Drugs, Health Technologies, Health Systems
Reimbursement Review
Cipaglucosidase Alfa (Pombiliti) With Miglustat (Opfolda)
Sponsor: Amicus Therapeutics Canada Inc.
Therapeutic area: Pompe disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
6MWD
6-minute walk distance
6MWT
6-minute walk test
AE
adverse event
ANCOVA
analysis of covariance
BiPAP
bilevel positive airway pressure
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
CrI
credible interval
DIC
deviance information criterion
EAMS
Early Access to Medicines Scheme
ERT
enzyme replacement therapy
FVC
forced vital capacity
GAA
acid alpha-glucosidase
GRADE
Grading of Recommendations Assessment, Development and Evaluation
GSGC
gait, stair, Gower manoeuvre, and chair
IAR
infusion-associated reaction
IOPD
infantile-onset Pompe disease
IPD
individual patient-level data
ITC
indirect treatment comparison
ITT
intention to treat
ITT-LOCF
intention-to-treat last observation carried forward
ITT-OBS
intention-to-treat observed
LOPD
late-onset Pompe disease
LS
least squares
MDC
Muscular Dystrophy Canada
MID
minimal important difference
ML-NMR
multilevel network meta-regression
MMRM
mixed model for repeated measures
MMT
manual muscle test
NMA
network meta-analysis
OLE
open-label extension
PROMIS
Patient-Reported Outcomes Measurement Information System
RCT
randomized controlled trial
rhGAA
recombinant human acid alpha-glucosidase
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
TEAE
treatment-emergent adverse event
TUG
Timed Up and Go
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Cipaglucosidase alfa for injection (Pombiliti), lyophilized powder, 105 mg/vial, IV infusion in combination with miglustat capsules (Opfolda), 65 mg, oral capsules |
Sponsor | Amicus Therapeutics Canada Inc. |
Indication | Cipaglucosidase alfa: Indicated in combination with the enzyme stabilizer Opfolda (65 mg miglustat capsule) for the treatment of adult patients with late-onset Pompe disease (acid alpha-glucosidase [GAA] deficiency) weighing ≥ 40 kg. Miglustat: An enzyme stabilizer indicated in combination with Pombiliti (cipaglucosidase alfa) for the treatment of adult patients with late-onset Pompe disease (acid alpha-glucosidase [GAA] deficiency) weighing ≥ 40 kg. Cipaglucosidase alfa must be used in combination with 65 mg miglustat capsules. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 14, 2025 (cipaglucosidase alfa) and April 10, 2025 (miglustat capsules) |
Recommended dose | Cipaglucosidase alfa: 20 mg/kg of body weight administered every other week as an IV infusion over approximately 4 hours. Miglustat: 260 mg (4 capsules of 65 mg) for patients weighing ≥ 50 kg; 195 mg (3 capsules of 65 mg) for patients weighing ≥ 40 kg to < 50 kg. |
GAA = acid alpha-glucosidase; NOC = Notice of Compliance.
Introduction
Pompe disease is a rare autosomal recessive disorder caused by pathogenic variants in the GAA gene, leading to dysfunctional acid alpha-glucosidase (GAA) enzymes.1 This defect allows glycogen accumulation, impairing cellular function and causing tissue damage.1,2 Patients with late-onset Pompe disease (LOPD) have 2% to 40% of normal enzyme activity, while those with infantile-onset Pompe disease (IOPD) have little to none.1,3 The diagnosis of Pompe disease can be a challenge because symptoms resemble those of other neuromuscular disorders.4 Diagnosis is typically confirmed with molecular testing and/or enzymatic analysis of blood samples; usually, both of these noninvasive tests are performed as part of the diagnostic process. In most cases, the combination of 2 pathogenic variants of the GAA gene, reduced enzyme activity, and the presence of a myopathic phenotype confirms a diagnosis of Pompe disease. Disease progression varies, with severity inversely correlated with residual enzyme activity and worsened by earlier symptom onset.3 Patients who were untreated with LOPD have a 95% likelihood of surviving for 5 years after diagnosis, dropping to a likelihood of 40% at 30 years after diagnosis.1 Enzyme replacement therapy (ERT) improves survival, although death often occurs before age 60, especially with early diaphragm involvement leading to respiratory failure. Clinical features range from slowly progressive myopathy to rapid progression with wheelchair and ventilator dependence. Respiratory muscle involvement, particularly of the diaphragm, is a hallmark of Pompe disease and a major cause of morbidity and mortality.1,3,4 A study using data from births between 1969 and 1996 in British Columbia estimated the incidence of Pompe disease to be 1 in 115,091.5 No updated prevalence or incidence data specific to Canada have been identified.
Treatment is currently focused on targeted, disease-modifying therapy as well as supportive, adjunctive interventions. In Canada, ERT is currently the standard of care for patients who are symptomatic and who have not yet reached “end stage” disease requiring 24 hours 7 days a week invasive ventilation. The first ERT, alglucosidase alfa, was approved by Health Canada in 2006. A newer form of recombinant human acid alpha-glucosidase (rhGAA), avalglucosidase alfa, was approved in 2021 for patients aged 1 year or older with LOPD. However, avalglucosidase alfa is not currently available on public plans in Canada.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of cipaglucosidase alfa, which is indicated in combination with the enzyme stabilizer Opfolda (miglustat), 65 mg capsule, for the treatment of adult patients with LOPD (GAA deficiency) weighing 40 kg or more.
Perspectives of Patient, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient group that responded to the review team’s call for input and from the clinical experts consulted by Canada’s Drug Agency (CDA-AMC) for the purpose of this review.
Patient Input
Input for this review was provided by Muscular Dystrophy Canada (MDC), a nonprofit that supports individuals with neuromuscular disorders. MDC gathered information through surveys, interviews, and the Pompe Canadian Journey Mapping Project. A total of 41 patients (24 males, 17 females) and 15 caregivers contributed, representing a significant portion of the estimated 60 people in Canada with Pompe disease. None had experience with the drug under review.
Patients identified 5 key quality of life impacts: mobility, strength, balance, and energy; breathing; mental health; daily activities; and effects on family and caregivers. Many struggled with tasks requiring strength or endurance, relying on mobility aids that posed accessibility challenges. Breathing difficulties led to poor sleep and reliance on respirators. Social isolation was common due to limited participation in activities, stigma, and illness concerns. High stress, anxiety, and depression were frequently reported, worsened by unpredictable symptoms and caregiver burden.
Diagnosing Pompe disease was often lengthy and frustrating, with most patients facing years of misdiagnoses, multiple tests, and specialty referrals before confirmation via muscle biopsy. Most received ERT and physiotherapy, though many experienced treatment delays due to equipment shortages or a lack of trained nurses. While health care teams managed funding applications for ERT, patients who applied independently faced long processes and occasional denials. Patients emphasized the need for more tolerable treatments with better disease control to maintain independence, reduce medical interventions, and improve quality of life. Key treatment priorities included improved strength and breathing, slowed disease progression without plateauing, and more convenient administration.
Clinician Input
Input From the Clinical Experts Consulted for This Review
The clinical experts consulted for this review indicated that the unmet needs of patients with LOPD would be new treatments that substantially reverse limb muscle and respiratory muscle weakness, rather than simply provide stabilization; earlier treatment to prevent substantial weakness; shorter infusion times; and the availability of additional therapies (currently, only 1 ERT is available in Canada). According to the clinical experts, patients would benefit from the availability of cipaglucosidase alfa with miglustat as an alternative treatment for patients, including those who have experienced adverse reactions to or no longer receive benefit from alglucosidase alfa. The clinical experts indicated that the patients best suited for cipaglucosidase alfa with miglustat would be those who are exhibiting symptoms from unequivocally diagnosed LOPD. The experts highlighted that in their local practice, they test for forced vital capacity (FVC), manual muscle strength and, sometimes, 6-minute walk distance (6MWD) using the 6-minute walk test (6MWT) (which is not always practical), along with other measures every 6 to 12 months. The experts agreed that a clinically meaningful response includes stabilization of the disease. The clinical experts indicated that treatment with cipaglucosidase alfa with miglustat should be discontinued if the patient experiences life-threatening or intolerable immune responses that cannot be overcome. The clinical experts noted that patients receiving cipaglucosidase alfa with miglustat should be under the care of a specialist with expertise in the management of Pompe disease (e.g., inherited metabolic disease specialists, medical geneticists, neuromuscular neurologists, and physiatrists).
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
Input was obtained from the drug programs through the reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a recommendation for cipaglucosidase alfa with miglustat:
relevant comparators
considerations for initiation of therapy
considerations for continuation or renewal of therapy
consideration for discontinuation of therapy
considerations for prescribing of therapy
care provision issues
system and economic issues.
The clinical experts provided advice on the potential implementation issues raised by the drug programs (refer to Table 4).
Clinical Evidence
Systematic Review
Description of Studies
The systematic review included 1 pivotal study. The PROPEL trial was a phase III, randomized, multicentre, double-blind study that evaluated the efficacy and safety of cipaglucosidase alfa plus miglustat compared with alglucosidase alfa plus placebo in adults with LOPD. The trial was conducted across 24 countries, including 2 sites in Canada, from December 2018 to December 2020. The study enrolled 123 participants who were currently receiving and had been receiving alglucosidase alfa for more than 24 months (ERT experienced) or had never received any ERT (ERT naive). Participants were randomized 2:1 to receive either cipaglucosidase alfa 20 mg/kg IV infusion plus miglustat 195 mg or 260 mg oral capsules, depending on body weight (n = 85), or alglucosidase alfa 20 mg/kg IV infusion plus placebo (n = 38) every 2 weeks for 52 weeks.
Overall, patients had a mean age of 47 years, and the mean age at diagnosis was 39 years. Most of the patients (77%) were ambulatory and 23% used assistive devices at baseline. In the ERT-experienced group, the average ERT treatment duration was 7.4 years and approximately 67% had 5 or more years of prior treatment with ERT.
Efficacy Results
Results for all efficacy outcomes are presented using the intention-to-treat (ITT) population without the outlier data from 1 patient (a patient who was ERT naive in the alglucosidase alfa plus placebo group). After the database was locked and treatment assignments were unblinded, the patient with outlier data were found to have baseline 6MWD results that were likely adversely affected by their pre-enrolment use of ostarine powder, an investigational anabolic steroid, that resulted in a clinically implausible change at week 52. Furthermore, this patient admitted to intentionally underperforming during the baseline assessment to enter the study. Given the patient’s admitted underperformance on the screening test and their clinically implausible 6MWD results (i.e., the observed change from baseline to 52 weeks was an increase of 355 m), all efficacy analyses are reported without the data from this patient.
6MWD at Week 52 (Primary Outcome)
The mean change from baseline to week 52 for 6MWD was 20.56 m (95% confidence interval [CI], 11.22 to 29.91) for the cipaglucosidase alfa with miglustat group and 8.02 m (95% CI, −5.71 to 21.74) for the alglucosidase alfa with placebo group. The least squares (LS) mean difference between treatment groups was 14.21 m (95% CI, −2.60 to 31.0). The results of the sensitivity analyses supported the primary analysis. Overall, 6MWD did not vary by any of the subgroups (i.e., ERT status, ERT duration, baseline 6MWD, and age group).
Sitting FVC (First Key Secondary Outcome)
The mean change from baseline to week 52 for percent predicted sitting FVC was −0.93% (95% CI, −2.29% to 0.42%) for the cipaglucosidase alfa with miglustat group and −3.95% (95% CI, −5.58% to −2.32%) for the alglucosidase alfa with placebo group. The LS mean difference between treatment groups was 2.66% (95% CI, 0.37% to 4.95%). The results of the sensitivity analyses supported the primary analysis. FVC did not vary by baseline 6MWD and age group. In the ERT-experienced subgroup, percent predicted sitting FVC showed stabilization over time, with a mean improvement of 0.05% (standard deviation [SD] = 5.84%) from baseline in the cipaglucosidase alfa with miglustat group, compared with −4.02% (SD = 5.01%) in the alglucosidase alfa with placebo group, resulting in an estimated treatment difference of 3.51% (95% CI, 1.03% to 5.99%). In the subgroup of patients on ERT for 5 years or more, the LS mean treatment difference was 3.71% (95% CI, 0.41% to 7.00%) in favour of cipaglucosidase alfa with miglustat. Interaction tests to determine whether results differed statistically by subgroup were not performed.
Manual Muscle Test Lower Extremity Score
The mean manual muscle test (MMT) lower extremity score increased by 1.56 (SD = 3.78) in the cipaglucosidase alfa plus miglustat group and 0.88 (SD = 2.58) in the alglucosidase alfa plus placebo group from baseline, resulting in an LS mean treatment difference of 0.96 (95% CI, −0.48 to 2.40).
6MWD at Week 26
At week 26, the mean 6MWD increased by 17.44 m (95% CI, 9.80 m to 25.0 m) in the cipaglucosidase alfa plus miglustat group and 9.19 m (95% CI, −0.20 m to 18.59 m) in the alglucosidase alfa plus placebo group from baseline to week 26. The LS mean treatment difference was 8.17 m (95% CI, −4.24 m to 20.57 m).
Patient-Reported Outcomes Measurement Information System
The Patient-Reported Outcomes Measurement Information System (PROMIS) Physical Function total score ranges from 20 to 100, with a higher score indicating better physical functioning. Minimal important differences (MIDs) of 2.4 (anchor-based) and 4.2 (distribution-based) have been reported for a clinically important improvement in physical function in patients with LOPD.
Compared with baseline, the PROMIS Physical Function score increased by a mean of 1.94 (95% CI, 0.31 to 3.57) in the cipaglucosidase alfa plus miglustat group, whereas the mean improvement was 0.19 (95% CI, −3.42 to 3.80) for the alglucosidase alfa plus placebo group. This translated to an LS mean treatment difference of 1.87 (95% CI, −1.51 to 5.25).
The PROMIS Fatigue total score ranges from 8 to 40, with lower scores indicating less fatigue. An MID for the PROMIS Fatigue instrument in patients with LOPD was not identified in the literature.
Compared with baseline, the PROMIS Fatigue score decreased by a mean of 2.02 (95% CI, −3.26 to −0.77) in the cipaglucosidase alfa with miglustat group, whereas the mean decreased by 1.67 (95% CI, −3.88 to 0.54) in the alglucosidase alfa with placebo group. This resulted in an LS mean treatment difference of 0.04 (95% CI, −2.12 to 2.20).
Percent Predicted 6MWD
The mean change from baseline to week 52 for the percent predicted 6MWD was 4.07% (95% CI, 2.56% to 5.59%) for the cipaglucosidase alfa with miglustat group and 1.58% (95% CI, −0.42% to 3.58%) for the alglucosidase alfa with placebo group. The LS mean difference was 2.38% (95% CI, −0.26% to 5.03%) between treatment groups.
Gower Manoeuvre
Gower manoeuvre is an individual functional test of the gait, stair, Gower manoeuvre, and chair (GSGC) composite test. Gower manoeuvre involves the patient lying down on the floor, then rising from the floor to get to a standing position. The time (in seconds) to perform the test is recorded. An MID for the Gower manoeuvre specific to patients with LOPD was not identified in the literature.
The mean changes from baseline for the Gower manoeuvre were −0.26 (95% CI, −1.74 to 1.22) and −2.19 (95% CI, −5.04 to 0.66) for the cipaglucosidase alfa with miglustat group and alglucosidase alfa with placebo group, respectively. The LS mean difference between treatment groups was 1.60 (95% CI, −1.48 to 4.68).
Timed Up and Go
The Timed Up and Go (TUG) test is a mobility test that assesses balance, gait speed, and functional ability. The TUG test measures the time a patient needs to rise from a chair, walk 3 m, turn around, walk back to the chair, and sit down, all at a regular pace. An MID for the TUG test specific to patients with LOPD was not identified in the literature.
The mean changes from baseline on the TUG test were −0.30 (95% CI, −2.24 to 1.65) for the cipaglucosidase alfa with miglustat group and −0.13 (95% CI, −1.11 to 0.85) for the alglucosidase alfa with placebo group. The LS mean difference between treatment groups was −0.47 (95% CI, −3.38 to 2.43).
MMT Total Score
The MMT total score was the sum of the lower extremity score and the upper extremity score. The total score ranged from 0 to 80, with lower scores indicating lower overall muscle strength. No MIDs for MMT scores specific to patients with LOPD were identified in the literature.
The mean changes from baseline for the MMT total score were 3.07 (95% CI, 1.66 to 4.48) and 1.41 (95% CI, −0.12 to 2.94) for the cipaglucosidase alfa with miglustat group and alglucosidase alfa with placebo group, respectively. The LS mean difference between treatment groups was 2.22 (95% CI, −0.09 to 4.53).
Harms Results
Adverse Events
A total of 118 of 123 patients (98.9%) experienced a treatment-emergent adverse event (TEAE) during the study. The overall incidence was similar between the cipaglucosidase alfa with miglustat group and the alglucosidase alfa with placebo group (95.3% and 97.4%, respectively). The most common TEAEs were falls, headaches, and nasopharyngitis. Most TEAEs were mild or moderate in severity.
Serious Adverse Events
Overall, 10 of 123 patients (8.1%) had a severe TEAE. Eight of 85 patients (9.4%) in the cipaglucosidase alfa with miglustat group reported 13 severe TEAEs (abdominal pain, enteritis, vomiting, chills, anaphylactic reaction, accidental overdose, fall, irregular heart rate, dyspnea, pruritus, urticaria, aortic aneurysm, and flushing), whereas 2 of 38 patients (5.3%) in the alglucosidase alfa with placebo group reported 3 severe TEAEs (diverticulitis, cerebrovascular accident, and glycosuria).
Withdrawals Due to Adverse Events
Two patients in the cipaglucosidase alfa with miglustat group withdrew from the study due to adverse events (AEs), 1 patient with an anaphylactic reaction and the other with chills, both deemed to be related to the study drug. In the alglucosidase alfa with placebo group, 1 patient withdrew due to a cerebrovascular accident unrelated to the study drug.
Mortality
No TEAEs leading to death were reported.
Notable Harms
Infusion-associated reactions (IARs) were reported in 21 of 85 patients (25%) in the cipaglucosidase alfa with miglustat group and in 10 of 38 patients (26%) in the alglucosidase alfa with placebo group. Two patients (1 in each group) reported 11 to 19 IARs and 1 patient in the cipaglucosidase alfa with miglustat group reported more 20 IARs. In the cipaglucosidase alfa with miglustat group, the most common IARs were dizziness, abdominal distension, headache, chills, diarrhea, dysgeusia, dyspnea, flushing, pruritus, pyrexia, and rash. In the alglucosidase alfa with placebo group, the most common IARs were nausea, fatigue, dizziness, and headache.
Critical Appraisal
Internal Validity
The PROPEL trial was a well-designed phase III, multicentre, double-blind, randomized placebo-controlled study assessing the efficacy and safety of cipaglucosidase alfa with miglustat versus alglucosidase alfa with placebo over 52 weeks in adult patients with LOPD. The trial used a 2:1 random allocation process that was generated by a computer algorithm and centrally managed to maintain allocation concealment. Blinding was effective and patients would not have inferred their treatment group due to the frequency of AEs. Adherence was high and protocol deviations were well documented. Missing data were handled through sensitivity analyses, and the results agreed with the primary analysis outcomes for the primary and key secondary outcomes. Outcome measures were validated and reliable, and the reported outcomes and analysis plan adhered to the study protocol.
External Validity
Overall, the patients in the PROPEL trial were deemed representative of the adult population with LOPD in Canada, although patients with more severe symptoms may not be properly represented, e.g., patients with a sitting FVC of less than 30% of the predicted value for healthy adults who do not require full ventilation support. However, the clinical experts considered the impact on the generalizability of results to be low, and the effects to be still applicable to the target population for reimbursement.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal randomized controlled trial (RCT) identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for outcomes considered most relevant to inform the expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.
Following the GRADE approach, the evidence from the RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for 6MWD and FVC were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts. The reference point for the certainty of evidence assessment for the PROMIS Physical Function was set according to the presence or absence of an important effect based on thresholds informed by the literature. Due to the lack of formal MID estimates for PROMIS Fatigue outcomes, severe TEAEs, and IARs, the targets of the certainty of evidence assessments were the presence or absence of any (non-null) effect for each outcome.
The selection of outcomes for the GRADE assessment was based on the sponsor’s summary of clinical evidence, consultation with clinical experts, and the input received from the patient group and public drug plans. The following are the outcomes that were assessed:
efficacy outcomes (6MWD and FVC)
health-related quality of life outcomes (PROMIS Physical Function and PROMIS Fatigue scores)
harms outcomes (severe TEAEs and IARs).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for the cipaglucosidase alfa plus miglustat group compared with the alglucosidase alfa plus placebo group.
Table 2: Summary of Findings for Cipaglucosidase Alfa With Miglustat vs. Alglucosidase Alfa With Placebo for Patients With Late-Onset Pompe Disease
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Alglucosidase alfa with placebo | Cipaglucosidase alfa with miglustat | Mean difference | |||||
Efficacy outcomes | |||||||
Change from baseline in 6MWD, distance in metres in 6MWT Follow-up: 52 weeks | 117 (1 RCT) | NR | 8.02 (−5.71 to 21.74) | 20.56 (11.22 to 29.91) | 14.21 (−2.60 to 31.02) | Lowa,b | Cipaglucosidase alfa plus miglustat may result in little or no clinically meaningful difference in 6MWD when compared with alglucosidase alfa plus placebo. |
Change from baseline in sitting FVC, % predicted Follow-up: 52 weeks | 121 (1 RCT) | NR | −3.95 (−5.58 to −2.32) | −0.93 (−2.29 to 0.42) | 2.66 (0.37 to 4.95) | Moderatec,d | Cipaglucosidase alfa plus miglustat likely results in little or no clinically meaningful difference in sitting FVC (% predicted) when compared with alglucosidase alfa plus placebo. |
Health-related quality of life | |||||||
Change from baseline in PROMIS Physical Function score Follow-up: 52 weeks | 121 (1 RCT) | NR | 1.94 (0.31 to 3.57) | 0.19 (−3.42 to 3.80) | 1.87 (−1.51 to 5.25) | Moderatee,d | Cipaglucosidase alfa plus miglustat likely results in little or no clinically meaningful difference in PROMIS Physical Function score compared with alglucosidase alfa plus placebo. |
Change from baseline in PROMIS Fatigue score Follow-up: 52 weeks | 122 (1 RCT) | NR | −2.02 (−3.26 to −0.77 | −1.67 (−3.88 to 0.54) | 0.04 (−2.12 to 2.20) | Moderatef,d | Cipaglucosidase alfa plus miglustat likely results in little or no difference in PROMIS Fatigue score compared with alglucosidase alfa plus placebo. The clinical magnitude of the effect is unclear.f |
Harms | |||||||
Patients with ≥ 1 severe TEAEs Follow-up: 52 weeks | 123 (1 RCT) | 3.6 (0.5 to 27.6) | 1 of 38 (2.6%)g | 8 of 85 (9.4%)g | 6.8% (−5.4% to 15.8%) | Lowh | Cipaglucosidase alfa plus miglustat may result in little or no difference in the occurrence of severe TEAEs compared with alglucosidase alfa plus placebo. The clinical magnitude of the effect is unclear.h |
Patients with any IARs Follow-up: 52 weeks | 123 (1 RCT) | 0.9 (0.5 to 1.8) | 10 of 38 (26.3%)g | 21 of 85 (24.7%)g | −1.6% (−18.4% to 15.1%) | Lowh | Cipaglucosidase alfa plus miglustat may result in little or no difference in the occurrence of IARs compared with alglucosidase alfa plus placebo. The clinical magnitude of the effect is unclear.h |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; CI = confidence interval; FVC = forced vital capacity; IAR = infusion-associated reaction; LOPD = late-onset Pompe disease; NR = not reported, PROMIS = Patient-Reported Outcomes Measurement Information System; RCT = randomized controlled trial; TEAE = treatment-emergent adverse event; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aRated down 1 level for serious imprecision. Based on clinical expert input, 20 m could be considered a clinically meaningful threshold for between-group difference. The 95% CI included the possibility of benefit and little to no difference.
bRated down 1 level for serious imprecision. The minimally important difference for 6MWD in patients with LOPD is very uncertain, based on the varying and wide ranges of minimally important differences reported in the literature and per clinical expert input.
cRated down 1 level for serious imprecision. Based on clinical expert input, 3% could be considered a clinically meaningful threshold for between-group difference. The 95% CI included the possibility of benefit and little to no difference.
dStatistical testing for this outcome was not adjusted for multiplicity in the trial and should be considered as supportive evidence.
eRated down 1 level for serious imprecision. Based on the relevant literature, 2.4 and 4.2 could be considered clinically meaningful thresholds. The 95% CI included the possibility of benefit and little to no difference.
fRated down 1 level for serious imprecision. No minimally important difference was found; therefore, the null effect was used. The 95% CI included the possibility of benefit and little to no difference.
gPresented as n (%).
hRated down 2 levels for very serious imprecision. There is a very small number of patients with events captured. No minimally important difference was found; therefore, the null effect was used. The 95% CI included the possibility of benefit and little to no difference.
Sources: PROPEL Clinical Study Report. Details included in the table are from the sponsor’s summary of clinical evidence.
Long-Term Extension Studies
Description of Studies
ATB200-07 is an ongoing, phase III, international, open-label extension (OLE) study to assess the long-term safety and tolerability of cipaglucosidase alfa and miglustat coadministration following 104 weeks of treatment (i.e., 52 weeks in the PROPEL trial and 52 weeks in the OLE study) in adult patients with LOPD. The efficacy of the drug combination was also assessed as a secondary objective. The efficacy and safety outcomes in the ATB200-07 trial were consistent with the PROPEL study. Patients enrolled in the OLE study who received cipaglucosidase alfa plus miglustat in the PROPEL trial continued on study treatment (continued-treatment group), and those receiving alglucosidase alfa with placebo were switched to cipaglucosidase alfa plus miglustat (treatment-switched group). No direct statistical comparisons between treatment groups were conducted and the results were descriptive only. Cipaglucosidase alfa plus miglustat was administered in the ATB200-07 trial in the same manner as the PROPEL trial. The first infusion visit in the OLE study was scheduled approximately 2 weeks after the last study visit of the PROPEL trial.
Efficacy Results
6MWD (In Metres)
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean 6MWD was 373.1 m (SD = 124.23 m) at baseline in the ATB200-07 OLE study, with a mean change from baseline of −2.0 m (95% CI, −9.1 m to 5.1 m) at week 52 of the OLE. The mean 6MWD in the treatment-switched group was 363.5 m (SD = 137.38 m) at baseline, with a mean change from baseline of −1.4 m (95% CI, −14.0 m to 11.1 m) at week 52.
Sitting FVC (Percent Predicted)
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean sitting FVC (percent predicted) was 69.1 (SD = 19.41) in the ATB200-07 OLE study at baseline and 67.9 (SD = 20.39) at week 52, with a mean change from baseline of −0.2 (95% CI, −1.9 to 1.6). The mean sitting FVC (percent predicted) in the treatment-switched group was 63.8 (SD = 19.63) at baseline and 64.2 (SD = 19.21) at week 52, with a mean change from baseline of 0.0 (95% CI, −1.8 to 1.8) at week 52.
MMT Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean MMT total score was 62.2 (SD = 8.33) at baseline in the ATB200-07 OLE study and 65.7 (SD = 8.41) at week 52, with a mean change from baseline of 3.2 (95% CI, 1.7 to 4.8). The mean MMT total score in the treatment-switched group was 62.7 (SD = 9.86) at baseline and 64.0 (SD = 10.93) at week 52, with a mean change from baseline of 1.4 (95% CI, −0.2 to 2.9).
PROMIS Physical Function Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean PROMIS Physical Function total score was 68.8 (SD = 12.81) at baseline in the ATB200-07 OLE study and 69.7 (SD = 12.98) at week 52, with a mean change from baseline of 0.6 (95% CI, −0.6 to 1.8). The treatment-switched group mean was 67.8 (SD = 16.74) at baseline and 67.4 (SD = 15.23) at week 52, with a mean change from baseline of −0.1 (95% CI, −2.5 to 2.2).
PROMIS Fatigue Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean PROMIS Fatigue total score was 19.9 (SD = 7.50) at baseline in the ATB200-07 OLE study and 20.1 (SD = 7.13) at week 52, with a mean change from baseline of 0.2 (95% CI, −1.1 to 1.6). The mean for the treatment-switched group was 19.3 (SD = 6.72) at baseline and 21.0 (SD = 6.80) at week 52, with a mean change from baseline of 1.5 (95% CI, −0.6 to 2.6).
Harms Results
No new safety signals were identified during the ATB200-07 OLE study, and commonly reported TEAEs associated with cipaglucosidase alfa plus miglustat treatment were consistent with the safety profile observed in the PROPEL trial. Most TEAEs were considered mild or moderate in severity. Overall, the safety profile of cipaglucosidase alfa plus miglustat observed during the ATB200-07 study was consistent with the PROPEL trial.
Critical Appraisal
The ATB200-07 study was designed as an OLE to assess the long-term safety and tolerability of cipaglucosidase alfa and miglustat coadministration in adult patients with LOPD. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and the reporting of safety parameters due to unblinded exposure to the study medication during the treatment period. The ATB200-07 study population for this interim analysis consisted of patients who took part in the PROPEL trial; therefore, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the OLE. Given that patients needed to complete the PROPEL trial before enrolling, the OLE study population is inherently enriched and introduces some selection bias for responders and those with a potentially lower risk for adverse effects.
Indirect Comparisons
Description of Studies
The sponsor conducted an indirect treatment comparison (ITC) using a network meta-analysis (NMA) framework to compare the efficacy of cipaglucosidase alfa with miglustat against avalglucosidase alfa and alglucosidase alfa in adult patients with LOPD. Given the absence of direct head-to-head clinical trials comparing all these treatments, the ITC was performed to estimate their relative effects based on available clinical trial data.
To address differences in study populations and baseline characteristics, the sponsor employed a multilevel network meta-regression (ML-NMR) approach. ML-NMR extends a standard NMA by integrating the individual patient-level data (IPD) where available (from the PROPEL trial) and adjusting for key effect modifiers, such as prior ERT exposure, age, and baseline function. This approach attempts to account for treatment-effect heterogeneity by modelling covariate distributions across studies, potentially improving the validity of indirect comparisons.
The ITC included data from 6 studies, 2 RCTs (PROPEL and COMET) and 4 single-arm studies, including OLE trials. The inclusion of the single-arm studies required statistical adjustments to match baseline characteristics with comparator arms in the RCTs. Two separate analyses were conducted:
Network A: Limited to the 2 RCTs (PROPEL and COMET) to ensure the comparability of treatment arms.
Network B: Included single-arm trials and OLEs to incorporate additional data, particularly from patients with ERT experience receiving avalglucosidase alfa.
The primary efficacy outcomes evaluated were the change from baseline in the 6MWD and FVC (percent predicted) at 52 weeks.
Efficacy Results
6MWD (Metres)
Network A (RCTs only): Cipaglucosidase alfa with miglustat was associated with a mean increase in 6MWD of 14.64 m (95% credible interval [CrI], 7.07 m to 22.31 m) compared with alglucosidase alfa, and cipaglucosidase alfa with miglustat was associated with a mean decrease in 6MWD of 10.02 m (95% CrI, −23.62 m to 4.00 m) compared with avalglucosidase alfa. Avalglucosidase alfa showed an increase of 24.66 m (95% CrI, 9.95 m to 39.55 m) versus alglucosidase alfa.
Network B (full evidence): Cipaglucosidase alfa with miglustat showed a 13.64 m (95% CrI, 8.73 m to 18.70 m) improvement versus alglucosidase alfa and a 28.93 m (95% CrI, 8.26 m to 50.11 m) increase versus avalglucosidase alfa.
FVC (Percent Predicted)
Network A (RCTs only): Cipaglucosidase alfa with miglustat showed a mean increase of 2.53 percentage points (95% CrI, 1.38 to 3.67) compared with alglucosidase alfa, and cipaglucosidase alfa with miglustat showed a mean decrease of 1.45 percentage points (95% CrI, −3.01 to 0.07) compared with avalglucosidase alfa. Avalglucosidase alfa demonstrated an improvement of 3.98 percentage points (95% CrI, 2.40 to 5.64) versus alglucosidase alfa.
Network B (full evidence): Cipaglucosidase alfa with miglustat increased FVC by 3.95 percentage points (95% CrI, 3.23 to 4.69) versus alglucosidase alfa and 2.88 percentage points (95% CrI, 1.07 to 4.71) versus avalglucosidase alfa.
Harms Results
The ITC did not include any formal analysis of safety outcomes.
Critical Appraisal
While the ML-NMR approach is a methodological strength, several limitations impact the robustness of the findings. The data extraction process for the updated systematic literature review (SLR) relied on a single reviewer, increasing the risk of bias or missing data. Furthermore, the selection of treatment-effect modifiers was not fully justified, excluding potentially relevant factors like weight, muscle damage, and ACE genotype. The analysis also relied on applying IPD-based adjustments to aggregate-level data from other trials. The sponsor provided overall Pearson residuals that suggested a good fit of the final ML-NMR model predictions to the observed study-level data. However, specific diagnostics were not available to assess the appropriateness or impact of the step involving the application of IPD-derived adjustments to the aggregate-level data from other trials. Additionally, inconsistency testing was not performed due to the network’s structure, limiting the ability to verify whether indirect comparisons aligned with direct evidence.
The inclusion of single-arm trials and OLEs in network B introduced heterogeneity, requiring matching techniques to align populations with appropriate comparator arms. This led to a notable shift in treatment rankings between network A (RCTs only) and network B (including single-arm studies). In network A, avalglucosidase alfa appeared more effective, whereas network B showed cipaglucosidase alfa plus miglustat to be superior. The difference suggests that ERT experience significantly influences treatment outcomes, but it also raises concerns about bias introduced by single-arm studies and the repeated use of data from comparators like alglucosidase alfa, which could artificially increase precision.
While the ITC provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings. The limited model diagnostics, missing inconsistency testing, reliance on reconstructed data, and variability in methodological choices introduce a high degree of uncertainty in the results.
Studies Addressing Gaps in the Evidence from the Systematic Review
Two studies were proposed by the sponsor as addressing gaps in the systematic review evidence, ATB200-02 and the UK Early Access to Medicines Scheme (EAMS) registry study.
ATB200-02 Trial
Description of Studies
ATB200-02 (N = 29) is an ongoing, open-label, phase I/II study evaluating the long-term (up to 48 months) efficacy of cipaglucosidase alglucosidase alfa plus miglustat. The ATB200-02 study was submitted to fill an evidence gap pertaining to the underrepresentation of patients who were ERT naive in the PROPEL trial and the common exclusion of nonambulatory patients from LOPD clinical trials. The efficacy outcomes assessed in the ATB200-02 study that were identified by the clinical experts as important to this review were 6MWD, percent predicted sitting FVC, and MMT total score. The study included 4 cohorts based on ERT experience and ambulatory status:
Cohort 1: ERT experience of 2 to 6 years and ambulatory
Cohort 2: ERT experience of 2 years or greater and nonambulatory
Cohort 3: ERT naive and ambulatory
Cohort 4: ERT experience of 7 years or greater and ambulatory.
Patients were excluded from the ATB200-02 study if they had previous use of any investigational therapy within 30 days or 5 treatment half-lives, a requirement for ventilatory support for 6 hours per day or greater (except the nonambulatory cohort), or a history of anaphylaxis to alglucosidase alfa and highly sustained anti-rhGAA antibodies (except the ERT-naive cohort). Across all groups, 90% of patients were between the ages of 18 and 64 years, and the mean duration of ERT for patients who were ERT experienced ranged from 5.1 to 10.6 years. Patients in both the ERT-experienced and ERT-naive groups exhibited a significant impact of Pompe disease at study entry, based on baseline 6MWD and percent predicted sitting FVC.
Efficacy Results
Six-Minute Walk Distance
Motor function was evaluated in all patients who were ambulatory using the 6MWT. At month 48, 88.9% of patients who were ERT experienced and 100% of patients who were ERT naive experienced an improvement in 6MWD from baseline. For patients who were ambulatory and ERT experienced (cohorts 1 and 4), mean and percent change from baseline were 20.7 m (95% CI, −57.6 m to 99.0 m) and 3.9%, respectively, at month 48. For patients who were ambulatory and ERT naive (cohort 3), mean and percent change from baseline were 52.2 m (95% CI, −21.9 m to 126.3 m) and 12.5%, respectively, at month 48.
Percent Predicted Sitting FVC
A meaningful change from baseline in percent predicted sitting FVC was defined as a 3% or greater change in points from baseline. At month 48, the mean change from baseline in percent predicted sitting FVC was 1.0 (95% CI, 5.7 to 7.7) for patients who were ambulatory and ERT experienced (cohorts 1 and 4) and 8.3 (95% CI, −9.2 to 6.7) for patients who were ambulatory and ERT naive.
For patients who were nonambulatory (cohort 2), percent predicted sitting FVC data were available for 2 patients who were nonambulatory and ERT experienced after 36 months and 1 patient at 48 months of follow-up. After 36 months of follow-up, 1 patient improved and the other worsened compared with baseline. The patient with available data after 48 months of follow-up was generally stable compared with baseline.
MMT Total Score
Muscle strength was evaluated in all cohorts using the MMT total score, where higher total scores indicate a reduced impact of disease on muscle function. For patients who were nonambulatory, the total score for MMT was based on the upper extremity score only. At month 48, mean change from baseline in MMT total score was 4.0 points (95% CI, 0.9 points to 7.1 points) for patients who were ambulatory and ERT experienced (cohorts 1 and 4) and −1.3 points (95% CI, −9.2 points to 6.7 points) for patients who were ambulatory and ERT naive.
Results at 48 months were not available for patients who were nonambulatory and ERT experienced (cohort 2), but at month 36, the change from baseline in MMT total score was −0.8 (95% CI, −17.8 to 16.3).
Harms Results
Overall, no unexpected safety events were observed during the extended treatment period of the ATB200-02 study. TEAEs leading to study withdrawal occurred in 2 patients: 1 patient in cohort 1 had diffuse large B-cell lymphoma, which the investigator assessed as unrelated to treatment, and 1 patient in cohort 2 had a drug-related TEAE of urticaria, considered to be an IAR. The incidence of IARs was similar between the ERT-experienced (48%) and ERT-naive (50%) cohorts.
Critical Appraisal
The ATB200-02 study was designed as an open-label phase I/II study to assess the long-term efficacy of cipaglucosidase alfa and miglustat coadministration in adult patients with LOPD. Although the ATB200-02 study was submitted to address the systematic review evidence gap pertaining to the exclusion of patients who were nonambulatory with LOPD from clinical trials, the small sample size makes it challenging to draw conclusions about long-term efficacy in this patient group. Efficacy data for pulmonary function and patient-reported outcomes in this group were limited to 1 and 2 patients, respectively. Additionally, statistical hypothesis testing was not part of the study design and there was no active comparator or placebo group. The open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and the reporting of safety parameters due to the unblinded exposure to the study medication during the treatment period.
UK EAMS Registry Study
Description of Studies
The UK EAMS registry study (N = 37) is a prospective, observational registry study evaluating the real-world safety and effectiveness of cipaglucosidase alfa plus miglustat in patients who were ERT experienced (2 years or more) with LOPD. This study was submitted to fill an evidence gap pertaining to a lack of real-world data in patients with LOPD. The reported efficacy outcomes that were identified by the clinical experts as important to this review were 6MWD and percent predicted sitting FVC. Harms results were not reported for this study. Compared with the PROPEL trial, at baseline, patients included in this registry study were slightly older (mean age of 53 years), with a slightly longer mean ERT duration (11.1 years), and lower 6MWD and percent predicted sitting FVC results.
Efficacy Results
Of all adults enrolled in the EAMS registry, 13 and 12 patients had both baseline and postbaseline assessments of 6MWD and percent predicted FVC, respectively; however, the time between these 2 assessments varied considerably, from 82 days to 1,401 days.
From baseline to the postbaseline visit, patients had a mean change from baseline in the postbaseline assessment in 6MWD of 10.2 m (SD = 33.9 m), and a mean change in percent predicted sitting FVC of 4.0% (SD = 9.1%).
Harms Results
Harms data were not reported for the UK EAMS registry study.
Critical Appraisal
At the time of submission, the available evidence was limited, with no comprehensive details on methods and results, which may impact the ability to sufficiently review and critically appraise the evidence and the robustness of the evidence and conclusions. Efficacy data were available for only one-quarter to one-third of patients which, in combination with a wide variation in time between baseline and postbaseline visits, limits the ability to draw firm conclusions about efficacy. Given that so few patients had both a baseline and after follow-up measurement, the mean changes observed were likely specific to a highly select group and may not be representative of the entire study population. Additionally, the lack of reported harms limits the ability to assess the safety of cipaglucosidase alfa plus miglustat in real-world clinical practice. Statistical hypothesis testing was not part of the study design and there was no active comparator or placebo group. The open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes. Additionally, because the EAMS study is based on data from a national UK registry, generalizability to the population of patients with LOPD living in Canada may be limited.
Conclusions
The evidence on the effects of cipaglucosidase alfa plus miglustat in adult patients with LOPD comprises 1 pivotal RCT comparing cipaglucosidase alfa plus miglustat with alglucosidase alfa plus placebo, 1 long-term extension study, 1 ITC, and 2 additional studies addressing gaps in evidence.
The outcomes considered by the clinical experts and the patient group to be critical for decision-making were 6MWD (to assess ambulatory function), percent predicted sitting FVC (to assess respiratory function), PROMIS Physical Function and PROMIS Fatigue scores (to assess health-related quality of life), and severe TEAEs and IARs (to assess harms).
The pivotal PROPEL study evaluated these outcomes and provided evidence that cipaglucosidase alfa plus miglustat compared with alglucosidase alfa plus placebo may result in no difference to minimal improvement in 6MWD. Additionally, cipaglucosidase alfa plus miglustat likely results in no difference to minimal improvement in percent predicted sitting FVC, PROMIS Physical Function score, and PROMIS Fatigue score when compared with alglucosidase alfa plus placebo.
The overall number of severe TEAEs was very small. The information comparing cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo suggests a numerical increase in the number of severe TEAEs, although the majority were unrelated to the study drug and the wide CIs indicate the finding was very imprecise. The number of patients with IARs was similar in the cipaglucosidase alfa plus miglustat group and alglucosidase alfa plus placebo group. Therefore, coadministration of cipaglucosidase alfa and miglustat was generally safe and well tolerated, with very little treatment discontinuation. No new safety signals were identified. No deaths were reported. Additionally, the incidence of IARs was similar between the treatment groups. All IARs were nonserious except 1 serious adverse event (SAE) of anaphylactic reaction in the cipaglucosidase alfa plus miglustat group. Two IARs (anaphylactic reaction and chills) in the cipaglucosidase alfa plus miglustat group led to study drug discontinuation. The overall safety profile of cipaglucosidase alfa plus miglustat was similar to alglucosidase alfa plus placebo.
The efficacy effects observed in the PROPEL trial generally remained stable across the OLE study to week 104 of treatment; no new safety signals were identified during the OLE study, and the commonly reported TEAEs associated with cipaglucosidase alfa plus miglustat treatment were consistent with the safety profile observed in the PROPEL trial. However, there is still uncertainty about the long-term effects of cipaglucosidase alfa combined with miglustat on wheelchair and ventilation dependency because these outcomes were not included in the submitted evidence.
The number of patients with available efficacy data in both the ATB200-02 study and UK EAMS registry (studies addressing gaps in evidence) was limited, which makes it challenging to draw meaningful conclusions.
While the ITC provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings. The limited model diagnostics, missing inconsistency testing, reliance on reconstructed data, and variability in methodological choices introduce a high degree of uncertainty in the results.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of cipaglucosidase alfa (Pombiliti) for injection, lyophilized powder, 105 mg per vial, IV infusion in combination with miglustat (Opfolda) 65 mg oral capsules for treatment in adult patients with LOPD or GAA deficiency.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
Pompe disease (also known as GAA deficiency or glycogen storage disease type II) is a rare, autosomal recessive disorder caused by pathogenic variants in the GAA gene, resulting in dysfunctional GAA enzymes.1 With Pompe disease, the defect in the enzyme allows glycogen to accumulate in cells, leading to impaired cellular function and tissue damage.1,2 Patients with LOPD have variable and reduced enzyme function (between 2% and 40% of normal), whereas patients with IOPD have minimal or no enzyme activity.1,3
The diagnosis of Pompe disease can be a challenge because symptoms resemble those of other neuromuscular disorders.4 Pompe disease might be suspected in children and adults who show progressive proximal limb weakness and significantly reduced FVC.1 Pompe disease is usually diagnosed with molecular testing and/or the enzymatic analysis of white blood cells or dried blood spots; however, in some cases, a biopsy of skin or muscle tissue can be performed and may show glycogen accumulation, but this method is more invasive. Gene sequencing is an important method to confirm a diagnosis and is both noninvasive and routinely available. In most cases, the combination of 2 pathogenic variants of the GAA gene, reduced enzyme activity, and the presence of a myopathic phenotype confirms a diagnosis of Pompe disease. A differential diagnosis might be necessary to distinguish Pompe disease from other myopathies using age at symptom onset, high creatine kinase levels, and absence of metabolic abnormalities (e.g., hypoglycemia, lactic acidosis, metabolic acidosis).
The rate of disease progression varies among patients, and disease severity is inversely correlated with residual GAA activity.3 Additionally, disease severity is associated with disease duration, and patients who have symptom onset at a younger age have more severe disease.1 It has been estimated that the 5-year postdiagnosis survival for patients with LOPD who were untreated is 95%, and 30-year postdiagnosis survival is 40%.1 It has been reported that patients treated with ERT have a mean age at death of fewer than 60 years,1 although this varies with rate of progression, extent of muscle involvement, and comorbidities.1 For instance, early involvement of the diaphragm is followed by respiratory failure and death during the second or third decade of life.1 In general, earlier diagnosis and treatment can improve outcomes. Patients with LOPD do not develop hypertrophic cardiomyopathy (a characteristic of IOPD), and clinical presentation can be at any age, even among individuals with the same genetic variant, indicating there are other factors that influence clinical outcomes.1 Clinical features vary from a slowly progressive myopathy, which might be preceded by an asymptomatic interval, to a much more rapid and progressive myopathy that results in wheelchair and ventilatory dependence and early death.1 It is also a common and unique feature of Pompe disease to have early involvement of the diaphragm and respiratory accessory muscles that is not observed with most other myopathies. This can lead to respiratory failure, which is a major cause of morbidity and mortality in patients with LOPD.1,3,4
The incidence of all Pompe disease (both LOPD and IOPD) has been estimated to be between 1 in 14,000 and 1 in 300,000, depending on geographic location and ethnicity,3 whereas the incidence of LOPD has been estimated to be 1.75 in 100,000 births.6 A study using data from births between 1969 and 1996 in British Columbia estimated the incidence of Pompe disease to be 1 in 115,091.5 It is expected that this is an underestimate of the true number of patients with LOPD in Canada because many would have been undiagnosed at the time of the study. No updated prevalence or incidence data specific to Canada have been identified.
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
According to the clinical experts consulted by CDA-AMC for this review, LOPD primarily affects skeletal muscle tissue, resulting in progressive weakness and respiratory insufficiency. Goals of treatment include the delay or prevention of respiratory failure; delay or prevention of invasive ventilation; delay or prevention of wheelchair dependence; improved or maintained mobility; maintenance of independence for self-care; maintenance of safety and prevention of injury; maintenance of employability; maintenance or improvement of quality of life, pain, and self-image; and minimization of treatment burden and the adverse effects of treatment.
The clinical experts stated that treatment is currently focused on targeted, disease-modifying therapy as well as nonspecific interventions. In Canada, ERT is currently the standard of care for patients who are symptomatic and who have not yet reached “end stage” disease requiring 24 hours 7 days a week invasive ventilation.
Guidelines exist for the treatment of LOPD, including recommendations from a Canadian expert panel7 on disease diagnosis and management and, most recently, an updated recommendation was issued by the European Pompe Consortium8 on the use of ERT in LOPD. The most important goals of the currently available forms of treatment are to stabilize and/or improve motor and respiratory function as well as to prevent disease progression. Treatment generally involves a multidisciplinary team of pediatricians, internists, neurologists, medical geneticists, respirologists, orthopedists, cardiologists, dieticians, physical therapists, and other health care professionals.
In terms of pharmacological interventions, ERT with rhGAA at a dose of 20 mg/kg per body weight every other week was advised by experts in Canada for patients who were symptomatic with LOPD whether or not they were ambulatory and received ventilation.7 The European Pompe Consortium also recommends ERT when patients show signs of disease including skeletal muscle weakness or respiratory muscle involvement.8 ERT is an IV-administered medication that helps reduce the accumulation of glycogen. The first ERT, Myozyme, was approved by Health Canada in 2006. A newer form of rhGAA, avalglucosidase alfa (Nexviazyme), was approved in 2021 for patients aged 1 year or older with LOPD. However, avalglucosidase alfa is not currently available on public plans in Canada.
Treatment goals with ERT should be defined before starting ERT and assessed regularly. The clinical experts reported that the most common response to ERT is a small improvement in FVC and distance walked in the 6MWT, followed by a long plateau of stabilization, followed by slow worsening in respiratory capacity and mobility. Although missing enzyme activity is replaced by ERT, it is insufficient for several reasons, including that the dosing interval leads to large amounts of time where there is no product exposure, the dose may be suboptimal, and the delivery into myocyte lysosomes is inadequate. Fiscal and time costs to patients and payers dampen interest in experimenting with alternative dosing regimens in adults.
Switching ERT may be considered if there is no indication that skeletal muscle and/or respiratory function have stabilized or improved during a minimum 12-month period and/or the patient has severe IARs that cannot be adequately managed.8 Criteria for cessation of treatment can vary but, generally, patients should be assessed for discontinuation if they experience severe untreatable IARs, high-neutralizing antidrug antibody titres, or if there is an insufficient response to therapy over a defined period of time (e.g., 2 years).7,8
Aside from ERTs, several supportive therapies are available to manage LOPD symptoms. This includes respiratory support using mechanical ventilation (i.e., bilevel positive airway pressure [BiPAP] or volume ventilators) through noninvasive or invasive techniques when needed during the night and/or periods of the day or during respiratory tract infections. The clinical expert stated that most patients use BiPAP when supine to maintain normal blood gas status. Ventilation is the primary supportive treatment because respiratory failure is the leading cause of morbidity and mortality.9 Physical therapy can help to strengthen skeletal and respiratory muscles. Mobility aids, including the use of canes or walkers, may be necessary. Eventually, some patients may require the use of a wheelchair. Orthopedic devices including braces may be recommended in some patients. Surgery may be required for certain orthopedic symptoms such as contractures or spinal deformity.4 The clinical expert stated that a high-protein diet has been shown to augment training-related strength gains and is routinely recommended to patients.
Drug Under Review
Key characteristics of cipaglucosidase alfa with miglustat are summarized in Table 3 with other treatments available for Pompe disease.
Cipaglucosidase alfa is indicated in combination with the enzyme stabilizer (65 mg miglustat capsule) for the treatment of adult patients with LOPD (GAA deficiency) weighing 40 kg or greater. The reimbursement request is as per the Health Canada indication. This drug combination is approved in the US and EU for the treatment of adult patients with LOPD.10,11
The recommended dose of cipaglucosidase alfa is 20 mg/kg of body weight administered every other week as an IV solution over approximately 4 hours. The recommended miglustat dose for patients weighing between 40 kg and 50 kg is 3 capsules of 65 mg (195 mg total) orally every other week or, for patients weighing 50 kg or greater, 4 capsules of 65 mg (260 mg total) orally every other week.
Cipaglucosidase alfa is an rhGAA that provides an exogenous source of GAA and degrades glycogen by catalyzing the hydrolysis of alpha-1,4 and alpha-1,6 glycosidic linkages of lysosomal glycogen. Miglustat is a pharmacokinetic enzyme stabilizer of cipaglucosidase alfa that binds selectively with cipaglucosidase alfa in blood during infusion, thereby stabilizing the conformation of cipaglucosidase alfa and minimizing the loss of enzyme activity while in circulation.
Table 3: Key Characteristics of Cipaglucosidase Alfa With Miglustat, Avalglucosidase Alfa, and Alglucosidase Alfa
Characteristic | Cipaglucosidase alfa with miglustat | Avalglucosidase alfa | Alglucosidase alfa |
|---|---|---|---|
Mechanism of action | Cipaglucosidase alfa: An rhGAA that provides an exogenous source of GAA and degrades glycogen by catalyzing the hydrolysis of alpha-1,4 and alpha-1,6 glycosidic linkages of lysosomal glycogen. Miglustat: A pharmacokinetic enzyme stabilizer of cipaglucosidase alfa that binds selectively with cipaglucosidase alfa in blood during infusion, thereby stabilizing the conformation of cipaglucosidase alfa and minimizing the loss of enzyme activity while in circulation. | An rhGAA provides an exogenous source of GAA and degrades glycogen by catalyzing the hydrolysis of alpha-1,4 and alpha-1,6 glycosidic linkages of lysosomal glycogen. | |
Indicationa | In combination with the enzyme stabilizer (65 mg miglustat capsule) for the treatment of adult patients with late-onset Pompe disease (GAA deficiency) weighing ≥ 40 kg. | For long-term treatment of patients with late-onset Pompe disease (GAA deficiency). | For use in patients with Pompe disease (GAA deficiency). |
Route of administration |
| IV infusion | IV infusion |
Recommended dose | Cipaglucosidase alfa: 20 mg/kg of body weight administered every other week as an IV infusion over approximately 4 hours. Miglustat: Taken orally every other week in adults aged 18 years and older, 195 mg (3 capsules of 65 mg) for patients weighing ≥ 40 kg to < 50 kg or 260 mg (4 capsules of 65 mg) for patients weighing ≥ 50 kg. | 20 mg/kg of body weight administered every other week | 20 mg/kg of body weight administered every other week |
Serious adverse effects or safety issues | Cipaglucosidase alfa:
Miglustat:
|
|
|
GAA = acid alpha-glucosidase; IgG = immunoglobulin G; rhGAA = recombinant human acid alpha-glucosidase.
aHealth Canada–approved indication.
Sources: Health Canada product monographs for cipaglucosidase alfa and miglustat,12,13 avalglucosidase alfa,14 and alglucosidase alfa.15
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Input for this review was submitted by 1 patient group, MDC. MDC is a nonprofit organization that provides programs, services, and advocacy to individuals affected by neuromuscular disorders. Information was gathered through a health care experience survey, semistructured virtual interviews, and patient insights from the Pompe Canadian Journey Mapping Project. A total of 41 patients (24 males, 17 females) with Pompe disease provided input, which MDC indicated is a notable sample, given there are approximately 60 individuals in total in Canada with Pompe disease; 15 caregivers or family members also contributed to the input. No patients who provided input had experience with the drug under review.
When patients were asked about their disease experience, 5 key themes were identified as having the greatest impact on quality of life. These identified themes, listed in order of frequency, were negative impacts on mobility, strength, balance, and energy levels; breathing; mental health; ability to participate in daily activities; and family and caregivers. Due to disease-associated challenges with movement and balance, many patients had trouble performing daily tasks requiring moderate strength or prolonged energy expenditure, such as house chores, sports, or playing with their children. Many patients use a walker or wheelchair for assistance with mobility, which they noted presents accessibility limitations in their daily life. Because muscle weakness also impacts breathing, patients frequently reported poor sleep quality and high levels of fatigue, with many requiring a respirator or BiPAP to sleep and waking frequently through the night. Patients also emphasized the impact of their disease on their relationships, noting feelings of social isolation due to a reduced ability to participate in social gatherings, societal stigma, and social avoidance to protect against illness. Many patients reported high levels of stress, as well as anxiety and depression, due to these significant impacts on their quality of life, which are compounded by unpredictable symptom onset and high perceived burden on caregivers.
The process for obtaining a diagnosis and treatment for Pompe disease was described by patients as frustrating and time-consuming. Most patients reported waiting years to receive an accurate diagnosis, with many first undergoing numerous diagnostic tests, referrals to different specialties, and misdiagnoses. Many patients were diagnosed with Pompe disease after undergoing muscle biopsy. Most patients had received ERT and physiotherapy as primary treatment for their Pompe disease; however, many noted delays in receiving treatment due to unavailable equipment or a lack of nurses trained to administer therapy. While most patients indicated that the cost of their ERT was covered and funding applications were managed by their health care teams, individuals who applied for their own funding reported lengthy application processes and occasional coverage denials.
Patients expressed a need for new, more tolerable treatments with superior disease control that minimize the impact of symptoms and prevent exacerbations. Effective therapies would help patients maintain their independence, reduce the frequency of serious medical interventions or hospitalizations, and improve their overall quality of life. Outcomes and features identified as important to patients in new treatments included improved strength and breathing function, slowed disease progression without a plateau effect, and a more convenient mode of administration.
Clinician Input
Input From the Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of LOPD.
Unmet Needs
The clinical experts agreed that adult patients are usually quite symptomatic by the time of LOPD diagnosis, and substantial reversal of myopathy is an unrealistic treatment goal for the foreseeable future. There continues to be disease progression despite ERT. Infusion times for ERT are lengthy, owing to the large protein load being infused. Most patients lose an entire day to receive treatment, with negative impacts on income and quality of life. Travel for work or leisure is harder. The cost of ERT is extremely high, reducing the likelihood that “stackable” disease-modifying treatments will be developed. Instead, alternative therapies currently in trials are primarily being investigated as standalone therapies.
One clinical expert indicated that a major unmet need is a treatment that substantially reverses the limb muscle and respiratory muscle weakness, rather than a therapy that simply provides stabilization. In addition, a treatment that results in long-term maintenance of stability, rather than stability followed by slow decline, is also needed. Having more than 1 treatment option will also be beneficial because this may be a partial solution to patients whose disease becomes refractory to the first treatment. Earlier treatment (or evidence for benefit from earlier treatment) is another unmet need to prevent the development of substantial weakness because, once this has developed, it is hard to reverse (and most patients are seen at a time when they already have substantial symptoms and signs of myopathy).
Place in Therapy
One clinical expert stated that cipaglucosidase alfa with miglustat is another form of ERT, and there is no rationale to treat a patient concurrently with 2 different ERT products. The clinical expert expects that cipaglucosidase alfa with miglustat would be an alternative treatment for patients. The available data support treating a newly diagnosed patient who is ERT naive with cipaglucosidase alfa with miglustat, avalglucosidase alfa, or alglucosidase alfa. Almost all patients diagnosed in adulthood are already symptomatic, meaning they qualify for treatment, according to Canadian guidelines. There is no newborn screening for Pompe disease yet in Canada, but it is available in other countries, leading to presymptomatic diagnosis of LOPD in many infants who screen positive. However, the definition of what constitutes whether a patient is diagnosed with newborn screening and is “symptomatic” is changing, with increasingly more sensitive measurements of muscle weakness being used, which are leading to the earlier start of treatment for LOPD.
The clinical experts stated there are advantages to having access to multiple products for LOPD treatment. About 5% of patients receiving alglucosidase alfa have problematic immune-mediated reactions to the product. Patients may react to 1 ERT product but not another. Furthermore, as experienced in Fabry disease, biologic manufacturing can be vulnerable to production issues, and shortages can lead to treatment interruptions. Having multiple products reduces the likelihood that patients will go without treatment.
Another clinical expert stated that cipaglucosidase alfa has basically the same mechanism of action as other current ERTs. The difference is the addition of miglustat, which acts as a stabilizer to increase the time that the enzyme remains in circulation without being cleared. As such, a clinician would not use multiple ERT therapies in combination. The clinical expert emphasized that the drug under review is not the first ERT for LOPD because alglucosidase alfa and avalglucosidase alfa are already available. However, it is the first that uses miglustat as a stabilizer.
The clinical expert felt that cipaglucosidase alfa plus miglustat would be used as a first-line treatment (i.e., as an option along with the other available ERT therapies). Given the shared mechanism of action, a clinician would not combine therapies, and there is no evidence for doing this. Cipaglucosidase alfa with miglustat would not necessarily be “reserved” for those who were intolerant to another ERT, but one could conceivably consider switching between first-line treatments if there is any specific contraindication or intolerance to 1 formulation.
Both clinical experts mentioned there is a plateau followed by a decline in efficacy over time in patients with LOPD receiving current ERTs, with some evidence that switching to another drug may regain some of the prior efficacy in those who start to experience decline on their initial ERT. Therefore, 1 clinical expert felt there may be a role for switching between ERT therapies in individuals whose disease initially stabilizes but then starts to decline on treatment. This clinical expert felt there is no rationale for patients trying another drug before cipaglucosidase alfa with miglustat because the alternative is simply another ERT.
Patient Population
The clinical experts agreed that a patient should have an unequivocal diagnosis of LOPD before treatment initiation. Usually, this would include both DNA sequencing of GAA showing biallelic pathogenic variants and an alpha-glucosidase enzyme assay (either white blood cell or bloodspot) showing deficiency in the affected range. If either of these test results is equivocal, then supportive and clarifying tests could include a muscle MRI scan showing a characteristic pattern, electromyography showing characteristic paravertebral myotonic discharges, Hex4 elevation, or a muscle biopsy showing glycogen accumulation.
The clinical experts noted that, currently, the indications to treat are clinically obvious, except for cases of extremely late diagnosis where a patient with LOPD has already advanced to continuous invasive ventilation. In this situation, the expected treatment benefits from ERT are much reduced. The current Canadian evidence-based guideline for LOPD7 is unclear on the appropriate treatment approach for this situation, owing to a lack of available evidence.
One clinical expert pointed out that the patients most likely to respond to cipaglucosidase alfa with miglustat are those who are experiencing clinical weakness, are ambulatory, and who may or may not be using noninvasive ventilation, as highlighted in the Canadian guideline.7 Treatment can still be offered to patients with more advanced disease, but specific treatment goals need to be considered and the decision becomes more individualized in discussion with the patient and family. Patients are generally identified when they develop symptoms and then would be evaluated by a neurologist who would identify weakness and order appropriate testing (e.g., creatine kinase levels, electromyography, muscle MRI, muscle biopsy, genetic testing, enzyme testing). LOPD is likely somewhat underdiagnosed because the symptoms themselves are quite nonspecific, particularly early in the disease course, and thus may be misattributed to other things (e.g., fatigue, mild weakness, mild dyspnea on exertion). Access to enzyme testing and genetic testing is improving, which improves the likelihood of diagnosis. In general, patients with less baseline disability (e.g., less weakness, better ambulatory status) often show a better response to ERT.
Assessing the Response Treatment
One clinical expert emphasized that the treatment response for adults with LOPD can be difficult to perceive on an individual basis. Modest lowering of creatine kinase and/or Hex4 biomarkers, and stabilization of 6MWT and FVC measurements constitute a realistic positive response to current ERT. Certain scales, like the Rotterdam Handicap Scale, can also be followed, with stability of scores considered a positive treatment response. Assessments should be performed every 6 to 12 months.
Another clinical expert added that, in most clinical practice settings, testing is performed on patients that includes the MMT, grip testing with a grip dynamometer, FVC measurements, and sometimes (because it can be impractical in many clinical settings), the 6MWT. One would look for a stabilization (or, in some cases, a slight improvement) in these parameters over time, rather than the expected slow decline over time. The clinical expert did not use any specific scales in their clinical practice.
Discontinuing Treatment
Discontinuation of treatment should be considered in cases of severe immune responses, including anaphylaxis, immune complex disease, or neutralizing antidrug antibodies. However, these AEs can sometimes be overcome, and ERT carefully resumed, with immune-tolerization protocols. The clinical expert pointed out that the Canadian guideline7 states that, “patients in whom respiratory dysfunction and/or skeletal myopathy continue to progress at the same rate despite the introduction of ERT should be considered candidates for cessation of therapy. Also, patients experiencing severe allergic reactions not amenable to standard therapy, a severe comorbid condition limiting lifespan, and noncompliance with infusions and recommended assessments are also indications to consider cessation of therapy in most of the jurisdictions of Canada” (p. 478).7
Practically, it can be challenging to determine whether the rate of progression has slowed or is the same as before treatment. This is a case-by-case decision made by the physician, patient, and family.
Prescribing Considerations
The clinical experts emphasized that it is important that the specialist have experience and training in the management of Pompe disease. A specialist is required for diagnosis, treatment, and monitoring because this is a rare disease. There are a few different specialties that may follow and treat LOPD (e.g., inherited metabolic disease specialists, medical geneticists, neurologists, and physiatrists). For neurologists, it would often be a neuromuscular neurologist, but this would depend highly on the setting (e.g., rural community). This can be done in a specialty clinic or community clinic, depending on the resources and expertise available.
Additional Considerations
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review process by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the 2 clinical experts consulted by CDA-AMC for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Avalglucosidase alfa is not reimbursed in any jurisdiction, and some jurisdictions do not reimburse alglucosidase alfa. | This is a comment from the drug programs to inform CDEC deliberations. |
Considerations for initiation of therapy | |
Patients needed a documented deficiency of GAA enzyme or GAA genotyping showing LOPD to be included in the trial. How is LOPD diagnosed in Canada? Is it standard to evaluate deficiency of GAA enzyme or use GAA genotyping? | In Canada, it is standard to test for GAA enzyme deficiency and use GAA genotyping to diagnose LOPD. A diagnosis of LOPD can usually be confirmed with GAA genotyping, but the clinical experts also confirmed the diagnosis can be made by identifying a GAA enzyme deficiency. All patients in Canada will undergo at least 1 of these investigations, and usually both. |
Patients were required to be ambulatory for the trial, with a sitting FVC ≥ 30% of the predicted value. Should reimbursement criteria reflect this? | The clinical experts stated they would treat patients with a sitting FVC of < 30% if they are not permanently ventilated. The FVC value does not determine whether patients should be treated. According to the Canadian guideline,7 patients who are not fully ventilated are eligible for ERT. |
The trial included patients who were ERT experienced (defined as currently receiving alglucosidase alfa at a recommended dose and regimen for at least 24 months), or ERT naive (have never received ERT). For patients who had been receiving standard ERT for fewer than 24 months, would you expect a full 24-month trial of alglucosidase alfa before considering switching to cipaglucosidase alfa and miglustat? | The clinical experts stated they often see small improvements in respiratory and motor function with ERT, and then a long plateau of about 5 years. Patients would then have a resumption of decline after approximately 5 to 7 years on ERT, at which time the clinical experts would determine that the patients were getting reduced benefit from ERT and consider switching from alglucosidase alfa to cipaglucosidase alfa and miglustat. The clinical experts also noted it is not necessary to do a trial of 1 ERT such as alglucosidase alfa before switching to cipaglucosidase alfa and miglustat, and that patients do not have to have a trial of 24 months of alglucosidase alfa before considering switching to cipaglucosidase alfa and miglustat. |
What is the standard of care for patients with LOPD in Canada? | The standard of care for patients with LOPD in Canada is ERT, and if ERT is not provided to patients in some jurisdictions, then patients would receive supportive care such as exercise, perhaps vitamin D supplementation, and ventilation. |
The proportion of patients who were ERT experienced in the trial was 77.2%. However, patients were not required to be unsuccessful on their current ERT to be eligible for the trial. Should patients switch to cipaglucosidase alfa and miglustat even if they are responding well to treatment with ERT? | The clinical experts noted that if patients are responding well to treatment with ERT (e.g., avalglucosidase alfa or alglucosidase alfa) they would not switch therapy to cipaglucosidase alfa and miglustat, and that patients should be switched to receive cipaglucosidase alfa and miglustat if they experience an IAR, anaphylaxis, or other SAE while receiving another ERT. |
Considerations for continuation or renewal of therapy | |
The primary end point of the trial was change in the 6MWD from baseline to week 52, and the key secondary end points were change in sitting FVC (% predicted) from baseline to week 52, change in the MMT lower extremity score from baseline to week 52, change in 6MWD from baseline to week 26, change in PROMIS Physical Function total score and PROMIS Fatigue score from baseline to week 52, and change in the GSGC total score from baseline to week 52. Are any of these end points used to monitor patients with LOPD in clinical practice in Canada? If not, how are patients monitored for therapeutic response? | One clinical expert stated that the change in 6MWD and in sitting FVC were monitored in practice in part because they were required to report these end points on each of their patients to their ministry. The clinical expert did not use the PROMIS instruments but did use the Short Form (36) Health Survey in practice. The other clinical expert shared that the 6MWT is not always practical because it is difficult to find the space to do a proper test in a neurology clinic. The clinical experts preferred to use the Gower manoeuvre test of the GSGC and the TUG test in practice. The clinical experts noted that patients should continue receiving ERT unless they experience life-threatening and unsurmountable anaphylactic reactions. |
Considerations for discontinuation of therapy | |
The discontinuation criteria recommended by CDEC for avalglucosidase alfa is that treatment with avalglucosidase alfa must be discontinued if the patient develops any of the following:
Should the discontinuation criteria for cipaglucosidase alfa and miglustat align with that of avalglucosidase alfa? | The clinical experts agreed that treatment with cipaglucosidase alfa and miglustat should be discontinued if the patient develops any of the following:
While the goal of treatment is to slow disease progression and avoid motor or respiratory function declining at a rate similar to the rate before therapy, being unsuccessful in reaching this goal does not mean the treatment is not working, i.e., function might have decreased at a faster rate without treatment. |
How would disease progression be defined in patients with LOPD? | Disease progression in patients with LOPD would be defined based on the following: worsening on the 6MWT, MMT, or respiratory function test (as measured by FVC); need for ventilation; or need for increasing ambulatory aids. |
Considerations for prescribing of therapy | |
Cipaglucosidase alfa requires administration by a trained health care professional to monitor and manage IARs and the potential for anaphylaxis. | This is a comment from the drug programs to inform CDEC deliberations. |
Care provision issues | |
Cipaglucosidase alfa needs to be administered in a clinic or hospital setting when first initiated to monitor for IARs, severe allergic reaction, and anaphylaxis. The sponsor states that cipaglucosidase alfa with miglustat can be administered at home by a trained health care professional after the evaluation of the risk for IARs. Patients in the PROPEL trial were eligible for home administration, conducted by a trained home-infusion nurse, after participating in the trial for 6 months. | This is a comment from the drug programs to inform CDEC deliberations. |
Patients must be monitored for development of severe infusion-associated reactions and anaphylaxis. | This is a comment from the drug programs to inform CDEC deliberations. |
Premedication with oral antihistamines, antipyretics, and/or corticosteroids may be required for potential infusion-associated reactions. | This is a comment from the drug programs to inform CDEC deliberations. |
System and economic issues | |
Given the rarity of LOPD, do you anticipate patients needing to travel to a larger centre to receive treatment? | Based on the clinical experts’ experience, patients have been able to receive infusion services from local hospitals in remote areas, and it has been working well for patients. |
Alglucosidase alfa was reviewed before the creation of pCPA, so it is unclear whether jurisdictions have confidential negotiated prices. | This is a comment from the drug programs to inform CDEC deliberations. |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; CDEC = Canadian Drug Expert Committee; ERT = enzyme replacement therapy; FVC = forced vital capacity; GAA = acid alpha-glucosidase; GSGC = gait, stair, Gower manoeuvre, and chair; IAR = infusion-associated reaction; LOPD = late-onset Pompe disease; MMT = manual muscle test; pCPA = pan-Canadian Pharmaceutical Alliance; PROMIS = Patient-Reported Outcomes Measurement Information System; SAE = severe adverse event; TUG = Timed Up and Go.
Clinical Evidence
The objective of this Clinical Review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of cipaglucosidase alfa for injection (Pombiliti), lyophilized powder, 105 mg per vial, IV infusion in combination with miglustat (Opfolda) 65 mg oral capsules for treatment in adult patients with LOPD (GAA deficiency). The focus will be placed on comparing cipaglucosidase alfa plus miglustat with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of cipaglucosidase alfa with miglustat is presented in 4 sections, with the CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The CDA-AMC assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal RCT identified in the systematic review (PROPEL)
1 long-term extension study
1 ITC
2 additional studies addressing gaps in evidence (i.e., real-world data studies).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
One pivotal trial (PROPEL) was identified from the sponsor submission. Characteristics of the PROPEL trial are summarized in Table 5.
Table 5: Details of Studies Included in the Systematic Review
Detail | PROPEL and ATB200-03 trials |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, DB, active-controlled, multicentre, global trial |
Locations | 62 sites in 24 countries (Argentina, Australia, Austria, Belgium, Bosnia and Herzegovina, Bulgaria, Canada, Denmark, France, Germany, Greece, Hungary, Italy, Japan, the Netherlands, New Zealand, Poland, Slovenia, South Korea, Spain, Sweden, Taiwan, UK, and the US) |
Patient enrolment dates |
|
Randomized (N) | 123 patients randomized 2:1 to cipaglucosidase alfa with miglustat (N = 85) or alglucosidase alfa (N = 38) |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Coadministration of cipaglucosidase alfa and miglustat:
|
Comparators | Coadministration of alglucosidase alfa and placebo:
|
Study duration | |
Screening phase | 30 days |
Treatment phase | 52 weeks |
Follow-up phase | 30 days |
Outcomes | |
Primary end point | Change in 6MWD (the distance walked in the 6MWT, in metres) from baseline to week 52, testing for superiority of the intervention versus alglucosidase alfa and placebo. |
Secondary and exploratory end points | Key secondary:
Other secondary:
Post hoc:
|
Publication status | |
Publications | Schoser et al. (2021)16 Kishnani et al. (2024)17 ClinicalTrials.gov: NCT03729362, NCT04138277 |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; DB = double-blind; ERT = enzyme replacement therapy; FVC = forced vital capacity; GAA = acid alpha-glucosidase; GSGC = gait, stair, Gower manoeuvre, and chair; LOPD = late-onset Pompe disease; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; MMT = manual muscle test; PGIC = Physician’s Global Impression of Change; PRO = patient-reported outcome; PROMIS = Patient-Reported Outcomes Measurement Information System; QMT = quantitative muscle testing; R-PAct = Rasch-built Pompe-specific activity scale; SGIC = Subject Global Impression of Change; SNIP = sniff nasal inspiratory pressure; TUG = Timed Up and Go; VAS = visual analogue scale; VC = vital capacity; SVC = sitting vital capacity.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
The primary objective of the PROPEL trial was to assess the efficacy of cipaglucosidase alfa and miglustat coadministration compared with alglucosidase alfa plus placebo on ambulatory function. The primary efficacy end point was the change from baseline to week 52 in 6MWD, measured by the distance walked in the 6MWT.
PROPEL16,17 was a phase III, double-blind, multicentre, active-controlled RCT in adult patients with LOPD (N = 123). The trial design is outlined in Figure 1 and consisted of a screening period of up to 30 days and a 52-week double-blind treatment period. The trial is closed; the database lock occurred on January 20, 2021.
The PROPEL trial was conducted at 62 sites in 24 countries, including 2 sites in Canada. Eligible patients were randomized in a 2:1 ratio using interactive response technology software to either the cipaglucosidase alfa plus miglustat group (n = 85) or the alglucosidase alfa plus placebo group (n = 38). Randomization was stratified by baseline 6MWD (75 m to < 150 m, 150 m to < 400 m, or ≥ 400 m) and previous ERT status (ERT experienced and ERT naive). Neither the patients nor study investigators were aware of the treatments received. Placebo was supplied to study sites as tablets matching the appearance of miglustat 65 mg tablets.
Figure 1: PROPEL (ATB200-03) Trial Design Schematic

ERT = enzyme replacement therapy; LOPD = late-onset Pompe disease; PO = by mouth.
Sources: ATB200-03 Clinical Study Report18 and the sponsor’s summary of clinical evidence.
Populations
Inclusion and Exclusion Criteria
Patients aged 18 years and older were eligible to participate in the PROPEL trial if they had a diagnosis of LOPD based on GAA enzyme deficiency testing or GAA genotyping results. A sitting FVC of 30% or higher of the predicted value for healthy adults and 2 valid 6MWTs (criteria described in Table 5) were required. Patients who were currently receiving and had been receiving alglucosidase alfa for more than 24 months (ERT experienced) or had never received any ERT (ERT naive) were eligible for inclusion. Patients that received any investigational treatments for Pompe disease other than alglucosidase alfa or gene therapy for Pompe disease were excluded. Patients were also ineligible if they were using ventilation support for more than 6 hours per day while awake or taking any prohibited medications (miglitol, miglustat, acarbose, or voglibose).
Interventions
Patients in the treatment group were administered cipaglucosidase alfa every 2 weeks in a 15 mg/mL powder concentrate for solution as a 4-hour IV infusion, at a dose of 20 mg/kg of body weight. Miglusat was administered 1 hour before the cipaglucosidase alfa infusion. The dose was 195 mg (3 × 65 mg oral capsules) for patients weighing from 40 kg to less than 50 kg and 260 mg (4 × 65 mg oral capsules) for patients weighing 50 kg or more.
Patients in the comparator group were administered alglucosidase alfa every 2 weeks in a 5 mg/mL powder concentrate for solution as a 4-hour IV infusion, at a dose of 20 mg/kg of body weight. The placebo was administered 1 hour before infusion with alglucosidase alfa.
Cipaglucosidase alfa and alglucosidase alfa appeared identical and were supplied in single-use, clear 20 mL glass vials. Administration was performed at a hospital or study site. After participation in the study for 6 months, patients were eligible for home infusions. Miglustat was supplied as white, hard capsules in 40 mL high-density polyethylene bottles and the placebo was matched to miglustat. Patients were required to fast for at least 2 hours before and 2 hours after receiving miglustat or placebo.
Patients who were ERT experienced continued to take alglucosidase alfa during the screening period; treatment with alglucosidase alfa was then replaced by the study drug (cipaglucosidase alfa with miglustat or alglucosidase alfa with placebo) on the same schedule without interruption (i.e., every 2 weeks).
Outcomes
A list of the efficacy end points assessed in this Clinical Review report is provided in Table 6, followed by descriptions of the outcome measures in Table 7. The summarized end points are based on outcomes included in the sponsor’s summary of clinical evidence as well as any outcomes identified as important to this review according to the clinical experts consulted and input from the patient group and public drug plans. Using the same considerations, the review team selected end points that were most relevant to inform expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Per the input received by the clinical experts, 6MWD (to assess motor function) and percent predicted sitting FVC (to assess respiratory function) were reflective of the treatment goals for LOPD and the results were assessed using GRADE. The PROMIS Physical Function score and PROMIS Fatigue score captured health-related quality of life and were also assessed using GRADE. The remaining outcomes were summarized as supportive information. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
Six-Minute Walk Distance
Change in 6MWD (defined as distance walked in the 6MWT in metres) from baseline to week 52 was the primary efficacy end point in the PROPEL trial and was identified as a key outcome by the clinical experts consulted. The 6MWD is a supervised, objective test used to assess ambulatory function involving the skeletal muscle, pulmonary, and cardiac systems, and motor function. Patients were instructed to walk (not run or jog) as far as possible for 6 minutes on a flat surface.
The validity and reliability of the 6MWT has been established, including in Pompe disease, where the 6MWT showed validity and responsiveness to change compared with the Rotterdam Handicap Scale and Rasch-built Pompe-specific activity instruments.19 No information on reliability was identified from the literature for patients with LOPD, although reliability has been established in various other conditions.20,21 In 1 review, the MIDs for 6MWT outcomes in patients with LOPD ranged from 2.27% to 8.11%, depending on the chosen method and disease severity, translating to a distance of 23.7 m to 57.2 m in the 6MWD.22 In a separate study, the between-group MID for 6MWT outcomes in patients with LOPD ranged from 0.35% to 7.47% points, equivalent to a distance of 2.18 m to 46.61 m and 1.97 m to 42.13 m for a male and a female, respectively, aged 50 years, with a height of 1.75 m and a weight of 80 kg.23
Based on input from the clinical experts, a 20 m change in the 6MWD was considered the MID for 6MWT outcomes in this review.
Table 6: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | PROPEL trial |
|---|---|---|
6MWD (distance walked in the 6MWT) | Change from baseline to week 52 | Primarya |
Sitting FVC (% predicted) | Change from baseline to week 52 | Key secondarya |
MMT lower extremity score | Change from baseline to week 52 | Key secondary |
6MWD | Change from baseline to week 26 | Key secondary |
PROMIS Physical Function total score | Change from baseline to week 52 | Key secondarya |
PROMIS Fatigue total score | Change from baseline to week 52 | Key secondarya |
Percent predicted 6MWD | Change from baseline to week 52 | Secondary |
Gower manoeuvre of the GSGC test | Change from baseline to week 52 | Secondary |
TUG test | Change from baseline to week 52 | Secondary |
MMT total score | Change from baseline to week 52 | Secondary |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; FVC = forced vital capacity; GRADE = Grading of Recommendations Assessment, Development and Evaluation; GSGC = gait, stair, Gower manoeuvre, and chair; MMT = manual muscle test; PROMIS = Patient-Reported Outcomes Measurement Information System; TUG = Timed Up and Go.
aOutcomes that were identified by the clinical experts as essential for decision-making and that were assessed using GRADE.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Sitting FVC
FVC in a sitting position was the first key secondary efficacy end point in the PROPEL trial and was identified as a key outcome by the clinical experts consulted by CDA-AMC. FVC is an objective pulmonary function test, defined as the total volume of air that can be forcibly exhaled after maximal inspiration. Normal FVC values range from 80% to 120%; a lower FVC value may indicate abnormal lung function. Acceptable validity has been demonstrated in patients with Pompe disease. No information on reliability and responsiveness was identified for patients with Pompe disease. The MIDs for upright seated FVC ranged from 2.47% to 4.83% points in patients with Pompe disease treated with ERTs.23 In a literature review, the majority of studies conducted in LOPD (6 out of 9) showed the relative change from baseline was higher or within the MID range (5% to 11%) established for idiopathic pulmonary fibrosis.24
Based on input from the clinical experts, 3% was considered the MID for sitting FVC in this review.
Manual Muscle Test
Manual muscle strength was assessed through a neurologic (physical) exam that uses the Medical Research Council scale (0 to 5 points, with 5 indicating normal function). The same rater and method were used as much as possible throughout the patient’s participation in the study. The following is a description of the relevant MMT scores:
MMT lower extremity score: The total score for MMT lower extremity strength was obtained by summing the test scores across the following 8 body parts: right and left hip flexion, right and left hip abduction, right and left knee flexion, and right and left knee extension (i.e., the lower extremity score equals the sum of the hip scores plus knee scores). The total lower extremity score ranged from 0 to 40, with lower scores indicating lower muscle strength.
MMT total score: The MMT total score was the sum of the lower extremity score and the upper extremity score. The total score ranged from 0 to 80, with lower scores indicating lower overall muscle strength.
MIDs for MMT scores specific to patients with LOPD were not identified in the literature.
Patient-Reported Outcomes Measurement Information System
Health-related quality of life was assessed using 2 PROMIS instruments.
The PROMIS Short Form v2.0 – Physical Function 20a consists of 20 items. The first 14 questions are scored on a scale of 1 to 5 with responses as follows: 1 = unable to do, 2 = with much difficulty, 3 = with some difficulty, 4 = with a little difficulty, and 5 = without any difficulty. The next 6 questions are scored on a scale of 1 to 5 and have the following responses: 1 = cannot do, 2 = quite a lot, 3 = somewhat, 4 = very little, and 5 = not at all. The total score ranges between 20 and 100, with a higher score indicating better physical functioning. The PROMIS Physical Function instrument has been validated in many conditions, including in Pompe disease.24,25 MIDs of 2.4 (anchor-based) and 4.2 (distribution-based) have been reported for a clinical important improvement in physical function in patients with LOPD.25 The review team used these MIDs (2.4 and 4.2) for the PROMIS Physical Function score in this review.
The PROMIS Short Form v1.0 – Fatigue 8a consists of 8 items. Six items are scored on a scale of 1 to 5 with responses as follows: 1 = not at all, 2 = a little bit; 3 = somewhat; 4 = quite a bit, and 5 = very much. Two questions are scored on a scale of 1 to 5 with responses as follows: 1 = never, 2 = rarely, 3 = sometimes, 4 = often, and 5 = always. The total score ranges between 8 and 40, with lower scores indicating less fatigue. An MID for the PROMIS Fatigue instrument in patients with LOPD was not identified in the literature.
Gower Manoeuvre
Gower manoeuvre is an individual functional test of the GSGC composite test. The Gower manoeuvre involves the patient lying down on the floor, then rising from the floor to get to a standing position. The time (in seconds) to perform the test is recorded. An MID for the Gower manoeuvre specific to patients with LOPD was not identified in the literature.
Timed Up and Go
The TUG test is a mobility test that assesses balance, gait speed, and functional ability. The TUG test measures the time a patient needs to rise from a chair, walk 3 m, turn around, walk back to the chair, and sit down, all at their regular pace. An MID for the TUG test specific to patients with LOPD was not identified in the literature.
Table 7: Summary of Efficacy Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Distance walked in the 6MWT in metres, defined as the 6MWD | A supervised test that measures the distance a patient can walk on a hard, flat surface over a 6-minute period. The 6MWT is a commonly used test to evaluate global function of organ systems involved in exercise, namely the heart, lungs, peripheral circulation, blood, nervous system, muscles, bones, and joints during walking, a self-paced activity. | Validity: In patients with a confirmed diagnosis of Pompe disease, 6MWT scores were significantly associated with better Rotterdam Handicap Scale and R-PAct scores.19 Reliability: No evidence was found for patients with Pompe disease. Responsiveness: Measure was found to be associated with Rotterdam Handicap Scale and R-PAct scores in patients with Pompe disease.19 | MIDs varied widely, depending on the method used and severity of Pompe disease. Based on data from the PROPEL trial, the MIDs for the 6MWT in patients with LOPD ranged from 2.27% to 8.11%, depending on the chosen method and disease severity, translating to 23.7 m to 57.2 m in the 6MWD.22 In a literature review, the between-group MID for the 6MWT in patients with LOPD ranged from 0.35% to 7.47% points, equivalent to 2.18 m to 46.61 m in the 6MWD for males and 1.97 m to 42.13 m for females, all aged 50 years, with a height of 1.75 m and weight of 80 kg.23 In a separate review, it was reported that 9 of 10 studies reported absolute changes from baseline lay within or more than the 6MWT MID of 24 m to 54 m in the 6MWD.24 The sponsor suggested a threshold of 6% improvement from baseline in the 6MWT, and the clinical experts validated a change of 20 m in 6MWD as the MID for this review. |
Sitting FVC | A measure of the volume of air that can be forcibly exhaled from the lungs after taking the deepest breath possible. Typically reported as the percentage of the volume predicted for a person of the same size, age, and sex. | Validity: Construct validity was demonstrated in patients with Pompe disease.19 Reliability: No information was identified for patients with Pompe disease. Responsiveness: No information was identified for patients with Pompe disease. | Between-group MIDs for upright seated FVC ranged from 2.47% to 4.83% points in patients with Pompe disease treated with ERTs.23 The sponsor suggested a threshold of 3% improvement from baseline, which was validated by the clinical experts for this review. |
MMT | An instrument testing 16 muscle groups or motions, scored on a scale of 0 to 5. Upper extremity MMT refers to shoulders and elbows; lower extremity MMT refers to hips and knees. | No evidence found for patients with Pompe disease. | An MID was not found for Pompe disease in the literature. |
PROMIS | A 7-domain patient-reported outcome of HRQoL measuring depression, anxiety, physical function, pain interference, fatigue, sleep disturbance, and the ability to participate in social roles and activities. | Validity: The PROMIS Physical Function instrument has been validated in many conditions, including in Pompe disease.25,26 Reliability: No evidence was found for patients with Pompe disease, although this is a common HRQoL instrument used in many conditions. Responsiveness: Responsiveness was reported for the Physical Function component of PROMIS.25 | MIDs of 2.4 (anchor-based) and 4.2 (distribution-based) have been reported for a clinically important improvement in PROMIS Physical Function in patients with LOPD.25 A relevant MID for PROMIS Fatigue was not found in the literature. |
GSGC Gower manoeuvre | One of 4 main motor performances on GCGC composite test. Gower manoeuvre assessed time to standing from laying. Scores for the test are from 1 (normal) to 7 points (unable to arise). | No evidence found for patients with Pompe disease. | A relevant MID was not found. |
TUG | A simple test that can be performed anywhere and consists of a patient getting up from a chair from the sitting to the bipedal position, walking 3 m, turning, returning, and sitting on the chair again. The total time taken to do the test is recorded in seconds. | No evidence found for patients with Pompe disease. | A relevant MID was not found. |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; ERT = enzyme replacement therapy; FVC = forced vital capacity; GSGC = gait, stair, Gower manoeuvre, and chair; HRQoL = health-related quality of life; LOPD = late-onset Pompe disease; MID = minimal important difference; MMT = manual muscle test; PROMIS = Patient-Reported Outcomes Measurement Information System; R-PAct = Rasch-built Pompe-specific activity; TUG = Timed Up and Go.
Harms
All AEs were measured in the PROPEL trial. AEs were defined as any untoward medical occurrence in a patient treated with a pharmaceutical product, biologic (at any dose), or medical device that did not necessarily have a causal relationship with the treatment. Therefore, an AE could be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medical product, whether or not considered related to the medical product.
A TEAE was defined as any AE that began after the first dose of the study drug. Patients experiencing AEs were followed up until their health returned to baseline status or stabilized.
A serious TEAE was defined as any TEAE that resulted in death, was life-threatening, required inpatient hospitalization or prolonged existing hospitalization, resulted in persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions (as decided by the investigator), or was a congenital anomaly or birth defect.
An IAR was defined as a disorder characterized by 1 or more adverse reactions to the infusion of pharmacological or biological substances. These reactions were classified into 2 major subtypes, immediate and late, according to the time interval between the infusion and the onset of an infusion-related AE.
Statistical Analysis
Sample Size and Power Calculation
The sample size calculation in the PROPEL trial was based on superiority testing of the primary efficacy end point of change from baseline to week 52 in the 6MWD. An effect size of 1.31 with a mean improvement of 9.7% (SD = 7.42%) in percent predicted 6MWD over 12 months with cipaglucosidase alfa plus miglustat compared with alglucosidase alfa alone was estimated based on data from the LOTS extension study.27 With a 1-sided significance level of 0.025 and an expected dropout rate of 10% (after randomization), 99 participants were required to achieve an overall 90% power to detect an effect size of 0.7 (53% of the estimated effect size of 1.31), accounting for uncertainty.
Statistical Testing
For all end points, the baseline and week 52 values were based on the average of the last 2 values obtained on or before the first dose date and at the week 52 or end of study visit (or end of trial visit), respectively. However, some patients may have had only 1 assessment of 6MWD at week 52. In this case, the single assessment was used as the week 52 value. As a result of COVID-19–related policies and restrictions, the week 52 visit (and/or the end of study or end of trial visit) could have been delayed. If that happened, the delayed visit assessment was still used for the analyses. Finally, for all percent predicted FVC outcomes, the following formula was used: percent predicted outcome equals (actual value of the outcome divided by predicted value of the outcome) multiplied by 100.
Primary Efficacy End Point Analysis
The primary efficacy end point (change in 6MWD from baseline to week 52) was analyzed using a mixed model for repeated measures (MMRM) on the intention-to-treat observed (ITT-OBS) population (described in Table 8). The model was adjusted for treatment, baseline 6MWD, age, height, weight, ERT status, sex, time, and treatment-by-time interaction. Time was used as a repeated measure, and an unstructured covariance approach was applied. A restricted maximum likelihood estimation and unstructured within-patient covariance structure was used for the analysis, with the Kenward-Roger approximation to estimate denominator degrees of freedom and adjust standard errors for the tests of fixed effects. If this model failed to converge, a compound symmetry covariance structure was used instead. The significance test between the treatment groups was based on the treatment comparison of LS means at week 52 and a P value was presented for this time point only.
Key Secondary and Other Secondary End Point Analyses
Each secondary end point was analyzed using an analysis of covariance (ANCOVA) model on the intention-to-treat last observation carried forward (ITT-LOCF) population. The model adjusted for treatment, baseline of response variable, age, height, weight (all as continuous covariates), ERT status, and sex. The use of the observed marginal distributions of the categorical variable was allowed rather than assuming a balance among the levels of the categorical variable.
Sensitivity Analyses
Sensitivity analyses performed in the PROPEL trial are provided in Table 8 and summarized here:
Nonparametric randomization-based covariance specified as the first sensitivity analysis for 6MWD and FVC: This nonparametric analysis was to be conducted if the primary parametric analyses failed to meet assumptions of normality. For this method, the ERT status (naive or experienced) was specified as strata, and baseline value, baseline age, sex, height, and weight were specified as covariates. The categorical variable (i.e., sex) was recoded as a numeric variable. Due to the potential for smaller ERT-naive stratum size, the weighted estimates were combined across strata before covariance adjustment. The covariance matrix calculated under the alternative hypothesis was used for a statistical test to compare between the treatment groups.
MMRM (primary end point) or ANCOVA (key secondary end points) incorporating the handling of the intercurrent events: This model was adjusted for the baseline value (continuous) and ERT status, as well as baseline age, sex, baseline height, and baseline weight.
ANCOVA applied to the per-protocol population: This model was adjusted for the baseline value (continuous) and ERT status, as well baseline age, sex, baseline height, and baseline weight to compare between the 2 treatment groups.
ANCOVA for percent predicted 6MWD: An ANCOVA using the change from baseline to week 52 in percent predicted 6MWD in which the percent predicted 6MWD was calculated using the Enright and Sherrill (1998) reference equations. This analysis was performed on observed data (ITT-OBS population) and the model was adjusted for baseline 6MWD (as a continuous variable) and ERT status to compare between the 2 treatment groups. The baseline age, sex, height, and weight were to be included as covariates in this ANCOVA analysis because they are already incorporated in the calculation of the percent predicted 6MWD values.
Randomization test applied to ANCOVA: An analysis using a randomization test applied to the test statistics obtained from the ANCOVA model and adjusted for baseline 6MWD (as a continuous variable), ERT status, baseline age, sex, baseline height, and baseline weight to compare between the 2 treatment groups. The P value for comparing between the 2 treatment groups was provided. This analysis was performed on the ITT-LOCF population.
Outlier data: One patient who was ERT naive reportedly had aberrant data points in the comparator group that turned out to be very influential outliers and distorted the mean change estimates calculated from the raw data. The data point from this patient alone accounted for approximately 56% of the mean change from baseline at week 52 in the alglucosidase alfa plus placebo group and inflated the variance in the comparator group to approximately 6 times that of the cipaglucosidase alfa plus miglustat group. A prespecified analysis of the primary efficacy outcome was performed that excluded the outlier data with externally studentized residuals greater than 3 in magnitude. In addition, all analyses of the efficacy end points were rerun excluding the data from this patient.
Multiple Testing Procedure
The primary and key secondary end points were analyzed in a hierarchical manner to control for familywise type I error at 0.025. The test for the primary end point was conducted first at the 1-sided 0.025 significance level and, if significant, the key secondary end points were tested in the order listed subsequently at the same significance level, each tested at the 1-sided alpha level of 0.025 (all measured at week 52): percent predicted sitting FVC, MMT lower extremity score, PROMIS Physical Function total score, and PROMIS Fatigue total score. If at any point the null hypothesis failed to be rejected, then that comparison and any other comparison below it could not be claimed as statistically significant on superiority, and subsequent analyses were conducted to assess for nominal statistically significance on superiority.
The hypothesis tests included in the other secondary end points (percent predicted 6MWD, Gower manoeuvre of the GSGC test, TUG test, and MMT total score) were not controlled for multiplicity, and each were tested at a 1-sided alpha level of 0.025.
Data Imputation Methods
An overview of missing data imputation methods for all outcomes and analyses in the PROPEL trial is provided in Table 8. For the primary analysis of the primary efficacy end point, 6MWD at week 52, the MMRM analysis used all available data without imputation for missing data, and the amount of missing data for 6MWD at week 52 was expected to be small (< 10%). Likewise, no baseline imputation was done for the first key secondary outcome of FVC.
For the multi-item end point (MMT total score), a patient could be missing data for either specific items or the entire assessment. If the baseline value was partially missing (patient was missing data for only specific items at baseline), the average value using all patients with nonmissing values for that item across the 2 treatment groups combined was used to replace the missing item score(s). If the baseline total score was completely missing, the average score using all patients with nonmissing total scores for the end point across the 2 treatment groups combined was used to replace the missing baseline total score.
Patients who permanently discontinued the assigned study drug were subsequently discontinued from the study. However, these patients were required to return for the end of study or end of trial visit. Efficacy assessments collected at the end of study or early-termination visit was used to replace the missing end point value at week 52 in the ITT-LOCF population, that is, the last available observation was carried forward to replace the missing value at week 52.
Table 8: Statistical Analysis of Efficacy End Points in the PROPEL Trial
End point | Statistical model | Analysis population | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|---|
Change from baseline to week 52 in 6MWD (primary end point) | MMRM | ITT-OBS | Fixed categorical effects: Treatment, time (i.e., visit), treatment-by-time interaction, ERT status, and sex. Fixed continuous covariates: Baseline 6MWD, baseline age, baseline weight, and baseline height. | Baseline value: Not needed. Postbaseline value: LOCF using EOS or ET visit data (those who permanently discontinued); no imputation was done for ITT-OBS analysis. |
|
FVC and all key secondary efficacy end points | ANCOVA | ITT-LOCF | Continuous covariate: Baseline value. Categorical covariate: ERT status (ERT naive vs. ERT experienced), baseline age, sex, baseline height, and baseline weight. | Baseline value: Not needed for FVC. For multicomponent and multi-item end points, the baseline value was the average value among all patients with nonmissing values for that item across all treatment groups combined (defined as partially missing), or the average score of all patients with nonmissing total scores for the end point across all treatment groups combined (defined as completely missing). Postbaseline value: LOCF using EOS or ET visit data (those who permanently discontinued); no imputation done for ITT-OBS analysis. |
|
Motor function tests (% predicted 6MWD, Gower manoeuvre, TUG test) | ANCOVA | ITT-LOCF | Similar to key secondary efficacy end points | Baseline value: For multicomponent and multi-item end points, the baseline value was the average value among all patients with nonmissing values for that item across all treatment groups combined (defined as partially missing), or the average score of all patients with nonmissing total scores for the end point across all treatment groups combined (defined as completely missing). Postbaseline value: LOCF using EOS or ET visit data (those who permanently discontinued); no imputation done for ITT-OBS analysis. | None |
Muscle strength test (MMT) | ANCOVA | ITT-LOCF | Similar to key secondary efficacy end points | Baseline value: For multicomponent and multi-item end points, the baseline value was the average value among all patients with nonmissing values for that item across all treatment groups combined (defined as partially missing), or the average score of all patients with nonmissing total scores for the end point across all treatment groups combined (defined as completely missing). Postbaseline value: LOCF using EOS or ET visit data (those who permanently discontinued); no imputation done for ITT-OBS analysis. | None |
PROs (PROMIS instruments) | ANCOVA | ITT-LOCF | Similar to key secondary efficacy end points | Baseline value: For multicomponent and multi-item end points, the baseline value was the average value among all patients with nonmissing values for that item across all treatment groups combined (defined as partially missing), or the average score of all patients with nonmissing total scores for the end point across all treatment groups combined (defined as completely missing). Postbaseline value: Total score calculated and prorated as average of nonmissing items multiplied by total number of items expected (if ≥ 50% of items available), total score not calculated and set to missing (ITT-OBS population) or imputed as indicated previously (ITT-LOCF population). | None |
6MWD = 6-minute walk distance; ANCOVA = analysis of covariance; EOS = end of study; ERT = enzyme replacement therapy; ET = early termination; FVC = forced vital capacity; ITT-LOCF = intention-to-treat last observation carried forward; ITT-OBS = intention-to-treat observed; MMRM = mixed model for repeated measures; MMT = manual muscle test; PP = per protocol (population); PP1 = population used for the per-protocol (sensitivity) analysis of 6MWD; PP2 = population used for the per-protocol (sensitivity) analysis of % predicted forced vital capacity; PRO = patient-reported outcome; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; TUG =Timed Up and Go; VAS = visual analogue scale; vs. = versus.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Subgroup Analyses
Based on the clinical expert opinion, the following prespecified subgroups for the primary (6MWD) and key secondary end point (percent predicted sitting FVC) were noted as relevant for this review:
ERT status (ERT experienced and ERT naive)
ERT duration (2 to < 3, 3 to < 5, and ≥ 5 years)
Baseline 6MWD (75 m to < 150 m, 150 m to < 400 m, and ≥ 400 m)
Age group (18 to < 35, 35 to < 50, 50 to < 65, and ≥ 65 years).
Analysis Populations
Full details of the analysis populations are provided in Table 9.
Table 9: Analysis Populations of the PROPEL Trial
Population | Definition | Application |
|---|---|---|
All randomized | All patients who were randomized. | None |
ITT-observeda | All patients who were randomized and received at least 1 dose of the study drug and analyzed according to planned treatment groups using all available observed data without imputation for missing postbaseline data (i.e., missing data at week 52 and at other visits were not replaced). | Baseline and demographic summaries and primary efficacy outcome |
ITT-LOCFa | All patients who were randomized and received at least 1 dose of the study drug, analyzed according to planned treatment groups with missing data replaced with the last available value from postbaseline results (i.e., LOCF replaced missing data at weeks 26, 38, and 52 with the last available end point value). | Key secondary and other secondary efficacy outcomes |
Those who did not complete the study | Patients who completed the week 52 visit and had both 6MWD and FVC assessments at week 52 (including make-up assessments or delayed visits), or those who completed week 52 visit and were able to do only 1 6MWT (instead of 2) at week 52. If a patient withdrew or was discontinued from the study and returned for the EOS visit, the patient was not considered to have completed the study. | Key secondary and other secondary efficacy outcomes |
PP1 population | Subset of the mITT population consisting of any patients in the mITT who did not have prespecified protocol deviations that were considered important. The PP population was analyzed according to the actual treatment received. | Primary efficacy outcome |
PP2 population | Subset of the mITT population consisting of any patients in the mITT who did not have prespecified protocol deviations that were considered important. The PP population was analyzed according to the actual treatment received. | Key secondary efficacy outcome (FVC only) |
Safety | Patients who received at least 1 dose of the study drug. Patients were analyzed according to the actual treatment received. | Safety data |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; EOS = end of study; FVC = forced vital capacity; ITT = intention to treat: ITT-LOCF = intention-to-treat last observation carried forward; ITT-OBS = intention-to-treat observed; LOCF = last observation carried forward; mITT = modified intention to treat; PP = per protocol.
aThe efficacy (ITT-OBS and ITT-LOCF) and safety populations included all patients who were randomized, with the exception of 2 patients in the alglucosidase alfa plus placebo group who were excluded from the ITT and safety populations because they were enrolled in the study but did not receive study treatment.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Patient Disposition
Table 10 summarizes the disposition for all patients enrolled in the PROPEL trial. Of 130 patients screened for inclusion into the study, 125 patients (85 ERT experienced and 40 ERT naive) were randomized. Five screened patients did not meet the study criteria (e.g., sitting FVC = 30% predicted value for healthy adults), and were not randomized. Two of the patients randomized to the alglucosidase alfa group were not dosed because genotyping did not confirm a diagnosis of Pompe disease. Among the 6 patients who discontinued from the study, 5 were in the cipaglucosidase alfa plus miglustat group and 1 was in the alglucosidase alfa group. Three of the 5 patients receiving cipaglucosidase alfa plus miglustat discontinued due to an AE (COVID-19–related pneumonia, withdrawn consent due to a serious TEAE of IAR anaphylactic event and IAR chills); the remaining patients discontinued due to unwillingness to travel to the study site during the COVID-19 pandemic. One patient in the alglucosidase alfa with placebo group discontinued due to a cerebrovascular accident AE that was considered by the investigator to be unrelated to study drug. The recruitment period was December 2018 to November 2019, and data collection was completed in December 2020.
Table 10: Summary of Patient Disposition From the PROPEL Trial Included in the Systematic Review
Patient disposition | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat | Alglucosidase alfa + placebo | |
Screened, N | 130 | |
Unsuccessful screening, n | 5 | |
Reason for unsuccessful screening, n | ||
Not meeting inclusion criteria | 5 | |
Randomized, N | 85 | 40 |
Randomized but not dosed, n | 0 | 2 |
Completed the study, n (%)a | 80 (94.1) | 37 (97.4) |
Discontinued from the study, n (%)a | 5 (5.9) | 1 (2.6) |
Reason for discontinuation, n (%) | ||
Adverse event | 0b | 1 (2.6) |
Withdrawal of consent by patient | 2 (2.4)c | 0 |
Investigator’s decision | 1 (1.2)d | 0 |
COVID-19 pandemic | 1 (1.2)e | 0 |
Other | 1 (1.2)f | 0 |
ITT-OBS and ITT-LOCF, N (%)a | 85 (100.0) | 38 (95.0) |
PP1, N (%)a | 77 (90.6) | 37 (97.4) |
PP2, N (%)a | 74 (87.1) | 33 (86.8) |
Safety, N (%)a | 85 (100.0) | 38 (100.0) |
IAR = infusion-associated reaction; ITT = intention to treat; ITT-LOCF = intention-to-treat last observation carried forward; ITT-OBS = intention-to-treat observed; PP1 = population used for the per-protocol (sensitivity) analysis of 6MWD; PP2 = population used for the per-protocol (sensitivity) analysis of % predicted forced vital capacity.
Note: Unsuccessful screening occurred for 5 patients who signed an informed consent form but did not meet study inclusion criteria and were therefore not randomly assigned.
aPercentages were based on the number of patients in each treatment group for the ITT population, which consisted of patients who were randomized and who received at least 1 dose of the study drug.
bThree patients discontinued cipaglucosidase alfa with miglustat due to AEs: Covid-19–related pneumonia (“other” was checked as reason), IAR (“withdrawal of consent by subject” was checked as reason), and IAR (“investigator’s decision” was checked as reason).
cOne patient withdrew consent due to an IAR that led to an anaphylactic event; 1 patient withdrew consent because they did not want to travel to the site.
dOne patient discontinued due to an IAR.
eOne patient withdrew, unwilling to travel to the site.
fOne patient discontinued due to Covid-19–related pneumonia.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
The baseline characteristics outlined in Table 11 are limited to those that are most relevant to this review or were felt to affect the outcomes or interpretation of the study results. The mean age of patients was 46.8 years (SD = 13.3 years), and the mean age of patients at diagnosis was 38.9 years (SD = 14.3 years). Body weight was slightly lower in the cipaglucosidase alfa plus miglustat group (mean = 73 kg) compared with the alglucosidase alfa plus placebo group (mean = 79 kg). The mean body mass index was comparable in the cipaglucosidase alfa plus miglustat group (25 kg/m2) compared with the alglucosidase alfa plus placebo group (27 kg/m2).
Baseline 6MWD was comparable in the intervention group (mean = 357.9 m; SD = 111.8 m) and the comparator group (mean = 350.1 m; SD = 119.8 m). A comparable proportion of patients in the cipaglucosidase alfa plus miglustat (mean = 97%) and alglucosidase alfa plus placebo group (mean = 96%) had baseline values for percent predicted FVC of 85% or more.
Ninety-five of 123 patients (77%) were ERT experienced and the remaining 38 patients (23%) were ERT naive. In the ERT-experienced group, average ERT treatment duration was 7.4 years (SD = 3.4 years) overall, 7.5 years (SD = 3.4 years) in the treatment group, and 7.1 years (SD = 3.6 years) in the comparator group. Overall, 67% of patients had 5 or more years of prior treatment with alglucosidase alfa (69% in the cipaglucosidase alfa with miglustat group and 63% in the alglucosidase alfa with placebo group).
One-half of patients had a history of falls before entering the study, and 23% patients used assistive devices at baseline. Twenty-one percent of patients in the cipaglucosidase alfa with miglustat group and 16% patients in the alglucosidase alfa with placebo group had a history of IARs. In terms of prior medications, the most common prior medications (≥ 10% patients) were vitamin D and analogues (29.3%), anilides (25.2%), propionic acid derivatives (25.2%), uncoded medications (17.1%), plain angiotensin-converting enzyme inhibitors (13%), proton pump inhibitors (11.4%), selective serotonin reuptake inhibitors (11.4%), and plain multivitamins (10.6%). Overall, more patients in the alglucosidase alfa with placebo group than in the cipaglucosidase alfa with miglustat group took preinfusion medications before the study (32% versus 19%, respectively), although patients in both groups took preinfusion medications at a comparable proportion during the study.
Table 11: Summary of Baseline Characteristics From the PROPEL Trial (ITT Population)
Characteristic | Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) |
|---|---|---|
Demographic characteristics | ||
Age at informed consent (years), mean (SD) | 47.6 (13.25) | 45.1 (13.30) |
Weight (kg), mean (SD) | 72.77 (14.70) | 79.04 (24.77) |
BMI (kg/m2), mean (SD) | 24.75 (4.54) | 26.66 (7.29) |
Sex, n (%) | ||
Female | 49 (57.6) | 18 (47.4) |
Male | 36 (42.4) | 20 (52.6) |
Race group,a n (%)b | ||
Asian | 2 (2.4) | 1 (2.6) |
American Indian or Alaska Native | 0 | 1 (2.6) |
Black or African American | 0 | 1 (2.6) |
Japanese | 2 (2.4) | 4 (10.5) |
Native Hawaiian or other Pacific Islander | 1 (1.2) | 0 |
White | 74 (87.1) | 30 (78.9) |
Other | 5 (5.9) | 1 (2.6) |
Disease characteristics | ||
Age at diagnosis (years), mean (SD) | 39.9 (13.82) | 36.9 (15.30) |
Age at first ERT dosec (years), mean (SD) | 40.8 (12.71) | 38.7 (15.14) |
ERT status, n (%) | ||
ERT-naive status | 20 (23.5) | 8 (21.1) |
ERT-experienced status | 65 (76.5) | 30 (78.9) |
ERT durationc,d | ||
Total (years), mean (SD) | 7.48 (3.38) | 7.14 (3.64) |
≥ 2 to < 3 years, n (%) | 4 (6.2) | 5 (16.7) |
≥ 3 to < 5 years, n (%) | 16 (24.6) | 6 (20.0) |
≥ 5 years, n (%) | 45 (69.2) | 19 (63.3) |
Medical history | ||
Using assistive devices at baseline, n (%) | 17 (20.0) | 11 (28.9) |
History of falls, n (%) | 44 (51.8) | 17 (44.7) |
History of IARs, n (%) | 18 (21.2) | 6 (15.8) |
Baseline 6MWDe | ||
Baseline 6MWD (metres), mean (SD) | 357.93 (111.84) | 350.14 (119.78) |
Baseline % predicted 6MWD (metres), mean (SD) | 57.82 (15.80) | 55.69 (17.18) |
Baseline 6MWD category 1, n (%) | ||
≥ 75 to < 150 m | 4 (4.7) | 4 (10.5) |
≥ 150 to < 400 m | 55 (64.7) | 22 (57.9) |
≥ 400 m | 26 (30.6) | 12 (31.6) |
Baseline pulmonary functionf | ||
Percent predicted sitting FVC, mean (SD) | 70.74 (19.57) | 70.04 (21.30) |
Prior medication (safety population) | ||
Patients who took any preinfusion medication before study,g n (%) | 16 (18.8) | 12 (31.6) |
Patients who took any preinfusion medication during study,h n (%) | 14 (16.5) | 5 (13.2) |
6MWD = 6-minute walk distance; BMI = body mass index; ERT = enzyme replacement therapy; FVC = forced vital capacity; IAR = infusion-associated reaction; ITT = intention to treat; SD = standard deviation.
aFrom original source.
bPatients may have chosen more than 1 racial category.
cFor patients who were ERT experienced only.
dERT duration was calculated as (date of informed consent minus date of first dose of ERT) divided by 365.25 and rounded to 1 decimal place.
eBaseline 6MWD was the average of the last 2 values obtained on or before the first dose date.
fBaseline for FVC was the average of the last 2 values obtained on or before the first dose date for each parameter.
gPremedication with onset dates before the first dose of the study drug.
hPreinfusion medications that started on or after the first dose of the study drug but before the last dose of the study drug.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
Table 12 summarizes the extent of the study drug exposure. Overall, the mean duration of exposure was similar, approximately 12 months, in both treatment groups. Adherence to dose and infusion was approximately 98% and similar in both groups.
Table 12: Summary of Drug Exposure in the PROPEL Trial (Safety Population)
Exposure | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) | |
Number of administered doses, mean (SD) | 25.7 (3.79) | 26.2 (1.99) |
Duration of treatment (months),a mean (SD) | 11.79 (1.80) | 11.97 (0.71) |
Duration of treatment (months),a n (%) | ||
≤ 3 | 1 (1.2) | 0 |
> 3 to ≤ 6 | 1 (1.2) | 0 |
> 6 to ≤ 9 | 2 (2.4) | 1 (2.6) |
> 9 to ≤ 12 | 19 (22.4) | 4 (10.5) |
> 12 | 62 (72.9) | 33 (86.8) |
Dose compliance (%),b mean (SD) | 98.62 (5.80) | 97.70 (6.04) |
Infusion compliance (%),c mean (SD) | 98.00 (5.21) | 97.82 (4.81) |
SD = standard deviation.
aDuration of treatment (months) equal (date of last dose minus date of first dose plus 1) divided by 30.4.
bDose compliance for alglucosidase alfa and cipaglucosidase alfa equals 100 multiplied by (total infusion dose administered in mg divided by total planned or intended infusion dose in mg); dose compliance for placebo and miglustat equals 100 multiplied by (total dose administered in mg divided by scheduled or planned dose in mg).
cCompliance for number of infusions (applicable for alglucosidase alfa and cipaglucosidase alfa only) equals 100 multiplied by (number of infusions administered divided by the number of infusions planned or intended), where the number of infusions planned was obtained as (patient’s last date in study while on treatment minus date of first infusion plus 14) divided by 14. Missed infusions due to policies related to COVID-19 were subtracted.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
The most common concomitant medications (≥ 10%) in the PROPEL trial were anilides (45.5% of patients), vitamin D and analogues (34.1%), influenza vaccines (30.9%), propionic acid derivatives (25.2%), uncoded medications (24.4%), proton pump inhibitors (15.4%), other antihistamines for systemic use (14.6%), plain angiotensin-converting enzyme inhibitors (13.8%), selective serotonin reuptake inhibitors (13.0%), glucocorticoids (11.4%), opioids in combination with nonopioid analgesics (10.6%), and plain multivitamins (10.6%).
Table 13 provides the distribution of concomitant medications in the treatment and comparator groups.
Table 13: Summary of Subsequent Treatment in the PROPEL Trial (Safety Population)
Variable | Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) |
|---|---|---|
Patients who took any medication, n (%) | 81 (95.3) | 37 (97.4) |
Most common concomitant medications (≥ 10%), n (%) | ||
Angiotensin-converting enzyme inhibitors | 10 (11.8) | 7 (18.4) |
Adrenergics in combination with corticosteroids or other drugs, excluding anticholinergics | 4 (4.7) | 4 (10.5) |
Angiotensin II receptor blockers | 3 (3.5) | 4 (10.5) |
Anilides | 40 (47.1) | 16 (42.1) |
Benzodiazepine derivatives | 2 (2.4) | 2 (5.3) |
Beta-blocking drugs | 2 (2.4) | 5 (13.2) |
Corticosteroids, moderately potent | 0 | 4 (10.5) |
Glucocorticoids | 11 (12.9) | 3 (7.9) |
Influenza vaccines | 27 (31.8) | 11 (28.9) |
Multivitamins, plain | 6 (7.1) | 7 (18.4) |
Opioids in combination with nonopioid analgesics | 6 (7.1) | 7 (18.4) |
Other antihistamines for systemic use | 10 (11.8) | 8 (21.1) |
Other lipid-modifying drugs | 9 (10.6) | 3 (7.9) |
Piperazine derivatives | 7 (8.2) | 4 (10.5) |
Progestogens and estrogens, fixed combinations | 6 (7.1) | 4 (10.5) |
Propionic acid derivatives | 30 (35.3) | 17 (44.7) |
Proton pump inhibitors | 13 (15.3) | 6 (15.8) |
Selective beta 2 adrenoreceptor agonists | 6 (7.1) | 5 (13.2) |
Selective serotonin reuptake inhibitors | 13 (15.3) | 3 (7.9) |
Uncoded | 19 (22.4) | 11 (28.9) |
Vitamin B12 | 6 (7.1) | 6 (15.8) |
Vitamin D and analogues | 26 (30.6) | 16 (42.1) |
Note: Concomitant medications are defined as medications with onset dates on or after the first dose of the study drug, or medications with onset dates before first dose of the study drug without a stop date, or medications with a stop date after first dose of the study drug. Medications starting after the last dose of the study drug are not considered concomitant medications.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Protocol Deviations
Overall, 67 of 85 patients (78.8%) in the cipaglucosidase alfa with miglustat group and 28 of 38 patients (73.7%) in the alglucosidase alfa with placebo groups experienced a major protocol deviation. Most major deviations were in the categories of study procedures (49.4% in the cipaglucosidase alfa with miglustat group and 47.7% in the alglucosidase alfa with placebo group), investigational product (41.2% in the cipaglucosidase alfa with miglustat group and 36.8% in the alglucosidase alfa with placebo group), and informed consent (22.4% in the cipaglucosidase alfa with miglustat group and 31.6% in the alglucosidase alfa with placebo group). Patients with protocol deviations due to the COVID-19 pandemic comprised 47 of 85 patients (55%) in the cipaglucosidase alfa with miglustat group compared with 19 of 38 patients (50%) in the alglucosidase alfa with placebo group.
Efficacy
The primary and key secondary efficacy outcomes from the PROPEL trial are summarized in Table 14. The remaining efficacy outcomes are summarized in Table 31 in Appendix 1.
Results for all efficacy outcomes are presented in this section using the ITT population without the outlier data from 1 patient (a patient who was ERT naive in the alglucosidase alfa plus placebo group). After the database was locked and treatment assignments were unblinded, the patient with outlier data were found to have baseline 6MWD results that were likely adversely affected by their pre-enrolment use of ostarine powder, an investigational anabolic steroid, that resulted in a clinically implausible change at week 52. Furthermore, this patient admitted to intentionally underperforming during the baseline assessment to enter the study. Given the patient’s admitted underperformance on the screening test and their clinically implausible 6MWD results (i.e., the observed change from baseline to 52 weeks was an increase of 355 m), all efficacy analyses have been reported without the data from this patient.
6MWD at Week 52 (Primary Outcome)
The mean change from baseline to week 52 in 6MWD was 20.56 m (95% CI, 11.22 m to 29.91 m) for the cipaglucosidase alfa with miglustat group and 8.02 m (95% CI, −5.71 m to 21.74 m) for the alglucosidase alfa with placebo group (ITT-OBS population). The LS mean difference between treatment groups in the 6MWD at week 52 was 14.21 m (95% CI, −2.60 m to 31.0 m), with a 1-sided P value of 0.048.
Because the primary end point (6MWD) did not meet statistical significance, all statistical testing in the end point hierarchy stopped. All key secondary end points were assessed for nominal statistical significance of superiority and should be viewed as supportive evidence for the overall effect within the trial.
The results of the sensitivity analyses supported the primary analysis and are otherwise not presented.
A summary of the subgroup analyses for 6MWD at week 52 is provided Table 32 in Appendix 1. Overall, 6MWD did not vary by any of the subgroups.
Sitting FVC (First Key Secondary Outcome)
The mean change from baseline to week 52 was −0.93% (95% CI, −2.29% to 0.42%) for the cipaglucosidase alfa with miglustat group and −3.95% (95% CI, −5.58% to −2.32%) for the alglucosidase alfa with placebo group. The LS mean difference between treatment groups was 2.66% (95% CI, 0.37% to 4.95%).
The results of the sensitivity analyses supported the primary analysis and are otherwise not presented.
A summary of the subgroup analyses for percent predicted sitting FVC is provided in Table 33 in Appendix 1. FVC did not vary by baseline 6MWD and age group. In the ERT-experienced subgroup, percent predicted sitting FVC showed stabilization over time, with a 0.05% mean improvement (SD = 5.84) from baseline in the cipaglucosidase alfa with miglustat group, compared with a mean 4.02% decrease (SD = 5.01%) in the alglucosidase alfa with placebo group, resulting in an estimate treatment difference of 3.51 (95% CI, 1.03 to 5.99) in favour of cipaglucosidase alfa with miglustat. In the subgroup of patients receiving ERT for 5 years or more, the LS mean treatment difference was 3.71% (95% CI, 0.41% to 7.00%). Interaction tests to determine whether results differed statistically by subgroup were not performed.
Table 14: Summary of Key Efficacy Results in the PROPEL Trial, Excluding Outlier
Variable | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) | |
6MWD in metres (ITT-OBS population) | ||
Number of patients contributing to the analysis, n (%) | 81 (95.30) | 36 (94.74) |
Baseline, mean (SD) | 376.41 (122.93) | 359.70 (137.36) |
Change from baseline to week 52, mean (95% CI) | 20.56 (11.22 to 29.91) | 8.02 (−5.71 to 21.74) |
Least squares mean difference (95% CI) | 14.21 (−2.60 to 31.02)a | Reference |
1-sided P value for superiority | 0.048 | Reference |
2-sided P value for superiority | 0.097 | Reference |
Sitting FVC, % predicted (ITT-LOCF population) | ||
Number of patients contributing to the analysis, n, (%) | 85 at baseline (100); 84 at week 52 (98.82) | 37 (97.37) |
Baseline, mean (SD) | 70.74 (19.57) | 69.68 (21.48) |
Change from baseline to week 52, mean (95% CI) | −0.93 (−2.29 to 0.42) | −3.95 (−5.58 to −2.32) |
Least squares mean difference (95% CI) | 2.66 (0.37 to 4.95)b | Reference |
Nominalc 1-sided P value for superiority | 0.012 | Reference |
Nominalc 2-sided P value for superiority | 0.023 | Reference |
6MWD = 6-minute walk distance; CI = confidence interval; ERT = enzyme replacement therapy; FVC = forced vital capacity; ITT-LOCF = intention-to-treat last observation carried forward; ITT-OBS = intention-to-treat observed; SD = standard deviation.
aA mixed model for repeated measures approach (using restricted maximum likelihood estimation) was used. The model included terms for treatment, baseline 6MWD, age, height, weight (all as continuous covariates), ERT status (ERT naive versus ERT experienced), sex, time, and treatment-by-time interaction. Time was used as a repeated measure, and an unstructured covariance approach was applied.
bAn analysis of covariance model was used. The model included terms for treatment, baseline response for each variable, age, height, weight (all as continuous covariates), ERT status (ERT naive versus ERT experienced), and sex. The last available postbaseline observation was used to replace the missing value at week 52.
cBecause the primary end point analysis showed no superiority of treatment, no further confirmatory conclusions are possible (statistically); therefore, the P values are nominal.
Source: ATB200-03 Clinical Study Report.18
MMT Lower Extremity Score
The mean MMT lower extremity score increased by 1.56 (SD = 3.78) in the cipaglucosidase alfa plus miglustat group and 0.88 (SD = 2.58) in the alglucosidase alfa plus placebo group from baseline, resulting in an LS mean treatment difference of 0.96 (95% CI, −0.48 to 2.40).
6MWD at Week 26
The mean on the 6MWD increased by 17.44 m (95% CI, 9.80 m to 25.08 m) in the cipaglucosidase alfa plus miglustat group and 9.19 m (95% CI, −0.20 m to 18.59 m) in the alglucosidase alfa plus placebo group from baseline to week 26. The LS mean treatment difference was 8.17 m (95% CI, −4.24 m to 20.57 m).
PROMIS Scores
The PROMIS Physical Function score increased by a mean of 1.94 (95% CI, 0.31 to 3.57) from baseline to week 52 in the cipaglucosidase alfa plus miglustat group, whereas the mean improvement was 0.19 (95% CI, −3.42 to 3.80) for the alglucosidase alfa plus placebo group. This translated to an LS mean treatment difference of 1.87 (95% CI, −1.51 to 5.25).
Compared with baseline, the PROMIS Fatigue score decreased by a mean of 2.02 (95% CI, −3.26 to −0.77) in the cipaglucosidase alfa with miglustat group, whereas the mean decreased by 1.67 (95% CI, −3.88 to 0.54) for the alglucosidase alfa with placebo group. This resulted in an LS mean treatment difference of 0.04 (95% CI, −2.12 to 2.20).
Percent Predicted 6MWD
The mean changes from baseline to week 52 for the percent predicted 6MWD was 4.07% (95% CI, 2.56% to 5.59%) for the cipaglucosidase alfa with miglustat group and 1.58% (95% CI, −0.42% to 3.58%) for the alglucosidase alfa with placebo group. The LS mean difference was 2.38% (95% CI, −0.26% to 5.03%) between treatment groups.
Gower Manoeuvre
The mean changes from baseline for the Gower manoeuvre were −0.26 (95% CI, −1.74 to 1.22) and −2.19 (95% CI, −5.04 to 0.66) for the cipaglucosidase alfa with miglustat and alglucosidase alfa with placebo groups, respectively. The mean difference between treatment groups was 1.60 (95% CI, −1.48 to 4.68).
Timed Up and Go
The mean changes from baseline in TUG was −0.30 (95% CI, −2.24 to 1.65) for the cipaglucosidase alfa with miglustat group and −0.13 (95% CI, −1.11 to 0.85) for the alglucosidase alfa with placebo group. The LS mean difference between treatment groups was −0.47 (95% CI, −3.38 to 2.43).
MMT Total Score
The mean changes from baseline for the MMT total score were 3.07 (95% CI, 1.66 to 4.48) and 1.41 (95% CI, −0.12 to 2.94) for the cipaglucosidase alfa with miglustat and alglucosidase alfa with placebo groups, respectively. The mean difference between treatment groups was 2.22 (95% CI, −0.09 to 4.53).
Harms
Table 15 summarizes the harms from the PROPEL trial.
Adverse Events
A total of 118 of 123 patients (98.9%) experienced a TEAE during the study. The overall incidence was similar between the cipaglucosidase alfa with miglustat and alglucosidase alfa with placebo groups (95.3% and 97.4%, respectively). The most common TEAEs were falls, headaches, and nasopharyngitis. Most TEAEs were mild or moderate in severity.
Serious Adverse Events
Overall, 9 of 123 patients (7.3%) had a severe TEAE. Eight patients (9.4%) in the cipaglucosidase alfa with miglustat group reported 11 serious TEAEs. One event (anaphylaxis) was considered by the investigator to be related to the study drug and the other events (abdominal pain, aortic aneurysm, bradycardia, contusion, enteritis, ilium fracture, removal of internal fixation, skin laceration, viral myosis, vomiting) were considered unrelated to cipaglucosidase alfa plus miglustat. One patient (2.6%) in the alglucosidase alfa with placebo group reported a cerebrovascular event that was unrelated to the study drug.
Withdrawals Due to AEs
Overall, 3 patients who were ERT experienced discontinued from the study due to an AE. Two patients in the cipaglucosidase alfa with miglustat group withdrew from the study due to an IAR, 1 with an anaphylactic reaction and the other with severe chills, both deemed to be related to the study drug. Both patients had high anti-rhGAA antibody titres. In the alglucosidase alfa with placebo group, 1 withdrew due to a cerebrovascular accident unrelated to the study drug.
Mortality
No TEAEs leading to death were reported.
Notable Harms
IARs were reported in 21 of 85 patients (25%) in the cipaglucosidase alfa with miglustat group and in 10 of 38 patients (26%) in the alglucosidase alfa with placebo group. Two patients (1 in each group) reported 11 to 19 IARs and 1 patient in the cipaglucosidase alfa with miglustat group reported more 20 IARs. In the cipaglucosidase alfa with miglustat group, the most common IARs were dizziness, abdominal distension, headache, chills, diarrhea, dysgeusia, dyspnea, flushing, pruritus, pyrexia, and rash. In the alglucosidase alfa with placebo group, the most common IARs were nausea, fatigue, dizziness, and headache. Most of the IARs were considered by the investigator to be mild or moderate in severity. Of the patients who reported IARs, 1 patient who was ERT experienced and treated with cipaglucosidase alfa had at least 1 positive anti-rhGAA immunoglobulin E result postbaseline.
Table 15: Summary of Harms Results in the PROPEL Trial (Safety Population)
TEAE | Cipaglucosidase alfa + miglustat n = 85 | Alglucosidase alfa + placebo n = 38 |
|---|---|---|
Most common TEAEs (≥ 10%), n (%) | ||
≥ 1 adverse event | 81 (95.3) | 37 (97.4) |
Fall | 25 (29.4) | 15 (39.5) |
Headache | 20 (23.5) | 9 (23.7) |
Nasopharyngitis | 19 (22.4) | 3 (7.9) |
Myalgia | 14 (16.5) | 5 (13.2) |
Arthralgia | 13 (15.3) | 5 (13.2) |
Nausea | 10 (11.8) | 8 (21.1) |
Back pain | 9 (10.6) | 7 (18.4) |
Diarrhea | 11 (12.9) | 4 (10.5) |
Urinary tract infection | 12 (14.1) | 2 (5.3) |
Fatigue | 8 (9.4) | 5 (13.2) |
Pain in extremity | 11 (12.9) | 2 (5.3) |
Oropharyngeal pain | 10 (11.8) | 2 (5.3) |
Severe TEAEs (by SOC), n (%) | ||
Patients with ≥ 1 severe TEAE | 8 (9.4) | 1 (2.6) |
Cardiac disorders | 1 (1.2) | 0 |
Gastrointestinal disorders | 1 (1.2) | 0 |
Immune system disorders | 1 (1.2) | 0 |
Infections and infestations | 1 (1.2) | 0 |
Injury, poisoning, and procedural complications | 2 (2.4) | 0 |
Nervous system disorders | 0 | 1 (2.6) |
Surgical and medical procedures | 1 (1.2) | 0 |
Vascular disorders | 1 (1.2) | 0 |
Patients who had a TEAE leading to study drug discontinuation, n (%) | ||
TEAE leading to study drug discontinuation | 2 (2.4) | 1 (2.6) |
Chills | 1 (1.2) | 0 |
Anaphylactoid reaction | 1 (1.2) | 0 |
Cerebrovascular accident | 0 | 1 (2.6) |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Adverse events of special interest, n (%) | ||
Patients who had any IARs | 21 (24.7) | 10 (26.3) |
Total number of IARs | 97 | 31 |
Cardiac disorders | 1 (1.2) | 0 |
Gastrointestinal disorders | 7 (8.2) | 3 (7.9) |
General disorders and administration site conditions | 6 (7.1) | 4 (10.5) |
Immune system disorders | 1 (1.2) | 0 |
Injury, poisoning, and procedural complications | 1 (1.2) | 0 |
Investigations | 2 (2.4) | 0 |
Musculoskeletal and connective tissue disorders | 1 (1.2) | 1 (2.6) |
Nervous system disorders | 10 (11.8) | 5 (13.2) |
Psychiatric disorders | 2 (2.4) | 1 (2.6) |
Respiratory, thoracic, and mediastinal disorders | 2 (2.4) | 1 (2.6) |
Skin and subcutaneous tissue disorders | 5 (5.9) | 2 (5.3) |
Vascular disorders | 3 (3.5) | 0 |
IAR = infusion-associated reaction; SOC = system organ class; TEAE = treatment-emergent adverse event.
Note: A TEAE was defined as any event that started on or after the first dose of respective study drug. Any adverse event that occurred after 30 days from the last dose of the study drug in the ATB200-03 trial and before the first dose of the study drug in the ATB200-07 trial was not counted as treatment emergent.
Sources: ATB200-03 Clinical Study Report.18 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
Internal Validity
The risk of bias due to randomization and allocation concealment was likely low. The randomization schedule was generated and administered centrally by Almac Clinical Technologies, independent of the sponsor’s project team. This centralized block randomization was stratified by baseline 6MWD and ERT status to reduce bias and increase the precision of statistical inference, and to allow various planned and unplanned subset analyses. However, randomization was not stratified by centre, although this was a multicentre trial, and it is unclear how this may have affected results. The patients, study sponsor, investigators, site personnel, and contracted research organizations involved in monitoring, data management, data analysis, or other aspects of the study were blinded from treatment assignment. A placebo matching miglustat was used for the control group. Unblinding deviation was reported in 1 patient in the intervention group and was unlikely to affect the results.
Demographic and disease characteristics were generally balanced between treatment groups. Although the proportions of male patients and the proportion of patients who took preinfusion medications before the study were both higher in the alglucosidase alfa with placebo group compared with the cipaglucosidase alfa with miglustat group, the clinical experts did not expect such imbalances would introduce bias to the study results.
There is likely a low risk of bias due to deviations from the assigned intervention. The clinical experts indicated no concerns with the concomitant treatments used in the trial; however, it is possible that imbalances in the treatments used could have an impact on subjective outcomes, such as the Gower manoeuvre and harms reporting.
It is likely there was a low risk of bias due to nonadherence to the study interventions. Adherence to dose and infusion was high and similar in both groups. The proportion of patients who discontinued participation in the trial was low in each treatment group (5.9% in the cipaglucosidase alfa with miglustat group and 2.6% in the alglucosidase alfa with placebo group).
It is likely there was a low risk of bias due to the measurement of study outcomes. When looking at the harms results, there were some imbalances in TEAEs between the treatment groups, particularly for falls, nausea, and urinary tract infections. However, the clinical experts expressed no concerns with the TEAEs reported in the study because they were all clinically manageable. There was no difference between treatment groups for diarrhea, which is known to be associated with miglustat.
The ITT-OBS population was used for the primary end point (6MWD) analysis. An ANCOVA model with ITT-LOCF for missing data at week 52 was used for the key secondary and other secondary end point analyses. None of these analyses used a true ITT population. However, only 5 patients (4%), 4 in the intervention group and 1 in the comparator group, had missing data for 6MWD and were not included in the analysis, which was unlikely to affect the results. For the first key secondary end point (sitting FVC) analyses, 1 patient in the comparator group was missing data, which was unlikely to affect the results. Sensitivity analyses were conducted for 6WMD and sitting FVC and were generally consistent with the primary analyses.
Key secondary and other secondary end points were analyzed according to a prespecified hierarchical order using a stepwise closed testing procedure to control the overall type I error rate. Because the primary end point analysis showed no superiority of treatment, no further confirmatory conclusions were possible (statistically). Therefore, any statistically significant P values on further outcome analyses (i.e., sitting FVC, percent predicted 6MWD, PROMIS instruments, MMT, Gower manoeuvre, TUG) were at high risk of type I error and should be considered nominal.
Subgroup analyses were prespecified but were underpowered, resulting in wide CIs, and were at high risk of type I error due to multiplicity of testing. Except for baseline 6MWD and ERT status, which were stratified in the randomization, the other subgroups were likely unbalanced with respect to characteristics between the groups, which would be expected to introduce bias. Therefore, the subgroup analyses should be considered supportive information on the overall effects. Also, interaction tests were not performed.
The sponsor proposed a clinically meaningful threshold of a 6% change from baseline in 6MWT results, and the clinical experts validated a change of 20 m in 6MWD as the MID for the 6MWT. However, varying and wide ranges of MIDs have been reported in the literature. Specifically, 1 study used data from the PROPEL trial and determined that the MIDs for the 6MWT in patients with LOPD ranged from 2.27% to 8.11%, depending on the chosen method and disease severity, translating to a change of 23.7 m to 57.2 m in the 6MWD.22 In a literature review, the between-group MID for 6MWT results in patients with LOPD ranged from 0.35% to 7.47% points, equivalent to a 6MWD of 2.18 m to 46.61 m for males and 1.97 m to 42.13 m for females, all aged 50 years, with a height of 1.75 m and a weight of 80 kg.23 In a separate review, it was reported that 9 of 10 studies on LOPD reported that the absolute changes from baseline in 6MWT results lay within or more than the absolute MID levels of 24 m to 54 m or a relative MID range of 5% to 11%.24 Therefore, the MID for the 6MWT is very uncertain.
External Validity
The PROPEL trial took place in 26 countries. Most patients were enrolled from Australia, France, and the US with very few patients enrolled in the 2 sites in Canada. The clinical experts did not express any concerns that differences in clinical practice, general medical care, reporting of AEs, and access to health care resources among the countries would confound the results. The clinical experts also confirmed that a diagnosis of LOPD in the PROPEL trial was made in a manner similar to clinical practice in Canada, namely, by identifying a GAA enzyme deficiency or through GAA genotyping. The dosing and administration of cipaglucosidase alfa with miglustat was consistent with the Health Canada–approved product monograph.
The clinical experts confirmed that the eligibility criteria for the PROPEL trial diverged somewhat from the Canadian guideline for patients eligible for ERT.7 Specifically, the PROPEL trial’s eligibility criteria were more restrictive. Patients with a sitting FVC of less than 30% of the predicted value for healthy adults or who could not perform two 6MWTs of at least 75 m were ineligible for the trial. However, in clinical practice, FVC predicted value and 6MWD would not determine whether a patient would be started on ERT. Rather, patients who are not fully ventilated would be eligible for ERT, as per the Canadian guideline.7 Therefore, a small number of patients with more severe symptoms, who might gain benefit with cipaglucosidase alfa plus miglustat, may have been excluded.
Despite the restrictive eligibility criteria, the clinical experts confirmed that the baseline characteristics of the PROPEL trial’s participants were representative of the patients they treat. The trial population was predominately white (85%), 2% were Asian, 1% were American Indian or Alaska Native, 1% were Black or African American, 5% were Japanese, 1% were Native Hawaiian or other Pacific Islander, and 5% were other races. One clinical expert explained there is a common variant for LOPD that is more prevalent in people from Europe, and all the patients this clinical expert has seen in clinical practice are white and have that variant.
The clinical experts noted that the primary outcome of 6MWD and first key secondary outcome of FVC were clinically relevant and both have available data for patients with LOPD, although some clinics may be restricted in their ability to conduct the tests. For instance, a clinic must have the time and physical space to perform a 6MWT. Two key outcomes of interest to the pharmacoeconomic analysis, long-term wheelchair use and ventilation dependency, were not assessed in the PROPEL trial. One clinical expert stated that important outcomes such as the use of mobility aids and the need for ventilation could not be realistically obtained in a trial such as PROPEL with a 1-year timeline conducted in patients with a rare disease and slow progression condition such as LOPD. The clinical experts indicated they do not use the PROMIS instruments in clinical practice.
While there may be some limitations to generalizability, as discussed here, the results of the PROPEL trial appear to be generalizable to clinical practice in Canada for treating patients with LOPD.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to inform expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group:28,29
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The reference points for the certainty of evidence assessment for 6MWD and FVC were set according to the presence or absence of an important effect based on thresholds informed by the clinical experts and suggested by the sponsor. The reference point for the certainty of evidence assessment for PROMIS Physical Function was set according to the presence or absence of an important effect based on thresholds informed by the literature. Due to the lack of formal MID estimates for PROMIS Fatigue score, severe TEAEs, and IARs, the targets of the certainty of evidence assessments were the presence or absence of any (non-null) effect for each outcome.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for cipaglucosidase alfa with miglustat versus alglucosidase alfa with placebo.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
ATB200-07 (NCT04138277) is an ongoing, phase III, international, OLE study to assess the long-term safety and tolerability of cipaglucosidase alfa and miglustat coadministration following 104 weeks of treatment (i.e., 52 weeks in the PROPEL trial and 52 weeks in the OLE study) in adult patients with LOPD. The efficacy of the drug combination was also assessed as a secondary objective.
To enrol in the OLE study, patients were required to have completed the pivotal study (ATB200-03; PROPEL). Additional inclusion and exclusion criteria for the OLE study were the same as for the PROPEL trial. Patients enrolled in the OLE study who received cipaglucosidase alfa plus miglustat in the PROPEL trial continued on study treatment, and those receiving alglucosidase alfa with placebo were switched to cipaglucosidase alfa plus miglustat. Of the 123 participants who completed the PROPEL trial, 117 (95%) enrolled in the OLE study. Two participants who did not complete the PROPEL trial also enrolled in the OLE study. Data presented in the current submission are from an interim analysis consisting of 119 participants with at least 52 weeks of data in the OLE study until the data cut-off on January 11, 2022.
Populations
Demographic and baseline characteristics for the OLE study were collected at week 52 of the PROPEL study, before enrolment in study ATB200-07. For some patients, baseline assessments were redone due to large gaps of time between week 52 of the PROPEL trial and the baseline visits for study ATB200-07. Demographic characteristics were generally similar between treatment groups in the population enrolled in the OLE study, including the outlier (described in Table 16).
Table 16: Summary of Baseline Characteristics in the ATB200-07 OLE Trial (Population Enrolled in the OLE Study)
Characteristics | Cipaglucosidase alfa + miglustat (N = 82) | Treatment switched (prior alglucosidase alfa + placebo) (N = 37) | Total (N = 119) |
|---|---|---|---|
Age, years, mean (SD) | 48.8 (13.47) | 46.0 (13.47) | 47.9 (13.48) |
Sex, n (%) | |||
Female | 48 (58.5) | 18 (48.6) | 53 (44.5) |
Male | 34 (41.5) | 19 (51.4) | 53 (44.5) |
Race,a n (%) | |||
Asian | 3 (3.7) | 1 (2.7) | 4 (3.4) |
Black or African American | 0 (0.0) | 1 (2.7) | 1 (0.8) |
Japanese | 2 (2.4) | 4 (10.8) | 6 (5.0) |
White | 71 (86.6) | 30 (81.1) | 101 (84.9) |
Other | 6 (7.3) | 1 (2.7) | 7 (5.9) |
Height (cm), mean (SD) | 171.2 (9.74) | 171.2 (11.32) | 171.2 (10.20) |
Weight (kg), mean (SD) | 73.31 (15.258) | 78.87 (26.801) | 75.06 (19.670) |
BMI (kg/m2), mean (SD) | 25.02 (4.847) | 26.48 (7.710) | 25.47 (5.882) |
BMI = body mass index; OLE = open-label extension; SD = standard deviation.
aFrom original source.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Intervention
Cipaglucosidase alfa plus miglustat was administered in the OLE study in the same manner as in the PROPEL trial. The first infusion visit in the OLE study was scheduled approximately 2 weeks after the last study visit of the PROPEL trial. Study drugs were administered every 2 weeks at the study site. After 3 months in the study without any moderate to severe IARs, patients may have been eligible for the administration of the study drug at their home by trained personnel because site visits and laboratory testing were impeded during the COVID-19 pandemic.
Outcomes
Efficacy outcomes in the OLE study were the same as in the PROPEL study; refer to Table 6 for a summary of outcomes identified as most important to this review. Assessments were performed at baseline, weeks 12 and 26, every 26 weeks thereafter, and at the end of study or end of treatment.
Statistical Analysis
No formal hypotheses were planned, and only descriptive statistics were presented. In general, where basic summary statistics were reported, continuous variables were summarized using descriptive statistics; categorical variables were summarized using number and percentage. For basic summaries involving the change from baseline, a 95% CI for the mean difference was provided.
Efficacy analyses were conducted on the following 3 study populations:
Patients who were enrolled in the OLE study, which represent changes from the PROPEL study from baseline through to week 104 (i.e., 52 weeks in the PROPEL study and 52 weeks in the ATB200-07 study).
OLE study full analysis set, which represents the OLE study alone and changes from baseline in the ATB200-07 study.
Population that switched treatments (patients treated with alglucosidase alfa plus placebo for 52 weeks of the pivotal study and switched to cipaglucosidase alfa plus miglustat for 52 weeks of the OLE study).
Safety analyses were performed on the integrated data from the PROPEL and ATB200-07 studies and presented by treatment group and overall. Safety data were summarized using descriptive statistics for continuous data and using counts and percentages for categorical data. Participants were expected to have baseline values for safety and efficacy assessments in the OLE study, so no rule for baseline imputation was needed. For the multicomponent and multi-item end points, if the baseline value was partially missing (patient was missing data for only specific items at baseline), the average value was calculated by using all patients with nonmissing values for that item to replace the missing item score. If the baseline total score was completely missing, the average score calculated by using all patients with nonmissing total scores for the end point combined was used to replace the missing baseline total score. Missing postbaseline values (whether efficacy or safety) were not imputed.
Results
Patient Disposition
Of the 123 patients (85 treated with cipaglucosidase alfa in combination with miglustat, 38 treated with alglucosidase alfa) who enrolled in the PROPEL trial, a total of 117 patients completed the study and then enrolled in in the ATB200-07 trial. Two additional patients did not complete the PROPEL trial but enrolled in the ATB200-07 study, resulting in a total of 119 patients (91 patients who were ERT experienced and 28 patients who were ERT naive). As of the data cut-off, 11 patients (9.2%) had discontinued prematurely from the study.
Table 17: Patient Disposition in the ATB200-07 OLE Study
Patient disposition | Cipaglucosidase alfa + miglustat (N = 85) | Treatment switched (prior alglucosidase alfa + placebo) (N = 40) | Total |
|---|---|---|---|
PROPEL ITT population | 85 | 38 | 123 |
Completed study, n (%) | 80 (94.1) | 37 (97.4) | 117 (95.1) |
PROPEL and ATB200-07 trial analysis populations | |||
OLE-enrolled population, n (%)a,b | 82 (96.5) | 37 (97.4) | 119 (96.7) |
Treatment-switched population, n (%)a | NA | 37 (97.4) | 37 (97.4) |
ATB200-07 trial standalone analysis populations | |||
OLE safety population, n (%)c | 81 (98.8) | 37 (100.0) | 118 (99.2) |
OLE full analysis population, n (%)c | 80 (97.6) | 36 (97.3) | 116 (97.5) |
ATB200-07 study | |||
Completed study, n (%)c,d | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Discontinued study prematurely, n (%)c | 7 (8.5) | 4 (10.8) | 11 (9.2) |
Reason for premature study discontinuation, n (%) | |||
Adverse event | 1 (1.2) | 2 (5.4) | 3 (2.5) |
Lost to follow-up | 1 (1.2) | 0 (0.0) | 1 (0.8) |
Withdrawal of consent by patient | 5 (6.1) | 1 (2.7) | 6 (5.0) |
Worsening of condition | 0 (0.0) | 1 (2.7) | 1 (0.8) |
ITT = intention to treat; NA = not applicable; OLE = open-label extension.
aPercentages are based on the number of patients in each treatment group in the PROPEL and ATB200-03 trial ITT populations.
bIncludes 2 patients who did not complete the PROPEL study for logistical reasons.
cPercentages are based on the number of patients enrolled in the ATB200-07 study.
dThis study is ongoing.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Exposure to Study Treatments
At the interim analysis data cut-off, 78 patients (91.8%) in the cipaglucosidase alfa plus miglustat group had received more than 24 months of treatment, with a maximum treatment duration of 35 months. A total of 34 patients (91.9%) in the treatment-switched group received more than 12 months of treatment with cipaglucosidase alfa plus miglustat in the ATB200-07 trial, with a maximum duration of treatment of 24 months. Overall mean treatment adherence in the OLE study was 98.6% across both treatment groups and ranged from 61% to 103%. Dose compliance was also high, with an overall mean of 98.5% for cipaglucosidase alfa and 98.4% for miglustat.
Across the pivotal study and the OLE study, the most common concomitant medications (taken by at least 20% of patients) were other viral vaccines (77.0%), anilides (54.1%), propionic acid derivatives (43.4%), influenza vaccines (39.3%), glucocorticoids (23.8%), and vitamin D and analogues (23.8%). More patients (20.0%) in the cipaglucosidase alfa plus miglustat continued-treatment group took preinfusion medications during treatment than in the treatment-switched group (13.5%) during the OLE study. The most common preinfusion medications taken during cipaglucosidase alfa plus miglustat treatment overall (pivotal study and OLE study) were anilides (9.0%) and glucocorticoids (8.2%).
Table 18: Patient Exposure Across the Entire Study Period (Safety Population)
Exposure | Cipaglucosidase alfa + miglustat (N = 85) | Treatment switched (prior alglucosidase alfa + placebo) (N = 37) |
|---|---|---|
Administered doses, mean (SD) | 57.9 (12.90) | 33.8 (10.80) |
Duration of treatment (months), mean, (SD) | 26.6 (5.85) | 15.4 (5.01) |
SD = standard deviation.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
Refer to Table 19 for a summary of efficacy results from the ATB200-07 OLE study. The efficacy results summarized subsequently are from the OLE study full analysis set population for the ATB200-07 study, excluding the outlier.
6MWD (In Metres)
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean 6MWD in the ATB200-07 trial at baseline was 373.1 m (SD = 124.23 m), with a mean change from baseline of −2.0 m (95% CI, −9.1 m to 5.1 m) at week 52 of the OLE study. The mean 6MWD for the treatment-switched group was 363.5 m (SD = 137.38 m) at baseline, with a mean change from baseline of −1.4 m (95% CI, −14.0 m to 11.1 m) at week 52.
Sitting FVC (Percent Predicted)
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean percent predicted sitting FVC was 69.1% (SD = 19.41%) for the ATB200-07 trial at baseline and 67.9% (SD = 20.39%) at week 52 of the OLE study, with a mean change from baseline of −0.2% (95% CI, −1.9% to 1.6%). The treatment-switched group mean percent predicted sitting FVC was 63.8% (SD = 19.63%) at baseline and 64.2% (SD = 19.21%) at week 52, with a mean change from baseline of 0.05 (95% CI, −1.8 to 1.8) at week 52.
MMT Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean MMT total score was 62.2 (SD = 8.33) for the ATB200-07 study at baseline and 65.7 (SD = 8.41) at week 52 of the OLE study, with a mean change from baseline of 3.2 (95% CI, 1.7 to 4.8). The treatment-switched group mean MMT total score was 62.7 (SD = 9.86) at baseline and 64.0 (SD = 10.93) at week 52, with a mean change from baseline of 1.4 (95% CI, −0.2 to 2.9).
PROMIS Physical Function Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean PROMIS Physical Function total score was 68.8 (SD = 12.81) in the ATB200-07 trial at baseline and 69.7 (SD = 12.98) at week 52, with a mean change from baseline of 0.6 (95% CI, −0.6 to 1.8). The treatment-switched group mean was 67.8 (SD = 16.74) at baseline and 67.4 (SD = 15.23) at week 52, with a mean change from baseline of −0.1 (95% CI, −2.5 to 2.2).
PROMIS Fatigue Total Score
For the cipaglucosidase alfa plus miglustat continued-treatment group, the mean PROMIS Fatigue total score was 19.9 (SD = 7.50) in the ATB200-07 trial at baseline and 20.1 (SD = 7.13) at week 52, with a mean change from baseline of 0.2 (95% CI, −1.1 to 1.6). The treatment-switched group mean was 19.3 (SD = 6.72) at baseline and 21.0 (SD = 6.80) at week 52, with a mean change from baseline of 1.5 (95% CI, −0.6 to 2.6).
Table 19: Summary of Efficacy Results in the ATB200-07 OLE Study (OLE-FAS Population, Excluding Outlier)
Variable | Cipaglucosidase alfa + miglustat (N = 80) | Treatment switched (prior alglucosidase alfa + placebo) (N = 35) |
|---|---|---|
6MWD in metres | ||
OLE study baseline, na (%) | 80 (100.0) | 35 (100.0) |
OLE study baseline, mean (SD) | 373.1 (124.23) | 363.5 (137.38) |
Week 52 of OLE study, n (%) | 74 (92.5) | 33 (94.3) |
Week 52 of OLE study, mean (SD) | 381.9 (126.30) | 360.0 (145.97) |
Change from OLE study baseline to week 52, mean (95% CI) | −2.0 (−9.1 to 5.1) | −1.4 (−14.0 to 11.1) |
Sitting FVC, % predicted | ||
OLE study baseline, nb (%) | 73 (91.3) | 31 (88.6) |
OLE study baseline, mean (SD) | 69.1 (19.41) | 63.8 (19.63) |
Week 52 of OLE study, n (%) | 70 (87.5) | 29 (82.9) |
Week 52 of OLE study, mean (SD) | 67.9 (20.39) | 64.2 (19.21) |
Change from OLE study baseline to week 52, mean (95% CI) | −0.2 (−1.9 to 1.6) | 0.0 (−1.8 to 1.8) |
MMT total score | ||
OLE-enrolled total population, n | 82 | 37 |
OLE study baseline, nb (%) | 81 (98.8) | 34 (91.9) |
OLE study baseline, mean (SD) | 62.2 (8.33) | 62.7 (9.86) |
Week 52 of OLE study, n (%) | 72 (87.8) | 33 (89.2) |
Week 52 of OLE study, mean (SD) | 65.7 (8.41) | 64.0 (10.93) |
Change from OLE study baseline to week 52, mean (95% CI) | 3.2 (1.7 to 4.8) | 1.4 (−0.2 to 2.9) |
PROMIS Physical Function score | ||
OLE study baseline, nb (%) | 79 | 35 |
OLE study baseline, mean (SD) | 68.8 (12.81) | 67.8 (16.74) |
Week 52 of OLE study, n (%) | 75 | 33 |
Week 52 of OLE study, mean (SD) | 69.7 (12.98) | 67.4 (15.23) |
Change from OLE study baseline to week 52, mean (95% CI) | 0.6 (−0.6 to 1.8) | −0.1 (−2.5 to 2.2) |
PROMIS Fatigue score | ||
OLE study baseline, nb (%) | 80 | 35 |
OLE study baseline, mean (SD) | 19.9 (7.50) | 19.3 (6.72) |
Week 52 of OLE study, n (%) | 74 | 33 |
Week 52 of OLE study, mean (SD) | 20.1 (7.13) | 21.0 (6.80) |
Change from OLE study baseline to week 52, mean (95% CI) | 0.2 (−1.1 to 1.6) | 1.5 (−0.6 to 2.6) |
6MWD = 6-minute walk distance; CI = confidence interval; FAS = full analysis set; FVC = forced vital capacity; MMT = manual muscle testing; OLE = open-label extension; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation.
aBaseline is the average of the last 2 values obtained on or before the first dose of the study drug in the ATB200-07 trial.
bBaseline is the last nonmissing value on or before the administration of the first dose of the study drug in the ATB200-07 trial.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Refer to Table 20 for a summary of harms data from the ATB200-07 study that were identified as most relevant for this review. Refer to Table 34 in Appendix 1 for a summary of all harms results from the ATB200-07 study. The safety population consisted of integrated data from the PROPEL and ATB200-07 studies and, therefore, harms results are for the entire 104-week study period.
A total of 84 patients (98.8%) in the cipaglucosidase alfa plus miglustat randomized group (defined as those who received the study drug in the PROPEL trial and may have continued into the ATB200-07 study) experienced at least 1 TEAE during either study. The most common TEAEs were fall (41.2%), headache (35.3%), and arthralgia (31.8%). SAEs were reported in 14 patients (16.5%), 1 of which was drug-related and resulted in discontinuation.
In the treatment-switched group (defined as those who received cipaglucosidase alfa plus miglustat in the ATB200-07 study after receiving alglucosidase alfa in the PROPEL trial), 36 patients (97.3%) experienced a TEAE, most commonly fall (35.1%), headache (29.7%), and arthralgia (27.0%). SAEs were reported in 6 patients (16.2%), 2 of whom discontinued treatment.
A total of 5 patients treated with cipaglucosidase alfa plus miglustat experienced an IAR TEAE leading to study drug discontinuation (2 during the PROPEL trial, 1 continuing treatment in the OLE study, and 2 in the treatment-switched population of the OLE study), 3 of which were considered serious IARs. All 5 of these patients tested positive for specific antidrug antibodies before their IAR TEAEs.
No TEAEs leading to death were reported in any treatment groups.
Table 20: Summary of Safety Results Across the Entire Study Period (104 Weeks) — PROPEL and ATB200-07 Trials (Safety Populations)
Adverse events | Cipaglucosidase alfa + miglustat (N = 85) | Treatment switched (prior alglucosidase alfa + placebo) (N = 37) |
|---|---|---|
Adverse events reported by ≥ 20% of patients, n (%) | ||
≥ 1 adverse event | 84 (98.8) | 36 (97.3) |
Fall | 35 (41.2) | 13 (35.1) |
Headache | 30 (35.3) | 11 (29.7) |
Arthralgia | 27 (31.8) | 10 (27.0) |
Nasopharyngitis | 24 (28.2) | 1 (2.7) |
Myalgia | 23 (27.1) | 7 (18.9) |
Back pain | 19 (22.4) | 5 (13.5) |
Pain in extremity | 17 (20.0) | 8 (21.6) |
Diarrhea | 17 (20.0) | 3 (8.1) |
SAEs reported by ≥ 5% of patients, n (%) | ||
Patients with ≥ 1 SAE | 14 (16.5) | 6 (16.2) |
Injury, poisoning, and procedural complications | 5 (5.9) | 1 (2.7) |
Patients who had any TEAE leading to study drug discontinuation, n (%)a | ||
Patients who stopped treatment due to adverse events | 3 (3.5) | 2 (5.4%) |
Urticaria | 1 (1.2) | 1 (2.7) |
Anaphylactic reaction | 0 | 1 (2.7) |
Anaphylactoid reaction | 1 (1.2) | 0 |
Chills | 1 (1.2) | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Adverse events of special interest, n (%) | ||
Patients with IAR TEAEsb | 27 (31.8) | 10 (27.0) |
Total number of IAR TEAEs | 148 | 46 |
IAR = infusion-associated reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aA TEAE was defined as any event that started on or after the first dose of respective study drug. Any adverse event that occurred after 30 days from the last dose of the study drug in the ATB200-03 trial and before the first dose of the study drug in the ATB200-07 trial was not counted as treatment-emergent.
bA patient experiencing the same TEAE multiple times was counted once for the corresponding system organ class or preferred term.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
The ATB200-07 study was designed as an OLE to assess the long-term safety and tolerability of cipaglucosidase alfa and miglustat coadministration in adult patients with LOPD. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and reporting of safety parameters due to unblinded exposure to the study medication during the treatment period.
The ATB200-07 study population for this interim analysis consisted of patients who took part in the PROPEL trial and therefore it is reasonable to expect that the same strengths and limitations related to generalizability apply to the OLE study. Given that patients needed to complete the PROPEL trial before enrolling, the OLE study population is inherently enriched and introduces some selection bias for responders and for those with a potentially lower risk for adverse effects.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
The sponsor conducted an ITC in the form of an NMA in support of the combination of cipaglucosidase alfa and miglustat treatment in adults with LOPD. No head-to-head trials are currently available comparing avalglucosidase alfa versus cipaglucosidase alfa plus miglustat, or avalglucosidase alfa versus cipaglucosidase alfa plus miglustat in patients with LOPD. Therefore, an ITC was done to obtain estimates on the relative effects of those treatments and to evaluate how those treatments and placebo compare with each other.1
Description of ITC
The sponsor submitted 1 ITC that included 2 related analyses. Evidence was identified through a systematic review and was synthesized through an ML-NMR.
Table 21: Study Selection Criteria and Methods for ITC Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Adult patients (≥ 18 years) with LOPD |
Intervention | Cipaglucosidase alfa plus miglustat |
Comparator |
|
Outcome |
|
Study designs |
|
Publication characteristics | Peer-reviewed article, conference abstract, conference paper, article in press, report |
Exclusion criteria | Not matching the previously described criteria |
Databases searched | Databases (search dates: September 14 to 15, 2022; update: August 2023):
Other sources Clinical trial registries (search date: September 13, 2022):
Conferences (2020 to 2023):
Grey literature:
|
Selection process |
|
Data extraction process | Single data extractor |
Quality assessment |
|
AAN = American Academy of Neurology; ACMG = American College of Medical Geneticists; CDER = Center for Drug Evaluation and Research; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; DARE = Database of Abstracts of Review of Effects; EAN = European Academy of Neurology; ICTRP = International Clinical Trials Registry Platform; ICNMD = International Congress on Neuromuscular Diseases; ITC = indirect treatment comparison; LOPD = late-onset Pompe disease; RoB = risk-of-bias; ROBINS-I = Risk of Bias in Nonrandomized Studies – of Interventions; SLR = systematic literature review; SSIEM = Society for the Study of Inborn Errors of Metabolism; WORLD = We’re Organizing Research on Lysosomal Diseases.
Sources: Shohet et al. (2024).31 Details included in the table are from the sponsor’s summary of clinical evidence.
ITC Design
Objectives
The objective of this NMA was to compare the efficacy of cipaglucosidase alfa with miglustat against avalglucosidase alfa and alglucosidase alfa in adult patients with LOPD.31
Study Selection Methods
The sponsor conducted an SLR from September 7 to 15, 2022, with an update in August 2023, to identify studies published up to July 31, 2023, that investigated the efficacy or safety of cipaglucosidase alfa plus miglustat, avalglucosidase alfa, or alglucosidase alfa in adults with LOPD. The eligibility criteria followed a population, intervention, comparison, and outcome framework: adult patients (≥ 18 years) with LOPD; interventions of cipaglucosidase alfa plus miglustat; comparators, including alglucosidase alfa, avalglucosidase alfa, or placebo or best supportive care; and reported outcomes of 6MWD in metres or FVC as a percentage of the predicted value. RTCs, nonrandomized clinical trials, single-arm, and OLE studies, as well as pertinent systematic reviews and meta-analyses, were considered. The sponsor searched multiple databases and trial registries, including MEDLINE, Embase, Cochrane CENTRAL, PROSPERO, ISRCTN.com, and others; conference proceedings and grey literature were also examined. In the original SLR, 2 reviewers independently screened citations, resolving discrepancies with a third reviewer, whereas the updated search was handled by a single reviewer. Potentially eligible papers underwent full-text review, and studies reporting 6MWD or FVC values were prioritized for possible inclusion in the indirect comparisons. Data extraction was performed by a single extractor using standardized forms, and the sponsor also had access to IPD from the PROPEL RCT for cipaglucosidase alfa plus miglustat.
Risk of bias in RCTs was assessed with the revised Cochrane risk-of-bias version 2 tool, while nonrandomized and single-arm studies were evaluated using the Risk of Bias in Nonrandomized Studies – of Interventions instrument. The sponsor excluded certain small or exploratory studies, such as the EMBASSY trial. Ultimately, the 2 efficacy end points — change from baseline in 6MWD and change in FVC (percent predicted) — were selected for quantitative synthesis. Both are widely recognized functional outcomes in LOPD trials, with 6MWD reflecting ambulatory and endurance capacity (often following standardized American Thoracic Society guidelines) and FVC capturing pulmonary function in either a sitting or upright position. Safety outcomes were identified during the SLR but were not formally pooled or analyzed in the ITC, so only efficacy results for 6MWD and FVC were carried forward into the final analyses.
To assess the heterogeneity of the studies identified by the SLR and studies subsequently becoming publicly available, a feasibility assessment was conducted. The assessment was based on the inclusion and exclusion criteria used in the literature review and referred to the comparability of study characteristics, populations, and outcome definitions. The aim was to determine the extent to which the study results could be combined into an ITC. The identified studies were compared with regard to the inclusion and exclusion criteria and baseline patient characteristics such as age, sex, ethnicity, previous ERT duration, baseline 6MWD, and baseline FVC. The outcomes of interest assessed for the ITC included change from baseline to week 52 in 6MWD (metres) and FVC (percent predicted). A publication was excluded from the analysis if it did not provide 6MWD or FVC measures.
ITC Analysis Methods
Overview and Justification for Statistical Method
For this ITC, the ML-NMR method was used,2-4 which is an extension to an NMA that accounts for the effect of study-level covariates. The sponsor reported that a standard NMA was not suitable because the 2 RCTs (PROPEL and COMET) forming the networks via the common comparator alglucosidase alfa differed with respect to previous treatment, and previous studies suggested ERT treatment is an effect modifier (i.e., factors that lead to a differential effect of the intervention).1 The sponsor noted that baseline covariates were balanced within each trial by randomization, on average (but may differ in distribution between trials). The sponsor has access to IPD for cipaglucosidase alfa plus miglustat in the PROPEL trial. The sponsor explained that because the COMET trial included only patients who were ERT naive, using an anchored matching-adjusted indirect comparison or simulated treatment comparison would lead to results to an NMA of the COMET trial and the ERT-naive subgroup from the PROPEL trial. Additionally, the sponsor aimed to incorporate data from multiple single-arm studies, which would not be possible in an unanchored matching-adjusted indirect comparison or unanchored simulated treatment comparison because they are applicable only to the simple pairwise scenario.1 Therefore, an ML-NMR was conducted, which is a generalization of a network meta-regression and can be applied to any connected network with any mixture of IPD and aggregate data.5
ML-NMR first fits a regression model at the individual patient level for studies where IPD exist, capturing how baseline covariates modify outcomes. This model is then applied to the aggregate-level data from other studies, integrating across the distribution of key covariates. As a result, ML-NMR can potentially account for relative treatment-effect estimates for a given target population by balancing baseline-effect modifiers that vary between studies.
Model Summary
The ML-NMR aggregate-level model is an integration of the individual-level model over the population in each trial. Briefly, an individual-level regression is fitted to IPD studies and a (numeric) integration of this regression is performed over the aggregate data studies. Refer to Shohet et al. (2024) for details.31
In this analysis, a Bayesian framework was chosen in which an ML-NMR had been implemented using Markov chain Monte Carlo sampling in Stan software.6 The sponsor considered the Bayesian framework to be suitable for performing ITCs that have a small number of trials and low sample sizes because this is common in cases of rare diseases and because informative priors, which reflect a prior belief of the possible values of the pooled relative effect and effects of covariates, can be chosen to alleviate the limitations of the small networks and low sample sizes of the trials included in the network. In random-effects models, the sponsor selected an informative prior for the between-study heterogeneity parameter tau.
For 6MWD and FVC, the 2 outcomes measured in the ITC, mean difference between treatments with associated 95% CrIs were calculated. The Bayesian probability that the treatment was superior (mean difference > 0) or inferior (mean difference < 0) to the reference treatment was also derived. Probabilities were interpreted to suggest strong evidence (probability < 0.1% or > 99.9%), some evidence (probability between 0.1% and 5% or between 95% and 99.9%), weak evidence (probability between 5% and 10% or 90% and 95%), and no evidence.
Statistical Models
The ML-NMR used fixed-effects models, which assumed a common treatment effect across all study settings, and any differences between observed effect sizes can be explained by the included effect modifiers or due to sampling error because the number of studies included in the network would not allow the estimation of between-study variability.
Two separate analyses were conducted, analysis A and analysis B:
In analysis A, only RCTs were included in the network (RCTs-only network).
In analysis B, all trials selected in the feasibility assessment were included, including single-arm trials (full evidence network). For this analysis, the single-arm study results were matched to the appropriate comparator arms of the comparative studies to allow for inclusion into the network.7 The inclusion of single-arm and OLE studies in network B meant data from patients who were ERT experienced and receiving avalglucosidase alfa were included. The was done to explore the impact of ERT experience on the relative effects of cipaglucosidase alfa versus avalglucosidase alfa. It was not possible to fully adjust for ERT experience based on RCT data only, because the COMET trial (the only other RCT besides the PROPEL study) with avalglucosidase alfa included only patients who were ERT naive.
Matching of Single-Arm Studies
Single-arm phase I/II and OLE trials were included into the network by matching them to a comparator arm of the comparative studies. The choice of the appropriate comparator arm was based on a matching criterion summing the distances between the (aggregate) baseline characteristics of the single-arm trial and the potential comparator arm. However, the consequence of such an approach is that the same comparison group has to be used multiple times across indirect comparisons, effectively artificially inflating the effect of comparator. Table 22 shows the matching of single-arm studies to an appropriate comparator arm and provides a justification for the choice of the comparator arm.
Table 22: Matching of Single-Arm Studies to Comparator Arms From RCTs
Single-arm trial and publication | Arm | Matched to | Comment |
|---|---|---|---|
NEO1 and NEO-EXT Dimachkie (2020) | Avalglucosidase alfa | Alglucosidase alfa arm of the PROPEL trial | Prior ERT duration was similar for these 2 trial arms:
|
COMET OLE Schoser (2022a) | Avalglucosidase alfa | Alglucosidase alfa arm of the COMET trial | Prior ERT duration was similar for these 2 trial arms:
|
ATB200-02 Schoser (2022b) | Cipaglucosidase alfa plus miglustat | Alglucosidase alfa arm of the PROPEL trial | Prior ERT duration was similar for these 2 trial arms:
|
PROPEL OLE Schoser (2023) | Cipaglucosidase alfa plus miglustat | Alglucosidase alfa arm of the PROPEL trial | Prior ERT duration was similar for these 2 trial arms:
|
ERT = enzyme replacement therapy; OLE = open-label extension; RCT = randomized controlled trial.
Sources: Shohet et al. (2024).31 Details included in the table are from the sponsor’s summary of clinical evidence.
Covariates and Distributions
The linear regression models that were part of the ML-NMR aimed to include all baseline covariates that were considered potential treatment-effect modifiers that could affect the outcomes of interest if reported in the studies. The baseline covariates included in the models were age (years), sex (percent male), baseline previous ERT duration (years), and baseline outcome values. Covariates were included into the model as main effects (prognostic factor) and interacting with treatment (treatment-effect modifier). Covariates were estimated for each treatment independently by specifying a separate regression model for each treatment, if data permitted, and for treatment classes with a regression model per treatment class, the latter approach assuming shared effect modifiers for treatments of the same class.
Suitable marginal distributions to draw the integration points from were chosen for these covariates, according to Table 23.
Table 23: Marginal Distributions for the Covariates
Covariate | Type | Marginal distribution |
|---|---|---|
Baseline age (years) | Continuous | Normal distribution |
Sex (% male) | Binary | Bernoulli distribution |
Baseline previous ERT duration (years) | Continuous | Gamma distribution (to account for skewness) |
Baseline 6MWD (metres) | Continuous | Normal distribution |
Baseline FVC (% predicted) | Percentage | Logit-normal distribution |
Visit time | Discrete | Poisson distribution |
6MWD = 6-minute walk test; FVC = forced vital capacity.
Sources: Shohet et al. (2024).31 Details included in the table are from the sponsor’s summary of clinical evidence.
Heterogeneity Assessment
The characteristics of patients at baseline in the included studies were first investigated and compared. All heterogeneity related to baseline age, sex, baseline previous ERT duration, baseline 6MWD, or baseline sitting FVC, and visit time had been incorporated in the model because these covariates mentioned are included as effect modifiers.
Consistency Assessment
Classic approaches for assessing inconsistency included the unrelated mean effects model12 and node-splitting models.13 However, in the star-shaped network, the statistical assessment of consistency was not allowed due to the lack of indirect and direct evidence for the same comparison.
Base Case and Scenarios
For each end point, the base-case scenario was defined as patient characteristics set to those of the PROPEL study, which was the target population of interest (Table 24).
Table 24: Base-Case Scenario Covariate Setting
Age | Percent male | Percent white | ERT duration (years) | 6MWD (metres) | FVC (% predicted) | Time (weeks) |
|---|---|---|---|---|---|---|
46.95 | 45.08 | 84.43 | 5.744 | 355.8 | 70.42 | 52 |
6MWD = 6-minute walk distance; ERT = enzyme replacement therapy; FVC = forced vital capacity.
Sources: Shohet et al.31 Details included in the table are from the sponsor’s summary of clinical evidence.
The following scenario analyses were done by varying previous ERT durations, while the other covariates were kept at the values of the base-case scenario:
ERT-naive scenario: Previous ERT duration of 0 years.
Short ERT duration scenario: Previous ERT duration of 2.5 years (25% quantile of the previous ERT duration distribution in the PROPEL trial).
Medium ERT duration scenario: Previous ERT duration of 5 years (median previous ERT duration in the PROPEL trial).
Long ERT duration scenario: Previous ERT duration of 9.2 years (75% quantile of the previous ERT duration distribution in the PROPEL trial).
Table 25: Indirect Comparison Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Multilevel NMR with Bayesian framework using fixed-effects and random-effects models. IPD from the PROPEL trial and aggregate-level data for the remaining trials were used. |
Priors | Informative priors reflecting a prior belief of the possible values of the pooled relative effect and effects of covariates. |
Assessment of model fit | DIC (all models) and the heterogeneity standard deviation tau (random-effects model only): Decrease of at least 5 points in the DIC-indicated heterogeneity. |
Assessment of consistency | Classic approaches for assessing inconsistency, such as unrelated mean effects models and node-splitting models, were not allowed in the star-shaped network due to lack of indirect and direct evidence for the same comparison, i.e., lack of closed loops in the network. |
Assessment of convergence | Gelman-Rubin statistic (i.e., R-hat) value ≤ 1.1 indicated convergence had been reached. |
Outcomes | Change from baseline in 6MWD (metres) and in FVC (% predicted) |
Follow-up time point | Week 52 |
Construction of nodes | As described subsequently |
Sensitivity and scenario analyses | ERT duration varied: ERT naive and short, medium, and long duration (0, 2.5, 5, and 9.2 years, respectively). |
Subgroup analysis | Not done |
Methods for pairwise meta-analysis | Not done |
6MWD = 6-minute walk distance; DIC = deviance information criterion; ERT = enzyme replacement therapy; FVC = forced vital capacity; IPD = individual patient-level data; NMR = network meta-regression.
Sources: Shohet et al. (2024).31 Details included in the table are from the sponsor’s summary of clinical evidence.
Results
Summary of Included Studies
Of the studies identified in the SLR, a feasibility assessment was done on 9 studies for inclusion in the NMA. The age across studies ranged from 42.6 to 51.6 years. Sex was not equally distributed (30% to 64% were male). There were differences in ERT status across studies: 4 studies were on patients who were ERT naive, while patients in 3 studies received ERT treatment for more than 5 years. The mean baseline 6MWD varied from 313 m to 450 m, and the mean baseline FVC varied from about 51% to 77% of predicted values. Refer to Shohet et al. (2024) for further details on study and patient characteristics.31
The observed differences in baseline characteristics across studies could affect the credibility of a standard ITC. However, the ML-NMR approach relaxed the assumption of balanced effect modifiers by adjusting for baseline covariates using the IPD from the PROPEL trial.
Three studies were excluded after the feasibility analysis: the LOTS and LOTS OLE trials were not used because the focus was on the indirect comparison of cipaglucosidase alfa plus miglustat with avalglucosidase alfa, to which the LOTS and the LOTS OLE trials did not contribute. The EMBASSY trial was not included in the ITC because of the short follow-up, small sample size, and exploratory nature of the study, leaving a total of 34 records from 6 studies for inclusion in the analysis.
Evidence Networks
The evidence networks are presented in Figure 2, including the network for analysis A (i.e., only the 2 RCTs, COMET and PROPEL) and the network for analysis B (comprising the 2 RCTs and the single-arm trials). Both 6MWD change from baseline and FVC change from baseline were reported in all studies included in the analysis; hence, the networks for the 2 end points were the same, as shown in Figure 2.
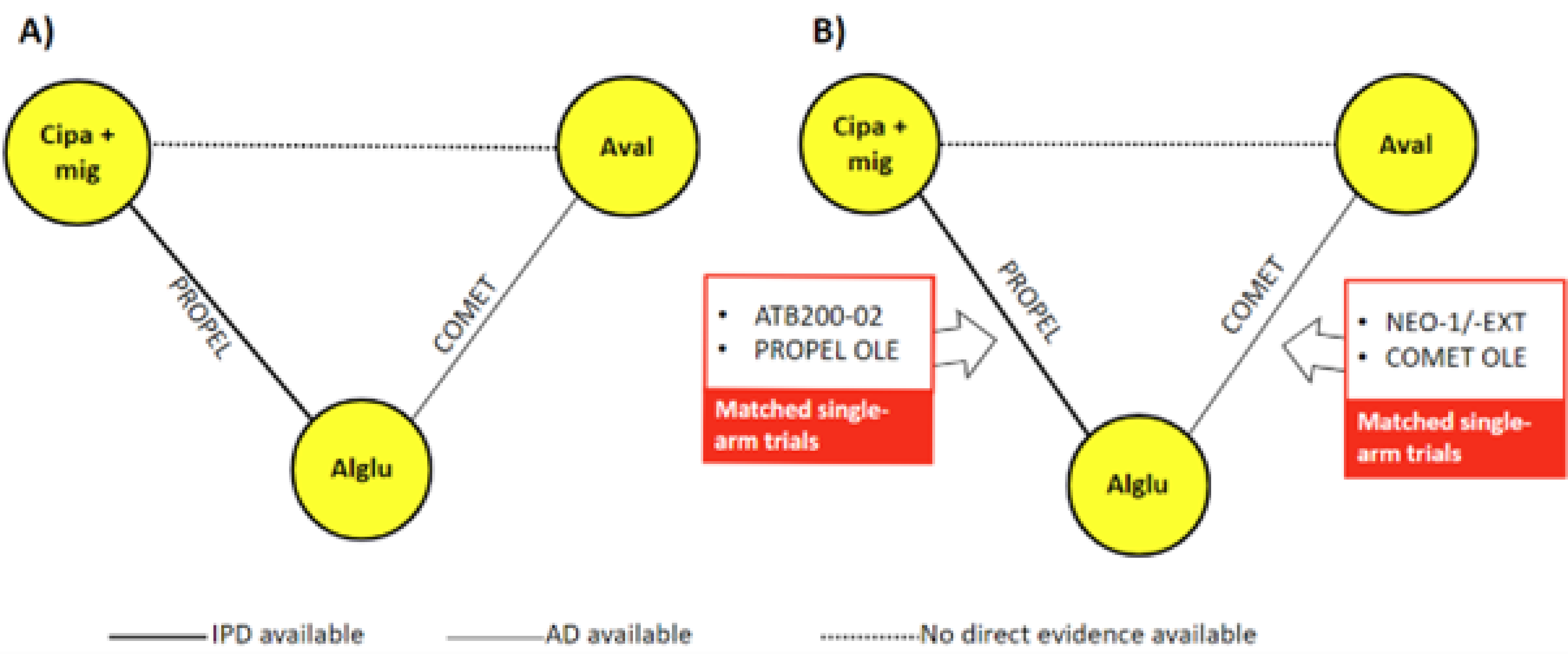
AD = aggregated data; Alglu = alglucosidase; Aval = avalglucosidase; Cipa = cipaglucosidase; IPD = individual patient-level data; Mig = miglustat; OLE = open-label extension; RCT = randomized controlled trial.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.
Efficacy
In this section, results from the model diagnostics are described first. The results of analysis A (RCTs-only network) and analysis B (including the full network) are shown subsequently.
Model Evaluation
The between-study SD tau was high when covariates were estimated for 2 treatment classes, suggesting more significant between-study heterogeneity. This was confirmed by the improved model fit for 6MWD in the random-effects model versus the fixed-effects model (deviance information criterion [DIC] decreased by > 5 points), but not for FVC (similar DICs). In contrast, between-study heterogeneity seemed low or accounted for in the models when the covariate effect was estimated separately for the 3 ERTs: tau was low, and the DICs were similar for both fixed-effects and random-effects models.
Based on this, B-FE-3 was chosen by the author of the study as the final model to analyze network B for both outcomes.31
Six-Metre Walk Distance
The analysis of network A (RCTs only) showed that cipaglucosidase alfa plus miglustat was associated with an increase in 6MWD (mean difference of 14.64 m; 95% CrI, 7.07 m to 22.31 m; Bayesian probability > 99.9%) compared with alglucosidase alfa, with a reduction in 6MWD of −10.02 m (95% CI, −23.62 m to 4.00 m; Bayesian probability = 91.8%) compared with avalglucosidase alfa, and avalglucosidase alfa was associated with an increase in 6MWD of 24.66 m (95% CI, 9.95 m to 39.55 m; Bayesian probability > 99.9%) compared with alglucosidase alfa.
The analysis of network B (full evidence) showed that cipaglucosidase alfa plus miglustat was associated with an increase in 6MWD compared with alglucosidase alfa and avalglucosidase alfa, with a mean difference of 13.64 m (95% CrI, 8.73 m to 18.70 m; Bayesian probability > 99.9%) and 28.93 m (95% CrI, 8.26 m to 50.11 m; Bayesian probability = 99.7%), respectively, and avalglucosidase alfa was associated with a reduction in 6MWD of −15.30 m (95% CrI, −35.33 m to 6.43 m; Bayesian probability = 92.8%) compared with alglucosidase alfa.
Forced Vital Capacity
The analysis of network A (RCTs only) showed that cipaglucosidase alfa plus miglustat was associated with an increase in percent predicted FVC of 2.53% (95% CrI, 1.38% to 3.67%; Bayesian probability > 99.9%) compared with alglucosidase alfa, with a reduction in FVC of −1.45% (95% CrI, −3.01% to 0.07%; Bayesian probability = 96.8%) compared with avalglucosidase alfa, and avalglucosidase alfa was associated with an increase in FVC of 3.98% (95% CrI, 2.40% to 5.64%; Bayesian probability > 99.9%) compared with alglucosidase alfa.
The analysis of network B (full evidence) showed that cipaglucosidase alfa plus miglustat was associated with an increase in FVC compared with alglucosidase alfa and avalglucosidase alfa of 3.95% (95% CI, 3.23% to 4.69%; Bayesian probability > 99.9%) and 2.88% (95% CI, 1.07% to 4.71%; Bayesian probability > 99.9%), respectively, but avalglucosidase alfa was no different in FVC (1.07%; 95% CI, −0.71% to 2.92%; Bayesian probability = 87.6%) compared with alglucosidase alfa.
Scenario Analysis
Scenario analyses were conducted with different durations of previous ERT (0 years [i.e., ERT naive], 2.5 years, 5 years, and 9.2 years). For both end points and in both networks, cipaglucosidase alfa plus miglustat became more favourable compared with alglucosidase alfa as the duration of previous ERT use increased. Compared with avalglucosidase alfa, however, the effect of increasing ERT duration was not so clear, especially between the 2 models. With network A (RCTs only), avalglucosidase alfa showed directional favourability versus cipaglucosidase alfa plus miglustat for both 6MWD and FVC. However, in all studies, network B (full evidence) demonstrated the opposite, indicating the importance of fully adjusting for ERT experience to determine the relative effects of these 2 treatments.
Harms
The ITC did not analyze any harms-related outcomes.
Critical Appraisal of ITC
The SLR conducted by the sponsor provides a good foundation for the ITC. However, the update and data extraction processes were conducted by a single reviewer, which may increase the risk of errors or missed information. This is a notable limitation because the robustness of ITC findings depends heavily on the comprehensiveness and accuracy of the data sources included.
The use of ML-NMR is a strength of the analysis because it allows for adjustment of treatment effects to address observed clinical heterogeneity. The adjustment for known treatment-effect modifiers, such as age, baseline 6MWD, and ERT duration, is another positive aspect. However, while these factors are widely recognized in the field, the sponsor did not provide explicit justification for their selection as treatment-effect modifiers. Other potential effect modifiers, such as weight, height, muscle damage, and angiotensin-converting enzyme genotype, should have been considered. Their omission, due to a lack of reporting, could lead to residual confounding and uncertainty in the results. Additionally, although adjustments were made for these factors, they may not fully mitigate the differences observed in these characteristics between the studies, and residual confounding could still result in some bias in the analyses.
An initial limitation identified during the review was the lack of reported diagnostics to assess the goodness of fit for the ML-NMR model, particularly concerning the application of IPD-derived adjustments to aggregate-level data. In response, the sponsor provided a Pearson residual analysis. This analysis compared the final ML-NMR model predictions with the observed study-level data for the key outcomes (6MWD and FVC) across all included studies. The results indicated that the absolute values of the Pearson residuals fell within the standard threshold of less than 2, suggesting an acceptable overall fit of the model to the data used in the network.
While this addresses the specific absence of residual analysis and increases transparency regarding overall model fit, other considerations remain. Specific diagnostics focused on validating the appropriateness or impact of the step involving the application of IPD-derived adjustments to aggregate data (such as postadjustment covariate balance assessments or sensitivity analyses of the adjustment model) were not provided. Additionally, other measures of model performance (e.g., R-squared values) were not reported. This absence of more detailed validation for the adjustment process itself reduces confidence in the specific methodology used to bridge evidence across disparate trial populations. Furthermore, the reliance on reconstructed aggregate-level data introduces potential bias, especially if covariates are inconsistently reported or misaligned across studies.
In network A, the effect of covariates was not estimated separately for either cipaglucosidase alfa with miglustat or avalglucosidase alfa. Instead, shared effect modifiers were assumed, requiring the extrapolation of the impact of previous ERT duration for avalglucosidase from the cipaglucosidase alfa with miglustat data. This strong assumption is a notable limitation because the COMET trial did not include patients who were ERT experienced, further complicating the analysis.
Network B attempted to address the gaps in network A by including single-arm trials and OLEs, which allowed for the inclusion of patients who were ERT experienced treated with avalglucosidase alfa. However, the inclusion of single-arm studies introduced considerable methodological heterogeneity. Single-arm trials are not anchored to a comparator, necessitating adjustments for both prognostic factors and treatment-effect modifiers. Even with matching methods, baseline differences and residual heterogeneity remain concerns. Additionally, the repeated use of comparator data, such as alglucosidase alfa, inflates precision estimates and may favour interventions that have more data points.
Further limitations include the lack of inconsistency testing because there were no closed loops in the networks, and the questionable transitivity assumption, particularly in network B, where single-arm studies were included. The use of informative priors for the random-effects models in network B was stated but not clearly justified, nor were other various priors tested in a sensitivity analysis, leaving uncertainty about the appropriateness of these priors. The data-driven approach to choosing fixed-effects versus random-effects models in network B was not applied consistently because network A used a fixed-effects model without a reported similar evaluation.
The increased residual heterogeneity and wider CrIs in network B underscore the challenges of incorporating single-arm trials and highlight the limitations of the ML-NMR approach in adjusting for underreported or unobserved effect modifiers and prognostic factors. The limited diagnostic information on numeric integration and the application of coefficients to aggregate data further complicates the interpretation of the results. While the sponsor’s use of an ML-NMR offers valuable insights, the methodological and reporting gaps identified here introduce substantial uncertainty, reducing the overall confidence in the findings.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Two studies were proposed by the sponsor as addressing gaps in the systematic review evidence, summarized in Table 27.
Table 26: Summary of Gaps in the Systematic Review Evidence
Study description | Evidence gap |
|---|---|
Open-label 48-month phase I/II study (ATB200-02) | Patients who were nonambulatory with LOPD are generally not included in clinical trials because these patients typically present with more severe symptoms. |
Patients who were ERT naive were underrepresented in the pivotal trial. | |
UK EAMS registry data | Real-world data in patients with LOPD is lacking. |
EAMS = Early Access to Medicines Scheme; ERT = enzyme replacement therapy; LOPD = late-onset Pompe disease.
Source: Details included in the table are from the sponsor’s summary of clinical evidence.
Table 27: Details of Studies Addressing Gaps in the Systematic Review Evidence
Detail | ATB200-02 trial | UK EAMS registry study |
|---|---|---|
Designs and populations | ||
Study design | Ongoing, open-label, phase I/II study evaluating the long-term (up to 48 months) efficacy of cipaglucosidase alfa plus miglustat | Prospective, observational registry study evaluating the real-world safety and effectiveness of cipaglucosidase alfa plus miglustat |
Enrolled, N | 29 | 37 |
Key inclusion criteria |
|
|
Key exclusion criteria |
| Not reported |
Drugs | ||
Intervention | Cipaglucosidase alfa: Administered every 2 weeks as a 4-hour IV infusion Miglustat: Administered orally 1 hour before cipaglucosidase alfa infusion, with fasting 2 hours before and 2 hours after miglustat uptake Dosages:
| Patients switched from standard of care treatment to IV cipaglucosidase alfa plus oral miglustat according to the same regimen as approved dose (and the PROPEL trial). |
Comparator(s) | No comparator or placebo | No comparator or placebo |
Outcomes | ||
End points | End points measured (over 48 months):
| End points measured:
|
Notes | ||
Publications | Byrne BJ, Schoser B, Kishnani PS et al. Long-term safety and efficacy of cipaglucosidase alfa plus miglustat in individuals living with Pompe disease: An open-label phase I/II study (ATB200-02). J Neurol. 2024 Apr;271(4):1787 to 1801. | Details included in the table are from the sponsor’s summary of clinical evidence. |
6MWD = 6-minute walk distance; CFB = change from baseline; EAMS = Early Access to Medicines Scheme; ERT = enzyme replacement therapy; FSS = Fatigue Severity Scale; FVC = forced vital capacity; GAA = acid alpha-glucosidase; IAR = infusion-associated reaction; LOPD = late-onset Pompe disease; PGIC = Physician’s Global Impression of Change; PK = pharmacokinetics; PRO = patient-reported outcome; MMT = manual muscle test; MRC = Medical Research Council; R-PAct = Rasch-built Pompe-specific activity scale; rhGAA = recombinant human acid alpha-glucosidase; RHS = Rotterdam Handicap Scale; SGIC = Subject Global Impression of Change.
aEnd point results available for this interim analysis.
Sources: ATB-200-02 Clinical Study Report.32 Details included in the table are from the sponsor’s summary of clinical evidence.
Study ATB200-02
Description
ATB200-02 (NCT02674565) is an ongoing, open-label, phase I/II study evaluating the long-term (up to 48 months) efficacy of cipaglucosidase alfa plus miglustat in adults with Pompe disease. This study includes patients who are nonambulatory with LOPD, who are typically excluded from clinical trials due to having more severe symptoms, and patients who are ERT naive due to underrepresentation in the pivotal trial.
The primary treatment period consisted of stage 1 (6 weeks) and stage 2 (12 weeks). Stage 3 was a 2-year treatment period and stage 4 is an ongoing long-term extension. The trial included 4 cohorts based on ERT experience, age, and ambulatory status:
Cohort 1: ERT experienced with 2 to 6 years of prior ERT with alglucosidase alfa every 2 weeks and ambulatory (6MWD of 200 m to 500 m) and an upright FVC of 30% to 80% of predicted normal value.
Cohort 2: ERT experienced with 2 years or more of prior ERT with alglucosidase alfa and nonambulatory (wheelchair dependence and the inability to walk unassisted).
Cohort 3: ERT naive and ambulatory (6MWD of 200 m to 500 m) and an upright FVC of 30% to 80% of predicted normal value.
Cohort 4: ERT experienced with 7 years or more of prior ERT with alglucosidase alfa every 2 weeks and ambulatory (6MWD of 75 m to 600 m) and an upright FVC of 30% to 85% of predicted normal value.
As shown in Figure 3, patients in cohort 1 received cipaglucosidase alfa as a single drug in ascending doses in stage 1. In stage 2, cohort 1 received cipaglucosidase alfa 20 mg/kg plus miglustat 130 mg or 260 mg for 3 consecutive doses each until an appropriate regimen had been determined. In stage 3, the additional cohorts of patients who were nonambulatory (cohort 2), patients who were ERT naive (cohort 3), and patients who were ERT experienced with 7 years or more of prior treatment with alglucosidase alfa (cohort 4) were enrolled. In stages 3 and 4, all cohorts received cipaglucosidase alfa 20 mg/kg coadministered with miglustat 260 mg.
Figure 3: Overview of ATB200-02 Study Design
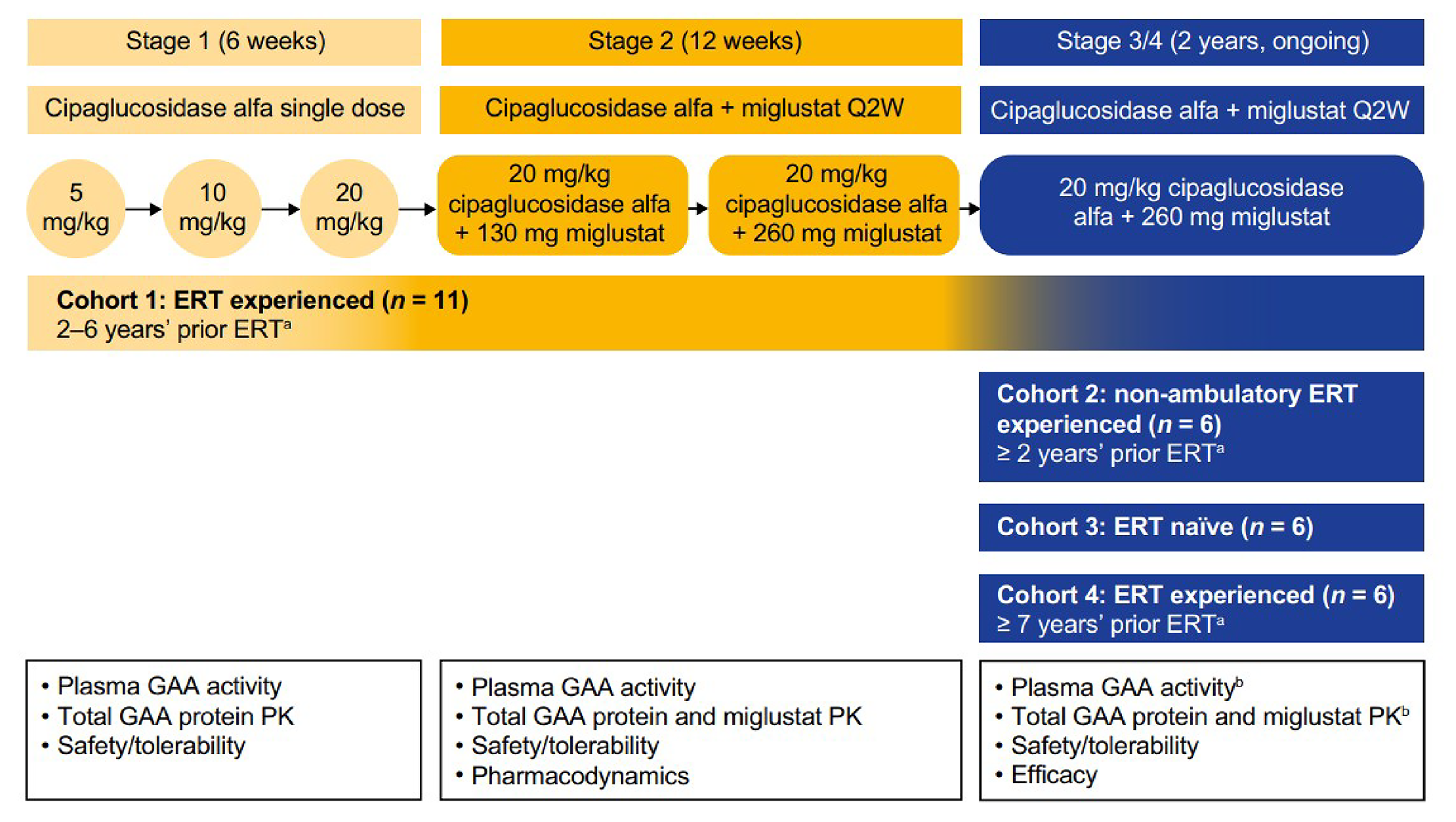
ERT = enzyme replacement therapy; GAA = acid alpha-glucosidase; PK = pharmacokinetics; Q2W = every 2 weeks.
Sources: ATB200-02 Clinical Study Report.32 Details included in the table are from the sponsor’s summary of clinical evidence.
Baseline Characteristics
Refer to Table 28 for a summary of the baseline characteristics of patients in the ATB200-02 study. Across all groups, 89.7% of patients were between the ages of 18 and 64 years. For patients who were ERT experienced (cohorts 1, 2, and 4), the mean duration of ERT ranged from 5.1 years (SD = 1.3 years) to 10.6 years (SD = 2.1 years), with an overall median dose of 20 mg/kg. The patients in both the ERT-experienced and ERT-naive groups exhibited a significant impact of Pompe disease at study entry, based on baseline 6MWD and percent predicted FVC.
Table 28: Summary of Baseline Characteristics of ATB200-02 Study Population
Characteristics | ERT experienced | ERT naive | ||
|---|---|---|---|---|
Cohort 1, 2 to 6 years before ERT (N = 11) | Cohort 2, nonambulatory ≥ 2 years before ERT (N = 6) | Cohort 4, ≥ 7 years before ERT (N = 6) | Cohort 3 (N = 6) | |
Age, years, mean (SD) | 49.4 (9.5) | 41.5 (18.1) | 40.8 (17.0) | 49.3 (15.1) |
Sex, male:female | 9:2 | 4:2 | 2:4 | 1:5 |
Race, white, n (%) | 8 (72.7) | 3 (50.0) | 5 (83.3) | 1 (16.7) |
Time since Pompe diagnosis, years, mean (SD) | 7.7 (5.1) | 15.9 (13.1) | 13.0 (4.3) | 5.2 (4.7) |
Time on alglucosidase alfa, years, mean (SD) | 5.1 (1.3) | 10.1 (4.8) | 10.6 (2.1) | NAa |
6MWD, % predicted, mean (SD) | 61.0 (13.43) | NA | 59.0 (21.44) | 67.8 (12.61) |
6MWD, metres, mean (SD) | 397.2 (96.80) | NA | 387.3 (161.28) | 396.0 (75.20) |
Sitting FVC, % predicted, mean (SD) | 52.6 (13.9) | 42.3 (28.2) | 65.3 (21.1) | 57.2 (20.8) |
MMT lower extremity score, mean (SD) | 31.8 (1.9) | NA | 27.3 (3.7) | 29.0 (1.7) |
6MWD = 6-minute walk distance; ERT = enzyme replacement therapy; FVC = forced vital capacity; MMT = manual muscle test; NA = not applicable; SD = standard deviation.
aOne patient who was ERT naive in cohort 3 received 1 dose of alglucosidase alfa > 6 months before study entry.
Sources: ATB-200-02 Clinical Study Report.32 Details included in the table are from the sponsor’s summary of clinical evidence.
Statistical Analysis
Study ATB200-02 did not involve any inferential statistics or hypothesis testing, and no formal sample size calculation was performed. However, a sample size of between 18 and 34 patients was considered by the sponsor to be adequate for this study. Continuous variables were summarized using descriptive statistics. Efficacy analyses were conducted in the efficacy population (patients who took at least 1 dose of 20 mg/kg cipaglucosidase alfa plus 260 mg miglustat coadministration in stage 3 and had both a baseline and at least 1 postbaseline assessment for any efficacy end point).
Results
Patient Disposition
A total of 29 patients were screened and 3 patients discontinued the study during stage 3 (in the first 2 years of treatment with cipaglucosidase alfa plus miglustat) before the data cut-off date. Discontinuations were due to AEs in 2 patients (6.9%) due to urticaria and diffuse B-cell lymphoma, and 1 patient (3.4%) withdrew consent due to the long commute to the study site. Because of the staggered enrolment dates, some patients who were still ongoing in the study had not yet reached the 48-month time point as of the data cut-off date.
Exposure to Study Treatment
Mean treatment duration during the study for patients who were ERT experienced was 51.8 months (SD = 21.46 months), 46.3 months (SD = 22.86 months), and 37.7 months (SD = 4.13 months) in cohorts 1, 2, and 4, respectively, and 54.7 months (SD = 12.14 months) for patients who were ERT naive in cohort 3. The mean treatment compliance rate was at least 95.6% in all stages of the study for both cipaglucosidase alfa and miglustat.
Prior medication use was similar across cohorts and stages except for alglucosidase alfa, which was predominantly used by all patients in the ERT-experienced cohorts. One of the 6 patients in the cohort who were ERT naive had received 1 dose of alglucosidase alfa more than 6 months before study entry, which was permitted in the Australia-specific study protocol.
Efficacy
Refer to Table 29 for a summary of the efficacy results for the pooled patients who were ambulatory and ERT experienced (i.e., cohort 1 and cohort 4), patients who were ambulatory and ERT naive (cohort 3), and patients who were nonambulatory and ERT experienced (cohort 2).
Six-Minute Walk Distance
Motor function was evaluated in all patients who were ambulatory using the 6MWT. At month 48, 88.9% of patients who were ERT experienced and 100% of patients who were ERT naive experienced an improvement in 6MWD from baseline. For patients who were ambulatory and ERT experienced (cohorts 1 and 4), mean and percent change from baseline in the 6MWD were 20.7 m (95% CI, −57.6 m to 99.0 m) and 3.9%, respectively, at month 48. For patients who were ambulatory and ERT naive (cohort 3), mean and percent change from baseline were 52.2 m (95% CI, −21.9 m to 126.3 m) and 12.5%, respectively, at month 48.
Sitting FVC (Percent Predicted)
A meaningful change from baseline in percent predicted sitting FVC was defined as a 3% or greater change in percentage points from baseline. The mean change from baseline in percent predicted sitting FVC was generally stable (no meaningful change) through to 48 months. At month 48, mean change from baseline in percent predicted sitting FVC was 1.0 (95% CI, 5.7 to 7.7) for patients who were ambulatory and ERT experienced (cohorts 1 and 4) and 8.3 (95% CI, −9.2 to 6.7) for patients who were ambulatory and ERT naive. For patients who were nonambulatory (cohort 2), percent predicted sitting FVC data were available for 2 patients who were nonambulatory and ERT experienced after 36 months and 1 patient at 48 months of follow-up. After 36 months of follow-up, 1 patient improved and the other worsened compared with baseline. The condition for the patient with available data after 48 months of follow-up was generally stable compared with baseline.
MMT Total Score
Muscle strength was evaluated in all cohorts using the MMT total score, where higher total scores indicate a reduced impact of disease on muscle function. For patients who were nonambulatory, the total score for MMT was based on the upper extremity score only. At month 48, mean change from baseline in MMT total score was 4.0 points (95% CI, 0.9 points to 7.1 points) for patients who were ambulatory and ERT experienced (cohorts 1 and 4) and −1.3 points (95% CI, −9.2 points to 6.7 points) for patients who were ambulatory and ERT naive.
Week 48 results were not available for patients who were nonambulatory and ERT experienced (cohort 2), but at month 36, the change from baseline in MMT total score was −0.8 (95% CI, −17.8 to 16.3).
Table 29: Overview of Efficacy Results in ATB200-02 Study (Efficacy Population)
Variable | Pooled ERT experienced and ambulatory, cohort 1 + cohort 4 (N = 16) | ERT naive and ambulatory, cohort 3 (N = 6) | ERT experienced and nonambulatory, cohort 2 (N = 6) |
|---|---|---|---|
6MWD in metres | |||
Baseline, n (%) | 16 (100.0) | 6 (100.0) | NR |
Baseline, mean (SD) | 393.5 (119.66) | 396.0 (75.20) | NR |
Change from baseline to month 48, n (%) | 9 (56.3) | 4 (66.7) | NR |
Change from baseline to month 48, mean (95% CI) | 20.7 (−57.6 to 99.0) | 52.2 (−21.9 to 126.3) | NR |
Sitting FVC, % predicted | |||
Baseline, n (%) | 16 (100.0) | 6 (100.0) | NR |
Baseline, mean (SD) | 57.4 (17.42) | 57.2 (20.84) | NR |
Change from baseline to month 48, n (%) | 6 (37.5) | 4 (66.7) | NR |
Change from baseline to month 48, mean (95% CI) | 1.0 (5.7 to 7.7) | 8.3 (1.1 to 15.4) | NR |
MMT total scorea | |||
Baseline, n (%) | 15 (93.8) | 5 (83.3) | 5a |
Baseline, mean (SD) | 65.1 (6.46) | 67.2 (3.70) | 18.4 (13.96)a |
Change from baseline to month 36, n (%) | 10 (62.5) | 4 (66.7) | 4a |
Change from baseline to month 36, mean (95% CI) | 3.6 (−0.1 to 7.3) | 3.5 (−1.3 to 8.3) | −0.8 (−17.8 to 16.3)a |
Change from baseline to month 48, n (%) | 8 (50.0) | 4 (66.7) | NR |
Change from baseline to month 48, mean (95% CI) | 4.0 (0.9 to 7.1) | −1.3 (−9.2 to 6.7) | NR |
6MWD = 6-minute walk distance; CI = confidence interval; ERT = enzyme replacement therapy; FVC = forced vital capacity; MMT = manual muscle test; NR = not reported; SD = standard deviation.
aFor patients who were nonambulatory, the total score for MMT was based on the upper extremity score only. The total MMT upper extremity score ranges from 0 to 40 based on all 8 muscle groups, which include right and left shoulder abduction, right and left shoulder adduction, right and left elbow flexion, and right and left elbow extension. Higher scores indicate less disease impact on muscle functions.
Sources: ATB-200-02 Clinical Study Report.32 Details included in the table are from the sponsor’s summary of clinical evidence.
Harms
Refer to Table 30 for a summary of harms results from the ATB200-02 study. All patients (100%) who received cipaglucosidase alfa and miglustat coadministration experienced TEAEs. The most frequently reported TEAEs (occurring in more than 40% of patients) for the overall population were falls, nasopharyngitis, diarrhea, headache, and arthralgia; most TEAEs were mild or moderate in severity. TEAEs leading to study withdrawal occurred in 2 patients: 1 patient in cohort 1 had diffuse large B-cell lymphoma, which the investigator assessed as unrelated to treatment, and 1 patient in cohort 2 had a drug-related TEAE of urticaria, considered to be an IAR. The incidence of IARs was similar between cohorts: ERT experienced (48%) and ERT naive (50%).
Table 30: Summary of Harms Results in ATB200-02 Study (Overall Population)
Harms | Pooled ERT-experienced cohorts 1, 2, and 4a (N = 23) | ERT-naive cohort 3 (N = 6) | Overall (N = 29) |
|---|---|---|---|
Patients with any TEAE, n (%) | 23 (100) | 6 (100) | 29 (100) |
Serious TEAEs, n (%) | 8 (35) | 4 (67) | 12 (41) |
TEAEs leading to study withdrawal, n (%) | 2 (9) | 0 (0) | 2 (7) |
IARs, n (%) | 10 (43) | 3 (50) | 13 (45) |
ERT = enzyme replacement therapy; IAR = infusion-associated reaction; TEAE = treatment-emergent adverse event.
aPooled data for ERT-experienced cohorts 1, 2, and 4 (includes both patients who were ambulatory and nonambulatory).
Sources: ATB-200-02 Clinical Study Report.32 Details included in the table are from the sponsor’s summary of clinical evidence.
Critical Appraisal
ATB200-02 was designed as an open-label phase I/II study to assess the long-term efficacy of cipaglucosidase alfa and miglustat coadministration in adult patients with LOPD. Although the ATB200-02 trial was submitted to address the systematic review evidence gap pertaining to the exclusion of patients who were nonambulatory with LOPD from clinical trials, the small sample size makes it challenging to draw conclusions about long-term efficacy in this patient group. Efficacy data for pulmonary function and patient-reported outcomes in this group were limited to 1 and 2 patients, respectively. Additionally, statistical hypothesis testing was not part of the study design and there was no active comparator or placebo group. The open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes and reporting of safety parameters due to unblinded exposure to the study medication during the treatment period.
UK EAMS Registry Study
Description
The UK EAMS study is a prospective, observational registry study evaluating the real-world safety and effectiveness of cipaglucosidase alfa plus miglustat in adults who were ERT experienced (at least 2 years) with LOPD. Before its marketing authorization, cipaglucosidase alfa plus miglustat was accepted into the UK EAMS in 2021, which enabled clinicians at 6 specialist centres in England and Wales to switch from standard of care treatment to cipaglucosidase alfa plus miglustat in 37 adults who were ERT experienced (2 years or greater) living with LOPD. This study aimed to provide evidence of treatment benefit in the real-world patient population.
Eligibility Criteria
A total of 45 adults received cipaglucosidase alfa plus miglustat under the EAMS program, of whom 37 consented to registry enrolment.
Intervention
Cipaglucosidase alfa plus miglustat was administered in the same manner as the approved dose.
Outcomes
Reported efficacy outcomes from the UK EAMS registry study that were identified by clinical experts as important to this review were 6MWD and percent predicted sitting FVC.
Participant Characteristics
At baseline, the mean age of the patients was 52.9 years (SD = 12.2 years), a slightly older population than that of the PROPEL trial. Patients had a mean ERT duration of 11.1 years (SD = 3.9 years), which was longer compared with the patients in the PROPEL study. The mean baselines for the 6MWD and percent predicted sitting FVC measures were 282.2 m (SD = 162.8 m) and 57.6% (SD = 24.8%), respectively, which were lower for adults enrolled in the EAMS registry than the results reported for patients in the PROPEL trial.
Statistical Analysis
Due to limited data availability and considerable variability in the time between the baseline and postbaseline assessments, formal hypothesis testing was not performed, and results were summarized using descriptive statistics only.
Results
Patient Disposition
A total of 45 patients were enrolled in the UK EAMS registry and received treatment during the program duration; 6 of those patients discontinued the EAMS program after receiving at least 1 administration of cipaglucosidase alfa plus miglustat. No new patients enrolled. The remaining 39 patients completed treatment and transitioned to the commercial product in November 2023.
Efficacy
The efficacy measures reported from the EAMS study were not necessarily from the same patients. Of all patients enrolled in the EAMS registry, 13 and 12 had both baseline and postbaseline assessments of 6MWD and percent predicted FVC, respectively. The time between baseline and first postbaseline visit varied considerably, from 82 to 1,401 days.
From the baseline to postbaseline visit, patients had a mean change from the baseline to the postbaseline assessment in 6MWD of 10.2 m (SD = 33.9 m), and a mean change in percent predicted sitting FVC of 4.0% (SD = 9.1%).
Harms
Harms data were not reported for this study.
Critical Appraisal
At the time of submission, the available evidence was limited, with no comprehensive details on methods and results, which may impact the ability to sufficiently review and critically appraise the evidence, as well as the robustness of evidence and conclusions. Efficacy data were available only for one-quarter to one-third of patients which, in combination with a wide variation in time between the baseline and postbaseline visits, limits the ability to draw firm conclusions about efficacy. Given that so few patients had both a baseline and after follow-up measurement, the mean changes observed were likely from a highly select group and may not be representative of the entire study population. Additionally, the lack of reported harms limits the ability to assess the safety of cipaglucosidase alfa plus miglustat in real-world clinical practice. Statistical hypothesis testing was not part of the study design and there was no active comparator or placebo group. This open-label design could bias the magnitude of treatment effect for subjective efficacy outcomes. Additionally, because the EAMS study is based on data from a national UK registry, generalizability to the population of patients with LOPD living in Canada may be limited.
Discussion
Summary of Available Evidence
One pivotal RCT, 1 long-term extension study, 1 ITC, and 2 additional studies addressing gaps in evidence submitted by the sponsor have been summarized in this report.
PROPEL was a phase III double-blind, randomized, 1-year study of cipaglucosidase alfa with miglustat compared with alglucosidase alfa with placebo in 95 patients who were ERT experienced and 28 patients who were ERT naive with LOPD. The main outcomes in the trial were change from baseline in 6MWD and percent predicted sitting FVC, which are commonly used end points used in LOPD trials. In addition, change from baseline in tests assessing motor function (Gower manoeuvre and TUG), muscle strength (MMT), and health-related quality of life (PROMIS) were also assessed. The trial population was predominately white (85%) with a mean age of 47 years. The majority of patients had more than 5 years of experience with ERT. Demographic characteristics, including baseline 6MWD and FVC, were generally similar between treatment groups and generally representative of the LOPD population in Canada.
Patients who completed the PROPEL trial were eligible to enrol in the ATB200-07 trial, an ongoing, phase III, international OLE study to assess the long-term safety and tolerability of cipaglucosidase alfa and miglustat coadministration following 104 weeks of treatment (i.e., 52 weeks in the PROPEL trial and 52 weeks in the OLE study) in adult patients with LOPD. The efficacy of the drug combination was also assessed as a secondary objective. Patients enrolled in the OLE study who received cipaglucosidase alfa plus miglustat in the PROPEL trial continued on study treatment, and those receiving alglucosidase alfa with placebo were switched to cipaglucosidase alfa plus miglustat. Cipaglucosidase alfa plus miglustat was administered in the OLE study in the same manner as in the PROPEL trial.
Two studies were proposed by the sponsor as addressing gaps in the systematic review evidence, ATB200-02 and the UK EAMS registry study. The ATB200-02 trial (N = 29) is an ongoing, open-label, phase I/II study evaluating the long-term (up to 48 months) efficacy of cipaglucosidase alfa plus miglustat. The ATB200-02 trial was submitted to fill an evidence gap pertaining to the underrepresentation of patients who were ERT naive in the PROPEL trial and the common exclusion of patients who were nonambulatory from LOPD clinical trials. The efficacy outcomes assessed in the ATB200-02 trial that were identified by the clinical experts as important to this review were 6MWD, percent predicted sitting FVC, and MMT total score. The study included 4 cohorts based on ERT experience and ambulatory status: ERT experienced (2 to 6 years) and ambulatory; ERT experienced (2 years or greater) and nonambulatory; ERT naive and ambulatory; and ERT experienced (7 years or greater) and ambulatory.
The UK EAMS registry study (N = 37) is a prospective, observational registry study evaluating the real-world safety and effectiveness of cipaglucosidase alfa plus miglustat in patients who were ERT experienced (2 years or greater) with LOPD. This study was submitted to fill an evidence gap pertaining to a lack of real-world data in patients with LOPD. The reported efficacy outcomes that were identified by the clinical experts as important to this review were 6MWD and percent predicted sitting FVC. Harms results were not reported for this study.
Interpretation of Results
Efficacy
LOPD is a rare autosomal recessive genetic disorder that primarily affects skeletal muscle tissue, resulting in progressive weakness and respiratory insufficiency. The patient group and clinical experts highlighted that key treatment goals for patients with LOPD include improved strength and breathing function, slowed disease progression without a plateau effect, and improved health-related quality of life. Motor function, respiratory function, and health-related quality of life were captured in the PROPEL trial by the assessment of efficacy end points, including 6MWD, FVC, and PROMIS Physical Function and PROMIS Fatigue scores. The clinical experts agreed with these considerations and emphasized that 6MWD and FVC were reliable and meaningful end points commonly used in clinical studies of patients with LOPD.
The results of the primary efficacy end point in the PROPEL trial — 6MWD at 52 weeks — demonstrated that cipaglucosidase alfa plus miglustat may have no difference to minimal improvement compared with alglucosidase alfa plus placebo in improving motor function. While the mean improvement of 20.56 m in 6MWD for the cipaglucosidase alfa with miglustat group represented a clinically meaningful group-level improvement, the mean improvement relative to alglucosidase alfa of 14.21 m did not reach the MID threshold for between-group difference. Based on the findings for 6MWD, 1 clinical expert said they would communicate to patients that cipaglucosidase alfa plus miglustat is a treatment option that is similar to alglucosidase alfa.
Statistical testing was stopped after cipaglucosidase alfa plus miglustat failed to show superiority compared with alglucosidase alfa plus placebo for the primary outcome. Therefore, FVC, PROMIS Physical Function, and PROMIS Fatigue results should be considered supportive.
Evidence on the first key secondary end point in the PROPEL trial — percent predicted sitting FVC at 52 weeks — was assessed as moderate (using GRADE) and suggested that cipaglucosidase alfa plus miglustat likely results in no to minimal additional effect on pulmonary function compared with alglucosidase alfa plus placebo. The certainty of evidence was lowered due to imprecision because the lower bound of the 95% CI for the difference in FVC included the possibility of little to no clinically significant benefit. Although the results for percent predicted sitting FVC demonstrated nominally statistically significant improvement with cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo, the mean difference did not meet the clinical threshold.
The ERT-experienced group in the PROPEL study had been receiving alglucosidase alfa for a mean duration of 7.4 years and were then switched to the study drug. In this subgroup, there was no clinically or significant difference in the 6MWD for cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo. There was a clinically and nominally statistically significant improvement in the percent predicted FVC for cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in the ERT-experienced subgroup. However, no interaction test was performed and the lower bound of the 95% CI for the difference in FVC included the possibility of little to no clinically significant benefit.
Similar to the evidence for FVC outcomes, evidence on the PROMIS Physical Function and PROMIS Fatigue scores was also considered moderate and suggested that cipaglucosidase alfa plus miglustat likely results in no to minimal additive effects on health-related quality of life at 52 weeks compared with alglucosidase alfa plus placebo. The PROMIS Physical Function results showed a numerically greater improvement in patients treated with cipaglucosidase alfa plus miglustat compared with alglucosidase alfa plus placebo, but this improvement was not clinically or nominally statistically significant. The PROMIS Fatigue scores showed similar improvement between patients treated with cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo. In the absence of an MID, the null effect was used.
The nominal findings of the other efficacy outcomes also suggested that cipaglucosidase alfa plus miglustat may have no to minimal effect on motor function (based on 6MWD at 26 weeks, percent predicted 6MWD, Gower manoeuvre, and TUG) or muscle strength (lower extremity MMT and total MMT) compared with alglucosidase alfa plus placebo.
The efficacy effects observed in the PROPEL trial generally remained stable across the OLE study to week 104 of treatment. For the patients who received cipaglucosidase alfa plus miglustat in the PROPEL trial and continued treatment in the OLE study, the effects on 6MWD, percent predicted sitting FVC, MMT, and PROMIS Physical Function were sustained through to week 104. The group of patients who were switched from alglucosidase alfa plus placebo to cipaglucosidase alfa plus miglustat during the OLE study showed slightly more variation across efficacy outcomes during the extension period. For the 6MWD and percent predicted sitting FVC, the treatment-switched group stabilized at a lower level in the OLE study compared with patients who received cipaglucosidase alfa plus miglustat in the PROPEL trial. Conversely, both MMT and PROMIS Physical Function scores in this group remained stable through to week 104. The PROMIS Fatigue scores demonstrated similar trends in both the continued-treatment and treatment-switched group, with scores worsening slightly through to week 104. However, there is still uncertainty about the long-term effects of cipaglucosidase alfa combined with miglustat on wheelchair and ventilation dependency because these outcomes were not included in the submitted evidence.
In the ATB200-02 study, the baseline 6MWDs were higher and the percentage predicted FVC in the sitting position were lower than in the PROPEL trial population at baseline. While both patients who were ambulatory and ERT experienced and ambulatory and ERT naive demonstrated improvements at month 48 in both 6MWD and percent predicted sitting FVC from baseline, patients who were ERT experienced with LOPD demonstrated substantially less improvement compared with patients who were ERT naive. However, the number of patients with available efficacy data in the ATB200-02 trial was limited, which makes it challenging to draw meaningful conclusions regarding long-term efficacy in this patient population.
The efficacy outcomes observed in the real-world data of the UK EAMS registry study demonstrated a smaller change from baseline in the 6MWD compared with the PROPEL trial, and a larger change from baseline in percent predicted sitting FVC. However, baseline 6MWD and percent predicted sitting FVC were lower in patients in the UK EAMS registry study compared with the PROPEL trial population baseline. Additionally, efficacy data were available only for one-quarter to one-third of registry patients and there was substantial variation in the time between the baseline and postbaseline visits, which limits the ability to draw firm conclusions about real-world efficacy.
The sponsor submitted an ITC that used an ML-NMR framework to adjust for differences in covariate distributions across studies. The ITC included 2 analyses: network A (comprising only RCTs) and network B (including additional single-arm studies and OLE studies). Network A was limited to 2 RCTs, PROPEL and COMET, and relied on grouping cipaglucosidase alfa plus miglustat and avalglucosidase alfa within the same treatment class due to insufficient data. This necessitated shared effect modifiers and the extrapolation of ERT duration effects for avalglucosidase alfa from those of cipaglucosidase alfa plus miglustat. Adjustments were made for known effect modifiers, including age, sex, prior ERT duration, baseline 6MWD, and baseline FVC. Network B expanded on this approach by including data from single-arm studies and OLE studies, allowing for the inclusion of patients who were ERT experienced treated with avalglucosidase alfa and requiring the use of matching techniques to align the single-arm data with appropriate comparator groups. While this broader evidence network provided an opportunity to analyze the impact of earlier ERT experience, it introduced significant methodological challenges, including potential biases from single-arm trial inclusion and repeated use of comparator data. Both networks relied on reconstructed aggregate data, with adjustments derived from the IPD in the PROPEL trial applied to aggregate-level data from other studies.
One main difference between the networks is the shift in the relative treatment effect of cipaglucosidase alfa plus miglustat versus avalglucosidase alfa. In network A, where both treatments were grouped into a single class, the 95% CrI included the null. However, in network B, which included additional data from patients who were ERT experienced, the treatment effect shifted to favour cipaglucosidase alfa plus miglustat. This difference may be attributed to the inclusion of single-arm trials in network B and the reliance on matching methods to adjust for covariate imbalances. While this allowed for the incorporation of ERT duration effects in network B, it also introduced potential biases due to methodological heterogeneity and other limitations associated with the matching of unanchored single-arm trials while only adjusting for treatment-effect modifiers. The differences between networks underscore the impact of methodological choices, data sources, and assumptions on the robustness of the findings.
The sponsor assessed model fit and convergence for the ML-NMR framework using DIC and R-hat statistics. However, diagnostics for the underlying IPD regression model and the application of coefficients to aggregate data were not explicitly reported, limiting transparency and confidence in the validity of these adjustments. Additional gaps include the lack of assessment of other important clinical outcomes (MMT lower extremity and total scores, PROMIS Physical Function and Fatigue scores, Gower manoeuvre results, TUG test results, and harms).
Harms
Based on evidence from the PROPEL trial, cipaglucosidase alfa plus miglustat was generally well tolerated. The most common AEs observed in the cipaglucosidase alfa plus miglustat group included falls, headache, and nasopharyngitis. None of these events were severe but falls were less common and nasopharyngitis was more common in the cipaglucosidase alfa plus miglustat group compared with the alglucosidase alfa plus placebo group. Most TEAEs reported for participants in each treatment group were mild or moderate in severity. According to the clinical experts, these AEs are not a critical concern and typically do not necessitate additional assessment.
The overall number of severe TEAEs was small. The information comparing cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo suggests a numerical increase in the number of severe TEAEs, although the effect had wide CIs that were considered very imprecise. The number of patients with IARs was similar in the cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo groups. All IARs were nonserious except 1 severe AE of anaphylactic reaction in the cipaglucosidase alfa plus miglustat group. Two IARs (anaphylactic reaction and chills) in the cipaglucosidase alfa plus miglustat group led to study drug discontinuation. No deaths were reported.
No new safety signals were identified during the ATB200-07 OLE trial, and the commonly reported TEAEs associated with cipaglucosidase alfa plus miglustat treatment were consistent with the safety profile observed in the PROPEL trial. Most TEAEs were considered mild or moderate in severity. Overall, the safety profile of cipaglucosidase alfa plus miglustat observed during the ATB200-07 trial was consistent with the PROPEL trial. Additionally, no unexpected safety events were observed during the extended treatment period of the ATB200-02 trial, and harms data were not reported in the UK EAMS study.
The clinical experts consulted noted that, overall, the safety profile of cipaglucosidase alfa plus miglustat was acceptable.
Other Considerations
Both cipaglucosidase alfa and alglucosidase alfa have the same mode and schedule of administration (every 2 weeks as a 4-hour IV infusion). In addition to cipaglucosidase alfa, patients must also take miglustat oral capsules 1 hour before the infusion and fast at least 2 hours before and 2 hours after taking miglustat. Despite the additional hassle and potential side effect of diarrhea, the clinical experts felt that taking miglustat every 2 weeks would be acceptable to patients.
The patient group and clinical experts emphasized the importance of new treatments for LOPD that slow disease progression without a plateau effect. Patients with LOPD have generally been shown to improve initially or stabilize with alglucosidase alfa during the first 2 to 5 years on treatment; however, this is followed by a plateau or steady decline in muscle strength, motor function, and respiratory function in many patients. The rate of decline differs across individuals, depending on disease severity, age of LOPD onset and diagnosis, muscle damage before starting ERT, and exercise levels.33-36 The clinical experts confirmed that this steady decline is often seen after an average of 5 to 7 years of receiving ERT; therefore, the patients enrolled in the PROPEL trial were not followed long enough (1 year in the trial plus 1 year in the OLE study) to assess whether the treatment trajectory and long-term sustained efficacy for cipaglucosidase alfa plus miglustat are similar to that for alglucosidase alfa.
Most patients (77%) were ambulatory and 23% used assistive devices at baseline. As LOPD progresses, patients often require increasing levels of mobility aids and increasing ventilatory support for breathing assistance. However, the PROPEL trial did not include the use of mobility aids and the need for ventilation as end points.
Conclusion
The evidence on the effects of cipaglucosidase alfa plus miglustat in adult patients with LOPD comprises 1 pivotal RCT comparing cipaglucosidase alfa plus miglustat with alglucosidase alfa plus placebo, 1 long-term extension study, 1 ITC, and 2 additional studies addressing gaps in evidence.
The outcomes considered by the clinical experts and patient groups to be critical for decision-making were 6MWD (to assess ambulatory function), percent predicted sitting FVC (to assess respiratory function), PROMIS Physical Function and PROMIS Fatigue scores (to assess health-related quality of life), and severe TEAEs and IARs (to assess harms).
The pivotal PROPEL study evaluated these outcomes and provided evidence that cipaglucosidase alfa plus miglustat compared with alglucosidase alfa plus placebo may result in no difference to minimal improvement in 6MWD. Additionally, cipaglucosidase alfa plus miglustat likely results in no difference to minimal improvement in sitting FVC (percent predicted), PROMIS Physical Function score, and PROMIS Fatigue score when compared with alglucosidase alfa plus placebo.
The overall number of severe TEAEs was very small. The information comparing cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo suggests a numerical increase in the number of severe TEAEs, although the majority were unrelated to the study drug and the wide CIs indicate the finding was very imprecise. The number of patients with IARs was similar in the cipaglucosidase alfa plus miglustat and alglucosidase alfa plus placebo groups. Therefore, coadministration of cipaglucosidase alfa and miglustat was generally safe and well tolerated, with very little treatment discontinuation. No new safety signals were identified. No deaths were reported. Additionally, the incidence of IARs was similar between the treatment groups. All IARs were nonserious except 1 SAE of anaphylactic reaction in the cipaglucosidase alfa plus miglustat group. Two IARs (anaphylactic reaction and chills) in the cipaglucosidase alfa plus miglustat group led to study drug discontinuation. The overall safety profile of cipaglucosidase alfa plus miglustat was similar to alglucosidase alfa plus placebo.
The efficacy effects observed in the PROPEL trial generally remained stable across the OLE study to week 104 of treatment, no new safety signals were identified during the OLE study, and commonly reported TEAEs associated with cipaglucosidase alfa plus miglustat treatment were consistent with the safety profile observed in the PROPEL trial. However, there is still uncertainty about the long-term effects of cipaglucosidase alfa combined with miglustat on wheelchair and ventilation dependency because these outcomes were not included in the submitted evidence.
The number of patients with available efficacy data in both the ATB200-02 study and UK EAMS registry (studies addressing gaps in evidence) was limited, which makes it challenging to draw meaningful conclusions.
While the ITC provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings. The limited model diagnostics, missing inconsistency testing, reliance on reconstructed data, and variability in methodological choices introduce a high degree of uncertainty in the results.
References
1.Hahn S. Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). Waltham (MA): UpToDate; 2020. Accessed 2025 Feb 27.
2.Genetic and Rare Diseases Information Center. Glycogen Storage Disease Type II. Bethesda (MD): National Institutes of Health; 2018: https://rarediseases.info.nih.gov/diseases/5714/glycogen-storage-disease-type-2. Accessed 2025 Feb 27.
3.Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288. PubMed
4.Cupler EJ, Berger KI, Leshner RT, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve. 2012;45(3):319-333. PubMed
5.Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105(1):e10. PubMed
6.Orphanet. Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection, January 2019, number 1: Diseases listed in alphabetical order [sponsor supplied reference]. 2019.
7.Tarnopolsky M, Katzberg H, Petrof BJ, et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. Can J Neurol Sci. 2016;43(4):472-485. PubMed
8.Schoser B, van der Beek N, Broomfield A, et al. Start, switch and stop (triple-S) criteria for enzyme replacement therapy of late-onset Pompe disease: European Pompe Consortium recommendation update 2024. Eur J Neurol. 2024;31(9):e16383. PubMed
9.Barba-Romero MA, Barrot E, Bautista-Lorite J, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol. 2012;54(8):497-507. PubMed
10.Amicus Therapeutics Announces Approval and Launch of New Pompe Disease Therapy in the European Union. Toronto (ON): Amicus Therapeutics Canada Inc; 2023: https://ir.amicusrx.com/node/22931/pdf. Accessed 2025 Feb 27.
11.Drug Trials Snapshots: POMBILITI. Silver Spring (MD): U.S. Food & Drug Administration; 2024: https://www.fda.gov/drugs/development-approval-process-drugs/drug-trials-snapshots-pombiliti. Accessed 2025 Feb 27.
12.Pombiliti® cipaglucosidase alfa for injection. Product Monograph [sponsor supplied reference].
13.Amicus Therapeutics Canada Inc. Opfolda® miglustat capsules [product monograph] [sponsor supplied reference]. 2024.
14.Avalglucosidase alfa for injection (Nexviazyme): Lyophilized Powder 100 mg/vial, intravenous infusion [product monograph]. Toronto, ON: Sanofi-aventis Canada Inc.; 2024: https://pdf.hres.ca/dpd_pm/00076711.PDF. Accessed 2025 Feb 11.
15.Alglucosidase alfa for injection (Myozyme): Lyophilized Powder, 50 mg vial, intravenous infusion [product monograph]. Toronto, ON: Sanofi-aventis Canada Inc.; 2024: https://pdf.hres.ca/dpd_pm/00078034.PDF. Accessed 2025 Feb 11.
16.Schoser B, Roberts M, Byrne BJ, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021;20(12):1027-1037. PubMed
17.Kishnani P, Byrne B., Claeys K., et al. Switching treatment to cipaglucosidase alfa plus miglustat positively affects patient-reported outcome measures in patients with late-onset Pompe disease. J Patient Rep Outcomes. 2024;8, 132. PubMed
18.Clinical study report: A Phase 3 Double-blind Randomized Study to Assess the Efficacy and Safety of Intravenous ATB200 Co-administered with Oral AT2221 in Adult Subjects with Late-Onset Pompe Disease Compared with Alglucosidase Alfa/Placebo. [CONFIDENTIAL internal sponsor’s report]. Amicus Therapeutics Canada Inc. 17 May 2021 [sponsor supplied reference].
19.Yuan M, Andrinopoulou E-R, Kruijshaar ME, et al. Positive association between physical outcomes and patient-reported outcomes in late-onset Pompe disease: a cross sectional study. Orphanet J Rare Dis. 2020;15:1-8. PubMed
20.van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab. 2012;107(3):456-461. PubMed
21.Bohannon RW, Crouch R. Minimal clinically important difference for change in 6-minute walk test distance of adults with pathology: a systematic review. J Eval Clin Pract. 2017;23(2):377-381. PubMed
22.Claeys KG, Kushlaf H, Raza S, et al. Minimal clinically important differences in six-minute walking distance in late-onset Pompe disease. Orphanet J Rare Dis. 2024;19(1):154. PubMed
23.Lika A, Andrinopoulou ER, van der Beek N, Rizopoulos D, van der Ploeg AT, Kruijshaar ME. Establishing how much improvement in lung function and distance walked is clinically important for adult patients with Pompe disease. Eur J Neurol. 2024;31(5):e16223. PubMed
24.Lachmann R, Schoser B. The clinical relevance of outcomes used in late-onset Pompe disease: can we do better? Orphanet J Rare Dis. 2013;8:160. PubMed
25.Kishnani PS, Shohet S, Raza S, et al. Validation of the Patient-Reported Outcomes Measurement Information System (PROMIS((R))) physical function questionnaire in late-onset Pompe disease using PROPEL phase 3 data. J Patient Rep Outcomes. 2024;8(1):13. PubMed
26.Harfouche M, Kishnani PS, Krusinska E, et al. Use of the patient-reported outcomes measurement information system (PROMIS(R)) to assess late-onset Pompe disease severity. J Patient Rep Outcomes. 2020;4(1):83. PubMed
27.van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396-1406. PubMed
28.Balshem H, Helfand M, Schunemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of clinical epidemiology. 2011;64(4):401-406. PubMed
29.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of clinical epidemiology. 2020;119:126-135. PubMed
30.Amicus Therapeutics Canada Inc. Clinical study report: Cipaglucosidase Alfa And Miglustat Study ATB200-07 A Phase 3 Open-Label Extension Study To Assess The Long-Term Safety And Efficacy Of Intravenous ATB200 Co-Administered With Oral At2221 In Adult Subjects With Late-Onset Pompe Disease. Data on File [sponsor supplied reference]. 2023.
31.Shohet S, Hummel N, Fu S, et al. Comparing the efficacy of cipaglucosidase alfa plus miglustat with other enzyme replacement therapies for late-onset Pompe disease: a network meta-analysis utilizing patient-level and aggregate data. J Comp Eff Res. 2024;13(10):e240045. PubMed
32.Clinical Study Report: ATB200-02. Interim Clinical Study Report [internal sponsor’s report]. Toronto (ON): Amicus Therapeutics Canada Inc; 2022.
33.Semplicini C, De Antonio M, Taouagh N, et al. Long-term benefit of enzyme replacement therapy with alglucosidase alfa in adults with Pompe disease: Prospective analysis from the French Pompe Registry. J Inherit Metab Dis. 2020;43(6):1219-1231. PubMed
34.Gutschmidt K, Musumeci O, Diaz-Manera J, et al. STIG study: real-world data of long-term outcomes of adults with Pompe disease under enzyme replacement therapy with alglucosidase alfa. J Neurol. 2021;268(7):2482-2492. PubMed
35.Kuperus E, Kruijshaar ME, Wens SC, et al. Long-term benefit of enzyme replacement therapy in Pompe disease: a 5-year prospective study. Neurology. 2017;89(23):2365-2373. PubMed
36.Papadimas GK, Anagnostopoulos C, Xirou S, et al. Effect of long term enzyme replacement therapy in late onset Pompe disease: A single-centre experience. Neuromuscul Disord. 2021;31(2):91-100. PubMed
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 31: Summary of Efficacy Results in the PROPEL Trial, Excluding Outlier, ITT-LOCF Population
Variable | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) | |
MMT lower extremity scorea | ||
Number of patients contributing to the analysis, n | 84 patients at baseline, 80 patients at week 52 | 37 patients at baseline, 34 patients at week 52 |
Baseline, mean (SD) | 27.96 (5.8) | 27.65 (6.2) |
Change from baseline to week 52, mean (95% CI) | 1.56 (0.72 to 2.40) | 0.88 (−0.02 to 1.78) |
Least squares mean difference (95% CI) | 0.96 (−0.48 to 2.40)b | Reference |
Nominalc 1-sided P value for superiority | 0.095 | Reference |
Nominalc 2-sided P value for superiority | 0.191 | Reference |
6MWD at week 26, distance in metres | ||
Number of patients contributing to the analysis, n | 85 patients at baseline, 73 patients at week 26 | 37 patients at baseline, 34 patients at week 26 |
Baseline, mean (SD) | 357.93 (11.43) | 350.95 (121.32) |
Change from baseline to week 26, mean (95% CI) | 17.44 (9.80 to 25.08) | 9.19 (−0.20 to 18.59) |
Least squares mean difference (95% CI) | 8.17 (−4.24 to 20.57)b | Reference |
Nominalc 1-sided P value for superiority | 0.097 | Reference |
Nominalc 2-sided P value for superiority | NR | Reference |
PROMIS Physical Function scored | ||
Number of patients contributing to the analysis, n | 84 | 37 |
Baseline, mean (SD) | 66.86 (12.26) | 67.97 (13.09) |
Change from baseline to week 52, mean (95% CI) | 1.94 (0.31 to 3.57) | 0.19 (−3.42 to 3.80) |
Least squares mean difference (95% CI) | 1.87 (−1.51 to 5.25)b | Reference |
Nominalc 1-sided P value for superiority | 0.138 | Reference |
Nominalc 2-sided P value for superiority | 0.276 | Reference |
PROMIS Fatigue scored | ||
Number of patients contributing to the analysis, n | 85 | 37 |
Baseline, mean (SD) | 22.26 (8.30) | 21.08 (6.10) |
Change from baseline to week 52, mean (95% CI) | −2.02 (−3.26 to −0.77) | −1.67 (−3.88 to 0.54) |
Least squares mean difference (95% CI) | 0.04 (−2.12 to 2.20)b | Reference |
Nominalc 1-sided P value for superiority | 0.515 | Reference |
Nominalc 2-sided P value for superiority | 0.970 | Reference |
6MWD, % predicted | ||
Number of patients contributing to the analysis, n | 85 | 37 |
Baseline, mean (SD) | 57.82 (15.8) | 56.03 (17.3) |
Change from baseline to week 52, mean (95% CI) | 4.07 (2.56 to 5.59) | 1.58 (−0.42 to 3.58) |
Least squares mean difference (95% CI) | 2.38 (−0.26 to 5.03)b | Reference |
Nominalc 1-sided P value for superiority | 0.038 | Reference |
Nominalc 2-sided P value for superiority | NR | Reference |
Gower manoeuvre, time in seconds | ||
Number of patients contributing to the analysis, n (%) | 61 | 27 patients at baseline, 25 patients at week 52 |
Baseline, mean (SD) | 10.84 (7.45) | 19.82 (25.21) |
Change from baseline to week 52, mean (95% CI) | −0.26 (−1.74 to 1.22) | −2.19 (−5.04 to 0.66) |
Least squares mean difference (95% CI) | 1.60 (−1.48 to 4.68)b | Reference |
Nominalc 1-sided P value for superiority | 0.848 | Reference |
Nominalc 2-sided P value for superiority | 0.305 | Reference |
TUG test, time in seconds | ||
Number of patients contributing to the analysis, n (%) | 75 | 32 patients at baseline, 31 patients at week 52 |
Baseline, mean (SD) | 12.88 (10.14) | 12.77 (8.86) |
Change from baseline to week 52, mean (95% CI) | −0.30 (−2.24 to 1.65) | −0.13 (−1.11 to 0.85) |
Least squares mean difference (95% CI) | −0.47 (−3.38 to 2.43)b | Reference |
Nominalc 1-sided P value for superiority | 0.374 | Reference |
Nominalc 2-sided P value for superiority | 0.748 | Reference |
Total MMT score | ||
Number of patients contributing to the analysis, n | 84 patients at baseline, 80 patients at week 52 | 34 |
Baseline, mean (SD) | 62.25 (8.24) | 62.35 (9.66) |
Change from baseline to week 52, mean (95% CI) | 3.07 (1.66 to 4.48) | 1.41 (−0.12 to 2.94) |
Least squares mean difference (95% CI) | 2.22 (−0.09 to 4.53)b | Reference |
Nominalc 1-sided P value for superiority | 0.030 | Reference |
Nominalc 2-sided P value for superiority | 0.059 | Reference |
6MWD = 6-minute walk distance; CI = confidence interval; ERT = enzyme replacement therapy;; ITT = intention to treat; LOCF = last observation carried forward; MMT = manual muscle test; NR = not reported; OR = odds ratio; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; TUG = Timed Up and Go.
aThe total score for the MMT lower extremity strength included the following 8 body parts: right and left hip flexion, right/left hip abduction, right and left knee flexion, and right and left knee extension. Total score ranged from 0 to 40, with lower scores indicating weaker muscle strength.
bAnalysis of covariance model was used. The model included terms for treatment, baseline of response variable, age, height, weight (all as continuous covariates), ERT status (ERT naive versus ERT experienced), and sex. The last available postbaseline observation was used to replace the missing value at week 52. The key secondary end points were in hierarchical order of importance.
cBecause the primary end point analysis showed no superiority of treatment, no further confirmatory conclusions are possible (statistically); therefore, the P values are nominal.
dThe score was calculated by summing up scores (1 to 5) across all items.
Source: ATB200-03 Clinical Study Report.18
Table 32: Subgroup Analyses for the Primary Efficacy Outcome (6MWD) in the PROPEL Trial, ITT-OBS Population, Excluding Outlier
Variable | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) | |
Subgroup analysis by ERT status | ||
ERT experienced | ||
Number of patients contributing to the analysis, n | 65 patients at baseline, 61 patients at week 52 | 30 patients at baseline, 29 patients at week 52 |
Baseline, mean (SD) | 346.94 (110.21) | 334.62 (114.02) |
Change from baseline to week 52, mean (SD) | 16.34 (39.46) | 0.70 (39.84) |
Least squares mean difference (95% CI) | 16.45 (−1.86 to 34.77)a | Reference |
1-sided P value for superiority | 0.039 | Reference |
2-sided P value for superiority | 0.078 | Reference |
ERT naive | ||
Number of patients contributing to the analysis, n | 20 | 7 |
Baseline, mean (SD) | 393.64 (112.39) | 420.94 (135.75) |
Change from baseline to week 52, mean (SD) | 33.44 (48.70) | 38.34 (29.32) |
Least squares mean difference (95% CI) | −6.55 (−48.19 to 35.08)a | Reference |
1-sided P value for superiority | 0.626 | Reference |
2-sided P value for superiority | 0.748 | Reference |
Subgroup analysis by ERT duration | ||
2 to < 3 years | ||
Number of patients contributing to the analysis, n | 4 | 5 |
Baseline, mean (SD) | 473.34 (96.17) | 405.84 (55.21) |
Change from baseline to week 52, mean (SD) | 10.28 (24.20) | 8.64 (50.37) |
Least squares mean difference (95% CI) | −13.02 (−62.50 to 36.46)a | Reference |
1-sided P value for superiority | 0.738 | Reference |
2-sided P value for superiority | NR | Reference |
3 to < 5 years | ||
Number of patients contributing to the analysis, n | 16 patients at baseline, 15 patients at week 52 | 6 |
Baseline, mean (SD) | 379.42 (108.23) | 350.62 (110.36) |
Change from baseline to week 52, mean (SD) | 26.54 (35.02) | −7.01 (16.54) |
Least squares mean difference (95% CI) | 42.80 (−0.25 to 85.85)a | Reference |
1-sided P value for superiority | 0.026 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 5 years | ||
Number of patients contributing to the analysis, n | 45 patients at baseline, 42 patients at week 52 | 19 |
Baseline, mean (SD) | 324.16 (103.33) | 310.83 (121.63) |
Change from baseline to week 52, mean (SD) | 13.28 (42.03) | 1.06 (43.56) |
Least squares mean difference (95% CI) | 14.70 (−9.17 to 38.58)a | Reference |
1-sided P value for superiority | 0.111 | Reference |
2-sided P value for superiority | NR | Reference |
Subgroup analysis by baseline 6MWD | ||
70 to < 150 m | ||
Number of patients contributing to the analysis, n | 4 | 4 |
Baseline, mean (SD) | 115.21 (28.86) | 129.96 (19.79) |
Change from baseline to week 52, mean (SD) | −7.93 (14.86) | −14.41 (40.73) |
Least squares mean difference (95% CI) | −41.33 (−628.24 to 545.59)a | Reference |
1-sided P value for superiority | 0.707 | Reference |
2-sided P value for superiority | NR | Reference |
150 to < 400 m | ||
Number of patients contributing to the analysis, n | 55 patients at baseline, 53 patients at week 52 | 21 patients at baseline, 20 patients at week 52 |
Baseline, mean (SD) | 315.62 (64.77) | 320.84 (59.26) |
Change from baseline to week 52, mean (SD) | 23.38 (49.79) | 7.93 (42.56) |
Least squares mean difference (95% CI) | 18.93 (−6.13 to 43.99)a | Reference |
1-sided P value for superiority | 0.068 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 400 m | ||
Number of patients contributing to the analysis, n | 26 patients at baseline, 24 patients at week 52 | 12 |
Baseline, mean (SD) | 484.77 (52.67) | 478.31 (63.61) |
Change from baseline to week 52, mean (SD) | 19.10 (22.59) | 15.65 (37.66) |
Least squares mean difference (95% CI) | 5.98 (−18.22 to 30.17)a | Reference |
1-sided P value for superiority | 0.307 | Reference |
2-sided P value for superiority | NR | Reference |
Subgroup analysis by age group | ||
18 to < 35 years | ||
Number of patients contributing to the analysis, n | 17 | 9 |
Baseline, mean (SD) | 417.59 (129.23) | 411.00 (148.09) |
Change from baseline to week 52, mean (SD) | 39.37 (47.00) | 24.22 (50.64) |
Least squares mean difference (95% CI) | 19.83 (−21.36 to 61.02)a | Reference |
1-sided P value for superiority | 0.165 | Reference |
2-sided P value for superiority | NR | Reference |
35 to < 50 years | ||
Number of patients contributing to the analysis, n | 27 patients at baseline, 25 patients at week 52 | 13 patients at baseline, 12 patients at week 52 |
Baseline, mean (SD) | 393.68 (76.72) | 351.86 (128.27) |
Change from baseline to week 52, mean (SD) | 17.24 (41.54) | −4.25 (45.19) |
Least squares mean difference (95% CI) | 19.83 (−21.36 to 61.02)a | Reference |
1-sided P value for superiority | 0.165 | Reference |
2-sided P value for superiority | NR | Reference |
50 to < 65 years | ||
Number of patients contributing to the analysis, n | 30 patients at baseline, 29 patients at week 52 | 12 |
Baseline, mean (SD) | 316.54 (112.61) | 324.50 (86.63) |
Change from baseline to week 52, mean (SD) | 20.09 (34.31) | 10.72 (29.35) |
Least squares mean difference (95% CI) | 9.00 (−13.47 to 31.46)a | Reference |
1-sided P value for superiority | 0.210 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 65 years | ||
Number of patients contributing to the analysis, n | 11 patients at baseline, 10 patients at week 52 | 3 |
Baseline, mean (SD) | 290.87 (81.82) | 272.67 (87.24) |
Change from baseline to week 52, mean (SD) | −1.71 (49.38) | −2.33 (16.75) |
Least squares mean difference (95% CI) | −3.55 (−64.51 to 57.42)a | Reference |
1-sided P value for superiority | 0.549 | Reference |
2-sided P value for superiority | NR | Reference |
6MWD = 6-minute walk distance; 6MWT = 6-minute walk test; CI = confidence interval; ERT = enzyme replacement therapy; ITT = intention to treat; NR = not reported; OBS = observed; SD = standard deviation.
Note: The primary end point is 6MWD (defined as distance walked in 6MWT in metres).
aMixed model for repeated measures approach (using restricted maximum likelihood estimation) was used. The model included terms for treatment, baseline 6MWD, age, height, weight (all as continuous covariates), ERT status (ERT naive versus ERT experienced), sex, time, and treatment-by-time interaction. Time was used as a repeated measure, and an unstructured covariance approach was applied.
Source: ATB200-03 Clinical Study Report.18
Table 33: Subgroup Analyses for the First Key Secondary Efficacy Outcome (Sitting FVC) in the PROPEL Trial, ITT-LOCF Population, Excluding Outlier
Variable | PROPEL trial | |
|---|---|---|
Cipaglucosidase alfa + miglustat (N = 85) | Alglucosidase alfa + placebo (N = 38) | |
Subgroup analysis by ERT status | ||
ERT experienced | ||
Number of patients contributing to the analysis, n | 65 patients at baseline, 64 patients at week 52 | 30 |
Baseline, mean (SD) | 67.85 (19.052) | 67.48 (20.993) |
Change from baseline to week 52, mean (SD) | 0.05 (5.84) | −4.02 (5.01) |
Least squares mean difference (95% CI) | 3.51 (1.03 to 5.99)a | Reference |
1-sided P value for superiority | 0.003 | Reference |
2-sided P value for superiority | 0.006 | Reference |
ERT naive | ||
Number of patients contributing to the analysis, n | 20 | 7 |
Baseline, mean (SD) | 80.15 (18.69) | 79.07 (22.58) |
Change from baseline to week 52, mean (SD) | −4.10 (6.53) | −3.64 (4.71) |
Least squares mean difference (95% CI) | −1.95 (−8.93 to 5.03)a | Reference |
1-sided P value for superiority | 0.717 | Reference |
2-sided P value for superiority | 0.566 | Reference |
Subgroup analysis by ERT duration | ||
2 to < 3 years | ||
Number of patients contributing to the analysis, n | 4 | 5 |
Baseline, mean (SD) | 67.63 (25.56) | 78.60 (19.06) |
Change from baseline to week 52, mean (SD) | 2.63 (4.75) | 4.40 (2.38) |
Least squares mean difference (95% CI) | 9.49 (−21.10 to 40.08)a | Reference |
1-sided P value for superiority | 0.157 | Reference |
2-sided P value for superiority | NR | Reference |
3 to < 5 years | ||
Number of patients contributing to the analysis, n | 16 patients at baseline, 15 patients at week 52 | 6 |
Baseline, mean (SD) | 70.94 (23.45) | 59.42 (16.89) |
Change from baseline to week 52, mean (SD) | 1.50 (5.09) | −1.08 (2.92) |
Least squares mean difference (95% CI) | 3.12 (−2.65 to 8.90)a | Reference |
1-sided P value for superiority | 0.133 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 5 years | ||
Number of patients contributing to the analysis, n | 45 patients at baseline, 42 patients at week 52 | 19 |
Baseline, mean (SD) | 66.77 (17.07) | 67.11 (22.22) |
Change from baseline to week 52, mean (SD) | −0.66 (6.11) | −4.84 (5.76) |
Least squares mean difference (95% CI) | 3.71 (0.41 to 7.00)a | Reference |
1-sided P value for superiority | 0.014 | Reference |
2-sided P value for superiority | NR | Reference |
Subgroup analysis by baseline 6MWD | ||
70 to < 150 m | ||
Number of patients contributing to the analysis, n | 4 | 4 |
Baseline, mean (SD) | 62.50 (7.08) | 67.00 (11.29) |
Change from baseline to week 52, mean (SD) | 4.50 (7.99) | −8.00 (8.05) |
Least squares mean difference (95% CI) | −11.52 (−188.40 to 165.36)a | Reference |
1-sided P value for superiority | 0.720 | Reference |
2-sided P value for superiority | NR | Reference |
150 to < 400 m | ||
Number of patients contributing to the analysis, n | 55 | 21 |
Baseline, mean (SD) | 68.93 (19.11) | 64.24 (21.89) |
Change from baseline to week 52, mean (SD) | −1.04 (6.27) | 4.00 (4.60) |
Least squares mean difference (95% CI) | 2.24 (−0.76 to 5.24)a | Reference |
1-sided P value for superiority | 0.070 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 400 m | ||
Number of patients contributing to the analysis, n | 26 patients at baseline, 25 patients at week 52 | 12 |
Baseline, mean (SD) | 75.85 (21.15) | 80.08 (20.64) |
Change from baseline to week 52, mean (SD) | −1.58 (5.70) | −2.50 (3.72) |
Least squares mean difference (95% CI) | 0.47 (−3.33 to 4.27)a | Reference |
1-sided P value for superiority | 0.400 | Reference |
2-sided P value for superiority | NR | Reference |
Subgroup analysis by age group | ||
18 to < 35 years | ||
Number of patients contributing to the analysis, n | 17 | 9 |
Baseline, mean (SD) | 83.50 (17.540) | 83.28 (21.42) |
Change from baseline to week 52, mean (SD) | 0.62 (6.81) | −4.94 (5.02) |
Least squares mean difference (95% CI) | 4.72 (−1.34 to 10.77)a | Reference |
1-sided P value for superiority | 0.060 | Reference |
2-sided P value for superiority | NR | Reference |
35 to < 50 years | ||
Number of patients contributing to the analysis, n | 27 | 13 |
Baseline, mean (SD) | 73.33 (19.28) | 73.42 (16.87) |
Change from baseline to week 52, mean (SD) | −1.07 (4.81) | −3.88 (5.44) |
Least squares mean difference (95% CI) | 2.67 (−0.92 to 6.26)a | Reference |
1-sided P value for superiority | 0.070 | Reference |
2-sided P value for superiority | NR | Reference |
50 to < 65 years | ||
Number of patients contributing to the analysis, n | 30 patients at baseline, 29 patients at week 52 | 12 |
Baseline, mean (SD) | 62.20 (18.04) | 60.83 (21.55) |
Change from baseline to week 52, mean (SD) | −0.95 (6.27) | −3.42 (4.68) |
Least squares mean difference (95% CI) | 0.31 (−3.84 to 4.45)a | Reference |
1-sided P value for superiority | 0.440 | Reference |
2-sided P value for superiority | NR | Reference |
≥ 65 years | ||
Number of patients contributing to the analysis, n | 11 | 3 |
Baseline, mean (SD) | 67.95 (17.14) | 48.00 (10.33) |
Change from baseline to week 52, mean (SD) | −2.95 (8.34) | −3.33 (5.01) |
Least squares mean difference (95% CI) | 3.41 (−10.01 to 16.82)a | Reference |
1-sided P value for superiority | 0.279 | Reference |
2-sided P value for superiority | NR | Reference |
6MWD = 6-minute walk distance; CI = confidence interval; ERT = enzyme replacement therapy; FVC = forced vital capacity; ITT = intention to treat; LOCF = last observation carried forward; NR = not reported; SD = standard deviation.
aAnalysis of covariance model was used. The model included terms for treatment, baseline of response variable, age, height, weight (all as continuous covariates), ERT status (ERT naive versus ERT experienced), and sex. The last available postbaseline observation was used to replace the missing value at week 52. The key secondary end points were in hierarchical order of importance.
Source: ATB200-03 Clinical Study Report.18
Table 34: Safety Results Across the Entire Study Period (104 Weeks) — PROPEL and ATB200-07 Trials (Safety Population)
Adverse events | Cipaglucosidase alfa + miglustat (N = 85) | Treatment switched (prior alglucosidase alfa + placebo) (N = 37) |
|---|---|---|
Most common adverse events, n (%) | ||
≥ 1 adverse event | 84 (98.8) | 36 (97.3) |
Fall | 35 (41.2) | 13 (35.1) |
Headache | 30 (35.3) | 11 (29.7) |
Arthralgia | 27 (31.8) | 10 (27.0) |
Nasopharyngitis | 24 (28.2) | 1 (2.7) |
Myalgia | 23 (27.1) | 7 (18.9) |
Back pain | 19 (22.4) | 5 (13.5) |
Pain in extremity | 17 (20.0) | 8 (21.6) |
Diarrhea | 17 (20.0) | 3 (8.1) |
Nausea | 16 (18.8) | 5 (13.5) |
Fatigue | 15 (17.6) | 6 (16.2) |
Oropharyngeal pain | 15 (17.6) | 2 (5.4) |
Musculoskeletal pain | 14 (16.5) | 3 (8.1) |
Urinary tract infection | 14 (16.5) | 3 (8.1) |
COVID-19 | 14 (16.5) | 3 (8.1) |
Pyrexia | 13 (15.3) | 2 (5.4) |
SAEs (by organ class), n (%) | ||
Patients with ≥ 1 SAE | 14 (16.5) | 6 (16.2) |
Cardiac disorders | 1 (1.2) | 1 (2.7) |
Eye disorders | 1 (1.2) | 0 |
Gastrointestinal disorders | 1 (1.2) | 1 (2.7) |
General disorders and administration site conditions | 0 | 1 (2.7) |
Hepatobiliary disorders | 0 | 1 (2.7) |
Immune system disorders | 1 (1.2) | 1 (2.7) |
Infections and infestations | 2 (2.4) | 1 (2.7) |
Injury, poisoning, and procedural complications | 5 (5.9) | 1 (2.7) |
Nervous system disorders | 1 (1.2) | 0 |
Respiratory, thoracic, and mediastinal disorders | 0 | 1 (2.7) |
Skin and subcutaneous tissue disorders | 0 | 1 (2.7) |
Surgical and medical procedures | 1 (1.2) | 0 |
Vascular disorders | 1 (1.2) | 1 (2.7) |
Patients who had any TEAE leading to study drug discontinuation, n (%)a | ||
Patients who stopped treatment due to adverse events | 3 (3.5) | 2 (5.4%) |
Urticaria | 1 (1.2) | 1 (2.7) |
Anaphylactic reaction | 0 | 1 (2.7) |
Anaphylactoid reaction | 1 (1.2) | 0 |
Chills | 1 (1.2) | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 0 |
Adverse events of special interest, n (%) | ||
Patients with IAR TEAEb | 27 (31.8) | 10 (27.0) |
Total number of IAR TEAEs | 148 | 46 |
IAR = infusion-associated reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Sources: ATB200-07 Clinical Study Report.30 Details included in the table are from the sponsor’s summary of clinical evidence.
Pharmacoeconomic Review
Abbreviations
6MWD
6-minute walk distance
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CUA
cost-utility analysis
ERT
enzyme replacement therapy
FVC
forced vital capacity
ICER
incremental cost-effectiveness ratio
LOPD
late-onset Pompe disease
PROMIS
Patient-Reported Outcomes Information System
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Cipaglucosidase alfa for injection (Pombiliti), lyophilized powder, 105 mg/vial, IV infusion in combination with miglustat capsules (Opfolda), 65 mg, oral capsules |
Indication | Cipaglucosidase alfa: Indicated in combination with the enzyme stabilizer Opfolda (65 mg miglustat capsule) for the treatment of adult patients with late-onset Pompe disease (acid alpha-glucosidase [GAA] deficiency) weighing ≥ 40 kg. Miglustat: An enzyme stabilizer indicated in combination with Pombiliti (cipaglucosidase alfa) for the treatment of adult patients with late-onset Pompe disease (acid alpha-glucosidase [GAA] deficiency) weighing ≥ 40 kg. Cipaglucosidase alfa must be used in combination with 65 mg miglustat capsules. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 14, 2025 (cipaglucosidase alfa) and April 10, 2025 (miglustat) |
Reimbursement request | As per indication |
Sponsor | Amicus Therapeutics Canada Inc. |
Submission history | No |
GAA = acid alpha-glucosidase; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis State transition microsimulation model |
Target population | Adult patients with LOPD |
Treatments | Cipaglucosidase alfa in combination with miglustat |
Dose regimens | Cipaglucosidase alfa: 20 mg/kg administered every other week Miglustat: 260 mg (4 tablets) for patients ≥ 50 kg, or 195 mg (3 tablets) for patients weighing from ≥ 40 kg to < 50 kg, taken 1 hour before cipaglucosidase alfa infusion |
Submitted prices | Cipaglucosidase alfa: $1,751.75 per 105 mg vial Miglustat: $46.04 per 65 mg tablet |
Submitted treatment cost | Cipaglucosidase alfa: $683,183 per patient annually Miglustat: $4,788.42 per patient annually Total treatment cost: $687,971 per patient annually |
Comparator | Alglucosidase alfa |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (approximately 23 years) |
Key data source | PROPEL trial for both cipaglucosidase alfa in combination with miglustat and alglucosidase alfa |
Submitted results | ICER = $3,347a per QALY gained vs. alglucosidase alfa (incremental costs: $3,094; incremental QALYs: 0.94) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; LOPD = late-onset Pompe disease; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
aCalculated by CDA-AMC using the results from the sponsor-submitted base case, which was conducted from a societal perspective, to reflect a health care system payer perspective.
Conclusions
Evidence from the PROPEL study suggests that cipaglucosidase alfa in combination with miglustat may result in no difference to minimal improvement in the 6-minute walk distance (6MWD) when compared with alglucosidase alfa plus placebo. In addition, cipaglucosidase alfa in combination with miglustat likely results in no difference to minimal improvement in percent predicted forced vital capacity (FVC), Patient-Reported Outcomes Information System (PROMIS) Physical Function score, and PROMIS Fatigue score when compared with alglucosidase alfa plus placebo. While the indirect treatment comparison provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings compared with alglucosidase alfa. There is uncertainty about how cipaglucosidase alfa in combination with miglustat compared with alglucosidase alfa will affect long-term wheelchair and ventilation dependence, and these outcomes were not included among the submitted evidence.
Canada’s Drug Agency (CDA-AMC) was unable to address several key limitations of the sponsor’s economic submission, including the aforementioned uncertainties in the comparative clinical data and also the uncertainties in the sponsor’s assumptions surrounding long-term effectiveness. Given poor flexibility with the submitted model, CDA-AMC could not estimate the incremental benefit accrued within the trial period and in the posttrial period. Therefore, the magnitude at which long-term assumptions drive the sponsor’s cost-effectiveness results is unclear. While the sponsor’s submitted model suggested a mortality improvement of 0.17 additional years of life for patients receiving cipaglucosidase alfa in combination with miglustat, and a gain of 1.04 quality-adjusted life-years (QALYs) attributed to mobility and wheelchair independence, this is not supported by the available clinical evidence reviewed. Therefore, it is highly uncertain whether any predicted clinical results from the submitted model will be realized in clinical practice. Together, the cost-effectiveness of cipaglucosidase alfa in combination with miglustat is unknown.
Patient Group and Drug Plan Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient group and drug plans that participated in the CDA-AMC review process. No clinician input was received for this review.
Patient input was received from Muscular Dystrophy Canada, which gathered information from 56 respondents; 41 respondents were patients with a confirmed diagnosis of Pompe disease and 15 were caregivers or family members of individuals with Pompe disease. The patient input noted that Pompe disease had a significant impact on all aspects of their daily lives (i.e., physical, social, and mental). All respondents noted that enzyme replacement therapy (ERT) is the primary treatment. Additionally, patients noted that when it comes to treatment options there is no choice, because only 1 treatment is available; therefore, there remains a need for additional therapies that reduce side effects, prevent exacerbations, and help patients maintain their independence. No participants reported experience with cipaglucosidase alfa in combination with miglustat.
The drug plan input received for this review raised concerns regarding comparators in jurisdictions where alglucosidase is not publicly funded. The drug plans had questions about how late-onset Pompe disease (LOPD) is diagnosed in Canada and whether the clinical trial inclusion criteria should reflect patients’ eligibility for ERT treatment. The drug plans additionally raised concerns regarding the travel logistics to receive treatment and the monitoring required for cipaglucosidase alfa in combination with miglustat, particularly when first initiated. Additionally, they inquired about how the reimbursement status of alglucosidase alfa may affect the budget impact of funding cipaglucosidase alfa in combination with miglustat.
Several of these concerns were addressed in the sponsor’s model:
The impact of Pompe disease on daily lives was explored through health-related quality of life outcomes incorporated in the sponsor’s model.
Reconstitution and administration costs associated with treatments were considered.
In addition, CDA-AMC addressed some of these concerns as follows:
The review team explored the budget impact of funding cipaglucosidase alfa in combination with miglustat in relation to the reimbursement status of alglucosidase alfa across pan-Canadian jurisdictions.
CDA-AMC was unable to address the following concerns raised from the input received:
The uncertainty associated with the long-term efficacy of cipaglucosidase alfa in combination with miglustat could not be addressed due to a lack of long-term data.
Economic Review
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Cipaglucosidase alfa in combination with miglustat is indicated for long-term treatment in adult patients with LOPD. The sponsor submitted a cost-utility analysis (CUA) that compared cipaglucosidase alfa plus miglustat with alglucosidase alfa for the treatment of adult patients with LOPD.1 The modelled population is in line with the Health Canada indication and is based on the patients enrolled in the PROPEL trial (although the majority of patients in the trial were ERT experienced).1
The recommended dose for cipaglucosidase alfa is 20 mg/kg administered every other week as an IV solution in combination with oral miglustat.2 Cipaglucosidase alfa is provided as a 105 mg single-use vial of lyophilized powder for IV infusion at a submitted price of $1,751.75 per vial. In the submitted model, the sponsor assumed the cost of cipaglucosidase alfa to be $648,134 per patient per year, assuming vial sharing and a mean patient weight of approximately 75 kg. The recommended dose of miglustat is 260 mg (4 tablets) for patients weighing 50 kg or greater, or 195 mg (3 tablets) for patients weighing from 40 kg to less than 50 kg, taken orally 1 hour before cipaglucosidase alfa infusion.2,3 Miglustat is provided in 65 mg oral tablets at a submitted price of $46.04 per tablet. In the sponsor’s analysis, miglustat was assumed to cost $4,788.42 per patient per year. Together, the total annual treatment cost of cipaglucosidase alfa in combination with miglustat was estimated to be $652,923 per patient.1 The sponsor assumed the cost of alglucosidase alfa to be $652,907 per patient per year.
The clinical outcomes of interest reported in the analysis were QALYs and life-years over a lifetime time horizon (i.e., approximately 23 years). Discounting (1.5% per annum) was applied to both costs and outcomes, and a 1-year cycle length was used.1 The sponsor’s base-case analysis was conducted from a societal perspective, with a public payer perspective included as a scenario analysis. The societal perspective included indirect costs associated with productivity loss by patients and caregivers.1 This report will focus on analyses conducted from the public health care payer perspective.
Model Structure
The sponsor submitted a state transition microsimulation model to track patients with LOPD. Within the model, patients could transition between 8 health states.1 Health states were based on level of mobility and/or respiratory support required and include “no support,” “intermittent mobility support,” “intermittent mobility AND intermittent, non-invasive respiratory support,” “intermittent, non-invasive respiratory support,” “wheelchair dependent,” “wheelchair dependent AND intermittent, non-invasive respiratory support,” “wheelchair AND invasive respiratory support dependent,” and “death” (Figure 1).1 Patients entered the model in the “no support” health state (i.e., not requiring respiratory or mobility support) and then moved through the health states as LOPD progressed.1
Patients entered the model, 1 at a time, with an assigned probabilistically sampled value for baseline 6MWD and FVC.1 Following treatment, a patient’s disease trajectory was individually tracked based on an assigned probabilistically sampled rate of change for the 6MWD and percent predicted FVC. Transitions between health states were based on prespecified threshold values in 6MWD and FVC percent predicted scores. It was assumed there was no consistent association between rate of mobility and respiratory decline.1 In the sponsor’s probabilistic base case, 30,000 patients were simulated and the expected costs and clinical effects of 6MWD and FVC percent predicted scores were calculated.
Model Inputs
The baseline characteristics of the average modelled patient was derived from PROPEL, a phase III randomized trial that compared cipaglucosidase alfa plus miglustat with alglucosidase alfa for the treatment of adult patients with LOPD.4 At baseline, the mean age of patients in the model was 46.8 years; 54.5% of patients were female and 45.5% were male.1 The baseline sitting percent predicted FVC was 70.5%.1 Baseline 6MWD was assumed to be correlated to percent predicted FVC, where a regression model indicating that a 1-unit increase in percent predicted FVC would correlate to an increase of 1.52 m in the 6MWD.1
Efficacy inputs (the initial change from baseline to year 1 in 6MWD and percent predicted FVC) were informed by the PROPEL trial for cipaglucosidase alfa plus miglustat compared with alglucosidase alfa. Absolute 6MWD outcomes from the PROPEL trial were converted to percent predicted values using an algorithm from Enright et al. and calculated based on sex, weight, age, and height.5 Weighted averages of male and female inputs were used to inform the algorithm’s coefficients.1 The change from year 1 to year 2 was informed by the PROPEL open-label extension trials for cipaglucosidase alfa in combination with miglustat and a French Pompe Registry published in 2020 for alglucosidase.6 Subsequent annual change (i.e., year 3 onward) assumed a 15% slower rate of long-term disease progression with cipaglucosidase alfa in combination with miglustat relative to alglucosidase alfa. This assumption was based on the greater improvement in 6MWD and percent predicted FVC observed in the PROPEL trial and was validated by the clinical experts consulted by the sponsor.
The thresholds used to inform the health state transitions were as follows:
patients with LOPD would require intermittent mobility support once they were unable to walk more than 250 m in the 6-minute walk test
wheelchair dependence occurred once a patient’s 6MWD fell below 75 m
intermittent respiratory support occurred once percent predicted FVC fell below 40%
dependence on respiratory support occurred when percent predicted FVC fell below 30%.
Threshold inputs were informed primarily by the clinical expert feedback received by the sponsor.
Background mortality was assumed to be equal to the mortality of the general population in Canada until patients required mobility and/or respiratory support.7 Hazard ratios informed by Gungor et al. (2013) were used to inform the increased risk of death once patients required mobility and respiratory support.8
Health state utility values were obtained from a vignette study by Hubig et al. (2023) where vignettes were developed for LOPD health states. Health state utility values of 0.61, 0.43, 0.36, 0.29, 0.11, 0.08, and −0.08 were used to inform the health state utility values of “no support,” “intermittent mobility support,” “intermittent, non-invasive respiratory support,” “intermittent mobility support AND intermittent non-invasive respiratory support,” “wheelchair dependent,” “wheelchair dependent AND intermittent, non-invasive respiratory support,” and “wheelchair AND invasive respiratory support dependent,” respectively. The sponsor used age-specific and sex-specific general population EQ-5D-3L utilities derived using methods from Ara and Brazier (2010) to adjust utility value by age and sex in the model.9,10
Based on treatment-related adverse events in the PROPEL study, which suggested that the incidence between treatment arms was similar, the sponsor excluded the cost and utilities of adverse events from the analysis.
The model included costs related to drug acquisition, treatment administration, and resource use for patient management (i.e., health state costs). Drug acquisition costs were calculated based on the simulated individual’s weight and recommended dosing schedule.1-3,11,12 The sponsor assumed vial sharing. Drug unit costs were informed by the sponsor and the Ontario Exceptional Access Program.1,13 It was assumed that all treatments would be administered every 2 weeks and that all patients would receive treatment in the hospital for the first 6 infusions. After the first 6 infusions, the sponsor assumed that the nurse fee for patients in a home-setting or clinic-setting would be covered by the sponsor. Given that miglustat is administered orally, no administration costs are incurred separately for miglustat. Health state costs were assumed to include patient management costs such as routine neurologist appointments, ventilation support (either invasive or noninvasive), and mobility support (i.e., wheelchair or home adjustments).1,14 Finally, patients were assumed to attend a regular annual follow-up visit with a neurologist.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 30,000 simulated patients (i.e., to address first-order uncertainty, which is the uncertainty around the natural variability within the population in aspects such as age, weight, baseline FVC, and so forth) and 5 second-order iterations (i.e., uncertainty around parameter estimates such as treatment efficacy and cost inputs). The deterministic and probabilistic results submitted by the sponsor were similar. The probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s health care payer perspective analysis, cipaglucosidase alfa in combination with miglustat was associated with more costs (incremental cost: $3,094), more QALYs (incremental QALYs: 0.924), and increased survival (incremental life-years: 0.17) compared with alglucosidase alfa (Table 8). Therefore, the incremental cost-effectiveness ratio (ICER) for cipaglucosidase alfa plus miglustat compared with alglucosidase alfa is $3,347 per QALY gained. The submitted model does not allow for any estimation of what proportion of the total incremental QALYs was accrued during or after the trial period (2 years) because it failed to run for shorter time horizons. The sponsor did not present separate subgroup results for patients who were ERT naive or ERT experienced because the Health Canada indication and reimbursement request are for the overall LOPD population.
Full disaggregated results of the sponsor’s economic evaluation are available in Table 8.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
Alglucosidase alfa | 12,418,503 | Reference | 7.057 | Reference | Reference |
Cipaglucosidase alfa in combination with miglustat | 12,421,597 | 3,094 | 7.981 | 0.924 | 3,347 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Notes: This reflects the analysis conducted from a public health care payer perspective. This analysis was calculated by CDA-AMC using the disaggregated results from the sponsor-submitted base-case analysis, which was conducted from a societal perspective (i.e., by excluding indirect costs).
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor submitted an analysis from a societal perspective that included additional indirect costs associated with productivity loss for patients and caregivers. In this analysis, relative to alglucosidase alfa, cipaglucosidase alfa in combination with miglustat was dominant (less costly, more QALYs).
Additionally, the sponsor conducted several scenario analyses to explore uncertainties in the analysis. All scenario analyses were conducted from a societal perspective. These analyses examined the impact of the following: alternative discount rates, alternative time horizons, the inclusion of avalglucosidase alfa as a comparator, alternative sources of utility values, capping mortality to 100 years, assuming an equal rate of long-term disease progression, and assuming no vial sharing. Generally, many scenario analyses showed results similar to the submitted societal perspective base-case analysis (i.e., cipaglucosidase alfa plus miglustat dominant over alglucosidase alfa) with some exceptions. An analysis exploring the impact of no vial sharing (ICER of $287,447) and capping mortality to 100 years (ICER of $161,248) had the largest impact on results. The analysis also included avalglucosidase alfa (not currently publicly funded in Canada) as a comparator. Cipaglucosidase alfa in combination with miglustat was associated with an ICER of $24,304 per QALY gained compared with avalglucosidase alfa, and alglucosidase alfa was dominated (no longer the reference treatment).
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative clinical effectiveness of cipaglucosidase alfa in combination with miglustat versus alglucosidase alfa is uncertain. To inform the clinical comparative efficacy in the pharmacoeconomic model, the sponsor used 6MWD and sitting percent predicted FVC data from the phase III PROPEL trial that compared cipaglucosidase alfa plus miglustat with alglucosidase alfa plus placebo in adults with LOPD. As noted in the CDA-AMC Clinical Review report, evidence from the PROPEL trial suggests that when compared with alglucosidase alfa plus placebo, cipaglucosidase alfa in combination with miglustat may result in no difference to minimal improvement in 6MWD, and likely results in no difference to minimal improvement in sitting percent predicted FVC, PROMIS Physical Function score, and PROMIS Fatigue score. While the indirect treatment comparison provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings. The lack of model diagnostics, missing inconsistency testing, reliance on reconstructed data, and variability in methodological choices introduce a high degree of uncertainty in the results. Additionally, as per the patient input received by CDA-AMC, there is a need for therapies that help patients to maintain independence. However, there remains uncertainty about how cipaglucosidase alfa in combination with miglustat will impact a patient’s long-term wheelchair and ventilation dependence, and these outcomes were not included in the submitted evidence.
CDA-AMC was unable to address this limitation.
The long-term effectiveness of cipaglucosidase alfa in combination with miglustat is uncertain. In the sponsor’s pharmacoeconomic submission, the efficacy of cipaglucosidase alfa in combination with miglustat was modelled based on change in 6MWD and sitting percent predicted FVC from 1 year to another, which would lead to wheelchair and ventilation dependence. While efficacy in the first 2 years of treatment with cipaglucosidase alfa in combination with miglustat was informed by data from the PROPEL and PROPEL open-label extension trials, long-term efficacy (i.e., year 3 and onward) was based on the assumption that patients on cipaglucosidase in combination with miglustat would experience a 15% slower rate of subsequent annual disease progression compared with alglucosidase alfa. Despite the sponsor’s submission of scenario analyses examining shorter time horizons of 2, 5, and 10 years, CDA-AMC was unable to validate these results because the model failed to run for shorter time horizons during review. As such, CDA-AMC was unable to validate the impact of extrapolations and long-term assumptions on the ICERs. CDA-AMC additionally notes that, in the absence of long-term data, there is uncertainty in the sponsor’s assumption that patients on cipaglucosidase alfa in combination with miglustat would experience a 15% slower rate of subsequent annual disease progression after year 3 compared with alglucosidase alfa. The clinical expert feedback received by CDA-AMC for this review indicated that, in the absence of long-term data, the duration of the benefit that patients receive from cipaglucosidase alfa in combination with miglustat is unknown. As an indirect result of these long-term assumptions, the economic model estimated independence and survival gains with treatment with cipaglucosidase alfa in combination with miglustat (approximately 1.04 QALYs attributed to ventilation or wheelchair independence and 0.17 additional years of life) (Table 8). However, there is no submitted evidence to support the comparative benefits in terms of wheelchair or ventilation independence or survival outcomes.
CDA-AMC was unable to address this limitation.
The submitted model lacked stability and poor modelling practices were employed. The sponsor-submitted base case was run using 30,000 first-order simulations and 5 second-order simulations. CDA-AMC noted that at these simulation counts, the model results were unstable, with the ICERs for cipaglucosidase alfa plus miglustat compared with alglucosidase alfa from 3 separate runs ranging from $51,056 to $167,374 (Table 9). As the addition of patients beyond 10,000 in the sponsor's stability test does not yield significant new information, CDA-AMC explored running the model using 10,000 patients with a higher second-order count. An initial value of 300 for the second-order count was considered to align with the number of second-order simulations submitted to the National Institute for Health and Care Excellence (NICE). However, CDA-AMC noted that the analyses conducted using 10,000 first-order simulations and 300 second-order simulations required more than 48 hours to complete and thus were excessively computationally burdensome, given the CDA-AMC runtime requirement (i.e., 8 hours). Based on the exploration by CDA-AMC of a lower second-order simulation count (n = 5) that could be completed within the CDA-AMC runtime requirement, the stability of the model remained uncertain.
Additionally, the sponsor used numerous IFERROR statements in the submitted model. IFERROR statements lead to situations in which the parameter value is automatically overwritten with an alternative value without alerting the user. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impossible because it remains unclear whether the model is running inappropriately by overriding errors. Best programming practices are such that any errors alert the user to a specific error. This, compounded with the previously noted model stability concerns, suggests that results should be treated with a high degree of caution. Finally, given poor flexibility with the submitted model, CDA-AMC could not estimate the incremental benefit accrued within the trial period and in the posttrial period (i.e., model fails to run with a time horizon shorter than 20 years). Therefore, the magnitude at which long-term assumptions drive the sponsor’s cost-effectiveness results is unclear.
CDA-AMC was unable to address this limitation. Due to the instability of the model and excessive computational resources required, the cost-effectiveness results of cipaglucosidase alfa in combination with miglustat should be viewed with a high degree of caution.
Analysis was based on publicly available list prices. The sponsor’s analysis was conducted using publicly available list prices for comparators. Thus, while the ICER for cipaglucosidase alfa in combination with miglustat was estimated to be low in the sponsor’s base-case analysis from a health care payer perspective, the ICERs were unstable. In jurisdictions in which the price of alglucosidase has been negotiated, the actual prices borne by payers will need to be considered.
CDA-AMC was unable to address this limitation.
Additionally, the following key assumptions were made by the sponsor and appraised by CDA-AMC (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
The incidence of treatment-related AEs was similar between the treatment arms in the PROPEL trial; treatment-related AEs were thus excluded from the economic model. | Reasonable. The clinical expert feedback received by CDA-AMC agreed that the incidence of treatment-related AEs is likely similar among patients who were ERT experienced. However, CDA-AMC notes that, numerically, more serious AE events occurred in patients treated with cipaglucosidase alfa plus miglustat compared with those treated with alglucosidase alfa plus placebo. However, there is low certainty related to these numbers due to wide confidence intervals and a very low number of events. |
Avalglucosidase alfa was excluded as a comparator. | Reasonable. The clinical expert feedback and drug plan input received by CDA-AMC noted that avalglucosidase alfa is not currently reimbursed or used in clinical practice in Canada. |
Reconstitution time and drug administration time for cipaglucosidase alfa plus miglustat were assumed to vary from the times for alglucosidase alfa. | Uncertain. The clinical expert feedback received by CDA-AMC noted that different ERTs are expected to have similar reconstitution and drug administration times. Additionally, the product monographs affirmed that similar times were required for reconstitution and drug administration. While the number of alglucosidase alfa vials required per infusion is higher than those for comparators, there is no available evidence quantifying whether such differences dramatically impact reconstitution or administration time. However, the differences assumed by the sponsor remained minimal and were thus unlikely to have a large impact on the overall cost-effectiveness results of cipaglucosidase alfa. |
AE = adverse event; CDA-AMC = Canada’s Drug Agency; ERT = enzyme replacement therapy.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
CDA-AMC was unable to address several key limitations within the sponsor’s submitted economic model, including uncertainty in the comparative clinical evidence and long-term efficacy estimates, as well as transparency and stability concerns for the model. These limitations prevented CDA-AMC from deriving a base-case estimate of the cost-effectiveness of cipaglucosidase alfa plus with miglustat compared with alglucosidase alfa for the treatment of adult patients with LOPD.
As noted in the CDA-AMC Clinical Review, the comparative evidence for cipaglucosidase alfa plus miglustat compared with alglucosidase is uncertain because the evidence from the PROPEL trial suggests that cipaglucosidase alfa in combination with miglustat may result in no difference to minimal improvement in the 6MWD and likely results in no difference to minimal improvement in the sitting percent predicted FVC, PROMIS Physical Function score, and PROMIS Fatigue score. Thus, CDA-AMC undertook a comparison of drug acquisition costs for cipaglucosidase alfa plus miglustat and alglucosidase alfa.
In the CDA-AMC cost comparison (based on public prices and no vial sharing), the annual drug acquisition costs for cipaglucosidase alfa in combination with miglustat based on the Health Canada–recommended dosage was $687,971 per patient. The annual drug acquisition cost for alglucosidase alfa was $655,442. The annual costs of each treatment, along with the difference in annual drug costs in comparison with cipaglucosidase alfa plus miglustat, and the price reduction needed to reach drug-cost neutrality across treatment options can be found in Table 5.
Table 5: CDA-AMC Cost Comparison Table
Treatment | Annual treatment cost ($) | Cipaglucosidase alfa in combination with miglustat, price reduction needed (%) | Difference in treatment cost ($) |
|---|---|---|---|
Cipaglucosidase alfa in combination with miglustat | 687,971 | NA | NA |
Alglucosidase alfa | 655,442 | 4.7 | 32,529 |
Avalglucosidase alfa | 622,670 | 9.5 | 65,301 |
CDA-AMC = Canada’s Drug Agency; NA = not applicable.
Note: The presented cost comparison analysis is based on publicly available list prices and on a patient weight of 75 kg.
Issues for Consideration
In 2022, avalglucosidase alfa (Nexviazyme) received a recommendation of reimburse with conditions from CDA-AMC for the long-term treatment of patients with LOPD; however, it is not currently funded by public drug plans because the pan-Canadian Pharmaceutical Alliance negotiations for avalglucosidase alfa did not conclude with an agreement.15,16 Should avalglucosidase alfa become reimbursed by jurisdictions, the cost-effectiveness of avalglucosidase alfa compared with cipaglucosidase alfa in combination with miglustat is highly uncertain, given the previously discussed model limitations and the limited and uncertain results from the indirect comparative evidence.
Alglucosidase alfa is an exceptional or restricted benefit in a few jurisdictions and a nonbenefit in the others. For jurisdictions that do not reimburse alglucosidase alfa, providing cipaglucosidase alfa in combination with miglustat may have a substantial budget impact (different from the impact in those jurisdictions that reimburse alglucosidase alfa in some capacity). Alglucosidase alfa was reviewed before the creation of the pan-Canadian Pharmaceutical Alliance, and it is unclear whether jurisdictions have confidential negotiated prices for this drug.
The clinical expert feedback received by CDA-AMC noted that the landscape for diagnosing Pompe disease is in flux due to the potential addition in Canada of screening for Pompe disease in newborns. The cost-effectiveness of currently available treatments for LOPD, together with treatment with cipaglucosidase alfa in combination with miglustat, to treat patients before or at early onset of the disease is unknown.
Overall Conclusions
Evidence from the PROPEL study suggests that cipaglucosidase alfa in combination with miglustat may result in no difference to minimal improvement in the 6MWD when compared with alglucosidase alfa plus placebo. In addition, cipaglucosidase alfa in combination with miglustat likely results in no difference to minimal improvement in percent predicted FVC, PROMIS Physical Function score, and PROMIS Fatigue score when compared with alglucosidase alfa plus placebo. While the indirect treatment comparison provides some comparative insights, several methodological and transparency gaps weaken confidence in the findings compared with alglucosidase alfa. There is uncertainty about how cipaglucosidase alfa plus miglustat compared with alglucosidase alfa will affect long-term wheelchair and ventilation dependence, and these outcomes were not included among the submitted evidence.
CDA-AMC was unable to address several key limitations of the sponsor’s economic submission, including the aforementioned uncertainties in the comparative clinical data but also the uncertainties in the sponsor’s assumptions surrounding long-term effectiveness. Given poor flexibility with the submitted model, CDA-AMC could not estimate the incremental benefit accrued within the trial period and in the posttrial period. Therefore, the magnitude at which long-term assumptions drive the sponsor’s cost-effectiveness results is unclear. While the sponsor’s submitted model suggested a mortality improvement of 0.17 additional years of life for patients receiving cipaglucosidase alfa in combination with miglustat, and a gain of 1.04 QALYs attributed to mobility and wheelchair independence, this is not supported by the available clinical evidence reviewed. Therefore, whether any predicted clinical results from the submitted model will be realized in clinical practice is highly uncertain. Together, the cost-effectiveness of cipaglucosidase alfa in combination with miglustat is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: POMBILITI™ (cipaglucosidase alfa) for use in combination with OPFOLDA™ (miglustat), doses and administration [internal sponsor's package]. Oakville (ON): Amicus Therapeutics Canada, Inc.; 2024.
2.PrPOMBILITI® (cipaglucosidase alfa for injection): Lyophilized Powder, 105 mg/vial, Intravenous infusion [product monograph]. Amicus Therapeutics Canada, Inc.; 2024.
3.PrOPFOLDA® (miglustat capsules): Capsules, 65 mg, oral [product monograph]. Amicus Therapeturics Canada, Inc.; 2024.
4.Schoser B, Roberts M, Byrne BJ, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021;20(12):1027-1037. doi:10.1016/S1474-4422(21)00331-8 PubMed
5.Enright PL, Sherrill DL. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med. 1998;158(5 Pt 1):1384-7. doi:10.1164/ajrccm.158.5.9710086 PubMed
6.Semplicini C, De Antonio M, Taouagh N, et al. Long-term benefit of enzyme replacement therapy with alglucosidase alfa in adults with Pompe disease: Prospective analysis from the French Pompe Registry. J Inherit Metab Dis. 2020;43(6):1219-1231. doi:10.1002/jimd.12272 PubMed
7.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Government of Canada; 2023. Accessed January 6, 2025. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
8.Gungor D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis. 2013;8:49. doi:10.1186/1750-1172-8-49 PubMed
9.Hubig L, Sussex AK, MacCulloch A, et al. Quality of Life with Late-Onset Pompe Disease: Qualitative Interviews and General Public Utility Estimation in the United Kingdom. J Health Econ Outcomes Res. 2023;10(1):41-50. doi:10.36469/001c.68157 PubMed
10.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-18. doi:10.1111/j.1524-4733.2010.00700.x PubMed
11.Sanofi Genzyme. PrMYOZYME® (alglucosidase alfa [Recombinant human acid alpha-glucosidase]). Lyophilized Powder, 50 mg vial, intravenous infusion [product monograph]. sanofi-aventis Canada Inc.; 2023.
12.Sanofi Genzyme. PrNEXVIAZYME™ (avalglucosidase alfa) for injection. Lyophilized Powder 100 mg/vial, intravenous infusion [product monograph]. sanofi-aventis Canada Inc.; 2021.
13.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed 2025 Jan 7. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
14.Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). Ontario Ministry of Health; 2023. Accessed 2025 Jan 9. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
15.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: Valglucosidase Alfa (Nexviazyme). Can J Health Technol. 2022;2(7). Accessed February 24, 2025. https://www.cda-amc.ca/sites/default/files/DRR/2022/SR0703%20Nexviazyme%20-%20CADTH%20Final%20Rec-meta.pdf
16.pan Canadian Pharmaceutical Alliance. Nexviazyme (avalglucosidase alfa). 2023. Accessed February 24, 2025. https://www.pcpacanada.ca/negotiation/21935
17.Canada's Drug Agency. CADTH Reimbursement Review. Avalglucosidase Alfa (Nexviazyme) Sponsor: Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc. Therapeutic area: Pompe disease [sponsor supplied reference]. 2022. https://www.cda-amc.ca/sites/default/files/DRR/2022/SR0703%20Nexviazyme%20combined.pdf
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 6: CDA-AMC Cost Comparison Table for LOPD
Treatment | Strength or concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($)a |
|---|---|---|---|---|---|---|
Cipaglucosidase alfa (Pombiliti) | 105 mg per vial | Vial | 1,751.7500a | 20 mg/kg every other week | 1,871.73 | 683,183 |
Miglustat (Opfolda) | 65 mg | Capsules | 46.0425a | ≥ 40 kg to < 50 kg: 195 mg every other week ≥ 50 kg: 260 mg every other week | 13.12 | 4,788 |
Cipaglucosidase alfa in combination with miglustat | 1,884.85 | 687,971 | ||||
Enzyme replacement therapies | ||||||
Alglucosidase alfa (Myozyme) | 5 mg/mL | 50 mg vial | 840.3100b | 20 mg/kg every other week | 1,795.73 | 655,442 |
Avalglucosidase alfa (Nexviazyme) | 10 mg/mL | 100 mg vial | 1,596.5890c | 20 mg/kg every other week | 1,705.94 | 622,670 |
CDA-AMC = Canada’s Drug Agency; LOPD = late-onset Pompe disease.
Notes: All prices are from the Ontario Exceptional Access Program (accessed January 2025), unless otherwise indicated, and do not include dispensing fees. Costs are based on a patient weight of 75 kg and assumes a 365-day year. No vial sharing is assumed.
aSponsor-submitted price.1
bOntario Drug Benefit Exceptional Access Program.13
cCDA-AMC review of avalglucosidase alfa.17
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing. | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity. | No | Refer to CDA-AMC critical appraisal. |
Model structure is adequate for decision problem. | No | Refer to CDA-AMC critical appraisal. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis). | No | Refer to CDA-AMC critical appraisal. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem. | No | Refer to CDA-AMC critical appraisal. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough detail). | No | Refer to CDA-AMC critical appraisal. |
CDA-AMC = Canada’s Drug Agency.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Cipaglucosidase alfa in combination with miglustat | Alglucosidase alfa |
|---|---|---|
Discounted LYs | ||
Total LYs | 18.19 | 18.01 |
Discounted QALYs | ||
Total | 7.98 | 7.05 |
No wheelchair use or respiratory support | 5.68 | 4.64 |
Intermittent mobility support | 1.81 | 1.87 |
Wheelchair dependent | 0.10 | 0.16 |
Intermittent respiratory support | 0.21 | 0.20 |
Intermittent mobility support and respiratory support | 0.25 | 0.29 |
Intermittent respiratory support and wheelchair dependent | −0.03 | −0.05 |
Wheelchair and respiratory support dependent | −0.03 | −0.05 |
Adverse event disutilities | 0.00 | 0.00 |
Discounted costs ($) | ||
Total costa | 12,421,597.18 | 12,418,503.42 |
No wheelchair use or respiratory support | 1,612.24 | 1,311.54 |
Intermittent mobility support | 40,445.48 | 41,407.72 |
Wheelchair dependent | 17,470.32 | 26,940.68 |
Intermittent respiratory support | 10,508.72 | 10,076.67 |
Intermittent mobility support and respiratory support | 26,762.99 | 30,562.37 |
Intermittent respiratory support and wheelchair dependent | 13,128.52 | 23,144.66 |
Wheelchair and respiratory support dependent | 181,266.77 | 263,436.34 |
Treatment costs | 12,129,962.28 | 12,021,183.64 |
Drug administration | 439.85 | 439.80 |
End of life | 0.00 | 0.00 |
Caregiver productivity loss | 0.00 | 0.00 |
Patient productivity loss | 0.00 | 0.00 |
Adverse events | 0.00 | 0.00 |
CDA-AMC = Canada’s Drug Agency; LY = life-year; QALY = quality-adjusted life-year.
aThis reflects the analysis conducted from the public health care payer perspective, calculated by CDA-AMC using the disaggregated results of the sponsor-submitted base case, which was conducted from the societal perspective (i.e., by excluding indirect costs).
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Table 9: Model Stability Results
Test number Number of first-order iterations × number of second-order iterations | Treatment | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
1. Sponsor-submitted base case (societal perspective) 30,000 × 5 | Alglucosidase alfa | 12,504,462 | 7.06 | Reference |
Cipaglucosidase alfa in combination with miglustat | 12,478,699 | 7.98 | Dominant | |
2. Sponsor-submitted scenario (health care payer perspective) 30,000 × 5 | Alglucosidase alfa | 11,506,028 | 6.691 | Reference |
Cipaglucosidase alfa in combination with miglustat | 11,539,009.42 | 7.444 | 43,776.73 | |
3. CDA-AMC calculated the health care payer perspective from the sponsor’s submitted societal perspectivea 30,000 × 5 | Alglucosidase alfa | 12,418,503 | 7.06 | Reference |
Cipaglucosidase alfa in combination with miglustat | 12,421,597 | 7.98 | 3,347.22 | |
4. CDA-AMC–run health care payer perspective, test 1 30,000 × 5 | Alglucosidase alfa | 12,701,847 | 7.7486 | Reference |
Cipaglucosidase alfa in combination with miglustat | 12,754,571 | 8.6480 | 58,620.46 | |
5. CDA-AMC–run health care payer perspective, test 2 30,000 × 5 | Alglucosidase alfa | 11,383,484 | 6.4671 | Reference |
Cipaglucosidase alfa in combination with miglustat | 11,515,467 | 7.1887 | 182,888.94 | |
6. CDA-AMC–run health care payer perspective, test 3 30,000 × 5 | Alglucosidase alfa | 11,247,477 | 6.1519 | Reference |
Cipaglucosidase alfa in combination with miglustat | 11,286,709 | 7.0390 | 44,226.92 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Notes: A test run with 10,000 first-order iterations and 300 second-order iterations failed to run within 8 hours (CDA-AMC procedural requirement). No changes were applied to the sponsor’s input values or assumptions for the test runs displayed previously.
aCalculated assuming no patient or caregiver productivity loss costs.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 10: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
CDA-AMC = Canada’s Drug Agency; CUA = cost-utility analysis; LOPD = late-onset Pompe disease.
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) to estimate the 3-year budget impact of reimbursing cipaglucosidase alfa in combination with miglustat for the treatment of adult patients with LOPD. The analysis was taken from the perspective of the public drug plan in Canada including the Non-Insured Health Benefits Program (NIHB) but excluding Quebec. The sponsor’s base case included drug acquisition costs only. A 3-year time horizon was used from 2025 to 2027 (with 2024 as the baseline year). The target population size was derived from claims-based data based on the MIDAS dataset from IQVIA. The sponsor used the average number of patients across all quarters per year to calculate the number of patients at baseline. Key inputs to the BIA are documented in Table 11.
Key assumptions to the BIA include:
Drug cost based on patient weight of approximately 75 kg and administration is every 2 weeks.
Alglucosidase alfa holds 100% of the market share in the reference scenario across all provinces.
Market share of cipaglucosidase alfa in combination with miglustat is based on sponsor’s internal market share estimates and clinical expert feedback. Assumed 100% of cipaglucosidase alfa in combination with miglustat market uptake is from alglucosidase alfa.
Table 11: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3) |
|---|---|
Target population | |
Total number of patients treated with Pompe disease across pan-Canadian jurisdictions | ██ / ██ / ██ |
Patients who were publicly reimbursed with Pompe disease, % | 72% |
Patients with LOPD, % | 75% |
Adherence to medication, % | 100% |
Number of patients eligible for drug under review | ██ / ██ / ██ |
Market uptake (3 years) | |
Uptake (reference scenario), % Cipaglucosidase alfa in combination with miglustat Alglucosidase alfa | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario), % Cipaglucosidase alfa in combination with miglustat Alglucosidase alfa | ██% / ██% / ██% ██% / ██% / ██% |
Cost of treatment (per patient, annually) | |
Cipaglucosidase alfa in combination with miglustat Alglucosidase alfa | $654,716.32 $654,701.13 |
BIA = budget impact analysis; CUA = cost-utility analysis; LOPD = late-onset Pompe disease.
Notes: Differences between the presented values between the BIA with the CUA are due to rounding for number of administrations a year. Drug costs presented were calculated assuming 26.07 administration a year.
Summary of the Sponsor’s BIA Results
In the sponsor’s base-case analysis, the estimated incremental budget impact of funding cipaglucosidase alfa in combination with miglustat for the treatment of adult patients with LOPD is $██ in year 1, $███ in year 2, and $███ in year 3. Therefore, the 3-year incremental budget impact is $350.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of claims-based approach to estimate market size introduces uncertainty with the anticipated budget impact of cipaglucosidase alfa in combination with miglustat. The sponsor estimated market size based on public claims data for the relevant comparators. However, there are several uncertainties associated with this approach. First, the sponsor noted that only partial data were available for 2021 and 2024, as such, an average number of patients from across all quarters per year were used to derive the number of patients at baseline. Due to the limitations with the data, this may overestimate or underestimate the number of patients modelled in the BIA.
Second, given that the MIDAS dataset from IQVIA reported data for all patients with Pompe disease across Canada, adjustments were required to exclude patients from Quebec and exclude the proportion of the cohort with infantile-onset Pompe disease. While the sponsor conducted these adjustments (i.e., assumed that the estimated proportion of patients likely to be from Quebec was equal the proportion of patients from Quebec relative to the population of the pan-Canadian jurisdiction and assumed that 75% of patients with Pompe disease have LOPD), there remains uncertainty in the final patient number estimate it is unclear if the claims data accurately captures data across Canada. For example, should the majority of the claims data be for pediatrics, then assuming 75% are patients with LOPD would be inappropriate.
Finally, drug plan feedback received by CDA-AMC noted that alglucosidase alfa is not reimbursed in all jurisdictions. As such, there is uncertainty introduced by estimating population size based on an unstable and potentially growing market based on claims.
CDA-AMC was unable to address the limitations of a claims-based approach of a budget impact; however, clinical expert feedback received by CDA-AMC noted the proposed estimates appear reasonable.
To explore the uncertainty associated with the different reimbursement status of alglucosidase alfa across Canada, CDA-AMC conducted scenario analysis where the budget impact of reimbursing cipaglucosidase alfa in combination with miglustat was considered for jurisdiction currently funding alglucosidase alfa (Ontario and British Columbia) separately from those who do not (or no information is available or confirmed).
Annual drug costs did not align with costs submitted in the CUA. In the sponsor’s BIA, the annual drug costs of cipaglucosidase alfa in combination with miglustat was expected to be $654,716.32 whereas the annual cost of alglucosidase alfa was $654,701.13. These values differed slightly from the annual costs used in the sponsor’s submitted CUA due to differences in administration assumptions used in the CUA and BIA analyses.
CDA-AMC aligned drug costs in the sponsor-submitted BIA and CUA, assuming 26 administrations per treatment a year.
Analysis was based on publicly available list prices. The sponsor’s analysis was conducted using publicly available list prices for comparators. The budget impact of reimbursing cipaglucosidase alfa in combination with miglustat may be higher if the price of comparators paid by the drug plans is lower.
CDA-AMC was unable to address this limitation.
CDA-AMC Reanalyses of the BIA
CDA-AMC revised the sponsor’s submitted analysis by modifying the treatment administration frequency used to inform the treatment costs in the analysis. The changes made to derive the CDA-AMC base case are described in Table 12.
Table 12: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Treatment administration frequency | 26.07 ( = 365 / 7 / 2) | 26 |
CDA-AMC base case | Reanalysis 1 | |
CDA-AMC = Canada’s Drug Agency.
The results of the CDA-AMC stepwise reanalysis are presented in summary format in Table 13 and a more detailed breakdown is presented in Table 14. In the CDA-AMC base-case analysis, the estimated incremental budget impact of funding cipaglucosidase alfa in combination with miglustat for the treatment of adult patients with LOPD was $53 in year 1, $120 in year 2, and $176 in year 3. Therefore, the 3-year incremental budget impact was $349.
Table 13: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 350 |
CDA-AMC base case (reanalysis 1) | 349 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty using the CDA-AMC base case (results are provided in Table 14):
Assessing the impact of reimbursing cipaglucosidase alfa in combination with miglustat in jurisdictions that currently fund alglucosidase alfa (i.e., Ontario and British Columbia) and those who do not (remaining jurisdictions, assuming no drug costs is currently being incurred).
Considering only 2 provinces confirmed to currently reimburse alglucosidase alfa, if cipaglucosidase alfa in combination with miglustat is reimbursed and adopted across all jurisdictions, the 3-year pan-Canadian budget impact can be expected to be $6,215,865.
Table 14: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 17,586,134 | ████████ | ████████ | ████████ | 53,769,231 |
New drug | 17,586,134 | ████████ | ████████ | ████████ | 53,769,582 | |
Budget impact | 0 | ██ | ███ | ███ | 350 | |
CDA-AMC base case | Reference | 17,537,953 | ████████ | ████████ | ████████ | 53,621,918 |
New drug | 17,537,953 | ████████ | ████████ | ████████ | 53,622,268 | |
Budget impact | 0 | 53 | 120 | 176 | 349 | |
CDA-AMC scenario analysis 1a: alglucosidase alfa reimbursed jurisdictions | Reference | 10,269,857 | ████████ | ████████ | ████████ | 31,482,082 |
New drug | 10,269,857 | ████████ | ████████ | ████████ | 31,482,287 | |
Budget impact | 0 | ██ | ██ | ███ | 205 | |
CDA-AMC scenario analysis 1b: alglucosidase alfa not reimbursed jurisdictions | Reference | 0 | ██ | ██ | ██ | 0 |
New drug | 0 | ███████ | ████████ | ████████ | 6,215,660 | |
Budget impact | 0 | ███████ | ████████ | ████████ | 6,215,660 | |
CDA-AMC scenario analysis 1c: pan-Canadian impact accounting for different reimbursement status of alglucosidase alfa across Canada | Reference | 10,269,857 | ████████ | ████████ | ████████ | 31,482,082 |
New drug | 10,269,857 | ████████ | ████████ | ████████ | 37,697,947 | |
Budget impact | 0 | ████████ | ████████ | ████████ | 6,215,865 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
Note: Scenario 1c results are simply the sum of the results from scenario 1a and scenario 1b.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.