Drugs, Health Technologies, Health Systems
Reimbursement Review
Bulevirtide (Hepcludex)
Sponsor: Gilead Sciences Canada, Inc.
Therapeutic area: Chronic hepatitis delta virus (HDV) infection
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AASLD
American Association for the Study of Liver Diseases
AE
adverse event
ALT
alanine aminotransferase
CDA-AMC
Canada's Drug Agency
CI
confidence interval
CrI
credible interval
DIC
deviance information criterion
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HBsAg
hepatitis B surface antigen
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
HCV
hepatitis C virus
HDV
hepatitis delta virus
HQLQ
Hepatitis Quality of Life Questionnaire
HRQoL
health-related quality of life
IQR
interquartile range
ITC
indirect treatment comparison
LLoQ
lower limit of quantification
LOCF
last observation carried forward
LS
least squares
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
PCR
polymerase chain reaction
PEG-IFN
pegylated interferon
RCT
randomized controlled trial
RNA
ribonucleic acid
RT-PCR
reverse transcription-polymerase chain reaction
SAE
serious adverse event
SD
standard deviation
SF-36
Short Form (36) Health Survey
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Bulevirtide (Hepcludex), 2 mg, powder for solution for injection, subcutaneous |
Sponsor | Gilead Sciences Canada, Inc. |
Indication | For the treatment of chronic hepatitis delta virus (HDV) infection in adults with compensated liver disease |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority |
NOC date | August 8, 2025 |
Recommended dose | 2 mg once daily administered by subcutaneous injection |
HDV = hepatitis delta virus; NOC = Notice of Compliance.
Introduction
Hepatitis delta is a rare, severe, and progressive liver disease caused by the hepatitis delta virus (HDV), an incomplete ribonucleic acid (RNA) virus that requires the hepatitis B virus (HBV) envelope to enter cells and replicate. HDV is considered the fastest progressing and most severe form of all viral hepatitis infections. HDV is a “satellite virus” and can only infect individuals with a concomitant HBV infection.1,2 Chronic HDV infection is defined as an infection lasting 6 months or more.3 The likelihood of progression to chronic hepatitis depends on whether the initial infection with HDV occurred by HBV and HDV coinfection or superinfection. While less than 5% of patients progress to a chronic infection after coinfection, HDV infection becomes chronic in more than 80% of individuals with HBV and HDV superinfection.4
A 2024 study from the Polaris Observatory found that the adjusted prevalence of HDV in the population infected with HBV in Canada is approximately 3.0%.5 These estimates are consistent with a retrospective study conducted using data collected from patients seen by physicians in the Canadian HBV Network (9 clinics in 6 provinces across Canada), which reported an estimated rate of 4.8% (95% confidence interval [CI], 4.3% to 5.3%).6 Given that the prevalence of chronic HBV in Canada is estimated at 0.66%7 and the HBV diagnosis rate is 70%,5 the prevalence of HDV (both chronic and acute) is estimated at between 0.020%5 and 0.032% %,6 which equates to 2 to 3.2 per 10,000 population.
Key signs and symptoms of chronic HDV infection can range from nonspecific symptoms to rapidly progressing hepatitis. Symptoms may include fever, fatigue, loss of appetite, nausea, vomiting, abdominal pain, dark urine, clay-coloured bowel movements, joint pain, and jaundice.4 Chronic hepatitis usually exacerbates any pre-existing liver disease associated with HBV.8 Recovery is unlikely once a person has an infection, with the likelihood of recovery decreasing as the disease progresses. Only 35% of patients with acute infection, and even less (9.96%) with chronic infection, recover.9 For those who do not recover at the early stages of the disease, HDV infection is associated with an accelerated progression to fibrosis, early liver decompensation with cirrhosis, and increased risk of hepatocellular carcinoma (HCC), leading to greater liver-related mortality compared to HBV or hepatitis C virus (HCV).10-12
Diagnosis of HDV generally involves 2 steps. The first step is detection of anti-HDV antibodies through an enzyme-linked immunosorbent assay to assess if a patient has ever been exposed to HDV. If the anti-HDV antibody test is positive, the patient is automatically tested for HDV RNA using reverse transcription-polymerase chain reaction (RT-PCR), to assess whether the HDV infection is active or chronic. Standard liver function tests are routinely performed if a patient already receives care for HBV; these assess the severity of a patient’s HBV and/or HDV disease. These liver function tests are done at initial diagnosis and to monitor treatment response.
According to the clinical experts consulted by Canada's Drug Agency (CDA-AMC) for this review, HDV occurs as a coinfection with HBV and is associated with a more aggressive liver disease, with a higher risk of cirrhosis, hepatic decompensation, and HCC. There are currently no approved treatments for chronic HDV in Canada.13-15
The European Association for the Study of the Liver and the American Association for the Study of Liver Diseases (AASLD)13-15 have indicated that pegylated interferon (PEG-IFN)-alpha has potential clinical benefit for patients with HBV and HDV coinfections. Of note, PEG-IFN-alpha is no longer used for HDV treatment in Canada due to its poor tolerability and limited accessibility. AASLD emphasized the importance of suppressing HBV replication in patients with chronic hepatitis D and elevated HBV DNA levels. Nucleoside (or nucleotide) analogues inhibit HBV DNA synthesis, but have no effect on HDV replication, which is independent of HBV. While guidelines vary on the use of nucleoside (or nucleotide) analogues for patients with HBV and HDV coinfections, they consistently highlight the lack of their efficacy against HDV.13-16
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of bulevirtide monotherapy, administered once daily by subcutaneous injection at a dose of 2 mg, for the treatment of chronic HDV infection in adults with compensated liver disease.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted by for the purpose of this review.
Patient Input
CDA-AMC received a joint patient input submission from 2 organizations, Liver Canada and BC Hepatitis Network. Patient perspectives were gathered via an online survey conducted between February 21 and March 8, 2025, using social media. The 6 patients who responded to the survey, ranged in age from their 30s to their 60s, and were living with HDV or caring for someone with HDV living in Ontario, Alberta, or British Columbia. The patient groups described how receiving a diagnosis of HDV and experiencing disease-specific symptoms, including general physical and psychological symptoms, substantially impacts patients’ health-related quality of life (HRQoL). PEG-IFN is used off-label in Canada for the treatment of HDV. Patients with experience with PEG-IFN treatment highlighted the associated intolerable side effects, frequent travel to clinics for injections, and frequent blood tests for monitoring purposes. Patient respondents indicated a preference for treatments that require fewer injections and blood tests.
Patient respondents described stabilization of their liver disease as an important treatment outcome. Stabilization refers to slowed progression to liver fibrosis, cirrhosis, and liver cancer; improvement in decompensation events; and reduced need for a liver transplant. According to the patient input received for this review, chronic HDV is most common among newcomers and immigrants to Canada, who face multiple barriers to timely diagnosis. According to the patient input, these individuals are less likely to be screened for viral hepatitis, face limited access to routine care, and are more likely to experience fear and stigma related to hepatitis. Together, these barriers place individuals at higher risk for late diagnosis and advanced viral hepatitis–related liver disease. Patient respondents identified reflex testing for HDV among individuals with HBV as an important unmet need.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted by CDA-AMC for this review identified significant unmet needs in the management of chronic HDV infection in Canada, emphasizing the absence of approved or reimbursed therapies. The clinical experts indicated that although PEG-IFN was previously used off-label for the treatment of HDV, it is no longer used in clinical practice due to its poor tolerability, limited accessibility, and lack of feasibility for routine use. The clinical experts emphasized that currently available therapies for hepatitis B, including nucleoside (or nucleotide) analogues, are ineffective against HDV infection, which remains the primary driver of liver damage in individuals with coinfections. As a result, patients with HDV experience progressive liver disease without access to an effective treatment option. The clinical experts highlighted the need for a targeted therapy to reduce HDV viral load, normalize alanine aminotransferase (ALT) levels, potentially stabilize or improve liver fibrosis, and mitigate the risk of HCC and liver decompensation. This need is particularly urgent due to the aggressive nature of HDV infection and the associated risk of rapid progression to cirrhosis and HCC. The clinical experts noted that early initiation of bulevirtide is recommended to help mitigate the risk of progression to advanced liver disease. The clinical experts stated that bulevirtide would be used in combination with nucleoside (or nucleotide) analogues, such as tenofovir or entecavir, which are used to manage the HBV coinfection but are ineffective against HDV. The clinical experts identified individuals with compensated cirrhosis as the most in need of bulevirtide treatment but emphasized that all patients with chronic HDV require treatment due to the potential for rapid disease progression.
The clinical experts noted that the key outcomes used to determine response to treatment with bulevirtide include virologic response (i.e., undetectable HDV RNA or reduction in HDV RNA levels), biochemical response (i.e., normalization of ALT levels), and stabilization or improvement in liver fibrosis. According to the clinical experts, long-term outcomes such as improvements in portal hypertension, reduced progression to cirrhosis, and lower rates of HCC would also be clinically beneficial but were not assessed in trials. The clinical experts indicated that bulevirtide treatment should generally be discontinued in cases of progression to decompensated liver disease requiring transplant or the development of HCC, which requires other therapy. However, they emphasized that treatment decisions for patients with decompensated liver disease and those with HCC should be made on a case by case basis, as discontinuing HDV therapy could potentially worsen their condition. The clinical experts noted that discontinuation can be considered in the event of severe adverse reactions, such as allergy or significant injection site reactions, or if the patient experiences barriers to adherence to the daily injectable therapy. The clinical experts indicated that diagnosis of HDV infection, prescribing of bulevirtide, and monitoring of treatment response should be managed by clinicians with specialized expertise in hepatology or infectious diseases, given the complexity of HDV infection and the need for accurate interpretation of HDV RNA and ALT levels. The clinical experts also noted that the optimal duration of bulevirtide therapy remains uncertain for those patients with hepatitis infection that responds more slowly to treatment. According to the clinical experts, underdiagnosis of HDV is a significant issue in practice, largely due to the limited availability of HDV RNA testing and lack of routine screening, posing a barrier to timely treatment initiation. The clinical experts noted that systematic screening is lacking, particularly in individuals with HBV infection receiving care in the primary care setting.
Clinician Group Input
Ten clinicians from the Canadian Hepatitis B Network provided input. The Canadian Hepatitis B Network is a collaborative organization of health care professionals and researchers from across Canada with an interest in advancing excellence in the care of patients with hepatitis B, research into hepatitis B, and education. The clinician group stated that there is an unmet need for improved therapies for HDV and HBV coinfection. There are currently no Health Canada–approved treatments for HDV infection. HDV causes the most severe form of viral hepatitis in humans. HDV requires the HBV surface (or envelope) protein to infect the liver, and therefore can only infect people who have HBV. Individuals can acquire HBV and HDV at the same time or HDV can superinfect a patient with underlying chronic hepatitis B infection because of a shared route of transmission. The drug under review, bulevirtide, is a novel antiviral drug specifically targeting the binding of HDV (and HBV) to the liver-specific cell surface bile acid receptor, NTCP. Based on input received from the clinician group, patients with HBV and HDV coinfection and positive test results for HDV RNA are most likely to respond to the drug under review.
Patients with HDV coinfection are at substantial risk of liver disease and cirrhosis within 5 years of diagnosis, regardless of any disease characteristics. Patients best suited for treatment with the drug under review would be identified based on the clinical judgment of expert specialists in hepatology or infectious disease as well as by testing for HDV RNA. The clinician group highlighted that outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials. A clinically meaningful response to treatment would be HDV RNA suppression, biochemical normalization, improvement in noninvasive fibrosis test results, and/or improvement in symptoms of liver disease decompensation (e.g., ascites, variceal bleeding). Based on input received from the clinician group, the criteria for discontinuing treatment include treatment failure, disease progression, and limited lifespan. The clinician group stated that the drug under review can be appropriately prescribed by a hepatology and infectious diseases specialist in a specialty clinic setting.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC reimbursement review process. The following were identified as key factors that could potentially impact CDA-AMC recommending bulevirtide:
relevant comparators
consideration for initiation of therapy
consideration for continuation or renewal of therapy
consideration of discontinuation of therapy
consideration for prescribing of therapy
generalizability
care provision issues
system and economic issues.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Systematic Review
Description of Studies
The sponsor-conducted systematic literature review (SLR) identified 1 pivotal, ongoing, phase III, randomized, open-label, parallel-group, multicentre trial (MYR301, N = 150). The primary objective of the MYR301 trial was to evaluate the efficacy and safety of bulevirtide at a dose of 2 mg for the treatment of chronic HDV infection in adults with compensated liver disease compared to delayed treatment with bulevirtide 10 mg after an observation period of 48 weeks. The secondary objectives of the trial included assessing the safety of bulevirtide and determining the optimal treatment duration. Patients were enrolled from 16 sites across 4 countries — Germany, Italy, Russia, and Sweden. The study includes a 4-week screening period, a 144-week treatment period, and a 96-week follow-up period. Following screening, patients were randomized using an electronic randomization system in a 1:1:1 ratio to receive delayed treatment with bulevirtide 10 mg after a 48-week observation period (N = 51), immediate treatment with bulevirtide 2 mg (N = 49), or immediate treatment with bulevirtide 10 mg (N = 50). This report primarily focuses on between-group comparisons of study end points up to week 48, specifically for bulevirtide at a dose of 2 mg, in line with the Health Canada indication. Data for the study group receiving immediate treatment with bulevirtide at a dose of 10 mg are not presented in this report because this dose is neither approved for use nor in use in clinical practice in Canada. Efficacy end points of interest to this review included combined response, undetectable HDV RNA or HDV RNA decrease of at least 2 log10 IU/mL from baseline, change in liver stiffness from baseline, ALT normalization, undetectable HDV RNA, liver-related clinical events, HRQoL, and safety outcomes. The primary and secondary end points of the MYR301 trial were analyzed using data up to the cut-off date of September 30, 2022, with the database locked on June 8, 2023.
The mean age of patients was higher in the bulevirtide 2 mg group (44 years [SD = 9.0 years]) than in the delayed treatment group (41 years [SD = 7.5 years]). The proportion of male patients was larger in the bulevirtide 2 mg group (61.2%) than in the delayed-treatment group (51.0%). Nearly one-half of patients (47.3%) had cirrhosis at the time of randomization; in all cases, cirrhosis was classified as Child-Pugh Class A. All patients in both the bulevirtide 2 mg and the delayed-treatment group had HDV genotype 1 at baseline. A larger proportion of patients in the bulevirtide 2 mg group had cirrhosis with a Child-Pugh score of 6 points (30.4% versus 20.8% in the delayed-treatment group), HBV genotype D (95.9% versus 86.3% in the delayed-treatment group), and HBV DNA greater than or equal to the lower limit of quantification (LLoQ) (67.3% versus 52.9% in the delayed-treatment group). A total of 65.3% in the bulevirtide 2 mg group and 62.7% in the delayed-treatment group received oral anti-HBV treatment during the MYR301 trial; 54.7% of these patients had initiated this treatment before baseline. In addition, 53.1% of patients in the bulevirtide 2 mg group and 62.7% of patients in the delayed-treatment group had previously received interferon treatment. Most of the disease characteristics in the delayed-treatment group were similar at randomization at week 48, with a few notable exceptions. Specifically, the mean serum ALT level was higher at randomization (102 U/L [SD = 61.9 U/L]) than at week 48 (82 U/L [SD = 51.1 U/L]) and the mean liver stiffness was 15.3 kPa (SD = 8.95 kPa) at randomization and 16.1 kPa (SD = 11.84 kPa) at week 48.
Efficacy Results
Combined Response
Combined response at week 48 was a primary end point in the MYR301 trial. A combined response is defined as the simultaneous fulfillment of 2 conditions: undetectable HDV RNA (less than the lower limit of detection) or a decrease of at least 2 log10 IU/mL from baseline, and ALT normalization. At week 48, 44.9% (95% CI, 30.7% to 59.8%) of patients in the bulevirtide 2 mg group compared to 2.0% (95% CI, 0.0% to 10.4%) of those in the delayed-treatment group achieved a combined response. The between-group difference in response rates was 42.9% (96% CI, 27.0% to 58.5%; P < 0.0001) in favour of the bulevirtide 2 mg group compared to the delayed-treatment group. The sensitivity analysis results, using the missing equals failure approach, were consistent with the primary analysis. Subgroup analyses of the primary end point, combined response at week 48, were conducted based on cirrhosis status. In the bulevirtide 2 mg group, the proportion of patients without cirrhosis who achieved a combined response was 53.8% (95% CI, 16.4% to 57.3%) compared to 34.8% (95% CI, 33.4% to 73.4%) of patients with cirrhosis. In the delayed-treatment group, 4.2% (95% CI, 0.1% to 21.1%) of patients with cirrhosis achieved a combined response compared to 0.0% (95% CI, 0.0% to 12.8%) of those without cirrhosis.
Undetectable HDV RNA at Weeks 24 and 48 After Scheduled End of Treatment
An undetectable HDV RNA at weeks 24 and 48 after scheduled end of treatment (sustained virologic response) was a secondary end point in the MYR301 trial; however, the results for this end point were not reported due to an insufficient follow-up period as the study is still ongoing.
Undetectable HDV RNA or HDV RNA Decrease by at Least 2 log10 IU/mL at Week 48
A decrease in HDV RNA of at least 2 log10 IU/mL or undetectable HDV RNA (virologic response) at week 48 was an additional secondary end point in the MYR301 trial. At Week 48, 73.5% (95% CI, 58.9% to 85.1%) of patients in the bulevirtide 2 mg group achieved a virologic response compared to 3.9% (95% CI, 0.5% to 13.5%) in the delayed-treatment group. The between-group difference in response rates was 69.5% (96% CI, 54.1% to 81.9%; P < 0.0001) in favour of the bulevirtide 2 mg group, compared to the delayed-treatment group. Subgroup analyses of virologic response were conducted based on cirrhosis status and concomitant treatment with HBV medication (ad hoc analysis). At week 48, 82.6% (95% CI, 61.2% to 95.0%) of the patients with cirrhosis in the bulevirtide 2 mg group and 8.3% (95% CI, 1.0% to 27.0%) of those in the delayed-treatment group had achieved a virologic response. Among patients without cirrhosis, the corresponding proportions were 65.4% (95% CI, 44.3% to 82.8%) and 0.0% (95% CI, 0.0% to 12.8%), respectively. At week 48, 78.1% (95% CI, 60.0% to 90.7%) of patients receiving concomitant HBV medication in the bulevirtide 2 mg group achieved a virologic response compared to 6.3% (95% CI, 0.8% to 20.8%) of those in the delayed-treatment group. Among the patients not receiving HBV medication, the proportions who achieved a virologic response were 64.7% (95% CI, 38.3% to 85.0%) and 0.0% (95% CI, 0.0% to 17.6%), respectively.
Change From Baseline in Liver Stiffness at Week 48
The change from baseline in liver stiffness assessed using FibroScan was a prespecified secondary end point for the week 48 analysis. At week 48, the least squares (LS) mean of change from baseline in liver stiffness was –3.06 kPa (95% CI, –4.67 kPa to –1.45 kPa) in the bulevirtide 2 mg group and 0.87 kPa (95% CI, −0.79 kPa to 2.53 kPa) in the delayed-treatment group. The between-group difference in LS means of change in liver stiffness was –3.93 (95% CI, –6.23 to –1.63; P = 0.0009) in favour of the bulevirtide 2 mg group. Subgroup analyses of the change in liver stiffness were conducted based on cirrhosis status. In the bulevirtide 2 mg group, the mean change from baseline in liver stiffness was –5.70 kPa (SD = 6.94 kPa) in patients with cirrhosis and –0.35 kPa (SD = 2.08 kPa) in patients without cirrhosis. In the delayed-treatment group, the mean change from baseline in liver stiffness was –0.20 kPa (SD = 4.00 kPa) in patients without cirrhosis and 1.78 kPa (SD = 10.1 kPa) in patients with cirrhosis.
Liver-Related Clinical Events
In the MYR301 trial, | █████████████ ███████████████ ███ ████████ ██ █ ███████ ██ ███ ███████████ █ ██ ██████ ██ ███ █ ███████ █████ █████████ ███ ██████████ ███████ ███ ███ ███ ██████████ ████ ███ ███████ ██████ █████████████ █ ███████ ████ █████████ ██ ███ ███████ █████████ █████ █████████ ███████████ █████ █ ████████ ███ ████ █████ █████████ ██ ███████████ ██ ██ ██████████.
Health-Related Quality of Life
The Hepatitis Quality of Life Questionnaire (HQLQ) was used to assess the quality of life of patients in the MYR301 trial. The HQLQ17 includes items from the Short Form (36) Health Survey (SF-36) and 4 hepatitis-specific health domain scores, including health distress, positive well-being, hepatitis-specific limitations, and hepatitis-specific health distress. Each domain is scored on a scale from 0 to 100, with higher scores indicating better HRQoL.
At week 24, compared to the delayed-treatment group, the LS mean differences in the bulevirtide 2 mg group were ███ (95% CI, ███ ██ ███) for the HQLQ physical component summary score, ███ (95% CI, ████ ██ ███) for the HQLQ mental component summary score, ████ (95% CI, ███ ██ ████) for the HQLQ health distress score, ███ (95% CI, ████ ██ ████) for the HQLQ positive well-being score| ███ (95% CI, ████ ██ ████) for the HQLQ hepatitis-specific limitations score, and ███ (95% CI, ████ ██ ████) for the HQLQ hepatitis-specific health distress score.
At week 48, compared to the delayed-treatment group, the LS mean differences in the bulevirtide 2 mg group were ███ (95% CI, ███ ██ ███) for the HQLQ physical component summary score, ███ (95% CI, ████ ██ ███) for the HQLQ mental component summary score, ███ (95% CI, ████ ██ ████) for the HQLQ health distress score, ███ (95% CI, ████ ██ ███) for the HQLQ positive well-being score, ███ (95% CI, ███ ██ ████) for the HQLQ hepatitis-specific limitations score, and ███ (95% CI, ███ ██ ████) for the HQLQ hepatitis-specific health distress score.
Undetectable HDV RNA
Undetectable HDV RNA (less than the LLoQ, target not detected) at week 48 was a key secondary end point in the MYR301 trial. At week 48, 12.2% (95% CI, 4.6% to 24.8%) of patients in the bulevirtide 2 mg group had undetectable HDV RNA compared to 0.0% (95% CI, 0.0% to 7.0%) of patients in the delayed-treatment group. The between-group difference in response rates was only calculated for the bulevirtide 10 mg group with the bulevirtide 2 mg group as the reference; this comparison is not relevant to this review. Subgroup analyses of the key secondary end point were conducted based on cirrhosis status and concomitant treatment with nucleoside (or nucleotide) analogues (ad hoc analysis). In the bulevirtide 2 mg group, 21.7% (95% CI, 7.5% to 43.7%) of patients with cirrhosis achieved undetectable HDV RNA, compared to 3.8% (95% CI, 0.1% to 19.6%) of those without cirrhosis. In the delayed-treatment group, 0.0% (95% CI, 0.0% to 14.2%) of patients with cirrhosis achieved an undetectable HDV RNA, compared to 0.0% (95% CI, 0.0% to 12.8%) of those without cirrhosis. Similarly, of the patients in the bulevirtide 2 mg group receiving concomitant HBV medication, 15.6% (95% CI, 5.3% to 32.8%) achieved undetectable HDV RNA compared to 5.9% (95% CI, 0.1% to 28.7%) of those not receiving HBV medication. In the delayed-treatment group, 0.0% of the patients achieved an undetectable HDV RNA, regardless of whether they were receiving concomitant HBV medication
ALT Normalization
ALT normalization at week 48 was a secondary end point in the MYR301 trial. At week 48, 51.0% (95% CI, 36.3% to 65.6%) of patients in the bulevirtide 2 mg group achieved ALT normalization compared to 11.8% (95% CI, 4.4% to 23.9%) of patients in the delayed-treatment group. The between-group difference in response rates was 39.3% (96% CI, 20.0% to 55.8%; P < 0.0001) in favour of the bulevirtide 2 mg group compared to the delayed-treatment group. Subgroup analyses of ALT normalization were conducted based on cirrhosis status and concomitant treatment with HBV medication. At week 48, 61.5% (95% CI, 40.6% to 79.8%) of patients without cirrhosis achieved ALT normalization compared to 39.1% (95% CI, 19.7% to 61.5%) of those with cirrhosis. In the delayed-treatment group, 7.4% (95% CI, 0.9% to 24.3%) of patients without cirrhosis achieved ALT normalization compared to 16.7% (95% CI, 4.7% to 37.4%) of patients with cirrhosis. Similarly, of patients in the bulevirtide 2 mg group who did not receive concomitant HBV medication, 52.9% (95% CI, 27.8% to 77.0%) achieved ALT normalization compared to 50.0% (95% CI, 31.9% to 68.1%) of those who received HBV medication. In the delayed-treatment group, 15.8% (95% CI, 3.4% to 39.6%) of patients who did not receive concomitant HBV medication achieved ALT normalization compared to 9.4% (95% CI, 2.0% to 25.0%) of those who received HBV medication.
Combined Response Over Time
The combined response at each study visit, an exploratory end point in the MYR301 trial, was analyzed exclusively using a missing equals failure approach. In contrast, the primary end point analysis incorporated a last observation carried forward (LOCF) approach to account for missing data due to COVID-19 pandemic-related restrictions at weeks 24 and 48. The combined response rates in the bulevirtide 2 mg group demonstrated consistent improvement over time: 34.7% (95% CI, 21.7% to 49.6%) at week 24, 44.9% (95% CI, 30.7% to 59.8%) at weeks 48 ███ ██, 55.1% (95% CI, 40.2% to 69.3%) at week 96, and 57.1% (95% CI, 42.2% to 71.2%) at week 144.
Virologic Response Over Time
The virologic response at each study visit, an exploratory end point in the MYR301 trial, was analyzed exclusively using a missing equals failure approach. The virologic response rates in the bulevirtide 2 mg group demonstrated improvement over time: 55.1% (95% CI, 40.2% to 69.3%) at week 24, 73.5% (95% CI, 58.9% to 85.1%) at week 48, █████ ████ ███ █████ ██ ██████ ██ ████ ██, 75.5% (95% CI, 61.1% to 86.7%) at week 96, and 73.5% (95% CI, 58.9% to 85.1%) at week 144.
Harms Results
Adverse Events
In the MYR301 trial, 83.7% of patients in the bulevirtide 2 mg group and 80.4% in the delayed-treatment group had experienced at least 1 adverse event (AE) by week 48. The most common AEs in the bulevirtide 2 mg group were headache (18.4% versus 0.0% in the delayed-treatment group), vitamin D deficiency (14.3% versus 25.5% in the delayed-treatment group), leukopenia (14.3% versus 19.6% in the delayed-treatment group), thrombocytopenia (10.2% versus 15.7% in the delayed-treatment group), and pruritus (12.2% versus 0.0% in the delayed-treatment group). The proportions of patients with grade 3 or higher AEs were 10.2% in the bulevirtide 2 mg group and 7.8% in the delayed-treatment group. Among patients in the bulevirtide 2 mg group, these grade 3 or higher AEs included depression (2.0% versus 0.0% in the delayed-treatment group), foot fracture (2.0% versus 0.0% in the delayed-treatment group), decreased neutrophil count (2.0% versus 0.0% in the delayed-treatment group), osteopenia (2.0% versus 0.0% in the delayed-treatment group), and thrombocytopenia (2.0% versus 5.9% in the delayed-treatment group).
At week 144, the proportion of patients who experienced at least 1 AE in the bulevirtide 2 mg group was 98.0%; 24.5% of patients in the bulevirtide 2 mg group experienced AEs that were grade 3 or higher. The most common AEs in the bulevirtide 2 mg group were vitamin D deficiency (44.9%), headache (20.4%), leukopenia (20.4%), thrombocytopenia (20.4%), lymphopenia (16.3%), and neutropenia (16.3%).
Serious Adverse Events
In the MYR301 trial, the proportions of patients who had experienced at least 1 serious adverse event (SAE) by week 48 were 4.1% in the bulevirtide 2 mg group and 2.0% in the delayed-treatment group. Most SAEs were experienced by 1 patient across the treatment groups. The SAEs in the bulevirtide 2 mg group included foot fracture (2.0% versus 0.0% in the delayed-treatment group), headache (2.0% versus 0.0% in the delayed-treatment group), hemiparesis (2.0% versus 0.0% in the delayed-treatment group), and depression (2.0% versus 0.0% in the delayed-treatment group). The SAEs in the delayed-treatment group included cholelithiasis (2.0% versus 0.0% in the bulevirtide 2 mg group) and COVID-19 (2.0% versus 0.0% in the bulevirtide 2 mg group).
At week 144, the proportion of patients who experienced at least 1 SAE in the bulevirtide 2 mg group was 6.1%, including esophageal varices (2.0%), foot fracture (2.0%), headache (2.0%), hemiparesis (2.0%), and depression (2.0%). Between weeks 48 and 144 (representing 96 weeks of treatment with bulevirtide at a dose of 10 mg), the proportion of patients who experienced at least 1 SAE in the delayed treatment group was 6.0%. These SAEs included COVID-19 (2.0%), urinary tract infection (2.0%), and plasma cell myeloma (2.0%).
Withdrawals Due to AEs
By week 144, there were no discontinuations due to treatment-related AEs in the MYR301 trial.
Mortality
By week 144, there were no deaths in either the bulevirtide 2 mg or the delayed-treatment group.
Notable Harms
In the MYR301 trial at week 48, 14.3% of patients in the bulevirtide 2 mg group and 9.8% in the delayed-treatment group had experienced at least 1 hepatic AE. The hepatic AEs in the bulevirtide 2 mg group included increased ALT (4.1% versus 7.8% in the delayed-treatment group), hyperbilirubinemia (4.1% versus 0.0% in the delayed-treatment group), increased blood bilirubin (4.1% versus 0.0% in the delayed-treatment group), increased aspartate aminotransferase (2.0% versus 5.9% in the delayed-treatment group), and hepatic pain (2.0% versus 0.0% in the delayed-treatment group). At week 144, the proportion of patients in the bulevirtide 2 mg group who had experienced at least 1 hepatic AE was 28.6%.
At week 48, 8.2% of patients in the bulevirtide 2 mg group and 5.9% in the delayed-treatment group had experienced at least 1 renal AE. The hepatic AEs in the bulevirtide 2 mg group included proteinuria (6.1% versus 3.9% in the delayed-treatment group) and urinary retention (2.0% versus 0.0% in the delayed-treatment group). At week 144, 98.0% of the patients in the bulevirtide 2 mg group had experienced at least 1 renal AE. At week 48, 10.2% of the patients in the bulevirtide 2 mg group versus 0.0% in the delayed-treatment group had experienced eosinophilia; at week 144, this proportion remained unchanged, at 10.2% of those in the bulevirtide 2 mg group. At week 48, 6.1% of patients in the bulevirtide 2 mg group versus 0.0% in the delayed-treatment group had experienced injection site reactions; at week 144, 20.4% of patients in the bulevirtide 2 mg group had experienced injection site reaction.
Critical Appraisal
Randomization in the MYR301 trial was performed using an appropriate methodology and stratification was prespecified. All efficacy and safety analyses were conducted using the full and safety analysis sets, respectively, and these analyses included all patients who were randomized and who had received at least 1 dose of bulevirtide after randomization. Some imbalances in baseline characteristics, likely due to small sample sizes, were observed; these may have hindered the achievement of true prognostic balance. Given the rarity of HDV and the challenges in recruiting patients into clinical trials, achieving large sample sizes is often not feasible. The open-label design of the MYR301 trial introduces a potential bias in the assessment of study outcomes; however, this bias was mitigated by blinding to treatment allocation the central laboratories employed for hematology and biochemistry assessment. Knowledge of the assigned treatment could have led to biases in the reporting and measurement of subjective outcomes, including patient-reported outcomes (e.g., HRQoL) and subjective AEs. However, the extent and direction of bias due to treatment knowledge is uncertain. Although there are always some concerns for risk of bias due to deviations from the intended interventions in open-label trials, there were relatively few protocol deviations, and these were balanced across the groups and therefore unlikely to have influenced the study results. Adherence to the interventions and study completion rates were generally high in the intervention groups of interest to this review, reducing concerns regarding deviations from the intended interventions that could have arisen due to the trial context.
According to FDA guidance,18 given that no drugs are currently approved for the treatment of HDV infection, a placebo-controlled trial is the preferred design for a phase III clinical trial. An alternative design could be a randomized controlled trial (RCT) in which patients are randomized to receive either the investigational drug (the immediate-treatment group) or placebo for a prespecified duration, followed by open-label treatment with the investigational drug for the deferred-treatment group.18 According to FDA guidance,18 a surrogate end point that provides evidence of both a decline in virologic replication and an improvement in associated liver inflammation, evident as a biochemical response, could reasonably predict clinical benefit. However, FDA guidance suggests that subsequent confirmation using clinical end points is required, and these should be collected as part of a long-term follow-up study. Based on the enrolled sample size, the MYR301 study was powered to test its primary and key secondary end points. Statistical analysis methods appear to be acceptable. Both interim analyses at weeks 24 and 48 were planned a priori. The risk of bias due to missing outcome data was considered to be low. The lack of adjustment for multiple comparisons for all but the primary and key secondary end points increases the risk of type I error, where false positives may be incorrectly identified as significant. Bulevirtide was favoured in the analysis, but there was uncertainty due to the small sample size, which increased the risk that prognostic balance was not achieved. The unadjusted comparisons and the decision not to stratify the analysis by cirrhosis due to the small sample size may introduce bias and reduce the accuracy of the estimated treatment effects. Subgroup analyses by cirrhosis status and prior use of nucleoside (or nucleotide) analogues were prespecified; however, they were descriptive and limited by small sample sizes. No definitive conclusions could be drawn about the effect of bulevirtide compared with delayed treatment on HRQoL due to risk of bias from assessors being aware of treatment assignments, as well as serious imprecision, with wide CIs that included both potential benefit and harm.
According to the clinical experts consulted by CDA-AMC, the inclusion and exclusion criteria of the MYR301 trial appropriately reflects the population with HDV who are eligible for bulevirtide therapy in clinical practice. While patients aged older than 65 years were excluded from the trial, the clinical experts emphasized that age alone should not preclude treatment with bulevirtide. The clinical experts noted that, in clinical practice, physicians are unlikely to delay treatment initiation for patients with HBV and HDV coinfection who present with advanced fibrosis, suggesting that earlier intervention may be clinically appropriate in such cases. Patients with HCV or uncontrolled HIV coinfection were excluded from the MYR301 trial. However, the clinical experts noted that in clinical practice, these coinfections can be sequentially managed, with HIV and HCV treated first, before initiation of HBV and HDV therapy. The clinical experts noted that although patients with creatinine clearance of less than 60 mL/min were excluded from the trial, those with moderate renal impairment may still be considered for treatment with appropriate monitoring. The clinical experts agreed that it is reasonable to expect that a significant proportion of patients treated for HDV in clinical practice will have cirrhosis, given that many are diagnosed at more advanced stages of the disease. The clinical experts also noted that patients with early-stage fibrosis (Meta-analysis of Histological Data in Viral Hepatitis [METAVIR] fibrosis stage 1 [F1] or fibrosis stage 2 [F2]) may be more likely to seek treatment to prevent disease progression. As a result, treatment in real-world settings may be initiated across a broader range of disease severity than is typically represented in clinical trials. Approximately 65% of patients in the MYR301 trial received concomitant oral anti-HBV therapy at baseline and this was balanced across the treatment groups. However, the clinical experts indicated that in clinical practice nearly all patients with HDV typically receive treatment with nucleoside (or nucleotide) analogues. The clinical experts noted that although HBV therapy may slow fibrosis progression, this is unlikely to affect the generalizability of the study, as improvements in both virologic and fibrosis outcomes were observed in patients who had previously received HBV therapy (54.7%) and progressed to advanced disease. According to the clinical experts, the surrogate outcomes used in the MYR301 trial — HDV RNA reduction or undetectability, ALT normalization, and changes in liver fibrosis — were appropriate for assessing treatment benefit. The clinical experts indicated that 48 weeks is a reasonable duration to assess efficacy in the MYR301 trial using these surrogate outcomes; however, longer-term follow-up would be required to assess hard clinical outcomes such as liver cancer, hepatic decompensation, or death, which are not expected to manifest within 1 year of treatment initiation. The MYR301 trial study population was drawn from several sites across 4 European countries, and no sites in Canada were included. The clinical experts indicated that overall, there are no major concerns with generalizing the findings from the trial to clinical settings in Canada.
Grading of Recommendations Assessment, Development and Evaluation Summary of Findings and Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group. The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: combined response, virologic response, change from baseline in liver stiffness, HRQoL, and SAEs.
For the GRADE summary of findings for bulevirtide 2 mg versus delayed treatment, refer to Table 2.
Table 2: Summary of Findings for Bulevirtide 2 mg vs. Delayed Treatment for Patients With HDV Infection in the MYR301 Trial
Outcome and follow-up | Patients (studies), N | Effect | Certainty | What happens |
|---|---|---|---|---|
Combined responsea | ||||
Proportion of patients with combined response at week 48 | 99b (1 RCT) |
| Moderated | Bulevirtide 2 mg likely results in a clinically important increase in the proportion of patients who achieve combined response at week 48 when compared with delayed treatment. |
Virologic responsee,f | ||||
Proportion of patients with virologic response at week 48 | 99b (1 RCT) |
| Moderateg | Bulevirtide 2 mg likely results in a clinically important increase in the proportion of patients who achieve virologic response at week 48 when compared with delayed treatment. |
Change from baseline in liver stiffnessf,h | ||||
Change from baseline in liver stiffness at week 48 | 99b (1 RCT) | LS mean (SE) kPa:
| Moderatei | Bulevirtide 2 mg likely results in an improvement in liver stiffness at week 48 when compared with delayed treatment. The clinical importance of the improvement is uncertain. |
HRQoLf | ||||
Change from baseline in HQLQ at week 24 | 99b (1 RCT) | Change from baseline in health distress score LS mean (SE):
Change from baseline in positive well-being score LS mean (SE):
Change from baseline in hepatitis-specific health distress score LS mean (SE):
| Very lowj | The evidence is very uncertain about the effect of bulevirtide 2 mg on HRQoL at week 24 when compared with delayed treatment. |
Change from baseline in HQLQ score at week 48 | 99b (1 RCT) | Change from baseline in health distress score LS mean (SE):
Change from baseline in positive well-being score LS mean (SE):
Change from baseline in hepatitis-specific health distress score LS mean (SE)
| Very lowj | The evidence is very uncertain about the effect of bulevirtide 2 mg on HRQoL at week 48 when compared with delayed treatment. |
Harms | ||||
Proportion of patients with serious adverse events at week 48 | 99b (1 RCT) |
| Very lowk | The evidence is uncertain about the effect of bulevirtide 2 mg on serious adverse events at week 48 when compared with delayed treatment. |
CI = confidence interval; CDA-AMC = Canada's Drug Agency; HDV = hepatitis delta virus; HQLQ = Hepatitis Quality of Life Questionnaire; HRQoL = health-related quality of life; LLoQ = lower limit of quantification; LS = least squares; MID = minimal important difference; NR = not reported; RCT = randomized controlled trial; RNA = ribonucleic acid; SE = standard error; vs. = versus.
Note: Study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aCombined response was defined as undetectable HDV RNA (less than the LLoQ, target not detected) or HDV RNA decrease ≥ 2 log10 IU/mL from baseline combined with ALT normalization.
bIn the MYR301 study, patients were randomized in a 1:1:1 ratio to receive delayed treatment with bulevirtide 10 mg (N = 51), immediate treatment with bulevirtide 2 mg (N = 49), or immediate treatment with bulevirtide 10 mg (N = 50). Data related to immediate treatment with bulevirtide 10 mg are not included in this report, as this dose has not been approved and is not used in clinical practice in Canada.
cDelayed treatment: Baseline at randomization to before the first dose of bulevirtide at week 48 or to early termination before week 48. From week 48 onwards, patients received bulevirtide 10 mg/day.
dRated down 1 level for serious study limitations. Evidence from 1 trial with small sample size, raising concerns that prognostic balance may not have been achieved and the effect could be overestimated. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 20% between the groups was identified as a threshold of clinical importance for this outcome by the clinical experts consulted by CDA-AMC.
eVirologic response was defined as undetectable HDV RNA or HDV RNA decrease ≥ 2 log10 IU/mL from baseline.
fIn the trial, statistical testing for this outcome was not adjusted for multiplicity. The results are considered as supportive evidence.
gRated down 1 level for serious study limitations. Evidence from 1 trial with small sample size, raising concerns that prognostic balance may not have been achieved and the effect could be overestimated. An empirically derived MID was not identified for the between-group difference for this outcome. A difference of 30% between the groups was identified as a threshold of clinical importance for this outcome by the clinical experts consulted by CDA-AMC.
hChange from baseline in liver stiffness was measured using FibroScan.
iRated down 1 level for serious study limitations. Evidence from 1 trial with small sample size, raising concerns that prognostic balance may not have been achieved and the effect could be overestimated. There is no established MID for this outcome, and the clinical experts consulted by CDA-AMC could not provide a threshold of important difference. In the absence of a known threshold, the null was used.
jRated down 1 level for serious risk of bias due to assessor knowledge of treatment assignment. Rated down 2 levels for very serious imprecision: CIs were wide and included potential for no difference and harm. There is no established MID for this outcome, and the clinical experts consulted by CDA-AMC could not provide a threshold of important difference. In the absence of a known threshold, the null was used.
kRated down 1 level for serious risk of bias due to assessor knowledge of treatment assignment. Rated down 2 levels for serious imprecision. The effect may be unstable as it is informed by few events. There is no established MID for this outcome, and the clinical experts consulted by CDA-AMC could not provide a threshold of important difference. In the absence of a known threshold, the null was used.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension study was submitted for this review.
Indirect Comparisons
Description of Studies
A sponsor-submitted indirect treatment comparison (ITC) evaluated bulevirtide 2 mg relative to other comparators, including PEG-IFN and best supportive care or nucleoside (or nucleotide) analogue therapy in patients with chronic HDV. The following outcomes were reported to address the objectives of the network meta-analysis (NMA): undetectable HDV RNA, undetectable HDV RNA or reduced by at least 2 log10 IU/mL (virologic response), combined response, ALT normalization at weeks 24 and 48, and AEs and treatment discontinuations due to AEs at week 48. The Bayesian NMA was conducted using both fixed-effects and random-effects models.
Efficacy Results
Combined Response
At week 48, results from the fixed-effects models suggested that bulevirtide 2 mg results in a favourable improvement in the combined response compared with both PEG-IFN (risk difference = ███; 95% credible interval [CrI], ██ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The corresponding random-effects models also indicated that bulevirtide 2 mg demonstrated a favourable improvement in the combined response compared with both PEG-IFN and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The point estimates were comparable across both the fixed- and random-effects models. Sensitivity analysis of the combined response was not feasible at week 48 due to limited number of available studies.
Virologic Response
At week 48, results from the fixed-effects models indicated that bulevirtide 2 mg demonstrated a favourable improvement in the virologic response compared with both PEG-IFN (risk difference = ███; 95% CrI, ██ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ██ ██ ██). However, the corresponding random-effects models yielded insufficient evidence to confirm a difference in the virologic response for the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. The point estimates were comparable across both the fixed-effects and random-effects models.
ALT Normalization
At week 48, results from the fixed-effects models indicated that treatment with bulevirtide 2 mg results in a favourable improvement in ALT normalization compared with both PEG-IFN (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The corresponding random-effects models yielded insufficient evidence to confirm a difference in the virologic response of patients in the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. The point estimates were comparable across both the fixed-effects and random-effects models.
Undetectable HDV RNA
At week 48, results from the fixed-effects models suggested that bulevirtide 2 mg results in a favourable improvement in achieving undetectable HDV RNA compared with the nucleoside (or nucleotide) analogue therapy group (risk difference = ███; 95% CrI, ██ ██ ███). However, there was insufficient evidence to confirm a difference in achieving undetectable HDV RNA in the bulevirtide 2 mg group compared with the PEG-IFN (risk difference = ███; 95% CrI, ████ ██ ██). The corresponding random-effects models yielded insufficient evidence to confirm a difference in achieving undetectable HDV RNA in the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups.
Harms Results
There was insufficient evidence to confirm a difference between bulevirtide 2 mg and PEG-IFN or nucleoside (or nucleotide) analogue therapy in the rates of AEs, SAEs, or withdrawals due to AEs.
Critical Appraisal
The clinical experts consulted for this review noted that, in the absence of approved treatments for HDV, some of the regimens included in the NMA, such as PEG-IFN and nucleoside (or nucleotide) analogues, were appropriate comparators for bulevirtide. Although PEG-IFN can be used off-label to treat HDV, its use in clinical practice has become uncommon due to significant adverse effects, a global shortage, and limited availability in Canada. The clinical experts also noted that nucleoside (or nucleotide) analogues, which most patients receive use to manage HBV, have no direct effect on HDV. The clinical experts observed that several comparators included in the NMA analyses are not relevant to the context of HDV treatment in Canada, including bulevirtide plus tenofovir or PEG-IFN combined with adefovir, ribavirin, tenofovir, or entecavir. The literature search for the NMA was last updated by the sponsor in December 2021. The included studies were published between 2006 and 2019, and the treatment landscape has since evolved. An outdated literature search with possible omission of relevant or unpublished evidence can result in bias. The feasibility assessment for this NMA revealed considerable heterogeneity among the studies included in the analyses. There was considerable variation in the study design, treatment duration and duration of follow-up, regimen dosing, which may represent a potential source of heterogeneity. Several important disease-specific characteristics were not reported in some studies in the network, which limits the ability to assess heterogeneity between the studies, and not all outcomes were reported across all of the studies. The patient populations in the studies also varied, particularly in baseline characteristics such as age, presence of cirrhosis, HDV RNA levels, ALT levels, and treatment history. The numerous sources of heterogeneity, which cannot be adjusted for in the NMA, compromise the underlying transitivity assumption that must be met to draw valid conclusions. The network of interconnected studies was constructed using nucleoside (or nucleotide) analogues or delayed treatment as a common comparator.
The network of evidence was sparse (i.e., there were few studies contributing to several comparisons); in many cases, there was only 1 study per link, which was insufficient to reliably estimate between-study variances. Bayesian fixed-effects models were used as a base-case analysis, with a random-effects model for the exploratory analysis. When heterogeneity is present, the CrIs of the fixed-effects model may be narrower and less conservative than the random-effects model, underestimating the uncertainty arising from between-study variation. The results of the random-effects models for most outcomes were affected by important imprecision, precluding drawing a conclusion about which treatment may be favoured. The risk-difference models employed for the meta-analyses may have limitations in this context; specifically, the results of risk-difference meta-analyses can be influenced by the nonreporting of zero-event outcomes. These models also typically demonstrate lower statistical power and produce more conservative CIs compared to relative measures such as risk ratios, particularly when event rates are low.20 No patient-reported quality of life data were evaluated, despite that HRQoL was considered an important end point for this review. The NMA evaluated the comparative safety of bulevirtide relative to PEG-IFN and nucleos(t)ide analogues; however, the wide CIs around these estimates indicate considerable uncertainty in the comparative safety outcomes.
Studies Addressing Gaps in the Evidence From the Systematic Review
SAVE-D Study
Description of Studies
SAVE-D was a multicentre, retrospective, real-world study (N = 244 patients) with the goal of addressing the gap in evidence on the effectiveness of bulevirtide treatment beyond 48 weeks in patients with cirrhosis with and without clinically significant portal hypertension. Patients with HDV-related cirrhosis who were starting bulevirtide monotherapy were consecutively enrolled in this study. Chronic HDV was defined as HDV RNA positivity for more than 6 months. Cirrhosis was defined either histologically (Meta-analysis of Histological Data in Viral Hepatitis [METAVIR] fibrosis stage 4 [F4]), noninvasively (liver stiffness of greater than 12.5 kPa), or clinically (nodular liver surface, splenomegaly, thrombocytopenia).
Efficacy Results
Of the patients who were enrolled, 244 (100%) had assessments available at baseline and 87 (36%) had assessments available at week 96.
Of the key efficacy results evaluated at week 96:
Virologic response (HDV RNA ≥ 2 log10 decline) rate was 79%.
Undetectable HDV RNA rate was 48%.
Biochemical response (ALT normalization < 40 U/L) rate was 64%.
Combined response (HDV RNA ≥ 2 log10 decline or undetectable, and ALT normalization) rate was 54%.
The 96-week cumulative incidences of de novo HCC and decompensation were 3.0% and 2.8%, respectively.
Harms Results
Bulevirtide-related AEs were relatively mild, with a median bile acid increase from 15 μmol (interquartile range [IQR] = 9 μmol to 32 μmol) at baseline to 36 μmol (IQR = 19 μmol to 66 μmol) at week 96 (all time points versus baseline, P < 0.001) and mild and transient pruritis experienced by 24 patients (10%).
Critical Appraisal
This was a single-arm real-world study without a comparator, which means that comparative effectiveness conclusions relative to usual care could not be drawn. Further, the single-arm design precludes definitive conclusions. Major limitations of the SAVE-D study, which increased the uncertainty around effect estimates, were the lack of investigation into possible missing outcome data between weeks 24 to 96, posing a high risk of bias; limited generalizability to Canada; and lack of reported HRQoL outcomes.
MYR203 Study
Description of Studies
The MYR203 study was a multicentre, open-label, randomized, comparative, parallel-arm phase II study to assess the efficacy and safety of bulevirtide in combination with PEG-IFN-alpha versus PEG-IFN-alpha alone in the treatment of patients with chronic HBV and HDV coinfection. Each treatment arm had 15 patients. The interventions relevant to this review were bulevirtide 2 mg by subcutaneous injection as monotherapy compared with PEG-IFN-alpha alone.
Efficacy Results
The primary outcome of the study was HDV RNA response at week 72 (24 weeks posttreatment), defined as an HDV RNA value of less than the lower level of detection, equal to 10 IU/mL. Secondary outcomes were measured at 24 and 48 weeks, comprising of HDV RNA response, ALT normalization, combined response (negative HDV RNA and ALT normalization), and HDV RNA levels (log10 scale). Liver fibrosis measured using FibroScan was an additional secondary end point.
At week 72 (24 weeks posttreatment), the primary efficacy end point of HDV RNA response was achieved by ███ ██████ ███ ███ ██ ██ ██████ ███ ███ ████ ███ ███ ████ ██ ██████ of the patients treated with PEG-IFN-alpha, and bulevirtide 2 mg, respectively. No patients (0 of 15 patients) treated with PEG-IFN-alpha only or with bulevirtide 2 mg only group achieved the HDV RNA response.
At week 48, secondary end points between PEG-IFN-alpha and bulevirtide 2 mg differed by end point. HDV RNA response was achieved by the same number of patients 2 of 15 (13.3%; 95% CI, 1.7% to 40.5%) treated with PEG-IFN-alpha and bulevirtide 2 mg. ALT normalization was achieved by 4 of 15 (26.7%; 95% CI, 7.8% to 55.1%) and 11 of 15 (73.3%; 95% CI, 44.9% to 92.2%) of the patients treated with PEG-IFN-alpha and bulevirtide 2 mg, respectively. The combined response (HDV RNA decline and ALT normalization) was achieved by 1 of 15 (6.7%; 95% CI, 0.2% to 31.9%) and 2 of 15 (13.3%; 95% CI, 1.7% to 40.5%) of the patients treated with PEG-IFN-alpha alone and bulevirtide 2 mg alone, respectively.
For liver stiffness, there were no statistically significant differences in change from baseline between the group receiving bulevirtide alone and the group receiving PEG-IFN-alpha at any time point.
Harms Results
There were no deaths in the study and none of the AEs were suspected unexpected serious adverse reactions.
Critical Appraisal
While the MYR203 study was an exploratory phase II trial, the very small sample sizes (baseline, N = 15 per group) with all patients recruited from Russia, limits the generalizability of findings to patients in Canada. There was a risk of bias of missing outcome data when measuring its primary end point (HDV RNA response at 72 weeks), with a decline in sample sizes for the comparator PEG-IFN-alpha (N = 7) and bulevirtide 2 mg (N = 9) groups. In addition, HRQoL outcomes were not reported.
Conclusions
HDV is a rare and severe condition that is recognized as the most aggressive form of viral hepatitis. At the time of this review, bulevirtide is the only approved treatment of HDV in adults with compensated liver disease. Input from both patients and clinicians on this review highlighted a significant unmet need for new treatments that effectively treat and prevent the progression of HDV. The pivotal MYR301 phase III trial examined the efficacy and safety of bulevirtide 2 mg for the treatment of patients with chronic HDV with compensated liver disease, compared to patients in the delayed-treatment group who received a 10 mg dose of bulevirtide following a 48-week observation period. The MYR301 trial demonstrated moderate certainty evidence that treatment with bulevirtide 2 mg likely results in a clinically meaningful increase in achieving combined response, virologic response, and improvement in liver stiffness at week 48, compared to delayed treatment. No definitive conclusion can be drawn regarding the effects of bulevirtide treatment on HRQoL due to the potential bias from assessor knowledge of treatment assignment, and imprecision in the data, making the direction of effects unclear. Bulevirtide 2 mg was generally well-tolerated, and no major safety concerns were identified in the MYR301 trial. Beyond week 48, there is no direct comparative evidence between bulevirtide and the relevant comparators.
The MYR203 phase II trial provided evidence on the efficacy and safety of bulevirtide 2 mg as monotherapy versus PEG-IFN-alpha-2a alone in patients with HDV who had compensated liver disease. However, due to the limited sample size, study design limitations, and exploratory nature of the study, definitive conclusions cannot be drawn. The SAVE-D study was a real-world study that provided evidence on the efficacy of bulevirtide for up to 96 weeks in patients with HDV-related cirrhosis with and without clinically significant portal hypertension. However, the single-arm design of the SAVE-D study limits the ability to assess comparative effectiveness relative to usual care and to infer causality. Key limitations of both studies include a risk of bias from missing outcome data that contributes to uncertainty in the effect estimates, limited generalizability to patients in Canada, and no reporting of HRQoL outcomes, which patients considered important.
The results of the sponsor-submitted ITC suggested that, compared with PEG-IFN and best supportive care, bulevirtide may offer potential benefit in achieving combined response at week 48. However, the interpretation of the ITC is limited by several factors, including methodological limitations, between-trial heterogeneity, a sparse network, and small sample sizes, which preclude definitive conclusions on the comparative effects of bulevirtide. In addition, PEG-IFN is no longer used for treatment of HDV in Canada due to poor tolerability and limited accessibility. Given the considerable uncertainty in the sponsor-submitted ITC, no definitive conclusions can be drawn regarding the safety of bulevirtide compared to PEG-IFN and best supportive care.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of bulevirtide 2 mg, administered once daily by subcutaneous injection, for the treatment of chronic HDV infection in adult patients with compensated liver disease.
Disease Background
The contents of this section were informed by materials submitted by the sponsor and input from clinical experts and have been validated and summarized by the review team.
Hepatitis delta is a rare, severe, and progressive liver disease caused by HDV, an incomplete RNA virus that requires the HBV envelope to enter cells and replicate. HDV is considered the fastest progressing and most severe form of all viral hepatitis infections. HDV is a “satellite virus” and can only infect individuals with a concomitant HBV infection.1,2 Chronic HDV infection is defined as an infection lasting 6 months or more.3 The likelihood of progression to chronic hepatitis depends on whether the initial infection with HDV occurred due to HBV and HDV coinfection or superinfection. While less than 5% of patients progress to a chronic infection after coinfection, HDV infection becomes chronic in more than 80% of individuals with HBV and HDV superinfection.4
A 2024 study from the Polaris Observatory found the adjusted prevalence of HDV in the HBV-infected population in Canada to be approximately 3.0%.5 These estimates are consistent with the estimated rate of 4.8% (95% CI, 4.3% to 5.3%) reported by a retrospective study conducted using patient data collected by physicians in the Canadian HBV Network (9 clinics in 6 provinces across Canada).6 Given that the prevalence of chronic HBV in Canada is approximately 0.66%7 and the HBV diagnosis rate is approximately 70%,5 the prevalence of HDV (chronic and acute) infection is estimated to be 0.020%5 to 0.032%,6 which equates to 2 to 3.2 per 10,000 population (includes both chronic and acute HDV).
Key signs and symptoms of chronic HDV infection can range from nonspecific symptoms to rapidly progressing hepatitis. Symptoms may include fever, fatigue, loss of appetite, nausea, vomiting, abdominal pain, dark urine, clay-coloured bowel movements, joint pain, and jaundice.4 Chronic hepatitis usually exacerbates any pre-existing liver disease associated with HBV.8 Recovery is unlikely, and the likelihood of recovery decreases as the disease progresses. Only 35% of patients with acute infection, and even less (9.96%) with chronic infection, recover.9 For those who do not recover at the early stages of the disease, HDV infection is associated with an accelerated progression to fibrosis, early liver decompensation with cirrhosis, and increased risk of HCC, leading to greater liver-related mortality compared to HBV or HCV.10-12
Diagnosis of HDV generally involves 2 steps. The first step is testing for anti-HDV antibodies through an enzyme-linked immunosorbent assay to assess if a patient has ever been exposed to HDV. If the anti-HDV antibody test is positive, meaning that the patient has been exposed to HDV, the patient is automatically tested for HDV RNA, using RT-PCR, to assess whether the HDV infection is active or chronic. Standard liver function tests are routinely performed to assess the severity of the HBV and HDV disease. These liver function tests are conducted at initial diagnosis and to monitor treatment response. Monitoring of response to HDV treatment involves several tests. Testing for HDV RNA through RT-PCR is required to determine whether to reinitiate treatment and to monitor treatment response. Bile acid tests are recommended to monitor for safety and assess adherence. All these monitoring tests are performed every 3 to 6 months. Both antibody and RNA tests are currently performed at the National Microbiology Laboratory in Winnipeg, Manitoba, and all other tests can be performed at outpatient labs or clinics.21
Standards of Therapy
The contents of this section were informed by materials submitted by the sponsor and input from clinical experts, and have been validated and summarized by the review team.
According to the clinical experts consulted by CDA-AMC for this review, HDV occurs as a coinfection with HBV and is associated with a more aggressive liver disease, with a higher risk of cirrhosis, hepatic decompensation, and HCC. There are currently no approved treatments for chronic HDV in Canada.13-15 The Canadian Association for the Study of the Liver, the Association of Medical Microbiologists and Infectious Disease Canada, the European Association for the Study of the Liver, and AASLD13-15 indicate that PEG-IFN-alpha has demonstrated some clinical benefit and is recommended as a potential treatment option for patients with HBV and HDV coinfection. AASLD further emphasizes the importance of suppressing HBV replication in individuals with chronic hepatitis D who have elevated HBV DNA levels. Nucleoside (or nucleotide) analogues are HBV polymerase inhibitors that primarily block HBV DNA synthesis, but do not directly suppress the production of the hepatitis B surface antigen (HBsAg), which is required by HDV for entry into hepatocytes.22 Although HDV requires coinfection with HBV, nucleoside (or nucleotide) analogues, which are effective against HBV, have no efficacy against HDV, as HDV replication is entirely independent of HBV replication.1,15 While Canadian and international guidelines vary in their recommendations on the use of nucleoside (or nucleotide) analogues in patients with HBV and HDV coinfections, they consistently emphasize the lack of efficacy of nucleoside (or nucleotide) analogues against HDV.13-16
The clinical experts indicated that, although there is limited evidence supporting the use of PEG-IFN-alpha for patients with HDV, response rates are generally low and the treatment is poorly tolerated. The clinical experts noted that, although PEG-IFN-alpha can be used off-label for the treatment of HDV, its use in clinical practice became uncommon due to significant adverse effects, a global shortage, and lack of availability in Canada.
According to the clinical experts consulted for this review, the therapeutic goals for chronic HDV infection include achieving undetectable or reduced HDV RNA, normalizing liver enzymes, improving liver function, and reducing the risk of decompensation, HCC, and liver-related mortality.
Drug Under Review
Key characteristics of bulevirtide are summarized in Table 3.
Bulevirtide has a Health Canada indication for the treatment of chronic HDV infection in adults with compensated liver disease, aligned with the reimbursement request. Bulevirtide has not been previously reviewed by CDA-AMC. The recommended dosage in adults is bulevirtide 2 mg once daily, administered by subcutaneous injection.23 The optimal treatment duration has not been established. The sponsor has proposed that treatment be continued for as long as it is associated with clinical benefit. In all patients, the underlying HBV infection is managed simultaneously as clinically appropriate.
The HDV virion is assembly-deficient, requiring an envelope provided by HBV to enter host cells; as such, HDV is assumed to enter hepatocytes through the same mechanism as HBV.24 The entry of both HBV and HDV into host cells requires the attachment of a surface protein found within the N-terminal myristoylated domain of the HBV envelope L-protein to NTCP, a hepatic bile salt transporter, followed by subsequent entry and membrane fusion.25,26 Bulevirtide is a 47-amino acid, N-terminal myristoylated, HBV-L protein–derived, synthesized lipopeptide that acts as a potent, highly selective entry inhibitor of HDV. Bulevirtide blocks the entry of HBV and HDV into hepatocytes by binding to and inactivating NTCP.23,26 By blocking the essential entry receptor, the de novo infection of liver cells is decreased, viral spread is inhibited, and the life cycle of HDV is disrupted. A reduction in the number of infected cells ultimately protects uninfected and newly formed hepatocytes from new infection and reinfection.1,27 In contrast to directly acting antivirals, where viral production must be significantly reduced before the achievement of biochemical remission, treatment with entry inhibitors reduces the plasma levels of HDV RNA, due to a decline in the number of infected, virus-producing hepatocytes in the liver.
The drug under review is recommended for reimbursement by the following health technology assessment agencies: the National Institute for Health and Care Excellence (NICE) (England), the Scottish Medicines Consortium (Scotland), and the Haute Autorité de Santé (France). Bulevirtide has not been recommended by the All Wales Medicines Strategy Group (Wales). It is currently under review by the Pharmaceutical Benefits Advisory Committee and the Medical Services Advisory Committee (Australia). Bulevirtide has been approved by the following regulatory agencies: the European Medicines Agency (European Union), the Therapeutic Goods Administration (Australia), and the Medicines and Healthcare products Regulatory Agency (UK). It was not approved by the FDA (US) due to concerns about the manufacture and delivery of the therapy.28
Table 3: Key Characteristics of Bulevirtide
Characteristic | Treatment |
|---|---|
Mechanism of action | Bulevirtide is a 47-amino acid, N-terminally myristoylated, HBV-L protein–derived, synthesized lipopeptide that acts as a potent, highly selective entry inhibitor of HDV. Bulevirtide blocks the entry of HBV and HDV into hepatocytes by binding to and inactivating NTCP. |
Indicationa | Bulevirtide is indicated for the treatment of chronic HDV infection in adults with compensated liver disease. |
Route of administration | SC |
Recommended dose | 2 mg once daily |
Serious adverse effects or safety issues | Severe acute exacerbations of HDV and HBV infections may occur after bulevirtide is discontinued. |
Other | The underlying HBV infection should be simultaneously managed according to current treatment guidelines. Tenofovir disoproxil fumarate or other nucleoside (or nucleotide) analogues were coadministered with bulevirtide in clinical studies. Close monitoring of HBV DNA levels is recommended. |
HBV = hepatitis B virus; HDV = hepatitis delta virus; SC = subcutaneous; SC = subcutaneous.
aHealth Canada indication.
Source: Gilead Sciences Canada Hepcludex clinical evidence and draft product monograph, Hepcludex (bulevirtide). Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
CDA-AMC received a joint patient input submission from 2 organizations, Liver Canada and BC Hepatitis Network.
Patient perspectives were gathered via an online survey conducted between February 21, 2025, and March 8, 2025, using social media. The 6 patients who responded to the survey ranged in age from their 30s to their 60s, and were living with HDV, in Ontario, Alberta, or British Columbia. Patients described how receiving a diagnosis of HDV and experiencing disease-specific symptoms, including general physical and psychological symptoms, substantially impacts their HRQoL. Caring for someone with HDV also leads to significant caregiver burden. All patients rated the importance of having access to new treatments for HDV as 5 out of 5 (where 5 is most important).
PEG-IFN is used off-label in Canada for the treatment of HDV. Patients with experience with PEG-IFN treatment highlighted the associated intolerable side effects, frequent need to travel to clinics for injections, and frequent blood tests for monitoring of response. Patients indicated a preference for treatments that require fewer injections and blood tests. One patient had experience with the drug under review through compassionate access from the manufacturer; they reported experiencing manageable side effects and rated their HRQoL at 4 out of 5 (where 5 is the best score), with improved liver test results and stabilization of liver disease.
Patients described stabilization of their liver disease as an important treatment outcome. Stabilization refers to slowed progression to liver fibrosis, cirrhosis, and liver cancer; improvement in decompensation events; and reduced need for a liver transplant. According to the patient input received for this review, chronic HDV is most common among newcomers and immigrants to Canada, who face multiple barriers to timely diagnosis. According to patient input, these individuals are less likely to be screened for viral hepatitis, face limited access to routine care, and are more likely to experience fear and stigma regarding hepatitis. Together, these barriers increase individuals’ risk for late diagnosis and advanced viral hepatitis-related liver disease. Patients identified reflex testing for HDV among individuals with HBV as an important unmet need. This diagnostic process occurs in 2 steps; the first step is an assay for anti-HDV antibodies, and if this is positive, the second step is a test for HDV RNA. This testing usually occurs in primary care, acute, or public health care settings or in hospitals. According to the patient input received, improving diagnosis rates allows earlier treatment initiation to stop the progression of liver disease before onset of liver cancer and/or liver failure.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place of the drug in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of chronic HDV infection.
Unmet Needs
The clinical experts consulted by CDA-AMC for this review identified significant unmet needs in the management of chronic HDV infection in Canada, emphasizing the absence of approved therapies or access to reimbursed therapies. The clinical experts indicated that although PEG-IFN was previously used off-label, it is no longer used in clinical practice due to its poor tolerability, limited accessibility, and lack of feasibility for routine use. The clinical experts emphasized that currently available therapies for hepatitis B, including nucleoside (or nucleotide) analogues, are ineffective against HDV infection, which remains the primary driver of liver damage in individuals with HBV and HDV coinfection. As a result, patients with chronic HDV experience progressive liver disease without access to an effective treatment option. The clinical experts highlighted the need for a targeted therapy to reduce HDV viral load, normalize ALT levels, potentially stabilize or improve liver fibrosis, and mitigate the risk of HCC and liver decompensation. This need is particularly urgent due to the aggressive nature of HDV infection and the associated risk of rapid progression to cirrhosis and HCC.
Place in Therapy
According to the clinical experts consulted, bulevirtide would be used as a first-line therapy upon diagnosis of active chronic HDV infection in patients with chronic hepatitis B. The clinical experts noted that bulevirtide acts directly on the HDV replication cycle by preventing viral entry into hepatocytes. According to the clinical experts consulted, patients with detectable HDV RNA and elevated ALT levels were identified as appropriate candidates for treatment, as they are at an increased risk of disease progression in the absence of effective intervention. The clinical experts noted that early initiation of bulevirtide is recommended to help mitigate the risk of progression to advanced liver disease. The clinical experts stated that bulevirtide would be used in combination with nucleoside (or nucleotide) analogues, such as tenofovir or entecavir, which would be used to manage the HBV coinfection, but which are ineffective against HDV. The clinical experts further noted that PEG-IFN is no longer a viable option for the treatment of HDV infection due to its poor tolerability, lack of reimbursement, and limited availability, and therefore would not be considered before bulevirtide.
Patient Population
The clinical experts consulted by CDA-AMC identified individuals with compensated HDV-related liver disease as appropriate candidates for bulevirtide, and those with advanced fibrosis or cirrhosis most in need. The clinical experts indicated that treatment should be based on objective markers such as detectable HDV RNA and elevated ALT levels, rather than symptoms. The clinical experts noted that treatment response is most likely to occur in patients who do not have an immunocompromised condition, have lower baseline HDV RNA levels, and exhibit minimal fibrosis. Conversely, this treatment may be less suitable for patients with barriers to adherence to daily injectable therapy. According to the clinical experts, underdiagnosis of HDV is a significant issue in practice, largely due to the limited availability of HDV RNA testing and lack of routine screening, which pose barriers to timely treatment initiation. The clinical experts noted that systematic screening is lacking, particularly for individuals with HBV who are receiving care in the primary care setting. According to the clinical experts, HDV RNA testing is currently conducted at the National Microbiology Laboratory. The clinical experts emphasized the need for initiatives to establish local access to anti-HDV antibody testing and, ideally, HDV RNA testing as well. The clinical experts noted that noninvasive fibrosis assessment is adequate for evaluation and that liver biopsy is not necessary.
Assessing the Response Treatment
The clinical experts noted that the key outcomes used to determine response to treatment with bulevirtide include virologic response (i.e., undetectable HDV RNA or reduction in HDV RNA levels), biochemical response (i.e., normalization of ALT levels), and stabilization or improvement in liver fibrosis. The clinical experts noted that long-term outcomes such as improvements in portal hypertension, reduced progression to cirrhosis, and lower rates of HCC would also be clinically beneficial but were not assessed in trials. The clinical experts indicated that HDV should be monitored regularly, with liver enzyme and function tests and HDV RNA testing every 3 months and noninvasive fibrosis assessments (e.g., FibroScan) annually. According to the clinical experts, clinicians generally see patients in the clinic every 6 to 12 months, with routine bloodwork conducted every 3 to 6 months and ultrasounds for HCC screening every 6 months. These intervals were deemed appropriate for assessing treatment response and guiding decisions regarding continuation of therapy. The clinical experts emphasized that consistent monitoring of response is essential, as patients with HDV remain at high risk of disease progression even if their HBV is well controlled.
Discontinuing Treatment
The clinical experts indicated that bulevirtide treatment should be discontinued in cases of progression to decompensated liver disease requiring transplant or the development of HCC requiring other therapy. The clinical experts noted that discontinuation can be considered in the event of severe adverse reactions, such as allergy or significant injection site reactions, or if the patient experiences barriers to adherence to the daily injectable therapy. The clinical experts also noted that treatment with bulevirtide could be stopped after a period of consolidation if HDV RNA becomes undetectable, and that emerging information suggests that longer treatment duration may support sustained viral suppression. However, the clinical experts highlighted that treatment discontinuation is generally unlikely unless prompted by patient refusal, death, or the need for liver transplant.
Prescribing Considerations
The clinical experts indicated that the diagnosis of HDV infection, prescribing of bulevirtide, and monitoring of response should be managed by clinicians with specialized expertise in hepatology or infectious diseases, given the complexity of HDV infection and the need for accurate interpretation of HDV RNA and ALT levels. The clinical experts noted that bulevirtide is used in combination with nucleoside (or nucleotide) analogues such as tenofovir or entecavir, which are part of standard hepatitis B management and do not require special prescribing restrictions. The clinical experts indicated that treatment could be administered in outpatient or community settings, and with proper instruction, patients may be able to self-administer the medication; however, clinical oversight by a specialist remains essential for safe and effective treatment.
Additional Considerations
According to the clinical experts consulted by CDA-AMC, chronic HDV infection is rare in Canada, affecting approximately 1% to 2% of individuals with chronic hepatitis B; however, it is associated with more severe liver disease and faster disease progression. The clinical experts highlighted the lack of effective treatment options beyond nucleoside (nucleotide) analogue therapy for HBV, noting that PEG-IFN is poorly tolerated, has limited availability, and is no longer considered a viable option. In the clinical experts’ opinion, bulevirtide addresses a significant unmet medical need for this high-risk population. Despite the potential benefit of bulevirtide treatment, the clinical experts pointed out several remaining challenges, including limited access to systematic HDV screening, gaps in linkage to care, and the burden of managing a medication that requires daily injections. The clinical experts highlighted the need to ensure that patients have reliable access to injection-related supplies such as syringes and saline and receive proper education on how to administer the treatment safely and effectively. The clinical experts also noted that the optimal duration of bulevirtide therapy remains uncertain for patients with HDV that responds more slowly to treatment.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Ten clinicians from the Canadian Hepatitis B Network provided input. The Canadian Hepatitis B Network is a collaborative organization of health care professionals and researchers from across Canada with an interest in advancing excellence in the care of patients with hepatitis B, research into hepatitis B, and education.
The clinician group stated that there is an unmet need for improved therapies for HDV and HBV coinfection. There are currently no treatments approved by Health Canada indicated for HDV. HDV causes the most severe form of viral hepatitis in humans. HDV requires the HBV surface (or envelope) protein to infect the liver and therefore can only infect people who have HBV. Patients with HDV are often young; yet they are at high risk of severe liver disease, liver failure, and liver cancer.
The clinician group stated that individuals can acquire HBV and HDV at the same time or HDV can superinfect a patient with underlying chronic HBV infection because of a shared route of transmission. The drug under review is a novel antiviral drug specifically targeting the binding of HDV (and HBV) to the liver-specific cell surface bile acid receptor, NTCP. Based on input received from the clinician groups, patients with HBV and HDV coinfection and who test positive for HDV RNA are most likely to benefit from treatment with the drug under review.
The clinician group noted that patients with HDV coinfections are at a high risk of liver disease and cirrhosis within 5 years of diagnosis, regardless of any disease characteristics. Patients best suited for treatment with the drug under review would be identified based on the clinical judgment of expert specialists in hepatology or infectious disease, as well as by testing for HDV RNA. No companion diagnostic test is required. The clinician group highlighted that outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials. A clinically meaningful response to treatment would be HDV RNA suppression, biochemical normalization, improvement in noninvasive fibrosis test results, and/or improvement in symptoms of liver disease decompensation (e.g., ascites, variceal bleeding).
Based on input received from the clinician group, suggested criteria for discontinuing treatment were treatment failure (indicated as no HDV RNA decline or ALT normalization), disease progression, and limited lifespan. The clinician group stated that the drug under review can be appropriately prescribed by a hepatology and infectious diseases specialist in a specialty clinic setting.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor performed a systematic review with PEG‑IFN‑alpha and best supportive care (nucleoside [or nucleotide] analogues) as comparators. Three open‑label RCTs were identified: MYR202, MYR203, and MYR301. The MYR202 trial used tenofovir 245 mg as a comparator for 48 weeks. The MYR203 trial used PEG‑IFN‑alpha 180 mcg as a comparator. In the MYR301 trial, treatment with bulevirtide 2 mg was compared with treatment with bulevirtide 10 mg that was delayed until after an observation period of 48 weeks (“delayed treatment”). Question for the clinical experts: What is the most relevant comparator(s) in clinical practice? | According to the clinical experts consulted by CDA‑AMC, there is currently no approved treatment for HDV. The clinical experts indicated that PEG-IFN and best supportive care alone (i.e., nucleoside [or nucleotide] analogues) are relatively appropriate comparators for bulevirtide. However, they noted that although PEG‑IFN can be used off-label to treat HDV, its use in clinical practice has become uncommon due to significant adverse effects, a global shortage, and limited availability in Canada. The clinical experts also noted that although nucleoside (or nucleotide) analogues are used to manage HBV, they have no direct antiviral activity against HDV. |
There are no treatment options approved by Health Canada for the treatment of chronic hepatitis D. Off-label PEG‑IFN‑alpha is not publicly funded across all jurisdictions. The jurisdictions that have criteria for coverage of hepatitis B do not specifically refer to hepatitis D. | Comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
HDV is a “satellite virus,” requiring an envelope, provided by HBV, to enter host cells; thus concomitant HBV infection is required. It is assumed that HDV enters hepatocytes through the same mechanism as HBV. In all patients, HBV infection should be managed simultaneously, as clinically appropriate. Diagnosis requires blood tests including standard tests (i.e., liver function tests) as well as tests that are conducted at the National Microbiology Laboratory in Winnipeg, Manitoba. The sponsor is suggesting that access to the required lab tests is widely available across Canada. Question for the clinical experts: Is there a specific stage of fibrosis that would determine eligibility for treatment? This would be a helpful parameter for CDEC to identify and highlight. Comment for CDEC: It is unclear if there would be excessive wait times for testing depending on location. | The clinical experts indicated that treatment initiation criteria based on fibrosis stage 2 (F2), commonly used in HBV or HCV management, may not be appropriate for HDV. Given the more aggressive and rapidly progressive nature of HDV infection, the clinical experts emphasized the importance of earlier intervention to stop viral replication and prevent disease progression. |
As per the indication, patients must have chronic hepatitis D, which by definition is an HDV infection lasting 6 months or longer. Question for the clinical experts: Would there be any circumstances where you would choose to treat HDV infection before the 6-month mark? The 3 trials included patients with anti-HCV antibodies but whose HCV RNA was negative. The trials also excluded patients with HIV infection. Question for the clinical experts: In the case of patients who have concomitant HDV and HCV (i.e., test positive for HDV RNA and HCV RNA), how would the treatment approach differ? Would treatments targeting each virus be administered concurrently? How would the treatment approach differ for patients with both HDV and HIV? Study participants were restricted to those aged between 18 and 65 years. Question for clinical experts: Would you offer treatment to patients outside of this age range? | According to the clinical experts, treatment for HDV may be initiated before the 6-month mark in patients with confirmed HDV RNA and elevated ALT levels, as the infection is typically chronic by the time patients are evaluated, having often been present but undiagnosed. For patients with HDV and HCV coinfections, the recommended approach is to first treat HCV using direct-acting antivirals, followed by initiation of HDV-directed treatment. For patients with HBV and HDV or HIV coinfections, HDV treatment would be introduced once HBV therapy is established, as this is typically already included in standard HIV treatment regimens. The clinical experts noted that although patients aged older than 65 years were excluded from the clinical trials, age alone should not preclude treatment with bulevirtide in clinical practice. Treatment decisions for older adults should be made on an individual basis, taking into account comorbidities, functional status, and patient preferences. A strict upper age limit should not be imposed, although treatment may not be appropriate for patients aged older than 80 years, particularly if the treatment burden outweighs the potential benefits. |
Question for the clinical experts: Given that there are no currently approved medications for the treatment of HDV infection, are there any off-label medications that would be required before patients could be eligible for treatment with bulevirtidey? | According to the clinical experts, there are no off-label medications that would be required before initiating treatment with bulevirtide. While patients are often already receiving treatment for HBV, this treatment is not considered a prerequisite for bulevirtide therapy. |
Question for the clinical experts: In cases where discontinuation has occurred and further monitoring shows relapse, would the patient be eligible for re-treatment? If yes, what would be the expected treatment duration? | According to the clinical experts, there is currently no evidence to guide decisions regarding re-treatment with bulevirtide following discontinuation and subsequent reversal of response. However, in cases where there was an initial response to treatment followed by a disease relapse after treatment discontinuation, re-treatment may be reasonable, though they emphasized the need for additional data to guide such decisions. |
The sponsor’s systematic review highlighted a special subgroup of interest, namely patients who are ineligible for PEG-IFN-alpha due to treatment failure, intolerance, or contraindication. In the potential scenario where PEG‑IFN‑alpha is a required prior therapy, would these patients be eligible for treatment with bulevirtide? Two of the trials excluded patients with a CrCl < 60 mL/min, and the drug product monograph also notes that the safety and efficacy of bulevirtide has not been evaluated in this population. Question for the clinical experts: Would you avoid bulevirtide treatment for patients who have moderate to severe renal impairment (CrCl < 60 mL/min)? | According to the clinical experts, patients with moderate renal impairment (e.g., CrCl approximately 60 mL/min) may still be considered for treatment with bulevirtide provided that their treatment is closely monitored and managed with caution. In cases of more severe renal impairment (e.g., CrCl of about 40 mL/min), greater caution would be necessary, and consultation with a nephrologist may be appropriate. According to the clinical experts, no off-label medications, including PEG-IFN, are required before initiating treatment with bulevirtide. PEG-IFN is neither available nor used in clinical practice in Canada. |
Considerations for continuation or renewal of therapy | |
Given the recommendation to continue to treat for as long as there is associated clinical benefit, what are the appropriate monitoring parameters for defining adequate response? The “combined treatment response” is defined as undetectable HDV RNA (HDV RNA < LLoD, where LLoD = 6 IU/mL) or decrease in HDV RNA by ≥ 2 log10 IU/mL from baseline, and ALT normalization (defined as an ALT value within the normal reference range). The sponsor considered efficacy outcomes as the proportion of patients with undetectable HDV RNA, ALT normalization or change in ALT levels, negative PCR results, and improved HRQoL. The trials used time points in 24‑week intervals for response assessments. Questions for CDEC:
| According to the clinical experts, continued benefit of bulevirtide treatment should be assessed using surrogate markers such as undetectable HDV RNA, at least 2 log10 IU/mL reduction in HDV RNA, and ALT normalization and longer-term indicators like improvement in fibrosis. While reductions in liver cancer and hepatic decompensation are important, these are not expected within 1 year of treatment initiation. Monitoring should occur regularly, with liver enzyme and function tests conducted every 1 to 3 months, HDV RNA tests every 3 months, and noninvasive fibrosis assessments (e.g., FibroScan) every 6 to 12 months. Clinicians typically see patients with HDV who are not receiving treatment in the clinic every 6 to 12 months, depending on the disease severity. Patients who are receiving treatment are generally monitored every 3 months to assess treatment response, check for adverse events, and support compliance. Routine bloodwork is conducted every 3 to 6 months, and ultrasounds for HCC screening every 6 months. Patients with partial or slow response should continue treatment given the progressive nature of HDV infection and the lack of available treatment options. |
Considerations for discontinuation of therapy | |
Question for the clinical experts: What would define loss of response or absence of clinical benefit? | According to the clinical experts, loss of response is defined as an initial virologic response, such as an undetectable HDV RNA level or at least 2 log10 reduction in HDV RNA, followed by viral rebound. This may be associated with issues related to treatment adherence, although the potential for antiviral resistance remains unclear. Discontinuation of therapy is not necessarily recommended in such cases, as it may trigger an HDV flare and lead to clinical deterioration. The clinical experts indicated that absence of clinical benefit is defined as less than 1 log10 reduction in HDV RNA at week 48 of treatment. |
Question for the clinical experts: If treatment is interrupted for any reason unrelated to ongoing HDV infection, can treatment be resumed? | According to the clinical experts, if treatment is interrupted for reasons unrelated to HDV, it should be resumed when appropriate with reassessment of HDV RNA, liver enzymes, and bilirubin levels, while routine monitoring should continue every 3 months as per typical protocol. |
Considerations for prescribing of therapy | |
Question for the clinical experts: Given that doses greater than 2 mg (i.e., 5 mg, 10 mg) were studied, would an increase in dose be considered in order to achieve clinical benefit in cases of treatment not meeting the primary outcomes? | According to the clinical experts, the benefits of increasing the bulevirtide dose to greater than 2 mg is not supported by current evidence. Phase III trial data have not demonstrated added benefit with higher doses of bulevirtide; treatment duration, rather than dose escalation, is likely more important for achieving clinical benefit. The CDA-AMC review team noted that the 5 mg and 10 mg doses are not under review by Health Canada and not of interest to this reimbursement review. |
Bulevirtide is administered by subcutaneous injection. | Comment from the drug plans to inform CDEC deliberations. |
It is recommended that a clinician experienced in the management of viral hepatitis prescribes and monitors bulevirtide treatment. Access to such clinicians may be limited in some areas. | Comment from the drug plans to inform CDEC deliberations. |
Question for the clinical experts: Are there circumstances where clinicians would prescribe bulevirtide in combination with PEG-IFN-alpha or with nucleoside (or nucleotide) analogues or with both? | The clinical experts consulted stated that bulevirtide would typically be used in combination with nucleoside (or nucleotide) analogues, such as tenofovir or entecavir, which are used to manage the HBV and have no effect on HDV. |
Generalizability | |
The mechanism by which HDV enters hepatocytes is assumed to be the same as for HBV; the mechanism of treatment is also the same. Questions for the clinical experts: Would this medication be used to treat HBV infection in the absence of HDV? Would this medication be used as prophylaxis for patients with HBV and/or HDV infection? | According to the clinical experts consulted, there are currently no data to support the use of bulevirtide for the treatment of HBV infection in the absence of HDV, nor as prophylaxis for patients with HBV and/or HDV infection. |
Care provision issues | |
Administration would require training, as it is given by subcutaneous injection, but it is not a prefilled syringe. It requires reconstitution. | Comment from the drug plans to inform CDEC deliberations. |
System and economic issues | |
Training in reconstitution and administration would be required. Who would cover the costs involved in this training? Who would cover the cost of supplies (needles and so on)? | The CDA-AMC review team noted that it is unclear who would cover these costs in practice. In the sponsor’s economic model, it was assumed that the costs of trainings would be covered by the public drug plans, and no assumptions were made regarding the cost of supplies. |
ALT = alanine aminotransferase; CDA-AMC = Canada's Drug Agency; CDEC = Canadian Drug Expert Committee; CrCl = creatinine clearance; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; HDV = hepatitis delta virus; HRQoL = health-related quality of life; LLoD = lower limit of detection; PCR = polymerase chain reaction; PEG-IFN = pegylated interferon; RCT = randomized controlled trial; RNA = ribonucleic acid.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of bulevirtide at a dose of 2 mg administered by subcutaneous injection in the treatment of chronic HDV infection in adult patients with compensated liver disease. The focus is on comparing bulevirtide to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of bulevirtide is presented in 3 sections with the CDA-AMC critical appraisal of the evidence at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs selected according to the sponsor’s systematic review protocol. The assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes indirect evidence from the sponsor. The third section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following are included in this review and appraised in this document:
1 pivotal RCT identified in systematic review
1 ITC
2 additional studies that address gaps in evidence.
Systematic Review
The contents of this section were informed by materials submitted by the sponsor, and have been validated and summarized by the review team.
Description of Study
Characteristics of the study included in the systematic review are summarized in Table 5.
Table 5: Details of the Study Included in the Systematic Review
Detail | MYR301 study |
|---|---|
Design and population | |
Study design | Phase III, randomized, open-label, multicentre study |
Locations | 16 sites from 4 countries, including Germany, Italy, Russia, and Sweden |
Patient enrolment dates | Start date: April 17, 2019 Cut-off date: September 30, 2022 End date: Ongoing |
Randomized | N = 150 Delayed treatment: N = 51 Bulevirtide 2 mg: N = 49 Bulevirtide 10 mg: N = 50 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Bulevirtide 2 mg: Bulevirtide at a dose of 2 mg SC, once daily for 144 weeks Bulevirtide 10 mg: Bulevirtide at a dose of 10 mg SC, once daily for 144 weeks |
Comparator(s) | Delayed treatment: Observation for 48 weeks followed by treatment with bulevirtide at a dose of 10 mg SC, once daily for 96 weeks |
Study duration | |
Screening phase | 4 weeks |
Treatment phase | Delayed-treatment group: 96 weeks Bulevirtide 2 mg and 10 mg: 144 weeks |
Follow-up phase | 96 weeks |
Outcomes | |
Primary end point | Combined response: Proportion of patients who achieved undetectable HDV RNA (less than the LLoD, where the LLoD < 6 IU/mL) or a ≥ 2 log10 IU/mL decline in HDV RNA, and ALT normalization at week 48 |
Secondary and exploratory end points | Key secondary:
Other secondary:
Additional secondary:
Exploratory:
|
Safety end points | The incidence of AEs, SAEs, special AEs of interest (hepatic AEs, renal AEs, eosinophilia, and injection site reaction). |
Publication status | |
Publications | Wedemeyer H, Aleman S, Brunetto MR, et al. A phase 3, randomized trial of bulevirtide in chronic hepatitis D. New England Journal of Medicine. 2023;389(1):22-32. doi:10.1056/NEJMoa2213429 ClinicalTrials.gov Identifier: NCT03852719 |
AE = adverse event; ALT = alanine aminotransferase; BLV = bovine leukemia virus; HBeAg = hepatitis B e-antigen; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCV = hepatitis C virus; HDV = hepatitis delta virus; HQLQ = Hepatitis Quality of Life Questionnaire; LLoD = lower limit of detection; PCR = polymerase chain reaction; PEG-IFN = pegylated interferon; RNA = ribonucleic acid; SAE = serious adverse event; SC = subcutaneous; ULN = upper limit of normal.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The MYR30119 trial is an ongoing, phase III, randomized, open-label, parallel-group, multicentre trial (Figure 1). The primary objective of the MYR301 trial was to evaluate the efficacy of bulevirtide at a dose of 2 mg for the treatment of HDV infection in adults with compensated liver disease, compared with delayed treatment with bulevirtide at a dose of 10 mg after a 48-week observation period. The secondary objectives of the trial included assessing the safety of bulevirtide and determining the optimal treatment duration. Patients were enrolled from 16 sites across 4 countries — Germany, Italy, Russia, and Sweden. The study includes a 4-week screening period, a 144-week treatment period, and a 96-week follow-up period.
During the screening period, patients were evaluated for study eligibility in accordance with the study protocol. Following screening, patients were randomized using an electronic randomization system in a 1:1:1 ratio to receive delayed treatment with bulevirtide 10 mg after a 48-week observation period of (N = 51), immediate treatment with bulevirtide 2 mg (N = 49), or immediate treatment with bulevirtide 10 mg (N = 50). Randomization was stratified based on the presence of liver cirrhosis (yes or no). This was an open-label study, with central laboratories blinded to actual treatment allocation (except the laboratories for pharmacokinetic, immunogenicity, NTCP polymorphism, and resistance tests). The primary and secondary end points of the MYR301 trial were analyzed using data up to the cut-off date of September 30, 2022, with the database locked on June 8, 2023.
Data for the study group receiving immediate treatment with bulevirtide at a dose of 10 mg are not presented in this report as this dose has not been approved and is not in use in clinical practice in Canada. This report primarily focuses on between-group comparisons of study end points up to week 48, specifically for bulevirtide at a dose of 2 mg, in line with the Health Canada indication. The use of bulevirtide at a dose of 10 mg as a comparator beyond this period was considered inappropriate based on input from the clinical experts consulted by CDA-AMC that the use of the 10 mg dose would not align with clinical practice as this dose has not been approved by Health Canada.
The systematic review conducted by the sponsor also identified 2 supportive phase II RCTs (MYR202 and MYR203), but these were ineligible for inclusion in this systematic review section, which is limited to pivotal trials, phase III clinical trials, and phase IV clinical trials. The CDA-AMC review team decided against summarizing the MYR202 study in this report because it does not address a gap in the pivotal RCT evidence. The MYR202 study assessed the efficacy of 3 doses of bulevirtide in combination with tenofovir versus observation on background therapy with tenofovir in patients with chronic hepatitis D. The MYR203 phase II trial is summarized in the Studies Addressing Gaps in the Systematic Review Evidence section.
Figure 1: Study Design of the MYR301 Trial
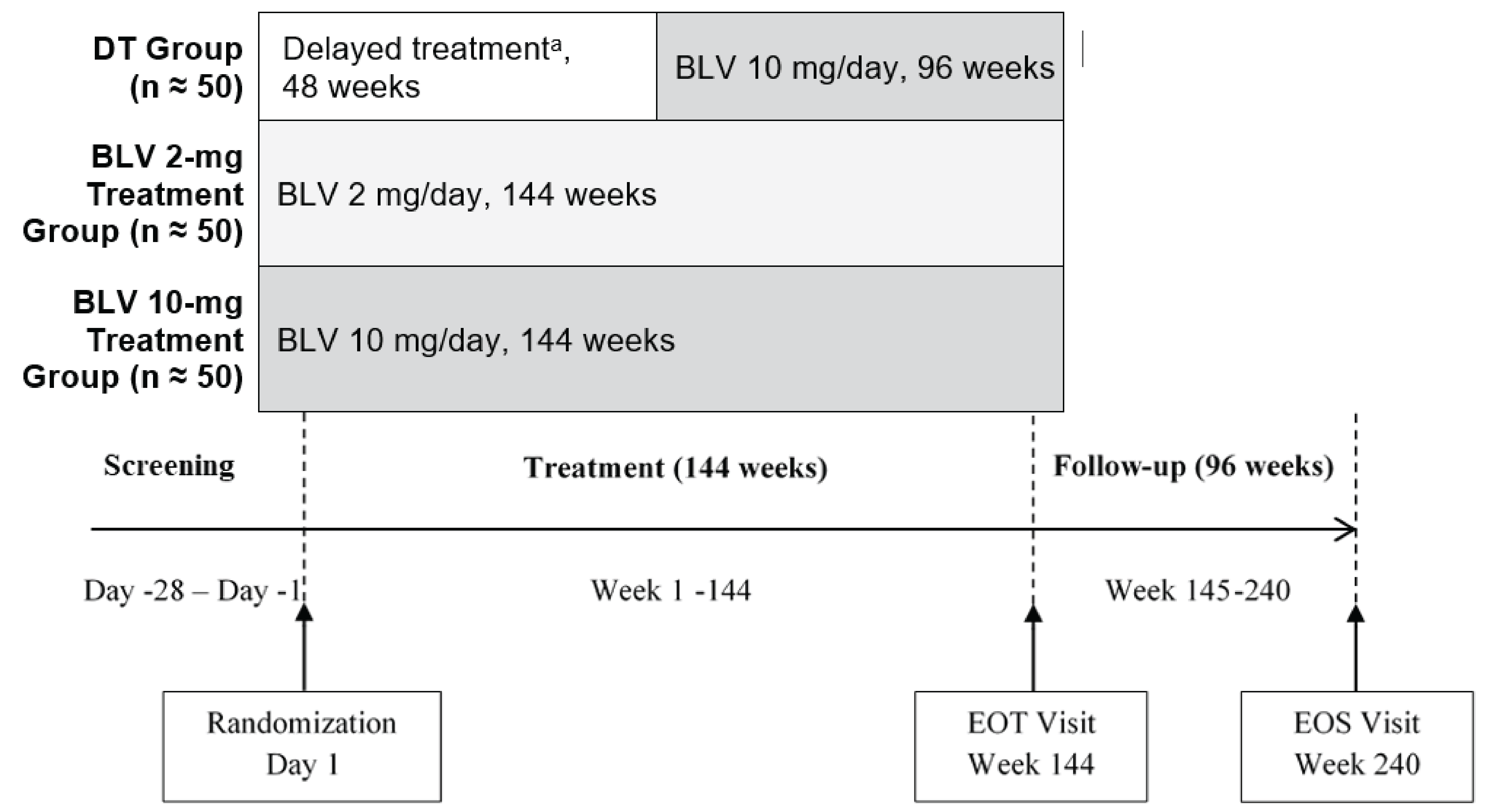
BLV = bulevirtide; DT = delayed treatment; EOT = end of treatment; EOS = end of study; HDV = hepatitis delta virus.
aPatients in the delayed-treatment group received bulevirtide 10 mg once daily for 96 weeks after receiving no treatment for HDV infection over a 48-week observation period.
Source: Clinical Study Report for MYR301.19 Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Patients were eligible for enrolment in the MYR301 trial if they were aged between 18 and 65 years and had chronic hepatitis D (i.e., positive serum anti-HDV antibody results or polymerase chain reaction [PCR]-detectable HDV RNA in serum or plasma for at least 6 months before screening). Additional eligibility criteria included positive PCR results for serum or plasma HDV RNA at screening; ALT levels greater than 1 times the upper limit of normal (ULN) but less than 10 times the ULN at screening; and serum albumin level greater than 28 g/L. Key exclusion criteria included decompensated cirrhosis, defined as a Child-Pugh hepatic insufficiency score greater than 7 points. Patients were also excluded from the MYR301 trial if they had current bleeding or ligation or a history of bleeding or ligation within the past 2 years. Additional exclusion criteria included coinfection with HCV or uncontrolled coinfection with HIV, a platelet count less than 60,000 cells/mm3, use of interferons within 6 months before screening, and creatinine clearance less than 60 mL/min.
Interventions
The MYR301 trial was designed to last for 240 weeks, including the 144-week treatment period and the 96-week off-treatment follow-up period.
In the bulevirtide 2 mg group, patients received bulevirtide 2 mg once daily via a single subcutaneous injection for 144 weeks.
In the delayed-treatment group, patients received bulevirtide 10 mg once daily, administered as 2 subcutaneous injections, for 96 weeks after a 48-week observation period with no active HDV treatment.
Bulevirtide was administered either by the patients at home or by designated personnel at the study site. Dose adjustments were not permitted in this study, and the use of rescue therapy was not applicable.
Concomitant Medications
Patients could receive nucleoside (or nucleotide) analogues at screening, during treatment, and during the follow-up period if required for the management of underlying chronic HBV infection, as indicated by current clinical practice guidelines.
Prohibited Medications
The following medications were prohibited in the MYR301 trial:
systemic glucocorticosteroids (inhaled glucocorticosteroids were allowed)
immunomodulatory drugs
antiviral drugs for HBV and/or HDV treatment, apart from permitted nucleoside (or nucleotide) analogues and rescue therapy with bulevirtide for hepatitis exacerbation in the posttreatment period
hematopoiesis-stimulating drugs
sulfasalazine
ezetimibe
cyclosporine
substrates of organic anion-transporting polypeptide
irbesartan
ritonavir.
Outcomes
A list of efficacy end points assessed in this Clinical Review report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review according to the clinical experts consulted for this review and input from patient and clinician groups and public drug plans. With this in mind, the review team selected end points considered to be most relevant in informing expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. The following efficacy end points were considered critical for expert committee deliberations and were assessed using GRADE: combined response; HDV RNA decrease of at least 2 log10 IU/mL or undetectable HDV RNA; change from baseline in liver stiffness measured using FibroScan; and change in HRQoL from baseline. The remaining end points (undetectable HDV RNA, ALT normalization, liver-related clinical events, combined response over time, and virologic response over time) were summarized as supportive information. SAEs were also assessed using GRADE.
Table 6: Outcomes Summarized From the MYR301 Study
Outcome measure | Time point | MYR301 study |
|---|---|---|
Combined response: Undetectable HDV RNA or decrease ≥ 2 log10 IU/mL from baseline and ALT normalization | Week 48 | Primarya |
Undetectable HDV RNA at weeks 24 and 48 after scheduled end of treatment (sustained virologic response) | Data not available | Secondary |
Undetectable HDV RNA | Week 48 | Key secondarya |
HDV RNA decrease ≥ 2 log10 IU/mL or undetectable HDV RNA (virologic response) | Week 48 | Additional secondary |
Change from baseline in liver stiffness measured using FibroScan | Week 48 | Secondary |
Change in quality of life assessed using the HQLQ | Weeks 24 and 48 | Exploratory |
ALT normalization (biochemical response) | Week 48 | Secondary |
Clinical events (decompensation, liver-related death, liver-related hospitalization) | At all postbaseline assessments up to week 144 | Exploratory |
Combined response over timeb | Weeks 24, 48, 72, and 96 | Exploratory |
Virologic response over timeb | Weeks 24, 48, 72, and 96 | Exploratory |
Safety outcomes: Incidence of AEs, SAEs, AEs of special interest (hepatic AEs, renal AEs, eosinophilia, and injection site reaction) | Weeks 48 and 144 | Exploratory |
AE = adverse event; ALT = alanine aminotransferase; HDV = hepatitis delta virus; HQLQ = Hepatitis Quality of Life Questionnaire; LLoQ = lower limit of quantification; RNA = ribonucleic acid; SAE = serious adverse event.
Note: Combined response was defined as undetectable HDV RNA (less than the LLoQ, target not detected) or HDV RNA decrease ≥ 2 log10 IU/mL from baseline combined with ALT normalization. Virologic response was defined as undetectable HDV RNA (less than the LLoQ, target not detected) or HDV RNA decrease ≥ 2 log10 IU/mL. ALT normalization was defined as an ALT level within the normal reference range, assessed according to central laboratory criteria (≤ 31 U/L for females and ≤ 41 U/L for males in Russia, ≤ 34 U/L for females and ≤ 49 U/L for males in all other locations).
aStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchical testing).
bThese end points were summarized because they were used to inform the health economics model.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The primary efficacy end point in the MYR301 trial was the combined response at week 48. Combined response was defined as meeting both of the following criteria: undetectable HDV RNA or decrease in HDV RNA of at least 2 log10 IU/mL from baseline (virologic response) and normalization of ALT levels (biochemical response).
Undetectable HDV RNA was defined as HDV RNA levels less than the LLoQ, with the HDV RNA target not detected.
Virologic response was defined as undetectable HDV RNA or decrease in HDV RNA of at least 2 log10 IU/mL from baseline.
ALT normalization was defined as an ALT level within the normal reference range, assessed according to central laboratory criteria (≤ 31 U/L for females and ≤ 41 U/L for males in Russia, and ≤ 34 U/L for females and ≤ 49 U/L for males in all other locations).
The change from baseline in liver stiffness was measured using FibroScan. FibroScan29 is a noninvasive imaging tool that uses transient elastography to measure liver stiffness to provide a reliable estimate of liver fibrosis. It is considered the standard of care in clinical practice and is widely used in various chronic liver diseases, including HBV, HCV, and HDV. Liver stiffness values are reported in kilopascals, with higher values indicating more advanced fibrosis or cirrhosis.
Health-Related Quality of Life
The MYR301 trial used 3 tools to assess quality of life — the EQ-5D, the Fatigue Severity Scale, and the HQLQ. The clinical experts consulted for this review identified HQLQ as the most relevant questionnaire for evaluating disease-specific elements of quality of life in patients with chronic hepatitis D.
The HQLQ17 (Table 7) includes both the SF-36 items and 4 hepatitis-specific health domain scores. These hepatitis-specific domains include health distress (4 items), positive well-being (4 items), hepatitis-specific limitations (3 items), and hepatitis-specific health distress (4 items). The SF-3630 is a self-administered questionnaire consisting of 36 items, which assess 2 main components: physical health and mental health. The physical component and the mental component summaries each contain 4 domains, which together evaluate the patient's overall HRQoL in relation to both physical and psychological aspects. Each domain is scored on a scale from 0 to 100, with higher scores indicating better HRQoL.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HQLQ | A generic and hepatitis-specific HRQoL measure | Validity: The known group validity of the HQLQ Version 2 was established in 2 RCTs of PEG-INF-alpha-2b used in the treatment of hepatitis C (n = 157).31 Patients with chronic hepatitis C scored worse on the generic scales than patients with other chronic conditions and worse than patients with good performance status. The physical functioning and bodily pain scales were associated with cirrhosis or high ALT levels31 Reliability: In a trial with 72 patients (90% with HCV) who completed the HQLQ Version 2 at baseline and 12 weeks after initiating treatment with direct-acting antiviral drugs, internal consistency reliability of the HQLQ Version 2 was demonstrated, with Cronbach alpha greater than 0.7; 12-week test-retest reliability was measured with an intraclass correlation coefficient greater than 0.7 for all SF-36 and hepatitis-specific subscales.17 Responsiveness: Responsiveness of the HQLQ Version 2 was compared with that of the SF-36 Version 2 based on the standardized response mean, mean ± SD, and percentage change stratified by improvement, status quo, or deterioration in health status.17 There was a significant change (P < 0.001) in improvement or deterioration in health condition, but not in status quo or stable health condition. HQLQ Version 2 and SF-36 Version 2 domain change scores were in agreement for improvement (57% vs. 53%), deterioration (31% vs. 30%), and stable health status (12% vs. 17%), respectively, by instrument.17 | There is no estimated MID for HQLQ. |
ALT = alanine aminotransferase; HCV = hepatitis C virus; HQLQ = Hepatitis Quality of Life Questionnaire; HRQoL = health-related quality of life; SF-36 = Short Form (36) Health Survey; MID = minimal important difference; RCT = randomized controlled trial; SD = standard deviation; vs. = versus.
Statistical Analysis
Sample Size Determination
Based on findings from the earlier, phase II MYR202 trial,32 the expected response rates of the primary end point of combined response at week 48 for bulevirtide at 2 mg and 10 mg doses were at least 45%, while the conservative estimate for the response rate in the delayed-treatment group was 8% or less. With a sample size of 47 patients per treatment group, a 2-sided Fisher exact test at a 0.04 significance level would provide 97.8% power to detect this difference in response rates between the bulevirtide 2 mg and 10 mg groups and the delayed-treatment group. The overall power of simultaneously rejecting both null hypotheses would be 95.6%.
Multiplicity Adjustment
An interim analysis at week 24 and a primary analysis at week 48 were conducted to evaluate treatment efficacy of bulevirtide based on the primary end point. To account for the repeated analysis, the overall 2-sided significance level of 0.05 was allocated across the 2 time points: 0.01 for week 24 and 0.04 for week 48. At each analysis, the bulevirtide 10 mg (hypothesis 1) group and the bulevirtide 2 mg (hypothesis 2) group were compared to the delayed-treatment group at the corresponding adjusted significance level. Under this hierarchical procedure, the second hypothesis was tested only if the first null hypothesis was rejected. The key secondary end point was tested only if the null hypothesis for the primary end point was rejected. All other analyses were conducted with no adjustments for multiple comparisons.
Statistical Testing
Combined Response at Week 48
The primary analysis of the primary end point, combined response at week 24 and week 48, compared the rate difference between the bulevirtide 2 mg, bulevirtide 10 mg, and delayed-treatment groups. The proportions of patients who achieved the combined response were analyzed separately for the bulevirtide 2 mg and bulevirtide 10 mg groups compared with the delayed-treatment group. The difference in response rates was estimated with a 99% exact unconditional confidence interval (CI), calculated using the score statistic. The P values from 2-sided Fisher exact tests were also provided. The comparison of bulevirtide 2 mg treatment versus delayed treatment was considered significant only if the comparison of bulevirtide 10 mg treatment versus delayed treatment was significant. Response rates for each group were also presented with Clopper-Pearson 95% CIs.
Due to the anticipated low number of patients with response in the delayed-treatment group, the analysis was not stratified by cirrhosis or other covariates.
Undetectable HDV RNA at Week 48
The key secondary end point of the MYR301 trial, undetectable HDV RNA at week 48, was analyzed using the same methods as used for the primary end point. This test was to be performed only if the 2 null hypotheses for the primary end point at week 48 were rejected.
ALT Normalization at Week 48
The secondary end point, ALT normalization at week 48, was analyzed using the same methods as the coprimary end point. Exact unconditional CIs based on scores for the proportion differences were presented, with corresponding 95% CIs. For the within-group proportions, Clopper-Pearson 95% CIs were calculated.
Change From Baseline in Liver Stiffness at Week 48
For the change in liver stiffness from baseline at week 48, an analysis of covariance model was used to compare the LS means between the bulevirtide 2 mg and delayed-treatment groups as well as the bulevirtide 10 mg and delayed-treatment groups. The model accounted for treatment, region, presence of cirrhosis, and baseline liver stiffness as covariates. Nominal P values and 95% CIs for the LS mean differences were provided, along with the LS mean for each treatment group.
Descriptive statistics for liver stiffness and its change from baseline was presented by treatment group. Plots of means (with SDs) of change from baseline over time, including subgroup analysis by cirrhosis status, were also presented.
Health-Related Quality of Life
In the MYR301 trial, the exploratory outcome HRQoL was measured by change from baseline in the HQLQ and results are expressed as the change from baseline. The clinical experts consulted by CDA-AMC for this review identified the HQLQ as the most relevant disease-specific tool for assessing the HRQoL of patients with chronic hepatitis D.
HQLQ was analyzed using mixed-effects models for repeated measures for change from baseline with treatment, region, presence of cirrhosis, visit, and treatment by visit interaction as fixed effects and the baseline value as a covariate. The model was fitted using the restricted maximum likelihood approach. The LS mean, standard error, 95% CIs, and P values were reported for each domain of the HQLQ tool.
Combined response and virologic response at each visit were exploratory end points in the MYR301 trial.
Subgroup Analysis
In the MYR301 trial, efficacy end points were evaluated in the full analysis set (FAS) population across the following prespecified subgroups using descriptive statistics:
cirrhosis status at randomization (presence versus absence)
concomitant HBV treatment (yes versus no).
Sensitivity Analysis
A sensitivity analysis was performed on the FAS for the primary (combined response) and key secondary (undetectable HDV RNA) end points, where assessments made outside of the time window defined by plus or minus 30 days from the planned time point were considered as missing, regardless of whether the time deviation was related to COVID-19.
Handling of Missing Values
The LOCF approach was used to impute missing values for the combined response and undetectable HDV RNA in cases where the missing data were related to COVID-19 for the primary efficacy parameter. For all other instances, a missing equals failure approach was applied, where missing values are imputed as patients who showed no response. For HQLQ, for the observed case analysis, missing values were not imputed and remained missing.
For virology data in the MYR301 trial, the following imputation rules were applied:
Values reported as less than the LLoQ with a note indicating “target not detected” were imputed as 0.
Values less than the LLoQ without a “target not detected” note were:
Imputed as one-half of the LLoQ if the LLoQ was not equal to the lower limit of detection
Imputed as 0 if the LLoQ equals the lower limit of detection.
Values greater than the upper limit of quantification were imputed as the upper limit of quantification.
Nonmeasurable data were considered missing.
For log10-transformed data, the following rules were applied:
A value of 0 was imputed as 0 if the limit of detection was greater than 1.
A value of 0 was imputed as the log10 of one-half the limit of detection if the limit of detection was less than 1.
Table 8: Statistical Analysis of Efficacy End Points in the MYR301 Study
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
MYR301 study | ||||
Combined response at week 48 |
| NR |
| Assessments made outside of the time window defined by ± 30 days from the planned time point were considered as missing, regardless of whether the time deviation was related to COVID-19. |
Undetectable HDV RNA at weeks 48 |
| NR |
| Assessments made outside of the time window defined by ± 30 days from the planned time point were considered as missing, regardless of whether the time deviation was related to COVID-19. |
Undetectable HDV RNA or decrease in HDV RNA by ≥ 2 log10 IU/mL from baseline at week 48 |
| NR | Missing equals failure approach | NR |
ALT normalization at week 48 |
| NR | Missing equals failure approach | NR |
Change from baseline in liver stiffness at week 48 | ANCOVA model | Treatment group, region, presence of cirrhosis, and baseline value | Missing equals failure approach | NR |
Change in baseline in HQLQ at week 48 | Mixed-effects model for repeated measures | Treatment, region, presence of cirrhosis, visit, and treatment by visit interaction as fixed effects and baseline value as a covariate | Observed case analysis | NR |
Combined response over time | Descriptive statistics | NR | Missing equals failure approach | NR |
Virologic response over time | Descriptive statistics | NR | NR | NR |
ALT = alanine aminotransferase; ANCOVA = analysis of covariance; HDV = hepatitis delta virus; NR = not reported; RNA = ribonucleic acid.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
Table 9: Analysis Populations of the MYR301 Study
Study | Population | Definition |
|---|---|---|
MYR301 | All randomized analysis set, N = 150 | All patients who were enrolled and randomized. |
Full analysis set (FAS), N = 150 | All patients randomized to the delayed-treatment and bulevirtide groups who received at least 1 dose of bulevirtide after randomization. | |
Safety analysis set (SAS), N = 150 | All patients randomized to the delayed-treatment and bulevirtide groups who received at least 1 dose of bulevirtide after randomization. |
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Summary of patient disposition is presented in Table 10.
A total of 183 patients were screened for the MYR301 trial, and 150 were randomized. Eighteen percent of patients were unsuccessful in meeting the screening criteria. By week 144, 149 patients had received bulevirtide treatment, including 49 patients in the bulevirtide 2 mg group and 50 patients in the delayed-treatment group who started bulevirtide 10 mg at week 48. By week 48, the proportion of patients who discontinued the study was similar in both the bulevirtide 2 mg and delayed-treatment groups, with a 2.0% discontinuation rate in each group. The reason for discontinuation was withdrawal of consent (2.0%) in the bulevirtide 2 mg group and pregnancy (2.0%) in the delayed-treatment group. By week 144, the proportion of patients who discontinued the study was higher in the bulevirtide 2 mg group (8.2%) than in the delayed-treatment group (3.9%). In the bulevirtide 2 mg group, the reasons for discontinuation included withdrawal of consent (6.1%) and pregnancy (2.0%). In the delayed-treatment group, the discontinuations were due to pregnancy (2.0%) and death (2.0%). None of the patients in the MYR301 trial discontinued the study treatment.
Table 10: Summary of Patient Disposition in the MYR301 Study
Patient disposition | MYR301 study | ||
|---|---|---|---|
Delayed treatmenta (N = 51) | Bulevirtide (2 mg) (N = 49) | Bulevirtide (10 mg) (N = 50) | |
Screened, N | 183 | ||
Reason for unsuccessful screening, n (%) | 33 (18.0) | ||
Randomized, N (%) | 51 | 49 | 50 |
Completed week 24 visit, n (%) | 50 (98.0) | 49 (100.0) | 47 (94.0) |
Completed week 48 visit, n (%) | 50 (98.0) | 48 (98.0) | 47 (94.0) |
Completed week 96 visit, n (%) | 49 (96.1) | 47 (95.9) | 47 (94.0) |
Completed week 144 visit, n (%) | 49 (96.1) | 45 (91.8) | 44 (88.0) |
Discontinued from study by week 48, n (%) | 1 (2.0) | 1 (2.0) | 3 (6.0) |
Reason for discontinuation, n (%) | |||
Withdrawal of consent | 0 (0) | 1 (2.0) | 2 (4.0) |
Physician decision | 0 (0) | 0 (0) | 1 (2.0) |
Pregnancy | 1 (2.0) | 0 (0) | 0 (0) |
Discontinued from study by week 144, n (%) | 2 (3.9) | 4 (8.2) | 6 (12.0) |
Reason for discontinuation, n (%) | |||
Withdrawal of consent | 0 (0) | 3 (6.1) | 5 (10.0) |
Pregnancy | 1 (2.0) | 1 (2.0) | 0 (0) |
Death | 1 (2.0) | 0 (0) | 0 (0) |
Physician decision | 0 (0) | 0 (0) | 1 (2.0) |
FAS, N | 51 | 49 | 50 |
SAS, N | 51 | 49 | 50 |
FAS = full analysis set.
Note: One patient in the bulevirtide 2 mg group completed week 144 of the study and withdrew consent on the same day.
aThe participants received bulevirtide at a dose of 10 mg from week 48 onwards.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
Patient characteristics at the time of randomization (outlined in Table 11), are limited to those that are most relevant to this review or most likely to affect the outcomes or interpretation of the study results. The mean age was higher in the bulevirtide 2 mg group (44 years [SD = 9.0 years]) than in the delayed-treatment group (41 years [SD = 7.5 years]). The proportion of male patients was larger in the bulevirtide 2 mg group (61.2%) than in the delayed-treatment group (51.0%). Nearly one-half of the patients (47.3%) had cirrhosis at the time of randomization; all the cases of cirrhosis were classified as Child-Pugh Class A. All the patients in both the bulevirtide 2 mg and delayed-treatment groups had HDV genotype 1 at baseline. A larger proportion of the patients in the bulevirtide 2 mg group had a Child-Pugh score of 6 points (30.4% versus 20.8% in the delayed-treatment group), HBV genotype D (95.9% versus 86.3% in the delayed-treatment group), and HBV DNA greater than or equal to the LLoQ (67.3% versus 52.9% in the delayed-treatment group). Mean ALT levels were comparable in the bulevirtide 2 mg group (108 U/L [SD = 62.5 U/L]) and the delayed-treatment group (102 U/L [SD = 61.9 U/L]). A total of 32 patients (65.3%) in the bulevirtide 2 mg group and 32 patients (62.7%) in the delayed-treatment group received oral anti-HBV treatment during the MYR301 trial; 54.7% had initiated treatment before baseline. In addition, 26 (53.1%) of the patients in the bulevirtide 2 mg group and 29 (62.7%) of the patients in the delayed-treatment group had previously received interferon treatment.
Table 11: Summary of Baseline Patient and Disease Characteristics in the MYR301 Study (FAS)
Characteristic | MYR301 study | |
|---|---|---|
Delayed treatmenta (N = 51) | Bulevirtide (2 mg) (N = 49) | |
Age (years), mean (SD) | 41 (7.5) | 44 (9.0) |
Sex, n (%) | ||
Female | 25 (49.0) | 19 (38.8) |
Male | 26 (51.0) | 30 (61.2) |
Race, n (%) | ||
Asian | 11 (21.6) | 8 (16.3) |
Black or African American | 0 (0) | 0 (0) |
White | 40 (78.4) | 41 (83.7) |
Body mass index, kg/m2 | ||
Mean (SD) | 25.3 (3.9) | 24.4 (3.1) |
Cirrhosis status at randomization, n (%) | ||
Presence | 24 (47.1) | 23 (46.9) |
Absence | 27 (52.9) | 26 (53.1) |
Baseline Child-Pugh score (category), n (%)c | ||
5 | ██ ██████ | ██ ██████ |
6 | ██ ██████ | ██ ██████ |
Child-Pugh Class A, n (%)c | 24 (100.0) | 23 (100.0) |
HDV genotype, n (%) | ||
Genotype HDV-1 | 51 (100.0) | 49 (100.0) |
Genotype HDV-5 | 0 (0) | 0 (0) |
Missing | 0 (0) | 0 (0) |
HBV genotype, n (%)d | ||
Genotype A | 2 (3.9) | 2 (4.1) |
Genotype D | 44 (86.3) | 47 (95.9) |
Genotype E | 0 (0) | 0 (0) |
Missing | 3 (5.9) | 0 (0) |
Unclassified | 2 (3.9) | 0 (0) |
HBV DNA (log10 IU/mL) | ||
Mean (SD) | 0.89 (0.99) | 1.30 (1.29) |
Baseline HBV DNA category, n (%) | ||
< LLoQ target not detected | ██ ██████ | ██ ██████ |
< LLoQ target detected | ██ ██████ | ██ ██████ |
≥ LLoQ | ██ ██████ | ██ ██████ |
Missing | ██ ██████ | ██ ██████ |
HDV RNA (log10 IU/mL) | ||
Mean (SD) | 5.08 (1.36) | 5.10 (1.19) |
HBeAg status, n (%) | ||
Positive | 4 (7.8) | 4 (8.2) |
Negative | 47 (92.2) | 45 (91.8) |
HBsAg (log10 IU/mL) | ||
Mean (SD) | 3.68 (0.46) | 3.67 (0.51) |
ALT (U/L) | ||
Mean (SD) | 102 (61.9) | 108 (62.5) |
Baseline creatinine clearance category (mL/min), n (%) | ||
≥ 60 to < 90 | ██ ██████ | ██ ██████ |
≥ 90 | ██ ██████ | ██ ██████ |
Baseline liver stiffness (kPa) | ||
Mean (SD) | 15.3 (8.95) | 14.0 (8.19) |
Baseline liver stiffness category (kPa), n (%) | ||
< 12 | ██ ██████ | ██ ██████ |
12 to 20 | ██ ██████ | ██ ██████ |
> 20 | ██ ██████ | ██ ██████ |
Previous interferon therapy, n (%) | ||
No | 22 (43.1) | 23 (46.9) |
Yes | 29 (56.9) | 26 (53.1) |
Concomitant HBV medication, n (%)e | ||
No | 19 (37.3) | 17 (34.7) |
Yes | 32 (62.7) | 32 (65.3) |
TDF- or TAF-containing concomitant HBV medication, n (%) | ||
No | ██ ██████ | ██ ██████ |
Yes | ██ ██████ | ██ ██████ |
ALT = alanine aminotransferase; FAS = full analysis set; HBeAg = hepatitis B e-antigen; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HDV = hepatitis delta virus; LLoQ = lower limit of quantification; RNA = ribonucleic acid; SD = standard deviation; TAF = tenofovir alafenamide; TDF = tenofovir disoproxil fumarate.
Notes: Baseline value was the last available value collected before or at the time of the first dose of bulevirtide in the bulevirtide 2 mg and bulevirtide 10 mg treatment groups and the last available value collected before or at randomization for the delayed-treatment group. For patients in the delayed treatment to bulevirtide 10-mg treatment group, baseline (i.e., value was the last available value collected before or at randomization for sex, race, and height, and the last available value collected before or at the time of the first dose of bulevirtide 10 mg at week 48 otherwise.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aPatients received bulevirtide at a dose of 10 mg from week 48 onwards.
bBody mass index = [weight (kg)/height (m)2] × 10,000.
cChild-Pugh score and class are presented only for patients with cirrhosis. Percentages are based on the number of patients with cirrhosis and available Child-Pugh scores at baseline.
dHBV genotyping was performed at the first positive test for HBV DNA, which could be postbaseline.
eAnti-HBV medications are defined as oral medications with preferred names containing terms of tenofovir, tenofovir alafenamide, tenofovir disoproxil fumarate, tenofovir disoproxil, entecavir, adefovir, lamivudine, telbivudine, and adefovir dipivoxil.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 12 shows a summary of key disease characteristics assessed at randomization and the reset baseline at week 48 for patients randomized to the delayed-treatment group following a 48-week observation period, before receiving the first dose of bulevirtide 10 mg. This treatment group consisted of 50 patients who received at least 1 dose of the study treatment after week 48. For most disease characteristics, the summary statistics remained similar at randomization and week 48, with a few notable exceptions. Specifically, the mean serum ALT level was higher at randomization (102 U/L [SD = 61.9 U/L]) compared to week 48 (82 U/L [SD = 51.1 U/L]) and the mean liver stiffness was 15.3 kPa (SD = 8.95 kPa) at randomization and 16.1 kPa (SD = 11.84 kPa) at week 48.
Table 12: Resetting Baseline for Delayed-Treatment Group at Week 48 (FAS)
Characteristic | Delayed treatment All randomized (N = 51) | Delayed treatment to bulevirtide (10 mg) (N = 50) |
|---|---|---|
HDV RNA (log10 IU/mL) | ||
Mean (SD) | 5.08 (1.358) | ██ ██████ |
HBV DNA (log10 IU/mL) | ||
Mean (SD) | ██ ██████ | ██ ██████ |
HBeAg status, n (%) | ||
Negative | ██ ██████ | ██ ██████ |
HBsAg (log10 IU/mL) | ||
Mean (SD) | 3.68 (0.465) | ██ ██████ |
ALT (U/L) | ||
Mean (SD) | 102 (61.9) | ██ ██████ |
Liver stiffness (kPa) | ||
Mean (SD) | 15.3 (8.95) | ██ ██████ |
ALT = alanine aminotransferase; FAS = full analysis set; HBeAg = hepatitis B e-antigen; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HDV = hepatitis delta virus; RNA = ribonucleic acid; SD = standard deviation.
Notes: Baseline value was the last available value collected before or at randomization for the delayed-treatment group.
For patients in the delayed-treatment group receiving bulevirtide 10 mg, the baseline (i.e., reset baseline) value was the last available value collected before or at randomization for HBeAg and HDV and HBV genotypes, the last available value collected before the first dose of bulevirtide at week 48 for ALT levels, the last available value collected before or at week 48 for liver stiffness, and the last available value collected before or at randomization otherwise.
Source: Clinical Study Report for MYR301.19
Exposure to Study Treatments
Table 13 summarizes the extent of exposure and adherence to bulevirtide by week 144 in the MYR301 trial. The mean duration of study treatment exposure was ██████ █████ (SD = █████ █████) in the bulevirtide 2 mg group from baseline to week 144 and █████ █████ (SD = █████ █████) in the delayed-treatment group from weeks 48 to 144. The mean adherence rate was similar in the bulevirtide 2 mg and delayed-treatment groups (96.75% versus 97.83%, respectively). In the MYR301 trials, █████ of patients in the bulevirtide 2 mg group and █████ in the delayed-treatment group missed at least 1 dose of bulevirtide by week 144.
Table 13: Summary of Patient Exposure and Treatment Adherence in the MYR301 Study (SAS)
Exposure | MYR301 study | |
|---|---|---|
Baseline to week 144 | Week 48 to week 144 | |
Bulevirtide (2 mg) (N = 49) | Delayed treatment to bulevirtide (10 mg) (N = 51) | |
Duration of exposure (weeks) | ||
Mean (SD) | ██ ██████ | ██ ██████ |
Dose intensity (mg/week) | ||
Mean (SD) | ██ ██████ | ██ ██████ |
Rate of adherence, % | ||
Mean (SD) | 96.75 (12.87) | 97.83 (11.45) |
Missed doses, mean (SD) | ██ ██████ | ██ ██████ |
SD = standard deviation; SAS = safety analysis set.
Notes: Total duration of bulevirtide exposure (week) = (last dose date of bulevirtide – first dose date of bulevirtide + 1)/7, regardless of any temporary interruptions.
Total dose administered (mg) = sum of all doses administered as reported in the participant diary, which took participant-reported missed doses into consideration – sponsor identified missed doses.
Dose intensity (mg/week) = total dose administered / total duration of exposure.
If the calculated adherence rate was greater than 100%, the result was set to 100%.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Protocol Deviations
The proportion of patients with any important protocol deviation was higher in the delayed-treatment group than the bulevirtide 2 mg group (████ versus ████, respectively). The most common types of protocol deviation in both groups were related to █████ ████ ██████████████ █████ ██ ███ ███████ █████████ █████ ██████ ████ ██ ███ ███████████ | ██ ███████ ██████ ██████ ██ ███████ ███ █████ ███████████ █████ ██ ███ ███████ █████████ █████ ██████ ████ ██ ███ ███████████ | ██ ███████ ███ ███ ██ ██████████ ███████████ █████ ██ ███ ███████ █████████ █████ ██████ ████ ██ ███ ███████████ | ██ ██████.
Efficacy
Efficacy results are summarized in the combined response rates in the bulevirtide 2 mg group, which demonstrated consistent improvement over time: 34.7% (95% CI, 21.7% to 49.6%) at week 24, 44.9% (95% CI, 30.7% to 59.8%) at weeks 48 ███ ██, 55.1% (95% CI, 40.2% to 69.3%) at week 96, and 57.1% (95% CI, 42.2% to 71.2%) at week 144.
Virologic Response Over Time
The virologic response at each study visit, an exploratory end point in MYR301, was analyzed exclusively using a missing equals failure approach. The virologic response rates in the bulevirtide 2 mg group demonstrated improvement over time: 55.1% (95% CI, 40.2% to 69.3%) at week 24, 73.5% (95% CI, 58.9% to 85.1%) at week 48, █████ ████ ███ █████ ██ ██████ ██ ████ ██, 75.5% (95% CI, 61.1% to 86.7%) at week 96, and 73.5% (95% CI, 58.9% to 85.1%) at week 144.
The results presented in Table 14 are for bulevirtide at a 2 mg dose compared to the delayed-treatment group, in accordance with the reimbursement request submitted by the sponsor.
Combined Response
The combined response at week 48 was a primary end point in the MYR301 trial. At week 48, 44.9% (95% CI, 30.7% to 59.8%) of patients in the bulevirtide 2 mg group and 2.0% (95% CI, 0.0% to 10.4%) of those in the delayed-treatment group achieved a combined response. The between-group difference in response rates was 42.9% (96% CI, 27.0% to 58.5%; P < 0.0001) in favour of the bulevirtide 2 mg group.
Missing data were minimal for the primary analysis. The sensitivity analysis results, using the missing equals failure approach, were consistent with the primary analysis. Subgroup analyses of the primary end point, combined response at week 48, were conducted based on cirrhosis status. In the bulevirtide 2 mg group, the proportion of patients without cirrhosis who achieved a combined response was 53.8% (95% CI, 16.4% to 57.3%) compared to 34.8% (95% CI, 33.4% to 73.4%) of patients with cirrhosis. In the delayed-treatment group, 4.2% (95% CI, 0.1% to 21.1%) of patients with cirrhosis achieved a combined response compared to 0.0% (95% CI, 0.0% to 12.8%) of those without cirrhosis.
Undetectable HDV RNA at Weeks 24 and 48 After Scheduled End of Treatment
An undetectable HDV RNA at weeks 24 and 48 after scheduled end of treatment (sustained virologic response) was a secondary end point in the MYR301 trial; however, the results for this end point were not reported due to an insufficient follow-up period as the study is still ongoing.
Undetectable HDV RNA or HDV RNA Decrease of at Least 2 log10 IU/mL at Week 48
A decrease in HDV RNA of at least 2 log10 IU/mL or undetectable HDV RNA (virologic response) at week 48 was an additional secondary end point in the MYR301 trial. At week 48, 73.5% (95% CI, 58.9% to 85.1%) of patients in the bulevirtide 2 mg group achieved a virologic response compared to 3.9% (95% CI, 0.5% to 13.5%) of patients in the delayed-treatment group. The between-group difference in response rates was 69.5% (96% CI, 54.1% to 81.9%; P < 0.0001) in favour of the bulevirtide 2 mg group. Missing data were minimal for the analyses.
Subgroup analyses for undetectable HDV RNA or HDV RNA decrease of at least 2 log10 IU/mL were conducted based on cirrhosis status and treatment with concomitant HBV medication (ad hoc analysis). At week 48, the proportion of patients with cirrhosis who achieved a virologic response was 82.6% (95% CI, 61.2% to 95.0%) in the bulevirtide 2 mg group and 8.3% (95% CI, 1.0% to 27.0%) in the delayed-treatment group. Among patients without cirrhosis, the corresponding proportions were 65.4% (95% CI, 44.3% to 82.8%) and 0.0% (95% CI, 0.0% to 12.8%), respectively. At week 48, 78.1% (95% CI, 60.0% to 90.7%) of patients receiving concomitant HBV medication in the bulevirtide 2 mg group achieved a virologic response compared to 6.3% (95% CI, 0.8% to 20.8%) of those in the delayed-treatment group. Among those who were not receiving HBV medication, the corresponding proportions were 64.7% (95% CI, 38.3% to 85.0%) and 0.0% (95% CI, 0.0% to 17.6%), respectively.
Change From Baseline in Liver Stiffness at Week 48
The change from baseline in liver stiffness was a prespecified secondary end point for the week 48 analysis. At week 48, the LS mean of change from baseline in liver stiffness was –3.06 kPa (95% CI, –4.67 kPa to –1.45 kPa) in the bulevirtide 2 mg group and 0.87 kPa (95% CI, −0.79 kPa to 2.53 kPa) in the delayed-treatment group. The between-group difference in LS means of change in liver stiffness was –3.93 (95% CI, –6.23 to –1.63; P = 0.0009) in favour of the bulevirtide 2 mg group. Missing data were minimal for the analysis.
Subgroup analyses of the change in liver stiffness were conducted based on cirrhosis status. At week 48, in the bulevirtide 2 mg group, the mean change from baseline in liver stiffness was –5.70 kPa (SD = 6.94 kPa) in patients with cirrhosis and –0.35 kPa (SD = 2.08 kPa) in patients without cirrhosis. In the delayed-treatment group, the mean change from baseline in liver stiffness was –0.20 kPa (SD = 4.00 kPa) in patients without cirrhosis and 1.78 kPa (SD = 10.1 kPa) in patients with cirrhosis.
Liver-Related Clinical Events
██ ███ ██████ ██████ | █████████████ ████████████████ ███ ██████████ ████ ███ ███████ ██████ ███ ████████ ██ | ███████ ██ ███ ███████████ | ██ ██████ ████ ███ | ███████ █████ ███████████████ ███ ██ ██████████ ████████ █████████████ | ███████ ████ █████████ ██ ███ ███████ █████████ █████ █████████ ███████████ █████ | ████████ ███ ████ █████ █████████ ██ ███████████ ██ ██ ██████████.
Health-Related Quality of Life
At week 24, compared to the delayed-treatment group, the LS mean differences in the bulevirtide 2 mg group were ███ (95% CI, ███ ██ ███) for the HQLQ physical component summary score, ███ (95% CI, ████ ██ ███) for the HQLQ mental component summary score, ████ (95% CI, ███ ██ ████) for the HQLQ health distress score, ███ (95% CI, ████ ██ ████) for the HQLQ positive well-being score, ███ (95% CI, ████ ██ ████) for the HQLQ hepatitis-specific limitations score, and ███ (95% CI, ████ ██ ████) for the HQLQ hepatitis-specific health distress score.
At week 48, compared to the delayed-treatment group, the LS mean differences in the bulevirtide 2 mg group were ███ (95% CI, ███ ██ ███) for the HQLQ physical component summary score, ███ (95% CI, ████ ██ ███) for the HQLQ mental component summary score, ███ (95% CI, ████ ██ ████) for the HQLQ health distress score, ███ (95% CI, ████ ██ ███) for the HQLQ positive well-being score, ███ (95% CI, ███ ██ ████) for the HQLQ hepatitis-specific limitations score, and ███ (95% CI, ███ ██ ████) for the HQLQ hepatitis-specific health distress score. Missing data were minimal for the analyses.
Undetectable HDV RNA
Undetectable HDV RNA (less than the LLoQ, target not detected) at week 48 was a key secondary end point in the MYR301 trial. At week 48, 12.2% (95% CI, 4.6% to 24.8%) of patients in the bulevirtide 2 mg group had undetectable HDV RNA compared to 0.0% (95% CI, 0.0% to 7.0%) in the delayed-treatment group. The between-group difference in response rates was calculated only for the bulevirtide 10 mg group, with the bulevirtide 2 mg group as the reference; however, this comparison is not relevant to this review. Missing data were minimal for the analysis.
Subgroup analyses of the key secondary end point were conducted based on cirrhosis status and concomitant treatment with nucleoside (or nucleotide) analogues (ad hoc analysis). In the bulevirtide 2 mg group, 21.7% (95% CI, 7.5% to 43.7%) of patients with cirrhosis achieved undetectable HDV RNA compared to 3.8% (95% CI, 0.1% to 19.6%) of those without cirrhosis. In the delayed-treatment group, 0.0% (95% CI, 0.0% to 14.2%) of patients with cirrhosis achieved an undetectable HDV RNA compared to 0.0% (95% CI, 0.0% to 12.8%) of those without cirrhosis. Similarly, 15.6% (95% CI, 5.3% to 32.8%) of patients in the bulevirtide 2 mg group receiving concomitant HBV medication achieved undetectable HDV RNA compared to 5.9% (95% CI, 0.1% to 28.7%) of those not receiving HBV medication. In the delayed-treatment group, no patients achieved an undetectable HDV RNA, regardless of concomitant treatment with HBV medication.
ALT Normalization
ALT normalization at week 48 was a secondary end point in the MYR301 trial. At week 48, 51.0% (95% CI, 36.3% to 65.6%) of patients in the bulevirtide 2 mg group achieved ALT normalization compared to 11.8% (95% CI, 4.4% to 23.9%) in the delayed-treatment group. The between-group difference in response rates was 39.3% (96% CI, 20.0% to 55.8%; P < 0.0001) in favour of the bulevirtide 2 mg group. Missing data were minimal for the analysis.
Subgroup analyses of ALT normalization were conducted based on cirrhosis status and concomitant treatment with HBV medication. In the bulevirtide 2 mg group, 61.5% (95% CI, 40.6% to 79.8%) of patients without cirrhosis achieved ALT normalization compared to 39.1% (95% CI, 19.7% to 61.5%) of patients with cirrhosis. In the delayed-treatment group, 7.4% (95% CI, 0.9% to 24.3%) of patients without cirrhosis achieved ALT normalization compared to 16.7% (95% CI, 4.7% to 37.4%) of patients with cirrhosis. Similarly, 52.9% (95% CI, 27.8% to 77.0%) of patients in the bulevirtide 2 mg group who did not receive concomitant HBV medication achieved ALT normalization, compared to 50.0% (95% CI, 31.9% to 68.1%) of those who received HBV medication. In the delayed-treatment group, 15.8% (95% CI, 3.4% to 39.6%) of patients who did not receive concomitant HBV medication achieved ALT normalization, compared to 9.4% (95% CI, 2.0% to 25.0%) of those who received HBV medication.
Combined Response Over Time
The combined response at each study visit, an exploratory end point in MYR301, was analyzed exclusively using a missing equals failure approach. In contrast, the primary end point analysis incorporated an LOCF approach to account for missing data due to COVID-19 at weeks 24 and 48. The combined response rates in the bulevirtide 2 mg group demonstrated consistent improvement over time: 34.7% (95% CI, 21.7% to 49.6%) at week 24; 44.9% (95% CI, 30.7% to 59.8%) at weeks 48 ███ ██; 55.1% (95% CI, 40.2% to 69.3%) at week 96; and 57.1% (95% CI, 42.2% to 71.2%) at week 144.
Table 14: Summary of Key Efficacy Results From the MYR301 Study (FAS)
Characteristic | Delayed treatmenta (N = 51) | Bulevirtide (2 mg) (N = 49) |
|---|---|---|
Combined responseb | ||
Week 24 | ||
Patients with response, n (%) | 0 (0.0) | 18 (36.7) |
95% CIc | 0.0 to 7.0 | 23.4 to 51.7 |
Response rate difference vs. control group, % (99% CId) | Reference | 36.7 (20.0 to 56.1) |
P valuee | Reference | < 0.0001 |
Week 48 | ||
Patients with response, n (%) | 1 (2.0) | 22 (44.9) |
95% CIc | 0.0 to 10.4 | 30.7 to 59.8 |
Response rate difference vs. control, % (96% CId) | Reference | 42.9 (27.0 to 58.5) |
P valuee | Reference | < 0.0001 |
Undetectable HDV RNA | ||
Week 24 | ||
Patients with response, n (%) | NR | 3 (6.1) |
95% CIc | NR | 1.3 to 16.9 |
Response rate difference vs. control, % (96% CId) | NR | Reference |
P valuee | NR | Reference |
Week 48 | ||
Patients with response, n (%) | 0 (0) | 6 (12.2) |
95% CIc | 0.0 to 7.0 | 4.6 to 24.8 |
Response rate difference vs. control, % (96% CId) | NR | Reference |
P valuee | NR | Reference |
Undetectable HDV RNA or HDV RNA decrease ≥ 2 log10 IU/mLc,e,f,g,h | ||
Week 24 | ||
Patients with response, n (%) | 2 (3.9) | 27 (55.1) |
95% CIc | 0.5 to 13.5 | 40.2 to 69.3 |
Response rate difference vs. control, % (95% CI) | Reference | 51.2 (34.4 to 65.9) |
P valuee | Reference | < 0.0001 |
Week 48 | ||
Patients with response, n (%) | 2 (3.9) | 36 (73.5) |
95% CIc | 0.5 to 13.5 | 58.9 to 85.1 |
Response rate difference vs. control, % (95% CI) | Reference | 69.5 (54.1 to 81.9) |
P valuee | Reference | < 0.0001 |
ALT normalization | ||
Week 24 | ||
Patients with response, n (%) | 3 (5.9) | 26 (53.1) |
95% CIc | 1.2 to 16.2 | 38.3 to 67.5 |
Response rate difference vs. control, % (95% CIf) | Reference | 47.2 (30.6 to 62.5) |
P valuee | Reference | < 0.0001 |
Week 48 | ||
Patients with response, n (%) | 6 (11.8) | 25 (51.0) |
95% CIc | 4.4 to 23.9 | 36.3 to 65.6 |
Response rate difference vs. control, % (95% CIf) | Reference | 39.3 (20.0 to 55.8) |
P valuee | Reference | < 0.0001 |
Change from baseline in liver stiffnessg,h,i,j | ||
Week 48 | ||
LS mean (SE), kPa | 0.87 (0.84) | –3.06 (0.81) |
95% CI | –0.79 to 2.53 | –4.67 to –1.45 |
LS means difference vs. contro, (95% CI) | Reference | –3.93 (–6.23 to –1.63) |
P valuee | Reference | 0.0009 |
Change from baseline in HQLQk | ||
Change from baseline in HQLQ physical component summary scorel | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Change from baseline in HQLQ mental component summary scorek,l,m | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Change from baseline in HQLQ health distress scorek,l,m | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Change from baseline in HQLQ positive well-being scorek,l,m | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Change from baseline in HQLQ hepatitis-specific limitations scale scorek,l,m | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Change from baseline in HQLQ hepatitis-specific health distress scale scorek,l,m | ||
Week 24 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Week 48 | ||
LS mean (SE) | █████████ | █████████ |
LS means difference vs. control, 95% CI | █████████ | █████████ |
P value | █████████ | █████████ |
Combined response over timek,m | ||
Week 24 | ||
Patients with response, n (%) | 0 (0.0) | 17 (34.7) |
95% CI | 0.0 to 7.0 | 21.7 to 49.6 |
Week 48 | ||
Patients with response, n (%) | 1 (2.0) | 22 (44.9) |
95% CI | 0.0 to 10.4 | 30.7 to 59.8 |
Week 72 | ||
Patients with response, n (%) | █████████ | █████████ |
95% CI | █████████ | █████████ |
Week 96 | ||
Patients with response, n (%) | NA | 27 (55.1) |
95% CI | NA | 40.2 to 69.3 |
Week 144 | ||
Patients with response, n (%) | NA | 28 (57.1) |
95% CI | NA | 42.2 to 71.2 |
Virologic response over timek,m | ||
Week 24 | ||
Patients with response, n (%) | 2 (3.9) | 27 (55.1) |
95% CI | 0.5 to 13.5 | 40.2 to 69.3 |
Week 48 | ||
Patients with response, n (%) | 2 (3.9) | 36 (73.5) |
95% CI | 0.5 to 13.5 | 58.9 to 85.1 |
Week 72 | ||
Patients with response, n (%) | █████████ | █████████ |
95% CI | █████████ | █████████ |
Week 96 | ||
Patients with response, n (%) | NA | 37 (75.5) |
95% CI | NA | 61.1 to 86.7 |
Week 144 | ||
Patients with response, n (%) | NA | 36 (73.5) |
95% CI | NA | 58.9 to 85.1 |
ALT = alanine aminotransferase; CI = confidence interval; FAS = full analysis set; HDV = hepatitis delta virus; HQLQ = Hepatitis Quality of Life Questionnaire; LLoQ = lower limit of quantification; LOCF = last observation carried forward; LS = least squares; NA = not applicable; NR = not reported; RNA = ribonucleic acid; SE = standard error; vs. = versus.
Note: Virologic response was defined as undetectable HDV RNA (less than the LLoQ, target not detected) or HDV RNA decrease ≥ 2 log10 IU/mL; ALT normalization was defined as an ALT level within the normal reference range, according to central laboratory criteria (≤ 31 U/L for females and ≤ 41 U/L for males in Russia, and ≤ 34 U/L for females and ≤ 49 U/L for males in all other locations).
aThe results for the delayed-treatment group were measured any time between baseline (at randomization) to before the patients in this treatment group received their first dose of bulevirtide at week 48 or early termination before week 48. From week 48 onwards, patients received bulevirtide 10 mg/day.
bCombined response was defined as undetectable HDV RNA (less than the LLoQ, target not detected) or HDV RNA decrease ≥ 2 log10 IU/mL from baseline combined with ALT normalization.
cThe 95% CI for each group was based on the Clopper-Pearson exact method.
dThe 99% and 96% exact unconditional CIs, based on the score statistic, were presented for the response rate difference at week 24 and week 48, respectively. For missing values related to COVID-19, the LOCF approach was used. Otherwise, the missing equals failure approach was used.
eThe P value was based on the Fisher exact test.
fThe analysis of the difference in the proportions of patients with response in the bulevirtide 10 mg group versus the bulevirtide 2 mg group was performed using Fisher exact test.
gFor response rate difference, the 95% exact unconditional CI based on the score statistic was presented. For missing values related to COVID-19, the LOCF approach was used. Otherwise, the missing equals failure approach was used.
hBaseline value was the last available value collected before or at the time of the first dose of bulevirtide for the bulevirtide 2 mg and 10 mg treatment groups. Baseline value was the last available value collected before or at randomization for the delayed-treatment group.
iThe LS mean, SE, 95% CIs, and P values were based on the analysis of covariance model for change from baseline with treatment group, region, presence of cirrhosis, and baseline value as covariates.
jLiver stiffness of 8.4 kPa at week 48 for 1 patient in the delayed-treatment group was added into the analysis.
kP value was not adjusted for multiple comparisons.
lThe baseline value was the last available value collected on or before first dose of study drug, or at or before randomization for the delayed-treatment group. The LS mean, SE, 95% CIs, and P values were based on the mixed-effects models for repeated measures for change from baseline with treatment, region, presence of cirrhosis, visit, and treatment by visit interaction as fixed effects and the baseline value as the covariate. The unstructured variance-covariance matrix was used. The Kenward-Roger method was used to estimate the degrees of freedom. Restricted maximum likelihoods were used to fit the model. Observed case: missing values remain missing.
mBaseline value was the last available value collected before or at the time of the first dose of bulevirtide for the bulevirtide 2 mg, bulevirtide 10 mg, and delayed-treatment groups, and the last available value collected before or at randomization for the delayed-treatment group. The 95% CI was based on Clopper-Pearson exact method. For missing values, the missing equals failure approach was used.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Virologic Response Over Time
The virologic response at each study visit, an exploratory end point in MYR301, was analyzed exclusively using a missing equals failure approach. The virologic response rates in the bulevirtide 2 mg group demonstrated improvement over time: 55.1% (95% CI, 40.2% to 69.3%) at week 24; 73.5% (95% CI, 58.9% to 85.1%) at week 48; █████ ████ ███ █████ ██ ██████ ██ ████ ██; 75.5% (95% CI, 61.1% to 86.7%) at week 96; and 73.5% (95% CI, 58.9% to 85.1%) at week 144.
Harms
The key harm results in the safety analysis set from the MYR301 trial are summarized in Table 15.
Table 15: Summary of Harms Results From the MYR301 Study (SAS)
Adverse events | Baseline to week 48 | Baseline to week 144 | |
|---|---|---|---|
Delayed treatment (N = 51) | Bulevirtide 2 mg (N = 49) | Bulevirtide 2 mg (N = 49) | |
TEAEs, n (%) | |||
Patients with ≥ 1 TEAEs | 41 (80.4) | 41 (83.7) | 48 (98.0) |
Grade 3 or higher | 4 (7.8) | 5 (10.2) | 12 (24.5) |
Thrombocytopenia | 3 (5.9) | 1 (2.0) | 1 (2.0) |
Depression | 0 (0) | 1 (2.0) | 1 (2.0) |
Foot fracture | 0 (0) | 1 (2.0) | 1 (2.0) |
Neutrophil count decreased | 0 (0) | 1 (2.0) | 1 (2.0) |
Osteopenia | 0 (0) | 1 (2.0) | 1 (2.0) |
Neutropenia | 2 (3.9) | 0 (0) | 1 (2.0) |
Leukopenia | 1 (2.0) | 0 (0) | 0 (0) |
TEAEs in ≥ 10% of patients in any group | |||
Vitamin D deficiency | 13 (25.5) | 7 (14.3) | 22 (44.9) |
Headache | 0 (0) | 9 (18.4) | 10 (20.4) |
Leukopenia | 10 (19.6) | 7 (14.3) | 10 (20.4) |
Thrombocytopenia | 8 (15.7) | 5 (10.2) | 10 (20.4) |
Lymphopenia | 4 (7.8) | 4 (8.2) | 8 (16.3) |
Neutropenia | 3 (5.9) | 2 (4.1) | 8 (16.3) |
Arthralgia | 0 | 3 (6.1) | 7 (14.3) |
Fatigue | 1 (2.0) | 5 (10.2) | 7 (14.3) |
Pruritus | 0 (0) | 6 (12.2) | 6 (12.2) |
ALT levels increased | 4 (7.8) | 2 (4.1) | 5 (10.2) |
Anemia | 3 (5.9) | 3 (6.1) | 5 (10.2) |
COVID-19 | 2 (3.9) | 1 (2.0) | 5 (10.2) |
Eosinophilia | 0 (0) | 5 (10.2) | 5 (10.2) |
Nasopharyngitis | 2 (3.9) | 4 (8.2) | 5 (10.2) |
SAEs, n (%) | |||
Patients with ≥ 1 SAE | 1 (2.0) | 2 (4.2) | 3 (6.1) |
Esophageal varices | 0 (0) | 0 (0) | 1 (2.0) |
Cholelithiasis | 1 (2.0) | 0 (0) | 0 (0) |
COVID-19 | 1 (2.0) | 0 (0) | 0 (0) |
Foot fracture | 0 (0) | 1 (2.0) | 1 (2.0) |
Headache | 0 (0) | 1 (2.0) | 1 (2.0) |
Hemiparesis | 0 (0) | 1 (2.0) | 1 (2.0) |
Depression | 0 (0) | 1 (2.0) | 1 (2.0) |
TEAE leading to premature discontinuation of study drug, n (%) | |||
Patients who withdrew from the study | 0 (0) | 0 (0) | 0 (0) |
Deaths, n (%) | |||
Patients who died | 0 (0) | 0 (0) | 0 (0) |
AEs of special interest, n (%) | |||
Hepatic AEs | 5 (9.8) | 7 (14.3) | 14 (28.6) |
Renal AEs | 3 (5.9) | 4 (8.2) | 5 (10.2) |
Eosinophilia | 1 (2.0) | 5 (10.2) | 5 (10.0) |
Injection site reactions | NR | 10 (20.4) | NR |
AE = adverse event; ALT = alanine aminotransferase; MedDRA = Medical Dictionary for Regulatory Activities; NR = not reported; SAE = serious adverse event; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
Notes: AEs were coded according to MedDRA Version 26.0.
The AEs were allocated to periods based on event onset dates. For the bulevirtide 2 mg treatment group, TEAEs began on or after the study drug initiation and continued up to 30 days after permanent discontinuation of the study drug or led to premature study drug discontinuation. For the delayed-treatment group, treatment-emergent events began on or after the randomization date and before week 48, or up to the study discontinuation date if the patients discontinued the study before the week 48 visit, or else began on or after the randomization date and before the bulevirtide initiation date.
Source: Clinical Study Report for MYR301.19 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Adverse Events
At week 48, the proportions of patients who experienced at least 1 AE were 83.7% in the bulevirtide 2 mg group and 80.4% in the delayed-treatment group in the MYR301 trial. The most common AEs in the bulevirtide 2 mg group were headache (18.4% versus 0.0% in the delayed-treatment group), vitamin D deficiency (14.3% versus 25.5% in the delayed-treatment group), leukopenia (14.3% versus 19.6% in the delayed-treatment group), pruritus (12.2% versus 0.0% in the delayed-treatment group), and thrombocytopenia (10.2% versus 15.7% in the delayed-treatment group). The proportions of patients with AEs of grade 3 or higher were 10.2% in the bulevirtide 2 mg group and 7.8% in the delayed-treatment group. Among the patients in the bulevirtide 2 mg group, these AEs included depression (2.0% versus 0.0% in the delayed-treatment group), foot fracture (2.0% versus 0.0% in the delayed-treatment group), decreased neutrophil count (2.0% versus 0.0% in the delayed-treatment group), osteopenia (2.0% versus 0.0% in the delayed-treatment group), and thrombocytopenia (2.0% versus 5.9% in the delayed-treatment group).
At week 144, the proportion of patients in the bulevirtide 2 mg group who experienced at least 1 AE was 98.0%, including 24.5% of patients who experienced grade 3 or higher AEs. At week 144, the most common AEs in the bulevirtide 2 mg group were vitamin D deficiency (44.9%), headache (20.4%), leukopenia (20.4%), thrombocytopenia (20.4%), lymphopenia (16.3%), and neutropenia (16.3%).
Serious Adverse Events
At week 48, the proportions of patients who experienced at least 1 SAE were 4.2% in the bulevirtide 2 mg group and 2.0% in the delayed-treatment group in the MYR301 trial. Most SAEs were experienced by 1 patient across the treatment groups. The SAEs in the bulevirtide 2 mg group included foot fracture (2.0% versus 0.0% in the delayed-treatment group), headache (2.0% versus 0.0% in the delayed-treatment group), hemiparesis (2.0% versus 0.0% in the delayed-treatment group), and depression (2.0% versus 0.0% in the delayed-treatment group). The SAEs in the delayed-treatment group included cholelithiasis (2.0% versus 0.0% in the bulevirtide 2 mg group) and COVID-19 (2.0% versus 0.0% in the buvelirtide 2 mg group).
At week 144, 6.1% of patients in the bulevirtide 2 mg group experienced at least 1 SAE, including esophageal varices (2.0%), foot fracture (2.0%), headache (2.0%), hemiparesis (2.0%), and depression (2.0%). Between weeks 48 and 144 (representing 96 weeks of treatment with bulevirtide at a dose of 10 mg), 6% of patients in the delayed-treatment group experienced at least 1 SAE; these SAEs included COVID-19 (2.0%), urinary tract infection (2.0%), and plasma cell myeloma (2.0%).
Withdrawals Due to AEs
By week 144, there were no AE-related discontinuations in the MYR301 trial.
Mortality
By week 144, there were no deaths in either the buvelirtide 2 mg or the delayed-treatment group.
Notable Harms
At week 48, 14.3% of patients in the bulevirtide 2 mg group and 9.8% of patients in the delayed-treatment group had experienced at least 1 hepatic AE. In the bulevirtide 2 mg group these hepatic AEs included increased ALT levels (4.1% versus 7.8% in the delayed-treatment group), hyperbilirubinemia (4.1% versus 0.0% in the delayed-treatment group), increased blood bilirubin (4.1% versus 0.0% in the delayed-treatment group), increased aspartate aminotransferase (2.0% versus 5.9% in the delayed-treatment group), and hepatic pain (2.0% versus 0.0% in the delayed-treatment group). At week 144, the proportion of patients in the bulevirtide 2 mg group experiencing at least 1 hepatic AE was 28.6%.
At week 48, the proportions of patients experiencing at least 1 renal AE were 8.2% in the bulevirtide 2 mg group and 5.9% in the delayed-treatment group. The renal AEs in the bulevirtide 2 mg group included proteinuria (6.1% versus 3.9% in the delayed-treatment group) and urinary retention (2.0% versus 0.0% in the delayed-treatment group). At week 144, the proportion of patients in the bulevirtide 2 mg group experiencing at least 1 renal AE was 10.2%.
At week 48, 10.2% of patients in the bulevirtide 2 mg group versus 0.0% in the delayed-treatment group had experienced eosinophilia. At week 144, 10.2% of patients in the bulevirtide 2 mg group had experienced eosinophilia.
At week 48, 6.1% of patients in the bulevirtide 2 mg group versus 0.0% in the delayed-treatment group had experienced injection site reactions. At week 144, 20.4% of patients in the bulevirtide 2 mg group had experienced injection site reactions.
Critical Appraisal
Internal Validity
Randomization in the MYR301 trial was performed using an appropriate methodology and stratification was prespecified. An electronic randomization system was used, which typically has a low risk of bias. All efficacy and safety analyses were conducted using the full and safety analysis sets, respectively, and these analyses included all patients who were randomized and who had received at least 1 dose of bulevirtide after randomization. Some imbalances in baseline characteristics, likely due to small sample sizes, were observed in the MYR301 trial. These may have hindered the achievement of true prognostic balance. Specifically, imbalances in baseline characteristics could complicate the interpretation of treatment effects, as observed outcomes may be influenced by these baseline differences rather than the treatment itself. Given the rarity of HDV and the challenges in recruiting patients into clinical trials, achieving large sample sizes is often not feasible. The open-label design of the MYR301 trial introduces a potential bias in the assessment of study outcomes; however, this bias was mitigated by blinding to treatment allocation the central laboratories employed for hematology and biochemistry assessment. Knowledge of the assigned treatment could have led to biases in the reporting and measurement of subjective outcomes, including patient-reported outcomes (e.g., HRQoL) and subjective AEs. Physician’s knowledge of their patients’ assigned treatments may influence how they manage their patients, while patients' awareness of their assigned treatment may impact their likelihood of continuing in the study, which could have led to bias in favour of the intervention; however, the extent of such bias cannot be determined. Although there are always some concerns for risk of bias due to deviations from the intended interventions in open-label trials, there were relatively few protocol deviations, and these were balanced across the groups and therefore unlikely to have influenced the study results. Adherence to the interventions and study completion rates were generally high in the intervention groups of interest to this review, reducing concerns regarding deviations from the intended interventions that could have arisen due to the trial context.
The objective of the MYR301 trial was to assess the efficacy and safety of bulevirtide at a dose of 2 mg compared to the delayed-treatment group, where patients received bulevirtide at a dose of 10 mg after a 48-week observation period. According to FDA guidance,18 given that no drugs are currently approved for the treatment of HDV infection, a placebo-controlled trial is the preferred design for a phase III clinical trial. An alternative design could be an RCT in which patients are randomized to receive either the investigational drug (the immediate-treatment group) or placebo for a prespecified duration, followed by open-label treatment with the investigational drug for the deferred-treatment group.18 In this case, effectiveness would be demonstrated by a significant early improvement among participants in the investigational group compared with those in the placebo control. It is worth noting that the proportion of patients with response in the delayed-treatment group was expected to be low, as the patients in this group did not receive HDV treatment and only about 60% received HBV treatment. The third treatment group in the trial consisted of patients who received bulevirtide at a dose of 10 mg. The data for this group are not included in this report, as this dosage of bulevirtide is neither approved nor used in clinical practice in Canada.
All patients randomized to the delayed-treatment and bulevirtide groups received at least 1 dose of bulevirtide after randomization. The primary end point in the MYR301 trial was a combined response, that is, undetectable serum HDV RNA (defined as less than the LLoQ, target not detected) or HDV RNA decrease of at least 2 log10 IU/mL, and ALT normalization. According to FDA guidance,18 a surrogate end point providing evidence of both a decline in virologic replication and an improvement in associated liver inflammation, as evident by biochemical response, could reasonably predict clinical benefit. However, FDA guidance suggests that subsequent confirmation using clinical end points is required and that these should be collected along with other long-term follow-up data. Although several liver-related clinical events were evaluated in the MYR301 trial, for example, cirrhosis development, decompensation of cirrhosis, and liver-related hospitalization, the study is ongoing and there has not been sufficient time to gather comprehensive data. As such, it remains uncertain whether the improvements observed in the surrogate end points evaluated in this trial will translate into meaningful benefits in clinical outcomes that patients consider to be important, or to what extent such benefits may be realized.
Based on the enrolled sample size, the study was powered to test its primary and key secondary end points. The analyses of primary and key secondary outcomes were conducted using the FAS population, which maintains randomization and minimizes the risk of bias by comparing groups that are randomized at baseline. Statistical analysis methods appear to be acceptable. Both interim analyses at weeks 24 and 48 were planned a priori. The lack of adjustment for multiple comparisons for all but the primary and key secondary end points increases the risk of type I error, where false positives may be incorrectly identified as statistically significant. Statistical analysis methods appear to be acceptable. Bulevirtide treatment outcomes were more favourable in the analysis with respect to a combined response, undetectable HDV RNA, virologic response, change in liver stiffness, and ALT normalization, but there was uncertainty in the true magnitude of the effect due to the small sample size, which increased the risk that prognostic balance was not achieved. The unadjusted comparisons and the decision not to stratify the analyses by cirrhosis due to the small sample size may introduce bias and reduce the accuracy of the estimated treatment effects. The long-term impact of treatment with bulevirtide 2 mg remains uncertain due to the relatively short 48-week duration of the comparison with usual care. A larger proportion of patients in the bulevirtide 2 mg group had a Child-Pugh score of 6 points (which is the borderline between Class A and Class B) compared to the delayed-treatment group. The clinical experts noted that this imbalance may increase the risk of AEs and disease progression in the bulevirtide 2 mg group; however, they highlighted that this should not impact the virological study outcomes. The clinical experts indicated that although liver stiffness was higher in the delayed-treatment group than in the bulevirtide 2 mg group, both at randomization and at week 48, this imbalance is unlikely to be clinically meaningful, as liver stiffness measurements can be affected by factors such as ALT levels and inflammation. Subgroup analyses by cirrhosis status and prior use of nucleoside (or nucleotide) analogues were prespecified; however, they were descriptive and were limited by small sample sizes, precluding any conclusions regarding the possibility of effect modification.
For the primary and key secondary end points, the LOCF approach was used to address missing values related to COVID-19, while in other cases, missing data were treated as treatment failures (patients with no response). The missing equals failure approach was considered a conservative assumption of missing data. In contrast, the LOCF approach, which requires that the value is constant from the time that the data are missing to the time point analyzed, may not reflect the true trajectory of the outcome. The sensitivity analyses were conducted using a nonresponder imputation approach, and the results were consistent with those of the primary analyses. The risk of bias due to missing outcome data in the MYR301 trial is considered to be low. Although the imputation methods may not be perfect, the proportion of missing data was low across the relevant treatment groups and therefore does not raise concerns regarding the validity of the results. Important protocol deviations were reported for 8.2% to 9.8% of patients across the 2 treatment groups, with a comparable proportion between groups, and all deviations were identified before database lock. All the randomized patients were treated and only 2% in both the delayed-treatment and bulevirtide 2 mg groups had discontinued the study prematurely by week 48. The clinical experts noted that this discontinuation rate is not high, given the once daily subcutaneous dosing regimen.
Several patient-reported outcomes were assessed in the MYR301 trial. However, the clinical experts considered the HQLQ tool important for this review as it is liver-specific, includes domains related to mental health and fatigue, and is better supported by data in populations with liver disease. The HQLQ has been validated in patients with hepatitis C, but not in those with hepatitis D, leading to uncertainty regarding what would constitute a clinically relevant between-group difference. No definitive conclusions could be drawn about the effect of bulevirtide compared with delayed treatment on HRQoL due to risk of bias from assessors being aware of treatment assignments, as well as serious imprecision, with wide CIs that included both potential benefit and harm. No estimated minimal important differences were provided for the scales presented by the sponsor, resulting in uncertainty regarding what would constitute a clinically meaningful between-group difference. The risk of bias due to missing outcome data are considered low, given the limited extent of missing data. The analysis method for HRQoL outcomes relied on the likely implausible assumption that data were missing at random, and no sensitivity analyses were conducted to assess robustness to different missingness mechanisms.
External Validity
According to clinical experts, the inclusion and exclusion criteria of the MYR301 trial appropriately reflect the population of patients with HDV who would be eligible for treatment with bulevirtide in clinical practice. Given the rarity of the disease, a small sample size was anticipated and a larger trial may be infeasible. This review assessed the efficacy and safety of bulevirtide at a dose of 2 mg, which is the dose submitted for Health Canada approval, compared with delayed treatment.
The screening failure rate was about 18%. The clinical experts considered this to be fairly inclusive because screening failure rates of between 15% and 40% are common in hepatitis trials. According to the clinical experts consulted by CDA-AMC, the inclusion and exclusion criteria in the MYR301 trial were generally appropriate for the clinical trial setting. The clinical experts noted that while certain exclusion criteria are considered typical for clinical trials, they do not always reflect real-world practice, where treatment decisions are often based on individual clinical judgment and which may include patients typically excluded from trials. For example, while patients aged older than 65 years were excluded from the trial, the clinical experts emphasized that age alone should not preclude treatment with bulevirtide in clinical practice. The clinical experts noted that treatment decisions for older adults should be individualized based on comorbidities, functional status, and patient preferences. However, they acknowledged that bulevirtide treatment may not be appropriate for patients aged, for example, 80 years and older, if the potential burden outweighs the benefits. The clinical experts also noted that the 6-month requirement for a positive anti-HDV antibody assay or HDV RNA PCR result was appropriate within the trial context to confirm chronic HDV infection. The clinical experts also noted that, in clinical practice, physicians are unlikely to delay treatment initiation for patients with HBV and HDV coinfection who present with advanced fibrosis, suggesting that earlier intervention may be clinically appropriate in such cases. The clinical experts indicated that while the ALT level requirement was appropriate as a trial inclusion criterion to reflect active disease, it should not restrict treatment in clinical practice, particularly for patients with advanced liver disease who may have ALT levels in the normal reference range. Patients with HCV or uncontrolled HIV coinfection were excluded from the MYR301 trial; the clinical experts noted that in clinical practice these coinfections can be sequentially managed, with the HIV and HCV infections treated before initiation of HBV and HDV therapy. The clinical experts also noted that although patients with creatinine clearance of less than 60 mL/min were excluded from the trial, those with moderate renal impairment could still be considered for treatment with appropriate monitoring.
The clinical experts agreed that it is reasonable to expect that a substantial proportion of patients treated for HDV in clinical practice will have cirrhosis, given that many are diagnosed at more advanced stages of the disease. The clinical experts also noted that patients with early-stage fibrosis (fibrosis stage 1 [F1] or fibrosis stage 2 [F2]) may be more likely to seek treatment to prevent disease progression. As a result, treatment in real-world settings may be initiated across a broader range of disease severity than is typically represented in clinical trials. According to the clinical experts, although most patients in the MYR301 trial were white, reflecting the European trial site populations, this does not limit the generalizability of the findings to the population in Canada, as the virologic characteristics and treatment response are not expected to differ meaningfully across racial or ethnic groups. Approximately 65% of patients in the MYR301 trial received concomitant oral anti-HBV therapy at baseline and this was balanced across groups. However, the clinical experts indicated that this proportion is lower than expected in clinical practice, where nearly all patients with HDV are typically treated with nucleoside (or nucleotide) analogues. The clinical experts noted that although HBV therapy may slow the progression of fibrosis, this is unlikely to affect the generalizability of the study, as improvements in both virologic and fibrosis outcomes were observed in patients who had previously received HBV therapy (54.7%) and progressed to advanced disease.
According to the clinical experts, the surrogate outcomes used in the MYR301 trial — HDV RNA reduction or undetectability, ALT normalization, and changes in liver fibrosis — were appropriate for assessing treatment benefit. The clinical experts indicated that 48 weeks is a reasonable duration to assess efficacy in the MYR301 trial using these surrogate outcomes; longer-term follow-up would be required to assess hard clinical outcomes such as liver cancer, hepatic decompensation, or death, which are not expected to manifest within 1 year of treatment initiation. The clinical experts also identified undetectable HDV RNA at weeks 24 and 48 after the scheduled end of treatment (sustained virologic response) as an important outcome. The results for this end point were not reported, as the study is ongoing and the data are not yet available. The clinical experts noted that these outcomes align with standard measures in clinical trials and serve as meaningful indicators of disease activity and progression. The clinical experts indicated that in patients with HDV, fibrosis improvement is typically observed over a longer time frame. According to the clinical experts, liver stiffness measurements may be influenced by ALT levels and should therefore be interpreted with caution, and the absence of fibrosis improvement at week 48 does not diminish the clinical relevance of virologic response or overall treatment benefit. The HRQoL outcome was not formally compared between groups, limiting the ability to draw definitive conclusions about this important measure; the results are provided as supportive evidence. Safety data for bulevirtide 2 mg were reported up to week 144, which the clinical experts considered sufficient given the advanced disease status of patients who are at increased risk for AEs.
The MYR301 trial study population was drawn from several sites across 4 European countries, with no sites in Canada included. Nevertheless, the clinical experts indicated that there are no major concerns with generalizing the findings from the trial to clinical settings in Canada.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal RCT identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:33,34
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from the RCT started as high certainty and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
The GRADE assessments included an evaluation of the main outcomes considered important by clinicians, patient groups, and committee members. Table 2 presents the GRADE summary of findings for bulevirtide 2 mg versus delayed treatment in patients with chronic HDV infection.
Long-Term Extension Studies
No long-term extension study was submitted for this review.
Indirect Evidence
The contents of this section were informed by materials submitted by the sponsor and have been validated and summarized by the review team.
Objectives for the Summary of Indirect Evidence
The sponsor-submitted NMA35 addressed the lack of head to head trial data comparing bulevirtide at a dose of 2 mg to the relevant comparators for the treatment of HDV. The indirect evidence is used to inform the pharmacoeconomic model.
The sponsor-submitted pairwise (direct) meta-analysis36 was not summarized in this report because it involves pooling data from trials that directly compare treatments, which is generally preferable but not aligned with the focus of this review. Specifically, the analysis included studies like MYR301 and MYR202. The MYR202 study was excluded from this review due to its irrelevance to the treatment comparisons under evaluation.
Description of Indirect Comparisons
The sponsor-submitted ITC included a systematic review and an NMA evaluating the efficacy and safety of bulevirtide at a dose of 2 mg as monotherapy relative to other treatments available for chronic hepatitis D. The primary aim of the NMA was to evaluate the relative efficacy and safety of bulevirtide 2 mg versus comparators, including PEG-IFN and best supportive care, for patients with chronic hepatitis D.
Based on the prespecified eligibility criteria outlined in Table 16, the sponsor conducted a systematic review to identify studies investigating the efficacy of interventions used in the management of chronic HDV infection. A systematic review, which was informed by a predetermined protocol, was conducted to identify RCTs that investigated bulevirtide 2 mg or other relevant therapeutic regimens for patients with chronic hepatitis D who have compensated liver disease. Relevant treatments include PEG-IFN-alpha and interferon, as well as nucleoside (or nucleotide) analogue monotherapy or in combination with any included intervention. The methods used to conduct the systematic review, extract data, and assess study quality are shown in Table 16. The literature search was conducted as of January 2021 and was extended beyond March 30, 2021. Multiple databases were searched from inception through December 2021 to identify clinical trials. Study screening, study selection, and data extraction were conducted by 2 independent reviewers. Any discrepancies between the 2 reviewers were resolved by a third reviewer. Only studies published in English were included in this review. The assessment of the risk of bias of the included RCTs was performed at the study level using the NICE checklist.37 The number of reviewers involved and whether they conducted assessments independently were not reported.
Table 16: Study Selection Criteria and Methods in the Sponsor-Submitted ITC
Characteristics | Indirect comparison |
|---|---|
Population | Adults with chronic hepatitis delta who have compensated liver disease |
Intervention | Bulevirtide, 2 mg once daily, administered by subcutaneous injection Pegylated interferon-alpha 180 mcg weekly subcutaneous or standard interferon-alpha |
Comparator |
|
Outcome |
|
Study designs | RCTs irrespective of blinding status |
Publication characteristics | No limits |
Exclusion criteria |
|
Databases searched |
|
Selection process | Two investigators independently screened all citations identified in the literature search and reviewed the full texts. Any differences in the studies selected by the 2 investigators were resolved by a third investigator. |
Data extraction process | Two investigators independently extracted data on study characteristics, interventions, patient characteristics, and outcomes for the study population of interest for the final list of selected eligible studies. Any discrepancies observed in the data extracted by the 2 data extractors were resolved through discussion. |
Quality assessment | The risk of bias assessment was performed at the study level using the NICE checklist. |
ALT = alanine aminotransferase; AASLD = American Association for the Study of Liver Diseases; CDA-AMC = Canada’s Drug Agency; CEA = cost-effectiveness analysis; CENTRAL = Cochrane Central Register of Controlled Trials; EASL = European Association for the Study of the Liver; ECCMID = European Congress for Clinical Microbiology and Infectious Diseases; G-BA = Gemeinsamer Bundesausschuss; HAS = Haute Autorité de Santé; HDV = hepatitis delta virus; HTA = health technology assessment; ICER = Institute for Clinical and Economic Review; ICTRP = International Clinical Trials Registry Platform; ISPOR = Professional Society for Health Economics and Outcomes Research; ITC = indirect treatment comparison; IQWiG = Institute for Quality and Efficiency in Health Care; NICE = National Institute for Health and Care Excellence; PBAC = Pharmaceutical Benefits Advisory Committee; RCT = randomized controlled trial, RNA = ribonucleic acid; SMC = Scottish Medicines Consortium.
Source: Sponsor-submitted direct meta-analysis36 and network meta-analysis.35 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
ITC Design
The sponsor submitted a systematic review and an NMA to evaluate the relative efficacy and safety of bulevirtide 2 mg as monotherapy compared to other available treatments for chronic HDV infection. The objective of the NMA was to evaluate the relative efficacy and safety of bulevirtide 2 mg as a monotherapy versus interferon (in particular, PEG-IFN) and best supportive care in patients with chronic HDV infection. Of note, PEG-IFN is an off-label treatment of chronic HDV infection in Canada.
All the NMA efficacy outcomes were assessed at 2 time points: week 24 and week 48. If a study did not report data at these specific time points, data from the closest available time point were used. For example, the analysis at week 24 included data reported within a time window from week 24 to week 26.
The following outcomes were reported to address the objectives of the NMA: undetectable HDV RNA; HDV RNA undetectable or reduced by at least 2 log10; combined response; ALT normalization at weeks 24 and 48; and AEs and treatment discontinuations due to AEs at week 48.
ITC Analysis Methods
For a summary of the analytical methods applied in the NMA, refer to Table 17.
Sponsor-Submitted NMA
NMA estimation was performed using Markov Chain Monte Carlo techniques, which require specifying prior distributions for model parameters. In all analyses, vague (noninformative) prior distributions were applied to the treatment-effect parameters, regardless of the model used.
The NMA was conducted within a generalized linear model framework, which has the flexibility of being able to accommodate the data structure in accordance with the guidelines of the NICE Decision Support Unit.38 For continuous outcomes, a generalized linear regression model with a normal likelihood and identity link was employed, using the PEG-IFN monotherapy group as the comparator in all comparisons. Treatment effects were reported as mean differences with 95% CrIs. For binary outcomes, a generalized linear regression model with a binomial likelihood and logit link was used, and relative treatment effects were expressed as odds ratios with 95% CrIs.
The Bayesian NMA was conducted using both fixed-effects and random-effects models. For the primary analysis, the standard vague priors recommended in the NICE Decision Support Unit Technical Support Document 238 were applied. In cases where convergence issues arose ( such as networks with several single-study connections, outcomes with low event rates, or clinical heterogeneity in study characteristics), random-effects NMAs et al.39 priors for between-study heterogeneity recommended by Turner were also explored. Choosing between fixed- and random-effects models was guided by the deviance information criterion (DIC).40 When the difference in the DICs of the 2 models exceeded 3, the model with the lower DIC was considered to provide a better fit and was chosen as the preferred model for inference. If the difference in the DICs was less than 3, the fixed-effects model was preferred due to the limited number of contributing studies and the alignment of its results with those observed in trial-level and direct meta-analysis findings.
Convergence was assessed by examining the relevant parameters across iterations, visualizing the histories of chains, checking for overlapping chains, and evaluating Brooks-Gelman-Rubin statistics. To mitigate numerical instability, particularly in studies with zero-event cells or small sample sizes, continuity corrections were applied by adding 0.5 to the numerator and 1 to the denominator.38 Studies with zero events in all arms that continued to impede convergence were excluded from the analysis. When convergence issues arose due to sparse data, single-study connections within the network, or clinical heterogeneity, random-effects NMAs incorporating informative priors were employed.39 If these adjustments did not adequately resolve the convergence issues, an alternative NMA based on the risk-difference model was conducted.41 For any pair of treatments with both direct and indirect evidence, statistical tests were used to evaluate the null hypothesis that there is no inconsistency between these sources of evidence.
Imputation of Missing Values
For dichotomous outcomes, when only the percentage of patients with the outcome was reported and the number analyzed was not specified, the number of patients randomized was used as the denominator to calculate the number of events, in accordance with the intention-to-treat principle.
For continuous outcomes, when SDs were not reported, imputation methods were employed to derive missing SDs from available standard errors or 95% CIs. Consistent with the approach used for dichotomous outcomes, if the number of patients analyzed was not provided, the number of patients randomized was used for analysis in line with the intention-to-treat principle.
Assessing Heterogeneity
Various parameters were used to assess the qualitative heterogeneity of the patients recruited across the trials contributing to the NMA (Table 19). These parameters included mean age, sex, trial design, previous interferon treatment, ALT values at baseline, cirrhosis status, and presence of genotype HDV-1. The various trial designs and methodological aspects were also compared. As part of a heterogeneity assessment, statistical heterogeneity was presented for each outcome by means of I2 statistics between available pairwise comparisons; according to the sponsor, an I2 statistic greater than 75% indicates high levels of heterogeneity in pairwise comparisons.
Table 17: NMA Analysis Methods
Methods | NMA method |
|---|---|
Analysis methods | The available evidence consists of a network of multiple RCTs involving treatments compared directly or indirectly or both; hence, it can be synthesized by means of a Bayesian framework. |
Priors | Standard vague priors as recommended by NICE were used for the primary analysis. In the case of convergence issues due to several single-study connections in networks, outcomes with low event rates, and clinical heterogeneity in study traits, random-effects NMAs using informative priors were also tested. |
Assessment of model fit | According to Spiegelhalter et al. (2002),40 a model with a deviance information criterion at least 3 points lower than that of another model is deemed to have the better fit. |
Assessment of consistency | Statistical tests for inconsistency are performed by comparing direct and indirect evidence for treatment pairs, with the difference (inconsistency) used to construct test statistics. |
Assessment of convergence | Convergence was assessed by visualizing the histories of the chains and of relevant parameters vs. the iteration number, overlapping histories, and the Brooks-Gelman-Rubin statistics. |
Outcomes |
|
Follow-up time points | Not reported |
Construction of nodes | The network of interconnected studies is formed by using NAT or delayed treatment as a common comparator. |
Sensitivity analyses |
|
Subgroup analysis | Not applicable |
ALT = alanine aminotransferase; HDV = hepatitis delta virus; NICE = National Institute for Health and Care Excellence; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; RCT = randomized controlled trial; RNA = ribonucleic acid; vs. = versus.
Source: Sponsor-submitted direct meta-analysis35technical report. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
NMA Results
Summary of Included Studies
Table 18 summarizes the 8 studies considered in the base-case of the NMA.
Table 18: Studies Considered for NMA Base-Case Analysis
Comparison | Study name | Treatment |
|---|---|---|
Bulevirtide 2 mg as monotherapy or in combination with TDF vs. PEG‑IFN, tenofovir, or delayed treatment |
| |
| ||
MYR20347 |
| |
MYR30119 |
| |
PEG-IFN vs. PEG-IFN plus nucleoside (or nucleotide) analogues or nucleoside (or nucleotide) analogue monotherapy | HIDIT-I48 |
|
HIDIT-II49 |
| |
Niro et al. (2006)50 |
| |
Abbas et al. (2016)51 |
|
NMA = network meta-analysis; PEG-IFN = pegylated interferon; p.o. = orally; SC = subcutaneous; TDF = tenofovir disoproxil fumarate; vs. versus.
Source: Sponsor-submitted NMA35 technical report. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
According to the sponsor, because standard interferon is no longer used in the UK for the treatment of hepatitis D, studies evaluating standard interferon were excluded from the base-case analysis. In addition, the NICE guideline for treatment of hepatitis B recommends a 48-week course of PEG-IFN-alpha-2a for individuals with HBV and HDV coinfections who exhibit significant fibrosis. However, a sensitivity analysis that included all standard interferon-alpha study results was conducted for all efficacy outcomes.
Studies Included in the NMA
An assumption was made in the NMA that nucleoside (or nucleotide) analogues, which are recommended for hepatitis B treatment and commonly used as concomitant medications in the treatment of hepatitis D (e.g., approximately 60% of patients in the MYR301 trial received concomitant antiviral treatment), have similar efficacy when used as monotherapy (tenofovir or adefovir) or in delayed treatment. Consequently, these treatments were grouped together under the common comparator, nucleoside (or nucleotide) analogue therapy, to create a combined network for analysis.
Table 19: Assessment of Homogeneity
Characteristics | Description and handling of potential effect modifiers with the NMA method |
|---|---|
Disease severity | There was notable variation in the baseline severity of hepatitis D infection across the studies. The proportion of patients with cirrhosis varied from 12.5% in the MYR201 study44,45 to 60.5% in the Niro et al. (2006) study.50 |
Treatment history | There was notable variation in treatment history, with the proportion of patients who had received prior interferon treatment ranging from 0% in the Abbas et al. (2016) study51 to 73.7% in the Niro et al. (2006) study.50 |
Trial eligibility criteria | Not reported |
Dosing of comparators | The comparators in the included studies involved various combinations of PEG-IFN-alpha-2a or PEG-IFN-alpha-2b with antiviral drugs. Specifically, PEG-IFN-alpha-2a was administered at 180 mcg/week subcutaneously in combination with adefovir (10 mg/day orally), tenofovir (245 mg/day orally), or entecavir (0.5 mg/day orally). PEG-IFN-alpha-2b was also dosed at 1.5 mcg/kg per week subcutaneously with ribavirin (800 mg/day orally). Interferon, recombinant interferon-alpha-2a, and recombinant interferon-alpha-2b were included in the sensitivity analysis, with doses of 5 MU, 9 MU, or 10 MU administered 3 times a week subcutaneously. |
Definitions of end points | The definition of HDV RNA being undetectable or reduced varied across studies, with thresholds ranging from a reduction of at least 2 log10 IU/mL to 3 log10 IU/mL. |
Timing of end point evaluation | All the efficacy outcomes were analyzed at 2 time points (i.e., week 24 and week 48). If a study did not report data at these specific time points, data from the closest available time point were used. |
Clinical trial setting | The included studies were conducted across diverse settings: MYR20144,45 and MYR20232,46 were single-centre studies conducted in Russia; MYR203 was a multicentre study that was also conducted in Russia; MYR30119 was a multicentre trial conducted across 5 countries; HIDIT-I48 and HIDIT-II49 were multicentre studies conducted in Germany and other international countries, respectively; Niro et al. (2006)50 was a multicentre study conducted in Italy; and Abbas et al. (2016)51 was a multicentre study conducted in Pakistan. |
Study design | The included studies differed in study phase (Ib/IIa, II, or III), and this information was not reported in 3 studies, i.e., HIDIT-I,48 Niro et al. (2006),50 and Abbas et al. (2016).51 |
HDV = hepatitis delta virus; NMA = network meta-analysis; PEG-IFN = pegylated interferon; RNA = ribonucleic acid.
Source: Sponsor-submitted NMA35 technical reports. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The studies included in the NMA demonstrated heterogeneity across multiple domains, including baseline patient characteristics, trial design, treatment duration, and disease severity. Results of the homogeneity assessment undertaken by the sponsor are in Table 19.
Patients’ mean age ranged from 26.7 years in the Abbas et al. (2016) study51 to 44.0 years in the Niro et al. (2006) study.50 The proportion of males ranged from 56.7% in the MYR203 study47 to 77.5% in the Abbas et al. (2016) study.51 Baseline HDV RNA levels varied, with the lowest observed in the MYR203,47 HIDIT-I,48 and Abbas et al. (2016)51 studies (0.0 IU/mL) and the highest in the Niro et al. (2006)50 and MYR20144,45 studies (7.9 log10 IU/mL and ███ █████ █████, respectively). Mean baseline ALT levels ranged from 79.0 U/L in the MYR203 study47 to 144.0 U/L in the Niro et al. (2006) study.50 Mean baseline HDV RNA levels varied from 0 log10 IU/mL in the MYR203,47 HIDIT-I,48 and Abbas et al. (2016)51 studies to ███ █████ ████ in the MYR201 study.44,45 Genotype 1 was the predominant HDV genotype across the included studies, although genotype data were not reported in the MYR20144,45 or Niro et al. (2006)50 studies.
The included studies differ in study phase (Ib/IIa, II, or III); this information was not reported in the HIDIT-I, Niro et al. (2006),50 or Abbas et al. (2016)51 studies. Four studies had an open-label design (MYR201,44,45 MYR202, MYR203,47 MYR30119) and 1 was a double-blind trial (HIDIT-II49); this information was not reported in the HIDIT-I,48 Niro et al. (2006),50 or Abbas et al. (2016)51 studies. Treatment duration ranged from 24 weeks (MYR20144,45 and MYR202 studies) to 96 weeks (HIDIT-II49 study), and the follow-up duration from 24 weeks in 6 trials to 96 weeks in 1 trial. Sample sizes ranged from 24 patients (MYR201 study44,45) to 150 patients (MYR301 study19). Figure 2, Figure 3, Figure 4, Figure 9, Figure 10, Figure 11, and Figure 12 illustrate the network diagrams for the base-case analysis of the NMA.
Figure 2: Master Evidence Network for the Base-Case NMA Analysis
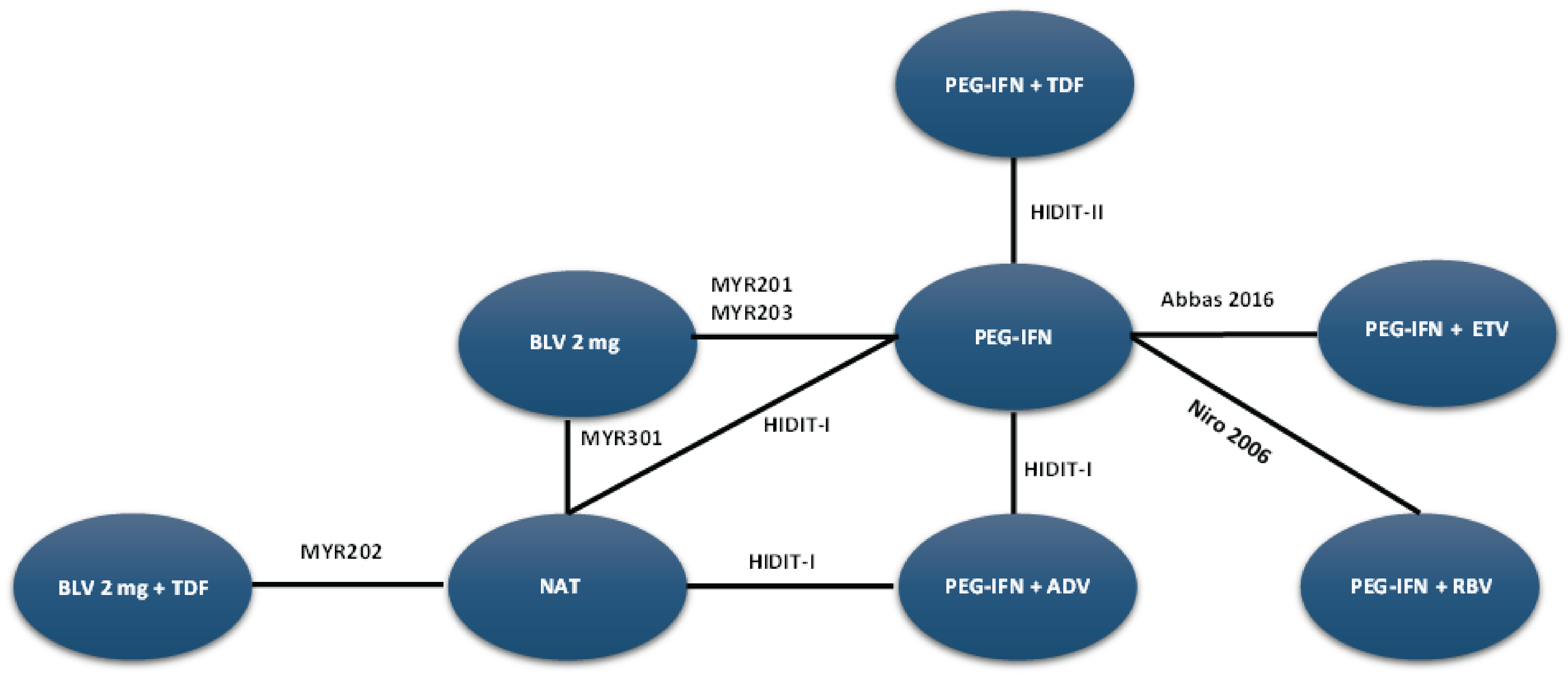
ADV = adefovir; BLV = bulevirtide; ETV = entecavir; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; RBV = ribavirin; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Figure 3: NMA Network Diagram of Combined-Response Outcome at Week 24
BLV = bulevirtide; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Figure 4: NMA Network Diagram of Combined-Response Outcome at Week 48
BLV = bulevirtide; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Assessment of Risk of Bias
According to the sponsor, the methods of randomization and allocation concealment were adequate in 7 trials but unclear in the HIDIT-1 trial.48 Only 1 study (HIDIT-II49) used an adequate double-blind setting for conducting the trial. Of the remaining trials, 3 were open label, and 3 did not report the blinding information.19,32,47 Most studies clearly reported patient withdrawals and employed appropriate analytical methods. The overall risk of bias was heterogeneous across the included studies, but none of the studies were considered to be outliers. Further, the risk of bias assessment did not identify any outlier high-risk study for the sensitivity assessment.
Efficacy Results
The Bayesian NMA was conducted using both fixed-effects and random-effects models. DIC was found to be comparable in the fixed-effects and random-effects models for all outcomes analyzed at all time points. For binomial outcomes (i.e., HDV RNA loss, HDV RNA loss or reduction by at least 2 log10 IU/mL), ALT normalization, and combined response, the standard odds ratio model did not converge properly, leading to abnormally wide CrIs due to several single-study connections in networks and a low event rate (zero events). The convergence issue was resolved by the risk-difference model; hence it was selected as the preferred model for the binomial outcomes.
The results of the NMA are presented for the bulevirtide 2 mg as monotherapy versus PEG-IFN-alpha-2 and best supportive care consisting of nucleoside (or nucleotide) analogue therapy at weeks 24 and 48 (Table 20). According to the clinical experts consulted, these are the only 2 comparators identified as relatively appropriate for bulevirtide in patients with chronic HDV infection, given the lack of approved HDV treatments in Canada.
Combined Response
At week 24, results from the fixed-effects models (base-case analysis) indicated that bulevirtide 2 mg demonstrated a favourable improvement in the combined response compared with both PEG-IFN (risk difference = ███; 95% CrI, ███ ██ ████ and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). However, the corresponding random-effects models yielded insufficient evidence to confirm a difference in the combined response for the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. The point estimates were comparable across both the fixed-effects and random-effects models, but the 95% CrIs were wider in the random-effects models. By keeping all treatments separate in the base-case model (no nucleoside [or nucleotide] analogue therapy assumption), the results of the sensitivity analysis were consistent with the results from the fixed-effects models.
At week 48, results from the fixed-effects models indicated that bulevirtide 2 mg demonstrated a favourable improvement in the combined response compared with both PEG-IFN (risk difference = ███; 95% CrI, ██ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The corresponding random-effects models also indicated that bulevirtide 2 mg demonstrated a favourable improvement in the combined response compared with both PEG-IFN and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The point estimates were comparable across both the fixed-effects and random-effects models. Sensitivity analysis of the combined response was not feasible at week 48 due to the limited number of available studies.
Virologic Response
At week 24, results from the fixed-effects models (base-case analysis) demonstrated that bulevirtide 2 mg resulted in a favourable improvement in the virologic response compared with nucleoside (or nucleotide) analogue therapy only (risk difference = ███; 95% CrI, ███ ██ ███).The corresponding random-effects models yielded insufficient evidence to confirm a difference in the virologic response for the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. By keeping all treatments separate in the base-case model (no nucleoside [or nucleotide] analogue therapy assumption), by excluding 2 studies based on the heterogeneity assessment, and by including 2 studies irrespective of HDV RNA reduction definition, the results of the sensitivity analyses were consistent with the results from the fixed-effects models.
At week 48, results from the fixed-effects models indicated that bulevirtide 2 mg demonstrated a favourable improvement in the virologic response compared with both PEG-IFN (risk difference = ███; 95% CrI, ██ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ██ ██ ██). However, the corresponding random-effects models yielded insufficient evidence to confirm a difference in the virologic response between the bulevirtide 2 mg group and the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. The point estimates were comparable across both the fixed-effects and random-effects models.
ALT Normalization
At week 24, results from the fixed-effects models indicated that bulevirtide 2 mg demonstrated a favourable improvement in ALT normalization compared with both PEG-IFN (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). Similarly, the corresponding random-effects models indicated a favourable improvement in ALT normalization compared with both PEG-IFN (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ██ ██ ███). The point estimates were comparable across both the fixed-effects and random-effects models. By keeping all treatments separate in the base-case model (no nucleoside [or nucleotide] analogue therapy assumption), the results of the sensitivity analyses were consistent with the results from both the fixed-effects and random-effects models.
At week 48, results from the fixed-effects models indicated that bulevirtide 2 mg demonstrated a favourable improvement in ALT normalization compared with both PEG-IFN (risk difference = ███; 95% CrI, ███ ██ ███) and nucleoside (or nucleotide) analogue therapy (risk difference = ███; 95% CrI, ███ ██ ███). The corresponding random-effects models yielded insufficient evidence to confirm a difference in the virologic response for the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. The point estimates were comparable across both the fixed-effects and random-effects models.
Undetectable HDV RNA
At week 24, both fixed-effects and random-effects models yielded insufficient evidence to confirm a difference in achieving undetectable HDV RNA in the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups. By keeping all treatments separate in the base-case model (no nucleoside [or nucleotide] analogue therapy assumption), including standard interferon studies in the base-case analysis, and removing 2 studies based on the heterogeneity assessment, the results of the sensitivity analyses were consistent with the results from both the fixed-effects and random-effects models.
At week 48, results from the fixed-effects models indicated that the bulevirtide 2 mg group demonstrated a favourable improvement in achieving undetectable HDV RNA compared with the nucleoside (or nucleotide) analogue therapy group (risk difference = ███; 95% CrI, ██ ██ ███). However, there was insufficient evidence to confirm a difference in achieving undetectable HDV RNA in the bulevirtide 2 mg group compared with the PEG-IFN group (risk difference = ███; 95% CrI, ████ ██ ██). The corresponding random-effects models yielded insufficient evidence to confirm a difference in achieving undetectable HDV RNA in the bulevirtide 2 mg group compared with the PEG-IFN or nucleoside (or nucleotide) analogue therapy groups.
Table 20: Summary of NMA Efficacy Outcomes of Bulevirtide
Comparator | Model | Combined response risk difference, % (95% CrI) | Virologic response risk difference, % (95% CrI) | ALT normalization risk difference, % (95% CrI) | Undetectable HDV RNA risk difference, % (95% CrI) |
|---|---|---|---|---|---|
Week 24 | |||||
PEG-IFN | Fixed-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ |
Random-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ | |
NAT | Fixed-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ |
Random-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ | |
Week 48 | |||||
PEG-IFN | Fixed-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ |
Random-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ | |
NAT | Fixed-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ |
Random-effects model | ██ ███ █ | ██ ███ █ | ██ ███ █ | ██ ███ █ | |
CrI = credible interval; HDV = hepatitis delta virus; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; RNA = ribonucleic acid.
Note: Results in bold reported 95% CrIs that excluded the null (redacted).
Source: Sponsor-submitted NMA35 reports. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Safety Outcomes
There was insufficient evidence to confirm differences between bulevirtide at a 2 mg dose and treatment with PEG-IFN or nucleoside (or nucleotide) analogue therapy in the rates of AEs, SAEs, or withdrawals due to AEs.
Table 21: Summary of NMA Results for Safety Outcomes of Bulevirtide at Week 48
Bulevirtide 2 mg vs. comparator | Any AE OR (95% CrI) | Any SAE Risk Difference (95% CrI) | AEs leading to study withdrawal OR (95% CrI) |
|---|---|---|---|
PEG-IFN | ██ ███ █ | ██ ███ █ | ██ ███ █ |
NAT | ██ ███ █ | ██ ███ █ | ██ ███ █ |
AE = adverse event; CrI = credible interval; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; OR = odds ratio; PEG-IFN = pegylated interferon; SAE = serious adverse event; vs. = versus.
Source: Sponsor-submitted NMA35 reports. Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal of NMA
The systematic review conducted to identify potentially relevant studies for the ITC was methodologically robust; the sponsor employed a comprehensive literature search strategy, conducted study selection and data extraction in duplicate, and provided a list of excluded studies along with justifications for their exclusion. However, the ITC report did not state whether the risk of bias and feasibility assessments were carried out by a single or by multiple assessors, which has implications for the risks for bias and errors in these processes. The study inclusion and exclusion criteria in this NMA were not provided, which reduced the potential to assess heterogeneity. The sponsor’s risk of bias assessment did not identify any studies with a high risk of bias, but the Cochrane Risk of Bias tool v.2.0 was not used as intended because the appraisals were performed at the study level, as this methodology does not take into account that risk of bias can vary depending on the effect estimate being evaluated, particularly for such domains as performance, detection, attrition, and reporting bias.52
The clinical experts consulted for this review noted that, in the absence of approved treatments for HDV, some regimens included in the NMA, such as PEG-IFN and nucleoside (or nucleotide) analogues, were relatively appropriate comparators for bulevirtide. Although PEG-IFN can be used off-label to treat HDV, its use in clinical practice has become uncommon due to significant adverse effects, a global shortage, and limited availability in Canada. The clinical experts also noted that nucleoside (or nucleotide) analogues are used to manage HBV but have no direct effect on HDV. The clinical experts observed that several comparators included in the NMA analyses are not relevant to the context of HDV treatment in Canada, including bulevirtide plus tenofovir or PEG-IFN combined with adefovir, ribavirin, tenofovir, or entecavir. The inclusion of these therapies in the network could increase heterogeneity; however, given that inclusion or exclusion criteria of the included trials were not reported, this could not be comprehensively assessed.
The literature search for the NMA was last updated by the sponsor in December 2021. The included studies were published between 2006 and 2019, and the treatment landscape has since evolved. An outdated literature search with the possible omission of relevant or unpublished evidence can result in bias. The feasibility assessment for this NMA revealed considerable heterogeneity among the studies in the analyses. Although the definitions of most end points were consistent across the trials, the definition of undetectable HDV RNA differed in 2 studies in the NMA. In addition, there was considerable variation in study design, treatment duration, duration of follow-up, and regimen dosing. These sources of heterogeneity cannot be adjusted for in the NMA. Although the I2 statistic was used to assess the heterogeneity, it does not fully capture the complexity of between-study variability, so the true potential for important between-study heterogeneity is uncertain.53 Several important disease-specific characteristics were not reported in some studies in the network, and the patient populations in the included studies varied, particularly in baseline characteristics such as age, presence of cirrhosis, HDV RNA levels, ALT levels, and treatment history. These numerous sources of heterogeneity compromise the underlying transitivity assumption that must be met to produce valid conclusions from the NMA. Not all outcomes were reported across all the studies included in the NMA. The network of interconnected studies was constructed using nucleoside (or nucleotide) analogues or delayed treatment as a common comparator. The clinical experts consulted for this review considered this approach relatively appropriate, as most patients with HBV and HDV coinfection receive nucleoside (or nucleotide) analogues for the management of HBV. The results of the sensitivity analysis that retained all treatments as separate in the base-case model (i.e., without the nucleoside [or nucleotide] analogue therapy assumption) were consistent with those from the fixed-effects and random-effects models for all outcomes at week 24; however, corresponding results were not reported for week 48.
The network of evidence was sparse (i.e., there were few studies contributing to several comparisons); in many cases, there was only 1 study per link, which is insufficient to reliably estimate between-study variances. Bayesian fixed-effects models were used as a base-case analysis, with a random-effects model for the exploratory analysis. When heterogeneity is present, the CrIs of the fixed-effects model may be narrower and less conservative than the random-effects model, underestimating the uncertainty arising from between-study variation. The results of the random-effects models for most outcomes were affected by important imprecision, precluding drawing a conclusion about which treatment may be favoured. In addition, the assumptions of the fixed-effects model (e.g., a single true effect size, or no between-study heterogeneity) are likely less realistic than those of the random-effects model (e.g., assumes variation in effect sizes across studies). The point estimates for efficacy outcomes were comparable in the fixed-effects and random-effects models. Statistical tests were used to evaluate the null hypothesis that there is no inconsistency between direct and indirect evidence for the same treatment comparisons. However, these tests may have limited power to detect incoherence when it exists. The sponsor noted that a common challenge with binomial outcomes was model nonconvergence, often due to single-study effects and the presence of zero-event data. As a result, risk-difference models were employed for the meta-analyses, but their use may be limited in this context; specifically, the results of risk-difference meta-analyses can be influenced by the nonreporting of zero-event outcomes. In addition, these models typically demonstrate lower statistical power and produce more conservative CIs compared to relative measures such as risk ratios, particularly when event rates are low.20
No patient-reported quality of life data were evaluated, despite that HRQoL was considered an important end point for this review. The wide CIs around the estimates of the safety of bulevirtide relative to PEG-IFN and nucleoside (or nucleotide) analogues indicate considerable uncertainty in the comparative safety outcomes.
Studies Addressing Gaps in the Systematic Review Evidence
The contents of this section were informed by materials submitted by the sponsor and have been validated and summarized by the review team.
Two studies submitted by the sponsor were summarized to address gaps in the systematic review evidence (Table 22). The SAVE‑D study was summarized to provide evidence of the effectiveness of bulevirtide beyond 48 weeks. The MYR203 study was summarized to provide direct evidence of the efficacy of bulevirtide monotherapy compared to IFN-alpha alone.
Of note, the sponsor also included real-world cohort evidence from the Canadian HBV Network registry as part of the submission; however, it is not summarized in this report. A poster of the study54 was submitted to CDA-AMC. The sponsor was unable to provide additional documentation in a format that allowed completion of a detailed review and appraisal of the data.
Table 22: Summary of Studies Addressing Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
To provide evidence of the effectiveness of bulevirtide treatment beyond 48 weeks in patients with cirrhosis with or without clinically significant portal hypertension | SAVE-D study:
| In the SAVE-D study, 244 patients with HDV-related cirrhosis received bulevirtide 2 mg/day monotherapy for up to 96 weeks. At week 96:
|
To demonstrate the clinical efficacy of bulevirtide 2 mg/day monotherapy compared with PEG-IFN-alpha alone | MYR203 study: A multicentre, open-label, randomized, comparative, parallel-arm phase II study | The MYR203 study is provided as part of an integrated 48-week summary of efficacy and an integrated 48-week summary of safety supporting the 2 mg dose of bulevirtide. At week 72:
At week 48:
|
ALT = alanine aminotransferase; CI = confidence interval; HCC = hepatocellular carcinoma; HDV = hepatitis delta virus; IQR = interquartile range; PEG-IFN = pegylated interferon; RNA = ribonucleic acid; vs. = versus.
Sources: Details included in the table are from Degasperi et al. (2025),55 and the MYR203 Clinical Study Report.
Description of Studies
This section summarizes the SAVE-D and MYR203 studies.
SAVE-D Study Design and Objectives
The aim of the SAVE-D study was to evaluate effectiveness, safety, and clinical outcomes of bulevirtide monotherapy in a large real-life cohort of patients with chronic hepatitis D and cirrhosis. Results were reported for up to 96 weeks. Details of the SAVE-D study are shown in Table 23.
Table 23: Details of the SAVE-D Study
Detail | SAVE-D study |
|---|---|
Design and population | |
Study design | Multicentre retrospective real-world study |
Enrolled, N | 244 |
Key inclusion criteria | Consecutive patients with HDV-related cirrhosis from 46 centres in Austria, France, Germany, Greece, and Italy. |
Key exclusion criteria | Not reported |
Drugs | |
Intervention | Bulevirtide 2 mg/day self-administered (subcutaneous injections) as monotherapy |
Comparator(s) | None |
Outcomes | |
Primary end point | Virologic response defined as HDV RNA undetectable or ≥ 2 log10 IU/mL decline vs. baseline at treatment week 48. |
Secondary end points |
|
Publications | Degasperi et al. (2025)55 |
ALT = alanine aminotransferase; EASL = European Association for the Study of the Liver; HCC = hepatocellular carcinoma; HDV = hepatitis delta virus; RNA = ribonucleic acid; vs. = versus.
Source: Degasperi et al. (2025).55
Populations
Consecutive patients with HDV-related cirrhosis starting bulevirtide 2 mg monotherapy between September 2019 and February 2023 were enrolled in this multicentre retrospective study. Patients were enrolled from centres in Austria, France, Germany, Greece, and Italy. Chronic HDV was defined as HDV RNA positivity for more than 6 months. Cirrhosis was defined either histologically (Meta-analysis of Histological Data in Viral Hepatitis [METAVIR] fibrosis stage 4 [F4]), noninvasively as liver stiffness measurement (LSM) greater than 12.5 kPa, or clinically (nodular liver surface, splenomegaly, thrombocytopenia). Exclusion criteria were not reported.
Interventions
Bulevirtide 2 mg was self-administered as subcutaneous injections every 24 hours.
Outcomes
Clinical and virologic responses were collected at treatment baseline and every 24 weeks thereafter. The primary and secondary end points are summarized in Table 23.
Statistical Analysis
Categorical variables were reported as frequencies (using percentages) while continuous variables were reported as medians (with range). To compare categorical variables, chi-square or Fisher exact tests were used, while continuous variables were analyzed using Student t tests, Mann-Whitney U tests, or Kruskal-Wallis tests, as appropriate. All tests were conducted as 2-sided using a significance level of 0.05.
For continuous variables that were assessed at multiple time points, repeated measures analysis of variance was used, and the Bonferroni correction was applied to address the issue of multiple testing. The Kaplan-Meier method was used to calculate cumulative incidences of liver-related events and mortality. Follow-up duration was measured from baseline to the date of a liver-related event, death, or latest follow-up.
Results
Patient Disposition
Patient baseline characteristics are summarized in Table 24. Median age was 49 years (IQR = 40 years to 58 years). Almost two-thirds (61%) of patients were males, 82% were white, 10% had HIV coinfection, and 94% had HDV genotype 1. Of the total cohort (N = 244), 95% had compensated cirrhosis Child-Pugh Class A in 24% of patients, while the remaining 11 patients had Child-Pugh Class B7 cirrhosis at the start of treatment with bulevirtide. Cirrhosis was diagnosed based on liver stiffness measurements in 158 patients (65%), clinical signs in 45 (18%), and histological findings in 41 (17%).
Most patients showed features of advanced liver disease in terms of severity of portal hypertension: 54% of patients had esophageal varices (37% on prophylaxis) and 15% had a history of previous decompensation (ascites 12%, bleeding 3%). Median spleen size was 15 cm (IQR = 12 cm to 17 cm), median platelet count was 94 multiplied by 103/mm3 (IQR = 67 × 103/mm3 to 145 × 103/mm3), and median liver stiffness was 18.3 kPa (IQR = 13.0 kPa to 26.3 kPa). Fourteen patients (6%) had active HCC at the start of bulevirtide treatment; 58% had a history of previous IFNa treatment; and 92% were receiving nucleoside (or nucleotide) analogue therapy for HBV (55% tenofovir disoproxil fumarate, 32% entecavir, 3% tenofovir alafenamide, 2% other).
Upon starting bulevirtide treatment, median ALT values were 80 U/L (IQR = 55 U/L to 130 U/L) and 8% of patients had ALT levels in the normal reference range. Median quantitative HBsAg values were 3.8 log10 IU/mL (IQR = 3.4 log10 IU/mL to 4.1 log10 IU/mL) and 93% of patients had HBV that was hepatitis B e-antigen negative. Median HDV RNA levels were 5.4 log10 IU/mL (IQR = 4.1 log10 IU/mL to 6.5 log10 IU/mL).
Table 24: Baseline Characteristics of Patients in the SAVE-D Study
Baseline variables | Overall (N = 244) |
|---|---|
Age (years), median (IQR) | 49 (40 to 58) |
Males, n (%) | 148 (61) |
White, n (%) | 201 (82) |
HDV genotype 1, n (%)a | 77 (94) |
HIV coinfection, n (%)b | 24 (10) |
Anti-HCV positive, n (%)c | 18 (7) |
BMI, median (IQR) kg/m2 | 25 (23 to 28) |
Child-Pugh Class A, n (%)d | 233 (95) |
Spleen diameter (cm), median (IQR) | 15 (12 to 17) |
Esophageal varices, n (%)e | 91 (54) |
Previous decompensation, n(%)f | 37 (15) |
History of HCC, n (%)g | 18 (7) |
Previous IFN treatment, n (%) | 142 (58) |
NUC treatment for HBV, n (%) | 224 (92) |
Liver stiffness (kPa), median (IQR) | 18.3 (13.0 to 26.3) |
Bilirubin (mg/dL), median (IQR) | 0.9 (0.6 to 1.4) |
AST (U/L), median (IQR) | 75 (54 to 113) |
ALT (U/L), median (IQR) | 80 (55 to 130) |
GGT (U/L), median (IQR) | 68 (39 to 114) |
Albumin (g/dL), median (IQR) | 3.9 (3.5 to 4.3) |
Creatinine (mg/dL), median (IQR) | 0.8 (0.7 to 0.9) |
Platelet count (103/mm3), median (IQR) | 94 (67 to 145) |
Bile acids (μmol/L), median (IQR) | 15 (9 to 32) |
HBsAg (log10 IU/mL), median (IQR) | 3.8 (3.4 to 4.1) |
HBeAg negative, n (%) | 227 (93) |
HBV DNA detectable, n (%)h | 52 (21) |
HDV RNA (log10 IU/mL), median (IQR) | 5.4 (4.1 to 6.5) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; BMI = body mass index; GGT = gammaglutamyltransferase; HBeAg = hepatitis B e-antigen; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; HDV = hepatitis delta virus; IFN = interferon; IQR = interquartile range; NUC = nucleoside (or nucleotide) analogue therapy; RNA = ribonucleic acid.
aAvailable for 82 patients.
bAll patients with undetectable HIV RNA.
cAll patients with undetectable HCV RNA.
dChild-Pugh A6 in 59 patients (24%); Child-Pugh B7 in 11 patients (5%).
eEsophagogastroduodenoscopy results were available for 169 patients (69%), of whom 62 (37%) received prophylaxis.
fAscites experienced by 30 patients (12%), bleeding experienced by 7 patients (3%).
gActive HCC experienced by 14 patients (6%).
hAccording to local laboratory assessments, median HBV DNA was 1.4 log10 IU/mL (IQR = 1.0 to 1.5 log10 IU/mL).
Source: Degasperi et al. (2025).55
Efficacy
Of the enrolled patients, 244 (100%) had assessments available at baseline, 242 (99%) at week 24, 209 (86%) at week 48, 164 (67%) at week 72, and 87 (36%) at week 96.
After initiating bulevirtide 2 mg/day monotherapy, median HDV RNA declined from 5.4 log10 IU/mL (IQR = 4.1 log10 IU/mL to 6.5 log10 IU/mL) at baseline to 2.4 log10 IU/mL (IQR = 0.7 log10 IU/mL to 3.8 log10 IU/mL) at week 96 (P < 0.001 for all time points versus baseline), with a median decline of 2.5 log10 IU/mL (1.3 log10 IU/mL to 3.7 log10 IU/mL) and 2.7 log10 IU/mL (1.6 log10 IU/mL to 4.1 log10 IU/mL) at weeks 48 and 96, respectively (Figure 5).
Figure 5: HDV RNA Levels During Bulevirtide Monotherapy in the SAVE-D Study
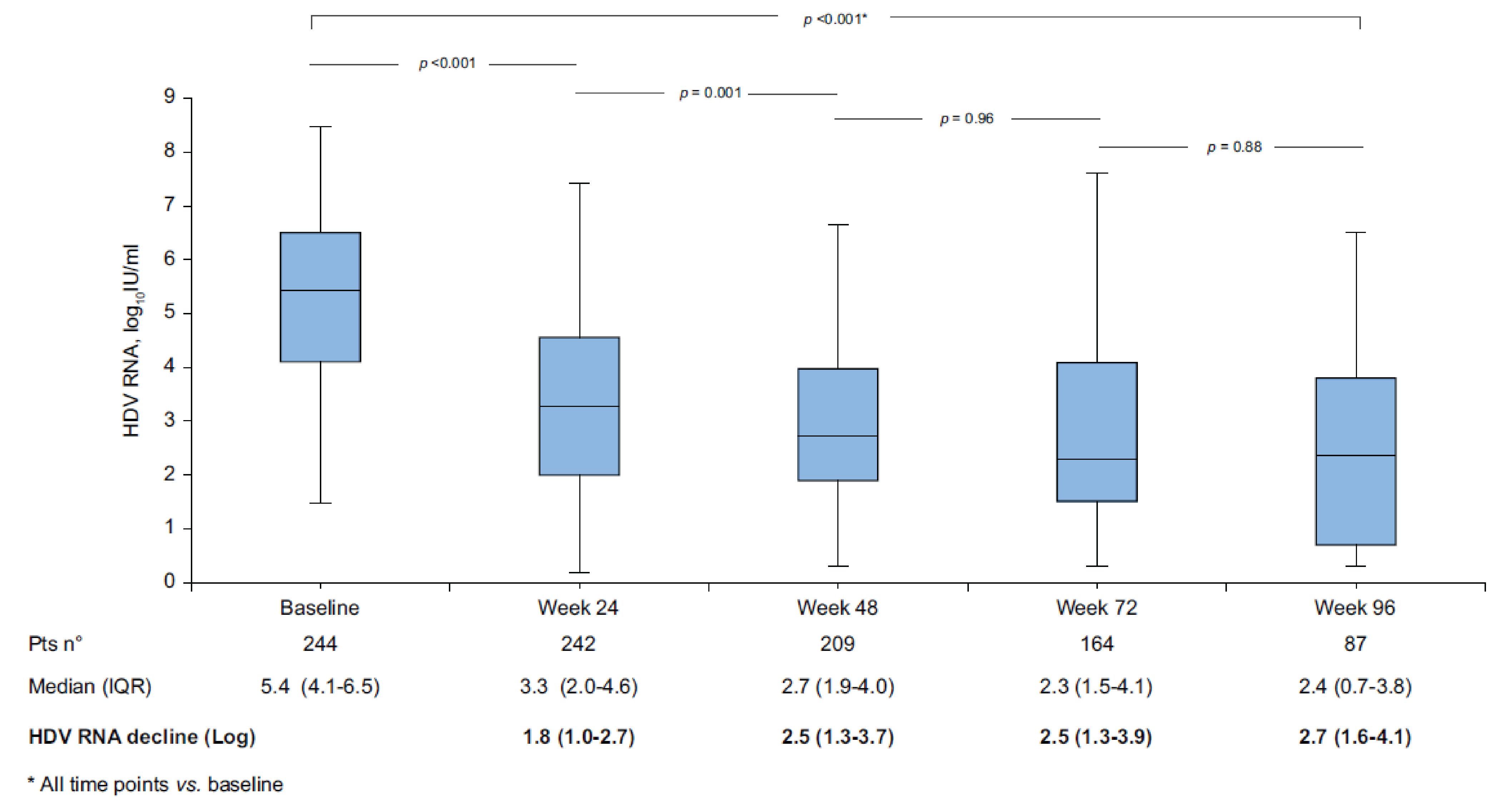
HDV = hepatitis delta virus; IQR = interquartile range; pts = points; RNA = ribonucleic acid; vs. = versus.
Source: Degasperi et al. (2025).55
Virologic response was achieved by 53% of patients at week 24, 65% at week 48, 67% at week 72, and 79% at week 96. Rates of HDV RNA undetectability at the same time points were 17%, 28%, 38%, and 48%, respectively (Figure 6).
Median ALT values declined from 80 U/L (IQR = 55 U/L to 130 U/L) at baseline to 39 U/L (IQR = 29 U/L to 53 U/L) at week 24 and stayed less than the ULN until week 96 (P < 0.001 for all time points versus baseline).
Biochemical response was achieved by 54% of patients at week 24, 61% at week 48, 61% at week 72, and 64% at week 96. For additional thresholds of biochemical response (ALT < 1.5 × ULN; ALT < 2.0 × ULN), rates of ALT normalization increased to 96% and 98%, respectively, at week 96 (Figure 7).
Rates of combined response were 33% at week 24, 44% at week 48, 46% at week 72, and 54% at week 96 (Figure 6). The proportion of patients with less than 1 log10 IU/mL HDV RNA decline from baseline progressively decreased over time, from 22% at week 24, to 19% at week 48, 14% at week 72, and 10% at week 96. Despite the suboptimal virologic response, a biochemical response was achieved by 33% of these patients at week 24 and 36% at week 48, while only 2 out of 8 patients (25%) with less than 1 log10 IU/mL decline at week 96 had ALT levels in the normal reference range. Overall, 31%, 30%, and 35% of patients with less than 1 log10 IU/mL HDV RNA decline had a greater than 50% decrease in ALT levels compared to baseline at weeks 24, 48, and 72, respectively.
Figure 6: Effectiveness of Bulevirtide Treatment in the SAVE-D Study
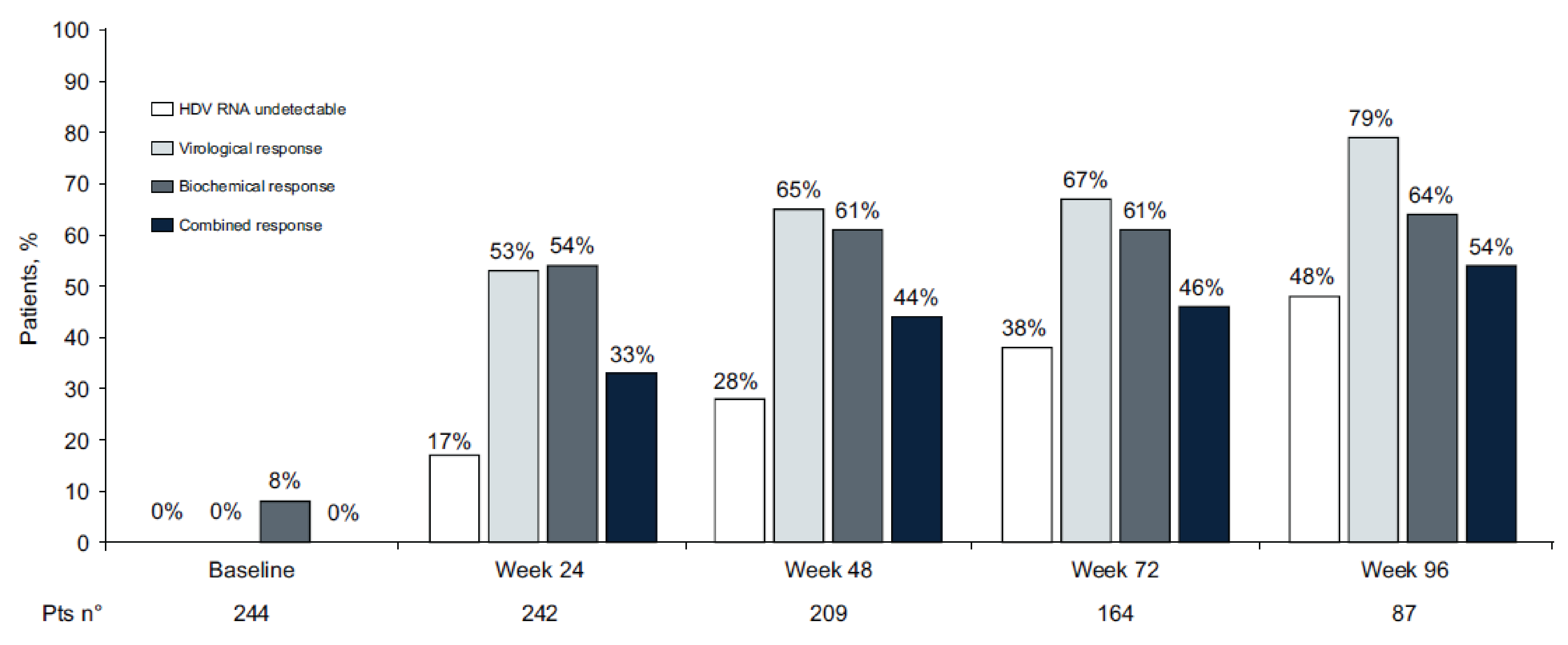
HDV = hepatitis delta virus; pts = points; RNA = ribonucleic acid.
Source: Degasperi et al. (2025).55
Figure 7: Achievement of ALT Normalization According to Different ALT Thresholds in the SAVE-D Study
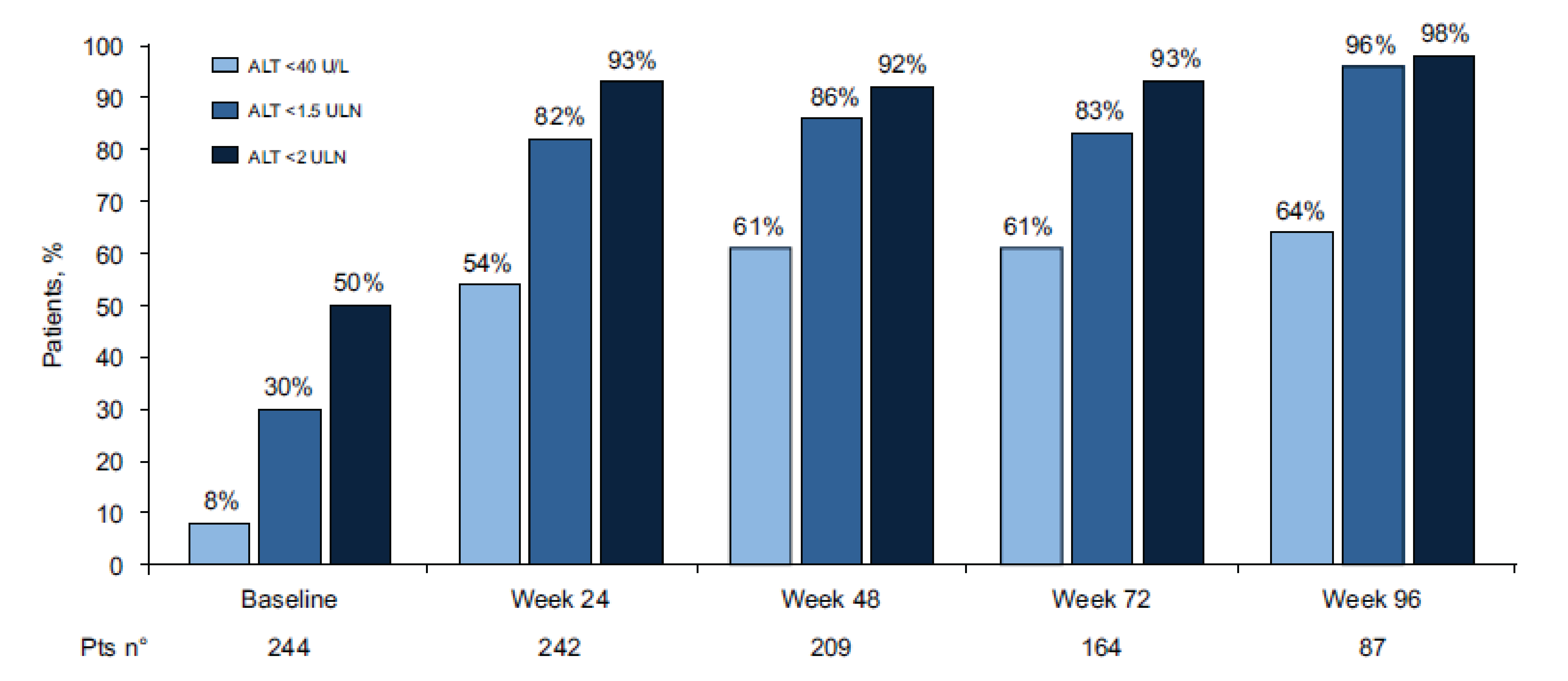
ALT = alanine aminotransferase; pts = points; ULN = upper limit of normal.
Source: Degasperi et al. (2025).55
Harms
During bulevirtide treatment, median bile acid levels increased from 15 μmol/L (IQR = 9 μmol/L to 32 μmol/L) at baseline to 37 μmol/L (IQR = 21 μmol/L to 73 μmol/L) at week 24, 41 μmol/L (IQR = 18 μmol/L to 82 μmol/L) at week 48, 39 μmol/L (IQR = 17 μmol/L to 66 μmol/L) at week 72, and 36 μmol/L (IQR = 19 μmol/L to 66 μmol/L) at week 96 (at all time points versus baseline, P < 0.001) (Figure 8). Mild and transient pruritus was experienced by 24 patients (10%); of the 24 patients, 19 (79%) experienced pruritus in the first 24 weeks of treatment. Bile acid levels at the time of pruritus onset were increased in all patients (37 μmol/L [IQR = 17 μmol/L to 94 μmol/L]), without any significant difference compared to patients without pruritus at the same time point. Injection site reactions were experienced by 7 patients (3%); a grade 3 maculopapular rash with mild eosinophilia experienced by 1 patient at treatment week 24 (and who had achieved a viral response at the time of event), leading to bulevirtide discontinuation with complete recovery.55
Figure 8: Serum Bile Acid Levels During Bulevirtide Monotherapy From Baseline up to 96 Weeks in the SAVE-D Study
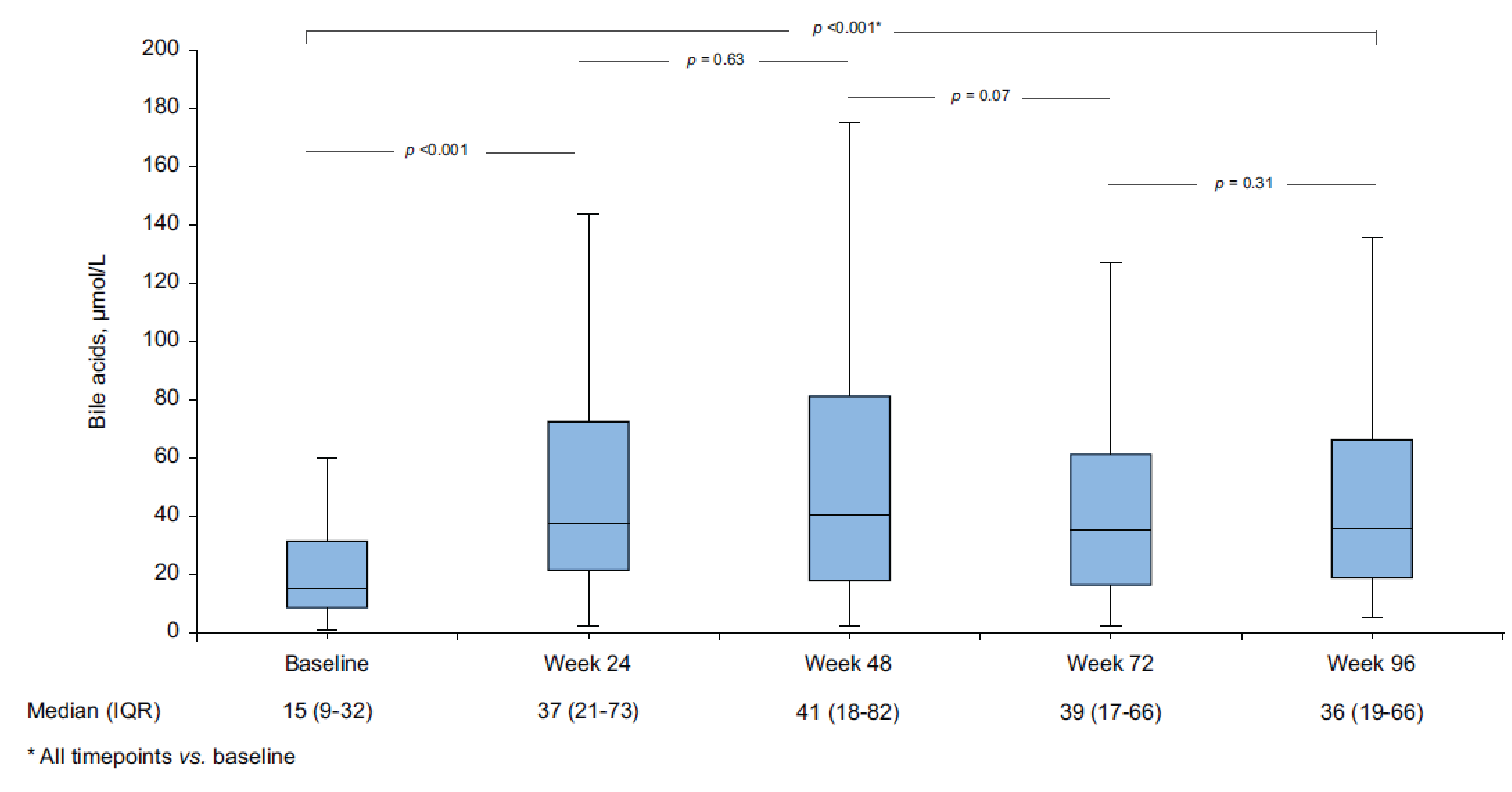
IQR = interquartile range; vs. = versus.
Source: Degasperi et al. (2025).55
MYR203 Study Design and Objectives
The MYR203 trial was a multicentre, open-label, randomized, comparative, parallel-arm phase II study to assess the efficacy of bulevirtide as a monotherapy or in combination with PEG-IFN-alpha or tenofovir compared with PEG-IFN-alpha alone. Patients were randomized into 6 treatment groups, 2 of which were considered relevant to the current review: group A, where patients received PEG-IFN-alpha 180 mcg, and group D, where patients received bulevirtide 2 mg. The study results were to be used to select the optimal treatment regimen for bulevirtide in the next phase.
Populations
Adults (aged 18 to 65 years) with chronic hepatitis B (hepatitis B e-antigen positive or negative) and presence of HBsAg and anti-HDV antibodies for at least 6 months before screening were eligible to participate in the study. Requirements included a positive test result for HDV RNA during screening and ALT levels greater than or equal to 1 times the ULN but less than 10 times the ULN. Patients were not included if they had received antiviral therapy to treat chronic hepatitis B, anticancer therapy, or immunomodulatory therapy during the previous 6 months or needed concomitant treatment with glucocorticosteroids and drugs with myelotoxic effect. Patients were also not included if they had HCV or HIV coinfections, an oncological disease, decompensated liver disease in the current or past medical history, signs of medication-induced or alcohol-induced liver impairment, or other medical conditions associated with chronic liver disease.
Interventions
Patients were randomly assigned in equal numbers to 1 of the 6 treatment groups: 2 mg bulevirtide plus PEG-IFN-alpha, 5 mg bulevirtide plus PEG-IFN-alpha, 10 mg (10 mg once daily) bulevirtide plus PEG-IFN-alpha, 10 mg (5 mg twice a day) bulevirtide plus tenofovir disoproxil fumarate, 2 mg bulevirtide alone, and PEG-IFN-alpha alone. All the groups received the assigned study treatment for 48 weeks, after which the durability of the viral response was assessed over a follow-up period of 24 weeks. This was an open-label study, where neither the patients nor the investigators were blinded to the assigned treatment.
Outcomes
The outcomes summarized from the MYR203 trial are presented in Table 25.
Table 25: Summary of MYR203 Study Outcomes
Outcome measure | Time point | MYR203 |
|---|---|---|
Undetectable HDV RNA or decrease in HDV RNA by ≥ 2 log10 IU/mL | At week 72 (primary); at weeks 24 and 48 (key secondary) | Key secondary |
Combined response: undetectable HDV RNA and ALT normalization | At weeks 24 and 48 | Key secondary |
ALT normalization | At weeks 24 and 48 | Key secondary |
The intensity of liver fibrosis based on results of transient elastography of liver | At week 48 | Other secondary |
ALT = alanine aminotransferase; HDV = hepatitis D virus; RNA = ribonucleic acid.
Note: tatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchical testing).
Source: Sponsor’s Summary of Clinical Evidence.
Statistical Analysis
Statistical models used along with the end point reported as found in Table 26. No adjustment factors, methods to handle missing data, or sensitivity analyses were performed.
Table 26: Statistical Analysis of Efficacy End Points in the MYR203 Study
End point | Statistical tests |
|---|---|
HDV RNA response | Fisher exact test |
Proportions | Fisher exact test |
Intensity of liver fibrosis | Summary statistics on intensity of liver fibrosis and Wilcoxon rank sum test results were used to compare change from baseline at weeks 48 and 72 between each of the arms B to F (groups B to F) vs. arm A (group A). |
Change in liver biopsy result | The number and percentage of patients with an improvement (decrease) or worsening (increase) of at least 1 point in the score are presented for fibrosis score and histological activity stage. |
HDV RNA levels and HBV DNA levels (on log10 scale) | Mixed-effects model for repeated measures with time point estimates |
HBV = hepatitis B virus; HDV = hepatitis delta virus; RNA = ribonucleic acid; vs. = versus.
Source: Sponsor’s Summary of Clinical Evidence.
Sample Size and Power Calculation
The primary end point was undetectable HDV RNA, assessed using PCR, at week 72, to justify the minimum size of the treatment groups.
The sample size calculation was carried out according to the method described by Chow et al.56 using the TrialSize package for the R programming language. The results from the 24-week treatment period in the MYR201 study were used: in the PEG-IFN-alpha therapy group (PEG-IFN-alpha 180 mcg for 48 weeks), the proportion of participants with undetectable HDV RNA, assessed using PCR, was 29% (2 of 7 participants), and in the combined-therapy group (bulevirtide 2 mg plus PEG-IFN-alpha 180 mcg for 24 weeks, followed by PEG-IFN-alpha 180 mcg for 24 weeks), the proportion of participants with undetectable HDV RNA, assessed using PCR, was 71% (5 of 7 participants). It was expected that 5 clinical study groups would demonstrate efficacy comparable to the MYR201 substudy combined-therapy results. Based on these assumptions and a significance level of 0.10, having a group size of 15 would make it possible to reach at least 60% power in the study.
Considering the exploratory nature of the study, the number of arms calculated could be considered sufficient to draw conclusions about the optimal dosing schedule to be used in clinical studies of the next phases.
Patient Disposition
Table 27: Patient Disposition in the MYR203 Study
Patient disposition | Group A PEG-IFN-alpha | Group D Bulevirtide 2 mg |
|---|---|---|
Randomized, N | 15 | 15 |
Received study medication, n (%) | 15 (100.0) | 15 (100.0) |
Completed the study, n (%) | 10 (66.7) | 13 (86.7) |
Premature withdrawal from the study,a n (%) | 5 (33.3) | 2 (13.3) |
Patient withdrew from study | 1 (20.0) | 1 (50.0) |
Pregnancy | 1 (20.0) | 0 (0) |
Loss to follow-up | 1 (20.0) | 1 (50.0) |
Adverse event | 2 (40.0) | 0 (0) |
Safety analysis set | 15 (100.0) | 15 (100.0) |
Full analysis set | 15 (100.0) | 15 (100.0) |
Per-protocol analysis set | 7 (46.7) | 10 (66.7) |
Pharmacokinetic analysis set — substudy | 0 (0) | 0 (0) |
Pharmacokinetic analysis set — main study | 0 (0) | 15 (100.0) |
PEG-IFN = pegylated interferon.
Notes: Percentages are based on the number of patients who were randomized within each treatment group except group A.
Patients are presented in the treatment groups as treated (i.e., actual treatment).
aPercentages are based on the number of patients who prematurely withdrew from each treatment group.
Source: MYR203 Clinical Study Report.
Patient Characteristics
Table 28: Patient Demographic and Baseline Characteristics in the MYR203 Study (SAS)
Characteristic | Group A PEG-IFN-alpha (N = 15) | Group D bulevirtide 2 mg (N = 15) |
|---|---|---|
Age, years | ||
n/nmiss | 15/0 | 15/0 |
Mean (SD) | 34.7 (7.1) | 42.0 (9.6) |
Median | 36.0 | 39.0 |
Q1, Q3 | ████ ██ | ████ ██ |
Minimum to maximum | 20 to 48 | 26 to 62 |
Sex, n (%) | ||
Female | 10 (66.7) | 4 (26.7) |
Male | 5 (33.3) | 11 (73.3) |
Race, n (%) | ||
American Indian or Alaska Native | ████ ██ | ████ ██ |
Asian | ████ ██ | ████ ██ |
Black or African American | ████ ██ | ████ ██ |
Native Hawaiian or Other Pacific Islander | ████ ██ | ████ ██ |
White | 14 (93.3) | 15 (100.0) |
Height, cm | ||
n/nmiss | 15/0 | 15/0 |
Mean (SD) | ████ ██ | ████ ██ |
Median | ████ ██ | ████ ██ |
Q1, Q3 | ████ ██ | ████ ██ |
Minimum to maximum | ████ ██ | ████ ██ |
Weight, kg | ||
n/nmiss | ████ ██ | ████ ██ |
Mean (SD) | ████ ██ | ████ ██ |
Median | ████ ██ | ████ ██ |
Q1, Q3 | ████ ██ | ████ ██ |
Minimum to maximum | ████ ██ | ████ ██ |
BMI | ||
n/nmiss | 15/0 | 15/0 |
Mean (SD) | ████ ██ | ████ ██ |
Median | 23.02 | 27.47 |
Q1, Q3 | ████ ██ | ████ ██ |
Minimum to maximum | 18.7 to 38.3 | 21.5 to 34.5 |
BMI categories, n (%) | ||
< 30 | ████ ██ | ████ ██ |
≥ 30 | ████ ██ | ████ ██ |
BMI = body mass index; n/nmiss = number of patients with evaluable data/number of patients with missing data; PEG-IFN = pegylated interferon; Q1 = first quartile; Q3 = third quartile; SAS = safety analysis set; SD = standard deviation.
Notes: Percentages are based on the number of patients in each treatment group.
Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: MYR203 Clinical Study Report.
Efficacy
Other Secondary Efficacy End Point: Degree of Liver Fibrosis
At baseline, the mean liver stiffness was 11.01 kPa (SD = 4.66 kPa) in the PEG-IFN-alpha group (group A) and 13.78 kPa (SD = 5.09 kPa) in the bulevirtide 2 mg group (group D). There were no statistically significant differences in the change from baseline between the group receiving bulevirtide alone (group D) and the PEG-IFN group (group A) at any time point. Similar results were observed when the analyses were performed on the per-protocol analysis set population.
Table 29: Key Efficacy Outcomes in the MYR203 Study (FAS)
Characteristic | Group A PEG-IFN-alpha (N = 15) | Group D bulevirtide 2 mg (N = 15) |
|---|---|---|
Primary efficacy variable: HDV RNA responsea,b,c,d,e | ||
Week 72 (24 weeks posttreatment) | ||
Number of patients in analysis, N | ████ ██ | ████ ██ |
Number of patients with virologic response, N | ████ ██ | ████ ██ |
Proportion of patients with virologic response, % (95% CI) | ████ ██ | ████ ██ |
Difference in proportions, n (95% CI) | Reference | NA |
P valuef | Reference | NA |
HDV RNA responsea,b,c,d,e | ||
Week 24 | ||
Number of patients in analysis, N | 15 | 15 |
Number of patients with virologic response, N | 1 | 2 |
Proportion of patients with virologic response, % (95% CI) | 6.7 (0.2 to 31.9) | 13.3 (1.7 to 40.5) |
Difference in proportions, n (95% CI) | Reference | 6.7 (−20.3 to 34.4) |
P valuef | Reference | 1.0000 |
Week 48 | ||
Number of patients in analysis, N | 15 | 15 |
Number of patients with virologic response, N | 2 | 2 |
Proportion of patients with virologic response, % (95% CI) | 13.3 (1.7 to 40.5) | 13.3 (1.7 to 40.5) |
Difference in proportions, n (95% CI) | Reference | 0.0 (−28.9 to 28.9) |
P valuef | Reference | 1.0000 |
ALT normalizationb,c,d,e,f,g | ||
Week 24 | ||
Number of patients in analysis, N | 15 | 14 |
Number of patients with ALT normalization, N | 0 | 9 |
Proportion of patients with ALT normalization, % (95% CI) | 0 (0.0 to 21.8) | 64.3 (35.1 to 87.2) |
Difference in proportions, n (95% CI) | Reference | 64.3 (33.9 to 87.2) |
P valuef | Reference | 0.0002 |
Week 48 | ||
Number of patients in analysis, N | 15 | 15 |
Number of patients with ALT normalization, N | 4 | 11 |
Proportion of patients with ALT normalization, % (95% CI) | 26.7 (7.8 to 55.1) | 73.3 (44.9 to 92.2) |
Difference in proportions, n (95% CI) | Reference | 46.7 (8.2 to 75.4) |
P valuef | Reference | 0.0268 |
Combined response (negative HDV RNA and ALT normalization)b,c,d,e,h | ||
Week 24 | ||
Number of patients in analysis, N | 15 | 15 |
Number of patients with combined response, N | 0 (0) | 2 |
Proportion of patients with combined response, % (95% CI) | 0 (0.0 to 21.8) | 13.3 (1.7 to 40.5) |
Difference in proportions, n (95% CI) | Reference | 13.3 (–10.1 to 40.5) |
P valuef | Reference | 0.4828 |
Week 48 | ||
Number of patients in analysis, N | 15 | 15 |
Number of patients with combined response, N | 1 | 2 |
Proportion of patients with combined response, % (95% CI) | 6.7 (0.2 to 31.9) | 13.3 (1.7 to 40.5) |
Difference in proportions, n (95% CI) | Reference | 6.7 (–20.3 to 34.4) |
P valuef | Reference | 1.0000 |
HDV RNA levels (log10 scale)e,i,j | ||
Week 24 | ||
Number of patients in analysis, N | 15 | 15 |
LS means (95% CI) | 4.447 (3.677 to 5.217) | 3.743 (2.970 to 4.515) |
Difference in LS means (95% CI) | Reference | −0.704 (–1.795 to 0.387) |
P valuef | Reference | 0.2027 |
Week 48 | ||
Number of patients in analysis, N | 15 | 15 |
LS means (95% CI) | 4.066 (3.175 to 4.957) | 3.162 (2.272 to 4.052) |
Difference in LS means (95% CI) | Reference | –0.904 (–2.164 to 0.355) |
P valuef | Reference | 0.1567 |
ALT = alanine aminotransferase; CI = confidence interval; HBsAg = hepatitis B surface antigen; HDV = hepatitis delta virus; FAS = full analysis set; LLoD = lower limit of detection; LS = least squares; NA = not available; PEG-IFN = pegylated interferon; RNA = ribonucleic acid; SD = standard deviation.
aHDV RNA response was defined as an HDV RNA value less than the LLoD, where LLoD = 10.
bProportions, as percentages, are based on the number of patients analyzed in each treatment group.
cCIs calculated using the Clopper-Pearson (exact) method for within-group proportions and exact unconditional method for difference in proportions.
dFisher exact test was used for the comparison of respective the bulevirtide group and PEG-IFN-alpha-2 only group.
eFor the FAS, patients were analyzed as randomized (i.e., planned treatment).
fP values were not adjusted for multiple testing using Bonferroni correction.
gALT normalization was defined as ALT value within the normal reference range.
hCombined response was defined as HDV RNA response and ALT normalization.
iA mixed-effects model for repeated measures was used to test null hypotheses of no difference compared to the control group.
jThe model included log10-transformed data for all postbaseline visits for the dependent variable and was adjusted for baseline HDV RNA (log10- transformed), treatment group, analysis visit, the interaction between treatment group and analysis visit.
Source: MYR203 Clinical Study Report.
Harms
For a summary of the key harms data in the MYR203 study, refer to Table 30.
Table 30: Key Harms Data in the MYR203 Study (SAS)
Characteristic | Group A PEG-IFN-alpha (N = 15) | Group D bulevirtide 2 mg (N = 15) |
|---|---|---|
Most common (≥ 10%) AEs by preferred term, n (%) | ||
Patients with any AE | 13 (86.7) | 15 (100.0) |
Life-threatening or disabling | 0 (0.0) | 0 (0.0) |
Total bile acids increased | 5 (33.3) | 12 (80.0) |
Neutropenia | 8 (53.3) | 3 (20.0) |
Leukopenia | 9 (60.0) | 4 (26.7) |
Thrombocytopenia | 8 (53.3) | 3 (20.0) |
ALT levels increased | 5 (33.3) | 8 (53.3) |
Aspartate aminotransferase increased | 5 (33.3) | 8 (53.3) |
GGT levels increased | 4 (26.7) | 4 (26.7) |
Influenza-like illness | 1 (6.7) | 0 (0) |
Lymphopenia | 5 (33.3) | 1 (6.7) |
Asthenia | 3 (20.0) | 0 (0) |
Hyperthermia | 7 (46.7) | 1 (6.7) |
Anemia | 3 (20.0) | 0 (0) |
Headache | 3 (20.0) | 1 (6.7) |
Hemoglobin decreased | 4 (26.7) | 1 (6.7) |
Reticulocytopenia | 5 (33.3) | 2 (13.3) |
Erythropenia | 4 (26.7) | 0 (0) |
Nausea | 4 (26.7) | 0 (0) |
Alopecia | 4 (26.7) | 0 (0) |
Rash | 3 (20.0) | 0 (0) |
Insomnia | 2 (13.3) | 1 (6.7) |
Monocytopenia | 3 (20.0) | 0 (0) |
Pruritus | 2 (13.3) | 1 (6.7) |
Respiratory tract infection | 1 (6.7) | 0 (0) |
Myalgia | 2 (13.3) | 0 (0) |
Irritability | 2 (13.3) | 0 (0) |
Somnolence | 2 (13.3) | 0 (0) |
Decreased appetite | 2 (13.3) | 0 (0) |
Menstrual disorder | 2 (13.3) | 0 (0) |
SAE by preferred term, n (%) | ||
Patients with any SAE | 0 (0) | 0 (0) |
Patients who stopped treatment due to AEs, n (%) | ||
Any AE leading to withdrawal of PEG-IFN | 2 (13.3) | NA |
Any AE leading to withdrawal of bulevirtide | NA | 0 (0) |
Any AE leading to withdrawal of tenofovir | NA | NA |
Deaths, n (%) | ||
Any AE leading to death | 0 (0) | 0 (0) |
AE = adverse event; ALT = alanine aminotransferase; GGT = gammaglutamyltransferase; MedDRA = Medical Dictionary for Regulatory Activities; NA = not available; PEG-IFN = pegylated interferon; SAE = serious adverse event; SAS = safety analysis set.
Notes: Adverse events are coded according to MedDRA Version 21.1.
Percentages are based on the number of patients in each treatment group.
For the SAS, patients were analyzed as treated (i.e., actual treatment).
Source: Table 35, Table 39, and Table 42, MYR203 Clinical Study Report.
Critical Appraisal
SAVE-D Study
Internal Validity
SAVE-D was a multicentre retrospective real-world study (n = 244 patients) with the goal of addressing the gap in evidence on the effectiveness of treatment with bulevirtide beyond 48 weeks in patients with cirrhosis with and without clinically significant portal hypertension. SAVE-D was a single-arm real-world study without a comparator, which means that conclusions could not be drawn about the effectiveness of bulevirtide treatment relative to usual care. Further, the single-arm design does not allow for causal conclusions.
The consecutive enrolment of new patients in the SAVE-D study helped minimize selection bias. The authors also correctly applied Bonferroni adjustment for multiple testing to reduce the risk of type I errors when comparing repeated measures analysis of variance for continuous variables at different time points. There was a potential risk of bias in the measurement of outcomes because HDV RNA quantification was performed locally using different assays with different limits of quantification and detection. This variability could impact the HDV RNA undetectability rates. As a function of the real-world, retrospective design, there was potential for underreporting of bulevirtide-related AEs and liver-related events. The SAVE-D study did not investigate missing outcome data, including possible informative censoring of patients between weeks 24, 48, 72, and 96. The sample sizes declined from 244 (100%) at baseline, to 87 (36%) at week 96, posing a high risk of bias due to missing outcome data, particularly after week 24. The open-label administration of bulevirtide meant that patients’ knowledge of their treatment could lead to more favourable outcomes and fewer SAEs. In addition, even though the AEs reported were relatively mild, patients who remained in the study represent a healthier subgroup who were better able to tolerate any AEs, potentially skewing the results. The authors reported confounders at baseline but did not assess time-varying exposure to other confounding viral conditions (e.g., HCV or HIV infection) after enrolment, which could impact concomitant treatment (e.g., nucleoside [or nucleotide] analogue) and study outcomes. Other statistical methods could have been used to adjust for time-varying confounders.
External Validity
Recruitment for SAVE-D occurred across 46 centres in Austria, France, Germany, Greece, and Italy, allowing generalizability to populations in these countries, but this limit generalizability to the patient population in Canada. Most of the study participants were white (82%), and 95% had cirrhosis classified as Child-Pugh Class A. The authors acknowledged the prevalence of the HDV-1 genotype in the European setting, which could limit generalizability to the context in Canada. The absence of comparators in this study meant there was no assessment of comparative effectiveness or causal inference of bulevirtide relative to usual care. Out of the outcomes patients deemed important, the SAVE-D study did not report HRQoL outcomes.
MYR203 Study
Internal Validity
The MYR203 study was an exploratory phase II trial that was susceptible to various risks of bias. The small sample sizes in each randomized group (n = 15), increased the risk that true prognostic balance was not achieved as was evidenced by the imbalance in baseline characteristics between groups, potentially leading to the overestimation of the beneficial effects of bulevirtide. A larger percentage of patients in the PEG-IFN-alpha group (33.3%) than in the bulevirtide alone group (13.3%) withdrew prematurely from the study. The relatively large percentage of missing data poses a risk of bias for this open-label study.
By week 72, there was a risk of bias due to missing outcome data evidence in a decline in patients remaining in the analysis set: 46% in the PEG-IFN-alpha group and 60% in the bulevirtide group alone. The sponsor used a missing equals failure approach that assumes that patients with missing data had a negative outcome. Sensitivity analyses were conducted using complete cases, which assumes that data are missing completely at random. The sponsor did not demonstrate that the 2 assumptions, missing equals failure and missing completely at random, are valid. Therefore, there is likely a risk of bias due to missing outcome data, the magnitude and direction of which are uncertain.
The open-label design can also pose a risk of bias in the measurement of AE outcomes. The authors did not apply any methods to adjust for multiple testing, increasing the likelihood of type I errors for statistically significant results. The small sample sizes also meant that the effect estimates for many end points were subject to high levels of uncertainty due to imprecision (with 95% CIs spanning the null).
External Validity
All the patients enrolled in the MYR203 study were from Russia, which limits the generalizability to patients in Canada. The exclusion of patients who had previously received antiviral therapy for the treatment of chronic hepatitis B and patients with HCV or HIV coinfections improved the internal validity but limited external validity. The comparator group used in this study was PEG-IFN, which is limited in use in Canada due to the adverse effects and drug availability issues, further limiting the generalizability of this study. HRQoL outcomes were not measured in the MYR203 trial despite this being an important outcome for patients with HDV infection. The use of very small sample sizes greatly limits the generalizability of the findings of bulevirtide as an effective therapy for HDV infection.
Discussion
Summary of Available Evidence
The MYR301 trial was an ongoing, phase III, randomized, open-label, parallel-group, multicentre study (N = 150). The objective of the MYR301 trial was to evaluate the efficacy and safety of bulevirtide at a dose of 2 mg for treating chronic HDV infection in adults with compensated liver disease, compared to delayed treatment with a 10 mg dose of bulevirtide following a 48-week observation period. The trial comprises a 4-week screening period, a 144-week treatment period, and a 96-week follow-up period. Patients were randomized to receive delayed treatment with bulevirtide 10 mg after an observation period of 48 weeks, immediate treatment with bulevirtide 2 mg, or immediate treatment with bulevirtide 10 mg. The mean age of patients was 42 years (SD = 8.4 years). Nearly one-half of the patients (47.3%) had cirrhosis at the time of randomization; all the cases of cirrhosis were classified as Child-Pugh Class A. During the MYR301 trial, 60.7% of patients received oral anti-HBV treatment and 56.0% of patients had a history of prior interferon treatment.
In the absence of direct comparative evidence of bulevirtide versus other relevant comparators, the sponsor conducted an NMA. The objective of the NMA was to evaluate the relative efficacy and safety of bulevirtide 2 mg as a monotherapy versus PEG-IFN and best supportive care, consisting of nucleoside (nucleotide) analogue therapy in patients with chronic HDV infection who had compensated liver disease, given the lack of approved treatments for HDV. Fixed-effects and random-effects models were applied within a Bayesian framework to assess combined response, virologic response, ALT normalization, and HDV RNA loss at weeks 24 and 48.
Two additional studies that address gaps in the evidence from the systematic review are summarized in this report. SAVE-D is a multicentre, retrospective, single-arm, real-world study (N = 244). The objective of the SAVE-D study was to evaluate the efficacy of treatment with bulevirtide 2 mg for up to 96 weeks in patients with HDV-related cirrhosis with and without clinically significant portal hypertension, and to explore predictors of treatment and the drug’s impact on hard outcomes. The study end points included virologic, biochemical, and combined responses, undetectable HDV RNA, suboptimal response, safety outcomes, liver-related complications, and harms. Most patients in the SAVE-D study presented with features of advanced liver disease in terms of severity of portal hypertension, with 54.0% experiencing esophageal varices and 15.0% with a history of previous decompensation. The MYR203 trial was a multicentre, open-label, randomized, comparative, parallel-arm phase II trial that provided direct comparative evidence for the efficacy and safety of bulevirtide 2 mg as monotherapy and PEG-IFN alone in the treatment of patients with chronic hepatitis D who had compensated liver disease, after 48 weeks of treatment. The study outcomes included HDV RNA response at weeks 24, 48 ███ ██, ALT normalization, combined response, liver fibrosis, and HDV RNA levels at weeks 24 and 48, and harms. The mean age of patients was 36.8 years (SD = 7.7 years). In the MYR203 trial, 45.6% of patients had a history of prior medication use, and 63.3% received concomitant medication during the study.
Interpretation of Results
Efficacy
HDV, a rare and rapidly progressing disease, is considered the most severe form of viral hepatitis. HDV occurs as a coinfection with HBV and is associated with a significantly higher risk of cirrhosis, hepatic decompensation, and HCC. There are currently no established consensus guidelines for the management and treatment of HDV infection in Canada. At present, bulevirtide is the only treatment specifically indicated for HDV treatment. This highlights the ongoing need for effective therapies that can both treat and prevent HDV infection. The current standard of care includes nucleoside (or nucleotide) analogues, which are effective against HBV but not HDV, and PEG-IFN-alpha, which can be used off-label for treating HDV. However, the clinical experts consulted for this review indicated that PEG-IFN-alpha is now rarely used in clinical practice due to its adverse effects, the global shortage, and limited availability in Canada. The optimal duration of bulevirtide therapy has not been established, and clinical experts have noted that the optimal duration remains uncertain for patients who demonstrate a slower response to treatment.
The evidence from the pivotal MYR301 trial addressed treatment outcomes deemed important by both patients and clinicians. Input from the patient group emphasized key priorities, namely slowing disease progression, reducing decompensation events and the need for a liver transplant, improving HRQoL, and having treatments that require fewer injections and blood tests. Similarly, input from the clinician group and clinical experts consulted by CDA-AMC highlighted that the most important treatment goals for patients with HDV are achieving HDV RNA suppression and biochemical normalization and stabilizing or improving liver fibrosis. The clinical experts noted that long-term outcomes such as improvements in portal hypertension, reduced progression to cirrhosis, lower rates of HCC, and liver-related mortality would also be clinically beneficial but were not measured in the MYR301 trial. Therefore, results from the analyses of the following outcomes measured in the MYR301 trial were summarized and assessed using GRADE: combined response, virologic response, change from baseline in liver stiffness, HRQoL, and SAEs. In addition, ALT normalization, undetectable HDV RNA, change from baseline in combined response, liver-related clinical events, change from baseline in virologic response were included in the report as supportive information. An undetectable HDV RNA after scheduled end of treatment (sustained virologic response) was identified by clinical experts as an important outcome; however, the results for this end point were not reported, as the study is still ongoing.
According to the clinical experts consulted for this review by CDA-AMC, although the use of a delayed-treatment group in patients with aggressive disease could raise ethical concerns, the trial design was deemed acceptable by regulatory authorities due to the absence of an established treatment for HDV at the time. The clinical experts also noted that the delayed-treatment group effectively served as the best available standard of care or placebo comparator for the MYR301 trial. The clinical experts indicated that the inclusion of a delayed-treatment group in the trial provided valuable insight into the natural progression of HDV and helped contextualize the benefits of early intervention. Data from the group receiving immediate treatment with bulevirtide 10 mg are not presented in this report because this dose is neither approved for use nor used in clinical practice in Canada. The focus of this report is on between-group comparisons of study end points up to week 48, specifically regarding the 2 mg bulevirtide dose, consistent with the Health Canada indication. The clinical experts indicated that using bulevirtide at the 10 mg dose as a comparator beyond week 48 was inappropriate because 10 mg is not a Health Canada–approved dose and is not expected to be used in clinical practice.
Evidence from the MYR301 trial demonstrated with moderate certainty that, compared with delayed treatment, treatment with bulevirtide 2 mg results in a clinically meaningful increase in achieving combined response at week 48. The point estimate for the between-group differences exceeded the 20% threshold the clinical experts had suggested, and the 95% CIs did not include trivial effects; this suggests favourable outcomes for bulevirtide 2 mg rather than for delayed treatment. The certainty of evidence was considered moderate due to imprecision, including potential for treatment overestimation due to the small sample size. Given the rarity of the disease, the small sample sizes observed in the MYR301 trial were expected and a larger trial may be infeasible. The clinical experts consulted considered the combined response an appropriate surrogate measure for assessing treatment benefit and noted that surrogate markers are routinely used in clinical trials, including those evaluating HDV therapies. According to the clinical experts, a reduction in HDV RNA levels of at least 2 log10 or achievement of undetectable HDV RNA (virologic response) are both considered indicative of a meaningful antiviral effect that may slow or halt disease progression. Normalization of ALT levels was also identified by the clinical experts as a clinically relevant biochemical marker of treatment response.
In addition to the combined response, individual component outcomes, virologic response and ALT normalization, were reported in the MYR301 trial, with both end points demonstrating a benefit of bulevirtide over delayed treatment at week 48. The point estimate for the between-group difference for the virologic response exceeded the threshold of 30% that the clinical experts had suggested, and the 95% CIs did not include trivial effects; these results favour treatment with bulevirtide 2 mg over delayed treatment. The certainty of evidence was considered moderate due to imprecision and there was potential for overestimating the treatment effect due to the small sample size. The secondary outcome in the MYR301 trial, the proportion of patients with ALT normalization at week 48, also supported the clinical benefit of bulevirtide over delayed treatment for chronic HDV infection. For undetectable HDV RNA at week 48, a secondary outcome in the trial, the between-group difference in response rates was calculated using the bulevirtide 2 mg group as the reference and the 10 mg group as the comparator; however, this comparison is not relevant to this review.
The clinical experts stated that measuring liver stiffness using FibroScan is the preferred method for assessing fibrosis in clinical practice. Improvement in fibrosis was identified by the clinical experts as a relevant and meaningful surrogate, although improvements require longer observation periods, from 1 to 3 years. Evidence from the MYR301 trial demonstrated with moderate certainty that, compared with delayed treatment, bulevirtide 2 mg likely results in improvement in liver stiffness at week 48. The clinical experts could not provide a threshold for a clinically meaningful between-group difference for this outcome. Subgroup analyses by cirrhosis status and prior use of nucleoside (or nucleotide) analogues were prespecified; however, the results were descriptive and limited by small sample sizes and should not be used to infer effect modification.
HRQoL is an important outcome to patients and clinicians and was assessed as an exploratory outcome using the HQLQ scale scores. The clinical experts noted that the HQLQ is a liver-specific instrument that captures aspects of mental health and fatigue and has been validated in the context of liver disease. For the HQLQ mental and physical component summary scores, the HQLQ positive well-being score, the HQLQ hepatitis-specific limitations score, and the HQLQ hepatitis-specific health distress score, there is very low-certainty evidence for a benefit when compared with delayed treatment at weeks 24 and 48. The very low certainty of evidence was attributed to risk of bias from assessors being aware of treatment assignments, as well as serious concerns for imprecision, with wide CIs that included both the potential for no difference and harm. There was insufficient information to allow for conclusions on the long-term efficacy of bulevirtide for patients with HDV. The clinical experts highlighted that long-term clinical end points such as reduced incidence of HCC or decreased episodes of hepatic decompensation are rarely captured within the assessment time frames of pivotal trials but are nevertheless important for long-term evaluation of treatment benefit.
Overall, there are no major concerns with the generalizability of the MYR301 trial results. According to the clinical experts consulted by CDA-AMC, patients enrolled in the trials were considered to have more severe disease than those typically seen in clinical practice. The clinical experts indicated that trial inclusion and exclusion criteria are more restrictive than the selection criteria that would typically be applied in clinical practice. For instance, patients aged older than 65 years, those with moderate renal impairment, or those with HCV or HIV coinfections, who would be eligible to receive bulevirtide in clinical practice, were excluded from the MYR301 trial. Clinical expert input also indicated that the proportion of patients with HBV and HDV coinfection who receive HBV therapy is higher in clinical practice than the proportion in the trial.
The aim of the sponsor-submitted ITC was to assess the efficacy and safety of bulevirtide 2 mg compared with best supportive care (nucleoside [or nucleotide] analogue therapy) and PEG-IFN, both of which were identified by the clinical experts as relatively appropriate comparators in the absence of approved treatments for HDV. The clinical experts indicated that while PEG-IFN was previously used, its use has largely declined in clinical practice due to poor tolerability, safety concerns (particularly in patients with advanced liver disease), and limited availability in Canada. The clinical experts also noted that nucleoside (or nucleotide) analogues are used to manage HBV but have no direct effect on HDV. According to the clinical experts consulted, several comparators included in the NMA analyses are not relevant to the context in Canada, including bulevirtide plus tenofovir or PEG-IFN combined with adefovir, ribavirin, tenofovir, or entecavir. The results of fixed-effects models suggested that bulevirtide showed potential advantage over PEG-IFN in achieving the combined response, virologic response, and ALT normalization at week 48. In contrast, the random-effects model showed benefit of bulevirtide over PEG-IFN only for the combined response. Similarly, compared to nucleoside (or nucleotide) analogue therapy, fixed-effects models indicated potential benefits of bulevirtide in achieving combined response, virologic response, ALT normalization, and undetectable HDV RNA at week 48, while the random-effects model demonstrated a benefit for combined response only. The CDA-AMC review team considered the results of the random-effects models to be more relevant, as the fixed-effects models assumed that effects are identical across studies, which is unlikely. Overall, substantial uncertainty remains regarding these relative effects due to several limitations: a large degree of clinical and methodological heterogeneity across studies, a sparse network, small sample sizes, imprecision, and assumptions made in the analysis that cannot be fully verified. No patient-reported quality of life data, which was considered an important end point for this review, were evaluated.
Two additional studies that addressed gaps in the evidence from the systematic review are summarized in this report: MYR203 and SAVE-D. However, due to the sample size and study design limitations of these studies and the exploratory nature of the MYR203 study, definitive conclusions cannot be drawn from them. The SAVE-D study was a single-arm study, which limits the ability to assess comparative effectiveness relative to usual care and for inferring causality. Key limitations of both studies were a risk of bias from missing outcome data that contribute to uncertainty in the effect estimates, limited generalizability to patients in Canada, and no reporting of HRQoL outcomes, which patients considered to be important.
Harms
According to the clinical experts consulted by CDA-AMC for this review, bulevirtide 2 mg was generally well-tolerated by the patients in the MYR301 trial, and no major safety concerns were identified. The clinical experts noted that most of the AEs would be expected and considered manageable in routine clinical practice. Common AEs, including lymphopenia, fatigue, eosinophilia, and injection site reactions, are considered manageable in clinical practice and are consistent with the underlying disease severity in the trial population, which included a high proportion of patients with cirrhosis. In particular, the experts noted that thrombocytopenia observed shortly after treatment initiation was likely a reflection of advanced liver disease rather than a direct effect of the drug. Overall, the proportion of patients in the MYR301 trial who experienced SAEs was low, with similar rates observed across treatment groups. The evidence is very uncertain about the effect of bulevirtide 2 mg on SAEs at week 48 when compared with delayed treatment. No treatment discontinuations due to AEs were reported by week 48. The clinical experts emphasized the importance of closely monitoring hepatic and renal function, particularly in light of the underlying comorbidities commonly present in the target patient population. The sponsor-submitted ITC evaluated the comparative safety of bulevirtide relative to PEG-IFN and nucleoside (or nucleotide) analogues; however, the wide CIs around these estimates indicate considerable uncertainty in the comparative safety outcomes.
Other Considerations
According to the clinical experts consulted for this review, HDV testing is not systematically conducted among individuals with HBV infection. The clinical experts emphasized the need for increased health care provider education and recommended the implementation of reflex testing in provincial or local laboratories when individuals test positive for HBsAg. The clinical experts also indicated that treatment with bulevirtide can be challenging for patients because of the need to reconstitute drugs and the associated supply requirements, which can be costly given the daily administration and chronic nature of the disease. At present, HDV testing is likely confined to hepatology specialists and is performed opportunistically rather than as part of routine screening. The clinical experts noted that initiating treatment with bulevirtide and monitoring response should be conducted by clinicians experienced in managing viral hepatitis because of the complexity of treatment, which involves daily injections, and the need for patient counselling; however, they acknowledged that access to such specialists may be difficult in certain regions, particularly rural areas.
Conclusion
HDV is a rare and severe condition that is recognized as the most aggressive form of viral hepatitis. At the time of this review, bulevirtide is the only approved treatment for HDV in adults with compensated liver disease. Input from both patients and clinicians highlighted a substantial unmet need for new treatments that effectively treat and prevent the progression of HDV. The pivotal MYR301 phase III trial compared the efficacy and safety of a 2 mg dose of bulevirtide in the treatment of patients with chronic HDV with compensated liver disease, with a 10 mg dose of bulevirtide following a 48-week observation period (the delayed-treatment group). The MYR301 trial demonstrated moderate certainty evidence that treatment with bulevirtide 2 mg likely results in a clinically meaningful increase in achieving combined response, virologic response, and improvement in liver stiffness at week 48 compared with delayed treatment. No definitive conclusion can be drawn regarding the effects of bulevirtide treatment on HRQoL due to the potential bias from assessor knowledge of treatment assignment, and imprecision in the data, making the direction of effects unclear. Bulevirtide 2 mg was generally well-tolerated, and no major safety concerns were identified in the MYR301 trial. There is no direct comparative evidence beyond week 48 between bulevirtide and the relevant comparators.
The MYR203 phase II trial provided evidence on the efficacy and safety of bulevirtide 2 mg as monotherapy versus PEG-IFN-alpha-2a alone in patients with HDV infection and compensated liver disease. However, due to the limited sample size, study design limitations, and its exploratory nature, definitive conclusions cannot be drawn from the MYR203 trial. The SAVE-D study was a real-world study with the objective of providing evidence on the efficacy of treatment with bulevirtide for up to 96 weeks in patients with HDV-related cirrhosis with and without clinically significant portal hypertension. However, the single-arm design of the SAVE-D study limits the ability to assess comparative effectiveness relative to usual care and to infer causality. Key limitations of both studies include a risk of bias from missing outcome data that contribute to uncertainty in the effect estimates, limited generalizability to patients in Canada, and no reporting of HRQoL outcomes, which were deemed important by patients.
The results of the sponsor-submitted ITC suggest that, compared with PEG-IFN and best supportive care, bulevirtide may have potential benefit in achieving combined response at week 48. However, the interpretation of the ITC is limited by several factors, including methodological limitations, between-trial heterogeneity, a sparse network, and small sample sizes, which preclude definitive conclusions on the comparative effects of bulevirtide. In addition, PEG-IFN is no longer used for HDV treatment in Canada due to its poor tolerability and limited accessibility. Given the considerable uncertainty in the sponsor-submitted ITC, no definitive conclusions can be drawn regarding the safety of bulevirtide compared to PEG-IFN and best supportive care.
References
1.Farci P, Anna Niro G. Current and future management of chronic hepatitis D. Gastroenterol Hepatol (N Y). 2018;14(6):342-351. PubMed
2.Koh C, Da BL, Glenn JS. HBV/HDV coinfection: A challenge for therapeutics. Clin Liver Dis. 2019;23(3):557-572. doi:10.1016/j.cld.2019.04.005 PubMed
3.Huang YH, Wu JC, Sheng WY, et al. Diagnostic value of anti-hepatitis D virus (HDV) antibodies revisited: a study of total and IgM anti-HDV compared with detection of HDV-RNA by polymerase chain reaction. J Gastroenterol Hepatol. 1998;13(1):57-61. doi:10.1111/j.1440-1746.1998.tb00546.x PubMed
4.Centers for Disease Control and Prevention. Hepatitis D questions and answers for health professionals. Accessed March, 2021. https://www.cdc.gov/hepatitis/hdv/hdvfaq.htm#ref12
5.Polaris Observatory Collaborators. Adjusted estimate of the prevalence of hepatitis delta virus in 25 countries and territories. J Hepatol. 2024;80(2):232-242. doi:10.1016/j.jhep.2023.10.043 PubMed
6.Osiowy C, Swidinsky K, Haylock-Jacobs S, et al. Molecular epidemiology and clinical characteristics of hepatitis D virus infection in Canada. JHEP Rep. 2022;4(5):100461. doi:10.1016/j.jhepr.2022.100461 PubMed
7.Gilead Sciences Canada Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Hepcludex (bulevirtide), 2mg, powder for solution for subcutaneous injection 2025.
8.Negro F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb Perspect Med. 2014;4(11):a021550. doi:10.1101/cshperspect.a021550 PubMed
9.Miao Z, Zhang S, Ou X, et al. Estimating the global prevalence, disease progression, and clinical outcome of hepatitis delta virus infection. J Infect Dis. 2020;221(10):1677-1687. doi:10.1093/infdis/jiz633 PubMed
10.Bockmann JH, Grube M, Hamed V, et al. High rates of cirrhosis and severe clinical events in patients with HBV/HDV co-infection: longitudinal analysis of a German cohort. BMC Gastroenterol. 2020;20(1):24. doi:10.1186/s12876-020-1168-9 PubMed
11.Grabowski J, Wedemeyer H. Hepatitis delta: immunopathogenesis and clinical challenges. Dig Dis. 2010;28(1):133-8. doi:10.1159/000282076 PubMed
12.Roulot D, Brichler S, Layese R, et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic hepatitis delta. J Hepatol. 2020;73(5):1046-1062. doi:10.1016/j.jhep.2020.06.038 PubMed
13.European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370-398. doi:10.1016/j.jhep.2017.03.021 PubMed
14.Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560-1599. doi:10.1002/hep.29800 PubMed
15.Coffin CS, Fung S, Alvarez F, et al. Management of hepatitis B virus infection: 2018 guidelines from the Canadian Association for the Study of the Liver and Association of Medical Microbiology and Infectious Disease Canada. Can Liver J. 2018;1(4):156-217. doi:10.3138/canlivj.2018-0008 PubMed
16.Da BL, Heller T, Koh C. Hepatitis D infection: from initial discovery to current investigational therapies. Gastroenterol Rep (Oxf). 2019;7(4):231-245. doi:10.1093/gastro/goz023 PubMed
17.Bertino G, Ragusa R, Corsaro LS, et al. Improvement of health-related quality of life and psychological well-being after HCV eradication with direct-acting antiviral agents. Real life setting data of an Italian cohort valued by Hepatitis Quality of Life Questionnaire (HQLQv2). Health Psychol Res. 2020;8(3):9450. doi:10.4081/hpr.2020.9450 PubMed
18.U.S. Food and Drug Administration. Chronic hepatitis D virus infection: Developing drugs for treatment guidance for industry. Draft guidance. 2019.
19.Gilead Sciences Inc. Clinical Study Report: MRO301. A multicenter, open-label, randomized phase 3 clinical study to assess efficacy and safety of bulevirtide in patients with chronic hepatitis delta [internal sponsor’s report]. April 17, 2024.
20.Bradburn MJ, Deeks JJ, Berlin JA, et al. Much ado about nothing: a comparison of the performance of meta-analytical methods with rare events. Stat Med. 2007;26(1):53-77. doi:10.1002/sim.2528 PubMed
21.Gilead Sciences Canada Inc. Sponsor's clinical summary report [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Hepcludex (bulevirtide), 2mg, powder for solution for subcutaneous injection. 2025.
22.Shah PA, Choudhry S, Reyes KJC, et al. An update on the management of chronic hepatitis D. Gastroenterol Rep (Oxf). 2019;7(6):396-402. doi:10.1093/gastro/goz052 PubMed
23.Gilead Sciences Canada Inc. Hepcludex (bulevirtide): Powder for solution for injection, 2 mg/vial, subcutaneous injection [product monograph]. August 8, 2025.
24.Lutgehetmann M, Mancke LV, Volz T, et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology. 2012;55(3):685-94. doi:10.1002/hep.24758 PubMed
25.Zhang Z, Urban S. Interplay between hepatitis D virus and the interferon response. Viruses. 2020;12(11)doi:10.3390/v12111334 PubMed
26.European Medicines Agency. Product information: Hepcludex (bulevirtide). 2025. Accessed by sponsor, no date provided. https://www.ema.europa.eu/en/documents/product-information/hepcludex-epar-product-information_en.pdf
27.European Medicines Agency. Assessment report: Hepcludex (bulevirtide). 2020. Accessed by sponsor, no date provided. https://www.ema.europa.eu/en/documents/assessment-report/hepcludex-epar-public-assessment-report_en.pdf.
28.Gilead Sciences Canada Inc. Gilead receives complete response letter from U.S. FDA for bulevirtide for the treatment of adults with hepatitis delta virus. October 27, 2022. Accessed July 7, 2025. https://www.gilead.com/company/company-statements/2022/gilead-receives-complete-response-letter-from-us-fda-for-bulevirtide-for-the-treatment-of-adults-with-hepatitis-delta-virus
29.Castera L, Forns X, Alberti A. Non-invasive evaluation of liver fibrosis using transient elastography. J Hepatol. 2008;48(5):835-47. doi:10.1016/j.jhep.2008.02.008 PubMed
30.Ware JE, Gandek B. Overview of the SF-36 Health Survey and the International Quality of Life Assessment (IQOLA) project. J Clin Epidemiol. 1998;51(11):903-912. doi:10.1016/S0895-4356(98)00081-X PubMed
31.Bayliss MS, Gandek B, Bungay KM, et al. A questionnaire to assess the generic and disease-specific health outcomes of patients with chronic hepatitis C. Qual Life Res. 1998;7(1):39-55. doi:10.1023/a:1008884805251 PubMed
32.Hepatera LLC. Clinical Study Report: MYR202. A multicenter, open-label, randomized clinical study to assess efficacy and safety of 3 doses of Myrcludex B for 24 weeks in combination with Tenofovir compared to Tenofovir alone to suppress HBV replication in patients with chronic hepatitis D [internal sponsor’s report]. December 18, 2020.
33.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
34.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
35.Gilead Sciences Inc. Network meta-analyses of pharmacological interventions in chronic hepatitis D [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Hepcludex (bulevirtide), 2mg, powder for solution for subcutaneous injection 2021.
36.Gilead Sciences Inc. Meta-analyses of pharmacological interventions in chronic hepatitis D [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Hepcludex (bulevirtide), 2mg, powder for solution for subcutaneous injection. 2022.
37.National Institute for Health and Care Excellence. NICE process and methods. Single technology appraisal and highly specialised technologies evaluation: User guide for company evidence submission template. January 8, 2015. Updated December 3, 2024. Accessed May 7, 2025. https://www.nice.org.uk/process/pmg24/chapter/clinical-effectiveness#quality-assessment-of-the-relevant-randomised-controlled-trials
38.Dias S, Welton NJ, Sutton AJ, et al. NICE Decision Support Unit technical support documents. NICE DSU technical support document 2: A generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. 2014.
39.Turner RM, Jackson D, Wei Y, et al. Predictive distributions for between-study heterogeneity and simple methods for their application in Bayesian meta-analysis. Stat Med. 2015;34(6):984-98. doi:10.1002/sim.6381 PubMed
40.Spiegelhalter DJ, Best NG, Carlin BP, et al. Bayesian measures of model complexity and fit. Journal of the Royal Statistical Society Series B: Statistical Methodology. 2002;64(4):583-639. doi:10.1111/1467-9868.00353
41.Warn DE, Thompson SG, Spiegelhalter DJ. Bayesian random effects meta-analysis of trials with binary outcomes: methods for the absolute risk difference and relative risk scales. Stat Med. 2002;21(11):1601-1623. doi:10.1002/sim.1189 PubMed
42.Abbas Z, Memon MS, Umer MA, et al. Co-treatment with pegylated interferon alfa-2a and entecavir for hepatitis D: A randomized trial. World J Hepatol. 2016;8(14):625-31. doi:10.4254/wjh.v8.i14.625 PubMed
43.Niro GA, Ciancio A, Gaeta GB, et al. Pegylated interferon alpha-2b as monotherapy or in combination with ribavirin in chronic hepatitis delta. Hepatology. 2006;44(3):713-720. doi:10.1002/hep.21296 PubMed
44.Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J Hepatol. 2016;65(3):490-8. doi:10.1016/j.jhep.2016.04.016 PubMed
45.MYR201, Randomized open-label substudy of daily Myrcludex B plus pegylated interferon-alpha-2a in patients with HBeAg negative chronic hepatitis B co-infected with hepatitis delta [sponsor supplied reference] 2017.
46.Wedemeyer H, Bogomolov P, Blank A, et al. Final results of a multicenter, open-label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with Tenofovir in patients with chronic HBV/HDV co-infection. J Hepatol. 2018;68:S3. doi:10.1016/S0168-8278(18)30224-1
47.Hepatera LLC. Clinical Study Report: MYR203. A multicentre, open-label, randomized, comparative, parallel-arm phase II study to assess efficacy and safety of Myrcludex B in combination with peginterferon alfa-2a versus peginterferon alfa-2a alone in patients with chronic viral hepatitis B with delta-agent [internal sponsor’s report]. December 17, 2020.
48.Wedemeyer H, Yurdaydìn C, Dalekos GN, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med. 2011;364(4):322-31. doi:10.1056/NEJMoa0912696 PubMed
49.Wedemeyer H, Yurdaydin C, Hardtke S, et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis. 2019;19(3):275-286. doi:10.1016/s1473-3099(18)30663-7 PubMed
50.Niro GA, Ciancio A, Gaeta GB, et al. Pegylated interferon alpha-2b as monotherapy or in combination with ribavirin in chronic hepatitis delta. Hepatology. 2006;44(3):713-20. doi:10.1002/hep.21296 PubMed
51.Abbas Z, Memon P, Umer M, et al. Co-treatment with pegylated interferon alfa-2a and entecavir for hepatitis D: A randomized trial. World J Hepatol. 2016;8:625. doi:10.4254/wjh.v8.i14.625 PubMed
52.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. doi:10.1136/bmj.l4898 PubMed
53.Guyatt G, Zhao Y, Mayer M, et al. GRADE guidance 36: updates to GRADE's approach to addressing inconsistency. J Clin Epidemiol. 2023;158:70-83. doi:10.1016/j.jclinepi.2023.03.003 PubMed
54.Poulin S, Sadler M, Doucette K, et al. Bulevirtide for the Treatment of Hepatitis Delta. Real-World Cohort from the Canadian HBV Network. Joint 2025 CDDW-CLM Conference. Poster Presentation [sponsor supplied reference] 2025;
55.Degasperi E, Anolli MP, Jachs M, et al. Real-world effectiveness and safety of bulevirtide monotherapy for up to 96 weeks in patients with HDV-related cirrhosis. J Hepatol. 2025;82(6):1012-1022. doi:10.1016/j.jhep.2024.12.044 PubMed
56.Chow S-C, Wang, H., & Shao, J. Sample size calculations in clinical research. 2nd ed. Chapman and Hall/CRC 2007.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Figure 9: Network Diagram of ALT Normalization at Week 24 in NMA
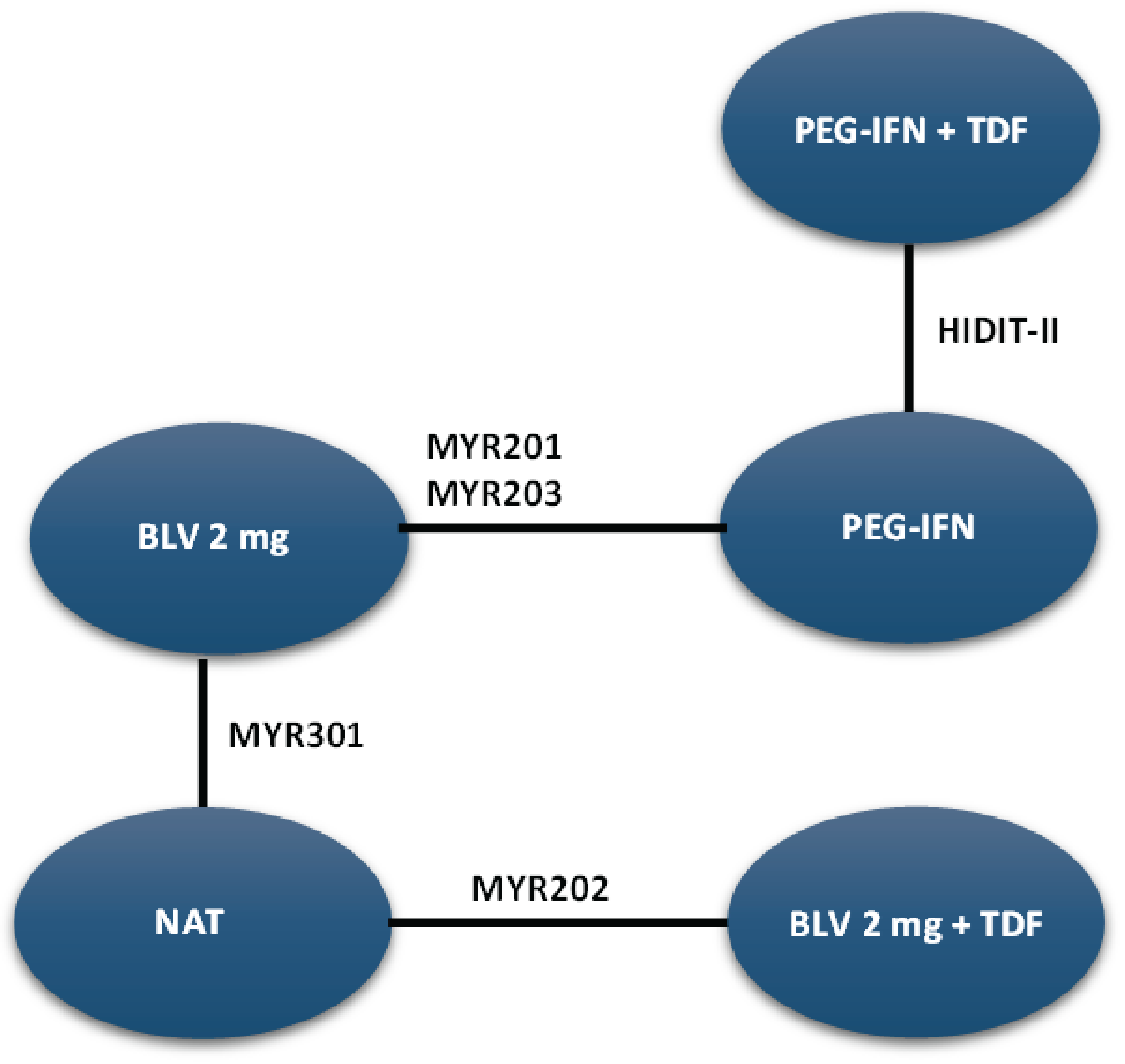
ALT = alanine aminotransferase; BLV = bulevirtide; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Figure 10: Network Diagram of ALT Normalization at Week 48 in NMA
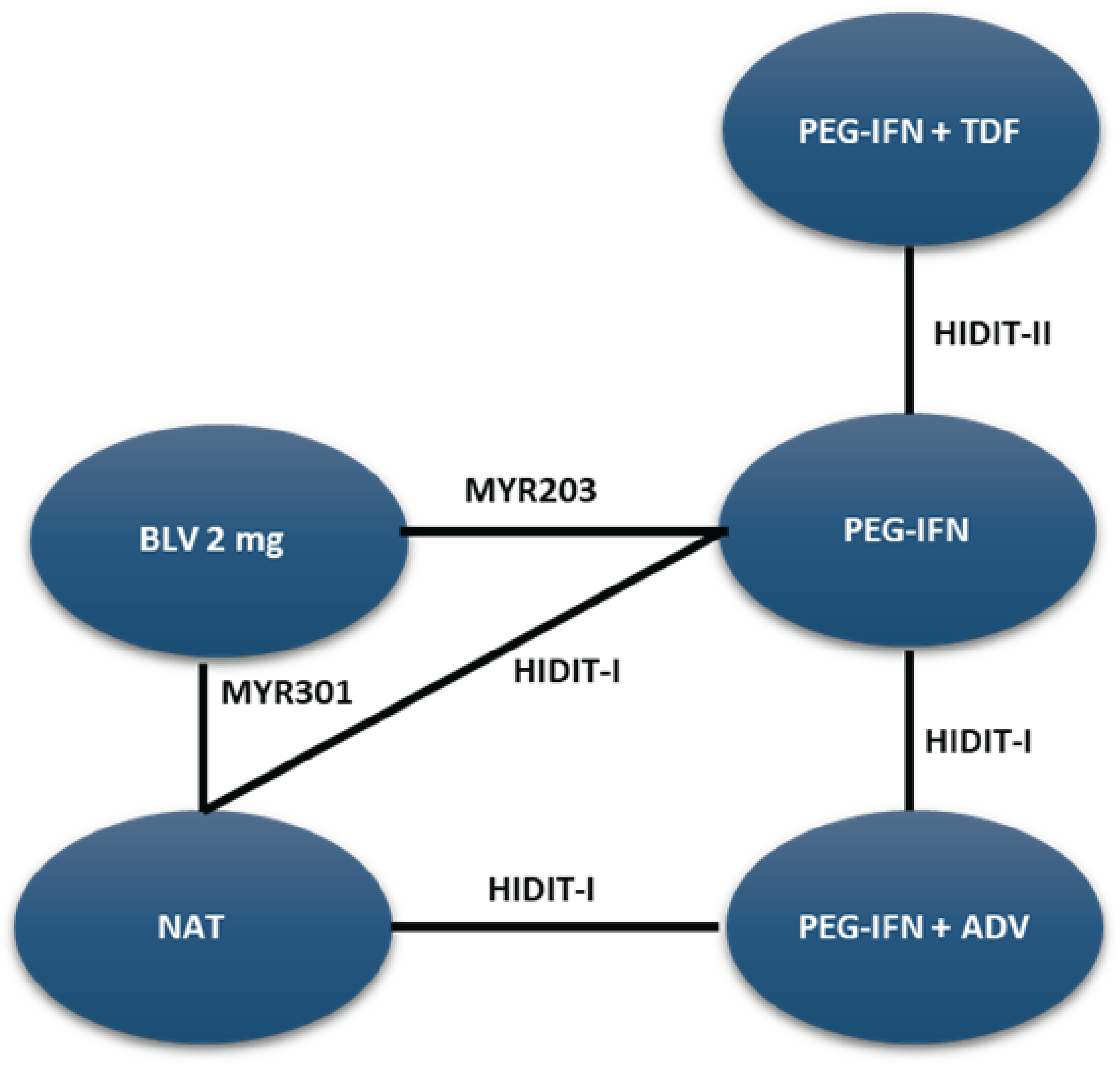
ALT = alanine aminotransferase; ADV = adefovir; BLV = bulevirtide; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Figure 11: Network Diagram of HDV RNA Loss Outcome at Week 24
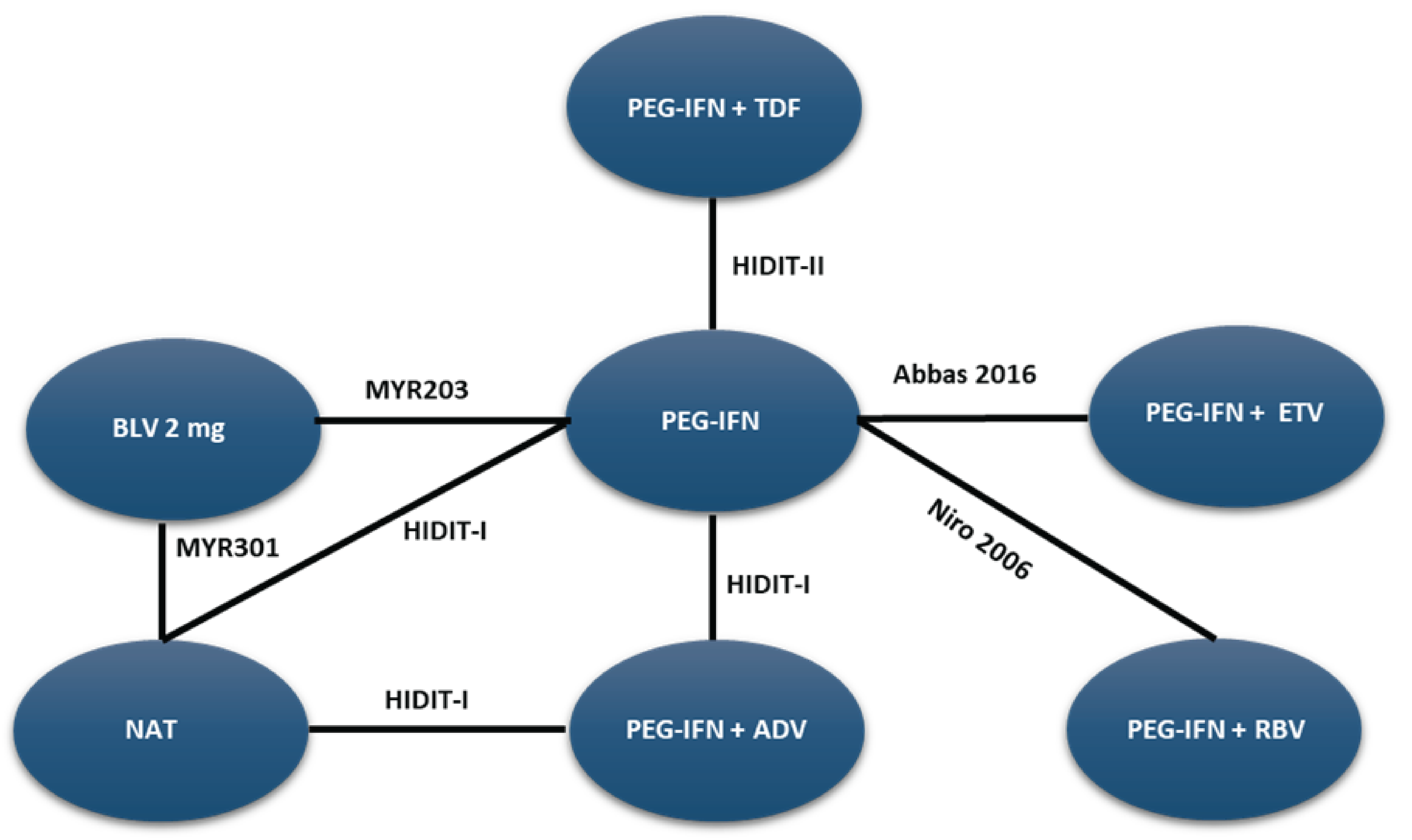
ADV = adefovir; BLV = bulevirtide; ETV = entecavir; HDV = hepatitis delta virus; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; RBV = ribavirin; RNA = ribonucleic acid; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Figure 12: Network Diagram of HDV RNA Loss Outcome at Week 48
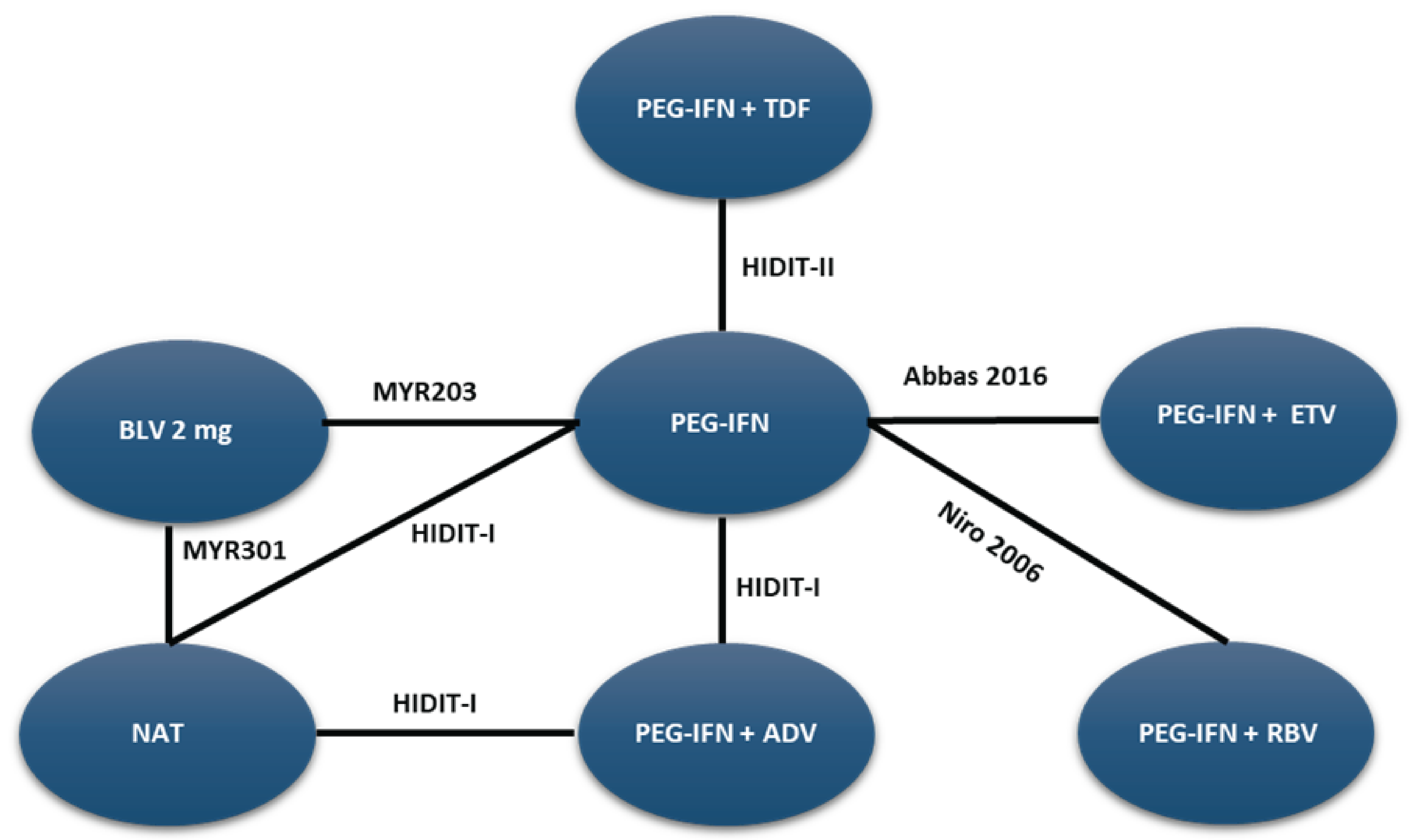
ADV = adefovir; BLV = bulevirtide; ETV = entecavir; HDV = hepatitis delta virus; NAT = nucleoside (or nucleotide) analogue therapy; NMA = network meta-analysis; PEG-IFN = pegylated interferon; RBV = ribavirin; RNA = ribonucleic acid; TDF = tenofovir.
Source: Sponsor-submitted NMA35 technical reports. Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
HDV
hepatitis delta virus
ICER
incremental cost-effectiveness ratio
PEG-IFN
pegylated interferon
QALY
quality-adjusted life-year
RNA
ribonucleic acid
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness and budget impact of bulevirtide compared to best supportive care for the treatment of chronic hepatitis delta virus (HDV) infection in adults with compensated liver disease.
Item | Description |
|---|---|
Drug product | Bulevirtide (Hepcludex), 2 mg, powder for solution for injection, subcutaneous |
Indication | For the treatment of chronic hepatitis delta virus (HDV) infection in adults with compensated liver disease |
Submitted price | $437.07 per 2 mg vial |
Health Canada approval status | NOC |
Health Canada review pathway | Priority |
NOC date | August 8, 2025 |
Reimbursement request | Per indication |
Sponsor | Gilead Sciences Canada, Inc. |
Submission history | Previously reviewed: No |
HDV = hepatitis delta virus; NOC = Notice of Compliance.
Summary
Bulevirtide is available as a powder for solution for subcutaneous injection (2 mg/mL).1 At the submitted price of $437.07 per vial, the annual cost of bulevirtide is expected to be $159,641 per patient, based on the Health Canada–recommended dosage.2
Comparative clinical efficacy in the economic analysis was derived from the MYR301 trial, which compared bulevirtide with a delayed-treatment group.3 Evidence submitted by the sponsor indicates that, in adult patients with HDV infection and compensated liver disease, bulevirtide is likely to achieve undetectable levels of HDV ribonucleic acid (RNA) or reduce HDV RNA by at least 2 log10 IU/mL from baseline, and normalize levels of alanine aminotransferase, compared with delayed treatment.
The results of the CDA-AMC base case suggest the following:
Bulevirtide is predicted to be associated with higher costs to the health care system than best supportive care (incremental costs = $1,297,909), primarily driven by increased costs associated with bulevirtide.
Bulevirtide is predicted to be associated with a gain of 4.01 life-years compared to best supportive care and may result in a gain of 3.38 quality-adjusted life-years (QALYs) compared to best supportive care.
The incremental cost-effectiveness ratio (ICER) of bulevirtide compared to best supportive care was $383,943 per QALY gained.
Approximately 99.1% of the predicted incremental benefits were accrued based on extrapolation where there are no trial data. The model assumed treatment efficacy would be maintained over a lifetime because patients who respond to treatment would receive life-long treatment. Given the uncertainty in the long-term efficacy of bulevirtide and the optimal duration of treatment, these uncertainties suggest that the CDA-AMC base-case analysis may overestimate the health benefits associated with bulevirtide.
CDA-AMC estimates that the budget impact of reimbursing bulevirtide for the treatment of chronic HDV infection in adults with compensated liver disease will be approximately $44,973,750 over the first 3 years of reimbursement compared to the amount currently spent on best supportive care. As there are no drug costs associated with best supportive care, this amount reflects the estimated expenditure for bulevirtide. The actual budget impact of reimbursing bulevirtide will depend on the negotiated drug price of bulevirtide and the number of people eligible for treatment.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis to estimate the cost-effectiveness of bulevirtide from the perspective of a public health care payer in Canada over a lifetime horizon (59.8 years). The modelled population comprised adults with compensated liver disease with the presence of viral HDV RNA as confirmed by blood tests, which is aligned with the Health Canada indication and was based on the participants in the MYR301 trial. The sponsor’s base-case analysis included costs related to drug acquisition, health state, adverse events, and monitoring.2 In the sponsor’s base case, bulevirtide was associated with higher costs (incremental costs = $1,403,673) and higher QALYs (incremental QALYs = 4.01) relative to pegylated interferon (PEG-IFN)-alpha. This resulted in an ICER for bulevirtide of $350,046 per QALY gained.2 Compared to PEG-IFN-alpha, approximately 98% of the incremental benefit was predicted to be accrued after the treatment duration of the MYR301 trial (maximum follow-up of 144 weeks). Additional information about the sponsor’s submission is summarized in Appendix 3.
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore uncertainty in a scenario analysis? |
|---|---|---|---|
The model does not adequately explore the relationship between treatment duration and long-term efficacy of bulevirtide. | The model does not explore the relationship between treatment discontinuation, viral or clinical rebound, and re-treatment. The model assumed that patients with full response and patients with suboptimal response would continue to receive treatment until discontinuation (i.e., 28.2% of patients with full response and 6.5% of patients with suboptimal response would still be receiving treatment at year 10). The optimal duration of treatment for patients with response remains unclear, and clinical expert feedback received by CDA-AMC noted that, upon discontinuation, viral and clinical rebound may occur and re-treatment may have to be initiated. For patients without response, treatment duration was assumed to be 144 weeks. The clinical expert feedback received by CDA‑AMC expressed that 72 weeks is likely more reflective practice in Canada. | CDA-AMC partly addressed this by changing the treatment duration for patients without response to 72 weeks. As nearly all (98.0%) of the incremental benefit was accrued after the treatment duration of the MYR301 trial, CDA-AMC cautions against interpreting the ICER because of the uncertainty related to the long-term efficacy of bulevirtide and the expected duration of therapy for patients with combined or suboptimal response. | A scenario analysis was conducted in which patients with no response discontinued treatment at 48 weeks. |
Patients within the same health state have different utility values based on their response status. Health state utility values further lacked face validity. | The sponsor’s submitted model applied different utility values within the same health state based on response status. This double counts the utility impact of responding to treatment. The sponsor’s utility values also appear to suggest that certain health states (i.e., noncirrhotic fibrosis) would have better quality of life than the general population, which lacks face validity. | CDA-AMC removed the additional 0.06 QALY increment for patients with a treatment response. Health state utility values were updated based on values reported in a study performed in Canada and a systematic review, both of which were deemed more valid based on clinical expert feedback.4,5 | No scenario analysis was conducted because the best available evidence was used in the CDA‑AMC base case. |
Natural history of disease underestimates disease mortality. | The transition of patients from compensated cirrhosis or decompensated cirrhosis to death health states was informed by the published literature.6,7 The clinical expert feedback received by CDA‑AMC indicated that these values did not align with their clinical experience. | Probabilities from compensated cirrhosis or decompensated cirrhosis to death informed by D’Amico et al.8 | No scenario analysis was conducted because best available evidence was used in the CDA‑AMC base case. |
Health care resource utilization is underestimated. | Training for reconstitution and self-administration for bulevirtide was assumed to involve 4 hours of nursing time. Clinical expert feedback obtained by CDA‑AMC indicated that 12 to 16 hours is more reflective of clinical practice. Health care utilization associated with cirrhosis was noted to also be underestimated. | CDA-AMC base case updated the health care resource utilization values based on the clinical expert feedback received. | No scenario analysis was conducted. |
Impact of treatment adherence is inadequately modelled. | The sponsor assumed adherence to bulevirtide to be 97.96%, which would lower drug acquisition cost. As the drug is dispensed as a full script, adherence is unlikely to impact the cost incurred by public payers. | CDA-AMC changed the adherence rate to bulevirtide to 100%. | No scenario analysis was conducted because the most reflective scenario was captured in the revised base case. |
General coding errors. | The hazard ratios for suboptimal response were calculated incorrectly. Additionally, transcription errors were identified in efficacy parameter estimates. | CDA-AMC corrected the formulas to calculate the hazard ratio for suboptimal response and inserted the correct efficacy parameters as reported in the MYR301 trial. | As this is a correction, no scenario analysis was conducted. |
PEG-IFN-alpha is unlikely to be a relevant comparator. | Feedback CDA-AMC received from patient groups, clinician groups, and drug plans and from the clinical experts consulted by CDA‑AMC all indicated that PEG-IFN-alpha is often unavailable for the treatment of HDV in Canada. | CDA-AMC removed PEG-IFN-alpha as a comparator in the main analysis. | Scenario analysis presented PEG-IFN as a comparator. |
CDA-AMC = Canada’s Drug Agency; HDV = hepatitis delta virus; ICER = incremental cost-effectiveness ratio; PEG-IFN = pegylated interferon; QALY = quality-adjusted life-year.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 4.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 7), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
Bulevirtide is predicted to be associated with additional health care costs compared to best supportive care (incremental costs = $1,297,909). This increase in health care spending results from drug acquisition costs associated with bulevirtide and additional monitoring costs (refer to Figure 1). Total health state costs associated with managing various fibrosis stages, cirrhosis, hepatocellular carcinoma (HCC), and liver transplant are lower for bulevirtide compared to best supportive care. Overall, taking into account the increased monitoring costs and the lower health state costs, the total nondrug costs were similar across both treatments.
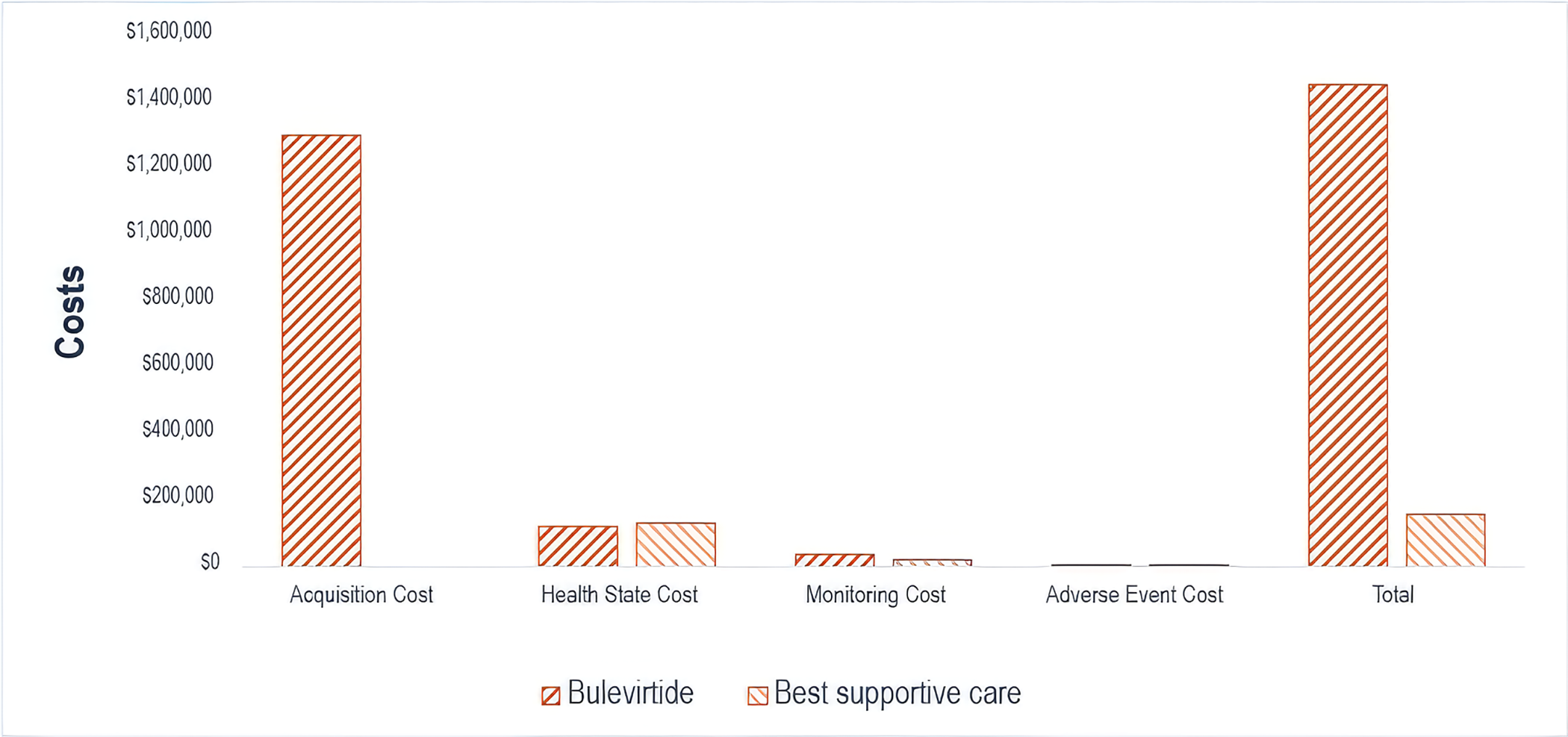
Impact on Health
Relative to best supportive care, bulevirtide is predicted to increase the amount of time a patient remains in noncirrhotic health states (i.e., fibrosis stage [F]0, F1, F2, and F3) by approximately 3.83 years more than best supportive care (refer to Figure 2). Considering the impact of treatment on both quality and length of life, bulevirtide is predicted to result in 3.38 additional QALYs per patient compared to best supportive care over the lifetime horizon. Approximately 99.1% of the predicted incremental benefit was accrued in the extrapolated period for which there are no trial data. Compared to best supportive care, bulevirtide was predicted to be associated with reduced clinical events, including a 19% reduction in decompensated cirrhosis, 19% reduction in liver transplants, and 16% reduction in liver-related mortality over the model lifetime horizon.
Figure 2: Impact of Bulevirtide vs. Best Supportive Care on Patient Health
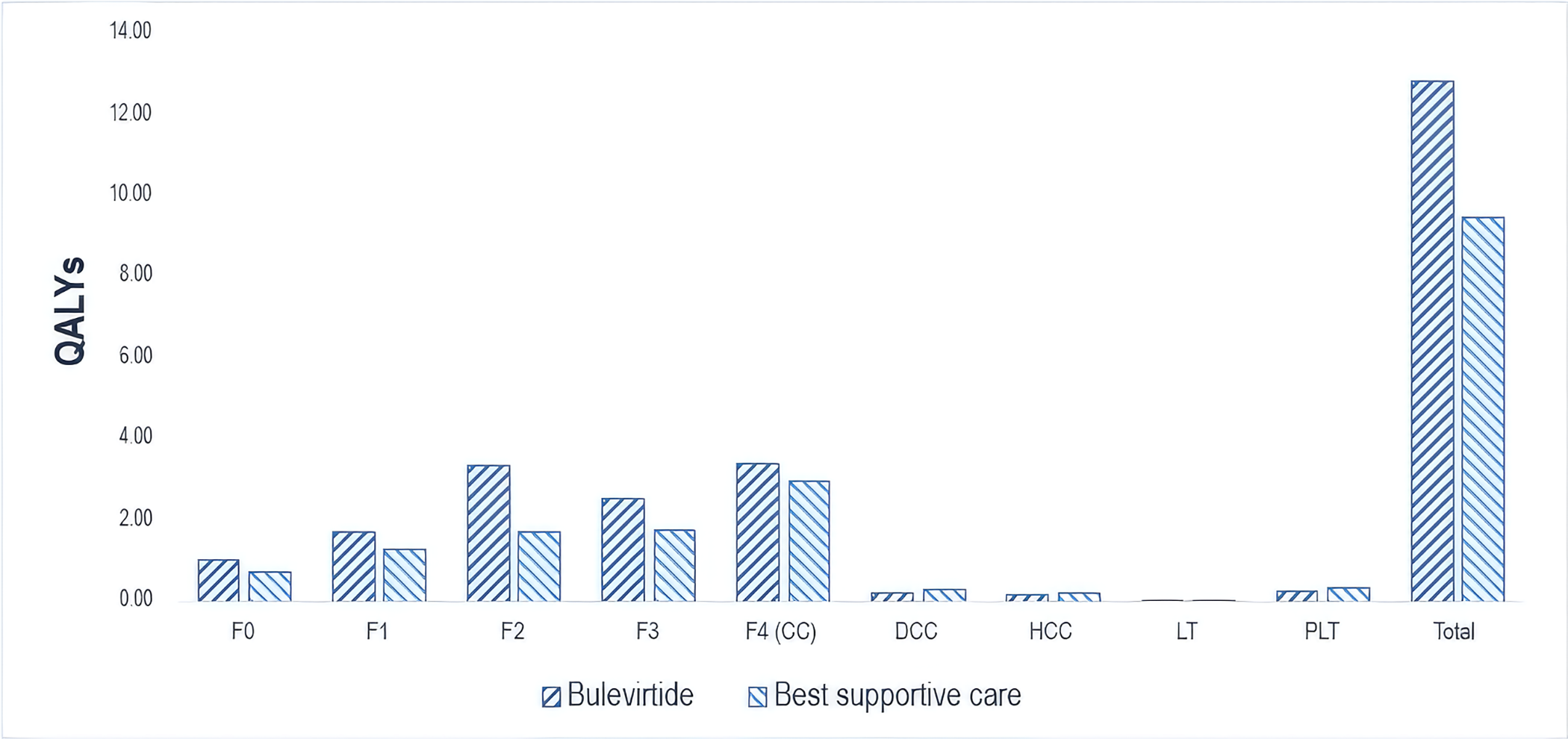
CC = compensated cirrhosis; DCC = decompensated cirrhosis; F0 = fibrosis stage 0; F1 = fibrosis stage 1; F2 = fibrosis stage 2; F3 = fibrosis stage 3; F4 = fibrosis stage 4; HCC = hepatocellular carcinoma; LT = liver transplant; PLT = post–liver transplant; QALY = quality-adjusted life-year; vs. = versus.
Overall Results
The results of the CDA-AMC base case suggest an ICER of $383,943 per QALY gained for bulevirtide compared to best supportive care (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | ICER vs. best supportive care ($/QALY) |
|---|---|---|---|
Best supportive care | $154,264 | 9.48 | Reference |
Bulevirtide | $1,452,173 | 12.86 | $383,943 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: Publicly available list prices were used for all comparators.
Uncertainty and Sensitivity
Uncertainty was explored through probabilistic analysis as well as through the scenario analyses outlined in Table 12 in Appendix 4. The submitted model assumes lifetime treatment duration for patients with response. As the major cost driver in the sponsor’s submitted model is the acquisition costs of bulevirtide, the model is sensitive to the expected duration of treatment. There is no evidence on the optimal treatment duration for patients with full or suboptimal response; the product monograph suggests treatment should be continued as long as associated with clinical benefit. The submitted model assumes that patients with response would continue to receive treatment all through the model’s lifetime horizon until discontinued based on a background discontinuation rate. It remains unclear how a shorter treatment duration for patients with full or suboptimal response may impact the ICER. While drug acquisition costs would decrease, there is lack of evidence on the clinical outcomes of patients with response after treatment discontinuation and, as such, the model does not account for possible re-treatment for patients who experience a disease relapse.
Uncertainty remains related to the impact of treatment adherence. While the CDA-AMC base case set adherence to 100% given that public payers are expected to cover the full cost of treatment, clinical expert feedback received by CDA‑AMC noted that adherence may be lower in clinical practice. As treatment efficacy informing the economic model was based on the adherence rate observed in the trial (i.e., mean adherence = 96.75%), it remains uncertain how a lower adherence rate in real practice may impact treatment effects and the predicted life-years or QALYs.
Summary of the Budget Impact
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year (2026 to 2028) budget impact of reimbursing bulevirtide for use in the Health Canada–indicated population. The sponsor assumed that the payer would be CDA‑AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of bulevirtide was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact and made changes to model parameters and assumptions in consultation with clinical experts to derive the CDA‑AMC base case (Appendix 5). CDA-AMC estimated that 352 patients would be eligible for treatment with bulevirtide over a 3-year period (year 1 = 73; year 2 = 125; year 3 = 154), of whom 282 are expected to receive bulevirtide (year 1 = 59; year 2 = 100; year 3 = 123). The estimated incremental budget impact of reimbursing bulevirtide compared with best supportive care (assumed to have no cost by the sponsor) is predicted to be approximately $45.0 million over the first 3 years. Clinical expert feedback received by CDA-AMC noted that there is likely to be a backlog of eligible patients. While the overall number of patients receiving treatment over the 3 years of the BIA model was a reasonable estimate, the distribution of patients receiving treatment may be higher in the first year than in subsequent years. Therefore, while the 3-year total budget impact is likely appropriate, the BIA model likely underestimates the budget impact in the first year and overestimates the budget impact in the second and third years.
Conclusion
Based on the CDA-AMC base case, bulevirtide would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $383,943 for each additional QALY gained. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 3; full details of the impact of price reductions on cost-effectiveness are presented in Table 11). There is uncertainty in the base-case analysis due to the lack of evidence to support model assumptions regarding duration of benefits beyond 144 weeks and the optimal duration of treatment. If the model extrapolation beyond 144 weeks has overestimated long-term efficacy, the ICER will increase. It remains unclear how a shorter duration of treatment beyond the model lifetime assumption for patients with a treatment response may impact the ICER. Although drug acquisition costs would be expected to decrease, the model does not adequately capture the relationship between treatment duration and efficacy as there is no evidence on clinical outcomes upon treatment discontinuation.
The incremental budget impact of reimbursing bulevirtide to the public drug plans in the first 3 years is estimated to be approximately $45.0 million. As the BIA assumed there is no additional drug acquisition costs associated with best supportive care, this also reflects the 3-year expenditure on bulevirtide.
Figure 3: Summary of the CDA-AMC Economic Analysis and Price Reduction
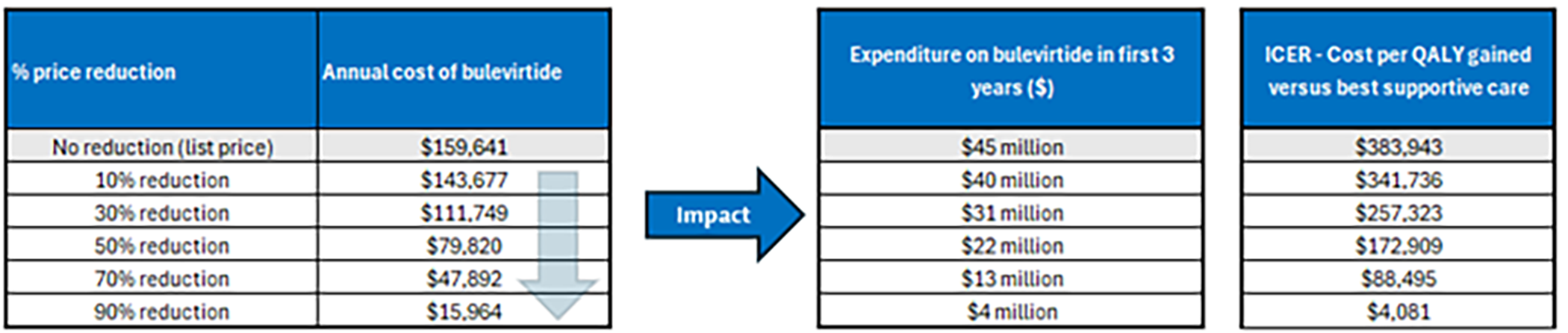
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; PEG-IFN-alpha = pegylated interferon-alpha; QALY = quality-adjusted life-year.
Note: Expenditure refers to only the drug cost of bulevirtide.
PEG-IFN-alpha was removed from consideration as a relevant comparator by CDA-AMC given that access to it is limited and it is rarely available for the treatment of HDV infection in Canada.
References
1.Gilead Sciences Canada Inc. PrHepcludex® (bulevirtide): Powder for solution for injection, 2 mg/vial (2 mg/mL when reconstituted), subcutaneous injection [DRAFT product monograph]. 2025. Updated April 25, 2025.
2.Gilead Sciences Canada Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: HEPCLUDEX® (bulevirtide), 2mg, powder for solution for subcutaneous injection [internal sponsor's package]. 2025.
3.Gilead Sciences, Inc. Clinical Study Report: A Multicenter, Open-label, Randomized Phase 3 Clinical Study to Assess Efficacy and Safety of Bulevirtide in Patients with Chronic Hepatitis Delta (MYR 301). April 2024.
4.Kaushik A, Dusheiko G, Kim C, et al. Understanding the Natural History of Chronic Hepatitis D: Proposal of a Model for Cost-Effectiveness Studies. PharmacoEconomics - Open. 2024;8(2):333-343. doi: doi:10.1007/s41669-023-00466-3 PubMed
5.Woo G, Tomlinson G, Yim C, et al. Health state utilities and quality of life in patients with hepatitis B. Can J Gastroenterol. 2012;26(7):445-51. doi: 10.1155/2012/736452 PubMed
6.Fattovich G. Natural history of hepatitis B. J Hepatol. 2003;39 Suppl 1:S50-8. doi: 10.1016/s0168-8278(03)00139-9 PubMed
7.H D, Fidler C, Harper. Mixed treatment comparison meta-analysis evaluating the relative efficacy of nucleos(t)ides for treatment of nucleos(t)ide-naive patients with chronic hepatitis B. Value in health: the journal of the International Society for Pharmacoeconomics and Outcomes Research. 2010;13(8). doi: 10.1111/j.1524-4733.2010.00777.x
8.D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217-31. doi: 10.1016/j.jhep.2005.10.013 PubMed
9.Ontario Go. Exceptional Access Program product prices. 2024.
10.CDA-AMC. Pharmacoeconomic Review Report TENOFOVIR ALAFENAMIDE (VEMLIDY). 2018. https://www.cda-amc.ca/sites/default/files/cdr/pharmacoeconomic/SR0537_Vemlidy_PE_Report.pdf
11.Alfaiate D, Clement S, Gomes D, Goossens N, Negro F. Chronic hepatitis D and hepatocellular carcinoma: A systematic review and meta-analysis of observational studies. J Hepatol. 2020;73(3):533-539. doi: 10.1016/j.jhep.2020.02.030 PubMed
12.Ek, V PG, Chrysanthos N, Hadziyannis E, Cholongitas E, Manesis. Longitudinal changes in serum HBV DNA levels and predictors of progression during the natural course of HBeAg-negative chronic hepatitis B virus infection. Journal of viral hepatitis. 2008;15(6). doi: 10.1111/j.1365-2893.2007.00957.x
13.Marcellin E, P, Gane E, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet (London, England). 2013;381(9865). doi: 10.1016/S0140-6736(12)61425-1 PubMed
14.Da BL, Heller T, Koh C. Hepatitis D infection: from initial discovery to current investigational therapies. Gastroenterol Rep (Oxf). 2019;7(4):231-245. doi: 10.1093/gastro/goz023 PubMed
15.C HS, Syu WJ, Sheen IJ, Liu HT, Jeng KS, Wu. Varied assembly and RNA editing efficiencies between genotypes I and II hepatitis D virus and their implications. Hepatology (Baltimore, Md). 2002;35(3). doi: 10.1053/jhep.2002.31777
16.Stoddard AM, Everhart JE, L DJ, et al. A prospective study of the rate of progression in compensated, histologically advanced chronic hepatitis C. Hepatology (Baltimore, Md). 2011;54(2). doi: 10.1002/hep.24370 PubMed
17.Fattovich G, Giustina G, Christensen E, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut. 2000;46(3):420-6. doi: 10.1136/gut.46.3.420 PubMed
18.K Z, Contag C, Whitaker E, Terrault. Spontaneous loss of surface antigen among adults living with chronic hepatitis B virus infection: a systematic review and pooled meta-analyses. The lancet Gastroenterology & hepatology. 2019;4(3). doi: 10.1016/S2468-1253(18)30308-X PubMed
19.Gish Robert P, G, Wong RJ, et al. Association of hepatitis delta virus with liver morbidity and mortality: A systematic literature review and meta-analysis. Hepatology (Baltimore, Md). 2024;79(5). doi: 10.1097/HEP.0000000000000642 PubMed
20.Wedemeyer H, Aleman S, Brunetto M, et al. Efficacy and safety of bulevirtide monotherapy given at 2 mg or 10 mg dose level once daily for treatment of chronic hepatitis delta: week 48 primary end point results from a phase 3 randomized, multicenter, parallel design study. Poster #GS006. Presented at The International Liver Congress (EASL): June 2022; London, UK.
21.MYR Pharmaceuticals. Clinical Study Report: A multicenter, open-label, randomized clinical study to assess efficacy and safety of 3 doses of Myrcludex B for 24 weeks in combination with Tenofovir compared to Tenofovir alone to suppress HBV replication in patients with chronic hepatitis D (MYR 202). September 2019.
22.MYR Pharmaceuticals. Clinical Study Report: A multicentre, open-label, randomized, comparative, parallel-arm phase II study to assess efficacy and safety of Myrcludex B in combination with peginterferon alfa-2a versus peginterferon alfa-2a alone in patients with chronic viral hepatitis B with delta-agent (MYR 203). December 2020.
23.A Multicenter, Open-label, Randomised Phase IIb Clinical Study to Assess Efficacy and Safety of Bulevirtide in Combination with Pegylated Interferon alfa-2a in Patients with Chronic Hepatitis Delta (MYR 204). 2021.
24.Canada S. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island (2018-2020). 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
25.Incorporated GS. Data on File. Meta-analyses of Health State Utilities in HCV and HBV. 2022.
26.Guertin JJ, R J, Feeny D, Tarride. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ: Canadian Medical Association journal = journal de l'Association medicale canadienne. 2018;190(6). doi: 10.1503/cmaj.170317
27.W SP, Ghushchyan. Preference-Based EQ-5D index scores for chronic conditions in the United States. Medical decision making: an international journal of the Society for Medical Decision Making. 2006;26(4). doi: 10.1177/0272989X06290495
28.Elsaid MI, Li Y, John T, Narayanan N, Catalano C, Rustgi VK. Economic and Health Care Burdens of Hepatitis Delta: A Study of Commercially Insured Adults in the United States. Hepatology. 2020;72(2):399-411. doi: 10.1002/hep.31055 PubMed
29.A E, Sahakyan Y, Everett K, et al. Hepatitis C Attributable Healthcare Costs and Mortality among Immigrants: A Population-Based Matched Cohort Study. Canadian journal of gastroenterology & hepatology. 2024;2024. doi: 10.1155/2024/5573068
30.Biondi J, J M, Estes C, et al. Cost-effectiveness modelling of birth and infant dose vaccination against hepatitis B virus in Ontario from 2020 to 2050. CMAJ open. 2023;11(1). doi: 10.9778/cmajo.20210284 PubMed
31.Information CIfH. Patient Cost Estimator. 2022. https://www.cihi.ca/en/patient-cost-estimator
32.Ontario Go. Schedule of Benefits for Physician Services. 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
33.Ontario Go. Schedule of Benefits for Laboratory Services. 2024. https://www.ontario.ca/page/ohip-schedule-benefits-and-fees#section-6
34.Cihi. Comprehensive ambulatory care classification system. 2022. https://www.cihi.ca/en/comprehensive-ambulatory-classification-system-cacs
35.Yan J, Xie S, Johnson JA, et al. Canada population norms for the EQ-5D-5L. Eur J Health Econ. 2024;25(1):147-155. doi: 10.1007/s10198-023-01570-1 PubMed
36.R, Del Ninno E, Rumi M, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136(5). doi: 10.1053/j.gastro.2009.01.052 PubMed
37.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1, by age and gender. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501.
38.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2022 to 2023. Accessed at: https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612.
39.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2021 to 2022. Accessed at: https://www.sac-isc.gc.ca/eng/1713194236054/1713194280612.
40.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2020 to 2021. Accessed at: https://www.sac-isc.gc.ca/eng/1645718409378/1645718500555.
41.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2019 to 2020. Accessed at: https://www.sac-isc.gc.ca/eng/1624462613080/1624462663098.
42.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2018 to 2019. Accessed at: https://www.sac-isc.gc.ca/DAM/DAM-ISC-SAC/DAM-HLTH/STAGING/texte-text/nihb-Annual_Report_2018-19_1589921777815_eng.pdf.
43.Adjusted estimate of the prevalence of hepatitis delta virus in 25 countries and territories. J Hepatol. 2024;80(2):232-242. doi: 10.1016/j.jhep.2023.10.043 PubMed
44.Roulot D, Brichler S, Layese R, et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic hepatitis delta. J Hepatol. 2020;73(5):1046-1062. doi: 10.1016/j.jhep.2020.06.038 PubMed
45.Sutherland, Greg, and Thy Dinh. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. Published in Canada.
46.Osiowy C, Swidinsky K, Haylock-Jacobs S, et al. Molecular epidemiology and clinical characteristics of hepatitis D virus infection in Canada. JHEP Rep. 2022;4(5):100461. doi: 10.1016/j.jhepr.2022.100461 PubMed
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in Table 4 have been deemed to be appropriate based on feedback from clinical experts and CDA‑AMC–participating public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: Cost Comparison for Chronic HDV
Treatment | Strength and/or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Bulevirtide (Hepcludex) | 2 mg | Single dose vial for SC injection | $437.0729 | 2 mg/mL daily | $437.07 | $159,640.88 |
Off-label treatment | ||||||
PEG-IFN-alpha | 180 mcg | Prefilled syringe | $436.08009 | Once weekly | $62.30 | $22,755 |
HDV = hepatitis delta virus; PEG-IFN = pegylated interferon; SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed June 2025), unless otherwise indicated, and do not include dispensing fees.
aOne year is assumed to be 365.25 days or 52.18 weeks.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA‑AMC review process.
Patient input was received from Liver Canada and the BC Hepatitis Network. The information was collected using an online survey between February 21 and March 8, 2025. Six adults in Canada aged between 30 and 69 years who were either a patient living with chronic HDV or a caregiver of such patient participated in the survey. Patients noted that this condition impacts their quality of life due to fatigue, lack of appetite, and other gastrointestinal symptoms, and impacts their social life and work life, and their emotional and psychological well-being. While PEG-IFN-alpha is noted as the only treatment option in Canada for patients with HDV, it is difficult to access with most provincial special access requests denied. Among patients who have experience with PEG‑IFN‑alpha, side effects were described as intolerable. One patient received access to bulevirtide through compassionate access from the manufacturer, and side effects were reported as minimal. Additional data from the poster Bulevirtide for the Treatment of Hepatitis Delta: Real-World Cohort from the Canadian HBV Network, presented by Dr. Sebastien Poulin and colleagues at the Canadian Liver Meeting in February 2025, indicate that bulevirtide was well-tolerated by a cohort of 13 patients in Canada, with most showing improvements in liver health and reduced HDV RNA.
Clinician group input was received from the Canadian Hepatitis B Network. The information was gathered via teleconference, email correspondence, and face-to-face meeting at the annual Canadian Liver Meeting. Patients with HDV-hepatitis B virus (HBV)‑coinfection have a significant risk of developing liver disease and cirrhosis within the initial 5 to 10 years of infection. PEG‑IFN‑alpha is an off-label treatment but is poorly tolerated, usually ineffective, and difficult to procure. PEG‑IFN-alpha is not recommended for patients with decompensated cirrhosis. It was noted that clinical end points, alanine aminotransferase normalization and viral suppression, are indicators of significant clinical benefits. Patients eligible for bulevirtide should be identified by specialists in hepatology or infectious disease, and diagnosis would require HDV RNA testing that is currently performed only at the National Reference Laboratory.
Input from CDA-AMC-participating drug plans inquired on the most relevant comparator in practice in Canada. It was noted that PEG‑IFN-alpha is covered as restricted benefit and, among plans that do fund this treatment, treatment is often limited to patients with hepatitis B. Questions were raised on how to define adequate response given the indication suggest treatment should be as long as there is associated clinical benefit and with concerns raised on criteria for treatment discontinuation. Plans noted that training is required on how to reconstitute and administer this product. Uncertainty remains to who would cover such costs along with the expected cost of supplies (e.g., needles).
Several of these concerns were addressed in the sponsor’s model:
The impact of chronic HDV infection on patient’s quality of life was captured through utility values measured through EQ‑5D which captures health domains including usual activities, pain and discomfort, and anxiety and depression.
Treatment efficacy within the submitted model was defined as combined response which considered both the end points of alanine aminotransferase normalization as well as viral suppression.
Training costs were included in the submitted model and assumed to be covered by public health care payers.
CDA-AMC addressed some of these concerns as follows:
Inaccessibility of PEG-IFN-alpha in Canada was addressed by removing PEG-IFN-alpha from the base-case analysis. As such, the reanalysis compared bulevirtide to best supportive care.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA‑AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA‑AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of bulevirtide, the sponsor provided a cost-utility analysis and a BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Bulevirtide (Hepcludex), subcutaneous injection (2 mg/mL) |
Submitted price of drug under review | Bulevirtide: $437.07 per 2 mg vial |
Regimen | 2 mg once daily subcutaneous injection |
Annual cost of drug under review | $159,640.88 per patient |
Model information2 | |
Type of economic evaluation | Cost-utility analysis Markov cohort model |
Treatment | Bulevirtide |
Included comparator(s) |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | Lifetime (59.8 years) |
Cycle length | Markov model: 24-weeks |
Modelled population(s) | Overall population: adults with compensated liver disease when the presence of viral HDV RNA has been confirmed by blood tests |
Characteristics of modelled population | Derived from the MYR301 trial with 56% males with a mean age of 42 years Baseline fibrosis distribution:
|
Model health states |
For additional information, refer to Model Structure. |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway | Natural history of disease:
Health state transitions:
|
Health-related utilities and disutilities |
|
Costs |
|
Summary of the submitted resultsa | |
Base case results | Bulevirtide was associated with an ICER of $350,046 per QALY gained compared to PEG-IFN-alpha (incremental costs = $1,403,673; incremental QALYs = 4.01).2 |
F0 = fibrosis stage 0; F1 = fibrosis stage 1; F2 = fibrosis stage 2; F3 = fibrosis stage 3; F4 = fibrosis stage 4; HBV = hepatitis B virus; HCV = hepatitis C virus; HDV = hepatitis delta virus; ICER = incremental cost-effectiveness ratio; NMA = network meta-analysis; PEG-IFN-alpha = pegylated interferon-alpha; QALY = quality-adjusted life-year; RNA = ribonucleic acid.
aScenarios were submitted by the sponsor. These had no meaningful impact on the estimated ICER.
Model Structure
Health states in the Markov model include fibrosis stages (i.e., F0, F1, F2, F3), compensated cirrhosis (F4), decompensated cirrhosis, HCC, liver transplant, post–liver transplant, and death (Figure 4).2 Patients who respond can progress or regress through F0 to F4 with the rate of progression reduced for both those with combined response and those with suboptimal response. Those who do not respond can progress through F0 to F4 and cannot regress. Patients with compensated cirrhosis can progress to decompensated cirrhosis. Patients in fibrosis or compensated cirrhosis health states can develop HCC. From the decompensated cirrhosis health state, patients can move to HCC or liver transplant while patients in HCC can move to liver transplant. Patients alive 1 year after liver transplant will move to post-liver transplant. Patients can experience liver-related death from F3 onwards. Patients in any model health state can transition to death related to background mortality.2
Figure 4: Markov Model Structure
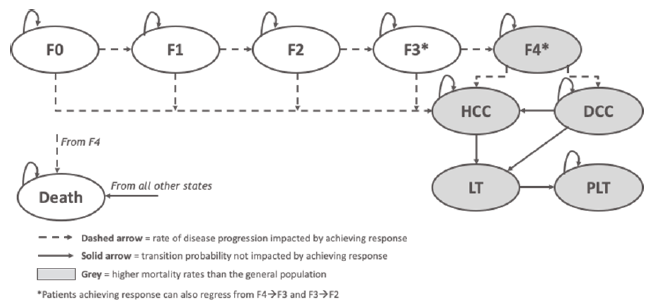
F0 = fibrosis stage 0; F1 = fibrosis stage 1; F2 = fibrosis stage 2; F3 = fibrosis stage 3; CC = compensated cirrhosis; DCC = decompensated cirrhosis; HCC = hepatocellular carcinoma; LT = liver transplant; PLT = post-liver transplant.
Source: Sponsor’s pharmacoeconomic submission2
Table 6: Summary of the Sponsor's Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/ QALY) |
|---|---|---|---|
Best supportive care | $142,661 | 8.99 | Reference |
PEG-IFN-alpha | $160,636 | 9.40 | $43,942 |
Bulevirtide | $1,564,310 | 13.41 | $350,046 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC Clinical Review found that, based on the MYR301 trial, treatment with bulevirtide 2 mg likely results in a clinically meaningful increase in achieving combined response and virologic response at week 48, compared to delayed treatment. No definitive conclusion can be drawn regarding the effects of bulevirtide treatment on health-related quality of life.
Key clinical efficacy inputs for bulevirtide were derived from the MYR301 trial. The sponsor used the proportion of combined response (i.e., defined as HDV RNA undetectability or HDV RNA > 2 log10 decline from baseline and alanine aminotransferase normalization) and suboptimal response (i.e., HDV RNA > 2 log10 decline from baseline) as reported in the MYR301 trial.2 Clinical outcomes reported for the delayed-treatment arm of MYR301 trial informed the efficacy of the best supportive care arm within the submitted model.2 Among those with combined response and those with suboptimal response who remained on treatment, treatment effects were assumed to persist over the patient’s lifetime. If patients discontinued treatment, disease progression would align with the expected natural history of HDV.2 Notably, based on the probabilistic results of the sponsor’s base-case analysis, approximately 98% of the incremental QALYs for bulevirtide were found to be accrued during the extrapolated period. Several sources of uncertainty related to the clinical efficacy inputs in the economic model were noted (refer to Key Issues of the Submitted Economic Evaluation). As such, the estimated incremental gain in life-years and QALYs predicted by the sponsor’s model for bulevirtide are highly uncertain.
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
Inability to explore the impact of treatment duration on the long-term comparative efficacy of bulevirtide: The long-term impact of bulevirtide remains uncertain as the comparative evidence between bulevirtide and the delayed-treatment group, informing the best supportive care arm of the model, is limited to 48-weeks. This reflects the duration of the comparative phase of the MYR301 trial.3 While there is clinical evidence up to 144 week, this is only for patients remaining on bulevirtide given the trial’s crossover design reassigned the delayed-treatment group to receive bulevirtide 10 mg after 48 weeks.3 Therefore, this precludes the ability to understand the comparative clinical benefits beyond 48 weeks. This is particularly of concern as nearly all (98.0%) of the incremental benefits were accrued after the treatment duration of MYR301 trial.
The duration in which patients remain on treatment could potentially impact the expected efficacy of bulevirtide; yet assumptions made by the sponsor on treatment duration and its impacts on clinical outcomes within the model are unlikely to reflect real-world practice. The product monograph states that the optimal treatment duration for bulevirtide is unknown with treatment continued as long as it is associated with clinical benefit.1 Within the sponsor’s submitted model, treatment duration for those who did not respond was assumed to be 144 weeks while those with response remained on treatment until discontinuation, with a constant discontinuation rate applied regardless of response status based on the discontinuation rates reported in MYR203 and MYR301 trials.2 According to clinical expert input received by CDA-AMC, the absence of clinical benefit (defined as less than 1 log10 reduction in HDV RNA) should be identified after 48 weeks of treatment.2 Physicians and patients would most likely decide to discontinue treatment between 48 and 72 weeks in such instances. It remains unclear what the optimal treatment duration would be for those with combined and suboptimal response. As those with combined and suboptimal response were assumed to remain on bulevirtide unless they had discontinued, the submitted model predicts that, after 5 years, 39.5% of those with response and 10.4% of those with suboptimal response would remain on treatment and, after 10 years 28.2% of those with response and 6.5% of those with suboptimal response would still be on treatment. Clinical expert feedback sought by CDA-AMC noted that for other hepatic therapies, such as obeticholic acid and nucleic acid, discontinuation criteria are present. However, given the potential risk of viral rebound for this condition that may lead to clinical deterioration, the optimal time point to discontinue therapy remains unclear. If the duration of therapy is shorter than modelled, drug acquisition costs would be expected to decrease although there is a lack of clinical evidence on the expected outcome of such patients. Clinical expert feedback received by CDA-AMC noted a possibility for viral and clinical rebound and, in cases where patients initially responded to treatment but experienced a disease relapse after discontinuing treatment, re-treatment may be reasonable although data are lacking to guide such decisions. The model does not explore the potential relationship between treatment discontinuation and viral or clinical rebound. The possibility of re-treatment with bulevirtide is also not modelled.
CDA-AMC partly addressed this limitation by revising the duration of treatment for those who did not respond to 72 weeks with a scenario analysis conducted at 48 weeks. Despite this correction, CDA-AMC cautions interpreting the revised ICER. The assumptions on treatment duration within the model are unlikely to reflect the expected use of bulevirtide in the settings in Canada and, as such, its associated clinical outcomes and potential costs (e.g., re-treatment).
Uncertainty in the model’s utility values: Utilities were sourced from an HBV-hepatitis C virus (HCV) meta-analysis in which those with combined response were assigned a utility increment of 0.06 within each health state.25 This therefore assumes patients who completely respond to treatment and occupy the same fibrosis stage would have a different quality of life than those who do not respond or who have suboptimal response in the same fibrosis stage. This was based on limited evidence as the meta-analysis that informed the sponsor’s estimates reported utility values separately by virological response and by fibrosis stage. These study authors did not report a utility difference by response status within the same fibrosis health state. Clinical expert feedback sought by CDA-AMC suggested that quality of life would be expected to differ by fibrosis stage; however, there is limited validity that those who respond would have substantially different quality of life compared to those who do not respond or those who have suboptimal response within the same fibrosis stage. The application of response-based utility values is expected to favour bulevirtide given bulevirtide has a higher response rate than best supportive care.
The selected health utilities further lacked face validity. The utility values for noncirrhotic fibrosis were higher than those reported for a general age-matched population in Canada.35 According to clinical expert input, this is implausible as patients, even with noncirrhotic fibrosis, would be expected to have a higher disease burden than the general population in Canada. While the sponsor adjusted utilities by applying an age-adjusted utility decrement, the health state utility estimates were still considered to be too high. For instance, patients with noncirrhotic fibrosis and compensated cirrhosis would have only a marginal reduction in quality of life (i.e., 91% and 85%, respectively). While published literature report heterogeneous utility values by health states, preference was toward using an estimate specific to Canada.
The CDA-AMC reanalysis assumed all patients within the same health state would have identical utility values, regardless of response status. Utility values for noncirrhotic and compensated cirrhosis health states were further revised based on the values reported in a study conducted in Canada by Woo et al. (2012).5 As there were no published utility values for patients with HDV for more advanced liver disease, clinical expert feedback obtained by CDA-AMC recommended to use patients with chronic HBV as a suitable proxy. As the revised utility values from Woo et al. were based on the health utility index instrument, utility weights also based on the same instrument for the compensated cirrhosis and noncirrhotic health states were further incorporated as part of the CDA-AMC base case for the sake of consistency across utility instruments.4 Of note, the revisions made by CDA-AMC still predicts an improvement in health-related quality of life despite the fact that the clinical review noted that no definitive conclusion can be drawn regarding the effects of bulevirtide treatment on health-related quality of life.
Disease-specific mortality underestimated: The sponsor conducted a literature search to inform disease progression using published literature on the natural history of chronic HDV. Specifically, the annual transition rates from compensated cirrhosis to death and from decompensated cirrhosis were 7.26% and 15.6%, respectively, based on a publication by Fattovich (2003)6 and Dakin et al. (2010).7 Clinical experts consulted by CDA-AMC noted that these mortality rates were implausibly low. CDA-AMC conducted a review of the literature to identify other publications that reported on the natural history of cirrhosis, and clinical experts consulted by CDA-AMC suggested revising these rates to those published by D’Amico et al. (2006).8
The CDA-AMC reanalysis revised transition probabilities based on the values published by D’Amico et al. (2006). D’Amico reported the annual rate from compensated cirrhosis to death (separately for stage 1 and 2 compensated cirrhosis) and decompensated cirrhosis to death (separately for stage 3 and 4 decompensated cirrhosis).8 As there was insufficient evidence to inform a weighted average of the transition probabilities (i.e., combined mortality based on stages 1 and 2; combined mortality based on stages 3 and 4), CDA-AMC selected the stage 2 value (3.4%) for compensated cirrhosis to death and stage 3 value (20.0%) for decompensated cirrhosis to death as these values also aligned with the mortality rates from another publication on the natural history of HDV.36
Health care resource utilization not reflective of expected clinical practice: The sponsor-submitted model used uncited HDV treatment guidelines and recommendations as well as expert opinion to inform off-treatment and on-treatment annual health care resource utilization.2 Based on input from clinical experts sought by CDA-AMC, the sponsor’s approach underestimated health care resource utilization. First, training for reconstitution and self-administration for bulevirtide was assumed to require 4 hours of nursing time, yet clinical experts indicated that 12 to 16 hours of nurse time is likely more reflective of clinical practice. Clinical experts further stated that, based on their experience with interferon treatments, patients have required assistance on treatment administration for at least a few weeks with some patients requiring long-term assistance. This is also expected for bulevirtide as it is not available as a prefilled syringe but would require reconstitution.
Clinical expert feedback obtained by CDA-AMC also noted that monitoring costs were not reflective of current treatment practices and were underestimated. Based on the health care resource utilization estimates used in the sponsor’s economic model, clinical experts stated that they would perform blood work at least 2 times more per year for patients in noncirrhotic fibrosis states and 3 times more per year for patients with compensated cirrhosis. They would provide double the number of follow-up appointments and perform a FibroScan once per year for all patients with HDV. They also stated that they expected patients to attend a hepatologist visit and perform DNA and RNA tests 4 times per year during the first year of treatment.
CDA-AMC revised nursing training time to 14 hours and updated health care utilization values based on clinical expert feedback (refer to Table 8).
Impact of treatment adherence to bulevirtide not fully captured within the model: The sponsor estimated a treatment adherence rate of 97.96% for bulevirtide based on the MYR301 trial.2 Clinical expert feedback sought by CDA-AMC noted that treatment adherence may be lower in the real-world setting as trial patients are highly motivated and more likely to adhere than patients in real-world practice. The sponsor’s model only captured the impact of nonadherence on treatment acquisition costs and did not fully consider the impact of nonadherence on treatment effectiveness. Revising the adherence rate in the model would not have any impact on the efficacy of treatment. As noted from clinical expert feedback received by CDA-AMC, poor adherence may lead to viral rebound. Savings from nonadherence rate (i.e., lowered treatment acquisition costs) may not be realized by public drug plans given that prescriptions are dispensed to cover a specified duration of therapy.
Owing to a lack of clinical data about the impact of treatment adherence on clinical outcomes, CDA-AMC was unable to assess the impact of changes in treatment adherence on the cost-effectiveness estimates. CDA-AMC revised treatment adherence to 100% given uncertainty as to whether savings on drug acquisition costs would be realized from nonadherence.
Poor modelling practices: The model incorrectly calculated the suboptimal response hazard ratios. The sponsor assumes that suboptimal response experience 0.5 times the benefit of combined response (𝑠𝑢𝑏𝑜𝑝𝑡𝑖𝑚𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 = 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 ∗ (1 + 0.5)).2 This formula does not derive estimates according to the sponsor’s intent (the correct formula should have been (𝑠𝑢𝑏𝑜𝑝𝑡𝑖𝑚𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 = 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 + (1 – 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅) ∗ (1 – 0.5)). The sponsor’s submitted model further incorrectly transcribed the combined and suboptimal response parameter values for bulevirtide and best supportive care from the MYR301 trial data.
CDA-AMC corrected the suboptimal response hazard ratio equation and updated the parameter values for combined and suboptimal response based on the values reported in the MYR301 trial. CDA-AMC did not validate the confidence interval values for suboptimal response given the sponsor applied an arbitrary 10% variation. As such, the probabilistic results from both the sponsor and CDA-AMC may not reflect the true parameter uncertainty associated with these parameter estimates.
Additional issues were identified but were not considered to be key issues:
Relevance of PEG-IFN-alpha as a comparator: Clinical experts expressed that PEG-IFN-alpha is not often available in clinical practice (outside of clinical trials) for the treatment of HDV in Canada. This is aligned with the feedback CDA-AMC received from patient groups, clinician groups, and drug plans.
In accordance with all the input received regarding the availability of PEG-IFN-alpha for patients with HDV infection, CDA‑AMC has removed PEG-IFN-alpha as a comparator.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 7). The impact of these changes, individually and collectively, is presented in Table 9.
Table 7: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Duration of bulevirtide treatment | Nonresponse treatment duration (144 weeks) | Nonresponse treatment duration (72 weeks) |
2. Utility values | Response QALY gain (0.06; 95% CI, −0.1 to 0.22) NC (0.91; 95% CI, 0.86 to 0.93) CC (0.85; 95% CI, 0.79 to 0.97) DCC (0.46; 95% CI, 0.41 to 0.51) HCC (0.52; 95% CI, 0.47 to 0.57) | Response QALY gain (0; 95% CI, 0 to 0) NC (0.87; 95% CI, 0.85 to 0.88) CC (0.81; 95% CI, 0.75 to 0.86) DCC (0.35; 95% CI, 0.31 to 0.40) HCC (0.49; 95% CI, 0.23 to 0.75) |
3. Natural history of disease transition rates | CC to death (7.26%) DCC to death (15.6%) | CC to death (3.4%) DCC to death (20%) |
4. Health care resource utilization | Self-administration instruction nurse time (4 hours) Health care utilization as per sponsor | Self-administration instruction nurse time (14 hours) Health care utilization (refer to Table 8) |
5. Treatment adherence | 97.96% | 100% |
6. Corrections | Suboptimal response: 𝑠𝑢𝑏𝑜𝑝𝑡𝑖𝑚𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 = 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 ∗ (1 + 0.5) Bulevirtide response:
Bulevirtide suboptimal response:
Best supportive care response:
Best supportive care suboptimal response:
| Suboptimal response: 𝑠𝑢𝑏𝑜𝑝𝑡𝑖𝑚𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 = 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅 + (1 – 𝑟𝑒𝑠𝑝𝑜𝑛𝑑𝑒𝑟 𝐻𝑅) ∗ (1 – 0.5) Bulevirtide response:
Bulevirtide suboptimal response:
Best supportive care response:
Best supportive care suboptimal response:
|
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
CC = compensated cirrhosis; CDA-AMC = Canada’s Drug Agency; CI = confidence interval; DCC = decompensated cirrhosis; HCC = hepatocellular carcinoma; HR = hazard ratio; NC = noncirrhotic; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
CDA-AMC was unable to resolve the issues with long-term efficacy estimates of bulevirtide and the expected duration of treatment for those who respond as there is no evidence. A large majority of the incremental clinical benefits are derived from the extrapolation period.
Table 8: Revisions to the Submitted Economic Evaluation — Health Care Utilization
Event | Treatment initiation | During treatment (frequency per year) | Off treatment (frequency per year) | |||
|---|---|---|---|---|---|---|
NC | CC | NC | CC | NC | CC | |
Hepatologist visit | 1 | 2 | 4 | 4 | 1 | 2 |
Outpatient visit | 1 | 1 | 1 | 2 | 1 | 2 |
FibroScan-liver biopsy | 1 | 1 | 1 | 1 | 1 | 1 |
HBV DNA test | 2 | 3 | 4 | 4 | 4 | 6 |
HDV RNA test | 2 | 3 | 4 | 6 | 2 | 3 |
Liver enzyme test | 2 | 6 | 4 | 9 | 4 | 6 |
Complete blood count | 2 | 6 | 4 | 9 | 4 | 6 |
Renal function test | 2 | 6 | 4 | 9 | 4 | 6 |
Bilirubin test, GGT, ALP test | 2 | 6 | 4 | 9 | 4 | 6 |
Ultrasound for HCC screening and surveillance | 1 | 1 | 2 | 2 | 2 | 2 |
Protime-INR | 2 | 6 | 2 | 9 | 2 | 6 |
anti-HDV IgG | 2 | 3 | 0 | 0 | 0 | 0 |
HBsAg | 2 | 3 | 2 | 3 | 2 | 3 |
Alpha-feto protein | 2 | 3 | 4 | 6 | 4 | 6 |
HCV Ab | 2 | 3 | 0 | 0 | 0 | 0 |
HIV Ab | 2 | 3 | 0 | 0 | 0 | 0 |
Hep A IgG | 2 | 3 | 0 | 0 | 0 | 0 |
ALP = alkaline phosphatase; CC = compensated cirrhosis; GGT = gammaglutamyltransferase; HBsAg = hepatitis B surface antigen; HBV = hepatitis B virus; HCC = hepatocellular carcinoma; HCV = hepatitis C virus; IgG = immunoglobulin G; HDV = hepatitis delta virus; hep A = hepatitis A virus; INR = International Normalized Ratio; NC = noncirrhotic; RNA = ribonucleic acid.
Table 9: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Best supportive care | $142,661 | 8.99 | Reference |
Bulevirtide | $1,564,310 | 13.41 | $321,710 | |
Sponsor’s base case (deterministic) | Best supportive care | $142,522 | 8.98 | Reference |
Bulevirtide | $1,525,977 | 13.25 | $323,908 | |
CDA-AMC reanalysis 1 | Best supportive care | $142,522 | 8.98 | Reference |
Bulevirtide | $1,382,217 | 12.73 | $330,945 | |
CDA-AMC reanalysis 2 | Best supportive care | $142,522 | 9.03 | Reference |
Bulevirtide | $1,525,977 | 13.06 | $343,899 | |
CDA-AMC reanalysis 3 | Best supportive care | $147,541 | 9.41 | Reference |
Bulevirtide | $1,575,884 | 13.74 | $330,158 | |
CDA-AMC reanalysis 4 | Best supportive care | $148,478 | 8.98 | Reference |
Bulevirtide | $1,547,187 | 13.25 | $327,479 | |
CDA-AMC reanalysis 5 | Best supportive care | $142,522 | 8.98 | Reference |
Bulevirtide | $1,554,850 | 13.25 | $330,668 | |
CDA-AMC reanalysis 6 | Best supportive care | $142,553 | 8.96 | Reference |
Bulevirtide | $1,490,595 | 13.01 | $332,701 | |
CDA-AMC base case: Reanalysis 1 + 2 + 3 + 4 + 5 + 6 (deterministic) | Best supportive care | $154,018 | 9.47 | Reference |
Bulevirtide | $1,426,792 | 12.76 | $386,652 | |
CDA-AMC base case Reanalysis 1 + 2 + 3 + 4 + 5 + 6 (probabilistic) | Best supportive care | $154,264 | 9.48 | Reference |
Bulevirtide | $1,452,173 | 12.86 | $383,943 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 10: Disaggregated Results of the CDA-AMC Probabilistic Base Case
Parameter | Bulevirtidea | Best supportive carea |
|---|---|---|
Discounted LYs | ||
Total | 16.53 | 12.52 |
F0 | 1.24 | 0.88 |
F1 | 2.08 | 1.57 |
F2 | 4.12 | 2.09 |
F3 | 3.08 | 2.15 |
F4 (CC) | 4.43 | 3.82 |
DCC | 0.71 | 0.92 |
HCC | 0.43 | 0.51 |
LT | 0.02 | 0.02 |
PLT | 0.42 | 0.56 |
Discounted QALYs | ||
Total | 12.86 | 9.48 |
F0 | 1.04 | 0.74 |
F1 | 1.74 | 1.32 |
F2 | 3.39 | 1.75 |
F3 | 2.54 | 1.79 |
F4 (CC) | 3.43 | 2.97 |
DCC | 0.24 | 0.31 |
HCC | 0.20 | 0.24 |
LT | 0.01 | 0.01 |
PLT | 0.26 | 0.35 |
Discounted costs | ||
Total cost | $1,452,173 | $154,264 |
Acquisition cost | $1,298,645 | $0 |
Health state cost | $118,827 | $132,573 |
Monitoring cost | $34,636 | $21,261 |
Adverse event cost | $65 | $430 |
CC = compensated cirrhosis; CDA-AMC = Canada’s Drug Agency; DCC = decompensated cirrhosis; F0 = fibrosis stage 0; F1 = fibrosis stage 1; F2 = fibrosis stage 2; F3 = fibrosis stage 3; F4 = fibrosis stage 4; HCC = hepatocellular carcinoma; LT = liver transplant; LY = life-year; PLT = post–liver transplant; QALY = quality-adjusted life-year.
Figure 5: Costs of Bulevirtide vs. Best Supportive Care by Health State
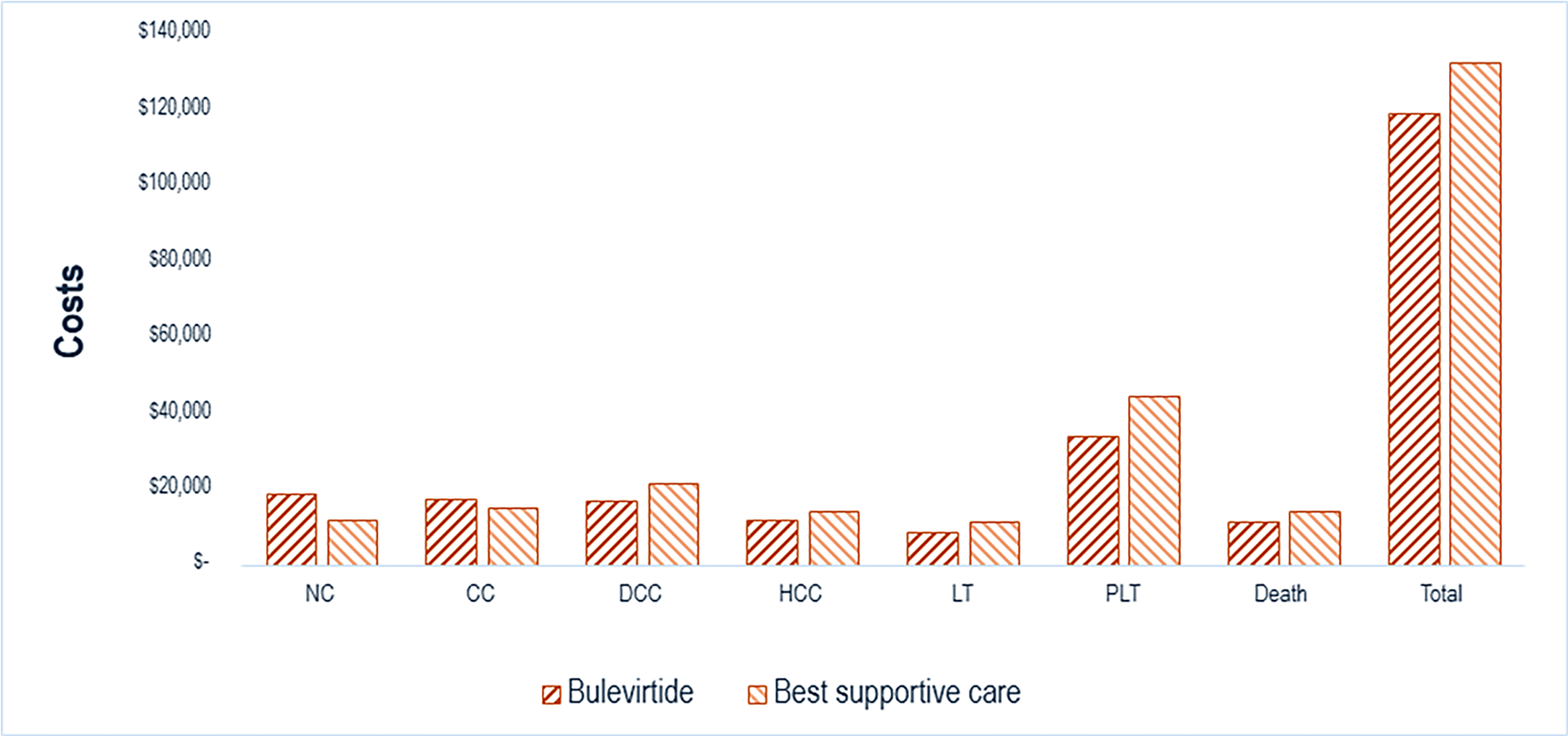
CC = compensated cirrhosis; DCC = decompensated cirrhosis; HCC = hepatocellular carcinoma; LT = liver transplant; NC = noncirrhotic; PLT = post-liver transplant; vs. = versus.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 11).
Table 11: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Annual cost ($) | ICERs for bulevirtide vs. best supportive care ($/QALY) | |
|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | |||
No price reduction | 437.07a | 159,641 | 321,710 | 383,943 |
10% | 393.36 | 143,677 | 287,178 | 341,736 |
20% | 349.66 | 127,713 | 252,646 | 299,529 |
30% | 305.95 | 111,749 | 218,115 | 257,323 |
40% | 262.24 | 95,785 | 183,583 | 215,116 |
50% | 218.54 | 79,820 | 149,051 | 172,909 |
60% | 174.83 | 63,856 | 114,519 | 130,702 |
70% | 131.12 | 47,892 | 79,987 | 88,495 |
80% | 87.41 | 31,928 | 45,445 | 46,288 |
90% | 43.71 | 15,964 | 10,923 | 4,081 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aSponsor’s submitted price for bulevirtide.2
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analyses to address uncertainty within the economic evaluation. The results are provided in Table 12. One scenario analysis was conducted applying treatment discontinuation at 48 weeks for nonresponse, and a second scenario analysis included PEG‑IFN‑alpha as a comparator.
Table 12: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | Best supportive care | $154,264 | 9.48 | Reference |
Bulevirtide | $1,452,173 | 12.86 | $383,943 | |
CDA-AMC scenario 1: Treatment discontinuation at 48 weeks for nonresponse | Best supportive care | $154,076 | 9.48 | Reference |
Bulevirtide | $1,412,067 | 12.73 | $386,766 | |
CDA-AMC scenario 1: PEG-IFN-alpha as comparator | Best supportive care | $154,004 | 9.48 | Reference |
PEG-IFN-alpha | $174,010 | 9.81 | $60,820 | |
Bulevirtide | $1,450,017 | 12.85 | $420,289 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; PEG-IFN = pegylated interferon; QALY = quality-adjusted life-year.
aProbabilistic analyses.
Issues for Consideration
According to clinician input received by CDA-AMC, diagnosis of HDV is currently performed by the National Reference Laboratory in Winnipeg. Clinical expert feedback received by CDA-AMC further notes that, while HDV testing is not systematically conducted among individuals with HBV infection, the introduction of bulevirtide may increase testing of HDV in patients with HBV infections.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing bulevirtide for the treatment of chronic HDV infection in adults with compensated liver disease. The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2029), with 2025 to 2026 as the base year.2 The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program.2 The sponsor estimated the eligible population using an epidemiological approach. The sponsor’s base case included drug acquisition costs. The market uptake for bulevirtide was estimated using internal forecasts.2 The key inputs to the BIA are documented in Table 13.
The sponsor estimated the 3-year incremental budget impact associated with reimbursing bulevirtide would be $58,470,125 (year 1 = $12,144,704; year 2 = $20,764,249; year 3 = $25,561,172).
Table 13: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/ year 2/ year 3 if appropriate) |
|---|---|
Target population | |
Population size (≥ 18 years; excluding Quebec) | |
Population growth | National average: 2.6% Payer-specific range: 1.1% to 3.3% |
Prevalence of chronic HBV | |
Patients diagnosed with HBV | 70%43 |
Patients with HBV tested for HDV | 15.0%/ 25.0%/ 30.0% |
Anti-HDV+ | National average: 3.4% Payer-specific range: 1.2% to 4.8% |
HDV RNA+ | 64.8% |
Patients with HDV eligible for treatment | 66.8%44 |
Patients eligible for public coverage | National average: 43.4%45 Payer-specific range: 31.2% to 100.0%45 |
Number of patients eligible for drug under review | 104/ 178/ 219 |
Market shares (reference scenario) | |
Bulevirtide | 0% |
PEG-IFN-alpha | 0% / 0% / 0% (BC, MB, PEI) 80% / 80% / 80% (AB, SK, ON, NB, NS, NL, NIHB) |
Best supportive care | 100% / 100% / 100% (BC, MB, PEI) 20% / 20% / 20% (AB, SK, ON, NB, NS, NL, NIHB) |
Market shares (new drug scenario) | |
Bulevirtide | 80% / 80% / 80% |
PEG-IFN-alpha | 0% / 0% / 0% (BC, MB, PEI) 16% / 16% / 16% (AB, SK, ON, NB, NS, NL, NIHB) |
Best supportive care | 20% / 20% / 20% (BC, MB, PEI) 4% / 4% / 4% (AB, SK, ON, NB, NS, NL, NIHB) |
Cost of treatment (per patient per year) | |
bulevirtide | $159,531.61 |
PEG-IFN-alpha | $20,145.60 |
Best supportive care | $0 |
AB = Alberta; BC = British Columbia; HBV = hepatitis B virus; HDV = hepatitis delta virus; MB = Manitoba; NB = New Brunswick; NIHB = Non-Insured Health Benefits; NL = Newfoundland and Labrador; NS = Nova Scotia; ON = Ontario; PEG-IFN-alpha = pegylated interferon-alpha; PEI = Prince Edward Island; RNA = ribonucleic acid; SK = Saskatchewan.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
PEG-IFN-alpha is unlikely to be available to patients with HDV infection: Market share for PEG‑IFN-alpha was assumed to be 80% in provinces where it was available and 0% in provinces where it was unavailable.2 Clinical expert feedback received by CDA‑AMC noted that this is not reflective of clinical practice in Canada given the reimbursement criteria with this therapy and supply shortages. This is further aligned with the feedback CDA-AMC received from patient groups, clinician groups, and drug plans noting challenges with accessing this therapy.
Given limited access of PEG-IFN-alpha for patients with HDV infection, CDA-AMC set the market share to be 0% in all provinces to reflect the feedback received.
Overestimation of the proportion of patients presenting who are anti-HDV positive: The proportion of patients that were HDV immunoglobulin G seropositive was based on a retrospective study conducted using data from patients of physicians in the CanHepB Network (from 8 clinics in 6 provinces across Canada).46 The anti-HDV positive proportions for Ontario, New Brunswick, Nova Scotia, Prince Edward Island, Newfoundland and Labrador, and the Non-Insured Health Benefits populations were estimated to be 4.8%. Clinical expert feedback received by CDA-AMC noted that this is likely an overestimate.
CDA-AMC revised the estimate to 3% anti-HDV positive as this better reflects clinical practice in Canada, according to clinical expert feedback.
Inappropriate approach to derive market size: The sponsor used a prevalence-based model in which the target population was derived by multiplying the overall eligible population size with prevalence measures. Clinical expert feedback received by CDA‑AMC noted that there is an existing backlog of patients diagnosed before the model’s base year who would be eligible in the first year for treatment while, in subsequent years, the population eligible for bulevirtide would reflect mostly newly diagnosed cases (i.e., incident population). As such, an incident-based BIA with a prevalent bolus would have been a more appropriate approach to derive the market size.
As this is a structural assumption to the sponsor’s BIA, CDA-AMC could not address this issue. Clinical experts consulted by CDA-AMC expressed that the overall number of patients treated over the 3 years of the BIA model was a reasonable estimate. As such, while the 3-year total budget impact is likely appropriate, caution is required to interpret the annual budget impact as it would be expected to be higher in the first year given the bolus cohort and lower in subsequent years to reflect the incident cohort.
Treatment duration not explicitly captured: As this is a prevalence-based model, discontinuation is not explicitly captured given that market shares are static (i.e., market share for bulevirtide is set to 80% across all 3 years). There remains uncertainty regarding the optimal duration of treatment, as noted previously. Clinical expert input received by CDA‑AMC suggested that treatment may be discontinued for those who do not respond between 48 to 72 weeks while uncertainty remains on how to implement the optimal treatment duration for those who respond.
CDA-AMC could not explore the impact of treatment duration on the expected budget impact. If discontinuation is expected within the first 3 years of treatment and discontinued patients are not replaced with an equal proportion of patients who are newly diagnosed, it would be expected that the budget impact is overestimated.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by making changes in model parameter values and assumptions, in consultation with clinical experts, as outlined in Table 14.
Table 14: Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. PEG-IFN-alpha market share | 80% for NL, NS, NB, ON, SK, AB and NIHB | 0% for NL, NS, NB, ON, SK, AB and NIHB |
2. Anti-HDV positive proportion | 4.8% for NL, PEI, NS, NB, ON and NIHB | 3% for NL, PEI, NS, NB, ON and NIHB |
CDA-AMC base case | ― | Reanalysis 1 + 2 |
AB = Alberta; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; HDV = hepatitis D virus; NB = New Brunswick; NIHB = Non-Insured Health Benefits; NL = Newfoundland and Labrador; NS = Nova Scotia; ON = Ontario; PEG-IFN-alpha = pegylated interferon-alpha; PEI = Prince Edward Island; SK = Saskatchewan.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 15. In the CDA-AMC base case, the 3‑year budget impact of reimbursing bulevirtide for the treatment of chronic HDV infection in adults with compensated liver disease was $44,973,750 (year 1 = $9,341,158; year 2 = $15,971,257; year 3 = $19,661,335).
Table 15: Summary of the Stepped Analysis of the CDA-AMC Base Case
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | $58,470,125 |
CDA-AMC reanalysis 1 | $63,814,947 |
CDA-AMC reanalysis 2 | $41,512,834 |
CDA-AMC base case: Reanalysis 1 + 2 | $44,973,750 |
CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments.
Table 16: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | $902,239 | $1,388,044 | $2,372,714 | $2,920,269 | $6,681,027 |
Bulevirtide | 0 | 0 | 0 | 0 | 00 | |
Comparators | $902,239 | $1,388,044 | $2,372,714 | $2,920,269 | $6,681,027 | |
New drug total | $902,239 | $13,532,748 | $23,136,963 | $28,481,441 | $65,151,152 | |
Bulevirtide | 0 | $13,255,139 | $22,662,420 | $27,897,387 | $63,814,947 | |
Comparators | $902,239 | $277,609 | $474,543 | $584,054 | $1,336,205 | |
Budget Impact | 0 | $12,144,704 | $20,764,249 | $25,561,172 | $58,470,125 | |
CDA-AMC base case | Reference total | 0 | 0 | 0 | 0 | 0 |
Bulevirtide | 0 | 0 | 0 | 0 | 0 | |
Best supportive care | 0 | 0 | 0 | 0 | 0 | |
New drug total | 0 | $9,341,158 | $15,971,257 | $19,661,335 | $44,973,750 | |
Bulevirtide | 0 | $9,341,158 | $15,971,257 | $19,661,335 | $44,973,750 | |
Best supportive care | 0 | 0 | 0 | 0 | 0 | |
Budget Impact | 0 | $9,341,158 | $15,971,257 | $19,661,335 | $44,973,750 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.