Drugs, Health Technologies, Health Systems
Reimbursement Review
Delgocitinib (Anzupgo)
Sponsor: Leo Pharma Inc.
Therapeutic area: Chronic hand eczema
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AD
atopic dermatitis
AE
adverse event
AESI
adverse event of special interest
CANN
Canadian Association of Neonatal Nurses
CDA-AMC
Canada’s Drug Agency
CFB
change from baseline
CHE
chronic hand eczema
CI
confidence interval
CrI
credible interval
DAO
Dermatology Association of Ontario
DIC
deviance information criterion
DLQI
Dermatology Life Quality Index
DSU
Decision Support Unit
ESC
Eczema Society of Canada
ESS
effective sample size
FAS
full analysis set
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HECSI
Hand Eczema Severity Index
HECSI-75
at least a 75% improvement in HECSI score from baseline
HECSI-90
at least a 90% improvement in HECSI score from baseline
HEIS
Hand Eczema Impact Scale
HESD
Hand Eczema Symptom Diary
HF-IGA
Hand and Foot Investigator’s Global Assessment
HRQoL
health-related quality of life
IGA-CHE
Investigator’s Global Assessment of Chronic Hand Eczema
IPD
individual patient data
ITC
indirect treatment comparison
ITT
intention to treat
JAK
Janus kinase
MAIC
matching-adjusted indirect comparison
MID
minimal important difference
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
NRS
Numerical Rating Scale
OR
odds ratio
PGA
Physician’s Global Assessment
PUVA
psoralen and UVA radiation
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
STAT
signal transducer and activator of transcription
TCS
topical corticosteroids
TEAE
treatment-emergent adverse event
WOCF
worst observation carried forward
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of the Application Submitted for Review
Item | Description |
|---|---|
Drug product, strength, formulation | Delgocitinib (Anzupgo), 20 mg/g, cream, topical |
Sponsor | Leo Pharma Inc. |
Indication | For the treatment of moderate to severe chronic hand eczema (CHE) in adults for whom topical corticosteroids are inadequate or are not advisable |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 25, 2025 |
Recommended dose | A thin layer of delgocitinib should be applied twice daily to the affected skin of the hands and wrists |
NOC = Notice of Compliance.
Introduction
Hand eczema is a painful, pruritic, inflammatory, noninfectious skin disease that primarily affects the hands and wrists.1-3 Chronic hand eczema (CHE) is defined by the European Society of Contact Dermatitis, as well as Canadian and German consensus-based guidelines, as hand eczema persisting for at least 3 months or recurring at least twice per year.1,4,5 Its severity is typically assessed through clinical signs, disease duration, treatment history, and impact on patient function.1,6
CHE has a multifactorial etiology, with both exogenous and endogenous triggers.2,5 Risk factors include a personal history of childhood eczema, asthma, or hay fever, as well as occupations involving frequent wet work or exposure to irritants and allergens, such as those in health care, food processing, and hairdressing.5,7-9 The pathophysiology involves dysregulated immune responses and a breakdown of skin barrier function.2,9 Activation of the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway contributes to inflammation and suppresses the expression of structural proteins such as filaggrin, leading to skin barrier impairment.2,9 Long-term exposure to environmental factors like mechanical irritation, extreme temperatures, or chemicals further exacerbates this dysfunction.10
The main symptoms of CHE include itch and pain, but people with CHE often also report dryness, cracking, thickening of the skin, and bleeding. These signs and symptoms fluctuate over time, with many people experiencing periodic flares.3,11 CHE imposes a substantial burden, contributing to impaired hand function, sleep disturbance, psychological distress, reduced work capacity, and negative social impacts.1,5,10,12
In Canada, the prevalence of CHE among adults is estimated at 6.2%, with a higher occurrence in females.13 In the Canada-based cohort of the international CHECK study, approximately 70% of patients were female (30% male), the median patient age was 41 years, and about 44% of patients had moderate to severe CHE.14,15
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups that responded to the Canada’s Drug Agency (CDA-AMC) call for input and from the clinical expert(s) consulted by CDA-AMC for the purpose of this review.
Patient Input
Two patient groups, the Eczema Society of Canada (ESC) and the Canadian Association of Neonatal Nurses (CANN), submitted input for this review. Another patient group, the Canadian Skin Patient Alliance, did not directly provide input for this submission but provided a letter of support for the patient submission developed and submitted by the ESC. The ESC group obtained information related to CHE through questionnaires and interviews with patients, caregivers, and health care professionals, as well as survey data on various topics (e.g., impact on quality of life, experience with treatments, patient journeys) from more than 3,000 people in Canada who live with eczema. CANN collected anonymous data through qualitative interviews with nurses conducted in July 2024 and obtained their perspectives of living and working with CHE. The group also conducted an online quantitative survey between October 23 and November 10, 2024, of its nursing members living with CHE (n = 27), which was distributed through CANN’s email communication list and established membership channels. All information was gathered in Canada.
Both patient groups highlighted a significant impairment to everyday living and quality of life due to CHE. The groups reported that people with CHE experienced negative impacts on sleep quality (due to pain), skin (burning, itching lesions, blisters, bleeding skin), work (loss of productivity, change in careers due to CHE), social interactions, fitness, intimacy, family life, and hygiene. CANN further noted that survey respondents reported itchy skin as the symptom that caused the greatest burden, followed by dry or chapped skin; dyshidrotic blisters; cracking skin or fissures; open wounds with bleeding; and painful, stinging, and throbbing hands. Both patient groups highlighted that current treatment regimens may not be sufficient and expressed concerns regarding long-term steroid use. In the survey conducted by CANN, 52% of the respondents reported that their current treatment and management regimen could not control or could only somewhat help them manage their hand eczema.
Both patient groups highlighted the need for a treatment that provided better disease management, including relief from itching, stinging, burning, and pain; improvement in skin disease; and reduction of flares. CANN further highlighted that people with CHE want to function normally, return to work, experience reduced severity of flares, and have decreased risk of blisters and open wounds. The ESC also noted that people with CHE were looking for treatments suitable for long-term use that could allow them to regain their confidence and self-esteem and improve their social life and quality of life. In both patient groups, those who had had the opportunity to be treated with delgocitinib noted positive experiences with the treatment. The ESC further highlighted that delgocitinib offered patients relief; was well tolerated, without stinging or burning; and was not greasy on the hands.
Clinician Input
Input From Clinical Experts Consulted for This Review
The experts identified a clear unmet need for effective, well-tolerated, nonsystemic therapies for adults with moderate to severe CHE, particularly for those who do not experience a response to topical corticosteroids (TCS) or who experience intolerance to systemic therapies such as alitretinoin due to side effects or contraindications. According to the experts, delgocitinib may fill this gap by offering a topical, nonsteroidal option that can be used earlier in the treatment pathway and may delay or prevent the need for systemic treatment.
The experts considered patients with moderate to severe CHE for whom first-line TCS has failed as the population most likely to benefit from delgocitinib. The patients least likely to benefit may include those with primarily irritant-driven eczema, those who would experience difficulty in adhering to the treatment regimen, or those with very limited disease that can be managed with intermittent topical steroids. The experts emphasized that treatment should begin once TCS have proven insufficient to achieve control of the disease, especially in patients with persistent symptoms affecting function or quality of life.
Response to treatment should be assessed based on improvements in visible skin signs, reduction in itching and pain, and enhanced ability to perform daily tasks, including work. Clinical tools such as the Investigator’s Global Assessment of Chronic Hand Eczema (IGA-CHE) and the Hand Eczema Severity Index (HECSI), along with patient-reported outcomes like the Dermatology Life Quality Index (DLQI) and symptom diaries, were viewed as useful for monitoring treatment response. The experts recommended discontinuing delgocitinib if no meaningful clinical improvement is observed within 8 to 12 weeks, or sooner if the disease worsens or adverse reactions occur.
Additional considerations raised by the experts included the importance of patient education on appropriate treatment application and expectations for treatment response. They also highlighted the potential role of delgocitinib in reducing the need for systemic immunosuppressants or biologics and its practical advantages for long-term management, especially for patients requiring maintenance therapy with a lower side-effect burden.
Clinician Group Input
Five clinician groups provided input for this review: the Dermatology Association of Ontario (DAO), the Atlantic Dermatology Group, the Canadian Dermatology Association’s Pharmacy and Therapeutics Advisory Board, the Saskatchewan Dermatology Association, and dermatologists practising in Canada with an interest in CHE. Information for this submission included input from 21 clinicians and was gathered through reviews of published literature, research and trial experience, clinical practice experience, and national and international meetings.
The input from clinician groups was generally consistent with the input from the clinical experts consulted for this review, emphasizing the ongoing unmet need for new topical prescription therapy options for CHE that would control the signs and symptoms of eczema, normalize quality of life, and reduce or abolish occupational impacts. The DAO group further emphasized the need to include therapies for long-term control of CHE that are safe, effective, and accessible. The clinician groups highlighted the limitations of current therapies, including adverse events (AEs) related to topical corticosteroid use such as skin atrophy, skin infections, pain, burning, fissures, reduced hand dexterity, and bleeding; limited efficacy and poor access to phototherapy; and contraindications related to alitretinoin use (e.g., pregnancy, breastfeeding). The DAO group noted that while topical calcineurin inhibitors like tacrolimus are sometimes used in mild cases of CHE when a steroid-sparing therapy is required due to AEs related to steroid use, they are not effective treatment alternatives for moderate and severe forms of CHE. The DAO group also highlighted several off-label systemic options for patients with CHE in cases when phototherapy and alitretinoin are not suitable, are not accessible, or fail to control the condition; these options include traditional immunosuppressants (i.e., methotrexate and cyclosporine), biologics (particularly ones used in atopic dermatitis [AD], such as dupilumab), and oral JAK inhibitors (i.e., upadacitinib). However, the group noted limitations related to these off-label options as well, including accessibility and cost (i.e., coverage criteria may make a biologic therapy inaccessible, or the co-pay cost to the patient may be a barrier), safety considerations (e.g., black box warnings and contraindications across the oral systemic therapies), and expected effectiveness (i.e., traditional immunosuppressants are not as effective in managing CHE as biologics or oral JAK inhibitors). The groups indicated that people involved in certain professions with frequent exposure to irritants and allergens — such as people working in health care, cleaning, and hairdressing — report higher incidence rates of CHE.
The DAO and the Canadian Dermatology Association’s Pharmacy and Therapeutics Advisory Board groups considered topical delgocitinib as a second-line treatment for patients with moderate to severe CHE (i.e., a Physician’s Global Assessment [PGA] score of 3 to 4) whose disease was refractory to TCS, or who experienced intolerance to TCS, or for whom TCS were contraindicated. The Atlantic Dermatology Group considered topical delgocitinib as a first-line or second-line treatment for adult patients with moderate to severe CHE for whom TCS are inadequate or inappropriate. The groups noted that delgocitinib is expected to cause a shift in the current treatment paradigm as a new targeted topical prescription therapy for CHE. The DAO group highlighted delgocitinib’s ease of access and treatment administration over phototherapy and noted that it did not require laboratory monitoring and had no contradictions (e.g., pregnancy, breastfeeding, or renal or hepatic insufficiency) as is the case for alitretinoin. Referring to the clinical trials, the groups also noted the efficacy and favourable safety profile of delgocitinib in patients with moderate to severe CHE. The clinician groups provided additional details on patient selection, noting that delgocitinib may be particularly appropriate for patients with moderate to severe CHE who have experienced an inadequate response to TCS or for whom TCS are not advisable or are contraindicated. The groups also noted that patients with mild CHE for whom TCS have not yet been used or whose CHE is being adequately managed by emollients, avoidance strategies, or TCS are less appropriate candidates for treatment with delgocitinib.
The clinician groups noted that an initial assessment of treatment response at approximately 3 to 6 months (around 16 weeks) after initiation would be reasonable. The DAO group further highlighted that both the timing and criteria used to assess response may vary among clinicians, depending on clinical practice wait times, availability of follow-up, and patient-specific factors. The clinician groups added that outcome measures are not formally standardized in routine clinical practice for CHE, and that clinicians typically rely on an informal PGA. The clinician groups agreed that reasons for discontinuation of delgocitinib may include inadequate or loss of efficacy, recurrent flares, disease worsening or nonresponse despite an adequate therapeutic trial, or intolerance and adverse effects.
Drug Program Input
The drug programs raised several questions related to the implementation of delgocitinib. First, they questioned the appropriateness of comparing delgocitinib to vehicle cream rather than other topicals like vitamin D3 derivatives (e.g., calcipotriol) or calcineurin inhibitors (e.g., tacrolimus, pimecrolimus). The clinical experts acknowledged this concern but emphasized that alitretinoin remains the only treatment with a Notice of Compliance in Canada for CHE, supporting the relevance of the chosen comparator despite regional differences in drug coverage.
Questions were also raised about the use of the IGA-CHE as the primary outcome measure rather than the more commonly used Eczema Area and Severity Index score. The experts clarified that the IGA-CHE and the EASI score are clinically comparable and can be used interchangeably in practice. They endorsed the use of the IGA-CHE in the trials.
The experts noted that delgocitinib is not approved for children, though interest in off-label use may arise. They referenced the existence of trials investigating related drugs in pediatric populations and pointed to systemic JAK inhibitors that already have pediatric indications.
Finally, the drug programs inquired about positioning delgocitinib in the treatment sequence — whether it should follow failure of topical steroids and/or nonsteroidal topicals like calcipotriol or tacrolimus. The experts indicated that delgocitinib would most often be used after the failure of TCS or intolerance to TCS but could also be considered when TCS are contraindicated. They did not suggest that systemic therapies like methotrexate must be attempted before delgocitinib is initiated.
Clinical Evidence
Systematic Review
Description of Studies
Three phase III randomized controlled trials (RCTs) evaluated the efficacy and safety of delgocitinib cream 20 mg/g in adults with moderate to severe CHE. Two vehicle-controlled trials, the DELTA 1 trial (N = 487) and the DELTA 2 trial (N = 473), assessed whether delgocitinib improved IGA-CHE treatment success and HECSI scores over 16 weeks compared to vehicle cream. An active-controlled trial, the DELTA FORCE trial (N = 513), compared delgocitinib to oral alitretinoin over 24 weeks in patients with severe CHE. The primary and key secondary outcomes included change in HECSI score, IGA-CHE treatment success, at least a 75% improvement in HECSI score from baseline (HECSI-75), at least a 90% improvement in HECSI score from baseline (HECSI-90), and patient-reported measures such as Hand Eczema Symptom Diary (HESD) itch and DLQI scores.
Across all trials, the baseline demographic and disease characteristics were generally well balanced between groups. The mean age of the participants ranged from 44 to 46 years, with most having had CHE symptoms for at least 4 to 6 years. The population was predominantly white (more than 90%) and slightly more than half were female. The baseline severity scores (e.g., HECSI, IGA-CHE) were consistent with moderate to severe CHE, and no notable imbalances were observed across treatment arms.
Efficacy Results
Vehicle-Controlled Studies
In the vehicle-controlled trials (DELTA 1 and DELTA 2 trials), delgocitinib cream 20 mg/g twice daily demonstrated superior efficacy over vehicle cream at week 16 across clinician-reported and patient-reported outcomes.
IGA-CHE treatment success was experienced by 64 of 325 patients (19.7%) treated with delgocitinib in the DELTA 1 trial versus 16 of 162 patients (9.9%) treated with vehicle cream (difference = 9.8%; 95% confidence interval [CI], 3.6 to 16.1) and by 91 of 313 patients (29.1%) treated with delgocitinib in the DELTA 2 trial compared to 11 of 159 patients (6.9%) treated with vehicle cream (difference = 22.2%; 95% CI, 15.8 to 28.5; P = 0.005).
More patients treated with delgocitinib experienced HECSI-75 than patients treated with vehicle cream (DELTA 1 trial: 49.2% [160 of 325] versus 23.5% [38 of 162]; DELTA 2 trial: 49.5% [155 of 313] versus 18.2% [29 of 159]). The same was true of HECSI-90 (DELTA 1 trial: 29.5% [96 of 325] versus 12.3% [20 of 162]; DELTA 2 trial: 31.0% [97 of 313] versus 8.8% [14 of 159]). The mean change from baseline (CFB) in HECSI score also favoured delgocitinib in both studies (DELTA 1 trial: difference = –35.2; 95% CI, –46.7 to –23.8; P < 0.001; DELTA 2 trial: difference = –45.5; 95% CI –56.4 to –34.6; P < 0.001).
A 4-point or greater reduction in HESD itch score was reported by 47.1% of patients treated with delgocitinib (152 of 323) in the DELTA 1 trial versus 23.0% of patients treated with vehicle cream (37 of 161). In the DELTA 2 trial, these values were 47.2% of patients treated with delgocitinib (146 of 309) versus 19.9% of patients treated with vehicle cream (31 of 156).
In terms of quality of life, an improvement in DLQI score of 4 points or greater was reported in 74.4% of patients treated with delgocitinib (227 of 305) in the DELTA 1 trial versus 50.0% of patients treated with vehicle cream (74 of 148) and in 72.2% of patients treated with delgocitinib (216 of 299) in the DELTA 2 trial versus 45.8% of patients treated with vehicle cream (70 of 153). The mean DLQI score improved by –7.6 and –7.0 points in the delgocitinib groups in the DELTA 1 and DELTA 2 trials, respectively, compared to –3.9 and –3.1 points in the vehicle cream groups. The mean difference was –3.6 (95% CI, –4.7 to –2.6) in the DELTA 1 trial and –3.9 (95% CI, –5.0 to –2.8) in the DELTA 2 trial. Hand Eczema Impact Scale (HEIS) scores also improved more with delgocitinib in the DELTA 1 trial (difference = –0.6; 95% CI, –0.8 to –0.5) and in the DELTA 2 trial (difference = –0.8; 95% CI, –1.0 to –0.6). All effects across these outcomes in the DELTA 1 and DELTA 2 trials were considered clinically meaningful.
Active-Controlled Study
In the active-controlled DELTA FORCE trial, delgocitinib demonstrated greater efficacy than oral alitretinoin 30 mg once daily. At week 12, IGA-CHE treatment success was experienced by 68 of 250 patients (27.2%) receiving delgocitinib versus 42 of 253 patients (16.6%) receiving alitretinoin (difference = 10.6%; 95% CI, 3.3 to 17.9; P = 0.004). The mean change CFB in HECSI score was –67.6 versus –51.5 at week 12 (difference = –16.1; 95% CI, –23.28 to –8.86; P < 0.001) and –69.6 versus –45.1 at week 24 (difference = –24.5; 95% CI, –32.55 to –16.36; P < 0.001), favouring delgocitinib. HECSI-90 was experienced by 96 of 249 patients (38.6%) receiving delgocitinib versus 65 of 250 patients (26.0%) receiving alitretinoin at week 12 (difference = 12.6%; 95% CI, 4.3 to 20.8; P = 0.003). While the observed differences in HESD itch score reduction of 4 points or more, in DLQI score, and in HEIS score were smaller and consistently favoured delgocitinib, they were judged to not be clinically meaningful.
These outcomes were exploratory and not adjusted for multiplicity.
Harms Results
Across the DELTA 1, DELTA 2, and DELTA FORCE trials, delgocitinib cream 20 mg/g twice daily demonstrated a favourable safety profile. In the vehicle-controlled studies (DELTA 1 and DELTA 2 trials), the proportion of patients experiencing at least 1 AE was similar between groups: 45.2% with delgocitinib versus 50.6% with vehicle cream in the DELTA 1 trial and 45.5% versus 44.7% in the DELTA 2 trial. Most AEs were mild to moderate. The most commonly reported AEs included COVID-19, nasopharyngitis, and headache.
In the active-controlled DELTA FORCE study, AEs occurred in 49.4% of patients treated with delgocitinib versus 76.1% of patients treated with oral alitretinoin. Some notable AEs occurred more frequently with alitretinoin, such as headache (32.4% versus 4.0%), nausea (5.7% versus 0.4%), dry skin (3.6% versus 1.2%), and hypercholesterolemia (3.6% versus 0%).
Serious AEs (SAEs) were infrequent across all studies: reported in no more than 2.0% of patients treated with delgocitinib in all trials, compared to up to 4.9% of patients treated with alitretinoin in the DELTA FORCE trial. No SAE led to major safety concerns or deaths. AEs leading to treatment discontinuation were also rare with delgocitinib (≤ 1.2% of patients) and notably higher with alitretinoin (10.1% of patients). No AEs of special interest (AESIs) occurred in the delgocitinib groups, and 1 patient in the alitretinoin group experienced a deep vein thrombosis. No deaths occurred in any of the trials.
Critical Appraisal
The vehicle-controlled trials (DELTA 1 and DELTA 2 trials) were methodologically robust, with low risk of bias across outcomes. Randomization, allocation concealment, and blinding were well executed, and outcome assessments used instruments that were overall validated (although some were validated within the sponsored study) and aligned with the trial objectives. Missing data were minimal and handled appropriately using standard imputation methods. In contrast, the DELTA FORCE trial employed an open-label design in which patients and treating physicians were aware of treatment allocation; however, efficacy assessments were conducted by blinded assessors to mitigate potential detection bias. Differential dropout rates between treatment arms (36% for alitretinoin versus 13% for delgocitinib) likely reflect differences in tolerability and clinical effects inherent to systemic versus topical products. However, this degree of imbalance raises concerns about attrition bias, particularly for longer-term and subjective outcomes, and the direction of this bias would likely favour delgocitinib.
The trials enrolled patients consistent with the Health Canada indication for delgocitinib, with baseline characteristics reflective of the intended population. However, the underrepresentation of racial and geographic diversity, the exclusion of patients with significant comorbidities, and the controlled trial setting may modestly limit generalizability. Nonetheless, the intervention and comparator regimens, background care, and outcomes assessed were aligned with clinical practice in Canada, supporting the overall applicability of findings to the real-world setting.
GRADE Summary of Findings and Certainty of the Evidence
The selection of outcomes for Grading of Recommendations Assessment, Development and Evaluation (GRADE) assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. Table 2 and Table 3 present the summary of findings for these outcomes.
Table 2: Summary of Findings for Delgocitinib Versus Vehicle Cream for Patients With CHE
Outcome and follow‑up | Patients (studies), N | Absolute effects | Certainty | What happens |
|---|---|---|---|---|
Severity of CHE | ||||
IGA-CHE treatment success Follow-up: 16 weeks | 959 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Higha | Delgocitinib results in a clinically meaningful increase in treatment success, as defined by the IGA-CHE score, compared to vehicle cream. |
HECSI-90 Follow-up: 16 weeks | 959 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Higha | Delgocitinib results in a clinically meaningful increase in patients experiencing a HECSI-90 response compared to vehicle cream. |
HECSI-75 Follow-up: 16 weeks | 959 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Higha | Delgocitinib results in a clinically meaningful increase in patients experiencing a HECSI-75 response compared to vehicle cream. |
HECSI, change from baseline Follow-up: 16 weeks | 959 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Highb | Delgocitinib results in a clinically meaningful reduction in HECSI score from baseline when compared to vehicle cream. |
Symptom reduction | ||||
HESD itch reduction ≥ 4 points Follow-up: 16 weeks | 949 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Higha | Delgocitinib results in a clinically meaningful increase in patients with an itch reduction of ≥ 4 points in the HESD score compared to vehicle cream. |
HRQoL | ||||
DLQI, change from baseline Follow-up: 16 weeks | 948 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Highc | Delgocitinib results in a clinically meaningful reduction of the DLQI score from baseline compared to vehicle cream. |
HEIS, change from baseline Follow-up: 16 weeks | 948 patients (2 RCTs) | DELTA 1 trial:
DELTA 2 trial:
| Highd | Delgocitinib results in a clinically meaningful reduction of the HEIS from baseline compared to vehicle cream. |
Harms | ||||
AEs Follow-up: 16 weeks | 960 patients (2 RCTs) | In the DELTA 1 trial, 45.2% of patients receiving delgocitinib experienced ≥ 1 AE, compared to 50.6% receiving vehicle cream. In the DELTA 2 trial, 45.5% of patients receiving delgocitinib experienced ≥ 1 AE, compared to 44.7% receiving vehicle cream. | Highe | Delgocitinib results in little to no difference in the number of patients with ≥1 AE compared to vehicle cream. The magnitude of meaningful effect is uncertain. |
SAEs Follow-up: 16 weeks | 960 patients (2 RCTs) | In the DELTA 1 trial, SAEs occurred in 1.8% of patients treated with delgocitinib and 1.9% of patients treated with vehicle cream. In the DELTA 2 trial, SAEs occurred in 1.6% of patients treated with delgocitinib and vehicle cream. | Moderatee | Delgocitinib likely results in little to no difference in the number of patients who experience ≥ 1 SAE compared to vehicle cream. The magnitude of meaningful effect is uncertain. |
AESI Follow-up: 16 weeks | 960 patients (2 RCTs) | No AESIs occurred in the DELTA 1 or DELTA 2 trials. | Moderatee | Delgocitinib likely results in little to no difference in the number of patients who experience ≥ 1 AESI compared to vehicle cream. The magnitude of meaningful effect is uncertain. |
AE = adverse event; AESI = adverse event of special interest; CHE = chronic hand eczema; CI = confidence interval; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; HRQoL = health-related quality of life; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aA clinically meaningful effect of at least 3 patients improved per 100 treated was established as the threshold for clinical importance; hence, the body of evidence was not rated down for imprecision. IGA-CHE treatment success was defined as a score of 0 or 1 with at least a 2-step improvement from baseline. Differences in effect estimates were detected between the DELTA 1 and DELTA 2 trials; however, this was likely explained by differences in patients’ baseline risk factors. Furthermore, estimates and CIs both showed a meaningful effect; hence, this outcome was not rated down for inconsistency.
bA minimal important difference of 20 was established as the threshold for clinical importance for the HECSI values; hence, the body of evidence was not rated down for imprecision.
cThe DLQI minimal important difference is 4 points.
dThe HEIS minimal important difference is 0.6 points.
eA threshold of clinical importance to determine the magnitude of effect was not possible to obtain. For SAEs and AESIs, the review team rated down the evidence by 1 level for imprecision because the magnitude of effect was uncertain and there were few or no events.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.16
Table 3: Summary of Findings for Delgocitinib Versus Alitretinoin for Patients With CHE
Outcome and follow‑up | Patients (studies), N | Absolute effects | Certainty | What happens |
|---|---|---|---|---|
Severity of CHE | ||||
IGA-CHE treatment success Follow-up: 12 weeks | 503 patients (1 RCT) |
| Moderatea,b | Delgocitinib likely results in a clinically meaningful treatment success, as defined by the IGA-CHE score, compared to alitretinoin. |
HECSI-90 Follow-up: 12 weeks | 499 patients (1 RCT) |
| Moderatea,b | Delgocitinib likely results in a clinically meaningful increase in patients experiencing a HECSI-90 response compared to alitretinoin. |
HECSI, change from baseline Follow-up: 12 weeks | 499 patients (1 RCT) |
| Moderatea,c | Delgocitinib likely results in a clinically meaningful reduction in HECSI score from baseline compared to alitretinoin. |
Symptom reduction | ||||
HESD itch score, change from baseline Follow-up: 12 weeks | 476 patients (1 RCT) |
| Moderatea,d | Delgocitinib likely results in little to no difference in the HESD itch score compared to alitretinoin. |
HRQoL | ||||
DLQI, change from baseline Follow-up: 12 weeks | 466 patients (1 RCT) |
| Moderatea,e | Delgocitinib likely results in little to no difference in the DLQI score compared to alitretinoin. |
HEIS, change from baseline Follow-up: 12 weeks | 468 patients (1 RCT) |
| Moderatea,f | Delgocitinib likely results in little to no difference in the HEIS score compared to alitretinoin. |
Harms | ||||
AEs Follow-up: 24 weeks | 513 patients (1 RCT) | AEs occurred in 49.4% of patients treated with delgocitinib and 76.1% of those treated with alitretinoin. The most common AEs in the alitretinoin group were headache (32.4%), nasopharyngitis (13.8%), lip dryness (3.2%), and nausea (5.7%), while the most frequent AEs in the delgocitinib group were nasopharyngitis (11.9%) and headache (4.0%). | Moderateg | Delgocitinib likely results in fewer AEs compared with alitretinoin, although the magnitude and clinical importance of this effect are uncertain. |
SAEs Follow-up: 24 weeks | 513 patients (1 RCT) | SAEs were reported in 2.0% of patients in the delgocitinib group and 4.9% in the alitretinoin group. All SAEs resolved by the end of the study, and none led to long-term harm. | Moderateg | Delgocitinib likely results in fewer SAEs compared to alitretinoin. The magnitude of this effect is uncertain. |
AESIs Follow-up: 24 weeks | 513 patients (1 RCT) | One patient (0.4%) in the alitretinoin group experienced an AESI: a postoperative deep vein thrombosis, which resolved. No AESIs were reported in the delgocitinib group. | Moderateg | Delgocitinib likely results in little to no difference in AESIs compared to alitretinoin. The magnitude of this effect is uncertain. |
AE = adverse event; AESI = adverse event of special interest; CHE = chronic hand eczema; CI = confidence interval; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; HRQoL = health-related quality of life; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; RCT = randomized controlled trial; SAE = serious adverse event; SE = standard error.
Notes: Study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level due to risk of bias in the DELTA FORCE study, an open-label study with outcome completion rates higher in the delgocitinib group (86.6%) than in the alitretinoin group (64.1%), with a differential dropout rate of approximately 22%. Discontinuations in the alitretinoin group were mainly due to AEs and lack of efficacy. Although sensitivity analyses were conducted, the missingness is likely related to the outcome.
bA clinically meaningful effect of at least 3 patients improved per 100 treated was established as the threshold for clinical importance; hence, the body of evidence was not rated down for imprecision. IGA-CHE success was defined as a score of 0 or 1 with at least a 2-step improvement from baseline.
cA minimal important difference of 20 was established as the threshold of clinical importance for the HECSI values; hence, the body of evidence was not rated down for imprecision.
dA 4-point or greater reduction in the HESD itch score was used as the threshold for a clinically meaningful result (minimal important difference). The effect estimate and CI do not include this threshold; there was precision in the result because it falls within a trivial or no-effect zone (between thresholds).
eA change of at least 4 points in the DLQI score was established as the threshold for a meaningful effect or minimal important difference. The effect estimate and CI do not include this threshold; there was precision in the result because it falls within a trivial or no-effect zone (between thresholds).
fFor the HEIS score, the minimal important difference was established as at least 0.6 points difference. The effect estimate and CI do not include this threshold; there was precision in the result because it falls within a trivial or no-effect zone (between thresholds).
gCertainty was rated down 1 level for imprecision due to an absence of effect estimates with CIs and a lack of established thresholds for clinical importance. These limitations preclude a high-certainty judgment.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.16
Long-Term Extension Studies
Description of Studies
The DELTA 3 trial was a phase III, open-label, multisite extension phase of the DELTA 1 and DELTA 2 trials and was conducted to assess the long-term safety and efficacy of twice-daily applications of delgocitinib cream 20 mg/g as needed (based on IGA-CHE score) in patients who completed 1 of the 2 pivotal phase III trials with delgocitinib cream 20 mg/g or vehicle cream. Together, the parent trials and the DELTA 3 extension trial provided 52 weeks of efficacy and safety data. The baseline visit (day 1, week 0) of this extension trial coincided with the end-of-treatment visit (week 16) of the parent trials.
Patients eligible for the DELTA 3 extension trial were those who completed the treatment period in the DELTA 1 or DELTA 2 parent trial. Participants who discontinued treatment with delgocitinib early, used rescue medication, or experienced any AE during participation in the parent trial that precluded further treatment with delgocitinib were not eligible to participate in the DELTA 3 trial. From baseline to week 36, patients were treated on an as-needed basis with delgocitinib cream 20 mg/g twice daily. At any time during the trial, if a patient had an IGA-CHE score of 2 or higher, the investigator dispensed delgocitinib cream and instructed the patient to start treatment with twice-daily applications. When the patient experienced an IGA-CHE score of 0 (clear) or 1 (almost clear), the patient could stop treatment.
The primary outcome of the DELTA 3 extension study was the number of treatment-emergent AEs (TEAEs) from baseline up to week 38. SAEs, AEs leading to discontinuation, and deaths were also reported. Long-term efficacy outcomes, including IGA-CHE score and HECSI score, were reported as secondary outcomes, and HESD itch score and DLQI score were reported as tertiary outcomes.
Patient Disposition
Of the 810 screened patients, 801 patients were eligible to participate in the DELTA 3 trial and were therefore included in the patient safety analysis. Among these 801 patients, 560 belonged to a previous delgocitinib group and 241 to a previous vehicle cream group. From the previous delgocitinib group, 88 patients (15.7%) discontinued from the extension study, as did 49 patients (20.3%) from the previous vehicle cream group. A total of 664 patients completed the extension treatment period: 472 (84.3%) from the previous delgocitinib group and 192 (79.7%) from the previous vehicle cream group.
Baseline Characteristics
The patient baseline characteristics in the DELTA 3 trial were similar to those of the parent trials. IGA-CHE and HECSI scores at parent trial baseline were similar between the treatment groups. There were some differences in terms of IGA-CHE score at extension trial baseline between the previous delgocitinib and previous vehicle cream groups. The median HECSI score, HESD itch and pain scores, HESD score, and DLQI score had decreased baseline values in both treatment groups compared to the parent trial baseline values. The distribution of CHE subtypes at parent trial baseline was similar between the groups, except for vesicular hand eczema (pompholyx): 11.3% versus 5.8% in the previous delgocitinib and previous vehicle cream groups, respectively.
Efficacy Results
In the DELTA 3 trial, efficacy outcomes were generally maintained or improved over 36 weeks of as-needed delgocitinib use. Among patients who had received delgocitinib in the parent trials, IGA-CHE treatment success was stable (24.6% at extension baseline; 30.0% at week 36). In those previously treated with vehicle cream, IGA-CHE success increased from 9.1% to 29.5% over the same period.
HECSI scores declined further in both groups during the first 16 weeks and then stabilized. The mean HECSI score fell from 23.9 to 14.8 in the former delgocitinib group and from 46.8 to 16.8 in the former vehicle cream group. Correspondingly, the percentage of patients experiencing a HECSI-75 response increased from 51.8% to 58.6% of patients in the former delgocitinib group and from 23.7% to 51.5% of patients in the former vehicle cream group. The percentage of patients experiencing a HECSI-90 response also improved, reaching 36.6% of patients in the former delgocitinib group and 35.7% of patients in the former vehicle cream group by week 36.
Symptom relief was sustained, with HESD itch reductions of 4 points or more experienced by 52.4% of patients previously treated with delgocitinib (versus 50.6% at baseline) and HESD pain reductions in 55.4% of patients previously treated with delgocitinib (versus 51.9% at baseline). Improvements were also seen in the former vehicle cream group (HESD itch reductions of 4 points or more: 26.3% to 41.3% of patients at DELTA 3 trial baseline to week 36; HESD pain reductions: 32.3% to 43.3% of patients at DELTA 3 trial baseline to week 36).
Patient-reported HEIS and DLQI scores showed modest continued improvement in the former delgocitinib group and early gains that stabilized in the former vehicle cream group, supporting durability of treatment benefit over the extension period.
Harms
Overall, 495 patients (61.8%) reported at least 1 AE, with similar rates among those previously treated with delgocitinib (61.1%) and those previously treated with vehicle cream (63.5%). The most common AEs (reported by ≥ 5% of patients) were COVID-19 and nasopharyngitis. While infection rates were slightly higher during on-treatment periods than during off-treatment periods, the difference was not clinically meaningful.
SAEs were infrequent (3.4%), with no major differences by treatment phase or prior treatment group. Nine patients permanently discontinued the study drug due to AEs (most of which were nonserious), and rates of AEs were comparable across groups. One AESI, eczema herpeticum, occurred during an on-treatment period in a patient with AD. Three deaths were reported, none of which were considered related to the study drug.
Critical Appraisal
The design of the open-label extension phase (DELTA 3 trial) may have biased the reporting of some end points because awareness of the study treatment received may have influenced the perception of improvement and/or harms by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation. As part of the eligibility criteria for the open-label extension phase, patients had to complete 1 of the prior studies, which may potentially allow for selection bias. Because all patients were taking delgocitinib during the open-label extension phase, there was no relevant randomized comparison group (for any active comparator of interest), which precludes causal conclusions.
Because the patients who took part in the DELTA 3 trial were originally from the DELTA 1 or DELTA 2 parent studies and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label extension study. For instance, because the participants were predominantly white (approximately 91%), the results from these trials may not be generalized to other racial groups who may be commonly seen at some centres in North America and Europe. One of the eligibility criteria in the extension phase was that patients who experienced any AE during participation in the parent trial that precluded further treatment with delgocitinib were not eligible to participate in the DELTA 3 trial. While this reflects, to some extent, what would be expected in clinical practice — patients who do not experience response to treatment or who discontinue treatment due to AEs are unlikely to subsequently continue with treatment — it also leads to a greater share of patients who tolerate and experience response to delgocitinib. As a result, the generalizability of the DELTA 3 trial to the overall population is uncertain.
Indirect Comparisons
Description of Studies
Due to limited head-to-head evidence, the sponsor submitted 2 indirect treatment comparisons (ITCs) to support the comparative efficacy and safety of delgocitinib for CHE. A Bayesian network meta-analysis (NMA) compared delgocitinib to phototherapy (psoralen and UVA radiation [PUVA]) and oral alitretinoin, using data from 7 RCTs, including the DELTA 1, DELTA 2, and DELTA FORCE trials. A matching-adjusted indirect comparison (MAIC) was also conducted to compare delgocitinib with dupilumab, using individual patient data (IPD) from the DELTA 1 and DELTA 2 trials and aggregate data from the LIBERTY-AD-HAFT trial, a trial involving patients with atopic hand and foot eczema.
Efficacy Results
In the NMA, delgocitinib demonstrated higher odds of achieving an IGA-CHE or PGA score of 0 or 1 end point response compared to vehicle cream (odds ratio [OR] = 2.92; 95% credible interval [CrI], 2.06 to 4.21), PUVA (OR = 2.73; 95% CrI, 1.43 to 5.25), and alitretinoin (OR = 1.88; 95% CrI, 1.23 to 2.93) at the primary end point. The results were consistent at the week 12 sensitivity analyses and across severity subgroups. Delgocitinib also showed improved odds of achieving HECSI-90 compared to vehicle cream (OR = 3.59; 95% CrI, 2.49 to 5.28) and alitretinoin (OR = 1.79; 95% CrI, 1.22 to 2.62). The MAIC found no significant difference between delgocitinib and dupilumab in achieving an IGA-CHE or Hand and Foot Investigator’s Global Assessment (HF-IGA) score of 0 or 1 (OR = 1.1; 95% CI, 0.3 to 3.4) or HECSI-90 (OR = 1.3; 95% CI, 0.4 to 4.9), with wide CIs reflecting imprecision.
Harms Results
The NMA indicated that delgocitinib was associated with lower odds of discontinuation due to AEs compared to vehicle cream (OR = 0.21; 95% CrI, 0.08 to 0.45), PUVA (OR = 0.09; 95% CrI, 0.03 to 0.32), and alitretinoin (OR = 0.09; 95% CrI, 0.04 to 0.21). These findings were consistent across severity-based sensitivity analyses. No harms data were included in the MAIC.
Critical Appraisal
The NMA was conducted using prespecified methods and included trials with a generally low risk of bias. However, key limitations include heterogeneity in outcome definitions (e.g., stricter criteria for IGA-CHE response in the DELTA trials), variation in disease severity and treatment duration across studies, and absence of closed loops, precluding formal consistency testing. Despite sensitivity analyses, residual concerns about effect modification remain. The network was sparse, particularly for phototherapy, and some outcomes (e.g., HECSI-90, cumulative response) could not be evaluated across all arms.
The MAIC used appropriate statistical methodology and adjusted for known effect modifiers (age, sex, race, HECSI score, CHE subtype). However, differences in underlying populations (CHE versus atopic hand and foot eczema), end point definitions, and the exclusion of harms limit its interpretability. The effective sample size (ESS) was reduced after matching, leading to wide CIs and increased uncertainty.
Overall, both ITCs provide exploratory comparative data, but the findings should be interpreted cautiously due to methodological and population-level differences.
Conclusions
Considering the totality of evidence, delgocitinib cream 20 mg/g is supported as a clinically effective and well-tolerated treatment option for adults with moderate to severe CHE, particularly among those who have not experienced response to TCS and are seeking nonsystemic alternatives. Across 3 RCTs and 1 long-term extension study, delgocitinib consistently demonstrated improvements — compared to vehicle cream — in outcomes that align with priorities identified by patients and clinicians, including visible skin improvement, symptom relief, and enhanced quality of life. Compared to oral alitretinoin, the only other approved treatment for severe CHE in Canada, delgocitinib showed comparable or superior efficacy in several important outcomes, with a more favourable safety profile and fewer systemic adverse effects.
Indirect comparisons provide additional context regarding the relative efficacy of delgocitinib. In an NMA, delgocitinib was favoured over both phototherapy and alitretinoin across key efficacy outcomes. In contrast, a MAIC against dupilumab showed no clear difference between treatments. These findings, while informative, should be interpreted with caution due to methodological limitations, including sparse data, residual confounding, and differences in outcome definitions across studies — all of which may bias the estimates or widen the uncertainty around treatment effects. The relevance of comparisons with PUVA therapy is limited because oral psoralen is no longer available in Canada. Similarly, the comparison with dupilumab has restricted applicability, given its current use is limited to patients who have not experienced adequate disease control with conventional systemic immunosuppressants.
Overall, delgocitinib addresses an important unmet need by offering an effective topical, nonsteroidal alternative for patients with persistent CHE. Uncertainties remain regarding its long-term efficacy and safety end points, as well as its position within the broader treatment framework, including its potential to delay or reduce the need for systemic therapies.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of delgocitinib 20 mg/g topical cream in the treatment of patients with moderate to severe CHE.
Disease Background
The contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the review team.
Hand eczema is a painful, pruritic, inflammatory, noninfectious skin disease of the hands and wrists.1-3 The European Society of Contact Dermatitis and consensus-based Canadian and German hand eczema treatment guidelines define CHE as an episode of hand eczema lasting at least 3 months or relapsing at least twice per year.1,4,5 The severity of CHE can be assessed based on clinical signs, disease duration, treatment history, and impact on patient function.1,6 The prevalence of CHE in adult living in Canada is estimated to be 6.2%, and it is more common in females, according to data from the Canada-based cohort in an international prospective patient survey (CHECK study).13 In this cohort, approximately 70% of patients were female; the median age was 41 years, and roughly 44% of patients reported having moderate to severe CHE.14,15
The signs and symptoms of CHE arise from complex interactions between skin and immune cells that lead to a cycle of proinflammatory signalling.2,9 The main symptoms of CHE include itch and pain. People with CHE may also experience dryness, cracking, thickened skin, and bleeding, and signs and symptoms can fluctuate in severity over time (i.e., people experiencing flares).3,11 Pan–JAK-STAT signalling plays a crucial role in the dysregulated inflammatory response seen in CHE.2,9 Activation of the JAK-STAT pathways leads to downregulation of antimicrobial peptides and structural proteins such as filaggrin, resulting in skin barrier dysfunction.2,17 With long-term exposure to environmental factors like mechanical irritation, hot or dry weather, water, or other irritants, there is added harm to the skin barrier.10 The etiology is multifactorial, involving triggers that could be exogenous and endogenous.2,5 As a result, risk factors for the development of CHE include having childhood eczema, asthma, and hay fever, as well as employment involving wet work or frequent exposure to irritants and allergens (occupations in areas such as health care, food processing, and hairdressing).5,7-9
CHE is associated with a considerable burden to patients arising from the signs and symptoms of the disease (e.g., itch, pain, bleeding), disfigurement and loss of function of the hands, impaired sleep, psychological distress, negative impact on social life, and reduced ability to work.1,5,10,12 A European study demonstrated that patients with hand eczema had significantly higher prevalence of clinical depression, anxiety disorder, and suicidal ideation compared with controls.18
Standards of Therapy
The contents within this section have been informed by materials submitted by the sponsor and by clinical expert input. The following has been summarized and validated by the review team.
In Canada, the management of CHE involves a stepwise therapeutic approach, combining nonpharmacologic strategies, topical therapies, and systemic agents for patients with more severe or refractory disease. Treatment typically begins with avoidance of irritants, protective hand care (e.g., gloves), and consistent use of emollients or moisturizers to repair skin barrier function.1,4,5,19 Pharmacologic interventions start with TCS (Figure 1), sometimes followed by topical calcineurin inhibitors such as tacrolimus or pimecrolimus for steroid-sparing purposes or in steroid-resistant cases.1,4,5,19 For patients with moderate to severe CHE that is not responsive to topicals, systemic therapy may be initiated, most commonly with oral alitretinoin — currently the only systemic agent approved by Health Canada for severe CHE. Biologic therapies for AD represent recent advancements in AD treatment (where dupilumab is the most accessible publicly reimbursed biologic), but while they are well tolerated, their mechanism of action is narrow because it targets only the type 2 inflammatory pathway associated with AD.20 Oral JAK inhibitors are also a relatively recent advancement in AD treatment with broad public coverage. However, their adoption is limited by safety considerations identified by Health Canada in certain patient groups (e.g., older adults, past or current smokers, patients with cardiovascular or malignancy risk factors, patients at risk for thrombotic events).21
Figure 1: Current Paradigm of Treatment for Patients With Moderate to Severe CHE
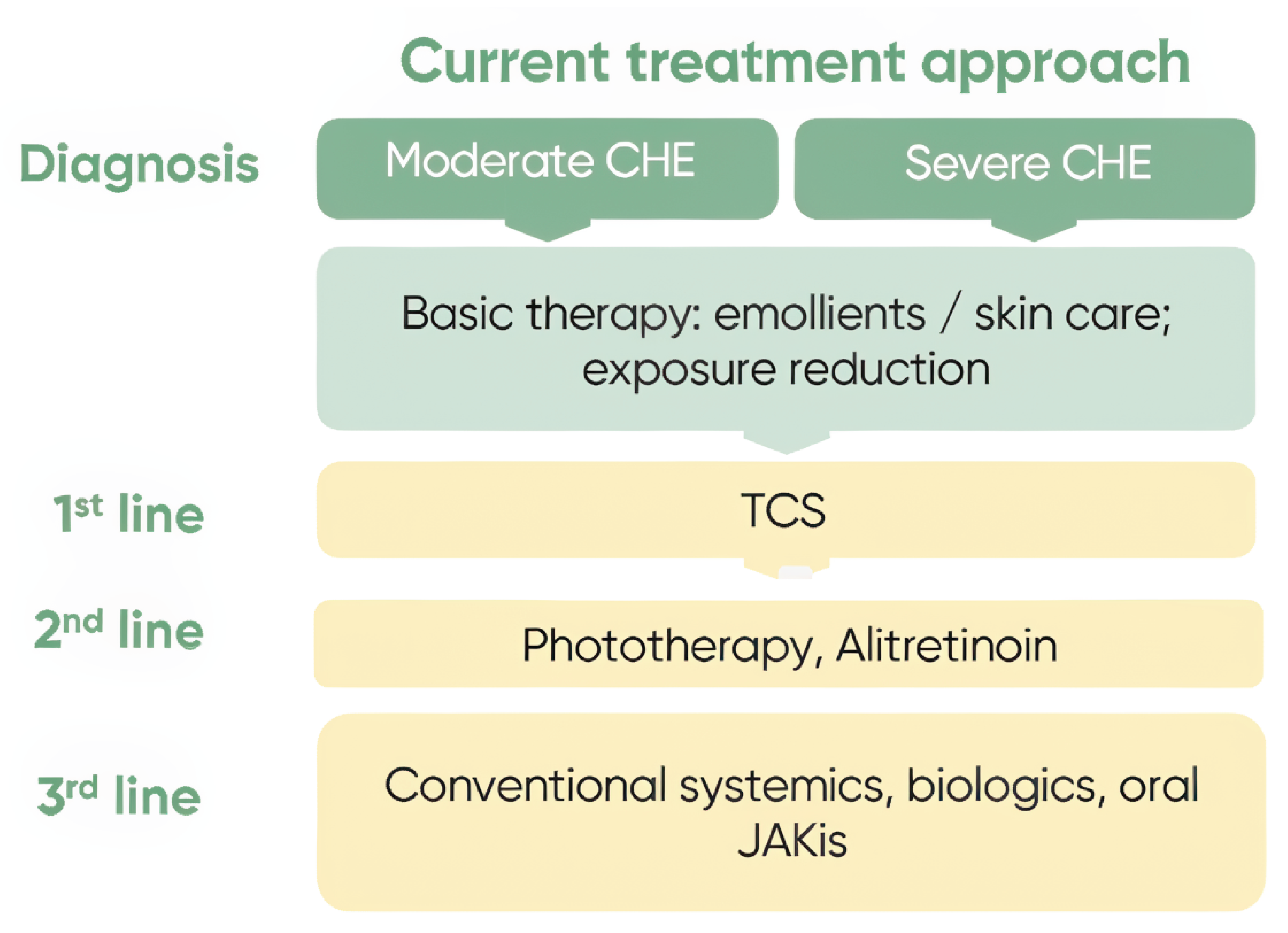
CHE = chronic hand eczema; JAKi = Janus kinase inhibitor; TCS = topical corticosteroids.
Source: Sponsor’s summary of clinical evidence.16
Input from clinical experts consulted for this review confirmed this treatment paradigm. They emphasized that TCS remain the mainstay of first-line pharmacologic therapy and that calcineurin inhibitors are used selectively, often limited by tolerability or cost. Experts noted that systemic therapies are generally reserved for patients with long-standing, severe CHE that is unresponsive to topical treatments and that alitretinoin is widely used but often associated with significant adverse effects (e.g., headache, mucocutaneous dryness, teratogenicity), limiting its tolerability and long-term use in some patients. They also indicated that while use of methotrexate, cyclosporine, mycophenolate, or azathioprine may be off-label, they are frequently prescribed and may require access through special authorization or use in consultation with a specialist. Phototherapy is used in some centres, though its availability is variable across Canada. Clinical experts indicated that psoralen, a photosensitizer that is part of PUVA therapy, is not available in Canada. Instead, centres in Canada use narrow-band UVA therapy, which is deemed less effective. Experts noted that dupilumab is currently not publicly available before unsuccessful treatment with methotrexate and cyclosporine.
According to clinical experts, the primary goals of therapy for CHE include rapid and sustained reduction in inflammation and itch, restoration of skin integrity, and improvement in health-related quality of life (HRQoL). Treatment success is also measured by its ability to reduce pain and sleep disturbance, maintain productivity (e.g., ability to work, particularly with hand-dependent tasks), and minimize psychological and social burden. Experts highlighted that long-term disease control with minimal side effects is critical, particularly for patients with frequent relapses or chronic symptoms. From both patient and clinician perspectives, improved tolerability, flexibility of use, and nonsystemic alternatives were cited as highly desirable features in new treatments.
Drug Under Review
Delgocitinib is a topical cream. Each gram of cream contains 20 mg of delgocitinib. The recommended dosing regimen of delgocitinib is as follows:22
A thin layer of delgocitinib should be applied twice daily to the affected skin of the hands and wrists.
Each affected area should be treated with delgocitinib twice daily until the skin is clear or almost clear.
In the event of recurrence of the signs and symptoms of CHE (flares), twice-daily treatment of the affected areas should be reinitiated as needed.
Delgocitinib is a JAK inhibitor that targets the underlying pathophysiology in CHE, specifically the JAK-STAT pathways. This is achieved by inhibiting the activity of all 4 members of the JAK family of enzymes, consisting of JAK1, JAK2, JAK3, and tyrosine kinase 2, thus blocking downstream signalling of multiple proinflammatory cytokines driving disease severity in CHE.22
Delgocitinib has been approved by Health Canada for the treatment of moderate to severe CHE, including the relief of pain and pruritus, in adults who have experienced inadequate response to TCS or for whom TCS are not advisable. The reimbursement request for delgocitinib is aligned with the anticipated Health Canada indication.
The key characteristics of delgocitinib are summarized in Table 4, along with those of other treatments available for CHE.
Table 4: Key Characteristics of Delgocitinib, Alitretinoin, and Dupilumab
Characteristic | Delgocitinib | Alitretinoin | Dupilumab |
|---|---|---|---|
Mechanism of action | Inhibitor of all 4 members of the JAK family (JAK1, JAK2, JAK3, and tyrosine kinase 2). | Unknown in CHE. Alitretinoin has demonstrated immunomodulatory and anti-inflammatory effects that are relevant to skin inflammation. | Monoclonal antibody that targets the IL‑4 receptor alpha, blocking IL‑4 and IL-13 signalling. This inhibits the T2‑type inflammatory cascade implicated in AD and has shown efficacy in hand eczema settings. |
Indicationa | For the treatment of moderate to severe chronic hand eczema (CHE) in adults for whom topical corticosteroids are inadequate or are not advisable. | For the treatment of severe chronic hand eczema refractory to high-potency topical corticosteroids in adults.a | Health Canada authorizes dupilumab for moderate to severe AD (for patients aged ≥ 6 months). While CHE is not an independent indication, dosing and safety are consistent with the AD approval, and off-label use in CHE is common. |
Route of administration | Topical cream | Oral capsules | Subcutaneous injection via prefilled syringe (for patients aged ≥ 6 months) or prefilled pen (for patients aged ≥ 2 years), given into thigh, abdomen (avoiding 5 cm around navel), or upper arm (if administered by caregiver) |
Recommended dose | 20 mg/g twice daily | 10 mg or 30 mg once daily | Adults or adolescents (aged ≥ 12 years): Load 600 mg (2 × 300 mg), then 300 mg q.2.w. Children aged 6 to 17 years: Weight-based (≥ 60 kg: load 600 mg, then 300 mg q.2.w.; 30 to < 60 kg: load 400 mg, then 200 mg q.2.w.; 15 to < 30 kg: load 600 mg, then 300 mg q.4.w.) For CHE, use adult or adolescent AD regimen. |
Serious adverse effects or safety issues | Preferable to avoid use during pregnancy as a precautionary measure. | Laboratory monitoring is required. Contraindicated in patients of childbearing potential, in pregnancy, and during breastfeeding. Contraindicated in patients with hepatic insufficiency, severe renal insufficiency, uncontrolled hypercholesterolemia, hypertriglyceridemia, hypothyroidism, hypervitaminosis A, benign intracranial hypertension, and use of tetracyclines. | Injection site reactions (e.g., pain, redness, swelling), conjunctivitis and other eye-related conditions (e.g., keratitis, blepharitis), hypersensitivity reactions (including rare cases of anaphylaxis and serum sickness–like reactions), upper respiratory tract infections, oral herpes, and headache. Dupilumab does not require routine laboratory monitoring. Use of live vaccines is not recommended during treatment. |
AD = atopic dermatitis; CHE = chronic hand eczema; IL = interleukin; JAK = Janus kinase; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
aHealth Canada–approved indication.
Sources: Product monographs of delgocitinib,22 alitretinoin,23 and dupilumab.24
Perspectives of Patients, Clinicians, and Drug Programs
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Two patient groups, the ESC and CANN, submitted input for this review. Another patient group, the Canadian Skin Patient Alliance, did not directly provide input for this submission but provided a letter of support for the patient submission developed and submitted by the ESC. The ESC group obtained information related to CHE through questionnaires and interviews with patients, caregivers and health care professionals, as well as survey data on various topics from more than 3,000 people living in Canada who have eczema (e.g., impact on quality of life, experience with treatments, patient journeys). CANN collected anonymous data through qualitative interviews with nurses conducted in July 2024 and obtained their perspectives of living and working with CHE. The group also conducted an online quantitative survey between October 23 and November 10, 2024, of its nursing members living with CHE (n = 27), which was distributed through CANN’s email communication list and established membership channels. All information was gathered in Canada.
Both patient groups highlighted a significant impairment to everyday living and quality of life due to CHE. The groups reported that people with CHE experienced negative impacts on sleep quality (due to pain), skin (burning, itching lesions, blisters, bleeding skin), work (loss of productivity, change in careers due to CHE), social interactions, fitness, intimacy, family life and hygiene. CANN further noted that survey respondents reported itchy skin as the symptom that caused the greatest burden, followed by dry or chapped skin; dyshidrotic blisters; cracking skin or fissures; open wounds with bleeding; and painful, stinging and throbbing hands. Both patient groups highlighted that current treatment regimens may not be sufficient and expressed concerns regarding long-term steroid use. In the survey conducted by CANN, 52% of the respondents reported that their current treatment and management regimen could not control or could only somewhat help them manage their hand eczema.
Both patient groups highlighted a need for a treatment that provided better disease management, including relief from itching, stinging, burning, and pain; improvement in skin disease; and reduction of flares. CANN further highlighted that people with CHE want to function normally, return to work, experience reduced severity of flares, and have decreased risk of blisters and open wounds. The ESC also noted that people were looking for treatments suitable for long-term use that could allow them to regain their confidence and self-esteem and improve their social life and quality of life. In both patient groups, those who had had the opportunity to be treated with delgocitinib noted positive experiences with the treatment. The ESC further highlighted that delgocitinib offered patients relief; was well tolerated, without stinging or burning; and was not greasy on the hands.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of CHE.
Unmet Needs
The experts identified significant unmet needs in the treatment of moderate to severe CHE. They noted that many currently available therapies, including alitretinoin and systemic immunosuppressants, are poorly tolerated, only moderately effective, or associated with significant toxicity. Topical calcineurin inhibitors were described as often ineffective, and high-potency TCS as unsuitable for long-term use due to safety concerns. The experts highlighted limited access to phototherapy across Canada and high costs of newer therapies like topical ruxolitinib. They considered that there is a need for an effective, safe, and accessible topical therapy with efficacy comparable to systemic agents, without their associated risks.
Place in Therapy
The experts indicated that delgocitinib would most likely be used after the failure of high-potency TCS. They suggested that delgocitinib could be used before the off-label systemic therapies currently used to treat CHE (e.g., immunosuppressants, dupilumab, JAK inhibitors), which carry systemic risks. Both experts emphasized a preference for topical therapies due to lower systemic exposure and ease of administration. They also noted that dupilumab is currently not available before unsuccessful treatment with methotrexate and cyclosporine. Hence, these immunosuppressants are expected to be displaced before delgocitinib can displace dupilumab or any JAK inhibitor. According to the experts, there is no evidence of JAK inhibitors being clinically superior to dupilumab.
The experts mentioned that delgocitinib can be expected to be used as monotherapy for patients whose disease is not controlled with corticosteroids or in combination with systemic treatments in more severe cases (e.g., in addition to alitretinoin). Overall, the experts considered that the introduction of delgocitinib may shift the treatment paradigm by reducing reliance on systemic agents, mainly alitretinoin, methotrexate, and cyclosporine.
Patient Population
The experts noted that CHE is not rare but that it disproportionately affects individuals in manual or caregiving professions. They identified people with functional impairment due to CHE, especially those whose work, sleep, or daily activities are impacted, as those most in need of intervention. They considered that people who are unable to use or unresponsive to corticosteroids or other topical agents, or for whom these treatments are contraindicated, could benefit most. Additionally, people with more recent disease onset and less lichenified lesions may respond better to treatment.
The experts indicated that diagnosing CHE can be challenging, with differential diagnoses including allergic contact dermatitis, psoriasis, and tinea manuum. They recommended detailed clinical history and examination, with patch testing in select cases. The experts acknowledged that misdiagnosis and underdiagnosis are likely, especially in underserved populations. While dermatologists were viewed as best suited to confirm diagnosis, the experts considered that family physicians could manage more straightforward cases.
The clinical experts considered that the people least suitable for delgocitinib would include those who found it difficult to adhere to treatment regimens, those with active infections or malignancies on the hands, and those with extensive warts (due to a theoretical risk of spreading).
Assessing the Response Treatment
The experts stated that response of CHE to treatment in clinical practice is usually assessed using clinician global assessment (e.g., IGA-CHE) and patient-reported outcomes such as symptom control and functional improvement. They noted that the HECSI is not commonly used in routine practice. A clinically meaningful response was considered to be IGA-CHE score of 0 or 1 (clear or almost clear) or a 75% improvement in the affected area. Return to function and patient satisfaction with symptom control (e.g., of itch, pain, and sleep) were also identified as important indicators.
The experts recommended assessing treatment response after 4 weeks and again at 12 weeks. They suggested that continued treatment should depend on these clinical targets being achieved or approached.
Discontinuing Treatment
The experts recommended discontinuation of delgocitinib in cases of complete lack of response after 12 weeks. They also cited failure to achieve an IGA-CHE score of 0 or 1 at 12 weeks, a lack of patient-reported improvement, or the emergence of adverse effects as appropriate reasons to reconsider or stop treatment.
Prescribing Considerations
The experts considered that delgocitinib may be initiated in either dermatology or primary care settings. While dermatologist confirmation of diagnosis may be required in some cases, the experts noted that both dermatologists and family physicians could prescribe and monitor treatment, particularly for less severe cases. They suggested this approach could help reduce referrals and wait times for dermatology care. Due to its topical formulation, delgocitinib was deemed suitable for community-based treatment and home use. However, the experts acknowledged ongoing regional disparities in access to dermatologists and advanced therapies, particularly in rural or economically disadvantaged areas.
The clinical experts also indicated that while delgocitinib is expected to be effective as monotherapy, combination with other therapies may be considered in specific cases.
Additional Considerations
The experts noted that CHE often affects visible areas, leading to stigma, social withdrawal, and reduced quality of life. They emphasized that equity-deserving populations may face additional barriers to diagnosis, access, and continuity of care. The experts considered that delgocitinib, as a safe, effective, and nonsystemic option, could reduce the use of harmful off-label systemic treatments and improve access to care for marginalized populations.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
Five clinician groups provided input for this review: the DAO, the Atlantic Dermatology Group, the Canadian Dermatology Association’s Pharmacy and Therapeutics Advisory Board, the Saskatchewan Dermatology Association, and dermatologists practising in Canada with an interest in CHE. Information for this submission included input from 21 clinicians and was gathered through reviews of published literature, research and trial experience, clinical practice experience, and national and international meetings.
The input from clinician groups was generally consistent with the input from the clinical experts consulted for this review, emphasizing the ongoing unmet need for new topical prescription therapy options for CHE that would control the signs and symptoms of eczema, normalize quality of life, and reduce or abolish occupational impacts. The DAO group further emphasized the need to include therapies for long-term control of CHE that are safe, effective, and accessible. The clinician groups highlighted the limitations of current therapies, including AEs related to topical corticosteroid use such as skin atrophy, skin infections, pain, burning, fissures, reduced hand dexterity, and bleeding; limited efficacy and poor access to phototherapy; and contraindications related to alitretinoin use (e.g., pregnancy, breastfeeding). The DAO group noted that while topical calcineurin inhibitors like tacrolimus are sometimes used in mild cases of CHE when a steroid-sparing therapy is required due to AEs related to steroid use, they are not effective treatment alternatives for moderate and severe forms of CHE. The DAO group also highlighted several off-label systemic options for patients with CHE in cases when phototherapy and alitretinoin are not suitable, are not accessible, or fail to control the condition; these options include traditional immunosuppressants (i.e., methotrexate and cyclosporine), biologics (particularly ones used in AD, such as dupilumab), and oral JAK inhibitors (i.e., upadacitinib). However, the group noted limitations related to these off-label options as well, including accessibility and cost (i.e., coverage criteria may make a biologic therapy inaccessible, or the co-pay cost to the patient may be a barrier), safety considerations (e.g., black box warnings and contraindications across the oral systemic therapies), and expected effectiveness (i.e., traditional immunosuppressants are not as effective in managing CHE as biologics or oral JAK inhibitors). The groups indicated that people involved in certain professions with frequent exposure to irritants and allergens — such as people working in health care, cleaning, and hairdressing — report higher incidence rates of CHE.
The DAO and the Canadian Dermatology Association’s Pharmacy and Therapeutics Advisory Board groups considered topical delgocitinib as a second-line treatment for patients with moderate to severe CHE (i.e., a PGA score of 3 to 4) whose disease was refractory to TCS, or who experienced intolerance to TCS, or for whom TCS were contraindicated. The Atlantic Dermatology Group considered topical delgocitinib as a first-line or second-line treatment for adult patients with moderate to severe CHE for whom TCS are inadequate or inappropriate. The groups noted that delgocitinib is expected to cause a shift in the current treatment paradigm as a new targeted topical prescription therapy for CHE. The DAO group highlighted delgocitinib’s ease of access and treatment administration over phototherapy and noted that it did not require laboratory monitoring and had no contradictions (e.g., pregnancy, breastfeeding, or renal or hepatic insufficiency) as is the case for alitretinoin. Referring to the clinical trials, the groups also noted the efficacy and favourable safety profile of delgocitinib in patients with moderate to severe CHE. The clinician groups provided additional details on patient selection, noting that delgocitinib may be particularly appropriate for patients with moderate to severe CHE who have experienced an inadequate response to TCS or for whom TCS are not advisable or are contraindicated. The groups also noted that patients with mild CHE for whom TCS have not yet been used or whose CHE is being adequately managed by emollients, avoidance strategies, or TCS are less appropriate candidates for treatment with delgocitinib.
The clinician groups noted that an initial assessment of response to treatment at approximately 3 to 6 months after initiation (i.e., around 16 weeks) would be reasonable. The DAO group further highlighted that both the timing of follow-up assessments and the criteria used to determine response may vary across clinicians, depending on clinical practice wait times, availability of follow-up, and patient-specific factors. The clinician groups added that outcome measures are not formally standardized in routine clinical practice for CHE, and that clinicians typically rely on an informal PGA. The clinician groups agreed that reasons for discontinuation of delgocitinib may include inadequate or loss of efficacy, recurrent flares, disease worsening or lack of response despite an adequate therapeutic trial, or intolerance and adverse effects.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Delgocitinib cream is compared to vehicle cream instead of other commonly used topical therapies for eczema (e.g., vitamin D3 derivatives [calcipotriol] and calcineurin inhibitors [tacrolimus and pimecrolimus]). Is it appropriate to compare delgocitinib to oral alitretinoin, given that not all jurisdictions include alitretinoin as a treatment option? | The experts acknowledged that while not all jurisdictions include oral alitretinoin on their formularies, such variation is common across treatments in Canada due to provincial differences in access (similar discrepancies occur with access to phototherapy and cyclosporine). Despite these access issues, the experts considered the comparison to alitretinoin appropriate, as it is currently the only drug with a Notice of Compliance for hand dermatitis. |
Considerations for initiation of therapy | |
Most eczema medications require patients to have a score ≥ 16 on the Eczema Area and Severity Index. The primary end point in the submitted studies was the IGA-CHE. IGA-CHE treatment success is defined as a score of 0 or 1 and a ≥ 2-step improvement from baseline at week 16. The studies for delgocitinib used the IGA-CHE score as the primary outcome instead of the Eczema Area and Severity Index score. Is this tool commonly used in clinical practice? Can the Physician’s Global Assessment be substituted? | Regarding the use of IGA-CHE as the primary outcome measure in delgocitinib trials, the experts indicated that IGA‑CHE is conceptually aligned with the Physician’s Global Assessment (PGA) commonly used in clinical practice. Both are clinician-rated global severity scales, and IGA-CHE was considered clinically reasonable and interpretable in the context of chronic hand eczema. The experts noted that the Eczema Area and Severity Index (EASI) is more commonly used in atopic dermatitis and is not routinely applied in chronic hand eczema. In clinical practice for CHE, formal composite scores such as EASI are not typically used; instead, clinicians generally rely on global assessment of disease severity, often using an informal PGA approach. Accordingly, the use of IGA-CHE as the primary outcome in the delgocitinib trials was considered appropriate and reflective of how response is assessed in routine CHE practice. |
Participants in the studies submitted were adults aged 18 years and older. Could experts comment on the use of delgocitinib in the pediatric population? Is there any clinical experience or evidence supporting the use of delgocitinib in children, especially for those seeking alternatives to topical steroids? | The experts indicated that delgocitinib has not yet been studied or approved for children. However, they noted ongoing studies with similar agents (e.g., topical ruxolitinib) in younger populations and acknowledged the potential for off-label use by clinicians. There are precedents for systemic JAK inhibitors (e.g., upadacitinib) being approved for juvenile indications, such as juvenile idiopathic arthritis. |
Should delgocitinib be reserved only for patients for whom topical steroids have failed or who experience intolerance to topical steroids? What is a reasonable time frame to define steroid treatment failure (e.g., 8 weeks)? Should failure of nonsteroidal treatments like calcipotriol or tacrolimus also be considered before initiating delgocitinib? Should systemic therapies such as methotrexate be tried before initiating treatment with delgocitinib? | The experts acknowledged that delgocitinib should generally be reserved for patients for whom topical corticosteroids have failed or who experience intolerance to topical corticosteroids. An 8-week duration was deemed a reasonable time frame to assess steroid treatment failure. The experts emphasized that the potency of the steroid used matters: patients for whom high-potency steroids have failed may not benefit from switching to topical nonsteroidal agents like tacrolimus, whereas the failure of low-potency steroids could justify such a switch. Regarding systemic therapies, the experts advised that methotrexate should not be used before delgocitinib, as this would bypass alitretinoin, which is a more logical and appropriate step in the treatment sequence. |
Considerations for prescribing of therapy | |
Should prescribing of delgocitinib be restricted to dermatologists? | The experts expressed that restricting prescribing to dermatologists may not be necessary, as family physicians often manage cases of hand dermatitis. They suggested that family doctors should be allowed to prescribe delgocitinib after topical corticosteroids have failed. However, they acknowledged that payers may impose restrictions to manage access and costs. |
Can delgocitinib be used in combination with a topical calcineurin inhibitor, biologics, and/or oral JAK inhibitor? | The experts recommended against using delgocitinib concurrently with topical calcineurin inhibitors. However, they supported its use alongside systemic agents, such as dupilumab or oral JAK inhibitors, especially in cases where a patient has residual hand dermatitis despite systemic therapy and topical steroids have already failed. |
Ruxolitinib (Opzelura) received a do not reimburse recommendation for atopic dermatitis. | This is a comment from the drug programs to inform CDEC deliberations. |
System and economic issues | |
Cost per 60 g tube: $636.98 Pan-Canadian budget impact: Year 1: –$5,891,445 Year 2: –$8,981,078 Year 3: –$12,169,822 Total, 3 years: –$27,042,344 | This is a comment from the drug programs to inform CDEC deliberations. |
CDEC = Canadian Drug Expert Committee; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; JAK = Janus kinase.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of delgocitinib 20 mg/g topical cream in the treatment of patients with CHE. The focus will be placed on comparing delgocitinib to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of delgocitinib is presented in 3 sections, with the CDA‑AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes the pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
3 pivotal studies (RCTs) identified in the systematic review
1 long-term extension study
2 ITCs.
Systematic Review
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The characteristics of the included studies are summarized in Table 6.
Table 6: Details of Studies Included in the Systematic Review
Variable | DELTA 1 | DELTA 2 | DELTA FORCE |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, double-blind, vehicle-controlled, parallel-group RCT | Phase III, assessor-blinded, active-controlled, parallel-group RCT | |
Locations | 53 sites in 6 countries: Canada, France, Germany, Italy, Poland, UK | 50 sites in 7 countries: Belgium, Canada, Denmark, Germany, Netherlands, Poland, Spain | 103 sites in 10 countries: Austria, Canada, France, Germany, Italy, Norway, Poland, Slovakia, Spain, UK |
Patient enrolment dates | Start date: May 10, 2021 End date: October 31, 2022 | Start date: May 25, 2021 End date: January 6, 2023 | Start date: June 15, 2022 End date: December 5, 2023 |
Randomized (N) | 487 Delgocitinib (n = 325) Vehicle cream (n = 162) | 473 Delgocitinib (n = 314) Vehicle cream (n = 159) | 513 Delgocitinib (n = 254) Alitretinoin (n = 259) |
Inclusion criteria |
|
| |
Exclusion criteria |
| ||
Drugs | |||
Intervention | Delgocitinib, 20 mg/g cream b.i.d. | Delgocitinib, 20 mg/g cream b.i.d. | Delgocitinib, 20 mg/g cream b.i.d. |
Comparator(s) | Vehicle cream b.i.d. | Vehicle cream b.i.d. | Alitretinoin, 30 mg capsule q.d. (option to reduce to 10 mg capsule q.d. during trial conduct) |
Study duration | |||
Screening phase | 4 weeks | 4 weeks | 4 weeks |
Treatment phase | 16 weeks | 16 weeks | 24 weeks |
Follow-up phase | 2 weeks or open-label extension | 2 weeks or open-label extension | 2 weeks |
Outcomes | |||
Primary end point | IGA-CHE treatment success at week 16 (i.e., score of 0 or 1 along with a ≥ 2-point improvement from baseline). | CFB in HECSI score at week 12 | |
Secondary and exploratory end points | Key secondary:
Secondary: Number of TEAEs per patient from baseline to week 16 (or week 18 for patients not participating in extension trial) Exploratory:
| Key secondary:
Secondary:
Exploratory:
| |
Publication status | |||
Publications | Bissonnette et al. (2024)25 ClinicalTrials.gov (NCT04871711)26 | Bissonnette et al. (2024)25 ClinicalTrials.gov (NCT04872101)27 | Gimenez-Arnau et al. (2025)28 ClinicalTrials.gov (NCT05259722) |
AD = atopic dermatitis; AE = adverse event; ALP = alkaline phosphatase; ALT= alanine transaminase; AST = aspartate transaminase; AUC = area under the curve; b.i.d. = twice a day; CFB = change from baseline; CHE = chronic hand eczema; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; IMP = investigational medicinal product; JAK = Janus kinase; PDAL = proximal daily activity limitations; q.d. = every day; RCT = randomized controlled trial; TCS = topical corticosteroids; TEAE = treatment-emergent adverse event; TESAE = treatment-emergent serious adverse event; TSQM= Treatment Satisfaction Questionnaire for Medication; ULN = upper limit of normal; VAS = visual analogue scale; WPAI-CHE = Work Productivity and Activity Impairment–Chronic Hand Eczema.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Three RCTs were identified and included in the clinical evidence for delgocitinib cream 20 mg/g: the DELTA 1, DELTA 2, and DELTA FORCE trials. These studies formed the pivotal evidence base for the sponsor’s submission, evaluating the efficacy, safety, and HRQoL impacts of delgocitinib in adults with moderate to severe CHE who had experienced inadequate response to TCS or for whom TCS were not advisable.
DELTA 1 and DELTA 2 Trials
The DELTA 1 and DELTA 2 trials were identically designed, double-blind, vehicle-controlled, parallel-group, phase III RCTs. Their primary objective was to evaluate the efficacy and safety of twice-daily topical delgocitinib cream versus vehicle cream in adults with moderate to severe CHE.
Both trials enrolled adults aged 18 years and older with a diagnosis of moderate to severe CHE (IGA-CHE score of 3 or 4) who had experienced inadequate response to TCS or for whom TCS were contraindicated. The DELTA 1 trial randomized 487 participants across 53 sites in 6 countries (including 10 sites in Canada); the DELTA 2 trial randomized 473 participants across 50 sites in 7 countries (also including 10 sites in Canada). Both trials used a 2:1 randomization ratio, stratified by baseline IGA-CHE severity (moderate versus severe) and geographic region (North America versus Europe). Randomization was conducted using an interactive response technology system. Patients applied delgocitinib cream 20 mg/g or matching vehicle cream twice daily for 16 weeks. The dosing aligns with the Health Canada product monograph; no other dosing regimens were evaluated.
Each study included a screening period of up to 4 weeks, allowing for washout from prior prohibited treatments and assessment of eligibility criteria, including adherence to nonmedicated skin care and presence of CHE flares (e.g., HESD itch score ≥ 4). Reasons for patients not proceeding beyond screening included unmet inclusion criteria or presence of an exclusion criterion, patient withdrawal, and missed visits. These aspects support external validity but may limit generalizability to patients who experience difficulty adhering to structured treatment regimens.
DELTA FORCE Trial
The DELTA FORCE trial was a phase III, assessor-blinded, open-label, active-controlled RCT designed to compare delgocitinib cream 20 mg/g twice daily with alitretinoin 30 mg once daily in adults with severe CHE (IGA-CHE score of 4). Adults aged 18 years or older with severe CHE who had experienced inadequate response to TCS or for whom TCS were contraindicated were included. A total of 513 patients were randomized across 103 sites in 10 countries, including 13 in Canada. Patients were randomized 1:1 to delgocitinib (n = 254) or alitretinoin (n = 259). Randomization was stratified by disease subtype (hyperkeratotic versus nonhyperkeratotic) and geographic region (North America versus Europe) via interactive response technology.
Delgocitinib cream 20 mg/g was applied twice daily. Alitretinoin 30 mg capsules were taken once daily, with a protocol-defined option to reduce to 10 mg in cases of AEs. The trial included a 4-week screening phase for washout, baseline symptom confirmation, and contraceptive adherence verification (for alitretinoin). The treatment period was 24 weeks, with primary efficacy outcomes assessed at week 12. Treatment could be discontinued early based on predefined response criteria or intolerance. To maintain blinding of efficacy assessments despite different treatment modalities, separate blinded assessors and unblinded investigators were used at each site.
Populations
Inclusion and Exclusion Criteria
Across the 3 pivotal trials (DELTA 1 trial: N = 487; DELTA 2 trial: N = 473; DELTA FORCE trial: N = 513), patients were adults (≥ 18 years) with a clinical diagnosis of CHE persisting for at least 3 months or relapsing at least twice in the past year. The DELTA 1 and DELTA 2 trials enrolled patients with moderate to severe CHE (IGA-CHE score of 3 or 4), whereas the DELTA FORCE trial included only those with severe disease (IGA-CHE score of 4), aligning with alitretinoin’s approved indication in Canada. All studies required documentation showing either that prior treatment with TCS had provided an inadequate response or that treatment with TCS was considered medically inadvisable, as defined in the protocol, and patients had to be able to be adherent to nonmedicated skin care and avoidance of known irritants or allergens. Key exclusion criteria included other dermatologic conditions on the hands (e.g., psoriasis or active AD), prior use of JAK inhibitors, or prior use systemic immunosuppressive therapy within 28 days before the baseline. The DELTA FORCE trial had additional exclusions to ensure alitretinoin safety, including hepatic or renal impairment, uncontrolled hyperlipidemia, and pregnancy-related contraindications. These criteria collectively defined a population with moderate to severe CHE unresponsive to standard first-line therapy, and the DELTA FORCE trial further restricted inclusion to a narrower, more medically complex subgroup.
Interventions
In all 3 trials (DELTA 1, DELTA 2, and DELTA FORCE trials), the investigational intervention was delgocitinib 20 mg/g cream, applied twice daily (approximately 12 hours apart) as a thin layer to affected areas of the hands and wrists. The cream was self-administered at home by participants and did not require any special device or training for application. The treatment duration was 16 weeks in the DELTA 1 and DELTA 2 trials and up to 24 weeks in the DELTA FORCE trial, depending on treatment response.
In the vehicle-controlled studies (DELTA 1 and DELTA 2 trials), patients were randomized 2:1 to receive either delgocitinib or vehicle cream, which was identical in appearance and texture to the active formulation. In the DELTA FORCE trial, a 1:1 randomization compared delgocitinib to oral alitretinoin 30 mg once daily, with dose reduction to 10 mg permitted in cases of AEs. The DELTA FORCE trial was assessor blinded only, due to the differing routes of administration; a double-dummy design was not used to avoid confounding treatment effects.
Concomitant use of nonmedicated emollients was permitted throughout all trials, provided they were not applied within 2 hours before or after delgocitinib application. Other topical therapies (e.g., corticosteroids, immunomodulators) and systemic therapies (e.g., corticosteroids, immunosuppressants, retinoids) were prohibited during the treatment period. Use of phototherapy, tanning beds, or bleach baths was also not allowed. Vaccinations and non–skin-related medications were permitted unless otherwise contraindicated.
Rescue treatment was allowed if symptoms became intolerable during the treatment or follow-up periods. Once rescue medication was initiated, patients were required to permanently discontinue study treatment and were not permitted to restart it. In the DELTA FORCE trial, alitretinoin was not permitted as a rescue treatment for patients in either arm.
Stopping criteria in all trials included the initiation of rescue medication, pregnancy, major AEs, or investigator-assessed lack of benefit. In the DELTA FORCE trial, alitretinoin dose reductions were allowed, but no upward titration was permitted. Dose reduction was most commonly implemented by week 8 due to adverse effects, though some reductions occurred earlier for clinical improvement or at investigator discretion.
Overall, all interventions were aligned with the anticipated product monograph for delgocitinib in Canada. Self-application of the topical treatment at home and the absence of required monitoring or laboratory assessments reflected the intended outpatient use in routine clinical practice.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 7, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical expert(s) consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, the review team selected the end points that were most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | DELTA 1 | DELTA 2 | DELTA FORCE |
|---|---|---|---|---|
IGA-CHE treatment success | At week 12 | NA | NA | Key secondarya |
At week 16 | Primarya | Primarya | NA | |
CFB in HECSI | At week 12 | NA | NA | Primarya |
At week 16 | Key secondarya | Key secondarya | NA | |
At week 24 | NA | NA | Key secondarya | |
HECSI-75 | At week 16 | Key secondarya | Key secondarya | Exploratory |
HECSI-90 | At week 12 | NA | NA | Key secondarya |
At week 16 | Key secondarya | Key secondarya | NA | |
HESD itch score reduction ≥ 4 points | At week 16 | Key secondarya | Key secondarya | NA |
CFB in HESD score | At week 16 | Key secondarya | Key secondarya | NA |
CFB in HESD itch score | At week 16 | Key secondarya | Key secondarya | Key secondary |
CFB in HEIS | At week 16 | Key secondarya | Key secondarya | Exploratory |
CFB in DLQI | At week 16 | Key secondarya | Key secondarya | Exploratory |
CFB = change from baseline; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; NA = not applicable.
aStatistical testing for these end points was adjusted for multiple comparisons (i.e., hierarchal testing).
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Investigator’s Global Assessment of Chronic Hand Eczema
IGA-CHE is a validated, clinician-reported outcome developed specifically for CHE trials by the sponsor29-31 and was used as the primary end point in the DELTA 1 and DELTA 2 trials at week 16 and as a key secondary end point in the DELTA FORCE trial at week 12. It employs a 5-point severity scale (0 = clear to 4 = severe) and requires a score of 0 or 1 plus at least a 2-step improvement from baseline to meet the definition of treatment success. Compared to traditional Investigator’s Global Assessment scales, IGA-CHE offers more stringent criteria — particularly for “almost clear” status — by excluding features like scaling, hyperkeratosis, or fissures. The tool evaluates global disease severity at a single time point based on visible signs, including erythema, lichenification, vesiculation, and edema. Testing from the DELTA 1 trial confirmed IGA-CHE’s reliability and construct validity, with a 2-step improvement established as a conservative threshold for meaningful clinical change. The IGA-CHE outcome measurement tool is described in Table 8, along with definitions of severity.
Table 8: Definition of Investigator’s Global Assessment of Chronic Hand Eczema
IGA-CHE severity | IGA-CHE score | Signs and intensity |
|---|---|---|
Clear | 0 | No signs of erythema, scaling, hyperkeratosis/lichenification, vesiculation, edema, or fissures |
Almost clear | 1 | Barely perceptible erythema No signs of scaling, hyperkeratosis/lichenification, vesiculation, edema, or fissures |
Mild | 2 | At least 1 of the following:
|
Moderate | 3 | At least 1 of the following:
|
Severe | 4 | At least 1 of the following:
|
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema.
Source: Sponsor’s clinical summary report.16
Hand Eczema Severity Index
The CFB in the HECSI score to week 12 served as the primary end point in the DELTA FORCE trial; the CFB to week 24 was assessed as a key secondary outcome. In the DELTA 1 and DELTA 2 trials, the percentage CFB in HECSI to week 16 was also a key secondary outcome, along with the proportions of patients experiencing HECSI-75 and HECSI-90 from baseline to week 16. In the DELTA FORCE trial, the proportion of patients experiencing HECSI-90 at week 12 was also a key secondary end point.
HECSI is a validated, clinician-assessed instrument used to measure both the severity and the extent of hand eczema.32 It evaluates 6 clinical signs — erythema, infiltration/papulation, vesicles, fissures, scaling, and edema — across 5 bilateral hand regions: fingertips, fingers (excluding tips), palms, backs of hands, and wrists. Each sign is rated using a 4-point severity scale (0 = none; 3 = severe), and the extent of involvement is captured using a 5-point area scale based on the percentage of the region affected. For each region, a score is generated by summing the severity scores for the 6 signs and multiplying by the area score. The total HECSI score is the sum of the 5 region scores, ranging from 0 (no disease) to 360 (most severe). A reduction in HECSI score reflects clinical improvement. The assessment is cross-sectional and based on disease status at the time of evaluation, including any new lesions on previously unaffected areas.
HECSI has been validated for use in clinical trials of CHE. In a study involving 12 physicians experienced in CHE assessment, the tool demonstrated excellent intrarater reliability (intraclass correlation coefficient = 0.90) and strong interrater reliability for absolute agreement (intraclass correlation coefficient, 0.79 to 0.84) and for consistency (intraclass correlation coefficient, 0.81 to 0.89).32 Estimates of the minimal important difference (MID) for within-group improvement vary in the literature, ranging from 8.3 to 41 points,33,34 depending on the anchor used. The lower estimate (8.3 points) was derived using a physician-rated global severity scale that captured nuanced clinical change and is considered more sensitive. The higher value (41 points) was based solely on the PGA, a more conservative tool that may underestimate subtle but clinically meaningful improvements. Analyses from the delgocitinib phase IIb trial (NCT03683719) estimated the between-group MID to be approximately 20 points; this value was used to define the noninferiority margin in the DELTA FORCE trial (noninferiority being a secondary hypothesis). Additionally, HECSI-75 has been proposed as a clinically relevant response threshold, consistent with response definitions used in AD trials (e.g., 75% improvement in Eczema Area and Severity Index).
A calculation for the assessment of the HECSI score is presented in Figure 2.
Figure 2: Calculation of the Total HECSI Score
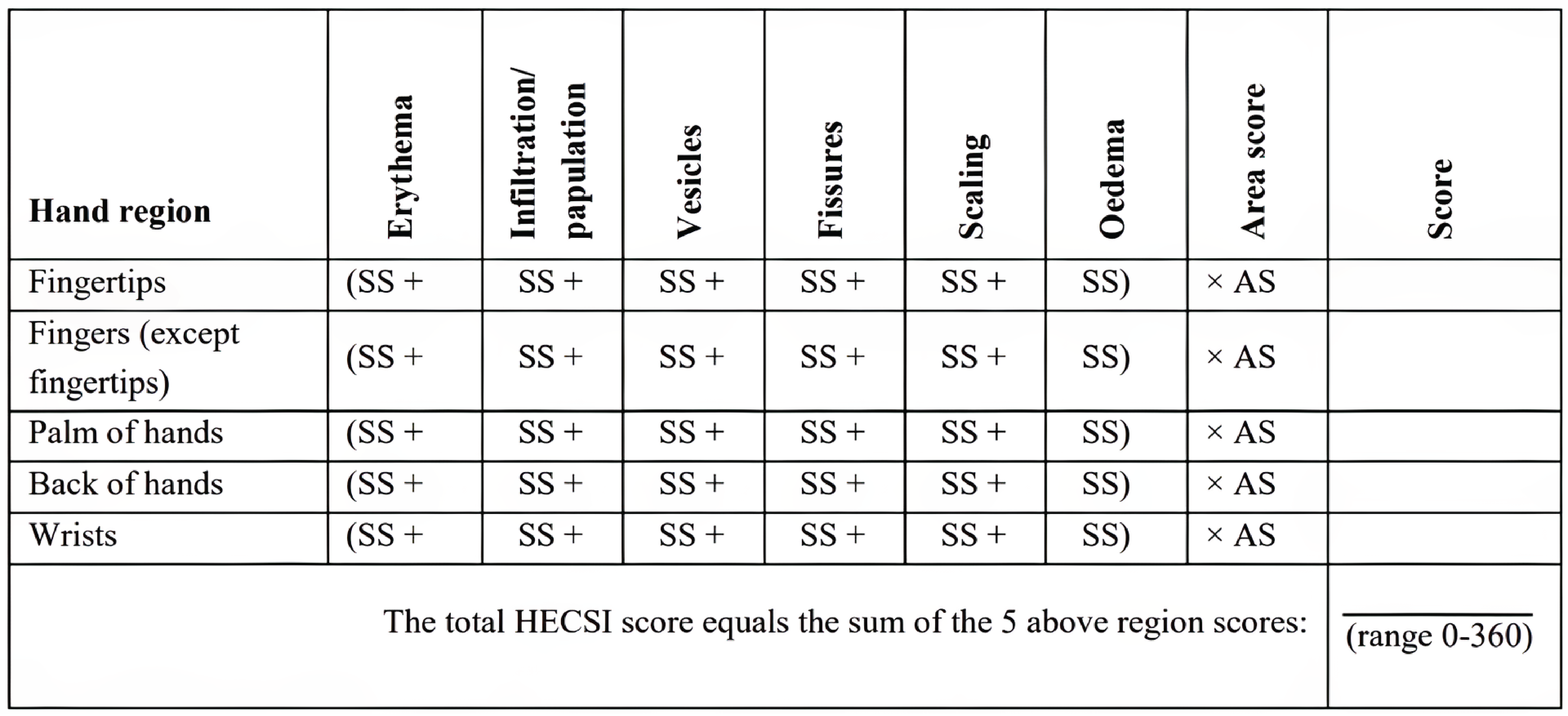
AS = area score; HECSI = Hand Eczema Severity Index; SS = severity score.
Source: Sponsor’s clinical summary report.16
Hand Eczema Symptom Diary
The CFB in HESD score (weekly average) to week 16 was a key secondary end point of the DELTA 1 and DELTA 2 trials. The CFBs in HESD itch score (weekly average) and HESD pain score (weekly average) to week 16 were key secondary outcomes of the DELTA 1 and DELTA 2 trials. The CFBs in HESD itch score (weekly average) and HESD pain score (weekly average) to week 12 were key secondary outcomes of the DELTA FORCE trial. The proportions of patients who experienced a reduction of at least 4 points in HESD, HESD itch, and HESD pain scores (weekly average) at week 16 were key secondary outcomes of the DELTA 1 and DELTA 2 trials.
The HESD is a patient-reported instrument developed by the sponsor and used in the phase IIb trial with delgocitinib (NCT03683719) and has been further revised for the current trials. Content validity of the HESD was confirmed through cognitive debriefing interviews with patients; psychometric validation activities using data from the phase IIb (NCT03683719) and DELTA 1 trials demonstrated construct validity, reliability, and the ability to detect change for the HESD itch score, HESD pain score, and overall HESD score. The HESD is a 6-item instrument designed to assess the severity of CHE signs and symptoms. Patients assessed the worst severity of 6 individual signs and symptoms of CHE (itch, pain, cracking, redness, dryness, and flaking) over the past 24 hours using an 11-point numeric rating scale with anchors of 0 (“no [symptom]”) and 10 (“severe [symptom]”). The overall HESD score is derived as an average of the 6 items. A decrease in score relates to an improvement in CHE signs and symptoms. Patients completed the HESD on a daily basis in an e-diary. An improvement of at least 4 points in the 7-day average HESD scores was considered a MID.35
Hand Eczema Impact Scale
The CFB in HEIS score to week 16 was a key secondary end point of the DELTA 1 and DELTA 2 trials. The CFB in HEIS score to week 12 and week 24 was an exploratory outcome of the DELTA FORCE trial.
The HEIS is a patient-reported instrument developed by the sponsor. The HEIS was used in the phase IIb trial with delgocitinib (NCT03683719) and validated based on these data, providing evidence of strong content and psychometric validity.36 The HEIS includes 9 items addressing the patient’s perception of the impact of hand eczema on their daily activities, embarrassment, frustration, sleep, work, and physical functioning over the past 7 days. Each item is scored on a 5-point scale (0 = not at all; 1 = a little; 2 = moderately; 3 = a lot; 4 = extremely). The HEIS score is the average of the 9 items. The highest possible score is 4, and a high score is indicative of a high impact. Six domain scores can be calculated for HEIS: proximal daily activity limitations (average of 3 items), embarrassment with the appearance of the hands (average of 2 items), frustration with CHE (1 item), sleep (1 item), work (1 item), and physical functioning (1 item). A decrease in score indicates an improvement in the impact of hand eczema on the patient’s quality of life. Anchor-based analyses suggested a within-group MID ranging from 0.90 to 1.35 points in HEIS score and a between-group MID of around 0.6 points in HEIS score.36
Dermatology Life Quality Index
The CFB in DLQI score to week 16 was a key secondary end point of the DELTA 1 and DELTA 2 trials. Additionally, the CFB in DLQI score to week 12 and week 24 was an exploratory outcome of the DELTA FORCE trial. The proportion of patients who experienced a reduction of at least 4 points in DLQI score at week 16 was a key secondary outcome of the DELTA 1 and DELTA 2 trials, and the proportion of patients who experienced a reduction of at least 4 points in DLQI score at week 12 and week 24 was a key secondary outcome of the DELTA FORCE trial.
The DLQI is a validated questionnaire with content specific to those with dermatologic conditions. It consists of 10 items addressing the patient’s perception of the impact of their skin disease on different aspects of their quality of life over the last week, such as dermatology-related symptoms and feelings, daily activities, leisure, work or school, personal relationships, and the treatment.37 Each item is scored on a 4-point Likert scale (0 = not at all/not relevant; 1 = a little; 2 = a lot; 3 = very much). The DLQI score is the sum of the 10 items (score ranging from 0 to 30); a high score is indicative of a poor quality of life. The threshold of an improvement of at least 4 points is aligned with the MID established in the literature for inflammatory skin diseases.38
The patient-reported outcome measures HEIS and DLQI used in this report are summarized in Table 9, along with their measurement properties and MIDs.
Table 9: Summary of Patient-Reported Outcome Measures and Their Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HEIS | The HEIS is a patient-reported instrument developed by the sponsor. The HEIS was used in the phase IIb trial with delgocitinib (NCT03683719) and validated based on these data, providing evidence of strong content and psychometric validity.36 The HEIS includes 9 items addressing a patient’s perception of the impact of hand eczema on their daily activities, embarrassment, frustration, sleep, work, and physical functioning over the past 7 days. Each item is scored on a 5-point scale (0 = not at all; 1 = a little; 2 = moderately; 3 = a lot; 4 = extremely). The HEIS score is the average of the 9 items. The highest possible score is 4, and a high score is indicative of a high impact. Six domain scores can be calculated for HEIS: PDAL (average of 3 items); embarrassment with the appearance of the hands (average of 2 items); frustration with CHE (1 item); sleep (1 item); work (1 item); and physical functioning (1 item). A decrease in score indicates an improvement in the impact of hand eczema on the patient’s quality of life.36 | Validity: Correlations with concurrent measures (0.66 to 0.87) and acceptable differences between severity groups (P < 0.001) indicated construct validity.36 Reliability: Internal consistency (Cronbach alpha ≥ 0.89) and test-retest reliability (ICC ≥ 0.79) results indicated acceptable reliability.36 Responsiveness: Large effect sizes for mean change scores in participants that improved and differences between groups indicated ability to detect change.36 | Anchor-based analyses suggested a within study-group MID ranging from 0.90 to 1.35 points in HEIS score and a between-group MID of around 0.6 points in HEIS score.36 |
DLQI | The DLQI is a validated questionnaire with content specific to those with dermatologic conditions. It consists of 10 items addressing the patient’s perception of the impact of their skin disease on different aspects of their quality of life over the last week, such as dermatology-related symptoms and feelings, daily activities, leisure, work or school, personal relationships, and the treatment.37 Each item is scored on a 4-point Likert scale (0 = not at all/not relevant; 1 = a little; 2 = a lot; 3 = very much). The DLQI score is the sum of the 10 items (score ranging from 0 to 30); a high score is indicative of a poor quality of life.39 | Validity: DLQI scores were correlated with other measures in observational studies, such as with disease severity, as measured by the clinician-reported HECSI (P < 0.001); with depressive symptoms measured by the Beck Depression Inventory II; and with prolonged sick leave and unemployment in patients with occupational hand eczema. This correlation established the instrument’s construct validity in CHE.39-41 Reliability: Internal consistency (acceptable Cronbach alpha > 0.70) and acceptable test-retest reliability threshold (ICC ≥ 0.75) were found from available literature.39 Responsiveness: Results related to the ability to detect changes were found to be limited and were mixed.39 | The threshold of an improvement of ≥ 4 points is aligned with the MID established in the literature for inflammatory skin diseases.38,39 |
CHE = chronic hand eczema; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HEIS = Hand Eczema Impact Scale; ICC = intraclass correlation coefficient; MID = minimal important difference; PDAL = proximal daily activity limitations.
Source: Sponsor’s clinical summary report.16
Statistical Analysis
Sample Size and Power Calculation
The DELTA 1 and DELTA 2 trials were designed to enrol approximately 470 and 450 patients, respectively, randomized in a 2:1 ratio to receive either delgocitinib cream or vehicle cream. These sample sizes were calculated to provide at least 99% power to detect a statistically significant difference in the primary outcome — IGA-CHE treatment success at week 16 — assuming response rates of 40% in the delgocitinib group and 10% in the vehicle cream group, using a 1-sided significance level of 2.5%. The assumptions for response rates were informed by findings from the phase IIb dose-ranging study (NCT03683719). The planned sample sizes also ensured sufficient safety data, including adequate exposure to delgocitinib over the study period, to support regulatory evaluation.
The DELTA FORCE trial was planned to randomize approximately 510 participants to receive either delgocitinib or alitretinoin (1:1 ratio). The sample size was powered to detect superiority of delgocitinib over alitretinoin with respect to the CFB in HECSI score at week 12, based on a 1-sided hypothesis test at a 2.5% significance level. The calculation assumed a between-group difference in mean HECSI CFB of 7.5 points, with a standard deviation (SD) of 30, yielding at least 80% power. Because limited data were available for alitretinoin using the HECSI scale, assumptions about treatment effect were guided by prior IGA-CHE–based outcomes from the phase IIb trial (NCT03683719). To achieve the target sample size, approximately 660 patients were expected to be screened.
Statistical Testing
For binary outcomes (e.g., IGA-CHE treatment success, HECSI-75, HECSI-90, HESD itch reduction ≥ 4 points), analyses across all 3 trials (DELTA 1, DELTA 2, and DELTA FORCE trials) used the Cochran-Mantel-Haenszel test, stratified by geographic region and either baseline IGA-CHE score (DELTA 1 and DELTA 2 trials) or hyperkeratotic versus nonhyperkeratotic subtype (DELTA FORCE trial). Missing data for binary outcomes were handled using imputation of nonresponse, and additional estimands (e.g., treatment policy, pandemic-modified composite) were employed in sensitivity analyses (refer to Table 10).
For continuous outcomes (e.g., CFB in HECSI scores, HESD scores, HEIS scores, and DLQI scores), data were analyzed using analysis of covariance models, adjusting for treatment group, region, baseline IGA-CHE severity, and the baseline value of the outcome measure. These models provided standard errors, 95% CIs, and P values. The sponsor used a composite estimand approach to account for intercurrent events (i.e., events that occur after treatment initiation and affect either the interpretation or the existence of the outcome data, such as treatment discontinuation, use of rescue therapy, or missing assessments), and worst observation carried forward (WOCF) imputation was applied for missing data after intercurrent events (refer to Table 10 and Table 11).
Across all 3 trials, the full analysis set (FAS) — defined as all participants who were randomized and exposed to treatment — was used for efficacy analyses.
Table 10: Statistical Analysis of Efficacy End Points for the DELTA 1, DELTA 2, and DELTA FORCE Trials
End point | Statistical model | Adjustment factors | Handling of missing dataa | Sensitivity analyses |
|---|---|---|---|---|
DELTA 1 and DELTA 2 | ||||
IGA-CHE treatment success | Cochran-Mantel-Haenszel test | Stratified by region and baseline IGA‑CHE score | Nonresponse imputation | Using different estimands and imputation methods:
|
CFB in HECSI | ANCOVA | Region, baseline IGA‑CHE score, and HECSI score baseline value | WOCF (including baseline value) | — |
HECSI-75 | Cochran-Mantel-Haenszel test | Stratified by region and baseline IGA-CHE score | Nonresponse imputation | — |
HECSI-90 | Cochran-Mantel-Haenszel test | Stratified by region and baseline IGA-CHE score | Nonresponse imputation | — |
HESD itch reduction ≥ 4 points | Cochran-Mantel-Haenszel test | Stratified by region and baseline IGA-CHE score | Nonresponse imputation | |
CFB in HEIS | ANCOVA | Region, baseline IGA‑CHE score, and HEIS score baseline value | WOCF (including baseline value) | |
CFB in DLQI | ANCOVA | Region, baseline IGA‑CHE score, and DLQI score baseline value | WOCF (including baseline value) | |
DELTA FORCE | ||||
CFB in HECSI | ANCOVA | Region, hyperkeratotic/nonhyperkeratotic subtype, and HECSI score baseline value | WOCF (including baseline value) | Using different estimands and imputation methods:
|
IGA-CHE treatment success | Cochran-Mantel-Haenszel test | Stratified by region and hyperkeratotic/nonhyperkeratotic subtype | Nonresponse imputation | |
HECSI-90 | Cochran-Mantel-Haenszel test | Stratified by region and hyperkeratotic/nonhyperkeratotic subtype | Nonresponse imputation | |
CFB in HESD itch score | ANCOVA | Region, hyperkeratotic/nonhyperkeratotic subtype, and HESD itch score baseline value | WOCF (including baseline value) | |
CFB in HEIS | ANCOVA | Region, hyperkeratotic/ nonhyperkeratotic subtype, and HEIS score baseline value | WOCF (including baseline value) | |
CFB in DLQI | ANCOVA | Region, hyperkeratotic/ nonhyperkeratotic subtype, and DLQI score baseline value | WOCF (including baseline value) | None |
ANCOVA = analysis of covariance; CFB = change from baseline; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; WOCF = worst observation carried forward.
aData after initiation of rescue treatments or permanent discontinuation of investigational medicinal product were treated as missing (primary and sensitivity analysis).
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Table 11: Definitions of Estimand and Imputation Methods
Analyses | Definition |
|---|---|
Estimands | |
Composite | The primary estimand used a composite strategy to handle IEs. With a composite strategy, the occurrence of an IE is a component of the end point. The occurrence of any IE led to assumed failure of the randomized treatment. For continuous end points, observed data after the IEs were imputed as nonresponse by using the WOCF (including baseline value), which is considered an unfavourable value. |
Pandemic-modified composite | The pandemic-modified composite strategy follows the composite strategy for IEs independent of the COVID-19 pandemic. However, data collected after the IE related to the COVID-19 pandemic were treated as missing and were imputed assuming they were MAR. |
Treatment policy | The treatment policy strategy attempts to quantify the effect of the randomized treatment, ignoring the occurrence of IEs. This policy reflects the intention-to-treat principle and is intended to measure the effectiveness of the treatment under conditions closer to real-world conditions, where patients will not adhere perfectly to the protocol when taking medication. Data collected for the end point of interest were used regardless of whether an IE occurred, except for the data on the permanent discontinuation of the IMP related to the COVID-19 pandemic, which were assumed MAR and imputed. |
Imputation methods | |
Nonresponse | Missing binary outcome assessments were imputed as nonresponse. |
MAR | Missing data or data treated as missing were imputed using multiple imputation, assuming data to be MAR within treatment groups. |
WOCF | Missing continuous outcomes were imputed as the worst observation (including baseline value). |
IE = intercurrent event; IMP = investigational medicinal product; MAR = missing at random; WOCF = worst observation carried forward.
Note: IEs include the initiation of rescue treatment, permanent discontinuation of the IMP independent of the COVID-19 pandemic, and permanent discontinuation of the IMP related to the COVID-19 pandemic.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Multiple Testing Procedures
Vehicle-Controlled Studies
In the DELTA 1 and DELTA 2 trials, a closed testing procedure with hierarchical tests, alpha splitting, and alpha recycling was used to control the overall type I error at a nominal 1-sided 2.5% level for the primary and key secondary end points. The statistical testing strategy was built on the principle that the IGA-CHE treatment success (IGA-CHE score of 0 or 1) superiority at week 16 had to be established before testing for additional benefits (key secondary end points) related to efficacy and HRQoL. The superiority of delgocitinib in the primary end point was tested at the overall 1-sided significance level of 2.5%. If a test was significant, the significance level was reallocated according to the weight and the direction of the arrows, as specified in Figure 3. Each of the hypotheses presented in Figure 3 were tested at their local significance level (alpha local). This process was to be repeated until no further tests were significant. The 1-sided (superiority) hypotheses were evaluated by deriving the 2-sided P value; the null hypothesis was to be rejected if the P value was smaller than 2 times the alpha and if the point estimate was in favour of the alternative hypothesis.
Figure 3: Closed Testing Procedure for Primary and Key Secondary End Points of the DELTA 1 and DELTA 2 Trials
DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; PDAL = proximal daily activity limitations; TS = treatment success defined as a score of 0 or 1 with at least a 2-step improvement from baseline.
Active-Controlled Study
A closed testing procedure with a hierarchical fixed-sequence design was used in the DELTA FORCE trial to control the overall type I error rate at a nominal 1-sided 2.5% level. The statistical testing strategy required that superiority of delgocitinib over alitretinoin in CFB in HECSI score at week 12 be demonstrated before any testing of subsequent key secondary end points related to efficacy and HRQoL could proceed. Hypotheses were tested sequentially according to a predefined order (Figure 4), with the significance level passed on to the next test only if the previous comparison was statistically significant. The final hypothesis in the sequence was the noninferiority of delgocitinib at week 24 with respect to CFB in HECSI score, using a noninferiority margin of 10 points, which was justified as a conservative value (less than half the MID) based on psychometric analyses from a prior phase IIb trial (NCT03683719). If noninferiority was confirmed, superiority was then formally tested.
Figure 4: Closed Testing Procedure for Primary and Key Secondary End Points of the DELTA FORCE Trial

AUC = area under the curve; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-90 = at least 90% improvement in Hand Eczema Severity Index score from baseline; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; TS = treatment success.
Source: DELTA FORCE Clinical Study Report.31
Analysis Populations
The analysis populations were the FAS (all participants randomized and exposed to the investigational product) and the safety analysis set in the DELTA 1, DELTA 2, and DELTA FORCE trials, as described in Table 12. The FAS can be considered the intention-to-treat (ITT) population.
Table 12: Analysis Populations of the DELTA 1, DELTA 2, and DELTA FORCE Trials
Study | Population | Definition | Application |
|---|---|---|---|
DELTA 1 and DELTA 2 | Full analysis set | All participants randomized and exposed to IMP | This set was used for all efficacy and HRQoL end points |
Safety analysis set | All participants exposed to IMP | This set was used for end points related to safety | |
DELTA FORCE | Full analysis set | All randomized participants, excluding some participants with who did not meet major inclusion criteria or to whom a major exclusion criterion applied | This set was used for all efficacy and HRQoL end points |
Safety analysis set | All participants exposed to the IMP | This set was used for end points related to safety |
HRQoL = health-related quality of life; IMP = investigational medicinal product.
Sources: DELTA 1, DELTA 2, and DELTA FORCE statistical analysis plans.42-44
Results
Patient Disposition
Patient disposition (Table 13) was generally balanced between arms in the DELTA 1 and DELTA 2 trials, but the DELTA FORCE trial showed a higher discontinuation rate in the alitretinoin group, mostly due to tolerability issues. While all studies achieved target sample sizes, the dropout was slightly higher in the DELTA FORCE trial.
Across all 3 trials, randomized sample sizes were achieved as planned, with no evidence of recruitment shortfalls. In the DELTA 1 trial (N = 487), completion rates were 93.8% in the delgocitinib group (20 of 325 patients [6.2%] discontinued) and 87.0% in the vehicle cream group (21 of 162 patients [13.0%] discontinued). In the DELTA 2 trial (N = 473), 93.0% of patients receiving delgocitinib completed the study (22 of 314 patients [7.0%] discontinued), compared with 76.7% in the vehicle cream group (37 of 159 [23.3%] discontinued). Withdrawal due to AEs and lack of efficacy were more common in the vehicle cream groups of both trials, but overall discontinuation rates remained modest.
In the DELTA FORCE trial (N = 513), completion rates diverged more substantially: 86.6% of patients in the delgocitinib group (34 of 254 patients [13.4%] discontinued) versus 64.1% in the alitretinoin group (93 of 259 patients [35.9%] discontinued). The most common reasons for early withdrawal in the alitretinoin group were AEs (24 of 259 patients [9.3%]) and lack of efficacy (26 of 259 patients [10.0%]). In contrast, only 2 of 254 patients (0.8%) in the delgocitinib arm discontinued due to AEs.
Table 13: Patient Disposition in the DELTA 1, DELTA 2, and DELTA FORCE Trials
Patient disposition | DELTA 1 | DELTA 2 | DELTA FORCE | |||
|---|---|---|---|---|---|---|
Delgocitinib 20 mg/g (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g (N = 314) | Vehicle cream (N = 159) | Delgocitinib 20 mg/g (N = 254) | Alitretinoin 30 mg (N = 259) | |
Screened, N | 566 | 557 | 693 | |||
Did not proceed beyond screening, n | 63 | 69 | 143 | |||
Reason for not proceeding beyond screening, n | ||||||
Inclusion criteria not met | 32 | 41 | NR | |||
Exclusion criteria met | 32 | 29 | NR | |||
Patient withdrew | 12 | 12 | NR | |||
Missed visit window | 1 | 3 | NR | |||
Personal reasons | 2 | 0 | NR | |||
Lost to follow-up | 1 | 0 | NR | |||
Randomized, N (%) | 325 (100) | 162 (100) | 314 (100) | 159 (100) | 254 (100) | 259 (100) |
Not exposed to IMP, N (%) | 0 | 0 | 1 (0.3) | 0 | 1 (0.4) | 12 (4.6) |
Discontinued from study, N (%) | 20 (6.2) | 21 (13.0) | 22 (7.0) | 37 (23.3) | 34 (13.4) | 93 (35.9) |
Reason for discontinuation, N (%) | ||||||
Patient withdrew | 11 (3.4) | 5 (3.1) | 10 (3.2) | 16 (10.1) | 15 (5.9) | 33 (12.7) |
Lack of efficacy | 5 (1.5) | 7 (4.3) | 6 (1.9) | 14 (8.8) | 8 (3.1) | 26 (10.0) |
Adverse events | 2 (0.6) | 6 (3.7) | 1 (0.3) | 6 (3.8) | 2 (0.8) | 24 (9.3) |
Lost to follow-up | 0 | 2 (1.2) | 2 (0.6) | 1 (0.6) | 5 (2.0) | 1 (0.4) |
Pregnancy | 0 | 0 | 2 (0.6) | 0 | 0 | 0 |
Other | 2 (0.6) | 1 (0.6) | 1 (0.3) | 0 | 3 (1.2) | 9 (3.5) |
FAS, N (%) | 325 (100) | 162 (100) | 313 (99.7) | 159 (100) | 250 (98.4) | 253 (97.7) |
Safety analysis set, N (%) | 325 (100) | 162 (100) | 313 (99.7) | 159 (100) | 253 (99.6) | 247 (95.4) |
FAS = full analysis set; IMP = investigational medicinal product; NR = not reported.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Baseline Characteristics
The baseline characteristics outlined in Table 14 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. No clinically meaningful imbalances were observed between arms within individual studies in terms of age, sex, race, region, or baseline disease severity. Overall, the populations enrolled align with the Health Canada indication for moderate to severe CHE, though the DELTA FORCE trial focused exclusively on severe disease.
In the DELTA 1 and DELTA 2 trials, the median patient age ranged from 42.0 to 46.0 years across arms, and there was a slight predominance of females (ranging from approximately 62% to 68% of patients across arms). Most participants in both trials (88.9% to 93.9%) were white, and most (approximately 80%) were recruited from European sites, with consistent representation from North America (approximately 20%). The median disease duration ranged from 4 to 6 years, and most patients (67% to 76%) had moderate CHE, as expected based on the inclusion criteria.
In the DELTA FORCE trial, which enrolled only patients with severe CHE (100% in the delgocitinib group and 99.6% in the alitretinoin group), the median age (44 to 46 years), the sex distribution (66% female; 34% male), and the racial composition (more than 92% white) were similar to the DELTA 1 and DELTA 2 trials. However, the geographic distribution differed, with more than 90% of participants recruited in Europe and fewer from North America (< 10%). Additionally, patients in the DELTA FORCE trial had slightly higher baseline HECSI scores (median of 80, versus 59 to 66) and lower median HESD itch and pain scores than in the DELTA 1 and DELTA 2 trials, consistent with these patients’ more severe clinical phenotype but possibly differing symptom burden.
Across all studies, the DLQI scores at baseline indicated moderate to severe impact on quality of life, with median scores ranging from 11.0 to 12.0. Almost all patients (> 99%) in the DELTA 1 and DELTA 2 trials had HESD itch scores of at least 4 at baseline; this percentage was lower in the DELTA FORCE trial (approximately 70%). Similarly, pain scores of 4 or higher were reported in 90% of patients in the DELTA 1 and DELTA 2 trials versus 66% in the DELTA FORCE trial. This difference may reflect variability in symptom reporting or subtype distribution.
The CHE subtypes also varied by study. In the DELTA 1 and DELTA 2 trials, atopic hand eczema and hyperkeratotic eczema were the most frequent diagnoses, whereas the DELTA FORCE trial included more patients with irritant contact dermatitis (approximately 29%) and fewer with atopic hand eczema (approximately 24%). Also, vesicular and hyperkeratotic hand eczema types were more common in the DELTA 2 study.
Table 14: Summary of Baseline Characteristics of the DELTA 1, DELTA 2, and DELTA FORCE Trials
Variable | DELTA 1 | DELTA 2 | DELTA FORCE | |||
|---|---|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g b.i.d. (N = 314) | Vehicle cream (N = 159) | Delgocitinib 20 mg/g b.i.d. (N = 254) | Alitretinoin 30 mg q.d. (N = 259) | |
Age (years), median (IQR) | 45.0 (32.0 to 55.0) | 42.5 (30.0 to 55.0) | 46.0 (34.0 to 57.0) | 42.0 (31.0 to 54.0) | 46.0 (34.0 to 56.0) | 44.0 (31.0 to 56.0) |
Sex, n (%) | ||||||
Female | 202 (62.2) | 104 (64.2) | 204 (65.0) | 108 (67.9) | 167 (65.7) | 167 (64.5) |
Male | 123 (37.8) | 58 (35.8) | 110 (35.0) | 51 (32.1) | 87 (34.3) | 92 (35.5) |
Race, n (%) | ||||||
Asian | 14 (4.3) | 5 (3.1) | 8 (2.5) | 7 (4.4) | 9 (3.6) | 5 (1.9) |
American Indian or Alaska | 1 (0.3) | 0 | 0 | 0 | 1 (0.4) | 2 (0.8) |
Black or African American | 3 (1) | 1 (0.6) | 2 (0.6) | 1 (0.6) | 1 (0.4) | 3 (1.2) |
Native Hawaiian or Other Pacific Islander | 0 | 0 | 1 (0.3) | 0 | 0 | 1 (0.4) |
White | 283 (87.1) | 144 (88.9) | 295 (93.9) | 146 (91.8) | 237 (93.3) | 240 (92.7) |
Multiple | 2 (0.6) | 1 (0.6) | 1 (0.3) | 3 (1.9) | 2 (0.8) | 0 |
Other or NR | 22 (6.8) | 11 (6.8) | 7 (2.2) | 2 (1.3) | 4 (1.6) | 8 (3.1) |
Ethnicity, n (%) | ||||||
Hispanic or Latino | 14 (4.3) | 4 (2.5) | 2 (0.6) | 5 (3.1) | 22 (8.7) | 14 (5.4) |
Not Hispanic or Latino | 292 (89.8) | 147 (90.7) | 310 (98.7) | 152 (95.6) | 226 (89.0) | 241 (93.1) |
NR | 19 (5.8) | 11 (6.8) | 2 (0.6) | 2 (1.3) | 6 (2.4) | 4 (1.5) |
Region, n (%) | ||||||
Europe | 260 (80.0) | 130 (80.2) | 250 (79.6) | 126 (79.2) | 229 (90.2) | 230 (88.8) |
North America | 65 (20.0) | 32 (19.8) | 64 (20.4) | 33 (20.8) | 25 (9.8) | 29 (11.2) |
Age at onset of CHE (years), median (IQR) | 33.0 (21.0 to 47.0) | 30.0 (20.0 to 46.0) | 35.0 (24.0 to 49.0) | 32.0 (21.0 to 46.0) | 37.5 (22.0 to 51.0) | 36.0 (22.0 to 49.0) |
Duration of CHE (years), median (IQR) | 6.0 (2.0 to 15.0) | 5.5 (2.0 to 15.0) | 4.0 (2.0 to 11.0) | 5.0 (2.0 to 12.0) | 4.0 (2.0 to 13.0) | 4.0 (2.0 to 10.0) |
IGA-CHE score, n (%) | ||||||
Mild | 0 | 0 | 0 | 0 | 0 | 1 (0.4)a |
Moderate | 218 (67.1) | 109 (67.3) | 239 (76.1) | 121 (76.1) | 0 | 0 |
Severe | 107 (32.9) | 53 (32.7) | 75 (23.9) | 38 (23.9) | 254 (100) | 258 (99.6) |
HECSI score | ||||||
n | 325 | 162 | 313 | 159 | 252 | 256 |
Median (IQR) | 66.0 (43.0 to 98.0) | 61.5 (38.0 to 105.0) | 59.0 (38.0 to 82.0) | 59.0 (40.0 to 90.0) | 79.5 (52.5 to 114.5) | 80.0 (52.0 to 119.0) |
DLQI score | ||||||
n | 321 | 158 | 310 | 159 | 233 | 242 |
Median (IQR) | 12.0 (9.0 to 17.0) | 12.0 (7.0 to 18.0) | 11.0 (7.0 to 17.0) | 11.0 (7.0 to 17.0) | 12.0 (8.0 to 17.0) | 12.0 (8.0 to 17.0) |
≥ 4, n (%) | 305 (95.0) | 148 (93.7) | 299 (96.5) | 153 (96.2) | NR | NR |
HESD itch score (weekly average) | ||||||
n | 324 | 162 | 312 | 157 | 240 | 244 |
Median (IQR) | 7.2 (5.9 to 8.3) | 7.6 (5.9 to 8.4) | 7.1 (5.9 to 8.0) | 7.1 (5.7 to 8.1) | 6.1 (3.8 to 8.0) | 6.4 (4.1 to 8.0) |
≥ 4, n (%) | 323 (99.7) | 161 (99.4) | 309 (99.0) | 156 (99.4) | 177 (69.7) | 185 (71.4) |
HESD pain score (weekly average) | ||||||
n | 324 | 162 | 312 | 157 | 240 | 244 |
Median (IQR) | 7.1 (5.7 to 8.3) | 7.2 (5.7 to 8.1) | 6.7 (5.3 to 8.0) | 6.7 (5.1 to 7.9) | 5.6 (3.1 to 7.6) | 6.1 (3.6 to 8.0) |
≥ 4, n (%) | 291 (89.8) | 149 (92.0) | 294 (94.2) | 141 (89.8) | 163 (64.2) | 176 (68.0) |
HESD score (weekly average) | ||||||
n | 324 | 162 | 312 | 157 | NR | NR |
Median (IQR) | 7.2 (6.1 to 8.4) | 7.3 (6.1 to 8.4) | 7.0 (6.0 to 8.0) | 7.1 (5.9 to 8.0) | NR | NR |
≥ 4, n (%) | 309 (95.4) | 156 (96.3) | 308 (98.7) | 153 (97.5) | NR | NR |
CHE subtype main diagnosis, n (%) | ||||||
Atopic hand eczema | 143 (44.0) | 74 (45.7) | 82 (26.1) | 46 (28.9) | 66 (26.0) | 57 (22.0) |
Allergic contact dermatitis | 51 (15.7) | 33 (20.4) | 27 (8.6) | 22 (13.8) | 58 (22.8) | 54 (20.8) |
Hyperkeratotic eczema | 57 (17.5) | 20 (12.3) | 86 (27.4) | 43 (27.0) | 31 (12.2) | 32 (12.4) |
Irritant contact dermatitis | 49 (15.1) | 26 (16.0) | 75 (23.9) | 38 (23.9) | 75 (29.5) | 76 (29.3) |
Vesicular hand eczema (pompholyx) | 25 (7.7) | 9 (5.6) | 44 (14.0) | 9 (5.7) | 22 (8.7) | 36 (13.9) |
Contact urticaria or protein contact dermatitis | 0 | 0 | 0 | 1 (0.6) | 0 | 0 |
NR | 0 | 0 | 0 | 0 | 2 (0.8) | 4 (1.5) |
b.i.d. = twice a day; CHE = chronic hand eczema; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; IQR = interquartile range; NR = not reported; q.d. = every day.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
aOne patient with mild CHE was included in the DELTA FORCE trial.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Exposure to Study Treatments
Vehicle-Controlled Studies
In both the DELTA 1 and DELTA 2 trials, exposure to delgocitinib cream was generally comparable to exposure to vehicle cream in terms of treatment duration, amount used, and adherence (table 15). In the DELTA 1 trial, the mean duration of exposure was 109.4 days (SD = 18.7 days) in the delgocitinib group and 103.9 days (SD = 26.6 days) in the vehicle cream group. The mean total amounts of the study drug applied were 134.2 g and 130.5 g in the delgocitinib and vehicle cream groups, respectively, with similar weekly usage throughout the 16-week treatment period. Results from the DELTA 2 trial showed slightly longer exposure in the delgocitinib group (108.9 days, versus 97.0 days in the vehicle cream group) and a small difference in total cream use (121.6 g versus 116.8 g). In both studies, adherence was high and similar between groups: more than 50% of patients in each arm missed less than 10% of scheduled applications, and around 16% to 23% of patients had perfect adherence.
Rescue medication (Table 16) use was infrequent and consistently lower in the delgocitinib arms. In the DELTA 1 trial, 2.2% of patients treated with delgocitinib required rescue therapy, compared with 4.3% in the vehicle cream group. In the DELTA 2 trial, the difference was more pronounced (1.0% versus 7.5%). TCS were the most common rescue therapies, and their use was defined in the protocol as requiring immediate discontinuation of the study treatment. Concomitant medication use was common across arms (approximately 70% to 80% of patients) and included nonsteroidal anti-inflammatory drugs, analgesics, antihistamines, and hormonal contraceptives. No imbalances were observed that would be expected to affect the interpretation of efficacy or safety.
Table 15: Patient Exposure in the DELTA 1 and DELTA 2 Trials
Exposure | DELTA 1 | DELTA 2 | ||
|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g b.i.d. (N = 313) | Vehicle cream (N = 159) | |
Duration of exposure | ||||
Total exposure time (patient‑days) | 97.3 | 46.1 | 93.3 | 42.23 |
Exposure time (patient‑days), mean (SD) | 109.4 (18.7) | 103.9 (26.6) | 108.9 (19.9) | 97.0 (33.6) |
Dosing | ||||
Total amount of drug used (g), mean (SD) | 134.2 (54.9) | 130.5 (63.3) | 121.6 (55.3) | 116.8 (64.5) |
Average weekly amount of drug used (g), mean (SD) | 8.5 (3.2) | 8.8 (3.8) | 7.7 (3.3) | 8.1 (3.3) |
Patients who missed application or had no compliance data, n (%) | ||||
No days with missed drug application | 74 (22.8) | 34 (21.0) | 50 (16.0) | 30 (18.9) |
> 0% and ≤ 10% of days with missed drug application | 164 (50.5) | 85 (52.5) | 159 (50.8) | 83 (52.2) |
> 10% and ≤ 20% of days with missed drug application | 48 (14.8) | 23 (14.2) | 39 (12.5) | 21 (13.2) |
> 20% and ≤ 30% of days with missed drug application | 17 (5.2) | 8 (4.9) | 18 (5.8) | 6 (3.8) |
> 30% of days with missed drug application | 22 (6.8) | 12 (7.4) | 47 (15.0) | 19 (11.9) |
b.i.d. = twice a day; SD = standard deviation.
Notes: A patient was considered as having missed a day of application of the investigational medicinal product when no drug was applied for that day. In cases of permanent discontinuation, only days missed before the permanent discontinuation were included. Treatment compliance was evaluated daily by the patient in the e-diary.
Sources: DELTA 1 and DELTA 2 Clinical Study Reports. 29,30
Table 16: Concomitant Medications and Rescue Treatment in the DELTA 1, DELTA 2, and DELTA FORCE Trials
Exposure | DELTA 1 | DELTA 2 | DELTA FORCE | |||
|---|---|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. | Vehicle cream | Delgocitinib 20 mg/g b.i.d. | Vehicle cream | Delgocitinib 20 mg/g b.i.d. | Alitretinoin 30 mg q.d. | |
Randomized participants | (N = 325) | (N = 162) | (N = 314) | (N = 159) | (N = 254) | (N = 259) |
Any concomitant medication, n (%) | 250 (76.9) | 129 (79.6) | 244 (77.7) | 110 (69.2) | 191 (75.2) | 207 (79.9) |
Most commona concomitant medications, n (%) | ||||||
Nonsteroidal anti-inflammatory and antirheumatic products | 51 (15.7) | 18 (11.1) | 63 (20.1) | 22 (13.8) | 48 (18.9) | 54 (20.8) |
Other analgesics and antipyretics | 47 (14.5) | 26 (16.0) | 45 (14.3) | 28 (17.6) | 32 (12.6) | 44 (17.0) |
Viral vaccines | 46 (14.2) | 21 (13.0) | 46 (14.6) | 15 (9.4) | 3 (1.2) | 2 (0.8) |
Systemic antihistamines | 47 (14.5) | 23 (14.2) | 31 (9.9) | 18 (11.3) | 35 (13.8) | 42 (16.2) |
Inhalants containing adrenergics | 37 (11.4) | 19 (11.7) | 23 (7.3) | 12 (7.5) | 20 (7.9) | 22 (8.5) |
Thyroid preparations | 29 (8.9) | 20 (12.3) | 27 (8.6) | 13 (8.2) | 19 (7.5) | 25 (9.7) |
Lipid-modifying agents | 31 (9.5) | 12 (7.4) | 33 (10.5) | 10 (6.3) | 24 (9.4) | 29 20 (7.7) |
Systemic hormonal contraceptives | 35 (10.8) | 12 (7.4) | 24 (7.6) | 17 (10.7) | 43 (16.9) | 69 (26.6) |
Rescue treatment, n (%) | (N = 325) | (N = 162) | (N = 313) | (N = 159) | (N = 254) | (N = 259) |
Any rescue treatment | 7 (2.2) | 7 (4.3) | 3 (1.0) | 12 (7.5) | 12 (4.7) | 21 (8.1) |
TCS | 4 (1.2) | 6 (3.7) | 2 (0.6) | 10 (6.3) | 11 (4.3) | 15 (5.8) |
Other dermatological preparations | 1 (0.3) | 2 (1.2) | 1 (0.3) | 1 (0.6) | 1 (0.4) | 5 (1.9) |
Corticosteroids for systemic use | 0 | 1 (0.6) | 2 (1.3) | 2 (0.4) | 0 | 1 (0.4) |
Antineoplastic and immunomodulating agents | 3 (0.9) | 1 (0.6) | 0 | 0 | 0 | 2 (0.8) |
Antihistamines for systemic use | 0 | 0 | 0 | 1 (0.6) | 0 | 2 (0.8) |
b.i.d. = twice a day; q.d. = every day; TCS = topical corticosteroids.
aAt least 10% of patients in any treatment group at Anatomical Therapeutic Chemical level 3.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Active-Controlled Study (DELTA FORCE Trial)
In the DELTA FORCE trial, differences in exposure patterns (Table 17) were evident between the delgocitinib and alitretinoin treatment arms.
The mean total amount of delgocitinib cream used during the total treatment period (16 weeks of continuous use, then as-needed dosing for remaining 8 weeks) was 207.8 g (SD = 93.1 g). The mean duration of exposure to delgocitinib was 149.7 days (SD = 34.4 days), corresponding to approximately 21 weeks. The mean duration of exposure to alitretinoin was 119.8 days (SD = 55.0 days), corresponding to approximately 17 weeks, with 52 participants (21.1%) reducing their alitretinoin dose from 30 mg to 10 mg. This discrepancy likely reflects higher discontinuation and dose reduction rates in the alitretinoin group. The overall treatment compliance was high and similar in both treatment groups of the DELTA FORCE trial, with 56.0% of patients in the delgocitinib group and 56.2% of patients in the alitretinoin group having at most 10% of days with missed treatment application or missing compliance data.
Rescue treatment use (Table 16) was again lower in the delgocitinib group (4.7%) than in the alitretinoin group (8.1%). The most frequently used class of rescue treatment was dermatological preparations, primarily TCS. Concomitant medication use was balanced (75.2% in the delgocitinib group versus 79.9% in the alitretinoin group), with frequent use of nonsteroidal anti-inflammatory drugs, analgesics, systemic antihistamines, and hormonal contraceptives. No major differences in co-interventions were observed that would affect the interpretation of treatment efficacy or safety.
Table 17: Patient Exposure in the DELTA FORCE Trial
Exposure | Delgocitinib 20 mg/g b.i.d. (N = 253) | Alitretinoin 30 mg q.d. (N = 247) |
|---|---|---|
Duration of exposure | ||
Total exposure time (patient-years) | NR | NR |
Exposure time (days), mean (SD) | 149.7 (34.4) | 119.8 (55.0) |
Dosing | ||
Total amount of drug used (g), mean (SD) | 207.8 (93.1) | NA |
Average weekly amount of drug used (g), mean (SD) | 9.6 (4.1) | NA |
Patients who missed administration or no compliance data until week 24,a n (%) | ||
No days with missed drug administration | 21 (10.0) | 13 (6.7) |
> 0% and ≤ 10% of days with missed drug administration | 117 (56.0) | 109 (56.2) |
> 10% and ≤ 20% of days with missed drug administration | 27 (12.9) | 28 (14.4) |
> 20% and ≤ 30% of days with missed drug administration | 20 (9.6) | 18 (9.3) |
> 30% of days with missed drug administration | 24 (11.5) | 26 (13.4) |
b.i.d. = twice a day; NA = not applicable; NR = not reported; q.d. = every day; SD = standard deviation.
Notes: Treatment compliance was evaluated daily from participant entries in the e-diary. A participant was considered as having missed a day’s administration when no drug was administered or if e-diary compliance data were missing on an on-treatment day. In cases of permanent discontinuation, only days missed before permanent discontinuation were included.
aAmong patients with less than 50% missing compliance data during period.
Source: DELTA FORCE Clinical Study Report.31
Efficacy
Vehicle-Controlled Studies
In both the DELTA 1 and DELTA 2 trials, treatment with delgocitinib cream 20 mg/g twice daily resulted in greater improvements in clinical and symptom-based outcomes at week 16 than treatment with vehicle cream (Table 18, Figure 5 and Figure 6).
The proportion of patients who experienced IGA-CHE treatment success at week 16 was higher with delgocitinib than with vehicle cream: 19.7% versus 9.9% in the DELTA 1 trial (absolute difference = 9.8%; 95% CI, 3.6% to 16.1%; P = 0.0055) and 29.1% versus 6.9% in the DELTA 2 trial (absolute difference = 22.2%; 95% CI, 15.8% to 28.5%; P < 0.0001).
Patient-reported improvement in itch, defined as a reduction of 4 points or more in the HESD itch score, also favoured delgocitinib. In the DELTA 1 trial, 47.1% of patients in the delgocitinib group reported this level of improvement, compared to 23.0% in the vehicle cream group (difference = 24.1%; 95% CI, 15.5% to 32.6%; P < 0.0001). A similar pattern was observed in the DELTA 2 trial (47.2% versus 19.9%; difference = 27.4%; 95% CI, 19.0% to 35.8%; P < 0.0001).
For clinician-assessed signs of hand eczema, HECSI-75 and HECSI-90 response rates were consistently higher in the delgocitinib groups. In the DELTA 1 trial, 49.2% of patients in the delgocitinib group experienced a HECSI-75 response and 29.5% experienced a HECSI-90 response, compared to 23.5% and 12.3% of patients in the vehicle cream group. The absolute differences were 25.7% (95% CI, 17.2% to 34.3%; P < 0.0001) for HECSI-75 and 17.2% (95% CI, 10.1% to 24.3%; P < 0.0001) for HECSI-90. The corresponding values in the DELTA 2 trial were 49.5% versus 18.2% for HECSI-75 (difference = 31.3%; 95% CI, 23.1% to 39.5%) and 31.0% versus 8.8% for HECSI-90 (difference = 22.2%; 95% CI, 15.4% to 29.0%), both with a P value less than 0.0001.
In terms of percentage CFB in HECSI score, delgocitinib demonstrated greater reductions than vehicle cream in both studies. The mean difference in the DELTA 1 trial was –35.2 percentage points (95% CI, –46.7 to –23.8 percentage points; P < 0.0001) and in the DELTA 2 trial was –45.5 percentage points (95% CI, –56.4 to –34.6 percentage points; P < 0.0001), favouring delgocitinib.
Table 18: Key Efficacy Outcomes of the DELTA 1 and DELTA 2 Trials
Variable | DELTA 1 | DELTA 2 | ||
|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g b.i.d. (N = 314) | Vehicle cream (N = 159) | |
IGA-CHE treatment success at week 16 | ||||
Patients contributing to the analysis, N | 325 | 162 | 313 | 159 |
Patients experiencing IGA-CHE treatment success at week 16, n (%) | 64 (19.7) | 16 (9.9) | 91 (29.1) | 11 (6.9) |
Difference, % (95% CI) | 9.8 (3.6 to 16.1) | 22.2 (15.8 to 28.5) | ||
P valuea | 0.0055 | < 0.0001 | ||
HECSI-90 at week 16a | ||||
Patients contributing to the analysis, N | 325 | 162 | 313 | 159 |
Patients experiencing HECSI-90 at week 16, n (%) | 96 (29.5) | 20 (12.3) | 97 (31.0) | 14 (8.8) |
Difference, % (95% CI) | 17.2 (10.1 to 24.3) | 22.2 (15.4 to 29.0) | ||
P value | < 0.0001 | < 0.0001 | ||
HECSI-75 at week 16a | ||||
Patients contributing to the analysis, N | 325 | 162 | 313 | 159 |
Patients experiencing HECSI-75 at week 16, n (%) | 160 (49.2) | 38 (23.5) | 155 (49.5) | 29 (18.2) |
Difference, % (95% CI) | 25.7 (17.2 to 34.3) | 31.3 (23.1 to 39.5) | ||
P value | < 0.0001 | < 0.0001 | ||
Change from baseline in HECSI at week 16b | ||||
Patients contributing to the analysis, N | 325 | 162 | 313 | 159 |
Change from baseline in HECSI at week 16 (percentage points), mean (SE) | –56.5 (3.4) | –21.2 (4.8) | –58.9 (3.2) | –13.4 (4.5) |
Difference (95% CI) | –35.2 (–46.7 to –23.8) | –45.5 (–56.4 to –34.6) | ||
P value | < 0.0001 | < 0.0001 | ||
HESD itch reduction ≥ 4 points at week 16a | ||||
Patients contributing to the analysis, N | 323 | 161 | 309 | 156 |
Patients experiencing HESD itch reduction ≥ 4 points at week 16, n (%) | 152 (47.1) | 37 (23.0) | 146 (47.2) | 31 (19.9) |
Difference, % (95% CI) | 24.1 (15.5 to 32.6) | 27.4 (19.0 to 35.8) | ||
P value | < 0.0001 | < 0.0001 | ||
b.i.d. = twice a day; CI = confidence interval; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; SE = standard error.
aBased on a Cochran-Mantel-Haenszel test stratified by region and baseline IGA-CHE score.
bBased on an analysis of covariance model adjusted for region, baseline IGA-CHE score, and tool score baseline value.
Sources: DELTA 1 and DELTA 2 Clinical Study Reports.29,30
Figure 5: IGA-CHE Treatment Success by Study Visits in the DELTA 1 Trial
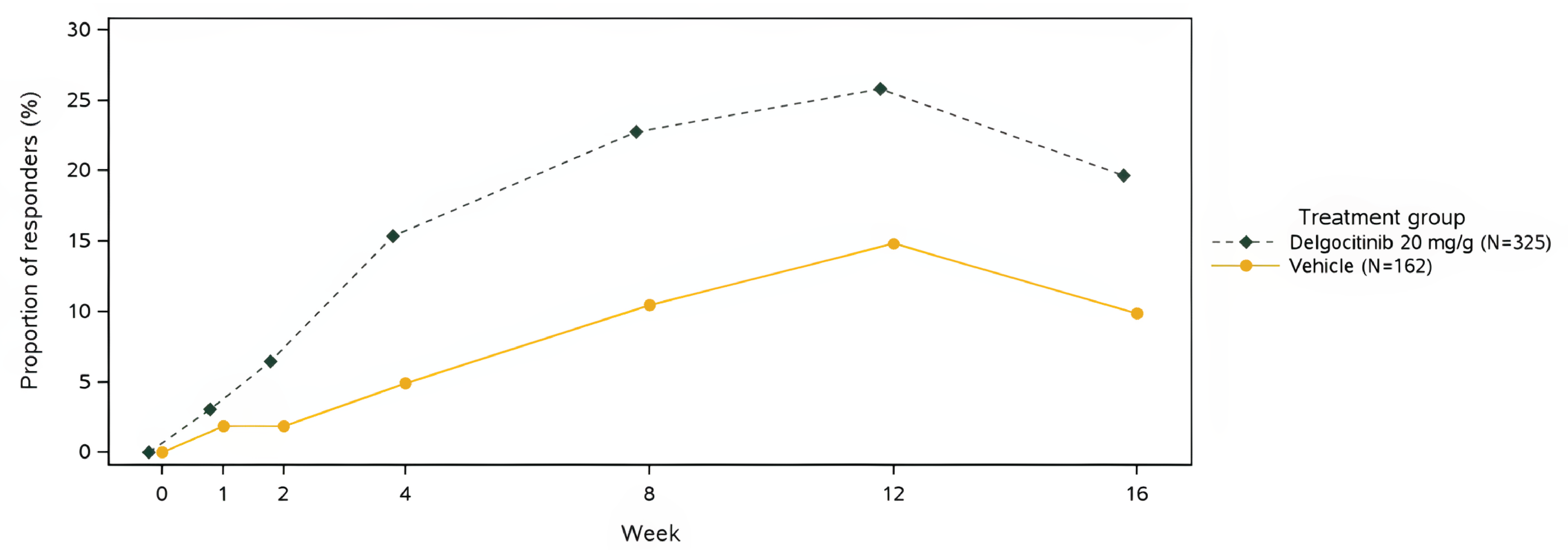
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema.
Notes: Composite estimand: Data considered to indicate nonresponse if observed after the initiation of rescue treatment or after permanent discontinuation of the investigational medicinal product. Missing data are imputed as nonresponse. Patients were considered to have experienced a response if they experienced an IGA-CHE score of 0 (clear) or 1 (almost clear) with at least a 2-step improvement from baseline.
Figure 6: IGA-CHE Treatment Success by Study Visits in the DELTA 2 Trial
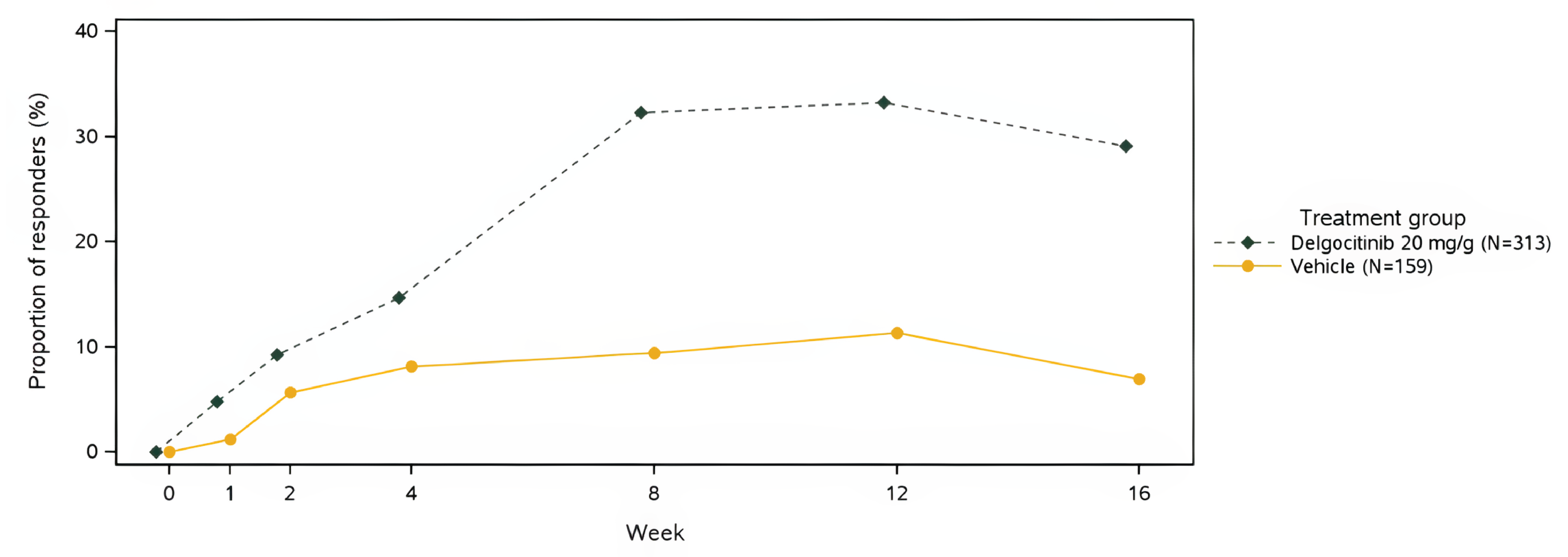
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema.
Notes: Composite estimand: Data considered to indicate nonresponse if observed after the initiation of rescue treatment or after permanent discontinuation of the investigational medicinal product. Missing data are imputed as nonresponse. Patients were considered to have experienced a response if they experienced an IGA-CHE score of 0 (clear) or 1 (almost clear) with at least a 2-step improvement from baseline.
Active-Controlled Study
In the DELTA FORCE trial (Table 19), delgocitinib cream 20 mg/g twice daily demonstrated greater clinical and symptom improvement than oral alitretinoin 30 mg once daily at week 12 in adults with severe CHE.
IGA-CHE treatment success at week 12 was experienced by 27.2% of patients treated with delgocitinib, compared to 16.6% with alitretinoin, corresponding to an absolute difference of 10.6% (95% CI, 3.31% to 17.87%; P = 0.004).
Improvements in HECSI score, a clinician-assessed measure of disease severity, were also greater with delgocitinib. At week 12, the mean CFB in HECSI score was –67.6 percentage points (standard error = 3.37) in the delgocitinib group and –51.5 (standard error = 3.36) in the alitretinoin group, resulting in a between-group difference of –16.1 points (95% CI, –23.28 to –8.86; P < 0.001). At week 24, this difference increased further to –24.5 points (95% CI, –32.55 to –16.36; P < 0.001), favouring delgocitinib.
The proportion of patients experiencing a HECSI-90 response at week 12 was also higher in the delgocitinib group (38.6%) than in the alitretinoin group (26.0%), for a difference of 12.6% (95% CI, 4.34% to 20.78%; P = 0.003). Additionally, the mean change in HESD itch score at week 12 was greater in the delgocitinib group (–3.0 points) than in the alitretinoin group (–2.4 points), with a mean difference of –0.7 (95% CI, –1.12 to –0.20; P = 0.005), indicating greater symptom relief with topical treatment.
Table 19: Key Efficacy Outcomes of the DELTA FORCE Trial
Variable | Delgocitinib 20 mg/g b.i.d. (N = 254) | Alitretinoin 30 mg q.d. (N = 259) |
|---|---|---|
IGA-CHE treatment success at week 12a | ||
Patients contributing to the analysis, N | 250 | 253 |
Patients experiencing IGA-CHE treatment success at week 12, n (%) | 68 (27.2) | 42 (16.6) |
Difference, % (95% CI) | 10.6 (3.31 to 17.87) | |
P value | 0.004 | |
Change from baseline in HECSI at week 12 | ||
Patients contributing to the analysis, N | 249 | 250 |
Change from baseline in HECSI at week 12, percentage points, mean (SE) | –67.6 (3.37) | –51.5 (3.36) |
Difference (95% CI) | –16.1 (–23.28 to –8.86) | |
P value | < 0.001 | |
Change from baseline in HECSI at week 24b | ||
Patients contributing to the analysis, N | 249 | 250 |
Change from baseline in HECSI at week 24, percentage points, mean (SE) | –69.6 (3.78) | –45.1 (3.77) |
Difference (95% CI) | –24.5 (–32.55 to –16.36) | |
P value | < 0.001 | |
HECSI-90 at week 12a | ||
Patients contributing to the analysis, N | 249 | 250 |
Patients experiencing HECSI-90 at week 12, n (%) | 96 (38.6) | 65 (26.0) |
Difference, % (95% CI) | 12.6 (4.34 to 20.78) | |
P value | 0.003 | |
Change from baseline in HESD itch score at week 12b | ||
Patients contributing to the analysis, N | 238 | 238 |
Change from baseline in HESD itch score at week 12, mean (SE) | –3.0 (0.22) | –2.4 (0.21) |
Difference (95% CI) | –0.7 (–1.12 to –0.20) | |
P value | 0.005 | |
b.i.d. = twice a day; CI = confidence interval; HECSI = Hand Eczema Severity Index; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; q.d. = every day; SE = standard error.
aBased on a Cochran-Mantel-Haenszel test stratified by region and hyperkeratotic or nonhyperkeratotic subtype.
bBased on an analysis of covariance model adjusted for region, hyperkeratotic or nonhyperkeratotic subtype, and tool score baseline value.
Source: DELTA FORCE Clinical Study Report. 31
In the DELTA FORCE trial, the results of all prespecified sensitivity analyses for the primary end point CFB in HECSI score at week 12 were consistent with the primary analysis results.
Health-Related Quality of Life
Vehicle-Controlled Studies
In both the DELTA 1 and DELTA 2 trials, treatment with delgocitinib was associated with greater improvements in quality-of-life outcomes than vehicle cream at week 16, as measured by the HEIS and the DLQI as shown in Table 20.
The mean CFB in HEIS score was –1.5 in both trials for the delgocitinib groups, compared to –0.8 and –0.6 in the vehicle cream groups in the DELTA 1 and DELTA 2 trials, respectively. The between-group difference in the DELTA 1 trial was –0.6 (95% CI, –0.8 to –0.5; P < 0.001) and in the DELTA 2 trial was –0.8 (95% CI, –1.0 to –0.6; P < 0.001), favouring delgocitinib.
A 4-point or greater improvement in DLQI, a commonly accepted threshold for meaningful improvement, was reported in 74.4% of patients treated with delgocitinib in the DELTA 1 trial, compared to 50.0% of those receiving vehicle cream (difference = 24.5%; 95% CI, 15.0% to 33.9%; P < 0.001). Similar results were observed in the DELTA 2 trial (72.2% versus 45.8%; difference = 26.4%; 95% CI, 17.0% to 35.9%; P < 0.001).
The mean CFB in DLQI scores was also more favourable in the delgocitinib groups: –7.6 versus –3.9 in the DELTA 1 trial (difference = –3.6; 95% CI, –4.7 to –2.6; P < 0.001) and –7.0 versus –3.1 in the DELTA 2 trial (difference = –3.9; 95% CI, –5.0 to –2.8; P < 0.001), reflecting clinically meaningful improvements in dermatology-related quality of life.
Table 20: Key Health-Related Quality of Life Outcomes of the DELTA 1 and DELTA 2 Trials, Week 16
Variable | DELTA 1 | DELTA 2 | ||
|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g b.i.d. (N = 314) | Vehicle cream (N = 159) | |
Change from baseline in DLQI | ||||
Patients contributing to the analysis, N | 321 | 158 | 310 | 159 |
Change from baseline in DLQI, mean (SE) | –7.6 (0.3) | –3.9 (0.4) | –7.0 (0.3) | –3.1 (0.5) |
Difference (95% CI) | –3.6 (–4.7 to –2.6) | –3.9 (–5.0 to –2.8) | ||
P value | < 0.001a | < 0.001a | ||
Change from baseline in HEIS score | ||||
Patients contributing to the analysis, N | 321 | 158 | 310 | 159 |
Change from baseline in HEIS score, mean (SE) | –1.5 (0.1) | –0.8 (0.1) | –1.5 (0.1) | –0.6 (0.1) |
Difference (95% CI) | –0.6 (–0.8 to –0.5) | –0.8 (–1.0 to –0.6) | ||
P value | < 0.001a | < 0.001a | ||
DLQI reduction ≥ 4 points | ||||
Patients contributing to the analysis, N | 305 | 148 | 299 | 153 |
Patients experiencing DLQI reduction ≥ 4 points, n (%) | 227 (74.4) | 74 (50.0) | 216 (72.2) | 70 (45.8) |
Difference, % (95% CI) | 24.5 (15.0 to 33.9) | 26.4 (17.0 to 35.9) | ||
P value | < 0.001b | < 0.001b | ||
b.i.d. = twice a day; CI = confidence interval; DLQI = Dermatology Life Quality Index; HEIS = Hand Eczema Impact Scale; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; SE = standard error.
aBased on an analysis of covariance model adjusted for region, baseline IGA-CHE score, and tool score baseline value.
bBased on a Cochran-Mantel-Haenszel test stratified by region and baseline IGA-CHE score.
Sources: DELTA 1 and DELTA 2 Clinical Study Reports.29,30
Active-Controlled Study
In the DELTA FORCE trial, delgocitinib resulted in greater improvements in HRQoL than alitretinoin at both week 12 and week 24. At week 24, the mean CFB in HEIS score was –1.3 with delgocitinib versus –0.8 with alitretinoin (difference = –0.5; 95% CI, –0.7 to –0.3; P < 0.001), and 71.2% of patients receiving delgocitinib experienced at least a 4-point improvement in DLQI compared to 47.6% with alitretinoin (difference = 23.6%; 95% CI, 14.8% to 32.5%; P < 0.001). The mean change in DLQI score was also greater with delgocitinib (–7.1 versus –4.6; difference = –2.5; 95% CI, –3.7 to –1.4; P < 0.001). The results were consistent at week 12, although all HRQoL outcomes were classified as exploratory with nominal P values due to lack of multiplicity adjustment.
Table 21: Key Health-Related Quality of Life Outcomes of the DELTA FORCE Trial
Variable | Week 12 | Week 24 | ||
|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 254) | Alitretinoin 30 mg q.d. (N = 259) | Delgocitinib 20 mg/g b.i.d. (N = 254) | Alitretinoin 30 mg q.d. (N = 259) | |
Change from baseline in HEIS score | ||||
Patients contributing to the analysis, N | 231 | 237 | 231 | 237 |
Change from baseline in HEIS score, mean (SE) | –1.4 (0.1) | –1.1 (0.1) | –1.3 (0.1) | –0.8 (0.1) |
Difference (95% CI) | –0.3 (–0.5 to –0.1) | –0.5 (–0.7 to –0.3) | ||
P value | 0.001a,b | < 0.001a,b | ||
Patients who experienced DLQI reduction ≥ 4 points | ||||
Patients contributing to the analysis, N | 219 | 229 | 219 | 229 |
Patients who experienced DLQI reduction ≥ 4 points, n (%) | 158 (72.1) | 129 (56.3) | 156 (71.2) | 109 (47.6) |
Difference, % (95% CI) | 15.8 (7.1 to 24.5) | 23.6 (14.8 to 32.5) | ||
P value | < 0.001b,c | < 0.001b,c | ||
Change from baseline in DLQI | ||||
Patients contributing to the analysis, N | 230 | 236 | 230 | 236 |
Change from baseline in DLQI, mean (SE) | –7.5 (0.5) | –5.8 (0.5) | –7.1 (0.5) | –4.6 (0.5) |
Difference (95% CI) | –1.8 (–2.8 to –0.7) | –2.5 (–3.7 to –1.4) | ||
P value | < 0.001a,b | < 0.001a,b | ||
b.i.d. = twice a day; CI = confidence interval; DLQI = Dermatology Life Quality Index; HEIS = Hand Eczema Impact Scale; q.d. = every day; SE = standard error.
aBased on an analysis of covariance model adjusted for region, hyperkeratotic or nonhyperkeratotic subtype, and tool score baseline value.
bExploratory end point; P value is considered nominal as no adjustments for multiple testing were conducted.
cBased on a Cochran-Mantel-Haenszel test stratified by region and hyperkeratotic or nonhyperkeratotic subtype.
Source: DELTA FORCE Clinical Study Report.31
Harms
The DELTA 1, DELTA 2, and DELTA FORCE trials conducted safety analyses based on the safety analysis set. AEs were coded using the Medical Dictionary for Regulatory Activities, version 24.0. In the DELTA 1 and DELTA 2 trials, AEs were evaluated from the time the patient signed the informed consent form until week 16 (or week 18 for patients not participating in the open-label extension trial [DELTA 3 trial]). In the DELTA FORCE trial, AEs were evaluated from the signing of the informed consent form until the end of the trial (defined as the last visit in the trial [the early termination visit, the nominal week 24 visit, or the last follow-up visit, whichever came last]). For all DELTA trials, AESIs were prespecified in the protocol and included the following: eczema herpeticum, deep vein thrombosis, and pulmonary embolism.
The key harms are described in Table 22.
Table 22: Key Harms Data in the DELTA 1, DELTA 2, and DELTA FORCE Trials
Adverse events | DELTA 1 | DELTA 2 | DELTA FORCE | |||
|---|---|---|---|---|---|---|
Delgocitinib 20 mg/g b.i.d. (N = 325) | Vehicle cream (N = 162) | Delgocitinib 20 mg/g b.i.d. (N = 314) | Vehicle cream (N = 159) | Delgocitinib 20 mg/g b.i.d. (N = 254) | Alitretinoin 30 mg q.d. (N = 259) | |
Most common adverse events (≥ 3% of patients in either group), n (%) | ||||||
≥ 1 adverse event | 147 (45.2) | 82 (50.6) | 143 (45.5) | 71 (44.7) | 125 (49.4) | 188 (76.1) |
COVID-19 | 35 (10.8) | 14 (8.6) | 36 (11.5) | 20 (12.6) | 5 (2.0) | 9 (3.6) |
Nasopharyngitis | 23 (7.1) | 14 (8.6) | 21 (6.7) | 10 (6.3) | 30 (11.9) | 34 (13.8) |
Headache | 9 (2.8) | 4 (2.5) | 19 (6.1) | 9 (5.7) | 10 (4.0) | 80 (32.4) |
Nausea | 3 (0.9) | 1 (0.6) | 0 | 0 | 1 (0.4) | 14 (5.7) |
Hand dermatitis | 1 (0.3) | 7 (4.3) | 3 (1.0) | 6 (3.8) | 2 (0.8) | 5 (2.0) |
Pharyngitis | 2 (0.6) | 0 | 3 (1.0) | 5 (3.1) | 4 (1.6) | 3 (1.2) |
Upper respiratory tract infection | 4 (1.2) | 1 (0.6) | 5 (1.6) | 0 | 6 (2.4) | 8 (3.2) |
Urinary tract infection | 2 (0.6) | 3 (1.9) | 4 (1.3) | 2 (1.3) | 1 (0.4) | 10 (4.0) |
Dry skin | 2 (0.6) | 1 (0.6) | 0 | 0 | 3 (1.2) | 9 (3.6) |
Erythema | 1 (0.3) | 0 | 1 (0.3) | 0 | 1 (0.4) | 9 (3.6) |
Lip dry | 0 | 0 | 0 | 0 | 0 | 8 (3.2) |
Hypercholesterolemia | 5 (1.5) | 3 (1.9) | 0 | 0 | 0 | 9 (3.6) |
Serious adverse events, n (%) | ||||||
Patients with ≥ 1 serious adverse events | 6 (1.8) | 3 (1.9) | 5 (1.6) | 3 (1.9) | 5 (2.0) | 12 (4.9) |
Adverse events leading to discontinuation of treatment, n (%) | ||||||
Patients who stopped treatment | 2 (0.6) | 6 (3.7) | 1 (0.3) | 5 (3.1) | 3 (1.2) | 25 (10.1) |
Headache | 0 | 0 | 0 | 0 | 0 | 11 (4.5) |
Nausea | 0 | 0 | 0 | 0 | 0 | 3 (1.2) |
Hand dermatitis | 0 | 2 (1.2) | 0 | 3 (1.9) | 0 | 2 (0.8) |
Deaths, n (%) | ||||||
Patients who died | 0 | 0 | 0 | 0 | 0 | 0 |
Adverse events of special interest, n (%) | ||||||
Patients who experienced any AESI | 0 | 0 | 0 | 0 | 0 | 1 (0.4) |
Deep vein thrombosis | 0 | 0 | 0 | 0 | 0 | 1 (0.4) |
b.i.d. = twice a day; q.d. = every day.
Sources: DELTA 1, DELTA 2, and DELTA FORCE Clinical Study Reports.29-31
Adverse Events
In the DELTA 1 and DELTA 2 trials, the incidence of any AE was similar between treatment groups. In the DELTA 1 trial, 45.2% of patients receiving delgocitinib experienced at least 1 AE, compared to 50.6% in the vehicle cream group. In the DELTA 2 trial, AEs occurred in 45.5% of patients in the delgocitinib group and 44.7% of patients in the vehicle cream group. Most AEs were mild or moderate in intensity. The most common AEs (occurring in ≥ 5% of patients in either group) were COVID-19 and nasopharyngitis. Headache occurred in 6.1% of patients treated with delgocitinib in the DELTA 2 trial, slightly higher than in the DELTA 1 trial (2.8%).
In the DELTA FORCE trial, the overall incidence of AEs was lower in the delgocitinib group (49.4%) than in the alitretinoin group (76.1%), despite a similar treatment duration of up to 24 weeks. The most frequently reported AEs in the alitretinoin group were headache (32.4%), nasopharyngitis (13.8%), and nausea (5.7%). In contrast, delgocitinib was associated with substantially lower rates of these events: headache (4.0%), nasopharyngitis (11.9%), and nausea (0.4%). Other AEs, such as dry skin, erythema, lip dryness, and hypercholesterolemia, occurred more frequently in the alitretinoin group.
Serious AEs
SAEs were infrequent across all 3 studies and balanced between treatment arms in the DELTA 1 and DELTA 2 trials. In the DELTA 1 trial, SAEs occurred in 1.8% of patients treated with delgocitinib and 1.9% of patients treated with vehicle cream. In the DELTA 2 trial, SAEs were reported in 1.6% and 1.9% of patients in the delgocitinib and vehicle cream groups, respectively. Most events were isolated, with no clear pattern, and all were resolved or resolving by the study’s end. No SAE led to major safety concerns.
In the DELTA FORCE trial, SAEs were more common in the alitretinoin group (4.9%) than in the delgocitinib group (2.0%). Most SAEs were single events, but 1 instance of increased blood potassium in the delgocitinib group was determined to be a lab artifact. No SAE in any group was associated with serious long-term harm.
Discontinuation Due to AEs
Treatment discontinuations due to AEs were rare with delgocitinib across all studies and more frequent with vehicle cream or alitretinoin. In the DELTA 1 trial, 0.6% of patients treated with delgocitinib discontinued treatment due to AEs, compared to 3.7% in the vehicle cream group. In the DELTA 2 trial, the corresponding rates were 0.3% and 3.1%. Hand dermatitis was the most common AE leading to discontinuation in the vehicle cream groups.
In the DELTA FORCE trial, 1.2% of patients in the delgocitinib group discontinued treatment due to AEs versus 10.1% in the alitretinoin group. Headache (4.5%) and nausea (1.2%) were the most frequently reported AEs leading to discontinuation in the alitretinoin group, consistent with its known safety profile.
AEs of Special Interest (Notable Harms)
No AESIs occurred in the DELTA 1 or DELTA 2 trial. In the DELTA FORCE trial, 1 patient (0.4%) in the alitretinoin group experienced an AESI: a postoperative deep vein thrombosis that resolved with treatment. No AESIs were reported in the delgocitinib group in any of the studies.
Mortality
No deaths were reported during the conduct of the DELTA 1, DELTA 2, or DELTA FORCE trials.
Critical Appraisal
Internal Validity
The DELTA 1 and DELTA 2 trials demonstrated good internal validity overall, with low risk of bias across most outcomes evaluated in this review. Randomization and allocation concealment were appropriately conducted using centralized methods. Baseline characteristics were well balanced across treatment arms, and the double-blind design minimized the risk of performance and detection bias for both clinician-assessed and patient-reported outcomes. The primary and key secondary outcomes were assessed using several outcome measures (e.g., IGA-CHE, HECSI, HESD, DLQI). The IGA-CHE and HEIS instruments were developed by the sponsor specifically for CHE and validated internally using analyses from the DELTA trials; however, these instruments lack external validation. In contrast, HECSI and DLQI are well-established, externally developed tools with demonstrated reliability and validity across dermatological conditions, including hand eczema. The HESD itch and pain scales, while based on widely used numeric rating scale formats, were developed by the sponsor and validated within the DELTA program; their stand-alone external validation is unclear. This blend of novel and established measures supports internal validity but limits the generalizability of some newer tools without broader external validation.
Statistical analyses followed the ITT principle. Key outcomes were adjusted for relevant covariates, and control for multiplicity was implemented through hierarchical testing. Data imputation for missing values was appropriate — nonresponse imputation for binary outcomes and mixed model for repeated measures or WOCF for continuous end points — with sensitivity analyses supporting the robustness of the findings. Although some patient-reported outcomes are prone to missingness, in the DELTA 1 and 2 trials the proportion of missing data was low and balanced across arms, and outcome measurement methods were considered valid. Few SAEs and AESIs were reported, leading to imprecision, but no concerns were raised about the validity of how these events were collected or adjudicated.
In contrast, the DELTA FORCE trial was associated with some concerns of risk of bias. Although randomization was adequately conducted and baseline characteristics were balanced, the trial was not double blinded; participants and treating physicians were aware of treatment assignment (topical versus oral), raising concerns about deviations from intended interventions, particularly in patient adherence and tolerability-driven dose reductions. While outcome assessors were blinded and outcome measures were objective and structured, the open-label design may have influenced the reporting of AEs and patient-reported outcomes. Importantly, more patients in the alitretinoin group (12 of 259 [4.6%]) did not receive the intervention than in the delgocitinib arm (1 of 254 [0.4%]). Furthermore, differential dropout was a concern, with 35.9% of patients in the alitretinoin arm discontinuing treatment, compared to 13.4% in the delgocitinib arm, mainly due to AEs and lack of efficacy. This imbalance in attrition introduces a risk of biased estimation of treatment effects (against alitretinoin and in favour of delgocitinib), particularly for later time points and subjective outcomes such as DLQI and HESD scores. Although ITT and per-protocol analyses were performed, and imputation methods (e.g., WOCF, nonresponse imputation) were applied consistently, the large differential in missing data may have influenced results.
No major issues were identified with statistical power, control for multiplicity, or interim analyses across the 3 trials. Subgroup analyses were prespecified and exploratory, with no statistical interaction testing reported. The clinical instruments used (e.g., DLQI, HECSI, IGA-CHE) were validated, and the MIDs applied for interpretation were appropriate and consistent with published literature.
External Validity
The DELTA 1, DELTA 2, and DELTA FORCE trials enrolled adult patients with moderate to severe CHE, consistent with the Health Canada–approved indication for delgocitinib. Baseline demographics, such as age, sex, and disease duration, were representative of patients seen in practice in Canada. However, the study populations were predominantly white and recruited primarily from European sites, with limited North American representation. Patients with significant comorbidities or recent systemic immunosuppressive therapy were excluded, potentially limiting generalizability to individuals with more complex disease. Importantly, the outcomes assessed — including IGA-CHE treatment success, HECSI, HESD, and DLQI — were clinically relevant, validated, and meaningful to patients and clinicians, with follow-up durations sufficient to capture short-term efficacy and harms (16 weeks in the DELTA 1 and DELTA 2 trials; 24 weeks in the DELTA FORCE trial), according to the clinical experts consulted by CDA-AMC.
The intervention (delgocitinib cream 20 mg/g twice a day) and comparator regimens were aligned with practice in Canada. The cream was applied at home without the need for special devices or training, supporting its use in real-world outpatient settings. In the DELTA FORCE trial, the comparator (oral alitretinoin 30 mg) was administered per Health Canada guidelines, though the irreversible dose reductions used in the trial may differ from real-world titration flexibility. Background care, including skin moisturizers, was standardized and similar to standards in Canada, although patients in trials likely received more frequent follow-up and monitoring. Overall, the trial findings are considered applicable to the intended population living in Canada, with modest limitations related to geography, trial intensity, and the exclusion of certain subgroups.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as “very uncertain.”
For RCTs: Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
For the GRADE assessments, findings from the DELTA 1 and DELTA 2 trials were considered together and summarized narratively per outcome because these studies were similar in population, interventions, design, and outcome measures.
Afterwards, the DELTA FORCE trial was assessed individually because it represents a different PICO (population, intervention, comparison, and outcome) question (i.e., delgocitinib versus alitretinoin).
Results of GRADE Assessments
Vehicle-Controlled Studies (Delgocitinib Versus Vehicle Cream)
Table 2 presents the GRADE summary of findings for delgocitinib compared to vehicle cream, based on the DELTA 1 and DELTA 2 trials. The certainty of the evidence was rated high for all efficacy outcomes. Although differences in effect estimates were observed between the DELTA 1 and DELTA 2 trials, these were likely attributable to differences in patients’ baseline risk profiles. Importantly, both the point estimates and CIs indicated a meaningful treatment effect; therefore, the review team did not rate down the effect estimate for inconsistency. The outcomes related to SAEs and AESIs were rated as moderate certainty, primarily due to the small number of events and lack of established thresholds for judging imprecision.
Active-Controlled Study (Delgocitinib Versus Alitretinoin)
Table 3 presents the GRADE summary of findings for delgocitinib compared to the active control, alitretinoin, based on the DELTA FORCE trial. For this body of evidence, the certainty for all outcomes was rated as moderate, due to study limitations related to risk of bias, particularly concerning deviations from intended interventions and missing outcome data. The results were considered precise, and no concerns were identified for other GRADE domains.
Long-Term Extension Studies
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
The DELTA 3 trial was a phase III, open-label, multisite extension phase of the DELTA 1 and DELTA 2 trials conducted to assess the long-term safety and efficacy of twice-daily applications of delgocitinib cream 20 mg/g as needed (based on IGA-CHE score) in patients who completed 1 of the 2 pivotal phase III trials with delgocitinib cream 20 mg/g or vehicle cream. The extension phase started in August 2021 and ended in September 2023. The DELTA 3 trial included a screening period of up to 4 weeks (week –4 to week 0) conducted during the parent DELTA 1 and DELTA 2 trials, followed by a treatment period of 36 weeks. Together, the parent trials and the extension trial (DELTA 3 trial) provided 52 weeks of efficacy and safety data. The baseline visit (day 1, week 0) of this extension trial coincided with the end-of-treatment visit (week 16) of the parent trials. During the treatment period, patients attended site visits every 4 weeks, until week 36. At the end of treatment, patients attended a safety follow-up of 2 weeks (week 36 to week 38).45 The efficacy end points included in this section are IGA-CHE treatment success, HECSI score at week 36, HECSI-75 at week 36, HECSI-90 at week 36, HESD itch reduction of at least 4 points at week 36, HESD pain reduction of at least 4 points at week 36, and HEIS score over time. Long-term harm outcomes (AEs, SAEs, AESIs) were also included in this report. The DELTA 3 study design is presented in Figure 7.
Figure 7: DELTA 3 Study Design
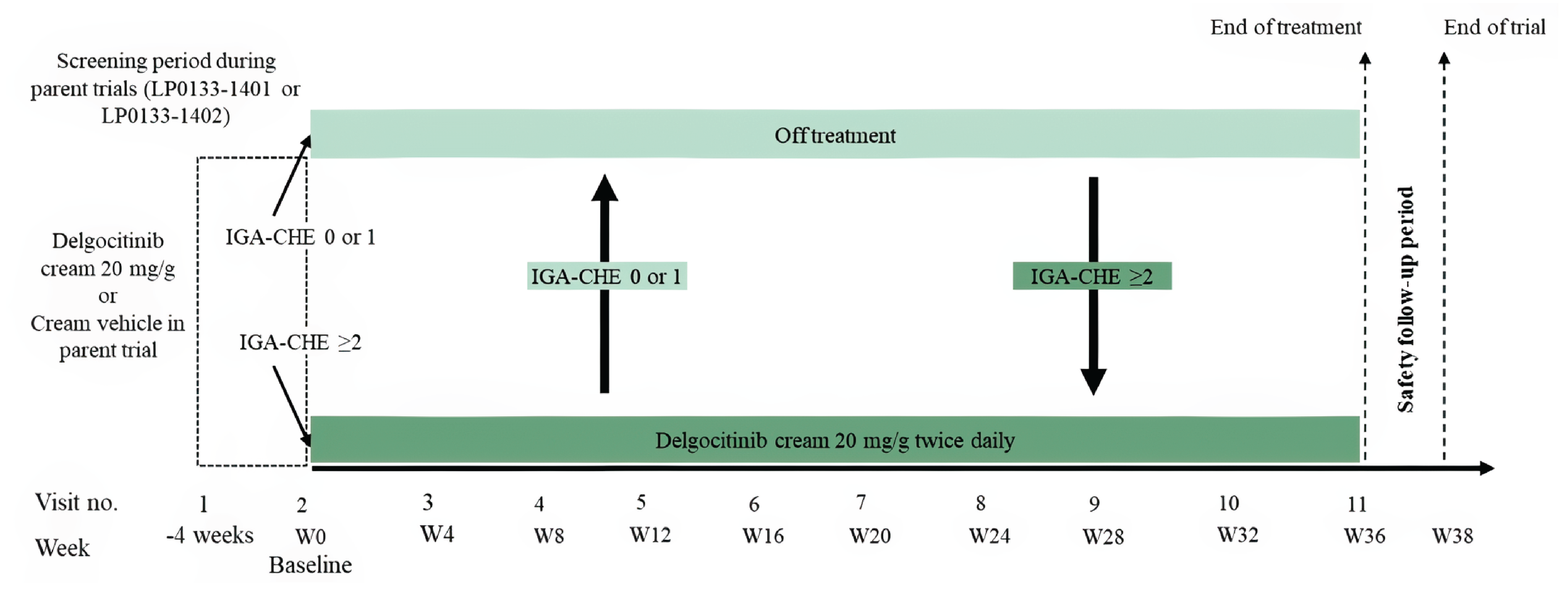
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; W = week.
Source: DELTA 3 Clinical Study Report.45
Populations
Patients eligible for the DELTA 3 extension trial were those who completed the treatment period and complied with the clinical trial protocol in the parent DELTA 1 or DELTA 2 trials. Participants who discontinued treatment with delgocitinib early, used rescue medication, or experienced any AE during participation in the parent trial that precluded further treatment with delgocitinib were not eligible to participate in the DELTA 3 trial.45
Interventions
From baseline to week 36, patients were treated on an as-needed basis with delgocitinib cream 20 mg/g twice a day. At any time during the trial, if a patient had an IGA-CHE score of 2 or more, the investigator dispensed delgocitinib cream and instructed the patient to start treatment with twice-daily applications. When the patient experienced an IGA-CHE score of 0 (clear) or 1 (almost clear), the investigator instructed the patient to stop treatment. If the patient experienced worsening of CHE signs and symptoms between scheduled visits, an unscheduled visit was planned as soon as possible to decide if treatment with delgocitinib should be started. Similarly, once symptoms resolved, the patient visited the study site as soon as possible (either at a planned visit or for an unscheduled visit) so that the investigator could assess if treatment should be stopped. Concomitant medications and procedures surrounding the use of rescue therapy during the DELTA 3 trial were the same as defined for the parent DELTA 1 and DELTA 2 trials.45
Outcomes
The primary outcome of the DELTA 3 extension study was the number of TEAEs from baseline up to week 38. TEAEs encompassed drug-related AEs, AEs categorized by severity, SAEs, AEs leading to discontinuation of the study drug, and death. Long-term efficacy outcomes, including IGA-CHE score and HECSI score, were reported as secondary outcomes; HRQoL outcomes, such as HESD itch score and DLQI score, were reported as tertiary outcomes for the extension phase.45
Statistical Analysis
The safety analysis set, defined as all enrolled patients, was used for the analysis of all end points for the open-label safety trial in the DELTA 3 trial. The overall safety profile was presented as the number and percentage of all participants treated with delgocitinib who reported TEAEs, related TEAEs, AESIs, serious TEAEs, and TEAEs leading to treatment and study discontinuation. AEs were allocated to treatment status (on or off treatment) based on the onset day of the AE. If the onset day was an on-treatment day, excluding days of treatment reinitiation, the AE was assigned to “on treatment.” If the onset day was a day of treatment reinitiation, the AE was assigned to “off treatment” if the AE originated from a safety assessment performed at the visit; otherwise, it was assigned to “on treatment.” If the onset day was an off-treatment day, the AE was assigned to “off treatment.” Additionally, the number and percentage of participants treated with delgocitinib reporting each TEAE experienced by 5% or more of the treated participants were summarized.
IGA-CHE, HECSI, HESD and DLQI scores were summarized based on observed data, with no imputation of missing values. Descriptive statistics were used to summarize all measures. These measures were grouped by parent trial treatment. For the proportion of patients experiencing an IGA-CHE score of 0 or 1, HECSI-75, HECSI-90, or a reduction in HESD itch or pain scores of at least 4 points from baseline in the parent trial, patients who discontinued treatment with the investigational medicinal product before week 36, who initiated rescue treatment, or who withdrew from the trial were imputed as not experiencing a response. Baseline measurements were defined as the latest available observation at or before the baseline visit (day 1), except for HECSI-75 or HECSI-90 and reduction in HESD itch or pain scores of at least 4 points, because the baseline values for these outcomes were derived from the baseline value in the parent trial (DELTA 1 or DELTA 2 trials).45
Results
Patient Disposition
Patient disposition for the DELTA 3 trial by parent trial treatment is presented in Table 23. Of the 810 patients screened from both trials, 801 patients were considered eligible to participate in the DELTA 3 trial and were therefore included in the patient safety analysis. From the DELTA 1 trial, 427 out of 487 patients (87.7%) were transferred to the extension trial, and from the DELTA 2 trial, 374 out of 473 patients (79.1%) were transferred to the extension trial. Among these 801 patients, 560 were included in the previous delgocitinib group and 241 in the previous vehicle cream group. Eighty-eight patients (15.7%) from the previous delgocitinib group and 49 patients (20.3%) from the previous vehicle cream group discontinued treatment. A total of 664 patients completed the treatment period, 472 (84.3%) from the previous delgocitinib group and 192 (79.7%) from the previous vehicle cream group. Among the 801 patients enrolled in the extension trial, 11 patients were enrolled in error despite not meeting the eligibility criteria (1 related to exclusion criteria and 10 related to inclusion criteria). After a safety evaluation of each incident, the sponsor allowed all 10 patients whose enrolment error related to inclusion criteria to continue the extension trial. The patient whose enrolment error related to exclusion criteria had withdrawn from the trial.45
Table 23: Summary of Patient Disposition From the DELTA 3 Trial
Patient disposition | Previous delgocitinib in DELTA 1 and DELTA 2 trials (N = 560) | Previous vehicle cream in DELTA 1 and DELTA 2 trials (N = 241) |
|---|---|---|
Screened, n | 810 | |
Did not proceed beyond screening, n (%) | 9 (1.1) | |
Reason for not proceeding beyond screening, n (%) | ||
Eligibility criteria not met | 6 (0.7) | |
Patient withdrew | 2 (0.2) | |
Lost to follow-up | 1 (0.1) | |
Assigned to treatment, n (%) | 560 (100.0) | 241 (100.0) |
Discontinued treatment, n (%) | 88 (15.7) | 49 (20.3) |
Reason for discontinuation, n (%) | ||
Lack of efficacy | 38 (6.8) | 17 (7.1) |
Withdrawal by patient | 35 (6.3) | 17 (7.1) |
Lost to follow-up | 4 (0.7) | 5 (2.1) |
Adverse event | 2 (0.4) | 5 (2.1) |
Pregnancy | 3 (0.5) | 1 (0.4) |
Death | 1 (0.1) | 0 (0) |
Other | 5 (0.9) | 4 (1.7) |
Completed treatment, n (%) | 472 (84.3) | 192 (79.7) |
Completed trial, n (%) | 469 (83.8) | 192 (79.7) |
Safety, n (%) | 560 (100.0) | 241 (100.0) |
Source: DELTA 3 Clinical Study Report.45 Details included in the table are from the sponsor’s summary of clinical evidence.16
Baseline Characteristics
The baseline characteristics outlined in Table 24 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The characteristics are similar to those of the parent trials. IGA-CHE and HECSI scores at parent trial baseline were similar between the treatment groups. There were some differences in terms of IGA-CHE score at extension trial baseline between the previous delgocitinib and previous vehicle cream groups. A higher proportion of patients previously treated with delgocitinib had IGA-CHE scores of clear to mild, and a higher proportion of patients previously treated with vehicle cream had IGA-CHE scores of moderate to severe. The median HECSI score, HESD itch and pain scores, HESD score, and DLQI score had decreased baseline values in both treatment groups compared to the parent trial baseline values. The baseline values in the previous delgocitinib group were lower than in the previous vehicle cream group for all the HRQoL outcomes in the extension phase. The distribution of CHE subtypes at parent trial baseline was similar between the groups, except for vesicular hand eczema (pompholyx), which was present in 11.3% versus 5.8% of patients in the previous delgocitinib and previous vehicle cream groups, respectively. The most frequently reported CHE subtype was atopic hand eczema (reported by approximately one-third of the trial population).45
Exposure to Study Treatments
In the DELTA 3 extension trial, the total patient-years of observation was higher in the group that had previously received delgocitinib in the parent trial (378.0 patient-years) than in the group that had received vehicle cream (157.6 patient-years). The total amount of investigational medicinal product used, as well as the weekly average amount (calculated over the entire treatment period), was similar in both groups. Notably, patients spent more time on treatment than off treatment (patient-years of observation on treatment: 407.5, versus 128.1 off treatment), both in the group previously treated with delgocitinib and the group previously treated with vehicle cream in the parent trial. Overall, the number and the duration of on-treatment periods were similar between both groups. However, among all patients entering the extended trial, despite having a similar mean number of on-treatment periods, the mean duration of on-treatment periods was shorter in those who had experienced IGA-CHE treatment success at extension trial baseline than in those who had not. There were no relevant differences in treatment compliance between patients who received delgocitinib and those who received vehicle cream in the parent trials.45
Table 24: Summary of Baseline Characteristics From the DELTA 3 Trial
Characteristic | Previous delgocitinib in DELTA 1 and DELTA 2 trials 20 mg/g b.i.d. (N = 560) | Previous vehicle cream in DELTA 1 and DELTA 2 trials (N = 241) |
|---|---|---|
Sex, n (%) | ||
Female | 355 (63.4) | 157 (65.1) |
Male | 205 (36.6) | 84 (34.9) |
Age (years), median (IQR) | 46.0 (34.0 to 56.0) | 44.0 (31.0 to 55.0) |
Race, n (%) | ||
Asian | 15 (2.7) | 9 (3.7) |
American Indian or Alaska Native | 1 (0.2) | 0 |
Black or African American | 3 (0.5) | 1 (0.4) |
Multiple | 2 (0.4) | 2 (0.8) |
Native Hawaiian or Other Pacific Islander | 0 | 0 |
White | 513 (91.6) | 219 (90.9) |
Other or not reported | 26 (4.6) | 10 (4.1) |
Geographic region, n (%) | ||
North America | 113 (20.2) | 49 (20.3) |
Europe | 447 (79.8) | 192 (79.7) |
Age at onset of CHE (years), median (IQR) | 35.0 (22.0 to 49.0) | 33.0 (21.0 to 46.0) |
Duration of CHE (years), median (IQR) | 5.0 (2.0 to 12.0) | 5.0 (2.0 to 12.0) |
HESD itch score (weekly average) at extended trial baseline | ||
Patients, n | 541 | 233 |
Score, median (IQR) | 2.71 (0.71 to 5.17) | 4.86 (2.57 to 7.00) |
Patients experiencing change of ≥ 4 points, n (%) | 215 (39.7) | 144 (61.8) |
HESD pain score (weekly average) at extended trial baseline | ||
Patients, n | 541 | 233 |
Score, median (IQR) | 2.14 (0.14 to 5.14) | 4.43 (2.00 to 6.86) |
Patients experiencing change of ≥ 4 points, n (%) | 195 (36.0) | 126 (54.1) |
HESD score (weekly average) at extended trial baseline | ||
Patients, n | 541 | 233 |
Score, median (IQR) | 2.79 (0.93 to 5.43) | 4.88 (3.00 to 6.95) |
Patients experiencing change of ≥ 4 points, n (%) | 222 (41.0) | 140 (60.1) |
DLQI score at extended trial baseline | ||
Patients, n | 557 | 240 |
Score, median (IQR) | 3.0 (1.0 to 6.0) | 7.0 (2.0 to 12.0) |
Patients experiencing change of ≥ 4 points, n (%) | 240 (43.1) | 155 (64.6) |
CHE subtype main diagnosis, N (%) | ||
Atopic hand eczema | 192 (34.3) | 89 (36.9) |
Allergic contact dermatitis | 74 (13.2) | 45 (18.7) |
Hyperkeratotic eczema | 120 (21.4) | 48 (19.9) |
Irritant contact dermatitis | 111 (19.8) | 45 (18.7) |
Vesicular hand eczema (pompholyx) | 63 (11.3) | 14 (5.8) |
b.i.d. = twice a day; CHE = chronic hand eczema; DLQI = Dermatology Life Quality Index; HESD = Hand Eczema Symptom Diary; IQR = interquartile range.
Note: Racial categories used in the table are as reported in the source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: DELTA 3 Clinical Study Report.45 Details included in the table are from the sponsor’s summary of clinical evidence.
Concomitant Medications and Co-Interventions
Table 25 provides a summary of concomitant medications and rescue treatments used among patients in the DELTA 3 trial.
The use of concomitant medications was comparable between the group treated with delgocitinib in the parent trial (79.1%) and the group treated with vehicle cream in the parent trial (79.3%). The concomitant medications most frequently reported were other analgesics and antipyretics (23.0%), anti-inflammatory and antirheumatic products (22.8%), and systemic antihistamines (22.8%), across all patients from the previous treatment groups.
The majority of patients did not use any rescue treatment during the trial: rescue treatment was used by 19 patients (3.4%) who received delgocitinib cream in the parent trial and 10 patients (4.1%) who received vehicle cream in the parent trial. Dermatologicals were the most frequently used class of rescue treatment, mainly driven by dermatological preparations of corticosteroids (used by 13 patients [2.3%] who received delgocitinib cream in the parent trial and 4 patients [1.7%] who received vehicle cream in the parent trial).
Table 25: Summary of Concomitant Medications and Rescue Treatment From the DELTA 3 Trial
Preferred term | All patients (N = 801) |
|---|---|
Most commona concomitant medications, n (%) | |
Other analgesics and antipyretics | 184 (23.0) |
Anti-inflammatory and antirheumatic products | 183 (22.8) |
Systemic antihistamines | 183 (22.8) |
Lipid-modifying agents | 89 (11.1) |
Inhalants containing adrenergics | 87 (10.9) |
Drugs for peptic ulcer and gastro-esophageal reflux disease | 83 (10.4) |
Systemic hormonal contraceptives | 81 (10.1) |
Thyroid preparations | 78 (9.7) |
Viral vaccines | 75 (9.4) |
Plain corticosteroids | 73 (9.1) |
Rescue treatment, n (%) | |
Any rescue treatment | 29 (3.6) |
aAt least 10% of patients in any treatment group at Anatomical Therapeutic Chemical level 3.
Source: DELTA 3 Clinical Study Report.45 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
The summary of key efficacy outcomes from the DELTA 3 extension trial is presented in Table 26.
IGA-CHE Treatment Success From Baseline up to Week 36
Among patients who received delgocitinib cream in the parent trial and subsequently enrolled in the DELTA 3 trial, the proportion of patients with an IGA-CHE score of 0 or 1 (i.e., IGA-CHE treatment success) was similar at all visits throughout the 36-week extension trial while on the “as-needed” dosing regimen (Figure 8). Specifically, 24.6% of these patients had experienced IGA-CHE treatment success at the baseline of the extension trial and 30.0% had by the end of the extension trial (week 36). Among patients who received vehicle cream in the parent trial, the proportion of patients who experienced IGA-CHE treatment success increased over time, rising from 9.1% at the extension trial baseline to 29.5% at week 36 of the extension trial.
Figure 8: IGA-CHE Score of 0 or 1 by Visit, Parent Trial Treatment, and Baseline IGA-CHE Treatment Success
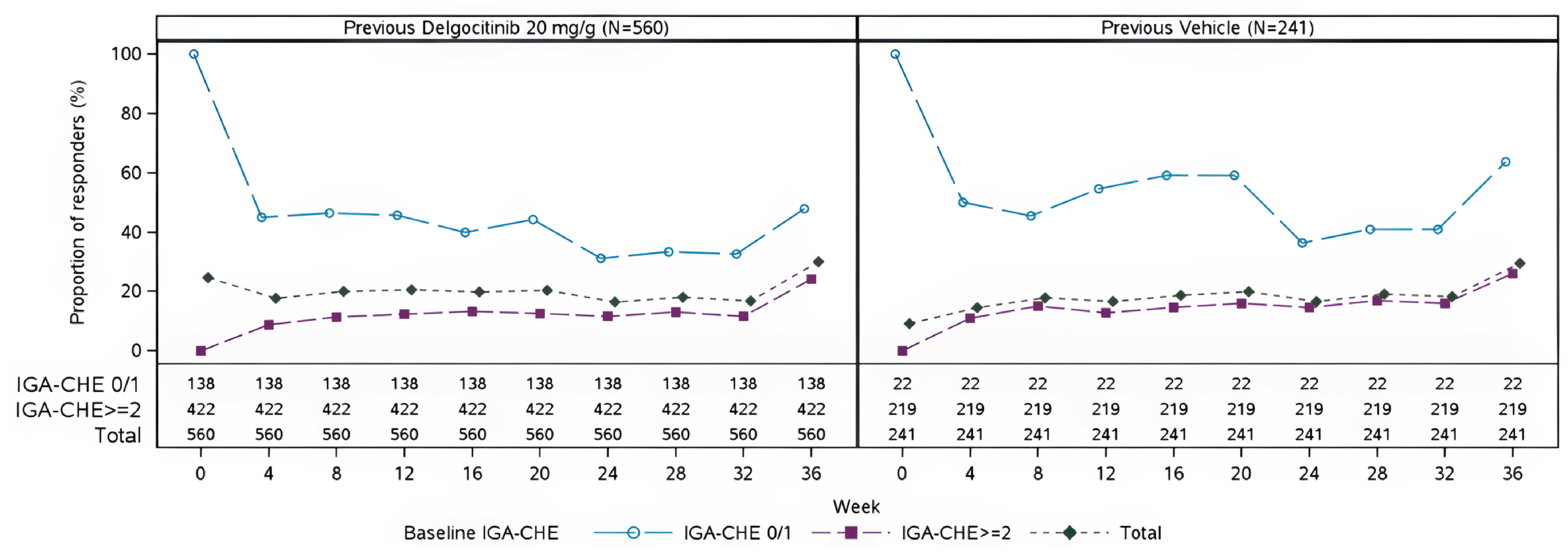
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema.
Notes: “Responders” in this figure indicates as patients who experienced an IGA-CHE score of 0 (clear) or 1 (almost clear). Patients who discontinued the investigational medicinal product, started rescue treatment, withdrew from the trial, or had missing data for other reasons are imputed as not experiencing response.
Source: DELTA 3 Clinical Study Report.45
HECSI Score at Week 36
A total of 472 patients who received delgocitinib cream and 192 patients who received vehicle cream in the parent trial had a week 36 visit. Among patients who received delgocitinib cream in the parent trial, the mean HECSI score further decreased within the first 16 weeks of the extension trial and then remained stable. The mean HECSI score for these patients was 23.9 (SD = 29.1) at the baseline of the extension trial and 14.8 (SD = 19.4) at week 36 of the extension trial. Similarly, among patients who received vehicle cream in the parent trial, the mean HECSI score decreased within the first 16 weeks of the extension trial and then remained stable. At the baseline of the extension trial, the mean HECSI score was 46.8 (SD = 46.0) for these patients; at week 36 of the extension trial, it was 16.8 (SD = 23.4).
HECSI-75 at Week 36
Among patients who received delgocitinib cream in the parent trial, the proportion of patients experiencing a HECSI-75 response in the extension trial was maintained over time. The proportion of patients who experienced a HECSI-75 response from the parent trial baseline was 51.8% at the baseline of the extension trial and 58.6% at week 36 of the extension trial. More specifically, in patients who had experienced IGA-CHE treatment success at the extension trial baseline, the proportion of experiencing a HECSI-75 response decreased within the first 4 weeks and then stabilized. In those who did not experience IGA-CHE treatment success at baseline, the proportion experiencing a HECSI-75 response increased within the first 16 weeks and then stabilized. Among patients who received vehicle cream in the parent trial, an increase in the proportion of patients experiencing a HECSI-75 response was observed within the first 4 weeks of the extension trial. The proportion of patients experiencing this response subsequently remained stable. The proportion of patients who experienced a HECSI-75 response from the baseline of the parent trial was 23.7% at the baseline of the extension trial and 51.5% at week 36 of the extension trial.
HECSI-90 at Week 36
Among patients who received delgocitinib cream in the parent trial, the proportion of patients who experienced a HECSI-90 response in the extension trial was maintained over time. The proportion of patients who experienced a HECSI-90 response from the parent trial baseline was 31.8% at the baseline of the extension trial and 36.6% at week 36 of the extension trial. More specifically, in patients who experienced IGA-CHE treatment success at the extension trial baseline, the proportion of patients who experienced a HECSI-90 response decreased within the first 4 weeks and then remained largely stable. In those who did not experience IGA-CHE treatment success at baseline, the proportion of patients experiencing a HECSI-90 response increased over time. Similarly, in patients who received vehicle cream in the parent trial, the proportion of patients who experienced a HECSI-90 response increased during the extension trial. The proportion of patients who experienced a HECSI-90 response from the baseline of the parent trial was 12.0% at extension trial baseline and 35.7% at week 36 of the extension trial.
HESD Itch Reduction of at Least 4 Points From Parent Trial Baseline at Week 36
Among patients who received delgocitinib cream in the parent trial, the proportion of patients experiencing a reduction of at least 4 points in HESD itch score from the parent trial baseline was maintained over time. The proportion of patients with a reduction of at least 4 points in HESD itch score from the parent trial baseline was 50.6% at the baseline of the extension trial and 52.4% at week 36 of the extension trial. Among patients who received vehicle cream in the parent trial, the proportion of patients experiencing a reduction of at least 4 points in HESD itch score from the parent trial baseline increased from 26.3% to 41.3% during the extension trial.
HESD Pain Reduction of at Least 4 Points From Parent Trial Baseline at Week 36
Among patients who received delgocitinib cream in the parent trial, the proportion of patients experiencing a reduction of at least 4 points in HESD pain score from the parent trial baseline was maintained over time. The proportion of patients with a reduction of at least 4 points in HESD pain score from the parent trial baseline was 51.9% at the baseline of the extension trial and 55.4% at week 36 of the extension trial. Among patients who received vehicle cream in the parent trial, the proportion of patients experiencing a reduction of at least 4 points in HESD pain score from the parent trial baseline increased from 32.3% to 43.3% during the extension trial.
HEIS Score Over Time
Among patients who received delgocitinib cream in the parent trial, the mean HEIS score slightly decreased (indicating improvement) over time in the extension trial. Among patients who received vehicle cream in the parent trial, the mean HEIS score decreased within the first 8 weeks of the extension trial and then remained stable (Figure 9).
Figure 9: HEIS Score by Visit, Parent Trial Treatment, and Baseline IGA-CHE Treatment Success — Observed Cases, Treatment Period (Safety Analysis Set)
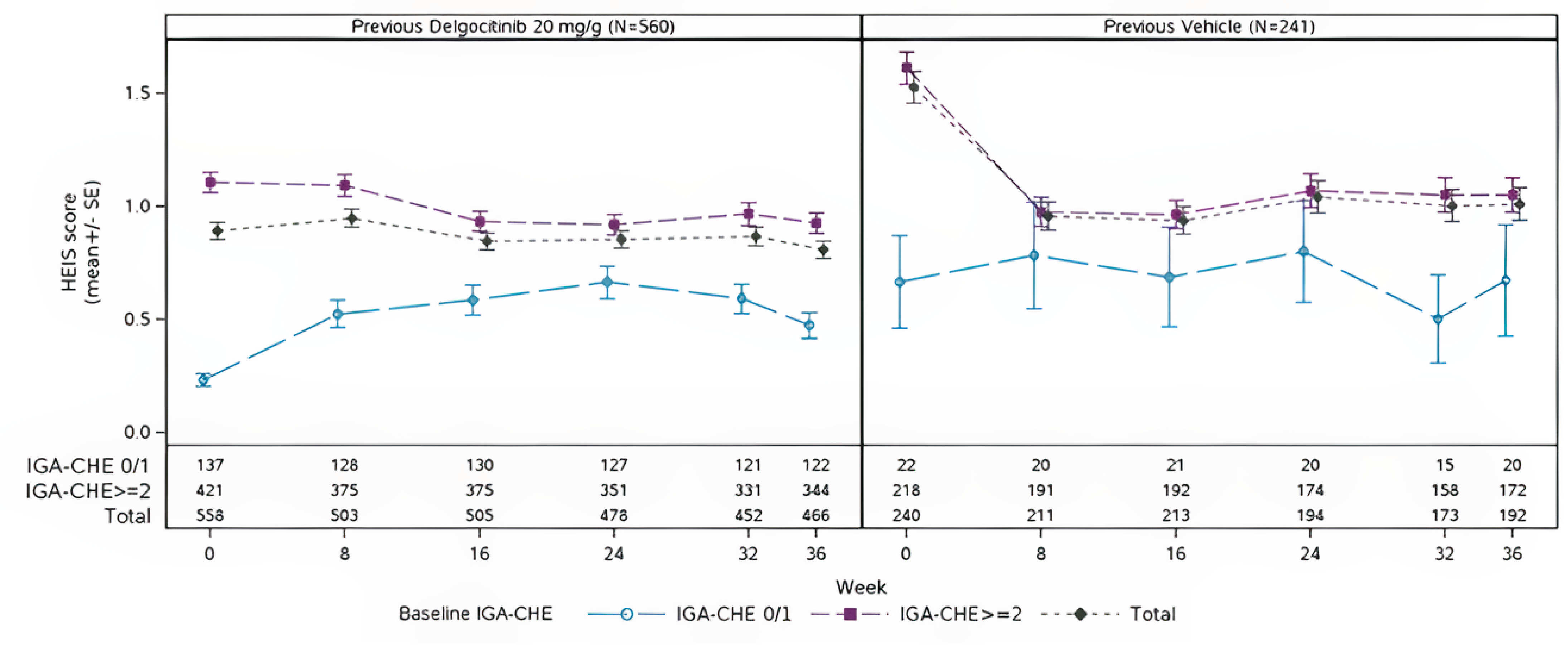
HEIS = Hand Eczema Impact Scale; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; SE = standard error.
Source: DELTA 3 Clinical Study Report.45
DLQI Score Over Time
Among patients who received delgocitinib cream in the parent trial, the mean DLQI score slightly decreased (indicating improvement) over time in the extension trial. Among patients who received vehicle cream in the parent trial, the mean DLQI score decreased within the first 8 weeks of the extension trial and then remained stable over time (Figure 10).
Figure 10: DLQI Score by Visit, Parent Trial Treatment, and Baseline IGA-CHE Treatment Success — Observed Cases, Treatment Period (Safety Analysis Set)
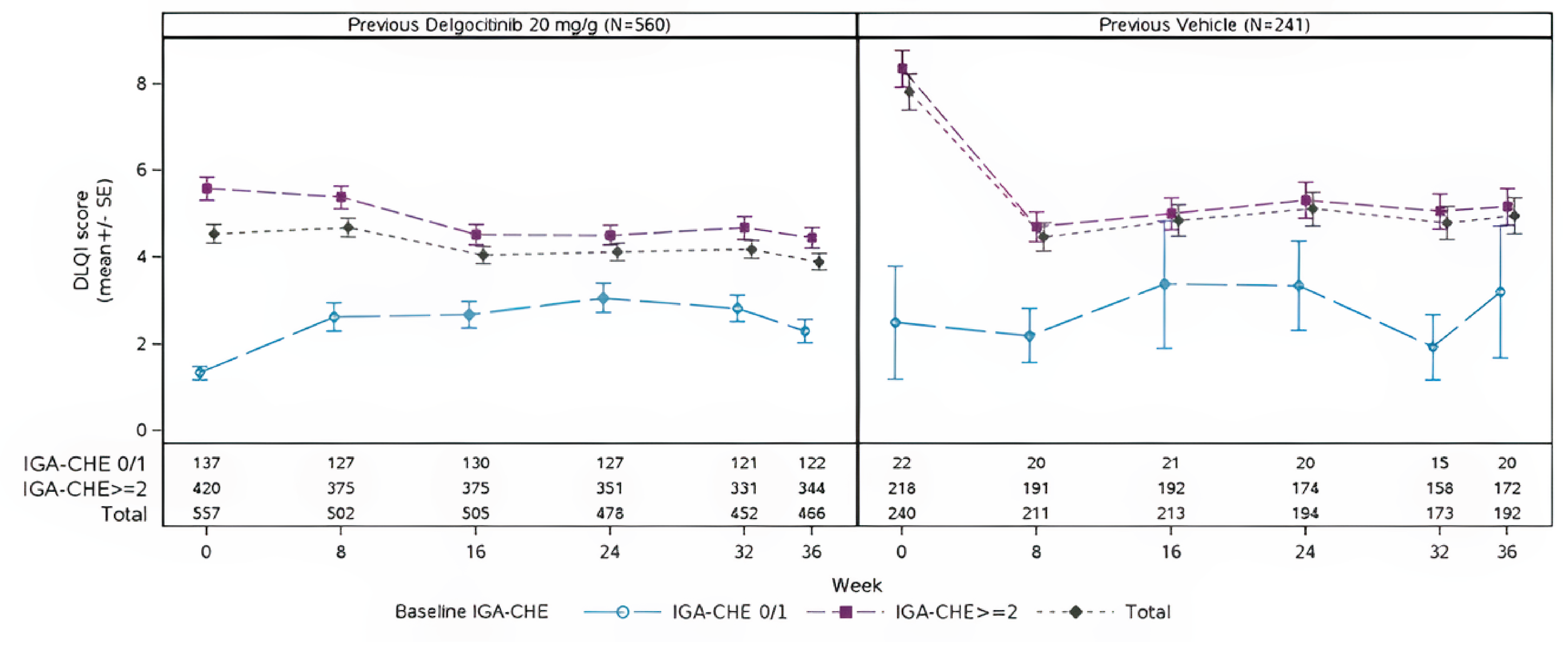
DLQI = Dermatology Life Quality Index; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; SE = standard error.
Source: DELTA 3 Clinical Study Report.45
Table 26: Summary of Key Efficacy Outcomes From the DELTA 3 Trial
Outcome | Previous delgocitinib 20 mg/g b.i.d. (N = 560) | Previous vehicle cream (N = 241) |
|---|---|---|
IGA-CHE treatment success at week 36 | ||
Patients contributing to the analysis, Na | 560 | 241 |
Extension trial baseline (week 0), n (%) | 138 (24.6) | 22 (9.1) |
Extension trial week 36, n (%) | 168 (30.0) | 71 (29.5) |
HECSI score at week 36 | ||
Patients contributing to the analysis at baseline, N | 560 | 241 |
Extension trial baseline (week 0), mean (SD) | 23.9 (29.1) | 46.8 (46.0) |
Patients contributing to the analysis at week 36, N | 472 | 192 |
Extension trial week 36, mean (SD) | 14.8 (19.4) | 16.8 (23.4) |
HECSI-75 at week 36 | ||
Patients contributing to the analysis, Na | 560 | 241 |
Extension trial baseline (week 0), n (%) | 290 (51.8) | 57 (23.7) |
Extension trial week 36, n (%) | 328 (58.6) | 124 (51.5) |
HECSI-90 at week 36 | ||
Patients contributing to the analysis, Na | 560 | 241 |
Extension trial baseline (week 0), n (%) | 178 (31.8) | 29 (12.0) |
Extension trial week 36, n (%) | 205 (36.6) | 86 (35.7) |
Patients with HESD itch score (weekly average) reduction of ≥ 4 points from parent trial baseline | ||
Patients contributing to the analysis, Na | 557 | 240 |
Extension trial baseline (week 0), n (%) | 282 (50.6) | 63 (26.3) |
Extension trial week 36, n (%) | 292 (52.4) | 99 (41.3) |
Patients with HESD pain score (weekly average) reduction of ≥ 4 points from parent trial baseline | ||
Patients contributing to the analysis, Na | 516 | 217 |
Extension trial baseline (week 0), n (%) | 268 (51.9) | 70 (32.3) |
Extension trial week 36, n (%) | 286 (55.4) | 94 (43.3) |
HEIS score change over time | ||
Patients contributing to the analysis at baseline, N | 558 | 240 |
Mean (SD) at baseline | 0.89 (0.88) | 1.52 (1.07) |
Median (IQR) at baseline | 0.56 (0.22 to 1.44) | 1.33 (0.56 to 2.44) |
Patients contributing to the analysis at week 36, N | 466 | 192 |
Mean (SD) at week 36 | 0.81 (0.82) | 1.01 (1.01) |
Median (IQR) at week 36 | 0.56 (0.11 to 1.22) | 0.67 (0.22 to 1.67) |
DLQI score change over time | ||
Patients contributing to the analysis at baseline, N | 557 | 240 |
Mean (SD) at baseline | 4.5 (5.0) | 7.8 (6.4) |
Median (IQR) at baseline | 3.0 (1.0 to 6.0) | 7.0 (2.0 to 12.0) |
Patients contributing to the analysis at week 36, N | 466 | 192 |
Mean (SD) at week 36 | 3.9 (4.1) | 5.0 (5.8) |
Median (IQR) at week 36 | 2.0 (1.0 to 6.0) | 3.0 (1.0 to 7.0) |
b.i.d. = twice a day; DLQI = Dermatology Life Quality Index; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HEIS = Hand Eczema Impact Scale; HESD = Hand Eczema Symptom Diary; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; IQR = interquartile range; SD = standard deviation.
aNumber of patients contributing to the analysis is the same for the baseline and week 36 analyses.
Source: DELTA 3 Clinical Study Report.45 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Table 27 and Table 28 present a summary of harms data from the DELTA 3 trial.
In total, 495 patients (61.8%) reported experiencing at least 1 AE, with the overall incidence of AE of any grade being comparable between the group previously treated with delgocitinib (61.1%) and the group previously treated with vehicle cream (63.5%) in the parent trials. The most common AEs (reported in ≥ 5% of patients) were COVID-19 and nasopharyngitis. There was a slightly higher rate of infections during on-treatment periods than during off-treatment periods, but this difference was not considered to be of clinical relevance. The rate of SAEs was low (3.4% in total), and similar rates were observed between on-treatment periods and off-treatment periods and between patients with different parent trial treatments. Nine patients discontinued the investigational medicinal product permanently or withdrew from the trial due to AEs, with similar rates observed between on-treatment periods and off-treatment periods and between patients with different parent trial treatments. All events leading to permanent discontinuation of the investigational medicinal product or withdrawal from the trial were nonserious, except for the fatal SAEs associated with 3 deaths. Regarding AESIs, 1 event of eczema herpeticum (neck and eyelids) was reported during an on-treatment period in a patient with a history of AD. Three deaths were reported during the trial. None of the deaths were assessed to be related to the investigational medicinal product by the investigator or the sponsor.
Table 27: Summary of Harms Results From the DELTA 3 Trial by Parent Trial Treatment (Safety Analysis Set)
AEs | Previous delgocitinib 20 mg/g b.i.d. (N = 560) | Previous vehicle cream (N = 241) |
|---|---|---|
Patients experiencing ≥ 1 AE, n (%) | 342 (61.1) | 153 (63.5) |
Patients experiencing AEs leading to withdrawal from trial or study drug discontinuation, n (%) | 3 (0.5) | 6 (2.5) |
Patients experiencing ≥ 1 SAE, n (%) | 19 (3.4) | 8 (3.3) |
Patients experiencing AEs leading to death, n (%) | 2 (0.4) | 1 (0.4) |
Most common AEs (reported in ≥ 5% of patients), n (%) | ||
COVID-19 | 95 (17.0) | 39 (16.2) |
Nasopharyngitis | 91 (16.3) | 37 (15.4) |
AE = adverse event; b.i.d. = twice a day; SAE = serious adverse event.
Source: DELTA 3 Clinical Study Report.45
Table 28: Summary of Harms Results From the DELTA 3 Trial by Treatment Status (Safety Analysis Set)
AEs | On treatment (N = 779) | Off treatment (N = 770) |
|---|---|---|
Patients experiencing ≥ 1 AE, n (%) | 443 (56.9) | 155 (20.1) |
Patients experiencing AEs leading to withdrawal from trial or study drug discontinuation, n (%) | 7 (0.9) | 2 (0.3) |
Patients experiencing ≥ 1 SAE, n (%) | 22 (2.8) | 7 (0.9) |
Patients experiencing AEs leading to death, n (%) | 1 (0.1) | 2 (0.3) |
Most common AEs (report in ≥ 5% of patients), n (%) | ||
COVID-19 | 110 (14.1) | 24 (3.1) |
Nasopharyngitis | 101 (13.0) | 37 (4.8) |
AEs of special interest, n (%) | ||
Eczema herpeticum | 1 (0.1) | — |
AE = adverse event; SAE = serious adverse event.
Source: DELTA 3 Clinical Study Report.45
Critical Appraisal
Internal Validity
The open-label extension phase design of the DELTA 3 trial may have biased the reporting of some end points because awareness of the study treatment received may have influenced the perception of improvement and/or harms by patients and clinicians, particularly for outcomes that are subjective in measurement and interpretation (e.g., HESD, HEIS, and DLQI scores and subjective AEs). The parent trial delgocitinib group would have lost anyone who experienced particular intolerance or nonresponse to delgocitinib, and the vehicle cream group might have lost patients with more severe disease who needed rescue medication and withdrew from the study. More than half the patients who initially experienced response (a baseline IGA-CHE score of 0 or 1) in both groups ceased to experience response by week 4 while off treatment in the as-needed regimen. Although a substantial proportion subsequently regained response upon reinitiation of delgocitinib (median 8 weeks; approximately 81% by the end of the treatment period), this on/off design introduces variability and possible regression to the mean, limiting the certainty with which durability of response can be interpreted.
Because all patients were taking delgocitinib during the open-label extension phase, there was no relevant randomized comparison group (i.e., for any active comparator of interest), which precludes causal conclusions. Because AEs were allocated to treatment status (on or off treatment) based on the onset day of the AE, the safety analysis loses the history and mixes new with longer-term treatment. While the assumption is that there will be no carryover effect, there may be misattribution of AEs in patients who had an event just after crossing over, especially from on treatment to off treatment. This issue raises concerns about undercounting or potentially missing a signal for AEs. In terms of HRQoL measures, 466 out of 558 patients (83.5%) from the parent trial delgocitinib group and 192 out of 240 patients (80%) from the parent trial vehicle cream group participated until week 36 for the HEIS score change measure, indicating 16.5% and 20% dropout rates in the previous delgocitinib group and previous vehicle cream group, respectively. The dropout rates were same for the DLQI score change measure. In terms of protocol deviations, 204 important deviations were reported up to the date of the data lock. Eleven patients were enrolled despite the violation of eligibility criteria, which may bias the trial results. The sponsor acknowledged that 14 important patient-level protocol deviations, which were not related to violation of eligibility criteria, were considered to impact the interpretability of the trial results for the individual patients. Additionally, the sponsor acknowledged 5 important site-level protocol deviations that were considered to impact the interpretability of the trial results, involving 34 patients in the extension phase. All these incidents of protocol deviation in different levels of the extension phase might have impacted the trial outcomes, though the direction of the potential impact is unclear.
External Validity
Because the patients who took part in the DELTA 3 trial were originally from the DELTA 1 or DELTA 2 parent studies and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label extension study. For instance, because the participants were predominantly white (approximately 91%), the results from these trials may not be generalized to other racial groups that may be commonly seen at some centres in North America and Europe. One of the eligibility criteria in the extension phase was that patients who experienced any AE during participation in the parent trial that precluded further treatment with delgocitinib were not eligible to participate in the DELTA 3 trial. This might have led to a greater share of patients who experience good tolerance of the drug being included in the extension phase, though the impact on the generalizability of the results will be small, considering only 6 patients were screened out on the basis of eligibility criteria.
Indirect Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
Although 1 head-to-head RCT (DELTA FORCE trial) has compared delgocitinib to alitretinoin — an agent currently used as second-line treatment for patients with moderate to severe CHE who do not experience response to TCS or for whom TCS are not appropriate — there remains a lack of direct comparative evidence between delgocitinib and other relevant therapeutic options. Phototherapy is commonly considered a second-line treatment option in both clinical guidelines and routine care for patients with moderate to severe CHE. Additionally, dupilumab (though not specifically approved for CHE) is frequently used off-label and is broadly reimbursed in Canada for the treatment of moderate to severe AD. According to the sponsor, this may be relevant in jurisdictions where access to phototherapy is limited or in jurisdictions where alitretinoin is not reimbursed or in patients who are ineligible for or for whom such therapies are contraindicated.
The objective of the systematic literature review (SLR) submitted by the sponsor was to identify any additional randomized trials directly comparing delgocitinib to other treatments used in practice in Canada.
Description of Indirect Comparisons
Given these gaps in direct evidence, ITCs were conducted to estimate the relative efficacy of delgocitinib versus phototherapy (PUVA) through an NMA incorporating randomized trial data and of delgocitinib versus dupilumab through a MAIC based on IPD from delgocitinib trials and aggregate data from a dupilumab study. These analyses aim to inform decision-making in the context of clinical practice in Canada and pharmacoeconomic modelling, where comparative evidence is needed to support the evaluation of treatment alternatives for moderate to severe CHE in adults.
An SLR was conducted by the sponsor that followed the PRISMA-P guidelines for systematic reviews and meta-analyses and adhered to the methodological standards outlined by the National Institute for Health and Care Excellence (NICE) technology appraisal process and the Cochrane Handbook.46-48 The search strategy, completed on October 8, 2024, included MEDLINE, Embase, and the Cochrane Library (including the Cochrane Database of Systematic Reviews and CENTRAL). To complement these electronic sources, pragmatic searches of grey literature were conducted, including reviews of bibliographies (e.g., SLRs, NMAs), clinical trial registries, conference proceedings, and health technology assessment submissions. Following de-duplication, titles and abstracts were independently screened by 2 experienced reviewers. Full-text articles were assessed for eligibility, and studies meeting predefined criteria were included in the SLR.
The eligibility criteria for the SLR were intentionally broad with respect to population, comparators, and outcomes to support potential submissions across different geographies. A narrower subset of evidence was then selected for the ITCs, as outlined in Table 29. For the purpose of this review, alitretinoin and phototherapy were considered the most relevant second-line comparators. Therefore, the ITC focused on studies involving these treatments and reporting outcomes aligned with those in the delgocitinib phase III program (DELTA 1, DELTA 2, and DELTA FORCE trials). Eligible outcomes included the proportion of patients experiencing an IGA-CHE or PGA score of 0 or 1 (end point response), IGA-CHE cumulative response, and HECSI-90 for efficacy, as well as discontinuation due to AEs and all-cause discontinuation for harms. Although the broader SLR included additional clinical and patient-reported outcomes such as HECSI-75, HECSI CFB, and DLQI CFB, these were not submitted in the ITC report because they were not used in the pharmacoeconomic analysis submitted by the sponsor.
Table 29: Study Selection Criteria and Methods for Indirect Comparisons
Characteristics | Indirect comparison |
|---|---|
Population | Adults (≥ 18 years) with moderate to severe CHEa |
Intervention | Delgocitinib: 20 mg/g, applied twice daily to the skin areas affected by CHE |
Comparator | Alitretinoin: 10 mg or 30 mg once daily Phototherapy (i.e., PUVA, UV light therapy) Cream vehicle or placebo |
Outcome | Efficacy outcomes:
For each of these efficacy outcomes, a base-case analysis was performed at 2 time points: the primary study end point and week 12 (if different from the primary study end point). Additionally, a base-case analysis was conducted at week 24 for IGA-CHE or PGA score of 0 or 1 cumulative response. Safety outcomes:
For all safety outcomes, the time points were set to the latest available in the included studies. |
Study designs | RCTs |
Publication characteristics | Studies published in English Clinical Study Reports (for delgocitinib only) |
Databases searched | Electronic bibliographical databases:
Grey literature:
|
Selection process | After removal of duplicate records, titles and abstracts were independently screened for inclusion by 2 experienced systematic reviewers, in a double-blind manner. Full texts were sought for all papers that met the inclusion criteria and were further assessed for inclusion by 2 independent reviewers. |
Data extraction process | Data extraction was performed by 1 reviewer and checked by a second reviewer. Details on study design, baseline population characteristics, interventions, time points, and outcomes of interest were extracted for each treatment arm from the included trials. |
Quality assessment | The quality of eligible trials was assessed by 1 reviewer and checked by a second reviewer using the Cochrane Risk of Bias tool. The Cochrane Risk of Bias tool addresses the following domains: random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other biases. For each domain, the risk of bias is graded as “low,” “high,” or “unclear” (if study information is insufficient to conclude).48 No studies were excluded based on study quality. |
AE = adverse event; CHE = chronic hand eczema; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; NMA = network meta-analysis; PGA = Physician’s Global Assessment; PUVA = psoralen and UVA radiation; RCT = randomized control trial; SLR = systematic literature review.
aThe target population for delgocitinib is adults with moderate to severe CHE. However, trials that recruited participants both ≥ 18 years and < 18 years were considered eligible.
Source: Delgocitinib NMA technical report.49
Indirect Comparison Analysis Methods
Two ITCs were conducted based on the evidence identified during the SLR:
an NMA to compare the relative efficacy of delgocitinib versus phototherapy
an anchored MAIC to compare the relative efficacy of delgocitinib versus dupilumab.
Due to the considerable heterogeneity between the delgocitinib and dupilumab trials identified, specifically that the dupilumab trial was conducted in a specific subgroup of patients with CHE (i.e., patients with atopic CHE subtype, rather than all forms of CHE), it was deemed not feasible to include dupilumab in the NMA. Thus, a MAIC was judged to be most appropriate. The following section describes, separately, the methods of each ITC in greater detail.
Network Meta-Analysis
Objectives
The objective of the NMA was to estimate — using a connected network of evidence derived from randomized trials — the relative efficacy and harms of delgocitinib compared to other relevant treatment options for adults with moderate to severe CHE who have not experienced response to, or are not candidates for, TCS.
Specifically, the NMA aimed to compare delgocitinib to alitretinoin, phototherapy (PUVA), or placebo or vehicle cream to inform the relative treatment effects within and across studies.
Outcomes of interest included both efficacy end points (e.g., IGA-CHE or PGA of 0 or 1, HECSI-90, and cumulative response) and AE end points (e.g., discontinuation due to AEs). The findings were intended to support the pharmacoeconomic evaluation and inform decision-making in the context in Canada, where direct evidence is limited.
Feasibility Assessment
Before conducting the NMA, a feasibility assessment was performed to determine potential sources of heterogeneity across the trials identified in the SLR and to ensure trial comparability for synthesis.
The similarity of the trial characteristics across the RCTs identified in the SLR evaluating delgocitinib or phototherapy was assessed qualitatively. The assessment of trial characteristics focused on the following:
study design characteristics (i.e., study phase, interventions, and eligibility criteria)
outcome reporting (definition and time points).
Additional steps were undertaken during the feasibility assessment, including assessment of connections, analysis of risk of unobserved variables, and consideration of specific assumptions. Specifically, several assumptions were made to indirectly compare delgocitinib to comparators of interest.
Assumptions Made in the Network Meta-Analysis
Based on the available evidence, the following key assumptions were made in the NMA:
IGA-CHE and PGA are sufficiently comparable to synthesize. While there are inherent differences between the PGA and IGA-CHE scale, notably an IGA-CHE score of 1 being more strict (barely perceptible erythema), for the purpose of estimating the relative efficacy of phototherapy compared to delgocitinib in an ITC anchored on alitretinoin, the sponsor assumed the scales to be comparable.
In the ALPHA trial, all missing patients were assumed not to experience response; therefore, it is appropriate to estimate the IGA-CHE or PGA data for the ALPHA trial using nonresponse imputation based on observed cases (i.e., participants with available data who experienced response).
Week 12 and week 16 outcomes are sufficiently similar to synthesize. The trials included in the NMAs reported results at distinct time points, ranging from 12 to 24 weeks. To facilitate comparison of different treatments in the NMAs, it was assumed that the data reported for efficacy outcomes at weeks 12 and 16 were unlikely to introduce statistically significant bias to treatment effect estimates. This assumption stemmed from the similar ORs of patients who experienced response at week 12 and week 16 in the delgocitinib trials.
Cream vehicle and placebo are sufficiently similar to synthesize in a single node. It was assumed that placebo and vehicle cream were clinically equivalent and, when these were reported in the same network, they were merged into a common placebo–vehicle cream node.
Baseline disease severity was not expected to be an effect modifier. The trials included in the NMAs recruited patients with different levels of CHE severity (i.e., moderate to severe versus severe only). To compare the efficacy of different treatment options, mixed populations (i.e., populations with both moderate and severe disease) were included in the base-case analysis, assuming that baseline severity was not an effect modifier.
Nonresponse imputation was used to harmonize outcome reporting across trials. The numbers of patients who experienced response in the ALPHA trial were presented as observed cases, meaning that the missing data in the ALPHA trial were not included in the outcome analyses. This method of treating missing data did not align with the DELTA trials, which used nonresponse imputation. To align with delgocitinib trials, the ITC authors reprocessed ALPHA trial data using nonresponse imputation, counting missing patients as patients who did not experience response (instead of excluding them). Of note, the ALPHA trial considered patients who received rescue therapy as experiencing response, whereas the delgocitinib trials considered these patients as not experiencing response; hence, it was not possible to align the methods of handling rescue therapy in the ALPHA trial and other trials.
Statistical Model
The NMAs were conducted using a Bayesian framework, consistent with guidance from the NICE Decision Support Unit (DSU).50 Analyses were implemented in WinBUGS using binomial logit models to estimate relative treatment effects for dichotomous outcomes. Markov chain Monte Carlo simulation methods were employed to generate posterior distributions. An initial burn-in of 20,000 iterations was followed by 50,000 sampling iterations across 3 chains. Convergence was assessed using trace plots and Brooks-Gelman-Rubin diagnostics, with shrink factors approaching 1 indicating convergence.51,52 The Brooks-Gelman-Rubin plots for each outcome are presented in the Results section.
Both fixed-effect and random-effects models were explored. Fixed-effect models were generally preferred due to the limited number of studies and the sparse connections within the network, which led to imprecise estimates under random-effects models. A baseline risk model was used to estimate absolute event rates for the delgocitinib arms using a random-effects approach. These models estimates were based on the mean and SD of the predictive distribution as described in NICE DSU Technical Support Document 5.50
Prior Distributions
Noninformative (vague) priors were used to ensure that the analysis results were primarily informed by observed trial data. Normal priors with a mean of 0 and a variance of 10,000 were applied for treatment effects, consistent with NICE DSU Technical Support Document 2.50 For the between-study heterogeneity parameter in the random-effects model, a uniform prior distribution ranging from 0 to 2 was used.
Model Fit
Model fit was evaluated using the deviance information criterion (DIC) and posterior mean residual deviance. The DIC combines a measure of model fit with a penalty for complexity (effective number of parameters). Models with lower DIC values are generally preferred, with a difference of more than 3 to 5 points considered meaningful. A posterior residual deviance close to the number of data points also supports adequate model fit. Results for the preferred models for each end point are presented in the Results section.
Homogeneity Assessment
A feasibility assessment was conducted to evaluate the assumption of clinical and methodological homogeneity across trials. This included review of study design characteristics, outcome definitions, timing of assessments, and baseline population characteristics. Minor sources of heterogeneity (e.g., estimand definitions, handling of intercurrent events) were addressed by standardizing imputation strategies and aligning outcome definitions where possible. Sensitivity analyses were conducted to assess the impact of baseline severity and the timing of outcome reporting.
Consistency Assessment
An assessment of consistency between the direct and indirect evidence was not applicable to this NMA as there were no closed loops in the networks.
Outcomes
The outcomes assessed in the NMA included the following:
Efficacy outcomes:
IGA-CHE or PGA score of 0 or 1 end point response (proportion of patients experiencing a score of 0 or 1 at a specified time point)
IGA-CHE score of 0 or 1 cumulative response (proportion of patients experiencing the score at any time point during the assessment period)
HECSI-90 (proportion of patients experiencing a HECSI-90 response).
Harms:
discontinuation due to AEs
all-cause discontinuation.
However, due to a lack of data relating to phototherapy, it was not possible to conduct an NMA for cumulative response or HECSI-90. In addition, all-cause discontinuation was excluded from the analysis due to inconsistencies within the network and between-trial heterogeneity in the handling of patients who experienced response early and the handling of treatment discontinuations.
Base-Case Analysis
The base-case analysis for efficacy outcomes included all patients with available data from studies that reported outcomes at their primary end point (typically week 12 or week 16). An additional analysis was performed using week 12 data only. For discontinuation due to AEs, the base case included all studies reporting treatment-level data at the end of treatment, ranging from week 16 to week 24 (refer to Table 30).
Table 30: Overview of the Analyses — Efficacy and Harms
Analysis | Description |
|---|---|
Efficacy outcomes | |
Primary end point analysis (base case) | All patients at primary end point, regardless of whether their disease was moderate or severe at baseline; results reported at weeks 12 and 16. |
Week 12 analysis (base case) | All patients, regardless of whether their disease was moderate or severe at baseline; results reported at week 12. |
Harms | |
End-of-treatment analysis (base case) | All patients, regardless of whether their disease was moderate or severe at baseline; results reported at the end of treatment (weeks 16 and 24). |
Source: Delgocitinib network meta-analysis report.49
Sensitivity Analysis
Sensitivity analyses were conducted to assess the potential influence of baseline disease severity (moderate versus severe CHE) and timing of outcome assessment on the estimated treatment effects (week 12 or week 16). These analyses followed the same methodological framework as the base-case analysis but applied to stratified populations and alternative time points.
For the primary efficacy outcome — IGA-CHE or PGA score of 0 or 1 response — 2 separate analyses were conducted: 1 restricted to patients with moderate CHE and another restricted to those with severe CHE. Each analysis included studies that reported outcomes at either the trial-defined primary end point (week 12 or week 16) or specifically at week 12. These analyses were intended to evaluate whether disease severity modified treatment effects and whether the results were robust across different assessment time points.
For the outcome of discontinuation due to AEs, a sensitivity analysis was performed among patients with severe CHE using outcome data reported at week 24. This analysis was intended to assess whether the risk of AE-related discontinuation varied by disease severity over a longer treatment duration.
A summary of the sensitivity analyses conducted, including population, outcome, and time point specifications, is provided in Table 31.
Analysis Set
The analysis set used in the NMA was dependent on the results reported by the authors of the individual studies (i.e., ITT population or FAS). Intercurrent events handling, including missing data and rescue therapy, were also different depending on the trials included. Further details on the analysis sets and imputation strategies used in the NMAs are provided in the Results section. A summary of the NMA methods is shown in Table 31.
Table 31: Summary of Network Meta-Analysis Methods
Methods | Description |
|---|---|
Analysis methods | The NMA was conducted using a Bayesian framework implemented in WinBUGS, with binomial logit models applied to dichotomous outcomes. Both fixed-effect and random-effects models were explored, though fixed-effect models were generally preferred due to the limited data and sparse network connections. A baseline risk model was used to estimate absolute event rates from the delgocitinib arms. The analyses estimated relative treatment effects as odds ratios or risk ratios with 95% credible intervals. |
Priors | Noninformative prior distributions for the parameters were estimated, with a mean of 0 and a variance of 10,000. Noninformative prior distributions for the random-effects between-study heterogeneity parameter were also used (approximately uniform (0,2)). |
Assessment of model fit | Model fit was assessed with the DIC and the posterior mean residual deviance. |
Assessment of consistency | Not applicable as there were no closed loops. |
Assessment of convergence | Burn-in consisted of 20,000 simulations, followed by 50,000 simulations across 3 chains to assess convergence. Convergence was confirmed using the Brook-Gelman-Rubin diagnostic, along with density, history, and autocorrelation plots. |
Outcomes |
|
Follow-up time points | IGA-CHE or PGA score of 0 or 1 (end point response): 12 or 16 weeks (primary end point) and 12 weeks (secondary end point) DAEs: last time point available (16 or 24 weeks) |
Construction of nodes | Treatment nodes were constructed as per the treatment regimens and the recommended dose. |
Sensitivity and subgroup analyses | Efficacy outcomes:
Harms:
|
CHE = chronic hand eczema; DAE = discontinuation due to adverse events; DIC = deviance information criterion; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; NMA = network meta-analysis; PGA = Physician’s Global Assessment.
Source: Delgocitinib NMA report.49
Results
Summary of Included Studies
The SLR identified 128 records comprising 44 unique trials. After considering heterogeneity for study design characteristics (namely, study phase, interventions, and eligibility criteria) and for outcome reporting, definition, and time points, 29 trials were excluded from the feasibility assessment, leaving 15 trials for further detailed assessment of study design and baseline characteristics. Of the 15 trials included in the feasibility assessment, 3 assessed the efficacy of delgocitinib compared with vehicle cream, and a fourth performed a head-to-head comparison between delgocitinib and alitretinoin. The remaining 11 trials reported data for 8 treatments for CHE positioned as second or later lines of therapy (alitretinoin, immersion PUVA, dupilumab, upadacitinib, abrocitinib, narrow-band UVB, ciclosporin, and azathioprine), as well as placebo.
Ultimately, 7 RCTs were identified for inclusion in the NMA (Table 32) based on potentially relevant global comparators (namely, vehicle cream, alitretinoin, and phototherapy) and outcomes: the DELTA 15 (NCT04871711), DELTA 225 (NCT04872101), Worm 202253 (NCT03683719), DELTA FORCE54 (NCT05259722), BACH55 (NCT00124475), HANDEL56 (NCT00817063), and ALPHA57 (ISRCTN80206075) trials.
Three trials (DELTA 1, DELTA 2, and Worm 2022 trials) compared delgocitinib to vehicle cream, whereas the DELTA FORCE trial compared delgocitinib to alitretinoin. The BACH and HANDEL trials compared alitretinoin to placebo, and the ALPHA trial compared alitretinoin to PUVA. The Worm 2022 trial53 is a dose-finding trial with 5 arms; however, only the vehicle cream and delgocitinib 20 mg arms were interventions of interest and were therefore eligible for inclusion in the feasibility assessment. The technical report submitted does not explicitly include a detailed risk of bias assessment for the included studies.
The outcomes measured in these studies include IGA-CHE or PGA score of 0 or 1 end point response, IGA-CHE score of 0 or 1 cumulative response, HECSI-90 end point response, and discontinuation due to AEs at various time points (Table 32). The 3 delgocitinib trials against vehicle cream (DELTA 1, DELTA 2, and Worm 2022 trials) reported outcomes at week 12 and week 16 for IGA-CHE or PGA score of 0 or 1 end point response, IGA-CHE score of 0 or 1 cumulative response, and HECSI-90 end point response, with discontinuation due to AEs noted at the end of treatment (week 16). The DELTA FORCE trial measured for IGA-CHE or PGA score of 0 or 1 end point response and HECSI-90 end point response at week 12, IGA-CHE score of 0 or 1 cumulative response at both week 12 and week 24, and discontinuation due to AEs at the end of treatment (week 24). Alitretinoin trials reported limited efficacy outcome data, with cumulative response assessed at week 12 and week 24 (BACH trial) and week 24 (HANDEL trial), and end point response assessed at week 12 (ALPHA trial). The 3 alitretinoin trials reported discontinuation due to AEs at the end of treatment (i.e., at week 24).
Table 32: Characteristics of Included Studies From the SLR
Trial | Intervention | Comparator | Time points of assessment | |||
|---|---|---|---|---|---|---|
IGA-CHE or PGA 0 or 1 end point response | IGA-CHE 0 or 1 cumulative response | HECSI-90 end point response | DAE | |||
DELTA 1 (N = 487) | Delgocitinib 20 mg/g (topical) | Vehicle cream | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) |
DELTA 2 (N = 484) | Delgocitinib 20 mg/g (topical) | Vehicle cream | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) |
Worm 2022 (N = 79) | Delgocitinib 20 mg/g (topical) | Vehicle cream | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) Week 12 (M,S) | Week 16 (M,S) |
DELTA FORCE (N = 503) | Delgocitinib 20 mg/g (topical) | Oral alitretinoin 30 mg | Week 12 (S) | Week 12 (S) Week 24 (S) | Week 12 (S) | Week 24 (S) |
ALPHA (N = 180) | PUVA (phototherapy) | Oral alitretinoin 30 mg | Week 12 (S) | NR | NR | Week 24 (S) |
BACH (N = 1,032) | Oral alitretinoin 30 mg | Placebo | NR | Week 12 (S) Week 24 (S) | NR | Week 24 (S) |
HANDEL (N = 64) | Oral alitretinoin 30 mg | Placebo | NR | Week 24 (S) | NR | Week 24 (S) |
CHE = chronic hand eczema; DAE = discontinuation due to adverse events; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; M = moderate; NR = not reported; PGA = Physician’s Global Assessment; PUVA = psoralen and UVA radiation; S = severe.
Notes: The BACH trial was included only in the IGA-CHE score of 0 or 1 cumulative response base-case analysis at week 24. The BACH trial was initially included in all base-case analyses for cumulative response. However, inconsistency was identified in the primary end point analysis and week 12 analysis; thus, both analyses were conducted again with the BACH trial excluded.
Source: Delgocitinib network meta-analysis report.49
Assessment of Homogeneity Among Trials Included in the NMA
Study Design
The studies included in the NMA were all RCTs. The DELTA FORCE trial was a phase III study, and the ALPHA trial was a phase IV study. The DELTA 1, DELTA 2, and Worm 2022 trials were phase III, double-blind, vehicle-controlled RCTs. The DELTA FORCE trial was open label, although outcome assessors were blinded to mitigate bias in evaluating CHE severity. The ALPHA trial was a double-blind, parallel-group RCT. The BACH and HANDEL studies were both double-blind, placebo-controlled RCTs evaluating oral alitretinoin. No studies were excluded based on design.
Disease Severity
The DELTA FORCE, ALPHA, BACH, and HANDEL trials included only patients with severe CHE. The DELTA 1, DELTA 2, and Worm 2022 trials included populations with both moderate and severe CHE. Sensitivity analyses in the NMA were stratified by disease severity to assess its impact on treatment effects.
Eligibility Criteria
All trials included adults with CHE of at least 3 months’ duration who had not experienced response to TCS or for whom TCS use was inappropriate. What was considered prior TCS failure varied widely (e.g., from 4 weeks up to 1 year). Use of systemic agents or phototherapy before the study was generally permitted with an appropriate washout period. The DELTA FORCE trial excluded patients previously treated with alitretinoin. The exclusion criteria included active skin infection and AD requiring treatment beyond the hands. The ALPHA trial excluded patients with known allergic contact dermatitis unless avoidance measures had been taken, but the DELTA FORCE trial allowed such patients.
Patient Demographic
Across all trials, the baseline demographics were generally comparable. The median patient age ranged from 40 to 50 years, and 40% to 60% of patients were female. More than 90% of participants were white. CHE subtypes varied: the DELTA FORCE trial predominantly included patients with atopic CHE, while the ALPHA trial enrolled primarily those with hyperkeratotic CHE.
Trial Population
The DELTA FORCE trial used an FAS that included all randomized participants, while the ALPHA trial used the ITT population. The Worm 2022, DELTA 1, and DELTA 2 trials used similar estimands for binary efficacy outcomes. However, missing data and intercurrent events were handled differently: the DELTA FORCE trial applied nonresponse imputation, whereas the ALPHA trial used observed case analysis at 12 weeks. To harmonize estimands for the NMA, ALPHA trial data were reanalyzed using nonresponse imputation. Patients who discontinued due to AEs were consistently reported. However, all-cause discontinuation was not analyzed due to differences in how the trials handled patients who experienced response early and how they handled rescue therapy (e.g., the ALPHA trial permitted continuation with added treatment; the DELTA FORCE trial treated rescue use as treatment failure).
Administration Form and Dosing of Comparators
Delgocitinib was administered topically at 20 mg/g, twice daily. Alitretinoin was administered orally at 30 mg daily, with optional dose reduction to 10 mg. PUVA therapy in the ALPHA trial consisted of twice-weekly sessions over 12 weeks. Vehicle comparators (placebo creams) were used in the DELTA 1, DELTA 2, and Worm 2022 trials.
Definitions of End Points
The primary efficacy end point was an IGA-CHE or PGA score of 0 or 1, defined as achieving a score of 0 (clear) or 1 (almost clear). Delgocitinib trials required an additional 2-point or greater improvement from baseline. Despite this stricter criterion, the end points were deemed sufficiently comparable for synthesis. Cumulative response and HECSI-90 were of interest but could not be evaluated across all arms due to missing data (e.g., HECSI-90 data were not available in the ALPHA trial). All-cause discontinuation was excluded due to inconsistencies in the handling of patients who experienced response. The cumulative response end points were reported but considered secondary to the more relevant end point results, in alignment with input from the clinical experts consulted by CDA-AMC. The cumulative response outcomes are not included in this report.
Timing of End Point Evaluation
The primary end point (IGA-CHE or PGA score of 0 or 1) was evaluated at week 12 for the ALPHA and DELTA FORCE trials. The DELTA 1, DELTA 2, and Worm 2022 trials assessed outcomes at week 16 (with additional reporting at week 12). Cumulative response could not be analyzed due to lack of data at comparable time points for PUVA. For discontinuation due to AEs, the last available time point was used (week 24 for both the ALPHA and DELTA FORCE trials).
Efficacy
IGA-CHE or PGA Score of 0 or 1 End Point Response
The evidence network for the NMA of IGA-CHE or PGA score of 0 or 1 end point response is shown in Figure 11. The network addressing the end point response at week 12 is the same (same studies and comparisons). The DELTA 1, DELTA 2, DELTA FORCE, Worm 2022, and ALPHA trials informed this end point. In these networks, the delgocitinib trials (DELTA 1, DELTA 2, DELTA FORCE, and Worm 2022 trials) used IGA-CHE to measure the efficacy of CHE treatments, whereas the ALPHA trial used PGA.
Figure 11: Network Diagram for IGA-CHE or PGA Score of 0 of 1 End Point Response — Primary End Point (Base Case)
IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; M = moderate chronic hand eczema; PGA = Physician’s Global Assessment; PUVA = psoralen and UVA radiation; S = severe chronic hand eczema.
Source: Delgocitinib network meta-analysis report.49
The total residual deviance and the DIC were similar for both fixed-effect and random-effects models; the difference in the DIC between models was less than 5 for all NMAs of this outcome. Therefore, no clear recommendation could be made for selecting one model over the other based on model fit statistics alone. Due to the small number of trials included in the network, the random-effects model showed wide 95% CrIs when estimating treatment effects compared to the fixed-effect model. Considering the sparsity of the network and the resulting imprecise treatment effect estimates generated by the random-effects model, the authors preferred the fixed-effect model for this outcome.
Table 33 presents the median OR and 95% CrIs for delgocitinib versus all treatments across different disease severity levels (sensitivity analyses 1 and 2) for the IGA-CHE or PGA score of 0 or 1 end point response, incorporating all primary end point analyses, and the same outcomes at 12 weeks.
Table 33: IGA-CHE or PGA Score of 0 or 1 End Point Response — Primary End Point Analysis
Delgocitinib versus comparator | Base case (all patients) | Sensitivity analysis 1 (severe disease) | Sensitivity analysis 2 (moderate disease) | |||
|---|---|---|---|---|---|---|
Fixed effects | Random effects | Fixed effects | Random effects | Fixed effects | Random effects | |
Primary end point analysisa | ||||||
Cream vehicle, median OR (95% CrI) | 2.92 (2.06 to 4.21) | ████ | 7.53 (3.22 to 21.03) | ████ | 3.34 (2.18 to 5.27) | ████ |
PUVA, median OR (95% CrI) | 2.73 (1.43 to 5.25) | ████ | 2.73 (1.43 to 5.28) | ████ | 2.74 (1.42 to 5.28) | ████ |
Alitretinoin, median OR (95% CrI) | 1.88 (1.23 to 2.93) | ████ | 1.89 (1.23 to 2.92) | ████ | 1.89 (1.23 to 2.94) | ████ |
Week 12 analysisb | ||||||
Cream vehicle, median OR (95% CrI) | 2.93 (2.07 to 4.23) | ████ | 3.51 (1.84 to 7.23) | ████ | 2.91 (1.97 to 4.42) | ████ |
PUVA, median OR (95% CrI) | 2.73 (1.43 to 5.25) | ████ | 2.73 (1.43 to 5.25) | ████ | 2.74 (1.42 to 5.26) | ████ |
Alitretinoin, median OR (95% CrI) | 1.89 (1.23 to 2.93) | ████ | 1.89 (1.22 to 2.92) | ████ | 1.88 (1.22 to 2.93) | ████ |
CrI = credible interval; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; OR = odds ratio; PGA = Physician’s Global Assessment; PUVA = psoralen and UVA radiation.
aPrimary end point analysis includes the primary end point for all relevant studies.
bWeek 12 analysis includes the week 12 end point for all relevant studies.
Source: Delgocitinib network meta-analysis report.49
Overall, all active treatments showed higher treatment response than vehicle cream. Across all analyses of this outcome, the results favoured delgocitinib over vehicle cream, PUVA, and alitretinoin. For the fixed-effect model, all results favoured delgocitinib. The results were generally consistent across all analyses of this outcome. However, for the primary end point sensitivity analysis assessing the population with severe disease, the OR median point estimates comparing delgocitinib to vehicle cream were noticeably higher (favouring delgocitinib) than the other estimates generated by the NMAs of this outcome.
HECSI-90 End Point Results
The HECSI-90 end point response base-case networks consist of 4 treatments reported in 5 studies for the primary end point analysis and week 12 analysis (Figure 12). For each network, sensitivity analyses were also conducted. All trials in these networks used IGA-CHE to measure the efficacy of CHE treatments.
Figure 12: Network Diagram for HECSI-90 End Point Response — Primary End Point and Week 12 End Point (Base Case)
HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; M = moderate chronic hand eczema; S = severe chronic hand eczema.
Source: Delgocitinib network meta-analysis report.49
The total residual deviance and the DIC were similar for both fixed-effect and random-effects models; the difference in the DIC between models was less than 5 points for all NMAs of this outcome. Therefore, no clear recommendation could be made to select one model over the other based on model fit statistics alone. Due to the small number of trials included in the network, there were wide 95% CrIs for the random-effects model, compared to the fixed-effect model, when estimating treatment effects. Considering the sparsity of the network and the resulting imprecise treatment effect estimates generated by the random-effects model, the fixed-effect model was preferred by the authors for this outcome.
The median ORs and 95% CrIs for delgocitinib versus all treatments across different disease severity levels and time points for the HECSI-90 end point response are presented in Table 34.
Table 34: HECSI-90 End Point Response
Delgocitinib versus treatment | Base case | Sensitivity analysis 1 | Sensitivity analysis 2 | |||
|---|---|---|---|---|---|---|
Fixed effects | Random effects | Fixed effects | Random effects | Fixed effects | Random effects | |
Primary end point analysisa | ||||||
Cream vehicle, median OR (95% CrI) | 3.59 (2.49 to 5.28) | ████ | 5.24 (2.73 to 11.06) | ████ | 3.00 (2.00 to 4.61) | ████ |
Alitretinoin, median OR (95% CrI) | 1.79 (1.22 to 2.62) | ████ | 1.79 (1.22 to 2.63) | ████ | 1.79 (1.22 to 2.63) | ████ |
Week 12 analysisb | ||||||
Cream vehicle, median OR (95% CrI) | 4.34 (3.02 to 6.39) | ████ | 5.10 (2.65 to 10.78) | ████ | 4.24 (2.81 to 6.59) | ████ |
Alitretinoin, median OR (95% CrI) | 1.79 (1.23 to 2.63) | ████ | 1.79 (1.22 to 2.63) | ████ | 1.79 (1.22 to 2.63) | ████ |
CrI = credible interval; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; OR = odds ratio.
aPrimary end point analysis includes the primary end point for all relevant studies.
bWeek 12 analysis includes week 12 end point for all relevant studies.
Source: Delgocitinib network meta-analysis report.49
Across all analyses of this outcome, the results favoured delgocitinib versus vehicle cream and alitretinoin. For the fixed-effect model, all results favoured delgocitinib. For the sensitivity analyses assessing the population with severe disease, the OR and relative risk median point estimates comparing delgocitinib to vehicle cream were noticeably higher (favouring delgocitinib) than for the other NMAs of this outcome.
Discontinuation Due to AEs
The discontinuation due to AEs base-case network consists of 4 treatments reported in 7 studies for the primary end point analysis (Figure 13). The second sensitivity analysis assessed discontinuation due to AEs at week 24. Based on the network plot presented, a closed loop was identified, and an inconsistency assessment was conducted. No significant inconsistency was found.
Figure 13: Network Diagram for Discontinuation Due to AEs — Primary End Point (Base Case)
AE = adverse event; M = moderate chronic hand eczema; PUVA = psoralen and UVA radiation; S = severe chronic hand eczema.
Source: Delgocitinib network meta-analysis report.49
In the model diagnostic measures for all analyses of discontinuation due to AEs, the total residual deviance and the DIC were similar for both fixed-effect and random-effects models; the difference in the DIC between models was less than 5 points for all NMAs of this outcome. Therefore, no clear recommendation was made to select one model over the other based on model fit statistics alone. Due to the small number of trials included in the network, wide 95% CrIs for the random-effects model were observed when estimating treatment effects compared to the fixed-effect model. Considering the sparsity of the network and the resulting imprecise treatment effect estimates generated by the random-effects model, the fixed-effect model was preferred by the authors for this outcome. The median ORs and CrIs for each comparison against delgocitinib are presented in Table 35.
Because only 2 interventions, reported in 3 trials (DELTA 1, DELTA 2, Worm 2022 trials), were included in discontinuation due to AEs sensitivity analysis 1 (moderate disease, week 16), a pairwise meta-analysis was performed to estimate the relative treatment effects. The results of this analysis are presented in Figure 14. A point estimate less than 1 favours delgocitinib.
Table 35: Discontinuation Due to AE Results for Delgocitinib — End of Treatment (Base Case and Sensitivity Analysis)
Delgocitinib versus treatment | Base case | Sensitivity analysis 2 | ||
|---|---|---|---|---|
Fixed effects | Random effects | Fixed effects | Random effects | |
Placebo or vehicle cream, median OR (95% CrI) | 0.21 (0.08 to 0.45) | ████ ████ | 0.22 (0.05 to 0.73) | ████ ████ |
PUVA, median OR (95% CrI) | 0.09 (0.03 to 0.32) | ████ ████ | 0.10 (0.02 to 0.41) | ████ ████ |
Alitretinoin, median OR (95% CrI) | 0.09 (0.04 to 0.21) | ████ ████ | 0.10 (0.02 to 0.29) | ████ ████ |
AE = adverse event; CrI = credible interval; OR = odds ratio; PUVA = psoralen and UVA radiation.
aIncludes all trials reporting the discontinuation due to AEs outcome at the end of treatment.
bSensitivity analysis 2 includes patients who had severe chronic hand eczema at baseline for trials reporting data at the week 24 end point.
Source: Delgocitinib network meta-analysis report.49
Figure 14: Discontinuation Due to AEs — Sensitivity Analysis 1 (Moderate Disease, Week 16)
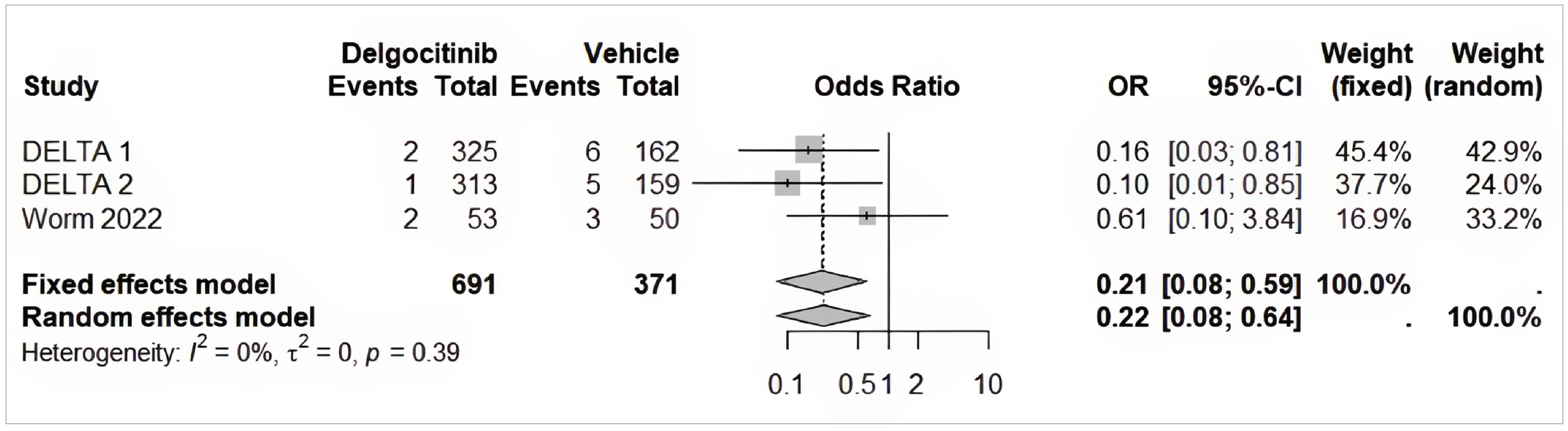
AE = adverse event; CI = confidence interval; OR = odds ratio.
Source: Delgocitinib network meta-analysis report.49
Across all analyses of discontinuation due to AEs, the results favoured delgocitinib versus vehicle cream or placebo, PUVA, and alitretinoin. For the fixed-effect model, all results significantly favoured delgocitinib. The pairwise meta-analysis for sensitivity analysis 1 supported the NMA results favouring delgocitinib over vehicle cream. The magnitude of the OR estimates was generally consistent across all analyses of this outcome.
Matched-Adjusted Indirect Comparison
Objectives
The objective of this MAIC was to estimate the relative efficacy of delgocitinib compared to dupilumab for the treatment of moderate to severe CHE in adults who have not experienced response to TCS or for whom TCS are not appropriate. As no head-to-head trials comparing these interventions were available, and in the absence of a common comparator arm across trials, an anchored MAIC was conducted using IPD from the DELTA 1 and DELTA 2 trials of delgocitinib and published aggregate data from the LIBERTY-AD-HAFT trial of dupilumab. The primary research question was whether delgocitinib is associated with a similar or greater probability of clinical response compared to dupilumab, as measured by the proportion of patients experiencing an IGA-CHE or HF-IGA score of 0 or 1 at week 16.
Study Selection Methods
Before conducting the ITC, a feasibility assessment was performed to determine potential sources of heterogeneity across the 44 trials identified in the SLR and to ensure trial comparability for synthesis. Five trials evaluated dupilumab and/or oral JAK inhibitors (abrocitinib and upadacitinib): the DUPSHE58 (NCT04512339), LIBERTY-AD-HAFT59 (NCT04417894), JADE DARE60 (NCT04345367), MEASURE UP 161 (NCT03569293), and MEASURE UP 261 (NCT03607422) trials.
The LIBERTY-AD-HAFT trial was the only study collecting data for a CHE subtype, specifically atopic hand and foot eczema, which reported data at relevant time points (week 16), with end point definitions similar or identical to the DELTA trials. Like the DELTA 1 and DELTA 2 trials, the LIBERTY-AD-HAFT trial targeted patients with moderate to severe disease, for which patient baseline characteristics were available. The availability of baseline characteristics for the population with moderate to severe disease means that sufficient adjustments could be made in a MAIC. Therefore, the LIBERTY-AD-HAFT trial was considered eligible for a MAIC to compare the efficacy of dupilumab and delgocitinib for the treatment of the AD subtype of moderate to severe CHE after 16 weeks of treatment in patients who did not benefit from TCS, who experienced intolerance to TCS, or who did not qualify for treatment with TCS.
Therefore, an anchored comparison between the DELTA 1 and 2 and LIBERTY-AD-HAFT trials through the common vehicle/placebo node was deemed to be feasible and the most appropriate analysis. Because an anchored approach was feasible, only effect modifiers (i.e., variables that impact the treatment effect) needed to be adjusted for in the analysis.25,62 No formal list of effect modifiers and the methods to obtain them is presented.
The MAIC was performed using the methodology in Signorovitch et al.62 OR was used for binary end points and response difference was used for the continuous end point as the parameters to assess efficacy outcomes. The ORs and the response differences for delgocitinib versus dupilumab were estimated using IPD from the DELTA 1 and DELTA 2 trials and published aggregate data from the LIBERTY-AD-HAFT trial. For the binary efficacy end points, rescue medication and missing values were imputed as nonresponse. For the continuous efficacy end point, missing values at week 16 due to rescue treatment, AEs, or lack of efficacy were imputed by WOCF. The exact same estimands for both the LIBERTY-AD-HAFT and DELTA 1 and 2 trials were used in these analyses. The results were presented in tables and forest plots, displaying the point estimates of the ORs and response differences along with their 95% CIs. All analyses were performed using SAS software version 9.4.63
Analysis Methods
The LIBERTY-AD-HAFT trial studied patients with AD, while the DELTA 1 and DELTA 2 trials included various subtypes of CHE. Thus, only the patients with AD as the primary subtype of eczema from the DELTA 1 and DELTA 2 trials were included in the comparison. After assessing trial similarities in terms of eligibility criteria, important cross-trial differences in patients’ baseline characteristics were identified. To adjust for these differences, using the IPD from the DELTA 1 and DELTA 2 trials, the baseline characteristics were weighted such that their weighted mean baseline characteristics matched those of the population of the LIBERTY-AD-HAFT trial. For an anchored MAIC, all variables considered important effect modifiers (i.e., sex, race, baseline disease severity [HECSI score]) were adjusted for and included in the logistic regression used to estimate propensity scores.62,64 Age was also adjusted for to further ensure comparability between the trials. Variables that were not considered important effect modifiers were excluded from the analysis to avoid unnecessary erosion of accuracy. The baseline variables that were reported by both trials and selected to match were age, sex, race (percentage of patients who were white), baseline HECSI score, and CHE subtype. Analyses of the efficacy outcomes were performed on the FAS population. DELTA 1 and DELTA 2 trial data were pooled and are referred to as 1 trial in the subsequent sections.
In addition to checking the distributions of covariates in each study, the ESS serves as a simple indicator of the extent of overlap between studies.65 ESS is calculated from the weights applied to the IPD.65 Large reductions in ESS and a small absolute ESS may indicate poor overlap between the IPD and aggregate data, potentially leading to instability.65 Thus, the ESS for the DELTA 1 and DELTA 2 trials, along with the baseline characteristics for the DELTA 1 and DELTA 2 trials after matching adjustment, were provided.
Outcomes Definitions
The primary end point for the analysis was the proportion of patients experiencing an IGA-CHE or HF-IGA score of 0 or 1 at week 16. There were slight differences between the scales used in the trials, which were discussed as part of the homogeneity assessment.59
HECSI-75, HECSI-90, percentage CFB in HECSI score, pruritus (itch), and pain were considered as secondary end points in the MAIC analyses. The definitions for HECSI-75, HECSI-90, and HECSI score were the same across studies, and all data were reported at week 16. For pruritus (itch) and pain response, the DELTA 1 and DELTA 2 trials used the HESD itch and pain scores, while the LIBERTY-AD-HAFT trial used the Hand and Foot Peak Pruritus/Pain Numerical Rating Scale (NRS) itch score. The HESD is a 6-item, patient-reported instrument designed to assess the severity of CHE signs and symptoms. Participants assess the worst severity of 6 signs and symptoms of CHE (itch, pain, cracking, redness, dryness, and flaking) over 24 hours (daily) using a 0 to 10 numeric rating scale with anchors of 0 (no [symptom]) and 10 (severe [symptom]). The Hand and Foot Peak Pruritus/Pain NRS is another patient-reported instrument for patients to rate their daily worst hand and foot itch or pain intensity on a scale ranging from 0 (no itch or pain) to 10 (worst imaginable itch or pain).59 The definition varied between scores: 10 points for HESD itch or pain score indicates severe symptoms, and 10 points for Hand and Foot Peak Pruritus/Pain NRS indicates the worst imaginable itch or pain. Because the scales are not comparable, itch and pain end points were not analyzed, and therefore no ITC was performed for this outcome.
Safety outcomes were not prioritized for this analysis. No sensitivity or subgroup analyses were performed.
A summary of the MAIC methods is presented in Table 36.
Table 36: Matching-Adjusted Indirect Comparison Methods
Methods | Description |
|---|---|
Analysis methods | Anchored MAIC:
|
Covariates used for propensity score weighting | Effect modifiers:
|
Outcomes |
|
Follow-up time points | Week 16 |
Construction of nodes | Treatment nodes were constructed as per the treatment regimens and the recommended dosing. A key assumption was that the vehicle and placebo treatments were sufficiently similar to be combined into a single node, which served as the anchor. |
Population | Only patients with the AD subtype of CHE were considered, including:
|
Sensitivity/subgroup analyses | NA |
AD = atopic dermatitis; CHE = chronic hand eczema; CI = confidence interval; ESS = effective sample size; FAS = full analysis set; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; IGA = Investigator’s Global Assessment; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; IMP = investigational medicinal product; IPD = individual patient data; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NA = not applicable; OR = odds ratio; RD = response difference.
Source: Delgocitinib MAIC technical report.66
Results
Studies and Homogeneity Assessment
Study design was similar between trials (Figure 15). Both trials were multicentre, double-blind RCTs in which the active treatment was compared with a placebo. For each of the trials, the method of administration of the placebo was identical to the method of administration of the active treatment: both active and placebo arms received topical treatment in the DELTA 1 and DELTA 2 trials and subcutaneous treatment in the LIBERTY-AD-HAFT trial.
The DELTA 1 and 2 trials and the LIBERTY-AD-HAFT trial were well matched in terms of mean age and percentage of patients for sex and race (percentage of patients who were white) after weighting the DELTA 1 and 2 trial population in the MAIC. However, the DELTA 1 and 2 trials only recruited adult patients (aged ≥ 18 years) with CHE, while the LIBERTY-AD-HAFT trial also recruited adolescents, and approximately 20% of patients in the trial were younger than 18 years. The baseline weighted mean age was similar between the trials.
Figure 15: Study Design of the Included Trials for the MAIC Analysis
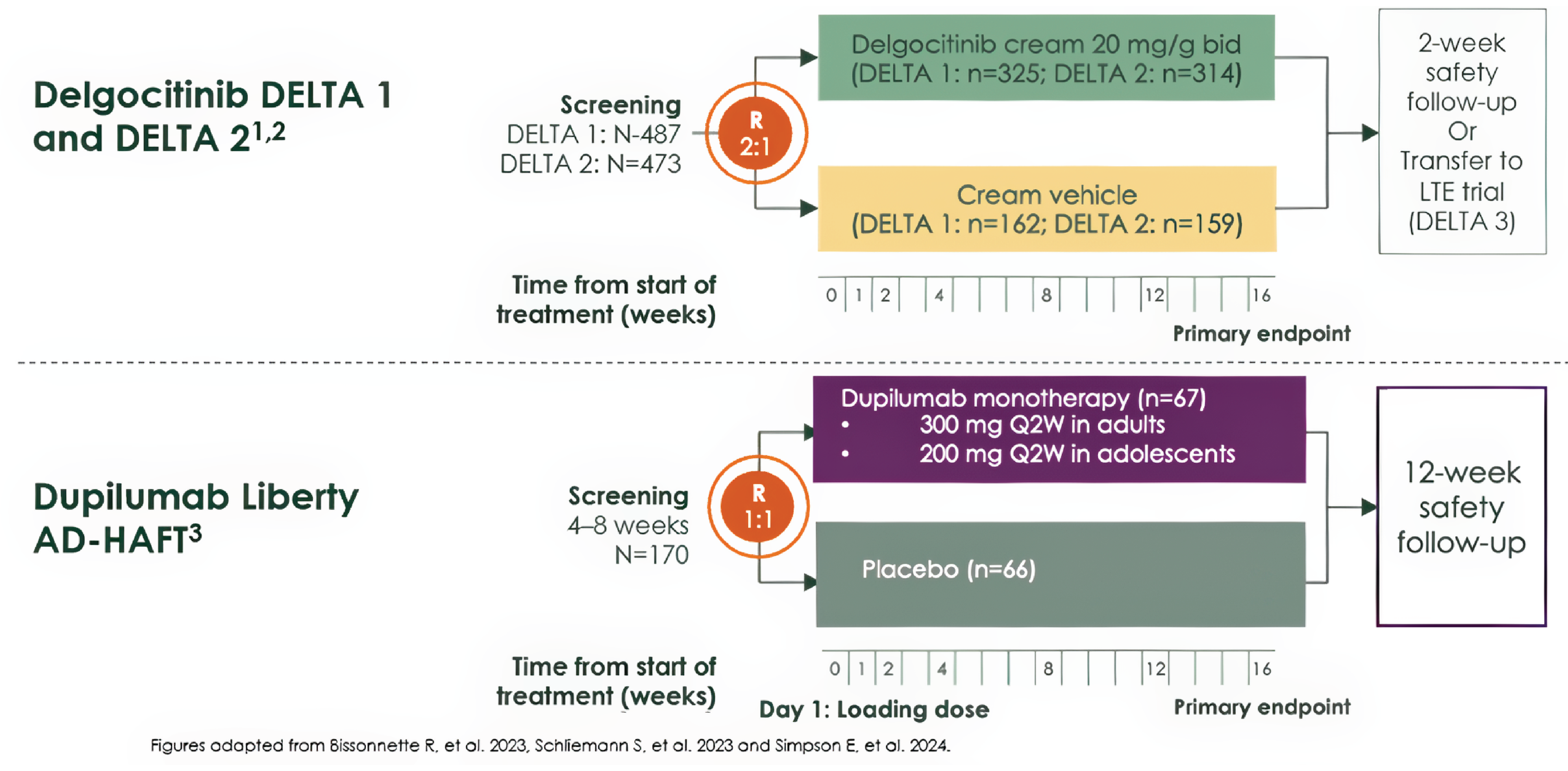
bid = twice a day; LTE = long-term extension; MAIC = matching-adjusted indirect comparison; R = randomization ratio; Q2W = every 2 weeks.
Source: Delgocitinib MAIC technical report.66
The DELTA 1 and 2 trials only recruited patients with CHE that had lasted at least 3 months or who had experienced relapses at least twice within a year. Participants needed an IGA-CHE score of 3 or 4, indicating moderate to severe disease, and a HESD itch score of at least 4. The LIBERTY-AD-HAFT trial recruited patients with AD affecting at least 2 anatomic areas on the hands and feet, with a diagnosis duration of at least 3 years for adults and 1 year for adolescents. Participants required an HF-IGA score of 3 or 4 and a Hand and Foot Peak Pruritus NRS score higher than 4 to be eligible.
The severity of hand eczema was adjusted for in the MAIC via matching of the baseline mean HECSI score of the DELTA 1 and 2 trial population to the LIBERTY-AD-HAFT trial population.
In the assessment of similitudes between studies’ populations, the LIBERTY-AD-HAFT trial recruited patients with hand and foot eczema, of whom almost three-quarters also had AD elsewhere. Although the analysis for the DELTA 1 and DELTA 2 trials were restricted to patients with atopic hand eczema as the primary CHE subtype, in the DELTA trials the presence of active AD in regions other than the hands and feet was an exclusion criterion. Only a small proportion of patients in the LIBERTY-AD-HAFT trial only had foot eczema (4 of 133).
In terms of treatment history, in the DELTA 1 and 2 trials, patients were required to have a documented recent history of experiencing inadequate response to treatment with TCS (i.e., at any time within 1 year before the screening visit) or of TCS being medically inadvisable. In the LIBERTY-AD-HAFT trial, patients were also required to have a history of their hand and foot dermatitis having an inadequate response to medium-potency to high-potency TCS within 6 months before screening or of use of TCS being inadvisable.
For the DELTA 1 and 2 trials, the FAS population included all patients randomized who were exposed to the investigational medicinal product; for the LIBERTY-AD-HAFT trial, exposure to the intervention was not specified as part of the FAS. In both the LIBERTY-AD-HAFT and DELTA 1 and 2 trials, for binary efficacy end points rescue medication and missing values were imputed as nonresponse. For the continuous efficacy end point, missing values at week 16 due to rescue treatment, AEs, or lack of efficacy were imputed by WOCF. The exact same estimands for both the LIBERTY-AD-HAFT and the DELTA 1 and 2 trials were used in these analyses.
The definition for HECSI-75, HECSI-90, and HECSI score were the same across the studies.
For the primary outcome, there were differences between the scales used in the trials, with a broader definition of the scale used in the LIBERTY-AD-HAFT trial (i.e., HF-IGA) than in the DELTA 1 and DELTA 2 trials (i.e., IGA-CHE). For the HF-IGA scale used in the LIBERTY-AD-HAFT trial, severity is stepped down if the extent of the lesions is limited. For instance, a patient could score a 2 based on the signs of definite erythema, scaling, lichenification, edema, vesicles, erosions, and fissures, but if the extent is limited then this could be stepped down to a 1. This meant that a patient with limited lesion extent might be classified as having milder disease severity (almost clear), even if they exhibited multiple symptoms. In contrast, the IGA-CHE scale used in the DELTA 1 and DELTA 2 trials required at least 1 slight but definite symptom from a specific set of symptoms (erythema, scaling, or hyperkeratosis/lichenification) and at least 1 symptom from another set of symptoms (scattered vesicles, barely palpable edema, or superficial fissures). This dual requirement meant a patient had to exhibit symptoms from both sets to qualify for a score of 2, thus setting a stricter criterion than the HF-IGA scale.
Patient Baseline Characteristics
The patient baseline characteristics for all the trials are shown in Table 37. After matching the selected baseline variables, the means of continuous baseline variables and percentages of categorical variables were identical between both delgocitinib and dupilumab. The ESS in the DELTA 1 and DELTA 2 trials after matching was 128 for the delgocitinib arm and 73 for the vehicle cream arm, which was over half of the unweighted sample size (56.8% in delgocitinib arm and 60.8% in vehicle cream arm), demonstrating a strong overlap between studies.65
Efficacy Results
The results for the ORs of the binary end points and the response differences of the continuous end point are presented in Table 38.
Table 37: Patient Baseline Characteristics Before and After Matching
Treatment arm | Baseline characteristics | LIBERTY-AD-HAFT baseline | DELTA 1 and 2 pooled baseline unweighted | DELTA 1 and 2 pooled baseline weighted |
|---|---|---|---|---|
Active treatment arm: DELTA 1 and 2 (delgocitinib) vs. LIBERTY-AD-HAFT (dupilumab) | Patients, N | 67 | 225a | 128b |
Sex, female/male (%) | 67.2/32.8 | 67.1/32.9 | 67.2/32.8 | |
Race, white (%) | 79.1 | 89.8 | 79.1 | |
Age in years (SD) | 35.8 (17.1) | 41.7 (14.8) | 35.8 (13.2) | |
HECSI score (SD) | 46.2 (14.4) | 71.5 (43.6) | 46.2 (26.6) | |
Control arm: DELTA 1 and 2 (vehicle cream) vs. LIBERTY‑AD-HAFT (placebo) | Patients, N | 66 | 120a | 73b |
Sex, male (%) | 42.4 | 38.3 | 42.4 | |
Race, white (%) | 80.3 | 88.3 | 80.3 | |
Age in years (SD) | 33.4 (17.7) | 39.1 (13.7) | 33.4 (10.6) | |
HECSI score (SD) | 47.4 (18.5) | 67.8 (41.4) | 47.4 (25.1) |
HECSI = Hand Eczema Severity Index; SD = standard deviation; vs. = versus.
aOnly patients with atopic dermatitis subtype of chronic hand eczema.
bEffective sample size after adjusted matching.
Source: Delgocitinib matching-adjusted indirect comparison technical report.66
Table 38: Summary of ITC Results for Efficacy End Points for Delgocitinib Versus Dupilumab at Week 16
End points | Binary end points | Continuous end points | ||
|---|---|---|---|---|
OR | 95% CI | RD | 95% CI | |
IGA-CHE or HF-IGA | 1.1 | (0.3 to 3.4) | NA | |
HECSI-90 | 1.3 | (0.4 to 4.9) | ||
HECSI-75 | 1.2 | (0.4 to 3.2) | ||
HECSI percent CFB | NA | 11.7 | (–9.2 to 32.7) | |
CFB = change from baseline; CI = confidence interval; HECSI = Hand Eczema Severity Index; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HF-IGA = Hand and Foot Investigator’s Global Assessment; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; ITC = indirect treatment comparison; NA = not applicable; OR = odds ratio; RD = response difference.
Source: Delgocitinib matching-adjusted indirect comparison technical report.66
Figure 16: OR of Achieving Binary Efficacy End Points for Delgocitinib (DELTA 1 and DELTA 2 Trials) Versus Dupilumab (LIBERTY-AD-HAFT Trial) at Week 16
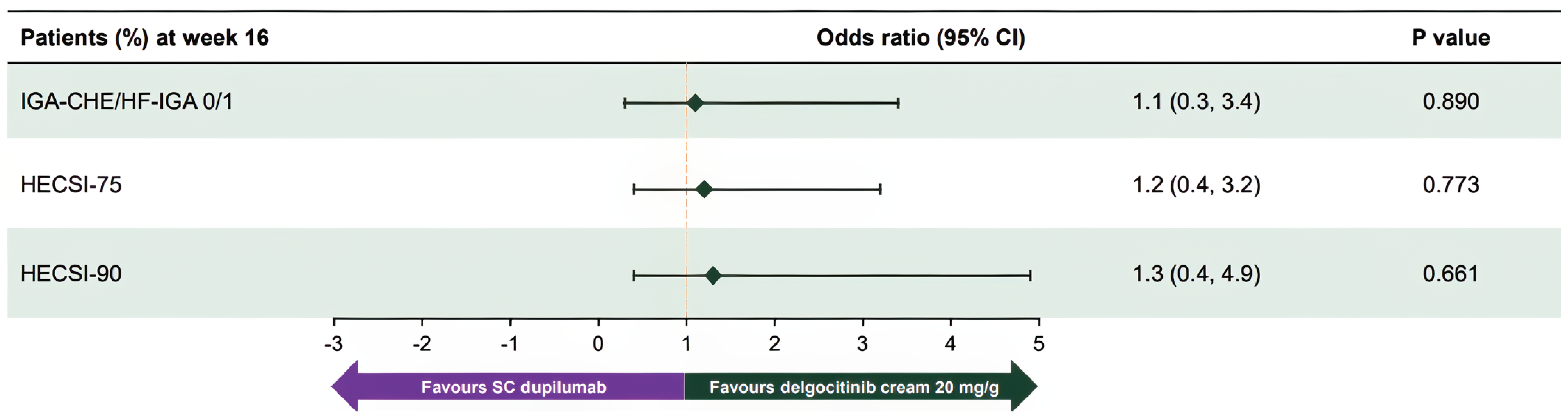
CI = confidence interval; HECSI-75 = at least a 75% improvement in Hand Eczema Severity Index score from baseline; HECSI-90 = at least a 90% improvement in Hand Eczema Severity Index score from baseline; HF-IGA = Hand and Foot Investigator’s Global Assessment; IGA-CHE = Investigator’s Global Assessment of Chronic Hand Eczema; OR = odds ratio; SC = subcutaneous.
Sources: Cohen et al. 2024;67 delgocitinib matching-adjusted indirect comparison technical report.66
Primary End Point IGA-CHE or HF-IGA Treatment Success
The MAIC results for the primary end point of IGA-CHE or HF-IGA treatment success for delgocitinib versus dupilumab at week 16 are presented in Table 38 and graphically depicted in a forest plot in Figure 16. Neither treatment was favoured (OR = 1.1; 95% CI, 0.3 to 3.4; P = 0.890).
Secondary End Point HECSI-90
The MAIC results for the secondary end point of HECSI-90 for delgocitinib versus dupilumab at week 16 are presented in Table 38 and graphically depicted in a forest plot in Figure 16. Neither treatment was favoured (OR = 1.3; 95% CI, 0.4 to 4.9; P = 0.661).
Secondary End Point HECSI-75
The MAIC results for the secondary end point of HECSI-75 for delgocitinib versus dupilumab at week 16 are presented in Table 38 and graphically depicted in a forest plot in Figure 16. Neither treatment was favoured (OR = 1.2; 95% CI, 0.4 to 3.2; P = 0.773).
Secondary End Point Percent CFB in HECSI
The MAIC results for the secondary end point of percent CFB in HECSI for delgocitinib versus dupilumab at week 16 are presented in Table 38 and Figure 17. Neither treatment was favoured (response difference = 11.7%; 95% CI, –9.2% to 32.7%; P = 0.273).
Figure 17: Response Difference for Continuous Efficacy End Point for Delgocitinib (DELTA 1 and DELTA 2 Trials) Versus Dupilumab (LIBERTY-AD-HAFT Trial) at Week 16
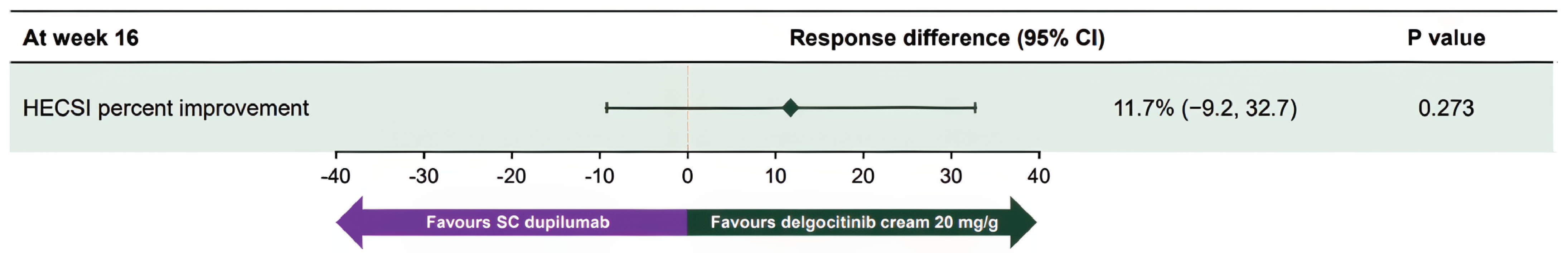
CI = confidence interval; HECSI = Hand Eczema Severity Index; SC = subcutaneous.
Sources: Cohen et al. 2024;67 delgocitinib matching-adjusted indirect comparison technical report.66
Harms
No harms end points were evaluated in the MAIC.
Critical Appraisal of ITCs
Network Meta-Analysis
The NMA was conducted using a prespecified protocol and SLR that followed accepted methodological standards (e.g., PRISMA-P, NICE DSU, Cochrane Handbook), which strengthens the transparency and reproducibility of the study selection. The included trials were all RCTs with generally low risk of bias in design, and no studies were excluded solely on methodological grounds. However, important differences across the trials raise concerns about the validity of combining evidence, particularly with respect to disease severity, end point definitions, and timing of assessments.
The primary efficacy outcome, IGA-CHE or PGA score of 0 or 1 (end point response), was reasonably comparable across studies but varied in stringency: delgocitinib trials required both an end point status of 0 or 1 and a larger than 2-point improvement from baseline, while other trials (e.g., ALPHA trial) used a simpler status-only definition. Although this was addressed in the justification for synthesis, it introduces uncertainty regarding consistency of measurement. Harm outcomes such as discontinuation due to AEs were included; however, all-cause discontinuation was excluded from the analysis due to irreconcilable differences in trial protocols and handling of response.
The network was sparse, with delgocitinib linked to phototherapy only via an indirect comparison through alitretinoin. Furthermore, the network lacked closed loops, precluding formal inconsistency testing. The assumption of transitivity (i.e., that indirect comparisons are valid based on similarity of included trials) was challenged by variation in trial populations (e.g., the ALPHA trial primarily enrolled patients with hyperkeratotic CHE, while the DELTA FORCE trial included more patients with atopic CHE), treatment duration, and analysis sets. Although a feasibility assessment was conducted and sensitivity analyses stratified by baseline severity were provided, it remains uncertain whether these adequately account for potential effect modification.
Statistical heterogeneity was not formally assessed due to the small number of studies per comparison, and the fixed-effect model was selected for most analyses, given the network sparsity. This choice is methodologically defensible but may underestimate uncertainty when between-study variability exists. Posterior estimates were generally precise, but CrIs for some comparisons (e.g., delgocitinib versus PUVA) remained wide, which could reflect uncertainty when considering the clinically meaningfulness of these effects.
While relevant comparators in Canada were included (alitretinoin, PUVA), dupilumab was excluded from the NMA due to a lack of connected data (although assessed in the MAIC described in this report). The follow-up durations were generally adequate (12 to 24 weeks), though longer-term comparative data, particularly for phototherapy and placebo, were limited. The exclusion of HECSI-75, DLQI, and other patient-reported outcomes due to lack of consistency across trials reduces the comprehensiveness of the NMA.
Overall, the NMA provides a useful but limited synthesis of the available comparative efficacy and safety data for delgocitinib in moderate to severe CHE. While the methods were generally appropriate and key comparators in Canada were included, the limited number of trials, the absence of closed loops, and the notable between-study differences in populations, outcome definitions, and analytic strategies introduce uncertainty.
Matching-Adjusted Indirect Comparison
The MAIC was conducted to compare delgocitinib to dupilumab in the absence of direct or network-connected trial data. The use of IPD from the DELTA 1 and DELTA 2 trials is a key strength, enabling reweighting to match the aggregate baseline characteristics of the LIBERTY-AD-HAFT trial of dupilumab. The matching variables were prespecified based on clinical relevance and available data, including disease severity, age, sex, CHE subtype, and CHE duration. However, there is no detailed description on how the effect modifiers were obtained (e.g., clinical input, exploration of literature); hence, residual confounding from unobserved or unreported effect modifiers remains a major limitation.
The primary outcome, IGA-CHE or HF-IGA score of 0 or 1 at week 16, was selected as a harmonized end point across the trials. Secondary outcomes (HECSI-75, HECSI-90, percent change in HECSI) were also aligned. While this common time point supports validity, there were key differences in population characteristics: the DELTA trials focused on patients with CHE that was unresponsive to TCS, whereas the LIBERTY-AD-HAFT trial enrolled patients with moderate to severe AD with hand involvement, which may not fully represent a population with CHE. The definition of hand involvement in the dupilumab trial and its relevance to a CHE diagnosis is unclear, which raises concerns about external validity to the CHE population.
The use of an anchored MAIC is appropriate given that both trials included placebo arms. However, the reweighting process led to a substantial reduction in ESS, particularly for subgroups such as hyperkeratotic CHE, increasing the uncertainty of estimates. The impact of this reduction on precision was reflected in wider CIs, particularly for HECSI outcomes.
AEs and discontinuation data were not included in the MAIC, limiting the assessment of harms. The exclusion of longer-term outcomes (e.g., week 24 or beyond) restricts conclusions on sustained treatment effects.
In summary, while the MAIC used appropriate statistical methodology and addressed a relevant comparator (dupilumab), its findings are limited by population differences, potential unmeasured confounding, reduced ESS, and lack of safety outcomes.
Discussion
Summary of Available Evidence
The clinical evidence base for delgocitinib cream 20 mg/g in adults with moderate to severe CHE comprises 3 phase III RCTs —the DELTA 1, DELTA 2, and DELTA FORCE trials — along with 1 long-term extension study (DELTA 3 trial) and 2 sponsor-submitted ITCs (an NMA and a MAIC).
The DELTA 1 and DELTA 2 trials were double-blind, vehicle-controlled trials that enrolled 487 and 458 participants, respectively, randomized 2:1 to delgocitinib or vehicle cream for 16 weeks. The DELTA FORCE trial was an assessor-blinded, active-controlled trial that enrolled 513 patients with severe CHE to receive either delgocitinib or oral alitretinoin (30 mg once daily) for 24 weeks. Across all 3 studies, efficacy outcomes included IGA-CHE treatment success, HECSI (absolute and percent change), HECSI-75 and HECSI-90, patient-reported symptoms (≥ 4-point reduction in HESD itch), and HRQoL measures (DLQI and HEIS). Safety outcomes included AEs, SAEs, and AESIs.
The DELTA 3 trial was a 36-week open-label extension study that enrolled patients completing the DELTA 1 or DELTA 2 trials to assess the long-term safety and sustained efficacy of delgocitinib. The primary objective was to evaluate long-term safety, while secondary outcomes included clinician-reported and patient-reported efficacy end points. The study included 801 patients and provided supportive data on the durability of response and the incidence of TEAEs with longer exposure to delgocitinib. During the extension period, patients received delgocitinib intermittently. Treatment was paused upon the patient experiencing sufficient clinical improvement and reinitiated if symptoms worsened.
Two ITCs were conducted to address evidence gaps related to the comparative efficacy of delgocitinib. The NMA assessed the comparisons of delgocitinib with alitretinoin and phototherapy using vehicle cream or placebo as the common comparator. A MAIC was also conducted to compare delgocitinib with dupilumab using patient-level data from the DELTA 1 and DELTA 2 trials and aggregate data from the LIBERTY-AD-HAFT trial of dupilumab. The NMA included discontinuation due to AEs as a safety outcome; the MAIC did not include any harms outcomes. Therefore, safety was only partially addressed through indirect evidence.
Participants across all trials were generally similar, with mean ages between 44 and 46 years, disease durations ranging from 4 to 6 years, and a slightly higher proportion of female participants. The majority of patients were recruited from European centres, with limited North American representation. Baseline severity scores (e.g., HECSI) were consistent across treatment arms, and no major imbalances were observed in demographic or disease characteristics. Inclusion criteria focused on moderate to severe CHE, and exclusion criteria (e.g., recent systemic immunosuppressant use) were consistent across studies, supporting comparability of the enrolled populations.
Interpretation of Results
Efficacy
Across the 3 phase III trials, delgocitinib cream demonstrated consistent improvements in both clinician-assessed and patient-reported outcomes in adults with moderate to severe CHE. In the vehicle-controlled trials (DELTA 1 and DELTA 2 trials), delgocitinib was associated with clinically meaningful improvements in IGA-CHE treatment success, HECSI (absolute and percent change), HECSI-75 and HECSI-90, HESD itch reduction, HEIS, and DLQI. In the active-controlled trial (DELTA FORCE trial), delgocitinib showed greater efficacy than oral alitretinoin in several outcomes, including higher rates of IGA-CHE treatment success and larger mean reductions in HECSI. Although delgocitinib was consistently favoured in HESD scores and patient-reported outcomes such as DLQI and HEIS, the magnitude of the difference was small and not considered clinically meaningful. These outcomes, particularly symptom relief and quality of life, were consistently identified as important by both patients and clinicians.
The magnitude of benefit observed was considered clinically relevant and supported by moderate-certainty to high-certainty evidence in the vehicle-controlled studies. In the DELTA FORCE trial, the certainty of evidence was rated moderate, due primarily to concerns related to open-label design and differential dropout, with a substantially higher discontinuation rate in the alitretinoin group. These limitations may have biased estimates, especially for outcomes evaluated later in the study and for subjective end points.
The single-arm, open-label extension study (DELTA 3 trial) provided additional evidence supporting the durability of the treatment effect. Patients received delgocitinib intermittently (pausing treatment upon improvement and resuming if symptoms returned), which led to fluctuations in disease severity over the 36-week period. Despite this, improvements in HECSI and DLQI were generally maintained. At the baseline of the extension trial, 24.6% of patients in the previous delgocitinib group experienced IGA-CHE treatment success, and 30.0% experienced IGA-CHE treatment success by the end of the extension trial (week 36). In the previous vehicle cream group, 9.1% of patients experienced IGA-CHE treatment success at the baseline of the extension trial and 29.5% by the end of the extension trial (week 36). Loss of response was observed in a proportion of patients who initially experienced response, which may be explained by regression to the mean due to inherent variability and does not suggest a general trend of increased drug resistance. Overall, this suggests that while some patients who had not experienced response at baseline experienced IGA-CHE treatment success during the extension (approximately 24%), prolonged use of delgocitinib did not result in a meaningful overall increase in the proportion of patients who experience response. Although the trial was uncontrolled (single arm) and subject to the limitations noted in this report, its findings are consistent with the randomized trials and suggest that benefits may extend beyond the 16-week to 24-week duration evaluated in the primary studies.
Indirect evidence from the sponsor-submitted NMA and MAIC also explored the relative efficacy of delgocitinib. The NMA suggested delgocitinib may be more effective than phototherapy and alitretinoin for key outcomes such as IGA-CHE treatment success and HECSI-90. The MAIC did not identify a meaningful and precise difference between delgocitinib and dupilumab. However, both analyses were limited by methodological concerns, including sparse data, residual confounding, and differences in outcome definitions, reducing confidence in the findings.
There were some concerns with the choice of comparators in both ITCs. According to experts, PUVA therapy is not provided in Canada due to unavailability of psoralen. Narrow-band UVA is available and is deemed less effective than PUVA by the clinical experts consulted for this review. This means the results of the indirect comparison are conservative when put in the context of Canada. Additionally, dupilumab was positioned as an alternative to delgocitinib in patients with CHE who have used TCS. However, public drug plan policies generally require the failure of at least 1 systemic immunosuppressant before reimbursement of dupilumab for AD. Although payer criteria may vary across jurisdictions, the clinical experts indicated that in practice in Canada conventional systemic therapies are generally trialled before initiation of dupilumab. Therefore, indirect comparisons of delgocitinib with conventional systemic immunosuppressants would have been most relevant, and although a MAIC with dupilumab was conducted, this analysis was limited by feasibility, methodological, and data constraints. Moreover, patients with AD may only be eligible for dupilumab if they have an Eczema Area and Severity Index score of 16 or more. Although payer criteria may vary across jurisdictions, the clinical experts indicated that most patients with CHE are unlikely to qualify because this score considers the totality of lesion coverage throughout the body.
Nonetheless, the availability of direct comparative data with alitretinoin enhances the robustness of the evidence base and provides important context for decision-making. Interest holders (patients and clinicians) have highlighted the need for safe, effective topical treatment options in CHE — particularly for patients who cannot tolerate systemic therapies — and delgocitinib appears to address this unmet need. Importantly, the outcomes most valued by patient groups (e.g., symptom relief, visible skin improvement, enhanced quality of life) were directly assessed in the trials and showed consistent improvement with delgocitinib. The alignment between patient priorities, clinical expectations, and observed efficacy strengthens the relevance of the findings. Nonetheless, key uncertainties remain, including the long-term comparative effectiveness of delgocitinib, its performance in more diverse and real-world populations, and how its use may impact the broader CHE treatment framework, particularly the need for or avoidance of systemic agents such as methotrexate or cyclosporine.
Harms
Delgocitinib cream was generally well tolerated, with a favourable safety profile in both the short-term and longer-term studies. In the 16-week and 24-week trials (DELTA 1, DELTA 2, and DELTA FORCE trials), the incidence of AEs was lower with delgocitinib than with oral alitretinoin, and most reported AEs were mild or moderate in severity. SAEs and AESIs were rare and comparable across groups. The long-term extension study (DELTA 3 trial), which followed patients for an additional 36 weeks, did not identify new safety signals, and the nature and frequency of AEs remained consistent with those observed in the core studies. Importantly, no cumulative toxicities or generalized drug resistance were reported with prolonged exposure to delgocitinib.
However, in the DELTA FORCE trial, the open-label design and differential dropout (higher in the alitretinoin group due to AEs) may have led to underestimation of harms associated with the comparator and limited the ability to assess relative tolerability over time. Additionally, of the 2 indirect comparisons submitted (NMA and MAIC), only the NMA included a safety outcome — discontinuation due to AEs — which limits the ability of these analyses to inform the broader comparative safety profile of delgocitinib. Although the product monograph does not identify major contraindications or organ-specific toxicities, the exclusion of patients with significant comorbidities or concurrent use of systemic immunosuppressants in the clinical trials reduces the generalizability of the safety data in terms of rare or delayed AEs in real-world settings.
Compared to other treatments used for moderate to severe CHE, delgocitinib offers a safety advantage due to its topical route of administration and minimal systemic absorption. Unlike systemic agents such as alitretinoin, methotrexate, or cyclosporine, delgocitinib does not carry risks of systemic toxicity or teratogenicity or require laboratory monitoring. This aligns with both clinical expert and patient group input, which emphasized the need for effective nonsystemic options that are better tolerated and do not interfere with work or daily activities. From the patient’s perspective, the lower burden of treatment and absence of systemic side effects are significant advantages that may improve adherence and overall quality of life.
Conclusion
Considering the totality of evidence, delgocitinib cream 20 mg/g is supported as a clinically effective and well-tolerated treatment option for adults with moderate to severe CHE, particularly in those who have not experienced response to TCS and are seeking nonsystemic alternatives. Across 3 RCTs and 1 long-term extension study, delgocitinib consistently demonstrated improvements — compared to vehicle cream — in outcomes that align with priorities identified by patients and clinicians, including visible skin improvement, symptom relief, and enhanced quality of life. Compared to oral alitretinoin, the only other approved treatment for severe CHE in Canada, delgocitinib showed comparable or superior efficacy in several important outcomes, with a more favourable safety profile and fewer systemic adverse effects.
Indirect comparisons provide additional context regarding the relative efficacy of delgocitinib. In an NMA, delgocitinib was favoured over both phototherapy and alitretinoin across key efficacy outcomes. In contrast, a MAIC against dupilumab showed no clear difference between treatments. These findings, while informative, should be interpreted with caution due to methodological limitations, including sparse data, residual confounding, and differences in outcome definitions across studies — all of which may bias the estimates or widen the uncertainty around treatment effects. The relevance of comparisons with PUVA therapy is limited because oral psoralen is no longer available in Canada. Similarly, the comparison with dupilumab has restricted applicability, given its current use is limited to patients who have not experienced adequate disease control with conventional systemic immunosuppressants.
Overall, delgocitinib addresses an important unmet need by offering an effective topical, nonsteroidal alternative for patients with persistent CHE. Uncertainties remain regarding its long-term efficacy and safety end points, as well as its position within the broader treatment framework, including its potential to delay or reduce the need for systemic therapies.
References
1.Lynde C, Guenther L, Diepgen TL, et al. Canadian hand dermatitis management guidelines. J Cutan Med Surg. 2010;14(6):267-84. doi: 10.2310/7750.2010.09094 PubMed
2.Lee GR, Maarouf M, Hendricks AK, Lee DE, Shi VY. Current and emerging therapies for hand eczema. Dermatol Ther. 2019;32(3):e12840. doi: 10.1111/dth.12840 PubMed
3.Menne T, Johansen JD, Sommerlund M, Veien NK, Danish Contact Dermatitis G. Hand eczema guidelines based on the Danish guidelines for the diagnosis and treatment of hand eczema. Contact Dermatitis. 2011;65(1):3-12. doi: 10.1111/j.1600-0536.2011.01915.x PubMed
4.Bauer A, Brans R, Brehler R, et al. S2k guideline diagnosis, prevention, and therapy of hand eczema. J Dtsch Dermatol Ges. 2023;21(9):1054-1074. doi: 10.1111/ddg.15179 PubMed
5.Thyssen JP, Schuttelaar MLA, Alfonso JH, et al. Guidelines for diagnosis, prevention, and treatment of hand eczema. Contact Dermatitis. 2022;86(5):357-378. doi: 10.1111/cod.14035 PubMed
6.Scalone L, Cortesi PA, Mantovani LG, et al. Clinical epidemiology of hand eczema in patients accessing dermatological reference centres: results from Italy. Br J Dermatol. 2015;172(1):187-95. doi: 10.1111/bjd.13220 PubMed
7.Apfelbacher C, Molin S, Weisshaar E, et al. Characteristics and provision of care in patients with chronic hand eczema: updated data from the CARPE registry. Acta Derm Venereol. 2014;94(2):163-7. doi: 10.2340/00015555-1632 PubMed
8.Meding B, Wrangsjo K, Jarvholm B. Fifteen-year follow-up of hand eczema: persistence and consequences. Br J Dermatol. 2005;152(5):975-80. doi: 10.1111/j.1365-2133.2005.06494.x PubMed
9.Dubin C, Del Duca E, Guttman-Yassky E. Drugs for the Treatment of Chronic Hand Eczema: Successes and Key Challenges. Ther Clin Risk Manag. 2020;16:1319-1332. doi: 10.2147/TCRM.S292504 PubMed
10.Agner T, Elsner P. Hand eczema: epidemiology, prognosis and prevention. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12. doi: 10.1111/jdv.16061 PubMed
11.Silverberg JI, Guttman-Yassky E, Agner T, et al. Chronic Hand Eczema Guidelines From an Expert Panel of the International Eczema Council. Dermatitis. 2021;32(5):319-326. doi: 10.1097/DER.0000000000000659 PubMed
12.Zalewski A, Krajewski PK, Szepietowski JC. Psychosocial Consequences of Hand Eczema-A Prospective Cross-Sectional Study. J Clin Med. 2023;12(17). doi: 10.3390/jcm12175741 PubMed
13.Apfelbacher C, Bewley A, Molin S, et al. Prevalence of Chronic Hand Eczema in adults: A cross-sectional multi-national study of over 60,000 respondents in the general population. Paper presented at: Congress of the European Society of Contact Dermatitis; 2024; Dresden, Germany.
14.Molin S, Fargnoli M, Crépy M, et al. Self-reported disease severity and treatment of Chronic Hand Eczema from the CHECK study - A multinational study in six countries. Paper presented at: Congress of the European Society of Contact Dermatitis 2024; Dresden, Germany.
15.LEO Pharma Inc. Non-interventional study report: LPCHE-2252 Chronic Hand Eczema epidemiology, Care, and Knowledge of real-life burden (CHECK) [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
16.Leo Pharma Inc. Sponsor's clinical summary report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Delgocitinib, cream 20 mg/g topical. 2025.
17.Diepgen TL, Andersen KE, Chosidow O, et al. Guidelines for diagnosis, prevention and treatment of hand eczema. J Dtsch Dermatol Ges. 2015;13(1):e1-22. doi: 10.1111/ddg.12510_1 PubMed
18.Dalgard FJ, Gieler U, Tomas-Aragones L, et al. The psychological burden of skin diseases: a cross-sectional multicenter study among dermatological out-patients in 13 European countries. J Invest Dermatol. 2015;135(4):984-991. doi: 10.1038/jid.2014.530 PubMed
19.LEO Pharma Inc. Consultations with Canadian Dermatologists [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
20.Haddad EB, Cyr SL, Arima K, McDonald RA, Levit NA, Nestle FO. Current and Emerging Strategies to Inhibit Type 2 Inflammation in Atopic Dermatitis. Dermatol Ther (Heidelb). 2022;12(7):1501-1533. doi: 10.1007/s13555-022-00737-7 PubMed
21.Health Canada. Canadian labelling for all JAK inhibitors to include risks of serious heart-related problems, fatal blood clots and cancer. 2022. Accessed August 12, 2024. https://recalls-rappels.canada.ca/en/alert-recall/canadian-labelling-all-jak-inhibitors-include-risks-serious-heart-related-problems
22.LEO Pharma Inc. Delgocitinib: Cream, 20 mg/g, Topical [draft product monograph].
23.Dr. Reddy's Laboratories, Inc. HANZEMA [product monograph]. June 18, 2018. Updated October 20, 2022. Accessed July 26, 2024. https://pdf.hres.ca/dpd_pm/00067982.PDF
24.Sanofi-aventis Canada Inc. DUPIXENT (dupilumab injection) [product monograph]. 2020.
25.Bissonnette R, Warren RB, Pinter A, et al. Efficacy and safety of delgocitinib cream in adults with moderate to severe chronic hand eczema (DELTA 1 and DELTA 2): results from multicentre, randomised, controlled, double-blind, phase 3 trials. Lancet. 2024;404(10451):461-473. doi: 10.1016/S0140-6736(24)01027-4 PubMed
26.LEO Pharma. NCT04871711: Efficacy and Safety of Delgocitinib Cream in Adults With Moderate to Severe Chronic Hand Eczema (DELTA 1) [sponsor supplied reference]. ClinicalTrials.gov; 2022. Accessed 2022-11-02. https://clinicaltrials.gov/study/NCT04871711?tab=history
27.LEO Pharma. NCT04872101: Efficacy and Safety of Delgocitinib Cream in Adults With Moderate to Severe Chronic Hand Eczema (DELTA 2) (DELTA 2) [sponsor supplied reference]. ClinicalTrials.gov; 2023. Accessed 2023-01-09. https://clinicaltrials.gov/study/NCT04872101?a=4
28.Gimenez-Arnau AM, Pinter A, Sondermann W, et al. Efficacy and safety of topical delgocitinib cream versus oral alitretinoin capsules in adults with severe chronic hand eczema (DELTA FORCE): a 24-week, randomised, head-to-head, phase 3 trial. Lancet. 2025;405(10490):1676-1688. doi: 10.1016/S0140-6736(25)00001-7 PubMed
29.LEO Pharma Inc. Clinical Trial Report: LP0133-1401 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
30.LEO Pharma Inc. Clinical Trial Report: LP0133-1402 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
31.LEO Pharma Inc. Clinical Trial Report: LP0133-1528 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
32.Held E, Skoet R, Johansen JD, Agner T. The hand eczema severity index (HECSI): a scoring system for clinical assessment of hand eczema. A study of inter- and intraobserver reliability. Br J Dermatol. 2005;152(2):302-7. doi: 10.1111/j.1365-2133.2004.06305.x PubMed
33.Oosterhaven JAF, Schuttelaar MLA. Responsiveness and interpretability of the Hand Eczema Severity Index. Br J Dermatol. 2020;182(4):932-939. doi: 10.1111/bjd.18295 PubMed
34.Yuksel YT, Agner T, Ofenloch R. New evidence on the minimal important change (MIC) for the Hand Eczema Severity Index (HECSI). Contact Dermatitis. 2021. doi: 10.1111/cod.13828 PubMed
35.Molin S, Larsen LS, Joensson P, et al. Development and Psychometric Validation of a Patient-Reported Outcome Measure to Assess the Signs and Symptoms of Chronic Hand Eczema: The Hand Eczema Symptom Diary (HESD). Dermatol Ther (Heidelb). 2024;14(3):643-669. doi: 10.1007/s13555-024-01114-2 PubMed
36.Weisshaar E, Yuksel YT, Agner T, et al. Development and Validation of a Patient-Reported Outcome Measure of the Impact of Chronic Hand Eczema on Health-Related Quality of Life: the Hand Eczema Impact Scale (HEIS). Dermatol Ther (Heidelb). 2024;14(11):3047-3070. doi: 10.1007/s13555-024-01267-0 PubMed
37.Cortesi PA, Scalone L, Belisari A, et al. Cost and quality of life in patients with severe chronic hand eczema refractory to standard therapy with topical potent corticosteroids. Contact Dermatitis. 2014;70(3):158-68. doi: 10.1111/cod.12130 PubMed
38.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. doi: 10.1159/000365390 PubMed
39.Barrett A, Hahn-Pedersen J, Kragh N, Evans E, Gnanasakthy A. Patient-Reported Outcome Measures in Atopic Dermatitis and Chronic Hand Eczema in Adults. Patient. 2019;12(5):445-459. doi: 10.1007/s40271-019-00373-y PubMed
40.Agner T, Andersen KE, Brandao FM, et al. Hand eczema severity and quality of life: a cross-sectional, multicentre study of hand eczema patients. Contact Dermatitis. 2008;59(1):43-7. doi: 10.1111/j.1600-0536.2008.01362.x PubMed
41.Cvetkovski RS, Zachariae R, Jensen H, Olsen J, Johansen JD, Agner T. Quality of life and depression in a population of occupational hand eczema patients. Contact Dermatitis. 2006;54(2):106-11. doi: 10.1111/j.0105-1873.2006.00783.x PubMed
42.LEO Pharma Inc. Statistical Analysis Plan: LP0133-1401 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2022.
43.LEO Pharma Inc. Statistical Analysis Plan: LP0133-1402 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
44.LEO Pharma Inc. Statistical Analysis Plan: LP0133-1528 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
45.LEO Pharma Inc. Clinical Trial Report: LP0133-1403. DELTA 3. A phase 3 extension trial of DELTA 1 and DELTA 2 to evaluate the long-term safety of a twice-daily treatment with delgocitinib cream 20 mg/g as needed for up to 36 weeks in adult subjects with chronic hand eczema (DELTA 3) [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
46.Moher D, Shamseer L, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4(1):1. doi: 10.1186/2046-4053-4-1 PubMed
47.Guide to the methods of technology appraisal. National Institute for Health and Care Excellence; 2013.
48.Higgins JPT, Green, S (editors). Cochrane handbook for systematic reviews of interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration; 2011. www.cochrane-handbook.org
49.Symmetron. Network meta-analysis to assess the efficacy and safety of treatments for moderate to severe chronic hand eczema [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
50.Dias S, Welton N, Sutton A, Ades A. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials. University of Sheffield; 2011. Accessed November 2024. www.nicedsu.org.uk
51.Tonin FS, Rotta I, Mendes AM, Pontarolo R. Network meta-analysis: a technique to gather evidence from direct and indirect comparisons. Pharm Pract (Granada). 2017;15(1):943. doi: 10.18549/PharmPract.2017.01.943 PubMed
52.Brooks SP, Gelman A. General Methods for Monitoring Convergence of Iterative Simulations. J Comput Graph Stat. 1998;7(4):434-455. doi: 10.1080/10618600.1998.10474787
53.Worm M, Thyssen JP, Schliemann S, et al. The pan-JAK inhibitor delgocitinib in a cream formulation demonstrates dose response in chronic hand eczema in a 16-week randomized phase IIb trial. Br J Dermatol. 2022;187(1):42-51. doi: 10.1111/bjd.21037 PubMed
54.Gimenez-Arnau A, Pinter A, Sondermann W, et al. DELTA FORCE trial: A 24-week head-to-head Phase 3 trial comparing the efficacy and safety of topical delgocitinib cream with oral alitretinoin capsules in adults with severe Chronic Hand Eczema. Paper presented at: European Academy of Dermatology and Venereology (EADV) Congress; 2024; Amsterdam, Netherlands.
55.Ruzicka T, Larsen FG, Galewicz D, et al. Oral alitretinoin (9-cis-retinoic acid) therapy for chronic hand dermatitis in patients refractory to standard therapy: results of a randomized, double-blind, placebo-controlled, multicenter trial. Arch Dermatol. 2004;140(12):1453-9. doi: 10.1001/archderm.140.12.1453 PubMed
56.Fowler JF, Graff O, Hamedani AG. A phase 3, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of alitretinoin (BAL4079) in the treatment of severe chronic hand eczema refractory to potent topical corticosteroid therapy. J Drugs Dermatol. 2014;13(10):1198-204. PubMed
57.ISRCTN. Comparing treatments for severe chronic hand eczema [sponsor supplied reference]. 2015. https://trialsearch.who.int/Trial2.aspx?TrialID=ISRCTN80206075
58.Voorberg AN, Kamphuis E, Christoffers WA, Schuttelaar MLA. Efficacy and safety of dupilumab in patients with severe chronic hand eczema with inadequate response or intolerance to alitretinoin: a randomized, double-blind, placebo-controlled phase IIb proof-of-concept study. Br J Dermatol. 2023;189(4):400-409. doi: 10.1093/bjd/ljad156 PubMed
59.Simpson EL, Silverberg JI, Worm M, et al. Dupilumab treatment improves signs, symptoms, quality of life, and work productivity in patients with atopic hand and foot dermatitis: Results from a phase 3, randomized, double-blind, placebo-controlled trial. J Am Acad Dermatol. 2024;90(6):1190-1199. doi: 10.1016/j.jaad.2023.12.066 PubMed
60.Bissonnette R, Worm M, Shi VY, et al. Efficacy of Abrocitinib and Dupilumab on Chronic Hand Eczema in Patients With Moderate-to-Severe Atopic Dermatitis: Results From the Phase 3 JADE DARE Study [sponsor supplied reference]. Paper presented at: European Academy of Dermatology and Venereology; 2022;
61.Simpson EL, Rahawi K, Hu X, et al. Effect of upadacitinib on atopic hand eczema in patients with moderate-to-severe atopic dermatitis: Results from two randomized phase 3 trials. J Eur Acad Dermatol Venereol. 2023;37(9):1863-1870. doi: 10.1111/jdv.19194 PubMed
62.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-7. doi: 10.1016/j.jval.2012.05.004 PubMed
63.SAS [software]. Version 9.04.01M7P080520. SAS Institute Inc; 2023.
64.Phillippo D.M., Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE University of Sheffield; 2016. http://www.nicedsu.org.uk
65.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Med Decis Making. 2018;38(2):200-211. doi: 10.1177/0272989X17725740 PubMed
66.Symmetron. Matching-adjusted indirect comparison to assess the efficacy of delgocitinib (DELTA 1/2) and dupilumab (LIBERTY-AD-HAFT) for moderate to severe chronic hand eczema. [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
67.Cohen DE, Bewley A, Wollenberg A, et al. Matching-adjusted indirect comparison of the efficacy of delgocitinib and dupilumab in the treatment of moderate to severe atopic hand eczema [sponsor supplied reference]. Paper presented at: European Academy of Dermatology and Venereology (EADV) Congress; 2024; Amsterdam, Netherlands.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CHE
chronic hand eczema
CUA
cost-utility analysis
ICER
incremental cost-effectiveness ratio
IGA-CHE
Investigator’s Global Assessment of Chronic Hand Eczema
JAK
Janus kinase
MAIC
matching-adjusted indirect comparison
NMA
network meta-analysis
QALY
quality-adjusted life-year
TCS
topical corticosteroids
Executive Summary
Item | Description |
|---|---|
Drug product | Delgocitinib (Anzupgo), 20 mg/g topical cream |
Indication | For the treatment of moderate to severe chronic hand eczema (CHE) in adults for whom topical corticosteroids are inadequate or are not advisable |
Submitted price | $636.98 per tube |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 25, 2025 |
Reimbursement request | Per indication |
Sponsor | Leo Pharma Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Economic Review
The objective of the economic review undertaken by Canada’s Drug Agency (CDA-AMC) is to review and critically appraise the pharmacoeconomic evidence submitted by the sponsor on the cost-effectiveness of delgocitinib compared to alitretinoin, phototherapy, and dupilumab for the treatment of moderate to severe chronic hand eczema (CHE) in adults for whom topical corticosteroids (TCS) are inadequate or are not advisable. The sponsor submitted a budget impact analysis (BIA) assessing the budgetary impact of reimbursing delgocitinib for the same population.
Summary
Delgocitinib is available as a topical cream (20 mg/g), commercialized in 60 g tubes. At the submitted price of $636.98 per tube, the cost per year of delgocitinib is expected to range from $637 to $5,096 per patient, based on the Health Canada–recommended dosing1 and the maximum consumption based on discontinuation rules assumed by the sponsor and clinical expert feedback.
Clinical efficacy in the economic analysis for delgocitinib versus alitretinoin and for delgocitinib versus phototherapy was derived from the DELTA 1,2 DELTA 2,2 DELTA 3,3 DELTA FORCE,4 and ALPHA5 trials, as well as the network meta-analysis (NMA) conducted by the sponsor. Evidence in the DELTA FORCE trial indicates that delgocitinib showed superior efficacy in terms of rates of Investigator’s Global Assessment of Chronic Hand Eczema (IGA-CHE) treatment success and improvements in the Hand Eczema Severity Index when compared with alitretinoin among adult patients with moderate to severe CHE, particularly those who have not experienced response to TCS and are seeking nonsystemic alternatives. The DELTA FORCE trial showed improvements in quality-of-life end points compared to alitretinoin; however, these were not considered clinically meaningful. The NMA suggested delgocitinib may be more effective than phototherapy and alitretinoin for key outcomes such as IGA-CHE treatment success and at least a 90% improvement in Hand Eczema Severity Index score from baseline. For delgocitinib versus dupilumab, clinical efficacy was informed by a sponsor-submitted matching-adjusted indirect comparison (MAIC), which did not identify a meaningful and precise difference between delgocitinib and dupilumab for the same outcomes. However, both the NMA and the MAIC were limited by methodological concerns, including sparse data, residual confounding, and differences in outcome definitions, reducing confidence in the findings.
The results of the CDA-AMC base case suggest the following:
Delgocitinib is predicted to be associated with higher costs to the health care system than alitretinoin (incremental costs = $1,862) and phototherapy (incremental costs = $4,174), primarily driven by increased costs associated with treatment acquisition.
Delgocitinib is not predicted to be associated with life-year gains compared to any comparator. Delgocitinib is predicted to be associated with a gain of 0.04 quality-adjusted life-years (QALYs) compared to alitretinoin and 0.05 QALYs compared to phototherapy, over a 30-year time horizon. These results are primarily driven by the incremental QALYs accrued in the response states.
The incremental cost-effectiveness ratio (ICER) of delgocitinib in the CDA-AMC base case was $47,393 per QALY gained compared to alitretinoin and $79,192 per QALY gained compared to phototherapy. More than 90% of the QALY differences between delgocitinib and the comparators were accrued in the extrapolated period.
The results of the CDA-AMC base case are highly uncertain due to the lack of comparative evidence for several model inputs and the lack of long-term clinical evidence. The estimated ICERs were highly sensitive to the assumptions around how delgocitinib is dispensed. The cost-effectiveness of delgocitinib compared to immunosuppressants and other relevant comparators excluded from the model is unknown. The cost-effectiveness of delgocitinib versus dupilumab is uncertain, given the limitations in the evidence presented and the uncertainties regarding delgocitinib’s place in therapy relative to dupilumab.
The sponsor-submitted BIA was associated with major limitations that precluded an accurate estimation of the budget impact.
Summary of the Submitted Economic Evaluation
The sponsor submitted a cost-utility analysis (CUA) to estimate the cost-effectiveness of delgocitinib from the perspective of a public health care payer in Canada over a 30-year horizon. The modelled population comprised adult patients with moderate to severe CHE, which is aligned with the proposed Health Canada indication, and was based on the participants in the DELTA 12 and DELTA 22 trials. The sponsor’s base-case analysis included costs related to drug acquisition and monitoring, health care usage, next-line treatment, and adverse events.
In the sponsor’s base case, delgocitinib was associated with the following incremental costs and QALYs relative to the different comparators:
Compared to alitretinoin: ICER = $6,335 per QALY gained (incremental costs = $247; incremental QALYs = 0.04)
Compared to phototherapy: ICER = $44,692 per QALY gained (incremental costs = $2,347; incremental QALYs = 0.05)
Compared to dupilumab: ICER = $217,165 saved per QALY lost (costs savings = –$72,705; QALY losses = –0.34)
Of the differences in QALYs estimated between delgocitinib and the comparators, more than 90% are estimated to have accrued beyond the period with available comparative clinical data (e.g., the DELTA FORCE trial had 24 weeks compared to alitretinoin; and the studies included in the NMA and the MAIC had 12 weeks compared to phototherapy and 16 weeks compared to dupilumab). Additional information about the sponsor’s submission is summarized in Appendix 3. Sequential analysis is of limited value for decision-making and not presented in this report because the comparative evidence across the different comparators is from different sources (NMA and MAIC).
CDA-AMC identified several key issues with the sponsor’s analysis (refer to Table 2; full details are provided in Appendix 4).
Table 2: Key Issues With the Sponsor’s Economic Submission
Issue | What evidence is there to inform this issue? | How was this issue addressed by CDA-AMC? | Did CDA-AMC explore the uncertainty in a scenario analysis? |
|---|---|---|---|
The economic model does not fully reflect the expected place in therapy, and relevant comparators were excluded. | Other off-label treatments for CHE, including upadacitinib, abrocitinib, methotrexate, cyclosporine, azathioprine, and mycophenolate mofetil, were not included in the economic model due to the lack of comparative data. According to clinical expert input, immunosuppressants are expected to be displaced before delgocitinib can displace dupilumab. Currently, access to dupilumab is generally conditional on the unsuccessful trial of ≥ 1 systemic immunosuppressant (e.g., methotrexate, cyclosporine). | CDA-AMC could not address this issue in the base case due to insufficient clinical evidence and exclusion from the model structure. | No scenario analysis was conducted. |
The comparative effectiveness of delgocitinib vs. comparators is uncertain, and the model assumptions may not reflect clinical practice in Canada. | Several model inputs were assumed to be the same across comparators, or their differences were based on naive comparisons across trials (e.g., relapse rates, probability of discontinuation, proportion of patients opting out of a successful treatment upon relapse), due to the absence of head-to-head clinical data. Additionally, the sponsor assumed that patients who experienced only minor response would switch treatment after 24 weeks. However, the clinical experts noted that, in clinical practice in Canada, such patients are recommended to switch treatment after 12 weeks. | CDA-AMC could not address this issue in the base case due to the absence of clinical evidence. | To explore some of the uncertainty around this issue, CDA-AMC conducted a scenario analysis in which patients who experienced only minor response to initial treatment at week 12 switch to next-line treatment. Comparison with dupilumab was excluded from the CDA-AMC base case and presented in scenario analysis. |
The approach used to estimate delgocitinib consumption underestimates drug acquisition costs. | Delgocitinib consumption was sourced from the DELTA trials6-8 and estimated based on the number of grams used. This method does not reflect clinical practice and does not account for wastage because topical creams are dispensed in tubes and delgocitinib stability is 12 months after opening. The drug acquisition costs for 50% of patients at model entry were excluded. | CDA-AMC estimated delgocitinib costs assuming a new tube is dispensed at the beginning of treatment and each time the cumulative consumption exceeds the tube volume or 12 months after opening, until the proportion of patients on treatment falls below 1% in the cohort. | To explore the maximum potential number of tubes dispensed, CDA-AMC explored a simplified approach assuming that the proportion of patients still on treatment in each of the mutually exclusive health states could be dispensed a maximum of 1 tube per cycle. |
The utility value in the next-line health state is uncertain. | The utility of patients under next-line treatment was the weighted average of all response states from the DELTA trials. This resulted in lower utilities than patients experiencing partial response to second-line treatment. The clinical experts indicated that it would be reasonable to expect that patients receiving next-line treatment would have a quality of life at least comparable to those experiencing partial response. | CDA-AMC could not address this issue in the base case due to the absence of clinical evidence. | To explore some uncertainty around this issue, CDA-AMC conducted a scenario analysis around next-line utility. |
The proportion of patients who, following disease relapse after an initial treatment response to alitretinoin and phototherapy, do not reinitiate the same therapeutic regimen is uncertain. | The sponsor sourced these proportions from the ALPHA trial,5 reporting 52.1% for alitretinoin and 95.6% for phototherapy (which is limited to 16 weeks). However, the clinical experts indicated that it is reasonable to expect that the majority of patients who experienced a successful response to a treatment would accept the same treatment option, unless they have experienced adverse events (more likely to occur with alitretinoin than with phototherapy). | CDA-AMC could not address this issue in the base case due to the absence of comparative clinical evidence. | To explore some uncertainty around this issue, CDA-AMC conducted a scenario analysis assuming 35% for both alitretinoin and phototherapy, based on clinical expert input. |
CDA-AMC = Canada’s Drug Agency; CHE = chronic hand eczema; vs. = versus.
Note: Full details of the issues identified by CDA-AMC are provided in Appendix 4.
CDA-AMC Assessment of Cost-Effectiveness
The CDA-AMC base case was derived by making changes to model parameter values and assumptions (refer to Table 7), in consultation with clinical experts. Detailed information about the CDA-AMC base case is provided in Appendix 4.
Impact on Health Care Costs
Delgocitinib is predicted to be associated with additional health care costs compared to alitretinoin (incremental costs = $1,862) and phototherapy (incremental costs = $4,174). In all comparisons, differences in health care spending were primarily owing to differences in drug acquisition cost (refer to Figure 1).
Impact on Health
Relative to alitretinoin and phototherapy, delgocitinib is expected to result in 0.04 and 0.05 additional QALYs per patient, respectively. The impact on health relative to immunosuppressants is unknown because they were not included in the cost-effectiveness analysis (refer to Table 8).
Overall Results
The results of the CDA-AMC base case suggest an ICER of $47,393 per QALY gained for delgocitinib compared to alitretinoin and an ICER of $79,192 per QALY gained compared to phototherapy (refer to Table 3). Additional details on the CDA-AMC base case are available in Appendix 4.
Figure 1: Impact of Delgocitinib Versus Comparators on Health Care Costs
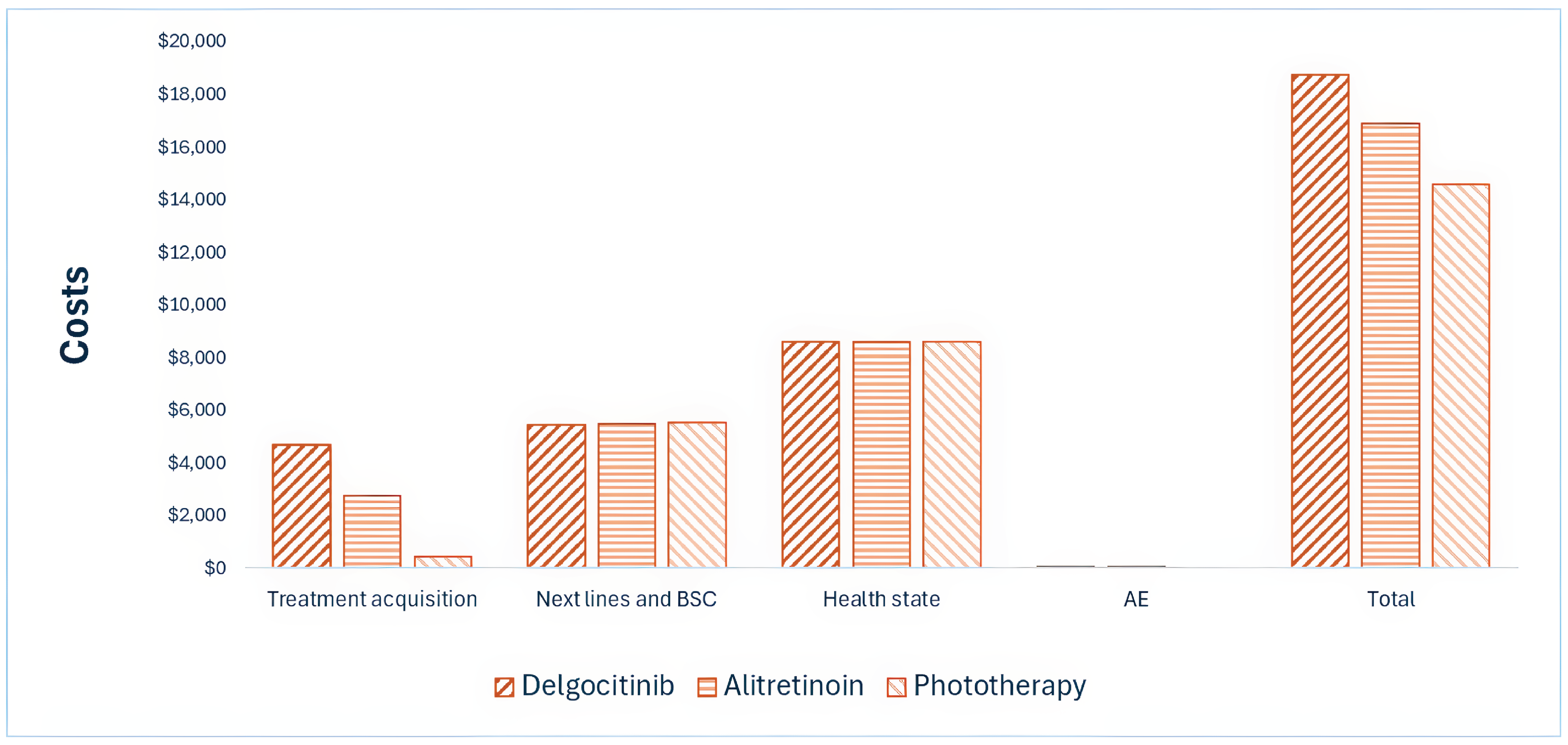
AE = adverse event; BSC = best supportive care.
Note: The impact on health care costs relative to immunosuppressants is unknown because they were not included in the cost-effectiveness analysis. Refer to Appendix 4 for full results.
Table 3: Summary of CDA-AMC Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Pairwise ICER ($/QALY) |
|---|---|---|---|
Delgocitinib vs. alitretinoin | |||
Alitretinoin | 16,880 | 15.71 | Reference |
Delgocitinib | 18,742 | 15.75 | 47,393 |
Delgocitinib vs. phototherapy | |||
Phototherapy | 14,568 | 15.70 | Reference |
Delgocitinib | 18,742 | 15.75 | 79,192 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Notes: Publicly available list prices were used for all comparators. The cost-effectiveness of delgocitinib relative to immunosuppressants is unknown because they were not included in the analysis.
Uncertainty and Sensitivity
Uncertainty was explored in scenario analyses as outlined in Table 2. Uncertainty around the consumption of delgocitinib had the largest impact on cost-effectiveness (refer to Table 11, Appendix 4). Based on the results of these analyses, the ICER for delgocitinib may range up to $97,524 per QALY gained compared to alitretinoin and up to $118,202 per QALY gained to compared to phototherapy. The scenario compared to dupilumab suggests an ICER of $221,499 saved per QALY lost compared to dupilumab because delgocitinib was less costly and less effective.
Summary of the Budget Impact
The sponsor submitted a BIA to estimate the 3-year (2025 to 2028) budget impact of reimbursing delgocitinib for use in patients with moderate to severe CHE who have experienced an inadequate response to TCS or for whom TCS are not advisable. The sponsor assumed that the payer would be a CDA-AMC–participating public drug plans and derived the size of the eligible population using an epidemiologic approach. The price of delgocitinib was aligned with the price included in the sponsor’s economic evaluation, while the prices of comparators were based on the publicly available list prices. Additional information pertaining to the sponsor’s submission is provided in Appendix 5.
CDA-AMC identified a number of issues with the sponsor’s estimated budget impact that could not be addressed. Although the sponsor estimates that reimbursing delgocitinib for use in the indicated population will be cost saving for the public drug plans, this result is highly uncertain and will depend on the consumption of delgocitinib in clinical practice, the average annual costs paid by the public drug plans for drug comparators and their subsequent treatments, the market uptake of delgocitinib among existing patients already being treated with systemic therapies, and how delgocitinib may displace other treatment options not included in the BIA. Whether there will be cost savings realized by the drug plans, and the extent of these savings, is highly uncertain.
Conclusion
Based on the CDA-AMC base case, delgocitinib would be considered cost-effective at the submitted price if the public health care system was willing to pay at least $47,393 per QALY gained compared to alitretinoin and $79,192 per QALY gained compared to phototherapy. If the public health care system is not willing to pay that amount, a price reduction should be considered (refer to Figure 2). Full details of the impact of price reductions on cost-effectiveness are presented in Table 10. Immunosuppressants were deemed relevant comparators for delgocitinib; however, the cost-effectiveness of delgocitinib relative to these comparators is unknown because they were not included in the analysis. Given the limitations with the clinical evidence and the uncertainty around dupilumab’s place in therapy, the results against dupilumab are limited.
The sponsor-submitted BIA was associated with major limitations that precluded an accurate estimation of the budget impact. Although CDA-AMC estimated a drug expenditure of approximately $22 million on delgocitinib over the first 3 years, this estimate may represent an upper limit given the optimistic market uptake projections (refer to Table 14 for alternative assumptions). Costs associated with other comparators could not be reliably estimated given that the submitted structure of the analysis precludes accurate assessment of costs and impact of subsequent therapies. The largest impact on the submitted BIA comes from any potential reduction in dupilumab use. Without accounting for its use as a subsequent therapy, the results of the analysis are misleading.
Figure 2: Summary of the CDA-AMC Economic Analysis and Price Reduction

CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Notes: The upper end of the annual cost of delgocitinib is presented (refer to Table 4 for full information). Expenditure includes only the drug cost of delgocitinib and refers to the simplified approach that assumes the same market uptake as submitted by the sponsor (refer to Table 14). The term “dominant” indicates that a drug costs less and provides more QALYs than a comparator. Immunosuppressants were deemed relevant comparators, but price reductions required against these comparators were not possible to estimate because they were not included in the cost-effectiveness analysis. Delgocitinib costs are higher than immunosuppressant costs (refer to Table 4).
References
1.LEO Pharma Inc. TRADENAME (delgocitinib): 20mg/g topical cream [product monograph].
2.Bissonnette R, Warren RB, Pinter A, et al. Efficacy and safety of delgocitinib cream in adults with moderate to severe chronic hand eczema (DELTA 1 and DELTA 2): results from multicentre, randomised, controlled, double-blind, phase 3 trials. Lancet. 2024;404(10451):461-473. doi: 10.1016/S0140-6736(24)01027-4 PubMed
3.LEO Pharma Inc. DELTA 3 and DELTA FORCE post-hoc analysis [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
4.Gimenez-Arnau A, Pinter A, Sondermann W, et al. DELTA FORCE trial: A 24-week head-to-head Phase 3 trial comparing the efficacy and safety of topical delgocitinib cream with oral alitretinoin capsules in adults with severe Chronic Hand Eczema [sponsor supplied reference]. Paper presented at: European Academy of Dermatology and Venereology (EADV) Congress; 2024; Amsterdam, Netherlands.
5.Wittmann M, Smith IL, Brown ST, et al. Alitretinoin versus phototherapy as the first-line treatment in adults with severe chronic hand eczema: the ALPHA RCT. Health Technol Assess. 2024;28(59):1-123. doi: 10.3310/TWQC0141 PubMed
6.LEO Pharma Inc. Clinical Trial Report: LP0133-1528 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
7.LEO Pharma Inc. Clinical Trial Report: LP0133-1401 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
8.LEO Pharma Inc. Clinical Trial Report: LP0133-1402 [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
9.Institut national d'excellence en sante et en services sociaux. INESSS Submission Guide for Drugs, Blood System Products and Medical Devices Related to the Administration of Drugs [sponsor supplied reference]. 2024. Accessed August 2024. https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Fiches_inscription/en/Submission_guidance_document.pdf
10.Ontario Ministry of Health. Schedule of Benefits - Physician Services Under the Health Insurance Act [sponsor supplied reference]. 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
11.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
12.Symmetron. Network meta-analysis to assess the efficacy and safety of treatments for moderate to severe chronic hand eczema [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
13.Symmetron. Matching-adjusted indirect comparison to assess the efficacy of delgocitinib (DELTA 1/2) and dupilumab (LIBERTY-AD-HAFT) for moderate to severe chronic hand eczema. [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
14.Worm M, Simpson EL, Thaci D, et al. Efficacy and Safety of Multiple Dupilumab Dose Regimens After Initial Successful Treatment in Patients With Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2020;156(2):131-143. doi: 10.1001/jamadermatol.2019.3617 PubMed
15.LEO Pharma Inc. Non-interventional study report: LPCHE-2252 Real-World trEatment & mAnagement of chronic hand eczema in cLinical practice (RWEAL) [CONFIDENTIAL data on file] [sponsor supplied reference]. 2024.
16.Statistics Canada. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island [sponsor supplied reference]. 2023. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
17.LEO Pharma Inc. Delgocitinib HTA04. DELTA trials analysis request to support decision-modelling of delgocitinib vs comparators for HTA Payer submission statistical appendix [CONFIDENTIAL data on file] [sponsor supplied reference]. 2023.
18.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-18. doi: 10.1111/j.1524-4733.2010.00700.x PubMed
19.Ara R, Brazier JE. Using health state utility values from the general population to approximate baselines in decision analytic models when condition-specific data are not available. Value Health. 2011;14(4):539-545. PubMed
20.Falk Hvidberg M, Hernandez Alava M. Catalogues of EQ-5D-3L Health-Related Quality of Life Scores for 199 Chronic Conditions and Health Risks for Use in the UK and the USA. Pharmacoeconomics. 2023;41(10):1287-1388. doi: 10.1007/s40273-023-01285-4 PubMed
21.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed November 22, 2024. https://www.formulary.health.gov.on.ca/formulary/
22.McKesson. McKesson Price List, August 2024 [sponsor supplied reference]. 2024.
23.Shoppers Drug Mart. Cerave moisturizing cream for normal to dry skin [sponsor supplied reference]. 2021. Accessed March 27, 2021. https://shop.shoppersdrugmart.ca/Shop/Categories/Health/Medicine-%26-Treatments/First-Aid/Skin-Relief/Eczema-Care-Moisturizing-Cream/p/BB_062600064363?variantCode=062600064363
24.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Ontario Ministry of Health; 2023. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
25.LEO Pharma Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TRADENAME (delgocitinib), 20mg/g topical cream. November 25, 2024.
26.Smith IL, Gilberts R, Brown S, et al. Comparison of ALitretinoin with PUVA as the first-line treatment in patients with severe chronic HAnd eczema (ALPHA): study protocol for a randomised controlled trial. BMJ Open. 2022;12(2):e060029. doi: 10.1136/bmjopen-2021-060029 PubMed
27.CADTH Reimbursement Review: Tralokinumab. Accessed July 14, 2025. https://www.cda-amc.ca/tralokinumab
28.CADTH Reimbursement Review: Lebrikizumab. Accessed July 14, 2025. https://www.cda-amc.ca/lebrikizumab
29.CADTH Reimbursement Review: Ruxolitinib. Accessed July 14, 2025. https://www.cda-amc.ca/ruxolitinib-1
30.CDA-AMC. Reimbursement Review: Nemolizumab. Accessed July 28, 2025. https://www.cda-amc.ca/nemolizumab-0
31.LEO Pharma Inc. LEO Pharma Inc. response to Canada's Drug Agency request for additional information regarding delgocitinib (ANZUPGO) review on June 6, 2025: additional information on product stability and product sizes to be marketed in Canada [internal additional sponsor's information]. June 6, 2025.
32.pan-Canadian Pharmaceutical Alliance. Dupixent (dupilumab). 2023. Accessed July 28, 2025. https://www.pcpacanada.ca/negotiation/22214
33.pan-Canadian Pharmaceutical Alliance. Cibinqo (abrocitinib). 2023. Accessed July 28, 2025. https://www.pcpacanada.ca/negotiation/21987
34.pan-Canadian Pharmaceutical Alliance. Rinvoq (upadacitinib). 2023. Accessed July 28, 2025. https://www.pcpacanada.ca/negotiation/21907
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
Table 4: Cost Comparison for the Treatment of Moderate to Severe CHE
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Delgocitinib (Anzupgo) | 20 mg/g | Topical cream 60 g per tube | 636.9800a | A thin layer should be applied twice daily to the affected skin of the hands and wrists until the skin is clear or almost clear. In the event of recurrence of the signs and symptoms of CHE (flares), twice-daily treatment of the affected areas should be re-initiated as needed. | 1.74 to 13.95 | 637 to 5,096b |
Recommended | ||||||
Retinoids | ||||||
Alitretinoin (Toctino) | 10 mg 30 mg | Capsule | 16.9869 | 30 mg once daily, dose may be reduced to 10 mg if unacceptable side effects | 1.30 to 15.63 | 476 to 5,708b,c |
Nondrug intervention | ||||||
Phototherapy | NA | NA | 7.8500d | 3 sessions per week | 0.26 to 3.09 | 94 to 1,130b |
Off-label systemic therapies | ||||||
Monoclonal antibody (indicated for AD) | ||||||
Dupilumab (Dupixent) | 200 mg/ 1.14 mL 300 mg/ 2 mL | Prefilled syringe Single-use pen or single-use syringe | 978.7000e 978.7000e | 600 mg as an initial dose, followed by 300 mg every 2 weeks, continuous use | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,425 Year 2+: 25,446 |
JAKi (indicated for AD) | ||||||
Abrocitinib (Cibinqo) | 50 mg 100 mg 200 mg | Tablet | 48.6667e 48.6667e 54.4667e | 100 or 200 mg once daily, minimum 12 weeks to continuous use | Year 1: 11.19 to 54.47 Year 2+: 48.67 to 54.47 | Year 1: 4,088 to 19,894 Year 2+: 17,776 to 19,894 |
Upadacitinib (Rinvoq) | 15 mg 30 mg | Extended-release tablet | 51.6810e 76.9600e | 15 or 30 mg, once daily, minimum 12 weeks to continuous use | Year 1: 11.89 to 76.96 Year 2+: 51.68 to 76.96 | Year 1: 4,341 to 28,110 Year 2+: 18,876 to 28,110 |
Immunosuppressants | ||||||
Azathioprine | 50 mg | Tablet | 0.5185 | 1.0 to 3.0 mg/kg per day, minimum 12 weeks and up to 6 monthsf | 0.24 to 1.30 | 189 to 473h |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7676 0.8657 1.6885 3.3792 | 2.5 to 5 mg/kg per day, minimum 12 weeks and up to 6 monthsf | 1.87 to 7.06 | 684 to 2,580g |
Methotrexate (generic) | 2.5 mg | Tablet | 0.2513 | 7.5 to 25 mg per week minimum 12 weeks to continuous usef | Year 1: 0.02 to 0.36 Year 2+: 0.11 to 0.36 | Year 1: 9 to 131 Year 2+: 39 to 131 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | 2,000 to 13,000 mg daily minimum 12 weeks to continuous usef | Year 1: 0.68 to 19.30 Year 2+: 2.97 to 19.30 | Year 1: 249 to 7,049 Year 2+: 1,085 to 7,049 |
AD = atopic dermatitis; CHE = chronic hand eczema; JAKi = Janus kinase inhibitor; NA = not applicable.
Notes: All prices are from the Ontario Drug Benefit Formulary (accessed May 2025), unless otherwise indicated, and do not include dispensing fees or markups. Annual period assumes 365.25 days, average weight of 76 kg for adults.9 Recommended dosages are based on respective product monographs for CHE or AD, unless otherwise indicated.
aSponsor’s submitted price.
bAnnual cost for treatments with intermittent use are presented as a range based on minimum and maximum utilization. The minimum consumption of delgocitinib assumes at least 1 tube (for cream) or discontinuation at week 4 due to no response (alitretinoin or phototherapy). Maximum weekly usage of cream was assumed to be 10 g per week as per clinical expert input, deemed reasonable to cover up to 75% surface of both hands. The maximum consumption assumes patients receive 12 weeks of initial treatment and due to partial response may continue treatment for another 12 weeks to achieve full response, then discontinue assuming full response was achieved (stopping rules assumed by the sponsor), and may experience relapse after 4 weeks, and reinitiate treatment that may last another 24 weeks following the same stopping rules.
cOne capsule of strength of 30 mg was assumed to reach a 30 mg once daily dosage.
dOntario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy.”10
eOntario Exceptional Access Program formulary list price (accessed May 2025).11
fRecommended dosage based on clinical expert input.
gDaily usage ranged from 190 mg to 380 mg. Costs were estimated assuming rounding up the number and combination of capsules to achieve recommended daily dosage.
hDaily usage ranged from 76 mg to 228 mg. Costs were estimated assuming rounding up the number and combination of capsules to achieve recommended daily dosage.
Appendix 2: Input Relevant to the Economic Review
Please note that this appendix has not been copy-edited.
This section is a summary of the input received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from the Canadian Association of Neonatal Nurses and Eczema Society of Canada (endorsed by the Canadian Skin Patient Alliance), primarily involving patients with CHE. Input was collected through interviews, questionnaires, and surveys in patients with CHE in Canada (i.e., nurses living with CHE). Input noted that current treatments, primarily TCS and occasionally oral steroids or phototherapy, often provide limited and temporary relief, with many expressing concerns about long-term side effects, cost, and inconvenience. Despite adherence to prescribed treatments, many patients continue to experience frequent, painful flares that interfere with daily functioning, work, and quality of life. There is a strong desire for a safe, effective, and long-term treatment that can reduce symptoms, improve disease control, and help patients regain normalcy in their lives. Some respondents had direct experience with delgocitinib through clinical trials and reported significant improvement, including complete symptom relief. They highlighted its effectiveness, ease of use, and better tolerability compared to previous treatments, noting improvements in daily functioning, ability to work, and overall quality of life.
Clinician input was received from Atlantic Dermatology Group, Canadian Dermatology Association and Dermatology Association of Ontario. Input noted that CHE care starts with avoidance strategies and emollients, followed by high-potency TCS as first-line treatment. If uncontrolled, patients may try phototherapy or alitretinoin, though both have limitations. Phototherapy is time-consuming, variably effective, and often inaccessible in rural areas. Alitretinoin has limited public coverage, requires lab monitoring, and poses safety concerns. Off-label systemic treatments are used when needed but pose safety and access challenges. Clinicians expect delgocitinib to fill a major treatment gap as a safe, effective, and well-tolerated topical option for adults with moderate to severe CHE who have not responded to, or cannot use TSC. It is anticipated to shift current practice by replacing or delaying the need for phototherapy, alitretinoin, or systemic therapies. Its use would reduce the burden of side effects, monitoring, and health care visits associated with systemic options. Delgocitinib is viewed as a preferred second-line therapy, offering targeted treatment with minimal systemic absorption and ease of use, making it suitable for general practice and not limited to specialist care.
Input from CDA-AMC–participating drug plans raised concerns about the use of a cream vehicle as the comparator rather than other topical therapies, and noted that alitretinoin, although included as a comparator, is not publicly funded in all provinces. Plans inquired about the commonly used instruments to assess the severity of CHE in clinical practice and questioned the potential use of delgocitinib in pediatric populations. They also asked whether prescribing should be restricted to dermatologists and whether delgocitinib would be eligible for use in combination with other treatments, such as topical calcineurin inhibitors or biologics.
Several of these concerns were addressed in the sponsor’s model:
Safety of delgocitinib was modelled by including the adverse event–related disutility.
Phototherapy and dupilumab were included as comparators in the economic model.
CDA-AMC addressed some of these concerns as follows:
IGA-CHE is used as the instrument to define treatment response in the CDA-AMC base case because the clinical experts noted that it is the only instrument that is used in clinical practice.
CDA-AMC was unable to address the following concerns:
The comparative effectiveness of delgocitinib versus off-label treatments (not included in the CUA) or in combination with other treatments is unknown and it lacks comparative clinical evidence.
Appendix 3: Summary of the Sponsor’s Submission
Please note that this appendix has not been copy-edited.
Summary of the Sponsor’s Economic Evaluation
For the pharmaceutical reviews program, clinical and economic information is submitted to CDA-AMC by the sponsor. The CDA-AMC health economics team reviews the submitted economic information and appraises the information in collaboration with clinical experts and the clinical review team to evaluate key assumptions, influential parameters, and the overall rigour of the economic submission. Based on what the team learns through this process, adjustments may be made to the sponsor’s model to produce the CDA-AMC base case. The CDA-AMC base case represents the team’s current understanding of the clinical condition, clinical evidence currently available, and best interpretation of the economic evidence based on the information provided.
For the review of delgocitinib, the sponsor provided a CUA and BIA. The sponsor’s economic submission is summarized in Table 5.
Table 5: Key Components of the Sponsor’s Economic Evaluation
Component | Description |
|---|---|
Treatment information | |
Drug under review | Delgocitinib (Anzupgo), topical cream (20 mg/g), supplied in 60 g laminated tubes |
Submitted price of drug under review | $636.98 per tube |
Regimen | A thin layer should be applied twice daily to the affected skin of the hands and wrists, until the skin is clear or almost clear. In the event of recurrence of the signs and symptoms of CHE (flares), twice-daily treatment of the affected areas should be re-initiated as needed. |
Weekly cost of drug under review | Per patient weekly drug consumption and costs vary by treatment response:6-8
|
Model information | |
Type of economic evaluation | CUA Markov model |
Treatment | Delgocitinib |
Included comparators |
|
Perspective | Publicly funded health care payer perspective |
Time horizon | 30 years |
Cycle length | 4 weeks |
Modelled population | Adult patients with moderate to severe CHE who have had an inadequate response to, or for whom TCS are not advisable. |
Characteristics of modelled population | Derived from the DELTA 1/2 trials2 (mean age: 44 years; sex = 64.4% female; 35.6% male), weight = 78.6 kg) |
Model health states |
For additional information, refer to Model Structure |
Data sources | |
Comparative efficacy |
|
Natural history and/or clinical pathway |
|
Health-related utilities and disutilities |
|
Costs |
|
Summary of the submitted results | |
Base-case results |
|
Scenario analysis results |
|
AE = adverse event; BSC = best supportive care; CHE = chronic hand eczema; CUA = cost-utility analysis; EQ-5D = European Quality of Life 5 Dimensions; JAKi = Janus kinase inhibitor; HECSI = Hand Eczema Severity Index; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; IGA = investigator global assessment; MAIC = matching-adjusted indirect comparison; NL = next-line; NMA = network meta-analysis; OR = odds ratio; QALY = quality-adjusted life-year; TCS = topical corticosteroids; WTP = willingness to pay.
Model Structure
The sponsor submitted a Markov cohort state-transition model with 7 health states, based on treatment response defined by the IGA-CHE scale and line of therapy. They are: full response (IGA 0/1), partial response (IGA 2), low response (IGA 3 with 1-point improvement), insufficient response (IGA 4 or IGA 3 without improvement), next-line treatment, best supportive care, and death. Patients with moderate to severe CHE who are refractory to TCS enter the model and receive initial treatment. Movement between states occurs in 4-week cycles and reflects response, treatment continuation, relapse, or transition to next-line treatment health state and then to best supportive care health state. Patients can discontinue treatment when achieving full response and initiate re-treatment when they subsequently relapse. Patients who discontinue treatment for any reason other than the achievement of full response or patients who are considered insufficient responders to treatment proceed to next-line treatments. Patients receiving next-line treatment can either continue with the next-line treatment or discontinue to best supportive care. From best supportive care, the only further transition patients can make is to death. Patients can die from any health state in the model at any time. Death is modelled as an absorbing state. The model accounts for treatment status (on/off), clinical response, associated costs, and health-related quality of life. A schematic of the model structure is presented in Figure 4.
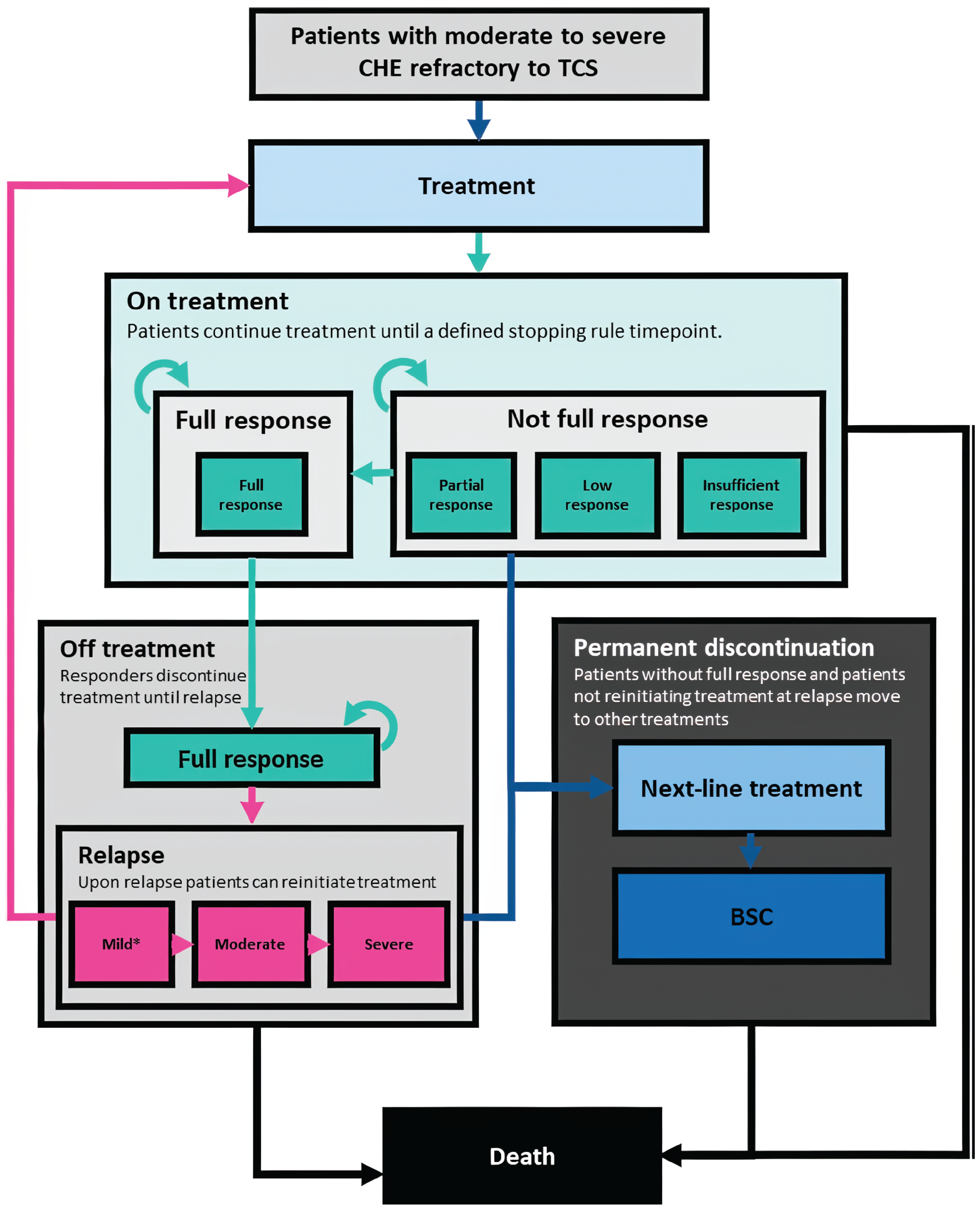
BSC = best supportive care; CHE = chronic hand eczema; TCS = topical corticosteroids.
Source: Sponsor’s pharmacoeconomic submission.25
Table 6: Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Total QALYs | Pairwise ICER ($ per QALY) |
|---|---|---|---|
Delgocitinib vs. alitretinoin | |||
Alitretinoin | 16,603 | 15.70 | Reference |
Delgocitinib | 16,851 | 15.74 | 6,335 |
Delgocitinib vs. phototherapy | |||
Phototherapy | 14,503 | 15.69 | Reference |
Delgocitinib | 16,851 | 15.74 | 44,692 |
Delgocitinib vs. dupilumab | |||
Dupilumab | 89,555 | 16.08 | Reference |
Delgocitinib | 16,851 | 15.74 | $217,165 saved per QALY lost |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Appendix 4: Additional Details of CDA-AMC Reanalyses
Please note that this appendix has not been copy-edited.
Clinical Data in the Economic Model
The CDA-AMC clinical review found that delgocitinib demonstrated superiority over both phototherapy and alitretinoin in terms of higher rates of IGA-CHE treatment success. However, the smaller differences favouring delgocitinib in terms of symptom scores and quality of life compared to alitretinoin were not judged to be clinically meaningful, and these outcomes were not included in the NMA compared to phototherapy. In contrast, a MAIC against dupilumab showed no clear difference between treatments in terms of treatment response. None of the indirect comparisons (NMA, MAIC) included safety outcomes. These findings from the NMA and MAIC, while informative, should be interpreted with caution due to methodological limitations, including sparse data, residual confounding, and differences in outcome definitions across studies. Overall, delgocitinib addresses an important unmet need by offering a topical, nonsteroidal alternative for patients with persistent CHE. Nonetheless, uncertainties remain regarding its long-term comparative effectiveness and safety and role within the broader treatment framework, including its impact on the use of systemic agents.
The economic model includes multiple outcomes along the patient pathway. However, only the outcome of full response after initial treatment is supported by comparative evidence across active comparators (from DELTA FORCE trial and the sponsor’s indirect treatment comparisons). All other probabilities were based in subgroup or post hoc analysis of the delgocitinib arm of DELTA trials (and assumed to be the same for the comparators) or based on naive comparisons across trials (e.g., probability of relapse after full response). Several other outcomes included in the pharmacoeconomic model such as relapse after full response, re-treatment after relapse, discontinuation after 12 weeks for those with partial and low response who continued treatment up to 24 weeks, the proportion of patients opting not to reinitiate treatment after relapse, and discontinuation from re-treatment were informed by naive comparisons. This approach introduces a risk of bias and limits the validity of these estimates for informing the pharmacoeconomic analysis. The model assumes a 30-year time horizon, while comparative clinical data are only available for 24 weeks for delgocitinib versus alitretinoin, 12 weeks for delgocitinib versus phototherapy, and 16 weeks for delgocitinib versus dupilumab. More than 90% of the QALY differences between delgocitinib and the comparators were accrued during the model extrapolated period.
Together, these concerns contribute to the uncertainty in the model results in the absence of long-term comparative evidence for several key model inputs. Thus, the CDA-AMC reanalysis remains subject to a high degree of uncertainty.
Pagebreak
Key Issues of the Submitted Economic Evaluation
CDA-AMC identified the following key issues with the sponsor’s analysis:
The economic model does not fully reflect expected place in therapy and some relevant comparators were not included. Clinical expert input obtained by CDA-AMC indicated that several treatments are used off-label for CHE in Canada, including upadacitinib, abrocitinib, methotrexate, cyclosporine, azathioprine, and mycophenolate mofetil. These treatments were not considered in the sponsor’s economic model due to the lack of comparative data, which limits the model’s relevance and generalizability to current clinical practice. Clinical expert input obtained by CDA-AMC emphasized a preference for topical therapies due to lower systemic exposure and ease of administration and indicated that delgocitinib could be used before off-label systemic therapies currently used to treat CHE (e.g., immunosuppressants, dupilumab, Janus kinase [JAK] inhibitors). Clinical expert input also noted that access to dupilumab currently is generally conditional to unsuccessful treatment with at least 1 systemic immunosuppressant prior (e.g., methotrexate, cyclosporine). Hence, these immunosuppressants are expected to be displaced before delgocitinib can displace dupilumab or any JAK inhibitors. Delgocitinib may also be considered for use in combination with systemic treatments in more severe cases (e.g., in addition to alitretinoin) despite the lack of evidence as a combination therapy. Overall, the experts considered that the introduction of delgocitinib may shift the treatment paradigm by reducing reliance on systemic agents, mainly affecting alitretinoin, methotrexate, and cyclosporine use.
CDA-AMC was unable to address this limitation in reanalysis due to lack of clinical evidence and inclusion of other comparators in the economic model. The cost-effectiveness of delgocitinib versus these other comparators excluded from the model remains unknown.
The comparative effectiveness of delgocitinib versus comparators is uncertain and model assumptions do not fully reflect clinical practice in Canada. First, among the parameters used to construct the patient pathway in the model, only the probability of achieving full response and disease severity after initial treatment was informed by comparative clinical data from the indirect treatment comparisons conducted by the sponsor. All other outcomes, including the probability of achieving full response during continued treatment, probability of relapse, probability of discontinuation (during continued treatment or during re-treatment), probability of choosing not to reinitiate treatment following relapse, were derived from subgroup or post hoc analysis of the delgocitinib arm of DELTA trials (and assumed to be the same for the comparators) or based on naive comparisons across trials (e.g., probability of relapse after full response). The CDA-AMC clinical review of the NMA found delgocitinib to be superior to alitretinoin and phototherapy in the probability of full response and in the improvement of disease severity after initial treatment (12 weeks). There is no evidence of a difference between delgocitinib and dupilumab in terms of treatment response effects, based on the MAIC. Adverse events were not included in the NMA or MAIC. The reliance on assumptions or noncomparative evidence for these key clinical events introduces uncertainty into the model. Second, the sponsor assumed that patients with low response would continue initial treatment up to 24 weeks. Clinical expert input varied and it was noted that in clinical practice in Canada, some clinicians may likely recommend switching treatments if patients present a low response after 12 weeks of treatment. Finally, there is an extensive extrapolation period between the time horizon of the economic model and the duration of the available clinical evidence. The model applies the efficacy inputs to a 30-year time horizon without any treatment waning, while the clinical evidence used to inform comparative efficacy spans only short-term follow-up periods. Specifically, the primary data included in the NMA12 and MAIC13 for the comparison with delgocitinib ranged from 12 to 24 weeks follow-up (24 weeks versus alitretinoin,4 12 weeks versus phototherapy, and 16 weeks versus dupilumab). As a result, more than 90% of the differences between delgocitinib and the comparators accrued during the model extrapolated period. Although DELTA 3 suggested that benefits may be sustained beyond the initial trial follow-up, this study does not provide long-term comparative evidence against active comparators included in the model. This extensive reliance on extrapolated outcomes further increases the uncertainty associated with the model’s results and limits the robustness of the cost-effectiveness estimates.
CDA-AMC was unable to address the lack of treatment waning in the submitted model, or the absence of comparative clinical evidence for several outcomes and long-term data.
CDA-AMC conducted a scenario analysis assuming patients with low response to initial treatment at week 12 would switch to next-line treatment.
Approach used to estimate delgocitinib consumption is uncertain and may underestimates drug acquisition costs. To calculate the drug acquisition cost for delgocitinib, the sponsor estimated consumption in grams based on patient responses from the DELTA trials.6-8 Specifically, gram-level consumption was calculated for each health state and then summed across all health states. The total consumption was multiplied by the cost per gram to estimate total drug costs and does not account for wastage (e.g., unused amounts of cream or expired tubes). This resulted in the dispensing of 4.6 tubes per patient over 30 years, on average, as estimated by the sponsor. However, this method does not reflect how typically topical treatments are dispensed in clinical practice according to input from the drug plans (i.e., per tube). Neither reflects the current delgocitinib stability recommended in the product monograph which instructs to “use within 12 months of first opening the tube and not passed the expiry date.”1 Thus, in clinical practice, a full tube is dispensed at the beginning to all patients starting treatment, and a new full tube dispensed to those that remain on treatment when cumulative use exceeds the tube volume (60 g) or when 12 months of first opening has passed, which CDA-AMC suggested as a more appropriate approach to calculate the base-case costs. CDA-AMC acknowledged that this method could overestimate consumption if not accounting for discontinuation and if patients continue to use topical creams beyond its recommended stability or expiry date. Clinical expert input noted that in clinical practice patients may use products well beyond the period recommended in the product monographs but only those with recurrent flares would require new tubes. Due to the structure and programming of the submitted model making the testing of alternative assumptions unfeasible within the time frame of this review, the sponsor suggested to cap the CDA-AMC base-case approach to calculate delgocitinib costs per tube to only where at least 1% of patients are still on treatment with delgocitinib, which in turn results in the dispensing of 8 tubes per patient, on average, over 30 years. This aimed to more closely reflect the dispensing of new tubes to only the proportion of the cohort still on treatment and mitigate overestimation of tubes attributed to the full cohort within the limitations of the submitted model. Although the estimated number of tubes dispensed over the time horizon by this approach falls within a valid range for a cohort model, CDA-AMC notes that there is still uncertainty in this approach. Based on the average weekly consumption provided by the sponsor from the DELTA trials (8.24 g to 8.78 g weekly across response states), those patients who remain on treatment would require more than half a tube of delgocitinib per 4-week cycle (i.e., 54% to 58.5% of a tube). The remaining volume of cream in the tube would not be sufficient to account for their consumption in the subsequent cycle, and a new tube could be dispensed for those still on treatment in each cycle. Therefore, in scenario analysis, CDA-AMC explored a simplified approach assuming that the proportion of patients still on treatment in each of the mutually exclusive health states could be dispensed a maximum of 1 tube per cycle, which resulted in a maximum consumption of 11.5 tubes per patient, on average, over 30 years.
Additionally, in the sponsor’s economic model, half-cycle correction was applied to adjust for the timing of transitions between health states within each cycle, which is a standard approach in state-transition modelling. However, at cycle 0, which is used to incur initial treatment costs and utilities, the formula used by the sponsor resulted in the sum of the proportion of patients in each health state not equalling 1. This violates a fundamental modelling principle where the total state membership distribution across health states must sum to 1 at any point in time. Most importantly, this programming choice underestimates drug acquisition costs at the beginning of the simulation for 50% of the patients entering the cohort model, with a larger effect when assuming the tube as the smallest dispensable unit. Finally, the sponsor used numerous IFERROR statements in the submitted model which makes thorough auditing of the sponsor’s model impossible, as it remains unclear whether the model is running inappropriately by overriding errors. This compounded with the limitation of the model in estimating the number of dispensed tubes and underestimation of state membership at entry suggests that results should be treated with a degree of caution.
CDA-AMC base case corrected the formula for half-cycle correction at model entry to ensure that the proportions of patients across all health states and cycles sum to 1. CDA-AMC was unable to address the use of IFERROR statements and notes that a thorough validation of the sponsor’s model was not possible.
CDA-AMC base case estimated delgocitinib drug acquisition costs according to the product monograph and assuming the tube as the smallest dispensable unit. The CDA-AMC base case incurred the costs of dispensing a tube at initiation to all patients starting treatment, and whenever the cumulative utilization of cream in the model exceeded 60 g or 12 months after opening of the previous tube, capped to count tubes only where at least 1% of patients are still on treatment with delgocitinib to mitigate overestimation due to the limitations of the model.
Alternatively, CDA-AMC scenario analysis explored a simplified approach assuming that the proportion of patients still on treatment in each of the mutually exclusive health states could be dispensed a maximum of 1 tube per cycle.
The utility value for next-line health state is uncertain. Utilities associated with different levels of treatment response were derived from pooled DELTA 12 and DELTA 22 trial, using Canadian value sets and adjusted overtime to the general population norms from the UK. The utility while in next-line treatment was calculated as a weighted average of the utilities from full response, partial, low, and insufficient response to initial treatment from the DELTA trials. Clinical experts indicated that it is reasonable to expect that patients receiving next-line treatment would have a quality of life at least comparable to those in partial response — given that the basket represents in its majority later lines of systemic therapies. This may favour delgocitinib results relative to alitretinoin and phototherapy, given that delgocitinib results in delay of next-line treatment because of the interaction of multiple outcomes in the model pathway. For example, the differences in the probability of relapse after full response and the differences in the proportion of patients opting not to re-treat with the same option, after a successful response – both informed by unadjusted naive comparisons. The sponsor’s base-case assumptions resulted in 0.04 and 0.05 incremental QALYs gained versus alitretinoin and phototherapy, respectively. This is equivalent to an extra 14 to 18 days of full health over a 30-year time horizon, with more than 90% of this benefit accruing in the extrapolated period. The CDA-AMC clinical review found a small but not clinically meaningful difference in quality of life versus alitretinoin, and this outcome was not included in the NMA versus phototherapy. Therefore, even uncertainties that have a small impact on overall utilities can be influential to the cost-effectiveness results when facing very small incremental gains across comparators.
Due the lack of evidence to inform this parameter and to explore the impact on overall cost-effectiveness, CDA-AMC scenario analysis assumed utility for next-line treatment to be equal to that of patient with partial response
The proportion of patients who, following relapse after an initial treatment response to alitretinoin and phototherapy, do not reinitiate the same therapeutic regimen is uncertain. In the economic model, 52.1% of patients in the alitretinoin arm, 95.6% in the phototherapy arm and ████ of patients in the delgocitinib arm would choose not to resume the same treatment upon relapse, despite having achieved full response to initial treatment with these options. These were based on naive comparisons of the ALPHA trial26 data and a post hoc analysis of the open-label extension trial DELTA 3. Clinical experts indicated that these proportions likely underestimate treatment reinitiation in clinical practice. Experts noted that it is reasonable to expected that the majority of patients who experienced a successful response to a treatment would accept the same treatment option — perceived as more than 2 thirds of patients post full response. Unless they have experienced adverse events, which are most likely to be experienced with alitretinoin than phototherapy. It was deemed unreasonable to expect that almost the entirety of patients who benefited from phototherapy would opt out of this treatment option.
To reflect clinical expert input, CDA-AMC conducted a scenario analysis assuming that 35% of patients in the alitretinoin arm would not reinitiate treatment following relapse. The proportions of patients in the phototherapy arm were assumed to be at least the same as those treated with alitretinoin.
CDA-AMC Reanalysis of the Economic Evaluation
The CDA-AMC base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts (refer to Table 7). The impact of these changes, individually and collectively, is presented in Table 8. Given the limitations in the indirect evidence presented and uncertainties regarding delgocitinib’s place in therapy relative to dupilumab, the comparison with dupilumab was excluded from the CDA-AMC base case and presented in scenario analysis.
Table 7: Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
1. Half-cycle correction | 50% of patients removed from initial distribution | Correction removed from initial distribution |
2. Delgocitinib costs estimation | Delgocitinib consumption was estimated based on the exact number of grams used | Assumed a new tube is dispensed at the beginning of the model and each time the cumulative consumption exceeds the tube volume (60 g) or 12 months after opening, whichever occurs first, limited to when there are ≥ 1% patients remaining on treatment |
CDA-AMC base case (health care payer perspective) | ― | Reanalysis 1 + 2 |
CDA-AMC = Canada’s Drug Agency.
Note: CDA-AMC was unable to resolve the issues related to the lack of comparative clinical evidence for model parameters and the exclusion of off-label comparators.
Table 8: Summary of the Stepped Analysis
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) vs. delgocitinib |
|---|---|---|---|---|
Sponsor base case | Delgocitinib | 16,775 | 15.73 | Reference |
Alitretinoin | 16,477 | 15.68 | 6,677 | |
Phototherapy | 14,330 | 15.67 | 42,952 | |
CDA-AMC reanalysis 1: half-cycle correction | Delgocitinib | 16,976 | 15.76 | Reference |
Alitretinoin | 16,746 | 15.71 | 5,121 | |
Phototherapy | 14,391 | 15.70 | 45,426 | |
CDA-AMC reanalysis 2: Delgocitinib drug costs per tube | Delgocitinib | 18,221 | 15.73 | Reference |
Alitretinoin | 16,477 | 15.68 | 38,998 | |
Phototherapy | 14,330 | 15.67 | 68,340 | |
CDA-AMC base case Reanalysis 1 + 2 (deterministic) | Delgocitinib | 18,847 | 15.76 | Reference |
Alitretinoin | 16,746 | 15.71 | 46,883 | |
Phototherapy | 14,391 | 15.70 | 78,317 | |
CDA-AMC base case Reanalysis 1 + 2 (probabilistic) | Delgocitinib | 18,742 | 15.75 | Reference |
Alitretinoin | 16,880 | 15.71 | 47,393 | |
Phototherapy | 14,568 | 15.70 | 79,192 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Notes: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments. Deterministic results are presented, unless otherwise indicated.
Table 9: Disaggregated Results of the CDA-AMC Base Case (Probabilistic)
Parameter | Delgocitinib | Alitretinoin | Phototherapy |
|---|---|---|---|
Discounted QALYs | |||
Total | 15.75 | 15.71 | 15.70 |
Baseline | 0.18 | 0.18 | 0.19 |
Full response | 0.22 | 0.13 | 0.08 |
Partial response | 0.16 | 0.07 | 0.06 |
Low response | 0.12 | 0.05 | 0.02 |
Insufficient response | 0.01 | 0.00 | 0.00 |
NL and BSC | 15.06 | 15.27 | 15.35 |
AE | 0.00 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 18,742 | 16,880 | 14,568 |
Treatment acquisition | 4,684 | 2,743 | 447 |
NL and BSC | 5,460 | 5,496 | 5,510 |
Health state | 8,578 | 8,598 | 8,611 |
AE | 20 | 43 | 0 |
AE = adverse event; BSC = best supportive care; CDA-AMC = Canada’s Drug Agency; NL = next-line; QALY = quality-adjusted life-year.
Price Reduction Analysis
CDA-AMC conducted price reduction analyses using the sponsor’s base case and the CDA-AMC base case (refer to Table 10).
Table 10: Results of the Price Reduction Analysis
Price reduction | Unit drug cost ($) | Annual cost ($) | ICERs for delgocitinib vs. alitretinoin ($/QALY) | ICERs for delgocitinib vs. phototherapy ($/QALY) | ||
|---|---|---|---|---|---|---|
Sponsor base case | CDA-AMC base case | Sponsor base case | CDA-AMC base case | |||
No price reduction | 637 | 5,096 | 6,335 | 47,393 | 44,692 | 79,192 |
10% | 573 | 4,586 | Dominant | 34,704 | 39,118 | 69,735 |
20% | 510 | 4,077 | Dominant | 22,016 | 33,544 | 60,279 |
30% | 446 | 3,567 | Dominant | 9,327 | 27,971 | 50,822 |
40% | 382 | 3,058 | Dominant | Dominant | 22,397 | 41,366 |
50% | 318 | 2,548 | Dominant | Dominant | 16,823 | 31,910 |
60% | 255 | 2,038 | Dominant | Dominant | 11,249 | 22,453 |
70% | 191 | 1,529 | Dominant | Dominant | 5,676 | 12,997 |
80% | 127 | 1,019 | Dominant | Dominant | 102 | 3,540 |
90% | 64 | 510 | Dominant | Dominant | Dominant | Dominant |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aSponsor’s submitted price for delgocitinib.25
Assessment of Uncertainty
CDA-AMC used the CDA-AMC base case to conduct scenario analysis that addresses uncertainty within the economic evaluation. Results are provided in Table 11.
Assumed patients with low response switch treatment after 12 weeks in delgocitinib, alitretinoin, and phototherapy arms, aligned with clinical expert input.
Assumed only 35% of patients who previously achieved full response with alitretinoin or phototherapy would opt out of treatments with these options upon relapse.
Assuming maximum number of tubes per patients remaining on treatment over the time horizon is 1 tube per cycle.
Assumed utility for next-line treatment to be at least equal to that of patient with partial response. The absolute differences in utility between full response and other health states (partial, low, and insufficient response) as found in the DELTA trials were maintained, as assumed by the sponsor.
Pairwise comparison to dupilumab (same CDA-AMC base case inputs)
Table 11: Results of CDA-AMC Scenario Analyses
Analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CDA-AMC base case | Delgocitinib | 18,847 | 15.76 | Reference |
Alitretinoin | 16,746 | 15.71 | 46,883 | |
Phototherapy | 14,391 | 15.70 | 78,317 | |
CDA-AMC scenario 1: Treatment duration for low response (12 weeks) | Delgocitinib | 17,657 | 15.73 | Reference |
Alitretinoin | 16,134 | 15.69 | 41,986 | |
Phototherapy | 14,318 | 15.68 | 75,431 | |
CDA-AMC scenario 2: Proportion of patients opting not to reinitiate initial treatment following relapse in alitretinoin and phototherapy arms | Delgocitinib | 18,847 | 15.76 | Reference |
Alitretinoin | 16,924 | 15.72 | 49,079 | |
Phototherapy | 14,427 | 15.71 | 89,253 | |
CDA-AMC scenario 3: Maximum number of tubes per cycle = 1 tube, for the % patients on treatment | Delgocitinib | 21,117 | 15.76 | Reference |
Alitretinoin | 16,746 | 15.71 | 97,524 | |
Phototherapy | 14,391 | 15.70 | 118,202 | |
CDA-AMC scenario 4: Utility values | Delgocitinib | 18,847 | 15.79 | Reference |
Alitretinoin | 16,746 | 15.74 | 47,101 | |
Phototherapy | 14,391 | 15.73 | 78,693 | |
CDA-AMC scenario 5: Pairwise comparison to dupilumab | Delgocitinib | 18,847 | 15.76 | Reference |
Dupilumab | 86,150 | 16.06 | 221,499 saved per QALY lost |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDeterministic analyses.
Issues for Consideration
Tralokinumab,27 lebrikizumab,28 and ruxolitinib29 have received “Do Not Reimburse” recommendations from CDA-AMC for the treatment of atopic dermatitis. Other off-label treatments for CHE in Canada include upadacitinib, abrocitinib, methotrexate, cyclosporine, azathioprine, and mycophenolate mofetil and were not included in the cost-effectiveness analysis. Nemolizumab is currently under review for the treatment of atopic dermatitis and may potentially be another systemic therapy for atopic dermatitis with off-label use to treat CHE.30
The current stability recommended in the product monograph for delgocitinib states “use within 12 months of first opening the tube and not passed the expiry date.”1 As such, each patient reinitiating treatment would require at least 1 tube of delgocitinib cream per year, even if the amount of drug used is less than the 60 g included in the tube. Although 15 g tubes are included in the current draft of the product monograph and could be used to reduce wastage or treatment costs after initial treatment, additional information31 provided by the sponsor during the review confirmed that the 15 g tube will not be marketed in Canada.
Clinical expert input received by CDA-AMC indicated that, in clinical practice, delgocitinib cream may be used in combination with other treatments (i.e., with systemic treatments). The cost-effectiveness of delgocitinib cream when used in combination with other treatments is unknown owing to the lack of clinical data. CDA-AMC notes the draft Health Canada monograph notes that delgocitinib cream interactions have not been evaluated.
Abrocitinib, upadacitinib, and dupilumab, which may be used off-label for CHE, have successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance (pCPA) for atopic dermatitis. Therefore, their actual reimbursed prices are likely lower than the publicly available list prices presented in Table 4.
Appendix 5: Budget Impact Analysis
Please note that this appendix has not been copy-edited.
Summary of the Submitted BIA
The sponsor submitted a BIA that estimated the expected incremental budgetary impact of reimbursing delgocitinib for the treatment of moderate to severe CHE in adults for whom TCS are inadequate or are not advisable.
The BIA was conducted from the perspective of public drug plan payers over a 3-year time horizon (2026 to 2028), with 2025 as the base year. The sponsor’s estimate reflects the aggregated results from the jurisdictional provincial budgets (excluding Quebec) as well as the Non-Insured Health Benefits Program. The sponsor estimated the eligible population using an epidemiological approach. The model assumed greatest displacement for alitretinoin and phototherapy (roughly 70% of their current market by year 3), followed by immunosuppressants (roughly 55%), JAK inhibitors (45%), then dupilumab (40%). In jurisdictions where alitretinoin is not funded, its shares were proportionally redistributed among other comparators based on their relative market shares. Market shares for the reference scenario were derived from the RWEAL study based, scaled to 100%. The sponsor’s base case included drug acquisition costs, dispensing fee, and wholesaler markups. The market uptake for delgocitinib was estimated based on assumptions. The key inputs to the BIA are documented in Table 12.
The sponsor estimated the 3-year budgetary savings associated with reimbursing delgocitinib would be $96,787,823 (year 1 = $19,713,218; year 2 = $34,759,783; year 3 = $42,314,821) when including dispensing fees, markups and phototherapy costs; and 3-year budgetary savings of $89,425,413 (year 1 = $18,184,067; year 2 = $32,134,293; year 3 = $39,107,053) without considering dispensing fees, markups, and phototherapy costs as part of the drug plan perspective.
Table 12: Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Adult population of Canada | 25,363,410 |
Public coverage | 26.4% |
Prevalence of CHE | 6.2% |
Percentage of patients with moderate to severe CHE | 44.1% |
Percentage of patients who have used TCS | 81.6% |
Percentage of patients who have failed TCS or for whom TCS are not advisable | 13.1% |
Percentage of patients who received NL therapies | 60.0% |
Eligible patients in base year | 11,736 |
Market shares (reference scenario) | |
Delgocitinib | 0% / 0% / 0% |
Alitretinoin | 10% / 10% / 10% |
Phototherapy | 15% / 15% / 15% |
Dupilumab | 20% / 20% / 20% |
Abrocitinib | 9% / 9% / 9% |
Upadacitinib | 11% / 11% / 11% |
Methotrexate | 23% / 23% / 23% |
Cyclosporine | 11% / 11% / 11% |
Market shares (new drug scenario) | |
Delgocitinib | 28% / 45% / 53% |
Alitretinoin | 5% / 4% / 3% |
Phototherapy | 9% / 7% / 5% |
Dupilumab | 16% / 14% / 12% |
Abrocitinib | 7% / 5% / 5% |
Upadacitinib | 8% / 7% / 6% |
Methotrexate | 17% / 12% / 11% |
Cyclosporine | 8% / 7% / 6% |
Cost of treatment (average weekly cost/average annual – per patient yeara) – source | |
Delgocitinib | $41.71 per week / $2,169 per year |
Alitretinoin | $48.18 per week/ $2,505 per year |
Phototherapy | $5.42 per week/ $282 per year |
Dupilumab | $487.68 per week/ $25,359 per year |
Abrocitinib | $381.01 per week/ $19,812 per year |
Upadacitinib | $361.52 per week / $18,799 per year |
Methotrexate | $0.46 per week / $24 per year |
Cyclosporine | $12.42 per week / $646 per year |
CHE = chronic hand eczema; NL = next-line; TCS = topical corticosteroids.
aWithout dispensing fees and markups, calculated as the sum of the weekly costs inputs over a year for those jurisdictions where the drug is funded.
Key Issues of the Submitted BIA
CDA-AMC identified several key issues to the sponsor’s analysis that have notable implications on the results of the BIA:
Annual drug costs are uncertain and cost savings may be overestimated. The sponsor used a predominantly prevalence-based model (i.e., incidence model with a prevalence bolus, in which > 95% patients are prevalent patients) and assumed average annual costs for all comparators from various methodologies and assumptions. In a prevalence-based model, it can be challenging to gather data on how long the individuals have had the condition before the start of the BIA. This could have a large influence on drug costs if the costs in the first year of treatment are substantially different from those in subsequent years (i.e., due to dose titration or changing treatment schedules).
For delgocitinib, the sponsor estimated weekly costs by multiplying the total grams of delgocitinib used in the first year of the sponsors CUA base case (205 g, which is equivalent to 3.4 tubes) by the delgocitinib price per gram, divided by the number of weeks per year. Then applied these same weekly costs uniformly across all 3 years of the BIA time horizon. This approach does not reflect how drug use varies over time (e.g., dose decreases after initial treatment) and how the drug is dispensed in clinical practice (per tube not per gram). This overestimates delgocitinib drug expenditure after year 1 and leads to inaccurate yearly budget estimations. According to the CDA-AMC CUA base case, the estimated average use per patient was 4 tubes in the first year, and only 1 tube in each of the second and third years. Furthermore, the BIA model as it is programmed by the sponsor does not appropriately apply year-specific costs to incident patients entering the model. For example, incident patients who entered the model in year 1 and continue treatment in year 2, are assigned costs equivalent to the costs of the first year on treatment. This is a nonissue if assuming average costs of a drug are constant throughout the 3 years. However, when assuming time-dependent costs, this can still slightly overestimate the yearly budget for delgocitinib.
For all other comparators included in the model, the average annual costs were estimated assuming various methodologies and assumptions. Specifically, continuous use for dupilumab and JAK inhibitors throughout the year, 15.8 weeks per year for cyclosporine and methotrexate, and 21 weeks per year for alitretinoin. Although, in theory, average annual costs for the comparators would be the more acceptable approach to estimate costs of the comparators in a prevalence-based model, in this specific model, these same comparators are also subsequent therapies to each other. If the expectation is a high rate of movement between the treatment options included in the BIA (i.e., due to discontinuation or treatment switch), the assumptions of a prevalence-based model limit the accuracy with which the model can estimate subsequent therapy costs. Using a prevalence-based model, the user must assume, every year, the percentage of patients who experience treatment failure and therefore require subsequent therapies. Then multiply this percentage by the average annual costs of the subsequent therapies in their treatment sequence. Treatment discontinuation and drug cost with subsequent treatments were not incorporated into the submitted BIA model which seems to overestimate the costs of comparators. For example, the majority of the cost savings estimated by the sponsor comes from the displacement of dupilumab. Using the average cost for dupilumab assumed by the sponsor in the BIA it results in a 3-year cost per patient of $76,077. While in the CDA-AMC CUA base case, the estimated costs of dupilumab over 3 years was $32,728 (plus $639 with subsequent treatments). Therefore, the costs of the comparators in the BIA may be overestimated, which in turn overestimates cost savings when delgocitinib displaces a comparator. A more accurate estimation for the costs of comparators would require a dynamic model that can track every patient entering and leaving the BIA, as well as when they switch therapies, their different rates of discontinuation and their different subsequent therapy costs.
CDA-AMC was unable to address the overestimation of costs for comparators with the submitted model structure and, therefore, the cost savings remain highly uncertain and likely overestimated.
Market shares in the reference scenario are uncertain and uptake of delgocitinib seem overly optimistic. The market shares in the reference scenario was informed by the RWEAL study15 and adjusted for internal market assumptions. The sponsor assumed the anticipated uptake of delgocitinib to be 28%, 45% and 53% in years 1, 2, and 3, respectively. However, clinical expert input obtained by CDA-AMC noted that the reference scenario market shares may not reflect clinical practice in Canada, and the uptake of delgocitinib seems overly optimistic. For instance, the current market share of dupilumab is expected to be at approximately 30% (which is higher than the 21% to 24% assumed by the sponsor). The combined market shares of alitretinoin, methotrexate, and cyclosporine sums to 44% as assumed by the sponsor. Delgocitinib is anticipated to replace primarily alitretinoin, methotrexate, and cyclosporine due to its avoidance of systemic side effects. Also, because current access to dupilumab is conditional on unsuccessful treatment with methotrexate and cyclosporine. Clinical expert input expects the market uptake of delgocitinib to reach up to 30 to 35% by year 3.
Additionally, prevalence-based models do not explicitly differentiate market uptake in incident patients versus prevalent patients. When a new therapy comes to market, it may displace current therapies if the treatment benefit is substantially improved. However, in some cases, there may be a reluctance to switch patients who are tolerating their current therapy; therefore, the main cohort of patients considered for new therapies would be incident patients. Further, if a drug has a high rate of discontinuation relative to its comparators, this will reduce its market share overtime. A more sophisticated approach (e.g., dynamic models) may be warranted to consider both a large prevalent and incident population with different and dynamic market shares and population sizes.
CDA-AMC could not adjust for this limitation in reanalyses due to a lack of data and due to the structure and programming of the submitted model making the testing of alternative assumptions unfeasible within the time frame of this review. Therefore, the market shares continue to be uncertain and uptake of delgocitinib continue to be overestimated.
Phototherapy costs are uncertain and included in the drug plan payer perspective: According to the Procedures for CDA-AMC Reimbursement Reviews, the BIA base case should be undertaken from the perspective of a pan-Canadian drug plan program. As such, costs relating to the use of phototherapy are not funded by jurisdictional drug plan budgets and should thus be excluded from the drug plan perspective. Additionally, the sponsor assumed that on average patients would only receive 1 course of phototherapy annually (3 session for 12 weeks totalling 36 sessions per year). This may be underestimating the costs of phototherapy. Clinical expert input elicited by CDA-AMC indicated that phototherapy usually has a very high repeat treatment rate if effective and those who have had previous complete response on phototherapy would go back on therapy upon relapse. The CDA-AMC cost-utility base case estimated approximately 58 sessions (19 weeks) in year 1 and none in the following. These were estimated without making any changes to the sponsor’s assumptions around stopping rules which included 95% of patients successfully treated with phototherapy opting out of re-treatment.
Costs associated with the use of phototherapy were excluded from the sponsor’s base case from the drug plan perspective.
Some relevant comparators were not included in the BIA: Clinical expert input obtained by CDA-AMC indicated that several treatments are used off-label for CHE in Canada, including azathioprine, and mycophenolate mofetil. These treatments were not considered in the sponsor’s budget impact model. Given that these comparators can have lower annual costs than delgocitinib (refer to Table 4), their exclusion from the BIA may be overestimating the estimated cost savings by the reimbursement of delgocitinib.
CDA-AMC was unable to incorporate these comparators in the reanalysis.
The price of drugs paid by public drug plans is uncertain: Analyses by both the sponsor and CDA-AMC are based on publicly available list prices for all comparators. Confidential negotiated prices are available for most comparators.32-34 Thus, the actual costs paid by the public drug plans for these drugs are unknown. Depending on the negotiated prices, the budgetary savings estimated due to reimbursing delgocitinib is uncertain.
CDA-AMC was unable to incorporate the presence of confidential negotiated prices in the reanalysis.
CDA-AMC Reanalyses of the BIA
CDA-AMC was unable to undertake a base case reanalysis. Due to the limitations associated with the sponsors approach the results are highly uncertain and no reliable budget impact can be derived from the submitted analysis. The expenditure on delgocitinib (year 1: $7,244,994; year 2: $11,836,254; year 3: $14,171,007) likely represents the upper limit over a 3 year period. This is because delgocitinib costs have been overestimated and market uptake is likely too high. The cost estimates applied to all to all other drugs in the analysis is too uncertain to estimate. A simplified approach to estimate drug expenditure with delgocitinib is provided in Table 14.
Table 13: Disaggregated Summary of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference total | 120,153,032 | 122,130,431 | 124,140,763 | 126,184,581 | 372,455,775 |
Delgocitinib | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 120,153,032 | 122,130,431 | 124,140,763 | 126,184,581 | 372,455,775 | |
New drug total | 120,153,032 | 102,417,212 | 89,380,980 | 83,869,760 | 275,667,952 | |
Delgocitinib | 0 | 7,899,318 | 12,905,358 | 15,451,148 | 36,255,824 | |
All other comparators | 120,153,032 | 94,517,895 | 76,475,622 | 68,418,612 | 239,412,128 | |
Budget Impact | 0 | –19,713,218 | –34,759,783 | –42,314,821 | –96,787,823 | |
Submitted base case without costs of markups, dispensing fees, and phototherapya | Reference total | 111,093,374 | 112,919,559 | 114,776,136 | 116,663,615 | 344,359,310 |
Delgocitinib | 0 | 0 | 0 | 0 | 0 | |
All other comparators | 111,093,374 | 112,919,559 | 114,776,136 | 116,663,615 | 344,359,310 | |
New drug total | 111,093,374 | 94,735,493 | 82,641,843 | 77,556,562 | 254,933,898 | |
Delgocitinib | 0 | 7,244,994 | 11,836,254 | 14,171,007 | 33,252,255 | |
All other comparators | 111,093,374 | 87,490,498 | 70,805,589 | 63,385,555 | 221,681,643 | |
Budget Impact | 0 | –18,184,067 | –32,134,293 | –39,107,053 | –89,425,413 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: The CDA-AMC reanalysis is based on the publicly available prices of the comparator treatments.
aObtained by unchecking the sponsor’s built-in option to include dispensing fees and markups, and setting the costs of phototherapy to zero (phototherapy market shares are maintained as the original values assumed by the sponsor).
Table 14: Drug Expenditure Estimation — Simplified Approach
Analysis | Scenario | BIA year | ||||
|---|---|---|---|---|---|---|
Year 0 | Year 1 | Year 2 | Year 3 | Total | ||
Number of patients in each year — pan-Canadian | ||||||
Treatment year | Year 0 | 11,736 | 194 | 197 | 201 | NA |
Year 1 | 0 | 11,736 | 194 | 197 | NA | |
Year 2 | 0 | 0 | 11,736 | 194 | NA | |
Year 3 | 0 | 0 | 0 | 11,736 | NA | |
Total | 11,736 | 11,930 | 12,127 | 12,328 | NA | |
Sponsor’s base case | ||||||
Delgocitinib market share | 0% | 28% | 45% | 53% | ||
Number of patients receiving delgocitinib in each treatment year | Year 1 | 0 | 3,340 | 2,117 | 1,076 | NA |
Year 2 | 0 | 0 | 3,340 | 2,117 | NA | |
Year 3 | 0 | 0 | 0 | 3,340 | NA | |
Total | 0 | 3,340 | 5,457 | 6,534 | 15,331 | |
Drug expenditure of delgocitinib ($) | 0 | 8,511,072 | 7,521,359 | 6,218,922 | 22,251,354 | |
CDA-AMC assumption | ||||||
Delgocitinib market share | 0% | 18% | 30% | 35% | NA | |
Number of patients receiving delgocitinib in each treatment year | Year 1 | 0 | 2,206 | 1,398 | 711 | NA |
Year 2 | 0 | 0 | 2,206 | 1,398 | NA | |
Year 3 | 0 | 0 | 0 | 2,206 | NA | |
Total | 0 | 2,206 | 3,604 | 4,315 | 10,125 | |
Drug expenditure of delgocitinib ($) | 0 | 5,620,519 | 4,966,935 | 4,106,835 | 14,694,290 | |
BIA = budget impact analysis.
Notes: Time-dependent cost of delgocitinib was applied in the calculation of drug expenditure, consistent with the CUA. The number of tubes assumed over the first 3 years was 4, 1, and 1, resulting in annual costs of $2,548, $637, and $636, respectively.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.