Drugs, Health Technologies, Health Systems
Reimbursement Review
Tofersen (Qalsody)
Sponsor: Biogen Canada Inc.
Therapeutic area: Amyotrophic lateral sclerosis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AE
adverse event
ALS
amyotrophic lateral sclerosis
ALSAQ-5
Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 items
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
ALS-PR
amyotrophic lateral sclerosis progression rate
ANCOVA
analysis of covariance
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CSF
cerebrospinal fluid
CTCAE
Common Terminology Criteria for Adverse Events
EAP
early access program
FSS
Fatigue Severity Scale
FVC
forced vital capacity
GMR
geometric mean ratio
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HHD
hand-held dynamometry
HR
hazard ratio
HRQoL
health-related quality of life
IQR
interquartile range
ITT
intention to treat
JRT
joint rank test
MAID
medical assistance in dying
MI
multiple imputation
MiToS
Milano-Torino Staging
mITT
modified intention to treat
MYMOP2
Measure Yourself Medical Outcome Profile 2
NfL
neurofilament light chain
NGS
next-generation sequencing
NPS
Net Promoter Score
OLE
open-label extension
PD
pharmacodynamic
PK
pharmacokinetic
pNfH
phosphorylated axonal neurofilament heavy chain
PT
preferred term
PV
permanent ventilation
QoL
quality of life
RCT
randomized controlled trial
RPSFTM
rank-preserving structural failure time model
SAE
serious adverse event
SD
standard deviation
SOC
system organ class
SVC
slow vital capacity
TSQM-9
Treatment Satisfaction Questionnaire for Medication-9 items
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Tofersen (Qalsody), 100 mg per 15 mL (6.7 mg/mL), solution for intrathecal injection |
Sponsor | Biogen Canada Inc. |
Indication | Qalsody (tofersen injection) is indicated for the treatment of adults with amyotrophic lateral sclerosis (ALS) associated with a mutation in the superoxide dismutase 1 (SOD1) gene |
Reimbursement request | Tofersen is to be used for the treatment of adults with ALS associated with a mutation in the SOD1 gene who:
|
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard |
NOC date | February 28, 2025 |
Recommended dose | Tofersen 100 mg per 15 mL injection |
ALS = amyotrophic lateral sclerosis; NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions.
Introduction
Amyotrophic lateral sclerosis (ALS) is a rare, progressive, and fatal neurodegenerative disease characterized by the loss of motor neurons. This leads to progressive muscle weakness, loss of function, and typically death from respiratory failure within 3 to 5 years of diagnosis. SOD1-ALS, an ultrarare genetic subtype caused by mutations in the SOD1 gene, affects an estimated 40 individuals in Canada, where the overall ALS prevalence is approximately 6.84 per 100,000 people. Key symptoms of ALS include muscle weakness and difficulties with mobility, speech, swallowing, and breathing, severely impacting daily activities and quality of life (QoL).
Current treatments for ALS, such as riluzole and edaravone, provide modest benefits in slowing ALS progression or extending survival for some patients. However, they do not address the underlying genetic cause of SOD1-ALS or significantly alter the disease course. Patients and clinicians emphasized a critical unmet need remains for disease-modifying therapies that can slow progression, preserve function, improve survival, and target the specific pathology of genetic forms of ALS like SOD1-ALS.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tofersen 100 mg per 15 mL (6.7 mg/mL), solution for intrathecal injection, for the treatment of adults with ALS associated with a mutation in the SOD1 gene.
Testing Procedure Considerations
According to the clinical experts consulted by the review team, testing for SOD1 mutations is currently performed as part of the standard of care for patients with ALS in Canada. The availability and accessibility of SOD1 mutation testing vary across jurisdictions. However, no substantial implementation barriers are anticipated from a testing perspective if tofersen were to be funded.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the call by Canada’s Drug Agency (CDA-AMC) for input and from clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
Patient input was submitted by the ALS Society of Canada, the ALS Action Canada Society, the ALS Society of Alberta, and the ALS Society of British Columbia. Information was gathered from patients and caregivers through an online survey, interviews, and focus groups. More than 20 respondents living with ALS had experience with tofersen.
The input noted that ALS severely impacts patients’ mobility, strength, daily activities, and emotional health. Key symptoms include muscle weakness, balance issues, cramping, and nerve pain, as well as difficulty speaking, swallowing, and breathing. Limited mobility hinders independence, affecting tasks like climbing stairs, carrying groceries, or standing for prolonged periods. Emotional tolls include fear of disease progression, anxiety about future independence, and the burden on loved ones. For those with familial ALS, the disease is deeply personal because many have seen its effects on loved ones, leading to significant emotional and psychological strain. Within a year of diagnosis, patients reported needing help with daily tasks like eating, walking, and bathing from caregivers. The input noted that patients with ALS, facing a progressive and fatal illness, often pursue all viable treatments, including clinical trials, alternative therapies, and off-label options. The input stated that in Canada, ALS treatments include riluzole and edaravone, which may provide some benefit but do not significantly alter disease progression. Furthermore, some patients indicated difficulty accessing edaravone due to a lack of private coverage, strict public funding criteria, out-of-pocket costs, and supply shortages. The input noted that neither therapy specifically addresses the SOD1 gene mutation in ALS.
Patients who had had experience with tofersen reported benefits including maintaining independence and delaying symptom onset, allowing them to maintain activities that were previously becoming difficult, and the ability to spend more time with loved ones. Patients noted that lumbar punctures to administer tofersen pose logistical challenges, because patients often needed to travel long distances to receive treatment, as well as physical challenges, with adverse effects such as headaches, migraines, nausea, and temporary incapacitation. The input stated that improved protocols, such as slow injections and localizing administration to nearby clinics, could enhance accessibility and patient experience. The input emphasized the importance of early and accessible genetic testing for timely intervention with tofersen. Respondents found their experiences with genetic testing to be efficient with timely results and minimal cost barriers. The most critical unmet needs according to the patient groups’ input include symptom reversal (i.e., muscle weakness, cramping, and fasciculations), the maintenance of mobility, function, and independence, the slowing of disease progression, increased survival, and improved QoL.
Clinician Input
Input From Clinical Experts Consulted for This Review
The information in this section is based on input received from a panel of 5 clinical specialists consulted by CDA-AMC for the purpose of this review.
According to the clinical experts consulted by CDA-AMC, the primary unmet need for patients with SOD1-ALS before the availability of tofersen was the lack of treatments capable of significantly altering the disease course. Existing therapies like riluzole and edaravone were considered to offer only modest effects, failing to halt or reverse functional decline, and they did not target the underlying genetic cause. The experts viewed a treatment targeting the mutant SOD1 protein as a critical gap. They also identified challenges such as geographical disparities in access to specialized ALS care, inconsistent access to timely and funded SOD1 genetic testing, and the resource implications of administering monthly intrathecal injections.
The clinical experts consulted view tofersen as a first-line, disease-modifying therapy for adults with symptomatic ALS confirmed to be caused by a pathogenic SOD1 mutation because it directly targets the underlying disease mechanism. They advised that treatment should be initiated as soon as an ALS specialist confirms the diagnosis, emphasizing that early initiation is critical to preserve motor neurons and maximize potential benefits. The experts suggested that riluzole and, where feasible, edaravone would typically be continued alongside tofersen for potential additive benefits, but patients should not be required to try these other drugs or not experience improvement with these other drugs before accessing tofersen.
The patient population identified by the experts as most likely to benefit from tofersen includes adults with a confirmed pathogenic SOD1 mutation who are symptomatic with ALS but still retain sufficient residual motor function. They noted that individuals with rapidly progressive phenotypes and younger individuals would have the greatest need. Conversely, the experts considered patients with very advanced disease (e.g., those whose anticipated survival < 6 months, those with an inability to tolerate a lumbar puncture) and presymptomatic carriers (outside of research settings) as least suitable for therapy.
In terms of assessing response to treatment, the experts indicated this is primarily done through clinical evaluation by an ALS specialist, typically every 3 months, focusing on monitoring the rate of functional decline for stabilization or the slowing of progression. Key clinical tools mentioned were manual muscle testing, patient-reported function, and respiratory assessment using forced vital capacity (FVC). While many end points used in the VALOR trial (slow vital capacity [SVC], hand-held dynamometry [HHD], specific quality-of-life scales, and biomarkers like cerebrospinal fluid [CSF] SOD1 protein and plasma neurofilament light chain [NfL]) are not standard tools for routine clinical decision-making in Canada, the experts noted that the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) is used during follow-up visits to measure score and monitor the change in the slope of ALSFRS-R, which the clinical experts identified as clinically meaningful. A clinically meaningful response, according to the experts, involves the stabilization or slowing of disease progression; any improvement in function is highly significant but not always the primary expectation. The patient’s and clinician’s perception of benefit relative to the burden of treatment is considered crucial.
Regarding discontinuing treatment, the clinical experts agreed that defining absolute objective criteria is difficult. Key reasons for discontinuation identified by the experts include patient preference due to a perceived lack of benefit or the burden of monthly treatment, intolerable or unmanageable adverse events (AEs) (such as severe neurologic inflammation or papilledema), the physical inability to perform the lumbar puncture, or reaching a very advanced, end-stage disease state where the potential for benefit is considered negligible. The need for permanent ventilation (PV) often prompts discussions about discontinuation. However, the experts noted that continued disease progression alone is usually not a trigger for stopping unless the disease becomes extremely advanced or the patient chooses to discontinue treatment.
Additional considerations highlighted by the experts included the need for diagnosis and treatment initiation to be led by neurologists or physiatrists with ALS expertise, though administration could be delegated to clinicians proficient in lumbar puncture. They noted that most injections occur in outpatient settings but acknowledged logistical challenges, particularly for rural and remote patients, and the need for sustainable resources. Periodic ophthalmologic examinations were advised due to risks of elevated intracranial pressure. The experts also underscored the need for expanded publicly funded genetic testing and research into less burdensome delivery systems and biomarker-guided dosing.
Clinician Group Input
One clinician group provided input for this review: The Canadian ALS Research Network, consisting of 14 clinicians. The submissions noted that ALS is a progressive neurodegenerative disease that leads to the degeneration of motor neurons in the brain and spinal cord. This results in severe weakness in limb, bulbar, and respiratory muscles, eventually causing the loss of autonomy and dependence on assistive devices like wheelchairs, feeding tubes, and ventilatory support. Most patients die from respiratory failure within 5 years of diagnosis. The experts noted that available ALS treatments include riluzole, which extends survival by about 3 months by targeting glutamate, and IV or oral edaravone, which may slow disease progression in specific patients. Sodium phenylbutyrate and ursodoxicoltaurine (Albrioza) was approved in 2022; however, it was withdrawn in 2024 after failing a phase III clinical trial. The input noted that these treatments provide only modest benefits and do not reverse the disease or halt its progression and there are currently no approved treatments specifically targeting hereditary ALS. Clinicians emphasized the urgent need for personalized, disease-modifying treatments. Ideal therapies would slow progression, improve QoL, target the root causes of ALS, and reduce the burden on caregivers. Given ALS’s complexity, treatments should be tailored to individual patients, prevent motor neuron degeneration, and incorporate precision medicine approaches.
The input stated that tofersen is best suited for patients with ALS with pathogenic or likely pathogenic SOD1 gene mutations and weakness linked to ALS, as determined by a specialist. Patients with uncertain SOD1 variants tied to the disease may also be eligible. Because there is no diagnostic biomarker for weakness related to ALS, a diagnosis is made based on a patient’s history, physical examination, and electrodiagnostic examination, and the exclusion of alternative diagnoses. Upon diagnosis, all patients with ALS should undergo genetic testing for common ALS-related genes, including SOD1. If an SOD1 mutation is identified, patients should be promptly considered for the drug under review. The input stated that tofersen would be used in combination with existing therapies, creating a multimodal approach that addresses multiple disease pathways. The input noted that there is no rationale for requiring patients to not experience improvement with other therapies before initiating tofersen, given the irreversible progression of ALS. The clinicians noted that a treatment response or failure for ALS is not precisely defined given that the primary goal of treatment is to slow the degeneration of motor neurons. Individual disease progression varies and tracking outcomes like slowed progression is challenging due to disease heterogeneity. An appropriate treatment strategy involves initiating the drug and monitoring the patient at regular intervals until care goals transition to a more palliative approach or the patient and physician decide to stop treatment based on an unfavourable risk-benefit assessment.
Drug Program Input
Drug programs submitted questions related to comparators, the initiation of therapy, the renewal of therapy, the discontinuation of therapy, the prescribing of therapy, and care provision issues.
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included part C of 1 pivotal phase III, randomized, double-blind, placebo-controlled study (the VALOR trial) and its multicentre, open-label, long-term extension (open-label extension [OLE]) study (the 23AS102 trial). Part C of the VALOR study (N = 108 randomized participants) assessed the efficacy, safety, and tolerability of tofersen 100 mg administered intrathecally compared to placebo over 28 weeks in adults with ALS and a confirmed SOD1 mutation. Randomization was 2:1 (tofersen [N = 72] to placebo [N = 36]). The primary end point was the change from baseline to week 28 in the ALSFRS-R total score in the modified intention-to-treat (mITT) population (N = 60), defined as those participants meeting prognostic enrichment criteria for faster disease progression. ALSFRS-R is a 12-item scale that assesses function in 4 domains: respiratory, bulbar, gross motor, and fine motor. Each item is rated on a scale of 0 to 4, generating an ALSFRS-R total score of 0 (maximum disability) to 48 (no disability). Secondary outcomes included changes in total SOD1 protein in CSF, NfL levels in plasma, SVC, HHD megascore, and time to death or PV. Participants who completed the VALOR trial could enrol in the OLE study to receive tofersen.
In the VALOR trial, the overall ITT population (N = 108) had a mean age of 49.2 years (standard deviation [SD] = 12.3 years); 42.6% of participants were female and 57.4% were male. Race was reported as Asian (8.3%), Black or African American (0.9%), white (63.9%), and “other” (0.9%); 25.9% of participants did not report their race. The mean ALSFRS-R total score at baseline was 37.1 (SD = 5.9). In the mITT faster-progressing subgroup (n = 60), the mean age was 49.7 years (SD = 14.3 years), and the mean baseline ALSFRS-R score was ████ ███████). Baseline plasma NfL levels in the ITT population were a mean of 96.9 pg/mL (SD = 84.2 pg/mL). The ████████ of participants in the ITT population had limb-onset ALS (placebo group = ███ lower and ███ upper; tofersen group = ███ lower and ███ upper).
In this section, only part C of the VALOR study is presented; the first 2 parts of the VALOR study (part A and part B) were dose-escalation trials conducted to assess the dose of tofersen to be used in part C. Patients who were enrolled in part A and part B of the VALOR study were not enrolled in part C of the study.
Efficacy Results
In the VALOR study’s randomized phase, the primary analysis — the change from baseline to week 28 in the ALSFRS-R total score in the mITT faster-progressing population (N = 60) — showed an adjusted mean difference between tofersen (−6.98 points from baseline) and placebo (−8.14 points from baseline) of 1.2 points (95% confidence interval [CI], −3.2 to 5.5 points; P = 0.9689; joint rank test [JRT] with multiple imputation [MI]). A post hoc analysis in the ITT population (N = 108), adjusting for baseline NfL, showed an adjusted mean difference in the ALSFRS-R score of 1.4 points (95% CI, −1.34 to 4.09 points; nominal P = 0.3218) favouring tofersen (−4.5 points from baseline) over placebo (−5.8 points from baseline).
Key secondary end points in the VALOR study at week 28 included the following:
Total CSF SOD1 protein (mITT population): Tofersen resulted in a geometric mean ratio (GMR) to baseline of 0.71 while placebo was 1.16. The difference in GMR for tofersen to placebo was 0.62 (95% CI, 0.49 to 0.78; nominal P < 0.0001).
Plasma NfL level (mITT population): Tofersen resulted in a GMR to baseline of 0.40 while placebo was 1.20. The difference in GMR for tofersen to placebo was 0.33 (95% CI, 0.25 to 0.45; nominal P < 0.0001).
Percent-predicted SVC (mITT population): The adjusted mean change from baseline was −14.31 percentage points for tofersen and −22.20 percentage points for placebo, an adjusted mean difference of 7.9 percentage points (95% CI, −3.5 to 19.3 percentage points; nominal P = 0.1755, analysis of covariance [ANCOVA] + MI).
HHD megascore (mITT population): The adjusted mean change from baseline was −0.34 for tofersen and −0.37 for placebo, an adjusted mean difference of 0.02 points (95% CI, −0.21 to 0.26 points; nominal P = 0.8390).
Number of and time to death or PV (mITT population): Few deaths or PV events occurred. There were 4 deaths or PV events in the tofersen group and 2 deaths or PV events in the placebo group; the hazard ratio (HR) was 1.39 (95% CI, 0.219 to 8.803).
Key end points in the OLE study (with a data cut-off date of February 28, 2023, ITT population from the VALOR study), comparing early-start tofersen (randomized to tofersen in the VALOR study) to delayed-start tofersen (randomized to placebo in the VALOR trial, then initiated with tofersen in the OLE study) included the following:
ALSFRS-R: At week 52, the adjusted mean difference was 3.5 points (95% CI, 0.4 to 6.7 points; nominal P = 0.0272) favouring the early-start group. At week 104, the difference was 3.7 points (95% CI, −0.7 to 8.2 points; nominal P = 0.1004).
Biomarkers: Reductions in CSF SOD1 protein and plasma NfL levels were sustained in the early-start group and observed in the delayed-start group after initiating tofersen. At week 104, plasma NfL GMR from baseline was 0.34 for the early-start group and 0.40 for the delayed-start group.
Percent-predicted SVC: At week 104, the adjusted mean difference was 9.7% favouring early-start tofersen.
Time to death or PV: The HR for the early-start group versus the delayed-start group was 0.47 (95% CI, 0.20 to 1.11).
Harms Results
In the VALOR study (safety population: N = 108), AEs were reported by 95.8% (69 of 72) of participants in the tofersen group and 94.4% (34 of 36) of participants in the placebo group. The most common AEs (≥ 20% in either group) were procedural pain (tofersen: 56.9%; placebo: 58.3%), headache (tofersen: 45.8%; placebo: 44.4%), pain in extremity (tofersen: 26.4%; placebo: 16.7%), a fall (tofersen: 23.6%; placebo: 41.7%), back pain (tofersen: 20.8%; placebo: 5.6%), and post–lumbar puncture syndrome (tofersen: 18.1%; placebo: 30.6%).
Serious adverse events (SAEs) occurred in 18.1% (13 of 72) of tofersen-treated participants and 13.9% (5 of 36) of placebo-treated participants during the VALOR study. The most frequently reported SAEs included dyspnea, pulmonary embolism, and aspiration pneumonia. Withdrawals due to AEs occurred in 5.6% (4 of 72) of participants in the tofersen group and 0% of those in the placebo group. One death (congestive cardiac failure, not considered treatment-related) occurred in the tofersen group during the VALOR study.
Notable harms included serious neurologic events reported in 5.6% (4 of 72) of tofersen-treated participants (lumbar radiculopathy, chemical meningitis, myelitis, and transverse myelitis) and none in the placebo group. Falls were less frequent in the tofersen group (23.6%) compared to the placebo group (41.7%). CSF abnormalities (e.g., increased white blood cell count, increased protein) were more common in the tofersen group (16.7%) than in the placebo group (2.8%).
In the OLE study (N = 104, with a data cut-off date of February 28, 2023), 99.0% (103 of 104) of participants experienced AEs. SAEs were reported by 46.2% (48 of 104) of participants. AEs leading to drug discontinuation occurred in 22.1% (23 of 104) of participants. A total of 18 (17.3%) deaths occurred during the VALOR and OLE studies. The safety profile in the OLE study was generally consistent with that of the VALOR study, with common AEs reflecting ALS progression or lumbar puncture procedures. Serious neurologic events like myelitis, radiculitis, aseptic meningitis, and papilledema continued to be observed.
Critical Appraisal
The internal validity of the VALOR trial was impacted by several factors. Critically, the trial did not meet its prespecified primary efficacy end point for ALSFRS-R at 28 weeks. Subsequent analyses, including those in the ITT population and those showing effects on biomarkers, relied on post hoc adjustments (e.g., for baseline NfL levels) or were exploratory due to the primary end point failure, increasing the risk of type I error. While methods of randomization and blinding were appropriately applied in the pivotal phase, baseline imbalances were noted, and the study was underpowered and likely had too short a duration for clinical end points, leading to imprecision in effect estimates. The reliance on surrogate biomarkers (CSF SOD1 and plasma NfL levels), though showing nominal statistically significant changes, means that their direct clinical meaningfulness remains unvalidated. The OLE study suffers from inherent limitations, including the lack of randomization and a concurrent control group, high participant attrition over time, and the potential for detection bias, which tempers confidence in the long-term findings.
Regarding external validity, the VALOR trial population was enriched for participants with faster disease progression and specific SOD1 mutations, which may limit the generalizability of the main findings to the broader, more heterogeneous SOD1-ALS population, including those with slower disease progression. While patients with a slower progression of ALS were enrolled, they were not part of the main analysis and, due to the disease nature and trial timeline, were not expected to show a difference. Participants were predominantly white, which is consistent with the known epidemiology of ALS. Furthermore, many of the outcome measures employed are primarily research tools, not routinely used for clinical decision-making in Canada, and the short 6-month blinded phase was insufficient to definitively assess effects on survival or functional measures. Finally, the evidence lacks comparison against standard therapeutic options in Canada.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for the Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
change from baseline to week 28 on the ALSFRS-R total score
GMR to baseline to week 28 on the total CSF SOD1 protein
GMR to baseline to week 28 on plasma NfL
change from baseline to week 28 on SVC percent predicted
change from baseline to week 28 on HHD megascore
time to death or PV
SAEs.
Table 2: Summary of Findings for Tofersen vs. Placebo for Patients With SOD1-ALS
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Tofersen | Difference | |||||
Change from baseline to week 28 on ALSFRS-R total score | |||||||
Change from baseline on ALSFRS-R total score (mITT population minus faster-progressing group) (more is better) Follow-up: Week 28 | 60 (1 RCT) | NA | −8.14 points | −6.98 points | 1.2 more points (3.2 fewer points to 5.5 more points) | Lowa,b | Tofersen may result in little or no clinically meaningful difference in the ALSFRS-R score compared to placebo. |
Change from baseline on ALSFRS-R total score (ITT population minus total patients post hoc analysis adjusted for baseline NfL levels) (more is better) Follow-up: Week 28 | 108 (1 RCT) | NA | −5.8 points | −4.5 points | 1.4 more points (1.34 fewer points to 4.09 more points) | Very lowa,c | The evidence is very uncertain about the effect of tofersen on the ALSFRS-R score when compared to placebo. |
GMR to baseline to week 28 on total CSF SOD1 protein | |||||||
Total CSF SOD1 protein GMR to baseline (mITT population minus faster-progressing group) (less is better) Follow-up: Week 28 | 60 (1 RCT) | 0.62 (0.49 to 0.78) | 1.16 | 0.71 (0.62 to 0.83) | NA | Moderated,e | Tofersen likely results in a decrease in CSF SOD1 protein GMR compared to placebo. |
GMR to baseline to week 28 on plasma NfL level | |||||||
Plasma NfL GMR to baseline (mITT population minus faster-progressing group) (less is better) Follow-up: Week 28 | 60 (1 RCT) | 0.33 (0.25 to 0.45) | 1.20 | 0.40 (0.33 to 0.48) | NA | Moderated,e | Tofersen likely results in a decrease in NfL GMR compared to placebo. |
Change from baseline to week 28 on percent-predicted SVC | |||||||
Change from baseline in percent-predicted SVC (mITT population minus faster-progressing group) (more is better) Follow-up: Week 28 | 60 (1 RCT) | NA | −22.20 percentage of predicted | −14.31 percentage of predicted (███████) | 7.9 more percentage of predicted (3.5 fewer points to 19.3 more points) | Lowb,f | Tofersen may result in little or no clinically meaningful difference in the SVC score compared to placebo. |
Change from baseline week to 28 on HHD megascore | |||||||
Change from baseline in HHD megascore (mITT population minus faster-progressing group) (more is better) Follow-up: Week 28 | 60 (1 RCT) | NA | −0.37 | −0.34 (███████) | 0.02 more points (0.21 fewer points to 0.26 more points) | Lowb,f | Tofersen may result in little or no clinically meaningful difference in the HHD score compared to placebo. |
Time to death or permanent ventilation | |||||||
Patients with an event of death or permanent ventilation (mITT population minus faster-progressing group) (less is better) Follow-up: Week 28 | 60 (1 RCT) | Hazard ratio = 1.39 (0.219 to 8.803) | 2 patients | 4 patients | NA | Very lowg | The evidence is very uncertain about the effect of tofersen on the proportion of patients experiencing death or permanent ventilation when compared to placebo. |
Median time to death or permanent ventilationh | 60 (1 RCT) | NA | NA | NA | NA | NA | NA |
Harms | |||||||
Patients with 1 or more serious adverse event (safety set) (less is better) Follow-up: Week 28 | 108 (1 RCT) | NA | 5 | 13 | NA | Very lowi | The evidence is very uncertain about the effect of tofersen on the proportion of patients experiencing serious adverse events when compared to placebo. |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; CSF = cerebrospinal fluid; GMR = geometric mean ratio; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HHD = hand-held dynamometry; ITT = intention to treat; MID = minimal important difference; mITT = modified intention to treat; NA = not available; NfL = neurofilament light chain; RCT = randomized controlled trial; SVC = slow vital capacity.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, the imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the Table 2 footnotes. When a published MID was available and judged applicable, that value was used as the threshold for imprecision. When no empirically validated MID was identified (or existing estimates were highly inconsistent), CDA-AMC used thresholds suggested by clinical experts consulted for this review. If the clinical experts were unable to suggest an MID, CDA-AMC defaulted to the null effect (no difference) as the threshold. The lack of an MID and the use of the null effect could result in a 1-level downgrade.
aRated down 1 level for serious imprecision. The MID for ALSFRS-R in patients with ALS is very uncertain, based on the varying and wide ranges of MIDs reported in the literature and per clinical expert input. The null effect (no difference) was therefore considered as a threshold for assessment. The 95% CI for the difference is wide and includes the possibility of potentially clinically meaningful benefit, no meaningful difference, and potential harm. The total sample (N = 60 mITT population; N = 108 ITT population) is below the optimal information size (< 400 for continuous outcomes). No further downgrade was assigned because larger studies may not be feasible in this ultrarare condition.
bRated down 1 level for study limitations. The primary clinical end point (ALSFRS-R in the mITT population) was not met; thus, the null hypothesis vs. placebo could not be rejected, imposing an elevated risk of type I error. Similarly, all subsequent results carry a high risk of level I error. Furthermore, the study was underpowered and inappropriately short for this clinical end point. In addition, baseline imbalances were noted in a number of possible treatment effect modifiers. This may have resulted in a bias of an undetermined direction, resulting in increased uncertainty in the outcome.
cRated down 2 levels for serious study limitations. The ALSFRS-R analysis in the ITT population was a post hoc analysis that included adjustment for baseline NfL; this was an amendment to the statistical analysis plan partway through the trial. This introduced a risk of analysis bias (due to data-driven analysis) and reduced confidence in this specific outcome. Furthermore, the study was underpowered and inappropriately short for this clinical end point. In addition, baseline imbalances were noted in a number of possible treatment effect modifiers beyond the NfL levels. This may have resulted in a bias of an undetermined direction.
dDid not rate down for imprecision. No MID was found; therefore, the null effect was used and the 95% CI included only the possibility of benefit. Despite the option to downgrade 1 point due to the lack of an important difference threshold, CDA-AMC opted not to due to several factors: mechanistic plausibility, large effect size, and the ultrarare nature of the condition.
eRated down 1 level for indirectness (surrogate outcomes). The effect estimates for the CSF SOD1 protein and plasma NfL reflect biochemical targets rather than patient-important or clinical outcomes. Neither marker has been formally validated as a surrogate that reliably predicts changes in survival, function (ALSFRS-R), or quality of life in SOD1-ALS. Consequently, the link from these biomarker shifts to outcomes that matter to patients remains uncertain, warranting a single-level downgrade for indirectness.
fRated down 1 level for serious imprecision. No MID was found; therefore, the null effect was used. The null effect (no difference) was considered as a threshold for assessment. The 95% CI for the difference is wide and includes the possibility of potentially clinically meaningful benefit, no meaningful difference, and potential harm. The size of the total sample (N = 60) is below the optimal information size. There was no further downgrade because larger studies may not be feasible in this ultrarare condition.
gRated down 2 levels for very serious imprecision. No minimally important difference was found; therefore, the null effect was used. The 95% CI included very wide possibilities of benefit and harm. The resulting hazard ratio is 1.39 with an extremely wide 95% CI of 0.22 to 8.80, spanning an 80% relative reduction to a 780% increase (i.e., clinically important benefit, no effect, and substantial harm are all plausible). For the composite “death or permanent ventilation” end point, the body of evidence comprised 6 total events in 108 participants (4 of 72 in the tofersen group vs. 2 of 36 in the placebo group). This is far below the optimal information size to assume adequate precision.
hThe median time to death could not be calculated because there were insufficient events during the trial.
IRated down 2 levels for very serious imprecision. For the serious adverse event outcome, there were only 18 events in 108 participants (13 of 72 in the tofersen group vs. 5 of 36 in the placebo group). That event count is orders of magnitude below the GRADE optimal information size benchmark of approximately 300 events to 500 events for a binary end point — the amount ordinarily needed to rule out a minimally important effect with adequate power. Any relative or absolute treatment effect would exhibit a very wide CI that would include the possibility of harm.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
None were submitted.
Indirect Comparisons
None were submitted.
Description of Studies: German and Italian Early Access Programs
Four real-world studies evaluated the effectiveness, safety, and patient-reported outcomes of tofersen treatment in patients with SOD1-ALS enrolled in an early access program (EAP). These included 3 studies from Germany (Wiesenfarth et al. [2024],1 Meyer et al. [2024],2 and Meyer et al. [2023]3) and 1 study from Italy (Sabatelli et al. [2024]).4 The studies included assessments of clinical progression, biomarkers, and health-related quality of life (HRQoL).
In all 4 studies, tofersen 100 mg was administered directly into the CSF on day 1, day 14, and day 28 as loading doses. Patients then received up to 16 maintenance doses at intervals of approximately 28 days (a minimum of 21 days). Across all EAP studies, the ALS progression rate (ALS-PR) was defined as ALSFRS-R points lost per month. In Meyer et al. (2024),2 patient-reported outcomes included the Measure Yourself Medical Outcome Profile 2 (MYMOP2), the Treatment Satisfaction Questionnaire for Medication–9 items (TSQM-9), and the Net Promoter Score (NPS). MYMOP2 allows patients to identify and rate the severity of their most troubling symptoms as assessed on a 7-point Likert scale (0 for “as good as it could be” to 6 for “as bad as it could be”). TSQM-9 evaluates treatment satisfaction on effectiveness, convenience, and overall satisfaction (scored 0 to 100). The NPS assesses the likelihood of recommending the treatment (range, –100 to 100), where scores above 0 indicate more promoters than detractors.
Wiesenfarth et al.1 followed 24 adult patients with SOD1-ALS over 12 months across 10 German centres. The median pretreatment ALSFRS-R score was 37.0 points (interquartile range [IQR], 29.8 to 41.8 points). Meyer et al. (2024)2 included 16 patients with SOD1-ALS, assessed for up to 18 months. The mean pretreatment ALSFRS-R score was 37.4 points (range, 7 to 46 points). Meyer et al. (2023)3 assessed the efficacy of tofersen among 6 patients with unique SOD1 mutations over a period of at least 5 months. One patient had an ALSFRS-R score of 1 point, and the remaining patients were considered to have high functional status, with ALSFRS-R scores ranging from 35 to 46 points at baseline. Sabatelli et al.4 reported on 27 of 42 (63.0%) enrolled patients living with SOD1-ALS, 17 (40.5%) of whom were included in efficacy analysis after exclusions and dropouts.
Efficacy
ALS Progression Rate
In Wiesenfarth et al.,1 the median ALSFRS-R score declined from 38.0 to 35.0, corresponding to a median ALS-PR of 0.11 points per month. Seventeen of 23 (73.9%) patients had slowed progression during treatment while 6 (26.1%) patients worsened. In Meyer et al. (2024),2 the ALS-PR decreased in 50% of patients with a mean change of –0.2 points per month. In Meyer et al. (2023),3 the ALS-PR decreased in 2 of 6 (33.3%) patients and no changes were observed in the remaining 4 (66.7%) patients. Sabatelli et al.4 observed a median ALS-PR reduction from 0.25 points per month pretreatment to 0.0 points per month after treatment.
Biomarkers
Wiesenfarth et al.1 reported a decreased mean serum NfL level from 78.0 pg/mL to 36.0 pg/mL and a CSF phosphorylated axonal neurofilament heavy chain (pNfH) level from 2,226 pg/mL to 1,151 pg/mL. Meyer et al. (2024)2 observed a 58% decrease in the mean serum NfL level in 15 of 16 (93.8%) patients. Meyer et al. (2023)3 reported 66% and 62% mean reductions in CSF and serum NfL, respectively. In 14 of 17 (82%) enrolled patients in the Sabatelli et al.4 study, the mean reduction in CSF NfL levels from baseline was 61% (range, 49% to 79%). The remaining 3 (17.6%) patients had unchanged or increased CSF NfL levels.
Health-Related Quality of Life
In Meyer et al. (2024),2 at baseline, the mean symptom severity on the MYMOP2 7-point scale was 3.8 points (n = 14); this decreased to a mean of 3.0 points at the last measured perception. From baseline to the last measured perception, MYMOP2 responses showed symptom improvement (defined as an improvement in at least 1 of the 2 target symptoms) in 10 of 14 (71.4%) patients and partial improvement (defined as improvement or stabilization in 1 symptom and deterioration of the other) in the remaining 6 (42.9%) patients. TSQM-9 scores were assessed in 15 (93.8%) patients with a mean global satisfaction score of 83 points (SD = 16 points). As assessed by the NPS, at 6 months of tofersen treatment, 12 of 15 (80%) patients were promoters of tofersen.
Harms
In Wiesenfarth et al.,1 common procedure-related side effects included back pain, headache, leg nerve pain, and dizziness. Two (8.7%) patients experienced SAEs possibly related to tofersen that occurred during the study; both patients stopped treatment voluntarily. There were no reported deaths during the observation period. Patients’ CSF changes indicated autoimmune inflammation in the central nervous system. Eleven of 15 (73%) patients experienced an increase in white blood cells in the CSF and 10 (66.7%) of these patients also experienced elevated protein levels. Nine of 10 (90%) patients showed immune protein production in the CSF.
In Sabatelli et al.,4 postinjection headaches occurred once in 4 (23.5%) patients. Seven (41.2%) patients reported limb pain, with pain following the path of a nerve from the spine to the arms or legs. Nine of 15 (60.0%) patients experienced increased white blood cell and protein levels in the CSF after tofersen therapy, indicative of drug-related spinal cord and nerve root inflammation. Two (22.2%) of these patients demonstrated clinical symptoms and responded to steroid treatment. Three (11.1%) of the 27 patients who began treatment with tofersen died soon after starting treatment and were not included in the analyses.
Harms results were not provided in the Meyer et al. studies.2,3
Critical Appraisal
The sponsor submitted real-world EAP cohort studies based on published reports. However, the absence of study protocols and statistical analysis plans limited the ability to fully assess study design, outcome measurement, and analytical methods.
The analyses were primarily descriptive and unadjusted for confounders, reducing internal validity and limiting the ability to infer treatment effects. Additionally, the lack of a comparator group makes it difficult to separate treatment effects from natural disease progression or external influences.
The lack of blinding, where both patients and clinicians were aware of the treatment administered, raises the potential for performance and detection biases, particularly in subjective measures such as ALSFRS-R and AE reporting.
The EAP studies were affected by small sample sizes, missing or incomplete data, and inconsistent reporting of outcomes, all of which undermine the reliability of progression slope estimates. For example, in the Italian study, 27 of 42 enrolled patients received treatment, 17 (40.5%) of whom had evaluable data. This substantial attrition raises the risk of survivor bias, where observed effects may reflect the characteristics of a selected subgroup rather than true treatment effectiveness.
ALSFRS-R slope estimates were based on 2 time points and assumed linear disease progression, which may not accurately reflect the typically nonlinear course of ALS.5,6 Therefore, these estimates may misrepresent true disease progression. The reliance on patient recall for prebaseline data introduces uncertainty because recall bias may lead to inaccurate reporting of disease onset and prior ALSFRS-R trajectory. In the Wiesenfarth et al. study, it was unclear whether observed ALSFRS-R changes reflected treatment response or were confounded by short-follow-up duration. Likewise, it is unclear whether any improvements in ALSFRS-R scores would be sustained over a longer period especially given that the trial’s enrichment strategy focused on patients with slower-progressing disease.
Although posttreatment reductions in ALSFRS-R decline were observed, the lack of a comparator group, small sample sizes, missing data, the potential for bias (particularly from confounding), and the reliance on slopes of ALSFRS-R scores from few time points mean that there was reduced certainty in the results from these studies regarding the effectiveness of tofersen.
The EAP studies were conducted in small patient cohorts from Germany and Italy, limiting confidence in the generalizability of the findings to the ALS population in Canada. Given the limited reporting of baseline characteristics and small sample sizes, it is unclear whether these study populations reflect the diversity and clinical profiles of patients in Canada. While the clinical experts consulted agreed that any trained clinician could administer the treatment under review, they emphasized that patients should remain under the care of an ALS specialist. The experts noted that access to multidisciplinary ALS care is more readily available to patients near more densely populated urban areas in Canada. Although efforts have been made to expand access in remote areas through telehealth and transport to specialized clinics, significant barriers to equitable care remain. As such, the centralized care settings in the reviewed studies may not fully reflect the realities of care delivery across Canada’s geographically dispersed health system.
Conclusions
Based on the 28-week data from the VALOR trial, the evidence for a clinically meaningful treatment effect of tofersen on functional decline, as measured by the ALSFRS-R tool, is of low certainty. Tofersen may result in little or no clinically meaningful difference compared to placebo in function outcomes, respiratory function, and muscle strength over this period. The effects of tofersen on the time to death or PV and on SAEs are very uncertain, due to the very limited number of events that occurred during the 28-week trial. Conversely, the VALOR study showed that tofersen likely reduces CSF SOD1 protein and plasma NfL levels, biomarkers of target engagement, and axonal injury with moderate certainty. However, these are surrogate end points and their direct translation to patient-centred clinical outcomes is not yet fully established.
The 28-week duration of the VALOR study randomized controlled trial (RCT) was likely too short to detect a meaningful treatment effect in ALS, including with tofersen. Longer-term data from the OLE and 4 real-world EAP cohort studies suggest slowed disease progression, or even stabilization in some patients, and potential survival benefits, especially with earlier initiation of tofersen. This evidence is primarily descriptive, noncomparative, and subject to a serious risk of bias and limited generalizability. Nonetheless, given the known natural history of ALS, which involves continuous functional decline, a reduction in the rate of ALSFRS-R decline or stabilization of function is unexpected and signals a potentially clinically important effect.
Patients with SOD1-ALS and their caregivers have expressed a strong need for treatments that can slow disease progression, maintain function and independence, and extend survival. While the biomarker changes witnessed with tofersen offer a mechanistic rationale and reductions in the rate of progression have been reported in real-world and longer-term extension studies within the context of the natural history of ALS, the currently available evidence suggests the potential for benefit but does not definitively establish that tofersen meets these expectations in a clinically significant way over the short term.
Key gaps in the evidence remain. There is a need for more robust, comparative data to assess the long-term clinical efficacy of tofersen, particularly regarding functional outcomes, QoL, and survival. Further research is needed to better understand the clinical relevance of the observed biomarker changes and their utility in predicting or monitoring individual patient response. Additionally, evidence in broader SOD1-ALS populations, including those with slower disease progression or different SOD1 mutation types less represented in the pivotal trial, is limited. The comparative effectiveness of tofersen against, or in addition to, a fully optimized standard of care has not been directly assessed.
While acknowledging the uncertainty in the results of the included studies, the clinical experts noted that tofersen might address some of the unmet needs in symptomatic patients with ALS with a mutation in the SOD1 gene as it is a disease-modifying therapy, and highlighted the observed reduction in the biomarkers, which supports target engagement and biological activity. The clinical experts also highlighted observed trends in slowing disease progression in the OLE and EAP studies.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of tofersen 100 mg per 15 mL (6.7 mg/mL), solution for intrathecal injection, for the treatment of adults with ALS associated with a mutation in the SOD1 gene.
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
ALS is a rare, progressive, and fatal neurodegenerative disease that causes the loss of upper and lower motor neurons and their axons within the cortex, brainstem, spinal cord, and peripheral nervous system.7,8 The loss of motor neurons leads to progressive loss of muscle mass, strength, and function in bulbar, respiratory, and limb muscles. Death typically occurs secondary to respiratory failure within 3 to 5 years of symptom onset.9,10 Although invasive ventilatory support is often required for people living with ALS to prolong life and/or maintain QoL, such interventions do not halt or slow the progressive decline in function.11
ALS may present in any anatomic region and spread throughout the body, with the progression pattern and speed being highly variable. Approximately 80% of people living with ALS experience limb-onset, with the remaining approximately 20% presenting with bulbar-onset or generalized symptoms.12-14 People with ALS experience progressive motor weakness, beginning focally and leading to complete paralysis. Fatigue, muscle weakness, and reduced motor function are common early symptoms in ALS. Early symptoms of ALS may also include weakness in the limbs, the loss of motor skills, muscle cramping or twitching, slurred speech, and difficulty swallowing.15-17 The middle stages of ALS typically present with widespread muscle paralysis, weakness in swallowing and breathing muscles, pseudobulbar affect, and falls. Weight loss and malnutrition are common in this stage of ALS, leading to a decreased QoL and shorter survival times.17 The late stages of ALS include paralysis of most voluntary muscles, leading to limited mobility and independent respiration.17
The etiology of ALS is multifactorial, but a hallmark of the disease is axonal injury and neurodegeneration that causes progressive weakness in all muscles within voluntary control. ALS is classified as familial (5% to 10% of ALS cases) or apparently sporadic (approximately 90% to 95% of ALS cases) based on the presence or absence of a known family history.18,19 Altogether, more than 15% of all ALS cases, familial and sporadic, are caused by a known genetic mutation.20-22 Currently, mutations in more than 50 genes have been identified as causing or being associated with ALS.23
SOD1-ALS is an ultrarare genetic subtype of ALS.7,24 Mutation(s) in the SOD1 gene lead to an accumulation of toxic SOD1 proteins via a gain of function mechanism. This causes the loss of neuromuscular junction viability and motor neuron dysfunction or death. SOD1-ALS, like all ALS, results in progressive weakness secondary to motor neuron degeneration; however, substantial interpatient variability exists regarding the rate of progression, which varies depending on the location of mutation in the SOD1 gene. More than 200 causative SOD1 mutations have been identified to date, with the majority following an autosomal-dominant inheritance pattern.25
The natural history of SOD1-ALS is variable and generally poorly characterized due to the rarity of the disease. The 2 largest natural history cohorts include a retrospective cohort study that reviewed records from 175 patients with SOD1-ALS across 15 institutions in North America26 and an international, retrospective observational study in 1,383 patients with SOD1-ALS.27 These studies describe a mean age of onset for SOD1-ALS that is consistent with that of the broad ALS population (aged approximately 49 to 50 years), though this can vary across individual SOD1 gene mutation types.26 The median survival and median disease duration in the SOD1-ALS population were reported as 2.7 years26 and 2.3 years,27 respectively. The median disease duration of 2.3 years in the SOD1-ALS population was significantly shorter than the comparator dataset of 12,622 patients with ALS, which had a median disease duration of 2.9 years (P < 0.001). However, intramutation variation exists, with disease durations ranging from less than a year to more than 20 years.26,27
ALS is a rare disease with a global prevalence estimated to be 4.42 per 100,000.28 A recent systematic review and meta-analysis study reports pooled prevalence rates (per 100,000 persons) and incidence rates (per 100,000 person-years) as 6.22 and 2.31 for Europe, 5.20 and 2.35 for North America, 3.41 and 1.25 for Latin America, 3.01 and 0.93 for Asian countries excluding Japan, and 7.96 and 1.76 for Japan, respectively.
In Canada, the estimated prevalence is 6.84 per 100,000, equating to approximately 2,800 Canadians living with ALS.
Most people who develop ALS are aged 40 years to 70 years, with familial ALS having a younger age of onset (40 to 60 years) compared to sporadic ALS (58 years to 63 years). Males have a higher risk of developing ALS with a male to female ratio of 1.2 to 1.5. Other risk factors include white and non-Hispanic ancestry.29
SOD1 mutations are estimated to be present in approximately 2% of all ALS cases.7,24 Therefore, there are an estimated 40 patients with ALS with a SOD1 mutation in Canada. In a dataset of 1,383 SOD1-ALS cases, the average age of onset was 48.9 years compared to 61.1 years in the total ALS dataset (n = 12,622).27 There was a slightly higher percentage of males with SOD1-ALS compared to females at 52.5% versus 47.4%, respectively; this is comparable to the total ALS population. The majority of people living with SOD1-ALS (70%) have a known family history.
As the disease progresses, it impacts the patient’s daily life, including walking, eating, and speaking. There is no cure for ALS and, eventually, all people with ALS will require enteral feedings, ventilatory support, and other invasive interventions to support survival. Although the progression rate is heterogeneous in ALS, all forms of the disease are severe and fatal.
Functional decline and loss of independence greatly impact QoL in people living with ALS. A recent systematic literature review investigating the burden of ALS confirmed that people living with ALS experienced poor QoL and loss of function, which deteriorated with disease progression. The burden of symptoms was high, with patients experiencing various debilitating symptoms; fatigue, depression, and pain were most frequently reported.30 In a survey of people living with ALS living in the EU5 (France, Germany, Italy, Spain, and the UK) and the US, QoL measured by the Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 items (ALSAQ-5) showed a stepwise worsening of HRQoL with disease progression.31 Depression is 1 of the most common sequelae associated with ALS, with previous studies reporting prevalence ranging 4% to 56%, depending on the assessment measure used.19 Suicidal ideation is exhibited at a higher rate in people living with ALS than in the general population.32 In 1 study of 71 people living with ALS, 39% expressed either passive or active suicidal ideation, with a higher risk of suicidality in patients with depressive symptoms and lower disability scores.32 Additionally, many people living with ALS choose to receive medical assistance in dying (MAID). In 2022 in Canada, neurologic conditions were reported as the main condition for receiving MAID by 12.6% of individuals.33 Of those, 18.5% of individuals had ALS, correlating to approximately 308 people with ALS receiving MAID in 2022 in Canada.
Stenson et al. (2021)34 analyzed data from the Adelphi ALS Disease Specific Programme from people with ALS (N = 142 [n = 32 SOD1-positive]) in France, Germany, Italy, Spain, the UK, and the US between June 2020 and March 2021. In this study, the average time to key disease milestones, such as the use of a walking aid, feeding tube, wheelchair, or ventilatory assistance, and monthly functional score decline were similar across genetic variants of ALS.34 For SOD1-ALS, the average time to the use of a walking aid, wheelchair, feeding tube, or ventilatory assistance was 13.8 months, 26.4 months, 30.3 months, and 25.4 months, respectively. The average decline of SVC from baseline through a 1.5-year follow-up is −2.7% points per month and is associated with meaningful clinical events in ALS.35 The rate of decline in ALSFRS-R varies widely but averages approximately 1 point per month.36,37
The functional decline and loss of independence in people living with ALS may also have a substantial impact on the QoL of caregivers. The existing literature suggests that nearly half of caregivers report high burden (physical burden, depression, anxiety and stress) associated with caring for a person living with ALS.38,39 Regardless of disease stage, most caregivers indicate that their own current health was worse than before assuming the ALS caregiver role. A real-world, point-in-time survey across key European markets and the US found that as individuals’ disease state progressed, their caregivers experienced a concurrent gradual decline in HRQoL.40
Studies have reported that up to 63% of patients with ALS receive no formal care (i.e., by a paid professional) and rely solely on family members and friends for informal care (i.e., by an unpaid family member, close relatives, friends, and/or neighbour).38,39,41,42 This contributes to a loss of income and increased absenteeism for caregivers and is a source of indirect costs for ALS. Caregiving accounts for more than half of the total ALS costs, with the typical number of hours devoted to caregiving reaching 130 hours per week (increased hours are needed for later stages of the disease).43,44
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
There is no single test to confirm a diagnosis of ALS. The diagnosis of ALS is based on clinical progression and the exclusion of alternative causes of signs and symptoms. Electromyography is used to measure the signals that run between the nerves and muscles and the electrical activity inside muscles to determine if there is a pattern consistent with ALS. Additional tests may include an MRI of the spinal cords and brain and a spinal tap or lumbar puncture to test the CSF. Blood tests and muscle biopsies can be used to exclude other disorders that mimic ALS. NfL level is an established marker of axonal injury and neurodegeneration in various diseases, including ALS. In ALS, higher levels of NfL have been associated with faster disease progression and shorter overall survival;45,46 however, NfL behaviour in slower-progressing disease, and the clinically asymptomatic phase of the disease, is not well characterized. Genetic testing can also help confirm a diagnosis of ALS and determine whether an ALS-associated genetic mutation is present.
The 2 most commonly used criteria for the diagnosis of ALS are the revised El Escorial criteria47 and the Awaji criteria.48 The revised El Escorial criteria published in 2000 defines ALS diagnosis as 1 of 3 categories based on involvement in different segments of the central nervous system.47
Possible ALS: This is a presence of upper and lower motor neuron signs in 1 body segment, or upper motor neuron signs in at least 2 segments, or lower motor neuron signs in a body segment above upper motor neuron signs.
Probable ALS: This is a presence of upper and lower motor neuron signs in at least 2 body segments, with upper motor neuron signs in a body segment above lower motor neuron signs.
Definite ALS: This is a presence of upper and lower motor neuron signs in at least 3 body segments.
The Awaji criteria, published in 2008, incorporates electromyography information with the clinical examination of the El Escorial criteria to enable earlier diagnosis and improve the diagnosis sensitivity.48,49
Further, the most recent Gold Coast criteria were created and published in 2020 to simplify and improve the diagnostic process.50 These criteria aim to establish a single clinical diagnostic entity rather than have different disease categories and include nonmotor and motor symptoms. Based on these criteria, a confirmed diagnosis of ALS requires all of the following:
progressive motor impairment documented by history or repeated clinical assessment, preceded by normal motor function
a presence of upper and lower motor neuron symptoms and signs in at least 1 limb or body region (bulbar, cervical, thoracic, and lumbosacral), or lower motor neuron symptoms and signs in at least 2 body segments
investigations that exclude other causes of neuron degeneration.
In 2020, the Canadian Best Practice Recommendations guidelines for the management of ALS were published.51 These guidelines recommend that the diagnosis of ALS should be confirmed by a neurologist or physiatrist with training and expertise in ALS and a patient should attend an ALS specialty clinic within 4 weeks of receiving a diagnosis.
In 2023, the ALS Genetic Testing and Counseling Guidelines Expert Panel published evidence-based consensus guidelines for ALS genetic testing and counselling.52 These guidelines recommend that all patients with ALS, regardless of family history, should be offered single-step genetic testing, consisting (at minimum) of a C9orf72 assay, along with sequencing of SOD1, FUS, and TARDBP and, ideally, all genes known to be definitively associated with ALS.
The 2024 European Academy of Neurology guidelines for the management of ALS used the GRADE methodology for the development of its guidelines.53 In people living with ALS caused by a SOD1 mutation, tofersen is strongly recommended as first-line treatment, with patient counselling on the potential for serious AEs. The guidelines also include a strong recommendation for lifelong riluzole treatment for all people living with ALS, from the time of diagnosis, in the absence of contraindications or AEs that preclude use. The use of edaravone (oral or IV) is not recommended (strong recommendation against) outside of the context of a clinical trial.
The 2020 Canadian Best Practice Recommendations guidelines for the management of ALS provide recommendations on the use of therapies that were available at the time.51 Riluzole has demonstrated a modest survival benefit of 3 months compared with placebo based on a meta-analysis of 4 RCTs. It is of expert consensus that riluzole should be started soon after the diagnosis of ALS. Edaravone is classified as probably effective in a select group of patients with ALS based on slowing progression of the ALSFRS-R score. The patients who have shown benefit with edaravone include those with disease duration of less than 2 years, FVC of more than 80%, all ALSFRS-R subcomponent scores being greater than 2, and a demonstrated steady decline in the ALSFRS-R score over a 3-month interval. It is of expert consensus that evidence of edaravone being beneficial at other stages of ALS has not been demonstrated. Of note, tofersen was not available at the time these guidelines were published.
Both the Canadian Best Practice Recommendations and the European Academy of Neurology guidelines recommend that people living with ALS be followed and treated by a multidisciplinary ALS team. Other treatment options available for all patients with ALS are symptomatic and intended to manage pain, cramps, excess salivation, spasticity, fatigue, pseudobulbar affect, depression, and insomnia that occur during the course of the disease. Nutritional management can include tactics such as altering food consistency and using nutritional supplements. The use of a percutaneous endoscopic gastrostomy may be used in advanced stages of the disease to help maintain nutrition. Respiratory management encompasses a large part of care for patients and includes preventive care (pneumococcal and flu vaccines), anticholinergic medications for sialorrhea (excessive salivation or drooling), manual cough assist, aspiration for excessive secretions, the management of acute infections, and noninvasive ventilation or tracheostomy for respiratory insufficiency and planning for withdrawal of care.
Drug Under Review
Key characteristics of tofersen are summarized in Table 3 with other treatments available for ALS.
Table 3: Key Characteristics of Tofersen, Riluzole, and Edaravone
Characteristic | Tofersen | Riluzole | Edaravone |
|---|---|---|---|
Mechanism of action | Antisense oligonucleotide that binds to SOD1 mRNA, leading to RNase H–mediated degradation of the mRNA and reduced synthesis of toxic SOD1 protein | The mode of action of riluzole is unknown, though its pharmacological properties include the following:
| The exact mechanism by which edaravone exerts therapeutic effects in ALS is not established (edaravone is a free radical scavenger, originally developed as a neuroprotective drug, but its specific action in ALS remains unclear). |
Indicationa | For the treatment of adults with amyotrophic lateral sclerosis (ALS) associated with a mutation in the superoxide dismutase 1 (SOD1) gene (approved with conditions – NOC/c) | May extend survival and/or time to tracheostomy in some patients with amyotrophic lateral sclerosis | Indicated for the treatment of patients with amyotrophic lateral sclerosis |
Route of administration | Intrathecal injection (via lumbar puncture) | Oral | IV or oral |
Recommended dosage | 100 mg intrathecally per dose. Begin with 3 loading doses at 14‑day intervals (day 0, day 14, day 28), then continue with a maintenance dose every 28 days. | 50 mg by mouth every 12 hours (twice daily) | IV infusion of 60 mg administered over a 60‑minute period, according to the following schedule:
Oral suspension 105 mg (5 mL) taken orally or via a feeding tube (nasogastric tube or percutaneous endoscopic gastrostomy tube) according to the following schedule:
|
Serious adverse effects or safety issues | Myelitis, radiculitis, aseptic meningitis, elevated intracranial pressure with papilledema | Hepatotoxicity; riluzole is contraindicated in patients who have hepatic disease or who have baseline transaminases > 3 times the upper limit of normal | Hypersensitivity reactions (redness, wheals, and erythema multiforme) and cases of anaphylaxis (urticaria, decreased blood pressure, and dyspnea) have been reported in spontaneous postmarketing reports. |
ALS = amyotrophic lateral sclerosis; mRNA = messenger ribonucleic acid; NOC/c = Notice of Compliance with Conditions; RNase H = ribonuclease H.
aHealth Canada–approved indication.
Sources: Product monographs for tofersen,54 edaravone,55 and riluzole.56
Testing Procedure Considerations
SOD1 Mutations
Mutations in the SOD1 gene are found in 10% to 20% of patients with familial ALS, as well as in 1% to 2% of patients without a family history of the disease (sporadic ALS).57
SOD1 mutations are identified by Sanger sequencing or next-generation sequencing (NGS), using DNA extracted from blood or saliva samples.58 While NGS can be used to identify both common and rare mutations on multiple genes, Sanger sequencing is considered the gold standard for single-gene sequencing and is commonly used to screen samples for mutations in known ALS- associated genes (e.g., SOD1, UBQLN2).59,60 It is also used to confirm mutations initially identified through NGS.60 Analyzing the vast amount of information generated by NGS requires bioinformatics expertise to produce interpretable and actionable results. On some occasions, NGS genetic testing may identify a variant of uncertain significance, where the pathogenicity of the identified variant is unclear, presenting a challenge in the interpretation of results. In such cases, geneticists typically use all available data (e.g., information provided by predictive software, a positive family history) to interpret the result.
A 2023 consensus guideline from the ALS Genetic Testing and Counseling Guidelines Expert Panel recommends that all individuals with ALS should be offered testing with an ALS gene panel that includes SOD1.52 According to the clinical experts consulted for this review, testing for SOD1 mutations is routinely performed as part of a diagnostic work-up for all patients with ALS in Canada. They also noted that, in most jurisdictions, SOD1 mutation testing is performed as part of a broader ALS gene panel that includes other genes (e.g., C9orf72). At the time of writing this report, an ALS genetic panel funded by Ionis Pharmaceuticals and performed by Prevention Genetics (using NGS with additional Sanger sequencing as necessary) is used in some facilities in the country.58,61 The long-term availability of this sponsor-funded testing is uncertain.
CDA-AMC considered the potential impacts of SOD1 mutation testing to ascertain eligibility for tofersen for adults with ALS, including those impacts to health systems, patients (including families and caregivers), and costs. No additional system-level impacts are anticipated if tofersen were to be funded. Key considerations and relevant information available from materials submitted by the sponsor, input from clinical experts (including the clinical expert panel consulted by the review team), and sources from the literature were validated by the review team when possible and are summarized in Table 4. Additional ethical considerations related to the testing procedure are detailed in the Ethics Review Report.
Neurofilament Light Chain
The CSF SOD1 protein level and NfL level were assessed as secondary end points in the VALOR trial included in this review62 and NfL level may be relevant to monitoring disease progression; therefore, they are discussed in this report. However, according to the clinical experts, although both CSF SOD1 protein and NfL levels may provide insight into disease progression, they are not currently used to guide clinical treatment decisions and therefore not routinely performed in clinical settings. Unlike SOD1 mutation testing, CSF SOD1 protein level and NfL level testing are not used to determine treatment eligibility for tofersen. As such, a detailed assessment of the considerations for their use, such as that presented in Table 4 for SOD1 mutation testing, was not conducted.
One clinical expert noted that clinics in Alberta have access to commercial NfL testing, which uses the single molecule array (Simoa) assay — the same method that was used in the VALOR trial to measure NfL levels in blood samples.63 The test costs about $200, although the clinical expert did not specify whether it is publicly reimbursed or requires out-of-pocket payment by patients; this could not be confirmed through other sources. NfL testing is not available through the public health systems in Ontario and Saskatchewan. The availability in other jurisdictions is unclear. The clinical experts highlighted that the limited availability of NfL testing is not expected to impact the implementation of tofersen, if it were to be funded.
Table 4: Considerations for SOD1 Mutation Testing for Establishing Treatment Eligibility With Tofersen in ALS
Consideration | Criterion | Available information |
|---|---|---|
Health system–related | Number of individuals in Canada expected to require the test (e.g., per year) | Based on ALS incidence in Canada, the clinical experts estimated that about 2,000 patients with ALS will be tested for SOD1 gene mutations per year. |
Availability and reimbursement status of the testing procedure in jurisdictions across Canada | According to the clinical experts, SOD1 mutation testing is generally available to patients in all jurisdictions either through in-house or out-of-jurisdiction testing. For example, in Nova Scotia65 and Alberta,64 testing is performed within the province, whereas in Ontario and Saskatchewan, samples are sent to the US for testing, as per the clinical experts. In Quebec66 and Manitoba,67 testing is available, although it is unclear whether it is performed locally or referred elsewhere. Specific information from other jurisdictions was not available. The clinical experts noted that SOD1 mutation testing is generally publicly funded across jurisdictions, though it may require preapproval when it is performed outside the jurisdiction. | |
Testing procedure as part of routine care | According to the clinical experts, testing for SOD1 mutations is currently performed as part of the standard of care for all patients with ALS in Canada. | |
Repeat testing requirements | The clinical experts indicated that testing for SOD1 mutations is performed at the time of diagnosis and does not need to be repeated. | |
Impacts on human and other health care resources by provision of the testing procedure | Given that several provinces across the country rely on out-of-country laboratories to conduct SOD1 mutation testing, there could be possible delays and logistical challenges related to coordination and the shipping of samples. However, because testing for SOD1 mutations is currently part of the standard of care for ALS, no additional impact on human and other health care resources is anticipated from testing as part of establishing treatment eligibility with tofersen. | |
Patient-related | Accessibility of the testing procedure in jurisdictions across Canada | Some patients and caregivers may face barriers to timely access to SOD1 mutation testing. The clinical experts highlighted that in some jurisdictions, preapproval requirements for out-of-province or out-of-country testing can take up to 4 weeks, delaying access to testing for SOD1 mutations. The need for pretest and post-test genetic counselling services could also pose some delays and system-level challenges. To help address this, ALS Canada has introduced a pilot program, National ALS Genetic Counsellor, to offer virtual genetic counselling services to all patients in Canada, regardless of their location.68 The program aims to connect patients with local genetics experts and genetic counsellors, which may help ensure timely access to SOD1 mutation testing for those requiring pretest counselling.68 The clinical experts also highlighted that patients living in rural and remote areas may face access barriers. For example, they may travel long distances for sample collection, and when phlebotomy services in the clinics are unavailable, the collection kits are shipped to the patient’s home, which can introduce delays in accessing the test. Because testing for SOD1 mutations is currently part of the standard of care for ALS, no additional impact on accessibility is anticipated if tofersen were to be funded. |
Expected turnaround times for the testing procedure | According to the clinical experts, the turnaround time for SOD1 gene mutation testing is typically 1 month; however, in local laboratories with limited testing capacity, this process may take up to 3 months. Because testing for SOD1 mutations is currently part of the standard of care, there is no additional impact on turnaround time anticipated as part of establishing treatment eligibility for tofersen. | |
Burden associated with the testing procedure for patients, families, and/or caregivers | According to the patient input for this review, SOD1 gene testing may cause emotional burdens on patients and their caregivers, such as those related to the risk of ALS in family members. If the results show a variant of uncertain significance, patients and caregivers may experience distress due to the uncertainty about the clinical implications of the finding and whether a targeted treatment would be available for them. However, because testing for SOD1 mutations is currently part of the standard of care for ALS, there is no additional burden to patients, families, and/or caregivers anticipated from the testing as part of establishing treatment eligibility for tofersen. | |
Clinical | Clinical utility and validity of the testing procedure | There is evidence showing the clinical utility of SOD1 mutation testing in guiding treatment decision-making for patients with ALS.69 The clinical experts noted that SOD1 mutation testing is highly accurate, with a low risk of missed diagnoses. Evidence suggests that routine screening for ALS-related pathogenic mutations can positively impact clinical care.20 Studies have demonstrated a high concordance between NGS and Sanger sequencing.60 A study in Canada observed 100% concordance in variants detected by an NGS panel, including SOD1 and those validated via Sanger sequencing.70,a |
Risks of harm associated with the testing procedure | Multigene NGS panels can identify incidental or secondary variants unrelated to ALS, which may lead to additional genetic consultation to help patients understand the clinical implications of these mutations.71 Blood sample collection poses minimal risks. Because testing for SOD1 mutations is currently part of the standard of care for ALS, there is no additional risk of harm associated with the testing as part of establishing treatment eligibility for tofersen. | |
Cost | Projected cost of the testing procedure | The sponsor estimated the cost of SOD1 mutation testing at $1,368.78 per test,72 while the clinical experts reported that the ALS testing panel at Prevention Genetics is approximately $1,600. |
ALS = amyotrophic lateral sclerosis; CDA-AMC = Canada’s Drug Agency; NGS = next-generation sequencing.
aCDA-AMC has not evaluated or critically appraised this evidence to determine its validity or reliability.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Patient input was submitted by the ALS Society of Canada, the ALS Action Canada Society, the ALS Society of Alberta, and the ALS Society of British Columbia. Information was gathered from patients and caregivers through an online survey, interviews, and focus groups. More than 20 respondents living with ALS had experience with tofersen.
The input noted that ALS severely impacts patients’ mobility, strength, daily activities, and emotional health. Key symptoms include muscle weakness, balance issues, cramping, and nerve pain, as well as difficulty speaking, swallowing, and breathing. Limited mobility hinders independence, affecting tasks like climbing stairs, carrying groceries, or standing for prolonged periods. Emotional tolls include the fear of disease progression, anxiety about future independence, and the burden on loved ones. For those with familial ALS, the disease is deeply personal because many have seen its effects on loved ones, leading to significant emotional and psychological strain. Within a year of diagnosis, patients reported needing help from caregivers with daily tasks like eating, walking, and bathing. The input noted that patients with ALS, facing a progressive and fatal illness, often pursue all viable treatments, including clinical trials, alternative therapies, and off-label options. The input stated that in Canada, ALS treatments include riluzole and edaravone; these may provide some benefit but they do not significantly alter disease progression. Furthermore, some patients indicated difficulty accessing edaravone due to a lack of private coverage, strict public funding criteria, out-of-pocket costs, and supply shortages. The input noted that neither therapy specifically addresses the SOD1 gene mutation in ALS.
Patients who have had experience with the drug under review reported benefits including maintaining independence and delaying symptom onset, allowing them to maintain activities that were previously becoming difficult and the ability to spend more time with loved ones. Patients noted that lumbar punctures to administer tofersen pose logistical challenges, because patients often needed to travel long distances to receive treatment, as well as physical challenges, with adverse effects such as headaches, migraines, nausea, and temporary incapacitation. The input stated that improved protocols, such as slow injections and localizing administration to nearby clinics, could enhance accessibility and patient experience. The input emphasized the importance of early and accessible genetic testing for timely intervention with tofersen. Respondents found their experiences with genetic testing to be efficient with timely results and minimal cost barriers. The most critical unmet needs according to the patient group input include symptom reversal (i.e., muscle weakness, cramping, and fasciculations), the maintenance of mobility, function, and independence, the slowing of disease progression, increased survival, and improved QoL.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). In addition, as part of the review of tofersen (Qalsody), a panel of 5 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with ALS associated with a mutation in the SOD1 gene, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
According to the clinical experts, before the availability of tofersen, the primary unmet need for patients with SOD1-ALS was the lack of treatments capable of significantly altering the disease course. Available therapies like riluzole and edaravone offered only modest effects, slowing progression slightly but failing to halt or reverse functional decline. All symptomatic SOD1 patients who were treated with riluzole and/or edaravone progress inexorably to respiratory failure and death. The main goals of treatment (ideally arresting disease progression, or at minimum significantly slowing the rate of functional decline) were largely unmet. Because riluzole and edaravone act through nonspecific neuroprotective mechanisms, a treatment targeting the pathogenic mutant SOD1 protein itself was viewed as a critical gap.
Further unmet needs and challenges were identified by the experts. Significant geographical disparities exist across Canada concerning access to specialized ALS care, timely diagnosis, genetic testing capabilities, and the necessary facilities and personnel for administering intrathecal tofersen. Patients in rural and remote areas face distinct barriers. Access to publicly funded, timely SOD1 genetic testing varies across provinces and territories, with some regions relying on testing conducted outside the province or country or on potentially unsustainable industry-sponsored panels. Additionally, the requirement for monthly intrathecal injections necessitates specific infrastructure and personnel, presenting potential resource challenges. The future development of intrathecal delivery devices was noted as a need. While the NfL level correlates with disease progression rate and is prognostic for disease progression in ALS, its utility in predicting an individual’s clinical outcomes is not fully established, routine accessibility is limited in Canada, and a direct correlation with individual clinical outcomes is not established enough to guide treatment decisions.
Place in Therapy
The clinical experts view tofersen as a first-line, disease-modifying therapy for adults with symptomatic ALS caused by a pathogenic SOD1 mutation. By lowering production of the toxic mutant SOD1 protein, tofersen directly targets the underlying disease mechanism. Clinical experts advise initiating treatment as soon as an ALS specialist confirms the diagnosis; current evidence does not support routine use in presymptomatic carriers outside research settings. Where tolerated and reimbursed, riluzole (and, when feasible, edaravone) will typically be continued alongside tofersen for potential additive benefit, though clinicians agree that patients should not be required to try or have an inadequate response to these drugs before accessing tofersen. Early initiation is considered critical to preserve motor neurons and maximize benefit, and experts expect the availability of tofersen to shift ALS care from merely slowing functional decline to the possibility of disease modification, altering both survival counselling and expectations for long-term stability.
Patient Population
According to the experts, the patients most likely to benefit are adults with a diagnosis of ALS and a confirmed pathogenic SOD1 mutation who are symptomatic and still retain sufficient residual motor function. The operationalization of the expectations of sufficient residual motor function would typically exclude patients who are in the final months of life or those permanently dependent on invasive ventilation. No meaningful differences in response are expected between SOD1 dominant and recessive inheritance types, and the clinical expert does not believe there is value in targeting certain pathogenic variants.
The experts noted that rapidly progressive phenotypes and younger individuals, who face the prospect of losing many life-years, would have the greatest need. Conversely, very advanced patients (e.g., anticipated survival < 6 months, an inability to tolerate a lumbar puncture) and presymptomatic carriers were considered least suitable for therapy. Universal SOD1 testing at ALS diagnosis was recommended, yet access remains inconsistent and often reliant on industry-sponsored programs, raising equity concerns; misdiagnosis was felt to be rare among specialists, though variants of uncertain significance require expert interpretation.
Assessing the Response Treatment
In clinical practice, the response to treatment is assessed primarily through clinical evaluation by an ALS specialist, usually every 3 months. However, timing may vary between 3 months and 6 months depending on disease severity, distance from clinic, and local ALS clinic capacity. Assessment focuses on monitoring the rate of functional decline, looking for stabilization or the slowing of progression. Key tools include manual muscle testing, patient-reported function, and respiratory assessment using FVC. Hard outcomes, including a delay to tracheostomy or death, remain the ultimate measures of effect, but accrue over longer periods.
The clinical experts noted that many end points used in the VALOR trial are not standard tools for routine clinical decision-making in Canada. While the ALSFRS-R scale score is often collected in clinics, clinical experts noted that its absolute score is mainly used for research or facilitating access to other therapies. For guiding tofersen management, they emphasized that it is the monitoring of the change in the slope of the ALSFRS-R that may be clinically meaningful. SVC is not the standard respiratory measure; FVC is preferred clinically, primarily to determine the need for ventilation support. HHD as well as QoL and fatigue scales (ALSAQ-5 and FSS) are considered research tools. Biomarkers and plasma NfL are not widely accessible or routinely used for monitoring response across most of Canada. The clinical experts were unable to provide a clear minimal important difference in these measures but emphasized that monitoring the change in the slope of ALSFRS-R can be clinically meaningful.
The experts noted that a clinically meaningful response to tofersen involves the stabilization or slowing of disease progression. Any improvement in function or strength is highly significant but not always the primary expectation. Changes in functional scales can be meaningful if they impact critical activities like swallowing or mobility. The patient’s and clinician’s perception of benefit, relative to the considerable burden of monthly intrathecal injections, is crucial in determining continued treatment value. The lack of functional improvement does not necessarily equate to treatment failure if the rate of decline has slowed. There are no set objective thresholds for minimum response; continuation is based on the overall clinical picture and patient preference.
Discontinuing Treatment
The clinical experts agreed that defining absolute objective discontinuation criteria is difficult due to disease variability and patient values. However, based on clinical experts’ experience, a number of factors commonly lead to cessation as described in the following paragraph.
Key reasons for discontinuation include patient preference due to a perceived lack of benefit, the significant burden of monthly travel and lumbar punctures, or a desire to shift focus entirely to palliative care. Intolerable or unmanageable AEs, such as severe neurologic inflammation (myelitis, radiculopathy) or papilledema leading to vision impairment despite treatment can necessitate stopping. The physical inability to perform the lumbar puncture procedure, due to the patient’s advanced immobility, inability to be positioned correctly, or severe spinal deformities, is another practical reason for discontinuation. Reaching a very advanced, end-stage disease state with minimal remaining neurologic function, where the potential for benefit is considered negligible compared to the treatment burden, often leads to discontinuation. The need for PV (either via tracheostomy or 24/7 noninvasive support) frequently prompts discussions about end-of-life care and treatment discontinuation, although it’s debated as a strict cut-off point because some function might persist. Importantly, continued disease progression alone is usually not a trigger for discontinuation unless the disease becomes extremely advanced or the patient chooses to stop.
Prescribing Considerations
The diagnosis of SOD1-ALS, the decision to initiate tofersen, and subsequent follow-up should be led by neurologists or physiatrists with ALS expertise who practice, ideally, within a multidisciplinary clinic but may also be at small satellite ALS clinics. Although the prescription remains a specialist responsibility, intrathecal administration can be delegated to any clinician proficient in lumbar puncture. These clinicians can include neurologists, anesthesiologists, interventional radiologists, emergency physicians, internists, or nurse practitioners, provided institutional protocols and resources permit. Most injections are performed in outpatient procedure suites, day surgery units, or community hospitals; a subset of patients may require fluoroscopic guidance, which necessitates access to specialized imaging facilities. Tofersen is given as 3 loading doses 14 days apart, followed by 100 mg maintenance doses every 28 days. The procedure itself does not demand intensive cardiorespiratory monitoring.
Patients should return to their ALS specialist roughly every 3 months for functional assessment, laboratory review, and AE surveillance. Because elevated intracranial pressure and papilledema have been reported, clinicians are advised to arrange periodic ophthalmologic examinations and to expedite evaluation if visual symptoms or optic disc edema are suspected. Riluzole, with or without edaravone, is generally continued for potential additive benefit, although some patients may forgo these drugs because of pill burden, intolerance, or reimbursement constraints.
Implementing monthly lumbar punctures poses significant logistical challenges. Provincial variation in access to ALS specialists, genetic testing, trained proceduralists, fluoroscopic suites, and hospital-based specialty pharmacies will influence how tofersen is delivered. Limited anesthesia support and long travel distances already restrict procedure capacity in some regions; several experts recommended hub-and-spoke or mobile outreach models to extend services to rural and remote patients. Ensuring sustainable staffing, infrastructure, and funding for recurrent intrathecal therapy is therefore a key consideration for health system planners.
Additional Considerations
The clinical expert panel underscored the need to expand publicly funded genetic testing infrastructure so that all newly diagnosed patients with ALS can be screened promptly for SOD1 mutations. Research into implantable intrathecal delivery systems and biomarker-guided dosing was considered a priority to reduce procedural burden and personalize treatment schedules.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One clinician group provided input for this review: The Canadian ALS Research Network, consisting of 14 clinicians. The submissions noted that ALS is a progressive neurodegenerative disease that leads to the degeneration of motor neurons in the brain and spinal cord. This results in severe weakness in limb, bulbar, and respiratory muscles, eventually causing loss of autonomy and dependence on assistive devices like wheelchairs, feeding tubes, and ventilatory support. Most patients die from respiratory failure within 5 years of diagnosis. The experts noted that available ALS treatments include riluzole, which extends survival by about 3 months by targeting glutamate, and IV or oral edaravone, which may slow disease progression in specific patients. Albrioza was approved in 2022; however, it was withdrawn in 2024 after failing a phase III clinical trial. The input noted that these treatments provide only modest benefits and do not reverse the disease or halt its progression and there are currently no approved treatments specifically targeting hereditary ALS. Clinicians emphasized the urgent need for personalized, disease-modifying treatments. Ideal therapies would slow progression, improve QoL, target the root causes of ALS, and reduce the burden on caregivers. Given the complexity of ALS, treatments should be tailored to individual patients, prevent motor neuron degeneration, and incorporate precision medicine approaches.
The input stated that tofersen is best suited for patients with ALS with pathogenic or likely pathogenic SOD1 gene mutations and weakness linked to ALS, as determined by a specialist. Patients with uncertain SOD1 variants tied to the disease may also be eligible. Because there is no diagnostic biomarker for weakness related to ALS, a diagnosis is made based on a patient’s history, physical examination, and electrodiagnostic examination, and the exclusion of alternative diagnoses. Upon diagnosis, all patients with ALS should undergo genetic testing for common ALS-related genes, including SOD1. If an SOD1 mutation is identified, patients should be promptly considered for the drug under review. The input stated that tofersen would be used in combination with existing therapies, creating a multimodal approach that addresses multiple disease pathways. The input noted that there is no rationale for requiring patients to have an inadequate response to other therapies before initiating tofersen, given the irreversible progression of ALS. The clinicians noted that a treatment response or failure for ALS is not precisely defined given that the primary goal of treatment is to slow the degeneration of motor neurons. Individual disease progression varies and tracking outcomes like slowed progression is challenging due to disease heterogeneity. An appropriate treatment strategy involves initiating the drug and monitoring the patient at regular intervals until care goals transition to a more palliative approach or the patient and physician decide to stop treatment based on an unfavourable risk-benefit assessment.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation question | Clinical expert response |
|---|---|
Relevant comparators | |
Is there any evidence to support the addition of riluzole and/or edaravone once a patient is stable on tofersen? | There is no direct evidence to confirm benefit or harm from triple therapy; however, experts believe there is no biological reason to withhold riluzole or edaravone and believe a modest additive benefit is plausible. In practice, most centres will continue riluzole — and, where funded and when tolerated, edaravone — alongside tofersen, while acknowledging that some patients or provinces may choose tofersen monotherapy to reduce pill burden or cost. |
Considerations for initiation of therapy | |
Ongoing measurement of SOD1 mutations and NFL is required to measure intermediate outcomes. Are these standard tests that can be undertaken in all provinces where patients are treated? | For SOD1 mutations, genetic testing is performed at diagnosis to confirm eligibility for treatments like tofersen. However, once a pathogenic SOD1 mutation is identified, ongoing or repeat measurement of the mutation itself is not considered necessary or standard practice because the genetic mutation does not change. The initial diagnostic testing for SOD1 mutations does face variability in access across Canada, with some provinces having in-house capabilities while others rely on out-of-province or out-of-country testing, sometimes facilitated by sponsored panels whose long-term availability is uncertain. Serial measurement of CSF SOD1 protein is not feasible in routine practice; the assay is research-only and unavailable in clinical laboratories in Canada, and clinicians do not consider it necessary for treatment monitoring. For NfL measurement, its availability as a standard test for ongoing monitoring is also inconsistent across Canadian provinces. While NfL assays are commercially available and used in some specialized clinics (e.g., in Alberta) to assess target engagement and as a prognostic marker, they are not routinely publicly funded or widely accessible in many other jurisdictions such as Ontario or Saskatchewan. Experts have indicated that while NfL measurement is a valuable research tool and can show target engagement, its individual-level predictive value for clinical outcomes or its utility in making ongoing treatment decisions (such as continuation or discontinuation of therapy) is an area of ongoing research where targeting normal levels is an intuitive goal. |
Can the drug be given again to patients who relapsed while off therapy due to side effects or discontinued for other reasons? | The experts noted that the reason for the initial stoppage is important; for instance, if it was due to temporary factors (like moving or pregnancy) or patient preference at the time, restarting could be considered. However, if the discontinuation was due to significant side effects, such as severe inflammatory reactions like myelitis or papilledema, the risks associated with re-exposure would need careful evaluation, and the potential outcome of restarting in such specific cases is considered unknown. The decision would likely involve a risk-benefit analysis in the context of the patient’s condition and the nature of the previous adverse event. |
Considerations for continuation or renewal of therapy | |
Despite evidence that tofersen had biological effects, the primary analysis at week 28 of the VALOR trial did not show a statistically significant difference between tofersen and placebo. What would be the length of time approved for treatment given these results? Is treatment to continue long term without any discontinuation parameters besides permanent ventilation? | Experts noted that a 6-month trial is too short for a gene-targeted therapy whose clinical effects emerge after biomarker normalization. Experts pointed to promising trends from the open-label extension and real-world use that supports sustained benefit over years. Regarding the approved length of treatment, the consensus among the consulted experts leans toward recommending long-term treatment approval without a predefined stop date, contingent on ongoing benefit perceived by the patient and clinician. Treatment continuation should generally occur as long as the benefits are deemed to outweigh the risks and burdens, particularly the monthly intrathecal injections. The clinical experts considered permanent invasive ventilation as a reasonable default stop point, although ultimate decisions should reflect patient preference and clinician judgment of ongoing benefit. Besides permanent ventilation (via tracheostomy or potentially 24/7 noninvasive ventilation), which is a common discontinuation point for other ALS therapies and was supported by some experts as a potential end point for tofersen, other parameters discussed include:
|
Considerations for discontinuation of therapy | |
What would be the criteria for discontinuation of therapy? | The committee discussed the following points as consideration for discontinuation:
|
Considerations for prescribing of therapy | |
Intrathecal administration requires special training and facilities. Is this available in all jurisdictions? Can any neurologist be trained to administer this medication? Does intrathecal administration require inpatient admission to hospital or administration in outpatient clinics? If yes, this has implications for which the budget or program would pay for the therapy. | Tofersen must be prescribed by an ALS-expert neurologist or physiatrist, but any clinician competent in LP (e.g., neurologist, anesthesiologist, interventional radiologist, many emergency physicians, some nurse practitioners) can give the dose after a brief orientation. Procedures are normally done in outpatient procedure rooms, day surgery units, or community hospitals; routine inpatient admission is unnecessary, which keeps the cost in the outpatient or hospital clinic budget rather than acute care beds. Minimal equipment is needed: an exam room, a sterile LP kit, and pharmacy cold-chain storage. Geographic disparities in procedural capacity and anesthesia support remain a bottleneck, so hub-and-spoke or mobile models may be needed for rural areas. |
Care provision issues | |
Who would be purchasing the drug? The hospital? A community pharmacy or specialty pharmacy? Specialty pharmacies would need a contract in place and this could affect the cost of acquiring the medication. | Distribution pathways are still being negotiated. Panellists expect most jurisdictions to route purchasing through hospital-based special services pharmacies or contracted specialty pharmacies that can manage cold-chain storage and controlled-access biologics; community retail pharmacies are viewed as unlikely partners. Final arrangements will influence the acquisition cost and may require provincial contracts with the sponsor. |
ALS = amyotrophic lateral sclerosis; CSF = cerebrospinal fluid; LP = lumbar puncture; NfL = neurofilament light chain.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of tofersen, intrathecal, 100 mg per 15 mL (6.7 mg/mL) initiated with 3 loading doses every 14 days and then as a maintenance dose every 28 days thereafter for the treatment of ALS associated with a mutation in the SOD1 gene. The focus has been placed on comparing tofersen to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of tofersen is presented in 2 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes additional studies that were considered to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document.
One RCT and 1 OLE study were identified in the systematic review. The OLE study was presented alongside the pivotal RCT because it had a large impact on the regulatory approval of the drug and was considered to provide important and pivotal complementary evidence to that of the RCT.
Four real-world studies (3 studies in Germany and 1 study in Italy) addressing gaps in evidence were also identified.
Systematic Review
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Detail | VALOR study | OLE study |
|---|---|---|
Designs and populations | ||
Study design | Randomized, double-blind, placebo-controlled, phase III study | Multicentre, open-label, long-term extension study |
Locations | 32 study sites in Australia, Belgium, Canada, Denmark, France, Germany, Italy, Japan, the UK, and the US | 32 study sites in Australia, Belgium, Canada, Denmark, France, Germany, Italy, Japan, the UK, and the US |
Patient enrolment dates | Start date: March 27, 2019 (Part C) End date: July 16, 2021 | Start date: March 8, 2017 End date: August 12, 2024 |
Randomized (N) | Tofersen N = 72 Placebo N = 36 | N = 135 N = 104 (VALOR study participants only) |
Inclusion criteria |
|
|
Exclusion criteria |
| — |
Drugs | ||
Intervention |
|
|
Comparator(s) | Placebo consisted of artificial CSF administered as an intrathecal bolus over 1 to 3 minutes, following the same dosing regimen as tofersen | — |
Study duration | ||
Screening phase | 4 weeks | — |
Treatment phase | 24 weeks | Up to 360 weeks |
Follow-up phase | 4 weeks to 8 weeks for patients who did not enrol or had delayed enrolment in the long‑term extension | 4 weeks |
Outcomes | ||
Primary end point | Change from baseline to week 28 in the ALSFRS-R total score in the mITT population | Incidence of AEs and SAEs |
Secondary and exploratory end points | Secondary end points
Exploratory end points
| Secondary end points
Exploratory end points
|
Publication status | ||
Publications | Miller et al. (2022a),73 Miller et al. (2022b),74 and Shaw et al. (2022)75 NCT02623699 | Miller et al. (2022b)74 and Shaw et al. (2022)75 NCT03070119 |
Ab = antibody; AE = adverse event; ALS = amyotrophic lateral sclerosis; ALSAQ-5 = Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 items: ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; anti-HBc = anti–hepatitis B core (antigen); anti-HBs = anti–hepatitis B surface (antigen); CGI-C = Clinical Global Impression–Change; CGI-S = Clinical Global Impressive–Severity of Illness; CNS = central nervous system; CSF = cerebrospinal fluid; ECG = electrocardiogram; FSS = Fatigue Severity Scale; HBsAg = hepatitis B surface antigen; HCV = hepatitis C virus; HHD = hand-held dynamometry; LP = lumbar puncture; mITT = modified intention to treat; MUNIX = motor unit number index; NfL = neurofilament light chain; OLE = open-label extension; PD = pharmacodynamic; PGI-C = Patient Global Impression–Change; PGI-S = Patient Global Impression–Severity of Illness; PK = pharmacokinetic; RNA = ribonucleic acid; SAE = serious adverse event; SF-36 = Short Form (36) Health Survey; SVC = slow vital capacity; WPAI = Work Productivity and Activity Impairment; ZBI = Zarit Burden Interview.
Sources: The VALOR trial Clinical Study Report76 and open-label extension Clinical Study Report.77 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The clinical trial program (Study 233AS101) for tofersen was conducted in a randomized, double-blind, placebo-controlled, 3-part, dose-escalation study consisting of a single ascending dose portion (part A of VALOR study), a multiple ascending dose portion (part B of VALOR study), and a pivotal portion (part C of VALOR study). Participants who completed Study 23AS101had the opportunity to be screened for the OLE study, Study 233AS102 (referred to as the OLE). The focus for this submission is on the VALOR study and its OLE study.
The VALOR study was a 28-week, phase III, randomized, double-blind, placebo-controlled study to examine the efficacy, safety, tolerability, pharmacokinetic (PK), and pharmacodynamic (PD) effects of tofersen administered by intrathecal injection in adults with ALS and a confirmed SOD1 mutation. Participants (N = 108) were randomized to receive tofersen 100 mg (3 loading doses once every 14 days and 5 maintenance doses once every 28 days) or placebo in a 2:1 (active to placebo) ratio via a centralized interactive response technology system. Randomization was stratified by 3 factors: whether a participant met prognostic enrichment criteria for rapid disease progression, whether a participant used edaravone at baseline, and whether a participant used riluzole at baseline.
The VALOR study was designed with a 6-month duration to offer the earliest opportunity to detect any impact of tofersen treatment. The study enrolled a broad population of adults with SOD1-ALS, with a primary analysis population (mITT population) comprising the subset of participants who met the prognostic enrichment criteria for faster disease progression (the “faster-progressing” subgroup) based on SOD1 mutation type and ALSFRS-R prerandomization slope; refer to Table 6 for more details on the criteria for the mITT population. The remaining participants were assigned to the non-mITT population or “slower-progressing” subgroup.
The primary objective of the VALOR study was to evaluate the clinical efficacy of tofersen administered to adult participants with ALS and a confirmed SOD1 mutation through genetic testing. The secondary objectives of the VALOR study were to evaluate the safety, tolerability, PD, and biomarker effects of tofersen administered to adults with SOD1-ALS.
The VALOR study was a total of 32 weeks to 36 weeks in duration, including a 4-week screening period, a 24-week treatment period (3 loading doses of tofersen or placebo 2 weeks apart, followed by 5 maintenance doses of tofersen or placebo 4 weeks apart), and a 4-week to 8-week follow-up period.
Study 102 was an OLE study with the primary objective of assessing the long-term safety and tolerability of tofersen in participants with ALS and a confirmed SOD1 mutation who completed part A, part B, or part C of Study 23AS101. The secondary objectives were to evaluate the PK, PD, and biomarker effects and the efficacy of tofersen. The data for the OLE study presented as follows focus on the participants who completed part C of the VALOR study. After completion of the VALOR trial, participants were eligible to enrol in the OLE study and could remain in the study to receive up to 90 maintenance doses of tofersen. Blinding of the VALOR trial’s treatment group was maintained in the OLE study. All participants enrolling in the OLE study received 3 loading doses at the start of the OLE study; participants who had received tofersen in the VALOR trial received 2 doses of tofersen on day 1 and day 29, and placebo on day 15.
The tofersen development program was prospectively designed to evaluate crossover from the VALOR trial to the OLE study. This enabled the comparison of early-start tofersen (participants who were randomized to tofersen in the VALOR trial and continued tofersen in the OLE study) versus delayed-start tofersen (participants who were randomized to placebo in the VALOR trial and had the opportunity to initiate tofersen in the OLE study approximately 6 months later). To protect the integrity of ongoing data collection in the OLE study, study participants, site staff, and the firewalled study team remained blinded to individual treatment assignments from the VALOR trial through to completion of the OLE study in August 2024. There were 2 reported data cut-offs for the OLE study: the first on January 16, 2022, which reported 52 weeks of tofersen use between the VALOR trial and the OLE study, and the second on February 28, 2023, which represented 104 weeks of tofersen use.
Populations
Inclusion and Exclusion Criteria
The VALOR study enrolled participants aged 18 years or older with a weakness attributable to ALS and a confirmed SOD1 mutation. The primary analysis population (the mITT population) consisted of participants considered more likely to experience faster disease progression during the study period (the enriched or faster-progressing subgroup) as compared to the other or slower-progressing subgroup, according to predefined criteria (the non-mITT population).
The prognostic enrichment criteria for rapid disease progression included participants who had 1 of the following 2 criteria:78,79
one of the following SOD1 mutations known to be associated with more rapid disease progression — p.Ala5Val, p.Ala5Thr, p.Leu39Val, p.Gly42Ser, p.His44Arg, p.Leu85Val, p.Gly94Ala, p.Leu107Val, or p.Val149Gly — and a prerandomization ALSFRS-R slope decline of 0.2 per month or more (calculated as [48 – baseline score] / time since symptom onset)
an SOD1 mutation other than those listed earlier with a prerandomization ALSFRS-R slope decline of 0.9 per month or more (calculated as [48 – baseline score] / time since symptom onset).
The criterion for all other eligible participants (the non-mITT population) was to have an SOD1 mutation other than those listed in the first bulleted point of the preceding bulleted list.
For participants who met the prognostic criteria for faster disease progression, an SVC of 65% or more of predicted value was required. All other eligible participants must have had an SVC of 50% or more of predicted value. Participants were permitted to be receiving concomitant riluzole and edaravone provided they were on stable doses for 30 days or more and 60 days or more, respectively.
After completion of the VALOR study, participants were eligible to enrol in the OLE study immediately, after a small break or no break at all.
Interventions
During the VALOR study, 100 mg of tofersen or placebo was administered 8 times (3 loading doses once every 2 weeks and 5 maintenance doses once every 4 weeks) by intrathecal administration. Before injection, approximately 10 mL of CSF was collected for analyses. A total volume of 15 mL of tofersen or placebo was administered over a 1-minute to 3-minute bolus injection.
The dose of 100 mg of tofersen was determined based on data from participants in part B (multiple ascending dose), cohort 5 to cohort 8, who were treated for 85 days. Safety analyses of these data suggested that all doses through 100 mg had been well tolerated, with a safety profile supportive of continued development of tofersen in SOD1-ALS participants. The selection of the 100 mg tofersen dose for part C was supported by PK and PD and exploratory efficacy analyses of data from participants in part A (single ascending dose) and part B (multiple ascending dose).
The participants who received placebo in the VALOR study received 100 mg of tofersen, 3 loading doses approximately once every 14 days (day 1, day 15, and day 29), and up to 90 maintenance doses approximately every 28 days, by intrathecal administration in the OLE study. The participants who received tofersen in the VALOR study received 100 mg of tofersen, 2 doses on day 1 and day 29, while receiving 1 dose of placebo on day 15 (to maintain blinding of the treatment group in the VALOR study), and up to 90 maintenance doses of tofersen approximately every 4 weeks, by intrathecal administration in the OLE study.
Participants taking riluzole at study entry must have been on a stable dose for at least 30 days before day 1, and the dose was expected to remain stable for the duration of the study. Participants taking concomitant edaravone at study entry must have initiated edaravone 60 days or more (2 treatment cycles) before day 1 and must have continued with the same dose regimen throughout the study. Edaravone may not have been administered on dosing days of this study.
Daily intake of vitamins and supplements must have been stabilized at least 14 days before day 1. Daily intake of creatine must have been 5 g or less, and vitamin E must have been 1,000 IU or less. Other concomitant medication for symptom management was at the investigator’s discretion.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, CDA-AMC selected end points that were considered to be most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized, placebo-controlled, efficacy end points were assessed using the GRADE tool. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | VALOR studya | OLE studyb |
|---|---|---|---|
Change from baseline in ALSFRS-R | 28 weeks | Primary | At weeks 52 and 104; secondary |
Change from baseline in CSF SOD1 proteinc | 28 weeks | Key secondaryd | At weeks 52 and 104; secondary |
Change from baseline in NfL in plasma | 28 weeks | Key secondaryd | At weeks 52 and 104; secondary |
Change from baseline in SVC | 28 weeks | Key secondaryd | At weeks 52 and 104; secondary |
Change from baseline in HHD megascore | 28 weeks | Key secondaryd | At weeks 52 and 104; secondary |
Time to death or permanent ventilation | 28 weeks | Key secondaryd | At weeks 52 and 104; secondary |
Change from baseline in ALSAQ-5 total score | 28 weeks | Exploratory | At weeks 52 and 104; exploratory |
Change from baseline in FSS total score | 28 weeks | Exploratory | At weeks 52 and 104; exploratory |
ALSAQ-5 = Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 items; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CSF = cerebrospinal fluid; FSS = Fatigue Severity Scale; HHD = hand-held dynamometry; ITT = intention to treat; mITT = modified intention to treat; NfL = neurofilament light chain; OLE = open-label extension; SVC = slow vital capacity.
aThe analysis population is the mITT population.
bThe analysis population is the ITT population.
cThe analysis population is the non-mITT population.
dStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchical testing).
Sources: The VALOR trial Clinical Study Report76 and open-label extension Clinical Study Report.77 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
Change from baseline through 28 weeks on the ALSFRS-R total score in the mITT population was assessed as the primary efficacy end point for the VALOR study and as a secondary end point in the OLE study. ALSFRS-R is a 12-item scale that assesses function in 4 domains: respiratory, bulbar, gross motor, and fine motor. Each item is rated on a scale of 0 to 4, generating an ALSFRS-R total score of 0 (maximum disability) to 48 (no disability). ALSFRS-R has been correlated with QoL, caregiver burden, and survival.36,80-82 In addition to its use in clinical practice, ALSFRS-R has been commonly used as a primary end point to assess function in daily activities in ALS clinical trials.83 Guidelines on the development of drugs to treat ALS published by the FDA and the European Medicines Agency both reference the ALSFRS-R as a suitable primary end point to assess effectiveness.84,85
The VALOR and OLE study protocols specified that the same qualified and trained ALSFRS-R rater should have consistently administered the ALSFRS-R for a participant to answer and remained blinded to both the participants’ treatment assignments and to the results of other assessments.
This measure has strengths, including ease of use and correlation with survival; however, notable limitations are documented regarding intraparticipant variability, the nonlinearity of the scale, and the lack of global standardized administration procedures, which have the potential to confound the evaluation of treatment effect in studies of relatively small size and duration.78,86-89
A minimal clinically important difference threshold has not been established for this measure, at least in part because of its ordinal nature. As with other ordinal scales, change in ALSFRS-R scores typically follow a nonlinear, S-shaped curve, where a 1-point change is not consistent across the scale. However, it is well recognized that even a small drop in a patient’s ALSFRS-R score can significantly inhibit functional abilities. For example, a 1-point drop can reflect the difference between being able to walk with assistance and being nonambulatory. A 1-point drop has also been associated with a 7% increase in the risk of death or tracheostomy.82 The natural history rate of decline in ALSFRS-R varies widely but on average, a 1-point decline per month on the population level is observed.36,37
Several ALS staging methods exist, each serving different purposes such as rehabilitation, rapid assessment, treatment comparison, biomarker analysis, and economic evaluation.90 ALSFRS-R scores are used to inform the Milano-Torino Staging (MiToS) functional staging method; it uses 6 stages, ranging from normal function (stage 0) to death (stage V). MiToS staging assesses loss of independence in 4 functional domains: movement (walking and self-care), swallowing, communication, and breathing. In MiToS staging, these 4 domains are values shown as either 0 (function still maintained) or 1 (functional loss). Functional loss is often defined as ALSFRS-R item scores of 1 or lower for item 6 (dressing/hygiene) or item 8 (walking) for movement, item 3 for swallowing, item 1 (speech) and item 4 (writing) for communication, and item 10 (dyspnea) or item 12 (respiratory insufficiency) for breathing. In some cases, a score of 2 or lower for item 12 (respiratory insufficiency) was used to define functional loss. The total number of domains with functional loss determines the MiToS stage.91,92
The King’s clinical staging system uses 5 clinical stages, ranging from symptom onset (stage I) to death (stage V). Though King’s clinical staging does not rely on the ALSFRS-R, its stages can be estimated from ALSFRS-R scores.90 In a study reanalyzing the clinical trial data of patients with ALS, both staging systems found higher stages occurring later in the disease, but the timing varied. King’s stage III most often aligned with MiToS stage I, while earlier King’s stages matched MiToS stage 0 or stage I. The study found that King’s staging offers more detail in early disease to mid-disease, reflecting clinical burden. MiToS provides better resolution in later stages, focusing on functional decline.90
Total CSF SOD1 Protein
Total CSF SOD1 protein levels were assessed as key secondary end points in the VALOR and OLE studies. Tofersen is an antisense oligonucleotide designed to bind and degrade SOD1 messenger ribonucleic acid to reduce the synthesis of SOD1 protein, which is thought to acquire toxic properties through a gain of function mechanism. Given that tofersen targets SOD1 messenger ribonucleic acid to reduce SOD1 protein synthesis, total CSF SOD1 protein was measured as a PD measure of indirect target engagement using a fit-for-purpose analytically validated commercial assay. Formal testing of this biomarker was performed in the slower-progressing population, dictating the sample size of this subgroup. Therefore, the primary end point in this slower-progressing population (the non-mITT population) was change from baseline in total CSF SOD1 protein.
NfL Levels
NfL levels in plasma were assessed as key secondary end points in the VALOR and OLE studies. NfL levels are established markers of axonal injury and neurodegeneration and are detectable in both CSF and blood.93 Elevated levels of pNfH and NfL are observed in blood and CSF in multiple neurodegenerative diseases, including ALS. Emerging data support NfL as a potential biomarker of ALS disease activity93-95 and treatment response.93 A fit-for-purpose assay was used to evaluate NfL and pNfH in plasma and CSF.
Slow Vital Capacity
Because respiratory failure is the most common cause of death from ALS, respiratory status is a critical parameter to monitor in patients living with ALS. Questions in the respiratory subdomain of the ALSFRS-R are relatively insensitive to change, thus highlighting the importance of incorporating a direct measure of respiratory function, such as vital capacity.35,96 SVC is the amount of air expelled from the lungs during a slow, gentle breath. SVC was selected as a secondary end point in the VALOR and OLE studies because it is considered less variable than FVC in patients with impaired breathing, spasticity, and/or significant bulbar dysfunction.35,96 Change in SVC over time has been shown to be predictive of respiratory failure, tracheostomy, or death in patients with ALS.35,96-99 Andrews et al.35 constructed a Cox proportional hazards regression model to evaluate time to key events. They concluded that a slowing in the rate of SVC decline by 1.5 percent predicted per month reduced the risk in first onset of respiratory insufficiency or death, first occurrence of tracheostomy or death, and death at any time after 6 months by 22%, 23%, and 23%, respectively (P < 0.001).35 The natural history of ALS suggests that the average decline of SVC is 2.7 percent predicted per month.35
Time to Death or PV
Recognizing the progressive and fatal nature of SOD1-ALS, time to death or PV and time to death were assessed as key secondary end points in the VALOR and OLE studies. PV was defined as 22 or more hours of mechanical ventilation (invasive or noninvasive) per day, for 21 or more consecutive days. The threshold for PV of 21 or more consecutive days was extended beyond that used in previous studies (e.g., ≥ 7 days to 10 days) to differentiate between PV and potential acute reversible illnesses (e.g., pneumonia) necessitating temporary ventilatory support.100,101 The definition is based solely on the duration of use and does not distinguish between invasive and noninvasive ventilation. Recognizing that standards of care for tracheostomy vary globally, the placement of a tracheostomy tube was not considered an event unless the threshold for ventilatory support was met.
Survival analysis necessitates studies of adequate size and duration. When designing VALOR, it was recognized that the short study duration (6 months) and intramutation heterogeneity in disease progression would make the detection of an effect on survival challenging. Therefore, data from the VALOR and OLE studies were integrated to enable the comparison of survival with early-start versus delayed-start tofersen as well as a comparison of the tofersen-treated population to the expected mutation-specific natural history.
Hand-Held Dynamometry
Loss of muscle strength is a hallmark of ALS. People living with ALS become progressively weaker over time, and strength loss is directly related to declining function.101 Weakness in ALS is relentlessly progressive, and increases in strength are inconsistent with the natural history of the disease.101,102 HHD is a reliable and reproducible quantitative measure of muscle strength decline in ALS.102
Quantitative muscle strength testing using HHD was incorporated as a key secondary end point in the VALOR and OLE studies. The HHD megascore was calculated by averaging z scores for 16 individual muscle groups in the upper and lower extremities (left and right shoulder flexion, elbow flexion, wrist extension, index finger abduction, thumb abduction, fifth digit abduction, knee extension, and ankle dorsiflexion). While assessment of muscle strength is highly clinically relevant, the location, severity, and progression of weakness can vary significantly between patients with ALS. The onset of weakness from ALS is often asymmetric, even focal. The composite HHD megascore is a single score across muscles, which may confound the ability of the score to detect a treatment effect on focal or asymmetric weakness, especially over the short term.
Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 Items
The ALSAQ-5 is a disease-specific patient self-reported health status questionnaire. The ALSAQ-5 contains 5 questions, each corresponding to 1 of the following 5 health-related dimensions: physical mobility, activities of daily living, eating and drinking abilities, communication, and emotional functioning. The questions are followed by 5 responses, with the raw scores ranging from 0 = never to 4 = always or cannot do at all. The score for each question was calculated as (raw score ÷ 4) × 100,103 ranging from 0 to 100, with lower scores representing better health-related status.
Fatigue Severity Scale
Fatigue is reported as 1 of the most bothersome and undertreated symptoms associated with ALS (Nicholson [2018]). The Fatigue Severity Scale (FSS) has been used to measure fatigue in a variety of conditions, including ALS.104-107 It is a 9-item self-reported questionnaire designed to assess fatigue across 3 domains: life participation, sleep, and daily activities. Higher scores are indicative of greater fatigue in everyday life.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ALSFRS-R | A 12-item clinician-administered questionnaire assessing daily living functionality in patients with ALS across 4 domains: bulbar, gross motor, fine motor, and respiratory. Scores range from 0 (absent function) to 4 (no impairment), summed for a total score of 0 to 48.108 | Validity: Strong correlation between the ALSFRS-R total score and the SIP (r = −0.72); weak correlation between its respiratory subscale and FVC (r = 0.33).108 Meta-analysis found a weak negative correlation between ALSFRS-R scores and CSF (r = –0.35) or blood (r = –0.32) NfL levels.109 Two studies found that CFA supported a 4-factor structure (bulbar, gross motor, fine motor, and respiratory) rather than a total score.110,111 In 1 study, CFA did not confirm a clear factor structure, indicating the ALSFRS-R may not reliably assess distinct functional areas.6 Rasch analysis found the total score invalid at the ordinal level due to multidimensionality.6 Weak to modest factor correlations suggest the ALSFRS-R represents a profile of 4 clinically relevant domain scores rather than a total score of disease severity.111 Reliability: Evidence of internal consistency (Cronbach alpha > 0.67) across all domains and total score (Cronbach alpha = 0.73).108 Test-retest reliability has been demonstrated across 3 trials (ICC > 0.93).112 There is substantial agreement for inter-rater reliability for the majority of items across 3 trials (Cohen kappa > 0.76).112 Responsiveness: No data were identified from the literature for this disease area. | For the total score In a study of ALS clinicians and researchers: A change of ≥ 20% in the decline of the ALSFRS-R score was clinically meaningful.36 Estimated MID in a study of patients with a motor neuron disease in the UK and Australia: There was a 3.8-point difference between groups over a 3-month time frame (1.27 points per month) using GROC as an anchor.5 Estimated MID in a study of patients with ALS at 1 US site: There was a 3.24-point difference between groups over an approximately 6-month time frame (0.57 points per month) using PGI-C as an anchor.5,6 |
ALSAQ-5 | A disease-specific, self-reported health status questionnaire with 5 questions assessing physical mobility, daily activities, eating and drinking, communication, and emotional functioning. Responses range from 0 (never) to 4 (always or cannot do at all). Scores range from 0 to 100; lower scores indicate better health status.103 | Validity: Evidence of correlation between the ALSAQ-5 items and their respective parent dimension on the ALSAQ-40 (r ≥ 0.79).103 Reliability: A systematic review found evidence of good test-retest reliability (r, 0.73 to 0.92) based on moderate quality evidence. There was a high degree of inter-relatedness among the ALSAQ-5 items (Cronbach alpha ≥ 0.72), indicative of internal consistency reliability.113 Responsiveness: Responsive to change in patients with a motor neuron disease who reported at follow-up that they felt “a little worse” for each item.114 | No MID was identified from the literature for this disease area. |
ALS = amyotrophic lateral sclerosis; ALSAQ-5 = Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 items; ALSAQ-40 = Amyotrophic Lateral Sclerosis Assessment Questionnaire–40 items; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CFA = confirmatory factor analysis; CSF = cerebrospinal fluid; FVC = forced vital capacity; GROC = global rating of change; ICC = intraclass correlation coefficient; MID = minimal important difference; NfL = neurofilament light chain PGI-C = Patient Global Impression–Change; SIP = Sickness Impact Profile.
Statistical Analysis
Primary End Point in the VALOR Study
The primary analysis of the primary efficacy end point, change from baseline to week 28 in the ALSFRS-R total score, was analyzed using the JRT methodology to account for mortality115 for the primary inference. JRT was conducted in conjunction with MI to handle withdrawals for reasons other than death. The joint rank allows a statistical test of the treatment effect on the ALSFRS-R total score while accounting for the truncation of data due to deaths. The JRT treats death as the worst outcome and assigns the lowest ranks to participants who have died. The JRT is very sensitive to slight shifts in the distribution of deaths and as a result, 1 death could potentially mask the overall treatment effect. Withdrawals were handled using an imputed score from MI. The analysis was only performed for the mITT population.
Sensitivity analysis of the primary end point using ANCOVA was also performed for the mITT population. The difference between treatment groups in the least squares mean change from baseline to day 197 was estimated using ANCOVA on the MI dataset, with corresponding standard errors and 95% CIs. The ANCOVA model included treatment group as a fixed effect and covariates for the ALSFRS-R total score, baseline disease duration since symptom onset, and the use of riluzole or edaravone. In addition, median changes from baseline to day 197 with corresponding 95% CIs were provided by treating death as worse than the median. The ranked scores were analyzed for each of the 100 MI complete datasets using an ANCOVA model with treatment included as a fixed effect and adjusted for the following covariates: baseline disease duration since symptom onset, baseline ALSFRS-R total score, and the use of riluzole or edaravone. The corresponding nominal P value from the ANCOVA model was presented as a sensitivity analysis.
Given the potential utility of NfL as a biomarker of ALS disease activity, baseline plasma NfL was added as a covariate in the ANCOVA model for the primary efficacy end point as a sensitivity analysis for the mITT population. This analysis was performed for the primary efficacy end point, change from baseline to week 28 in the ALSFRS-R total score, based on both JRT methodology as used in the primary efficacy analysis and on the ANCOVA model based on MI imputed datasets. This analysis was also performed for the non-ITT population as a secondary analysis.
Secondary End Points in the VALOR Study
Unless otherwise stated, all statistical tests for primary and secondary efficacy end points were 2-sided, with overall type I error control at the 5% statistical significance level.
The analyses of secondary end points were based on a sequential testing procedure in the order of the rank of secondary end points as follows:
change from baseline (i.e., ratio) to week 28 (day 197) in total CSF SOD1 protein
change from baseline (i.e., ratio) to week 28 (day 197) in NfL in plasma
change from baseline to week 28 (day 197) in SVC
change from baseline to week 28 (day 197) in HHD megascore to assess muscle strength, as measured by the HHD device
time to death or PV, which is defined as the time to the earliest occurrence of 1 of the following events:
death
PV (≥ 22 hours of mechanical ventilation [invasive or noninvasive] per day for ≥ 21 consecutive days)
time to death.
Details for the statistical testing of the secondary end points are described in Table 9.
Exploratory End Points in the VALOR Study
The change from baseline for the exploratory end points ALSAQ-5, EQ-5D-5L, and FSS were analyzed using the ANCOVA model based on MI imputed datasets.
Efficacy End Points in the OLE Study
The intention-to-treat (ITT) population was used to analyze the change from baseline for ALSFRS-R, CSF SOD1 protein, NfL levels, SVC, and HHD using an ANCOVA model with MI. Details on the statistical testing are provided in Table 9.
In addition to the survival analyses comparing early-start to delayed-start tofersen from the VALOR and OLE study participants and recognizing the potential for convergence, a rank-preserving structural failure time model (RPSFTM) analysis was performed on measures of survival (death; death or PV; and death, PV, or withdrawal due to disease progression) to compare what was observed in the early-start tofersen participants with what would be expected to have occurred in the delayed-start (placebo) group had they not switched to tofersen in the OLE study. RPSFTM is a method used to adjust for treatment switching in trials with survival outcomes. It uses counterfactual survival (survival when only placebo treatment would have been used) and assumes that, at randomization, the counterfactual survival distribution for the investigational and placebo arms are identical.116 Given the 6-month duration of the placebo control in the study and the limitations of available natural history datasets to appropriately match participants in the VALOR study and serve as a control, this analysis provided a way of comparing survival in tofersen-treated participants with a longer-term control. Time to death analyses were also performed in participants from the phase I and phase II period of Study 23AS101 (part A and part B) who received at least 1 dose of tofersen 100 mg in either Study 23AS101 (part B) or the OLE study (N = 43).
Sample Size and Power Calculation
The VALOR trial aimed to randomize 99 participants with approximately 66 participants receiving tofersen 100 mg and approximately 33 participants receiving placebo in a 2:1 ratio.
Given the high unmet medical need and natural history of SOD1-ALS, the tofersen development program aimed to identify the earliest opportunity to detect a clinically meaningful benefit. The primary end point, change from baseline to week 28 in the total ALSFRS-R score, was powered based on those patients who met the enrichment criteria for faster progression, the mITT population, and was only formally tested in this population. The duration (6 months) and sample size (n = 60 in the primary analysis population, the mITT population) of the VALOR study were selected based on the results of the VALOR study, part B,93 and a randomized, controlled clinical trial of arimoclomol,100 wherein SOD1 mutation carriers who received placebo declined rapidly on the ALSFRS-R scale within 6 months. Specifically, data from 12 participants across these datasets who matched the mITT population criteria (according to SOD1 mutation type and prerandomization ALSFRS-R slope) informed the assumption for decline on the ALSFRS-R scale over 6 months in the placebo group (24.7-point decline in the placebo group; i.e., a 3.83 points per month decline [SD = 20.39; i.e., 3.166 points per month]) that was used to calculate the sample size. Assumptions for overall survival incorporated into the sample size estimate (82% in the placebo group and 90% in the tofersen group) were taken from survival estimates in the literature for A5V mutation carriers, the most prevalent fast-progressing mutation type enrolled that is consistently associated with a median survival (based on Kaplan-Meier) of 1.2 years or less.26,27 With the aforementioned assumptions, the study was powered at 84% with a 2-sided alpha of 0.05.
To mitigate potential risks of the relatively short study duration and small sample size, delayed-start analyses at later time points (e.g., week 52 and week 104) were prospectively planned via the integration of data from the VALOR and OLE studies.
The slower-progressing population, the non-mITT population, was not expected to experience a sufficient enough decline on clinical function in the placebo arm over 6 months to be able to detect separation between treatment groups; this population was included in the study design for ethical reasons. For this non-mITT population, the primary focus was the pharmacodynamic biomarker end point of total CSF SOD1 protein concentration. Because total CSF SOD1 protein levels are not correlated with disease progression, the sample size for this subgroup was determined based on the ability to detect changes in this biomarker, rather than changes in clinical outcomes. A sample size of 26 participants in the treated group and 13 participants in the placebo group for the population outside the mITT population would provide 97% power to detect a 25% reduction in total CSF SOD1 protein from baseline in the treated group, with an assumed SD of 0.216 (natural log scale), compared to the placebo group.
Subgroup Analyses
Subgroup analysis was performed for the primary efficacy end point and for the following secondary end points: the change from baseline in the percentage predicted for SVC, the change from baseline in the HHD megascore, and the ratio to baseline for each of total CSF SOD1 protein and plasma NfL values for each of the mITT, non-mITT, and ITT populations. The prespecified subgroups were as follows:
sex (female or male)
baseline disease duration since symptom onset by tertiles (tertiles to be defined separately for each of mITT, non-mITT, and overall ITT populations) (median values may be used instead of tertiles if there are insufficient data)
baseline NfL plasma level (tertiles to be defined separately for each of mITT, non-mITT, and overall ITT populations) (median values may be used instead of tertiles if there are insufficient data)
site of onset (bulbar, other site of onset); if a patient had onset at multiple sites and 1 of the sites is bulbar, the patient was to be included under the bulbar category for subgroup analysis
geographic region (North America, Europe, or Asia Pacific); Canada was included under North America
riluzole or edaravone use (note that if edaravone use was limited to < 5 patients within the relevant population [e.g., the mITT population], then riluzole and edaravone use were to be combined so that there were only 2 levels).
Each subgroup category was analyzed separately using the ANCOVA model based on MI for the mITT, non-mITT, and ITT populations. No P values were available for the ITT population.
There were 2 additional predefined disease progression subgroups specified for each of the 5 aforementioned end points: participants with a prerandomization ALSFRS-R slope decline of at least 0.9 points per month, and a baseline plasma NfL level above or below the median.
The prerandomization ALSFRS-R slope was incorporated into the enrichment criteria based on experience from the dexpramipexole EMPOWER study, which demonstrated that participants with a prerandomization ALSFRS-R slope decline of at least 0.9 points per month maintained a mean postrandomization slope decline of at least 0.9 points per month at 6 months, 9 months, and 12 months.101 However, nonlinear progression on the ALSFRS-R scale, with periods of stable disease preceded or followed by periods of rapid decline,78,117 can limit the utility of the prerandomization ALSFRS-R slope as a marker of active disease progression at a specific point in time (e.g., baseline) as well as the prognostic value over time.
The utility of NfL as a mechanism to control for heterogeneity was not fully appreciated when the VALOR trial was designed. Accumulating literature supporting NfL as a prognostic marker of disease progression led the sponsor to prespecify alternative disease progression subgroups defined according to baseline plasma NfL levels as part of the statistical analysis plan before the NfL final database lock, whereby participants with a baseline plasma NfL level above the median comprised the faster-progressing subgroup and participants with a level below the median comprised the slower-progressing subgroup. Recognizing that adjustment for baseline NfL as a continuous covariate provides greater precision to the estimated treatment difference than categorical subgrouping according to the median, sensitivity analyses for ALSFRS-R incorporating baseline plasma NfL as a covariate in the ANCOVA model were also prespecified in the mITT and non-mITT populations as part of the statistical analysis plan before the VALOR trial’s final database lock. Due to the prognostic value of baseline NfL, analyses in the ITT population adjusting for baseline plasma NfL levels are the most robust and appropriate analyses to account for baseline disease heterogeneity.
Analysis Populations
The overall ITT population for the VALOR and OLE studies was defined as all participants who were randomized and had received at least 1 dose of the study treatment. The safety population was defined as all participants who were randomized or enrolled and had received 1 dose in the study period.
In the VALOR study, the primary analysis of clinical function at week 28 was evaluated in the mITT population, which comprised the subset of the population who met prognostic enrichment criteria for rapid disease progression and who were randomized and had received at least 1 dose of study treatment. At the time of study design, the prerandomization ALSFRS-R slope and SOD1 mutation type were thought to be appropriate tools to control for disease heterogeneity at baseline. These criteria were intended to identify participants more likely to experience faster disease progression during the study period. This enriched mITT population informed the sample size calculation and served as the primary analysis population for the formal testing of the primary and key secondary end points for the VALOR trial.
The population outside the mITT population was the non-mITT population (i.e., all other eligible participants who were randomized and had received at least 1 dose of the study treatment). This population was also referred to as the slower-progressing subgroup; they were expected to decline more slowly over the study period, thus offering less opportunity to demonstrate a difference between groups. The slower-progressing population was not expected to experience a sufficient enough decline on clinical function in the placebo arm over 6 months to be able to detect separation between treatment groups. However, given that total CSF SOD1 protein does not correlate with disease progression, formal testing of this biomarker was performed in the slower-progressing population, dictating the sample size of this subgroup.
Analyses using the mITT population, the non-mITT population, and the overall ITT population were performed according to the treatment assignment at the time of randomization.
Secondary efficacy and PD or biomarker end points were analyzed and tested for the mITT population based on the sequential testing procedure. Formal statistical testing in the non-mITT population was based on total CSF SOD1 protein only. No formal statistical testing was performed for the overall ITT population in the VALOR trial.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model (primary analysis) | Adjustment factors | Handling of missing data | Sensitivity analyses | Secondary analyses |
|---|---|---|---|---|---|
VALOR study | |||||
Change from baseline in ALSFRS-R | JRT + MI (mITT population) | Baseline disease duration since symptom onset, baseline ALSFRS-R total score, and use of riluzole or edaravone | MI |
|
|
Change from baseline in CSF SOD1 protein | ANCOVA + MI (non‑mITT population) | Baseline value, baseline disease duration since symptom onset, and use of riluzole or edaravone | MI | — | ANCOVA + MI (mITT population) |
Change from baseline in NfL in plasma | ANCOVA + MI (mITT population) | Baseline value, baseline disease duration since symptom onset, and use of riluzole or edaravone | MI | — | ANCOVA + MI (non-mITT population) |
Change from baseline in SVC | JRT + MI (mITT population) | Baseline disease duration since symptom onset, baseline ALSFRS-R total score, and use of riluzole or edaravone | MI |
| ANCOVA + MI (non-mITT population) |
Change from baseline in HHD megascore | ANCOVA + MI (mITT population) | Baseline disease duration since symptom onset, baseline ALSFRS-R total score, and use of riluzole or edaravone | MI | — | ANCOVA + MI (non-mITT population) |
Time to death or permanent ventilation | Stratified log-rank test and Cox proportional hazards model (mITT population) | — | — | — |
|
Time to death | Stratified log-rank test and Cox proportional hazards model (mITT population) | — | — | — |
|
OLE study | |||||
Change from baseline in ALSFRS-R | ANCOVA + MI | Prior treatment group from VALOR study, baseline value, and disease duration since symptom onset | MI | Inclusion of baseline plasma NfL as a covariate in ANCOVA (non-mITT population) | — |
Change from baseline in CSF SOD1 protein | ANCOVA + MI | — | MI | — | — |
Change from baseline in NfL in plasma | ANCOVA + MI | — | MI | — | — |
Change from baseline in SVC | ANCOVA + MI | Prior treatment group from VALOR study, baseline value, baseline ALSFRS-R, and disease duration since symptom onset | MI | Inclusion of baseline plasma NfL as a covariate in ANCOVA (non-mITT population) | — |
Change from baseline in HHD megascore | ANCOVA + MI | Prior treatment group from VALOR study, baseline value, and disease duration since symptom onset | MI | Inclusion of baseline plasma NfL as a covariate in ANCOVA (non-mITT population) | — |
Time to death or permanent ventilation | Stratified log-rank test and Cox proportional hazards model | — | — | — | RPSFTM |
Time to death | Stratified log-rank test and Cox proportional hazards model | — | — | — | RPSFTM |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ANCOVA = analysis of covariance; CSF = cerebrospinal fluid; HHD = hand-held dynamometry; ITT = intention to treat; JRT = joint rank test; MI = multiple imputation; mITT = modified intention to treat; NfL = neurofilament light chain; OLE = open-label extension; RPSFTM = rank-preserving structural failure time model; SVC = slow vital capacity.
Sources: The VALOR trial statistical analysis plan118 and the open-label extension statistical analysis plan.119 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 10: Analysis Populations of VALOR and OLE Studies
Study | Population | Definition | Application |
|---|---|---|---|
VALOR | ITT | All participants who were randomized and had received at least 1 dose of study treatment | Efficacy analyses |
mITT | Participants who met the prognostic enrichment criteria for rapid disease progression | Efficacy analyses | |
Non-mITT | Participants in the ITT who were not included in the mITT | Efficacy analyses | |
Safety | All participants who were randomized and had received at least 1 dose of study treatment (i.e., the overall ITT population). Participants were analyzed under the actual treatment they received. | Safety analyses | |
OLE | ITT | All participants who were enrolled and had received at least 1 dose of study treatment | Clinical function |
Safety | All participants who enrolled and had received at least 1 dose of study treatment in the OLE study (i.e., the overall ITT population of participants) | Safety analyses |
ITT = intention to treat; mITT = modified intention to treat; OLE = open-label extension.
Sources: The VALOR trial Clinical Study Report76 and the open-label extension Clinical Study Report.77 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
A total of 60 participants were included in the mITT population, 48 participants were included in the non-mITT population, and 108 participants were included in the ITT population.
Participants were screened at 32 study sites in 10 countries, with 108 participants randomized. All randomized participants were dosed. The majority of study participants were randomized in the US (n = 55 [50.9%] participants), with the remainder randomized in Canada (n = 18 [16.7%] participants); Belgium and Japan (n = 7 [6.5%] participants each); France, Germany, Italy, and the UK (n = 5 [4.6%] participants each); and Denmark (n = 1 [0.9%] participant). Overall, the tofersen and placebo treatment groups were well matched in terms of participant disposition, demographics, and baseline disease characteristics.
A total of 108 participants were enrolled and dosed, and 97 participants completed the study. Treatment discontinuations and study withdrawals were similar in the tofersen group compared to the placebo group: 3 (8.3%) participants in the placebo group and 9 (12.5%) participants in the tofersen group discontinued treatment, and 3 (8.3%) participants in the placebo group and 8 (11.1%) participants in the tofersen group were withdrawn from the study. The discrepancy between treatment discontinuation and study withdrawal in the tofersen group was due to 1 participant who discontinued treatment as the result of an AE (pulmonary embolism) but continued and completed the study. Treatment discontinuations and study withdrawals were more common in the enriched (the mITT population) subgroup. The most common reason for the discontinuation of study treatment was disease progression (n = 5 [4.6%] participants), all of which occurred in the enriched faster-progressing subgroup, followed by AEs (n = 3 [2.8%] participants). One participant (in the enriched subgroup) died 114 days after being randomized to tofersen 100 mg. The cause of death was reported as congestive cardiac failure, and the investigator assessed the event to be unrelated to tofersen.
There was overlap across definitions for faster progressors, but there were a number of examples where this was not the case. For example, there were participants who were defined as “faster-progressing” based on the protocol-defined criteria (mutation + prerandomization ALSFRS-R slope) who were progressing more slowly (≤ 0.9 points per month) during the run-in period and/or had a plasma NfL level below the median, indicative of slower disease progression. Similarly, there were participants defined as “slower-progressing” who were actually progressing much more quickly leading into the study, as illustrated by their run-in slope and/or baseline plasma NfL level.
Baseline NfL levels were more strongly correlated with longitudinal change in the ALSFRS-R tool (Spearman correlation coefficient = −0.59; P = 0.0003) than was the prerandomization ALSFRS-R slope decline (Spearman correlation coefficient = 0.32; P = 0.0071) in the placebo participants in the VALOR trial. Accordingly, before analysis of data from the January 16, 2022, data cut-off of the VALOR trial and the 12-month OLE study data, the integrated statistical analysis plan was updated to include covariate adjustment for baseline levels of plasma NfL. Adjusting for baseline NfL as a continuous covariate more accurately accounts for baseline disease heterogeneity and thus permitted analyses in the larger ITT population (n = 108), increasing power compared to analyses in disease progression subgroups.
Ninety-five of 108 (88%) participants randomized in the VALOR trial went on to receive tofersen 100 mg in the OLE study. Participants who discontinued during the VALOR trial were not eligible to enter the OLE study. One participant from the tofersen group and 1 participant from the placebo group did not enrol in the OLE study. A total of 63 participants were included from the tofersen group (early-start tofersen), and 32 participants were included from the placebo group (delayed-started tofersen).
At the time of the January 16, 2022, interim data cut-off (52-week data), 18 (50%) participants in the delayed-start (placebo) group and 49 (68%) participants in the early-start group remained ongoing in the OLE study.
At the time of the February 28, 2023, interim data cut-off (104-week data), 16 (44.4%) participants in the delayed-start (placebo) group and 44 (61.1%) participants in the early-start group remained ongoing in the OLE study. The most common reasons for discontinuing the OLE study were death (n = 10 [14%] participants in the early-start tofersen group and n = 7 [19%] participants in the delayed-start tofersen group) and disease progression (n = 7 [10%] participants in the early-start tofersen group and n = 5 [14%] participants in the delayed-start tofersen group).
Table 11: Summary of Patient Disposition From the VALOR Study in the Systematic Review
Participant disposition | mITT population (N = 60) | Non-mITT population (N = 48) | ITT population (N = 108) | |||
|---|---|---|---|---|---|---|
Placebo (n = 21) | Tofersen (n = 39) | Placebo (n = 15) | Tofersen (n = 33) | Placebo (n = 36) | Tofersen (n = 72) | |
Screened, N | 162 | |||||
Reason for screening failure, n | ||||||
Inclusion criteria not met | ██ ██████ | |||||
Exclusion criteria met | ██ ██████ | |||||
Noncompliance | ██ ██████ | |||||
Other | ██ ██████ | |||||
Randomized, N | 21 | 39 | 15 | 33 | 36 | 72 |
Participants who received ≥ 1 dose, n (%) | 21 (100) | 39 (100) | 15 (100) | 33 (100) | 36 (100) | 72 (100) |
Participant who died | ██████ | ██████ | ██████ | ██████ | 0 | 1 (1.4) |
Participants who completed study treatment | ██████ | ██████ | ██████ | ██████ | 33 (91.7) | 63 (87.5) |
Participants who completed study | ██████ | ██████ | ██████ | ██████ | 33 (91.7) | 64 (88.9) |
Participants who completed study but missed 1 or more doses | ██████ | ██████ | ██████ | ██████ | ██████ | ██████ |
Participants who completed week 32 | ██████ | ██████ | ██████ | ██████ | ██████ | ██████ |
Discontinued from study treatment, n (%) | ██████ | ██████ | ██████ | ██████ | 3 (8.3) | 9 (12.5) |
Reason for discontinuation, n (%) | ||||||
Adverse events | ██████ | ██████ | ██████ | ██████ | 0 | 3 (4.2) |
Lost to follow-up | ██████ | ██████ | ██████ | ██████ | 0 | 0 |
Consent withdrawn | ██████ | ██████ | ██████ | ██████ | 1 (2.8) | 1 (1.4) |
Investigator decision | ██████ | ██████ | ██████ | ██████ | 0 | 0 |
Death | ██████ | ██████ | ██████ | ██████ | 0 | 1 (1.4) |
Disease progression | ██████ | ██████ | ██████ | ██████ | 2 (5.6) | 3 (4.2) |
Other | ██████ | ██████ | ██████ | ██████ | 0 | 1 (1.4) |
Discontinued from study, n (%) | ██████ | ██████ | ██████ | ██████ | 3 (8.3) | 8 (11.1) |
Reason for discontinuation, n (%) | ||||||
Adverse events | ██████ | ██████ | ██████ | ██████ | 0 | 2 (2.8) |
Lost to follow-up | ██████ | ██████ | ██████ | ██████ | 0 | 0 |
Consent withdrawn | ██████ | ██████ | ██████ | ██████ | 1 (2.8) | 2 (2.8) |
Investigator decision | ██████ | ██████ | ██████ | ██████ | 0 | 0 |
Death | ██████ | ██████ | ██████ | ██████ | 0 | 1 (1.4) |
Disease progression | ██████ | ██████ | ██████ | ██████ | 2 (5.6) | 3 (4.2) |
ITT population, N (%) | ██████ | ██████ | ██████ | ██████ | 36 (100.0) | 72 (100.0) |
mITT population, N (%) | ██████ | ██████ | ██████ | ██████ | 21 (58.3) | 39 (54.2) |
Non-mITT population, N (%) | ██████ | ██████ | ██████ | ██████ | 15 (41.7) | 33 (45.8) |
Per-protocol population, N (%) | ██████ | ██████ | ██████ | ██████ | 20 (55.6) | 34 (47.2) |
PK population, N (%) | 21 (100.0) | 39 (100.0) | 15 (100.0) | 33 (100.0) | 36 (100.0) | 72 (100.0) |
Safety population, N (%) | 21 (100.0) | 39 (100.0) | 15 (100.0) | 33 (100.0) | 36 (100.0) | 72 (100.0) |
ITT = intention to treat; mITT = modified intention to treat; PK = pharmacokinetic.
Sources: The VALOR trial Clinical Study Report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
Most participants in the VALOR trial were white (63.9%), followed by those who were Asian (8.3%). A large proportion of participants did not report their race and ethnicity (25.9%) due to confidentiality regulations. The percentage of males and females was similar across treatment groups and populations, with more males enrolled overall (males = 57.4%; females = 42.6%), which is reflective of the ALS population. The mean participant body mass index was 26.75 kg/m2. Participant ages ranged from 23 years to 78 years, with an overall older mean participant age in the placebo group (51.2 years) compared with the tofersen group (48.1 years); this difference was driven by a difference in the enriched subgroup, where the mean age was higher in the placebo group (54.0 years) compared with the tofersen group (47.3 years). Other demographic parameters were generally balanced between treatment groups and populations. Demographic characteristics in participants with baseline plasma NfL levels of less than the median or at the median or greater in the ITT population were also balanced between treatment groups.
Baseline and disease characteristics were balanced across treatment groups for the use of riluzole and/or edaravone and characteristics related to the stage of disease, including time from onset, the total ALSFRS-R score, and SVC. However, baseline plasma NfL levels were approximately 15% to 25% higher in the tofersen group compared to the placebo group. Consistently, the rate of decline on the ALSFRS-R tool from screening to day 15 (an approximate 42-day period) was higher in the tofersen group compared to the placebo group.
Medical history was consistent with that anticipated in this population. In general, similar percentages of participants between treatment groups reported medical history conditions.
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Table 12: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | mITT population (N = 60) | Non-mITT population (N = 48) | ITT population (N = 108) | |||
|---|---|---|---|---|---|---|
Placebo (n = 21) | Tofersen (n = 39) | Placebo (n = 15) | Tofersen (n = 33) | Placebo (n = 36) | Tofersen (n = 72) | |
Demographic | ||||||
Age categories, years, (%) | 21 (100) | 39 (100) | 15 (100) | 33 (100) | 36 (100) | 72 (100) |
18 to < 35 | █████ | █████ | █████ | █████ | 2 (5.6) | 10 (13.9) |
35 to < 50 | █████ | █████ | █████ | █████ | 15 (41.7) | 32 (44.4) |
50 to < 65 | █████ | █████ | █████ | █████ | 14 (38.9) | 21 (29.2) |
≥ 65 | █████ | █████ | █████ | █████ | 5 (13.9) | 9 (12.5) |
Age (years), mean (SD) | 54.0 (12.16) | 47.3 (14.30) | 47.3 (9.79) | 49.0 (10.49) | 51.2 (11.57) | 48.1 (12.64) |
Sex | 21 (100) | 39 (100) | 15 (100) | 33 (100) | 36 (100) | 72 (100) |
Female | 10 (47.6) | 17 (43.6) | 7 (46.7) | 12 (36.4) | 17 (47.2) | 29 (40.3) |
Male | 11 (52.4) | 22 (56.4) | 8 (53.3) | 21 (63.6) | 19 (52.8) | 43 (59.7) |
Ethnicity | █████ | █████ | █████ | █████ | 36 (100) | 72 (100) |
Hispanic or Latino | █████ | █████ | █████ | █████ | 1 (2.8) | 4 (5.6) |
Not Hispanic or Latino | █████ | █████ | █████ | █████ | 28 (77.8) | 47 (65.3) |
Not reported | █████ | █████ | █████ | █████ | 7 (19.4) | 21 (29.2) |
Race | █████ | █████ | █████ | █████ | 36 (100) | 72 (100) |
American Indian or Alaska Native | █████ | █████ | █████ | █████ | 0 | 0 |
Asian | █████ | █████ | █████ | █████ | 4 (11.1) | 5 (6.9) |
Black or African American | █████ | █████ | █████ | █████ | 0 | 1 (1.4) |
White | █████ | █████ | █████ | █████ | 25 (69.4) | 44 (61.1) |
Not reported | █████ | █████ | █████ | █████ | 7 (19.4) | 21 (29.2) |
Other | █████ | █████ | █████ | █████ | 0 | 1 (1.4) |
Height (cm), mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Weight (kg), mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
BMI (kg/m2), mean (SD) | 27.98 (6.187) | 26.65 (6.404) | 26.61 (7.035) | 26.15 (4.633) | 27.41 (6.491) | 26.42 (5.629) |
Disease characteristics | ||||||
Mutation type, n (%) | ||||||
p.Ile114Thr | █████ | █████ | █████ | █████ | █████ | █████ |
p.Ala5Val | █████ | █████ | █████ | █████ | █████ | █████ |
p.Gly94Cys | █████ | █████ | █████ | █████ | █████ | █████ |
p.His47Arg | █████ | █████ | █████ | █████ | █████ | █████ |
Site of onset, n (%) | ||||||
Bulbar | █████ | █████ | █████ | █████ | █████ | █████ |
Lower limbs | █████ | █████ | █████ | █████ | █████ | █████ |
Upper limbs | █████ | █████ | █████ | █████ | █████ | █████ |
Respiratory | █████ | █████ | █████ | █████ | █████ | █████ |
Multiple sites | █████ | █████ | █████ | █████ | █████ | █████ |
Time from symptom onset (months), median (range) | 8.3 (2.4 to 21.3) | 8.3 (1.7 to 18.5) | 39.6 (11.8 to 103.2) | 35.5 (3.9 to 145.7) | 14.6 (2.4 to 103.2) | 11.4 (1.7 to 145.7) |
ALSFRS-R prerandomization slope, median (range) | █████ (−4.91 to −0.42) | █████ (−8.30 to −0.39) | █████ (−0.84 to −0.02) | █████ (−0.77 to 0.00) | █████ (−4.91 to −0.02) | █████ (−8.30 to 0.00) |
ALSFRS-R baseline total score, mean (SD) | 35.4 (5.66) | 36.0 (6.40) | 39.9 (5.09) | 38.1 (5.13) | 37.3 (5.81) | 36.9 (5.91) |
ALSFRS-R run-in slope, screening to day 15, mean (SD) | −1.3 (3.91) | −1.8 (2.47) | 0.1 (1.87) | −0.1 (1.34) | −0.7 (3.25) | −1.0 (2.19) |
Percent-predicted SVC at baseline, mean (SD) | 83.7 (17.87) | 80.3 (14.22) | 87.1 (14.82) | 84.2 (19.02) | 85.13 (16.53) | 82.1 (16.59) |
Plasma NfL at baseline (pg/mL), mean (SD) | 127.3 (94.4) | 146.2 (82.6) | 37 (29.5) | 47.6 (41.8) | 89.7 (86.5) | 100.4 (82.8) |
Medical history | ||||||
History, n (%) | ||||||
Hypertension | █████ | █████ | █████ | █████ | █████ | █████ |
Anxiety | █████ | █████ | █████ | █████ | █████ | █████ |
Tonsillectomy | █████ | █████ | █████ | █████ | █████ | █████ |
Seasonal allergy | █████ | █████ | █████ | █████ | █████ | █████ |
Depression | █████ | █████ | █████ | █████ | █████ | █████ |
Drug hypersensitivity | █████ | █████ | █████ | █████ | █████ | █████ |
Muscle spasms | █████ | █████ | █████ | █████ | █████ | █████ |
Insomnia | █████ | █████ | █████ | █████ | █████ | █████ |
Appendectomy | █████ | █████ | █████ | █████ | █████ | █████ |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; BMI = body mass index; ITT = intention to treat; mITT = modified intention to treat; NfL = neurofilament light chain; NR = not reported; SD = standard deviation; SVC = slow vital capacity.
Sources: The VALOR trial Clinical Study Report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Over the course of the VALOR study ████% of participants received █ doses of study treatment. Participants randomized to tofersen each received a mean cumulative dose of tofersen of █████ mg. The mean (SD) continuous time on study was █████ (█████) days. There was no imbalance of exposure to study treatment between the mITT, non-mITT, and ITT populations. The number of missed and delayed doses was low across treatment groups and populations.
Table 13: Summary of Patient Exposure to Tofersen in VALOR Study
Exposure | mITT population | Non-mITT population | ITT population | |||
|---|---|---|---|---|---|---|
Placebo (n = 21) | Tofersen (n = 39) | Placebo (n = 15) | Tofersen (n = 33) | Placebo (n = 36) | Tofersen (n = 72) | |
Number of doses received per patient, n (%) | ||||||
1 | █████ | █████ | █████ | █████ | █████ | █████ |
2 | █████ | █████ | █████ | █████ | █████ | █████ |
3 | █████ | █████ | █████ | █████ | █████ | █████ |
4 | █████ | █████ | █████ | █████ | █████ | █████ |
5 | █████ | █████ | █████ | █████ | █████ | █████ |
6 | █████ | █████ | █████ | █████ | █████ | █████ |
7 | █████ | █████ | █████ | █████ | █████ | █████ |
8 | █████ | █████ | █████ | █████ | █████ | █████ |
Study drug compliance, n (%) | ||||||
Mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
< 80% | █████ | █████ | █████ | █████ | █████ | █████ |
80% to < 90% | █████ | █████ | █████ | █████ | █████ | █████ |
90% to 100% | █████ | █████ | █████ | █████ | █████ | █████ |
Cumulative tofersen dose (mg), mean | █████ | █████ | █████ | █████ | █████ | █████ |
Time on study (days), mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
Time to treatment discontinuation (days), mean (SD) | █████ | █████ | █████ | █████ | █████ | █████ |
ITT = intention to treat; mITT = modified intention to treat; SD = standard deviation.
Sources: The VALOR trial Clinical Study Report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 14: Tofersen Exposure in the OLE Study for Participants in the VALOR Trial
Exposure | Tofersen (N = 104) |
|---|---|
Number of 100 mg doses of tofersen received per participant, n (%) | |
1 to 5 | ██ ██████ |
6 to 10 | ██ ██████ |
11 to 15 | ██ ██████ |
16 to 20 | ██ ██████ |
21 to 25 | ██ ██████ |
26 to 30 | ██ ██████ |
31 to 35 | ██ ██████ |
36 to 40 | ██ ██████ |
41 to 45 | ██ ██████ |
46 to 50 | ██ ██████ |
51 to 55 | ██ ██████ |
Number of 100 mg doses of tofersen, mean (SD) | ██ ██████ |
Cumulative dose received (mg), mean (SD) | ██ ██████ |
Duration of exposure (weeks), mean (SD) | ██ ██████ |
Participants years (incidence rates per 100 participant-years) | ██ ██████ |
OLE = open-label extension; SD = standard deviation.
Sources: Common Technical Document - 2.7.4 Summary of Clinical Safety.122 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Concomitant Medications and Cointerventions
The use of concomitant medications was similar across treatment groups and was not expected to confound the interpretation of data.
The proportion of VALOR trial participants who received concomitant nondrug therapies was █████ ███████ in the tofersen group and █████ ███████ in the placebo group.
Overall, 61.8% of participants used riluzole and 8.3% of participants used edaravone before and/or upon entry into the study, and ████% and ███% continued usage during the study period, respectively. The use of riluzole and edaravone was similar across treatment groups and populations.
███ participant in the tofersen arm of the mITT population received concomitant riluzole postbaseline that was considered as disallowed because the participant was not on a stable dose for at least 30 days before day 1. No changes to riluzole or edaravone use or dosage were reported by any participants during the study. No protocol deviations for changes of riluzole or edaravone use were reported during the study.
A similar proportion of participants in the tofersen group (███%; ████ of 72) and placebo group (███%; ████ of 36) took disallowed concomitant medications. Disallowed concomitant medication use included medications that were not stabilized for the length of time outlined in the protocol before day 1 as part of the protocol inclusion and exclusion criteria and were all reported in the enriched subgroup only.
Table 15: Concomitant Riluzole and Edaravone Use in VALOR Study
Participant disposition | mITT population (N = 60) | Non-mITT population (N = 48) | ITT population (N = 108) | |||
|---|---|---|---|---|---|---|
Placebo (n = 21) | Tofersen (n = 39) | Placebo (n = 15) | Tofersen (n = 33) | Placebo (n = 36) | Tofersen (n = 72) | |
Prior riluzole use, n (%) | 13 (61.9) | 25 (64.1) | 9 (60.0) | 20 (60.6) | 22 (61.1) | 45 (62.5) |
Current riluzole use, n (%) | 13 (61.9) | 25 (64.1) | 9 (60.0) | 20 (60.6) | 22 (61.1) | 45 (62.5) |
Prior edaravone use, n (%) | 1 (4.8) | 2 (5.1) | 2 (13.3) | 4 (12.1) | 3 (8.3) | 6 (8.3) |
Current edaravone use, n (%) | 1 (4.8) | 1 (2.6) | 2 (13.3) | 4 (12.1) | 3 (8.3) | 5 (6.9) |
Current riluzole and edaravone use, n (%) | 1 (4.8) | 1 (2.6) | 2 (13.3) | 4 (12.1) | 3 (8.3) | 5 (6.9) |
ITT = intention to treat; mITT = modified intention to treat.
Sources: Appendix 16.2.4 and Appendix 16.2.5 of the VALOR trial Clinical Study Report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Amyotrophic Lateral Sclerosis Functional Rating Scale-–Revised
Primary Analysis
In the mITT population, a non–statistically significant difference of 1.2 favouring tofersen was observed in ALSFRS-R change from baseline at week 28 (a −6.98-point decline in the tofersen group and a −8.14-point decline in the placebo group; JRT plus MI P = 0.9689). The results from the ANCOVA plus MI sensitivity analysis support the results from the JRT plus MI analysis (P = 0.5998).
Week 52
When adjusting for NfL levels at baseline in the ITT population, nominally statistically significant differences were observed between delayed-start (placebo) and early-start participants. Placebo (delayed-start) participants experienced a greater decline of a 3.5-point difference (95% CI, 0.4 points to 6.7 points; P = 0.0272) on the ALSFRS-R tool than did the early-start participants from the VALOR trial’s baseline to week 52.
Additional sensitivity analyses support the results in the ITT population at week 52.
Week 104
This slower decline in early-start participants persisted at week 104. Compared to delayed-start (placebo) participants, the early-start participants demonstrated an adjusted mean difference of 3.7 (95% CI, −0.7 to 8.2; ANCOVA + MI P = 0.1004). Analysis results are consistent across different populations and disease progression subgroups (faster and slower progressors and above or below the median NfL level).
Table 16: Change in ALSFRS-R Total Score From VALOR and OLE Studies (ITT Population)
Variable | VALOR study | OLE study | ||
|---|---|---|---|---|
Placebo (n = 36) | Tofersen (n = 72) | Delayed-start (placebo) tofersen (n = 36) | Early-start tofersen (n = 72) | |
Change from baseline on ALSFRS-R total score to week 28 | ||||
Adjusted means | −6.2 | −4.1 | NA | NA |
Adjusted mean difference (95% CI) | 2.1 (−0.33 to 4.54) | NA | ||
P value (ANCOVA + MI) | 0.0904 | NA | ||
Change from baseline on ALSFRS-R total score to week 52 | ||||
Adjusted means | NA | NA | −9.5 | −6.0 |
Adjusted mean difference (95% CI) | NA | 3.5 (0.4 to 6.7) | ||
P value (ANCOVA + MI) | NA | 0.0272 | ||
Change from baseline on ALSFRS-R total score to week 104 | ||||
Adjusted means (original MI approach) | NA | −13.2 | −9.5 | |
Adjusted mean difference (95% CI) | NA | 3.7 (−0.7 to 8.2) | ||
P value (ANCOVA + MI) | NA | 0.1004 | ||
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ANCOVA = analysis of covariance; CI = confidence interval; ITT = intention to treat; MI = multiple imputation; NA = not applicable; OLE = open-label extension.
Sources: Common Technical Document - 2.5 Clinical Overview.120,121,123 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
SOD1 Protein Concentrations in the CSF
Tofersen treatment resulted in reductions of total CSF SOD1 protein that were apparent 8 weeks after treatment initiation and sustained over time. In the mITT population, a reduction in total CSF SOD1 protein was observed at week 28 in the tofersen group compared to the placebo group (with a difference in GMR for tofersen to placebo of 38% and nominal P < 0.0001), with a mean change of ██████ ng/mL.
In the non-mITT population, reductions were also observed in total CSF SOD1 protein at week 28 in the tofersen group compared to the placebo group (with a difference in GMR for tofersen to placebo of 26% and nominal P = 0.0007), with a mean change of ██████ ng/mL.
Figure 1: Tofersen Effect on ALSFRS-R Total Score at Week 104 (VALOR and OLE Studies, ITT Population)
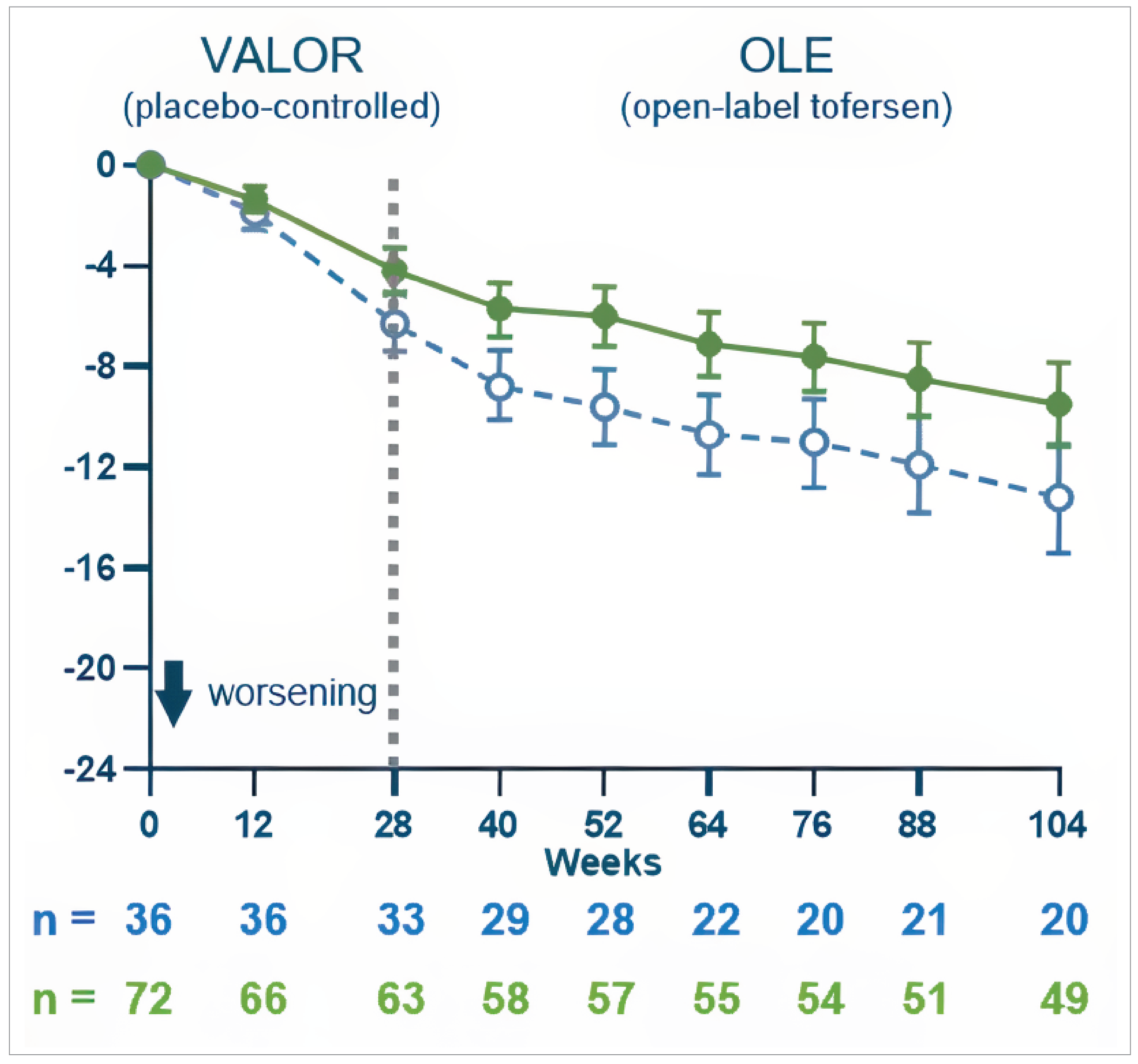
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ANCOVA = analysis of covariance; ITT = intention to treat; LSM = least squares mean; NfL = neurofilament light chain; OLE = open-label extension.
Notes: Baseline was defined as the day 1 value before starting the study drug and presented as day 1. If day 1 value was missing, the nonmissing value (including the screening visit) closest to and before the first dose was used as the baseline value.
Multiple imputation that included treatment group, the use of riluzole or edaravone, baseline plasma NfL, and the relevant baseline and postbaseline values for the end point was used for missing data.
For participants who were not Japanese, the Global ALSFRS-R was used. For Japanese participants, the Japanese (Ohashi et al.) ALSFRS-R was used except for Q5a and Q11, where the Japanese-translated Global ALSFRS-R was used. A positive change indicated an improvement.
LSM were obtained from the ANCOVA model with treatment included as a fixed effect and adjusted for the following covariates: baseline plasma NfL level, baseline ALSFRS-R total score, and the use of riluzole or edaravone.
Sources: Common Technical Document - 2.7.3 Summary of Clinical Efficacy.120,121,124 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Week 52 and Week 104
In participants who had previously received tofersen in the VALOR trial (the early-start tofersen group), 52 weeks of total tofersen treatment during the VALOR and OLE studies maintained the previously lowered total CSF SOD1 protein levels, with a ██% decrease from the OLE study baseline levels at week 24 (GMR to baseline). In participants who had received placebo in the VALOR trial (the delayed-start tofersen group), 24 weeks of newly initiated treatment with open-label tofersen reduced total CSF SOD1 protein levels by ██% (GMR to baseline). At week 52, the percentage reduction (GMR) from baseline was 21% (95% CI, 4% to 35%) and 33% (95% CI, 21% to 42%) for the delayed-start and early-start tofersen groups, respectively. Similar reductions were seen at week 104, with reductions of ██% (95% CI, █████████) in the delayed-start group and ██% (95% CI, █████████) in the early-start group. Variability in the percentage reduction is likely influenced by assay variability, which can vary up to 15.6%. Analyses by subgroups and the overall observed data analyses support these results.
Figure 2: Tofersen Effect on SOD1 Concentration in CSF at 104 Weeks (VALOR and OLE Studies, ITT Population)
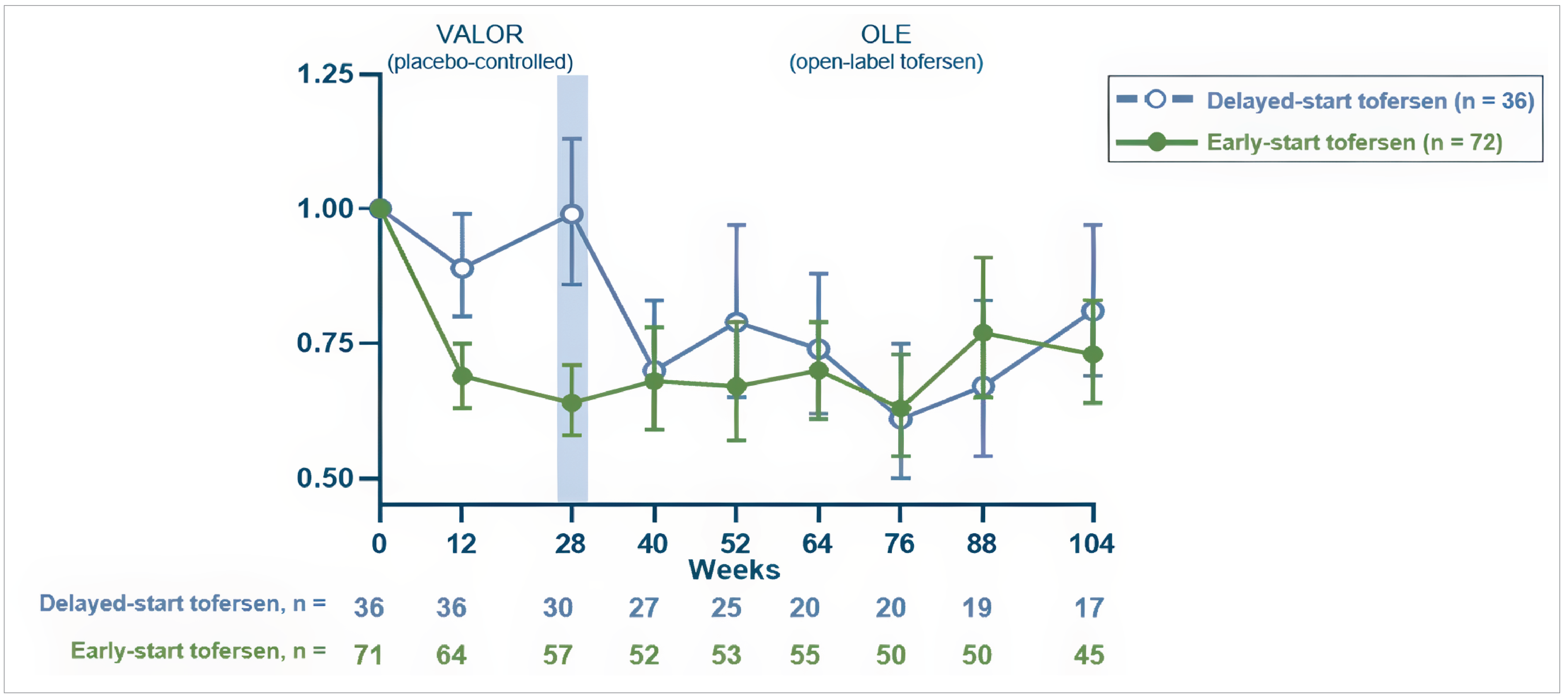
ANCOVA = analysis of covariance; CSF = cerebrospinal fluid; ITT = intention to treat; LLOQ = lower limit of quantitation; OLE = open-label extension.
Notes: Baseline was defined as day 1 value before starting the study drug. If the day 1 value was missing, the nonmissing value (including screening visit) closest to and before the first dose was used as the baseline value.
The LLOQ was 15.6 ng/mL. Values below the limit of quantitation were set to half of LLOQ in calculations.
Multiple imputation including treatment group, the use of riluzole or edaravone, and the relevant baseline and postbaseline values for the end point was used for missing data.
The analysis was based on the ANCOVA model with natural log transformed data. The model included covariates for the corresponding baseline value (i.e., log value, and the use of riluzole or edaravone).
Sources: Common Technical Document - 2.7.2 Summary of Clinical Pharmacology Studies.120,121,125 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Plasma NfL
Plasma NfL levels declined through approximately week 16, and the reductions were sustained over time, suggesting that tofersen administration reduced axonal injury and neurodegeneration. In the mITT population, a reduction in plasma NfL levels from baseline to week 28 of 67% (a difference in GMR for tofersen to placebo of 0.33; nominal P < 0.0001) was observed. Despite lower levels at baseline in the non‑mITT population, reductions in plasma NfL levels from baseline to week 28 of 48% (a difference in GMR for tofersen to placebo of 0.52; nominal P < 0.0001) were observed. In the ITT population, a reduction in plasma NfL levels was also observed at week 28 in the tofersen group compared to placebo (a difference in GMRs for tofersen to placebo of 0.40; post hoc nominal P < 0.0001).
Week 52 and Week 104
At week 52, the percentage reduction (GMR) from baseline was 41% (95% CI, 26% to 54%) and 51% (95% CI, 42% to 60%) for the delayed-start and early-start tofersen groups, respectively. These reductions in plasma NfL persisted and continued to decline at week 104, with levels of 60% and 66% for the delayed-start and early-start tofersen groups, respectively. Consistent reductions in CSF NfL were observed out to 104 weeks, indicating that tofersen robustly reduces axonal injury and neurodegeneration, with no evidence of attenuation of effect over time.
Figure 3: Tofersen Effect on NfL in Plasma at Week 104 (VALOR and OLE Studies, ITT Population)
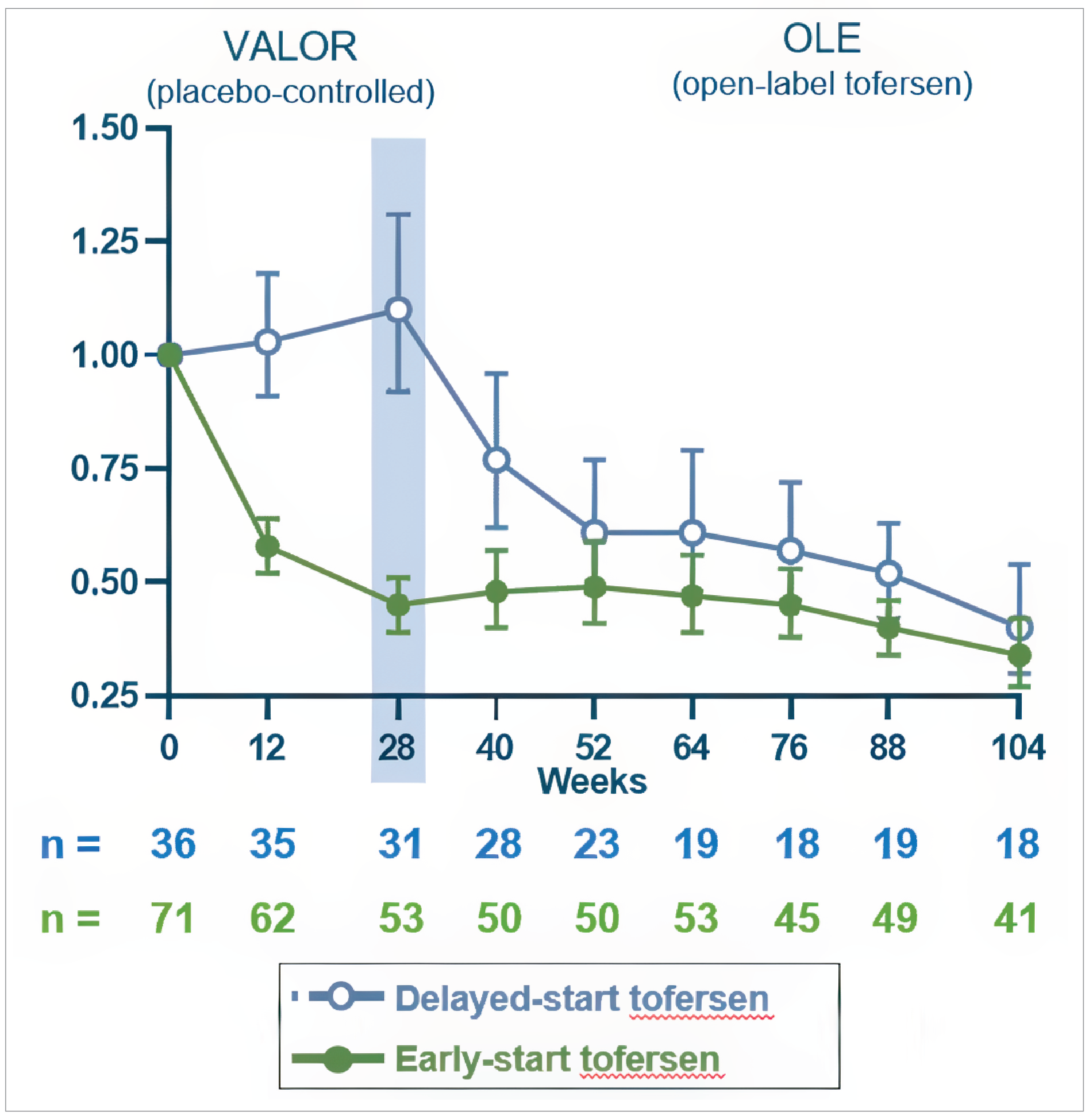
ANCOVA = analysis of covariance; ITT = intention to treat; LLOQ = lower limit of quantitation; MI = multiple imputation; NfL = neurofilament light chain; OLE = open-label extension.
Notes: Baseline was defined as the day 1 value before starting the study drug. If the day 1 value was missing, the nonmissing value (including screening visit) closest to and before the first dose was used as the baseline value.
The LLOQ was 4.9 pg/mL. Values below the limit of quantitation were set to half of LLOQ in calculations.
MI that included treatment group, the use of riluzole or edaravone, and the relevant baseline and postbaseline values for the end point was used for missing data. An extreme value of > 477 pg/mL was set to missing and was imputed with MI in the ANCOVA analysis.
The analysis was based on the ANCOVA model with natural log transformed data. The model included covariates for the corresponding baseline value (i.e., log value, and the use of riluzole or edaravone).
Sources: Common Technical Document - 2.7.2 Summary of Clinical Pharmacology Studies.120,121,125 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Respiratory Function: SVC
Change from baseline to week 28 in the percent-predicted SVC in the mITT population showed a numerically lesser decline in the tofersen group compared to the placebo group, with a 7.9% predicted treatment difference (95% CI, −3.5% to 19.3%; ANCOVA + MI: nominal P = 0.1755 ). The sensitivity and secondary analyses also support these results.
Some differences were observed between subgroups in the analyses based on sex, disease duration since symptom onset, site of onset, geographic region, and riluzole or edaravone use, although the majority showed an effect in favour of tofersen.
On the inclusion of baseline plasma NfL as a covariate in the imputation model for the ITT population and the replacement of both disease duration since symptom onset and baseline ALSFRS-R total score with baseline plasma NfL in the ANCOVA model, the treatment difference was 8.5% predicted at week 28 (95% CI, 1.81% to 15.15%; ANCOVA + MI: nominal P = 0.0128).
Week 52 and Week 104
During the OLE study, in the ITT population, participants in the placebo (delayed-start) arm experienced a nominally statistically significant greater decline from baseline to week 52 on percent-predicted SVC than the participants in the early-start arm (9.2 percent-predicted treatment difference; 95% CI, 1.7 to 16.6; P = 0.0159). Notably in the placebo (delayed-start) group, stabilization in SVC appeared after week 40, approximately 12 weeks after the initiation of tofersen and the time point at which maximum biological activity would be expected. The trends are consistently in favour of tofersen across different populations and disease progression subgroups.
Many participants in the placebo group who initiated tofersen in the OLE study fell below the initial VALOR trial’s SVC inclusion criteria eligibility thresholds during the 6 months of the VALOR trial. Of the 36 participants who were randomized to the placebo group in the VALOR trial, 32 enrolled in the OLE study, and ███ of these had missing baseline assessments for SVC. ██████ of the 30 (██%) participants in the OLE study with nonmissing baseline assessments had an SVC of less than 65 percent predicted at baseline of the OLE study; 7 (████%) participants had SVC of less than 50 percent predicted.
Week 104
The effects on respiratory strength seen at earlier time points were sustained at 104 weeks, with a 9.7 percent-predicted difference in favour of the early-start tofersen group.
Andrews et al. found that a slowing in the rate of SVC decline by 1.5 percent-predicted per month reduced the risk in first onset of respiratory insufficiency or death, first occurrence of tracheostomy or death, and death at any time after 6 months by 22%, 23%, and 23%, respectively (P < 0.001).35 As such, the 8.5, 9.2, and 9.7 percent-predicted differences observed between treatment groups at 28 weeks, 52 weeks, and 104 weeks, respectively, are highly clinically relevant in this population.
Results were consistent across different subgroups and covariates. In the early-start group, ████% of participants experienced improvement in percent-predicted SVC at 104 weeks and ███% of delayed-start participants experienced an improvement.
Figure 4: Tofersen Effect on Respiratory Function to Week 104 (VALOR and OLE Studies, ITT Population) [Redacted]

Time to Death or PV
In the VALOR trial, in the primary analysis of the mITT population, few deaths (n = 1 due to congestive cardiac failure in the tofersen group; n = 0 in the placebo group) or PV events (n = [███%] in the tofersen group; n = [███%] in the placebo group) occurred during the study. The median time to death or PV could not be estimated due to the small number of events observed. The estimated proportion of participants with an event of death or PV by week 28 was █████ in the tofersen group and █████ in the placebo group.
No events of death or PV occurred in the non-mITT population.
As of January 16, 2022, all participants enrolled in the VALOR trial had the opportunity for at least 1 year of follow-up. There was an apparent reduction in the risk of death or PV (HR = 0.36; 95% CI, 0.137 to 0.941) in participants who initiated tofersen early compared to those with the delayed-start group.
As of February 28, 2023, all participants enrolled in the VALOR trial had had the opportunity for at least 2 years of follow-up (median opportunity for follow-up = 3.4 years; range, 2.2 to 3.9 years). Despite the duration of follow-up in the OLE study, there was a limited number of death-equivalent events and therefore, the median time to death or PV could not be estimated. The reduction in the risk of death or PV was 53% in the early-start group compared with the delayed-start group in the ITT population (HR = 0.47; 95% CI, 0.20 to 1.11).
These data provide early evidence of a prolongation of event-free survival with earlier tofersen initiation in the overall population. In the faster-progressing subgroup, the median time to death or PV was reached in both treatment groups, enabling the estimation of the extension of event-free survival associated with early-start tofersen (approximately ███ years). Consistently, the median time to death, PV, or withdrawal due to disease progression was approximately ███ years longer in the early-start, faster-progressing tofersen group than in the delayed-start group.
RPSFTM analysis indicate that had the delayed-start group remained on placebo, the risk of death or PV would have been reduced by ██% in the early-start group compared with the delayed-start group (HR ████; 95% CI, (█████-█████).
Amyotrophic Lateral Sclerosis Assessment Questionnaire–5 Items
The ALSAQ-5 is disease-specific, patient self-reported, health status questionnaire. The ALSAQ-5 contains 5 questions, each corresponding to 1 of the following 5 health-related dimensions: physical mobility, activities of daily living, eating and drinking abilities, communication, and emotional functioning. The total score ranges from 0 to 100, with a lower score representing better health-related status.
During the VALOR study, a numerical difference between treatment groups in the ALSAQ-5 scores favoured tofersen over placebo. Analysis of the ALSAQ-5 total scores indicated less worsening from baseline to week 28 in the tofersen group (least squares mean change from baseline in the mITT population = 9.98; in the non-mITT population = 1.32) compared to the placebo group (Least squares mean change from baseline in the mITT population: 15.57; in the non-mITT population: 2.95).
In the ITT population with NfL as a covariate, a ████-point treatment difference favouring tofersen was observed in the ALSAQ-5 total score change from baseline at week 28 (95% CI, ██████, ████; nominal P = 0.1848).
In the OLE study, the baseline plasma NfL was included in the analysis model when analyzing the change from baseline in the ITT population for all quality-of life measures. This trend of slowing decline in the ALSAQ-5 score was continued in the OLE study for the early-start tofersen group compared with the delayed-start tofersen group. At week 52 in the ITT population, there was a −10.3-point change (95% CI; −17.33 points to −3.20 points) from baseline for the early-start tofersen group compared with the delayed-start tofersen group (nominal P = 0.0044). At week 104, there was a ████-point (95% CI, ██████, ████) change from baseline for the early-start tofersen group compared with the delayed-start tofersen group (nominal P = ██████).
Fatigue Severity Scale
The FSS is a self-reported questionnaire designed to assess fatigue. It consists of 9 questions using a 7-point Likert scale ranging from strongly disagree to strongly agree. The scores from each question were totalled, with lower scores indicating less fatigue in everyday life.
During the VALOR study, a trend favouring tofersen was observed in FSS scores for the mITT population, suggesting less fatigue. In the ITT population analysis where baseline NfL was adjusted, a −2.4-point treatment difference favouring tofersen was observed in the FSS total score change from baseline at week 28 (95% CI, −7.45 to 2.55 points; nominal P = 0.3365).
A numerical, non–statistically significant trend of less fatigue was observed in the early-start tofersen group in the OLE. At week 52 in the ITT population, there was a −3.8-point difference (95% CI, −9.03 to 1.38 points) in change from baseline on the FSS with the early-start group compared with the delayed-start group (nominal P = 0.1493). Consistent with some of the earlier analysis time points, results on the FSS were the 1 instance in which effects favoured the delayed-start group. At week ███, there was a ████-point change (95% CI, ██████ ████) from baseline for the early-start tofersen group compared with the delayed-start tofersen group (nominal P = ██████). Additionally, at week ███ in the ITT population, ████% of early-start participants and ████% of delayed-start participants achieved stabilization or improvement in FSS.
Table 17: Summary of Key Efficacy Results From Studies Included in the Systematic Review
Variable | VALOR study | |
|---|---|---|
Placebo (n = 36) | Tofersen (n = 72) | |
Change from baseline to week 28 on ALSFRS-R total score | ||
Faster-progressing subgroup – mITT population | ||
N | 21 | 39 |
Adjusted mean | −8.14 | −6.98 |
Adjusted mean difference (95% CI) | 1.2 (−3.2 to 5.5) | |
P value (joint rank + MI) | 0.9689 | |
Nominal P value (ANCOVA + MI) | 0.5998 | |
Slower-progressing subgroup – non-mITT population | ||
N | 15 | 33 |
Adjusted mean | −2.73 | −1.33 |
Adjusted mean difference (95% CI) | 1.4 (−1.1 to 3.9) | |
Nominal P value (ANCOVA + MI) | 0.2726 | |
Post hoc analyses with baseline plasma NfL as a covariate – ITT population | ||
N | 36 | 72 |
Adjusted mean | −5.8 | −4.5 |
Adjusted mean difference (95% CI) | 1.4 (−1.34 to 4.09) | |
Nominal P value (ANCOVA + MI) | 0.3218 | |
GMR to baseline to week 28 on total CSF SOD1 protein | ||
Faster-progressing subgroup – mITT population | ||
N | 21 | 39 |
Adjusted GMR to baseline | 1.16 | 0.71 |
Difference in GMR (95% CI) | 0.62 (0.49 to 0.78) | |
Nominal P value (ANCOVA + MI) | < 0.0001 | |
Slower-progressing subgroup — non-mITT population | ||
N | 15 | 33 |
Adjusted GMR to baseline | 0.81 | 0.60 |
Difference in GMR (95% CI) | 0.74 (0.63 to 0.88) | |
P value (ANCOVA + MI) | 0.0007 | |
GMR to baseline to week 28 on plasma NfL | ||
Faster-progressing subgroup — mITT population | ||
N | 21 | 39 |
Adjusted GMR to baseline | 1.20 | 0.40 |
Difference in GMR (95% CI) | 0.33 (0.25 to 0.45) | |
Nominal P value (ANCOVA + MI) | < 0.0001 | |
Slower-progressing subgroup — non-mITT population | ||
N | 15 | 33 |
Adjusted GMR to baseline | 0.95 | 0.50 |
Difference in GMR (95% CI) | 0.52 (0.43 to 0.63) | |
P value (ANCOVA + MI) | ███████ | |
Change from baseline to week 28 on percent-predicted SVC | ||
Faster-progressing subgroup — mITT population | ||
N | 21 | 39 |
Adjusted mean | −22.20 | −14.31 |
Adjusted mean difference (95% CI) | 7.9 (−3.5 to 19.3) | |
Nominal P value (joint rank + MI) | 0.3233 | |
Nominal P value (ANCOVA + MI) | 0.1755 | |
Slower-progressing subgroup — non-mITT population | ||
N | 15 | 33 |
Adjusted mean | −4.90 | −0.26 |
Adjusted mean difference (95% CI) | 4.6 (−1.2 to 10.5) | |
Nominal P value (ANCOVA + MI) | 0.1210 | |
Change from baseline to week 28 on HHD megascore | ||
Faster-progressing subgroup — mITT population | ||
N | 21 | 39 |
Adjusted mean | −0.37 | −0.34 |
Adjusted mean difference (95% CI) | 0.02 (−0.21 to 0.26) | |
Nominal P value (ANCOVA + MI) | 0.8390 | |
Slower-progressing subgroup — non-mITT population | ||
N | 15 | 33 |
Adjusted mean | −0.18 | −0.09 |
Adjusted mean difference (95% CI) | 0.09 (−0.08 to 0.26) | |
Nominal P value (ANCOVA + MI) | 0.2832 | |
ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ANCOVA = analysis of covariance; CI = confidence interval; CSF = cerebrospinal fluid; GMR = geometric mean ratio; HHD = hand-held dynamometry; ITT = intention to treat; MI = multiple imputation; mITT = modified intention to treat; NfL = neurofilament light chain; SVC = slow vital capacity.
Source: The VALOR trial Clinical Study Report.76 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
The safety population was used for the analyses of the safety data in the VALOR and OLE studies. AEs and SAEs were measured and recorded throughout the study period. A treatment-emergent AE or SAE was defined as any AE or SAE with an onset date and time that was on or after the first dose of study drug or any pre-existing condition that had worsened in severity after the first dose of study drug. All AEs were assessed to determine if the event met the criteria for an SAE, the relationship to the study drug, and the severity of the event. Severity was assessed using the Common Terminology Criteria for Adverse Events (CTCAE) grading system where grade 1 is mild, grade 2 is moderate, grade 3 is severe or medically significant, grade 4 is life-threatening, and grade 5 is death related to an AE.
The safety discussion focuses on key elements of the safety experience with tofersen in the VALOR and OLE studies as of the February 28, 2023, data cut-off. An overview of the key safety data is presented in Table 18. AEs were experienced by the majority of participants with most AEs being mild or moderate in severity and not resulting in discontinuation of treatment. The incidence of AEs between tofersen and placebo was similar in the VALOR study and safety signals remained consistent in the OLE study. The most common AEs were procedural pain, headache, pain in an extremity, a fall, back pain, and post–lumbar puncture syndrome. The incidence of SAEs was higher in the tofersen group compared with the placebo group during the VALOR trial. The most frequently reported SAEs were dyspnea, pulmonary embolism, aspiration pneumonia, dehydration, and atelectasis; they represent the morbidity of ALS. Select neurologic SAEs were also noted, including papilledema, neuralgia, myelitis, radiculitis, and aseptic or chemical meningitis; however, they occurred in fewer than 5% of participants each. AEs related to lumbar puncture were experienced by the majority of participants.
Adverse Events
During the VALOR study, the incidence of AEs was similar between the tofersen group (69 of 72; 95.8%) and the placebo group (34 of 36; 94.4%) in the safety population. The most frequently reported (≥ 50%) AEs by system organ class (SOC) were injury, poisoning, and procedural complications; nervous system disorders; and musculoskeletal and connective tissue disorders. Within those SOCs, the most frequently reported (≥ 20% in either treatment group) AEs by preferred term (PT) were procedural pain, headache, pain in extremity, a fall, back pain, and post–lumbar puncture syndrome.
A few differences in the rates of AEs between the tofersen and placebo groups were observed. At the SOC level, there was a higher rate (≥ 10%) of investigations in the tofersen group compared to the placebo group, whereas injury, poisoning, and procedural complications; gastrointestinal disorders; respiratory, thoracic, and mediastinal disorders; and infections and infestations were all at least 10% higher in the placebo group compared to the tofersen group. By PT level, the tofersen group had a higher incidence (≥ 10%) of pain in extremity, back pain, and fatigue compared with the placebo group. The placebo group had a higher incidence (≥ 10%) of a fall, post–lumbar puncture syndrome, nasopharyngitis, and diarrhea compared with the tofersen group.
The majority of these AEs were mild to moderate in severity (CTCAE grade 1 or grade 2). There was a higher incidence of AEs that were CTCAE grade 3 or higher in the tofersen group (12 of 72; 16.7%) compared with the placebo group (4 of 36; 11.1%).
There was 1 of 72 (1.4%) participants in the tofersen group who experienced a CTCAE grade 4 AE (aspiration) and 1 of 72 (1.4%) participants in the tofersen group who experienced a CTCAE grade 5 AE (congestive cardiac failure).
In the OLE study, based on the data cut-off on February 28, 2023, 103 of 104 (99.0%) participants experienced AEs. Many of the commonly reported AEs were consistent with events occurring in the natural history of ALS, common events in the general population, or events secondary to the lumbar puncture procedure. Most AEs were mild to moderate in severity. The most common AEs (i.e., reported in > 15% of participants) were ████ ██ ██████████ ██████████ █████████ ██████████ █████ █████ ████ █████ ████ ██ ██████████ ███████████ █████████ ████████ ███ ███████ ██████████ ███████ ████ ██████ █████.
Serious Adverse Events
During the VALOR study, in the safety population, the incidence of SAEs was higher in the tofersen group (13 of 72; 18.1%) compared with the placebo group (5 of 36; 13.9%). The most frequently reported SAEs by SOC (≥ 4%) were respiratory, thoracic, and mediastinal disorders; general disorders and administration site conditions; and nervous system disorders. The most frequently reported SAEs by PT (≥ 2%) were dyspnea, pulmonary embolism, aspiration pneumonia, dehydration, and atelectasis.
During the VALOR and OLE studies, a total of 48 of 104 (46.2%) participants reported at least 1 SAE. The most frequently reported SAEs by SOC (≥ 7%) were respiratory, thoracic, and mediastinal disorders; infections and infestations; gastrointestinal disorders; and nervous system disorders.
Respiratory complications and pulmonary embolism are common causes of morbidity in the ALS population.
Withdrawals Due to Adverse Events
During the VALOR trial, in the safety population, the incidence of AEs that led to discontinuation of the study treatment were only reported in participants in the tofersen group (4 of 72; 5.6%) and included the following AEs by PT: congestive cardiac failure, myelitis, chemical meningitis, and pulmonary embolism (each 1 of 72 [1.4%]). The participant with pulmonary embolism discontinued study treatment from week 12 to week 24 but continued in the study; all other participants who discontinued the study treatment withdrew from the study.
AEs that led to withdrawal from the study occurred in the tofersen group only (3 of 72; 4.2%). AEs that led to withdrawal from the study included congestive cardiac failure, myelitis, and chemical meningitis (each 1 of 72 [1.4%]).
During the VALOR and OLE studies, 23 of 104 (22.1%) patients experienced an AE that led to drug discontinuation and 22 of 104 (21.2%) patients experienced an AE that led to study withdrawal.
Mortality
One participant in the tofersen group (1 of 72; 1.4%) died during the VALOR trial. This participant was in the enriched subgroup (1 of 39; 2.6%) and experienced a CTCAE grade 5 SAE of congestive cardiac failure resulting in death on study day 114; it was not considered treatment-related.
During the VALOR and OLE studies, a total of 18 of 104 (17.3%) patients died. The reasons for death by SOC included respiratory, thoracic, and mediastinal disorders, cardiac disorders, infections and infestations, nervous system disorders, and general disorders and administration site conditions that align with the common causes of mortality in the ALS population.
Notable Harms
There were no AEs of special interest specified for the VALOR trial; however, AEs of note were reported. The PTs for AEs of note (serious neurologic events, falls, and CSF abnormalities) were identified by medical review before unblinding the study. Overall, in the safety population, the incidence of AEs of note was lower in the tofersen group (██ of 72 participants [████%]) compared with the placebo group (██ of 36 participants [████%]).
Serious neurologic events were reported in 4 of 72 (5.6%) patients. Three events occurred in the enriched subgroup and 1 event occurred in the “other” subgroup. These events included lumbar radiculopathy, chemical meningitis, myelitis, and transverse myelitis.
In the safety population, the number of participants with an AE was lower in the tofersen group (17 of 72; 23.6%) compared with the placebo group (15 of 36; 41.7%).
The number of participants with AEs of CSF abnormalities was higher in the tofersen group (12 of 72; 16.7%) compared with the placebo group (1 of 36; 2.8%). In the tofersen group, AEs of CSF abnormalities by PT included an increased CSF white blood cell count, increased CSF protein, pleocytosis, an increased CSF cell count, and an abnormal CSF test. In the placebo group, AEs of CSF abnormalities by PT included increased CSF protein.
There were no SAEs of note in the falls or CSF abnormalities categories.
Table 18: Summary of Harms Results From Studies Included in the Systematic Review
Adverse event | VALOR study | OLE study | |
|---|---|---|---|
Placebo (n = 36) | Tofersen (n = 72) | Tofersen 100 mg (n = 104) | |
Most common adverse events, n (%) | |||
≥ 1 adverse event | 34 (94.4) | 69 (95.8) | ██ ██████ |
Injury, poisoning, and procedural complications | ██ ██████ | ██ ██████ | ██ ██████ |
Procedural pain | 21 (58.3) | 41 (56.9) | ██ ██████ |
Fall | 15 (41.7) | 17 (23.6) | ██ ██████ |
Post–lumbar puncture syndrome | 11 (30.6) | 13 (18.1) | ██ ██████ |
Ligament sprain | 2 (5.6) | 4 (5.6) | ██ ██████ |
Nervous system disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Headache | 16 (44.4) | 33 (45.8) | ██ ██████ |
Paresthesia | 6 (16.7) | 6 (8.3) | ██ ██████ |
Dizziness | 3 (8.3) | 4 (5.6) | ██ ██████ |
Muscle contractions, involuntary | 1 (2.8) | 4 (5.6) | ██ ██████ |
Neuralgia | 0 | 4 (5.6) | ██ ██████ |
Musculoskeletal and connective tissue disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Pain in extremity | 6 (16.7) | 19 (26.4) | ██ ██████ |
Back pain | 2 (5.6) | 15 (20.8) | ██ ██████ |
Arthralgia | 2 (5.6) | 10 (13.9) | ██ ██████ |
Myalgia | 2 (5.6) | 10 (13.9) | ██ ██████ |
Muscle spasms | 2 (5.6) | 5 (6.9) | ██ ██████ |
Muscular weakness | 4 (11.1) | 4 (5.6) | ██ ██████ |
Musculoskeletal pain | 2 (5.6) | 4 (5.6) | ██ ██████ |
Musculoskeletal stiffness | 0 | 4 (5.6) | ██ ██████ |
Neck pain | 4 (11.1) | 4 (5.6) | ██ ██████ |
General disorders and administration site conditions | ██ ██████ | ██ ██████ | ██ ██████ |
Fatigue | 2 (5.6) | 12 (16.7) | ██ ██████ |
Pain | 0 | 7 (9.7) | ██ ██████ |
Pyrexia | 1 (2.8) | 3 (4.2) | ██ ██████ |
Gastrointestinal disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Nausea | 6 (16.7) | 9 (12.5) | ██ ██████ |
Constipation | 4 (11.1) | 6 (8.3) | ██ ██████ |
Salivary hypersecretion | 1 (2.8) | 4 (5.6) | ██ ██████ |
Abdominal distension | 2 (5.6) | 2 (2.8) | ██ ██████ |
Diarrhea | 5 (13.9) | 1 (1.4) | ██ ██████ |
Respiratory, thoracic, and mediastinal disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Cough | 1 (2.8) | 5 (6.9) | ██ ██████ |
Dyspnea | 5 (13.9) | 4 (5.6) | ██ ██████ |
Infections and infestations | ██ ██████ | ██ ██████ | ██ ██████ |
Upper respiratory tract infection | 2 (5.6) | 5 (6.9) | ██ ██████ |
Nasopharyngitis | 7 (19.4) | 2 (2.8) | ██ ██████ |
Urinary tract infection | 2 (5.6) | 2 (2.8) | ██ ██████ |
Investigations | ██ ██████ | ██ ██████ | ██ ██████ |
CSF white blood cell count, increased | 0 | 7 (9.7) | ██ ██████ |
CSF protein, increased | 1 (2.8) | 6 (8.3) | ██ ██████ |
Skin and subcutaneous tissue disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Psychiatric disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Anxiety | 3 (8.3) | 4 (5.6) | ██ ██████ |
Insomnia | 3 (8.3) | 3 (4.2) | ██ ██████ |
Metabolism and nutrition disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Renal and urinary disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Eye disorders | ██ ██████ | ██ ██████ | ██ ██████ |
Vascular disorders | ██ ██████ | ██ ██████ | ██ ██████ |
SAEs, n (%)a | |||
Participants with ≥ 1 SAE | 5 (13.9) | 13 (18.1) | ██ ██████ |
Respiratory, thoracic, and mediastinal disorders | 4 (11.1) | 5 (6.9) | ██ ██████ |
Pulmonary embolism | 1 (2.8) | 3 (4.2) | ██ ██████ |
Pneumonia, aspiration | 0 | 2 (2.8) | ██ ██████ |
Acute respiratory failure | 0 | 1 (1.4) | ██ ██████ |
Aspiration | 0 | 1 (1.4) | ██ ██████ |
Respiratory failure | 0 | 1 (1.4) | ██ ██████ |
Chronic respiratory failure | 0 | 0 | ██ ██████ |
Respiratory arrest | 0 | 0 | ██ ██████ |
Atelectasis | 1 (2.8) | 0 | ██ ██████ |
Dyspnea | 2 (5.6) | 0 | ██ ██████ |
Acute respiratory distress syndrome | 0 | 0 | ██ ██████ |
Hypoxia | 0 | 0 | ██ ██████ |
Pneumothorax | 0 | 0 | ██ ██████ |
General disorders and administration site conditions | 0 | 3 (4.2) | ██ ██████ |
Hypothermia | 0 | 1 (1.4) | ██ ██████ |
Impaired self-care | 0 | 1 (1.4) | ██ ██████ |
Respiratory complication associated with device | 0 | 1 (1.4) | ██ ██████ |
Nervous system disorders | 0 | 3 (4.2) | ██ ██████ |
Loss of consciousness | 0 | 1 (1.4) | ██ ██████ |
Lumbar radiculopathy | 0 | 1 (1.4) | ██ ██████ |
Myelitis, transverse | 0 | 1 (1.4) | ██ ██████ |
Intracranial pressure, increased | 0 | 0 | ██ ██████ |
Injury, poisoning, and procedural complications | 0 | 2 (2.8) | ██ ██████ |
Fibula, fracture | 0 | 1 (1.4) | ██ ██████ |
Meningitis, chemical | 0 | 1 (1.4) | ██ ██████ |
Cardiac disorders | 0 | 1 (1.4) | ██ ██████ |
Cardiac failure, congestive | 0 | 1 (1.4) | ██ ██████ |
Cardiorespiratory arrest | 0 | 0 | ██ ██████ |
Gastrointestinal disorders | 0 | 1 (1.4) | ██ ██████ |
Dysphagia | 0 | 0 | ██ ██████ |
Fecaloma | 0 | 1 (1.4) | ██ ██████ |
Infections and infestations | 0 | 2 (2.8) | ██ ██████ |
Pneumonia, aspiration | 0 | 1 (1.4) | ██ ██████ |
Myelitis | 0 | 1 (1.4) | ██ ██████ |
COVID-19 | 0 | 0 | ██ ██████ |
Pneumonia | 0 | 0 | ██ ██████ |
Septic shock | 0 | 0 | ██ ██████ |
Vascular disorders | 0 | 1 (1.4) | ██ ██████ |
Deep vein thrombosis | 0 | 1 (1.4) | ██ ██████ |
Renal and urinary disorders | 0 | 0 | ██ ██████ |
Musculoskeletal and connective tissue disorders | 0 | 0 | ██ ██████ |
Hepatobiliary disorders | 0 | 0 | ██ ██████ |
Surgical and medical procedures | 0 | 0 | ██ ██████ |
Eye disorders | 0 | 0 | ██ ██████ |
Neoplasms — benign, malignant, and unspecified (including cysts and polyps) | 0 | 0 | ██ ██████ |
Psychiatric disorders | 0 | 0 | ██ ██████ |
Metabolism and nutrition disorders | 1 (2.8) | 0 | ██ ██████ |
Dehydration | 1 (2.8) | 0 | ██ ██████ |
Participants who stopped treatment due to adverse events, n (%) | |||
Events leading to drug withdrawal | 0 | 4 (5.6) | ██ ██████ |
Events leading to study withdrawal | 0 | ██ ██████ | ██ ██████ |
Events leading to drug interruption | 0 | 3 (4.2) | ██ ██████ |
Deaths, n (%) | |||
Number of patients who died | 0 | 1 (1.4) | ██ ██████ |
Cardiac failure, congestive | 0 | 1 (1.4) | ██ ██████ |
Respiratory failure | 0 | 0 | ██ ██████ |
Respiratory arrest | 0 | 0 | ██ ██████ |
Cardiac arrest | 0 | 0 | ██ ██████ |
Cardiorespiratory arrest | 0 | 0 | ██ ██████ |
Pneumonia, aspiration | 0 | 0 | ██ ██████ |
Pulmonary sepsis | 0 | 0 | ██ ██████ |
Septic shock | 0 | 0 | ██ ██████ |
Amyotrophic lateral sclerosis | 0 | 0 | ██ ██████ |
Sudden death | 0 | 0 | ██ ██████ |
CSF = cerebrospinal fluid; CTCAE = Common Terminology Criteria for Adverse Events; NR = not reported; OLE = open-label extension; SAE = serious adverse event.
aEach participant was counted once at the maximum CTCAE grade.
bRelated as assessed by the investigator.
cRelated to lumbar puncture as assessed by the investigator.
Sources: The VALOR trial Clinical Study Report76 and Common Technical Document - 2.7.4 Summary of Clinical Safety.120-122 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
The VALOR trial was a multicentre, randomized, double-blind, placebo-controlled, phase III study. It used appropriate methods for randomization, including centralized computer-generated treatment assignment, an identical placebo comparator using artificial CSF, and comprehensive blinding of investigators, participants, and outcome assessors during the 28-week pivotal phase. These elements likely minimized selection, performance, and detection biases, and represent strengths of the study design.
However, several limitations impact the internal validity of the VALOR trial. The statistical analysis plan was amended partway through the trial to include baseline plasma NfL levels as a covariate and to define progression subgroups based on NfL. Although the primary JRT remained unchanged, all subsequent ANCOVA analyses, including those in the ITT population, relied on this post hoc adjustment, which may have introduced analysis bias and favoured the detection of benefit. This approach raises concerns for analysis bias for several reasons. First, making such changes after trial commencement can lead to data-driven decisions that selectively favour the detection of a treatment effect. Second, if baseline NfL levels (presumably a known prognostic factor) is strongly associated with the ALSFRS-R outcome, its inclusion as a covariate could mean that observed effects are influenced more by this prognostic adjustment than by a true treatment effect, potentially overestimating any benefit of tofersen.
At baseline, notable imbalances in characteristics between groups were present, particularly in the primary analysis population (the mITT population). Participants in the placebo group were, on average, older than those in the tofersen group (54.0 years versus 47.3 years, respectively). In addition, the slower-progressing I114T mutation, which is often linked to a more indolent course but whose carriers were included in this faster-progressing cohort if they exhibited a steep prerandomization ALSFRS-R slope decline (≥ 0.9 points per month), was numerically more common in the placebo arm. This imbalance could have potentially diluted an observable treatment effect if these participants with the I114T mutation in the placebo group subsequently progressed more slowly than anticipated based on their entry slope. Conversely, participants in the tofersen group had 15% to 25% higher baseline NfL levels and a steeper ALSFRS-R decline during the run-in period. Because higher NfL levels and faster prebaseline decline predict poorer prognosis, these combined imbalances likely biased the primary comparison against detecting a benefit for tofersen. Because higher NfL levels and faster functional decline are associated with a worse prognosis, these imbalances likely biased results against detecting a treatment effect for tofersen.
Participant attrition during the blinded phase was low and balanced at approximately 11% between groups, which likely limits the overall magnitude of attrition bias. However, the nature of missingness (e.g., whether data were missing not at random) remains a consideration; if reasons for withdrawal differed systematically between groups in relation to outcomes, bias could still have been introduced. MI was used to handle missing data, incorporating clinically relevant covariates. Sensitivity analyses supported the adequacy of the imputation methods, assuming data were missing at random or missing completely at random. Nevertheless, in the OLE study, retention dropped considerably, with only 61% of early-start participants and 44% of delayed-start participants remaining by week 104, raising concerns about informative missingness and the potential for significant attrition bias in the long-term data.
The study was underpowered for detecting clinically meaningful differences. The VALOR trial had a small sample size and was designed based on an assumption of a very large 20-point difference in ALSFRS-R score over 6 months, an assumption derived from only 12 historical cases. The actual observed CI for the primary ALSFRS-R end point was wide and encompassed both clinically meaningful benefit and harm, reflecting serious imprecision. As well, survival outcomes were based on too few events to yield reliable estimates. Furthermore, the clinical experts noted that the duration of the trial was likely insufficient to detect a treatment effect.
Following the failure to demonstrate a statistically significant effect on the primary outcome, all subsequent analyses were considered exploratory, and no adjustments were made for multiplicity. This increases the risk of false-positive findings and limits the interpretability of secondary outcomes.
The ALSFRS-R tool, while widely used, has its limitations. These encompass potential multidimensionality of the scale and the absence of a well-defined minimally important difference, which complicates interpretation in small, short-duration trials. The scale's utility is further challenged by the nonlinear nature of functional decline in ALS, meaning that calculating a consistent monthly rate of change can be inappropriate and may not reflect the true disease trajectory. Clinicians emphasize that — given the progressive nature of ALS as described by its natural history — a clinically important effect would be a slowing of the monthly rate of decline (slope) rather than an absolute point difference at a fixed time point. However, the VALOR study did not prespecify or analyze slope of change as an end point, so any potential disease-modifying effect on the rate of decline was neither captured nor powered for evaluation. While statistically significant reductions in CSF SOD1 protein and plasma NfL levels were observed, these biomarkers were unvalidated surrogates for clinical outcomes, and their statistical significance was nominal in nature due to failure on the primary end point, contributing to indirectness.
The OLE study’s data were nonrandomized and subject to potential confounding over time. Further, the OLE study lacked randomization and a concurrent control group, and blinding was inherently compromised by the open-label design. The study maintained blinding to the original VALOR trial’s treatment assignment, potentially providing some mitigation to the limitations associated with the open-label design of the OLE study. However, adverse effects associated with intrathecal administration and inflammatory responses in the CSF in the original trial may have revealed treatment allocation in certain groups of patients, potentially introducing detection bias in subjective outcomes. The high dropout rate in the OLE study further limits the reliability of long-term outcome comparisons. These limitations all compound the interpretation of results that already lack causal inference value.
External Validity
The study enrolled adults with genetically confirmed SOD1 mutations causing ALS, representing the relevant target population. There is also heterogeneity within SOD1-ALS, with some mutations linked to faster progression than others, creating uncertainty around generalizing results across all mutation types. In addition, the VALOR trial deliberately enriched the primary analysis set for rapid progressors by requiring either specific high-risk mutations or a steep prerandomization ALSFRS-R slope. While this strategy improved statistical efficiency, it further narrows external validity; the treatment effect observed in this highly selected, fast-progressing cohort may not translate to patients with slower-progressing or clinically heterogeneous SOD1-ALS encountered in routine practice. While patients with a slower progression of ALS were enrolled, they were not part of the main analysis and, due to the disease nature and trial timeline, were not expected to show a difference. Participants were predominantly white, with limited representation of other ethnic groups common in Canada; however, this is consistent with the known epidemiology of ALS. Most participants had relatively preserved function at baseline, including SVC greater than 50 to 65 percent-predicted and the ability to ambulate. Standard trial exclusions, including significant comorbidities and the imminent need for ventilation, mean that findings may not apply to the more complex cases often encountered in practice in Canada.
Tofersen was administered intrathecally, requiring repeated lumbar punctures — 3 loading doses followed by monthly maintenance. This invasive method requires significant resources. While the procedure could potentially be performed by any clinician trained and comfortable with intrathecal administration, the expert panel emphasized that patients must be under the ongoing care of an ALS specialist. This requirement for specialist oversight, coupled with the procedural demands, likely limits accessibility to specialized ALS clinics or academic hospitals, which may not be readily available or feasible in many parts of Canada. In its feedback to this report, the sponsor noted that some patients are currently accessing tofersen through Health Canada’s Special Access Program in various practices, including small Maritime practices and community neurology practices. This route of administration contrasts with more accessible oral or IV ALS therapies. The use of placebo as a comparator limits understanding of the comparative efficacy against the prescribed options in Canada (riluzole and edaravone). However, allowing background therapy (riluzole and edaravone allowed) was appropriate and likely reflects clinical practice in Canada where the clinical experts noted that most patients will receive riluzole and edaravone. Nonetheless, only a small proportion of participants ended up receiving both (around 6% of the study participants), while the majority received riluzole (approximately 60%).
Outcomes evaluated included ALSFRS-R, SVC, survival (death or PV), HHD, and patient-reported outcomes like ALSAQ-5 and FSS. Clinical experts consulted for this review indicated that while the ALSFRS-R score is often collected in clinics in Canada, its primary use is for research or facilitating access to other therapies rather than for the routine guidance of tofersen management. They did note that monitoring the change in the slope of ALSFRS-R can be clinically meaningful. Other measures used in the VALOR study, such as SVC (with FVC being preferred clinically), HHD, and specific quality-of-life scales like ALSAQ-5 and FSS, are generally considered research tools and not standard for routine clinical decision-making in Canada. Biomarkers such as SOD1 protein and NfL levels were used to evaluate the extent to which tofersen achieved target engagement and impacted neurodegeneration, respectively. However, the clinical experts noted that they have a limited role in the management of patients and they are not part of routine care in Canada and currently lack established clinical utility. The long-term follow-up in the OLE study allowed for a broader understanding of disease progression and survival but was limited by the absence of a concurrent control group and increasing dropout rates over time.
Participants were relatively young, with a median symptom duration of 11 months and a mean ALSFRS-R score of 37 at baseline, indicating early disease. Rapid progression genotypes, such as A5V and I114T (if they presented with an even steeper prerandomization ALSFRS-R decline), were overrepresented whereas slower progression mutations, older individuals, and those with significant respiratory compromise or comorbidities were largely excluded. Thus, generalizability is strongest for younger, fitter individuals with rapidly progressing SOD1-ALS under the care of specialized clinics.
Although background therapy in the VALOR trial was aligned with Canadian standards and most participants received riluzole, fewer than 8% of participants used edaravone. Thus, the additive benefit of tofersen in fully optimized background therapy remains uncertain. The study’s outcomes are meaningful, but the 6-month blinded phase may have been too short to detect definitive effects on survival end points. The long-term OLE study data provide additional insight, but without randomization or control, these results must be interpreted cautiously.
Furthermore, an important evidence gap is the lack of information on the comparative efficacy of tofersen against the current standard of care (riluzole + edaravone).
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, the GRADE tool was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.126,127
“High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word ‘likely’ for evidence of moderate certainty (e.g., ‘X intervention likely results in Y outcome’).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word ‘may’ for evidence of low certainty (e.g., ‘X intervention may result in Y outcome’).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as ‘very uncertain.’”
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for tofersen versus placebo.
Long-Term Extension Studies
Long-term extension studies were included in the main systematic review section.
Indirect Evidence
None was submitted by the sponsor.
Studies Addressing Gaps in the Systematic Review Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
The safety and efficacy of tofersen have been assessed in patients enrolled in a German and Italian EAP (Table 19). These 2 prospective observational studies provide additional insights into the safety and efficacy profile of tofersen in a real-world setting.
Table 19: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Uncertain clinical benefit of tofersen as measured by the ALSFRS-R at week 28 due to missed primary end point in the VALOR study. | German and Italian EAP | Patients experienced a reduction in ALS-PR compared to prebaseline levels. A decrease in NfL levels indicates that tofersen may be effective for patients with ALS associated with a mutation in the SOD1 gene. |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; ALS-PR = amyotrophic lateral sclerosis progression rate; EAP = early access program; NfL = neurofilament light chain.
Sources: Meyer et al. (2023),3 Meyer et al. (2024),2 Sabatelli et al. (2024),4 and Wiesenfarth et al. (2024).1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Description of Studies: German and Italian EAPs
Four real-world studies, including 3 studies from Germany (Wiesenfarth et al.,1 Meyer et al., [2024],2 and Meyer et al. [2023]3) and 1 study from Italy (Sabatelli et al.4). The studies evaluated the effectiveness, safety, and patient-reported outcomes of tofersen treatment in patients with SOD1-ALS and included assessments of clinical progression, biomarkers, and HRQoL.
Interventions
In all 4 studies, tofersen 100 mg was administered directly into the CSF on day 1, day 14, and day 28 as loading doses. Patients then received up to 16 maintenance doses at intervals of approximately 28 days (with a minimum of 21 days).
Outcomes
Across all of the EAP studies, ALS-PR was defined as points of ALSFRS-R lost per month. ALS-PR was calculated by the following: the ALSFRS-R score at first application minus the ALSFRS-R score at last visit divided by months between the 2 visits. The prebaseline progression rate was calculated as 48 minus the ALSFRS-R score at first application divided by months between disease onset and the first application. Two classifications of ALS-PR were used in the Meyer et al. (2023)3 study. One classification consisted of slower-progressing ALS (< 0.5 ALSFRS-R decline per month), intermediate-progressing ALS (≥ 0.5 and ≤ 1.0 ALSFRS-R decline per month), and faster-progressing ALS (> 1.0 ALSFRS-R decline per month) based on arbitrary groups used in a previous ALS study.128 A second classification included a distinction between faster-progressing ALS (≥ 0.9 ALSFRS-R decline per month) and slower-progressing ALS (< 0.9 ALSFRS-R decline per month), a threshold informed by the EMPOWER study in which patients with a prerandomization decline of 0.9 points or more per month maintained this rate at 6 months, 9 months, and 12 months postrandomization.3
In Meyer et al. (2024),2 patient-reported outcomes included the MYMOP2, the TSQM-9, and the NPS. MYMOP2 allows patients to identify and rate the severity of their most troubling symptoms as assessed on a 7-point Likert scale (0 for “as good as it could be” to 6 for “as bad as it could be”). TSQM-9 evaluates patients’ satisfaction with their treatment in terms of effectiveness, convenience, and overall satisfaction with a total score that can range from 0 to 100 (a higher total score equates to greater satisfaction). The NPS measures how likely patients are to recommend the treatment to others, with a score that ranges from –100 to 100. A score greater than 0 indicates that more patients are promoters than detractors, reflecting overall support for the treatment.
Statistical Analysis
In Wiesenfarth et al.,1 CSF NfL and pNfH levels were assessed in a hierarchical manner. No other adjustments for multiple testing were conducted. Change over time was analyzed using the Wilcoxon signed rank test with baseline defined as the first tofersen application. Analysis of variance was performed for multiple comparisons. All statistical analyses were performed at a 2-sided level of alpha of 0.05. In Sabatelli et al.,4 the Wilcoxon signed rank test compared disease progression rates before and after treatment using decline in ALSFRS-R points per month. CSF NfL concentrations were measured in all patients at baseline and every 12 weeks. Descriptive statistics were used (frequency, mean, median, and ranges) for both Meyer et al. studies.2,3
Results
Patient Disposition
The cohort study by Wiesenfarth et al.1 evaluated the effects of tofersen 100 mg on clinical and safety outcomes in 24 patients with ALS across 10 centres in Germany over 12 months. All patients had a mutation in the SOD1 gene. Eligibility required no contraindications to lumbar puncture; prior patients from the VALOR and OLE studies were excluded. Half of the patients were female and half male (n = 12 in each group). Patients were diagnosed with ALS at a median of 11.7 months (IQR, 3.6 to 28.5 months) before study inclusion. At the time of the first tofersen dose, the median age of participants was 53.0 years (IQR, 41.5 to 61.0 years). The median ALFRS-R score at the first study visit was 37.0 points (IQR, 29.8 to 41.8 points). The median progression rate from disease onset to first visit, as measured by the ALS-PR, was a median of 0.41 points lost per month (IQR, 0.20 to 0.83 points).
Meyer et al. (2024)2 assessed clinical and patient-reported outcomes in 16 patients with ALS with an SOD1 mutation receiving tofersen treatment in the German EAP study for up to 18 months (mean = 11 months). Seven of these patients were also included in the Wiesenfarth et al. study. The mean age of participants was 53.4 years (range, 32 to 71 years), and 62.5% of patients were female and 37.5% were male. The mean pretreatment ALSFRS-R score was 37.4 points (range, 7 to 46 points).
Meyer et al. (2023)3 assessed efficacy outcomes in 6 patients with ALS with 1 of 3 unique SOD1 mutations (c.272A > C, c.346C > G, and c.396_399dup) for up to 8 months (mean = 6.5 months). One patient had an ALSFRS-R score of 1, with invasive ventilation and profound loss of motor function. The remaining patients were considered to have high functional status, with ALSFRS-R scores ranging from 35 to 46 points at baseline. Of the 6 patients in the study, 4 (66.7%) patients were female, and 2 (33.3%) patients were male. The mean age of participants was 53.3 years (range, 39 to 60 years). Disease duration at baseline varied from 16 months to 105 months (median = 34 months). At baseline, motor function ranged from mild distal limb paresis to complete tetraplegia, with 1 (17%) patient requiring invasive ventilation and feeding support.
In Sabatelli et al.,4 27 of 42 (64.3%) enrolled patients with SOD1-ALS began treatment with tofersen. Eleven (26.2%) patients declined treatment due to slow disease progression and treatment burden; 4 (9.5%) patients were ineligible due to advanced disease requiring ventilatory support. Of the 27 patients who began treatment with tofersen, 17 (63.0%) were included in the efficacy results. Five (18.5%) patients were excluded due to incomplete data, and 2 (7.4%) patients discontinued the study. Three (11.1%) patients died within the first 6 months of treatment.
Efficacy
ALS Progression Rate
In the Wiesenfarth et al.1 study, the median ALSFRS-R score at baseline was 38.0 points (IQR, 32.0 to 42.0 points); this declined (worsened) to 35.0 points (IQR, 29.0 to 42.0 points) by the last tofersen dose. This corresponded to an ALS-PR of 0.11 points per month (IQR, –0.09 to 0.32 points per month). Seventeen (73.9%) patients experienced slower ALS progression rates during treatment compared to prebaseline (median = 0.0 per month; IQR, −0.31 to 0.18 per month). Six (26.1%) patients had faster ALS progression rates during treatment compared to prebaseline (median = 0.98 per month; IQR, 0.32 to 1.81 per month). Patients with faster progression had shorter disease duration before treatment, higher ALSFRS-R baseline scores, and higher baseline NfL and pNfH levels. They also had shorter follow-up, receiving fewer tofersen doses.
In Meyer et al. (2024),2 the ALS-PR was reduced in 8 of 16 (50%) patients and unchanged in the remaining 8 (50%) patients with a mean change of –0.2 points per month (range, 0 to –1.1 points per month).
In Meyer et al. (2023),3 the ALS-PR decreased in 2 (33.3%) patients whereas no changes in the ALSFRS-R score were observed in the 4 (66.7%) patients with slowest ALS-PR or a very low ALSFRS-R value at baseline.
In Sabatelli et al.,4 the median pretreatment ALS-PR was 0.25 points per month (IQR, 0.13 to 0.62 points per month), which declined to a median of 0.0 points per month (IQR, –0.10 to 0.20 points per month) posttreatment. This represented a median monthly change of –0.20 points per month (IQR, –0.62 to –0.04 points per month).
Biomarkers
In Wiesenfarth et al.,1 the median serum NfL level decreased from 78.0 pg/mL (IQR, 37.0 pg/mL to 147.0 pg/mL [n = 23]) at baseline to 36.0 pg/mL (IQR, 22.0 pg/mL to 65.0 pg/mL [n = 23]) at the last tofersen dose. The median pNfH in CSF level decreased from 2,226 pg/mL (IQR, 1,061 pg/mL to 6,138 pg/mL [n = 18]) at baseline to a median of 1,151 pg/mL (IQR, 521 pg/mL to 2,360 pg/mL [n = 18]) at last tofersen dose.
In Meyer et al. (2024),2 serum NfL levels decreased in 15 of 16 (93.8%) patients. The mean serum NfL level decreased by 58% (range, – 92% to 27%) from 62 pg/mL before tofersen treatment to 23 pg/mL during tofersen treatment.
In the 6 patients in the Meyer et al. (2023) study,3 mean CSF NfL levels decreased by 66% (range, 52% to 86%) and mean serum NfL levels decreased by 62% (range, –36% to –84%) over the course of treatment.
In 14 of 17 (82%) enrolled patients in Sabatelli et al.,4 the mean reduction in CSF NfL levels from baseline was 61% (range, 49% to 79%). Of the remaining 3 (17.6%) patients, 1 (5.9%) patient had unchanged CSF NfL levels, and 2 (11.8%) patients had increased CSF NfL levels.
Health-Related Quality of Life
In Meyer et al. (2024),2 at baseline, the mean symptom severity on the MYMOP2 7-point scale was 3.8 (n = 14), which decreased to a mean of 3.0 at patients’ last measured perception. From baseline to last measured perception, MYMOP2 responses showed symptom improvement (defined as an improvement in at least 1 of the 2 target symptoms) in 10 of 14 (71.4%) patients and partial improvement (defined as improvement or stabilization in 1 symptom and deterioration of the other) in the remaining 4 (42.9%) patients. TSQM-9 scores were assessed in 15 (93.8%) patients with a mean global satisfaction score of 83 (SD = 16) and the convenience of intrathecal administration had a mean score of 50 (SD = 27). As assessed by the NPS, at 6 months of tofersen treatment, 12 of 15 (80%) patients were promoters of tofersen.
Harms
In Wiesenfarth et al.,1 common procedure-related side effects included back pain, headache, leg nerve pain, and dizziness. Two (8.7%) patients experienced SAEs possibly related to tofersen that occurred during the study; both of these patients stopped treatment voluntarily. One patient developed autoimmune spinal cord and nerve root inflammation after 6 doses. Another patient experienced short-term leg weakness after each of their first 3 doses. Symptoms resolved in both cases; the former required immune-targeted treatment. There were no reported deaths during the observation period. Patients' CSF changes indicated autoimmune inflammation in the central nervous system. Eleven of 15 (73%) patients experienced an increase in white blood cells in the CSF ranging from 8 leukocytes/μL to 56 leukocytes/μL. Ten (66.7%) of these patients also experienced elevated protein levels. Nine of 10 (90%) patients showed immune protein (immunoglobulin M, immunoglobulin G, or immunoglobulin A) production in the CSF.
In Sabatelli et al.,4 postinjection headaches occurred once in 4 (23.5%) patients. Seven (41.2%) patients reported limb pain, with pain following the path of a nerve from the spine to the arms or legs. Nine of 15 (60.0%) patients experienced increased white blood cell and protein levels in the CSF after tofersen therapy, indicative of drug-related spinal cord and nerve root inflammation. Two (22.2%) of these patients demonstrated clinical symptoms and responded to steroid treatment. Three (11.1%) of the 27 patients who began treatment with tofersen died soon after starting treatment and were not included in the analyses.
Harms results were not provided in the Meyer et al. studies.2,3
Critical Appraisal
Internal Validity
The sponsor submitted data from EAP cohort studies based on available publications. Study protocols and statistical analysis plans were not provided, limiting the ability to comprehensively assess the studies’ design and methodology — especially outcome measurement — and statistical analyses.
The analyses were primarily descriptive and not adjusted for confounders, which substantially limits internal validity and the ability to infer treatment effects. The lack of a comparator group further impairs the ability to distinguish treatment effects from natural disease progression or other external influences.
The lack of blinding, where both patients and clinicians were aware of the treatment administered, raises the potential for performance and detection biases, particularly in subjective measures such as ALSFRS-R and AE reporting.
The EAP studies were affected by small sample sizes missing or incomplete data, and the inconsistent reporting of outcomes, all of which undermine the reliability of progression slope estimates. For example, in the Italian study, 27 of 42 enrolled patients received treatment, 17 (40.5%) of whom had evaluable data. This substantial attrition raises the risk of survivor bias, where observed effects may reflect characteristics of a selected subgroup rather than true treatment effectiveness.
ALSFRS-R slope estimates were based on pre-post comparisons using 2 time points, assuming linear decline. This assumption may not reflect the true disease trajectory because ALS progression and ALSFRS-R decline are often nonlinear and can vary within patients, accelerating at some stages and plateauing at others.5,6 Therefore, these estimates may misrepresent true disease progression. The reliance on patient recall for prebaseline data introduces uncertainty because recall bias may lead to the inaccurate reporting of disease onset and the prior ALSFRS-R trajectory. In the Wiesenfarth et al. study, it was unclear whether observed ALSFRS-R changes reflected treatment response or were confounded by a short follow-up duration. Likewise, it is unclear whether any improvements in ALSFRS-R scores would be sustained over a longer period, particularly in the context of the non–rapidly progressing disease.
Although posttreatment reductions in ALSFRS-R decline were observed, the lack of a comparator group, small sample sizes, missing data, the potential for bias (particularly from confounding), and the reliance on slopes of ALSFRS-R scores from few time points reduced certainty in the results from these studies regarding the effectiveness of tofersen.
External Validity
The reviewed studies were conducted in small samples of patients in Germany and Italy, with sample sizes ranging from 6 to 24 patients. It is difficult to determine based on the small cohort sizes and limited reporting of characteristics whether these patient populations are representative or transferrable to patients with ALS in Canada. Additionally, access to multidisciplinary ALS care and specialized centres, which were available to patients in the studies, may not reflect accessibility for patients with ALS in Canada given the geographically dispersed and diverse ALS population in Canada. While the clinical experts consulted agreed that any trained clinician could administer the treatment under review, they emphasized that patients should remain under the care of an ALS specialist. The experts noted that access to multidisciplinary ALS care is more readily available to patients near major centres in Canada. Although efforts have been made to expand access in remote areas through telehealth and transport to specialized clinics, significant barriers to equitable care remain. As such, the centralized care settings in the reviewed studies may not fully reflect the realities of care delivery across Canada’s geographically dispersed health system.
Discussion
Summary of Available Evidence
The primary evidence for tofersen comes from the VALOR study (part C), a phase III, randomized, double-blind, placebo-controlled trial with 108 adult participants with SOD1-ALS (60 enriched high progressors), and its ongoing OLE (Study 102). The VALOR trial had a 28-week placebo-controlled period. Supportive real-world evidence is emerging from EAP studies in Germany and Italy involving patients with SOD1-ALS treated with tofersen.
Despite appropriate randomization and blinding procedures, the total sample was small (N = 60 for the primary end point) and enriched for fast progressors. No head-to-head trials versus the dual standard of care (riluzole + edaravone) exist and the evidence base therefore consists of this single pivotal program, augmented by the uncontrolled OLE study and real-world observations.
The blinded portion of the VALOR trial failed to demonstrate a statistically significant benefit on the prespecified primary functional outcome (the ALSFRS-R score at week 28), and all downstream clinical end points remain exploratory. The strongest numeric signals favour tofersen on biomarkers (CSF SOD1 and plasma NfL levels) but these are unvalidated surrogates. Longer-term, open-label data suggest a trend toward slower functional decline and improved survival relative to delayed-start patients, yet the absence of concurrent control, high attrition, and potential informative missingness temper confidence in these findings.
Interpretation of Results
Efficacy
The VALOR trial did not meet its primary efficacy end point because the change from baseline to week 28 in the ALSFRS-R total score in the predefined mITT population showed a non–statistically significant difference of 1.2 points favouring tofersen over placebo (JRT + MI: P = 0.9689). The study was potentially underpowered and had an insufficient duration for this end point, and baseline imbalances in the mITT population (e.g., older age and higher prevalence of slower-progressing mutations in the placebo group versus a higher baseline NfL level and faster prebaseline decline in the tofersen group) may have influenced this outcome, with some imbalances having likely biased results against detecting a treatment effect for tofersen while other imbalances were noted to have potentially biased the results in favour of tofersen.
Despite not meeting the primary end point, tofersen demonstrated numerically large effects on biomarkers. In the VALOR trial’s mITT population, tofersen led to a 38% reduction in total CSF SOD1 protein (nominal P < 0.0001) and a 67% reduction in the plasma NfL level (nominal P < 0.0001) compared to placebo at week 28. Similar reductions were observed in the non-mITT (slower-progressing) population and the overall ITT population. These biomarker changes were sustained through the OLE study up to 104 weeks, suggesting a successful and durable target engagement (SOD1 reduction) and a reduction in axonal injury (NfL reduction). However, these are surrogate markers and their direct correlation with individual clinical benefit requires further empirical validation, although higher NfL levels are associated with faster progression and shorter survival.
Analyses of clinical outcomes from the OLE study, which were presented by the sponsor as supportive evidence of disease-slowing activity for tofersen, showed trends favouring the early initiation of tofersen. At week 52, the early-start tofersen group showed a 3.5-point smaller decline on ALSFRS-R (95% CI, 0.4 to 6.7 points) when compared to the delayed-start tofersen group. These trends persisted at week 104, with a 3.7-point difference in ALSFRS-R (95% CI, −0.7 to 8.2 points) and a 9.7 percent-predicted difference in SVC between the early-start and delayed-start tofersen groups. Although these OLE study findings are encouraging, they must be interpreted within the high uncertainty associated with method and limitations. These include open-label design, the lack of a concurrent control group for the long-term comparison, the potential for detection bias, and significant participant attrition by week 104 (61% of participants in the early-start group and 44% of participants in the delayed-start group were remaining at that point).
There is no definitive evidence of a causal relationship between tofersen and clinical benefits in terms of function, quality-of-life improvements, survival, or symptoms improvements. Available evidence is exploratory in nature and thus suffers from a potential likelihood of high type I error (false-positive) and is best used for hypothesis generation to be tested in a subsequent confirmatory trial. Additional evidence beyond the 6 months of the phase III trial is descriptive in nature and lacks any comparison group.
Discussions on the clinical relevance of the results are inherently challenging because causality is difficult to establish. While the primary end point in the VALOR trial — an adjusted mean difference of 1.2 points on the ALSFRS-R scale at 28 weeks — did not reach statistical significance, its clinical importance requires careful consideration. A 1.2-point change is less than most published estimates for a minimal important difference, which typically range from 3 to 4 points. However, given the relentlessly progressive nature of SOD1-ALS, where a continuous functional decline is expected, even a modest difference favouring tofersen, such as the 1.2 points seen in the VALOR trial, in a short-duration trial could be a signal of potential biological activity that may become more pronounced over time.
The argument for clinical meaningfulness could be strengthened by the longer-term data from the OLE study. The observed difference of 3.7 points on the ALSFRS-R scale at 104 weeks between the early-start and delayed-start groups falls within the range considered to be clinically important. In the context of the disease's natural history, where patients typically lose approximately 1 point per month on the ALSFRS-R scale, a 3.7-point difference is potentially clinically meaningful. This suggests that after 2 years, patients who started tofersen early were functioning at a level that the delayed-start group had been at nearly 4 months prior. For a patient with ALS, such a difference could translate into a significant delay in the loss of critical functions like walking, swallowing, or independent breathing, thereby preserving HRQoL and independence for longer. Nonetheless, it is important to note that the comparison in the OLE study is not a true control and does not establish any sort of causal inference. This means that much of the speculation around the clinical meaningfulness of the observed results is, inherently, highly uncertain.
The clinical experts consulted on this review expressed that tofersen is likely effective for patients with SOD1-ALS, despite the limitations associated with the evidence. They noted the drug’s mechanism of action and the observed reduction in the biomarkers, which supports target engagement and biological activity. The clinical experts also highlighted observed trends in slowing disease progression in the OLE and EAP studies. The EAP studies suggest that tofersen may slow disease progression, with some patients showing stabilization or even improvement in the ALS-PR measure. These findings point to a potential treatment benefit, though variability in responses with wide IQRs highlight that not all patients experience the same degree of effect and some experienced worsening. Notably, the clinical experts emphasized that some patients in these studies exhibited improvement in their ALSFRS-R scores, which is a very unlikely scenario given the natural history of ALS, where continuous decline is expected. Moreover, the clinical experts reported real-world clinical experiences of patients experiencing meaningful clinical benefits from the treatment in practice settings.
The experts felt that — given the high unmet need in patients with ALS, coupled with the totality of information regarding the biologic rationale —the mechanism of action, the biomarker changes in the pivotal trial, and the slower progression observed in the OLE and EAP studies support the potential benefit of tofersen. Although the OLE and EAP studies lack a control or comparison group, making causal inference more challenging,126,127,129 the observed outcomes appear to exceed what would be expected in untreated disease, lending further support for the potential clinical relevance of the findings.130 However, it is crucial to acknowledge the significant heterogeneity inherent in ALS, even within SOD1-ALS, which makes comparisons to natural history data or pretreatment trajectories subject to considerable uncertainty and potential biases.
The available evidence, particularly from the controlled phase of the VALOR trial, is not conclusive in demonstrating clinical benefit. The Health Canada Notice of Compliance with conditions status for tofersen underscores this uncertainty and highlights the regulatory expectation for further confirmatory studies to definitively establish clinical benefit. Therefore, while a potential for benefit is suggested by biomarker effects and trends in uncontrolled data, confirmed clinical efficacy awaits more robust evidence.
Also, it is worth noting that the sponsor is currently conducting the phase III ATLAS trial (which is specified in the qualifying notice from Health Canada), and based on CDA-AMC assessment, CDA-AMC believes that the criterion for a time-limited recommendation could be met because a small subset of patients in the ATLAS trial reflect the same indication that CDA-AMC is currently reviewing. Specifically, those who receive placebo in the presymptomatic phase and then transition to active treatment upon development of symptoms appear to match the target population for the current review and would be covered under the current indication approved by Health Canada. The sponsor opted not to participate in the time-limited recommendation pathway and noted that the ongoing ATLAS trial enrols presymptomatic SOD1 mutation carriers, which does not reflect the approved indication for symptomatic SOD1-ALS, and that the subset of participants who convert to symptomatic ALS is limited to approximately ██ patients and the study is not powered to confirm efficacy in this group.
Harms
AEs were common and largely related to lumbar puncture procedures (headache, back pain, post–lumbar puncture syndrome) or ALS progression (falls, respiratory events). Serious neurologic inflammatory events (e.g., aseptic meningitis, radiculitis, papilledema) were infrequent (< 5 %) but clinically relevant given the chronic dosing schedule. One death occurred during the blinded phase; otherwise, mortality differences were too small for conclusions. The open-label dataset reports a ██% death rate over 3 years. Overall, the safety profile appears acceptable in specialized centres experienced with intrathecal therapy, but real-world rates of procedure-related complications and raised intracranial pressure remain uncertain.
Conclusion
Based on the 28-week data from the VALOR trial, the evidence for a clinically meaningful treatment effect of tofersen on functional decline, as measured by the ALSFRS-R, is of low certainty. Tofersen may result in little or no clinically meaningful difference compared to placebo in function outcomes, respiratory function, and muscle strength over this period. The effects of tofersen on the time to death or PV and on SAEs are very uncertain, due to the very limited number of events that occurred during the 28-week trial. Conversely, the VALOR study showed that tofersen likely reduces CSF SOD1 protein and plasma NfL levels, biomarkers of target engagement, and axonal injury, with moderate certainty. However, these are surrogate end points and their direct translation to patient-centred clinical outcomes is not yet fully established.
The 28-week duration of the VALOR study RCT was likely too short to detect a meaningful treatment effect in ALS, including with tofersen. Longer-term data from the OLE study and the 4 real-world EAP cohort studies suggest slowed disease progression, or even stabilization in some patients, and potential survival benefits, especially with earlier initiation of tofersen. This evidence is primarily descriptive, noncomparative, and subject to a serious risk of bias and limited generalizability. Nonetheless, given the known natural history of ALS, which involves continuous functional decline, a reduction in the rate of ALSFRS-R decline, or stabilization of function, is unexpected and signals a potentially clinically important effect.
Patients with SOD1-ALS and their caregivers have expressed a strong need for treatments that can slow disease progression, maintain function and independence, and extend survival. While the biomarker changes observed with tofersen offer a mechanistic rationale and reductions in the rate of progression have been reported in real-world and longer-term extension studies within the context of the natural history of ALS, the currently available evidence suggests the potential for benefit but does not definitively establish that tofersen meets these expectations in a clinically significant way over the short term.
Key gaps in the evidence remain. There is a need for more robust, comparative data to assess the long-term clinical efficacy of tofersen, particularly regarding functional outcomes, QoL, and survival. Further research is needed to better understand the clinical relevance of the observed biomarker changes and their utility in predicting or monitoring individual patient response. Additionally, evidence in broader SOD1-ALS populations, including those with slower disease progression or different SOD1 mutation types that were less represented in the pivotal trial, is limited. The comparative effectiveness of tofersen against, or in addition to, a fully optimized standard of care has not been directly assessed.
While acknowledging the uncertainty in the results of the included studies, the clinical experts noted that tofersen might address some of the unmet needs in symptomatic patients with ALS with a mutation in the SOD1 gene as it is a disease-modifying therapy, and highlighted the observed reduction in the biomarkers, which supports target engagement and biological activity. The clinical experts also highlighted observed trends in slowing disease progression in the OLE and EAP studies.
References
1.Wiesenfarth M, Dorst J, Brenner D, et al. Effects of tofersen treatment in patients with SOD1-ALS in a “real-world” setting - a 12-month multicenter cohort study from the German early access program. EClinicalMedicine. 2024;69:102495. doi:10.1016/j.eclinm.2024.102495 PubMed
2.Meyer T, Schumann P, Weydt P, et al. Clinical and patient-reported outcomes and neurofilament response during tofersen treatment in SOD1-related ALS-A multicenter observational study over 18 months. Muscle Nerve. 2024;70(3):333-345. doi:10.1002/mus.28182 PubMed
3.Meyer T, Schumann P, Weydt P, et al. Neurofilament light-chain response during therapy with antisense oligonucleotide tofersen in SOD1-related ALS: Treatment experience in clinical practice. Muscle Nerve. 2023;67(6):515-521. doi:10.1002/mus.27818 PubMed
4.Sabatelli M, Cerri F, Zuccarino R, et al. Long-term treatment of SOD1 ALS with tofersen: a multicentre experience in 17 patients. J Neurol. 2024;271(8):5177-5186. doi:10.1007/s00415-024-12437-7 PubMed
5.Boddy SL, Simpson RM, Walters SJ, et al. Estimating the minimum important difference in the ALSFRS-R-instrument in people living with MND. Amyotroph Lateral Scler Frontotemporal Degener. 2025;26(3-4):249-258. doi:10.1080/21678421.2024.2447916 PubMed
6.Young CA, Chaouch A, McDermott CJ, et al. Improving the measurement properties of the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R): deriving a valid measurement total for the calculation of change. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(3-4):400-409. doi:10.1080/21678421.2024.2322539 PubMed
7.Bunton-Stasyshyn RK, Saccon RA, Fratta P, Fisher EM. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist. 2015;21(5):519-29. doi:10.1177/1073858414561795 PubMed
8.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi:10.1186/1750-1172-4-3 PubMed
9.ALS Association. Stages of ALS. Accessed February 11, 2025. https://www.als.org/understanding-als/stages
10.Wolf J, Safer A, Wöhrle JC, et al. Factors predicting one-year mortality in amyotrophic lateral sclerosis patients--data from a population-based registry. BMC Neurol. 2014;14:197. doi:10.1186/s12883-014-0197-9 PubMed
11.Turner MR, Faull C, McDermott CJ, Nickol AH, Palmer J, Talbot K. Tracheostomy in motor neurone disease. Pract Neurol. 2019;19(6):467-475. doi:10.1136/practneurol-2018-002109 PubMed
12.Logroscino G, Traynor BJ, Hardiman O, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385-90. doi:10.1136/jnnp.2009.183525 PubMed
13.Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum (Minneap Minn). 2014;20(5 Peripheral Nervous System Disorders):1185-207. doi:10.1212/01.CON.0000455886.14298.a4
14.Moura MC, Novaes MR, Eduardo EJ, Zago YS, Freitas Rdel N, Casulari LA. Prognostic Factors in Amyotrophic Lateral Sclerosis: A Population-Based Study. PLoS One. 2015;10(10):e0141500. doi:10.1371/journal.pone.0141500 PubMed
15.Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377(2):162-172. doi:10.1056/NEJMra1603471 PubMed
16.National Institute of Neurological Disorders and Stroke (NIND). Amyotrophic Lateral Sclerosis (ALS) [sponsor supplied reference]. Accessed October 07, 2024. https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
17.Muscular Dystrophy Association. Amyotrophic Lateral Sclerosis (ALS): Stages of ALS [sponsor supplied reference]. Accessed October 07, 2024. https://www.mda.org/disease/amyotrophic-lateral-sclerosis/signs-and-symptoms/stages-of-als
18.Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623-7. doi:10.1136/jnnp.2010.224501 PubMed
19.Zarei S, Carr K, Reiley L, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171. doi:10.4103/2152-7806.169561 PubMed
20.Shepheard SR, Parker MD, Cooper-Knock J, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(5):510-518. doi:10.1136/jnnp-2020-325014 PubMed
21.Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. 2018;30(2):252-258. doi:10.1007/s11825-018-0185-3 PubMed
22.Akçimen F, Lopez ER, Landers JE, et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet. 2023;24(9):642-658. doi:10.1038/s41576-023-00592-y PubMed
23.Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019;13:1310. doi:10.3389/fnins.2019.01310 PubMed
24.Müller K, Brenner D, Weydt P, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. 2018;89(8):817-827. doi:10.1136/jnnp-2017-317611 PubMed
25.ALSoD. SOD1 Gene Summary [sponsor supplied reference]. Accessed October 07, 2024. https://alsod.ac.uk/output/gene.php/SOD1
26.Bali T, Self W, Liu J, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88(2):99-105. doi:10.1136/jnnp-2016-313521 PubMed
27.Opie-Martin S, Iacoangeli A, Topp SD, et al. The SOD1-mediated ALS phenotype shows a decoupling between age of symptom onset and disease duration. Nat Commun. 2022;13(1):6901. doi:10.1038/s41467-022-34620-y PubMed
28.Xu L, Liu T, Liu L, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. 2020;267(4):944-953. doi:10.1007/s00415-019-09652-y PubMed
29.Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015;7:181-93. doi:10.2147/clep.S37505 PubMed
30.Forsythe A, Vieweg DC, Marshall S, Tomaras D, Kim E. Pnd110 Symptoms, Delayed Diagnosis and Quality of Life (Qol) Deterioration - Exploring the Burden of Amyotrophic Lateral Sclerosis (Als). Value Health. 2020;23(Suppl 1):S280-S281. doi:10.1016/j.jval.2020.04.1000
31.Stenson K, Fecteau TE, O'Callaghan L, et al. Health-related quality of life across disease stages in patients with amyotrophic lateral sclerosis: results from a real-world survey. J Neurol. 2024;271(5):2390-2404. doi:10.1007/s00415-023-12141-y PubMed
32.Verschueren A, Kianimehr G, Belingher C, et al. Wish to die and reasons for living among patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(1-2):68-73. doi:10.1080/21678421.2018.1530265 PubMed
33.Health Canada. Fourth Annual Report on Medical Assistance in Dying in Canada 2022. October 2023. Accessed April 28, 2025. https://www.canada.ca/content/dam/hc-sc/documents/services/medical-assistance-dying/annual-report-2022/annual-report-2022.pdf
34.Stenson K, Cochrane T, O'Callaghan L, et al. Variation in age of onset and disease progression among genetic subsets of Amyotrophic Lateral Sclerosis (ALS) patients: Results from a real-world point-in-time survey [sponsor supplied reference]. Paper presented at: 32nd International Symposium on ALS/MND; Dec 7-10 2021; Northhampton, UK.
35.Andrews JA, Meng L, Kulke SF, et al. Association Between Decline in Slow Vital Capacity and Respiratory Insufficiency, Use of Assisted Ventilation, Tracheostomy, or Death in Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2018;75(1):58-64. doi:10.1001/jamaneurol.2017.3339 PubMed
36.Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler. 2010;11(1-2):178-80. doi:10.3109/17482960903093710 PubMed
37.Thakore NJ, Lapin BR, Pioro EP. Trajectories of impairment in amyotrophic lateral sclerosis: Insights from the Pooled Resource Open-Access ALS Clinical Trials cohort. Muscle Nerve. 2018;57(6):937-945. doi:10.1002/mus.26042 PubMed
38.de Wit J, Bakker LA, van Groenestijn AC, et al. Caregiver burden in amyotrophic lateral sclerosis: A systematic review. Palliat Med. 2018;32(1):231-245. doi:10.1177/0269216317709965 PubMed
39.Lillo P, Mioshi E, Hodges JR. Caregiver burden in amyotrophic lateral sclerosis is more dependent on patients' behavioral changes than physical disability: a comparative study. BMC Neurol. 2012;12:156. doi:10.1186/1471-2377-12-156 PubMed
40.Stenson K, Agnese W, de Silva S, et al. POSB356 Health-Related Quality of Life across King's and Mitos Stages Among Amyotrophic Lateral Sclerosis Patients: Results from a Real-World Point-in-Time Survey. Value Health. 2022;25(1):S229-S230. doi:10.1016/j.jval.2021.11.1123
41.Schepelmann K, Winter Y, Spottke AE, et al. Socioeconomic burden of amyotrophic lateral sclerosis, myasthenia gravis and facioscapulohumeral muscular dystrophy. J Neurol. 2010;257(1):15-23. doi:10.1007/s00415-009-5256-6 PubMed
42.Larkindale J, Yang W, Hogan PF, et al. Cost of illness for neuromuscular diseases in the United States. Muscle Nerve. 2014;49(3):431-8. doi:10.1002/mus.23942 PubMed
43.Nonoyama ML, McKim DA, Road J, et al. Healthcare utilisation and costs of home mechanical ventilation. Thorax. 2018;(73):644-651. doi:10.1136/thoraxjnl-2017-211138 PubMed
44.López-Bastida J, Perestelo-Pérez L, Montón-Alvarez F, Serrano-Aguilar P, Alfonso-Sanchez JL. Social economic costs and health-related quality of life in patients with amyotrophic lateral sclerosis in Spain. Amyotroph Lateral Scler. 2009;10(4):237-43. doi:10.1080/17482960802430781 PubMed
45.Lu CH, Macdonald-Wallis C, Gray E, et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247-57. doi:10.1212/wnl.0000000000001642 PubMed
46.Vacchiano V, Mastrangelo A, Zenesini C, et al. Plasma and CSF Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: A Cross-Sectional and Longitudinal Study. Front Aging Neurosci. 2021;13:753242. doi:10.3389/fnagi.2021.753242 PubMed
47.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-9. doi:10.1080/146608200300079536 PubMed
48.Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis:a systematic review. Arch Neurol. 2012;69(11):1410-6. doi:10.1001/archneurol.2012.254 PubMed
49.de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497-503. doi:10.1016/j.clinph.2007.09.143 PubMed
50.Turner MR. Diagnosing ALS: the Gold Coast criteria and the role of EMG. Pract Neurol. 2022;22(3):176-178. doi:10.1136/practneurol-2021-003256 PubMed
51.Shoesmith C, Abrahao A, Benstead T, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ. 2020;192(46):E1453-E1468. doi:10.1503/cmaj.191721 PubMed
52.Roggenbuck J, Eubank BHF, Wright J, Harms MB, Kolb SJ. Evidence-based consensus guidelines for ALS genetic testing and counseling. Ann Clin Transl Neurol. 2023;10(11):2074-2091. doi:10.1002/acn3.51895 PubMed
53.Van Damme P, Al-Chalabi A, Andersen PM, et al. European Academy of Neurology (EAN) guideline on the management of amyotrophic lateral sclerosis in collaboration with European Reference Network for Neuromuscular Diseases (ERN EURO-NMD). Eur J Neurol. 2024;31(6):e16264. doi:10.1111/ene.16264 PubMed
54.Biogen Canada Inc. Qalsody (tofersen): injection solution, 100 mg / 15 mL (6.7 mg/mL), for intrathecal use [product monograph]. 2025. Accessed May 30, 2025. https://pdf.hres.ca/dpd_pm/00078733.PDF
55.Mitsubishi Tanabe Pharma America, Inc. Radicava (edaravone): solution, 30mg/100mL (0.3mg/mL), intravenous administration [product monograph]. March 25, 2021. Accessed May 30, 2025. https://pdf.hres.ca/dpd_pm/00060555.PDF
56.Sanofi-aventis Canada Inc. Rilutek (riluzole): film-coated tablets, 50 mg [product monograph]. May 11, 2010. Accessed May 30, 2025. https://pdf.hres.ca/dpd_pm/00010806.PDF
57.Berdyński M, Miszta P, Safranow K, et al. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep. 2022;12(1):103. doi:10.1038/s41598-021-03891-8 PubMed
58.Prevention Genetics. Amyotrophic Lateral Sclerosis (ALS) Panel. Accessed November 06, 2025. https://www.preventiongenetics.com/tests/amyotrophic-lateral-sclerosis-als-panel
59.Udine E, Jain A, van Blitterswijk M. Advances in sequencing technologies for amyotrophic lateral sclerosis research. Mol Neurodegener. 2023;18(1):4. doi:10.1186/s13024-022-00593-1 PubMed
60.Pecoraro V, Mandrioli J, Carone C, Chiò A, Traynor BJ, Trenti T. The NGS technology for the identification of genes associated with the ALS. A systematic review. Eur J Clin Invest. 2020;50(5):e13228. doi:10.1111/eci.13228 PubMed
61.Prevention Genetics. Ionis Amyotrophic Lateral Sclerosis (ALS) Testing Program. Accessed April 28, 2025. https://www.preventiongenetics.com/sponsoredTesting/Ionis_ALS
62.Biogen. Clinical Study Report: 233AS101. A Study to Evaluate the Efficacy, Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of BIIB067 Administered to Adult Subjects with Amyotrophic Lateral Sclerosis and Confirmed Superoxide Dismutase 1 Mutation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2022.
63.Everett WH, Bucelli RC. Tofersen for SOD1 ALS. Neurodegener Dis Manag. 2024;14(5):149-160. doi:10.1080/17582024.2024.2402216 PubMed
64.Alberta Precision Laboratories. Amyotrophic Lateral Sclerosis (Synonyms: ALS, SOD1, C9ORF72, Lou Gehrig Disease). 2022. Accessed April 28, 2025. https://www.albertahealthservices.ca/webapps/labservices/indexAPL.asp?id=8520&tests=&zoneid=1&details=true
65.IWK Health. Clinical Genomics: Testing Menu. Accessed April 28, 2025. https://iwkhealth.ca/sites/default/files/2024-08/IWK%20CGL%20Test%20Menu%20-%20with%20restrictions%20and%20gene%20content.pdf
66.McGill University Health Centre. List of testing done out of Québec. 2025. Accessed April 28, 2025. https://muhc.ca/sites/default/files/docs/m-Labs/list-testing-done-out-quebec-2025-02-03.pdf
67.Shared Health Manitoba. Lab Information Manual. Familial amyotrophic lateral sclerosis (ALS) - SOD1 gene - (B). Accessed April 28, 2025. https://apps.sbgh.mb.ca/labmanual/test/view?seedId=13445
68.ALS Society of Canada. ALS Canada announces first-of-its-kind National ALS Genetic Counsellor with funding from TD Bank Group. 2024. Accessed May 07, 2025. https://als.ca/news/als-canada-announces-first-of-its-kind-national-als-genetic-counsellor-with-funding-from-td-bank-group/
69.Meyer T, Schumann P, Grehl T, et al. SOD1 gene screening in ALS - frequency of mutations, patients' attitudes to genetic information and transition to tofersen treatment in a multi-center program. Amyotroph Lateral Scler Frontotemporal Degener. 2025;26(1-2):162-171. doi:10.1080/21678421.2024.2401131 PubMed
70.Farhan SMK, Dilliott AA, Ghani M, et al. The ONDRISeq panel: custom-designed next-generation sequencing of genes related to neurodegeneration. NPJ Genom Med. 2016;1:16032. doi:10.1038/npjgenmed.2016.32 PubMed
71.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565-74. doi:10.1038/gim.2013.73 PubMed
72.Biogen. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
73.Miller TM, Cudkowicz ME, Genge A, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-1110. doi:10.1056/NEJMoa2204705 PubMed
74.Miller T, Cudkowicz ME, Genge A, et al. Evaluating Efficacy and Safety of Tofersen in Adults with SOD1-ALS: Results from the Phase 3 VALOR Trial and Open-Label Extension [presentation slides]. Paper presented at: ENCALS (2022) European Network for the Cure of Amyotrophic Lateral Sclerosis - 20th Meeting; June 1-3, 2022; Edinburgh, UK. Accessed April 28, 2025. https://www.mndassociation.org/media/796
75.Shaw P, Miller T, Cudkowicz M, et al. Tofersen in adults with SOD1-ALS: phase 3 VALOR trial and open-label extension results. J Neurol Neurosurg Psychiatry. 2022;93(9):e2.208. doi:10.1136/jnnp-2022-abn2.6
76.Biogen. A Study to Evaluate the Efficacy, Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of BIIB067 Administered to Adult Subjects with Amyotrophic Lateral Sclerosis and Confirmed Superoxide Dismutase 1 Mutation. Clinical Study Report. Part C only [sponsor supplied reference]. April 12, 2022.
77.Biogen. An Extension Study to Assess the Long-Term Safety, Tolerability, Pharmacokinetics, and Effect on Disease Progression of BIIB067 Administered to Previously Treated Adults with Amyotrophic Lateral Sclerosis Caused by Superoxide Dismutase 1 Mutation. Interim 2 [sponsor supplied reference]. May 09, 2022.
78.Proudfoot M, Jones A, Talbot K, Al-Chalabi A, Turner MR. The ALSFRS as an outcome measure in therapeutic trials and its relationship to symptom onset. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(5-6):414-25. doi:10.3109/21678421.2016.1140786 PubMed
79.Hamidou B, Marin B, Lautrette G, et al. Exploring the diagnosis delay and ALS functional impairment at diagnosis as relevant criteria for clinical trial enrolment. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(7-8):519-527. doi:10.1080/21678421.2017.1353098 PubMed
80.Gordon PH, Cheng B, Salachas F, et al. Progression in ALS is not linear but is curvilinear. J Neurol. 2010;257(10):1713-7. doi:10.1007/s00415-010-5609-1 PubMed
81.Gordon PH. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013;4(5):295-310. doi:10.14336/ad.2013.0400295 PubMed
82.Kaufmann P, Levy G, Thompson JL, et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology. 2005;64(1):38-43. doi:10.1212/01.Wnl.0000148648.38313.64 PubMed
83.Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. 2014;13(11):1127-1138. doi:10.1016/s1474-4422(14)70129-2 PubMed
84.European Medicines Agency. Guideline on clinical investigation of medicinal products for the treatment of amyotrophic lateral sclerosis (ALS). November 19, 2015. Accessed April 28, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-amyotrophic-lateral-sclerosis_en.pdf
85.U.S. Food and Drug Administration. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry. September 2019. Accessed April 28, 2025. https://www.fda.gov/media/130964/download
86.Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology. 2016;86(9):808-12. doi:10.1212/wnl.0000000000002251 PubMed
87.Bakers JNE, de Jongh AD, Bunte TM, et al. Using the ALSFRS-R in multicentre clinical trials for amyotrophic lateral sclerosis: potential limitations in current standard operating procedures. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23(7-8):500-507. doi:10.1080/21678421.2021.2016838 PubMed
88.Franchignoni F, Mora G, Giordano A, Volanti P, Chiò A. Evidence of multidimensionality in the ALSFRS-R Scale: a critical appraisal on its measurement properties using Rasch analysis. J Neurol Neurosurg Psychiatry. 2013;84(12):1340-5. doi:10.1136/jnnp-2012-304701 PubMed
89.van Eijk RPA, de Jongh AD, Nikolakopoulos S, et al. An old friend who has overstayed their welcome: the ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22(3-4):300-307. doi:10.1080/21678421.2021.1879865 PubMed
90.Fang T, Al Khleifat A, Stahl DR, et al. Comparison of the King's and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):227-232. doi:10.1080/21678421.2016.1265565 PubMed
91.Chio A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(1):38-44. doi:10.1136/jnnp-2013-306589 PubMed
92.Corcia P, Beltran S, Lautrette G, Bakkouche S, Couratier P. Staging amyotrophic lateral sclerosis: A new focus on progression. Rev Neurol (Paris). 2019;175(5):277-282. doi:10.1016/j.neurol.2018.09.017 PubMed
93.Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109-119. doi:10.1056/NEJMoa2003715 PubMed
94.Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130-139. doi:10.1002/ana.25276 PubMed
95.Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88(24):2302-2309. doi:10.1212/wnl.0000000000004029 PubMed
96.Pinto S, de Carvalho M. Correlation between Forced Vital Capacity and Slow Vital Capacity for the assessment of respiratory involvement in Amyotrophic Lateral Sclerosis: a prospective study. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(1-2):86-91. doi:10.1080/21678421.2016.1249486 PubMed
97.Baumann F, Henderson RD, Morrison SC, et al. Use of respiratory function tests to predict survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(1-2):194-202. doi:10.3109/17482960902991773 PubMed
98.Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10(5-6):310-23. doi:10.3109/17482960802566824 PubMed
99.Traynor BJ, Zhang H, Shefner JM, Schoenfeld D, Cudkowicz ME. Functional outcome measures as clinical trial endpoints in ALS. Neurology. 2004;63(10):1933-5. doi:10.1212/01.wnl.0000144345.49510.4e PubMed
100.Benatar M, Wuu J, Andersen PM, et al. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology. 2018;90(7):e565-e574. doi:10.1212/wnl.0000000000004960 PubMed
101.Cudkowicz ME, van den Berg LH, Shefner JM, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12(11):1059-67. doi:10.1016/s1474-4422(13)70221-7 PubMed
102.Shefner JM, Liu D, Leitner ML, et al. Quantitative strength testing in ALS clinical trials. Neurology. 2016;87(6):617-24. doi:10.1212/wnl.0000000000002941 PubMed
103.Jenkinson C, Fitzpatrick R. Reduced item set for the amyotrophic lateral sclerosis assessment questionnaire: development and validation of the ALSAQ-5. J Neurol Neurosurg Psychiatry. 2001;70(1):70-3. doi:10.1136/jnnp.70.1.70 PubMed
104.Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol. 1989;46(10):1121-3. doi:10.1001/archneur.1989.00520460115022 PubMed
105.McElhiney MC, Rabkin JG, Gordon PH, Goetz R, Mitsumoto H. Prevalence of fatigue and depression in ALS patients and change over time. J Neurol Neurosurg Psychiatry. 2009;80(10):1146-9. doi:10.1136/jnnp.2008.163246 PubMed
106.Rabkin JG, Gordon PH, McElhiney M, Rabkin R, Chew S, Mitsumoto H. Modafinil treatment of fatigue in patients with ALS: a placebo-controlled study. Muscle Nerve. 2009;39(3):297-303. doi:10.1002/mus.21245 PubMed
107.Ramirez C, Piemonte ME, Callegaro D, Da Silva HC. Fatigue in amyotrophic lateral sclerosis: frequency and associated factors. Amyotroph Lateral Scler. 2008;9(2):75-80. doi:10.1080/17482960701642502 PubMed
108.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169(1-2):13-21. doi:10.1016/s0022-510x(99)00210-5 PubMed
109.Shahim P, Norato G, Sinaii N, et al. Neurofilaments in Sporadic and Familial Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Genes (Basel). 2024;15(4):16. doi:10.3390/genes15040496 PubMed
110.Bacci ED, Staniewska D, Coyne KS, et al. Item response theory analysis of the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised in the Pooled Resource Open-Access ALS Clinical Trials Database. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(3-4):157-67. doi:10.3109/21678421.2015.1095930 PubMed
111.Bakker LA, Schroder CD, van Es MA, Westers P, Visser-Meily JMA, van den Berg LH. Assessment of the factorial validity and reliability of the ALSFRS-R: a revision of its measurement model. J Neurol. 2017;264(7):1413-1420. doi:10.1007/s00415-017-8538-4 PubMed
112.Cedarbaum JM, Stambler N. Performance of the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) in multicenter clinical trials. J Neurol Sci. 1997;152 Suppl 1:S1-9. doi:10.1016/s0022-510x(97)00237-2 PubMed
113.Zahir F, Hanman A, Yazdani N, et al. Assessing the psychometric properties of table 19 measures in individuals with amyotrophic lateral sclerosis: a systematic review. Qual Life Res. 2023;32(9):2447-2462. doi:10.1007/s11136-023-03377-2 PubMed
114.Jenkinson C, Fitzpatrick R, Swash M, Jones G. Comparison of the 40-item Amyotrophic Lateral Sclerosis Assessment Questionnaire (ALSAQ-40) with a short-form five-item version (ALSAQ-5) in a longitudinal survey. Clin Rehabil. 2007;21(3):266-72. doi:10.1177/0269215506071123 PubMed
115.Berry JD, Miller R, Moore DH, et al. The Combined Assessment of Function and Survival (CAFS): a new endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(3):162-8. doi:10.3109/21678421.2012.762930 PubMed
116.White IR, Babiker AG, Walker S, Darbyshire JH. Randomization-based methods for correcting for treatment changes: examples from the Concorde trial. Stat Med. 1999;18(19):2617-34. doi:10.1002/(sici)1097-0258(19991015)18:19<2617::aid-sim187>3.0.co;2-e PubMed
117.Ramamoorthy D, Severson K, Ghosh S, et al. Identifying patterns in amyotrophic lateral sclerosis progression from sparse longitudinal data. Nat Comput Sci. 2022;2(9):605-616. doi:10.1038/s43588-022-00299-w PubMed
118.Biogen. Statistical Analysis Plan - Study 233AS101 Part C [sponsor supplied reference]. August 14, 2021.
119.Biogen. Statistical Analysis Plan - 233AS102 [sponsor supplied reference]. February 02, 2022.
120.Biogen. Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use [internal sponsor's package]. 2025.
121.Biogen. CADTH reimbursement review sponsor summary of clinical evidence of Qalsody (tofersen) for the treatment of amyotrophic lateral sclerosis (ALS) associated with a mutation in the superoxide dismutase 1 (SOD1) gene [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
122.Biogen. Common Technical Document - 2.7.4 Summary of Clinical Safety [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
123.Biogen. Common Technical Document - 2.5 Clinical Overview [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
124.Biogen. Common Technical Document - 2.7.3 Summary of Clinical Efficacy [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
125.Biogen. Common Technical Document - 2.7.2 Summary of Clinical Pharmacology Studies [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use. 2025.
126.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
127.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
128.Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66(2):265-7. doi:10.1212/01.wnl.0000194316.91908.8a PubMed
129.Canada’s Drug Agency. Methods Guide for Health Technology Assessment. 2025. Accessed April 28, 2025. https://www.cda-amc.ca/sites/default/files/MG%20Methods/MG0030-Quantitative-Methods-Manual_Mar%202025.pdf
130.Hamad AA, Alkhawaldeh IM, Nashwan AJ, Meshref M, Imam Y. Tofersen for SOD1 amyotrophic lateral sclerosis: a systematic review and meta-analysis. Neurol Sci. 2025;17:17. doi:10.1007/s10072-025-07994-2 PubMed
131.Alirezaei Z, Pourhanifeh MH, Borran S, Nejati M, Mirzaei H, Hamblin MR. Neurofilament light chain as a biomarker, and correlation with magnetic resonance imaging in diagnosis of CNS-related disorders. Mol Neurobiol. 2020;57:469-491. doi:10.1007/s12035-019-01698-3 PubMed
132.Benatar M, Macklin EA, Malaspina A, et al. Prognostic clinical and biological markers for amyotrophic lateral sclerosis disease progression: validation and implications for clinical trial design and analysis. EBioMedicine. 2024;108:105323. doi:10.1016/j.ebiom.2024.105323 PubMed
133.Witzel S, Frauhammer F, Steinacker P, et al. Neurofilament light and heterogeneity of disease progression in amyotrophic lateral sclerosis: development and validation of a prediction model to improve interventional trials. Transl Neurodegener. 2021;10(1):31. doi:10.1186/s40035-021-00257-y PubMed
134.Witzel S, Statland JM, Steinacker P, et al. Longitudinal course of neurofilament light chain levels in amyotrophic lateral sclerosis-insights from a completed randomized controlled trial with rasagiline. Eur J Neurol. 2024;31(3):e16154. doi:10.1111/ene.16154 PubMed
135.Liu Y, Atkinson RA, Fernandez-Martos CM, et al. Changes in TDP-43 expression in development, aging, and in the neurofilament light protein knockout mouse. Neurobiol Aging. 2015;36(2):1151-9. doi:10.1016/j.neurobiolaging.2014.10.001 PubMed
136.Thouvenot E, Demattei C, Lehmann S, et al. Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur J Neurol. 2020;27(2):251-257. doi:10.1111/ene.14063 PubMed
137.Halbgebauer S, Steinacker P, Verde F, et al. Comparison of CSF and serum neurofilament light and heavy chain as differential diagnostic biomarkers for ALS. J Neurol Neurosurg Psychiatry. 2022;93(1):68-74. doi:10.1136/jnnp-2021-327129 PubMed
138.Gagliardi D, Rizzuti M, Masrori P, et al. Exploiting the role of CSF NfL, CHIT1, and miR-181b as potential diagnostic and prognostic biomarkers for ALS. J Neurol. 2024;271(12):7557-7571. doi:10.1007/s00415-024-12699-1 PubMed
139.Witzel S, Huss A, Nagel G, et al. Population-Based Evidence for the Use of Serum Neurofilaments as Individual Diagnostic and Prognostic Biomarkers in Amyotrophic Lateral Sclerosis. Ann Neurol. 2024;96(6):1040-1057. doi:10.1002/ana.27054 PubMed
140.Khalil M, Teunissen CE, Lehmann S, et al. Neurofilaments as biomarkers in neurological disorders - towards clinical application. Nat Rev Neurol. 2024;20(5):269-287. doi:10.1038/s41582-024-00955-x PubMed
141.Verde F, Otto M, Silani V. Neurofilament Light Chain as Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2021;15:679199. doi:10.3389/fnins.2021.679199 PubMed
142.Benatar M, Ostrow LW, Lewcock JW, et al. Biomarker Qualification for Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: Theory and Practice. Ann Neurol. 2024;95(2):211-216. doi:10.1002/ana.26860 PubMed
143.University of Ulm. NCT02306590: Efficacy, Safety and Tolerability of High Lipid and Calorie Supplementation in Amyotrophic Lateral Sclerosis. ClinicalTrials.gov; 2019. Accessed April 15, 2025. https://www.clinicaltrials.gov/study/NCT02306590
144.Esselin F, De la Cruz E, Hirtz C, et al. Repeated neurofilament light chain measurements did not capture Riluzole therapeutic effect in amyotrophic lateral sclerosis patients. CNS Neurosci Ther. 2022;28(10):1532-1538. doi:10.1111/cns.13894 PubMed
145.Paganoni S, Macklin EA, Hendrix S, et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med. 2020;383(10):919-930. doi:10.1056/NEJMoa1916945 PubMed
146.Center for Drug Evaluation Research. Integrated review. Qalsody (tofersen) injection, solution. Company: Biogen, Inc. Application No.: 215887. Approval date: 04/25/2023 (FDA approval package). U.S. Food and Drug Administration (FDA); 2022. Accessed April 15, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/215887Orig1s000IntegratedR.pdf
147.Committee for Medicinal Products for Human Use. Assessment report: Qalsody (tofersen) (European public assessment report). European Medicines Agency; 2024. Accessed April 15, 2025. https://www.ema.europa.eu/en/documents/assessment-report/qalsody-epar-public-assessment-report_en.pdf
148.Benatar M, McDermott C, Turner MR, van Eijk RPA. Rethinking phase 2 trials in amyotrophic lateral sclerosis. Brain. 2024;06:06. doi:10.1093/brain/awae396
149.Carroll E, Scaber J, Huber KVM, et al. Drug repurposing in amyotrophic lateral sclerosis (ALS). Expert Opin Drug Discov. 2025;20(4):447-464. doi:10.1080/17460441.2025.2474661 PubMed
150.Massachusetts General Hospital. HEALEY ALS Platform Trial. 2023. Accessed April 13, 2025. https://www.massgeneral.org/assets/MGH/pdf/neurology/als/GeneralPlatform.June2023.pdf
151.UK MND Research Institute. EXPErimental Medicine Route to Success in Amyotrophic Lateral Sclerosis: EXPERTS-ALS. 2023. Accessed April 13, 2025. https://www.experts-als.uk/about-experts-als
152.Elman LB, McCluskey L. Post TW, ed. Diagnosis of amyotrophic lateral sclerosis and other forms of motor neuron disease. UptoDate; 2024. Accessed March 28, 2025. https://www.uptodate.com/
Appendix 1: Use of NfL
Please note that this appendix has not been copy-edited.
What Is NfL?
NfL is a protein found in protective nerve fibres (myelinated axons) that enters the blood and CSF after nerve damage or degeneration.131 Elevated levels of NfL are observed in neurologic disorders including ALS, Alzheimer disease, frontotemporal dementia, Huntington’s disease, and multiple sclerosis.132 NfL levels are detected in CSF, as well as blood, serum, or plasma samples.131
Use as a Diagnostic, Prognostic, or Response Biomarker
There is a large body of evidence demonstrating the relevance of NfL in supporting ALS diagnosis, disease progression rate, and survival.133-138 Neurofilaments are not specific to ALS and cannot be used alone to confirm a diagnosis,139 however, NfL levels can support the diagnostic process.11 The literature suggests that NfL displays diagnostic accuracy in distinguishing patients with ALS from controls, with high sensitivity and specificity.109,140 CSF NfL levels in ALS exceed those found in in most neurologic diseases, assisting with differentiating it from other motor neuron diseases.141 The literature supports the use of NfL as a prognostic marker, suggesting that baseline plasma or serum NfL levels may predict future decline and survival in patients with ALS.142 A meta-analysis found that higher NfL levels in the blood or CSF were weakly correlated with lower (worse) ALFSRS-R scores and moderately correlated with faster disease progression (Table 8).109 Higher NfL levels in the blood were also associated with shorter survival duration (time from symptom onset to permanent assisted ventilation, tracheostomy, or death).45,46,109
NfL shows promise as a response biomarker in ALS, though evidence remains limited. NfL levels have dropped with high-fat, high-calorie supplements in 1 ALS trial (the LIPCAL-ALS trial)143 and the utility of neurofilaments have been demonstrated in trials in other motor neuron diseases such as spinal muscular atrophy.142 The lack of strong evidence for NfL as a response biomarker in ALS may be due to the absence of highly effective treatments. Notably, NfL levels did not decrease in patients treated with riluzole144 nor was a reduction in a related biomarker pNfH observed in the phase II trial for sodium phenylbutyrate- ursodoxicoltaurine (Albrioza).142,145 It is still unclear whether these results reflect the limited effectiveness of the therapies or the low sensitivity of NfL to drugs with different mechanisms of action.142
Age has been found to be a potential confounder when interpreting NfL levels.139 While most studies found little to no correlation between age and CSF or serum NfL levels in patients with ALS, age is known to correlate with NfL levels in healthy individuals and those with other neurologic conditions. This is likely due to the significant NfL increase in ALS from rapid motor neuron degeneration, surpassing the mild axonal loss due to aging.141 Furthermore, NfL behaviour in slower-progressing and clinically asymptomatic phase of disease is not well characterized.121 The clinical expert consulted for this review also noted uncertainty about whether patients with low NfL levels often linked to slower disease progression would benefit from a given treatment. Therefore, it remains unclear if NfL levels can reliably predict treatment response in all patients with ALS.
Surrogate Marker of Clinical Benefit
The FDA integrated review of tofersen146 conducted in 2023 recognized “NfL concentrations as a surrogate end point reasonably likely to predict clinical benefit in patients with SOD1-ALS.” The review considered the prognostic value of plasma NfL in ALS progression and survival and the correlation between NfL reduction and a decrease in clinical decline as measured by the ALSFRS-R.146 The European Medicines Agency assessment report of tofersen in 2024 noted that (based on their consultation with a Scientific Advisory Group on Neurology) NfL is not yet a validated surrogate marker for measuring treatment effectiveness in ALS.147 However, they acknowledged that NfL is increasingly recognized as an important biomarker in neurodegenerative diseases used to assess the PD effects of treatments. The group noted that uncertainties remain regarding the timeline between NfL changes and clinical outcomes because clinical benefits from fluid biomarker changes may take years to confirm. Furthermore, the European Medicines Agency highlights the absence of a standardized NfL threshold to define a clinically meaningful effect beyond survival benefit.147
Use in Clinical Trials
Biomarkers like NfL are increasingly recognized as tools to assist with developing therapies, strengthening biological reasoning, and selecting drug candidates for phase III trials.148 The literature states that a significant reduction in NfL following an experimental treatment is a promising indicator that may better inform phase II advancement decisions than traditional clinical outcome measures like ALSFRS-R.148 To date, such outcome measures have been poorly predictive of phase III outcomes in ALS clinical trials.148 There is growing interest in leveraging NfL for faster drug screening, where a predefined reduction in mean NfL levels could guide “go” or “no-go” decisions for advancing to definitive trials.149 Unlike traditional trials testing 1 treatment at a time, platform trials evaluate multiple treatments simultaneously, reducing the time between identifying promising treatments and testing them in patients with ALS. Such endeavours include the HEALEY ALS Platform Trial in the US, led by the Healey & AMG Center at Massachusetts General Hospital.150 The HEALEY ALS Platform Trial has 70 sites in the US and are enrolling 160 to 240 patients per treatment regimen.150 Also, the EXPErimental Medicine Route to Success in Amyotrophic Lateral Sclerosis project in the UK,149 is using Bayesian modelling to evaluate up to 3 repurposed ALS drugs at a time.151 It aims to detect a 30% or greater NfL reduction (“go”) or ineffectiveness (“no-go”) within 3 to 6 months, with 30 to 75 patients per treatment. While it cannot confirm clinical benefit, treatments showing significant NfL reduction can be prioritized for phase III trials, leveraging NfL as a potential surrogate marker of clinical impact.149
Use in Clinical Settings
Although the use of NfL levels are becoming more common in clinical trials, their use in the clinical setting for the diagnosis of ALS is still uncertain.152 Recent improvements in immunoassay technology now allow highly sensitive measurement of NfL in the blood, enabling accurate differentiation between patients with ALS and controls.109 The clinical experts consulted for this review noted that such tests for NfL are not widely available in Canada. They stated that there is no current evidence to support their use for individual clinical decisions as they have only been evaluated using aggregate data.
Next Steps
NfL, although not ALS-specific, has been shown in the literature to be a potential marker of ALS disease progression and survival.133-138 Evidence supports using NfL as a diagnostic, prognostic, and potential response biomarker in the general ALS population.142 While it may not yet universally qualify as a surrogate end point,147 NfL can guide go/no-go decisions in phase II trials, as are being undertaken in the US and the UK to help prioritize which treatments should proceed to more intensive testing.151 It should be noted that the clinical experts consulted for this reviews noted that NfL currently plays a limited role in the clinical management of patients, is not routinely used in practice in Canada, and lacks established clinical utility at this time.
Importantly, current literature suggests that establishing an internationally agreed-upon definition of a significant reduction in biomarkers such as NfL is crucial for future studies.148 Caution is warranted when generalizing NfL data from individuals with fast-progressing, highly penetrant SOD1 mutations to other genetic subtypes. NfL trajectories may differ by mutation, and a rise in NfL alone may not reliably predict disease onset in at-risk individuals.142 Also, it is still unclear whether initiating a treatment after signs of axonal degeneration but before clinical symptoms is sufficient enough to alter the disease course.142 Future strategies may require biomarkers that detect earlier cellular or molecular dysfunction to guide even more timely interventions.142
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ALS
amyotrophic lateral sclerosis
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
ICER
incremental cost-effectiveness ratio
MiToS
Milano-Torino Staging
QALY
quality-adjusted life-year
Economic Review
The objective of the economic review is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of tofersen (in conjunction with background therapy) compared to background therapy alone for the treatment of SOD1 amyotrophic lateral sclerosis (ALS) in adults. This indication is in line with the sponsor’s requested reimbursement population.
Item | Description |
|---|---|
Drug product | Tofersen (Qalsody), 100 mg per 15 mL, solution for intrathecal injection |
Indication | Qalsody (tofersen injection) is indicated for the treatment of adults with amyotrophic lateral sclerosis (ALS) associated with a mutation in the superoxide dismutase 1 (SOD1) gene |
Submitted price | $28,370.68 per vial |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard |
NOC date | February 28, 2025 |
Reimbursement request | For the treatment of adults with ALS associated with a mutation in the SOD1 gene who:
|
Sponsor | Biogen Canada Inc. |
Submission history | Previously reviewed: No |
ALS = amyotrophic lateral sclerosis; NOC/c = Notice of Compliance with conditions.
Key Messages
Tofersen is available as 100 mg per 15 mL vials for intrathecal injection. At the submitted price of $28,370.68 per vial, the annual cost of tofersen is expected to be $425,560 per patient in the first year of treatment and $368,819 in subsequent years, based on the Health Canada–recommended dosage.
Comparative clinical efficacy in the economic analysis was derived from the VALOR trial (part C), which compared tofersen to placebo. Evidence submitted by the sponsor indicates that tofersen may result in little or no clinically meaningful difference compared to placebo for change in the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R) score.
The results of the Canada’s Drug Agency (CDA-AMC) base case suggest:
Tofersen plus background therapy will be associated with higher costs to the health care system than background therapy alone (incremental costs = $1,205,885), primarily driven by increased costs associated with tofersen.
Tofersen plus background therapy will be associated with a gain of 1.93 life-years compared to background therapy alone. When the impact on health-related quality of life is also considered, tofersen plus background therapy may result in a gain of 0.76 quality-adjusted life-years (QALYs) compared to background therapy alone.
The incremental cost-effectiveness ratio (ICER) of tofersen plus background therapy compared to background therapy alone was $1,586,423 per QALY gained. The estimated ICER is highly uncertain because of the uncertainty in the comparative efficacy, economic model structure, and assumptions used in the sponsor’s societal analysis.
Given the identified limitations with the submitted model structure, the economic analysis may not accurately assess the impact on patient health and health care resources. Thus, the cost-effectiveness estimates are highly uncertain.
Results from the analyses from both the sponsor and CDA-AMC suggest that, with substantial price reductions (i.e., 90%), the ICER for tofersen plus background therapy remains greater than $250,000 per QALY gained. There are 2 reasons for this result. This is partly due to more non–drug-related costs associated with tofersen plus background therapy. This is further due to the predicted improvement in life expectancy associated with tofersen, which results in both additional drug acquisition costs and non–drug-related costs (drug administration, monitoring, and the management of side effects) during this prolonged survival time. Even if drug acquisition costs of tofersen are substantially lowered, there will still be high costs to the health care system as tofersen treatment is associated with background therapy costs and costs pertaining to drug administration, monitoring, and the management of side effects (approximately $51,000).
The sponsor submitted a base-case analysis conducted from the societal perspective. Due to the lack of robust evidence to support this approach, CDA-AMC was unable to consider a societal perspective.
CDA-AMC estimates that the budget impact of reimbursing tofersen plus background therapy for the treatment of adult patients with SOD1-ALS will be approximately $36,169,911 over the first 3 years of reimbursement compared to the amount currently spent on background therapy alone, with an estimated expenditure of $37,446,934 on tofersen plus background therapy over this period. The actual budget impact of reimbursing tofersen will depend on the number of people eligible for treatment.
While genetic testing for SOD1 mutations is currently performed as part of standard of care for patients with ALS, clinical expert feedback received by CDA-AMC indicated that there are access differences across jurisdictions that may impact the feasibility of accessing tofersen. The budget impact analysis (BIA) assumes all patients receive genetic testing. However, if there are access issues associated with genetic testing, the budget impact may be overestimated.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of tofersen plus background therapy from the perspective of a public drug plan payer in Canada and from a societal perspective over a lifetime horizon (10 years). The modelled population comprised adult patients with SOD1-ALS, which is aligned with the Health Canada indication and reimbursement request. This population was based on the participants in part C of the VALOR trial. The sponsor’s base-case analysis included costs related to drug acquisition, monitoring and administration, disease management, and adverse event (AE) management. The sponsor’s societal base case included additional indirect costs. In the sponsor’s base case, which adopted a health care payer perspective, tofersen plus background therapy was associated with incremental costs of $1,177,401 and 0.76 incremental QALYs relative to background therapy alone. This resulted in an ICER of $1,556,441 per QALY gained. From a societal perspective, the ICER was $2,291,756 per QALY gained. Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4). A revised base case was therefore developed.
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The comparative efficacy of tofersen plus background therapy compared to background therapy alone is uncertain. | Based on the primary 28-week data from the VALOR trial (part C), tofersen may result in little or no clinically meaningful difference compared to placebo for functional decline as measured by ALSFRS-R, respiratory function, and muscle strength. There remains significant uncertainty in the comparative effectiveness of tofersen plus background therapy vs. background therapy alone. | This issue could not be addressed. | No scenario analysis was conducted due to the lack of alternative evidence. |
The submitted economic model is not reflective of clinical practice. | The sponsor-submitted model is based on the MiToS staging system, which is not used in clinical practice because it fails to appropriately capture the clinical presentation of patients. | This issue could not be addressed. | No scenario analysis was conducted because alternatives (i.e., King’s staging) were deemed similarly inappropriate based on clinical input received by CDA-AMC. |
The drug cost of tofersen is underestimated. | Based on the Health Canada product monograph, year 1 of tofersen treatment should consist of 15 administrations if assuming a 365-day year.1 | CDA-AMC assumed 3 administrations in cycle 1 as a proxy to correct for the number of administrations in year 1 of tofersen treatment. | No scenario analysis was conducted because best evidence was used in the CDA-AMC base case. |
Relevant considerations were missing from the societal perspective analysis. | Productivity loss was not considered in the societal perspective. ALS can impact working-age patients and/or caregivers as suggested in a 2014 Canadian study examining the economic burden of ALS.14 It was noted that annual indirect costs (i.e., loss of wages) for patients with ALS and family members providing care was approximately $56,821 (2014 values).14 | The CDA-AMC base case focused on a health care payer perspective and omitted societal costs due to the high degree of uncertainty. | No scenario analysis was conducted. |
Indirect costs included in the societal perspective analysis are uncertain. | Indirect costs associated with the health states were informed by Ploug et al.; these values lacked face validity and could not be validated.2 | The CDA-AMC base case focused on a health care payer perspective and omitted societal costs due to the high degree of uncertainty. | No scenario analysis was conducted. |
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CDA-AMC = Canada’s Drug Agency; MiToS = Milano-Torino Staging.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 4.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 6), in consultation with clinical experts. Detailed information about the base case is provided in Appendix 4.
Impact on Health Care Costs
Tofersen plus background therapy is expected to be associated with additional health care costs compared to background therapy alone (incremental costs = $1,205,885). This increase in health care spending results from drug acquisition costs associated with tofersen (refer to Figure 1). Although they constitute a small amount of the relative total, treatment with tofersen also results in an additional $8,738 to the health system specific to drug administration, monitoring, and the management of tofersen side effects.
Figure 1: Impact of Tofersen Plus Background Therapy vs. Background Therapy Alone on Health Care Costs
vs. = versus.
Note: Results presented are from the health care payer perspective; please refer to Appendix 4 for more results.
Impact on Health
Relative to background therapy alone, tofersen plus background therapy is expected to increase overall survival by 1.93 years (refer to Figure 2). Considering the impact of treatment on both quality and length of life, tofersen plus background therapy is expected to result in 0.76 additional QALYs per patient compared to background therapy alone over the lifetime horizon.
Figure 2: Impact of Tofersen Plus Background Therapy vs. Background Therapy on Patient Health
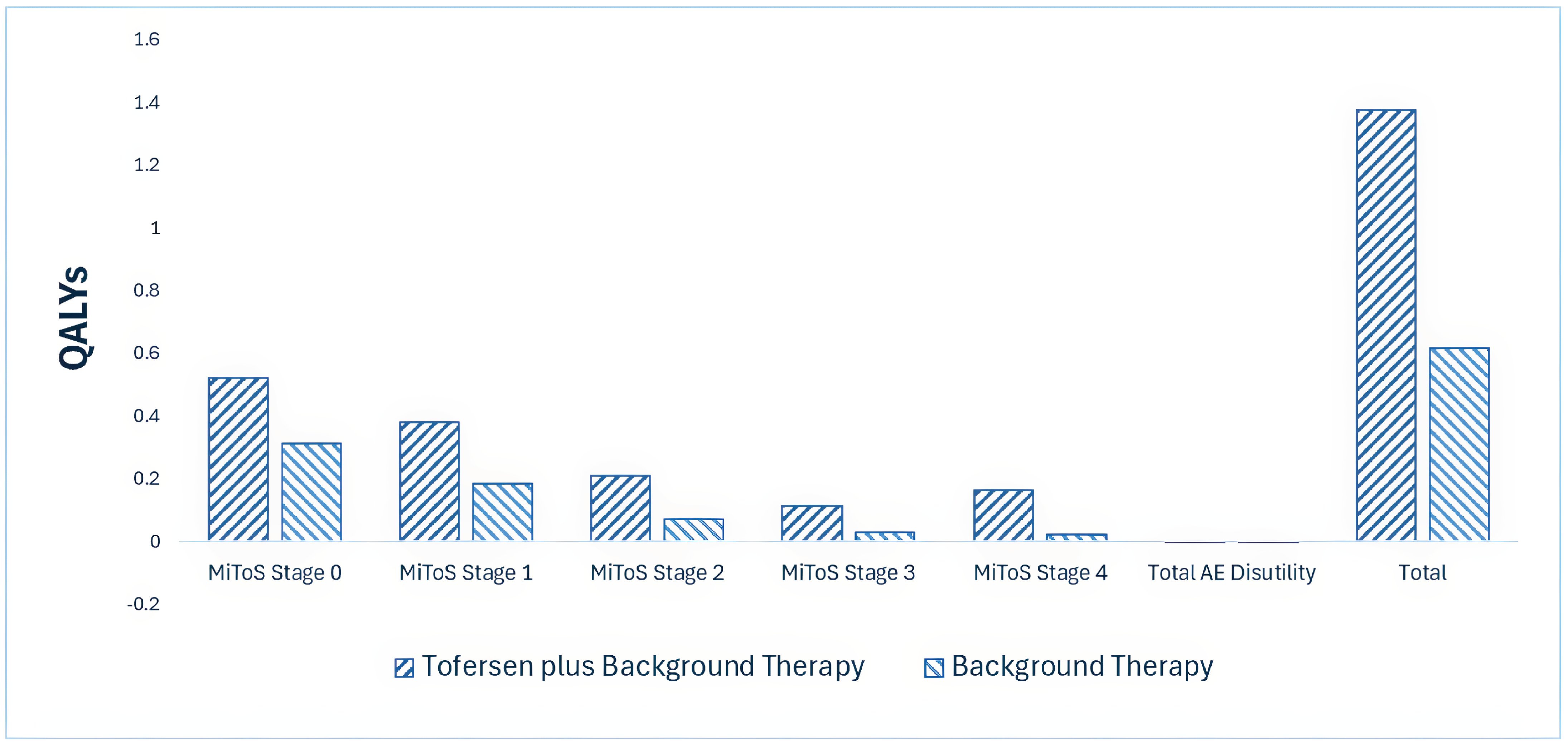
AE = adverse event; MiToS = Milano-Torino Staging; QALY = quality-adjusted life-year; vs. = versus.
Note: Results presented are from the health care payer perspective; please refer to Appendix 4 for more results.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $1,586,423 per QALY gained for tofersen plus background therapy compared to background therapy alone (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. background therapy ($/QALY) |
|---|---|---|---|
Publicly funded health care payer perspective | |||
Background therapy | 39,585 | 0.617 | Reference |
Tofersen plus background therapy | 1,245,469 | 1.377 | 1,586,423 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
aIncludes costs related to indirect costs (i.e., transport to appointments and hospital visits, transport modifications, home modifications, supportive devices, testing, hospital visits, and care home and assisted living) in addition to all costs included in the publicly funded health care payer perspective.
Pagebreak
Summary of the Budget Impact
The sponsor submitted a BIA to estimate the 3-year (2026 to 2028) budget impact of reimbursing tofersen for the treatment of adult patients with SOD1-ALS. The sponsor assumed that the payer would be CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The unit price of tofersen was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 3.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA-AMC base case (Appendix 5). CDA-AMC estimated that 104 patients would be eligible for treatment with tofersen plus background therapy over a 3-year period (year 1 = 34 patients; year 2 = 35 patients; year 3 = 35 patients). The estimated incremental budget impact of reimbursing tofersen is expected to be approximately $36,169,911 over the first 3 years, with an expected expenditure of $37,446,934 on tofersen plus background therapy. The actual budget impact will depend on the number of people eligible for treatment and the uptake of tofersen.
Conclusion
Based on the CDA-AMC base case, tofersen plus background therapy would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $1,586,423 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 9). The estimated cost-effectiveness of tofersen plus background therapy compared to background therapy alone is uncertain due to uncertainty in the comparative efficacy of tofersen compared to background therapy alone and limitations with the submitted economic model because it did not reflect clinical practice. In the base-case analyses from the sponsor and CDA-AMC, approximately 98% of the incremental QALYs for tofersen plus background therapy were found to be accrued during the extrapolated period of the VALOR trial. This clinical benefit should be interpreted with caution given the lack of data to confirm this modelled benefit.
The budget impact of reimbursing tofersen plus background therapy to the public drug plans in the first 3 years is estimated to be approximately $36.2 million. The 3-year expenditure on tofersen (i.e., not accounting for current expenditure on comparators) is estimated to be $37.4 million. The true budget impact of reimbursing tofersen plus background therapy will depend on the confidential negotiated prices of available therapies.
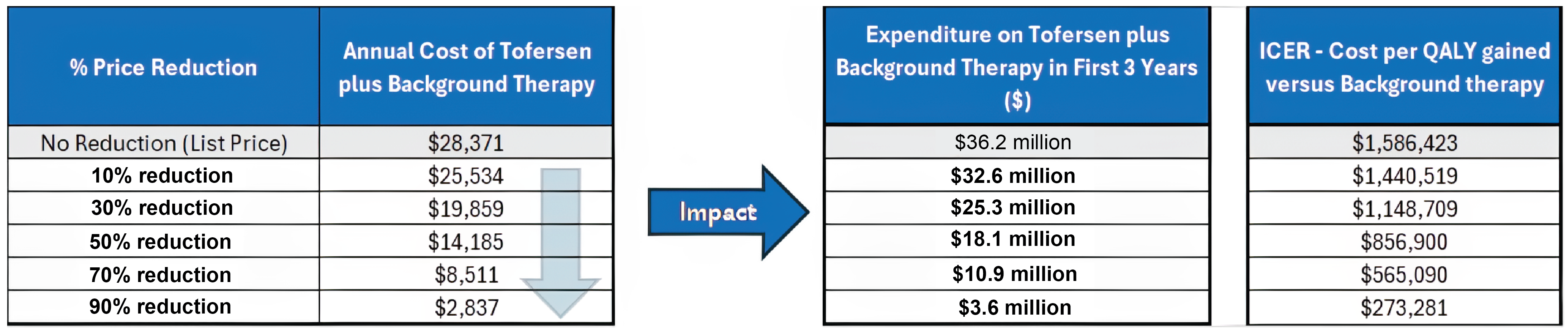
References
1.Biogen Canada Inc. Qalsody (tofersen injection): solution, 100 mg / 15 mL (6.7 mg/mL), for intrathecal use [product monograph]. 2023. Updated February 28, 2025.
2.Ploug U, Savic N, Mellor J, Bonar K. EE514 Treatment-Related and Non-Treatment-Related Out-of-Pocket Costs by Amyotrophic Lateral Sclerosis Disease Stage: A Cross-Sectional Patient Survey and Retrospective Chart Review. Value Health. 2022;25(12 Suppl):S157. doi:10.1016/j.jval.2022.09.756
3.Biogen Canada Inc. Drug Reimbursement Review sponsor submission: Qalsody (tofersen injection), solution, 100 mg/15 mL (6.7 mg/mL), for intrathecal use [internal sponsor's package]. 2025.
4.Ontario Ministry of Health. Exceptional Access Program (EAP). Accessed March 25, 2025. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
5.Thakore NJ, Lapin BR, Kinzy TG, Pioro EP. Deconstructing progression of amyotrophic lateral sclerosis in stages: a Markov modeling approach. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(7-8):483-494. doi:10.1080/21678421.2018.1484925 PubMed
6.Opie-Martin S, Iacoangeli A, Topp SD, et al. The SOD1-mediated ALS phenotype shows a decoupling between age of symptom onset and disease duration. Nat Commun. 2022;13(1):6901. doi:10.1038/s41467-022-34620-y PubMed
7.Moore A, Young CA, Hughes DA. Health utilities and costs for motor neurone disease. Value Health. 2019;22(11):1257-1265. doi:10.1016/j.jval.2019.05.011 PubMed
8.National Institute for Health and Care Excellence (NICE). Ponesimod for treating relapsing–remitting multiple sclerosis (Technology appraisal guidance TA767) [sponsor supplied reference]. 2022. https://www.nice.org.uk/guidance/ta767
9.Stenson K, Agnese W, de Silva S, et al. POSB356 Health-related quality of life across King's and MitoS stages among amyotrophic lateral sclerosis patients: results from a real-world point-in-time survey. Value Health. 2022;25(1):S229-S230. doi:10.1016/j.jval.2021.11.1123
10.Ontario Ministry of Health. Ontario drug benefit formulary/comparative drug index. Accessed April 3, 2025. https://www.formulary.health.gov.on.ca/formulary/
11.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (February 14, 2025 (effective March 3, 2025)). 2025. Accessed April 7, 2025. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
12.Rampersaud YR, Power JD, Perruccio AV, et al. Healthcare utilization and costs for spinal conditions in Ontario, Canada - opportunities for funding high-value care: a retrospective cohort study. Spine J. 2020;20(6):874-881. doi:10.1016/j.spinee.2020.01.013 PubMed
13.Biogen Canada Inc. Biogen Canada Inc. response to May 6, 2025 request for additional information regarding Qalsody (tofersen) CDA-AMC review [internal additional sponsor's information]. May 13, 2025.
14.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender [sponsor supplied reference]. 2024. https://doi.org/10.25318/1710000501-eng
15.CADTH. CADTH Reimbursement Review: Edaravone Oral Suspension (Radicava) [sponsor supplied reference]. Can J Health Technol. 2023;3(2):1-77. doi:10.51731/cjht.2023.583
16.Miller TM, Cudkowicz ME, Genge A, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099-1110. doi:10.1056/NEJMoa2204705 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and CDA-AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans
Table 4: Cost Comparison for ALS Associated With an SOD1 Gene
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Tofersen (Qalsody) | 100 mg/ 15 mL | Vial | 28,370.6776a | Loading: 100 mg on days 0, 14, and 28 Maintenance: 100 mg every 28 days thereafter | Year 1: 1,165.92 Year 2+: 1,010.46 | Year 1: 425,560c Year 2+: 368,819c |
Background Therapy | ||||||
Edaravone (Radicava) | 105 mg/ 5 mL | Oral suspension | $189.5200/ mL |
| Year 1: 347.89 Year 2 + 337.50 | Year 1: 126,978 Year 2+: 123,188 |
30 mg/ 100mL | Solution for IV infusion | $947.6000 per 2 × 30 mg bags |
| |||
Riluzole | 50 mg | Tablet | 7.3630 | 50 mg every 12 hours | 14.73 | 5,375 |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2025), unless otherwise indicated, and do not include dispensing fees. All costs assume 365 days annually, unless otherwise noted.
aSponsor submitted3
bExceptional access program4
cAnnual drug costs calculated assuming thirteen 28-day cycles (i.e., 364 days)
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA‑AMC review process.
Patient input was received from ALS Action Canada Society, ALS Society of Alberta, ALS Society of British Columbia, and ALS Society of Canada. Information from patients or caregivers in Canada was gathered via interviews and surveys. Patient input noted that ALS impacts many aspects of life including daily functionality and emotional well-being. Commonly reported physical symptoms include muscle weakness leading to difficulties in speaking, swallowing, and breathing; difficulty with balance; cramping; pain and fatigue. Patient input noted that while 2 therapies are currently available for ALS in Canada (i.e., riluzole and edaravone), they do not significantly alter disease progression. Of patients with experience with tofersen, many noted stabilizations in their ALS symptoms and maintenance of daily functioning. Many patients further commented on the logistical and physical challenges of tofersen treatment, including the burden of travel and adverse events associated with treatment injection. Lastly, patient input noted the importance of early genetic testing that is essential for prompt intervention with treatments such as tofersen.
Clinician group input was received from The Canadian ALS Research Network. Clinician input noted that the only currently available disease-modifying treatments in Canada are riluzole and edaravone (oral and IV). However, these treatments only provide modest benefits in slowing disease progression therefore additional treatment options are needed. Clinician input noted that “treatment response” is difficult to define for ALS as the goal of treatment is to slow the degeneration of motor neurons. Treatment with tofersen could be considered until the focus of care shifts to palliative and supportive treatment.
Input from CDA-AMC–participating drug plans inquired if there were standard tests that could be used to measure outcomes associated with SOD1 mutations and neurofilament light chain. They further questioned what criteria would be used to inform discontinuation of therapy and if patients could be re-treated with tofersen if they previously discontinued treatment. Lastly, due to the intrathecal administration of tofersen, the drug plants inquired if there were any specific administration requirements that should be considered.
Several of these concerns were addressed in the sponsor’s model:
The impact of ALS on patient’s quality of life was captured through utility values measured through EQ‑5D which captures domains of mobility, usual activities, self-care, pain/discomfort, and anxiety/depression.
Adverse events such as limb pain, back pain, radiculitis, and myelitis were considered in the economic analysis.
The submitted model included the option to consider genetic testing costs.
CDA-AMC was unable to validate that the following concerns were adequately captured:
While transport to appointments/hospital visits and transport modification were part of the cost parameters captured under the model’s societal perspective, CDA—AMC could not validate the provided source.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA‑AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of tofersen, the sponsor provided a cost-utility analysis and a budget impact analysis. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Tofersen (Qalsody), intrathecal solution, (100 mg/ 15 mL) |
Submitted price of drug under review | $28,370.6776 per vial |
Regimen | Loading: 100 mg on days 0, 14, and 28 Maintenance: 100 mg every 28 days thereafter |
28-day cycle cost of drug under review | $56,741.36 in cycle 1a $28,370.68 in cycle 2+ |
Model information | |
Type of economic evaluation | Cost-utility analysis Markov model |
Treatment | Tofersen as a monotherapy or with background therapy |
Included comparator | Background therapy defined as riluzole and/or edaravone |
Perspective | Publicly funded health care payer perspective Societal perspective |
Time horizon | Lifetime (10 years) |
Cycle length | 4 weeks |
Modelled population | Adult patients with SOD1-ALS |
Characteristics of modelled population | Derived from Part C of the VALOR trial:
|
Model health states | Health states defined by MiToS staging system as follows:
For additional information, refer to Model Structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs included in the model |
|
Summary of the submitted results | |
Base case results |
|
Scenario analysis resultsb |
|
AE = adverse events; ALS = amyotrophic lateral sclerosis HR = hazard ratio; ICER = incremental cost-effectiveness ratio; MiToS = Milano-Torino functional scaling; OLE = open-label extension; QALY = quality-adjusted life-years.
aSponsor assumes that cycle 1 only has 2 doses of tofersen.
bResults of scenario analyses that had a meaningful impact on the estimated ICER compared to the sponsor’s base case. Additional scenarios were submitted that had no meaningful impact on the estimated ICER.
Model Structure
The sponsor submitted a Markov model with 6 health states based on the MiToS staging system (i.e., Stage 0, Stage 1, Stage 2, Stage 3, Stage 4, and death; Figure 4). Patients entered the model based on baseline distributions derived from the VALOR (Part C) trial. At each cycle, patients could either remain in the same health state, progress to a more severe health state, or die. In the base-case analysis, patients could not experience regression (i.e., could not transition to a less severe health state).
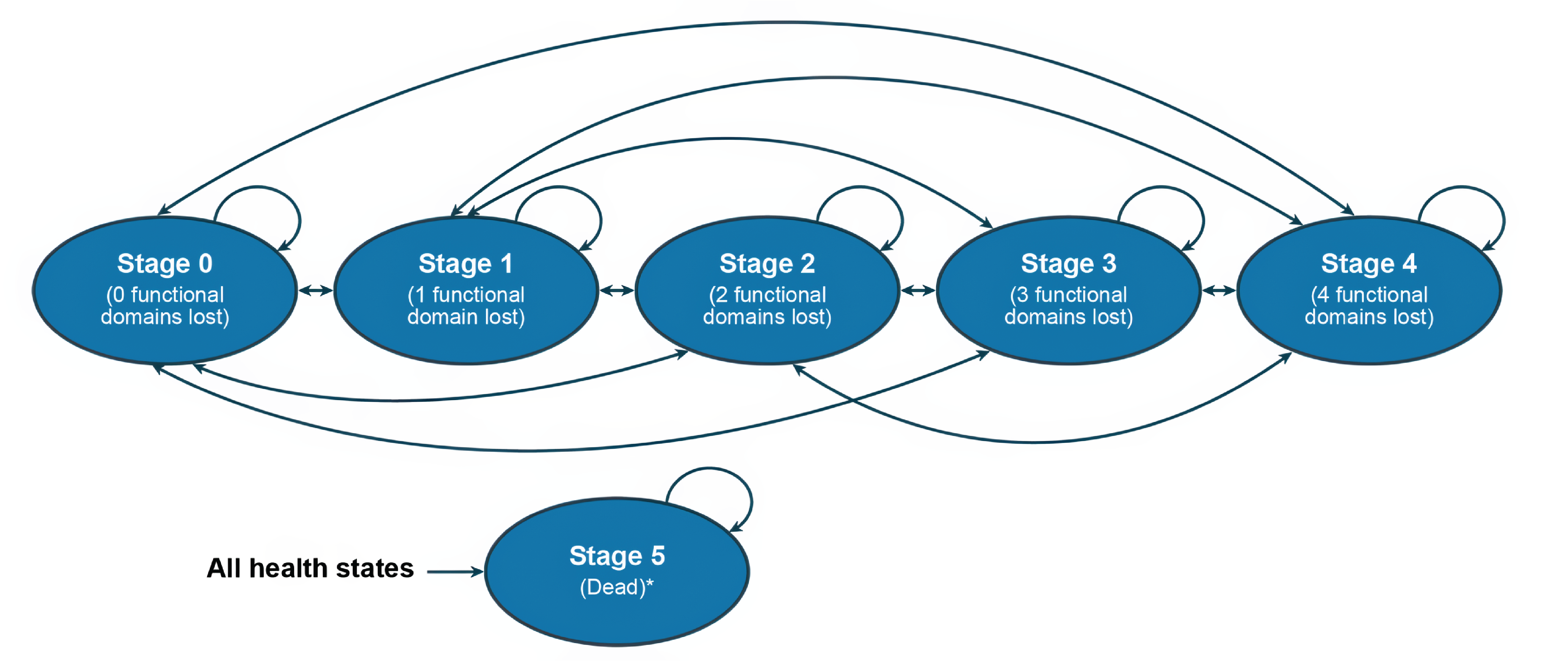
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC clinical review found that based on the primary 28-week data from Part C of the VALOR trial, tofersen in addition to background therapy may result in little or no clinically meaningful difference compared to placebo for functional decline as measured by ALSFRS-R, with low certainty. Similar results were found for little to no clinically meaningful difference for respiratory function and muscle strength. Conversely, tofersen likely results in a reduction in cerebral spinal fluid SOD1 protein and plasma neurofilament light chain (NfL) levels with moderate certainty in these findings; however, these surrogate outcomes and their direct translation to patient-perceived clinical benefit is not yet fully established.
Clinical efficacy of background therapy alone was informed by using 3-month transition probabilities reported by Thakore et al. based on the PRO-ACT database.5 Comparative efficacy for the economic evaluations were obtained from Part C of the VALOR trial. Hazard ratios (HRs) were derived from time-to-event data, defined as the time from baseline to the first time that a patient progresses by at least 1 stage on the MiToS scale.3 Based on the deterministic results of the sponsor’s case analysis, approximately 98% of the incremental QALYs for tofersen plus background therapy were found to be accrued during the extrapolated period.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The comparative efficacy of tofersen compared to background therapy alone is uncertain. Comparative efficacy in the model was informed from the VALOR (Part C) trial and open-label extension study where a HR for tofersen plus background therapy versus background therapy alone was derived and applied to background therapy transition probabilities. As noted in the CDA-AMC clinical review, based on the primary 28-week data from the VALOR (Part C) trial, tofersen may result in little or no clinically meaningful difference compared to placebo for functional decline as measured by ALSFRS-R. Similarly, tofersen may result in little to no clinically meaningful difference for respiratory function and muscle strength. There remains significant uncertainty in the comparative effectiveness of tofersen plus background therapy versus background therapy alone. While the longer-term data from the open-label extension and early access programs suggest trends toward slowed disease progression and potential survival benefits with earlier initiation of tofersen, this evidence is primarily descriptive and lacks concurrent randomized controls.
The magnitude of benefit associated with tofersen plus background therapy versus background therapy along is highly uncertain. CDA-AMC was unable to address this issue.
The submitted economic model is not reflective of clinical practice. In the base-case analysis, the sponsor submitted a Markov model with 6 health states based on the MiToS staging system. The sponsor also provided the option to inform health states based on the King’s staging. The MiToS system uses 6 stages and assesses the complete loss of independence in 4 functional domains (i.e., swallowing, walk/self-care, communicating, and breathing). CDA-AMC acknowledges the complexity of modelling a disease such as ALS and, while MiToS staging is based on ALSFRS-R scores, clinical expert feedback received by CDA-AMC noted neither the MiToS staging nor the King’s staging systems are used in clinical practice as these staging systems do not appropriately capture the clinical presentation of patients. For example, although MiToS Stage 1 is defined as the loss of independence in 1 domain, clinical expert input received by CDA-AMC noted broad variability within each Stage in terms of patient’s quality of life and disease prognosis. Even if a domain is scored as “loss of independence in one domain,” due to the variability in patients and their clinical presentation, 1 patient may still be considered to have early/mid ALS and have a reasonable quality of life whereas another patient in the same MiToS Stage 1 may be more mid/severe ALS and have a considerable different quality of life. Informing an economic model on a staging system that is not reflective of clinical practice and that has considerable variability within health states increases uncertainty and may fail to appropriately estimate total benefit and health care costs associated with tofersen plus background therapy across a patient’s lifetime.
CDA-AMC was unable to address this issue.
The drug cost of tofersen is underestimated. In the sponsor’s submitted economic model, it was estimated that tofersen would be associated with 14 administrations in the first year and 13 administrations in subsequent years, respectively. The annual drug cost of tofersen calculated by the sponsor for year 1 was $397,189, with subsequent years costing $368,819. Based on the Health Canada product monograph, tofersen “treatment should be initiated with three (3) loading doses administered at 14-day intervals (Day 0, Day 14, Day 28)” and “maintenance dose should be administered every 28 days thereafter (Day 56, Day 84, etc.).”1 While CDA-AMC acknowledges that the loading doses occur in a span of 29 days and this therefore may cause discrepancies in a model using a 28-day cycle length, based on the product monograph, the first year of tofersen treatment should consist of 15 administrations if assuming a 365-day year. Therefore, the sponsor underestimated the drug cost of tofersen in the first year of treatment.
Due to the constraints of the model’s 28-day cycle length, CDA-AMC assumed 3 administrations in Cycle 1 as a proxy to correct for the number of expected administrations in year 1 of tofersen treatment.
The following issues only apply from a societal perspective:
Important societal perspective considerations were missing from the analysis. The sponsor submitted an analysis from a societal perspective, which included indirect costs and caregiver disutilities. CDA-AMC noted that the inclusion of these elements is appropriate; however additional indirect costs such as those associated with productivity loss may also be important to consider. Clinical expert feedback noted that patients with ALS and their caregivers can include those of working age; particularly those with SOD1 mutation which were noted too often have earlier onset of disease compared to their sporadic counterparts. In a 2014 Canadian study examining the economic burden of ALS, it was noted that annual indirect costs (i.e., loss of wages) for patients with ALS and family members providing care was approximately $56,821 (CAD 2014).14 CDA-AMC notes that there are limitations with the Gladman et al. study such as study design and small sample size and thus productivity costs should be taken with caution, however, this study highlights the existent of a relationship between ALS and productivity loss. As such, improvement in quality of life and stabilization of disease that may impact a patient’s or caregiver’s ability to work may impact the cost-effectiveness of tofersen under a societal perspective. CDA-AMC notes that the exclusion of productivity costs may biases results against tofersen if the efficacy benefit of tofersen versus background therapy is realized.
In the absence of more robust data and lack of flexibility to incorporate it into the model, CDA-AMC was unable to address this issue.
Indirect costs included in the societal perspective analysis are uncertain. In the submitted analysis, the sponsor only considered indirect costs associated with the health states as informed by Ploug et al.2 There are several uncertainties associated with the values used to inform these indirect costs. First, Ploug et al. was a cross-sectional patient survey and retrospective chart review looking at treatment-related and non–treatment-related out-of-pocket costs associated with ALS in Europe and UK.2 In the published poster, non–treatment-related out-of-pocket costs were noted to include “transport to appointments/hospital visits, transport modifications, home modifications, supportive devices, testing, hospital visits, and care home/assisted living.” In an additional information request, CDA-AMC asked the sponsor to provide supplemental materials describing the study’s methodology. The sponsor responded that they were unable to acquire this material.13 As such, in the absence of more granular information, it remains unclear if these costs are applicable to the Canadian setting and/or the appropriateness of its inclusion into the economic model’s societal perspective. For example, patient input received by CDA-AMC commented on the logistical challenges of tofersen treatment. Without a detailed breakdown of what is captured by ‘transport to appointments/hospital visits’, there remains uncertainty if this consideration has been appropriately captured in the analysis.
CDA-AMC additionally notes that the indirect costs associated with each MiToS stage lack face validity. For example, MiToS staging 1 ($14,885) and 3 ($5,952) were associated with significantly more costs than those of MiToS stage 2 ($130) or 4 ($404). Without context as to the amount within each cost categories that makes up the overall costs for each staging, the decrease in indirect costs associated with stage 2 and 4 fail to correlate with the severity of increasing demands associated with increasing MiToS staging.
CDA-AMC was unable to address this issue. The impact of tofersen plus background therapy on costs and outcomes related to the societal perspective is highly uncertain due to a lack of evidence and uncertainties associated with the chosen model inputs.
Pagebreak
Additional issues were identified but were not considered to be key issues:
Exclusion of treatment waning for tofersen is uncertain. In the sponsor’s analysis, tofersen treatment waning was excluded. This assumption may bias results in favour of tofersen as patients maintain their benefit relative to background therapy alone. Clinical expert feedback received by CDA-AMC noted that, in the absence of long-term data informing sustained benefit, tofersen is likely to become less effective over time; however, it is unclear when that warning would be observed.
CDA-AMC was unable to address this issue given the lack of long-term data on the use of tofersen. CDA-AMC notes that the inclusion of treatment waning is expected to increase the ICER associated with tofersen plus background therapy compared to background therapy alone, however its inclusion is not anticipated to impact the overall interpretation of the cost-effectiveness of tofersen plus background therapy.
Adverse events may be underestimated. AEs included in the sponsor’s submitted analysis included limb pain, back pain, radiculitis, and myelitis. The probability of experiencing an AE was informed by the VALOR (Part C) trial for both tofersen plus background therapy and background therapy alone. Clinical expert feedback received by CDA-AMC noted that these included AEs were reasonable, but additional AEs associated with tofersen such as optic neuropathies due to raised intracranial pressure were not captured. Although not captured in the trial, AEs due to raised intracranial pressure such as optic neuropathies were noted to be of large concern and may require involvement of other specialists to treat.
In the absence of more robust data, CDA-AMC was unable to address this issue. Exclusion of tofersen specific AEs may bias results in favour of tofersen, however they are not expected to change the overall results of the cost-effectiveness of tofersen plus background therapy.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 6). The impact of these changes, individually and collectively, is presented in Table 7.
Table 6: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case (applied to both the health care payer and special perspective) | ||
1. Number of tofersen administration in year 1 | 14 | 15 |
CDA-AMC base case | ― | Reanalysis 1 |
CDA-AMC = Canada’s Drug Agency.
Note: CDA-AMC was unable to resolve the issues with comparative clinical efficacy, model structure, and indirect costs associated with the societal perspective.
Table 7: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (health care payer perspective; probabilistic) | Background Therapy | 39,507 | 0.618 | Reference |
Tofersen plus Background Therapy | 1,216,908 | 1.374 | 1,556,441 | |
Sponsor’s base case (societal payer perspective; probabilistic) | Background Therapy | 46,560 | 0.525 | Reference |
Tofersen plus Background Therapy | 1,230,334 | 1.076 | 2,151,535 | |
CDA-AMC reanalysis 1 | Background Therapy | 39,922 | 0.628 | Reference |
Tofersen plus Background Therapy | 1,232,025 | 1.347 | 1,658,220 | |
CDA-AMC base case (health care payer perspective; probabilistic) Reanalysis 1 | Background Therapy | 39,585 | 0.617 | Reference |
Tofersen plus Background Therapy | 1,245,469 | 1.377 | 1,586,423 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated. CDA-AMC presented stepped analysis were performed on the sponsor’s health care payer perspective scenario unless otherwise stated.
Table 8: Disaggregated Results of the CDA-AMC
Parameter | Tofersen plus background therapy | Background therapy |
|---|---|---|
Discounted LYs | ||
Total | 3.125 | 1.199 |
MiToS Stage 0 | 0.736 | 0.442 |
MiToS Stage 1 | 0.797 | 0.388 |
MiToS Stage 2 | 0.586 | 0.202 |
MiToS Stage 3 | 0.346 | 0.086 |
MiToS Stage 4 | 0.661 | 0.082 |
Discounted QALYs | ||
Total | 1.377 | 0.617 |
MiToS Stage 0 | 0.520 | 0.313 |
MiToS Stage 1 | 0.378 | 0.185 |
MiToS Stage 2 | 0.208 | 0.072 |
MiToS Stage 3 | 0.113 | 0.028 |
MiToS Stage 4 | 0.163 | 0.020 |
AE Disutility | −0.004 | −0.001 |
Discounted costs ($) | ||
Total | 1,245,469 | 39,585 |
Healthstate Costs | 63,133 | 22,767 |
MiToS Stage 0 | 8,629 | 5,177 |
MiToS Stage 1 | 18,917 | 9,213 |
MiToS Stage 2 | 14,467 | 4,994 |
MiToS Stage 3 | 6,197 | 1,538 |
MiToS Stage 4 | 14,923 | 1,846 |
Treatment Costs | 1,182,336 | 16,817 |
Drug Acquisition | 1,173,471 | 16,563 |
Administration Costs | 7,415 | 82 |
Monitoring Costs | 267 | 0 |
Adverse Event Costs | 1,183 | 173 |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; LY = life-year; MiToS = Milano-Torino Staging; QALY = quality-adjusted life-year.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case under the health care payer perspective (refer to Table 9).
Results from both the sponsor’s and the CDA-AMC analyses suggest that even with substantial price reduction (i.e., 90%), the ICER for tofersen plus background therapy remains above $250,000 per QALY gained. There are 2 reasons for this result. This is partially due to more non–drug-related costs associated with tofersen plus background therapy. This is further due to the predicted improvement in life expectancy associated with tofersen, which results in both additional drug acquisition costs and non–drug-related costs (drug administration, monitoring and management of side effects) during this prolonged survival time. Specifically, in the deterministic CDA-AMC reanalysis, tofersen was associated with $42,149 in drug acquisition cost and $8,738 in non–drug-related costs. As such, even if unit drug cost were reduced, tofersen plus background therapy would remain associated with significant costs to the health care system compared those taking background therapy alone.
Table 9: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Annual cost ($, year 1/year 2+) | ICERs for tofersen plus background therapy vs. background therapy ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 28,371a | 425,560 / 368,819 | 1,556,442 | 1,586,423 |
10% | 25,534 | 383,004 / 331,937 | 1,412,290 | 1,439,233 |
20% | 22,697 | 340,448 / 295,055 | 1,268,138 | 1,292,043 |
30% | 19,859 | 297,892 / 258,173 | 1,123,987 | 1,144,853 |
40% | 17,022 | 255,336 / 221,291 | 979,835 | 997,662 |
50% | 14,185 | 212,780 / 184,409 | 835,683 | 850,472 |
60% | 11,348 | 170,224 / 147,528 | 691,532 | 703,282 |
70% | 8,511 | 127,668 / 110,646 | 547,380 | 556,092 |
80% | 5,674 | 85,112 / 73,764 | 403,229 | 408,901 |
90% | 2,837 | 42,556 / 36,882 | 259,077 | 261,711 |
100% | 0 | 0 / 0 | 114,925 | 114,521 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; vs. = versus; QALY = quality-adjusted life-year.
Note: Presented price reductions were derived from the health care payer perspective analyses.
aSponsor’s submitted price for tofersen.3
Issues for Consideration
While genetic testing for SOD1 mutations is currently performed as part of standard of care for patients with ALS, clinical expert feedback received by CDA-AMC indicated that there are access differences across jurisdictions that may impact the feasibility of accessing tofersen.
Clinical expert feedback noted that there may be logistical issues with accessing tofersen (especially for patients living in rural and remote areas) due to the need for lumbar puncture for administration. Access may be increased if more health care professionals who are competent in lumbar puncture are trained to administer tofersen. Until then, for patients who reside in rural and remote communities, accessing centres that are capable of administrating tofersen may be associated with additional travel burdens that are not captured under the sponsor’s submitted societal perspective.
Clinical expert feedback flagged that if treatment with tofersen can stabilize a patient’s disease, then such patients may live longer and continue to require treatment. While the CDA-AMC BIA was unable to address this issue, increased life expectancy would mean that the number of individuals eligible for treatment could grow over time. The budget impact of reimbursing tofersen plus background therapy may therefore increase in the long term.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing tofersen for the treatment of adult patients with SOD1-ALS.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. The market uptake for tofersen was estimated using market research conducted by the sponsor. The key inputs to the BIA are documented in Table 11.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing tofersen for the treatment of adult patients with SOD1-ALS would be $35,267,763 (year 1 = $12,044,982; year 2 = $11,473,706; year 3 = $11,749,075).
Table 10: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
pan-Canada population (18+; 2023) | 25,410,90414 |
Prevalence of ALS | 0.00684%15 |
% of SOD1-ALS | 2%16 |
% eligible for tofersen | 100% |
% eligible for public coverage | 90% |
Annual population growth | 2.4% |
Number of patients eligible for drug under review | 34 / 35 / 35 |
Market shares (reference scenario) | |
Tofersen plus background therapy | 0% / 0% / 0% |
Background therapy | 100% / 100% / 100% |
Market shares (new drug scenario) | |
Tofersen plus background therapy | 90% / 90% / 90% |
Background therapy | 10% / 10% / 10% |
Cost of treatment (per patient per year) | |
Tofersen plus background therapy a | Initial: $410,894 Maintenance: $382,524 |
Background therapy a | $13,705 |
ALS = amyotrophic lateral sclerosis.
aCost of background therapy was calculated as an average of the annual cost of treatment in the initial year and maintenance year
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
The drug cost of tofersen is underestimated. As noted in the key issue section for the sponsor’s submitted economic model, based on the Health Canda product monograph treatment with tofersen would result in 15 administrations in year 1. As such, the sponsor underestimated the drug cost of tofersen in the first year of treatment but calculating results using 14 administrations.
CDA-AMC used 15 administrations in year 1 of tofersen treatment.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 11.
Table 11: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Tofersen Year 1 Costs | 14 | 15 |
CDA-AMC base case | ― | Reanalysis 1 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 13 and a more detailed breakdown is presented in Table 14. In the CDA-AMC base case, the 3-year budget impact of reimbursing tofersen plus background therapy for adult patients with SOD1-ALS was $36,169,911 (year 1 = $12,905,338; year 2 = $11,494,354; year 3 = $11,770,219).
Table 12: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 35,267,763 |
CDA-AMC base case: Reanalysis 1 | 36,169,911 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
Table 13: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 450,976 | 461,800 | 472,883 | 484,232 | 1,418,914 |
Tofersen plus background therapy | 0 | 0 | 0 | 0 | 0 | |
Background therapy | 450,976 | 461,800 | 472,883 | 484,232 | 1,418,914 | |
New drug total | 450,976 | 12,506,782 | 11,946,589 | 12,233,307 | 36,686,677 | |
Tofersen plus background therapy | 0 | 12,905,338 | 11,494,354 | 11,770,219 | 36,169,911 | |
Background therapy | 450,976 | 46,180 | 47,288 | 48,423 | 141,891 | |
Budget Impact | 0 | 12,044,982 | 11,473,706 | 11,749,075 | 35,267,763 | |
CDA-AMC base case | Reference total | 450,976 | 461,800 | 472,883 | 484,232 | 1,418,914 |
Tofersen plus background therapy | 0 | 0 | 0 | 0 | 0 | |
Background therapy | 450,976 | 461,800 | 472,883 | 484,232 | 1,418,914 | |
New drug total | 450,976 | 13,367,138 | 11,967,237 | 12,254,451 | 37,588,825 | |
Tofersen plus background therapy | 0 | 13,320,958 | 11,919,949 | 12,206,028 | 37,446,934 | |
Background therapy | 450,976 | 46,180 | 47,288 | 48,423 | 141,891 | |
Budget Impact | 0 | 12,905,338 | 11,494,354 | 11,770,219 | 36,169,911 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
Ethics Review
Abbreviations
ALS
amyotrophic lateral sclerosis
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
CDA-AMC
Canada’s Drug Agency
EAP
early access program
EUnetHTA
European Network for Health Technology Assessment
OLE
open-label extension
SOD1-ALS
amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene
Summary
Amyotrophic lateral sclerosis (ALS) is a rare, debilitating, and ultimately fatal neurodegenerative disease. Patients with ALS experience progressive muscle weakness and loss of function and often die within 3 to 5 years of diagnosis. ALS associated with a mutation in the SOD1 gene (SOD1-ALS) is an ultrarare form of ALS.
Tofersen is the first SOD1-ALS treatment proposed to address the underlying genetic cause of SOD1-ALS and to significantly alter the disease course. This drug was approved by Health Canada with conditions. It is administered through intrathecal injections every 28 days.
This Ethics Review was informed by reviewing relevant literature as well as patient group, clinician group, clinical expert, and drug program input gathered during this review. Ethical considerations identified include the following.
Diagnosis, treatment, and experiences of ALS: The initial time to diagnosis is lengthy given the lack of an ALS diagnostic marker, frequent misdiagnosis, and delayed referrals to neurologists. Diagnosing SOD1-ALS is associated with additional challenges, including geographic disparities in access to genetic testing, delayed access to pretesting genetic counselling, and the existence of SOD1 mutations of undetermined significance. Current treatments offer modest benefits and do not treat SOD1-ALS specifically. Patients who have ALS and their caregivers experience physical, emotional, and financial burdens associated with the disease. Patients typically have progressive difficulty with mobility, speech, swallowing, performing tasks of daily living, and breathing. Patients and caregivers can experience profound distress, fear, and anxiety associated with disease progression, the loss of independence, and the impacts of ALS on family members. Patients and caregivers also incur expenses associated with purchasing medical equipment, formal care support, travel, and home modifications, and often lose their income because they are unable to work.
Clinical and economic evidence used in the evaluation of tofersen: The pivotal phase III VALOR trial failed to meet its primary efficacy end point, and although there were meaningful differences in the 2 biomarkers used as secondary end points, their clinical meaningfulness is unvalidated. No studies have assessed tofersen’s comparative effectiveness relative to the current standard of care and, although the VALOR trial’s open-label extension (OLE) study and the findings from the early access program (EAP) studies suggested trends toward slowed disease progression, significant methodological limitations temper confidence in their findings. Further, several patient groups that likely would be encountered in clinical practice in Canada were underrepresented or excluded from trial populations, including older patients, patients from racialized groups, patients diagnosed with slower-progressing SOD1 mutations, and patients who were more medically complex. Collectively, the clinical uncertainties associated with the evaluation of tofersen complicate decision-making by patients, clinicians, and health systems. These uncertainties are heightened for groups that were underrepresented or excluded from the trial populations.
Clinical use and implementation of tofersen: Treatment with tofersen also will entail several access challenges because tofersen must be prescribed and monitored by ALS specialists in tertiary care centres mainly located in large urban centres. Although access could be improved if patients received their monthly intrathecal injections in local settings, some patients would continue to experience access challenges. The use of tofersen may pose novel risks for patients compared to the current standard of care, including serious neurologic adverse events and risks associated with its intrathecal mode of delivery. Patients living with ALS and their caregivers, as well as clinicians, hold enhanced expectations and hopes for disease-modifying therapies because current treatments offer only modest benefits. These expectations, and the ongoing unmet needs of patients with ALS, should be accounted for in considering the potential benefits, harms, and evidentiary uncertainties associated with tofersen.
Health systems: The introduction of tofersen may pose health resource usage and system integration challenges, especially due to the resource-intensive nature of intrathecal delivery and the need to monitor and manage serious adverse events. The introduction of tofersen may require ongoing data collection and postmarket evaluation as well as the development of genetic testing infrastructure to support ongoing delivery and monitoring.
Objective and Research Questions
The objective of this Ethics Review is to identify and describe ethical considerations associated with the use of tofersen for the treatment of adults with ALS associated with a mutation in the SOD1 gene, including considerations related to the disease context, the evidentiary basis, the use of tofersen, and the impact on health systems.
To address this objective, this review addresses the following research questions:
What ethical considerations arise in the context of tofersen, including considerations related to diagnosis, treatment, and outcomes?
What ethical considerations arise in relation to the evidence (e.g., clinical and economic data) used to evaluate tofersen?
What ethical considerations arise in relation to the use of tofersen for patients, their caregivers, and their clinicians?
What are the ethical considerations for health systems related to tofersen?
Methods
Guiding questions identified in the European Network for Health Technology Assessment (EUnetHTA) Model 3.0, Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment (ECHTA),2 drove the identification of ethical considerations relevant to the use of tofersen in the treatment of ALS associated with a mutation in the SOD1 gene in this Ethics Review. These guiding questions were organized to respond to the research questions and to investigate ethical considerations related to:
patients living with ALS, including those with ALS associated with a mutation in the SOD1 gene, and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges or burdens related to diagnosis or clinical care; and factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms, and value of tofersen (i.e., ethical considerations in relevant clinical trials, including their representativeness, the choice of outcome measures, the appropriateness of the analytical methods and models used for all population groups, and ethical considerations related to the data or assumptions in the economic evaluation)
the use of tofersen, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, and society, as well as considerations related to access to this therapy
the uptake of tofersen in health systems, including considerations related to the distribution of health care resources.
Review of Project Inputs
A single reviewer collected and considered input from 7 main sources of data related to ethical considerations relevant to the research questions guiding this Ethics Review. The reviewer considered the following sources:
a review of evidence from a search of published literature
the sponsor submission, including noting relevant information and external references or sources relevant to each of the research questions driving this report
clinician group input received from The Canadian ALS Research Network
patient input received from the ALS Action Canada Society, the ALS Society of Alberta, the ALS Society of British Columbia, and the ALS Society of Canada
drug program input received from drug programs participating in the Canada’s Drug Agency (CDA-AMC) reimbursement review process
discussion with clinical experts (n = 5 clinical experts directly engaged over the course of this reimbursement review), including through 3 consultation meetings involving 2 experts and 1 panel meeting involving 5 experts (during each of these meetings, the reviewer asked the clinical experts targeted questions related to ethical considerations corresponding to the research questions driving this report; all the clinical experts are practising neurologists and all had experience treating patients with ALS and with tofersen)
engagement with clinical and economic reviewers and the testing procedure considerations section in the clinical review to identify domains of ethical interest arising from their respective reviews and to identify relevant questions and sources to further pursue in this report.
Details on the Published Literature Search
Literature Search Methods
An information specialist conducted a literature search on key resources, including MEDLINE via Ovid, The Philosopher’s Index via Ovid, and the Cumulative Index to Nursing and Allied Health Literature (CINAHL) via EBSCO. A targeted Google Scholar search was also performed. The search strategy comprised using both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was tofersen. Search filters were applied to limit retrieval to citations related to ethical concepts or considerations, equity concepts or considerations, or qualitative studies.
An additional targeted search was conducted in the same databases using the search concepts “SOD1” and “amyotrophic lateral sclerosis.” For this search, focused terms related to ethical concepts or considerations, equity concepts or considerations, or qualitative studies were used.
No filters were applied to the searches conducted in The Philosopher’s Index. Duplicates were removed by manual deduplication in EndNote. Retrieval was limited to the English language. The searches were completed on March 24, 2025. Updated literature searches were completed on April 24, 2025.
Literature Screening and Selection
A single reviewer screened literature in 2 stages. In the first stage, the reviewer screened the titles and abstracts of the retrieved citations and identified and retrieved articles for full-text review if their titles or abstracts identified ethical considerations, or provided normative analyses (i.e., focusing on “what ought to be” through argumentation) or empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experiences, incidence, diagnosis, treatment, or outcomes of ALS in general and the experiences, incidence, diagnosis, treatment, or outcomes of ALS associated with a mutation in the SOD1 gene, or related to the evidence on, the use of, or implications of, tofersen for the treatment of adults with ALS associated with a mutation in the SOD1 gene. In the second stage, the same reviewer reviewed full-text publications categorized as “retrieve.” The reviewer included texts that included substantive information meeting the aforementioned criteria. Additionally, the reviewer retrieved and reviewed select sources drawn from relevant bibliographies, relevant key concepts, and consultation with experts or other reviewers using these selection criteria.
Data Analysis
The 4 research questions driving this review guided the collection, coding, and thematic analysis of data. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations from the literature and from relevant project inputs. In the initial coding phase, the reviewer read the publications and input sources for ethical content (e.g., claims related to potential harms, benefits, equity, justice, and resource allocation as well as ethical issues in the evidentiary basis). The reviewer coded the identified claims related to ethical content using methods of qualitative description.3 In the second coding phase, the reviewer identified major themes and subcodes through repeated readings of the data,3 and summarized them in thematic categories within each guiding domain or research question. The reviewer noted if the ethical content did not fit into these categories or into the domains outlined in the research questions, or if there were discrepancies or conflicts between the ethical considerations or values identified between project sources or within thematic categories. Data analysis was iterative, and the reviewer used themes identified in the literature, in project inputs, and during consultations with clinical experts to further refine and reinterpret the ethical considerations identified. Finally, the reviewer thematically organized and described the data according to the 4 research questions and domains driving this Ethics Review. The results, limitations, and conclusions of this analysis are described in the following sections.
Results
Key Ethical Considerations
Experiences, Diagnosis, and Treatment of ALS
Disease Burden
ALS is a rare, debilitating, progressive, and ultimately fatal neurodegenerative disease characterized by the deterioration and death of motor neurons within the brain and spinal cord.4,5 This leads to progressive muscle weakness and eventual loss of motor function.6,7 Death is usually due to respiratory failure and typically occurs within 3 years to 5 years of symptom onset.8,9
There are 2 main types of ALS: familial ALS (i.e., ALS with a known family history, which accounts for 5% to 10% of ALS cases) and apparently sporadic ALS (i.e., ALS with no known family history, which accounts for 90% to 95% of ALS cases).10,11 More than 15% of all ALS cases are caused by a known genetic mutation.12,13 SOD1 mutations are one of the most common and are estimated to account for approximately 2% of all ALS cases.14 In Canada, it is estimated that 2,800 people are living with ALS and 40 people are living with SOD1-ALS, and that 1 to 2 people will be newly diagnosed with SOD1-ALS annually.
Patients living with ALS experience impacts on their physical, emotional, social, and financial well-being. Physically, patients often develop difficulty with walking and standing and therefore require assistive devices, including walkers and power wheelchairs, to assist with mobility. They will also face difficulties in performing tasks of daily living and require assistance with eating, drinking, and transitioning from lying to sitting, and with speech and swallowing. They will face difficulties with breathing and often require the use of nonventilatory support and eventually permanent ventilation.10 Muscle cramping, nerve pain, and fatigue are also common. Many patients have reported experiencing these difficulties within a year of diagnosis. A clinical expert indicated that, in general, patients with SOD1-ALS deteriorate more rapidly than patients with other types of ALS.
Patients living with ALS also experience impacts on their emotional and social well-being. In the patient group input and published literature, it was emphasized that patients can experience profound emotional distress and fear associated with disease progression, and anxiety and grief related to their loss of independence and the impacts of ALS on family members.15 Depression is common and some patients report thoughts of suicidal ideation.15-19 For patients diagnosed with SOD1-ALS, the genetic nature of the disease often intensifies their emotional distress. Patients frequently fear their relatives may be at risk and, for those with children, they fear they have passed on this variant to their children and that they may develop ALS.20 In the patient group input, it also was emphasized that patients often experienced becoming increasingly socially isolated as it becomes more difficult for them to communicate and pursue activities they once enjoyed.
The financial burden of ALS is also significant because patients incur both direct and nonmedical costs and a loss of income.21,22 Direct medical costs include the purchase of medical equipment, medications, additional therapies, and professional caregiving services.22 Direct nonmedical costs include travel expenses, home modifications, and informal care involving family and nonprofessional caregivers.22 Most patients become unable to work, leading to a loss of income. In the patient group input, it was reported that patients find the costs associated with ALS overwhelming.
Caregivers, who are frequently spouses, children, or close family members of patients, also experience impacts on their physical, emotional, social, and financial well-being. In the patient group input and published literature, the physical demands of caregiving, such as assisting patients with medical care and activities of daily living, were identified as strenuous, exhausting, and increasingly challenging as the patient becomes more debilitated.16 Emotionally, caregivers frequently experience feelings of helplessness, grief, anxiety, and emotional exhaustion due to witnessing the patient decline and because of caregiving’s relentless demands.16 Levels of depression among caregivers is also high.23 However, caregiving can have positive impacts on caregivers’ emotional well-being because it can bring meaning to their lives, strengthen bonds with patients, and contribute to developing resilience.16,24,25 Caregiving also places considerable demands on caregivers’ time. It is estimated that caregivers who are family members can spend upward of 11 hours per day caring for the patient.25 Caregivers also often experience financial hardships because many must either reduce their work hours or leave their jobs to care for patients.21
Diagnostic Challenges and Disparities
The length of time it takes to first diagnose a patient with ALS has been identified as a major challenge because it delays treatment initiation.26 In Canada, the time to diagnosis varies considerably, with Saskatchewan having the longest time to diagnosis (27 months) and Nova Scotia the shortest time to diagnosis (15.1 months).21 The lengthy time to diagnosis arises because there is no diagnostic marker for ALS — instead, diagnosis is based on patient history, physical examination, electrodiagnostic examination, and the exclusion of alternative diagnoses. There is a high rate of misdiagnoses by nonspecialist physicians, and referral to neurologists is often delayed.21,22 The delay in time to diagnosis is also associated with disparities based on geography and race. The published literature and clinical experts indicated that patients living in rural or remote areas are diagnosed at later ages and stages than those living in urban areas due to less access to care.27 Research also has demonstrated that patients who are from racialized groups experience significant delays in diagnosis as compared to people who are white.26,28 Chen et al. (2024)28 identified that in the US context, there was a 64% longer diagnostic delay for patients who are Black as compared to patients who are white. As a result, patients from racialized groups are often likely to have poorer outcomes — including having their survival rate negatively impacted.26,28
The diagnosis of SOD1-ALS is associated with additional challenges. There are geographic disparities associated with access to genetic testing for the SOD1 gene; patients living in rural and remote areas often need to travel to access genetic testing because the laboratories are associated with ALS specialist clinics that are typically located in urban centres. The clinical experts, however, indicated that genetic testing could be done in local laboratories, which could improve access for patients in rural and remote areas, although processes would need to be implemented to facilitate testing because it is an unfamiliar process for most local laboratories in Canada. In the testing procedure considerations section in the clinical review, additional challenges associated with accessing genetic testing and the processing of results are identified.
Additionally, patients can experience psychological impacts because of undergoing genetic testing. In the patient group input and published literature, it was highlighted that some patients who undergo genetic testing experience significant psychological distress.29 Additionally, if patients are the only person in their families to undergo genetic testing, they can experience distress over whether to disclose their results to family members and to discuss pursuing genetic testing with other family members.29 However, the patient group input highlights how some patients who receive a confirmed diagnosis for SOD1-ALS experience this as psychologically positive because it gives them an increased sense of clarity and control in connection with life-planning — including pursuing treatment options, rather than living in uncertainty. Given the potential psychological impacts of genetic testing, it has been recommended that patients should be offered a choice in terms of what information they would like to receive — a course of action that supports patient autonomy.30
Finally, diagnosing SOD1-ALS is also challenged by the existence of mutations of undetermined significance. As indicated by a clinical expert, although there are many known pathological SOD1 mutations that confirm the diagnosis of SOD1-ALS, there also are SOD1 mutations of undetermined significance that make confirming an SOD1-ALS diagnosis more complicated. In these circumstances, it was recommended that the geneticist interpreting the results use all data that are available to them, including predictive software and a positive family history. When these uncertainties arise, genetic counsellors and physicians will need to discuss them with patients and navigate these uncertainties in terms of their implications for clinical care.
Current Treatments
In Canada, 2 approved treatments currently available for patients with ALS are riluzole and edaravone. There are no approved therapies specifically for the treatment of SOD1-ALS presently available in Canada. Both riluzole and edaravone are administered orally, although edaravone also can be administered intravenously. Each has been shown to have modest benefits in slowing disease progression or extending survival time for some but neither addresses SOD1-ALS’s underlying genetic cause or significantly alters its course. In the patient group input, it was reported that some patients and their caregivers could not definitively say whether they had experienced slowed disease progression due to using riluzole or edaravone, though some patients believed they might have experienced slowed disease progression because of using edaravone. The clinical experts indicated that there is differential access to these treatments because their cost is out of reach for patients who live in jurisdictions that do not provide public coverage and for those without private coverage.
Ethics of Evidence and Evaluation of Tofersen
One pivotal trial, its OLE study, and 4 real-world publications associated with 2 EAPs were considered in connection with this submission. The VALOR trial was a pivotal 28-week, phase III, randomized, double-blind, placebo-controlled trial that evaluated tofersen administered by intrathecal injection in adults with ALS and a confirmed SOD1 mutation. The VALOR trial’s primary objective was to assess tofersen’s clinical efficacy and its secondary objective was to evaluate tofersen’s safety, tolerability, pharmacodynamics, and biomarker effects. The primary objective of the VALOR trial’s OLE study was to assess tofersen’s longer-term safety and tolerability in participants who had completed the VALOR trial. Its treatment phase lasted up to 360 weeks. Four publications deriving from patients enrolled in German and Italian EAPs were prospective observational studies that assessed tofersen’s safety and efficacy in patients enrolled in these EAPs.
Uncertainties in Clinical Effectiveness
As highlighted in the clinical review, there are several evidentiary uncertainties associated with the evidence used to assess tofersen that raise questions about tofersen’s clinical effectiveness for patients with a confirmed diagnosis of SOD1-ALS and its cost-effectiveness. Crucially, the VALOR trial failed to meet its primary efficacy end point — namely, the change from baseline to week 28 in the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) scale — which indicates tofersen may not result in a clinically meaningful difference in function at 28 weeks. Two secondary end points (cerebrospinal fluid SOD1 protein levels and plasma neurofilament light-chain levels) demonstrated statistically significant differences at 28 weeks; however, because these end points were based on biomarkers, their correlation with direct clinical benefit remains unvalidated at this time.
The comparative effectiveness of tofersen against, or in addition to, the current standard of care (riluzole and/or edaravone) has not been assessed. Therefore, it is unclear whether tofersen is more efficacious than riluzole and/or edaravone or if tofersen in combination with riluzole and/or edaravone provides an additive benefit. Tofersen’s longer-term safety and effectiveness are also uncertain. While the findings from the OLE study and the EAPs’ studies suggested trends toward slowed disease progression and potential improved survival, their clinical meaningfulness is tempered for several reasons. The OLE study did not include a concurrent control group and there was significant attrition. The EAPs’ studies relied primarily on descriptive statistics and were not adjusted for confounders, which limits the ability to infer treatment effects; they were not blinded, which introduces the possibility of performance and detection biases, especially in connection with subjective measures such as ALSFRS-R and adverse event reporting; and they relied on patient recall. The need for additional longer-term evidence was reiterated by Health Canada’s Notice of Compliance with conditions pending the results of additional studies verifying tofersen’s clinical benefit.
Additionally, the ALSFRS-R scale and the 2 biomarkers, the main outcome measures used in the VALOR trial, the OLE study, and the EAPs’ studies, are primarily research tools rather than tools used in clinical practice. While clinical experts indicated that patients would consider the ALSFRS-R score to be meaningful because it focuses on key activities of daily living, such as stair climbing and cutting food, the meaningfulness of impacts on biomarkers and the translation to patient benefit remains uncertain.
Underrepresentation of Patient Groups
Although the VALOR trial enrolled adults with a confirmed SOD1 mutation and therefore represented the overarching relevant target population for tofersen, there was under-representation and the exclusion of several patient groups that likely would be encountered in clinical practice in Canada. Demographically, participants in the VALOR trial reflected the relatively younger demographic for patients with ALS and most participants were white (63.9%), whereas other racialized groups were minimally represented (Asian [8.3%], Black or African American [0.9%], and “other” [0.9%]), or their representation was unknown because a significant number of participants did not report their race (25.9%).Additionally, the VALOR trial largely excluded patients with slower-progressing variants and those who were more medically complex due to significant respiratory compromise or comorbidities. The under-representation and exclusion of these patient groups implies further uncertainties in tofersen’s clinical efficacy and safety for these groups.
Collectively, the evidentiary uncertainties associated with tofersen’s clinical effectiveness has implications for clinical care and health system decision-making. For patients, it means they could be offered treatment based on limited evidence of tofersen’s clinical effectiveness and safety. For clinicians, the uncertainties associated with tofersen’s short-term and long-term effectiveness and comparative value complexifies their efforts to assess tofersen’s risk-benefit profile — a complexity that must be discussed with patients during the informed consent process. These decisions and conversations should take into account the enhanced uncertainties and evidence limitations for those groups underrepresented in the trials. The uncertainties in the clinical data also complicate the economic assessment of tofersen’s value and therefore, tofersen’s cost-effectiveness is also uncertain. This has implications for the assessment of the value of tofersen for health systems investment.
Ethical Considerations in the Use of Tofersen
Tofersen is administered using intrathecal injections and consists of a 100 mg dose. Treatment begins with 3 biweekly loading doses followed by maintenance doses administered once every 28 days thereafter. Treating patients with tofersen requires ongoing monitoring by an ALS specialist of patients’ clinical status and the benefits and harms they are experiencing. The potential clinical use of tofersen raises ethical considerations associated with access to treatment, potential harms associated with tofersen, and patient expectations of tofersen.
Access to Treatment
Treatment with tofersen is typically provided by ALS specialists (i.e., ALS neurologists and physiatrists) in tertiary care centres that are mainly located in large urban centres. All patients with ALS will face challenges in travelling to these centres for treatment; however, these challenges would be exacerbated for patients living in rural or remote locations given the distance they need to travel to access care. The clinical experts indicated, however, that access to treatment could be improved if patients received their monthly intrathecal injections at local outpatient clinics or community hospitals. Doing so is possible because intrathecal injections do not need to be administered by an ALS specialist but can be administered by clinicians who are competent in lumbar puncture (e.g., neurologists, anesthesiologists, interventional radiologists, many emergency physicians, some nurse practitioners). Further, the serious side effects associated with tofersen are not immediate and likely would be caught during a follow-up visit with an ALS specialist. However, the initial prescribing of tofersen and the ongoing monitoring of patients will require ALS specialists; therefore, travel for specialist care is likely for many patients to sustain treatment with tofersen. In the patient group input, it was highlighted that patients would prefer to be treated locally.
While administering tofersen in local outpatient clinics and community hospitals would likely improve access to treatment for many patients, some patients would continue to experience access challenges. This includes patients who are unable to independently access treatment beds given their compromised physical condition (e.g., patients who are dependent on electric wheelchairs for mobility and who will require a Hoyer lift to transfer them to the treatment bed, and patients who require fluoroscopic guidance to be administered tofersen, will likely need to continue to travel to ALS specialist clinics). Some patients may not be able to access treatment at all due to their physical condition or if they find travel of any kind too burdensome. Diagnostic and outcome disparities for racialized and systemically marginalized groups are likely to persist without further supports. Moreover, patients with limited or no caregiver support will also experience access challenges with tofersen treatment because patients likely require caregivers’ assistance to travel to appointments.
Potential Harms Associated With Tofersen
Tofersen notably has a different mechanism of action than current standard of care therapies (i.e., riluzole and/or edaravone); it aims to treat the underlying genetic cause of SOD1-ALS and may introduce novel risks or harms compared to the standard of care. As identified in the Clinical Review Report, tofersen can cause serious neurologic adverse events resulting from inflammation of the central nervous system (i.e., myelitis, radiculitis, aseptic or chemical meningitis, increased cranial pressure, and papilledema). The clinical experts indicated that the observed frequency of the serious neurologic adverse events in the VALOR trial (5.6%) was significant, although they also indicated most are reversible, and that treatments are being developed to manage many of these serious adverse events and have demonstrated some improvement.
The intrathecal injections used to deliver tofersen introduce additional risks as compared to the oral or IV modes of administration associated with current therapies. Intrathecal injections are an invasive method that can have life-threatening complications.31 Complications include problems with the catheter such as migration, laceration, occlusion or disconnection, human error such as miscalculating the catheter’s content, and cerebrospinal fluid leaks caused by the ongoing dural opening at the catheter insertion site or by a perforated catheter — which, if severe, constitutes an emergency because leaks may cause subdural hematomas that can be life-threatening.31 The clinical experts noted that patients may decline tofersen treatment because they experience the side effects to be intolerable and indicated that they may initiate discussions to discontinue treatment if patients experience unmanageable serious adverse events.
Patient and Clinician Expectations of Tofersen
Tofersen has been proposed as a disease-modifying therapy that can address the underlying genetic cause of SOD1-ALS and significantly alter the disease course, including slowed disease progression and potentially disease stabilization or even improvements in function. Patients living with ALS, their caregivers, and clinicians hold enhanced expectations and hopes for disease-modifying therapies for ALS, especially given that current treatments offer only modest benefits. These expectations, and the ongoing unmet needs of patients with ALS for therapies that can halt or reverse disease progression, should be accounted for in considering the potential benefits, harms, and evidentiary uncertainties associated with tofersen to support balanced decision-making for patients, caregivers, and clinicians.
Health Systems Considerations
Alongside the challenges related to health systems decision-making given the evidentiary uncertainties associated with tofersen, the funding and implementation of tofersen raises additional potential challenges and considerations associated with health resource usage and system integration. The intrathecal mode of treatment delivery, as compared to the oral mode of delivery associated with riluzole and edaravone, will be more resource intensive. The administration of tofersen requires trained proceduralists, treatment rooms, lumbar puncture kits, and potentially fluoroscopic suites and/or Hoyer lifts. There also will be increased demands on clinicians to administer tofersen, on ALS specialists to monitor patients for side effects and their treatment response on an ongoing basis, and on the pharmacies responsible for managing the tofersen supply. Further, there will likely be an increased demand for MRIs to assess patients with myelitis or radiculitis — 2 of the serious adverse events associated with tofersen — and for ophthalmologic examinations given the risk of blindness associated with papilledema. These increased demands will be sustained over time given the ongoing monthly injections, especially if the use of tofersen leads to prolonged survival. Some health systems may not be equipped to meet these increased resource demands and, therefore, disparities could arise in the ability to deliver care across health care organizations in Canada.
Two potential additional health resource usage considerations relate to postmarket data and genetic testing infrastructure. Given the lack of long-term safety and efficacy data and data assessing tofersen’s effectiveness, postmarket data collection may help address some of the evidentiary uncertainties. This will require additional infrastructure to implement, monitor, and sustain. Additionally, as indicated in the testing procedure considerations section of the clinical review, many provinces rely on out-of-province or out-of-country genetic testing, which can lead to delays in accessing the results of genetic testing and, therefore, initiating treatment. Enhancing local capacity to support genetic testing, including engaging genetic counsellors, will also require increased health system resources and capacity.
Limitations
Very little published literature discusses ethical considerations related to the use of tofersen for the treatment of SOD1-ALS given both the rarity of the disease and the novelty of the drug under review. This does not imply that ethical considerations in the context of tofersen for SOD1-ALS are absent. Drawing on input received during this reimbursement review, including patient group, clinician group, and drug program input, and discussion with clinical experts, as well as engagement with the clinical, pharmacoeconomic, and TPA review teams, provides a more comprehensive understanding of the ethical considerations related to the use of tofersen for the treatment of SOD1-ALS. It is possible that more direct engagement (e.g., through direct interviews) with patients, their caregivers, their family members, and decision-makers on their specific experiences with SOD1-ALS and/or tofersen would offer additional relevant ethical considerations or domains of analysis.
Conclusion
This report examined the ethical considerations associated with the use of tofersen for the treatment of adults with a confirmed diagnosis of SOD1-ALS by drawing on input from patient groups, clinician groups, provincial drug programs, direct engagement with 5 clinical experts, a review of published literature, and engagement with the CDA-AMC review team. ALS is a severe disease and there are no treatments specifically for SOD1-ALS. Tofersen is the first SOD1-ALS therapy proposed to treat the underlying genetic cause of SOD1-ALS and to significantly alter the disease course.
There is a lengthy initial time to diagnosis for patients with ALS, and especially those patients from racialized or underserved groups. The diagnosis of SOD1-ALS is further challenged by geographic disparities in access to genetic testing, and the existence of SOD1 mutations of undetermined significance. Patients who have ALS and their caregivers experience physical, emotional, and financial burdens associated with the disease, including severe challenges related to tasks of daily living, profound distress, and expenses associated with purchasing medical equipment and supports.
The VALOR trial assessed the efficacy, safety, and tolerability of tofersen administered to adults with a confirmed SOD1-ALS diagnosis. The trial failed to meet its primary efficacy end point and although there were meaningful differences in the 2 biomarkers used as secondary end points, their clinical meaningfulness was unvalidated. Though the VALOR trial’s OLE study and the findings from the EAPs’ studies suggested trends toward slowed disease progression, significant methodological limitations temper confidence in their findings. No studies have assessed tofersen’s comparative effectiveness relative to the current standard of care. Further, several patient groups that likely would be encountered in clinical practice in Canada were underrepresented or excluded from the trials. Collectively, the uncertainties associated with the evaluation of tofersen complicates decision-making by patients, clinicians, and health systems. These uncertainties are heightened for groups that were underrepresented or excluded from the trial populations.
Treatment with tofersen will entail several access challenges because tofersen must be prescribed and monitored by ALS specialists in tertiary care centres. Although access could be improved if patients received their monthly intrathecal injections in local settings, some patients would continue to experience access challenges. The use of tofersen may pose several risks for patients as compared to the current standard of care, including serious neurologic adverse events and risks associated with its intrathecal mode of delivery. The context of enhanced expectations and hope for effective treatments for ALS should be accounted for in clinical decision-making regarding the potential benefits, harms, and evidentiary uncertainties associated with tofersen.
The implementation of tofersen may introduce health resource usage and system integration challenges, especially due to the resource-intensive nature of the intrathecal mode of delivery and the need to monitor and manage serious adverse events. Ongoing data collection and postmarket evaluation may address some of the evidentiary uncertainties associated with tofersen to facilitate health system decision-making but will require further health system infrastructure. The equitable implementation of tofersen may also require increased capacity of local laboratories and genetic counsellors to support increased testing requirements for SOD1 gene variants, including the potential for increased demands for predictive testing.
References
1.EUnetHTA. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0. 2016. Accessed November 4, 2025. https://web.archive.org/web/20241116160013/https://www.eunethta.eu/wp-content/uploads/2018/03/HTACoreModel3.0-1.pdf
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37:e17. doi:10.1017/S0266462320002275 PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-40. doi:10.1002/1098-240x(200008)23:4<334::aid-nur9>3.0.co;2-g PubMed
4.Bunton-Stasyshyn RK, Saccon RA, Fratta P, Fisher EM. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist. 2015;21(5):519-29. doi:10.1177/1073858414561795 PubMed
5.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi:10.1186/1750-1172-4-3 PubMed
6.Logroscino G, Traynor BJ, Hardiman O, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385-90. doi:10.1136/jnnp.2009.183525 PubMed
7.Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum (Minneap Minn). 2014;20(5 Peripheral Nervous System Disorders):1185-207. doi:10.1212/01.CON.0000455886.14298.a4
8.Wolf J, Safer A, Wöhrle JC, et al. Factors predicting one-year mortality in amyotrophic lateral sclerosis patients--data from a population-based registry. BMC Neurol. 2014;14:197. doi:10.1186/s12883-014-0197-9 PubMed
9.ALS Association. Stages of ALS. Accessed Feburary 11, 2025. https://www.als.org/understanding-als/stages
10.Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623-7. doi:10.1136/jnnp.2010.224501 PubMed
11.Zarei S, Carr K, Reiley L, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171. doi:10.4103/2152-7806.169561 PubMed
12.Shepheard SR, Parker MD, Cooper-Knock J, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(5):510-518. doi:10.1136/jnnp-2020-325014 PubMed
13.Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. 2018;30(2):252-258. doi:10.1007/s11825-018-0185-3 PubMed
14.Dharmadasa T, Henderson RD, Talman PS, et al. Motor neurone disease: progress and challenges. Med J Aust. 2017;206(8):357-362. doi:10.5694/mja16.01063 PubMed
15.McCluskey L. Amyotrophic Lateral Sclerosis: ethical issues from diagnosis to end of life. NeuroRehabilitation. 2007;22(6):463-72. doi:10.3233/NRE-2007-22609 PubMed
16.Johnson S, Alonso B, Faulkner K, et al. Quality of Life Perspectives of People With Amyotrophic Lateral Sclerosis and Their Caregivers. Am J Occup Ther. 2017;71(3):7103190010p1-7103190010p7. doi:10.5014/ajot.2017.024828
17.Matuz T, Birbaumer N, Hautzinger M, Kübler A. Coping with amyotrophic lateral sclerosis: an integrative view. J Neurol Neurosurg Psychiatry. 2010;81(8):893-8. doi:10.1136/jnnp.2009.201285 PubMed
18.Mukherjee D. Recognizing Psychological Diversity in People with End-Stage Amyotrophic Lateral Sclerosis. AMA J Ethics. 2015;17(6):530-4. doi:10.1001/journalofethics.2015.17.6.nlit2-1506 PubMed
19.Rabkin JG, Goetz R, Factor-Litvak P, et al. Depression and wish to die in a multicenter cohort of ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(3-4):265-73. doi:10.3109/21678421.2014.980428 PubMed
20.Eisen A, Krieger C. Ethical considerations in the management of amyotrophic lateral sclerosis. Prog Neurobiol. 2013;110:45-53. doi:10.1016/j.pneurobio.2013.05.001 PubMed
21.Hodgkinson VL, Lounsberry J, Mirian A, et al. Provincial Differences in the Diagnosis and Care of Amyotrophic Lateral Sclerosis. Can J Neurol Sci. 2018;45(6):652-659. doi:10.1017/cjn.2018.311 PubMed
22.Wong W. Managed care considerations to improve health care utilization for patients with ALS. Am J Manag Care. 2023;29(7 Suppl):S120-S126. doi:10.37765/ajmc.2023.89388 PubMed
23.Vitaliano PP, Zhang J, Scanlan JM. Is caregiving hazardous to one's physical health? A meta-analysis. Psychol Bull. 2003;129(6):946-72. doi:10.1037/0033-2909.129.6.946 PubMed
24.Weisser FB, Bristowe K, Jackson D. Experiences of burden, needs, rewards and resilience in family caregivers of people living with Motor Neurone Disease/Amyotrophic Lateral Sclerosis: A secondary thematic analysis of qualitative interviews. Palliat Med. 2015;29(8):737-45. doi:10.1177/0269216315575851 PubMed
25.Cipolletta S, Amicucci L. The family experience of living with a person with amyotrophic lateral sclerosis: a qualitative study. Int J Psychol. 2015;50(4):288-94. doi:10.1002/ijop.12085 PubMed
26.Raymond J, Nair T, Gwathmey KG, Larson T, Horton DK, Mehta P. Racial Disparities in the Diagnosis and Prognosis of ALS Patients in the United States. J Racial Ethn Health Disparities. 2024:1-7. doi:10.1007/s40615-024-02099-6 PubMed
27.Hart AA, Swenson A, Narayanan NS, Simmering JE. Rurality modifies the association between symptoms and the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(5-6):517-527. doi:10.1080/21678421.2024.2315185 PubMed
28.Chen S, Carter D, Brockenbrough PB, Cox S, Gwathmey K. Racial disparities in ALS diagnostic delay: a single center's experience and review of potential contributing factors. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(1-2):112-118. doi:10.1080/21678421.2023.2273361 PubMed
29.Spencer D, Polke J, Campbell J, Houlden H, Radunovic A. 'Outcomes of genetic testing in the London MND Center: the importance of achieving timely results and correlations to family history'. Amyotroph Lateral Scler Frontotemporal Degener. 2024;25(7-8):737-742. doi:10.1080/21678421.2024.2370808 PubMed
30.Meyer T, Schumann P, Grehl T, et al. SOD1 gene screening in ALS - frequency of mutations, patients' attitudes to genetic information and transition to tofersen treatment in a multi-center program. Amyotroph Lateral Scler Frontotemporal Degener. 2025;26(1-2):162-171. doi:10.1080/21678421.2024.2401131 PubMed
31.Delhaas EM, Huygen F. Complications associated with intrathecal drug delivery systems. BJA Educ. 2020;20(2):51-57. doi:10.1016/j.bjae.2019.11.002 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.