Drugs, Health Technologies, Health Systems
Reimbursement Review
Guselkumab (Tremfya)
Sponsor: Janssen Inc.
Therapeutic area: Ulcerative colitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
5-ASA
5-aminosalicylate
ADT
advanced therapy
AE
adverse event
CD
Crohn disease
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
Crl
credible interval
FAS
full analysis set
GI
gastrointestinal
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
IL
interleukin
ITC
indirect treatment comparison
MID
minimal important difference
MMS
modified Mayo score
NMA
network meta-analysis
OR
odds ratio
RCT
randomized controlled trial
SAS
safety analysis set
SC
subcutaneous
TEAE
treatment-emergent adverse event
TNF
tumour necrosis factor
UC
ulcerative colitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Drug product | Induction: Guselkumab (Tremfya) 200 mg/20 mL vial of sterile solution administered through IV infusion. Maintenance: Guselkumab (Tremfya) 100 mg/1 mL pre-filled syringe, 100 mg/1 mL patient-controlled injector, 200 mg/2 mL pre-filled syringe, and 200 mg/2 mL pre-filled pen administered by subcutaneous injection. |
Sponsor | Janssen Inc. |
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | September 3, 2025 |
Recommended dose | Induction: The recommended induction dosage is 200 mg of Tremfya administered by IV infusion over a period of at least 1 hour at week 0, week 4, and week 8. Maintenance: The recommended maintenance dosage is 100 mg of Tremfya administered by subcutaneous injection at week 16 and every 8 weeks thereafter after completion of induction dosing. A dose of 200 mg administered by subcutaneous injection at week 12 and every 4 weeks thereafter may be considered for patients who do not show adequate therapeutic benefit to Tremfya, or according to clinical judgment. |
NOC = Notice of Compliance.
Introduction
Inflammatory bowel disease (IBD) is a group of diseases characterized by chronic recurrent, progressive inflammation of the gastrointestinal (GI) tract. There are 2 main types of IBD: Crohn disease (CD) and ulcerative colitis (UC). UC is a chronic disease characterized by inflammation and ulcers in the mucosal layer of the large intestine (colon), typically beginning at the rectum (anus), progressing upward and, in some cases, affecting the entire colon. The cause of UC remains uncertain, but a combination of genetic and environmental factors contributes to immune dysregulation and upregulation in response to micro-organisms in the GI tract. Patients with UC present with the following symptoms: blood in the stool with mucus, frequent diarrhea, loss of appetite, and tenesmus (strong urge to use the bathroom without necessarily having a bowel movement), in addition to abdominal pain, rectal bleeding, and weight loss. UC generally develops in young adulthood and persists throughout life, marked by periods of spontaneous remission and relapse. UC has a worldwide annual incidence rate of 1.2 to 20.3 cases per 100,000 people and a prevalence of 7.6 to 246.0 cases per 100,000 people. The prevalence for UC in Canada in 2023 was estimated to be 414 per 100,000. It is estimated that 32% to 46% of people in Canada with UC have moderate disease, and 13% to 14% have severe disease.
UC can be further classified in clinical practice based on severity: mild, moderate, or severe disease, depending on the specific index score used (Truelove and Witts Severity Index, Mayo score, or the Montreal classification). While most patients have a mild to moderate disease course, about 10% to 15% experience an aggressive course of UC. Relapse is common, with the cumulative risk of relapse being 70% to 80% at 10 years. Achieving endoscopic healing earlier may be associated with a reduced risk of future colectomy. The chronic nature of UC has a considerable impact on patients’ health-related quality of life (HRQoL), including psychological, physical, sexual, and social domains of HRQoL due to the chronicity of symptoms such as urgency and frequency and incontinence.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of guselkumab (Tremfya) 200 mg for induction, administered by IV infusion, followed by guselkumab 100 mg for maintenance, administrated by subcutaneous (SC) injection, for the treatment of adults with moderately to severely active UC.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient groups that responded to the call for input by Canada’s Drug Agency (CDA-AMC) and by the clinical experts consulted by CDA-AMC for the purpose of this review.
Patient Input
Two patient groups, Crohn’s and Colitis Canada and the Gastrointestinal Society, provided input for this review of guselkumab for the treatment of UC. The patient input from Crohn’s and Colitis Canada was gathered from an online survey conducted in 2022. The patient input from the Gastrointestinal Society was based on responses to questionnaires collected in 2024, 2023, 2022, 2020, 2018, and 2015, and 1 focus group conducted in 2022.
When asked what UC-related complications patients were experiencing currently or had experienced within the past year, the most frequently reported complications cited by respondents in the Crohn’s and Colitis Canada input were mental health and stress (65%), followed by joint inflammation and arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), and skin conditions and malnutrition and weight loss, both of which were reported by around 30% of respondents. In the Gastrointestinal Society input, diarrhea, bowel urgency, incontinence, abdominal pain, fever, rectal bleeding, and nausea were reported by the respondents as the common symptoms of UC. Patients from the Gastrointestinal Society input added that sustained remission or treatment response was more important than relieving any 1 symptom.
In the 2020 and 2024 surveys conducted by the Gastrointestinal Society, 33% and 29% of respondents believed their IBD was not well controlled by their current medications. While the Gastrointestinal Society indicated that corticosteroids are helpful to reduce inflammation in moderate to severe cases, it also emphasized that these medications are appropriate for short-term treatment only because they are generally not well tolerated and can have potentially serious side effects. Half of the respondents in the Crohn’s and Colitis Canada input indicated that systemic steroids are or were a burden in the management of UC, particularly among those with moderate to severe forms of UC, and among women. According to patients in the Crohn’s and Colitis Canada input who indicated managing medication use is important, having enough treatment options, understanding side effects of treatment, and minimizing steroid use were the most important aspects in the management of UC. No patients from either patient group had experience with the drug under review.
Clinician Input
Input From Clinical Experts Consulted by CDA-AMC
The clinical experts indicated that the treatment goals for patients with moderately to severely active UC is to induce and maintain clinical and endoscopic remission. The experts noted that first-line therapy typically includes oral mesalamine (a 5-aminosalicylate [5-ASA]) plus corticosteroids, which is used for rapid achievement of clinical remission. Corticosteroids are not used in maintenance therapy and are avoided when used as a recurrent “rescue” medication, given the wide array of adverse effects common with their use. The experts noted that although mesalamine is effective for some patients, it is ineffective in those with more severe disease and, therefore, these patients require an alternative advanced therapy (ADT) to maintain remission. The experts noted that the challenge with currently available therapies for UC in Canada is that a portion of patients will not respond to ADT and, among the patients who experience an initial, most experience loss of response after a period of symptom relief. As such, in many patients, it is necessary to escalate the dose and try several types of therapy to maintain response and meet longer-term treatment goals. The clinical experts pointed out that multiple drug failures and ongoing progressive disease activity may lead to adverse consequences, including surgery to remove the entire colon. The experts noted there is also a gap in available oral therapies, as most current therapies are administered intravenously or subcutaneously. The clinical experts indicated that a clear sequence of medications that is optimal to treat moderate to severe UC is not yet established. In the clinical experts’ opinion, guselkumab should be available as a first-line treatment option, and experiencing failure with a previous therapy should not be a criterion for accessing the drug. The clinical experts indicated that the patients best suited for guselkumab would be those with moderately to severely active UC. The experts highlighted that patients would be identified based on clinical judgment and an examination that included a review of a patient’s history of symptoms, a biomarker evaluation, an endoscopic assessment, and a histopathological evaluation. The clinical experts indicated that in clinical practice, a combination of clinical scoring systems (e.g., full Mayo and partial Mayo scores), endoscopic outcomes, histopathological evaluation, and patient-reported HRQoL scores are used to determine whether a patient is experiencing a response or disease progression on treatment. The experts noted that patients starting a new ADT should have a clinical follow-up assessment within 14 to 16 weeks of initiating therapy. The clinical experts indicated that treatment with guselkumab should be discontinued if patients are primary nonresponders, and if patients experience disease progression (e.g., ongoing symptoms or symptom escalation) while on maintenance therapy.
Clinician Group Input
No clinician group input was received by CDA-AMC for this review.
Drug Program Input
Input was obtained from the drug programs that participate in the CDA-AMC Reimbursement Review process. The following were identified as key factors that could potentially impact the implementation of a CDA-AMC recommendation for guselkumab:
considerations for initiation of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy.
The clinical experts consulted by CDA-AMC provided advice on the potential implementation issues raised by the drug programs (refer to Table 4).
Clinical Evidence
Systematic Review
Description of Studies
Two pivotal phase III double-blind randomized controlled trials (RCTs) submitted by the sponsor have been summarized in this report.
The QUASAR induction (N = 701) and maintenance (N = 568) studies met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the induction study was to assess the efficacy and safety of guselkumab 200 mg IV as induction therapy versus matching placebo administered at weeks 0, 4, and 8 in adults with moderately to severely active UC. The study enrolled patients who have experienced an inadequate response or are unable to tolerate conventional therapy (i.e., 6-mercaptopurine, azathioprine, or corticosteroids) or ADT (i.e., tumour necrosis factor [TNF] antagonists, vedolizumab, or tofacitinib). Patients were required to have a modified Mayo score (MMS) of 4 to 9, a Mayo rectal bleeding score of 1 or more, and a Mayo endoscopy subscore of 2 or more. Patients were eligible for the maintenance study if they had experienced a clinical response to guselkumab or placebo treatment at weeks 12 or 24. The objective of the maintenance study was to assess the efficacy and safety of the guselkumab SC injection maintenance regimens (100 mg or 200 mg) versus matching placebo in patients who achieved clinical response with guselkumab IV induction. Patients received guselkumab 200 mg every 4 weeks, guselkumab 100 mg every 8 weeks, or placebo every 4 weeks starting at week 0 through week 44. In general, the approved Health Canada indication and reimbursement request aligned with both study populations, except that the indication is not limited to patients who have experienced an inadequate response or are unable to tolerate conventional therapy or ADT. The outcomes most relevant to the CDA-AMC review included the primary outcome of clinical remission, and secondary outcomes of endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance only), and Inflammatory Bowel Disease Questionnaire (IBDQ) remission, a disease-specific HRQoL measure defined as an IBDQ score of 170 or greater. For both studies, the key baseline characteristics were generally balanced between treatment groups. Patients in the induction study were predominantly white (72.5%) and male (56.9%). The median age was 39.0 years (range, 18 to 79 years), and the majority of patients were from Eastern Europe (42.2%), followed by rest of world (37.5%) and Asia (20.3%). The mean duration of disease was 7.5 years, and the mean baseline MMS was 6.9, with 64.5% of patients with severely active disease (i.e., MMS 7 to 9) and 47.8% presenting with extensive disease. At baseline, 43.1% of patients were taking corticosteroids, 20.8% were taking immunomodulatory drugs, and 72.5% were taking oral 5-ASAs, with generally similar proportions of patients across the treatment groups. Among all patients, 93.2% experienced an inadequate response or intolerance to corticosteroids and/or 6-mercaptopurine or azathioprine or experienced corticosteroid dependence. Overall, 49.1% of patients had a history of experiencing ADT failure (i.e., treatment with an anti–TNF alpha, vedolizumab, or tofacitinib) and 50.9% had a history of experiencing ADT nonfailure. At induction baseline for the patients in the maintenance study, 40% were taking corticosteroids and 22.2% were taking immunomodulatory drugs (6-mercaptopurine, azathioprine, or methotrexate), 57.7% did not have a history of experiencing an inadequate response or intolerance to ADT (of these, 94.2% were naive to ADT), and 42.3% had experienced a prior inadequate response or intolerance to ADT. At maintenance baseline, the mean MMS was 2.5; 34.2% of patients were in clinical remission and 39.1% achieved endoscopic healing.
Efficacy Results
For the induction study, the primary analysis of efficacy outcomes was conducted in the FAS, which included patients with a baseline MMS of 5 to 9 who had received at least 1 (partial or complete) dose of the study intervention. For the maintenance study, the primary analysis of efficacy outcomes was performed in the randomized FAS among treated patients who had an MMS of 5 to 9 at induction baseline. The following results are from the primary analyses.
Clinical Remission (Primary Outcome)
A greater proportion of patients in the guselkumab group (22.6%) compared with the placebo group (7.9%) achieved clinical remission at week 12, with an estimated adjusted between-group difference of 14.9% (95% confidence interval [CI], 9.9 to 19.9; P value < 0.001). The results for clinical remission were consistent across the 2 prespecified ADT-related subgroups of interest (i.e., patients who were ADT-naive versus those who had experienced ADT failure) and were in favour of guselkumab compared with placebo, with an adjusted between-group difference of 20.0% (95% CI, 11.6 to 28.3) and 8.8% (95% CI, 3.4 to 14.3), respectively.
A greater proportion of patients in the guselkumab 100 mg (45.2%) and guselkumab 200 mg (50.0%) groups compared with the placebo group (18.9%) achieved clinical remission at week 44, with an estimated adjusted between-group difference of 25.2% (95% CI, 16.4 to 33.9; P value < 0.001) and 29.5% (95% CI, 20.9 to 38.1; P value < 0.001), respectively. The results for clinical remission were consistent across the prespecified ADT-related subgroups of interest (ADT-naive versus failure of ADT) in favour of guselkumab 100 mg and 200 mg compared with placebo, with an adjusted between-group difference of 24.3% (95% CI, 12.0, 36.5) and 28.8% (16.5% to 41.1), respectively.
Endoscopic Healing
A greater proportion of patients in the guselkumab group (26.8%) compared with the placebo group (11.1%) had endoscopic healing at week 12, with an estimated adjusted between-group difference of 16.0% (95% CI, 10.5 to 21.4; P value < 0.001). The results for clinical remission were consistent across the prespecified subgroups of interest, ADT-naive and failure of ADT, in favour of guselkumab compared with placebo (data not shown).
A greater proportion of patients in the guselkumab 100 mg (49.5%) and guselkumab 200 mg (51.6%) groups compared with the placebo group (18.9%) had endoscopic healing at week 44 of the maintenance study, with an estimated adjusted between-group difference of 29.5% (95% CI, 20.7 to 38.3; P value < 0.001) and 31.1% (95% CI, 22.5 to 39.8; P value < 0.001), respectively.
Clinical Response
Overall, 77.2% (n = 325) of patients achieved a clinical response at week 12 or week 24. Among the patients who were not in clinical response at week 12 with IV guselkumab and received SC guselkumab treatment (n = 125), 55.2% (n = 69) achieved a clinical response at week 24. A greater proportion of patients in the guselkumab 200 mg group (61.5%) compared with the placebo group (27.9%) experienced clinical response at week 12, with an estimated adjusted between-group difference of 33.8% (95% CI, 26.9 to 40.7; P value < 0.001).
A greater proportion of patients in the guselkumab 100 mg (77.7%) and guselkumab 200 mg (74.7%) groups compared with the placebo group (43.2%) continued to experience clinical response at week 44, with an estimated adjusted between-group difference of 33.6% (95% CI, 24.5 to 42.7; P value < 0.001) and 30.7% (95% CI, 21.5 to 40.0; P value < 0.001), respectively.
Corticosteroid-Free Clinical Remission
A greater proportion of patients in the guselkumab 100 mg (45.2%) and guselkumab 200 mg (48.9%) groups compared with the placebo group (18.4%) achieved clinical remission and were corticosteroid-free at week 44 of the maintenance study, with an estimated adjusted between-group difference of 25.7% (95% CI, 17.0 to 34.5; P value < 0.001) and 29.0% (95% CI, 20.5 to 37.6; P value < 0.001), respectively.
HRQoL Measured by IBDQ-Defined Remission
A greater proportion of patients in the guselkumab group (51.3%) compared with the placebo group (29.6%) achieved IBDQ remission at week 12, with an estimated adjusted between-group difference of 21.9% (95% CI, 14.9 to 29.0; P value < 0.001).
A greater proportion of patients in the guselkumab 100 mg (64.4%) and guselkumab 200 mg (64.2%) groups compared with the placebo group (37.4%) achieved IBDQ remission at week 44 of the maintenance study, with an estimated adjusted between-group difference of 26.3% (95% CI, 16.8 to 35.7; P value < 0.001) and 25.9% (95% CI, 16.5 to 35.4; P value < 0.001), respectively.
Subgroup and Sensitivity Analyses of Key Secondary Outcomes
For both studies, the results for endoscopic healing, clinical response, corticosteroid-free clinical remission (maintenance only), and IBDQ remission were consistent across the prespecified ADT-related subgroups of interest (ADT-naive and failure of ADT) in favour of guselkumab compared with the placebo group (data not shown). For both studies, the results of the sensitivity analyses were consistent with the those of the primary analyses.
Harms Results
In the induction study, 49.4% and 49.3% of patients reported at least 1 treatment-emergent adverse event (TEAE) in the guselkumab and placebo groups, respectively. Over the 12-week treatment period, the most frequently reported TEAEs in the guselkumab and placebo groups were anemia (5.0% and 6.8%, respectively), COVID-19 (5.0% and 4.3%), headache (3.1% and 2.9%), and colitis ulcerative (2.4% and 8.2%). Of these TEAEs, a numerically higher proportion of UC was reported in patients taking placebo. In the maintenance study, 64.5%, 70.0%, and 68.2% of patients reported at least 1 TEAE in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively. Over the 44-week treatment period, the most frequently reported TEAEs in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups were UC (9.1%, 13.2%, and 29.7%, respectively) and COVID-19 (12.9%, 9.5%, and 14.1%). Of these TEAEs, a numerically higher proportion of colitis ulcerative was reported in patients taking placebo. In the induction study, 2.9% and 7.1% of patients reported at least 1 serious TEAE in the guselkumab and placebo groups, respectively. In the maintenance study, 2.7%, 6.3%, and 0.5% of patients reported at least 1 serious TEAE in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively. In the induction study, 1.7% and 3.9% of patients reported at least 1 TEAE leading to discontinuation of treatment in the guselkumab and placebo groups, respectively. In the maintenance study, 3.8%, 2.6%, and 6.8% of patients reported at least 1 TEAE leading to discontinuation of treatment in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively. In the induction study, 1 and 2 deaths were reported in the guselkumab and placebo groups, respectively. There were no deaths reported in the maintenance study. In the induction study, the incidence of notable TEAEs, which included infections and serious infections, were similar between groups. Infections were reported in approximately 15% of patients and serious infections were reported in approximately less than 1% of patients in both groups. In the maintenance study, the incidence of notable infections and serious infections was also similar between groups. Infections were reported in approximately 32% of patients in all groups, and serious infections were reported in approximately 1% of patients in the guselkumab groups and 0 patients in the placebo group. No cases of active tuberculosis were reported in either study.
Critical Appraisal
Both the induction and maintenance studies were randomized, double-blind, placebo-controlled, parallel-group trials. Randomization and allocation concealment for both studies were performed using appropriate methodology via an interactive web response system. Randomization stratification was prespecified and was based on relevant prognostic factors, i.e., whether ADT had failed previously, region, and concomitant use of corticosteroids at baseline for the induction study, and clinical remission status at maintenance baseline, concomitant use of corticosteroids at maintenance baseline, and induction treatment for the maintenance study. Overall, the baseline demographic and disease characteristics appeared to be reasonably balanced between treatment groups in both studies. For the induction study, patients were eligible and enrolled if they had a baseline MMS of 4 to 9, and those with a baseline MMS of 4 were capped at 5% or less. However, according to the sponsor, the statistical analysis plan was amended per “health authority request” (on April 26, 2022, after patient enrolment) for the primary efficacy and safety analyses to include randomized and treated patients with a baseline MMS of 5 to 9. As a result, a total of 34 patients with a baseline MMS of 4 were excluded from the FAS primary efficacy and safety analyses, which could have compromised randomization. In general, the CDA-AMC review team and the clinical experts consulted by CDA-AMC did not consider this exclusion to have an important impact on study results because the patient characteristics appeared to be balanced between the treatment groups, and the findings of the supplementary analyses that included those with an MMS of 4 were consistent with the primary analyses. Overall, the statistical methods used in both studies were appropriate. The studies were powered sufficiently for primary and key secondary outcomes between the treatment groups. The subgroup analyses were likely underpowered to identify subgroup differences. An appropriate method for adjusting for multiplicity was used for the primary and secondary outcomes, but there was no multiplicity control for IBDQ remission and the subgroup analyses. The sponsor noted that based on feedback from the FDA, IBDQ remission was not included in the US-specific hierarchal testing procedure. Rates of study discontinuation were generally low in both studies and similar between groups although, in the induction study, the rate in the placebo group (12.9%) was higher than the guselkumab group (5.5%), mostly attributed to withdrawal by patients. Nonresponder imputation was used for missing data in the primary analysis, and preplanned sensitivity analyses were done under different assumptions, which were considered appropriate by the CDA-AMC review team.
In general, the study populations align with the approved Health Canada indication and reimbursement request, except that the indication is not limited to patients who have experienced an inadequate response or an ability to tolerate ADT or conventional therapy. The clinical experts did not consider the broader indication to affect the generalizability of the findings from the induction and maintenance studies. The dosing and administration of guselkumab were consistent with the approved product monograph. According to the clinical experts, the patient eligibility criteria and baseline characteristics of both studies were generalizable to adults with moderately to severely active UC in the Canadian setting. There were 8 and 6 patients from Canada included in the induction and maintenance studies, respectively. There was a relatively high rate of unsuccessful screenings in the induction study (269 out of 1,005 patients were screened out), mainly due to not meeting eligibility criteria. The clinical experts did not regard this as a factor that might influence the generalizability of the studies’ results. The studies included outcomes that were important to patients and clinicians, including clinical remission, clinical response, corticosteroid-free clinical remission, and HRQoL.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform the deliberations of the CDA-AMC expert committee, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty-of-evidence assessment for clinical remission, endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission, and HRQoL measured via IBDQ remission were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts consulted for this review.
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
remission and response (clinical remission, endoscopic healing, clinical response and maintenance of clinical response, and corticosteroid-free clinical remission)
HRQoL outcome (IBDQ remission).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for guselkumab versus placebo.
Table 2: Summary of Findings for Guselkumab vs. Placebo for Patients With Moderately to Severely Active Ulcerative Colitis — Induction and Maintenance Studies
Outcome and follow-up | Patients, N (studies) | Absolute effects (95% CI) | Certainty | What happens | ||||
|---|---|---|---|---|---|---|---|---|
Placebo | Guselkumab | Difference | ||||||
200 mg IV | 100 mg SC | 200 mg SC | ||||||
Clinical remission (FAS population) | ||||||||
Proportion of patients with clinical remission Follow-up: 12 weeks (induction study) | 701 | 79 per 1,000 | 226 per 1,000 | NA | NA | 149 more per 1,000 (99 more to 199 more) | Higha | Guselkumab induction treatment results in a clinically important increase in clinical remission at 12 weeks when compared with placebo. |
Proportion of patients with clinical remission Follow-up: 44 weeks (maintenance study) | 568 | 189 per 1,000 | NA | 452 per 1,000 | 500 per 1,000 | 100 mg SC: 252 more per 1,000 (164 more to 339 more) 200 mg SC: 295 more per 1,000 (209 more to 381 more) | Higha | Guselkumab maintenance treatment results in a clinically important increase in clinical remission at 44 weeks when compared with placebo. |
Endoscopic healing (FAS population) | ||||||||
Proportion of patients with endoscopic healing Follow-up: 12 weeks (induction study) | 701 | 111 per 1,000 | 268 per 1,000 | NA | NA | 160 more per 1,000 (105 more to 214 more) | Highb | Guselkumab induction treatment results in an increase in endoscopic healing at 12 weeks when compared with placebo. |
Proportion of patients with endoscopic healing Follow-up: 44 weeks (maintenance study) | 568 | 189 per 1,000 | NA | 495 per 1,000 | 516 per 1,000 | 100 mg SC: 295 more per 1,000 (207 more to 383 more) 200 mg SC: 311 more per 1,000 (225 more to 398 more) | Highc | Guselkumab maintenance treatment results in a clinically important increase in endoscopic healing at 44 weeks when compared with placebo. |
Clinical response (FAS population) | ||||||||
Proportion of patients with clinical response Follow-up: 12 weeks (induction study) | 701 | 279 per 1,000 | 615 per 1,000 | NA | NA | 338 more per 1,000 (269 more to 407 more) | Higha | Guselkumab induction treatment results in a clinically important increase in clinical response at 12 weeks when compared with placebo. |
Proportion of patients with clinical response Follow-up: 44 weeks (maintenance study) | 568 | 432 per 1,000 | NA | 777 per 1,000 | 747 per 1,000 | 100 mg SC: 336 more per 1,000 (245 more to 427 more) 200 mg SC: 307 more per 1,000 (215 more to 400 more) | Higha | Guselkumab maintenance treatment results in a clinically important increase in clinical response at 44 weeks when compared with placebo. |
Corticosteroid-free clinical remission (FAS population) | ||||||||
Proportion of patients with corticosteroid-free clinical remission Follow-up: 44 weeks (maintenance study) | 568 | 184 per 1,000 | NA | 452 per 1,000 | 489 per 1,000 | 100 mg SC: 257 more per 1,000 (170 more to 345 more) 200 mg SC: 290 more per 1,000 (205 more to 376 more) | Highd | Guselkumab maintenance treatment results in a clinically important increase in corticosteroid-free clinical remission at 44 weeks when compared with placebo. |
IBDQ remission (FAS population) | ||||||||
Proportion of patients with IBDQ remission Follow-up: 12 weeks (induction study) | 701 | 296 per 1,000 | 513 per 1,000 | NA | NA | 219 more per 1,000 (149 more to 290 more) | Higha | Guselkumab induction treatment results in a clinically important increase in IBDQ remission at 12 weeks when compared with placebo. |
Proportion of patients with IBDQ remission Follow-up: 44 weeks (maintenance study) | 568 | 374 per 1,000 | NA | 644 per 1,000 | 642 per 1,000 | 100 mg SC: 263 more per 1,000 (168 more to 357 more) 200 mg SC: 259 more per 1,000 (165 more to 354 more) | Higha | Guselkumab maintenance treatment results in a clinically important increase in IBDQ remission at 44 weeks when compared with placebo. |
CI = confidence interval; FAS = full analysis set; IBDQ = Inflammatory Bowel Disease Questionnaire; NA = not applicable; RCT = randomized controlled trial; SC = subcutaneous; vs. = versus.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aFor clinical remission, clinical response, and IBDQ remission, between-group absolute risk differences of 7.5% and 10% (75 and 100 fewer or more events per 1,000 patients) at 12 and 44 weeks, respectively, were clinically important, according to the clinical experts. The point estimate and entire CI exceeded the threshold.
bFor endoscopic healing at 12 weeks, the clinical experts could not determine a minimal important difference, so the target of the certainty appraisal was any effect. The point estimate and entire CI exceeded the null in favour of guselkumab.
cFor endoscopic healing at 44 weeks, a between-group absolute risk difference of 10% (100 fewer or more events per 1,000 patients) was clinically important, according to the clinical experts. The point estimate and entire CI exceeded the threshold.
dFor corticosteroid-free clinical remission at 44 weeks, a between-group absolute risk difference of 15% (150 fewer or more events per 1,000 patients) was clinically important, according to the clinical experts. The point estimate and entire CI exceeded the threshold.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Comparisons
One network meta-analysis (NMA) was submitted by the sponsor to inform the pharmacoeconomic model and fill gaps in the comparative evidence for other treatments of interest for moderately to severely active UC.
Description of Studies
The sponsor undertook a feasibility assessment to ascertain the extent of clinical heterogeneity across the trials identified in the systematic literature review. Trial design characteristics, patient eligibility criteria, baseline patient characteristics, outcome characteristics (i.e., definitions and methods of reporting outcomes), and placebo response were considered sources of clinical heterogeneity and explored in the feasibility assessment. Based on the feasibility assessment, an NMA was used for the outcomes of clinical remission and clinical response for induction and maintenance treatments (1-year analysis). The NMA assessed the induction and maintenance treatment effects of guselkumab versus adalimumab, infliximab, tofacitinib, upadacitinib, mirikizumab, vedolizumab, etrasimod, ozanimod, and ustekinumab for the treatment of adults with moderately to severely active UC. Some comparators were not relevant to Canadian public payers (e.g., risankizumab, filgotinib) and have therefore been excluded from the Clinical Review. A Bayesian NMA was conducted as the primary analysis of outcomes using an ordinal regression model to estimate odds ratios (ORs) and 95% credible intervals (CrIs). Random-effects NMA results were summarized for both induction and maintenance therapy based on 2 distinct patient populations: those who had experienced the failure of ADT and those who had not experienced ADT failure. The model fit of the induction results was adjusted by a baseline response due to placebo, but it was not possible to fit the same model in the maintenance phase (1-year analysis); therefore, the results presented are unadjusted.
Efficacy Results
The results for clinical remission at induction in the ADT nonfailure population did not favour any particular treatment (i.e., 95% CrIs crossed the null), and the results for the ADT failure population favoured upadacitinib versus guselkumab 200 mg. For clinical remission at maintenance in the ADT nonfailure population, the results favoured guselkumab 200 mg versus adalimumab and golimumab, and the results for the ADT failure population showed no difference between treatments. The results for clinical response at induction in the ADT nonfailure population favoured guselkumab 200 mg versus golimumab and adalimumab, and the results for the ADT failure population favoured guselkumab versus adalimumab. For clinical response at maintenance in the ADT nonfailure population, the results favoured guselkumab 100 mg and 200 mg versus adalimumab, and the results for the ADT failure population showed no difference between treatments.
There were several notable sources of heterogeneity for potential effect modifiers across the included studies. These included trial design characteristics, patient eligibility criteria, baseline patient characteristics, and definitions and methods of reporting outcomes. In addition, beyond the induction phase, the sponsor noted that although placebo is a common comparator across the majority of the included trials, placebo groups across re-randomized studies may not be equivalent due to variation in the treatments received during the preceding induction phase. The magnitude and direction of potential bias due to the heterogeneity of the outcome estimates cannot be predicted.
Harms Results
Harms were not assessed in the NMA.
Critical Appraisal
The methods used to conduct the systematic literature review and NMA were prespecified with an a priori protocol and used appropriate criteria to search databases, select studies, extract data, and assess the risk of bias of the included studies. Selection bias is expected to be low, given the comprehensiveness of the searches and methods for study selection. The NMA included relevant outcomes identified by the CDA-AMC team (clinical remission and clinical response); however, clinical and patient-relevant outcomes, such as endoscopic healing, corticosteroid-free clinical remission, HRQoL, and harms, were not included in the comparisons. The majority of comparators within the evidence network were informed by a single study, resulting in a star-shaped network and limiting certainty in the assumptions of the analysis.
Heterogeneity was noted across the studies, and while the meta-regression random-effects model accounts for some source of heterogeneity, there is a risk of the exchangeability assumption being validated. Specifically, potential effect modifiers across the included studies that could not be controlled for. Identified variables of concern included trial design characteristics, patient eligibility criteria, baseline patient characteristics, and outcome characteristics (i.e., definitions and methods of reporting outcomes). Variations in placebo effect estimates across the studies support these concerns about heterogeneity as well as concerns about possible violations of the assumptions of transitivity for the NMA. In addition, beyond the induction phase, the sponsor noted that although placebo is considered to be a common comparator across the majority of included trials, placebo arms across re-randomized trials may not be equivalent due to variation in the treatments received during the preceding induction phase. In particular, carryover effects observed due to previous treatments are likely to vary based upon treatment mechanism, potentially constituting a substantial source of cross-trial heterogeneity, which is evidenced in part by variations in baseline risk across included trials.
The specified model, while referred to as ordinal regression, is a multinomial regression that removes correlations between the outcomes of response and remission and estimates an OR for each outcome individually against no response or remission. Furthermore, the model structures the data such that the response outcome used a definition that is different from the one used in the clinical trial setting, where it is defined as response without remission. This redefinition introduces a core issue in that the response outcome is defined conditionally on a separate outcome that can occur at a future date; thus, whether an improvement in response rate due to treatment is a benefit or a harm becomes unclear. Ultimately, the interpretation of the ordinal ORs when the model assumptions are violated can create a misleading impression of how the outcomes and treatments are related. Due to the limitations in the NMA, the relative treatment effects of guselkumab versus other relevant comparators is uncertain and there is no conclusive evidence of a preferred treatment, based on the reported results.
Studies Addressing Gaps in the Evidence From the Systematic Review
No additional studies were submitted by the sponsor.
Conclusions
Evidence from 2 phase III double-blind RCTs (QUASAR induction and maintenance studies) reported on outcomes that were important to both patients and clinicians. The studies showed a high certainty of evidence that treatment with guselkumab results in a clinically meaningful increase in clinical remission, endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance only), and HRQoL via IBDQ remission at 12 weeks (induction therapy) and 44 weeks (maintenance therapy) compared with placebo in adults with moderately to severely active UC. Although the approved Health Canada indication is for a population that is broader than the patient populations enrolled in the studies (i.e., patients who had experienced an inadequate response or were unable to tolerate ADT or conventional therapy), the clinical experts consulted by CDA-AMC did not consider the indication to affect the generalizability of the findings. No new safety signals were identified, and the safety of guselkumab was consistent with the known safety profile of the drug. Due to limitations of the indirect treatment comparison (ITC), mostly attributed to the heterogeneity across studies, no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of guselkumab (Tremfya) 200 mg for induction, administered by IV infusion, followed by guselkumab 100 mg for maintenance, administered by SC injection, for the treatment of adults with moderately to severely active UC.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
IBD is a group of diseases characterized by chronic recurrent, progressive inflammation of the GI tract.3 There are 2 main types of IBD: CD and UC. UC is a chronic disease characterized by inflammation and ulcers in the mucosal layer of the large intestine (colon), typically beginning at the rectum (anus), progressing upward and, in some cases, affecting the entire colon.4-6 The cause of UC remains uncertain, but a combination of genetic and environmental factors contributes to immune dysregulation and upregulation in response to micro-organisms in the GI tract.7 Patients with UC present with the following symptoms: blood in the stool with mucus, frequent diarrhea, loss of appetite, and tenesmus (strong urge to use the bathroom without necessarily having a bowel movement), in addition to abdominal pain, rectal bleeding, and weight loss.8-10 Although UC principally affects the GI tract, extraintestinal manifestations may also occur, such as arthritis.11 There is no notable difference in the incidence of UC among male and female patients.12 UC generally develops in young adulthood13 and persists throughout life, marked by periods of spontaneous remission and relapse.14 UC has a worldwide annual incidence rate of 1.2 to 20.3 cases per 100,000 people and a prevalence of 7.6 to 246.0 cases per 100,000 people.8 The prevalence for UC in Canada in 2023 was estimated to be 414 per 100,000.15 It is estimated that 32% to 46% of the people in Canada with UC have moderate disease, and 13% to 14% have severe disease.16
Although the risk of mortality from UC itself is low, the disease is associated with an increased risk of other complications (e.g., respiratory diseases, colorectal cancer, lymphoma, and skin cancer) that result in increased mortality compared with the general population.17 About 30% to 60% of UC patients first present with isolated proctitis (involvement is limited to the rectum).18,19 Patients with proctitis are prone to proximal extension, which is associated with more colon involved in active disease, higher colectomy rates, increased need for ADT, and higher hospitalization rates than patients who start with extensive colitis.18,20,21 Among patients with isolated proctitis who are untreated for 1 year, the relapse rate is between 47% and 86%.22
UC can be further classified in clinical practice based on severity: mild, moderate, or severe disease, depending on the specific index score used (Truelove and Witts severity index, Mayo score, or the Montreal classification).23 While most patients have a mild to moderate disease course, about 10% to 15% experience an aggressive course of UC.13 Relapse is common, with the cumulative risk of relapse being 70% to 80% at 10 years.13 Achieving endoscopic healing earlier may be associated with a reduced risk of future colectomy.13 The chronic nature of UC has a considerable impact on patients’ HRQoL, including psychological, physical, sexual, and social domains of HRQoL due to the chronicity of symptoms such as urgency and frequency, and incontinence.24,25 The medical and surgical treatments for UC (e.g., colectomies) and their potential accompanying complications can also negatively impact HRQoL and productivity.26-30 Individuals with UC are at greater risk of comorbid anxiety, depression, and impaired social interactions.24,25,31,32 Patients with UC frequently report fatigue and sleep disturbance, as well as an inability to perform regular daily routines such as jobs or domestic chores.27,33-35 Furthermore, the disease can impact the patients’ caregivers and family, workplace, and community.17
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CDA-AMC review team.
The clinical experts consulted by CDA-AMC noted that short- and long-term treatment priorities for adults with moderately to severely active UC are to induce and maintain clinical remission (i.e., improve symptoms) and mucosal healing, reduce the use of corticosteroids, minimize adverse events (AEs), and improve HRQoL. The current standard of care for first-line treatment is oral corticosteroids but, due to their adverse effects, they are used only for inducing remission.36,37 To maintain remission, thiopurines (e.g., azathioprine and 6-mercaptopurine), 5-ASAs, anti-TNF therapy, or vedolizumab can be used.36,37 If 5-ASAs, corticosteroids, or thiopurines are ineffective or not tolerated, ADTs are considered (e.g., TNF antagonists, Janus kinase inhibitors, interleukin [IL]-23 inhibitors). However, most Canadian drug plans require unsuccessful steroid tapering with azathioprine or 6-mercaptopurine before a patient with moderately to severely active UC can be eligible for a biologic. As such, ADTs are typically not used for first-line maintenance of steroid-induced remission.37
For patients in whom 5-ASAs, corticosteroids, or thiopurines have not induced or maintained remission or are not tolerated, ADTs are used. Anti-TNF therapy followed by vedolizumab was recommended for second- and third-line induction and maintenance of remission by the Toronto Consensus guidelines in 2015 (which are the most recent Canadian guidelines for the treatment of UC).36,37 However, clinical practice has evolved since then, and the clinical experts consulted by CDA-AMC noted that early introduction of effective ADT is important for patient benefit, particularly for avoiding the adverse effects of recurrent or prolonged courses of steroids. The clinical experts and sponsor noted there are currently no Canadian guidelines to assist with the sequencing of ADTs; therefore, the choice of which ADT to prescribe is dependent on the patient’s disease characteristics, comorbidities, and preferences. Patients who experience treatment failure or loss of response or are intolerant to 1 advanced treatment would be offered a different drug, potentially in a different medication class with a different mechanism of action. If these therapies fail to keep a patient’s UC in complete remission, colectomy (removal of all or a part of the colon) may be required.
Drug Under Review
Key characteristics of guselkumab and other treatments available for moderately to severely active UC in adult patients are summarized in Table 3.
The reimbursement criteria requested for guselkumab is for the treatment of adult patients with moderately to severely active UC. In general, the reimbursement request aligns with the approved Health Canada indication, except that the indication is not limited to patients who have experienced an inadequate response or are unable to tolerate conventional therapy or ADT. Other Health Canada indications for guselkumab included plaque psoriasis and psoriatic arthritis; guselkumab received a CADTH recommendation for reimbursement with conditions for both indications.
The recommended induction dosage for UC is 200 mg of guselkumab IV administered by IV infusion over a period of at least 1 hour at week 0, week 4, and week 8. The recommended maintenance dosage for UC is 100 mg of guselkumab administered by SC injection at week 16 and every 8 weeks thereafter after completion of induction dosing. A dose of 200 mg administered by SC injection at week 12 and every 4 weeks thereafter may be considered for patients who do not show adequate therapeutic benefit to guselkumab, or according to clinical judgment.
Guselkumab is a human immunoglobulin G1 lambda monoclonal antibody that selectively binds to the p19 subunit of IL-23,38 which is a cytokine implicated in IBD pathogenesis because it drives intestinal inflammation.39 Guselkumab blocks the binding of extracellular IL-23 to the cell-surface IL-23 receptor, inhibiting IL-23–mediated intracellular signalling, activation, and production of inflammatory cytokines involved in IBD pathogenesis, such as IL-17A, IL-17F, and IL-22.40
Table 3: Key Characteristics of Guselkumab and Main Comparators
Drug name (brand) | Mechanism of action | Indicationa | Route of administration and recommended dose | Serious adverse effects and/or safety issues |
|---|---|---|---|---|
Drug under review | ||||
Guselkumab (Tremfya)38 | Blocks the binding of extracellular IL‑23 to the cell‑surface IL‑23 receptor, inhibiting IL‑23–mediated intracellular signalling, activation, and production of inflammatory cytokines. | For the treatment of adult patients with moderately to severely active UC. | Initiation: 200 mg IV administered by IV infusion over a period of at least 1 hour at week 0, week 4, and week 8. Maintenance: 100 mg administered by SC injection at week 16 and every 8 weeks thereafter. |
|
Comparators | ||||
S1P receptor modulators | ||||
Ozanimod (Zeposia)41 | S1P receptor modulator. Binds to the S1P1 receptors on lymphocytes, preventing egress from lymph nodes. It may reduce lymphocyte migration into the CNS and intestine. | For the treatment of adult patients with moderately to severely active UC who had an inadequate response, loss of response, or were intolerant to either conventional therapy or a biologic drug. | Initiation: 0.23 mg orally once daily on days 1 to 4, then 0.46 mg orally once daily on days 5 to 7. Maintenance: 0.92 mg orally once daily. | Malignancies, particularly of the skin. Initiation of ozanimod may result in transient reductions in heart rate and atrioventricular delays. |
Etrasimod (Velsipity)42 | Selective S1P receptor modulator. It may reduce lymphocyte migration into inflammation sites and reduce cytokine response. | For the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response or loss of response or were intolerant to either conventional therapy or an advanced treatment. | 2 mg orally, once daily. |
|
Anti-TNF biologics | ||||
Adalimumab (Humira)43 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. |
|
| Serious infections, malignancies, and neurologic events. The most common adverse reaction in rheumatoid arthritis patients treated with Humira was injection site reactions. |
Adalimumab biosimilars (Abrilada, Amgevita, Hulio, Hyrimoz)44-47 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. |
|
| Serious infections (pneumonia), malignancies, and neurologic events. |
Adalimumab biosimilars (Hadlima, Idacio, Simlandi, Yuflyma)48-51 | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. | For the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response to conventional therapy, including corticosteroids and/or azathioprine or 6‑MP, or who are intolerant to such therapies. | 160 mg in week 0, 80 mg in week 2, then 40 mg every other week thereafter as monotherapy or in combination with conventional therapies. Administered by SC injection. | Serious infections (pneumonia), malignancies, and neurologic events. |
Golimumab (Simponi)52 | Anti-TNF. Human monoclonal antibody that binds with p55 or p75 human TNF receptors. | For the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response to or have medical contraindications for conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6‑MP, for inducing and maintaining clinical response, inducing clinical remission, achieving sustained clinical remission in induction responders, or improving endoscopic appearance of the mucosa during induction. | 200 mg administered by SC injection at week 0, followed by 100 mg at week 2 and then 50 mg every 4 weeks thereafter. For maintenance, a dose 100 mg every 4 weeks can be considered at the discretion of the treating physician. | Upper respiratory tract infection. |
Infliximab (Remicade)53 | Anti-TNF. IgG1 kappa monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors. |
| IV infusion 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter, for the treatment of adult and pediatric patients (aged ≥ 6 years). Doses up to 10 mg/kg may be used in some adult patients. | Infections and malignancies. |
Anti-TNF. IgG1 kappa monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors. |
| Adults and pediatric patients (aged ≥ 6 years): IV infusion 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter. Doses up to 10 mg/kg may be used. | Infections and malignancies. | |
Anti-integrin | ||||
Vedolizumab (Entyvio)57 | IgG1 monoclonal antibody. Binds to the human alpha 4 beta 7 integrin, acting as a gut-selective anti-inflammatory biologic. | For the treatment of adult patients with moderately to severely active UC who have experienced an inadequate response or loss of response or were intolerant to either conventional therapy or infliximab, a TNF alpha antagonist. | 300 mg administered by IV infusion at 0, 2, and 6 weeks and then every 8 weeks thereafter. The SC maintenance dose is 108 mg every 8 weeks. | Infections and malignancies. |
IL-12 and IL-23 inhibitors | ||||
Mirikizumab (Omvoh)58 | Humanized IgG4 monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human IL‑23 cytokine to inhibit its interaction with the IL‑23 receptor. | For the treatment of adult patients with moderately to severely active ulcerative colitis who have experienced an inadequate response or loss of response or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. | Induction: 300 mg infused IV for at least 30 minutes at week 0, week 4, and week 8. If patients do not have adequate therapeutic response at week 12, consider extended inducted dosing of 300 mg IV at weeks 12, 16, and 20. Maintenance: 200 mg given as 2 consecutive SC injections of 100 mg each every 4 weeks after completion of induction dosing. | Upper respiratory tract infection, headache, and site injection reactions (e.g., rash, rash maculo-papular, rash popular, and rash pruritic). |
Ustekinumab (Stelara)59 | Human IgG1 monoclonal antibody. Neutralizes cellular responses mediated by IL‑12 and IL‑23. | For the treatment of adult patients with moderately to severely active UC who have had experienced an inadequate response or loss of response or were intolerant to either conventional therapy or a biologic or have medical contraindications to such therapies. | Single weight-based IV infusion (approximating 6 mg/kg) followed by a 90 mg SC dose 8 weeks later, then 90 mg SC every 8 weeks thereafter for maintenance. In patients with low inflammatory burden, a single IV dose followed 8 weeks later by 90 mg SC, then every 12 weeks thereafter, may be considered at the discretion of the treating physician. | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. |
JAK inhibitors | ||||
Selective JAK inhibitor. Blocks several cytokine pathways and lymphocyte activation. | For the treatment of adult patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to either conventional UC therapy or a TNF alpha inhibitor. | 10 mg orally b.i.d. for induction for at least 8 weeks and 5 mg given b.i.d. for maintenance. Depending on therapeutic response, 10 mg b.i.d. may also be used for maintenance in some patients. However, the lowest effective dose possible should be used for maintenance therapy to minimize adverse effects. | A Health Canada warning indicated an increased risk of thromboses (pulmonary and deep vein thrombosis) and death, and increased risk of serious infection, including herpes zoster infections. Of note, tofacitinib is not recommended in combination with biological UC therapies or with potent immunosuppressants such as azathioprine and cyclosporine. | |
Upadacitinib (Rinvoq)64 | Selective JAK inhibitor. Demonstrates activity against JAK1, JAK2, JAK3, and TYK2. | For the treatment of adult patients with moderately to severely active UC who have experienced prior treatment failure, i.e., experienced an inadequate response or loss of response or intolerance to at least 1 conventional and/or biologic therapy. | Induction: 45 mg orally once daily for 8 weeks. Maintenance: 15 mg orally once daily. For some patients, such as those with refractory, severe, or extensive disease, a maintenance dose of 30 mg once daily may be appropriate. The lowest effective dose needed to maintain response should be used. For patients aged ≥ 65 years, the only recommended maintenance dose is 15 mg once daily. | Upper respiratory tract infection. Of note, upadacitinib should not be used in combination with other JAK inhibitors, immunomodulating biologics (e.g., biologic DMARDs), or with potent immunosuppressants such as azathioprine, 6-MP, and cyclosporine. |
6-MP = 6-mercaptopurine; AE = adverse event; b.i.d. = twice a day; CNS = central nervous system; CV = cardiovascular; DMARD = disease-modifying antirheumatic drug; IgG1 = immunoglobulin G1; IL = interleukin; ITC = indirect treatment comparison; JAK = Janus kinase; PRES = posterior reversible encephalopathy syndrome; S1P = sphingosine 1-phosphate; S1P1 = sphingosine 1-phosphate receptor subtype 1; SC = subcutaneous; TNF = tumour necrosis factor; TYK2 = tyrosine kinase 2; UC = ulcerative colitis.
Notes: All the comparators in this table were included in the ITC as well as pharmacoeconomic analyses. Adalimumab was included in the ITC, but it was not broken down by adalimumab biosimilars versus the reference biologic drug. Infliximab was included in the ITC, but it was not broken down by infliximab biosimilars versus reference biologic drug. Tofacitinib was included in the ITC, but it was not broken down by tofacitinib generic versus the reference biologic drug.
aHealth Canada–approved indication.
Sources: Product monographs.41-64
Perspectives of Patients, Clinicians, and Drug Programs
The full patient group submissions received are available in the consolidated patient group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Two patient groups, Crohn’s and Colitis Canada and the Gastrointestinal Society provided input for guselkumab for the treatment of UC. Patient input for Crohn’s and Colitis Canada was gathered from an online survey conducted in 2022, including responses from 1,706 patients in Canada with CD or UC and their caregivers. A total of 354 patients from this survey identified as having moderate to severe UC. Patient input for the Gastrointestinal Society was based on responses to questionnaires conducted in 2024, 2023, 2022, 2020, 2018, and 2015, and 1 focus group conducted in 2022. A total of 514 respondents from Canada completed the 2024 survey about the unmet needs of individuals living with IBD, along with a follow-up survey focusing on opinions regarding biologics and biosimilars completed by 55 respondents. A total of 7 respondents (nationality not indicated) and 54 respondents with IBD from Canada completed the 2023 and 2022 surveys, respectively. For the 2020 survey, 145 respondents, most of whom had IBD (some had other inflammatory conditions), completed the survey on biosimilars, whereas 579 respondents completed the survey regarding the unmet needs of IBD. A total of 432 respondents with IBD from Canada completed the 2018 survey on the unmet needs in IBD, and 423 respondents from Canada with IBD, including CD and UC, completed the 2015 survey on biologics and biosimilars (then called subsequent-entry biologics).
When asked what UC-related complications patients were experiencing currently or had experienced within the past year, the most frequently reported complications cited by respondents in the Crohn’s and Colitis Canada input were mental health and stress (65%), followed by joint inflammation and arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), and skin conditions and malnutrition and weight loss, both of which were reported by around 30% of respondents. Other complications reported by the respondents in this input included strictures, adhesions (scar tissue), bowel obstruction, eye inflammation, perianal or anal fistulas and abscesses, internal (or intra-abdominal) fistulas or abscesses, stricture, ankylosing spondylitis (arthritis of the spine), liver conditions, and cancer. On the other hand, diarrhea, bowel urgency, incontinence, abdominal pain, fever, rectal bleeding, and nausea were reported by the respondents in the Gastrointestinal Society input as the common symptoms of UC. Respondents from this input also reported a profound effect on an individual’s life — physically, emotionally, and socially, both at home and at school or in the workplace. Respondents also indicated that the severity of the disease could fluctuate, making it necessary to go through routine testing, reassessments, and medication changes. The Gastrointestinal Society input further added that UC is particularly difficult for children and young adults because it often affects a person’s sense of self. Patients from the Gastrointestinal Society input added that sustained remission or treatment response was more important than relieving any 1 symptom.
In the 2022 survey conducted by Crohn’s and Colitis Canada, most patients reported using a combination of medications to manage their disease, with systemic steroids (79%), sulfasalazine and 5-ASAs (76%), and biologics (57%) being most common among those with UC, followed by immunomodulators (45%), antibiotics (42%), and nonsystemic steroids (38%). In the 2020 and 2024 surveys conducted by the Gastrointestinal Society, 33% and 29% of respondents believed their IBD was not well controlled by their current medications. Among the surveyed patients in the Crohn’s and Colitis Canada input, more than half (56%) of the respondents believed that different treatment options could make them feel better. Both patient groups emphasized that patients affected by UC need access to medications that work and can mitigate symptoms. The Gastrointestinal Society input further elaborated the importance of having a variety of options, keeping in mind that patients experience different responses to various medications and, in some cases, stop experiencing a response to medications after using them for some time.
While the Gastrointestinal Society indicated that corticosteroids are helpful for reducing inflammation in moderate to severe cases, it also emphasized that these medications are appropriate for short-term treatment only because they are generally not well tolerated and can have potentially serious side effects. Half of the respondents in the Crohn’s and Colitis Canada input indicated that systemic steroids are or were a burden in the management of UC, particularly among those with moderate to severe forms of UC, and among women. According to patients in the Crohn’s and Colitis Canada input who indicated that managing medication use is important, having enough treatment options, understanding the side effects of treatments, and minimizing steroid use were the most important aspects in the management of UC.
No patients from either patient group had experience with the drug under review. However, the Gastrointestinal Society reported conducting several patient surveys on biologics and biosimilars across several disease areas. In 1 of the surveys conducted by this group, 63% of respondents reported symptom reduction while receiving a biologic and 23% reported confirmed remission. In the 2024 survey on IBD conducted by the Gastrointestinal Society, 30% of respondents indicated they had no preferred route of administration and 41% preferred a daily oral medication; monthly injections were less preferred.
Clinician Input
Input From the Clinical Experts Consulted by CDA-AMC
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of moderately to severely active UC.
Unmet Needs
The clinical experts indicated that the treatment goals for patients with moderately to severely active UC is to induce and maintain clinical and endoscopic remission. The experts noted that first-line therapy typically includes oral mesalamine (a 5-ASA) plus corticosteroids, which are used for rapid achievement of clinical remission. Corticosteroids are not used in maintenance therapy and are avoided for use as a recurrent “rescue” medication, given the wide array of adverse effects common to their use. The experts noted that although mesalamine is effective for some patients, it is ineffective in those with more severe disease and, therefore, these patients require an alternative ADT to maintain remission. The experts noted that the challenge with currently available therapies for UC in Canada is that a portion of patients will not respond to ADT and, among the patients who experience a response initially, most experience a loss of response after a period of symptom relief. As such, in many patients, it is necessary to escalate the dose and try several types of therapy to maintain response and meet longer-term treatment goals. The clinical experts pointed out that multiple drug failures and ongoing progressive disease activity may lead to adverse consequences, including surgery to remove the entire colon. The experts noted there is also a gap in available oral therapies, given that most current therapies are administered intravenously or subcutaneously.
Place in Therapy
The clinical experts indicated that a clear sequence of medications that is optimal to treat moderate to severe UC is not yet established. In the clinical experts’ opinion, guselkumab should be available as a first-line treatment option, and failure of previous therapy should not be a criterion to access the drug. They noted that corticosteroids would commonly be used to induce response or remission before the initiation of guselkumab, but corticosteroids have recognized limitations and are not an option for long-term maintenance therapy.
Patient Population
The clinical experts agreed that the patients best suited for guselkumab would be those with moderately to severely active UC. The experts highlighted that patients would be identified based on clinical judgment and an examination that included a review of a patient’s history of symptoms, a biomarker evaluation, an endoscopic assessment, and a histopathological evaluation.
Assessing the Response Treatment
The clinical experts indicated that in clinical practice, a combination of clinical scoring systems (e.g., full Mayo and partial Mayo scores), endoscopic outcomes, histopathological evaluation, and patient-reported HRQoL scores is used to determine whether a patient is experiencing a response or disease progression on treatment. They also noted that biomarkers (most commonly fecal calprotectin) are used to monitor ongoing treatment response and maintenance therapy. The experts noted that patients starting a new ADT should have a clinical follow-up assessment within 14 to 16 weeks of initiating therapy. This could include biomarker evaluation in addition to the clinical assessment.
Discontinuing Treatment
The clinical experts indicated that treatment with guselkumab should be discontinued if patients are primary nonresponders, and if patients experience disease progression (e.g., ongoing symptoms or symptom escalation) while on maintenance therapy. The experts noted that disease progression while on guselkumab maintenance therapy would not necessarily mean the medication needs to be discontinued, and that further clinical assessment would be required to determine whether discontinuation is warranted. They also noted that anaphylactic reactions would necessitate discontinuation of the medication.
Prescribing Considerations
The clinical experts indicated that patients receiving guselkumab should be under the care of a gastroenterologist or general internist with experience in the treatment of IBD. They noted it would be reasonable for patients to receive the therapy in any clinical setting, including outpatient.
Clinician Group Input
No clinician group input was received by CDA-AMC for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC Reimbursement Review processes by identifying issues that may impact their ability to implement a recommendation. Their implementation questions and the corresponding responses from the clinical experts consulted by CDA-AMC are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The sponsor conducted a systematic review, ultimately identifying 3 trial records, each being a part of the QUASAR trial program: induction study 1, induction study 2, and the maintenance study. These were double-blind, placebo-controlled, parallel-group, multicentre studies. The study drug was not compared with a relevant comparator because placebo was used. Comparator drugs include ustekinumab, mirikizumab, golimumab, vedolizumab, upadacitinib, adalimumab, infliximab, tofacitinib, ozanimod, and etrasimod, of which most were included in the indirect comparison studies. Of note, the sponsor is currently conducting the ASTRO trial to evaluate the efficacy of guselkumab SC induction compared with placebo in participants with moderately to severely active UC. | This was a comment from the drug plans to inform CDEC deliberations. |
Not all relevant comparators are currently listed under drug plans. Some recommendations are recent, and some negotiations are ongoing, and this does also vary by jurisdiction. | This was a comment from the drug plans to inform CDEC deliberations. |
Considerations for initiation of therapy | |
The studies included adult patients with moderately to severely active UC, defined as a baseline modified Mayo score of 4 to 9, and a Mayo rectal bleeding subscore of ≥ 1. Question for the clinical experts: Is this the most appropriate scoring tool for identifying eligible patients for treatment with guselkumab in current clinical practice? | The clinical experts indicated that the Mayo score is an appropriate tool to identify eligible patients for treatment with guselkumab and is commonly used in clinical practice when assessing patients with UC. |
The studies excluded patients with a presence or history of fistula. Question for the clinical experts: Would you avoid use of guselkumab in these patients in your clinical practice? Are there any other exclusions listed in the studies that you would identify as contraindications to treatment with guselkumab? | The clinical experts noted they would not use guselkumab in patients with a presence or history of fistula. They indicated that once a patient has developed fistulizing disease, they are no longer classified as having UC. |
ADT is considered to be treatment with 1 or more TNF alpha antagonists or vedolizumab or tofacitinib at a dose approved for the treatment of UC. Question for the clinical experts: Do you feel that the prior treatments listed in the previously mentioned inclusion criteria are appropriate prior therapies to require, in the real-world setting, before a patient is eligible for the initiation of guselkumab therapy? | In the clinical experts’ opinion, guselkumab should be available as a first-line treatment option for patients with UC because the induction and maintenance trials included patients who were ADT-naive and those who had experienced ADT failure. They also confirmed that the list of prior ADTs (i.e., TNF alpha antagonists, vedolizumab, or tofacitinib) are appropriate and currently available treatment options for UC. |
Consider alignment with reimbursement criteria of recently reviewed comparators (i.e., Omvoh, Rinvoq, Zeposia). | This was a comment from the drug plans to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Consider alignment with renewal criteria of recently reviewed comparators (i.e., Omvoh, Rinvoq, Zeporia). | This was a comment from the drug plans to inform CDEC deliberations. |
Considerations for discontinuation of therapy | |
The study defined loss of response as no longer satisfying the definition of clinical response. Clinical response considered a range of improvements with the minimally important difference being a ≥ 3‑point decrease in either the Mayo score or the partial Mayo score. Question for the clinical experts: Does your definition of loss of response requiring a change in therapy align with this? | The clinical experts indicated that a 2- to 3‑point or higher change in the partial Mayo score would be a reasonable definition for loss of response. |
Consider alignment with stopping criteria of recently reviewed comparators (i.e., Omvoh, Rinvoq, Zeposia). | This was a comment from the drug plans to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
Induction: Recommended dose of 200 mg IV infusion at weeks 0, 4, and 8. Maintenance: 1. Recommended dose of 100 mg SC at week 16 and every 8 weeks thereafter. 2. A dose of 200 mg SC at week 12 and every 4 weeks thereafter may be considered for patients who do not experience adequate therapeutic benefit to guselkumab, or according to clinical judgment. Note for CDEC: It would be helpful for drug plans to have some defined conditions for considering the 200 mg maintenance option. With the sponsor noting its current ASTRO study for SC induction with guselkumab, it is possible that the recommended induction dosing will change. Question for the clinical experts: Are you able to clarify or define cases that would prompt you to prescribe the 200 mg maintenance dosing option? Participants experiencing a loss of response to the 100 mg dose in the maintenance arm were eligible to receive 200 mg dosing beginning between maintenance weeks 8 and 32. Considering this, would this be an option chosen only in patients with inadequate benefit following induction, or would this also be an option if a patient were to begin experiencing a loss of effect on the maintenance dosage of 100 mg every 8 weeks? Is there a time limit to increasing the dose? | The clinical experts noted that the option to use the 200 mg maintenance dose should be at the discretion of the treating physician, and noted the following 2 examples that would prompt them to prescribe the 200 mg maintenance dose option:
|
The induction dosing will need to be administered by a trained health care professional in a hospital setting or infusion clinic. Training will be required for patients or caregivers administering the maintenance dosing. | This was a comment from the drug plans to inform CDEC deliberations. |
Access to gastroenterologists can be limited in some areas. | This was a comment from the drug plans to inform CDEC deliberations. |
Consider alignment with prescribing criteria of recently reviewed comparators (i.e., Omvoh, Rinvoq, Zeposia). | This was a comment from the drug plans to inform CDEC deliberations. |
System and economic issues | |
Given there are several biologics, including biosimilars, currently listed on drug plans as options for UC, it is difficult to estimate how much of the market will be displaced by guselkumab. For CDEC consideration: Given there are 2 maintenance dosing options, the price impact would be significant if taking 100 mg every 8 weeks compared with 200 mg every 4 weeks. It is unclear whether the sponsor would make provisions for comparable pricing, regardless of the option prescribed. | This was a comment from the drug plans to inform CDEC deliberations. |
There are current PLAs in place for other biologics for UC, and there are negotiations in the works for additional biologics as well. | This was a comment from the drug plans to inform CDEC deliberations. |
ADT = advanced therapy; CDEC = Canadian Drug Expert Committee; PLA = product listing agreement; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
Clinical Evidence
The objective of this CDA-AMC Clinical Review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of guselkumab 200 mg for induction, administered by IV infusion, followed by 100 mg of guselkumab for maintenance, administered by SC injection, for the treatment of adults with moderately to severely active UC. The focus will be placed on comparing guselkumab with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of guselkumab is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section using the GRADE approach follows the critical appraisal of the evidence. The second section would typically include long-term extension studies; however, none were submitted by the sponsor. The third section includes indirect evidence from the sponsor. The fourth section would typically include additional studies to address important gaps in the systematic review evidence; however, none were submitted by the sponsor.
Included Studies
Clinical evidence from the following is included in the CDA-AMC review and appraised in this document:
2 pivotal trials identified in the systematic review
1 ITC.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5. The QUASAR trial consists of 3 separate studies conducted under a single protocol: a phase II induction dose-ranging study, a phase III induction study,1 and a phase III maintenance study.2 For the purpose of this review, the phase III induction (referred to herein as the induction study) and maintenance studies are summarized. The phase II dose-ranging study was not included in this review because some of the doses did not match the recommended dose for the approved indication, the sample size was substantially smaller than the phase III study sample size, and the outcomes between the studies overlapped. It should be noted that clinical responders from both the phase II and phase III induction studies were included in the maintenance study. The study designs of the induction and maintenance studies are depicted in Figure 1. The focus of the approved Health Canada indication and reimbursement request is generally aligned with the phase III induction and maintenance study populations, except that the indication is not limited to patients who have experienced an inadequate response or are unable to tolerate ADT or conventional therapy.
Induction
The QUASAR phase III induction study is a randomized, double-blind, parallel-group, multicentre trial that aimed to evaluate the efficacy and safety of guselkumab 200 mg IV as induction therapy versus matching placebo administered at weeks 0, 4, and 8 in adults with moderately to severely active UC.1 Using a permuted block design, enrolled patients were randomly assigned via an interactive web response system in a 3:2 ratio (N = 736 patients from 240 sites) to receive guselkumab 200 mg IV (N = 421) or placebo IV (N = 280). There were 8 patients included from 4 Canadian sites. Randomization was stratified by ADT failure status (yes or no), region (Eastern Europe, Asia, or rest of world), and concomitant use of corticosteroids at baseline (yes or no). The trial included an 8-week screening phase and a treatment phase; patients could become eligible for the maintenance study if they had demonstrated a clinical response to guselkumab or placebo treatment at week 12 or week 24. At the follow-up phase, nonresponders at week 24 stopped treatment and had a safety follow-up 12 weeks after their last intervention dose. The date of the last observation recorded as part of the database lock was January 12, 2023.
Maintenance
The QUASAR phase III maintenance study is a randomized, double-blind, parallel-group, multicentre trial that aimed to evaluate the efficacy and safety of guselkumab SC injection maintenance regimens (100 mg or 200 mg) versus matching placebo in patients who experienced a clinical response with guselkumab IV induction treatment in either the phase II or phase III induction study.2 Patients were randomized via an interactive web response system in a 1:1:1 ratio to guselkumab 200 mg (N = 190) every 4 weeks, guselkumab 100 mg (N = 188) every 8 weeks, or placebo (N = 190) every 4 weeks starting at week 0 and continuing through week 44. There were 6 patients included from 3 Canadian sites. Patients were allocated to an intervention group using permuted block randomization stratified by clinical remission status at maintenance baseline (yes or no), concomitant use of corticosteroids at maintenance baseline (yes or no), and induction treatment: guselkumab 400 mg IV, guselkumab 200 mg IV, and placebo IV or guselkumab 200 mg IV (placebo crossover). The date of the last observation recorded as part of the database lock for the maintenance study was September 19, 2023. Patients who completed the safety and efficacy evaluations (including the required endoscopy procedure) at week 44 and who, in the opinion of the investigator, might benefit from continued study intervention, entered the long-term extension of the maintenance study.
Table 5: Details of Studies Included in the Systematic Review
Detail | QUASAR phase III induction study | QUASAR maintenance study |
|---|---|---|
Designs and populations | ||
Study design | Phase III, randomized (3:2), double-blind, placebo-controlled, parallel-group, multicentre | Phase III, randomized (1:1:1), double-blind, placebo-controlled, parallel-group, multicentre |
Locations | 240 centres across 32 countries or territories that included Eastern Europe (41.4%), Asia (20.5%), and rest of world (38.0%). There were 8 patients across 4 sites in Canada | 254 sites across 32 countries or territories that included Eastern Europe (47.2%), Asia (20.5%), and rest of world (32.3%). There were 6 patients across 3 sites in Canada |
Patient enrolment dates | Start date: September 26, 2019 End date: Ongoing (data cut-off date for current analysis: January 12, 2023) | Start date: September 26, 2019 End date: Ongoing (data cut-off date for current analysis: September 19, 2023) |
Randomized (N) | 736 randomized patients (701 randomized patients with a modified Mayo score of 5 to 9):
| 846 randomized patients (805 treated patients with a modified Mayo score 5 to 9):
Nonrandomized:
|
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Interventions and comparators | Guselkumab 200 mg IV q.4.w. or placebo IV q.4.w. administered at weeks 0, 4, and 8 | Guselkumab 200 mg SC q.4.w., guselkumab 100 mg SC q.8.w., or placebo SC q.4.w. starting at week 0 through week 44:
Randomized patients with loss of clinical response were eligible to receive a single blinded dose adjustment to guselkumab 200 mg SC q.4.w., beginning at a scheduled visit between week 8 and week 32 |
Study duration | ||
Screening phase | Approximately 8 weeks | None (patients were screened approximately 8 weeks before the induction study) |
Treatment phase | Clinical responders: Treated up to week 12 followed by entry into maintenance study Week 12 clinical nonresponders: Treated up to induction week 24 followed by entry into the maintenance study if they experienced a response or stopped treatment if they did not experience a response | Treated up to week 44 |
Follow-up phase | Clinical responders: Immediate transition into maintenance study Induction week 12 clinical nonresponders: Nonresponders at induction week 24 stopped treatment and had a safety follow-up of 12 weeks after their last intervention dose | Entry into the maintenance LTE study at week 44 Patients who discontinued treatment at or before week 44 had a safety follow-up of 12 weeks after their last intervention dose |
Outcomes | ||
Primary end point | Clinical remission at week 12 | Clinical remission at week 44 |
Secondary and exploratory end points | Major secondary:
| Major secondary:
|
Publication status | ||
Publications | Peyrin-Biroulet L, Allegretti JR, Rubin DT, et al. Guselkumab in patients with moderately to severely active ulcerative colitis: QUASAR Phase 2b induction study. Gastroenterology. 2023;165(6):1443 to 1457. | |
Clinical trial number EudraCT number | NCT04033445 2018 to 004002 to 25 | |
EQ VAS = EQ visual analogue scale; IBDQ = Inflammatory Bowel Disease Questionnaire; LTE = long-term extension; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
aAdvanced therapy is considered to be treatment with 1 or more TNF alpha antagonists or vedolizumab or tofacitinib at a dose approved for the treatment of UC.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 1: Study Design of QUASAR Induction and Maintenance Trials
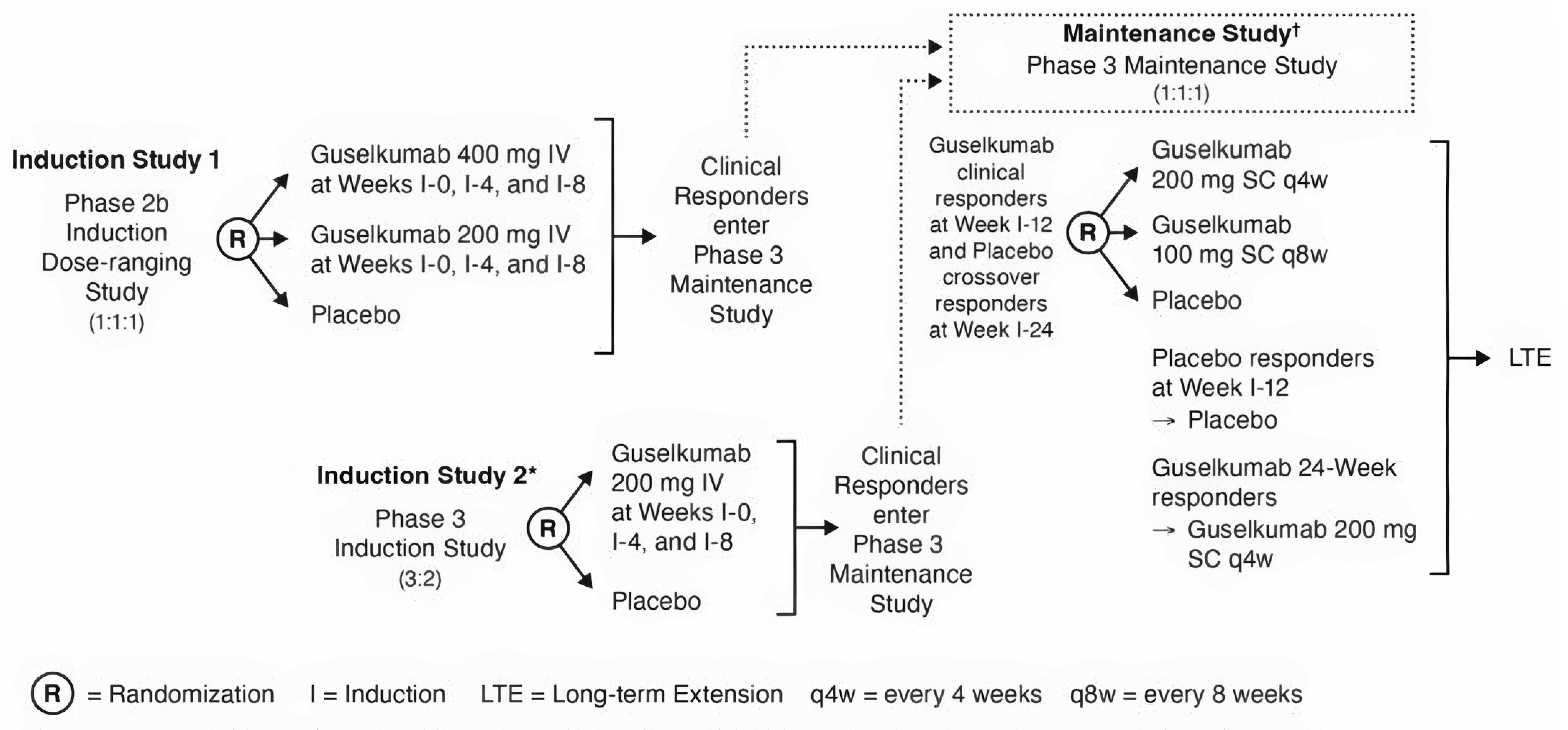
I = induction; LTE = long-term extension; q4w = every 4 weeks; q8w = every 8 weeks; R = randomization.
†Placebo responders at week I-12 and guselkumab 24-week responders entered the maintenance study but did not undergo re-randomization.
*Induction study 2 will not begin until the phase III guselkumab induction dose is selected based on an interim analysis of induction study 1.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
A detailed description of the key inclusion and exclusion criteria for the QUASAR induction and maintenance studies is in Table 5.
Induction Study
Eligible patients were adults aged 18 years or older with moderately to severely active UC who had demonstrated an inadequate response or inability to tolerate conventional therapy (i.e., 6-mercaptopurine, azathioprine, or corticosteroids) or ADT (i.e., TNF antagonists, vedolizumab, or tofacitinib). Patients were required to have an MMS of 4 to 9, a Mayo rectal bleeding score of 1 or more, and a Mayo endoscopy subscore of 2 or more. Patients were excluded if they were hospitalized for treatment of UC, were likely to require a colectomy within 12 weeks of baseline, had required any surgery within the 2 months before screening for active GI bleeding, peritonitis, intestinal obstruction, or intra-abdominal or pancreatic abscess requiring surgical drainage, or any other conditions possibly modifying the evaluation of benefit from the study intervention treatment.
Maintenance Study
Patients were eligible if they had demonstrated a clinical response to induction guselkumab IV or placebo at week 12 or 24. Patients who initiated or increased the dose of a UC-specific medication (or a restricted or prohibited medication) during the induction studies were prohibited from entering the maintenance study.
Interventions
Induction Study
Guselkumab was supplied as a 100 mg/mL sterile liquid in a single-dose prefilled syringe. Placebo was supplied as a 1 mL sterile liquid in a single-dose syringe assembled in the same way as for guselkumab. The study intervention was administered intravenously at the study site over a period of no less than 1 hour and not more than 2 hours (including the flush). From week 0 through week 8, patients were randomized to either guselkumab 200 mg IV at weeks 0, 4, and 8 (i.e., 3 IV doses) or matching placebo IV.
At week 12, all patients were evaluated for clinical response. Further study intervention administration was determined by the participant's clinical response status (using the Mayo endoscopy subscore assigned by the local endoscopist) at week 12 using the following procedure:
Guselkumab and placebo clinical responders at week 12 entered the maintenance study.
Patients initially randomized to placebo who were not in clinical response at week 12 crossed over to guselkumab and received 3 doses of guselkumab 200 mg IV at weeks 12, 16, and 20.
At week 24, patients who were not in clinical response at week 12 were re-evaluated for clinical response and entry into the maintenance study. This included patients initially randomized to placebo who were not in clinical response at week 12 who then crossed over to guselkumab induction 200 mg IV and achieved clinical response at week 24.
Maintenance Study
Guselkumab and placebo SC was supplied in the same manner as described in the induction study. Randomized patients received 1 of the 3 following interventions:
Guselkumab 100 mg SC every 8 weeks starting at week 4 and continuing through week 44 (1 injection at each visit). To maintain the blind, patients received 1 placebo SC injection plus 1 injection of 100 mg guselkumab or 2 placebo SC injections at alternating visits.
Guselkumab 200 mg SC every 4 weeks at week 0 and continuing through week 44 (2 injections of 100 mg at each visit).
Placebo SC every 4 weeks starting at week 0 and continuing through week 44. To maintain the blind, patients received 2 placebo injections at each visit.
Patients who were randomized and met predefined criteria for loss of clinical response were eligible to receive a single blinded dose adjustment to guselkumab 200 mg SC every 4 weeks beginning at a scheduled visit between week 8 and week 32. In addition, guselkumab induction responders at week 24 and induction placebo responders at week 12 from both induction studies entered the maintenance study but were not randomized. Patients remained on their assigned study intervention through week 44.
Concomitant Therapies
Patients who were receiving oral 5-ASA compounds, oral corticosteroids, 6-mercaptopurine, azathioprine, or methotrexate for the treatment of UC at baseline (i.e., week 0) were required to maintain a stable dose through week 44, except for corticosteroids. Oral corticosteroids were to be maintained at baseline doses during induction, and all patients were required to taper oral corticosteroids at week 0 of the maintenance study, unless this was medically not feasible.
Outcomes
A list of efficacy end points assessed in this Clinical Review is provided in Table 6, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence as well as any outcomes identified as important to this review, according to the clinical experts consulted by CDA-AMC and the input received from the patient groups and public drug plans. Using the same considerations, the review team selected end points that were considered most relevant to inform the expert committee deliberations and finalized this list in consultation with members of the expert committee. All summarized efficacy end points were assessed using GRADE. A description of the efficacy outcome measures and their measurement properties that were used in both the induction and maintenance studies are presented in Table 7.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | Induction study | Maintenance study |
|---|---|---|---|
Clinical remission | Induction: At week 12 Maintenance: At week 44 | Primarya | Primarya |
Endoscopic healing | Induction: At week 12 Maintenance: At week 44 | Secondarya | Secondarya |
Clinical response and maintenance of clinical response | Induction: At week 12 Maintenance: At week 44 | Secondarya | Secondarya |
Corticosteroid-free clinical remission | Maintenance: At week 44 | Secondarya | Secondarya |
IBDQ-defined remission | Induction: At week 12 Maintenance: At week 44 | Secondary | Secondary |
IBDQ = Inflammatory Bowel Disease Questionnaire.
aStatistical testing was performed hierarchically based on a US-specific testing procedure.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Clinical Remission
The primary outcome of clinical remission for the induction study at week 12 and maintenance study at week 44 was defined as a Mayo score stool frequency subscore of 0 or 1 that had not increased from induction baseline, a rectal bleeding subscore of 0, and an endoscopy subscore of 0 or 1 with no friability present on the endoscopy. The Mayo score instrument, which was administered by an investigator, is composed of 4 parts: rectal bleeding, stool frequency, physician assessment, and endoscopy appearance. Each part is rated from 0 to 3, giving a total score of 0 to 12, with a higher score indicative of more severe symptoms. The validity, reliability, responsiveness, and minimal important difference (MID) of the Mayo score is summarized in Table 7, although the findings are not specific to patients with UC.
Endoscopic Healing
The secondary outcome of endoscopic healing for the induction study at week 12 and maintenance study at week 44 was defined as a Mayo endoscopy subscore of 0 or 1 with no friability present on the endoscopy. The endoscopic findings were assessed by the investigator (i.e., local endoscopist) during the endoscopy procedure and by a central reader reviewing a video of the endoscopy.
Clinical Response
The secondary outcome of clinical response for the induction study at week 12 was defined as a decrease from induction baseline in the MMS of 30% or more and 2 points or more, with a decrease of 1 point or more from induction baseline in the rectal bleeding subscore or a rectal bleeding subscore of 0 or 1. The MMS is the Mayo score without the Physician’s Global Assessment subscore and was calculated as the sum of the stool frequency, rectal bleeding, and endoscopy subscores. The total score ranges from 0 to 9. An MMS of 5 to 9 and 7 to 9 indicate moderate and severe active disease, respectively. Maintenance of clinical response for the maintenance study was defined as clinical response at week 44 among patients in clinical response at maintenance baseline. The validity, reliability, responsiveness, and MID of the MMS are summarized in Table 7.
Corticosteroid-Free Clinical Remission
The secondary outcome of corticosteroid-free clinical remission for the maintenance study at week 44 was defined as clinical remission (as defined previously) without any use of corticosteroids for at least 8 weeks before assessment.
IBDQ-Defined Remission
The secondary outcome of IBDQ remission at week 12 in the induction study and week 44 in the maintenance study was used to assess disease-specific HRQoL, defined as a total IBDQ score of 170 or greater. The IBDQ consists of a 32-item list that is subdivided into 4 dimensions: systemic symptoms, bowel symptoms, emotional function, and social function. Total scores range from 32 to 224, with a higher score indicating better HRQoL. The IBDQ has been shown to have good internal consistency and test–retest reliability and responsiveness to change in IBD. Available studies have suggested that an improvement of 30 points from baseline or an improvement of at least 15 points versus placebo may constitute an MID.
Harms Outcomes
The assessment of safety was based on the proportion of patients experiencing 1 or more TEAEs, serious TEAEs, TEAEs leading to discontinuation, and deaths. TEAEs were coded using the Medical Dictionary for Regulatory Activities. Notable AEs identified by the sponsor and considered important by the clinical experts included malignancy and active tuberculosis or opportunistic infections.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Mayo score | A disease-specific physician-measured and patient-reported score that includes the following components: rectal bleeding (0 to 3), stool frequency (0 to 3), PGA (0 to 3), and endoscopy findings (0 to 3). A total score ranges from 0 to 12 for the full Mayo score with a higher score indicating more severe disease. An adapted Mayo score is calculated by subtracting the PGA from the full Mayo score. A partial Mayo score is a noninvasive 9-point system consisting of rectal bleeding, stool frequency, and PGA but without an endoscopy component. | Validity: Construct validity of the full Mayo score was demonstrated by a strong correlation with the patient’s assessment of disease activity (rho = 0.71 at week 12).65 A strong correlation was found between the partial and total Mayo scores (rho = 0.97 at weeks 4 and 8).66 Construct validity of the Mayo endoscopic subscore was supported by a strong correlation with the total Mayo score (Spearman rho = 0.97), the Riley histologic score (r = 0.55), and the Rubin histologic score (r = 0.60).67 Reliability and responsiveness: The endoscopic subscore was found to have moderate-to-substantial inter-rater agreement (range, 0.45 to 0.75). It was also found to be responsive to change over time with treatment.65,67-69 | Clinical response: ≥ 3 points decrease in the Mayo score or the partial Mayo score.65 Clinical remission: Clinical remission is indicated by a total Mayo score of ≤ 2 points, with or without an individual subscore of < 1.65,70 |
MMS | This is a modified version of the Mayo score. In the MMS, the definition of an ES of 1 no longer includes mucosal friability and the PGA is excluded. The components of the MMS include:
Scale components are scored on a 4-point scale from 0 to 3, with a score of 0 indicative of normal and a higher score indicative of more severe symptoms. The MMS is a sum of the Mayo stool frequency, rectal bleeding, and ES, giving a maximum score of 9. | Validity: In a cross-sectional survey of 2,608 patients with UC and their treating gastroenterologist, increases in the MMS were associated with increased odds of adverse outcomes, including a current flare (OR = 1.52; SE = 0.10), a higher number of flares in the past year (OR = 1.17; SE = 0.03), deterioration in clinical status (OR = 1.48; SE = 0.10), and a higher patient-reported overall WPAI score of 6.94 (SE = 0.888).71 A 1‑point increase in the MMS was associated with a 0.02‑unit decrease in the EQ-5D and a 2.73‑point decrease in the SIBDQ.71 Reliability and responsiveness: No studies of the reliability and responsiveness of the MMS were identified. | Evidence of an MID for the MMS in patients with UC was not identified. However, a value of ≤ 1 indicates remission for the MMS. In the context of UC, clinical remission is marked by a total Mayo score of ≤ 2 points.65,70 A clinical response is indicated by a ≥ 3 points decrease in the Mayo score or the partial Mayo score, demonstrating a meaningful reduction in disease severity.65 |
IBDQ | The IBDQ is a disease-specific questionnaire used to assess disease-specific HRQoL in patients with IBD.72 The IBDQ is a 32-item Likert‑based questionnaire divided into 4 dimensions:
Responses are graded on a scale from 1 (worse situation) to 7 (best situation). Total IBDQ score ranges between 32 and 224, with higher scores representing better HRQoL. Scores ranging from 170 to 190 are indicative of remission. | Validity: The emotional function dimension of the IBDQ was correlated with the Rand questionnaire (r = –0.76; P < 0.001); while the systemic symptoms dimension of the IBDQ was not correlated to change in the disease activity index (r = 0.036; P = 0.442) and patients’ global rating of change in emotional function was moderately correlated to the emotional function dimension (r = 0.52; P < 0.001) and the bowel symptom dimension (r = 0.42; P = 0.003) of the IBDQ.72 The IBDQ was found to detect changes in the social and emotional state of patients.73 Reliability and responsiveness: The IBDQ was considered reliable in 2 studies through an evaluation of internal consistency (Cronbach alpha of 0.7) and test–retest assessment (ICC = 0.9 to 0.99 or Pearson r ≥ 0.8). The IBDQ was also shown to be responsive to change in patients with IBD (P < 0.05).74,75 | While some sources suggest that an increase of 15 to 32 points may be considered a clinically relevant improvement in HRQoL for patients with CD and UC, evidence from clinical trials suggests that a change of more than 30 points is associated with clinical benefits and an improvement of 15 points or greater vs. placebo is required among patients with IBD, including those with UC.76-81 An IBDQ total score of ≥ 170 signifies disease remission, reflecting a significant improvement in the patient’s quality of life.76 |
CD = Crohn disease; ES = endoscopic score; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation; MID = minimal important difference; MMS = modified Mayo score; OR = odds ratio; PGA = Physician’s Global Assessment; SE = standard error; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; UC = ulcerative colitis; WPAI = Work Productivity and Activity Impairment questionnaire; vs. = versus.
Statistical Analysis
The statistical analysis of efficacy outcomes in the induction and maintenance studies is summarized in Table 8.
Sample Size and Power Calculation
Induction Study
The sample size for the phase III induction study was based on achieving 90% power to detect a treatment difference in the primary efficacy evaluation of clinical remission at week 12 using a 2-sided chi-square test and 0.05 significance level. Assuming an 8% clinical remission in the placebo group and 20% in the guselkumab group (assumed rates were based on data from the CNTO1275UCO3001 ustekinumab UC induction study and the mirikizumab phase II UC study), enrolling 148 patients in the placebo group and 222 patients in the 200 mg guselkumab group (370 patients in total, with a 2:3 placebo to guselkumab randomization ratio) was sufficient to achieve the prespecified statistical power.
To provide a sufficient number of patients for the primary analysis population in the maintenance study, it was estimated that a total sample size of approximately 1,000 patients across the phase II induction study and the phase III induction study was required and that the sample size for the phase III induction study would be at least 560 patients in the primary analysis population.
Maintenance Study
Assuming a 25% clinical remission rate at week 44 for placebo and 45% for each of the guselkumab treatment groups (assumed rates were based on data from the CNTO1275UCO3001 ustekinumab UC maintenance study), 118 patients in each randomized group (354 patients in total) would provide 90% power to reach a (2-sided) significance level of 0.05 for the primary outcome of clinical remission. However, the targeted number in the primary analysis population was increased to 484 patients due to the need to power at least 90% for the major secondary outcomes included in this report. The actual number of patients in the primary analysis population of this study depended on the number of guselkumab clinical responders at induction weeks 12 and 24 from the phase II and phase III induction studies.
Statistical Testing and Multiple Testing Procedures
Induction Study
The proportion of patients in clinical remission at week 12 was summarized within the treatment group, along with the associated 95% CIs. The adjusted treatment difference between treatment groups, incorporating Cochran-Mantel-Haenszel (CMH) weights and the corresponding 95% CI were also calculated. Primary outcome testing was conducted using a 2-sided CMH test, stratified by ADT failure status (yes or no) and baseline concomitant corticosteroid use (yes or no). The comparison between guselkumab and placebo was controlled at a 2-sided significance level of 0.05. Categorical data analyses, including chi-square tests, CMH chi-square tests, and logistic regression, were employed as appropriate to compare the proportions of patients achieving the specified outcomes. For continuous response parameters measured at multiple postbaseline visits, a mixed model for repeated measures was used, while an analysis of variance or analysis of covariance was applied when only 1 postbaseline visit was involved. The statistical testing strategy of the major secondary outcomes was the same as that used for the primary outcome analysis.
The hypothesis testing strategy adjusted for multiple testing by employing a fixed-sequence testing procedure to control for multiplicity across the primary and major secondary outcomes at a 2-sided 0.05 significance level. This means that after testing the primary outcome (clinical remission), the major secondary outcomes (endoscopic healing and clinical response) were tested in a specific order at the same significance level. If any outcome in this sequence was not significant, all subsequent tests were also considered not significant, and their corresponding P values were treated as nominal. Notably, the order of testing outcomes differed between the global and US strategies due to regional preferences. For example, IBDQ remission at week 12 was excluded from the US-specific testing procedure based on feedback from the FDA. For this submission, the US-specific testing procedure was presented because it was used in the Health Canada submission.
Maintenance Study
The proportion of patients achieving the primary and each key secondary outcome was summarized by treatment group, along with the associated 95% CIs. The adjusted treatment difference between treatment groups, incorporating CMH weights, and the corresponding 95% CIs were also presented. For all statistical comparisons of the primary and key secondary outcomes, a 2-sided CMH test was used, stratified by clinical remission status at maintenance baseline (based on the final endoscopy score, yes or no) and induction dose treatment. Similar to the induction study, the US-specific multiple testing procedure was used, starting with guselkumab 200 mg for clinical remission at week 44, to control the overall type I error rate at the (2-sided) 0.05 level for the primary and major secondary outcomes, except for IBDQ remission. The order of testing was as follows: clinical remission, endoscopic healing, maintenance of clinical response, and corticosteroid-free clinical remission.
Data Imputation
In both studies, any missing data for the outcomes were treated using nonresponder imputation (e.g., not in clinical remission). The patients who had an ostomy or colectomy, a prohibited change in UC medication, or discontinued the study intervention due to a lack of efficacy, an AE of worsening UC, or other reasons, except for reasons related to COVID-19 (excluding COVID-19 infection) or the regional crisis in Russia and Ukraine, were considered to have not experienced the desired outcomes.
Subgroup Analysis
The prespecified subgroups of interest for both studies included patients who were ADT-naive and those who had experienced ADT failure. A subgroup analysis for both studies was conducted using the rate difference and 95% CIs, adjusted with CMH weights, incorporating treatment group, ADT failure status, and concomitant corticosteroid use as baseline factors. Missing values were imputed as nonresponders.
Sensitivity Analyses
The sensitivity analyses performed for the primary and secondary outcomes are summarized in Table 8.
Analysis Populations
The analysis populations are summarized in Table 9. The efficacy and safety analyses were based on the FAS and safety analysis set (SAS), respectively, which were defined as all randomized patients with an MMS of 5 to 9 who received at least 1 (partial or complete) dose of the study intervention. For the maintenance study, the FAS and SAS included patients with an MMS of 5 to 9 who were randomized and treated in the maintenance study.
Table 8: Statistical Analysis of Efficacy Outcomes
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Induction study | ||||
Clinical remission Endoscopic healing Clinical response IBDQ remission | Weighted CMH test of relative difference in proportions, adjusted for the stratification factors | Stratification factors | Imputed as nonresponder | Sensitivity analysis 1: Tipping point with exhaustive scenarios Sensitivity analysis 2: Tipping point based on multiple imputation with Bernoulli draws Sensitivity analysis 3: Multiple imputation Sensitivity analysis 4: Exclusion of patients whose data cannot be verified by source data |
Maintenance study | ||||
Clinical remission Endoscopic healing Maintenance of clinical response IBDQ remission Corticosteroid-free clinical remission | Weighted CMH test of relative difference in proportions, adjusted for the stratification factors | Stratification factors | Imputed as nonresponder | Sensitivity analysis 1: Tipping point Sensitivity analysis 2: Multiple imputation Sensitivity analysis 3: Exclusion of patients whose data cannot be verified by the source data due to major disruption |
CMH = Cochran-Mantel-Haenszel; IBDQ = Inflammatory Bowel Disease Questionnaire.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 9: Analysis Populations of Induction and Maintenance Studies
Study | Population | Definition | Application |
|---|---|---|---|
Induction study | FAS and SAS (N = 701) | Randomized patients with an MMS of 5 to 9 who received at least 1 (partial or complete) dose of the study intervention | Primary efficacy and safety analyses |
Maintenance study | Randomized FAS and SAS (N = 568) | Patients with an MMS of 5 to 9 at induction baseline who were randomized and treated in the maintenance study | Primary efficacy and safety analyses |
FAS = full analysis set; MMS = modified Mayo score; SAS = safety analysis set.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Patient disposition for the induction and maintenance studies is summarized in Table 10 and Table 11, respectively.
Induction Study
Of the 1,005 patients screened, 736 were randomized in 240 centres across 32 countries or territories. The main reasons for unsuccessful screening were not reported. Of the 736 randomized patients, 702 had a baseline MMS of 5 to 9 and 1 patient was randomized to placebo at week 0 but was not treated because of inadequate venous access for administration of the study intervention and protocol-specified laboratory assessments. The FAS, which is the primary efficacy analysis set, comprised 701 treated patients with a baseline MMS of 5 to 9. In total, 62 patients (8.8%) in the FAS discontinued treatment. Treatment discontinuation before week 12 occurred in 18 patients (4.3%) in the guselkumab group and 24 patients (8.6%) in the placebo group. The most common reasons for treatment discontinuation for both groups were similar and consisted of AEs (2.3%) and withdrawal by participant (2.4%). In total, 20 patients discontinued the study treatment after week 12, with the most common reason being AEs (3.4%) for both groups. A total of 398 patients (94.5%) in the guselkumab group and 244 patients (87.1%) in the placebo group completed study participation.
Through week 12, a similar proportion of patients in each group had major protocol deviations (10.0% in the guselkumab group and 9.3% in the placebo group), with the most common reasons in both groups being not satisfying the study entry criteria (total of 5.6%) and “other” (total of 3.9%).
Maintenance Study
The maintenance study enrolled 846 patients, 568 of whom comprised the FAS primary analysis population of patients who had an MMS of 5 to 9 at induction baseline and who were randomized and treated. In the randomized FAS, treatment discontinuation before week 40 occurred in 20 patients (10.6%) in the guselkumab 100 mg group, 22 patients (11.6%) in the guselkumab 200 mg group, and 26 patients (13.7%) in the placebo group. The most common reasons for treatment discontinuation were similar across groups and consisted of AEs (5.1%) and withdrawal by participant for reasons other than AEs (3.3%).
Through week 44, a similar proportion of patients in each group had major protocol deviations (8.5% in the guselkumab 100 mg group, 6.3% in the guselkumab 200 mg group, and 8.4% in the placebo group), with the most common reason being “other” (total of 6.3%) in all groups.
Table 10: Summary of Patient Disposition for the Induction Study
Patient disposition | Guselkumab 200 mg IV | Placebo |
|---|---|---|
Screened, N | 1,005 | |
Reason for unsuccessful screening, n (%) | NR | NR |
Randomized, N | 736 | |
Randomized and treated, N | 735 | |
Randomized and treated with a modified Mayo score of 5 to 9 (FAS), N | 421 | 280 |
Patients who completed study participation, n (%) | 398 (94.5) | 244 (87.1) |
Patients entering the maintenance study, n (%) | 348 (82.7) | 202 (72.1) |
Patients not entering the maintenance study who continued to the safety follow-up visit, n (%) | 50 (11.9) | 42 (15.0) |
Discontinued study treatment before week 12, n (%) | 18 (4.3) | 24 (8.6) |
Reason for treatment discontinuation, n (%) | ||
Adverse events | 7 (NR) | 9 (NR) |
Withdrawal by patient | 6 (NR) | 11 (NR) |
Discontinued from study, n (%) | 23 (5.5) | 36 (12.9) |
Reason for discontinuation, n (%) | ||
Lost to follow-up | 1 (0.2) | 2 (0.7) |
Withdrawal by patient | 11 (2.6) | 19 (6.8) |
Death | 0 (0.0) | 2 (0.7) |
Other | 11 (2.6) | 13 (4.6) |
COVID-19-related | 0 (0.0) | 1 (0.4) |
Other major disruption | 0 (0.0) | 2 (0.7) |
SAS, N | 421 | 280 |
FAS = full analysis set; NR = not reported; SAS = safety analysis set.
Sources: QUASAR induction Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 11: Summary of Patient Disposition for the Maintenance Study
Patient disposition | Guselkumab 100 mg | Guselkumab 200 mg | Placebo | ||
|---|---|---|---|---|---|
Enrolled, N | 846 | ||||
Randomized and treated, N | 599 | ||||
Randomized and treated with a modified Mayo score of 5 to 9 (FAS), N | 188 | 190 | 190 | ||
Completed study through week 44, n (%) | 178 (NR) | 177 (NR) | 181 (NR) | ||
Patients entered long-term extension phase, n (%) | 165 (87.8) | 164 (86.3) | 149 (78.4) | ||
Patients not entering long-term extension phase at week 44, n (%) | 13 | 13 | 32 | ||
Discontinued study treatment before week 40, n (%) | 20 (10.6) | 22 (11.6) | 26 (13.7) | ||
Reason for discontinuation, n (%) | |||||
Adverse events | 7 (NR) | 10 (NR) | 12 (NR) | ||
Withdrawal by patient | 4 (2.1) | 7 (3.7) | 8 (4.2) | ||
Discontinued study before week 44, n (%) | 10 (5.3) | 13 (6.8) | 9 (4.7) | ||
Lost to follow-up | 1 (0.5) | 1 (0.5) | 0 (0.0) | ||
Withdrawal by patient | 7 (3.7) | 9 (4.8) | 10 (5.3) | ||
Death | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
Other | 1 (0.5) | 0 (0.0) | 3 (1.6) | ||
SAS, N | 186 | 190 | 192a | ||
FAS = full analysis set; NA = not applicable; NR = not reported; SAS = safety analysis set; SC = subcutaneous.
aTwo patients who were randomized to the guselkumab 100 mg SC group received only placebo at week 0 (these 2 patients discontinued the study intervention before their first scheduled guselkumab dose at week 4). They were therefore included in the placebo SC treatment group for the safety analyses.
Sources: QUASAR maintenance Clinical Study Report.2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics for the induction and maintenance studies are outlined in Table 12 and Table 13, respectively, and are limited to those that were considered most relevant to this review or were felt to affect the outcomes or interpretation of the study results.
Induction Study
The baseline characteristics were generally similar between treatment groups. Patients were predominantly white (72.5%) and male (56.9%). The median age was 39.0 years (range, 18 to 79 years), and the median weight was 70.6 kg. The majority of patients in the FAS were from Eastern Europe (42.2%), followed by rest of world (37.5%) and Asia (20.3%). The mean duration of disease was 7.5 years, and the mean baseline MMS was 6.9, with 64.5% of patients with severely active disease (i.e., MMS of 7 to 9) and 47.8% presenting with extensive disease. At baseline, 43.1% of patients were taking corticosteroids, 20.8% were taking immunomodulatory drugs, and 72.5% were taking oral 5-ASAs, with generally similar proportions of patients across the treatment groups. Among all patients, 93.2% experienced an inadequate response or intolerance to corticosteroids and/or 6-mercaptopurine or azathioprine or experienced corticosteroid dependence. Overall, 49.1% of patients had a history of experiencing ADT failure (i.e., anti–TNF alpha, vedolizumab, or tofacitinib) and 50.9% had a history of experiencing ADT nonfailure.
Maintenance Study
Baseline characteristics in the randomized FAS were generally similar between treatment groups. At induction baseline, the majority of patients were white (73.2%) and male (54.8%). The median age was 39.0 years (range, 18 to 79 years), and the median weight was 70.0 kg. The majority of these patients were from Eastern Europe (48.4%), followed by rest of world (32.6%) and Asia (19.0%). At induction baseline, the median UC disease duration was 5.4 years, the mean MMS was 6.9 (63.9% had a score indicating severe disease, i.e., an MMS of 7 to 9), and the proportion of patients with a Mayo endoscopy subscore of 3 (severe) was 66.4%. At induction baseline, 40% of patients were taking corticosteroids and 22.2% were taking immunomodulatory drugs (6-mercaptopurine, azathioprine, or methotrexate), 57.7% did not have a history of experiencing an inadequate response or intolerance to ADT (of these, 94.2% were naive to ADT), and 42.3% had experienced a prior inadequate response or intolerance to ADT (of these, 42.5% had experienced the failure of ≥ 2 ADT classes). At maintenance baseline, the mean MMS was 2.5, 34.2% of patients were in clinical remission, and 39.1% achieved endoscopic healing.
Table 12: Summary of Baseline Characteristics for the Induction Study — FAS Population
Characteristic | Placebo (N = 280) | Guselkumab 200 mg (N = 421) |
|---|---|---|
Demographic characteristics | ||
Mean age, years (SD) | 39.8 (13.43) | 41.0 (13.90) |
Sex, n (%) | ||
Female | 119 (42.5) | 183 (43.5) |
Male | 161 (57.5) | 238 (56.5) |
Region, n (%) | ||
Asia | 58 (20.7) | 84 (20.0) |
Eastern Europe | 117 (41.8) | 179 (42.5) |
Rest of world | 105 (37.5) | 158 (37.5) |
Race, n (%) | ||
Asian | 62 (22.1) | 88 (20.9) |
White | 205 (73.2) | 303 (72.0) |
Mean weight, kg (SD) | 71.83 (17.032) | 72.87 (16.713) |
UC-related characteristics | ||
Mean UC disease duration, years (SD) | 7.09 (6.545) | 7.80 (7.728) |
Limited to left side of colon, n (%) | 133 (47.5) | 233 (55.3) |
Mean Mayo score (SD) | 9.2 (1.34) | 9.1 (1.36) |
Moderate severity (6 ≤ Mayo score ≤ 10), n (%) | 229 (81.8) | 346 (82.2) |
Severe (Mayo score > 10), n (%) | 51 (18.2) | 75 (17.8) |
Mean modified Mayo score (SD) | 6.9 (1.07) | 6.9 (1.13) |
Mean partial Mayo score (SD) | 6.5 (1.21) | 6.4 (1.22) |
Moderate (endoscopy subscore = 2), n (%) | 100 (35.7) | 125 (29.7) |
Severe (endoscopy subscore = 3), n (%) | 180 (64.3) | 296 (70.3) |
Absence of extraintestinal manifestation, n (%) | 250 (89.3) | 361 (85.7) |
Mean CRP, mg/L (SD) | 8.3 (11.66)a | 9.0 (12.40)a |
Abnormal CRP (> 3 mg/L), n (%) | 160 (57.6)a | 248 (59.6)a |
Mean fecal calprotectin, mg/kg (SD) | 2,709.2 (4,018.92)a | 3,422.5 (5,174.90)a |
Abnormal fecal calprotectin (> 250 mg/kg), n (%) | 225 (88.9)a | 333 (90.0)a |
Mean albumin, g/L (SD) | 43.0 (4.07) | 42.9 (4.08) |
CRP = C-reactive protein; FAS = full analysis set; SD = standard deviation; UC = ulcerative colitis.
aSome patients were missing data; as a result, the total N does not match.
Sources: QUASAR induction Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 13: Summary of Baseline Characteristics for the Maintenance Study — Randomized FAS Population
Characteristic | Placebo (N = 190) | Guselkumab 100 mg (N = 188) | Guselkumab 200 mg (N = 190) |
|---|---|---|---|
Demographic characteristics at induction baseline | |||
Mean age (SD) | 41.2 (13.58) | 40.3 (13.00) | 40.6 (14.66) |
Sex, n (%) | |||
Female | 81 (42.6) | 86 (45.7) | 90 (47.4) |
Male | 109 (57.4) | 102 (54.3) | 100 (52.6) |
Region, n (%) | |||
Asia | 30 (15.8) | 36 (19.1) | 42 (22.1) |
Eastern Europe | 102 (53.7) | 85 (45.2) | 88 (46.3) |
Rest of world | 58 (30.5) | 67 (35.6) | 60 (31.6) |
Race, n (%) | |||
Asian | 34 (17.9) | 38 (20.2) | 44 (23.2) |
White | 142 (74.7) | 139 (73.9) | 135 (71.1) |
Mean weight (SD) | 73.56 (16.997) | 70.68 (16.757) | 70.94 (16.624) |
UC-related characteristics at maintenance baseline | |||
Mean Mayo score (SD) | 3.3 (2.01) | 3.2 (1.91) | 3.2 (1.85) |
Mean modified Mayo score (SD) | 2.5 (1.57) | 2.6 (1.51) | 2.5 (1.50) |
Mean partial Mayo score (SD) | 1.7 (1.34) | 1.6 (1.18) | 1.6 (1.19) |
Endoscopic healing: Yes, n (%) | 68 (35.8) | 75 (39.9) | 79 (41.6) |
Endoscopic remission (endoscopic normalization): Yes, n (%) | 39 (20.5) | 41 (21.8) | 47 (24.7) |
Clinical remission, n (%) | 59 (31.1) | 66 (35.1) | 69 (36.3) |
IBDQ remission, n (%) | 142 (75.5) | 134 (71.3) | 128 (67.7) |
Mean CRP, mg/L (SD) | 3.5 (5.13) | 4.6 (11.19) | 3.4 (6.61) |
Abnormal CRP (> 3 mg/L), n (%) | 63 (33.2) | 63 (33.5) | 56 (29.5) |
Mean fecal calprotectin, mg/kg (SD) | 1,143.2 (2,683.43)a | 1,292.3 (2,953.34)a | 942.4 (1,577.16)a |
Abnormal fecal calprotectin (> 250 mg/kg), n (%) | 102 (54.3)a | 101 (54.6)a | 97 (51.9)a |
Mean albumin, g/L (SD) | 45.9 (3.08)a | 45.9 (3.40) | 45.4 (3.40) |
CRP = C-reactive protein; FAS = full analysis set; IBDQ = Inflammatory Bowel Disease Questionnaire; SC = subcutaneous; SD = standard deviation; UC = ulcerative colitis.
aSome patients were missing data; as a result, the total N does not match. Please refer to the maintenance study Clinical Study Report for the exact N.
Sources: QUASAR maintenance Clinical Study Report.2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Induction Study
In the FAS, all patients received at least 1 (partial or complete) dose of the study intervention. A total of 290 patients who were not in clinical response at week 12 received guselkumab between week 12 and week 24 as follows: the patients who received placebo IV crossed over to guselkumab 200 mg IV (165 patients), and the patients who received guselkumab 200 mg IV crossed over to guselkumab 200 mg SC (125 patients).
Maintenance Study
Patients in the randomized SAS received the study intervention (up to the dose adjustment) as follows:
Guselkumab 100 mg SC every 8 weeks: 186 patients received guselkumab 100 mg, with a median cumulative dose of 500.0 mg
Guselkumab 200 mg SC every 4 weeks: 190 patients received guselkumab 200 mg, with a median cumulative dose of 2,200.0 mg
Placebo SC: 192 patients
Concomitant Medications
Induction Study
At baseline, 612 patients (87.3%) in the FAS were receiving 1 or more concomitant medications for UC with similar proportions of patients across both treatment groups. At baseline, a total of 302 patients (43.1%) were receiving oral corticosteroids, 146 patients (20.8%) were receiving immunomodulatory drugs (i.e., azathioprine, 6-mercaptopurine, or methotrexate), and 508 patients (72.5%) were receiving oral 5-ASAs, with generally similar proportions of patients in both guselkumab treatment groups. Concomitant UC medications were to be maintained at a stable dose throughout the study.
Maintenance Study
For patients who were receiving oral corticosteroids on entry into the maintenance study, the investigator was to begin tapering the daily dose of corticosteroids at week 0. Other UC-specific therapies (i.e., oral 5-ASA compounds, 6-mercaptopurine, azathioprine, or methotrexate) were to be maintained at stable doses through week 44 (without initiation or increase in dose) unless investigator judgment required that the therapy be discontinued or the dose reduced because of toxicity or medical necessity. Tapering of the daily dose of corticosteroids was allowed to be paused for patients who met clinical flare criteria.
Efficacy
A summary of the key efficacy results for both studies is in Table 14. For the induction study, the primary analysis of efficacy outcomes was conducted in the FAS, which included patients who received at least 1 (partial or complete) dose of the study intervention who had a baseline MMS of 5 to 9. For the maintenance study, the primary analysis of the efficacy outcomes was performed in the randomized FAS among treated patients who had an MMS of 5 to 9 at induction baseline.
Clinical Remission (Primary Outcome)
Induction Study
A greater proportion of patients in the guselkumab group (22.6%) compared with the placebo group (7.9%) experienced clinical remission at week 12, with an estimated adjusted between-group difference of 14.9% (95% CI, 9.9 to 19.9; P value < 0.001).
Subgroup Analyses
The results for clinical remission were consistent across the prespecified ADT-related subgroups of interest (i.e., patients who were ADT-naive versus those who had experienced ADT failure) in favour of guselkumab compared with placebo, with an adjusted between-group difference of 20.0% (95% CI, 11.6 to 28.3) and 8.8% (95% CI, 3.4 to 14.3), respectively.
Maintenance Study
A greater proportion of patients in the guselkumab 100 mg (45.2%) and guselkumab 200 mg (50.0%) groups compared with the placebo group (18.9%) experienced clinical remission at week 44, with an estimated adjusted between-group difference of 25.2% (95% CI, 16.4 to 33.9; P value < 0.001) and 29.5% (95% CI, 20.9 to 38.1; P value < 0.001), respectively.
Subgroup Analyses
The results for clinical remission were consistent across the 2 prespecified ADT-related subgroups of interest (i.e., patients who were ADT-naive versus those who had experienced ADT failure) in favour of guselkumab 100 mg and 200 mg compared with placebo, with an adjusted between-group difference of 24.3% (95% CI, 12.0 to 36.5) and 28.8% (16.5% to 41.1), respectively.
Endoscopic Healing
Induction Study
A greater proportion of patients in the guselkumab group (26.8%) compared with the placebo group (11.1%) had endoscopic healing at week 12, with an estimated adjusted between-group difference of 16.0% (95% CI, 10.5 to 21.4; P value < 0.001).
Maintenance Study
A greater proportion of patients in the guselkumab 100 mg (49.5%) and guselkumab 200 mg (51.6%) groups compared with the placebo group (18.9%) had endoscopic healing at week 44 of the maintenance study, with an estimated adjusted between-group difference of 29.5% (95% CI, 20.7 to 38.3; P value < 0.001) and 31.1% (95% CI, 22.5 to 39.8; P value < 0.001), respectively.
Clinical Response
Induction Study
Overall, 77.2% of patients (n = 325) achieved clinical response at week 12 or week 24; among patients who were not in clinical response at week 12 to IV guselkumab and received SC guselkumab treatment (n = 125), 55.2% (n = 69) achieved clinical response at week 24. A greater proportion of patients in the guselkumab group (61.5%) compared with the placebo group (27.9%) experienced clinical response at week 12, with an estimated adjusted between-group difference of 33.8% (95% CI, 26.9 to 40.7; P value < 0.001).
Maintenance Study
A greater proportion of patients in the guselkumab 100 mg (77.7%) and guselkumab 200 mg (74.7%) groups compared with the placebo group (43.2%) maintained clinical response at week 44, with an estimated adjusted between-group difference of 33.6% (95% CI, 24.5 to 42.7; P value < 0.001) and 30.7% (95% CI, 21.5 to 40.0; P value < 0.001), respectively.
Corticosteroid-Free Clinical Remission
Maintenance Study
A greater proportion of patients in the guselkumab 100 mg (45.2%) and guselkumab 200 mg (48.9%) groups compared with the placebo group (18.4%) achieved clinical remission and were corticosteroid-free at week 44, with an estimated adjusted between-group difference of 25.7% (95% CI, 17.0 to 34.5; P value < 0.001) and 29.0% (95% CI, 20.5 to 37.6; P value < 0.001), respectively.
HRQoL Measured by IBDQ Remission
Induction Study
A greater proportion of patients in the guselkumab group (51.3%) compared with the placebo group (29.6%) achieved IBDQ remission at week 12, with an estimated adjusted between-group difference of 21.9% (95% CI, 14.9 to 29.0; P value < 0.001).
Maintenance Study
A greater proportion of patients in the guselkumab 100 mg (64.4%) and guselkumab 200 mg (64.2%) groups compared with the placebo group (37.4%) achieved IBDQ remission at week 44 of the maintenance study, with an estimated adjusted between-group difference of 26.3% (95% CI, 16.8 to 35.7; P value < 0.001) and 25.9% (95% CI, 16.5 to 35.4; P value < 0.001), respectively.
Subgroup Analyses of Key Secondary Outcomes
For both studies, the results for endoscopic healing, clinical response, corticosteroid-free clinical remission (maintenance only), and IBDQ remission were consistent across the 2 prespecified ADT-related subgroups of interest (i.e., patients who were ADT-naive versus those who had experienced ADT failure) and were in favour of guselkumab compared with placebo (data not shown).
Sensitivity Analyses
For both studies, the results of sensitivity analyses for clinical remission, endoscopic healing, clinical response, corticosteroid-free clinical remission (maintenance only), and IBDQ remission were consistent with the primary analysis results at week 12 of the induction study and week 44 of the maintenance study, respectively.
Table 14: Summary of Key Efficacy Results for the Induction and Maintenance Studies — FAS Population
Variable | Induction study | Maintenance study | |||
|---|---|---|---|---|---|
Placebo IV (N = 280) | Guselkumab 200 mg IV (N = 421) | Placebo SC (N = 190) | Guselkumab 100 mg SC (N = 188) | Guselkumab 200 mg SC (N = 190) | |
Clinical remission at week 12 (induction study) and week 44 (maintenance study) | |||||
Patients with clinical remission, n (%) | 22 (7.9) | 95 (22.6) | 36 (18.9) | 85 (45.2) | 95 (50.0) |
95% CI for treatment proportion,a % | 4.7 to 11.0 | 18.6 to 26.6 | 13.4 to 24.5 | 38.1 to 52.3 | 42.9 to 57.1 |
Adjusted treatment difference, % (95% CI)b | 14.9 (9.9 to 19.9) | Reference | 25.2 (16.4 to 33.9) | 29.5 (20.9 to 38.1) | |
P valuec | < 0.001 | Reference | < 0.001 | < 0.001 | |
Endoscopic healing at week 12 (induction study) and week 44 (maintenance study) | |||||
Patients with endoscopic healing, n (%) | 31 (11.1) | 113 (26.8) | 36 (18.9) | 93 (49.5) | 98 (51.6) |
95% CI for treatment proportion,a (%) | 7.4 to 14.7 | 22.6 to 31.1 | 13.4 to 24.5 | 42.3 to 56.6 | 44.5 to 58.7 |
Adjusted treatment difference, % (95% CI)b | 16.0 (10.5 to 21.4) | Reference | 29.5 (20.7 to 38.3) | 31.1 (22.5 to 39.8) | |
P valuec | < 0.001 | Reference | < 0.001 | < 0.001 | |
Clinical response at week 12 (induction study) and maintenance of clinical response at week 44 (maintenance study) | |||||
Patients with clinical response, n (%) | 78 (27.9) | 259 (61.5) | 82 (43.2) | 146 (77.7) | 142 (74.7) |
95% CI for treatment proportion,a (%) | 22.6 to 33.1 | 56.9 to 66.2 | 36.1 to 50.2 | 71.7 to 83.6 | 68.6 to 80.9 |
Adjusted treatment difference, % (95% CI)b | 33.8 (26.9 to 40.7) | Reference | 33.6 (24.5 to 42.7) | 30.7 (21.5 to 40.0) | |
P valuec | < 0.001 | Reference | < 0.001 | < 0.001 | |
Corticosteroid-free clinical remission at week 44 | |||||
Patients with corticosteroid-free clinical remission, n (%) | NA | NA | 35 (18.4) | 85 (45.2) | 93 (48.9) |
95% CI for treatment proportion,a (%) | NA | NA | 12.9 to 23.9 | 38.1 to 52.3 | 41.8 to 56.1 |
Adjusted treatment difference, % (95% CI)b | NA | Reference | 25.7 (17.0 to 34.5) | 29.0 (20.5 to 37.6) | |
P valuec | NA | Reference | < 0.001 | < 0.001 | |
IBDQ remission at week 12 (induction study) and week 44 (maintenance study) | |||||
Patients with IBDQ remission, n (%) | 83 (29.6) | 216 (51.3) | 71 (37.4) | 121 (64.4) | 122 (64.2) |
95% CI for treatment proportion,a (%) | 24.3 to 35.0 | 46.5 to 56.1 | 30.5 to 44.2 | 57.5 to 71.2 | 57.4 to 71.0 |
Adjusted treatment difference, % (95% CI)b | 21.9 (14.9 to 29.0) | Reference | 26.3 (16.8 to 35.7) | 25.9 (16.5 to 35.4) | |
P valuec,d | < 0.001 | Reference | < 0.001 | < 0.001 | |
ADT = advanced therapy; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; FAS = full analysis set; IBDQ = Inflammatory Bowel Disease Questionnaire; SC = subcutaneous.
Note: Includes only patients with a modified Mayo score of 5 to 9 at induction baseline.
aThe 95% CIs for the proportion of patients meeting the end point in each treatment group were based on the normal approximation confidence limits.
bThe adjusted treatment difference and CIs were based on the Wald statistic with CMH weight.
cFor the induction study, the P values were based on the CMH chi-square test, stratified by ADT failure status (yes or no) and concomitant use of corticosteroids at baseline (yes or no). For the maintenance study, the P values were based on a CMH chi-square test, stratified by clinical remission status at maintenance baseline (yes or no) and induction treatment (guselkumab 400 mg IV, guselkumab 200 mg IV, placebo IV crossover to guselkumab 200 mg IV).
dIBDQ remission was not adjusted for multiplicity.
Sources: QUASAR induction and maintenance Clinical Study Reports.1,2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
A summary of harms reported in the SASs in the induction and maintenance studies in active tuberculosis was reported in both studies in Table 15 and Table 16, respectively.
Adverse Events
In the induction study, 49.4% and 49.3% of patients reported at least 1 TEAE in the guselkumab and placebo groups, respectively. Over the 12-week treatment period, the most frequently reported TEAEs in the guselkumab and placebo groups were anemia (5.0% and 6.8%, respectively), COVID-19 (5.0% and 4.3%), headache (3.1% and 2.9%), and UC (2.4% and 8.2%). Of these TEAEs, a numerically higher proportion of UC was reported in patients receiving placebo compared with those receiving guselkumab.
In the maintenance study, 64.5%, 70.0%, and 68.2% of patients reported at least 1 TEAE in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively. Over the 44-week treatment period, the most frequently reported TEAEs in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups were colitis ulcerative (9.1%, 13.2%, and 29.7%, respectively) and COVID-19 (12.9%, 9.5%, and 14.1%). Of these TEAEs, a numerically higher proportion of colitis ulcerative was reported in patients receiving placebo compared with those receiving guselkumab.
Serious Adverse Events
In the induction study, 2.9% and 7.1% of patients reported at least 1 serious TEAE in the guselkumab and placebo groups, respectively.
In the maintenance study, 2.7%, 6.3%, and 0.5% of patients reported at least 1 serious TEAE in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively.
Withdrawals Due to Adverse Events
In the induction study, 1.7% and 3.9% of patients reported at least 1 TEAE leading to discontinuation of treatment in the guselkumab and placebo groups, respectively.
In the maintenance study, 3.8%, 2.6%, and 6.8% of patients reported at least 1 TEAE leading to discontinuation of treatment in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups, respectively.
Mortality
In the induction study, 1 and 2 deaths were reported in the guselkumab and placebo groups, respectively. There were no deaths reported in the maintenance study.
Notable Harms
In the induction study, the incidence of notable TEAEs, which included infections and serious infections, were similar between groups. Infections were reported in approximately 15% of patients and serious infections were reported in less than 1% of patients in both groups.
Table 15: Summary of Harms Results for the Induction Study — SAS Population
AEs through week 12 | Placeboa (N = 280) | Guselkumab 200 mg IVa (N = 421) |
|---|---|---|
Patients with ≥ 1 TEAE, n (%) | 138 (49.3) | 208 (49.4) |
Most common TEAEs (≥ 3% of any treatment group), n (%) | ||
Anemia | 19 (6.8) | 21 (5.0) |
COVID-19 | 12 (4.3) | 21 (5.0) |
Headache | 8 (2.9) | 13 (3.1) |
Colitis ulcerative | 23 (8.2) | 10 (2.4) |
Patients with ≥ 1 serious TEAE, n (%) | 20 (7.1) | 12 (2.9) |
Patients with ≥ 1 TEAEs leading to discontinuation of treatment, n (%) | 11 (3.9) | 7 (1.7) |
TEAEs within 1 hour of infusion, n (%) | 1 (0.4) | 6 (1.4) |
Deaths, n (%) | 2 (0.7) | 1 (0.2) |
Patients with ≥ 1 notable harms, n (%) | ||
Infectionsb | 43 (15.4) | 67 (15.9) |
Serious infectionsb | 1 (0.4) | 3 (0.7) |
AE = adverse event; MedDRA = Medical Dictionary for Regulatory Activities; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
aIncludes data up to week 12 for patients who received treatment at week 12, and all data through the final safety visit for patients who did not receive treatment at week 12.
bInfections were defined as any AE that was coded to the MedDRA “infections and infestations” system organ class.
Sources: QUASAR induction Clinical Study Report.1 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the maintenance study, the incidence of notable infections and serious infections were also similar between groups. Infections were reported in approximately 32% of patients in all groups, and serious infections were reported in approximately 1% of patients in the guselkumab groups and 0 patients in the placebo group. No cases of active tuberculosis were reported in either study.
Table 16: Summary of Harms Results for the Maintenance Study — Randomized SAS Population
AEs through week 44 | Placebo (N = 192) | Guselkumab 100 mg SC (N = 186) | Guselkumab 200 mg SC (N = 190) |
|---|---|---|---|
Patients with ≥ 1 TEAE, n (%) | 131 (68.2) | 120 (64.5) | 133 (70.0) |
Most common TEAEs (≥ 3% of any treatment group), n (%) | |||
Colitis ulcerative | 57 (29.7) | 17 (9.1) | 25 (13.2) |
COVID-19 | 27 (14.1) | 24 (12.9) | 18 (9.5) |
Arthralgia | 13 (6.8) | 8 (4.3) | 15 (7.9) |
Upper respiratory tract infection | 8 (4.2) | 6 (3.2) | 13 (6.8) |
Headache | 12 (6.3) | 7 (3.8) | 8 (4.2) |
Pyrexia | 5 (2.6) | 7 (3.8) | 9 (4.7) |
Nasopharyngitis | 9 (4.7) | 8 (4.3) | 7 (3.7) |
Anemia | 5 (2.6) | 4 (2.2) | 6 (3.2) |
Abdominal pain | 4 (2.1) | 5 (2.7) | 7 (3.7) |
Back pain | 5 (2.6) | 8 (4.3) | 2 (1.1) |
Injection site reaction | 0 (0.0) | 0 (0.0) | 7 (3.7) |
Patients with ≥ 1 serious TEAE, n (%) | 1 (0.5) | 5 (2.7) | 12 (6.3) |
Patients with ≥ 1 TEAEs leading to discontinuation of treatment, n (%) | 13 (6.8) | 7 (3.8) | 5 (2.6) |
Deaths, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Patients with ≥ 1 notable harm, n (%) | |||
Infectionsa | 63 (32.8) | 59 (31.7) | 59 (31.1) |
Serious infectionsa | 0 (0.0) | 1 (0.5) | 2 (1.1) |
AE = adverse event; SAS = safety analysis set; SC = subcutaneous; TEAE = treatment-emergent adverse event.
Note: Includes only patients with a modified Mayo score of 5 to 9 at induction baseline.
aInfections were defined as any AE coded to the Medical Dictionary for Regulatory Activities “infections and infestations” system organ class.
Sources: QUASAR maintenance Clinical Study Report.2 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Both the induction and maintenance studies were randomized, double-blind, placebo-controlled, parallel-group trials. Randomization and allocation concealment for both studies were performed using appropriate methodology via an interactive web response system. Randomization stratification was prespecified and was based on relevant prognostic factors, i.e., ADT failure status, region, and concomitant use of corticosteroids at baseline for the induction study and clinical remission status at maintenance baseline, concomitant use of corticosteroids at maintenance baseline, and induction treatment for the maintenance study. Overall, the baseline demographic and disease characteristics appeared to be reasonably balanced between treatment groups in both studies.
For the induction study, patients were eligible and enrolled if they had a baseline MMS of 4 to 9, and those with a baseline MMS of 4 were capped at 5% of the trial population or less. However, according to the sponsor, the statistical analysis plan was amended per “health authority request” (on April 26, 2022, after patient enrolment) for the primary efficacy and safety analyses to include randomized and treated patients with a baseline MMS of 5 to 9. As a result, a total of 34 patients with a baseline MMS of 4 were excluded from the FAS primary efficacy and safety analyses, which could have compromised randomization. In general, the CDA-AMC review team and the clinical experts consulted by CDA-AMC did not consider this exclusion to have had an important impact on study results because the patient characteristics appeared to be balanced between the treatment groups, and the findings of the supplementary analyses that included those with an MMS of 4 were consistent with the primary analyses.
In both studies, the double-blind approach of masking patients and investigators (including the outcome assessors) to treatment allocation from the time of random assignment until the time of unblinding, per the study protocols, was appropriate. The study drugs were identical in physical appearance and packaging. Some efficacy outcomes (clinical remission and clinical response) were composite outcomes that included Mayo subscores of stool frequency and rectal bleeding, which were reported by patients. The HRQoL outcome assessed with IBDQ was also a patient-reported measure. Although these subjective (components of) outcomes may be influenced by knowledge of treatment assignment, the double-blind design of the trials likely mitigated this risk.
Both studies used placebo as the comparator instead of an ADT for UC that is relevant to clinical practice in Canada. The clinical expert consulted by CDA-AMC noted that relevant pharmacotherapies for patients with moderately to severely active UC in clinical practice in Canada include adalimumab, golimumab, infliximab, mirikizumab, ozanimod, tofacitinib, upadacitinib, ustekinumab, and vedolizumab. The sponsor submitted evidence regarding the comparative effectiveness that is summarized in the ITC section of the current report.
In the induction study, most patients received 1 or more concomitant medications for UC, including oral corticosteroids, immunomodulatory drugs, and oral 5-ASAs. The use of these concomitant drugs was generally similar between groups and, according to the clinical experts, unlikely to have confounded the study results.
Overall, the statistical methods used in both studies were appropriate. The studies were powered based on their primary and key secondary outcomes between the treatment groups. The subgroup analyses were likely underpowered to identify subgroup differences. An appropriate method for adjusting for multiplicity was used for the primary and secondary outcomes, but there was no multiplicity control for IBDQ remission and the subgroup analyses. The sponsor noted that, based on feedback from the FDA, IBDQ remission was not included in the US-specific hierarchical testing procedure.
Rates of study discontinuation were generally low in both studies and similar between groups although, in the induction study, the rate in the placebo group (12.9%) was higher than in the guselkumab group (5.5%), mostly attributed to withdrawal by patients. Nonresponder imputation was used for missing data in the primary analysis, and preplanned sensitivity analyses were done under different assumptions. Nonresponder imputation is a method in which missing data are believed to occur randomly and are unrelated to observed or unobserved variables. However, this assumption is often unrealistic and, if violated, the imputed values may be biased, especially when differences between groups are pronounced. However, the between-group differences did not appear pronounced in these trials and appropriate methods (e.g., tipping point and multiple imputation) for the sensitivity analyses were used, which confirmed that the results of the studies remained robust to the differential discontinuations between groups.
External Validity
In general, the population requested for reimbursement aligns with the approved Health Canada indication, except that the indication is not limited to patients who have experienced an inadequate response or are unable to tolerate ADT or conventional therapy. The clinical experts did not consider the broader indication to affect the generalizability of the findings from the induction and maintenance studies. The dosing and administration of guselkumab were consistent with the approved product monograph.
Most patients enrolled in both studies (41% to 47%) were from Eastern Europe, with the remainder distributed across Asia (21%) and rest of world (32% to 38% across Western Europe, North America, Latin America, Australia, Israel, and New Zealand). There were 8 and 6 patients from Canada included in the induction and maintenance studies, respectively. Also, there was a relatively high rate of unsuccessful screenings in the induction study (269 out of 1,005 patients were screened out), mainly due to patients not meeting eligibility criteria. The clinical experts did not regard these factors as influencing the generalizability of the studies’ results.
According to the clinical experts, the patient eligibility criteria and baseline characteristics of both studies were generalizable to adults with moderately to severely active UC in the clinical setting in Canada.
The studies included outcomes that were important to patients and clinicians, including clinical remission, clinical response, corticosteroid-free clinical remission, and HRQoL.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform the CDA-AMC expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:36,82
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate — The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited — The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate — The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty-of-evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
For the GRADE assessments, the induction and maintenance studies were assessed individually because they had different treatment regimens and durations and different time points for assessing outcomes.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for guselkumab versus placebo.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following was summarized and validated by the CDA-AMC review team.
Objectives for the Summary of Indirect Evidence
The aim of this section is to summarize and critically appraise 1 sponsor-submitted NMA that was used to inform the pharmacoeconomic model and to fill gaps in the comparative evidence for guselkumab versus other treatments of interest for moderately to severely active UC.
Description of Indirect Comparison
The systematic literature search and study selection criteria for the NMA are summarized in Table 17.
Indirect Treatment Comparison Design
Objectives
The objective of the sponsor-submitted NMA was to compare the treatment effects of guselkumab versus other relevant comparators for the treatment of adults with moderately to severely active UC in induction and maintenance. In this setting, maintenance outcomes evaluated response at 1 year in all enrolled patients (i.e., not the response in the subpopulation of induction responders). The protocol of the systematic review and NMA was a priori registered in PROSPERO (CRD42023466248).
Table 17: Study Selection Criteria and Methods for NMA Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adults and select adolescents (aged ≥ 16 years) with moderately to severely active ulcerative colitis (as defined by study) Subgroups of interest:
|
Intervention | The following therapies alone or in combination with conventional therapy Biologics:
JAK inhibitors:
Sphingosine 1-phosphate receptor modulator:
|
Comparator | Active comparator or placebo |
Outcome | Efficacy outcomes (as defined by study):
Safety outcomes:
Quality of life outcomes:
|
Study designs | Randomized, double-blinded or open-label, placebo- and active-controlled, parallel-group trials evaluating efficacy and safety in phase II, III, or IV |
Publication characteristics | Searches were restricted to published clinical trials |
Exclusion criteria | Irrelevant intervention or a nonphase II, III, or IV RCT |
Databases searched | Ovid MEDLINE, Ovid MEDLINE Epub Ahead of Print, In-Process & Other Non-Indexed Citations and Daily, Ovid Embase, Ovid EBM Reviews - Cochrane Central Register of Controlled Trials, Ovid EBM Reviews - Cochrane Database of Systematic Reviews |
Selection process | Identified articles were screened by 2 independent reviewers |
Data extraction process | Data from the included records were extracted by a single reviewer and then validated by a second independent reviewer |
Quality assessment | The risk-of-bias assessment of included trials was performed using the NICE Single Technology Appraisal Evidence Submission checklist for assessment of risk of bias in RCTs.83 Assessments of risk of bias were performed by 1 reviewer and validated by a second independent reviewer to verify accuracy |
ADT = advanced therapy; EBM = Evidence-Based Medicine; IL = interleukin; JAK = Janus kinase; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; PROMIS-29 = 29-item Patient-Reported Outcomes Measurement Information System questionnaire; RCT = randomized controlled trial; TNF = tumour necrosis factor.
aClinical remission is measured as a stool frequency subscore of 0 or 1, a rectal bleeding subscore of 0, and an endoscopy subscore of 0 or 1 with no friability present on the endoscopy, where the stool frequency subscore has not increased from induction baseline.
bClinical response is measured as a decrease from induction baseline in the modified Mayo score by ≥ 30% and ≥ 2 points, with either a ≥ 1-point decrease from baseline in the rectal bleeding subscore or a rectal bleeding subscore of 0 or 1. The modified Mayo score is a 3-component (stool frequency, rectal bleeding, and endoscopy subscores) Mayo score without the Physician’s Global Assessment.
Sources: NMA technical report.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Study Selection Methods
A systematic literature search was conducted on July 27, 2023, using the criteria described in Table 17. The review included trials investigating treatments in adults with moderately to severely active UC treated with biologics, Janus kinase inhibitors, or sphingosine 1-phosphate receptor modulators. The following efficacy outcomes of interest for this CDA-AMC review that were reported were clinical remission and clinical response assessed at the induction and maintenance treatment periods. The identified clinical trials were assessed for eligibility against predefined inclusion and exclusion criteria. Two reviewers independently screened titles and abstracts and relevant full-text citations for inclusion. Discrepancies were resolved through consensus or by a third reviewer. Data extraction and a quality assessment of the included studies were performed by 1 reviewer and validated by a second reviewer, and discrepancies were resolved through consensus or by a third reviewer. The assessment of the risk of bias of the included studies was performed using the National Institute for Health and Care Excellence Single Technology Appraisal Evidence Submission checklist for assessing the risk of bias in RCTs.
ITC Analysis Methods
Details of the NMA analysis methods are in Table 18. An NMA feasibility assessment was undertaken to ascertain the extent of clinical heterogeneity across the trials identified in the systematic literature review. Trial design characteristics, patient eligibility criteria, baseline patient characteristics, outcome characteristics (i.e., definitions and methods of reporting outcomes), and placebo response were considered sources of clinical heterogeneity and explored in the feasibility assessment. Based on the feasibility assessment, an NMA was used for the outcomes of clinical remission and clinical response for induction and maintenance (1-year analysis).
Studies were included in the NMA if they reported outcomes of interest for patients receiving doses approved in Europe, the US, Canada, and Japan. Only studies with a minimum follow-up duration of 38 weeks were included in the maintenance analysis. NMA outcomes included clinical remission and clinical response, assessed at the induction and maintenance treatment periods. For the induction analyses, the trial’s primary time point was used. For the maintenance analyses, the time point closest to 52 weeks (and later than 24 weeks) was used. The goal of the NMA was to inform the comparative effectiveness of guselkumab across a broad range of available comparators; however, given that some comparators were not relevant to Canadian public payers (e.g., risankizumab, filgotinib) they have been excluded from the Clinical Review.
A Bayesian NMA was conducted as the primary analysis of outcomes. Clinical remission and clinical response were analyzed together as an ordinal outcome using an inverse logit multivariate regression model for ordered categorical data. Within the ordinal model, it was assumed that all patients achieving remission also achieved response; thus, the response was redefined to be response without remission. In the induction setting, ordinal ORs were estimated from a random-effects model that adjusted for a baseline response rate estimated from the QUASAR placebo group. The 1-year maintenance analyses were based on an unadjusted random-effects model. In both cases, informative prior distributions were used based on recommendations in Turner et al.85,86 All effect estimates were accompanied with 95% CrIs.
Fixed-effect NMAs were also conducted for maintenance time points but are not included in the report. All analyses were performed using 4 unique sets of starting values and were based on at least 20,000 iterations with a burn-in of 20,000. Convergence was monitored quantitatively using 4 chains and the latest implementation Gelman-Rubin diagnostic (Rhat) to capture nonconvergence from stationary but nonoverlapping chains, overlapping nonstationary chains, chains with heavy tails, and chains with different variances. Samples were considered to have converged if the Rhat was 1.05 or less. This was assessed for all parameters used in each model. Effective sample size and Monte Carlo standard errors were also assessed to ensure sufficient postconvergence samples were taken to support inference. If the rank-normalized effective sample size was greater than 400 (i.e., 100 per chain) then additional samples were taken to ensure the Monte Carlo standard error was small enough to allow for stable estimates to at least 1 decimal place.
Table 18: NMA Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian network meta-analyses (random effects and fixed effects) were conducted. |
Priors | Informative prior distributions were assigned in all analyses for both unadjusted and meta-regression risk-adjusted models. |
Assessment of model fit | Model selection was based on a holistic review that also considered the extent of known cross-trial heterogeneity and the plausibility of estimates. |
Assessment of inconsistency | Assessment of inconsistency consisted of a global test alongside loop-specific tests. These were conducted using the node splitting and treatment by design interaction models as implemented in the netmeta package. |
Assessment of convergence | Convergence was monitored quantitatively using the latest implementation Gelman-Rubin diagnostic (Rhat) based on 4 chains. |
Outcomes | NMA outcomes included clinical remission and clinical response assessed at induction and 1-year maintenance. |
Follow-up time points | For the induction analyses, the trial’s primary time point was used. For the maintenance analyses, the time point closest to 52 weeks (and later than 24 weeks) was used. |
Construction of nodes | Each treatment (i.e., intervention or comparator) was represented by a node (or circle) and comparisons between treatments were shown by lines linking the nodes. |
Sensitivity analyses | Sensitivity analyses applying imputation methods that do not account for delayed responders were conducted for maintenance (1-year) outcomes. Sensitivity analyses were planned to assess the impact of heterogeneity identified in the feasibility assessment, including differences in the definition of moderately to severely active UC based on the modified Mayo score. |
Subgroup analysis | Subgroup NMAs were conducted for 2 distinct patient populations: ADT failure and ADT nonfailure. |
ADT = advanced therapy; NMA = network meta-analysis; UC = ulcerative colitis.
Sources: NMA technical report.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Subgroup NMAs (induction and 1-year analyses) were conducted for 2 distinct patient populations: ADT failure and ADT nonfailure. For the 1-year NMAs, trial data were normalized to mimic a type 1 treat-through trial design as best as possible. This normalization approach is termed the 1-year “with delayed responders” analysis. Analyses applying imputation methods that do not account for delayed responders were also conducted, and these analyses were used to inform the economic model.
Results of the NMA
Summary of Included Studies
The search identified ████ unique RCTs, of which ████ trials were excluded from the NMA feasibility assessment; ████ due to unapproved treatment regimens and ███ █████ because it recruited a patient subgroup not of interest (i.e., | █████ including Japanese patients from a broader etrasimod trial). A total of ██ trials were included in the NMA, of which ████ trials were phase III, ███ trials were phase II, and ███ trials were phase IIb or III. All trials were conducted in multicentre settings. ███ █████ did not explicitly report phase; however, its design and objectives aligned with those of a phase III trial. The following therapies were included in the NMA: adalimumab,87-90 infliximab,91-93 tofacitinib,94-96 and upadacitinib97-99 were each investigated by ███ trials; mirikizumab100,101 and vedolizumab88,102,103 were investigated by ███ trials each; etrasimod,104-106 ozanimod,100,101 ustekinumab,107 and guselkumab,108 were each examined by ███ trials.
Assessment of homogeneity is summarized in Table 19, and a summary of the included RCTs is in Table 20. ████████ ██████████████████████████████████████ had evidence available for both induction and maintenance trial periods, while ██████████████████████████████████████ and █████ ██████████████████████████████████████████████ had data for only induction and only maintenance periods, respectively. All trials included in the NMA were double-blind, comparative, randomized trials. Only ███ trials ██████ were head-to-head comparisons of 2 active therapies, ███ ████████ assessed a biologic and immunosuppressant combination therapy regimen against biologic monotherapy, and the remaining trials utilized a placebo comparator. A total of ██████ trials employed responder re-randomization, ███ trials █████████████████████████ were treat-through design, ████ trials ██████████████ were induction only, and ███ ████████ was response conditional (i.e., patients continued active therapy if they experienced a response to treatment at assessment).
A summary of the baseline patient characteristics across the trials included in the NMA is in Table 21. The QUASAR trial program included patients aged 18 years and older with a baseline MMS score of 5 to 9, a rectal bleeding subscore of at least 1, and a Mayo endoscopic subscore of at least 2. Patients in the QUASAR trial must also have experienced the failure of a biologic, advanced, or conventional therapy before inclusion and have at least 20 cm of colon affected by the disease process. There was no prespecified minimum disease duration for the QUASAR trial. Additionally, endoscopy and histological confirmation was performed as part of trial enrolment for QUASAR trial eligibility. Overall, both age requirements and endoscopic and histological confirmation were relatively similar between the comparator studies and the QUASAR trial. Disease duration, baseline Mayo score, prior failure of biologic, advanced, or conventional therapies, and extent of disease varied across comparator trials, as follows:
The QUASAR trial and ██████ other studies ████████████ excluded patients who had been previously exposed to a biologic therapy targeting IL-12 and/or IL-23.
A majority of trials (including QUASAR) employed the same definition of clinical remission predicated on the total Mayo score, while █████ trials █████████ used the MMS, ███ trials ██████ used the adapted Mayo score, ███ █████ ████ used the partial Mayo score ██████ and 3-component Mayo score. ██ Specific aspects of the definition used for clinical remission were assessed as well, including requirements for the endoscopic subscore, total Mayo score, stool subscore, rectal bleeding subscore, and an aggregate requirement for all subscores. A total of █████ trials had criteria identical to the QUASAR trial, while minor variations were noted in the remaining █████ trials. █████████████████████████████████████████████████████
A total of █████ trials used the same clinical response definition as the QUASAR trial. ███████████████████████████████████████████████████████. Trials were additionally evaluated based on specific aspects of the clinical response definition, including the type of Mayo scoring system used, the overall Mayo score requirement, and the rectal bleeding subscore requirement. None of the trials reported an absolute Mayo score or endoscopic subscore requirement.
Most trials reported induction periods ranging from 6 to 14 weeks, with maintenance periods that typically measured outcomes around 52 weeks but had a large amount of variance in timing.
Table 19: Assessment of Homogeneity for the NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | A total of █████ trials reported baseline mean Mayo score, ranging from ████ ██ ████. Disease duration varied across trials. |
Treatment history | QUASAR and ███ other studies ███████████████████ excluded patients who had been previously exposed to a biologic therapy targeting IL-12 and/or IL-23 such as guselkumab, ustekinumab, and mirikizumab. The QUASAR study also excluded patients who were treated with natalizumab or drugs that deplete T cells or B cells within 12 months of trial start, investigational drugs up to 4 weeks before trial start, apheresis within 2 weeks of trial start, or receipt of a fecal microbiota transplant within 12 weeks of trial start.1,2,109 ████ ███ ██████████ ████████ employed these specific criteria, while other trials shared only a few criteria with the QUASAR study. |
Trial eligibility criteria | Overall, both age requirements and endoscopic and histological confirmation were relatively similar between comparator trials and the QUASAR study. Specifically, ███ ███ ███ ████████████ required patients to undergo endoscopic confirmation before trial enrolment and ███ trials ███████████████████████████████████████████████ did not report whether histological confirmation was required. Disease duration, baseline Mayo score, prior failure of biologic, advanced, or conventional therapies, and extent of disease varied across comparator trials. |
Definitions of end points | Clinical response was defined in the QUASAR trial as a decrease from baseline in the Mayo score of at least 3 points and 30% or more, with an accompanying decrease in the rectal bleeding score of more than 1 point or an absolute rectal bleeding score of 0 or 1.1,2,109 A total of █████ trials matched the QUASAR trial’s definition for clinical response. ███████████████████████████████████████████████████████.Trials were additionally evaluated based on specific aspects of the clinical response definition, including the type of Mayo scoring system used, the overall Mayo score requirement, and the rectal bleeding subscore requirement. None of the trials reported an absolute Mayo score or endoscopic subscore requirement. The definition of clinical remission used in the QUASAR trial was a total Mayo score of 2 or less, with no subscore greater than 1.1,2,109 A majority of trials employed the same definition of clinical remission predicated on the total Mayo score, while █████ trials ███████████ used the modified Mayo score, ███ trials ███████ used the adapted Mayo score, ███ █████ ████ used the partial Mayo score ██████ and 3-component Mayo score.███████ Specific aspects of the definition used for clinical remission were assessed as well, including requirements for the endoscopic subscore, total Mayo score, stool subscore, rectal bleeding subscore, and an aggregate requirement for all subscores. A total of █████ trials had criteria identical to those of the key trial, while variations were noted in the remaining █████ trials. █████████████████████████████████████████████████████ |
Study design | █████ trials were phase III or IIIb, █████ trials were phase II, and ████ trials were phase IIb or III. All trials were conducted in multicentre settings. Summary-level data were available for all included trials, and individual patient data were available for █████ trials. |
Outcomes of interest and time points reported across trials | The outcomes of interest for quantitative analysis were clinical response and clinical remission at both induction and maintenance. The time point of assessment varied across trials; the QUASAR trial assessed both outcomes at week 12 for induction and at week 44 for maintenance. Competitor trials reported induction periods ranging from 6 to 14 weeks, with maintenance periods typically around 52 weeks. Some trials, such as GEMINI 1 and PURSUIT-SC, had a 6-week induction, while the HIBISCUS I and II, SELECTION, and True North trials each had a 10-week induction. The VARSITY trial had a 14-week induction, and the ULTRA 1 trial had multiple induction points at 2, 4, 6, and 8 weeks. Maintenance periods were generally around 52 weeks, with some trials, such as PURSUIT‑J, reporting maintenance periods of up to 60 weeks and some as short as 30 weeks. Several trials did not report specific time points for either induction or maintenance, such as 16T‑MC‑AMAC and VISIBLE 1. |
IL = interleukin; NMA = network meta-analysis.
Sources: NMA technical report.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 20: Summary of Study Characteristics Included in the NMA
Trial name | Phase | Blinding | Trial duration | Treatment design | Trial period | Treatment arms | Number randomized |
|---|---|---|---|---|---|---|---|
QUASAR | IIb and III | Double-blind | Induction: 12 weeks; maintenance: 60 weeks | RR | Induction | Placebo | 105 |
Guselkumab 200 mg week 0, 4, 8 | 101 | ||||||
Guselkumab 400 mg week 0, 4, 8 | 107 | ||||||
Maintenance | Placebo | 190 | |||||
Guselkumab 200 mg q.4.w. | 190 | ||||||
16T-MC-AMAC | II | Double-blind | 52 weeks | RR | Maintenance | Placebo | 13 |
Mirikizumab 200 mg q.4.w. | 47 | ||||||
ACT-1 | III | Double-blind | Induction: 8 weeks; maintenance: 30 to 54 weeks | TT | Induction and maintenance | Placebo | 121 |
Infliximab 5 mg/kg week 0, 2, 6, then q.8.w. | 121 | ||||||
ACT-2 | III | Double-blind | Induction: 8 weeks; maintenance: 30 weeks | TT | Induction and maintenance | Placebo | 123 |
Infliximab 5 mg/kg week 0, 2, 6, then q.8.w. | 121 | ||||||
ELEVATE UC 12 | III | Double-blind | 16 weeks | TT | Induction | Placebo | 116 |
Etrasimod 2 mg q.d. | 238 | ||||||
ELEVATE UC 52 | III | Double-blind | 56 weeks | TT | Maintenance | Placebo | 144 |
Etrasimod 2 mg q.d. | 289 | ||||||
GEMINI 1 | III | Double-blind | Induction: 6 weeks; maintenance: 52 weeks | RR | Induction | Placebo | 149 |
Vedolizumab 300 mg at weeks 0, 2, 6 | 225 | ||||||
Maintenance | Placebo | 126 | |||||
Vedolizumab 300 mg q.8.w. | 122 | ||||||
HIBISCUS I | III | Double-blind | 14 weeks | TT | Induction | Placebo | 72 |
Adalimumab 160 mg at week 0, 80 mg at week 2, then 40 mg q.2.w. | 142 | ||||||
HIBISCUS II | III | Double-blind | 14 weeks | TT | Induction | Placebo | 72 |
Adalimumab 160 mg at week 0, 80 mg at week 2, then 40 mg q.2.w. | 143 | ||||||
Japic CTI-060298 | III | Double-blind | Induction: 8 weeks; maintenance: 38 weeks | TT | Induction | Placebo | 104 |
Infliximab 5 mg/kg at weeks 0, 2, 6, then q.8.w. | 104 | ||||||
Jiang 2015 | NR | Double-blind | 30 weeks | TT | Induction and maintenance | Placebo | 41 |
Infliximab 5 mg/kg at weeks 0, 2, 6, then q.8.w. | 41 | ||||||
LUCENT-1 | III | Double-blind | Induction: 12 weeks | RR | Induction | Placebo | 294 |
Mirikizumab 300 mg at weeks 0, 4, 8 | 868 | ||||||
LUCENT-2 | III | Double-blind | Induction: 12 weeks; maintenance: 52 weeks | RR | Maintenance | Placebo | 179 |
Mirikizumab 200 mg q.4.w. | 365 | ||||||
OASIS | II | Double-blind | 14 weeks | Induction only study | Induction | Placebo | 54 |
Etrasimod 2 mg q.d. | 50 | ||||||
OCTAVE 1 | III | Double-blind | 8 weeks | RR | Induction | Placebo | 122 |
Tofacitinib 10 mg b.i.d. | 476 | ||||||
OCTAVE 2 | III | Double-blind | 8 weeks | RR | Induction | Placebo | 112 |
Tofacitinib 10 mg b.i.d. | 429 | ||||||
OCTAVE Sustain | III | Double-blind | 52 weeks | RR | Maintenance | Placebo | 198 |
Tofacitinib 5 mg b.i.d. | 198 | ||||||
Tofacitinib 10 mg b.i.d. | 197 | ||||||
PURSUIT-J | III | Double-blind | Induction: 6 weeks; maintenance: 60 weeks | RR | Maintenance | Placebo | 31 |
Golimumab 100 mg q.4.w. | 32 | ||||||
PURSUIT-M | III | Double-blind | 54 weeks | RR | Maintenance | Placebo | 154 |
Golimumab 100 mg q.4.w. | 151 | ||||||
PURSUIT-SC | II and III | Double-blind | 6 weeks | RR | Induction | Placebo | 251 |
Golimumab 200 mg at week 0 and 100 mg at week 2 | 253 | ||||||
Sandborn 2012 | II | Double-blind | 12 weeks | Induction only study | Induction | Placebo | 48 |
Tofacitinib 10 mg b.i.d. | 33 | ||||||
TOUCHSTONE | II | Double-blind | Induction: 8 weeks; maintenance: 32 weeks | Response conditional (no randomization) | Induction and maintenance | Placebo | 65 |
Ozanimod 1 mg q.d. | 67 | ||||||
True North | III | Double-blind | Induction: 10 weeks; maintenance: 52 weeks | RR | Induction | Placebo | 216 |
Ozanimod 0.92 mg q.d. | 429 | ||||||
Maintenance | Placebo | 227 | |||||
Ozanimod 0.92 mg q.d. | 230 | ||||||
U-ACCOMPLISH | III | Double-blind | 8 weeks | RR | Induction | Placebo | 174 |
Upadacitinib 45 mg q.d. | 341 | ||||||
U-ACHIEVE SS1 part 1 and part 2 | III | Double-blind | 8 weeks | RR | Induction | Placebo | 46 |
Upadacitinib 45 mg q.d. | 123 | ||||||
U-ACHIEVE SS2; U-ACHIEVE induction | III | Double-blind | 8 weeks | RR | Induction | Placebo | 154 |
Upadacitinib 45 mg q.d. | 319 | ||||||
U-ACHIEVE SS3; U-ACHIEVE maintenance | III | Double-blind | 52 weeks | RR | Maintenance | Placebo | 149 |
Upadacitinib 15 mg q.d. | 148 | ||||||
Upadacitinib 30 mg q.d. | 154 | ||||||
UC SUCCESS | III | Double-blind | 16 weeks | Induction only study | Induction | Infliximab 5 mg/kg at weeks 0, 2, 6, 14 | 77 |
Infliximab 5 mg/kg + azathioprine 2.5 mg/kg | 78 | ||||||
ULTRA 1 | III | Double-blind | 8 weeks | Induction only study | Induction | Placebo | 130 |
Adalimumab 160 mg at week 0, 80 mg at week 2, then 40 mg q.2.w. | 130 | ||||||
ULTRA 2 | III | Double-blind | Induction: 8 weeks; maintenance: 52 weeks | RR | Induction | Placebo | 246 |
Adalimumab 160 mg at week 0, 80 mg at week 2, then 40 mg q.2.w. | 248 | ||||||
Maintenance | Adalimumab 40 mg q.2.w. (week 8 responders) | 123 | |||||
UNIFI | III | Double-blind | 52 weeks | RR | Induction | Placebo | 319 |
Ustekinumab 6 mg/kg | 322 | ||||||
Maintenance | Placebo | 175 | |||||
Ustekinumab 90 mg q.8.w. | 176 | ||||||
VARSITY | IIIb | Double-blind | 68 weeks | TT | Induction and maintenance | Adalimumab 160 mg at week 0, 80 mg at week 0, then 40 mg q.2.w. | 374 |
Vedolizumab 300 mg at weeks 0, 2, 6 | 367 | ||||||
VEGA | II | Double-blind | 50 weeks | TT | Induction and maintenance | Golimumab 200 mg at week 0, 100 mg at week 2, then 100 mg q.4.w. | 72 |
Guselkumab 200 mg at weeks 0, 4, 8, then 100 mg q.8.w. | 71 | ||||||
VISIBLE 1 | III | Double-blind | 68 weeks | RR | Maintenance | Placebo | 56 |
Vedolizumab 108 mg q.2.w. | 106 | ||||||
Vedolizumab 300 mg q.8.w. | 54 |
b.i.d. = twice a day; NMA = network meta-analysis; NR = not reported; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.d. = once a day; RR = responder re-randomization; SS = substudy; TT = treat through.
Sources: NMA technical report.84 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
████████████████████████ | ████████████████████████ |
|---|---|
████████████████████████ | ████████████████████████ |
████████████████████████ | ████████████████████████ |
Results
The results for random-effects models for the relevant comparators are summarized for clinical remission and response. In the induction setting, the model was adjusted for baseline risk but not for the maintenance phase (1-year analysis) because of a lack of multistudy connections. Inconsistency analyses indicated concordance between direct and indirect evidence across the conducted NMAs. In general, the authors noted that the risk-of-bias assessment of the included trials was reported to be of good quality. However, the clinical experts noted heterogeneity in the inclusion and exclusion criteria and outcomes definitions and timing.
Induction Analysis
Network diagrams for studies reporting clinical response and clinical remission for the induction analysis in the ADT nonfailure and failure populations are presented in Figure 2 and Figure 3, respectively. For the population of patients who had not experienced ADT failure, the network consisted of ████ treatment nodes informed by ████ RCTs. Most connections between treatment nodes were informed by a single trial; however, some connections were informed by up to ███ trials. For the ADT failure population, the network consisted of ████ treatment nodes informed by ████ RCTs, and most connections between treatment nodes were informed by a single trial.
Clinical remission: In the ADT nonfailure population, no comparators were statistically significantly more effective than guselkumab 200 mg (i.e., 95% CrIs crossed the null) or vice versa (Figure 4). In the ADT failure population, the results favoured upadacitinib versus guselkumab 200 mg (Figure 5).
Clinical response: In the ADT nonfailure population, no comparators were statistically significantly more effective than guselkumab 200 mg, and the results favoured guselkumab versus golimumab and adalimumab (Figure 6). In the ADT failure population, no comparators were statistically significantly more effective than guselkumab, and the results favoured guselkumab versus adalimumab (Figure 7).
Maintenance Analysis
Network diagrams for studies reporting clinical response and clinical remission for the maintenance analysis in the ADT nonfailure and failure populations are presented in Figure 8 and Figure 9, respectively. For the ADT nonfailure population, the network consisted of ████ treatment nodes informed by ████ trials, and most connections between treatment nodes were informed by single trials. For the ADT failure population, the network consisted of ████ treatment nodes, and all connections between treatment nodes were informed by single trials.
Clinical remission: In the ADT nonfailure population, no comparators were statistically significantly more efficacious than guselkumab 100 mg or 200 mg (i.e., 95% CrIs crossed the null), and the results favoured guselkumab 200 mg versus adalimumab, and golimumab (Figure 10). In the ADT failure population, no comparators were statistically significantly more efficacious than guselkumab 100 mg or 200 mg or vice versa (Figure 11).
Clinical response: In the ADT nonfailure population, no comparators were statistically significantly more efficacious than guselkumab 100 mg or 200 mg (i.e., 95% CrIs crossed the null), and the results favoured both doses of guselkumab versus adalimumab (Figure 12). In the ADT failure population, no comparators were statistically significantly more efficacious than guselkumab 100 mg or 200 mg or vice versa (Figure 13).
Sensitivity analyses: The results of the sensitivity analyses for clinical remission and clinical response in the maintenance phase, which did not account for delayed responders, were consistent with the primary analyses (data not shown).












Critical Appraisal of NMA
The methods used to conduct the systematic literature review and NMA were prespecified with an a priori protocol and used appropriate criteria to search databases, select studies, extract data, and assess the risk of bias of the included studies. Selection bias is expected to be low, given the comprehensiveness of the searches and methods for study selection.
The NMA included relevant outcomes identified by the CDA-AMC team (clinical remission and clinical response); however, other clinically and patient-relevant outcomes such as endoscopic healing, corticosteroid-free clinical remission, HRQoL, and harms were not included in the comparisons.
The majority of comparators within the evidence network were informed by a single study, resulting in a star-shaped network and limiting certainty in the assumptions of the analysis. The authors of the sponsor’s NMA report noted that the results of the inconsistency analysis indicated broad concordance between the direct and indirect evidence for both clinical remission and clinical response. Because most nodes were informed by only 1 or 2 trials and, in several trials, patients were re-randomized, comparisons were underpowered, which contributed to wide CrIs in the analyses. In addition, infliximab and golimumab were not compared with guselkumab in the analysis of the population of patients who have experienced ADT failure due to a lack of data, which represents a gap in the available indirect evidence, given the shared place in therapy for UC.
Substantial heterogeneity was noted across the studies, and while the meta-regression random-effects model accounts for some sources of heterogeneity, there is a risk of the exchangeability assumption being violated. Specifically, potential effect modifiers across the included studies that could not be controlled for included trial design characteristics, patient eligibility criteria, baseline patient characteristics, and outcome characteristics (i.e., definitions and methods of reporting outcomes). Variations in the placebo effect estimates across studies support these concerns about heterogeneity as well as concerns about possible violations of the assumptions of transitivity for the NMA.
In addition, beyond the induction phase, the authors noted that although placebo is considered to be a common comparator across the majority of the included trials, placebo arms across re-randomized trials may not be equivalent due to variation in the treatments received during the preceding induction phase. In particular, the carryover effects observed due to previous treatments are likely to vary based on treatment mechanism, potentially constituting a substantial source of cross-trial heterogeneity, which is evidenced in part by variations in baseline risk across the included trials. Variations in the timing of the measured Mayo scores that informed the response and remission outcomes also introduce biases that could not be appropriately accounted for by the NMA methods. In general, the magnitude and direction of potential bias due to the heterogeneity of the outcome estimates cannot be predicted. The risk-of-bias assessment at the outcome level and its potential impact on the NMA effect estimates was not explicitly assessed or discussed, and no sensitivity analyses were conducted to examine the influence of studies with a high risk of bias on relative treatment effects, although the authors of the NMA stated that, in general, the trials were found to be of good quality.
Efforts were made in the NMA analysis to reduce heterogeneity by stratifying the patient population into ADT failure and ADT nonfailure subgroups. While this approach provides a more accurate estimate of efficacy within those populations, it does not provide an overall estimate of efficacy across the entire indicated population. Consequently, neither subgroup estimate of efficacy is representative of overall efficacy in the Canadian context.
The specified model, while referred to as an ordinal regression, is a multinomial regression that removes correlations between the outcomes of response and remission and estimates an OR for each outcome individually against no response or remission. Furthermore, the model restructures the data such that the response outcome used a definition that is different from the one used in the clinical trial setting, where it is defined as response without remission. This redefinition introduces a core issue in that the response outcome is defined conditionally on a separate outcome that can occur at a future date; thus, whether an improvement in response rate due to treatment is a benefit or a harm becomes unclear. Ultimately, the interpretation of the ordinal OR when the model assumptions are violated can create a misleading impression of how the outcomes and treatment are related.136 Due to the limitations in the NMA, the relative treatment effects of guselkumab versus other relevant comparators is uncertain and there is no conclusive evidence of a preferred treatment, based on the reported results.
Studies Addressing Gaps in the Systematic Review Evidence
No studies addressing gaps in the pivotal phase III evidence were submitted by the sponsor.
Discussion
Summary of Available Evidence
Two pivotal phase III, randomized, double-blind trials and 1 ITC submitted by the sponsor have been summarized in this report.
The QUASAR induction (N = 701) and maintenance (N = 568) studies met the inclusion criteria for the systematic review conducted by the sponsor. The objective of the induction study was to assess the efficacy and safety of guselkumab 200 mg IV as induction therapy versus matching placebo administered at weeks 0, 4, and 8 in adults with moderately to severely active UC. The study enrolled patients who had experienced an inadequate response or failure to tolerate conventional therapy (i.e., 6-mercaptopurine, azathioprine, or corticosteroids) or ADT (i.e., TNF antagonists, vedolizumab, or tofacitinib). Patients were required to have an MMS of 4 to 9, a Mayo rectal bleeding score of 1 or more, and a Mayo endoscopy subscore of 2 or more. Patients were eligible for the maintenance study if they had demonstrated a clinical response to guselkumab or placebo treatment at weeks 12 or 24. The objective of the maintenance study was to assess the efficacy and safety of guselkumab SC injection maintenance regimens (100 mg or 200 mg) versus matching placebo in patients who achieved clinical response with guselkumab IV induction. Patients received guselkumab 200 mg every 4 weeks, guselkumab 100 mg every 8 weeks, or placebo every 4 weeks starting at week 0 through week 44. In general, the approved Health Canada indication and reimbursement request aligned with both study populations, except that the indication is not limited to patients who have experienced an inadequate response or are unable to tolerate ADT or conventional therapy. The outcomes most relevant to the CDA-AMC review included the primary outcome of clinical remission and secondary outcomes of endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance only), and HRQoL measured via IBDQ remission. For both studies, key baseline characteristics were generally balanced between treatment groups. Patients in the induction study were predominantly white (72.5%) and male (56.9%). The median age was 39.0 years (range, 18 to 79 years), and the majority of patients were from Eastern Europe (42.2%) followed by rest of world (37.5%) and Asia (20.3%). The mean duration of disease was 7.5 years, and the mean baseline MMS was 6.9, with 64.5% of patients presenting with severely active disease (i.e., an MMS of 7 to 9) and 47.8% with extensive disease. At baseline, 43.1% of patients were taking corticosteroids, 20.8% were taking immunomodulatory drugs, and 72.5% were taking oral 5-ASAs, with generally similar proportions of patients across the treatment groups. Among all patients, 93.2% experienced an inadequate response or intolerance to corticosteroids and/or 6-mercaptopurine or azathioprine or experienced corticosteroid dependence, 49.1% had a history of experiencing ADT failure (i.e., anti–TNF alpha, vedolizumab, or tofacitinib), and 50.9% had a history of nonfailure of ADT. At induction baseline for the patients in the maintenance study, 40% were taking corticosteroids and 22.2% were taking immunomodulatory drugs (6-mercaptopurine, azathioprine, or methotrexate), 57.7% did not have a history of experiencing an inadequate response or intolerance to ADT (of these, 94.2% were naive to ADT), and 42.3% had experienced a prior inadequate response or intolerance to ADT. At maintenance baseline, the mean MMS was 2.5, 34.2% of patients were in clinical remission, and 39.1% achieved endoscopic healing. For both studies, the primary analysis of efficacy and safety outcomes were conducted in the FAS, which included patients who received at least 1 (partial or complete) dose of the study intervention who had a baseline MMS of 5 to 9.
In the absence of direct comparative evidence of guselkumab versus relevant comparators of interest, the sponsor conducted an NMA. The NMA assessed the induction and maintenance treatment effects of guselkumab versus adalimumab, infliximab, tofacitinib, upadacitinib, mirikizumab, vedolizumab, etrasimod, ozanimod, and ustekinumab for the treatment of adults with moderately to severely active UC. Some comparators were not relevant to Canadian public payers (e.g., risankizumab, filgotinib) and have therefore been excluded from the Clinical Review. Outcomes of interest included clinical remission and clinical response. Bayesian NMAs were conducted using ordinal regression models to estimate ORs and 95% CrIs. Random-effects NMA results for both induction and maintenance therapies were summarized based on 2 distinct patient populations, ADT failure and ADT nonfailure.
Interpretation of Results
The evidence from the QUASAR induction and maintenance studies addressed treatment outcomes noted to be important by both patients and clinicians. The patient group inputs indicated that clinical remission, clinical response, corticosteroid-free clinical remission, and HRQoL are important to them. Similarly, the clinical experts consulted by the review team indicated that the unmet needs of patients with moderately to severely active UC would be new treatments that induce and maintain clinical and endoscopic remission.
Efficacy
Both the induction and maintenance studies supported a clinically meaningful improvement of guselkumab over placebo for clinical remission, endoscopic healing, clinical response, corticosteroid-free clinical remission (maintenance therapy only), and IBDQ remission at 12 weeks (induction therapy) and 44 weeks (maintenance therapy) (100 mg and 200 mg dosages) in adults with moderately to severely active UC. For clinical remission at 12 and 44 weeks, the estimated between-group difference was 14.9% (95% CI, 9.9 to 19.9) and 25.2% (95% CI, 16.4 to 33.9) for the 100 mg dose, and 29.5% (95% CI, 20.9 to 38.1) for the 200 mg dose, respectively. For the GRADE assessment, the clinical experts consulted by CDA-AMC suggested a clinically important threshold of 7.5% and 10% for the between-group absolute risk difference at 12 and 44 weeks, respectively. Based on these thresholds, there was a high certainty of evidence for a clinically important increase in clinical remission at both time points. For endoscopic healing at 12 and 44 weeks, the estimated between-group difference was 16.0% (95% CI, 10.5 to 21.4) and 29.5% (95% CI, 20.7 to 38.3) for the 100 mg dose and 31.1% (95% CI, 22.5 to 39.8) for the 200 mg dose, respectively. The clinical experts suggested a clinically important threshold of 10% for the between-group absolute risk difference at 44 weeks but could not determine a threshold at 12 weeks; therefore, the target-of-certainty appraisal was any effect (i.e., crossed the null). Based on the 10% threshold, there was a high certainty of evidence for a clinically important increase in endoscopic healing at 44 weeks. For clinical response at 12 weeks and maintenance of clinical response at 44 weeks, the estimated between-group difference was 33.8% (95% CI, 26.9 to 40.7) and 33.6% (95% CI, 24.5 to 42.7) for the 100 mg dose and 30.7% (95% CI, 21.5 to 40.0) for the 200 mg dose, respectively. Using the same thresholds as those used for clinical remission, as suggested by the clinical experts, there was a high certainty of evidence for a clinically important increase in clinical response at 12 weeks and in maintenance of clinical response at 44 weeks. For corticosteroid-free clinical remission at 44 weeks, the estimated between-group difference was 25.7% (95% CI, 17.0 to 34.5) for the 100 mg dose and 29.0% (95% CI, 20.5 to 37.6) for the 200 mg dose. For the GRADE assessment, the clinical experts suggested a clinically important threshold of 15% in the between-group absolute risk difference at 44 weeks. Based on this threshold, there was a high certainty of evidence for a clinically important increase in corticosteroid-free clinical remission at 44 weeks. For IBDQ remission at 12 and 44 weeks, the estimated between-group difference was 21.9% (95% CI, 14.9 to 29.0) and 26.3% (95% CI, 16.8 to 35.7) for the 100 mg dose and 25.9% (95% CI, 16.5 to 35.4) for the 200 mg dose. Using the same thresholds as those used for clinical remission, as suggested by the clinical experts, there was a high certainty of evidence for a clinically important increase in IBDQ remission at both time points. The results for all primary and secondary outcomes listed were consistent across the prespecified subgroups of interest (i.e., ADT-naive and ADT failure), in favour of guselkumab.
Based on the sponsor-submitted NMA, the results for clinical remission at induction in the ADT nonfailure population did not favour any particular treatment (i.e., 95% CrIs crossed the null), and the results for the ADT failure population favoured only upadacitinib versus guselkumab 200 mg. For clinical remission at maintenance in the ADT nonfailure population, the results favoured guselkumab 200 mg versus adalimumab and golimumab, and the results for the ADT failure population did not favour any treatments. The results for clinical response at induction in the ADT nonfailure population favoured guselkumab 200 mg versus golimumab and adalimumab, and the results for the ADT failure population favoured guselkumab versus adalimumab. For clinical response at maintenance in the ADT nonfailure population, the results favoured guselkumab 100 mg and 200 mg versus adalimumab, and the results for the ADT failure population did not favour any treatments. However, there were several notable sources of heterogeneity for potential effect modifiers across the included studies that create uncertainty in the NMA results. These included trial design characteristics, patient eligibility criteria, baseline patient characteristics, and definitions and methods of reporting outcomes. In addition, beyond the induction phase, the authors noted that although placebo is a common comparator across the majority of the included trials, placebo groups across re-randomized studies may not be equivalent due to variation in the treatments received during the preceding induction phase. The magnitude and direction of potential bias due to heterogeneity on outcome estimates cannot be predicted. Additionally, the interpretation of the ordinal OR when the model assumptions are violated can create a misleading impression of how the outcomes and treatments are related. It should also be noted that the NMA results were based on the ADT failure and ADT nonfailure subgroups and not on the overall population, which is the focus of the approved Health Canada indication. Due to these limitations in the NMA, no definitive conclusions could be drawn on the relative treatment effects of guselkumab induction and maintenance therapies versus other relevant comparators for the treatment of moderately to severely active UC.
Harms
In both the induction and maintenance studies, most patients reported at least 1 TEAE. During the 12-week induction treatment period, the most frequently reported TEAEs in the guselkumab and placebo groups were anemia, COVID-19, headache, and UC. Of these TEAEs, a numerically higher proportion of UC was reported in patients taking placebo. Over the 44-week maintenance treatment period, the most frequently reported TEAEs in the guselkumab 100 mg, guselkumab 200 mg, and placebo groups were UC and COVID-19, with a numerically higher proportion of UC being reported in patients taking placebo. In both studies, serious TEAEs, TEAEs leading to discontinuation of treatment, and death were infrequent and similar across the treatment groups; no deaths were reported in the maintenance study. In both studies, the incidence of notable harms, which included infections and serious infections, was similar between groups, and serious infections were infrequent. No cases of active tuberculosis were reported in either study. The clinical experts indicated that the incidence of these TEAEs is expected with guselkumab and would be manageable for many patients.
The sponsor-submitted NMA did not include harms; therefore, no conclusions could be drawn on the relative safety of guselkumab versus other relevant comparators.
Conclusions
Evidence from 2 phase III double-blind RCTs (QUASAR induction and maintenance studies) reported on outcomes that were important to both patients and clinicians. The studies showed a high certainty of evidence that treatment with guselkumab results in a clinically meaningful increase in clinical remission, endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance only), and HRQoL via IBDQ remission at 12 weeks (induction therapy) and 44 weeks (maintenance therapy), compared with placebo in adults with moderately to severely active. Although the approved Health Canada indication is broader than the patient populations enrolled in the studies (i.e., patients who had experienced an inadequate response or were unable to tolerate ADT or conventional therapy), the clinical experts consulted by CDA-AMC did not consider the indication to affect the generalizability of the findings. No new safety signals were identified, and the safety of guselkumab was consistent with the drug’s known safety profile. Due to the limitations of the ITC, mostly attributed to the heterogeneity across studies, no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators.
References
1.Janssen Research & Development LLC. Clinical Study Report: Induction Study 2, CNTO1959UCO3001. A Phase 2b/3, Randomized, Double-blind, Placebo-controlled, Parallel-group, Multicenter Protocol to Evaluate the Efficacy and Safety of Guselkumab in Participants with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. February 8, 2024.
2.Janssen Research & Development LLC. Clinical Study Report: Maintenance Study, CNTO1959UCO3001, week 44. A Phase 2b/3, Randomized, Double-blind, Placebo-controlled, Parallel-group, Multicenter Protocol to Evaluate the Efficacy and Safety of Guselkumab in Participants with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. February 20, 2024.
3.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12 Suppl 1:S3-9. doi: 10.1097/01.mib.0000195385.19268.68 PubMed
4.Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625-9. doi: 10.1056/nejm198712243172603 PubMed
5.Stenson WF. Inflammatory bowel diseaes. In: Goldman I, Bennett JC, eds. Cecil Textbook of Medicine. 21st ed. WB Saunders Co; 2000:722-729.
6.Gastrointestinal Society, Canadian Society of Intestinal Research. Inflammatory Bowel Disease. 2023. Accessed October 23, 2023. https://badgut.org/information-centre/a-z-digestive-topics/inflammatory-bowel-disease/
7.Kaplan GG, Ng SC. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 2017;152(2):313-321 e2. doi: 10.1053/j.gastro.2016.10.020 PubMed
8.Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713-25. doi: 10.1056/NEJMra1102942 PubMed
9.Travis SP, Stange EF, Lémann M, et al. European evidence-based consensus on the management of ulcerative colitis: current management. J Crohns Colitis. 2008;2(1):24-62. doi: 10.1016/j.crohns.2007.11.002 PubMed
10.Rubin DT, Sninsky C, Siegmund B, et al. International Perspectives on Management of Inflammatory Bowel Disease: Opinion Differences and Similarities Between Patients and Physicians From the IBD GAPPS Survey. Inflamm Bowel Dis. 2021;27(12):1942-1953. doi: 10.1093/ibd/izab006 PubMed
11.Greuter T, Vavricka SR. Extraintestinal manifestations in inflammatory bowel disease - epidemiology, genetics, and pathogenesis. Expert Rev Gastroenterol Hepatol. 2019;13(4):307-317. doi: 10.1080/17474124.2019.1574569 PubMed
12.Crohn’s and Colitis Foundation of Canada. The impact of inflammatory bowel disease in Canada 2012 Final Report and Recommendations. 2012. Accessed May 10, 2023. http://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/ccfc-ibd-impact-report-2012.pdf
13.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural History of Adult Ulcerative Colitis in Population-based Cohorts: A Systematic Review. Clin Gastroenterol Hepatol. 2018;16(3):343-356.e3. doi: 10.1016/j.cgh.2017.06.016 PubMed
14.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-94. doi: 10.1053/j.gastro.2011.01.055 PubMed
15.Crohn’s and Colitis Canada. Impact of inflammatory bowel disease in Canada. 2023. Accessed August 8, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2023-IBD-Report-English-LR.pdf
16.Schreiber S, Panes J, Louis E, Holley D, Buch M, Paridaens K. National differences in ulcerative colitis experience and management among patients from five European countries and Canada: an online survey. J Crohns Colitis. 2013;7(6):497-509. doi: 10.1016/j.crohns.2012.07.027 PubMed
17.Crohn’s and Colitis Canada. Impact of inflammatory bowel disease in Canada. 2018. Accessed October 5, 2022. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf
18.Torres J, Billioud V, Sachar DB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis as a progressive disease: the forgotten evidence. Inflamm Bowel Dis. 2012;18(7):1356-63. doi: 10.1002/ibd.22839 PubMed
19.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389(10080):1756-1770. doi: 10.1016/s0140-6736(16)32126-2 PubMed
20.Roda G, Narula N, Pinotti R, et al. Systematic review with meta-analysis: proximal disease extension in limited ulcerative colitis. Aliment Pharmacol Ther. 2017;45(12):1481-1492. doi: 10.1111/apt.14063 PubMed
21.Ungar B, Kopylov U. Long-term outcome of ulcerative proctitis. United European Gastroenterol J. 2020;8(8):847-848. doi: 10.1177/2050640620948795 PubMed
22.Regueiro MD. Diagnosis and treatment of ulcerative proctitis. J Clin Gastroenterol. 2004;38(9):733-40. doi: 10.1097/01.mcg.0000139178.33502.a3 PubMed
23.Al Hashash J, Regueiro M. Medical management of low-risk adult patients with mild to moderate ulcerative colitis. UpToDate; 2023. Accessed October 23, 2024. https://www.uptodate.com
24.Armuzzi A, Liguori G. Quality of life in patients with moderate to severe ulcerative colitis and the impact of treatment: A narrative review. Dig Liver Dis. 2021;53(7):803-808. doi: 10.1016/j.dld.2021.03.002 PubMed
25.Benchimol EI, Bernstein CN, Bitton A, et al. The Impact of Inflammatory Bowel Disease in Canada 2018: A Scientific Report from the Canadian Gastro-Intestinal Epidemiology Consortium to Crohn's and Colitis Canada. J Can Assoc Gastroenterol. 2019;2(Suppl 1):S1-S5. doi: 10.1093/jcag/gwy052 PubMed
26.Casellas F, Arenas JI, Baudet JS, et al. Impairment of health-related quality of life in patients with inflammatory bowel disease: a Spanish multicenter study. Inflamm Bowel Dis. 2005;11(5):488-96. doi: 10.1097/01.mib.0000159661.55028.56 PubMed
27.Armuzzi A, Tarallo M, Lucas J, et al. The association between disease activity and patient-reported outcomes in patients with moderate-to-severe ulcerative colitis in the United States and Europe. BMC Gastroenterol. 2020;20(1):18. doi: 10.1186/s12876-020-1164-0 PubMed
28.Sarid O, Slonim-Nevo V, Schwartz D, et al. Differing Relationship of Psycho-Social Variables with Active Ulcerative Colitis or Crohn's Disease. Int J Behav Med. 2018;25(3):341-350. doi: 10.1007/s12529-018-9712-5 PubMed
29.Ghosh S, Sanchez Gonzalez Y, Zhou W, et al. Upadacitinib Treatment Improves Symptoms of Bowel Urgency and Abdominal Pain, and Correlates With Quality of Life Improvements in Patients With Moderate to Severe Ulcerative Colitis. J Crohns Colitis. 2021;15(12):2022-2030. doi: 10.1093/ecco-jcc/jjab099 PubMed
30.Peyrin-Biroulet L, Germain A, Patel AS, Lindsay JO. Systematic review: outcomes and post-operative complications following colectomy for ulcerative colitis. Aliment Pharmacol Ther. 2016;44(8):807-16. doi: 10.1111/apt.13763 PubMed
31.Rubin DT, Dubinsky MC, Panaccione R, et al. The impact of ulcerative colitis on patients' lives compared with other chronic diseases: a patient survey. Dig Dis Sci. 2010;55(4):1044-52. doi: 10.1007/s10620-009-0953-7 PubMed
32.Becker HM, Grigat D, Ghosh S, et al. Living with inflammatory bowel disease: A Crohn's and Colitis Canada survey. Can J Gastroenterol Hepatol. 2015;29(2):77-84. doi: 10.1155/2015/815820 PubMed
33.Sikirica M, Lynch J, Hoskin B, Kershaw J, Wild R, Lukanova R. P0412 Humanistic burden and impact of the disease in moderate to severe patients with Crohn’s disease (CD) and ulcerative colitis (UC): findings from a cross sectional study in the US and France, Germany, Italy, Spain, UK (EU5). United Eur Gastroenterol J. 2020;8(S8):348. doi: 10.1177/2050640620927345
34.Burisch J, Zhao M, Odes S, et al. P860 The Costs of Inflammatory Bowel Diseases in High-Income Settings. J Crohns Colitis. 2023;17(Supplement_1):i983-i983. doi: 10.1093/ecco-jcc/jjac190.0990
35.Ruiz-Casas L, Evans J, Rose A, et al. The LUCID study: living with ulcerative colitis; identifying the socioeconomic burden in Europe. BMC Gastroenterol. 2021;21(1):456. doi: 10.1186/s12876-021-02028-5 PubMed
36.Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035-1058.e3. doi: 10.1053/j.gastro.2015.03.001 PubMed
37.de Léséleuc L, McGolrick D. Appropriate pharmacotherapy for inflammatory bowel disease. (Technology Review: Optimal Use 360 Report). CADTH; 2019. Accessed October 21, 2025. https://www.cda-amc.ca/sites/default/files/hta-he/ho0003-he0018-ibd-ou360.pdf
38.Janssen Inc. Tremfya (guselkumab). Data on file [sponsor supplied reference]. 2024. Accessed July 31, 2024.
39.Bourgonje AR, Ungaro RC, Mehandru S, Colombel JF. Targeting the Interleukin 23 Pathway in Inflammatory Bowel Disease. Gastroenterology. 2024. doi: 10.1053/j.gastro.2024.05.036 PubMed
40.Navarro-Compan V, Puig L, Vidal S, et al. The paradigm of IL-23-independent production of IL-17F and IL-17A and their role in chronic inflammatory diseases. Front Immunol. 2023;14:1191782. doi: 10.3389/fimmu.2023.1191782 PubMed
41.Bristol-Myers Squibb Canada. Zeposia (ozanimod hydrochloride): 0.23 mg, 0.46 mg, and 0.92 mg, oral capsules [product monograph; sponsor supplied reference]. October 2, 2020. Updated August 28, 2024. Accessed August 11, 2023. https://www.bms.com/assets/bms/ca/documents/productmonograph/ZEPOSIA_EN_PM.pdf
42.Pfizer Canada ULC. Velsipity (etrasimod): 2 mg etrasimod (as etrasimod L-arginine) oral tablets [product monograph; sponsor supplied reference]. January 30, 2024.
43.AbbVie Corporation. Humira (adalimumab): 50 mg/mL or 100 mg/mL, sterile solution, for subcutaneous injection [product monograph]. September 24, 2004. Updated September 16, 2022. Accessed August 10, 2023. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf
44.Amgen Canada Inc. Amgevita (adalimumab): 50 mg/mL sterile solution in prefilled pen for subcutaneous injection; or 50 mg/mL sterile solution in a pre-filled SureClick autoinjector for subcutaneous injection [product monograph]. November 4, 2020. Updated September 9, 2022. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00067468.PDF
45.BGP Pharma ULC. Hulio (adalimumab injection): 50 mg/mL sterile solution for subcutaneous injection [product monograph]. November 24, 2020. Updated July 20, 2023. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00071858.PDF
46.Pfizer Canada ULC. Abrilada (adalimumab injection): 50 mg/mL sterile solution in prefilled pen for subcutaneous injection; 50 mg/mL sterile solution in prefilled glass syringe for subcutaneous injection; or 50 mg/mL sterile solution glass vial for subcutaneous injection [product monograph]. January 14, 2021. Updated November 14, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00068228.PDF
47.Sandoz Canada Inc. Hyrimoz (adalimumab injection): 50 mg/mL sterile solution in prefilled pen for subcutaneous injection; or 50 mg/mL sterile solution in a pre-filled autoinjector for subcutaneous injection [product monograph]. November 4, 2020. Updated October 11, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00067757.PDF
48.Merck Canada Inc. Hadlima (adalimumab injection): 40 mg in 0.8 mL sterile solution in pre-filled syringe for subcutaneous injection; Hadlima PushTouch (adalimumab injection): 40 mg in 0.8 mL sterile solution in auto-injector for subcutaneous injection [product monograph]. March 13, 2019. Updated November 26, 2020. Accessed August 21, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/HADLIMA-PM_E.pdf
49.Celltrion Healthcare Canada Limited. Yuflyma (adalimumab injection): 100 mg/mL sterile solution in pre-filled syringe for subcutaneous injection; 100 mg/mL sterile solution in pre-filled syringe with Needle guard for subcutaneous injection; or 100 mg/mL sterile solution in pre-filled pen (auto-injector) for subcutaneous injection [product monograph] December 24, 2021. Updated February 3, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00069429.PDF
50.Fresenius Kabi Canada Ltd. Idacio (adalimumab injection): 40 mg/0.8 mL sterile solution in either autoinjector, prefilled syringe, or vial, for subcutaneous injection [product monograph]. October 30, 2020. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00058541.PDF
51.JAMP Pharma Corporation. Simlandi (adalimumab injection): 100 mg/mL sterile solution in pre-filled syringe for subcutaneous injection [product monograph]. January 05, 2022. Updated December 09, 2022. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00068814.PDF
52.Janssen Inc. Simponi (golimumab): 50 mg/0.5 mL or 100 mg/1.0 mL, single-use sterile solution in a SmartJect autoinjector or pre-filled syringe, for subcutaneous injection; Simponi I.V. (golimumab): 50 mg/4.0 mL, sterile solution in single-use vials, for intravenous infusion [product monograph]. January 15, 2018. Accessed August 11, 2023. https://pdf.hres.ca/dpd_pm/00043422.PDF
53.Janssen Inc. Remicade (infliximab): 100 mg/vial, sterile lyophilized powder for solution, for intravenous injection [product monograph]. August 4, 2017. Accessed August 11, 2023. https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF
54.Amgen Canada Inc. Avsola (infliximab): 100 mg/vial, sterile lyophilized powder for solution, for intravenous infusion [product monograph]. March 12, 2020. Updated May 17, 2022. Accessed August 17, 2023. https://pdf.hres.ca/dpd_pm/00065940.PDF
55.Organon Canada Inc. Renflexis (infliximab for injection): 100 mg/vial, sterile lyophilized powder for solution, for intravenous infusion [product monograph]. December 1, 2017. Updated February 9, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00069528.PDF
56.Pfizer Canada Inc. Inflectra (infliximab for Injection): 100 mg/vial, lyophilized powder for solution [product monograph]. November 7, 2017. Accessed August 11, 2023. https://pdf.hres.ca/dpd_pm/00042037.PDF
57.Takeda Canada Inc. Entyvio (vedolizumab): 300 mg / vial, sterile powder for solution for infusion [product monograph]. March 22, 2016. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00034239.PDF
58.Eli Lilly Canada Inc. Omvoh (mirikizumab): 100 mg/mL solution, in prefilled pens and prefilled syringes for subcutaneous injection or 20 mg/mL solution, in vial for intravenous infusion [product monograph]. July 20, 2023. Accessed September 19, 2023. https://pdf.hres.ca/dpd_pm/00071771.PDF
59.Janssen Inc. Stelara (ustekinumab): 45 mg/0.5 mL 90 mg/1.0 mL, sterile solution in single-use pre-filled syringe, for subcutaneous injection; Stelara I.V. (ustekinumab): 130 mg/26 mL, sterile solution in single-use vial, intravenous infusion [product monograph]. December 12, 2008. Updated January 5, 2023. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00069002.PDF
60.Pfizer Canada ULC. Xeljanz (tofacitinib citrate): 5 mg and 10 mg, oral tablets; Xeljanz XR (tofacitinib citrate): 11 mg extended-release oral tablets [product monograph; sponsor supplied reference]. April 16, 2014. Updated August 11, 2023. Accessed August 21, 2023. https://pdf.hres.ca/dpd_pm/00071982.PDF
61.Pharmascience Inc. pms-Tofacitinib (tofacitinib): 5 mg oral tablets [product monograph]. November 29, 2021. Updated August 23, 2022. Accessed August 15, 2023. https://pdf.hres.ca/dpd_pm/00069222.PDF
62.Taro Pharmaceuticals Inc. Taro-Tofacitinib (tofacitinib tablets): 5 mg and 10 mg, oral tablets [product monograph]. January 18, 2021. Updated July 13, 2022. Accessed August 18, 2023. https://pdf.hres.ca/dpd_pm/00068405.PDF
63.Auro Pharma Inc. Auro-Tofacitinib (tofacitinib): 5 mg and 10 mg, oral tablets [product monograph]. August 19, 2022. Accessed September 22, 2023. https://pdf.hres.ca/dpd_pm/00068455.PDF
64.AbbVie Corporation. Rinvoq (upadacitinib): 15 mg, 30 mg, or 45 mg, extended-release oral tablets [product monograph; sponsor supplied reference]. October 31, 2023. Accessed December 11, 2023. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/RINVOQ_PM_EN.pdf
65.Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14(12):1660-6. doi: 10.1002/ibd.20520 PubMed
66.Dhanda AD, Creed TJ, Greenwood R, Sands BE, Probert CS. Can endoscopy be avoided in the assessment of ulcerative colitis in clinical trials? Inflamm Bowel Dis. 2012;18(11):2056-62. doi: 10.1002/ibd.22879 PubMed
67.Mohammed Vashist N, Samaan M, Mosli MH, et al. Endoscopic scoring indices for evaluation of disease activity in ulcerative colitis. Cochrane Database Syst Rev. 2018;1(1):CD011450. doi: 10.1002/14651858.CD011450.pub2 PubMed
68.Levesque B, Pola S, King D, et al. Tu1119 Responsiveness of Central Endoscopic Assessment of Disease Activity Using the Modified Mayo Clinic Score in Ulcerative Colitis. Gastroenterology. 2013;144(5). doi: 10.1016/s0016-5085(13)62835-8
69.Walsh AJ, Ghosh A, Brain AO, et al. Comparing disease activity indices in ulcerative colitis. J Crohns Colitis. 2014;8(4):318-25. doi: 10.1016/j.crohns.2013.09.010 PubMed
70.Higgins PD, Schwartz M, Mapili J, Zimmermann EM. Is endoscopy necessary for the measurement of disease activity in ulcerative colitis? Am J Gastroenterol. 2005;100(2):355-61. doi: 10.1111/j.1572-0241.2005.40641.x PubMed
71.Naegeli AN, Hunter T, Dong Y, et al. Full, Partial, and Modified Permutations of the Mayo Score: Characterizing Clinical and Patient-Reported Outcomes in Ulcerative Colitis Patients. Crohns Colitis 360. 2021;3(1):otab007. doi: 10.1093/crocol/otab007 PubMed
72.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-10. PubMed
73.Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-9. doi: 10.1097/00054725-200405000-00014 PubMed
74.Chen X-L, Zhong L-h, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177. doi: 10.1186/s12955-017-0753-2 PubMed
75.Alrubaiy L, Rikaby I, Dodds P, Hutchings HA, Williams JG. Systematic review of health-related quality of life measures for inflammatory bowel disease. J Crohns Colitis. 2015;9(3):284-92. doi: 10.1093/ecco-jcc/jjv002 PubMed
76.Irvine EJ, Feagan B, Rochon J, et al. Quality of life: a valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Canadian Crohn's Relapse Prevention Trial Study Group. Gastroenterology. 1994;106(2):287-96. doi: 10.1016/0016-5085(94)90585-1 PubMed
77.Pallis AG, Vlachonikolis IG, Mouzas IA. Assessing health-related quality of life in patients with inflammatory bowel disease, in Crete, Greece. BMC Gastroenterol. 2002;2:1. doi: 10.1186/1471-230x-2-1 PubMed
78.Gregor JC, McDonald JW, Klar N, et al. An evaluation of utility measurement in Crohn's disease. Inflamm Bowel Dis. 1997;3(4):265-76. PubMed
79.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-7. doi: 10.1097/00005176-199904001-00003 PubMed
80.Irvine EJ. Quality of life of patients with ulcerative colitis: past, present, and future. Inflamm Bowel Dis. 2008;14(4):554-65. doi: 10.1002/ibd.20301 PubMed
81.LeBlanc K, Mosli MH, Parker CE, MacDonald JK. The impact of biological interventions for ulcerative colitis on health-related quality of life. Cochrane Database Syst Rev. 2015;2015(9):CD008655. doi: 10.1002/14651858.CD008655.pub3 PubMed
82.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
83.Centre for Reviews and Dissemination. Systematic Reviews: CRD’s guidance for undertaking reviews in health care. University of York; 2008. Accessed September 28, 2022. https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf
84.Janssen Inc. Efficacy of guselkumab versus available biologic therapies for the treatment of moderate to severe ulcerative colitis: A systematic review and network meta-analysis. Technical Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya/Tremfya I.V. (guselkumab) 200 mg/2mL solution for subcutaneous Injection, 100 mg/1mL solution for subcutaneous injection, and 200 mg/20 mL solution for intravenous infusion. October 8, 2024.
85.Dias S AA, Welton N, Jansen J, Sutton A. Network Meta‐Analysis for Decision Making. John Wiley & Sons Ltd; 2018.
86.Turner RM, Jackson D, Wei Y, Thompson SG, Higgins JP. Predictive distributions for between-study heterogeneity and simple methods for their application in Bayesian meta-analysis. Stat Med. 2015;34(6):984-98. doi: 10.1002/sim.6381 PubMed
87.Sandborn WJ, Colombel JF, D'Haens G, et al. One-year maintenance outcomes among patients with moderately-to-severely active ulcerative colitis who responded to induction therapy with adalimumab: subgroup analyses from ULTRA 2. Aliment Pharmacol Ther. 2013;37(2):204-13. doi: 10.1111/apt.12145 PubMed
88.Sands BE, Peyrin-Biroulet L, Loftus EV, Jr., et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N Engl J Med. 2019;381(13):1215-1226. doi: 10.1056/NEJMoa1905725 PubMed
89.Hoffmann-La Roche. NCT02163759: A Study Copmparing the Efficacy and Safety of Etrolizumab With Adalimumab and Placebo in Participants with Moderate to Severe Ulcerative Colitis (UC) in Participants Naive to Tumor Necrosis Factor (TNF) Inhibitors (HIBISCUS I). ClinicalTrials.gov; 2021. Accessed October 21, 2025. https://clinicaltrials.gov/study/NCT02163759
90.Hoffmann-La Roche. NCT02171429: A Study Comparing the Efficacy and Safety of Etrolizumab With Adalimumab and Placebo in Participants with Moderate to Severe Ulcerative Colitis (UC) in Participants Naive to Tumor Necrosis Factor (TNF) Inhibitors (HIBISCUS II). 2021. Accessed October 21, 2025. https://clinicaltrials.gov/study/NCT02171429
91.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-76. doi: 10.1056/NEJMoa050516 PubMed
92.Kobayashi T, Suzuki Y, Motoya S, et al. First trough level of infliximab at week 2 predicts future outcomes of induction therapy in ulcerative colitis-results from a multicenter prospective randomized controlled trial and its post hoc analysis. J Gastroenterol. 2016;51(3):241-51. doi: 10.1007/s00535-015-1102-z PubMed
93.Jiang XL, Cui HF, Gao J, Fan H. Low-dose Infliximab for Induction and Maintenance Treatment in Chinese Patients With Moderate to Severe Active Ulcerative Colitis. J Clin Gastroenterol. 2015;49(7):582-8. doi: 10.1097/MCG.0000000000000319 PubMed
94.Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017;376(18):1723-1736. doi: 10.1056/NEJMoa1606910 PubMed
95.Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616-24. doi: 10.1056/NEJMoa1112168 PubMed
96.Sandborn WJ, Lawendy N, Danese S, et al. Safety and efficacy of tofacitinib for treatment of ulcerative colitis: final analysis of OCTAVE Open, an open-label, long-term extension study with up to 7.0 years of treatment. Aliment Pharmacol Ther. 2022;55(4):464-478. doi: 10.1111/apt.16712 PubMed
97.Vermeire S, Danese S, Zhou W, et al. Efficacy and safety of upadacitinib maintenance therapy for moderately to severely active ulcerative colitis in patients responding to 8 week induction therapy (U-ACHIEVE Maintenance): overall results from the randomised, placebo-controlled, double-blind, phase 3 maintenance study. Lancet Gastroenterol Hepatol. 2023;8(11):976-989. doi: 10.1016/S2468-1253(23)00208-X PubMed
98.Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022;399(10341):2113-2128. doi: 10.1016/S0140-6736(22)00581-5 PubMed
99.Sandborn WJ, Ghosh S, Panes J, et al. Efficacy of Upadacitinib in a Randomized Trial of Patients With Active Ulcerative Colitis. Gastroenterology. 2020;158(8):2139-2149 e14. doi: 10.1053/j.gastro.2020.02.030 PubMed
100.Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N Engl J Med. 2016;374(18):1754-62. doi: 10.1056/NEJMoa1513248 PubMed
101.Sandborn WJ, Feagan BG, D'Haens G, et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2021;385(14):1280-1291. doi: 10.1056/NEJMoa2033617 PubMed
102.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699-710. doi: 10.1056/NEJMoa1215734 PubMed
103.Sandborn WJ, Baert F, Danese S, et al. Efficacy and Safety of Vedolizumab Subcutaneous Formulation in a Randomized Trial of Patients With Ulcerative Colitis. Gastroenterology. 2020;158(3):562-572 e12. doi: 10.1053/j.gastro.2019.08.027 PubMed
104.Sandborn WJ, Peyrin-Biroulet L, Zhang J, et al. Efficacy and Safety of Etrasimod in a Phase 2 Randomized Trial of Patients With Ulcerative Colitis. Gastroenterology. 2020;158(3):550-561. doi: 10.1053/j.gastro.2019.10.035 PubMed
105.Feagan B, Peyrin-Biroulet L, Sandborn W, et al. MP289 Etrasimod 2mg Once daily as treatment for Biologic/Janus Kinase Inhibitor-Naive and -Experienced Patients with Moderately to Severely Active Ulcerative Colitis: Subgroup Analysis from the Phase 3 Elevate UC 52 and Elevate UC 12 Trials. United European Gastroenterol J. 2022;10(S8):388-390. doi: 10.1002/ueg2.1229
106.Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. 2023;401(10383):1159-1171. doi: 10.1016/S0140-6736(23)00061-2 PubMed
107.Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2019;381(13):1201-1214. doi: 10.1056/NEJMoa1900750 PubMed
108.CADTH. Reimbursement review: guselkumab (Tremfya). Updated February 27, 2023. Accessed July 4, 2023. https://www.cadth.ca/guselkumab
109.Janssen Research & Development LLC. Clinical Study Report: Induction Study 1, CNTO1959UCO3001. A Phase 2b/3, Randomized, Double-blind, Placebo-controlled, Parallel-group, Multicenter Protocol to Evaluate the Efficacy and Safety of Guselkumab in Participants with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. August 1, 2022.
110.David T. Rubin JRA, Bruce E. Sands, Kuan-Hsiang Huang, Mary Kavalam, Matthew Germinaro, Rebecca WIlson, Hongyan Zhang, Emese Mihaly, Tadakazu Hisamatsu, Axel Dignass, Julian Panes. The Effect of Guselkumab Induction Therapy in patients with Moderately to Severely Active Ulcerative Colitis: QUASAR Phase 2b Induction Results at Week 12 by prior Inadequate Reponse or Intolerance to Advanced Therapy. 2022.
111.Dignass A, Rubin D, Bressler B, et al. OP23 The efficacy and safety of guselkumab induction therapy in patients with moderately to severely active Ulcerative Colitis: Phase 2b QUASAR Study results through week 12. J Crohns Colitis. 2022;16(Supplement_1):i025-i026. doi: 10.1093/ecco-jcc/jjab232.022
112.Feagan BG, Rubin DT, Danese S, et al. Efficacy of Vedolizumab Induction and Maintenance Therapy in Patients With Ulcerative Colitis, Regardless of Prior Exposure to Tumor Necrosis Factor Antagonists. Clin Gastroenterol Hepatol. 2017;15(2):229-239 e5. doi: 10.1016/j.cgh.2016.08.044 PubMed
113.Schreiber S, Feagan B, Peyrin-Biroulet L, et al. OP07 Exploring disease control by combining clinical, biological, and health-related quality of life remission with endoscopic improvements among Ulcerative Colitis patients treated with filgotinib: A post-hoc analysis from the SELECTION trial. J Crohns Colitis. 2022;16(Supplement_1):i007-i008. doi: 10.1093/ecco-jcc/jjab232.006
114.Dotan I, Feagan BG, Taliadouros V, et al. Efficacy of Filgotinib in Patients with Ulcerative Colitis by Line of Therapy in the Phase 2b/3 SELECTION Trial. J Crohns Colitis. 2023;17(8):1207-1216. doi: 10.1093/ecco-jcc/jjad039 PubMed
115.Feagan BG, Danese S, Loftus EV, Jr., et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397(10292):2372-2384. doi: 10.1016/S0140-6736(21)00666-8 PubMed
116.Siegmund B, Axelrad J, Pondel M, et al. DOP43 Rapidity of ozanimod-induced symptomatic response and remission in patients with moderately to severely active Ulcerative Colitis: Results from the induction period of True North. J Crohns Colitis. 2022;16(Supplement_1):i092-i093. doi: 10.1093/ecco-jcc/jjab232.082
117.Sands BE, Jain A, Ahmad HA, et al. P376 Efficacy of ozanimod in vedolizumab-exposed patients with ulcerative colitis: a phase 3 True North post hoc analysis. J Crohns Colitis. 2023;17(Supplement_1):i507-i508. doi: 10.1093/ecco-jcc/jjac190.0506
118.Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257-65 e1-3. doi: 10.1053/j.gastro.2011.10.032
119.Feagan BG, Peyrin-Biroulet L, Sandborn WJ, et al. S734 Etrasimod 2mg Once Daily as Treatment for Patients With Moderately to Severely Active Ulcerative Colitis: Topline and Subgroup Analysis From ELEVATE UC 52 and ELEVATE UC 12. Am J Gastroenterol. 2022;117(10S):e516-e517. doi: 10.14309/01.ajg.0000859576.23012.a3
120.Sandborn WJ, Vermiere S, Peyrin-Biroulet L, et al. OP086 Etrasimod 2MG Once Daily as Treatment for Moderately to Severely Ulcerative Colitis: Reults from the Phase 3 ELVEATE UC 52 and ELEVATE UC 12 Trials. United European Gastroenterol J. 2022;10(S8):67-69. doi: 10.1002/ueg2.12293
121.Risankizumab Induction Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Efficacy and Safety in the Randomized Phase 3 INSPIRE Study. Gastroenterol Hepatol (N Y). 2023;19(12 Suppl 9):9-10. PubMed
122.Panaccione R, Louis E, Colombel JF, et al. P593 Efficacy of Risankizumab in Patients With Moderately to Severely Active Ulcerative Colitis by Prior Advanced Therapy Failure and Mechanism of Action: Post Hoc Analysis of the INSPIRE and COMMAND Phase 3 Studies. J Crohns Colitis. 2024;18(Supplement_1):i1146-i1148. doi: 10.1093/ecco-jcc/jjad212.0723
123.Navabi S, D’Haens G, Samaan KH, et al. P469 Effect of mirikizumab on clinical and endoscopic outcomes based on prior advanced therapy failure in patients with moderately to severely active ulcerative colitis. J Crohns Colitis. 2023;17(Supplement_1):i598-i598. doi: 10.1093/ecco-jcc/jjac190.0599
124.D'Haens G, Dubinsky M, Kobayashi T, et al. Mirikizumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2023;388(26):2444-2455. doi: 10.1056/NEJMoa2207940 PubMed
125.Sandborn WJ, Sands BE, Vermeire S, et al. Modified Mayo score versus Mayo score for evaluation of treatment efficacy in patients with ulcerative colitis: data from the tofacitinib OCTAVE program. Therap Adv Gastroenterol. 2022;15:17562848221136331. doi: 10.1177/17562848221136331 PubMed
126.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):85-95; quiz e14-5. doi: 10.1053/j.gastro.2013.05.048 PubMed
127.Panaccione R, Ghosh S, Middleton S, et al. Combination therapy with infliximab and azathioprine is superior to monotherapy with either agent in ulcerative colitis. Gastroenterology. 2014;146(2):392-400 e3. doi: 10.1053/j.gastro.2013.10.052 PubMed
128.Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780-7. doi: 10.1136/gut.2010.221127 PubMed
129.Feagan BG, Sands BE, Sandborn WJ, et al. Guselkumab plus golimumab combination therapy versus guselkumab or golimumab monotherapy in patients with ulcerative colitis (VEGA): a randomised, double-blind, controlled, phase 2, proof-of-concept trial. Lancet Gastroenterol Hepatol. 2023;8(4):307-320. doi: 10.1016/S2468-1253(22)00427-7 PubMed
130.Sandborn WJ, Ferrante M, Bhandari BR, et al. Efficacy and Safety of Mirikizumab in a Randomized Phase 2 Study of Patients With Ulcerative Colitis. Gastroenterology. 2020;158(3):537-549 e10. doi: 10.1053/j.gastro.2019.08.043 PubMed
131.Louis E, Panaccione R, Parkes G, et al. OP06 Risankizumab Maintenance Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Efficacy and Safety in the Randomised Phase 3 COMMAND Study. J Crohns Colitis. 2024;18(Supplement_1):i10-i12. doi: 10.1093/ecco-jcc/jjad212.0006
132.Hibi T, Imai Y, Senoo A, Ohta K, Ukyo Y. Efficacy and safety of golimumab 52-week maintenance therapy in Japanese patients with moderate to severely active ulcerative colitis: a phase 3, double-blind, randomized, placebo-controlled study-(PURSUIT-J study). J Gastroenterol. 2017;52(10):1101-1111. doi: 10.1007/s00535-017-1326-1 PubMed
133.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96-109 e1. doi: 10.1053/j.gastro.2013.06.010 PubMed
134.Hoffmann-La Roche. NCT02163759: A Study Comparing the Efficacy and Safety of Etrolizumab With Adalimumab and Placebo in Participants with Moderate to Severe Ulcerative Colitis (UC) in Participants Naive to Tumor Necrosis Factor (TNF) Inhibitors (HIBISCUS I). 2021. Accessed October 21, 2025. https://clinicaltrials.gov/study/NCT02163759
135.Sandborn WJ, D'Haens GR, Reinisch W, et al. Guselkumab for the Treatment of Crohn's Disease: Induction Results From the Phase 2 GALAXI-1 Study. Gastroenterology. 2022;162(6):1650-1664 e8. doi: 10.1053/j.gastro.2022.01.047 PubMed
136.Williams R. Understanding and interpreting generalized ordered logit models. J Math Sociol. 2016;40(1):7-20. doi: 10.1080/0022250x.2015.1112384
Pharmacoeconomic Review
Abbreviations
ADT
advanced therapy
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CUA
cost-utility analysis
NMA
network meta-analysis
QALY
quality-adjusted life-year
UC
ulcerative colitis
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Guselkumab (Tremfya), solution for injection |
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 3, 2025 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Previously reviewed: Yes Indication: Tremfya (guselkumab injection) is indicated for the treatment of adult patients with active psoriatic arthritis. Tremfya can be used alone or in combination with a cDMARD (e.g., methotrexate) Recommendation date: November 17, 2022 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Tremfya (guselkumab injection) is indicated for the treatment of adult patients with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy Recommendation date: February 21, 2018 Recommendation: Reimburse with clinical criteria and/or conditions |
cDMARD = conventional disease-modifying antirheumatic drug; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by a Markov model |
Target population | Adult patients with moderately to severely active UC who experience an inadequate response or failure to tolerate conventional therapya (ADT-naive subgroup) or ADTb (ADT‑experienced subgroup) |
Treatment | Guselkumab |
Dose regimen | 200 mg by IV in week 0, week 4, and week 8 followed by 100 mg by SC injection at week 16 and 100 mg every 8 weeks thereafter. A dose of 200 mg administered by SC injection at week 12 and every 4 weeks thereafter may be considered for patients who do not show adequate therapeutic benefit to guselkumab, or according to clinical judgment. |
Submitted price | Guselkumab: $3,059.74 per 100 mg/mL prefilled syringe or patient-controlled injector Guselkumab: $3,059.74 per 200 mg/2 mL prefilled syringe or prefilled pen Guselkumab: $3,059.74 per 200 mg/20 mL vial for IV infusion |
Submitted treatment cost | $32,252 in year 1 and $29,995 annually thereafter, assuming 49.7% of patients receive the lower maintenance dose (i.e., 100 mg every 8 weeks) |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (58 years) |
Key data sources | Sponsor-submitted NMA in which the efficacy for guselkumab was informed by the QUASAR trial |
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results | There is insufficient clinical evidence to justify a price premium for guselkumab relative to currently available treatments for moderately to severely active UC who have experienced an inadequate response or failure to tolerate conventional therapy or ADT |
ADT = advanced therapy; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; QALY= quality-adjusted life-year; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
aConventional therapy is assumed to comprise azathioprine, 6-mercaptopurine, methotrexate, 5-aminosalicylate, prednisone, and budesonide.
bADT is assumed to comprise anti-TNFs (adalimumab, infliximab, and golimumab), vedolizumab, and tofacitinib.
cNot included as a comparator in the ADT-experienced subgroup due to a lack of clinical data.
Conclusions
The CDA-AMC Clinical Review found that, compared with placebo, treatment with guselkumab results in a clinically meaningful increase in clinical remission, endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance therapy only), and health-related quality of life at 12 weeks of induction therapy and 44 weeks of maintenance therapy in adults with moderately to severely active ulcerative colitis (UC). Although the proposed health Canada indication is for a broader patient population than the populations enrolled in the studies (i.e., patients who have experienced an inadequate response or were unable to tolerate conventional therapy or advanced therapy [ADT]), the clinical experts consulted by CDA-AMC did not consider the proposed indication to affect the generalizability of the findings. No new safety signals were identified and the safety of guselkumab was consistent with the known safety profile the drug. Due to limitations of the indirect treatment comparison, mostly attributed to heterogeneity across studies, no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators.
In the sponsor’s base case, guselkumab was dominated by comparators. In the ADT-naive population, guselkumab was dominated by ustekinumab and upadacitinib (greater costs: $12,641 to $27,749; fewer quality-adjusted life-years [QALYs]: 0.09 to 0.10); in the ADT-experienced population, guselkumab was dominated by ustekinumab, etrasimod, and ozanimod (greater costs: $586 to $23,076; fewer QALYs: 0.09 to 0.19). According to the CDA-AMC Clinical Review, the comparative efficacy of guselkumab versus other ADTs is uncertain. Given that no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators, there is insufficient evidence to suggest that total treatment costs for guselkumab should be higher than other currently available biologics used in the treatment of moderately to severely active UC.
Input Relevant to the Economic Review
This section is a summary of the feedback received from patient groups and drug plans that participate in the CDA-AMC review process.
Patient group input was received from the Gastrointestinal Society and Crohn’s and Colitis Canada. Information from the Gastrointestinal Society was collected from respondents who have experience with inflammatory bowel disease through surveys, interviews, a focus group, conversations, a patient round table, and social media. Information from Crohn’s and Colitis Canada was collected from caregivers and patients in Canada who have experience with Crohn disease and UC. Input specific to UC noted that most patients reported having used a combination of medications to manage their UC that included systemic steroids, sulfasalazine and 5-aminosalicylates, biologics, immunomodulators, antibiotics, and nonsystemic steroids. Immunomodulators were more commonly used in patients with severe disease. The input noted that the majority of patients with moderate to severe UC continue to experience symptoms with current treatment options and that quality of life could be greatly improved if patients were to experience remission. No patients had experience with guselkumab.
No clinician group input was received for this review.
The drug plan input identified ustekinumab, mirikizumab, golimumab, vedolizumab, upadacitinib, adalimumab, infliximab, tofacitinib, ozanimod, and etrasimod as relevant comparators. However, the plans noted that not all relevant comparators are currently reimbursed, and funding varies by jurisdiction. It was noted that guselkumab will need to be administered by a trained health care professional in a hospital setting or infusion clinic in the induction phase and training will be required for patients or caregivers administering guselkumab in the maintenance phase. The drug plans noted there are several biologics, including biosimilars, currently reimbursed for UC; as such, it is difficult to estimate how much of the market will be displaced by guselkumab.
Several of these concerns were addressed in the sponsor’s model:
The impact of disease and treatment on patient’s health-related quality of life was captured with utility values.
The costs of administration during the induction period for guselkumab were included in the sponsor’s model.
Because golimumab and infliximab were not included as comparators in the sponsor’s pharmacoeconomic base case in the ADT-experienced population, and because training costs for subcutaneous administration for guselkumab in the maintenance phase were also not included in the sponsor’s base case, the comparative clinical efficacy of guselkumab versus comparators is uncertain; CDA-AMC was unable to address this concern.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) assessing guselkumab compared with adalimumab, conventional therapy (i.e., 6-mercaptopurine, azathioprine, or corticosteroids), etrasimod, golimumab, infliximab, mirikizumab, ozanimod, tofacitinib, upadacitinib, ustekinumab, and vedolizumab for adult patients with moderately to severely active UC who have experienced an inadequate response or failure to tolerate conventional therapy (ADT-naive subgroup) or ADT (i.e., tumour necrosis factor antagonists, vedolizumab, or tofacitinib) (ADT-experienced subgroup).1 The modelled population was aligned with the population enrolled in the QUASAR trial.2 However, the modelled population is narrower than the proposed Health Canada indication and reimbursement request, which do not restrict the use of guselkumab to those who have experienced an inadequate response or failure to tolerate conventional therapy or ADT.3
Guselkumab is available as a 100 mg/mL solution for injection in either a prefilled syringe or patient-controlled injector, as a 200 mg/2 mL solution for injection in a prefilled syringe or prefilled pen, and as a 200 mg/20 mL vial for IV infusion at a submitted price of $3,059.74.1 The recommended dosage, per the proposed product monograph, is 200 mg by IV in week 0, week 4, and week 8 followed by 100 mg by subcutaneous injection at week 16 and 100 mg every 8 weeks thereafter.3 The product monograph notes that a dose of 200 mg administered by subcutaneous injection at week 12 and 200 mg administered every 4 weeks thereafter may be considered for patients who do not show adequate therapeutic benefit to guselkumab or according to clinical judgment.3 Assuming 49.7% of patients received the lower maintenance dose (i.e., 100 mg every 8 weeks) and assuming wastage of unused product (i.e., no vial sharing), the sponsor estimated drug acquisition costs for guselkumab to be $32,252 in year 1 and $29,995 annually thereafter. Comparator costs ranged from $1,617 (for conventional therapy) to $37,060 (for vedolizumab) in year 1 and $1,370 (for conventional therapy) to $32,616 (for vedolizumab) annually in the maintenance period.
The clinical outcomes modelled were clinical response and clinical remission. The model simulated life-years, QALYs, and costs for each treatment over a lifetime time horizon (58 years), discounted at a rate of 1.5% per annum. The analysis was undertaken from the perspective of the Canadian public health care payer.
Model Structure
The sponsor’s analysis included a short-term induction phase and a longer-term maintenance phase (Appendix 2, Figure 1 and Figure 2), with the same model structure for both the ADT-naive subgroup and ADT‑experienced subgroup. Patients entered the induction phase and received guselkumab or a comparator. The length of induction differed across treatments (range, 8 to 12 weeks). At the end of the induction period, patients could experience either a response or remission and receive the same treatment during maintenance therapy, not experience a response to therapy and transition to another ADT, or transition to the death health state.
Following the induction phase, patients who had experienced remission or a response entered the maintenance phase in the corresponding health state and remained on their initial treatment. Each cycle (2‑week cycle length), a proportion of patients in these states was assumed to be at risk of experiencing a loss of response. The sponsor assumed that patients who do not experience a response to initial treatment or who experience the loss of the initial response will try up to 2 lines of subsequent therapy, with a portion of patients transitioning to conventional therapy each cycle. Patients who transition to conventional therapy may transition to the first surgical health state, after which they could experience surgical remission or complications. Patients who experienced surgical complications could receive a second surgery, after which they were assumed to not experience any further complications. All patients who undergo surgery were assumed to remain in the postsurgery health states for the remainder of the model’s time horizon. Patients could transition to death from any health state.
Model Inputs
Baseline patient characteristics in the model were aligned with those of the QUASAR induction trial (ADT‑naive subgroup: aged 39.6 years, weight of 71.4 kg, 57.1% male; ADT-experienced subgroup: aged 42.2 years, weight of 72.2 kg, 58.1% male).2 Patient weight was used to calculate treatment costs for therapies with weight-based dosing.
Transition probabilities between health states were derived from the sponsor-submitted network meta-analyses (NMAs).4 A constant risk of loss of response was assumed throughout the maintenance period to extrapolate beyond the trial period. The probabilities of undergoing surgery and postsurgery complications were derived from the literature.5,6 The rate of serious infections was obtained from the QUASAR trial for guselkumab and conventional therapy and from published sources for all other ADTs.7-16 Discontinuation of ADT was assumed to occur only due to a loss of response; it was assumed no patients discontinued ADT during the induction period.
The sponsor incorporated age- and sex-specific mortality risk in the model,17 as well as an increased risk of mortality for patients in the surgery health states compared with the general population in Canada (standardized mortality ratio = 1.3).18
Health state utility values were sourced from published literature and a utility decrement was included for serious infections.19-21
The costs considered in the submitted economic model included those associated with drug acquisition and administration, health care resource use, surgery, and serious infections (i.e., adverse events). Drug acquisition costs, including for guselkumab, were obtained from the 2024 McKesson Price list,22 and the recommended dosages were based on the Health Canada–approved monograph for each treatment. For treatments in which both a low dose and a high dose for maintenance are specified in the product monograph (i.e., guselkumab, adalimumab, infliximab, golimumab, vedolizumab, ustekinumab, tofacitinib, and upadacitinib), the proportion of patients receiving a low dose versus a high dose was based on the clinical expert input elicited by the sponsor and, for golimumab and upadacitinib, on the CADTH review of upadacitinib for rheumatoid arthritis.23 Administration costs for IV treatments were sourced from Tam et al.24 Infusion times for guselkumab were sourced from the QUASAR trial; comparator infusion times were sourced from their respective product monographs.10,11,25,26 For simplicity, the cost of concomitant therapies (e.g., immunomodulators) was not considered. Health care resource use frequencies were sourced primarily from the UK;27-29 costs were obtained from various sources.30-32 The costs of serious infections were derived from a previous CADTH submission and published literature.23,33
Summary of Sponsor’s Economic Evaluation Results
The base-case analyses were run probabilistically (3,000 iterations); the sensitivity analyses were conducted deterministically. The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
The submitted analysis was based on publicly available prices of the comparator treatments. Results from the base case of the submitted economic evaluation are presented in Table 3 and Table 4. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 2.
In the sponsor’s submitted base case for the ADT-naive subgroup, the expected total costs and QALYs over a lifetime horizon for guselkumab were $301,857 and 14.44, respectively (Table 3). In the sponsor’s submitted base case for the ADT‑experienced subgroup, the expected total costs and QALYs over a lifetime horizon for guselkumab were $282,610 and 13.62, respectively (Table 4). In both subgroups, guselkumab was dominated (i.e., guselkumab is more costly and less effective than at least 1 comparator). In the ADT‑naive subgroup, guselkumab was dominated by ustekinumab biosimilar and upadacitinib (greater costs: $27,749 and $12,641, respectively; fewer QALYs: 0.10 and 0.09, respectively). In the ADT‑experienced subgroup, guselkumab was dominated by ustekinumab biosimilar, etrasimod, and ozanimod (greater costs: $20,506, $23,076, and $586, respectively; fewer QALYs: 0.09, 0.17, and 0.19, respectively). In both subgroups, guselkumab had a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. Results were driven largely by drug acquisition costs and QALYs gained in the “remission” health state (refer to Table 6 and Table 7 for disaggregated results for the ADT‑naive and ADT‑experienced subgroups, respectively).
Table 3: Summary of the Sponsor’s Economic Evaluation Results, ADT-Naive Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 199,462 | 12.95 | Reference |
Tofacitinib generic | 250,774 | 14.13 | 43,412 vs. conventional therapy |
Ustekinumab biosimilar | 274,108 | 14.54 | 57,597 vs. tofacitinib generic |
Vedolizumab | 322,702 | 14.75 | 228,537 vs. ustekinumab biosimilar |
Dominated | |||
Etrasimod | 261,911 | 14.24 | Extendedly dominated by ustekinumab biosimilar |
Adalimumab biosimilar | 260,536 | 14.07 | Dominated by tofacitinib generic |
Golimumab | 270,937 | 14.22 | Dominated by etrasimod |
Ozanimod | 273,041 | 14.10 | Dominated by tofacitinib generic, golimumab, and etrasimod |
Infliximab biosimilar | 275,128 | 14.29 | Dominated by ustekinumab biosimilar |
Upadacitinib | 289,216 | 14.53 | Dominated by ustekinumab biosimilar |
Mirikizumab | 299,973 | 14.36 | Dominated by ustekinumab biosimilar |
Guselkumab | 301,857 | 14.44 | Dominated by ustekinumab biosimilar and upadacitinib |
ADT = advanced therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Table 4: Summary of the Sponsor’s Economic Evaluation Results, ADT-Experienced Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/ QALY) |
|---|---|---|---|
Conventional therapy | 190,783 | 12.34 | Reference |
Etrasimod | 259,534 | 13.79 | 47,653 vs. conventional therapy |
Upadacitinib | 309,742 | 14.41 | 80,037 vs. etrasimod |
Dominated | |||
Adalimumab biosimilar | 248,060 | 13.19 | Extendedly dominated by tofacitinib generic, etrasimod, and upadacitinib |
Tofacitinib generic | 248,217 | 13.40 | Extendedly dominated by etrasimod and upadacitinib |
Ozanimod | 282,023 | 13.81 | Extendedly dominated by upadacitinib |
Ustekinumab biosimilar | 262,103 | 13.71 | Dominated by etrasimod |
Vedolizumab | 270,932 | 13.34 | Dominated by tofacitinib generic, ustekinumab biosimilar, and etrasimod |
Mirikizumab | 274,933 | 13.52 | Dominated by ustekinumab biosimilar and etrasimod |
Guselkumab | 282,610 | 13.62 | Dominated by ustekinumab biosimilar, etrasimod, and ozanimod |
ADT = advanced therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted deterministic scenario analyses encompassing considerations such as alternative time horizons, discount rates, utility values, comparators in the ADT-experienced population, induction and maintenance response rates, the inclusion of a delayed response period, the exclusion of subsequent therapies, and the exclusion of adverse events. The results of the sponsor’s scenario analyses in both subgroups showed that guselkumab remained dominated (i.e., guselkumab is more costly and less effective than at least 1 comparator).
The sponsor conducted scenario analyses from a societal perspective for both the ADT-naive and ADT‑experienced subgroups. These analyses included additional costs associated with loss of productivity. Similar to the sponsor’s base-case analysis using a health care payer perspective, guselkumab was dominated in both the ADT‑naive and ADT‑experienced subgroups.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The comparative clinical efficacy and safety of guselkumab compared with other biologics is uncertain: There was no direct head-to-head evidence comparing guselkumab with other biologics used to treat UC in Canada. To inform the comparative efficacy estimate in the pharmacoeconomic analysis, the sponsor conducted NMAs that assessed the short-term comparative efficacy of guselkumab versus biologics in ADT-naive and ADT-experienced populations separately. According to the CDA-AMC Clinical Review, guselkumab was not statistically significantly better than any active comparators for: clinical remission in the induction analysis of the ADT nonfailure and ADT failure populations, clinical remission in the maintenance analysis of the ADT failure population, and clinical response in the maintenance analysis of the ADT failure population. As well, for clinical remission in the induction analysis of the ADT failure population, upadacitinib was favoured over guselkumab. For clinical remission in the maintenance analysis of the ADT nonfailure population, clinical response in the induction analysis of the ADT nonfailure and ADT failure populations, and clinical response in the maintenance analysis of the ADT nonfailure population, guselkumab was favoured over some comparators. Therefore, the NMA results indicate that guselkumab may be no different than, superior to, or inferior to some comparators, depending on the population and the outcome examined. Uncertainty regarding incremental benefit is demonstrated in the scatterplot results for guselkumab versus comparators (Appendix 2, Figure 3 and Figure 4), which highlight that, for most comparisons, guselkumab could lead to incremental benefit or harm. However, this conclusion is limited because, according to the CDA-AMC Clinical Review, no definitive conclusions could be drawn on the relative treatment effects of guselkumab versus other relevant comparators for the treatment of moderately to severely active UC. This was due to notable sources of heterogeneity for potential effect modifiers across the included studies (including trial design characteristics, patient eligibility criteria, baseline patient characteristics, and the definitions and methods for reporting outcomes). Additionally, the comparative safety of guselkumab relative to other ADTs is unknown because safety was not assessed in the sponsor’s NMAs.
Given the lack of direct evidence and limitations with the sponsor’s NMAs, the comparative efficacy and cost-effectiveness of guselkumab relative to other therapies currently reimbursed for adult patients with moderately to severely active UC who have experienced an inadequate response or failure to tolerate conventional therapy or ADT is highly uncertain. As such, it is uncertain whether guselkumab provides a net benefit relative to other therapies that are currently funded. CDA-AMC was unable to address this limitation in reanalyses.
Comparators were not aligned between the pharmacoeconomic analysis and the budget impact analysis (BIA): In the sponsor’s pharmacoeconomic analysis, conventional therapy was included as a comparator for guselkumab. According to the clinical expert feedback received for this review, given the number of existing biologic treatments available for UC, the proportion of patients who would receive conventional therapy as opposed to an advanced treatment is expected to be low. As such, the relevancy of conventional therapy as a comparator is highly uncertain. The uncertainty in the relevance of conventional therapy as a comparator is further highlighted in the sponsor’s BIA, which did not include conventional therapy as a comparator. As such, the BIA implies that the entry of guselkumab will not impact the proportion of patients receiving conventional therapy, meaning that conventional therapy will not be displaced by guselkumab.
Additionally, infliximab and golimumab were not included as comparators in the pharmacoeconomic analysis for the ADT-experienced population due to a lack of clinical data in this population.1 In the sponsor’s BIA, infliximab has a market share of 15%, 13%, and 11% in years 1, 2, and 3, respectively, indicating that it is available and being used in the ADT-experienced population. Infliximab’s relevance as a comparator in the ADT-experienced population was confirmed by the clinical expert feedback received by CDA-AMC for this review. While golimumab’s market share in the BIA was small, it was identified by the clinical experts and in the drug plan input as being a relevant comparator in the ADT-experienced population.
In a scenario where conventional therapy is not a comparator, guselkumab would continue to be dominated by ustekinumab and upadacitinib in an ADT-naive population, and by ustekinumab, etrasimod, and ozanimod in an ADT-experienced population. Therefore, although the relevance of conventional therapy as a comparator is uncertain, excluding such therapy is not expected to change the overall conclusions regarding the cost-effectiveness of guselkumab.
In the absence of evidence, the relative effectiveness of guselkumab versus infliximab and golimumab is unknown. Based on the sponsor’s submitted price for guselkumab and the public list prices for comparators, drug acquisition costs for guselkumab are expected to be higher than for infliximab and golimumab (Table 5). Therefore, the inclusion of infliximab and golimumab in the ADT-experienced analysis is unlikely to change the overall conclusions regarding the cost-effectiveness of guselkumab (i.e., guselkumab is expected to remain dominated by ustekinumab, etrasimod, and ozanimod).
The proportion of patients receiving high versus low dosing for guselkumab and comparators is uncertain: In the sponsor’s pharmacoeconomic analysis for guselkumab and comparators, the sponsor assumed that a proportion of patients would receive the low or high dose of a given treatment, which was meant to reflect dose escalations and de-escalations. The proportion of people receiving guselkumab 100 mg every 8 weeks versus 200 mg every 4 weeks was informed by the QUASAR trial and the opinion of the sponsor’s clinical expert.1,2 The distribution of patients across the low and high doses for comparator regimens was based on the opinion of the sponsor’s clinical expert and a previous CADTH review.1,23 This approach is uncertain for several reasons.
Firstly, the proportion of patients on the high-dose treatment differed between the pharmacoeconomic analysis and the BIA for guselkumab, despite both citing the same sources of information. In the ADT-naive population, 49.7% and 47.8% of patients received guselkumab 200 mg every 4 weeks in the CUA and BIA, respectively. In the ADT-experienced population, 50.3% and 53.3% of patients received guselkumab 200 mg every 4 weeks in the CUA and BIA, respectively. While the clinical expert feedback for this review noted it is reasonable to assume that half of all patients will be treated with high-dose guselkumab in clinical practice, the inconsistencies between the sponsor’s CUA and BIA must be noted because the proportions influence drug acquisition costs.
Secondly, in some instances, the proportions of patients using high versus low doses for comparators did not align with the clinical expert feedback received by CDA-AMC for this review. For example, for adalimumab, the experts predicted 50% of those in the ADT-naive population would receive the high dose rather than the 30% assumed by the sponsor; for vedolizumab, the experts predicted 50% of those in the ADT-naive population would receive the high dose rather than the 30% assumed by the sponsor and, for upadacitinib, the experts predicted 80% of those in the ADT-naive population would receive the high dose rather than the 44% assumed by the sponsor. Finally, the clinical expert feedback received by CDA-AMC for this review indicated some use of off-label dosing for some comparators. For golimumab, the experts indicated that approximately 10% of ADT-experienced patients would receive 100 mg every 2 weeks. For ustekinumab, the experts indicated that most patients, irrespective of ADT experience, would receive 90 mg every 4 weeks. Therefore, there is uncertainty in the dosing for comparator treatments, introducing uncertainty in comparator costs.
In the ADT-naive population, the overall conclusions regarding the cost-effectiveness of guselkumab are unlikely to change if the doses for comparators were adjusted. If a greater proportion of patients used the high dose for adalimumab, vedolizumab, and upadacitinib, guselkumab would remain dominated by ustekinumab. If all patients receiving ustekinumab received the 90 mg every 4 weeks dosage, guselkumab would remain dominated by upadacitinib. If both steps are combined, then guselkumab remains off the frontier because it would be extendedly dominated by tofacitinib.
In the ADT-experienced population, the overall conclusions regarding the cost-effectiveness of guselkumab are unlikely to change if the doses for comparators were adjusted. If all patients receiving ustekinumab received the 90 mg every 4 weeks dosage, then guselkumab would remain dominated by etrasimod and ozanimod.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently.
The sponsor’s model had additional issues: During the review process, additional issues were identified with the sponsor’s model but these were not prioritized, given the uncertainty in the comparative clinical evidence. For example, the lack of long-term evidence available, the remission utility value implying that those in remission with UC have a better quality of life than the general public in Canada, the assumption of no vial sharing, and uncertainty in the impact of subsequent therapy on efficacy and costs.
Addressing these limitations would not resolve the uncertainty in the clinical evidence that precludes drawing conclusions regarding the comparative safety and efficacy of guselkumab versus comparators.
Issues for Consideration
CADTH previously reviewed guselkumab for plaque psoriasis and psoriatic arthritis, both of which received a recommendation to reimburse with clinical criteria and/or conditions.34,35 In both reviews, the Canadian Drug Expert Committee recommended that guselkumab be reimbursed with the condition that the drug plan cost for guselkumab not exceed the drug plan cost of treatment with the least costly biologic reimbursed for the treatment of the respective conditions.34,35 In both reviews, the sponsor-submitted price for guselkumab was aligned with the submitted price for this review.34,35
Ustekinumab biosimilars, adalimumab biosimilar, infliximab biosimilar, vedolizumab, golimumab, ozanimod, tofacitinib, mirikizumab, and upadacitinib have successfully completed negotiations with the pan-Canadian Pharmaceutical Alliance for UC and are listed on public formularies;36 etrasimod is currently under active negotiation.36 Therefore, the price paid by the drug plans for comparators may be lower than the price incorporated in the sponsor’s pharmacoeconomic model.
According to the drug plan input received for this review, coverage of UC treatments varies across drug plans, and not all drug plans may cover all of the treatments considered in the sponsor’s analysis.
The sponsor’s proposed indication for guselkumab does not restrict use to patients who have experienced an inadequate response or failure to tolerate conventional therapy or ADT (i.e., the target population of the CUA). The clinical expert input received by CDA-AMC indicated that patients who had not been previously treated with conventional therapy would likely not be considered for treatment with guselkumab; however, the cost-effectiveness among patients who are naive to conventional therapy is unknown, owing to a lack of clinical data.
Overall Conclusions
The CDA-AMC Clinical Review found that, compared with placebo, treatment with guselkumab results in a clinically meaningful increase in clinical remission, endoscopic healing, clinical response and maintenance of clinical response, corticosteroid-free clinical remission (maintenance only), and health-related quality of life at 12 weeks of induction therapy and 44 weeks of maintenance therapy in adults with moderately to severely active UC. Although the proposed health Canada indication is for a broader patient population than the populations enrolled in the studies (i.e., patients who have experienced an inadequate response or were unable to tolerate conventional therapy or ADT), the clinical experts consulted by CDA-AMC did not consider the proposed indication to affect the generalizability of the findings. No new safety signals were identified and the safety of guselkumab was consistent with the known safety profile of the drug. Due to the limitations of the indirect treatment comparison, mostly attributed to heterogeneity across studies, no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators.
In the sponsor’s base case, guselkumab was dominated by comparators (in the ADT-naive population, by ustekinumab and upadacitinib; in the ADT-experienced population, by ustekinumab, etrasimod, and ozanimod). According to the CDA-AMC Clinical Review, the comparative efficacy of guselkumab versus other ADTs is uncertain. Given that no conclusions can be drawn on the relative efficacy and safety of guselkumab versus other relevant comparators, there is insufficient evidence to suggest the total treatment costs for guselkumab should be higher than other available biologics currently used in the treatment of moderately to severely active UC.
References
1.Janssen Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya/Tremfya I.V. (guselkumab) 200 mg/2mL solution for subcutaneous Injection, 100 mg/1mL solution for subcutaneous injection, and 200 mg/20 mL solution for intravenous infusion. October 21, 2024.
2.Janssen Research & Development LLC. Clinical Study Report: Maintenance Study, CNTO1959UCO3001, week 44. A Phase 2b/3, Randomized, Double-blind, Placebo-controlled, Parallel-group, Multicenter Protocol to Evaluate the Efficacy and Safety of Guselkumab in Participants with Moderately to Severely Active Ulcerative Colitis [internal sponsor's report]. February 20, 2024.
3.Janssen Inc. Tremfya (guselkumab injection): 100 mg/1 mL or 200 mg/2 mL in a pre-filled syringe, or 200 mg/2 mL pre-filled pen, sterile solution for injection; Tremfya One-Press (guselkumab injection): 100 mg/1 mL patient-controlled injector, sterille solution for injection; Tremfya I.V. (guselkumab injection): 200 mg/20 mL sterile solution in single-use vial, for intravenous Infusion [product monograph]. November 10, 2017. Updated September 3, 2025.
4.Janssen Inc. Efficacy of guselkumab versus available biologic therapies for the treatment of moderate to severe ulcerative colitis: A systematic review and network meta-analysis. Technical Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya/Tremfya I.V. (guselkumab) 200 mg/2mL solution for subcutaneous Injection, 100 mg/1mL solution for subcutaneous injection, and 200 mg/20 mL solution for intravenous infusion. October 8, 2024.
5.Briggs A, Claxton K, Sculpher M. Decision Modelling for Health Economic Evaluation. Oxford University Press; 2006.
6.Causey MW, Stoddard D, Johnson EK, et al. Laparoscopy impacts outcomes favorably following colectomy for ulcerative colitis: a critical analysis of the ACS-NSQIP database. Surg Endosc. 2013;27(2):603-9. doi: 10.1007/s00464-012-2498-7 PubMed
7.AbbVie Corporation. Humira (adalimumab): 50 mg/mL or 100 mg/mL, sterile solution, for subcutaneous injection [product monograph; sponsor supplied reference]. September 24, 2004. Accessed January 23, 2019.
8.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-76. doi: 10.1056/NEJMoa050516 PubMed
9.Janssen Inc. Simponi (golimumab): 50 mg / 0.5 mL or 100 mg / 1.0 mL, single-use sterile solution in a SmartJect autoinjector or pre-filled syringe, for subcutaneous injection; Simponi I.V. (golimumab): 50 mg / 4.0 mL, sterile solution in single-use vials, for intravenous infusion [product monograph; sponsor supplied reference]. November 6, 2018.
10.Takeda Canada Inc. Entyvio (vedolizumab): 300 mg / vial, sterile powder for solution for infusion, or 108 mg / 0.68 mL, solution for injection in pre-filled syringe or pen [product monograph; sponsor supplied reference]. January 29, 2015. Updated November 17, 2023. https://assets-dam.takeda.com/image/upload/v1702414583/legacy-dotcom/siteassets/en-ca/home/what-we-do/our-medicines/product-monographs/entyvio/ENTYVIO-PM-(EN).pdf
11.Janssen Inc. Stelara (ustekinumab): 45 mg/0.5 mL 90 mg/1.0 mL, sterile solution in single-use pre-filled syringe, for subcutaneous injection; Stelara I.V. (ustekinumab): 130 mg/26 mL, sterile solution in single-use vial, intravenous infusion [product monograph; sponsor supplied reference]. December 12, 2008. Updated January 5, 2023.
12.CADTH. Drug reimbursement review: mirikizumab (Omvoh) for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy, a biologic treatment, or a Janus kinase (JAK) inhibitor [sponsor supplied reference]. 2023. Updated February 9, 2024. https://www.cadth.ca/mirikizumab
13.Pfizer Canada ULC. Xeljanz (tofacitinib citrate): 5 mg or 10 mg, oral tablets; Xeljanz XR (tofacitinib citrate): 11 mg extended-release oral tablets [product monograph; sponsor supplied reference]. April 16, 2014. Updated May 9, 2022.
14.AbbVie Corporation. Rinvoq (upadacitinib): 15 mg, 30 mg, or 45 mg, extended-release oral tablets [product monograph; sponsor supplied reference]. December 23, 2019. Updated July 5, 2024. https://pdf.hres.ca/dpd_pm/00073108.PDF
15.Bristol-Myers Squibb Canada. Zeposia (ozanimod hydrochloride): 0.23 mg, 0.46 mg, and 0.92 mg, oral capsules [product monograph; sponsor supplied reference]. October 2, 2020. Updated August 28, 2024.
16.Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet. 2023;401(10383):1159-1171. doi: 10.1016/S0140-6736(23)00061-2 PubMed
17.Statistics Canada. Life Tables, Canada, Provinces and Territories (Tables: 84-537-X) [sponsor supplied reference]. 2019. https://www150.statcan.gc.ca/n1/en/catalogue/84-537-X
18.Jess T, Gamborg M, Munkholm P, Sorensen TI. Overall and cause-specific mortality in ulcerative colitis: meta-analysis of population-based inception cohort studies. Am J Gastroenterol. 2007;102(3):609-17. doi: 10.1111/j.1572-0241.2006.01000.x PubMed
19.Woehl A, Hawthorne AB, McEwan P. The Relation Between Disease Activity, Quality of Life and Health Utility In Patients With Ulcerative Colitis. Gut. 2008;57(Suppl 1):A1-A172.
20.Arseneau KO, Sultan S, Provenzale DT, et al. Do patient preferences influence decisions on treatment for patients with steroid-refractory ulcerative colitis? Clin Gastroenterol Hepatol. 2006;4(9):1135-42. doi: 10.1016/j.cgh.2006.05.003 PubMed
21.Stevenson M, Archer R, Tosh J, et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess. 2016;20(35):1-610. doi: 10.3310/hta20350 PubMed
22.Janssen Inc. McKesson Price List (Ontario) [sponsor supplied reference]. 2024.
23.CADTH. Drug Reimbursement Review pharmacoeconomic report: upadacitinib (Rinvoq) for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to or intolerance to methotrexate. March 2020. Accessed by sponsor, no date provided. https://www.cda-amc.ca/sites/default/files/cdr/pharmacoeconomic/sr0614-rinvoq-pharmacoeconomic-review-report.pdf
24.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. doi: 10.3747/co.20.1223 PubMed
25.Janssen Inc. Remicade (infliximab): 100 mg/vial, sterile lyophilized powder for solution, for intravenous injection [product monograph; sponsor supplied reference]. August 31, 2018.
26.Eli Lilly Canada Inc. Omvoh (mirikizumab): 100 mg/mL solution, in prefilled pens and prefilled syringes for subcutaneous injection or 20 mg/mL solution, in vial for intravenous infusion [product monograph; sponsor supplied reference]. July 20, 2023. https://pdf.hres.ca/dpd_pm/00071771.PDF
27.Tsai HH, Punekar YS, Morris J, Fortun P. A model of the long-term cost effectiveness of scheduled maintenance treatment with infliximab for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2008;28(10):1230-9. doi: 10.1111/j.1365-2036.2008.03839.x PubMed
28.Sandborn WJ, Reinisch W, Yan S, et al. Infliximab Reduces Ulcerative Colitis-Related Hospitalizations Requiring High-Dose Corticosteroids. American College of Gastroenterology, Las Vegas, Nevada, USA [sponsor supplied reference]. 2016.
29.Samuel S, Ingle SB, Dhillon S, et al. Cumulative incidence and risk factors for hospitalization and surgery in a population-based cohort of ulcerative colitis. Inflamm Bowel Dis. 2013;19(9):1858-66. doi: 10.1097/MIB.0b013e31828c84c5 PubMed
30.Ontario Ministry of Health. Schedule of Benefits for Physician Services, effective July 24, 2023 [sponsor supplied reference]. 2023. https://www.ontario.ca/page/ohip-schedule-benefits-and-fees
31.Sharara N, Adam V, Crott R, Barkun AN. The costs of colonoscopy in a Canadian hospital using a microcosting approach. Can J Gastroenterol. 2008;22(6):565-70. doi: 10.1155/2008/854984 PubMed
32.Coward S, Heitman SJ, Clement F, et al. Ulcerative colitis-associated hospitalization costs: a population-based study. Can J Gastroenterol Hepatol. 2015;29(7):357-62. doi: 10.1155/2015/627370 PubMed
33.Dubois L, Vogt KN, Davies W, Schlachta CM. Impact of an outpatient appendectomy protocol on clinical outcomes and cost: a case-control study. J Am Coll Surg. 2010;211(6):731-7. doi: 10.1016/j.jamcollsurg.2010.07.017 PubMed
34.CADTH. Drug Reimbursement Expert Review Committee final recommendation: guselkumab (Tremfya — Janssen Inc.). CADTH; 2018 Feb 21. Accessed July 4, 2022. https://www.cadth.ca/sites/default/files/cdr/complete/SR0530_Tremfya_complete_Feb-23-18.pdf
35.CADTH. Reimbursement recommendation: guselkumab (Tremfya). Can J Health Technol. 2022;2(12). doi: 10.51731/cjht.2022.517
36.pan-Canadian Pharmaceutical Alliance. Brand Name Drug Negotiations Status. 2025. Accessed January 31, 2025. https://www.pcpacanada.ca/negotiations
37.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed November 15, 2024. https://www.formulary.health.gov.on.ca/formulary/
38.CADTH. Reimbursement recommendation: etrasimod (Velsipity). Can J Health Technol. 2024;4(8). doi: 10.51731/cjht.2024.963
39.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Exceptional Access Program (EAP). 2024. Accessed November 15, 2024. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
40.CADTH. Drug Reimbursement Expert Review Committee draft recommendation: mirikizumab (Omvoh - Eli Lilly Canada). 2023. Accessed October 17, 2025. https://www.cadth.ca/sites/default/files/DRR/2023/SR0773%20Omvoh%20-%20Draft%20CADTH%20Recommendation_For%20posting%20August%2017%2C%202023.pdf
41.Government of Saskatchewan. Saskatchewan Drug Plan: search formulary. 2024. Accessed May 6, 2024. https://formulary.drugplan.ehealthsask.ca/SearchFormulary
42.Statistics Canada. Table 17-10-0005-01: Population estimates on July 1, by age and gender [sponsor supplied reference]. 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
43.Coward S, Benchimol EI, Bernstein CN, et al. Forecasting the Incidence and Prevalence of Inflammatory Bowel Disease: A Canadian Nationwide Analysis. Am J Gastroenterol. 2024. doi: 10.14309/ajg.0000000000002687 PubMed
44.Benchimol EI, Manuel DG, Guttmann A, et al. Changing age demographics of inflammatory bowel disease in Ontario, Canada: a population-based cohort study of epidemiology trends. Inflamm Bowel Dis. 2014;20(10):1761-9. doi: 10.1097/MIB.0000000000000103 PubMed
45.Melesse DY, Lix LM, Nugent Z, et al. Estimates of Disease Course in Inflammatory Bowel Disease Using Administrative Data: A Population-level Study. J Crohns Colitis. 2017;11(5):562-570. doi: 10.1093/ecco-jcc/jjw201 PubMed
46.Siffledeen J, Singh S, Shulman SM, et al. Effect of Suboptimal Disease Control on Patient Quality of Life: Real-World Data from the Observational IBD-PODCAST Canada Trial. Dig Dis Sci. 2024;69(5):1636-1648. doi: 10.1007/s10620-024-08313-z PubMed
47.IQVIA. PharmaStat [sponsor supplied reference]. Accessed September 2024. www.iqvia.com
48.Janssen Inc. Stelara (ustekinumab): 45 mg/0.5 mL 90 mg/1.0 mL, sterile solution in single-use pre-filled syringe, for subcutaneous injection; Stelara I.V. (ustekinumab): 130 mg/26 mL, sterile solution in single-use vial, intravenous infusion [product monograph; sponsor supplied reference]. December 12, 2008. Updated August 14, 2024.
49.Gagnon-Arpin I, Chen W, Leaver C. Understanding the Gap 2.0: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. The Conference Board of Canada; 2022. Accessed January 30, 2025. https://www.conferenceboard.ca/wp-content/uploads/2022/10/understanding-the-gap-2.0_2022.pdf
50.Janssen Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya/Tremfya I.V. (guselkumab) 200 mg/2mL solution for subcutaneous Injection, 100 mg/1mL solution for subcutaneous injection, and 200 mg/20 mL solution for intravenous infusion. October 21, 2024.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 5: CDA-AMC Cost Comparison Table for Moderately to Severely Active UC
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Guselkumab (Tremfya) | 100 mg/mL | Prefilled syringe, prefilled pen, or autoinjector for SC injection | 3,059.7400a | Induction: 200 mg by IV in weeks 0, 4, and 8 Maintenance: 100 mg by SC injection at week 16 and 100 mg every 8 weeks thereafter or 200 mg administered by SC injection at week 12 and every 4 weeks thereafter | Year 1: 63.02 to 109.28 Year 2+: 54.64 to 109.28 | Year 1: 23,016 to 39,913 Year 2+: 19,957 to 39,913 |
200 mg/20 mL | Vial for IV infusion | 3,059.7400a | ||||
S1P receptor agonists | ||||||
Etrasimod (Velsipity) | 2 mg | Tablet | 43.1000b | 2 mg daily | 43.10 | 15,742 |
Ozanimod (Zeposia) | 0.23 mg 0.46 mg 0.92 mg | Capsule | 68.4929 68.4929 68.4932 | Induction: 0.23 mg daily on days 1 to 4, 0.46 mg daily on days 5 to 7 Maintenance: 0.92 daily | 68.49 | 25,017 |
Biologics | ||||||
Adalimumab (Humira) | 20 mg/0.2 mL 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 397.0500c 794.1000c | Induction: 160 mg at week 0, then 80 mg at week 2 Maintenance: 40 mg every other week | Year 1: 65.42 Year 2+: 56.72 | Year 1: 23,880 Year 2+: 20,703 |
Adalimumab biosimilars | 20 mg /0.4 mL 40 mg/ 0.4 mL 40 mg/0.8 mL 80 mg/ 0.8 mL | Prefilled syringe or autoinjector for SC injection | 235.6350 471.2700 471.2700 942.5400 | Induction: 160 mg at week 0, then 80 mg at week 2 Maintenance: 40 mg every other week | Year 1: 38.82 Year 2+: 33.66 | Year 1: 14,172 Year 2+: 12,287 |
Golimumab (Simponi) | 50 mg/0.5 mL 100 mg/1 mL | Prefilled syringe or autoinjector for SC injection | 1,555.1700c 1,555.1700c | Induction: 200 mg at week 0, then 100 mg at week 2 Maintenance: 50 mg every 4 weeks | Year 1: 61.93 Year 2+: 55.54 | Year 1: 22,606 Year 2+: 20,273 |
Infliximab (Remicade) | 100 mg | Vial for IV infusion | 987.5600c | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 84.07 Year 2+: 70.54 | Year 1: 30,685 Year 2+: 25,747 |
Infliximab biosimilar (Inflectra) | 100 mg | Vial Powder for IV infusion | 525.0000 | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 44.69 Year 2+: 37.50 | Year 1: 16,313 Year 2+: 13,688 |
Infliximab biosimilar (Avsola, Renflexis) | 100 mg | Vial Powder for IV infusion | 493.0000 | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Year 1: 41.96 Year 2+: 35.21 | Year 1: 15,318 Year 2+: 12,853 |
Mirikizumab (Omvoh) | 300 mg/15 mL | Vial for IV infusion | 2,374.6600d | Induction: 300 mg IV infusion at weeks 0, 4, and 8 Maintenance: 200 mg SC injection every 4 weeks | 68.49 | 25,000 |
100 mg/mL | Autoinjector pen for SC injection or prefilled syringe for SC injection | 1,187.3300d | ||||
Ustekinumab (Stelara) | 130 mg/26.0 mL | Vial for IV infusion | 2,080.0000e | Induction (IV infusion): 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereafter Maintenance (SC injection): 90 mg every 8 weeks | Year 1: 86.53 Year 2+: 82.02 | Year 1: 31,584 Year 2+: 29,937 |
45 mg/0.5 mL 90 mg/1.0 mL | Prefilled syringe for SC injection | 4,593.1400 | ||||
Ustekinumab (Jamteki, Wezlana) | 45 mg/0.5 mL 90 mg/mL | Prefilled syringe or vial for SC injection | 2,755.8840 2,755.8840 | Induction (IV infusion): 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereafter Maintenance (SC injection): 90 mg every 8 weeks | Year 1: 58.53 Year 2+: 49.21 | Year 1: 21,362 Year 2+: 17,962 |
130 mg/26 mL | Vial for IV infusion | 1,248.0000 | ||||
Vedolizumab (Entyvio) | 300 mg | Vial for IV infusion | 3,571.9500c | Induction: 300 mg at weeks 0, 2, and 6 Maintenance: 300 mg every 8 weeks | Year 1: 72.40 Year 2+: 60.75 | Year 1: 26,425 Year 2+: 22,173 |
Vedolizumab SC (Entyvio) | 108 mg/0.68 mL | Prefilled syringe or pen for SC injection | 892.9800c | Induction: 300 mg by IV infusion at weeks 0 and 2 Maintenance: 108 mg by SC injection every 2 weeks | Year 1: 74.73 Year 2+: 60.75 | Year 1: 27,276 Year 2+: 22,173 |
JAK inhibitors | ||||||
Tofacitinib (generics) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | Induction: 10 mg twice daily for at least 8 weeks Maintenance: 5 mg or 10 mg twice daily | Year 1: 13.82 to 23.96 Year 2+: 11.98 to 23.96 | Year 1: 5,046 to 8,750f Year 2+: 4,375 to 8,750f |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Extended-release tablet | 51.6810 76.9600 101.8100 | Induction: 45 mg once daily for 8 weeks Maintenance: 15 mg or 30 mg once daily | Year 1: 59.37 to 80.77 Year 2+: 51.68 to 76.96 | Year 1: 21,671 to 29,482 Year 2+: 18,864 to 28,090 |
b.i.d. = twice a day; JAK = Janus kinase; S1P = sphingosine 1-phosphate; SC = subcutaneous; UC = ulcerative colitis.
Notes: All prices are from the Ontario Drug Benefit Formulary (accessed November 15, 2024),37 unless otherwise indicated, and do not include dispensing fees. Annual period assumes 365.25 days. Average weight is assumed to be 75 kg. Recommended doses are based on the respective product monographs.
aSponsor’s submitted price.1
bEtrasimod CADTH Reimbursement Review.38
cOntario Exceptional Access Program.39
dMirikizumab CADTH Reimbursement Review.40
eSaskatchewan Drug Plan formulary.41
fAssumes the CDA-AMC participating drug plans would reimburse the lowest-cost option (i.e., patients who require 10 mg b.i.d. would receive 2 × 5 mg b.i.d.). If drug plans reimburse the higher-cost 10 mg tablet, the annual cost of generic tofacitinib would be $15,455 per patient (daily cost: $42.34).
Appendix 2: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Figure 1: Decision Tree Model Structure
M = maintenance; UC = ulcerative colitis; w/o = without.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 3: Scatterplot for Guselkumab vs. Comparators — ADT‑Naive Subgroup
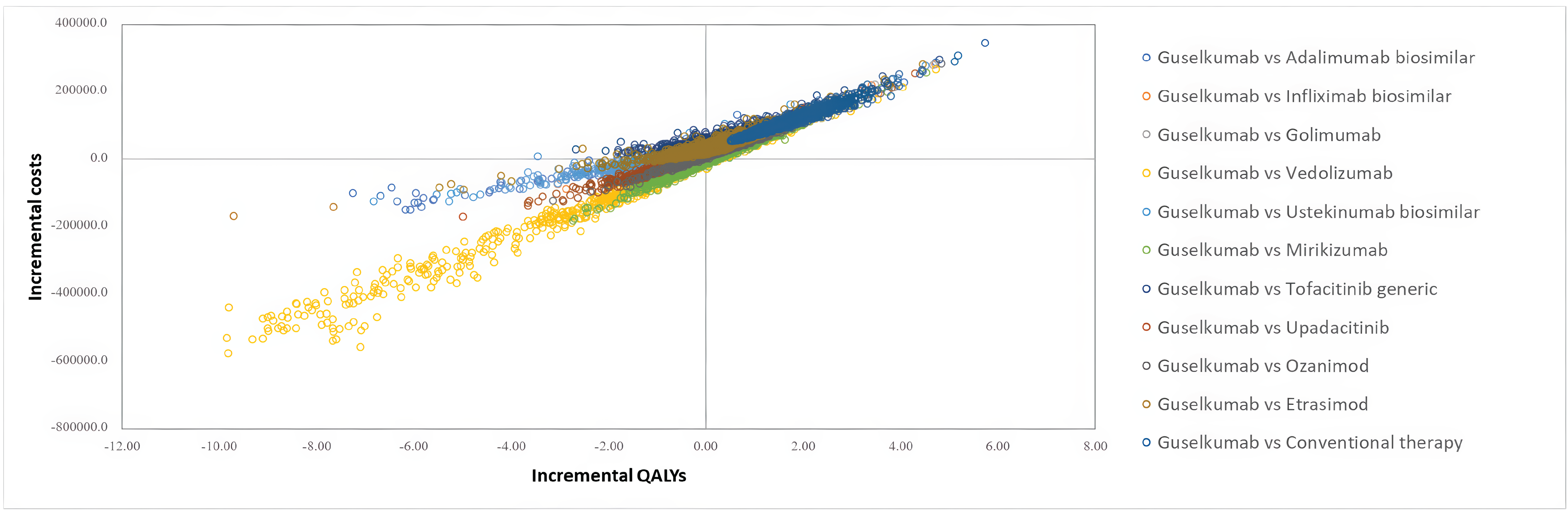
QALY = quality-adjusted life-year; vs = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 4: Scatterplot for Guselkumab vs. Comparators — ADT‑Experienced Subgroup
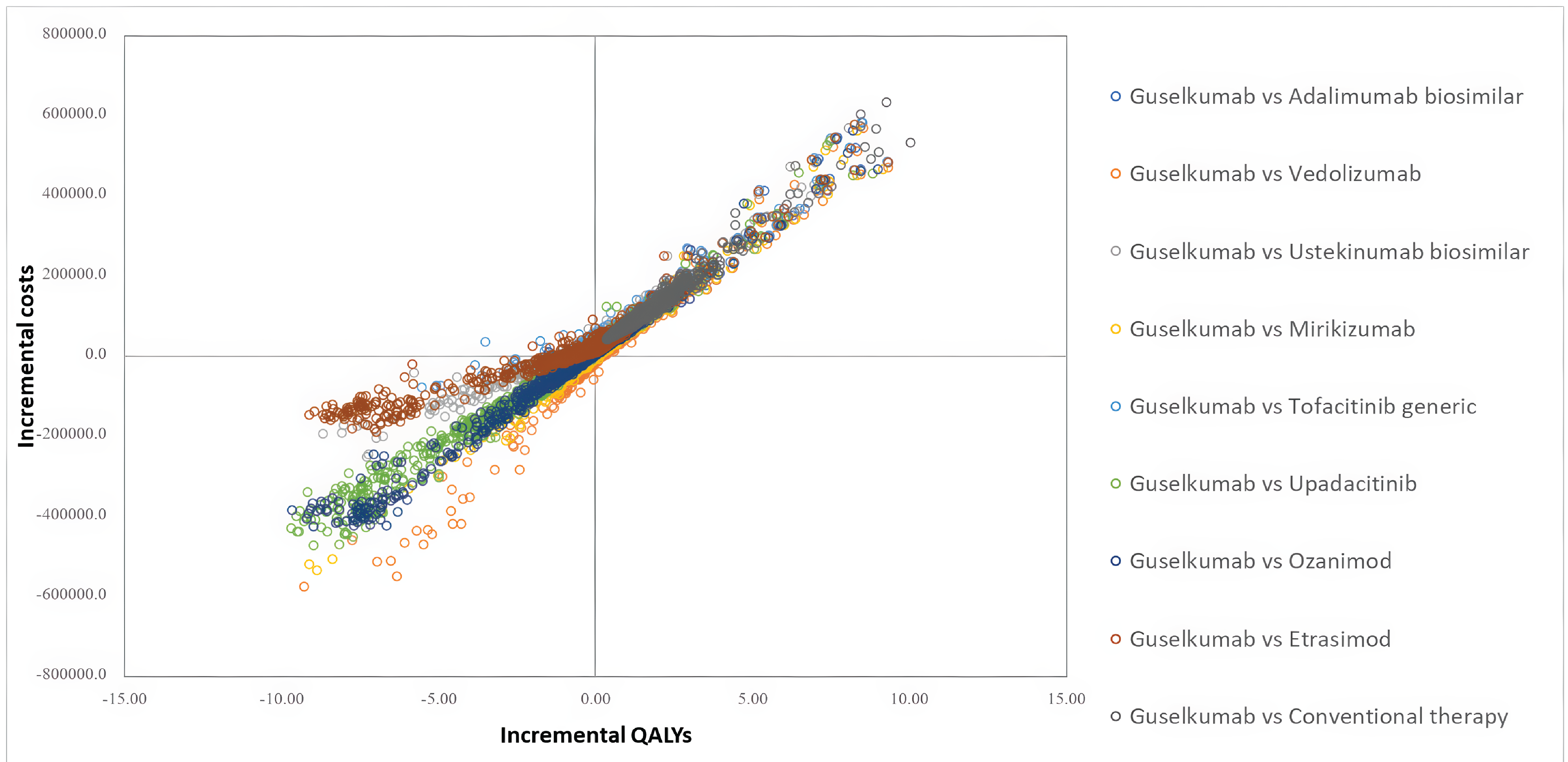
ADT = advanced therapy; QALY = quality adjusted life-year; vs = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Table 6: Disaggregated Summary of the Sponsor’s Economic Evaluation Results, ADT‑Naive Subgroup
Parameter | GUS | ADAa | IFXa | GOL | VDZ | USTa | MKZ | TFCb | UPA | OZA | ETR | CT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | ||||||||||||
Total | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 | 31.02 |
Remission | 3.33 | 2.43 | 2.86 | 2.85 | 3.81 | 3.51 | 2.88 | 2.61 | 3.33 | 2.54 | 3.04 | 0.25 |
Response | 0.53 | 0.65 | 0.72 | 0.53 | 0.84 | 0.60 | 0.90 | 0.59 | 0.81 | 0.57 | 0.33 | 0.21 |
Active UC | 23.62 | 24.24 | 23.84 | 23.99 | 22.93 | 23.40 | 23.69 | 24.14 | 23.38 | 24.23 | 24.02 | 26.28 |
First surgery | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.10 |
Post first surgery remission | 1.42 | 1.47 | 1.44 | 1.45 | 1.38 | 1.41 | 1.42 | 1.46 | 1.40 | 1.47 | 1.45 | 1.67 |
Post first surgery complications | 1.84 | 1.93 | 1.87 | 1.90 | 1.79 | 1.82 | 1.85 | 1.91 | 1.82 | 1.92 | 1.89 | 2.26 |
Second surgery | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
Post second surgery remission | 0.18 | 0.20 | 0.19 | 0.19 | 0.18 | 0.18 | 0.19 | 0.19 | 0.18 | 0.20 | 0.19 | 0.24 |
Discounted QALYs | ||||||||||||
Total | 14.44 | 14.07 | 14.29 | 14.22 | 14.75 | 14.54 | 14.36 | 14.13 | 14.53 | 14.10 | 14.24 | 12.95 |
Remission | 2.90 | 2.11 | 2.49 | 2.48 | 3.31 | 3.05 | 2.50 | 2.27 | 2.90 | 2.21 | 2.65 | 0.22 |
Response | 0.40 | 0.50 | 0.55 | 0.41 | 0.64 | 0.46 | 0.68 | 0.45 | 0.62 | 0.43 | 0.25 | 0.16 |
Active UC | 9.43 | 9.67 | 9.51 | 9.58 | 9.15 | 9.34 | 9.45 | 9.64 | 9.33 | 9.67 | 9.59 | 10.49 |
First surgery | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 |
Post first surgery remission | 1.01 | 1.05 | 1.03 | 1.04 | 0.98 | 1.01 | 1.02 | 1.05 | 1.00 | 1.05 | 1.04 | 1.19 |
Post first surgery complications | 0.63 | 0.66 | 0.64 | 0.65 | 0.61 | 0.62 | 0.63 | 0.65 | 0.62 | 0.65 | 0.64 | 0.77 |
Second surgery | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Post second surgery remission | 0.13 | 0.14 | 0.14 | 0.14 | 0.13 | 0.13 | 0.13 | 0.14 | 0.13 | 0.14 | 0.14 | 0.17 |
AEs | –0.10 | –0.10 | –0.11 | –0.10 | –0.11 | –0.11 | –0.10 | –0.10 | –0.11 | –0.10 | –0.10 | –0.09 |
Discounted costs ($) | ||||||||||||
Total | 301,857 | 260,536 | 275,128 | 270,937 | 322,702 | 274,108 | 299,973 | 250,774 | 289,216 | 273,041 | 261,911 | 199,462 |
Tx acquisition | 156,518 | 111,139 | 121,850 | 123,260 | 177,861 | 129,755 | 154,215 | 102,216 | 144,993 | 124,302 | 114,762 | 36,990 |
Index tx | 71,069 | 24,141 | 35,707 | 36,665 | 94,778 | 44,810 | 68,569 | 15,282 | 60,036 | 37,228 | 28,294 | 1,186 |
Sub tx 1 | 27,175 | 27,507 | 27,344 | 27,451 | 26,452 | 27,040 | 27,228 | 27,533 | 27,056 | 27,564 | 27,419 | 0 |
Sub tx 2 | 26,648 | 26,979 | 26,816 | 26,921 | 25,930 | 26,512 | 26,700 | 27,003 | 26,530 | 27,034 | 26,890 | 0 |
CT | 31,625 | 32,512 | 31,984 | 32,223 | 30,701 | 31,393 | 31,718 | 32,398 | 31,371 | 32,475 | 32,158 | 35,804 |
Tx admin | 1,154 | 506 | 6,880 | 505 | 2,969 | 716 | 828 | 507 | 498 | 507 | 505 | 0 |
Disease management | 130,456 | 134,514 | 132,052 | 132,937 | 127,485 | 129,487 | 131,150 | 133,781 | 129,380 | 134,182 | 132,678 | 147,840 |
Surgery | 8,898 | 9,153 | 9,002 | 9,072 | 8,645 | 8,833 | 8,926 | 9,121 | 8,825 | 9,143 | 9,051 | 10,088 |
AEs | 4,831 | 5,223 | 5,344 | 5,162 | 5,743 | 5,317 | 4,854 | 5,149 | 5,520 | 4,907 | 4,916 | 4,544 |
ADA = adalimumab; admin = administration; AE = adverse event; CT = conventional therapy; ETR = etrasimod; GOL = golimumab; GUS = guselkumab; IFX = infliximab; MKZ = mirikizumab; OZA = ozanimod; sub = subsequent; TFC = tofacitinib; tx = treatment; UC = ulcerative colitis; UPA = upadacitinib; UST = ustekinumab; VDZ = vedolizumab.
aBiosimilar.
bGeneric.
Source: Sponsor’s pharmacoeconomic submission.1
Table 7: Disaggregated Summary of the Sponsor’s Economic Evaluation Results, ADT‑Experienced Subgroup
Parameter | GUS | ADAa | VDZ | USTa | MKZ | TFCb | UPA | OZA | ETR | CT |
|---|---|---|---|---|---|---|---|---|---|---|
Discounted LYs | ||||||||||
Total | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 | 29.74 |
Remission | 2.55 | 1.92 | 2.10 | 2.78 | 2.49 | 2.16 | 4.08 | 2.42 | 2.91 | 2.55 |
Response | 0.68 | 0.27 | 0.48 | 0.65 | 0.49 | 0.58 | 1.00 | 1.40 | 0.69 | 0.68 |
Active UC | 23.14 | 23.98 | 23.67 | 22.96 | 23.35 | 23.54 | 21.57 | 22.63 | 22.81 | 23.14 |
First surgery | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.09 | 0.08 | 0.09 | 0.09 | 0.09 |
Post first surgery remission | 1.37 | 1.44 | 1.41 | 1.36 | 1.38 | 1.40 | 1.27 | 1.34 | 1.35 | 1.37 |
Post first surgery complications | 1.73 | 1.84 | 1.80 | 1.72 | 1.75 | 1.78 | 1.58 | 1.70 | 1.71 | 1.73 |
Second surgery | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
Post second surgery remission | 0.17 | 0.18 | 0.18 | 0.17 | 0.17 | 0.17 | 0.15 | 0.16 | 0.17 | 0.17 |
Discounted QALYs | ||||||||||
Total | 13.62 | 13.19 | 13.34 | 13.71 | 13.52 | 13.40 | 14.41 | 13.81 | 13.79 | 13.62 |
Remission | 2.22 | 1.67 | 1.82 | 2.42 | 2.17 | 1.88 | 3.55 | 2.10 | 2.53 | 2.22 |
Response | 0.52 | 0.21 | 0.36 | 0.50 | 0.37 | 0.44 | 0.76 | 1.06 | 0.53 | 0.52 |
Active UC | 9.24 | 9.58 | 9.46 | 9.17 | 9.33 | 9.41 | 8.62 | 9.04 | 9.11 | 9.24 |
First surgery | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.04 | 0.03 | 0.04 | 0.04 | 0.04 |
Post first surgery remission | 0.98 | 1.03 | 1.01 | 0.97 | 0.99 | 1.00 | 0.91 | 0.96 | 0.97 | 0.98 |
Post first surgery complications | 0.59 | 0.62 | 0.61 | 0.58 | 0.60 | 0.61 | 0.54 | 0.58 | 0.58 | 0.59 |
Second surgery | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Post second surgery remission | 0.12 | 0.13 | 0.13 | 0.12 | 0.12 | 0.12 | 0.11 | 0.12 | 0.12 | 0.12 |
AEs | –0.09 | –0.10 | –0.10 | –0.10 | –0.09 | –0.10 | –0.11 | –0.09 | –0.09 | –0.09 |
Discounted costs ($) | ||||||||||
Total | 282,610 | 248,060 | 270,932 | 262,103 | 274,933 | 248,217 | 309,742 | 282,023 | 259,534 | 190,783 |
Tx acquisition | 141,959 | 102,811 | 126,343 | 122,410 | 133,425 | 105,419 | 177,540 | 144,012 | 120,983 | 35,743 |
Index tx | 51,472 | 9,783 | 34,603 | 32,381 | 42,328 | 13,576 | 93,218 | 55,496 | 31,987 | 922 |
Sub tx 1 | 30,045 | 30,710 | 30,289 | 29,895 | 30,200 | 30,409 | 27,965 | 29,379 | 29,515 | 0 |
Sub tx 2 | 29,406 | 30,068 | 29,650 | 29,257 | 29,562 | 29,770 | 27,350 | 28,758 | 28,889 | 0 |
CT | 31,036 | 32,251 | 31,800 | 30,878 | 31,335 | 31,665 | 29,007 | 30,380 | 30,592 | 34,821 |
Tx admin | 1,207 | 566 | 1,515 | 769 | 883 | 560 | 518 | 541 | 544 | 0 |
Disease management | 126,039 | 130,751 | 129,133 | 125,231 | 127,107 | 128,420 | 118,180 | 124,276 | 124,707 | 140,858 |
Surgery | 8,747 | 9,096 | 8,970 | 8,701 | 8,831 | 8,929 | 8,164 | 8,561 | 8,626 | 9,812 |
AEs | 4,657 | 4,837 | 4,972 | 4,993 | 4,687 | 4,889 | 5,340 | 4,633 | 4,674 | 4,370 |
ADA = adalimumab; admin = administration; AE = adverse event; CT = conventional therapy; ETR = etrasimod; GUS = guselkumab; MKZ = mirikizumab; OZA = ozanimod; sub = subsequent; TFC = tofacitinib; tx = treatment; UC = ulcerative colitis; UPA = upadacitinib; UST = ustekinumab; VDZ = vedolizumab.
aBiosimilar.
bGeneric.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 3: Submitted BIA and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 8: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
ADT = advanced therapy; BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Summary of Sponsor’s BIA
In the submitted BIA, the sponsor assessed the budget impact of reimbursing guselkumab for adult patients with moderately to severely active UC who are eligible for ADT. The BIA was undertaken from the perspective of a public payer in Canada over a 3-year time horizon (2025 to 2027) using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits Program. The sponsor’s analysis included drug acquisition costs. Key inputs to the BIA are documented in Table 9.
The sponsor’s BIA included the following key assumptions:
Only patients who are eligible for ADT are included in the target population.
No adult patients with moderately to severely active UC who are eligible for ADT are currently treated with conventional therapy.
Previous claims-based data for ADTs with at least 1 claim and an approved indication for UC are representative of the current proportion of patients with public drug plan coverage.
The proportion of patients treated with the high-dose maintenance regimen for applicable comparators in the real-world is reflective of the clinical expert input received by the sponsor.
The proportion of patients treated with the high-dose maintenance regimen for guselkumab in the real-world is reflective of the randomization split in the QUASAR clinical trial.
The proportion of vedolizumab IV versus the vedolizumab subcutaneous maintenance dose is 80% versus 20%, respectively.
The introduction of guselkumab to the Canadian market is not expected to increase the total number of UC patients eligible for treatment with ADTs, nor take market share from patients only receiving conventional therapy.
If a biosimilar or generic product is available on public drug plans, the brand name therapy has no market share.
Table 9: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Target population | ||
Age category | Aged 18 to 64 years | Aged 65+ years |
Proportion of patients in each age category | 76.8%42 | 23.2%42 |
Prevalence | ||
Prevalence growth | 2.36%43 | 0.27%43 |
Proportion of patients with moderate to severe disease | 32.44%45 | |
Proportion of diagnosed patients who are eligible for ADT | 59.21%46 | |
Proportion of patients who are ADT-naive vs. ADT-experienced | ADT-naive = 53.33%; ADT-experienced = 46.67% | |
Proportion of the population with public drug plan coverage | 54.95%47 | |
Number of patients eligible for drug under review | 14,264 / 15,100 / 15,986 | |
Market uptake (3 years) | ||
Uptake (reference scenario) Ustekinumab biosimilar Adalimumab biosimilar Infliximab biosimilar Vedolizumab Golimumab Ozanimod Tofacitinib generic Mirikizumab Upadacitinib Etrasimod | ADT-naive 1.4% / 1.5% / 1.6% 0.8% / 0.7% / 0.6% 27.1% / 28.2% / 29.1% 55.2% / 52.7% / 49.1% 0.3% / 0.3% / 0.2% 0.2% / 0.3% / 0.3% 0.7% / 0.6% / 0.4% 2.4% / 2.9% / 3.4% 11.7% / 12.5% / 14.9% 0.2% / 0.3% / 0.3% | ADT-experienced 9.2% / 9.8% / 9.2% 5.4% / 4.5% / 3.6% 14.6% / 13.2% / 11.2% 14.1% / 13.8% / 13.0% 0.3% / 0.2% / 0.1% 0.2% / 0.2% / 0.3% 0.6% / 0.5% / 0.3% 8.7% / 9.9% / 10.5% 46.8% / 47.4% / 51.5% 0.2% / 0.2% / 0.3% |
Uptake (new drug scenario) Guselkumab Ustekinumab biosimilar Adalimumab biosimilar Infliximab biosimilar Vedolizumab Golimumab Ozanimod Tofacitinib generic Mirikizumab Upadacitinib Etrasimod | ADT-naive 0.3% / 2.4% / 3.7% 1.4% / 1.5% / 1.5% 0.8% / 0.7% / 0.6% 27.0% / 27.5% / 28.1% 55.0% / 51.5% / 47.3% 0.3% / 0.3% / 0.2% 0.2% / 0.3% / 0.3% 0.7% / 0.6% / 0.4% 2.4% / 2.8% / 3.3% 11.6% / 12.2% / 14.4% 0.2% / 0.3% / 0.3% | ADT-experienced 1.6% / 6.6% / 9.4% 9.1% / 9.2% / 8.3% 5.3% / 4.2% / 3.3% 14.3% / 12.3% / 10.1% 13.9% / 12.9% / 11.8% 0.3% / 0.2% / 0.1% 0.2% / 0.2% / 0.2% 0.6% / 0.5% / 0.3% 8.6% / 9.3% / 9.5% 46.0% / 44.3% / 46.7% 0.2% / 0.2% / 0.2% |
Annual per-patient cost of treatmenta | ||
Guselkumab Ustekinumab biosimilar Adalimumab biosimilar Infliximab biosimilar Vedolizumab Golimumab Ozanimod Tofacitinib generic Mirikizumab Upadacitinib Etrasimod | Year 1 = $31,423; year 2+ = $29,904 Year 1 = $18,734; year 2+ = $17,615 Year 1 = $18,216; year 2+ = $17,073 Year 1 = $18,967; year 2+ = $17,860 Year 1 = $34,145; year 2+ = $30,523 Year 1 = $20,995; year 2+ = $20,217 Year 1 = $24,932; year 2+ = $24,932 Year 1 = $7,189; year 2+ = $6,910 Year 1 = $30,871; year 2+ = $30,871 Year 1 = $26,171; year 2+ = $24,192 Year 1 = $15,688; year 2+ = $15,688 | |
ADT = advanced therapy; NA = not applicable; vs. = versus.
Note: Branded ustekinumab, branded adalimumab, branded infliximab, branded tofacitinib, risankizumab, and conventional therapy were included but were assumed to have no market share.
aThe following proportion of patients are assumed to receive the high-dose maintenance regimen in the ADT-naïve population: guselkumab (47.8%), ustekinumab biosimilar (95%), adalimumab biosimilar (30%), infliximab biosimilar (30%), vedolizumab (30%), golimumab (100%), ozanimod (NA), tofacitinib generic (44%), mirikizumab (NA), upadacitinib (44.4%), etrasimod (NA). In the ADT-experienced population, the following proportion of patients received the high-dose maintenance regimen: guselkumab (53.3%), ustekinumab biosimilar (95%), adalimumab biosimilar (50%), infliximab biosimilar (50%), vedolizumab (50%), golimumab (100%), ozanimod (NA), tofacitinib generic (75%), mirikizumab (NA), upadacitinib (75%), etrasimod (NA). NA indicates that only 1 maintenance dose is specified in the product monograph.
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the 3-year budget impact of reimbursing guselkumab for adult patients with moderately to severely active UC who are eligible for ADT will be $10,194,289 (year 1: $863,116; year 2: $3,649,218; year 3: $5,681,955).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Drug acquisition costs are highly uncertain: In the sponsor’s submitted BIA, annual treatment costs were calculated based on the weighted average of the proportion of patients treated with a “low” versus “high” maintenance dose for applicable treatments (i.e., when a higher maintenance dose is specified in the product monograph [i.e., guselkumab, ustekinumab, adalimumab, infliximab, vedolizumab, golimumab, tofacitinib, and upadacitinib]). With the exception of ozanimod, mirikizumab, and etrasimod, guselkumab and all other comparator product monographs specify an increased maintenance dose. For comparator treatments, the sponsor estimated the proportion of patients receiving the higher maintenance dose based on clinical expert opinion. First, the sponsor assumed that in the ADT-naive subgroup, 30% of patients receive the higher maintenance dose of adalimumab (40 mg weekly) and vedolizumab (300 mg every 4 weeks) and 44% receive the higher maintenance dose of upadacitinib (30 mg daily). Clinical expert input commented that in clinical practice, approximately 50% of patients receive the higher maintenance dose of adalimumab and vedolizumab and approximately 80% for upadacitinib. Therefore, the proportion of patients receiving higher maintenance doses for comparator regimens may be higher than estimated by the sponsor.
Second, the sponsor assumed that among patients treated with golimumab, 100% receive 100 mg every 4 weeks and 0% received 50 mg every 4 weeks. Clinical expert input commented that in the ADT-experienced subgroup, approximately 10% of patients are treated with 100 mg golimumab every 2 weeks in clinical practice. Similarly, the sponsor assumed that 95% of all patients treated with ustekinumab received 90 mg every 8 weeks. However, clinical expert input noted that almost all patients in Canada are treated with 90 mg every 4 weeks. Therefore, higher maintenance doses may be used in clinical practice for golimumab and ustekinumab than estimated by the sponsor. CDA-AMC notes that the alternative maintenance dosages suggested by clinical experts consulted by CDA-AMC for this review exceed the doses specified in the respective product monograph.9,48
Lastly, both the sponsor and CDA-AMC analyses are based on publicly available list prices for all comparators. Because adalimumab, vedolizumab, ustekinumab, tofacitinib, upadacitinib, infliximab, and golimumab have gone through negotiations with the pan-Canadian Pharmaceutical Alliance, the prices paid by public drug plans are not known.
CDA-AMC conducted a scenario analysis in which the dose mix suggested by the clinical experts consulted by CDA‑AMC was adopted. In that scenario, the following parameters were assumed:
95% of patients treated with ustekinumab were treated with 90 mg every 4 weeks and the remaining 5% were treated with 90 mg every 8 weeks
50% of patients treated with adalimumab were treated with 40 mg weekly and 50% were treated with 40 mg every 2 weeks
in the ADT-experienced population only, 10% of patients treated with golimumab were treated with 100 mg every 2 weeks and 90% were treated with 100 mg every 4 weeks
50% of patients treated with vedolizumab were treated with 300 mg every 4 weeks and 50% were treated with 300 mg every 8 weeks
75% of patients treated with upadacitinib were treated with 30 mg daily and 25% were treated with 15 mg daily.
In its reanalysis, CDA-AMC was unable to adjust for the prices paid by public drug plans.
The full Health Canada population was not modelled: The sponsor’s proposed indication is for adults with moderately to severely active UC; this indication does not restrict the use of guselkumab to patients who are eligible for ADT. However, the sponsor’s epidemiologic approach assumes that only patients eligible for ADT will be treated with guselkumab. The clinical expert input confirmed that guselkumab will likely be used only among patients who are eligible for ADT and found the sponsor’s estimate of the proportion of patients with moderate to severe UC who are eligible for ADT to be reasonable (59.21%). However, the inclusion of this restricts the size of the eligible population to a population smaller than the sponsor’s proposed indication.
CDA-AMC conducted a scenario analysis that captured the full proposed Health Canada indication (i.e., for the treatment of all adult patients with moderately to severely active UC, rather than only patients who are eligible for ADT).
The proportion of patients eligible for public drug coverage is uncertain: The sponsor estimated that 54.95% of patients in both the 18 to 64 years old cohort and 65 and older cohort had public drug coverage based on previous claims-based data for ADTs with at least 1 claim and an approved indication for UC.47 CDA-AMC was unable to validate this proportion, adding uncertainty to this estimate. As well, CDA-AMC notes that the sponsor may have underestimated the proportion of patients eligible for public drug coverage specifically for adults 65 and older as published literature estimates the proportion of patients to be eligible for public drug coverage aged 65 and older in Canada as 98.8%.49
CDA-AMC conducted a scenario analysis that assumed the proportion of patients aged 65 and older eligible for public drug coverage was 98.8%.
The market uptake of guselkumab may be underestimated: The sponsor’s submitted BIA indicated that guselkumab would result in a market uptake in the ADT-naive subgroup of 0.3%, 2.4%, and, 3.7% in year 1, year 2, and year 3, respectively, and 1.6%, 6.6%, and, 9.4% in year 1, year 2, and year 3, respectively, in the ADT-experienced subgroup.50 Market uptake was estimated based on the sponsor’s internal forecasting and guselkumab was assumed to capture market shares proportionally across all comparators. However, CDA-AMC obtained clinical expert feedback indicating that the market uptake of both subgroups may be underestimated and that the uptake of guselkumab may be faster than assumed by the sponsor. Experts anticipate the majority of guselkumab’s market capture to be taken from vedolizumab in the ADT-naive subgroup due to the 2 treatments having a comparable safety profile. In the ADT-experienced subgroup, experts noted that guselkumab will likely hold a higher market share than ustekinumab in years 2 and 3 of the BIA as ustekinumab is rarely used as a front-line therapy and thus guselkumab will likely supplant ustekinumab.
CDA-AMC did not adjust for this limitation in reanalysis owing to a lack of alternative data.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
The market share estimates in the reference scenario do not align with clinical expectations: In the sponsor’s submitted BIA, the sponsor assumed that ozanimod held a small market share (under 0.4% in both the ADT-naive and ADT-experienced subgroups). CDA-AMC obtained clinical expert feedback that suggested the ozanimod is not used in clinical practice and should likely have a market share of 0.0%. It was noted that the market share captured by ozanimod in the sponsor’s BIA would likely be transplanted by etrasimod. CDA-AMC notes that assuming etrasimod captures the entire market share held by ozanimod in both the reference and new drug scenarios has minimal impact (i.e., less than a 0.5% increase) on the sponsor’s submitted base case.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
CDA-AMC Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA (i.e., dose mix, market shares, and market uptake), the sponsor’s submitted base case was maintained. CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the sponsor’s base case (results are provided in Table 10):
aligning the eligible population size with the proposed Health Canada indication (i.e., not restricting to those eligible for ADT)
aligning the dose mix for comparator treatments with clinical expert input
aligning the proportion of patients aged 65 and older eligible for public drug coverage with published literature.
Table 10: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 363,416,836 | 355,681,293 | 375,801,522 | 398,150,062 | 1,129,632,877 |
New drug | 363,416,836 | 356,544,409 | 379,450,740 | 403,832,018 | 1,139,827,166 | |
Budget impact | 0 | 863,116 | 3,649,218 | 5,681,955 | 10,194,289 | |
CDA-AMC scenario analysis 1: Full population for the proposed Health Canada indication | Reference | 613,770,656 | 600,706,183 | 634,687,015 | 672,431,216 | 1,907,824,415 |
New drug | 613,770,656 | 602,163,890 | 640,850,139 | 682,027,408 | 1,925,041,436 | |
Budget impact | 0 | 1,457,707 | 6,163,123 | 9,596,191 | 17,217,021 | |
CDA-AMC scenario analysis 2: Dose mix | Reference | 387,610,677 | 384,723,912 | 407,073,674 | 430,599,120 | 1,222,396,706 |
New drug | 387,610,677 | 385,277,520 | 409,316,948 | 433,975,407 | 1,228,569,875 | |
Budget impact | 0 | 553,608 | 2,243,274 | 3,376,287 | 6,173,169 | |
CDA-AMC scenario analysis 3: Public drug coverage | Reference | 432,668,800 | 423,459,188 | 447,413,487 | 474,020,719 | 1,777,562,194 |
New drug | 432,668,800 | 424,486,777 | 451,758,092 | 480,785,416 | 1,789,699,086 | |
Budget impact | 0 | 1,027,589 | 4,344,605 | 6,764,697 | 12,136,892 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@cda-amc.ca.