Drugs, Health Technologies, Health Systems
Reimbursement Review
Fecal Microbiota (Rebyota)
Sponsor: Ferring Canada Inc.
Therapeutic area: Clostridioides difficile infection, prevention
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Clinical Review
Abbreviations
AE
adverse event
CDA-AMC
Canada’s Drug Agency
CDI
Clostridioides difficile infection
Cdiff32
Clostridioides difficile Health-Related Quality of Life
CI
confidence interval
CrI
credible interval
DB
double blind
FAS
full analysis set
FMT
fecal microbiota transplant
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBS
irritable bowel syndrome
ICU
intensive care unit
ITT
intention to treat
LOCF
last observation carried forward
MCID
minimal clinically important difference
mITT
modified intention to treat
OL
open label
PP
per protocol
PSS
primary safety set
rCDI
recurrent Clostridioides difficile infection
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SF-36
36-Item Short Form Survey
SOC
standard of care
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Fecal microbiota (Rebyota), 150 mL, rectal suspension |
Sponsor | Ferring Inc. |
Indication | For the prevention of recurrence of Clostridioides difficile infection (CDI) in adults following antibiotic treatment for recurrent CDI. Fecal microbiota (Rebyota) is not indicated for the treatment of CDI. |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 5, 2025 |
Recommended dose | The recommended dose is 1 single dose of 150 mL microbiota suspension by rectal administration from 24 hours to 72 hours after the last dose of antibiotics. An additional treatment may be administered in the event of a CDI recurrence. |
CDI = Clostridioides difficile infection; NOC = Notice of Compliance.
Introduction
Clostridioides difficile infection (CDI) is a spore-forming, toxin-producing, Gram-positive anaerobe that colonizes the gut, frequently in association with antibiotic therapy.1 Its hallmark symptom is profuse watery diarrhea (≥ 3 loose stools in 24 hours), although severity ranges from mild diarrhea to fulminant colitis with systemic toxicity and shock.2 CDI is a leading cause of health care–associated diarrhea, contributing to significant morbidity and mortality.3 Recurrent CDI (rCDI) occurs when symptoms return, often 2 to 8 weeks after treatment, and affects up to 25% of patients within 30 days.1 Diagnosis is based on clinical presentation and stool testing.4 CDI has a significant impact on health-related quality of life (HRQoL), and affects physical, mental, social, and professional functioning.5 Patients report symptoms like fatigue; weight loss; abdominal pain; and frequent, urgent bathroom use; alongside psychological distress, which includes anxiety, depression, and social isolation.6
Based on prevalence estimates from Levy et al.,7 the projected number of CDI cases in Canada in 2024 was approximately 47,000. In 2021, health care–associated CDI (i.e., CDI that has been acquired in a health care facility) rates were 3.54 infections per 10,000 patient days in Canada.8,9 CDI severity increases with age, as seen in a 2002 Quebec outbreak in which infection rates were 10 times higher in patients aged 65 years or older than in younger patients. CDI is associated with a substantial increase in the risk of mortality, with reported 30-day, all-cause mortality rates that range from 6.1% in Germany to 11.4% in the US.10-15 In Canada, the mortality rate within 30 days of a CDI diagnosis in 2021 was 2.3 per 100 cases.8
The Health Canada–approved indication for fecal microbiota (Rebyota) is for the prevention of a recurrence of CDI in adults after antibiotic treatment for rCDI, which is in line with the sponsor’s reimbursement request. Of note, the sponsor requests reimbursement to prevent rCDI as early as the first recurrence. The recommended dose of fecal microbiota (Rebyota) is 1 single dose of 150 mL microbiota suspension administered by rectal administration. Additional treatment may be administered in the event of CDI recurrence.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of the fecal microbiota (Rebyota) 150 mL rectal suspension for the prevention of a recurrence of CDI in adults after antibiotic treatment for rCDI.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of input provided by the patient and clinician groups that responded to the Canada's Drug Agency (CDA-AMC) call for input and from clinical experts consulted for the purpose of this review.
Patient Input
Patient input was submitted by the Gastrointestinal Society, the Peggy Lillis Foundation, and the Canadian Digestive Health Foundation for this review, with data gathered from patient questionnaires, interviews, social media posts, emails, and personal stories. The total number of patients who contributed to the input was not reported. According to the input, CDI is a debilitating condition characterized primarily by severe diarrhea, often accompanied by symptoms like dehydration, fever, abdominal pain, and fatigue. It can lead to life-threatening complications, including recurrent infections, sepsis, and, in extreme cases, the need for colon removal. Certain populations, such as those aged 65 years or older or individuals on prolonged antibiotics, are at higher risk. The input noted that with the chronic nature and high recurrence rate, the impact of CDI extends beyond physical symptoms, profoundly affecting patients’ mental health, with feelings of shame, anxiety, and even post-traumatic stress disorder. The condition disrupts daily life, requiring isolation to prevent transmission and placing significant burdens on patients and caregivers, and their ability to work or socialize.
The patient input stated that current treatments for CDI include antibiotics like vancomycin, metronidazole, and fidaxomicin. However, metronidazole has limited effectiveness for patients with severe CDI and has significant side effects. Despite the availability of these drugs, they can destabilize gut microbiota, contributing to the cycle of recurrence, according to the input. The input also stated that probiotics and prebiotics may be helpful in improving the balance of the microbiome, but current research has not yet provided evidence of their benefit in CDI management. As such, there is a need for treatments that adequately manage symptoms, are curative, and prevent CDI recurrence, the input noted.
Patients who had experience with the treatment under review reported significant benefits, citing ease of access, minimal side effects, and effective relief from rCDI. Many expressed the wish that it had been offered earlier in their treatment journey. Among the respondents who contributed to the input, fecal microbiota (Rebyota) was preferred over vancomycin because of its simpler administration — it is delivered once via enema or colonoscopy without the need for bowel preparation — and its ability to address microbiome dysbiosis, reducing vulnerability to future recurrences. Patients highlighted its simplicity and efficiency; the procedure takes about 30 minutes and causes only minor side effects, such as slight cramping.
Clinician Input
Input From Clinical Experts Consulted for This Review
The information in this section is based on input received from a panel of 3 clinical specialists consulted by CDA-AMC for the purpose of this review.
The clinical experts highlighted the need for accessible, effective, and well-tolerated therapies to prevent rCDI in patients who have a multirecurrent condition, despite appropriate antibiotic treatment, and in whose quality of life is substantially impacted by the condition. As such, the clinical experts recommended the use of fecal microbiota (Rebyota) in patients who have experienced at least 2 recurrences, at least 1 of which occurred despite a prolonged vancomycin taper or pulse regimen. In patients who experience a first recurrence, the condition may improve spontaneously. However, the natural disease trajectory becomes more complex in patients who have experienced at least 2 recurrences, and the risk of further recurrences becomes incrementally higher. Fecal microbiota (Rebyota) will likely be used in clinical practice in patients who have concomitant inflammatory bowel disease (IBD) or immunocompromising conditions, despite the fact that these patients have been underrepresented in fecal microbiota (Rebyota) studies, according to the clinical experts.
Fecal microbiota (Rebyota) is very similar to the product that is being administered through conventional fecal microbiota transplant (FMT), the clinical experts noted. However, fecal microbiota (Rebyota) relies on a single administration, whereas FMT programs routinely offer repeated doses over 1 week. The clinical experts indicated that although FMT is effective at preventing rCDI, it is not readily available to many patients in Canada. Issues surrounding limited access to FMT include the lack of public or private funding, the infrastructures and level of expertise required to run an FMT program, and challenges related to the establishment and maintenance of stool banks for FMT. Fecal microbiota (Rebyota) would be regulated in terms of stability, reliability, and quality standards, and it is expected to remove the onus of managing stool banks from FMT providers. As such, the commercial version of fecal microbiota (Rebyota) could potentially increase access to treatment in Canada, which the clinical experts viewed as an acceptable trade-off, despite the single administration. In addition, the clinical experts made the CDA-AMC clinical review team aware of the possibility that conventional FMT programs could be shut down by Health Canada when a commercial product becomes available. Health Canada notified CDA-AMC that clinicians have been advised that, although fecal microbiota (Rebyota) has been authorized, it is not yet marketed. Therefore, no changes will be made to the current interim policy outlined in the guidance document Fecal Microbiota Therapy Used in the Treatment of Clostridioides difficile Infection Not Responsive to Conventional Therapies. Once fecal microbiota (Rebyota) becomes marketed, Health Canada will explore options for transitioning away from the interim policy, with further details to be provided in due course.
The experts indicated that the increased availability of fecal microbiota (Rebyota) may cause a shift in the current treatment paradigm, as long as health care systems can provide the funding, infrastructures, and resources related to administration of the product.
The clinical experts suggested that prescribing and treatment administration of fecal microbiota (Rebyota) should be limited to clinicians and health care teams with expertise in the management of patients with rCDI and, ideally, in providing FMT, which would optimize the selection of patients who may benefit the most from treatment with fecal microbiota (Rebyota). In addition, the experts emphasized that both fecal microbiota (Rebyota) and conventional FMT are not risk-free and require a thorough informed consent process, as is the standard of care (SOC) for any intervention. The clinical experts highlighted the importance of follow-up after fecal microbiota (Rebyota) administration. Experience from clinical practice suggests that symptoms at 1 month are most predictive of long-term prognosis. The experts do not expect that all patients will go back to their pre-CDI baseline bowel habits; the goal is to reduce symptoms to a reasonable level and achieve a better quality of life while avoiding chronic antibiotic use.
Clinician Group Input
One clinician group, the Canadian Antimicrobial Resistance Alliance, provided input for this review. The input stated that fecal microbiota (Rebyota) demonstrates efficacy and safety in patients with rCDI infection and offers a first-in-class live therapeutic option. The input noted that fecal microbiota (Rebyota) will improve antimicrobial stewardship because it reduces reliance on prolonged courses of vancomycin or fidaxomicin. The treatment under review would be most appropriate for patients who have experienced at least 2 CDI recurrences, particularly those in high-risk groups, such as hospitalized patients, older adults, patients with severe disease, and those with immunocompromised conditions, the input stated.
Drug Program Input
The drug programs provide input on each drug being reviewed through CDA-AMC reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. For this review, the drug plans provided questions pertaining mainly to considerations for the initiation and prescribing of therapy. These questions were addressed by the clinical experts consulted for this review, and their responses are provided in Table 3.
Clinical Evidence
Systematic Review
Description of Studies
The 1 study reviewed — the PUNCH CD3 study (n = 289) — was a phase III, multicentre, double-blind (DB), placebo-controlled, randomized trial designed to evaluate the efficacy and safety of fecal microbiota (Rebyota) in the prevention of recurrence of CDI in adult patients after antibiotic treatment for rCDI. Patients could enter the trial if they had at least 1 recurrence after a primary episode and had received at least 1 round of oral antibiotic therapy. Fecal microbiota (Rebyota) was administered as 1 single-dose enema.
The primary outcome was the recurrence of CDI over 8 weeks, which was defined as the presence of CDI diarrhea, with a stool test positive for the presence of C. difficile toxin in the 8 weeks after administration of the study enema. The primary efficacy analysis was performed using a Bayesian hierarchical model, which formally incorporated data on the treatment effect of fecal microbiota (Rebyota) from the randomized, placebo-controlled, phase IIb PUNCH CD2 study. To account for potential heterogeneity between populations in the 2 trials, a dynamic borrowing approach was used. This method allowed for data to be borrowed from the previous trial when response rates were similar.
A sustained clinical response at 6 months in patients who had previously achieved treatment success at 8 weeks was assessed as the secondary end point in the study. HRQoL was an exploratory outcome in the studies and was assessed using the C. difficile Health-Related Quality-of-Life Questionnaire (Cdiff32) Questionnaire, a validated, disease-specific assessment tool that indicates how a person is affected by CDI-related physical, mental, and social health concerns. The total and domain scores of the Cdiff32 Questionnaire range from 0 to 100, and lower scores denote a greater negative impact on quality of life. A minimal clinically important difference (MCID) of 10 points, using a distribution-based approach, has been suggested in the literature.
Efficacy Results
Response to Treatment at 8 Weeks
For the primary outcome, the mean success rate was 70.4% in the fecal microbiota (Rebyota) group and 58.1% in the placebo group. The between-group difference in mean success rate was 12.3% (95% credible interval [CrI, 1.4% to 23.3%), based on posterior estimates from the Bayesian hierarchical model.
The between-group difference for treatment success was 5.4% (95% confidence interval [CI], −9.4% to 20.2%; █████████) in patients who had no more than 3 prior CDI episodes (or 2 CDI recurrences) and 16.1% (95% CI; −5.4% to 37.7%; █████████) in patients who had more than 3 prior CDI episodes (or more than 2 CDI recurrences). In patients who received vancomycin alone, the between-group difference was 1.9% (95% CI; −20.3% to 24.0%; █████████) in patients who were treated for no more than 14 days and 12.8% (95% CI; −3.5% to 29.1%; █████████) in patients who were treated for more than 14 days.
Among patients who achieved treatment success at 8 weeks after treatment with fecal microbiota (Rebyota), the increase in the proportion of patients with a sustained clinical response at 6 months, compared to placebo, may not be clinically important, based on a between-group difference of 1.5% ████ ███ ████ ██ █████ █████████.
Health-Related Quality of Life
HRQoL was assessed as an exploratory outcome. HRQoL was not tested statistically and the results are considered to be supportive evidence. Results suggest that treatment with fecal microbiota (Rebyota) may result in little to no difference in the change from baseline in HRQoL, measured with the Cdiff32 Questionnaire, over 8 weeks, compared to placebo. The mean between-group difference in change from baseline at 8 weeks in Cdiff32 Questionnaire score was 5.71 ████ ███ █████ ██ ██████.
The sponsor provided an additional exploratory analysis of HRQoL data in patients who were deemed responders at 8 weeks. In both the main study population and the responder patients, the between-group differences reported in overall Cdiff32 Questionnaire scores crossed the null and were modest, especially relative to the large within-group improvements seen in both patients receiving fecal microbiota (Rebyota) and those receiving placebo. Although statistical significance was achieved for the mental domain summary in responder patients, these domain-level results are from exploratory analyses without multiplicity adjustment and from restricted responder subsets, increasing the risk of chance findings. In addition, limited details were provided for proper appraisal of the findings; this precluded formal assessments of the magnitude of effect and clinical relevance.
Stool Testing: Fecal Microbial Composition
Fecal sequencing analysis suggests that the microbiome composition of patients who experienced treatment success changed from baseline at all time points (P < 0.001 for within-group change from baseline using the generalized Wald test). The change in microbiome composition after treatment was characterized by an increase in Bacteroidia-class bacteria and Clostridia-class bacteria and a decrease in Gammaproteobacteria-class bacteria and Bacilli-class bacteria.
Health Care Resource Use
Hospitalizations and intensive care unit (ICU) admissions were reported as harms outcomes for patients who were randomized to active treatment with fecal microbiota (Rebyota), but ██ ██████ was reported for patients who were randomized to placebo. In the PUNCH CD3 study, a total of ██ patients in the fecal microbiota (Rebyota) group required hospitalization for rCDI during the 8-week follow-up after blinded treatment, as did ██ patients during the 8-week follow-up after open-label (OL) enema. A total of 2 patients in the fecal microbiota (Rebyota) group required ICU admission during the 8-week follow-up after blinded treatment. No patient experienced major complications of CDI after OL enema in this treatment group.
Harms Results
A relatively high proportion of patients receiving fecal microbiota (Rebyota) in the PUNCH CD3 study (70%) experienced at least 1 adverse event (AE), compared with 60% in the placebo group. AEs were numerically more frequent in patients who received 2 fecal microbiota (Rebyota) enemas █████ than in those who received only 1 fecal microbiota (Rebyota) enema ███. The most common treatment-emergent adverse events (TEAEs) were related to gastrointestinal disorders, and included diarrhea, abdominal pain, nausea, and abdominal distention.
Serious adverse events (SAEs) were relatively uncommon in patients receiving a fecal microbiota (Rebyota) enema (8%) or placebo (7%), but they were also numerically more frequent in patients who received 2 fecal microbiota (Rebyota) enemas ████ than in patients who received only 1 fecal microbiota (Rebyota) enema ████. Two deaths were reported during the trial in patients who received fecal microbiota (Rebyota), but they were not related to the study drug or the enema procedure, according to the sponsor. The sponsor reported no events of toxic megacolon or colonic perforation; however, 1 patient in the fecal microbiota (Rebyota) group experienced septic shock and underwent an emergency colectomy to deal with major complications of a new CDI.
Overall, the clinical experts indicated that the harms profile of fecal microbiota (Rebyota) did not raise any new safety signals or any particular safety concerns. As with most clinical trials, the study was not powered to detect infrequent AEs or designed to detect those with a lag time. The clinical experts indicated that there may be long-term, unintended effects related to the manipulation of a patient’s microbiome, such as the risk of developing a range of conditions over time; the impact of this is currently unknown.
Critical Appraisal
Although the randomization methods were appropriate for reducing the risk of bias in the randomization process, more patients in the fecal microbiota group than in the placebo group had experienced 2 or more CDI recurrences. This could have introduced a possible risk of bias of uncertain direction or magnitude, but it may have directionally favoured placebo.
Because patients who experienced treatment failure at 8 weeks could receive OL fecal microbiota (Rebyota), the interpretation of end points tested beyond 8 weeks is challenged by the fact that a proportion of patients in the placebo group received fecal microbiota (Rebyota).
There is uncertainty surrounding the interpretation of the findings. Because no MCID was reported in the literature as a threshold for treatment success, the presence of an important effect was informed by the clinical experts consulted for this review. The point estimate (12.3%) suggests the presence of a clinically important effect; however, the lower bound of the CrI (1.4%) is consistent with a trivial effect that would not be considered clinically important for patients. On the Cdiff32 Questionnaire, the magnitude of the findings was not considered clinically meaningful by the clinical experts consulted for this review. The MCID of 10 points estimated in the literature used a distribution-based approach, so it may not fully reflect a change that is important for patients. Further, the MCID was suggested for within-group changes, rather than for between-group differences. In addition, there is a risk of bias because of missing outcome data at 6 months; relatively few patients in both groups were available for HRQoL assessments, and missing data were imputed using the last observation carried forward (LOCF), which relies on assumptions about the trajectory of the end point that may not be plausible.
A sustained clinical response at 6 months was measured in patients with a response at 8 weeks; there is a risk of bias in the measurement of this end point, as it is unclear whether prognostic balance would be maintained in this subpopulation.
Input from clinical experts suggested that findings from the PUNCH CD3 study may be generalizable to patients considered, overall, to be at lower risk for CDI recurrences than patients typically seen in clinical practice, who would have the greatest unmet need and be the best candidates to receive fecal microbiota (Rebyota). This was based on the low number of patients with previous CDI recurrences in the trial, as well as the exclusion of patients with relevant comorbidities. In addition, it is uncertain whether patients previously received an appropriate antibiotic regimen for the treatment of the qualifying CDI episode. The trial could not inform on the effectiveness and safety of fecal microbiota (Rebyota) relative to other currently used therapies for rCDI, such as conventional FMT.
GRADE Summary of Findings and Certainty of the Evidence
For pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach was used to assess the certainty of the evidence for outcomes considered most relevant to expert committee deliberations, and a final certainty rating was determined as outlined by the GRADE Working Group.16,17
In accordance with the GRADE approach, evidence from the PUNCH CD3 study started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
The selection of outcomes for GRADE was based on the sponsor’s Summary of Clinical Evidence, consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
treatment success rate
sustained clinical response among patients who previously achieved treatment success
HRQoL (Cdiff32 Questionnaire)
SAEs.
Table 2 presents the GRADE summary of findings for fecal microbiota (Rebyota) versus placebo.
Table 2: Summary of Findings for Fecal Microbiota (Rebyota) vs. Placebo for Patients With Recurrent CDI
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Fecal microbiota (Rebyota) | Difference | |||||
Clinical response | |||||||
Treatment success rate Follow-up: 8 weeks | 262 (1 RCT) | NR | Mean: 58.1% | Mean (95% CrI): 70.4% █████ | Mean (95% CrI): 12.3% (1.4% to 23.3%) | Moderatea | Fecal microbiota (Rebyota) likely results in a clinically important increase in treatment success rate over 8 weeks, compared to placebo |
Proportion of patients with sustained clinical response among those who previously achieved treatment success Follow-up: 6 months | 179 (1 RCT) | NR | 906 per 1,000 patients | 921 per 1,000 patients (CrI NR) | █████ | Lowb | Fecal microbiota (Rebyota) may result in little to no clinically important difference in the proportion of patients with a sustained clinical response over 6 months, among those who previously achieved treatment success, compared to placebo |
HRQoL | |||||||
Mean change from baseline in Cdiff32 Questionnaire score (from 0 [worst] to 100 [best]) Follow-up: 8 weeks | 262 (1 RCT) | NA | █████ | █████ | 5.71 █████ | Moderatec | Fecal microbiota (Rebyota) may result in little to no clinically important difference in HRQoL, measured with the Cdiff32 Questionnaire, over 8 weeks, compared to placebo |
Mean change from baseline in Cdiff32 Questionnaire score (from 0 [worst] to 100 [best]) Follow-up: 6 months | 262 (1 RCT) | NA | █████ | █████ | █████ | Very lowd | The evidence is very uncertain about the effect of fecal microbiota (Rebyota) on HRQoL at 6 months, compared to placebo |
Harms | |||||||
Patients with ≥ 1 SAE Follow-up: 6 months | 267 (1 RCT) | NR | 83 per 1,000 patients | 69 per 1,000 patients (CrI NR) | █████ (NR) | Lowe | Fecal microbiota (Rebyota) may result in little to no clinically important difference in the proportion of patients with ≥ 1 SAEs over 6 months, compared to placebo |
CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; CI = confidence interval; CrI = credible interval; HRQoL = health-related quality of life; NA = not applicable; NR = not reported; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation; vs. = versus.
Note: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
aTreatment success: Rated down 1 level for imprecision. The presence of an important effect was informed by the clinical expert consulted for this review. The point estimate (12.3%) suggests the presence of a clinically important effect; however, the lower bound of the CrI (1.4%) is consistent with an effect that would not be considered clinically important for patients.
bSustained clinical response: Rated down 1 level for imprecision. The presence of an important effect was informed by the clinical experts consulted for this review. The point estimate (1.5%) suggests a trivial effect, which would not be considered clinically important for patients; however, the lower bound ███████ and upper bound ███████ of the CrI could be consistent with either a clinically important decrease or increase, respectively, in sustained clinical response.
Rated down 1 level for study limitation. There likely was a loss of prognosis balance in the subpopulation of patients analyzed, as it consisted of patients who previously achieved treatment success at 8 weeks.
cHRQoL: Rated down 1 level for imprecision. The Cdiff32 Questionnaire was assessed as an exploratory outcome. Change from baseline in Cdiff32 Questionnaire score was not tested statistically in the trial, and should be considered to be supportive evidence. Although the point estimate for the between-group difference suggests little to no clinically important difference, based on a suggested MCID of 10 points, the upper bound of the 95% CI suggests the possibility of a clinically important difference between fecal microbiota (Rebyota) and placebo. The MCID of 10 points suggested in the literature for this instrument is, however, uncertain, because it was estimated using a distribution-based approach. Because distribution-based methods rely on the statistical properties of the data, they may not always reflect clinically meaningful changes from the perspectives of patients. This MCID was also estimated for within-group changes, rather than between-group differences.
dHRQoL: Rated down 1 level for serious imprecision. Although the point estimate for the between-group difference suggests little to no clinically important difference, based on a suggested MCID of 10 points, the lower bound of the 95% CI suggests a clinically important detriment with fecal microbiota (Rebyota) compared to placebo. The MCID of 10 points suggested in the literature for this instrument is uncertain, as it was estimated using a distribution-based approach. Because distribution-based methods rely on statistical properties of the data, they may not always reflect clinically meaningful changes from the perspectives of patients. In addition, this MCID was estimated for within-group changes, rather than between-group differences.
Rated down 2 levels for very serious study limitations. Few patients completed the 6-month assessments. The last observation carried forward was used to impute missing outcome data, but this approach relies on assumptions about the missing data that are unlikely to be realistic. Further, some patients in the placebo group had received open-label fecal microbiota (Rebyota). The Cdiff32 Questionnaire was assessed as an exploratory outcome. Statistical testing for the Cdiff32 Questionnaire was not adjusted for multiplicity in the trial and should be considered to be supportive evidence.
eHarms: Rated down 1 level for imprecision because the between-group effect estimate was based on few events, and rated down 1 level for study limitation because some patients in the placebo group received active treatment with open-label fecal microbiota (Rebyota).
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Long-Term Extension Studies
No long-term extension studies were submitted by the sponsor for this review.
Indirect Comparisons
No indirect treatment comparison were submitted by the sponsor for this review. According to the sponsor, there are no comparators to fecal microbiota (Rebyota) in Canada for the preventive treatment of rCDI after antibiotic treatment for rCDI. In clinical practice, treatment options include long-term vancomycin administered in a taper or pulse regimen and conventional FMT.
Studies Addressing Gaps in the Evidence From the Systematic Review
PUNCH CD2 Study
Description of Study
The PUNCH CD2 study (N = 133) was a phase II, prospective, multicentre, DB, randomized, placebo-controlled trial designed to assess the efficacy and safety of fecal microbiota (Rebyota) for rCDI and to establish a treatment regimen for subsequent confirmation in the PUNCH CD3 study. The PUNCH CD2 study included adults (aged ≥ 18 years) with rCDI who had experienced at least 2 CDI recurrences that required at least 2 rounds of SOC oral antibiotic therapy or who had 2 severe CDI episodes that resulted in hospitalization. Of the 150 patients enrolled, 133 were randomized into 1 of 3 groups: group A (N = 45), group B (N = 44), and group C (N = 44). Patients were randomized in a 1:1:1 ratio to receive 2 fecal microbiota (Rebyota) enemas (group A), 2 doses of placebo (group B), or 1 dose of fecal microbiota (Rebyota) followed by 1 dose of placebo (group C). Doses were administered 7 days apart. If patients experienced a CDI recurrence in the 8 weeks after treatment, they could opt for either SOC treatment or a second OL fecal microbiota (Rebyota) course. This report primarily focuses on group B and group C, which align with Health Canada’s proposed recommended 1-dose fecal microbiota (Rebyota) regimen.
Efficacy and Harms Results
Efficacy results in the intention-to-treat (ITT) population showed that 8 weeks after treatment completion, treatment success rates were 43.2% (19 of 44 patients) in group B and 56.8% (25 of 44 patients) in group C, for a difference of 13.6% (95% CI, █████ ██ █████; P = 0.201). The 25th percentile for time to recurrence, estimated using the Kaplan-Meier method, was 8.0 days in both group B (95% CI, ██ ██ ████) and group C (95% CI, ██ ██ ████). The combined treatment success rate (defined as no C. difficile–associated diarrhea and no need for re-treatment or fecal transplant by day 56 after the last study enema) of all patients in the OL portion was █████ ███████. Additionally, HRQoL was assessed using the 36-Item Short Form Survey (SF-36). Baseline values were comparable between groups, and both treatment groups showed a numerical increase in mean scores for all SF-36 components from baseline to week 8. Between-group differences were not reported. Safety was assessed up to 24 months after treatment. TEAEs occurred in 86.4% of group B and 78.6% of group C. For TEAEs, anemia, nausea, flatulence, hematochezia, pyrexia, pneumonia, falls, and dyspnea were more frequent in group C than in group B. SAEs were reported in 36.4% and 35.7% of patients in group B and group C, respectively, but none were considered to be related to treatment, according to the sponsor. Deaths occurred in ████ (group B) and █████ (group C) of patients, primarily related to pre-existing conditions.
Critical Appraisal
The PUNCH CD2 study provided additional data on CDI recurrence, HRQoL (SF-36), and long-term safety up to 24 months. The primary efficacy analysis (ITT) did not reject the null hypothesis, and secondary comparisons proceeded without alpha adjustment, increasing the risk of false positives in the secondary analysis results. Because of the small sample sizes across groups, there is an increased risk that prognostic balance was not achieved (as evidenced by imbalances across groups by sex and prior hospitalization for CDI). As such, it is possible that the observed effects were either overestimated or underestimated. The study is limited by the lack of placebo control beyond 8 weeks of treatment; as such, results beyond 8 weeks are confounded by OL fecal microbiota (Rebyota) use in the placebo group. In the analysis of SF-36 scores, there is a risk of bias because of missing outcome data. Data were missing for up to 30% of patients across groups at 8 weeks. These data were imputed using the LOCF, a single-imputation method that relies on the assumption that HRQoL remains constant over time after the last available assessment, which is likely not reasonable. Immunocompromised patients were excluded from the PUNCH CD2 study, raising concerns about pathogen transmission. Most patients in the trial (89.8%) used vancomycin, which aligns with clinical practice; fidaxomicin is rarely used to treat rCDI in Canada.
PUNCH CD3 OLS Study
Description of Study
The PUNCH CD3 OLS study (N = 698) was a phase III, prospective, multicentre, OL, single-arm study. The objective of the study was to evaluate the safety, tolerability, and effectiveness of fecal microbiota (Rebyota) as single and repeat administrations in a broader population of patients than in previous trials, which had excluded patients with conditions such as Crohn disease, ulcerative colitis, IBD, irritable bowel syndrome (IBS), and immunodeficiency.
Efficacy Results
Of 676 patients in the primary efficacy analysis, 499 patients (73.8%) achieved treatment success in the 8 weeks after the first dose of fecal microbiota (Rebyota), which was defined the absence of CDI diarrhea during the 8 weeks after the first dose. Similar treatment success rates were observed for most demographic subgroups, including sex, race, ethnic group, site geography, number of prior CDI episodes, and Cdiff32 Questionnaire score; however, the treatment effect appeared to be of smaller magnitude in patients aged 65 years and older.
A total of 151 patients experienced a recurrence of CDI after the first dose and, of these, 121 patients received a second dose of fecal microbiota (Rebyota). Treatment success was then achieved in 67 (55.4%) of those patients.
Among patients who completed the 6-month follow-up, the sustained clinical response rate of fecal microbiota (Rebyota) among patients who previously achieved treatment success was 91.0%.
The mean (standard deviation [SD]) change from screening to 6 months in the Cdiff32 Questionnaire scores was █████ █████████
Harms Results
Of the 697 patients included in the safety population, ███ ███████ patients reported AEs after fecal microbiota (Rebyota) administration. These were mostly mild (20.4%) to moderate ███████ in severity, and the most frequently reported AEs included gastrointestinal disorders and infections.
Critical Appraisal
The single-arm nature of the PUNCH CD3 OLS does not allow for causal conclusions to be drawn regarding the efficacy and safety of fecal microbiota (Rebyota). Having knowledge of the treatment received can introduce bias in the assessment of subjective outcomes. Although the selection criteria allowed for a broader population, the number of patients with Crohn disease, ulcerative colitis, IBD, IBS, or immunodeficiency enrolled in the study was small. Therefore, the generalizability of the findings to patients who have these coexisting comorbidities remains uncertain.
Feuerstadt et al. Study
A retrospective study by Feuerstadt et al. (2023)19 (N = 94) assessed the effectiveness and safety of fecal microbiota (Rebyota) in adults with rCDI using broad eligibility criteria to reflect real-world conditions, including those with comorbidities such as IBS, IBD, or immunodeficiency. The study enrolled patients in 5 study sites in the US from 2015 to 2019. Patients could receive up to 4 fecal microbiota (Rebyota) enemas (2 treatment courses of 2 doses each), per physician discretion. No lab confirmation of CDI was required, there were no exclusion criteria, and data were retrospectively collected via chart review. The mean age of the patients was 59.8 years, 44.7% were 65 years or older, and 72.3% were female.
Sixty-six of the 94 (70.2%) patients in the full analysis set (FAS) achieved treatment success (defined as no CDI recurrence in the 8 weeks after the final treatment dose for the qualifying event) at week 8, 58 (87.9%) of whom maintained a sustained clinical response for 6 months after treatment. TEAEs occurred in 40 of 64 patients (62.5%) in the primary safety set (PSS), 17.2% of which were deemed to be product-related by the investigators and 4.7% of which were deemed to be procedure-related. Common TEAEs in the PSS included abdominal pain (14.1%), diarrhea (14.1%), and urinary tract infection (10.9%). SAEs occurred in 12.5% of patients; the 1 death (1.6%), due to multiorgan failure, was unrelated to treatment, according to the investigators.
The study included broad eligibility criteria but the noncomparative nature of its design does not allow for causal conclusions to be drawn regarding the efficacy and harms of fecal microbiota (Rebyota). Subjective outcome reporting may be biased and dosing could have exceeded Health Canada’s 1-dose recommendation.
Lee et al. Study
The summary by Lee et al. included pooled safety data from 5 prospective studies, incorporating 3 phase II studies (the PUNCH CD, PUNCH CD2, and PUNCH OL studies) and 2 phase III studies (the PUNCH CD3 and PUNCH CD3 OLS studies). These studies evaluated the cumulative safety of fecal microbiota (Rebyota) using standardized manufacturing practices and evolving pathogen screening to ensure safety.
All 5 trials enrolled adults aged at least 18 years with rCDI who had received antibiotics for their CDI episode before study treatment. Dosing regimens varied, with patients receiving either a single or 2 doses of fecal microbiota (Rebyota) and/or placebo, administered 7 ± 2 days apart. Four trials allowed OL treatment if CDI recurrence was confirmed in the 8 weeks after the initial course. The full population comprised 978 patients who received at least 1 dose of fecal microbiota (Rebyota) and 83 patients who received only placebo. At baseline, the pooled trial data showed that a higher proportion of patients in the fecal microbiota (Rebyota) group were 65 years or older than in the placebo group (48.2% versus 37.3%). Additionally, more patients in the fecal microbiota (Rebyota) group (78.0%), the placebo plus fecal microbiota (Rebyota) group (83.3%), and the fecal microbiota (Rebyota) plus OL fecal microbiota (Rebyota) group (79.6%) had experienced 3 or more CDI episodes before trial entry than those in the placebo group (68.7%).
The primary outcome measured the number of patients who experienced TEAEs related to fecal microbiota (Rebyota) and/or to the administration procedure; TEAEs were defined as AEs occurring on or after the day of treatment. The most common TEAEs were gastrointestinal disorders, and most were mild or moderate in severity. Serious TEAEs were similar in the placebo (60.2%) and fecal microbiota (Rebyota) 1-dose (66.4%) groups, but higher in blinded or any fecal microbiota (Rebyota) groups (68.8%). Eighteen deaths were reported, all in patients who received fecal microbiota (Rebyota); however, none were considered to be related to the treatment.
Limitations included heterogeneity in study protocols, varied dosing regimens, and differences in patient populations, which affected internal validity. OL treatments could introduce bias in the reporting of subjective AEs. Pooling data from multiple trials may obscure specific interactions or effects, impacting safety outcome interpretations.
Khanna et al. Study
The sponsor provided additional data from a retrospective cohort study by Khanna et al. (2025).20 In this study of 196 adults in the US with rCDI who received fecal microbiota (Rebyota) between July 2023 and August 2024, the overall treatment success rate at 8 weeks was 83%. In the overall population, most patients (n = 136) had at least 3 prior CDI recurrences. Success rates were consistent across subgroups of patients categorized by the number of prior recurrences. The study, however, had important limitations. Baseline patient characteristics were limited; no detailed information was provided on the exact number of prior CDI episodes, comorbidities, or the antibiotic regimen used (other than product name), preventing assessment of the impact of the characteristics on outcomes. The analyses did not adjust for confounding factors, lacked a control arm, and could have been subject to selection bias because of its nonrandomized design. Differences in health care access and the predominance of treatment administration in infusion centres in the southern and western US may further limit the generalizability of the findings. As such, the results should be considered to be supportive evidence.
Conclusions
In patients with rCDI, findings of moderate certainty from the PUNCH CD3 study suggest that treatment with fecal microbiota (Rebyota) likely results in a clinically important increase in the treatment success rate over 8 weeks compared to placebo. Findings were obtained in a population of patients who were, however, at a lower risk of experiencing CDI recurrences than those who have the greatest unmet need and who would benefit the most from treatment with fecal microbiota (Rebyota), according to the clinical experts. This population in need would include patients who experienced at least 2 recurrences despite an appropriate antibiotic treatment that consisted of a vancomycin taper or pulse regimen. Findings from supplementary, prespecified subgroup analyses created uncertainty as to whether the overall effect can be generalized to patients with fewer than 2 recurrences or who received vancomycin for 14 days or less. Although the generalizability of the study population to patients who would be candidates for fecal microbiota (Rebyota) in clinical practice is uncertain, it appears plausible that the benefits of treatment could be maintained in a population at higher risk of recurrence. Most patients who previously achieved treatment success at 8 weeks with either fecal microbiota (Rebyota) or placebo were able to maintain a sustained clinical response at 6 months; therefore, fecal microbiota (Rebyota) may be no more beneficial for maintaining response over time than placebo. Based on results from exploratory analyses, fecal microbiota (Rebyota) may result in little to no difference in HRQoL, measured with the Cdiff32 Questionnaire, over 8 weeks compared to placebo. HRQoL results at 6 months were too uncertain to draw a definite conclusion. In addition, the evidence was insufficient to draw any conclusions about the effect of fecal microbiota (Rebyota) relative to placebo on hospitalizations or ICU admissions.
A relatively high proportion of patients in the PUNCH CD3 study experienced AEs, most notably relating to gastrointestinal disorders. AEs were numerically more frequent in patients who received 2 doses of fecal microbiota (Rebyota) than in patients who received only 1 fecal microbiota (Rebyota) enema, and were also numerically more frequent than in patients who received placebo. However, fecal microbiota (Rebyota) appeared to be well tolerated, with relatively few SAEs. The clinical experts noted that although treatment with fecal microbiota (Rebyota) is not without risks, the overall harms profile did not raise any particular safety signals. However, the long-term, unintended effects of manipulating a patient’s microbiome are currently unknown.
The trial could not inform on the effectiveness and safety of fecal microbiota (Rebyota) relative to other currently used therapies for rCDI, such as conventional, multidose FMT. Whether patients in the 2 groups previously received an appropriate antibiotic regimen of vancomycin taper or pulse for the treatment of the qualifying CDI episode is unknown. The relevance of using placebo as a comparator is therefore uncertain, as the CDI treatment in the study was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice. Four additional studies were assessed in the fecal microbiota (Rebyota) review, but could not inform on important gaps in the evidence.
The clinical experts indicated that although effective, FMT is an intervention with limited access and, therefore, it is not readily available to many patients in Canada. A commercial, regulated stool-derived product such as fecal microbiota (Rebyota) can potentially increase access to treatment, which was viewed by the clinical experts as an appropriate trade-off to its single administration. The experts indicated that fecal microbiota (Rebyota) may, therefore, cause a shift in the current treatment paradigm, as long as the health care systems can provide the infrastructures and resources to address prescribing considerations and administration of the product.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of the rectal administration of fecal microbiota (Rebyota), 150 mL, in the prevention of CDI recurrence in adult patients after antibiotic treatment for rCDI.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
CDI is a spore-forming, toxin-producing, and Gram-positive anaerobic bacterium that typically colonizes the human intestinal tract after the normal gut flora has been disrupted, frequently in association with antibiotic therapy.1 Profuse watery diarrhea, characterized by 3 or more loose stools in 24 hours, is the hallmark symptom of CDI.2 However, patients can experience a range of symptoms, from mild diarrhea to severe disease or fulminant colitis, which includes significant systemic toxic effects and shock.2 CDI is a leading infectious cause of health care–associated diarrhea and a major cause of morbidity and mortality among hospitalized patients.3 Recurrent СDІ is characterized by the return of symptoms, often 2 to 8 weeks after they have resolved with appropriate treatment. Up to 25% of patients experience recurrent CDΙ in the 30 days after treatment.1 The diagnosis of CDI relies on clinical presentation and is confirmed with stool testing.4
CDI has a significant impact on patients' HRQoL, affecting physical, mental, social, and professional functioning both during infection and after infection resolution.5 Patients have reported symptoms such as disturbed sleep, fatigue, weight loss, appetite loss, abdominal pain, and frequent, urgent bathroom use, along with unpleasant odours. Beyond these physical effects, CDI and the fear of recurrence can have a profound mental impact, with patients experiencing anxiety, depression, isolation, stress, anger, and embarrassment related to the loss of bowel control.6
In Canada, health care–associated CDI rates (CDI that has been acquired in a health care facility) were 3.54 infections per 10,000 patient days in 2021.8,9 CDI is also a concern in community settings, with approximately 25% of CDI cases being community-acquired, but this rate is potentially underestimated in the literature.6,21-27 Using the estimates from the study by Levy et al.,7 the sponsor estimates that the prevalence of CDI in Canada in 2024 was approximately 47,000 cases. CDI frequency and severity increase with age, as demonstrated by a 2002 Quebec outbreak in which the rate of CDI among patients aged 65 years or older was 10 times higher than that observed in younger patients.28 CDI significantly increases the risk of mortality, with 30-day all-cause mortality rates ranging from 6.1% to 11.4% across various countries.10-15 In Canada, the Canadian Antimicrobial Resistance Surveillance System monitors attributable mortality using surveillance data from select hospitals. The overall mortality rate in the 30 days after diagnosis in 2021 was 2.3 per 100 cases.8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
The clinical experts indicated that most patients with a first CDI episode will respond to antibiotic treatment with vancomycin or fidaxomicin, and noted that rCDI will occur in approximately 15% of patients. Patients who experience a first recurrence may have their condition improve spontaneously after treatment with oral vancomycin or fidaxomicin. However, the natural disease trajectory becomes more complex in patients who have experienced at least 2 recurrences, and the risk of experiencing further recurrences becomes incrementally higher. According to the Association of Medical Microbiology and Infectious Disease Canadian guidelines,27 a 10-day to 14-day vancomycin regimen is considered the treatment of choice for a first recurrence of CDI, whereas a prolonged vancomycin taper or pulse regimen (in which the dose is gradually decreased [tapered] and/or given intermittently [pulse]) is recommended for the treatment of a second recurrence of CDI.27 Although antibiotic therapy is targeted at the infection itself, it also negatively impacts the gut microbiome. Therefore, the Association of Medical Microbiology and Infectious Disease guidelines27 recommend FMT as a treatment option for subsequent recurrences after a vancomycin taper. FMT is expected to address the underlying disease process by repopulating and restoring the composition and diversity of the gut microbiome.
The main goal of treatment is to achieve a complete and sustained clinical response. Not all patients are expected to return to their pre-CDI baseline bowel habits, according to the clinical experts; the goal is to reduce symptoms to a reasonable level and achieve a better quality of life. Measures can be taken to reduce the risk of subsequent CDI, including avoiding unnecessary antibiotics, reducing immunosuppression when relevant, and preventing reinfection from ambient spores in the home environment.
Drug Under Review
The mechanism of action for fecal microbiota (Rebyota) is believed to involve repopulation and restoration of the composition and diversity of the gut microbiome to suppress C. difficile outgrowth and CDI recurrence.29 The Health Canada–approved indication is for the prevention of recurrence of CDI in adults after antibiotic treatment for rCDI that is in line with the sponsor’s reimbursement request. Of note, the sponsor requests reimbursement to prevent rCDI as early as the first recurrence.
The recommended dose of fecal microbiota (Rebyota) is 1 single dose of 150 mL of microbiota suspension by rectal administration. Additional treatment may be administered in the event of a CDI recurrence.29 It is recommended that the treatment be administered from 24 hours to approximately 72 hours after the last dose of antibiotics. It should not be administered during antibiotic treatment for CDI. Patients should not take any oral antibiotic therapy for up to 8 weeks after administration of fecal microbiota (Rebyota) unless directed by their physician.29
FMT is the only preventive option for rCDI after antibiotic treatment; however, it is not an approved treatment by Health Canada. Health Canada issued interim guidance for FMT that classified it as a biologic drug.30 Its use across Canada is highly heterogenous, with limited access, and it requires significant time from specialists.31
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
Patient input was submitted by the Gastrointestinal Society, the Peggy Lillis Foundation, and the Canadian Digestive Health Foundation for this review. Input from the Gastrointestinal Society was gathered mainly from patient questionnaires and interviews, whereas input from the Peggy Lillis Foundation and Canadian Digestive Health Foundation was collected via emails from patients, social media posts, and personal patient stories. The total number of patients who contributed to the input was not reported. According to the input, CDI is a debilitating condition characterized primarily by severe diarrhea, and often accompanied by symptoms like dehydration, fever, abdominal pain, and fatigue. It can lead to life-threatening complications, including recurrent infections, sepsis, and, in extreme cases, the need for colon removal. Certain patients, such as those aged 65 years or older or individuals on prolonged antibiotics, are at higher risk. The input noted that with the chronic nature and high recurrence rate of CDI, the impact extends beyond physical symptoms, profoundly affecting patients’ mental health, with feelings of shame, anxiety, and even post-traumatic stress disorder. The condition disrupts daily life, requiring isolation to prevent transmission and placing significant burdens on patients, caregivers, and their ability to work or socialize.
The patient input stated that current treatments for CDI include antibiotics like vancomycin, metronidazole, and fidaxomicin. However, metronidazole has limited effectiveness for severe cases and has significant side effects. Despite the availability of drugs, the input noted that such treatments can destabilize gut microbiota, contributing to the cycle of recurrence. The input stated that probiotics and prebiotics may be helpful in improving the balance of the microbiome, but current research has not yet provided evidence of their benefit in CDI management. As such, the input noted, there is a need for treatments that adequately manage symptoms, are curative, and prevent CDI recurrence.
Patients who had experience with the treatment under review reported significant benefits, citing ease of access, minimal side effects, and effective relief from rCDI. Many wish it were offered earlier in their treatment journey. Among the respondents, fecal microbiota (Rebyota) was preferred over vancomycin because of its simpler administration — it is delivered once via enema or colonoscopy without the need for bowel preparation — and its ability to address microbiome dysbiosis, reducing vulnerability to future recurrences. Patients highlighted its simplicity and efficiency, with the procedure taking about 30 minutes and causing only minor side effects, such as slight cramping.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the review of fecal microbiota (Rebyota), a panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations in which there are gaps in the evidence that could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
The clinical experts indicated that most patients with a first CDI episode will respond to antibiotic treatment with vancomycin or fidaxomicin, and noted that rCDI will occur in approximately 15% to 20% of patients after a first CDI episode. According to the literature, however, up to 25% of patients may experience rCDI after antibiotic treatment, and after the second recurrence, the risk of recurrence increases substantially, to up to 50%.27 Indeed, the clinical experts confirmed that patients who experience a first recurrence may have their condition improve spontaneously after treatment with vancomycin or fidaxomicin; however, the natural disease trajectory becomes more complex in patients who have experienced at least 2 recurrences, and the risk of experiencing further recurrences and more severe disease becomes incrementally higher.
The clinical experts indicated that in clinical practice, FMT is considered effective at preventing rCDI in patients who have experienced at least 2 recurrences; however, FMT is not readily available to many patients in Canada. Issues surrounding limited access to FMT are multifactorial, according to the clinical experts. The lack of public or private funding for FMT programs has resulted in a heavy dependence on physicians and research grants. Additionally, the establishment and maintenance of an FMT program requires significant infrastructure and specialized expertise. Finally, there are challenges in setting up and sustaining stool banks for FMT, as criteria to become a stool donor are strict and donors are usually volunteers, which makes them difficult to retain. In addition, the clinical experts made the CDA-AMC clinical review team aware of the possibility that conventional FMT programs could be shut down by Health Canada when a commercial product (i.e., fecal microbiota [Rebyota]) becomes available. Health Canada notified CDA-AMC that clinicians were advised that, although fecal microbiota (Rebyota) has been authorized, it is not yet marketed. Therefore, no changes will be made to the current interim policy outlined in the guidance document: Fecal Microbiota Therapy Used in the Treatment of Clostridioides difficile Infection Not Responsive to Conventional Therapies. Once fecal microbiota (Rebyota) becomes marketed, Health Canada will explore options for transitioning away from the interim policy, with further details to be provided in due course.
Therefore, the clinical experts highlighted the need for accessible, effective, and well-tolerated therapies to prevent rCDI, especially in patients who have experienced at least 2 recurrences despite appropriate antibiotic treatment. Patients with the greatest unmet needs are those who have a multirecurrent condition with a high risk of further recurrences, those who are not able to stop an antibiotic taper or pulse regimen, and those who have a substantially impacted quality of life from the condition.
Place in Therapy
The clinical experts recommended the use of fecal microbiota (Rebyota) in patients who have experienced at least 2 recurrences despite appropriate antibiotic treatment, as is the case for FMT, because the natural trajectory becomes more severe at this stage of the disease.
The clinical experts highlighted the need for patients to first receive an appropriate antibiotic regimen for the treatment of rCDI, namely vancomycin administered in a taper or pulse regimen over several weeks. A prolonged vancomycin taper or pulse regimen is considered the treatment of choice for a second rCDI27 and should be used before the initiation of treatment with fecal microbiota (Rebyota), according to the experts. Although antibiotic therapy is targeted at the acute, active infection, it also negatively impacts the gut microbiome. Fecal microbiota (Rebyota) is expected to address the underlying disease process by repopulating and restoring the composition and diversity of the gut microbiome.
According to the clinical experts, fecal microbiota (Rebyota) is similar to the product that is being administered through conventional FMT. However, an important difference between fecal microbiota (Rebyota) and conventional FMT resides in the number of doses administered per treatment. Whereas fecal microbiota (Rebyota) relies on a single administration as the total regimen for preventing each rCDI, FMT may routinely offer repeated doses over 1 week. With such multidose regimens, experience from clinical practice and data from the literature suggests that FMT may achieve clinical response rates of up to more than 90%.27 Therefore, the clinical experts showed concern regarding the potential lower magnitude of effect with fecal microbiota (Rebyota), as they felt that its single administration may lead to less successful outcomes than a series of doses upfront. In addition, conventional FMT offers flexibility in the mode of administration, as delivery can be delivered by enema, oral capsules, or, in some cases, colonoscopy. There is currently no specific guidance for dosing frequency, volume, or mode of delivery for conventional FMT; protocols vary in FMT clinics across Canada.
However, the main advantages that were noted with fecal microbiota (Rebyota) included the fact that the product administered would be regulated in terms of stability, reliability, and high-quality standards, and that it is expected to remove the onus of managing stool banks from FMT providers. As such, a commercial version of fecal microbiota (Rebyota) can potentially increase access to treatment in Canada. Accessibility to FMT is a major issue, according to the clinical experts, who viewed the use of the single-administration, commercially manufactured enema to be an acceptable trade-off. In addition, as previously mentioned, there is a possibility that conventional FMT programs may be shut down when a commercial product becomes available. Therefore, the experts indicated that fecal microbiota (Rebyota) may cause a shift in the current treatment paradigm through increased availability, provided that health care systems can provide the necessary funding, infrastructure, and resources required for product administration and that there are no interruptions in the supply chain.
Patient Population
Patients who would be suited for treatment with fecal microbiota (Rebyota) are those who have experienced at least 2 recurrences despite having tried an appropriate antibiotic treatment with a prolonged vancomycin taper or pulse regimen. Patients should have typical presentations of multiple episodes of rCDI, have no other gastrointestinal issues, and should be able to retain and have no contraindication to enema infusions. The clinical experts emphasized the importance of ensuring that patients are stable and do not have evidence of ongoing, active inflammation of the bowel when they receive a fecal microbiota (Rebyota) enema.
The clinical experts indicated that they would use fecal microbiota (Rebyota) in patients who have concomitant IBD, a population that was underrepresented in the fecal microbiota (Rebyota) studies. The presence of concomitant IBD poses a challenge because of the vicious cycle of dysbiosis related to IBD and the inability to decrease the inflammatory response to CDI to the desired level. In addition, FMT may trigger IBD flare in some patients. Cautions should also be used when providing fecal microbiota (Rebyota) to patients with severe immunocompromising disease, but the clinical experts did not consider this to be a contraindication. Patients who cannot receive an enema because of anatomic issues should not receive fecal microbiota (Rebyota) and should be referred to a conventional FMT program that uses orally administered capsules.
Assessing the Response to Treatment
The clinical experts indicated that response to treatment is entirely based on clinical assessment, which focuses on self-reported resolution or diminution of symptoms to a level that is acceptable for patients to maintain their quality of life.
Stool testing is not routinely performed or recommended after CDI treatment, as most patients with rCDI remain colonized by C. difficile after recovering. Similarly, the experts indicated that there would be no role for testing stools for C. difficile after fecal microbiota (Rebyota), unless a recurrence is suspected.
The clinical experts highlighted the importance of follow-up after fecal microbiota (Rebyota) administration. Experience from clinical practice suggests that symptoms at 1 month are most predictive of long-term prognosis. The experts expect that not all patients will revert to their pre-CDI baseline bowel habits; the goal is to reduce symptoms to a reasonable level and achieve a better quality of life.
Per the Health Canada indication, a second dose of fecal microbiota (Rebyota) is only recommended in the case of a recurrence. Most recurrences would typically be expected to occur 4 weeks to 8 weeks after treatment. If a recurrence occurs, patients should receive a new antibiotic course of vancomycin, using a taper or pulse regimen, to get immediate symptom relief and reduce active inflammation before any additional administration of fecal microbiota (Rebyota) or FMT is considered. In the context of a conventional FMT program, the clinical experts indicated that after a recurrence, they would not consider administering a second round of single-dose FMT using the same mode of administration. The experts would consider repeating FMT either with a more aggressive treatment approach (e.g., multiple doses) or with a different route of administration (e.g., capsules or colonoscopy) after the vancomycin taper or pulse regimen. This would, however, not apply to fecal microbiota (Rebyota) because the recommended dosage is a single-dose enema.
Prescribing Considerations
The clinical experts suggested that prescribing and treatment administration of fecal microbiota (Rebyota) should be limited to clinicians and health care teams with expertise in the management of patients with rCDI and, ideally, in providing FMT. The experts emphasized the need to optimize the selection of patients who may benefit the most from treatment with fecal microbiota (Rebyota). For example, the experts noted that there can be many issues with diagnosis, specifically, overdiagnosis of rCDI in patients who are colonized with C. difficile and have other conditions that cause diarrhea. In addition, the experts emphasized that both fecal microbiota (Rebyota) and FMT are not risk-free and require a thorough informed consent process. Therefore, providers should be well aware of the risks and alternatives when discussing treatment options with patients. The risks associated with the interventions can include the following.
Acute procedure-related risks: Although enemas are generally gentle procedures, they carry a low risk of perforation related to the introduction of a catheter, especially if the bowel wall is already weakened because of inflammation. This can be minimized by not performing FMT on patients who have evidence of ongoing, active inflammation of the colon. There is also a risk of translocation of bacteria and other microbes into the bloodstream during the procedure, which has the potential of causing bacteremia, particularly in patients with severe immunodeficiency and medically complex conditions.
Risks related to donor stools: There is a risk of transmission of various infections from the stool donor to the receiving patient. The clinical experts indicated that stool donor recruitment in FMT programs is performed under strict criteria. Laboratory and questionnaire screenings occur regularly to ensure that donors are healthy and to test for different pathogens. However, no donor screening program can eliminate the risk of all infections, including emerging infectious diseases that may not yet be discovered.
Long-term, unintended effects of manipulating the gut microbiota: The clinical experts indicated that there is hypothesis-generating evidence surrounding the role of the intestinal microbiome in human health in general. Changing a patient’s microbiome may affect the risk of developing a range of conditions over time, the impact of which is currently unknown.
Fecal microbiota (Rebyota) could be administered in an outpatient or inpatient setting, according to the clinical experts.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
One clinician group, the Canadian Antimicrobial Resistance Alliance, provided input for this review. In the opinion of the clinician group, fecal microbiota (Rebyota) demonstrates efficacy and safety in patients with rCDI infection, offering a first-in-class live therapeutic option. The input noted that fecal microbiota (Rebyota) will improve antimicrobial stewardship. In the opinion of the clinician group, it reduces reliance on prolonged courses of vancomycin or fidaxomicin. The input stated that the treatment under review would be most appropriate for patients who have experienced at least 2 CDI recurrences, particularly for high-risk groups such as hospitalized patients, older adults, and patients with severe disease or immunocompromised conditions.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by for this review are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The PUNCH CD2 and PUNCH CD3 studies were both placebo-controlled. There were no active comparators. The PUNCH CD3-OLS study was a single-arm study. Is placebo the most appropriate comparator? The sponsor states that “antibiotics treat the infection thus are not considered comparators.” Is this accurate? | The clinical experts felt that placebo was appropriate in the context of a clinical trial, as long as patients were receiving an appropriate therapy for the number of recurrences, which could not be confirmed, however, as the PUNCH CD3 study did not report the type of antibiotic regimen used (e.g., whether patients with more than 1 recurrence of CDI received a taper or pulse regimen). |
Current guidelines indicate a second (or subsequent) recurrence should be treated with a tapered vancomycin regimen. Is this what currently done in clinical practice? | The clinical experts indicated that this is what is being done in clinical practice. Shorter courses of antibiotics, even up to 21 days, would not be sufficient to manage multiple episodes of rCDI, according to the experts. |
Considerations for initiation of therapy | |
The PUNCH CD2 study is a phase IIb RCT that enrolled 133 patients with at least 2 recurrences of CDI or 2 episodes of CDI requiring hospitalization. The PUNCH CD3 study is a phase III RCT that enrolled 289 patients with at least 1 recurrence of CDI or 2 episodes of CDI requiring hospitalization. The PUNCH CD3-OLS study is a phase III open-label study that enrolled 698 patients with at least 1 recurrence of CDI or 2 episodes of CDI requiring hospitalization. Canadian, US, and European guidelines state that FMT may be considered at the second recurrence.
| The clinical experts agree that the place in therapy of fecal microbiota (Rebyota) should be aligned with the guidelines, and that the treatment should be available to patients who have experienced at least 2 recurrences. In addition, the clinical experts emphasized that a vancomycin taper/pulse regimen should be used before considering fecal microbiota (Rebyota), as it can resolve recurrences in a majority of patients who experience multiple episodes of rCDI. Also, the administration of stool-derived products, such as fecal microbiota (Rebyota) and FMT, carries some potential risks. Adequate patient selection is considered important. The clinical experts did not consider the history of hospitalization as relevant to decision-making. The main treatment goal is to improve long-term quality of life, which applies to both inpatients and outpatients. The clinical experts noted that patients can be safely managed outside of the hospital setting. |
In the PUNCH CD2 and PUNCH CD3 studies, patients were stratified by prior antibiotic use. Very few patients received fidaxomicin before treatment. The best evidence supports fidaxomicin for the first recurrence of CDI. The sustained clinical cure rate for rCDI after 30 days is approximately 70% with fidaxomicin and approximately 55% with vancomycin. Were participants potentially undertreated before placebo treatment, and does this inappropriately favour the fecal microbiota (Rebyota) product? | According to the clinical experts, although there is opportunity to do better with the early use of fidaxomicin, this reflects the current reality in clinical practice. Fidaxomicin is an expensive drug that is currently not accessible to most patients for the treatment of a first episode or first recurrence of CDI. However, as the PUNCH CD3 study did not report the type of antibiotic regimen used (e.g., whether patients received a taper or pulse regimen), whether patients were receiving an appropriate therapy for the number of recurrences in the study could not be confirmed. |
The treatment success rate is about 75% to 80%, meaning a number of patients will require an additional dose.
| The clinical experts indicated that some conventional FMT programs routinely provide several administrations of FMT per patient as a treatment plan. This is based on prior experience with FMT, which provides evidence of increased effectiveness with the number of doses. The clinical experts expressed concern regarding the potential lower magnitude of effect with the administration of only 1 dose of fecal microbiota (Rebyota) as the total regimen for preventing a rCDI. Indeed, per the Health Canada indication, a second dose of fecal microbiota (Rebyota) is only recommended in the case of a recurrence; however, the clinical experts indicated that in the context of an FMT program, they would not consider administering a second round of single-dose FMT using the same mode of administration after a recurrence. Most rCDI would typically be expected occur 4 weeks to 8 weeks after treatment. If a recurrence occurs, patients should receive a new antibiotic course of vancomycin with a taper or pulse regimen to get immediate symptom relief and reduce active inflammation before any additional administration of fecal microbiota (Rebyota) or FMT is considered. The window outside of which an episode of CDI is considered a new infection would be a matter of clinical judgment. The experts mentioned that an unprovoked recurrence of CDI after 6 months to 12 months may be considered a new episode. The treatment sequence would remain the same. |
Considerations for discontinuation of therapy | |
|
|
Considerations for prescribing of therapy | |
Should fecal microbiota (Rebyota) be prescribed only by an infectious diseases specialist or gastroenterologist? | The clinical experts agreed that the prescribing and administration of fecal microbiota (Rebyota) should be limited to clinicians and health care teams with expertise in the management of patients with rCDI and, ideally, in providing FMT. This will allow optimization of the selection of patients who may benefit the most from treatment with fecal microbiota (Rebyota), with the hope of limiting the overdiagnosis of rCDI in patients who are colonized with C. difficile and have other causes of diarrhea. In addition, the experts emphasized that both fecal microbiota (Rebyota) and FMT are not risk-free and that providers should be well aware of the risks and alternatives when discussing treatment options with patients. |
Fecal microbiota (Rebyota) is administered rectally by a nurse or clinician, so requires clinician time and expertise. No anesthesia or colonoscopy is required. Do you foresee that this will cause challenges for patients living in rural areas? | The clinical experts noted that limiting administration to clinicians and health care teams with expertise in the management of patients with rCDI and in providing FMT may cause challenges for patients living in rural areas at first; however, capacity can be built and the procedure is relatively simple. |
Fecal microbiota (Rebyota) needs to be kept in an ultracold freezer at −60ºC to −90ºC (–76ºF to –130ºF) until its expiration or in a regular fridge for up to 5 days (including the 24-hour thawing period). It must be thawed in the fridge for 24 hours before use. Keeping in mind that the availability of ultracold freezers is limited in the hospital and/or health authority structure and almost nonexistent in community pharmacies, making distribution a challenge, how does the sponsor intend to distribute this product? | The sponsor noted that a PSP will be offered to support specialty pharmacy dispensing, among other services. In association with the PSP, fecal microbiota (Rebyota) will be warehoused and openly distributed by a third-party logistics provider located in Canada. Fecal microbiota (Rebyota) will be stored in an ultracold freezer until a pharmacy places an order. Fecal microbiota (Rebyota) and ancillary supplies (i.e., administration kit) will be shipped directly to the dispensing pharmacy with validated cold chain packaging, based on the pharmacy’s product management capabilities, for just-in-time delivery to the administration site (e.g., hospital or clinic). The validated pack out container can maintain the ultracold temperature for 72 hours. The product is also stable at 2°C to 8°C (36°F to 46°F) for up to 5 days (including the 24-hour thawing period). |
Generalizability | |
The guidelines discuss the potential role of FMT in refractory CDI, but this is a rare situation with limited published evidence. Does the evidence show there is a place for fecal microbiota (Rebyota) in refractory CDI? What is the real-world experience with FMT in patients with refractory disease? | The clinical experts noted that genuine refractory CDI is rare, and explained that what is thought to be a refractory CDI is usually another condition that was misdiagnosed. Evidence regarding the effect of FMT in patients with refractory CDI is limited, and the effect of fecal microbiota (Rebyota) is unknown. |
Care provision issues | |
How soon after administration would diarrhea be considered to have rendered the treatment insufficient? | The clinical experts indicated that this is an area of uncertainty. One clinical expert explained that in their FMT program, the product should be retained in the bowel for a minimum of 20 minutes. However, there is no evidence on the correlation between longer retention and improved outcomes. Minor diarrhea and changes in stool patterns is common immediately after FMT and should be monitored for progression in the subsequent days (beyond 48 hours to 72 hours). Recurrences can occur very soon after FMT (in the first few days) or weeks afterward. It depends on the individual’s pattern of disease and risk factors for recurrence. |
CDI = Clostridium difficile infection; FMT = fecal microbiota transplant; PSP = Patient Support Program; rCDI = recurrent C. difficile infection; RCT = randomized controlled trial.
Clinical Evidence
The objective of this Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of rectally administered fecal microbiota (Rebyota), 150 mL, in the prevention of CDI recurrence in adult patients after antibiotic treatment for rCDI. The focus will be placed on comparing fecal microbiota (Rebyota) to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of fecal microbiota (Rebyota) is presented in 4 sections, with a CDA-AMC critical appraisal of the evidence included at the end of each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected in accordance with the sponsor’s systematic review protocol. CDA-AMC assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. No long-term extension study or indirect evidence was submitted by the sponsor. The last section includes additional studies that were considered by the sponsor to address important gaps in the systematic review evidence.
Included Studies
Clinical evidence from the following is included in the review and appraised in this document:
1 pivotal study or RCT identified in the systematic review
3 additional studies addressing gaps in the evidence.
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of the Study
One study was identified and included in the systematic review. The PUNCH CD3 study was a phase III, multicentre, DB, placebo-controlled, randomized trial designed to evaluate the efficacy and safety of fecal microbiota (Rebyota) in the prevention of recurrences of CDI in adult patients after antibiotic treatment for rCDI. The primary outcome was the recurrence of CDI in the 8 weeks after treatment. The primary efficacy analysis was performed using a Bayesian hierarchical model, which formally incorporated data on the treatment effect of fecal microbiota (Rebyota) from the prior randomized, placebo-controlled, phase IIb PUNCH CD2 study.
At the time of enrolment, patients were to receive antibiotic treatment for rCDI, chosen at the discretion of the investigator. A 24-hour to 72-hour washout period was required at the end of antibiotic treatment, before administration of the study drug. Patients were then randomized in a 2:1 ratio to receive a single dose of either fecal microbiota (Rebyota) or placebo enema at the beginning of the 8-week DB treatment phase. Randomization was performed using an interactive response technology and was stratified by the antibiotic therapy used at screening (vancomycin alone, vancomycin in combination, fidaxomicin, or other). Patients, investigators, and study-site personnel were blinded to the randomization assignment.
Two interim analyses were performed by an independent statistical analysis committee. The trial was to be stopped early for success at either of the interim analyses if the posterior probability of superiority exceeded 0.99943, based on a Pocock group sequential design that controlled the type I error rate, without borrowing, at 0.00125 (1-sided).
After the primary outcome assessment at 8 weeks, patients who had a CDI recurrence could receive an additional OL fecal microbiota (Rebyota) enema. Follow-up continued for a total of 6 months.
The characteristics of the included study are summarized in Table 4.
Table 4: Details of the Study Included in the Systematic Review
Detail | PUNCH CD3 study |
|---|---|
Designs and populations | |
Study design | Pivotal, phase III, prospective, multicentre, DB, randomized, placebo-controlled trial |
Locations | 44 sites in Canada (5 sites) and the US |
Patient enrolment dates | Start date: July 31, 2017 |
Randomized (N) | 289 patients
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 1 blinded fecal microbiota (Rebyota) enema |
Comparator | 1 blinded placebo enema |
Concomitant intervention | Option to receive an open-label fecal microbiota (Rebyota) enema for patients with documented treatment failure at 8 weeks |
Study duration | |
Screening phase | 1 visit Study treatment was administered in the 21 days after the screening visit |
Run-in phase | Antibiotics were administered for a minimum of 10 consecutive days, followed by a 24-hour to 72-hour washout period, before administration of the study treatment |
Treatment phase | 8 weeks |
Follow-up phase | 6 months |
Outcomes | |
Primary end point | Recurrence of CDI in the 8 weeks after blinded treatment |
Secondary and exploratory end points | Secondary:
Safety end points:
Other efficacy end points:
|
Publication status | |
Publications |
Clinicaltrials.gov: NCT03244644 |
AE = adverse event; ATLAS = age, temperature, leucocytes, albumin, systemic antibiotics; CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; DB = double-blind; IBD = inflammatory bowel disease; IBS = irritable bowel syndrome; ICU = intensive care unit; IP = investigative product; rCDI = recurrent C. difficile infection; TEAE = treatment-emergent adverse event; VRE = vancomycin-resistant enterococci.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
The PUNCH CD3 study design is illustrated in Figure 1.
Figure 1: PUNCH CD3 Study Design
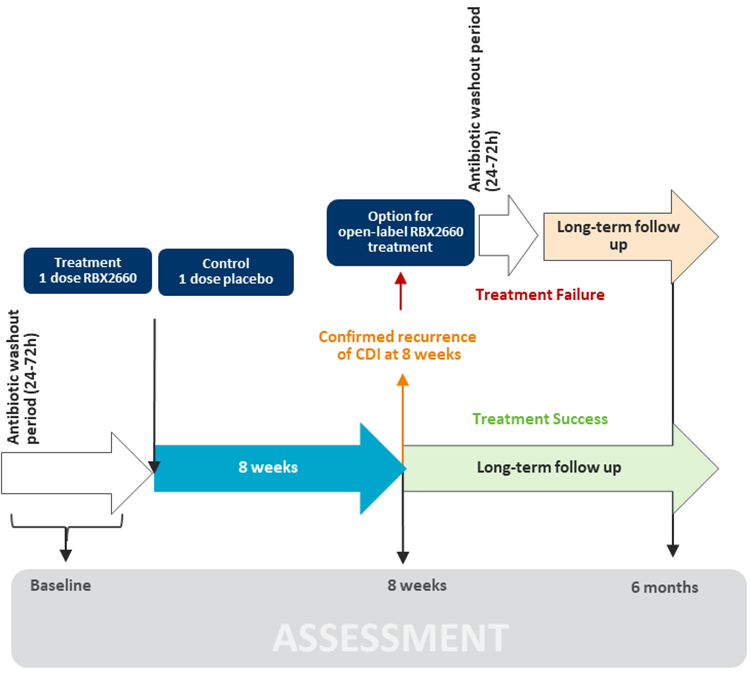
CDI = Clostridium difficile infection; RBX2660 = fecal microbiota (Rebyota).
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the figure are from the sponsor’s Summary of Clinical Evidence.
Populations
Inclusion and Exclusion Criteria
Patients aged 18 years and older were eligible for the PUNCH CD3 trial if they had a documented recurrence of CDI, which was defined in the trial as at least 1 recurrence after a primary episode (with at least 1 round of SOC oral antibiotic therapy) or at least 2 episodes of severe CDI resulting in hospitalization in the previous year. Participants had to have received a positive stool test for the presence of toxigenic C. difficile in the previous 30 days, and had to be taking or to have just been prescribed antibiotics to control CDI-related diarrhea at the time of enrolment.
Patients were excluded from the trial if they had a known history of refractory CDI or experienced continued CDI diarrhea despite appropriate antibiotic treatment. The receipt of prior FMT, CDI vaccine, or monoclonal antibodies was prohibited, as was the use of an antibiotic for a condition other than CDI. Other key exclusion criteria included a wide range of comorbidities — such as IBD (e.g., ulcerative colitis, Crohn disease, or microscopic colitis), IBS, celiac disease, short gut syndrome or motility disorders, current colostomy, recent intra-abdominal surgery, chronic diarrhea or diarrhea caused by something other than CDI — or a compromised immune system (e.g., HIV infection, inherited or primary immune disorder, or immunodeficiency or immunosuppression due to a medical condition or medication). Finally, the concomitant use of systemic steroids exceeding 20 mg of prednisone or prednisone-equivalent a day was not permitted.
Interventions
Patients received either a single-dose fecal microbiota (Rebyota) enema or a matching-administration placebo enema. Both study treatments were administered by a qualified and trained health care professional.
Nondietary probiotics, as well as oral vancomycin, metronidazole, fidaxomicin, rifaximin, nitazoxanide, and IV immunoglobulin, were not allowed during the 8-week DB assessment, unless they were newly prescribed by a treating investigator during the course of the study as a result of rCDI diagnosis.
All patients who had a treatment failure at 8 weeks were eligible to receive an OL dose of fecal microbiota (Rebyota). Treatment failure was defined as the presence of CDI diarrhea in patients with a positive stool test for the presence of a C. difficile toxin in the 8 weeks after administration of a study enema.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 5, followed by descriptions of the outcome measures. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence, any outcomes identified as important to this review by the clinical experts consulted for this review, and input from patient and clinician groups and public drug plans. Using the same considerations, CDA-AMC selected end points that were considered to be most relevant to expert committee deliberations and finalized this list of end points in consultation with members of the expert committee. Recurrence of CDI (treatment success), loss of sustained clinical response, HRQoL (measured using the Cdiff32 Questionnaire), and SAEs were assessed using GRADE. Fecal microbial composition and ICU admissions were included as supportive evidence but were not assessed using GRADE.
Table 5: Outcomes Summarized From the Study Included in the Systematic Review
Outcome measure | Time point | PUNCH CD3 study |
|---|---|---|
Clinical response | ||
Recurrence of CDI (treatment success) | 8 weeks | Primarya,b |
Loss of sustained clinical response | 6 months | Secondarya,b |
HRQoL | ||
Cdiff32 Questionnaire | 8 weeks and 6 months | Exploratorya |
Stool testing | ||
Fecal microbial composition | 8 weeks and 6 months | Exploratory |
Health care resource use | ||
ICU admissions | 8 weeks and 6 months | Exploratory (safety) |
CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; HRQoL = health-related quality of life; ICU = intensive care unit.
aOutcomes included in the GRADE assessment.
bStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Clinical Response
Recurrence of CDI: The primary outcome in the PUNCH CD3 study was the recurrence of CDI within 8 weeks. Recurrence of CDI in the study was synonymous with treatment failure, which was defined as the presence of CDI diarrhea, with a positive stool test for the presence of a C. difficile toxin in the 8 weeks after administration of a study enema.
Patients were followed for recurrence via study follow-up visits at 1 week, 4 weeks, and 8 weeks (± 3 days) and via phone assessments at week 2, week 3, and week 6 (± 3 days) after the administration of a study enema. Recurrences could also be identified during unscheduled visits.
Sustained clinical response: A sustained clinical response in patients who previously achieved treatment success was the secondary outcome in the PUNCH CD3 study. The outcome was defined as the absence of CDI diarrhea for 8 weeks after administration of a study enema, which was also labelled a treatment success for the presenting CDI recurrence, in addition to having no new CDI episodes through 6 months.
Patients were followed for recurrence via phone assessment at 6 months (± 14 days). Recurrences could also be identified via unscheduled visits.
Health-Related Quality of Life
Cdiff32 Questionnaire: HRQoL was assessed in the PUNCH CD3 study using the Cdiff32 Questionnaire, a disease-specific HRQoL assessment tool that includes domains related to physical, mental, and social health.38,39 The self-administered questionnaire comprises 32 items; patients provided responses to each question on a 5-point Likert scale, which indicated how that patient was affected by CDI-related concerns in the previous 7 days. Total and domain scores on the Cdiff32 Questionnaire range from 0 (worst score) to 100 (best score), and responses to the questions are aggregated and rescaled so that lower scores denote a greater negative impact on quality of life. An MCID of 10 points using a distribution-based approach has been suggested in the literature.40 The MCID was estimated for within-group changes, rather than between-group differences. The measurement properties of the Cdiff32 Questionnaire are summarized in Table 7. The recall period of this questionnaire was not reported.
Patients completed the Cdiff32 Questionnaire at screening, and 1 week (± 3 days), 4 weeks (± 3 days), 8 weeks (± 3 days), 3 months (± 14 days), and 6 months (± 14 days) after administration of a study enema.
Fecal Microbiota (Rebyota) Composition
Stool samples were collected by patients at home in prelabelled containers to assess changes in fecal microbial composition 4 weeks, 8 weeks, 3 months, and 6 months after blinded study treatment.
Harms
The safety analysis included AEs, SAEs, AEs leading to study discontinuation, and mortality. In addition, the number of ICU admissions through 8 weeks was reported as a safety outcome. AEs of special interest included septic shock, toxic megacolon, colonic perforation, and emergency colectomy.
Patients were followed for harms outcomes via study follow-up visits at 1 week (± 3 days), 4 weeks (± 3 days), and 8 weeks (± 3 days) after the administration of a study enema and via phone assessments at 2 weeks (± 3 days), 3 weeks (± 3 days), 6 weeks (± 3 days), 3 months (± 14 days) and 6 months (± 14 days). Recurrences could also be identified during enema administration and unscheduled visits.
Table 6: Summary of Outcome Measure and Its Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Cdiff32 Questionnaire score | A 32-item, self-administered, HRQoL questionnaire with domains related to the physical, mental, and social health of patients with CDI.39 | Validity: In the ECOSPOR III trial, among patients with rCDI, correlations with the EQ-5D-5L index score were weak to moderate (r = 0.27 to 0.36). The questionnaire differentiated between age groups (< 65 years vs. ≥ 65 years) and between patients with recurrent vs. nonrecurrent CDI (effect size = 0.38 to 0.43).40 In a study of patients with primary CDI or rCDI, there was a strong correlation with physical and mental components of the SF - 36 (r = 0.67 to 0.91). The questionnaire is more sensitive than the SF - 36 in detecting quality-of-life differences between patients with primary CDI and rCDI.39 Reliability: Good internal consistency was demonstrated in the ECOSPOR III trial and in a study of patients with primary CDI or rCDI (Cronbach alpha > 0.80).39,40 Responsiveness: Improvement from baseline to week 8 was seen in patients who did not experience a recurrence in the ECOSPOR III trial (effect size = 0.75 to 1.02).40 | In the ECOSPOR III trial, among patients with rCDI, an MCID of 10 points was suggested, based on half of the baseline SD.40 The MCID was estimated for within-group changes, rather than between-group differences. |
CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; MCID = minimal clinically important difference; MID = minimal important difference; rCDI = recurrent C. difficile infection; SD = standard deviation; SF-36 = 36-Item Short Form Health Survey; vs. = versus.
Statistical Analysis
A summary of the statistical analyses is presented in Table 7.
Sample Size and Power Calculation
The sample size calculation used a Pocock spending function, taking into account 2 interim analyses for futility, and assumed a treatment success rate of 69% for fecal microbiota (Rebyota) and 47% for placebo. Based on these considerations, a sample size of 240 patients, randomized in a 2:1 ratio to fecal microbiota (Rebyota) or placebo, was determined to provide sufficient statistical power (> 90%, nominal 2.5% type I error rate) to detect a statistically significant effect in the primary end point of CDI recurrence at 8 weeks at either of the 2 interim analyses or at the end-of-trial analysis. Up to an additional 30 patients were enrolled to allow for a 10% loss-to-follow-up rate, for a total of approximately 270 patients.
Statistical Test or Model: Bayesian Borrowing
The primary efficacy analysis of CDI recurrence at 8 weeks was performed using a Bayesian hierarchical model, which formally incorporated data on the treatment effect of fecal microbiota (Rebyota) from the prior randomized, placebo-controlled, phase IIb PUNCH CD2 study. The protocol-specified decision rule was to declare study success if the posterior probability of superiority exceeded 0.99943 at the first 2 interim analyses and exceeded 0.9993275 at the final analysis. This threshold was selected to control the type I error rate, without borrowing at a 1-sided 0.00125 alpha, accounting for multiple interim analyses.18 Additionally, superiority was also evaluated using a lower threshold, set at 0.97706 in the Statistical Analysis Plan, which corresponded to a less demanding 1-sided alpha of 0.025.18 The final secondary threshold used was adjusted to 0.9750338 to reflect the actual number of patients and events observed in the trial.18 This is the relevant threshold used for the treatment comparison. The sponsor reported that this approach was taken in agreement with the FDA during recruitment to the phase III PUNCH CD3 trial, and is consistent with the FDA’s guidance on Complex Innovative Trial Designs for Drugs and Biological Products.41
Details regarding the PUNCH CD2 study can be found in the previous section on Clinical Evidence, which includes additional studies considered to address important gaps in the systematic review evidence. As the PUNCH CD3 study was not originally designed to use a Bayesian hierarchical model to dynamically borrow information from the PUNCH CD2 study, some differences between the protocols for the 2 trials existed. To account for potential heterogeneity between the populations in the 2 trials, a dynamic borrowing approach was used, which aimed to ensure that an appropriate amount of data was borrowed from the PUNCH CD2 study. The dynamic aspect of the borrowing meant that the more similar the response rates were between the 2 trials, the more previous data were borrowed. Differences between the 2 protocols included the following.
Differences in populations: Patients enrolled in the PUNCH CD2 study had to have experienced at least 2 recurrences of CDI, whereas patients in the PUNCH CD3 study had to have experienced at least 1 recurrence, according to the inclusion criteria for the respective trials.
Differences in the intervention received and follow-up duration: The PUNCH CD2 study investigated a dosing regimen in which 2 doses were administered 1 week apart, with a follow-up period from the first dose of 9 weeks instead of 8 weeks.
Differences in the analysis populations: The planned primary analysis for the PUNCH CD2 study was based on the ITT population, which included all randomized patients, regardless of whether they completed their assigned study treatment. In the PUNCH CD3 study, however, the primary analysis was based on the modified intention-to-treat (mITT) population, which included only randomized patients who successfully received blinded treatment and excluded patients who discontinued the study before the primary efficacy end point assessment of treatment success at 8 weeks (if the reason for exit was not related to CDI symptoms).
In the final Bayesian hierarchical analysis of the primary end point, the sponsor addressed these differences by borrowing data from only the 1-dose fecal microbiota (Rebyota) group and the placebo group in the PUNCH CD2 study, as this was the dosing scheme used in the PUNCH CD3 study and is the recommended dose in the proposed indication. The mITT analysis population was used in the Bayesian hierarchical model, and the PUNCH CD3 study definition of the analysis population was applied to the PUNCH CD2 study efficacy data. In addition, the follow-up period in the PUNCH CD2 study was restricted to 8 weeks from the first dose.
Sensitivity Analyses
Analysis population in the Bayesian hierarchical analysis: Sensitivity analyses of the primary end point were performed using alternate analysis populations. The primary efficacy analysis was repeated using the ITT and per protocol (PP) populations; the ITT analysis served as the conservative analysis, as it considered that all patients who exited either study before the 8-week efficacy assessment experienced treatment failures, regardless of treatment failure documentation.
Additional sensitivity analyses of the primary end point were performed using different analysis populations from the PUNCH CD2 study, which included the ITT population and a population of 1 patient who was exposed to fecal microbiota (Rebyota) but did not receive the full enema, to assess the sensitivity of the results to the borrowing of historical data from the previous PUNCH CD2 study.
Testing of the primary end point without Bayesian borrowing: The primary efficacy analysis was repeated without the borrowing of historical data from the PUNCH CD2 study using the mITT, ITT, and PP populations. In addition, alternate testing (i.e., Pearson’s chi-square test), using data only from the PUNCH CD3 study, was used to test the null hypothesis that the response rate in the treatment group is equal to that of the control group. Two-sided 95% CIs for the difference in response rate between arms were calculated using a normal distribution approximation. This sensitivity analysis was conducted using the mITT, ITT, and PP populations.
Additional assessments of the primary outcome using Kaplan-Meier estimates: An analysis of time to CDI recurrence after completion of the assigned blinded treatment was performed using the Kaplan-Meier procedure. The median duration was reported in days and weeks using the ITT, mITT, and PP populations. The Kaplan-Meier estimate of the median time in days and the associated 95% CI were presented for each treatment group. Between-group comparisons were performed using the stratified log-rank test, based on the stratification factor at randomization (antibiotic therapy used at screening). The corresponding hazard ratio of the treatment effect, along with the 95% CI, was calculated using a Cox proportional hazards model, with treatment and the stratification factor at randomization as explanatory variables. Time to CDI recurrence was defined as the number of days from enema administration to first assessment indicating CDI recurrence for patients who were deemed to have a treatment failure. All patients whose assessment of treatment failure was indeterminate or who discontinued the study for any reason before the assessment of efficacy were censored at the last assessment date at or before week 8. For the analysis of time to CDI recurrence within 8 weeks, all patients considered to have a treatment success were censored at the date of their 8-week assessment. For the analysis of time to CDI recurrence through 6 months, all patients considered to have achieved a sustained clinical response were censored at the date of their 6-month assessment.
Additional assessments of the primary outcome using multivariate logistic regression: Sensitivity of the primary efficacy end point was assessed in a multivariate logistic regression that adjusted for possible interactions of the following covariates with treatment group [wording from original source]: age (< 65 years, ≥ 65 years), sex (female, male), race group (white, nonwhite), ethnicity (Hispanic-Latino, not Hispanic-Latino), site geography (outside the US, Eastern US, Southern US, Northern US, Western US), randomization strata (i.e., antibiotic treatment received at the qualifying event), and number of previous episodes of CDI recurrence at baseline. This analysis was performed on all analysis populations that had at least 20 patients in each subgroup available for analysis.
Secondary End Points of the Studies and Multiple Testing Procedure
The secondary efficacy end point was a sustained clinical response throughout the remainder of the 6-month follow-up period. This end point aimed to evaluate whether patients without a recurrence of CDI 8 weeks after fecal microbiota (Rebyota) treatment remained disease-free after 6 months. A hierarchical, closed-testing procedure was used for the primary end point and for the secondary end point of loss of sustained clinical response at 6 months. As such, analysis of the secondary end point was to be performed only if statistical significance was demonstrated for the primary efficacy end point. A Pearson’s chi-square test was used to assess the null hypothesis that the response rate in the treatment group is equal to that of the control group. The secondary end point was tested at a 2-sided significance level of 0.05. Two-sided 95% CIs for the difference in response rate between arms were calculated using a normal distribution approximation. In addition, an analysis of time to CDI occurrence was preformed using the Kaplan-Meier procedure, where between-group comparisons were preformed using the log-rank test.
Data Imputation Methods
No imputations were used in the primary or secondary end point analyses. Patients exiting the study before the 8-week efficacy assessment were excluded from the analysis that used the mITT population (primary analysis population), and were considered to have experienced treatment failure, regardless of treatment failure documentation in the analysis using the ITT population (sensitivity analysis).
A LOCF analysis was used for evaluation of Cdiff32 Questionnaire scores.
Subgroup Analyses
Subgroup analyses were performed for the primary end point based on the following patient characteristics and baseline variables [wording from original source]:
age (< 65 years, ≥ 65 years)
sex (female, male)
race group (white, nonwhite)
ethnicity (Hispanic-Latino, not Hispanic-Latino)
site geography (outside the US, Eastern US, Southern US, Northern US, and Western US)
number of previous episodes of CDI recurrence at baseline
vancomycin use duration for the qualifying CDI episode.
Subgroup efficacy analyses were prespecified and were performed using the Cochran-Mantel-Haenszel test for the mITT, ITT, and PP populations. Whether the comparability of the treatment arms was checked and whether multiplicity was taken into account were not reported.
Table 7: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Recurrence of CDI within 8 weeks of blinded treatment | Bayesian hierarchical model | None | None |
|
Loss of sustained clinical response through 6 months after blinded treatment | Pearson’s chi-square test and Kaplan-Meier procedure | None | None | None |
Cdiff32 Questionnaire scores at screening, at 1-week, 4-week, and 8-week assessment visits, and at 3 months and 6 months | Descriptive | None | LOCF | None |
Patient fecal microbial composition at screening, at 4 weeks and 8 weeks, and at 3 months and 6 months | Descriptive | None | None | None |
CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; ITT = intention to treat; LOCF = last observation carried forward; PP = per protocol.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Analysis Populations
The analysis populations in the PUNCH CD3 study are described in Table 8.
Table 8: Analysis Populations of the PUNCH CD3 Study
Population | Definition | Application |
|---|---|---|
Intention to treat | All randomized patients. | Primary end point as a sensitivity analysis, and secondary end point. |
Modified intention to treat | All randomized patients who successfully received blinded treatment, excluding patients who withdrew before treatment; patients in whom treatment was attempted but not completed; and patients who discontinued the study before evaluation of treatment failure or success for the primary end point (if the reason for exit is not related to CDI symptoms). | Primary, secondary, and exploratory end points. |
Per protocol | All patients who successfully received blinded treatment and were analyzed according to the treatment they received, excluding patients with documented deviations from inclusion or exclusion criteria and patients who exited before the 8-week efficacy evaluation (if the reason for exit was not related to CDI symptoms). | Primary, secondary, and exploratory end points. |
Safety analysis population | The population of randomized patients in whom any blinded treatment was attempted or completed. | Safety end points. |
CDI = Clostridium difficile infection.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Results
Patient Disposition
Of the 320 screened patients in the PUNCH CD3 study, 31 failed screening and 289 were randomized in the study. Historical data were borrowed from ██ patients who participated in the PUNCH CD2 study. A total of 41 patients in the fecal microbiota (Rebyota) group and 24 patients in the placebo group opted to receive an OL second study treatment with fecal microbiota (Rebyota) after a documented treatment failure at 8 weeks.
Overall, discontinuations and reasons for discontinuations appeared to be balanced between treatment groups. Details of patient disposition are presented in Table 9. Only a few patients who received DB study treatment discontinued before the 8-week primary efficacy assessment (3 patients in the fecal microbiota [Rebyota] group and no patients in the placebo group).
Table 9: Summary of Patient Disposition in the PUNCH CD3 Study
Patient disposition | Fecal microbiota (Rebyota) | Placebo |
|---|---|---|
Screened, N | 320 | |
Screening failure, n (%) | 31 (9.7) | |
Reason for screening failure, n (%) | NR | |
Total randomized, N | 289 | |
Randomized in each treatment arm, N | 193 | 96 |
Patients from the PUNCH CD2 study, N | ██ | ██ |
Received double-blind treatment, n (%) | 180 (93.3) | 87 (90.6) |
Not treated, n (%) | 13 (6.7) | 9 (9.4) |
Received an open-label second study treatment with fecal microbiota (Rebyota) after ≥ 8 weeks, n (%) of treated patients | 41 (22.8) | 24 (27.6) |
Discontinued from the study at any time, n (%) | 21 (10.8) | 12 (12.5) |
Reason for discontinuation, n (%) | ||
Adverse event | 2 (1.0) | 0 |
Death | 2 (1.0) | 0 |
Investigator withdrawal | 3 (1.6) | 3 (3.1) |
Lost to follow-up | 3 (1.6) | 1 (1.0) |
Withdrawal by patient | 6 (3.1) | 5 (5.2) |
Other | 5 (2.6) | 3 (3.1) |
Completed the study, n (%) | 159 (82.4) | 75 (78.1) |
Completed the 8-week assessment, n (%) | 177 (91.7) | 87 (90.6) |
Completed the 6-month assessment, n (%) | 126 (65.3) | 53 (55.2) |
ITT population, N | 193 | 96 |
mITT population, N | 177 | 85 |
PP population, N | 167 | 78 |
Safety analysis population, N | 180 | 87 |
ITT = intention to treat; mITT = modified intention to treat; NR = not reported; PP = per protocol.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
The baseline characteristics that are most relevant to this review or that may affect the interpretation of the study results are outlined in Table 10.
The mean (SD) age was ████ ███████ years in patients who received fecal microbiota (Rebyota) and ████ ███████ years in patients who received placebo, with patients ranging in age from ██ ██ ██ years. A total of ██ patients █████ in the fecal microbiota (Rebyota) group and ██ patients █████ in the placebo group were at least 65 years of age. There were more females █████ than males █████ in the study, and most █████ patients were classified as white. The majority of patients in the study █████ received vancomycin alone to treat the qualifying CDI episode. The mean (SD) duration of the qualifying CDI episode was ████ ███████ days in the fecal microbiota (Rebyota) group and ████ ███████ days in the placebo group.
Overall, most baseline characteristics appeared to be balanced between treatment groups in the PUNCH CD3 study; however, there was imbalance between groups in the total number of previous CDI episodes, which was identified as a relevant prognosis factor by the clinical experts. In the PUNCH CD3 study, patients who received fecal microbiota (Rebyota) appeared to have more severe conditions, based on the number of previous CDI episodes (███ of these patients had experienced 2 episodes and ███ had experienced more than 3 episodes) than patients who received placebo (███ of these patients had experienced 2 episodes and ███ had experienced more than 3 episodes).
Table 10: Summary of Baseline Characteristics in the PUNCH CD3 Study ████
Characteristic | Fecal microbiota (Rebyota) N = ███ | Placebo N = ██ |
|---|---|---|
Age, years | ||
Mean (SD) | ████ ████ ███ | ████ ████ ███ |
Median (minimum to maximum) | ████ ████ ███ | ████ ████ ███ |
Age group, n (%) | ||
< 65 years | ████ ████ ███ | ████ ████ ███ |
≥ 65 years | ████ ████ ███ | ████ ████ ███ |
Sex, n (%) | ||
Female | ████ ████ ███ | ████ ████ ███ |
Male | ████ ████ ███ | ████ ████ ███ |
Race, n (%) | ||
American Indian or Alaska Native | ████ ████ ███ | ████ ████ ███ |
Asian | ████ ████ ███ | ████ ████ ███ |
Black or African American | ████ ████ ███ | ████ ████ ███ |
Native Hawaiian or Other Pacific Islander | ████ ████ ███ | ████ ████ ███ |
White | ████ ████ ███ | ████ ████ ███ |
Multiple | ████ ████ ███ | ████ ████ ███ |
Other | ████ ████ ███ | ████ ████ ███ |
Charlson Comorbidity Index at screening | ||
< 3 | ████ ████ ███ | ████ ████ ███ |
≥ 3 | ████ ████ ███ | ████ ████ ███ |
Total number of CDI episodes before receiving DB study treatment, n (%) | ||
2 episodes (1 recurrence) | ████ ████ ███ | ████ ████ ███ |
3 episodes (2 recurrences) | ████ ████ ███ | ████ ████ ███ |
> 3 episodes (> 2 recurrences) | ████ ████ ███ | ████ ████ ███ |
ATLAS score for the most recent qualifying CDI episode | ||
Mean (SD) | ████ ████ ███ | ████ ████ ███ |
Median (minimum to maximum) | ████ ████ ███ | ████ ████ ███ |
Duration of the most recent qualifying CDI episode (days) | ||
Mean (SD) | ████ ███████ | ████ ███████ |
Median (minimum to maximum) | ████ ███████ | ████ ███ ███ |
Antibiotic used at screening (for the treatment of the most recent qualifying CDI episode), n (%) | ||
Vancomycin alone | ████ ████ ███ | ████ ████ ███ |
Vancomycin in combination | ████ ████ ███ | ████ ████ ███ |
Fidaxomicin | ████ ████ ███ | ████ ████ ███ |
Other | ████ ████ ███ | ████ ████ ███ |
C. difficile test results for qualifying CDI episodes, n (%) | ||
C. difficile PCR positive | ████ ████ ███ | ████ ████ ███ |
C. difficile EIA positive | ████ ████ ███ | ████ ████ ███ |
C. difficile LAMP positive | ████ ████ ███ | ████ ████ ███ |
GDH positive | ████ ████ ███ | ████ ████ ███ |
Toxin A and toxin B positive | ████ ████ ███ | ████ ████ ███ |
Other test positive | ████ ████ ███ | ████ ████ ███ |
Hospitalization in relation to the most recent qualifying CDI episode, n (%) | ||
Yes | ████ ████ ███ | ████ ████ ███ |
No | ████ ████ ███ | ████ ████ ███ |
Unknown | ████ ████ ███ | ████ ████ ███ |
Cdiff32 Questionnaire score at screening | ||
Mean (SD) | ████ ████ ███ | ████ ████ ███ |
Median (minimum to maximum) | ████ ████ ███ | ████ ████ ███ |
ATLAS = age, temperature, leucocytes, albumin, systemic antibiotics; CDI = Clostridium difficile infection; Cdiff32 = C. difficile Health-Related Quality of Life; DB = double blind; EIA = enzyme immunoassay; GDH = glutamate dehydrogenase; LAMP = loop-mediated isothermal amplification; PCR = polymerase chain reaction; SD = standard deviation.
Note: Racial categories used in the table are as reported in the original source and may not align with Canada's Drug Agency inclusive language guidelines.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Exposure to study treatment is outlined in Table 11. Data were reported for the safety population (i.e., patients in whom any blinded treatment was attempted or completed).
Of the 267 patients in the safety population who received blinded study treatment successfully in the blinded phase of the study, 180 patients received fecal microbiota (Rebyota) and 87 patients received placebo. In addition, 41 patients (22.8%) in the fecal microbiota (Rebyota) group and 24 patients (27.6%) in the placebo group who were deemed to experience treatment failure during the DB period, based on the 8-week efficacy assessment, opted to receive a second enema during the OL phase (and all of these patients received fecal microbiota [Rebyota]).
Table 11: Summary of Patient Exposure in the PUNCH CD3 Study (Safety Population)
Exposure | Fecal microbiota (Rebyota) N = 180 | Placebo N = 87 |
|---|---|---|
Number of patients receiving only 1 dose of blinded fecal microbiota (Rebyota) or placebo, n (%) | 139 (77.2) | 63 (72.4) |
Number of patients who received open-label fecal microbiota (Rebyota) enemas (i.e., 2 doses of fecal microbiota [Rebyota]; or 1 dose of placebo and 1 dose of fecal microbiota [Rebyota]), n (%) | 41 (22.8) | 24 (27.6) |
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Efficacy
Detailed efficacy results are presented in Table 12 for the primary outcome and in Table 13 for the secondary and exploratory outcomes.
Treatment Response After 8 Weeks
Primary Analysis
The mean success rate after 8 weeks was 70.4% in patients in the fecal microbiota (Rebyota) group and 58.1% in patients in the placebo group, based on posterior estimates from the Bayesian hierarchical model in the mITT population. This primary analysis in the study resulted in a between-group difference of 12.3% (95% CrI, 1.4% to 23.3%). The magnitude of the findings was considered to be clinically meaningful by the clinical experts consulted for this review, based on the point estimate. The posterior probability of superiority was 0.986354, which exceeded the secondary threshold of 0.9750338 that controlled the type I error rate at a 1-sided level of 0.025. This indicates a 98.6% probability of statistical superiority of fecal microbiota (Rebyota), compared to placebo, at a 2-sided alpha level of 0.05.
Sensitivity Analyses
Alternate analysis populations: Sensitivity analyses of the primary outcome, based on alternate analysis populations, yielded consistent results, aligned with those from the main analysis, with a between-group difference of 12.5% (95% CrI, 1.6% to 23.3%) in the ITT population and of 13.7% (95% CrI, 2.4% to 25.1%) in the PP population.
Analysis without Bayesian borrowing: In the sensitivity analysis without Bayesian borrowing, the mean success rate after 8 weeks was 71.2% in patients in the fecal microbiota (Rebyota) group and 62.4% in patients in the placebo group, yielding a between-group difference of 8.8% ████ ██ █████ █████ █████████ in the mITT population. Results obtained in the ITT and PP populations suggested consistent findings. Overall, results aligned with those from the main analysis, suggesting an effect bordering on clinically important, with imprecision related to the potential for little to no difference, based on the lower bound of the 95% CrI or CI.
Time-to-event analysis: In the comparison of fecal microbiota (Rebyota) with placebo for time to CDI recurrence, the hazard ratio was ████ ████ ██ █████ █████ ████████ █████████ in the mITT population. Results for time to CDI recurrence through 8 weeks are presented as Kaplan-Meier curves in Figure 2. ███████ ████████ ██ ███ ███ ███ ██ ███████████ █████████ ██████████ ████████.
Subgroup Analyses
Subgroup analyses identified by the clinical experts that are particularly relevant in the context of rCDI included number of prior CDI episodes (≤ 3 episodes; > 3 episodes) and vancomycin use duration for qualifying CDI episode in patients who received vancomycin alone (≤ 14 days; > 14 days). The between-group difference for treatment success was 5.4% (95% CI, −9.4% to 20.2%; ████████) in patients who had experienced 3 or fewer CDI episodes and 16.1% (95% CI, −5.4% to 37.7%; ████████) in patients who had experienced more than 3 CDI episodes. In patients who received vancomycin alone, the between-group difference was 1.9% (95% CI, −20.3% to 24.0%; ████████) in patients who were treated for 14 or fewer days and 12.8% (95% CI, −3.5% to 29.1%; ████████) in patients who were treated for more than 14 days.
The results of the subgroup analyses were, overall, aligned with those from the main analysis in terms of direction. The magnitude of effect appeared to be greater in patients who had experienced more than 3 CDI episodes and in those who received treatment with vancomycin for more than 14 days; however, the number of patients in each subgroup was small and, therefore, the CIs crossed the null.
Response to OL Fecal Microbiota (Rebyota) Enema
Fecal microbiota (Rebyota) group: Of the 41 patients in the fecal microbiota (Rebyota) group who were deemed to have experienced treatment failure at the primary efficacy assessment at 8 weeks and went on to receive a second, OL enema of fecal microbiota (Rebyota), 22 patients achieved treatment success 8 weeks after the second enema, resulting in a treatment success rate of 53.7%.
Placebo group: Of the 24 patients in the placebo group who were deemed to have experienced treatment failure at the primary efficacy assessment at 8 weeks and went on to receive an OL enema of fecal microbiota (Rebyota), 15 patients achieved treatment success 8 weeks after the second enema, resulting in a treatment success rate of 62.5%.
Sustained Clinical Response After 6 Months
In the mITT population, 116 of 126 patients (92.1%) in the fecal microbiota (Rebyota) group who achieved previous treatment success maintained a sustained clinical response after 6 months, compared with 48 of 53 patients (90.6%) in the placebo group. This resulted in a between-group difference of 1.5% ████ ██ █████ █████ █████████. Results obtained in the ITT and PP populations suggested consistent findings. The magnitude of the between-group difference based on the point estimate was not considered clinically meaningful by the clinical experts consulted for this review.
HRQoL (Cdiff32 Questionnaire)
In the mITT population, nearly all patients in the fecal microbiota (Rebyota) and placebo groups, respectively, provided baseline ██████ ███ ██████ and 8-week ██████ ███ ██████ assessments. At 6 months, assessments were available for a respective █████ and █████ of patients. The mean (SD) change from baseline through 8 weeks in Cdiff32 Questionnaire score was █████ ████████ in patients in the fecal microbiota (Rebyota) group and █████ ████████ in patients in the placebo group, yielding a between-group difference of ████ ████ ██ ██████ ██████. The mean (SD) change from baseline through 6 months was █████ ████████ in patients in the fecal microbiota (Rebyota) group and █████ ████████ in patients in the placebo group, yielding a between-group difference of ████ ███████ ██████. The magnitude of the findings was not considered clinically meaningful by the clinical experts consulted for this review.
The sponsor provided an additional exploratory analysis of HRQoL data for patients who were deemed to be responders at 8 weeks. ██ ████ ███ ████ █████ ██████████ ███ ███ █████████ █████████ ███ █████████████ ███████████ ████████ ██ ███████ ███████ ██████ ███████ ███ ████ ███ ████ ███████ ██████████ ████████ ██ ███ █████ ████████████ ███ ██████████ █████████ ██ █████████ ███████ ███████ ████ ████████ ███ ██████ ███████████ ███ ██████ ██████████ ██ ███ █████████ ██ ██████ ███ ████████ █████████ ██ ███ █████████
Stool Testing (Fecal Microbial Composition)
Fecal sequencing analysis suggested that the microbiome composition of patients experiencing treatment success changed from baseline over all time points (P < 0.001 for within-group change from baseline using the generalized Wald test). Results are presented in Figure 3. The change in microbiome composition after treatment was characterized by an increase in Bacteroidia-class and Clostridia-class bacteria and a decrease in Gammaproteobacteria-class and Bacilli-class bacteria.
Health Care Resource Use
Hospitalizations and ICU Admissions
Both hospitalizations and ICU admissions were reported as harms outcomes in the PUNCH CD3 study. Results are described for patients who were ██████████ ██ ██████ █████████ ████ █████ ██████████ ██████████ ████████ ██ ██████ ███ ████████ ███ ████████ ███ ████ ██████████ ██ ████████
█████ ██ █ ████████ ██ ███ █████ ██████████ █████████ █████ ████████ ████████████████ ███ ██ ████ ██████ ██████ █████████ ██ ███████ ██████████ ███ █ ████████ ██████ ██████ █████████ ██ ██ ██████ ██ ████ █████████ ██████
A total of 2 patients in the fecal microbiota group (Rebyota) required ICU admission during the 8-week follow-up of blinded treatment. Of these, 1 patient experienced septic shock as a major complication of new CDI and underwent emergency colectomy, and another patient was admitted to the ICU because of the severity of a new CDI event. No patient experienced major complications of CDI after OL enema in this treatment group.
Table 12: Summary of Key Efficacy Results in the PUNCH CD3 Study — Primary Outcome (mITT Population)
Variable | Fecal microbiota (Rebyota) N = 177 | Placebo N = 85 |
|---|---|---|
Primary outcome of treatment success after 8 weeks (posterior estimates from the Bayesian hierarchical model) | ||
mITT population, N | 177 | 85 |
Number of patients from the PUNCH CD2 study | ████ | ████ |
Posterior probability of superioritya | 0.986354 | |
Mean success rate (SD) | 70.4% █████ | 58.1% █████ |
95% CrI | █████ ████ | █████ ████ |
Mean treatment effect (SD) | 12.3% █████ | |
95% CrI | 1.4 to 23.3 | |
Sensitivity analyses of the primary outcome (alternate analysis populations) | ||
ITT population, N | ████ | ████ |
Number of patients from the PUNCH CD2 study | ████ | ████ |
Mean success rate (SD) | 69.1% █████ | 56.7% █████ |
95% CrI | █████ ████ | █████ ████ |
Mean treatment effect (SD) | 12.5% █████ | |
95% CrI | 1.6 to 23.3 | |
PP population, N | 167 | 78 |
Number of patients from the PUNCH CD2 study | ████ | ████ |
Mean success rate (SD) | 70.9% █████ | 57.2% █████ |
95% CrI | █████ ████ | █████ ████ |
Mean treatment effect (SD) | 13.7% █████ | |
95% CrI | 2.4 to 25.1 | |
Sensitivity analysis of the primary outcome (without Bayesian borrowing) | ||
mITT population, N | 177 | 85 |
Treatment success, n (%) | 126 (71.2) | 53 (62.4) |
Difference | 8.8 | |
95% CI | −3.4 to 21.1 | |
P valueb | 0.1502 | |
ITT population, N | ████ | ████ |
Patients included in the analysis, N | 180 | 87 |
Treatment success, n (%) | 126 (70.0) | 53 (60.9) |
Difference | 9.1 | |
95% CI | −3.2 to 21.3 | |
P valueb | 0.1390 | |
PP population, N | 167 | 78 |
Treatment success, n (%) | 120 (71.9) | 48 (61.5) |
Difference | 10.3 | |
95% CI | −2.5 to 23.1 | |
P valueb | 0.1051 | |
Sensitivity analysis of the primary outcome (time to CDI recurrence through 8 weeks) | ||
mITT population, N | ██ ██████ | ██ ██████ |
Patients with CDI recurrence, n (%) | ██ ██████ | ██ ██████ |
Time to CDI recurrence in days, median (minimum to maximum) | ██ ██████ | ██ ██████ |
Hazard ratio (95% CI) | ████ ██████ █████ | |
Log-rank P value | ████ ██████ █████ | |
Subgroup analyses of the primary outcome (in the mITT population) | ||
≤ 3 prior CDI episodes (≤ 2 recurrences), N | ██ ██████ | ██ ██████ |
Patients with treatment success, n (%) | ██ ██████ | ██ ██████ |
Difference | ██ ██████ | |
95% CI | ██ ██████ | |
P valueb | ██ ██████ | |
> 3 prior CDI episodes (> 2 recurrences), N | ██ ██████ | ██ ██████ |
Patients with treatment success, n (%) | ██ ██████ | ██ ██████ |
Difference | ██ ██████ | |
95% CI | █████ ████ | |
P valueb | ██ ██████ | |
≤ 14 days of vancomycin use, N | ██ ██████ | ██ ██████ |
Patients with treatment success, n (%) | ██ ██████ | ██ ██████ |
Difference | ██ ██████ | |
95% CI | ██████ ████ | |
P valueb | ██ ██████ | |
> 14 days of vancomycin use, N | ██ ██████ | ██ ██████ |
Patients with treatment success, n (%) | ██ ██████ | ██ ██████ |
Difference | ██ ██████ | |
95% CI | █████ ████ | |
P valueb | ██ ██████ | |
CDI = Clostridium difficile infection; CI = confidence interval; CrI = credible interval; ITT = intention to treat; mITT = modified intention to treat; PP = per protocol; SD = standard deviation.
aThe threshold used to evaluate the estimated posterior probability of superiority was 0.9750338.
bP values are obtained from Pearson’s chi-square test.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 13: Summary of Key Efficacy Results in the PUNCH CD3 Study — Secondary and Exploratory Outcomes (mITT Population)
Variable | Fecal microbiota (Rebyota) N = 177 | Placebo N = 85 |
|---|---|---|
Sustained clinical response after 6 months (secondary outcome in the study) | ||
mITT population, N | 126 | 53 |
Sustained clinical response, n (%) | 116 (92.1) | 48 (90.6) |
Difference | 1.5 | |
95% CI | █████ ████ | |
P valuea,b | █████ ████ | |
ITT population, N | 126 | 53 |
Sustained clinical response, n (%) | 116 (92.1) | 48 (90.6) |
Difference | 1.5 | |
95% CI | █████ ████ | |
P valuea,b | █████ ████ | |
PP population, N | 120 | 48 |
Sustained clinical response, n (%) | 110 (91.7) | 43 (89.6) |
Difference | 2.1 | |
95% CI | █████ ████ | |
P valuea,b | █████ ████ | |
Cdiff32 Questionnaire scores (mITT population) | ||
At baseline | ||
Number of patients contributing to the analysis | █████ ████ | █████ ████ |
Mean score (SD) | █████ ████ | █████ ████ |
At 8 weeks | ||
Number of patients contributing to the analysis | █████ ████ | █████ ████ |
Mean score (SD) | █████ ████ | █████ ████ |
Change from baseline, mean (SD) | █████ ████ | █████ ████ |
Between-group difference, mean (95% CI) | 5.71 █████ ████ | |
At 6 months | ||
Number of patients contributing to the analysis | █████ ████ | █████ ████ |
Mean score (SD) | █████ ████ | █████ ████ |
Change from baseline, mean (SD) | █████ ████ | █████ ████ |
Between-group difference, mean (95% CI) | █████ ████ | |
Cdiff32 = Clostridium difficile Health-Related Quality of Life; CI = confidence interval; ITT = intention to treat; mITT = modified intention to treat; PP = per protocol; SD = standard deviation.
aP values are obtained from Pearson’s chi-square test.
bStatistical testing for the secondary end point was controlled for multiple comparisons.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Figure 2: Kaplan-Meier Curve of ████ ███████████ [Redacted]

CDI = Clostridium difficile infection; mITT = modified intention to treat.
Source: Clinical Trial Report for PUNCH CD3 study.18
Figure 3: Fecal Microbiota (Rebyota) Composition
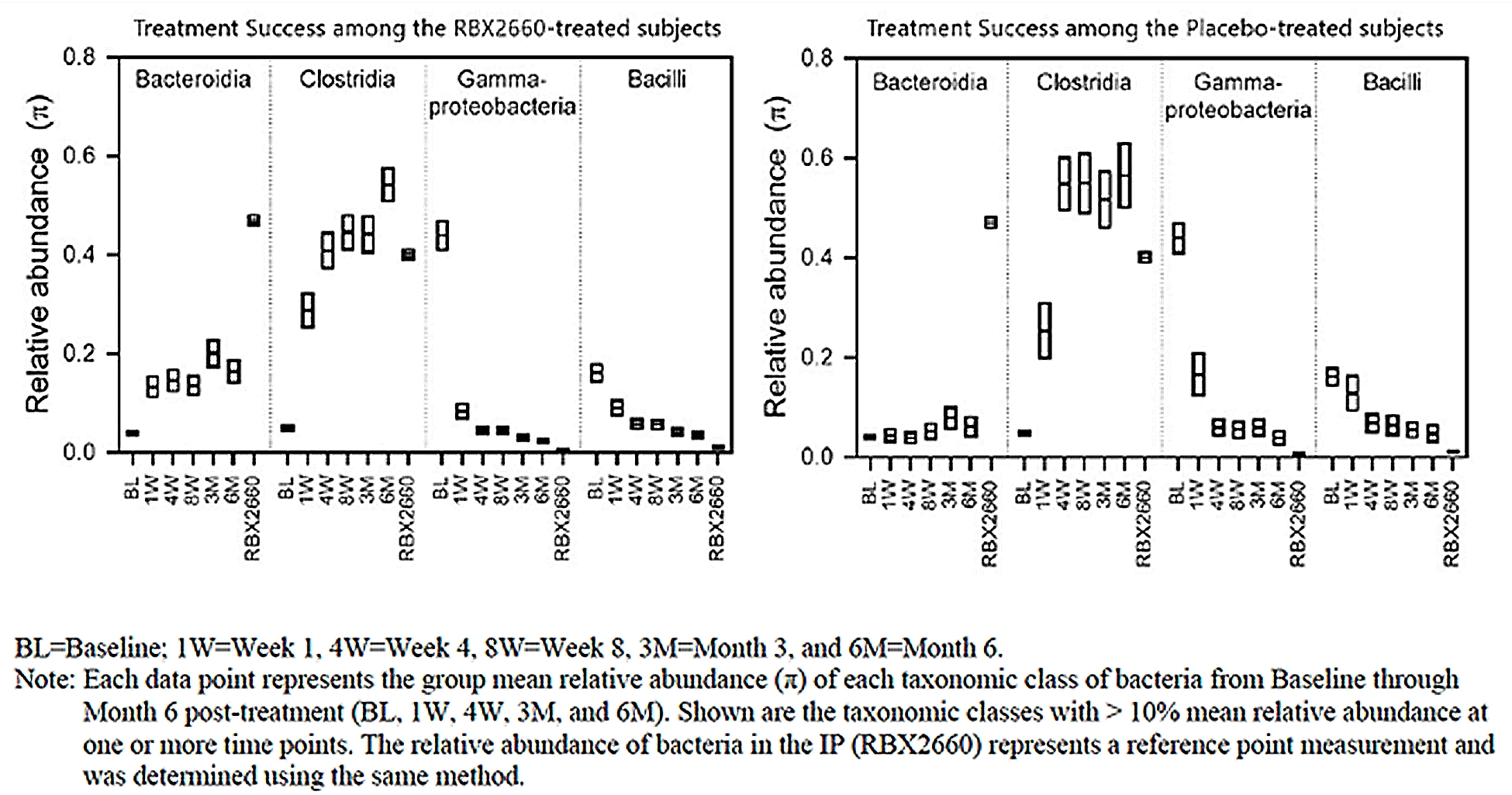
IP = investigational product; RBX2660 = fecal microbiota Rebyota.
Source: Clinical Trial Report for PUNCH CD3 study.18
Harms
Data for harms outcomes included in the review are presented in Table 14 and in Table 15.
Adverse Events
In the PUNCH CD3 study, 70.0% of 180 patients in the fecal microbiota (Rebyota) group and 59.8% of 87 patients in the placebo group experienced at least 1 AE.
Because patients in the study could receive up to 2 fecal microbiota (Rebyota) enemas, AEs were also reported according to the number of fecal microbiota (Rebyota) enemas received. This allowed for the description of potential additional harms in patients who received 2 fecal microbiota (Rebyota) enemas. In patients randomized to placebo who received no fecal microbiota (Rebyota) enema, 36 of 63 patients (57.1%) experienced at least 1 AE. In patients who were randomized to fecal microbiota (Rebyota), 94 of 139 patients (67.6%) who received only 1 blinded fecal microbiota (Rebyota) enema experienced at least 1 AE, as did 32 of 41 patients (78.0%) who received 1 blinded plus 1 OL fecal microbiota (Rebyota) enemas.
The most common TEAEs in the study included diarrhea, abdominal pain, nausea, and abdominal distension.
Serious Adverse Events
A total of 8.3% of patients in the fecal microbiota (Rebyota) group and 6.9% of patients in the placebo group experienced at least 1 SAE.
In patients randomized to placebo who received no fecal microbiota (Rebyota) enema, ██ ██ ████████ ██████ experienced at least 1 SAE. In patients who were randomized to fecal microbiota (Rebyota), ██ ███ ████████ ██████ who received only 1 blinded fecal microbiota (Rebyota) enema experienced at least 1 SAE, as did ██ ██ ████████ ████ ██ who received 1 blinded plus 1 OL fecal microbiota (Rebyota) enema.
The most common treatment-emergent SAEs in the study were not reported.
Withdrawals Due to Adverse Events
███ ████████ ██ ███ █████ ██████████ █████████ █████ ████████████ ███ █████ ███ ██ ████ ███ ██████ ███ ██████████ ██ ████ █████ ███ ████████. No patient in the placebo group withdrew from the study due to AEs.
Mortality
Of the 2 deaths were reported in the study, both were in patients in the fecal microbiota (Rebyota) group. The causes of death were reported as multimorbidity and cardiorespiratory arrest related to pre-existing conditions. None of the events were reported as being related to the study drug or enema procedure, according to the investigators.
Notable Harms
AEs of special interest in the study included septic shock, toxic megacolon, colonic perforation, and emergency colectomy. One patient in the fecal microbiota (Rebyota) group experienced septic shock as a major complication of new CDI and underwent emergency colectomy. The sponsor reported no events of toxic megacolon or colonic perforation. No event of bacteremia, considered a notable harm by the clinical experts consulted for this review, was reported during the study.
Table 14: Summary of Harms Results in the PUNCH CD3 Study (Safety Population)
Adverse events | Fecal microbiota (Rebyota) N = 180 | Placebo N = 87 |
|---|---|---|
Most common adverse events, n (%) | ||
≥ 1 treatment-emergent adverse event | 126 (70.0) | 52 (59.8) |
Serious adverse events, n (%) | ||
≥ 1 treatment-emergent serious adverse event | 15 (8.3) | 6 (6.9) |
Patients who stopped treatment because of treatment-emergent adverse events, n (%) | ||
Patients who stopped the study | █████ | █████ |
Diarrhea | █████ | █████ |
Deaths, n (%) | ||
Patients who died | 2 (1.1) | 0 |
Fatal event of multimorbidity | 1 (0.6) | 0 |
Fatal event of cardiorespiratory arrest | 1 (0.6) | 0 |
Adverse events of special interest, n (%) | ||
Hospitalization for rCDI | █████ | █████ |
rCDI = recurrent Clostridium difficile infection.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 15: Most Commonly Reported Harms Results in the PUNCH CD3 Study (Safety Population)
Adverse events | Fecal microbiota (Rebyota) 1 blinded enema N = 139 (group B) | Fecal microbiota (Rebyota) 1 blinded + 1 OL enema N = 41 (group D) | Placebo N = 63 (group A) |
|---|---|---|---|
Most common adverse events, n (%) | |||
≥ 1 treatment-emergent adverse event | 94 (67.6) | 32 (78.0) | 36 (57.1) |
Diarrhea | 28 (20.1) | 10 (24.4) | 12 (19.0) |
Abdominal pain | 26 (18.7) | 5 (12.2) | 5 (7.9) |
Nausea | 15 (10.8) | 1 (2.4) | 3 (4.8) |
Abdominal distention | 8 (5.8) | 2 (4.9) | 3 (4.8) |
Serious adverse events, n (%) | |||
≥ 1 treatment-emergent serious adverse event | █████ | █████ | █████ |
Most commonly reported serious adverse events | █████ | █████ | █████ |
OL = open label.
Source: Clinical Trial Report for PUNCH CD3 study.18 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
Internal Validity
Study Design
Patients were allocated to treatment groups using appropriate methodology (i.e., an interactive response technology), and randomization was stratified by a relevant and clinically important potential prognostic factor (i.e., antibiotic therapy used at screening).
Patient Population
Overall, most baseline characteristics appeared to be balanced between treatment groups in the PUNCH CD3 study; however, there was an imbalance between groups in the total number of previous CDI episodes (more patients in the fecal microbiota (Rebyota) group than in the placebo group had > 3 prior episodes). This was identified as a relevant prognostic factor by the clinical experts; patients who have fewer recurrences may have their condition improve spontaneously, whereas the natural disease trajectory becomes more severe in patients who have experienced at least 2 recurrences. In the PUNCH CD3 study, patients who received fecal microbiota (Rebyota) appeared to have more severe conditions, based on the number of previous CDI episodes, than patients who received placebo. The direction of the bias is uncertain, although it may have favoured placebo.
Interventions
All patients who experienced treatment failure at 8 weeks were eligible to receive an OL dose of fecal microbiota (Rebyota). This is not expected to affect the assessment of outcomes at 8 weeks; however, it limits the interpretation of outcomes assessed at time points beyond 8 weeks (i.e., sustained clinical response, HRQoL, fecal microbial composition, and health care resource use, all of which were assessed at 6 months). Because a proportion of patients in each treatment group will have received active treatment with fecal microbiota (Rebyota), the direction of the bias would most likely be toward the null.
The relevant concomitant medications reported were not expected to have a clinically meaningful impact on disease course or on treatment response, according to the clinical experts.
Outcome Measures
HRQoL was assessed as an exploratory outcome using the Cdiff32 Questionnaire, which is a validated, disease-specific instrument that is, however, not routinely used in clinical practice. Patient-reported assessment of diarrhea and its impact on HRQoL may be viewed as somewhat subjective, as interpretation of bowel movement habits and diarrhea may be different from 1 individual to another. This could result in a risk of bias; a proportion of patients received OL fecal microbiota (Rebyota) and, thus, were aware of the treatment that they had received, which could have influenced their responses. The MCID of 10 points suggested in the literature for this instrument is uncertain, as it was estimated using a distribution-based approach (half SD approach). Because distribution-based methods rely on statistical properties of the data, they may not always reflect clinically meaningful changes from the perspective of patients. Thus, an anchor-based approach would have been preferred. In addition, this MCID was intended to inform a within-group change; therefore, the appropriateness of applying the same threshold to a between-group difference is unclear.
Statistical Analysis
In the primary efficacy analysis performed using a Bayesian hierarchical model, the posterior probability of superiority exceeded the final secondary threshold, but not the primary, protocol-specified threshold.
No sensitivity analysis of the Bayesian hierarchical approach was performed focusing specifically on the impact of the priors that were used from the PUNCH CD2 study. The best practice in a Bayesian analysis setting would be to rerun the Bayesian model with alternative priors to test the impact on the results. This may be evaluated to a degree in the frequentist sensitivity analysis; however, interpretation of the findings is limited by the fact that several additional factors that differ between a frequentist and Bayesian approach may also potentially explain some of the differences. Results from the frequentist sensitivity analysis, which used no data from the PUNCH CD2 study, were, overall, aligned with those from the Bayesian analysis. Although the results from the frequentist analysis were not statistically significant, estimates from both the Bayesian approach and the frequentist approach appear to suggest an effect bordering on clinically important, with imprecision related to the potential for little to no difference, based on the lower bound of the 95% CrI or CI.
The PUNCH CD3 study used a hierarchical, closed-testing procedure to control the overall type I error rate for the primary end point and for the secondary end point of loss of sustained clinical response at 6 months.
Prespecified subgroup analyses included the number of previous episodes of CDI recurrence at baseline and the duration of vancomycin use for the qualifying CDI episode, which were considered to be particularly relevant to the management of rCDI by the clinical experts. Whether multiplicity was taken into account was not reported. In addition, the subgroups were small, effects were imprecise, and there were no tests for subgroup by treatment interactions. Although the point estimate for the between-group difference appears larger in patients who had more than 3 prior CDI episodes and in those who received treatment with vancomycin for more than 14 days, the 95% CIs for the estimated effects in each group overlap as a result of imprecision. As such, credible conclusions about effect modification cannot be made, and findings from subgroup analyses should be viewed as supplementary.
No imputations were used in the primary or secondary end point analyses. Patients exiting the study before the 8-week efficacy assessment were excluded from the analysis that used the mITT population (primary analysis population), and were considered to have experienced treatment failure regardless of treatment failure documentation in the analysis using the ITT population (sensitivity analysis). Because results using the ITT population were aligned with those of the main analysis using the mITT population, the risk of bias related to missing outcome data for these end points can be considered low. However, an LOCF analysis was used for the evaluation of the Cdiff32 Questionnaire scores. This is not the approach of choice for HRQoL, as this imputation method hinges on the assumption that patients’ HRQoL will remain stable from the time of their last available assessment, which may not be realistic. No information was provided by the sponsor to justify this assumption. As such, there is risk of bias related to missing outcome data for this end point. In addition, the HRQoL analysis population at 6 months had substantial attrition, introducing uncertainty and precluding a definite conclusion.
As with most clinical trials, the studies were not powered to detect infrequent AEs or those with a lag time.
External Validity
Population
Patients with a wide range of comorbidities were excluded from the trial, including IBD (e.g., ulcerative colitis, Crohn disease, or microscopic colitis), IBS, celiac disease, short gut syndrome or motility disorders, chronic diarrhea, and a compromised immune system (e.g., HIV infection, inherited or primary immune disorders, or immunodeficiency or immunosuppression due to a medical condition or medication). Patients were also excluded from the trial if they had a known history of refractory CDI or experienced continued CDI diarrhea despite appropriate antibiotic treatment. Therefore, findings from the PUNCH CD3 study may not be generalizable to these patients. However, the clinical experts noted that fecal microbiota (Rebyota) is likely to be given to patients who have these concomitant conditions in clinical practice, in alignment with the Health Canada indication, despite the fact that the balance between the benefits and risks may differ and the fact that the effectiveness of the intervention has not been studied in these subpopulations.
The Health Canada indication is aligned with the trial and does not require a specific number of recurrences to be eligible to receive fecal microbiota (Rebyota). The clinical experts, however, indicated that patients included in the trial could be considered, overall, to be at a lower risk for CDI recurrences than patients typically seen in clinical practice, who would have the greatest unmet need and be the best candidates to receive fecal microbiota (Rebyota). One of the main reasons identified for this was the inclusion of patients who had only 1 recurrence of a CDI, who accounted for 29.5% of patients in the fecal microbiota (Rebyota) group and 38.5% of patients in the placebo group. Patients who experience a first recurrence may have their condition improve spontaneously and maintain a sustained response. However, the natural disease trajectory becomes more severe in patients who have experienced at least 2 recurrences, and the risk of experiencing further recurrences becomes incrementally higher.
The clinical experts emphasized that fecal microbiota (Rebyota) should be reserved for patients who experienced at least 2 recurrences and who had an unprovoked relapse after a vancomycin taper or pulse regimen. In the PUNCH CD3 study, although the majority of patients received vancomycin for the treatment of the qualifying episode, no details on the specific regimen used were provided (e.g., whether patients received a prolonged taper or pulse treatment). Therefore, it is uncertain whether the population from the study is consistent in that regard with patients who would be candidates to receive fecal microbiota (Rebyota) in clinical practice.
Intervention and Comparator
The administration of fecal microbiota (Rebyota) in the trial was consistent with the Health Canada recommended dosage (i.e., 1 single-dose enema administered 24 hours to 72 hours after the last dose of antibiotics, with a possible additional treatment in the event of a CDI recurrence).
Because the PUNCH CD3 study included a placebo control group, there is no direct evidence comparing fecal microbiota (Rebyota) to other currently used therapies for rCDI, such as a prolonged vancomycin taper or pulse regimen or conventional FMT. This absence restricts the ability of the trial to inform on the effectiveness and safety assessments relative to other treatment options available.
According to the clinical experts, fecal microbiota (Rebyota) can be considered very similar to the product that is being administered through conventional FMT, in that it is a prepared stool-derived product. However, an important difference between fecal microbiota (Rebyota) and FMT resides in the number of doses administered per treatment. Fecal microbiota (Rebyota) is indicated as a single administration. However, the administration regimen in FMT programs may routinely consist of a series of enemas over a 1-week period. With such regimens, experience from clinical practice and data from the literature suggests, FMT may achieve clinical response rates of up to more than 90%.27 In addition, the clinical experts mentioned that in the case of a recurrence, they would consider repeating FMT either with a more aggressive treatment approach or using a different route of administration (e.g., capsules or colonoscopy) after a course of taper or pulse vancomycin. Therefore, the administration regimen in the trial was not representative of the way stool-derived products are currently being used in clinical practice with conventional FMT, according to the experts. There are currently no specific guidance for dosing frequency, volume, or mode of delivery for conventional FMT; protocols vary in FMT clinics across Canada.
The PUNCH CD3 study specified that study treatment was to be administered in the 21 days after the screening visit, which, according to the clinical experts, seemed too short to allow for settling of the acute inflammation in the gut after a CDI episode. Active inflammation in the gut could impede the uptake of the microbiota in the enema, which could have impacted the results in both groups. In clinical practice, clinicians would wait for a longer period of time, sometimes up to 6 weeks or more, before proceeding with administration of the stool-derived enemas in conventional FMT programs.
Outcomes
The primary outcome in the PUNCH CD3 study was the recurrence of CDI within 8 weeks, which was considered appropriate to assess the efficacy of fecal microbiota (Rebyota), according to the clinical experts. The experts considered this to be representative of routine clinical practice in Canada, which focuses on self-reported resolution or a diminution of symptoms to a level that is acceptable for patients to maintain their quality of life. This was also consistent with patient and clinician input, all of which highlighted the importance of preventing CDI recurrences and adequately managing symptoms to improve HRQoL.
Follow-Up
The follow-up duration of 8 weeks for assessment of the primary outcome was considered appropriate by the clinical experts. In clinical practice, symptoms at 1 month are considered the most predictive of long-term prognosis. As such, the experts felt that an 8-week time point would capture most relapses. Nevertheless, although treatment is likely to last in the long-term, evidence beyond the follow-up duration of the studies is limited. The clinical experts indicated that there may be long-term, unintended effects of manipulating a patient’s microbiome, such as affecting the risk of developing a range of conditions over time, the impact of which is currently unknown.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for outcomes considered most relevant to expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.16,17
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word likely for evidence of moderate certainty (e.g., X intervention likely results in Y outcome).
Low certainty: Our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect. We use the word may for evidence of low certainty (e.g., X intervention may result in Y outcome).
Very low certainty: We have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of the effect. We describe evidence of very low certainty as very uncertain.
In accordance with the GRADE approach, evidence from the PUNCH CD3 study started as high-certainty evidence and could be rated down for concerns related to study limitations (which refer to internal validity or risk of bias), indirectness, imprecision of effects, and publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null.
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for fecal microbiota (Rebyota) and placebo. The target of the certainty of evidence assessment was the presence or absence of an important effect, based on thresholds identified in the literature. In the absence of such thresholds, the presence or absence of an important effect was informed by the clinical experts consulted for this review.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
No long-term extension studies were submitted by the sponsor.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
No indirect evidence was submitted by the sponsor or reviewed by the CDA-AMC team.
The sponsor deemed that there were no comparators to fecal microbiota (Rebyota) in Canada for the preventive treatment of rCDI after antibiotic treatment for rCDI.
The appropriateness of this assumption is limited by the fact that patients who experience rCDI in clinical practice would not be left untreated, according to the clinical experts consulted for this review. Most notably, treatment options include long-term vancomycin administered in a taper or pulse regimen, as well as FMT. The CDA-AMC review team acknowledged that the use of FMT is generally restricted to trial settings in Canada; however, Health Canada has exempted the use of FMT to treat patients with CDI who are not responsive to conventional therapies from this restricted use. In addition, there is evidence in the literature pertaining to the effectiveness of various FMT programs,27 although these are not directly funded by CDA-AMC participating drug plans.
As a result, the efficacy and safety of fecal microbiota (Rebyota) compared with any other therapies used in clinical practice to prevent rCDI is unknown.
Studies Addressing Gaps in the Systematic Review Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
An overview of the studies addressing gaps in the systematic review evidence submitted by the sponsor, including a summary of key results, is provided in Table 16.
Table 16: Summary of Gaps in the Systematic Review Evidence
Evidence gap | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Included additional efficacy outcomes: SF-36 and time to recurrence of CDI. Long-term safety outcomes, up to 24 months. | PUNCH CD2 study: A phase II, prospective, multicentre, double-blind, randomized, placebo-controlled trial. | The difference in rate of treatment success between 1 dose of fecal microbiota (Rebyota) and 1 dose of placebo (group C) vs. 2 doses of placebo (group B) was 13.6% ████ ███ ████ ██ ████; P = 0.201). The time by which 25% of patients had a CDI recurrence was 8.0 days in both group B (95% CI, 5 to 16 days) and group C (95% CI, 5 to 32 days). Both group B and group C showed increases in mean SF-36 scores from baseline to week 8. Rates of SAEs were similar in group B and group C (█████ ███ █████), with no relationship to treatment, according to the investigators. Deaths █████ ███ ██████ in both groups, mostly due to pre-existing conditions, with no link to treatment or procedure, according to investigators. |
Included patients with comorbidities commonly seen among patients with CDI, such as IBS, IBD, and immunocompromising conditions. | PUNCH CD3 OLS study: A phase III, prospective, multicentre, OL, single-arm study. | Of the 676 patients in the primary efficacy analysis population (mITT), 499 (73.8%) patients reported treatment success in the 8 weeks after the first dose of fecal microbiota (Rebyota). A total of ███ ███████ patients reported TEAEs after fecal microbiota (Rebyota) administration. TEAEs were mostly mild ███████ to moderate (██████ in severity, and related to pre-existing conditions and/or CDI ██████ ███ █████ respectively). |
Feuerstadt et al. (2023):19 A retrospective, multicentre, OL, single-arm study. | In the primary safety set, 82.8% of treated patients responded at 8 weeks, 88.7% of whom had a sustained response through 6 months. Most TEAEs were mild or moderate in severity. | |
Integrated safety analysis | Lee et al.42 Includes 3 phase II trials (PUNCH CD, PUNCH CD2, PUNCH OL studies) and 2 phase III trials (PUNCH CD3 study and an interim data cut of PUNCH CD3-OLS study) of fecal microbiota (Rebyota). | Across 5 trials, 978 participants received at least 1 dose of fecal microbiota (Rebyota) and 83 participants received placebo only. TEAEs occurred in 60.2% of participants in the placebo group and 66.4% of those in the fecal microbiota (Rebyota) group. Abdominal pain, nausea, and flatulence were more frequent in the fecal microbiota (Rebyota) group. Most TEAEs were mild or moderate and were linked to pre-existing conditions. No infections were traced to fecal microbiota (Rebyota), and life-threatening TEAEs were rare (3.0% of patients). |
CDI = Clostridium difficile infection; IBD = inflammatory bowel disease; IBS = irritable bowel syndrome; mITT = modified intention to treat; OL = open-label; SAE = serious adverse event; SF-36 = 36-Item Short Form Health Survey; TEAE = treatment-emergent adverse event; vs. = versus.
Sources: Clinical Trial Reports for PUNCH CD3 OLS and PUNCH CD2 studies and Feuerstadt et al. (2023).19,43,44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.45
PUNCH CD2 Study
Description of Study
The PUNCH CD2 study was conducted to assess the efficacy and safety of fecal microbiota (Rebyota) and to establish a treatment regimen of fecal microbiota (Rebyota) for subsequent confirmation in the PUNCH CD3 study. The PUNCH CD2 study was a phase II, prospective, multicentre, DB, randomized, placebo-controlled trial and has been summarized to provide evidence on additional efficacy outcomes (i.e., quality of life, assessed with the SF-36, and time to recurrence of CDI), as well as long-term safety outcomes (i.e., follow-up of 24 months for SAEs).
Populations
As in the pivotal trial, the PUNCH CD2 study included adult patients aged 18 years or older with rCDI. The PUNCH CD2 study included patients with more rCDI episodes than the PUNCH CD3 study, which required patients to have had 2 recurrences of CDI and at least 2 rounds of SOC oral antibiotic therapy or at least 2 severe CDI episodes resulting in hospitalization. Overall, both studies included similar inclusion and exclusion criteria, so the study populations were similar, as shown in Table 4.
Interventions
Adult patients with rCDI were randomized in a 1:1:1 ratio to receive a treatment course consisting of 2 doses of fecal microbiota (Rebyota) (group A), 2 doses of placebo (group B), or 1 dose of fecal microbiota (Rebyota) followed by 1 dose of placebo (group C). Randomization was stratified by antibiotic therapy used at screening. After antibiotic therapy for the qualifying CDI episode and a 24-hour to 48-hour antibiotic washout period, patients received blinded treatment. In each treatment group, doses were administered 7 ± 2 days apart. If a patient experienced a confirmed CDI recurrence in the 8 weeks after administration of the first course of treatment, they could choose to receive either SOC treatment or a second OL treatment course of fecal microbiota (Rebyota) of up to 2 doses, administered 7 ± 2 days apart.
Antibiotic therapy before the OL treatment course was at the investigator’s discretion, and was followed by a 24-hour to 48-hour antibiotic washout period, if taken. If a patient received OL fecal microbiota (Rebyota), follow-up requirements restarted from the last OL fecal microbiota (Rebyota) dose, in alignment with the same schedule as required for the blinded part of the trial.
To reflect the Health Canada recommended administration of 1 dose of fecal microbiota (Rebyota), this report will focus on the results of group B and group C in the PUNCH CD2 trial.
Outcomes
In the PUNCH CD2 study, both efficacy and safety were assessed during in-person site visits for symptoms 1 week, 4 weeks, and 8 weeks after the last assigned enema and during weekly (week 2, week 3, week 5, week 6, and week 7) and monthly (month 3, month 6, month 12, and month 24) phone calls; as well, patient diaries were reviewed at baseline and week 1. If a patient received treatment during the OL phase of the study, follow-up visits, phone calls, and completion of a new posttreatment patient diary occurred, in alignment with the same schedule as in the blinded phase.
Treatment Success
Treatment success was defined as the absence of C. difficile–associated diarrhea without the need for re-treatment with C. difficile anti-infective therapy or fecal transplant 56 days after administration of the last assigned study enema. Because the PUNCH CD2 study used a dosing regimen of 2 doses given 1 week apart, the follow-up period after the first dose was effectively 9 weeks. Furthermore, in the PUNCH CD2 study, treatment failure consisted of 4 criteria:
The presence of CDI diarrhea, with or without other CDI symptoms, occurring less than 8 weeks after administration of the last assigned trial dose
A positive stool test for C. difficile
The need for re-treatment for CDI
No other cause for CDI symptoms had been determined.
Some patients were considered to have experienced treatment failure by the study investigator because of a suspected CDI recurrence, despite not meeting all 4 criteria. These patients were classified as having an indeterminate response and were considered to have experienced treatment failure for efficacy analyses. Additionally, some patients were classified as experiencing treatment failure and offered OL treatment after only 1 blinded study treatment. These instances were recorded as protocol deviations, but the patients were still considered to have experienced treatment failure in the efficacy analysis.
Time to CDI Recurrence
Time to CDI recurrence was defined as the number of days from the second enema administration to the first assessment indicating recurrence for patients who met the criteria for failed treatment defined for the primary efficacy end point.
Quality of Life
The SF-36 scale was used to identify changes to quality of life after study treatment. The scale assesses 8 health concepts: physical functioning, bodily pain, role limitations due to physical health problems, role limitations due to personal or emotional problems, emotional well-being, social functioning, energy/fatigue, and general health perceptions. It also includes a single item that provides an indication of perceived change in health. Each item is scored from 0 to 100, with a higher score indicating a more favourable health state. Results were reported for the physical component and mental component summary scores.
Harms
Data related to AEs and SAEs were systematically collected during in-office visits and during phone calls after the last enema for up to 24 months.
Statistical Analysis
To demonstrate 80% treatment success with 2 doses of fecal microbiota (Rebyota) versus 40% with placebo, and to evaluate 1 dose of fecal microbiota (Rebyota) and 1 dose of placebo, 105 patients were required (to provide power of 90% at a type I error of 0.05). An additional 12 patients were enrolled to account for a 10% dropout rate, totalling 117 patients (39 per group). After protocol changes and the addition of antibiotic use as a stratification factor, the randomization schedule was updated, bringing the total to 132 patients (44 per group).
The primary efficacy analysis of group A versus group B and the first 2 secondary efficacy analyses of group C versus group B and group A versus group C were completed on the ITT, mITT, and PP populations (defined in a manner similar to that used in the PUNCH CD3 trial, with ITT being the primary analysis). A hierarchal testing method was used to assess treatment success. Comparisons followed a closed hierarchical testing method at a 2-sided alpha of 0.05. If group A and group B differed significantly, group C was compared to group B and, if significant, group A was compared to group C. However, the null hypothesis was not rejected for the primary efficacy analysis, and the proposed comparison of group C to group B commenced without alpha level adjustment. Patients who did not complete treatment or discontinued before 8 weeks after administration of the last dose were considered to have experienced treatment failure.
Time to CDI recurrence was analyzed using the Kaplan-Meier method, with median duration reported in days (and weeks) for both the ITT and PP populations. Estimates were provided for each scheduled CDI recurrence assessment time point (weekly up to week 8). Group comparisons between group B and group C were conducted using the log-rank test.
For SF-36 end points, changes from baseline were summarized using descriptive statistics by treatment arms using the ITT population. LOCF was used to impute missing postbaseline data.
Results
Patient Disposition
A total of 150 patients were enrolled into the PUNCH CD2 trial, with 133 patients randomized into group A (N = 45), group B (N = 44), and group C (N = 44) (Table 17). Of these, ██ ███████ in group B and ██ ███████ in group C entered the OL period after week 8. A total of ███████ patients in group B and ██ ███████ patients in group C discontinued the study for various reasons, including death █████ ██ ██████, other reasons █████ ██ █████, patient withdrawal █████ ██ █████, loss to follow-up █████ ██ ███████ or investigator withdrawal ███ ██ █████.
Table 17: Patient Disposition in the PUNCH CD2 Study
Patient disposition | Group A (2 doses of fecal microbiota [Rebyota]) | Group B (2 doses of placebo) | Group C (1 dose of fecal microbiota [Rebyota] and 1 dose of placebo) |
|---|---|---|---|
Screened, N | 150 | ||
Screening failure, N | 17 | ||
Randomized, N | 45 | 44 | 44 |
Entered open-label period, N (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Discontinued the study, N (%) | ██ ██████ | ██ ██████ | ██ ██████ |
Reason for discontinuation, N (%) | |||
Death | ██ ██████ | ██ ██████ | ██ ██████ |
Investigator withdrawal | ██ ██████ | ██ ██████ | ██ ██████ |
Loss to follow-up | ██ ██████ | ██ ██████ | ██ ██████ |
Patient withdrawal | ██ ██████ | ██ ██████ | ██ ██████ |
Other | ██ ██████ | ██ ██████ | ██ ██████ |
ITT, N (%) | 45 (100.0) | 44 (100.0) | 44 (100.0) |
mITT, N (%) | 40 (88.9) | 43 (97.7) | 38 (86.4) |
PP, N (%) | 28 (62.2) | 31 (70.5) | 24 (54.5) |
Safety analysis set, N (%) | 42 (93.3) | 44 (100.0) | 42 (95.5) |
ITT = intention to treat; mITT = modified intention to treat; PP = per protocol.
Sources: Clinical Trial Report for PUNCH CD2 study.43 Details included in the table are from the sponsor’s Summary of Clinical Evidence.45
Baseline Characteristics
The demographic characteristics were mostly balanced across treatment groups and were in line with those of patients in the PUNCH CD3 study, as shown in Table 9. Most patients in group B and group C were female (68.2% and 57.1%, respectively), white (█████ and 95.2%, respectively), and used vancomycin at screening (90.9% and █████, respectively). Compared with the PUNCH CD3 study, ████ ████████ ██ █████ ███ ███ ████ ████████████ ███ ████ ███ ███ ██████████ ██ ██████████ ███ ██████████ ██████ ██████ ██████ ██ █████ █ ███████ ██ █ ██ ███████ ███████ █ ███████ ██████████ ██ ████████ ██ █████ ███ ███ █████ ███ ███ ███████████ █ ██ ████ ███████████ ██ ████ ██ ████ ███████████ ███ ███ ████████ ██ █████ ████
Exposure to Study Treatments
All patients in group B (100%; n = 44) and all but 1 patient in group C (███████) received at least 1 enema during the blinded treatment portion of the trial. A total of ██ ███████ patients in group B and ██ ███████ patients in group C received their second blinded dose. During the OL portion, ██ ██ ██ enrolled ███████ patients originally in group B and ██ of ██ ███████ patients originally in group C received 2 fecal microbiota (Rebyota) enemas; the remaining patients received 1 enema.
Efficacy
A summary of efficacy results can be found in Table 18.
Treatment Success
In the ITT population, the treatment success rate 8 weeks after completion of the study treatment was 43.2% (19 of 44 patients) for group B and 56.8% (25 of 44 patients) for group C, with a difference of 13.6% (95% CI, ████ ██ ████; P = 0.201). During the OL phase, the treatment success rate among patients originally randomized to group B and group C was █████ ███ ██ ██ █████████ and █████ ██ ██ ██ █████████, respectively. The combined treatment success of all patients in the OL portion was █████ ███ ██ ██ █████████.
Time to CDI Recurrence
In the ITT population, the time at which ███ of the patients had a recurrence was ███ days in both group B (███ ███ █ ██ ██) and group C (███ ███ █ ██ ██).
Quality of Life
At baseline, all patients in group B and 95% of those in group C completed SF-36 assessments. At week 8, assessments were available for 70% and 73% of patients in group B and group C, respectively. Overall, both group B and group C showed an increase in mean scores for all SF-36 components from baseline to week 8. Between-group differences in the change from baseline were not reported.
Table 18: Summary of Efficacy Outcomes in the PUNCH CD2 Study
Outcome | ITT | mITT | PP | |||
|---|---|---|---|---|---|---|
Treatment success (blinded portion) | Group B (N = 44) | Group C (N = 44) | Group B (N = 43) | Group C (N = 38) | Group B (N = 31) | Group C (N = 24) |
Success, n (%) | 19 (43.2) | 25 (56.8) | 19 (44.2) | 25 (65.8) | 18 (58.1) | 21 (87.5) |
Failure, n (%) | 25 (56.8) | 19 (43.2) | 24 (55.8) | 13 (34.2) | 13 (41.9) | 3 (12.5) |
Differencea (95% CI)b | 13.6 ██████████ | 21.6 ██████████ | 29.4 (7.6 to 51.3) | |||
P valuec,d | 0.201 | 0.051 | 0.017 | |||
Time to CDI recurrencee | Group B (N = 44) | Group C (N = 44) | ||||
Time to recurrence (25th percentile), days (95% CI) | ██████████ | ██████████ | ||||
P valued | ██████████ | |||||
SF-36 (ITT) | Group B (N = 44) | Group C (N = 44) | ||||
Physical component score, mean (SD) | ||||||
Baseline | ██████████ | ██████████ | ||||
Week 8 | ██████████ | ██████████ | ||||
Change from baseline | ██████████ | ██████████ | ||||
Mental component score, mean (SD) | ||||||
Baseline | ██████████ | ██████████ | ||||
Week 8 | ██████████ | ██████████ | ||||
Change from baseline | ██████████ | ██████████ | ||||
Treatment success (open-label portion; ITT) | Group B (N = 24) | Group C (N = 14) | ||||
Success, n (%) | ██████████ | ██████████ | ||||
Failure, n (%) | ██████████ | ██████████ | ||||
CDI = Clostridium difficile infection; CI = confidence interval; ITT = intention to treat; mITT = modified intention to treat; NE = not estimable; PP = per protocol; SD = standard deviation; SF-36 = 36-Item Short Form Health Survey.
Note: Treatment success in the PUNCH CD2 study was defined as the absence of C. difficile–associated diarrhea without the need for re-treatment with C. difficile anti-infective therapy or fecal transplant 56 days after administration of the last assigned study enema. Patients in group B received 1 dose of fecal microbiota (Rebyota) followed by 1 dose of placebo; patients in group C received 2 doses of placebo.
aDifference in the percentage of treatment success between the treatment groups 8 weeks after treatment completion.
bTwo-sided 95% CI using normal approximation for the difference in percentages between treatments.
cThe P value from Pearson's chi-square test is for the difference between the treatment groups with respect to percentage of treatment successes. Indeterminate response was treated as a failure for the purpose of analysis.
dThe null hypothesis was not rejected for the primary analysis and statistical comparisons continued without adjustment for multiple comparisons.
eTime to CDI recurrence is the number of days from the second enema administration to the first assessment indicating recurrence. Patients who did not complete the assigned study treatment were censored at day 0. Patients who discontinued before 56 days after treatment completion were censored at the date of discontinuation. Patients whose treatment was considered a success were censored at day 56.
Source: Clinical Trial Report for PUNCH CD2 study.43 Details included in the table are from the sponsor’s Summary of Clinical Evidence.45
Harms
In the PUNCH CD2 study, 86.4% of patients in group B and 78.6% in group C reported TEAEs during both the blinded and OL periods. From the start of the blinded phase through to 24 months after the last study enema or until OL treatment, █████ of group B and █████ of group C reported TEAEs. Of the most common TEAEs reported during this period (with at least 5% of patients in either arm), the following events were reported more frequently in group C than group B: ██████ █████ ███ ██████ ████████████ █████ ███ ██████ ██████ █████ ███ ██████ ██████████ █████ ███ ██████ ████████████ █████ ███ ██████ ███████ ██████ ███ ██████ █████████ █████ ███ ██████ ████ █████ ███ ██████ ███ ███████ █████ ███ █████. Overall, █████ of group B and █████ of group C reported SAEs, none of which were deemed by the investigators to be related to the treatment. ███ ███████ ██████ ██ █████ █ ███ ████████████ upon CDI recurrence within 8 weeks of blinded treatment and there were no ICU admissions. ████ ██████ ██████ occurred in group B and ███████ in group C, with most attributed to pre-existing conditions, and none were considered by the investigator to be related to the treatment under review or the enema procedure.
Critical Appraisal
The PUNCH CD2 study provided additional data on CDI recurrence, HRQoL assessed by the SF-36, as well as long-term safety data through the OL period, up to 24 months. Although the randomization methods appeared to be appropriate for limiting risk of bias in the randomization process, the small sample sizes in the study groups increased the risk that prognostic balance was not achieved (evidenced by imbalances in groups by sex and prior hospitalization for CDI). As such, it is possible that the observed effects were either overestimated or underestimated. The null hypothesis of the primary outcome of treatment success between group A and group B was not rejected in the primary efficacy analysis population (ITT), and the proposed secondary comparisons commenced without alpha level adjustment, violating the closed hierarchical testing method. In fact, continued testing without adjustments led to an increased risk of false positives in subsequent comparisons, such as for the primary end point in the PP population. The study is limited by the lack of placebo control beyond 8 weeks of treatment; results beyond 8 weeks are confounded by the use of OL fecal microbiota (Rebyota) in the placebo group. In the analysis of SF-36 scores, there is a risk of bias because of missing outcome data. Data were missing for up to ███ of patients across groups at 8 weeks. These data were imputed using the LOCF, a single-imputation method that relies on the assumption that HRQoL remained constant over time after the last available assessment, which is likely not reasonable. In terms of generalizability, patients with immunodeficiency, IBD, IBS, or celiac disease were excluded from the trial, which may raise concerns regarding the potential for pathogen transmission in such patient populations. A large majority of patients ███████ were using vancomycin at screening, which aligns with clinical practice, the clinical experts explained, because fidaxomicin is not commonly used to treat rCDI in Canada. As in the PUNCH CD3 study, the lack of comparative data limited the trial's ability to provide insight into the effectiveness and safety of fecal microbiota (Rebyota) compared to other available treatment options. In the PUNCH CD2 study, patients were required to have at least 2 recurrences of CDI in the trial; as such, the study population reflects that which the clinical experts indicated would be treated with fecal microbiota (Rebyota) in clinical practice.
PUNCH CD3 OLS Study
Description of Study
The PUNCH CD3 OLS study was a phase III, prospective, multicentre, OL, single-arm study. The objective of the study was to evaluate the safety, tolerability, and effectiveness of fecal microbiota (Rebyota) as single and repeat administrations in a population of patients with conditions that were excluded from previous trials. Specifically, this trial included patients with conditions such as IBS, IBD, and immunodeficiency.44
Populations
Key inclusion criteria included:
age 18 years or older.
medical record documentation of either
a current diagnosis or history of rCDI, determined by the treating physician, or
at least 2 previous episodes of severe CDI resulting in hospitalization
currently taking or newly prescribed antibiotics to control CDI-related diarrhea at the time of enrolment
agreement not to take oral vancomycin, metronidazole, fidaxomicin, rifaximin, nitazoxanide, bezlotoxumab, or IV immunoglobulin before the 8-week follow-up assessment, unless newly prescribed by a treating investigator during the course of the trial as a result of rCDI diagnosis.
Key exclusion criteria included:
a known history of refractory CDI
continued CDI diarrhea despite receiving a course of antibiotics prescribed for CDI treatment
receipt of systemic antibiotic therapy for a condition other than CDI
prior participation in a clinical trial in which fecal microbiota (Rebyota) was received
FMT in the previous 6 months
FMT with an associated SAE related to the FMT product or procedure
receipt of the CDI monoclonal antibody bezlotoxumab in the year before trial enrolment
disease symptoms (diarrhea) caused by a confirmed intestinal pathogen other than C. difficile.
Interventions
At the time of enrolment in the PUNCH CD3 OLS study, patients were on active antibiotic treatment or had been prescribed antibiotics to control rCDI symptoms. Therefore, patients who had already completed a prescribed course of antibiotics to treat rCDI could not be considered for enrolment unless they had another recurrence requiring antibiotics.
Patients were treated with OL fecal microbiota (Rebyota), which was administered as a single dose. Trial treatment was completed in the 24 hours to 72 hours after completion of antibiotic treatment (i.e., antibiotic washout). Patients were eligible to receive a second dose of fecal microbiota (Rebyota) if the first dose was deemed to have failed. The use of antibiotics before the second dose was at the discretion of the investigator. If antibiotics were prescribed to control symptoms, the antibiotics were to be discontinued 24 hours to 72 hours before the administration of fecal microbiota (Rebyota).
Outcomes
The primary objective was to evaluate the safety and tolerability of fecal microbiota (Rebyota) in patients with rCDI. The primary safety end point was the number of patients with a TEAE related to treatment and/or to the administration procedure.
Other safety end points included the number of AEs per patient; the timing of attributable TEAEs; the duration of TEAEs; the relatedness of TEAEs; the severity of TEAEs; the causality of TEAEs to fecal microbiota (Rebyota), the administration procedure, C. difficile, or a prior condition; and number of each of the major complications of CDI reported up to 8 weeks after treatment.
Assessment of the secondary objectives included the following efficacy end points:
recurrence of CDI up to 8 weeks after treatment
loss of a sustained clinical response up to 6 months after treatment
change from baseline in Cdiff32 Questionnaire score up to 8 weeks and up to 6 months after treatment.
Statistical Analysis
For continuous variables, descriptive statistics were generated. For discrete and/or categorical variables, the number and percentage of nonmissing patients were generated. Descriptive statistics were provided for all patients.
Assessment of the primary safety end point was performed on all patients exposed to fecal microbiota (Rebyota) (safety population), and included descriptive statistics. The safety population was used to summarize all AE data, unless otherwise specified. Safety data were summarized separately, using the same approach, for the first dose and, if applicable, the second dose.
Assessment of secondary efficacy objectives for the analysis of treatment success included descriptive statistics and was performed on the mITT population, defined as all enrolled patients who successfully received treatment, but excluding patients in whom treatment was attempted but not completed and patients who died, withdrew consent, or discontinued the trial before the 8-week efficacy end point evaluation, if the reason for exit was not related to CDI symptoms. To assess the sensitivity of the end points, efficacy analyses were repeated using the ITT population (i.e., all enrolled patients, excluding those who failed screening) and the PP population (i.e., patients who successfully received treatment and were analyzed according to the treatment they received, excluding patients with documented deviations from the inclusion or exclusion criteria and/or patients who exited the study before the 8-week efficacy evaluation, if the reason for exit was not related to CDI symptoms).
Results
Patient Disposition
Patient disposition in the PUNCH CD3 OLS study is summarized in Table 19. A total of 793 patients were screened, of whom 698 were enrolled and 697 were treated. Nine percent (n = 63) of treated patients discontinued the study, mostly because of loss to follow-up or withdrawal by the patient.
Table 19: Patient Disposition in the PUNCH CD3 OLS Study
Patient disposition | Total |
|---|---|
Screened, N | 793 |
Screening failure, N (%) | 95 (12) |
Enrolled (ITT), N | 698 |
Not treated | 1 (0.1) |
Discontinued the study (ITT), N (%) | ██ █████ |
Reason for discontinuation (ITT), N (%) | ██ █████ |
Adverse event | ██ █████ |
Death | ██ █████ |
Investigator withdrawal | ██ █████ |
Loss to follow-up | ██ █████ |
Withdrawal by patient | ██ █████ |
Other | ██ █████ |
ITT, N | 698 |
mITT, N | 676 |
PP, N | ██ ██████ |
Safety analysis set, N | 697 |
ITT = intention to treat; mITT = modified intention to treat; PP = per protocol.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Baseline Characteristics
Baseline characteristics of patients in the PUNCH CD3 OLS study are summarized in Table 20. The median age was ████ years (range, ██ ██ ██); 69.9% of patients were female; 93.8% of patients were white; 73.0% of patients had at least 3 prior CDI episodes; 84.6% of patients had received vancomycin to treat the CDI; 10.6% of patients had ulcerative colitis, Crohn disease, or IBD; and 20.2% of patients had immunocompromising conditions.
Table 20: Baseline Characteristics in the PUNCH CD3 OLS Study
Characteristics | Fecal microbiota (Rebyota) |
|---|---|
Safety population (N = 697) | |
Demographics | |
Age | |
Median, years (range) ≥ 65 years, n (%) | █████████ 338 (48.5) |
Female, n (%) | 487 (69.9) |
Male, n (%) | 210 (30.1) |
Race, n (%) | |
American Indian or Alaska Native | 1 (0.1) |
Asian | 7 (1.0) |
Black or African American | 21 (3.0) |
Native Hawaiian or Other Pacific Islander | 0 (0.0) |
White | 654 (93.8) |
Other | 10 (1.4) |
Multiple | 4 (0.6) |
Number of previous CDI episodes, n (%)a | |
2 | 186 (26.7) |
3 | 268 (38.5) |
≥ 4 | 239 (34.3) |
Average duration of qualifying CDI episodes | |
Days, mean (SD) | █████████ |
Enrolling diagnostic test, n (%)b | |
PCR | 432 (62.0) |
EIA | 75 (10.8) |
GDH | 50 (7.2) |
Otherc | 65 (9.3) |
Treatment administered for most recent qualifying CDI episode, n (%) | |
Prescribed antibiotics | █████████ |
Othera | █████████ |
Nonea | █████████ |
Most recent antibiotic received, n (%) | |
Vancomycin | 590 (84.6) |
Fidaxomicin | 99 (14.2) |
Rifaximin | 7 (1.0) |
Charlson Comorbidity Index at screening | |
Mean (SD) | █████████ |
< 3, n (%) | 329 (47.2) |
≥ 3, n (%) | 368 (52.8) |
ITT population (N = 698) | |
Inflammatory bowel disease, n (%) | |
Ulcerative colitis | █████████ |
Crohn disease | █████████ |
Inflammatory bowel disease | █████████ |
Immunocompromising conditions, n (%) | |
Malignant tumoursd | █████████ |
Other medical historye | █████████ |
AIDS or HIV | █████████ |
Concomitant immunocompromising medications, n (%)f | |
Corticosteroids | █████████ |
Noncorticosteroids | █████████ |
CDI = Clostridioides difficile infection; EIA = enzyme immunoassay; GDH = glutamate dehydrogenase; ITT = intention to treat; PCR = polymerase chain reaction; SD = standard deviation.
aNumber of CDI episodes recorded was incomplete for 4 participants.
bEnrolling tests could be used in combination.
cCategory other for enrolling diagnostic tests included medical record documentation of toxin A and toxin B and loop-mediated isothermal amplification (LAMP).
dStandardized Medical Dictionary for Regulatory Activities (MedDRA) query of malignant tumours.
eOther medical history includes the preferred terms of end stage renal disease, renal failure, asplenia, HIV, and HIV infection; the high-level term of hemoglobinopathies congenital; and the high-level group term of immunodeficiency syndromes.
fImmunocompromising medications included corticosteroids and systemic immunosuppressive medications.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Exposure to Study Treatments
Of the 697 patients in the safety population, all 697 (100%) patients successfully received 1 dose of fecal microbiota (Rebyota). An additional 121 patients successfully received a second dose, for a total of 818 doses administered during the trial.
Efficacy
Treatment Success
Treatment success was defined as the absence of CDI diarrhea in the 8 weeks after completion of the first dose. Of the 676 patients in the primary efficacy analysis population (mITT), 499 (73.8%) patients were reported to have treatment success in the 8 weeks after the first dose of fecal microbiota (Rebyota) (Table 21). Similar treatment success rates were observed for most demographic subgroups, including sex, race, ethnic group, site geography, number of prior CDI episodes, and Cdiff32 Questionnaire score; however, the treatment effect appeared to be of a smaller magnitude in patients aged65 years and older.
Table 21: Treatment Success in the 8 Weeks After the First Treatment in the PUNCH CD3 OLS Study
Outcome | ITT N = 698 | mITT N = 676 | PP N = 634 |
|---|---|---|---|
Treatment success, n (%) | █████████ | 499 (73.8) | █████████ |
Treatment failure, n (%) | █████████ | 151 (22.3) | █████████ |
Indeterminate, n (%) | █████████ | 26 (3.8) | █████████ |
ITT = intention to treat; mITT = modified intention to treat; PP = per protocol.
Notes: Percentage is calculated using the number of patients in the column as the denominator.
The treatment outcomes were based on Endpoint Adjudication Committee determination.
Treatment success is defined as the absence of CDI diarrhea up to 8 weeks after completion of the first dose. Treatment failure is defined as the presence of CDI diarrhea in the 8 weeks after administration of a dose, which includes a positive stool test for C. difficile (determined by the central laboratory). If neither of the definitions was met, the treatment outcome was considered indeterminate.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
In the mITT population, a total of 121 patients experienced a recurrence of CDI after the first dose and received a second dose. Of those 121 patients, 67 patients (55.4%) achieved treatment success (Table 22).
Table 22: Treatment Success in the 8 Weeks After the Second Treatment in the PUNCH CD3 OLS Study
Outcome | ITT N = 698 | mITT N = 676 |
|---|---|---|
Patients who received 2 doses of fecal microbiota (Rebyota), N | ████████ | 121 |
Treatment success, n (%) | ████████ | 67 (55.4) |
Treatment failure, n (%) | ███████ | 29 (24.0) |
Indeterminate, n (%) | ███████ | 25 (20.7) |
ITT = intention to treat; mITT = modified intention to treat.
Notes: Percentage is calculated using the number of patients who received 2 doses of fecal microbiota (Rebyota) as the denominator.
Treatment success is defined as absence of CDI diarrhea up to 8 weeks after completion of the second dose.
The treatment outcomes are based on the investigator's determination.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Sustained Clinical Response
Among patients who completed the 6-month follow-up, the sustained clinical response rate of fecal microbiota (Rebyota) among patients who previously achieved treatment success was 91.0% (Table 23).
Table 23: Sustained Clinical Response Rate up to 6 Months After the First Dose in the PUNCH CD3 OLS Study
Response | ITT N = 698 | mITT N = 676 | PP N = 634 |
|---|---|---|---|
Patients with treatment success in the 8 weeks after the first dose, N | ████████ | 499 | ████████ |
Sustained clinical response, n (%)a | ████████ | 454 (91.0) | ████████ |
ITT = intention to treat; mITT = modified intention to treat; PP = per protocol.
Note: Percentage is calculated using the number of patients with treatment success in the 8 weeks after the first treatment dose as the denominator.
aSustained clinical response is defined as treatment success for the presenting CDI recurrence, with no new CDI episodes for more than 8 weeks after completion of the first dose during the 6 months of follow-up.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
HRQoL (Cdiff32 Questionnaire Score)
The mean (SD) change from screening to postdose assessment in Cdiff32 Questionnaire scores, ██████████████████████████ ███████████████ (Table 24).
Table 24: Summary of Cdiff32 Questionnaire Scores by Visit After the First Dose in the PUNCH CD3 OLS Study
Visit | mITT (N = 676) | ||
|---|---|---|---|
Screening | Postdose | Change | |
Screening | |||
N | ███████████ | ███████████ | ███████████ |
Mean (SD) | ███████████ | ███████████ | ███████████ |
First dose at week 8 | |||
Na | ███████████ | ███████████ | ███████████ |
Mean (SD) | ███████████ | ███████████ | ███████████ |
First dose at month 6 | |||
Na | ███████████ | ███████████ | ███████████ |
Mean (SD) | ███████████ | ███████████ | ███████████ |
Cdiff32 = Clostridioides difficile infection Health-Related Quality of Life; mITT = modified intention to treat; SD = standard deviation.
aN is the number of patients with both screening and postdose measurements.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Harms
A total of 697 patients were included in the safety population. Of these, ██████████ patients reported TEAEs after fecal microbiota (Rebyota) administration. TEAEs were ██████████████████████████████████ in severity and related to pre-existing conditions and/or CDI (█████████████ respectively) (Table 25 and Table 26).
Table 25: Summary of Harms in the PUNCH CD3 OLS Study (Safety Population)
TEAEs during the overall trial period, n (%) | Total (N = 697) |
|---|---|
All TEAEs | █████████ |
TEAEs by maximum severity | |
Mild | █████████ |
Moderate | █████████ |
Severe | █████████ |
Potentially life-threatening | █████████ |
TEAEs leading to patient withdrawal from the trial | █████████ |
TEAEs leading to death | █████████ |
All serious TEAEs | █████████ |
Serious TEAEs by maximum severitya | |
Mild | █████████ |
Moderate | █████████ |
Severe | █████████ |
Potentially life-threatening | █████████ |
Serious TEAEs leading to patient withdrawal from the trial | █████████ |
Serious TEAEs leading to death | █████████ |
TEAE = treatment-emergent adverse events.
Notes: Percentage is calculated using the number of patients in the column heading as the denominator.
TEAEs are defined as any adverse events occurring on or after the first dose.
aBoth patients and events are by maximum severity per patient.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Table 26: Details of Harms in the PUNCH CD3 OLS Study (Safety Population)
MedRA system organ class and preferred term, n (%) | Within 8-week follow-up period N = 697 | After 8-week follow-up period N = 697 |
|---|---|---|
Most common TEAEs occurring in ≥ 5% of patients within and after 8 weeks of the first dose | ||
TEAEs | █████ | █████ |
Gastrointestinal disorders | █████ | █████ |
Diarrhea | █████ | █████ |
Abdominal pain | █████ | █████ |
Nausea | █████ | █████ |
Abdominal distension | █████ | █████ |
General disorders and administration-site conditions | █████ | █████ |
Infections and infestations | █████ | █████ |
Investigations | █████ | █████ |
Nervous system disorders | █████ | █████ |
Most common TEAEs occurring in ≥ 5% of patients within and after 8 weeks of the second dose | ||
TEAEs | █████ | █████ |
Gastrointestinal disorders | █████ | █████ |
Diarrhea | █████ | █████ |
Nausea | █████ | █████ |
Abdominal pain | █████ | █████ |
General disorders and administration-site conditions | █████ | █████ |
Infections and infestations | █████ | █████ |
Urinary tract infection | █████ | █████ |
Investigations | █████ | █████ |
Metabolism and nutrition disorders | █████ | █████ |
Musculoskeletal and connective tissue disorders | █████ | █████ |
Nervous system disorders | █████ | █████ |
Vascular disorders | █████ | █████ |
MedDRA = Medical Dictionary for Regulatory Activities; TEAE = treatment-emergent adverse events.
Notes: Percentage is calculated using the number of patients in the column heading as the denominator.
TEAEs are defined as any adverse events occurring on or after the first dose.
TEAEs counted in the second treatment period are reported with an onset date on or after the date of the second dose during the 8-week time frame.
Source: Clinical Trial Report for PUNCH CD3-OLS study.44 Details included in the table are from the sponsor’s Summary of Clinical Evidence.
Critical Appraisal
The single-arm nature of the PUNCH CD3 OLS study does not allow for causal conclusions to be drawn regarding the efficacy or safety of fecal microbiota (Rebyota). Given the absence of a randomized comparator, it is not possible to determine the extent to which the observed responses can be attributed to fecal microbiota (Rebyota) or to the natural history of disease. A patient’s awareness of the treatment received can introduce bias in outcomes such as HRQoL and subjective harms outcomes. Although the selection criteria allowed for a broader population, the number of patients with Crohn disease, ulcerative colitis, IBD, and immunodeficiency was small. Therefore, the generalizability of the findings to patients who have these coexisting comorbidities is uncertain.
Feuerstadt et al. Study
A retrospective study by Feuerstadt et al. (2023)19 assessed the efficacy and safety of fecal microbiota (Rebyota) administered to adult patients with rCDI, using broad eligibility criteria to reflect real-world conditions. The study provides evidence on the effectiveness and safety of fecal microbiota (Rebyota) in patients with common rCDI comorbidities, such as IBS, IBD, and immunodeficiency.
Populations
The study enrolled adult patients with rCDI who received fecal microbiota (Rebyota) across 5 study sites in the US from November 2015 to September 2019. Eligibility was determined by the treating physician, in accordance with guidance from the FDA enforcement discretion policy46 to include patients with CDI that is unresponsive to standard therapies. A C. difficile lab diagnosis was not required, and there were no exclusion criteria. The treating physician decided on whether to administer 1 or 2 doses of fecal microbiota (Rebyota) before the intervention. Patients with rCDI after the first dose could receive a second dose. The maximum number of doses per patient was 4 (2 treatment courses of 2 doses each).
Outcomes
The primary end point was the number of patients with TEAEs, defined as AEs that occurred after treatment with fecal microbiota (Rebyota). Other safety end points included TEAEs of special interest, the relatedness and severity of TEAEs, and the onset or worsening of chronic conditions. Secondary objectives assessed the efficacy of fecal microbiota (Rebyota) in preventing rCDI in the 8 weeks after treatment and the sustained response up to 6 months. Treatment success was defined as the absence of CDI recurrence in the 8 weeks after administration of fecal microbiota (Rebyota). Sustained clinical response was defined as no documented on-study CDI event in the 6 months after the final dose of fecal microbiota (Rebyota) administered for the qualifying CDI event.
Statistical Analysis
Patient data were retrospectively captured by site staff chart review and recorded using an electronic database using a prespecified protocol. The FAS (N = 94) comprised all enrolled patients that met the eligibility criteria. The PSS (N = 64) included patients who were naive to fecal microbiota (Rebyota) treatment and who had continuous medical records for 6 months after treatment administration.
Results
In the FAS, the mean age was 59.8 years (SD = 20.3 years), 44.7% of patients were aged 65 years or older, and 72.3% were female. A total of 47.9% of patients had a comorbid condition, with the most common including gastroesophageal reflux disease (47.9%), IBS (17.0%), diverticulitis (14.9%), and gastritis (11.7%).
In the FAS, 66 of 94 patients (70.2%) achieved treatment success at week 8 after their first fecal microbiota (Rebyota) treatment, with 74.4% and 66.7% of patients who received 1 or 2 doses, respectively, experiencing treatment success at week 8. Of the 66 patients who experienced treatment success at week 8, 58 (87.9%) showed a sustained clinical response for 6 months after treatment. In the PSS, 53 of 64 patients (82.8%) experienced treatment success, of whom 47 (88.7%) demonstrated a sustained clinical response at 6 months.
In the PSS, 40 of 64 patients (62.5%) experienced any TEAE, 17.2% of which were deemed to be related to fecal microbiota (Rebyota) and 4.7% of which were deemed related to the procedure, according to the investigators. The most common TEAEs included abdominal pain (14.1%), diarrhea (14.1%), and urinary tract infection (10.9%). A total of 12.5% of patients experienced an SAE, and 1 patient (1.6%) died during the study analysis period from multiorgan failure; none of these events were deemed to be related to the treatment or procedure, according to the investigators.
Critical Appraisal
The retrospective analysis evaluated a patient population with broad eligibility criteria, which included patients with various comorbidities and immunocompromising conditions. As only a publication was provided for this retrospective study, the review team was unable to perform a thorough evaluation of the methods and the reporting of the analyses. The reporting of harms and subjective measures (such as symptoms) may be biased by patient knowledge of the treatment received. The noncomparative nature of the study design does not allow for causal conclusions to be drawn regarding the efficacy and harms of fecal microbiota (Rebyota). Patients were eligible to receive up to 4 doses of fecal microbiota (Rebyota) over the course of 2 treatments, which is more than the Health Canada recommendation of 1 dose of fecal microbiota (Rebyota). As such, the results may not be generalizable to the Health Canada recommended dose.
Lee et al. Study
Description of Study
The summary by Lee et al.42 included pooled safety data from 5 prospective studies of patients exposed to at least 1 study treatment, including 3 phase II studies (the PUNCH CD, PUNCH CD2, and PUNCH CD3 OLS studies), and 2 phase III studies (the PUNCH CD3 study and an interim data cut of the PUNCH CD3-OLS study). These studies evaluated the cumulative safety of fecal microbiota (Rebyota) using standardized manufacturing practices and evolving pathogen screening to ensure safety.
Population
All trials enrolled adults aged at least 18 years with rCDI who had received antibiotics for their CDI episode before study treatment. Dosing regimens varied among trials, with patients receiving either a single dose or 2 doses of fecal microbiota (Rebyota) and/or placebo, administered 7 ± 2 days apart. Four trials allowed OL treatment with fecal microbiota (Rebyota) if a CDI recurrence was confirmed during the 8 weeks after the initial treatment course. The full summary population comprised 978 patients who received at least 1 dose of fecal microbiota (Rebyota) and 83 patients who received only placebo. At baseline, the pooled trial data showed that a higher proportion of patients in the fecal microbiota (Rebyota) group were aged 65 years or older than in the placebo group (48.2% versus 37.3%). Additionally, more patients in the fecal microbiota (Rebyota) group (78.0%), the placebo plus fecal microbiota (Rebyota) group (83.3%), and the fecal microbiota (Rebyota) plus OL fecal microbiota (Rebyota) group (79.6%) had experienced 3 or more CDI episodes before trial entry than in the placebo group (68.7%).
Outcomes
The primary outcome was the number of patients who experienced a TEAE related to fecal microbiota (Rebyota) and/or the administration procedure that occurred on or after the day of treatment. Additional safety objectives included the number of AEs per patient, the occurrence of TEAEs of special interest (per the study definition), the relation between TEAEs and the treatment, the severity of TEAEs, and the onset of new chronic conditions or the worsening of existing conditions after treatment.
Results
The most common TEAEs across all treatment groups and courses were in the gastrointestinal disorders system organ class, and included diarrhea, abdominal pain, and nausea. Most of the events were mild or moderate in severity. Serious TEAEs were similar in the placebo (60.2%) and fecal microbiota (Rebyota) 1-dose (66.4%) groups, but higher in the blinded or any fecal microbiota (Rebyota) groups (68.8%). The majority of serious TEAEs were unrelated to fecal microbiota (Rebyota) treatment. A total of 18 deaths were reported in the clinical trials, all in patients who received fecal microbiota (Rebyota), although none were considered to be related to the treatment.
Critical Appraisal
Evidence from the pooled safety data suggests acceptable tolerability of fecal microbiota (Rebyota) in a diverse cohort of patients from 5 prospective trials, although a small proportion received placebo only (N = 83). The long-term harms data from these trials are consistent, with gastrointestinal disorders being the most common TEAE. However, it is important to note that the pooled data were not designed to assess the safety of fecal microbiota (Rebyota) in a statistically rigorous manner. Limitations include potential heterogeneity in the study protocols, variations in dosing regimens, and differences in patient populations that may have affected the internal validity of the safety results. Furthermore, the OL treatment allowed in some trials could introduce bias, particularly in the reporting of subjective AEs if participants believed the treatment was beneficial. Additionally, the pooling of data from multiple trials may obscure specific interactions or effects evident in individual studies, impacting the overall interpretation of safety outcomes.
Khanna et al. Study
The sponsor provided additional data from a retrospective cohort study by Khanna et al. (2025).20 In this study of 196 adults in the US with rCDI who received fecal microbiota (Rebyota) between July 2023 and August 2024, the overall treatment success rate at 8 weeks was 83%. In the overall population, most patients (n = 136) had experienced at least 3 prior CDI recurrences. Success rates were consistent in the subgroups of the number of prior recurrences. The study, however, had important limitations. Baseline patient characteristics were limited, with no detailed information on the exact number of prior CDI episodes, comorbidities, or the antibiotic regimen used other than the product name, preventing the assessment of their impact on outcomes. The analyses did not adjust for confounding factors, lacked a control arm, and may have been subject to selection bias because of the nonrandomized study design. Differences in health care access and the predominance of treatment in infusion centres in the southern and western US may further limit the generalizability of the findings. As such, the results should be considered to be supportive evidence.
Discussion
Summary of Available Evidence
One study was identified in systematic review: the PUNCH CD3 study (n = 289), which was a phase III, multicentre, DB, placebo-controlled, randomized trial designed to evaluate the efficacy and safety of fecal microbiota (Rebyota) in preventing recurrences of CDI in adult patients after antibiotic treatment for rCDI. The primary outcome was the recurrence of CDI at 8 weeks.
Input from clinical experts suggested that findings from the PUNCH CD3 trial may be generalizable to patients considered, overall, to be at a lower risk for CDI recurrences than patients typically seen in clinical practice who would have the greatest unmet need and be the best candidates to receive fecal microbiota (Rebyota). This was based on the low number of previous CDI recurrences in the trial, as well as on the exclusion of patients with relevant comorbidities. In addition, it is unknown whether patients in both groups previously received an appropriate antibiotic regimen of vancomycin taper or pulse for the treatment of the qualifying CDI episode. The relevance of using placebo as a comparator is, therefore, uncertain, as the CDI treatment in the study was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice.
Additional studies assessed in the fecal microbiota (Rebyota) review to address important gaps in the evidence included the following:
The PUNCH CD2 study (N = 133) is a phase II, prospective, multicentre, DB, randomized, placebo-controlled trial that assessed the efficacy and safety of fecal microbiota (Rebyota) in adult patients aged 18 years or older with rCDI. Patients were randomized into 1 of 3 groups after a course of antibiotics: 2 doses of fecal microbiota (Rebyota) (group A); 2 doses of placebo (group B); or 1 dose of each (group C). The study focused on treatment success (no CDI recurrence in the 8 weeks after treatment), time to CDI recurrence, quality-of-life changes measured with the SF-36 scale, and long-term safety outcomes (with follow-up for AEs up to 24 months).
The PUNCH CD3 OLS study (N = 698) was a phase III, prospective, multicentre, OL, single-arm study. The objective of the study was to evaluate the safety, tolerability, and effectiveness of fecal microbiota (Rebyota) as single and repeat administrations in a population of patients with conditions that were excluded from previous trials, such as Crohn disease, ulcerative colitis, IBD, IBS, and immunodeficiency.
Feuerstadt et al. (2023)19 (N = 94) conducted a retrospective study on the efficacy and safety of fecal microbiota (Rebyota) in adult patients with rCDI, using broad eligibility criteria to mirror real-world conditions. The study assessed the effectiveness and safety of fecal microbiota (Rebyota) in patients with comorbidities like IBS, IBD, and immunodeficiency.
Lee et al.42 assessed pooled safety data from 5 prospective studies of patients exposed to at least 1 study treatment, including 3 phase II studies (the PUNCH CD, PUNCH CD2, and PUNCH CD3 OLS studies) and 2 phase III studies (the PUNCH CD3 study and an interim data cut of the PUNCH CD3-OLS study).
Interpretation of Results
Efficacy
CDI is a spore-forming, toxin-producing, Gram-positive anaerobe that colonizes the gut,1 causing profuse, watery diarrhea. Severity can reach fulminant colitis with systemic toxicity and shock2 and, as such, CDI contributes to significant morbidity and mortality.3 Based on prevalence estimates, the projected number of CDI cases in Canada in 2024 was approximately 47,000.7 With symptoms such as fatigue, weight loss, abdominal pain, frequent urgent bathroom use, and psychological distress, CDI significantly impacts HRQoL.5 Up to 25% of patients may experience rCDI after antibiotic treatment, and the risk of recurrence increases substantially, to up to 50%, after the second recurrence.27 Indeed, the natural disease trajectory becomes more complex in patients who have experienced at least 2 recurrences, after which the risk of experiencing further recurrences becomes incrementally higher. Therefore, there is a need for accessible, effective, and well-tolerated therapies to prevent rCDI in patients who have a multirecurrent condition despite appropriate antibiotic treatment and whose condition has a substantial impact on quality of life.
The primary outcome in the PUNCH CD3 study was treatment response after 8 weeks. Results suggest that a higher proportion of patients is likely to achieve treatment success at 8 weeks after fecal microbiota (Rebyota) than after placebo. The between-group difference in the mean success rate was 12.3% (95% CrI, 1.4% to 23.3%) over 8 weeks. No MCID was reported in the literature as a threshold for treatment success or for between-group differences, so the presence of an important effect was informed by the clinical experts consulted for this review. The point estimate suggests the presence of a clinically important effect; however, the lower bound of the CrI is consistent with a trivial effect, which would not be considered clinically important for patients.
The clinical experts noted that patients included in the trial were likely to be at a lower risk for CDI recurrence than patients typically seen in clinical practice who would have the greatest unmet need and be the best candidates to receive fecal microbiota (Rebyota). One of the reasons for this is the inclusion of patients who had only 1 recurrence of CDI, who accounted for █████ of patients in the fecal microbiota (Rebyota) group and █████ of patients in the placebo group. Patients who experience a first recurrence may have their condition improve spontaneously and may maintain a sustained response without active treatment. However, the natural disease trajectory becomes more severe in patients who have experienced at least 2 recurrences, and the risk of experiencing further recurrences becomes incrementally higher. Therefore, the clinical experts emphasized that fecal microbiota (Rebyota) should be reserved for patients who have experienced at least 2 recurrences and who have had a relapse after a vancomycin taper or pulse regimen. Results of the prespecified subgroup analyses of treatment response were mostly consistent with the main analysis; however, for patients who experienced 2 or fewer recurrences and who were treated with vancomycin for 14 days or less, the results suggested less benefit with fecal microbiota (Rebyota). Although credible effect modification could not be inferred from these analyses, the results raise uncertainty about whether the magnitude of benefit observed in the PUNCH CD3 study can be generalized to these groups of patients. However, it appears plausible, based on experience in clinical practice and supplementary findings from the PUNCH CD3 study, that the benefits of treatment would be maintained in a population of patients who have a more severe condition. In the PUNCH CD3 study, although the majority of patients received vancomycin for the treatment of the qualifying episode, no detail was reported regarding the specific regimen used (e.g., whether patients received a prolonged taper or pulse vancomycin treatment, per recommendations from the Canadian guidelines). Short courses of antibiotics, even up to 21 days (as the trial’s protocol required), are unlikely to be sufficient to manage multiple episodes of rCDI, according to the clinical experts. The relevance of using placebo as a comparator is, therefore, uncertain, as the CDI treatment in the study was not consistent with the treatment that patients would receive in clinical practice. Thus, the generalizability of the results from patients in the study population to patients who would be candidate to receive fecal microbiota (Rebyota) in clinical practice is uncertain.
The primary analysis was based on posterior estimates from the Bayesian hierarchical model in the mITT population. The posterior probability of superiority exceeded the final secondary threshold, but not the primary, protocol-specified threshold. Sensitivity analyses of the primary outcome based on alternate analysis populations yielded consistent results. Findings from a frequentist sensitivity analysis without Bayesian borrowing were, overall, consistent and suggest that results from the Bayesian approach are likely driven by the informative priors used based on the phase II PUNCH CD2 trial.
There was no clinically important difference between fecal microbiota (Rebyota) and placebo in sustained clinical response at 6 months among patients who previously achieved treatment success at 8 weeks. In addition, the evidence was insufficient to draw any conclusions about the effect of fecal microbiota (Rebyota) relative to placebo on hospitalizations and ICU admissions.
HRQoL was assessed as an exploratory outcome using the Cdiff32 Questionnaire. The study was not designed to test for differences in HRQoL, and findings should be interpreted with this consideration. The results did not suggest HRQoL benefits with fecal microbiota (Rebyota). The between-group difference in the change from baseline in HRQoL was less than the suggested MCID of 10 points. There was uncertainty because of imprecision and the literature-based MCID, which was estimated using a distribution-based approach. As such, it is unclear whether the estimated MCID would reflect a clinically meaningful change from the perspectives of patients. This MCID was also estimated for within-group changes, rather than between-group differences. The evidence for change from baseline HRQoL at 6 months was very uncertain, owing to imprecision and an important risk of bias due to missing outcome data. In addition, some patients in the placebo group had also received active treatment with OL fecal microbiota (Rebyota).
Fecal sequencing analysis suggests that the microbiome composition of patients experiencing treatment success changed from baseline over all time points. The change in microbiome composition after treatment was characterized by an increase in Bacteroidia-class and Clostridia-class bacteria and a decrease in Gammaproteobacteria-class and Bacilli-class bacteria.
Additional studies were assessed in the fecal microbiota (Rebyota) review to address important gaps in the evidence. In the PUNCH CD2 study, efficacy results of treatment success at 8 weeks and median time to CDI recurrence between treatment group B versus treatment group C align with the results of the PUNCH CD3 study. However, the primary outcome's null hypothesis between group A and group B was not rejected. In the PUNCH CD3 OLS single-arm study, efficacy results suggested a treatment success at 8 weeks that aligns with the results of the PUNCH CD3 study. In the absence of a randomized comparator, however, it is not possible to draw causal conclusions regarding the efficacy of fecal microbiota (Rebyota) from this study. In addition, the number of patients with Crohn disease, ulcerative colitis, IBD, IBS, and immunodeficiency enrolled in the study was small; thus, the generalizability of the findings to patients who have these coexisting comorbidities remains uncertain. Feuerstadt et al. (2023)19 showed that a similar proportion of real-world patients, including those with comorbidities (IBS, IBD, immunodeficiency) achieved treatment success at week 8 after fecal microbiota (Rebyota) treatment; however, the design of this study did not allow for causal conclusions to be drawn regarding the efficacy of fecal microbiota (Rebyota), and patients could receive up to 4 doses of fecal microbiota (Rebyota) — 2 courses of 2 doses each — which exceeds Health Canada’s 1-dose recommendation and could, therefore, lead to an overestimation of treatment response.
Because the PUNCH CD3 study included a placebo control group, there is no direct evidence to inform the effectiveness and safety of fecal microbiota (Rebyota) relative to other currently used therapies for the prevention of rCDI, particularly FMT, in patients who have received an appropriate antibiotic regimen of prolonged vancomycin taper or pulse. In addition, no indirect evidence was submitted. The absence of comparative evidence, especially against conventional, multidose FMT, was considered a limitation, as experience from clinical practice and data from the literature suggest that FMT may achieve clinical response rates of up to more than 90%.27 As a result, however, the comparative efficacy and safety of fecal microbiota (Rebyota) is unknown. It is worth noting, however, that the RCTs investigating FMT are heterogeneous; many studies included small patient numbers, and trials varied by inclusion and exclusion criteria, donor type, mode of delivery, number of doses, length of follow-up, and volume per dose. The response rates ranged from 44% up to more than 90%, and it appears that patients may require multiple FMTs to achieve a clinical response rate of more than 80%.27
Harms
More than two-thirds of patients receiving fecal microbiota (Rebyota) experienced at least 1 AE during the PUNCH CD3 study. AEs were numerically more frequent in patients who received 2 doses of fecal microbiota (Rebyota) than in patients who received only 1 fecal microbiota (Rebyota) dose. The most common TEAEs were related to gastrointestinal disorders (i.e., diarrhea, abdominal pain, nausea, and abdominal distention).
SAEs were relatively uncommon, but were also numerically more frequent in patients who received 2 doses of fecal microbiota (Rebyota) than in patients who received only 1 fecal microbiota (Rebyota) dose. The 2 deaths reported during the trial were in patients who received fecal microbiota (Rebyota), but were not related to the study drug or enema procedure, according to the sponsor. One patient in the fecal microbiota (Rebyota) group experienced septic shock as a major complication of new CDI and underwent emergency colectomy. The sponsor reported no events of toxic megacolon or colonic perforation. Findings from the phase II, placebo-controlled PUNCH CD2 study, as well as from the single-arm PUNCH CD3 OLS study, were consistent with those from the pivotal trial. However, because of the absence of a randomized comparator, the PUNCH CD3 OLS study did not allow for causal conclusions to be drawn regarding the safety of fecal microbiota (Rebyota).
Overall, the clinical experts indicated that the harms profile of fecal microbiota (Rebyota) did not raise any new safety signals or any particular safety concern. As with most clinical trials, the study was not powered to detect infrequent AEs or those with a lag time. The clinical experts indicated that there may be long-term, unintended effects of manipulating a patient’s microbiome, such as affecting the risk of developing a range of conditions over time, the impact of which is currently unknown. In addition, the experts emphasized that fecal microbiota (Rebyota) is not risk-free and, therefore, shared decision-making is required so that patients are informed about the balance of potential benefits and risks before they decide whether to proceed with fecal microbiota (Rebyota) treatment.
Other Considerations
The clinical experts highlighted the need for accessible, effective, and well-tolerated therapies to prevent rCDI. Patients with the greatest unmet needs are those who have a severe, multirecurrent condition; those who are not able to stop an antibiotic taper or pulse regimen; and those in whom the condition has a substantial impact on quality of life.
The clinical experts indicated that in clinical practice, FMT is effective in these patients at preventing rCDI; however, FMT is not readily available to many patients in Canada. Issues currently surrounding the limited access to FMT, according to the clinical experts, include the lack of public or private funding for FMT (which leaves FMT programs heavily dependant on physicians and research grants); the infrastructures and level of expertise that are required to create and administer an FMT program; and the challenges involved in establishing and maintaining stool banks for FMT (criteria to become a stool donor are strict and donors are essentially volunteering, which makes them difficult to retain).
In addition, the clinical experts made the CDA-AMC clinical review team aware of the possibility that conventional FMT programs could be shut down by Health Canada when a commercial product, (i.e., fecal microbiota [Rebyota]) becomes available. Health Canada notified CDA-AMC that clinicians were advised that, although fecal microbiota (Rebyota) has been authorized, it is not yet marketed. Therefore, no changes will be made to the current interim policy outlined in the guidance document: Fecal Microbiota Therapy Used in the Treatment of Clostridioides difficile Infection Not Responsive to Conventional Therapies. Once fecal microbiota (Rebyota) becomes marketed, Health Canada will explore options for transitioning away from the interim policy, with further details to be provided in due course.
According to the clinical experts, fecal microbiota (Rebyota) can be considered very similar to the product that is being administered through conventional FMT. However, whereas fecal microbiota (Rebyota) relies on a single administration, FMT programs may routinely offer repeated doses over 1 week. A commercial, regulated, stool-derived product such as fecal microbiota (Rebyota) can potentially increase access to treatment, which was viewed by the clinical experts as an acceptable trade-off to its single administration. The experts indicated that fecal microbiota (Rebyota) may, therefore, cause a shift in the current treatment paradigm, provided that health care systems can offer the necessary funding, infrastructures, and resources for product administration and that there are no interruptions in the supply chain.
Conclusion
In patients with rCDI, moderate-certainty findings from the PUNCH CD3 study suggest that treatment with fecal microbiota (Rebyota) likely results in a clinically important increase in the treatment success rate over 8 weeks, compared to placebo. Findings were obtained in a population that was, however, at a lower risk for CDI recurrences than patients who have the greatest unmet need and would benefit the most from treatment with fecal microbiota (Rebyota), according to the clinical experts. This population at need would include patients who have experienced at least 2 recurrences despite an appropriate antibiotic treatment consisting of a vancomycin taper or pulse regimen. Findings from supplementary, prespecified subgroup analyses raise uncertainty as to whether the overall effect can be generalized to patients who have experienced fewer than 2 recurrences or who have received vancomycin for 14 days or less. Although the generalizability of results from patients in the study population to patients who would be candidates to receive fecal microbiota (Rebyota) in clinical practice is uncertain, it appears plausible that the benefits of treatment could be maintained in a population of patients who have a higher risk of recurrence. Most patients who previously achieved treatment success at 8 weeks with either fecal microbiota (Rebyota) or placebo were able to maintain a sustained clinical response at 6 months; therefore, there may be no added benefit with fecal microbiota (Rebyota) for maintaining response over time compared to placebo. Based on results from exploratory analyses, fecal microbiota (Rebyota) may result in little to no difference in HRQoL, measured with the Cdiff32 Questionnaire, over 8 weeks compared to placebo. HRQoL results at 6 months were too uncertain to draw a definite conclusion. In addition, the evidence was insufficient to draw any conclusions about the effect of fecal microbiota (Rebyota) relative to placebo on hospitalizations and ICU admissions.
A relatively high proportion of patients in the PUNCH CD3 study experienced AEs, most notably related to gastrointestinal disorders. AEs were numerically more frequent in patients who received 2 doses of fecal microbiota (Rebyota) than in patients who received only 1 fecal microbiota (Rebyota) dose and in patients who received placebo. However, fecal microbiota (Rebyota) appeared to be well tolerated, with relatively few SAEs. The clinical experts noted that although treatment with fecal microbiota (Rebyota) is not without risks, the overall harms profile did not raise any particular safety signals. However, the long-term, unintended effects of manipulating a patient’s microbiome are currently unknown.
The trial could not inform on the effectiveness and safety of fecal microbiota (Rebyota) relative to other currently used therapies for rCDI, such as conventional, multidose FMT. Whether patients in the 2 groups previously received an appropriate antibiotic regimen of vancomycin taper or pulse for the treatment of the qualifying CDI episode is unknown. The relevance of using placebo as a comparator is therefore uncertain, as the CDI treatment in the study was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice. Four additional studies were assessed in the fecal microbiota (Rebyota) review, but could not inform on important gaps in the evidence.
The clinical experts indicated that although effective, FMT is an intervention with limited access and, therefore, it is not readily available to many patients in Canada. A commercial, regulated, stool-derived product such as fecal microbiota (Rebyota) can potentially increase access to treatment, which was viewed by the clinical experts as an appropriate trade-off to its single administration. The experts indicated that fecal microbiota (Rebyota) may, therefore, cause a shift in the current treatment paradigm, as long as health care systems can provide the infrastructures and resources related to prescribing considerations and administration of the product.
References
1.Lamont JT, Bakken JS, Kelly CP. Clostridioides difficile infection in adults: Epidemiology, microbiology, and pathophysiology. UpToDate; January 17, 2023. Accessed January 7, 2025. www.uptodate.com
2.Lamont JT, Kelly CP, Bakken JS. Clostridioides difficile infection in adults: Clinical manifestations and diagnosis. UpToDate; October 18, 2024. Accessed January 7, 2025. www.uptodate.com
3.McDonald LC, Kutty PK, Kaplan SL. Clostridioides difficile infection: Prevention and control. UpToDate; February 22, 2024. Accessed January 7, 2025. www.uptodate.com
4.Mada PK, Alam MU. Clostridioides difficile infection. Internet. StatPearls Publishing; 2024. Accessed May 21, 2024. https://www.ncbi.nlm.nih.gov/books/NBK431054/
5.Armstrong EP, Malone DC, Franic DM, Pham SV, Gratie D, Amin A. Patient Experiences with Clostridioides difficile Infection and Its Treatment: A Systematic Literature Review. Infect Dis Ther. 2023;12(7):1775-1795. doi: 10.1007/s40121-023-00833-x PubMed
6.Slimings C, Armstrong P, Beckingham WD, et al. Increasing incidence of Clostridium difficile infection, Australia, 2011-2012. Med J Aust. 2014;200(5):272-6. doi: 10.5694/mja13.11153 PubMed
7.Levy AR, Szabo SM, Lozano-Ortega G, et al. Incidence and Costs of Clostridium difficile Infections in Canada. Open Forum Infect Dis. 2015;2(3):ofv076. doi: 10.1093/ofid/ofv076 PubMed
8.Health Infobase. Canadian Antimicrobial Resistance Surveillance System (CARSS): Antimicrobial resistance. Government of Canada. Updated November 24, 2023. Accessed February 3, 2025. https://health-infobase.canada.ca/carss/amr/results.html?ind=08
9.Public Health Agency of Canada. Canadian Antimicrobial Resistance Surveillance System Report. 2022. Accessed April 7, 2024. https://www.canada.ca/content/dam/phac-aspc/documents/services/publications/drugs-health-products/canadian-antimicrobial-resistance-surveillance-system-report-2022/canadian-antimicrobial-resistance-surveillance-system-report-2022.pdf
10.Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Heguy A, Nault V, Valiquette L. Clinical and Healthcare Burden of Multiple Recurrences of Clostridium difficile Infection. Clin Infect Dis. 2016;62(5):574-580. doi: 10.1093/cid/civ958 PubMed
11.Olsen MA, Stwalley D, Demont C, Dubberke ER. Clostridium difficile infection increases acute and chronic morbidity and mortality. Infect Control Hosp Epidemiol. 2019;40(1):65-71. doi: 10.1017/ice.2018.280 PubMed
12.Lubbert C, Zimmermann L, Borchert J, Horner B, Mutters R, Rodloff AC. Epidemiology and recurrence rates of Clostridium difficile infections in Germany: a secondary data analysis. Infect Dis Ther. 2016;5(4):545-554. doi: 10.1007/s40121-016-0135-9 PubMed
13.Granata G, Petrosillo N, Adamoli L, et al. Prospective Study on Incidence, Risk Factors and Outcome of Recurrent Clostridioides difficile Infections. J Clin Med. 2021;10(5):1127. doi: 10.3390/jcm10051127 PubMed
14.Larrainzar-Coghen T, Rodriguez-Pardo D, Fernandez-Hidalgo N, et al. Secular trends in the epidemiology of Clostridium difficile infection (CDI) at a tertiary care hospital in Barcelona, 2006-2015: A prospective observational study. Anaerobe. 2018;51:54-60. doi: 10.1016/j.anaerobe.2018.04.002 PubMed
15.Du T, Choi KB, Silva A, et al. Characterization of Healthcare-Associated and Community-Associated Clostridioides difficile Infections among Adults, Canada, 2015-2019. Emerg Infect Dis. 2022;28(6):1128-1136. doi: 10.3201/eid2806.212262 PubMed
16.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
17.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
18.Rebiotix Inc. Clinical Study Report: 2017-01 PUNCH CD3. A Phase 3 Prospective, Randomized, Double-Blinded, Placebo-Controlled Clinical Study to Evaluate the Efficacy and Safety of Rebiotix RBX2660 (microbiota suspension) for the Prevention of Recurrent Clostridium difficile Infection [internal sponsor's report]. March 17, 2021.
19.Feuerstadt P, Harvey A, Yoho DS, Garcia-Diaz JB, Knapple WL, Bancke L. Retrospective Analysis of the Safety and Efficacy of Fecal Microbiota, Live-jslm (REBYOTA) Administered Under Enforcement Discretion to Patients With Clostridioides difficile Infection. Open Forum Infect Dis. 2023;10(5):ofad171. doi: 10.1093/ofid/ofad171 PubMed
20.Khanna S, Seo S, Yang M, et al. Characteristics and Real-World Outcomes of Patients Treated with Fecal Microbiota, Live-jslm (RBL) for the Prevention of Recurrent Clostridioides difficile Infection. Infect Dis Ther. 2025;14(4):793-802. doi: 10.1007/s40121-025-01130-5 PubMed
21.Dubberke ER, Butler AM, Yokoe DS, et al. Multicenter study of Clostridium difficile infection rates from 2000 to 2006. Infect Control Hosp Epidemiol. 2010;31(10):1030-7. doi: 10.1086/656245 PubMed
22.European Centre for Disease Prevention and Control (ECDC). Clostridioides (Clostridium) difficile infections: Annual Epidemiological Report for 2016-2017. 2022. Accessed November 4, 2025. https://www.ecdc.europa.eu/sites/default/files/documents/clostridioides-clostridium-difficile-infections.pdf
23.Gastmeier P, Weitzel-Kage D, Behnke M, Eckmanns T. Surveillance of Clostridium difficile-associated diarrhoea with the German nosocomial infection surveillance system KISS (CDAD-KISS). Int J Antimicrob Agents. 2009;33 Suppl 1:S19-23. doi: 10.1016/s0924-8579(09)70011-1 PubMed
24.Public Health Agency of Canada. Canadian Nosocomial Infection Surveillance Program (CNISP): Summary Report of Healthcare Associated Infection (HAI), Antimicrobial Resistance (AMR) and Antimicrobial Use (AMU) Surveillance Data from January 1, 2013 to December 31, 2017. 2019. Accessed November 5, 2025. https://www.canada.ca/content/dam/canada/public-health/services/publications/science-research-data/summary-report-healthcare-associated-infection-antimicrobial-resistance-antimicrobial-use-surveillance-data-2013-2017/CNISP-2013-2017-Report-EN-WEB-v2.pdf
25.Gupta A, Ananthakrishnan AN. Economic burden and cost-effectiveness of therapies for Clostridiodes difficile infection: a narrative review. Therap Adv Gastroenterol. 2021;14:17562848211018654. doi: 10.1177/17562848211018654 PubMed
26.Zanichelli V, Garenc C, Villeneuve J, et al. Increased Community-Associated Clostridioides difficile Infections in Quebec, Canada, 2008-2015(1). Emerg Infect Dis. 2020;26(6):1291-1294. doi: 10.3201/eid2606.190233 PubMed
27.Loo VG, Davis I, Embil J, et al. Association of Medical Microbiology and Infectious Disease Canada treatment practice guidelines for Clostridium difficile infection. J Assoc Med Microbiol Infect Dis Can. 2018;3(2):71-92. doi: 10.3138/jammi.2018.02.13
28.Pepin J, Valiquette L, Cossette B. Mortality attributable to nosocomial Clostridium difficile-associated disease during an epidemic caused by a hypervirulent strain in Quebec. CMAJ. 2005;173(9):1037-42. doi: 10.1503/cmaj.050978 PubMed
29.Ferring Inc. Rebyota (fecal microbiota): 150 mL rectal suspension [draft product monograph]. 2024.
30.Health Canada. Guidance Document: Fecal Microbiota Therapy Used in the Treatment of Clostridioides difficile Infection Not Responsive to Conventional Therapies. 2022. Accessed November 4, 2025. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/regulation-fecal-microbiota-therapy-treatment-difficile-infections-2022-09-26.pdf
31.Quay T, Walter M. Fecal Microbiota Therapy in Canada: An Environmental Scan. (Environmental scan no. 95). CADTH; 2020. Accessed November 4, 2025. https://www.cadth.ca/sites/default/files/es/es0341-fecal-microbiota-therapy-in-canada.pdf
32.Khanna S, Assi M, Lee C, et al. Efficacy and Safety of RBX2660 in PUNCH CD3, a Phase III, Randomized, Double-Blind, Placebo-Controlled Trial with a Bayesian Primary Analysis for the Prevention of Recurrent Clostridioides difficile Infection. Drugs. 2022;82(15):1527-1538. doi: 10.1007/s40265-022-01797-x PubMed
33.Blount KF, Papazyan R, Ferdyan N, et al. Microbiome and Metabolome Restoration After Administration of Fecal Microbiota, Live-jslm (REBYOTA) for Preventing Recurrent Clostridioides difficile Infection. J Infect Dis. 2025;231(6):e1022-e1033. doi: 10.1093/infdis/jiae418 PubMed
34.Feuerstadt P, Crawford CV, Tan X, et al. Fecal Microbiota, Live-jslm for the Prevention of Recurrent Clostridioides difficile Infection: Subgroup Analysis of PUNCH CD2 and PUNCH CD3. J Clin Gastroenterol. 2024;58(8):818-824. doi: 10.1097/MCG.0000000000001947 PubMed
35.Garey KW, Dubberke ER, Guo A, et al. Effect of Fecal Microbiota, Live-Jslm (REBYOTA [RBL]) on Health-Related Quality of Life in Patients With Recurrent Clostridioides difficile Infection: Results From the PUNCH CD3 Clinical Trial. Open Forum Infect Dis. 2023;10(8):ofad383. doi: 10.1093/ofid/ofad383 PubMed
36.Tillotson G, Archbald-Pannone L, Johnson S, et al. Microbiota-Based Live Biotherapeutic RBX2660 for the Reduction of Recurrent Clostridioides difficile Infection in Older Adults With Underlying Comorbidities. Open Forum Infect Dis. 2023;10(1):ofac703. doi: 10.1093/ofid/ofac703 PubMed
37.Feuerstadt P, Allegretti JR, Dubberke ER, et al. Efficacy and Health-Related Quality of Life Impact of Fecal Microbiota, Live-jslm: A Post Hoc Analysis of PUNCH CD3 Patients at First Recurrence of Clostridioides difficile Infection. Infect Dis Ther. 2024;13(1):221-236. doi: 10.1007/s40121-023-00907-w PubMed
38.Garey KW, Feuerstadt P, Dubberke ER, Guo A, Tillotson GS. Effect of fecal microbial transplantation on Clostridioides difficile infection: dysbiosis, metabolites and health related quality of life. Open Forum Infect Dis. 2023;10(3):ofad113. doi: 10.1093/ofid/ofad113 PubMed
39.Garey KW, Aitken SL, Gschwind L, et al. Development and Validation of a Clostridium difficile Health-related Quality-of-Life Questionnaire. J Clin Gastroenterol. 2016;50(8):631-7. doi: 10.1097/mcg.0000000000000473 PubMed
40.Lapin B, Garey KW, Wu H, et al. Validation of a health-related quality of life questionnaire in patients with recurrent Clostridioides difficile infection in ECOSPOR III, a Phase 3 randomized trial. Clin Infect Dis. 2023;76(3):e1195-e1201. PubMed
41.U.S. Food and Drug Administration. Interacting with the FDA on complex innovative trial designs for drugs and biological products: Guidance for industry. 2020. Accessed November 4, 2025. https://www.fda.gov/media/130897/download
42.Lee C, Louie T, Bancke L, et al. Safety of fecal microbiota, live-jslm (REBYOTA™) in individuals with recurrent Clostridioides difficile infection: data from five prospective clinical trials. Therap Adv Gastroenterol. 2023;16:17562848231174277. doi: 10.1177/17562848231174277 PubMed
43.Rebiotix Inc. Clinical Study Report: 2014-01. A Phase 2B Prospective, Randomized, Double-blinded, Placebo-controlled Clinical Study Demonstrating the Efficacy and Safety of Rebiotix RBX2660 (microbiota suspension) for the Prevention of Recurrent Clostridium difficile Infection [internal sponsor's report]. March 26, 2020.
44.Rebiotix Inc. Clinical Study Report: 2019-01 PUNCH CD3-OLS. A Phase 3 Open-Label Clinical Study to Evaluate the Safety and Tolerability of Rebiotix RBX2660 (microbiota suspension) in Subjects with Recurrent Clostridioides difficile Infection [internal sponsor's report]. July 16, 2024.
45.Ferring Inc. Reimbursement Review Sponsor Summary of Clinical Evidence Template: Rebyota (fecal microbiota) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rebyota (fecal microbiota): 150 mL rectal suspension. January 3, 2025.
46.U.S. Food and Drug Administration. Guidance for Industry: Enforcement Policy Regarding Investigational New Drug Requirements for Use of Fecal Microbiota for Transplantation To Treat Clostridium difficile Infection Not Responsive to Standard Therapies; Availability. 2013. Accessed January 30, 2025. https://www.federalregister.gov/documents/2013/07/18/2013-17223/guidance-for-industry-enforcement-policy-regarding-investigational-new-drug-requirements-for-use-of
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Additional post hoc survival analyses conducted on PUNCH-CD2 data were used to inform the pharmacoeconomic model, the results of which are described below.
PUNCH CD2 ITT Survival Analysis
As noted in Table 27, in a post hoc survival analysis in the ITT population, ████████ ███████████ █████████ ████ ████ ██ ██████ █ ███ ██ ███████ █████ █ ████ ███ █████ █████ ██ █████ █ ████ █ ████████ ███████████ ████ ████████ For Arm A, ███ █████ █████ ████████ ██ █████ ██ ████ █ ██████ ███ ██ ██ █████████ █████████ ██ █ ████████ ███████████ ████████ By month 12, ███ ██████████ █████ ████████ ███ ██ ███ █████████ ██ █████████ ███████ ██ █ ████████ ███████████ ██ █████████ █████████ ██████ ██████ █ ███████ ███████ ██ █████ ██ █████ ███████████ ███ ████████ ██████████████
Table 27: Post Hoc Survival Analysis (ITT Population)
Arm | Time (weeks) | Number at risk | Number of events | Survival (SE) | 95% CI |
|---|---|---|---|---|---|
A | 6 | ██ | ██ | █████████ | █████████ |
12 | ██ | ██ | █████████ | █████████ | |
B | 12 | ██ | ██ | █████████ | █████████ |
24 | ██ | ██ | █████████ | █████████ | |
C | 6 | ██ | ██ | █████████ | █████████ |
CI = confidence interval; ITT = intention to treat; SE = standard error.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.45
mITT and SP Survival Analysis
In the modified intended to treat (mITT) and safety population (SP) designs, ███ ████ ██████████ ████████ ██ ██ ███ ███ ██████ ████ ████████ ███ ████ ███████ ████████ ██ ████ ████████
PP Survival Analysis
As show in Table 28, in the per protocol design, ████████ ███████████ █████████ ████ ████ ██ █████ ██ ████ █████ █████████ ██ █████ █ ███ █████ ███ █████ █ ███ █ ████ ███ ██████ █████ ██ █████ ██ ███ █████ ██ ████████████.
Table 28: Post Hoc Survival Analysis (PP Population)
Arm | Time (weeks) | Number at risk | Number of events | Survival (SE) | 95% CI |
|---|---|---|---|---|---|
A | 6 | ██ | ██ | █████████ | █████████ |
12 | ██ | ██ | █████████ | █████████ | |
B | 12 | ██ | ██ | █████████ | █████████ |
C | 6 | ██ | ██ | █████████ | █████████ |
CI = confidence interval; PP = per protocol; SE = standard error.
Source: Details included in the table are from the sponsor’s Summary of Clinical Evidence.45
In the PUNCH-CD2 study, long-term follow-up data were collected via monthly phone assessments (months 3, 6, 12, and 24), which may not fully capture changes in patients’ health status between assessments. This could have led to an underreporting of events in the survival analysis. A large proportion of patients discontinued the study (█████████████ ███ ██ █████ ██ █████ ████ ██ █████████. This can introduce bias, as patients who withdrew may have been in worse health, which could artificially inflate the survival probability. The combination of high dropout rates, potential underreporting of events, and censoring bias limits the reliability of the results of the survival analysis.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CDA-AMC
Canada's Drug Agency
CDI
Clostridioides difficile infection
FMT
fecal microbiota transplant
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LY
life-year
QALY
quality-adjusted life-year
rCDI
recurrent Clostridioides difficile infection
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Fecal microbiota (Rebyota), 150 mL suspension for enema |
Indication | Fecal microbiota (Rebyota) is indicated for the prevention of recurrence of CDI in adults following antibiotic treatment for recurrent CDI Fecal microbiota is not indicated for the treatment of CDI |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 5, 2025 |
Reimbursement request | As per indication |
Sponsor | Ferring Inc. |
Submission history | No |
CDI = Clostridioides difficile infection; NOC = Notice of Compliance.
Table 2: Summary of the Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with at least 1 rCDI resolved following antibiotic treatment |
Treatment | Fecal microbiota (Rebyota) after prior antibiotic treatment (i.e., vancomycin, fidaxomicin) |
Dose regimen | Single 150 mL dose via rectal administration |
Submitted price | $9,125.00 per 150 mL liquid suspension |
Submitted treatment cost | $9,125.00 per treatment course |
Comparator | No preventive therapy after prior antibiotic treatment (i.e., vancomycin, fidaxomicin) |
Perspective | Publicly funded health care payer Societal (including patient productivity loss and travel costs) |
Outcomes | QALYs, LYs |
Time horizon | 5 years |
Key data sources | PUNCH CD3 and PUNCH open-label studies |
Submitted results | Publicly funded health care payer perspective: fecal microbiota (Rebyota) dominates no preventive therapy ($2,135 less costly and 0.14 QALYs more effective) Societal perspective: fecal microbiota (Rebyota) dominates no preventive therapy ($8,592 less costly and 0.14 QALYs more effective) |
Key limitations |
|
CDA-AMC reanalysis results |
|
CDA-AMC = Canada's Drug Agency; CDI = Clostridioides difficile infection; FMT = fecal microbiota transplant; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; rCDI = recurrent C. difficile infection; vs. = versus; WTP = willingness to pay.
Conclusions
Evidence from the PUNCH CD3 trial suggests that fecal microbiota (Rebyota) likely leads to a clinically important improvement in treatment success rate over 8 weeks compared to placebo for the prevention of the recurrence of Clostridioides difficile infection (CDI) in adults after antibiotic treatment for recurrent CDI (rCDI). According to the Canada's Drug Agency (CDA-AMC) Clinical Review Report, most patients who previously achieved treatment success at 8 weeks with either fecal microbiota (Rebyota) or placebo were able to maintain a sustained clinical response at 6 months; therefore, there may be no added benefit with fecal microbiota (Rebyota) on this outcome compared to placebo. Based on results from exploratory analyses, fecal microbiota (Rebyota) may result in little to no difference in health-related quality of life (HRQoL) over 8 weeks compared to placebo, and HRQoL results at 6 months were too uncertain to draw a definite conclusion. In addition, the evidence was insufficient to draw any conclusions on the effect of fecal microbiota (Rebyota) relative to placebo on hospitalizations or intensive care unit (ICU) admissions. The trial could not inform on the effectiveness or safety of fecal microbiota (Rebyota) relative to other currently used therapies for rCDI, such as conventional, multidose fecal microbiota transplant (FMT), nor did the sponsor submit indirect evidence to supplement the gap in comparative evidence. Whether patients in the 2 groups previously received an appropriate antibiotic regimen of vancomycin taper-pulse for the treatment of the qualifying CDI episode is unknown. The relevance of using placebo as a comparator is, therefore, uncertain, as the CDI treatment in the study was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice.
CDA-AMC undertook reanalyses to address several key limitations in the sponsors submission, including the assumption that a sustained response does not change after 6 months; the application of more appropriate rates for death and colectomy occurring in the rCDI health state; the alignment of antibiotic distributions for no preventive therapy with what patients received in the PUNCH CD3 trial; the addition of administration costs for fecal microbiota (Rebyota); and the application of more appropriate assumptions to estimate productivity losses. Results from the CDA-AMC base case suggest that the incremental cost-effectiveness ratio (ICER) for fecal microbiota (Rebyota) versus no preventive therapy is $203,679 per quality-adjusted life-year (QALY) gained (incremental costs = $6,328; incremental QALYs = 0.03) for the health care payer perspective and $136,782 per QALY gained (incremental costs = $4,547; incremental QALYs = 0.03) for the societal perspectives. At the sponsor’s submitted price, fecal microbiota (Rebyota) would require price reductions of 53% or 32%, (from $9,125 per dose to $4,289 or $6,205 per dose) to be considered cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained for the public health care payer and societal perspectives, respectively.
The CDA-AMC base case maintained some benefit for fecal microbiota (Rebyota) that has not been clinically proven, including maintained differences in sustained response between fecal microbiota (Rebyota) and no preventive therapy and impacts on rCDI-related mortality and colectomy. In a scenario in which there is no difference in sustained response after 6 months between fecal microbiota (Rebyota) and no preventive therapy, the ICER for fecal microbiota (Rebyota) versus no preventive therapy increased to $476,035 per QALY gained and $407,736 per QALY gained in the health care payer and societal perspectives, respectively. This could be clinically plausible, given the CDA-AMC Clinical Review Report conclusion that there may be no added benefit with fecal microbiota (Rebyota) compared to placebo in terms of sustained response at 6 months among patients who achieved treatment success at 8 weeks. Additionally, in a scenario in which the mortality and colectomy benefit associated with fecal microbiota (Rebyota) was removed, which is consistent with the clinical evidence provided in the PUNCH CD3 trial, the ICER for fecal microbiota (Rebyota) versus no preventive therapy increased to $212,188 per QALY gained and $158,901 per QALY gained in the health care payer and societal perspectives, respectively. Therefore, the CDA-AMC base case may represent an optimistic reimbursement context, should these longer-term benefits associated with fecal microbiota (Rebyota) not be realized.
CDA-AMC was unable to address uncertainty related to the homogenous rCDI health state, which was more representative of an acute recurrence of CDI than the spectrum of rCDI symptoms and severity that patients with rCDI may experience. Because it is likely that rCDI is less severe in the overall patient population, such patients will incur fewer costs (both health care and indirect) than modelled by the sponsor, the CDA-AMC base case is likely conservative in terms of overall cost-effectiveness of fecal microbiota (Rebyota).
As well, CDA-AMC was unable to address the lack of comparative efficacy between fecal microbiota (Rebyota) and FMT. Given that there are no available cost estimates for FMT, there is insufficient evidence for a price premium for fecal microbiota (Rebyota) over FMT.
Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was provided by 3 groups: the Gastrointestinal Society, the Peggy Lillis Foundation, and the Canadian Digestive Health Foundation. Input was gathered through questionnaires, interviews, and email testimonials of patients and caregivers (119 patients and 48 caregivers living in Canada), with participants indicating the significant mental, physical, psychological, and life-threatening impacts of CDI. Patients reported the use of antibiotics, such as vancomycin, fidaxomicin, and metronidazole, to treat and cure CDI. Although these treatments were noted by patients to aid in the resolution of symptoms, they were also noted to possibly have a negative impact the gut microbiome. Participants also indicated that CDI can become recurrent, at which point treatment access becomes imperative, because treatment options are limited to a vancomycin taper-pulse regimen and FMT. The patient groups indicated that treatments that can provide stabilization of the microbiome and the desired alleviation of symptoms and improvement in quality of life. Patient experience with fecal microbiota (Rebyota) indicated improved rates of treatment success (i.e., prevention of rCDI) with minimal side effects.
No clinician input was received for this review.
The drug plans expressed concerns about the use of placebo as a comparator in the PUNCH CD3 trial, the use of no preventive therapy as the comparator in the pharmacoeconomic evaluation (rather than long-term oral vancomycin), the variability in coverage of current antibiotic treatments, the expected place in therapy of fecal microbiota (Rebyota), and the misalignment of the sponsor’s pharmacoeconomic evaluation with current clinical practice guidelines (i.e., the 2018 Association of Medical Microbiology and Infectious Disease guidelines, and, specifically, the use of FMT for patients after the second recurrence and antibiotic dosing). The plans expressed implementation concerns regarding the use of fecal microbiota (Rebyota) in rural settings related to limited health care resources, including the clinician time and expertise required for enema administration. The drug plans also had questions regarding the provision of additional doses of fecal microbiota (Rebyota) upon CDI recurrence. Additionally, the plans noted that the provision of fecal microbiota (Rebyota) for the first recurrence of CDI may have a substantial budget impact.
Several of these concerns were addressed in the sponsor’s model:
Quality of life was incorporated in the sponsor’s model using EQ-5D values sourced from literature.
Prevention of rCDI was the primary outcome incorporated into the model.
CDA-AMC addressed some of these concerns as follows:
Administration cost was considered.
A scenario analysis was included to consider a second dose of fecal microbiota (Rebyota) upon recurrence.
CDA-AMC was unable to address the following concerns raised in the patient and drug program input:
The use of placebo as a comparator could not be addressed.
Economic Review
Economic Evaluation
Summary of the Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis that compared fecal microbiota (Rebyota) to no preventive therapy for the prevention of recurrence of CDI in adults after antibiotic treatment for rCDI.1 The modelled population was consistent with the PUNCH CD3, PUNCH CD2, and PUNCH open-label clinical trials and was aligned with the Health Canada indication.
Fecal microbiota (Rebyota) is available in a 150 mL liquid suspension containing microbiota created from healthy human stool.2 The recommended dose of fecal microbiota (Rebyota) is 1 single dose of 150 mL microbiota suspension rectally administered 24 hours to 72 hours after the last dose of antibiotics.2 At the submitted price of $9,125 per 150 mL liquid suspension, the sponsor estimated total fecal microbiota (Rebyota) treatment costs to be $9,125 per patient.1 Patients were treated with fecal microbiota (Rebyota) after antibiotics (94% received vancomycin 125 mg 4 times daily for 2 weeks1 and 6% received fidaxomicin 200 mg twice daily for 10 days)3,4 or antibiotics and no preventive therapy (69% received a vancomycin taper-pulse regimen, 25% received vancomycin 125 mg 4 times daily for 2 weeks,1 and 7% received fidaxomicin 200 mg twice daily for 10 days).5
The clinical outcomes of interest were QALYs and life-years (LYs) over a 5-year time horizon.6 A discount rate of 1.5% was applied to both costs and outcomes.7 Two base-case perspectives were presented: 1 from the perspective of the publicly funded health care payer and 1 from the societal perspective.6 The societal perspective included indirect costs related to patient productivity losses and transportation costs.6
Model Structure
The sponsor submitted a Markov model with an 8-week cycle length that consisted of 5 health states (i.e., rCDI, absence of CDI after rCDI, ileostomy, ileostomy reversal, and death) and 1 colectomy tunnel state (i.e., rCDI to colectomy to ileostomy) (Figure 1).6 All patients enter the model in the rCDI health state and, after 1 round of antibiotic treatment, they either received 1 dose of fecal microbiota (Rebyota) or no preventive therapy.6 Upon receiving treatment, patients who experience treatment success at 8 weeks, defined as the absence of CDI, could transition to the absence-of-CDI-after-rCDI health state.6 Patients who do not experience treatment success could remain in the rCDI health state and receive subsequent treatment with antibiotics.6 After subsequent antibiotic treatment, patients could either experience subsequent treatment success and transition to the absence-of-CDI-after-rCDI health state or they could continue to not experience treatment success and remain in the rCDI health state.6 From the rCDI health state, patients could require colectomy and transition to the colectomy health state or they could die. Patients who enter the colectomy tunnel state will remain there for 1 cycle and transition to the ileostomy health state or the death health state.6 Patients who transition to the ileostomy health state can transition to the ileostomy-reversal health state, remain in the ileostomy health state, or die.6 Patients who transition to the absence-of-CDI-after-rCDI health state could experience sustained response, characterized by the absence of CDI, and remain in the absence-of-CDI-after-rCDI health state. Patients who do not experience a sustained response could transition to the rCDI state or die.6
Model Inputs
The baseline characteristics used to inform the model were based on the safety population of the double-blind, randomized, placebo-controlled, phase III PUNCH CD3 clinical trial (69.1% of participants were female and mean age was 60.1 years).8
The primary measure of efficacy that informed the transition from rCDI to the absence-of-CDI-after-rCDI health state was the probability of treatment success at 8 weeks in the PUNCH CD3 trial, defined as the absence of CDI at 8 weeks.8 Primary results from the PUNCH CD3 trial reported that 70.4% and 58.1% of patients in the fecal microbiota (Rebyota) and placebo arms, respectively, achieved an absence of rCDI after 8 weeks.8 The probability of a sustained response, which was used to determine who remained in the absence-of-CDI-after-rCDI health state, and the probability of a transition back to the rCDI health state were derived from the secondary end points of the PUNCH CD3 trial and a post hoc analysis of the PUNCH CD open-label extension studies.8,9 Of patients who experienced treatment success after 8 weeks with fecal microbiota (Rebyota) and no preventive therapy in the PUNCH CD3 trial, 92.1% and 90.6%, respectively, had a sustained response at 6 months.8 Sustained responses at 12 months and 24 months were estimated to be ████% and ████%, respectively, for fecal microbiota (Rebyota) and 88.2% and 81.9%, respectively, for placebo in a post hoc survival analysis of patients who achieved a primary response and a sustained response at 8 weeks and 6 months in the PUNCH CD2 trial.9 The transition probability of patients in the rCDI health state to the colectomy health state was informed by the literature.10 All patients who underwent a colectomy and did not die were assumed to undergo ileostomy. The transition from the ileostomy health state to the ileostomy-reversal health state was informed by the literature.11
Background mortality was informed by all-cause mortality from Statistics Canada for all health states.6,12 The rCDI and colectomy health states assumed an additional mortality risk based on the literature.6 Mortality for the rCDI health state was informed by a US retrospective comparative study, which showed that patients with CDI had a 10.9% higher annual all-cause mortality risk than people in the general population in 2010 to 2012.13 For the colectomy state, mortality was informed by a US retrospective study that evaluated the colectomy postoperative mortality rate.14
The sponsor assumed that utilities for the absence-of-CDI-after-rCDI and ileostomy-reversal health states would be equal to those in the general population (i.e., 0.842).6,15 The utilities for all other health states (i.e., 0.420 for rCDI, 0.536 for colectomy, and 0.700 for ileostomy) were sourced from the literature.15-17
From the public payer perspective, the sponsor included costs related to drug acquisition, medical care (i.e., health care resource use), and terminal care. Drug-acquisition costs for fecal microbiota (Rebyota) were based on the sponsor’s submitted price.1 Antibiotic drug costs were obtained from the Ontario Drug Benefit Formulary (i.e., vancomycin)18 and the Ontario Exceptional Access Program (i.e., fidaxomicin).19 Medical costs included health care resource use (i.e., hospitalizations, sepsis-related hospitalizations, ICU stays, emergency department visits, general practitioner visits, specialists visits, nurse visits, and stool tests); the frequency of use was informed by multiple literature sources and sponsor-elicited clinician input.20-22 Costs for health care resource use were informed by the Ontario Schedule of Benefits for Physician Services, the Canadian Institute of Health Information, the Ontario Nurse Collective Agreement, and the literature.22-25 Surgery-related costs (i.e., colectomy and ileostomy) were informed by the literature, with ileostomy reversal assumed to be equivalent in cost to colectomy.26,27 Terminal-care costs were informed by a Canadian comparative study that examined costs incurred over 6 months in a palliative care program in rural and urban areas.28 All costs were reported in 2024 Canadian dollars and, when applicable, costs were inflated or discounted using the Bank of Canada’s inflation calculator.7
From the societal perspective, the sponsor additionally included indirect costs, which were associated with travel expenses and lost productivity hours for a patient, based on a human capital approach. Travel expenses were calculated for the number of general practitioner and specialist visits associated with each health state. The average travel distance was informed by the 2021 Canadian Digital Health Survey and an analysis that evaluated travel to and from gastroenterology appointments at a clinic in Ontario for general practitioner and specialist visits, respectively.29,30 Total cost per trip was calculated using the aforementioned average distance, the prescribed rate per kilometre determined by the Canada Revenue Agency, and parking costs, which were informed by the University Health Network parking rates.31,32 Productivity loss was calculated based on wages lost from the number of days absent from work. Lost wages were calculated from the average hourly wage in Canada for people 25 years and older, the average number of hours worked per day, and the employment rate for individuals 25 years and older, all informed by Statistics Canada.33-35 The proportion and number of days patients with active and resolved rCDI who would be absent from work was informed by an observational, cross-sectional study in which patients with self-reported CDI in the US completed a quality-of-life survey.36
Summary of the Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (2,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar; the probabilistic findings presented here.
Base-Case Results
From the publicly funded health care payer perspective, fecal microbiota (Rebyota) dominated no preventive therapy (fecal microbiota [Rebyota] was associated with an incremental QALY gain of 0.14 and a cost savings of $2,135) over the five-year time horizon (Table 3).6 Fecal microbiota (Rebyota) had a 87% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained.
In the sponsor’s analysis conducted from the societal perspective, fecal microbiota (Rebyota) also dominated no preventive therapy (fecal microbiota [Rebyota] was associated with an incremental QALY gain of 0.14 and cost savings of $8,592) over the five-year time horizon (Table 3).6 Fecal microbiota (Rebyota) had a 91% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained.
In both the health care payer and societal perspective, 33% of incremental QALYs gained were observed during the trial period. In both the health care payer and societal perspective, fecal microbiota (Rebyota) was associated with a 0.07 LY gain compared with no preventive therapy.
Full disaggregated results are presented in Table 10 in Appendix 3. Results were driven by drug-acquisition costs associated with fecal microbiota (Rebyota), which were entirely offset by medical costs associated with the management of CDI (Table 10). The key cost driver was medical costs, with the majority of those attributed to hospitalization costs in the rCDI health state. Medical costs associated with the rCDI health state contributed 85% of the medical cost savings associated with fecal microbiota (Rebyota). From the societal perspective, indirect costs also contributed to a large proportion (76%) of the cost savings associated with fecal microbiota (Rebyota) compared with no preventive therapy. From both the health care payer and societal perspectives, the incremental QALY benefits observed for fecal microbiota (Rebyota) were entirely driven by patients spending more time in the absence-of-CDI-after-rCDI health state (Table 10). Scatterplots in the sponsor’s base case indicated the uncertainty in the finding that fecal microbiota (Rebyota) is less costly than no preventive therapy; in 743 of 2,000 iterations in the public health care payer base case, fecal microbiota (Rebyota) was associated with incremental costs (Figure 2 in Appendix 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no preventive treatment ($/QALY) | |
|---|---|---|---|---|---|---|
Results from the publicly funded health care payer perspective | ||||||
No preventive therapy | 33,486 | Reference | 3.62 | Reference | Reference | |
Fecal microbiota (Rebyota) | 31,350 | −2,135 | 3.76 | 0.14 | Dominant (less costly, more effective) | |
Results from the societal perspective | ||||||
No preventive therapy | 51,998 | Reference | 3.63 | Reference | Reference | |
Fecal microbiota (Rebyota) | 43,406 | −8,592 | 3.77 | 0.14 | Dominant (less costly, more effective) | |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.6
Sensitivity and Scenario Analyses Results
Scenario analyses conducted by the sponsor included subgroup analyses of patients younger than 65 years and 65 years or older, treatment of rCDI as early as the first recurrence, an additional dose of fecal microbiota (Rebyota) for patients who experience a recurrence, and alternative assumptions on discounting and mortality. All scenario analyses for the Canadian health care payer and societal perspectives had conclusions similar to the sponsor’s base case; that is, fecal microbiota (Rebyota) remained dominant or cost-effective compared to no preventive therapy.
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
The comparative efficacy of fecal microbiota (Rebyota) is uncertain. In the sponsor’s model, the primary efficacy outcome (treatment success) was informed by the fecal microbiota (Rebyota) and placebo arms of the PUNCH CD3 trial, with the placebo arm representing the current standard of care (i.e., no preventive therapy after treatment of the recurrence with antibiotics).6,37 According to the CDA-AMC Clinical Review Report, there was moderate certainty that treatment with fecal microbiota (Rebyota) would result in a clinically important increase in the treatment success rate over 8 weeks compared to placebo. The sponsor’s submitted base case attributed different antibiotic treatment distributions to the fecal microbiota (Rebyota) arm (94% received vancomycin 125 mg 4 times daily for 2 weeks1 and 6% received fidaxomicin 200 mg twice daily for 10 days) and the no preventive therapy arm (69% received a vancomycin taper-pulse regimen, 25% received vancomycin 125 mg 4 times daily for 2 weeks,1 and 7% received fidaxomicin 200 mg twice daily for 10 days), based on the antibiotic treatment observed in the PUNCH CD3 trial and sponsor-elicited clinical expert opinion, respectively.3,8 As noted in the sponsor’s pharmacoeconomic report and the clinical group input received by CDA-AMC, and corroborated by clinical expert feedback obtained by CDA-AMC, a vancomycin taper-pulse regimen is commonly used for patients who experience 2 or more recurrences of CDI.38 However, the CDA-AMC Clinical Review Report noted that it is unknown whether patients in both groups received an appropriate antibiotic regimen of vancomycin taper-pulse for the treatment of the qualifying CDI episode. Therefore, the relevance of using placebo as a comparator is uncertain, as the CDI treatment in the PUNCH CD3 trial was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice.38 Consequently, it is uncertain whether the efficacy of no preventive therapy in clinical practice is equivalent to the placebo arm of the PUNCH CD3 study.
Additionally, in the sponsor’s model, if patients did not achieve treatment success after the first cycle, patients were assumed to receive subsequent antibiotic treatment upon recurrence.6 The efficacy of the subsequent antibiotic treatment was derived from a subgroup of patients who experienced 3 or more recurrences in the placebo arm of the PUNCH CD3 trial.3 However, in the model, the mix of subsequent antibiotic treatment was based on sponsor-elicited clinician opinion, rather than the PUNCH CD3 trial.5 Because the efficacy is derived from the PUNCH CD3 trial, it is inappropriate to attribute a different antibiotic treatment mix for costs.
As well, conventional multidose FMT, which the clinical experts indicated is currently being used for the treatment of rCDI in Canada, was not included as a comparator in the sponsor’s submitted model.6 As noted in the CDA-AMC Clinical Review Report, no direct or indirect evidence was submitted to evaluate fecal microbiota (Rebyota) relative to other currently used therapies for the prevention of rCDI, including FMT. The lack of conventional FMT is a major limitation, as literature suggests that FMT can achieve clinical response rates of more than 90% (response rates range from 44% up to more than 90%, and it appears that patients may require multiple FMTs to achieve clinical response rates of more than 80%).38
In its reanalysis, CDA-AMC revised the distribution of antibiotic treatment for the no preventive therapy arm to reflect the antibiotic treatment distribution in the PUNCH CD3 trial.37 Similarly, the distribution of subsequent antibiotic treatment was changed to reflect that in the PUNCH CD3 trial.37
CDA-AMC was unable to address the lack of FMT as a comparator in the model because of the gap in evidence. Similarly, CDA-AMC was unable to address limitations regarding whether the placebo arm represented the current standard of care.
The long-term clinical efficacy of fecal microbiota (Rebyota) is uncertain. Long-term efficacy in the sponsor’s economic model was based on the proportion of patients who experienced a sustained response after treatment success at 6 months, 12 months, and 24 months.6 The sustained response at 6 months with fecal microbiota (Rebyota) and no preventive therapy was informed by the PUNCH CD3 trial (92.1% and 90.6%, respectively).8 The sustained response at 12 months and 24 months was informed by the sponsor’s survival analysis, which was based on a subgroup of patients who experienced treatment success in the PUNCH CD2 trial.3 Among the patients who were treated with 1 dose of fecal microbiota (Rebyota) in the PUNCH CD2 trial, there was only 1 observation of 1 recurrence at 6 months, which translated to a sustained response of ████%.3 The sustained response observed for fecal microbiota (Rebyota) at 6 months from the PUNCH CD2 trial was used as a proxy for the 12-month and 24-month sustained response in the sponsor’s base case.3 Patients treated with placebo in the PUNCH CD2 trial were observed to experience recurrences at both the 12-month and 24-month time points, which translated to sustained responses of 88.2% and 81.9%, respectively.3 This approach to estimating long-term efficacy is inappropriate, as the efficacy values incorporated were from different patient populations; the patients enrolled in the PUNCH CD2 trial were not the same as the patients enrolled in the PUNCH CD3 trial. This is highlighted by the lack of face validity in the approach, as the sustained response at 6 months in the PUNCH CD2 trial, based on the sponsor’s survival analysis, was higher than the sustained response at 6 months in the PUNCH CD3 trial. According to clinical expert feedback received by CDA-AMC for this review, there is no reason why the sustained response would be higher at 12 months and 24 months for fecal microbiota (Rebyota).
According to the CDA-AMC Clinical Review Report, the sponsor’s post hoc survival analysis has several methodological limitations, including a lack of reporting on the number of patients who were censored at various follow-up times, and limitations stemming from the small sample size and very few events. Finally, according to the CDA-AMC Clinical Review Report, no conclusions can be drawn regarding the sustained response beyond 6 months, and the conclusion regarding this outcome at 6 months in patients who achieved previous treatment success at 8 weeks was that there was no clinically important difference between fecal microbiota (Rebyota) and placebo. Therefore, there may be no added benefit with fecal microbiota (Rebyota) in terms of sustained response at 6 months.
In its reanalysis, CDA-AMC assumed that, after 6 months, the probability of recurrence would not change.
Additionally, in alignment with the conclusion of the Clinical Review Report, there may be no added sustained response benefit for fecal microbiota (Rebyota) compared to no preventive therapy at 6 months. CDA-AMC conducted a scenario analysis in which the sustained response for fecal microbiota (Rebyota) and no preventive therapy were the same at 6 months and beyond.
Model structure does not reflect clinical expectations. In the sponsor’s pharmacoeconomic model, patients with rCDI can remain in the rCDI health state or transition to 1 of the following health states: absence of CDI after rCDI, colectomy, or death.6 This model structure assumes that all patients with rCDI who do not experience treatment success, surgery, or death will have a similar experience. Clinical expert feedback obtained by CDA-AMC indicated that patients with rCDI are not homogenous and may experience a different severity of symptoms and different impacts of CDI. For example, some patients may not revert to their pre-CDI baseline of bowel habits, whereas chronic vancomycin suppression therapy can mitigate symptoms associated with an episode of CDI in other patients. As well, some patients may experience different severities of CDI, from mild diarrhea to fulminant colitis with systemic toxicity and stock.39 Although patients will have different experiences during and after the occurrence of an rCDI episode, the sponsor’s model does not account for heterogeneity within the rCDI health state. The implications of heterogeneity in health states have been well documented in the literature.40 The heterogeneity of the rCDI health state has implications for costs and quality of life associated with the health state, as outlined in subsequent limitations.
CDA-AMC was unable to address the homogenous nature of the rCDI health state in the sponsor’s model. Because patients who are treated with fecal microbiota (Rebyota) spend less time in the rCDI health state than those treated with no preventive therapy, if the rCDI health state is reflective of a poorer quality of life and higher costs than the average patient with rCDI, this approach will favour fecal microbiota (Rebyota).
Utilities do not meet face validity and are associated with uncertainty. Because of limitations in the sponsor’s model structure stemming from heterogeneity in the rCDI health state (refer to previous model structure limitation), the utility value assigned to the rCDI health state is likely not reflective of all patients in that state. The utility value of the rCDI health state was informed by a retrospective study conducted in the UK with an observation period of 2012 to 2014, in which the EQ-5D estimate of 64 patients with rCDI and 64 patients with a first CDI episode who were hospitalized was reported to be 0.42 ± 0.29.16 According to clinical expert feedback, corroborated by the sponsor’s pharmacoeconomic report, not all patients with rCDI will be hospitalized and they will have different experiences with severity of disease and the potential application of chronic vancomycin suppression therapy. Thus, the assumption that all patients in the rCDI health state will experience a utility value similar to that in patients who are hospitalized with rCDI does not meet face validity. If the utility estimate of the rCDI health state is underestimated compared to the true utility experienced by patients with rCDI, there would be fewer incremental QALYs for fecal microbiota (Rebyota), as these patients would spend less time in the rCDI health state than those who received no preventive therapy.
Additionally, the assumption that patients who experience treatment success return to baseline utility, compared to the general population, does not meet face validity. Clinical expert feedback obtained by CDA-AMC indicated that patients who experience treatment success may not return to their pre-CDI baseline bowel habits and may suffer from lingering mental health impacts, such as posttraumatic stress disorder, as noted in patient group input. If the utility value in the absence-of-CDI-after-rCDI health state is lower than assumed by the sponsor, this would result in fewer incremental QALYs for fecal microbiota (Rebyota), because patients who receive fecal microbiota (Rebyota) would spend more time in that state than those who receive no preventive therapy.
The sponsor’s submitted model derived utility benefits based on the time spent in each health state and reported an incremental QALY benefit of 0.14, which represents approximately 2 quality-adjusted months gained over 5 years with fecal microbiota (Rebyota).6 HRQoL was reported in the PUNCH CD3 trial, based on C. difficile Health-Related Quality-of-Life (Cdiff32) Questionnaire scores.37 As noted in the CDA-AMC Clinical Review Report, fecal microbiota (Rebyota) may result in little to no difference in HRQoL compared to placebo at 8 weeks, and results were too uncertain to draw any conclusions at 6 months.37 Therefore, the incremental QALY benefit finding suggesting that fecal microbiota (Rebyota) will result in an improvement in quality of life is uncertain.
CDA-AMC was unable to address this limitation because of limitations in the sponsor’s model structure. If utilities were lower than assumed by the sponsor for the absence-of-CDI-after-rCDI health state and higher than assumed by the sponsor for the rCDI state, as expected based on clinical feedback received by CDA-AMC for this review, this would bias results in favour of fecal microbiota (Rebyota).
Medical costs are associated with uncertainty and do not reflect clinical practice. Because of limitations in the sponsor’s model structure stemming from heterogeneity in the rCDI health state (refer to previous model structure limitation), the health care resource costs assigned to the rCDI health state are likely not reflective of all patients in that state. The sponsor relied on published literature and clinical expert opinion to assign and validate health care resource use for each acute case of recurrent CDI (i.e., the published literature cited by the sponsor estimates the proportion of patients that require health care resources when experiencing a recurrent episode of CDI).20-22 As outlined previously, as the rCDI health state is heterogenous and includes people with an acute episode of recurrent CDI and those who are receiving chronic vancomycin suppression therapy to manage their chronic rCDI, the assumption that costs associated with an acute occurrence of an episode of recurrent CDI will be similar in all patients with rCDI is uncertain. According to clinical expert feedback, health care resource use during an acute episode can vary; some patients will only require a vancomycin taper-pulse regimen to achieve treatment success, whereas others may require surgery and an ICU stay to combat fulminant CDI and septic shock. This is in contrast to people in the general population with rCDI, as the costs associated with an acute episode of rCDI is drastically different than in patients whose disease is well controlled on chronic vancomycin suppression therapy (who would be expected to require minimal health care resource use to manage their rCDI). As well, according to the CDA-AMC Clinical Review Report, based on data from the PUNCH CD3 trial, there was insufficient evidence to draw any conclusions about the effect of fecal microbiota (Rebyota), relative to placebo, on hospitalizations and ICU admissions. As such, whether fecal microbiota (Rebyota) will lead to reductions in medical costs, and to what extent, is uncertain based on the available clinical evidence.
CDA-AMC was unable to address this limitation because of limitations in the sponsor’s model structure. The sponsor’s approach likely favours fecal microbiota (Rebyota); if rCDI health state costs were lower, patients receiving no preventive therapy would accrue fewer health state costs because those who receive no preventive therapy spend more time in the rCDI state than those who receive fecal microbiota (Rebyota).
Some outcomes assumed by the sponsor were not reflective of the clinical trial evidence and did not align with clinical practice in Canada. The sponsor’s submitted model included an additional mortality risk associated with the rCDI and colectomy health states, along with background all-cause mortality, with the rates of additional mortality risk being sourced from the literature.1,13,14 The rCDI health state mortality risk was based on a US study of the one-year all-cause probability of mortality attributed to patients with CDI compared to those without CDI.13 The study did not stratify the patient population by the number of recurrences previously experienced by the patient, but concluded that a 10.9% increased mortality risk exists for patients with CDI.13 The application of mortality due to rCDI is uncertain because mortality was not a clinical trial outcome in the PUNCH CD3 trial and there was no evidence from the PUNCH CD3 or PUNCH CD2 studies, or the PUNCH open-label extension study, that fecal microbiota (Rebyota) would generate a mortality benefit compared to no preventive therapy. More specifically, the 2 deaths reported over the trial’s duration in patients who received fecal microbiota (Rebyota) were not related to the study drug or the enema procedure, according to the sponsor; no deaths were observed in the placebo arm.37,41,42 According to clinical expert feedback obtained by CDA-AMC for this review, rCDI should have almost no mortality, because people with rCDI will receive chronic vancomycin suppression therapy unless they experience delayed diagnosis and treatment. Although clinical expert feedback acknowledged that rCDI could result in a morality risk, the experts indicated that it is uncertain whether fecal microbiota (Rebyota) would result in a decrease in mortality risk because of the absence of evidence. As well, clinical expert feedback indicated that a 10.9% annual mortality risk is not representative of clinical practice in Canada, where expected mortality associated with rCDI is very low. In 2023, the Public Health Agency of Canada reported on CDI-attributable mortality, and estimated it to be between 1.2% to 2.6% between 2017 to 2021.43 This was deemed by clinical experts to be more representative of rCDI mortality risk.
In addition to mortality stemming from having rCDI, the sponsor’s model derived a 0.2 incremental LY gain for fecal microbiota (Rebyota) in surgical health states (i.e., colectomy, ileostomy, and ileostomy reversal), representing approximately 2 months gained with fecal microbiota (Rebyota) compared with no preventive therapy. As noted in the CDA-AMC Clinical Review Report, there was 1 reported colectomy in the fecal microbiota (Rebyota) arm of the PUNCH CD3 trial and no colectomy events in the placebo arm.37 As such, there is no clinical evidence to support lower rates of colectomy with fecal microbiota (Rebyota). Further, there is no clinical evidence that fecal microbiota (Rebyota) would be associated with an incremental LY gain related to the prevention of colectomy.37 Clinical expert feedback obtained by CDA-AMC indicated that fecal microbiota (Rebyota) could reduce the proportion of patients who will experience a subsequent recurrence, based on the difference in treatment success, and that it is clinically plausible that reductions in recurrence could result in fewer colectomies, as colectomy can be an rCDI complication. However, based on clinical expert feedback, the probability of colectomy in the sponsor’s model (7.3% annually) was higher than would be expected in clinical practice in Canada, as colectomies are very rare. A 2020 retrospective cohort study conducted in Ontario showed that the proportion of patients with community-onset and hospital-onset rCDI who required a colectomy was 0.4% and 1.3%, respectively.44 Additionally, a 2017 population-based cohort study conducted in Ontario showed that the risk of colectomy for patients with community-onset rCDI was 0.6%.45 Thus, the assumption that patients with rCDI in Canada would undergo a colectomy at a rate similar to patients in the US may not be applicable, based on clinical expert feedback and studies conducted in Canada.
To estimate the mortality risk associated with colectomy, the sponsor used a US retrospective study of 30-day mortality in patients with CDI who underwent a colectomy (35.2%).14 This approach assumes that the rate of death during the 30 days after the procedure is constant for 8 weeks after the procedure, which may have limited face validity, as a Canadian study that showed a similar 30-day mortality rate (33%) had a median time to death of 8 days, and that patients who did not die were discharged after a mean of 24 days after colectomy.46
In its reanalysis, CDA-AMC revised the annual mortality risk for the rCDI health state to the 2021 value for the Public Health Agency of Canada’s CDI-attributable mortality risk.43 As well, to align with the care paradigm in Canada, CDA-AMC conducted a reanalysis in which the proportion of patients who transition from the rCDI to the colectomy health state is 0.7% annually, based on a weighted average of patients with community-onset and hospital-onset rCDI who required a colectomy.44 In addition, CDA-AMC adjusted the 8-week mortality risk for the colectomy health state to reflect the 30-day mortality risk.14,43
To explore the possibility that fecal microbiota (Rebyota) does not result in a mortality or colectomy benefit, which is consistent with findings from the PUNCH CD3 trial, CDA-AMC conducted a scenario analysis in which mortality risk and colectomy risk were both removed from the rCDI health state.
Alternative dosing of fecal microbiota (Rebyota) was not included in the sponsor’s base case. The sponsor’s base case only evaluated the administration of 1 dose of fecal microbiota (Rebyota) upon recurrence of CDI (aligned with the product monograph).2 Clinical expert feedback noted that, based on clinical experience with FMT, more than 1 dose of FMT may be administered per treatment course in FMT programs. Although the clinical experts acknowledged that a only single dose of fecal microbiota (Rebyota) is indicated per recurrence, if permitted or available, there may be cases in which multiple doses of fecal microbiota (Rebyota) per recurrence would be administered. Therefore, whether multiple doses of fecal microbiota (Rebyota) would be administered per treatment course, should this be permitted, is uncertain, especially because the PUNCH CD3 trial only investigated the administration of a single dose; however, 2 doses of fecal microbiota (Rebyota) were studied in 1 treatment arm of the PUNCH CD2 trial.
Additionally, the sponsor’s base-case analysis did not evaluate the administration of further doses of fecal microbiota (Rebyota) upon subsequent recurrences, which is permitted, according to the fecal microbiota (Rebyota) product monograph.2 However, the sponsor did submit a scenario analysis that explored the impact of using fecal microbiota (Rebyota) to treat subsequent recurrences after a patient has already received fecal microbiota (Rebyota) for a prior recurrence. According to clinical expert feedback received by CDA-AMC for this review, if patients with rCDI experience a relapse in symptoms after fecal microbiota (Rebyota) treatment, the subsequent use of fecal microbiota (Rebyota) would be considered.
To address uncertainty in the expected use of fecal microbiota (Rebyota), 2 scenario analyses were conducted: in the first, patients received 2 doses of fecal microbiota (Rebyota) for their first recurrence in the model (off-label dosing), with the assumption that the efficacy is equivalent to 1 dose of fecal microbiota (Rebyota) (in the absence of available evidence for 2 doses); and, in the second, patients were able to receive an additional treatment course of fecal microbiota (Rebyota) for their next recurrence, aligned with the sponsor’s submitted scenario analysis.
The calculated productivity loss is likely inappropriate. To calculate productivity loss, the sponsor used a human capital approach that involved wage, employment, and loss of working hours due to illness.1 The employment rate was derived from Statistics Canada and was for individuals 25 years and older.35 This is inappropriate, as the cited employment rate of the general population 25 years and older of Canada is not expected to reflect the patient population of rCDI. In the PUNCH CD3 trial, the median age at baseline was 63 years, and 54.3% of patients were 65 years or older.37 This is corroborated by published literature and by clinical expert feedback, which indicated that rCDI disproportionately affects patients 65 years and older.10,37,38As such, the group of patients 25 years and older is not reflective of the expected patient population who will receive fecal microbiota (Rebyota).6,34
Additionally, the homogenous nature of the rCDI health state may introduce uncertainty to productivity loss, as the model assumes that wages and working time of the general population is reflective of that in patients with rCDI. This assumption may not be appropriate, as patients with rCDI can experience a range of severity, and the disease may be well controlled with vancomycin suppression therapy, which will impact the patient’s ability to work. The proportion of patients unable to work because of rCDI was informed by an observational, cross-sectional study of patients in the US with an active or previous CDI diagnosis, in which it was reported that 47.2% and 25.5% of patients with an active or previous CDI diagnosis, respectively, were unable to work.36 The heterogenous nature of rCDI, as highlighted in the previous limitation, makes the likelihood that 47.2% of patients with rCDI would be unable to work uncertain, as the observational, cross-sectional study did not stratify patients by severity of disease. Although clinical expert feedback aligned with the proportion of patients with active CDI who are unable to work, it indicated that it is unreasonable to assume that patients with resolved CDI, in which their symptoms are under control, would be unable to work; however, given the possible post-CDI effects on bowel habits and mental health, the proportion of patients with resolved CDI who are unable to work may be plausible.
The sponsor used the number of days away from work because of illness associated with the absence-of-CDI-after-rCDI and the rCDI health states to calculate the lost productivity. The sponsor’s cited evidence demonstrated that patients were unable to fulfill their professional responsibilities for a mean of 118.0 days and a median of 60.0 days during infection, and for a mean 310.0 days and a median of 62.5 days after CDI infection.36 As such, the sponsor assumed that among patients who were off work, they would be off work for 56 days per 8-week cycle for both the rCDI and absence-of-CDI-after-rCDI health states, which represents every day for 8 weeks.36 This is inappropriate, as most people do not work every day for 56 days straight. The average usual hours for people living in Canada in 2024 was 37.6 hours per week.47 Given the sponsor’s estimate of 7.36 hours worked per day (informed by Statistics Canada33), this translates to 5.1 days lost per week, or 40.9 days lost per 8-week cycle if patients were unable to work over an 8-week time period. Additionally, the studied population informing time off work may not be representative of the patient population that will receive fecal microbiota (Rebyota), as baseline characteristics indicated that ████% of patients were older than 50 years, ████% of patients were female, and the number of days unable to work was not stratified by the number of recurrences, so patients who experienced only 1 CDI episode could be included.
CDA-AMC revised the employment rate and accounted for retirees in its reanalysis by assuming that the proportion of patients 65 years and older (54.3%) from the PUNCH CD3 trial37 was multiplied by the 65-years-and-older employment rate (14.4%) and the proportion of patients younger than 65 years (45.7%) from the PUNCH CD3 trial37 was multiplied by the 25-years-and-older employment rate (62.4%), sourced from Statistics Canada in 2024,35 to determine the proportion of patients with rCDI who are employed.
Additionally, CDA-AMC revised the number of days off per 8-week model cycle to 40.9 days (i.e., assuming a 5.1-day work week), based on the usual average hours worked by people in Canada.33,47 To explore the impacts of productivity loss, CDA-AMC conducted a scenario analysis in which the proportion of patients with resolved rCDI who would be unable to work was revised to 0%.
Administration costs were not considered in the model. The sponsor’s submitted model does not consider the administration costs for fecal microbiota (Rebyota). This assumption was deemed to be inappropriate, based on clinical expert feedback received for this review, which indicated that FMT involves the administration of an enema by a health care provider (physician or a nurse under physician supervision), nursing support, a private room with an attached washroom, housekeeping time, and personal protective equipment (gloves, gowns, and masks). As such, it is inappropriate to assume that the administration of fecal microbiota (Rebyota) will be $0.
Additionally, the sponsor did not include transportation costs associated with the administration of fecal microbiota (Rebyota) in the societal perspective. This may be inappropriate, as patients in the model are expected to accrue transportation costs when visiting general practitioners and specialists, whereas transportation costs associated with fecal microbiota (Rebyota) were excluded, which will favour the fecal microbiota (Rebyota) arm.
In its reanalysis, CDA-AMC derived a cost for fecal microbiota (Rebyota) administration using a bottom-up approach, based on clinical expert feedback on administration costs (Table 14).
Additionally, the key assumptions outlined in Table 4 were made by the sponsor and have been appraised by CDA-AMC.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Patient baseline characteristics in the sponsor’s model were represented by the safety population of the PUNCH CD3 trial. | Uncertain. The sponsor arbitrarily incorporated baseline characteristics from the safety population of the PUNCH CD3 trial, rather than the ITT population. The CDA-AMC base-case reanalysis maintains the sponsor’s assumption. |
AEs were not included in the sponsor’s model, as they were assumed to not have an impact on the economic model results. | Uncertain. As noted in the CDA-AMC Clinical Review Report, treatment-emergent AEs were higher in the fecal microbiota (Rebyota) group than in the placebo group. Clinical experts also indicated that there may be long-term, unintended effects of manipulating a patient’s microbiome, which could affect the risk of developing a range of conditions over time, the impact of which is currently unknown. As AE management was not captured in the model, total costs and possible utility detriments associated with fecal microbiota (Rebyota) may be underestimated. |
Patients in the ileostomy-reversal health state were assumed to have utilities that reflect those in the general population. | Inappropriate. Clinical expert feedback indicated that patients who have been treated for rCDI may not return to their baseline quality of life. |
AE = adverse event; CDA-AMC = Canada's Drug Agency; ITT = intention to treat; rCDI = recurrent Clostridioides difficile infection.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
The CDA-AMC base case was derived by making changes to model parameter values and assumptions, in consultation with clinical experts. CDA-AMC undertook reanalyses that addressed the limitations of the sponsor’s model, as summarized in Table 5. CDA-AMC was unable to address the following key limitations of the sponsor’s submitted model: the lack of FMT as a comparator; limitations regarding the placebo arm’s representativeness of the current standard of care; the homogenous nature of the rCDI health state; the lack of face validity of utility estimates; and the uncertainty associated with medical costs for rCDI.
Table 5: CDA-AMC Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Ileostomy indirect costs for no preventive therapy | Calculated based on ileostomy-reversal indirect costs | Corrected based on the ileostomy indirect costs |
2. Sponsor transcription error | No preventive therapy sustained a response at 24 months of 81.3% | No preventive therapy sustained a response at 24 months of 81.9% |
Changes to derive the CDA-AMC base case | ||
1. Sustained response at 12 months and 24 months and transition probability from the absence-of-CDI-after-rCDI to the rCDI health states | Fecal microbiota (Rebyota) Sustained response
Probability of recurrence
No preventive therapy Sustained response
Probability of recurrence:
| Fecal microbiota (Rebyota) Sustained response
Probability of recurrence
No preventive therapy Sustained response
Probability of recurrence:
|
2. rCDI mortality | 10.9% annually | 2.2% annually |
3. Proportion of the rCDI health state transitioning to the colectomy health state | 7.3% annually | 0.7% annually |
4. Colectomy mortality | 55.5% per 8-week cycle | 35.2% per 8-week cycle |
5. No preventive therapy antibiotic treatment distribution | Vancomycin taper-pulse regimen: 24.5% Vancomycin standard regimen: 68.9% Fidaxomicin: 6.6% | Vancomycin taper-pulse regimen: 0.0% Vancomycin standard regimen: 93.6% Fidaxomicin: 6.4% |
6. Administration of fecal microbiota (Rebyota) | $0.00 | $447.12 (refer to Table 14) |
Additional changes only applied to the societal perspective analysis | ||
7. Productivity loss |
|
|
CDA-AMC base case from the societal perspective | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 + 7 |
CDA-AMC base case from the publicly funded health care payer perspective | ― | Reanalysis 1 + 2 + 3 + 4 + 5 + 6 |
CDA-AMC = Canada's Drug Agency; rCDI = recurrent Clostridioides difficile infection.
The CDA-AMC base case from the public health care payer perspective estimated that fecal microbiota (Rebyota) was $6,328 more expensive and yielded 0.03 more QALYs than no preventive therapy (Table 6). Based on the CDA-AMC reanalysis, fecal microbiota (Rebyota) was associated with an ICER of $203,679 per QALY gained compared to no preventive therapy (Table 6). Fecal microbiota (Rebyota) had a 33% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. The stepped analysis is presented in Table 11.
From the societal perspective, the CDA-AMC base-case analysis estimated that fecal microbiota (Rebyota) was $4,547 more expensive and yielded 0.03 more QALYs than no preventive therapy (Table 6). Based on the CDA-AMC reanalysis, fecal microbiota (Rebyota) was associated with an ICER of $136,782 per QALY gained compared to no preventive therapy (Table 6). Fecal microbiota (Rebyota) had a 37% probability of being cost-effective at a WTP threshold of $50,000 per QALY gained. The stepped analysis is presented in Table 12.
For both public health care payer and societal perspectives, the CDA-AMC base-case results were similar, albeit the societal perspective ICER was lower, given the increased productivity from no preventive therapy. The key cost driver was treatment-acquisition costs, as they comprised 100% of the incremental costs attributed to fecal microbiota (Rebyota) (Table 13).
As in the sponsor’s base case, the incremental QALY gain in both perspectives was driven by patients spending more time in the absence-of-CDI-after rCDI health state when treated with fecal microbiota (Rebyota) compared to no preventive therapy (Table 13). In the CDA-AMC base case, the majority (70%) of the incremental QALY gains were observed within the trial period. Scatterplots of the CDA-AMC base-base results highlight the uncertainty associated with an incremental benefit of fecal microbiota (Rebyota) compared to no preventive therapy, because in 40% and 38% of the iterations in the public health care payer and societal perspectives, respectively, fecal microbiota (Rebyota) was associated with no benefit or with a QALY detriment compared to no preventive therapy (refer to Figure 3 and Figure 4).
Table 6: Summary of the CDA-AMC Economic Evaluation Results (Probabilistic)
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. no preventive treatment ($/QALY) |
|---|---|---|---|---|---|
Results from the publicly funded health care payer perspective | |||||
No preventive therapy | 29,986 | Reference | 3.80 | Reference | Reference |
Fecal microbiota (Rebyota) | 36,314 | 6,328 | 3.83 | 0.03 | 203,679 |
Results from the societal perspective | |||||
No preventive therapy | 38,194 | Reference | 3.81 | Reference | Reference |
Fecal microbiota (Rebyota) | 42,742 | 4,547 | 3.85 | 0.03 | 136,782 |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Scenario Analysis Results
CDA-AMC undertook price-reduction analyses for the public health care payer and societal perspectives. In the CDA-AMC base case, to be considered cost-effective at a WTP threshold of $50,000 per QALY gained in the health care payer and societal perspectives, the price of fecal microbiota (Rebyota) would need to be reduced by 53%, corresponding to $4,288.75 per dose, or 32%, corresponding to $6,205.00 per dose, respectively (Table 7).
Table 7: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost ($) | ICERs for fecal microbiota (Rebyota) vs. no preventive therapy ($/QALY) | |||
|---|---|---|---|---|---|
Price reduction | $ | Sponsor base case (health care payer) | Sponsor base case (societal) | CDA-AMC reanalysis (health care payer) | CDA-AMC reanalysis (societal) |
No price reduction | 9,125.00 | Dominant | Dominant | 203,679 | 136,782 |
10% | 8,212.50 | Dominant | Dominant | 174,308 | 109,334 |
20% | 7,300.00 | Dominant | Dominant | 144,937 | 81,886 |
30% | 6,387.50 | Dominant | Dominant | 115,566 | 54,439 |
40% | 5,475.00 | Dominant | Dominant | 86,195 | 26,991 |
50% | 4,562.50 | Dominant | Dominant | 56,824 | Dominant |
60% | 3,650.00 | Dominant | Dominant | 27,453 | Dominant |
70% | 2,737.50 | Dominant | Dominant | Dominant | Dominant |
80% | 1,825.00 | Dominant | Dominant | Dominant | Dominant |
90% | 912.50 | Dominant | Dominant | Dominant | Dominant |
CDA-AMC = Canada's Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CDA-AMC conducted several scenario analyses on the CDA-AMC base case to explore the remaining sources of uncertainty (results are shown in Table 15). If subsequent doses of fecal microbiota (Rebyota) are permitted upon recurrence after fecal microbiota (Rebyota) treatment, which is aligned with product monograph dosing, the ICER is expected to increase to $257,843 and $256,069 per QALY gained for the health care payer and societal perspectives, respectively. If fecal microbiota (Rebyota) does not result in a benefit in terms of colectomy or mortality compared with no preventive therapy, then the ICER is also expected to increase (to $212,188 and $158,901 per QALY gained for the health care payer and societal perspectives, respectively). Finally, in a scenario that assumes that there is no difference in sustained response between fecal microbiota (Rebyota) and no preventive therapy after 6 months, in line with the CDA-AMC Clinical Review Report, the ICER increases to $476,035 and $407,736 per QALY gained in the health care payer and societal perspectives, respectively.
In the scenario in which people with resolved rCDI all return to work, the ICERs were lower than those in the CDA-AMC base case; however, the scenario analysis remained not cost-effective at a WTP of $50,000 per QALY gained.
Issues for Consideration
Generalizability of the PUNCH CD3 trial results: As noted in the CDA-AMC Clinical Review Report, patients with some comorbidities (e.g., Crohn disease, ulcerative colitis, inflammatory bowel disease, irritable bowel syndrome, and immunocompromised conditions) were excluded from the PUNCH CD3 trial.37 Patients with comorbidities were permitted in the PUNCH open-label extension study,37,42 but the proportion of such patients was small, limiting the generalizability of the findings and the certainty of evidence for patients with comorbid conditions.
Storage of fecal microbiota (Rebyota): Fecal microbiota (Rebyota) must be stored in an ultracold freezer, at a temperature of −90°C to −60°C (–76ºF to –130ºF), or stored in the refrigerator at 2°C to 8°C (36°F to 46°F) for up to 5 days.2 There may be implementation issues with storage because of these temperature requirements. Costs and considerations related to storage and implementation were not considered in the economic evaluation of either the sponsor or CDA-AMC.
Availability of fidaxomicin: Public reimbursement of fidaxomicin is jurisdiction-dependent. Currently, British Columbia, Alberta, Saskatchewan, Ontario, New Brunswick, Nova Scotia, and Prince Edward Island provide public coverage of fidaxomicin through the Exceptional Access Program or the Special Access Program.19,48-50 However, this is subject to change, as fidaxomicin is currently undergoing a CDA-AMC nonsponsored reimbursement review.51
Confidential price for comparators: Analyses by the sponsor and the CDA-AMC are based on publicly available list prices for most comparators. Actual costs paid by public drug plans are unknown, and any potential confidential rebates are not reflected in the analyses.
Overall Conclusions
Evidence from the PUNCH CD3 trial suggests that fecal microbiota (Rebyota) likely leads to a clinically important improvement in the treatment success rate over 8 weeks, compared to placebo, for the prevention of the recurrence of CDI in adults after antibiotic treatment for rCDI. According to the CDA-AMC Clinical Review Report, most patients who achieved treatment success at 8 weeks with either fecal microbiota (Rebyota) or placebo were able to maintain a sustained clinical response at 6 months; therefore, there may be no added benefit with fecal microbiota (Rebyota) for this outcome, compared to placebo. Based on results from exploratory analyses, fecal microbiota (Rebyota) may result in little to no difference in HRQoL over 8 weeks, compared to placebo, and HRQoL results at 6 months were too uncertain to lead to a definite conclusion. In addition, the evidence was insufficient to conclude anything about the effect of fecal microbiota (Rebyota) relative to placebo on hospitalizations and ICU admissions. The trial could not inform on the effectiveness or safety of fecal microbiota (Rebyota) relative to other currently used therapies for rCDI, such as conventional, multidose FMT, nor did the sponsor submit indirect evidence to supplement the gap in the comparative evidence. Whether patients in the 2 groups previously received an appropriate antibiotic regimen of vancomycin taper-pulse for the treatment of the qualifying CDI episode is unknown. The relevance of using placebo as a comparator is, therefore, uncertain, as the CDI treatment in the study was not consistent with recommendations from the Canadian guidelines or with the treatment that patients would receive in clinical practice.
CDA-AMC undertook reanalyses to address several key limitations in the sponsors submission, including the assumption that a sustained response does not change after 6 months, the application of more appropriate rates for death and colectomy in the rCDI health state, the alignment of antibiotic distributions for no preventive therapy with those received in the PUNCH CD3 trial, the addition of administration costs for fecal microbiota (Rebyota), and the application of more appropriate assumptions to estimate productivity losses. Results from the CDA-AMC base case suggest that the ICER for fecal microbiota (Rebyota) versus no preventive therapy is $203,679 per QALY gained (incremental costs = $6,328; incremental QALYs = 0.03) for the health care payer perspective and $136,782 per QALY gained (incremental costs = $4,547; incremental QALYs = 0.03) for the societal perspectives. At the sponsor’s submitted price, fecal microbiota (Rebyota) would require price reductions of 53% or 32% (from $9,125 per dose to $4,289 or $6,205 per dose) to be considered cost-effective at a WTP threshold of $50,000 per QALY gained for the public health care payer and societal perspectives, respectively.
The CDA-AMC base case maintained some benefit for fecal microbiota (Rebyota) that has not been clinically proven, including maintained differences in sustained responses between fecal microbiota (Rebyota) and no preventive therapy and impacts on rCDI-related mortality and colectomy. In a scenario in which there is no difference in sustained response after 6 months between fecal microbiota (Rebyota) and no preventive therapy, the ICER for fecal microbiota (Rebyota) versus no preventive therapy increased to $476,035 and $407,736 per QALY gained in the health care payer and societal perspectives, respectively. This could be clinically plausible, given the conclusion in the CDA-AMC Clinical Review Report that there may be no added benefit with fecal microbiota (Rebyota), compared to placebo, in terms of sustained response at 6 months among patients who achieved treatment success at 8 weeks. Additionally, in a scenario in which the mortality and colectomy benefit associated with fecal microbiota (Rebyota) was removed, which is consistent with the clinical evidence provided in the PUNCH CD3 trial, the ICER for fecal microbiota (Rebyota) versus no preventive therapy increased to $212,188 and $158,901 per QALY gained in the health care payer and societal perspectives, respectively. Therefore, the CDA-AMC base case may represent an optimistic reimbursement context, should these longer-term benefits associated with fecal microbiota (Rebyota) not be realized.
CDA-AMC was unable to address the uncertainty related to the homogenous rCDI health state, which was more representative of an acute recurrence of CDI than the spectrum of rCDI symptoms and severities that patients with rCDI may experience. As its likely that the overall patient population with rCDI has less severe disease and will incur fewer costs (both health care and indirect) than modelled by the sponsor, the CDA-AMC base case is likely conservative in terms of the overall cost-effectiveness of fecal microbiota (Rebyota).
As well, CDA-AMC was unable to address the lack of comparative efficacy of fecal microbiota (Rebyota) and FMT. Given that there are no available cost estimates for FMT, there is insufficient evidence for a price premium for fecal microbiota (Rebyota) over FMT.
References
1.Ferring Inc. Drug Reimbursement Review sponsor submission: Rebyota (fecal microbiota): 150 mL rectal suspension [internal sponsor's package]. January 3, 2025.
2.Ferring Inc. Rebyota (fecal microbiota): 150 mL rectal suspension [product monograph]. March 5, 2025.
3.Ferring. Data on file: RBX2660 2014-01 Final Clinical Study Report [sponsor supplied reference]. 2020.
4.Rebiotix Inc. Clinical Study Report: 2017-01. A Phase 3 Prospective, Randomized, Double-Blinded, Placebo-Controlled Clinical Study to Evaluate the Efficacy and Safety of Rebiotix RBX2660 (microbiota suspension) for the Prevention of Recurrent Clostridium difficile Infection [internal sponsor's report] March 17, 2021.
5.Ferring. Data on file [sponsor supplied reference]. 2021.
6.Ferring Inc. Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rebyota (fecal microbiota): 150 mL rectal suspension [internal sponsor's package]. January 3, 2025.
7.Bank of Canada. Inflation calculator [sponsor supplied reference]. 2023. Accessed December 2024. https://www.bankofcanada.ca/rates/related/inflation-calculator/
8.Khanna S, Assi M, Lee C, et al. Efficacy and Safety of RBX2660 in PUNCH CD3, a Phase III, Randomized, Double-Blind, Placebo-Controlled Trial with a Bayesian Primary Analysis for the Prevention of Recurrent Clostridioides difficile Infection. Drugs. 2022;82(15):1527-1538. doi: 10.1007/s40265-022-01797-x PubMed
9.Ferring. Data on file [sponsor supplied reference]. 2021.
10.Feuerstadt P, Boules M, Stong L, et al. Clinical complications in patients with primary and recurrent Clostridioides difficile infection: A real-world data analysis. SAGE Open Med. 2021;9:2050312120986733. doi: 10.1177/2050312120986733 PubMed
11.Neal MD, Alverdy JC, Hall DE, Simmons RL, Zuckerbraun BS. Diverting loop ileostomy and colonic lavage: an alternative to total abdominal colectomy for the treatment of severe, complicated Clostridium difficile associated disease. Ann Surg. 2011;254(3):423-7; discussion 427-9. doi: 10.1097/SLA.0b013e31822ade48 PubMed
12.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island [sponsor supplied reference]. 2022. Accessed December 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401
13.Olsen MA, Stwalley D, Demont C, Dubberke ER. Clostridium difficile infection increases acute and chronic morbidity and mortality. Infect Control Hosp Epidemiol. 2019;40(1):65-71. doi: 10.1017/ice.2018.280 PubMed
14.Peprah D, Chiu AS, Jean RA, Pei KY. Comparison of Outcomes Between Total Abdominal and Partial Colectomy for the Management of Severe, Complicated Clostridium Difficile Infection. J Am Coll Surg. 2019;228(6):925-930. doi: 10.1016/j.jamcollsurg.2018.11.015 PubMed
15.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. doi: 10.1503/cmaj.170317 PubMed
16.Wilcox MH, Ahir H, Coia JE, et al. Impact of recurrent Clostridium difficile infection: hospitalization and patient quality of life. J Antimicrob Chemother. 2017;72(9):2647-2656. PubMed
17.Bartsch SM, Curry SR, Harrison LH, Lee BY. The potential economic value of screening hospital admissions for Clostridium difficile. Eur J Clin Microbiol Infect Dis. 2012;31(11):3163-71. doi: 10.1007/s10096-012-1681-z PubMed
18.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023. Accessed March 1, 2025. https://www.formulary.health.gov.on.ca/formulary/
19.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Exceptional Access Program (EAP). 2023. Accessed Oct 23, 2024. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
20.Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Heguy A, Nault V, Valiquette L. Clinical and Healthcare Burden of Multiple Recurrences of Clostridium difficile Infection. Clin Infect Dis. 2016;62(5):574-580. doi: 10.1093/cid/civ958 PubMed
21.Rodrigues R, Barber GE, Ananthakrishnan AN. A Comprehensive Study of Costs Associated With Recurrent Clostridium difficile Infection. Infect Control Hosp Epidemiol. 2017;38(2):196-202. doi: 10.1017/ice.2016.246 PubMed
22.Levy AR, Szabo SM, Lozano-Ortega G, et al. Incidence and Costs of Clostridium difficile Infections in Canada. Open Forum Infect Dis. 2015;2(3):ofv076. doi: 10.1093/ofid/ofv076 PubMed
23.Ontario Ministry of Health. Schedule of Benefits: Physician Services Under the Health Insurance Act [sponsor supplied reference]. 2024. https://www.ontario.ca/files/2024-08/moh-schedule-benefit-2024-08-30.pdf
24.Ontario Nurses Association. Collective Agreement [sponsor supplied reference]. 2024. https://www.ona.org/wp-content/uploads/20250331_hospitalcentralagreementenglish.pdf
25.Canadian Institute for Health Information. Hospital spending. Focus on the emergency department. CIHI; 2020. Accessed by sponsor, no date provided. https://www.cihi.ca/sites/default/files/document/hospital-spending-highlights-2020-en.pdf
26.Ostomy Canada Society. Ostomy Supply Reimbursement in Ontario. 2024. Accessed December 20, 2024. https://www.ostomycanada.ca/ostomy-supply-reimbursement-in-ontario/
27.Coward S, Heitman SJ, Clement F, et al. Ulcerative colitis-associated hospitalization costs: a population-based study. Can J Gastroenterol Hepatol. 2015;29(7):357-62. doi: 10.1155/2015/627370 PubMed
28.Dumont S, Jacobs P, Turcotte V, Turcotte S, Johnston G. Palliative care costs in Canada: A descriptive comparison of studies of urban and rural patients near end of life. Palliat Med. 2015;29(10):908-17. doi: 10.1177/0269216315583620 PubMed
29.Simms N, Beausejour W, Gheorghiu B. The environmental benefits of virtual care utilization in Canada: An analysis of travel distance avoided and associated carbon reductions as reported in the Canada Health Infoway Canadian Digital Health Survey 2021: What Canadians Think. Longwoods; 2022. Accessed by sponsor, no date provided. https://www.longwoods.com/content/26820/the-environmental-benefits-of-virtual-care-utilization-in-canada-an-analysis-of-travel-distance-avo
30.Galts C, Anvari S, Kim A, Leontiadis G, Armstrong D. Sustainable practice in gastroenterology: travel-related CO(2) emissions for gastroenterology clinic appointments in Canada. J Can Assoc Gastroenterol. 2025;8(1):7-12. doi: 10.1093/jcag/gwae049 PubMed
31.University Health Network. Parking at UHN. 2024. Accessed by sponsor, no date provided. https://www.uhn.ca/corporate/Directions/Pages/parking.aspx
32.Canada Revenue Agency (CRA). Reasonable per-kilometre allowance. Updated May 31, 2024. Accessed October 23, 2024. https://www.canada.ca/en/revenue-agency/services/tax/businesses/topics/payroll/benefits-allowances/automobile/automobile-motor-vehicle-allowances/reasonable-kilometre-allowance.html
33.Statistics Canada. Table: 14-10-0320-02. Average usual hours and wages by selected characteristics, monthly, unadjusted for seasonality [sponsor supplied reference]. 2021. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410032002&pickMembers%5B0%5D=3.4&cubeTimeFrame.startMonth=07&cubeTimeFrame.startYear=2021&referencePeriods=20210701%2C20210701
34.Statistics Canada. Table: 14-10-0060-01. Retirement age by class of worker, annual. Statistics Canada. 2024. Updated January 5, 2024. Accessed December 19, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410006001
35.Statistics Canada. Table: 14-10-0020-01. Unemployment rate, participation rate and employment rate by educational attainment, annual. Statistics Canada. 2024. Updated January 5, 2024. Accessed December 19, 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1410002001
36.Lurienne L, Bandinelli PA, Galvain T, Coursel CA, Oneto C, Feuerstadt P. Perception of quality of life in people experiencing or having experienced a Clostridioides difficile infection: a US population survey. J Patient Rep Outcomes. 2020;4(1):14. doi: 10.1186/s41687-020-0179-1 PubMed
37.Rebiotix Inc. Clinical Study Report: 2017-01. A Phase 3 Prospective, Randomized, Double-Blinded, Placebo-Controlled Clinical Study to Evaluate the Efficacy and Safety of Rebiotix RBX2660 (microbiota suspension) for the Prevention of Recurrent Clostridium difficile Infection [internal sponsor's report]. March 17, 2021.
38.Loo VG, Davis I, Embil J, et al. Association of Medical Microbiology and Infectious Disease Canada treatment practice guidelines for Clostridium difficile infection. J Assoc Med Microbiol Infect Dis Can. 2018;3(2):71-92. doi: 10.3138/jammi.2018.02.13
39.Blount KF, Papazyan R, Ferdyan N, et al. Microbiome and Metabolome Restoration After Administration of Fecal Microbiota, Live-jslm (REBYOTA(R)) for Preventing Recurrent Clostridioides difficile Infection. J Infect Dis. 2024. doi: 10.1093/infdis/jiae418 PubMed
40.Zaric GS. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-96. doi: 10.1177/0272989X03256883 PubMed
41.Rebiotix Inc. Clinical Study Report: 2014-01. A Phase 2B Prospective, Randomized, Double-blinded, Placebo-controlled Clinical Study Demonstrating the Efficacy and Safety of Rebiotix RBX2660 (microbiota suspension) for the Prevention of Recurrent Clostridium difficile Infection [internal sponsor's report]. March 26, 2020.
42.Rebiotix Inc. Clinical Study Report: 2019-01. A Phase 3 Open-Label Clinical Study to Evaluate the Safety and Tolerability of Rebiotix RBX2660 (microbiota suspension) in Subjects with Recurrent Clostridioides difficile Infection [internal sponsor's report]. July 16, 2024.
43.Canadian, Nosocomial, Infection, Surveillance, Program1*. Healthcare-associated infections and antimicrobial resistance in Canadian acute care hospitals, 2017-2021. Can Commun Dis Rep. 2023;49(5):235-252. doi: 10.14745/ccdr.v49i05a09 PubMed
44.Pereira JA, McGeer A, Tomovici A, Selmani A, Chit A. The Clinical Burden of Clostridioides difficile in Ontario, Canada. Open Forum Infect Dis. 2020;7(2):ofz523. doi: 10.1093/ofid/ofz523 PubMed
45.Nanwa N, Sander B, Krahn M, et al. A population-based matched cohort study examining the mortality and costs of patients with community-onset Clostridium difficile infection identified using emergency department visits and hospital admissions. PLoS One. 2017;12(3):e0172410. doi: 10.1371/journal.pone.0172410 PubMed
46.Lee DY, Chung EL, Guend H, Whelan RL, Wedderburn RV, Rose KM. Predictors of mortality after emergency colectomy for Clostridium difficile colitis: an analysis of ACS-NSQIP. Ann Surg. 2014;259(1):148-56. doi: 10.1097/SLA.0b013e31828a8eba PubMed
47.Statistics Canada. Table: 14-10-0043-01. Average usual and actual hours worked in a reference week by type of work (full- and part-time), annual. 2025. Accessed April 8, 2025. https://doi.org/10.25318/1410004301-eng
48.Government of Saskatchewan. Saskatchewan Drug Plan: search formulary. 2023. Accessed March 2025. https://formulary.drugplan.ehealthsask.ca/SearchFormulary
49.B. C. Government. BC PharmaCare formulary search. 2023. Accessed March 2025. https://pharmacareformularysearch.gov.bc.ca
50.Government of Alberta. Interactive drug benefit list. 2023. Accessed March 2025. https://idbl.ab.bluecross.ca/idbl/load.do
51.Canada's Drug Agency. Reimbursement Review: fidaxomicin (Dificid) for Clostridium difficile infection. 2025. Accessed April 8, 2025. https://www.cda-amc.ca/fidaxomicin
52.Sharma A, Leal J, Kim J, Pearce C, Pillai DR, Hollis A. The Cost of Contact Precautions: A Systematic Analysis. Can J Infect Control. 2020;35(4):163-169. doi: 10.36584/cjic.2020.004.03.163.169
53.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (June 29, 2023 (effective July 24, 2023)). 2023. Accessed March 2025. https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf
54.Quay T, Walter M. Fecal Microbiota Therapy in Canada: An Environmental Scan. (Environmental scan no. 95). 2020. Accessed by sponsor, no date provided. https://www.cadth.ca/sites/default/files/es/es0341-fecal-microbiota-therapy-in-canada.pdf
55.Island Health. Questions about Preferred Accommodation: Private and Semi-Private Rooms. 2014. Accessed November 10, 2025. https://www.islandhealth.ca/sites/default/files/2018-06/hospital-accommodation-brochure.pdf
56.Ferring Inc. Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rebyota (fecal microbiota): 150 mL rectal suspension [internal sponsor's package]. January 3, 2025.
57.Provincial Infection Control Network of British Columbia (PICNet). Clostridium difficile Infection (CDI) Surveillance Report, Quarter 1 and Quarter 2, 2011/2012. 2012. Accessed November 6, 2025. https://www.picnet.ca/uploads/files/surveillance/CDI%20Surveillance%20Report%20semiannual%202011-2012%20Q1-2.pdf
58.Public Health Disease Surveillance System. Manitoba Monthly Surveillance Unit Report, Reported up to January 31, 2012. Manitoba Health – Public Health Branch; 2012. Accessed November 10, 2025. https://web.archive.org/web/20131001165716/http://www.gov.mb.ca/health/publichealth/surveillance/scd/jan12.pdf
59.Canadian Nosocomial Infection Surveillance Program. Clostridium difficile Associated Diarrhea in Acute-Care Hospitals Participating in CNISP: November 1, 2004 to April 30, 2005: final report. Public Health Agency of Canada 2007. Accessed March 24, 2025. https://www.phac-aspc.gc.ca/nois-sinp/pdf/c-difficile_cnisp-pcsin-eng.pdf
60.Kelly CP. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect. 2012;18 Suppl 6:21-7. doi: 10.1111/1469-0691.12046 PubMed
61.Statistics Canada. Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000) [sponsor supplied reference]. 2022. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701
62.Health Infobase. Canadian Antimicrobial Resistance Surveillance System (CARSS). Government of Canada. Updated November 24, 2023. Accessed by sponsor, no date provided. https://health-infobase.canada.ca/carss/amr/results.html?ind=08
63.Pereira JA, McGeer A, Tomovici A, Selmani A, Chit A. Incidence and economic burden of Clostridioides difficile infection in Ontario: a retrospective population-based study. CMAJ Open. 2020;8(1):E16-E25. doi: 10.9778/cmajo.20190018 PubMed
64.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed March 2025. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CDA-AMC Cost Comparison for Clostridioides difficile Infection
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Course cost ($)a |
|---|---|---|---|---|---|---|
Fecal microbiota (Rebyota) | 150 mL | Liquid suspension | 9,125.0000 | One dose of 150 mL fecal microbiota (Rebyota) | Not applicable | 9,125 |
Antibiotics | ||||||
Vancomycin (generic) | 125 mg 250 mg | Oral tablet | 5.1800 10.3600 | 125 to 500 mg administered orally every 6 to 8 hours for 7 to 10 days Taper-pulse: 125 qid for 14 days; 125 mg tid for 7 days; 125 mg BID for 7 days; 125 mg once daily for 7 days; and every 2 to 3 days for 2 to 8 weeks | Standard: 15.54 to 82.88 Taper-pulse: 24.17 to 290.08 | Standard: 109 to 829 Taper-pulse: 532 to 653 |
Fidaxomicin (Dificid) | 200 mg | Oral tablet | 94.6000b | 200 mg twice daily for 10 days | 189.20 | 1,892 |
CDA-AMC = Canada’s Drug Agency.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2025), unless otherwise indicated, and do not include dispensing fees.18
aCourses: Fecal microbiota (Rebyota) is one time administration; Vancomycin standard was calculated for 125 mg every 8 hours for 7 days to 500 mg every 6 hours per 10 days; Vancomycin taper-pulse was calculated for the duration accordingly to the duration of 49 to 91 days; fidaxomicin was calculated for twice a day per 10 days.38
bDrug price sourced from Ontario Exception Access Program (accessed March 2025).19
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to clinical efficacy key limitation. |
Model has been adequately programmed and has sufficient face validity | No | Refer to transition probability of rCDI to colectomy health state key limitation. |
Model structure is adequate for decision problem | No | Refer to model structure key limitation. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to corrections made to sponsor’s model. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | For some model parameters, the sponsor arbitrarily incorporated uncertainty (i.e., Treatment success, age, etc.) using a standard deviation equal to a 90 percentile in a normal or beta distribution. This is unlikely to reflect the true uncertainty around the model’s parameters. Additionally, more appropriate uncertainty parameters from the PUNCH CD3 study could have been applied. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | One transcription error (i.e., post hoc survival analysis) and 1 calculation error were found in the sponsor’s model. Additionally, some citations could not be identified in the model or pharmacoeconomic report (e.g., terminal-care costs). |
rCDI = recurrent Clostridioides difficile infection.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
CDI = Clostridioides difficile infection; rCDI = recurrent Clostridioides difficile infection.
Source: Sponsor’s pharmacoeconomic submission.6
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Fecal microbiota (Rebyota) | No preventive therapy |
|---|---|---|
Discounted LYs | ||
Total | 4.53 | 4.46 |
Absence of CDI | 4.12 | 3.86 |
rCDI | 0.36 | 0.54 |
Colectomy | 0.004 | 0.006 |
Ileostomy | 0.004 | 0.006 |
Ileostomy reversal | 0.04 | 0.05 |
Discounted QALYs | ||
Total | 3.77 | 3.63 |
Absence of CDI | 3.58 | 3.35 |
rCDI | 0.15 | 0.23 |
Colectomy | 0.002 | 0.003 |
Ileostomy | 0.003 | 0.004 |
Ileostomy reversal | 0.032 | 0.043 |
Discounted costs ($) | ||
Total | 42,951 | 51,629 |
Acquisition | 9,518 | 592 |
Administration | 0 | 0 |
Subsequent treatment | 603 | 1,347 |
Medical | 20,928 | 19,474 |
Indirect | 11,903 | 18,474 |
CDI = Clostridioides difficile infection; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; rCDI = recurrent Clostridioides difficile infection; QALY = quality-adjusted life-year.
Note: Indirect costs are only relevant to the societal perspective.
Source: Sponsor’s pharmacoeconomic submission.6
Figure 2: Incremental Cost vs. Incremental QALYs (Public Health Care Payer Perspective)
CE = cost-effectiveness; PSA = probabilistic sensitivity analysis; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.6
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of CDA-AMC Base Case
Table 11: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results (Public Health Care Payer Perspective)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | No preventive therapy | 33,486 | 3.62 | Reference |
Fecal microbiota (Rebyota) | 31,350 | 3.76 | Dominant | |
Sponsor’s corrected base case | No preventive therapy | 32,830 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 31,067 | 3.77 | Dominant | |
CDA-AMC reanalysis 1 | No preventive therapy | 32,325 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 37,529 | 3.70 | 97,921 | |
CDA-AMC reanalysis 2 | No preventive therapy | 32,446 | 3.75 | Reference |
Fecal microbiota (Rebyota) | 30,677 | 3.86 | Dominant | |
CDA-AMC reanalysis 3 | No preventive therapy | 31,622 | 3.68 | Reference |
Fecal microbiota (Rebyota) | 30,182 | 3.81 | Dominant | |
CDA-AMC reanalysis 4 | No preventive therapy | 32,785 | 3.66 | Reference |
Fecal microbiota (Rebyota) | 31,042 | 3.79 | Dominant | |
CDA-AMC reanalysis 5 | No preventive therapy | 32,184 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 30,864 | 3.77 | Dominant | |
CDA-AMC reanalysis 6-administration | No preventive therapy | 32,830 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 31,514 | 3.77 | Dominant | |
CDA-AMC base case (1 + 2 + 3 + 4 + 5 + 6) | No preventive therapy | 29,996 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 36,087 | 3.84 | 185,213 | |
CDA-AMC base case (1 + 2 + 3 + 4 + 5 + 6) (probabilistic) | No preventive therapy | 29,986 | 3.80 | Reference |
Fecal microbiota (Rebyota) | 36,314 | 3.83 | 203,679 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Table 12: Summary of the Stepped Analysis of the CDA-AMC Reanalysis Results (Societal Perspective)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | No preventive therapy | 51,998 | 3.63 | Reference |
Fecal microbiota (Rebyota) | 43,406 | 3.77 | Dominant | |
Sponsor’s corrected base case | No preventive therapy | 51,042 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 42,978 | 3.77 | Dominant | |
CDA-AMC reanalysis 1 | No preventive therapy | 50,318 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 52,269 | 3.70 | 36,709 | |
CDA-AMC reanalysis 2 | No preventive therapy | 51,299 | 3.75 | Reference |
Fecal microbiota (Rebyota) | 42,873 | 3.86 | Dominant | |
CDA-AMC reanalysis 3 | No preventive therapy | 50,193 | 3.68 | Reference |
Fecal microbiota (Rebyota) | 42,225 | 3.81 | Dominant | |
CDA-AMC reanalysis 4 | No preventive therapy | 51,019 | 3.66 | Reference |
Fecal microbiota (Rebyota) | 42,967 | 3.79 | Dominant | |
CDA-AMC reanalysis 5 | No preventive therapy | 50,397 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 42,775 | 3.77 | Dominant | |
CDA-AMC reanalysis 6 -administration | No preventive therapy | 51,042 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 43,425 | 3.77 | Dominant | |
CDA-AMC reanalysis 7 | No preventive therapy | 40,514 | 3.64 | Reference |
Fecal microbiota (Rebyota) | 36,071 | 3.77 | Dominant | |
CDA-AMC base case (1 + 2 + 3 + 4 + 5 + 6 + 7) | No preventive therapy | 37,989 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 42,569 | 3.84 | 139,269 | |
CDA-AMC base case (1 + 2 + 3 + 4 + 5 + 6 + 7) (probabilistic) | No preventive therapy | 38,194 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 42,742 | 3.85 | 136,782 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CDA-AMC reanalysis is based on publicly available prices of the comparator treatments. The results of all steps are presented deterministically unless otherwise indicated, while the cumulative CDA-AMC base case is always presented both deterministically and probabilistically.
Table 13: Disaggregated Summary of the CDA-AMC Economic Evaluation Results (Deterministic)
Parameter | Fecal microbiota (Rebyota) | No preventive therapy |
|---|---|---|
Discounted LYs | ||
Total | 4.68 | 4.67 |
Absence of CDI | 4.18 | 4.10 |
rCDI | 0.49 | 0.56 |
Colectomy | 0.00 | 0.00 |
Ileostomy | 0.00 | 0.00 |
Ileostomy reversal | 0.01 | 0.01 |
Discounted QALYs | ||
Total | 3.84 | 3.81 |
Absence of CDI | 3.63 | 3.57 |
rCDI | 0.21 | 0.24 |
Colectomy | 0.00 | 0.00 |
Ileostomy | 0.00 | 0.00 |
Ileostomy reversal | 0.01 | 0.01 |
Discounted costs ($) | ||
Total (health care payer) | 36,087 | 29,996 |
Total (societal) | 42,569 | 37,989 |
Acquisition | 9,518 | 393 |
Administration | 447 | 0 |
Subsequent treatment | 751 | 947 |
Medical | 25,372 | 28,657 |
Indirect | 6,482 | 7,993 |
CDA-AMC = Canada’s Drug Agency; CDI = Clostridioides difficile infection; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; rCDI = recurrent Clostridioides difficile infection; QALY = quality-adjusted life-year.
Note: Indirect costs are only relevant to the societal perspective.
Figure 3: CDA-AMC Base-Case Incremental Cost vs. Incremental QALYs (Public Health Care Payer Perspective)
CDA-AMC = Canada’s Drug Agency; CE = cost-effectiveness; PSA = probabilistic sensitivity analysis; QALY = quality-adjusted life-year; vs. = versus.
Figure 4: CDA-AMC Base-Case Incremental Costs vs. Incremental QALYs (Societal Perspective)
CDA-AMC = Canada’s Drug Agency; CE = cost-effectiveness; PSA = probabilistic sensitivity analysis; QALY = quality-adjusted life-year; vs. = versus.
Table 14: Bottom-Up Approach to the Cost of Fecal Microbiota (Rebyota) Administration
Component | Item | Time/quantity | Unit cost | Cost |
|---|---|---|---|---|
Doctor administration | Gastroenterology consultationa | 1 visit | $157 per visit | $157 |
Nursing assistance | RN wageb | 2 hours | $42 per hour | $84 |
Private room with dedicated bathroom | Room | 1 | $195c | $195 |
Housekeeping | Housekeeper | 30 minutes | $21.08 per hour52 | $10.54 |
Personal protective equipmentd | Gloves | 1 pair of gloves | $0.10 per unit52 | $0.20 |
Gown | 1 gown | $0.42 per unit52 | $0.42 | |
Total | $447.12 | |||
CDA-AMC = Canada’s Drug Agency; RN = registered nurse.
Note: All time and quantity for admin were based on clinical expert feedback received by CDA-AMC.
aSourced from the Ontario Schedule of benefits for physician services.53
bBased on a pan-Canadian median wage of $42 per hour.33
cBased on the minimum cost from publicly available single patient bed upgrades for hospitals that currently provide fecal microbiota transplant54,55
dSourced from literature.52
Scenario Analyses
Table 15: Scenario Analyses Conducted on the CDA-AMC Base Case (Probabilistic)
Stepped analysis | Treatment | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CDA-AMC base case (health care payer) | No preventive therapy | 29,986 | 3.80 | Reference |
Fecal microbiota (Rebyota) | 36,314 | 3.83 | 203,679 | |
CDA-AMC base case (societal) | No preventive therapy | 38,194 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 42,742 | 3.85 | 136,782 | |
CDA-AMC scenario analysis 1: additional dose for next recurrence (health care payer) | No preventive therapy | 30,337 | 3.80 | Reference |
Fecal microbiota (Rebyota) | 39,223 | 3.83 | 257,843 | |
CDA-AMC scenario analysis 1: additional dose for next recurrence (societal) | No preventive therapy | 37,879 | 3.79 | Reference |
Fecal microbiota (Rebyota) | 45,738 | 3.82 | 256,069 | |
CDA-AMC scenario analysis 2: 2 doses for first recurrence (health care payer) | No preventive therapy | 30,183 | 3.80 | Reference |
Fecal microbiota (Rebyota) | 45,750 | 3.83 | 538,859 | |
CDA-AMC scenario analysis 2: 2 doses for first recurrence (societal) | No preventive therapy | 38,131 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 51,921 | 3.85 | 419,593 | |
CDA-AMC scenario analysis 3: no mortality or colectomy benefit (health care payer-deterministic)a | No preventive therapy | 29,741 | 3.84 | Reference |
Fecal microbiota (Rebyota) | 35,853 | 3.87 | 212,188 | |
CDA-AMC scenario analysis 3: no mortality or colectomy benefit (societal-deterministic)a | No preventive therapy | 37,817 | 3.84 | Reference |
Fecal microbiota (Rebyota) | 42,394 | 3.87 | 158,901 | |
CDA-AMC scenario analysis 4: Patient with resolved CDI at work (societal - deterministic)a | No preventive therapy | 37,989 | 3.81 | Reference |
Fecal microbiota (Rebyota) | 41,749 | 3.84 | 114,333 | |
CDA-AMC scenario analysis 5: same sustained response (health care payer - deterministic)b | No preventive therapy | 28,200 | 3.83 | Reference |
Fecal microbiota (Rebyota) | 36,087 | 3.84 | 476,035 | |
CDA-AMC scenario analysis 5: same sustained response (societal - deterministic)c | No preventive therapy | 35,814 | 3.83 | Reference |
Fecal microbiota (Rebyota) | 42,569 | 3.84 | 407,736 |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aModel did not produce probabilistic results for this scenario, therefore, only deterministic results are presented.
bIn this scenario the probabilistic results were highly unstable, therefore only deterministic results are presented.
cDeterministic results are presented as the sponsor’s model was unable to produce stable probabilistic results (i.e., probabilistic results varied significantly from deterministic results).
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 16: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
CDA-AMC identified several key limitations in the sponsor’s analysis:
CDA-AMC conducted reanalyses including: increasing the proportion of patients who are covered under public drug plans; revising the antibiotic treatment distribution; and adjusting the market uptake of fecal microbiota (Rebyota). CDA-AMC reanalyses suggest that reimbursing fecal microbiota (Rebyota) for the prevention of rCDI in adults following antibiotic treatment for rCDI would be associated with a budget impact of $39,889,069 over 3 years (year 1: $13,122,859; year 2: $13,295,593; year 3: $13,470,617). |
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) to assess the budget impact of reimbursing fecal microbiota (Rebyota) for the prevention of rCDI in adults following antibiotic treatment for rCDI.56 The BIA was conducted from the perspective of a Canadian public drug plan payer over a 3-year time horizon (January 2026 to December 2028).
A case based approach was taken to determine the number of patients eligible for fecal microbiota (Rebyota) using data from a study conducted in Canada that estimated the number of cases based on the mean rate of infections from the Canadian Nosocomial Infection Surveillance Program between 2011 to 2012 and assumed a ratio for the number of community-acquired CDI cases.22 The prevalence-based study determined the number of hospital-acquired CDI cases by multiplying the rate of CDI per 10,000 bed-days and the total number of bed-days in each jurisdiction sourced from the 2011 to 2012 Canadian Nosocomial Infection Surveillance Program, Provincial Infection Control Network, and Manitoba Health.57-59 The number of community-acquired CDI cases was estimated to be a ratio of hospital- to community-acquired CDI cases observed in Manitoba between 2005 to 2006.58 Informed by Levy et al., the sponsor stratified the total CDI cases into number of recurrences based on data from other published literature.22,60 Subsequently, the sponsor extrapolated the number of CDI cases based on the population growth in Canada to estimate the number of CDI cases in 2025.61 It was assumed that the prevalence of CDI cases is similar among jurisdictions and that the prevalence of CDI was maintained from 2012 to 2024. Of the estimated rCDI cases, the sponsor assumed that only 70% of patients would be covered by a public drug plan.56 The sponsor compared a reference scenario where patients are treated with antibiotics (i.e., vancomycin and fidaxomicin), to a new drug scenario in which a proportion of patients who are also treated with antibiotics would receive fecal microbiota (Rebyota). In the new drug scenario, patients who were treated with fecal microbiota (Rebyota) and no preventive therapy were assumed to avoid a recurrence based on the treatment success observed in the PUNCH CD3 study. Market uptake in the new drug scenario was derived from the sponsor’s internal projections. Key inputs to the BIA are documented in Table 17.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Estimated number of CDI cases in 2012 Proportion of cases that are primary CDI cases Proportion of primary CDI cases that experience a first recurrence Proportion of secondary CDI cases that experience a third recurrence Proportion of tertiary CDI cases that experience a fourth or more recurrence Population growth rate Patients covered by public plans | 37,93322 72.9%60 25%60 4560 65%60 1.29%61 70%56 |
Number of patients eligible for drug under reviewa | 6,176 / 6,258 / 6,340 |
Market uptake (3 years) | |
Uptake (reference scenario) Fecal microbiota (Rebyota) No preventive therapy | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Fecal microbiota (Rebyota) No preventive therapy | 1.72% / 6.30% / 11.86% 98.28% / 94.70% / 88.14% |
Cost of treatment (per patient, per course) | |
Fecal microbiota (Rebyota) Fecal microbiota (Rebyota) treatment Antibiotic treatment No preventive therapy | $9,517.60 $9,125.00 $392.60b $591.69b |
CDI = Clostridioides difficile infection.
aThe number of patients in 2025 was calculated based on the stratification of recurrences from the number of CDI cases in 2012 and the percentage of cases that relapse, the proportion of recurrent cases relative to the total population of Canada in 2012, and the growth rate of Canada with the assumption that the prevalence of recurrent cases will remain constant.56
bProportions of antibiotic treatments for fecal microbiota (Rebyota) (Vancomycin 125 mg twice daily 2 weeks: 93.6%; fidaxomicin: 6.4%) and no preventive therapy (Vancomycin taper-pulse: 68.92%; Vancomycin 125 mg twice daily 2 weeks: 24.51%; fidaxomicin: 6.57%).
Summary of the Sponsor’s BIA Results
The sponsor estimated that funding fecal microbiota (Rebyota) for the prevention of rCDI in adults following antibiotic treatment for rCDI would be associated with a cumulative budget impact of $10,960,596 (Year 1: $938,433; Year 2: $3,462,885; Year 3: $6,559,278).
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The total number of CDI cases is uncertain. The sponsor’s submitted BIA estimated the number of total cases using a case based approach informed by a prevalence study conducted in Canada.22 The prevalence study estimated the number of CDI cases in Canada in 2012 based on the prevalence of hospital-acquired CDI and applied a ratio for community-acquired CDI. There is significant uncertainty regarding the total number of CDI cases, as the rate of CDI per 1,000 patient admissions (2012: 4.5; 2021: 3.94) and CDI per 10,000 patient days (2012: 6.4; 2021: 5.05) has fluctuated throughout the years.43 Clinical expert feedback received for this review also indicated that prevalence and incidence of CDI cases is not consistent over time.59,62 Therefore, whether the number of cases in 2012 is representative of the number of cases in 2024, or over the next 3 years, is uncertain.
Additionally, the number of community-acquired CDI cases in the aforementioned prevalence study, which informed the sponsor’s BIA, is based on a 2005 ratio of hospital versus community-acquired CDI cases (2005: 64.2 to 35.8) in Manitoba.58 While the 2021-updated incident rates of CDI43 are numerically lower than the published literature in 2012, the proportion of community-acquired CDI cases across the jurisdictions in Canada is uncertain and the true Canada-wide hospital- to community-acquired CDI ratio may not be reflective of Manitoba in 2005. If the proportion of community-acquired CDI cases compared to hospital-acquired CDI cases has changed, then the total number of CDI cases estimated by the Levy et al. study would change. A retrospective analysis conducted in Ontario, Canada between 2005 and 2015 estimated that 50.9% of CDI cases were hospital-acquired, which differs from the proportion of Manitoba in 2005.63 However, there is uncertainty if the ratio of hospital- versus community-acquired in Ontario between 2005 and 2015 is reflective of the remainder of Canada.
CDA-AMC was unable to validate the estimated total number of CDI cases due to the gaps in evidence. While there appears to be a numerical decrease in the rates of CDI, there is uncertainty regarding the number of community-acquired versus hospital-acquired CDI cases.
The number of patients who experience a relapse is likely overestimated. To estimate the number of CDI cases eligible for fecal microbiota (Rebyota), the sponsor started with the total number of cases in 2012 from Levy et al.22 (37,933) and then removed 72.9% of cases, which were all assumed to be primary cases. Based on the 27.1% of remaining cases, the sponsor estimated the proportion that were first, second or 3+ recurrences, and then summed these to determine the total number of recurrent cases. The sponsor used published literature to determine the number of patients who experience a relapse given a primary infection for each province (where the proportion of patients expected to experience a first recurrence given a primary infection was 25%; the proportion of patients for a second recurrence given a first recurrence was 45%, and a third recurrence given a second recurrence was 65%).60 When the total number of patients who experienced a relapse was expressed as a proportion (i.e., the sum of the recurrences divided by the total number of CDI cases, primary and recurrent), the sponsor’s approach resulted in an estimate that 32% of CDI cases were recurrent cases. However, this value is not aligned with the sponsor’s Canada-wide estimate that 27.1% of patients would experience a relapse. There is therefore uncertainty associated with the sponsor’s approach to stratifying patients based on the number of recurrences they have experienced as when calculating the total number of patients with CDI using the recurrence stratification, the sponsor’s model estimates that the total number of patients with CDI is 5% greater than their cited published literature (model: 39,700 versus Levy et al. study: 37,933).
CDA-AMC conducted a scenario analysis that adjusted the percentage of patients in each province who experience a relapse to be reflective of the Levy et al. study.22 Additionally, in this scenario, as the sponsor’s model does not distinguish the treatment that each patient received based on the number of relapses previously experienced, CDA-AMC assumed equal patient proportions for each recurrence stratification.
The public coverage rate assumed by the sponsor is likely underestimated. The sponsor assumed that 70% of patients with rCDI across Canada would be eligible for coverage by a public drug plan.6 This estimate was informed by the Levy et al. study,22 where 70% of CDI cases were in patients aged older than 65. The sponsor then assumed that 100% of those over 65 would have public drug plan coverage whereas among the 30% of eligible patients who were 65 and younger, 0% would have public drug plan coverage. This is inappropriate as it is unlikely that all patients with CDI who are younger than 65 would not be covered by a public drug plan and because the proportion over 65 may not represent the patient population who are eligible to receive fecal microbiota (Rebyota).
In reanalysis, CDA-AMC revised the proportion of patients who are over and under 65 to align with the PUNCH CD3 safety analysis (aligned with the baseline characteristics in the sponsor’s pharmacoeconomic analysis). In those who are 65 years and over, CDA-AMC maintained the sponsor’s assumption that the coverage rate would be 100%. In those who are under 65, the coverage rate was informed by jurisdiction-specific rates of coverage between 25 and 64 years.64
Market uptake of fecal microbiota (Rebyota) is associated with uncertainty. The sponsor assumed that if fecal microbiota (Rebyota) was fully funded by the publicly funded drug plans, it would be associated with a market uptake of 1.72%, 6.30%, and 11.86%, for year 1, year 2, and year 3, respectively, based on the sponsor’s internal projections.56 Clinical expert feedback obtained by CDA-AMC noted that the sponsor’s estimated market uptake did not reflect clinical expectations as fecal microbiota (Rebyota) is considered to be an accessible product that can be administered in multiple health care settings. Therefore, clinical expert feedback indicated that it is expected that the uptake of fecal microbiota (Rebyota) is likely to be higher than estimated by the sponsor. Additionally, clinical expert feedback noted that the proportion of patients that will use fecal microbiota (Rebyota) would be dependent on the number of recurrences experienced by the patient; however, this approach was not considered by the sponsor.
In reanalysis, CDA-AMC revised the market uptake of fecal microbiota (Rebyota) to 20% across all 3 years, in accordance with clinical expert feedback.
Treatment mix of antibiotics for no preventive therapy is uncertain and likely inappropriate. Similar to the pharmacoeconomic analysis, the sponsor’s BIA base case assumes different proportions of antibiotics for the treatment of the recurrence before patients receiving fecal microbiota (Rebyota) (vancomycin 125 mg 4 times daily for 2 weeks:1 94%; fidaxomicin 200 mg twice daily for 10 days: 6%) versus no preventive therapy (vancomycin taper-pulse: 69%; vancomycin 125 mg 4 times daily for 2 weeks:1 25%; fidaxomicin 200 mg twice daily for 10 days: 7%). Discussions with clinical experts indicated that patients would receive similar proportions of prior antibiotic treatment to treat the recurrence regardless of whether patients were to receive subsequent fecal microbiota (Rebyota) treatment. Additionally, the sponsor assumed that patients would avoid subsequent recurrences based on the efficacy of fecal microbiota (Rebyota) and no preventive therapy from the PUNCH CD3 study. As the antibiotic treatment received would impact the efficacy of fecal microbiota (Rebyota) and no preventive therapy, it is likely inappropriate to assume a different antibiotic treatment mix that is not reflective of the PUNCH CD3 study.
In reanalysis, CDA-AMC revised the antibiotic treatment distribution for no preventive therapy to align with the treatment mix from the PUNCH CD3 study.
Poor modelling practices were employed. The sponsor’s submitted BIA model did not allow for the simultaneous viewing of results for each jurisdiction or the direct calculation of pan-Canadian costs for the reference and new drug scenarios. Additionally, the baseline year for the reference scenario, new drug scenario, and subsequent budget impact was not calculated for the pan-Canadian results.
CDA-AMC was unable to report the baseline year due to the sponsor’s model. Reference and new drug scenarios were obtained using a sponsor’s scenario analysis where the intended modification was removed, allowing for the calculation of the desired reference and new drug scenario.
CDA-AMC Reanalyses of the BIA
The CDA-AMC base-case reanalyses revised the proportion of patients covered by a public drug plan, adjusted the antibiotic treatment distribution for no preventive therapy, and revised the market uptake of fecal microbiota (Rebyota).
Table 18: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Public drug plan coverage | All provinces: 70% | British Columbia: 100% Alberta: 100% Saskatchewan: 100% Manitoba: 100% Ontario: 70% Nova Scotia: 100% New Brunswick: 66% Prince Edward Island: 63% Newfoundland and Labrador: 64% |
2. No preventive therapy antibiotic treatment mix | Vancomycin taper-pulse: 24.5% Vancomycin standard: 68.9% Fidaxomicin: 6.6% | Vancomycin taper-pulse: 0% Vancomycin standard: 93.6% Fidaxomicin: 6.4% |
3. Market uptake of fecal microbiota (Rebyota) | Year 1: 1.72% Year 2: 6.3% Year 3: 11.6% | Year 1: 20% Year 2: 20% Year 3: 20% |
CDA-AMC base-case | 1 + 2 + 3 | |
CDA-AMC = Canada’s Drug Agency; CDI = Clostridioides difficile infection; rCDI = recurrent Clostridioides difficile infection.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in and a more detailed breakdown is presented in. Similar to the pharmacoeconomic analysis, FMT was not included as a comparator in the sponsor’s BIA. As noted previously in this report, FMT is commonly used to treat rCDI. However, as FMT is not reimbursed by a public drug plan, the influence of FMT on the budget impact from a public drug plan perspective is null.
Based on the CDA-AMC reanalysis, the budget impact of funding fecal microbiota (Rebyota) for the prevention of recurrence of CDI in adults following antibiotic treatment for recurrent CDI is $39,889,069 3-year time horizon (year 1: $13,122,859; year 2: $13,295,593; year 3: $13,470,617). CDA-AMC reanalyses are based on publicly available list prices for all comparators.
Table 19: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 10,960,596 |
CDA-AMC reanalysis 1 | 13,153,471 |
CDA-AMC reanalysis 2 | 11,237,772 |
CDA-AMC reanalysis 3 | 32,417,012 |
CDA-AMC base case | 39,889,069 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analyses to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 20):
Assumed 100% public coverage.
Removed the rCDI stratification.
Table 20: Detailed Breakdown of the CDA-AMC Reanalyses of the BIAa
Stepped analysis | Scenario | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|
Submitted base case | Reference | 3,654,280 | 3,702,573 | 3,751,510 | 11,108,363 |
New drug | 4,592,713 | 7,165,458 | 10,310,788 | 22,068,960 | |
Budget impact | 938,433 | 3,462,885 | 6,559,278 | 10,960,596 | |
CDA-AMC base case | Reference | 2,910,040 | 2,948,345 | 2,987,157 | 8,845,542 |
New drug | 16,032,899 | 16,243,938 | 16,457,774 | 48,734,611 | |
Budget impact | 13,122,859 | 13,295,593 | 13,470,617 | 39,889,069 | |
CDA-AMC scenario analysis 1: 100% public coverage | Reference | 3,399,592 | 3,444,211 | 3,489,420 | 10,333,223 |
New drug | 18,730,087 | 18,975,919 | 19,224,998 | 56,931,003 | |
Budget impact | 15,330,495 | 15,531,708 | 15,735,578 | 46,597,781 | |
CDA-AMC scenario analysis 2: No rCDI stratification | Reference | 2,478,153 | 2,510,747 | 2,543,774 | 7,532,673 |
New drug | 13,653,409 | 13,832,989 | 14,014,948 | 41,501,347 | |
Budget impact | 11,175,256 | 11,322,242 | 11,471,175 | 33,968,673 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency; rCDI = recurrent Clostridioides difficile infection.
aThe sponsor’s submitted BIA model did not permit presentation of the baseline year and subsequently this is not reported by CDA-AMC.
Ethics Review
Abbreviations
CDA-AMC
Canada’s Drug Agency
CDI
Clostridioides difficile infection
CrI
credible interval
FMT
fecal microbiota transplant
rCDI
recurrent Clostridioides difficile infection
Summary
Clostridioides difficile infection (CDI) is a bacterial infection that can cause severe diarrhea, significantly impacting patients’ health-related quality of life and increasing mortality risk. Recurrent CDI (rCDI) occurs in up to 25% of patients following initial treatment, with the likelihood of further recurrences increasing over time. This report describes ethical considerations regarding the use of fecal microbiota (Rebyota) to prevent recurrence of CDI in adults following antibiotic treatment for rCDI.
Fecal microbiota (Rebyota) is the first commercial fecal microbiota–based therapy to be approved by Health Canada for any condition, including rCDI. Unlike conventional fecal microbiota transplant (FMT), which depends on patient-identified donors and local program infrastructure, fecal microbiota (Rebyota) is delivered to clinical providers as a standardized, ready-to-use enema.
This Ethics Review report was informed by relevant literature as well as patient group, clinician group, clinical expert, and drug program input. Ethical considerations identified in this review include those related to the following:
Diagnosis, treatment, and experiences of rCDI: rCDI is a highly burdensome condition that can significantly diminish quality of life, disrupt daily functioning, and cause prolonged psychological distress for both patients and caregivers. Each recurrence increases the risk of future episodes, trapping individuals in a cycle of worsening illness and fear of relapse. Those most affected by rCDI — including older adults, individuals who are immunocompromised, and people experiencing systemic barriers to care — often face compounding medical and structural vulnerabilities. While fecal microbiota–based therapies have demonstrated effectiveness in preventing recurrence, access to conventional FMT in Canada has been limited due to regulatory, logistical, and funding constraints, leaving many patients without timely and equitable access to a therapeutic option that has the potential to restore gut stability and reduce long-term suffering.
Clinical and economic evidence used in the evaluation of fecal microbiota (Rebyota): Moderate-certainty findings from the pivotal PUNCH CD3 trial suggest that treatment with fecal microbiota (Rebyota) likely results in a clinically important increase in treatment success over 8 weeks compared to placebo, but the study design raised important evidentiary and ethical concerns. The absence of an active comparator (particularly conventional FMT) limited the trial’s ability to assess relative effectiveness, while the inclusion of lower-risk patients (i.e., patients who had only experienced a single recurrence of CDI), exclusion of populations likely to receive fecal microbiota (Rebyota) in practice, and lack of clarity around prior antibiotic regimens limit the generalizability of the study findings to routine clinical practice. These limitations create uncertainty about the benefits and risks of treatment for those most affected by rCDI, complicating informed consent, clinical decision-making, and health system decision-making.
Clinical use and implementation of fecal microbiota (Rebyota): While fecal microbiota (Rebyota) may improve access to microbiota-based therapies, its potential implementation also raises important ethical considerations related to patient safety, informed consent, and appropriate prescribing. The absence of long-term safety data on both conventional FMT and fecal microbiota (Rebyota) creates challenges in fully informing patients of potential risks. There is also a risk of misdiagnosis of rCDI and subsequent inappropriate prescribing. While prescribing could be limited to specialists with expertise in rCDI and experience with fecal microbiota–based therapies, this could raise concerns regarding equitable access across geographic settings. In addition, as a standardized, single-dose enema, fecal microbiota (Rebyota) may not be suitable for all patients and may limit provider flexibility in selecting appropriate delivery modalities or tailoring dosing regimens to individual needs.
Health systems: Fecal microbiota (Rebyota) may introduce both opportunities and challenges for health system integration. While it may help to alleviate some of the logistical burdens associated with donor screening and stool preparation, persistent geographic and health infrastructural barriers may continue to limit access, particularly for rural and remote populations. Current reliance on a single US-based manufacturer introduces the risk that supply disruptions could interrupt access to an otherwise effective therapy, raising ethical concerns about treatment continuity and equitable care.
Objective and Research Questions
The objective of this Ethics Review was to identify and describe ethical considerations associated with the use fecal microbiota (Rebyota) to prevent recurrence of CDI in adults following antibiotic treatment for rCDI, including considerations related to the disease context, evidentiary basis, use of fecal microbiota (Rebyota), and impact on health systems.
To address this objective, this review addresses the following research questions:
What ethical considerations arise in the context of rCDI in adults, including considerations related to diagnosis, treatment, and outcomes?
What ethical considerations arise in relation to the evidence (e.g., clinical and economic data) used to evaluate fecal microbiota (Rebyota)?
What ethical considerations arise in relation to the use of fecal microbiota (Rebyota) for patients, their caregivers, and their clinicians?
What are the ethical considerations for health systems related to fecal microbiota (Rebyota)?
Methods
Guiding questions identified in the EUnetHTA Core Model 3.0 Ethical Analysis domain1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment (ECHTA)2 drove the identification of ethical considerations relevant to the use of fecal microbiota (Rebyota) for the treatment of rCDI in this Ethics Review. These guiding questions are organized to respond to the research questions and to investigate ethical considerations related to:
the patients living with rCDI and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges or burdens related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms, and value of fecal microbiota (Rebyota) (i.e., ethical considerations in relevant clinical trials, including their representativeness, the choice of outcome measures, the appropriateness of the analytical methods and models used to all population groups; ethical considerations related to the data or assumptions in the economic evaluation)
the use of fecal microbiota (Rebyota), including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, and society, as well as considerations related to access to this therapy
The uptake of fecal microbiota (Rebyota) in health systems, including considerations related to the distribution of health care resources.
Review of Project Inputs
One reviewer collected and considered input from 7 main sources of data related to ethical considerations relevant to the research questions guiding this Ethics Review. The reviewer considered the following sources:
evidence from a search of published literature
the sponsor submission, including relevant information and external references or sources relevant to each of the research questions driving this report
clinician group input received from the Canadian Antimicrobial Resistance Alliance
patient input received from the Peggy Lillis Foundation, the GI Society, and the Canadian Digestive Health Foundation
drug program input received from drug programs participating in the Canada’s Drug Agency (CDA-AMC) reimbursement review process
discussion with clinical experts (n = 3) directly engaged by CDA-AMC over the course of this reimbursement review, including through the clinical and economic consultation meetings involving 2 experts, and the panel meeting involving 3 experts; during each of these meetings, clinical experts were asked targeted questions related to ethical considerations corresponding to the research questions driving this report (all clinical experts were practising infectious disease specialists with experience treating patients with rCDI using conventional FMT)
engagement with clinical and economic reviewers to identify domains of ethical interest arising from their respective reviews and to identify relevant questions and sources to further pursue in this report.
Details on the Published Literature Search
Literature Search Methods
An information specialist conducted a literature search on key resources including MEDLINE via Ovid and Philosopher’s Index via Ovid. A targeted Google Scholar search was also performed. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Rebyota or fecal microbiota transplant and Clostridioides difficile.
Search filters were applied to limit retrieval to citations related to ethical concepts or considerations or equity concepts or considerations. No filters were applied to the searches conducted in Philosopher’s Index. Duplicates were removed by manual deduplication in EndNote. Retrieval was limited to the English language. The search was completed on February 26, 2025.
Literature Screening and Selection
One reviewer screened literature in 2 stages. First, the reviewer screened the titles and abstracts of the retrieved citations and identified and retrieved articles for full-text review if their titles or abstracts identified ethical considerations, or provided normative analyses (i.e., focusing on “what ought to be” through argumentation), or empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the experiences, incidence, diagnosis, treatment, or outcomes of rCDI, or related to the evidence on, use of, or implications of fecal microbiota (Rebyota) for patients with rCDI. In the second stage, the same reviewer reviewed full-text publications categorized as “retrieve.” The reviewer included texts that included substantive information meeting the aforementioned criteria. Additionally, the reviewer retrieved and reviewed select sources drawn from relevant bibliographies, relevant key concepts, and consultation with experts, or other reviewers using the selection criteria listed previously.
Data Analysis
The 4 research questions driving this review guided the collection, coding, and thematic analysis of data. The reviewer conducted 2 iterative cycles of coding and analysis to abstract, identify, and synthesize relevant ethical considerations from the literature and from relevant project inputs. In the initial coding phase, the reviewer read the publications and input sources for ethical content (e.g., claims related to potential harms, benefits, equity, justice, and resource allocation, as well as ethical issues in the evidentiary basis). The reviewer coded the identified claims related to ethical content using methods of qualitative description.3 In the second coding phase, the reviewer identified major themes and subcodes through repeated readings of the data3 and summarized them into thematic categories within each guiding domain or research question. The reviewer noted if the ethical content did not fit into these categories or into the domains outlined in the research questions, or if there were discrepancies or conflicts between the ethical considerations or values identified between project sources or within thematic categories. The data analysis was iterative, and the reviewer used themes identified in the literature, in project inputs, and during consultations with clinical experts to further refine and reinterpret the ethical considerations identified. Finally, the reviewer thematically organized and described the data according to the 4 research questions and domains driving this Ethics Review. The results, limitations, and conclusions of this analysis are described in the following sections.
Results
Key Ethical Considerations
Diagnosis, Treatment, and Experiences of rCDI
C. difficile is a spore-forming, toxin-producing, gram-positive anaerobic bacterium that colonizes the intestinal tract following disruption of normal gut flora, often due to antibiotic use.4 CDI is characterized by profuse watery diarrhea, defined as 3 or more loose stools within 24 hours, although severity ranges from mild diarrhea to fulminant colitis with systemic toxicity and shock.5 rCDI occurs when symptoms return within 2 to 8 weeks after treatment, leading to a cycle of infection that becomes increasingly difficult to manage. Up to 25% of patients experience recurrence within 30 days,4 and each recurrence increases the likelihood of future episodes, making rCDI a chronic and worsening condition for many individuals.
Although anyone can develop CDI, certain populations face disproportionately higher risks of severe disease, prolonged hospitalization, and recurrence. Patient group input identified adults older than 65 years, individuals undergoing chemotherapy or immunosuppressive therapy, those receiving prolonged antibiotic treatment, and people taking proton pump inhibitors as particularly vulnerable to CDI and rCDI. Socioeconomic and systemic factors further contribute to disparities in disease burden. Research from the US suggests that racialized individuals (e.g., those who are Black, Hispanic, Asian, Pacific Islanders, or from multiple ethnic backgrounds) and those with lower socioeconomic status (as defined by income, employment, education, and housing quality) are more likely to experience severe CDI, longer hospital stays, and recurrent infections.6-9 These disparities are likely driven by social determinants of health, including structural racism, food insecurity, housing instability, and barriers to health care access.10 In Canada, similar inequities exist, with clinical experts emphasizing that individuals in rural and remote communities may face additional challenges accessing specialized care, including limited access to providers who are familiar with current clinical guidelines for managing CDI and rCDI. These access challenges may lead to inappropriate treatment options and an increased risk of recurrence.
CDI represents a significant public health concern due to its infectious nature and potential for outbreaks in health care and community settings. While CDI is well known as a leading cause of health care–associated diarrhea,11 provincial data suggests that between 22% and 56% of cases in Canada are community-acquired.12 In their 2022 report, the Canadian Antimicrobial Resistance Surveillance System identified a 30-day attributable mortality of 2.2% among all individuals with health care–acquired or community-acquired CDI.13
Diagnosis
Accurately diagnosing rCDI can be challenging, and misdiagnosis is a known risk. Clinical experts noted that some providers may assume recurrence when a patient presents with watery diarrhea soon after completing antibiotic therapy for CDI. However, not all posttreatment diarrhea is due to CDI, and some individuals may instead be experiencing other gastrointestinal conditions with similar symptoms. Even when providers test for the presence of C. difficile, a positive result does not necessarily confirm rCDI, as C. difficile colonization can continue to appear in the stool for several weeks following antibiotic treatment.14 As such, clinical experts stressed that, particularly among providers who may be less familiar with CDI, a positive test result can be misleading and may discourage consideration of alternative diagnoses. In such cases, patients may receive unnecessary or inappropriate treatment, delaying proper care while exposing patients to avoidable risks.
Clinical experts also cautioned that some patients’ condition may be mistakenly labelled as refractory to antibiotics when they were being treated for unconfirmed CDI. Without confirmatory testing for CDI, these patients may be subjected to repeated antibiotic courses or unnecessary FMT, not only delaying appropriate care but also exposing them to avoidable risks, including antibiotic resistance and disruption of gut microbiota.
Disease Burden
Living with rCDI can be physically exhausting, emotionally isolating, and profoundly disruptive to daily life. Individuals with CDI face uncontrollable diarrhea, painful abdominal cramping, fatigue, and disrupted sleep due to frequent nighttime bathroom use.15 Patient group input described an overwhelming sense of powerlessness, as individuals plan their lives around access to a bathroom and avoid public spaces for fear of incontinence. Some individuals withdraw entirely from social interactions due to shame, embarrassment, or concerns about body odour. The fear of transmitting the infection to loved ones adds another layer of anxiety and guilt, particularly for individuals in close-contact environments like shared housing or caregiving settings.
Because CDI is a highly infectious disease, many patients adopt rigorous, time-consuming cleaning regimens to try to prevent household transmission.15 Those with access to multiple bathrooms often isolate themselves from family members,15 while individuals in single-bathroom homes or communal living settings may struggle to implement adequate infection control. These precautions, while necessary, can feel both physically and emotionally draining.15
Patient group input emphasized that individuals with multiple recurrences may also live in constant fear that a single round of antibiotics — whether for a minor infection, dental work, or surgery — will trigger another episode. Some patients described life after CDI as similar to “living with PTSD,” feeling trapped in a cycle of anxiety and hypervigilance, especially when health care providers may fail to acknowledge the ongoing risk of rCDI when prescribing antibiotics.
The burden of rCDI extends beyond the individual to caregivers, who must often provide emotional, logistical, and physical support. For those caring for individuals with multiple recurrences, the strain can be particularly severe, requiring constant monitoring, assistance with hygiene management, and adherence to strict infection control measures.
Current Treatment Options
Treatment for CDI involves 2 interrelated goals: controlling active infection and preventing future recurrence. Most individuals who experience an initial episode of CDI recover with antibiotic therapy alone. However, approximately 25% of patients experience a recurrence following successful treatment of their initial infection.4
For individuals with multiple recurrences of CDI (i.e., 2 or more recurrences), treatment becomes more complex. Canadian guidelines recommend that a second recurrence of active infection be managed with a prolonged taper or pulse regimen of vancomycin.14 While this may be an effective treatment for active infection, repeated courses of antibiotics can further disrupt the gut microbiome, limiting the body’s ability to restore a stable and protective microbial environment. As a result, those with multiple recurrences often face a worsening disease trajectory, with each episode increasing the likelihood of future recurrence.4
To address the complications of a depleted microbial environment, both Canadian and US guidelines recommend the use of fecal microbiota–based therapy for individuals with multiple recurrences of CDI.14,16 Unlike antibiotics, which work to control active infection, fecal microbiota–based therapies are preventive interventions designed to restore microbial diversity and support a more resilient gut environment to reduce the likelihood of future recurrence. To do this, stool from a healthy donor is screened for a variety of infectious microbes, processed, and then delivered to the recipient through 1 of many administration modalities, including enema, colonoscopy, nasogastric tube, or oral capsules. Canadian guidelines suggest that it is possible patients will achieve greater clinical response with multiple administrations.14 However, a recent Cochrane review determined that there is too little evidence to support conclusive statements regarding differences in safety or efficacy according to mode or frequency of administration.17 As such, choice is often influenced by factors such as patient preference, clinical considerations (e.g., anatomic needs or limited success with previous administration modalities), and the infrastructure and resources available at the administering centre.
Accessing Fecal Microbiota–Based Therapies
Although there are some risks associated with fecal microbiota–based therapy, it is increasingly seen as a safe and effective treatment option in the context of rCDI prevention.17 Nonetheless, patient access has been limited in Canada. Two overarching challenges impact access in Canada: regulatory complexity surrounding fecal microbiota–based therapy and infrastructural and logistical burdens associated with existing access pathways.
At present, Health Canada classifies the fecal matter used in fecal microbiota–based therapies as biologic drugs under the Food and Drugs Act.18 As biologics, fecal microbiota–based therapies must meet stringent requirements around product safety, manufacturing, and clinical efficacy.18 While these requirements align with standards for other biologics, they may also introduce challenges bringing new fecal microbiota–based therapies to market, given the complexity of demonstrating safety, purity, and potency within inherently variable stool-derived microbiota. Although Health Canada has recognized the clinical value of conventional FMT (as a form of fecal microbiota–based therapy) to rCDI since at least 2015,18 no commercial products were available in Canada until fecal microbiota (Rebyota) received market authorization in March 2025.
Health Canada has permitted the use of conventional FMT under an interim policy exemption since 2015, provided specific regulatory safeguards are consistently met.18 This exemption has allowed the therapy to be used while recognizing its investigational nature and maintaining stringent donor screening and safety standards.18 Importantly, conventional FMT allows for clinical flexibility that can be tailored to individual patient needs. Clinical experts noted that current FMT programs may offer 1 or more administration modalities (i.e., enema, oral capsule, colonoscopy) and that dosing regimens can easily be adjusted based on patient need.
Despite this exemption, clinical experts indicated that access and logistical challenges have persisted. Clinical providers must independently manage the recruitment, screening, and retention of qualified stool donors, which is costly and time-consuming. Due to stringent eligibility criteria, frequent retesting requirements, reliance on voluntary donor participation, and ongoing stigma surrounding stool donorship, the number of qualified donors remains limited.19,20 Additionally, because stool must be sourced from a single donor known to the patient or treating provider,18 patients may feel pressure to identify suitable donors from their family or social network. This expectation can create stress or embarrassment given the intimate and stigmatized nature of what is being donated.19 These factors contribute to substantial delays for patients, particularly if repeated treatments or alternative administration modalities become necessary but are unavailable with the same FMT program. Additional barriers may include infrastructural requirements such as specialized laboratory equipment for stool screening, trained personnel, and dedicated clinical spaces for administration.
Ethics of Evidence and Evaluation of Fecal Microbiota (Rebyota)
Trial Information
The PUNCH CD3 trial was a phase III, double-blind, placebo-controlled randomized study (n = 289) evaluating the efficacy and safety of fecal microbiota (Rebyota) (delivered as a single-dose enema) in preventing rCDI following antibiotic treatment for rCDI. The primary outcome was treatment response at 8 weeks, with follow-up extending to 6 months. While clinical experts noted that the between-group difference of 12.3% (favouring fecal microbiota [Rebyota] over placebo) suggests a clinically meaningful effect, they also highlighted the broad credible interval (CrI) (95% CrI, 1.4 to 23.3), which introduces uncertainty regarding the magnitude of benefit. Clinical experts further noted that the lower boundary of this CrI falls below what would generally be considered clinically meaningful to both clinicians and patients.
All trial participants received antibiotic therapy before fecal microbiota (Rebyota) administration to treat the active infection, as is standard practice. However, the PUNCH CD3 study did not specify the type of vancomycin regimen used (e.g., standard course versus prolonged taper or pulse regimen), making it unclear whether the trial population aligned with real-world candidates for FMT in Canada. Given that FMT is typically reserved for patients who have experienced multiple recurrences after a vancomycin taper or pulse regimen, this omission creates uncertainty about whether the trial results apply to the higher-risk patients (e.g., those who have had 2 or more recurrences) most likely to receive fecal microbiota (Rebyota) in practice.
The decision to omit conventional FMT as a comparator raises ethical concerns. While the clinical experts suggested that a placebo comparator was appropriate in the context of a clinical trial, some authors have argued that excluding conventional FMT from trials evaluating new microbiota–based therapies is no longer ethically justified, given its well-established efficacy in preventing rCDI.21 The absence of conventional FMT in the PUNCH CD3 trial, or as part of a sponsor-driven indirect treatment comparison, is therefore difficult to justify. While regulatory restrictions on conventional FMT in the US and Canada may have contributed to the omission of conventional FMT as a comparator in the trial, they would not have precluded the inclusion of an indirect treatment comparison comparing fecal microbiota (Rebyota) to conventional FMT.
Underrepresentation and Generalizability
While the PUNCH CD3 trial provided important evidence on fecal microbiota (Rebyota) for preventing rCDI, several factors limit its generalizability to real-world clinical practice in Canada.
Exclusion of key patient populations: The trial excluded patients with inflammatory bowel disease, irritable bowel syndrome, celiac disease, motility disorders, immunocompromised status, and refractory CDI. While these exclusions reduce confounding, they also mean that many patients who would receive fecal microbiota (Rebyota) in practice were not represented.
Inclusion of lower-risk patients: A significant portion of trial participants had only 1 recurrence of CDI (29.5% in the active arm and 38.5% in the placebo arm), despite clinical guidelines recommending fecal microbiota–based therapies only for patients with at least 2 recurrences.14,16 As the risk of recurrence increases substantially following a second recurrence, this inclusion may underrepresent the higher-risk patients who most need fecal microbiota–based therapies, including fecal microbiota (Rebyota).
Limited demographic diversity and unclear inclusion of systemically marginalized populations: The PUNCH CD3 trial population was disproportionately white (92%), raising concerns about whether the findings are generalizable to racialized groups. It is unclear to what extent other systemically marginalized groups — such as those with low incomes, housing insecurity, or limited access to health care — were included. This lack of demographic diversity and uncertain representation of social diversity raises concerns about generalizability, particularly given the growing evidence that gut microbiota are shaped by social and environmental conditions such as diet, food security, housing stability, and differential patterns of antibiotic use.10 These factors are often linked to structural inequities and my influence both the development of CDI and rCDI, as well as the effectiveness of microbiota–based therapies like fecal microbiota (Rebyota).
The exclusion or underrepresentation of populations who may be disproportionately affected by CDI and rCDI means that many patients could be offered treatment based on limited or uncertain evidence of safety and effectiveness for their specific circumstances. This has implications for both clinical care and health system decision-making. For clinicians, it complicates efforts to assess the risk-benefit profile of fecal microbiota (Rebyota) for patients from underrepresented or equity-deserving groups, which may constrain their ability to support fully informed consent. For health systems, these evidence gaps introduce uncertainty about the value of investing in fecal microbiota (Rebyota) for populations most at risk of severe or recurrent infection. Addressing these limitations in future trials or through postmarket surveillance may be necessary to ensure that fecal microbiota (Rebyota) is evaluated in the diverse populations that will receive them in real-world settings.
Ethical Considerations in the Use of Fecal Microbiota (Rebyota)
Fecal microbiota (Rebyota) is a standardized, Health Canada–approved, fecal microbiota–based therapy for rCDI that offers an alternative to conventional FMT currently permitted under Health Canada’s 2015 exemption. It consists of a single-dose, ready-to-use enema containing human fecal microbiota derived from rigorously screened, anonymous donors. Clinical experts viewed the potential addition of fecal microbiota (Rebyota) to public formularies as a positive step in rCDI care. While they suggested there is little difference in the therapeutic mechanism between conventional FMT and commercial products like fecal microbiota (Rebyota), increased availability of commercial products may significantly reduce the logistical barriers associated with conventional FMT.22-24 By shifting responsibility for donor recruitment, stool screening, and product manufacturing from FMT programs to commercial manufacturers, clinical experts suggested that commercial therapies like fecal microbiota (Rebyota) could potentially improve patient access, reduce provider workload, enhance treatment consistency, and alleviate liability risks currently borne by clinical providers.24 However, ethical questions related to informed consent, patient autonomy, and long-term safety remain relevant.
Patient Autonomy and Informed Consent
Unlike conventional FMT, which typically involves stool from known donors selected by patients or their providers, fecal microbiota (Rebyota) relies exclusively on anonymous donors chosen and screened by the sponsor. While this approach may reduce patient and provider burdens associated with recruitment and screening, it significantly limits patient choice and knowledge regarding the origin and composition of the microbiota they receive. Patients have no opportunity to select donors based on characteristics that may be important to them, such as dietary habits, lifestyle factors, or health history, all of which can influence microbiota composition.10,25 For patients with certain religious, cultural, or personal beliefs, receiving microbiota from an unknown source without any control or insight into these donor attributes can raise meaningful concerns or discomfort.25 While not all patients will consider donor identity to be relevant to their care, for those who do, this lack of transparency and choice may compromise their ability to fully weigh treatment options, understand potential implications of the therapy, and make decisions aligned with their personal values and preferences.
As with conventional FMT, the long-term effects of introducing new microbial communities into the gut through fecal microbiota (Rebyota) remain poorly understood, posing further challenges to informed consent and patient autonomy. Although clinical experts were satisfied with the short-term safety profile of fecal microbiota (Rebyota), they flagged that altering the gut microbiome may have downstream effects on neurologic, metabolic, or immune system functions, even if current research is not yet able to predict or quantify these risks.22,26 As such, while patients may understand the short-term benefit of preventing another recurrence of CDI, they may lack sufficient information to fully assess the longer-term risks.22,27 Literature indicates that patients often struggle to weigh these unknowns, especially when faced with ongoing illness and limited treatment options.22 Ensuring consent procedures clearly communicate both known benefits and unresolved risks is critical to supporting patient autonomy and preventing misconceptions about the safety and efficacy of fecal microbiota (Rebyota).
Additionally, as with all biotherapeutic products, fecal microbiota (Rebyota) carries a theoretical risk of transmitting antibiotic resistance genes from donor stool.26 While Health Canada and the FDA require manufacturers to screen donor stool for multidrug-resistant organisms (e.g., Escherichia coli, vancomycin-resistant enterococci, and methicillin-resistant Enterobacteriaceae), current screening methods may fail to detect novel resistance pathways or emerging antimicrobial threats.26 The risk of developing antibiotic resistance is not unique to fecal microbiota (Rebyota), as conventional FMT carries the same risk. Further, the repeated and potential long-term use of antibiotics to treat active rCDI can also lead to antimicrobial resistance. Nonetheless, as with uncertainties related to donor anonymity and long-term microbiome impacts, this residual uncertainty about antibiotic resistance highlights the importance of clear patient education and informed consent processes that acknowledge the limits of current knowledge.
Potential for Misuse
Fecal microbiota (Rebyota) is intended to improve access to fecal microbiota–based therapies, but concerns remain about its potential misuse in clinical practice. As described previously, diagnosing rCDI can be challenging. Some providers may mistakenly assume recurrence when a patient presents with watery diarrhea soon after treatment, without confirmatory testing or consideration of alternative diagnoses. This can be further complicated by the fact that the presence of C. difficile does not necessarily indicate active infection, especially in patients with overlapping gastrointestinal symptoms. In such cases, patients may receive fecal microbiota (Rebyota) unnecessarily, exposing them to avoidable risks and contributing to inappropriate health care spending.
Separate from diagnostic uncertainty, there is also concern about the potential for off-label prescribing. Clinical experts noted that while some off-label use of conventional FMT may already occur (e.g., for ulcerative colitis or other gastrointestinal disorders), such use is likely limited by the logistical complexity of sourcing donor stool and preparing the product. The introduction of a ready-made, commercially available product may lower the threshold for prescribing outside of approved indications.24 Without strong safeguards in place, this could lead to increased use in clinical scenarios where evidence of safety and efficacy is limited or lacking, potentially exposing patients to unknown risks and undermining informed decision-making.
To mitigate these risks, clinical experts recommended that fecal microbiota (Rebyota) only be prescribed by specialists with expertise in rCDI and familiarity with fecal microbiota–based therapies, such as infectious disease specialists and gastroenterologists. While this approach could help ensure accurate diagnosis and appropriate patient selection, clinical experts acknowledged that it may also create geographic access challenges for patients in rural and remote areas, where these specialists are less available.
Potential Limitations to Access
While fecal microbiota (Rebyota) may improve access to fecal microbiota–based therapies by alleviating logistical and administrative burdens, its regulatory approval of a single-dose, enema-based formulation introduces new limitations. Unlike conventional FMT, which allows for flexible delivery modalities and individualized dosing regimens, fecal microbiota (Rebyota) offers a standardized product intended to standardize care across settings. Clinical experts emphasized, however, that this standardization may not suit all patients. Individuals with anatomic constraints preventing enema use, those who could benefit from multidose treatment, or patients with specific preferences related to donor identity or delivery modality may be excluded from treatment unless conventional FMT remains available.25,28
Standardization, while important for regulatory approval and manufacturing consistency, may inadvertently constrain both patient and provider autonomy. The process of transforming fecal microbiota–based therapies into standardized products often reflects industrial and regulatory priorities (e.g., consistency in formulation, administration, and storage) but may fail to accommodate the biological and experiential diversity of patients living with rCDI.23 In this context, limiting treatment options to a single product formulation risks narrowing the clinical tools available for tailoring care to individual needs and values. While other modalities may eventually be granted market authorization by Health Canada, access to these options remains limited in Canada.
Clinical experts expressed concern that the introduction of fecal microbiota (Rebyota) could eventually prompt Health Canada to sunset the current exemption permitting conventional FMT. If this were to occur, the range of delivery modalities and dosing flexibility currently offered through conventional FMT programs could be lost, potentially restricting access for individuals who may benefit from customized treatment.
Health Systems Considerations
Persistent Barriers to Access and System Integration
Although fecal microbiota (Rebyota) may address several supply-side challenges associated with conventional FMT (e.g., donor screening, stool preparation, and product manufacturing), its integration into the health care systems in Canada presents ongoing challenges. These challenges are not solely logistical, but are also structural, raising ethical concerns around equitable access, sustainability, and long-term service delivery.
Reimbursement remains a key issue. Clinical experts indicated that it is presently unclear whether the introduction of fecal microbiota (Rebyota) will include specific billing codes for provider time spent administering the product. Without specific billing codes or reimbursement pathways for providers administering the product, there is a risk that access may be restricted to sites with capacity to absorb these costs. Additionally, existing FMT programs may have been designed around a single administration modality (i.e., colonoscopy, oral capsule, or enema). The enema-only format of fecal microbiota (Rebyota) may not align with these established infrastructures or patient preferences, requiring significant changes in clinical workflow that could slow uptake.
Even if fecal microbiota (Rebyota) is recommended for public reimbursement, its impact may be limited by the uneven distribution of FMT programs across the country. As access to FMT programs remains concentrated in urban, academic centres, patients in rural and remote regions may still face barriers to treatment due to geographic disparities in specialist access. It is possible that these challenges may be alleviated as providers become more familiar with the product and can teach their patients to self-administer at home, but this is not currently the case.
Additionally, because fecal microbiota (Rebyota) is currently manufactured in the US, access in Canada is dependent on international supply chains at this time. The potential for cross-border transit issues raises concerns about long-term supply stability. While no immediate supply concerns were raised by the clinical experts, maintaining a stable and reliable source of fecal microbiota (Rebyota) will require careful monitoring of trade and regulatory developments. One potential strategy to mitigate reliance on international supply chains would be the development of publicly funded regional or national stool banks. Such facilities could help ensure a stable domestic supply of fecal microbiota–based therapies, streamline donor screening, and reduce reliance on US manufacturers.29
Long-Term Monitoring to Help Address Uncertainties
The ethical considerations surrounding fecal microbiota (Rebyota) and conventional FMT, including the unknown systemic risks of introducing a new microbial community into a patient’s gut, warrant ongoing monitoring.22,26 Experts noted that comprehensive long-term data collection would improve informed consent processes, strengthen clinical decision-making, and ensure that emerging risks or benefits are identified and addressed as FMT-based therapies become more widely adopted.
The clinical experts and the published literature suggested that a national registry tracking both short-term and long-term patient outcomes should be established, particularly as commercial fecal microbiota–based therapies (like Rebyota) are introduced into broader clinical use.21,22,24,26,30 Given ongoing uncertainties about real-world effectiveness — especially for patient populations excluded from the PUNCH CD3 trial — and the unknown long-term impacts of gut microbiota transfer, a registry could support more informed, evidence-based system-level decision-making over time. The standardized nature of fecal microbiota (Rebyota) may facilitate consistent data collection for registry purposes, although any such system would need to carefully consider how to mitigate patient privacy and data governance challenges.
Limitations
Little published literature discusses ethical considerations related to the use of fecal microbiota (Rebyota) for the prevention of rCDI in adults. Input received during this reimbursement review (including patient group, clinician group, and drug program input and discussion with clinical experts, as well as engagement with the clinical and pharmacoeconomic review teams) provided a more comprehensive understanding of the ethical considerations related to the use of fecal microbiota (Rebyota) for the treatment of rCDI. It is possible that more direct engagement (e.g., through direct interviews) with patients, their caregivers, their family members, and decision-makers on their specific experiences with rCDI and/or fecal microbiota (Rebyota) would offer additional relevant ethical considerations or domains of analysis.
Conclusion
This report examined ethical considerations surrounding the use of fecal microbiota (Rebyota) for the prevention of rCDI in adults, drawing on input from patient groups, clinician groups, provincial drug programs, direct engagement with 3 clinical experts, and a review of published literature. rCDI imposes significant physical, emotional, and social burdens on affected individuals. Those experiencing multiple recurrences face significant disruptions to daily life and an elevated risk of serious complications, including hospitalization and death. These burdens are disproportionately experienced by populations already facing structural disadvantage, including older adults, individuals who are immunocompromised, those living in rural or remote areas, and people facing other systemic barriers to care.
While fecal microbiota–based therapies have demonstrated efficacy in preventing rCDI, conventional FMT has remained difficult to access in Canada due to regulatory restrictions, health infrastructure, and the logistical challenges of donor recruitment and screening. If recommended for reimbursement, it is possible that fecal microbiota (Rebyota) can help mitigate some of these barriers by shifting the responsibility for donor screening and stool preparation from providers to manufacturers. However, challenges related to geographic access, delivery modality, and tailored dosing are likely to persist, particularly for patients who require more than a single dose, cannot receive treatment via enema, or prefer a known donor.
Moderate-certainty findings from clinical trial data suggest that treatment with fecal microbiota (Rebyota) likely results in a clinically important increase in treatment success over 8 weeks compared to placebo. However, key limitations in the PUNCH CD3 trial — such as the lack of comparison to conventional FMT, the inclusion of lower-risk patients, and the exclusion of patients most likely to receive fecal microbiota (Rebyota) in clinical practice — raise questions about its applicability to routine clinical care. Additionally, as with conventional FMT broadly, the long-term systemic harms of fecal microbiota (Rebyota) remain unknown, presenting challenges for ensuring fully informed consent. Finally, reliance on a single US-based manufacturer raises ethical concerns about treatment continuity and equitable care, given that any supply disruption could limit patient access to this therapy.
References
1.EUnetHTA. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0. 2016. Accessed November 4, 2025. https://web.archive.org/web/20241116160013/https://www.eunethta.eu/wp-content/uploads/2018/03/HTACoreModel3.0-1.pdf
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37:e17. doi: 10.1017/S0266462320002275 PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-40. doi: 10.1002/1098-240x(200008)23:4<334::aid-nur9>3.0.co;2-g PubMed
4.Khanna S, Assi M, Lee C, et al. Efficacy and safety of RBX2660 in PUNCH CD3, a Phase III, randomized, double-blind, placebo-controlled trial with a Bayesian primary analysis for the prevention of recurrent Clostridioides difficile infection. Drugs. 2022;82(15):1527-1538. doi: 10.1007/s40265-022-01797-x PubMed
5.Blount KF, Papazyan R, Ferdyan N, et al. Microbiome and Metabolome Restoration After Administration of Fecal Microbiota, Live-jslm (REBYOTA) for Preventing Recurrent Clostridioides difficile Infection. J Infect Dis. 2025;231(6):e1022-e1033. doi: 10.1093/infdis/jiae418 PubMed
6.Argamany JR, Delgado A, Reveles KR. Clostridium difficile infection health disparities by race among hospitalized adults in the United States, 2001 to 2010. BMC Infect Dis. 2016;16(1):454. doi: 10.1186/s12879-016-1788-4 PubMed
7.Young EH, Strey KA, Lee GC, Carlson TJ, Koeller JM, Reveles KR. Clostridioides difficile Infection Treatment and Outcome Disparities in a National Sample of United States Hospitals. Antibiotics (Basel). 2022;11(9):1203. doi: 10.3390/antibiotics11091203 PubMed
8.Lee JM, Zhou AY, Ortiz-Gratacos NM, Al Isso A, Tan KK, Abdul-Mutakabbir JC. Examining the impact of racial disparities on Clostridioides difficile infection outcomes at a Southern California academic teaching hospital. Infect Control Hosp Epidemiol. 2023;44(11):1-5. doi: 10.1017/ice.2023.84 PubMed
9.Scaria E, Powell WR, Birstler J, et al. Neighborhood disadvantage and 30-day readmission risk following Clostridioides difficile infection hospitalization. BMC Infect Dis. 2020;20(1):762. doi: 10.1186/s12879-020-05481-x PubMed
10.Reveles KR, Strey KA, Abdul-Mutakabbir JC, Mendoza VM, Carreno JJ. Infectious Inequity: How the Gut Microbiome and Social Determinants of Health May Contribute to Clostridioides difficile Infection Among Racial and Ethnic Minorities. Clin Infect Dis. 2023;77(Suppl 6):S455-S462. doi: 10.1093/cid/ciad586 PubMed
11.Smits WK, Lyras D, Lacy DB, Wilcox MH, Kuijper EJ. Clostridium difficile infection. Nat Rev Dis Primers. 2016;2:16020. doi: 10.1038/nrdp.2016.20 PubMed
12.Xia Y, Tunis MC, Frenette C, et al. Epidemiology of Clostridioides difficile infection in Canada: A six-year review to support vaccine decision-making. Can Commun Dis Rep. 2019;45(7-8):191-211. doi: 10.14745/ccdr.v45i78a04 PubMed
13.Public Health Agency of Canada. Canadian Antimicrobial Resistance Surveillance System (CARSS) Report 2022. 2022. Accessed April 4, 2024. https://www.canada.ca/en/public-health/services/publications/drugs-health-products/canadian-antimicrobial-resistance-surveillance-system-report-2022.html
14.Loo VG, Davis I, Embil J, et al. Association of Medical Microbiology and Infectious Disease Canada treatment practice guidelines for Clostridium difficile infection. J Assoc Med Microbiol Infect Dis Can. 2018;3(2):71-92. doi: 10.3138/jammi.2018.02.1
15.Armstrong EP, Malone DC, Franic DM, Pham SV, Gratie D, Amin A. Patient Experiences with Clostridioides difficile Infection and Its Treatment: A Systematic Literature Review. Infect Dis Ther. 2023;12(7):1775-1795. doi: 10.1007/s40121-023-00833-x PubMed
16.Peery AF, Kelly CR, Kao D, et al. AGA Clinical Practice Guideline on Fecal Microbiota-Based Therapies for Select Gastrointestinal Diseases. Gastroenterology. 2024;166(3):409-434. doi: 10.1053/j.gastro.2024.01.008 PubMed
17.Minkoff NZ, Aslam S, Medina M, et al. Fecal microbiota transplantation for the treatment of recurrent Clostridioides difficile (Clostridium difficile). Cochrane Database Syst Rev. 2023;4(4):CD013871. doi: 10.1002/14651858.CD013871.pub2 PubMed
18.Health Canada. Guidance document: Fecal microbiota therapy used in the treatment of Clostridioides difficile infection not responsive to conventional therapies. 2020. Accessed March 20, 2025. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/regulation-fecal-microbiota-therapy-treatment-difficile-infections.html
19.Quay T, Walter M. Fecal microbiota therapy in Canada: an environmental scan. (Environmental scan no. 95). CADTH; 2020. Accessed March 20, 2025. https://www.cda-amc.ca/sites/default/files/es/es0341-fecal-microbiota-therapy-in-canada.pdf
20.Guilfoyle J, Considine J, Bouchoucha SL. Faecal microbiota transplantation and the patient experience: A systematic review. J Clin Nurs. 2021;30(9-10):1236-1252. doi: 10.1111/jocn.15625 PubMed
21.Kelly CR, Kahn SA. Is it unethical to conduct placebo-controlled trials of faecal microbiota transplantation for recurrent Clostridioides difficile infection? Lancet Gastroenterol Hepatol. 2020;5(5):432-433. doi: 10.1016/S2468-1253(20)30043-1 PubMed
22.Ma Y, Liu J, Rhodes C, Nie Y, Zhang F. Ethical Issues in Fecal Microbiota Transplantation in Practice. Am J Bioeth. 2017;17(5):34-45. doi: 10.1080/15265161.2017.1299240 PubMed
23.Wolf-Meyer MJ. Normal, Regular, and Standard: Scaling the Body through Fecal Microbial Transplants. Med Anthropol Q. 2017;31(3):297-314. doi: 10.1111/maq.12328 PubMed
24.Hota SS, Poutanen SM. Microbiome-based therapeutics for Clostridioides difficile infection: helpful solutions or unclear cocktails? Lancet Infect Dis. 2023;23(9):999-1000. doi: 10.1016/s1473-3099(23)00484-x PubMed
25.Bokek-Cohen Y, Ravitsky V. Cultural and Personal Considerations in Informed Consent for Fecal Microbiota Transplantation. Am J Bioeth. 2017;17(5):55-57. doi: 10.1080/15265161.2017.1299241 PubMed
26.Khoruts A, Hoffmann DE, Palumbo FB. The Impact of Regulatory Policies on the Future of Fecal Microbiota Transplantation. J Law Med Ethics. 2019;47(4):482-504. doi: 10.1177/1073110519897726 PubMed
27.Mikail M, O'Doherty KC, Poutanen SM, Hota SS. Ethical implications of recruiting universal stool donors for faecal microbiota transplantation. Lancet Infect Dis. 2020;20(3):e44-e49. doi: 10.1016/S1473-3099(19)30569-9 PubMed
28.American Gastroenterological Association. Retention of access to conventional fecal microbiota transplantation for complicated and vulnerable patients with C. difficile infection. 2024. Accessed March 20, 2025. https://aga-fileuploader-bucket.s3.us-east-2.amazonaws.com/Aga-Files/Multisociety%20Letter%20on%20FMT_20241008.pdf
29.Panchal P, Budree S, Scheeler A, et al. Scaling Safe Access to Fecal Microbiota Transplantation: Past, Present, and Future. Curr Gastroenterol Rep. 2018;20(4):14. doi: 10.1007/s11894-018-0619-8 PubMed
30.Huss J. Fecal Transplant Bioethics: Beyond Chicken Little. Am J Bioeth. 2017;17(5):48-50. doi: 10.1080/15265161.2017.1299254 PubMed
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca