Drugs, Health Technologies, Health Systems
Reimbursement Review
Rozanolixizumab (Rystiggo)
Sponsor: UCB Canada Inc.
Therapeutic area: Generalized myasthenia gravis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AChEI
acetylcholinesterase inhibitor
AChR
acetylcholine receptor
AE
adverse event
CI
confidence interval
CDA-AMC
Canada’s Drug Agency
EMG
electromyography
ESS
estimated sample size
FAS
full analysis set
gMG
generalized myasthenia gravis
GRADE
Grading of Recommendations Assessment, Development and Evaluation
HRQoL
health-related quality of life
IgG
immunoglobulin G
IMP
investigational medicinal product
IST
immunosuppressive therapy
IVIg
IV immunoglobulin
J2R
jump to reference
LRP4
lipoprotein-related protein 4
LS
least squares
MAIC
matching-adjusted indirect comparison
MAR
missing at random
MDC
Muscular Dystrophy Canada
MG
myasthenia gravis
MG-ADL
Myasthenia Gravis Activities of Daily Living
MGC
Myasthenia Gravis Composite
MGFA
Myasthenia Gravis Foundation of America
MG-QoL15r
revised 15-item Myasthenia Gravis Quality of Life
MID
minimally important difference
MuSK
muscle-specific tyrosine kinase
NMA
network meta-analysis
NSIST
nonsteroidal immunosuppressive therapy
OLE
open-label extension
OR
odds ratio
pCPA
pan-Canadian Pharmaceutical Alliance
PICO
population, intervention, comparator, and outcome
PLEX
plasma exchange
QMG
Quantitative Myasthenia Gravis
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SCIg
subcutaneous immunoglobulin
SD
standard deviation
SE
standard error
SLR
systematic literature review
SSQ
single simple question
TEAE
treatment-emergent adverse event
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on the Application Submitted for Review
Item | Description |
|---|---|
Drug product | Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use |
Sponsor | UCB Canada Inc. |
Indication | For the treatment of adult patients with gMG who are anti-acetylcholine receptor or anti-muscle-specific tyrosine kinase inhibitor antibody positive |
Reimbursement request | As an add-on therapy for the treatment of adult patients with gMG who are either AChR antibody-positive or MuSK antibody-positive and for whom symptoms persist despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | Original NOC received: March 28, 2025 Revised NOC received: April 25, 2025 |
Recommended dose | Administered as an SC infusion using an infusion pump at a rate of up to 20 mL/hour once weekly for 6 weeks The weekly recommended dose varies by body weight category:
Subsequent treatment cycles are administered according to clinical evaluation. The frequency of treatment cycles may vary by patient. |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; CDA-AMC = Canada’s Drug Agency; gMG = generalized myasthenia gravis; MuSK = muscle-specific tyrosine kinase; NDS = new drug submission; NOC = Notice of Compliance; NSIST = nonsteroidal immunosuppressive therapy; SC = subcutaneous; TBC = to be confirmed.
Introduction
Myasthenia gravis (MG) is a rare, chronic, autoimmune neuromuscular disease in which antibodies against the neuromuscular junction disrupt neuromuscular transmission, resulting in localized or generalized skeletal muscle weakness.1-3 The Myasthenia Gravis Foundation of America (MGFA) stratifies people with MG into 5 functional classes based on clinical features and symptom severity. The classification ranges from class I (ocular weakness only) to class V (intubation, with or without mechanical ventilation [e.g., myasthenic crisis], except when employed during routine postoperative management). Classes II, III, and IV represent patients with mild, moderate, and severe generalized weakness involving nonocular muscles, respectively.4 Patients experience a variety of symptoms, including fatigue, droopy eyelids, diplopia, neck weakness, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and upper and/or lower limb weakness.3 There are 2 clinical forms of MG: ocular (in which weakness is limited to the eyelids and extraocular muscles) and generalized (in which weakness involves a variable combination of ocular, bulbar, limb, and respiratory muscles).4 Based on serology, 85% of patients with generalized MG (gMG) are acetylcholine receptor (AChR) antibody-positive, 8% are muscle-specific tyrosine kinase (MuSK) antibody-positive, 1% are lipoprotein-related protein 4 (LRP4) antibody-positive, and the remaining 6% are seronegative (i.e., no detectable antibodies).5 Globally, the incidence of MG varies from 4 cases per million person-years to 30 cases per million person-years, and the prevalence ranges from 150 cases per million to 200 cases per million.6 In Canada, the incidence and prevalence of gMG are estimated at 23 per 1 million person-years and 26.3 per 100,000, respectively.7 The mortality rate of MG has been reported to be from 0.06 per million person-years to 0.89 per million person-years.8 The symptoms of gMG occur unpredictably and fluctuate in nature, intensity, and severity on a day-to-day basis throughout a patient’s life, requiring intervention or treatment change; this is known as MG exacerbation.3,9 The unpredictable exacerbation and myasthenic crisis, in combination with a variety of symptoms, result in a chronic disease with significant burden that negatively affects patients’ quality of life.10,11
There are currently no Canadian guidelines for the treatment of gMG. The MGFA international consensus guidelines for the management of MG12 were updated in 2020 and are now the most recent guidelines.14 According to these guidelines and clinical experts in Canada, the goal of treatment for patients with gMG is to reduce disease symptoms as well as adverse effects of MG therapy and allow the patient to function and work normally, with good health-related quality of life (HRQoL).12,13 Other treatment goals include avoiding MG exacerbations and myasthenic crises, minimizing hospitalizations and intensive care unit admissions, and reducing the numbers and doses of therapies (especially corticosteroids) required for symptom control.13 The MGFA international consensus guidelines for the management of MG recommend thymectomy in young (i.e., aged 18 years to 50 years) patients with gMG early in the disease course to improve clinical outcomes and minimize immunotherapy requirements, hospitalization, and disease exacerbations.14 However, thymectomy is an elective procedure, and not all patients with gMG will be candidates; candidacy is based on age and ability to tolerate limited postoperative respiratory function.14
Conventional therapy for all patients with gMG generally begins with acetylcholinesterase inhibitors (AChEIs). However, AChEIs can worsen the symptoms of MuSK antibody-positive MG; therefore, these may not be used in all such patients.12 If AChEI therapy alone provides insufficient symptom relief, immunosuppressive therapy (IST) with a corticosteroid, such as prednisone, is administered.12 In patients who do not respond to corticosteroids or have significant comorbidities (such that long-term corticosteroid treatment is contraindicated), or in whom doses of corticosteroids cannot be tapered, treatment with nonsteroidal immunosuppressive therapies (NSISTs) (such as azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, or methotrexate) may be initiated, either alone or in combination with corticosteroids.12 It can take several months to years, depending on the NSIST, for the drug to produce a clinically relevant effect and reduce a patient’s gMG symptoms.1 While patients wait for NSIST treatment to take effect, they may experience MG exacerbations and/or myasthenic crises that require the acute use of IV immunoglobulin (IVIg), subcutaneous immunoglobulin (SCIg), or plasma exchange (PLEX).12 If patients continue to experience gMG symptoms, the dose may be increased or switched to an alternative NSIST.
It is estimated that 15% to 40% of patients will continue to experience symptoms despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs.15-17 Patients with AChR antibody-positive gMG whose symptoms persist despite conventional therapy would be eligible for treatment with rituximab, chronic IVIg or SCIg, and/or chronic PLEX.12,14 These patients would also qualify for treatment with efgartigimod alfa, as per the recommendation by Canada’s Drug Agency (CDA-AMC).18,19 Patients who have refractory gMG — defined as inadequate symptom control after an adequate trial of 2 or more ISTs, either in combination or as monotherapy over the past 12 months (or at least 1 IST and PLEX or IVIg at least every 3 months over the past 12 months) — would also be eligible for treatment with eculizumab, as per the CDA-AMC recommendation.19 However, the pCPA negotiations for eculizumab concluded without agreement; as a result, this drug is not listed on any public drug program formulary, and very few patients are able to access it.
Currently, there are no targeted therapies approved by Health Canada for the treatment of MuSK antibody-positive gMG. The same conventional therapies used for patients with AChR antibody-positive gMG are applied in patients with MuSK antibody-positive gMG.12 However, patients with MuSK antibody-positive gMG are less responsive to AChEIs and frequently intolerant to pyridostigmine at conventional doses.20 Patients with MuSK antibodies typically respond well to corticosteroids and NSISTs, but tend to remain dependent on corticosteroids, despite concomitant therapy with NSISTs.20 For patients whose symptoms persist despite treatment with corticosteroids and NSISTs, options include rituximab and PLEX; IVIg is usually less effective.20 Rituximab is recommended by international consensus guidance for patients with MuSK antibody-positive gMG who have an unsatisfactory response to initial immunotherapy.21 That is in contrast to patients with AChR antibody-positive gMG, in whom rituximab is only considered if patients do not respond to or tolerate other immunotherapies.21 Eculizumab and efgartigimod alfa are not approved by Health Canada or reimbursed by public drug programs for the treatment of MuSK antibody-positive gMG.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of rozanolixizumab 140 mg/mL solution for subcutaneous (SC) injection (280 mg in 2 mL in single-dose glass vials) in the treatment of gMG in adult patients.
Perspectives of Patients, Clinicians, and Drug Programs
The information in this section is a summary of the input provided by the patient and clinician groups who responded to our call for input and from clinical expert(s) consulted for the purpose of this review.
Patient Input
There was 1 patient group submission. The submission was from Muscular Dystrophy Canada, which collected input through surveys and semistructured virtual interviews, for a total sample of 194 patients.
Patients reported issues with productivity, fatigue, low energy, quality of life, mental health, respiratory health, mobility and strength, independence, relationships and social participation, eyes and vision, speech, and swallowing. Respondents stated that the impact of gMG extended beyond physical symptoms, affecting their quality of life and well-being as well as that of their families.
Outcomes of importance to patients include a decrease in the number of exacerbations, a reduction in medication side effects, maintenance of independence, and a reduction in the number of serious hospital admissions.
With respect to their experience with currently available therapies, a key theme that emerged was patients’ negative experiences with steroids. Patients also reported that conventional treatments require a long time to take effect, and that treatment pathways can involve a lot of trial and error.
Clinician Input
Input From Clinical Experts Consulted for This Review
The clinical expert consulted for this review noted numerous needs that are not being met by currently available therapies: 10% of patients are refractory to all currently available treatments; immunosuppressants have delayed effects; and harms are associated with conventional therapies, notably corticosteroids. The clinical expert noted that refractory patients are most likely to require hospitalization or costly and more involved rescue therapies, such as IVIg or PLEX, on a more chronic basis.
The clinical expert views rozanolixizumab as add-on therapy to be used in a manner similar to IVIg and efgartigimod, specifically in patients who have tried glucocorticoids and/or a steroid-sparing drug and had an inadequate response, or in whom steroids could not be tapered or side effects were intolerable. The clinical expert also noted the potential for rozanolixizumab to act as bridging therapy while a patient waits for the typically delayed effects of immunosuppressants (i.e., several months or more after initiation).
The clinical expert believed that the patients most likely to respond to rozanolixizumab would be the type included in the pivotal trial, namely those who are MuSK antibody-positive or AChR antibody-positive and who continue to be symptomatic (based on Myasthenia Gravis Activities of Daily Living [MG-ADL] score) despite trials of first-line therapy.
According to the clinical expert, response to treatment would be assessed, including a patient’s subjective response (symptoms) and the treating clinician’s observation of functional capacity. Treatment would be discontinued for side effects or lack of response. The clinical expert would suggest a trial of 3 months to 6 months before discontinuing for lack of efficacy. Treatment discontinuation may also be considered if the patient determines that their treatment goals have been achieved based on the response.
Clinician Group Input
Input was received from 1 clinician group: the Neuromuscular Disease Network for Canada. This response included responses from 5 clinicians.
The clinician group was generally in agreement with the views of the clinical expert consulted with respect to unmet need and patients most likely to respond to treatment. The group also agreed that key outcomes are MG-ADL and Quantitative Myasthenia Gravis (QMG) scores.
The clinicians did not indicate whether they had experience using rozanolixizumab; however, they did note that it is likely to replace standard immunoglobulin therapies.
Drug Program Input
Input was obtained from the drug programs that participate in the reimbursement review process. The key factors identified that could potentially affect the implementation of a CDA-AMC recommendation for rozanolixizumab were:
relevant comparators
considerations for initiation of therapy
considerations for prescribing of therapy
system and economic issues.
The clinical expert provided details on questions from the drug plans. Please refer to Table 4 for more details.
Clinical Evidence
Systematic Review
Description of Studies
MycarinG was a phase III, sponsor-funded, double-blind, randomized, controlled study. Eligible patients were adults aged 18 years and older with AChR antibody-positive or MuSK antibody-positive gMG (i.e., MGFA disease class II to IVa), an MG-ADL score greater than or equal to 3 (with score of at least 3 for nonocular symptoms), and a QMG score of greater than or equal to 11; patients also had to be under consideration by the investigator for additional treatment, such as IVIg or PLEX. The study began enrolling patients in June 2019 and concluded in October 2021, with a final data cut-off date of September 17, 2021. A total of 200 patients were enrolled and randomized in a 1-to-1-to-1 ratio to receive 6 weekly SC infusions of rozanolixizumab 10 mg/kg, rozanolixizumab 7 mg/kg, or matching placebo. The recommended dose under review by Health Canada is 7 mg/kg; this dose is the focus of this review. The study spanned 81 sites across 17 countries, with 4 sites in Canada. The total duration of study participation for all patients was up to approximately 18 weeks, including a screening period of up to 4 weeks, a 6-week treatment period, and an 8-week observation period. Patients who completed the 6-week treatment period and 8-week observation period had the opportunity to roll over into the MG0004 trial, an open-label extension (OLE) study where the long-term safety, tolerability, and efficacy of rozanolixizumab was measured in patients with gMG over 52 weeks of weekly chronic treatment. The MG0004 trial was terminated in 2021 and replaced by the MG0007 trial, an ongoing, OLE study consisting of 6-week treatment cycles based on MG worsening. Patients could roll over from the MycarinG trial or the MG0004 trial directly into the MG0007 trial.
Patients in the pivotal study were aged 52 years (standard deviation [SD] = 16 years) on average, and the majority (61%) were female. Most patients were MGFA class IIa or IIb (39%) or class IIIa or IIIb (57%) at baseline. The majority of patients (83%) were AChR antibody-positive; 9% were MuSK antibody-positive.
Efficacy Results
The outcomes determined to be of importance — based on clinical expert consultations and input received from patient and clinician groups and public drug plans — are discussed herein. Additional outcome data are available in Appendix 1 (Table 36).
MG-ADL Score
The primary end point was change from baseline to day 43 in MG-ADL score (range, 0 to 24, with higher scores indicating more severe symptoms). From baseline mean MG-ADL scores of 8.4 (SD = 3.8) in the rozanolixizumab group and 8.4 (SD = 3.4) in the placebo group, the least squares (LS) mean changes from baseline were –3.22 (standard error [SE] = 0.480) and –0.65 (SE = 0.363). The LS mean difference in change from baseline was –2.586 (95% confidence interval [CI], –4.091 to –1.249; P < 0.001), favouring rozanolixizumab. Results from the sensitivity analysis were consistent with those from the main analysis. Overall, compared with placebo, treatment with rozanolixizumab resulted in consistently greater decreases from baseline in MG-ADL score at day 43 across all subgroups, except for the subgroups with low numbers of patients. Forty-five patients (68.2%) in the rozanolixizumab group had an MG-ADL response with at least a 2-point improvement at day 43 versus 19 patients (28.4%) in the placebo group, with a between-group difference of 39.8% (95% CI, 24.2% to 55.4%).
The sponsor also reported data from a post hoc subgroup analysis for the ██ patients in the rozanolixizumab group and ██ patients in the placebo group who had 2 or more prior MG-specific therapies. From a mean (SD) baseline score of ███ █████ in the rozanolixizumab group and ███ █████ in the placebo group, the LS mean (SE) changes from baseline to day 43 in MG-ADL scores were ██████ ███████ for rozanolixizumab and ██████ ███████ for placebo, with an LS mean difference between groups of ██████ █████████ ██████ ██ ███████ In this subgroup, the numbers of responders with at least a 2-point improvement in MG-ADL at day 43 were ██ █████% and ███████ in the rozanolixizumab and placebo groups, respectively.
QMG Score
The QMG score can range from 0 to 39, with higher scores indicating more severe impairment. From mean baseline scores of 15.4 (SD = 3.7) in the rozanolixizumab group and 15.8 (SD = 3.5) in the placebo group, the LS mean changes from baselines were –5.598 (SE = 0.679) in the rozanolixizumab group and –1.915 (SE = 0.685) in the placebo group. The LS mean between-group difference in change from baseline was –3.483 (95% CI, 5.614 to –1.584; P < 0.001) favouring rozanolixizumab. Results for various sensitivity analyses were consistent with the overall analysis of change from baseline to day 43 in QMG score.
The sponsor also reported subgroup analyses of QMG scores by baseline antibody status for the 59 patients in the rozanolixizumab group and the 51 patients in the placebo group who were AChR antibody-positive as well as for the 4 patients in the rozanolixizumab group and 7 patients in the placebo group who were MuSK antibody-positive. In the AChR antibody-positive subgroup, the LS mean changes from baseline to day 43 were –4.660 (SE = 1.605) in the rozanolixizumab group and –1.189 (SE = 1.575), in the placebo group for an LS mean difference between groups of –3.471 (97.5% CI, –5.433 to –1.510). In the MuSK antibody-positive subgroup, the LS mean changes from baseline to day 43 were –10.276 (SE = 3.490) in the rozanolixizumab group and –2.662 (SE = 2.710) in the placebo group, for an LS mean difference between groups of –7.614 (97.5% CI, –16.291 to 1.062).
The sponsor also reported data from a post hoc subgroup analysis of the ██ patients in the rozanolixizumab group and ██ patients in the placebo group who had 2 or more prior MG-specific therapies. From a mean (SD) baseline score of ████ █████ in the rozanolixizumab group and ████ █████ in the placebo group, the mean (SD) change from baseline to day 43 in QMG scores with rozanolixizumab was ██████ ███████ and for placebo ██████ ███████ for an LS mean difference between groups of ██████ ██████ ███ ██████ ██ ████████.
MG-QoL15r Score
Scores for the revised 15-Item Myasthenia Gravis Quality of Life (MG-QoL15r) can range from 0 to 30, with higher scores indicating a more severe impact of disease on HRQoL. From mean baselines of 15.7 (SD = 7.7) in the rozanolixizumab group and 15.0 (SD = 6.4) in the placebo group, the LS mean changes from baseline were –4.0 (SE = 6.1) in the rozanolixizumab group compared to –1.3 (SE = 4.3) in the placebo group. The LS mean between-group difference in change from baseline was –2.245 (95% CI, –4.096 to –0.394) favouring rozanolixizumab.
Myasthenia Gravis Composite Score
The Myasthenia Gravis Composite (MGC) score can range from 0 to 50, with higher scores indicating more severe impairment. The LS mean changes from baseline were –5.23 (SE = 0.828) with rozanolixizumab and –1.47 (SE = 0.722) with placebo. The LS mean between-group difference in change from baseline was –3.901 (95% CI, –6.634 to –1.245; P < 0.001) favouring rozanolixizumab. Results of the sensitivity analyses were consistent with those of the overall analysis.
Harms Results
Adverse Events
Overall, the numbers of patients who experienced adverse events (AEs) were 52 patients (81.3%) in the rozanolixizumab group and 45 patients (67.2%) in the placebo group. The most common AEs (i.e., experienced by 10% or more of patients in either group) for rozanolixizumab versus placebo, respectively, were diarrhea (25.0% versus 13.4%), pyrexia (12.5% versus 1.5%), and headache (45.3% versus 19.4%).
Serious AEs
A comparable number of patients in the rozanolixizumab (5 patients [7.8%]) and placebo groups (6 patients [9.0%]) reported serious adverse events (SAEs). The only SAE reported in more than 1 study patient per treatment group was MG crisis, which occurred in 0 patients in the rozanolixizumab group and in 2 patients (3.0%) in the placebo group.
Withdrawals Due to AEs
The incidence of AEs leading to permanent discontinuation of the study drug was similar in both groups, with 2 patient withdrawals (3.1%) in the group receiving rozanolixizumab 7 mg/kg group and 2 patient withdrawals (3.0%) in the group receiving placebo.
Mortality
There were no deaths in the study.
Notable Harms
Infection was identified as a notable harm for this review. Infections or infestations occurred in 10 patients (15.6%) in the rozanolixizumab group and in 13 patients (19.4%) in the placebo group.
Critical Appraisal
The MycarinG trial was relatively well conducted, with adequate allocation concealment and steps taken to maintain blinding. With the exception of MG-ADL responses and MG-QoL15r outcomes, all efficacy outcomes were controlled for multiplicity, reducing the risk of type I error. Although there were some imbalances in baseline characteristics between groups, the clinical expert did not believe these were of clinical relevance.
With respect to external validity, the clinical expert believed that the population of patients in the MycarinG trial was generalizable to the population of patients who would be expected to use the drug in Canada. Although the sponsor is seeking reimbursement for patients whose symptoms persist despite treatment with AChEIs, corticosteroids, and/or NSISTs, the pivotal study did not restrict enrolment based on prior treatment. However, most patients (96%) had received at least 1 prior gMG-specific therapy before the trial. The sponsor provided data from a post hoc subgroup analysis of patients who had received 2 prior therapies; however, limited conclusions can be drawn from these data because of the inherent limitations in post hoc analyses.
GRADE Summary of Findings and Certainty of the Evidence
For the pivotal studies and randomized controlled trials (RCTs) identified in the sponsor’s systematic review, Grading of Recommendations Assessment, Development and Evaluation (GRADE) was used to assess the certainty of the evidence for the outcomes considered most relevant to inform CDA-AMC expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group.22,23
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (i.e., internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, or publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and its location relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds identified in the literature and supported by the clinical expert for the change from baseline to day 43 in MG-ADL and QMG scores; the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for MG-ADL responders; and the presence or absence of any (non-null) effect for the change from baseline to day 43 in MG-QoL15r scores and for notable harms (i.e., infections and infestations).
Table 2: Summary of Findings for Rozanolixizumab vs. Placebo for Patients With gMG
Outcome and follow-up | Patients (studies), N | Relative effect (95% CI) | Absolute effects (95% CI) | Certainty | What happens | ||
|---|---|---|---|---|---|---|---|
Placebo | Rozanolixizumab | Difference | |||||
MG-ADL | |||||||
Change from baseline in MG-ADL (scale from 0 points to 24 points; higher scores indicate more severe symptoms) Follow-up: day 43 | 127 (1 RCT) | NA | –0.65 | –3.22 (0.480) | –2.586 (–4.091 to | Moderatea | Rozanolixizumab likely results in a clinically important improvement in MG-ADL scores compared to placebo. |
Patients achieving response, n (%) (response defined as at least a 2-point improvement in MG-ADL) Follow-up: day 43 | 133 (1 RCT) | OR = 5.765 (2.100 to 14.882) | 28 per 100 | 68 per 100 | 39.8 more per 100 (24.2 per 100 to 55.4 per 100 more) | Moderateb | Rozanolixizumab likely results in a clinically important increase in the number of MG-ADL responders compared to placebo. |
MG-QoL – 15r | |||||||
LS mean change from baseline to day 43 in MG-QoL15r score (from 0 to 30, with higher scores indicating more severe impact of disease on HRQoL) | 133 (1 RCT) | NA | –2.1 | –4.4 (0.9) | –2.245 (–4.365 to | Moderatec | Rozanolixizumab likely results in an improvement in MG-QoL15r scores compared to placebo. The clinical significance of this improvement is not known. |
QMG | |||||||
Mean change from baseline to day 43 in QMG score (from 0 to 39, with higher scores indicating more severe impairment) | 127 (1 RCT) | NA | –0.89 | –4.22 (0.574) | –3.483 (–5.614 to | Moderated | Rozanolixizumab likely results in a clinically important improvement in QMG scores compared to placebo. |
Harms | |||||||
Infections and infestations Follow-up: to 8 weeks after the final dose | 133 (1 RCT) | 19 per 100 | 16 per 100 (NR) | 4 fewer per 100 (17 fewer per 100 to 9 more per 100) | Lowe | Rozanolixizumab may result in little to no difference in the risk of infection compared to placebo. | |
CI = confidence interval; gMG = generalized myasthenia gravis; HRQoL = health-related quality of life; LS = least squares; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MID = minimally important difference; NA = not applicable; NR = not reported; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; RCT = randomized clinical trial; vs. = versus.
Notes: Study limitations (which refer to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, and publication bias were considered when assessing the certainty of the evidence. All serious concerns in these domains that led to the rating down of the level of certainty are documented in the table footnotes.
Results for MG-ADL response and change from baseline in MG-QoL15r were not adjusted for multiplicity and should be considered as supportive evidence.
aRated down 1 level for serious imprecision. The point estimate suggests a clinically important benefit; however, the upper bound of the 95% CI crossed the MID found in the literature. The literature-based MID was estimated for within-group effects; input from the clinical expert consulted by the review team indicated that a between-group difference smaller than 2 points was not likely to be clinically important.
bRated down 1 level for serious imprecision. There is a small sample size and the number of events raising concern for prognostic imbalance and the potential that the effect may be overestimated.
cRated down 1 level for serious imprecision. The null was used as the threshold, and the point estimate suggests benefit; however, the upper bound of the 95% CI includes a value that most reasonable individuals would agree is unimportant.
dRated down 1 level for serious imprecision. The point estimate suggests a clinically important benefit; however, the upper bound of the 95% CI crossed the MID found in the literature.
eRated down 2 levels for very serious imprecision. The points estimate suggests little to no difference, but the 95% CI includes potential for both benefit and harm.
Source: Sponsor’s Summary of Clinical Evidence24 and response to a request for information.25
The selection of outcomes for GRADE assessment was based on the sponsor’s Summary of Clinical Evidence,24 consultation with clinical experts, and input received from patient and clinician groups and public drug plans. The following list of outcomes was finalized in consultation with expert committee members:
MG-ADL score (change from baseline to day 43; responders [i.e., patients with at least a 2-point improvement from baseline in MG-ADL])
QMG score (change from baseline to day 43)
MG-QoL15r score (change from baseline to day 43)
notable harms (infections and infestations).
Long-Term Extension Studies
Description of Studies
Results from 2 OLEs — the MG0004 trial (NCT04124965; data cut-off date of September 1, 2021) and the MG0007 trial (NCT04650854; data cut-off date of July 8, 2022) — were reviewed. The MG0007 trial had been ongoing for approximately 1.5 years at the date of the data cut-off for interim analysis. Results for the 7 mg/kg group only are summarized to align with the reimbursement request.
MG0004 Trial
MG0004 was a phase III, multicentre, randomized, OLE study of the MycarinG (MG0003) trial to investigate the long-term safety, tolerability, and efficacy of rozanolixizumab (using a weekly dosing regimen for 52 weeks) in adult patients with gMG who were experiencing moderate to severe symptoms and under consideration for IVIg or PLEX therapy, indicating they were in need of additional therapeutic intervention. Patients were randomized to 2 different treatment arms in a 1-to-1 ratio to receive SC rozanolixizumab at either 7 mg/kg or 10 mg/kg. The primary safety end points were the occurrence of adverse events leading to the permanent withdrawal of study medication. Other safety end points included the occurrence of AEs requiring special monitoring (i.e., potential Hy’s law, defined as aspartate aminotransferase or alanine aminotransferase > 3 times the upper limit of normal [ULN], total bilirubin > 2 times the ULN, and alkaline phosphatase < 2 times the ULN, with no other explanation for the biochemical abnormality), vital signs, electrocardiogram assessments, and clinical laboratory findings. Patients who experienced disease worsening (e.g., an increase of 2 points on the MG-ADL or 3 points on the QMG between 2 consecutive visits) were considered for rescue therapy. If they received IVIg or PLEX during the study, treatment with rozanolixizumab was discontinued or paused for a minimum of 2 weeks, but patients continued with visits, as per the schedule of assessments; after that, they continued to receive rozanolixizumab at the discretion of the investigator. Following the temporary discontinuation of study medication, patients restarted at the same dose of rozanolixizumab as previously. Patients at a dose level of 7 mg/kg rozanolixizumab could restart at 10 mg/kg rozanolixizumab at the discretion of the investigator.
Of the 71 patients enrolled, 35 patients were randomized to the rozanolixizumab 7 mg/kg group. The mean age was 50.6 years (SD = 14.2 years). More than half of all patients were female (54.3%). Most patients (29 patients [82.9%]) permanently discontinued the study during the COVID-19 pandemic. One patient (2.9%) permanently discontinued the study due to TEAEs before the COVID-19 pandemic, and 2 patients (5.7%) did so for this reason during the pandemic. Most patients permanently discontinued the study to transition to the MG0007 study (i.e., 25 patients [71.4%] in the rozanolixizumab 7 mg/kg group).
The mean duration of the study medication by first dose received was 22.93 weeks. There was no treatment nonadherence, incorrect treatment, or incorrect dosing. Beyond 18 weeks, the numbers of patients steadily decreased; this decrease was mainly due to the 53 patients (74.6%) who discontinued the study to transition to the MG0007 trial.
MG0007 Trial
The MycarinG (MG0007) trial was a 2-arm, phase III, randomized, OLE study that replaced the MG0004 trial to evaluate the long-term safety, tolerability, and efficacy of repeated 6-week treatment cycles of rozanolixizumab based on MG worsening in adult patients with gMG. Worsening of disease was defined as worsening of gMG symptoms (e.g., an increase of 2 points on the MG-ADL or 3 points on the QMG) between 2 consecutive visits. Patients were randomized to receive an initial, fixed, 6-week treatment cycle of rozanolixizumab at 7 mg/kg or 10 mg/kg once weekly, followed by an observation period that began after the last dose of that treatment cycle. Eligible patients from the MG0004 trial who completed at least 6 scheduled visits in the treatment period could move directly into the observation period in the MG0007 trial. In the case of worsening MG symptoms, patients underwent another 6 weeks of treatment followed by an observation period. Rescue therapy was given as per conventional therapy and at the discretion of the investigator. Patients who continued to experience moderate to severe symptoms despite treatment with rozanolixizumab could be treated IVIg, SCIg, PLEX or plasmapheresis, or IV corticosteroids as rescue therapy (at higher doses than previous oral doses). Patients treated with rescue therapies were withdrawn from the study.
Of the 157 patients enrolled, 79 patients received rozanolixizumab 7 mg/kg. The mean age was 52.7 years (SD = 15.7 years). More than half of patients were female (55.7%). A total of 16 patients (20.3%) treated with rozanolixizumab permanently discontinued the study during the COVID-19 pandemic; the most common reason for study discontinuation was TEAEs (in 8 patients [10.1%]), followed by withdrawal by patient (in 5 patients [6.3%]).
Of the 79 patients who received rozanolixizumab 7 mg/kg in cycle 1, 18 patients (22.8%) had only 1 treatment cycle, and 43 patients (54.4%) continued to receive rozanolixizumab 7 mg/kg in subsequent cycles. Sixteen patients (20.5%) switched to rozanolixizumab 7 mg/kg in subsequent cycles (5 patients switched at cycle 2; 3 patients switched at cycle 4; 2 patients switched at cycle 5; 2 patients switched at cycle 7; 3 patients switched at cycle 2 and switched back at cycle 3; and 1 patient switched at cycle 3 and switched back at cycle 4). Five patients (6.3%) in the rozanolixizumab 7 mg/kg group received rescue medication (4 of these patients received immunoglobulins [1 of whom continued treatment with efgartigimod alfa] and the fifth patient received methylprednisolone sodium succinate); 2 patients (2.5%) required a rescue procedure (i.e., PLEX).
Efficacy Results
Change in MG-ADL
MG0004 trial: Changes from baseline in MG-ADL score showed a stable trend up to week 33; study participant numbers declined steadily over time. The maximum mean reduction from baseline to week 33 was 3.1 points (week 13, n = 30) for the rozanolixizumab 7 mg/kg group. When assessed by autoantibodies subgroup, a consistent reduction in MG-ADL scores was observed from baseline in patients who were MuSK antibody-positive up to week 25. The greatest reduction (improvement) from baseline was 2.4 points; this was observed at week 5 (n = 5). The smallest reductions (improvements) from baseline were 1.6 points, observed at week 9 (n = 5) and sustained at week 13 (n = 5), and 1.3 points at week 21 (n = 3). For patients who were AChR antibody-positive, the greatest reduction (improvement) from baseline to week 29 was 3.4 points (n = 25, week 13). Between weeks 29 and 52, there was a consistent change (improvement) in MG-ADL scores from baseline, ranging from –4.2 points (week 37, n = 5) to –2.0 points (week 49, n = 3).
MG0007 trial: The number of participants declined across cycles, from 79 participants at cycle 1 to 11 participants at cycle 6. Within each cycle, the mean change from baseline ranged from –3.0 to –4.3 points depending on the cycle. When assessed by antibodies subgroup, a consistent reduction (improvement) in MG-ADL scores was observed from baseline at day 43, with repeated cyclic treatments for both MuSK antibody-positive (n = 8; cycles 1 to 4) and AChR antibody-positive (n = 62; cycles 1 to 6) patients; however, sample sizes steadily declined within each cycle. For patients who were MuSK antibody-positive, the mean change from baseline ranged from –6.5 points (n = 8; cycle 1) to –3.8 points (n = 3; cycle 3). For patients who were AChR antibody-positive, the mean change from baseline ranged from –4.0 points (n = 6; cycle 6) to –2.8 points (n = 41; cycle 2).
Change in QMG
MG0004 trial: Changes from baseline showed a stable trend over time to week 52; study participant numbers steadily declined over time. The maximum mean reductions from baseline up to week 29 were 5.4 points (week 29; n = 11) for the AChR antibody-positive subgroup and 6.0 points for the MuSK antibody-positive subgroup (week 25; n = 3). The sample sizes in both groups steadily declined over time (n ≤ 10).
MG0007 trial: The sample size declined from 79 patients at cycle 1 to 11 patients at cycle 6. The mean change from baseline ranged from –4.1 points to –6.4 points across cycles. A consistent improvement in QMG scores was observed with repeated cyclic treatment from baseline at day 43 when assessed by subgroups: MuSK antibody-positive (from cycles 1 to 4) and AChR antibody-positive (from cycles 1 to 5). However, sample sizes declined over time with less than or equal to 10 patients in any subgroup.
Change in MGC
MG0004 trial: Changes from baseline showed a consistent trend to week 52; study participant numbers declined steadily over time. A consistent change from baseline up to weeks 25 and 29 was observed when assessed by MuSK and AChR antibodies, respectively. The maximum mean reductions were 7.0 points (week 25; n = 15) from baseline to week 29 for the AChR antibody-positive subgroup (week 25; n = 15) and 3.6 points (week 5; n = 5) from baseline up to week 25 for the MuSK antibody-positive subgroup. The sample sizes declined over time across the subgroups.
MG0007 trial: The sample size declined over time, from 79 patients at cycle 1 to 11 patients at cycle 4. The mean change from baseline ranged from –6.1 points to –9.6 points across cycles. A consistent improvement from baseline in MGC scores was observed at day 43, with repeated cyclic treatment when assessed by antibody subgroups.
Change in MG-QoL15r
MG0004 trial: The mean MG-QoL15r score at baseline was 14.4 points. An improvement in HRQoL was observed. The maximum mean reduction from baseline to week 33 was 5.1 points (week 21, n = 20).
MG0007 trial: Quality of life for patients with MG was an exploratory outcome. The sample size declined over time, from 79 patients at cycle 1 to 11 patients at cycle 4. The mean change from baseline ranged from –2.2 points to –6.1 points across cycles (Table 23).
Harms Results
Adverse Events
MG0004 trial: Seventy-six percent of the patients in the rozanolixizumab 7 mg/kg group experienced any AE. The most common AEs (20% of patients or more) were nervous system disorders (36.0%), gastrointestinal disorders (26.0%), infections and infestations (26.0%), investigations (22.0%), and musculoskeletal and connective tissue disorders (20.0%).
MG0007 trial: Sixty-eight patients (69.4%) in the rozanolixizumab 7 mg/kg group experienced any AE. Nervous system disorders (36.7%), infections and infestations (34.7%), gastrointestinal issues (24.5%), and general site-administration issues (27.6%) were the most reported.
Serious AEs
MG0004 trial: SAEs were reported in 7 patients (14.0%). The only SAE occurring in more than 1 patient was nervous system disorders (n = 3, 6.0%).
MG0007 trial: SAEs were reported in 9 patients (9.2%). The SAEs occurring in more than 1 patient were nervous system disorders (n = 3, 3.1%) and infections and infestations (n = 2; 2.0%).
Withdrawals due to AEs
MG0004 trial: A total of 4 patients (8.0%) experienced AEs that led them to discontinue from the study. Three of these patients (75.0%) experienced MG, while 1 patient (25.0%) experienced congestive cardiac failure. In patients who temporarily discontinued rozanolixizumab (n = 12; 24.0%), the main reasons were decreased blood immunoglobulin G (IgG) and hypogammaglobulinemia.
MG0007 trial: A total of 6 patients (6.1%) permanently discontinued the study. Two patients (33.3%) had AEs with preferred term MG, while 1 patient each reported AEs of adrenal disorder, pneumonia, tendon disorder, tenosynovitis, retroperitoneal neoplasm, thymoma, and subacute cutaneous lupus erythematosus. In patients who temporarily discontinued rozanolixizumab, the main reasons were decreased blood IgG, hypogammaglobulinemia, and COVID-19.
Mortality
MG0004 trial: There were no AEs leading to death in this study.
MG0007 trial: One death was reported due to a fatal AE (pneumonia).
Critical Appraisal
The patients who were enrolled in the MycarinG pivotal trial were the ones entering the OLEs (i.e., studies MG0004 and MG0007). The MG0004 and MG0007 studies were limited by their noncomparative, open-label study designs. A lack of a control group precludes causal statements about benefit and harm versus any comparator. The open-label, nonblinded nature of the study may increase the risk of bias in determining the magnitude of the subjective safety outcomes and all efficacy end points because these were subjective (e.g., MG-ADL, QMG, MG-QoL15r, and MGC scores); the lack of blinding may influence patients’ expectations of the treatment. The direction and magnitude of these potential biases remain unclear. Concomitant treatments were intended to remain stable within treatment cycles but could be adjusted between cycles. These additional treatments could confound the relationship between rozanolixizumab and the efficacy and harm outcomes; however, the degree of impact on the results cannot be predicted. Efficacy results were assessed by MG-specific antibody subgroups; however, these results should be interpreted with caution due to the small sample sizes (especially in the subgroup of patients who were MuSK antibody-positive). Baseline MG-ADL, QMG, and MGC scores indicated higher disease severity in patients entering Study MG0007, potentially suggesting a selection bias. Patients in Study MG0007 were allowed to switch between the 7 mg/kg and 10 mg/kg groups, based on investigator’s discretion, before the start of each subsequent cycle of treatment. Therefore, it is difficult to differentiate the effect of the 7 mg/kg dose (which is the focus of the reimbursement request) from that of the 10 mg/kg dose on the efficacy outcomes. There is a high risk of attrition bias, given that the number of patients contributing to the analyses declined steadily over time.
Indirect Comparisons
The submission included a network meta-analysis (NMA) and matching-adjusted indirect comparison (MAIC). The comparator treatments included in the NMAs were zilucoplan, efgartigimod alfa, eculizumab, IVIg, PLEX, rituximab, and ravulizumab; of these, results from the comparisons with efgartigimod, eculizumab, IVIg, PLEX, and rituximab were included in the review. The comparator treatments included in the MAIC submission were efgartigimod and IVIg or PLEX.
Description of Studies
The study selection methods were the same for the NMA and the MAIC. Briefly, a clinical systematic literature review (SLR) based on studies identified from database searches from inception to May 1, 2023, was performed to inform both the NMA and MAIC. Results from the SLR were then filtered by distinct populations, interventions, comparators, and outcomes (PICOs) for the NMA or MAIC as part of the feasibility assessment.
NMA Design
Homogeneity in the NMA network was assessed by visual inspection of the distribution of baseline characteristics for the trials comprising the network, as well as the time point at which study outcomes were reported. Plot digitization at 12 weeks was carried out to obtain data points from published figures. The NMAs were performed using a Bayesian approach with noninformative priors, and fixed-effects models were used. Change from baseline outcomes were assessed at the 12-week time point (± 2 weeks) using only phase III studies in the primary analysis, and the results were presented with estimates for the treatment effects of each intervention relative to placebo as the reference treatment. Relative treatment effects (MG-ADL responders, defined as patients with an improvement of 3 points or more in score) were presented as odds ratios (ORs); continuous treatment effects (i.e., changes in baseline MG-ADL scores) were reported as mean differences. Analysis was conducted in the overall and refractory populations (which were defined according to the RAISE trial). The sensitivity analyses were conducted assessing the inclusion of different time points of reporting outcomes as well as differences in study design (i.e., phase II versus phase III studies).
MAIC Design
Before carrying out a feasibility assessment, 2 clinical experts ranked the relative importance of all baseline characteristics based on their impacts on outcomes. The feasibility assessment consisted of comparing the relevant trials for each comparison in terms of their baseline characteristics and inclusion and exclusion criteria. In cases of differences in these criteria, a subset of patients from the MycarinG trial was used to match the comparator trial. If feasibility was confirmed, the 2 studies were matched using a propensity score weighting method. A comparison of all potential analysis scenarios was presented to knowledge leaders, and the base case was selected according to specific criteria. The comparisons of rozanolixizumab versus efgartigimod (i.e., the ADAPT trial) were modelled using an anchored MAIC, and the results for rozanolixizumab versus IVIg (i.e., the Barth et al. [2011] study) were modelled using an unanchored MAIC. Continuous outcomes (i.e., changes from baseline in MG-ADL, MGC, MG-QoL, and QMG scores) were modelled using linear regression, with results presented as mean differences. Binary outcomes (i.e., 2-point or 3-point improvements in MG-ADL or QMG) were modelled using logistic regression, with results presented as ORs.
Efficacy Results
Network Meta-Analysis
Heterogeneity was observed throughout the NMA network in disease severity, treatment history (where reported), trial eligibility criteria, placebo response, the definition of MG-ADL responders, the timing of end point evaluation, study designs, and baseline characteristics. The majority of patients enrolled in the trials were AChR antibody-positive, female, and had MGFA class II to IV gMG. The duration of disease ranged from 6.9 years to 10.3 years. MuSK antibody status was reported in 2 trials (i.e., in the MycarinG trial [12% of patients] and the ADAPT trial [4% of patients]). Study durations ranged from 12 weeks to 48 weeks. The study network for the primary analysis showed that all included studies compared treatments to placebo, and each node in the network consisted of a single study.
The NMA primary analysis results for rozanolixizumab 7 mg/kg indicated ██████████ ██ ██████ ███ █████████ ███ ███████████ ████ ██ █████████ ██ ███ ████ ████ ███ █████ ███ ███ ████████. Rozanolixizumab 7 mg/kg was ████████ ████ ███████ ███ ██████ ██████████.
Matching-Adjusted Indirect Comparison
There were some differences identified between the MycarinG trial (rozanolixizumab) and the ADAPT trial (efgartigimod). Most notably, there were differences in the minimum MG-ADL scores required for enrolment. In addition, the ADAPT trial required patients to be on stable doses of gMG therapy, whereas the MycarinG trial required patients to be under consideration for additional therapy. There were also differences noted between the inclusion criteria for the MycarinG trial and the Barth et al. (2011) trial (IVIg), most notably that the Barth et al. (2011) trial was an active-controlled trial that did not require a specific MGFA class diagnosis or MG-ADL baseline score for enrolment, whereas the MycarinG trial was placebo-controlled and required weight and MG-ADL thresholds as well as AChR antibody-positive or MuSK antibody-positive status.
Results of the primary analysis for rozanolixizumab versus efgartigimod indicated that at 6 weeks, the results were ███████ ████ ██ █████████ ███ ███ █████████ ████ ███ █████ for the outcomes of change from baseline in MG-ADL, MGC, MG-QoL, or QMG scores. The ████ ███████ ████ ████ █████████ for the outcomes of 2-point or 3-point improvements in MG-ADL at 4 weeks or 3-point improvements in QMG at 4 weeks.
Results from the primary analysis of rozanolixizumab versus IVIg indicated that at 2 weeks and 4 weeks, ███ ████ ██████████ ███████ ███████████████ ███ ████ ███ ███ ██████ ████ ████████ ██ ███ ██████ ████████ ███████████████ ███████ ███ ███████ ███ ███ ██████████ ████ ███████ ████ ██ █████████ ███ ███ █████████ ████ ███ ██████.
Harms Results
Harms outcomes were not analyzed as part of the indirect comparisons.
Critical Appraisal
Some limitations of the SLR include the fact that the search was run only until 2023; therefore, the review may miss more recent publications of comparators. In addition, the quality assessment was carried out at the level of the trial, which might not capture the fact that risks of some domains of bias (e.g., attrition bias) can vary by outcome. According to the clinical expert, the NMA included comparators that are relevant to the Canadian context; the outcome was of interest to clinicians. However, data from some relevant comparators, such as IVIg and rituximab, were not available in the primary analysis of the NMA, and additional limitations in the sensitivity analyses do not allow conclusions to be drawn regarding these comparators. Likewise, results from all comparators were not available in the submission for the MAIC.
There are important sources of heterogeneity in the NMA network that have clinical relevance and affect the certainty of the results. While all trials enrolled patients with gMG (class II to IV), there were differences between trials in the refractory statuses of the enrolled patients that were not accounted for in the analyses. For example, the trials for eculizumab and rituximab generally enrolled refractory and newly diagnosed patients, respectively; the trial for zilucoplan included refractory patients; and the trial for rozanolixizumab required patients to be under consideration for additional therapy. In addition, MuSK antibody status was reported in only 2 of the 6 included trials, and the trials in the network used MG-ADL thresholds ranging from 3 points to 6 points. Sensitivity analyses were conducted, but these do not address the heterogeneity concerns. Taken together, these could represent clinically meaningful differences in patient disease status and affect confidence that the transitivity assumption underpinning the NMA was met.
With regard to the MAICs, while the clinical expert consulted for the review noted that the list of known prognostic- and/or effect-modifying variables used for weighting in both MAICs was comprehensive, not all baseline characteristics were reported before and after weighting; therefore, it is not known whether there were other potential sources of heterogeneity in the trial populations remaining after weighting. Weighting controlled for the differences in the reported baseline characteristics for the anchored MAIC comparing rozanolixizumab to efgartigimod. However, the estimated sample size (ESS) was considerably smaller than the sample sizes of the 2 trials prematching. This suggests that a small proportion of the patient population may be driving the results; therefore, the findings could be unstable. This also suggests that there remains uncertainty in the results for the comparison of rozanolixizumab versus efgartigimod. The comparison of rozanolixizumab versus IVIg was carried out using an unanchored MAIC that relied on the assumption that all possible prognostic factors and treatment-effect modifiers are controlled for — an assumption that is largely considered impossible to meet.26 The scenario used in the current MAIC did not include all baseline characteristics in the weighting process, resulting in a high risk of residual confounding. Therefore, confidence in the results is highly uncertain. Furthermore, there are important study differences that were not controlled for by the weighting process, such as the lack of placebo comparator in the Barth et al. (2011) trial, the timing of the primary end point, and the proportion of MuSK patients. Taken as a whole, conclusions about the efficacy of rozanolixizumab versus IVIg are challenging to draw.
The indirect evidence, as a whole, is also subject to some limitations that affect generalizability. First, the study population of the MycarinG trial included patients who were AChR antibody-positive or MuSK antibody-positive. To improve similarity to the efgartigimod trial population, only the patients who were MuSK antibody-negative, AChR antibody-positive from the MycarinG trial were included in the unanchored MAIC for rozanolixizumab versus efgartigimod (i.e., a subset of the full trial population). This adversely affects the generalizability of those results to the population of patients with gMG who are MuSK antibody-positive. Furthermore, the results of the MAIC were assessed as early as 2 weeks to 6 weeks; the clinical expert noted that this is early to assess treatment response and might lead to not capturing maximal treatment response. Lastly, information about long-term comparative efficacy and harms is unavailable.
Studies Addressing Gaps in the Evidence From the Systematic Review
The sponsor did not submit any studies addressing gaps.
Conclusions
One pivotal, sponsor-funded, multinational (81 sites, including 4 in Canada), phase III, double-blind RCT was included in this report. The MycarinG trial randomized 200 patients with AChR antibody-positive or MuSK antibody-positive gMG in a 1-to-1-to-1 ratio to 1 of 2 doses of rozanolixizumab (i.e., 7 mg/kg or 10 mg/kg) or to placebo, administered as weekly SC infusions over a 6-week treatment course. The rozanolixizumab 7 mg/kg dose is the dose being sought for Health Canada approval; therefore, this dose is the focus of this report. After 43 days, rozanolixizumab likely resulted in a clinically significant improvement in MG-ADL scores compared to placebo; this was the primary outcome of this study. Treatment with rozanolixizumab also likely resulted in a clinically significant increase in the number of MG-ADL responders and QMG scores after 43 days compared to placebo. Rozanolixizumab also likely improved MG-QoL15r scores after 43 days compared to placebo; however, the clinical significance of this improvement is not known. A post hoc subgroup analysis of patients with 2 or more prior treatments suggested a ███████ █████████ ██ ███████ on MG-ADL and QMG in this group. The number of patients was small, and the analysis was not preplanned; these factors limit the conclusions that can be drawn. Infection was a notable harm identified for this review; however, there was no indication of increased infection risk at the 7 mg/kg dose of rozanolixizumab. Long-term evidence was limited to data from 2 open-label, noncomparative extension studies (i.e., studies MG0004 and MG0007). These studies suggested that the benefits of rozanolixizumab may be maintained for up to 52 weeks. However, definitive conclusions were hindered by the lack of control groups and the risk of bias due to open-label design as well as by substantial attrition. No additional safety concerns were identified in the extension studies.
The submitted NMA was limited by likely intransitivity and imprecision, which introduce uncertainty about the comparative efficacy of rozanolixizumab versus eculizumab or efgartigimod at 12 weeks of follow-up. ██ ████████ ████ █████████ ████ ████████████ ███ ███████████████ ███ ████ ███████ ███████ ██ █ ██ █ █████ ██ ██████████. These findings were uncertain due to the potential for residual confounding, small ESS, and some imprecision. The unanchored MAIC comparing IVIg to rozanolixizumab had important limitations that limited the ability to draw conclusions. Both MAICs are limited in generalizability because the timing of maximal treatment response might not have been captured. Information about long-term comparative efficacy and harms is unavailable, and comparison to some relevant treatments (e.g., rituximab) is lacking.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of rozanolixizumab (Rystiggo) 140 mg/mL solution for SC injection (280 mg in 2 mL in single-dose glass vials) for use in the treatment of gMG in adult patients.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
MG is a rare, chronic, autoimmune, neuromuscular disease in which antibodies against the neuromuscular junction disrupt neuromuscular transmission, resulting in localized or generalized skeletal muscle weakness.1-3 The weakness is due to an antibody-mediated, immunologic attack directed at proteins in the postsynaptic membrane of the neuromuscular junction (i.e., AChRs or receptor-associated proteins).27 MG can be categorized based on disease severity. The MGFA stratifies people with MG into 5 functional classes based on clinical features and symptom severity. The classifications range from class I (ocular weakness only) to class V (intubation, with or without mechanical ventilation [e.g., during myasthenic crisis], except when employed during routine postoperative management). Classes II, III, and IV represent patients with mild, moderate, and severe generalized weakness involving nonocular muscles, respectively.4 Patients experience a variety of symptoms, including fatigue, droopy eyelids, diplopia, neck weakness, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and upper and/or lower limb weakness.3
There are 2 clinical forms of MG: ocular (weakness is limited to the eyelids and extraocular muscles) and generalized (weakness involves a variable combination of ocular, bulbar, limb, and respiratory muscles).4 Patients with gMG who are AChR antibody-positive account for 85% of cases; patients who are MuSK antibody-positive account for 8% of cases; patients with LRP4 antibodies account for 1% of cases; and patients in the remaining 6% of cases are seronegative.5 A gMG diagnosis is typically made by a neurologist based on signs and symptoms in combination with laboratory, electrophysiologic, and imaging tests.5 Because AChR antibodies are present in 80% to 85% of patients with gMG,2,3 clinicians will typically conduct the serologic test for AChR antibodies first, followed by tests for MuSK and LRP4 antibodies if a patient is negative for AChR antibodies.16 Both serologic and electrophysiological testing are available in Canada.
Globally, the incidence of MG varies from 4 cases per million person-years to 30 cases per million person-years, and the prevalence ranges from 150 cases per million to 200 cases per million.6 In Canada, the incidence and prevalence of gMG are estimated at 23 per 1 million person-years and 26.3 per 100,000, respectively.7 The mortality rate of MG has been reported to range from 0.06 per million person-years to 0.89 per million person-years.8 Approximately 80% to 90% of patients with gMG are AChR antibody-positive,28,29 while 5% to 10% are MuSK antibody-positive.20,30,31
The symptoms of gMG occur unpredictably and fluctuate in nature, intensity, and severity on a day-to-day basis throughout a patient’s life, requiring intervention or treatment change; this is known as MG exacerbation.3,9 An MG exacerbation can deteriorate into a myasthenic crisis — a severe life-threatening manifestation in which patients experience sudden respiratory failure requiring emergency intubation or noninvasive ventilation, supportive enteral feeding, and intensive care unit management.12,32,33 Myasthenic crisis occurs in approximately 15% to 20% of patients with MG, usually within the first 2 to 3 years of the disease course.32 Patients with MuSK antibody-positive gMG experience more frequent and severe exacerbations and myasthenic crises compared to patients with AChR antibody-positive gMG.34,35 Unpredictable exacerbations and myasthenic crises, in combination with a variety of symptoms, lead to a chronic disease with significant burden that negatively affects a patient’s quality of life.10,11
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the review team.
There are currently no Canadian guidelines for the treatment of gMG. The MGFA international consensus guidelines for the management of MG12 were updated in 2020 and are now the most recent guidelines.14 According to these guidelines and clinical experts in Canada, the goal of treatment for patients with gMG is to reduce disease symptoms as well as the adverse effects of MG therapy and allow the patient to function and work normally, with good HRQoL.12,13 Other treatment goals include avoiding MG exacerbations and myasthenic crises, minimizing hospitalizations and intensive care unit admissions, and reducing the numbers and doses of therapies (especially corticosteroid use) required for symptom control.13
The MGFA international consensus guidelines for the management of MG recommend thymectomy in young patients (i.e., aged 18 years to 50 years) with gMG early in the disease course to improve clinical outcomes and minimize immunotherapy requirements, hospitalization, and disease exacerbations.14 However, thymectomy is an elective procedure, and not all patients with gMG will be candidates; candidacy is based on age and ability to tolerate limited postoperative respiratory function.14 Conventional therapy for all patients with gMG generally begins with AChEIs; however, AChEIs can worsen the symptoms of MuSK antibody-positive gMG. As a result, it may not be used in all patients with MuSK antibody-positive gMG.12 A maximal response to corticosteroids typically occurs within 2 months to 6 months of initiating treatment, at which time a slow tapering of prednisone should occur to avoid adverse effects associated with long-term steroid use.1 In patients who do not respond to corticosteroids or have significant comorbidities (such that long-term corticosteroid treatment is contraindicated), or in whom doses of corticosteroids cannot be tapered, treatment with NSISTs, such as azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine, tacrolimus, or methotrexate may be initiated, either alone or in combination with corticosteroids.12 It can take several months to years, depending on the NSIST, for the drug to produce a clinically relevant effect and reduce a patient’s gMG symptoms.1 The majority of these conventional treatments for gMG are publicly reimbursed without restrictions across Canada, with some exceptions.
While patients wait for NSIST treatment to take effect, they may experience MG exacerbations and/or myasthenic crises requiring the acute use of IVIg, SCIg, or PLEX.12 If patients continue to experience gMG symptoms, the dose may be increased or the patient may switch to an alternative NSIST. The majority of patients are able to control their disease using conventional therapy; however, 15% to 40% of patients will continue to experience symptoms despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs.15-17
Patients with AChR antibody-positive gMG whose symptoms persist despite conventional therapy would be eligible for treatment with rituximab, chronic IVIg or SCIg, and/or chronic PLEX.12,14 Clinicians consulted for this submission and clinical experts consulted by CDA-AMC for previous reimbursement reviews in the gMG disease area have indicated that accessing public reimbursement for off-label rituximab for patients with AChR antibody-positive gMG can be challenging in some jurisdictions because rituximab is not indicated for gMG.18 These patients would also qualify for treatment with efgartigimod alfa, as per the CDA-AMC recommendation.18,19 Patients who have refractory gMG (defined as inadequate symptom control after an adequate trial of 2 or more ISTs, either in combination or as monotherapy over the past 12 months, or at least 1 IST and PLEX or IVIg at least every 3 months over the past 12 months) would also be eligible for treatment with eculizumab, as per the CDA-AMC recommendation.19 However, because the pCPA negotiations for eculizumab concluded without an agreement, it is not listed on any public drug program formulary; therefore, very few patients are able to access this therapy.
While the conventional therapies used in patients with MuSK antibody-positive gMG are generally the same as those for patients with AChR antibody-positive gMG, there are no targeted therapies currently approved by Health Canada for the treatment of MuSK antibody-positive gMG, and the responsiveness of patients with MuSK antibody-positive gMG to specific therapies differs from that of patients with AChR antibody-positive gMG.12 Patients with MuSK antibody-positive gMG are less responsive to AChEIs and are frequently intolerant to pyridostigmine at conventional doses.20 Patients with MuSK antibodies typically respond well to corticosteroids and NSISTs, but tend to remain dependent on corticosteroids, despite concomitant therapy with NSISTs.20 For patients whose symptoms persist despite treatment with corticosteroids and NSISTs, options include rituximab and PLEX; IVIg is usually less effective.20 The most robust evidence for the use of rituximab comes from a prospective blinded study of 55 patients with MuSK antibody-positive MG in which 58% of patients receiving rituximab achieved minimal manifestation status and required only low-dose NSIST (compared with 16% of patients who did not receive rituximab).36 Other evidence includes prospective and retrospective studies showing positive outcomes for patients with MuSK antibody-positive gMG when treated with rituximab.37-42 Therefore, rituximab is recommended by international consensus guidance for patients with MuSK antibody-positive gMG who have an unsatisfactory response to initial immunotherapy.21 This is in contrast to patients with AChR antibody-positive gMG, for whom rituximab is considered only if they do not respond to or tolerate other immunotherapies.21 However, as stated earlier, access to off-label rituximab is challenging, and there is variable reimbursement across the provinces. Eculizumab and efgartigimod alfa are not approved by Health Canada or reimbursed by public drug programs for the treatment of MuSK antibody-positive gMG.
Drug Under Review
Key characteristics of rozanolixizumab — along with those of other treatments available for adults with gMG — are summarized in Table 3. Rozanolixizumab is a humanized immunoglobulin G4 monoclonal antibody that decreases serum IgG concentration by inhibiting the binding of IgG to the neonatal fragment crystallizable receptor, a receptor that normally protects IgG from intracellular degradation and recycles IgG back to the cell surface. By the same mechanism, rozanolixizumab decreases the concentration of pathogenic IgG autoantibodies associated with gMG.
Rozanolixizumab is supplied as a 280 mg per 2 mL (i.e., 140 mg/mL) single-dose vial. The recommended dosage (based on patients’ body weight; proposed dosage is 7 mg/kg) is administered as a solution for SC infusion using an infusion pump at a rate of 20 mL/hour once weekly for 6 weeks. Only a health care professional should prepare and infuse the drug. The sponsor indicated that there will be an optional patient support program providing patient education in addition to health care professional support for the administration of rozanolixizumab at program infusion clinics or in patients’ homes.
Table 3: Key Characteristics of Rozanolixizumab, Pyridostigmine, IST, IVIg, Rituximab, Eculizumab, and Efgartigimod Alfa for the Treatment of Patients With gMG
Characteristic | Rozanolixizumab | Pyridostigmine | IST (e.g., corticosteroids, steroid-sparing drugs, rituximab) | IVIg | Eculizumab | Efgartigimod alfa |
|---|---|---|---|---|---|---|
Mechanism of action | Monoclonal antibody that decreases serum IgG concentration by inhibiting the binding of IgG to neonatal FcRn | Cholinergic drug that acts primarily by inhibiting cholinesterase | Suppression of production of AChR antibodies | Immunosuppressant that suppresses B-cells and T-cells | Monoclonal antibody that specifically binds to the complement protein C5 with high affinity | Human IgG1 antibody crystallizable fragment engineered for increased affinity to FcRn |
Indicationa | For the treatment of adult patients with gMG who are AChR antibody-positive or MuSK antibody-positive | For symptomatic treatment of MG | NA | NA | Adult patients with gMG who are AChR antibody-positive and refractory to treatment | For the treatment of adult patients with gMG who are AChR antibody-positive |
Route of administration | SC infusion | Oral | Oral or IV | IV | IV | IV |
Recommended dose | 20 mL/hour once weekly for 6 weeks through an infusion pump The weekly recommended dose varies by body weight category:
Subsequent treatment cycles are administered according to clinical evaluation. The frequency of treatment cycles may vary by patient. | 60 mg to | Various | 1 g/kg to | 900 mg weekly for the first 4 weeks, followed by 1,200 mg for the fifth dose 1 week later, then | 10 mg/kg once weekly for 4 weeks. In patients weighing ≥ 120 kg, the recommended dose is 1,200 mg (3 vials) per infusion. Subsequent administration of treatment is based on clinical evaluation. The frequency of treatment cycles may vary by patient. |
Serious adverse effects or safety issues | Infections, headache, diarrhea, nausea, pyrexia | Respiratory (increased bronchial secretions), gastrointestinal (nausea, vomiting, increased peristalsis), musculoskeletal, dermatologic (urticaria, rash), miosis, diaphoresis, weakness, allergic reactions | Infections, infusion reactions | Infusion reactions | Infections, including serious meningococcal infections | Upper respiratory tract infections |
AChR = acetylcholine receptor; FcRn = neonatal fragment crystallizable receptor; gMG = generalized myasthenia gravis, IgG = immunoglobulin G; IST = immunosuppressive therapy; IVIg = IV immunoglobulin; MG = myasthenia gravis, MuSK = muscle-specific kinase; NA = not applicable.
aHealth Canada–approved indication.
Sources: Product monographs for rozanolixizumab (draft),43 pyridostigmine,44 prednisone,45 azathioprine,46 cyclosporine,47 mycophenolate,48 tacrolimus,49 rituximab,50 eculizumab,51 and efgartigimod alfa.52
Rozanolixizumab received Health Canada approval and is indicated for the treatment of adult patients with gMG who are AChR or MuSK antibody-positive. The reimbursement request is for rozanolixizumab as an add-on therapy for the treatment of adult patients with gMG who are either AChR or MuSK antibody positive and for whom symptoms persist despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs. The drug has not been previously reviewed by CDA-AMC.
Perspectives of Patients, Clinicians, and Drug Programs
The full patient and clinician group submissions received are available in the consolidated patient and clinician group input document for this review on the project website.
Patient Group Input
This section was prepared by the review team based on the input provided by patient groups.
A single patient group submission was received from Muscular Dystrophy Canada (MDC). MDC supports people affected by muscular dystrophies and related muscle diseases in Canada. Its mission is to enhance the lives of those affected by neuromuscular disorders by providing support through all stages of disease progression so individuals with the disease have the tools to navigate the challenges. MDC has neuromuscular service support staff in all provinces in Canada who provide front-line support to thousands of people in Canada affected by neuromuscular disorders. These staff members work directly with patients and families to address barriers, share education materials, enhance life skills, embrace inclusion, network, and provide the support needed to improve overall well-being and quality of life for patients and their family members.
The neuromuscular service support staff identified and contacted adults living with MG to participate in a survey and semistructured virtual interviews. The survey was shared with members through e-blasts, personalized invites, and online patient groups in Canada. MDC conducted an MG Canadian Journey Mapping Project that consisted of 1-hour interviews, round table sessions, surveys, and HRQoL measures (i.e., EQ visual analogue scale, EQ-5D, MG-ADL, and MG-QoL). Information was collected from 194 individuals from all provinces in Canada (117 females and 77 males) with a confirmed gMG diagnosis who were aged 22 years to 78 years. Additionally, 20 people in Canada (16 females and 4 males) aged 34 years to 68 years living with gMG provided input on their knowledge of the drug under review and their everyday experiences with MG. A qualitative descriptive approach, employing a technique of constant comparison, was used to produce a thematic analysis.
The analysis identified 7 key issues that were frequently reported by adults living with MG. Participants consistently reported significant impacts on productivity; fatigue, energy levels, quality of sleep; respiratory health; mobility and strength; independence; relationships and social participation; vision, speech, and swallowing. They also conveyed that the impact of MG extended beyond physical symptoms, affecting their mental health, quality of life, and families’ well-being.
Four main themes were observed when participants were asked about their experiences with currently available treatments to manage MG: negative experiences with steroids; experiences with conventional treatments taking a long time to take effect; experience with trial and error; and experience with IVIg.
With regard to improved outcomes, patient respondents identified several aspects of their condition over which they wanted more control. These included decreased intensity of exacerbations, reduced medication side effects, maintenance of independence, and fewer serious hospital admissions. They additionally noted that current medications seemed to decrease the number of exacerbations, but did not have an impact on overall quality of life. Factors such as therapies with low invasiveness, minimal hospital visits, low risk of side effects, convenience, duration and frequency of treatment, and financial impacts were highly valued by both patients and caregivers when evaluating different therapies. Respondents stated that HRQoL was a key priority compared to the convenience of a drug. They also stated a desire for newer treatments to provide relief from constant discomfort, target respiratory weakness and overall fatigue, reduce the financial burden, offer convenient treatment administration, produce a quicker response to the treatment, and refrain from causing other health complications. It was noted that when switching to a different therapy, respondents would consider its potential side effects versus those of their current medications, ease of access to the new treatment, and whether it would be covered by private or provincial insurance.
All respondents had undergone diagnostic blood testing, and many had undergone single-fibre electromyography (EMG) to confirm diagnosis. Significant difficulties in getting diagnosed were reported by 85% of respondents. The early findings of the Canadian MG Journey Mapping Project suggested that it can take 7 years from the time of the first bothersome symptom to diagnosis, with the range up to 23 years. Most respondents found the testing process to be cost-effective but lengthy, with many missed opportunities due to delays in diagnosis, misdiagnosis (i.e., with stroke or Bell palsy), or diagnosing difficulties (e.g., long turnaround times for results, negative test results), resulting in incurred costs. Around 25% of respondents had received their MG diagnosis as a part of a medical event or hospitalization and reported a smooth experience with the testing procedure.
Clinician Input
Input From Clinical Experts Consulted for This Review
All CDA-AMC review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by a clinical specialist with expertise in the diagnosis and management of gMG.
Unmet Needs
The clinical expert consulted on this review noted numerous needs that are not being met by currently available treatments. First, the clinical expert noted that about 10% of patients are refractory to all currently available treatments. Because many of the current immunosuppressants (such as azathioprine and mycophenolate) take many months before their effect (or lack thereof) is known, patients can have a prolonged course of treatment without significant benefit, facing disability for years due to their gMG symptoms. The clinical expert noted that part of the reason for this delayed effect is that traditional therapies do not target the specific disease pathways known to be involved in gMG; rather, these drugs are designed as general immunosuppressants. The clinical expert added that these refractory patients are the ones most likely to require hospitalization or more costly and involved rescue therapies, such as IVIg and PLEX, on a chronic basis.
Additionally, the clinical expert noted that new treatment options are needed due to the harms associated with current options, especially glucocorticoids, which have numerous adverse effects and should be avoided in patients with pre-existing conditions often seen in older adults, such as diabetes, hypertension, osteoporosis, and infection. The clinical expert noted that tolerability issues can extend to younger, otherwise healthy patients as well due to cosmetic or other side effects, such as weight gain.
Finally, the clinical expert noted that better formulations are important because treatments such as IVIg and PLEX are resource-intensive, requiring either a hospital infusion clinic (for IVIg) or specialized centres and the insertion of a central line (for PLEX). In summary, the clinical expert noted that treatments that target disease-specific mechanisms, are associated with minimal side effects, and can be administered at home are lacking for patients with gMG.
Place in Therapy
The clinical expert noted that rozanolixizumab would likely not be used as a first-line treatment; rather, it would likely be an add-on therapy, similar to the way IVIg is used and the way efgartigimod alfa has fit into practice. The clinical expert added that in this situation, patients would have likely tried either glucocorticoids and/or a steroid-sparing drug (such as azathioprine or mycophenolate) and found that the clinical response was inadequate, steroids could not be tapered, or side effects could not be tolerated. The clinical expert noted that in this case, rozanolixizumab would be added, while glucocorticoids and/or a steroid-sparing drug would also be continued. The clinical expert also noted that rozanolixizumab could be used as bridging therapy, given that many of the drugs used have delayed onset (i.e., many months); therefore, rozanolixizumab would provide an effective treatment while patients are waiting for the effects of the others to begin. Patients could perhaps even use rozanolixizumab temporarily for a short course (weeks or months), then taper their use as these immunosuppressants begin to work. The clinical expert noted that, due to its unique mechanism —shared only with efgartigimod alfa — rozanolixizumab can be used in conjunction with and in addition to pyridostigmine, glucocorticoids, and traditional steroid-sparing immunosuppressants.
Although rozanolixizumab could provide another option for patients who are intolerant or have contraindications to other treatments, it is unlikely to be reserved only for those patients, according to the clinical expert. The clinical expert did not view rozanolixizumab as likely to shift the current treatment paradigm, but did note the importance of having a variety of treatment options available because not all patients will tolerate a given drug and because patients will likely have different preferences for modes of administration (such as IV versus SC).
For numerous practical and financial reasons, it would be appropriate and reasonable, according to the clinical expert, to require patients to have attempted other therapies before moving on to rozanolixizumab. The clinical expert noted that the currently available treatment paradigm works well for a large percentage of patients with respect to both tolerability and efficacy; therefore, it is appropriate to begin with these. Given that newer therapies like rozanolixizumab are likely to be more expensive, the clinical expert noted that it would not be feasible to use them in the first line; however, they added that there is a large unmet need among patients who have inadequate response to the current treatment paradigm, cannot continue due to tolerability issues or contraindications (such as diabetes, liver disease, or age), or require bridging therapy.
Patient Population
The clinical expert believed that the patients who are most appropriate for treatment with rozanolixizumab would be similar to those included in the pivotal trial: adults with gMG who have either AChR or MuSK antibodies and significant symptoms (i.e., the trial used an MG-ADL score where at least 3 symptoms were non ocular). Patients with purely ocular symptoms were not included in the pivotal trial; therefore, according to the clinical expert, rozanolixizumab would not be best suited for this group until more evidence is available. The clinical expert noted that the patients most in need of rozanolixizumab would be adult patients with gMG with positive antibodies who continue to be symptomatic (with significant MG-ADL scores) despite trials of first-line therapies, such as glucocorticoid and/or steroid-sparing immunosuppressants. The clinical expert added that this would depend on symptom severity, usually determined using the MG-ADL score, and that patients should be MGFA class II to IV. These patients would be identified by their neurologists, according to the clinical expert, through clinical assessments, patient-reported outcomes, and investigations. For example, a patient would need to be diagnosed with gMG based on symptoms, signs, antibody testing, and electrophysiological testing (i.e., repetitive nerve stimulation, single-fibre EMG). The clinical expert noted that once the diagnosis is established and the patient is confirmed to have AChR or MuSK antibodies, the most appropriate patients for rozanolixizumab would be identified based on their ongoing symptoms (including MG-ADL scores) and signs, as determined by their treating neurologist. The clinical expert noted that repetitive nerve stimulation and EMG should be performed by experienced neuromuscular neurologists, and that underdiagnosis or overdiagnosis occurs mainly when a patient is diagnosed by a non-neurologist and lacks supporting investigations. The clinical expert noted that, beyond the presence of antibodies and significant symptoms, it is not possible to determine which patients with gMG might respond (or not) to rozanolixizumab. The clinical expert added that although a patient who responds to IVIg or PLEX might be expected to be more likely to respond to rozanolixizumab, given the similar mechanisms, this has not been shown in clinical trials or in practice; therefore, in their opinion, this cannot be used to predict response to rozanolixizumab.
Assessing the Response to Treatment
At each visit, a thorough history is taken to assess the patient’s subjective experience regarding their symptoms, whether they have improved or not, and whether there has been functional improvement, according to the clinical expert. The most commonly used scale in clinical trials and in practice, according to the clinical expert, is the MG-ADL. The physician will also examine the patient at each clinical visit for objective evidence of disease activity, including ptosis, eye movement restrictions, nasal speech, facial weakness, limb weakness (including fatigability), or signs of shortness of breath. Another objective outcome, according to the clinical expert, may be QMG score, which is commonly used in clinical trials; however, many neurologists would likely not use this in clinical practice. According to the clinical expert, less commonly used assessments to follow patients in clinical practice would be repetitive nerve stimulation and single-fibre EMG; formal swallowing assessments or forced vital capacity to assess respiratory muscle strength may also be performed. The clinical expert noted that, overall, objective improvement in neurologic symptoms and signs are what they are looking for, as well as a reduction in the need for rescue or add-on therapies (for example, being able to taper off IVIg, stop PLEX, or taper prednisone). The clinical expert also noted that they look for objective improvements in the degree of abnormality on examination, but also that this is not as easy to quantify (for example, whether they observe eye movement abnormalities or the degree of ptosis). The clinical expert also noted that they grade muscle strength on a scale from 0 to 5 and look for improvements in this.
Discontinuing Treatment
The clinical expert noted that it would be appropriate to discontinue treatment if the patient did not improve or worsened with treatment, noting in addition that the exact duration of treatment required to determine that there is no response is not clear, but should become clearer with clinical use. Additionally, the clinical expert noted that there are cases where patients are unable to tolerate a treatment due to side effects, adding that it is difficult to specify the frequency, type, or severity of side effects that might trigger discontinuation because these vary from patient to patient. Another situation in which discontinuation would be appropriate, according to the clinical expert, would be 1 in which patients respond well to therapy and reach their treatment goal. The reasons to switch to a different therapy would be similar to those for discontinuing, according to the clinical expert.
Prescribing Considerations
The clinical expert noted that rozanolixizumab could, in theory, be administered in any number of settings, including hospital infusion centres, community infusion centres, or even the patient’s home, adding that it may even be possible for patients to self-administer. It would be most appropriate for patients to be diagnosed and treated by experienced neuromuscular neurologists to confirm the diagnosis and determine that rozanolixizumab is the most appropriate treatment, according to the clinical expert. However, ongoing monitoring could be performed by a community or non-neuromuscular neurologist (or another specialist, depending on the community and access to specialists, such as in internal medicine), under the guidance of a neuromuscular neurologist.
Clinician Group Input
This section was prepared by the review team based on the input provided by clinician groups.
A single clinician group input was received from Neuromuscular Disease Network for Canada. The network launched in January 2000, bringing together Canada’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease.
The clinicians who contributed to this submission have experience treating individuals with gMG, including experience with standards of care for gMG and with drugs such as ravulizumab, eculizumab, efgartigimod alfa, and rozanolixizumab. Information was gathered from 5 clinicians through one-to-one discussions with the lead author as well as through group discussions.
The clinician group emphasized some of the unmet needs of standard treatment for gMG, pointing out that current treatments can be associated with transient effectiveness, long wait times before benefits are experienced, and side effects; in addition, treatment is not effective for all patients. Another unmet need is the lack of treatment for patients who are MuSK antibody-positive.
The group noted that the patients more likely to respond to rozanolixizumab were those with AChR or MuSK antibodies; those with double seronegative status may also respond, but further research is required. The group also highlighted that patients with impending MG crisis require rapid intervention, whereas those whose symptoms are restricted to ocular muscles may not require such rapid intervention. The patients best suited for treatment would be identified by clinician judgment supplemented by assessments using the MG-ADL scale and other scales that reflect disease severity, such as QMG score, the Myasthenia Gravis Impairment Index, and single simple question (SSQ). In the absence of such assessments, antibody testing would be required. Additionally, cluster antibodies to both AChR and MuSK may be present in those who are double seronegative; this may cause issues in diagnosis and lead to the need for retesting, which may take weeks.
The clinician group recommended that when assessing patients’ response to therapy, scales such as the MG-ADL, QMG score, Myasthenia Gravis Impairment Index, and SSQ at 2 and 4 weeks are required; after that, the assessment is based on the patient’s status. In terms of a clinically meaningful response to treatment used in clinical trials, a score of 2 or more points on the MG-ADL or 3 or more points on the QMG was noted. For the SSQ, the clinician group noted that levels greater than 72% indicated general satisfaction. They also suggested that clinicians should consider discontinuing treatment in patients who do not respond.
Generally accepted treatments (i.e., IVIg and SCIg) that are effective in patients with MG place a significant burden on Canada’s health care system, and supplies can be at risk in situations such as a pandemic. Therefore, the clinician group highlighted the benefit of an SC route of admission, which reduces the need to rely on infusion centres. Lastly, they suggested that rozanolixizumab had the potential to replace standard immunoglobulin therapies.
Drug Program Input
The drug programs provide input on each drug being reviewed through the reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted for this review are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert responses |
|---|---|
Relevant comparators | |
For patients with AChR antibody-positive gMG, if both Vyvgart and rozanolixizumab were available through public reimbursement, which drug would be preferred and why? | The clinical expert noted that the choice between efgartigimod alfa and rozanolixizumab would largely come down to patient preference. They did not expect a clear advantage of 1 over the other. |
If both rituximab and rozanolixizumab were available through public reimbursement, which drug would be preferred for patients with MuSK antibody-positive gMG? | The clinical expert noted that although efficacy-wise, there are no data to necessarily prefer 1 over the other, rituximab is associated with significant safety issues that might make it the less favourable of these 2 options. Rituximab also does not have approval for the treatment of MuSK antibody-positive gMG. |
If you had access to rituximab through public reimbursement, would it be used as a first-line treatment in patients with AChR or MuSK? If so, in what clinical situations? | The clinical expert noted that rituximab would not be a first-line therapy for patients who are AChR antibody-positive. The clinical expert noted that among patients who are MuSK antibody-positive, rituximab might be a first-line therapy in those who are more severely affected (i.e., MG-ADL score of 5 or higher), such as those with swallowing difficulties, or at least might be used early if there is limited benefit from glucocorticoids, IVIg, or PLEX. |
What is the prevalence of patients with MuSK antibody-positive gMG in your practice? | The clinical expert estimated that patients with gMG who are MuSK antibody-positive make up about 5% of their practice. |
Does rozanolixizumab meet an unmet need for adult patients with AChR antibody-positive or MuSK antibody-positive gMG who have symptoms despite conventional treatment? | The clinical expert noted that rozanolixizumab meets an unmet need for patients who are refractory to or intolerant of other therapies. |
Considerations for initiation of therapy | |
Patients enrolled in the MycarinG trial (Study MG0007) were required to meet the following criteria:
The primary end point was change from baseline to day 43 in MG-ADL score. Do the scores in the criteria align with what you would observe in practice of initiating treatment? For CDA-AMC, should the initiation criteria align with the inclusion criteria? | The clinical expert noted that yes, these scoring results listed in the inclusion criteria for the MycarinG trial are consistent with the patients they would encounter in their practice. The clinical expert noted that it would be reasonable for the initiation criteria to align closely with the inclusion criteria, with the exception of a requirement for the patient to be considered for additional treatment, such as IVIg and or PLEX. This is because there may be patients who do not respond to glucocorticoid and/or another NSIST, but may not be suitable for IVIg or PLEX and may still benefit from rozanolixizumab. |
1. If patients with AChR antibody-positive gMG experience treatment failure with Vyvgart or rituximab, would they be eligible for rozanolixizumab? 2. If patients with MuSK antibody-positive gMG experience treatment failure on rituximab, would they be eligible for rozanolixizumab? | The clinical expert believed that yes, patients who are AChR antibody-positive and experience treatment failure with efgartigimod or rituximab should be eligible for treatment with rozanolixizumab. The clinical expert believed that yes, patients who are MuSK antibody-positive and experience treatment failure on rituximab should be eligible for treatment with rozanolixizumab. |
Under what conditions can a patient restart treatment with rozanolixizumab? | The clinical expert noted that they would observe the patient’s response and treat when needed. The goal is to avoid re-treating for minor symptoms, but also to intervene before the patient deteriorates too much. |
Considerations for prescribing of therapy | |
Rozanolixizumab is administered through a short (< 18-minute) SC injection using an infusion pump and a single-dose vial once weekly for 6 weeks. Vyvgart is administered through IV over 1 hour once weekly for 4 doses. Does the shorter infusion time and SC administration of rozanolixizumab influence your choice of therapy? | The clinical expert believed that they would prefer a shorter infusion time and a SC route of administration because these use fewer health care resources and are preferred by patients. |
Considering that intrathecal administration requires special training and facilities, are there any scenarios where administration at a clinic would be preferred over at-home treatment? | The clinical expert noted that generally, administration at a clinic would not be preferred over home administration, with the lone exception of the first dose, where the clinician may wish to observe the patient in case they have a reaction. |
For CDA-AMC, regarding consistency with the prescribing criteria associated with other drugs reviewed by CDA-AMC in the same therapeutic space, should the prescribing criteria align with those of Vyvgart? | The clinical expert believed that yes, it would be reasonable for the criteria to align with those of Vyvgart, with the addition of an indication for MuSK antibody-positive gMG (which Vyvgart does not have). |
System and economic issues | |
Concerns regarding anticipated budget impacts and sustainability:
| For information. |
Additional costs to be considered (other than related to care provision, as detailed here): The sponsor indicated that it would offer an optional patient support program to provide patient education as well as health care professional support for the administration of rozanolixizumab at program infusion clinics or in patients’ homes. | For information. |
Presence of confidential negotiated prices for comparators: Vyvgart is currently under negotiation with pCPA. | pCPA has concluded negotiations (date concluded: December 13, 2024). |
AChR = acetylcholine receptor; CDA-AMC = Canada’s Drug Agency; gMG = generalized myasthenia gravis; IVIg = IV immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; MuSK = muscle-specific tyrosine kinase; pCPA = pan-Canadian Pharmaceutical Alliance; PLEX = plasma exchange; QMG = Quantitative Myasthenia Gravis; SC = subcutaneous.
Clinical Evidence
The objective of this clinical review report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of rozanolixizumab 140 mg/mL solution for injection for SC use in the treatment of gMG in adults. The focus will be on comparing rozanolixizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of rozanolixizumab is presented in 4 sections, with our critical appraisal of the evidence included at the end of each. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. Our assessment of the certainty of the evidence in this first section, using the GRADE approach, follows the critical appraisal of the evidence. The second section includes sponsor-submitted, long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following are included in the review and appraised in this document:
1 pivotal study
2 long-term extension studies
4 indirect treatment comparisons (2 NMAs and 2 MAICs).
Systematic Review
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
The objective of the MycarinG study was to evaluate the efficacy, safety, and tolerability of rozanolixizumab in patients with gMG. The MycarinG study was a phase III, randomized, double-blind, placebo-controlled study. Eligible patients were adults aged 18 years and older with AChR antibody-positive or MuSK antibody-positive gMG (i.e., MGFA disease class II to IVa), an MG-ADL score of 3 or higher (with a score of at least 3 from nonocular symptoms), a QMG score of 11 or more, and under consideration by the investigator for additional treatment, such as IVIg or PLEX.
The study began enrolling patients in June 2019 and concluded in October 2021, with a final data cut-off date of September 17, 2021. A total of 200 patients were enrolled and randomized in a 1-to-1-to-1 ratio to receive 6 weekly SC infusions of rozanolixizumab 10 mg/kg, rozanolixizumab 7 mg/kg, or matching placebo. Rozanolixizumab was dosed by body weight range based on a patient’s weight as measured at the screening or baseline visit (Table 7). Dose group identifiers of 7 mg/kg and 10 mg/kg were averages used to differentiate the weekly doses per body weight between each treatment group. Note that the recommended dosage under review by Health Canada is 7 mg/kg; therefore, this dose is the focus of this report.
Table 5: Details of Studies Included in the Systematic Review
Detail | MG0003 (MycarinG trial) |
|---|---|
Designs and populations | |
Study design | Multicentre, randomized, double-blind, placebo-controlled, phase III study |
Locations | 81 sites in 17 countries: Belgium, Canada (4 sites), Czech Republic, Denmark, France, Georgia, Germany, Hungary, Italy, Japan, Poland, Russian Federation, Serbia, Spain, Taiwan, UK, US |
Patient enrolment dates | Start date: June 3, 2019 End date: October 26, 2021 |
Randomized (N) | N = 200 Rozanolixizumab 7 mg/kg, n = 66 Rozanolixizumab 10 mg/kg, n = 67 Placebo, n = 67 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Rozanolixizumab (7 mg/kg) through SC infusion once weekly for 6 weeks Rozanolixizumab (10 mg/kg) through SC infusion once weekly for 6 weeks |
Comparators | Placebo through SC infusion once weekly for 6 weeks |
Durations of study phases | |
Screening phase | 1 day to 28 days |
Treatment phase | 6 weeks |
Follow-up phase | 8 weeks of observation At the conclusion of the observation period, all study patients had the option to enrol in a separate extension study (the MG0004 trial or MG0007 trial), provided they met the extension study inclusion criteria. |
Outcomes | |
Primary end point | Change from baseline to day 43 in MG-ADL score |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Publication status | |
Publications | Bril et al. (2023)53 |
Clinical trial record number | NCT03971422 Sponsor-provided Clinical Study Report54 |
AChR = acetylcholine receptor; FcRn = neonatal Fc receptor; gMG = generalized myasthenia gravis; IMP = investigational medicinal product; IVIg = IV immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MGFA = Myasthenia Gravis Foundation of America; MGII = Myasthenia Gravis Impairment Index; MuSK = muscle-specific tyrosine kinase; NCT = national clinical trial number; PGI-C = Patient Global Impression of Change; PGI-S = Patient Global Impression of Severity; PLEX = plasma exchange; PRO = patient-reported outcome; QMG = Quantitative Myasthenia Gravis; SC = subcutaneous; TEAE = treatment-emergent adverse event.
aTreatment-free periods varied by medication.
Source: Sponsor’s Clinical Study Report for the MycarinG trial (NCT03971422).54
Randomization was stratified by MG-specific autoantibody (i.e., MuSK antibody-positive or AChR antibody-positive). The study spanned 81 sites across 17 countries, with 4 sites in Canada. The total duration of study participation for all patients was up to approximately 18 weeks, including a screening period of up to 4 weeks, a 6-week treatment period, and an 8-week observation period. Patients who completed the 6-week treatment period and 8-week observation period had the opportunity to roll over into the MG0004 trial — an OLE study in which the long-term safety, tolerability, and efficacy of rozanolixizumab was measured in patients with gMG over 52 weeks of weekly chronic treatment. The MG0004 trial was terminated in 2021 and replaced by the MG0007 trial, an ongoing OLE study consisting of 6-week treatment cycles based on MG worsening. Patients could roll over from the MycarinG trial or MG0004 trial directly into the MG0007 trial. A schematic diagram of the study design is presented in Figure 1.
Populations
Inclusion and Exclusion Criteria
Patients in the MycarinG study had to have been diagnosed with AChR antibody-positive or MuSK antibody-positive gMG. Patients were required to have an MG-ADL score of at least 3 (with at least 3 points coming from ocular symptoms) and a QMG score of at least 11 at screening and baseline. Patients must have been considered for additional treatment, such as IVIg or PLEX. A male patient could participate if they agreed to use contraception during the treatment period and for 90 days following the last dose; a female patient could participate if they were not pregnant, planning to become pregnant, or breastfeeding. Patients who underwent thymectomy within 6 months before baseline or had a thymoma at any time that required chemotherapy and/or radiotherapy were excluded. Patients with a history of meningococcal disease were also excluded. Treatment with rituximab in the 6 months before baseline and treatment with rituximab in the 12 months before baseline without returning to normal B-cell range was prohibited for participating patients.
Figure 1: Study Design of the MycarinG Trial
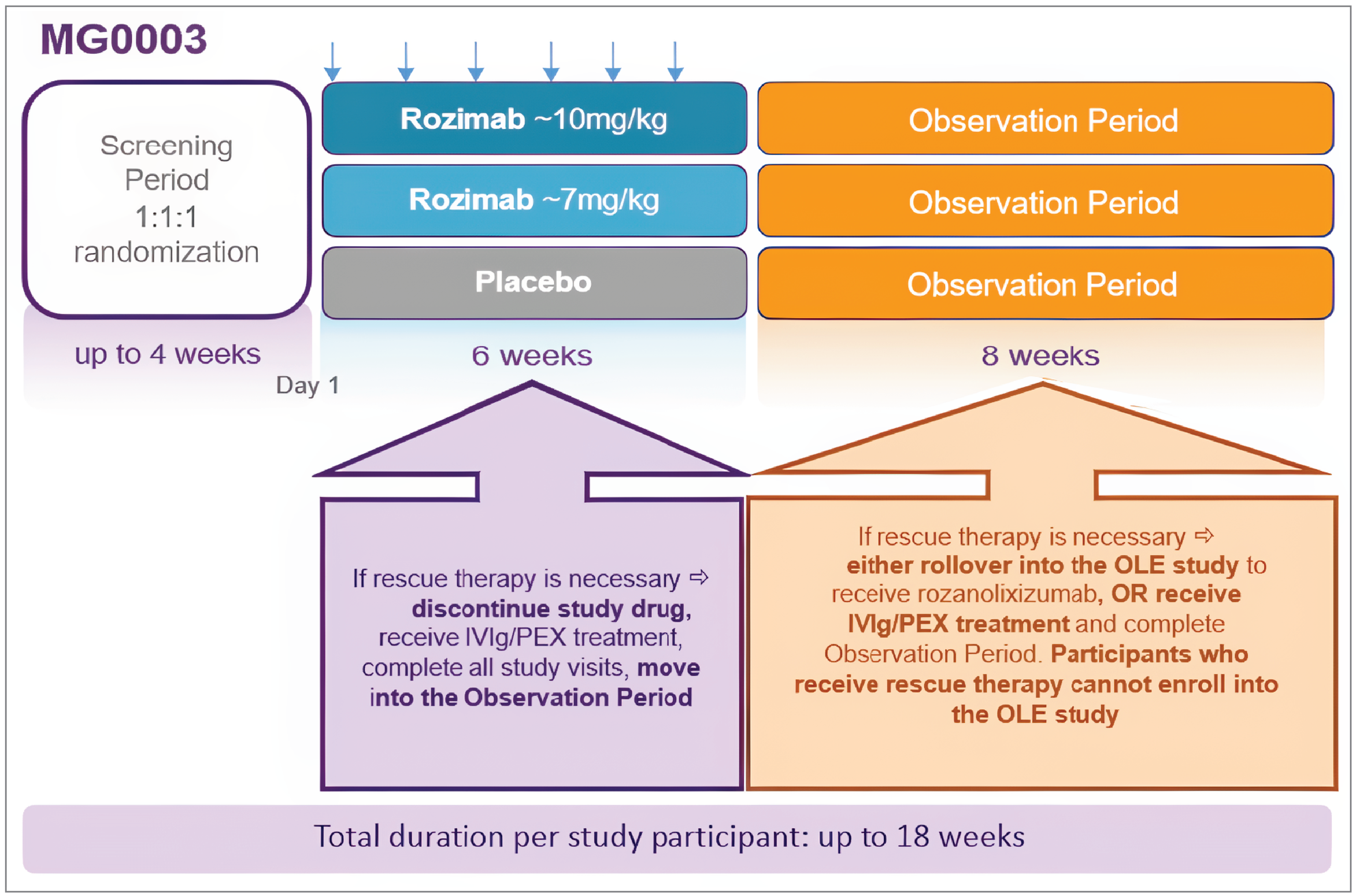
IVIg = IV immunoglobulin G; OLE = open-label extension; PEX = plasma exchange; Rozimab = rozanolixizumab.
Source: Sponsor’s Clinical Study Report for the MycarinG trial (NCT03971422), Figure 3-3.54
Interventions
Randomized patients received rozanolixizumab 7 mg/kg weekly, rozanolixizumab 10 mg/kg weekly, or placebo administered through SC infusion for a total of 6 weeks in addition to their current gMG treatments. Doses were administered into the abdomen by study personnel at study centres, with an infusion rate up to 20 mL/hour. Rozanolixizumab was dosed by body weight range based on a patient’s weight measured at the screening or baseline visit. Dose group identifiers of 7 mg/kg and 10 mg/kg were averages used to differentiate the weekly doses per body weight between each treatment group. An interactive response technology was used to assign eligible study patients to treatment regimens based on a predetermined production randomization and/or packaging schedule.
Rescue therapy for the study consisted of IVIg or PLEX. Study patients who experienced disease worsening (e.g., an increase of 2 points on the MG-ADL or 3 points on the QMG between 2 consecutive visits) were considered for rescue therapy at the discretion of the investigator. Study patients who received rescue therapy during the treatment period were to complete any subsequent visit(s) and move into the observation period.
If IgG levels dropped to less than 1g/L, the study drug was temporarily discontinued. If IgG levels were greater than or equal to 1 g/L and less than 2 g/L, and a patient experienced a persistent or recurrent, nonserious infection, rozanolixizumab treatment could be temporarily discontinued. Temporary treatment discontinuation due to low IgG levels could be a trigger to unblind the treatment assignment of the specific study to both the patient and study site personnel. In that case, infusions continued, but were given as mock infusions with only placebo, irrespective of prior study drug designation. In the event of an emergency, it was possible to determine to which treatment group and dose the study patient had been allocated by consulting the interactive response technology.
Details of the permitted concomitant medications are in Table 6. Concomitant conventional therapy was allowed if stable for a predefined period before baseline (except for AChEIs, for which stable dosage was not required). The dosages of permitted concomitant medications (except for AChEIs and corticosteroids) needed to be stable during each 6-week treatment cycle. Every effort should have been made to maintain a stable dosage during the first 8 weeks of each observation period. Dose adjustments were allowed between cycles.
Table 6: Permitted Concomitant Medications in the MycarinG Trial
Permitted medications | Dose | Comment |
|---|---|---|
Oral corticosteroids (e.g., prednisolone) | No specific requirements | Stable for 4 weeks before baselinea |
Methotrexate | ≤ 30 mg/week | Treated for previous 6 months and on a stable dosage for 2 months before baselinea |
Mycophenolate mofetil | ≤ 3 g/day | Treated for previous 6 months and on a stable dosage for 2 months before baselinea |
Cyclosporineb | ≤ 5 mg/kg/day for unmodified ≤ 4 mg/kg/day for modified (i.e., microemulsion) | Treated for previous 6 months and on a stable dosage for 2 months before baselinea |
Azathioprine | ≤ 3 mg/kg/day | Treated for previous 6 months and on a stable dosage for 2 months before baselinea |
Cholinesterase inhibitors | ≤ 600 mg pyridostigmine/day | Stable dosage not required; dose held on mornings with efficacy outcome assessmentsa |
Tacrolimusc | ≤ 5 mg/day | Treated for previous 6 months and on a stable dosage for 2 months before baselinea |
The following concomitant medications and/or treatments were prohibited during the study:
all biologics, including rituximab
cyclophosphamide
pimecrolimus
IPP-201101 (Lupuzor)
immunoadsorption
vinca alkaloids (vincristine, vinblastine).
Outcomes
A list of efficacy end points assessed in this clinical review report is provided in Table 7, followed by descriptions of the outcome measures. Properties of the outcome measures are in Table 8. Summarized end points are based on outcomes included in the sponsor’s Summary of Clinical Evidence24 as well as on any outcomes identified as important to this review, according to the clinical expert(s) consulted for this review and input from patient and clinician groups and public drug plans. Using the same considerations, we selected end points that were considered most relevant to inform expert committee deliberations and finalized this list in consultation with members of the committee. All summarized efficacy end points were assessed using GRADE. Select notable harms outcomes considered important for informing expert committee deliberations were also assessed using GRADE.
The clinical expert noted that the MG-ADL and QMG scores are used in clinical practice to assess patients. These tend to be used in tandem, given that the MG-ADL is a patient-reported score while the QMG score is obtained through clinical assessment of the patient. The MG-QoL15r score was included to reflect the fact that gMG has a significant impact on HRQoL; this instrument is specifically designed to assess patients with gMG, whereas more generic instruments were used to assess HRQoL in the pivotal study. Finally, the MGC score was not assessed using GRADE, given that there is considerable overlap between this instrument and the MG-ADL instrument. However, because MGC is still an important instrument in assessing patients with gMG, the results for MGC were included in the clinical report as supportive information. Due to the brevity of the trial and the fact that patients received only a single treatment course, it was decided that the time point of interest for all assessments would be 43 days, or the end of the treatment period. Infections and infestations are mentioned as a warning on the proposed product monograph and included in the assessment as potential harms of special interest.
Table 7: Outcomes Summarized From the Studies Included in the Systematic Review
Outcome measure | Time point | MycarinG trial |
|---|---|---|
Change in MG-ADL score | From baseline to day 43 | Primarya |
Percentage of patients achieving MG-ADL response | From baseline to day 43 | Secondary |
Change in QMG score | From baseline to day 43 | Secondarya |
Change in MGC score | From baseline to day 43 | Secondarya |
Change in MG-QoL15r score | From baseline to day 43 | Exploratory |
MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; QMG = Quantitative Myasthenia Gravis.
aStatistical testing for these end points was adjusted for multiple comparisons.
Source: Sponsor’s Summary of Clinical Evidence.24
MG-ADL Score
Change from baseline to day 43 in MG-ADL score was the primary end point of the MycarinG trial. The MG-ADL is a brief, 8-item, interviewer- or self-administered, patient-reported outcome assessment designed to evaluate MG symptom severity.55 Two questions pertain to ocular functions, 3 to oropharyngeal functions, 1 to respiratory function, and 2 to extremity functions. Each of the 8 items is scored on a scale of 0 points (normal) to 3 points (most severe); the total score is the sum of the 8 individual scores, ranging from 0 to 24. Higher scores are associated with more severe symptoms.55 A 2-point change in MG-ADL score is considered clinically meaningful; this was the threshold used to indicate a response in the responder analysis.56 This 2-point change threshold was based on a receiver operator characteristic curve method from an observational study of patients with MG managed at the discretion of the treating physician.56 The sponsor and review team were unable to identify literature-based, between-group minimally important difference (MID) estimates. In consultation with clinical experts, the review team considered that a difference between groups of fewer than 2 points was not likely to be clinically important. For MG-ADL response, the clinical expert consulted by the review team suggested that a between-group difference of at least 20% may be considered clinically important.
MGC Score
The MGC is a 10-item PRO scale that has been used to measure the clinical status of patients with MG, both in the practice setting and in clinical studies, to evaluate treatment response. The 10 items and corresponding response scale scores are weighted and totalled. The total score is the sum of the 10 individual scores and ranges from 0 to 50. Higher scores indicate more severe impairment due to the disease.57,58 A study of patients with MG under routine care found that a 3-point change in this assessment is considered clinically meaningful and is associated with the best sensitivity and specificity, based on a receiver operator curve method.59 In the MycarinG study, the change from baseline to day 43 in MGC score was assessed as a secondary outcome.
QMG Score
The QMG is a standardized and validated quantitative strength PRO scoring system that was developed specifically for MG.60 The scoring system consists of 13 individual assessments, each scored on a scale from 0 points (normal) to 3 points (most severe). The total score is the sum of the individual scores, ranging from 0 to 39. Higher scores are representative of more severe impairment.60 A change in QMG score of 3 points or more may be considered clinically meaningful in a typical clinical study population of patients with MG.60,61 In the MycarinG study, change from baseline to day 43 (visit 10) in QMG score was assessed as a secondary outcome.62
MG-QoL15r Score
The MG-QoL15r is a 15-item, self-administered PRO scale designed to assess quality of life in patients with MG.63 The 15 items relate to physical, social, and psychological aspects of well-being and are associated with corresponding response scales, each scored on a scale from 0 points to 2 points. The total score is the sum of the 15 individual item scores, and ranges from 0 to 30. Higher scores indicate more severe impact of the disease on aspects of the patient’s life.63,64 A literature-based MID has not yet been determined,65 and the review team was not able to estimate what between-group difference might be considered clinically important. The change from baseline to week 12 in MG-QoL15r score was assessed as an exploratory outcome.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
MG-ADL | The MG-ADL is a brief, 8-item, interviewer-administered PRO scale designed to evaluate MG symptom severity.55 Two questions pertain to ocular functions, 3 to oropharyngeal functions, 1 to respiratory function, and 2 to extremity functions. Each of the 8 items is scored on a scale from 0 points (normal) to 3 points (most severe), and the total score is the sum of the 8 individual scores, ranging from 0 to 24. Higher scores are associated with more severe symptoms of MG.55 | Validity: The validity of the MG-ADL was assessed in a study of 87 patients with a confirmed MG diagnosis based on clinical, serologic, and electrodiagnostic testing. In this study, the correlation between MG-ADL score and physician impression of change between visits was strong (r = 0.70; P < 0.0001).56 Reliability: Test-retest in 20 patients demonstrated a reliability coefficient of 93.7% among 20 patients, with the lower bound of the 95% CI at 87.3%, when tested twice within 1 week.56 Responsiveness: The MG-ADL was assessed at 2 visits; the mean improvement in score in patients who improved, based on the gold standard, was 3.88 (SD = 2.7).56 | A 2-point improvement in MG-ADL score is a threshold that optimally indicates clinical improvement at the level of the individual (within-group MID) for patients with MG.56 |
MG-QoL15r | The MG-QoL15r is a 15-item, self-administered PRO scale designed to assess quality of life in patients with MG.63 The 15 items relate to physical, social, and psychological aspects of well-being and are associated with corresponding response scales, each scored on a scale of 0 points to 2 points. The total score is the sum of the 15 individual item scores and ranges from 0 to 30. Higher scores indicate more severe impact of the disease on aspects of the patient’s life.63,64 | Validity: The MG-QoL15 has been previously validated in a study of 175 patients.63,66 The MG-QoL15r was developed based on an analysis of scores from 1,362 MG-QoL15 surveys completed by 954 patients seen in 19 participating clinics in various international jurisdictions, including Canada.64 The study demonstrated that the MG-QoL15r had slightly improved clinometric properties and face and content validity relative to the MG-QoL15.64 A study of 872 patients with autoimmune MG found that construct validity was demonstrated for the MG-QoL15r with QMG (r = 0.550), MG-ADL (r = 0.701), and MGC (r = 0.635). For discriminant validity, the MG-QoL15r scores differed among patients based on their MGFA classification and MGC scores.67 Reliability: The test-retest reliability coefficient for the MG-QoL15 was 98.6%, with a lower bound on a 95% CI equal to 97.5%. Scores varied by 3 points or less 87% of the time and by 4 points or less 94% of the time.66 In a study of 872 patients with autoimmune MG, internal consistency reliability of the MG-QoL15r was demonstrated by the Cronbach alpha of 0.93.67 Responsiveness: The correlation between MGC score changes and MG-QoL15 score changes for patients in the prospective scale validation trial was 0.53 (95% CI, 0.41 to 0.65; P < 0.0001).66 There was no loss of sensitivity with fewer choices in the MG-QoL15r vs. the MG-QoL15.64 In a study of 872 patients with autoimmune MG, the r between changes in MG-QoL15r and QMG after treatment was 0.423.67 | A MID has not yet been determined.65 |
QMG | The QMG is a standardized, validated, quantitative strength scoring system that was developed specifically for MG.60 The scoring system consists of 13 individual assessments, each scored on a scale of 0 points (normal) to 3 points (most severe). The total score is the sum of the individual scores, ranging from 0 to 39. Higher scores represent more severe impairment.60 | Validity: A study of 135 patients aged 18 years and older with a diagnosis of MG and worsening weakness requiring a change in therapy demonstrated a positive association between the QMG and the MGFA scale, the MG-QoL15, patient serological status, and objective markers of neuromuscular function.68 Construct validity was assessed through correlations with the MMT (r = 0.69 in 303 patients69 and r = 0.73 in 53 patients).70 Reliability: Internal consistency assessed through Cronbach alpha value was 0.74 for the QMG, demonstrating an acceptable threshold (n = 251).71,72 Test-retest reliability was studied in 209 stable patients assessed 2 weeks apart. The intraclass correlation coefficient for the total scores was 0.88 (95% CI, 0.85 to 0.91).71,72 A study of 9 patients, 5 with MG and 4 without, found that at the 95% confidence level (1.96 SD), QMG scores do not differ from the observed values by more than ± 2.63 units (1.96 SD × 1.342).60 Responsiveness: A study analyzing data from 53 patients with a clinical diagnosis of MG calculated that a mean difference between the improved or worsened and unchanged group was 2.32 points. Dividing this by the SD of the unchanged group (1.60 points) provided an index of responsiveness of 1.45.70 | A change in the QMG score of 3 points or more may be considered clinically meaningful in a typical clinical study population of patients with MG (between-group MID).60,61 |
MGC | The MGC is a 10-item scale that has been used to measure the clinical status of patients with MG, both in the practice setting and in clinical studies, to evaluate treatment response. The 10 items and corresponding response scale scores are weighted and totalled. The total score is the sum of the 10 individual scores and ranges from 0 to 50. Higher scores on the MGC indicate more severe impairment due to the disease.57,58 | Validity: The validity of the MGC was assessed in a study of 38 patients aged 17 years and older with a clinical diagnosis of MG made by an MG specialist and almost always confirmed by serologic and electrodiagnostic testing.59 In this study, the correlation between MGC scores and other MG-specific scales was moderate to strong. At visit 1, the total MGC score had a correlation of 0.68 (95% CI, 0.59 to 0.75) with the MG-QoL15 total score; 0.85 (95% CI, 0.77 to 0.90) with the MG-ADL total score; and 0.80 (95% CI, 0.72 to 0.86) with the MG-MMT total score. Nearly identical correlations were observed at visit 2.59 Reliability: The test-retest reliability coefficient was 98%, with a lower 95% CI of 97%, indicating excellent test-retest reliability.59 Responsiveness: No evidence identified. | A study of patients with MG under routine care found that a 3-point change in this assessment is considered clinically meaningful (within-group MID) and associated with the best sensitivity and specificity based on a ROC curve method.59 |
CI = confidence interval; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MG-MMT = Myasthenia Gravis Manual Muscle Test; MG-QoL15 = 15-item Myasthenia Gravis Quality of Life; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MID = minimally important difference; PRO = patient-reported outcome; r = Pearson correlation coefficient; ROC = receiver operating characteristic; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Source: Additional information provided by the sponsor.73
Statistical Analysis
The primary end point of the MycarinG study was change from baseline to day 43 in MG-ADL score. The total score calculated after single-item imputation was used to calculate change from baseline in summaries and for efficacy analyses. The primary analysis was performed on the randomized set under the hypothetical and treatment policy strategy to handle intercurrent events (primarily rescue therapy and treatment or study discontinuation). For patients who required rescue therapy before week 43, data at and after the use of rescue therapy were considered as missing. Patients discontinuing treatment due to TEAEs continued to be included up to day 43. The statistical model was a stage-wise, mixed model for repeated measures analysis of covariance, with treatment group, baseline MG-ADL score, region, stratification factor(s) (including MuSK antibody status [positive or negative] and /or AChR antibody status [positive or negative]), and treatment group by day (i.e., interaction term) as fixed factors and study patients as random effects. The mixed model for repeated measures included days 8, 15, 22, 29, 36, and 43.
The model utilized an unstructured covariance pattern for the repeated measures. If the model did not converge using the unstructured pattern, then an autoregressive covariance structure was used. The Kenward-Roger approximation was used to estimate the denominator degrees of freedom. For each stage (interim and final), the LS means of each treatment group and the LS mean differences between each dose group and placebo were reported for day 43 (visit 10), along with the corresponding 2-sided 97.5% CIs and P values. The study followed a 2-stage group sequential adaptive design using combination tests based on the inverse-normal method of combining independent stage-wise P values. Given that the trial was not stopped for futility at interim analysis at the end of the trial, the combined P value (2-sided) for the 2 stages was based on the stage-wise, inverse-normal combination of independent stage-wise P values using the Lehmacher and Wassmer method. The analysis produced multiplicity-adjusted 95% CIs. Through use of the inverse-normal combination function together with the critical value Zalpha/2, the lower bound of the 2-sided 95% repeated CI for a given treatment arm was found as the smallest value for which the multiplicity-adjusted combination test yields nonrejection at a 2-sided alpha of 5%. The upper bound of the CI was found analogously.
Sample Size and Power Calculation
This study consisted of 2 stages, with a formal interim analysis at the end of stage 1. It was proposed that the interim analysis be conducted when approximately 90 eligible study patients had been treated and were evaluable for the primary end point (i.e., approximately 30 study patients per dose group in stage 1). If 2 treatment doses were selected, then the conditional power associated with the higher treatment effect was used to determine the stage 2 sample size. Conditional power was calculated as described in formula 7.2 of Wassmer and Brannath (2016).74
If 2 doses were considered for stage 2, then this formula would be applied with the conditional error divided by 2 to account for multiplicity. Depending upon the selection of 1 or 2 doses after stage 1, a further 60 — and up to a maximum of 150 — eligible study patients would be randomized in stage 2 of the study. Thus, the total sample size of the study could have ranged between 150 patients and 240 patients if the study was not futile at stage 1. This would provide 90% power.
Multiple Testing Procedure
The statistical analysis of the primary efficacy and selected secondary efficacy end points (i.e., changes from baseline in MG-ADL, MGC, and QMG scores as well as changes from baseline in patient-reported outcomes of muscle weakness and fatigue, physical fatigue, and bulbar symptoms scores) accounted for multiplicity and controlled the family-wise type I error rate at a 2-sided alpha level of 0.05 by using a parallel gatekeeping testing procedure with a truncated Hochberg test for each of the 6 type I error families (corresponding to the primary end point and the 5 secondary end points). The hypotheses were mapped into 2 sets so that hypotheses within each set corresponded to the same rozanolixizumab dose. Serial restrictions were applied so that the end points could be tested only for each dose, if all previous end points for that dose were significant.
For family 1 (the primary end point hypotheses corresponding to the pairwise comparisons of each dose versus placebo), the Hochberg truncation parameter was set to 0, which was equivalent to using the Bonferroni approach, in which the type I error was split equally between rozanolixizumab dose levels of 7 mg/kg and 10 mg/kg, such that each dose level was tested at a 2-sided alpha level of 0.025. In comparison, for families 2 to 5, the Hochberg truncation parameter was set to 0.2; and for the final family, the truncation parameter was 1, such that the standard Hochberg test was used. The scenarios for the sequential procedure began with the evaluation of the primary efficacy end point.
Changes from baseline in MGC and MG-QoL15r were uncontrolled for multiplicity.
Data Imputation Methods
Data imputations methods are elaborated in Table 9.
Subgroup Analyses
The primary and continuous secondary efficacy end points were evaluated for subgroups of interest, including stratification factors: MG-specific autoantibodies, AChR antibodies (positive or negative), and MuSK antibodies (positive or negative). Note that as stratification factors, the AChR antibodies (positive or negative) and MuSK antibodies (positive or negative) in the subgroup analysis were based on the values from the MG-specific autoantibody assessment taken at baseline. Historical AChR antibody status (positive or negative) and historical MuSK antibody status (positive or negative) were also examined in the subgroup analysis; in this case, baseline AChR antibody status (positive or negative) and baseline MuSK antibody status (positive or negative) were replaced by historical AChR antibody status (positive or negative) and historical MuSK antibody status (positive or negative).
The sponsor also performed various post hoc subgroup analyses that included patients who had received at least 2 prior MG-specific therapies versus those who had not.
All subgroup analyses were descriptive; no statistical testing of treatment-by-subgroup interactions or of treatment effects within subgroups was carried out. No subgroup analysis was performed for safety variables. Subgroup analyses were performed only for subgroups where there were at least 5 study patients in each subgroup level.
Sensitivity Analyses
To check the assumptions around the estimand in the (primary) analysis of the primary end point, the following sensitivity analyses were performed:
hypothetical and treatment policy strategy on full analysis set (FAS)
hypothetical and treatment policy strategy with a jump-to-reference (J2R) approach on missing data (used to verify the robustness of the missing at random [MAR] approach)
hypothetical and treatment policy strategy based on the subgroup of the random set that received all 6 SC doses
hypothetical and treatment policy strategy based on the subgroup of the FAS, excluding confirmed COVID-19 cases.
Additionally, there were 2 supplementary analyses:
composite strategy on the random set, a trimmed mean approach in which data assumed to be missing not at random were used to impute missing MG-ADL scores with the worst score of the MG-ADL
treatment policy strategy based on the random set, in which missing MG-ADL scores were handled by maximum likelihood estimation under the MAR assumption.
Secondary Outcomes of the Studies
Each of the continuous secondary end points (i.e., changes from baseline in QMG score, MGC score, MG-QoL15r, and patient-reported outcomes related to MG symptoms) was assessed using the same statistical approach as for the primary end point. Sensitivity analyses of secondary end points were conducted using the same statistical approach as for the primary end point. Binary secondary end points (i.e., MG-ADL responders) were assessed using a logistic mode with factors of treatment group and baseline MG-ADL score and stratification factors (i.e., MuSK and AChR antibody status [positive or negative]). No further imputation was used. The analysis of MG-QoL15r was a post hoc analysis based on the review team’s request for additional information. No adjustment was made to the P value to account for multiple testing or the sequential adaptive design.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Change in MG-ADL score | MMRM ANCOVA |
| Missing data were imputed using maximum likelihood estimation (MAR assumption) |
|
Percentage of patients achieving MG-ADL response | Logistic regression model |
| Missing data were imputed using NRI | NR |
Change in MGC score | MMRM ANCOVA |
| Missing data were imputed using maximum likelihood estimation (MAR assumption) |
|
Change in QMG score | MMRM ANCOVA |
| Missing data were imputed using maximum likelihood estimation (MAR assumption) |
|
Change in MG-QoL15r score | MMRM ANCOVA |
| Missing data were imputed using maximum likelihood estimation (MAR assumption) | NR |
ANCOVA = Analysis of Covariance; FAS = full analysis set; J2R = jump to reference; MAR = missing at random; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MMRM = mixed model for repeated measures; NR = not reported; NRI = nonresponse imputation; QMG = Quantitative Myasthenia Gravis; RS = randomized set; SC = subcutaneous.
Source: Sponsor’s Summary of Clinical Evidence.24
Analysis Populations
Table 10: Analysis Populations
Population | Definition | Application |
|---|---|---|
Enrolled set | All study patients who signed the informed consent form | Patient disposition |
RS | All study patients who were randomized, using the treatment assigned instead of the actual treatment received | Study population and characteristics, secondary and exploratory efficacy analyses |
SS | All randomized study patients who received at least 1 dose of IMP, analyzed according to the actual treatment the patients received | Safety analyses |
Full analysis set | All study patients in the RS who had a baseline and least 1 postbaseline MG-ADL measurement | Study population and characteristics, all efficacy analyses |
IMP = investigational medicinal product; MG-ADL = Myasthenia Gravis Activities of Daily Living; RS = randomized set; SS = safety set.
Source: Sponsor’s Clinical Study Report for the MycarinG trial (NCT03971422), Section 6.1.4.62
Results
Patient Disposition
There were 23 patients (35%) in the rozanolixizumab group and 25 patients (37%) in the placebo group who discontinued the study. Most discontinued due to the need for rescue medication during the observation period (19 patients [29%] in the rozanolixizumab group and 18 patients [27%] in the placebo group). Most rolled over into the MG0004 trial or the MG0007 trial. A greater proportion of patients in the placebo group (n = 5; 7.5%) compared to the rozanolixizumab group (n = 1; 1.5%) discontinued the study due to lack of efficacy.
Table 11: Summary of Patient Disposition
Patient disposition | MycarinG trial | |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 66) | Placebo (N = 67) | |
Screened, N | 300 | |
Screen failures, N (%) | 100 (33.3) | |
Reason for screening failure, n (%) | ||
Adverse event | 8 (2.7) | |
Ineligibility | 86 (28.7) | |
Withdrawal by patient or parent/guardian | 5 (1.6) | |
Other | 1 (0.3) | |
Randomized, N (%) | 66 (100) | 67 (100) |
Discontinued from study, n (%) | 23 (34.8) | 25 (37.3) |
Reason for discontinuation, n (%) | ||
Adverse events | 2 (3.0) | 2 (3.0) |
Lack of efficacy | 1 (1.5) | 5 (7.5) |
Lost to follow-up | 1 (1.5) | 0 |
Other | 19 (28.8) | 18 (26.9) |
Due to COVID-19 pandemic | 1 (1.5) | 0 |
Mandatory withdrawal and roll over to the MG0004 trial | 8 (12.1) | 7 (10.4) |
Mandatory withdrawal and roll over to the MG0007 trial | 6 (9.1) | 10 (14.9) |
RS, N | 66 | 67 |
FAS, N | 66 | 67 |
Safety, N | 64 | 67 |
FAS = full analysis set; RS = randomized set.
Note: Mandatory withdrawal and rollover to the MG0004 trial or the MG0007 trial referring to patients requiring rescue therapy in the MG0003 trial observation period. Another 13 study participants rolled over to the MG0004 trial (6 patients) or the MG0007 trial (7 patients) after completion of the treatment period and during the observation period (data on file); the reasons provided for discontinuation for these study participants were lack of efficacy (6 patients), worsening of symptoms (5 patients), adverse event (1 patient), and other (1 patient).
Source: Sponsor’s Clinical Study Report for the MycarinG trial (NCT03971422), Section 7.1, Tables 7-1, 1.1.1.54
Baseline Characteristics
The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or that were believed to affect the outcomes or interpretation of the study results. Patients in the pivotal study were aged 52 years (SD = 16 years), on average, and the majority (61%) were female. Most patients were MGFA class IIa or IIb (39%) or class IIIa or IIIb (57%) at baseline. At baseline, the majority of patients were AChR antibody-positive (83%); 9% were MuSK antibody-positive. Notable differences between groups included baseline autoantibody status, given that there were fewer patients in the rozanolixizumab group who were MuSK antibody-positive than in the placebo group (6% versus 12%) and more patients in the rozanolixizumab group who were AChR antibody-positive than in the placebo group (85% versus 79%). Other baseline characteristics for which there were notable differences between groups included duration of disease, the proportion of patients who had experienced myasthenia crisis in the past, MGFA class at screening, and gender.
Table 12: Summary of Baseline Characteristics From Studies Included in the Systematic Review
Characteristic | MycarinG trial | |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 66) | Placebo (N = 67) | |
Age (years)a | ||
Mean (SD) | 53.2 (14.7) | 50.4 (17.7) |
Median | 52.0 | 51.0 |
Minimum to maximum | 22 to 89 | 18 to 85 |
Age category, n (%) | ||
≤ 18 years | 0 | 1 (1.5) |
19 years to < 65 years | 49 (74.2) | 50 (74.6) |
≥ 65 years | 17 (25.8) | 16 (23.9) |
Sex, n (%) | ||
Male | 27 (40.9) | 20 (29.9) |
Female | 39 (59.1) | 47 (70.1) |
Weight (kg) | ||
Mean (SD) | 79.56 (25.52) | 80.80 (22.57) |
Median | 78.00 | 80.00 |
Minimum to maximum | 37.7 to 154.2 | 39.7 to 150.5 |
Weight category (kg), n (%) | ||
< 50 | 7 (10.6) | 4 (6.0) |
50 to < 70 | 19 (28.8) | 16 (23.9) |
70 to < 100 | 26 (39.4) | 35 (52.2) |
≥ 100 | 14 (21.2) | 12 (17.9) |
Racial group, n (%)b | ||
Asian | 9 (13.6) | 5 (7.5) |
Black | 0 | 1 (1.5) |
Native Hawaiian or other Pacific Islander | 0 | 1 (1.5) |
White | 41 (62.1) | 46 (68.47) |
Missing | 16 (24.2) | 14 (20.9) |
MG-ADL score | ||
Mean (SD) | 8.4 (3.8) | 8.4 (3.4) |
Median | 8.0 | 8.0 |
Minimum to maximum | 3 to 18 | 3 to 16 |
MG-ADL group, n (%) | ||
≥ 5 | 55 (83.3) | 57 (85.1) |
< 5 | 11 (16.7) | 10 (14.9) |
QMG score | ||
Mean (SD) | 15.4 (3.7) | 15.8 (3.5) |
Median | 15.0 | 15.0 |
Minimum to maximum | 9 to 27 | 11 to 23 |
MGFA class at screening, n (%) | ||
Class IIa | 13 (19.7) | 11 (6.4) |
Class IIb | 16 (24.2) | 12 (17.9) |
Class IIIa | 21 (31.8) | 28 (41.8) |
Class IIIb | 13 (19.7) | 13 (19.4) |
Class IVa | 3 (4.5) | 2 (3.0) |
Class IVb | 0 | 1 (1.5) |
Thymectomy, n (%) | ||
Yes | 32 (48.5) | 31 (46.3) |
No | 34 (51.5) | 36 (53.7) |
Myasthenia crisis in the past, n (%) | ||
Yes | 19 (28.8) | 23 (34.3) |
No | 46 (69.7) | 44 (65.7) |
Missing | 1 (1.5) | 0 |
Duration of disease (years) | ||
Mean (SD) | 6.877 (6.799) | 9.418 (9.348) |
Median | 5.280 | 6.790 |
Minimum to maximum | 0.14 to 33.09 | 0.14 to 48.94 |
Age at initial MG diagnosis (years) | ||
Mean (SD) | 46.6 (16.0) | 41.4 (19.1) |
Median | 46.0 | 38.0 |
Minimum to maximum | 13 to 83 | 12 to 79 |
Historical autoantibody status, n (%)a | ||
AChR-positive | 60 (90.9) | 59 (88.1) |
MuSK-positive | 5 (7.6) | 8 (11.9) |
Baseline autoantibody status, n (%)b | ||
AChR-positive | 56 (84.8) | 53 (79.1) |
MuSK-positive | 4 (6.1) | 8 (11.9) |
AChR = acetylcholine receptor; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; MuSK = muscle-specific tyrosine kinase; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
aAChR and MuSK autoantibody status are captured from the Confirmatory (Historical) Diagnostic Tests for Primary Condition case report form.
bAChR and MuSK autoantibody status are captured from baseline visit.
Source: Sponsor’s Summary of Clinical Evidence.24
Exposure to Study Treatments
The mean durations of treatment were similar between the rozanolixizumab group (35.0 days; SD = 5.8 days) and placebo group (35.1 days; SD = 4.1 days) (Table 13).
All study patients received at least 1 infusion (excluding mock infusions). A total of 52 patients (81.3%) in the rozanolixizumab 7 mg/kg group and 56 patients (83.6%) in the placebo group received all 6 infusions (excluding mock infusions). Mock infusions using placebo were given to reduce unblinding potential when IgG levels dropped to less than 1g/L. No study patients in the placebo group needed mock infusions, whereas 3 total infusions in 2 patients were required in the 7 mg/kg group.
Table 13: Summary of Patient Exposure From Studies Included in the Systematic Review
Exposure | MycarinG trial | |
|---|---|---|
Rozanolixizumab (N = 64) | Placebo (N = 67) | |
Duration, mean days (SD) | 35.0 (5.8) | 35.1 (4.1) |
Duration, median days | 36.0 | 36.0 |
Range, days | 1 to 43 | 16 to 41 |
Adherence, n (%) received all 6 infusions | 52 (81.3%) | 56 (83.6%) |
SD = standard deviation.
Source: Sponsor’s Clinical Study Report for MycarinG (NCT03971422), Section 11.1, Table 6.1.1.54
In the FAS, almost all patients (198 patients [99.0%]) reported using at least 1 prior medication, defined as medications that started before the first administration of the study drug. Almost all patients (193 patients [96.5%]) had received any prior gMG medication, defined as medications given for an indication related to gMG, including corticosteroids, immunosuppressants, and parasympathomimetics, while 165 patients (82.5%) had received any prior gMG medication, excluding AChEIs.
According to the sponsor, the use of any rescue medication occurred in 3 patients (4.5%) who received placebo compared to 1 patient (1.5%) who received rozanolixizumab 7 mg/kg. It is not clear why these numbers are lower than those reported in the disposition, in which the number of patients reported under “mandatory rollover” (i.e., requiring rescue therapy) into the extensions is n = 31 across the 2 groups. All patients received immunoglobulin as rescue medication. Patients requiring IVIg or PLEX rescue therapy during the treatment period proceeded to the 8-week observation period after completing the assessments in the MycarinG trial. Patients requiring rescue therapy during the observation period had the option to roll over to the OLE studies (i.e., the MG0004 trial, later replaced with the MG0007 trial), provided they met the inclusion criteria, or to discontinue the study to receive IVIg or PLEX.
Table 14: gMG Medications Started Before Dosing With Rozanolixizumab and Continued Afterward (FAS)
Exposure | MycarinG trial | |
|---|---|---|
Rozanolixizumab (N = 64) | Placebo (N = 67) | |
Any prior therapy, n (%) | 65 (98.5) | 67 (100) |
Any prior MG-specific therapy (including AChEIs), n (%) | 63 (95.5) | 64 (95.5) |
Any prior MG-specific therapy (excluding AChEIs), n (%) | 50 (75.8) | 53 (79.1) |
Any AChEI, n (%) | 55 (83.3) | 61 (91.0) |
Any steroid, n (%) | 43 (65.2) | 40 (59.7) |
Any NSIST, n (%) | 32 (48.5) | 35 (52.2) |
Any IVIg or PLEX treatment | 12 (18.2) | 6 (9.0) |
1 class of conventional therapy, n (%) | ||
AChEI only | 14 (21.2) | 13 (19.4) |
Steroid only | 17 (25.8) | 16 (23.9) |
NSIST only | 6 (9.1) | 11 (16.4) |
2 classes of conventional therapy, n (%) | ||
Steroid + NSIST | 24 (36.4) | 21 (31.3) |
Steroid + AChEI | 36 (54.5) | 37 (55.2) |
NSIST + AChEI | 25 (37.9) | 34 (50.7) |
All 3 classes of conventional therapy, n (%) | 20 (30.3) | 23 (34.3) |
AChEI = acetylcholinesterase inhibitor; FAS = full analysis set; IVIg = IV immunoglobulin; MG = myasthenia gravis; NSIST = nonsteroidal immunosuppressive therapy; PLEX = plasma exchange.
Sources: MycarinG trial (NCT03971422) Post Hoc Data Tables 2022-RLZ-04, Table 2.5.1, Table 2.5.2.54
Efficacy
The outcomes determined to be of importance based on consultation with clinical experts — and the input received from patient and clinician groups and public drug plans — are discussed herein. Additional outcome data are available in Appendix 1.
MG-ADL Score
The primary end point was change from baseline to day 43 in MG-ADL score (range, 0 to 24; higher scores indicate more severe symptoms). From baseline mean MG-ADL scores of 8.4 (SD = 3.8) in the rozanolixizumab group and 8.4 (SD = 3.4) in the placebo group, the LS mean changes from baseline were –3.370 (SE = 0.486) and –0.784 (SE = 0.488), respectively, for an LS mean difference between groups (rozanolixizumab minus placebo) of –2.586 (95% CI, –4.091 to –1.249; P < 0.001) (Table 15).
The results from the sensitivity analyses using J2R for imputation of missing scores, including study participants who received all 6 doses of the study drug and excluding patients with confirmed COVID-19, were consistent with those from the main analysis. Supportive analyses using the composite strategy and the treatment policy strategy for handling intercurrent events also had results that were consistent with the primary analysis. Overall, compared with placebo, treatment with rozanolixizumab resulted in consistently greater decreases from baseline in MG-ADL score at day 43 across all subgroups (Figure 2), except for the subgroups with low numbers of patients. The sponsor also reported data from a post hoc subgroup analysis of the ██ patients in the rozanolixizumab group and ██ patients in the placebo group who had 2 or more prior MG-specific therapies. From a mean (SD) baseline score of ███ █████ in the rozanolixizumab group and ███ █████ in the placebo group, the LS mean (SE) change from baseline to day 43 in MG-ADL scores with rozanolixizumab was ██████ ███████ and for placebo ██████ ███████ for an LS mean difference between groups of ██████ ██████ ███ ██████ ██ ███████.
With respect to MG-ADL response (i.e., at least a 2-point improvement), at day 43, there were 45 responders in the rozanolixizumab group (68.2%) and 19 in the placebo group (28.4%). The OR for rozanolixizumab versus placebo was 5.765 (95% CI, 2.100 to 14.882; P < 0.001) favouring rozanolixizumab. The absolute difference between groups in the proportion of patients with a response was 39.8% (95% CI, 24.2 to 55.4%) (Table 15). ███ ███████ ████ ████████ ████ ████ █ ████ ███ ████████ ████████ ██ ███ ██ ████████ ██ ███ ███████████████ █████ ███ ██ ████████ ██ ███ ███████ █████ ███ ███ █ ██ ████ █████ ███████████ ██████████ ██ ███ ███ ███ ██████ ██ ██████████ ██ ███ ███████████████ █████ ███ ██ ████████ ███████ ███ ██ ███ ███████ █████ ███ █ ████████ ████████.
Figure 2: Forest Plot of MG-ADL Change From Baseline to Day 43 (Visit 10) by Subgroups (Randomized Set)
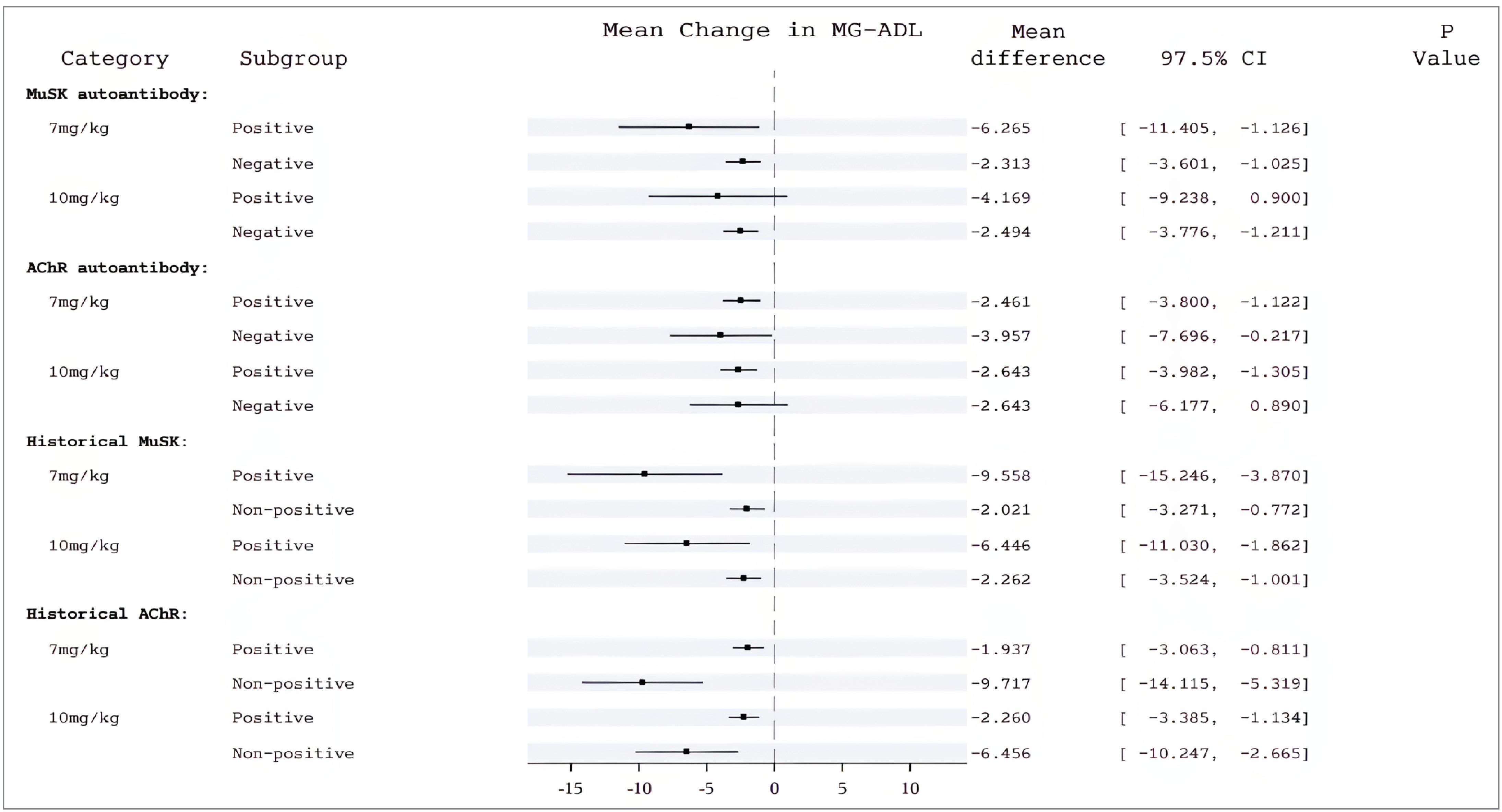
AChR = acetylcholine receptor; CI = confidence interval; MG-ADL = Myasthenia Gravis Activities of Daily Living; MuSK = muscle-specific tyrosine kinase.
Source: Sponsor Summary of Clinical Evidence.24
QMG Score
QMG scores range from 0 to 39, with higher scores indicating more severe impairment. From mean baseline scores of 15.4 (SD = 3.7) in the rozanolixizumab group and 15.8 (SD = 3.5) in the placebo group, the LS mean changes from baseline were –5.598 (SE = 0.679) in the rozanolixizumab group and –1.915 (SE = 0.685) in the placebo group (Table 15). The between-group difference in change from baseline was –3.483 (95% CI, 5.614 to –1.584; P < 0.001), favouring rozanolixizumab. Results for the sensitivity analyses using the J2R (for imputation of missing scores), the composite strategy, and the COVID-19 composite strategy were consistent with those for the overall analysis of change from baseline to day 43 in QMG score.
The sponsor also reported results for subgroup analyses of QMG scores by baseline antibody status for the 59 patients in the rozanolixizumab group and the 51 patients in the placebo group who were AChR antibody-positive and for the 4 patients in the rozanolixizumab group and 7 patients in the placebo group who were MuSK antibody-positive. In the subgroup of patients who were AChR antibody-positive, the LS mean changes from baseline to day 43 were –4.660 (SE = 1.605) in the rozanolixizumab group and –1.189 (SE = 1.575) in the placebo group for an LS mean difference between groups of –3.471 (97.5% CI, –5.433 to –1.510). In the subgroup of patients who were MuSK antibody-positive, the LS mean changes from baseline to day 43 were –10.276 (SE = 3.490) in the rozanolixizumab group and –2.662 (SE = 2.710) in the placebo group, for an LS mean difference between groups of –7.614 (97.5% CI, –16.291 to 1.062). The sponsor also reported data from a post hoc subgroup analysis of the ██ patients in the rozanolixizumab group and ██ patients in the placebo group who had 2 or more prior MG-specific therapies. From a mean (SD) baseline score of ████ █████ in the rozanolixizumab group and ████ █████ in the placebo group, the mean (SD) changes from baseline to day 43 in QMG scores with rozanolixizumab were – █████ ███████ and for placebo ██████ ███████ for a LS mean difference between groups of ██████ ██████ ███ ██████ ██ ████████.
MG-QoL15r Score
MG-QoL15r scores range from 0 to 30, with higher scores indicating a more severe impact of disease on HRQoL. From mean baseline scores of 15.7 (SD = 7.7) in the rozanolixizumab group and 15.0 (SD = 6.4) in the placebo group, the LS mean changes from baseline were –4.4 (SE = 0.9) in the rozanolixizumab group and –2.1 (SE = 1.0) in the placebo group (Table 15). The LS mean between-group difference in change from baseline was –2.245 (95% CI, –4.096 to –0.394), favouring rozanolixizumab.
MGC Score
MGC scores range from 0 to 50, with higher scores indicating more severe impairment. The LS mean changes from baseline were –5.23 (SE = 0.828) in the rozanolixizumab group and –1.47 (SE = 0.722) in the placebo group. The LS mean between-group difference in change from baseline was –3.901 (95% CI, –6.634 to –1.245; P < 0.001), favouring rozanolixizumab (Table 15). Results of sensitivity analyses using the J2R approach (for imputation of missing scores), the composite strategy (for handling intercurrent events), and the COVID-19 hypothetical strategy were consistent with those of the overall analysis.
Table 15: Summary of Key Efficacy Results From Studies Included in the Systematic Review
Variable | Rozanolixizumab 7 mg/kg (N = 66) | Placebo (N = 67) |
|---|---|---|
Change from baseline to day 43 in MG-ADL score | ||
Number of patients contributing to the analysis | 65 | 62 |
LS mean (SE) | –3.22 (0.480) | –0.65 (0.363) |
Treatment group difference vs. control (95% CI) | –2.586 (–4.091 to –1.249) | |
P value | < 0.001 | |
MG-ADL responders at day 43 | ||
Number of patients contributing to the analysis | 66 | 67 |
n (%) | 45 (68.2) | 19 (28.4) |
OR vs placebo (95% CI) | 5.765 (2.100 to 14.882) | |
RD vs. placebo (95% CI), % | 39.8 (24.2 to 55.4) | |
P valuea | < 0.001 | |
Change from baseline to day 43 in QMG score | ||
Number of patients contributing to the analysis | 65 | 62 |
LS mean (SE) | –4.22 (0.574) | –0.89 (0.525) |
Treatment group difference vs. control (95% CI) | –3.483 (–5.614 to –1.584) | |
P value | < 0.001 | |
Change from baseline to day 43 in MGC score | ||
Number of patients contributing to the analysis | 65 | 62 |
LS mean (SE) | –5.23 (0.828) | –1.47 (0.722) |
Treatment group difference vs. control (95% CI) | –3.901 (–6.634 to –1.245) | |
P value | < 0.001 | |
Change from baseline in MG-QoL15r score | ||
Number of patients contributing to the analysis | 66 | 67 |
LS mean (SE) | –4.4 (0.9) | –2.1 (1.0) |
Treatment group difference vs. control (95% CI) | –2.245 (–4.096 to –0.394) | |
P value | NR | |
CI = confidence interval; LS = least squares; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; NR = not reported; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; RD = risk difference; SE = standard error; vs. = versus.
aThis P value has not been adjusted for multiple testing.
Source: Sponsor’s Clinical Study Report for the MycarinG trial (NCT03971422), Section 8.54
Harms
Refer to Table 16 for harms data.
Adverse Events
Overall, the number of study patients who experienced at least 1 AE was 52 patients (81.3%) in the rozanolixizumab group and 45 patients (67.2%) in the placebo group (Table 16). The most common AEs (experienced by 10% of patients or more in either group), for rozanolixizumab versus placebo, respectively, were diarrhea (25.0% versus 13.4%), pyrexia (12.5% versus 1.5%), and headache (45.3% versus 19.4%).
Serious AEs
SAEs were reported in a comparable number of study patients in the rozanolixizumab group (5 patients [7.8%]) and placebo group (6 patients [9.0%]) (Table 16). The only serious TEAEs reported in more than 1 study patient per treatment group was MG crisis, which occurred in 0 patients in the rozanolixizumab group and in 2 patients (3.0%) in the placebo group.
Withdrawals Due to AEs
Incidences of AEs leading to permanent discontinuation of the study drug were reported in similar proportions between groups (i.e., 2 patients [3.1%] in the rozanolixizumab group and 2 patients [3.0%] in the placebo group) (Table 16).
Mortality
There were no deaths in the study.
Notable Harms
Infection was identified as a notable harm for this review. Infections and infestations occurred in 10 patients (15.6%) in the rozanolixizumab group and 13 patients (19.4%) in the placebo group (Table 16).
Table 16: Summary of Harms Results From Studies Included in the Systematic Review
Adverse events | Rozanolixizumab (N = 64) | Placebo (N = 67) |
|---|---|---|
Adverse events | ||
Patients with > 1 adverse event | 52 (81.3) | 45 (67.2) |
Specific events that occurred in ≥ 1 patient, any group, n (%) | ||
Gastrointestinal disorders | 21 (32.8) | 16 (23.9) |
Diarrhea | 16 (25.0) | 9 (13.4) |
Nausea | 5 (7.8) | 5 (7.5) |
Vomiting | 2 (3.1) | 1 (1.5) |
Abdominal pain upper | 3 (4.7) | 2 (3.0) |
General disorders and administration-site conditions | 16 (25.0) | 13 (19.4) |
Pyrexia | 8 (12.5) | 1 (1.5) |
Chest pain | 2 (3.1) | 0 |
Injury, poisoning, and procedural complications | 5 (7.8) | 5 (7.5) |
Fall | 0 | 3 (4.5) |
Musculoskeletal and connective tissue disorders | 15 (23.4) | 9 (13.4) |
Arthralgia | 4 (6.3) | 2 (3.0) |
Myalgia | 2 (3.1) | 1 (1.5) |
Muscle spasms | 3 (4.7) | 1 (1.5) |
Nervous system disorders | 37 (57.8) | 21 (31.3) |
Headache | 29 (45.3) | 13 (19.4) |
Myasthenia gravis | 3 (4.7) | 3 (4.5) |
Somnolence | 1 (1.6) | 3 (4.5) |
Renal and urinary disorders | 1 (1.6) | 2 (3.0) |
Respiratory, thoracic, and mediastinal disorders | 4 (6.3) | 4 (6.0) |
Oropharyngeal pain | 0 | 1 (1.5) |
Skin and subcutaneous tissue disorders | 6 (9.4) | 4 (6.0) |
Rash | 3 (4.7) | 0 |
Vascular disorders | 7 (10.9) | 1 (1.5) |
Hypertension | 5 (7.8) | 0 |
Serious adverse events | ||
Patients with ≥ 1 serious adverse event, n (%) | 5 (7.8) | 6 (9.0) |
Gastritis | 1 (1.6) | 0 |
Vomiting | 1 (1.6) | 0 |
COVID-19 pneumonia | 0 | 1 (1.5) |
Thoracic vertebral fracture | 0 | 1 (1.5) |
Arthralgia | 1 (1.6) | 0 |
Muscular weakness | 0 | 1 (1.5) |
Myasthenia gravis | 1 (1.6) | 1 (1.5) |
Myasthenia gravis crisis | 0 | 2 (3.0) |
Seizure | 1 (1.6) | 0 |
Patients who stopped treatment due to adverse events, n (%) | ||
Patients who stopped treatment | 2 (3.1) | 2 (3.0) |
Joint-related signs and symptoms | 1 (1.6) | 0 |
Arthralgia | 1 (1.6) | 0 |
Headaches | 1 (1.6) | 0 |
Myasthenia gravis | 0 | 1 (1.5) |
Myasthenia gravis crisis | 0 | 1 (1.5) |
Mortality | ||
Patients who died | 0 | 0 |
Notable harms | ||
Infections and infestations | 10 (15.6) | 13 (19.4) |
Nasopharyngitis | 1 (1.6) | 3 (4.5) |
Oral herpes | 0 | 0 |
Urinary tract infection | 2 (3.1) | 4 (6.0) |
Source: Sponsor’s Summary of Clinical Evidence.24
Critical Appraisal
Internal Validity
The study used adequate methods for randomization and to maintain allocation concealment. There were some numerical differences between groups in baseline characteristics. Notable differences included baseline autoantibody status, given that there were fewer patients in the rozanolixizumab group who were MuSK antibody-positive than in the placebo group (6% versus 12%), and more patients in the rozanolixizumab group who were AChR antibody-positive than in the placebo group (85% versus 79%). Other baseline characteristics involving notable differences between groups included duration of disease, the proportion of patients who had experienced myasthenia crisis in the past, MGFA class at screening, and gender. There were also some differences in the background therapies used for MG, including the fact that there were fewer patients in the rozanolixizumab group versus the placebo group who had used or were using AChEIs (83% versus 91%) and more patients in the rozanolixizumab group compared to the placebo group who had used or were using IVIg or PLEX (18% versus 9%). The clinical expert consulted on this review did not believe that imbalances in these observed characteristics would systematically favour the rozanolixizumab group. Full prognostic balance is not likely to be reached with a sample size as small as this, despite an adequate randomization process; therefore, it seems likely that the imbalances are the result of chance. The small sample is expected, given the rare nature of the condition.
A matched placebo was used to maintain blinding throughout the study. Unblinding occurred only in the event of an emergency. While it is possible for patients or clinicians to become unblinded as a result of known harms, there is no evidence that this occurred. Exposure and adherence to rozanolixizumab and placebo were similar. Concomitant conventional therapies (aside from AChEIs and corticosteroids) were to remain stable during the 6-week treatment cycle. Few patients required rescue therapies during the 6-week treatment period (4.5% in the placebo group and 1.5% in the rozanolixizumab group). The data for these patients — and for those discontinuing the study medication due to AEs — were considered missing (2 patients per group) in the primary analysis estimand.
The sponsor selected a hierarchical testing procedure to account for multiplicity and appears to have followed this procedure. The multiple testing procedure was extensive, covering both doses of rozanolixizumab in the trial as well as several efficacy outcomes across the interim and final analysis. Multiplicity-adjusted 95% CIs were provided to align with the testing procedure. The outcomes of interest for this review that were not multiplicity controlled were the MG-ADL responder analysis and the change from baseline in MG-QoL15r. The findings for these outcomes are at increased risk of type I error (i.e., erroneously excluding the null hypothesis) and can be considered supportive of the other end points.
A large proportion of patients withdrew from the study; however, there was no clear difference in withdrawals between the rozanolixizumab (35%) and placebo (37%) groups. The vast majority of these withdrawals were due to patients being rolled into the extension due to use of rescue medications during the observation period. Therefore, these patients had completed the treatment phase and were assessed as part of the efficacy analysis. As a result, there are relatively few missing data for these efficacy outcomes. For example, for the primary outcome, change from baseline in MG-ADL, data were available for 127 patients out of an original intention-to-treat population of 133. Nevertheless, there were some missing data for the analysis of the primary outcome and other outcomes. For the primary outcome and other analyses of change from baseline, missing data were implicitly imputed under the MAR assumption. While the plausibility of this assumption cannot be verified, the proportion of missing data was small. In addition, numerous sensitivity analyses, including with different handling of intercurrent events (e.g., the J2R approach), supported the robustness of the primary analyses. For the analysis of MG-ADL response, patients with missing data (primarily due to use of rescue therapy or withdrawal due to AE) were considered non-responders. This is unlikely to have had a major impact, given that the frequency of these events was low and balanced across groups.
All outcome measures (MG-ADL, QMC, MGC, and MG-QoL15r) are validated in patients with MG. These outcomes were patient- or clinician-reported; however, the blinded nature of the trial and low likelihood of unblinding suggests that there is low risk of bias in the measurement of the outcomes.
Subgroup analyses were intended to demonstrate consistency across the study population. A post hoc subgroup of patients with 2 or more prior treatments was presented in the sponsor’s Summary of Clinical Evidence.24 The post hoc subgroup analyses have limitations. Examples include the risk of selective reporting (due to lack of prespecification); the possibility that the randomization is not fully upheld, given the lack of stratification by this variable; a reduction in the sample size; and lack of multiplicity control.
External Validity
The reimbursement request from the sponsor is to restrict coverage for rozanolixizumab to those patients whose symptoms persist despite treatment with conventional therapy, including AChEIs, corticosteroids, and/or NSISTs. Prior treatment with these conventional therapies does not appear to have been a requirement for enrolment in the pivotal trial; however, it appears that most patients (96%) had been exposed to prior gMG-specific therapy before the trial. If AChEIs are not included, then the percentage of patients with prior therapy drops to 77%. Nevertheless, despite there not being a requirement for prior MG-directed therapy, most patients had been exposed to treatment for their condition. The clinical expert consulted by the review team suggested that the eligibility criteria related to the MG-ADL and QMG score would be sufficient to demonstrate that the patients had persistent symptoms. What is not known is what percentage of these patients would be considered treatment-refractory, because that population was neither sought nor defined in the pivotal trial. The sponsor did provide a post hoc subgroup analysis that reported on the subgroup of patients who had at least 2 prior MG-specific therapies; however, post hoc analyses are useful for generating hypotheses, not for testing them. The reimbursement request includes patients with either AChR or MuSK antibodies. The population of patients who were MuSK antibody-positive was small (n = 12 in the groups assessed), which reduces the confidence in the results for this population. However, the proportion of patients who were MuSK antibody-positive aligned with the disease prevalence in the MG population, and subgroup analyses suggested that the effect seen in the full population was consistent in this group.
Outcomes were measured at 43 days, which is a relatively short time period. The longer-term treatment effect can be assessed only within the OLE period. Moreover, the MycarinG trial did not provide evidence versus other available treatments for gMG.
GRADE Summary of Findings and Certainty of the Evidence
Methods for Assessing the Certainty of the Evidence
For the pivotal studies and RCTs identified in the sponsor’s systematic review, GRADE was used to assess the certainty of the evidence for the outcomes considered most relevant to inform the CDA-AMC expert committee deliberations, and a final certainty rating was determined, as outlined by the GRADE Working Group:22,23
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. We use the word “likely” for evidence of moderate certainty (e.g., “X intervention likely results in Y outcome”).
Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. We use the word “may” for evidence of low certainty (e.g., “X intervention may result in Y outcome”).
Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. We describe evidence of very low certainty as “very uncertain.”
Following the GRADE approach, evidence from RCTs started as high-certainty evidence and could be rated down for concerns related to study limitations (which refers to internal validity or risk of bias), inconsistency across studies, indirectness, imprecision of effects, or publication bias.
When possible, certainty was rated in the context of the presence of an important (nontrivial) treatment effect; if this was not possible, certainty was rated in the context of the presence of any treatment effect (i.e., the clinical importance is unclear). In all cases, the target of the certainty of evidence assessment was based on the point estimate and where it was located relative to the threshold for a clinically important effect (when a threshold was available) or to the null. The target of the certainty of evidence assessment was the presence or absence of an important effect based on thresholds identified in the literature and supported by the clinical expert for the change from baseline to day 43 in MG-ADL and QMG scores; the presence or absence of an important effect based on thresholds informed by the clinical expert consulted for this review for MG-ADL responders; and the presence or absence of any (non-null) effect for the change from baseline to day 43 in MG-QoL15r scores and for notable harms (infections and infestations).
Results of GRADE Assessments
Table 2 presents the GRADE summary of findings for rozanolixizumab versus placebo.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Description of Studies
Two OLEs, the MG0004 trial (NCT04124965; data cut-off date of September 1, 2021) and the MG0007 trial (NCT04650854; data cut-off date of July 8, 2022) are summarized in this section. The MG0007 trial had been ongoing for approximately 1.5 years at the date of the data cut-off for the interim analysis. Results for the 7 mg/kg group only are summarized, given that the reimbursement request is for the rozanolixizumab 7 mg/kg dose.
MG0004 Trial
This was a phase III, multicentre, randomized, OLE of the MycarinG (MG0003) trial (NCT03971422) to investigate the long-term safety, tolerability, and efficacy of rozanolixizumab (weekly dosing regimen for 52 weeks) in adult patients with gMG who were experiencing moderate to severe symptoms and under consideration for IVIg or PLEX therapy, indicating a need for additional therapeutic intervention. Patients were randomized to 2 different treatment arms in a 1-to-1 ratio to receive SC rozanolixizumab (7 mg/kg or 10 mg/kg). For any patient who enrolled in the MG0004 trial, the final visit in the MycarinG trial (i.e., visit 14) served as the first visit in the MG0004 trial (i.e., visit 1). The primary safety end points were the occurrence of TEAEs and TEAEs leading to permanent withdrawal of study medication. Other safety end points included the occurrence of AEs requiring special monitoring (i.e., potential Hy’s law, defined as aspartate aminotransferase or alanine aminotransferase > 3 times the ULN, total bilirubin > 2 times the ULN, and alkaline phosphatase < 2 times the ULN, with no other explanation for the biochemical abnormality), vital signs, electrocardiogram assessments, and clinical laboratory findings.
MG0007 Trial
This was a phase III, 2-arm, randomized, OLE of the MycarinG trial to evaluate the long-term safety, tolerability, and efficacy of repeated 6-week treatment cycles of rozanolixizumab based on MG worsening in adult patients with gMG. Worsening of disease was defined as worsening of gMG symptoms (e.g., an increase of 2 points on the MG-ADL or 3 points on the QMG scale) between 2 consecutive visits. This OLE study replaced the MG0004 trial and provided the opportunity for patients who had participated in the MycarinG trial and MG0004 trial to benefit from long-term rozanolixizumab treatment based on their MG symptoms. Patients were randomized to receive an initial, fixed, 6-week treatment cycle of rozanolixizumab (7 mg/kg or 10 mg/kg) once weekly, followed by an observation period that began after the last dose of that treatment cycle. Eligible patients from the MG0004 trial who completed at least 6 scheduled visits in the treatment period could move directly into the observation period in the MG0007 trial. In the case of worsening MG symptoms, patients underwent another 6 weeks of treatment followed by an observation period. The dose could be adjusted to 7 mg/kg or 10 mg/kg at the beginning of each treatment cycle based on the investigator’s discretion.
Figure 3: Study Design for the MycarinG Trial (Study MG0003) and the Rollover Into the MG0004 Trial and MG0007 Trial
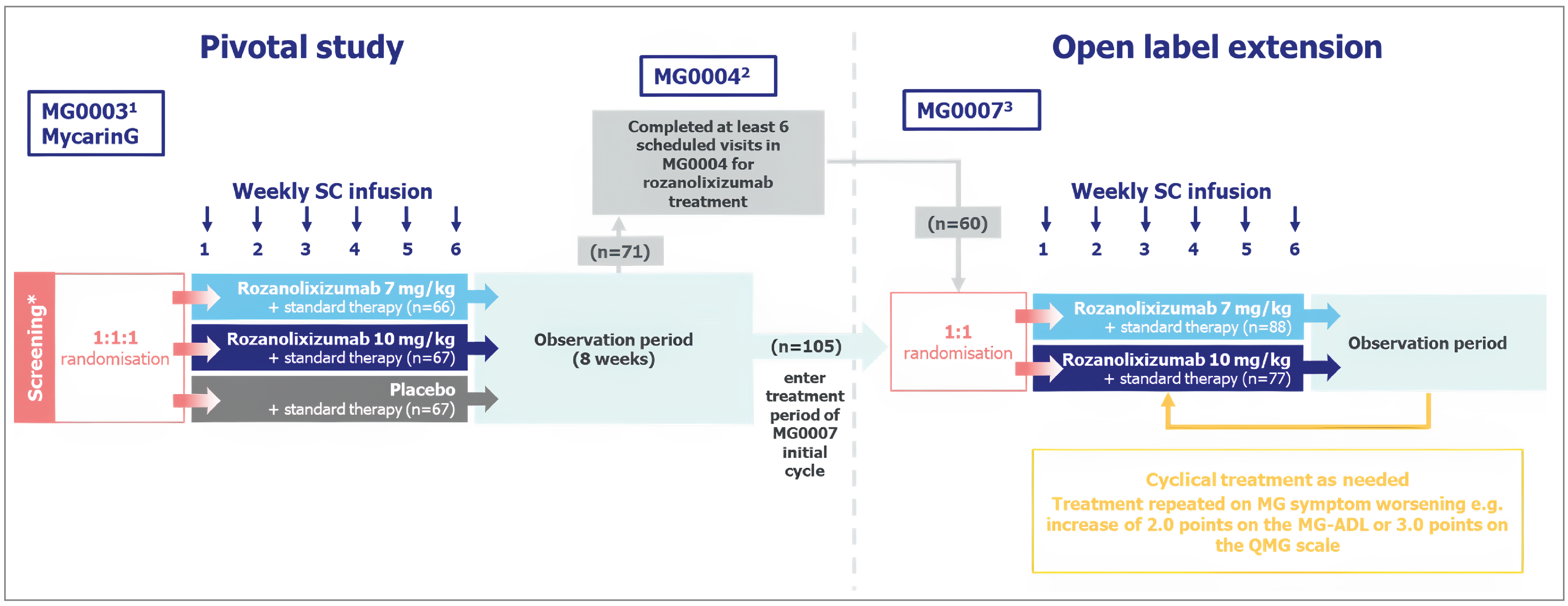
AChR = acetylcholine receptor; CS = corticosteroid; gMG = generalized myasthenia gravis; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MuSK = muscle-specific tyrosine kinase; NSIST = nonsteroid immunosuppressant; QMG = Quantitative Myasthenia Gravis; SC = subcutaneous.
Notes: Adults with moderate to severe gMG and autoantibodies against AChR or MuSK who required additional therapy.
Permitted background therapy included concomitant conventional treatment for gMG (i.e., standard therapy), such as CS and NSISTs. In MycarinG, patients were required to have been on concomitant conventional treatment for the previous 6 months and on a stable dosage for 2 months before baseline.
Sources: MycarinG study protocol;54 MG0004 study protocol;75 MG0007 study protocol.62
Populations
MG0004 Trial
Patients were eligible for enrolment in the MG0004 trial if they met the inclusion criteria for the MycarinG trial at the time of enrolment (refer to Table 5 for a list of inclusion and exclusion criteria). They must also have completed the observation period of the MycarinG trial or required (but not received) rescue therapy (i.e., IVIg or PLEX) during the observation period of the MycarinG trial. Key exclusion criteria included impending or myasthenic crisis and severe weakness affecting the oropharyngeal or respiratory muscles.
MG0007 Trial
Patients were eligible for enrolment in the MG0007 trial if they completed the MycarinG trial, required but did not receive rescue therapy during the observation period for the MycarinG trial, or completed at 6 visits in the MG0004 trial. Key exclusion criteria included impending or myasthenic crisis and severe weakness affecting the oropharyngeal or respiratory muscles.
Interventions
MG0004 Trial
Patients were randomized in a 1-to-1 ratio to receive 1 of 2 doses of rozanolixizumab (i.e., 7 mg/kg or 10 mg/kg) on a weekly basis over a 52-week treatment period. Rozanolixizumab was administered as an SC infusion. The specific dose levels of rozanolixizumab were administered based on patient body weight. After the 52-week treatment period, patients entered an 8-week observation period, with the maximum study duration per patient being 60 weeks.
Rescue therapy consisted of IVIg or PLEX. Patients who experienced disease worsening (e.g., an increase of 2 points on the MG-ADL scale or 3 points on the QMG scale between 2 consecutive visits) may have been considered for rescue therapy at the discretion of the investigator. If the patient received IVIg or PLEX as rescue therapy during the study, treatment with rozanolixizumab must have been discontinued or paused for a minimum of 2 weeks, but patients continued with visits as per the schedule of assessments, after which they may have continued to receive rozanolixizumab at the discretion of the investigator. This 2-week period may have been extended at the discretion of the investigator, but for no longer than 6 weeks. Following the temporary discontinuation of study medication, patients must have restarted at the same dose of rozanolixizumab as previously. Patients at a dose level of 7 mg/kg rozanolixizumab could have been restarted at 10 mg/kg rozanolixizumab at the discretion of the investigator.
MG0007 Trial
Patients were randomized in a 1-to-1 ratio to receive 1 of 2 doses of SC rozanolixizumab (i.e., 7 mg/kg or 10 mg/kg), similar to the MG0004 trial. Rozanolixizumab was administered as an SC infusion. The permitted concomitant medications (refer to Table 5) for the treatment of MG in the MG0007 trial were also similar to those permitted in the MG0004 trial.
Rescue therapy was given as per conventional therapy and at the discretion of the investigator. Patients who continued to experience moderate to severe symptoms despite treatment with rozanolixizumab may have been treated with IVIg, SCIg, PLEX or plasmapheresis, or IV corticosteroids (at a higher dose than the previous oral dose) as rescue therapy. Patients who were treated with rescue therapy were withdrawn from the MG0007 trial.
Outcomes
MG0004 Trial
The 2 primary safety end points were the occurrence of TEAEs and the occurrence of TEAEs leading to permanent withdrawal from the study. The secondary objective was to evaluate the long-term efficacy of rozanolixizumab in patients with gMG. Several secondary end points were assessed from baseline to each scheduled assessment. The results for relevant secondary end points, (changes in MG-ADL, MGC, and QMG scores) and the exploratory end point (changes in MG-QoL15r scores) are presented in this report.
MG0007 Trial
The 2 primary safety end points and other safety end points were the same as those in the MG0004 trial (mentioned previously). Several secondary efficacy end points were assessed from baseline to day 43 during each treatment cycle. Results of relevant secondary and exploratory end points, similar to the MG0004 trial, are presented in this report. Results of other relevant secondary and exploratory end points, such as patient-reported outcomes, MG-ADL responder rates, and treatment-free survival, can be found in Appendix 1 (Table 37, Table 38 and Table 39).
Statistical Analysis
MG0004 Trial
For continuous variables, descriptive statistics were presented, which included the number of patients with available measurements (i.e., n), mean, SD, median, minimum, and maximum. For categorical variables, the number and percentage of patients in each category were presented. Baseline values were defined as the last available values before or on the same date (and same time, if time was collected for the individual assessment) of the first administration of the study treatment in the MG0004 trial. Scheduled or unscheduled measurements could be used as the baseline value. If MG-ADL, MG-QoL15r, or QMG scores were missing, these were imputed with the average score across the remaining items at the specific visit. Missing MGC scores were not imputed. If 5 items or more were missing on the MG-QoL15r, a total score was not calculated.
MG0007 Trial
Continuous and categorical variables were analyzed similarly to the MG0004 trial. For safety analyses, data were summarized by dose levels of rozanolixizumab at the time of the event or measurement; for efficacy analyses, data were summarized by the dose first received in the study as well as by dose level of rozanolixizumab received in each treatment cycle. For analyses conducted by entire study, baseline values were defined as the last available values before or on the same date of the first administration of the study treatment; scheduled or unscheduled measurements could be used as the baseline value. For the analyses done by study cycle, baseline values were defined as the last available values before or on the same date (and same time, if time was collected for the individual assessment) of the first administration of the study treatment at each cycle (i.e., baseline [day 1]) value for that cycle. Missing data were handled similarly to the MG0004 trial.
Definitions of the analysis sets that were considered in the MG0004 trial and the MG0007 trial are presented in Table 17.
Results
Baseline Characteristics
MG0004 Trial
Out of a total of 71 patients, 35 patients were randomized to the rozanolixizumab 7 mg/kg group, with a mean age of 50.6 years (SD = 14.2) (Table 18). More than half were female. The mean weight and body mass index of patients in the rozanolixizumab 7 mg/kg group were 83.1 kg (SD = 23.4) and 28.3 kg/m2 (SD = 6.8), respectively. Of note, the number of patients in the body weight category of less than 50 kg was low (2 patients [5.7%]).
Table 17: Definitions of Analysis Sets in Study MG0004 and Study MG0007
Analysis set | Study MG0004 | Study MG0007 |
|---|---|---|
ES | Consisted of all patients who signed the informed consent | Consisted of all patients who signed the informed consent form |
RS | Consisted of all patients who were randomized, using the treatment assigned instead of the actual treatment received | NA |
FAS | NA | Consisted of all patients in the ES who were randomized in this study or in the MG0004 trial; patients who enrolled from the MG0004 trial utilized their last assigned dose level from the MG0004 trial as their dose in the MG0007 trial |
SS | Consisted of all randomized patients who received at least 1 dose of the study treatment in this study; analysis of this set was conducted according to the treatment patients actually received in the MG0004 trial, and was used for efficacy, demographic, PK, PD, and safety analyses | Consisted of all patients in the FAS who received at least 1 dose of the study treatment in the MG0007 trial |
ES = enrolled set; FAS = full analysis set; NA = not applicable; PD = pharmacodynamic; PK = pharmacokinetic; RS = randomized set; SS = safety set.
Source: Sponsor’s Summary of Clinical Evidence.24
MG0007 Trial
Out of a total of 157 patients, 79 patients were randomized to the 7 mg/kg group, with a mean age of 52.7 years (SD = 15.7) (Table 18). More than half were female. The mean weight and body mass index for patients in the rozanolixizumab 7 mg/kg group were 82.4 kg (SD = 22.3) and 28.0 kg/m2 (SD = 6.0), respectively. Of note, similar to the MG0004 trial, the number of patients in the body weight category of less than 50 kg was low (4 patients [5.1%]).
Table 18: Summary of Baseline Demographics and Disease Characteristics in Study MG0004 (Randomized Set) and Study MG0007 (Safety Set)
Characteristics | Study MG0004 | Study MG0007 |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 35) | Rozanolixizumab 7 mg/kg (N = 79) | |
Age (years), mean (SD) | 50.6 (14.2) | 52.7 (15.7) |
Female, n (%) | 19 (54.3) | 44 (55.7) |
Weight (kg), mean (SD) | 83.1 (23.4) | 82.4 (22.3) |
BMI (kg/m2), mean (SD) | 28.3 (6.8) | 28.0 (6.0) |
Body weight categories (kg), n (%) | ||
< 50 | 2 (5.7) | 4 (5.1) |
≥ 50 to < 70 | 8 (22.9) | 20 (25.3) |
≥ 70 to < 100 | 17 (48.6) | 36 (45.6) |
≥ 100 | 8 (22.9) | 19 (24.1) |
Racial group, n (%) | ||
Asian | 4 (11.4) | 8 (10.1) |
Black | 2 (5.7) | 1 (1.3) |
Native Hawaiian or other Pacific Islander | 0 | 0 |
White | 17 (48.6) | 53 (67.1) |
Missinga | 12 (34.3) | 17 (21.5) |
Regions, n (%) | ||
Asia (excluding Japan) | 0 | 6 (7.6) |
Europe | 15 (42.9) | 45 (57.0) |
Japan | 4 (11.4) | 2 (2.5) |
North America | 16 (45.7) | 26 (32.9) |
MG-ADL score, mean (SD) | 8.4 (3.6) | 15.4 (7.6) |
MGC score, mean (SD) | 15.0 (7.3) | 15.4 (7.6) |
QMG score, mean (SD) | 15.2 (5.1) | 14.4 (5.1) |
MG-specific autoantibody at OLE baseline, n (%) | ||
AChR-positive | 26 (74.3) | 62 (78.5) |
MuSK-positiveb | 5 (14.3) | 7 (8.9) |
Total IgG (g/L), mean (SD) | 9.1 (3.2) | 8.8 (2.8) |
AChR = acetylcholine receptor; BMI = body mass index; IgG = immunoglobulin G; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MuSK = muscle-specific tyrosine kinase; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Note: All characteristics presented in this table refer to values at the Study MG0004 baseline or Study MG0007 baseline.
aRace and ethnicity were prohibited to collect in Canada and France.
bPatients in the MG0007 trial were considered to be MuSK-antibody negative if MuSK was less than 0.05 nmol/L at the baseline visit.
Sources: MG0004 Clinical Study Report (2022), Table 3.1.1 and Table 3.1.2;75 MG0007 Clinical Study Report (2022), Table 3.1.1.3 and Table 3.1.2.62
Patient Disposition
MG0004 Trial
Seventy-one patients in the MG0004 trial entered from the MycarinG trial, almost all during the COVID-19 pandemic (i.e., 63 patients [88.7%]). Of these, 35 patients were randomized to the rozanolixizumab 7 mg/kg group, and 36 patients were randomized to the 10 mg/kg group (refer to Table 19) (data not presented for 10 mg/kg). Overall, 5 patients (14.3%) completed the study. Most patients (29 patients [82.9%]) permanently discontinued the study during the COVID-19 pandemic. One patient (2.9%) permanently discontinued the study due to a TEAE before the COVID-19 pandemic, and 2 patients (5.7%) permanently discontinued due to TEAEs during the pandemic. Most permanently discontinued the study to transition to the MG0007 study (25 patients [71.4%] in the 7 mg/kg group). One patient (2.9%) permanently discontinued the study with the primary reason considered as patient choice.
MG0007 Trial
All 165 patients in the FAS entered the MG0007 trial during the COVID-19 pandemic. Of these, 88 patients were assigned to the rozanolixizumab 7 mg/kg group (52 patients from the MycarinG trial and 36 patients from the MG0004 trial), and 77 patients were assigned to the rozanolixizumab 10 mg/kg group (53 patients from the MycarinG trial and 24 patients from the MG0004 trial) in cycle 1. Overall, 157 patients received rozanolixizumab in their first cycle: 79 patients received 7 mg/kg, and 78 patients received 10 mg/kg (data not presented for 10 mg/kg) (refer to Table 19). At the data cut-off date of July 8, 2022, no patients had completed the study, which was ongoing for most. A total of 16 patients (20.3%) treated with rozanolixizumab permanently discontinued the study during the COVID-19 pandemic; the most common reasons for study discontinuation were TEAEs (in 8 patients [10.1%]), followed by “withdrawal by patient” (in 5 patients [6.3%]).
Exposure to Study Treatments
MG0004 Trial
The mean duration of the study medication by first dose received was 22.93 weeks (Table 20). All patients in the safety set received at least 2 infusions. The mean number of infusions received was 21.7 (SD = 13.0). There was no treatment nonadherence, incorrect treatment, or incorrect dosing. Beyond 18 weeks, the numbers of patients decreased steadily; this was mainly due to the 53 patients (74.6%) who discontinued the study to transition to the MG0007 trial. More than 50% of the patients had greater than 3 months of exposure (27 patients out of 35 patients [77.1%]). In the rozanolixizumab 7 mg/kg group, 5 patients of 35 patients switched to 10 mg/kg (patients who switched doses for a single week only were not counted); of those, 2 patients subsequently switched back to 7 mg/kg. Ten patients in the 7 mg/kg group had an exposure of greater than 200 days; 1 patient who switched from 7 mg/kg to 10 mg/kg had an exposure of greater than 200 days. Taking the dose changes during the study into account, exposure to the 7 mg/kg dose was higher than exposure to the 10 mg/kg dose, and as the study continued, more patients received the 7 mg/kg dose.
Disposition | Study MG0004 | Study MG0007 |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 35) | Rozanolixizumab 7 mg/kg (N = 79) | |
Started study,a n (%) | 35 (100) | 79 (100) |
Pre–COVID-19 | 4 (11.4) | 0 |
During COVID-19 | 31 (88.6) | 79 (100) |
Completed study,b n (%) | 5 (14.3) | 0c |
Pre–COVID-19 | 0 | NA |
During COVID-19 | 5 (14.3) | 0 |
Permanently discontinued from the study, n (%) | 30 (85.7) | 16 (20.3) |
Pre–COVID-19, n (%) | 1 (2.9) | NA |
AEs | 1 (2.9) | NA |
During COVID-19, n (%) | 29 (82.9) | 16 (20.3) |
AEs | 2 (5.7) | 8 (10.1) |
Lack of efficacy | 0 | 1 (1.3) |
Lost to follow-up | 0 | 1 (1.3) |
Withdrawal by patient | 1 (2.9) | 5 (6.3) |
Other | 26 (74.3) | 1 (1.3) |
Transitioned to the MG0007 trial, n (%) | 25 (71.4) | NA |
Patient received rescue medication, n (%) | 0 | 1 (1.3) |
AE = adverse event; NA = not applicable.
Note: The COVID-19 period was based on the start, completed, and discontinuation dates relative to the pandemic cut-off date (i.e., start date of March 20, 2020). For the MG0004 trial, the study extended from the before–COVID-19 period to the during–COVID-19 period; a post–COVID-19 period did not apply to the MG0004 trial.
aDefined as signing informed consent.
bIn the MG0004 trial, this was defined as having completed the treatment and observation period; in the MG0007 trial, this was defined as having completed all phases of the study, including the observation period and the end-of-study visit.
cAt the data cut-off date of July 8, 2022 (as per the MG0007 trial interim Clinical Study Report), no patients had completed the study; the majority of patients were ongoing.
Sources: MG0004 Clinical Study Report (2022), Table 1.3.1;75 MG0007 Clinical Study Report (2022), Table 1.3.1.1.62
In the rozanolixizumab 7 mg/kg group, 4 patients (11.4%) received immunoglobulins as rescue medication; of these 4 patients, 1 rolled over into the MG0007 trial, 2 discontinued due to TEAEs, and 1 discontinued due to pregnancy. No patients in the MG0004 trial required a PLEX procedure during the study.
MG0007 Trial
Overall, 545 treatment cycles in 157 patients were included in the safety set. The median number of treatment cycles was 3.0 (range, 1 to 8), while the average number of treatment cycles per patient year was 4.31. A total of 79 patients (50.3%) received rozanolixizumab 7 mg/kg in cycle 1 (Table 20). Of these, 18 patients (22.8%) had only 1 treatment cycle, and 43 patients (54.4%) continued to receive rozanolixizumab 7 mg/kg in subsequent cycles. Sixteen patients (20.5%) switched to rozanolixizumab 7 mg/kg in subsequent cycles (5 patients switched at cycle 2; 3 patients switched at cycle 4; 2 patients switched at cycle 5; 2 patients switched at cycle 7; 3 patients switched at cycle 2 and switched back at cycle 3; and 1 patient switched at cycle 3 and switched back at cycle 4).
All patients who received rescue therapies discontinued the MG0007 trial. Five patients (6.3%) in the rozanolixizumab 7 mg/kg group received rescue medication (4 patients received immunoglobulins [1 of whom continued treatment with efgartigimod alfa] and 1 patient received methylprednisolone sodium succinate); 2 patients (2.5%) required a rescue procedure (i.e., PLEX).
Table 20: Patient Exposure To Rozanolixizumab in Study MG0004 (Safety Set) and Study MG0007 (Safety Set)
Rozanolixizumab exposure | Study MG0004 | Study MG0007 |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 35) | Rozanolixizumab 7 mg/kg (N = 79) | |
Duration (weeks),a mean (SD) | 22.93 | 16.68 (9.804) |
# of treatment cycles, median | NAb | 3.0 |
# of treatment cycles per patient per year | NAb | 4.22 |
# of infusions received,b mean (SD) | 21.7 (13.0) | 19.0 (11.21) |
≥ 3 months’ exposure | 27 (77.1) | 49 (62.0) |
≥ 6 months’ exposure | 13 (37.1) | 23 (29.1) |
≥ 9 months’ exposure | 6 (17.1) | 0 |
NA = not applicable; SD = standard deviation.
aDuration of rozanolixizumab by first dose received.
bRozanolixizumab was not administered in cycles; it was administered on a weekly basis over a 52-week treatment period. At the end of the 52-week treatment period in the MG0004 trial, study patients entered an 8-week observation period.
Sources: Sponsor’s Clinical Study Report for MG0004, Table 5.1.2;75 Clinical Study Report for MG0007, Table 5.1.2, Table 5.1.3.62
Efficacy
Change in MG-ADL
MG0004 Trial
Changes from baseline in MG-ADL score showed a stable trend up to week 33; participant numbers steadily declined over time. The maximum mean reduction from baseline to week 33 was 3.1 points (week 13; n = 30) for the rozanolixizumab 7 mg/kg group (Table 21). When assessed by autoantibodies subgroup, a consistent reduction from baseline in MG-ADL scores was observed in patients who were MuSK antibody-positive up to week 25. The greatest reduction (improvement) from baseline was 2.4 points, which was observed at week 5 (n = 5). The smallest reductions (improvements) from baseline were 1.6 points, observed at week 9 (n = 5) and sustained at week 13 (n = 5), and 1.3 points at week 21 (n = 3). For patients who were AChR antibody-positive, the greatest reduction (improvement) from baseline up to week 29 was 3.4 points (n = 25; week 13). From week 29 to 52, there was a consistent change (improvement) in MG-ADL scores from baseline, ranging from –4.2 points (week 37; n = 5) to –2.0 points (week 49; n = 3).
MG0007 Trial
Baseline MG-ADL scores and changes from baseline to day 43 in MG-ADL scores for the 6 treatment cycles are summarized in Table 22. Baseline values were defined as the last available value before or on the same date of the first administration of the IMP at each cycle (i.e., baseline [day 1]) value for that cycle. The number of participants declined across cycles, from 79 at cycle 1 to 11 at cycle 6. Within each cycle, the mean change from baseline ranged from –3.0 points to –4.3 points, depending on the cycle. When assessed by antibody subgroup, a consistent reduction from baseline in MG-ADL scores was observed at day 43, with repeated cyclic treatments for patients who were MuSK antibody-positive (n = 8; cycles 1 to 4) and for those who were AChR antibody-positive (n = 62; cycles 1 to 6); however, sample sizes steadily declined within each cycle. For patients who were MuSK antibody-positive, the mean change from baseline ranged from –6.5 points (n = 8; cycle 1) to –3.8 points (n = 3; cycle 3). For patients who were AChR antibody-positive, the mean change from baseline ranged from –4.0 points (n = 6; cycle 6) to –2.8 points (n = 41; cycle 2).
Change in QMG
MG0004 Trial
Changes from baseline showed a stable trend over time to week 52; study participant numbers steadily declined (Table 21). The maximum mean reductions from baseline to week 29 were 5.4 points for the subgroup of patients who were AChR antibody-positive (week 29; n = 11) and 6.0 points for the subgroup of patients who were MuSK antibody-positive (week 25; n = 3).
MG0007 Trial
Baseline QMG scores and changes from baseline at day 43 in QMG scores for the 6 treatment cycles are summarized in Table 22. Baseline values were defined as the last available value before or on the same date of first administration of IMP at each cycle value (i.e., [day 1]) for that cycle. The sample size declined from 79 at cycle 1 to 11 at cycle 6. The mean change from baseline ranged from –4.1 points to –6.4 points across cycles. Consistent improvements in QMG scores were observed with repeated cyclic treatment from baseline to day 43 when assessed for participants who were MuSK antibody-positive (cycles 1 to 4) and AChR antibody-positive (cycles 1 to 5).
Change in MGC
MG0004 Trial
Changes from baseline showed a consistent trend to week 52; study participant numbers declined steadily over time (Table 21). A consistent change from baseline to weeks 21 and 29 was observed when assessed by MuSK and AChR antibodies, respectively. The maximum mean reduction from baseline to week 29 was 7.0 points (week 25; n = 15) for patients who were AChR antibody-positive. The maximum mean reduction from baseline to week 25 was 3.6 points (week 5; n = 5) for patients who were MuSK antibody-positive.
MG0007 Trial
Baseline MGC scores and changes from baseline to day 43 in MGC scores for the 6 treatment cycles are summarized in Table 22. Baseline values were defined as the last available value before or on the same date of first administration of IMP at each cycle (i.e., baseline value [day 1]) for that cycle. The sample size declined over time, from 79 patients at cycle 1 to 11 patients at cycle 4. The mean change from baseline ranged from –6.1 points to –9.6 points across cycles. A consistent improvement in MGC scores was observed from baseline to day 43, with repeated cyclic treatment when assessed by antibody subgroups.
Change in MG-QoL15r
MG0004 Trial
Baseline QMG scores and changes from baseline to day 43 in QMG scores for the 6 treatment cycles are summarized in Table 22. The mean MG-QoL15r score at baseline was 14.4 for the rozanolixizumab 7 mg/kg group. An improvement in HRQoL was observed. The maximum mean change from baseline to week 33 was –5.1 points (week 21; n = 20).
MG0007 Trial
Quality of life for patients with MG was an exploratory outcome. The sample size declined over time, from 79 patients at cycle 1 to 11 patients at cycle 4. The mean change from baseline ranged from –2.2 points to –6.1 points across cycles (Table 22).
Table 21: Change in MG-ADL, QMG, MGC, and MG-QoL15r Scores From Baseline Over Time for Patients in the Rozanolixizumab 7 mg/kg Group (Safety Set) — Study MG0004
Change from baseline to treatment week | Change in MG-ADL (N = 35) | Change in QMG (N = 35) | Change in MGC (N = 35) | Change in MG-QoL15r (N = 35) | |
|---|---|---|---|---|---|
From baseline to treatment week 13 | n | 30 | 30 | 30 | 28 |
Mean (SD) | –3.1 (3.4) | –2.6 (4.2) | –4.8 (5.5) | –4.1 (5.0) | |
From baseline to treatment week 21 | n | 20 | 20 | 20 | 20 |
Mean (SD) | –3.0 (3.4) | –4.0 (4.7) | –5.3 (6.9) | –5.1 (5.3) | |
From baseline to treatment week 29 | n | 13 | 13 | 13 | 12 |
Mean (SD) | –2.8 (2.1) | –5.4 (3.6) | –5.1 (5.5) | –3.9 (4.3) | |
From baseline to treatment week 33 | n | 10 | 10 | 9 | 7 |
Mean (SD) | –3.0 (2.8) | –4.9 (4.8) | –4.6 (5.1) | –3.7 (2.4) | |
From baseline to treatment week 52 | n | 5 | 5 | 5 | NA |
Mean (SD) | –3.8 (3.1) | –4.6 (2.9) | –2.6 (1.3) | NA | |
From baseline to observation week 60 | n | 7 | 7 | 7 | 6 |
Mean (SD) | –0.3 (2.1) | –0.9 (2.4) | 1.7 (3.7) | –2.0 (4.0) | |
MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Garvis Quality of Life; NA = not applicable; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Note: Not all time points are shown. Baseline was defined as the last available value before or on the same date of the first administration of the study treatment in Study MG0004. Total MG-ADL score ranged from 0 to 24, with a higher score indicating more severe disability. QMG scores ranged from 0 to 39, with a higher score indicating more severe disability. MGC scores ranged from 0 to 50, with a higher score indicating more severe disease.
Source: Sponsor’s MG0004 Clinical Study Report (2022), tables 6.1.1, 6.2.1, 6.3.1, and 6.7.1.75
Table 22: Change in MG-ADL, QMG, MGC, and MG-QoL15r Scores From Baseline to Day 43 During Each Treatment Cycle for the Rozanolixizumab 7 mg/kg Group (Safety Set) — Study MG0007
Change from baseline to day 43 for various patient-reported outcomes | ||||||
|---|---|---|---|---|---|---|
Details | Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 5 | Cycle 6 |
Change in MG-ADL scores | ||||||
Sample size at baseline | N = 79 | N = 54 | N = 41 | N = 31 | N = 24 | N = 11 |
Baseline, mean | 8.4 | 7.9 | 8.0 | 7.6 | 8.2 | 9.1 |
Baseline, SD | 4.2 | 3.6 | 4.0 | 3.1 | 2.6 | 4.1 |
Baseline, median | 8.0 | 7.0 | 6.0 | 7.0 | 7.5 | 9.0 |
Baseline, minimum to maximum | 0 to 17 | 1 to 19 | 2 to 20 | 3 to 14 | 4 to 14 | 3 to 18 |
Sample size at day 43 | N = 73 | N = 50 | N = 35 | N = 29 | N = 16 | N = 7 |
Mean change from baseline to day 43 | –3.6 | –3.0 | –3.4 | –4.2 | –3.3 | –4.3 |
Median change from baseline to day 43 | –3.0 | –3.0 | –3.0 | –3.0 | –3.0 | –5.0 |
Change in QMG scores | ||||||
Sample size at baseline | N = 79 | N = 54 | N = 40 | N = 31 | N = 24 | N = 11 |
Baseline, mean | 14.4 | 14.7 | 14.8 | 15.0 | 14.6 | 15.5 |
Baseline, SD | 5.1 | 4.5 | 6.2 | 5.3 | 4.2 | 6.9 |
Baseline, median | 14.0 | 14.0 | 14.5 | 16.0 | 16.0 | 17.0 |
Baseline, minimum to maximum | 3 to 24 | 6 to 27 | 3 to 39 | 4 to 25 | 7 to 21 | 5 to 29 |
Sample size at day 43 | N = 72 | N = 49 | N = 35 | N = 29 | N = 16 | N = 7 |
Mean change from baseline to day 43 | –4.4 | –4.1 | –5.1 | –5.9 | –4.3 | –6.4 |
Median change from baseline to day 43 | –3.5 | –3.0 | –4.0 | –5.0 | –4.2 | –8.0 |
Change in MGC scores | ||||||
Sample size at baseline | N = 79 | N = 54 | N = 40 | N = 31 | N = 24 | N = 11 |
Baseline, mean | 15.4 | 14.7 | 15.4 | 14.1 | 15.0 | 17.4 |
Baseline, SD | 7.6 | 6.6 | 8.0 | 6.1 | 5.3 | 7.9 |
Baseline, median | 15.0 | 13.0 | 25.0 | 14.0 | 16.0 | 18.0 |
Baseline, minimum to maximum | 2 to 34 | 1 to 34 | 5 to 42 | 5 to 29 | 5 to 26 | 7 to 34 |
Sample size at day 43 | N = 72 | N = 50 | N = 35 | N = 29 | N = 16 | N = 7 |
Mean change from baseline to day 43 | –7.3 | –6.1 | –7.0 | –7.4 | –5.7 | –9.6 |
Median change from baseline to day 43 | –6.5 | –7.0 | –7.0 | –6.0 | –7.0 | –10.0 |
Change in MG-QoL15r scores | ||||||
Sample size at baseline | N = 79 | N = 54 | N = 41 | N = 31 | N = 24 | N = 11 |
Baseline, mean | 14.9 | 12.9 | 14.1 | 13.5 | 16.4 | 18.8 |
Baseline, SD | 6.8 | 7.0 | 7.4 | 7.0 | 7.8 | 6.9 |
Baseline, median | 15.5 | 12.0 | 12.0 | 13.0 | 17.5 | 20.0 |
Baseline, minimum to maximum | 0 to 29 | 1 to 29 | 2 to 30 | 2 to 27 | 1 to 29 | 8 to 28 |
Sample size at day 43 | N = 74 | N = 49 | N = 34 | N = 29 | N = 16 | N = 7 |
Mean change from baseline to day 43 | –3.8 | –2.2 | –3.5 | –4.7 | –4.2 | –6.1 |
Median change from baseline to day 43 | –3.0 | –1.0 | –2.5 | –1.0 | –2.0 | –4.0 |
IMP = investigational medicinal product; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Garvis Quality of Life; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Notes: Study participants were grouped according to the actual dose level received within the study cycle. Baseline values were defined as the last available value before or on the same date of the first administration of IMP in each cycle (i.e., baseline [day 1]) value for that cycle.
The total MG-ADL score ranges from 0 to 24, with a higher score indicating more severe disability. QMG scores range from 0 to 39, with a higher score indicating more severe disability. MGC scores range from 0 to 50, with a higher score indicating more severe disease. MG-QoL15r scores range from 0 to 30, with a higher score indicating more severe disability.
Source: Sponsor’s clinical evidence report for Study MG0007 (2022), Tables 6.1.1, 6.2.1, 6.3.1, and 6.5.1.62
Harms
Refer to Table 23 for harms data.
In the MG0004 trial, the incidence of TEAEs was reported by preferred term in greater than 2 patients by the most recent dose. In the MG0007 trial, the incidence of TEAEs was reported by preferred term in greater than or equal to 5% of patients in any treatment group by the most recent dose.
Adverse Events
MG0004 Trial
Seventy-six percent of the patients in the 7 mg/kg group experienced any AE (refer to Table 23). The most common AEs (experienced by 20% of patients or more) were nervous system disorders (36.0%), gastrointestinal disorders (26.0%), infections and infestations (26.0%), investigations (22.0%), and musculoskeletal and connective tissue disorders (20.0%).
MG0007 Trial
Sixty-eight patients (69.4%) in the 7 mg/kg group experienced any AE (refer to Table 23). Nervous system disorders (36.7%), infections and infestations (34.7%), gastrointestinal issues (24.5%), and general site-administration issues (27.6%) were the most reported.
Serious AEs
MG0004 Trial
SAEs were reported in 7 patients (14.0%). The only SAE occurring in more than 1 patient was nervous system disorders (n = 3; 6.0%).
MG0007 Trial
SAEs were reported in 9 patients (9.2%). The SAEs that occurred in more than 1 patient were nervous system disorders (n = 3; 3.1%) and infections and infestations (n = 2; 2.0%).
Withdrawals Due AEs
MG0004 Trial
A total of 4 patients (8.0%) experienced AEs that led them to discontinue from the study. Three of these patients (75.0%) experienced MG, while 1 patient (25.0%) experienced congestive cardiac failure. In patients who temporarily discontinued rozanolixizumab (n = 12; 24.0%), the main reasons were decreased blood IgG and hypogammaglobulinemia.
MG0007 Trial
A total of 6 patients (6.1%) permanently discontinued the study. Two patients (33.3%) had AEs with preferred term MG, while 1 patient each reported AEs of adrenal disorder, pneumonia, tendon disorder, tenosynovitis, retroperitoneal neoplasm, thymoma, and subacute cutaneous lupus erythematosus. In patients who temporarily discontinued rozanolixizumab, the main reasons were decreased blood IgG, hypogammaglobulinemia, and COVID-19.
Table 23: Summary of Harms Results From Long-Term Extension Studies
Adverse events | MG0004 trial | MG0007 trial |
|---|---|---|
Rozanolixizumab 7 mg/kg (N = 50)a | Rozanolixizumab 7 mg/kg (N = 98)a | |
Any AE | 38 (76.0) | 68 (69.4) |
Gastrointestinal disorders | 13 (26.0) | 24 (24.5) |
Diarrhea | 6 (12.0) | 15 (15.3) |
Nausea | 4 (8.0) | 8 (8.2) |
Abdominal pain | 0 | 8 (8.2) |
General disorders and administration-site conditions | 11 (22.0) | 27 (27.6) |
Pyrexia | 4 (8.0) | 7 (7.1) |
Infections and infestations | 13 (26.0) | 34 (34.7) |
COVID-19 | 0 | 10 (10.2) |
Nasopharyngitis | 2 (4.0) | 5 (5.1) |
Upper respiratory tract Infection | 5 (10.0) | 3 (3.1) |
Injury, poisoning, or procedural complications (fall) | 3 (6.0) | 9 (9.2) |
Investigations | 11 (22.0) | 12 (12.2) |
Blood immunoglobulin G decrease | 6 (12.0) | 6 (6.1) |
Musculoskeletal and connective tissue disorders | 10 (20.0) | 18 (18.4) |
Arthralgia | 0 | 5 (5.1) |
Back pain | 2 (4.0) | 0 |
Muscular weakness | 3 (6.0) | 0 |
Nervous system disorders | 18 (36.0) | 36 (36.7) |
Headaches | 15 (30.0) | 31 (31.6) |
Myasthenia gravis | 3 (60.0) | 4 (4.1) |
Skin and subcutaneous tissue disorders | 6 (12.0) | 15 (15.3) |
Vascular disorders | 4 (8.0) | 1 (1.0) |
Patients with serious AEs | ||
Patients with serious AEs | 7 (14.0) | 9 (9.2) |
Cardiac disorders | 1 (2.0) | 0 |
Endocrine disorders | NR | 1 (1.0) |
Eye disorders | 1 (2.0) | 0 |
Infections and infestations | 0 | 2 (2.0) |
Investigations | 1 (2.0) | 0 |
Musculoskeletal and connective tissue disorders | 1 (2.0) | 1 (1.0) |
Neoplasms (benign, malignant, and unspecified [including cysts and polyps]) | 0 | 1 (1.0) |
Nervous system disorders | 3 (6.0) | 3 (3.1) |
Skin and subcutaneous tissue disorders | 0 | 1 (1.0) |
Surgical and medical procedures | 0 | 1 (1.0) |
Discontinuations due to AEs | ||
Patients who discontinued from the study due to AEs | 4 (8.0) | 6 (6.1) |
Patients who permanently discontinued rozanolixizumab due to AEs | 3 (6.0) | 6 (6.1) |
Patients who temporarily discontinued rozanolixizumab due to AEs | 12 (24.0) | 14 (14.3) |
All deathsb (AEs leading to death) | 0 | 1 (1.0) |
Potential Hy’s Lawc | 0 | 0 |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; NR = not reported; TBL = total bilirubin; ULN = upper limit of normal.
aIn the MG0004 trial, the incidence of AEs was reported by preferred term in greater than 2 patients by the most recent dose; in the MG0007 trial, the incidence of TEAEs was reported by preferred term in greater than or equal to 5% of patients in any treatment group by the most recent dose.
bAll deaths was based on rozanolixizumab total screened and refers to all deaths occurring in both the MG0004 trial and the MG0007 trial.
cDefined as AST or ALT greater than 3 times the ULN, TBL greater than 2 times the ULN, and ALP less than 2 times the ULN in both the MG0004 trial and the MG0007 trial, with no alternative explanation for the biochemical abnormality (i.e., without waiting for any additional etiologic investigations to conclude).
Sources: MG0004 Clinical Study Report (2022), Table 8.1.1, Table 8.1.2, Table 8.1.12, Table 8.1.14, Table 8.1.19;75 MG0007 Clinical Study Report (2022), Table 8.1.1, Table 8.1.2, Table 8.1.12. Table 8.1.14, Table 8.1.19;62 sponsor’s Summary of Clinical Evidence.24
Mortality
MG0004 Trial
There were no AEs leading to death in this study.
MG0007 Trial
One death was reported due to fatal AEs (pneumonia).
Critical Appraisal
Internal Validity
The MG0004 and MG0007 trials were limited by their single-arm (noncomparative), open-label designs. A lack of a control group precludes the ability to make causal statements about benefits and harms versus any comparator. The open-label, nonblinded nature of the study may increase the risk of bias in determining the magnitude of the subjective safety outcomes and all efficacy end points because these were subjective (e.g., MG-ADL, QMG, MG-QoL15r, and MGC scores); the lack of blinding may influence patients’ expectations of the treatment. The direction and magnitude of these potential biases remains unclear. Concomitant treatments were intended to remain stable within treatment cycles but could be adjusted between cycles. These additional treatments could confound the relationship between rozanolixizumab and the efficacy and harms outcomes; however, the degree of impact on the results cannot be predicted. Efficacy results were assessed by MG-specific antibody subgroups; however, these results should be interpreted with caution due to the small sample sizes (especially in the MuSK antibody-positive subgroup). Of the participants who rolled over to the MG0007 trial from the pivotal trial, 35 patients had received placebo in that study and were first exposed to rozanolixizumab in the MG0007 trial. One study participant received placebo in the MG0003 trial and rolled over into the MG0004 trial, but did not receive treatment and subsequently rolled over into the MG0007 trial, with first exposure to rozanolixizumab. Baseline MG-ADL, QMG, and MGC scores indicated higher disease severity of patients entering the MG0007 trial, potentially suggesting selection bias. Patients in the MG0007 trial were allowed to switch between the 7 mg/kg and 10 mg/kg groups, based on investigator’s discretion, before the start of each subsequent cycle of treatment. Therefore, it is difficult to differentiate the effect of the 7 mg/kg dose (which is the focus of the reimbursement request) from that of the 10 mg/kg dose on efficacy outcomes. There is a high risk of attrition bias, given that the number of patients contributing to the analyses declined steadily over time.
External Validity
The patients who were enrolled in the pivotal trial (i.e., the MycarinG trial) were the ones entering the OLEs; therefore, it is reasonable to expect similar validity concerns to those highlighted for the pivotal trial.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the review team.
Objectives for the Summary of Indirect Evidence
A review of the indirect evidence was required because the pivotal trial did not contain direct comparative evidence of rozanolixizumab relative to an active comparator. Furthermore, an appraisal of the indirect evidence was needed because evidence from the NMA was incorporated into the sponsor’s pharmacoeconomic model.
Description of Indirect Comparisons
The submission included an NMA and a MAIC. The comparator treatments included in the NMAs were zilucoplan, efgartigimod alfa, eculizumab, IVIg, PLEX, rituximab, and ravulizumab. Of these, efgartigimod, eculizumab, IVIg, PLEX, and rituximab were included in the review; zilucoplan was not an approved treatment at the time of the submission, and ravulizumab was not deemed to be a relevant comparator because the Canadian Drug Expert Committee had recommended that it not be reimbursed for gMG. Trials with IVIg, PLEX, and rituximab were not part of the network for the primary analysis of the NMA. However, while zilucoplan and ravulizumab were not included as comparators in the NMA, information from the placebo arms would have been incorporated into the NMA. The comparator treatments included in the MAIC were efgartigimod, eculizumab, IVIg or PLEX, ravulizumab, and zilucoplan. Of these, comparisons between rozanolixizumab and efgartigimod, as well as rozanolixizumab and IVIg or PLEX, were included in the submission. Of note, the rozanolixizumab 7 mg/kg dosage was included in the appraisal, given that this is the only indication under review by Health Canada.
Study Selection Methods
The study selection methods were the same for the NMA and the MAIC. Briefly, a clinical SLR informed both the NMA and MAIC and consisted of broader PICOs (described in Table 24). Databases were searched from inception to May 1, 2023. Conference proceedings were also searched manually from 2017 to 2023 (except those indexed in Embase). Bibliographic searching of SLRs and meta-analyses was also conducted, and ClinicalTrials.gov as well as the European Union clinical trials register were explored for trial data. The articles from the SLR were then filtered according to the PICO criteria for the NMA and MAIC described in Table 25.
Table 24: SLR Informing the NMA and MAIC
Characteristics | SLR inclusion criteria |
|---|---|
Population | Adult patients (aged ≥ 18 years) of any gender or race with MG as the primary disease |
Intervention |
|
Comparator |
|
Outcomes | Efficacy:
Safety and tolerability:
|
Study designs |
|
Language | English language |
Publication time frame | No restriction (databases searched from inception to May 1, 2023) |
Databases searched | Databases searched:
Conference proceedings searched:
|
Selection process | Two independent reviewers screened all citations against predefined eligibility criteria. Any discrepancies in their decisions were resolved by a third reviewer. The full-text publications of all citations of potential interest were then screened for inclusion by 2 independent reviewers. Studies meeting the eligibility criteria at the second screening stage were extracted. At the full-text screening stage, if there was lack of clarity on whether the publication met the eligibility criteria, these citations were excluded. |
Data extraction process | Two reviewers extracted data from all included studies into a data extraction table and any discrepancies were resolved by a third reviewer. When more than 1 publication was identified describing a single trial, the data were compiled into a single entry in the data extraction table. |
Quality assessment | Assessment of the quality of reporting of trials were carried out by 2 independent reviewers followed by reconciliation of the differences between the 2 reviewers by a third independent reviewer. Critical appraisal of the included RCTs was conducted using comprehensive assessment criteria based on the recommendations in the NICE manufacturer’s template (NICE STA 2015).76 |
MAIC = matching-adjusted indirect comparison; MG = myasthenia gravis; MG-ADL = MG Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; nRCT = non randomized controlled trial; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial; SLR = systematic literature review; STA = single-technology appraisal.
Source: Sponsor’s Summary of Clinical Evidence24 and SLR Technical Report.77
Details of the study selection and methods for the NMA and MAIC are provided in Table 25. The submission did not provide a rationale for the differences between the list of comparators and the list of outcomes for the MAIC relative to the NMA.
Table 25: Study Selection Criteria and Methods for the NMA and the MAIC
Characteristics | NMA | MAIC |
|---|---|---|
Population | Generalized MG | |
Intervention | Rozanolixizumab (7 mg/kg) | |
Comparator |
|
|
Outcome |
|
|
Study designs | RCTs | |
Publication characteristics | English language publications from any country | |
Exclusion criteria | None | |
IVIg = IV immunoglobulin; MAIC = matching-adjusted indirect comparison; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MSE = minimal symptom expression; NMA = network meta-analysis; PLEX = plasma exchange; QMG = quantity myasthenia gravis; RCT = randomized control trial.
Source: Sponsor’s Summary of Clinical Evidence.24
NMA Design
Objectives
The objective of the NMA was to evaluate the relative effectiveness of rozanolixizumab compared to other treatments used for the management of AChR antibody-positive or MuSK antibody-positive gMG.
Analysis Methods
Full details of the NMA analysis methods are presented in Table 26. Briefly, the submission did not provide a rationale for the choice to conduct an NMA. Where the results of RCTs formed part of 1 evidence network and were deemed sufficiently similar for each population of interest, these were synthesized by NMA. Homogeneity was assessed by visual inspection of the distribution of baseline characteristics for the trials comprising the network, as well as the time point at which study outcomes were reported. Plot digitization at a commonly reported time point (12 weeks) was carried out to provide missing evidence in the analysis. The submission provided illustrations of the constructed networks and noted that networks usually contained 1 trial per direct comparison and did not contain closed loops of more than 1 study; therefore, inconsistency could not be assessed. The submission did not provide further details on whether there was any pooling of estimates based on different dosing or routes of administration before constructing the networks.
The NMAs were performed using a Bayesian approach with noninformative priors. Both fixed- and random-effects models were considered; however, the submission noted that the fixed-effect models were used because heterogeneity could not be estimated for random-effects models, due to the small number of trials in each network. Model selection was done using the deviance information criterion, with a difference of 5 points considered meaningful. The submission did not provide further details on how models were chosen. Changes from baseline outcomes were assessed at the 12-week (± 2 weeks) time point using only phase III studies in the primary analysis. Additional outcome data were provided by digitization of published figures using the Digitize It website.
The results of the NMA were presented with estimates for treatment effects of each intervention relative to placebo as the reference treatment. The posterior distributions of relative treatment effects are summarized by the median and 95% credible intervals, which were constructed from the 2.5th and 97.5th percentiles of the posterior distributions. Credible intervals not overlapping 1.0 were considered statistically significant. For binary outcomes, the NMA was performed based on the proportion of patients experiencing the event of interest and using a regression model with a binomial likelihood and logit link, or random distribution with a normal likelihood and natural scale link. Relative treatment effects were presented as ORs. For continuous outcomes, the NMA was performed on the outcome of mean change from baseline using a normal likelihood and identity link. Relative treatment effects were expressed as mean differences in the change from baseline.
Table 26: NMA Analysis Methods
Methods | Description |
|---|---|
Analysis methods | A Bayesian approach with fixed-effects and random-effects models considered:
|
Priors | Noninformative prior distributions were used for the model parameter(s). |
Assessment of model fit | The DIC was used to compare the goodness-of-fit of competing models. A difference in DIC of about 5 points was considered meaningful. |
Assessment of consistency | Before the actual NMA, homogeneity was assessed by visually inspecting the distribution of baseline characteristics for the trials comprising the network and considering the time point at which study outcomes were reported. The submission noted that there was a plan to test the consistency between direct and indirect comparisons; however, no closed loops of more than 1 trial connecting different interventions existed. Therefore, it was not possible to assess consistency. |
Assessment of convergence | Not reported |
Outcomes |
|
Follow-up time points |
|
Construction of nodes | Not explicitly described in the submission. |
Sensitivity analyses | To assess the robustness of the results, 5 sensitivity analyses including Phase 2 studies and different time points were conducted |
Subgroup analysis | Not reported |
Methods for pairwise meta-analysis | Due to the very small number of studies accounting for the final analysis network, there was insufficient power to perform meta-regression. |
DIC = deviance information criterion; MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis.
Source: Sponsor’s Summary of Clinical Evidence24 and NMA Technical Report.78
Sensitivity Analyses
Per the submission, sensitivity analyses were conducted assessing the inclusion of different time points for reporting outcomes as well as differences in study design (i.e., phase II versus phase III studies). The sensitivity analyses consisted of comparing the studies at their respective primary end points, including phase II studies. Additional outcome data were provided by digitization of the study figures from publications using the Digitize It website. Details of the sensitivity analyses are presented in Table 27.
Table 27: Definition and Methods for the Primary and Sensitivity Analyses in the NMA
Scenario | Definition | Methods |
|---|---|---|
Outcome: MG-ADL responder | ||
Primary analysis | Analysis at 12 weeks ± 2 weeks with phase III data | The NMA was performed based on the proportion of patients experiencing the event of interest using a regression model with a binomial likelihood and logit link. FEMs were preferred over REMs because the networks consisted of 1 trial per comparison. Noninformative priors were used. |
Sensitivity 1 | Analysis at 12 weeks ± 2 weeks with phase III and phase II data | |
Outcome: Change from baseline in MG-ADL | ||
Primary analysis | Analysis at 12 weeks ± 2 weeks with phase III trials only | The NMA was performed based on the mean change from baseline in the outcome and the corresponding SEs using model with a normal likelihood and identity link. FEMs were preferred over REMs because the networks consisted of 1 trial per comparison. Noninformative priors were used. |
Sensitivity 1 | Analysis at 12 weeks ± 2 weeks with both phase III and phase II trials | |
Sensitivity 2 | Analysis at 12 weeks ± 2 weeks with both phase III and phase II trials and adding in trials reporting data at their primary end point if it was other than 12 weeks | |
Sensitivity 3 | Analysis at the time at which the primary end point was evaluated with phase III trials only | |
Sensitivity 4 | Analysis at the time at which the primary end point was evaluated with phase II and phase III trials | |
Sensitivity 5 | Analysis at week 4 (digitized time point) | Plots presenting the data were digitized to obtain mean change from baseline and SE. The NMA was performed based on the mean change from baseline in the outcome and the corresponding SE using a model with a normal likelihood and identity link. FEMs were preferred over REMs because the networks consisted of 1 trial per comparison. Noninformative priors were used. |
FEM = fixed-effects model; MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis; REM = random-effects model; SE = standard error.
Source: Sponsor’s Summary of Clinical Evidence24 and the NMA Technical report.78
MAIC Design
Objectives
The objective of the MAIC was to assess the comparative efficacy of rozanolixizumab versus comparators used for the treatment of gMG. The comparators analyzed in the MAIC included rozanolixizumab, zilucoplan, efgartigimod, ravulizumab, eculizumab, and IVIg; the analyses provided in the submission were for rozanolixizumab versus efgartigimod and rozanolixizumab versus IVIg.
Analysis Methods
A systematic literature search was undertaken to provide potential comparators for the MAIC. These were rescreened using the PICOs provided in Table 25. Before carrying out a feasibility assessment, the relative importance of all the baseline characteristics, based on their impacts on the outcomes, was ranked by 2 clinical experts. The submission provided details on the rankings from each expert. The feasibility assessment consisted of comparing the relevant trials for each comparison in terms of their baseline characteristics and inclusion and exclusion criteria. In cases of differences in the inclusion and exclusion criteria, a subset of patients from the rozanolixizumab trial was used to match the comparator trial. The definitions of outcomes were also matched, to the extent possible. The submission did not provide details on the thresholds used to determine similarity or dissimilarity.
If feasibility was confirmed, the 2 studies were matched using a propensity score weighting method. A comparison of all potential analysis scenarios was presented to knowledge leaders, and the base case was selected on the basis of the following criteria:
The scenario should have included as many of the important patient characteristics in the weighting process as feasible, ideally for all effect modifiers.
Estimated sample size (ESS) should be sufficiently large (defined as more than 30 patients).
The distribution of weights should be uniform. There should be fewer patients who obtain weights close to 0 or weights considered to be very high (i.e., > 5). The submission did not provide details on whether a threshold for the number of patients with extreme weights was used to determine uniformity of the distribution.
The baseline characteristics used in the weighting were presented before and after the weighting process, and distributions of weights were provided in the submission. The comparisons of rozanolixizumab versus efgartigimod and were modelled using an anchored MAIC, and the results for rozanolixizumab versus IVIg were modelled using an unanchored MAIC. Continuous outcomes were modelled using linear regression, with results presented as mean differences. Binary outcomes were modelled using logistic regression, with results presented as ORs. The submission did not provide details on a specific method (e.g., Signorovitch et al.) used in the MAIC analysis.
Results of the SLR Informing the NMA and the MAIC
Summary of Included Studies
The SLR yielded a total of 12,449 references after deduplication, a total of 1,043 references after title and abstract screening, and 80 studies (represented by 302 publications) after full-text screening. The results of the National Institute for Health and Care Excellence quality assessment indicated that 5% of the studies were considered to have an unclear risk of bias in the statistical analysis; 18% had an unclear risk of bias in outcome selection and reporting; 14% had an unclear risk of bias in withdrawals; 3% were at high risk and 5% at unclear risk of bias in blinding; 4% had unclear risk of bias in baseline characteristics; and 11% had unclear risk of bias in randomization. The remaining studies had low risk of bias for the aforementioned domains.
Results of the NMA
Summary of Included Studies
A total of 47 studies were included in the NMA. After the feasibility assessment for the NMA was completed, a further 35 studies were excluded (because, among other reasons, 18 studies did not have any interventions of interest; 13 studies did not have any outcomes of interest; 2 studies were not connected to any network; 1 study had a crossover design; and 1 study was a phase IV study with outcomes at week 2). A total of 12 studies identified by the submission as having sufficient overlap between baseline characteristics were included in the NMAs; no formal definition was provided for “sufficient overlap.” A total of 6 studies were phase III studies and were included in the primary analysis for the NMA.
Details of the assessment of homogeneity are in Table 28. Briefly, heterogeneity was observed throughout the NMA network in disease severity, treatment history (where reported), trial eligibility criteria, placebo response, the definition of MG-ADL responders, the timing of end point evaluation, study designs, and baseline characteristics.
The baseline characteristics of the trials included in the NMA are in Table 29. Of note, the majority of the patients enrolled in the trials were AChR-antibody positive, female, and had class II to IV gMG. The duration of disease ranged from 6.9 years to 10.3 years. MuSK antibody status was reported in 2 trials (the MycarinG trial [12% of patients] and the ADAPT trial [4% of patients]). Study duration ranged from 12 weeks to 48 weeks.
Table 28: Assessment of Homogeneity for the NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Heterogeneity was noted in the gMG disease severities included in the trials. For example, the REGAIN trial79 (eculizumab comparator) enrolled patients with gMG who were refractory, defined as having received treatment with ≥ 2 immunosuppressive therapies or ≥ 1 immunosuppressive therapy with IVIg or PLEX given at least 4 times per year, for 12 months, without symptom control. The RINOMAX trial80 (rituximab comparator) enrolled patients whose gMG onset was less than 12 months before inclusion. NMA analyses were carried out for MG-ADL response in refractory patients as well as the overall population; the submission defined refractoriness as per the RAISE trial81 (zilucoplan comparator) but did not subset other trials in the network that enrolled nonrefractory patients for the analysis of refractory patients. |
Treatment history | Heterogeneity was noted in the treatment histories available in the submission. Of note, the proportion of patients with previous NSIST treatments ranged from 48.5% to 74%, where explicitly reported in 2 studies, and reporting of treatment history or medications was inconsistent across studies. Of note, some studies reported treatments at baseline only, while others reported any prior use of gMG medication(s). |
Trial eligibility criteria | Heterogeneity was noted across studies in the inclusion and exclusion criteria. The minimum MG-ADL scores required for enrolment ranged from 3 points or more to 6 points or more, and the minimum QMG scores (required in 3 studies) ranged from 6 points to 12 points. Two studies required minimum body weight thresholds of 35 kg or more and 40 kg or more, while other studies did not require weight thresholds. Four out of 6 studies required AChR antibody-positive gMG. The REGAIN trial79 (eculizumab comparator) required patients to meet criteria for refractory gMG, and the MycarinG trial required patients to be under consideration for additional treatment, such as IVIg or PLEX. |
Dosing of comparators | Reported in Table 29. |
Placebo response | The submission noted that heterogeneity was observed between placebo response rates across trials for the MG-ADL responder outcome, but did not provide further specifics. |
Definitions of end points | Standardized scores were used as end points. Heterogeneity was noted in the threshold considered to be an MG-ADL responder across studies; this ranged from a 1-point to 3-point change from baseline. |
Timing of end point evaluation | Heterogeneity was noted in the timing of the primary end point evaluation, ranging from 4 weeks to 26 weeks. For change from baseline outcomes, studies reporting data at 12 weeks ± 2 weeks were utilized in the primary analysis, and plot digitization was performed for the studies not reporting the data at this time point. Further sensitivity analyses were performed comparing studies at their respective primary end point as well (i.e., week 26, week 16, and so on); sensitivity analyses are described in Table 27. |
Withdrawal frequency | Not reported |
Clinical trial setting | Not reported |
Study design | Heterogeneity was noted in the study designs, with both phase II and III trials included in the NMA. The prespecified primary analysis was conducted using only phase III studies; sensitivity analyses were conducted using phase II and III studies (described in Table 27). |
Baseline characteristics | Heterogeneity was noted in some key baseline characteristics that were possible effect measure modifiers across studies. Of note, the baseline MG-ADL score ranged from 5.1 to 10.3, and the duration of gMG disease ranged from 6.9 years to 10.1 years. Certain baseline characteristics, such as history of gMG crises and MuSK antibody status, were not reported for all studies. |
AChR = acetylcholinesterase receptor; gMG = generalized myasthenia gravis; IVIg = IV immunoglobulin; MG-ADL = Myasthenia Gravis Activities of Daily Living; MuSK = muscle-specific tyrosine kinase; NMA = network meta-analysis; NSIST = nonsteroidal immunosuppressive therapy; PLEX = plasma exchange.
Source: Sponsor’s Summary of Clinical Evidence24 and NMA Technical Report.78
Table 29: Summary of Phase III Studies Included in the NMA Primary Analysis
Study characteristics | Population key inclusion criteria | Intervention | Outcomes of interest | Key baseline characteristics (treatment arm) |
|---|---|---|---|---|
MycarinG trial (rozanolixizumab) | ||||
Multicentre, randomized, double-blind, placebo-controlled, phase III study N = 200 Sites: Belgium, Canada, Czech Republic, Denmark, France, Georgia, Germany, Hungary, Italy, Japan, Poland, Russian Federation, Serbia, Spain, Taiwan, UK, US Duration: up to 18 weeks |
| Rozanolixizumab (7 mg/kg) through SC infusion once weekly for 6 weeks Permitted cotherapies:
| Primary: change from baseline to day 43 in MG-ADL score Other secondary:
|
|
ADAPT trial (efgartigimod) | ||||
Multicentre, randomized, double-blind, placebo-controlled, phase III trial N = 167 Sites: Europe, Japan, North America Duration: 26 weeks |
|
|
|
|
CHAMPION MG trial (ravulizumab) | ||||
Multicentre, randomized, double-blind, placebo-controlled, phase III trial Sites: Asia-Pacific, Europe, Japan, North America N = 175 Duration: 26 weeks |
|
| Primary: change from baseline in MG-ADL total score at week 26 Other secondary:
|
|
RAISE trial (zilucoplan) | ||||
Multicentre, randomized, double-blind, placebo-controlled, phase III trial Sites: Europe, Japan, North America N = 174 Duration: 12 weeks |
|
| Primary: change from baseline in MG-ADL score at week 12 Other secondary:
|
|
REGAIN trial (eculizumab) | ||||
Multicentre, randomized, double-blind, placebo-controlled, phase III trial N = 126 Sites: Asia, Europe, Latin America, North America Duration: 26 weeks |
|
| Primary: change in MG-ADL total score from baseline to week 26 Other secondary:
|
|
RINOMAX trial (rituximab) | ||||
Randomized, double-blind, placebo-controlled, phase III trial N = 25 Sites: Sweden Duration: 48 weeks |
|
| Primary: proportion of patients with QMG score ≤ 4 and prednisolone ≤ 10 mg/d at week 16 Other secondary:
|
|
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholinesterase receptor; gMG = generalized myasthenia gravis; IVIg = IV immunoglobulin; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL = Myasthenia Gravis Quality of Life; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; MGFA = Myasthenia Gravis Foundation of America; MSE = minimal symptom expression; MuSK = muscle-specific kinase; NeuroQoL = Quality of Life in Neurological Disorders; QMG = Quantitative Myasthenia Gravis; NA = not available; NR = not reported; NSIST = nonsteroidal immunosuppressive therapy; PLEX = plasma exchange; vs. = versus.
aMSE was defined as an MG-ADL score of 0 or 1 without rescue therapy.
bTreatment-refractory was defined as treatment for at least 1 year with 2 or more of the following therapies: prednisone, azathioprine, mycophenolate, cyclosporine, cyclophosphamide, methotrexate, tacrolimus, rituximab, eculizumab, or other corticosteroids; or a history of treatment with at least 1 of the preceding therapies for 1 year or more and requiring chronic PLEX, IVIg, or subcutaneous immunoglobulin at least every 3 months for the 12 months before enrolment.
Sources: Sponsor’s NMA Technical Report;78 additional information provided by the sponsor;73 the ADAPT trial;82 the CHAMPION MG trial;83 the RAISE trial;81 the REGAIN trial;79 and the RINOMAX trial.80
Results
The same study network was applicable to the analysis for MG-ADL responders and change from baseline in MG-ADL. The study network for the primary analysis is in Figure 4. Briefly, all included studies compared treatments to placebo, and each node in the network consisted of a single study. The only loop in the NMA was formed by rozanolixizumab 7 mg/kg and rozanolixizumab 10 mg/kg (not appraised in this review).
Figure 4: NMA Network for MG-ADL Responder Outcome and Change From Baseline in MG-ADL — Overall Population
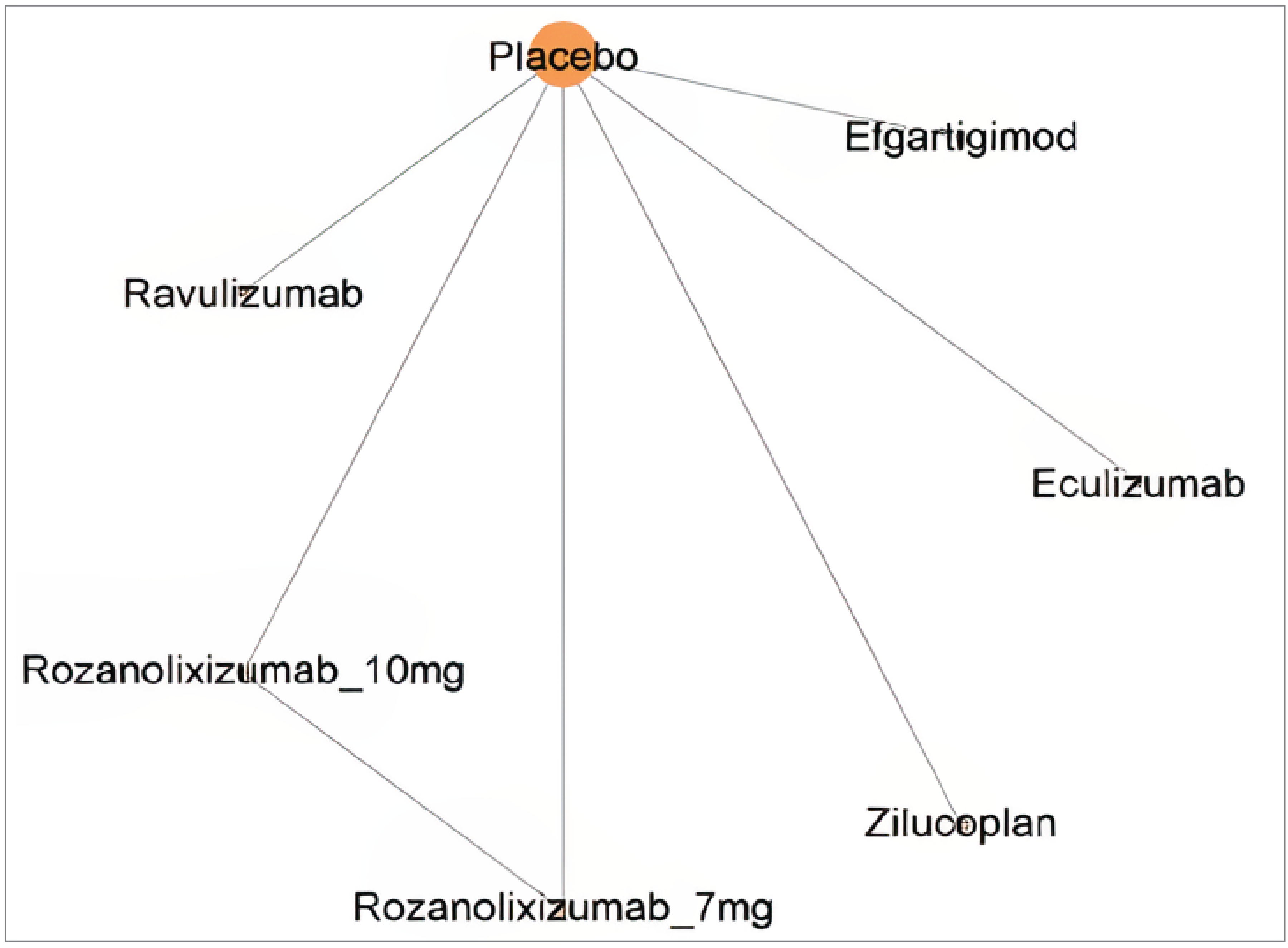
MG-ADL = Myasthenia Gravis Activities of Daily Living; NMA = network meta-analysis.
Source: Sponsor’s NMA Technical Document.78
Results from the relevant comparators included in the primary analysis of the NMA are in Table 30, and the NMAs used in the sensitivity analyses are in Appendix 1. Briefly, results for rozanolixizumab 7 mg/kg versus efgartigimod and versus eculizumab ████ ███████ ████ ██ █████████ ██ ███ █████████ ████ ███ ███████ ███ ███ ████████. Rozanolixizumab 7 mg/kg was ████████ ████ placebo for MG-ADL responders. The results of the sensitivity analyses were broadly similar to those of the primary analyses, indicating ██ █████████ ██ ███ █████████ ████ ███ █████ ████████████ █████ ████ ███████. The results of the primary analysis in the refractory population (defined using the definition of the RAISE trial and described in Table 29) were very similar to the results of the primary analysis in the overall population.
Table 30: NMA Primary Analysis Results (12 Weeks ± 2 Weeks) — Overall Population
Detail | Rozanolixizumab 7 mg/kg | |
|---|---|---|
MG-ADL respondera OR (95% CI) | Change from baseline in MG-ADL MD (95% CI) | |
Efgartigimod | ████ ██████ █████ | ████ ███████ █████ |
Eculizumabb | ████ ██████ █████ | ████ ███████ █████ |
Placebob,c | ████ ██████ ██████ | █████ ███████ █████ |
CI = confidence interval; CrI = credible interval; OR = odds ratio; MD = mean difference; MG-ADL = Myasthenia Gravis Activities of Daily Living; NA = not applicable; NMA = network meta-analysis.
aDefined as an improvement of at least 3 points in MG-ADL at the study end point.
bIncludes digitized data for the MG-ADL change from baseline outcome.
cIncludes digitized data for the MG-ADL responder outcome.
Source: Sponsor’s Summary of Clinical Evidence24 and the NMA Technical Report.78
Results of the MAIC
Summary of Included Studies
The SLR yielded a total of 73 studies, 67 of which were excluded from the MAIC analysis because the interventions were not of interest. This left 7 studies informing the MAIC. Relevant comparisons included in the submission were for rozanolixizumab (the MycarinG trial) versus efgartigimod (the ADAPT trial) in patients with gMG and for rozanolixizumab (the MycarinG trial) versus IVIg (the Barth et al. [2011] trial) in patients with gMG. Details of the trials, as well as comparisons of the inclusion and exclusion criteria, are in Table 31. Briefly, some differences were identified between the MycarinG trial and the ADAPT trial. Most notably, there were differences in the minimum MG-ADL scores required for enrolment; in addition, the ADAPT trial required patients to be on stable doses of gMG therapy, while the MycarinG trial required patients to be under consideration for additional therapy. There were also differences noted between the inclusion criteria for the MycarinG trial versus the Barth et al. (2011) trial, most notably that Barth et al. was an active-controlled trial that did not require a specific MGFA class diagnosis or MG-ADL baseline score for enrolment, whereas the MycarinG trial was placebo-controlled, had required weight and MG-ADL thresholds, and required AChR or MuSK antibody-positive status.
Results
The MycarinG Trial Versus the ADAPT Trial
For the MAIC comparing MycarinG and ADAPT, to improve similarity to the efgartigimod trial population, only the patients from the MycarinG trial who were MuSK antibody-negative and AChR antibody-positive were included in the unanchored MAIC for rozanolixizumab versus efgartigimod (i.e., a subset of the full trial population). The baseline characteristics included in matching, both before and after matching, are presented in Table 32. Of the 5 scenarios run during the weighting procedure, scenario 1, which matched on all the selected baseline characteristics, was used in the primary analysis.
Table 31: Comparability Assessment for MAIC — ADAPT, MycarinG, and Barth et al. (2011) Studies
Characteristic | ADAPT trial | MycarinG trial | Barth et al. (2011) trial |
|---|---|---|---|
Trial design | Phase III, double-blind, placebo-controlled, multisite RCT | Phase III, double-blind, placebo-controlled, multisite RCT | Randomized, active-controlled, phase IV trial |
N | 129 | 200 | 41 |
Key inclusion criteria |
|
|
|
Key exclusion criteria |
|
|
|
Intervention | Efgartigimod 10 mg/kg, administered as 4 infusions per cycle, 1 infusion per week with ≥ 5 weeks of follow-up in between | Rozanolixizumab 7 mg/kg through SC infusion once weekly for 6 weeks | IVIg or PLEX (1.0 plasma volume exchanges with 5% albumin replacement fluid) |
Outcome(s) | Primary: Proportion of patients who are AChR antibody-positive with MG-ADL response (≥ 2-point improvement sustained ≥ 4 consecutive weeks, with first improvement by week 4 of the cycle) Other secondary:
| Primary: change from baseline to day 43 in MG-ADL score Other secondary:
| Primary: change in QMG from baseline to day 14 after full treatment Other secondary:
|
Study duration | 26 weeks | Up to 18 weeks | 60 days |
AChR = acetylcholinesterase receptor; FcRn = neonatal fragment crystallizable receptor; gMG = generalized myasthenia gravis; IM = intramuscular; IMP = investigational medicinal product; IgA = Immunoglobulin A; IVIg = IV immunoglobulin; MAIC = matching-adjusted indirect comparison; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MGFA = Myasthenia Gravis Foundation of America; MuSK = muscle-specific kinase; PLEX = plasma exchange; PRO = patient-reported outcome; QMG = Quantitative Myasthenia Gravis; RCT = randomized controlled trial; RNS = repetitive nerve stimulation; SC = subcutaneous; SFEMG = single-fibre EMG.
Sources: Sponsor’s Summary of Clinical Evidence;24 the ADAPT trial;82 the Barth et al. (2011) trial.84
Results of the primary analysis for rozanolixizumab versus efgartigimod are presented in Table 33. Briefly, at 6 weeks, the results indicated ██████████ ████ ███████ ████ ██ █████████ ██ ███ █████████ ████ ███ ██████ ███ ███ ███████.
Table 32: Baseline Characteristics of the MycarinG Study and ADAPT Study — Prematching and Postmatching
Parameter | ADAPT trial (N = 129) | MycarinG trial before matching (N = 164) | MycarinG trial after matching | Included in base case |
|---|---|---|---|---|
QMG at baseline (SD) | 15.6 (4.8) | 15.6 (3.6) | 15.60 | Yes |
MG-ADL at baseline (SD) | 8.8 (2.3) | 8.2 (3.3) | 8.80 | Yes |
MG duration (SD) | 9.3 (8.2) | 8.4 (8.9) | 9.30 | Yes |
Any NSIST at baseline (%) | 60 | 50 | 60 | Yes |
Any steroid at baseline (%) | 75 | 62 | 75 | Yes |
Prior thymectomy (%) | 58 | 40 | 58 | Yes |
MGC at baseline (SD) | 18.4 (5.7) | 16.0 (6.4) | 18.35 | Yes |
Age (SD) | 46.9 (15.4) | 51.8 (16.7) | 46.93 | Yes |
Sex (% female) | 67 | 60 | 67 | Yes |
Race (% white) | 85 | 70 | 85 | Yes |
Baseline MG-QoL15r score (SD) | 16.2 (5.9) | 15.4 (6.9) | NR | No |
MAIC = matching-adjusted indirect comparison; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MG-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; NR = not reported; NSIST = nonsteroidal immunosuppressive therapy; QMG = Quantitative Myasthenia Gravis; SD = standard deviation.
Sources: Sponsor’s MAIC Technical Report;85 additional information provided by the sponsor.25,73
Table 33: Results of the MAIC Analysis for Rozanolixizumab vs. Efgartigimod at 4 Weeks or 6 Weeks1
Outcomea | Rozanolixizumab 7 mg/kg | |
|---|---|---|
ESS | Adjusted MD or OR (95% CI)b | |
Change from baseline in QMG (MD) | ██ | █████ ███████ █████ |
3-point improvement in QMG (OR) | ██ | ████ ██████ █████ |
Change from baseline in MG-ADL (MD) | ██ | █████ ███████ █████ |
2-point improvement in MG-ADL (OR) | ██ | ████ ██████ █████ |
3-point improvement in MG-ADL (OR) | ██ | ████ ██████ █████ |
Change from baseline in MGC (MD) | ██ | █████ ███████ █████ |
Change from baseline in MG-QoL15r (MD) | ██ | ████ ███████ █████ |
CI = confidence interval; ESS = estimated sample size; MAIC = matching-adjusted indirect comparison; MD = mean difference; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGC = Myasthenia Gravis Composite; MQ-QoL15r = revised 15-item Myasthenia Gravis Quality of Life; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; vs. = versus.
aChange from baseline results were reported at 6 weeks; results for 2-point or 3-point improvements were reported at 4 weeks.
bResults were reported for rozanolixizumab vs. efgartigimod; negative MD and OR greater than 1 both favour rozanolixizumab.
Source: Sponsor’s MAIC Technical Report.85
The MycarinG Trial Versus the Barth et al. (2011) Trial
The submission did not specify whether the study population in the MycarinG trial was subset to match the inclusion criteria for the Barth et al. (2011) trial. The baseline characteristics, both before and after matching, are presented in Table 34. Briefly, differences were noted between the rozanolixizumab 7 mg/kg group and the IVIg or PLEX group in the proportions of patients with prior thymectomy, corticosteroid at baseline, AChR antibodies, and azathioprine at baseline. After matching, notable differences remained in the proportion of patients with azathioprine at baseline and the proportion of patients who were MuSK antibody-positive; MGFA class after matching was not available.
Table 34: Baseline Characteristics in the MycarinG and Barth et al. (2011) Studies — Prematching and Postmatching
Parameter | Barth et al. (2011) trial (N = 41) | MycarinG trial before matching (N = 66)a | MycarinG trial after matching | Included in base case |
|---|---|---|---|---|
QMG at baseline, mean (SD) | 14.3 (4.0) | 15.4 (3.7) | 14.3 | Yes |
MG duration, years, mean (SD) | 5.9 (7.5) | 6.9 (6.8) | 5.9 | Yes |
Prior thymectomy (%) | 32 | 48 | 32 | Yes |
CS at baseline (%) | 34 | 64 | 34 | Yes |
AChR antibody-positive (%) | 68 | 85 | 68 | Yes |
Prior IVIg (%) | 22 | 18 | 22 | Yes |
Prior PLEX (%) | 10 | 0 | NR | No |
Azathioprine at baseline (%) | 15 | 26 | 27 | No |
Mycophenolate at baseline (%) | 5 | 12 | 9 | No |
MuSK antibody-positive (%) | 5 | 6 | 15 | No |
Age, years, mean (SD) | 57.0 (18.0) | 53.2 (14.7) | 52.4 | No |
Sex (% female) | 58 | 59 | 54 | No |
MGFA grade 3 or less, (%) | 94 | 57 | NR | No |
AChR = acetylcholine receptor; CS = corticosteroid; IVIg = IV immunoglobulin; MAIC = matching-adjusted indirect comparison; MG = myasthenia gravis; MGFA = Myasthenia Gravis Foundation of America; MuSK = muscle-specific tyrosine kinase; NR = not reported; PLEX = plasma exchange; QMG = Quantitative Myasthenia Gravis.
aPatients receiving rozanolixizumab 7 mg/kg.
Sources: Sponsor’s MAIC Technical Report;85 additional information provided by the sponsor.25,73
Results of the primary analysis are presented in Table 35. Briefly, at 2 weeks and 4 weeks, the mean difference between rozanolixizumab and IVIg for change from baseline in QMG scores ████████ ███████████████ ██████; the results for QMG responders indicated ██████████ ████ ██ █████████ ██ ███ ██████████ ████ ███ █████.
Table 35: Results of the MAIC Analysis for Rozanolixizumab vs. IVIg at 2 Weeks and 4 Weeks
Outcome | Rozanolixizumab 7 mg/kg | |
|---|---|---|
ESS | Adjusted MD or OR (95% CI) | |
Change from baseline in QMG at 4 weeks | ██ | █████ ███████ ██████ |
QMG respondera at 2 weeks | ██ | ████ ██████ █████ |
CI = confidence interval; ESS = estimated sample size; MAIC = matching-adjusted indirect comparison; MD = mean difference; OR = odds ratio; QMG = Quantitative Myasthenia Gravis; vs. = versus.
aThe submission specified a minimal clinically important difference for QMG of 2 points in patients with a baseline QMG score from 0 to 16, and 3 points for patients with a baseline QMG score of greater than or equal to 16.
Source: Sponsor’s MAIC Technical Report.85
Critical Appraisal
The body of evidence for the indirect treatment comparison contained an NMA and a MAIC informed by an SLR. The search included a comprehensive list of databases. The screening, data extraction, and quality assessment of studies were generally conducted according to accepted methods. Some limitations of the SLR include the fact that the search was run only until 2023; therefore, it might have missed more recent publications on comparators. In addition, the quality assessment was carried out at the level of the trial, which might not capture the fact that risks of some domains of bias (e.g., attrition bias) can vary by outcome. According to the clinical expert, the NMA included comparators that are relevant to the Canadian context, and the outcome was of interest to clinicians. However, data about some relevant comparators, such as IVIg and rituximab, were not available in the primary analysis of the NMA, and additional limitations in the sensitivity analyses do not allow conclusions to be drawn regarding these comparators. Likewise, the MAIC comparison to efgartigimod or IVIg was relevant, as were the outcomes included in the analysis. The submission noted that clinical expert input was solicited to help rank the list of characteristics for weighting in the MAICs; the clinical expert consulted for this review noted that the list of comparators was comprehensive. However, results from all comparators were not available in the submission; there were no details on why fewer comparators were analyzed in the MAIC compared to the NMA when both analyses used results from the same SLR. Furthermore, some trials included in the SLR had an unclear risk of bias in certain domains, which may increase the uncertainty in the reported results.
There are important sources of heterogeneity in the NMA network that have clinical relevance and affect the certainty of the results. While all trials enrolled patients with gMG class II to IV, there were differences between trials in the refractory status of the enrolled patients, and these were not accounted for in the analyses. For example, the trials for eculizumab and rituximab generally enrolled patients who were refractory and newly diagnosed, respectively; the trial for zilucoplan included patients who were refractory; and the trial for rozanolixizumab required patients to be under consideration for additional therapy. In addition, MuSK antibody status was reported in only 2 of the 6 included trials, and the trials in the network used MG-ADL thresholds ranging from 3 points to 6 points. The clinical expert consulted for this review noted that both disease severity and MuSK antibody status are treatment-effect modifiers and that the MG-ADL cut-off range spanned both generally mild and generally moderate gMG disease. Taken together, these could represent clinically meaningful differences in patient disease status. Certain sources of heterogeneity, such as study design and the timing of end point evaluation, were addressed by including only phase III trials in the primary analysis and using plot digitization to extract data from time points not reported in the published study figure. While digitization of results is a generally accepted practice, there is increased room for error compared to directly collecting numerical results from study publications. The sensitivity analyses conducted do not address the heterogeneity concerns; the network for sensitivity 2 included IVIg, which could be used chronically or as a rescue therapy; the network for sensitivity 3 included primary outcome reporting at any time point rather than a single time point; the network for sensitivity 4 repeated this, with the addition of phase II studies; and sensitivity 5 was assessed at 4 weeks. Taken as a whole, these limitations affect the confidence that the transitivity assumption underpinning the NMA was met.
Both MAICs are, likewise, subject to limitations. In general, a large number of comparators were included in the SLR and could be included in the MAIC. However, only comparisons of rozanolixizumab with IVIg and efgartigimod were provided, and it is not known whether comparisons with other treatments were infeasible or whether the results influenced the decision to exclude other comparators from the submission. While the clinical expert consulted for the review noted that the list of known prognostic and/or effect-modifying variables used for weighting in both MAICs was comprehensive, not all baseline characteristics were reported before and after weighting; therefore, it is not known whether there were other potential sources of heterogeneity in the trial populations remaining after weighting. Weighting controlled for the differences in the reported baseline characteristics for the anchored MAIC comparing rozanolixizumab to efgartigimod (i.e., the ADAPT trial). However, residual confounding is possible due to unidentified or unknown effect-modifying variables, and the ESS was considerably smaller than the sample sizes of the 2 trials prematching, suggesting that a small proportion of the patient population may be driving the results and that the findings could be unstable. This suggests that there remains uncertainty in the results for the comparison of rozanolixizumab to efgartigimod.
The comparison of rozanolixizumab to IVIg was carried out using an unanchored MAIC, the methodology of which is subject to important limitations. Unanchored MAICs rely on the assumption that all possible prognostic factors and treatment-effect modifiers are controlled for — an assumption that is largely considered impossible to meet.26 The scenario used in the current MAIC did not include all baseline characteristics in the weighting process, resulting in a high risk of residual confounding. Therefore, confidence in the results is highly uncertain. Furthermore, there are important study differences that were not controlled for by the weighting process, such as the lack of a placebo comparator in the Barth et al. (2011) trial, the timing of the primary end point, and the proportion of patients with MuSK antibodies. Taken as a whole, conclusions about the efficacy of rozanolixizumab versus IVIg are challenging to draw.
The indirect evidence as a whole is also subject to some limitations that affect generalizability. First, the clinical expert consulted for this review noted that rituximab would be a viable comparator to rozanolixizumab in the Canadian setting; however, information comparing to rituximab to rozanolixizumab was not available for any of the indirect comparison primary analyses. In addition, the study population of the MycarinG trial, which included patients who were AChR antibody-positive or MuSK antibody-positive, was subset to include only patients who were MuSK antibody-negative and AChR antibody-positive to match the inclusion criteria of the ADAPT trial. This affects the generalizability of those results to the population of patients who have MuSK antibody-positive gMG. The clinical expert consulted for this review noted that MuSK status affects both choice of treatment and treatment response. Furthermore, the results of the MAIC were assessed as early as 2 weeks to 6 weeks, which the clinical expert noted is early to assess treatment response and might not capture maximal response. Lastly, long-term information about comparative efficacy and harms is unavailable.
Summary
The submission included a body of indirect evidence consisting of 2 NMAs and 2 MAICs. The primary analyses for the NMAs were conducted in phase III trials and included evidence for eculizumab and efgartigimod; other comparators included in the network (ravulizumab and zilucoplan) were not included in the review because ravulizumab is not recommended to reimburse in gMG, zilucoplan was not approved for gMG at the time of the review, and rituximab was not included in the network for the primary analysis. The disease severity of patients included in the trials was a source of considerable heterogeneity and an important treatment-effect modifier not controlled for in the NMA. The trials for eculizumab and rituximab generally enrolled refractory and newly diagnosed patients, respectively; the trial for zilucoplan included refractory patients; and the trial for rozanolixizumab required patients to be under consideration for additional therapy. Furthermore, the trials in the network used MG-ADL thresholds ranging from 3 points to 6 points and QMG thresholds ranging from 6 points to 12 points (in 3 studies). The clinical expert consulted for this review noted that disease severity is a treatment-effect modifier and noted that the MG-ADL cut-off range spanned both generally mild and generally moderate gMG disease. Taken together, these differences could represent clinically meaningful differences in patient disease status. This limitation, along with limitations in the NMA methodology, affect confidence that the transitivity assumption underpinning the NMA was met; therefore, conclusions on the comparative efficacy of rozanolixizumab versus eculizumab or efgartigimod could not be drawn on the basis of this evidence.
The MAICs included comparisons of rozanolixizumab to efgartigimod (the ADAPT trial, a 26-week, phase III, double-blind, placebo-controlled, multisite RCT comparing efgartigimod to placebo in patients with class II to IV gMG) and rozanolixizumab to IVIg (the Barth et al. [2011] trial, a 60-day, randomized, active-controlled trial comparing IVIg with PLEX in patients with gMG requiring a change in therapy). The results for MG-ADL responders and change from baseline in MG-ADL from the anchored MAIC for efgartigimod and rozanolixizumab suggest ██████████ ██ █████████ ██████, similar to the results from the NMA; however, these results were subject to uncertainty due to limitations such as the potential for residual confounding, small ESS, and imprecision. The unanchored MAIC comparing IVIg to rozanolixizumab was limited considerably by the fact that not all prognostic or effect-modifying factors were controlled, given that not all baseline characteristics were included in the weighting; and differences remained in the proportion of patients with MuSK antibodies, which are effect modifiers, as per the clinical expert consulted for this review. Therefore, conclusions about the comparative efficacy of rozanolixizumab and IVIg cannot be drawn. Both MAICs are also subject to limitations around generalizability with regard to the timing of the analysis (2 weeks to 6 weeks) because the results may not capture maximal treatment response. Information about long-term comparative efficacy and harms is unavailable.
Studies Addressing Gaps in the Systematic Review Evidence
There were no studies identified.
Discussion
Summary of Available Evidence
One pivotal, phase III, sponsor-funded, multinational (81 sites, including 4 in Canada), double-blind RCT was the focus of the systematic review portion of this report. The MycarinG study randomized 200 patients with gMG in a 1-to-1-to-1 ratio to weekly SC infusions of 1 of 2 different doses of rozanolixizumab (7 mg/kg or 10 mg/kg) or matching placebo over a treatment course of 6 weeks, followed by an 8-week observation period. The rozanolixizumab 7 mg/kg dose is the dose under review by Health Canada; therefore, it was the focus of this report. The primary outcome was the change from baseline to day 43 in MG-ADL scores. Secondary outcomes of interest for this review included the percentage of patients achieving an MG-ADL response (i.e., an improvement from baseline in MG-ADL score of at least 2 points) at day 43 and the change from baseline to day 43 in QMG score. Patients who completed the 6-week treatment period and 8-week observation period in the MycarinG trial had the opportunity to roll over into the MG0004 trial, an OLE study in which the long-term safety, tolerability, and efficacy of rozanolixizumab was measured in patients with gMG over 52 weeks of weekly chronic treatment. The MG0004 trial was terminated in 2021 and replaced by the MG0007 trial, an ongoing OLE study consisting of 6-week treatment cycles based on MG worsening. Patients could roll over from the MycarinG trial or the MG0004 trial into the MG0007 trial directly.
Patients in the pivotal study were aged 52 years on average (SD = 16 years), and the majority (61%) were female. Most patients were MGFA class IIa or IIb (39%) or class IIIa or IIIb (57%) at baseline. At baseline, the majority of patients were AChR antibody-positive (83%), while 9% were MuSK antibody-positive.
There are no trials comparing rozanolixizumab to other potential comparators for gMG, and indirect evidence from the NMA was included in the pharmacoeconomic model; therefore, an appraisal of the indirect evidence was undertaken. The submission included a body of indirect evidence consisting of 2 relevant NMAs and 2 MAICs. The primary analyses for the NMA were conducted in phase III trials and included evidence from eculizumab and efgartigimod; other comparators included in the network (ravulizumab and zilucoplan) were not included in the review because ravulizumab is not recommended to reimburse in gMG, zilucoplan was not approved for gMG at the time of the review, and rituximab was not included in the network for the NMA primary analysis. The MAICs included comparisons of rozanolixizumab with efgartigimod (i.e., the ADAPT trial, a 26-week, phase III, double-blind, placebo-controlled, multisite RCT comparing efgartigimod to placebo in patients with class II to IV gMG) and rozanolixizumab with IVIg (i.e., the Barth et al. [2011] trial, a 60-day, phase IV, randomized, active-controlled trial comparing IVIg with PLEX in patients with gMG who require a change in therapy).
Interpretation of Results
Efficacy
Although rozanolixizumab is indicated for gMG, the sponsor-requested reimbursement criteria is for patients with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy with AChEIs, NSISTs, or corticosteroids. Although prior treatment with 1 of these conventional therapies was not an inclusion criterion for the MycarinG trial, nearly all patients (96%) had received at least 1 conventional therapy. However, only 32% of patients had received all 3 classes of conventional therapy. It is unclear from the reimbursement request how many conventional therapies a patient should try before moving on to rozanolixizumab; however, the clinical expert consulted by the review team noted that they would not necessarily expect patients to have tried all 3 classes, given that there may be patients with contraindications or who are unable to tolerate corticosteroids or immunosuppressants, each of which can have significant side effects. It is also unclear whether the number of prior therapies affects response to therapy with rozanolixizumab, given that the only subgroup analyses performed were post hoc, and such analyses are typically not used to draw definitive conclusions. Results from this post hoc subgroup analysis suggest that the magnitude of the between-group difference is smaller in the subgroup of patients with 2 or more prior therapies, while for the QMG score, the between-group difference seems more similar between the subgroup of patients who had tried 2 or more prior therapies and the subgroup that had not. Therefore, because almost all patients in the MycarinG trial had tried at least 1 conventional therapy, we can draw conclusions about the efficacy and harms of rozanolixizumab in patients who have tried 1 therapy, but no more.
Treatment with rozanolixizumab likely elicited a clinically important improvement in MG-ADL scores and QMG scores after a 43-day treatment period. The clinical expert consulted on this review emphasized the importance of each instrument in assessing patients with gMG, noting that MG-ADL is a patient-reported outcome that clinicians would use to follow subjective assessments from patients about their symptoms, while the QMG is an objective measure that is administered by clinicians to assess patients (however, it would be used less often than the MG-ADL). The clinical expert was less familiar with the MG-QoL15r instrument used to assess HRQoL, and the sponsor was unable to find a MID in the literature; therefore, although there appeared to be a statistically significant improvement in this instrument during the pivotal trial, the clinical significance of this improvement is not known. Patient input would suggest that HRQoL is a key outcome in this disease. The population of patients who were MuSK antibody-positive in the MycarinG trial was small (n = 12 in the groups assessed), which reduces the confidence in the results for this population. However, the proportion of patients who were MuSK antibody-positive aligned with the disease prevalence in the MG population, and subgroup analyses suggested that the effect seen in the full population was consistent in this group.
The patients in the MycarinG trial were treated for only 6 weeks, and the outcomes were assessed at the end of this treatment period. As a result, the RCT phase of this pivotal trial provides only very limited information about the longer-term efficacy and harms of rozanolixizumab. There were long-term extensions to the MycarinG trial; however, these were single-arm studies, which makes it challenging to draw causal conclusions about the effects of rozanolixizumab. Despite this, in the MG0004 trial, the improvements in MG-ADL scores appeared to be maintained through 52 weeks. (That said, by that time, there were only 5 patients remaining in the rozanolixizumab 7 mg/kg group from the original intention-to-treat set of 35 patients.) Similar results were seen for the MG0007 trial extension: efficacy results were maintained across treatment cycles, but steady attrition resulted in a small sample as follow-up reached 52 weeks. As a result, the findings are at risk of attrition bias. Additionally, these extensions were open label; therefore, bias may have been introduced by patients knowing which group they were assigned to, and patient-reported outcomes, such as MG-ADL, are particularly prone to this type of bias.
Appraisal of the indirect treatment comparisons showed that the treatment history, disease severity, and antibody status of patients included in the trials were sources of considerable heterogeneity that were not controlled for in the NMA. Furthermore, the trials in the network enrolled both refractory and newly diagnosed patients, used MG-ADL thresholds ranging from 3 points to 6 points, used QMG thresholds ranging from 6 points to 12 points (3 studies), and differed in their placebo responses. These limitations affect the confidence that the transitivity assumption underpinning the NMA was met. Added to imprecision in the effect estimates, this limits the ability to draw conclusions about the comparative efficacy of rozanolixizumab versus eculizumab or efgartigimod at 12 weeks of follow-up. The anchored MAIC for efgartigimod and rozanolixizumab was subject to limitations, such as considerably smaller ESSs than the trials, and a lack of information on baseline characteristics not included in weighting. Overall, the findings from the MAIC comparing efgartigimod to rozanolixizumab suggest that the effect of these treatments at 4 weeks to 6 weeks of follow-up ███ ██ ███████; however, there remains uncertainty in the results. The unanchored MAIC comparing IVIg to rozanolixizumab was limited considerably by the fact that not all prognostic or effect-modifying factors were included in the weighting, and differences remained in the proportion of patients with MuSK antibodies. Therefore, conclusions about the comparative efficacy of rozanolixizumab and IVIg cannot be drawn. Both MAICs are also subject to limitations around generalizability with regard to the timing of the analysis (2 weeks to 6 weeks) because the results may not capture maximal treatment response. Lastly, information about long-term comparative efficacy and safety is unavailable.
Harms
Patient input suggests that the side effects associated with the medications used to treat gMG are an important issue. Patients noted that this would be a key consideration for them when deciding whether to switch from 1 therapy to another. Patients seemed to be most concerned about the side effects associated with corticosteroids, which is not surprising, considering the large number of harms associated with the use of this class of drugs. Although patients did not comment specifically on AChEIs, this class of drugs is also known for its side effects; however, these tend to tolerability issues (e.g., nausea, vomiting, diarrhea), in contrast to the more serious and potentially permanent adverse effects associated with corticosteroids, such as impaired bone health and fractures. When assessing the harms associated with rozanolixizumab, it is important to note that in the pivotal trial, harms were assessed after 6 weeks; this is clearly not a sufficient follow-up period or duration of treatment to assess the risk for longer-term harms associated with its use. Nevertheless, the data reported for harms over this relatively short follow-up period suggest no obvious tolerability or safety issues with this novel drug. A similar number of patients (3% in each group) stopped treatment due to an AE. In addition, there were similar numbers of patients who had SAEs (5 patients [8%] in the rozanolixizumab group and 6 patients [9%] in the placebo group). Headache was numerically more common in patients treated with rozanolixizumab than with placebo (45% versus 19% of patients), as was diarrhea (25% versus 13%); however, as noted, these adverse effects did not appear to result in issues with patients continuing with therapy. There were 5 patients (8%) treated with rozanolixizumab who reported an AE of hypertension compared to 0 in the placebo group; however, there were no events of hypertension reported as SAEs. Due to its mechanism of action and based on the information available on the product monograph for rozanolixizumab, infections were identified as the notable harm of interest for this review.
There was no indication from the 7 mg/kg dose in the pivotal trial of an increased risk of infection or serious infection with rozanolixizumab compared to placebo. There were numerically fewer infection- and infestation-related AEs with rozanolixizumab compared to placebo (16% versus 19%), and there was no pattern indicating increased risk of any specific infections with rozanolixizumab. However, there was some indication of an increased risk of infections and infestations at the higher dose (10 mg/kg), given that these were reported in 30% of patients. Although approval is not being sought for this higher dose, this increased risk is noted in the product monograph. There was no indication of any issues with infections during the extension phase.
Harms were not analyzed in the indirect evidence comparison; therefore, comparative evidence on harms for rozanolixizumab is lacking.
Conclusion
One pivotal, sponsor-funded, multinational (81 sites, including 4 in Canada), phase III, double-blind RCT was included in this report. The MycarinG trial randomized 200 patients with AChR antibody-positive or MuSK antibody-positive gMG in a 1-to-1-to-1 ratio to 1 of 2 doses of rozanolixizumab (7 mg/kg or 10 mg/kg) or to placebo, administered as weekly SC infusions over a 6-week treatment course. The rozanolixizumab 7 mg/kg dose is the dose being sought for Health Canada approval; therefore, this dose is the focus of this report. After 43 days, rozanolixizumab likely resulted in a clinically significant improvement in MG-ADL scores compared to placebo; this was the primary outcome of this study. Treatment with rozanolixizumab also likely resulted in a clinically significant increase in the number of MG-ADL responders and QMG scores after 43 days compared to placebo. Rozanolixizumab also likely improved MG-QoL15r scores after 43 days compared to placebo; however, the clinical significance of this improvement is not known. A post hoc subgroup analysis of patients with 2 or more prior treatments suggested ████████ █████████ ██ ███████ on MG-ADL and QMG in this group. The number of patients was small, and the analysis was not preplanned, limiting the conclusions that can be drawn. Infection was a notable harm identified for this review; however, there was no indication of increased infection risk at the 7 mg/kg dose of rozanolixizumab. Long-term evidence was limited by open-label, noncomparative extension studies (the MG0004 and MG0007 trials). These studies suggested that the benefits of rozanolixizumab may be maintained for up to 52 weeks; however, the ability to draw definitive conclusions was limited by the lack of a control group, risk of bias due to the open-label design, and substantial attrition. No additional safety concerns were identified in the extension studies.
The submitted NMA was limited by likely intransitivity and imprecision, introducing uncertainty about the comparative efficacy of rozanolixizumab versus eculizumab or efgartigimod at 12 weeks of follow-up. An anchored MAIC suggested that efgartigimod and rozanolixizumab ███ ████ ███████ ███████ at 4 weeks to 6 weeks of follow-up. These findings were uncertain due to the potential for residual confounding, small ESS, and some imprecision. The unanchored MAIC comparing IVIg to rozanolixizumab had important limitations that limited the ability to draw conclusions. Both MAICs are limited in generalizability because the timing of maximal treatment response might not have been captured. Information about long-term comparative efficacy and harms is unavailable, and comparison to some relevant treatments (e.g., rituximab) is lacking.
References
1.Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren J. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30. doi: 10.1038/s41572-019-0079-y PubMed
2.Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res. 2016;5. doi: 10.12688/f1000research.8206.1
3.Li Y, Arora Y, Levin K. Myasthenia gravis: newer therapies offer sustained improvement. Cleve Clin J Med. 2013;80(11):711-21. doi: 10.3949/ccjm.80a.13044 PubMed
4.Bird SJ. Shefner JM, Goddeau RD, eds. Clinical manifestations of myasthenia gravis. UpToDate; 2024. Accessed September 12, 2024. http://www.uptodate.com
5.Bird SJ. Shefner JM, Goddeau RD, eds. Diagnosis of myasthenia gravis. UpToDate; 2022. Accessed September 12, 2024. http://www.uptodate.com
6.Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. J Clin Med. 2021;10(11). doi: 10.3390/jcm10112235 PubMed
7.Bubuioc AM, Kudebayeva A, Turuspekova S, Lisnic V, Leone MA. The epidemiology of myasthenia gravis. J Med Life. 2021;14(1):7-16. doi: 10.25122/jml-2020-0145 PubMed
8.Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:46. doi: 10.1186/1471-2377-10-46 PubMed
9.Schroeter M, Thayssen G, Kaiser J. Myasthenia Gravis - Exacerbation and Crisis. Neurology International Open. 2018;2:E10-E15. https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0043-118441.pdf
10.The Centre for International Economics. The cost to patients and the community of myasthenia gravis. 2013. Accessed November 15, 2023. https://www.touchneurology.com/wp-content/uploads/sites/3/2018/06/www.thecie.com_.au_wp-content_uploads_2014_06_Final-report_Economic-Impact-of-Myasthenia-Gravis-08112013.pdf
11.Law N, Davio K, Blunck M, Lobban D, Seddik K. The Lived Experience of Myasthenia Gravis: A Patient-Led Analysis. Neurol Ther. 2021;10(2):1103-1125. doi: 10.1007/s40120-021-00285-w PubMed
12.Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: Executive summary. Neurology. 2016;87(4):419-25. doi: 10.1212/WNL.0000000000002790 PubMed
13.Sussman J, Farrugia ME, Maddison P, Hill M, Leite MI, Hilton-Jones D. Myasthenia gravis: Association of British Neurologists' management guidelines. Pract Neurol. 2015;15(3):199-206. doi: 10.1136/practneurol-2015-001126 PubMed
14.Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021;96(3):114-122. doi: 10.1212/WNL.0000000000011124 PubMed
15.Tran C, Biswas A, Mendoza M, Katzberg H, Bril V, Barnett C. Performance of different criteria for refractory myasthenia gravis. Eur J Neurol. 2021;28(4):1375-1384. doi: 10.1111/ene.14675 PubMed
16.Eversana. Rozanolixizumab for gMG Clinician Interview Report [sponsor submitted reference]. 2024.
17.CADTH. CADTH Reimbursement Review: Efgartigimod Alfa (Vyvgart) [sponsor submitted reference]. 2024. https://www.cda-amc.ca/sites/default/files/DRR/2024/SR0782REC-Vyvgart-meta.pdf
18.CADTH. Efgartigimod Alfa (VYVGART) Draft Reimbursement Recommendation [sponsor submitted reference]. 2023.
19.CADTH. Eculizumab (SOLIRIS) Final Reimbursement Recommendation [sponsor submitted reference]. 2020.
20.Rodolico C, Bonanno C, Toscano A, Vita G. MuSK-Associated Myasthenia Gravis: Clinical Features and Management. Front Neurol. 2020;11. doi: 10.3389/fneur.2020.00660 PubMed
21.Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2020. doi: 10.1212/WNL.0000000000011124 PubMed
22.Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401-6. doi: 10.1016/j.jclinepi.2010.07.015 PubMed
23.Santesso N, Glenton C, Dahm P, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. J Clin Epidemiol. 2020;119:126-135. doi: 10.1016/j.jclinepi.2019.10.014 PubMed
24.UCB Canada Inc. (UCB). Rozanolixizumab as an add-on therapy for the treatment of adult patients with gMG who are either AChR or MuSK antibody positive and for whom symptoms persist despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs. [internal sponsor's summary of clinical evidence report]. In: Drug Reimbursement Review sponsor submission: rozanolixizumab (Rystiggo), 140mg/mL solution for subcutaneous injection (280mg in 2mL). UCB Canada; 2024.
25.UCB Canada response to October 8, 2024 CADTH request for additional information regarding rozanolixizumab CADTH review (part 2) [internal additional sponsor's information]. UCB Canada; 2024.
26.NICE Decision Support Unit. NICE DSU technical support document 18: methods for population-adjusted indirect comparisons in submissions to NICE. 2016. Accessed November 8, 2024. https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf
27.Bird SJ. Shefner JM, Goddeau RD, eds. Overview of the treatment of myasthenia gravis. UpToDate; 2023. Accessed September 11, 2024. http://www.uptodate.com
28.Wang L, Zhang Y, He M. Clinical predictors for the prognosis of myasthenia gravis. BMC Neurol. 2017;17(1):77. doi: 10.1186/s12883-017-0857-7 PubMed
29.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116(11):2843-54. doi: 10.1172/JCI29894 PubMed
30.Lazaridis K, Tzartos SJ. Autoantibody Specificities in Myasthenia Gravis; Implications for Improved Diagnostics and Therapeutics. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.00212 PubMed
31.Borges LS, Richman DP. Muscle-Specific Kinase Myasthenia Gravis. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.00707 PubMed
32.Claytor B, Cho S-M, Li Y. Myasthenic crisis. Muscle Nerve. 2023;68(1):8-19. doi: https://doi.org/10.1002/mus.27832 PubMed
33.Shanker PS, Ramizuddin K. Review on: Myasthenia Gravis and Telithromycin-Myasthenia Crisis. IOSR Journal of Dental and Medical Sciences. 2014;13(7):67-97. https://www.iosrjournals.org/iosr-jdms/papers/Vol13-issue7/Version-5/N013756797.pdf
34.Cao M, Koneczny I, Vincent A. Myasthenia Gravis With Antibodies Against Muscle Specific Kinase: An Update on Clinical Features, Pathophysiology and Treatment. Front Mol Neurosci. 2020;13. doi: 10.3389/fnmol.2020.00159 PubMed
35.Nelke C, Stascheit F, Eckert C, et al. Independent risk factors for myasthenic crisis and disease exacerbation in a retrospective cohort of myasthenia gravis patients. J Neuroinflammation. 2022;19(1). doi: 10.1186/s12974-022-02448-4 PubMed
36.Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: Multicenter blinded prospective review. Neurology. 2017;89(10):1069-1077. doi: 10.1212/WNL.0000000000004341 PubMed
37.Beecher G, Anderson D, Siddiqi ZA. Rituximab in refractory myasthenia gravis: Extended prospective study results. Muscle Nerve. 2018;58(3):452-455. doi: 10.1002/mus.26156 PubMed
38.Anderson D, Phan C, Johnston WS, Siddiqi ZA. Rituximab in refractory myasthenia gravis: a prospective, open‐label study with long‐term follow‐up. Ann Clin Transl Neurol. 2016;3(7):552-555. doi: 10.1002/acn3.314 PubMed
39.Cortés‐Vicente E, Rojas‐Garcia R, Díaz‐Manera J, et al. The impact of rituximab infusion protocol on the long‐term outcome in anti‐MuSK myasthenia gravis. Ann Clin Transl Neurol. 2018;5(6):710-716. doi: 10.1002/acn3.564 PubMed
40.Topakian R, Zimprich F, Iglseder S, et al. High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol. 2019;266(3):699-706. doi: 10.1007/s00415-019-09191-6 PubMed
41.Afanasiev V, Demeret S, Bolgert F, Eymard B, Laforêt P, Benveniste O. Resistant myasthenia gravis and rituximab: A monocentric retrospective study of 28 patients. Neuromuscul Disord. 2017;27(3):251-258. doi: 10.1016/j.nmd.2016.12.004 PubMed
42.Tandan R, Hehir MK, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve. 2017;56(2):185-196. doi: 10.1002/mus.25597 PubMed
43.U. C. B. Canada Inc. Rozanolixizumab Product Monograph (Draft) [sponsor submitted reference]. 2023.
44.Bausch Health Canada Inc. MESTINON (pyridostigmine bromide) [sponsor submitted reference]. 2019. Accessed July 3.
45.Apotex Inc. APO-PREDNISONE (prednisone tablets) [sponsor submitted reference]. 2015.
46.Apotex Inc. APO-AZATHIOPRINE (azathioprine tablets) [sponsor submitted reference]. 2023.
47.Novartis Pharmaceuticals Canada Inc. NEORAL (cyclosporine capsules) [sponsor submitted reference]. 2023. Accessed February 23.
48.Accord Healthcare Inc. ACH-Mycophenolate (mycophenolte mofetil tablets) [sponsor submitted reference]. 2022.
49.Accord Healthcare Inc. ACH-Tacrolimus (tacrolimus capsule) [sponsor submitted reference]. 2023.
50.Hoffman-La Roche Limited. RITUXAN (rituximab for injection) [sponsor submitted reference]. 2023.
51.Alexion Pharma GmbH. SOLIRIS (eculizumab) Product Monograph [sponsor submitted reference]. 2021. https://pdf.hres.ca/dpd_pm/00060546.PDF
52.argenx B. V. VVYGART (efgartigimod alfa) Product Monograph [sponsor submitted reference]. 2023. https://pdf.hres.ca/dpd_pm/00073416.PDF
53.Bril V, Drużdż A, Grosskreutz J, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 2023;22(5):383-394. doi: 10.1016/s1474-4422(23)00077-7 PubMed
54.MycarinG (MG0003) Clinical Study Report [sponsor submitted reference]. 2022.
55.Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology. 1999;52(7):1487-9. doi: 10.1212/wnl.52.7.1487 PubMed
56.Muppidi S, Wolfe GI, Conaway M, Burns TM. MG-ADL: still a relevant outcome measure. Muscle Nerve. 2011;44(5):727-31. doi: 10.1002/mus.22140 PubMed
57.Benatar M, Sanders DB, Burns TM, et al. Recommendations for myasthenia gravis clinical trials. Muscle Nerve. 2012;45(6):909-17. doi: 10.1002/mus.23330 PubMed
58.Sadjadi R, Conaway M, Cutter G, Sanders DB, Burns TM. Psychometric evaluation of the myasthenia gravis composite using Rasch analysis. Muscle Nerve. 2012;45(6):820-5. doi: 10.1002/mus.23260 PubMed
59.Burns TM, Conaway M, Sanders DB. The MG Composite: A valid and reliable outcome measure for myasthenia gravis. Neurology. 2010;74(18):1434-40. doi: 10.1212/WNL.0b013e3181dc1b1e PubMed
60.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769-72. doi: 10.1111/j.1749-6632.1998.tb11015.x PubMed
61.Katzberg HD, Barnett C, Merkies IS, Bril V. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve. 2014;49(5):661-5. doi: 10.1002/mus.23988 PubMed
62.UCB. MG0007 Clinical Study Report [sponsor submitted reference]. 2022.
63.Burns TM, Grouse CK, Conaway MR, Sanders DB. Construct and concurrent validation of the MG-QOL15 in the practice setting. Muscle Nerve. 2010;41(2):219-26. doi: 10.1002/mus.21609 PubMed
64.Burns TM, Sadjadi R, Utsugisawa K, et al. International clinimetric evaluation of the MG-QOL15, resulting in slight revision and subsequent validation of the MG-QOL15r. Muscle Nerve. 2016;54(6):1015-1022. doi: 10.1002/mus.25198 PubMed
65.Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring Clinical Treatment Response in Myasthenia Gravis. Neurol Clin. 2018;36(2):339-353. doi: 10.1016/j.ncl.2018.01.006 PubMed
66.Burns TM, K. GC, I. WG, R. CM, B. SD. The MG-QOL15 for following the health-related quality of life of patients with myasthenia gravis. Muscle Nerve. 2011;43(1):14-8. doi: 10.1002/mus.21883 PubMed
67.Luo Y, Dong X, Peng Y, et al. Evaluation of outcome measures for myasthenia gravis subgroups. J Clin Neurosci. 2021;91:270-275. doi: 10.1016/j.jocn.2021.07.020 PubMed
68.Barnett C, Katzberg H, Nabavi M, Bril V. The quantitative myasthenia gravis score: comparison with clinical, electrophysiological, and laboratory markers. J Clin Neuromuscul Dis. 2012;13(4):201-5. doi: 10.1097/CND.0b013e31824619d5 PubMed
69.Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann N Y Acad Sci. 2003;998:440-4. doi: 10.1196/annals.1254.057 PubMed
70.Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker-Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology. 2005;64(11):1968-70. doi: 10.1212/01.Wnl.0000163988.28892.79 PubMed
71.Barnett C, Merkies IS, Katzberg H, Bril V. Psychometric Properties of the Quantitative Myasthenia Gravis Score and the Myasthenia Gravis Composite Scale. J Neuromuscul Dis. 2015;2(3):301-311. doi: 10.3233/jnd-150082 PubMed
72.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-905. doi: 10.1007/s11136-012-0344-y PubMed
73.UCB Canada response to October 8, 2024 CADTH request for additional information regarding rozanolixizumab CADTH review [internal additional sponsor's information]. UCB Canada; 2024.
74.Wassmer G, Brannath W. Group Sequential and Confirmatory Adaptive Designs in Clinical Trials. Springer Series in Pharmaceutical Statistics. 2016. doi: 10.1007/978-3-319-32562-0
75.MG0004 Clinical Study Report [sponsor submitted reference]. 2022.
76.National Institute for Health and Care Excellence. National Institute for Health and Care Excellence (NICE): Technology Appraisals. 2015. Accessed November 7, 2024. https://www.nice.org.uk/Media/Default/About/accreditation/accreditation-decisions/NICE-STA-%20Renewal_Final%20accreditation_report.pdf
77.A systematic literature review of clinical evidence in Myasthenia Gravis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. Prepared for UCB Canda by Parexel; September 2023.
78.Network meta-analysis of zilucoplan and rozanolixizumab versus competitors for the treatment of generalized myasthenia gravis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. Prepared for UCB Canada by Parexel; January 2024.
79.Howard JF, Jr., Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976-986. doi: 10.1016/S1474-4422(17)30369-1 PubMed
80.Piehl F, Eriksson-Dufva A, Budzianowska A, et al. Efficacy and Safety of Rituximab for New-Onset Generalized Myasthenia Gravis: The RINOMAX Randomized Clinical Trial. JAMA Neurol. 2022;79(11):1105-1112. doi: 10.1001/jamaneurol.2022.2887 PubMed
81.Howard JF, Jr., Bresch S, Genge A, et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. 2023;22(5):395-406. doi: 10.1016/S1474-4422(23)00080-7 PubMed
82.Howard JF, Jr., Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-536. doi: 10.1016/S1474-4422(21)00159-9 PubMed
83.Vu T, Meisel A, Mantegazza R, et al. Terminal Complement Inhibitor Ravulizumab in Generalized Myasthenia Gravis. NEJM Evid. 2022;1(5):EVIDoa2100066. doi: 10.1056/EVIDoa2100066
84.Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. 2011;76(23):2017-23. doi: 10.1212/WNL.0b013e31821e5505 PubMed
85.Comparing the efficacy of zilucoplan and rozanolixizumab versus competitors for the treatment of generalized myasthenia gravis through matched-adjusted indirect comparison [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. Prepared for UCB Canada by Parexel; March 2024.
Appendix 1: Detailed Outcome Data
Please note that this appendix has not been copy-edited.
Table 36: Other Efficacy Outcomes Reported in the Pivotal Study
Outcomes | Rozanolixizumab 7 mg/kg (N = 66) | Placebo (N = 67) |
|---|---|---|
MG symptoms PRO muscle weakness fatigability score change from baseline to day 43 | ||
Number of patients contributing to the analysis | 65 | 62 |
Baseline, mean (SE) | −18.89 (3.197) | −6.14 (2.310) |
LS mean (SE) | −23.029 (3.034) | −10.588 (3.034) |
Treatment group difference vs. control (95% CI) | −12.441 (−21.804, −4.089) | |
P value | < 0.001 | — |
MG symptoms PRO physical fatigue score change from baseline to day 43 | ||
Number of patients contributing to the analysis | 65 | 62 |
Baseline, mean (SD) | −16.10 (2.817) | −7.53 (2.304) |
LS mean (SE) | −19.287 (3.046) | −10.637 (3.051) |
Treatment group difference vs. control (95% CI) | −8.650 (−18.058, −0.134) | |
P value | 0.012 | — |
MG symptoms PRO bulbar muscle weakness score change from baseline to day 43 | ||
Number of patients contributing to the analysis | 65 | 62 |
Baseline, mean (SD) | −13.69 (2.382) | −2.26 (2.124) |
LS mean (SE) | −14.839 (2.406) | −3.519 (2.397) |
Treatment group difference vs. control (95% CI) | −11.320 (−18.958, −4.998) | |
P value | < 0.001 | — |
Use of rescue medication | ||
Any rescue medication, n (%) | 1 (1.5) | 3 (4.5)a |
During treatment period, n (%) | 0 | 3 (4.5) |
During observation period, n (%) | 1 (1.5) | 1 (1.5) |
Minimal symptom expression at any time during treatment and observation periods | ||
n (%) | 66 | 67 |
Yes, n (%) | 17 (25.8) | 2 (3.0) |
EQ-5D-5L VAS change from baseline to day 43 | ||
Baseline, n | 65 | 66 |
Mean (SD) | 57.8 (16.5) | 54.4 (19.2) |
Day 43, n | 65 | 66 |
Mean (SD) | 70.2 (19.9) | 60.0 (18.5) |
Change from baseline day 43, n | 64 | 65 |
Mean (SD) | 12.2 (19.9) | 6.1 (18.2) |
MGII change from baseline to day 43 in MycarinG | ||
Baseline, n | 55 | 53 |
Mean (SD) | 36.3 (15.2) | 35.6 (12.4) |
Day 43, n | 49 | 48 |
Mean (SD) | 23.2 (14.6) | 32.6 (13.8) |
Change from baseline to day 43, n | 49 | 47 |
Mean (SD) | −12.4 (16.5) | −3.4 (10.4) |
CI = confidence interval; LS = least squares; MGII = Myasthenia Gravis Impairment Index; PRO = patient-reported outcome; SD = standard deviation; SE = standard error; VAS = visual analogue scale; vs. = versus.
Note: Minimal symptom expression was defined as an MG-ADL total score of 0 or 1, at any time during the treatment and observation periods.
Source: Clinical Study Report for MycarinG.54
Table 37: Observed MG-ADL Responder Rates at Day 43 During Each Treatment Cycle in the MG0007 Trial (Safety Set)
Observed MG-ADL responder rates | Rozanolixizumab 7 mg/kg (N = 79) | |
|---|---|---|
Cycle 1 | n | 73 |
Responders, n (%) | 54 (74.0) | |
Cycle 2 | n | 50 |
Responders, n (%) | 33 (66.0) | |
Cycle 3 | n | 35 |
Responders, n (%) | 27 (77.1) | |
Cycle 4 | n | 29 |
Responders, n (%) | 25 (86.2) | |
Cycle 5 | n | 16 |
Responders, n (%) | 12 (75.0) | |
Cycle 6 | n | 7 |
Responders, n (%) | 4 (57.1) | |
Cycle 7 | n | 5 |
Responders, n (%) | 3 (60.0) | |
MG-ADL = Myasthenia Gravis Activities of Daily Living.
Note: Percentages were based on the number of patients with nonmissing data at each visit in the safety set. Total MG-ADL scores range from 0 to 24 with a higher score indicating more severe disability; MG-ADL responder at a visit was defined as having at least a 2.0-point improvement (decrease) from baseline. Baselines were defined as the last available value before or on the same date of first administration of the study treatment at each cycle (i.e., baseline [day 1]) value for that cycle. Patients were grouped according to the actual dose level received.
Source: MG0007 Clinical Study Report (2022), Table 6.3.1.62
Table 38: Changes in PRO Scores for MG Symptoms From Baseline to Day 43 During Each Treatment Cycle in the MG0007 Trial (Safety Set)
Change in MG Symptoms PRO | Rozanolixizumab 7 mg/kg (N = 79) | |
|---|---|---|
Muscle weakness fatigabilitya | ||
From baseline to Day 43 during cycle 1 | n | 74 |
Mean (SD) | −16.9 (20.0) | |
From baseline to Day 43 during cycle 2 | n | 50 |
Mean (SD) | −15.0 (19.2) | |
From baseline to Day 43 during cycle 3 | n | 34 |
Mean (SD) | −13.7 (14.7) | |
From baseline to Day 43 during cycle 4 | n | 29 |
Mean (SD) | −18.5 (15.9) | |
From baseline to Day 43 during cycle 5 | n | 16 |
Mean (SD) | −20.8 (19.9) | |
From baseline to Day 43 during cycle 6 | n | 7 |
Mean (SD) | −19.4 (24.4) | |
From baseline to Day 43 during cycle 7 | n | 5 |
Mean (SD) | −16.1 (9.9) | |
Physical fatigueb | ||
From baseline to Day 43 during cycle 1 | n | 74 |
Mean (SD) | −15.9 (17.9) | |
From baseline to Day 43 during cycle 2 | n | 50 |
Mean (SD) | −15.3 (21.4) | |
From baseline to Day 43 during cycle 3 | n | 34 |
Mean (SD) | −17.3 (17.6) | |
From baseline to Day 43 during cycle 4 | n | 29 |
Mean (SD) | −15.2 (20.4) | |
From baseline to Day 43 during cycle 5 | n | 16 |
Mean (SD) | −20.2 (18.4) | |
From baseline to Day 43 during cycle 6 | n | 7 |
Mean (SD) | −22.9 (28.9) | |
From baseline to Day 43 during cycle 7 | n | 5 |
Mean (SD) | −17.3 (16.2) | |
Bulbar muscle weaknessc | ||
From baseline to Day 43 during cycle 1 | n | 74 |
Mean (SD) | −12.7 (18.9) | |
From baseline to Day 43 during cycle 2 | n | 50 |
Mean (SD) | −11.5 (16.8) | |
From baseline to Day 43 during cycle 3 | n | 34 |
Mean (SD) | −15.0 (18.1) | |
From baseline to Day 43 during cycle 4 | n | 29 |
Mean (SD) | −12.0 (15.8) | |
From baseline to Day 43 during cycle 5 | n | 16 |
Mean (SD) | −15.4 (20.0) | |
From baseline to Day 43 during cycle 6 | n | 7 |
Mean (SD) | −17.6 (21.2) | |
From baseline to Day 43 during cycle 7 | n | 5 |
Mean (SD) | −14.0 (15.9) | |
MG = myasthenia gravis; PRO = patient-reported outcome; SD = standard deviation.
Note: Patients were grouped according to the actual dose level received within the study cycle. Baseline values were defined as the last available value before or on the same date of first administration of the study treatment at each cycle (i.e., baseline [day 1]) value for that cycle.
aThe muscle weakness fatigability scale score ranges from 0 to 100, with a higher result indicating more frequent and severe symptoms.
bThe physical fatigue scale score ranges from 0 to 100, with a higher result indicating more frequent and severe symptoms.
cThe bulbar muscle weakness scale score ranges from 0 to 100, with a higher result indicating more frequent and severe symptoms.
Source: MG0007 Clinical Study Report (2022), Table 6.4.1, Table 6.4.3, Table 6.4.5.62
Table 39: Survival Analysis of Treatment-Free Intervals in the MG0007 Trial (Safety Set)
Treatment-free interval | Rozanolixizumab 7 mg/kg (N = 79) | |
|---|---|---|
Between Cycles 1 and 2 | n | 79 |
Median (days) (95% CI) | 64.0 (50.0, 86.0) | |
% censored | 22.8 | |
Between Cycles 2 and 3 | n | 54 |
Median (days) (95% CI) | 57.0 (43.0, 67.0) | |
% censored | 20.4 | |
Between Cycles 3 and 4 | n | 41 |
Median (days) (95% CI) | 37.0 (35.00, 50.0) | |
% censored | 29.3 | |
Between Cycles 4 and 5 | n | 31 |
Median (days) (95% CI) | 35.0 (30.0, 47.0) | |
% censored | 25.8 | |
CI = confidence interval.
Note: Patients who did not commence a subsequent cycle were censored at the date of their last assessment within the study cycle. Patients were grouped according to the actual dose level received in the preceding study cycle.
Source: MG0007 Clinical Study Report (2022), Table 6.11.2.62
Table 40: Results of NMA Sensitivity Analyses
Detail | Rozanolixizumab 7 mg/kg | |
|---|---|---|
MG-ADL responder OR (95% CI) | Change from baseline in MG-ADL MD (95% CI)1 | |
Sensitivity 1a | ||
Efgartigimodb | ████ ██████ █████ | ████ ███████ █████ |
Eculizumabb | ████ ██████ █████ | ████ ███████ █████ |
Placebob,c | ████ ██████ ██████ | █████ ███████ █████ |
Sensitivity 2d | ||
Efgartigimodb | ██ | █████ ███████ █████ |
Eculizumabb | ██ | ████ ███████ █████ |
Placebob | ██ | █████ ███████ █████ |
Sensitivity 3e | ||
Efgartigimodb | ██ | ████ ███████ █████ |
Eculizumabb | ██ | █████ ███████ █████ |
Placebob | ██ | █████ ███████ ██████ |
Sensitivity 4f | ||
Efgartigimodb | ██ | ████ ███████ █████ |
Eculizumabb | ██ | ████ ███████ █████ |
Placebob | ██ | █████ ███████ ██████ |
Sensitivity 5g | ||
Efgartigimod | ██ | ████ ███████ █████ |
Eculizumab | ██ | ████ ███████ █████ |
Placebo | ██ | █████ ███████ ██████ |
CI = confidence interval; MD = mean difference; MG-ADL = Myasthenia Gravis Activities of Daily Living; NR = not reported; OR = odds ratio.
aIncluded Phase II and III trials.
bIncluded digitized data in the trial for that comparator for the change from baseline in MG-ADL.
cIncluded digitized data in the trial for that comparator, for the MG-ADL responders outcome.
dIncluded phase II, III studies reporting at 10 weeks ± 2 weeks, as well as studies reporting the primary end point at any other time.
eIncluded phase III studies reporting on the primary end point at any time point.
fIncluded phase II or III studies reporting on the primary end point at any time point.
gIncluded phase II or III studies reporting on the most commonly reported time point (4 weeks).
Source: details included in the table are from the sponsor’s NMA Technical Report.78
Pharmacoeconomic Review
Abbreviations
AChR
acetylcholine receptor
AE
adverse event
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CrI
credible interval
gMG
generalized myasthenia gravis
ICER
incremental cost-effectiveness ratio
IVIg
IV immunoglobulin
MAIC
matching-adjusted indirect comparison
MDC
Muscular Dystrophy Canada
MG
myasthenia gravis
MG-ADL
Myasthenia Gravis Activities of Daily Living
MGFA
Myasthenia Gravis Foundation of America
MuSK
muscle-specific tyrosine kinase
NMA
network meta-analysis
pCPA
pan-Canadian Pharmaceutical Alliance
PLEX
plasma exchange
QALY
quality-adjusted life-year
QMG
Quantitative Myasthenia Gravis
SC
subcutaneous
SCIg
subcutaneous immunoglobulin
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Rozanolixizumab (Rystiggo), 140 mg/mL solution for subcutaneous injection |
Indication | For the treatment of adult patients with gMG who are AChR or MuSK antibody positive |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 25, 2025 |
Reimbursement request | As an add-on therapy for the treatment of adult patients with gMG who are either AChR antibody-positive or MuSK antibody-positive and for whom symptoms persist despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs |
Sponsor | UCB Canada Inc. |
Submission history | None |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; gMG = generalized myasthenia gravis; MuSK = muscle-specific tyrosine kinase; NOC = Notice of Compliance; NSIST = nonsteroidal immunosuppressive therapy.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with AChR antibody-positive or MuSK antibody-positive gMG for whom symptoms persist despite conventional therapy with AChEIs, corticosteroids, and/or NSISTs |
Treatment | Rozanolixizumab (Rystiggo) plus conventional therapy |
Dose regimen | Administered weekly for 6 weeks. Subsequent treatment cycles are based on clinical evaluation and may vary by patient. Doses are: Body weight ≥ 35 to < 50 kg: 280 mg Body weight ≥ 50 kg to < 70: 420 mg Body weight ≥ 70 to < 100 kg: 560 mg Body weight ≥ 100 kg: 840 mg |
Submitted price | Rozanolixizumab 280 mg/2 mL single-dose vial: $12,260 |
Submitted treatment cost | $436,956 per year, assuming 2.97 treatment cycles per year and a patient weighing ≥ 70 kg and < 100 kg |
Comparators |
|
Perspective | Publicly funded health care payer in Canada |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (52.5 years) |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CDA-AMC reanalysis results |
|
AChR = acetylcholine receptor; AChEI = acetylcholinesterase inhibitor; CDA-AMC = Canada’s Drug Agency; gMG = generalized myasthenia gravis; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; IVIg = IV immunoglobulin; LY = life-year; MGFA = Myasthenia Gravis Foundation of America; MuSK = muscle-specific tyrosine kinase; NMA = network meta-analysis; NSIST = nonsteroidal immunosuppressive therapy; PLEX = plasma exchange; QALY = quality-adjusted life-year; SCIg = subcutaneous immunoglobulin.
aConventional therapy is defined as consisting of 12.5% each of prednisone, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, methotrexate, pyridostigmine, and cyclophosphamide.
Conclusions
In adult patients with Myasthenia Gravis Foundation of America (MGFA) class II to IVa acetylcholine receptor (AChR) antibody-positive or muscle-specific tyrosine kinase (MuSK) antibody-positive generalized myasthenia gravis (gMG), evidence from the MycarinG trial demonstrated that compared to placebo, treatment with rozanolixizumab resulted in a statistically significant improvement in change from baseline to day 43 in Myasthenia Gravis Activities of Daily Living (MG-ADL) score. Based on the clinical review team’s appraisal of the MycarinG trial, treatment with rozanolixizumab likely resulted in a clinically important improvement in MG-ADL score and an increase in the number of patients with at least a 2-point improvement in MG-ADL score compared to placebo at day 43. In the absence of direct evidence comparing rozanolixizumab to other active comparators currently used as add-on therapies for the treatment of adults with gMG (i.e., chronic IV immunoglobulin [IVIg] or subcutaneous immunoglobulin [SCIg], chronic plasma exchange [PLEX], rituximab, eculizumab, or efgartigimod alfa), the sponsor submitted a network meta-analysis (NMA) as well as naive comparisons. Input from these sources informed the economic model regarding the relative probability of response to therapy (defined as a ≥ 3-point improvement in MG-ADL score, a ≥ 3-point improvement in QMG score, or a ≥ 5-point improvement in Quantitative Myasthenia Gravis [QMG] score, depending on comparator) and extent of response (the proportion of responders estimated to have a 3-point to 4-point improvement in MG-ADL score versus a ≥ 5-point improvement) among comparators. Response to therapy was applied at different time points in the model, depending on the comparator. The Clinical Review by Canada’s Drug Agency (CDA-AMC) concluded that the sponsor’s NMA had several limitations (i.e., sparse evidence networks, imprecision of estimates, and heterogeneity in patient and study characteristics) that precluded the drawing of conclusions on the comparative efficacy of rozanolixizumab relative to its add-on comparators.
CDA-AMC was unable to address limitations with the sponsor’s submitted model as well as uncertainty related to the comparative clinical data and long-term efficacy of rozanolixizumab compared to other add-on therapies used in addition to conventional therapy in adults with AChR antibody-positive or MuSK antibody-positive gMG and persistent symptoms despite conventional therapy. As such, CDA-AMC was unable to derive a more reliable base-case estimate of the cost-effectiveness of rozanolixizumab. Results of the sponsor’s base-case sequential analysis suggest that rozanolixizumab plus conventional therapy is associated with an incremental cost-effectiveness ratio (ICER) of $2,676,135 per quality-adjusted life-year (QALY) gained (incremental cost = $21,998; incremental QALYs = 0.008) compared to efgartigimod alfa plus conventional therapy. Based on this analysis, a price reduction of approximately 87.5% would be required for rozanolixizumab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. This reduces the unit price to $1,533 from $12,260 per 280 mg vial (in turn reducing the annual drug acquisition cost to $54,620 from $436,956 per patient). This analysis likely underestimates the ICER, given that the sponsor’s model was programmed to assume treatment effects were maintained beyond treatment discontinuation. There is no robust evidence to support this assumption regarding maintenance of effect. CDA-AMC could not revise treatment effects, but undertook scenario analyses (using the submitted price of rozanolixizumab) increasing the duration of treatment to match the duration of benefit, in which the ICER increased to $9,682,589 per QALY gained relative to efgartigimod alfa plus conventional therapy due to increased drug acquisition costs. As such, a greater price reduction may be required to account for the assumptions used by the sponsor. The cost-effectiveness of rozanolixizumab in patients not studied in the MycarinG trial (e.g., those with MGFA class I, IVb, or V gMG, those not meeting the MG-ADL and QMG inclusion thresholds, or those not under consideration for additional treatment with IVIg or PLEX) is unknown.
The results of the economic model were based on efficacy inputs obtained from the sponsor’s submitted NMA and naive comparisons. CDA-AMC notes that the relative effects obtained from the NMA are highly unreliable due to the analysis limitations, and the naive comparisons used for the effectiveness parameters of some comparators in the model are highly uncertain. While the sponsor’s base case suggests differences in treatment benefit for adults with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy, these results will be realized only if the numerical differences observed in the NMA and naive comparisons occur in clinical practice and lead to meaningful improvement for patients. Clinical expert feedback suggested that rozanolixizumab and efgartigimod alfa may have similar efficacy.
Patient, Clinician, and Drug Plan Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, clinician groups, and drug plans that participated in the CDA-AMC review process.
Patient input was received from Muscular Dystrophy Canada (MDC), which conducted an online survey, interviews, round tables, and health-related quality of life measures to produce a qualitative thematic analysis. In total, its submission included data from 194 individuals with a confirmed diagnosis of gMG. Respondents were from all provinces of Canada. Among them, 20 respondents were specifically asked to provide input on their hopes and expectations for rozanolixizumab.
MDC identified the following themes regarding MG symptoms: loss of productivity; fatigue; energy levels and quality of sleep; respiratory health; mobility and strength; independence; relationships and social participation; and vision, speech, and swallowing. Themes identified regarding current treatments included negative experiences with steroids, the slow onset of medication effects, and a feeling of trial and error with medications. Patients reported experience with prednisone, pyridostigmine, azathioprine, mycophenolate mofetil, IVIg, and thymectomy; they reported difficulty accessing rituximab. IVIg was reported by several patients as being effective or helpful, but patients also said it was time consuming, with the effects wearing off too quickly. In terms of treatment gaps, MDC reported themes of patients wanting decreased intensity of exacerbations and side effects, maintenance of independence, and less serious hospital admissions. Patients were reported as stating that their current medications seemed to decrease the number of exacerbations but did not affect their overall quality of life. MDC emphasized the need for improved treatment options. No responding patients had experience with rozanolixizumab.
Clinician input was received from the Neuromuscular Disease Network for Canada. The clinician group stated that the current treatment landscape for MG includes symptomatic therapy (e.g., acetylcholinesterase inhibitors), short-term rescue immunotherapy (e.g., PLEX and IVIg), and long-term immunosuppressive therapy (e.g., corticosteroids and nonsteroidal immunosuppressants). They noted that most of these therapies are off-label for gMG, require long treatment periods before benefit is observed, are not effective for all patients, and are associated with burdensome or intolerable side effects. The goal of gMG treatment was noted to be the achievement of complete remission, pharmacological remission, or minimal manifestation status (i.e., asymptomatic or no disease-related functional limitation), with minimal adverse events (AEs). When outlining which patients would be best suited for treatment with rozanolixizumab, the clinician group noted that patients with AChR antibody-positive or MuSK antibody-positive gMG. Efficacy may also be possible in patients with double seronegative status; however, this has not yet been established. Of particular concern were patients who are getting worse rapidly or have an impending MG crisis; these patients require a fast-acting therapy, such as rozanolixizumab. The clinician group also noted that rozanolixizumab would be an excellent option for patients who are candidates for, or are intolerant to, IVIg or SCIg therapy; that Fc receptor inhibitors, such as rozanolixizumab, are likely to replace immunoglobulin therapies; and that the subcutaneous (SC) administration of rozanolixizumab would be an advantage in avoiding the use of specialized infusion centres.
The drug plans noted that rozanolixizumab is the first Fc receptor inhibitor providing targeted therapy for both AChR antibody-positive and MuSK antibody-positive patients with worsening gMG symptoms despite conventional treatment. It has a shorter infusion time compared to efgartigimod alfa and an SC route of administration rather than IV. Drug plans questioned whether the prescribing criteria should be aligned with those for efgartigimod alfa, which is under active negotiation with the pan-Canadian Pharmaceutical Alliance (pCPA).
Several of these concerns were addressed in the sponsor’s model:
Model health states were based on improvements to the MG-ADL scale, which considers many of the symptoms mentioned by patients.
The sponsor’s model includes all comparators currently or potentially to be reimbursed soon for patients with gMG for whom symptoms persist despite conventional therapy.
The sponsor’s model included quality of life and mortality impacts for chronic corticosteroid use.
CDA-AMC was unable to address the following concern raised from this input:
a lack of robust comparative efficacy and safety data between zilucoplan and other add-on therapies to conventional therapy.
Economic Review
Economic Evaluation
Summary of the Sponsor’s Economic Evaluation
Overview
Rozanolixizumab (Rystiggo) is indicated for the treatment of adult patients with gMG who are either AChR antibody positive or MuSK antibody positive. The reimbursement request is for rozanolixizumab as an add-on therapy for the treatment of adult patients with gMG who are either AChR antibody-positive or MuSK antibody-positive and for whom symptoms persist despite conventional therapy with acetylcholinesterase inhibitors, corticosteroids, and/or nonsteroidal immunosuppressive therapies.1 CDA-AMC accepted a deviation request from the sponsor to focus the economic model on the reimbursement request population. The sponsor submitted a cost-utility analysis of rozanolixizumab plus conventional therapy compared with conventional therapy alone and to conventional therapy in combination with chronic IVIg or SCIg, chronic PLEX, rituximab, eculizumab, or efgartigimod alfa. Conventional therapy was defined as a mixed treatment basket consisting of 12.5% each of prednisone, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, methotrexate, pyridostigmine, and cyclophosphamide.
The sponsor submitted a new pharmacoeconomic model and report in October 2024. The inputs and results that follow are reflective of these new files.
Rozanolixizumab is available as a solution for SC infusion (280 mg in 2 mL single-dose vials).2 The recommended dosages are 6 weekly infusions of 280 mg for patients weighing 35 kg to less than 50 kg, 420 mg for patients weighing 50 kg to less than 70 kg, 560 mg for patients weighing 70 kg to less than 100 kg, and 840 mg for patients weighing more than 100 kg. Subsequent cycles of treatment should be based on clinical evaluation and may vary by patient. At the submitted price of $12,260.28 per 280 mg vial, the cost per 6-week course ranges from $73,562 to $220,685, depending on patient weight. Assuming a mean of 2.97 treatment courses per year (i.e., 17.8 infusions), as reported in pooled data from the MycarinG trial and the MG0004 and MG0007 extension studies,3 and a mean patient weight of 81 kg, the mean annual cost of treatment is $436,956. Mean annual drug acquisition costs for the comparators, assuming an 81 kg patient, were as follows: conventional therapy, $1,537; chronic IVIg or SCIg, $85,017; eculizumab, $728,136; rituximab, $19,008; efgartigimod alfa, $447,456; and chronic PLEX, $31,725.
The clinical outcomes of interest were life-years and QALYs. The sponsor adopted a lifetime time horizon (i.e., 52.5 years), with the analysis conducted from the perspective of a publicly funded health care payer. Future costs and benefits were discounted at a rate of 1.5% per year, and the model cycle length was 2 weeks. Owing to the short cycle length, a half-cycle correction was not used.
Model Structure
The sponsor submitted a Markov model with 6 health states: uncontrolled on high-dose steroids and immunosuppressant therapies, 2 response-based states (a 3-point to 4-point change or a ≥ 5-point change from baseline in MG-ADL total score), acute exacerbation requiring rescue therapy, myasthenic crisis, and death (Figure 1). Patients entered the model in the uncontrolled state. Up until the response assessment time point, which differed by treatment, patients could achieve a response defined as a minimum 3-point reduction from baseline in MG-ADL total score and move into 1 of the 2 response health states, based on their probability of having achieved a 3-point to 4-point or greater than or equal to 5-point improvement in MG-ADL score. After the response assessment time point, patients who were not in a response state were assumed to discontinue non–conventional therapy treatment and remain in the uncontrolled health state.
Model Inputs
The baseline population characteristics used to inform the model were based on the MycarinG trial, a randomized, double-blind, placebo-controlled, multicentre, adaptive trial that included patients aged 18 years and older (mean = 51.8 years) who had AChR antibody-positive or MuSK antibody-positive gMG that was MGFA class II to IVa at screening and an MG-ADL score of at least 3, with at least 3 points from nonocular symptoms (mean = 8.3), a QMG score of at least 11 (mean = 15.6), and who had been considered for treatment with additional therapy, such as IVIg or PLEX, by the investigator.4 Patients in the MycarinG trial were 61.5% female, 39.5% male, and had a mean body weight of 81.2 kg.1
The primary measure of efficacy in the model was the probability of treatment response at the assessment time points. Response rates — with response defined as at least a 3-point improvement in MG-ADL score for rozanolixizumab (73%), eculizumab (54%), efgartigimod alfa (71%), and conventional therapy (34%) — were derived from a sponsor-conducted NMA, the results of which were applied at different assessment time points in the economic model (e.g., 10 weeks for efgartigimod, 26 weeks for eculizumab), with conventional therapy assumed equivalent to placebo. Response rates for IVIg or SCIg (51%) and PLEX (57%) were derived from a 2011 trial5 in which response was defined as an improvement of at least 3.5 units on the QMG score, measured at 14 days. Response for rituximab (35%) was derived from the Beat MG study,6 in which it was defined as an improvement of at least 5 points on the QMG at 52 weeks.
Patients who responded before the assessment time point transitioned into 1 of the 2 response states. For patients responding to rozanolixizumab, eculizumab, or efgartigimod, 29% were assumed to have a 3-point to 4-point improvement in MG-ADL (mean MG-ADL improvement = 3.46), while 71% had a greater than or equal to 5-point improvement (mean MG-ADL improvement = 7.58), based on the distribution of patients responding to zilucoplan (used as a proxy) in the RAISE-XT extension study7 and the mean change from baseline in MG-ADL score across treatment groups (zilucoplan and conventional therapy) for responding patients who achieved a 3-point to 4-point or greater than or equal to 5-point improvement in MG-ADL score, respectively. Among patients responding to rituximab, chronic IVIg or SCIg, PLEX, or conventional therapy, 44% were assumed have a 3-point to 4-point MG-ADL improvement, while 56% had a greater than or equal to 5-point improvement, with the same mean improvements in score.
Patients who did not respond by the assessment time point were assumed to discontinue their add-on therapy, if applicable, and remain in the uncontrolled state on conventional therapy alone. Patients who responded were assumed to remain on their add-on therapy, if applicable, for a maximum of 2 years, with benefits continuing to accrue for the remainder of the time horizon until exacerbation, myasthenic crisis, or death.
In each cycle, patients could experience clinical events (i.e., myasthenic exacerbations or crises). Annual rates depended on whether the patient was in a response state (0.1179 for exacerbation; 0.0117 for crisis) or an uncontrolled state (0.6515 for exacerbation; 0.0117 for crisis).8,9 Following an exacerbation, 16.8% of patients were assumed to worsen to having a crisis, based on the number of patients requiring ventilation by day 15 after an acute exacerbation in an IVIg study.10 Before the assessment time point, 35% of patients having an exacerbation were assumed to re-enter the uncontrolled health state and discontinue their add-on therapy,10 if applicable, with the rest assumed to re-enter 1 of the 2 response health states in the original proportion assigned to their specific therapy. After the assessment time point, all patients experiencing an exacerbation were assumed to either worsen to crisis (16.8%) or discontinue their add-on therapy, if applicable, and enter the uncontrolled state (83.2%). Given that the only serious AE with an incidence of 5% or more in the MycarinG trial was headaches — and that severe headaches within the trial were mostly managed by over-the-counter medications — AEs were not considered in the model.1 Mortality within the response, uncontrolled, and exacerbation health states was assumed to be equal to that of the general population, while patients in myasthenic crisis had a 5% annual risk of death within the 2-week model cycle.9
Utility values were derived using MG-ADL score as a predictor of EQ-5D using a repeated measures regression model of UK crosswalk utilities from pooled treatment arms of the MycarinG trial.11 Mean utility value by model health state is reported in Table 9. Utilities were further adjusted to consider age- and sex-related disutilities using a UK-based regression algorithm.12 Patients entering the greater than or equal to 5-point MG-ADL improvement state did not accrue the greater utility value associated with that state until after the assessment time point for their treatment. A disutility of –0.20 was applied for 14 days for patients experiencing an exacerbation, while those in crisis experienced a disutility of –0.72 for 28 days.9 Additionally, disutilities associated with chronic steroid use were assigned to patients in the uncontrolled and 3-point to 4-point MG-ADL improvement states, assuming that patients in these states used high-dose (disutility = 0.18) and low-dose (disutility = 0.07) corticosteroids, respectively.
Costs in the model included drug acquisition, administration, vaccination, routine care, clinical event management costs, and end-of-life costs. All patients were assumed to receive conventional therapy, with equal proportions assumed to use prednisone, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, methotrexate, cyclophosphamide, and pyridostigmine. The acquisition costs of the drugs used for conventional therapy were taken from the Ontario Drug Benefit Formulary,13 as was that of rituximab, while those of IVIg or SCIg were based on an annual report for Atlantic Canada in 2022 to 2023.14 The acquisition cost of PLEX was taken from the literature,15 and costs of eculizumab and efgartigimod were based on prices submitted to CDA-AMC for their respective reimbursement reviews.9,16 An administration cost17 was applied for each nonblood product that required IV infusion (i.e., rituximab, efgartigimod, eculizumab), assuming a 1-hour infusion time, while PLEX and IVIg or SCIg were associated with a cost that assumed 3-hour infusions. Rozanolixizumab was associated with a cost for the first SC administration, based on the median hourly wage of a registered nurse in Canada, with 15 minutes of nursing time for subsequent administrations, given the assumption that providers would require less time for subsequent infusions.18 Four percent of patients receiving eculizumab were assumed to require a meningococcal vaccine.16,19
Routine care costs included GP visits, other health care professional visits, outpatient hospital visits, emergency department visits, hospital stays, and intensive care unit stays, based on frequencies reported in the literature8,20,21 and Canadian cost sources.22-27 Costs for managing corticosteroid use were derived from the submission to CDA-AMC for efgartigimod alfa;9 these were $12,197 annually for patients in the uncontrolled state, $5,309 annually for patients in the 3-point to 4-point MG-ADL improvement state, and $0 for patients in the greater than or equal to 5-point MG-ADL health state. Overall annual health care resource use costs, excluding exacerbations and crises, totalled $21,831 for patients in the uncontrolled state, $11,266 for patients in the CFB 3-point to 4-point MG-ADL state, and $5,956 for patients in the greater than or equal to 5-point MG-ADL improvement state. Costs for exacerbations and myasthenic crises were $7,014 and $18,022 per event, respectively, for patients not receiving chronic IVIg or SCIg; $4,318 and $17,831 per event, respectively, for those receiving chronic IVIg or SCIg; and $8,035 and $24,547 per event, respectively, for patients receiving chronic PLEX. The model assumed a 1-time end-of-life cost of $11,405.28
Summary of the Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (500 iterations) for the base-case and scenario analyses and are presented here. The probabilistic and deterministic results were similar except where noted. The sponsor’s model lacked reliability when reporting sequential analysis results; all sequential results reported herein have been checked and recalculated by CDA-AMC, where needed. All results are based on publicly available list prices.
Base-Case Results
In sequential analysis, the sponsor’s probabilistic base case reported that rozanolixizumab plus conventional therapy was associated with an ICER of $2,676,135 per QALY gained (incremental cost = $21,998; incremental QALYs = 0.008) compared to efgartigimod alfa plus conventional therapy (Table 3). Rozanolixizumab had a 0% chance of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. CDA-AMC is unable to report the proportion of incremental benefit within the trial period (as opposed to the extrapolated period) due to differences in the time points at which benefits are accrued for treatments within the model. At the end of the time horizon, CDA-AMC calculated that approximately 1.1% of patients on rozanolixizumab remained alive. Disaggregated results (Table 10) indicate that the main QALY driver was time spent in the controlled versus uncontrolled health state, while the main cost drivers were treatment costs as well as health care resource use costs. Rozanolixizumab was not predicted to have a substantial impact on extending life.
Deterministic results were similar, with the exception that efgartigimod alfa was dominated by rozanolixizumab, leading to a sequential ICER for rozanolixizumab plus conventional therapy of $1,622,629 (incremental cost = $544,985; incremental QALY = 0.34) compared to rituximab plus conventional therapy. The difference between the deterministic and probabilistic analysis appears to be driven primarily by the assumption that all patients will receive the number of vials of add-on therapy appropriate to the mean patient body weight, rounded up, which disproportionately affects the cost of efgartigimod alfa.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 655,680 | 10.60 | Reference |
Rituximab plus conventional therapy | 676,040 | 10.68 | 262,458 vs. conventional therapy |
Efgartigimod alfa plus conventional therapy | 1,256,334 | 11.00 | 1,776,088 vs. rituximab plus conventional therapy |
Rozanolixizumab plus conventional therapy | 1,278,332 | 11.01 | 2,676,135 vs. efgartigimod alfa plus conventional therapy |
Dominated treatments | |||
Chronic PLEX plus conventional therapy | 888,741 | 10.78 | Extendedly dominated by mix of rituximab and efgartigimod |
Chronic IVIg or SCIg plus conventional therapy | 894,483 | 10.73 | Dominated by chronic PLEX |
Eculizumab plus conventional therapy | 2,101,292 | 10.84 | Dominated by rozanolixizumab |
ICER = incremental cost-effectiveness ratio; IVIg = IV immunoglobulin; PLEX = plasma exchange; QALY = quality-adjusted life-year; SCIg = subcutaneous immunoglobulin; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor presented deterministic scenario analyses considering alternate inputs for the discount rate and time horizon, allowing vial-sharing, and assuming 3.4 cycles of rozanolixizumab per year, consistent with the number of cycles initiated per year in the MycarinG trial rather than the number of cycles completed. All scenario analyses were rerun by CDA-AMC because the sponsor’s submitted report contained insufficient detail to calculate sequential ICERs. In these scenarios, the ICERs for rozanolixizumab were similar to those of the sponsor’s deterministic base case; however, the treatments on the efficacy frontier differed, with the exception of the scenario assuming 3.4 cycles of rozanolixizumab were used per year. This scenario resulted in an ICER of $4,387,400 per QALY gained compared to efgartigimod alfa.
The sponsor conducted a scenario analysis from a societal perspective. This deterministic analysis included additional costs associated with work hours lost by the patient and the cost of caregiver hours spent caring for a patient with gMG. When considering the sequential results of this analysis (as rerun by CDA-AMC), rozanolixizumab was associated with an ICER of $1,630,558 per QALY gained compared to rituximab, similar to the sponsor’s deterministic base-case analysis using a health care payer perspective (ICER = $1,622,629 per QALY gained compared to rituximab).
CDA-AMC Appraisal of the Sponsor’s Economic Evaluation
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The full Health Canada–indicated population was not modelled: The sponsor-submitted model reflected the cost-effectiveness of rozanolixizumab plus conventional therapy in patients with MGFA class II to class IVa AChR antibody-positive or MuSK antibody-positive gMG with an MG-ADL score of at least 3 for nonocular symptoms and a QMG score of at least 11, with effectiveness informed by the MycarinG trial. The MycarinG trial excluded patients with MGFA class I, class IVb, and class V gMG, as well as patients who did not meet the MG-ADL and QMG cut-offs.4 Given that the Health Canada indication is not restricted by MGFA class, MG-ADL score, or QMG score or by patients having persistent symptoms despite conventional therapy,29 as in the sponsor’s reimbursement request,1 the modelled population is narrower than the Health Canada–indicated population. Clinical expert feedback obtained by CDA-AMC indicated that, while efficacy has not been established in patients beyond those meeting the inclusion criteria of the MycarinG trial, clinicians are likely to consider using rozanolixizumab beyond the population studied.
CDA-AMC was unable to address this limitation owing to a lack of clinical data.
Relative clinical efficacy is highly uncertain: Efficacy in the economic model is based on the proportion of patients who respond to treatment. Because there have been no head-to-head trials comparing the efficacy of rozanolixizumab versus other add-on comparators (i.e., rituximab, chronic IVIg or SCIg, chronic PLEX, efgartigimod alfa, eculizumab), the sponsor conducted an NMA.30 However, the NMA was associated with substantial limitations, including sparse evidence networks, short-term data, and heterogeneity in time points of assessment, disease severity (e.g., mean MG-ADL score at baseline), refractory disease status, and antibody status. The CDA-AMC Clinical Review noted that the heterogeneity in patient and study characteristics within the NMA affected the resulting confidence in whether the transitivity assumption was met; thus, it is unclear if the comparative results were valid. Additionally, the results lacked precision, as evidenced by wide credible intervals (CrIs). For example, in the overall population responder analysis, the odds ratio for rozanolixizumab 7 mg/kg versus placebo was 5.17 (95% CrI, 2.39 to 11.61), while that of efgartigimod alfa versus placebo was 4.74 (95% CrI, 2.27 to 10.30). Given the limitations with the NMA, the CDA-AMC clinical report noted that conclusions could not be drawn on the comparative efficacy of rozanolixizumab relative to the other treatments included in the NMA. While these were not used to inform the economic model, the sponsor also submitted matching-adjusted indirect comparisons (MAICs) comparing rozanolixizumab to efgartigimod alfa and to IVIg. The CDA-AMC Clinical Review noted that substantial limitations were associated with these MAICs, rendering the results uncertain; however, the anchored MAIC comparing rozanolixizumab to efgartigimod alfa suggested similarity, with no advantage for 1 treatment over the other.
In the submitted model, the proportion of patients responding to rituximab, chronic IVIg or SCIg, or chronic PLEX were not derived from the sponsor’s NMA, but were instead based on naive comparison, with response defined as the proportion of patients achieving an improvement of at least 5 points on the QMG scale by 52 weeks for rituximab, and the proportion of patients achieving at least a 3.5-point improvement on the QMG scale by 2 weeks for chronic IVIg or SCIg and chronic PLEX. Given the direct use of clinical trial data, with no adjustment or approaches taken to account for differences in patient characteristics, it is not possible to determine if any observed differences in treatment response between the therapies are due solely to the treatment or, rather, to bias or confounding factors (e.g., differences in study populations, definitions of outcomes, or study designs). As such, conclusions also cannot be drawn regarding the relative efficacy of these 3 comparators to each other or to those included in the sponsor’s NMA.
Finally, the proportion of patients assumed to have a greater than or equal to 5-point MG-ADL improvement rather than a 3-point to 4-point improvement were informed by the zilucoplan RAISE-XT extension study, with modelled patients receiving rozanolixizumab, eculizumab, or efgartigimod alfa assumed to have the same response distribution as those receiving zilucoplan, while those receiving rituximab, chronic IVIg or SCIg, chronic PLEX, or conventional therapy alone were assumed to have the same response distribution as those receiving placebo. Given that patients in the RAISE trial and RAISE-XT extension study were not the same as those in the MycarinG trial, and that zilucoplan was not included as a comparator in the submitted model, the use of zilucoplan and placebo as proxies for modelled response distributions of the included comparators is highly uncertain.
CDA-AMC was unable to adequately address the uncertainty associated with comparative effectiveness estimates derived from either the submitted NMA or naive comparisons within the model. Long-term comparative efficacy is unknown.
Comparator relevance varies by serotype: The indication for rozanolixizumab is for the treatment of adults with AChR antibody-positive or MuSK antibody-positive gMG, while the sponsor’s reimbursement request is for rozanolixizumab as an add-on therapy for the treatment of AChR antibody-positive or MuSK antibody-positive gMG in patients with persistent symptoms,4 While efgartigimod alfa is indicated for the treatment of AChR antibody-positive gMG31 and received a positive reimbursement recommendation with conditions,32 it is not a comparator of interest for the subpopulation of patients with MuSK antibody-positive gMG. Additionally, ravulizumab and zilucoplan, should these become funded, will also be treatment options only for patients with AChR antibody-positive gMG. In contrast, rituximab, while not indicated for any serotype of gMG, is more frequently used for patients with MuSK antibody-positive gMG, according to the clinical expert input received by CDA-AMC. The sponsor’s model did not include the option to consider cost-effectiveness within specific serotype subpopulations.
CDA-AMC was unable to address this limitation in reanalysis. The cost-effectiveness of rozanolixizumab compared to relevant comparators is likely to differ between gMG serotype subpopulations.
Discontinuation assumptions were inappropriate: The sponsor assumed that after the assessment time point, any patient who experienced an exacerbation or crisis discontinued their add-on therapy (if applicable) and returned to the uncontrolled state, regardless of the initial state in which they experienced the exacerbation or crisis. Clinical expert opinion obtained by CDA-AMC did not find this plausible, noting that patients would typically have their event treated and return to their previous treatment and health state. The sponsor also assumed that any patient who discontinues their add-on therapy due to lack of response or clinical event (e.g., exacerbation or crisis) remains on standard of care alone for the remainder of their lives; however, clinical expert input obtained by CDA-AMC noted that patients who discontinue 1 add-on therapy would switch to another.
Additionally, the sponsor assumed that all patients who are still receiving their add-on therapy (i.e., did not discontinue due to lack of efficacy or to having an exacerbation or crisis) will discontinue after 2 years of therapy and, as a result, stop accruing drug acquisition costs. Yet patients are assumed to continue experiencing the improved MG-ADL score and, as a result, the improved quality of life and reduced risk of exacerbation or crisis associated with their add-on therapy for the remainder of the time horizon (i.e., up to 50 more years), in the absence of an exacerbation, crisis, or death. According to clinical expert opinion obtained by CDA-AMC, this is not a reasonable assumption; most patients who discontinue a therapy would not be expected to retain its benefit over a very long-term. As such, the submitted model substantially underestimates drug costs for all add-on therapies and substantially overestimates their benefits after treatment discontinuation.
Due to the structure of the model, CDA-AMC was unable to return patients to their previous health state after exacerbation or crisis, assign a subsequent therapy upon drug discontinuation, or implement a gradual return to baseline quality of life status for patients who discontinued their add-on therapy. In a scenario analysis, CDA-AMC deactivated the 2-year stopping rule to ensure that patients receiving the benefit of their add-on therapies were still accruing the costs associated with them.
Assumptions regarding corticosteroid use were inappropriate: The sponsor assumed that 100% of patients in the uncontrolled health state used high-dose corticosteroids; that 100% of patients in the 3-point to 4-point MG-ADL improvement state used low-dose corticosteroids; and that patients in the greater than or equal to 5-point MG-ADL improvement state did not use corticosteroids at any dose. However, the clinical expert input obtained by CDA-AMC did not consider this to be plausible, noting that while reduction in steroid use is a treatment goal, many patients would remain on these drugs despite reductions in MG-ADL score. Change in corticosteroid use was not an outcome of the MycarinG trial, which expected patients to remain on stable doses of conventional therapy treatments, including corticosteroids, unless medically indicated. As such, the MycarinG trial did not demonstrate an association between rozanolixizumab and reduction in corticosteroid use. At baseline, 65% of patients in the MycarinG trial were using concomitant corticosteroids, suggesting that fewer than 100% of patients with uncontrolled gMG in clinical practice would be expected to be on corticosteroids, high dose or otherwise (refer to the CDA-AMC clinical report).
The sponsor assumed utility decrements associated with steroid use in a similar manner to that of the submission to CDA-AMC for efgartigimod alfa,33 derived from observational studies conducted in Sweden, the UK, and the US of patients with lupus or any condition requiring chronic corticosteroid use.34,35 As noted within the CDA-AMC review of that submission, generalizability, confounding, and partial double-counting of health-related quality of life effects are an issue when applying decrements derived from these sources to patients with gMG and when assuming that such decrements are lessened or no longer apply if a patient experiences an MG-ADL score improvement. Similarly, the sponsor assumed annual costs associated with corticosteroid use consistent with those used in the efgartigimod alfa submission to CDA-AMC.33 Limitations associated with the sponsor’s assumptions around differential costs are similar to those of differential utility decrements due to corticosteroid use; these are subject to generalizability concerns, confounding, and partial double-counting of impact, in addition to the issue that reductions in the proportion of use and/or dosing of corticosteroids were not found in the clinical trials of either rozanolixizumab or efgartigimod alfa.4,36
To explore uncertainty in the magnitude of effect that potential reductions in corticosteroid use would have on patient quality of life and health care costs, as well as uncertainty in the proportion of patients using corticosteroids at baseline and the proportion who could reduce such usage due to the use of add-on therapies to conventional therapy, CDA-AMC conducted a scenario removing the additional costs and utility decrements associated with the assumption of changes in corticosteroid use.
Relative costs of efgartigimod alfa and rozanolixizumab may be inappropriate: The sponsor’s model assumed that all patients receive a dose of their add-on therapy consistent with the mean patient body weight reported in the MycarinG trial (i.e., 81 kg), assuming wastage of excess medication in vials. While mean patient weight varied probabilistically from this mean based on the standard deviation reported in the MycarinG trial, all patients within each iteration received a dose appropriate for the single weight generated with wastage assumed. As such, the sponsor’s results may overestimate the treatment cost of add-on therapies, particularly in the deterministic analysis; in clinical practice, patients would receive the required number of vial sizes based on their respective weight, rather than each patient receiving the dose appropriate for the mean weight of the population, rounded up. Based on the available vial sizes and recommended dosages of rozanolixizumab and efgartigimod alfa (refer to Table 6), and the mean body weight and standard deviation reported in the MycarinG trial, the sponsor’s submitted base case of 500 iterations resulted in all patients on rozanolixizumab receiving 2 280 mg vials of rozanolixizumab and 69% of patients on efgartigimod alfa receiving 3 400 mg vials. However, this dose distribution is inconsistent with the recommended doses for the distribution of patient weights reported in the MycarinG trial (refer to the Clinical Review). When considering the placebo and rozanolixizumab 7 mg/kg groups from the trial, 8% of patients weighed less than 50 kg and would need 1 280 mg vial of rozanolixizumab; 72% of patients weighed 50 kg to less than 100 kg and would need 2 vials; and 20% weighed 100 kg or more and would need 3 vials. Efgartigimod alfa has a recommended dose of 10 mg/kg and is available in 400 mg vials with a maximum dose of 1,200 mg; assuming the body weight distribution reported in the MycarinG trial (simplified to assume that no patients weighed less than 40 kg and that one-third of the patients who weighed 70 kg to 100 kg weighed ≤ 80 kg), approximately 50% of patients would require 2 vials of efgartigimod alfa per dose, while the remaining 50% would require 3 vials. This estimate is consistent with the distribution of doses derived from the ADAPT trial, in which 60% of patients required 2 vials of efgartigimod alfa,9,37 when accounting for the difference in mean body weight between trials. As such, the sponsor’s model may overestimate the drug acquisition cost of efgartigimod alfa relative to that of rozanolixizumab.
CDA-AMC conducted a scenario analysis in which patients receiving efgartigimod alfa received 2.5 vials per dose (weighted average of 50% requiring 2 vials per dose while the rest would require 3 vials per dose). Patients receiving rozanolixizumab were assumed to receive 2.1 vials per dose (weighted average of 8% of patients requiring 1 vial per dose, 72% requiring 2 vials per dose, and 20% requiring 3 vials per dose).
Model lacked transparency and reliability: The sponsor’s submitted model included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automated overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical because it remains unclear whether the model is running appropriately. Additionally, after running a probabilistic analysis, the model returned some inputs (e.g., drug acquisition prices) to default settings. As such, additional runs of the probabilistic analysis could be based on unintended inputs if not thoroughly reviewed each time.
Furthermore, the submitted model had instances in which parameters or programming were not incorporated as described in the submitted technical report. For example, the sponsor’s submitted report describes mortality due to myasthenic crisis as a 5% risk per event (i.e., 5% of patients die within the 2-week model cycle when experiencing a crisis, consistent with the source of that assumption33); however, the implementation of this assumption resulted in a 5% annual risk of mortality in the crisis state, corresponding to a risk of approximately 0.2% per 2-week cycle spent in the crisis state. It was also unclear if and how the model incorporated uncertainty into parameters such as response rates, given that the rates and odds ratios reported in the parameters sheet did not appear to match those used to calculate transition probabilities in the model’s engine. Additionally, the sponsor indicated that the submitted model was intended to use odds ratios reported in the overall population responder analysis of the NMA to inform the modelled efficacy of rozanolixizumab, efgartigimod alfa, eculizumab, and conventional therapy. The revised model submitted in October 2024 instead utilized odds ratio inputs from the refractory population responder analysis.
Finally, the model did not reliably report sequential analyses due to errors in how the determination of extended dominance was implemented.
In scenario analyses, CDA-AMC corrected mortality in the model such that all patients having a myasthenic crisis had a 5% risk of death within the 2-week cycle of their crisis. All sequential results in this report have been confirmed by CDA-AMC and corrected if necessary. Substantial uncertainty remains in the validity and reliability of the modelled results, given the programming issues noted previously.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CDA-AMC (refer to Table 4).
Pagebreak
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CDA-AMC comment |
|---|---|
Background therapies used in the MycarinG trial represent conventional therapy. | Uncertain. The MycarinG trial compared rozanolixizumab to placebo rather than to conventional therapy. Instead, patients within both groups could receive conventional therapy therapies, including AChEIs (stable dose not required), oral CS (stable for 4 weeks before baseline), azathioprine, ciclosporin, methotrexate, mycophenolate mofetil, and/or tacrolimus (all received for the previous 6 months and at a stable dose for 2 months before baseline).4 Because this concomitant conventional therapy was typically held stable, rozanolixizumab was not compared to any individual or combination conventional therapy, given that it would be used in clinical practice (i.e., altering doses or adding medications to suit patients’ current symptoms or other needs). As such, the cost-effectiveness of rozanolixizumab compared to customizable conventional therapy is uncertain. |
All patients use a single drug within the conventional therapy basket in equal proportions. | Inappropriate, but unlikely to affect the results. Patients with gMG are not equally likely to use any 1 of the AChEIs, CSs, or NSISTs included within the conventional therapy category, and many are on more than 1 at a time, as demonstrated in the baseline MG medications reported in the MycarinG trial.4 However, because conventional therapy was assumed to be the same between all comparators, patients did not discontinue conventional therapy; and because there were minimal differences in life-years between comparators, changes to the cost of conventional therapy are not expected to affect the model results. |
Utility values of model health states were derived with MG-ADL score as a predictor of EQ-5D using a repeated measures regression model of UK crosswalk utilities from pooled treatment arms of the MycarinG trial. | Uncertain. The resulting utility values for each MG-ADL score were substantially lower than those reported in the literature for EQ-5D–based utilities by MG-ADL score derived from other gMG trials38,39 or by MGFA class.40 Given these health state utility values, when considering the 0.72 disutility associated with having a myasthenic crisis, all patients in crisis were assumed to have a utility of substantially less than 0, indicating that patients would view the time spent in crisis as being worse than death. The impact of potentially underestimated utility weights for the main model health states on results is uncertain. |
AEs are excluded from the model. | Uncertain but acceptable. The sponsor noted its intention to consider any serious AE with an incidence ≥ 5% in the model, given that it was assumed that only serious AEs would incur costs to the health care system. Due to the safety profile of the included comparators, only headaches (in approximately 6% of patients) met this inclusion criterion for rozanolixizumab. It was also assumed that such headaches would be treated with over-the-counter medications for a duration of 2 days to 4 days. Therefore, the cost of treatment for this AE was not included in the model. In addition, a disutility for severe headaches was not applied. Clinical expert opinion obtained by CDA-AMC expressed that the AEs observed in the MycarinG trial and extension study were considered to be manageable and comparable to those associated with other available treatments for gMG. |
Eculizumab is included as a comparator. | Reasonable, but irrelevant. According to the current CDA-AMC Procedures for Reimbursement Reviews,41 comparators should include treatments that are currently reimbursed by at least 1 participating drug plan for the indication under review or treatments that have received a recommendation in favour of reimbursement. Although eculizumab meets these criteria (i.e., CDEC recommended reimbursing eculizumab with conditions),42 negotiations with the pCPA were unsuccessful;43 thus, eculizumab is not currently funded by any participating plan. As such, CDA-AMC considered eculizumab to be an irrelevant comparator at the time of this review and removed it from scenario analyses. This change has no impact on the results because eculizumab was dominated in the sponsor’s base case and remained so in all CDA-AMC scenarios. |
Ravulizumab is excluded as a comparator. | Reasonable, but uncertain. The initial CDA-AMC review of ravulizumab for AChR antibody-positive gMG resulted in a Do Not List recommendation.44 Therefore, the criteria for inclusion as a comparator (outlined in the CDA-AMC Procedures for Reimbursement Reviews) were not met at the time the sponsor submitted the request for rozanolixizumab.41 However, during this review, a resubmission of ravulizumab resulted in a draft recommendation to list with conditions.45 As such, it is uncertain whether ravulizumab will become a funded comparator for the treatment of gMG before the potential funding of rozanolixizumab. The submitted model did not have an option to include ravulizumab. |
AChEI = acetylcholinesterase inhibitor; AChR = acetylcholine receptor; AE = adverse event; CDA-AMC = Canada’s Drug Agency; CDEC = Canadian Drug Expert Committee; CS = corticosteroid; gMG = generalized myasthenia gravis; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; NSIST = nonsteroidal immunosuppressive therapy; pCPA = pan-Canadian Pharmaceutical Alliance.
CDA-AMC Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations that CDA-AMC identified with the sponsor’s economic submission, CDA-AMC was unable to derive a more reliable estimate of the cost-effectiveness of rozanolixizumab as an add-on therapy for the treatment of adult patients with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy. CDA-AMC notes that the sponsor’s analysis remains uncertain. The estimates of relative effect obtained from the sponsor’s NMAs, as well as the naive comparisons informing effectiveness within the model, represent the largest sources of uncertainty. The outputs of the sponsor’s base case suggest differences in treatment benefits between therapies for the add-on treatment of AChR antibody-positive and MuSK antibody-positive gMG. These results will be realized only if the numerical differences observed in the NMA and naive comparisons are deemed valid.
Scenario Analysis Results
To explore uncertainty in some of the assumptions in the model, CDA-AMC conducted scenario analyses removing the 2-year stopping rule (rozanolixizumab ICER = $9,682,589 per QALY gained relative to efgartigimod alfa); removed utility decrements and extra costs associated with corticosteroid use (rozanolixizumab ICER = $15,991,013 per QALY gained relative to efgartigimod alfa); revised efgartigimod and rozanolixizumab dosages to be based on the patient weight distribution reported in the MycarinG trial (rozanolixizumab was dominated by efgartigimod alfa); assumed a 5% mortality rate per myasthenic crisis (rozanolixizumab ICER = $2,075,184 per QALY gained relative to efgartigimod alfa); and combined all 4 of the aforementioned issues (rozanolixizumab was dominated by efgartigimod alfa) (Table 11). Additionally, CDA-AMC undertook price analyses based on the sponsor’s submitted base case to explore the price reduction required to obtain an ICER for rozanolixizumab beneath the threshold of $50,000 per QALY gained. These analyses were conducted on the probabilistic base case. To be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained, the price of rozanolixizumab would need to be reduced by 87.5%, corresponding to approximately $1,532 per 280 mg vial (Table 5).
Table 5: CDA-AMC Price Reduction Analyses
Analysis | Unit drug cost per 280 mg vial | ICERs for rozanolixizumab vs. comparators ($/QALY) |
|---|---|---|
Price reduction | ($) | Sponsor’s base case |
No price reduction | 12,260 | 2,676,135 vs. efgartigimod alfa |
10% | 11,034 | 1,592,542 vs. rituximab |
20% | 9,808 | 1,386,907 vs. rituximab |
30% | 8,582 | 1,181,273 vs. rituximab |
40% | 7,356 | 975,638 vs. rituximab |
50% | 6,130 | 770,003 vs. rituximab |
60% | 4,904 | 564,369 vs. rituximab |
70% | 3,678 | 358,734 vs. rituximab |
80% | 2,452 | 173,665 vs. conventional therapy |
90% | 1,226 | 6,600 vs. conventional therapy |
CDA-AMC = Canada’s Drug Agency; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Issues for Consideration
Differing budget holders: While the use of rozanolixizumab would be associated with additional costs from the perspective of both the drug plan payer in Canada as well as the overall health care payer perspective, the reimbursement of rozanolixizumab for gMG may reduce the use of blood products (IVIg, SCIg, and PLEX), which may result in some savings for alternate budget holders. A potential reduction in the use of blood products is also relevant, given ongoing reports of blood and plasma shortages in Canada.46
Zilucoplan is currently under review for gMG: At the time of this review, zilucoplan is also under review by CDA-AMC for the treatment of AChR antibody-positive gMG.47 It is uncertain whether zilucoplan will become a funded comparator for the treatment of gMG before the potential funding of rozanolixizumab. The submitted model did not have an option to include zilucoplan.
Anticipated patent expiration of eculizumab: Negotiations with the pCPA for eculizumab for gMG concluded without an agreement.43 The patent for eculizumab is expected to expire in March 2027.48 If eculizumab biosimilars become available and are considered clinically equivalent to eculizumab, rozanolixizumab is unlikely to remain less costly than eculizumab biosimilars.
Overall Conclusions
Evidence from the MycarinG trial demonstrated that in adult patients with MGFA class II to IVa AChR antibody-positive or MuSK antibody-positive gMG, compared to placebo, treatment with rozanolixizumab resulted in a statistically significant improvement in change from baseline to day 43 in MG-ADL score. Based on the clinical review team’s appraisal of the MycarinG trial, treatment with rozanolixizumab likely resulted in a clinically important improvement in MG-ADL score and an increase in the number of patients with at least a 2-point improvement in MG-ADL score compared to placebo at day 43. In the absence of direct evidence comparing rozanolixizumab to other active comparators currently used as add-on therapies for the treatment of adults with gMG (i.e., chronic IVIg or SCIg, chronic PLEX, rituximab, eculizumab, and efgartigimod alfa), the sponsor submitted an NMA as well as naive comparisons to inform the economic model regarding the relative probability of response to therapy (defined as a ≥ 3-point improvement in MG-ADL score, a ≥ 3-point improvement in QMG score, or a ≥ 5-point improvement in QMG score, depending on the comparator) and extent of response (i.e., the proportion of responders estimated to have a 3-point to 4-point improvement in MG-ADL score versus a ≥ 5-point improvement) between comparators. Response to therapy was applied at different time points in the model depending on the comparator. The CDA-AMC Clinical Review concluded that the sponsor’s NMA had several limitations (i.e., sparse evidence networks, imprecision of estimates, and heterogeneity in the patient and study characteristics) that precluded the drawing of conclusions on the comparative efficacy of rozanolixizumab relative to its add-on comparators.
CDA-AMC was unable to address limitations with the sponsor’s submitted model as well as uncertainty related to the comparative clinical data and long-term efficacy of rozanolixizumab versus other add-on therapies used in addition to conventional therapy in adults with AChR antibody-positive or MuSK antibody-positive gMG and persistent symptoms despite conventional therapy. As such, CDA-AMC was unable to derive a more reliable base-case estimate of the cost-effectiveness of rozanolixizumab. Results of the sponsor’s base-case sequential analysis suggest that rozanolixizumab plus conventional therapy is associated with an ICER of $2,676,135 per QALY gained (incremental cost = $21,998; incremental QALYs = 0.008) compared to efgartigimod alfa plus conventional therapy. Based on this analysis, a price reduction of approximately 87.5% would be required for rozanolixizumab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained, reducing the unit price to $1,533 from $12,260 per 280 mg vial (and reducing the annual drug acquisition cost to $54,620 from $436,956 per patient). This analysis likely underestimates the ICER, given that the sponsor’s model was programmed to assume that treatment effects were maintained beyond treatment discontinuation. There is no robust evidence to support this assumption regarding maintenance of effect. CDA-AMC could not revise treatment effects, but undertook scenario analyses (using the submitted price of rozanolixizumab) increasing the duration of treatment to match the duration of benefit; in these analyses, the ICER increased to $9,682,589 per QALY gained relative to efgartigimod alfa plus conventional therapy due to increased drug acquisition costs. As such, a greater price reduction may be required to account for the assumptions used by the sponsor. The cost-effectiveness of rozanolixizumab in patients not studied in the MycarinG trial (e.g., those with MGFA class I, IVb, or V gMG, those not meeting the MG-ADL and QMG inclusion thresholds, and those not under consideration for additional treatment with IVIg or PLEX) is unknown.
The results of the economic model were based on efficacy inputs obtained from the sponsor’s submitted NMA and naive comparisons. CDA-AMC notes that the relative effects obtained from the NMA are highly unreliable due to the analysis limitations, and that the naive comparisons used for the effectiveness parameters of some comparators in the model are highly uncertain. While the sponsor’s base case suggests differences in treatment benefit for adults with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy, these results will be realized only if the numerical differences observed in the NMA and naive comparisons occur in clinical practice and lead to meaningful improvement for patients. The clinical expert feedback suggested that rozanolixizumab and efgartigimod alfa may have similar efficacy.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. UCB Canada; January 2024.
2.Myfortic (mycophenolic acid): 180 mg nd 360 mg enteric-coated tablets [product monograph]. Novartis Pharmaceuticals Canada Inc.; 2005. Accessed 12 Nov 2024. https://pdf.hres.ca/dpd_pm/00064471.PDF
3.Vissing J, Drużdż A, Grosskreutz J, Habib AA, Mantegazza R, et al. Rozanolixizumab responder and minimal symptom expression rates in generalised MG: Pooled Phase 3 and extension studies. EAN 2023, Budapest, Hungary; 1–4 July 2023. 2023.
4.Bril V, Druzdz A, Grosskreutz J, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 2023;22(5):383-394. doi: 10.1016/S1474-4422(23)00077-7 PubMed
5.Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. 2011;76(23):2017-2023. PubMed
6.Nowak RJ, Coffey CS, Goldstein JM, et al. Phase 2 Trial of Rituximab in Acetylcholine Receptor Antibody-Positive Generalized Myasthenia Gravis: The BeatMG Study. Neurology. 2021;98(4):e376-89. doi: 10.1212/wnl.0000000000013121 PubMed
7.Ucb. Data on file. MG0010 (RAISE) CSR [sponsor submitted reference]. 2022.
8.Abuzinadah AR, Alanazy MH, Butt NS, Barohn RJ, Dimachkie MM. Exacerbation Rate in Generalized Myasthenia Gravis and Its Predictors. Eur Neurol. 2021;84(1):43-48. doi: 10.1159/000512077 PubMed
9.CDA-AMC. Clinical and Pharmacoeconomic Combined Report. Efgartigimod alfa (Vyvgart). April 2024 [sponsor submitted reference]. 2024. https://www.cadth.ca/sites/default/files/DRR/2024/SR0782-Vyvgart.pdf
10.Gajdos P, Tranchant C, Clair B, et al. Treatment of Myasthenia Gravis Exacerbation With Intravenous Immunoglobulin: A Randomized Double-blind Clinical Trial. Arch Neurol. 2005;62(11):1689-1693. doi: 10.1001/archneur.62.11.1689 PubMed
11.Ucb. Data on file. MG0003 EQ-5D UK Crosswalk Utility Scores [sponsor submitted reference]. 2024.
12.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-18. doi: 10.1111/j.1524-4733.2010.00700.x PubMed
13.Ontario Ministry of Health. ODB Formulary/Comparative Drug Index. Effective from February 29, 2024 [sponsor submitted reference]. 2024. https://www.formulary.health.gov.on.ca/formulary/
14.Nova Scotia Provincial Blood Coordinating Team. Atlantic Canada Annual IVIG and SCIG FY 2022-2023 Report. July 2023 [sponsor submitted reference]. 2023. https://0-nsleg--edeposit-gov-ns-ca.legcat.gov.ns.ca/deposit/b10625884_2022-2023.pdf
15.Furlan JC, Barth D, Barnett C, Bril V. Cost-minimization analysis comparing intravenous immunoglobulin with plasma exchange in the management of patients with myasthenia gravis. Muscle Nerve. 2016;53(6):872-876. PubMed
16.CDA-AMC. Pharmacoeconomic Report: Eculizumab (Soliris). December 2020 [sponsor submitted reference]. 2020. https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0605-soliris-mg-pharmacoeconomic-review-report.pdf
17.Tam VC, Ko YJ, Mittmann N, Cheung MC, Kumar K. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
18.Job Bank. Registered Nurse (R.N.) in Canada. Prevailing wages in Canada. Date modified: 2024-01-23 [sponsor submitted reference] (2024).
19.Howard JF, Jr., Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976-986. doi: 10.1016/s1474-4422(17)30369-1 PubMed
20.Harris L, Graham S, MacLachlan S, Exuzides A, Jacob S. Healthcare resource utilization by patients with treatment-refractory myasthenia gravis in England. J Med Econ. 2019;22(7):691-697. doi: 10.1080/13696998.2019.1592180 PubMed
21.Phillips G, Abreu C, Goyal A, et al. Real-World Healthcare Resource Utilization and Cost Burden Assessment for Adults With Generalized Myasthenia Gravis in the United States. Front Neurol. 2022;12:809999. doi: 10.3389/fneur.2021.809999 PubMed
22.CIHI. Care in Canadian ICUs. 2016. Accessed March 20, 2024. https://secure.cihi.ca/free_products/ICU_Report_EN.pdf
23.CIHI. Hospital spending: Focus on the emergency department. 2020. Accessed March 20, 2024. https://www.cihi.ca/sites/default/files/document/hospital-spending-highlights-2020-en.pdf
24.CIHI. Cost of a Standard Hospital Stay [sponsor submitted reference]. 2023. https://www.cihi.ca/en/indicators/cost-of-a-standard-hospital-stay
25.CIHI. Hospital stays in Canada [sponsor submitted reference]. 2023. https://www.cihi.ca/en/hospital-stays-in-canada
26.Government of Alberta. Alberta Interactive Health Data Application [sponsor submitted reference]. Accessed January 2024. http://www.ahw.gov.ab.ca/IHDA_Retrieval/ihdaData.do
27.Ontario Ministry of Health. Schedule of Benefits. Physicians Services Under the Health Insurannce Act (February 20, 2024 (effective April 1, 2024)) [sponsor submitted reference]. 2024. https://www.ontario.ca/files/2024-04/moh-schedule-benefit-2024-02-20.pdf
28.CIHI. Patient Cost Estimator (2021-2022) [sponsor submitted reference]. 2022. https://www.cihi.ca/en/patient-cost-estimator
29.UCB Canada Inc. Rozanolixizumab Product Monograph (Draft) [sponsor submitted reference]. 2023.
30.Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. UCB Canada; January 2024.
31.Argenx B. V. Vyvgart (efgartigimod alfa for injection) [sponsor submitted reference]. 2023.
32.CADTH. Efgartigimod Alfa (VYVGART) Reimbursement Recommendation [sponsor submitted reference]. 2024.
33.CADTH. CADTH Reimbursement Review: Efgartigimod Alfa (Vyvgart) [sponsor submitted reference]. 2024.
34.Bexelius C, Wachtmeister K, Skare P, Jonsson L, Vollenhoven R. Drivers of cost and health-related quality of life in patients with systemic lupus erythematosus (SLE): a Swedish nationwide study based on patient reports. Lupus. 2013;22(8):793-801. doi: 10.1177/0961203313491849 PubMed
35.Sullivan PW, Ghushchyan VH, Globe G, Sucher B. Health-related quality of life associated with systemic corticosteroids. Qual Life Res. 2017;26(4):1037-1058. doi: 10.1007/s11136-016-1435-y PubMed
36.Howard JF, Jr., Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526-536. doi: 10.1016/s1474-4422(21)00159-9 PubMed
37.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rozanolixizumab (Rystiggo), 140 mg/mL, solution for injection (280 mg in 2 mL in single-dose glass vials) for SC use. UCB Canada; January 2024.
38.Dewilde S, Qi CZ, Phillips G, Iannazzo S, Janssen MF. Association Between Myasthenia Gravis-Activities of Daily Living (MG-ADL) and EQ-5D-5L Utility Values: The Additional Effect of Efgartigimod on Utilities. Adv Ther. 2023;40(4):1818-1829. doi: 10.1007/s12325-023-02437-w PubMed
39.Grimson FK, S; Pannullo, F. Health-related quality of life in generalised myasthenia gravis: The relationship between Myasthenia Gravis Activities of Daily Living and EQ-5D-5L in the RAISE study. presented at: ISPOR Europe 2023; 12–15 November 2023 2023; Copenhagen, Denmark. https://www.ispor.org/docs/default-source/euro2023/isporeuropegrimsonco128129087-pdf.pdf?sfvrsn=9dabc855_0
40.Barnett C, Bril V, Bayoumi AM. EQ-5D-5L and SF-6D health utility index scores in patients with myasthenia gravis. Eur J Neurol. 2019;26(3):452-459. doi: 10.1111/ene.13836 PubMed
41.CDA-AMC. Procedures for CADTH Reimbursement Reviews. May 2024 [sponsor submitted reference]. 2024. https://cadth.ca/sites/default/files/Drug_Review_Process/CADTH_Drug_Reimbursement_Review_Procedures.pdf
42.CADTH. Eculizumab (SOLIRIS) Final Reimbursement Recommendation [sponsor submitted reference]. 2020.
43.pan-Canadian Pharmaceutical Alliance. Soliris (eculizumab): Myasthenia Gravis. 2022. Accessed October 10, 2024. https://www.pcpacanada.ca/negotiation/21306
44.CDA-AMC. Reimbursement Recommendation. Ravulizumab (Ultomiris). August 2023 [sponsor submitted reference]. 2023. https://www.cadth.ca/sites/default/files/DRR/2023/SR0765Ultomiris%20-%20Confidential%20Final%20CADTH%20Recommendation%20August%2024%2C%202023%20revised.pdf
45.CDA-AMC. Ravulizumab resubmission. 2024. Accessed November 22, 2024. https://www.cda-amc.ca/ravulizumab-4
46.Pasieka C. Canadian Blood Services needs thousands more donors to roll up their sleeves. CBC; 2023. Accessed November 12, 2024. https://www.cbc.ca/news/canada/toronto/canadian-blood-services-needs-thousands-more-canadians-to-roll-up-their-sleeves-1.6879312
47.CDA-AMC. Zilucoplan submission. 2024. Accessed 18 Oct 2024. https://www.cda-amc.ca/zilucoplan
48.Health Canada. Patent Register. Accessed 2024 Nov 12. https://pr-rdb.hc-sc.gc.ca/pr-rdb/index-eng.jsp
49.Vissing J, Druzdz A, Grosskreutz J, et al. Rozanolixizumab responder and minimal symptom expression rates in generalised MG: Pooled Phase 3 and extension studies [sponsor submitted reference]. 2023.
50.Exceptional Access Program (EAP). Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2024. Accessed 2024 Oct 5. http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx
51.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2024. Accessed 2024 Oct 5. https://www.formulary.health.gov.on.ca/formulary/
52.Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of Myasthenia Gravis. Neurol Clin. 2018;36(2):311-337. doi: 10.1016/j.ncl.2018.01.011 PubMed
53.DeltaPA. IQVIA; 2023. Accessed 2024 Oct 4. https://www.iqvia.com/
54.Saskatchewan Drug Plan: search formulary. 2024. Accessed 2024 Oct 05. https://formulary.drugplan.ehealthsask.ca/SearchFormulary
55.Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol. 2011;11:97. doi: 10.1186/1471-2377-11-97 PubMed
56.Melzer N, Ruck T, Fuhr P, Gold R, Hohlfeld R, et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol. 2016;263:1473-1494. PubMed
57.Bank of Canada. Inflation calculator. 2024. Accessed 2024 Oct 05. https://www.bankofcanada.ca/rates/related/inflation-calculator/
58.CADTH. CADTH Drug Reimbursement Review Pharmacoeconomic Report: Eculizumab (SOLIRIS) [sponsor submitted reference]. 2020. https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0605-soliris-mg-pharmacoeconomic-review-report.pdf
59.Bubuioc AM, Kudebayeva A, Turuspekova S, Lisnic V, Leone MA. The epidemiology of myasthenia gravis. J Med Life. 2021;14(1):7-16. doi: 10.25122/jml-2020-0145 PubMed
60.Spillane J, Higham E, Kullmann DM. Myasthenia gravis. BMJ. 2012;345:e8497. doi: 10.1136/bmj.e8497 PubMed
61.Rodolico C, Bonanno C, Toscano A, Vita G. MuSK-Associated Myasthenia Gravis: Clinical Features and Management. Front Neurol. 2020;11. doi: 10.3389/fneur.2020.00660 PubMed
62.Lazaridis K, Tzartos SJ. Autoantibody Specificities in Myasthenia Gravis; Implications for Improved Diagnostics and Therapeutics. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.00212 PubMed
63.Eversana. Data on file. Rozanolixizumab for gMG Clinician Interview Report [sponsor submitted reference]. 2024.
64.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. The Conference Board of Canada; 2017. Accessed May 13, 2024. https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326
65.Adelphi Real World. Adelphi MG DSP I – EU Weight tables [sponsor submitted reference]. 2024.
66.Government of Canada. Population estimates on July 1st, by age and sex [sponsor submitted reference]. 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501
67.Government of Canada. First National and Inuit Health Branch: Non-Insured Health Benefits Program Annual Report 2021/2022 [sponsor submitted reference]. 2023. https://www.sac-isc.gc.ca/eng/1683039690813/1683039973755
68.The Conference Board of Canada. Understanding the Gap 2.0: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage [sponsor submitted reference]. 2022.
Appendix 1: Cost Comparison Table
Please note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CDA-AMC Cost Comparison Table for Complement Inhibitors Indicated for the Treatment of Generalized Myasthenia Gravis
Treatment | Strength or concentration | Form | Price | Recommended dosage | Daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Rozanolixizumab (Rystiggo) | 140 mg/mL | 280 mg vial solution for SC injection | $12,260.2760a | Dose weekly for 6 weeks. Dose for patients weighing:
Subsequent treatment cycles are based on clinical evaluation and may vary by patient. | 598.16 to 1,794.48a | $218,478 to 655,434a |
Eculizumab (Soliris) | 10 mg/mL | 300 mg single-use vial | 6,675.3000b | Loading: 900 mg weekly for 4 weeks, then 1,200 mg for the fifth dose 1 week later Maintenance: 1,200 mg every 2 weeks thereafter | First year: 1,943.78 Subsequent years: 1,907.23 | First year: 709,966 Subsequent years: 696,615 |
Efgartigimod alfa (Vyvgart) | 20 mg/mL | 400 mg vial Solution for IV use | 7,900.0000c | Loading: 10 mg/kg (max 1,200 mg) weekly for 4 weeks. Subsequent treatment cycles are based on clinical evaluation and may vary by patient | 816.71 to 1,225.07b | 298,304 to 447,456c |
Ravulizumab (Ultomiris) | 10 mg/mL 100 mg/mL 100 mg/mL | 30 mL 3 mL 11 mL Solution for IV infusion | 7,282.1500d 7,282.1500d 26,701.2000d | Loading dose at weeks 0, then maintenance dose at week 2 and every 8 weeks thereafter based on weight as follows:d ≥ 40 kg to < 60 kg:
≥ 60 kg to < 100 kg:
≥ 100 kg:
| First year: 1,410.04 to 1,700.02 Subsequent years: 1,300.38 to 1,560.46 | First year: 515,017 to 620,933 Subsequent years: 474,965 to 569,958 |
Zilucoplan (Zilbrysq) | 40 mg/mL | 16.6 mg 23.0 mg 32.4 mg Prefilled syringe for SC injection | $650.2718e $900.9790e $1,269.2052e | For patients weighing:
| 650.27 to 1,269.21 | 237,512 to 463,577 |
SC = subcutaneous.
aPrice is as submitted by sponsor.1 Daily and annual costs assume an average of 2.97 6-week courses per year.49 Cost per 6-week course is $73,562 for patients weighing 35 to < 50 kg, $147,123 for patients weighing 50 to < 100 kg, and $220,685 for patients weighing 100 or more kg.
bAlberta formulary, accessed in August 2024. Note that eculizumab is not funded for generalized myasthenia gravis.
cPrice is as submitted for the CDA-AMC review of Vyvgart. Daily and annual costs assume an average of 4.72 4-week courses per year.9
dOntario Drug Benefit Exceptional Access Program, accessed in August 2024.50 Note that ravulizumab is not funded for generalized myasthenia gravis at the time of this review.
ePrice is as submitted for the CDA-AMC review of Zilbrysq.47
Table 7: CDA-AMC Cost Comparison Table for Other Treatments for Generalized Myasthenia Gravis
Treatment | Strength or concentration | Form | Price ($) | Recommended dosagea | Average daily cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Other biologics | ||||||
Rituximab (biosimilars) | 10 mg/mL | 10 mL 50 mL Vial for IV infusion | 297.0000 1,485.0000 | 375 mg/m2 weekly for 4 doses | NA | Cost per course: 8,316 |
Alternate dosing: 1 g, followed by 1 g 2 weeks later, and then every 6 months | First year: 31.90 Subsequent years: 16.26 | First year: 11,652 Subsequent years: 5,940 | ||||
Glucocorticoids | ||||||
Prednisone (Winpred, generics) | 1 mg 5 mg 50 mg | Tablet | 0.1276 0.0220 0.1735 | Initiate at 10 to 20 mg/day, increase by 5 mg/day per week until stable remission (target 1 mg/kg/day) | 0.04 to 0.31 | 16 to 112 |
Alternate dosing: Initiate at 60 to 80 mg/day, then taper after improvement | 0.21 to 0.31 | 77 to 112 | ||||
Immunosuppressive drugs | ||||||
Azathioprine (generics) | 50 mg | Tablet | 0.5185 | Initiate at 50 mg/day for 5 days, and then, escalate to 2.5 to 3 mg/kg/dayd | 2.07 to 2.59 | 757 to 976 |
Cyclophosphamide (Procytox, generics) | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 500 mg/m2 to 1,000 mg/m2 every month for 6 months | NA | Cost per course: 52 to 103 |
500 mg 1,000 mg 2000 mg | IV vial, powder for injection | 101.7100b 184.3600b 339.2000b | NA | Cost per course: 1,106 to 2,035 | ||
Cyclosporine (generics) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7526 0.7870 1.5350 3.0720 | Starting dose: 100 mg twice daily Target dose: 5 to 6 mg/kg/day in 2 divided doses, adjust for serum trough level of 75 to 150 ng/mL | 12.29 to 15.34 | 4,489 to 5,606 |
Methotrexate (generic, Metoject SC) | 2.5 mg 10 mg | Tablet | 0.2513 2.7983c | 10 mg to 20 mg/week, orally or SC | 0.14 to 0.29 | 52 to 105 |
20 mg/2 mL 50 mg/2 mL | Vial for injection | 8.9200 12.5000 | 1.27 to 2.55 | 465 to 930 | ||
10mg/0.2mL 12.5mg/0.25mL 15 mg/0.3 mL 17.5mg/0.35 mL 20mg/0.4mL 22.5mg/0.45mL 25mg/0.5mL | Prefilled syringe for SC use | 16.3020 17.1600 16.3800 16.0000 17.5000 17.5000 19.5000 | 2.29 to 3.18 | 835 to 1,160 | ||
Mycophenolate mofetil (generics) | 250 mg | Capsule | 0.3712 | 1,000 mg twice daily | 2.97 | 1,084 |
500 mg | Tablet | 0.7423 | 2.98 | 1,087 | ||
Mycophenolate Sodium (generics) | 180 mg 360 mg | Enteric Tablet | 0.9989 1.9977 | 720 mg twice dailye | 7.99 | 2,917 |
Tacrolimus (generics) | 0.5 mg 1 mg 5 mg | Capsule | 1.0146 1.2978 6.4993 | 3 to 5 mg per dayf | 3.89 to 6.50 | 1,421 to 2,372 |
Cholinesterase inhibitors | ||||||
Pyridostigmine (Mestinon, generics) | 60 mg | Tablet | 0.2673 | 60 mg to 120 mg every 3 to 8 hours while awake | 0.53 to 3.20 | 195 to 1,172 |
180 mg | SR tablet | 1.3919 | 180 to 540 mg once or twice daily | 1.39 to 8.35 | 508 to 3,050 | |
Blood products | ||||||
IV immunoglobulin | 10,591 per exacerbationg | |||||
Plasma exchange | 7,784 per exacerbationg | |||||
SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed in October 2024),51 unless otherwise indicated, and do not include dispensing fees. All cost calculations for drugs with weight or body surface area-based dosing were calculated using the mean body surface area of 1.8 m2 and mass of 80 kg. Drug wastage was included.
aDosing is from a review by Farmakidis et al., unless otherwise indicated.52
bDelta PA database wholesale prices (accessed in October 2024).53
cSaskatchewan Drug Plan formulary (accessed in October 2024).54
dAzathioprine dosing was obtained from published literature.55
eMyfortic product monograph, dose indicated for the prophylaxis of organ rejection in patients receiving allogeneic renal transplants, previously confirmed with clinical experts as also use for generalized myasthenia gravis.2,9
fTacrolimus dose reported for patients with therapy-refractory myasthenia gravis in Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurologic Society.56
gThe cost of IV immunoglobulin and plasma exchange, totalling $8,277 and $6,084 respectively in 2014 dollars was for rescue therapy and included cost of blood products and hospital costs and was inflated to 2024 dollars by CDA-AMC.15,57 Due to confidential prices of immunoglobulin products and plasma exchange, chronic treatment cost is unknown.
Appendix 2: Submission Quality
Please note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Refer to key limitations noted previously. The sequential analysis used inappropriate methodology in determining extended dominance; sometimes leading to inappropriate reporting of sequential results. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to key limitations noted previously regarding uncertainty in the NMA results (i.e., the wide credible intervals). Some parameters were not implemented in the way described by the sponsor (e.g., mortality associated with myasthenic crisis). |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The model sometimes lacked clarity on how parameters varied within the probabilistic analyses. Model further incorporated assumptions on the maintenance of treatment effect after drug discontinuation that could not be explored. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The model sometimes lacked adequate labelling and clarity regarding what parameters were active and how these were varied within the probabilistic analyses. The submitted report included insufficient detail to determine sequential ICERs without rerunning scenario analyses. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Please note that this appendix has not been copy-edited.
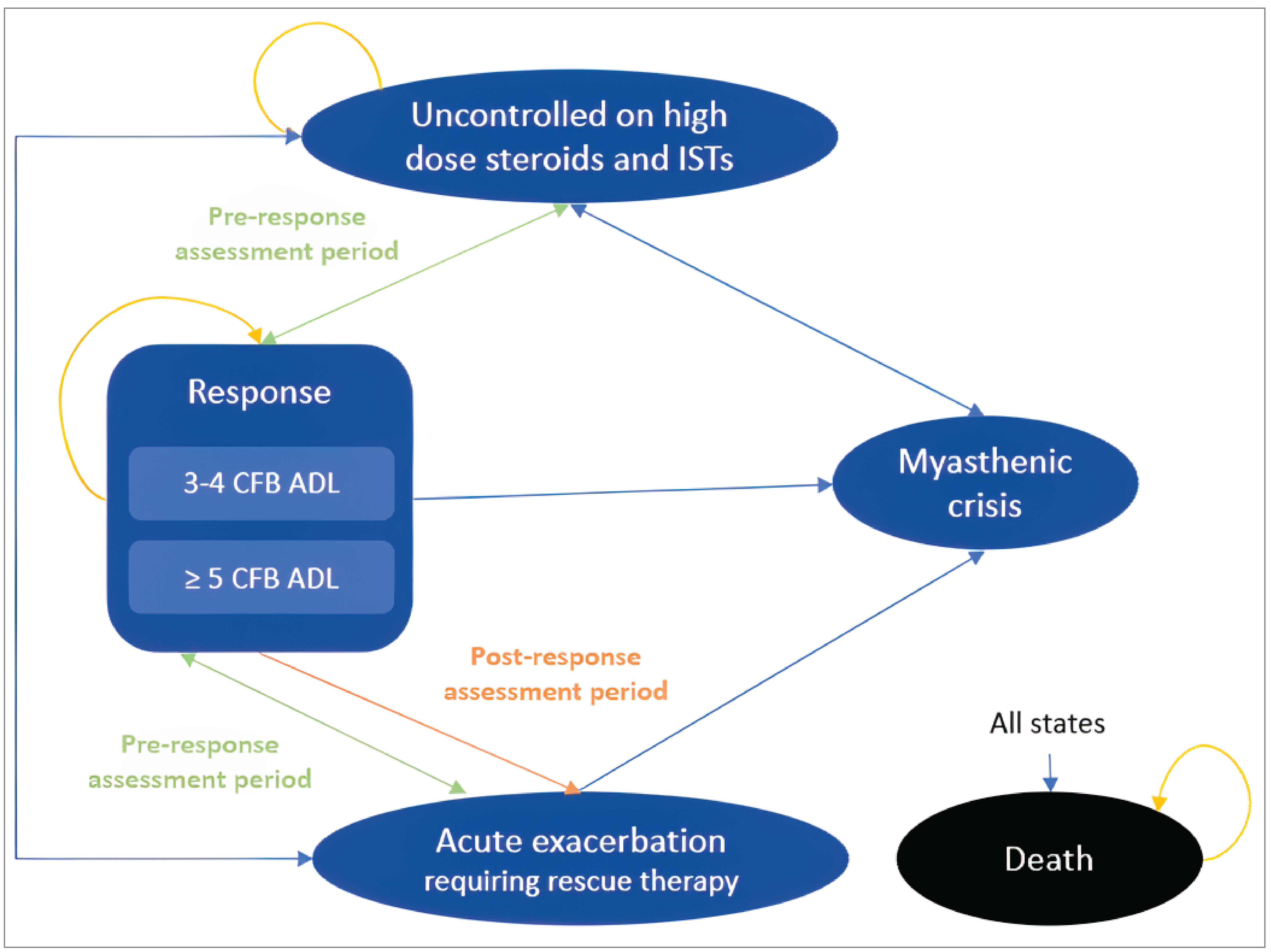
ADL = Activities of Daily Living; CFB = change from baseline; IST = immunosuppressive therapy.
Note: Activities of Daily Living in this figure refers to the Myasthenia Gravis Activities of Daily Living scale.
Source: Sponsor’s pharmacoeconomic submission.1
Table 9: Model Input Parameters by Health State
Health state | MG-ADL score | Utility valuea | Proportion of patients receiving CS | Annual cost of managing CS use | Annual rate of exacerbation | Annual rate of crisis |
|---|---|---|---|---|---|---|
Uncontrolled, any time point | 8.30 | 0.475 | 100% high-dose CS | $12,197 | 0.651 | 0.0117 |
Response (CFB ≥ 3 in MG-ADL), before assessment time point | 4.84 | 0.551 | 100% low-dose CS | $5,309 | 0.118 | 0.0117 |
CFB 3 to 4 in MG-ADL, after assessment time point | 4.84 | 0.551 | ||||
CFB ≥ 5 in MG-ADL after assessment time point | 0.72 | 0.642 | 0% receiving CS | $0 |
CFB = change from baseline; CS = corticosteroid; MG-ADL = Myasthenia Gravis Activities of Daily Living.
aUtility values do not include disutilities associated with corticosteroid use, where all patients in the uncontrolled state received a −0.1750 disutility, while those in the CFB 3 to 4 MG-ADL response state received a −0.0700 disutility, based on their assumed use of high-dose and low-dose corticosteroids, respectively. Additionally, all patients receiving IVIg or PLEX experience a −0.1200 disutility during cycles within which it is administered either as a chronic or rescue therapy.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation (Probabilistic)
Parameter | Rozanolixizumab + conventional therapy | Conventional therapy alone | Rituximab + conventional therapy | PLEX + conventional therapy | IVIg or SCIg + conventional therapy | Efgartigimod alfa + conventional therapy |
|---|---|---|---|---|---|---|
Discounted LYs | ||||||
Total | 25.09 | 25.08 | 25.08 | 25.08 | 25.08 | 25.09 |
Uncontrolled | 19.64 | 21.95 | 21.36 | 20.66 | 21.04 | 19.75 |
Response | 4.85 | 2.47 | 3.08 | 3.80 | 3.40 | 4.73 |
Exacerbation | 0.51 | 0.55 | 0.54 | 0.53 | 0.54 | 0.51 |
Myasthenic crisis | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 |
Discounted QALYs | ||||||
Total | 11.01 | 10.60 | 10.67 | 10.78 | 10.73 | 11.00 |
Uncontrolled | 8.19 | 9.23 | 8.97 | 8.65 | 8.82 | 8.25 |
Response | 2.88 | 1.43 | 1.77 | 2.20 | 1.98 | 2.82 |
Exacerbation | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 | 0.08 |
Myasthenic crisis | −0.14 | −0.15 | −0.15 | −0.15 | −0.15 | −0.14 |
Discounted costs ($) | ||||||
Total | 1,278,332 | 655,680 | 676,040 | 888,741 | 894,483 | 1,256,334 |
Resource use – uncontrolled | 427,252 | 477,637 | 464,752 | 629,360 | 640,901 | 429,855 |
Resource use – response | 36,267 | 20,491 | 25,536 | 36,692 | 30,109 | 35,338 |
Resource use – exacerbation | 92,099 | 100,548 | 98,372 | 110,425 | 60,045 | 92,538 |
Resource Use – myasthenic crisis | 44,954 | 48,602 | 47,660 | 55,824 | 46,749 | 45,145 |
Treatment costs | 670,722 | 1,362 | 32,680 | 49,401 | 109,639 | 646,418 |
End-of-life costs | 7,039 | 7,040 | 7,040 | 7,040 | 7,040 | 7,039 |
IVIg = IV immunoglobulin; LY = life-year; PLEX = plasma exchange; QALY = quality-adjusted life-year; SCIg = subcutaneous immunoglobulin.
Note: Results for eculizumab have been omitted for brevity (refer to Table 4).
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CDA-AMC Reanalyses and Sensitivity Analyses of the Economic Evaluation
Please note that this appendix has not been copy-edited.
Detailed Results of the CDA-AMC Base Case
CDA-AMC did not conduct a reanalysis of the sponsor’s base case.
Scenario Analyses
Table 11: Summary of CDA-AMC Scenario Analysis Results (Probabilistic)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case | |||
Conventional therapy | 655,680 | 10.60 | Reference |
Rituximab conventional therapy | 676,040 | 10.68 | 262,458 vs. conventional therapy |
Efgartigimod alfa plus conventional therapy | 1,256,334 | 11.00 | 1,776,088 vs rituximab + conventional therapy |
Rozanolixizumab plus conventional therapy | 1,278,332 | 11.01 | 2,676,135 vs efgartigimod + conventional therapy |
Dominated treatments | |||
Chronic PLEX plus conventional therapy | 888,741 | 10.78 | Extendedly dominated by mix of rituximab and efgartigimod |
Chronic IVIg or SCIg plus conventional therapy | 894,483 | 10.73 | Dominated by chronic PLEX |
Eculizumab plus conventional therapy | 2,101,292 | 10.84 | Dominated by rozanolixizumab |
CDA-AMC scenario 1: Removal of 2-year stopping rule | |||
Conventional therapy | 659,218 | 10.58 | Reference |
Rituximab conventional therapy | 729,208 | 10.66 | 892,530 vs. conventional therapy |
Chronic PLEX plus conventional therapy | 1,005,568 | 10.77 | 2,465,856 vs rituximab + conventional therapy |
Efgartigimod alfa plus conventional therapy | 2,671,050 | 10.97 | 8,450,274 vs chronic PLEX + conventional therapy |
Rozanolixizumab plus conventional therapy | 2,807,162 | 10.98 | 9,682,589 vs efgartigimod + conventional therapy |
Dominated treatments | |||
Chronic IVIg or SCIg plus conventional therapy | 1,157,692 | 10.71 | Dominated by chronic PLEX |
CDA-AMC scenario 2: Removal of extra costs and utility decrements associated with corticosteroid use | |||
Conventional therapy | 383,444 | 10.83 | Reference |
Rituximab plus conventional therapy | 409,341 | 10.91 | 361,690 vs. conventional therapy |
Efgartigimod alfa plus conventional therapy | 997,772 | 11.21 | 1,931,473 vs. rituximab + conventional therapy |
Rozanolixizumab plus conventional therapy | 1,028,426 | 11.21 | 15,991,013 vs. efgartigimod + conventional therapy |
Dominated treatments | |||
Chronic PLEX plus conventional therapy | 626,380 | 11.01 | Extendedly dominated by mix of rituximab and efgartigimod |
Chronic IVIg or SCIg plus conventional therapy | 630,507 | 10.96 | Dominated by chronic PLEX |
CDA-AMC scenario 3: Efgartigimod alfa and rozanolixizumab dosing based on patient weight distribution | |||
Conventional therapy | 656,624 | 10.58 | Reference |
Rituximab plus conventional therapy | 676,946 | 10.66 | 263,681 vs. conventional therapy |
Efgartigimod alfa plus conventional therapy | 1,189,389 | 11.00 | 1,498,475 vs. rituximab + conventional therapy |
Dominated treatments | |||
Chronic PLEX plus conventional therapy | 887,021 | 10.77 | Extendedly dominated by mix of rituximab and efgartigimod |
Chronic IVIg or SCIg plus conventional therapy | 895,518 | 10.71 | Dominated by chronic PLEX |
Rozanolixizumab plus conventional therapy | 1,315,090 | 10.98 | Dominated by efgartigimod |
CDA-AMC scenario 4: 5% mortality per myasthenic crisis | |||
Conventional therapy | 605,873 | 9.80 | Reference |
Rituximab plus conventional therapy | 627,237 | 9.90 | 216,776 vs. conventional therapy |
Chronic PLEX plus conventional therapy | 822,691 | 10.04 | 1,423,267 vs. rituximab + conventional therapy |
Efgartigimod alfa plus conventional therapy | 1,201,944 | 10.28 | 1,588,554 vs. chronic PLEX + conventional therapy |
Rozanolixizumab plus conventional therapy | 1,231,932 | 10.29 | 2,075,184 vs. efgartigimod + conventional therapy |
Dominated treatments | |||
Chronic IVIg or SCIg plus conventional therapy | 833,486 | 9.97 | Dominated by chronic PLEX |
CDA-AMC scenario 5: Combination of CDA-AMC scenarios 1 through 4 | |||
Conventional therapy | 356,955 | 10.05 | Reference |
Rituximab plus conventional therapy | 432,915 | 10.14 | 849,744 vs. conventional therapy |
Chronic PLEX plus conventional therapy | 697,559 | 10.26 | 2,056,996 vs. rituximab + conventional therapy |
Efgartigimod alfa plus conventional therapy | 2,174,300 | 10.51 | 5,987,648 vs. chronic PLEX + conventional therapy |
Dominated treatments | |||
Chronic IVIg or SCIg plus conventional therapy | 849,939 | 10.20 | Dominated by chronic PLEX |
Rozanolixizumab plus conventional therapy | 2,650,307 | 10.51 | Dominated by efgartigimod |
ICER = incremental cost-effectiveness ratio; IVIg = IV immunoglobulin; PLEX = plasma exchange; QALY = quality-adjusted life-year; SCIg = subcutaneous immunoglobulin; SOC = standard of care; vs. = versus.
Appendix 5: Submitted Budget Impact Analysis and CDA-AMC Appraisal
Please note that this appendix has not been copy-edited.
Table 12: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of the Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the expected incremental budget impact of reimbursing rozanolixizumab plus conventional therapy for the treatment of adults with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy with acetylcholinesterase inhibitors, corticosteroids, and/or nonsteroidal immunosuppressive therapies.37 The BIA was conducted from the perspective of drug plans across Canada over a 3-year time horizon (October 2025 through September 2028, with October 2024 through September 2025 as the base year). The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets, excluding Quebec, as well as the Non-Insured Health Benefits Program (NIHB). The sponsor’s base case included drug acquisition costs only. Market shares in the reference scenario were derived from sponsor-obtained clinical expert opinion and previous BIAs reviewed by CDA-AMC.33,58 Market uptake of rozanolixizumab was estimated based on internal sponsor forecasts. Rozanolixizumab was assumed to displace all comparators in proportion to their reference scenario market share and was not assumed to increase the number of patients eligible for treatment. An additional analysis representing the budget impact of reimbursing rozanolixizumab for the entire Health Canada–indicated population was also submitted but this analysis was broader than the final indication approved by Health Canada and will not be further covered within this report. Key inputs to the BIA are documented in Table 13.
Summary of the Sponsor’s BIA Results
Results for the sponsor’s analyses suggested that the reimbursement of rozanolixizumab for adults with AChR antibody-positive or MuSK antibody-positive gMG with persistent symptoms despite conventional therapy would be associated with an incremental cost of $21,001,257 in year 1, $36,598,504 in year 2, and $55,291,188 in year 3, for a 3-year incremental budgetary impact of $112,890,948.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 where appropriate) |
|---|---|
Target population | |
CDA-AMC participating adult population | 26,899,939 / 27,343,661 / 27,787,384a |
Prevalence of gMG | 32 per 100,00059 |
Proportion of patients with gMG who are AChR-Ab+ | 85%60 |
Proportion of patients with gMG who are MuSK-Ab+ | |
Proportion patients with gMG for whom symptoms persist despite CT | 25%63 |
Proportion patients who are eligible for public coverage | 65.6%64 |
Number of patients eligible for drug under review | 1,285 / 1,306 / 1,327 |
Market shares (3 years, reference scenario) | |
Rozanolixizumab plus CT | 0% / 0% / 0% |
CT | 27.5% / 26.5% / 26.5% |
Chronic IVIG/SCIG plus CT | 33.0% / 31.5% / 31.0% |
Chronic PLEX plus CT | 26.0 / 24.5% / 24.0% |
Rituximab plus CT | 3.5% / 3.5% / 3.5% |
Eculizumab plus CT | 1.0% / 1.0% / 1.0% |
Efgartifimod alfa plus CT | 9.0% / 13.0% / 14.0% |
Market shares (3 years, new drug scenario) | |
Rozanolixizumab plus CT | 4.5% / 8.0% / 12.0% |
CT | 26.3% / 24.4% / 23.3% |
Chronic IVIG/SCIG plus CT | 31.5% / 29.0% / 27.3% |
Chronic PLEX plus CT | 24.8% / 22.5% / 21.1% |
Rituximab plus CT | 3.3% / 3.2% / 3.1% |
Eculizumab plus CT | 1.0% / 0.9% / 0.9% |
Efgartifimod alfa plus CT | 8.6% / 12.0% / 12.3% |
Cost of treatment (per patient, per year) | |
Rozanolixizumab plus CT | $434,347 |
CT | $1,372 |
Chronic IVIG/SCIG plus CT | $85,272 |
Chronic PLEX plus CT | $8,668 |
Rituximab plus CT | $20,677 |
Eculizumab plus CT | $722,766 |
Efgartifimod alfa plus CT | $359,337 |
AChR-Ab+ = anti-acetylcholine receptor antibody-positive; CDA-AMC = Canada’s Drug Agency; CT = conventional therapy; gMG = generalized myasthenia gravis; IVIG = IV immunoglobulin; MuSK-Ab+ = muscle-specific kinase antibody-positive; N/A = not applicable; PLEX = plasma exchange; SCIG = subcutaneous immunoglobulin.
aSum of the adult populations (18+ years) of all provinces except for Quebec, plus the client population of NIHB who are 20+ years of age. The base year and years 1 through 3 population estimates for all provinces were linearly forecast from 2018 to 2023 Statistics Canada population estimates, while that of the NIHB was linearly forecast from 2018 through 2022 NIHB annual reports.
CDA-AMC Appraisal of the Sponsor’s BIA
CDA-AMC identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Reference scenario market shares are uncertain: According to clinical expert input obtained by CDA-AMC, as well as expert input obtained by the sponsor,63 the sponsor’s estimate that approximately 25% of patients receive chronic PLEX is inconsistent with clinical practice in Canada. The sponsor-obtained clinical expert input estimated that 1% to 2% of the overall AChR antibody-positive or MuSK antibody-positive gMG population were receiving PLEX, while 10% to 15% of the refractory AChR antibody-positive gMG population were doing so. Instead of 25% of the patient population receiving PLEX, clinical expert input obtained by CDA-AMC indicated that more patients would receive rituximab considering that the reimbursement request includes patients with MuSK+ gMG, as well as IVIg and conventional therapy alone.
Additionally, the sponsor assumed that 1% of the reimbursement request population would receive eculizumab. While the CDA-AMC reimbursement review for eculizumab for the treatment of adults with refractory gMG resulted in a recommendation to reimburse with conditions,42 negotiations with pCPA were not successful.43 As such, the public plans do not currently fund eculizumab for the treatment of gMG and thus eculizumab is not a comparator of interest when assessing the budget impact of publicly reimbursing rozanolixizumab.
In reanalysis, CDA-AMC halved the proportion of patients receiving PLEX plus conventional therapy in the reference scenario, increased the proportion of patients receiving rituximab plus conventional therapy to 5%, and divided the remainder between IVIg or SCIg plus conventional therapy and conventional therapy alone. Additionally, CDA-AMC assumed a 0% market share for eculizumab and proportionally redistributed its original share to the remaining comparators. These changes also affected the new drug scenario.
Drug plan payer perspective: According to the Procedures for Reimbursement Reviews,41 the BIA base case should be from the perspective of a pan-Canadian drug plan program. As such, costs relating to the use of blood products (i.e., IVIg or SCIg, PLEX) are not funded by jurisdictional drug plan budgets and should thus be excluded from the drug plan perspective. As the sponsor’s submission did not include the option to switch to a health care payer perspective where other cost categories such as administration were included, CDA-AMC was unable to present both a drug plan payer and a health care payer perspective. Of note, due to a lack of publicly accessible pricing for blood products, CDA-AMC was unable to independently validate the sponsor’s estimated costs for IVIg or SCIg and PLEX.
In reanalysis, costs associated with the use of blood products were excluded. If the inclusion of the estimated acquisition costs of blood products is considered useful, CDA-AMC scenario 1 reintroduces blood product costs. CDA-AMC could not undertake a robust analysis from the health care payer perspective due to the exclusion of relevant costs, such as administration costs, that are expected to differ by treatment.
Distribution of patients’ body weights: The sponsor’s BIA assumed a distribution of rozanolixizumab doses consistent with body weights reported in EU weight tables for patients with gMG,65 estimating that 2.8% of patients weighed less than 50 kg and would receive 1 280 mg vial, 96% weighed between 50 to < 100 kg and would receive 2 vials, and 1.2% weighed 100 kg of more and would receive 3 vials. As these weights are based on EU data, these may not be generalizable to the population of patients with gMG in Canada. The submitted BIA also assumed that for efgartigimod alfa, 60% of patients would weigh less than or equal to 80 kg and would therefore receive 2 400mg vials per dose, while the remaining 40% weighed more than 80 kg and would receive 3 vials, based on the distribution of patient weights reported in CDA-AMC review of efgartigimod alfa, derived from the ADAPT trial.33,36 According to the EU weight tables provided by the sponsor, less than 30% of patients would require 3 vials of efgartigimod alfa per dose. As such, the submitted BIA effectively assumes a different patient population depending on which therapy is chosen, which is inappropriate.
CDA-AMC reanalyses assumed a distribution of patient body weights consistent with those reported in the 7 mg/kg rozanolixizumab and placebo groups of the MycarinG trial when deriving the cost of rozanolixizumab (i.e., ≥ 35 to < 50 kg: 8.3%, ≥ 50 to < 70 kg: 26.3%, ≥ 70 to < 100 kg: 45.9%, ≥ 100 kg: 19.5%). As the mean body weight reported in MycarinG was higher than that reported in the ADAPT trial, the proportion of patients requiring 3 vials of efgartigimod alfa was increased to 50%. All other therapies were flat-dosed (rituximab, eculizumab, PLEX), or wastage was not assumed when using the mean body weight reported in the MycarinG trial (IVIg) to calculate costs or were equally applied to all treatment groups (conventional therapy), thus adjusting patient weight assumptions for these other comparators had no effect on BIA results.
The NIHB population was inappropriately calculated: The sponsor calculated the total population of CDA-AMC-participating drug plans by adding the population of the provinces,66 excluding Quebec, to the population of NIHB clients.67 NIHB clients living within the borders of a province are counted within provincial population data as reported by Statistics Canada; thus, the NIHB population was in effect double counted within the sponsor’s analysis. Additionally, NIHB clients residing within Ontario who are aged < 25 years or ≥ 65 years are eligible for reimbursement by Ontario Drug Benefit and thus should be counted as Ontario Drug Benefit Formulary clients (and included in the Ontario population estimates) rather than NIHB clients for the purposes of modelling the budget impact of reimbursing rozanolixizumab. Finally, the NIHB reports its client population by age in groups of 5 years.67 In determining the adult population of NIHB clients, the sponsor summed all age categories from 20 to 65+ years. As such, the sponsor excluded NIHB clients who are ages 18 or 19 from the adult population.
CDA-AMC did not adjust for this limitation in reanalysis. The impact on pan-Canadian model results is expected to be minimal.
Proportion of patients who are publicly funded was inappropriately estimated: The sponsor assumed that 65.6% of patients would be eligible for public funding of their gMG therapy, based on the proportion of the overall population of Canada who are eligible for public plan funding as reported by The Conference Board of Canada in 2022.68 This estimate does not account for differences in eligibility rates across jurisdictions.
CDA-AMC did not adjust the model to reflect public funding eligibility rates within each jurisdiction. The impact on pan-Canadian model results is expected to be minimal, however the impact on individual jurisdiction model results may be substantial, depending on how far the eligibility rates within that jurisdiction deviate from 65.6%.
CDA-AMC Reanalyses of the BIA
CDA-AMC reanalyses focused on the reimbursement requested population. CDA-AMC revised the sponsor’s submitted analysis by assuming eculizumab has 0% of the public market, by redistributing half the market share assigned to chronic PLEX to other comparators, by excluding the cost of blood products from the analysis,, and by altering the distribution of patient body weights used to estimate dosing for rozanolixizumab and efgartigimod alfa to be consistent with weights reported in the MycarinG trial. The changes applied to derive the CDA-AMC base case are described in Table 14.
Table 14: CDA-AMC Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CDA-AMC value or assumption |
|---|---|---|
Changes to derive the CDA-AMC base case | ||
1. Reference scenario public market shares |
|
|
2. Drug plan perspective | Blood product costs included. | Blood product costs excluded. |
3. Patient weight distribution | Rozanolixizumab: derived from patients with gMG in EU Efgartigimod alfa: derived from CDA-AMC review, which was based on ADAPT trial | Rozanolixizumab: Derived from MycarinG trial Efgartigimod alfa: Proportion requiring 3 vials increased to be 50%, based on weight differences between MycarinG and ADAPT trials |
CDA-AMC base-case | Reimbursement request population: 1 + 2 + 3 | |
CDA-AMC = Canada’s Drug Agency; IVIg = IV immunoglobulin; PLEX = plasma exchange; SCIg = subcutaneous immunoglobulin.
The results of the CDA-AMC step-wise reanalysis are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16. In adults with AChR antibody-positive or MuSK antibody-positive gMG whose symptoms persist despite conventional therapy, CDA-AMC reanalyses suggest that the reimbursement of rozanolixizumab in combination with conventional therapy will be associated with a 3-year incremental budgetary cost of $132,461,365 (year 1: $24,638,709, year 2: $42,950,784, year 3: $64,871,872).
Table 15: Summary of the CDA-AMC Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total ($) Reimbursement request population |
|---|---|
Submitted base case | 112,890,948 |
CDA-AMC reanalysis 1: reference scenario market shares | 113,698,368 |
CDA-AMC reanalysis 2: drug plan perspective | 121,969,381 |
CDA-AMC reanalysis 3: patient weight distribution | 121,314,453 |
CDA-AMC base case | 132,461,365 |
CDA-AMC = Canada’s Drug Agency.
CDA-AMC conducted the following scenario analysis to address remaining uncertainty, using the CDA-AMC base case (results are provided in Table 16):
Including the acquisition costs of blood products.
Table 16: Detailed Breakdown of the CDA-AMC Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $62,740,603 | $91,290,834 | $109,708,276 | $115,633,808 | $379,373,520 |
New drug | $62,740,603 | $112,292,091 | $146,306,779 | $170,924,995 | $492,264,469 | |
Budget impact | $0 | $21,001,257 | $36,598,504 | $55,291,188 | $112,890,948 | |
CDA-AMC base case | Reference | $17,242,266 | $46,563,098 | $67,004,858 | $73,090,424 | $203,900,645 |
New drug | $17,242,266 | $71,201,807 | $109,955,642 | $137,962,295 | $336,362,010 | |
Budget impact | $0 | $24,638,709 | $42,950,784 | $64,871,872 | $132,461,365 | |
CDA-AMC scenario analysis 1: blood product costs included | Reference | $60,258,251 | $89,981,033 | $108,991,821 | $115,031,399 | $374,262,504 |
New drug | $60,258,251 | $112,665,935 | $148,583,648 | $174,870,354 | $496,378,188 | |
Budget impact | $0 | $22,684,902 | $39,591,827 | $59,838,955 | $122,115,684 |
BIA = budget impact analysis; CDA-AMC = Canada’s Drug Agency.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.