procedure, treatment duration tends to be shorter than 2 years. As noted by the expert, the benefit in preparation for surgery with respect to the reduction of the size of the uterus and fibroids is achieved after 3 months. If wait times are longer than the 3 months, benefit can be maintained by longer use of the treatment.
RGX-E2-NETA would be used in a community setting, acute care hospital, or specialty clinic. While diagnosis and initial prescription of the drug would be performed by an obstetrician-gynecologist, the expert noted that follow-up management and monitoring may be done by a primary care physician.
Clinician Group Input
No input was received from clinician groups for this review.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, initiation of therapy, continuation or renewal of therapy, and system and economic issues. The clinical expert consulted provided input on the potential implementation issues raised by the drug programs in Table 4.
Clinical Evidence
Systematic Review
Description of Studies
Two phase III, double-blind, randomized controlled trials (RCTs) — the LIBERTY 1 trial (N = 388) and the LIBERTY 2 trial (N = 382) — of female patients aged 18 to 50 years with a confirmed diagnosis of UFs and HMB (n = 128 and 126, respectively) assessed the efficacy and safety of RGX-E2-NETA (40 mg RGX, 1 mg E2, and 0.5 mg NETA) once daily compared to placebo (n = 128 and n = 129, respectively) at 24 weeks.12,13 Efficacy was measured using the primary end point of MBL volume responder analyses, defined as patients who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL volume. Other clinically relevant outcomes included improvement in anemia, pain, symptom severity, and HRQoL.
The LIBERTY 1 and LIBERTY 2 trials were mostly identical in design, with the main differences being the countries where the studies took place and the order of statistical testing of secondary outcomes. The mean age across the studies was approximately 42 years. In both studies, the proportion of patients who were Black was lower in the RGX-E2-NETA groups than in the placebo groups (46.1% versus 51.2% in the LIBERTY 1 trial; 49.6% versus 57.4% in the LIBERTY 2 trial). Conversely, the proportion of patients who were white was higher in the RGX-E2-NETA groups than in the placebo groups (50.0% versus 44.1% in the LIBERTY 1 trial; 46.4% versus 38.0% in the LIBERTY 2 trial). Proportions of other races and ethnicities were generally balanced between treatment groups and across the studies. Mean hemoglobin concentration was similar across the treatment groups in both studies; however, anemia (i.e., hemoglobin concentration of less than 12 g/dL) was slightly more prevalent in the placebo groups than in the RGX-E2-NETA groups.
Efficacy Results
Responder Rate for Change in MBL Volume
Response to treatment was defined as having an MBL volume of less than 80 mL and a 50% or greater reduction from baseline in MBL volume over the preceding 35 days of treatment as measured using the alkaline hematin method.
In the LIBERTY 1 trial, 73.4% (95% confidence interval [CI], 64.9% to 80.9%) of patients who received RGX-E2-NETA responded to treatment versus 18.9% (95% CI, 12.5% to 26.8%) of patients who received placebo. The treatment difference in the responder rate for change in MBL volume at week 24 was 54.5% (95% CI, 44.3% to 64.7%; P < 0.0001) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, 71.2% (95% CI, 62.4% to 79.0%) of patients who received RGX-E2-NETA responded to treatment compared to 14.7% (95% CI, 9.1% to 22.0%) of patients who received placebo. The treatment difference in the responder rate for change in MBL volume at week 24 was 56.5% (95% CI, 46.6% to 66.5%; P < 0.0001) in favour of RGX-E2-NETA.
Improvement of Anemia
For the subset of patients who had a hemoglobin level of 10.5 g/dL or lower at baseline, response was defined as an increase of 2 g/dL or more at week 24.
In the LIBERTY 1 trial, 15 patients (50.0%) and 5 patients (21.7%) in the RGX-E2-NETA and placebo groups, respectively, experienced an increase of 2 g/dL or more in their hemoglobin levels. The treatment difference in the proportion of patients who experienced improvement in anemia at week 24 was 28.3% (95% CI, 3.7% to 52.8%; P = 0.0377) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, 19 patients (61.3%) and 2 patients (5.4%) in the RGX-E2-NETA and placebo groups, respectively, experienced an increase of 2 g/dL or more in their hemoglobin levels. The treatment difference in the proportion of patients who experienced an improvement in anemia at week 24 was 55.9% (95% CI, 37.3% to 74.5%; P < 0.0001) in favour of RGX-E2-NETA.
Improvement of Pain
For the subset of patients who had a maximum numerical rating scale (NRS) score of 4 points or greater during the 35 days before randomization, response to treatment was defined as a maximum NRS score of 0 points or 1 point for UF-associated pain over the preceding 35 days of treatment.
In the LIBERTY 1 trial, 33 patients (39.3%) and 11 patients (11.6%) in the RGX-E2-NETA and placebo groups, respectively, experienced a maximum NRS score of 0 points or 1 point for UF-associated pain. The treatment difference in the proportion of patients who experienced an improvement in pain to a maximum NRS score of 0 point or 1 point was 27.7% (95% CI, 15.4% to 40.0%; P < 0.0001) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, 34 patients (36.6%) and 17 patients (17.9%) in the RGX-E2-NETA and placebo groups, respectively, experienced a maximum NRS score of 0 points or 1 point for UF-associated pain. The treatment difference in the proportion of patients who experienced improvement in pain to a maximum NRS score of 0 point or 1 point was 18.7% (95% CI, 6.2% to 31.1%; P = 0.0056) in favour of RGX-E2-NETA.
Change in Uterine Fibroid Symptom Quality of Life Questionnaire Symptom Severity Scale Score and HRQoL Total Score
The Uterine Fibroid Symptom Quality of Life questionnaire (UFS-QoL) is a disease-specific instrument used to assess symptom severity and HRQoL in patients with UFs over the preceding month.14 The UFS-QoL Symptom Severity Scale (SSS) consists of 8 items rated on a 5-point Likert scale ranging from “not at all” to “a very great deal.” The HRQoL component includes 29 items rated on a 5-point scale ranging from “none of the time” to “all of the time.” Scores for these SSS and HRQoL subscales are linearly transformed to scales ranging from 0 points to 100 points. Higher SSS scores indicate more severe UF symptoms, while higher HRQoL total scores indicate better quality of life.
In the LIBERTY 1 trial, the least squares mean (LSM) changes from baseline to week 24 in UFS-QoL SSS scores were –30.9 points (95% CI, –36.1 points to –25.8 points) and –10.5 points (95% CI, –15.5 points to –5.5 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in LSM change from baseline to week 24 in the UFS-QoL SSS score was –20.4 points (95% CI, –27.1 to –13.7 points) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, the LSM changes from baseline to week 24 in UFS-QoL SSS scores were –36.1 points (95% CI, –41.2 points to –31.0 points) and –13.7 points (95% CI, –18.8 points to –8.5 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in LSM change from baseline to week 24 in UFS-QoL SSS score was –22.4 points (95% CI, –29.4 points to –15.4 points) in favour of RGX-E2-NETA.
In the LIBERTY 1 trial, the LSM changes from baseline to week 24 in UFS-QoL HRQoL total score were 38.0 points (95% CI, 32.4 points to 43.5 points) and 12.8 points (95% CI, 7.5 points to 18.2 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL HRQoL total score was 25.1 points (95% CI, 17.9 points to 32.3 points) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, the LSM changes from baseline to week 24 in UFS-QoL HRQoL total score were 37.8 points (95% CI, 32.9 points to 42.6 points) and 13.8 points (95% CI, 8.9 points to 18.8 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL HRQoL total score was 23.9 points (95% CI, 17.2 points to 30.7 points) in favour of RGX-E2-NETA.
Harms Results
In the LIBERTY 1 trial, 61.7% of patients who received RGX-E2-NETA and 66.1% of patients who received placebo had a treatment-emergent adverse event (TEAE). Headache (10.9% of patients in the RGX-E2-NETA group versus 15.0% of patients in the placebo group) and hot flush (10.9% of patients in the RGX-E2-NETA group versus 7.9% of patients in the placebo group) were the most common TEAEs. In the LIBERTY 2 trial, 60.3% of patients who received RGX-E2-NETA and 58.9% of patients who received placebo had a TEAE. Headache (8.7% of patients in the RGX-E2-NETA group versus 11.6% of patients in the placebo group) was the most common TEAE. In general, the types and frequencies of TEAEs were similar between the treatment groups for both studies and across studies.
In the LIBERTY 1 trial, 7 patients (5.5%) who received RGX-E2-NETA reported 10 serious adverse events (SAEs), and 2 patients (1.6%) who received placebo reported 2 SAEs. In the LIBERTY 2 trial, 1 patient (0.8%) who received RGX-E2-NETA reported 1 SAE, and 4 patients (3.1%) who received placebo reported 5 SAEs.
In the LIBERTY 1 trial, 7 patients (5.5%) who received RGX-E2-NETA stopped treatment due to 12 TEAEs, and 5 patients (3.9%) who received placebo stopped treatment due to 13 TEAEs. In the LIBERTY 2 trial, 3 patients (2.4%) who received RGX-E2-NETA stopped treatment due to 3 TEAEs, and 6 patients (4.7%) who received placebo stopped treatment due to 7 TEAEs.
There were no deaths reported in either study.
The treatment differences in the percentage change from baseline to week 24 in BMD of the lumbar spine were –0.41% (95% CI, –1.16% to 0.34%) in the LIBERTY 1 trial and –0.44% (95% CI, –1.23% to 0.34%) in the LIBERTY 2 trial.
Critical Appraisal
In both trials, a lower proportion of patients were Black and a higher proportion were white in the RGX-E2-NETA groups versus the placebo groups (race and ethnicity are risk factors for developing UFs),15 and mean MBL volume was higher in the RGX-E2-NETA group versus the placebo group. While the baseline imbalances could have biased the study results (i.e., the direction and magnitude of bias are unknown), the clinical expert consulted on this review did not expect the imbalances to meaningfully affect the interpretation of the results. There was a potential risk of bias in the outcomes for improvement in anemia and pain, and the treatment effects were notably different between the studies for both. The reason for the inconsistency was unclear, but it may have had to do with the smaller number of patients contributing data (the outcomes used subpopulations) and/or a loss of prognostic balance. Patients who had anemia at baseline or new anemia during the study started or continued iron therapy. While the iron supplementation protocol was consistent across the treatment groups and studies, it is unknown if improvement in anemia was a result of iron therapy or reduced MBL; information on changes in the amount of iron throughout the studies was not available to clarify this. Bias may also have arisen from the missing data (5% to 30%) for the anemia, pain, UFS-QoL, and BMD outcomes (explanations for why these data were missing were not available); this reduces certainty in the results. Considering the large difference in efficacy between the RGX-E2-NETA and placebo groups for the primary outcome, patients may have been able to infer which treatment they received in the study, which would have affected the subjective outcomes (i.e., pain, UFS-QoL, and harms).
According to the clinical expert, 2 eligibility criteria that do not reflect clinical practice were exclusions based on an upper age limit (patients had to be adults in the premenopausal stage) and receiving gynecological surgery or ablation procedures for UFs within the 6 months before treatment. Moreover, the definition of HMB used in the clinical trials, while acceptable for the setting, is not used in practice. Instead, HMB is identified through clinical assessment and described as AUB that interferes with a patient’s HRQoL and activities of daily living. Furthermore, although the study end points inform on outcomes of importance to patients (i.e., pain, bulk symptoms, and HRQoL), these are not used in practice. Rather than quantifying treatment effect by measuring MBL, clinicians assess patient satisfaction with how well the treatment is addressing their disease symptoms and whether the patient can participate in daily activities without interference from HMB and UFs. The 24-week study duration was deemed acceptable in terms of meeting the clinical end points; however, the clinical expert does not expect most patients to use RGX-E2-NETA as a long-term treatment. Most would use it instead as a bridging therapy to surgery or menopause. The expert indicated that there are rare instances in which treatment beyond 24 months may be warranted, such as in patients whose disease responds well to treatment without intolerable TEAEs; however, it is noted in the Health Canada product monograph that due to the risk of irreversible bone loss, use of the drug should be limited to 24 months.16 Should the drug be used for the longer term, regular BMD scans would be important to ensure that the patient is not experiencing clinically important bone loss. If this develops, discontinuation of the drug should be considered.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.17,18
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect, informed by the clinical expert consulted for this review, for responder rate for change in MBL volume, improvement of anemia, improvement of pain, change in UFS-QoL SSS score and HRQoL total score, and the presence or absence of any (non-null) effect for change in BMD at the lumbar spine.
For the GRADE assessments, findings from the LIBERTY 1 trial and the LIBERTY 2 trial were assessed together per outcome because these studies were similar in population, intervention, design, and outcome measures.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with a clinical expert, and the input received from patient groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
responder rate for change in MBL volume
improvement in anemia
improvement in pain
change in UFS-QoL (SSS score and HRQoL total score)
change in BMD.
Table 2: Summary of Findings for RGX-E2-NETA Versus Placebo for Patients With HMB Associated With UFs
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
MBL response | ||||
Proportion of patients who experienced an MBL volume of < 80 mL and a ≥ 50% reduction from baseline MBL volume, n (95% CI) Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| High | RGX-E2-NETA results in a clinically important increase in the proportion of patients who experience an MBL response when compared with placebo. |
Anemia improvement | ||||
Proportion of patients who experienced an increase in hemoglobin level of > 2 g/dL from baseline in the subset of patients with a hemoglobin level of ≤ 10.5 g/dL at baseline, n (95% CI) Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| Lowa | RGX-E2-NETA may result in a clinically important increase in the proportion of patients who experience improvement in anemia when compared with placebo. |
Pain improvement | ||||
Proportion of patients who experienced a maximum NRS score of ≤ 1 for UF-associated pain over the last 35 days of treatment within the subset of patients who had a maximum pain score of ≥ 4 during the 35 days before randomization, n (95% CI) Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| Lowb | RGX-E2-NETA may result in a clinically important increase in the proportion of patients who experience an improvement in pain when compared with placebo. |
UFS-QoL | ||||
UFS-QoL SSS score (0 [best] to 100 [worst]) LSM change from baseline, points Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| Moderatec | RGX-E2-NETA likely results in a clinically important reduction in UFS-QoL SSS score when compared with placebo. |
UFS-QoL HRQoL total score (100 [best] to 0 [worst]) LSM change from baseline, points Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| Moderated | RGX-E2-NETA likely results in a clinically important increase in UFS-QoL HRQoL total score when compared with placebo. |
Harms | ||||
BMD for lumbar spine percentage change from baseline (95% CI), % Follow-up: 24 weeks |
| LIBERTY 1 trial
LIBERTY 2 trial
| Lowe | RGX-E2-NETA may result in a greater proportion of patients who experience a reduction in BMD when compared with placebo. The clinical relevance of the decrease is uncertain. |
BMD = bone mineral density; CI = confidence interval; HMB = heavy menstrual bleeding; HRQoL = health-related quality of life; LSM = least squares mean; MBL = menstrual blood loss; NRS = numerical rating scale; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for study limitations because this outcome uses a subpopulation, and prognostic balance may have been lost. Not rated down for inconsistency because, although the magnitude of the between-group difference differs across the 2 studies, the point estimate of the between-group difference was considered clinically meaningful in each, according to the clinical expert consulted for the review. Rated down 1 level for imprecision because there was no known threshold for a clinically important effect, and the presence of an important effect was informed by the clinical expert consulted for this review. The point estimates in both studies suggest the presence of a clinically important effect; however, the lower bound of the 95% CI for the LIBERTY 1 trial is consistent with an effect that would not be considered clinically important for patients.
bRated down 1 level for study limitations because this outcome uses a subpopulation, and prognostic balance may have been lost. Not rated down for inconsistency because, although the magnitude of the between-group difference differs across the 2 studies, the point estimate of the between-group difference was considered clinically meaningful in each, according to the clinical expert consulted for the review. Rated down 1 level for imprecision because there was no known threshold for a clinically important effect, and the presence of an important effect was informed by the clinical expert consulted for this review. The point estimates in both studies suggest the presence of a clinically important effect; however, the lower bound of the 95% CIs for the LIBERTY 1 trial and the LIBERTY 2 trial are consistent with an effect that would not be considered clinically important for patients.
cRated down 1 level for study limitations because data were missing for 24% and 19% of patients in the RGX-E2-NETA and placebo groups, respectively, in the LIBERTY 1 trial, and for 23% and 26% of patients, respectively, in the LIBERTY 2 trial. Not rated down for imprecision because there was no known threshold for a clinically important effect, and the presence of an important effect was informed by the clinical expert consulted for this review. The point estimates in both studies suggest the presence of a clinically important effect, and although the lower bound of the 95% CIs for the LIBERTY 1 trial and the LIBERTY 2 trial are consistent with an effect that would not be considered clinically important for patients, the review team did not rate down because the lower bound of the CIs likely do not considerably cross the minimum threshold for a clinically important effect. Analysis of this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
dRated down 1 level for study limitations because data were missing for 24% and 19% of patients in the RGX-E2-NETA and placebo groups, respectively, in the LIBERTY 1 trial, and for 23% and 26% of patients, respectively, in the LIBERTY 2 trial. Not rated down for imprecision because there was no known threshold for a clinically important effect, and the presence of an important effect was informed by the clinical expert consulted for this review; the point estimates and lower bound of the 95% CIs in both studies suggest the presence of a clinically important effect. Analysis of this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
eRated down 1 level for study limitations because data were missing for 22% and 20% of patients in the RGX-E2-NETA and placebo groups, respectively, in the LIBERTY 1 trial, and for 25% and 26% of patients, respectively, in the LIBERTY 2 trial. Not rated down for indirectness because the follow-up was limited to 24 weeks, which may be insufficient to detect clinically important BMD loss; however, the clinical expert expects that most patients would not use RGX-E2-NETA for long-term treatment. Rated down 1 level for imprecision because there was no known threshold for a clinically important effect; the point estimate suggested a decrease with RGX-E2-NETA, while the 95% CI included the potential for no difference and an increase. Analysis of this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19
Long-Term Extension Studies
Description of Studies
The LIBERTY extension study was a single-arm, phase III, 28-week, open-label extension (OLE) study of patients who had completed either pivotal trial. In the OLE study, the long-term efficacy and safety of RGX-E2-NETA were evaluated for an additional 28 weeks (i.e., after the 24 weeks of treatment during either parent study, the LIBERTY 1 trial or LIBERTY 2 trial).
The LIBERTY randomized withdrawal study (RWS) was a double-blind, phase III, placebo-controlled, 52-week study enrolling those who had completed the LIBERTY extension study and whose disease had responded to treatment at the week 48 visit (i.e., the patient had experienced an MBL volume of less than 80 mL and a 50% or greater reduction from the pivotal study baseline MBL). Patients were rerandomized in a 1-to-1 ratio to receive either oral RGX-E2-NETA or placebo once daily for up to 52 weeks. For patients whose HMB recurred during the LIBERTY RWS (i.e., MBL volume ≥ 80 mL), blinded treatment was stopped, and re-treatment with open-label RGX-E2-NETA was offered.
Efficacy Results
LIBERTY Extension Trial
At week 52 of the LIBERTY extension trial, 87.7% of patients (95% CI, 81.7% to 92.3%) who had received RGX-E2-NETA after the parent study baseline (i.e., the RGX-E2-NETA group) were responders, while 75.6% of patients (95% CI, 68.3% to 82.0%) who had received placebo in the parent studies, but switched to RGX-E2-NETA (i.e., placebo-to-RGX-E2-NETA group) during the OLE study, were responders.
For the anemia outcome (i.e., patients whose hemoglobin level was ≤ 10.5 g/dL at baseline and who experienced an increase of ≥ 2 g/dL), 23 patients of 39 patients (59.0%) in the RGX-E2-NETA group and 16 patients of 38 patients (42.1%) in the placebo-to-RGX-E2-NETA group experienced an improvement in anemia at week 52.
At week 52, the LSMs for the UFS-QoL SSS score were –37.3 points (SE = 2.5 points) in the RGX-E2-NETA group and –35.0 points (SE = 2.5 points) in the placebo-to-RGX-E2-NETA group. The LSMs for the UFS-QoL HRQoL total score were 40.4 points (SE = 2.5 points) in the RGX-E2-NETA group and 39.0 points (SE = 2.6 points) in the placebo-to-RGX-E2-NETA group.
LIBERTY RWS
At week 76, 78.4% of patients (95% CI, 69.3% to 85.1%) in the rerandomized RGX-E2-NETA group and 15.1% of patients (95% CI, 8.9% to 22.8%) in the rerandomized placebo group responded to treatment. The difference in responder rates between treatment groups was 63.4% (95% CI, 52.9% to 73.9%) in favour of RGX-E2-NETA.
At week 104, 69.8% of patients (95% CI, 59.7% to 77.8%) in the rerandomized RGX-E2-NETA group and 11.8% of patients (95% CI, 6.3% to 19.0%) in the rerandomized placebo group responded to treatment. The difference in responder rates between treatment groups was 58.0% (95% CI, 47.0% to 69.1%) in favour of RGX-E2-NETA.
At week 76, for the UFS-QoL SSS score, patients had changes from rerandomization baseline (i.e., week 52 of total study treatment) of 2.2 points (standard deviation [SD] = 18.2 points) in the rerandomized RGX-E2-NETA group and 14.1 points (SD = 21.1 points) in the rerandomized placebo group. For the UFS-QoL HRQoL total score, patients had changes from rerandomization baseline of –0.8 points (SD = 16.8 points) in the rerandomized RGX-E2-NETA group and –9.1 points (SD = 22.4 points) in the rerandomized placebo group.
At week 104, for the UFS-QoL SSS score, patients had changes from rerandomization baseline (i.e., week 52 of total study treatment) of 0.1 points (SD = 15.9 points) in the rerandomized RGX-E2-NETA group and –4.3 points (SD = 42.2 points) in the rerandomized placebo group. For the UFS-QoL HRQoL total score, patients had changes from rerandomization baseline of 1.1 points (SD = 15.3 points) in the rerandomized RGX-E2-NETA group and 8.2 points (SD = 42.1 points) in the rerandomized placebo group.
Harms Results
The LIBERTY Extension Trial
During the OLE study, 54.6% of patients in the RGX-E2-NETA group and 62.8% of patients in the placebo-to-RGX-E2-NETA group experienced at least 1 TEAE. Nasopharyngitis (6.1%) was the most common TEAE in the RGX-E2-NETA group, while hot flush (7.9%), headache (6.7%), and hypertension (6.1%) were the most common TEAEs in the placebo-to-RGX-E2-NETA group.
Overall, 1 patient (0.6%) in the RGX-E2-NETA group and 11 patients (6.7%) in the placebo-to-RGX-E2-NETA group experienced at least 1 SAE. No SAE was reported by more than 2 patients.
Two patients (1.2%) in the RGX-E2-NETA group and 9 patients (5.5%) in the placebo-to-RGX-E2-NETA group stopped treatment due to a TEAE. No TEAE leading to treatment discontinuation was reported by more than 1 patient.
There were no deaths in the OLE study.
At the end of the LIBERTY extension trial (i.e., 52 weeks of study treatment), the LSM percentage changes in BMD at the lumbar spine were –0.80 g/cm2 (95% CI, –1.36 g/cm2 to –0.25 g/cm2) for the RGX-E2-NETA group and –0.78 g/cm2 (95% CI, –1.32 g/cm2 to –0.23 g/cm2) for the placebo-to-RGX-E2-NETA group.
LIBERTY RWS
In the LIBERTY RWS, 58.6% of patients in the rerandomized RGX-E2-NETA group and 64.3% of patients in the rerandomized placebo group experienced at least 1 TEAE. Nasopharyngitis (11.2%) was the most common TEAE in the rerandomized RGX-E2-NETA group, while nasopharyngitis (10.7%), hot flush (7.1%), dysmenorrhea (7.1%), hypertension (5.4%), and upper respiratory tract infection (5.4%) were the most common TEAEs in the rerandomized placebo group.
Overall, 2 patients (1.7%) in the rerandomized RGX-E2-NETA group and 2 patients (1.8%) in the rerandomized placebo group experienced at least 1 SAE. No SAE was reported by more than 1 patient.
Two patients (1.7%) in the rerandomized RGX-E2-NETA group and 3 patients (2.7%) in the rerandomized placebo group stopped treatment due to a TEAE. No TEAE leading to treatment discontinuation was reported by more than 1 patient.
There were no deaths in the LIBERTY RWS.
At the end of the LIBERTY RWS (i.e., 104 weeks of study treatment), the LSM percentage changes in BMD at the lumbar spine were 0.81 g/cm2 (95% CI, 0.20 g/cm2 to 1.41 g/cm2) in the rerandomized RGX-E2-NETA group and 0.10 g/cm2 (95% CI, –0.52 g/cm2 to 0.71 g/cm2) in the rerandomized placebo group. The LSM percentage between-group difference was 0.71 g/cm2 (95% CI, –0.13 g/cm2 to 1.55 g/cm2).
Critical Appraisal
There is the possibility of selection bias in the LIBERTY extension study because eligible patients had to have successfully completed either the LIBERTY 1 trial or the LIBERTY 2 trial; thus, patients were more likely to be those who had responded to treatment and did not experience intolerable TEAEs. The single-arm, OLE study was noncomparative, preventing conclusions from being made about the benefits or harms attributable to RGX-E2-NETA versus any comparator. In addition, the open-label design could bias the assessment of subjective, patient-reported outcomes (i.e., UFS-QoL and harms). The lack of data beyond week 52 in the OLE study makes it challenging to draw conclusions on the long-term sustainability of the treatment effect. Treatment in the LIBERTY RWS was double-blinded, which is important, given the subjective nature of the patient-reported outcomes; however, there is the risk of unblinding of the treatment received due to the large difference in efficacy results between the active and placebo groups. In both extension studies, the reasons for missing data for each outcome were not available; as such, the effect of these missing data remains uncertain. In the LIBERTY RWS, there is a risk of bias due to missing outcome data, particularly at longer follow-up times for the UFS-QoL SSS score, HRQoL total score, and BMD.
Because patients who were enrolled in the extension studies must have successfully completed the parent trials, the extension studies have the same generalizability issues. Of note, the clinical expert confirmed that patients receiving RGX-E2-NETA would not be restricted by an upper age limit; rather, they would be restricted by status — that is, being in the premenopausal stage or having recent or planned gynecological surgery or procedures for UFs would not preclude patients from using the drug. The efficacy outcomes, while relevant to patients and practice, are not used in clinical settings (aside from measuring hemoglobin), therefore, the results in real-world clinical settings may differ from those observed in the extension studies.
Indirect Comparisons
The sponsor submitted a systematic review with a network meta-analysis (NMA), a type of indirect treatment comparison (ITC), to estimate the efficacy and safety of RGX-E2-NETA compared with LA (with or without add-back) for the treatment of HMB associated with UFs in people in the premenopausal stage.20
An ITC was required because of a lack of studies directly comparing RGX-E2-NETA with LA (with and without add-back) (the only authorized treatment for UFs in the Canadian setting).
To support the NMA, the sponsor:
conducted a systematic literature review (SLR) to identify RCTs to estimate the relative efficacy and safety of RGX-E2-NETA versus LA (with or without add-back)
performed an NMA to assess the relative efficacy and safety of RGX-E2-NETA versus LA (without add-back) for the treatment of HMB associated with UFs in people in the premenopausal stage (a comparison of RGX-E2-NETA to LA with add-back was considered infeasible by the sponsor).
Outcomes of interest for the NMA included effectiveness, TEAEs, and tolerability. The specific effectiveness outcomes were MBL resolution and UFS-QoL (HRQoL total score and SSS score). TEAEs included hot flushes and headaches. Tolerability outcomes were discontinuations due to TEAEs.
Of 228 reports identified in the literature, 31 trials were included in the SLR, and 12 of these were included in the NMA.
Treatments included in the NMA to establish a network connection between RGX-E2-NETA and LA were ulipristal acetate (UPA) 5 mg, UPA 10 mg, and mifepristone (MFP) 10 mg. Although UPA 5 mg, UPA 10 mg, and MFP 10 mg are not considered relevant comparators, these were maintained in the NMA to establish network connections between RGX-E2-NETA and LA (without add-back). These comparators were excluded from the main results despite being retained to establish a network connection between RGX-E2-NETA and LA.
Efficacy Results
MBL Resolution
For the comparison of RGX-E2-NETA versus LA (without add-back), the point estimate for the relative risk (RR) for MBL resolution favoured RGX-E2-NETA; however, the 95% credible interval (CrI) was wide and included the possibility that either RGX-E2-NETA or LA (without add-back) could be favoured ███ █ █████ ███ ████ ████ ██ █████. A sensitivity analysis included a study with a different outcome definition of MBL. Overall, it reached the same conclusion as the base case.
Uterine Fibroid Symptom Quality of Life Questionnaire
Point estimates of the mean differences (MDs) in UFS-QoL HRQoL total scores and UFS-QoL SSS scores favoured LA (without add-back), but were lower than the estimated clinically meaningful threshold of 15 points suggested by the clinical expert. The 95% CrIs crossed the line of equivalence, which suggested that either RGX-E2-NETA or LA (without add-back) could be favoured; however, not all values within the 95% CIs may be clinically important: UFS-QoL total score MD of █████ ██████ ████ ████ █████ ██ ████ ███████ and UFS-QoL symptom severity MD of ████ ██████ ████ ████ █████ ██ █████ ███████.
Two sensitivity analyses were performed for the UFS-QoL HRQoL total score and UFS-QoL SSS score by first excluding 2 studies (with < 10% of patients who were Black), then excluding a study with high heterogeneity. The conclusions drawn from the sensitivity analyses were similar to those drawn from the base-case analysis, favouring LA (without add-back).
Harms Results
Hot Flushes
RGX-E2-NETA was associated with a lower risk of hot flushes compared with LA (without add-back), with an RR of ████ ████ █ ██ █████. A sensitivity analysis was conducted that included studies beyond 1 month after the end of treatment. This sensitivity analysis was consistent with the base case.
Headaches
For the outcome of headaches, the RR estimate was imprecise, suggesting that the direction of the effects are inconclusive. Comparing RGX-E2-NETA and LA (without add-back), the point estimate for the RR for headaches favoured LA (without add-back); however, the 95% CrI was wide and included the possibility that either RGX-E2-NETA or LA (without add-back) could be favoured ███ █ ███████ ███ ████ ████ ██ █████████. A sensitivity analysis conducted for the time point estimate of headaches found the estimate consistent with the base case, with a similarly high level of imprecision.
Discontinuation Due to TEAEs
The point estimate for the RR of the tolerability outcome, discontinuation due to TEAEs, favoured RGX-E2-NETA with a high degree of imprecision, with an RR of ████ ████ ████ ████ ██ █████. The wide 95% CrI that crosses the null also suggests that either RGX-E2-NETA or LA (without add-back) could be favoured. No sensitivity analysis was reported for this outcome.
Critical Appraisal
A systematic review of published literature and grey literature was conducted to identify studies for the NMA. Even though a prespecified protocol for the SLR and NMA could not be obtained, the CDA-AMC review team appraised the submitted SLR using the criteria from A MeaSurement Tool to Assess systematic Reviews 2.21 Overall, the searches were comprehensive, and the methods for data extraction and risk of bias appraisal were adequate to reduce the risk of error and bias in these procedures. Although study selection was undertaken by 2 independent reviewers with consensus, the rationale for why certain studies were excluded from the networks was inconsistent. Of the 31 studies identified through the SLR, 12 were retained for the NMAs. Although certain studies of comparators not used in Canada were included in the networks to allow connections between RGX-E2-NETA and LA, other studies with the same comparators were excluded (e.g., Bagaria [2009], comparing placebo and MFP 10 mg, was included to connect the network, whereas other studies, including a study with an MFP 10 mg treatment group, were excluded because the treatments were not of interest). The selective inclusion of studies in the network may result in risk of bias due to missing results in the syntheses, the magnitude and direction of which cannot be predicted.
A Bayesian NMA included some outcomes (i.e., effectiveness, safety, and tolerability), but not all. Outcomes deemed relevant by patients and clinicians that were not included in the constructed networks were the efficacy outcomes of improvement in anemia and pain and the harms outcomes of BMD. BMD as a safety outcome was not included due to lack of comparative data. As such, this NMA cannot inform on the comparative effects of RGX-E2-NETA and LA (without add-back) for these outcomes.
There are limitations to both the fixed-effects and random-effects models used in this NMA. The models were selected based on better model fit (i.e., smaller deviance information criterion). Given that most outcomes were modelled with fixed effects, we expect that the true magnitude of uncertainty was underestimated. Because the effect estimates for these end points were already affected by important imprecision, reliance on the results of the unselected models would not have changed the review team’s conclusions about the estimated effects.
A key limitation of this NMA is the fact that the clinically relevant comparator, LA, did not contain add-back. While hormonal add-back is a part of RGX-E2-NETA, according to the sponsor, LA with add-back was excluded from the NMA due to feasibility reasons. This means that RGX-E2-NETA compared with LA (without add-back) does not reflect how the actual comparator is prescribed in Canada.
The sponsor found that RGX-E2-NETA had a reduced RR of hot flushes compared with LA. All other efficacy, safety, and tolerability outcomes crossed the null, with wide CrIs. These suggest that either RGX-E2-NETA or LA (without add-back) could be favoured.
There were other notable limitations of the overall NMA, given that the assumptions of exchangeability, similarity of outcomes, and consistency (coherence) could not be satisfied.
Overall, due to the absence of a comparison of RGX-E2-NETA versus LA (with add-back), the high degree of imprecision represented by RR or MD CrIs that can favour either RGX-E2-NETA or LA (without add-back), and the inability to satisfy the key assumptions required for an NMA (i.e., exchangeability, homogeneity, and consistency), definitive conclusions about the relative treatment effects of RGX-E2-NETA versus LA (with add-back) cannot be drawn based on this NMA.
Studies Addressing Gaps in the Evidence From the Systematic Review
There were no studies addressing gaps in the systematic review evidence submitted by the sponsor.
Conclusions
Evidence from 2 phase III, double-blind RCTs (the LIBERTY 1 trial and the LIBERTY 2 trial) suggests that patients aged 18 to 50 years with confirmed UFs and HMB (defined as an MBL volume of 160 mL or greater in 1 cycle or 80 mL or greater per cycle for 2 cycles) who received RGX-E2-NETA once a day for 24 weeks were more likely to experience treatment response for HMB compared to those who received placebo. Patients receiving RGX-E2-NETA may also have clinically meaningful improvements in anemia, UF-associated pain, symptom severity, and HRQoL (as measured using the UFS-QoL). However, the evidence for these outcomes was uncertain due to an increased risk of bias, lack of known thresholds for a clinically meaningful effect, and missing data. Longer-term evidence suggested that treatment effects were maintained for reduction in MBL and the patient-reported outcomes of symptom severity and HRQoL (with up to 104 weeks of study treatment), as well as for improvements in anemia (with up to 52 weeks of study treatment). Limitations with the study designs and numerous discontinuations increase the uncertainty of the longer-term results. It is also likely that patients who experienced benefits without intolerable harms remained in the studies longer. Results from the ITC of RGX-E2-NETA versus LA (without add-back) indicated that either treatment could be favoured for efficacy outcomes. Additionally, LA with add-back was not included in the ITC, and definitive conclusions about its treatment effect relative to that of RGX-E2-NETA could not be drawn. The incidence and types of harms were generally balanced and similar between the treatment groups in the pivotal studies and across the studies. Longer-term evidence did not indicate new safety signals; however, expert opinion noted that assessment of long-term bone health is important to inform decisions regarding prolonged treatment with RGX-E2-NETA. The Health Canada product monograph notes that use of RGX-E2-NETA should be limited to 24 months due to the risk of bone loss; expert opinion indicated that use in clinical practice would be short-term.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of oral RGX-E2-NETA (40 mg RGX, 1 mg E2, and 0.5 mg NETA) for the treatment of HMB associated with UFs in patients in the premenopausal stage.
Disease Background
The contents in this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
UFs, also known as myomas or leiomyomas, are benign neoplasms of uterine smooth muscle and are the most common tumours in females.1 Patients with UFs can present with 1 predominant fibroid or with clusters of many fibroids.22 The exact etiology of UFs is unclear, but estrogen and progesterone play an important role in UF initiation and growth.1 UFs are typically diagnosed in people between menarche and menopause. The main risk factors are younger age at menarche, late onset of menopause, African ancestry, and a child-bearing history of few or no children.1
A large proportion of patients with UFs are asymptomatic.5 A common symptom is AUB, particularly HMB, which is reported by 50% to 80% of patients with symptomatic UFs in surveys and trials.2,3 Infertility, abdominal bloating, constipation, increased urinary frequency, incontinence, dyspareunia, anemia, and fatigue are among other symptoms, all of which can affect patients’ HRQoL.1 It has been reported that the risk of poor mental health outcomes is higher among patients with diagnosed UFs, particularly those who experience pain-related symptoms or undergo hysterectomy, compared to those without diagnosed UF.23 For research purposes, HMB is defined as more than 80 mL of blood loss during each menstrual cycle and based on direct measurement of MBL using the alkaline hematin method.4 However, from a clinical practice point of view, it is defined as a volume that interferes with the patient's quality of life, including physical, social, and emotional aspects. Self-reported HMB can be inaccurate because it is based on the patient's perception of volume. It should be noted that some patients think their heavy volume is “normal.”24
Prevalence estimates of UFs vary widely depending on the study methods applied and the characteristics of the populations assessed. A large, international, internet-based survey was conducted by Zimmermann et al. (2009) in which 21,746 females across 8 countries participated and provided patient-reported UF prevalence data. It included 2,500 females aged 15 to 49 years from Canada, among whom the self-reported prevalence of diagnosed UFs was 5.5%.5 An online survey of people in Canada published by Laberge et al. (2016) that included 9,413 females aged 20 to 49 years found a self-reported, physician-diagnosed UF prevalence of 4.1%. The authors noted that scores for all HRQoL domains were lower among females with moderate or severe UF symptoms than in those with mild UF symptoms. Additionally, the authors identified responses that were consistent with moderate or severe UFs in females who made no report of physician-diagnosed UF; some of these females may be experiencing substantial burden due to undiagnosed UFs.25
Diagnosis of UFs is based on a pelvic examination and the finding of an enlarged, mobile uterus with an irregular contour. Diagnosis can be confirmed through pelvic ultrasonography. Ultrasonography is the most widely used diagnostic tool to confirm UFs due to its availability, ease of use, utility in visualizing genital tract structures, and cost-effectiveness.15
Standards of Therapy
The contents in this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
According to the literature and the clinical expert consulted on this review, the overall goals of treatment are to reduce menstrual bleeding, allow for activities of daily living, increase hemoglobin levels, address pain and bulk symptoms, and improve HRQoL.6,7 Other reasons noted by the clinical expert for using treatment to reduce the size of UFs include enabling less invasive surgery approaches, delaying surgery, reducing the need for surgery until menopause (when symptoms tend to subside), and reducing the impact of UFs on fertility. Treatment can include surgical, procedural, and pharmacological options. As per the expert, the choice of treatment depends on the clinical presentation (i.e., symptoms, severity, and size and location of UFs), the patient’s age, and the patient’s goals (e.g., desire to preserve fertility and/or the uterus).
For patients who do not desire fertility, hysteroscopic resection or myomectomy of submucosal UFs may be options. Each has benefits and drawbacks.6 Patients with UFs that are not submucosal are offered treatments to improve bleeding, but these do not reduce the size of the UFs. First-line pharmacological options include combined estrogen-progestin contraceptives, progestin-releasing intrauterine systems, and tranexamic acid (for patients who wish to avoid hormonal contraceptives or to treat only when symptoms occur). However, the use of hormonal contraceptives for treating UFs is off-label, and HMB is often inadequately controlled with hormonal contraceptives alone.8 For patients whose disease does not respond to first-line treatments, GոRΗ antagonists and agonists may be used with or without add-back therapies (i.e., E2 and NETA), which are used to mitigate hypoestrogenic side effects and protect the endometrium.9 Some medications, such as RGX-E2-NETA, contain add-back therapy in the combination. GnRH agonists (e.g., LA) are often used as short-term, preoperative therapies (i.e., for 3 months to 6 months) with concomitant iron therapy to reduce the size of UFs and improve anemia before surgery.10 Some caveats with the use of GnRH agonists are the initial flare effect (i.e., symptom exacerbation), which is not seen with GոRΗ antagonists, and the fact that once the drug is stopped, UF regrowth often occurs within 3 months.7 Uterine artery embolization can treat HMB and bulk symptoms, but does not remove UFs and is associated with ovarian dysfunction and the need for reintervention (rates between 14% and 38% within 5 years).11 Progesterone receptor modulators are generally not available in most countries for treating fibroids, and use is restricted due to increased risk of hepatic toxicity. Hysterectomy is the only definitive treatment for UFs, but it is associated with significant morbidity and mortality; therefore, it is a last-line option for patients who have persistent symptoms despite other treatments.7
For patients who desire fertility with submucosal UFs, hysteroscopic myomectomy is minimally invasive and can relieve symptoms. Myomectomy allows for the uterus to be retained, but is associated with a higher risk of blood loss than hysterectomy.7 In addition, UFs have a 15% recurrence rate after myomectomy; risk varies with age, number of fibroids, uterine size, and childbirth after the procedure.7 Patients with UFs that are not submucosal may be offered laparoscopic or open abdominal myomectomy. Drug treatments are associated with adverse effects when used for long durations, and symptoms quickly return when treatment is discontinued.
Before September 2020, UPA was a hormonal drug indicated for the treatment of UFs. It was associated with morphological changes in the endometrium and an increased risk of rare but serious liver injury, and it was subsequently withdrawn from the Canadian market.26-28 With UPA no longer available, physicians continue to treat UFs with tranexamic acid, GոRΗ agonists and antagonists (with add-back therapy), hormonal contraceptives, medical procedures, and surgery, as previously described.9
Drug Under Review
Key characteristics of RGX-E2-NETA are summarized in Table 3 along with those of other drug treatments available for patients with HMB associated with UFs who wish to maintain fertility, preserve the uterus, and reduce symptoms while avoiding surgery or other procedures, as suggested by the sponsor.
Myfembree is a combination of RGX, E2, and NETA. RGX is a nonpeptide, GnRH receptor antagonist that binds competitively to GnRH receptors in the anterior pituitary gland, reducing the secretion of luteinizing hormone and follicle-stimulating hormone.16 This reduction of follicle-stimulating and luteinizing hormone concentrations limits the production of estrogen and progesterone, respectively, reducing the bleeding associated with UFs and the pain associated with endometriosis. E2 is an agonist of nuclear estrogen receptor subtypes with organ-specific effects. Exogenously administered E2 in RGX-E2-NETA may alleviate symptoms, such as BMD loss and vasomotor symptoms, that can occur due to a decrease in circulating estrogen concentrations from RGX alone. NETA is a synthetic progestin that acts as an agonist of progesterone receptors and reduces the estrogen-induced risk of endometrial hyperplasia.
The recommended dose is 1 tablet of RGX-E2-NETA to be taken orally once daily.16 It is recommended that the administration of RGX-E2-NETA be initiated as early as possible after the onset of menses, but no later than 5 days after menses have started. The Health Canada product monograph states that use of RGX-E2-NETA should be limited to 24 months due to the risk of continued, irreversible bone loss.
RGX-E2-NETA is indicated in women in the premenopausal stage for the management of HMB associated with UFs and for the management of moderate to severe pain associated with endometriosis.16 RGX and E2 have been reviewed by CDA-AMC as individual products and in combination, such as E2 with progesterone. However, the combination RGX-E2-NETA has not been reviewed before.
The sponsor’s reimbursement request is for the management of HMB associated with UFs in women in the premenopausal stage.
Table 3: Key Characteristics of RGX-E2-NETA and Leuprolide Acetate
Characteristic | RGX-E2-NETA | Leuprolide acetate for depot suspension |
|---|---|---|
Mechanism of action | RGX is a nonpeptide GnRH receptor antagonist that reduces FSH and LH (and consequently reduces estrogen and progesterone levels). E2 is an agonist of the nuclear estrogen receptor that may alleviate symptoms due to decreased estrogen, and NETA is a synthetic progestin that reduces the estrogen-induced risk of endometrial hyperplasia. | A synthetic nonapeptide analogue of naturally occurring GnRH. The analogue possesses greater potency than the natural hormone. When administered as indicated, leuprolide acetate acts as a potent inhibitor of gonadotropin production. Leuprolide acetate exerts specific action on the pituitary gonadotrophs and the human reproductive tract, reducing the likelihood of secondary adverse effects such as gynecomastia, thromboembolism, edema, and liver and gallbladder involvement. |
Indicationa | In premenopausal women for the management of heavy menstrual bleeding associated with UFs and for the management of moderate to severe pain associated with endometriosis | Concomitant administration with iron therapy for the preoperative hematologic improvement in patients of reproductive age with anemia caused by UFs Various other indications |
Route of administration | Oral | IM to provide continuous sustained release of leuprolide for 1 month |
Recommended dosage | 1 tablet of Myfembree (40 mg RGX, 1 mg E2, 0.5 mg NETA) daily; the recommended duration is up to 24 months. | For UFs, 3.75 mg monthly with concomitant daily oral iron therapy for the preoperative hematologic improvement of patients with leiomyomas and iron deficiency anemia caused by excessive uterine bleeding; the recommended duration is up to 3 months. Various dosages for other indications |
Serious adverse effects or safety issues | Increased risk of thrombotic or thromboembolic disorders, including pulmonary embolism, deep vein thrombosis, stroke, and myocardial infarction, especially in women already at increased risk for these events. Consider discontinuing treatment if hair loss becomes a concern. Contraindicated in patients who:
| During the early phase of therapy, sex steroids temporarily rise higher than baseline because of the physiologic effect of the drug, particularly when given in the follicular phase of the menstrual cycle, causing the “flare effect.” Therefore, an increase in clinical signs and symptoms may be observed during the initial days of therapy. However, these will dissipate with continued therapy at adequate doses, or when the drug is given later in the luteal phase. Contraindicated in patients who are or may become pregnant and in patients who have undiagnosed abnormal vaginal bleeding. There is potential for reduction in BMD when leuprolide acetate is used without add-back therapy. |
BMD = bone mineral density; E2 = estradiol; FSH = follicle-stimulating hormone; GnRH = gonadotropin-releasing hormone; IM = intramuscular; LH = luteinizing hormone; NETA = norethindrone acetate; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; UF = uterine fibroid.
aHealth Canada–approved indication.
Source: Product monographs for Myfembree and Lupron Depot.16,29
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received 1 patient group input submission from CANFib. The organization provides a forum for females in Canada with UFs to discuss their symptoms, talk to peers, find out more about UFs, and discuss navigation of the Canadian medical system while learning more about the treatments and medications approved by Health Canada for UFs. CANFib also works with Canadian pharmaceutical outreach, researchers, insurance companies, and specialists to push for new ideas that become future treatments and solutions.
CANFib conducted a survey in 2023. Respondents included 315 females with UFs. The patient group noted that 59% of the respondents had to wait from 7 months to more than 13 months to visit their health care provider. Based on the survey results, a new symptom or a current medication that was not working were the top reasons to return to the specialist. Additionally, pain was the top reason (n = 266) for seeking medical help, followed by excessive bleeding (n = 249); these symptoms led 73% of respondents to take at least 2 days off work per month. CANFib explained that 261 respondents reported consulting the internet for information on UF relief; that 84% of those who consulted medical professionals felt they left the consultation still “uninformed” about the condition; and that 86% felt they were not given the full list of Health Canada–approved treatments and medications. CANFib highlighted that because the top reason to return to a specialist was ineffectiveness of medications — and given the long wait times from diagnosis to treatment and for appointments — more options and earlier treatment will benefit patients and reduce the need for follow-up visits and surgery.
CANFib also conducted group discussions with 32 females with UFs who had experience with RGX-E2-NETA. Among these participants, 31 reported being “very happy” about the effects of RGX-E2-NETA on their symptoms, while 1 reported feeling “indifferent.” Two had been able to work again. Two out of the 32 respondents had access to RGX-E2-NETA through extended insurance coverage, while 30 respondents had to pay out of pocket for the medication; 3 respondents said they could or would continue to afford the medication, even for a lengthy period. CANFib added that RGX-E2-NETA enabled the females in the discussion group to lead full lives, spend time with their families, care for children and/or parents, show up at work, and function without debilitating pain, heavy blood loss, or ongoing depression.
CANFib explained that, based on member discussions, most females reported primary symptoms of heavy bleeding and pain; other common events are lost time at work, inability to function within a family unit, a diminished social or professional life, and eventual depression. The discussion group described the constant need for open access to a bathroom at work or at home. Members described how their relationships suffered from the immense pressure of feeling as though they must carry on as if they do not have symptoms.
Regarding currently available treatments, CANFib noted that although there are treatments and medications available to treat UFs, there is no cure other than surgical hysterectomy. According to the organization, desirable outcomes include access to an effective treatment that does not require a 3-month dosage commitment, regaining confidence, and being able to enjoy family, social, and professional lives.
This patient group submission also included comments from the incoming Society of Obstetricians and Gynecologists of Canada president and an endorsement from the Women’s Health Coalition.
Clinician Input
Input From the Clinical Expert Consulted for this Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of HMB associated with UFs.
Unmet Needs
According to the clinical expert consulted on this review, patients seek new therapies that can adequately and rapidly address symptoms without adverse effects, improve HRQoL, offer simpler administration, with quick resolution upon discontinuation.
Place in Therapy
Based on input from the clinical expert, first-line treatments for AUB can include nonsteroidal anti-inflammatory drugs, hormonal contraceptives (including progestins and levonorgestrel intrauterine systems), tranexamic acid, and iron repletion as further investigations take place to identify the cause of the AUB. Once a diagnosis of UFs (with HMB) is confirmed, RGX-E2-NETA would be a treatment option in the absence of endometrial pathology, depending on the symptoms, severity, and location of the UFs and on the patient’s goals. The clinical expert expects that RGX-E2-NETA will be used in practice to complement and facilitate less invasive medical procedures and surgical approaches, such as hysteroscopic procedures and fertility-preserving surgical approaches. As per the expert, RGX-E2-NETA may be used as a bridge treatment in patients who plan to have gynecological surgery within 3 months (to elevate hemoglobin and enable less invasive surgical methods), want to delay surgery for fertility, or anticipate menopause within a couple of years.
Patient Population
The clinical expert indicated that patients receiving RGX-E2-NETA would be adults in the premenopausal stage, given that the symptoms of UFs tend to diminish after menopause. Additionally, the patients most likely to respond to treatment with RGX-E2-NETA include those with HMB associated with UFs and bulk symptomatology secondary to UFs. The expert also noted that patients with anemia secondary to severe AUB, severe pain, and ureteric obstruction by UFs are most in need of treatment.
The patients best suited for treatment with RGX-E2-NETA would be identified based on clinical history, physical examination, and investigations (e.g., Papanicolaou test, biopsy, complete blood count, pregnancy testing, and sonohysterography hysteroscopy). As part of confirming a diagnosis, endometrial and cervical pathology must be ruled out based on the patient’s age and risk factors.
Assessing the Response to Treatment
As per the clinical expert, the main outcomes used to assess treatment response in practice include qualitative clinical assessments for a reduction in primary symptomatology (i.e., reduced HMB), reduction in pain and bulk symptoms, return to normal function (e.g., activities of daily living, work), and fertility success, if this is the patient’s goal. Clinicians also focus on the patient’s satisfaction with the treatment rather than on quantitative measures, such as MBL volume. However, physicians may measure hemoglobin levels with the goal of increasing them to normal values in patients experiencing anemia. The expert explained that although the outcomes used in clinical trials may be informative, these are generally not used in practice (aside from measuring and improving hemoglobin levels).
Discontinuing Treatment
The clinical expert stated that RGX-E2-NETA should be discontinued upon successful surgery or procedure, plans for pregnancy, menopause, a meaningful decline in BMD, or intolerable adverse effects. Although the Health Canada product monograph states that use of RGX-E2-NETA should be limited to 24 months due to the risk of continued, irreversible bone loss, the expert suggested that treatment beyond 2 years may be considered if the patient is maintaining their goals for symptom improvement and has a healthy BMD. However, if RGX-E2-NETA is used to treat HMB and anemia in anticipation of a planned surgery or medical procedure, the treatment duration tends to be less than 2 years. As noted by the expert, the benefit in preparation for surgery with respect to the reduction of the size of the uterus and fibroids is achieved after 3 months. If wait times are longer than 3 months, benefit can be maintained by longer use of the treatment.
Prescribing Considerations
RGX-E2-NETA would be used in a community setting, acute care hospital, or specialty clinic. While diagnosis and initial prescription of the drug would be performed by an obstetrician-gynecologist, the expert noted that follow-up management and monitoring may be done by a primary care physician.
Clinician Group Input
No input was received from clinician groups for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect the programs’ ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor selected 2 identical studies (the LIBERTY 1 trial and the LIBERTY 2 trial) from a systematic literature review. Both trials studied RGX-E2-NETA versus placebo. There was an ITC to leuprolide plus add-back therapy, which is used off-label to treat this condition when surgery is not an option. Lupron (leuprolide) is a regular benefit in most jurisdictions and has restricted use criteria in the other jurisdictions. It is also a slow-release injectable, whereas RGX-E2-NETA includes RGX as a daily oral formulation. Outside of off-label use of leuprolide plus add-back hormonal therapy, would the following off-label drugs also be appropriate comparators (in addition to add-back therapy)?
| According to the clinical expert consulted for this review, nafarelin, buserelin, goserelin, and degarelix are not commonly used because these do not have a Health Canada indication for the treatment of UFs; in addition, some have associated adverse effects that patients wish to avoid. The expert stated that goserelin may be used instead of leuprolide acetate for patients who cannot receive intramuscular injections (e.g., patients who are on anticoagulants or those with diseases affecting coagulation, such as von Willebrand disease); however, in general, none of the treatments would be considered comparators for RGX-E2-NETA. |
Of note, Myfembree is a fixed-dose, oral tablet containing all 3 drugs (RGX, E2, and NETA). The LIBERTY trials used 2 separate tablets, RGX and E2-NETA (Activelle). The sponsor has confirmed that a study was conducted to demonstrate the bioequivalence of the fixed-dose tablet compared to the coadministration of RGX and E2-NETA tablets. Are there any concerns with this approach? | The clinical expert had no concerns with the approach used in the trial (i.e., RGX available as a tablet separate from the combined E2-NETA) versus how RGX-E2-NETA is available commercially (i.e., as a single combined tablet). |
Considerations for initiation of therapy | |
The LIBERTY studies required that diagnosis of UFs be confirmed by a transvaginal ultrasound. At least 1 UF must be verified to meet at least 1 of the following criteria:
Considering diagnostic imaging, although the LIBRERTY studies required transvaginal ultrasound, would 1 of the following 3 tests be acceptable for meeting diagnostic criteria: hysteroscopy, ultrasonography, or MRI? Do you agree with these requirements for access to the medication? | The clinical expert noted that of the 3 imaging methods listed, ultrasonography (specifically sonohysterography) is the preferred method because of its accuracy. Hysteroscopy may be an option, but it is less preferred because it does not show fibroids located outside the uterus. MRI is rarely used because ultrasound methods are better for imaging UFs and less costly. The expert also emphasized the importance of UF location in diagnosing the severity of disease and choosing a treatment; therefore, the criteria used in the trial are reasonable. |
Patients aged 18 to 50 years in the premenopausal stage were included. Would patients outside this age range and in the premenopausal stage be eligible for treatment? | The clinical expert stated that patients aged 18 years and older and in the premenopausal stage would be eligible for treatment with RGX-E2-NETA. |
Given that the use of hormonal contraceptives to treat UFs is considered off-label in Canada, are there any appropriate prior therapies that should be required? | As per the Society of Obstetricians and Gynaecologists of Canada’s clinical practice guidelines for abnormal uterine bleeding in premenopausal women,30 nonhormonal drugs (e.g., nonsteroidal anti-inflammatory drugs and antifibrinolytics) can be used to treat HMB that is cyclic or has predictable timing. Combined hormonal contraceptives, progestins, depot medroxyprogesterone acetate, and levonorgestrel-releasing intrauterine systems are hormonal drugs that can address HMB. The clinical expert stated that these are reasonable options for treating AUB or for treating HMB associated with UFs. |
Considerations for continuation or renewal of therapy | |
Response is shown by improvement in bleeding, including experiencing amenorrhea and anemia, where applicable. The primary end point in the LIBERTY studies was defined by the percentage of participants who experienced an MBL of < 80 mL and ≥ 50% reduction from baseline MBL over the last 35 days of the treatment. Measurement of MBL in the real-world setting is not practical. How will MBL reduction be monitored and assessed? Given that persons with HMB associated with UFs often have iron deficiency anemia requiring treatment, a key secondary end point in the studies was the proportion of patients with a hemoglobin level ≤ 10.5 g/dL at baseline who experienced an increase of ≥ 2 g/dL from baseline at week 24. This increase is in line with the expected increase in hemoglobin following the transfusion of 2 units of red blood cells. Is it possible that a patient would meet a significant increase in hemoglobin independent of meeting the specified MBL reduction? For CDEC consideration: Consider whether renewal criteria might allow for the renewal of drug eligibility for patients with HMB who have demonstrated an increase in hemoglobin compared to baseline. | As per the clinical expert, exact quantification of MBL (such as by using the alkaline hematin method) is not performed in clinical practice, where MBL reduction is monitored through clinical assessment based on improvements reported by the patient and the return of hemoglobin levels to normal. Furthermore, the focus of treatment is to have a reduction in MBL that the patient is satisfied with, and for hemoglobin levels to improve such that the HMB and anemia no longer affect the patient’s HRQoL or activities of daily living. Because the goals of treatment differ from the outcomes in a clinical trial, it can be difficult to know if a patient has met the significant increase in hemoglobin independent of meeting the specified MBL reduction. |
System and economic issues | |
When estimating the costs of comparative treatments, the sponsor assumed that the treatment unit cost of leuprolide plus add-back (Activelle) equates only to the cost of leuprolide because Activelle is not publicly funded. Given that Activelle is not publicly funded, are there situations where patients who wish to be treated with leuprolide plus add-back would replace the Activelle with a different hormonal alternative that is covered? This cost may not be captured and would affect the BIA. For CDEC consideration: For jurisdictions that list leuprolide as an open or free benefit, it may not be possible to estimate the proportion of leuprolide usage that is attributable to patients in the premenopausal stage with HMB and UF. | The clinical expert confirmed that the add-back therapy does not have to be Activelle, but can be another similar treatment that is covered by insurance. In previous clinical studies, standard add-back has been NETA 5 mg, which the expert noted is less expensive than Activelle. |
AUB = abnormal uterine bleeding; BIA = budget impact analysis; CDEC = Canadian Drug Expert Committee; E2 = estradiol; E2-NETA = estradiol and norethindrone acetate; GnRH = gonadotropin-releasing hormone; HMB = heavy menstrual bleeding; HRQoL = health-related quality of life; ITC = indirect treatment comparison; LHRH = luteinizing hormone-releasing hormone; MBL = menstrual blood loss; NETA = norethindrone acetate; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; UF = uterine fibroid.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of oral RGX-E2-NETA in the treatment of HMB associated with UFs in patients in the premenopausal stage. The focus will be placed on comparing RGX-E2-NETA to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of RGX-E2-NETA is presented in 4 sections, with our critical appraisal of the evidence included at the end of each. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section is meant to include additional studies considered by the sponsor to address important gaps in the systematic review evidence; however, there were no such studies.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
2 pivotal RCTs identified in the systematic review
2 extension studies
1 ITC.
Systematic Review
The contents in this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Description of Studies
Two phase III, double-blind, placebo-controlled RCTs were included in the systematic review: the LIBERTY 1 trial (N = 388) and the LIBERTY 2 trial (N = 382).12,13 Characteristics of the included studies are summarized in Table 5. The studies were mostly identical in design and were conducted to evaluate the efficacy and safety of RGX-E2-NETA in patients in the premenopausal stage for the management of HMB associated with UFs. The main differences between the studies were the countries where the studies took place and the order of statistical testing of secondary outcomes. The studies started with a screening period of up to 13 weeks to confirm eligibility through diagnosis of UFs, validation of HMB, assessment of BMD by dual-energy X-ray absorptiometry (DXA), and endometrial biopsies. Patients were randomized to 24 weeks of treatment, after which they could enrol directly into the 28-week OLE study followed by the 52-week RWS; those who did not were followed for safety monitoring for 30 days.31,32 Although each study had 3 treatment groups, only the RGX-E2-NETA groups (n = 128 in the LIBERTY 1 trial, n = 126 in the LIBERTY 2 trial) and placebo groups (n = 128 in the LIBERTY 1 trial, n = 129 in the LIBERTY 2 trial) are described in this report. The group receiving RGX plus delayed estradiol and norethindrone acetate (E2-NETA) was included in the LIBERTY trials to provide an assessment of the requirement for E2 and NETA to mitigate the adverse effects of RGX monotherapy on BMD loss and vasomotor symptoms. Results are not reported herein because RGX monotherapy is not available in Canada.
Table 5: Details of Studies Included in the Systematic Review
Detail | LIBERTY 1 trial | LIBERTY 2 trial |
|---|---|---|
Designs and populations | ||
Study design | Phase III, double-blind, placebo-controlled RCT | |
Locations | 80 sites in 6 countries (Brazil, Italy, Poland, South Africa, UK, and US) | 99 sites in 8 countries (Belgium, Brazil, Chile, Czechia, Hungary, Poland, South Africa, and US) |
Patient enrolment dates | Start date: April 4, 2017 End date: August 24, 2020 | Start date: June 14, 2017 End date: September 16, 2020 |
Randomized (N) | N = 388
| N = 382
|
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention |
| |
Comparators |
| |
Study duration | ||
Screening phase | Up to 13 weeks | |
Treatment phase | 24 weeks | |
Follow-up phase | Entry into a 28-week OLE study (the LIBERTY extension trial) or 30-day safety monitoring for patients who did not continue in the OLE study | |
Outcomes | ||
Primary end point | Proportion of patients in the RGX-E2-NETA group vs. the placebo group who experienced an MBL volume of < 80 mL and a ≥ 50% reduction from baseline in MBL volume over the preceding 35 days of treatment, as measured using the alkaline hematin method | |
Secondary and exploratory end points | Main secondary:a
Safety:
| |
Publication status | ||
Publications | Al-Hendy et al. (2021),33 Stewart et al. (2022),34 and NCT0304973535 | Al-Hendy et al. (2021),33 Stewart et al. (2022),34 and NCT0310308736 |
BMD = bone mineral density; BPD = bleeding and pelvic discomfort; DXA = dual-energy X-ray absorptiometry; E2 = estradiol; E2-NETA = estradiol and norethindrone acetate; HMB = heavy menstrual bleeding; HRQoL = health-related quality of life; MBL = menstrual blood loss; NETA = norethindrone acetate; NRS = numerical rating scale; OLE = open-label extension; RCT = randomized controlled trial; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; TEAE = treatment-emergent adverse event; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
aMain secondary end points included key secondary end points and those deemed by the sponsor to be most relevant to the reimbursement review.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Eligibility criteria were consistent between the studies. Eligible patients included females in the premenopausal stage aged 18 to 50 years with regularly occurring menstrual periods of 14 or fewer days’ duration with a cycle of 21 days to 38 days. Patients had to have had a confirmed UF diagnosis by transvaginal ultrasound, HMB associated with UFs (defined as MBL of ≥ 160 mL during 1 menstrual cycle or ≥ 80 mL per cycle during 2 cycles), and no significant endometrial pathology on biopsy. Individuals were not eligible if they had ultrasound readings demonstrating other pathology that could be responsible for or contributing to HMB; if they had known, rapidly enlarging UFs; if they had undergone gynecological surgery or ablation procedures for UFs within the 6 months before screening; if they had a history of or current osteoporosis, other metabolic bone disease, hyperparathyroidism, hyperprolactinemia, hyperthyroidism, anorexia nervosa, or low traumatic or atraumatic fracture; or if they had a history of medication use to treat BMD loss (other than calcium and vitamin D preparations).
Interventions
Patients were randomized 1 to 1 to 1 to receive oral RGX-E2-NETA (40 mg RGX, 1 mg E2, and 0.5 mg NETA), RGX plus delayed E2 and NETA, or matching placebo once daily in a fasted state for 24 weeks. Randomization was stratified by geographic region (North America versus rest of the world) and by mean screening MBL using the alkaline hematin method (< 225 mL versus ≥ 225 mL).
In the LIBERTY studies, patients received RGX with commercially available E2-NETA in a copackaged configuration. The Health Canada–approved, commercialized formulation of RGX-E2-NETA is a fixed-dose combination tablet consisting of all 3 drugs. According to the sponsor, a bioequivalence study was conducted that demonstrated bioequivalence between the fixed-dose combination tablet and the coadministration of a RGX 40 mg tablet and an E2-NETA tablet containing 1 mg of E2 and 0.5 mg of NETA.37
Supplementation with calcium and/or vitamin D was permitted in the studies, but was not required. Patients with a hemoglobin level of less than 8.0 g/dL were excluded from the studies. During screening, patients with microcytic iron deficiency anemia (defined as a hemoglobin level ≥ 8 g/dL and ≤ 10 g/dL), a mean corpuscular volume below the lower limit of normal, and low serum iron and ferritin were started on iron therapy, which was continued during the studies. Patients who entered the screening period on iron therapy could continue it during the studies. Patients who developed new microcytic iron deficiency anemia during the studies (defined as a hemoglobin level ≤ 10 g/dL), a mean corpuscular volume below the lower limit of normal, and low serum iron and ferritin were started on iron therapy. If the hemoglobin level was less than 10 g/dL and the mean corpuscular volume was below the lower limit of normal, a ferritin and iron level was reported through the central lab.
Prohibited medications included drugs affecting the hypothalamic-pituitary-gonadal axis (e.g., GnRH analogues, estrogens, and progestins), drugs to treat BMD (e.g., bisphosphonates, specified antiepileptic drugs, glucocorticoids), drugs to treat blood loss (e.g., anticoagulants), and drugs with the potential to affect RGX absorption (e.g., P-glycoprotein inducers and moderate and strong P-glycoprotein inhibitors).
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures in Table 7. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review, according to the clinical expert consulted for this review and input from patient groups and public drug plans. Using the same considerations, we selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing the expert committee deliberations were also assessed using GRADE.
The following considerations went into the selection of the outcomes that were summarized in the report and assessed using GRADE:
improvement of symptoms (e.g., MBL, anemia, pain) and HRQoL, which were important to the patient group that provided input and to the clinical expert consulted on this review
measurement of MBL using the alkaline hematin method, which has been used as a primary end point in clinical trials assessing HMB.38
Table 6: Outcomes Summarized From the LIBERTY 1 and LIBERTY 2 Trials
Outcome measure | Time point | LIBERTY 1 trial and LIBERTY 2 trial |
|---|---|---|
Proportion of patients in the RGX-E2-NETA group vs. the placebo group who experienced an MBL volume of < 80 mL and ≥ 50% reduction from baseline MBL volume, as measured using the alkaline hematin method | Last 35 days of treatment | Primarya |
Proportion of patients with a hemoglobin level of 10.5 g/dL or lower at baseline who experienced an increase of ≥ 2 g/dL | From baseline to week 24 | Key secondarya |
Proportion of patients who experienced a maximum NRS score ≤ 1 for UF-associated pain in the subset of patients with a maximum pain score ≥ 4 and completed the daily e-diary during the 35 days before randomization | Last 35 days of treatment | Key secondarya |
Change in the UFS-QoL SSS score | From baseline to week 24 | Secondary |
Change in the UFS-QoL HRQoL total score | From baseline to week 24 | Secondary |
Safety | From first dose of study drug to follow-up visit (30 days after last dose of study drug) | Safety |
Percentage change in BMD, as assessed by DXA | From baseline to week 24 | Safety |
BMD = bone mineral density; DXA = dual-energy X-ray absorptiometry; HRQoL = health-related quality of life; MBL = menstrual blood loss; NRS = numerical rating scale;RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Sponsor’s Summary of Clinical Evidence.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Responder Rate and Change in MBL Volume
Responder rate was defined as the percentage of participants in the RGX-E2-NETA group versus the placebo group who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from baseline in MBL volume over the last 35 days of treatment as measured using the alkaline hematin method. The effects of RGX-E2-NETA and placebo on reducing HMB associated with UFs were also measured by the percentage change from baseline to week 24 in MBL volume.
Baseline MBL was defined as the average MBL during the 2 screening menstrual cycles used to assess the inclusion criteria before the date of the first dose of the study drug. The MBL during the final study month was defined as the total MBL during the last 35 days on treatment. To ensure collection of all feminine products used during that menstrual cycle, an interval of up to 35 days for measurement of the primary end point was selected to accommodate patients who continued to have cyclic bleeding on study treatment and whose natural cycles were at the upper end of the normal cycle duration range.
MBL volume was analyzed using the alkaline hematin method, which is the gold standard for objective measurement of blood loss using feminine sanitary products. It has been used as a primary end point in clinical trials assessing HMB.4,39 Used feminine products are pummelled in a 5% sodium hydroxide solution to convert hemoglobin to alkaline hematin, and the resulting hematin absorbance is compared against calibration curves coming from a diluted venous blood sample from the same patient.39
The upper limit of normal MBL has been suggested to be 80 mL.40 Based on the literature, a clinically meaningful change in MBL has been estimated to be a 22% reduction for patients with HMB, as measured using the alkaline hematin method.41 This estimate was calculated based on a receiver-operating characteristic curve using data from a multicentre, double-blind, placebo-controlled RCT of a medication for HMB that included some patients who had UFs.41
Improvement in Anemia
A baseline hemoglobin concentration of 10.5 g/dL or lower is below the lower limit of normal for females (i.e., 12 g/dL).42 An increase of 2 g/dL or greater is expected following the transfusion of 2 units of red blood cells; the transfusion is expected to result in a clinically meaningful improvement in hemoglobin concentration.43,44 The proportion of patients who had an increase in hemoglobin level of at least 2 g/dL at week 24 was reported for the subset of patients who had a baseline hemoglobin value of 10.5 g/dL or lower. Clinical laboratory testing (including for ferritin and iron) was conducted at every visit (i.e., every 4 weeks).
NRS for UF-Associated Pain
Daily e-diary entries included assessments of UF-associated pain during the preceding 24 hours using an 11-point NRS (ranging from 0 to 10, where higher scores indicated worse pain). The maximum NRS score for pain at week 24 or end-of-treatment was calculated as the maximum NRS score during the last 35 days on study treatment. A minimal important difference (MID) for the NRS was not identified in the literature.
Uterine Fibroid Symptom Quality of Life Questionnaire
The UFS-QoL is a disease-specific instrument used to assess symptom severity and HRQoL in patients with UFs during the preceding month.14 The SSS consists of 8 items rated on a 5-point Likert scale ranging from “not at all” to “a very great deal.” The HRQoL component of the instrument includes 29 items rated on a 5-point scale ranging from “none of the time” to “all of the time.” Higher SSS scores indicate more severe UF symptoms, while higher HRQoL total scores indicate better quality of life. Scores for the SSS and HRQoL subscales are linearly transformed to a scale ranging from 0 points to 100 points:
Symptom severity raw scores were transformed using the following formula: actual raw score minus lowest possible raw score, divided by total possible raw score range, multiplied by 100.
Raw scores for scales other than symptom severity were transformed using the following formula: highest possible score minus actual raw score, divided by total possible raw score range, multiplied by 100.
The sponsor did not identify any literature-based MID estimate for this end point; however, a 20-point or greater decrease in the SSS has been associated with patients feeling “much better.”45
Patients completed the UFS-QoL at their baseline, week 12, and week 24 visits.
Safety
Safety outcomes included the proportion of patients who reported TEAEs (graded according to the Common Terminology Criteria for TEAEs), SAEs, withdrawals from treatment due to TEAEs, and deaths. TEAEs were collected from when the first dose of the study drug was administered until the follow-up visit approximately 30 days after the last dose of study drug (or upon initiation of another investigational drug or hormonal therapy affecting the hypothalamic-pituitary-gonadal axis or surgical intervention for UFs, whichever occurred first). Based on clinical expert opinion, loss of BMD at 24 months was deemed relevant to the current review.
Bone Mineral Density
The percentage change from baseline to week 24 in BMD at the lumbar spine (i.e., L1 to L4) was assessed using DXA. RGX monotherapy suppresses sex hormones through GnRH receptor antagonism, resulting in low estrogen levels, leading to declines in BMD — an important determinant for fracture risk.46 To assess the effect of adding E2-NETA to RGX, BMD was assessed throughout the LIBERTY clinical program.
Patients were assessed using the same DXA apparatus at each site with the same scan mode. The central radiology laboratory collected and evaluated the scans for acceptability and assessed BMD as per the imaging charter. DXA scans were completed at the screening, week 12, and week 24 visits.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Alkaline hematin method for the measurement of MBL | Considered the gold standard for the objective measurement of MBL using feminine sanitary products.4 Used feminine products are pummelled in a 5% sodium hydroxide solution that converts hemoglobin to alkaline hematin, and the resulting hematin absorbance is compared against calibration curves coming from a diluted venous blood sample from the same patient.4 | The alkaline hematin technique is a validated method of measuring MBL. It uses Always Ultra sanitary products that contain superabsorbent polymers.4 Recovery of blood, linearity, and intra-assay variation was assessed over a range of simulated menstrual fluid volumes applied to these sanitary products. Because of the variable percentage of blood in menstrual fluid, recovery was tested using simulated fluid with 10%, 25%, 50%, and 100% blood.4 The lower limit of reliable detection and the effect of storing used products for up to 4 weeks at 15°C to 20°C, 4°C, and –20°C before analyses were determined.4 Ninety percent recovery was reproducibly achieved for up to 30 mL applied volume at all tested, simulated menstrual fluid compositions, except at low volume or high dilution equivalent to < 2 mL whole blood.4 HMB (menorrhagia) is defined as MBL of > 80 mL per cycle.4 | No MID has been estimated. |
UFS-QoL SSS | A patient-reported, UF-specific questionnaire evaluating the severity of symptoms associated with UFs. The SSS consists of 8 items, each reported on a 5-point Likert scale ranging from “not at all” to “a very great deal.”14 The symptom severity item scores are summed and transformed into a 0-point to 100-point scale, with higher scores indicating more severe UF symptoms.14 | Concurrent and divergent validity:
Test-retest reliability and internal consistency:
Responsiveness:
| No MID has been estimated. |
UFS-QoL HRQoL total score | A patient-reported, UF-specific questionnaire evaluating the impact of UFs on HRQoL. The questionnaire consists of 29 items within 6 subscales: concern, activities, energy or mood, control, self-consciousness, and sexual function. Each item is reported on a 5-point Likert scale ranging from “none of the time” to “all of the time.” The subscales are summed from the corresponding items. To calculate the HRQoL total score, the value of each subscale is summed (not individual items) and transformed into a 0-point to 100-point scale, with higher scores indicating better HRQoL.14 | Concurrent and divergent validity:
Test-retest reliability and internal consistency:
Responsiveness:
| No MID has been estimated. |
HMB = heavy menstrual bleeding; HRQoL = health-related quality of life; MBL = menstrual blood loss; MID = minimal important difference; SD = standard deviation; SF-36 = Short Form (36) Health Survey; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
Source: Request to sponsor for additional information.48
Statistical Analysis
Statistical analyses are summarized in Table 8.
Sample Size and Power Calculation
Assuming a 20% dropout rate, approximately 130 patients in the RGX-E2-NETA group and 130 patients in the placebo group would provide at least 99% power at a 2-sided 0.05 significance level to detect a 30% difference in responder rates between the RGX-E2-NETA group and the placebo group for the primary end points for each of the LIBERTY 1 trial and the LIBERTY 2 trial.49 The assumed responder rate of 25% for the placebo group was within the range of the responder rates observed from similar phase III trials involving UFs.50 The sample size and power calculations were based on a chi-square test.
Statistical Testing
The primary efficacy end point (i.e., the proportion of patients in the RGX-E2-NETA group versus placebo group who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL volume over the preceding 35 days of treatment, as measured using the alkaline hematin method) was analyzed using a Cochran-Mantel-Haenszel test statistic for proportions and was stratified by the baseline mean MBL volume using the alkaline hematin method and geographic region. The between-group difference in responder rate, its 2-sided 95% CI, and P value were estimated using stratum-adjusted Cochran-Mantel-Haenszel proportions. Details on the statistical testing of secondary outcomes are summarized in Table 8.
Multiple Testing Procedure
A gatekeeping, mixed-sequence testing procedure was applied to maintain the family-wise type I error rate for the primary end point and 7 key secondary end points in the LIBERTY 1 trial and LIBERTY 2 trial (Figure 1 and Figure 2, respectively). The studies were considered positive if the treatment effects for the primary end points were statistically significant with a 2-sided P value of less than 0.05. If the P value for the primary end point (i.e., family 1, MBL responders) was less than 0.05, the first 4 key secondary end points (i.e., family 2, amenorrhea, percentage change in MBL, UFS-QoL bleeding and pelvic discomfort, and anemia responders or NRS responders) were tested sequentially in a similar manner. If the 2-sided P value was less than 0.05 for all of the first 5 end points tested, then the remaining 3 key secondary end points (i.e., family 3, NRS responders or anemia responders, percentage change in UF volume, and percentage change in uterine volume) were tested using the Hochberg step-up procedure.
From the Hochberg procedure, P values were calculated for the 3 end points and ranked from smallest to largest. The end point corresponding to the largest P value was tested first. If it was less than 0.05, then no further testing occurred, and it was concluded that all 3 end points were significant. Otherwise, the end point corresponding to the second largest P value was tested, and if the P value was less than 0.025, then no further testing occurred, and it was concluded that the end points corresponding to the middle and smallest P values were significant. Otherwise, the end point with the smallest P value was tested, and if the P value was less than 0.0167, then no further testing occurred, and it was concluded that only the end point with the smallest P value was significant. Otherwise, all 3 end points did not pass the statistical significance criterion at the 0.05 level.
The difference between the 2 sequences was that in the LIBERTY 1 trial, the fourth key secondary end point tested was the proportion of patients with a hemoglobin level of 10.5 g/dL or lower at baseline who experienced an increase of greater than or equal to 2 g/dL (part of family 2), and the fifth key secondary end point tested was the proportion of patients who experienced a maximum NRS score of less than or equal to 1 for UF-associated pain in the subset of patients with a maximum pain score of greater than or equal to 4 who had completed the daily e-diary during the 35 days before randomization (part of family 3), whereas in the LIBERTY 2 trial, these end points were reversed in sequence. According to the sponsor, the change in the order of hierarchical testing was made on the basis of the results of the LIBERTY 1 trial, before unblinding and data analysis in the LIBERTY 2 trial.
All other end points were assessed at a nominal 2-sided alpha of 0.05 without adjusting for multiple comparisons. Comparative statistics (i.e., 95% CIs and P values) were provided for RGX-E2-NETA versus placebo for all other secondary efficacy end points.
Data Imputation Methods
Details for how missing data were handled are summarized in Table 8. For the primary end point, imputations for missing or incomplete measures of responder status were based on duration of treatment exposure, adherence with feminine product collection compared to what was reported in the e-diary, adherence with e-diary entry, and reasons for no feminine product collection. A mixed-effects model for repeated measures was used to predict the percentage change in MBL volume from baseline through the fixed effects associated with covariates (i.e., the stratification factors of baseline MBL volume and geographic region, visit, treatment, and visit by treatment interaction) and random effects (from the individual patient). An unstructured variance-covariance matrix was assumed for each patient. Data imputation methods for the other outcomes of interest are summarized in Table 8.
Subgroup Analyses
Prespecified subgroup analyses for the primary end point included geographic region, baseline MBL volume, age, race or ethnicity, and baseline body mass index. Based on clinical expert opinion, none of the prespecified subgroups were deemed relevant to the review because these were not likely to identify a subpopulation of patients who might benefit more from treatment or to inform reimbursement decisions.
Sensitivity Analyses
Several sensitivity analyses of the primary end point were conducted to test the impacts of the following scenarios:
Unvalidated feminine product use: This scenario analysis was conducted using the week 24 or end-of-treatment, validated MBL volume (excluding unvalidated products).
Missing data due to inadequate collection of feminine products: No imputation was implemented for patients with less than 100% feminine product adherence and both a reported MBL volume less than 80 mL and a 50% or greater reduction from baseline.
Early discontinuation: Patients who discontinued the study drug during the first 4 weeks for any reason — or between week 4 and week 12 due to a TEAE, surgery or other intervention for HMB, reported lack of efficacy, or bleeding complaints — were considered as nonresponders, while all other patients had their responder status defined using data from the week 24 or end-of-treatment assessment period using the last observation carried forward method.
Duration of treatment exposure: The analysis used the “completers population,” defined as patients in the modified intention-to-treat (mITT) population who completed 24 weeks of study treatment.
Protocol deviations: A per-protocol population analysis was used.
Different imputation method for missing data: Using the mixed-effects model for imputing missing or partially missing MBL volumes at week 24 or end-of-treatment, a multiple imputation approach was used. An arbitrary missing pattern was assumed using Markov chain Monte Carlo imputation to generate a monotone missing pattern for the observed longitudinal MBL volume values. Imputation was performed separately by randomized treatment group, given the distinct bleeding patterns among the treatment groups. Normalizing transformations were applied to the statistics estimated from each imputed dataset before Rubin’s combination rules were applied. This combined estimation and statistical test accounted for the additional variability due to imputation to provide a robust assessment of the treatment effect.
Figure 1: LIBERTY 1 Trial Mixed-Sequence Testing Procedure for Primary and Key Secondary End Points
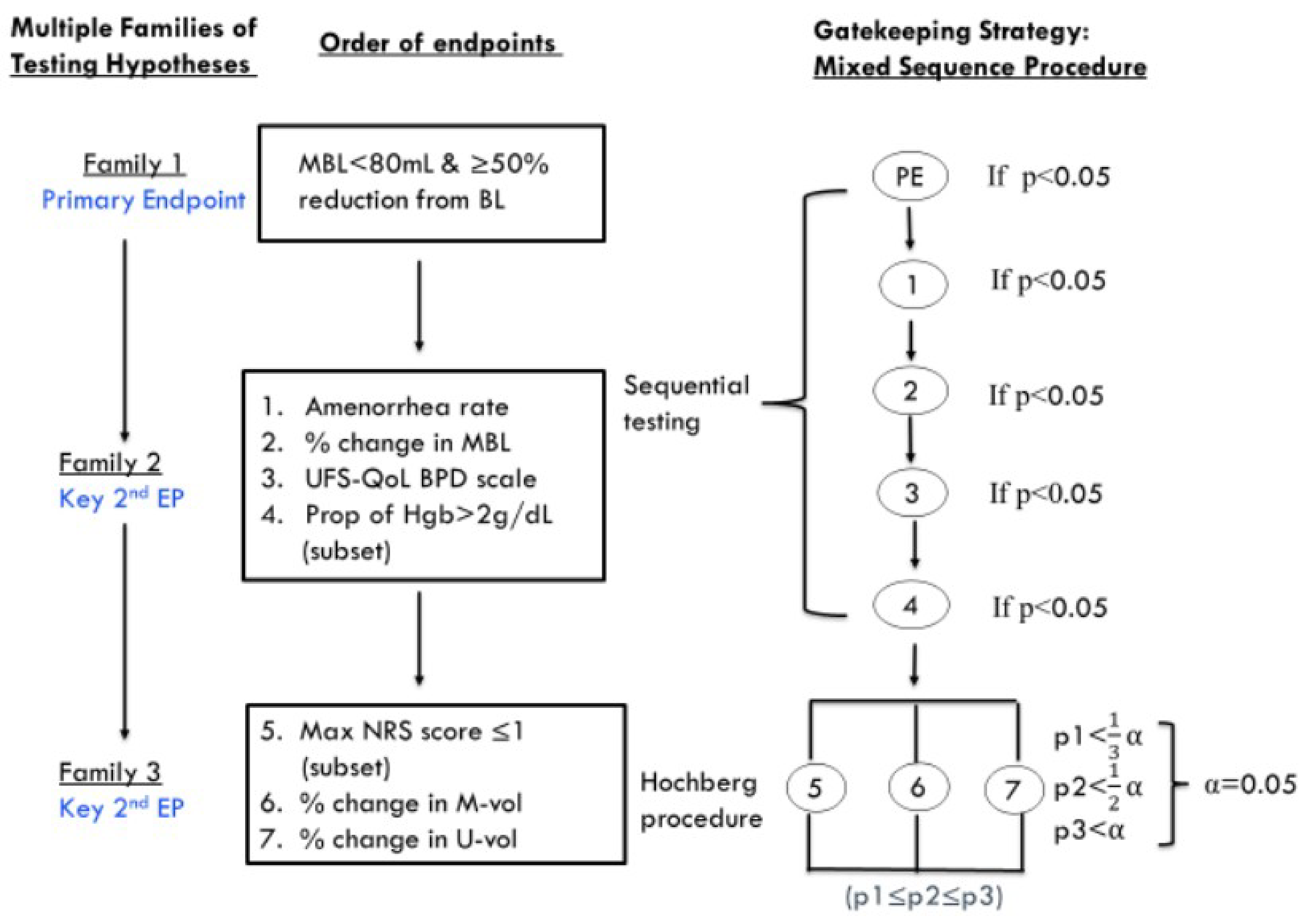
BL = baseline; BPD = bleeding and pelvic discomfort; EP = end point; Hgb = hemoglobin; MBL = menstrual blood loss; M-vol = myoma volume; NRS = numerical rating scale; PE = primary end point; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; U-vol = uterine volume.
Source: LIBERTY 1 trial and LIBERTY 2 trial Statistical Analysis Plan.49
Figure 2: LIBERTY 2 Trial Mixed-Sequence Testing Procedure for Primary and Key Secondary End Points
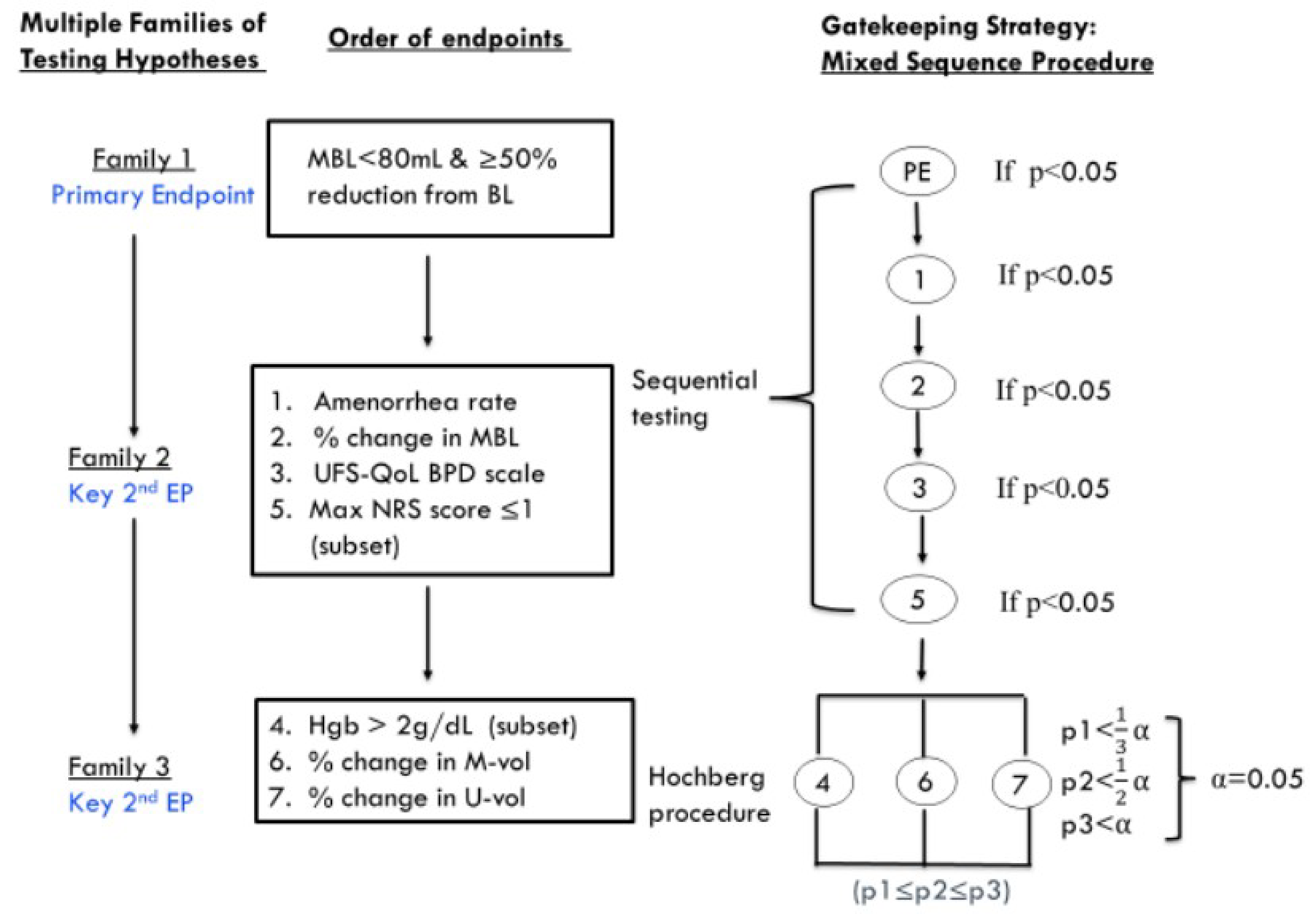
BL = baseline; BPD = bleeding and pelvic discomfort; EP = end point; Hgb = hemoglobin; max = maximum; MBL = menstrual blood loss; M-vol = myoma volume; NRS = numerical rating scale; PE = primary end point; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; U-vol = uterine volume.
Source: LIBERTY 1 trial and LIBERTY 2 trial Statistical Analysis Plan.49
Table 8: Statistical Analysis of Efficacy End Points for the LIBERTY 1 and LIBERTY 2 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end point: proportion of patients in the RGX-E2-NETA group vs. the placebo group who experienced an MBL volume of < 80 mL and ≥ 50% reduction from baseline MBL volume over the last 35 days of treatment, as measured using the alkaline hematin method (responder ratea) | CMH | Stratified by the randomization stratification factors: geographic region (North America vs. rest of world) and baseline MBL volume (< 225 mL vs. ≥ 225 mL) | Responder rate Patients who had a treatment exposure of < 4 weeks were imputed as nonresponders. For patients who had a treatment exposure of ≥ 4 weeks:
For imputing missing data, an MMRM of MBL volume at multiple time points was fitted to predict percentage change in MBL volume through fixed effects for the covariates (stratification factors, visit, treatment, and visit by treatment interaction) and a random effect for individual patient. | 3 sensitivity analyses evaluated the impact of using different data-handling rules:
3 sensitivity analyses assessed the potential impact of unvalidated feminine product use:
|
Proportion of patients with a hemoglobin level of 10.5 g/dL or lower at baseline who experienced an increase of ≥ 2 g/dL from baseline at week 24 | CMH | Same as primary end point | No imputation | None |
Proportion of patients who experienced a maximum NRS score ≤ 1 for UF-associated pain in the subset of patients with a maximum pain score ≥ 4 and who completed the daily e-diary during the 35 days before randomization | CMH | Same as primary end point | The maximum score was calculated as the maximum of all nonmissing scores. | None |
Change from baseline to week 24 in UFS-QoL SSS score | MMRM | Treatment, visit, randomization stratification factors, and treatment-by-visit interactions were included as fixed effects. Baseline value was included as a covariate. An unstructured variance-covariance matrix was assumed. | If < 50% of the scale items were missing, the scale was retained using the mean scale score of the items present. If ≥ 50% of the items were missing, no scale score was calculated; the subscale score was considered missing. | None |
Change from baseline to week 24 in UFS-QoL HRQoL total score | MMRM | Treatment, visit, randomization stratification factors, and treatment-by-visit interactions were included as fixed effects. Baseline value was included as a covariate. An unstructured variance-covariance matrix was assumed. | If < 50% of the scale items were missing, the scale was retained using the mean scale score of the items present. If ≥ 50% of the items were missing, no scale score was calculated; the subscale score was considered missing. | None |
CMH = Cochran-Mantel-Haenszel; FP = feminine product; HRQoL = health-related quality of life; MBL = menstrual blood loss; MMRM = mixed-effects model for repeated measures; NRS = numerical rating scale; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
aDefined as the proportion of patients who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL volume over the preceding 35 days of treatment.
bDefined as patients who had an e-diary entry rate of at least 70%, no more than 3 consecutive days of bleeding with product use, and no more than 5 days’ total of bleeding or spotting with product use reported in the e-diary over the week 24 or end-of-treatment visit window.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports, LIBERTY 1 trial and LIBERTY 2 trial Statistical Analysis Plan, and sponsor’s Summary of Clinical Evidence.12,13,19,49 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The analyzed populations are defined in Table 9.
Table 9: Analysis Populations of the LIBERTY 1 and LIBERTY 2 Trials
Study | Population | Definition | Application |
|---|---|---|---|
LIBERTY 1 and LIBERTY 2 trials | mITT | All randomized patients who received any amount of study drug | Efficacy analyses unless otherwise specified |
PP | Patients from the mITT population who did not have any of the specified subset of important protocol deviations | Sensitivity analysis of the primary efficacy end point | |
Safety | Same as mITT | Safety analyses unless otherwise specified |
mITT = modified intention to treat; PP = per protocol.
Source: Sponsor’s Summary of Clinical Evidence.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition is summarized in Table 10. In the LIBERTY 1 trial, 1,891 of 2,279 individuals were screened out; in the LIBERTY 2 trial, 2,517 of 2,899 individuals were screened out. The most common reasons for being screened out included not meeting the eligibility criteria (80.1% of patients in the LIBERTY 1 trial and 83.2% of patients in the LIBERTY 2 trial) and withdrawn consent (12.0% of patients in the LIBERTY 1 trial and 10.1% of patients in the LIBERTY 2 trial). In the LIBERTY 1 trial, 128 patients were randomized to each of the RGX-E2-NETA and placebo groups. The proportions of patients who discontinued from the study were similar between the groups, with 21.9% leaving the RGX-E2-NETA group and 17.2% leaving the placebo group. The most common reason for discontinuation from either treatment group was withdrawal by the patient (7.8% and 5.5% in the RGX-E2-NETA and placebo groups, respectively), with all reasons being generally balanced. Fewer patients in the RGX-E2-NETA group (60.9%) than the placebo group (68.5%) enrolled in the LIBERTY OLE study. In the LIBERTY 2 trial, 126 patients were randomized to receive RGX-E2-NETA, and 129 patients were randomized to receive placebo. A similar proportion of patients discontinued from either group: 18.3% discontinued from RGX-E2-NETA and 20.9% discontinued from placebo. The most common reasons for discontinuation were withdrawal by patient (10.3% of patients and 4.7% of patients in the RGX-E2-NETA and placebo groups, respectively) and loss to follow-up (3.2% of patients and 5.4% of patients in the RGX-E2-NETA and placebo groups, respectively). A greater proportion of patients in the RGX-E2-NETA group (67.5%) than in the placebo group (59.7%) enrolled in the LIBERTY OLE study.
Across the studies, patient disposition was generally similar, except for the proportions of patients who enrolled in the LIBERTY OLE study (as described previously).
Table 10: Summary of Patient Disposition From the LIBERTY 1 and LIBERTY 2 Trials
Patient disposition | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA | Placebo | RGX-E2-NETA | Placebo | |
Screened, N | 2,279 | 2,899 | ||
Screened out | 1,891 (83.0) | 2,517 (86.8) | ||
Reason for screening out, n (%) | ||||
Did not meet criteria | 1,531 (81.0) | 2,093 (83.2) | ||
Withdrew consent | 226 (12.0) | 253 (10.1) | ||
Adverse event | 2 (0.1) | 0 | ||
Other | 132 (7.0) | 171 (6.8) | ||
Randomized, N | 128 | 128 | 126 | 129 |
Completed study, n (%) | 100 (78.1) | 105 (82.0) | 102 (81.0) | 102 (79.1) |
Discontinued from study, n (%) | 28 (21.9) | 22 (17.2) | 23 (18.3) | 27 (20.9) |
Reason for discontinuation, n (%) | ||||
Withdrawal by patient | 10 (7.8) | 7 (5.5) | 13 (10.3) | 6 (4.7) |
Adverse event | 7 (5.5) | 5 (3.9) | 2 (1.6) | 6 (4.7) |
Lack of efficacy | 4 (3.1) | 3 (2.3) | 2 (1.6) | 1 (0.8) |
Lost to follow-up | 1 (0.8) | 5 (3.9) | 4 (3.2) | 7 (5.4) |
Protocol deviation | 1 (0.8) | 0 | 1 (0.8) | 1 (0.8) |
Pregnancy | 0 | 1 (0.8) | 0 | 1 (0.8) |
Other | 5 (3.9) | 1 (0.8) | 1 (0.8) | 5 (3.9) |
Enrolled in LIBERTY OLE study, n (%) | 78 (60.9) | 87 (68.0) | 85 (67.5) | 77 (59.7) |
mITT, N (%) | 128 (100.0) | 127a (99.2) | 125b (99.2) | 129 (100.0) |
PP, N (%) | 119 (93.0) | 121 (94.5) | 113 (89.7) | 122 (94.6) |
Safety, N (%) | 128 (100.0) | 127a (99.2) | 126c (100.0) | 129 (100.0) |
E2-NETA = estradiol and norethindrone acetate; mITT = modified intention to treat; NETA = norethindrone acetate; OLE = open-label extension; PP = per protocol; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate.
aOne patient who was randomized to the placebo group did not receive the study drug and was excluded from the mITT and safety populations.
bOne patient was excluded from the RGX-E2-NETA group of the mITT population because the patient was randomized in error before eligibility was confirmed and did not receive treatment.
cOne patient was randomized to RGX plus delayed E2-NETA (not reported here) but received RGX-E2-NETA. This patient was included in the RGX-E2-NETA group for safety analyses.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
Baseline characteristics are summarized in Table 11 and are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. All patients were female, and the mean age across the studies was approximately 42 years. For both studies, the proportion of patients who were Black was lower in the RGX-E2-NETA groups than in the placebo groups (46.1% versus 51.2% in the LIBERTY 1 trial; 49.6% versus 57.4% in the LIBERTY 2 trial). Conversely, the proportion of patients who were white was higher in the RGX-E2-NETA groups than in the placebo groups (50.0% versus 44.1% in the LIBERTY 1 trial; 46.4% versus 38.0% in the LIBERTY 2 trial). Proportions of other races and ethnicities were generally balanced between treatment groups and across the studies. Baseline mean MBL volume ranged from 218.8 mL to 239.4 mL in the LIBERTY 1 trial and from 211.8 mL to 246.7 mL in the LIBERTY 2 trial; volumes were higher in the RGX-E2-NETA groups than in the placebo groups. Mean hemoglobin concentration was similar across the treatment groups in both studies. However, more patients in the RGX-E2-NETA group (32.1%) of the LIBERTY 1 trial had a hemoglobin concentration of less than 10.5 g/dL compared to the placebo group (22.0%), and fewer patients in the RGX-E2-NETA group (30.5%) had a hemoglobin concentration from 10.5 g/dL to 12 g/dL compared to the placebo group (44.1%). The proportions of patients with a hemoglobin concentration of less than 10 g/dL were generally similar between treatment groups in the LIBERTY 2 trial; however, there were more patients in the RGX-E2-NETA group (36.8%) with a hemoglobin concentration of 12 g/dL or more compared to the placebo group (27.1%). The proportion of patients reporting a baseline maximum NRS score of 4 points or more was lower in the RGX-E2-NETA group (65.6%) than in the placebo group (74.8%) in the LIBERTY 1 trial.
Table 11: Summary of Baseline Characteristics From the LIBERTY 1 and LIBERTY 2 Trials — mITT Population
Characteristic | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA (N = 128) | Placebo (N = 127) | RGX-E2-NETA (N = 125) | Placebo (N = 129) | |
Demographic characteristics | ||||
Age (years), mean (SD) | 42.5 (5.0) | 42.2 (5.7) | 42.4 (5.4) | 41.8 (5.3) |
Race and ethnicity, n (%) | ||||
American Indian or Alaska Native | 2 (1.6) | 1 (0.8) | 0 | 1 (0.8) |
Asian | 0 | 2 (1.6) | 0 | 1 (0.8) |
Black | 59 (46.1) | 65 (51.2) | 62 (49.6) | 74 (57.4) |
White | 64 (50.0) | 56 (44.1) | 58 (46.4) | 49 (38.0) |
Other race or ethnicity | 2 (1.6) | 1 (0.8) | 1 (0.8) | 3 (2.3) |
Multiple races or ethnicities | 1 (0.8) | 2 (1.6) | 1 (0.8) | 0 |
Not reported | 0 | 0 | 3 (2.4) | 1 (0.8) |
Geographic region, n (%) | ||||
North America | 98 (76.6) | 98 (77.2) | 93 (74.4) | 96 (74.4) |
Rest of world | 30 (23.4) | 29 (22.8) | 32 (25.6) | 33 (25.6) |
Clinical characteristics | ||||
BMI (kg/m2), mean (SD) | 31.4 (7.6) | 32.3 (7.5) | 31.0 (6.6) | 32.1 (7.6) |
BMD (g/cm2) lumbar spine, mean (SD) | 1.2 (0.2) | 1.2 (0.2) | 1.2 (0.2) | 1.2 (0.2) |
MBL | ||||
Mean volume (mL) (SD) | 239.4 (180.3) | 218.8 (125.0) | 246.7 (186.0) | 211.8 (129.9) |
< 225 mL, n (%) | 84 (65.6) | 85 (66.9) | 80 (64.0) | 86 (66.7) |
≥ 225 mL, n (%) | 44 (34.4) | 42 (33.1) | 45 (36.0) | 43 (33.3) |
Hemoglobin concentration | ||||
Mean (g/dL) (SD) | 11.2 (1.6) | 11.4 (1.4) | 11.3 (1.5) | 11.1 (1.6) |
< 8 g/dL, n (%) | 2 (1.6) | 0 | 1 (0.8) | 5 (3.9) |
≥ 8 g/dL to < 10.5 g/dL, n (%) | 39 (30.5) | 28 (22.0) | 38 (30.4) | 41 (31.8) |
≥ 10.5 g/dL to < 12 g/dL, n (%) | 39 (30.5) | 56 (44.1) | 40 (32.0) | 48 (37.2) |
≥ 12 g/dL, n (%) | 48 (37.5) | 43 (33.9) | 46 (36.8) | 35 (27.1) |
Maximum NRS score for UF-associated pain, n (%) | ||||
< 4 | 43 (33.6) | 31 (24.4) | 30 (24.0) | 31 (24.0) |
≥ 4 | 84 (65.6) | 95 (74.8) | 93 (74.4) | 95 (73.6) |
Missing | 1 (0.8) | 1 (0.8) | 2 (1.6) | 3 (2.3) |
UFS-QoL SSS score, mean (SD) | 55.7 (20.5) | 61.1 (19.0) | 59.9 (22.1) | 60.1 (19.6) |
UFS-QoL HRQoL total score, mean (SD) | 23.0 (18.6) | 22.2 (19.3) | 24.9 (22.4) | 24.9 (20.6) |
BMD = bone mineral density; BMI = body mass index; HRQoL = health-related quality of life; MBL = menstrual blood loss; mITT = modified intention to treat; NRS = numerical rating scale; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SD = standard deviation; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Patient exposure to study treatments is summarized in Table 12. Durations of treatment exposure were similar across the studies, ranging from 22.0 weeks to 23.3 weeks (SD = 6.2 weeks to 7.4 weeks). Because patients received RGX (or its placebo) and E2-NETA (or its placebo) as separate pills, adherence was calculated based on each component. Adherence to the study drug ranged from 97.8% to 100.9% (SD = 5.1% to 25.8%) across the studies.
Table 12: Summary of Patient Exposure in the LIBERTY 1 and LIBERTY 2 Trials — Safety Population
Exposure | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA (N = 128) | Placebo (N = 127) | RGX-E2-NETA (N = 126) | Placebo (N = 129) | |
Duration (weeks), mean (SD) | 22.0 (7.4) | 23.3 (6.5) | 22.7 (6.6) | 23.0 (6.2) |
Adherence (%), mean (SD)a | ||||
RGX (or placebo of RGX) | 100.1 (13.2) | 100.9 (25.7) | 97.8 (5.5) | 98.7 (10.6) |
E2-NETA (or placebo of E2-NETA) | 100.1 (13.2) | 100.9 (25.8) | 97.9 (5.1) | 98.7 (10.5) |
E2-NETA = estradiol and norethindrone acetate; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol-norethindrone acetate; SD = standard deviation.
aPatients received RGX with commercially available E2-NETA in a copackaged configuration, or placebo of RGX and placebo of E2-NETA. To determine adherence from pill counts, each component was counted separately (i.e., RGX or placebo of RGX, and E2-NETA or placebo of E2-NETA).
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19
Concomitant Medications
Concomitant medications reported in more than 5% of patients in any treatment group are summarized in Table 13. More than 94% of patients in any treatment group reported some use of concomitant medication during the LIBERTY 1 trial and the LIBERTY 2 trial. There was an imbalance in ibuprofen use across groups in the LIBERTY 1 trial. Fifty-nine percent of patients in the RGX-E2-NETA group and 72.4% in the placebo group used ibuprofen during the study.
Table 13: Summary of Concomitant Medications From the LIBERTY 1 and LIBERTY 2 Trials — Safety Population
Concomitant medication | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA (N = 128) | Placebo (N = 127) | RGX-E2-NETA (N = 126) | Placebo (N = 129) | |
Received concomitant treatment, n (%) | 125 (97.7) | 120 (94.5) | 120 (95.2) | 122 (94.6) |
Concomitant treatment, n (%)a | ||||
Ibuprofen | 76 (59.4) | 92 (72.4) | 80 (63.5) | 81 (62.8) |
Ferrous sulphate | 36 (28.1) | 32 (25.2) | 35 (27.8) | 35 (27.1) |
Paracetamol | 23 (18.0) | 26 (20.5) | 22 (17.5) | 32 (24.8) |
Iron | 22 (17.2) | 18 (14.2) | 25 (19.8) | 23 (17.8) |
Cholecalciferol | 13 (10.2) | 15 (11.8) | 10 (7.9) | 12 (9.3) |
Hydrochlorothiazide | 13 (10.2) | 9 (7.1) | 7 (5.6) | 7 (5.4) |
Vitamins NOS | 11 (8.6) | 22 (17.3) | 13 (10.3) | 16 (12.4) |
Vitamin D NOS | 9 (7.0) | 19 (15.0) | 16 (12.7) | 15 (11.6) |
Lisinopril | 9 (7.0) | 4 (3.1) | 0 | 0 |
Naproxen sodium | 8 (6.3) | 10 (7.9) | 7 (5.6) | 5 (3.9) |
Ascorbic acid | 8 (6.3) | 5 (3.9) | 3 (2.4) | 4 (3.1) |
Ferric sodium gluconate complex | 7 (5.5) | 5 (3.9) | 1 (0.8) | 2 (1.6) |
Phentermine | 7 (5.5) | 3 (2.4) | 2 (1.6) | 2 (1.6) |
Naproxen | 6 (4.7) | 15 (11.8) | 9 (7.1) | 7 (5.4) |
Loratadine | 3 (2.3) | 5 (3.9) | 7 (5.6) | 4 (3.1) |
Vitamin B12 NOS | 3 (2.3) | 2 (1.6) | 3 (2.4) | 11 (8.5) |
Amoxicillin | 2 (1.6) | 8 (6.3) | 5 (4.0) | 2 (1.6) |
Calcium carbonate | 2 (1.6) | 7 (5.5) | 2 (1.6) | 1 (0.8) |
Tramadol | 2 (1.6) | 2 (1.6) | 7 (5.6) | 4 (3.1) |
Metronidazole | 2 (1.6) | 2 (1.6) | 1 (0.8) | 7 (5.4) |
NOS = nitric oxide supplement; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate.
aReported in more than 5% of patients.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Protocol Deviations
In the mITT population in the LIBERTY 1 trial, 31.3% of patients in the RGX-E2-NETA group and 28.3% of patients in the placebo group had an important protocol deviation. In both groups, the most common protocol deviation was for missing key study procedures (18.0% in the RGX-E2-NETA group and 18.9% in the placebo group), such as missed endometrial biopsy (9.4% and 5.5%, respectively) and DXA not performed (1.6% and 6.3%, respectively). Aside from those described, the frequencies of reported deviations were generally balanced between the treatment groups.
From the mITT population in the LIBERTY 2 trial, 30.4% of patients in the RGX-E2-NETA group and 31.0% of patients in the placebo group had an important protocol deviation. In both groups, the most common protocol deviation was for missing key study procedures (17.6% in the RGX-E2-NETA group and 22.5% in the placebo group). Missed endometrial biopsy (12.0% and 11.6%, respectively) was the most frequently reported procedure. The frequencies of reported deviations were generally balanced between the treatment groups.
Efficacy
Key efficacy results are summarized in Table 14.
Responder Rate for Change in MBL Volume
Response to treatment was defined as having an MBL volume of less than 80 mL and a 50% or greater reduction from baseline in MBL volume over the preceding 35 days of treatment as measured using the alkaline hematin method.
In the LIBERTY 1 trial, 73.4% of patients (95% CI, 64.9% to 80.9%) who received RGX-E2-NETA compared to 18.9% of patients (95% CI, 12.5% to 26.8%) who received placebo responded to treatment. The treatment difference in the responder rate for change in MBL volume at week 24 was 54.5% (95% CI, 44.3% to 64.7%; P < 0.0001) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, 71.2% of patients (95% CI, 62.4% to 79.0%) who received RGX-E2-NETA compared to 14.7% of patients (95% CI, 9.1% to 22.0%) who received placebo responded to treatment. The treatment difference in the responder rate for change in MBL volume at week 24 was 56.5% (95% CI, 46.6% to 66.5%; P < 0.0001) in favour of RGX-E2-NETA.
When looking at the individual components that made up the response definition, in both studies, similar proportions of patients met each of the criteria necessary for a response.
Results of the 6 sensitivity analyses were similar to and supported those of the primary analysis for both the LIBERTY 1 trial and the LIBERTY 2 trial.
Improvement of Anemia
For the subset of patients who had a hemoglobin level of 10.5 g/dL or lower at baseline, response was defined as an increase of 2 g/dL or more at week 24.
In the LIBERTY 1 trial, of the 30 patients and 23 patients who were in the subset from the RGX-E2-NETA and placebo groups, respectively, 15 patients (50.0%) and 5 patients (21.7%), respectively, experienced an increase of 2 g/dL or more in their hemoglobin levels. The treatment difference in the proportion of patients who experienced an improvement in anemia at week 24 was 28.3% (95% CI, 3.7% to 52.8%; P = 0.0377) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, of the 31 patients and 37 patients who were in the subset from the RGX-E2-NETA and placebo groups, respectively, 19 patients (61.3%) and 2 patients (5.4%), respectively, experienced an increase of 2 g/dL or more in their hemoglobin levels. The treatment difference in the proportion of patients who experienced an improvement in anemia at week 24 was 55.9% (95% CI, 37.3% to 74.5%; P < 0.0001) in favour of RGX-E2-NETA.
Improvement of Pain
For the subset of patients who had a maximum NRS score of 4 points or greater during the 35 days before randomization, response to treatment was defined as a maximum NRS score of 0 points or 1 point for UF-associated pain over the last 35 days of treatment in this subset.
In the LIBERTY 1 trial, of the 84 patients and 95 patients who were in the subset from the RGX-E2-NETA and placebo groups, respectively, 33 patients (39.3%) and 11 patients (11.6%) patients, respectively, experienced a maximum NRS score of 0 points or 1 point for UF-associated pain. The treatment difference in the proportion of patients who experienced an improvement in pain to a maximum NRS score of 0 points or 1 point was 27.7% (95% CI, 15.4% to 40.0%; P < 0.0001) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, of the 93 patients and 95 patients who were in the subset from the RGX-E2-NETA and placebo groups, respectively, 34 patients (36.6%) and 17 patients (17.9%), respectively, experienced a maximum NRS score of 0 points or 1 point for UF-associated pain. The treatment difference in the proportion of patients who experienced an improvement in pain to a maximum NRS score of 0 points or 1 point was 18.7% (95% CI, 6.2% to 31.1%; P = 0.0056) in favour of RGX-E2-NETA.
Change in UFS-QoL SSS Score
In the LIBERTY 1 trial, the LSM changes from baseline to week 24 in UFS-QoL SSS score were –30.9 points (95% CI, –36.1 to –25.8 points) and –10.5 points (95% CI, –15.5 points to –5.5 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL SSS score was –20.4 points (95% CI, –27.1 points to –13.7 points) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, the LSM changes from baseline to week 24 in UFS-QoL SSS score were –36.1 points (95% CI, –41.2 points to –31.0 points) and –13.7 points (95% CI, –18.8 points to –8.5 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL SSS score was –22.4 points (95% CI, –29.4 to –15.4 points) in favour of RGX-E2-NETA.
Change in the UFS-QoL HRQoL Total Score
In the LIBERTY 1 trial, the LSM changes from baseline to week 24 in UFS-QoL HRQoL total score were 38.0 points (95% CI, 32.4 points to 43.5 points) and 12.8 points (95% CI, 7.5 points to 18.2 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL HRQoL total score was 25.1 points (95% CI, 17.9 points to 32.3 points) in favour of RGX-E2-NETA.
In the LIBERTY 2 trial, the LSM changes from baseline to week 24 in UFS-QoL HRQoL total score were 37.8 points (95% CI, 32.9 points to 42.6 points) and 13.8 points (95% CI, 8.9 points to 18.8 points) for patients in the RGX-E2-NETA and placebo groups, respectively. The treatment difference in the LSM change from baseline to week 24 in UFS-QoL HRQoL total score was 23.9 points (95% CI, 17.2 points to 30.7 points) in favour of RGX-E2-NETA.
Table 14: Summary of Key Efficacy Results From the LIBERTY 1 and LIBERTY 2 Trials — mITT Population
Variable | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA (N = 128) | Placebo (N = 127) | RGX-E2-NETA (N = 125) | Placebo (N = 129) | |
Proportion of patients who experienced an MBL volume of < 80 mL and a ≥ 50% reduction from baseline MBL volume | ||||
Responders, n | 94 | 24 | 89 | 19 |
Proportion of responders (95% CI)a | 73.4 (64.9 to 80.9) | 18.9 (12.5 to 26.8) | 71.2 (62.4 to 79.0) | 14.7 (9.1 to 22.0) |
Treatment group difference vs. control, % (95% CI)b | 54.5 (44.3 to 64.7) | 56.5 (46.6 to 66.5) | ||
P valuec,d | < 0.0001 | < 0.0001 | ||
Patients with MBL volume of < 80 mL over the last 35 days of treatment, n (%) | 97 | 34 | 92 | 25 |
Proportion of patients (95% CI)a | 75.8 (67.4 to 82.9) | 26.8 (19.3 to 35.4) | 73.6 (65.0 to 81.1) | 19.4 (13.0 to 27.3) |
Treatment group difference vs. control, % (95% CI)e | 49.0 (38.3 to 59.7) | 54.2 (43.9 to 64.5) | ||
Patients with ≥ 50% reduction from baseline over the last 35 days of treatment, n (%) | 101 | 28 | 96 | 28 |
Proportion of patients (95% CI)a | 78.9 (70.8 to 85.6) | 22.1 (15.2 to 30.3) | 76.8 (68.4 to 83.9) | 21.7 (14.9 to 29.8) |
Treatment group difference vs. control, % (95% CI)e | 56.9 (46.8 to 67.0) | 55.1 (44.8 to 65.4) | ||
Proportion of patients who experienced an increase of > 2 g/dL from baseline at week 24 in the subset of patients with a hemoglobin level of ≤ 10.5 g/dL at baseline | ||||
Number of patients contributing to the week 24 analysis, n (%) | 30 (23.4) | 23 (18.1) | 31 (24.8) | 37 (28.7) |
Responder rate, n (%) | 15 (50.0) | 5 (21.7) | 19 (61.3) | 2 (5.4) |
Treatment group difference vs. control, % (95% CI)e | 28.3 (3.7 to 52.8) | 55.9 (37.3 to 74.5) | ||
P valued,f | 0.0377 | < 0.0001 | ||
Proportion of patients who experienced a maximum NRS score of ≤ 1 for UF-associated pain over the last 35 days of treatment in the subset of patients with a maximum pain score of ≥ 4 during the 35 days before randomization | ||||
Number of patients with a maximum NRS score ≥ 4 at baseline, n (%) | 84 (65.6) | 95 (74.8) | 93 (74.4) | 95 (73.6) |
Number of patients who experienced maximum NRS score ≤ 1, n (%)a | 33 (39.3) | 11 (11.6) | 34 (36.6) | 17 (17.9) |
Treatment group difference vs. control, % (95% CI)e | 27.7 (15.4 to 40.0) | 18.7 (6.2 to 31.1) | ||
P valuec,d | < 0.0001 | 0.0056 | ||
Change from baseline to week 24 in UFS-QoL SSS score | ||||
Number of patients contributing to the week 24 analysis, n (%) | 97 (75.8) | 103 (81.1) | 96 (76.8) | 95 (73.6) |
Baseline (points), LSM (95% CI) | 55.1 (51.3 to 58.9) | 60.3 (56.5 to 64.1) | 59.1 (55.3 to 63.0) | 59.2 (55.4 to 63.0) |
Week 24 (points), LSM (95% CI) | 23.4 (19.1 to 27.8) | 49.2 (44.9 to 53.4) | 21.7 (17.0 to 26.4) | 45.1 (40.4 to 49.8) |
Change from baseline (points), LSM (95% CI) | –30.9 (–36.1 to –25.8) | –10.5 (–15.5 to –5.5) | –36.1 (–41.2 to –31.0) | –13.7 (–18.8 to –8.5) |
Treatment group difference vs. control (points), LSM (95% CI)g | –20.4 (–27.1 to –13.7) | –22.4 (–29.4 to –15.4) | ||
P valueg,h | < 0.0001 | < 0.0001 | ||
Change from baseline to week 24 in UFS-QoL HRQoL total score | ||||
Number of patients contributing to the week 24 analysis, n (%) | 97 (75.8) | 103 (81.1) | 96 (76.8) | 95 (73.6) |
Baseline (points), LSM (95% CI) | 37.2 (33.4 to 41.0) | 33.5 (29.7 to 37.3) | 38.9 (34.9 to 43.0) | 37.3 (33.3 to 41.3) |
Week 24 (points), LSM (95% CI) | 74.0 (69.3 to 78.7) | 44.9 (40.2 to 49.5) | 78.7 (73.8 to 83.5) | 51.0 (46.1 to 55.9) |
Change from baseline (points), LSM (95% CI) | 38.0 (32.4 to 43.5) | 12.8 (7.5 to 18.2) | 37.8 (32.9 to 42.6) | 13.8 (8.9 to 18.8) |
Treatment group difference vs. control (points), LSM (95% CI)i | 25.1 (17.9 to 32.3) | 23.9 (17.2 to 30.7) | ||
P valueh,i | < 0.0001 | < 0.0001 | ||
CI = confidence interval; HRQoL = health-related quality of life; LSM = least squares mean; MBL = menstrual blood loss; mITT = modified intention to treat; NRS = numerical rating scale; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
aBased on exact binomial 95% CI (Clopper-Pearson).
bAdjusted 95% CI adjusted for baseline MBL volume (< 225 mL, ≥ 225 mL) and geographic region (North America, rest of world).
cP value based on Cochran-Mantel-Haenszel test stratified by baseline MBL volume (< 225 mL or ≥ 225 mL) and geographic region (North America or rest of world).
dP value has been adjusted for multiple testing.
eThe 95% CI for difference is based on the normal approximation.
fP value is based on Cochran-Mantel-Haenszel test stratified by baseline MBL volume (< 225 mL, ≥ 225 mL).
gThe LSM and P value are based on a mixed-effect model, with treatment, visit, region, baseline MBL, and treatment-by-visit interaction included as fixed effects. The multiple visits for each patient were the repeated measures as random effect within each patient and an unstructured covariance. The transformed score ranges from 0 to 100 based on a Likert scale (none of the time, a little of the time, some of the time, most of the time, and all the time). Lower scores indicate lower symptom severity, and higher scores indicate greater symptom severity.
hP value has not been adjusted for multiple testing.
iThe LSM and P value are based on a mixed-effect model, with treatment, visit, region, baseline MBL, and treatment-by-visit interaction included as fixed effects. The multiple visits for each patient were the repeated measures as random effect within each patient and an unstructured covariance. The transformed score ranges from 0 to 100 based on a Likert scale (none of the time, a little of the time, some of the time, most of the time, and all the time). Higher scores are indicative of better HRQoL (high score = good).
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Harms are summarized in Table 15.
Adverse Events
In the LIBERTY 1 trial, 61.7% of patients who received RGX-E2-NETA and 66.1% of patients who received placebo had a TEAE. Headache (10.9% of patients in the RGX-E2-NETA group versus 15.0% of patients in the placebo group) and hot flush (10.9% of patients in the RGX-E2-NETA group versus 7.9% of patients in the placebo group) were the most common TEAEs.
In the LIBERTY 2 trial, 60.3% of patients who received RGX-E2-NETA and 58.9% of patients who received placebo had a TEAE. Headache (8.7% of patients in the RGX-E2-NETA group versus 11.6% of patients in the placebo group) was the most common TEAE.
In general, the types and frequencies of TEAEs were similar between the treatment groups for both studies and across studies.
Serious Adverse Events
In the LIBERTY 1 trial, 7 patients (5.5%) who received RGX-E2-NETA reported 10 SAEs (ankle fracture, avulsion fracture, hematemesis, hypothyroidism, menorrhagia, pelvic pain, rhabdomyolysis, uterine leiomyoma, uterine myoma expulsion, and vitreous detachment), and 2 patients (1.6%) who received placebo reported 2 SAEs (acute psychosis and pneumonia).
In the LIBERTY 2 trial, 1 patient (0.8%) who received RGX-E2-NETA reported 1 SAE (cholecystitis), and 4 patients (3.1%) who received placebo reported 5 SAEs (anemia, necrotising fasciitis, radius fracture, road traffic accident, and syncope).
Withdrawals Due to Adverse Events
In the LIBERTY 1 trial, 7 patients (5.5%) who received RGX-E2-NETA stopped treatment due to 12 TEAEs (asthenia, paresthesia, metrorrhagia, increased alanine aminotransferase, anxiety, fatigue, headache, hot flush, irritability, menorrhagia, muscle twitching, and vomiting), and 5 patients (3.9%) who received placebo stopped treatment due to 13 TEAEs (asthenia, paresthesia, alopecia, constipation, depressed mood, muscular weakness, pelvic pain, pollakiuria, pruritus [generalized], urticaria, vitreous floaters, vulvovaginal burning sensation, and vulvovaginal pruritus).
In the LIBERTY 2 trial, 3 patients (2.4%) who received RGX-E2-NETA stopped treatment due to 3 TEAEs (metrorrhagia, breast cyst, and mood swings), and 6 patients (4.7%) who received placebo stopped treatment due to 7 TEAEs (anemia, arthralgia, gastrointestinal pain, muscle spasms, necrotising fasciitis, pain in extremity, and vaginal discharge).
Mortality
There were no deaths reported in either study.
Notable Harms
According to the clinical expert consulted on this review, loss of BMD was noted as a TEAE of special interest for treatment with RGX-E2-NETA. In the LIBERTY 1 trial, the LSM changes from baseline to week 24 in BMD at the lumbar spine were –0.36% (95% CI, –0.93% to 0.22%) among patients who received RGX-E2-NETA and 0.05% (95% CI, –0.52% to 0.62%) among patients who received placebo. The treatment difference between RGX-E2-NETA and placebo in mean change from baseline to week 24 in BMD at the lumbar spine was –0.41% (95% CI, –1.16% to 0.34%).
In the LIBERTY 2 trial, the LSM changes from baseline to week 24 in BMD at the lumbar spine were –0.13% (95% CI, –0.71% to 0.46%) among patients who received RGX-E2-NETA and 0.32% (95% CI, –0.26% to 0.89%) among patients who received placebo. The treatment difference between RGX-E2-NETA and placebo in mean change from baseline to week 24 in BMD at the lumbar spine was –0.44% (95% CI, –1.23% to 0.34%).
Table 15: Summary of Harms Results From the LIBERTY 1 and LIBERTY 2 Trials — Safety Population
Adverse event | LIBERTY 1 trial | LIBERTY 2 trial | ||
|---|---|---|---|---|
RGX-E2-NETA (N = 128) | Placebo (N = 127) | RGX-E2-NETA (N = 126) | Placebo (N = 129) | |
Most common TEAEs, n (%)a | ||||
Patients with ≥ 1 TEAE | 79 (61.7) | 84 (66.1) | 76 (60.3) | 76 (58.9) |
Headache | 14 (10.9) | 19 (15.0) | 11 (8.7) | 15 (11.6) |
Hot flush | 14 (10.9) | 10 (7.9) | 7 (5.6) | 5 (3.9) |
Hypertension | 7 (5.5) | 0 | 5 (4.0) | 4 (3.1) |
Irritability | 5 (3.9) | 0 | 1 (0.8) | 0 |
Nausea | 4 (3.1) | 6 (4.7) | 6 (4.8) | 10 (7.8) |
Anemia | 4 (3.1) | 6 (4.7) | 2 (1.6) | 8 (6.2) |
Nasopharyngitis | 2 (1.6) | 5 (3.9) | 4 (3.2) | 6 (4.7) |
Urinary tract infection | 1 (0.8) | 5 (3.9) | 2 (1.6) | 3 (2.3) |
Upper respiratory tract infection | 1 (0.8) | 3 (2.4) | 6 (4.8) | 7 (5.4) |
Cough | 1 (0.8) | 7 (5.5) | 0 | 4 (3.1) |
SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 7 (5.5) | 2 (1.6) | 1 (0.8) | 4 (3.1) |
Patients who stopped treatment due to TEAEs, n (%) | ||||
Patients who stopped | 7 (5.5) | 5 (3.9) | 3 (2.4) | 6 (4.7) |
Deaths, n (%) | ||||
Patients who died | 0 | 0 | 0 | 0 |
TEAEs of special interest, n (%) | ||||
BMD for lumbar spine at week 24 (g/cm2) | ||||
Number of patients contributing to analysis, n (%) | 100 (78.1) | 102 (80.3) | 95 (75.4) | 95 (73.6) |
Baseline, LSM (95% CI) | 1.19 (1.15 to 1.22) | 1.24 (1.21 to 1.27) | 1.23 (1.19 to 1.26) | 1.24 (1.20 to 1.27) |
Week 24 percentage change from baseline, LSM (95% CI) | –0.36 (–0.93 to 0.22) | 0.05 (–0.52 to 0.62) | –0.13 (–0.71 to 0.46) | 0.32 (–0.26 to 0.89) |
Treatment group difference vs. control, LSM (95% CI) | –0.41 (–1.16 to 0.34) | –0.44 (–1.23 to 0.34) | ||
BMD = bone mineral density; CI = confidence interval; LSM = least squares mean; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SAE = serious adverse event; TEAE = treatment-emergent adverse event; vs. = versus.
aOccurring in at least 5% of patients in any treatment group.
Source: LIBERTY 1 trial and LIBERTY 2 trial clinical study reports and sponsor’s Summary of Clinical Evidence.12,13,19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
In the LIBERTY 1 and LIBERTY 2 trials, randomization was performed using an interactive web response system and stratified by geographic region and mean screening MBL volume. Patients, investigators, and sponsor staff were blinded to treatment, and matching placebo was used. Based on baseline characteristics, there was a slight imbalance in the proportions of patients who were Black and white between the treatment groups for both studies (i.e., there was a lower proportion of patients who were Black and a higher proportion of patients who were white in the RGX-E2-NETA groups versus the placebo groups). Race and ethnicity are risk factors for developing UFs, and the incidence of UFs and occurrence of clinically relevant UFs are higher in patients who are Black.15 The mean MBL volume was higher in the RGX-E2-NETA group than in the placebo group for both studies. It is possible that the baseline imbalances could have biased the study results; however, the direction and magnitude of bias are unknown. The clinical expert consulted on this review did not expect the imbalances to meaningfully affect the interpretation of the results.
Of the patients who were randomized, 17% to 22% of patients discontinued from the studies; the most frequently reported reasons were patient withdrawal, TEAEs, and loss to follow-up. Sensitivity analyses tested the primary outcome using different data-handling methods, and the results were similar to and supportive of those for the primary analysis. The anemia and pain end points were based on subsets of patients who met the low hemoglobin and high pain criteria, respectively, at baseline, and whose disease improved (per the trial definitions). These criteria reduced the number of patients who contributed data to the anemia improvement outcome (21% and 27% of patients in the LIBERTY 1 trial and the LIBERTY 2 trial, respectively) and the pain improvement outcome (70% and 74% of patients, respectively). The magnitudes of the treatment effects were notably different between the treatment groups; however, the reasons for these differences are not clear. It may have been due to the small number of patients contributing data and/or because randomization was not stratified for these variables. As a result, there is an increased risk of bias due to a loss of prognostic balance, which increases the uncertainty in these results. For the UFS-QoL outcomes, data were missing for 24% and 19% of patients in the RGX-E2-NETA and placebo groups, respectively, in the LIBERTY 1 trial, and for 23% and 26% of patients, respectively, in the LIBERTY 2 trial. These end points were analyzed using a mixed-effects model for repeated measures, which assumes that data were missing at random and that patients for whom data were missing would continue to change similarly to those with ongoing data — an assumption that is not verifiable. Moreover, there were no sensitivity analyses to inform on how results may differ under different assumptions about the missing data. Data were also missing for the anemia, pain, and BMD outcomes (5% to 30%), leading to greater uncertainty in these results than in the MBL response. Considering the large difference in efficacy between the RGX-E2-NETA and placebo groups for the primary outcome, patients may have been able to infer which treatment they received in the study. If so, this would affect subjective outcomes like pain, UFS-QoL, and harms. The reasons for missing data for each outcome were not available for review. As a result, it is difficult to pinpoint the extent of bias and how it may have affected the interpretation of the results.
Both the diagnosis of UFs and the treatment response, defined by the primary end point, were centrally reviewed. According to the FDA, MBL volume (measured using the alkaline hematin method) is an established primary end point in clinical trials for assessing HMB.38 Patients who had anemia during screening were identified, and they started (or continued their pre-existing) iron therapy. While the iron supplementation protocol was consistent across the treatment groups and studies, it is unknown if improvement in anemia was a result of iron therapy or reduced MBL; information on changes in the amount of iron throughout the studies was not available to clarify this. The incidences of different types of anemia as a TEAE were lower in the RGX-E2-NETA groups versus the placebo groups in both studies (4 patients versus 6 patients, respectively, experienced anemia in the LIBERTY 1 trial, and 4 patients versus 11 patients, respectively, experienced anemia in the LIBERTY 2 trial). Pain is subjective and variable among individuals, which can make it challenging to interpret changes in this outcome; however, the variability is expected to be similar across treatment groups. There is evidence of validity, reliability, and responsiveness for the UFS-QoL in patients with UFs. There is a low risk of bias in the measurement of study outcomes that are objective or centrally reviewed. However, for the pain, UFS-QoL, and harms outcomes, there is some risk of bias due to their subjective nature and because patients may have been able to infer what treatment they were receiving based on the large difference in the primary efficacy end point between the RGX-E2-NETA and placebo groups.
The statistical testing procedure used in the studies was acceptable. For the UFS-QoL symptom severity and HRQoL total scores, multiplicity was not controlled for in the studies; as a result, there is an increased risk of type I error. While these end points are considered supportive evidence, the results align with those for the other end points. Furthermore, the clinical expert indicated that the subgroups from the studies were not likely to identify subpopulations of patients who would benefit more from treatment; the review team agreed that results from the subgroups would not meaningfully inform reimbursement decisions.
External Validity
Based on study eligibility criteria, the clinical expert confirmed that the diagnostic methods used in the studies were consistent with those used in practice in Canada. However, the expert noted that patients would be excluded based not on an upper age limit, but on whether they were adults in the premenopausal stage. Additionally, receiving gynecological surgery or ablation procedures for UFs within the 6 months before or after treatment would not be reasons to exclude a patient from receiving RGX-E2-NETA in practice. Patients must have had HMB (i.e., MBL of at least 160 mL during 1 cycle or at least 80 mL per cycle for 2 menstrual cycles) to be included; the clinical expert stated that this was an acceptable definition for a clinical trial setting, but clarified that it is not used in clinical practice. HMB is identified through clinical assessment and described as AUB that interferes with a patient’s HRQoL and daily activities. Based on the baseline characteristics, the study populations adequately represent patients who could receive RGX-E2-NETA in Canada. Approximately three-quarters of patients across the studies were from the US, and none of the trial sites were in Canada. However, the expert felt that this did not meaningfully limit generalizability to practice in Canada.
The expert indicated no concerns with the types or usage of concomitant medications in the studies. It was noted that, in practice, clinicians would recommend that patients receiving GnRH agonists be on calcium and vitamin D supplements; however, the use of these supplements was not expected to influence the study results. For the purposes of a drug reimbursement review, placebo is not relevant to clinical practice; thus, the LIBERTY trials cannot inform on the efficacy and safety of RGX-E2-NETA relative to acceptable comparator treatments. Indirect comparison of relevant treatments for HMB associated with UFs is discussed further in the indirect evidence section of this report.
The study outcomes are not used in practice (aside from hemoglobin testing for anemia); however, the clinical expert confirmed that these were reasonable for a clinical trial setting and were the most important outcomes to address for the disease. Input from the patient group submission identified bleeding, anemia, pain, and bulk symptoms as important to treat, and noted that improved HRQoL is also important. Rather than quantifying treatment effect by measuring MBL, clinicians assess patient satisfaction with how well the treatment is addressing their symptoms and whether the patient is able to participate in daily activities without interference from HMB and UFs.
The 24-week study duration was deemed acceptable for meeting the clinical end points, based on expert opinion. Although the clinical expert does not expect most patients to use RGX-E2-NETA as a long-term treatment (rather as a short-term bridging therapy until surgery), they indicated that there are rare instances where treatment beyond 24 months may be warranted to maintain symptom improvement, such as in patients who are approaching menopause or whose disease responds well to treatment without intolerable TEAEs, including significant bone loss. It is noted in the Health Canada product monograph that the use of RGX-E2-NETA should be limited to 24 months due to the risk of continued and irreversible bone loss. There were no data available for this review to evaluate the use of the drug beyond 24 months.16 The expert noted that, should the drug be used long-term, regular BMD scans would be important to ensure the patient is not experiencing a clinically important decline in BMD; if so, discontinuation should be considered.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations. A final certainty rating was determined as outlined by the GRADE Working Group:17,18
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (referring to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, or publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect, informed by the clinical expert consulted for this review, for responder rate for change in MBL volume, improvement of anemia, improvement of pain, change in UFS-QoL SSS score and HRQoL total score, and the presence or absence of any (non-null) effect for change in BMD at the lumbar spine.
For the GRADE assessments, findings from the LIBERTY 1 trial and LIBERTY 2 trial were assessed together per outcome because these studies were similar in population, intervention, design, and outcome measures.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for RGX-E2-NETA versus placebo.
Long-Term Extension Studies
The contents in this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the review team.
Description of Studies
This section contains 2 studies: the LIBERTY extension trial (a 28-week OLE study involving patients who had completed either pivotal trial) and the LIBERTY RWS (a 52-week study involving those who had completed the LIBERTY extension trial).31,32
The objectives of the LIBERTY extension trial were to evaluate the long-term efficacy and safety of RGX-E2-NETA for up to 52 weeks (including the 24 weeks of treatment during either of the parent studies — i.e., the LIBERTY 1 trial or LIBERTY 2 trial). It was a multinational, single-arm, phase III, OLE study that enrolled eligible patients who had completed participation in 1 of the parent studies. The core study was divided into 2 periods: an open-label treatment phase (28 weeks) and a safety follow-up period (30 days). The LIBERTY extension trial was conducted at 150 sites in 9 countries (Belgium, Brazil, Chile, Czechia, Hungary, Italy, Poland, South Africa, and the US).
Patients with UFs who completed the LIBERTY extension trial and whose disease responded to treatment at the week 48 visit (i.e., patients who demonstrated an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL in the pivotal study) could enrol in the LIBERTY RWS, an international, phase III, double-blind, placebo-controlled RWS of RGX-E2-NETA. Patients were randomized 1 to 1 to receive either oral RGX-E2-NETA or placebo once daily for up to 52 weeks. Randomization was stratified by geographic region (Europe, Latin America, North America, and rest of world), duration of RGX-E2-NETA exposure before randomization (28 weeks or 52 weeks), and pivotal study baseline MBL volume (< 225 mL or ≥ 225 mL). The study was designed to establish the long-term efficacy of RGX-E2-NETA at week 76 (24 weeks after randomization in this study) as a primary end point and at week 104 as a secondary end point. Additionally, this study aimed to investigate the effect of re-treatment with RGX-E2-NETA. The study was conducted at 92 sites in 9 countries (Brazil, Chile, Czechia, Hungary, Italy, Poland, South Africa, and the US).
Populations
Participants were eligible for the LIBERTY extension trial if they completed 24 weeks of study drug treatment in either the LIBERTY 1 trial or the LIBERTY 2 trial. To be eligible for the LIBERTY RWS, patients had to complete the LIBERTY extension trial and have disease that responded to treatment. In both studies, patients were not expected to undergo gynecological surgery or ablation procedures for UFs within the respective study periods, including during the safety follow-up periods. Additionally, participants had to have a negative urine pregnancy test at the baseline visits and agree to continue to use acceptable nonhormonal contraceptive methods consistently during the studies and for at least 30 days after the last dose of the study drug.
Patients were excluded if they had undergone myomectomy, ultrasound-guided laparoscopic radiofrequency ablation, or any other surgical procedure for UFs; uterine artery embolization; MRI-guided, focused ultrasound; or endometrial ablation for AUB at any time during the parent study. Finally, any patient with any contraindication to treatment with E2-NETA, or who met a withdrawal criterion in the parent study, was excluded. For the LIBERTY extension trial, an additional exclusion criterion was a BMD z score of less than –2.0 or a 7% or greater decrease in BMD from the parent study baseline at the lumbar spine, total hip, or femoral neck, based on the parent study week 24 DXA assessment.
The baseline demographic and clinical characteristics of patients enrolled in the LIBERTY extension trial were consistent across pivotal trial treatment groups (Table 16). Approximately 70% of the population was enrolled in North America (i.e., US). Patients who were Black represented approximately 50% of those enrolled.
Table 16: Summary of Baseline Characteristics From the LIBERTY Extension Trial (mITT Population)
Characteristic | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | |
Demographic characteristics | ||
Age (years), mean (SD) | 42.6 (5.1) | 41.9 (5.4) |
Race and ethnicity, n (%) | ||
American Indian or Alaska Native | 2 (1.2) | 1 (0.6) |
Asian | 0 | 0 |
Black | 69 (42.3) | 88 (53.7) |
White | 85 (52.1) | 71 (43.3) |
Multiple races or ethnicities | 2 (1.2) | 1 (0.6) |
Other race or ethnicity | 2 (1.2) | 2 (1.2) |
Not reported | 3 (1.8) | 1 (0.6) |
Geographic region, n (%) | ||
North America | 113 (69.3) | 117 (71.3) |
Rest of the world | 50 (30.7) | 47 (28.7) |
Clinical characteristics | ||
BMI (kg/m2), mean (SD) | 31.4 (7.0) | 32.6 (7.5) |
BMD (g/cm2) lumbar spine, mean (SD) | 1.2 (0.2) | 1.2 (0.2) |
MBL | ||
Volume (mL), mean (SD) | 248.7 (196.7) | 216.0 (123.8) |
< 225 mL, n (%) | 107 (65.6) | 109 (66.5) |
≥ 225 mL, n (%) | 56 (34.4) | 55 (33.5) |
Hemoglobin concentration (g/dL), mean (SD) | 11.4 (1.5) | 11.2 (1.5) |
UFS-QoL SSS score, mean (SD) | 56.6 (20.9) | 61.3 (19.2) |
UFS-QoL HRQoL total score, mean (SD) | 40.4 (21.4) | 32.9 (20.4) |
BMD = bone mineral density; BMI = body mass index; HRQoL = health-related quality of life; MBL = menstrual blood loss; mITT = modified intention to treat; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SD = standard deviation; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The baseline (week 52 of the clinical program) demographics and clinical characteristics of patients rerandomized in the LIBERTY RWS were similar across treatment groups (Table 17).
Table 17: Summary of Baseline Characteristics of Patients Enrolled in the LIBERTY RWS (mITT Population)
Characteristic | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA (N = 115) | PBO (N = 113) | |
Demographic characteristics | ||
Age (years), mean (SD) | 43.3 (5.6) | 44.2 (4.4) |
Race or ethnicity, n (%) | ||
American Indian or Alaska Native | 1 (0.9) | 0 |
Asian | 1 (0.9) | 1 (0.9) |
Black | 49 (42.6) | 57 (50.4) |
White | 63 (54.8) | 52 (46.0) |
Multiple races or ethnicities | 0 | 0 |
Other race or ethnicity | 1 (0.9) | 2 (1.8) |
Not reported | 0 | 1 (0.9) |
Geographic region, n (%) | ||
North America | 74 (64.3) | 73 (64.6) |
Rest of the world | 41 (35.7) | 40 (35.4) |
Clinical characteristics | ||
BMI (kg/m2) | ||
Pivotal study baseline, mean (SD) | 30.5 (6.9) | 31.1 (6.5) |
RWS baseline, mean (SD) | 30.6 (6.7) | 31.4 (6.5) |
BMD (g/cm2) lumbar spine | ||
Pivotal study baseline, mean (SD) | 1.2 (0.2) | 1.2 (0.2) |
RWS baseline, mean (SD) | 1.2 (0.2) | 1.2 (0.1) |
MBL | ||
Pivotal study baseline volume (mL), mean (SD) | 224.5 (134.1) | 210.3 (124.2) |
< 225 mL, n (%) | 73 (63.5) | 76 (67.3) |
≥ 225 mL, n (%) | 42 (36.5) | 37 (32.7) |
RWS baseline volume (mL), mean (SD) | 8.7 (30.9) | 6.4 (20.1) |
Hemoglobin concentration (g/dL) | ||
Pivotal study baseline, mean (SD) | 11.4 (1.5) | 11.4 (1.7) |
RWS baseline, mean (SD) | 13.0 (1.4) | 12.8 (1.3) |
UFS-QoL SSS score | ||
Pivotal study baseline, mean (SD) | 59.2 (19.3) | 60.0 (19.7) |
RWS baseline, mean (SD) | 14.6 (18.3) | 18.4 (22.7) |
UFS-QoL HRQoL total score | ||
Pivotal study baseline, mean (SD) | 37.2 (20.4) | 37.9 (20.3) |
RWS baseline, mean (SD) | 86.1 (18.6) | 81.1 (22.0) |
BMD = bone mineral density; BMI = body mass index; HRQoL = health-related quality of life; MBL = menstrual blood loss; mITT = modified intention to treat; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SD = standard deviation; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Note: Baseline values were defined by the pivotal study baseline value unless stated otherwise, and percentages were based on the total number of patients in each parent study treatment group or total.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Interventions
LIBERTY Extension Trial
All patients received RGX 40 mg, E2 1 mg, and NETA 0.5 mg for 28 weeks.
LIBERTY RWS
Patients received 1 of the following blinded oral study treatments for 52 weeks:
RGX 40 mg, E2 1.0 mg, and NETA 0.5 mg
RGX placebo plus E2-NETA placebo.
The blinding procedure was the same as that used in the LIBERTY 1 trial and LIBERTY 2 trial. For patients whose HMB recurred during the LIBERTY RWS (i.e., MBL volume ≥ 80 mL), blinded treatment was stopped, and re-treatment with open-label RGX-E2-NETA was offered.
Outcomes
The outcomes in the LIBERTY extension trial and LIBERTY RWS are outlined in Table 18 and Table 19, respectively, and are measured as described in the main report.
Table 18: Outcomes Summarized From the LIBERTY Extension Trial
Outcome measure | Time point | LIBERTY extension trial |
|---|---|---|
Proportion of patients who experienced or maintained an MBL < 80 mL and a ≥ 50% reduction from baseline in MBL as measured using the alkaline hematin method (responder rate) | From parent study baseline up to last 35 days of treatment (up to week 52) | Primary efficacy |
Proportion of patients with a hemoglobin level of ≤ 10.5 g/dL at baseline who experienced an increase of ≥ 2 g/dL | From parent study baseline to week 52 | Secondary efficacy |
Change in UFS-QoL SSS score and HRQoL total score | From parent study baseline to week 52 | Secondary efficacy |
Safety | From first dose administration until the follow-up visit approximately 30 days after the last dose of the study drug | Safety |
HRQoL = health-related quality of life; MBL = menstrual blood loss; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 19: Outcomes Summarized From the LIBERTY RWS
Outcome measure | Time point | LIBERTY RWS |
|---|---|---|
Proportion of patients who maintained an MBL volume of < 80 mL as measured using the alkaline hematin method (i.e., sustained responder rate) | At week 76 (24 weeks of the randomized treatment period) | Primary efficacy |
Proportion of patients who maintained an MBL volume < 80 mL | At week 104 | Key secondary efficacy |
Proportion of patients whose disease responded (i.e., MBL volume < 80 mL) to re-treatment with RGX-E2-NETA during the re-treatment period among patients in the placebo group whose MBL volume had returned to ≥ 80 mL | During the 52-week randomized treatment period | Other secondary efficacy |
Change in UFS-QoL SSS score and HRQoL total score | From week 52 or baseline to week 104 | Other secondary efficacy |
Safety | From first dose administration until the follow-up visit approximately 30 days after the last dose of the study drug | Safety |
Percentage change in BMD at the lumbar spine (L1 to L4) as assessed by DXA | From week 52 or baseline to week 104 and from the pivotal study baseline to week 104 | Safety |
BMD = bone mineral density; DXA = dual-energy X-ray absorptiometry; HRQoL = health-related quality of life; MBL = menstrual blood loss; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Statistical Analysis
LIBERTY Extension Trial
Sample Size and Power Calculation
Because this was an OLE study, the sample size was determined by the number of patients who had completed either parent study (i.e., the LIBERTY 1 trial or LIBERTY 2 trial) and were eligible and willing to participate in this study. It was estimated that approximately 600 patients (i.e., 75% of the 780 patients who were randomized into the parent studies) would participate in the OLE study. Of note, the number of patients included those who completed the parent studies in the delayed RGX-E2-NETA arm that is not presented in this summary.
Statistical Testing
Only descriptive statistics were reported for the LIBERTY extension trial. The handling of missing data was performed as described for the parent studies, except that the last observation carried forward approach was used for the latest nonmissing MBL volume to determine response status. No sensitivity analysis was conducted.
Subgroup Analyses
In the LIBERTY extension trial, subgroup analyses of the primary efficacy end point were performed to assess whether the response rate was consistent across clinically important subgroups, including geographic region, MBL volume, age, race or ethnicity, UF volume, uterine volume, BMI, maximum NRS score for UF-associated pain, history of prior pregnancy, alcohol use, and smoking history at baseline and pivotal study baseline. Based on clinical expert opinion, none of the prespecified subgroups were deemed relevant to the review because these subgroups were not likely to identify a subpopulation of patients who might benefit more from treatment or to inform reimbursement decisions.
Analysis Populations
The analysis sets used in the LIBERTY extension trial are described in Table 20.
Table 20: Analysis Populations of the LIBERTY Extension Trial
Study | Population | Definition | Application |
|---|---|---|---|
LIBERTY extension trial | Extension study population | All patients who enrolled and received any amount of open-label study drug in the OLE study. | Used for all efficacy analyses unless otherwise specified. Efficacy analyses were performed by treatment groups as randomized in the parent studies. |
Extension safety population | All enrolled patients who received any amount of open-label study drug in the OLE study. Any patient who received at least 1 dose of RGX was considered an RGX patient, consistent with analysis in the parent studies. | Used for all safety analyses unless otherwise specified. Safety data were analyzed by parent study treatment groups according to the actual treatment received (not the randomized treatment). |
OLE = open-label extension; RGX = relugolix.
Source: LIBERTY extension study Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
LIBERTY RWS
The statistical analyses used in LIBERTY RWS are outlined in Table 21.
Table 21: Statistical Analysis of Efficacy End Points of the LIBERTY RWS
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Primary end point: proportion of patients who maintained an MBL volume of < 80 mL as measured using the alkaline hematin method (i.e., sustained responder rate) | Mantel-Haenszel test and Kaplan-Meier method | Stratified by the randomization stratification factors: pivotal study baseline MBL volume (< 225 vs. ≥ 225 mL), geographic region (North America vs. other), and duration of RGX exposure before randomization (28 weeks vs. 52 weeks) | FP collection If observed MBL volume was:
No FP collection If the reason for no FP collection was:
| The following were performed if the data in the IWRS and eCRF for stratification factors differed by > 5%:
|
Time to MBL volume ≥ 80 mL | Kaplan-Meier method | Hazard ratio stratified by the randomization stratification factors: pivotal study baseline MBL volume (< 225 mL vs. ≥ 225 mL) and duration of exposure to RGX (28 weeks vs. 52 weeks) | Not reported | Not performed |
Percentage change in MBL volume; proportion of patients who experienced an MBL volume < 80 mL during re-treatment with RGX-E2-NETA during the re-treatment period | Descriptive statistics | None | Not reported | Not performed |
Proportion of patients who maintained an MBL volume < 80 mL | Same as primary end point | Same as primary end point | Not reported | Not performed |
Change in UFS-QoL SSS score and HRQoL total score | Descriptive statistics | None | If < 50% of the scale items were missing, then the scale was retained using the mean scale score of the present item. If ≥ 50% of the items were missing, then no scale score could be calculated, and the subscale score was be considered missing. | Not performed |
eCRF = electronic case report form; FP = feminine product; FPRR = feminine product return rate; HRQoL = health-related quality of life; IWRS = interactive web response system; MBL = menstrual blood loss; mITT = modified intention to treat; PP = per protocol; RGX = relugolix; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire; vs. = versus.
aThe week 76 completers population is defined as patients in the mITT population who completed 24 weeks of treatment.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
Given that the LIBERTY RWS was an extension of the LIBERTY extension trial, the sample size of this study was based on the number of patients who had completed either pivotal study (N = 780 patients) and participated in the OLE study (N = 600 patients, assuming a 20% dropout rate at 6 months from the pivotal studies). Assuming that the proportion of responders at week 52 in the OLE study would be 60%, it was estimated that approximately 360 patients would be randomized to this study.
With 360 patients, the study would have at least 90% power to detect a difference of 20% or greater between RGX-E2-NETA and placebo for the primary end point. The following assumptions were used to determine the sample size for this study:
2-sided type I error rate: 0.05
randomization: 1 to 1
responder rate for the placebo group: 40%
responder rate for the RGX-E2-NETA group: 60%
dropout rate: 20%.
Multiple Testing Procedure
In the LIBERTY RWS, to test whether RGX-E2-NETA was statistically superior to placebo for the primary efficacy end point and 3 key secondary end points, a fixed-sequence testing procedure was applied to maintain the family-wise type I error rate. Under this testing procedure, the primary end point was first tested at a 2-sided 0.05 significance level. If the P value for the primary end point was less than 0.05, then the 3 key end points would be tested sequentially: time to relapse, sustained responder rate at week 104, and amenorrhea rate at week 76.
Subgroup Analyses
In the LIBERTY RWS, subgroup analyses of the primary efficacy end point were performed to assess whether the response rate was consistent across clinically important subgroups, including geographic region, MBL volume, duration of exposure to RGX-E2-NETA before randomization, age, race or ethnicity, UF volume, uterine volume, body mass index, and maximum NRS score for UF-associated pain. Based on clinical expert opinion, none of the prespecified subgroups were deemed relevant to the review because the subgroups were not likely to identify a subpopulation of patients who might benefit more from treatment or to inform reimbursement decisions.
Analysis Populations
The analysis sets used in the LIBERTY RWS trial are described in Table 22.
Table 22: Analysis Populations of the LIBERTY RWS
Study | Population | Definition | Application |
|---|---|---|---|
LIBERTY RWS | mITT | All patients randomized to treatment who had taken at least 1 dose of the study treatment (i.e., RGX or its placebo and E2-NETA or its placebo) | Used for efficacy analyses unless otherwise specified. Efficacy analyses were performed by the treatment group as randomized. |
PP | Patients in the mITT population who had no relevant important protocol deviations, defined as a subset of all important protocol deviations | Used for supportive analysis of the primary efficacy end point. PP was not to be analyzed if this population comprised > 95% or < 50% of the mITT population. | |
Safety | All randomized patients who received at least 1 dose of study treatment (any patient who received at least 1 dose of RGX during the randomized treatment period was considered an RGX patient) | Used for safety analyses unless otherwise specified. Safety data were analyzed by treatment groups according to the actual treatment received (not the randomized treatment). | |
Re-treatment | Patients in the mITT population from both treatment groups whose MBL volume reached ≥ 80 mL during the randomized treatment period and who started re-treatment | Used for selected efficacy and safety data. |
E2-NETA = estradiol and norethindrone acetate; mITT = modified intention to treat; PP = per protocol; RGX = relugolix; RWS = randomized withdrawal study.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Summaries of patient disposition in the LIBERTY extension trial and LIBERTY RWS are provided in Table 23 and Table 24, respectively. Of note, each pivotal study contributed approximately 50% of the patients enrolled in the LIBERTY extension trial. Patients who were randomized in the LIBERTY RWS included those who had received RGX-E2-NETA for 52 weeks (i.e., 24 weeks in 1 of the pivotal studies and 28 weeks in the LIBERTY extension trial); had been randomized to placebo in the pivotal studies and then received 28 weeks of open-label RGX-E2-NETA in the LIBERTY extension trial; or had received RGX monotherapy for 12 weeks in the pivotal study in the RGX-E2-NETA group and open-label RGX-E2-NETA for 28 weeks in the LIBERTY extension trial. Thus, although patients were randomized to the placebo group, they had at least some exposure to RGX-E2-NETA in a previous study. Of note, this placebo group contained patients who received open-label RGX-E2-NETA as rescue therapy after experiencing relapsing HMB during the LIBERTY RWS.
Exposure to Study Treatments
Exposures to open-label and randomized treatments are summarized in Table 25 and Table 26, respectively. In the LIBERTY extension trial, most patients in the RGX-E2-NETA-to-RGX-E2-NETA group received treatments for up to 52 weeks from the start of the pivotal trials, and most patients in the placebo-to-RGX-E2-NETA group were exposed for more than 24 to 36 weeks, which is reflective of patients’ transition into open-label RGX-E2-NETA after the pivotal trials.
Table 23: Patient Disposition in the LIBERTY Extension Trial
Patient disposition | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA | PBO to RGX-E2-NETA | |
Enrolled, N | 477 | |
From the LIBERTY 1 trial, n | 244a | |
From the LIBERTY 2 trial, n | 233a | |
Parent study treatment assignment, N | 163 | 164 |
Discontinued from study, n (%) | 30 (18.4) | 42 (25.6) |
Reason for discontinuation, n (%) | ||
Adverse event | 5 (3.1) | 9 (5.5) |
Protocol deviation | 2 (1.2) | 2 (1.2) |
Lost to follow-up | 7 (4.3) | 10 (6.1) |
Withdrawal by patient | 9 (5.5) | 11 (6.7) |
Lack of efficacy | 2 (1.2) | 5 (3.0) |
Pregnancy | 0 | 0 |
Other | 5 (3.1) | 5 (3.0) |
Extension study population | 163 | 164 |
Extension safety population | 163 | 164 |
E2-NETA = estradiol and norethindrone acetate; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate.
aNumbers included patients from the delayed E2-NETA groups, which are not summarized in this report.
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31
Table 24: Patient Disposition in the LIBERTY RWS
Patient disposition | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA | PBO | |
Enrolled, N | 229 | |
Randomized, N | 115 | 114 |
Discontinued from study, n (%) | 26 (22.6) | 27 (23.7) |
Reason for discontinuation, n (%) | ||
Adverse event | 4 (3.5) | 3 (2.6) |
Protocol deviation | 0 | 1 (0.9) |
Lost to follow-up | 6 (5.2) | 4 (3.5) |
Withdrawal by patient | 6 (5.2) | 8 (7.0) |
Lack of efficacy | 1 (0.9) | 1 (0.9) |
Pregnancy | 0 | 1 (0.9) |
Other | 9 (7.8) | 9 (7.9) |
mITT, N | 115 | 113 |
PP, N | 112 | 100 |
Safety, N | 116 | 112 |
Re-treatment population, N | 27 | 88 |
mITT = modified intention to treat; PBO = placebo; PP = per protocol; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study.
Note: In the LIBERTY RWS, 1 patient was randomized in error and did not receive any treatment. In addition, 1 patient was randomized to placebo but received a dose of RGX-E2-NETA. This patient was considered an RGX-E2-NETA patient in the safety population.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 25: Summary of Patient Exposure to Study Drug in the LIBERTY Extension Trial (Safety Population)
Exposure | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | |
Cumulative duration to any study drugs (weeks), mean (SD)a | 51.8 (6.7) | 50.4 (8.9) |
Cumulative duration to RGX-E2-NETA (weeks), mean (SD)a | 51.8 (6.7) | 24.7 (8.8) |
PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SD = standard deviation.
aIn the LIBERTY extension trial, cumulative duration and adherence are defined as a cumulative summary of exposure throughout the 52-week treatment period (parent study and extension study).
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the LIBERTY RWS, most patients in the RGX-E2-NETA group (59.5%) were exposed to the study treatment for more than 48 weeks during the randomized treatment period, with an average exposure of 40.1 weeks (SD = 18.0 weeks). In contrast, the majority of patients in the placebo group (83.0%) discontinued placebo and were exposed to placebo for 16.9 weeks (SD = 12.2 weeks) (Table 26). Patients in both arms who had a return of their MBL volume to 80 mL or greater during the randomized treatment period could receive open-label RGX-E2-NETA as rescue medication. The mean re-treatment period was generally shorter in the RGX-E2-NETA group than in the placebo group (i.e., 27.6 weeks [SD = 12.8 weeks] versus 35.4 weeks [SD = 10.3 weeks], respectively). Most of the patients (86.4%) in the placebo group were exposed to re-treatment for more than 24 weeks due to return of HMB (MBL ≥ 80 mL) early in the LIBERTY RWS, reflecting the initiation of re-treatment within 1 month to 2 months after stopping treatment with RGX-E2-NETA at week 52 or baseline. Adherence to the study drug was high (> 98%) throughout the clinical program.
Table 26: Summary of Patient Exposure to the Study Drug in the LIBERTY RWS (Safety Population)
Exposure | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA N = 116 (treatment) N = 27 (re-treatment) | PBO N = 112 (treatment) N = 88 (re-treatment) | |
Treatment duration during treatment period (weeks), mean (SD) | 40.1 (18.0) | 16.9 (12.2) |
Treatment duration during re-treatment period (weeks), mean (SD) | 27.6 (12.8) | 35.4 (10.3) |
PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SD = standard deviation.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
In the LIBERTY extension trial, a total of 464 patients (97.5%) received concomitant medications during the study, including 160 patients (98.2%) in the RGX-E2-NETA-to-RGX-E2-NETA group and 160 patients (97.6%) in the placebo-to-RGX-E2-NETA group. In the former group, the most frequently reported concomitant medications were vitamin and iron supplements and analgesics, with 61.3% of patients reporting the use of ibuprofen, 30.1% reporting the use of ferrous sulphate, 19.0% reporting the use of acetaminophen, and 17.2% reporting the use of iron. Additionally, the proportions of patients who reported using vitamin D and cholecalciferol were 12.9% and 10.4%, respectively. In the placebo-to-RGX-E2-NETA group, the most frequently reported concomitant medications were vitamin and iron supplements and analgesics, with 70.7% of patients reporting the use of ibuprofen, 30.5% reporting the use of ferrous sulphate, 25.0% reporting the use of acetaminophen, 17.1% reporting the use of iron, and 17.1% reporting the use of vitamins. Additionally, the proportions of patients who reported the use of vitamin D and cholecalciferol were 14.6% and 12.2%, respectively.
In the LIBERTY RWS, a total of 220 patients (96.5%) received concomitant medications during the randomized treatment period, including 109 patients (94.0%) in the RGX-E2-NETA group and 111 patients (99.1%) in the placebo group. The reported concomitant medications reflect vitamin and iron supplements, analgesics, and antihypertensive medications. Iron and hydrochlorothiazide were reported more frequently in the RGX-E2-NETA group, while ibuprofen, vitamins (vitamin nitric oxide supplement, vitamin D nitric oxide supplement, and vitamin B12 nitric oxide supplement), ranitidine, and ketoprofen were reported more frequently in the placebo group.
Subsequent Treatment
In the LIBERTY RWS, patients who experienced an MBL volume of 80 mL or more during the randomized, double-blinded phase with either RGX-E2-NETA or placebo entered the re-treatment period with open-label RGX-E2-NETA. More specifically, 27 patients (23.3%) in the RGX-E2-NETA group and 88 patients (78.6%) in the placebo group received re-treatment with open-label RGX-E2-NETA.
Efficacy
LIBERTY Extension Trial
Responder Rate Over the Preceding 35 Days of Treatment
As presented in Table 27, and consistent with the pivotal trials (in which 73% and 71% of participants on RGX-E2-NETA from the LIBERTY 1 and LIBERTY 2 trials, respectively, responded to treatment at week 24), 87.7% of participants who continued to be treated with RGX-E2-NETA for more than 52 weeks responded to treatment. Moreover, a response rate (75.6%) similar to that reported in the pivotal studies was observed for patients who were originally assigned to the placebo group and received open-label RGX-E2-NETA for 28 weeks in the LIBERTY extension trial. The proportions of patients meeting the criteria for the individual components of the composite primary end point (MBL < 80 mL over the last 35 days of treatment and ≥ 50% reduction in MBL from pivotal baseline over the last 35 days of treatment) were similar (87.1% and 89.0%, respectively, in the RGX-E2-NETA-to-RGX-E2-NETA group, and 75.6% and 81.1%, respectively, in the placebo-to-RGX-E2-NETA group). Results from the prespecified subgroups were consistent with those of the primary analysis.
Table 27: Responder Rate at Week 52 in the LIBERTY Extension Trial (Study Population)
Variable | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | |
Week 52 | ||
Responder rate, na | 143 | 124 |
Proportion of responders (95% CI)b | 87.7 (81.7 to 92.3) | 75.6 (68.3 to 82.0) |
Patients with MBL < 80 mL over the last 35 days of treatment, n | 142 | 124 |
Proportion of patients (95% CI)b | 87.1 (81.0 to 91.8) | 75.6 (68.3 to 82.0) |
Number of patients with ≥ 50% reduction in MBL from pivotal baseline over the last 35 days of treatment, n | 145 | 133 |
Proportion of patients (95% CI)b | 89.0 (83.1 to 93.3) | 81.1 (74.3 to 86.8) |
CI = confidence interval; MBL = menstrual blood loss; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate.
aResponders were patients who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from pivotal trial baseline over the last 35 days of treatment.
bBased on exact binomial 95% CI (Clopper-Pearson).
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 28: Summary of Secondary Outcomes in the LIBERTY Extension Trial (Study Population)
Variable | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | |
Proportion of patients with a hemoglobin level ≤ 10.5 g/dL at baseline who experienced an increase of ≥ 2 g/dL | ||
Number of evaluable patients, N | 39 | 38 |
Patients, n (%) | 23 (59.0) | 16 (42.1) |
PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate.
Source: Al-Hendy (2022).51
Proportion of Patients With a Hemoglobin Level of 10.5 g/dL or Lower at Baseline Who Experienced an Increase of 2 g/dL or More at Week 52
There were 39 patients (23.9%) in the RGX-E2-NETA-to-RGX-E2-NETA group and 38 patients (23.2%) in the placebo-to-RGX-E2-NETA group who had a hemoglobin concentration of 10.5 g/dL or lower at pivotal study baseline, representing the cohort of patients who had symptomatic anemia (Table 28). Patients in both pivotal study groups — the RGX-E2-NETA-to-RGX-E2-NETA group (23 patients [59.0%]) and the placebo-to-RGX-E2-NETA group (16 patients [42.1%]) — experienced an increase of more than 2 g/dL at week 52.
Change From Baseline to Week 52 in UFS-QoL SSS Score and HRQoL Total Score
In the RGX-E2-NETA-to-RGX-E2-NETA group, the LSM SSS score changed from baseline by –36.9 points (standard error [SE] = 2.2 points) at week 24; this reduction was maintained through week 52 (–37.3 points; SE = 2.5 points) (Table 29). In the placebo-to-RGX-E2-NETA group, the LSM SSS scores changed from baseline by –10.8 points (SE = 2.1 points) at week 24. After the initiation of open-label RGX-E2-NETA, scores changed, with an LSM reduction in SSS score of –35.0 points (SE = 2.5 points) at week 52.
Over the 52-week treatment, the RGX-E2-NETA group experienced an LSM improvement of 40.4 points (SE = 2.5 points) in UFS-QoL HRQoL total score, which was consistent with the LSM change from baseline score observed at week 24 (40.8 points; SE = 2.2 points) (Table 29). The improvement in overall HRQoL was similar (LSM change = 39.0 points [SE = 2.6 points] at week 52) in the placebo group upon receiving open-label RGX-E2-NETA for 28 weeks.
Table 29: Change From Baseline to Week 52 in UFS-QoL Symptoms Severity Score and HRQoL Total Score (Extension Study Population)
Variable | LIBERTY extension trial | |
|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | |
Week 52 | ||
UFS-QoL SSS score, n | 129 | 120 |
UFS-QoL SSS score, LSM (SE) CFBa | –37.3 (2.5) | –35.0 (2.5) |
HRQoL total score, n | 129 | 120 |
HRQoL total score, LSM (SE) CFBb | 40.4 (2.5) | 39.0 (2.6) |
CFB = change from baseline; HRQoL = health-related quality of life; LSM = least squares mean; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SE = standard error; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
aThe transformed score ranges from 0 points to 100 points. Higher scores indicate greater symptom severity. For the change from baseline, a negative score indicates improvement.
bThe transformed score ranges from 0 points to 100 points. Higher scores indicate better HRQoL. For the change from baseline, a positive score indicates improvement.
Source: LIBERTY extension trial Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,31 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
LIBERTY RWS
Sustained Responder Rate Through Week 76 and Week 104
At week 76, more patients in the RGX-E2-NETA group than in the placebo group (78.4% versus 15.1%; P < 0.0001) experienced a sustained treatment response after maintaining an MBL volume of less than 80 mL, as demonstrated in Table 30. Similar results were observed at week 104. Results from the sensitivity analyses and prespecified subgroup analyses were consistent with the primary analysis.
Table 30: Sustained Responder Rate at Week 76 and Week 104 in the LIBERTY RWS (mITT Population)
Variable | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA (N = 115) | PBO (N = 113) | |
Week 76 | ||
Sustained responder rate (%)a (95% CI) | 78.4 (69.3 to 85.1) | 15.1 (8.9 to 22.8) |
Treatment group difference vs. control (%) (95% CI) | 63.4 (52.9 to 73.9) | |
P valueb | < 0.0001 | |
Week 104 | ||
Sustained responder rate (%)a (95% CI) | 69.8 (59.7 to 77.8) | 11.8 (6.3 to 19.0) |
Treatment group difference vs. control (95% CI) | 58.0 (47.0 to 69.1) | |
P valueb | < 0.0001 | |
CI = confidence interval; MBL = menstrual blood loss; mITT = modified intention to treat; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; vs. = versus.
aThe sustained responder rate at week 76 was defined as the proportion of patients who maintained an MBL volume of less than 80 mL through week 76. It was calculated as the Kaplan-Meier estimate of the cumulative probability of an MBL volume of less than 80 mL while on randomized treatment through week 76.
bBased on the stratified test statistics through log-log transformation of the difference in survival curve at a fixed time point stratified by pivotal study baseline MBL volume (< 225 mL, ≥ 225 mL) and duration of prior exposure to RGX (28 weeks, 52 weeks). Aside from the end points listed in the testing order, P values (if provided) were not adjusted for multiplicity; thus, the values are at a nominal level of 0.05.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Proportion of Patients Who Responded (MBL Volume < 80 mL) to Re-Treatment With RGX-E2-NETA Among Those Whose MBL Volume Had Returned to 80 mL or Greater
As presented earlier, most patients (81%) who were rerandomized to the placebo group had their MBL volume return to 80 mL or greater. Following re-treatment with open-label RGX-E2-NETA, nearly all patients (97.8%) experienced treatment response within 2 menstrual cycles (Table 31). A smaller proportion of patients (23%) experienced disease relapse in the RGX-E2-NETA group rather than in the placebo group; most of these occurrences were single episodes of bleeding that did not recur with re-treatment.
Change From LIBERTY RWS Baseline to Week 104 in UFS-QoL SSS Score and HRQoL Total Score
As presented in Table 32, UFS-QoL SSS scores remained relatively stable from week 52 to week 104 in the RGX-E2-NETA group. For patients who received placebo, cessation of treatment was accompanied by the return of symptoms, as indicated by the higher SSS scores at week 64 (i.e., 12 weeks following rerandomization); these improved at subsequent time points as patients received re-treatment with RGX-E2-NETA.
HRQoL total scores remained relatively stable in the RGX-E2-NETA group. However, in the placebo group, HRQoL scores decreased at week 64 compared to week 52 or baseline because patients transitioned from RGX-E2-NETA to placebo. Upon reinitiation of RGX-E2-NETA, HRQoL total scores improved to a level similar to that of the group that had continued RGX-E2-NETA.
Table 31: Proportion of Patients Who Responded to Re-Treatment in the LIBERTY RWS (mITT Population)
Variable | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA (N = 115) | PBO (N = 113) | |
Patients who started re-treatment | 26 | 89 |
Patients who experienced treatment response when retreated, n (%)a | 25 (96.2) | 87 (97.8) |
(95% CI)b | (80.4 to 99.9) | (92.1 to 99.7) |
CI = confidence interval; MBL = menstrual blood loss; mITT = modified intention to treat; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study.
aResponders were patients with an MBL volume of less than 80 mL during the re-treatment period. MBL volume was calculated as the sum of MBL volume from all feminine products (validated, unauthorized, and unvalidated) assessed using the alkaline hematin method at each visit.
bBased on exact binomial 95% CI (Clopper-Pearson).
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 32: Change From Baseline to Week 104 in UFS-QoL SSS Score and HRQoL Total Score (mITT population)
Variable | LIBERTY RWS | |
|---|---|---|
RGX-E2-NETA (N = 115) | PBO (N = 113) | |
Week 64 | ||
Evaluable patients, n | 101 | 76 |
CFB in UFS-QoL SSS score, mean (SD)a | 4.0 (17.7) | 25.0 (28.8) |
CFB in HRQoL total score, mean (SD)b | –0.7 (15.2) | –17.4 (28.5) |
Week 76 | ||
Evaluable patients, n | 86 | 21 |
CFB in UFS-QoL SSS score, mean (SD)a | 2.2 (18.2) | 14.1 (21.1) |
CFB in HRQoL total score, mean (SD)b | –0.8 (16.8) | –9.1 (22.4) |
Week 104 | ||
Evaluable patients, n | 65 | 8 |
CFB in UFS-QoL SSS score, mean (SD)a | 0.1 (15.9) | –4.3 (42.2) |
CFB in HRQoL total score, mean (SD)b | 1.1 (15.3) | 8.2 (42.1) |
CFB = change from baseline; HRQoL = health-related quality of life; mITT = modified intention to treat; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SD = standard deviation; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
aThe transformed score ranges from 0 points to 100 points. Higher scores indicate greater symptom severity. For the change from baseline, a negative score indicates improvement.
bThe transformed score ranges from 0 points to 100 points. Higher scores indicate better HRQoL. For the change from baseline, a positive score indicates improvement.
Source: LIBERTY RWS Clinical Study Report and sponsor’s Summary of Clinical Evidence.19,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
Key harms results are presented in Table 33. Headaches and hot flushes remained the most common TEAEs. In the LIBERTY extension trial, the proportion of patients with 1 or more TEAEs was numerically smaller among patients who remained on RGX-E2-NETA compared with those who transitioned from placebo. In the LIBERTY RWS, a numerically larger proportion of patients reported 1 or more TEAEs in the placebo group compared with the RGX-E2-NETA group.
SAEs were reported infrequently in the LIBERTY extension trial and the LIBERTY RWS. In the LIBERTY extension trial, SAEs were reported for 1 patient (0.6%) in the RGX-E2-NETA-to-RGX-E2-NETA group and 11 patients (6.7%) in the placebo-to-RGX-E2-NETA group during the 28-week open-label period. The SAE in the RGX-E2-NETA group was a uterine hemorrhage, and it led to the patient leaving the study. In the placebo-to-RGX-E2-NETA group, 2 patients with an SAE of menorrhagia, 1 patient with metrorrhagia, and 1 patient with atrial fibrillation withdrew from the study.
In the LIBERTY RWS, SAEs were reported for 2 patients (1.7%) in the RGX-E2-NETA group and 2 patients (1.8%) in the placebo group. The 2 SAEs in the RGX-E2-NETA group were related to breast cancer and anemia, and led to study discontinuation; the SAEs in the placebo group included 1 case of transient global amnesia, which led to study discontinuation, and 1 case of myxoid liposarcoma; the latter patient remained on treatment.
No deaths occurred during either study. No pregnancies were reported in the LIBERTY extension trial; 1 participant in the placebo group of the LIBERTY RWS became pregnant, while no pregnancies were reported in the RGX-E2-NETA group. A respective 1.2% and 5.5% of patients in the RGX-E2-NETA-to-RGX-E2-NETA and placebo-to-RGX-E2-NETA groups discontinued the LIBERTY extension trial due to TEAEs. One patient (1.7%) from the RGX-E2-NETA group and 3 patients (2.7%) from the placebo group in the LIBERTY RWS discontinued the treatment due to a TEAE.
The changes from baseline in lumbar spine BMD for the LIBERTY extension trial and LIBERTY RWS trial are presented in Table 34. In the LIBERTY extension trial, the mean percentage change in BMD at the lumbar spine was –0.8% in the RGX-E2-NETA group, which was similar to the placebo group that transitioned to RGX-E2-NETA (–0.8%). In both treatment groups of the LIBERTY RWS, the percentage change in BMD was generally stable, with minimal changes over 1 year. The difference in mean percentage change from placebo was 0.7% (95% CI, –0.1% to 1.6%).
Table 33: Summary of Harms in the LIBERTY Extension Trial (Safety Population) and the LIBERTY RWS (Safety Population)
Adverse events | LIBERTY extension triala | LIBERTY RWSb | ||
|---|---|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to RGX-E2-NETA (N = 164) | RGX-E2-NETA (N = 116) | PBO (N = 112) | |
Most common TEAEs (≥ 3%), n (%) | ||||
Patient with ≥ 1 TEAE | 89 (54.6) | 103 (62.8) | 68 (58.6) | 72 (64.3) |
Nasopharyngitis | 10 (6.1) | 8 (4.9) | 13 (11.2) | 12 (10.7) |
Pelvic pain | 7 (4.3) | 2 (1.2) | 3 (2.6) | 4 (3.6) |
Headache | 6 (3.7) | 11 (6.7) | 8 (6.9) | 5 (4.5) |
Back pain | 5 (3.1) | 3 (1.8) | 5 (4.3) | 5 (4.5) |
Dizziness | 5 (3.1) | 3 (1.8) | 1 (0.9) | 3 (2.7) |
Fatigue | 5 (3.0) | 1 (0.6) | 3 (2.6) | 2 (1.8) |
Hot flush | 4 (2.5) | 13 (7.9) | 2 (1.7) | 8 (7.1) |
Urinary tract infection | 3 (1.8) | 3 (1.8) | 1 (0.9) | 4 (3.6) |
Depression | 3 (1.8) | 5 (3.0) | 1(0.9) | 2(1.8) |
Abdominal pain lower | 2 (1.2) | 0 | 2 (1.7) | 4 (3.6) |
Anxiety | 2 (1.2) | 1 (0.6) | 1 (0.9) | 4 (3.6) |
Breast tenderness | 2 (1.2) | 0 | 2 (1.7) | 5 (4.5) |
Bronchitis | 2 (1.2) | 1 (0.6) | 0 | 5 (4.5) |
Dysmenorrhea | 2 (1.2) | 0 | 2 (1.7) | 8 (7.1) |
Hypertension | 2 (1.2) | 10 (6.1) | 2 (1.7) | 6 (5.4) |
Sinusitis | 1 (0.6) | 1 (0.6) | 4 (3.4) | 0 |
Anemia | 0 | 5 (3.0) | 3 (2.6) | 0 |
Arthralgia | 0 | 3 (1.8) | 4 (3.4) | 2 (1.8) |
Breast pain | 0 | 1 (0.6) | 4 (3.4) | 0 |
Menorrhagia | 0 | 2 (1.2) | 0 | 5 (4.5) |
Upper respiratory tract infection | 0 | 3 (1.8) | 2 (1.7) | 6 (5.4) |
Cellulitis | NR | NR | 4 (3.4) | 0 |
SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 1 (0.6) | 11 (6.7) | 2 (1.7) | 2 (1.8) |
Anemia | 0 | 1 (0.6) | 1 (0.9) | 0 |
Atrial fibrillation | 0 | 1 (0.6) | NR | NR |
Cholecystitis, acute | 0 | 1 (0.6) | NR | NR |
Appendicitis | 0 | 2 (1.2) | NR | NR |
Road traffic accident | 0 | 1 (0.6) | NR | NR |
Blood pressure increase | 0 | 1 (0.6) | NR | NR |
Intervertebral disc protrusion | 0 | 1 (0.6) | NR | NR |
Nephrolithiasis | 0 | 1 (0.6) | NR | NR |
Menorrhagia | 0 | 2 (1.2) | NR | NR |
Metrorrhagia | 0 | 1 (0.6) | NR | NR |
Ovarian cyst rupture | 0 | 1 (0.6) | NR | NR |
Uterine hemorrhage | 1 (0.6) | 0 | NR | NR |
Breast cancer, stage II | NR | NR | 1 (0.9) | 0 |
Myxoid liposarcoma | NR | NR | 0 | 1 (0.9) |
Transient global amnesia | NR | NR | 0 | 1 (0.9) |
Patients who stopped treatment due to TEAEs, n (%) | ||||
Patients who stopped | 2 (1.2) | 9 (5.5) | 2 (1.7) | 3 (2.7) |
Atrial fibrillation | 0 | 1 (0.6) | NR | NR |
Haemangioma of liver | 0 | 1 (0.6) | NR | NR |
Depression | 1 (0.6) | 0 | 0 | 1 (0.9) |
Endometrial hyperplasia | 0 | 1 (0.6) | NR | NR |
Menometrorrhagia | 0 | 1 (0.6) | NR | NR |
Menorrhagia | 0 | 2 (1.2) | NR | NR |
Metrorrhagia | 0 | 1 (0.6) | NR | NR |
Uterine hemorrhage | 1 (0.6) | 0 | NR | NR |
Acne | 0 | 1 (0.6) | NR | NR |
Alopecia | 0 | 1 (0.6) | NR | NR |
Hot flush | 0 | 1 (0.6) | NR | NR |
Hypertension | 0 | 1 (0.6) | NR | NR |
Anemia | NR | NR | 1 (0.9) | 0 |
Blood pressure increase | NR | NR | 1 (0.9) | 0 |
Diarrhea | NR | NR | 1 (0.9) | 0 |
Hyperhidrosis | NR | NR | 1 (0.9) | 0 |
Migraine | NR | NR | 1 (0.9) | 0 |
Muscle spasms | NR | NR | 1 (0.9) | 0 |
Pharyngitis, streptococcal | NR | NR | 1 (0.9) | 0 |
Phlebitis, superficial | NR | NR | 0 | 1 (0.9) |
Transient global amnesia | NR | NR | 0 | 1 (0.9) |
Deaths, n (%) | ||||
Patients who died | 0 | 0 | 0 | 0 |
LSM = least squares mean; NR = not reported; OLE = open-label extension; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aIn the LIBERTY extension trial, the harms are presented for the OLE study period.
bIn the LIBERTY RWS, the harms are presented for the 52-week cumulative treatment period, which included the randomized treatment and re-treatment periods.
cThe LSM percentage change is calculated from baseline to week 52 in the LIBERTY extension trial, and from week 52 or baseline to week 104 in the LIBERTY RWS.
Source: LIBERTY extension trial and LIBERTY RWS clinical study reports and sponsor’s Summary of Clinical Evidence.19,31,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 34: Changes From Baseline in Lumbar Spine BMD at Week 52 in the LIBERTY Extension Trial (Safety Population) and at Week 104 in the LIBERTY RWS (Safety Population)
Variable | LIBERTY extension trial | LIBERTY RWS | ||
|---|---|---|---|---|
RGX-E2-NETA to RGX-E2-NETA (N = 163) | PBO to (N = 164) | RGX-E2-NETA (N = 126) | PBO (N = 129) | |
Baseline (LIBERTY extension trial) and week 52 or RWS baseline (LIBERTY RWS) | ||||
Patients, n | 163 | 164 | 112 | 111 |
LSM (95% CI), % | 1.19 (1.17 to 1.22) | 1.25 (1.22 to 1.28) | 1.19 (1.16 to 1.22) | 1.20 (1.17 to 1.23) |
Week 52 (LIBERTY extension trial) or week 104 (LIBERTY RWS) | ||||
Patients, n | 132 | 120 | 79 | 79 |
LSM percentage change (95% CI), % | –0.80 (–1.36 to –0.25) | –0.78 (–1.32 to –0.23) | 0.81 (0.20 to 1.41) | 0.10 (–0.52 to 0.71) |
Difference in LSM percentage change from placebo (95% CI), % | NA | NA | 0.71 (–0.13 to 1.55) | NA |
BMD = bone mineral density; CI = confidence interval; LSM = least squares mean; NA = not applicable; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RWS = randomized withdrawal study.
Source: LIBERTY extension trial and LIBERTY RWS clinical study reports and sponsor’s Summary of Clinical Evidence.19,31,32 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
LIBERTY Extension Trial
Patients eligible for the LIBERTY extension trial must have successfully completed either the LIBERTY 1 trial or the LIBERTY 2 trial. Therefore, patients who continued in the OLE study were more likely to be those who had responded to treatment and did not experience intolerable TEAEs. As a result, the results may not be generalizable to all patients who may be treated with RGX-E2-NETA in clinical practice. The single-arm OLE study was noncomparative; this prevents conclusions from being made about the benefits or harms attributable to RGX-E2-NETA versus any comparator. In addition, the open-label designs could bias the assessment of subjective, patient-reported outcomes (i.e., UFS-QoL and harms). The lack of data beyond week 52 in the OLE study makes it challenging to draw conclusions about the long-term sustainability of the treatment effect.
In the single-arm OLE trial, all patients in the RGX-E2-NETA-to-RGX-E2-NETA group received 28 weeks of RGX-E2-NETA (resulting in a total of 52 weeks of active treatment for those who had received RGX-E2-NETA in the parent studies). The Health Canada product monograph recommends limiting treatment to 2 years due to the risk of continued and irreversible bone loss.16 According to the clinical expert consulted for this review, when RGX-E2-NETA is used as a preoperative treatment, patients are not likely to receive the drug for more than 2 years. Instead, treatment would be for a few months in preparation for surgery. The expert noted that if a patient is meeting their treatment goals without intolerable TEAEs or clinically significant bone loss, then treatment beyond 2 years could be an option; however, these would be rare cases. With extended treatment, BMD measurements at regular intervals would be necessary to ensure that a patient is not experiencing bone loss.
Up to 22% of patients discontinued the study. The reasons for missing data for each outcome were not available. As such, the effect of the missing data and its magnitude remains uncertain. Although up to 98% of patients had at least 1 concomitant medication, the clinical expert consulted by CDA-AMC had no concern about the impact of these medications on the treatment results.
LIBERTY RWS
No concerns were identified with respect to the methods used for randomization or allocation concealment. There were some minor demographic differences between the groups at baseline; however, the disease-related characteristics appeared balanced. The clinical expert agreed that any baseline discrepancies were unlikely to bias the findings. A fixed-sequence testing procedure was applied to maintain the family-wise type I error rate for the primary efficacy end point as well as the 3 key secondary end points. Although similar proportions of patients in the placebo group (24%) and RGX-E2-NETA group (23%) discontinued, it is unclear if the treatment groups remained balanced throughout the study. The study was double blind, which is important given the subjective nature of the patient-reported outcomes; however, there is the risk of unblinding due to the large difference in efficacy results between the active and placebo groups. There was evidence of validity, reliability, and responsiveness for both the UFS-QoL SSS score and the HRQoL total score. There were imbalances in the use of concomitant medications. For example, there were higher levels of iron, hydrochlorothiazide, metformin, and amlodipine in the RGX-E2-NETA group compared to the placebo group. However, the clinical expert did not think these imbalances would meaningfully affect the study results. There is a risk of bias due to missing outcome data, particularly at longer follow-up times (i.e., 76 weeks and 104 weeks), for the UFS-QoL SSS score and the HRQoL total score. At 76 weeks, data were missing for 25% of patients in the RGX-E2-NETA group and for 81% of patients in the placebo group. At 104 weeks, data were missing for 43% of patients in the RGX-E2-NETA group and for 93% of patients in the placebo group. Similarly, there is a risk of bias due to missing data for BMD in the lumbar spine at 104 weeks, given that data were missing for 37% to 39% of patients across groups.
External Validity
LIBERTY Extension Trial
Patients who were enrolled in the LIBERTY extension trial had completed the parent trials; therefore, the trial has the same generalizability issues as the parent trials. Although no study sites were located in Canada, the clinical expert did not have concerns with generalizing based on geography. The clinical expert noted that older age or having undergone recent gynecological surgery or ablation procedures for UFs would not exclude a patient from being treated with RGX-E2-NETA. Because these groups were not represented in the study, the results may not be generalizable to these patients. The efficacy outcomes assessed in the study were of importance to patients, but the study outcomes (aside from measuring hemoglobin) are not used in the clinical setting (e.g., MBL is not quantified using the alkaline hematin method; pain and HRQoL are not measured using instruments). The patient population of the OLE study may not be reflective of the broader, more heterogeneous population in terms of demographic and clinical characteristics, particularly given that only a subset of patients in the parent trials continued to the OLE study. Therefore, the results in a real-world clinical setting may differ from those observed in the OLE study.
LIBERTY RWS
There were no study sites in Canada, but the clinical expert did not have concerns about generalizing based on geography. However, the results may not be generalizable to patients with conditions that excluded participants from the study, such as having undergone medical or surgical procedures for UFs, having a weight that exceeded the limit of the DXA scanner (or a condition that precluded an adequate DXA measurement), or having or developing a contraindication to treatment with E2 or NETA (e.g., breast cancer or estrogen-dependent neoplasia). Additionally, the race and ethnicity proportions in the study may not reflect those of the broader population of patients in Canada. In that case, the results might not be generalizable, particularly for the ethnic groups in which UFs tend to be more clinically significant.
Indirect Evidence
The contents in this section have been informed by materials submitted by the sponsor and by clinical expert input. The following information has been summarized and validated by the review team.
The sponsor submitted a systematic review with an NMA, a type of ITC, to estimate the efficacy and safety of RGX-E2-NETA compared with LA with or without add-back for the treatment of HMB associated with UFs in people in the premenopausal stage.20
Objectives for the Summary of Indirect Evidence
An ITC was required because of a lack of studies directly comparing RGX-E2-NETA with LA with or without add-back (the only authorized treatment for UFs in the Canadian setting).
Description of Indirect Comparisons
Objectives
To support the NMA, the sponsor:
conducted an SLR to identify RCTs to estimate the relative efficacy and safety of RGX-E2-NETA versus LA (with or without add-back)
performed an NMA to assess the relative efficacy and safety of RGX-E2-NETA versus LA (without add-back) for the treatment of HMB associated with UFs in people in the premenopausal stage.
SLR Methods
Table 35 shows the study selection criteria and key aspects of the methods for the SLR. No predetermined protocol was identified for the SLR. The patient population of interest was people in the premenopausal stage with a confirmed diagnosis of symptomatic UFs.
An initial search was conducted from inception of the databases until February 7, 2022. An update was performed on December 10, 2024.
According to the sponsor, an SLR was conducted following Cochrane guidance.52 The search strategy was peer-reviewed using the Peer Review of Electronic Search Strategies checklist.53 Searches were performed for clinical trials of RGX-E2-NETA, LA, and several other historical comparators in MEDLINE and Embase through the Ovid platform. Historical comparators were treatments no longer in use in Canada.
A search of grey literature was conducted on Clinicaltrials.gov, the WHO International Clinical Trials Registry Platform, the European Union Clinical Trial Registry, Health Canada’s clinical trial databases, Google, and Google Scholar. Hand searches of reference lists of included trials and key SLRs were conducted during screening. Unpublished data from conference abstracts were included from the American College of Obstetricians and Gynecologists and the European Society of Human Reproduction and Embryology.
Two independent reviewers performed the study selection process, which comprised title and abstract screening and full-text article review. Discrepancies were resolved by consensus or a third reviewer. For inclusion in the NMAs, studies in the SLR had to meet the study selection criteria outlined in Table 35.
Data were extracted by a single reviewer using a predefined data grid. This information was validated by a second reviewer. A third reviewer resolved discrepancies to ensure the accuracy and integrity of the data.
Study-level risk of bias was assessed based on the Cochrane risk of bias tool for randomized trials54 by 2 reviewers, and discrepancies were resolved through discussion. This tool addresses random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other potential sources of bias. Each domain was rated as being at “low,” “high,” or “unclear” risk of bias.
Outcomes of interest for the NMA included effectiveness, TEAEs, and tolerability. The specific effectiveness outcomes were MBL resolution and UFS-QoL symptom severity and HRQoL total score. TEAEs included hot flushes and headaches. Tolerability outcomes were discontinuations due to TEAEs.
Of 228 reports identified in the literature, 31 trials were retained from the SLR, and 12 of these were included in the NMA.
Table 35: Study Selection Criteria and Methods for ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Patients in the premenopausal stage with a confirmed diagnosis of symptomatic UFs |
Intervention | RGX-E2-NETA |
Comparator | Pharmacological treatments GnRH agonists:
SPRM:
Control:
|
Outcome | Efficacy:
Safety:
Tolerability:
|
Study designs | RCTs |
Publication characteristics | Published and unpublished |
Exclusion criteria |
|
Sources searched | Main literature search:
Grey literature search:
Hand searches of reference lists of included trials and key SLRs identified during screening |
Selection process | Two independent researchers screened articles based on:
|
Data extraction process |
|
Quality assessment | 2 independent reviewers performed risk of bias assessments at the study level using the 2011 version of the Cochrane risk of bias tool for randomized trials. Discrepancies were resolved through discussion. |
AE = adverse event; BMD = bone mineral density; GA = goserelin acetate; GnRH = gonadotropin hormone-releasing hormone; HRQoL = health-related quality of life; ITC = indirect treatment comparison; LA = leuprolide acetate; MBL = menstrual blood loss; MFP = mifepristone; RCT = randomized controlled trial; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SLR = systematic literature review; SPRM = selective progesterone receptor modulator; SSS = Symptom Severity Scale; TEAE = treatment-emergent adverse event; UF = uterine fibroid; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
Source: Details included in the table are from the sponsor’s ITC Technical Report.20
NMA Feasibility Appraisal
The sponsor conducted a feasibility assessment by evaluating the studies’ comparators, network connectivity, similarity of outcome definitions, time points assessed, baseline characteristics, NMA assumptions, and risk of bias.
Of the 31 trials included in the SLR, 12 were retained because these allowed a connection between RGX-E2-NETA and LA (without add-back). The NMA included UPA 5 mg, UPA 10 mg, and MFP 10 mg as treatments to establish a network connection between RGX-E2-NETA and LA (without add-back). MFP was excluded from the NMA because it is not indicated for the treatment of UFs in Canada; nor is it used for UF-related symptoms. UPA was withdrawn from the Canadian market in 2020 because it was associated with morphological changes in the endometrium and associated with increased risk of a rare but serious liver injury.27,28,55 These comparators were excluded from the main results despite being retained to establish a network connection between RGX-E2-NETA and LA (without add-back).
The efficacy outcomes included were MBL resolution, amenorrhea, and change from baseline in UFS-QoL SSS scores and UFS-QoL HRQoL total scores.
Safety outcomes included were hot flushes and headaches. According to the sponsor’s NMA technical report, networks could not be constructed for the safety outcomes of BMD loss, hypertension, fatigue, or nausea involving RGX-E2-NETA and LA.
ITC Analysis Methods
Bayesian NMAs were conducted for each outcome, as outlined in Table 36.
No prespecified statistical analysis plan was identified for the NMA.
Table 36: Analysis Methods in ITC 1
Methods | Description |
|---|---|
Analysis methods |
|
Priors |
|
Assessment of model fit |
|
Homogeneity |
|
Assessment of consistency |
|
Assessment of convergence |
|
Outcomes | Efficacy:
Safety:
Tolerability:
|
Follow-up time points | MBL resolution LIBERTY 1 and LIBERTY 2 trials:
PEARL 1 and PEARL 2 trials:
Hot flushes, headaches LIBERTY 1 and LIBERTY 2 trials:
PEARL 1 and PEARL 2 trials:
PEARL 4 trial:
VENUS 1 trial:
VENUS 2 trial:
Discontinuation due to TEAEs PEARL 1, PEARL 2, and PEARL 4 trials:
VENUS 1 and VENUS 2 trials:
|
Construction of nodes | Nodes in each network were constructed based on experimental or control drugs, with separate nodes for different dosages of UPA and MFP. For networks in efficacy and safety outcomes, the nodes comprised:
|
Sensitivity analyses | For each of the outcomes, sensitivity analyses could have been conducted for outcome definition, similarity assessment, and statistical homogeneity and inconsistency assessment. Time point was assessed for hot flushes and headaches. |
Subgroup analysis | Not performed |
Methods for pairwise meta-analysis | Not reported |
DIC = deviance information criterion; EOT = end of treatment; HRQoL = health-related quality of life; LA = leuprolide acetate; MBL = menstrual blood loss; MFP = mifepristone; NMA = network meta-analysis; PBO = placebo; PSRF = potential scale reduction factor; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SLR = systematic literature review; SSS = Symptom Severity Scale; TEAE = treatment-emergent adverse event; UFS-QoL = Uterine Fibroid Symptoms Quality of Life questionnaire; UPA = ulipristal acetate.
aThe cumulative dose of LA administered after 3 months is equivalent for 3.75 mg per month and 11.75 mg every 3 months; both are reported as LA.
Source: Details included in the table are from the sponsor’s SLR and NMA report.20
Results of the ITC
Summary of Included Studies
A summary of the included studies is presented in Table 37. Networks for all outcomes included nodes for RGX-E2-NETA, LA 3.75 mg, UPA 5 mg, UPA 10 mg, and placebo:
The NMA for MBL included data from 4 studies (the LIBERTY 1, LIBERTY 2, PEARL 1, and PEARL 2 trials).
The NMA for UFS-QoL (HRQoL total score and symptom severity) included data from 7 studies (the LIBERTY 1, LIBERTY 2, MYOMEX, VENUS 1, VENUS 2, PEARL 2, and PEARL 4 trials).
The NMA for hot flushes included data from 5 studies (the LIBERTY 1, LIBERTY 2, PEARL 2, PEARL 4, and VENUS 1 trials).
The NMA for headaches included 5 studies (the LIBERTY 1, LIBERTY 2, PEARL 1, PEARL 2, and PEARL 4 trials).
The NMA for discontinuation due to adverse events included 8 studies (the LIBERTY 1, LIBERTY 2, ASTEROID 2, PEARL 1, PEARL 2, PEARL 4, VENUS 1, and VENUS 2 trials).
CDA-AMC selected outcomes that were considered most relevant to inform the expert committee deliberations and finalized this list of end points in consultation with members of the expert committee.
A sample network for MBL resolution is provided in Figure 3. The networks for UFS-QoL HRQoL total score, UFS-QoL SSS score, headaches, hot flushes, and discontinuations due to adverse events contained the same connections; however, the number of patients contributing to each node and the number of studies contributing to each comparison sometimes differed.
Figure 3: Evidence Network Diagram for MBL Resolution
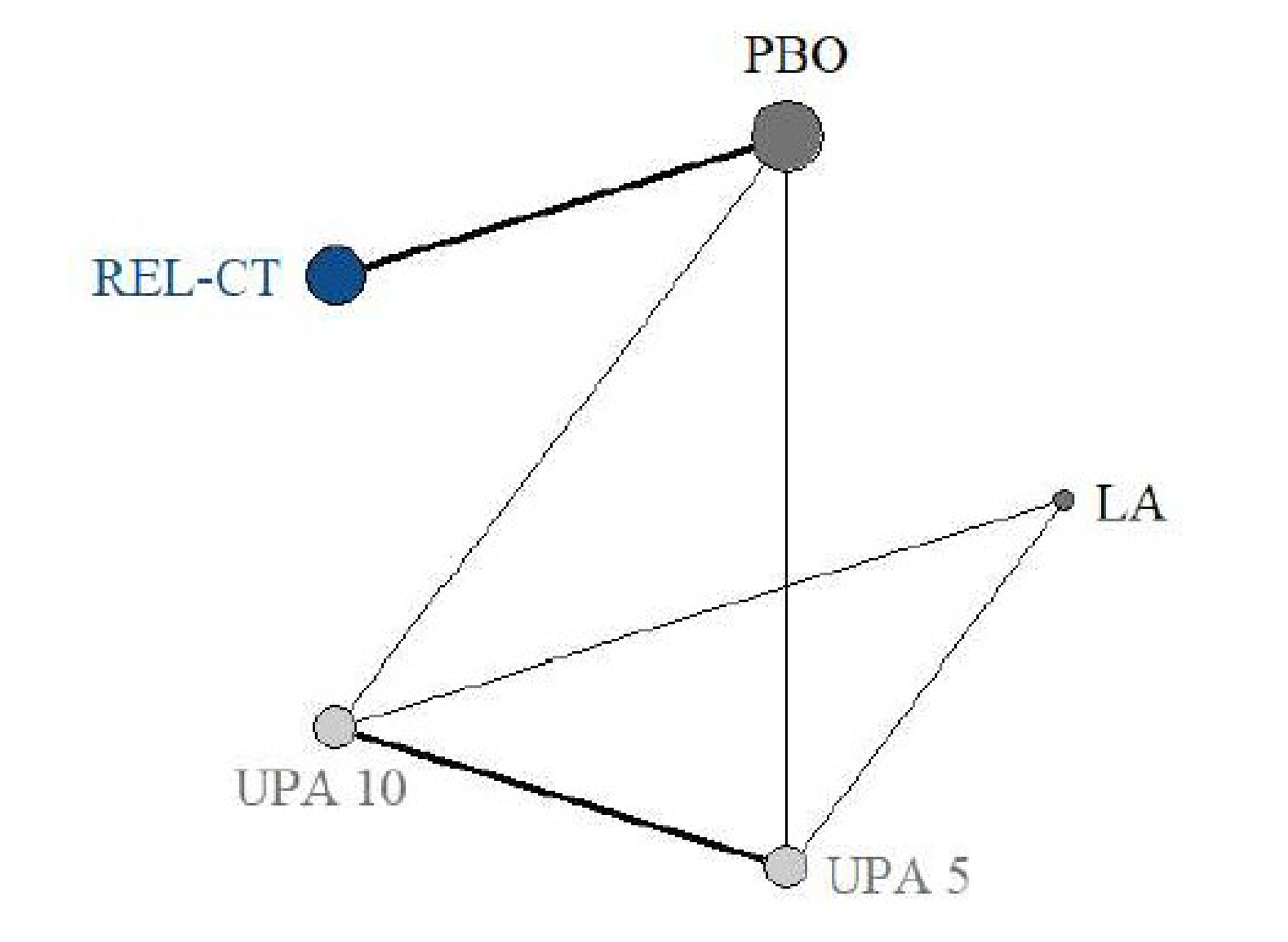
LA = leuprolide acetate; PBO = placebo; REL-CT = relugolix combination therapy; UPA 5 = ulipristal acetate 5 mg; UPA 10 = ulipristal acetate 10 mg.
Source: Myfembree A Systematic Review and Network Meta-Analysis Report.20
Table 37: Summary of Studies Included in the NMA — Reported Definitions for Outcomes
Study | Interventions | N | Efficacy outcomes | Safety outcomes | Tolerability outcomes |
|---|---|---|---|---|---|
LIBERTY 1 trial | PBO RGX-E2-NETA | 127 128 | MBL resolution MBL volume < 80 mL and ≥ 50% reduction from baseline to the last 35 days of treatment at week 24 UFS-QoL HRQoL total score UFS-QoL SSS score | Hot flushes, headaches, all EOT at 24 weeks | Discontinuation due to AEs at 24 weeks |
LIBERTY 2 trial | PBO RGX-E2-NETA | 129 125 | |||
PEARL 1 trial | PBO UPA 5 mg UPA 10 mg | 48 94 93 | MBL resolution PBAC score < 75, summed over the 28 days, at week 13 UFS-QoL HRQoL total score UFS-QoL HRQoL SSS score | Hot flushes not reported Headaches: up to 4 weeks after EOT, at 13 weeks | Discontinuation due to AEs included at 13 weeks |
PEARL 2 trial | LA 3.75 mg UPA 5 mg UPA 10 mg | 92 93 95 | Hot flushes, headaches up to 4 weeks after EOT, at 13 weeks | ||
PEARL 4 trial | UPA 5 mg UPA 10 mg | 216 207 | Excluded in the base case (included in sensitivity analysis) MBL resolution UFS-QoL HRQoL total score UFS-QoL SSS score | Hot flushes, headaches up to 1 week after EOT, at 12 weeks | |
ASTEROID 2 trial | PBO UPA 5 mg | 20 74 | Not reported | Not reported | Discontinuation due to AEs included at 12 weeks |
Irahara (2019) trial | PBO UPA 5 mg UPA 10 mg | 22 22 25 | Not reported | Hot flushes excluded in the base case; included in a sensitivity analysis for time point up to 12 weeks after EOT (at 12 weeks) Headaches not reported | Not reported |
VENUS 1 trial | PBO UPA 5 mg UPA 10 mg | 56 53 48 | Not reported UFS-QoL HRQoL total score UFS-QoL SSS score | Hot flushes, EOT at 12 weeks Headaches not reported | Discontinuation due to AEs included at 12 weeks |
VENUS 2 trial | PBO UPA 5 mg UPA 10 mg | 113 162 157 | Excluded in the base case, included in a sensitivity analysis for time point up to 2 menstrual cycles after EOT at 12 weeks | ||
MYOMEX trial | LA UPA 5 mg | 23 26 | MBL resolution not reported UFS-QoL HRQoL total score UFS-QoL SSS score | Not reported | Not reported |
AE = adverse event; EOT = end of treatment; LA = leuprolide acetate; MBL = menstrual blood loss; NMA = network meta-analysis; PBAC = pictorial blood loss assessment chart; PBO = placebo; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptoms Quality of Life questionnaire; UPA = ulipristal acetate.
Source: Myfembree A Systematic Review and Network Meta-Analysis Report.20
Assessment of Similarity
Table 38 outlines an assessment of similarity across studies.
Table 38: Assessment of Similarity for Studies in the NMAs
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Mean age at baseline
Ethnicity
Mean fibroid volume at baseline
Mean and median uterine volume at baseline
Mean MBL at baseline
|
Dosing of comparators |
(The cumulative dose administered after 3 months is equivalent for 3.75 mg monthly and 11.75 mg every 3 months; both are reported as LA.) |
Route of administration |
|
Definitions of end points | Two definitions were provided for MBL resolution, based on either of the following:
HRQoL was reported using the UFS-QoL in the LIBERTY 1 and LIBERTY 2 trials and in the MYOMEX, PEARL 2, VENUS 1, and VENUS 2 trials:
Safety outcomes of hot flushes and headaches had to be assessed in a manner consistent with the LIBERTY 1 and 2 trials:
For discontinuation due to AEs, no time frame restriction was applied; therefore, assessment times were:
|
Timing of end point evaluation | End point evaluation by study occurred at EOT and varied by study as described here. LIBERTY 1 and LIBERTY 2 trials:
Safety outcomes (hot flushes, headaches):
Discontinuation due to AEs:
|
AE = adverse event; CDA-AMC = Canada’s Drug Agency; EOT = end of treatment; HRQoL = health-related quality of life; IM = intramuscular; LA = leuprolide acetate; MBL = menstrual blood loss; NMA = network meta-analysis; PBAC = pictorial blood loss assessment chart; SC = subcutaneous; SD = standard deviation; SSS = Symptom Severity Scale; UFS-QoL = Uterine Fibroid Symptom Quality of Life questionnaire.
aLiu (2017) was excluded from the CDA-AMC evaluation of the NMA because the study did not provide clinically meaningful outcomes.
Source: Myfembree A Systematic Review and Network Meta-Analysis Report.20
Risk of Bias of the Included Studies
A Cochrane risk of bias assessment was provided by the sponsor. In 17% of the studies (2 out of 12 studies), the risk of bias was high due to allocation concealment, and in 1 of these 2 studies, the risk of bias was also high for performance (i.e., blinding of participants). The only 2 outcomes involving these studies were amenorrhea (Irahara [2019]) and all-cause discontinuation (Liu [2017]). The remaining 10 out of 12 included studies had either some concerns or low risk of concerns.
Efficacy Results
The estimated treatment effects of each outcome were quantified as RR with a 95% CrI (Table 39) or as MD with 95% CrI (Table 40).
███████ | | ███████ ██ ████ ████| | ██| | ███ ███ |
|---|---|---|---|
████████ | |||
███ ██████████ | || | ████ | █████ ██ █████ |
██████ | |||
███ ███████ | || | ████ | █████ ██ █████ |
█████████ | || | ██████ | █████ ██ ████ |
████████████ | |||
███████████████ ███ ██ █████ | || | ████ | ██████ █████ |
CrI = credible interval; LA = leuprolide acetate; MBL = menstrual blood loss; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; RR = relative risk; TEAE = treatment-emergent adverse event.
aSample sizes in each study are not reported.
bFor MBL, RR of less than 1 favours LA. For hot flushes, headaches, and discontinuation due to TEAEs, RR of less than 1 favours RGX-E2-NETA.
Source: Myfembree A Systematic Review and Network Meta-Analysis Report.20
███████ | | ███████ ██ ████ ████| | ██| | ███ ███ |
|---|---|---|---|
████████ | |||
███████ █████ █████ █████ | || | █████ | ███████ ██ █████ |
███████ ███████ ████████ | || | ████ | ██████ ██ ██████ |
CrI = credible interval; HRQoL = health-related quality of life; LA = leuprolide acetate; MD = mean difference; UFS-QoL = Uterine Fibroid Symptoms Quality of Life questionnaire.
aSample sizes in each study are not reported.
bFor UFS-QoL HRQoL total score, a negative MD favours LA. For UFS-QoL symptom severity, a positive MD favours LA.
Source: Myfembree A Systematic Review and Network Meta-Analysis Report.20
MBL Resolution
For the comparison of RGX-E2-NETA versus LA (without add-back), the point estimate for the RR for MBL favoured RGX-E2-NETA; however, the 95% CrI was wide and included the possibility that either RGX-E2-NETA or LA (without add-back) could be favoured ███ █ █████ ███ ████ ████ ██ █████.
A sensitivity analysis was conducted for the outcome definition of MBL resolution by including the PEARL 4 trial, which had a different outcome definition than the LIBERTY 1 and LIBERTY 2 trials. The results of this sensitivity analysis were consistent with those of the base-case analysis, with a wide 95% CrI, and included the possibility that either RGX-E2-NETA or LA (without add-back) could be favoured ███ █ █████ ███ ████ ████ ██ █████.
The sponsor reported I2 values of less than 50% for all direct and indirect comparisons of MBL resolution. For the comparison of UPA 5 mg versus UPA 10 mg for MBL resolution, I2 was 33.5%.
Consistency of MBL resolution could not be assessed because this outcome did not have direct and indirect comparisons.
Uterine Fibroid Symptoms Quality of Life
In the absence of a published MID, expert opinion was considered to estimate a clinically meaningful threshold of 15 points. Based on this value, the MDs between RGX-E2-NETA and LA for the UFS-QoL HRQoL total and SSS scores likely do not meet the threshold; therefore, these are likely not different than one another.
Both point estimate MDs of the UFS-QoL HRQoL total and SSS scores favoured LA (without add-back); however, the magnitude of the MDs did not reach a clinically important threshold of 15, as suggested by the clinical expert. The 95% CrIs crossed the line of equivalence, suggesting that either RGX-E2-NETA or LA (without add-back) could be favoured:
███████ █████ ██████ ██ ██ █████ ██████ ████ ████ ██████ ██ ████ ████████████████ ███████ █████████ ██ ██ ████ ██████ ████ ████ █████ ██ █████ █████████
For UFS-QoL HRQoL total score, 2 sensitivity analyses were performed. The first excluded 2 studies in which less than 10% of patients were Black (the PEARL 2 and PEARL 4 studies), and the second excluded the PEARL 4 study, which showed a treatment effect in the opposite direction from that of the other studies (i.e., the PEARL 1, VENUS 1, and VENUS 2 studies).
The conclusions drawn from the sensitivity analyses were similar to those drawn from the base-case analysis, favouring LA (without add-back). The 95% CrIs crossed the line of equivalence, suggesting that either RGX-E2-NETA or LA (without add-back) could be favoured:
███████ █████ █████ ████████ ██ █ ███████ ████ █ ███ ████████ ███ ████ ███████ █████ ██████ ████ ████ ██████ ██ ████ ████████████████ █████ █████ ████████ ██ █████ ████ ████ ███████████████ █████ ██████ ████ ████ ██████ ██ ████ █████████
For the UFS-QoL SSS score, 2 sensitivity analyses were conducted using the same approach as that used for the UFS-QoL HRQoL total score. First, the authors excluded 2 studies in which less than 10% of patients were Black (i.e., the PEARL 2 and PEARL 4 studies); the second excluded the PEARL 4 study, which showed a treatment effect in the opposite direction of the other studies (i.e., the PEARL 1, VENUS 1, and VENUS 2 studies).
The conclusions drawn from the sensitivity analyses were similar to those drawn from the base-case analysis, favouring LA (without add-back); however, the magnitude of the MDs did not reach the clinically important threshold of 15. The 95% CrIs crossed the line of equivalence, suggesting that either RGX-E2-NETA or LA (without add-back) could be favoured:
███████ ███████ ████████ █████ ████████ ██ █ ███████ ████ █ ███ ████████ ███ ████ ███████ ████ ██████ ████ █████ ██████ ██ █████ ████████████████ ███████ ████████ █████ ████████ ██ █████ ████ ████ ███████████████ ████ ██████ ████ █████ █████ ██ █████ █████████
Key differences across studies were reported in UFS-QoL scores with an I2 greater than or equal to 50%:
UFS-QoL (HRQoL total score): UPA 5 mg to UPA 10 mg, with an I2 of 72.7%
UFS-QoL score (symptom severity): UPA 5 mg to UPA 10 mg, with an I2 of 59.1%.
Consistency was assessed for 1 node only for the following outcomes using the node-splitting analysis:
For UFS-QoL HRQoL total score, consistency was assessed between comparators UPA 10 mg and LA, with a P value of 0.61.
For UFS-QoL SSS score, consistency was assessed between comparators UPA 10 mg and LA, with a P value of 0.55.
Harms Results
Hot Flushes
RGX-E2-NETA was associated with a lower risk of hot flushes than LA (without add-back), with an RR of | ████ ████ █ ██ ██████ A sensitivity analysis was conducted that included studies beyond 1 month after the end of treatment. The results of this sensitivity analysis were consistent with those of the base-case analysis, with an RR of ████ ████ ████ ████ ██ ██████
Headaches
For the outcome of headaches, the RR estimate was imprecise, suggesting that the direction of the effects are inconclusive. The point estimate for the risk ratio for headaches favoured LA without add-back; however, the 95% CrI was wide, including the potential that either treatment could be favoured ███ █ ███████ ███ ███ ████ ██ ██████████
A sensitivity analysis was conducted by including a trial (the VENUS 2 study) with a different time point estimate (i.e., reporting headaches for up to 2 menstrual cycles after the end of treatment). The results of this analysis were consistent with those of the base-case analysis, with a similarly high level of imprecision. This sensitivity analysis suggests that either RGX-E2-NETA or LA (without add-back) could be favoured ███ █ ███████ ███ ████ ████ ██ █████████
For the safety outcomes, in the comparison of UPA 5 mg versus UPA 10 mg, heterogeneity was low (I2 = 0% for hot flushes and I2 = 6.9% for headaches).
Discontinuation Due to TEAEs
The RR estimate of the tolerability outcome discontinuation due to TEAEs favoured RGX-E2-NETA with a high degree of imprecision, with an RR of ████ ████ ████ ████ ██ █████. The 95% CrI also includes values that suggest that either RGX-E2-NETA or LA (without add-back) could be favoured. No sensitivity analysis was reported for this outcome.
For discontinuations due to TEAEs, heterogeneity between RGX-E2-NETA and placebo was low (I² = 18.6%). Consistency between UPA 10 mg and placebo was supported (P = 0.35).
Critical Appraisal of ITC
A systematic review of published and grey literature was conducted to identify studies for the NMA. A prespecified protocol for the SLR and NMA could not be obtained. As such, it is unclear whether the analyses were preplanned and whether the reported analyses were selected based on their results (i.e., direction and/or magnitude of effect). The CDA-AMC review team appraised the submitted SLR using the criteria from A MeaSurement Tool to Assess systematic Reviews 2.21
Overall, the searches were comprehensive, and the methods for data extraction and risk of bias appraisal were adequate to reduce the risk of error and bias in these procedures. Although study selection was undertaken by 2 independent reviewers with consensus, there was inconsistent rationale for why certain studies were excluded from the networks. Of the 31 studies identified through the SLR, 12 studies were retained for the NMAs. Although certain studies of comparators not used in Canada were included in the networks to allow for connections between RGX-E2-NETA and LA, other studies with the same comparators were excluded. For example, Bagaria (2009) — comparing placebo and MFP 10 mg — was included to connect the network, whereas other studies that included a treatment group receiving MFP 10 mg were excluded because the treatments were not of interest. The selective inclusion of studies in the network may result in risk of bias due to missing results in the syntheses; the magnitude and direction of this bias cannot be predicted.
A Bayesian NMA included some outcomes, but not all, including effectiveness, safety, and tolerability. Outcomes deemed relevant by patients and clinicians that were not included in the constructed networks were efficacy outcomes of improvements in anemia and reduction in pain, and the harms outcomes of BMD. BMD as a safety outcome was not included due to lack of comparative data. As such, this NMA cannot inform on the comparative effects of RGX-E2-NETA versus LA for these outcomes.
There are limitations to both fixed-effects and random-effects models, both of which were used in this NMA. Models were selected based on better fit (i.e., a smaller deviance information criterion). Fixed-effects models were used for outcomes such as MBL resolution, hot flushes, headaches, and discontinuation due to TEAEs. Random-effects models with informative priors56 were used for the UFS-QoL outcomes (i.e., HRQoL total and SSS scores). The selection of informative priors in this context was appropriate. Using fixed-effects models, the width of the 95% CrI may underestimate the uncertainty arising from the between-study heterogeneity because treatment effects between studies are assumed to be identical. Using random-effects models, accounting for between-study heterogeneity can lead to wider CrIs and greater uncertainty in estimating the effect sizes. Given that most outcomes were modelled with fixed effects, we expect that the true magnitude of the uncertainty was underestimated. Because the effect estimates for these end points were already affected by important imprecision, reliance on the results of the unselected models would not have changed the review team’s conclusions about the estimated effects.
A key limitation of this NMA is that the clinically relevant comparator LA did not contain add-back. Long-term use of LA is typically given with add-back to mitigate the hypoestrogenic effects of BMD loss and vasomotor symptoms. While hormonal add-back is a part of RGX-E2-NETA, according to the sponsor, LA with add-back was excluded from the NMA due to feasibility reasons. This means that RGX-E2-NETA compared with LA (without add-back) does not reflect how the actual comparator is prescribed in Canada.
The efficacy outcome of MBL, the safety outcome of headaches, and the tolerability outcome of discontinuation due to TEAEs crossed the null, with very wide CrIs. These results suggest that either RGX-E2-NETA or LA (without add-back) could be favoured, rendering the NMAs inconclusive for the outcomes.
For the HRQoL outcome measured using the UFS-QoL, the point estimates of the UFS-QoL HRQoL total score MD and UFS-QoL SSS score MD did not exceed an estimated 15-point threshold of clinical meaningfulness; both favoured LA (without add-back). The 95% CrI for both total score MD and SSS score MD also crossed the line of equivalence, suggesting that either RGX-E2-NETA or LA (without add-back) could be favoured. Another limitation of relying on the UFS-QoL was that the time point of measurement was unclear and could have differed across studies. Although the sponsor has stated that the effect of RGX-E2-NETA is relatively stable after 4 weeks of treatment, no supportive evidence was provided for the intervention or for the stability of the treatment effect for the comparator. In addition, there was insufficient detail on how stable treatment effect is defined across studies. This makes comparisons of HRQoL across studies more difficult. There were other notable limitations of the overall NMA because the assumptions of exchangeability, similarity of outcomes, and consistency (coherence) could not be satisfied.
The exchangeability (transitivity) assumption was likely not satisfied. First, the sponsor did not report a methodology on how effect modifiers were identified. The reported effect modifiers, such as ethnicity, fibroid volume, and MBL at baseline, were not reported across all included trials. Because the number of studies was small (i.e., < 10 studies per comparison), meta-regression methods were not feasible. As such, consistency in these effect modifiers across the included trials could not be comprehensively assessed. If reported, sensitivity analyses could not remove the 4 studies in which less than 10% of patients were Black because the studies were essential for connecting the nodes in the network. Second, the location of UFs, which directly affects MBL resolution, is an important effect modifier that was not assessed by the sponsor, according to the clinical expert consulted for this review. Location of UFs is graded using the International Federation of Gynecology and Obstetrics (FIGO) classification system. In this system, submucosal myomas are FIGO type 0, 1, or 2, and intramural myomas are FIGO type 3, 4, or 5.15
Potential imbalances in the distribution of effect modifiers among the included trials — such as location of UFs across studies (not reported), mean age of trial participants, and the percentage of patients who are Black — can cause bias and reduce the validity of the NMA.57
Definitions of the relevant outcomes were variable, as reported for MBL resolution (Table 37). MBL used both objective alkaline hematin methods and a partially subjective pictorial blood loss assessment chart. The time points of outcome evaluation were heterogeneous (Table 37). The longer follow-up times in the LIBERTY 1 and LIBERTY 2 trials, in which assessment occurred in the last 35 days of treatment at week 24, could have facilitated a different MBL resolution than all the other studies, in which MBL resolution was evaluated at 12 weeks or 13 weeks.
The sponsor reported statistical heterogeneity using I2, a quantification of the proportion of variation in posterior estimates due to among-study differences,58 which was low overall across studies (< 50%). The largest magnitude of heterogeneity was reported for the outcomes of UFS-QoL (HRQoL total score and SSS score) for UPA 5 mg versus UPA 10 mg, which are no longer available in Canada. In the absence of additional information (e.g., estimated effects from the individual contributing studies), more nuanced appraisals of potential heterogeneity could not be undertaken. Therefore, the importance of such heterogeneity is uncertain.59
The consistency (also known as coherence) assumption could not be assessed for all outcomes. The networks were sparse, consisting of many comparisons from a total of 12 studies, with a small number of studies contributing to each node. Consistency was assessed for outcomes using the node-splitting approach for the outcomes of UFS-QoL (HRQoL total score and SSS score) and discontinuation due to TEAEs. Consistency could not be assessed for MBL resolution in the absence of direct and indirect evidence. The sponsor’s reliance on statistical significance is insufficient to demonstrate the consistency assumption, given that statistical tests for incoherence have low power and may fail to detect incoherence when it is present.60
Risk of bias assessment at the study level was evaluated using the Cochrane risk of bias tool for randomized trials.54 In 17% of the studies (2 out of 12 studies), the risk of bias was high due to selection bias (allocation concealment); in 1 of these 2 studies, the risk of bias was high in performance (blinding of participants). The sponsor conducted sensitivity analyses removing studies with high risk of bias; therefore, the networks for the outcomes included in the current review did not include any studies with high risk of bias.
Notably, risk of bias was assessed at the study level rather than at the level of the reported effects (i.e., for each outcome of interest). This methodology ignores the fact that risk of bias can vary depending on the effect estimate being evaluated, particularly for such domains as performance, detection, attrition, and reporting bias. As such, the risk of bias reported by the sponsor for each study may not apply universally to MBL resolution, UFS-QoL HRQoL total score, UFS-QoL SSS score, or harms and tolerability outcomes.
Overall, because of the absence of an RGX-E2-NETA versus LA (with add-back) comparison, the high degree of imprecision represented by RR or MD CrIs that can favour either RGX-E2-NETA or LA (without add-back), and uncertainty related to the key assumptions required for an NMA (i.e., exchangeability, homogeneity, and consistency), definitive conclusions on the relative treatment effects of RGX-E2-NETA versus LA (without add-back) cannot be drawn based on this NMA.
Studies Addressing Gaps in the Systematic Review Evidence
There were no studies addressing gaps in the systematic review evidence submitted by the sponsor.
Discussion
Summary of Available Evidence
The evidence included in this review consisted of 2 pivotal studies identified through the sponsor’s systematic review (the LIBERTY 1 trial and the LIBERTY 2 trial), 2 extension studies (the LIBERTY extension trial and the LIBERTY RWS), and 1 sponsor-submitted ITC. There were no additional studies addressing gaps in the evidence submitted by the sponsor.
The LIBERTY 1 trial (N = 388) and LIBERTY 2 trial (N = 382) were phase III, double-blind, placebo-controlled RCTs evaluating the efficacy and safety of RGX-E2-NETA (40 mg RGX, 1 mg E2, and 0.5 mg NETA) (n = 128 and n = 126 in the LIBERTY 1 and LIBERTY 2 trials, respectively) once daily compared to placebo (n = 128 and n = 129, respectively) at 24 weeks. Eligible patients were females aged 18 to 50 years with a confirmed diagnosis of UFs and HMB (defined as an MBL volume of 160 mL or greater during 1 menstrual cycle or 80 mL or greater per cycle for 2 cycles), no gynecological surgery or procedure within 6 months before treatment or following enrolment, and no history or current metabolic bone disease or treatment for BMD loss. The primary outcome was MBL volume response, defined as patients who experienced an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL volume. Other clinically relevant outcomes included improvements in anemia, pain, symptom severity, and HRQoL. The mean age across the studies was approximately 42 years. Baseline clinical characteristics were generally balanced between the groups, apart from slight imbalances in race and ethnicity (with a higher proportion of patients who were Black and a lower proportion of patients who were white in the placebo groups for both studies), MBL (between treatment groups for both studies), and pain scores (between treatment groups in the LIBERTY 1 trial).
The LIBERTY extension trial (N = 327) was a single-arm, phase III, 28-week OLE study involving patients who had completed either pivotal trial. In the OLE study, the long-term efficacy and safety of RGX-E2-NETA were evaluated for an additional 28 weeks (after the 24 weeks of treatment during either parent study). The LIBERTY RWS was a double-blind, phase III, placebo-controlled study enrolling patients who had completed the LIBERTY extension trial and responded to treatment. The LIBERTY RWS rerandomized patients 1 to 1 to receive either RGX-E2-NETA or placebo once daily for up to 52 weeks. For patients whose HMB recurred during the LIBERTY RWS (i.e., MBL volume ≥ 80 mL), blinded treatment was stopped, and re-treatment with open-label RGX-E2-NETA was offered. MBL response, improvement in anemia (in the LIBERTY extension trial only), and UFS-QoL scores were the main efficacy outcomes of interest. Baseline characteristics and imbalances were generally similar to those reported for the pivotal studies.
Due to the lack of evidence directly comparing RGX-E2-NETA with relevant therapies for the treatment of HMB associated with UFs, an ITC was summarized and appraised. An NMA was used to assess the relative efficacy and safety of RGX-E2-NETA versus LA (without add-back) for MBL response, UFS-QoL HRQoL total and SSS scores, safety, and tolerability. Other treatments (i.e., UPA and MFP) were used to establish the network connection between RGX-E2-NETA and LA; however, UPA and MFP were excluded in the comparison (because UPA has been withdrawn from the market, and MFP is not indicated for the treatment of UFs in Canada). Comparison to LA with add-back, which is used in clinical practice when LA is given for more than 3 months, was not possible because relevant studies that could connect to the networks were not available.
Interpretation of Results
Efficacy
UFs are a common type of uterine tumour and are associated with HMB, anemia, pain, and bulk symptomatology that interfere with HRQoL and daily living.1 The overall goals of treatment are to reduce menstrual bleeding and other symptoms and increase hemoglobin levels to improve HRQoL and allow for uninterrupted activities of daily living.6,7 Input from patients emphasized the need for safe and effective options that address the goals of treatment and do not require 3-month dosage commitments.
In clinical practice, HMB is defined as MBL that interferes with a person’s HRQoL. It is not quantified as it is in clinical trials (i.e., as more than 80 mL of blood loss during each menstrual cycle). After 24 weeks of treatment in the pivotal studies, high-certainty evidence demonstrated that more patients experienced disease response (i.e., an MBL volume of less than 80 mL and a 50% or greater reduction from baseline MBL volume) in the RGX-E2-NETA group compared to the placebo group. This treatment effect (between-group difference in the LIBERTY 1 trial = 54.5%; 95% CI, 44.3% to 64.7%; between-group difference in the LIBERTY 2 trial = 56.5%; 95% CI, 46.6% to 66.5%) was considered clinically meaningful, according to expert opinion, and statistically significant.
Because treatment continued for an additional 28 weeks in the OLE study, responder rates increased in the group of patients who continued to receive RGX-E2-NETA (87.7%; 95% CI, 81.7% to 92.3%); patients who switched from placebo to active treatment experienced disease response, but to a lesser degree (75.6%; 95% CI, 68.3% to 82.0%). However, the study lacked a randomized comparator, which precludes the ability to draw causal conclusions; and patients in the OLE study represented those who stayed on treatment and did not discontinue or have intolerable TEAEs. Responder rates in the LIBERTY RWS (in which patients were rerandomized) were similar to those observed in the pivotal studies at week 76 (63.4%; 95% CI, 52.9% to 73.9%) and week 104 (58.0%; 95% CI, 47.0% to 69.1%); however, only patients who had demonstrated a treatment response in the LIBERTY extension trial were eligible to enrol in the RWS. When RGX-E2-NETA was compared to LA (without add-back) in the NMA, the RR favoured the former; however, the results were inconclusive due to imprecision (i.e., wide 95% CrIs indicated that either treatment could be favoured). In addition, important methodological limitations prevent firm conclusions from being drawn regarding the comparative efficacy of the 2 treatments.
Improvement in anemia was assessed in a subset of patients whose baseline hemoglobin level was 10.5 g/dL or lower and who experienced an increase of more than 2 g/dL from baseline. In the pivotal studies, a greater proportion of patients who received RGX-E2-NETA compared to placebo experienced an improvement in anemia. The responder rate was notably lower in the LIBERTY 1 trial (between-group difference of 28.3%; 95% CI, 3.7% to 52.8%) compared to the LIBERTY 2 trial (between-group difference of 55.9%; 95% CI, 37.3% to 74.5%); and although it was unclear why, the difference could have been caused by the small number of patients contributing to the analysis and the loss of prognostic balance across treatment groups stemming from the use of a subpopulation in both studies. While the treatment effect of RGX-E2-NETA versus placebo in both studies was considered clinically meaningful, based on expert opinion, in the LIBERTY 1 trial, the lower bound of the 95% CI suggests that the treatment effect may not be clinically meaningful to patients. The evidence supporting this outcome was of low certainty due to the study limitations and imprecision previously described. The magnitudes of the responder rates in the OLE study were similar to those observed in the LIBERTY 2 trial. Improvement in anemia was not assessed in the LIBERTY RWS or the ITC.
Improvement in pain was evaluated in a subset of patients whose maximum pain NRS score was 4 points or greater before treatment and who experienced a maximum pain NRS score of 0 points or 1 point for UF-associated pain at the end of the study. Low-certainty evidence from the pivotal studies indicated that a greater proportion of patients who received RGX-E2-NETA compared to placebo experienced an improvement in UF-associated pain. The responder rate was higher in the LIBERTY 1 trial (between-group difference of 27.7%; 95% CI, 15.4% to 40.0%) compared to the LIBERTY 2 trial (between-group difference of 18.7%; 95% CI, 6.2% to 31.1%). As with the anemia improvement outcome, the results for pain improvement were based on a subpopulation in which prognostic balance may have been lost, thereby increasing the risk of bias. Although the treatment effect was considered clinically meaningful, the lower bound of the 95% CI in the LIBERTY 2 trial may include an effect that is not considered meaningful to patients; thus, the GRADE was rated down for imprecision. Improvements in pain were not assessed in the extension study or the ITC.
UF-associated HMB, whose symptoms contribute to the decreased HRQoL that patients experience, was highlighted as an important unmet need in both the patient group input and the clinical expert input. In the pivotal studies, patients who received RGX-E2-NETA compared to placebo showed a greater decline in symptom severity and increase in HRQoL, as measured using the UFS-QoL. The certainty of evidence for both outcomes was moderate due to the risk of bias arising from missing data (19% to 26% across the studies). Although there is no known threshold for a clinically important effect, the clinical expert indicated that the treatment effect was likely to be clinically meaningful to patients. The magnitude of effect in the OLE study was similar to that seen in the RGX-E2-NETA group from the pivotal studies for both change in symptom severity and HRQoL. In the LIBERTY RWS, patients randomized to RGX-E2-NETA continued to maintain the improvements they had gained from active treatment in the OLE study, while patients randomized to placebo showed an increase in symptom severity and a decline in HRQoL. Over time in the LIBERTY RWS, symptom severity and HRQoL tended to improve for the placebo group; however, there were increasing amounts of missing data with longer follow-up times, increasing the risk of bias for these outcomes. When RGX-E2-NETA was compared to LA (without add-back) in the NMA, the point estimate for the RR favoured LA for both the UFS-QoL SSS and HRQoL total scores; however, the 95% CrIs crossed the line of equivalence, suggesting that either treatment could be favoured. Significant methodological limitations prevent firm conclusions from being drawn regarding the comparative efficacy of the 2 treatments.
The ITC results indicated that either treatment could be favoured; imprecision and methodological limitations with the ITC preclude the ability to draw conclusions about the direction of treatment effect. The clinical expert noted that there are practical benefits associated with the use of RGX-E2-NETA, such as ease of administration (i.e., combined oral pills versus intramuscular injections that require separate oral add-back therapy), timing (LA must be timed carefully to prevent symptom flare), and quicker loss of effect once the drug is stopped (compared to a 3-month commitment with the LA injection). The expert was of the opinion that both treatments are expected to be used for similar purposes in practice (that is, for short-term use to address UF-associated HMB). As per the expert, RGX-E2-NETA would be used as a preoperative treatment to reduce the size of UFs, reduce HMB, and improve low hemoglobin in anticipation of surgery. The expert explained that doing so can facilitate less invasive surgical or medical procedures and reduce the risks of surgery (e.g., significant blood loss). RGX-E2-NETA could also be used to avoid surgery or as a bridging therapy before menopause, when symptoms tend to subside. The Health Canada product monograph states that the use of RGX-E2-NETA should be limited to 24 months due to the risk of irreversible bone loss. While treatment beyond 24 months may be an option for patients whose disease is responding well to treatment and who are not experiencing clinically significant bone loss, these are expected to be rare cases. In such instances, regular BMD assessments would be necessary to monitor bone health and inform decisions about continuing or stopping treatment.
The sponsor indicated that RGX-E2-NETA would be used in patients who wish to maintain fertility, preserve their uterus, and reduce their symptoms while avoiding surgery or other procedures. Patients in the studies were not expected to undergo gynecological surgeries or procedures within 6 months of enrolment. Considering the expected and current use of RGX-E2-NETA in practice (as per the clinical expert) and the fact that outcomes for fertility, uterus preservation, and avoidance of surgical or medical procedures were not investigated, evidence from the pivotal studies may not fully inform on the sponsor’s suggested place in therapy.
Harms
In the LIBERTY 1 and LIBERTY 2 trials, up to two-thirds of patients in each treatment group experienced at least 1 TEAE; in general, the types and frequences of TEAEs were similar between the treatment groups in either study and across the studies. The clinical expert consulted for this review indicated no concerns with the safety profile of RGX-E2-NETA and stated that TEAEs would generally be manageable for patients. Reports of SAEs and patients withdrawing from treatment due to TEAEs were low across the studies. There were no deaths.
TEAEs in the LIBERTY extension trial and the LIBERTY RWS were similar to those of the pivotal studies. There did not appear to be any new safety signals with continued use of RGX-E2-NETA. SAEs and withdrawals due to TEAEs were generally low across the studies. There were no deaths.
Based on the ITC results, RGX-E2-NETA was associated with a lower risk of hot flushes, but not headaches, compared to LA (without add-back). LA without add-back is known to exacerbate vasomotor symptoms (including hot flushes) and is a reason for having add-back included in RGX-E2-NETA. The observed difference may reflect the effect of add-back therapy in RGX-E2-NETA; thus, the difference in effect between the drugs in the ITC may not be relevant. Results comparing discontinuations due to TEAEs were uncertain due to imprecision, indicating that either treatment could be favoured for these outcomes. Because RGX-E2-NETA and LA (with or without add-back) have not been directly compared in a robust RCT — and the sponsor-submitted ITC did not include LA with add-back — it is not possible to say with uncertainty that the drugs have the same safety or efficacy. The indirect evidence available for review also had methodological issues, and the key assumptions of the NMAs could not be satisfied, preventing definitive conclusions from being made.
According to the clinical expert consulted on this review, loss of BMD was noted as a TEAE of special interest for treatment with RGX-E2-NETA. In both pivotal studies, there was a numerically larger decrease in BMD among patients who received RGX-E2-NETA compared to placebo after 24 weeks of treatment. With no known clinically meaningful threshold, it is unclear how significant the change in BMD is. Decreases in BMD were numerically larger at the end of the LIBERTY extension trial, suggesting that bone loss may continue with longer treatment times; conversely, BMD measures in the rerandomized RGX-E2-NETA group were numerically greater compared to the rerandomized placebo group in the LIBERTY RWS. The reason for the rebound is unclear, and the wide 95% CI for the between-group difference that includes 0 suggests the possibility that there is no difference between the treatment groups. Limitations in the study designs and missing data amplify the uncertainty in these estimates. The clinical expert stated that to mitigate BMD decline, preventive strategies for bone health are implemented with these types of drugs. Furthermore, it would be important to monitor BMD with prolonged use of RGX-E2-NETA; however, most patients are expected to use the treatment for a short duration (months rather than years). Bone loss may be less of a concern if treatment remains short.
Conclusion
Evidence from 2 phase III, double-blind RCTs (the LIBERTY 1 trial and the LIBERTY 2 trial) suggests that patients aged 18 to 50 years with confirmed UFs and HMB (defined as an MBL volume of 160 mL or greater in 1 cycle or 80 mL or greater per cycle for 2 cycles) who received RGX-E2-NETA once a day for 24 weeks were more likely to experience treatment response for HMB compared to those who received placebo. Patients receiving RGX-E2-NETA may also have clinically meaningful improvements in anemia, UF-associated pain, symptom severity, and HRQoL (as measured using the UFS-QoL). However, the evidence for these outcomes was uncertain due to an increased risk of bias, lack of known thresholds for a clinically meaningful effect, and missing data. Longer-term evidence suggested that treatment effects were maintained for reduction in MBL and the patient-reported outcomes of symptom severity and HRQoL (with up to 104 weeks of study treatment), as well as for improvements in anemia (with up to 52 weeks of study treatment). Limitations with the study designs and numerous discontinuations increase the uncertainty of the longer-term results. It is also likely that patients who experienced benefits without intolerable harms remained in the studies longer. Results from the ITC of RGX-E2-NETA versus LA (without add-back) indicated that either treatment could be favoured for efficacy outcomes. Additionally, LA with add-back was not included in the ITC, and definitive conclusions on its treatment effect relative to that of RGX-E2-NETA could not be drawn. The incidence and types of harms were generally balanced and similar between the treatment groups in the pivotal studies and across the studies. Longer-term evidence did not indicate new safety signals; however, expert opinion noted that assessment of long-term bone health is important to inform decisions regarding prolonged treatment with RGX-E2-NETA. The Health Canada product monograph notes that use of RGX-E2-NETA should be limited to 24 months due to the risk of bone loss; expert opinion indicated that use in clinical practice would be short-term.
References
1.Wise LA, Laughlin-Tommaso SK. Epidemiology of Uterine Fibroids: From Menarche to Menopause. Clin Obstet Gynecol. 2016;59(1):2-24. doi: 10.1097/GRF.0000000000000164 PubMed
2.Pron G, Cohen M, Soucie J, Garvin G, Vanderburgh L, Bell S. The Ontario Uterine Fibroid Embolization Trial. Part 1. Baseline patient characteristics, fibroid burden, and impact on life. Fertil Steril. 2003;79(1):112-119. doi: 10.1016/S0015-0282(02)04539-9 PubMed
3.Soliman AM, Margolis MK, Castelli-Haley J, Fuldeore MJ, Owens CD, Coyne KS. Impact of uterine fibroid symptoms on health-related quality of life of US women: evidence from a cross-sectional survey. Curr Med Res Opin. 2017;33(11):1971-1978. doi: 10.1080/03007995.2017.1372107 PubMed
4.Magnay JL, Nevatte TM, Dhingra V, O'Brien S. Menstrual blood loss measurement: validation of the alkaline hematin technique for feminine hygiene products containing superabsorbent polymers. Fertil Steril. 2010;94(7):2742-6. doi: 10.1016/j.fertnstert.2010.03.061 PubMed
5.Zimmermann A, Bernuit D, Gerlinger C, Schaefers M, Geppert K. Prevalence, symptoms and management of uterine fibroids: an international internet-based survey of 21,746 women. BMC Womens Health. 2012;12:6. doi: 10.1186/1472-6874-12-6 PubMed
6.Stewart EA. Uterine fibroids (leiomyomas): Treatment overview. UpToDate; 2024. Accessed February 6, 2025. https://www.uptodate.com
7.Vilos GA, Allaire C, Laberge P-Y, et al. The Management of Uterine Leiomyomas. J Obstet Gynaecol Can. 2015;37(2):157-178. doi: 10.1016/s1701-2163(15)30338-8 PubMed
8.Sayed GH, Zakherah MS, El-Nashar SA, Shaaban MM. A randomized clinical trial of a levonorgestrel-releasing intrauterine system and a low-dose combined oral contraceptive for fibroid-related menorrhagia. Int J Gynaecol Obstet. 2011;112(2):126-30. doi: 10.1016/j.ijgo.2010.08.009 PubMed
9.Tulandi T, Casper R. Management of Uterine Fibroids After the Withdrawal of Fibristal. J Obstet Gynaecol Can. 2021;43(7):797-799. doi: 10.1016/j.jogc.2020.12.002 PubMed
10.Laberge PY, Murji A, Vilos GA, Allaire C, Leyland N, Singh SS. Guideline No. 389-Medical Management of Symptomatic Uterine Leiomyomas - An Addendum. J Obstet Gynaecol Can. 2019;41(10):1521-1524. doi: 10.1016/j.jogc.2019.01.010 PubMed
11.Management of Symptomatic Uterine Leiomyomas: ACOG Practice Bulletin, Number 228. Obstet Gynecol. 2021;137(6):e100-e115. doi: 10.1097/aog.0000000000004401 PubMed
12.Myovant Sciences GmbH. Clinical Study Report: MVT-601-3001. LIBERTY 1: An International Phase 3 Randomized, Double-Blind, Placebo-Controlled Efficacy and Safety Study to Evaluate Relugolix Co-Administered with and without Low-Dose Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. January 31, 2020.
13.Myovant Sciences GmbH. Clinical Study Report: LIBERTY 2: An International Phase 3 Randomized, Double-Blind, Placebo-Controlled Efficacy and Safety Study to Evaluate Relugolix Co-Administered with and without Low-Dose Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. January 31, 2020.
14.Spies JB, Coyne K, Guaou Guaou N, Boyle D, Skyrnarz-Murphy K, Gonzalves SM. The UFS-QOL, a new disease-specific symptom and health-related quality of life questionnaire for leiomyomata. Obstet Gynecol. 2002;99(2):290-300. doi: 10.1016/s0029-7844(01)01702-1 PubMed
15.Stewart EA, Laughlin-Tommaso SK. Uterine fibroids (leiomyomas): Epidemiology, clinical features, diagnosis, and natural history. UpToDate; 2024. Accessed February 6, 2025. https://www.uptodate.com
16.Pfizer Canada ULC. Myfembree (relugolix, estradiol, and norethindrone acetate): 40 mg/1 mg/0.5 mg oral tablets [product monograph]. October 13, 2023.
17.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
18.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
19.Knight Therapeutics Inc. Myfembree (Relugolix, estradiol, and norethindrone acetate) for the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. 2023.
20.Knight Therapeutics Inc. Myfembree (40 mg Relugolix, 1 mg Estradiol and 0.5 mg Norethindrone Acetate Tablets) for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids in Premenopausal Women: A Systematic Review and Network Meta-Analysis Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. December 2024.
21.Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. doi: 10.1136/bmj.j4008 PubMed
22.Bulun SE. Uterine fibroids. N Engl J Med. 2013;369(14):1344-55. doi: 10.1056/NEJMra1209993 PubMed
23.Chiuve SE, Huisingh C, Petruski-Ivleva N, Owens C, Kuohung W, Wise LA. Uterine fibroids and incidence of depression, anxiety and self-directed violence: a cohort study. J Epidemiol Community Health. 2022;76(1):92-99. doi: 10.1136/jech-2020-214565 PubMed
24.Kaunitz AM. Abnormal uterine bleeding in nonpregnant reproductive age patients: Terminology, evaluation, and approach to diagnosis. UpToDate; 2024. Accessed February 6, 2025. https://www.uptodate.com/
25.Laberge PY, Vilos GA, Vilos AG, Janiszewski PM. Burden of symptomatic uterine fibroids in Canadian women: a cohort study. Curr Med Res Opin. 2016;32(1):165-75. doi: 10.1185/03007995.2015.1107534 PubMed
26.Government of Canada. Fibristal (ulipristal acetate tablets, 5 mg) - Voluntary Withdrawal in Canada due to Risk of Drug-Induced Liver Injury. 2020. Accessed by sponsor, no date provided. https://recalls-rappels.canada.ca/en/alert-recall/fibristal-ulipristal-acetate-tablets-5-mg-voluntary-withdrawal-canada-due-risk-drug
27.Health Canada. Summary Safety Review - Fibristal (5 mg ulipristal acetate) - Assessing the potential risk of rare but serious liver injury. 2018. Accessed by sponsor, no date provided. https://hpr-rps.hres.ca/reg-content/summary-safety-review-detail.php?lang=en&linkID=SSR00210
28.Murji A, Whitaker L, Chow TL, Sobel ML. Selective progesterone receptor modulators (SPRMs) for uterine fibroids. Cochrane Database Syst Rev. 2017;4:CD010770. doi: 10.1002/14651858.CD010770.pub2 PubMed
29.Abbvie Corporation. Product monograph including patient medication information PrLUPRON DEPOT® leuprolide acetate for depot suspension 3.75 mg/syringe (1-Month slow release), 7.5 mg/syringe (1-Month slow release), 11.25 mg/syringe (3-Month slow release), 22.5 mg/syringe (3-Month slow release), 30 mg/syringe (4-Month slow release) pre-filled dual-chamber syringe containing sterile lyophilized microspheres intramuscular injection [sponsor supplied reference]. 2021.
30.Singh S, Best C, Dunn S, Leyland N, Wolfman WL. No. 292-Abnormal Uterine Bleeding in Pre-Menopausal Women. J Obstet Gynaecol Can. 2018;40(5):e391-e415. doi: 10.1016/j.jogc.2018.03.007 PubMed
31.Myovant Sciences GmbH. Clinical Study Report: MVT-601-3003. LIBERTY EXTENSION: An International Phase 3 Open-Label, Single-Arm, Long-Term Efficacy and Safety Extension Study to Evaluate Relugolix Co-Administered with Low-Dose Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. 2020.
32.Myovant Sciences GmbH. Clinical Study Report: MVT-601-035, LIBERTY Randomized Withdrawal Study. An International Phase 3 Double-Blind, Placebo-Controlled, Randomized Withdrawal Study of Relugolix Co-Administered with Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. December 1, 2021.
33.Al-Hendy A, Lukes AS, Poindexter AN, 3rd, et al. Treatment of Uterine Fibroid Symptoms with Relugolix Combination Therapy. N Engl J Med. 2021;384(7):630-642. doi: 10.1056/NEJMoa2008283 PubMed
34.Stewart EA, Lukes AS, Venturella R, et al. Relugolix Combination Therapy for Uterine Leiomyoma-Associated Pain in the LIBERTY Randomized Trials. Obstet Gynecol. 2022;139(6):1070-1081. doi: 10.1097/AOG.0000000000004787 PubMed
35.Myovant Sciences GmbH. NCT03049735: LIBERTY 1: Efficacy & Safety Study of Relugolix in Women With Heavy Menstrual Bleeding Associated With Uterine Fibroids. ClinicalTrials.gov. Updated April 19, 2022. Accessed by sponsor, no date provided. https://clinicaltrials.gov/study/NCT03049735
36.Myovant Sciences GmbH. NCT03103087: LIBERTY 2: Efficacy & Safety Study of Relugolix in Women With Heavy Menstrual Bleeding Associated With Uterine Fibroids. ClinicalTrials.gov. Updated April 20, 2022. Accessed by sponsor, no date provided. https://clinicaltrials.gov/study/NCT03103087
37.Knight Therapeutics Inc. 2.7.1 Summary of Biopharmaceutical Studies and Associated Analytical Methods [sponsor supplied reference]. 2022.
38.Center for Drug Evaluation and Research. Summary review(s). Myfembree (relugolix, estradiol, and norethindrone acetate) tablets, for oral use. Company: Sumitomo Pharma Switzerland GmbH. Application No.: 214846. Approval date: 05/24/2021 (FDA approval package). U.S. Food and Drug Administration (FDA); 2021. Accessed April 5, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214846Orig1s000SumR.pdf
39.Hallberg L, Nilsson L. Determination of Menstrual Blood Loss. Scand J Clin Lab Invest. 1964;16:244-8. PubMed
40.Hallberg L, Hogdahl AM, Nilsson L, Rybo G. Menstrual blood loss--a population study. Variation at different ages and attempts to define normality. Acta Obstet Gynecol Scand. 1966;45(3):320-51. doi: 10.3109/00016346609158455 PubMed
41.Lukes AS, Muse K, Richter HE, Moore KA, Patrick DL. Estimating a meaningful reduction in menstrual blood loss for women with heavy menstrual bleeding. Curr Med Res Opin. 2010;26(11):2673-8. doi: 10.1185/03007995.2010.526098 PubMed
42.Turner J, Parsi M, Badireddy M. Anemia. StatPearls Publishing LLC.; 2025. Accessed April 10, 2025. https://www.ncbi.nlm.nih.gov/books/NBK499994/
43.Bachowski G, Borge D, Brunker P, et al. A Compendium of Transfusion Practice Guidelines. American Red Cross; Report No.: 3rd Edition [sponsor supplied reference]. 2017.
44.Man L, Tahhan HR. Body surface area: a predictor of response to red blood cell transfusion. J Blood Med. 2016;7:199-204. doi: 10.2147/JBM.S105063 PubMed
45.Coyne KS, Margolis MK, Bradley LD, Guido R, Maxwell GL, Spies JB. Further validation of the uterine fibroid symptom and quality-of-life questionnaire. Value Health. 2012;15(1):135-42. doi: 10.1016/j.jval.2011.07.007 PubMed
46.Osuga Y, Enya K, Kudou K, Tanimoto M, Hoshiai H. Oral Gonadotropin-Releasing Hormone Antagonist Relugolix Compared With Leuprorelin Injections for Uterine Leiomyomas: A Randomized Controlled Trial. Obstet Gynecol. 2019;133(3):423-433. doi: 10.1097/AOG.0000000000003141 PubMed
47.Harding G, Coyne KS, Thompson CL, Spies JB. The responsiveness of the uterine fibroid symptom and health-related quality of life questionnaire (UFS-QOL). Health Qual Life Outcomes. 2008;6:99. doi: 10.1186/1477-7525-6-99 PubMed
48.Knight Therapeutics Inc. Knight Therapeutics Inc. response to Canada's Drug Agency request for additional information regarding Myfembree review on March 3, 2025 [internal additional sponsor's information]. 2025.
49.Myovant Sciences GmbH. Statistical Analysis Plan: MVT-601-3001 and 3002, LIBERTY 1 and LIBERTY 2. An International Phase 3 Double-Blind, Placebo-Controlled, Randomized Withdrawal Study of Relugolix Co-Administered with Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. June 14, 2019.
50.Stewart E, Owens C, Duan WR, Gao J, Chwalisz K, Simon JA. Elagolix Alone and With Add-Back Decreases Heavy Menstrual Bleeding in Women With Uterine Fibroids. Obstet Gynecol. 2017;129(5):87. doi: 10.1097/01.AOG.0000514923.67415.29
51.Al-Hendy A, Lukes AS, Poindexter AN, 3rd, et al. Long-term Relugolix Combination Therapy for Symptomatic Uterine Leiomyomas. Obstet Gynecol. 2022;140(6):920-930. doi: 10.1097/AOG.0000000000004988 PubMed
52.The Cochrane Collaboration. Cochrane Handbook for Systematic Reviews of Interventions [sponsor supplied reference]. 2022. http://handbook-5-1.cochrane.org/
53.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-6. doi: 10.1016/j.jclinepi.2016.01.021 PubMed
54.Higgins JP, Altman DG, Gotzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. doi: 10.1136/bmj.d5928 PubMed
55.Donnez J, Donnez O, Matule D, et al. Long-term medical management of uterine fibroids with ulipristal acetate. Fertil Steril. 2016;105(1):165-173 e4. doi: 10.1016/j.fertnstert.2015.09.032 PubMed
56.Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JPT. Predicting the extent of heterogeneity in meta-analysis, using empirical data from the Cochrane Database of Systematic Reviews. Int J Epidemiol. 2012;41(3):818-827. PubMed
57.Jansen JP, Naci H. Is network meta-analysis as valid as standard pairwise meta-analysis? It all depends on the distribution of effect modifiers. BMC Med. 2013;11:159. doi: 10.1186/1741-7015-11-159 PubMed
58.Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 7. Rating the quality of evidence--inconsistency. J Clin Epidemiol. 2011;64(12):1294-302. doi: 10.1016/j.jclinepi.2011.03.017 PubMed
59.Deeks JJ, Higgins JPT, Altman DG, McKenzie JE, Veroniki AAe. Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JPT, Thomas J, Chandler J, et al., eds. Cochrane Handbook for Systematic Reviews of Interventions version 65. Cochrane; 2024. Accessed April 23, 2025. https://training.cochrane.org/handbook/current/chapter-10
60.Chaimani A, Caldwell DM, Li T, Higgins JPT, Salanti G. Chapter 11: Undertaking network meta-analyses [last updated October 2019]. In: Higgins JPT, Thomas J, Chandler J, et al., eds. Cochrane Handbook for Systematic Reviews of Interventions version 65. Cochrane; 2024. Accessed August 6, 2025. https://training.cochrane.org/handbook/current/chapter-11
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
E2-NETA
estradiol and norethindrone acetate
HMB
heavy menstrual bleeding
ITC
indirect treatment comparison
NETA
norethindrone acetate
NMA
network meta-analysis
RGX
relugolix
RGX-E2-NETA
relugolix-estradiol–norethindrone acetate
UF
uterine fibroid
Economic Review
The objective of this economic review is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost and budget impact of relugolix-estradiol–norethindrone acetate (RGX-E2-NETA) compared to leuprolide acetate plus estradiol and norethindrone acetate (E2-NETA) (in which E2-NETA serves as add-back therapy to counteract the effects of estrogen suppression) for the management of heavy menstrual bleeding (HMB) associated with uterine fibroids (UFs) in patients in the premenopausal stage. The current review does not include the use of RGX-E2-NETA for the treatment of endometriosis. Additional information about the sponsor’s submission is summarized in Appendix 2.
Item | Description |
|---|---|
Drug product | Relugolix-estradiol–norethindrone acetate, 40 mg/1 mg/0.5 mg oral tablets |
Indication | In premenopausal women for the management of heavy menstrual bleeding associated with uterine fibroids and for the management of moderate to severe pain associated with endometriosisa |
Submitted price | $9.00 per tablet |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 22, 2023 |
Reimbursement request | For the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women |
Sponsor | Knight Therapeutics Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
aIndication is as worded in the product monograph.1
Key Messages
RGX-E2-NETA is available as oral tablets containing 40 mg of relugolix (RGX), 1 mg of estradiol, and 0.5 mg of norethindrone acetate (NETA). At the submitted price of $9.00 per tablet, the annual cost of RGX-E2-NETA for the management of HMB associated with UFs is expected to be $3,285 per patient, based on the Health Canada–recommended dosage.
Based on the results of the sponsor’s base case, and despite inconclusive evidence on clinical similarities between comparators, RGX-E2-NETA is expected to be associated with lower costs to the health care system (including drug acquisition costs) compared to leuprolide acetate plus add-back (incremental savings = $1,385). This lower cost is driven by reduced costs associated with drug acquisition.
Canada’s Drug Agency (CDA-AMC) estimates that the incremental budget impact of reimbursing RGX-E2-NETA for the management of HMB associated with UFs in patients in the premenopausal stage will be an increase of approximately $7,620,729 over the first 3 years of reimbursement compared to the amount currently spent on comparators, with an estimated expenditure of $49,083,074 on RGX-E2-NETA over this period. The actual budget impact of reimbursing RGX-E2-NETA is uncertain and will depend on the proportion of patients who would otherwise undergo watchful waiting without active treatment.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-minimization analysis comparing RGX-E2-NETA to leuprolide acetate plus E2-NETA from the perspective of a public drug plan payer in Canada over a 1-year time horizon.2 The modelled population comprised patients in the premenopausal stage and experiencing HMB associated with UFs and who wish to receive a pharmacological treatment allowing them to maintain fertility, preserve their uterus, and reduce symptoms while avoiding surgery or procedures. This population is narrower than the Health Canada–indicated population. The sponsor’s base-case analysis included costs related to drug acquisition only. A scenario analysis was submitted that further incorporates drug administration costs.
In the sponsor’s base case, RGX-E2-NETA was associated with incremental savings of $1,385 per patient relative to the cost of leuprolide acetate plus E2-NETA, with E2-NETA assigned a cost of $0 to public payers. Additional information about the sponsor’s submission is summarized in Appendix 2.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 3).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The clinical similarity of RGX-E2-NETA to relevant comparators is highly uncertain. | There have been no head-to-head trials comparing RGX-E2-NETA to relevant comparators. The results of the sponsor’s submitted NMA are highly uncertain, owing to wide credible intervals that include effect estimates both in favour of and against RGX-E2-NETA compared to other treatments, the absence of leuprolide acetate plus add-back as a comparator, and unsatisfied key assumptions of exchangeability, homogeneity, and consistency. | CDA-AMC could not address uncertainty in the comparative clinical evidence. Given the lack of direct evidence for RGX-E2-NETA versus relevant comparators and limitations with the sponsor’s NMA, it is uncertain whether RGX-E2-NETA provides a clinical benefit greater than that offered by any of the currently available treatments for the management of HMB associated with UFs in patients in the premenopausal stage. | No scenario analysis was conducted because of a lack of robust clinical data. |
The population considered is narrower than the reimbursement request and the Health Canada indication. | The population is not aligned with the Health Canada indication or the sponsor’s reimbursement request and may not fully represent how RGX-E2-NETA is expected to be used in clinical practice, as indicated by drug plans and clinical experts. | CDA-AMC could not address this difference in population; however, the issue may be negligible because it is not expected to change the choice of relevant comparators. | Given that the choice of relevant comparators was unlikely to change (and no cost differences would be expected), no scenario analyses were conducted. |
Some comparators were not considered. | Clinical expert opinion indicated that NETA 5 mg is also used as add-back therapy in combination with leuprolide acetate to treat endometriosis. Leuprolide acetate 11.25 mg every 3 months may also be used in patients requiring long-term leuprolide acetate plus add-back therapy. | Because NETA 5 mg is not funded by most public drug plans, the cost of leuprolide acetate plus add-back to public drug plans is equivalent to the cost of leuprolide acetate alone, as assumed by the sponsor. | The cost of leuprolide acetate plus NETA 5 mg combination therapy was added to the cost comparison table in Appendix 1 for jurisdictions that fund NETA 5 mg. |
The sponsor’s scenario analysis may overestimate the administration costs associated with comparators. | The sponsor assumed that there would be administration costs for leuprolide acetate injections. However, according to clinical expert opinion obtained by CDA-AMC, such costs are likely overestimated. | Administration costs may vary across jurisdictions and are likely lower, on average, than estimated; however, this is not expected to have an impact on the interpretation of the economic results. | No scenario analyses were conducted. |
The sponsor’s analyses rely on publicly accessible list prices and do not reflect existing confidential prices potentially negotiated by public plans. | Leuprolide acetate may be available to public plans at a confidential price; thus, the current unit cost paid by public drug plans may be lower than the submitted prices. | CDA-AMC was unable to incorporate the presence of confidential negotiated prices in its reanalysis. | No scenario analyses were conducted. Therefore, whether there will be cost savings (and the extent of any savings realized by the drug plans) is uncertain. |
CDA-AMC = Canada’s Drug Agency; HMB = heavy menstrual bleeding; NETA = norethindrone acetate; RGX-E2-NETA = relugolix-estradiol–norethindrone acetate; UF = uterine fibroid.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 3.
CDA-AMC Assessment of Cost
Based on the CDA-AMC clinical review, the sponsor’s submitted indirect treatment comparison (ITC) comparing RGX-E2-NETA to leuprolide acetate without add-back indicated that either treatment could be favoured for efficacy outcomes, and that limitations with the ITC preclude conclusions about the direction of treatment effect. Leuprolide acetate plus add-back was not included in the network meta-analysis (NMA); thus, definitive conclusions about its treatment effect relative to RGX-E2-NETA could not be made. Despite inconclusive evidence on clinical similarity between comparators, the sponsor adequately captured relative drug acquisition costs to public drug plan payers. Clinical expert input obtained by CDA-AMC agreed with the sponsor that leuprolide acetate plus add-back would be the most relevant comparator. Given that the add-back options most frequently used in Canada (i.e., E2-NETA and NETA 5 mg) are rarely funded by public drug plans, the cost of leuprolide acetate plus add-back to public payers in most jurisdictions remains equivalent to the cost of leuprolide acetate alone, regardless of the add-back chosen. Thus, CDA-AMC did not perform a reanalysis.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing RGX-E2-NETA for the management of HMB associated with UFs in patients in the premenopausal stage. The sponsor assumed that payers would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of RGX-E2-NETA was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 4.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that approximately 50,000 patients per year would be eligible for treatment with RGX-E2-NETA over a 3-year period (year 1 = 49,407; year 2 = 50,208; year 3 = 52,241). The estimated incremental budget impact of reimbursing RGX-E2-NETA is expected to be approximately $7,620,729 over the first 3 years, with an expected expenditure of $49,083,074.
Conclusions
No comparative evidence exists for RGX-E2-NETA versus leuprolide acetate plus add-back. However, based on the CDA-AMC clinical review of the sponsor’s submitted NMA comparing RGX-E2-NETA to leuprolide acetate without add-back, either treatment could be favoured for efficacy outcomes. Nonetheless, limitations of the ITC preclude conclusions about the direction of treatment effect. Given the uncertainty in the clinical evidence, there is insufficient evidence to suggest that RGX-E2-NETA should be priced higher than leuprolide acetate. At the submitted price, RGX-E2-NETA is expected to be less costly than leuprolide acetate plus add-back for the treatment of HMB associated with UFs in patients in the premenopausal stage. However, confidential pricing agreements may exist for leuprolide acetate, and a price reduction for RGX-E2-NETA may be required, such that no additional costs are incurred by the health care system.
The budget impact of reimbursing RGX-E2-NETA to the public drug plans in the first 3 years is estimated to be approximately $7.6 million. The 3-year expenditure on RGX-E2-NETA (i.e., not accounting for current expenditure on comparators) is estimated to be $49.1 million. The estimated budget impact will depend on the proportion of patients not currently receiving funded therapies who may switch to RGX-E2-NETA (i.e., a higher proportion switching would increase the budget impact of RGX-E2-NETA), the average duration of therapy for all comparators (i.e., a shorter duration would decrease the budget impact of RGX-E2-NETA), and the dose frequency of leuprolide acetate.
References
1.Pfizer Canada ULC. Myfembree (relugolix, estradiol, and norethindrone acetate): 40 mg/1 mg/0.5 mg oral tablets [product monograph]. October 13, 2023.
2.Knight Therapeutics Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. February 4, 2025.
3.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed April 14, 2025. https://www.formulary.health.gov.on.ca/formulary/
4.IQVIA. DeltaPA. 2025. Accessed Apr 14, 2025. https://www.iqvia.com/
5.B. C. Government. BC PharmaCare formulary search. Accessed April 14, 2025. https://pharmacareformularysearch.gov.bc.ca
6.Abbvie Corporation. Lupron Depot (leuprolide acetate): pre-filled dual-chamber syringe containing sterile lyophilized microspheres, intramuscular injection, for 1 month, 3 month, or 4 month slow release [product monograph]. November 26, 1986. Updated March 19, 2024. Accessed April 14, 2025. https://pdf.hres.ca/dpd_pm/00075000.PDF
7.Knight Therapeutics Inc. Myfembree (40 mg Relugolix, 1 mg Estradiol and 0.5 mg Norethindrone Acetate Tablets) for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids in Premenopausal Women: A Systematic Review and Network Meta-Analysis Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. December 2024.
8.Myovant Sciences GmbH. Clinical Study Report: MVT-601-035. An International Phase 3 Double-Blind, Placebo-Controlled, Randomized Withdrawal Study of Relugolix Co-Administered with Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. December 1, 2021.
9.Myovant Sciences GmbH. Clinical Study Report: MVT-601-3001. LIBERTY 1: An International Phase 3 Randomized, Double-Blind, Placebo-Controlled Efficacy and Safety Study to Evaluate Relugolix Co-Administered with and without Low-Dose Estradiol and Norethindrone Acetate in Women with Heavy Menstrual Bleeding Associated with Uterine Fibroids [internal sponsor's report]. January 31, 2020.
10.Osuga Y, Enya K, Kudou K, Tanimoto M, Hoshiai H. Oral Gonadotropin-Releasing Hormone Antagonist Relugolix Compared With Leuprorelin Injections for Uterine Leiomyomas: A Randomized Controlled Trial. Obstet Gynecol. 2019;133(3):423-433. doi: 10.1097/AOG.0000000000003141 PubMed
11.Ontario Ministry of Health. Schedule of Benefits - Physician Services Under the Health Insurance Act Code G372 (February 20, 2024, Effective, April 1, 2024) [sponsor supplied reference]. 2024. Accessed September 20, 2024. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
12.Verity Pharmaceuticals Inc. Zeulide Depot (Leuprolide acetate for depot suspension): 3.75 mg [1-Month] and 22.5 mg [3-Month], lyophilized powder (suspension after reconstitution with diluent), for intramuscular injection [product monograph]. July 16, 2019. Updated January 8, 2024. Accessed April 14, 2025. https://pdf.hres.ca/dpd_pm/00074213.PDF
13.Health Canada. Patent register. 2025. Accessed April 14, 2025. https://pr-rdb.hc-sc.gc.ca/pr-rdb/index-eng.jsp
14.Statistics Canada. Table: 17-10-0057-01. Projected population, by projection scenario, age and sex, as of July 1 (x 1,000); Projection scenario M3: medium-growth. 2024. Accessed September 20, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
15.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2022 to 2023. 2023. Accessed July 2024. https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612
16.Zimmermann A, Bernuit D, Gerlinger C, Schaefers M, Geppert K. Prevalence, symptoms and management of uterine fibroids: an international internet-based survey of 21,746 women. BMC Womens Health. 2012;12:6. doi: 10.1186/1472-6874-12-6 PubMed
17.Strand T, Kives S, Leyland N, Ashkenas J, Janiszewski P, Thiel J. Treatment Choices in a National Cohort of Canadian Women With Symptomatic Uterine Fibroids. J Obstet Gynaecol Can. 2020;42(12):1475-1482 e2. doi: 10.1016/j.jogc.2020.06.012 PubMed
18.The Conference Board of Canada. Understanding the Gap 2.0 A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [sponsor supplied reference]. 2022.
19.Knight Therapeutics Inc. MYFEMBREE (Relugolix, Estradiol, and Norethindrone Acetate Tablets) for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids in Premenopausal Women, in Canada: KOL Survey [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. 2024.
20.Knight Therapeutics Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Myfembree (relugolix, estradiol, and norethindrone acetate), 40 mg/1 mg/0.5 mg, oral tablet. February 4, 2025.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table, and as such, the table may not represent the actual costs to public drug plans
Table 3: Cost Comparison for the Treatment of HMB Associated With UFs
Treatment | Strength and/or concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Relugolix-estradiol–norethindrone (Myfembree) | 40 mg/1 mg/0.5 mg | Tablet | 9.0000a | One tablet daily | 9.00 | 3,285 |
Leuprolide acetate plus estradiol/norethindrone | ||||||
Leuprolide acetate (Lupron Depot) | 3.75 mg 11.25 mg | Prefilled syringe | 389.1300 1,159.5200 | 3.75 mg IM once per month or 11.25 mg once every 3 monthsb | 12.79 12.70 | 4,670 4,638 |
Estradiol/norethindrone (Activelle) | 1 mg/0.5 mg | Tablets | 2.8400c | One tablet daily | 2.84 | 1,037 |
Leuprolide acetate 3.75 mg monthly plus estradiol/norethindrone regimen cost | 15.63 | 5,706 | ||||
Portion of regimen cost funded by public drug plans | 12.79 | 4,670 | ||||
Leuprolide acetate plus norethindrone | ||||||
Leuprolide acetate (Lupron Depot) | 3.75 mg 11.25 mg | Prefilled syringe | 389.1300 1,159.5200 | 3.75 mg IM once per month or 11.25 mg once every 3 monthsb | 12.79 12.70 | 4,670 4,638 |
Norethindrone (Norlutate) | 5 mg | Tablet | 1.9496d | 5 mg dailye | 1.95 | 712 |
Leuprolide acetate 3.75 mg monthly plus norethindrone regimen cost, in jurisdictions funding norethindrone | 14.73 | 5,382 | ||||
Leuprolide acetate 3.75 mg monthly plus norethindrone regimen cost, in jurisdictions which do not fund norethindrone | 12.79 | 4,670 | ||||
CDA-AMC = Canada’s Drug Agency; HMB = heavy menstrual bleeding; IM = intramuscular; UF = uterine fibroid.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed April 2025),3 unless otherwise indicated, and do not include dispensing fees.
aSponsor’s submitted price.2
bAccording to clinical expert opinion obtained by CDA-AMC, some patients with HMB associated with UFs receive leuprolide acetate 11.25 mg every 3 months if being treated over a long term. The expert noted this to be rare compared to those receiving leuprolide acetate 3.75 mg monthly.
cIQVIA DeltaPA wholesale list price (accessed April 2025).4
dBased on British Columbia Pharmacare Formulary maximum price covered ($2.1056 per 5 mg tablet), after removal of standard 8% markup.5 Norethindrone acetate 5 mg is not listed on most public formularies.
eLeuprolide acetate 3.75 mg monthly and 11.25 mg every 3 months are indicated for the initial management of endometriosis and for management of recurrence of symptoms, in combination with norethindrone acetate 5 mg daily. According to clinical expert opinion obtained by CDA-AMC, this regimen is also used off-label for patients with UFs, including those with HMB.6
Appendix 2: Additional Details of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Table 4: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Relugolix-estradiol–norethindrone acetate (Myfembree), oral tablets (40 mg/1 mg/0.5 mg) |
Submitted price of drug under review | $9.0000 per tablet |
Regimen | One 40 mg/1 mg/0.5 mg tablet daily |
Annual cost of drug under review | $3,285 per patient |
Model information | |
Type of economic evaluation | Cost-minimization analysis |
Treatment | RGX-E2-NETA |
Included comparator | Leuprolide acetate plus E2-NETA |
Perspective | Publicly funded health care payer |
Time horizon | One year |
Modelled population | Patients in the premenopausal stage with HMB associated with UFs who wish to receive a pharmacological treatment for UFs (assumed to be 100% female, 0% male; mean age not specified) |
Model health states | NA |
Data sources | |
Comparative efficacy |
|
Resource use and costs |
|
Summary of the submitted results | |
Base-case results | RGX-E2-NETA was associated with lower costs compared to leuprolide acetate plus E2-NETA (incremental savings of $1,385 per patient). |
Scenario analysis results | In a scenario analysis that included administration costs, RGX-E2-NETA was associated with lower costs than leuprolide acetate plus E2-NETA (incremental savings of $1,891 per patient). |
E2 = estradiol; HMB = heavy menstrual bleeding; NA = not applicable; NETA = norethindrone acetate, NMA = network meta-analysis; RGX = relugolix, UF = uterine fibroid.
Table 5: Summary of the Sponsor’s Cost Analysis
Drug | Unit drug cost ($) | Annual drug cost ($) | Incremental total costs vs. RGX-E2-NETA ($) |
|---|---|---|---|
RGX-E2-NETA | 9.0000a | 3,285 | Reference |
Leuprolide acetate plus E2-NETA | 389.1300bc | 4,670 | 1,385 |
E2 = estradiol; NETA = norethindrone acetate; RGX = relugolix; vs. = versus.
Note: Assumes a 365-day year and that leuprolide acetate injections occur monthly.
aSponsor’s submitted price.2
bOntario Drug Benefit Formulary list price for leuprolide acetate (Lupron) 3.75 mg.3
cE2-NETA is not funded by public drug plans, and thus the cost associated with it is assumed to be $0. The sponsor did not consider NETA 5 mg daily as an alternate add-back option. As it is also not funded by most public drug plans, its cost to most public payers can also be assumed to be $0. For jurisdictions (e.g., British Columbia) that fund NETA 5 mg, refer to Table 3 in Appendix 1 to find the total cost of such therapy.
Appendix 3: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
Assuming similar clinical efficacy and safety for RGX-E2-NETA compared to leuprolide acetate plus E2-NETA, the sponsor submitted a cost-minimization analysis comparing drug acquisition costs for a population aligned with the sponsor’s suggested place in therapy (i.e., patients in the premenopausal stage experiencing HMB associated with UF, who wish to receive a pharmacological treatment for UF, allowing them to maintain fertility, preserve their uterus, and to reduce symptoms while avoiding surgery). Based on the CDA-AMC clinical review of the sponsor-submitted NMA, the assumption of comparable clinical efficacy and safety between RGX-E2-NETA and leuprolide acetate without add-back is highly uncertain, with a high degree of imprecision where results could favour either RGX-E2-NETA or leuprolide acetate without add-back, unsatisfied key assumptions necessary for an NMA, and the absence of leuprolide acetate plus add-back (E2-NETA or otherwise) as a comparator. Overall, the CDA-AMC clinical review reported that definitive conclusions on the relative treatment effects between RGX-E2-NETA and leuprolide acetate plus add-back could not be drawn based on the submitted NMA. The sponsor further justified the appropriateness of similar clinical effects between RGX-E2-NETA and leuprolide acetate plus E2-NETA based on a clinical trial conducted in Japan.10 This study reported that RGX monotherapy demonstrated noninferiority in terms of HMB and a more rapid effect on menstrual bleeding than leuprolide acetate monotherapy, however, this study was not submitted as part of the clinical evidence package and was thus not reviewed by CDA-AMC. The sponsor subsequently inferred that, because RGX is similar to leuprolide acetate based on this trial, then RGX-E2-NETA is also similar to leuprolide acetate plus E2-NETA.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The assumption of clinical similarity is uncertain: The sponsor’s assumption of clinical similarity was based on the results of their submitted NMA,7 which compared RGX-E2-NETA to leuprolide acetate without add-back, as well as an assumption that because a clinical trial of RGX reported similarity in outcomes compared to leuprolide acetate, therefore RGX-E2-NETA would be similar to leuprolide acetate plus E2-NETA.10 Overall, the CDA-AMC clinical review noted that due to the high degree of imprecision in credible intervals in both relative risk and mean difference that could favour either RGX-E2-NETA or leuprolide acetate without add-back within the NMA, uncertainty related to key assumptions required for an NMA (exchangeability, homogeneity, and consistency) and the absence of a comparison between RGX-E2-NETA and leuprolide acetate plus add-back, definitive conclusions on the relative treatment effects between RGX-E2-NETA and leuprolide acetate plus add-back could not be drawn.
Given the lack of direct evidence and limitations with the sponsor’s NMA, whether RGX-E2-NETA has similar clinical effects (i.e., benefits and harms) as other treatments used for patients in the premenopausal stage who are experiencing HMB associated with UFs is uncertain. CDA-AMC was unable to address this limitation.
The population considered is narrower than the reimbursement request: The sponsor’s reimbursement request is for the management of HMB associated with UFs in patients in the premenopausal stage, consistent with the relevant portion of the Health Canada indication.1 However, the population considered in the economic analysis was patients in the premenopausal stage who are experiencing HMB associated with UFs and who wish to receive a pharmacological treatment allowing them to maintain fertility, preserve their uterus, and to reduce symptoms while avoiding surgeries or procedures, which is consistent with the sponsor’s expected place in therapy for RGX-E2-NETA.2 The target population of the economic analysis was therefore narrower than the reimbursement request. When considering the sponsor’s reimbursement request, all treatments used for the treatment of HMB associated with UFs could be considered comparators (such as oral contraceptives, tranexamic acid, progestins, watchful waiting, or surgical procedures), while the economic analysis only considers leuprolide acetate plus add-back to be a comparator. In clinical practice, clinical expert input obtained by CDA-AMC expected that RGX-E2-NETA will be used as a bridge treatment in patients planning gynecological surgery to elevate hemoglobin levels and enable less invasive surgical methods, to delay surgery for fertility reasons, or to bridge patients to menopause if expected within a few years. This place in therapy was noted to be primarily met by leuprolide acetate plus add-back in current practice, and thus leuprolide acetate plus add-back is the most relevant comparator and the one most likely to be displaced by RGX-E2-NETA.
CDA-AMC was unable to address this limitation, however, it is unlikely to have a substantial impact on the cost comparison results, given the expected place in therapy for RGX-E2-NETA.
Some potential comparators were not considered: The sponsor’s analysis considered leuprolide acetate 3.75 monthly plus E2-NETA as the comparator to RGX-E2-NETA. According to clinical expert input obtained by CDA-AMC, while other treatments are used for the treatment of HMB associated with UFs (e.g., oral contraceptives, tranexamic acid, progestins), most of these would have already been tried before patients received either leuprolide acetate plus add-back or RGX-E2-NETA, and thus are unlikely to be displaced by the potential funding of RGX-E2-NETA. Clinical expert input obtained by CDA-AMC also identified elagolix with or without add-back as a therapy that may be used off-label for the treatment of HMB associated with UFs but noted that it was unlikely to be substantially displaced by the funding of RGX-E2-NETA and is not currently funded by public plans. As such, the clinical expert input obtained by CDA-AMC was consistent with the sponsor’s assumption that leuprolide acetate plus add-back is the relevant comparator to RGX-E2-NETA. In terms of add-back options, clinician input obtained by CDA-AMC considered norethindrone 5 mg tablets used in combination with leuprolide acetate to also be relevant. Of note, norethindrone 5 mg is less expensive than E2-NETA (Activelle) per day, however, it is not funded by most public drug plans, and thus the cost borne by public payers for leuprolide acetate plus norethindrone would still be equivalent to the cost of leuprolide acetate alone, as the sponsor assumed for leuprolide acetate plus E2-NETA. Finally, when considering patients who are expected to use leuprolide acetate plus add-back over a long period of time, clinical expert input obtained by CDA-AMC indicated that some may receive leuprolide acetate 11.25 mg every 3 months rather than leuprolide acetate 3.75 mg monthly, however, this was also estimated to affect only a small proportion of patients and thus did not lead to a CDA-AMC reanalysis.
CDA-AMC did not perform a base-case reanalysis to include other possible comparators. To allow for a cost comparison to RGX-E2-NETA in jurisdictions which fund norethindrone 5 mg tablets (e.g., British Columbia), CDA-AMC included the cost of leuprolide acetate plus norethindrone in Table 3 of Appendix 1. Similarly, the cost of leuprolide acetate 11.25 mg every 3 months is included in Table 3.
Scenario analysis may overestimate administration costs: The sponsor provided a scenario analysis which included administration costs associated with monthly leuprolide acetate injections, including a physician visit ($38.35) and injection fee ($3.89) per administration.11 According to clinical expert opinion obtained by CDA-AMC, patients visiting clinics which do not have a full-time nurse on staff may receive their injections from their physician, who may also assess the patient alongside giving them their injection and thus bill the public payer for a physician visit. However, in clinics which do have nursing staff, most leuprolide acetate injections will occur without seeing a physician, and thus billing for a physician visit will not occur. Additionally, patients receiving leuprolide acetate injections over long periods of time may receive injections every 3 months rather than monthly. As such, the sponsor likely overestimated the average administration cost per patient.
In the absence of evidence of the frequency at which patients receive leuprolide acetate injections from a nurse rather than from a physician, CDA-AMC did not conduct a reanalysis of this scenario. The cost of leuprolide acetate administration is likely lower than estimated by the sponsor.
Confidential pricing agreements: The sponsor’s analysis incorporated pricing available from the Ontario Drug Benefit Exceptional Access Program for leuprolide acetate. However, a confidential pricing agreement may exist for leuprolide acetate. Therefore, the submitted price of RGX-E2-NETA may require a price reduction to avoid incurring additional costs relative to its comparators.
CDA-AMC was unable to address this limitation in reanalysis because the existence and magnitude of negotiated prices are unknown.
CDA-AMC Cost Analysis
Based on the CDA-AMC clinical review, the sponsor’s submitted NMA comparing RGX-E2-NETA to leuprolide acetate without add-back indicated that either treatment could be favoured for efficacy outcomes. Leuprolide acetate plus add-back was not included in the NMA, and thus definitive conclusions on its treatment effect relative to RGX-E2-NETA could not be made. However, clinical expert input obtained by CDA-AMC agreed with the sponsor that leuprolide acetate plus add-back was the most relevant comparator. As the add-back options most frequently used in Canada (E2-NETA, NETA 5 mg) are rarely funded by public drug plans, the cost of leuprolide acetate plus add-back to public payers in most jurisdictions remains equivalent to the cost of leuprolide acetate alone, regardless of the add-back chosen, and thus CDA-AMC did not perform a reanalysis.
Issues for Consideration
The recommended duration of therapy for leuprolide acetate for preoperative hematologic improvement in patients of reproductive age with anemia caused by UFs is up to 3 months, to be taken with iron.6 Leuprolide acetate plus NETA 5 mg is indicated for endometriosis6 but is also used off-label for UF, according to clinical expert opinion. The recommended initial duration of therapy for leuprolide acetate plus NETA 5 mg is 6 months, with the potential for an additional 6-month re-treatment.6 The maximum recommended duration of therapy for RGX-E2-NETA is 24 months.1 Clinical expert input obtained by CDA-AMC indicated that treatment with either leuprolide acetate plus add-back or RGX-E2-NETA may be considered beyond 2 years of therapy if the patient is maintaining the goals of symptom improvement, such as in patients who are approaching menopause or whose disease responds well to treatment without intolerable adverse events, including significant bone mineral density loss. However, this expert input also noted that as these treatments are frequently used in anticipation of planned surgical procedures, treatment duration is often shorter than 2 years.
An alternate brand of leuprolide acetate (Zeulide) is available and funded as a general benefit in some jurisdictions (e.g., Ontario) at a list price of $304 per 3.75 mg dose, compared to $389 for the Lupron Depot brand.3 Zeulide branded leuprolide acetate is not interchangeable with the Lupron brand and is only indicated for prostate cancer.12 However, should the Zeulide brand of leuprolide acetate 3.75 mg be used for the treatment of HMB associated with UF, or should 1 or more generics become available (as leuprolide acetate is no longer subject to patent protection13), the annual cost of leuprolide acetate therapy would be lower than estimated.
E2-NETA and NETA 5 mg are generally not funded by public drug payers in Canada. As such, most patients who require add-back therapy currently pay for it out of pocket or with private drug plan coverage. The funding of RGX-E2-NETA may relieve financial burden for patients with HMB associated with UFs who are enrolled in public drug plans.
Appendix 4: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing RGX-E2-NETA for the management of HMB associated with UFs in patients in the premenopausal stage who wish to maintain fertility/preserve their uterus, or are ineligible for surgery or procedure, or who wish to avoid surgery or procedure. This population is narrower than the Health Canada indication and reimbursement request, which are for the management of HMB associated with UFs in patients in the premenopausal stage.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits (NIHB) Program. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. The market uptake for RGX-E2-NETA was estimated using the sponsor’s projected market penetration rate (detail not provided). The key inputs to the BIA are documented in Table 6.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing RGX-E2-NETA for the treatment of HMB associated with UFs in patients in the premenopausal stage who wish to preserve their uterus would be $3,997,556 (year 1 = $690,994; year 2 = $1,263,093; year 3 = $2,043,469).
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Number of women aged 18 to 49 (pan-Canada)a,b | |
Prevalence of UF | 5.5%16 |
Proportion with UFs who have HMB | 59.8%16 |
Proportion of patients with HMB due to UFs treated with hysterectomy only (and thus excluded) | 13.9%17 |
Percentage of patients covered by public drug plan | 25.1%18 |
Number of patients eligible for drug under review | 49,407/50,208/52,241 |
Market shares (reference scenario)c | |
RGX-E2-NETA | ||%/||%/||% |
Leuprolide acetate plus E2-NETA | ██%/██%/██% |
Tranexamic acid | ██%/██%/██% |
Oral contraceptive | ██%/██%/██% |
Levonorgestrel IUS | ██%/██%/██% |
Danazol | ||%/||%/||% |
Progestins | ██%/██%/██% |
Watchful waiting | ██%/██%/██% |
Market shares (new drug scenario)d | |
RGX-E2-NETA | 5%/9%/14% |
Leuprolide acetate plus E2-NETA | ██%/██%/██% |
Tranexamic acid | ██%/██%/██% |
Oral contraceptive | ██%/██%/██% |
Levonorgestrel IUS | ██%/██%/██% |
Danazol | ||%/||%/||% |
Progestins | ██%/██%/██% |
Watchful waiting | ██%/██%/██% |
Cost of treatment (per patient per year)e | |
RGX-E2-NETA | $3,276 |
Leuprolide acetate plus E2-NETA | $5,059 |
Tranexamic acid | $315 |
Oral contraceptive | $101 |
Levonorgestrel IUS | $70 |
Danazol | $925 |
Progestins | $766 |
Watchful waiting | $0 |
CDA-AMC = Canada’s Drug Agency; E2 = estradiol; HMB = heavy menstrual bleeding; IUS = intrauterine system; NETA = norethindrone acetate; NIHB = Non-Insured Health Benefits; NMA = network meta-analysis; RGX = relugolix; UF = uterine fibroid.
aThe pan-Canada population includes that of 9 provinces (excluding Quebec) as well as NIHB clients. NIHB clients were subtracted from the population totals of the respective provinces in which they reside.
bCDA-AMC notes that not all people in Canada with a uterus are women, and that not all women have a uterus. Statistics Canada does not currently report population projections including other genders, nor by sex assigned at birth. However, as the affected proportion of the population is expected to be relatively small, and that the inclusion of women without uteruses is expected to be at least partially offset by the exclusion of people with uteruses who do not identify as women, the impact of using women as a proxy for people with a uterus is expected to have minimal impact on BIA results.
cBased on a survey of 7 gynecologists and obstetricians practising in Canada.19
dBased on projected market penetration rates (analysis not provided), and the assumption that RGX-E2-NETA would only displace leuprolide acetate plus E2-NETA and watchful waiting, which the sponsor reports as validated by 9 clinical experts in Canada (details not provided).20
eCost for RGX-E2-NETA was based on the sponsor’s submitted price, other comparators were based on Ontario Drug Benefit list prices, with the exception of progestins, which were based on the British Columbia Drug Formulary list price, and E2-NETA, which was assumed to have a cost of $0 to public plans.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The dosing of leuprolide acetate was inconsistent with the submitted cost comparison: In its submitted cost comparison, the sponsor assumed that leuprolide acetate 3.75 mg would be given every month (i.e., 12 times per year), as recommended in the product monograph.6 However, in the submitted BIA, the sponsor assumed patients would receive leuprolide acetate once every 28 days (i.e., 13.04 times per year). The dosing of leuprolide acetate is therefore inconsistent between the sponsor’s analyses.
For consistency with the submitted cost comparison, CDA-AMC assumed in reanalysis that leuprolide acetate 3.75 mg would be given 12 times per year rather than 13. In jurisdictions where the usual practice is to give leuprolide acetate every 4 weeks, the corrected sponsor’s base case would be representative of the expected budget impact.
The average duration of therapy may be overestimated: According to clinical expert input obtained by CDA-AMC, while some patients may receive RGX-E2-NETA or leuprolide acetate plus add-back over a long term, many will be treated with these therapies as a bridge to surgical procedures, and are thus unlikely to require treatment with them for multiple years. The assumption that all patients receiving RGX-E2-NETA or leuprolide acetate plus add-back will continue to do so for the entire time horizon of the BIA likely overestimates the duration of therapy that would be seen in clinical practice.
CDA-AMC was unable to adjust for this limitation due to the prevalence-based structure of the submitted model. The incremental budget impact of reimbursing RGX-E2-NETA may be less than estimated, as the costs associated with it as well as with leuprolide acetate plus add-back would be reduced relative to watchful waiting.
The proportion of patients receiving watchful waiting may be overestimated: The sponsor assumed that ██% of eligible patients would undergo watchful waiting in the reference scenario, and that ██% of RGX-E2-NETA uptake would come from patients who would otherwise undergo watchful waiting. This estimate was based on an average of 7 clinical expert estimates obtained by the sponsor, of whom 4 experts estimated that ||% or ██% of patients would undergo watchful waiting. Clinical expert input obtained by CDA-AMC suggested that all patients meeting the eligibility criteria for RGX-E2-NETA would receive some form of active therapy in the reference scenario, consistent with the sponsor’s experts who estimated ||% of patients would undergo watchful waiting. CDA-AMC acknowledges that clinical practice may vary, that specialists are more likely to actively treat patients within their specialty and that specialists are more likely to treat patients more severely affected by their condition than nonspecialized health care providers. Additionally, CDA-AMC notes that some patients may be receiving unfunded treatments (e.g., elagolix) in the reference scenario, at no cost to public drug payers. These patients could switch to RGX-E2-NETA if funded, which would have similar effect on the budget impact as patients switching from watchful waiting.
CDA-AMC conducted a scenario analysis assuming that only 10% of patients in the reference scenario undergo watchful waiting, and that only 30% of the market uptake of RGX-E2-NETA comes from watchful waiting.
The NIHB population was inappropriately calculated: While the sponsor appropriately removed NIHB clients from the populations of the provincial jurisdictions to avoid double counting, the sponsor did not adjust the population reported in the 15-to-19-year age category to remove NIHB clients aged 15 to 17 years, who would not be eligible for RGX-E2-NETA. Additionally, NIHB clients residing within Ontario who are aged < 25 years or ≥ 65 years are eligible for reimbursement by the Ontario Drug Benefit program and thus should be counted as Ontario Drug Benefit program clients (and included in the Ontario population estimates) rather than NIHB clients for the purposes of modelling the budget impact of reimbursing RGX-E2-NETA. The sponsor removed all NIHB clients aged 15 to 49 years living within Ontario from the eligible Ontario population. In doing so, the sponsor failed to consider that patients under 25 years of age would primarily be reimbursed by Ontario rather than NIHB.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on the pan-Canadian model results is expected to be minimal.
One further issue was identified but was not considered to be a key issue as it would unlikely impact the BIA results for any individual jurisdictions. The target population of the BIA (patients with HMB associated with UFs who are in the premenopausal stage and who wish to maintain fertility/preserve their uterus, or are ineligible for surgery or procedure, or who wish to avoid surgery or procedure) is narrower than the reimbursement request and relevant Health Canada indication (patients with HMB associated with UFs who are in the premenopausal stage). However, the sponsor’s analysis excludes only those patients who will be treated with hysterectomy alone from eligibility of the target population and does not remove those who will receive some form of drug therapy in addition to their hysterectomy, which is consistent with clinical expert opinion obtained by CDA-AMC that RGX-E2-NETA will be used as a bridge to surgical treatment. As clinical expert opinion obtained by CDA-AMC did not anticipate that RGX-E2-NETA would alter the pattern of hysterectomy or other surgical procedures for UFs beyond the effects of leuprolide acetate plus add-back on such patterns, CDA-AMC did not consider this to be a key limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 7. CDA-AMC corrected an error in the submitted BIA where the number of claims per year for each product was rounded down, leading to small underestimates in the estimated average annual drug cost for all comparators.
Table 7: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Correction to annual drug costs | Number of claims per year rounded down | Number of claims per year was not rounded |
1. Dose frequency of leuprolide acetate | Leuprolide acetate 3.75 mg given every 28 days | Leuprolide acetate given monthly |
CDA-AMC base case | ― | Reanalysis 1 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; NIHB = Non-Insured Health Benefits.
Note: CDA-AMC was unable to resolve the issues with the expected duration of therapy and the NIHB population miscalculated.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 8 and a more detailed breakdown is presented in Table 9. In the CDA-AMC base case, the 3-year budget impact of reimbursing RGX-E2-NETA for the management of HMB associated with UFs in patients in the premenopausal stage was $7,620,729 (year 1 = $1,317,080; year 2 = $2,408,203; year 3 = $3,895,446).
Table 8: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $3,997,556 |
Corrected sponsor’s base case | $4,011,284 |
CDA-AMC base case: Reanalysis 1 (with correction) | $7,620,729 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to explore uncertainty in the estimated budget impact of reimbursing RGX-E2-NETA. The results are provided in Table 9.
Only 10% of eligible patients were assumed to undergo watchful waiting in the reference case, and 30% of the market uptake of RGX-E2-NETA was assumed to come from watchful waiting.
Table 9: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | $73,807,025 | $75,047,991 | $76,253,738 | $79,294,310 | $230,596,040 |
RGX-E2-NETA | $0 | $0 | $0 | $0 | $0 | |
All other comparators | $73,807,025 | $75,047,991 | $76,253,738 | $79,294,310 | $230,596,040 | |
New drug total | $73,807,025 | $75,738,985 | $77,516,831 | $81,337,780 | $234,593,596 | |
RGX-E2-NETA | $0 | $8,452,765 | $15,459,375 | $25,002,957 | $48,915,096 | |
All other comparators | $73,807,025 | $67,286,220 | $62,057,457 | $56,334,823 | $185,678,500 | |
Budget impact | $0 | $690,994 | $1,263,093 | $2,043,469 | $3,997,556 | |
CDA-AMC base case | Reference total | $68,947,747 | $70,106,799 | $71,232,926 | $74,074,293 | $215,414,018 |
RGX-E2-NETA | $0 | $0 | $0 | $0 | $0 | |
All other comparators | $68,947,747 | $70,106,799 | $71,232,926 | $74,074,293 | $215,414,018 | |
New drug total | $68,947,747 | $71,423,880 | $73,641,129 | $77,969,739 | $223,034,748 | |
RGX-E2-NETA | $0 | $8,481,792 | $15,512,463 | $25,088,819 | $49,083,074 | |
All other comparators | $68,947,747 | $62,942,088 | $58,128,666 | $52,880,920 | $173,951,674 | |
Budget impact | $0 | $1,317,080 | $2,408,203 | $3,895,446 | $7,620,729 | |
CDA-AMC scenario analyses | ||||||
Scenario 1: Fewer patients receiving watchful waiting | Reference total | $70,551,944 | $71,737,562 | $72,889,443 | $75,799,054 | $220,426,058 |
New drug total | $70,551,944 | $71,860,524 | $73,113,603 | $76,162,270 | $221,136,397 | |
Budget impact | $0 | $122,962 | $224,160 | $363,217 | $710,339 | |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; E2 = estradiol; NETA = norethindrone acetate; RGX = relugolix.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.