Drugs, Health Technologies, Health Systems
Reimbursement Review
Dupilumab (Dupixent)
Sponsor: Sanofi-Aventis Canada Inc.
Therapeutic area: Prurigo nodularis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CSPA
Canadian Skin Patient Alliance
CSR
Clinical Study Report
DLQI
Dermatology Life Quality Index
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IGA PN-S
Investigator’s Global Assessment Prurigo Nodularis–Stage
IL
interleukin
LS
least squares
MID
minimal important difference
NRS
numerical rating scale
PN
prurigo nodularis
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
TEAE
treatment-emergent adverse event
WI-NRS
Worst Itch Numerical Rating Scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), 300 mg single-use prefilled syringe or pen administered by SC injection |
Sponsor | Sanofi-Aventis Canada Inc. (Sanofi) |
Indication | For the treatment of adult patients with moderate-to-severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Dupilumab can be used with or without topical corticosteroids. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 12, 2023 |
Recommended dose | Initial dose of 600 mg (two 300 mg SC injections), followed by 300 mg every 2 weeks |
NOC = Notice of Compliance; PN = prurigo nodularis; SC = subcutaneous.
Introduction
Prurigo nodularis (PN) is a chronic and rare inflammatory skin disease that presents with numerous symmetrically distributed, pruritic, hyperkeratotic nodules, papules, and plaques.1 PN is associated with a neuronal sensitization to itch, which leads to development of an itch-scratch cycle.2 Continuous intense pruritus and scratching in PN culminates in the formation of crusted or excoriated, hyperkeratotic, light- to bright-red nodules or plaques with hyperpigmented margins that may number from a few to hundreds of lesions.3,4 The burden of disease is significant as patients experience debilitating signs and symptoms that negatively affect their health-related quality of life (HRQoL), sleep, and mental health.5 While the exact prevalence and incidence rates of PN are not known, the estimated prevalence rates reported in the literature from the UK, Poland, and the US range from 5.82 cases per 100,000 individuals to 70 cases per 100,000 individuals.6-8
The clinical experts consulted by Canada’s Drug Agency (CDA-AMC) for this review noted that PN is 1 of the most challenging dermatologic conditions to manage due to its severe, chronic pruritus. The existing topical and off-label systemic treatments are often unable to control the disease and/or may be associated with significant toxicity. The initial treatment of PN includes a medium- to high-potency topical corticosteroid (TCS) to reduce inflammation and alleviate pruritus. Intralesional corticosteroid injections may also be used, as well as other symptomatic therapies, such as topical calcineurin inhibitors (TCIs), topical anesthetics, and oral antihistamines. For patients with moderate to severe PN, topical therapies often do not adequately control symptoms of PN and are associated with adverse effects such as skin thinning and systemic corticosteroid absorption. Phototherapy may be effective for some patients; however, it requires multiple clinic visits each week, and is inaccessible to the majority of patients due to the shortage of phototherapy equipment in many jurisdictions. Off-label systemic immunosuppressant therapies (e.g., methotrexate and cyclosporine) that are used for patients with severe or refractory PN have limited evidence of efficacy, are not suitable for many patients who are older or have comorbidities, and pose safety concerns when used over the long term. The clinical experts consulted for this review identified a need for highly effective, disease-modifying, systemic PN therapies that provide sustained relief, are safe for long-term use, and are convenient and accessible.
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg subcutaneous (SC) injections in the treatment of moderate to severe PN in adults whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups who responded to our call for input and from clinical experts consulted for the purpose of this review.
Patient Input
CDA-AMC received input from the Canadian Skin Patient Alliance (CSPA) for this submission. The CSPA is a national charitable organization that works to improve the health and well-being of people across Canada affected by skin, hair, and nail conditions through collaboration, advocacy, and education. Information for this submission was gathered via an online survey conducted from September 12 to November 29, 2024. The survey had 9 respondents (8 patients and 1 caregiver), and 2 of the respondents had experience with the drug under review. The CSPA highlighted that PN is far more than a skin condition; it affects patients’ psychological well-being, social interactions, day-to-day functioning, sleep, and body image. Constant pruritus and discomfort in particular can lead to feelings of frustration, irritability and embarrassment. Survey respondents identified a range of life areas affected by their PN diagnosis, including family and intimate relationships, sex life, work life, mental health, social life, daily activities, sleep, self-esteem, and finance. The CSPA explained that caregivers are also affected, often witnessing their loved ones endure emotional pain, insecurity, and social withdrawal.
When asked about the aspects of a new PN treatment that would be most important to patients, respondents to the CSPA survey identified effectiveness, affordability, and lack of adverse effects. Other less-important aspects included the treatment being easy to take or apply and conducive to the patient’s schedule. The caregiver who responded to the survey disclosed that the cost of medication is the most important aspect of a new treatment for them. Many respondents expressed frustration and emotional distress from trying multiple therapies with little to no improvement. The CSPA highlighted that the respondents strongly agreed that they would be interested in a new treatment for PN.
The CSPA added that 2 patients reported trying dupilumab in the past but both patients found there to be “no change” in their condition. The treatment protocol followed by these 2 patients or prescribed by their providers, how they adhered to it, and if it aligned with the recommendations under review remained unknown according to the CSPA. The CSPA noted the small sample size of survey respondents with experience with dupilumab and the resulting challenges of drawing conclusions from their feedback. Dupilumab is an expensive biologic medication, and our survey results revealed that financial costs can be a barrier to accessing medications.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical experts consulted for this review identified a need for highly effective systemic drugs that directly address the underlying immune dysregulation and neuronal sensitization driving PN, control symptoms of PN, produce sustained disease remission, and prevent the recurrence of new nodules. In addition, there is a need for treatments that are safe for both short- and long-term use in all age groups and ensure sustained relief without accumulating risks. Therapies that effectively address the HRQoL concerns of patients, particularly with respect to the emotional and psychological toll of chronic pruritus and disfiguring lesions, are also required. Last, there is an unmet need for convenient treatment options that do not require regular monitoring or frequent clinic visits and reduce the burden on patients.
According to the clinical experts, dupilumab (and other biologics) are expected to redefine the treatment approach for PN, potentially becoming a first-line systemic treatment for moderate to severe PN in eligible patients. Dupilumab directly targets key cytokines involved in type 2 inflammation, which plays a major role in PN pathogenesis. The experts indicated that dupilumab may be used as monotherapy or it can be combined with a TCS, intralesional corticosteroid injections, phototherapy (if available), and other symptomatic treatments, such as antihistamines. The clinical experts stated that it would be appropriate that patients first try traditional therapies, such as a TCS, with or without intralesional corticosteroid injections, to manage mild to moderate PN. However, for patients with more severe, chronic, or refractory PN, the use of dupilumab would be justified when standard therapies fail to provide adequate relief, ensuring that patients benefit from a targeted therapy aimed at the underlying disease.
Based on input from the clinical experts, dupilumab is most suitable for patients with moderate to severe or refractory PN, particularly those with persistent pruritus that significantly affects HRQoL and who have chronic or widespread nodules that have not responded to conventional treatments. Dupilumab would be suitable for patients with comorbidities (e.g., kidney disease, liver disease, hepatitis, or HIV) for whom other systemic drugs are contraindicated. It may also benefit patients with a history of type 2 inflammatory diseases, such as atopic dermatitis or asthma. The clinical experts indicated that dupilumab is less suitable for patients with mild PN or those with mild symptoms who can manage with conventional treatments.
The experts indicated that response to treatment in clinical practice is assessed based on a reduction in disease severity, pruritus intensity, and HRQoL. Disease severity is often assessed by lesion count and scoring systems such as the Investigator’s Global Assessment Prurigo Nodularis–Stage (IGA PN-S), with a clinically meaningful response being a reduction to an IGA PN-S score of 0 or 1 (i.e., 5 or fewer lesions). Itch intensity (worst itch) may be assessed by patients’ subjective reporting of improvement (e.g., “good to excellent” improvement in itch) or measured by a numerical rating scale (NRS) such as the Worst Itch Numerical Rating Scale (WI-NRS), with a reduction of at least 4 points considered clinically important (on a scale of 0 to 10). Additionally, improvement in overall quality of life as measured by the Dermatology Life Quality Index (DLQI) is an important indicator, particularly as PN has a severe effect on sleep and social well-being.
The experts indicated that the initial treatment response for a biologic would usually be assessed after 6 months. When deciding whether to discontinue treatment with dupilumab, the clinical experts would evaluate whether the patient had achieved the desired reduction in lesion count and itch intensity, noting that not all patients may meet specific response criteria (i.e., 5 or fewer lesions, or a 4-point reduction on the WI-NRS) but may still continue to receive therapy, particularly if no other effective treatment options are available. Additional factors for discontinuation may include serious adverse events (SAEs) or the emergence of comorbidities requiring other treatments, but the primary focus should be on achieving clinical milestones related to disease severity and symptom control.
Both clinical experts consulted for this review agreed that, because of the complexity of diagnosing PN, it is essential that treatment with dupilumab be initiated by a dermatology specialist. PN has a broad differential that requires the clinician to rule out other potential causes. PN is a clinical diagnosis that cannot be reliably diagnosed through biopsy alone as dermatopathologists rely on clinical correlation for pathologic diagnosis, and many conditions appear similar under histology. The clinical experts stated that dermatologists are best equipped to maximize treatment outcomes of dupilumab by optimizing the use of concurrent conventional therapies, and they noted that academic (hospital) and community dermatologists are qualified to diagnose PN and prescribe dupilumab.
Clinician Group Input
No clinician group submitted input to CDA-AMC.
Drug Program Input
Refer to Table 4 for the summary of drug program input.
Clinical Evidence
Systematic Review
Description of Studies
Two randomized, double-blind, placebo-controlled, multicentre, parallel-group, phase III studies were included in the systematic review.9,10 The LIBERTY-PN PRIME and LIBERTY-PN PRIME2 trials (hereafter referred to as the PRIME and PRIME2 trials) evaluated the efficacy and safety of dupilumab in adult patients (aged 18 to 80 years) with PN who were inadequately controlled on topical prescription therapies or for whom those therapies were not advisable. Patients were eligible for enrolment in the trials if they had a total of at least 20 PN lesions on both legs, and/or both arms, and/or trunk at the screening visit and on day 1 (randomization); a history of a failed 2-week course of a medium- to superpotent TCS or when those therapies were not advisable; and an average WI-NRS score of at least 7 points in the 7 days before randomization.
In the PRIME and PRIME2 studies, 151 and 160 patients, respectively, were randomized (1:1) to receive either dupilumab 300 mg every 2 weeks (with a 600 mg loading dose), or matching placebo, by SC injection for 24 weeks. Concurrent use of a low- to medium-potency TCS or TCI was allowed if patients were receiving these treatments before the study.
The primary outcome in both studies was the proportion of patients with improvement (reduction) in the WI-NRS score of at least 4 points from baseline to 24 weeks in the PRIME study, and from baseline to 12 weeks in the PRIME2 study. The WI-NRS is a patient-reported outcome consisting of a single item rated on a scale from 0 (“no itch”) to 10 (“worst imaginable itch”). The analysis was based on the 7-day average of daily WI-NRS scores over the previous week. Key secondary end points included the proportion of patients with an IGA PN-S score of 0 or 1 at week 24 (for both studies). The IGA PN-S is a clinician-reported assessment of the stage of the disease using a 5-point scale as follows: 0 (clear; no nodules); 1 (almost clear; ≤ 5 nodules), 2 (mild; 6 to 19 nodules), 3 (moderate; 20 to 99 nodules) to 4 (severe; ≥ 100 nodules). Another secondary outcome was the change from baseline to week 24 in the DLQI total score. The DLQI is a patient-reported outcome that assesses the impact of skin disease on patients’ HRQoL over the previous week. It includes 10 items that cover symptoms, leisure activities, work, school or holiday time, personal relationships including intimate activities, the adverse effects of treatment, and emotional reactions to having a skin disease. Overall scoring ranges from 0 to 30, with a high score indicative of a poor HRQoL.
The patients enrolled in the 2 trials had similar characteristics, with a mean age per treatment group ranging from 46.7 years (standard deviation [SD] = 15.2) to 51.1 years (SD = 15.8). Most patients were female (62.2% to 69.3%) and the minority were male (30.7% to 37.8%). The mean duration of PN was between 5.4 years (SD = 6.9) and 6.0 years (SD = 7.6), and less than half (36.8% to 48.8%) of patients enrolled had a history of atopy. The majority of patients were on stable doses of a TCS or TCI (56.1% to 62.7%) at baseline, and all but 1 patient had used topical therapies in the past. Most patients had also received systemic therapies for PN, of which antihistamines (46.2% to 60.0%), corticosteroids (11.5% to 22.7%) and nonsteroidal immunosuppressant drugs (13.2% to 25.6%) were the most common. ███ ████████ ██ ████████ ███ ██████████ ████████ █████████████ ██████████████ ███████████ █████████████ ██ ███████████ ██ █████ █████ ██ █████ ██ ███████. Based on IGA PN-S scores, more patients (60.5% to 72.0%) had moderate PN than severe PN (28.0% to 39.5%), and the mean WI-NRS score at baseline ranged from 8.3 (SD = 1.1) to 8.6 (SD = 0.9).
Efficacy Results
In the PRIME study, 60.0% versus 18.4% of patients in the dupilumab and placebo group, respectively, reported at least a 4-point reduction in WI-NRS scores from baseline to week 24. The between-group difference was 42.7% (95% confidence interval [CI], 27.8% to 57.7%) favouring dupilumab versus placebo (P < 0.0001) (Table 2). At week 24, the results of the PRIME2 study also favoured dupilumab, with 57.7% versus 19.5% of patients meeting the WI-NRS response criteria (difference = 42.6%; 95% CI, 29.1% to 56.1%; P < 0.0001 for dupilumab versus placebo). At week 12, the between-group differences in the proportion of patients with at least a 4-point reduction in WI-NRS score was 29.2% (95% CI, 14.5% to 43.8%; P = 0.0003 [not controlled for multiple comparisons]), and 16.8% (95% CI, 2.3% to 31.2%; P = 0.0216) in the PRIME and PRIME2 studies, respectively.
The proportions of patients in the PRIME study with an IGA PN-S score of 0 (clear) or 1 (almost clear) at week 24 were reported for 48.0% of patients in the dupilumab group and 18.4% of patients in the placebo group, with a between-group difference of 28.3% (95% CI, 13.4% to 43.2%; P = 0.0004) favouring dupilumab. The results were similar in the PRIME2 study, in which 44.9% and 15.9% of patients met the IGA PN-S response criteria in the dupilumab and placebo groups, respectively. The between-group difference was 30.8% (95% CI, 16.4% to 45.2%; P < 0.0001).
The change from baseline in DLQI total scores to week 24 favoured the dupilumab group versus the placebo group in both studies. The PRIME study reported a least squares (LS) mean difference of −6.2 points (95% CI, −8.3 to −4.1; P < 0.0001), and the PRIME2 study reported an LS mean difference of −6.4 points (95% CI, −8.4 to −4.4; P < 0.0001) for dupilumab versus placebo. In patients with PN, a 4.0-point change in DLQI (range, 3.5 to 6.5) was estimated to represent a meaningful between-group change.11
Harms Results
Based on the pooled data from the PRIME and PRIME2 studies, 97 patients (63.8%) who received dupilumab and 89 patients (56.7%) who received placebo reported at least 1 treatment-emergent adverse event (TEAE) over the 24-week treatment period. The most common events were accidental overdose ███ █████ ██████ ██████ headache (5.3% versus 5.7%), nasopharyngitis (3.9% versus 1.9%), and neurodermatitis (2.6% versus 7.0%) in the dupilumab versus placebo groups, respectively.
Seven patients (4.6%) in the dupilumab group experienced an SAE compared with 12 patients (7.6%) in the placebo group. No patients died during the trials and none of the patients who received dupilumab stopped treatment due to TEAEs, whereas in the placebo group, 4 patients (2.5%) discontinued therapy for this reason.
Conjunctivitis was identified as an important adverse event (AE), and the product monograph contains a warning for these AEs. In the PRIME study, ████ of patients in each group experienced 1 or more TEAE related to conjunctivitis, whereas in the PRIME2 study, ████ of patients in the dupilumab group and ██ of patients in the placebo group had at least 1 of these AEs. No severe conjunctivitis AEs were reported in either trial. The evidence was too uncertain to draw conclusions on the impact of dupilumab on the frequency of conjunctivitis-related AEs.
Critical Appraisal
The CDA-AMC review team did not identify any concerns with the methods used to randomize patients, conceal allocation, or maintain blinding in either study. There were some minor demographic differences between groups at baseline, but the clinical experts consulted for this review agreed that any baseline discrepancies were unlikely to bias the findings. However, more patients in the placebo group dropped out or stopped the study drug than in dupilumab group, making it unclear if the treatment groups remained balanced throughout the study.
Based on the patient and clinician input received, the outcomes assessed were clinically relevant. No major limitations were identified in the statistical analysis of the WI-NRS or IGA PN-S data; however, some of the DLQI data that may affect the change-from-baseline results were missing. In the PRIME study, 24-week DLQI data were missing for ██ ███ ███ and were imputed for ███ ███ ███ of patients in the dupilumab and placebo groups, respectively. For the PRIME2 trial, ██ ███ ██ of patients had missing DLQI data at week 24, and ███ ███ ███ of patients had data imputed in the dupilumab and placebo groups, respectively. DLQI data after patients received rescue treatment or missing data after treatment discontinuation due to lack of efficacy were imputed using the worst observation carried forward. Missing data due to other reasons were imputed using multiple imputation methods. Given the magnitude and differential nature of the missing and imputed data in the dupilumab and placebo groups, the potential for bias cannot be ruled out. No supplementary analyses were available to test other imputation strategies for missing data, which may have improved confidence in the DLQI findings.
Of the key efficacy outcomes included in this report, only 1 was not part of the hierarchical statistical testing procedure to control for multiple comparisons. In the PRIME study, the proportion of patients with at least a 4-point improvement in the WI-NRS score at 12 weeks was not controlled for multiple comparisons. No statistical inferences can therefore be drawn on these findings and they should be viewed as supportive evidence only.
The clinical experts consulted for this review agreed that the characteristics of the patients enrolled were generally consistent with those of patients with moderate to severe PN living in Canada who would be eligible for dupilumab. As background therapy, 61% and 56% of patients in the PRIME and PRIME2 studies, respectively, were on stable doses of a TCS or TCI at the start of the trial. All but 1 of the patients enrolled had previously received topical therapies and most patients (71% and 63% in the PRIME and PRIME2 studies, respectively) had received prior systemic therapies to manage their PN. Prior systemic therapies included nonsteroidal immunosuppressants (e.g., cyclosporine, methotrexate, or thalidomide) in 17% and 24% of patients in the PRIME and PRIME2 studies, respectively. The clinical experts agreed that it would be reasonable to generalize the trial results to anticipated clinical practice (i.e., first-line systemic therapy for patients with moderate to severe PN whose disease was not adequately controlled with topical prescription therapies or when those therapies were not advisable). Given the poor efficacy and safety concerns with off-label systemic immunosuppressants, the clinical experts did not recommend that patients be required to try these therapies before initiating dupilumab.
The clinical experts noted that PN is most prevalent in older patients, who often have comorbidities; however, the trials excluded patients with comorbid conditions such as neuropathy or psychiatric conditions, renal disease, immunodeficiency, a history of malignancy, substance use disorder, or infection. The safety and efficacy of dupilumab in these patients is therefore unclear. The trials were 24 weeks in duration, which the clinical experts consulted for this review agreed was a reasonable time frame to show initial treatment response. However, the longer-term efficacy and safety are uncertain.
Based on clinical expert feedback, off-label systemic therapies may be used to manage patients with PN whose disease is not adequately controlled with topical therapies. A sponsor-conducted feasibility assessment concluded that an indirect treatment comparison was likely to provide biased treatment-effect estimates when comparing dupilumab with other systemic therapies due to the low-quality evidence available. How dupilumab compares with other systemic therapies remains unknown. Nemolizumab is currently under review by CDA-AMC for moderate to severe PN. CDA-AMC has not issued a final recommendation for nemolizumab and, therefore, based on CDA-AMC procedures, it was not considered to be a relevant comparator for the present review of dupilumab.
GRADE Summary of Findings and Certainty of the Evidence
For the randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.12.13 Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The thresholds used for the presence or absence of an important effect were based on clinical expert opinion for the WI-NRS and IGA PN-S end points, and on the literature for the DLQI end point. For conjunctivitis, the presence or absence of any non-null effect was used.
For the GRADE assessments, findings from PRIME and PRIME2 were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.14 The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from the patient group and public drug plans. The following list of outcomes was finalized in consultation with expert committee members: proportion of patients with at least a 4-point improvement in WI-NRS, proportion of patients with an IGA PN-S score of 0 or 1, the change from baseline in the DLQI total score, and conjunctivitis-related AEs (Table 2).
Long-Term Extension Studies
No long-term extension studies were submitted.
Indirect Comparisons
No indirect comparisons were submitted.
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor submitted 5 studies to address sponsor-identified gaps in the pivotal evidence. These included retrospective observational studies by Paganini et al. (2024),16 Richter et al. (2023),17 Selvaraj et al. (2023),18 and Chiricozzi et al. (2024)19 to address the lack of long-term data from the PRIME and PRIME2 trials, and 1 retrospective cohort study by Georgakopoulous et al. (2021)20 to address the lack of long-term data for patients living in Canada. Full texts were available for 2 studies only.16,17
Table 2: Summary of Findings for Dupilumab vs. Placebo for Patients With Moderate to Severe PN
Outcome and follow-up | Patients, N (studies) | Effect | Certainty | What happens |
|---|---|---|---|---|
Pruritus NRS ≥ 4-point reduction | ||||
Proportion of patients with ≥ 4-point reduction from baseline in WI-NRSa Follow-up: 24 weeks | 251 (2 RCTs) | PRIME study:
PRIME2 study:
| High | Dupilumab results in a clinically important increase in the proportion of patients with at least a 4-point improvement in WI-NRS score at 24 weeks compared with placebo |
Proportion of patients with ≥ 4-point reduction from baseline in WI-NRSa Follow-up: 12 weeks | 251 (2 RCTs) | PRIME study:
PRIME2 study:
| Moderateb | Dupilumab likely results in a clinically important increase in the proportion of patients with at least a 4-point improvement in WI-NRS score at 12 weeks compared with placebo |
IGA PN-S score of 0 or 1 | ||||
Proportion of patients with IGA PN-S score of 0 or 1c Follow-up: 24 weeks | 251 (2 RCTs) | PRIME study:
PRIME2 study:
| High | Dupilumab results in a clinically important increase in the proportion of patients with an IGA PN-S score of 0 or 1 compared with placebo |
Change in DLQI total score | ||||
DLQI score (0 [best] to 30 [worst]) LS mean change from baselined Follow-up: 24 weeks | 296 (2 RCTs) | PRIME study
PRIME2 study
| Moderatee | Dupilumab likely results in a clinically important improvement in the change in the DLQI total score compared with placebo |
Harms | ||||
Proportion of patients with conjunctivitis-related AEsf Follow-up: 24 weeks | N (2 RCTs) | PRIME study: ████ ████ ███ ████ PRIME2 study: ████ ██ ███ █ ███ █ | Very lowg | The evidence is very uncertain about the effect of dupilumab on the proportion of patients with 1 or more conjunctivitis-related AEs compared with placebo |
AE = adverse event; aRD = adjusted risk difference; CI = confidence interval; DLQI = Dermatology Life Quality Index; DUP = dupilumab; GRADE = Grading of Recommendations Assessment, Development and Evaluation; HRQoL = health-related quality of life; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; LS = least squares; NRS = numerical rating scale; PBO = placebo; PN = prurigo nodularis; RCT = randomized controlled trial; SE = standard error; WI-NRS = Worst Itch Numerical Rating Scale; vs. = versus.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aThe WI-NRS is a patient-reported, single-item, daily, 11-point scale. The scale is used by patients to rate their worst itch severity over the past 24 hours, with 0 indicating “no itch” and 10 indicating “worst itch imaginable.” Based on clinical expert input, the threshold for a clinically important between-group difference was 20% (200 per 1,000) for the proportion of patients with at least a 4-point reduction from baseline.
bThe WI-NRS end point at 12 weeks was rated down 1 level due to serious limitations due to imprecision, as the lower bound of the 95% CIs for the between-group differences of both studies include the possibility of no clinically important difference. Although the point estimate of the PRIME2 study is below the clinically relevant threshold of 20%, had the 2 studies been pooled, the overall effect estimate would likely exceed the threshold. The CDA-AMC review team therefore did not rate it down due to inconsistency. The 12-week time point in the PRIME study was not controlled for the type I error rate for multiple comparisons and should be interpreted as supportive data.
cThe IGA PN-S measures the investigator’s global assessment of the patient’s stage of PN using a 5-point scale as follows: 0 (clear; no nodules); 1 (almost clear; ≤ 5 nodules), 2 (mild; 6 to 19 nodules), 3 (moderate; 20 to 99 nodules) to 4 (severe; ≥ 100 nodules). Based on clinical expert input, the threshold for a clinically important between-group difference was 15% (150 per 1,000) for the proportion of patients with an IGA PN-S score of 0 or 1. The lower bound of the 95% CI for the PRIME study included the possibility of no clinically important difference between treatments. However, the review team did not rate down the outcome for imprecision because that the GRADE guidance states that the threshold should be “appreciable crossed” to warrant downgrading and that pooling results across the 2 trials may have shifted the lower bound of the 95% CI closer to the threshold of clinical importance.
dThe DLQI is a patient-reported, 10-item, HRQoL questionnaires that covers 6 domains (symptoms and feelings, daily activities, leisure, work and school, personal relationships, and treatment) over the “last week.” The total score ranges from 0 (no impact of skin disease on quality of life) to 30 (maximum impact on quality of life). A minimal important difference of 4 points for the DLQI was selected as the threshold for a clinically important between-group difference based on the literature and clinical expert input.
eThe change from baseline in DLQI was rated down 1 level due to serious study limitations due to missing data.
fConjunctivitis-related AEs included the following events: conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis, blepharitis, dry eye, eye irritation, eye pruritus, increased lacrimation, eye discharge, foreign body sensation in eyes, photophobia, xerophthalmia, ocular hyperemia, and conjunctival hyperemia.
gConjunctivitis-related AEs were rated down 2 levels due to serious imprecision because the CIs for the between-groups differences include the possibility of no difference, benefit (fewer harms), or increased harms. Rated down 1 level due to indirectness, as the sample size and duration of treatment were insufficient to detect uncommon AEs. In addition, the clinical experts consulted for this review stated that, because dermatologists may not have sufficient expertise to distinguish between eye disorders with a similar presentation, the reported conjunctivitis-related AEs may be flawed.
Sources: Clinical Study Report for PRIME,9 Clinical Study Report for PRIME2,10 and additional data supplied by the sponsor.15
The study by Paganini et al. (2024)16 was a retrospective, cross-sectional, observational, chart review of 16 patients with PN and moderate to severe atopic dermatitis who received dupilumab at a single centre in Rome, Italy. The proportion of patients with an IGA PN score of 0 or 1 was 77% by week 16, 83% by week 52, and 100% by week 84. The proportion of patients with at least a 4-point improvement in the itch NRS was 91% by week 6, 100% by week 16, 83% by week 52, and 100% by week 84. The proportion of patients with at least a 4-point improvement in the DLQI was 92% by week 16, 83% by week 52, and 67% by week 84.
The study by Richter et al. (2023)17 was a retrospective, single-centre case series of 10 patients with chronic PN who received dupilumab at a single centre in, Zurich, Switzerland. The average and maximum itch NRS scores both showed a 54% decrease, and the DLQI score decreased by 42% reduction over 1 year.
In both studies, the observational and retrospective nature of the studies limits the ability to draw inferences on the effects of dupilumab. Because the patients were aware they were receiving active treatment, their expectations of treatment may have influenced reporting of subjective outcomes. Given the small sample sizes, the results may not be broadly generalizable.
Conclusions
Based on 2 clinical trials in adults with moderate to severe PN who were inadequately controlled on topical prescription therapies or for whom those therapies were not advisable, dupilumab demonstrated a statistically significant and clinically important improvement versus placebo in pruritus intensity, and in the clearance or near clearance of PN nodules, in terms of the proportion of responders for the WI-NRS and the IGA PN-S at 24 weeks. Based on the change from baseline in the DLQI total score, dupilumab likely improves HRQoL versus placebo; however, this finding was of limited value due to missing data.
While TEAEs were commonly reported by patients in both the dupilumab and placebo groups, the proportion of patients who stopped therapy due to AEs was low. Conjunctivitis was identified as an important AE; however, the evidence was too uncertain to draw conclusions on the impact of dupilumab on the frequency of conjunctivitis-related AEs.
Because the evidence was drawn from 2 placebo-controlled RCTs that were 24 weeks in duration, the longer-term efficacy and safety are uncertain. While the sponsor submitted 5 longer-term retrospective observational studies, the lack of control groups, other biases associated with nonrandomized studies, and small sample sizes mean these studies do not fill the evidence gap.
Introduction
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg SC injections for the treatment of moderate to severe PN in adults whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
PN is a chronic and rare inflammatory skin disease with numerous symmetrically distributed, pruritic, hyperkeratotic nodules, papules, and plaques.1 The dome-shaped, firm, pruritic nodules can range in size from a few millimetres to several centimetres in size and can be flesh-coloured, brown, or black. The lesions are usually distributed symmetrically on the extensor surfaces of the arms and legs and on the trunk. The number of nodules can range from a few to several hundred.3 PN is associated with a neuronal sensitization to pruritus, which leads to the development of an itch-scratch cycle.2 PN increases the activation of mediators involved in type 2 inflammation, including T cells, mast cells, granulocytes, dendritic cells, and eosinophils within the dermis.4 These cell types are involved in the regulation of type 2 inflammation through release of tryptase, histamines, interleukin, neuropeptides, and prostaglandins.4 Continuous, intense pruritus and scratching in PN culminates in the formation of crusted or excoriated, hyperkeratotic, light- to bright-red nodules or plaques with hyperpigmented margins.4
Although the exact cause of PN is unknown, some of the predisposing factors include neurologic, dermatologic, systemic, and psychiatric diseases, which are associated with severe pruritus. РΝ may occur in patients with chronic pruritus and almost half of all patients with PN have a history of atopic dermatitis.3 ΡN can occur in all age groups and has no sex preferences, but it appears to be more common in older adults and people who are African American.3 An estimated point prevalence in the UK has shown lower prevalence in males.13
The burden of disease with PN is significant, as patients experience signs and symptoms that negatively affect their HRQoL, sleep, and mental health.5 The highest burdens reported by patients with PN included itch, visibility of skin lesions, bleeding of skin lesions, impact on everyday life activities, sleep disturbance, psychological consequences, and pain.21 An increased risk of mental health disorders, including depression (most common), anxiety, substance use disorder, personality trait disorder, bipolar disorder, post-traumatic stress disorder, childhood or adult-onset attention-deficit/hyperactivity disorder, eating disorder, and schizophrenia disorders, has been reported among patients in Canada with PN.22 Comorbid medical conditions reported among patients in Canada with PN include hypertension, malignancy (including nonmelanoma skin cancer), type 2 diabetes, thyroid disease, chronic kidney disease, fibromyalgia, inflammatory arthritis, irritable bowel syndrome, and osteoporosis.22
While the exact prevalence and incidence rates of PN are not known, prevalence rates reported in the literature based on estimates from the UK, Poland, and the US range from 5.82 cases per 100,000 individuals to 70 per 100,000 individuals.6-8
The diagnosis of РN is based on the history of pruritus and observation of skin lesions such as nodules and excoriations during a clinical exam.3
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the review team.
According to the 2 clinical experts consulted for this review, the primary goals of therapy for patients with PN are to reduce the severity of pruritus, treat existing lesions (reduce both the number and size of nodules), prevent the formation of new lesions, and improve HRQoL while minimizing the adverse effects of treatment. In Canada, there are no formal practice guidelines for the management of PN; however, international guidelines and published literature describe various medications and procedures for the treatment of PN using a stepwise and multimodal approach, considering factors such as the patient’s age, comorbidities, severity of PN, HRQoL, concomitant medications, and the treatment side-effect profile.23-26 Acknowledging the lack of a standardized approach to the treatment of PN, guideline recommendations generally include emollients for PN of any severity; topical medications (e.g., corticosteroids, calcineurin inhibitors, and capsaicin), intralesional corticosteroids, and H1 antihistamines as initial-tier treatment options; and various systemic medications (e.g., methotrexate, cyclosporine, gabapentinoids, thalidomide, and dupilumab) and UV light phototherapy as later-tier options (noting that patients may enter the treatment ladder at any tier, based on clinical presentation). Several medication options detailed in the guidelines are off-label and the evidence to support their efficacy is limited (e.g., small clinical trials, observational studies or case reports, or data from non-PN pruritic conditions).24-26 In Canada, dupilumab is currently the only approved treatment for patients with PN.
When describing the current treatment paradigm for PN in Canada, the experts noted that clinicians rely on broader dermatology guidelines and expert opinion to inform treatment decisions. In current practice, PN would be initially managed with a high-potency TCS to reduce inflammation and alleviate pruritus, but this may lead to skin thinning and other adverse effects with prolonged use. Other initial treatment options include a TCI, topical anesthetics, and oral antihistamines; however, these treatments may not provide sufficient relief of pruritus and/or may cause adverse effects. The clinical experts described intralesional corticosteroid injections as often effective for flattening nodules and reducing pruritus, but noted that this option is limited by adverse effects (e.g., pain and skin atrophy) and an inability to address multiple lesions. Other therapies include topical capsaicin, which may have limited effectiveness and cause burning sensations with initial application, and UV light treatment (phototherapy), which can be effective but has limitations in terms of access and patients’ ability to adhere to treatment. Systemic immunosuppressants may be used off label for severe or treatment-resistant disease. These include cyclosporine and methotrexate, which may be effective in reducing inflammation and pruritus. However, the clinical experts consulted for this review noted that these medications have limited efficacy and are associated with significant toxicity that limits long-term use. Due to safety concerns, off-label systemic immunosuppressants may not be suitable for older patients who often have comorbidities or those with polypharmacy. Other medications that may be considered include gabapentin, pregabalin, carbamazepine, doxepin, mirtazapine, mycophenolate mofetil, and low-dose naltrexone. These medications also have little evidence to support their use and are associated with adverse effects. The clinical experts highlighted that PN is difficult to treat; the current conventional treatment options provide only partial and/or short-term symptomatic relief, and their use is often limited by adverse effects, safety risks, and feasibility issues. The clinical experts expressed need for highly effective, disease-modifying, systemic PN therapies that provide sustained relief, are safe for long-term use, and are convenient and accessible.
Drug Under Review
Dupilumab was approved by Health Canada for the treatment of adult patients with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.27 The sponsor’s reimbursement request is aligned with the approved Health Canada indication.28 Dupilumab is the only Health Canada–approved treatment for adults with PN in Canada. It is available as a 300 mg per 2 mL prefilled syringe or pen for SC injection. The product monograph recommends an initial dose of 600 mg (2 injections of 300 mg) followed by 300 mg given every 2 weeks.27
Dupilumab is a recombinant human immunoglobin G4 monoclonal antibody that inhibits interleukin (IL)-4 and IL-13 signalling by specifically binding to the IL-4Ralpha subunit, which is shared by the IL-4 and IL-13 receptor complexes.28 Dupilumab inhibits IL-4 signalling via the type I receptor (IL-4Ralpha/gamma chain), and both IL-4 and IL-13 signalling through the type II receptor (IL-4Ralpha/IL-13Ralpha). IL-4 and IL-13 are key type 2 cytokines involved in atopic disease.28
Dupilumab is the first Health Canada–approved therapy specifically indicated for PN that targets the underlying cause of PN.
In addition to the treatment of moderate to severe PN, dupilumab has approved indications for patients with atopic dermatitis, severe asthma, chronic rhinosinusitis, and eosinophilic esophagitis. Dupilumab has been previously reviewed by CDA-AMC for atopic dermatitis and asthma and is currently under review for chronic rhinosinusitis with nasal polyposis and chronic obstructive pulmonary disease.
The key characteristics of dupilumab for the treatment of PN are summarized in Table 3.
Table 3: Key Characteristics of Dupilumab
Characteristic | Dupilumab |
|---|---|
Mechanism of action | Inhibition of interleukin-4 and interleukin-13 |
Indicationa | Treatment of adult patients with moderate-to-severe prurigo nodularis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable |
Route of administration | Subcutaneous |
Recommended dose | 600 mg initial dose, followed by 300 mg given every 2 weeks |
Serious adverse effects or safety issues | Hypersensitivity reactions, serious systemic eosinophilia, conjunctivitis and keratitis |
aHealth Canada–approved indication.
Source: Dupilumab product monograph.27
Perspectives of Patients, Clinicians, and Drug Programs
The full patient group submission received is available in the consolidated patient and clinician group input document for this review on the project website.
No clinician groups provided input for this review.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
The CSPA supplied input for this submission. The CSPA is a national charitable organization that works to improve the health and well-being of people across Canada affected by skin, hair, and nail conditions through collaboration, advocacy, and education. Information for this submission was gathered via an online survey conducted from September 12 to November 29, 2024. The survey had 9 respondents (8 patients and 1 caregiver), and 2 respondents had experience with the drug under review.
Based on the patient group input, most of the respondents (5 of 9) were from Ontario. Five respondents provided their age; all were aged over 35 years, with more than half aged over 55 years. Five respondents who answered the disease-history question indicated that they had PN for less than 5 years. Three respondents reported having severe PN and 1 respondent rated their PN as moderate in severity: no respondent reported their PN as mild.
The CSPA highlighted that PN is far more than a skin condition; it affects patients’ psychological well-being, social interactions, day-to-day functioning, sleep, and body image. Persistent and severe itching (pruritus) is the predominant symptom, which significantly disrupts daily activities and sleep. The visible nodules and scars may lead to embarrassment and reduced self-esteem. The chronic nature of PN can result in emotional distress, anxiety, and depression. According to the CSPA survey, all patients reported itchy skin, itchy bumps (nodules), burning or stinging skin, scratching, pain, and hyperpigmentation (dark spots). Three patients (75%) reported skin symptoms of scarring because of PN. One patient (25%) reported experiencing adverse effects of flares and hypopigmentation (light spots).
Regarding the aspects of life affected by a PN diagnosis, survey respondents reported that family relationships, intimate relationships, sex life, work life, mental health, social life, daily activities, sleep, self-esteem, and finances were all effected. Regarding impact on daily activities, 25% of respondents reported missing work 5 to 10 times monthly due to PN, 25% reported missing work 1 to 5 times monthly, and 50% reported not missing work due to PN.
The CSPA explained that caregivers are also affected, often witnessing their loved ones endure emotional pain, insecurity, and social withdrawal. The psychological burden on caregivers was reported as being potentially immense, as they provide ongoing emotional support and encouragement, while often feeling helpless themselves.
According to the CSPA, many patients reported limited success with existing therapies, highlighting the need for new and more effective treatments. CSPA survey respondents reported trying multiple treatments to manage their PN, such as a TCS, a TCI, topical capsaicin, oral antihistamines and methotrexate, phototherapy, and medical cannabis. The respondents emphasized the limited efficacy of these treatments. The CSPA added that 2 patients who reported trying dupilumab in the past found there to be “no change” in their condition. The CSPA noted the small sample size of survey respondents with experience with dupilumab and resulting difficulty drawing conclusions on dupilumab’s efficacy. Many respondents expressed frustration and emotional distress from trying multiple therapies that provided little to no improvement. The CSPA explained that, when asked about aspects of a new PN treatment that are most important to patients, survey respondents indicated effectiveness, affordability, and lack of adverse effects. Other less-important aspects reported were the treatment being easy to take or apply and being conducive to the patient’s schedule. According to the patient group input, in addition to managing itch, psychological and social relief are the motivations underlying these desired outcomes. With a treatment that offers tangible improvement and restores a sense of normalcy, patients anticipate a significant enhancement in quality of life, self-confidence, and social participation.
Regarding the adverse effects of current treatments, CSPA survey respondents reported experiencing a racing heart, skin irritation, nausea, vomiting, hypopigmentation, and hyperpigmentation. Only 1 respondent shared that they stopped treatment for PN because of adverse effects, and 3 respondents reported stopping treatment because of a lack of efficacy. The CSPA noted that 4 of 5 respondents strongly agreed that they would be interested in a new treatment for PN and that they wished a better PN treatment option was available.
Survey respondents also shared that they have experienced financial challenges that have affected their access to PN treatments, and the caregiver who responded to the survey disclosed that the cost of medication is the most important aspect of a new treatment for them. Currently, no treatments indicated for PN are funded by public drug plans in Canada.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of PN.
Unmet Needs
The clinical experts consulted for this review identified a need for safe and effective therapies to treat PN that do not have a significant treatment burden. One critical gap is for highly effective systemic drugs that directly address the underlying immune dysregulation and neuronal sensitization driving PN, rather than just providing generalized immunosuppression or some symptomatic management. No currently available treatment can permanently cure or halt the progression of the disease. While many therapies can temporarily alleviate symptoms, relapses are common after the cessation of treatment, leading to a cyclical pattern of flare-ups. This underscores the need for more durable therapeutic solutions that can sustain disease remission and prevent the recurrence of new nodules. In addition, treatment resistance can occur over time, leading to refractoriness, where patients who initially show improvements later experience recurrent symptoms or worsening disease. This necessitates a constant adjustment of therapy and highlights the limitations of current treatment options in achieving long-term disease control. Moreover, existing treatments do not effectively address the HRQoL concerns of patients, particularly with respect to the emotional and psychological toll of chronic pruritus and disfiguring lesions.
The clinical experts stated there is a need for treatments that are safe for both short- and long-term use to ensure sustained relief without accumulating risks. This includes treatments that are suitable for all age groups, particularly older patients, who represent a significant portion of the PN population and are more vulnerable to adverse effects of treatment. The clinical experts noted that existing off-label systemic immunosuppressants are often not viable options for patients with common comorbidities, including chronic liver disease, chronic kidney disease, HIV, and hepatitis; the treatments available for these patients are therefore limited. In addition, there is a need for treatments that are safe during pregnancies, allowing those with PN to receive effective therapy without risk to their fetus. Last, there is an unmet need for convenient treatment options that do not require regular monitoring or frequent clinic visits, reducing the burden on patients. Rather than topical therapies that can be messy and cumbersome to apply, oral therapies that require frequent dosing, or phototherapy that requires multiple appointments per week, there is a growing need for long-acting formulations or alternative delivery systems that could simplify treatment regimens and improve overall adherence.
The experts stated that, at present, there are no Health Canada–approved treatments that fully meet these needs. The lack of targeted, safe, and accessible therapies leaves many patients with chronic, debilitating symptoms and few viable options, highlighting the urgent need for novel treatments that can address the immune-mediated, neurogenic, and quality of life aspects of PN, while ensuring safety across diverse patient populations.
Place in Therapy
According to the clinical experts consulted for this review, dupilumab (and other biologics) are expected to redefine the treatment approach for PN, potentially becoming a first-line systemic treatment for moderate to severe PN in eligible patients. The availability of dupilumab may allow for earlier escalation to disease-modifying systemic therapy. Dupilumab directly targets IL-4 and IL-13, key cytokines involved in type 2 inflammation, which plays a major role in PN pathogenesis. The clinical experts noted that the targeted mechanism allows dupilumab to be used without the risks associated with broad immunosuppression. Dupilumab may be used as monotherapy for many patients, or it can be combined with a TCS, intralesional corticosteroid injections, phototherapy (if available), and other symptomatic treatments, such as antihistamines. The clinical experts stated that, in their experience, methotrexate may be used as a short-term bridging therapy in patients with severe PN at the start of dupilumab treatment, and is tapered and discontinued once dupilumab takes effect.
The clinical experts agreed that it would be appropriate that patients first try traditional therapies, such as a TCS with or without intralesional corticosteroid injections, to manage mild to moderate cases of PN. However, for patients with more severe, chronic, or refractory PN, the use of dupilumab would be justified when these standard therapies fail to provide adequate relief. For many patients with PN, off-label systemic immunosuppressants are contraindicated, making dupilumab a reasonable first-line systemic treatment for this cohort. Given the poor efficacy and safety concerns of off-label systemic immunosuppressants, the clinical experts did not recommend that all patients be required to try these therapies before initiating dupilumab.
Patient Population
According to the clinical experts consulted for this review, dupilumab is most suitable for patients with moderate to severe or refractory PN, particularly those with persistent pruritus that significantly affects HRQoL and who have chronic or widespread nodules that have not responded to conventional treatments. Dupilumab would be suitable for patients with comorbidities (e.g., kidney disease, liver disease, hepatitis, or HIV) for whom other systemic drugs are contraindicated. It may also benefit patients with a history of type 2 inflammatory diseases, such as atopic dermatitis or asthma.
The clinical experts indicated that dupilumab is not suitable for patients with mild PN or those with mild symptoms who can manage with conventional treatments. The primary method of identifying suitable patients is clinical judgment, based on the number of lesions, and focusing on symptom severity (itch severity), and HRQoL.
The clinical experts emphasized that diagnosis of PN should be made through a thorough clinical evaluation by a dermatologist who has the expertise to distinguish PN from other pruritic conditions (e.g., eczema, psoriasis). A skin biopsy is not required for an accurate diagnosis.
Assessing the Response to Treatment
In clinical practice, the experts indicated that response to treatment is assessed based on a reduction in disease severity, itch intensity, and HRQoL. Disease severity is often assessed by lesion count and scoring systems such as the IGA PN-S, with a clinically meaningful response being a reduction to an IGA PN-S score of 0 or 1 (i.e., 5 or fewer lesions). Itch intensity may be assessed by patients’ subjective reporting of improvement (e.g., a “good to excellent” improvement in itch) or with an NRS such as the WI-NRS, with a reduction of at least 4 points considered clinically important. Improvements in quality of life (e.g., on the DLQI) are also important indicators, particularly as PN severely affects sleep and social well-being.
Treatment response is typically assessed at regular intervals, with a follow-up every 3 to 6 months when active, and up to 1 year if disease is controlled (with or without therapy). The experts indicated that the initial treatment response for a biologic would usually be assessed after 6 months. While clinical outcomes in practice may overlap with those used in clinical trials, practical applications focus more on symptom relief and functional improvements. A clinically meaningful response would include a reduction in lesions and/or a substantial reduction in pruritus and improvement in daily life, with variations in how these outcomes are prioritized across physicians.
Discontinuing Treatment
When deciding whether to discontinue treatment with dupilumab, the clinical experts stated they evaluate whether the patient had achieved the desired clinical outcomes within a 6-month time frame. Specifically, a clinically important response would be a decrease in the lesion count from 20 or more at the start of therapy to 5 or fewer, or if worst-itch severity decreased by at least 4 points out of 10 on the WI-NRS. The experts emphasized that some patients may not meet these specific thresholds, but if they had experienced some improvement and wished to continue, then therapy would not be stopped. In patients with a partial response to therapy, decisions to stop treatment would consider whether any other effective treatment options were available. Moreover, a patient-centred approach to treatment and patient-reported “happiness” are considered when deciding whether to continue therapy. The experts also noted that pruritus may improve before lesions or vice versa, and both may not always show simultaneous improvement. If neither outcome is achieved, discontinuation due to a lack of improvement would be considered. Additional factors for discontinuation may include SAEs or the emergence of comorbidities requiring other treatments, but the primary focus should be on achieving clinical milestones related to disease severity and symptom control.
Prescribing Considerations
Both clinical experts agreed that due to the complexity of diagnosing PN, it is essential that treatment with dupilumab be initiated by a dermatology specialist. PN is both underdiagnosed and overdiagnosed by nondermatology providers. PN has a broad differential that requires the clinician to rule out other potential causes. Patients with PN can also have other underlying diagnoses. As such, treatment with dupilumab for PN should be limited to dermatologists. PN is a clinical diagnosis that cannot be reliably diagnosed through biopsy alone as dermatopathologists rely on clinical correlation for pathologic diagnosis, and many conditions appear similar under histology. The clinical experts stated that dermatologists are best equipped to maximize dupilumab treatment outcomes by optimizing the use of concurrent conventional therapies, and they noted that academic (hospital) and community dermatologists are qualified to diagnose PN and prescribe dupilumab.
Clinician Group Input
No clinician groups submitted input for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CDA-AMC for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Before dupilumab there were no Health Canada–approved targeted systemic therapies, only off-label therapies. The comparators used are “best supportive care,” which includes:
Question: Are there other drugs (including off-label) with similar clinical effects as dupilumab in the treatment of PN which should be included as relevant comparators? | The clinical experts agreed that emollients and TCSs with intralesional corticosteroid injections are currently the best supportive care for patients with PN. Although the experts stated that off-label immunosuppressants are used in clinical practice, they lack evidence to support their use and are often ineffective. In addition, short- and longer-term safety concerns with methotrexate or cyclosporin limit their use, especially considering that PN is more common in older patients who often have comorbidities. |
Question: Nemolizumab is also being reviewed for PN. Should dupilumab and nemolizumab be considered in the same line of therapy for PN? | The clinical experts agreed that dupilumab and nemolizumab would be considered in the same line of therapy for patients with PN. |
Considerations for initiation of therapy | |
According to the sponsor, a “diagnosis of PN is clinically determined and is largely one of exclusion. A PN diagnosis is typically based on the presence of core symptoms.” “The workup may include history of comorbidities, possible skin biopsy, laboratory investigations, or radiological investigations.” Question: When should a biopsy be required to confirm a diagnosis of PN? | The clinical experts noted that PN is a clinical diagnosis, and if PN is diagnosed by a dermatologist, a biopsy is not required. PN on biopsy can be similar to other chronic pruritic or excoriating disorders and biopsy results are not entirely specific to PN. |
The PRIME and PRIME2 trials inclusion criteria required:
Question: Should patients have to meet all these inclusion criteria listed to be eligible for dupilumab? | The experts agreed that the inclusion criteria listed were reasonable initiation criteria for reimbursement of dupilumab. Diagnosis of PN by a dermatologist is important as PN may be misdiagnosed or underdiagnosed by other practitioners. The clinical experts commented that, at the time patient eligibility is determined, a specific itch-score requirement may not be necessary, as the criterion of severe itching would be suitable, given that patients with moderate to severe PN would be experiencing severe itching. However, clinical experts acknowledged that documenting a WI-NRS score at baseline may be useful to allow for quantitative follow-up in determining clinical improvement in pruritus. The number of lesions and WI-NRS criteria were consistent with moderate to severe PN. The experts stated that all patients should have tried topical therapies before starting dupilumab or nemolizumab, particularly considering that a TCS would likely be prescribed by family physicians before patients can access a dermatologist. |
Question: Is the WI-NRS score commonly used in practice? | The clinical experts indicated that patients’ subjective reporting of the severity of pruritus is usually used to assess itch intensity. The WI-NRS, while not routinely used, could be incorporated into practice. They stated the WI-NRS would have value as part of reimbursement initiation and renewal criteria, as it provides a numerical measure of pruritus intensity. |
The sponsor suggested the following reimbursement criteria: Initiation condition For the treatment of adult patients with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable defined as:
Renewal condition Patient must exhibit reduced pruritus from baseline, reduced lesion count and improved overall HRQoL as reported by the patient (e.g., sleep disturbance, emotional status) Prescribing conditions: Dupilumab should be prescribed by physicians experienced in the treatment of PN. The initiation conditions requested by the sponsor do not include any baseline scoring; however, renewal conditions include reduced pruritus from baseline, reduced lesion count, and improved HRQoL as reported by the patient (e.g., sleep disturbance, emotional status). Question: Should a baseline score for 1 or both of pruritus (WI-NRS or other) and/or HRQoL be required? | The clinical experts agreed that a baseline WI-NRS score of 7, and an IGA PN-S score of at least 3 (i.e., ≥ 20 PN lesions) were reasonable criteria for initiation of dupilumab therapy. |
The Health Canada indication for dupilumab in PN and reimbursement requested is for adults only. Dupilumab is indicated in pediatric individuals with other medical conditions (asthma, atopic dermatitis, and eosinophilic esophagitis). Question: Would you anticipate dupilumab being used in pediatric individuals with PN? Question: Should pediatric individuals be excluded from dupilumab funding for the treatment of PN? | The experts stated that PN is rare in pediatric patients. At present there are no phase III studies in pediatric patients, and until these data are available, dupilumab should not be approved or reimbursed for these patients. |
Question: Should individuals be required to have an inadequate response to off-label oral immunosuppressant therapies (such as methotrexate) before becoming eligible for dupilumab? | The experts emphasized that off-label systemic immunosuppressants have low efficacy and significant toxicity, which limits their long-term use. As most patients with PN are older and have other comorbidities and polypharmacy, methotrexate and cyclosporin are often not suitable treatments. Dupilumab offers targeted immunosuppression that addresses the underlying disease process and is generally safe for older patients. A requirement to use off-label systemic immunosuppressants before dupilumab would delay effective treatment of many patients. |
Question: Is phototherapy an appropriate alternative for the treatment of PN? Question: Is phototherapy readily available in all areas of Canada? Question: If CDEC determines phototherapy is an appropriate alternative for the treatment of PN, should individuals be required to have an inadequate response to phototherapy (if accessible) before being eligible for dupilumab? | The experts noted that there are significant barriers to access for phototherapy, and wait lists to access this treatment can be substantial. They also noted that the treatment burden is high, as phototherapy requires patients to visit the clinic 3 times a week, which is not feasible for most patients. While it may be effective for some patients, long-term efficacy is an issue. The experts stated patients should not be required to try phototherapy before being eligible for dupilumab. |
The PRIME and PRIME2 studies excluded individuals with PN secondary to medications or medical conditions. Question: Should individuals with secondary PN be excluded from funding? | The experts stated that patients with chronic pruritus associated with medications or other comorbid conditions, but who did not meet the criteria for PN, would not be eligible for dupilumab. Only patients with idiopathic PN or PN related to renal failure, atopic dermatitis, or other underlying conditions would be treated with dupilumab. |
With regard to consistency of initiation criteria with other drugs, the drug programs note that nemolizumab is currently under review by CDA-AMC for the treatment of moderate to severe PN. | This was a comment from the drug programs to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
The requested renewal conditions include reduced pruritus from baseline, reduced lesion count and improved HRQoL as reported by the patient (e.g., sleep disturbance, emotional status). Question: Should patients be required to see an improvement in all 3 areas (pruritus, lesion count, and HRQoL)? Or would improvement in 1 of the 3 areas (pruritus, lesion count, or HRQoL) be sufficient to determine the response? Question: Should response to therapy be based on the patient’s subjective response (i.e., improvement in HRQoL and/or itch), or should an objective measure (scoring tool) be used to determine the response? | The clinical experts stated that improvement in pruritus symptoms and number of lesions are the key considerations for continuation of therapy. The experts considered an itch response to be at least a 4-point improvement in WI-NRS from baseline. One expert indicated an IGA PN-S score of 0 or 1 (≤ 5 lesions) could be used as a criterion for improvement in the number of lesions, but the other expert stated that either an IGA PN-S score of 0 or 1, or a 2-point improvement from baseline, would be appropriate. While both pruritus and lesion-count outcomes are important, the experts agreed that patients should not be required to fulfill both criteria to continue treatment. Patients with a partial response should not be prohibited from receiving dupilumab, given that no other treatment options may be available, and even an incomplete response to 1 or both criteria may be considered important by patients. The clinical experts commented that assessment for treatment renewal (i.e., response to treatment) should occur after 4 to 6 months of therapy. They noted that improvement in lesions may be delayed, relative to relief of pruritus, and that at least 6 months may be needed for patients to show full treatment effects. Typically, in practice, patients are assessed after the first 6 months of therapy and then annually. The clinical experts agreed that for patients who do not maintain a good response to therapy (as previously outlined), treatment with dupilumab should be discontinued. |
The primary end point measured in the PRIME and PRIME2 trials was the proportion of patients with a ≥ 4-point reduction in WI-NRS from baseline at week 24. Question: If an objective measure should be used to determine response and the WI-NRS is commonly used in clinical practice, would a reduction of 4 or more points in the WI-NRS score be an appropriate renewal parameter? | The experts agreed that 4-point improvement in the WI-NRS from baseline would be a reasonable renewal criterion for dupilumab. |
The key secondary end point of the PRIME and PRIME2 trials was an IGA PN-S of 0 to 1 at 24 weeks, with other end points including change from baseline in DLQI, Skin Pain NRS, HADS, and Sleep NRS. Question: Would IGA PN-S, DLQI, Skin Pain NRS, HADS, or Sleep NRS be more appropriate tools than WI-NRS to determine response to therapy? | The clinical experts stated that improvement in pruritus symptoms and number of lesions are the key considerations for continuation of therapy. |
Question: Once the initial response to dupilumab is determined, how often would patients likely be assessed for ongoing response? Question: Should the improvements initially noted in the first 24 weeks of dupilumab be maintained indefinitely? | Patients who start on dupilumab will typically be seen by the dermatologist for a first assessment of response to treatment after 4 to 6 months, preferably 6 months. For those responding well to treatment at that assessment, the next scheduled assessment would be about 1 year later. Dupilumab has shown good longer-term efficacy in other dermatologic conditions, but for patients with PN, the trial data are limited to 24 weeks in duration. |
With regards to consistency of renewal criteria with other drugs, the drug programs note that nemolizumab is currently under review by CDA-AMC for the treatment of moderate to severe PN. | This was a comment from the drug programs to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
The sponsor’s reimbursement request states “dupilumab should be prescribed by physicians experienced in the treatment of PN.” Question: What types of specialists may be “experienced in the treatment of PN”? Question: Would all dermatologists be considered “experienced in the treatment of PN”? Question: If a patient is unable to access a physician experienced in the treatment of PN, are there other practitioners who could manage this condition? | Both clinical experts agreed that due to the complexity of diagnosing PN, it is essential that treatment with dupilumab be initiated by a dermatology specialist. PN has a broad differential that requires the clinician to rule out other potential causes. As such, treatment with dupilumab for PN should be limited to dermatologists as these specialists are best equipped to maximize dupilumab treatment outcomes by optimizing the use of concurrent conventional therapies. Academic (hospital) and community dermatologists are well equipped to diagnose PN and prescribe dupilumab. |
Question: Should individuals be eligible for the use of dupilumab in combination with other treatments for PN (i.e., off-label methotrexate or nemolizumab)? | The experts stated dupilumab would not be used in combination with nemolizumab or cyclosporin, but may be used with methotrexate in patients with severe PN, particularly in the first few months of dupilumab therapy. Dupilumab would be used concurrently with emollients, TCSs, intralesional corticosteroid injections, and potentially phototherapy (if accessible). |
Dupilumab is indicated (and funded in many jurisdictions) for atopic dermatitis and asthma. Additionally, dupilumab is currently under review for nasal polyps. Question: If an individual has multiple medical conditions that may be treated with dupilumab (and is funded by drugs plans; i.e., atopic dermatitis or asthma), should reimbursement of multiple biologics be considered for the individual? Alternatively, should the individual be provided funding for only 1 biologic that treats both conditions? For instance, if the patient is being treated for PN with dupilumab and another product (e.g., mepolizumab) for asthma, should they be eligible for funding of both? Or should the individual be provided funding for only 1 product which is indicated in the treatment of both conditions? | The experts indicated that dupilumab would not be used in combination with other biologics. If a patient had multiple conditions, clinicians would try to find 1 biologic that could be used to manage both conditions. |
With regard to consistency of prescribing criteria with other drugs, the drug programs note that nemolizumab is currently under review by CDA-AMC for the treatment of moderate to severe PN. | This was a comment from the drug programs to inform CDEC deliberations. |
Generalizability | |
Is the efficacy and safety of dupilumab as observed in the adult clinical trials generalizable to pediatric patients? | The clinical experts stated that PN is rare in children. Based on their clinical practice, they had no comments on the use of dupilumab in pediatric patients with PN. |
System and economic issues | |
The budget impact may be more substantial if off-label oral immunotherapies (e.g., methotrexate) are not required to be trialled before initiation of dupilumab for PN. | This was a comment from the drug programs to inform CDEC deliberations. |
Dupilumab has successfully gone through price negotiations for asthma and atopic dermatitis. | This was a comment from the drug programs to inform CDEC deliberations. |
CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; NRS = numerical rating scale; PN = prurigo nodularis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; WI-NRS = Worst Itch Numerical Rating Scale.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of dupilumab 300 mg SC injection in the treatment of moderate to severe PN in adults whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The focus is on comparing dupilumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of dupilumab is presented in 2 sections, with a critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. An assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes additional studies to address gaps in the systematic review evidence.
No long-term extension studies or indirect treatment comparisons were submitted by the sponsor.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
2 RCTs identified in the systematic review
5 observational studies to address gaps in the evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The follow material has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | PRIME | PRIME2 |
|---|---|---|
Designs and populations | ||
Study design | Multicentre, randomized, double-blind, placebo-controlled, parallel-group, phase III trial | |
Locations | 63 sites in 8 countries (US, Argentina, Mexico, Mainland China, Japan, Russian Federation, Republic of Korea, France) | 57 sites in 11 countries (Canada, Chile, France, Hungary, Italy, Portugal, Republic of Korea, Spain, Taiwan, UK, US) |
Patient enrolment dates | Start date: December 2019 End date: February 2022 Data cut-off: ██ ████████ ███ | Start date: January 2020 End date: November 2021 Data cut-off: ██ ██████ ████ |
Randomized (N) | 151 | 160 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Dupilumab 600 mg (2 SC injections of 300 mg) followed by 300 mg by SC injection q.2.w. | Same as PRIME |
Comparator | Matched placebo SC injections | Same as PRIME |
Study duration | ||
Screening phase | 2 to 4 weeks | 2 to 4 weeks |
Treatment phase | 24 weeks | 24 weeks |
Follow-up phase | 12 weeks | 12 weeks |
Outcomes | ||
Primary end point | Pruritus improvement measured by the proportion of patients with a ≥ 4-point reduction in WI-NRS from baseline at week 24 | Pruritus improvement measured by the proportion of patients with a ≥ 4-point reduction in WI-NRS from baseline at week 12 |
Key secondary end points |
|
|
Other secondary end points |
|
|
Other end points |
|
|
Publication status | ||
Publications | Yosipovitch et al. (2023)29 Clinicaltrials.gov: NCT0418333530 | Yosipovitch et al. (2023)29 Clinicaltrials.gov: NCT0420267931 |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; HADS = Hospital Anxiety and Depression Scale; IGA PN-A = Investigator’s Global Assessment Prurigo Nodularis–Activity; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; IST = immunosuppressant; NRS = numerical rating scale; PN = prurigo nodularis; q.2.w = every 2 weeks; SC = subcutaneous; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; WI-NRS = Worst Itch Numerical Rating Scale.
a████ ████████ ███ ███ ███████ ████████ ████ ████████ ███ ████ ███ ███ █████ █████ ███ ██████ ███ ████ ███ ███ ██████ ██████ ███ ████████ █████████ ███ ███ ██ █████████ █████ ██████ ████ ██ ████████ █████ ███ ██ ████████ █████ ███████ ███████████ ██ ███ ███████ █████████ ██████.
bPatients with comorbid conditions associated with PN were allowed to enrol in the trials; however, a cap was instituted such that no more that 60% of patients had a history of atopy (of whom only 10% in this subgroup could have active mild atopic dermatitis). Patients with moderate to severe atopic dermatitis were excluded.
cFailure was defined as patients who were unable to achieve and/or maintain remission and low disease activity (similar to an IGA PN-S score of ≤ 2 [≤ 19 nodules]) despite treatment with a daily regimen of a medium- to super-potent TCS (with or without a TCI as appropriate), applied for at least 14 days, or for the maximum duration recommended by the product prescribing information, whichever is shorter.
Sources: Clinical Study Report for PRIME,9 Clinical Study Report for PRIME2,10 and the sponsor’s Summary of Clinical Evidence.32
The primary objective of both the PRIME and PRIME2 trials was to evaluate the efficacy and safety of dupilumab in adult patients with PN who were inadequately controlled on topical prescription therapies or when those therapies were not advisable. Both trials were designed as international, randomized, double-blind, placebo-controlled, multicentre, parallel-group, phase III studies that enrolled 151 patients (PRIME) and 160 patients (PRIME2).
Enrolled patients were centrally randomized (1:1) using interactive response technology to receive either dupilumab 300 mg every 2 weeks (with a 600 mg loading dose) or matching placebo. Randomization was stratified by a documented history of atopy (atopic or nonatopic), stable use of a TCS or TCI (yes or no), and country or territory code. The studies included a screening period of 2 to 4 weeks (to determine stable regimens for TCS and TCI use), a 24-week treatment period, and a 12-week post-treatment follow-up period. Patients in both trials were enrolled from North and South America, Europe and Asia. The PRIME2 study included 6 study sites in Canada.
The Clinical Study Reports include information collected up to ████████ ███ ████ for PRIME and ██████ ███ ████ for PRIME2, when the last patients enrolled had completed the end of treatment visit. In the PRIME and PRIME2 studies, ██ ████████ █████ ███ ██ ████████ ██████ █████████████ ████ ███████ ███████████ ██ ███ ███████ █████████ █████. The study design of the trials is shown in Figure 1.
Figure 1: Study Design of the PRIME and PRIME2 Clinical Trials
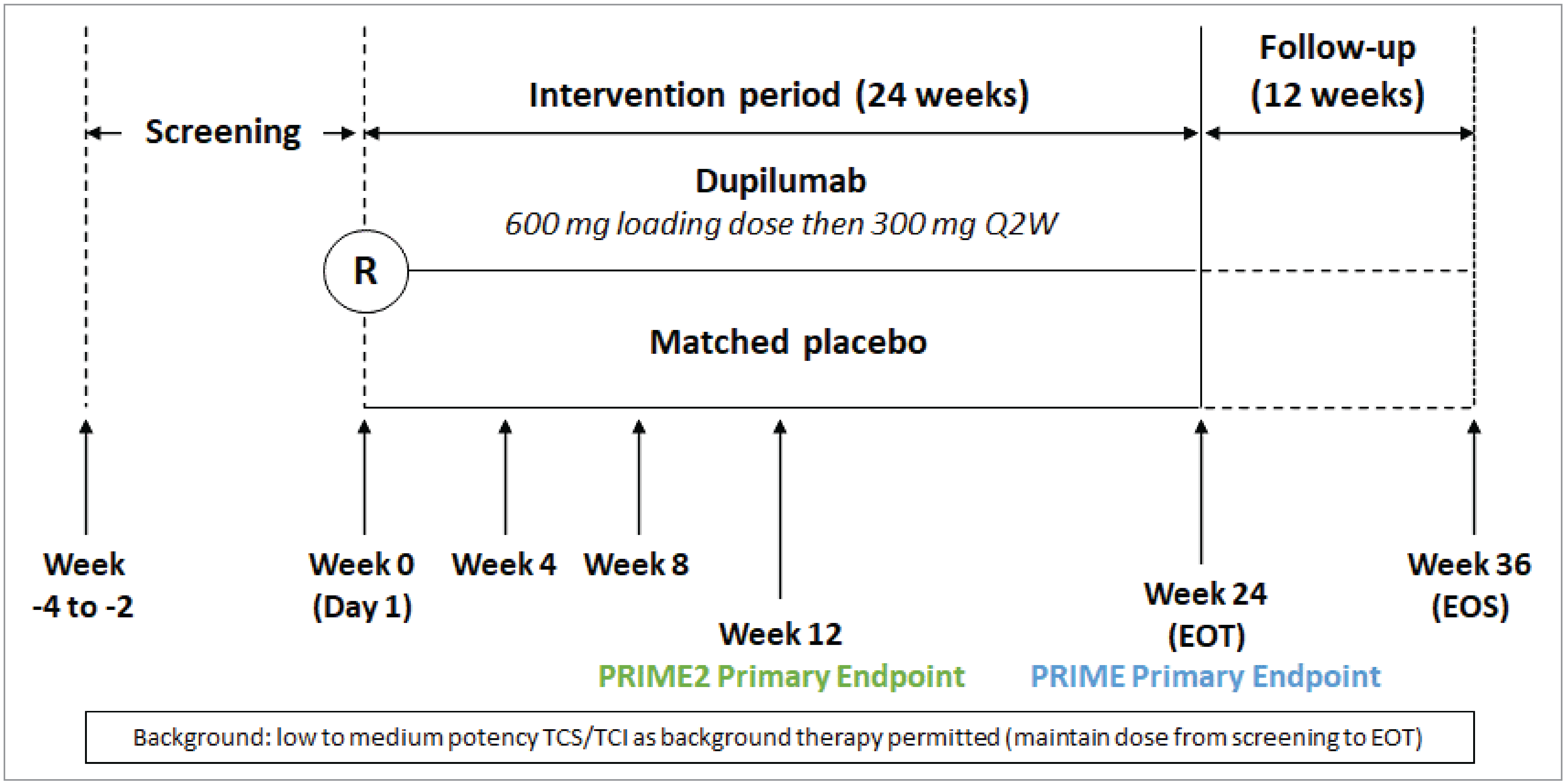
EOS = end of study; EOT = end of treatment; PN = prurigo nodularis; Q2W = every 2 weeks; R = randomization; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
Notes: In the PRIME2 study the primary end point was measured at week 12, whereas in the PRIME study it was measured at week 24. Background therapy with a low- to medium-potency TCS or TCI were permitted at stable doses from screening to the EOT.
Source: Sponsor’s Summary of Clinical Evidence.32
Populations
The inclusion and exclusion criteria for PRIME and PRIME2 trials were identical. The studies enrolled adult patients (18 to 80 years) with a clinical diagnosis of PN whose disease was inadequately controlled on topical prescription therapies or for whom those therapies were not advisable (Table 5). Patients were required to have at least 20 PN lesions in total on both legs, and/or both arms, and/or trunk at the screening visit and on day 1; a history of failure of a 2-week course of a medium- to super-potent TCS or that made TCS not medically advisable; and an average WI-NRS score of at least 7 points in the 7 days before day 1 (randomization). Patients were also required to be adherent to topical emollient administration, applying moisturizer once or twice daily for at least 5 out of 7 consecutive days immediately before day 1.
The study population included patients with underlying systemic medical conditions known to be associated with PN; however, a cap was instituted such that no more than 60% of enrolled patients could have a history of atopy. The total number of patients with active mild atopic dermatitis in this atopic subgroup was capped at 10%, while patients with moderate to severe atopic dermatitis were excluded.
Patients were also excluded if they had other skin morbidities that may interfere with the assessment of the study outcomes, PN secondary to medications or to other medical conditions, severe concomitant illness, active chronic or acute infection (except HIV infection) requiring treatment with systemic antibiotics, antivirals, antiprotozoals, or antifungals during screening or the 2 weeks before the screening visit.
Interventions
Both studies were double-blinded, with patients receiving either dupilumab 600 mg initial loading dose (2 injections of 300 mg) followed by 300 mg every 14 days (± 3 days) by SC injection, or matched placebo by SC injection. The study drug or placebo was supplied as 2 mL prefilled syringes. The investigator or delegate trained the patient (or caregiver) on how to prepare and inject the study drug and administered the first injection, whereas the patient (or caregiver) administered the second injection under supervision. The patient or caregiver administered subsequent injections.
Patients were required to apply moisturizers once or twice daily throughout the study, and those on a stable regimen of a low- to medium-potency TCS or TCI at screening could continue these therapies during the 24-week treatment period. Patients who were on a stable regimen of a high-potency or super-potent TCS were to decrease TCS potency to medium during the screening and study-drug treatment periods. Occlusion was not allowed during the trial.
Dermatological preparations of a high-potency or super-potent TCS or TCI were available as rescue medications at the discretion of the investigator if medically necessary (i.e., to control intolerable PN symptoms).
The concomitant use of nonsedating antihistamines was allowed during the study if they were used to treat other conditions besides atopic dermatitis or PN. Patients on stable doses of antidepressants could continue these therapies provided they had been on stable doses for at least 3 months before screening.
Concomitant use of the following treatments were prohibited during the study, and the study drug was stopped in any patients who received these treatments: systemic immunosuppressive drugs (e.g., systemic corticosteroids, cyclosporine, mycophenolate mofetil, interferon gamma, Janus kinase inhibitors, azathioprine, methotrexate, hydroxychloroquine, dapsone, sulfasalazine, and colchicine); other monoclonal antibodies that are biological modifiers; phototherapy (including tanning beds); naltrexone or other opioid antagonists; gabapentin; pregabalin; and thalidomide. Patients had to stop these treatments at least 4 weeks before the screening visit. Other prohibited medications included cell-depleting drugs (e.g., rituximab) within 6 months of screening, and omalizumab and other immunomodulatory biologics within 5 months or 5 half-lives of screening. Intralesional corticosteroid injections or cryotherapy, sedating antihistamines, and nonsedating antihistamines (when used specifically for the treatment of pruritus due to atopic dermatitis and PN) were prohibited during the trial, but patients who received these therapies did not need to discontinue the study drug.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important by the clinical experts consulted for this review and input from patient groups and public drug plans. Using the same considerations, we selected end points that were considered most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Select notable harms outcomes considered important to expert committee deliberations were also assessed using GRADE.
Pruritus intensity was identified as the symptom that had the greatest impact on patients. The proportion of patients with at least a 4-point improvement in the WI-NRS was selected as a clinically relevant end point by the clinical experts consulted for this review: it was the primary end point in both trials, and was used to inform the pharmacoeconomic model. Disease severity, focusing on the clearance of lesions (i.e., IGA PN-S score of 0 or 1) was also identified in patient group and clinical expert input. As PN may have a substantial impact on patients’ HRQoL, the change from baseline in DLQI was selected as key end point. The clinical experts indicated that dupilumab is associated with conjunctivitis-related AEs and it is an important treatment-related harm.
As a supplementary end point, the change and percent change from baseline to week 24 in the weekly average WI-NRS score and trajectory over time are summarized in Appendix 1 (Table 20, Figure 2 and Figure 3) at the request of members of the expert committee. While patients indicated sleep is often affected by the itching associated with PN, the instrument used to assess sleep in the clinical trials had limitations. In both studies, patients’ sleep quality was measured using an NRS from 0 to 10, where 0 indicated the worst possible sleep and 10 the best possible sleep. Based on clinical expert input, Sleep NRS results were likely confounded by other factors that may affect sleep and would not be sufficient to assess the impact of PN symptoms on sleep disturbance. For this reason, Sleep NRS data have not been included in this report.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | PRIME | PRIME2 |
|---|---|---|---|
Key outcomes (assessed using GRADE) | |||
Proportion of patients with reduction in WI-NRS of ≥ 4 points from baseline | To 24 weeks | Primarya | Key secondarya |
Proportion of patients with reduction in WI-NRS of ≥ 4 points from baseline | To 12 weeks | Secondary | Primarya |
Proportion of patients with reduction in skin lesion numbers to an IGA PN-S score of 0 or 1 | At week 24 | Key secondarya | Key secondarya |
Change from baseline in DLQI | At week 24 | Secondarya | Secondarya |
Supplementary outcome (not assessed with GRADE) | |||
Change and percent change from baseline to week 24 in weekly average WI-NRS score | To 24 weeks | Secondaryb | Secondaryb |
DLQI = Dermatology Life Quality Index; GRADE = Grading of Recommendations Assessment, Development and Evaluation; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; WI-NRS = Worst Itch Numerical Rating Scale.
aStatistical testing for these end points was adjusted for multiple comparisons and are reported in the order of hierarchal testing.
bOnly the percent change from baseline in the WI-NRS was part of the statistical testing hierarchy and adjusted for multiple comparisons.
Sources: Clinical Study Report for PRIME,9 Clinical Study Report for PRIME2,10 and the sponsor’s Summary of Clinical Evidence.32
Pruritus
The primary outcome in both studies was the proportion of patients with an improvement (reduction) in the WI-NRS of at least 4 points from baseline (Table 7). In the PRIME study, the primary end point was measured at week 24, with the week 12 analyses used as a secondary outcome without control for multiple comparisons. In the PRIME2 study the primary end point was measured at week 12, with the 24-week analysis used as a multiple comparison–controlled key secondary end point. The PRIME and PRIME2 studies originally had identical efficacy outcomes that were controlled for type I error for multiple comparisons. However, the results of the PRIME2 study, which became available while the results for the PRIME study were ongoing, showed that the benefit of dupilumab treatment continued to increase beyond week 12 and to at least week 24. Consequently, the PRIME study protocol and statistical analysis plan were updated before the study database lock such that the primary efficacy outcome was assessed at week 24 and not at week 12 (protocol amendment 03, October 2021). The percent change from baseline in the WI-NRS at week 24 was a secondary outcome and was part of the hierarchical statistical testing procedure for both studies. Data for the absolute change from baseline in the WI-NRS over time was also reported.
A 4-point change in WI-NRS (range, 3.0 to 4.5) in patients with PN was estimated to represent a minimal important difference (MID) for within-patient improvement.33-35 Based on clinical expert input, the threshold for a clinically important between-group difference was 20% for the proportion of patients with at least a 4-point reduction from baseline.
Patients completed the WI-NRS daily and rated the intensity of their worst pruritus over the past 24 hours using this scale. Both daily and weekly WI-NRS scores were calculated (i.e., the weekly score was the average of daily nonmissing scores within the week window of each trial week). The analyses presented were based on the 7-day average of daily WI-NRS scores over the previous week.
Disease Severity
Key secondary end points in both trials included the proportion of patients with an IGA PN-S score of 0 or 1 at week 24 (Table 7). While no MID was reported in the literature, the clinical experts consulted for this review considered a change from a score of 3 or 4 to a score of 0 or 1 to be clinically important. Based on this input, the threshold for a clinically important between-group difference was 15% for the proportion of patients with an IGA PN-S score of 0 or 1.
Health-Related Quality of Life
HRQoL was assessed based on the change from baseline in the DLQI score to week 24 as a secondary outcome in both studies (controlled for multiple comparisons) (Table 7). The sponsor stated that there is evidence to support the validity of the DLQI in clinical practice and clinical trials, including evidence of its validity, reliability, and responsiveness in patients with PN.11,36-40 In patients with PN, a 4-point change in DLQI (range, 3.5 to 6.5) was estimated to represent a meaningful between-group change.11
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
WI-NRS | A single-item, patient-reported scale based on a 11-point rating scale where 0 = “no itch” to 10 = “worst imaginable itch,” the WI-NRS is based on a patient’s report of their worst itch (intensity of itch) over the past 24 hours. Both daily and weekly WI-NRS scores were calculated. A lower score on the WI-NRS indicates improvement.41 | The psychometric properties of the WI-NRS were specifically evaluated in a secondary analysis of the PRIME and PRIME2 trials using pooled data from the ITT patient population (N = 311) with PN.42,43 One other study reported psychometric data from patients with multiple different pruritic conditions, including PN.41 Validity: Content validity was assessed through in-depth 1:1 interviews with 20 patients.42 The WI-NRS questions, recall period, and response scale were easy to understand and relevant for patients.42 Convergent and discriminant validity was supported by moderate to strong correlations between WI-NRS scores and those of similar constructs (Skin Pain NRS, Sleep NRS; absolute r range, 0.34 to 0.73) and weaker correlations with less-related measures (PAS, IGA PN-S, HADS; absolute r range, 0.06 to 0.32).42 The WI-NRS was able to distinguish between different disease severity levels and QoL effects defined by PGIS, IGA PN-S, and DLQI scores at baseline and week 12.42 In another study, convergent validity with the DLQI was low (Spearman correlation coefficient = 0.17) in the subgroup with PN.41 Reliability: Adequate test-retest reliability of the WI-NRS was observed in stable patients (according to PGIS) between screening and baseline (ICC = 0.72).42 This is corroborated by earlier psychometric analyses of WI-NRS that reported ICCs of 0.78 and 0.96 in patients with PN.33,34 Another study reported moderate agreement for test-retest reliability (weighted kappa = 0.64) in patients with PN.41 Responsiveness: The WI-NRS was sensitive to change as moderate to strong correlations were observed for change in WI-NRS scores and change in Skin Pain NRS, Sleep NRS, DLQI total score, and PGIS, and with PGIC from baseline to week 12 (absolute r range, 0.40 to 0.76).42 The WI-NRS was also sensitive to changes in disease and QoL-related outcomes in another psychometric analysis.34 | Thresholds for meaningful within-patient improvement in the WI-NRS in patients with PN were determined using anchor and distribution-based approaches. Using PGIS and PGIC as anchors, it was concluded that a clinically meaningful improvement threshold on the WI-NRS was 4 points (range, 3.0 to 4.5), which was also supported by distribution methods.42 Earlier psychometric analyses have reported MIDs of ≥ 3.8-point improvement in the WI-NRS in patients with PN (corresponding to approximately 40% improvement) and 2.3 to 4.5 points, corresponding to a 30% to 60% improvement in WI-NRS scores.33,34 |
IGA PN-S | A clinician-reported assessment of the number of palpable pruriginous nodules in a representative area, the IGA PN-S instrument uses a 5-point Likert scale ranging from 0 to 4 where 0 = “clear”; no nodules, 1 = “almost clear”; rare palpable nodules (≤ 5 nodules), 2 = “mild”; few palpable nodules (6 to 19 nodules), 3 = “moderate”; many palpable nodules (20 to 100 nodules), and 4 = “severe”; abundant palpable nodules (> 100 nodules). A lower score indicates improvement. | Validation of the IGA CPG stage and IGA CNPG stage (which is identical to the IGA PN-S) was completed in 187 consecutive patients with chronic prurigo.44 Validity: Correlations between the IGA CPG stage, IGA CNPG stage, and IGA CPG activity) were statistically significant (P < 0.001). Correlation between IGA CPG stage and IGA CNPG stage was strong (Kendall t = 0.62) and with IGA CPG activity was moderate (t = 0.468). Convergent validity showed strong correlations between IGA CPG stage and PAS item 2 (t = 0.794; P < 0.01), total counted number of all pruriginous lesions (t = 0.729, P < 0.01), moderate correlation with NRS worst 24 hours (t = 0.343; P < 0.01), and weak correlation with DLQI (t = 0.265; P < 0.01). IGA CPNG stage showed weak correlation with NRS worst 24 hours and DLQI (t = 0.162 to 0.231) and moderate correlation with PAS (t = 0.328 to 0.485). Reliability: Intrarater test-retest reliability was evaluated using 5 experienced dermatologist raters who completed the 3 IGA instruments for 8 randomly chosen patients twice within 1 hour. Results were IGA CPG stage (ICC = 0.915) and IGA CNPG stage (ICC = 0.874). Congruence among raters was good for both IGA CPG stage and IGA CNPG stage (Kendall W = 0.747 for both). Responsiveness: No evidence was identified. | No MID has been identified in adult patients with PN. |
DLQI | The DLQI is a patient self-reported, dermatology-specific HRQoL instrument validated for dermatology patients ≥ 16 years of age and used in more than 36 skin conditions.38,39 It comprises 10 items that assess patient perception (7-day recall) of the impact of their skin disease on 5 different aspects of HRQoL (dermatology-related symptoms and feelings; daily activities and leisure; school and work performance; personal relationships; treatment). Each item is scored on a 4-point Likert scale where 0 = “not at all/not relevant,” 1 = “only a little,” 2 = “quite a lot,” and 3 = “very much.” Total DLQI score is the sum of the 10 items (0 to 30 points) with a higher score indicating greater impairment of a patient’s HRQoL (i.e., 0 to 1 = no effect, 2 to 5 = small effect, 6 to 10 = moderate effect, 11 to 20 = very large effect, 21 to 30 = extremely large effect). | The psychometric and measurement properties of the DLQI were evaluated in patients with PN (N = 311) based on the pooled ITT patient population from the PRIME and PRIME2 trials.11,37 Validity: Content validity was assessed through in-person interviews with 19 patients.37 Patients found the items, recall period, and response scale easy to understand and relevant. Convergent validity of DLQI total scores was shown by moderate correlations (absolute r range: 0.33 to 0.50) between scores expected to be closely related (e.g., WI-NRS, Skin Pain NRS, Sleep NRS, PGIS, EQ-5D pain/discomfort items, EQ VAS, HADS A and D). Divergent validity was supported at baseline with lower correlations (absolute r range: 0.03 to 0.22) for scores that were less closely related (e.g., IGA PN-A, IGA PN-S, and PAS).11 Reliability: Adequate test-retest reliability for DLQI total scores was observed using the PGIS (ICC range: 0.64 to 0.87) and good to excellent internal consistency at baseline (Cronbach alpha = 0.88).11 Responsiveness: The responsiveness of DLQI total scores was shown by moderate to strong correlations (absolute r = 0.30 to 0.50 and > 0.50, respectively) with PGIS, PGIC, WI-NRS, Skin Pain NRS, Sleep NRS, and EQ VAS change scores except for correlations at baseline with EQ VAS.11 | Within-patient change thresholds for DLQI total scores were derived using an anchor-based approach, with PGIS and PGIC as anchors and time points week 12 and week 24. At week 12, the MID for DLQI total score in patients with PN ranged from −9.00 to −10.00 (using PGIS improved 1 category and PGIC moderately better), whereas at week 24, the MID ranged from −8.00 to −10.00.11 This exceeded the distribution-based values of 0.5 SD = 3.50 and SEM = 4.22. These values also exceed the MID for the DLQI of 2.24 to −3.10 recommended for interpreting results for patients with chronic idiopathic urticaria.40 For patients with PN, Stander et al. (2024)11 estimated the within-patient improvement threshold for DLQI total score was 9.0 points. A 4.0-point change in DLQI (range, 3.5 to 6.5) indicated a between-group meaningful change.11 |
DLQI = Dermatology Life Quality Index; EQ VAS = EQ visual analogue scale; HADS = Hospital Anxiety and Depression Scale; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; IGA = Investigator’s Global Assessment; IGA CNPG = Investigator’s Global Assessment Chronic Nodular Prurigo; IGA CPG = Investigator’s Global Assessment Chronic Prurigo; IGA PN-A = Investigator’s Global Assessment Prurigo Nodularis–Activity; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; ITT = intention to treat; MID = minimal important difference; NRS = numerical rating scale; PAS = Prurigo Activity and Severity Score; PGIC = Physician’s Global Impression of Change; PGIS = Physician’s Global Impression of Severity; PN = prurigo nodularis; QoL = quality of life; SD = standard deviation; SEM = standard error of the mean; WI-NRS = Worst Itch Numerical Rating Scale.
Note: The table was provided by the sponsor (and validated by the CDA-AMC review team.
Source: Sponsor’s Summary of Clinical Evidence.32
Statistical Analysis
The primary outcome in both studies was the proportion of patients with an improvement (reduction) in weekly average WI-NRS by at least 4 points from baseline to week 24 (the PRIME trial) or week 12 (the PRIME2 trial). This end point was analyzed using a Cochran-Mantel-Haenszel test adjusted for the randomization stratification factors (i.e., history of atopy, stable use of a TCS or TCI, and region). A treatment policy and composite estimand was used to address intercurrent events. Data from patients who stopped the study drug before week 24, including data collected after discontinuation, were used in the analysis (i.e., treatment policy estimand). Patients who used prohibited or rescue medications, or who had missing data, were analyzed as nonresponders, as per the composite estimand. Three supplementary analyses were conducted for the primary end point, as described in Table 8. The proportion of patients with an IGA PN-S score of 0 or 1 at week 24 was analyzed using the same methods applied to the WI-NRS end point.
The change from baseline in DLQI was analyzed using an analysis of covariance (ANCOVA) model with intervention group, stratification factors, baseline antidepressant use, and relevant baseline measurement as covariates. Data from patients who took prohibited or rescue medications was set to “missing” after the medication usage, and the worst postbaseline value on or before the time of medication use was used to impute the missing end point values. For patients who stopped the study drug due to lack of efficacy, but did not use prohibited medications, all data after discontinuation was included in the analysis (with the worst outcome carried forward if data were missing). For patients who stopped treatment due to other reasons, all postdiscontinuation data were included in the analysis but with a multiple imputation approach to impute missing data.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Supplementary analyses |
|---|---|---|---|---|
Proportion of patients with reduction in WI-NRS by ≥ 4 points from baseline to week 24 or to week 12 Proportion of patients with an IGA PN-S 0 or 1 score at week 24 | Binary response end point analyzed using the CMH test Categorical and ordinal data summarized using counts and percentages of patients; OR and response rate differences with 95% CIs and P values provided | Documented history of atopy (atopic or nonatopic), stable use of a TCS or TCI (yes or no), region (countries combined) and baseline antidepressant use (yes or no) | Patients with missing data at week 24 or week 12 were considered nonresponders |
|
Change from baseline in DLQI at week 24a | Continuous end point analyzed using ANCOVA Statistical inference obtained from all imputed data by an ANCOVA model were combined using the Rubin rule Continuous data summarized by number of observations, mean (SD), and median (range) | Intervention group, documented history of atopy (atopic or nonatopic), stable use of a TCS or TCI (yes or no), region (countries combined) and baseline antidepressant use (yes or no), and baseline DLQI measurement as covariates | Efficacy data after rescue treatment or missing data after treatment discontinuation due to lack of efficacy were imputed by WOCF and other missing data were imputed by MI | Not reported |
ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DLQI = Dermatology Life Quality Index; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; MI = multiple imputation; OR = odds ratio; SD = standard deviation; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; WI-NRS = Worst Itch Numerical Rating Scale; WOCF = worst observation carried forward.
aThe same methods were used to analyze the absolute and percent change from baseline to week 24 in the weekly average WI-NRS score (a supplementary outcome for this review).
Sources: Clinical Study Report for PRIME,9 Clinical Study Report for PRIME2,10 and the sponsor’s Summary of Clinical Evidence.32
Sample Size and Power Calculation
A sample size of 150 patients per study was estimated to provide 90% power to detect a 28% difference in the primary outcome (proportion of patients with ≥ 4-point reduction in WI-NRS at week 12) between dupilumab and placebo with a Fisher exact test at a 2-sided level of 0.05, assuming response rates of 39% and 11%, respectively.29 The magnitude of effect was based on the effect of dupilumab in patients with atopic dermatitis in the SOLO1 (AD-1334) and SOLO2 (AD-1416) trials.45 Because the sample size assumed a 15% dropout rate during 12 weeks of treatment, the target was to randomize 75 patients per treatment group, with a cap of up to 10% of patients in the atopic population having active mild atopic dermatitis.
Procedure to Control for Multiple Comparisons
Two prespecified statistical hierarchical testing procedures were used to control the type I error for multiple comparisons; 1 for the US and US reference countries and the other for European and all other countries, including Japan. For both statistical hierarchies, hierarchical testing was applied to the primary, key secondary, and other selected end points for the comparison between dupilumab and placebo to control the overall type I error rate at a 2-sided significance level of 0.05. The comparisons of dupilumab versus placebo were tested based on the hierarchical order:
In US and US reference countries:
Proportion of participants with improvement (reduction) in WI-NRS of ≥ 4 from baseline to week 12
Proportion of participants with improvement (reduction) in WI-NRS of ≥ 4 from baseline to week 24
Proportion of participants with IGA PN-S score of 0 or 1 at week 24
Proportion of participants with both an improvement (reduction) in WI-NRS of ≥ 4 from baseline to week 24 and an IGA PN-S score of 0 or 1 at week 24
Proportion of participants with IGA PN-S score of 0 or 1 at week 12
Percent change from baseline in WI-NRS at week 24
Change from baseline in HRQoL, as measured by the DLQI to week 24
Change from baseline in Skin Pain NRS to week 24
Change from baseline in Sleep NRS to week 24
Change from baseline in Hospital Anxiety and Depression Scale total score to week 24
In all other countries (including European Union and European Union reference countries, as well as China and Japan):
Proportion of participants with improvement (reduction) in WI-NRS of ≥ 4 from baseline to week 12
Proportion of participants with improvement (reduction) in WI-NRS of ≥ 4 from baseline to week 24
Proportion of participants with IGA PN-S score of 0 or 1 at week 24
Percent change from baseline in WI-NRS at week 24
Proportion of participants with IGA PN-S score of 0 or 1 at week 12 (PRIME2 only)
Change from baseline in the DLQI to week 24
Change from baseline in Skin Pain NRS to week 24
Change from baseline in Sleep NRS to week 24 (PRIME2 only)
Change from baseline in Hospital Anxiety and Depression Scale total score to week 24
The sponsor focused on the results reported in the hierarchical order corresponding with that used for European reference countries, consistent with the review of the data by Health Canada. For both the PRIME and PRIME2 studies, the outcome was considered positive if the primary outcome achieved statistical significance. Each subsequent outcome was tested and considered statistically significant only if the prior tested outcome was statistically significant. For outcomes tested outside of the statistical hierarchy, nominal P values were provided.
Subgroup Analyses
Both studies analyzed subgroups for the primary outcome to assess the homogeneity of the treatment effect across various populations. The prespecified subgroups were: age group (< 65 years, ≥ 65 years); gender; region; territory; race; ethnicity; baseline weight (< 60 kg, 60 kg to < 90 kg, ≥ 90 kg); baseline body mass index (< 25 kg/m2, 25 kg/m2 to < 30 kg/m2, ≥ 30 kg/m2); participants without a current diagnosis of atopic dermatitis; history of atopy (atopic or nonatopic); stable use of a TCS or TCI (yes or no); antidepressant use (yes or no) at baseline; and baseline IGA PN-S score of moderate versus severe (3 versus 4). Of these subgroups, the sponsor identified atopy, atopic dermatitis, topical therapies, and baseline IGA PN-S as subgroups most relevant to this review. Logistic regression models were used to test the interaction between the treatment and the subgroup factor. The subgroup analyses were not controlled for type I error for multiple comparisons.
Analysis Populations
In the PRIME and PRIME2 studies, all efficacy outcomes were based on the intention-to-treat population, which included all randomized patients analyzed according to the intervention group allocated by randomization regardless of whether the study drug was used. The safety population included all patients randomly assigned to study intervention who took at least 1 dose of the study intervention. Patients were analyzed according to the intervention they actually received.
Results
Patient Disposition
The PRIME study screened 200 patients, of whom 151 (75.5%) were randomized. In the dupilumab group, ████ of patients discontinued from the study, compared with █████ in the placebo group (Table 9). The main reason for discontinuation was withdrawal by the patient (4.0% and 13.2% and in the dupilumab and placebo groups, respectively). Fewer patients in the dupilumab group discontinued the study drug early compared with the placebo group (1.3% versus 21.1%, respectively).
In the PRIME2 study, 221 patients were screened, and 160 patients (72.4%) were randomized. During the trial, ████ of patients in the dupilumab group and █████ of patients in the placebo group discontinued the study early. Withdrawal by patient was the most common reason for discontinuation (6.4% and 25.6% for the dupilumab and placebo groups, respectively). Over the 24-week treatment period, 2.6% of patients in the dupilumab group and 30.5% of patients in the placebo group stopped the study drug early.
Table 9: Summary of Patient Disposition in the PRIME and PRIME2 Studies
Patient disposition | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab | Placebo | Dupilumab | Placebo | |
Screened, N | 200 | 221 | ||
Screening failures, N (%) | 49 (24.5) | 61 (27.6) | ||
Reason for screening failure, N (%)a | ||||
Not meeting inclusion criteriab | 18 (9.0) | 17 (7.7) | ||
Met exclusion criteria | ██ ██████ | 44 (19.9) | ||
Randomized, N (%) | 75 | 76c | 78d | 82 |
Discontinued from study, N (%) | █████ | ██ ██████ | █████ | ██ ██████ |
Reason for discontinuation, N (%) | ||||
Adverse events | 0 (0) | 1 (1.3) | 0 (0) | 2 (2.4) |
Poor adherence to protocol | 0 (0) | 0 (0) | 0 (0) | 1 (1.2) |
Withdrawal by patient | 3 (4.0) | 10 (13.2) | 5 (6.4) | 21 (25.6) |
Discontinued study drug, N (%) | 1 (1.3) | 16 (21.1) | 2 (2.6) | 25 (30.5) |
Reason for discontinuation, N (%) | ||||
Adverse events | 0 | 3 (3.9) | 0 | 2 (2.4) |
Lack of efficacy | 0 | 5 (6.6) | 1 (1.3) | 11 (13.4) |
Poor adherence to protocol | 1 (1.3) | 0 | 0 | 2 (2.4) |
Withdrawal by patient | 0 | 8 (10.5) | 1 (1.3) | 10 (12.2) |
ITT, N | 75 | 76 | 78 | 82 |
Safety, N | 75 | 75 | 77 | 82 |
ITT = intention-to-treat.
aA patient could have had more than one criterion not met for inclusion or exclusion.
bThe most common reason in both studies was patients did not have an average WI-NRS score of at least 7 in the week before randomization (6% and 4% of screened patients in PRIME and PRIME2, respectively).
cOne patient in PRIME was randomized to placebo but not treated due to patient’s decision.
dOne patient in PRIME2 was randomized to dupilumab but not treated due to patient’s decision.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Baseline Characteristics
The baseline characteristics outlined in Table 10 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
The patients enrolled in the 2 trials had similar characteristics, with a mean age per treatment group ranging from 46.7 years (SD = 15.2) to 51.1 years (SD = 15.8). Most patients were female (62.2% to 69.3%) and the minority were male (30.7% to 37.8%). The mean duration of PN was between 5.4 years (SD = 6.9) and 6.0 years (SD = 7.6), and less than half of patients enrolled had a history of atopy (36.8% to 48.8%). The majority of patients were on stable doses of a TCS or TCI (56.1% to 62.7%), with 9.0% to 12.0% of patients receiving antidepressants at baseline. Based on the IGA PN-S score, more patients had moderate PN (60.5% to 72.0%) than severe PN (28.0% to 39.5%), and the mean WI-NRS score at baseline ranged from 8.3 (SD = 1.1) to 8.6 (SD = 0.9).
There were some minor differences between groups within each study in the population demographics (age, sex, and race) and comorbid hypertension and diabetes, but overall, the disease-related characteristics appeared to be similar across the dupilumab and placebo groups.
Table 10: Summary of Baseline Characteristics in the PRIME and PRIME2 Studies (ITT Population)
Characteristic | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Age (years), mean (SD) | 49.2 (17.4) | 51.1 (15.8) | 51.0 (15.8) | 46.7 (15.2) |
Male, n (%) | 23 (30.7) | 28 (36.8) | 26 (33.3) | 31 (37.8) |
Female, n (%) | 52 (69.3) | 48 (63.2) | 52 (66.7) | 51 (62.2) |
Race, n (%) | ||||
American Indian or Alaska Native | 3 (4.0) | 2 (2.6) | 0 (0) | 1 (1.2) |
Asian | 29 (38.7) | 25 (32.9) | 25 (32.1) | 27 (32.9) |
Black or African American | 8 (10.7) | 3 (3.9) | 3 (3.8) | 5 (6.1) |
Multiple | 0 (0) | 0 (0) | 1 (1.3) | 0 (0) |
Native Hawaiian or other Pacific Islander | 0 (0) | 0 (0) | 1 (1.3) | 0 (0) |
White | 35 (46.7) | 45 (59.2) | 48 (61.5) | 48 (58.5) |
Not reported | 0 (0) | 1 (1.3) | 0 (0) | 1 (1.2) |
Weight (kg), mean (SD) | 75.2 (17.3) | 71.4 (17.0) | 73.9 (17.5) | 75.0 (19.7) |
BMI (kg/m2), mean (SD) | 28.1 (6.3) | 26.4 (5.8) | 26.9 (5.9) | 27.0 (5.9) |
Age at onset of PN (years), mean (SD) | 43.9 (18.2) | 46.2 (16.2) | 46.1 (16.5) | 41.7 (15.3) |
Duration of PN (years),a mean (SD) | 6.01 (7.55) | 5.40 (6.21) | 5.36 (6.90) | 5.48 (6.97) |
History of atopy, n (%)b | ||||
Atopic | 33 (44.0) | 28 (36.8) | 34 (43.6) | 40 (48.8) |
Ongoing mild atopic dermatitis | 4 (5.3) | 2 (2.6) | 2 (2.6) | 5 (6.1) |
Nonatopic | 42 (56.0) | 48 (63.2) | 44 (56.4) | 42 (51.2) |
Stable TCS or TCI use, n (%)c | 47 (62.7) | 45 (59.2) | 44 (56.4) | 46 (56.1) |
Antidepressant use at baseline, n (%) | 9 (12.0) | 9 (11.8) | 7 (9.0) | 8 (9.8) |
WI-NRS, mean (SD) | 8.6 (0.9) | 8.3 (1.1) | 8.5 (1.0) | 8.5 (1.0) |
WI-NRS, median (range) | 8.7 (7 to 10) | 8.3 (2 to 10) | 8.5 (7 to 10) | 8.5 (3 to 10)d |
IGA PN-S, n (%)e | ||||
≤ 2 (clear to mild) | 0 | 0 | 0 | 0 |
3 (moderate) | 54 (72.0) | 53 (70.7) | 49 (62.8) | 49 (60.5) |
4 (severe) | 21 (28.0) | 22 (29.3) | 29 (37.2) | 32 (39.5) |
IGA PN-A, mean (SD) | 3.3 (0.6) | 3.3 (0.6) | 3.4 (0.6) | 3.4 (0.6) |
Skin Pain NRS, mean (SD) | 7.2 (2.5) | 7.2 (2.3) | 7.3 (2.4) | 7.1 (2.5) |
Skin Pain NRS, median (range) | 7.9 (0 to 10) | 7.8 (0 to 10) | 8.0 (0 to 10) | 7.7 (0 to 10) |
Sleep NRS, mean (SD) | 4.4 (2.4) | 4.3 (2.2) | 4.4 (2.3) | 4.2 (2.5) |
Sleep NRS, median (range) | 4.2 (0 to 9) | 4.4 (0 to 9) | 4.2 (0 to 9) | 3.9 (0 to 10) |
Number of lesions from PAS, n (%) | ||||
0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
1 to 19 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
20 to 100 | 54 (72.0) | 52 (69.3) | 47 (60.3) | 52 (63.4) |
> 100 | 21 (28.0) | 23 (30.7) | 31 (39.7) | 30 (36.6) |
DLQI, mean (SD) | 17.8 (7.1) | 15.7 (7.3) | 18.2 (6.5) | 18.2 (7.0) |
Comorbid conditions associated with PN, n (%)f | 48 (64.0) | 40 (52.6) | 41 (52.6) | 48 (58.5) |
Thyroid hypofunction disorder | 7 (9.3) | 8 (10.5) | 7 (9.0) | 5 (6.1) |
Diabetes | 13 (17.3) | 5 (6.6) | 5 (6.4) | 8 (9.8) |
Depressive disorder | 5 (6.7) | 4 (5.3) | 6 (7.7) | 7 (8.5) |
Anxiety symptoms | 6 (8.0) | 2 (2.6) | 3 (3.8) | 4 (4.9) |
Lipid metabolism disorder | 4 (5.3) | 4 (5.3) | 4 (5.1) | 7 (8.5) |
Vascular hypertensive disorders | 22 (29.3) | 20 (26.3) | 10 (12.8) | 22 (26.8) |
Dermatitis and eczema | 7 (9.3) | 6 (7.9) | 6 (7.7) | 6 (7.3) |
BMI = body mass index; DLQI = Dermatology Life Quality Index; IGA PN-A = Investigator’s Global Assessment Prurigo Nodularis–Activity; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; ITT = intention to treat; NRS = Numeric Rating Scale; PAS = prurigo activity score; PN = prurigo nodularis; SD = standard deviation; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; WI-NRS = Worst Itch Numeric Rating Scale.
Note: A low score indicates a good outcome for WI-NRS (range, 0 to 10), IGA PN-S (range, 0 to 4), IGA PN-A (range, 0 to 4), Skin Pain NRS (range, 0 to 10), and DLQI (range, 0 to 30); a high score indicates good outcome for Sleep NRS (range, 0 to 10).
aDerived as (year of randomization − year of first diagnosis of PN) + (month of randomization − month of first diagnosis of PN)/12.
bDefined as having a medical history of atopic dermatitis, allergic rhinitis or rhinoconjunctivitis, asthma, food allergy, or eosinophilic esophagitis.
cStable regimen for TCSs is defined as maintaining the same medicine (a low- to medium-potency TCS) and maintaining the same frequency of treatment (once or twice daily) used from 2 weeks before screening. Stable regimen for TCIs is defined as maintaining the same medicine and treatment frequency (once or twice daily) used from 2 weeks before screening.
dTwo patients in the placebo group had a major protocol violation as they did not have an average WI-NRS score of at least 7 at baseline. This included 1 patient with a score of 3 and 1 patient with a score of 6.2.
eBaseline IGA PN-S score was missing for 1 patient in the placebo group of both studies.
fOnly comorbid conditions occurring in ≥ 8.0% of patients in any treatment arm have been included.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
With regards to prior therapies, all but 1 patient enrolled in the PRIME and PRIME2 trials had prior experience with topical therapies (Table 11). Most patients had also received systemic therapies for PN, of which antihistamines (46.2% to 60.0%), corticosteroids (11.5% to 22.7%) and nonsteroidal immunosuppressant drugs (13.2% to 25.6%) were most common. ███ ████████ ██ ████████ ███ ██████████ ████████ █████████████ ██████████████ ███████████ ████████████ ██ ███████████ ██ █████ █████ ██ █████ ██ ███████.
Table 11: Summary of Prior Treatments Used in the PRIME and PRIME2 Studies (ITT Population)
Characteristic | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Prior topical medications for PN, n (%) | ||||
Any topical medication | 74 (98.7) | 76 (100) | 78 (100) | 82 (100) |
TCS | 74 (98.7) | 75 (98.7) | 77 (98.7) | 80 (97.6) |
TCI | 9 (12.0) | 12 (15.8) | 6 (7.7) | 8 (9.8) |
Prior systemic medications for PN, n (%) | ||||
Any systemic medication | 53 (70.7) | 52 (68.4) | 49 (62.8) | 52 (63.4) |
Antihistamines | 45 (60.0) | 44 (57.9) | 36 (46.2) | 40 (48.8) |
Corticosteroids | 17 (22.7) | 13 (17.1) | 9 (11.5) | 15 (18.3) |
Nonsteroidal ISTs | 16 (21.3) | 10 (13.2) | 20 (25.6) | 18 (22.0) |
Gabapentinoids | 5 (6.7) | 2 (2.6) | 0 (0) | 1 (1.2) |
Opioid antagonists | 2 (2.7) | 2 (2.6) | 2 (2.6) | 1 (1.2) |
Antidepressants | 1 (1.3) | 2 (2.6) | 10 (12.8) | 13 (15.9) |
Prior procedures for PN, n (%) | ||||
███ ██████ | ██ ██████ | █████ | ██ ██████ | ██ ██████ |
█████████ | ██████ | █████ | █████ | █████ |
█████████ | █████ | █████ | ██████ | █████ |
████████ | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
IST = immunosuppressant; ITT = intention to treat; PN = prurigo nodularis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Exposure to Study Treatments
The mean duration of treatment was higher in the dupilumab than in the placebo groups in the PRIME (█████ ████ ██████ █████ ████) and PRIME2 studies (█████ ██████ █████ ████), although the median duration was the same across all treatment groups (█████ ████) (Table 12). Adherence to the study drug, background TCS, and emollient therapies were generally high in all groups.
Fewer patients in the dupilumab groups received rescue therapies than in the placebo groups of the 24-week treatment period. In the PRIME and PRIME2 studies, 6.7% versus 21.1%, and 7.7% versus 24.4% of patients in the dupilumab and placebo groups, respectively, received topical rescue treatment (i.e., a high-potency or super-potent TCS). Other topical therapies the patients received during the trials are summarized in Table 13.
Table 12: Summary of Patient Exposure in the PRIME and PRIME2 Studies (Safety Population)
Exposure (up to week 24) | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 75) | Dupilumab (N = 77) | Placebo (N = 82) | |
Cumulative total (patient-years) | █████ | █████ | █████ | █████ |
Duration (days) | ||||
Mean (SD) | █████ | █████ | █████ | █████ |
Median (range) | █████ | █████ | █████ | █████ |
Overall adherence to study drug, n (%) | ||||
Patients with < 80% | █████ | █████ | █████ | █████ |
Patients with ≥ 80% | █████ | █████ | █████ | █████ |
Overall adherence to a background TCS or TCI (up to week 24), n (%) | ||||
Total N | █████ | █████ | █████ | █████ |
Patients with < 80% | █████ | █████ | █████ | █████ |
Patients with ≥ 80% | █████ | █████ | █████ | █████ |
Overall adherence to background emollient therapy (up to week 24), n (%) | ||||
Patients with < 80% | █████ | █████ | █████ | █████ |
Patients with ≥ 80% | █████ | █████ | █████ | █████ |
Use of rescue therapy (up to week 24), n (%)a | 5 (6.7) | 16 (21.1) | 6 (7.7) | 20 (24.4) |
High-potency or super-potent TCS | █████ | █████ | █████ | █████ |
TCI | █████ | █████ | █████ | █████ |
ITT = intention to treat; SD = standard deviation; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid.
aITT population.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Table 13: Summary of Concomitant Medication Use in the PRIME and PRIME2 Studies (ITT Population)
Concomitant medication by anatomic or therapeutic class | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Any concomitant medication | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Dermatologicals | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Emollients and protectives | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Corticosteroids, dermatological preparations | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Other dermatological preparations | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antipruritics (e.g., antihistamines, anesthetics) | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antiacne preparations | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antifungals for dermatologic use | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Preparations for wounds and ulcers | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Medicated dressings | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antibiotics and chemotherapeutics | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antiseptics and disinfectants | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
Antipsoriatics | ██ ██ | ██ ██ | ██ ██ | ██ ██ |
ITT = intention to treat.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Efficacy
Pruritus
In the PRIME study for the primary outcome, 60.0% versus 18.4% of patients in the dupilumab and placebo group, respectively, reported at least a 4-point reduction in WI-NRS scores from baseline to week 24. All analyses presented were based on the 7-day average of daily WI-NRS scores over the previous week. The between-group difference was 42.7% (95% CI, 27.8% to 57.7%) favouring dupilumab versus placebo (P < 0.0001) (Table 14). At week 12, the proportion of responders was 44.0% versus 15.8%, with a between-group difference of 29.2% (95% CI, 14.5% to 43.8%; P = 0.0003, not controlled for multiple comparisons) for dupilumab versus placebo.
At week 24 in the PRIME2 study, the WI-NRS responder results also favoured dupilumab with 57.7% versus 19.5% of patients reporting at least a 4-point reduction in WI-NRS score from baseline (difference = 42.6%; 95% CI, 29.1% to 56.1%; P < 0.0001) for dupilumab versus placebo. For the primary outcome in the PRIME2 study, WI-NRS responder results measured at week 12, 37.2% versus 22.0% of patients in the dupilumab and placebo groups, respectively, achieved at least a 4-point reduction in the WI-NRS, with a between-group difference of 16.8% (95% CI, 2.3% to 31.2%; P = 0.0216).
In both trials, the supplementary analyses (as observed, hybrid and tipping point analyses) for handling of missing data of the proportion of patients with WI-NRS response at week 24 reported results that were comparable to the results of the primary analyses. The sponsor identified 4 clinically important subgroups based on a history of atopy (yes or no), current diagnosis of atopic dermatitis (no), stable use of a TCS or TCI (yes or no) and baseline IGA PN-S score of 3 (moderate) or 4 (severe). The sponsor also provided post hoc analyses based on prior use of systemic immunosuppressants, and prior nonsteroidal systemic immunosuppressants (yes or no). The results of these subgroup analyses were generally consistent with those of the overall population. For all subgroup analyses, the P values for treatment by subgroup interaction were greater than 0.05.
The percent change from baseline to week 24 in the weekly average WI-NRS score favoured dupilumab versus placebo in both studies (Appendix 1, Table 20). The PRIME study reported an LS mean difference of −26.7% (95% CI, −38.4% to −14.9%; P < 0.0001) and the PRIME 2 study reported a −23.2% difference (95% CI, −33.8% to −12.5%; P < 0.0001) versus placebo.
Disease Severity
The proportion of patients with an IGA PN-S score of 0 or 1 at week 24 was a key secondary outcome in both studies (Table 14). In the PRIME study 48.0% of patients in dupilumab group and 18.4% of patients in the placebo group met the response criteria, with a between-group difference of 28.3% (95% CI, 13.4% to 43.2%; P = 0.0004) favouring dupilumab. The results were similar in the PRIME2 study, in which 44.9% and 15.9% of patients met the IGA PN-S response criteria in the dupilumab and placebo groups, respectively. The between-group difference was 30.8% (95% CI, 16.4% to 45.2%; P < 0.0001).
Table 14: Summary of Pruritus and Disease Severity Scale Results From the PRIME and PRIME2 Studies (ITT Population)
Outcomesa, b | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Proportion of patients with WI-NRS reduction by ≥ 4 points from baseline to week 24 | ||||
n (%) | 45 (60.0) | 14 (18.4) | 45 (57.7) | 16 (19.5) |
Percent difference vs. placebo (95% CI) | 42.7 (27.8 to 57.7) | 42.6 (29.1 to 56.1) | ||
OR (95% CI) | 6.5 (2.78 to 15.41) | 9.0 (3.56 to 22.66) | ||
P value | < 0.0001 | < 0.0001 | ||
Proportion of patients with WI-NRS reduction by ≥ 4 points from baseline to week 12 | ||||
n (%) | 33 (44.0) | 12 (15.8) | 29 (37.2) | 18 (22.0) |
Percent difference vs. placebo (95% CI) | 29.2 (14.5 to 43.8) | 16.8 (2.3 to 31.2) | ||
OR (95% CI) | 4.3 (1.86 to 9.77) | 2.3 (1.08 to 5.00) | ||
P value | 0.0003c | 0.0216 | ||
Proportion of patients with IGA PN-S 0 or 1 score at week 24 | ||||
n (%) | 36 (48.0) | 14 (18.4) | 35 (44.9) | 13 (15.9) |
Percent difference vs. placebo (95% CI) | 28.3 (13.4 to 43.2) | 30.8 (16.4 to 45.2) | ||
OR (95% CI) | 4.0 (1.81 to 8.98) | 4.4 (2.02 to 9.55) | ||
P value | 0.0004 | < 0.0001 | ||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis–Stage; ITT = intention to treat; OR = odds ratio; WI-NRS: Worst Itch Numeric Rating Scale; vs. = versus.
Notes: A low score indicates a good outcome for WI-NRS (range, 0 to 10), and IGA PN-S (range, 0 to 4). Patients who received prohibited medications or procedures and/or rescue medications that impacted efficacy before the time point were considered nonresponders, and patients with missing data at the time point were considered nonresponders.
aOutcomes are presented in the order of the statistical testing hierarchy for European Union and European Union reference countries. The P value for all outcomes have been adjusted for multiple testing except for in the PRIME study, the proportion of patients with WI-NRS reduction by ≥ 4 points from baseline to week 12.
bFor binary end points the CMH response rate difference and OR for binary variables derived from the Mantel-Haenszel estimator. The CMH test was performed on the association between the responder status and intervention group adjusted by documented history of atopy (atopic or nonatopic), stable use of topical corticosteroids or topical calcineurin inhibitors (yes or no), region, and baseline antidepressant use (yes or no).
cP value reported has not been adjusted for multiple testing (i.e., nominal).
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Health-Related Quality of Life
The change from baseline in DLQI total score to week 24 favoured the dupilumab group versus the placebo group in both studies (Table 15). The PRIME study reported an LS mean difference of −6.2 points (95% CI, −8.3 to −4.1; P < 0.0001), and the PRIME2 study reported an LS mean difference of −6.4 points (95% CI, −8.4 to −4.4; P < 0.0001) for dupilumab versus placebo.
Table 15: Change From Baseline in DLQI Total Score to Week 24 From the PRIME and PRIME2 Studies (ITT Population)
Outcomesa | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Baseline mean (SD) | 17.81 (7.06) | 15.68 (7.32) | 18.15 (6.47) | 18.23 (6.98) |
Week 24 mean (SD) | 6.11 (5.30)b | 11.09 (9.36)b | 6.61 (6.07)c | 12.89 (8.29)c |
LS mean change from baseline (SE) | −11.97 (1.02)b | −5.77 (1.05)b | −13.16 (1.21)c | −6.77 (1.18)c |
LS mean difference vs. placebo (95% CI) | −6.19 (−8.34 to −4.05)b | −6.39 (−8.42 to −4.36)c | ||
P value | < 0.0001 | < 0.0001 | ||
CI = confidence interval; DLQI = Dermatology Life Quality Indes; ITT = intention to treat; LS = least squares; SD = standard deviation; SE = standard error; vs. = versus.
Notes: A low score indicates a good outcome for DLQI (range, 0 to 30). Patients who received prohibited medications or procedures and/or rescue medications that affected efficacy before the time point were considered nonresponders, and patients with missing data at the time point were considered as nonresponders.
aAn analysis of covariance model was fitted with intervention group, documented history of atopy (atopic or nonatopic), stable use of topical corticosteroids or topical calcineurin inhibitors (yes or no), region (countries combined) and baseline antidepressant use (yes or no), and baseline DLQI measurement as covariates. Results are reported as LS mean change from baseline with SE and LS mean difference (95% CI). The P values have been adjusted for multiple testing.
bIn the PRIME study, 24-week data in the dupilumab group were imputed for ████ patients (███; █████ missing patients). In the placebo group the 24-week data were based on █████ patients (missing ████ patients, █████), with data imputed for █████ patients (███).
cIn the PRIME2 study, 24-week data in the dupilumab group were based on █████ patients (missing ████ patient, █████) and outcomes were imputed for ████ patients (███). In the placebo group, 24-week data were based on █████ patients (missing for ████ patients, █████) and imputed for █████ patients (███).
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Harms
Refer to Table 16 for harms data. The sponsor provided a pooled safety analysis for the PRIME and PRIME2 studies that was submitted to Health Canada.46 For the pooled safety analysis, the cumulative duration of treatment exposure was ████ ███ ████ person-years in the dupilumab (N = 152) and placebo (N = 157) groups, respectively.
Adverse Events
Based on the pooled data from the PRIME and PRIME2 studies, 97 patients (63.8%) who received dupilumab and 89 patients (56.7%) who received placebo reported at least 1 TEAE over the 24-week treatment period. The most common events were accidental overdose ███ ██████ ██████ headache (5.3% versus 5.7%), nasopharyngitis (3.9% versus 1.9%), and neurodermatitis (2.6% versus 7.0%) in the dupilumab versus placebo groups, respectively.
Serious Adverse Events
Seven patients (4.6%) in the dupilumab group experienced an SAE compared with 12 patients (7.6%) in the placebo group. Table 16 lists the specific SAEs that occurred, and, with 1 exception, all were reported in a single patient only. Two patients in the placebo group experienced acute myocardial infarction.
Withdrawals due to Adverse Events
None of the patients who received dupilumab stopped treatment due to AEs, whereas in the placebo group, 4 patients discontinued therapy (2.5%).
Mortality
No deaths were reported during the trials.
Notable Harms
Conjunctivitis was identified as an important AE that may be associated with dupilumab. In the PRIME study, ████ of patients in each group experienced 1 or more TEAE related to conjunctivitis, whereas in the PRIME2 study, ████ of patients in the dupilumab group and ██ of patients in the placebo group, had a conjunctivitis-related TEAE. No conjunctivitis AEs led to study-treatment discontinuation and no severe conjunctivitis AEs were reported in either trial.
The AEs of special interest, as defined in the study protocols for PRIME and PRIME2, included systemic hypersensitivity reactions, anaphylaxis, helminthic infections, severe conjunctivitis or blepharitis, keratitis, clinically symptomatic eosinophilia, pregnancy, significant alanine aminotransferase elevation, or symptomatic overdose with the study drug. Of these events, 0 and 1 (1.3%) patient in the dupilumab and placebo groups, respectively, of the PRIME study, and 1 patient (1.3%) in each group of the PRIME2 study experienced a systemic hypersensitivity reaction. No other patients in either study reported the other AEs of special interest.
The frequency of infections or infestations, serious or severe infections, injection-site reactions, and skin infections (excluding herpetic infections), were generally similar in the dupilumab and placebo groups in both studies (Table 17). Musculoskeletal and connective tissue disorders were reported in █████ versus █████ and ████ versus ████ of patients in the dupilumab versus placebo groups in the PRIME and PRIME2 studies, respectively.
Table 16: Summary of Harms Results From Studies Included in the Systematic Review (Pooled Safety Population)
Adverse events | Dupilumab (N = 152) | Placebo (N = 157) |
|---|---|---|
Most common adverse events, n (%)a | ||
Patients with ≥ 1 TEAE | 97 (63.8) | 89 (56.7) |
Accidental overdose | ██ ██████ | ██ ██████ |
Headache | 8 (5.3) | 9 (5.7) |
Nasopharyngitis | 6 (3.9) | 3 (1.9) |
Increased blood creatine phosphokinase | ██ ██████ | ██ ██████ |
Dizziness | ██ ██████ | ██ ██████ |
Neurodermatitis | 4 (2.6) | 11 (7.0) |
Diarrhea | 4 (2.6) | 1 (0.6) |
Conjunctivitis | 3 (2.0) | 1 (0.6) |
Conjunctivitis allergic | 3 (2.0) | 1 (0.6) |
Injection-site reaction | 3 (2.0) | 2 (1.3) |
Eczema | ██ ██████ | ██ ██████ |
Myalgia | ██ ██████ | ██ ██████ |
COVID-19 | 1 (0.7) | 5 (3.2) |
Injection-site pain | 1 (0.7) | 5 (3.2) |
Folliculitis | ██ ██████ | ██ ██████ |
Serious adverse events, n (%)b | ||
Patients with ≥ 1 SAE | 7 (4.6) | 12 (7.6) |
Description: (all events reported in 1 patient only, except if noted otherwise) | COVID-19 pneumonia, pelvic inflammatory disease, acute pyelonephritis, lipoma, papillary thyroid cancer, uterine leiomyoma, asthma, interstitial lung disease, musculoskeletal chest pain | COVID-19, sepsis, cutaneous T-cell lymphoma, Hodgkin disease, large granular lymphocytosis, cauda equina syndrome, AMI (2 patients), duodenal ulcer perforation, inflammatory bowel disease, mesenteritis, acute cholecystitis, neurodermatitis, rotator cuff syndrome, alcohol poisoning |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped treatment | 0 (0) | 4 (2.5) |
Hodgkin disease | 0 (0) | 1 (0.6) |
████████ █████ ███████████ | ██ ██████ | ██ ██████ |
Neurodermatitis | 0 (0) | 1 (0.6) |
Urticaria | 0 (0) | 1 (0.6) |
Deaths, n (%) | ||
Patients who died | 0 (0) | 0 (0) |
AMI = acute myocardial infarction; TEAE = treatment-emergent adverse event; SAE = serious adverse event.
Notes: Patients may have experienced ≥ 1 SAE. TEAEs are reported at the preferred term level.
aFrequency of TEAEs of ≥ 2% in either treatment group.
bFrequency of SAEs was 1 occurrence per patient except for acute myocardial infarction, which occurred in 2 patients in the placebo group
Source: 2.7.4 Common Technical Document: Summary of Clinical Safety.46
Table 17: Summary of Notable Harms in the PRIME and PRIME2 Studies (Safety Population)
Adverse events | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 75) | Dupilumab (N = 77) | Placebo (N = 82) | |
Conjunctivitis-related adverse events, n (%) | ||||
Conjunctivitis-related AEs, n (%)a | █████ | █████ | █████ | █████ |
RD vs. placebo (95% CI) | █████ | █████ | █████ | █████ |
Other notable harms, n (%) | ||||
Infections and infestations (SOC) | █████ | █████ | █████ | █████ |
Severe or serious infections | █████ | █████ | █████ | █████ |
Injection-site reactions | █████ | █████ | █████ | █████ |
Skin infection (excluding herpetic infections) | █████ | █████ | █████ | █████ |
Musculoskeletal and connective tissue disorders (SOC) | █████ | █████ | █████ | █████ |
AE = adverse event; CI = confidence interval; NE = not estimable; RD = risk difference; SOC = system organ class; vs. = versus.
aConjunctivitis-related AEs included the following terms: conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis, blepharitis, dry eye, eye irritation, eye pruritus, increased lacrimation, eye discharge, foreign body sensation in eyes, photophobia, xerophthalmia, ocular hyperemia, and conjunctival hyperemia. RD 95% CIs of the proportion (incidence) by Wald confidence limits.
Sources: Clinical Study Report for PRIME,9 Clinical Study Report for PRIME2,10 and additional data supplied by the sponsor.15
Critical Appraisal
Internal Validity
The CDA-AMC review team did not identify any concerns with the methods used to randomize or conceal allocation in either study. There were some minor demographic differences between groups at baseline, but the disease-related factors appeared to be well balanced. The clinical experts consulted for this review agreed that any baseline discrepancies were unlikely to bias the findings. However, because more patients in the placebo group dropped out or stopped the study drug compared with those in dupilumab group, it is unclear if the treatment groups remained balanced throughout the study. The studies were double-blind, which is important given the subjective nature of the outcomes measured. No major differences in the frequency of AEs were apparent, and the studies were not at risk of unblinding due to known, frequent AEs.
Based the patient and clinician input received, the outcomes assessed were relevant. Specifically, pruritus associated with PN has the greatest impact on patients and their HRQoL. Evidence from patients with PN support WI-NRS validity, reliability, and a 4-point MID for within-patient improvement.33,34,42 The clinical experts agreed that a 4-point improvement estimated for patients with PN is a clinically relevant threshold of change. Based on clinical expert input, the threshold for a clinically important between-group difference was 20% for the proportion of patients with at least a 4-point reduction from baseline.
Disease severity based on lesion count was also clinically relevant, and the clinical experts agreed that a reduction from at least 20 lesions (i.e., an IGA PN-S score of 3 or 4) at baseline to 5 lesions or fewer (i.e., an IGA PN-S score of 0 or 1) represented a clinically important improvement, although no MID is estimated in the literature. Based on clinical expert input, the threshold for a clinically important between-group difference was 15% for the proportion of patients with an IGA PN-S score of 0 or 1.
The DLQI is often used in clinical trials and its validity and reliability in patients with PN are supported by the evidence. For within-patient change, the MID estimates in the literature range from −8.0 to −10.0 points in patients with PN.11 An MID of 4 points for the DLQI has been estimated for a between-group meaningful change in patients with PN.11 An MID of 4 points for the DLQI was selected as the threshold for a clinically important between-group difference based on the literature and clinical expert input.
The primary and secondary binary outcomes were analyzed using a stratified Cochran-Mantel-Haensel test, with a treatment policy and composite estimand to address intercurrent events. All data from patients who stopped the study drug before week 24, including data collected after discontinuation, were used in the analysis (i.e., treatment policy estimand). Patients who used prohibited or rescue medications, or who had missing data, were analyzed as nonresponders, according to the composite estimand. The CDA-AMC review team agreed these methods were acceptable, and no major concerns that the imputation methods would bias the findings were identified. The supplementary analyses of different strategies to handle missing data showed similar results, which suggests the findings of the primary analysis were robust.
The CDA-AMC review team identified imbalances in missing data that may affect the DLQI change from baseline results. In the PRIME study, 24-week DLQI data were missing for ██ and ██, and were imputed for ███ and ███ of patients in the dupilumab and placebo groups, respectively. For the PRIME2 trial, DLQI data for ██ and ██ patients were missing at week 24, and ███ and ███ of patients had data imputed in the dupilumab and placebo groups, respectively. For these analyses, data were imputed using the worst observation carried forward after patients received rescue treatment or discontinued treatment due to lack of efficacy. Missing data due to other reasons were imputed by multiple imputation. Given the magnitude and differential nature of the missing and imputed data, the potential for bias cannot be ruled out. No supplementary analyses were available to test other imputation strategies for missing DLQI data, which may have improved confidence in the DLQI findings.
Of the key efficacy outcomes included in this report, only 1 measure was excluded from the hierarchical statistical testing procedure to control the type I error for multiple comparisons. In the PRIME study, the proportion of patients with at least a 4-point improvement in WI-NRS at 12 weeks was not controlled for multiple comparisons and no statistical inferences can be drawn on these findings.
The trials were 24 weeks in duration, which the clinical experts consulted for this review agreed was a reasonable time frame to show initial treatment response. However, the longer-term efficacy is unclear.
External Validity
The patients enrolled in the 2 trials were, on average, aged 50 years, with a diagnosis of PN for 5 years. Most patients (71% in the PRIME trial and 62% in the PRIME2 trial) had PN rated as moderate in severity, with the remainder of PN cases rated as severe. The mean WI-NRS score at baseline ranged from 8.3 (SD = 1.1) to 8.6 (SD = 0.9), which the clinical experts described as indicative of severe pruritus. As background therapy, 61% of patients in the PRIME study and 56% of patients in the PRIME2 study were on stable doses of a TCS or TCI at the start the trials. In clinical practice, the experts consulted for this review stated that concurrent use of a medium-potency TCS (for general application), with lesion-specific application of higher-potency TCS, would continue in patients who received dupilumab until their symptoms were resolved. The clinical experts also noted that intralesional corticosteroids or possibly phototherapy (if accessible) could be used concurrently with dupilumab, but these treatments were prohibited during the trial.
In the PRIME trial, 99% of patients had previously received topical therapy, 69% systemic therapy, 17.2% nonsteroidal immunosuppressants (e.g., cyclosporine, methotrexate, or thalidomide), and 2% antidepressants. In the PRIME2 trial, 100% of patients had received topical therapy, 63% systemic therapy, 23.8% nonsteroidal immunosuppressants (e.g., cyclosporine, methotrexate, or thalidomide), and 14.4% antidepressants. The experts noted that the treatments used in the trials were applicable for decision-making. While the majority of patients in both trials had previously received systemic therapy, the clinical experts consulted for this review stated that it would be reasonable to generalize the trial results to anticipated clinical practice (i.e., first-line systemic therapy for patients with moderate to severe PN). Given the poor efficacy and safety concerns associated with off-label systemic immunosuppressants, the clinical experts consulted did not recommend that patients be required to try these therapies before initiating dupilumab.
The clinical experts noted that PN is most prevalent in older patients, who often have comorbidities. However, the trials excluded patients with concurrent conditions (e.g., neuropathy or psychiatric conditions, renal disease, immunodeficiency, history of malignancy, substance use disorder, or infection). Although the safety and efficacy of dupilumab in patients with comorbidities are unclear, the clinical experts expressed no major safety concerns given the safety profile of dupilumab of patients with other skin diseases. The trials also excluded patients with moderate to severe atopic dermatitis, which is common in patients with PN, but the clinical experts did not expect that dupilumab efficacy or safety would be different in patients with and without atopic dermatitis. Overall, the clinical experts consulted for this review agreed that the characteristics of the patients enrolled were consistent with those with moderate to severe PN living in Canada who would be eligible for dupilumab.
Based on clinical expert feedback, off-label systemic therapies may be used to manage patients with PN whose disease is not adequately controlled with topical therapies. Based on the results of a sponsor conducted feasibility assessment, the sponsor concluded that an indirect treatment comparison was likely to provide biased treatment-effect estimates for the comparison of dupilumab and other systemic therapies due to the low-quality evidence available. Therefore, it remains unknown how dupilumab compares to other systemic therapies. Nemolizumab is currently under review by CDA-AMC for moderate to severe PN. CDA-AMC has not issued a final recommendation for nemolizumab and, therefore, based on CDA-AMC procedures, it was not considered a relevant comparator for the present review of dupilumab.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:12.13
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The thresholds used for the presence or absence of an important effect was based on clinical expert opinion regarding the WI-NRS and IGA PN-S end points, and on the literature for the DLQI end point. For conjunctivitis, the presence or absence of any non-null effect was used.
For the GRADE assessments, findings from PRIME and PRIME2 were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.14
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for dupilumab versus placebo.
Dupilumab results in a clinically important increase in the proportion of patients with at least a 4-point improvement in WI-NRS score at 24 weeks compared with placebo (high certainty).
Dupilumab likely results in a clinically important increase in the proportion of patients with at least a 4-point improvement in WI-NRS score at 12 weeks compared with placebo (moderate certainty due to serious imprecision).
Dupilumab results in a clinically important increase in the proportion of patients with an IGA PN-S score of 0 or 1 compared with placebo (high certainty).
Dupilumab likely results in a clinically important improvement in the change in the DLQI total score compared with placebo. (moderate certainty due to serious study limitations due to missing data).
The evidence is very uncertain about the effect of dupilumab on the proportion of patients with 1 or more conjunctivitis-related AEs compared with placebo (very low certainty due to indirectness and serious imprecision).
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
The sponsor submitted 5 studies to address gaps in the pivotal evidence. These include retrospective observational studies by Paganini et al. (2024),16 Richter et al. (2023),17 Selvaraj et al. (2023),18 and Chiricozzi et al. (2024)19 to address the lack of long-term data from the PRIME and PRIME2 trials, and 1 retrospective cohort study by Georgakopoulous et al. (2021)20 to address both a potential concern of the lack of generalizability of the results of the included trials to patients in Canada and the lack of long-term data.
In this section, studies by Paganini et al. (2024)16 and Richter et al. (2023)17 are summarized in Table 18 and Table 19. The data for Selvaraj et al. (2023)18 are only available in abstract form, and the results of Chiricozzi et al. (2024)19 and Georgakopoulous et al. (2021)20 are only reported in a letter to the editor. Because of the lack of complete information regarding methods and results, the summary of these 3 studies is presented in Appendix 2 and not included in this section.
Table 18: Summary of Study by Paganini et al. (2024)
Detail | Description |
|---|---|
Evidence gap | Lack of long-term data |
Study design |
|
Population |
|
Interventions | Dupilumab 600 mg SC at week 0 and then 300 mg SC every 2 weeks. |
Key findings | Effectiveness:
Harms data: only 1 patient developed an episode of conjunctivitis |
Limitations |
|
DLQI = Dermatology Life Quality Index; IGA PN-S = Investigator’s Global Assessment Prurigo Nodularis-Scale; NRS = Numerical Rating Scale; PN = prurigo nodularis; SC = subcutaneous; SD = standard deviation
Sources: Paganini et al. (2024)16 and sponsor’s Summary of Clinical Evidence.32
Table 19: Summary of Study by Richter et al. (2023)
Detail | Description |
|---|---|
Evidence gap | Lack of long-term data |
Study design |
|
Population |
|
Interventions |
|
Key findings | Effectiveness:
Harms: After an average of 59.9 weeks of treatment, the reported adverse effects were headache (n = 2; 20%), transient eosinophilia (n = 1; 10%), and vertigo (n = 1; 10%). |
Limitations |
|
CI = confidence interval; DLQI = Dermatology Life Quality Index; NRS = numerical rating scale; PAS = Prurigo Activity and Severity Scale; PN = prurigo nodularis; SC = subcutaneous.
Notes: The PAS is used to objectively establish the type, number, distribution, size, represented body area, and activity in terms of percentage of lesions with excoriations or crusts in patients with PN. It can be filled out by experienced clinicians. The PAS score range is 0 to 11, with higher scores indicative of worsening.
Sources: Richter et al. (2023)17 and sponsor’s Summary of Clinical Evidence.32
Discussion
Summary of Available Evidence
The evidence was based on 2 double-blind RCTs (PRIME and PRIME2) that evaluated the efficacy and safety of dupilumab in adult patients (aged 18 to 80 years) with PN who were inadequately controlled on topical prescription therapies or when those therapies were not advisable. At baseline, the patients enrolled were required to have at least 20 PN lesions, a 7-day average WI-NRS score of at least 7 points, and a history of failure of a 2-week course of a medium- to superpotent TCS, or when those therapies were not advisable. In the PRIME and PRIME2 studies, 151 and 160 patients, respectively, were randomized (1:1) to receive either dupilumab 300 mg SC every 2 weeks (with a 600 mg loading dose) or matching placebo, for 24 weeks. Concurrent use of a low- to medium-potency TCS or TCI was allowed if patients were receiving these treatments before the study. The key outcomes were the proportion of patients with at least a 4-point improvement in the WI-NRS score (the primary outcome for PRIME and PRIME2 studies), the proportion of patients with IGA PN-S score of 0 or 1 and the change from baseline in DLQI.
The patients enrolled in the 2 trials had similar characteristics, with a mean age per treatment group ranging from 46.7 years (SD = 15.2) to 51.1 years (SD = 15.8). Most patients were female (62.2% to 69.3%) and the minority were male (30.7% to 37.8%). The mean duration of PN was between 5.4 years (SD = 6.9) and 6.0 years (SD = 7.6), and less than half of patients enrolled had a history of atopy (36.8% to 48.8%). The majority of patients were on stable doses of a TCS or TCI (56.1% to 62.7%) at baseline, and all but 1 patient had used topical therapies in the past. Based on IGA PN-S scores, more patients had moderately severe PN (60.5% to 72.0%) than severe PN (28.0% to 39.5%), and the mean WI-NRS score at baseline ranged from 8.3 (SD = 1.1) to 8.6 (SD = 0.9).
The sponsor submitted 5 observational studies that provided longer-term effectiveness. These studies included 9 to 64 patients with PN who received dupilumab for up to 52 weeks20 84 weeks,16 104 weeks,18,19 or 116.7 weeks.17 Two studies were available as full-text publications.16,17
No long-term extension studies or indirect treatment comparisons were submitted for this review.
Interpretation of Results
Efficacy
In both studies, the key outcomes (≥ 4-point decrease in WI-NRS, IGA PN-S score of 0 or 1, or change in DLQI) after 24 weeks of treatment were statistically significantly in favour of dupilumab versus placebo. The clinical experts consulted for this review concluded that the effect of dupilumab on pruritus intensity was clinically important, as both studies showed between-group differences that exceeded the 20% MID selected by the experts. Specifically, the PRIME and PRIME2 studies reported between-group differences of 42.7% (95% CI, 27.8% to 57.7%) and 42.6% (95% CI, 29.1% to 56.1%), respectively, for the proportions of patients in the dupilumab versus placebo groups with at least a 4-point improvement in the WI-NRS score at 24 weeks. While the WI-NRS results at 12 weeks also favoured dupilumab versus placebo, because the 95% CI in both studies included the possibility of no clinically important difference between groups, the results at this time point were rated as moderately certain evidence due to imprecision in the GRADE. The experts indicated that a lower percentage of responders at 12 weeks was not unexpected, given their experience with dupilumab, which indicates that more that 12 weeks may be needed to see the full effects of therapy. Supplementary data on the change from baseline in WI-NRS at 24 weeks also favoured dupilumab versus placebo.
The effects of dupilumab on the clearance of PN nodules also demonstrated statistically significant and clinically important improvements versus placebo at 24 weeks. The between-group differences in the proportion of patients with an IGA PN-S score of 0 (clear) or 1 (almost clear) at week 24 were 28.3% for the PRIME study and 30.8% for the PRIME2 study, favouring dupilumab. The point estimates of both studies exceeded the 15% MID selected by the clinical experts, and were rated as high-certainty evidence based on the GRADE.
Overall, there were no major sources of bias identified by the CDA-AMC review team in either study, except for issues with missing data for the DLQI analyses. Both studies showed LS mean differences versus placebo in the change from baseline in DLQI that were statistically and clinically significant, based on the MID of 4 points reported in the literature. The trials reported LS mean differences in the change from baseline to week 24 in the DLQI total score of −6.2 points and −6.4 points for the PRIME and PRIME2 studies, respectively. However, given the higher withdrawal rate in the placebo group versus the dupilumab group, the evidence was rated as moderately certain on the GRADE. Missing DLQI data after patients received rescue treatment, or missing data after treatment discontinuation due to lack of efficacy were imputed using the worst observation carried forward method. Missing data due to other reasons were imputed using multiple imputation methods. Given the magnitude and differential nature of the missing and imputed data in the dupilumab and placebo groups, the potential for bias cannot be ruled out. Moreover, no supplementary analyses were provided to test other imputation strategies, which may have improved confidence in the DLQI findings.
Based on clinical expert feedback, off-label systemic therapies may be used to manage patients with PN whose disease is not adequately controlled with topical therapies. The clinical experts consulted for this review noted that high-quality evidence is lacking for these off-label systemic therapies. There was neither RCT evidence supporting the efficacy of other potential comparators (e.g., antihistamines, immunosuppressants, or antidepressants) nor comparative analyses between dupilumab and those treatments. Based on the results of a sponsor conducted feasibility assessment, the sponsor concluded that an indirect treatment comparison was likely to provide biased treatment-effect estimates for the comparison of dupilumab versus other systemic therapies due to the low-quality evidence available, the lack of standardized outcome measures, the small sample sizes, and the heterogeneity across potential studies. Therefore, it remains unknown how dupilumab compares to other systemic therapies. Nemolizumab is also under review by CDA-AMC for moderate to severe PN, and the experts agreed that nemolizumab will be used in the same place in therapy as dupilumab. Because CDA-AMC has not issued a final recommendation for nemolizumab, and following CDA-AMC procedures, nemolizumab was not considered a comparator for this review and no comparative efficacy or safety data were submitted by the sponsor.
The clinical experts agreed that the characteristics of the patients enrolled were generally consistent with those with moderate to severe PN living in Canada who would be eligible for dupilumab. All but 1 of the patients enrolled had previously received topical therapies and most patients (63% to 71%) had received systemic therapies to manage their PN. As background therapy, 56% to 61% of patients were on stable doses of a TCS or TCI at the start of the trial. The clinical experts noted that PN is most prevalent in older patients, who often have comorbidities; however, the trials excluded patients with comorbid conditions such as neuropathy or psychiatric conditions, renal disease, immunodeficiency, history of malignancy, substance use disorder, or infection. Although the safety and efficacy of dupilumab in patients with comorbidities are unclear, the clinical experts had no major safety concerns given the safety profile of dupilumab observed in patients with other skin diseases.
The trials were 24 weeks in duration, which the clinical experts consulted for this review agreed was a reasonable time frame to show initial treatment response. However, the longer-term efficacy is uncertain. Five observational studies were summarized in this report to address this evidence gap, with the duration of dupilumab treatment ranging from 52 to 117 weeks across these studies. The studies had serious limitations, including the lack of a comparator group, small sample sizes, lack of blinding, retrospective reporting, and incomplete reporting of information. Due to these limitations, the long-term efficacy of dupilumab in patients with PN remains unclear.
Harms
TEAEs were common in both the dupilumab and placebo groups (63.8% and 56.7%, respectively), but few patients stopped treatment due to AEs (0% and 2.5%), and SAEs were relatively infrequent (4.6% and 7.6%) based on pooled data from the PRIME and PRIME2 studies. The most common events in the dupilumab versus placebo groups, respectively, were accidental overdose ██████ ██████ headache (5.3% versus 5.7%), nasopharyngitis (3.9% versus 1.9%), and neurodermatitis (2.6% versus 7.0%).
The clinical experts consulted for this review identified conjunctivitis as a key safety issue for dupilumab, and the product monograph contains a warning for such AEs. In patients with PN, the frequency of conjunctivitis (4.0%) was the same in both treatment groups of the PRIME study and 6.5% and 0% in the dupilumab and placebo groups, respectively, of the PRIME2 study. No conjunctivitis-related AEs led to study treatment discontinuation and no severe conjunctivitis-related AEs were reported in either trial. These clinical trial data were rated as very low in certainty according to a GRADE assessment and insufficient to conclusively inform on the relative incidence of conjunctivitis-related AEs in patients with PN.
The treatment duration was 24 weeks in both studies, and the sample size was limited to 152 patients who received dupilumab. No extension studies were submitted to provide longer-term safety data, and the 5 observational studies the sponsor submitted had serious limitations and therefore did not address this evidence gap. Given the relatively short duration, limited sample size, and study exclusion criteria, the longer-term safety of dupilumab in patients with PN is unclear. However, because dupilumab has been approved in Canada for other indications since 2017, there is substantial treatment experience with it in clinical practice and dupilumab’s safety profile in other populations is known.
Conclusion
Based on 2 clinical trials in adults with moderate to severe PN who were inadequately controlled on topical prescription therapies or for whom those therapies were not advisable, dupilumab demonstrated a statistically significant and clinically important improvement versus placebo in pruritus intensity, and in the clearance or near clearance of PN nodules, in terms of the proportion of responders for the WI-NRS and the IGA PN-S at 24 weeks. Based on the change from baseline in the DLQI total score, dupilumab likely improves HRQoL versus placebo; however, this finding was of limited value due to missing data.
While TEAEs were commonly reported by patients in both the dupilumab and placebo groups, the proportion of patients who stopped therapy due to AEs was low. Conjunctivitis was identified as an important AE; however, the evidence was too uncertain to draw conclusions on the impact of dupilumab on the frequency of conjunctivitis-related AEs.
Because the evidence was drawn from 2 placebo-controlled RCTs that were 24 weeks in duration, the longer-term efficacy and safety are uncertain. While the sponsor submitted 5 longer-term retrospective observational studies, the lack of control groups, other biases associated with nonrandomized studies, and small sample sizes mean these studies do not fill the evidence gap.
References
1.Huang AH, Canner JK, Khanna R, et al. Real-world prevalence of prurigo nodularis and burden of associated diseases. J Invest Dermatol. 2020;140(2):480-483.e4. doi:10.1016/j.jid.2019.07.697 PubMed
2.Pereira MP, Steinke S, Zeidler C, et al. European academy of dermatology and venereology European prurigo project: expert consensus on the definition, classification and terminology of chronic prurigo. J Eur Acad Dermatol Venereol. 2018;32(7):1059-1065. doi:10.1111/jdv.14570 PubMed
3.Watsky K. Post TW, ed. Prurigo nodularis. UpToDate; 2024. Accessed December 24, 2024. http://www.uptodate.com
4.Zeidler C, Tsianakas A, Pereira M, et al. Chronic prurigo of nodular type: a review. Acta Derm Venereol. 2018;98(2):173-179. doi:10.2340/00015555-2774 PubMed
5.Wong D. Prurigo nodularis. Can Dermatol Today. 2021;2(4):27-33.
6.Ryczek A, Reich A. Prevalence of prurigo nodularis in Poland. Acta Derm Venereol. 2020;100(10):1-4. doi:10.2340/00015555-3518 PubMed
7.Wongvibulsin S, Sutaria N, Williams KA, et al. A nationwide study of prurigo nodularis: disease burden and healthcare utilization in the United States. J Invest Dermatol. 2021;141:2530-3e1(5p.). doi:10.1016/j.jid.2021.02.756
8.Morgan CL, Thomas M, Ständer S, et al. Epidemiology of prurigo nodularis in England: a retrospective database analysis. Br J Dermatol. 2022;doi:10.1111/bjd.21032 PubMed
9.Sanofi Recherche & Développement. Clinical Study Report: EFC16459. A randomized, double blind, placebo-controlled, multi-center, parallel group study to evaluate the efficacy and safety of dupilumab in patients with prurigo nodularis who are inadequately controlled on topical prescription therapies or when those therapies are not advisable (LIBERTY-PN PRIME). Database lock December 09, 2021 [internal sponsor's report] February 23, 2022.
10.Sanofi Recherche & Développement. Clinical Study Report: EFC16460. A randomized, double blind, placebo-controlled, multi-center, parallel group study to evaluate the efficacy and safety of dupilumab in patients with prurigo nodularis who are inadequately controlled on topical prescription therapies or when those therapies are not advisable (LIBERTY-PN PRIME2). Database lock: September 27, 2021 [internal sponsor's report] February 24, 2022.
11.Stander S, Kim BS, Guillemin I, et al. Estimating meaningful change thresholds for Skin Pain-Numeric Rating Scale, Sleep-Numeric Rating Scale and Dermatology Life Quality Index in patients with prurigo nodularis. J Eur Acad Dermatol Venereol. 2024;38(7):1401-1409. doi:10.1111/jdv.19800 PubMed
12.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi:10.1016/j.jclinepi.2010.07.015 PubMed
13.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi:10.1016/j.jclinepi.2019.10.014 PubMed
14.Popay J, Roberts HF, Sowden AG, et al. Guidance on the conduct of narrative synthesis in systematic reviews. a product from the ESRC methods programme, version1. Lancaster University; 2006. Accessed February 28, 2023. https://www.lancaster.ac.uk/media/lancaster-university/content-assets/documents/fhm/dhr/chir/NSsynthesisguidanceVersion1-April2006.pdf
15.Sanofi-Aventis Canada Inc. Sanofi-Aventis Canada Inc. response to February 21, 2025 Canada's Drug Agency request for additional information regarding Dupixent (dupilumab) PN CDA-AMC review: conjunctivitis AEs, validity of outcomes [internal additional sponsor's information]. February 28, 2025.
16.Paganini C, Talamonti M, Maffei V, et al. Dupilumab for treatment of prurigo nodularis: Real-life effectiveness for up to 84 weeks. J Clin Med. 2024;13(3):878. doi:10.3390/jcm13030878 PubMed
17.Richter C, Hafner J, Schuermann M, et al. Dupilumab for chronic prurigo: case series on effectiveness, safety, and quality of life. Dermatology. 2023;239(5):811-817. doi:10.1159/000531708 PubMed
18.Selvaraj KJ, Stewart T, Frew JW. Maintenance of response to dupilumab in prurigo nodularis: a retrospective cohort study. JAAD International. 2023;11:143-144. doi:10.1016/j.jdin.2023.02.006 PubMed
19.Chiricozzi A, Gori N, Ippoliti E, et al. Long-term therapeutic response to dupilumab in patients affected by prurigo nodularis: a real-world retrospective study. J Eur Acad Dermatol Venereol. 2024;38(10):e892-e895. doi:10.1111/jdv.19980 PubMed
20.Georgakopoulos JR, Croitoru D, Felfeli T, et al. Long-term dupilumab treatment for chronic refractory generalized prurigo nodularis: a retrospective cohort study. J Am Acad Dermatol. 2021;85(4):1049-1051. doi:10.1016/j.jaad.2021.02.038 PubMed
21.Pereira MP, Zeidler C, Wallengren J, et al. Chronic nodular prurigo: a European cross-sectional study of patient perspectives on therapeutic goals and satisfaction. Acta Derm Venereol. 2021;101(2):adv00403. doi:10.2340/00015555-3726 PubMed
22.Taghaddos D, Savinova I, Abu-Hilal M. Clinical characteristics and treatment outcomes of prurigo nodularis: a retrospective study. J Cutan Med Surg. 2024;28(2):141-145. doi:10.1177/12034754241227808 PubMed
23.Kowalski EH, Kneiber D, Valdebran M, et al. Treatment-resistant prurigo nodularis: challenges and solutions. Clin Cosmet Investig Dermatol. 2019;12:163-172. doi:10.2147/CCID.S188070 PubMed
24.Ständer S, Pereira MP, Berger T, et al. IFSI-guideline on chronic prurigo including prurigo nodularis. Itch. 2020;5(4):e42. doi:10.1097/itx.0000000000000042
25.Elmariah S, Kim B, Berger T, et al. Practical approaches for diagnosis and management of prurigo nodularis: United States expert panel consensus. J Am Acad Dermatol. 2021;84(3):747-760. doi:10.1016/j.jaad.2020.07.025 PubMed
26.Satoh T, Yokozeki H, Murota H, et al. 2020 guidelines for the diagnosis and treatment of prurigo. J Dermatol. 2021;48(9):e414-e431. doi:10.1111/1346-8138.16067 PubMed
27.Sanofi-Aventis Canada Inc. Dupixent (duilumab): 300 mg single-use syringe (300 mg/2 mL); 300 mg single-use pen (300 mg/2 mL); 200 mg single-use syringe (200 mg/1.14 mL); 200 mg single-use pen (200 mg/1.14 mL) solution for injection [product monograph] September 6, 2024.
28.Sanofi-Aventis Canada Inc. Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 200 mg and 300 mg solution for injection [internal sponsor's package]. December 20, 2024.
29.Yosipovitch G, Mollanazar N, Stander S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29(5):1180-1190. doi:10.1038/s41591-023-02320-9 PubMed
30.Sanofi. NCT04183335: Study of dupilumab for the treatment of prurigo nodularis, inadequately controlled on topical prescription therapies or when those therapies are non advisable (LIBERTY-PN-PRIME). ClinicalTrials.gov. Accessed December 3, 2022. https://clinicaltrials.gov/ct2/show/NCT04183335?term=dupilumab&cond=Prurigo+Nodularis&draw=2&rank=1
31.Sanofi. NCT042022679: Study of dupilumab for the treatment of patients with prurigo nodularis, inadequately controlled on topical prescription therapies or when those therapies are not advisable (PRIME2). Clinicaltrials.gov. Accessed December 3, 2022. https://clinicaltrials.gov/ct2/show/NCT04202679?term=dupilumab&cond=Prurigo+Nodularis&draw=2&rank=2
32.Sanofi-Aventis Canada Inc. Sponsor summary of clinical evidence: Dupixent (dupilumab injection) for the treatment of adult patients with moderate-to-severe prurigo nodularis (PN) whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 200 mg and 300 mg solution for injection. Sanofi-Aventis Canada Inc; December, 2024.
33.Ständer S, Zeidler C, Pereira M, et al. Worst itch numerical rating scale for prurigo nodularis: a psychometric evaluation. J Eur Acad Dermatol Venereol. 2022;36(4):573-581. doi:10.1111/jdv.17870 PubMed
34.Kimel M, Zeidler C, Kwon P, et al. Validation of psychometric properties of the Itch Numeric Rating Scale for pruritus associated with prurigo nodularis: a secondary analysis of a randomized clinical trial. JAMA Dermatol. 2020;156(12):1354-1358. doi:10.1001/jamadermatol.2020.3071 PubMed
35.Riepe C, Osada N, Reich A, et al. Minimal clinically important difference in chronic pruritus appears to be dependent on baseline itch severity. Acta Derm Venereol. 2019;99(13):1288-1290. doi:10.2340/00015555-3332 PubMed
36.Chernyshov PV. The evolution of quality of life assessment and use in dermatology. Dermatology. 2019;235(3):167-174. doi:10.1159/000496923 PubMed
37.Bahloul D, Thomas RB, Rhoten S, et al. 43531 Validation of the Dermatology Life Quality Index (DLQI) in prurigo nodularis (PN) based on clinical studies of dupilumab in adults with PN. J Am Acad Dermatol. 2023;89(3):AB103. doi:10.1016/j.jaad.2023.07.415
38.Basra MK, Fenech R, Gatt RM, et al. The Dermatology Life Quality Index 1994-2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159(5):997-1035. doi:10.1111/j.1365-2133.2008.08832.x PubMed
39.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-80. doi:10.1111/j.1087-0024.2004.09113.x PubMed
40.Shikiar R, Harding G, Leahy M, et al. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health Qual Life Outcomes. 2005;3:36. doi:10.1186/1477-7525-3-36 PubMed
41.Storck M, Sandmann S, Bruland P, et al. Pruritus Intensity scales across Europe: a prospective validation study. J Eur Acad Dermatol Venereol 2021;35(5):1176-1185. doi:10.1111/jdv.17111 PubMed
42.Kwatra SG, Yosipovitch G, Kim B, et al. Worst Itch Numeric Rating Scale for prurigo nodularis: a secondary analysis of 2 randomized clinical trials. JAMA Dermatology. 2024;160(8):813-821. doi:10.1001/jamadermatol.2024.1634 PubMed
43.Bahloul D, Thomas RB, Rhoten S, et al. 42899 Validation of the Worst-Itch Numeric Rating Scale (WI-NRS) in prurigo nodularis (PN) based on clinical studies of dupilumab in adults with PN. J Am Acad Dermatol. 2023;89(3):AB103. doi:10.1016/j.jaad.2023.07.417
44.Zeidler C, Pereira MP, Augustin M, et al. Investigator's global assessment of chronic prurigo: a new instrument for use in clinical trials. Acta Derm Venereol. 2021;101(2):adv00401. doi:10.2340/00015555-3701 PubMed
45.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335-2348. doi:10.1056/NEJMoa1610020 PubMed
46.Sanofi. Common Technical Document section 2.7.4: summary of clinical safety [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab) 200 mg and 300 mg solution for injection. March 10. 2022.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 20: Change and Percentage Change From Baseline in Weekly Average WI-NRS at Week 24 — the PRIME and PRIME2 Studies (ITT Population)
Outcomea | PRIME | PRIME2 | ||
|---|---|---|---|---|
Dupilumab (N = 75) | Placebo (N = 76) | Dupilumab (N = 78) | Placebo (N = 82) | |
Baseline mean (SD) | █████ | █████ | █████ | █████ |
Week 24 mean (SD) | █████ | █████ | █████ | █████ |
Change from baseline: LS mean (SE) | █████ | █████ | █████ | █████ |
Change difference: LS mean difference vs. placebo (95%CI) | █████ | █████ | ||
P value | █████ | █████ | ||
% change from baseline: LS mean (SE) | −48.89 (5.61)b | −22.22 (5.74)b | −59.34 (6.39)c | −36.18 (6.21)c |
% change difference: LS mean difference vs. placebo (95% CI) | −26.67 (−38.44 to −14.90)b | −23.16 (−33.81 to −12.51)c | ||
P value | < 0.0001d | < 0.0001d | ||
CI = confidence interval; ITT = intention-to-treat; LS = least squares; SD = standard deviation; SE = standard error; WI-NRS = Worst Itch Numerical Rating Scale.
Note: Reduction in WI-NRS score (range 0 to 10) indicates an improvement.
aAn ANCOVA model was fitted with intervention group, documented history of atopy (atopic or nonatopic), stable use of a TCS or TCI (yes or no), region (countries combined) and baseline antidepressant use (yes or no), and baseline WI-NRS measurement as covariates. Results are reported as LS mean absolute change and percent change from baseline with SE and LS mean difference in the absolute change and percent change from baseline (95% CI). The P values for the percent change from baseline, but not the absolute change, have been adjusted for multiple testing. The absolute change from baseline results should be interpreted as supportive data.
bIn the PRIME study, 24-week data in the dupilumab group were imputed for 8 patients (11%; no missing patients). In the placebo group the 24-week data were based on 66 patients (missing 9 patients, 12%), with data imputed for 22 patients (29%).
cIn the PRIME2 study, 24-week data in the dupilumab group were based on 76 patients (missing 2 patients, 3%) and outcomes were imputed for 10 patients (13%). In the placebo group, 24-week data were based on 74 patients (missing for 8 patients, 10%) and imputed for 25 patients (30%).
dThe P values for the percent change from baseline have been adjusted for multiple testing.
Sources: Clinical Study Report for PRIME9 and Clinical Study Report for PRIME2.10
Figure 2: Least Squares Mean Change From Baseline in WI-NRS Over Time — the PRIME Study (ITT population)
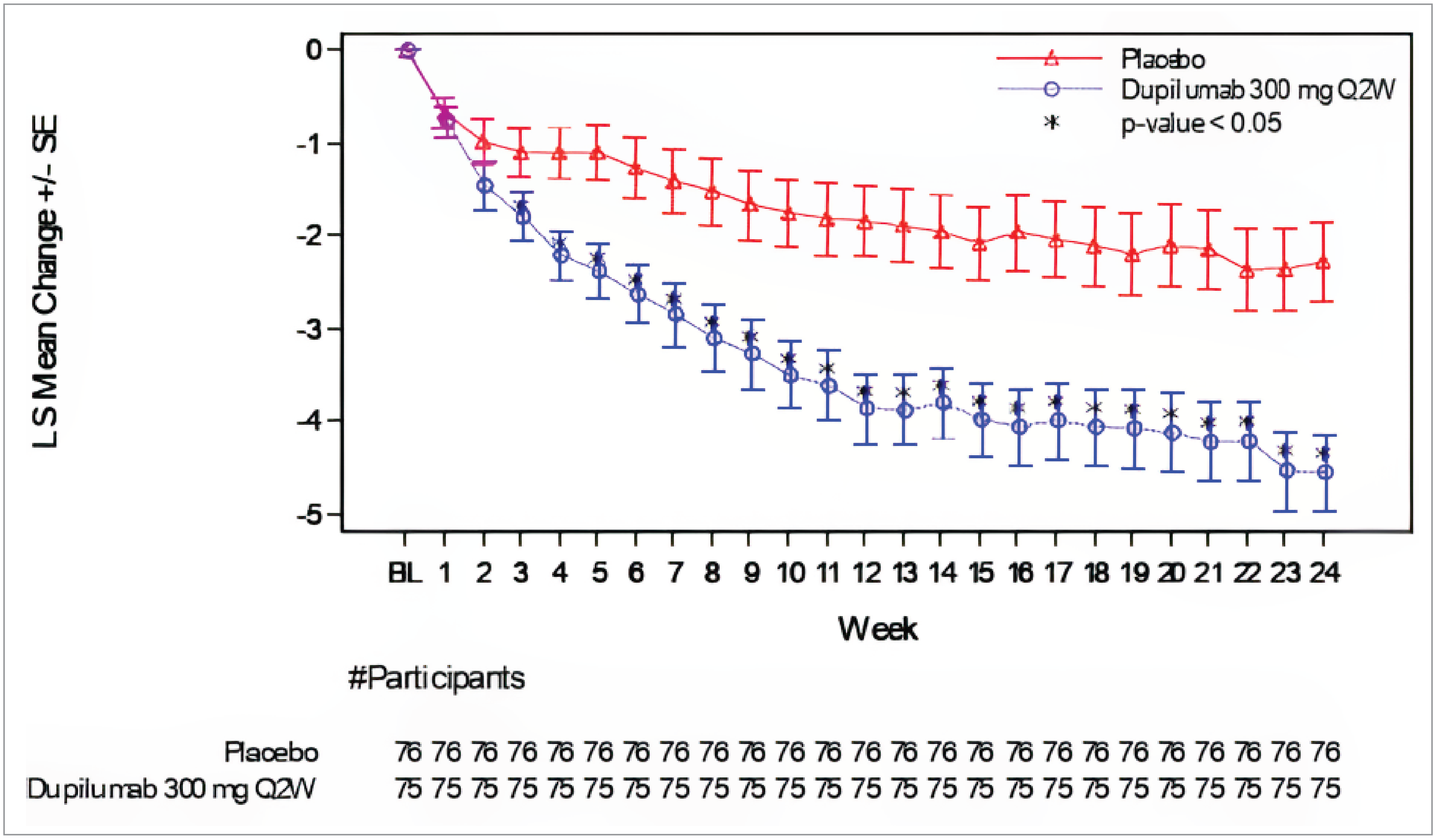
ITT = intention to treat; LS = least squares; Q2W = every 2 weeks; SE = standard error; WI-NRS = Worst Itch Numerical Rating Scale.
Source: Clinical Study Report for PRIME.9
Figure 3: Least Squares Mean Change From Baseline in WI-NRS Over Time — the PRIME2 Study (ITT Population)
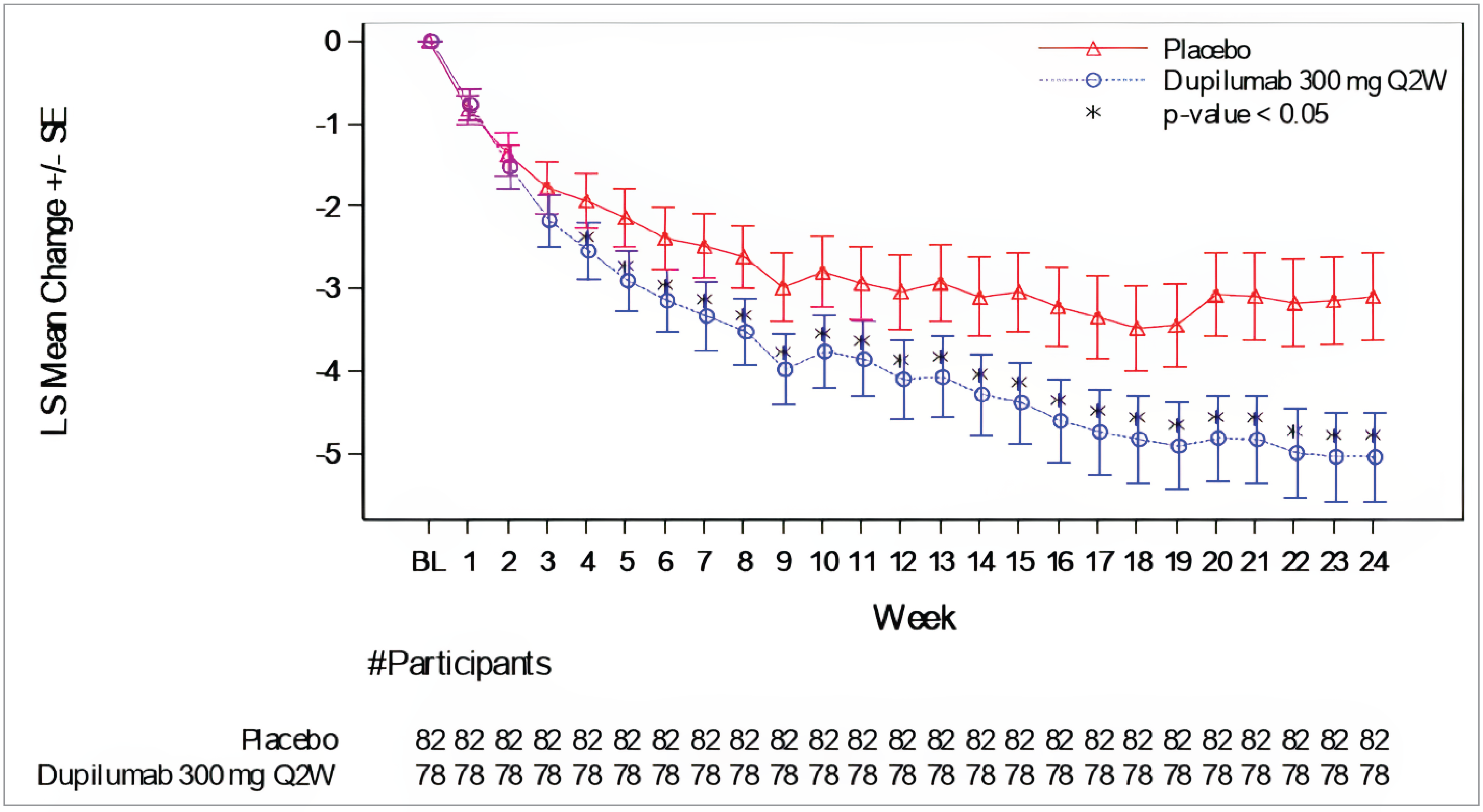
ITT = intention to treat; LS = least squares; SE standard error; WI-NRS = Worst Itch Numerical Rating Scale.
Source: Clinical Study Report for PRIME2.10
Appendix 2: Summary of Other Studies
Please note that this appendix has not been copy-edited.
Table 21: Summary of Study by Selvaraj et al. (2023)
Detail | Description |
|---|---|
Evidence gap | Lack of long-term data |
Study design |
|
Population | Adult patients (N = 21), mean age of 55 years, 10 females and 11 males, who received dupilumab monotherapy for PN for a minimum of 52 weeks from June 2020 to November 2022 Patients receiving a TCS, intralesional corticosteroids, oral corticosteroids, or systemic immunosuppressant drugs (such as methotrexate or azathioprine) were excluded |
Interventions | Dupilumab, 600 mg SC at week 0, then 300 mg SC every 2 weeks, SC |
Key findings | Effectiveness:
No patients reported infectious adverse events, and no dupilumab associated ocular inflammation was identified |
Limitations |
|
DLQI = Dermatology Life Quality Index; N = number; NRS = numeric rating scale; PN = prurigo nodularis; SD = standard deviation; TCS = topical corticosteroid.
Sources: Abstract of Selvaraj et al. (2023)18 and Sponsor’s Summary of Clinical Evidence.32
Table 22: Summary of Study by Chiricozzi et al. (2024)
Detail | Description |
|---|---|
Evidence gap | Lack of long-term data |
Study design |
|
Population | Adult patients (N = 64) with chronic PN Specific inclusion and exclusion criteria were not reported |
Interventions | Dupilumab, dosage not reported. |
Key findings | Effectiveness:
Harms data: 9 AEs including arthralgias, telogen effluvium, conjunctivitis or ocular disturbances, injection-site reaction, acute respiratory failure, oral candida infection, and asthenia were reported |
Limitations |
|
AE = adverse event; DLQI = Dermatology Life Quality Index; IGA CPN-S = Investigator’s Global Assessment Chronic Prurigo Nodularis–Stage; NRS = numeric rating scale; PN = prurigo nodularis; SD = standard deviation.
Sources: Chiricozzi et al. (2024)19 and sponsor’s Summary of Clinical Evidence.32
Table 23: Summary of Study by Georgakopoulous et al. (2021)
Detail | Description |
|---|---|
Evidence gap | Lack of generalizability to patients living in Canada and lack of long-term data |
Study design |
|
Population | Adult patients (N = 19) with chronic PN and IGA score of 3 or 4 at baseline who were treated with dupilumab at 2 tertiary care academic centres in Canada |
Interventions | Dupilumab, 300 mg; no further details were provided |
Effectiveness:
Harms data: AEs reported by 7 (36.8%) patients by week 52, including 5 (26.3%) patients with dupilumab-induced ocular surface disease, 1 (5.3%) patient with an injection-site reaction, and 1 (5.3%) patient with arthralgias | |
Limitations |
|
AE = adverse event; IGA = Investigator’s Global Assessment; PN = prurigo nodularis.
Sources: Georgakopoulous et al. (2021)20 and sponsor’s Summary of Clinical Evidence.32
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CDA-AMC
Canada’s Drug Agency
DLQI
Dermatology Life Quality Index
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
WI-NRS
Worst Itch Numeric Rating Scale
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), solution for subcutaneous injection (300 mg per 2 mL [150 mg/mL] single-use prefilled syringe or pen administered by subcutaneous injection) |
Indication | Treatment of adult patients with moderate to severe prurigo nodularis (PN) whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Dupixent can be used with or without topical corticosteroids. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 12, 2023 |
Reimbursement request | As per indication |
Sponsor | Sanofi-Aventis Canada Inc. |
Submission history | Previously reviewed: Yesa Indication: Treatment of patients aged 6 months and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable, according to the Health Canada indication Recommendation date: October 13, 2023 Recommendation: Reimburse with clinical criteria and/or conditions Indication: For the treatment of patients aged 12 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable and/or who are refractory to or ineligible for systemic immunosuppressant therapies (i.e., due to contraindications, intolerance, or need for long-term treatment) Recommendation date: April 22, 2020 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Adult patients with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable Recommendation date: June 27, 2018 Recommendation: Do not reimburse |
NOC = Notice of Compliance; PN = prurigo nodularis.
aDupilumab has been reviewed and received a reimburse with conditions recommendation for the treatment severe asthma with a type 2 or eosinophilic phenotype or oral corticosteroid–dependent asthma.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by Markov model |
Target population | Adult patients with moderate to severe PN, aligned with the PRIME and PRIME2 clinical trials |
Treatment | Dupilumab plus BSC |
Dose regimen | Initial dose of 600 mg, followed by 300 mg every other week |
Submitted price | Dupilumab: $978.70 per prefilled syringe |
Submitted treatment cost | First year: $26,425 per patient Subsequent years: $25,446 per patient |
Comparator | BSC is defined as a basket of therapies, consisting of low- to medium-potency topical corticosteroids and calcineurin inhibitors. |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (50 years) |
Key data sources | PRIME and PRIME2 clinical trials |
Submitted results | ICER = $74,305 per QALY gained versus BSC ($74,942 in incremental costs and 1.01 incremental QALYs) |
Key limitations |
|
CDA-AMC reanalysis results |
|
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; PN = prurigo nodularis; QALY = quality-adjusted life-year.
Conclusions
A Clinical Review of the PRIME and PRIME2 trials undertaken by Canada’s Drug Agency (CDA-AMC) found that, in adults with moderate to severe prurigo nodularis (PN) who were inadequately controlled on topical prescription therapies, dupilumab demonstrated a significant and clinically important decrease in pruritus intensity and a reduction in PN nodules compared to placebo. The trial data collected on the Dermatology Life Quality Index (DLQI) suggested that, to a moderate degree of certainty, dupilumab likely improved patient-reported quality of life compared to placebo. No serious safety concerns were raised. Although conjunctivitis was identified as an important adverse event (AE), the evidence was too uncertain to draw conclusions about the impact of dupilumab on the frequency of conjunctivitis-related AEs. Long-term efficacy and safety (beyond the 24-week trial follow-up) is uncertain. Although observational studies are available, the CDA-AMC Clinical Review identified serious methodological limitations that prevented drawing any definitive conclusions.
CDA-AMC identified several limitations with the sponsor’s submitted pharmacoeconomic analysis. These included adopting utility-waning assumptions (informed by sponsor-sought expert opinion) that disproportionately lowered utilities experienced by nonresponders on BSC, overestimation of disease management costs, and use of adherence to adjust treatment costs of dupilumab only.
It its reanalysis, CDA-AMC addressed some of the limitations in the sponsor’s analysis by setting the utility to be identical to baseline at 24 weeks for initial nonresponders and after 6 months for patients discontinuing treatment, assuming no hospitalization for nonresponders, and assuming 100% adherence to dupilumab. Results of the CDA-AMC reanalysis align with those of the sponsor’s submitted analysis, indicating that dupilumab was not a cost-effective treatment option in the indicated population at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) gained compared to best supportive care (BSC). In the CDA-AMC base case, dupilumab plus BSC was associated with an incremental cost-effectiveness ratio (ICER) of $138,915 per QALY gained ($122,220 in incremental costs and 0.88 incremental QALYs) compared with BSC alone in the patient population aligned with the PRIME trials. A price reduction of approximately 61% would be required for dupilumab to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. This would reduce the price of dupilumab from $979 to $384 per 300 mg single-use prefilled syringe. With this price reduction, the annual per-patient drug acquisition costs for dupilumab would be approximately $10,393 in the first year and $9,626 in subsequent years. Drug acquisition costs represented the majority of the total costs associated with dupilumab. All (100%) of the incremental QALYs gained for dupilumab plus BSC were derived after the randomized, double-blinded period of 24 weeks of the PRIME and PRIME2 trials.
Clinical expert input received by CDA-AMC for this review indicated that immunosuppressants (such as methotrexate and cyclosporin) are used in clinical practice to manage patients with PN. The comparative effectiveness and cost-effectiveness of dupilumab relative to these therapies for moderate to severe PN is unknown.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Canadian Skin Patient Alliance, which collected the perspectives of caregivers and patients aged 35 years and older with PN through an online survey across Canada. Patients with PN reported experiencing pruritus, nodules (i.e., itchy bumps), skin scarring, hyperpigmentation, hypopigmentation, and a negative impact on psychological health. Patients experiencing persistent and severe itching reported that it disrupted their ability to carry out daily activities, work, and sleep. The presence of visible modules and scars also negatively affected their social interactions and mental health. Caregivers reported experiencing a psychological burden and feeling helpless. Patients with PN described experience with a variety of treatments, which included topical corticosteroids (TCSs), topical calcineurin inhibitors (TCIs), immunosuppressants such as methotrexate and cyclosporin, narrowband UV-B phototherapy, psoralen plus UV-A photochemotherapy, topical capsaicin, oral antihistamines, intralesional corticosteroids, topical vitamin D derivatives, thalidomide, tricyclic depressants, anticonvulsants. and medical cannabis. Respondents expressed frustration and emotional distress from trying multiple therapies with limited efficacies. Two patients had experience with dupilumab, and neither reported any improvement in their condition. The patient input described AEs experienced by respondents; however, it was unclear whether these events were linked to dupilumab, other treatments, or both. The reported side effects included racing heart, skin irritation, nausea, vomiting, hypopigmentation, and hyperpigmentation. Patients reported discontinuing treatment due to AEs and lack of efficacy. Treatment goals described by patients included improved effectiveness in managing itch, quality of life, and safety profile.
No clinician input was received for this review.
The drug plan input noted concerns with whether prior therapy with immunosuppressants or phototherapy would be required before initiation of dupilumab for PN, which suggested uncertainty regarding the anticipated place in therapy. The plans flagged a potentially substantial budget impact if immunosuppressants would not be required to be trialled before dupilumab. The drug plan input also highlighted concerns with potential use of dupilumab in pediatric population and for multiple conditions given that it is funded in many jurisdictions for indications other than PN.
Two of these concerns were addressed in the sponsor’s model:
Quality of life was incorporated in the sponsor’s model by use of EQ-5D data collected in the PRIME trials.
Health outcomes values by patients (such as management of itch) were considered.
CDA-AMC was unable to address concerns about the cost-effectiveness of dupilumab compared to immunosuppressants and phototherapy because of the lack of submitted clinical data, and the cost-effectiveness of dupilumab when used in combination with other treatments (aside from topicals) for PN.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of dupilumab plus BSC compared to BSC alone for the treatment of adult patients with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.1 The modelled population was aligned with those used in the PRIME and PRIME2 trials,2,3 which enrolled patients for whom a 2-week course of a medium- to super-potent TCS had failed or when TCSs were not medically advisable. The modelled population was aligned with the Health Canada indication and reimbursement request.
Two strengths of dupilumab are available (200 mg per 1.14 mL [175 mg/mL] and 300 mg per 2 mL [150 mg/mL]) in prefilled syringes for self-administration.4 The recommended dose of dupilumab is an initial dose of 600 mg (2 injections of 300 mg each), followed by 300 mg every other week.4 The sponsor estimated that 2 injections of dupilumab would be used to administer the loading dose and 5 injections would be used to administer the maintenance dose over the first 12 weeks. The sponsor estimated that, following this, 6 injections of dupilumab would be used to administer the maintenance dose over 12 weeks. At the submitted price of $978.70 per 300 mg injection,1 the sponsor estimated the per-patient annual drug acquisition cost of dupilumab to be $26,425 in the first year of treatment and $25,446 in subsequent years. Treatment costs for dupilumab were adjusted by an assumed patient-adherence rate of 88.5%. BSC consisted of a low- to medium-potency TCS (such as hydrocortisone, betamethasone, and clobetasone) and TCIs (such as tacrolimus). The sponsor estimated an average cost of $20 for 100 g of TCSs and $327 for 100 g of TCIs, with prices per gram for each topical therapy obtained from the Ontario Drug Benefit Formulary.5 The sponsor assumed that 6 tubes of TCSs and 2 tubes of TCIs would be used every 12 weeks. Based on these assumptions, the sponsor estimated that the annual per-patient treatment costs to be $531 and $2,834 for mild to moderate TSCs and TCIs, respectively.
The analysis was undertaken from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and QALYs) were estimated over a lifetime time horizon of 50 years and discounted at an annual rate of 1.5%. The cycle length was 12 weeks.
Model Structure
The sponsor submitted a pharmacoeconomic model that included a decision tree (Figure 1) and a Markov state transition model (Figure 2). The decision tree assessed treatment response at 24 weeks and categorized patients into responders and nonresponders using data from the PRIME and PRIME2 trials.2,3 Patients entered the Markov model into health states based on their responses. The Markov model simulated the movement of patients though 3 mutually exclusive health states: “Response,” “No response” and “Death,” over the lifetime horizon. Responders entered the “Response” health state and continued with initial treatment. Nonresponders entered the “No response” health state and switched to BSC. Patients could remain in their assigned health states or experience the possibility of transitioning to the “No response” state in each cycle. Patients on dupilumab who transitioned to nonresponder status discontinued treatment. The model included 13 tunnel states to track duration since treatment discontinuation for up to 3 years and captured time-dependent changes in utility for nonresponders. Patients could transition to the “Death” state from either of the 2 other health states at any time.
Model Inputs
The baseline characteristics in the model were derived from the PRIME and PRIME2 trials (mean age of 49.5 years, 35% male, mean weight of 73.9 kg).2,3
Clinical efficacy inputs (i.e., response and all-cause discontinuation rates) were derived using data from the PRIME and PRIME2 trials.2,3 In the trials, treatment response was defined as a reduction in the Worst Itch Numeric Rating Scale (WI-NRS) score of 4 points or greater at week 24 (in the PRIME trial) or week 12 (in PRIME2 trial) compared to baseline.2,3 Response rates for dupilumab plus BSC and BSC alone were estimated based on the primary analysis method in the PRIME and PRIME2 trials. These rates informed the distribution of patients on dupilumab plus BSC and BSC alone into “Response” and “No response” health states in the beginning of the Markov model phase. The transition probability from responder status to nonresponder status in the Markov model was informed by treatment discontinuation and sponsor’s assumptions on treatment waning. Discontinuation rates for dupilumab plus BSC and BSC alone were derived using data from the PRIME and PRIME2 trials,2,3 and reflected treatment discontinuation for any reason. The sponsor assumed this was a proxy for loss of response to treatment. The sponsor also applied an additional treatment-waning effect in each cycle, which increased the transition of patients from the “Response” to the “No response” health state at a higher rate for BSC compared to dupilumab. Treatment waning modelled for the BSC arm was based on clinical expert feedback obtained in the CDA-AMC Pharmacoeconomic Review of dupilumab for atopic dermatitis.6 The proportion of initial responders to dupilumab who became nonresponders was informed by the sponsor’s assumptions. The sponsor assumed that treatment did not affect mortality risks. Age- and sex-specific mortality rates were based on Statistics Canada life tables for the general population.7
The sponsor included the impact of AEs, such as allergic conjunctivitis, injection-site reactions, skin infections, oral herpes, and infectious conjunctivitis using data pooled from the PRIME and PRIME2 trials.2,3 The costs and disutility associated with AEs were calculated for dupilumab plus BSC and BSC, and applied in each cycle. The cost of treating an AE was assumed to be equivalent to the cost of 1 general physician visit.8 Disutilities associated with each AE were sourced from published literature.9-11 The estimated cost and disutility were weighted by the proportion of patients experiencing each AE in the PRIME and PRIME2 trials.2,3
Health-state utility values for “Response” and “No response” were derived at baseline, week 12, and week 24 using patient-level EQ-5D-5L data from both arms of the PRIME and PRIME2 trials.2,3 The value set used to derive utility scores was not specified in the sponsor’s submission. The sponsor estimated utility scores using mixed linear models that controlled for several variables, such as age, sex, baseline utility score, baseline DLQI total score, postbaseline DLQI total score, and WI-NRS score using a forward selection process. The EQ-5D-5L scores, which assessed health using 5 levels, were converted to EQ-5D-3L scores, which uses 3 levels and an algorithm published by van Hout (2022).12 Utilities were also adjusted for age-related decrements using an approach described by Ara et al. (2017).13 The sponsor modelled an increase in utility at week 12 for all patients compared to baseline. Responders experienced a further increase in utilities at week 24. For nonresponders, a decrease in utility was modelled, with decrements applied at 6, 12, 24, and 48 months based on feedback received from sponsor-sought clinical experts. Patients not responding on BSC alone experienced a greater decrease in utility compared to patients on dupilumab plus BSC. By 48 months, nonresponders on BSC returned to baseline, while those on dupilumab plus BSC retained a utility that was 9.15% higher than baseline.
The economic model included costs associated with drug acquisition, treatment administration, and disease management. Drug acquisition costs related to dupilumab, BSC, and rescue medications were included. The cost of rescue medications was estimated for patients on dupilumab plus BSC and BSC alone using data on frequency of use in the PRIME and PRIME2 trials,2,3 relevant acquisition costs, and an assumed treatment duration of 2 weeks for most medications. Prices were obtained from the Ontario Drug Benefits Formulary.5 Drug administration costs included the cost of training to self-inject delivered by a registered nurse. This was assumed to be the equivalent of the hourly wage of a registered nurse and applied as a 1-time upfront cost in the decision-tree phase of the model.14 Disease management costs included the cost of visits to health care professionals (such as a primary care physician, outpatient dermatologist, psychologist, and allergist), emergency department visits, hospitalization, laboratory test for blood counts, and phototherapy sessions. Costs were obtained from the Ontario Schedule of Benefits and Canadian Institute of Health Information.15-17 The frequency of resource use by response status was based on sponsor-sought expert opinion. Notably, the sponsor assumed 1 hospitalization per year for nonresponders based on sponsor-sought expert feedback on barriers to access to primary care physicians. The sponsor also estimated that nonresponders would have 45.7 phototherapy sessions annually based on published literature.18
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (500 iterations). The results of the deterministic and probabilistic results were similar; the results of the probabilistic analyses are presented in the following section. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base case, dupilumab plus BSC was more effective (1.01 incremental QALYs) and $74,942 more costly compared to BSC alone, resulting in an ICER of $74,305 per QALY gained (Table 3). Dupilumab plus BSC was not associated with any life-years gained compared to BSC alone. At a WTP threshold of $50,000 per QALY gained, there was a 11% probability of dupilumab plus BSC being cost-effective compared with BSC alone. The submitted analysis is based on the publicly available prices for all drug treatments.
All (100%) of the incremental QALYs gain for dupilumab plus BSC accrued after the randomized, double-blinded period of 24 weeks of the PRIME and PRIME2 trials. In the decision-tree phase, total QALYs were driven by utility estimates rather than effectiveness. The decision-tree phase did not contribute to incremental QALYs. The incremental QALYs observed in the Markov model phase were derived from the response and nonresponse rates at 24 weeks. Incremental costs were driven primarily by the higher drug acquisition costs for dupilumab ($117,216 in incremental costs), which was offset by reduced disease management costs for nonresponders ($42,938 in cost savings). Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3, Table 11.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($ per QALY) |
|---|---|---|---|---|---|
BSC | 345,090 | Reference | 15.46 | Reference | Reference |
Dupilumab plus BSC | 420,031 | 74,942 | 16.47 | 1.01 | 74,305 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario and sensitivity analyses, which included exploring alternative discount rates, time horizons, and response criteria (defined as an improvement in the WI-NRS score of ≥ 4 points and a reduction in the Investigator’s Global Assessment Prurigo Nodularis–Stage score of ≥ 1). Additional scenarios involved restricting patient populations to those with a starting WI-NRS score of 7 or greater, a history of atopic dermatitis or atopy, and prior use of immunosuppressants. Adopting a 0% discount rate decreased the ICER to $72,047 per QALY gained. When the patient population was restricted to those with a history of atopic dermatitis, the ICER increased to $88,799 per QALY gained. The results of a 1-way sensitivity analysis showed that the ICER was sensitive to variations in baseline utility and utility for responders. When the baseline utility was increased or the responder utility for dupilumab patients was decreased by 10% of the mean value, the ICER increased to $125,595 per QALY gained and $118,654 per QALY gained, respectively.
The sponsor included a scenario analysis that included additional costs associated with productivity losses for patients. The sponsor included per-patient estimated earnings lost due to time incurred from visits to health care professionals (such as a primary care physician, outpatient dermatologist, psychologist, and allergist), laboratory tests, phototherapy, emergency department visits, and hospitalization. The sponsor assumed that a visit to an emergency department and hospitalization were the equivalent of losing 1 day and 4.2 days of earnings, respectively. Adopting this perspective decreased the ICER to $24,833 per QALY gained.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Long-term treatment efficacy is uncertain: The CDA-AMC Clinical Review of the PRIME and PRIME2 trials found that dupilumab provided a significant and clinically meaningful benefit to patients compared to BSC. The randomized, double-blinded period of these studies was 24 weeks. The Clinical Review also found that, while some observational studies support the durability of treatment effects, serious methodological concerns precluded drawing definitive conclusions about long-term treatment efficacy. The entirety of the sponsor’s estimated incremental QALYs are derived from the Markov model, which begins after 24 weeks. The lack of strong evidence to support the duration of the treatment response beyond the trial period, coupled with the model’s reliance on extrapolation and an assumption of treatment-effectiveness beyond 24 weeks, adds uncertainty to the results of the economic analysis.
CDA-AMC could not address this limitation due to the lack of other sources of long-term treatment-efficacy data.
Utility waning for nonresponders is uncertain: During its review of the sponsor-submitted pharmacoeconomic model, CDA-AMC noted that the sponsor implemented utility-waning assumptions for nonresponders that were not described in the technical report. CDA-AMC requested clarification of the modelled utility-waning inputs, including the rationale and data sources used to derive them. In response, the sponsor referenced sponsor-sought clinical expert input that suggested dupilumab would extend the quality of life of patients who discontinue treatment after initially responding. However, modelling a utility higher than the baseline value after discontinuation implies that patients are still receiving some benefit from treatment. This appears to lack face validity because, by the time patients discontinue treatment, the benefits from treatment would have largely ceased, and it is reasonable to assume that utility would return to baseline. It was also observed that, in the model, nonresponders on dupilumab plus BSC did not return to baseline utility but instead maintained a higher utility than baseline over the lifetime horizon. In contrast, nonresponders on BSC returned to baseline after 2 years. This higher utility for dupilumab nonresponders was applied not only to those who discontinued treatment after initial response but also to those who never responded in the first place (i.e., at 24 weeks). It also lacks face validity for initial nonresponders (who experienced no treatment benefit in the first place) to have a higher utility than baseline, further weakening the rationale for this assumption. The sponsor also cited the results of a regression analysis based on data from the PRIME study to support the modelled difference in utility between dupilumab and BSC nonresponders. However, the sponsor did not submit this analysis as part of its submission of clinical evidence, preventing CDA-AMC from appraising and reviewing this information within the review timelines. As such, there were no clinical data to support the sponsor’s modelled utility-waning assumptions beyond sponsor-sought expert opinion. The sponsor’s modelled utility assumptions for nonresponders overestimated the QALYs associated with dupilumab plus BSC, resulting in a lower ICER that favours dupilumab.
CDA-AMC notes that the National Institute for Health and Care Excellence appraisal of dupilumab for the treatment of adult patients with PN raised similar concerns about the sponsor’s assumptions regarding treatment waning. The appraisal was conducted, and its results were published (on March 13, 2024)21 before the economic evaluation was submitted to CDA-AMC (on December 20, 2024).
In a reanalysis, CDA-AMC assumed that initial nonresponders (i.e., patients who did not respond to treatment in the first place) would return to baseline utility at week 24. For patients who transitioned to nonresponder status after initially responding, CDA-AMC assumed the utility would resume to baseline within 6 months of discontinuing treatment.
Relevant comparators are excluded: The sponsor’s submitted base case compared dupilumab plus BSC to BSC alone. However, according to clinical expert input received by CDA-AMC for this review, as well as published literature,19 patients with PN may be treated with immunosuppressants such as methotrexate and cyclosporin.19 The exclusion of immunosuppressants from the sponsor’s analysis is problematic because it fails to assess the cost-effectiveness of dupilumab among all relevant treatment options.
CDA-AMC notes that the sponsor requested a deviation from the CDA-AMC pharmacoeconomic requirements to exclude immunosuppressants from the economic model. Although this request was approved by CDA-AMC on the basis that an indirect treatment comparison was not feasible for the indicated population, the sponsor was advised that the exclusion of relevant comparators could be noted as a limitation of its submission. The sponsor’s decision to exclude all relevant comparators is problematic because it does not fully capture the comparative cost-effectiveness of dupilumab. The cost-effectiveness of dupilumab compared to existing therapies for the indicated population remains unknown.
CDA-AMC could not address this limitation due to the lack of data on comparative effectiveness. At publicly available list prices, dupilumab is more costly than immunosuppressants (Appendix 1).
Disease management costs for nonresponders have been overestimated: The sponsor assumed that nonresponders would require 1 hospitalization per year while responders would have none, and cited lack of access to a primary care physician as the reason. Clinical expert input obtained for this review noted that, while PN patients without access to primary care may visit walk-in clinics or hospital emergency departments, inpatient admissions are uncommon for patients with PN. The cost associated with hospitalization in the model reflected the cost incurred by the public health care payer for inpatient hospital admission.22 The sponsor’s assumptions about cost and frequency of receiving care overestimated the disease management cost for nonresponders. This introduced a bias in estimated costs that favoured dupilumab.
In a reanalysis, CDA-AMC assumed zero hospitalizations for nonresponders based on expert feedback that inpatient admissions are uncommon for patients with PN.
Use of adherence to adjust drug costs is not appropriate: The sponsor adjusted the total treatment cost of dupilumab by assuming a treatment adherence rate of █████████ sourced from the SINUS-52 trial.23 The sponsor’s approach effectively assumed that only █████████ of the dupilumab injections would be used and underestimated the treatment cost of dupilumab. This consideration of adherence is problematic because, once the injections are dispensed, the full cost is incurred by public drug plans regardless of actual usage.
In a reanalysis, a treatment adherence rate of 100% was assumed.
Poor modelling practices were employed: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical. It remains unclear whether the model is running inappropriately by overriding errors.
CDA-AMC was unable to address the use of IFERROR statements in the model and notes that a thorough validation of the sponsor’s model was not possible.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patients experience adverse events at the same risk as observed in the PRIME trials over the lifetime horizon. | Inappropriate. Due to the lack of long-term clinical data, it is unknown if the risk of adverse events will remain constant. CDA-AMC evaluated the impact of excluding adverse events, which had a minor impact on the cost-effectiveness of dupilumab. |
The sponsor assumed that patients experience an increase in utility at week 12 compared to baseline, even though response is assessed at week 24. | Inappropriate. The sponsor artificially introduced earlier benefits for responders. Given the response rate is higher for dupilumab plus BSC compared with BSC alone in the Markov model phase, the sponsor’s approach overestimated the QALYs gained for dupilumab plus BSC more so than for BSC alone, making dupilumab appear more cost-effective by lowering the ICER. |
The sponsor assumed that both all-cause discontinuation and treatment waning reflects loss of response. | Inappropriate. Modelling both loss of response and all-cause discontinuation as a proxy of loss of response leads to double-counting the decline in effectiveness of treatment. Because patients on BSC transition from the “Response” to “No response” health state at a faster rate than those receiving dupilumab, this introduces a bias that favours the relative effectiveness of dupilumab. CDA-AMC explored the impact of excluding all-cause discontinuation rates and found it had minimal impact on cost-effectiveness results. |
The sponsor assumed that a single 1-hour training session with a registered nurse is sufficient and that no additional training or support from health care professionals would be needed to administer dupilumab. | Uncertain. If additional support from health care professionals would be required to administer dupilumab subcutaneous injections, the drug administration costs may have been underestimated. |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CDA-AMC undertook a stepped analysis, sequentially incorporating each adjustment outlined in Table 5 into the sponsor’s model to demonstrate the impact of each change. These included setting utilities for nonresponders at week 24 to be identical to baseline utilities, and utilities for discontinuing patients to be identical to baseline after 6 months, assuming no hospitalization for nonresponders and assuming 100% adherence to dupilumab. The results of the CDA-AMC reanalyses are summarized in Table 6.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Utility for nonresponders | Decrements to utility applied at 6, 12, 24, and 48 months | Utility for nonresponders at week 24 set to be identical to baseline Utility for discontinuing patients to be identical to baseline after 6 months |
2. Frequency of hospitalization for nonresponders | 1 | 0 |
3. Adherence to dupilumab | ██████ | 100% |
CDA-AMC base case | Reanalysis 1 + 2 + 3 | |
CDA-AMC = Canada’s Drug Agency.
In the CDA-AMC base case, dupilumab plus BSC was associated with an ICER of $138,915 per QALY gained compared with BSC alone ($122,220 in incremental costs and 0.88 incremental QALYs) (Table 6). Incremental costs were driven by higher drug acquisition costs ($131,471 in incremental costs) (Table 14). All of the incremental QALYs gained for dupilumab plus BSC were derived after the randomized, double-blinded period of 24 weeks of the PRIME and PRIME2 trials. At a WTP threshold of $50,000 per QALY gained, there was a 0.04% probability of dupilumab plus BSC being cost-effective compared with BSC alone. The submitted analysis is based on the publicly available prices for all drug treatments.
Table 6: Summary of the Stepped Analysis of CDA-AMC Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | BSC | 345,090 | 15.46 | Reference |
Dupilumab plus BSC | 420,031 | 16.47 | 74,305 | |
CDA-AMC reanalysis 1 | BSC | 346,053 | 15.47 | Reference |
Dupilumab plus BSC | 419,703 | 16.32 | 86,548 | |
CDA-AMC reanalysis 2 | BSC | 151,788 | 15.50 | Reference |
Dupilumab plus BSC | 258,078 | 16.47 | 108,985 | |
CDA-AMC reanalysis 3 | BSC | 346,053 | 15.50 | Reference |
Dupilumab plus BSC | 433,033 | 16.47 | 89,184 | |
CDA-AMC base case (Reanalysis 1 + 2 + 3) | BSC | 151,788 | 15.47 | Reference |
Dupilumab plus BSC | 271,408 | 16.32 | 140,571 | |
CDA-AMC base case (Reanalysis 1 + 2 + 3) (probabilistic) | BSC | 151,837 | 15.44 | Reference |
Dupilumab plus BSC | 274,057 | 16.32 | 138,915 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Scenario Analysis Results
CDA-AMC undertook price-reduction analyses based on the sponsor’s results and the CDA-AMC base case (Table 7). The CDA-AMC base case suggests that a price reduction of approximately 61% would be required for dupilumab plus BSC to be considered cost-effective at a WTP threshold of $50,000 per QALY relative to BSC alone. This would reduce the price of dupilumab from $979 to $384 per 300 mg single-use prefilled syringe. With this price reduction, the annual per-patient drug acquisition costs for dupilumab would be approximately $10,393 in the first year and $9,626 in subsequent years.
Table 7: CDA-AMC Price Reduction Analyses
Price reduction | Unit drug cost ($) | ICERs for dupilumab plus BSC versus BSC ($ per QALY) | |
|---|---|---|---|
Sponsor base case | CDA-AMC reanalysis | ||
No price reduction | 979 | 74,305 | 138,915 |
10% | 880 | Dominant | 124,294 |
20% | 783 | Dominant | 109,673 |
30% | 685 | Dominant | 95,053 |
40% | 587 | Dominant | 80,432 |
50% | 489 | Dominant | 65,811 |
60% | 391 | Dominant | 51,190 |
70% | 294 | Dominant | 36,570 |
80% | 196 | Dominant | 21,949 |
90% | 98 | Dominant | 7,328 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Issues for Consideration
CDA-AMC has previously reviewed and recommended dupilumab for several conditions, including for the treatment of patients aged 6 months and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.24,25 The cost-effectiveness results of these evaluations may not be directly comparable to those in the current review because of differences in target populations, model structures, clinical effectiveness parameters, health-state utility values, and cost inputs. The pan-Canadian Pharmaceutical Alliance concluded negotiations with a letter of intent for dupilumab for adults with atopic dermatitis, and the drug is under negotiation for pediatric patients with atopic dermatitis.26 As such, dupilumab has a confidential negotiated price, and is currently funded by jurisdictional formularies.27-30 The CDA-AMC reanalyses are based on the publicly available price of dupilumab, which may be different than the confidential price and may influence the results of the cost-effectiveness and budget impact analyses (BIA).
Nemolizumab is currently being reviewed by CDA-AMC for the treatment of moderate to severe PN.31 The expert input obtained for this review noted that dupilumab and nemolizumab are likely to be positioned similarly in the treatment pathway of PN. The experts also noted that the 2 drugs have different mechanisms of action, and clinicians may switch treatment between them if patients have an inadequate response to 1 or the other. The cost-effectiveness of dupilumab relative to nemolizumab is unknown because of the lack of direct and indirect comparative clinical evidence. It is also unknown how switching between dupilumab and nemolizumab would affect total costs and health outcomes.
The sponsor assumed that only 300 mg single-use prefilled syringes would be used in the treatment of patients with PN. Dupilumab is available in 2 formulations (200 mg and 300 mg syringes);4 however, the cost-utility analysis and BIA considered only 1 formulation (300 mg syringe) to estimate treatment costs. Although the other formulation is priced similiarly, it contains a lower dose (200 mg) of dupilumab rather than the recommended dose for PN. If the lower-dose formulation is used to administer the initial dose in clinical practice, the treatment costs incurred by a public drug plan may be higher than modelled.
Overall Conclusions
The CDA-AMC clinical review of the PRIME and PRIME2 trials found that, in adults with moderate to severe PN who were inadequately controlled on topical prescription therapies, dupilumab resulted in a significant and clinically important decrease in pruritus intensity and a reduction in PN nodules compared to placebo. The trial data collected through the DLQI suggested, to a moderate degree of certainty, that dupilumab likely improved patient-reported quality of life compared to placebo. No serious safety concerns were raised. Although conjunctivitis was identified as an important AE, the evidence was too uncertain to draw conclusions about the impact of dupilumab on the frequency of conjunctivitis-related AEs. Long-term efficacy and safety (beyond the 24-week trial follow-up) are uncertain. Although observational studies are available, the CDA-AMC Clinical Review identified serious methodological limitations that prevented any definitive conclusions.
CDA-AMC identified several limitations with the sponsor’s pharmacoeconomic analysis. This included adopting utility-waning assumptions informed by sponsor-sought expert opinion that disproportionately lowered utilities experienced by nonresponders on BSC, overestimated disease management costs, and used treatment adherence to adjust treatment costs of dupilumab only.
CDA-AMC undertook reanalyses to address some of the limitations in the sponsor’s analysis, including setting the utility to be identical to baseline at 24 weeks for initial nonresponders and after 6 months for patients discontinuing treatment, assuming no hospitalization for nonresponders, and assuming 100% adherence to dupilumab. Results of the CDA-AMC reanalysis align with those of the sponsor’s submitted analysis, indicating that dupilumab was not a cost-effective treatment option in the indicated population at a WTP threshold of $50,000 per QALY gained compared to BSC. In the CDA-AMC base case, dupilumab plus BSC was associated with an ICER of $138,915 per QALY gained compared to BSC alone in the patient population aligned with those of the PRIME trials ($122,220 in incremental costs and 0.88 incremental QALYs). A price reduction of approximately 61% would be required for dupilumab to be considered cost-effective at a WTP threshold of $50,000 per QALY gained. This would reduce the price of dupilumab from $979 to $384 per 300 mg single-use prefilled syringe. With this price reduction, the annual per-patient drug acquisition costs for dupilumab would be approximately $10,393 in the first year and $9,626 in subsequent years. Drug acquisition accounted for the majority of the total costs associated with dupilumab. All (100%) of the incremental QALYs gained for dupilumab plus BSC accrued after the randomized, double-blinded period of 24 weeks of the PRIME and PRIME2 trials.
Clinical expert input received by CDA-AMC for this review indicated that immunosuppressants (such as methotrexate and cyclosporin) are used to manage patients with PN in clinical practice. The comparative effectiveness and cost-effectiveness of dupilumab relative to these therapies for moderate to severe PN are unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 300 mg single-use syringe (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL) for subcutaneous injection. Toronto (ON): Sanofi-aventis Canada Inc; 2025 Mar 11.
2.Sanofi Research and Development. [CONFIDENTIAL] Clinical Study Report for EFC16459. A randomized, double blind, placebo-controlled, multi-center, parallel group study to evaluate the efficacy and safety of dupilumab in patients with prurigo nodularis who are inadequately controlled on topical prescription therapies or when those therapies are not advisable (LIBERTY-PN PRIME). Database lock December 09, 2021 (Data cut-off November 12, 2021).
3.Sanofi Research and Development [CONFIDENTIAL} Clinical Study Report for EFC16460. A randomized, double blind, placebo-controlled, multi-center, parallel group study to evaluate the efficacy and safety of dupilumab in patients with prurigo nodularis who are inadequately controlled on topical prescription therapies or when those therapies are not advisable (LIBERTY-PN PRIME2). Database lock: September 27, 2021 (Data cut-off August 30, 2021).
4.Sanofi-aventis Canada Inc. Dupixent (duilumab injection) 200 mg and 300 mg Product Monograph. Date of Initial Authorization: November 30, 2017. Date of Revision: September 6, 2024.
5.Ontario Ministry of H. Ontario Drug Benefit Formulary/Comparative Drug Index. https://www.formulary.health.gov.on.ca/formulary/
6.Cda AMC, CDA-AMC. CDA-AMC dupilumab AD - Adolescent indication. 2020. https://www.cda-amc.ca/sites/default/files/cdr/pharmacoeconomic/sr0636-dupixent-pharmacoeconomic-review-report.pdf
7.StatsCan. Statistics C. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island. 2020-2022. 2024. https://www150.statcan.gc.ca/n1/en/catalogue/84-537-X
8.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. Accessed 2025 Mar 11, https://www.formulary.health.gov.on.ca/formulary/
9.Retzler J, Grand TS, Domdey A, Smith A, Romano Rodriguez M. Utility elicitation in adults and children for allergic rhinoconjunctivitis and associated health states. Qual Life Res. Sep 2018;27(9):2383-2391. doi:10.1007/s11136-018-1910-8 PubMed
10.Boye KS, Matza LS, Walter KN, Van Brunt K, Palsgrove AC, Tynan A. Utilities and disutilities for attributes of injectable treatments for type 2 diabetes. The European Journal of Health Economics. 2011/06/01 2011;12(3):219-230. doi:10.1007/s10198-010-0224-8
11.Matza LS, Kim KJ, Yu H, et al. Health state utilities associated with post-surgical Staphylococcus aureus infections. Eur J Health Econ. Aug 2019;20(6):819-827. doi:10.1007/s10198-019-01036-3 PubMed
12.van Hout B, Janssen MF, Feng Y-S, et al. Interim Scoring for the EQ-5D-5L: Mapping the EQ-5D-5L to EQ-5D-3L Value Sets. Value Health. 2012/07/01/ 2012;15(5):708-715. doi:https://doi.org/10.1016/j.jval.2012.02.008
13.Ara R, Brazier JE. Using health state utility values from the general population to approximate baselines in decision analytic models when condition-specific data are not available. Value Health. Jun 2011;14(4):539-45. doi:10.1016/j.jval.2010.10.029 PubMed
14.Island H. British Columbia Nurses Union. Salary Information. https://www.islandhealth.ca/sites/default/files/careers/documents/BC-nurses-salary-benefits.pdf
15.Government of Ontario MoH, Long-Term C, Government of Ontario MoH, Long-Term C. Schedule of Benefits and Fees. Government of Ontario, Ministry of Health and Long-Term Care. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/
16.Ontario Ministry of H. Schedule of Benefits for Laboratory Services. 2020. http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf
17.Cihi. Patient Cost Estimator | CIHI. https://www.cihi.ca/en/patient-cost-estimator
18.Adrus E. NB-UVB Phototherapy Demonstrates Efficacy, Leads to Complete Response in Prurigo Nodularis. Dermatology Times. https://www.dermatologytimes.com/view/nb-uvb-phototherapy-demonstrates-efficacy-leads-to-complete-response-in-prurigo-nodularis
19.Ständer S, Pereira MP, Berger T, et al. IFSI-guideline on chronic prurigo including prurigo nodularis. Itch. 2020;5(4)
20.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 300 mg single-use syringe (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL) for subcutaneous injection. Toronto (ON): Sanofi-aventis Canada Inc; 2025 Mar 11.
21.NICE. Dupilumab for treating moderate to severe prurigo nodularis (TA955). 2024. Accessed 2024 Mar 20. www.nice.org.uk/guidance/ta955
22.CIHI. Cost of a Standard Hospital Stay. Accessed March 11, 2025, https://yourhealthsystem.cihi.ca/hsp/inbrief?lang=en#!/indicators/015/cost-of-a-standard-hospital-stay/;mapC1;mapLevel2;/
23.sanofi. Clinical Study Report. DUPILUMAB/SAR231893/REGN668. EFC14280. SINUS-52. 15.1 DEVIATIONS DATA. 2018.
24.CDA-AMC Canadian Drug Expert Committee (CDEC) final recommendation: Dupilumab for patients aged 6 years and older with AD. CDA-AMC; 2023. Accessed 2025 Mar 11. https://www.cda-amc.ca/dupilumab-5
25.CDA-AMC Canadian Drug Expert Committee (CDEC) final recommendation: Dupilumab for patients aged 12 years and older with AD. CDA-AMC; 2023. Accessed 2025 Mar 11. https://www.cda-amc.ca/dupilumab-0
26.Alliance P-CP. PCPA negotiations for AD. Accessed March 11, 2025, https://www.pcpacanada.ca/negotiation/22496
27.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed 2025 Mar 11. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
28.Saskatchewan Drug Plan: search formulary. Accessed March 5, 2025, https://formulary.drugplan.ehealthsask.ca/SearchFormulary
29.Government BC. BC PharmaCare formulary search. Accessed 2025 Mar 5, https://pharmacareformularysearch.gov.bc.ca
30.Government of A. Interactive drug benefit list. Accessed 2025 Mar 5, https://idbl.ab.bluecross.ca/idbl/load.do
31.Drug Reimbursement Review pharmacoeconomic report: Nemolizumab (Nemluvio) for Prurigo nodularis. CDA-AMC; 2025. Accessed 2025 Mar 11. https://www.cda-amc.ca/nemolizumab
32.Cyclosporin: capsules, 10, 25, 50 and 100 mg [product monograph]. Novartis Pharmaceuticals Canada Inc; 2017. chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.novartis.com/ca-en/sites/novartis_ca/files/neoral_scrip_e.pdf
33.Methotrexate: tablets, 2.5 mg [product monograph]. Pfizer Canada Inc; 2024. https://pdf.hres.ca/dpd_pm/00043044.PDF
34.Klejtman T, Beylot-Barry M, Joly P, et al. Treatment of prurigo with methotrexate: a multicentre retrospective study of 39 cases. J Eur Acad Dermatol Venereol. Mar 2018;32(3):437-440. doi:10.1111/jdv.14646 PubMed
35.Wiznia LE, Callahan SW, Cohen DE, Orlow SJ. Rapid improvement of prurigo nodularis with cyclosporine treatment. J Am Acad Dermatol. 2018;78(6):1209-1211. doi:10.1016/j.jaad.2018.02.024 PubMed
36.Siepmann D, Luger TA, Ständer S. Antipruritic effect of cyclosporine microemulsion in prurigo nodularis: results of a case series. J Dtsch Dermatol Ges. Nov 2008;6(11):941-6. doi:10.1111/j.1610-0387.2008.06745.x PubMed
37.StatsCan. Statistics C. Population estimates on July 1st, by age and sex. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
38.Morgan CL, Thomas M, Ständer S, et al. Epidemiology of prurigo nodularis in England: a retrospective database analysis. Br J Dermatol. Jan 26 2022;doi:10.1111/bjd.21032 PubMed
39.Pereira MP, Zeidler C, Wallengren J, et al. Chronic Nodular Prurigo: A European Cross-sectional Study of Patient Perspectives on Therapeutic Goals and Satisfaction. Acta Derm Venereol. Feb 17 2021;101(2):adv00403. doi:10.2340/00015555-3726 PubMed
40.INESSS. Dupixent Atopic Dermatitis (12 years of age and older). Available at: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Juillet_2020/Dupixent_Ado_2020_06.pdf.
41.Sutherland, al e. The Conference Board of Canada: Understanding the Gap. A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. 2017;
42.Ontario Drug Benefit e-formulary. Available at: https://www.formulary.health.gov.on.ca/formulary.
43.Bahloul D, Hudson R, Balogh O, et al. Prevalence, incidence and treatment patterns of prurigo nodularis in England: a retrospective database analysis. Br J Dermatol. 2024;191(4):548-555. doi:10.1093/bjd/ljae207 PubMed
44.Huang AH, Canner JK, Khanna R, Kang S, Kwatra SG. Real-World Prevalence of Prurigo Nodularis and Burden of Associated Diseases. J Invest Dermatol. 2020/02/01/ 2020;140(2):480-483.e4. doi:https://doi.org/10.1016/j.jid.2019.07.697
45.Murota H, Arima K, Yoshida T, Fujita H. Disease burden and treatment satisfaction in patients with prurigo nodularis in Japan. J Dermatol. Feb 2024;51(2):223-233. doi:10.1111/1346-8138.17045 PubMed
46.Information CIfH. Pan-Canadian Prescription Drug Data Landscape. CIHI; 2024. Accessed 2025 Mar 19. https://www.cihi.ca/sites/default/files/document/pan-canadian-prescription-drug-data-landscape-report-en.pdf
47.Information CIfH. Pharmaceutical Data Tool. 2025 Mar 19, https://www.cihi.ca/en/pharmaceutical-data-tool?utm_medium=email&utm_source=crm&utm_campaign=pharmaceutical-data-tool-jan-2025&utm_content=pharmaceutical-data-tool-en&mkt_tok=Mjg3LVZLSS04NjEAAAGYM28a6QKN8nG-ZKxao72em_vQadc22GyLcun7vPboQLwfwKCL6g5CezPzEGZ_HG1oQFvmhIqDyGFqAOfmqVbdW25f_y4XYhMyF4JiUw
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison Table for Prurigo Nodularis
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Dupilumab (Dupixent) | 300 mg/2 mL 200 mg/1.14 mL | Prefilled syringe or pen | 978.7000a 978.7000b | Initial dose of 600 mg. 300 mg given every 2 weeks | First year: 72.59 Subsequent year: 69.91 | First year: 26,512 Subsequent year: 25,534 |
CDA-AMC = Canada’s Drug Agency.
Note: Annual costs assume 365.25 days for all comparators. Dose and formulation is obtained from product monograph.4
aSponsor-submitted price.1
bPrice obtained from the Ontario Exceptional Access Program, accessed March 11, 2025.27
Table 9: CDA-AMC Cost Comparison Table for Systemic Off-Label Treatments for Prurigo Nodularis
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Immunosuppressant | ||||||
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | 5 to 25 mg per weeka | 0.07 to 0.36 | 26 to 131 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7526 0.8657 1.6885 3.3792 | 2 to 5 mg per kg per dayb | 5.07 to 11.82 | 1,850 to 4,317 |
CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
Notes: Annual costs assume 365.25 days and weight of 70 kg. All prices are from the Ontario Drug Benefit Formulary (accessed March 11, 2025),8 unless otherwise indicated, and do not include dispensing fees. Formulation information was obtained from respective product monographs.32,33 Dosage was obtained from published literature,34-36 and validated by clinical expert feedback. The clinical expert input also noted that patients with PN are typically treated with immunosuppressants for a year.
aMulticentre retrospective study of prurigo cases treated with methotrexate.34
bA chart review of patients with prurigo nodularis treated with cyclosporine.35,36
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | No | Refer to key limitation on “Relevant comparators are excluded.” |
Model has been adequately programmed and has sufficient face validity. | No | Refer to key limitation on “Poor modelling practices were employed.” |
Model structure is adequate for decision problem. | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | Utility-waning assumptions based on sponsor-sought expert input were hard-coded in the backend worksheet. Although the sponsor estimated range around the mean by assuming a 10% variation for utility inputs in the absence of published validity, this was not done for the inputs informing utility waning in the submitted model. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | The uncertainty around utility-waning assumptions was not captured, which limited the ability to assess their impact on the probabilistic cost-effectiveness results. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details). | No | There were several instances of misalignment between the technical report and pharmacoeconomic model, which prompted CDA-AMC to request clarification regarding efficacy inputs and results. The model also included a treatment-specific utility-waning feature that was not described in the technical report. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Model Structure — Decision Tree
BSC = best supportive care; PN = prurigo nodularis.
aPatients had at least 20 prurigo nodularis lesions at baseline, which corresponds to moderate to severe on the Investigator's Global Assessment – Prurigo Nodularis scale.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of Sponsor’s Economic Evaluation Results
Parameter | Dupilumab plus BSC | BSC |
|---|---|---|
Discounted LYs | ||
Total | 26.43 | 26.43 |
Life-years in decision tree | 0.46 | 0.46 |
Life-years in Markov model | 25.96 | 25.96 |
Responder | 4.64 | 0.14 |
Nonresponder | 21.32 | 25.83 |
Discounted QALYs | ||
Total | 16.47 | 15.46 |
QALYs in decision tree | 0.32 | 0.32 |
Disutilities in decision tree | −0.00088 | −0.00020 |
QALYs in Markov model | 16.17 | 15.16 |
Disutilities in Markov model | −0.02 | −0.01 |
By health state | ||
Response | 3.74 | 0.11 |
Nonresponse | 12.44 | 15.04 |
Responder Disutilities | −0.01 | 0.00 |
Nonresponder Disutilities | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 420,031 | 345,090 |
Decision tree | ||
Drug acquisition | 14,276 | 1,553 |
Dupilumab | 12,723 | 0 |
BSC | 1,553 | 1,553 |
Rescue medication | 4 | 90 |
Drug administration | 44 | 0 |
Disease management | 4,404 | 4,404 |
Adverse events | 14 | 2 |
Markov model | ||
Drug acquisition | 196,087 | 92,462 |
Responder – dupilumab | 104,492 | 0 |
Responder – BSC | 15,616 | 468 |
Responder – rescue medication | 39 | 27 |
Nonresponder – BSC | 71,769 | 86,917 |
Nonresponder – rescue medication | 4,170 | 5,050 |
Drug administration | 0 | 0 |
Disease management | 204,969 | 246,467 |
Responder | 1,484 | 44 |
Nonresponder | 203,485 | 246,423 |
Adverse events | 234 | 111 |
Responder | 143 | 1 |
Nonresponder | 91 | 110 |
BSC = best supportive care; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 12: Disaggregated Summary of the CDA-AMC Economic Evaluation Results
Parameter | Dupilumab plus BSC | BSC |
|---|---|---|
Discounted LYs | ||
Total | 26.42 | 26.42 |
Life-years in decision tree | 0.46 | 0.46 |
Life-years in Markov model | 25.96 | 25.96 |
Responder | 4.67 | 0.14 |
Nonresponder | 21.29 | 25.82 |
Discounted QALYs | ||
Total | 16.31 | 15.43 |
QALYs in decision tree | 0.32 | 0.32 |
Disutilities in decision tree | −0.00088 | −0.00019 |
QALYs in Markov model | 16.02 | 15.13 |
Disutilities in Markov model | −0.02 | −0.01 |
By health state | ||
Response | 3.75 | 0.12 |
Nonresponse | 12.27 | 15.01 |
Responder Disutilities | −0.01 | 0.00 |
Nonresponder Disutilities | −0.01 | −0.01 |
Discounted costs ($) | ||
Total | 274,057 | 151,837 |
Decision tree | ||
Drug acquisition | 14,276 | 1,553 |
Dupilumab | 12,723 | 0 |
BSC | 1,553 | 1,553 |
Rescue medication | 4 | 90 |
Drug administration | 44 | 0 |
Disease management | 1,011 | 1,011 |
Adverse events | 14 | 2 |
Markov model | ||
Drug acquisition | 134,454 | 469 |
Responder – dupilumab | 118,748 | 0 |
Responder – BSC | 15,706 | 469 |
Responder – rescue medication | 39 | 27 |
Nonresponder – BSC | 71,664 | 86,902 |
Nonresponder – rescue medication | 4,164 | 5,049 |
Drug administration | 0 | 0 |
Disease management | 48,155 | 56,627 |
Responder | 1,493 | 45 |
Nonresponder | 46,662 | 56,583 |
Adverse events | 232 | 107 |
Responder | 144 | 1 |
Nonresponder | 88 | 106 |
BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; LY = life-year; PN = prurigo nodularis; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
Summary of Sponsor’s Budget Impact Analysis
The submitted BIA assessed the expected budgetary impact of reimbursing dupilumab for the treatment of adult patients with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.20 The BIA was undertaken from the perspective of the Canadian public drug plans at base year (2025) and over a three-year time horizon (2026 to 2028). The sponsor’s pan-Canadian estimates reflected the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 14.
The sponsor estimated the number of patients eligible for dupilumab using an epidemiologic approach with data obtained from published literature and sponsor’s clinical experts input.37-41 The sponsor narrowed the population in each jurisdiction to adults with PN who seek treatment but are uncontrolled on topical therapy and eligible for advanced therapy.37-40 The sponsor also restricted the number of patients to those eligible for coverage by public drug plan.41 Comparators included immunosuppressants (such as methotrexate and cyclosporin) and BSC. BSC was assumed to consist of a basket of TCSs and TCIs, and have zero cost to the drug plans. The cost of treatment with immunosuppressants was calculated based on the price and required dose of methotrexate and cyclosporin, and assuming 50% utilization of each drug. Prices for methotrexate and cyclosporin were obtained from Ontario Drug formulary,42 and dosing was informed by sponsor-sought clinical experts. The sponsor also included the cost of folic acid based on sponsor-sought expert feedback that it reduces the toxicity of methotrexate. Dispensing fees and mark-ups were not included. Market share assumptions were based on sponsor’s internal estimates and opinion of clinical experts. The sponsor assumed that market share of dupilumab would be captured by approximately ███ from immunosuppressants and ███ from BSC.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Percentage of adult | 81.32% |
Prevalence of PN | 0.03% |
Percentage of patients treated | 90.00% |
Proportion of patients uncontrolled on topical therapy | |
Proportion of patients eligible for advanced therapy | 75.00% |
Proportion of patients eligible for drug plan coverage | 48.42% |
Number of patients eligible for drug under review | |
Market uptake (3 years) | |
Uptake (reference scenario) BSC Immunosuppressants | █████████ █████████ |
Uptake (new drug scenario) Dupilumab BSC Immunosuppressants | █████████ █████████ █████████ |
Cost of treatment (per patient, per year) | |
Dupilumab First year Subsequent year | $26,425 $25,446 |
BSC Immunosuppressants | $0 $1,194 |
BSC = best supportive care; PN = prurigo nodularis.
Summary of the Sponsor’s BIA Results
The sponsor estimated that reimbursing dupilumab for the treatment of adult patients with PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable to be $54,350,257 over the first 3 years (Year 1: $9,959,875; Year 2: $19,745,355; Year 3: $24,645,027).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Prevalence is uncertain: The sponsor adopted a prevalence of 3 per 10,000 persons using a 2022 study by Morgan et al., that estimated the prevalence rate using a retrospective analysis of medical records from the Clinical Practice Research Datalink in England.38 However, there is considerable uncertainty associated with prevalence rates in published literature. For example, another retrospective database analysis study reported a prevalence of 8.8 per 10,000 persons living in England.43 Furthermore, as noted by the sponsor, the estimated prevalence rate among individuals living in the US was reported to be 7.2 per 10,000 persons.44 This suggests that the adopted prevalence rate may have underestimated the number of eligible patients.
Given the lack of Canada-specific data, CDA-AMC adopted a prevalence rate of 8.3 per 10,000 persons in reanalysis based on a meta-analysis.45 In scenario analysis, CDA-AMC explored the impact of adopting a prevalence of 5 per 10,000 based on expert input received for this review.
Number of eligible patients were underestimated: The sponsor estimated the number of eligible patients by restricting the number of PN patients to those uncontrolled on topical therapy and then, further to those eligible for advanced therapy. These inputs were based on sponsor’s internal market research and sponsor’s assumption. The data used to estimate the proportion of patients uncontrolled on topical therapy was not provided, which prevented CDA-AMC from validating the estimate. Moreover, the proportion of patients eligible for advanced therapy was informed by an assumed acceptance of dupilumab by prescribing physicians. The clinical expert feedback received for this review noted that all patients with moderate to severe disease and uncontrolled on topical therapy would be eligible for treatment with dupilumab. According to expert input, approximately 60% to 70% of patients are uncontrolled on topical therapy.
In reanalysis, CDA-AMC estimated that 45.4% of PN patients would have moderate to severe disease and uncontrolled on topical therapy.45 This estimate was based on the distribution of patients across disease severity in published literature and clinical expert feedback.
The proportion of patients covered by public drug plans is uncertain: The sponsor derived jurisdiction-specific coverage rate for patients aged between 18 to 64 years using data on the proportion of patients enrolled in the report by Sutherland et al., (2017).41 According to clinical expert input obtained for this review, the proportion of patients with moderate to severe PN on disability tend to be higher than the general population. Many jurisdictions have support programs available to help cover the cost of treatments for patients with low-income, including those on disability.46,47
In scenario analysis, CDA-AMC explored the impact of deriving coverage rate for patients aged 18 to 64 years using data on the proportion of patients eligible for coverage with public drug plans.41 CDA-AMC retained the sponsor’s assumption that patients aged 18 to 24 years would have the same public coverage as adults aged 25 to 64 years. This increased the average pan-Canadian coverage rate weighted by population size of each jurisdiction to 70%.
Market share of dupilumab may have been underestimated: The sponsors assumed that dupilumab would have a market share of ███ in year 1, ███ in year 2 and ███ by year 3, if it is reimbursed. The clinical expert feedback received by CDA-AMC for this review anticipated a higher uptake of dupilumab in clinical practice. The expert input noted that physicians have familiarity with prescribing dupilumab for other indications such as atopic dermatitis. As such, physicians would be more inclined to prescribe dupilumab for PN. Given this, if dupilumab is reimbursed, its uptake is expected to be rapid in clinical practice.
In scenario analysis, CDA-AMC assumed that the market uptake of dupilumab would be 60%, 70% and 80% in years 1, 2, and 3, respectively, based on clinical expert feedback.
Market capture is uncertain: The market share capture of dupilumab is highly uncertain due to evolving treatment landscape for PN. The sponsor assumed that market share of dupilumab would be captured from BSC (consisting of a TCS and a TCI but incurring no cost to plans) and immunosuppressants (i.e., methotrexate and cyclosporine). However, new treatments are emerging in this space. In particular, nemolizumab is currently under review for patients with moderate to severe PN.31 If new treatments entering this space change prescribing pattens, the market capture may differ significantly from the sponsor’s assumptions.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analyses by adopting a prevalence rate of 8.3 per 10,000 persons, and restricting eligibility criteria to those with moderate to severe disease and uncontrolled on topical therapy (Table 15).
Table 15: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CDA-AMC base case | ||
1. Prevalence of PN | 0.03% | 0.083% |
2. Eligibility criteria | Proportion of patients uncontrolled on topical therapy (██████) and proportion of patients eligible for advanced therapy (75.00%) | Proportion of patients with moderate to severe PN (64.9%) and proportion of patients uncontrolled on topical therapy (70.0%) Eligibility by advanced therapy was excluded. This was done by assuming 100% patients were eligible for advanced therapy |
CDA-AMC base case | Reanalysis 1 + 2 | |
CDA-AMC = Canada’s Drug Agency; PN = prurigo nodularis.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 18.
Based on the CDA-AMC base case, the budget impact associated with the reimbursement of dupilumab for the treatment of adult patients with moderate to severe PN whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable increased to $186,941,572 (year 1: $34,257,697, year 2: $67,915,552, year 3: $84,768,323).
Table 16: Summary of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 54,350,257 |
CDA-AMC reanalysis 1 | 137,953,251 |
CDA-AMC reanalysis 2 | 73,650,475 |
CDA-AMC base case | 186,941,572 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 18):
Adopting a lower prevalence (i.e., 5 per 10,000) based on expert input.
Increasing market share of dupilumab (i.e., 60% in year 1, 70% in year 2 and 80% in year 3) based on expert feedback.
Deriving coverage rate for patients aged 18 to 64 years using data on the proportion of patients eligible for coverage with public drug plans.
Table 17: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 308,582 | 311,647 | 314,744 | 317,873 | 944,264 |
New drug | 308,582 | 10,271,522 | 20,060,099 | 24,962,900 | 55,294,521 | |
Budget impact | 0 | 9,959,875 | 19,745,355 | 24,645,027 | 54,350,257 | |
CDA-AMC base case | Reference | 1,061,388 | 1,071,932 | 1,082,584 | 1,093,347 | 3,247,863 |
New drug | 1,061,388 | 35,329,629 | 68,998,136 | 85,861,670 | 190,189,435 | |
Budget impact | 0 | 34,257,697 | 67,915,552 | 84,768,323 | 186,941,572 | |
CDA-AMC scenario analysis 1: prevalence of 5 per 10,000 | Reference | 639,391 | 645,742 | 652,159 | 658,642 | 1,956,544 |
New drug | 639,391 | 21,282,909 | 41,565,142 | 51,723,897 | 114,571,949 | |
Budget impact | 0 | 20,637,167 | 40,912,983 | 51,065,255 | 112,615,405 | |
CDA-AMC scenario analysis 2: increased market share of dupilumab | Reference | 1,061,388 | 1,071,932 | 1,082,584 | 1,093,347 | 3,247,863 |
New drug | 1,061,388 | 69,587,326 | 79,250,134 | 91,254,203 | 240,091,663 | |
Budget impact | 0 | 68,515,394 | 78,167,549 | 90,160,856 | 236,843,800 | |
CDA-AMC scenario analysis 3: deriving coverage rate based on proportion eligible | Reference | 1,525,700 | 1,540,855 | 1,556,167 | 1,571,637 | 4,668,660 |
New drug | 1,525,700 | 50,784,799 | 99,181,773 | 123,422,333 | 273,388,904 | |
Budget impact | 0 | 49,243,943 | 97,625,606 | 121,850,696 | 268,720,245 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.